User login
Should healthy people take a multivitamin?
No. There is no scientific basis for recommending vitamin-mineral supplements to the healthy population.
(This commentary deals only with healthy people in the general US population. There are well-established guidelines for the use of supplements in pregnant and lactating women, infants, and individuals with a wide variety of health conditions.)
TAKE A PILL, OR EAT A HEALTHIER DIET?
The Dietary Guidelines for Americans1 reported that the US population consumes insufficient amounts of green leafy vegetables, fresh fruits, whole grains, and fiber and excessive amounts of refined carbohydrates, saturated fat, and sodium. This may result in inadequate intake of some nutrients. (The term “inadequate” intake is being used to differentiate this situation from “deficiency,” which is rare in the general population.)
So the real question is, Should we pop a vitamin pill every day and forget about it, or try to eat a healthier diet?
To answer that question, consider this: no supplement trial has ever been able to reproduce the health benefits of eating adequate amounts of fresh fruits and vegetables.
One reason is that natural foods contain far more compounds than the few we know about and can put in a supplement pill. For example, vegetables contain hundreds of antioxidant compounds, many perhaps acting synergistically, while so far we have been able to identify and isolate only a handful.
Second, nutrients have different health effects depending on the host’s conditions. A calcium supplement will not increase bone mineral density unless accompanied by regular, weight-bearing exercise that stimulates bone accretion.
This is why most supplement trials have shown disappointing results. A recent National Institutes of Health state-of-the-science conference on multivitamin-mineral supplements2 concluded that there is no consistent evidence that single-vitamin or multivitamin supplements help in preventing a wide range of diseases studied.
‘AT LEAST IT WON’T HURT’ MAY NOT BE TRUE
In spite of the lack of evidence, many will go on taking supplements, with the argument that “at least it won’t hurt.” They should be reminded that several supplement trials had to be stopped prematurely due to unexpected adverse effects.
In the Selenium and Vitamin E Cancer Prevention Trial (SELECT),3 which evaluated supplementation to prevent prostate cancer, the group receiving vitamin E had more cases of prostate cancer than controls, and the group taking selenium had more diabetes cases. While these differences were not statistically significant, they were of enough concern to stop the trial.
A meta-analysis of vitamin E trials4 showed a slight increase in the rate of all-cause mortality in those receiving the active supplement.
The bottom line: the evidence that supplements “won’t hurt” is even more limited than the evidence for their efficacy, because trials are usually not designed to address safety outcomes.
TELL YOUR PATIENTS THE THINGS THEY DO NOT WANT TO HEAR
Unfortunately, this means you have to tell your patients all the things they do not want to hear: cut the ice cream, eat more broccoli, exercise regularly. But because of their position of authority and credibility, physicians can play a crucial role in helping the US population improve its dietary and lifestyle habits.
The key is to introduce and support minor but sustained changes in the diet and physical activity. For example, we have shown that simply reducing consumption of caloric beverages (soft drinks) can result in significant weight loss in overweight adults, without any other dietary intervention.5
The other key is of course to modify the obesogenic environment we live in. Only by creating conditions that facilitate healthy eating and regular activity will we have a significant impact on public health.
- United States Department of Agriculture Center for Nutrition Policy and Promotion. Report of the Dietary Guidelines Advisory Committee on the Dietary Guidelines for Americans, 2010. www.cnpp.usda.gov/DGAs2010-DGACReport.htm. Accessed 8/25/2010.
- Huang HY, Caballero B, Chang S, et al. The efficacy and safety of multivitamin and mineral supplement use to prevent cancer and chronic disease in adults: a systematic review for a National Institutes of Health state-of-the-science conference. Ann Intern Med 2006; 145:372–385.
- Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009; 301:39–51.
- Miller ER, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med 2005; 142:37–46.
- Chen L, Appel LJ, Loria C, et al. Reduction in consumption of sugar-sweetened beverages is associated with weight loss: the PREMIER trial. Am J Clin Nutr 2009; 89:1299–1306.
No. There is no scientific basis for recommending vitamin-mineral supplements to the healthy population.
(This commentary deals only with healthy people in the general US population. There are well-established guidelines for the use of supplements in pregnant and lactating women, infants, and individuals with a wide variety of health conditions.)
TAKE A PILL, OR EAT A HEALTHIER DIET?
The Dietary Guidelines for Americans1 reported that the US population consumes insufficient amounts of green leafy vegetables, fresh fruits, whole grains, and fiber and excessive amounts of refined carbohydrates, saturated fat, and sodium. This may result in inadequate intake of some nutrients. (The term “inadequate” intake is being used to differentiate this situation from “deficiency,” which is rare in the general population.)
So the real question is, Should we pop a vitamin pill every day and forget about it, or try to eat a healthier diet?
To answer that question, consider this: no supplement trial has ever been able to reproduce the health benefits of eating adequate amounts of fresh fruits and vegetables.
One reason is that natural foods contain far more compounds than the few we know about and can put in a supplement pill. For example, vegetables contain hundreds of antioxidant compounds, many perhaps acting synergistically, while so far we have been able to identify and isolate only a handful.
Second, nutrients have different health effects depending on the host’s conditions. A calcium supplement will not increase bone mineral density unless accompanied by regular, weight-bearing exercise that stimulates bone accretion.
This is why most supplement trials have shown disappointing results. A recent National Institutes of Health state-of-the-science conference on multivitamin-mineral supplements2 concluded that there is no consistent evidence that single-vitamin or multivitamin supplements help in preventing a wide range of diseases studied.
‘AT LEAST IT WON’T HURT’ MAY NOT BE TRUE
In spite of the lack of evidence, many will go on taking supplements, with the argument that “at least it won’t hurt.” They should be reminded that several supplement trials had to be stopped prematurely due to unexpected adverse effects.
In the Selenium and Vitamin E Cancer Prevention Trial (SELECT),3 which evaluated supplementation to prevent prostate cancer, the group receiving vitamin E had more cases of prostate cancer than controls, and the group taking selenium had more diabetes cases. While these differences were not statistically significant, they were of enough concern to stop the trial.
A meta-analysis of vitamin E trials4 showed a slight increase in the rate of all-cause mortality in those receiving the active supplement.
The bottom line: the evidence that supplements “won’t hurt” is even more limited than the evidence for their efficacy, because trials are usually not designed to address safety outcomes.
TELL YOUR PATIENTS THE THINGS THEY DO NOT WANT TO HEAR
Unfortunately, this means you have to tell your patients all the things they do not want to hear: cut the ice cream, eat more broccoli, exercise regularly. But because of their position of authority and credibility, physicians can play a crucial role in helping the US population improve its dietary and lifestyle habits.
The key is to introduce and support minor but sustained changes in the diet and physical activity. For example, we have shown that simply reducing consumption of caloric beverages (soft drinks) can result in significant weight loss in overweight adults, without any other dietary intervention.5
The other key is of course to modify the obesogenic environment we live in. Only by creating conditions that facilitate healthy eating and regular activity will we have a significant impact on public health.
No. There is no scientific basis for recommending vitamin-mineral supplements to the healthy population.
(This commentary deals only with healthy people in the general US population. There are well-established guidelines for the use of supplements in pregnant and lactating women, infants, and individuals with a wide variety of health conditions.)
TAKE A PILL, OR EAT A HEALTHIER DIET?
The Dietary Guidelines for Americans1 reported that the US population consumes insufficient amounts of green leafy vegetables, fresh fruits, whole grains, and fiber and excessive amounts of refined carbohydrates, saturated fat, and sodium. This may result in inadequate intake of some nutrients. (The term “inadequate” intake is being used to differentiate this situation from “deficiency,” which is rare in the general population.)
So the real question is, Should we pop a vitamin pill every day and forget about it, or try to eat a healthier diet?
To answer that question, consider this: no supplement trial has ever been able to reproduce the health benefits of eating adequate amounts of fresh fruits and vegetables.
One reason is that natural foods contain far more compounds than the few we know about and can put in a supplement pill. For example, vegetables contain hundreds of antioxidant compounds, many perhaps acting synergistically, while so far we have been able to identify and isolate only a handful.
Second, nutrients have different health effects depending on the host’s conditions. A calcium supplement will not increase bone mineral density unless accompanied by regular, weight-bearing exercise that stimulates bone accretion.
This is why most supplement trials have shown disappointing results. A recent National Institutes of Health state-of-the-science conference on multivitamin-mineral supplements2 concluded that there is no consistent evidence that single-vitamin or multivitamin supplements help in preventing a wide range of diseases studied.
‘AT LEAST IT WON’T HURT’ MAY NOT BE TRUE
In spite of the lack of evidence, many will go on taking supplements, with the argument that “at least it won’t hurt.” They should be reminded that several supplement trials had to be stopped prematurely due to unexpected adverse effects.
In the Selenium and Vitamin E Cancer Prevention Trial (SELECT),3 which evaluated supplementation to prevent prostate cancer, the group receiving vitamin E had more cases of prostate cancer than controls, and the group taking selenium had more diabetes cases. While these differences were not statistically significant, they were of enough concern to stop the trial.
A meta-analysis of vitamin E trials4 showed a slight increase in the rate of all-cause mortality in those receiving the active supplement.
The bottom line: the evidence that supplements “won’t hurt” is even more limited than the evidence for their efficacy, because trials are usually not designed to address safety outcomes.
TELL YOUR PATIENTS THE THINGS THEY DO NOT WANT TO HEAR
Unfortunately, this means you have to tell your patients all the things they do not want to hear: cut the ice cream, eat more broccoli, exercise regularly. But because of their position of authority and credibility, physicians can play a crucial role in helping the US population improve its dietary and lifestyle habits.
The key is to introduce and support minor but sustained changes in the diet and physical activity. For example, we have shown that simply reducing consumption of caloric beverages (soft drinks) can result in significant weight loss in overweight adults, without any other dietary intervention.5
The other key is of course to modify the obesogenic environment we live in. Only by creating conditions that facilitate healthy eating and regular activity will we have a significant impact on public health.
- United States Department of Agriculture Center for Nutrition Policy and Promotion. Report of the Dietary Guidelines Advisory Committee on the Dietary Guidelines for Americans, 2010. www.cnpp.usda.gov/DGAs2010-DGACReport.htm. Accessed 8/25/2010.
- Huang HY, Caballero B, Chang S, et al. The efficacy and safety of multivitamin and mineral supplement use to prevent cancer and chronic disease in adults: a systematic review for a National Institutes of Health state-of-the-science conference. Ann Intern Med 2006; 145:372–385.
- Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009; 301:39–51.
- Miller ER, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med 2005; 142:37–46.
- Chen L, Appel LJ, Loria C, et al. Reduction in consumption of sugar-sweetened beverages is associated with weight loss: the PREMIER trial. Am J Clin Nutr 2009; 89:1299–1306.
- United States Department of Agriculture Center for Nutrition Policy and Promotion. Report of the Dietary Guidelines Advisory Committee on the Dietary Guidelines for Americans, 2010. www.cnpp.usda.gov/DGAs2010-DGACReport.htm. Accessed 8/25/2010.
- Huang HY, Caballero B, Chang S, et al. The efficacy and safety of multivitamin and mineral supplement use to prevent cancer and chronic disease in adults: a systematic review for a National Institutes of Health state-of-the-science conference. Ann Intern Med 2006; 145:372–385.
- Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009; 301:39–51.
- Miller ER, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med 2005; 142:37–46.
- Chen L, Appel LJ, Loria C, et al. Reduction in consumption of sugar-sweetened beverages is associated with weight loss: the PREMIER trial. Am J Clin Nutr 2009; 89:1299–1306.
Finding the right target for treating Alzheimer disease

The process may be relatively easy for the few diseases in which an etiologic agent fulfills Koch’s postulates. Otherwise, potential targets for therapeutic intervention are selected via several approaches.
Observational studies may suggest risk factors, and a number of molecular techniques may be used to identify specific cell types, up-regulated genes, or overexpressed potential critical mediators within diseased tissues. The latter approach was used, in part, in the successful development of therapies for rheumatoid arthritis that block tumor necrosis factor (TNF) and interleukin 6 (IL-6).
Once a potential target is found, molecular biologists using current tools of drug development—gene chip analysis, molecular modeling, proteomic scanning, and hybridoma-based synthesis—can produce a small molecule or biologic product to block the effect or expression of nearly any molecule or pathway (although a successful therapy cannot always be developed easily).
But sometimes, potential markers of disease pathogenesis are actually embers of the pathologic process rather than flames driving the disease.
For example, consider tuberculosis. Granulomas are typical of Mycobacterium tuberculosis infection. Suppose we didn’t know about mycobacteria but assumed instead it was the granuloma per se that was the actual agent of disease. It might then be reasonable to try to treat tuberculosis with the anti-TNF agents, since they can prevent the development of mature granulomas. But that approach would lead to uncontrolled M tuberculosis infection, not resolution of clinical tuberculosis.
Although not entirely analogous to an infectious disease, Alzheimer disease is another case in point. On page 689 in this issue of the Journal, Dr. David S. Geldmacher discusses how the amyloid plaques of Alzheimer disease may be the footprint but not the actual cause of the disorder. This hypothesis would explain the failure of attempts at treating Alzheimer disease by attacking this presumed pathogenic target and, if true, would require starting anew to find the right target.
The process may be relatively easy for the few diseases in which an etiologic agent fulfills Koch’s postulates. Otherwise, potential targets for therapeutic intervention are selected via several approaches.
Observational studies may suggest risk factors, and a number of molecular techniques may be used to identify specific cell types, up-regulated genes, or overexpressed potential critical mediators within diseased tissues. The latter approach was used, in part, in the successful development of therapies for rheumatoid arthritis that block tumor necrosis factor (TNF) and interleukin 6 (IL-6).
Once a potential target is found, molecular biologists using current tools of drug development—gene chip analysis, molecular modeling, proteomic scanning, and hybridoma-based synthesis—can produce a small molecule or biologic product to block the effect or expression of nearly any molecule or pathway (although a successful therapy cannot always be developed easily).
But sometimes, potential markers of disease pathogenesis are actually embers of the pathologic process rather than flames driving the disease.
For example, consider tuberculosis. Granulomas are typical of Mycobacterium tuberculosis infection. Suppose we didn’t know about mycobacteria but assumed instead it was the granuloma per se that was the actual agent of disease. It might then be reasonable to try to treat tuberculosis with the anti-TNF agents, since they can prevent the development of mature granulomas. But that approach would lead to uncontrolled M tuberculosis infection, not resolution of clinical tuberculosis.
Although not entirely analogous to an infectious disease, Alzheimer disease is another case in point. On page 689 in this issue of the Journal, Dr. David S. Geldmacher discusses how the amyloid plaques of Alzheimer disease may be the footprint but not the actual cause of the disorder. This hypothesis would explain the failure of attempts at treating Alzheimer disease by attacking this presumed pathogenic target and, if true, would require starting anew to find the right target.
The process may be relatively easy for the few diseases in which an etiologic agent fulfills Koch’s postulates. Otherwise, potential targets for therapeutic intervention are selected via several approaches.
Observational studies may suggest risk factors, and a number of molecular techniques may be used to identify specific cell types, up-regulated genes, or overexpressed potential critical mediators within diseased tissues. The latter approach was used, in part, in the successful development of therapies for rheumatoid arthritis that block tumor necrosis factor (TNF) and interleukin 6 (IL-6).
Once a potential target is found, molecular biologists using current tools of drug development—gene chip analysis, molecular modeling, proteomic scanning, and hybridoma-based synthesis—can produce a small molecule or biologic product to block the effect or expression of nearly any molecule or pathway (although a successful therapy cannot always be developed easily).
But sometimes, potential markers of disease pathogenesis are actually embers of the pathologic process rather than flames driving the disease.
For example, consider tuberculosis. Granulomas are typical of Mycobacterium tuberculosis infection. Suppose we didn’t know about mycobacteria but assumed instead it was the granuloma per se that was the actual agent of disease. It might then be reasonable to try to treat tuberculosis with the anti-TNF agents, since they can prevent the development of mature granulomas. But that approach would lead to uncontrolled M tuberculosis infection, not resolution of clinical tuberculosis.
Although not entirely analogous to an infectious disease, Alzheimer disease is another case in point. On page 689 in this issue of the Journal, Dr. David S. Geldmacher discusses how the amyloid plaques of Alzheimer disease may be the footprint but not the actual cause of the disorder. This hypothesis would explain the failure of attempts at treating Alzheimer disease by attacking this presumed pathogenic target and, if true, would require starting anew to find the right target.
Alzheimer disease prevention: Focus on cardiovascular risk, not amyloid?
Efforts to modify the relentless course of Alzheimer disease have until now been based on altering the production or clearance of beta-amyloid, the protein found in plaques in the brains of patients with the disease. Results have been disappointing, possibly because our models of the disease—mostly based on the rare, inherited form—may not be applicable to the much more common sporadic form.
Ely Lilly’s recent announcement that it is halting research into semagacestat, a drug designed to reduce amyloid production, only cast further doubt on viability of the amyloid hypothesis as a framework for effective treatments for Alzheimer disease.
Because of the close association of sporadic Alzheimer disease with vascular disease and type 2 diabetes mellitus, increased efforts to treat and prevent these conditions may be the best approach to reducing the incidence of Alzheimer disease.
This article will discuss current thinking of the pathophysiology of Alzheimer disease, with special attention to potential prevention and treatment strategies.
THE CANONICAL VIEW: AMYLOID IS THE CAUSE
The canonical view is that the toxic effects of beta-amyloid are the cause of neuronal dysfunction and loss in Alzheimer disease.
Beta-amyloid is a small peptide, 38 to 42 amino acids long, that accumulates in the extracellular plaque that characterizes Alzheimer pathology. Small amounts of extracellular beta-amyloid can be detected in the brains of elderly people who die of other causes, but the brains of people who die with severe Alzheimer disease show extensive accumulation of plaques.
The amyloid precursor protein is cleaved by normal constitutive enzymes, leaving beta-amyloid as a fragment. The beta-amyloid forms into fibrillar aggregations, which further clump into the extracellular plaque. Plaques can occur in the normal aging process in relatively low amounts. However, in Alzheimer disease, through some unknown trigger, the immune system appears to become activated in reference to the plaque. Microglial cells—the brain’s macrophages—invade the plaque and trigger a cycle of inflammation. The inflammation and its by-products cause local neuronal damage, which seems to propagate the inflammatory cycle to an even greater extent through a feed-forward loop. The damage leads to metabolic stress in the neuron and collapse of the cytoskeleton into a neurofibrillary tangle. Once the neurofibrillary tangle is forming, the neuron is probably on the path to certain death.
This pathway might be interrupted at several points, and in fact, much of the drug development world is working on possible ways to do so.
GENETIC VS SPORADIC DISEASE: WHAT ARE THE KEY DIFFERENCES?

Although the autosomal dominant form of the disease accounts for probably only 1% or 2% of all cases of Alzheimer disease, most animal models and hence much of the basic research and drug testing in Alzheimer disease are based on those dominant mutations. The pathology—the plaques and tangles—in Alzheimer disease in older adults is identical to that in younger adults, but the origins of the disease may not be the same. Therefore, the experimental model for one may not be relevant to the other.
In the last several years, some have questioned whether the amyloid hypothesis applies to all Alzheimer disease.1,2 Arguments go back to at least 2002, when Bishop and Robinson in an article entitled “The amyloid hypothesis: Let sleeping dogmas lie?”3 criticized the hypothesis and suggested that the beta-amyloid peptide appeared to be neuroprotective, not neurotoxic, in most situations. They suggested we await the outcome of antiamyloid therapeutic trials to determine whether the amyloid hypothesis truly explains the disorder.
The antiamyloid trials have now been under way for some time, and we have no definitive answer. Data from the phase II study of the monoclonal antibody agent bapineuzumab suggests there might be some small clinical impact of removing amyloid from the brain through immunotherapy mechanisms, but the benefits thus far are not robust.
COULD AMYLOID BE NEUROPROTECTIVE?
A pivotal question might be, “What if sick neurons made amyloid, instead of amyloid making neurons sick?” A corollary question is, “What if the effect were bidirectional?”
It is possible that in certain concentrations amyloid is neurotoxic, but in other concentrations, it actually facilitates neuronal repair, healing, and connection.
REDUCING METABOLIC STRESS: THE KEY TO PREVENTION?
If our current models of drug therapy are not effective against sporadic Alzheimer disease, perhaps focusing on prevention would be more fruitful.
Consider diabetes mellitus as an analogy. Its manifestations include polydipsia, polyuria, fatigue, and elevated glucose and hemoglobin A1c. Its complications are cardiovascular disease, nephropathy, and retinopathy. Yet diabetes mellitus encompasses two different diseases—type 1 and type 2—with different underlying pathophysiology. We do not treat them the same way. We may be moving toward a similar view of Alzheimer disease.
Links have been hypothesized between vascular risks and dementia. Diabetes, hypertension, dyslipidemia, and obesity might lead to dementia in a process abetted by oxidative stress, endothelial dysfunction, insulin resistance, inflammation, adiposity, and subcortical vascular disease. All of these could be targets of intervention to prevent and treat dementia.4
Instead of a beta-amyloid trigger, let us hypothesize that metabolic stress is the initiating element of the Alzheimer cascade, which then triggers beta-amyloid overproduction or underclearance, and the immune activation damages neurons. By lessening metabolic stress or by preventing immune activation, it may, in theory, be possible to prevent neurons from entering into the terminal pathway of tangle formation and cell death.
LINKS BETWEEN ALZHEIMER DISEASE AND DIABETES
Rates of dementia of all causes are higher in people with diabetes. The strongest effect has been noted in vascular dementia, but Alzheimer disease was also found to be associated with diabetes.5 The Framingham Heart Study6 found the association between dementia and diabetes was significant only when other risk factors for Alzheimer disease were minimal: in an otherwise healthy population, diabetes alone appears to trigger the risk for dementia. But in a population with a lot of vascular comorbidities, the association between diabetes and dementia is not as clear. Perhaps the magnitude of the risk is overwhelmed by greater cerebrovascular and cardiovascular morbidity.
A systematic review7 supported the notion that the risk of dementia is higher in people with diabetes, and even raised the issue of whether we should consider Alzheimer disease “type 3 diabetes.”
Testing of the reverse hypothesis—diabetes is more common in people with Alzheimer disease—also is supportive: diabetes mellitus and even impaired fasting glucose are approximately twice as common in people with Alzheimer disease than in those without.8 Fasting blood glucose levels increase steadily with age, but after age 65, they are higher in people with Alzheimer disease than in those without.
Glucose has some direct effects on brain metabolism that might explain the higher risk. Chronic hyperglycemia is associated with excessive production of free radicals, which leads to reactive oxygen species. These are toxic to neuronal membranes as well as to mitochondria, where many of the reactive oxygen species are generated. Free radicals also facilitate the inflammatory response.
We also see greater neuronal and mitochondrial calcium influx in the presence of hyperglycemia. The excess calcium interferes with mitochondrial metabolism and may trigger the cascade of apoptosis when it reaches critical levels in neurons.
Chronic hyperglycemia is also associated with increased advanced glycation end-products. These are toxic molecules produced by the persistent exposure of proteins to high sugar levels and may be facilitated by the presence of reactive oxygen species that catalyze the reactions between the sugars and the peptides. Glycation end-products are commonly recognized as the same as those occurring during browning of meat (the Maillard reaction).
Hyperglycemia also potentiates neuronal damage from ischemia. Animal experiments show that brain infarction in the presence of hyperglycemia results in worse damage than the same degree of ischemia in the absence of hyperglycemia. Hyperglycemia may exaggerate other blows to neuronal function such as those from small strokes or microvascular ischemia.
AN ALTERNATIVE TO THE AMYLOID HYPOTHESIS: THE ‘MITOCHONDRIAL CASCADE HYPOTHESIS’
Swerdlow and Khan9 have proposed an alternative to the amyloid hypothesis as the cause of Alzheimer disease, known as the “mitochondrial cascade hypothesis.” According to this model, as we age we accumulate more wear-and-tear from oxidative mitochondrial damage, especially the accumulation of toxins leading to reduced cell metabolic activity. This triggers the “3-R response”:
Reset. When toxins alter cell metabolism, neurons try to repair themselves by manufacturing beta-amyloid, which is a “repair-and-reset” synaptic signaling molecule that reduces energy production. Under the lower energy state, beta-pleated sheets develop from beta-amyloid, which aggregate and form amyloid plaque.
Remove. Many cells undergo programmed death when faced with oxidative stress. The first step in neuronal loss is reduced synaptic connections and, hence, losses in neuronal communication. This results in impaired cognition.
Replace. Some cells that are faced with metabolic stress re-enter the cell cycle by undergoing cell division. Neurons, however, are terminally postmitotic and die if they try to divide: by synthesizing cell division proteins, duplicating chromosomes, and reorganizing the complex internal structure, the cell cannot work properly and cell division fails. In the mitochondrial cascade hypothesis, neurofibrillary tangles result from this attempted remodeling of the cytoskeletal filaments, furthering neuronal dysfunction.
ALZHEIMER DISEASE AND STROKE: MORE ALIKE THAN WE THOUGHT?
Although historically clinicians and researchers have tried to distinguish between Alzheimer disease and vascular dementia, growing evidence indicates that the two disorders overlap significantly and that the pathologies may be synergistic.
Alzheimer disease has been hypothesized as being a vascular disorder.10 It shares many of the risk factors of vascular disease, and preclinical detection of Alzheimer disease is possible from measurements of regional cerebral perfusion. Cerebrovascular and neurodegenerative pathology are parallel in Alzheimer disease and vascular disease.
Pure Alzheimer disease and vascular disease are two ends of a pathologic continuum.11 At one end is “pure” Alzheimer disease, in which patients die only with histologic findings of plaques and neurofibrillary tangles. This form may occur only in patients with the autosomal dominant early-onset form. At the other end of the spectrum are people who have serious vascular disease, multiple strokes, and microvascular ischemia and who die demented but with no evidence of the plaques and tangles of Alzheimer disease.
Between these poles is a spectrum of overlapping pathology that is either Alzheimer disease-dominant or vascular disease-dominant, with varying degrees of amyloid plaque and evidence of microvascular infarcts. Cerebral amyloid angiopathy (the accumulation of beta-amyloid in the wall of arteries in the brain) bridges the syndromes.12 In some drug studies that attempted removing amyloid from the brain, vascular permeability was altered, resulting in brain edema.
Along the same lines as Kalaria’s model,11 Snowden et al13 found at autopsy of aged Catholic nuns that for some the accumulation of Alzheimer pathology alone was insufficient to cause dementia, but dementia was nearly universal in nuns with the same burden of Alzheimer pathology commingled with vascular pathology.
DOES INFLAMMATION PLAY A ROLE?
The inflammatory state is a recognized risk factor for Alzheimer disease, but the clinical data are mixed. Epidemiologic evidence is strong: patients who regularly take nonsteroidal anti-inflammatory drugs (NSAIDs) or steroids for chronic, systemic inflammatory diseases (eg, arthritis) have a 45% to 60% reduced risk for Alzheimer disease.14,15
However, multiple clinical trials in patients with Alzheimer disease have failed to show a benefit of taking anti-inflammatory drugs. One preliminary report suggested that indomethacin (Indocin) might offer benefit, but because of gastrointestinal side effects its usefulness in an elderly population is limited.
Diabetes and inflammation are also closely linked: hyperinsulinemia is proinflammatory, promoting the formation of reactive oxygen species, inhibiting the degradation of oxidized proteins, and increasing the risk for lipid per-oxidation. Insulin acts synergistically with endotoxins to raise inflammatory markers, eg, proinflammatory cytokines and C-reactive protein.16
It is possible that anti-inflammatory drugs may not work in Alzheimer disease because inflammation in the brain is mediated more by microglial cells than by prostaglandin pathways. In Alzheimer disease, inflammation is mediated by activated microglial cells, which invade plaques with their processes; these are not evident in the diffuse beta-amyloid-rich plaques seen in typical aging. The trigger for their activation is unclear, but the activated microglial cells and the invasion of plaques are seen in transgenic mouse models of Alzheimer disease, and activation is seen when beta-amyloid is injected into the brain of a healthy mouse.17
Activated microglial cells enlarge and their metabolic rate increases, with a surge in the production of proteins and other protein-mediated inflammatory markers such as alpha-antichymotrypsin, alpha-antitrypsin, serum amyloid P, C-reactive protein, nitric oxide, and proinflammatory cytokines. It is unlikely that it is healthy for cells to be exposed to these inflammatory products. Some of the cytokines are now targets of drug development for Alzheimer disease, and agents targeting these pathways have already been developed for connective tissue diseases.
In a controversial pilot study, Tobinick et al18 studied the use of etanercept (Enbrel), an inhibitor of tumor necrosis factor-alpha (an inflammatory cytokine). They injected etanercept weekly into the spinal canal in 15 patients with mild to severe Alzheimer disease, for 6 months. Patients improved in the Mini-Mental State Examination by more than two points during the study. Patent issues surrounding use of this drug in Alzheimer disease may delay further trials.
Thiazolidinediones block microglial cell activation
The reactive microglial phenotype can be prevented in cell culture by peroxisome proliferator-activated receptor (PPAR) gamma agonists. These include the antidiabetic thiazolidinediones such as pioglitazone (Actos), troglitazone (Rezulin), and rosiglitazone (Avandia), and indomethacin and other NSAIDs.
Using a Veterans Administration database of more than 142,000 patients, Miller et al19 retrospectively found that patients who took a thiazolidinedione for diabetes had a 20% lower risk of developing Alzheimer disease compared with users of insulin or metformin (Glucophage).
However, rosiglitazone showed no benefit against Alzheimer disease in a large clinical trial,20 but this may be because it is rapidly cleared from the brain. Pioglitazone is not actively exported from the brain, so it may be a better candidate, but pharmaceutical industry interest in this agent is low because its patent will soon expire.
Fish oil is another PPAR-gamma agonist, and some studies indicate that eating fish may protect against developing Alzheimer disease; it may also be therapeutic if the disease is present. Double-blind controlled studies have not been carried out and likely will not because of patent issues: the costs of such studies are high, and the potential payback is low.
ESTROGEN: PROTECTIVE OR NOT?
Whether taking estrogen is a risk factor or is protective has not yet been determined. Estrogen directly affects neurons. It increases the number of dendritic spines, which are associated with improved memory. Meta-analyses suggest that hormone replacement therapy reduces the risk of dementia by about one-third. 21,22 Both positive and negative prospective studies exist, but all are complicated by serious methodologic flaws.23,24
Combined analysis of about 7,500 women from two double-blind, randomized, placebo-controlled trials of the Women’s Health Initiative Memory Study found that the risks of dementia and mild cognitive impairment were increased by hormone replacement therapy. The hazard ratio for dementia was found to be 1.76 (P < .005), amounting to 23 new cases of dementia per 10,000 prescriptions annually.25
Patient selection may account for the conflicting results in different studies. Epidemiologic studies consisted mostly of newly postmenopausal women and those who were being treated for symptoms of vasomotor instability. In contrast, the Women’s Health Initiative enrolled only women older than 65 and excluded women with vasomotor instability. Other studies indicate that the greatest cognitive improvements with hormone therapies are seen in women with vasomotor symptoms.
WHICH RISK FACTORS CAN WE CONTROL?
In summary, some of the risk factors for Alzheimer disease can be modified if we do the following.
Aggressively manage diabetes and cardiovascular disease. Vascular risk factors significantly increase dementia risk, providing good targets for prevention: clinicians should aggressively help their patients control diabetes, hypertension, and hyperlipidemia.26 However, aggressive control of hypertension in a patient with already-existing dementia may exacerbate the condition, so caution is warranted.
Optimize diet. Dietary measures include high intake of antioxidants (which are especially high in brightly colored and tart-flavored fruits and vegetables) and polyunsaturated fats.26 Eating a Mediterranean-type diet that includes a high intake of cold-water ocean fish is recommended. Fish should not be fried: the high temperatures may destroy the omega-3 fatty acids, and the high fat content may inhibit their absorption.
Weigh the risks and benefits of estrogen. Although estrogen replacement therapy for postmenopausal women has had mixed results for controlling dementia, it appears to be clinically indicated to control vasomotor symptoms and likely does not increase the risk of dementia for newly menopausal women. Risks and benefits should be carefully weighed for each patient.
Optimize exercise. People who are physically active in midlife have a lower risk of Alzheimer disease.27 Those who adopt new physical activity late in life may also gain some protective or restorative benefit.28
Many measures, such as taking anti-inflammatory or antihypertensive drugs, probably have a very small incremental benefit over time, so it is difficult to measure significant effects during the course of a typical clinical trial.
Clinicians are already recommending actions to reduce the risk of dementia by focusing on lowering cardiovascular risk. Hopefully, as these actions become more commonly practiced as lifelong habits in those reaching the age of risk for Alzheimer disease, we will see a reduced incidence of that devastating and much-feared illness.
- Castellani RJ, Lee HG, Zhu X, Nunomura A, Perry G, Smith MA. Neuropathology of Alzheimer disease: pathognomic but not pathogenic. Acta Neuropathol 2006; 111:503–509.
- Geldmacher DS. Alzheimer’s pathogenesis: are we barking up the wrong tree? Pract Neurol 2006( 4):14–15.
- Bishop GM, Robinson SR. The amyloid hypothesis: let sleeping dogmas lie? Neurobiol Aging 2002; 23:1101–1105.
- Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol 2009; 66:1210–1215.
- Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia 1996; 39:1392–1397.
- Akomolafe A, Beiser A, Meigs JB, et al. Diabetes mellitus and risks of developing Alzheimer disease: results from the Framingham Study. Arch Neurol 2006; 63:1551–1555.
- Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol 2006; 5:64–74.
- Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004; 53:474–481.
- Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses 2004; 63:8–20.
- de la Torre JC. Vascular basis of Alzheimer’s pathogenesis. Ann NY Acad Sci 2002; 977:196–215.
- Kalaria R. Similarities between Alzheimer’s disease and vascular dementia. J Neurol Sci 2002; 203–204:29–34.
- Prada CM, Garcia-Alloza M, Betensky RA, et al. Antibody-mediated clearance of amyloid-beta peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J Neurosci 2007; 27:1973–1980.
- Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 1997; 277:813–817.
- McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology 1996; 47:425–432.
- Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology 1997; 48:626–632.
- Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol 2004; 3:169–178.
- Bamberger ME, Landreth GE. Inflammation, apoptosis, and Alzheimer’s disease. Neuroscientist 2002; 8:276–283.
- Tobinick E, Gross H, Weinberger A, Cohen H. TNF-alpha modulation for treatment of Alzheimer’s disease: a 6-month pilot study. MedGenMed 2006; 8:25.
- Miller DR, Fincke BG, Davidson JE, Weil JG. Thiazolidinedione use may forestall progression of Alzheimer’s disease in diabetes patients. Alzheimer’s & Dementia: Journal of the Alzheimer’s Association 2006(2 suppl July):S148.
- Gold M, Alderton C, Zvartau-Hind M, et al. Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord 2010; 30:131–146.
- Yaffe K, Sawaya G, Lieberburg I, Grady D. Estrogen therapy in postmenopausal women: effects on cognitive function and dementia. JAMA 1998; 279:688–695.
- Nelson HD, Humphrey LL, Nygren P, Teutsch SM, Allan JD. Postmenopausal hormone replacement therapy: scientific review. JAMA 2002; 288:872–881.
- LeBlanc ES, Janowsky J, Chan BK, Nelson HD. Hormone replacement therapy and cognition: systematic review and meta-analysis. JAMA 2001; 285:1489–1499.
- Hogervorst E, Williams J, Budge M, Riedel W, Jolles J. The nature of the effect of female gonadal hormone replacement therapy on cognitive function in post-menopausal women: a meta-analysis. Neuroscience 2000; 101:485–512.
- Shumaker SA, Legault C, Kuller L, et al; Women’s Health Initiative Memory Study. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA 2004; 291:2947–2958.
- Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol 2009; 66:1210–1215.
- Etgen T, Sander D, Huntgeburth U, Poppert H, Förstl H, Bickel H. Physical activity and incident cognitive impairment in elderly persons: the INVADE study. Arch Intern Med 2010; 170:186–193.
- Heyn P, Abreu BC, Ottenbacher KJ. The effects of exercise training on elderly persons with cognitive impairment and dementia: a meta-analysis. Arch Phys Med Rehabil 2004; 85:1694–1704.
Efforts to modify the relentless course of Alzheimer disease have until now been based on altering the production or clearance of beta-amyloid, the protein found in plaques in the brains of patients with the disease. Results have been disappointing, possibly because our models of the disease—mostly based on the rare, inherited form—may not be applicable to the much more common sporadic form.
Ely Lilly’s recent announcement that it is halting research into semagacestat, a drug designed to reduce amyloid production, only cast further doubt on viability of the amyloid hypothesis as a framework for effective treatments for Alzheimer disease.
Because of the close association of sporadic Alzheimer disease with vascular disease and type 2 diabetes mellitus, increased efforts to treat and prevent these conditions may be the best approach to reducing the incidence of Alzheimer disease.
This article will discuss current thinking of the pathophysiology of Alzheimer disease, with special attention to potential prevention and treatment strategies.
THE CANONICAL VIEW: AMYLOID IS THE CAUSE
The canonical view is that the toxic effects of beta-amyloid are the cause of neuronal dysfunction and loss in Alzheimer disease.
Beta-amyloid is a small peptide, 38 to 42 amino acids long, that accumulates in the extracellular plaque that characterizes Alzheimer pathology. Small amounts of extracellular beta-amyloid can be detected in the brains of elderly people who die of other causes, but the brains of people who die with severe Alzheimer disease show extensive accumulation of plaques.
The amyloid precursor protein is cleaved by normal constitutive enzymes, leaving beta-amyloid as a fragment. The beta-amyloid forms into fibrillar aggregations, which further clump into the extracellular plaque. Plaques can occur in the normal aging process in relatively low amounts. However, in Alzheimer disease, through some unknown trigger, the immune system appears to become activated in reference to the plaque. Microglial cells—the brain’s macrophages—invade the plaque and trigger a cycle of inflammation. The inflammation and its by-products cause local neuronal damage, which seems to propagate the inflammatory cycle to an even greater extent through a feed-forward loop. The damage leads to metabolic stress in the neuron and collapse of the cytoskeleton into a neurofibrillary tangle. Once the neurofibrillary tangle is forming, the neuron is probably on the path to certain death.
This pathway might be interrupted at several points, and in fact, much of the drug development world is working on possible ways to do so.
GENETIC VS SPORADIC DISEASE: WHAT ARE THE KEY DIFFERENCES?
Although the autosomal dominant form of the disease accounts for probably only 1% or 2% of all cases of Alzheimer disease, most animal models and hence much of the basic research and drug testing in Alzheimer disease are based on those dominant mutations. The pathology—the plaques and tangles—in Alzheimer disease in older adults is identical to that in younger adults, but the origins of the disease may not be the same. Therefore, the experimental model for one may not be relevant to the other.
In the last several years, some have questioned whether the amyloid hypothesis applies to all Alzheimer disease.1,2 Arguments go back to at least 2002, when Bishop and Robinson in an article entitled “The amyloid hypothesis: Let sleeping dogmas lie?”3 criticized the hypothesis and suggested that the beta-amyloid peptide appeared to be neuroprotective, not neurotoxic, in most situations. They suggested we await the outcome of antiamyloid therapeutic trials to determine whether the amyloid hypothesis truly explains the disorder.
The antiamyloid trials have now been under way for some time, and we have no definitive answer. Data from the phase II study of the monoclonal antibody agent bapineuzumab suggests there might be some small clinical impact of removing amyloid from the brain through immunotherapy mechanisms, but the benefits thus far are not robust.
COULD AMYLOID BE NEUROPROTECTIVE?
A pivotal question might be, “What if sick neurons made amyloid, instead of amyloid making neurons sick?” A corollary question is, “What if the effect were bidirectional?”
It is possible that in certain concentrations amyloid is neurotoxic, but in other concentrations, it actually facilitates neuronal repair, healing, and connection.
REDUCING METABOLIC STRESS: THE KEY TO PREVENTION?
If our current models of drug therapy are not effective against sporadic Alzheimer disease, perhaps focusing on prevention would be more fruitful.
Consider diabetes mellitus as an analogy. Its manifestations include polydipsia, polyuria, fatigue, and elevated glucose and hemoglobin A1c. Its complications are cardiovascular disease, nephropathy, and retinopathy. Yet diabetes mellitus encompasses two different diseases—type 1 and type 2—with different underlying pathophysiology. We do not treat them the same way. We may be moving toward a similar view of Alzheimer disease.
Links have been hypothesized between vascular risks and dementia. Diabetes, hypertension, dyslipidemia, and obesity might lead to dementia in a process abetted by oxidative stress, endothelial dysfunction, insulin resistance, inflammation, adiposity, and subcortical vascular disease. All of these could be targets of intervention to prevent and treat dementia.4
Instead of a beta-amyloid trigger, let us hypothesize that metabolic stress is the initiating element of the Alzheimer cascade, which then triggers beta-amyloid overproduction or underclearance, and the immune activation damages neurons. By lessening metabolic stress or by preventing immune activation, it may, in theory, be possible to prevent neurons from entering into the terminal pathway of tangle formation and cell death.
LINKS BETWEEN ALZHEIMER DISEASE AND DIABETES
Rates of dementia of all causes are higher in people with diabetes. The strongest effect has been noted in vascular dementia, but Alzheimer disease was also found to be associated with diabetes.5 The Framingham Heart Study6 found the association between dementia and diabetes was significant only when other risk factors for Alzheimer disease were minimal: in an otherwise healthy population, diabetes alone appears to trigger the risk for dementia. But in a population with a lot of vascular comorbidities, the association between diabetes and dementia is not as clear. Perhaps the magnitude of the risk is overwhelmed by greater cerebrovascular and cardiovascular morbidity.
A systematic review7 supported the notion that the risk of dementia is higher in people with diabetes, and even raised the issue of whether we should consider Alzheimer disease “type 3 diabetes.”
Testing of the reverse hypothesis—diabetes is more common in people with Alzheimer disease—also is supportive: diabetes mellitus and even impaired fasting glucose are approximately twice as common in people with Alzheimer disease than in those without.8 Fasting blood glucose levels increase steadily with age, but after age 65, they are higher in people with Alzheimer disease than in those without.
Glucose has some direct effects on brain metabolism that might explain the higher risk. Chronic hyperglycemia is associated with excessive production of free radicals, which leads to reactive oxygen species. These are toxic to neuronal membranes as well as to mitochondria, where many of the reactive oxygen species are generated. Free radicals also facilitate the inflammatory response.
We also see greater neuronal and mitochondrial calcium influx in the presence of hyperglycemia. The excess calcium interferes with mitochondrial metabolism and may trigger the cascade of apoptosis when it reaches critical levels in neurons.
Chronic hyperglycemia is also associated with increased advanced glycation end-products. These are toxic molecules produced by the persistent exposure of proteins to high sugar levels and may be facilitated by the presence of reactive oxygen species that catalyze the reactions between the sugars and the peptides. Glycation end-products are commonly recognized as the same as those occurring during browning of meat (the Maillard reaction).
Hyperglycemia also potentiates neuronal damage from ischemia. Animal experiments show that brain infarction in the presence of hyperglycemia results in worse damage than the same degree of ischemia in the absence of hyperglycemia. Hyperglycemia may exaggerate other blows to neuronal function such as those from small strokes or microvascular ischemia.
AN ALTERNATIVE TO THE AMYLOID HYPOTHESIS: THE ‘MITOCHONDRIAL CASCADE HYPOTHESIS’
Swerdlow and Khan9 have proposed an alternative to the amyloid hypothesis as the cause of Alzheimer disease, known as the “mitochondrial cascade hypothesis.” According to this model, as we age we accumulate more wear-and-tear from oxidative mitochondrial damage, especially the accumulation of toxins leading to reduced cell metabolic activity. This triggers the “3-R response”:
Reset. When toxins alter cell metabolism, neurons try to repair themselves by manufacturing beta-amyloid, which is a “repair-and-reset” synaptic signaling molecule that reduces energy production. Under the lower energy state, beta-pleated sheets develop from beta-amyloid, which aggregate and form amyloid plaque.
Remove. Many cells undergo programmed death when faced with oxidative stress. The first step in neuronal loss is reduced synaptic connections and, hence, losses in neuronal communication. This results in impaired cognition.
Replace. Some cells that are faced with metabolic stress re-enter the cell cycle by undergoing cell division. Neurons, however, are terminally postmitotic and die if they try to divide: by synthesizing cell division proteins, duplicating chromosomes, and reorganizing the complex internal structure, the cell cannot work properly and cell division fails. In the mitochondrial cascade hypothesis, neurofibrillary tangles result from this attempted remodeling of the cytoskeletal filaments, furthering neuronal dysfunction.
ALZHEIMER DISEASE AND STROKE: MORE ALIKE THAN WE THOUGHT?
Although historically clinicians and researchers have tried to distinguish between Alzheimer disease and vascular dementia, growing evidence indicates that the two disorders overlap significantly and that the pathologies may be synergistic.
Alzheimer disease has been hypothesized as being a vascular disorder.10 It shares many of the risk factors of vascular disease, and preclinical detection of Alzheimer disease is possible from measurements of regional cerebral perfusion. Cerebrovascular and neurodegenerative pathology are parallel in Alzheimer disease and vascular disease.
Pure Alzheimer disease and vascular disease are two ends of a pathologic continuum.11 At one end is “pure” Alzheimer disease, in which patients die only with histologic findings of plaques and neurofibrillary tangles. This form may occur only in patients with the autosomal dominant early-onset form. At the other end of the spectrum are people who have serious vascular disease, multiple strokes, and microvascular ischemia and who die demented but with no evidence of the plaques and tangles of Alzheimer disease.
Between these poles is a spectrum of overlapping pathology that is either Alzheimer disease-dominant or vascular disease-dominant, with varying degrees of amyloid plaque and evidence of microvascular infarcts. Cerebral amyloid angiopathy (the accumulation of beta-amyloid in the wall of arteries in the brain) bridges the syndromes.12 In some drug studies that attempted removing amyloid from the brain, vascular permeability was altered, resulting in brain edema.
Along the same lines as Kalaria’s model,11 Snowden et al13 found at autopsy of aged Catholic nuns that for some the accumulation of Alzheimer pathology alone was insufficient to cause dementia, but dementia was nearly universal in nuns with the same burden of Alzheimer pathology commingled with vascular pathology.
DOES INFLAMMATION PLAY A ROLE?
The inflammatory state is a recognized risk factor for Alzheimer disease, but the clinical data are mixed. Epidemiologic evidence is strong: patients who regularly take nonsteroidal anti-inflammatory drugs (NSAIDs) or steroids for chronic, systemic inflammatory diseases (eg, arthritis) have a 45% to 60% reduced risk for Alzheimer disease.14,15
However, multiple clinical trials in patients with Alzheimer disease have failed to show a benefit of taking anti-inflammatory drugs. One preliminary report suggested that indomethacin (Indocin) might offer benefit, but because of gastrointestinal side effects its usefulness in an elderly population is limited.
Diabetes and inflammation are also closely linked: hyperinsulinemia is proinflammatory, promoting the formation of reactive oxygen species, inhibiting the degradation of oxidized proteins, and increasing the risk for lipid per-oxidation. Insulin acts synergistically with endotoxins to raise inflammatory markers, eg, proinflammatory cytokines and C-reactive protein.16
It is possible that anti-inflammatory drugs may not work in Alzheimer disease because inflammation in the brain is mediated more by microglial cells than by prostaglandin pathways. In Alzheimer disease, inflammation is mediated by activated microglial cells, which invade plaques with their processes; these are not evident in the diffuse beta-amyloid-rich plaques seen in typical aging. The trigger for their activation is unclear, but the activated microglial cells and the invasion of plaques are seen in transgenic mouse models of Alzheimer disease, and activation is seen when beta-amyloid is injected into the brain of a healthy mouse.17
Activated microglial cells enlarge and their metabolic rate increases, with a surge in the production of proteins and other protein-mediated inflammatory markers such as alpha-antichymotrypsin, alpha-antitrypsin, serum amyloid P, C-reactive protein, nitric oxide, and proinflammatory cytokines. It is unlikely that it is healthy for cells to be exposed to these inflammatory products. Some of the cytokines are now targets of drug development for Alzheimer disease, and agents targeting these pathways have already been developed for connective tissue diseases.
In a controversial pilot study, Tobinick et al18 studied the use of etanercept (Enbrel), an inhibitor of tumor necrosis factor-alpha (an inflammatory cytokine). They injected etanercept weekly into the spinal canal in 15 patients with mild to severe Alzheimer disease, for 6 months. Patients improved in the Mini-Mental State Examination by more than two points during the study. Patent issues surrounding use of this drug in Alzheimer disease may delay further trials.
Thiazolidinediones block microglial cell activation
The reactive microglial phenotype can be prevented in cell culture by peroxisome proliferator-activated receptor (PPAR) gamma agonists. These include the antidiabetic thiazolidinediones such as pioglitazone (Actos), troglitazone (Rezulin), and rosiglitazone (Avandia), and indomethacin and other NSAIDs.
Using a Veterans Administration database of more than 142,000 patients, Miller et al19 retrospectively found that patients who took a thiazolidinedione for diabetes had a 20% lower risk of developing Alzheimer disease compared with users of insulin or metformin (Glucophage).
However, rosiglitazone showed no benefit against Alzheimer disease in a large clinical trial,20 but this may be because it is rapidly cleared from the brain. Pioglitazone is not actively exported from the brain, so it may be a better candidate, but pharmaceutical industry interest in this agent is low because its patent will soon expire.
Fish oil is another PPAR-gamma agonist, and some studies indicate that eating fish may protect against developing Alzheimer disease; it may also be therapeutic if the disease is present. Double-blind controlled studies have not been carried out and likely will not because of patent issues: the costs of such studies are high, and the potential payback is low.
ESTROGEN: PROTECTIVE OR NOT?
Whether taking estrogen is a risk factor or is protective has not yet been determined. Estrogen directly affects neurons. It increases the number of dendritic spines, which are associated with improved memory. Meta-analyses suggest that hormone replacement therapy reduces the risk of dementia by about one-third. 21,22 Both positive and negative prospective studies exist, but all are complicated by serious methodologic flaws.23,24
Combined analysis of about 7,500 women from two double-blind, randomized, placebo-controlled trials of the Women’s Health Initiative Memory Study found that the risks of dementia and mild cognitive impairment were increased by hormone replacement therapy. The hazard ratio for dementia was found to be 1.76 (P < .005), amounting to 23 new cases of dementia per 10,000 prescriptions annually.25
Patient selection may account for the conflicting results in different studies. Epidemiologic studies consisted mostly of newly postmenopausal women and those who were being treated for symptoms of vasomotor instability. In contrast, the Women’s Health Initiative enrolled only women older than 65 and excluded women with vasomotor instability. Other studies indicate that the greatest cognitive improvements with hormone therapies are seen in women with vasomotor symptoms.
WHICH RISK FACTORS CAN WE CONTROL?
In summary, some of the risk factors for Alzheimer disease can be modified if we do the following.
Aggressively manage diabetes and cardiovascular disease. Vascular risk factors significantly increase dementia risk, providing good targets for prevention: clinicians should aggressively help their patients control diabetes, hypertension, and hyperlipidemia.26 However, aggressive control of hypertension in a patient with already-existing dementia may exacerbate the condition, so caution is warranted.
Optimize diet. Dietary measures include high intake of antioxidants (which are especially high in brightly colored and tart-flavored fruits and vegetables) and polyunsaturated fats.26 Eating a Mediterranean-type diet that includes a high intake of cold-water ocean fish is recommended. Fish should not be fried: the high temperatures may destroy the omega-3 fatty acids, and the high fat content may inhibit their absorption.
Weigh the risks and benefits of estrogen. Although estrogen replacement therapy for postmenopausal women has had mixed results for controlling dementia, it appears to be clinically indicated to control vasomotor symptoms and likely does not increase the risk of dementia for newly menopausal women. Risks and benefits should be carefully weighed for each patient.
Optimize exercise. People who are physically active in midlife have a lower risk of Alzheimer disease.27 Those who adopt new physical activity late in life may also gain some protective or restorative benefit.28
Many measures, such as taking anti-inflammatory or antihypertensive drugs, probably have a very small incremental benefit over time, so it is difficult to measure significant effects during the course of a typical clinical trial.
Clinicians are already recommending actions to reduce the risk of dementia by focusing on lowering cardiovascular risk. Hopefully, as these actions become more commonly practiced as lifelong habits in those reaching the age of risk for Alzheimer disease, we will see a reduced incidence of that devastating and much-feared illness.
Efforts to modify the relentless course of Alzheimer disease have until now been based on altering the production or clearance of beta-amyloid, the protein found in plaques in the brains of patients with the disease. Results have been disappointing, possibly because our models of the disease—mostly based on the rare, inherited form—may not be applicable to the much more common sporadic form.
Ely Lilly’s recent announcement that it is halting research into semagacestat, a drug designed to reduce amyloid production, only cast further doubt on viability of the amyloid hypothesis as a framework for effective treatments for Alzheimer disease.
Because of the close association of sporadic Alzheimer disease with vascular disease and type 2 diabetes mellitus, increased efforts to treat and prevent these conditions may be the best approach to reducing the incidence of Alzheimer disease.
This article will discuss current thinking of the pathophysiology of Alzheimer disease, with special attention to potential prevention and treatment strategies.
THE CANONICAL VIEW: AMYLOID IS THE CAUSE
The canonical view is that the toxic effects of beta-amyloid are the cause of neuronal dysfunction and loss in Alzheimer disease.
Beta-amyloid is a small peptide, 38 to 42 amino acids long, that accumulates in the extracellular plaque that characterizes Alzheimer pathology. Small amounts of extracellular beta-amyloid can be detected in the brains of elderly people who die of other causes, but the brains of people who die with severe Alzheimer disease show extensive accumulation of plaques.
The amyloid precursor protein is cleaved by normal constitutive enzymes, leaving beta-amyloid as a fragment. The beta-amyloid forms into fibrillar aggregations, which further clump into the extracellular plaque. Plaques can occur in the normal aging process in relatively low amounts. However, in Alzheimer disease, through some unknown trigger, the immune system appears to become activated in reference to the plaque. Microglial cells—the brain’s macrophages—invade the plaque and trigger a cycle of inflammation. The inflammation and its by-products cause local neuronal damage, which seems to propagate the inflammatory cycle to an even greater extent through a feed-forward loop. The damage leads to metabolic stress in the neuron and collapse of the cytoskeleton into a neurofibrillary tangle. Once the neurofibrillary tangle is forming, the neuron is probably on the path to certain death.
This pathway might be interrupted at several points, and in fact, much of the drug development world is working on possible ways to do so.
GENETIC VS SPORADIC DISEASE: WHAT ARE THE KEY DIFFERENCES?
Although the autosomal dominant form of the disease accounts for probably only 1% or 2% of all cases of Alzheimer disease, most animal models and hence much of the basic research and drug testing in Alzheimer disease are based on those dominant mutations. The pathology—the plaques and tangles—in Alzheimer disease in older adults is identical to that in younger adults, but the origins of the disease may not be the same. Therefore, the experimental model for one may not be relevant to the other.
In the last several years, some have questioned whether the amyloid hypothesis applies to all Alzheimer disease.1,2 Arguments go back to at least 2002, when Bishop and Robinson in an article entitled “The amyloid hypothesis: Let sleeping dogmas lie?”3 criticized the hypothesis and suggested that the beta-amyloid peptide appeared to be neuroprotective, not neurotoxic, in most situations. They suggested we await the outcome of antiamyloid therapeutic trials to determine whether the amyloid hypothesis truly explains the disorder.
The antiamyloid trials have now been under way for some time, and we have no definitive answer. Data from the phase II study of the monoclonal antibody agent bapineuzumab suggests there might be some small clinical impact of removing amyloid from the brain through immunotherapy mechanisms, but the benefits thus far are not robust.
COULD AMYLOID BE NEUROPROTECTIVE?
A pivotal question might be, “What if sick neurons made amyloid, instead of amyloid making neurons sick?” A corollary question is, “What if the effect were bidirectional?”
It is possible that in certain concentrations amyloid is neurotoxic, but in other concentrations, it actually facilitates neuronal repair, healing, and connection.
REDUCING METABOLIC STRESS: THE KEY TO PREVENTION?
If our current models of drug therapy are not effective against sporadic Alzheimer disease, perhaps focusing on prevention would be more fruitful.
Consider diabetes mellitus as an analogy. Its manifestations include polydipsia, polyuria, fatigue, and elevated glucose and hemoglobin A1c. Its complications are cardiovascular disease, nephropathy, and retinopathy. Yet diabetes mellitus encompasses two different diseases—type 1 and type 2—with different underlying pathophysiology. We do not treat them the same way. We may be moving toward a similar view of Alzheimer disease.
Links have been hypothesized between vascular risks and dementia. Diabetes, hypertension, dyslipidemia, and obesity might lead to dementia in a process abetted by oxidative stress, endothelial dysfunction, insulin resistance, inflammation, adiposity, and subcortical vascular disease. All of these could be targets of intervention to prevent and treat dementia.4
Instead of a beta-amyloid trigger, let us hypothesize that metabolic stress is the initiating element of the Alzheimer cascade, which then triggers beta-amyloid overproduction or underclearance, and the immune activation damages neurons. By lessening metabolic stress or by preventing immune activation, it may, in theory, be possible to prevent neurons from entering into the terminal pathway of tangle formation and cell death.
LINKS BETWEEN ALZHEIMER DISEASE AND DIABETES
Rates of dementia of all causes are higher in people with diabetes. The strongest effect has been noted in vascular dementia, but Alzheimer disease was also found to be associated with diabetes.5 The Framingham Heart Study6 found the association between dementia and diabetes was significant only when other risk factors for Alzheimer disease were minimal: in an otherwise healthy population, diabetes alone appears to trigger the risk for dementia. But in a population with a lot of vascular comorbidities, the association between diabetes and dementia is not as clear. Perhaps the magnitude of the risk is overwhelmed by greater cerebrovascular and cardiovascular morbidity.
A systematic review7 supported the notion that the risk of dementia is higher in people with diabetes, and even raised the issue of whether we should consider Alzheimer disease “type 3 diabetes.”
Testing of the reverse hypothesis—diabetes is more common in people with Alzheimer disease—also is supportive: diabetes mellitus and even impaired fasting glucose are approximately twice as common in people with Alzheimer disease than in those without.8 Fasting blood glucose levels increase steadily with age, but after age 65, they are higher in people with Alzheimer disease than in those without.
Glucose has some direct effects on brain metabolism that might explain the higher risk. Chronic hyperglycemia is associated with excessive production of free radicals, which leads to reactive oxygen species. These are toxic to neuronal membranes as well as to mitochondria, where many of the reactive oxygen species are generated. Free radicals also facilitate the inflammatory response.
We also see greater neuronal and mitochondrial calcium influx in the presence of hyperglycemia. The excess calcium interferes with mitochondrial metabolism and may trigger the cascade of apoptosis when it reaches critical levels in neurons.
Chronic hyperglycemia is also associated with increased advanced glycation end-products. These are toxic molecules produced by the persistent exposure of proteins to high sugar levels and may be facilitated by the presence of reactive oxygen species that catalyze the reactions between the sugars and the peptides. Glycation end-products are commonly recognized as the same as those occurring during browning of meat (the Maillard reaction).
Hyperglycemia also potentiates neuronal damage from ischemia. Animal experiments show that brain infarction in the presence of hyperglycemia results in worse damage than the same degree of ischemia in the absence of hyperglycemia. Hyperglycemia may exaggerate other blows to neuronal function such as those from small strokes or microvascular ischemia.
AN ALTERNATIVE TO THE AMYLOID HYPOTHESIS: THE ‘MITOCHONDRIAL CASCADE HYPOTHESIS’
Swerdlow and Khan9 have proposed an alternative to the amyloid hypothesis as the cause of Alzheimer disease, known as the “mitochondrial cascade hypothesis.” According to this model, as we age we accumulate more wear-and-tear from oxidative mitochondrial damage, especially the accumulation of toxins leading to reduced cell metabolic activity. This triggers the “3-R response”:
Reset. When toxins alter cell metabolism, neurons try to repair themselves by manufacturing beta-amyloid, which is a “repair-and-reset” synaptic signaling molecule that reduces energy production. Under the lower energy state, beta-pleated sheets develop from beta-amyloid, which aggregate and form amyloid plaque.
Remove. Many cells undergo programmed death when faced with oxidative stress. The first step in neuronal loss is reduced synaptic connections and, hence, losses in neuronal communication. This results in impaired cognition.
Replace. Some cells that are faced with metabolic stress re-enter the cell cycle by undergoing cell division. Neurons, however, are terminally postmitotic and die if they try to divide: by synthesizing cell division proteins, duplicating chromosomes, and reorganizing the complex internal structure, the cell cannot work properly and cell division fails. In the mitochondrial cascade hypothesis, neurofibrillary tangles result from this attempted remodeling of the cytoskeletal filaments, furthering neuronal dysfunction.
ALZHEIMER DISEASE AND STROKE: MORE ALIKE THAN WE THOUGHT?
Although historically clinicians and researchers have tried to distinguish between Alzheimer disease and vascular dementia, growing evidence indicates that the two disorders overlap significantly and that the pathologies may be synergistic.
Alzheimer disease has been hypothesized as being a vascular disorder.10 It shares many of the risk factors of vascular disease, and preclinical detection of Alzheimer disease is possible from measurements of regional cerebral perfusion. Cerebrovascular and neurodegenerative pathology are parallel in Alzheimer disease and vascular disease.
Pure Alzheimer disease and vascular disease are two ends of a pathologic continuum.11 At one end is “pure” Alzheimer disease, in which patients die only with histologic findings of plaques and neurofibrillary tangles. This form may occur only in patients with the autosomal dominant early-onset form. At the other end of the spectrum are people who have serious vascular disease, multiple strokes, and microvascular ischemia and who die demented but with no evidence of the plaques and tangles of Alzheimer disease.
Between these poles is a spectrum of overlapping pathology that is either Alzheimer disease-dominant or vascular disease-dominant, with varying degrees of amyloid plaque and evidence of microvascular infarcts. Cerebral amyloid angiopathy (the accumulation of beta-amyloid in the wall of arteries in the brain) bridges the syndromes.12 In some drug studies that attempted removing amyloid from the brain, vascular permeability was altered, resulting in brain edema.
Along the same lines as Kalaria’s model,11 Snowden et al13 found at autopsy of aged Catholic nuns that for some the accumulation of Alzheimer pathology alone was insufficient to cause dementia, but dementia was nearly universal in nuns with the same burden of Alzheimer pathology commingled with vascular pathology.
DOES INFLAMMATION PLAY A ROLE?
The inflammatory state is a recognized risk factor for Alzheimer disease, but the clinical data are mixed. Epidemiologic evidence is strong: patients who regularly take nonsteroidal anti-inflammatory drugs (NSAIDs) or steroids for chronic, systemic inflammatory diseases (eg, arthritis) have a 45% to 60% reduced risk for Alzheimer disease.14,15
However, multiple clinical trials in patients with Alzheimer disease have failed to show a benefit of taking anti-inflammatory drugs. One preliminary report suggested that indomethacin (Indocin) might offer benefit, but because of gastrointestinal side effects its usefulness in an elderly population is limited.
Diabetes and inflammation are also closely linked: hyperinsulinemia is proinflammatory, promoting the formation of reactive oxygen species, inhibiting the degradation of oxidized proteins, and increasing the risk for lipid per-oxidation. Insulin acts synergistically with endotoxins to raise inflammatory markers, eg, proinflammatory cytokines and C-reactive protein.16
It is possible that anti-inflammatory drugs may not work in Alzheimer disease because inflammation in the brain is mediated more by microglial cells than by prostaglandin pathways. In Alzheimer disease, inflammation is mediated by activated microglial cells, which invade plaques with their processes; these are not evident in the diffuse beta-amyloid-rich plaques seen in typical aging. The trigger for their activation is unclear, but the activated microglial cells and the invasion of plaques are seen in transgenic mouse models of Alzheimer disease, and activation is seen when beta-amyloid is injected into the brain of a healthy mouse.17
Activated microglial cells enlarge and their metabolic rate increases, with a surge in the production of proteins and other protein-mediated inflammatory markers such as alpha-antichymotrypsin, alpha-antitrypsin, serum amyloid P, C-reactive protein, nitric oxide, and proinflammatory cytokines. It is unlikely that it is healthy for cells to be exposed to these inflammatory products. Some of the cytokines are now targets of drug development for Alzheimer disease, and agents targeting these pathways have already been developed for connective tissue diseases.
In a controversial pilot study, Tobinick et al18 studied the use of etanercept (Enbrel), an inhibitor of tumor necrosis factor-alpha (an inflammatory cytokine). They injected etanercept weekly into the spinal canal in 15 patients with mild to severe Alzheimer disease, for 6 months. Patients improved in the Mini-Mental State Examination by more than two points during the study. Patent issues surrounding use of this drug in Alzheimer disease may delay further trials.
Thiazolidinediones block microglial cell activation
The reactive microglial phenotype can be prevented in cell culture by peroxisome proliferator-activated receptor (PPAR) gamma agonists. These include the antidiabetic thiazolidinediones such as pioglitazone (Actos), troglitazone (Rezulin), and rosiglitazone (Avandia), and indomethacin and other NSAIDs.
Using a Veterans Administration database of more than 142,000 patients, Miller et al19 retrospectively found that patients who took a thiazolidinedione for diabetes had a 20% lower risk of developing Alzheimer disease compared with users of insulin or metformin (Glucophage).
However, rosiglitazone showed no benefit against Alzheimer disease in a large clinical trial,20 but this may be because it is rapidly cleared from the brain. Pioglitazone is not actively exported from the brain, so it may be a better candidate, but pharmaceutical industry interest in this agent is low because its patent will soon expire.
Fish oil is another PPAR-gamma agonist, and some studies indicate that eating fish may protect against developing Alzheimer disease; it may also be therapeutic if the disease is present. Double-blind controlled studies have not been carried out and likely will not because of patent issues: the costs of such studies are high, and the potential payback is low.
ESTROGEN: PROTECTIVE OR NOT?
Whether taking estrogen is a risk factor or is protective has not yet been determined. Estrogen directly affects neurons. It increases the number of dendritic spines, which are associated with improved memory. Meta-analyses suggest that hormone replacement therapy reduces the risk of dementia by about one-third. 21,22 Both positive and negative prospective studies exist, but all are complicated by serious methodologic flaws.23,24
Combined analysis of about 7,500 women from two double-blind, randomized, placebo-controlled trials of the Women’s Health Initiative Memory Study found that the risks of dementia and mild cognitive impairment were increased by hormone replacement therapy. The hazard ratio for dementia was found to be 1.76 (P < .005), amounting to 23 new cases of dementia per 10,000 prescriptions annually.25
Patient selection may account for the conflicting results in different studies. Epidemiologic studies consisted mostly of newly postmenopausal women and those who were being treated for symptoms of vasomotor instability. In contrast, the Women’s Health Initiative enrolled only women older than 65 and excluded women with vasomotor instability. Other studies indicate that the greatest cognitive improvements with hormone therapies are seen in women with vasomotor symptoms.
WHICH RISK FACTORS CAN WE CONTROL?
In summary, some of the risk factors for Alzheimer disease can be modified if we do the following.
Aggressively manage diabetes and cardiovascular disease. Vascular risk factors significantly increase dementia risk, providing good targets for prevention: clinicians should aggressively help their patients control diabetes, hypertension, and hyperlipidemia.26 However, aggressive control of hypertension in a patient with already-existing dementia may exacerbate the condition, so caution is warranted.
Optimize diet. Dietary measures include high intake of antioxidants (which are especially high in brightly colored and tart-flavored fruits and vegetables) and polyunsaturated fats.26 Eating a Mediterranean-type diet that includes a high intake of cold-water ocean fish is recommended. Fish should not be fried: the high temperatures may destroy the omega-3 fatty acids, and the high fat content may inhibit their absorption.
Weigh the risks and benefits of estrogen. Although estrogen replacement therapy for postmenopausal women has had mixed results for controlling dementia, it appears to be clinically indicated to control vasomotor symptoms and likely does not increase the risk of dementia for newly menopausal women. Risks and benefits should be carefully weighed for each patient.
Optimize exercise. People who are physically active in midlife have a lower risk of Alzheimer disease.27 Those who adopt new physical activity late in life may also gain some protective or restorative benefit.28
Many measures, such as taking anti-inflammatory or antihypertensive drugs, probably have a very small incremental benefit over time, so it is difficult to measure significant effects during the course of a typical clinical trial.
Clinicians are already recommending actions to reduce the risk of dementia by focusing on lowering cardiovascular risk. Hopefully, as these actions become more commonly practiced as lifelong habits in those reaching the age of risk for Alzheimer disease, we will see a reduced incidence of that devastating and much-feared illness.
- Castellani RJ, Lee HG, Zhu X, Nunomura A, Perry G, Smith MA. Neuropathology of Alzheimer disease: pathognomic but not pathogenic. Acta Neuropathol 2006; 111:503–509.
- Geldmacher DS. Alzheimer’s pathogenesis: are we barking up the wrong tree? Pract Neurol 2006( 4):14–15.
- Bishop GM, Robinson SR. The amyloid hypothesis: let sleeping dogmas lie? Neurobiol Aging 2002; 23:1101–1105.
- Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol 2009; 66:1210–1215.
- Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia 1996; 39:1392–1397.
- Akomolafe A, Beiser A, Meigs JB, et al. Diabetes mellitus and risks of developing Alzheimer disease: results from the Framingham Study. Arch Neurol 2006; 63:1551–1555.
- Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol 2006; 5:64–74.
- Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004; 53:474–481.
- Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses 2004; 63:8–20.
- de la Torre JC. Vascular basis of Alzheimer’s pathogenesis. Ann NY Acad Sci 2002; 977:196–215.
- Kalaria R. Similarities between Alzheimer’s disease and vascular dementia. J Neurol Sci 2002; 203–204:29–34.
- Prada CM, Garcia-Alloza M, Betensky RA, et al. Antibody-mediated clearance of amyloid-beta peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J Neurosci 2007; 27:1973–1980.
- Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 1997; 277:813–817.
- McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology 1996; 47:425–432.
- Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology 1997; 48:626–632.
- Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol 2004; 3:169–178.
- Bamberger ME, Landreth GE. Inflammation, apoptosis, and Alzheimer’s disease. Neuroscientist 2002; 8:276–283.
- Tobinick E, Gross H, Weinberger A, Cohen H. TNF-alpha modulation for treatment of Alzheimer’s disease: a 6-month pilot study. MedGenMed 2006; 8:25.
- Miller DR, Fincke BG, Davidson JE, Weil JG. Thiazolidinedione use may forestall progression of Alzheimer’s disease in diabetes patients. Alzheimer’s & Dementia: Journal of the Alzheimer’s Association 2006(2 suppl July):S148.
- Gold M, Alderton C, Zvartau-Hind M, et al. Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord 2010; 30:131–146.
- Yaffe K, Sawaya G, Lieberburg I, Grady D. Estrogen therapy in postmenopausal women: effects on cognitive function and dementia. JAMA 1998; 279:688–695.
- Nelson HD, Humphrey LL, Nygren P, Teutsch SM, Allan JD. Postmenopausal hormone replacement therapy: scientific review. JAMA 2002; 288:872–881.
- LeBlanc ES, Janowsky J, Chan BK, Nelson HD. Hormone replacement therapy and cognition: systematic review and meta-analysis. JAMA 2001; 285:1489–1499.
- Hogervorst E, Williams J, Budge M, Riedel W, Jolles J. The nature of the effect of female gonadal hormone replacement therapy on cognitive function in post-menopausal women: a meta-analysis. Neuroscience 2000; 101:485–512.
- Shumaker SA, Legault C, Kuller L, et al; Women’s Health Initiative Memory Study. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA 2004; 291:2947–2958.
- Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol 2009; 66:1210–1215.
- Etgen T, Sander D, Huntgeburth U, Poppert H, Förstl H, Bickel H. Physical activity and incident cognitive impairment in elderly persons: the INVADE study. Arch Intern Med 2010; 170:186–193.
- Heyn P, Abreu BC, Ottenbacher KJ. The effects of exercise training on elderly persons with cognitive impairment and dementia: a meta-analysis. Arch Phys Med Rehabil 2004; 85:1694–1704.
- Castellani RJ, Lee HG, Zhu X, Nunomura A, Perry G, Smith MA. Neuropathology of Alzheimer disease: pathognomic but not pathogenic. Acta Neuropathol 2006; 111:503–509.
- Geldmacher DS. Alzheimer’s pathogenesis: are we barking up the wrong tree? Pract Neurol 2006( 4):14–15.
- Bishop GM, Robinson SR. The amyloid hypothesis: let sleeping dogmas lie? Neurobiol Aging 2002; 23:1101–1105.
- Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol 2009; 66:1210–1215.
- Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia 1996; 39:1392–1397.
- Akomolafe A, Beiser A, Meigs JB, et al. Diabetes mellitus and risks of developing Alzheimer disease: results from the Framingham Study. Arch Neurol 2006; 63:1551–1555.
- Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol 2006; 5:64–74.
- Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004; 53:474–481.
- Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses 2004; 63:8–20.
- de la Torre JC. Vascular basis of Alzheimer’s pathogenesis. Ann NY Acad Sci 2002; 977:196–215.
- Kalaria R. Similarities between Alzheimer’s disease and vascular dementia. J Neurol Sci 2002; 203–204:29–34.
- Prada CM, Garcia-Alloza M, Betensky RA, et al. Antibody-mediated clearance of amyloid-beta peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J Neurosci 2007; 27:1973–1980.
- Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 1997; 277:813–817.
- McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology 1996; 47:425–432.
- Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology 1997; 48:626–632.
- Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol 2004; 3:169–178.
- Bamberger ME, Landreth GE. Inflammation, apoptosis, and Alzheimer’s disease. Neuroscientist 2002; 8:276–283.
- Tobinick E, Gross H, Weinberger A, Cohen H. TNF-alpha modulation for treatment of Alzheimer’s disease: a 6-month pilot study. MedGenMed 2006; 8:25.
- Miller DR, Fincke BG, Davidson JE, Weil JG. Thiazolidinedione use may forestall progression of Alzheimer’s disease in diabetes patients. Alzheimer’s & Dementia: Journal of the Alzheimer’s Association 2006(2 suppl July):S148.
- Gold M, Alderton C, Zvartau-Hind M, et al. Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord 2010; 30:131–146.
- Yaffe K, Sawaya G, Lieberburg I, Grady D. Estrogen therapy in postmenopausal women: effects on cognitive function and dementia. JAMA 1998; 279:688–695.
- Nelson HD, Humphrey LL, Nygren P, Teutsch SM, Allan JD. Postmenopausal hormone replacement therapy: scientific review. JAMA 2002; 288:872–881.
- LeBlanc ES, Janowsky J, Chan BK, Nelson HD. Hormone replacement therapy and cognition: systematic review and meta-analysis. JAMA 2001; 285:1489–1499.
- Hogervorst E, Williams J, Budge M, Riedel W, Jolles J. The nature of the effect of female gonadal hormone replacement therapy on cognitive function in post-menopausal women: a meta-analysis. Neuroscience 2000; 101:485–512.
- Shumaker SA, Legault C, Kuller L, et al; Women’s Health Initiative Memory Study. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA 2004; 291:2947–2958.
- Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol 2009; 66:1210–1215.
- Etgen T, Sander D, Huntgeburth U, Poppert H, Förstl H, Bickel H. Physical activity and incident cognitive impairment in elderly persons: the INVADE study. Arch Intern Med 2010; 170:186–193.
- Heyn P, Abreu BC, Ottenbacher KJ. The effects of exercise training on elderly persons with cognitive impairment and dementia: a meta-analysis. Arch Phys Med Rehabil 2004; 85:1694–1704.
KEY POINTS
- Vascular risk factors clearly increase the risk of Alzheimer disease and can be addressed. However, controlled trials in patients with hypertension or with dyslipidemia have had negative results.
- Risk is lower with a diet high in antioxidants and polyunsaturated fatty acids.
- Estrogen therapy has had mixed results in observational studies, mostly hinting at lower risk. However, a randomized trial of hormone replacement therapy in late life indicated a higher risk of dementia with estrogen.
- Physical activity in midlife and in late life was associated with a lower risk of Alzheimer disease in observational studies. Controlled trials were not so positive, but the benefits of exercise may be slowly cumulative.
Timeliness of treatment is more important than choice of reperfusion therapy
Reperfusion therapy decreases morbidity and mortality rates in patients with ST-segment elevation myocardial infarction (MI). Primary percutaneous coronary intervention (PCI) is preferred over fibrinolytic therapy as a reperfusion strategy when the delay in the time to treatment is short and the patient presents to a high-volume, well-equipped center with expert interventional cardiologists.
Compared with fibrinolytic therapy in randomized clinical trials, primary PCI produces higher rates of infarct artery patency, complete reperfusion (grade 3 by the criteria of the Thrombolysis in Myocardial Infarction [TIMI] study), and access-site bleeding. It also produces lower rates of recurrent ischemia, reinfarction, emergency repeat revascularization procedures, intracranial hemorrhage, and death.1 If performed early and successfully, primary PCI also greatly decreases the rates of complications of ST-elevation MI that result from longer ischemic times or unsuccessful fibrinolytic therapy, allowing earlier hospital discharge and resumption of daily activities. Primary PCI is also the best reperfusion option in patients who present late after the onset of symptoms and in patients with cardiogenic shock, and it is the only option in patients who have contraindications to fibrinolytic therapy because of bleeding risk.
However, most hospitals do not have PCI capability. Two options at these hospitals are to transfer the patient to a PCI center quickly for primary PCI or to keep the patient on site and give fibrinolytic therapy, with its limitations. Earlier trials suggested that the transfer strategy was superior, but they had limitations: most patients received streptokinase, an inferior fibrinolytic agent, and door-to-door-to-balloon times were rapid, averaging only 95 minutes because of excellent logistical protocols and careful patient selection.2 Most importantly, rescue PCI and routine PCI were seldom performed in patients receiving fibrinolytics, so fibrinolytic therapy was being tested as monotherapy.
In the real world, however, treatment delays are much longer, and fibrinolytic therapy has evolved into a strategy that includes crossover to rescue PCI or routine PCI. Therefore, the initial trials of transfer for primary PCI do not reflect current practice. In fact, recent registry data suggest that prehospital fibrinolytic therapy followed by early angiography is superior to primary PCI because most patients can be treated within 2 hours of symptom onset; they also suggest that on-site fibrinolytic therapy followed by early angiography is equal in efficacy to primary PCI as long as rescue PCI and routine PCI can be performed.3,4
The most important modifiable predictor of outcome in ST-elevation MI is the time to treatment, a biological truth that continues to be supported by clinical evidence despite ideologic arguments by some interventional cardiology enthusiasts who claim that primary PCI is always superior to the fibrinolytic strategy, regardless of delays.
SURPRISINGLY, OUTCOMES WERE WORSE WITH FACILITATED PCI
It made sense, then, to conclude that the perfect strategy for hospitals without PCI capability would be a combined strategy of immediate fibrinolytic therapy to decrease the time delay associated with organizing PCI, and rapid transfer for immediate PCI to improve the limited reperfusion rates associated with fibrinolytic therapy.
Surprisingly, though, randomized trials found worse outcomes with this “facilitated PCI” strategy.5
Again, limitations in trial design might explain the lack of benefit in the trials. Inadequate anticoagulant and antiplatelet therapy were given to the fibrinolytic patients, and primary PCI patients had relatively short treatment delays, with many patients enrolled at hospitals with PCI capability.
PROGRESS IN REPERFUSION THERAPY
Great strides have been made in reperfusion therapy in recent years. Adjunctive therapy with clopidogrel (Plavix) and enoxaparin (Lovenox) has been shown to improve outcomes with fibrinolytic therapy. Bivalirudin (Angiomax) and stents have improved primary PCI’s performance. Reducing bleeding complications has become a clinical priority, with increasing emphasis on adjusting some drug doses according to renal function and using the radial artery for cardiac catheterization.
The American College of Cardiology initiative, “Door-to-Balloon (D2B): An Alliance for Quality,” focused much attention on organizing in-hospital systems of care for primary PCI, thus increasing the national rate of achieving a door-to-balloon time within 90 minutes from 50% to over 75% in patients who presented to hospitals with PCI capability.6
The American Heart Association has launched “Mission: Lifeline,” a national campaign to organize prehospital systems of care with their program,7 working within communities to address their unique needs, resources, and barriers to implementing systems of care for ST-elevation MI. The key aspect of this effort is to help geographic regions develop local solutions, an explicit recognition that there is no one-size-fits-all solution. Early triage by emergency medical services, rapid diagnosis with prehospital electrocardiography, destination and interhospital transfer protocols, and prehospital activation of the cardiac catheterization laboratory can greatly streamline emergency care and decrease treatment delays for primary PCI.
FOR OUTLYING HOSPITALS, A PHARMACOINVASIVE STRATEGY
So what about hospitals without PCI capability that cannot routinely transfer patients to a hospital with PCI capability within 90 minutes?
Lessons learned from the experiences with immediate PCI, rescue PCI, and facilitated PCI have evolved into the “pharmacoinvasive strategy.” Patients with ST-elevation MI are treated as rapidly as possible with a bolus of a fibrinolytic drug, eg, tenecteplase (TNKase) or reteplase (Retavase), and are also given aspirin, clopidogrel, and enoxaparin. Then, they are rapidly transferred to a PCI-capable hospital so that emergency PCI can be performed if reperfusion is not clinically apparent or if the patient develops pulmonary edema or cardiogenic shock. If the clinical signs suggest that reperfusion has been achieved (relief of chest pain, rapid resolution of ST-segment elevation, bursts of accelerated idioventricular rhythm), coronary angiography (and PCI, if indicated) can be performed within 3 to 24 hours of fibrinolytic therapy. This time frame allows the initial fibrinolytic effect to dissipate, while the antiplatelet and anticoagulant drugs achieve therapeutic levels.
Today, the goal is to treat every patient with the best reperfusion strategy available, given the limitations in resources and the geographic location of some centers, and to maximize the possibility of sustained patency of the infarct-related artery by implanting a stent, even if it takes several hours and transfer to another hospital to perform PCI.8 The pharmacoinvasive strategy of rapid administration of fibrinolytic therapy followed by PCI within 24 hours would be practical in most hospitals without PCI capability where treatment delays prohibit performance of primary PCI within 90 minutes of first medical contact.9
THE ‘STREAM’ TRIAL IS UNDER WAY
As proof of concept, the Strategic Reperfusion Early After Myocardial Infarction (STREAM) trial is enrolling 2,000 patients with ST-elevation MI presenting within 3 hours of symptom onset if primary PCI is not feasible within 60 minutes of first medical contact.10 Patients will be randomized to either of the following:
- Receive prehospital therapy with tenecteplase, aspirin, clopidogrel, and enoxaparin and undergo cardiac catheterization in 6 to 24 hours (or rescue PCI if reperfusion fails within 90 minutes of fibrinolysis)
- Undergo primary PCI performed according to local guidelines.
The primary measure of efficacy will be the composite rate of death, cardiogenic shock, heart failure, and reinfarction at 30 days. Measures of safety include the rates of ischemic stroke, intracranial hemorrhage, and major nonintracranial bleeding.
- Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet 2003; 361:13–20.
- Dalby M, Bouzamondo A, Lechat P, Montalescot G. Transfer for primary angioplasty versus immediate thrombolysis in acute myocardial infarction: a meta-analysis. Circulation 2003; 108:1809–1814.
- Danchin N, Coste P, Ferrières J, et al; FAST-MI Investigators. Comparison of thrombolysis followed by broad use of percutaneous coronary intervention with primary percutaneous coronary intervention for ST-segment-elevation acute myocardial infarction: data from the French registry on Acute ST-elevation Myocardial Infarction (FASTMI). Circulation 2008; 118:268–276.
- Lambert L, Brown K, Segal E, Brophy J, Rodes-Cabau J, Bogaty P. Association between timeliness of reperfusion therapy and clinical outcomes in ST-elevation myocardial infarction. JAMA 2010; 303:2148–2155.
- Keeley EC, Boura JA, Grines CL. Comparison of primary and facilitated percutaneous coronary interventions for ST-elevation myocardial infarction: quantitative review of randomised trials. Lancet 2006; 367:579–588.
- Krumholz HM, Bradley EH, Nallamothu BK, et al. A campaign to improve the timeliness of primary percutaneous coronary intervention: Door-to-Balloon: An Alliance for Quality. JACC Cardiovasc Interv. 2008; 1:97–104.
- Jacobs AK, Antman EM, Faxon DP, Gregory T, Solis P. Development of systems of care for ST-elevation myocardial infarction patients: executive summary. Circulation 2007; 116:217–230.
- Kushner FG, Hand M, Smith SC, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2009 Focused Updates: ACC/AHA Guidelines for the Management of Patients With ST-Elevation Myocardial Infarction (updating the 2004 Guideline and 2007 Focused Update) and ACC/AHA/SCAI Guidelines on Percutaneous Coronary Intervention (updating the 2005 Guideline and 2007 Focused Update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2009; 120:2271–2306.
- Bates ER, Nallamothu BK. Commentary: the role of percutaneous coronary intervention in ST-segment-elevation myocardial infarction. Circulation 2008; 118:567–573.
- Armstrong PW, Gershlick A, Goldstein P, et al; STREAM Steering Committee. The Strategic Reperfusion Early After Myocardial Infarction (STREAM) study. Am Heart J 2010; 160:30–35.
Reperfusion therapy decreases morbidity and mortality rates in patients with ST-segment elevation myocardial infarction (MI). Primary percutaneous coronary intervention (PCI) is preferred over fibrinolytic therapy as a reperfusion strategy when the delay in the time to treatment is short and the patient presents to a high-volume, well-equipped center with expert interventional cardiologists.
Compared with fibrinolytic therapy in randomized clinical trials, primary PCI produces higher rates of infarct artery patency, complete reperfusion (grade 3 by the criteria of the Thrombolysis in Myocardial Infarction [TIMI] study), and access-site bleeding. It also produces lower rates of recurrent ischemia, reinfarction, emergency repeat revascularization procedures, intracranial hemorrhage, and death.1 If performed early and successfully, primary PCI also greatly decreases the rates of complications of ST-elevation MI that result from longer ischemic times or unsuccessful fibrinolytic therapy, allowing earlier hospital discharge and resumption of daily activities. Primary PCI is also the best reperfusion option in patients who present late after the onset of symptoms and in patients with cardiogenic shock, and it is the only option in patients who have contraindications to fibrinolytic therapy because of bleeding risk.
However, most hospitals do not have PCI capability. Two options at these hospitals are to transfer the patient to a PCI center quickly for primary PCI or to keep the patient on site and give fibrinolytic therapy, with its limitations. Earlier trials suggested that the transfer strategy was superior, but they had limitations: most patients received streptokinase, an inferior fibrinolytic agent, and door-to-door-to-balloon times were rapid, averaging only 95 minutes because of excellent logistical protocols and careful patient selection.2 Most importantly, rescue PCI and routine PCI were seldom performed in patients receiving fibrinolytics, so fibrinolytic therapy was being tested as monotherapy.
In the real world, however, treatment delays are much longer, and fibrinolytic therapy has evolved into a strategy that includes crossover to rescue PCI or routine PCI. Therefore, the initial trials of transfer for primary PCI do not reflect current practice. In fact, recent registry data suggest that prehospital fibrinolytic therapy followed by early angiography is superior to primary PCI because most patients can be treated within 2 hours of symptom onset; they also suggest that on-site fibrinolytic therapy followed by early angiography is equal in efficacy to primary PCI as long as rescue PCI and routine PCI can be performed.3,4
The most important modifiable predictor of outcome in ST-elevation MI is the time to treatment, a biological truth that continues to be supported by clinical evidence despite ideologic arguments by some interventional cardiology enthusiasts who claim that primary PCI is always superior to the fibrinolytic strategy, regardless of delays.
SURPRISINGLY, OUTCOMES WERE WORSE WITH FACILITATED PCI
It made sense, then, to conclude that the perfect strategy for hospitals without PCI capability would be a combined strategy of immediate fibrinolytic therapy to decrease the time delay associated with organizing PCI, and rapid transfer for immediate PCI to improve the limited reperfusion rates associated with fibrinolytic therapy.
Surprisingly, though, randomized trials found worse outcomes with this “facilitated PCI” strategy.5
Again, limitations in trial design might explain the lack of benefit in the trials. Inadequate anticoagulant and antiplatelet therapy were given to the fibrinolytic patients, and primary PCI patients had relatively short treatment delays, with many patients enrolled at hospitals with PCI capability.
PROGRESS IN REPERFUSION THERAPY
Great strides have been made in reperfusion therapy in recent years. Adjunctive therapy with clopidogrel (Plavix) and enoxaparin (Lovenox) has been shown to improve outcomes with fibrinolytic therapy. Bivalirudin (Angiomax) and stents have improved primary PCI’s performance. Reducing bleeding complications has become a clinical priority, with increasing emphasis on adjusting some drug doses according to renal function and using the radial artery for cardiac catheterization.
The American College of Cardiology initiative, “Door-to-Balloon (D2B): An Alliance for Quality,” focused much attention on organizing in-hospital systems of care for primary PCI, thus increasing the national rate of achieving a door-to-balloon time within 90 minutes from 50% to over 75% in patients who presented to hospitals with PCI capability.6
The American Heart Association has launched “Mission: Lifeline,” a national campaign to organize prehospital systems of care with their program,7 working within communities to address their unique needs, resources, and barriers to implementing systems of care for ST-elevation MI. The key aspect of this effort is to help geographic regions develop local solutions, an explicit recognition that there is no one-size-fits-all solution. Early triage by emergency medical services, rapid diagnosis with prehospital electrocardiography, destination and interhospital transfer protocols, and prehospital activation of the cardiac catheterization laboratory can greatly streamline emergency care and decrease treatment delays for primary PCI.
FOR OUTLYING HOSPITALS, A PHARMACOINVASIVE STRATEGY
So what about hospitals without PCI capability that cannot routinely transfer patients to a hospital with PCI capability within 90 minutes?
Lessons learned from the experiences with immediate PCI, rescue PCI, and facilitated PCI have evolved into the “pharmacoinvasive strategy.” Patients with ST-elevation MI are treated as rapidly as possible with a bolus of a fibrinolytic drug, eg, tenecteplase (TNKase) or reteplase (Retavase), and are also given aspirin, clopidogrel, and enoxaparin. Then, they are rapidly transferred to a PCI-capable hospital so that emergency PCI can be performed if reperfusion is not clinically apparent or if the patient develops pulmonary edema or cardiogenic shock. If the clinical signs suggest that reperfusion has been achieved (relief of chest pain, rapid resolution of ST-segment elevation, bursts of accelerated idioventricular rhythm), coronary angiography (and PCI, if indicated) can be performed within 3 to 24 hours of fibrinolytic therapy. This time frame allows the initial fibrinolytic effect to dissipate, while the antiplatelet and anticoagulant drugs achieve therapeutic levels.
Today, the goal is to treat every patient with the best reperfusion strategy available, given the limitations in resources and the geographic location of some centers, and to maximize the possibility of sustained patency of the infarct-related artery by implanting a stent, even if it takes several hours and transfer to another hospital to perform PCI.8 The pharmacoinvasive strategy of rapid administration of fibrinolytic therapy followed by PCI within 24 hours would be practical in most hospitals without PCI capability where treatment delays prohibit performance of primary PCI within 90 minutes of first medical contact.9
THE ‘STREAM’ TRIAL IS UNDER WAY
As proof of concept, the Strategic Reperfusion Early After Myocardial Infarction (STREAM) trial is enrolling 2,000 patients with ST-elevation MI presenting within 3 hours of symptom onset if primary PCI is not feasible within 60 minutes of first medical contact.10 Patients will be randomized to either of the following:
- Receive prehospital therapy with tenecteplase, aspirin, clopidogrel, and enoxaparin and undergo cardiac catheterization in 6 to 24 hours (or rescue PCI if reperfusion fails within 90 minutes of fibrinolysis)
- Undergo primary PCI performed according to local guidelines.
The primary measure of efficacy will be the composite rate of death, cardiogenic shock, heart failure, and reinfarction at 30 days. Measures of safety include the rates of ischemic stroke, intracranial hemorrhage, and major nonintracranial bleeding.
Reperfusion therapy decreases morbidity and mortality rates in patients with ST-segment elevation myocardial infarction (MI). Primary percutaneous coronary intervention (PCI) is preferred over fibrinolytic therapy as a reperfusion strategy when the delay in the time to treatment is short and the patient presents to a high-volume, well-equipped center with expert interventional cardiologists.
Compared with fibrinolytic therapy in randomized clinical trials, primary PCI produces higher rates of infarct artery patency, complete reperfusion (grade 3 by the criteria of the Thrombolysis in Myocardial Infarction [TIMI] study), and access-site bleeding. It also produces lower rates of recurrent ischemia, reinfarction, emergency repeat revascularization procedures, intracranial hemorrhage, and death.1 If performed early and successfully, primary PCI also greatly decreases the rates of complications of ST-elevation MI that result from longer ischemic times or unsuccessful fibrinolytic therapy, allowing earlier hospital discharge and resumption of daily activities. Primary PCI is also the best reperfusion option in patients who present late after the onset of symptoms and in patients with cardiogenic shock, and it is the only option in patients who have contraindications to fibrinolytic therapy because of bleeding risk.
However, most hospitals do not have PCI capability. Two options at these hospitals are to transfer the patient to a PCI center quickly for primary PCI or to keep the patient on site and give fibrinolytic therapy, with its limitations. Earlier trials suggested that the transfer strategy was superior, but they had limitations: most patients received streptokinase, an inferior fibrinolytic agent, and door-to-door-to-balloon times were rapid, averaging only 95 minutes because of excellent logistical protocols and careful patient selection.2 Most importantly, rescue PCI and routine PCI were seldom performed in patients receiving fibrinolytics, so fibrinolytic therapy was being tested as monotherapy.
In the real world, however, treatment delays are much longer, and fibrinolytic therapy has evolved into a strategy that includes crossover to rescue PCI or routine PCI. Therefore, the initial trials of transfer for primary PCI do not reflect current practice. In fact, recent registry data suggest that prehospital fibrinolytic therapy followed by early angiography is superior to primary PCI because most patients can be treated within 2 hours of symptom onset; they also suggest that on-site fibrinolytic therapy followed by early angiography is equal in efficacy to primary PCI as long as rescue PCI and routine PCI can be performed.3,4
The most important modifiable predictor of outcome in ST-elevation MI is the time to treatment, a biological truth that continues to be supported by clinical evidence despite ideologic arguments by some interventional cardiology enthusiasts who claim that primary PCI is always superior to the fibrinolytic strategy, regardless of delays.
SURPRISINGLY, OUTCOMES WERE WORSE WITH FACILITATED PCI
It made sense, then, to conclude that the perfect strategy for hospitals without PCI capability would be a combined strategy of immediate fibrinolytic therapy to decrease the time delay associated with organizing PCI, and rapid transfer for immediate PCI to improve the limited reperfusion rates associated with fibrinolytic therapy.
Surprisingly, though, randomized trials found worse outcomes with this “facilitated PCI” strategy.5
Again, limitations in trial design might explain the lack of benefit in the trials. Inadequate anticoagulant and antiplatelet therapy were given to the fibrinolytic patients, and primary PCI patients had relatively short treatment delays, with many patients enrolled at hospitals with PCI capability.
PROGRESS IN REPERFUSION THERAPY
Great strides have been made in reperfusion therapy in recent years. Adjunctive therapy with clopidogrel (Plavix) and enoxaparin (Lovenox) has been shown to improve outcomes with fibrinolytic therapy. Bivalirudin (Angiomax) and stents have improved primary PCI’s performance. Reducing bleeding complications has become a clinical priority, with increasing emphasis on adjusting some drug doses according to renal function and using the radial artery for cardiac catheterization.
The American College of Cardiology initiative, “Door-to-Balloon (D2B): An Alliance for Quality,” focused much attention on organizing in-hospital systems of care for primary PCI, thus increasing the national rate of achieving a door-to-balloon time within 90 minutes from 50% to over 75% in patients who presented to hospitals with PCI capability.6
The American Heart Association has launched “Mission: Lifeline,” a national campaign to organize prehospital systems of care with their program,7 working within communities to address their unique needs, resources, and barriers to implementing systems of care for ST-elevation MI. The key aspect of this effort is to help geographic regions develop local solutions, an explicit recognition that there is no one-size-fits-all solution. Early triage by emergency medical services, rapid diagnosis with prehospital electrocardiography, destination and interhospital transfer protocols, and prehospital activation of the cardiac catheterization laboratory can greatly streamline emergency care and decrease treatment delays for primary PCI.
FOR OUTLYING HOSPITALS, A PHARMACOINVASIVE STRATEGY
So what about hospitals without PCI capability that cannot routinely transfer patients to a hospital with PCI capability within 90 minutes?
Lessons learned from the experiences with immediate PCI, rescue PCI, and facilitated PCI have evolved into the “pharmacoinvasive strategy.” Patients with ST-elevation MI are treated as rapidly as possible with a bolus of a fibrinolytic drug, eg, tenecteplase (TNKase) or reteplase (Retavase), and are also given aspirin, clopidogrel, and enoxaparin. Then, they are rapidly transferred to a PCI-capable hospital so that emergency PCI can be performed if reperfusion is not clinically apparent or if the patient develops pulmonary edema or cardiogenic shock. If the clinical signs suggest that reperfusion has been achieved (relief of chest pain, rapid resolution of ST-segment elevation, bursts of accelerated idioventricular rhythm), coronary angiography (and PCI, if indicated) can be performed within 3 to 24 hours of fibrinolytic therapy. This time frame allows the initial fibrinolytic effect to dissipate, while the antiplatelet and anticoagulant drugs achieve therapeutic levels.
Today, the goal is to treat every patient with the best reperfusion strategy available, given the limitations in resources and the geographic location of some centers, and to maximize the possibility of sustained patency of the infarct-related artery by implanting a stent, even if it takes several hours and transfer to another hospital to perform PCI.8 The pharmacoinvasive strategy of rapid administration of fibrinolytic therapy followed by PCI within 24 hours would be practical in most hospitals without PCI capability where treatment delays prohibit performance of primary PCI within 90 minutes of first medical contact.9
THE ‘STREAM’ TRIAL IS UNDER WAY
As proof of concept, the Strategic Reperfusion Early After Myocardial Infarction (STREAM) trial is enrolling 2,000 patients with ST-elevation MI presenting within 3 hours of symptom onset if primary PCI is not feasible within 60 minutes of first medical contact.10 Patients will be randomized to either of the following:
- Receive prehospital therapy with tenecteplase, aspirin, clopidogrel, and enoxaparin and undergo cardiac catheterization in 6 to 24 hours (or rescue PCI if reperfusion fails within 90 minutes of fibrinolysis)
- Undergo primary PCI performed according to local guidelines.
The primary measure of efficacy will be the composite rate of death, cardiogenic shock, heart failure, and reinfarction at 30 days. Measures of safety include the rates of ischemic stroke, intracranial hemorrhage, and major nonintracranial bleeding.
- Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet 2003; 361:13–20.
- Dalby M, Bouzamondo A, Lechat P, Montalescot G. Transfer for primary angioplasty versus immediate thrombolysis in acute myocardial infarction: a meta-analysis. Circulation 2003; 108:1809–1814.
- Danchin N, Coste P, Ferrières J, et al; FAST-MI Investigators. Comparison of thrombolysis followed by broad use of percutaneous coronary intervention with primary percutaneous coronary intervention for ST-segment-elevation acute myocardial infarction: data from the French registry on Acute ST-elevation Myocardial Infarction (FASTMI). Circulation 2008; 118:268–276.
- Lambert L, Brown K, Segal E, Brophy J, Rodes-Cabau J, Bogaty P. Association between timeliness of reperfusion therapy and clinical outcomes in ST-elevation myocardial infarction. JAMA 2010; 303:2148–2155.
- Keeley EC, Boura JA, Grines CL. Comparison of primary and facilitated percutaneous coronary interventions for ST-elevation myocardial infarction: quantitative review of randomised trials. Lancet 2006; 367:579–588.
- Krumholz HM, Bradley EH, Nallamothu BK, et al. A campaign to improve the timeliness of primary percutaneous coronary intervention: Door-to-Balloon: An Alliance for Quality. JACC Cardiovasc Interv. 2008; 1:97–104.
- Jacobs AK, Antman EM, Faxon DP, Gregory T, Solis P. Development of systems of care for ST-elevation myocardial infarction patients: executive summary. Circulation 2007; 116:217–230.
- Kushner FG, Hand M, Smith SC, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2009 Focused Updates: ACC/AHA Guidelines for the Management of Patients With ST-Elevation Myocardial Infarction (updating the 2004 Guideline and 2007 Focused Update) and ACC/AHA/SCAI Guidelines on Percutaneous Coronary Intervention (updating the 2005 Guideline and 2007 Focused Update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2009; 120:2271–2306.
- Bates ER, Nallamothu BK. Commentary: the role of percutaneous coronary intervention in ST-segment-elevation myocardial infarction. Circulation 2008; 118:567–573.
- Armstrong PW, Gershlick A, Goldstein P, et al; STREAM Steering Committee. The Strategic Reperfusion Early After Myocardial Infarction (STREAM) study. Am Heart J 2010; 160:30–35.
- Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet 2003; 361:13–20.
- Dalby M, Bouzamondo A, Lechat P, Montalescot G. Transfer for primary angioplasty versus immediate thrombolysis in acute myocardial infarction: a meta-analysis. Circulation 2003; 108:1809–1814.
- Danchin N, Coste P, Ferrières J, et al; FAST-MI Investigators. Comparison of thrombolysis followed by broad use of percutaneous coronary intervention with primary percutaneous coronary intervention for ST-segment-elevation acute myocardial infarction: data from the French registry on Acute ST-elevation Myocardial Infarction (FASTMI). Circulation 2008; 118:268–276.
- Lambert L, Brown K, Segal E, Brophy J, Rodes-Cabau J, Bogaty P. Association between timeliness of reperfusion therapy and clinical outcomes in ST-elevation myocardial infarction. JAMA 2010; 303:2148–2155.
- Keeley EC, Boura JA, Grines CL. Comparison of primary and facilitated percutaneous coronary interventions for ST-elevation myocardial infarction: quantitative review of randomised trials. Lancet 2006; 367:579–588.
- Krumholz HM, Bradley EH, Nallamothu BK, et al. A campaign to improve the timeliness of primary percutaneous coronary intervention: Door-to-Balloon: An Alliance for Quality. JACC Cardiovasc Interv. 2008; 1:97–104.
- Jacobs AK, Antman EM, Faxon DP, Gregory T, Solis P. Development of systems of care for ST-elevation myocardial infarction patients: executive summary. Circulation 2007; 116:217–230.
- Kushner FG, Hand M, Smith SC, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2009 Focused Updates: ACC/AHA Guidelines for the Management of Patients With ST-Elevation Myocardial Infarction (updating the 2004 Guideline and 2007 Focused Update) and ACC/AHA/SCAI Guidelines on Percutaneous Coronary Intervention (updating the 2005 Guideline and 2007 Focused Update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2009; 120:2271–2306.
- Bates ER, Nallamothu BK. Commentary: the role of percutaneous coronary intervention in ST-segment-elevation myocardial infarction. Circulation 2008; 118:567–573.
- Armstrong PW, Gershlick A, Goldstein P, et al; STREAM Steering Committee. The Strategic Reperfusion Early After Myocardial Infarction (STREAM) study. Am Heart J 2010; 160:30–35.
Combined reperfusion strategies in ST-segment elevation MI: Rationale and current role
Effective and rapid reperfusion is crucial in patients with acute ST-segment elevation myocardial infarction (MI). The preferred strategy for reperfusion—when it can be performed in a timely fashion at an experienced facility—is primary percutaneous coronary intervention (PCI), which produces outcomes superior to those of pharmacologic thrombolysis.1
Unfortunately, in the United States about half of patients present to hospitals that do not have PCI capability,2 and in one analysis, 91% of transferred patients had a door-to-balloon time greater than the recommended 90 minutes, with a mean of 152 minutes.3 (In this case, the door-to-balloon time was the time that elapsed between entry into the first hospital and inflation of the PCI balloon at the second hospital.)
In situations such as these, a combined approach may be appropriate, with thrombolysis delivered by paramedics or at a local facility, followed by transfer to a PCI facility and performance of PCI within a few hours. However, this is feasible only if standardized community-based or regional protocols for prompt transfer and reperfusion are in place.
In this paper we discuss the rationale and the clinical data behind several approaches to combined reperfusion, as well as experiences with community-based care protocols.
WITHIN 3 HOURS OF SYMPTOM ONSET, THROMBOLYSIS IS AS GOOD AS PCI
The PRAGUE-2 Trial
In the randomized PRAGUE-2 trial,4 patients with ST-elevation MI who presented to a non-PCI facility had better outcomes if they were transferred promptly for PCI (median door-to-balloon time 97 minutes), as opposed to receiving local therapy with streptokinase. However, for patients presenting within 3 hours of symptom onset, the mortality rates were comparable with either strategy.4
See the glossary of clinical trial names below
The CAPTIM trial
In the CAPTIM trial,5 patients who presented within 2 hours of symptom onset and who were randomized to receive prehospital thrombolysis had outcomes similar to those of patients treated with primary PCI, despite a short door-to-balloon time (82 minutes).
The Vienna STEMI Registry
In the Vienna STEMI Registry,6 the mortality rates with primary PCI and with thrombolysis were similar when patients presented within 2 hours of symptom onset. However, as the time from symptom onset increased, primary PCI appeared to offer an increasing survival benefit compared with thrombolysis.
Comments: Thrombolysis is effective mostly in the first 2 to 3 hours, with some benefit up to 12 hours
Previous studies have shown that the sooner thrombolysis is given after symptom onset, the more effective it is. If it is given within an hour of symptom onset, the relative reduction in the mortality rate is 50% and the absolute reduction is 6.5% compared with no reperfusion therapy. If it is started in the second hour, the absolute reduction in the mortality rate drops to 4%, and a lesser benefit extends to patients presenting up to 12 hours after symptom onset.7 This time-dependent benefit is due to the fact that very early reperfusion of the occluded coronary artery may lead to full recovery of ischemic tissue and thus prevent necrosis. In addition, thrombolysis in the first 2 hours is highly efficacious in lysing a fresh thrombus.
These data support the current guidelines of the American College of Cardiology (ACC) and the American Heart Association (AHA), which state no preference for either thrombolytic therapy or PCI in ST-elevation MI if the presentation is less than 3 hours after symptom onset.8
Of note, in the CAPTIM trial and in the Vienna STEMI Registry, rescue PCI was available and was in fact used after thrombolysis in about 25% of patients, which might have contributed to the benefit of early thrombolysis.

PRIMARY PCI MAY NOT BE SUPERIOR IF TRANSFER TIME IS LONG
Another time-related factor to consider is the PCI-related delay, ie, the theoretical difference between the expected time from first medical contact to balloon inflation (if the patient undergoes primary PCI) and the time from first medical contact to the start of thrombolytic therapy (if the patient undergoes primary thrombolysis).
A meta-analysis of 13 trials comparing PCI and thrombolysis showed that a PCI-related delay of more than 60 minutes might negate the potential advantage of primary PCI over immediate thrombolysis in terms of deaths.9
This observation has been further refined by data from the National Registry of Myocardial Infarction.10 In this analysis, patient factors, including age, duration of symptoms, and infarct location, significantly affected the point at which the PCI-related delay negated the survival advantage of primary PCI. The survival advantage of primary PCI was lost more rapidly—with a PCI-related delay as short as 40 minutes—in patients who presented sooner, were younger, or had anterior MI. Primary PCI maintained its survival advantage even with a PCI-related delay longer than 100 minutes in older patients or patients with nonanterior MI presenting more than 3 hours after symptom onset. Given that median door-to-balloon times in the United States may exceed 150 minutes when transfer is involved, 3 primary PCI may be no better than primary thrombolysis in transferred patients who present early or who have large infarcts.
Although these results were derived from a post hoc analysis of a registry and the delay times reported were sometimes inaccurate, they suggest that both the PCI-related delay time and patient characteristics should be considered when selecting a reperfusion strategy. Thrombolytic therapy before and in conjunction with primary PCI was considered a potential solution to these concerns.
In addition, while the benefit of any reperfusion strategy depends on the time of presentation, the loss in benefit by later presentation is less pronounced with primary PCI than with thrombolysis, making thrombolysis less attractive in later presentations (> 3 hours).11
Also, while thrombolytic therapy in patients older than 75 years was associated with a lower mortality rate compared with no therapy in a large Swedish registry,12 this benefit was less striking than in younger patients. A meta-analysis of thrombolysis trials failed to show a similar benefit in patients over age 75 vs younger patients,13 whereas primary PCI remained effective and superior to thrombolysis in the elderly, with more absolute reduction in mortality rates in the elderly subgroup than with younger patients. 14 This makes thrombolysis less attractive in the elderly, either as a stand-alone therapy or in conjunction with PCI. Studies of combined thrombolysis and PCI included very few patients over age 75.15–17
THREE COMBINATION REPERFUSION STRATEGIES
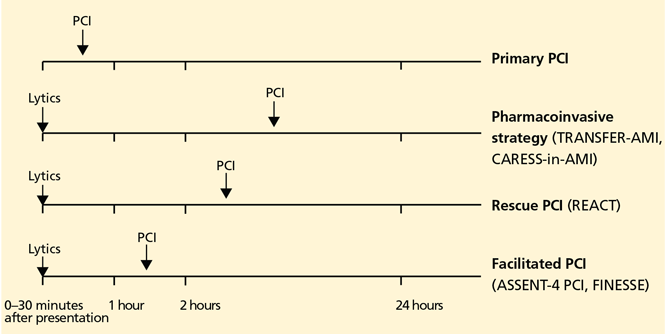
Facilitated PCI is a strategy of thrombolysis immediately followed by PCI, with a planned door-to-balloon time of 90 to 120 minutes.
Pharmacoinvasive therapy means giving thrombolysis at a non-PCI facility and then promptly and systematically transferring the patient to a PCI facility, where PCI is performed 2 to 24 hours after the start of thrombolytic therapy, regardless of whether thrombolysis results in successful reperfusion. 15 Thus, the time to PCI is longer than with facilitated PCI. Facilitated PCI addresses the value of pretreatment with thrombolytics or glycoprotein IIb/IIIa inhibitors in patients otherwise eligible for primary PCI, whereas pharmacoinvasive therapy addresses the value of routine early PCI after thrombolysis in patients who are not eligible for primary PCI.16
Rescue PCI refers to PCI that is performed urgently if thrombolysis fails, failure being defined as persistent hemodynamic or electrical instability, persistent ischemic symptoms, or failure to achieve at least a 50% to 70% resolution of the maximal ST-segment elevation 90 minutes after the infusion is started.
FACILITATED PCI: NEGATIVE RESULTS IN CLINICAL TRIALS
ASSENT-4 PCI trial
In the ASSENT-4 PCI trial,18 patients receiving full thrombolytic therapy before PCI had a higher rate of in-hospital death, bleeding, and cardiovascular events at 90 days than patients treated with primary PCI.
This trial recruited patients arriving at hospitals with or without PCI capability. The door-to-balloon time was about 110 minutes in both groups, which might not have been prolonged enough to show a benefit from a timely addition of thrombolysis. In addition, antiplatelet therapy was limited in these patients: glycoprotein IIb/IIIa inhibitors were not given, and clopidogrel (Plavix) was not appropriately preloaded, and this might have offset the potential benefit of early PCI. In fact, data suggest that platelet activation and aggregation are heightened after thrombolysis, 21–23 and that glycoprotein IIb/IIIa antagonists can inhibit these effects.23
The FINESSE trial
In the FINESSE trial,19 patients were randomized to undergo primary PCI, to undergo PCI facilitated (ie, preceded) by abciximab (Reo-Pro), or to undergo PCI facilitated by half-dose reteplase (Retavase) and full-dose abciximab. Despite a median door-to-balloon time of 132 minutes, the three strategies were associated with similar rates of death, heart failure, or ischemic outcome at 90 days. Even though the dosage of heparin was weight-adjusted, more major bleeding events occurred with the facilitated strategies.
Comments: Some subgroups may still benefit from facilitated PCI
The results of ASSENT-4 PCI and FINESSE led to the conclusion that PCI facilitated by full-dose thrombolysis should be avoided, and called into question the value of PCI facilitation using glycoprotein IIb/IIIa inhibitors with or without half-dose thrombolytic therapy.
However, subgroup analyses of these trials identified some subgroups that may benefit from a facilitated strategy. In ASSENT-4 PCI, 45% of patients were enrolled at PCI hospitals with a minimal PCI-related delay time. These patients had the worst outcome with the facilitated strategy. In contrast, patients who had a short time from pain onset to thrombolysis (2 to 3 hours) and who were given prehospital thrombolysis had a trend toward better outcomes with facilitated PCI.24 And in FINESSE, 60% of patients were enrolled at centers with PCI capability. Analysis of a small subgroup of patients with a Thrombolysis in Myocardial Infarction study (TIMI) risk score of 3 or greater presenting to non-PCI hospitals within 4 hours of symptom onset suggested a potential reduction of ischemic events with the facilitated strategy in these patients.25
Thus, for patients seen in the first 2 to 3 hours after symptom onset, immediate thrombolysis is recommended if PCI will likely be delayed, with or without plans for subsequent early PCI. “Time is muscle,” especially during the first 3 hours.
PHARMACOINVASIVE STRATEGY: GOOD RESULTS IN HIGH-RISK PATIENTS
A number of randomized studies during the last 10 years have examined the value of a pharmacoinvasive strategy.15,16,26–29
The TRANSFER-AMI trial
The TRANSFER-AMI trial15 randomized 1,059 patients with high-risk ST-elevation MI (ie, anterior or high-risk inferior) at non-PCI centers to undergo either pharmacoinvasive care, ie, full-dose tenecteplase (TNKase) with immediate transfer for PCI or standard care, ie, tenecteplase with transfer for rescue PCI if the patient had persistent ST-segment elevation, chest pain, or hemodynamic instability.15 The goal was to perform PCI within 6 hours of thrombolysis, and the median time to PCI was 3.9 hours (range 2–6 hours). In the standard-care group, 35% of patients needed to be transferred for rescue PCI. Unlike in the ASSENT-4 trial, over 80% of patients received aggressive antiplatelet therapy with both 300 mg of clopidogrel and glycoprotein IIb/IIIa inhibitors.
The rate of cardiovascular events at 30 days was significantly lower with pharmacoinvasive therapy than with standard care and rescue PCI (11% vs 17%, P = .004). This difference was driven by lower rates of recurrent ischemia, reinfarction, and heart failure.
The CARESS-in-AMI study
The CARESS-in-AMI study16 found a similar improvement in ischemic outcomes in 600 patients with high-risk ST-elevation MI arriving at non-PCI centers if they had received pharmacoinvasive therapy. Patients received half-dose reteplase and abciximab and were randomized either to be immediately transferred for PCI (median time to PCI 2.25 hours) or to be transferred only if they had persistent ST-segment elevation or clinical deterioration.16 The event rate was low with pharmacoinvasive therapy, comparable to that achieved in primary PCI trials.
Interestingly, no significant increase was seen in the risk of major and minor bleeding in these two trials despite the use of a femoral approach for PCI in over 80% of the cases; this is probably due to the delays between thrombolytic administration and PCI and to the use of a highly fibrin-specific thrombolytic agent and adjusted-dose heparin.
Meta-analysis of pharmacoinvasive trials
A meta-analysis29 of studies of systematic early PCI (mainly with stenting) within 24 hours of thrombolysis showed a reduction in the rates of mortality and reinfarction with this strategy, without an increase in the risk of major or intracranial bleeding.30 In contrast to the results of the trials of facilitated PCI, a pharmacoinvasive strategy improved outcomes in these trials because the delay between thrombolysis and PCI was more than 2 hours, ie, long enough to prevent bleeding complications, and because most patients randomized in these trials presented within 2 to 3 hours of symptom onset, when the time to reperfusion is critical. After 3 hours, the PCI-mediated myocardial salvage is less time-dependent. Moreover, trials of pharmacoinvasive strategy used aggressive antiplatelet therapy with clopidogrel and glycoprotein IIb/IIIa inhibitors.
Comment: Pharmacoinvasive strategy in the guidelines
These results and those of the subgroup analysis from the FINESSE trial suggest that patients with high-risk ST-elevation MI treated at non-PCI hospitals have better outcomes without an increase in major bleeding events when given thrombolysis and then immediately transferred for routine PCI, rather than being transferred only if reperfusion fails.
Hence, the 2009 update of the ACC/AHA guidelines31 gives a class IIa recommendation for transferring patients with anterior ST-elevation MI or high-risk inferior ST-elevation MI treated with thrombolysis to a PCI-capable facility where PCI is performed as part of a pharmacoinvasive or rescue strategy soon after thrombolysis.
This strategy has been particularly studied in patients younger than 75 years presenting with high-risk types of ST-elevation MI early (< 3 hours) after symptom onset. If not at high risk, the patient may be transferred to a PCI facility after receiving thrombolysis or observed in the initial facility (class IIb recommendation). Consideration should be given to starting anticoagulant and antiplatelet therapy before and during transfer—ie, 300 mg of clopidogrel before transfer for PCI and glycoprotein IIb/IIIa inhibitor therapy during PCI.
The European Society of Cardiology (ESC) guidelines32 recommend early routine angiography 3 to 24 hours after successful thrombolysis. This time window was selected to avoid PCI during the prothrombotic period in the first few hours after thrombolysis and to minimize the risk of reocclusion with PCI delays of more than 24 hours (class IIa recommendation).
Larger randomized trials are still needed to establish whether the pharmacoinvasive strategy confers a survival benefit, to determine its usefulness in low-risk inferior or lateral ST-elevation MI, and to further refine the time window when PCI is both safe and beneficial after thrombolysis.33
RESCUE PCI REDUCES MORTALITY RATES
Rescue PCI is the most accepted form of thrombolysis-PCI combination.
The REACT trial
The REACT trial20 showed that rescue PCI performed at a mean of 4.5 hours after failed thrombolysis reduces the rate of adverse cardiovascular events by more than 50% at 6 to 12 months and reduces the 5-year mortality rate by more than 50% compared with conservative management.20 As in the pharmacoinvasive strategy, aggressive antiplatelet regimens were used in the REACT trial.
A meta-analysis of rescue PCI trials
A meta-analysis of rescue PCI trials34 confirmed these results, showing a reduction in heart failure and reinfarction and a trend toward a lower mortality rate with rescue PCI.34 After thrombolysis, 40% of patients do not achieve grade 3 TIMI flow, which explains why in modern clinical trials 30% of patients treated with thrombolysis require rescue PCI.5,15,16,35
For patients with high-risk ST-elevation MI, current ACC/AHA guidelines assign a class IIa recommendation to rescue PCI.31
WHEN PATIENTS WITH ST-ELEVATION MI PRESENT TO A NON-PCI HOSPITAL
Transfer for primary PCI vs thrombolysis at the non-PCI hospital
The DANAMI-2 trial36 found that immediate transfer for PCI was superior to onsite thrombolytic therapy, as measured by a reduction in the rate of ischemic events (composite of death, myocardial infarction, or stroke at 30 days): 8.5% vs 14.2% (P < .001). There were no deaths during transfer.3
The PRAGUE-2 trial4 showed similar results for patients presenting 3 to 12 hours after symptom onset (30-day mortality rate 6% with immediate transfer vs 15.3% with on-site thrombolysis, P < .002), whereas patients presenting within 3 hours of symptom onset had a similar mortality rate with either therapy.4
Comment. These trials showed that transfer for primary PCI is superior to thrombolytic therapy when performed in a timely fashion. However, they were done in countries with established transfer networks and short distances between community hospitals and PCI centers, with a PCI-related delay of only 44 minutes and a door-to-balloon time of 90 minutes despite transfer. The large-scale application of this prompt transfer policy is not practical in most regions in the United States. Thus, a strategy of local thrombolysis followed by routine early transfer for routine or rescue PCI seems warranted when the door-to-balloon time or the PCI-related delay time is expected to be too long.
Experiences with community-based systems of care and prehospital thrombolysis
In Minnesota, Henry et al37 developed a PCI-based treatment system and an integrated transfer program for ST-elevation MI involving 30 hospitals within 210 miles of the Minneapolis Heart Institute. Participating hospitals were divided into two zones: zone 1 hospitals were within 60 miles, and zone 2 facilities were between 60 and 210 miles from the Heart Institute. Zone 2 patients received half-dose tenecteplase (if thrombolytic therapy was not contraindicated) in anticipation of a lengthy transfer time.
The median door-to-balloon time for zone 1 patients was 95 minutes (interquartile range 82 and 116 minutes) and for zone 2 patients 120 minutes (interquartile range 100 and 145 minutes). The diagnosis of ST-elevation MI was made by the emergency department physician, who activated the system with a phone call. The patient was then directly transferred to the catheterization laboratory, most often by helicopter.
The in-hospital death rate for patients who presented to the PCI center and for patients in zones 1 and 2 was similarly low (about 5%).37
In France, the FAST-MI registry,17 which collected outcome data for different reperfusion strategies, found that thrombolysis yielded in-hospital and midterm results that were comparable to those of primary PCI. Of note, thrombolysis was started early after symptom onset (about 2 hours), and was started in the ambulance in two-thirds of cases. Nearly all patients underwent a pharmacoinvasive strategy that combined thrombolysis with coronary angiography and PCI within 24 hours of symptom onset. These findings suggest that timely thrombolysis followed by semiurgent transfer for PCI is an alternative to primary PCI for patients presenting to hospitals with no PCI capability, and that this alternative offers similar benefit to that of primary PCI.
Five centers in the United States have reported their experience with half-dose thrombolysis in the prehospital setting (in the field or during transfer) or at a non-PCI hospital, followed by prompt transfer to a PCI facility. In this registry of almost 3,000 patients,38 patients treated with thrombolysis had better outcomes than patients directly transferred for primary PCI, with a significantly lower 30-day mortality rate (3.8% vs from 6.4%), and no increase in bleeding.38,39 The mean door-to-balloon time was long (168 minutes in the primary PCI group and 196 minutes in the thrombolysis-PCI group), which might explain the benefit achieved with prompt thrombolysis.
CARDIOGENIC SHOCK
Patients presenting with left ventricular cardiogenic shock derive a large mortality benefit from revascularization, whether they are transferred or directly admitted to a PCI center. 40 Moreover, in the SHOCK registry, patients with predominant right ventricular cardiogenic shock had an in-hospital mortality rate similar to that of patients with predominant left ventricular cardiogenic shock, and revascularization (PCI or surgical revascularization) was associated with a strikingly lower mortality rate in both groups.41
Thus, all patients with left or right cardiogenic shock should be revascularized on an emergency basis, either surgically or percutaneously.
While trials of pharmacoinvasive therapy excluded patients with cardiogenic shock,15,16 thrombolytic therapy was associated with improved outcomes in the drug-therapy group of the SHOCK trial and in hypotensive patients randomized in the early thrombolysis trials.13 Thus, the ACC/AHA guidelines recommend thrombolytic therapy before transfer if a patient presents in shock within 3 to 6 hours of onset of the MI and delays in transport and intervention are anticipated.8
PUTTING IT ALL TOGETHER: MANAGEMENT STRATEGIES
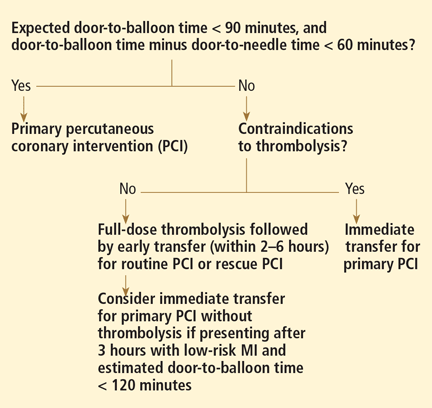
If an effective transfer system is in place, primary PCI not preceded by thrombolytic therapy or glycoprotein IIb/IIIa inhibitor therapy is the preferred approach, according to ACC/AHA and ESC guidelines.31,32 Giving thrombolytics immediately before PCI is harmful and thus should be avoided when the expected door-to-balloon time is 90 minutes or less.
All hospitals (whether or not they offer PCI) and regional emergency medical services should participate in a community-based system of care for ST-elevation MI, with protocols for expeditious transfer as defined and coordinated by the American Heart Association initiative “Mission: Lifeline.” In addition, a system of field triage and direct transport to the catheterization laboratory of a PCI facility after field activation significantly reduces door-to-balloon times and improves outcomes.42
If such a system is not in place, then a pharmacoinvasive strategy seems best: ie, local full-dose thrombolysis (if not contraindicated) followed by transfer to a PCI facility and routine performance of PCI 2 to 6 hours after thrombolysis—in conjunction with aggressive early dual oral antiplatelet therapy and “downstream” glycoprotein IIb/IIIa inhibition. This approach is associated with outcomes similar to those of primary PCI.15–17,37
Prehospital thrombolysis delivered by paramedics and followed by early transfer to a PCI facility has been associated with further reduction in mortality rates compared with in-hospital thrombolysis (as in the Swedish registry43), and a reduction in death rate comparable to that of primary PCI in patients presenting early. This is an adequate strategy in regions where such a system can be established.5,17,38,43,44
Patients presenting more than 3 to 4 hours after symptom onset, older patients, and patients with lower-risk MI or a higher risk of bleeding may still be suited for primary PCI even when the door-to-balloon time is 90 to 120 minutes, as stated by the European guidelines,32 or when the PCI-related delay time is as long as 100 minutes. 10 On the other hand, while the ACC/AHA guidelines recognize that in these patients the mortality advantage of primary PCI vs thrombolytic therapy is maintained with more prolonged door-to-balloon times, they nevertheless state that the focus should be on developing systems of care to increase the number of patients with access to primary PCI in less than 90 minutes rather than extending the acceptable window for door-to-balloon time.
In conclusion, for patients presenting with ST-elevation MI who cannot undergo timely primary PCI, the best approach seems to be prehospital thrombolysis delivered by paramedics or local thrombolysis at the non-PCI hospital followed by transferring the patient and performing PCI within a few hours. This is especially important in patients with high-risk ST-elevation MI who present early after symptom onset, when the extent of myocardial necrosis associated with delayed primary PCI is largest.
In addition, every community should develop a coordinated transfer strategy between non-PCI and PCI hospitals.
- Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet 2003; 361:13–20.
- Waters RE, Singh KP, Roe MT, et al. Rationale and strategies for implementing community-based transfer protocols for primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. J Am Coll Cardiol 2004; 43:2153–2159.
- Chakrabarti A, Krumholz HM, Wang Y, Rumsfeld JS, Nallamothu BK; National Cardiovascular Data Registry. Time-to-reperfusion in patients undergoing interhospital transfer for primary percutaneous coronary intervention in the U.S: an analysis of 2005 and 2006 data from the National Cardiovascular Data Registry. J Am Coll Cardiol 2008; 51:2442–2443.
- Widimský P, Budesínský T, Vorác D, et al; ‘PRAGUE’ Study Group Investigators. Long distance transport for primary angioplasty vs immediate thrombolysis in acute myocardial infarction. Final results of the randomized national multicentre trial—PRAGUE-2. Eur Heart J 2003; 24:94–104.
- Steg PG, Bonnefoy E, Chabaud S, et al; Comparison of Angioplasty and Prehospital Thrombolysis in Acute Myocardial infarction (CAPTIM) Investigators. Impact of time to treatment on mortality after prehospital fibrinolysis or primary angioplasty: data from the CAPTIM randomized clinical trial. Circulation 2003; 108:2851–2856.
- Kalla K, Christ G, Karnik R, et al; Vienna STEMI Registry Group. Implementation of guidelines improves the standard of care: the Viennese registry on reperfusion strategies in ST-elevation myocardial infarction (Vienna STEMI registry). Circulation 2006; 113:2398–2405.
- Boersma E, Maas AC, Deckers JW, Simoons ML. Early thrombolytic treatment in acute myocardial infarction: reappraisal of the golden hour. Lancet 1996; 348:771–775.
- Antman EM, Anbe DT, Armstrong PW, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines; Canadian Cardiovascular Society. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of Patients with Acute Myocardial Infarction). Circulation 2004; 110:e82–e292.
- Nallamothu BK, Bates ER. Percutaneous coronary intervention versus fibrinolytic therapy in acute myocardial infarction: is timing (almost) everything? Am J Cardiol 2003; 92:824–826.
- Pinto DS, Kirtane AJ, Nallamothu BK, et al. Hospital delays in reperfusion for ST-elevation myocardial infarction: implications when selecting a reperfusion strategy. Circulation 2006; 114:2019–2025.
- Boersma E; Primary Coronary Angioplasty vs Thrombolysis Group. Does time matter? A pooled analysis of randomized clinical trials comparing primary percutaneous coronary intervention and in-hospital fibrinolysis in acute myocardial infarction patients. Eur Heart J 2006; 27:779–788.
- Stenestrand U, Wallentin L; Register of Information and Knowledge About Swedish Heart Intensive Care Admissions (RIKS-HIA). Fibrinolytic therapy in patients 75 years and older with ST-segment-elevation myocardial infarction: one-year follow-up of a large prospective cohort. Arch Intern Med 2003; 163:965–971.
- Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group. Lancet 1994; 343:311–322.
- Grines CL, Browne KF, Marco J, et al. A comparison of immediate angioplasty with thrombolytic therapy for acute myocardial infarction. The Primary Angioplasty in Myocardial Infarction Study Group. N Engl J Med 1993; 328:673–679.
- Cantor WJ, Fitchett D, Borgundvaag B, et al; TRANSFER-AMI Trial Investigators. Routine early angioplasty after fibrinolysis for acute myocardial infarction. N Engl J Med 2009; 360:2705–2718.
- Di Mario C, Dudek D, Piscione F, et al; CARESS-in-AMI (Combined Abciximab RE-teplase Stent Study in Acute Myocardial Infarction) Investigators. Immediate angioplasty versus standard therapy with rescue angioplasty after thrombolysis in the Combined Abciximab REteplase Stent Study in Acute Myocardial Infarction (CARESS-in-AMI): an open, prospective, randomised, multicentre trial. Lancet 2008; 371:559–568.
- Danchin N, Coste P, Ferrières J, et al; FAST-MI Investigators. Comparison of thrombolysis followed by broad use of percutaneous coronary intervention with primary percutaneous coronary intervention for ST-segment-elevation acute myocardial infarction: data from the French registry on Acute ST-elevation Myocardial Infarction (FAST-MI). Circulation 2008; 118:268–276.
- Assessment of the Safety and Efficacy of a New Treatment Strategy with Percutaneous Coronary Intervention (ASSENT-4 PCI) investigators. Primary versus tenecteplase-facilitated percutaneous coronary intervention in patients with ST-segment elevation acute myocardial infarction (ASSENT-4 PCI): randomised trial. Lancet 2006; 367:569–578.
- Ellis SG, Tendera M, de Belder MA, et al; FINESSE Investigators. Facilitated PCI in patients with ST-elevation myocardial infarction. N Engl J Med 2008; 358:2205–2217.
- Carver A, Rafelt S, Gershlick AH, Fairbrother KL, Hughes S, Wilcox R; REACT Investigators. Longer-term follow-up of patients recruited to the REACT (Rescue Angioplasty Versus Conservative Treatment or Repeat Thrombolysis) trial. J Am Coll Cardiol 2009; 54:118–126.
- Rasmanis G, Vesterqvist O, Gréen K, Edhag O, Henriksson P. Evidence of increased platelet activation after thrombolysis in patients with acute myocardial infarction. Br Heart J 1992; 68:374–376.
- Gurbel PA, Serebruany VL, Shustov AR, et al. Effects of reteplase and alteplase on platelet aggregation and major receptor expression during the first 24 hours of acute myocardial infarction treatment. GUSTO-III Investigators. Global Use of Strategies to Open Occluded Coronary Arteries. J Am Coll Cardiol 1998; 31:1466–1473.
- Coulter SA, Cannon CP, Ault KA, et al. High levels of platelet inhibition with abciximab despite heightened platelet activation and aggregation during thrombolysis for acute myocardial infarction: results from TIMI (thrombolysis in myocardial infarction) 14. Circulation 2000; 101:2690–2695.
- Ross AM, Huber K, Zeymer U, et al. The impact of place of enrollment and delay to reperfusion on 90-day post-infarction mortality in the ASSENT-4 PCI trial: assessment of the safety and efficacy of a new treatment strategy with percutaneous coronary intervention. JACC Cardiovasc Interv 2009; 2:925–930.
- Herrmann HC, Lu J, Brodie BR, et al; FINESSE Investigators. Benefit of facilitated percutaneous coronary intervention in high-risk ST-segment elevation myocardial infarction patients presenting to nonpercutaneous coronary intervention hospitals. JACC Cardiovasc Interv 2009; 2:917–924.
- Scheller B, Hennen B, Hammer B, et al; SIAM III Study Group. Beneficial effects of immediate stenting after thrombolysis in acute myocardial infarction. J Am Coll Cardiol 2003; 42:634–641.
- Fernandez-Avilés F, Alonso JJ, Castro-Beiras A, et al; GRACIA (Grupo de Análisis de la Cardiopatía Isquémica Aguda) Group. Routine invasive strategy within 24 hours of thrombolysis versus ischaemiaguided conservative approach for acute myocardial infarction with ST-segment elevation (GRACIA-1): a randomised controlled trial. Lancet 2004; 364:1045–1053.
- Le May MR, Wells GA, Labinaz M, et al. Combined angioplasty and pharmacological intervention versus thrombolysis alone in acute myocardial infarction (CAPITAL AMI study). J Am Coll Cardiol 2005; 46:417–424.
- Bøhmer E, Hoffmann P, Abdelnoor M, Arnesen H, Halvorsen S. Efficacy and safety of immediate angioplasty versus ischemia-guided management after thrombolysis in acute myocardial infarction in areas with very long transfer distances results of the NORDISTEMI (NORwegian study on DIstrict treatment of ST-elevation myocardial infarction). J Am Coll Cardiol 2010; 55:102–110.
- Wijeysundera HC, You JJ, Nallamothu BK, Krumholz HM, Cantor WJ, Ko DT. An early invasive strategy versus ischemia-guided management after fibrinolytic therapy for ST-segment elevation myocardial infarction: a meta-analysis of contemporary randomized controlled trials. Am Heart J 2008; 156:564–572,572.e1–e2.
- Kushner FG, Hand M, Smith SC, et al. 2009 focused updates: ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction (updating the 2004 guideline and 2007 focused update) and ACC/AHA/SCAI guidelines on percutaneous coronary intervention (updating the 2005 guideline and 2007 focused update) a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2009; 54:2205–2241.
- Van de Werf F, Bax J, Betriu A, et al. Management of acute myocardial infarction in patients presenting with persistent ST-segment elevation: the Task Force on the Management of ST-Segment Elevation Acute Myocardial Infarction of the European Society of Cardiology. Eur Heart J 2008; 29:2909–2945.
- Mukherjee D, Moliterno DJ. The timely coupling of mechanical revascularization following thrombolysis for myocardial infarction. Cardiology 2007; 107:337–339.
- Wijeysundera HC, Vijayaraghavan R, Nallamothu BK, et al. Rescue angioplasty or repeat fibrinolysis after failed fibrinolytic therapy for ST-segment myocardial infarction: a meta-analysis of randomized trials. J Am Coll Cardiol 2007; 49:422–430.
- The GUSTO Angiographic Investigators. The effects of tissue plasminogen activator, streptokinase, or both on coronary-artery patency, ventricular function, and survival after acute myocardial infarction. N Engl J Med 1993; 329:1615–1622.
- Andersen HR, Nielsen TT, Rasmussen K, et al; DANAMI-2 Investigators. A comparison of coronary angioplasty with fibrinolytic therapy in acute myocardial infarction. N Engl J Med 2003; 349:733–742.
- Henry TD, Sharkey SW, Burke MN, et al. A regional system to provide timely access to percutaneous coronary intervention for ST-elevation myocardial infarction. Circulation 2007; 116:721–728.
- Denktas AE, Athar H, Henry TD, et al. Reduced-dose fibrinolytic acceleration of ST-segment elevation myocardial infarction treatment coupled with urgent percutaneous coronary intervention compared to primary percutaneous coronary intervention alone results of the AMICO (Alliance for Myocardial Infarction Care Optimization) Registry. JACC Cardiovasc Interv 2008; 1:504–510.
- Smalling RW. Ischemic time: the new gold standard for ST-segment elevation myocardial infarction care. J Am Coll Cardiol 2009; 54:2154–2156.
- Hochman JS, Sleeper LA, White HD, et al; SHOCK Investigators. Should We Emergently Revascularize Occluded Coronaries for Cardiogenic Shock. One-year survival following early revascularization for cardiogenic shock. JAMA 2001; 285:190–192.
- Jacobs AK, Leopold JA, Bates E, et al. Cardiogenic shock caused by right ventricular infarction: a report from the SHOCK registry. J Am Coll Cardiol 2003; 41:1273–1279.
- Pedersen SH, Galatius S, Hansen PR, et al. Field triage reduces treatment delay and improves long-term clinical outcome in patients with acute ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. J Am Coll Cardiol 2009; 54:2296–2302.
- Björklund E, Stenestrand U, Lindbäck J, Svensson L, Wallentin L, Lindahl B. Pre-hospital thrombolysis delivered by paramedics is associated with reduced time delay and mortality in ambulance-transported real-life patients with ST-elevation myocardial infarction. Eur Heart J 2006; 27:1146–1152.
- The European Myocardial Infarction Project Group. Prehospital thrombolytic therapy in patients with suspected acute myocardial infarction. N Engl J Med 1993; 329:383–389.
Effective and rapid reperfusion is crucial in patients with acute ST-segment elevation myocardial infarction (MI). The preferred strategy for reperfusion—when it can be performed in a timely fashion at an experienced facility—is primary percutaneous coronary intervention (PCI), which produces outcomes superior to those of pharmacologic thrombolysis.1
Unfortunately, in the United States about half of patients present to hospitals that do not have PCI capability,2 and in one analysis, 91% of transferred patients had a door-to-balloon time greater than the recommended 90 minutes, with a mean of 152 minutes.3 (In this case, the door-to-balloon time was the time that elapsed between entry into the first hospital and inflation of the PCI balloon at the second hospital.)
In situations such as these, a combined approach may be appropriate, with thrombolysis delivered by paramedics or at a local facility, followed by transfer to a PCI facility and performance of PCI within a few hours. However, this is feasible only if standardized community-based or regional protocols for prompt transfer and reperfusion are in place.
In this paper we discuss the rationale and the clinical data behind several approaches to combined reperfusion, as well as experiences with community-based care protocols.
WITHIN 3 HOURS OF SYMPTOM ONSET, THROMBOLYSIS IS AS GOOD AS PCI
The PRAGUE-2 Trial
In the randomized PRAGUE-2 trial,4 patients with ST-elevation MI who presented to a non-PCI facility had better outcomes if they were transferred promptly for PCI (median door-to-balloon time 97 minutes), as opposed to receiving local therapy with streptokinase. However, for patients presenting within 3 hours of symptom onset, the mortality rates were comparable with either strategy.4
See the glossary of clinical trial names below
The CAPTIM trial
In the CAPTIM trial,5 patients who presented within 2 hours of symptom onset and who were randomized to receive prehospital thrombolysis had outcomes similar to those of patients treated with primary PCI, despite a short door-to-balloon time (82 minutes).
The Vienna STEMI Registry
In the Vienna STEMI Registry,6 the mortality rates with primary PCI and with thrombolysis were similar when patients presented within 2 hours of symptom onset. However, as the time from symptom onset increased, primary PCI appeared to offer an increasing survival benefit compared with thrombolysis.
Comments: Thrombolysis is effective mostly in the first 2 to 3 hours, with some benefit up to 12 hours
Previous studies have shown that the sooner thrombolysis is given after symptom onset, the more effective it is. If it is given within an hour of symptom onset, the relative reduction in the mortality rate is 50% and the absolute reduction is 6.5% compared with no reperfusion therapy. If it is started in the second hour, the absolute reduction in the mortality rate drops to 4%, and a lesser benefit extends to patients presenting up to 12 hours after symptom onset.7 This time-dependent benefit is due to the fact that very early reperfusion of the occluded coronary artery may lead to full recovery of ischemic tissue and thus prevent necrosis. In addition, thrombolysis in the first 2 hours is highly efficacious in lysing a fresh thrombus.
These data support the current guidelines of the American College of Cardiology (ACC) and the American Heart Association (AHA), which state no preference for either thrombolytic therapy or PCI in ST-elevation MI if the presentation is less than 3 hours after symptom onset.8
Of note, in the CAPTIM trial and in the Vienna STEMI Registry, rescue PCI was available and was in fact used after thrombolysis in about 25% of patients, which might have contributed to the benefit of early thrombolysis.
PRIMARY PCI MAY NOT BE SUPERIOR IF TRANSFER TIME IS LONG
Another time-related factor to consider is the PCI-related delay, ie, the theoretical difference between the expected time from first medical contact to balloon inflation (if the patient undergoes primary PCI) and the time from first medical contact to the start of thrombolytic therapy (if the patient undergoes primary thrombolysis).
A meta-analysis of 13 trials comparing PCI and thrombolysis showed that a PCI-related delay of more than 60 minutes might negate the potential advantage of primary PCI over immediate thrombolysis in terms of deaths.9
This observation has been further refined by data from the National Registry of Myocardial Infarction.10 In this analysis, patient factors, including age, duration of symptoms, and infarct location, significantly affected the point at which the PCI-related delay negated the survival advantage of primary PCI. The survival advantage of primary PCI was lost more rapidly—with a PCI-related delay as short as 40 minutes—in patients who presented sooner, were younger, or had anterior MI. Primary PCI maintained its survival advantage even with a PCI-related delay longer than 100 minutes in older patients or patients with nonanterior MI presenting more than 3 hours after symptom onset. Given that median door-to-balloon times in the United States may exceed 150 minutes when transfer is involved, 3 primary PCI may be no better than primary thrombolysis in transferred patients who present early or who have large infarcts.
Although these results were derived from a post hoc analysis of a registry and the delay times reported were sometimes inaccurate, they suggest that both the PCI-related delay time and patient characteristics should be considered when selecting a reperfusion strategy. Thrombolytic therapy before and in conjunction with primary PCI was considered a potential solution to these concerns.
In addition, while the benefit of any reperfusion strategy depends on the time of presentation, the loss in benefit by later presentation is less pronounced with primary PCI than with thrombolysis, making thrombolysis less attractive in later presentations (> 3 hours).11
Also, while thrombolytic therapy in patients older than 75 years was associated with a lower mortality rate compared with no therapy in a large Swedish registry,12 this benefit was less striking than in younger patients. A meta-analysis of thrombolysis trials failed to show a similar benefit in patients over age 75 vs younger patients,13 whereas primary PCI remained effective and superior to thrombolysis in the elderly, with more absolute reduction in mortality rates in the elderly subgroup than with younger patients. 14 This makes thrombolysis less attractive in the elderly, either as a stand-alone therapy or in conjunction with PCI. Studies of combined thrombolysis and PCI included very few patients over age 75.15–17
THREE COMBINATION REPERFUSION STRATEGIES
Facilitated PCI is a strategy of thrombolysis immediately followed by PCI, with a planned door-to-balloon time of 90 to 120 minutes.
Pharmacoinvasive therapy means giving thrombolysis at a non-PCI facility and then promptly and systematically transferring the patient to a PCI facility, where PCI is performed 2 to 24 hours after the start of thrombolytic therapy, regardless of whether thrombolysis results in successful reperfusion. 15 Thus, the time to PCI is longer than with facilitated PCI. Facilitated PCI addresses the value of pretreatment with thrombolytics or glycoprotein IIb/IIIa inhibitors in patients otherwise eligible for primary PCI, whereas pharmacoinvasive therapy addresses the value of routine early PCI after thrombolysis in patients who are not eligible for primary PCI.16
Rescue PCI refers to PCI that is performed urgently if thrombolysis fails, failure being defined as persistent hemodynamic or electrical instability, persistent ischemic symptoms, or failure to achieve at least a 50% to 70% resolution of the maximal ST-segment elevation 90 minutes after the infusion is started.
FACILITATED PCI: NEGATIVE RESULTS IN CLINICAL TRIALS
ASSENT-4 PCI trial
In the ASSENT-4 PCI trial,18 patients receiving full thrombolytic therapy before PCI had a higher rate of in-hospital death, bleeding, and cardiovascular events at 90 days than patients treated with primary PCI.
This trial recruited patients arriving at hospitals with or without PCI capability. The door-to-balloon time was about 110 minutes in both groups, which might not have been prolonged enough to show a benefit from a timely addition of thrombolysis. In addition, antiplatelet therapy was limited in these patients: glycoprotein IIb/IIIa inhibitors were not given, and clopidogrel (Plavix) was not appropriately preloaded, and this might have offset the potential benefit of early PCI. In fact, data suggest that platelet activation and aggregation are heightened after thrombolysis, 21–23 and that glycoprotein IIb/IIIa antagonists can inhibit these effects.23
The FINESSE trial
In the FINESSE trial,19 patients were randomized to undergo primary PCI, to undergo PCI facilitated (ie, preceded) by abciximab (Reo-Pro), or to undergo PCI facilitated by half-dose reteplase (Retavase) and full-dose abciximab. Despite a median door-to-balloon time of 132 minutes, the three strategies were associated with similar rates of death, heart failure, or ischemic outcome at 90 days. Even though the dosage of heparin was weight-adjusted, more major bleeding events occurred with the facilitated strategies.
Comments: Some subgroups may still benefit from facilitated PCI
The results of ASSENT-4 PCI and FINESSE led to the conclusion that PCI facilitated by full-dose thrombolysis should be avoided, and called into question the value of PCI facilitation using glycoprotein IIb/IIIa inhibitors with or without half-dose thrombolytic therapy.
However, subgroup analyses of these trials identified some subgroups that may benefit from a facilitated strategy. In ASSENT-4 PCI, 45% of patients were enrolled at PCI hospitals with a minimal PCI-related delay time. These patients had the worst outcome with the facilitated strategy. In contrast, patients who had a short time from pain onset to thrombolysis (2 to 3 hours) and who were given prehospital thrombolysis had a trend toward better outcomes with facilitated PCI.24 And in FINESSE, 60% of patients were enrolled at centers with PCI capability. Analysis of a small subgroup of patients with a Thrombolysis in Myocardial Infarction study (TIMI) risk score of 3 or greater presenting to non-PCI hospitals within 4 hours of symptom onset suggested a potential reduction of ischemic events with the facilitated strategy in these patients.25
Thus, for patients seen in the first 2 to 3 hours after symptom onset, immediate thrombolysis is recommended if PCI will likely be delayed, with or without plans for subsequent early PCI. “Time is muscle,” especially during the first 3 hours.
PHARMACOINVASIVE STRATEGY: GOOD RESULTS IN HIGH-RISK PATIENTS
A number of randomized studies during the last 10 years have examined the value of a pharmacoinvasive strategy.15,16,26–29
The TRANSFER-AMI trial
The TRANSFER-AMI trial15 randomized 1,059 patients with high-risk ST-elevation MI (ie, anterior or high-risk inferior) at non-PCI centers to undergo either pharmacoinvasive care, ie, full-dose tenecteplase (TNKase) with immediate transfer for PCI or standard care, ie, tenecteplase with transfer for rescue PCI if the patient had persistent ST-segment elevation, chest pain, or hemodynamic instability.15 The goal was to perform PCI within 6 hours of thrombolysis, and the median time to PCI was 3.9 hours (range 2–6 hours). In the standard-care group, 35% of patients needed to be transferred for rescue PCI. Unlike in the ASSENT-4 trial, over 80% of patients received aggressive antiplatelet therapy with both 300 mg of clopidogrel and glycoprotein IIb/IIIa inhibitors.
The rate of cardiovascular events at 30 days was significantly lower with pharmacoinvasive therapy than with standard care and rescue PCI (11% vs 17%, P = .004). This difference was driven by lower rates of recurrent ischemia, reinfarction, and heart failure.
The CARESS-in-AMI study
The CARESS-in-AMI study16 found a similar improvement in ischemic outcomes in 600 patients with high-risk ST-elevation MI arriving at non-PCI centers if they had received pharmacoinvasive therapy. Patients received half-dose reteplase and abciximab and were randomized either to be immediately transferred for PCI (median time to PCI 2.25 hours) or to be transferred only if they had persistent ST-segment elevation or clinical deterioration.16 The event rate was low with pharmacoinvasive therapy, comparable to that achieved in primary PCI trials.
Interestingly, no significant increase was seen in the risk of major and minor bleeding in these two trials despite the use of a femoral approach for PCI in over 80% of the cases; this is probably due to the delays between thrombolytic administration and PCI and to the use of a highly fibrin-specific thrombolytic agent and adjusted-dose heparin.
Meta-analysis of pharmacoinvasive trials
A meta-analysis29 of studies of systematic early PCI (mainly with stenting) within 24 hours of thrombolysis showed a reduction in the rates of mortality and reinfarction with this strategy, without an increase in the risk of major or intracranial bleeding.30 In contrast to the results of the trials of facilitated PCI, a pharmacoinvasive strategy improved outcomes in these trials because the delay between thrombolysis and PCI was more than 2 hours, ie, long enough to prevent bleeding complications, and because most patients randomized in these trials presented within 2 to 3 hours of symptom onset, when the time to reperfusion is critical. After 3 hours, the PCI-mediated myocardial salvage is less time-dependent. Moreover, trials of pharmacoinvasive strategy used aggressive antiplatelet therapy with clopidogrel and glycoprotein IIb/IIIa inhibitors.
Comment: Pharmacoinvasive strategy in the guidelines
These results and those of the subgroup analysis from the FINESSE trial suggest that patients with high-risk ST-elevation MI treated at non-PCI hospitals have better outcomes without an increase in major bleeding events when given thrombolysis and then immediately transferred for routine PCI, rather than being transferred only if reperfusion fails.
Hence, the 2009 update of the ACC/AHA guidelines31 gives a class IIa recommendation for transferring patients with anterior ST-elevation MI or high-risk inferior ST-elevation MI treated with thrombolysis to a PCI-capable facility where PCI is performed as part of a pharmacoinvasive or rescue strategy soon after thrombolysis.
This strategy has been particularly studied in patients younger than 75 years presenting with high-risk types of ST-elevation MI early (< 3 hours) after symptom onset. If not at high risk, the patient may be transferred to a PCI facility after receiving thrombolysis or observed in the initial facility (class IIb recommendation). Consideration should be given to starting anticoagulant and antiplatelet therapy before and during transfer—ie, 300 mg of clopidogrel before transfer for PCI and glycoprotein IIb/IIIa inhibitor therapy during PCI.
The European Society of Cardiology (ESC) guidelines32 recommend early routine angiography 3 to 24 hours after successful thrombolysis. This time window was selected to avoid PCI during the prothrombotic period in the first few hours after thrombolysis and to minimize the risk of reocclusion with PCI delays of more than 24 hours (class IIa recommendation).
Larger randomized trials are still needed to establish whether the pharmacoinvasive strategy confers a survival benefit, to determine its usefulness in low-risk inferior or lateral ST-elevation MI, and to further refine the time window when PCI is both safe and beneficial after thrombolysis.33
RESCUE PCI REDUCES MORTALITY RATES
Rescue PCI is the most accepted form of thrombolysis-PCI combination.
The REACT trial
The REACT trial20 showed that rescue PCI performed at a mean of 4.5 hours after failed thrombolysis reduces the rate of adverse cardiovascular events by more than 50% at 6 to 12 months and reduces the 5-year mortality rate by more than 50% compared with conservative management.20 As in the pharmacoinvasive strategy, aggressive antiplatelet regimens were used in the REACT trial.
A meta-analysis of rescue PCI trials
A meta-analysis of rescue PCI trials34 confirmed these results, showing a reduction in heart failure and reinfarction and a trend toward a lower mortality rate with rescue PCI.34 After thrombolysis, 40% of patients do not achieve grade 3 TIMI flow, which explains why in modern clinical trials 30% of patients treated with thrombolysis require rescue PCI.5,15,16,35
For patients with high-risk ST-elevation MI, current ACC/AHA guidelines assign a class IIa recommendation to rescue PCI.31
WHEN PATIENTS WITH ST-ELEVATION MI PRESENT TO A NON-PCI HOSPITAL
Transfer for primary PCI vs thrombolysis at the non-PCI hospital
The DANAMI-2 trial36 found that immediate transfer for PCI was superior to onsite thrombolytic therapy, as measured by a reduction in the rate of ischemic events (composite of death, myocardial infarction, or stroke at 30 days): 8.5% vs 14.2% (P < .001). There were no deaths during transfer.3
The PRAGUE-2 trial4 showed similar results for patients presenting 3 to 12 hours after symptom onset (30-day mortality rate 6% with immediate transfer vs 15.3% with on-site thrombolysis, P < .002), whereas patients presenting within 3 hours of symptom onset had a similar mortality rate with either therapy.4
Comment. These trials showed that transfer for primary PCI is superior to thrombolytic therapy when performed in a timely fashion. However, they were done in countries with established transfer networks and short distances between community hospitals and PCI centers, with a PCI-related delay of only 44 minutes and a door-to-balloon time of 90 minutes despite transfer. The large-scale application of this prompt transfer policy is not practical in most regions in the United States. Thus, a strategy of local thrombolysis followed by routine early transfer for routine or rescue PCI seems warranted when the door-to-balloon time or the PCI-related delay time is expected to be too long.
Experiences with community-based systems of care and prehospital thrombolysis
In Minnesota, Henry et al37 developed a PCI-based treatment system and an integrated transfer program for ST-elevation MI involving 30 hospitals within 210 miles of the Minneapolis Heart Institute. Participating hospitals were divided into two zones: zone 1 hospitals were within 60 miles, and zone 2 facilities were between 60 and 210 miles from the Heart Institute. Zone 2 patients received half-dose tenecteplase (if thrombolytic therapy was not contraindicated) in anticipation of a lengthy transfer time.
The median door-to-balloon time for zone 1 patients was 95 minutes (interquartile range 82 and 116 minutes) and for zone 2 patients 120 minutes (interquartile range 100 and 145 minutes). The diagnosis of ST-elevation MI was made by the emergency department physician, who activated the system with a phone call. The patient was then directly transferred to the catheterization laboratory, most often by helicopter.
The in-hospital death rate for patients who presented to the PCI center and for patients in zones 1 and 2 was similarly low (about 5%).37
In France, the FAST-MI registry,17 which collected outcome data for different reperfusion strategies, found that thrombolysis yielded in-hospital and midterm results that were comparable to those of primary PCI. Of note, thrombolysis was started early after symptom onset (about 2 hours), and was started in the ambulance in two-thirds of cases. Nearly all patients underwent a pharmacoinvasive strategy that combined thrombolysis with coronary angiography and PCI within 24 hours of symptom onset. These findings suggest that timely thrombolysis followed by semiurgent transfer for PCI is an alternative to primary PCI for patients presenting to hospitals with no PCI capability, and that this alternative offers similar benefit to that of primary PCI.
Five centers in the United States have reported their experience with half-dose thrombolysis in the prehospital setting (in the field or during transfer) or at a non-PCI hospital, followed by prompt transfer to a PCI facility. In this registry of almost 3,000 patients,38 patients treated with thrombolysis had better outcomes than patients directly transferred for primary PCI, with a significantly lower 30-day mortality rate (3.8% vs from 6.4%), and no increase in bleeding.38,39 The mean door-to-balloon time was long (168 minutes in the primary PCI group and 196 minutes in the thrombolysis-PCI group), which might explain the benefit achieved with prompt thrombolysis.
CARDIOGENIC SHOCK
Patients presenting with left ventricular cardiogenic shock derive a large mortality benefit from revascularization, whether they are transferred or directly admitted to a PCI center. 40 Moreover, in the SHOCK registry, patients with predominant right ventricular cardiogenic shock had an in-hospital mortality rate similar to that of patients with predominant left ventricular cardiogenic shock, and revascularization (PCI or surgical revascularization) was associated with a strikingly lower mortality rate in both groups.41
Thus, all patients with left or right cardiogenic shock should be revascularized on an emergency basis, either surgically or percutaneously.
While trials of pharmacoinvasive therapy excluded patients with cardiogenic shock,15,16 thrombolytic therapy was associated with improved outcomes in the drug-therapy group of the SHOCK trial and in hypotensive patients randomized in the early thrombolysis trials.13 Thus, the ACC/AHA guidelines recommend thrombolytic therapy before transfer if a patient presents in shock within 3 to 6 hours of onset of the MI and delays in transport and intervention are anticipated.8
PUTTING IT ALL TOGETHER: MANAGEMENT STRATEGIES
If an effective transfer system is in place, primary PCI not preceded by thrombolytic therapy or glycoprotein IIb/IIIa inhibitor therapy is the preferred approach, according to ACC/AHA and ESC guidelines.31,32 Giving thrombolytics immediately before PCI is harmful and thus should be avoided when the expected door-to-balloon time is 90 minutes or less.
All hospitals (whether or not they offer PCI) and regional emergency medical services should participate in a community-based system of care for ST-elevation MI, with protocols for expeditious transfer as defined and coordinated by the American Heart Association initiative “Mission: Lifeline.” In addition, a system of field triage and direct transport to the catheterization laboratory of a PCI facility after field activation significantly reduces door-to-balloon times and improves outcomes.42
If such a system is not in place, then a pharmacoinvasive strategy seems best: ie, local full-dose thrombolysis (if not contraindicated) followed by transfer to a PCI facility and routine performance of PCI 2 to 6 hours after thrombolysis—in conjunction with aggressive early dual oral antiplatelet therapy and “downstream” glycoprotein IIb/IIIa inhibition. This approach is associated with outcomes similar to those of primary PCI.15–17,37
Prehospital thrombolysis delivered by paramedics and followed by early transfer to a PCI facility has been associated with further reduction in mortality rates compared with in-hospital thrombolysis (as in the Swedish registry43), and a reduction in death rate comparable to that of primary PCI in patients presenting early. This is an adequate strategy in regions where such a system can be established.5,17,38,43,44
Patients presenting more than 3 to 4 hours after symptom onset, older patients, and patients with lower-risk MI or a higher risk of bleeding may still be suited for primary PCI even when the door-to-balloon time is 90 to 120 minutes, as stated by the European guidelines,32 or when the PCI-related delay time is as long as 100 minutes. 10 On the other hand, while the ACC/AHA guidelines recognize that in these patients the mortality advantage of primary PCI vs thrombolytic therapy is maintained with more prolonged door-to-balloon times, they nevertheless state that the focus should be on developing systems of care to increase the number of patients with access to primary PCI in less than 90 minutes rather than extending the acceptable window for door-to-balloon time.
In conclusion, for patients presenting with ST-elevation MI who cannot undergo timely primary PCI, the best approach seems to be prehospital thrombolysis delivered by paramedics or local thrombolysis at the non-PCI hospital followed by transferring the patient and performing PCI within a few hours. This is especially important in patients with high-risk ST-elevation MI who present early after symptom onset, when the extent of myocardial necrosis associated with delayed primary PCI is largest.
In addition, every community should develop a coordinated transfer strategy between non-PCI and PCI hospitals.
Effective and rapid reperfusion is crucial in patients with acute ST-segment elevation myocardial infarction (MI). The preferred strategy for reperfusion—when it can be performed in a timely fashion at an experienced facility—is primary percutaneous coronary intervention (PCI), which produces outcomes superior to those of pharmacologic thrombolysis.1
Unfortunately, in the United States about half of patients present to hospitals that do not have PCI capability,2 and in one analysis, 91% of transferred patients had a door-to-balloon time greater than the recommended 90 minutes, with a mean of 152 minutes.3 (In this case, the door-to-balloon time was the time that elapsed between entry into the first hospital and inflation of the PCI balloon at the second hospital.)
In situations such as these, a combined approach may be appropriate, with thrombolysis delivered by paramedics or at a local facility, followed by transfer to a PCI facility and performance of PCI within a few hours. However, this is feasible only if standardized community-based or regional protocols for prompt transfer and reperfusion are in place.
In this paper we discuss the rationale and the clinical data behind several approaches to combined reperfusion, as well as experiences with community-based care protocols.
WITHIN 3 HOURS OF SYMPTOM ONSET, THROMBOLYSIS IS AS GOOD AS PCI
The PRAGUE-2 Trial
In the randomized PRAGUE-2 trial,4 patients with ST-elevation MI who presented to a non-PCI facility had better outcomes if they were transferred promptly for PCI (median door-to-balloon time 97 minutes), as opposed to receiving local therapy with streptokinase. However, for patients presenting within 3 hours of symptom onset, the mortality rates were comparable with either strategy.4
See the glossary of clinical trial names below
The CAPTIM trial
In the CAPTIM trial,5 patients who presented within 2 hours of symptom onset and who were randomized to receive prehospital thrombolysis had outcomes similar to those of patients treated with primary PCI, despite a short door-to-balloon time (82 minutes).
The Vienna STEMI Registry
In the Vienna STEMI Registry,6 the mortality rates with primary PCI and with thrombolysis were similar when patients presented within 2 hours of symptom onset. However, as the time from symptom onset increased, primary PCI appeared to offer an increasing survival benefit compared with thrombolysis.
Comments: Thrombolysis is effective mostly in the first 2 to 3 hours, with some benefit up to 12 hours
Previous studies have shown that the sooner thrombolysis is given after symptom onset, the more effective it is. If it is given within an hour of symptom onset, the relative reduction in the mortality rate is 50% and the absolute reduction is 6.5% compared with no reperfusion therapy. If it is started in the second hour, the absolute reduction in the mortality rate drops to 4%, and a lesser benefit extends to patients presenting up to 12 hours after symptom onset.7 This time-dependent benefit is due to the fact that very early reperfusion of the occluded coronary artery may lead to full recovery of ischemic tissue and thus prevent necrosis. In addition, thrombolysis in the first 2 hours is highly efficacious in lysing a fresh thrombus.
These data support the current guidelines of the American College of Cardiology (ACC) and the American Heart Association (AHA), which state no preference for either thrombolytic therapy or PCI in ST-elevation MI if the presentation is less than 3 hours after symptom onset.8
Of note, in the CAPTIM trial and in the Vienna STEMI Registry, rescue PCI was available and was in fact used after thrombolysis in about 25% of patients, which might have contributed to the benefit of early thrombolysis.
PRIMARY PCI MAY NOT BE SUPERIOR IF TRANSFER TIME IS LONG
Another time-related factor to consider is the PCI-related delay, ie, the theoretical difference between the expected time from first medical contact to balloon inflation (if the patient undergoes primary PCI) and the time from first medical contact to the start of thrombolytic therapy (if the patient undergoes primary thrombolysis).
A meta-analysis of 13 trials comparing PCI and thrombolysis showed that a PCI-related delay of more than 60 minutes might negate the potential advantage of primary PCI over immediate thrombolysis in terms of deaths.9
This observation has been further refined by data from the National Registry of Myocardial Infarction.10 In this analysis, patient factors, including age, duration of symptoms, and infarct location, significantly affected the point at which the PCI-related delay negated the survival advantage of primary PCI. The survival advantage of primary PCI was lost more rapidly—with a PCI-related delay as short as 40 minutes—in patients who presented sooner, were younger, or had anterior MI. Primary PCI maintained its survival advantage even with a PCI-related delay longer than 100 minutes in older patients or patients with nonanterior MI presenting more than 3 hours after symptom onset. Given that median door-to-balloon times in the United States may exceed 150 minutes when transfer is involved, 3 primary PCI may be no better than primary thrombolysis in transferred patients who present early or who have large infarcts.
Although these results were derived from a post hoc analysis of a registry and the delay times reported were sometimes inaccurate, they suggest that both the PCI-related delay time and patient characteristics should be considered when selecting a reperfusion strategy. Thrombolytic therapy before and in conjunction with primary PCI was considered a potential solution to these concerns.
In addition, while the benefit of any reperfusion strategy depends on the time of presentation, the loss in benefit by later presentation is less pronounced with primary PCI than with thrombolysis, making thrombolysis less attractive in later presentations (> 3 hours).11
Also, while thrombolytic therapy in patients older than 75 years was associated with a lower mortality rate compared with no therapy in a large Swedish registry,12 this benefit was less striking than in younger patients. A meta-analysis of thrombolysis trials failed to show a similar benefit in patients over age 75 vs younger patients,13 whereas primary PCI remained effective and superior to thrombolysis in the elderly, with more absolute reduction in mortality rates in the elderly subgroup than with younger patients. 14 This makes thrombolysis less attractive in the elderly, either as a stand-alone therapy or in conjunction with PCI. Studies of combined thrombolysis and PCI included very few patients over age 75.15–17
THREE COMBINATION REPERFUSION STRATEGIES
Facilitated PCI is a strategy of thrombolysis immediately followed by PCI, with a planned door-to-balloon time of 90 to 120 minutes.
Pharmacoinvasive therapy means giving thrombolysis at a non-PCI facility and then promptly and systematically transferring the patient to a PCI facility, where PCI is performed 2 to 24 hours after the start of thrombolytic therapy, regardless of whether thrombolysis results in successful reperfusion. 15 Thus, the time to PCI is longer than with facilitated PCI. Facilitated PCI addresses the value of pretreatment with thrombolytics or glycoprotein IIb/IIIa inhibitors in patients otherwise eligible for primary PCI, whereas pharmacoinvasive therapy addresses the value of routine early PCI after thrombolysis in patients who are not eligible for primary PCI.16
Rescue PCI refers to PCI that is performed urgently if thrombolysis fails, failure being defined as persistent hemodynamic or electrical instability, persistent ischemic symptoms, or failure to achieve at least a 50% to 70% resolution of the maximal ST-segment elevation 90 minutes after the infusion is started.
FACILITATED PCI: NEGATIVE RESULTS IN CLINICAL TRIALS
ASSENT-4 PCI trial
In the ASSENT-4 PCI trial,18 patients receiving full thrombolytic therapy before PCI had a higher rate of in-hospital death, bleeding, and cardiovascular events at 90 days than patients treated with primary PCI.
This trial recruited patients arriving at hospitals with or without PCI capability. The door-to-balloon time was about 110 minutes in both groups, which might not have been prolonged enough to show a benefit from a timely addition of thrombolysis. In addition, antiplatelet therapy was limited in these patients: glycoprotein IIb/IIIa inhibitors were not given, and clopidogrel (Plavix) was not appropriately preloaded, and this might have offset the potential benefit of early PCI. In fact, data suggest that platelet activation and aggregation are heightened after thrombolysis, 21–23 and that glycoprotein IIb/IIIa antagonists can inhibit these effects.23
The FINESSE trial
In the FINESSE trial,19 patients were randomized to undergo primary PCI, to undergo PCI facilitated (ie, preceded) by abciximab (Reo-Pro), or to undergo PCI facilitated by half-dose reteplase (Retavase) and full-dose abciximab. Despite a median door-to-balloon time of 132 minutes, the three strategies were associated with similar rates of death, heart failure, or ischemic outcome at 90 days. Even though the dosage of heparin was weight-adjusted, more major bleeding events occurred with the facilitated strategies.
Comments: Some subgroups may still benefit from facilitated PCI
The results of ASSENT-4 PCI and FINESSE led to the conclusion that PCI facilitated by full-dose thrombolysis should be avoided, and called into question the value of PCI facilitation using glycoprotein IIb/IIIa inhibitors with or without half-dose thrombolytic therapy.
However, subgroup analyses of these trials identified some subgroups that may benefit from a facilitated strategy. In ASSENT-4 PCI, 45% of patients were enrolled at PCI hospitals with a minimal PCI-related delay time. These patients had the worst outcome with the facilitated strategy. In contrast, patients who had a short time from pain onset to thrombolysis (2 to 3 hours) and who were given prehospital thrombolysis had a trend toward better outcomes with facilitated PCI.24 And in FINESSE, 60% of patients were enrolled at centers with PCI capability. Analysis of a small subgroup of patients with a Thrombolysis in Myocardial Infarction study (TIMI) risk score of 3 or greater presenting to non-PCI hospitals within 4 hours of symptom onset suggested a potential reduction of ischemic events with the facilitated strategy in these patients.25
Thus, for patients seen in the first 2 to 3 hours after symptom onset, immediate thrombolysis is recommended if PCI will likely be delayed, with or without plans for subsequent early PCI. “Time is muscle,” especially during the first 3 hours.
PHARMACOINVASIVE STRATEGY: GOOD RESULTS IN HIGH-RISK PATIENTS
A number of randomized studies during the last 10 years have examined the value of a pharmacoinvasive strategy.15,16,26–29
The TRANSFER-AMI trial
The TRANSFER-AMI trial15 randomized 1,059 patients with high-risk ST-elevation MI (ie, anterior or high-risk inferior) at non-PCI centers to undergo either pharmacoinvasive care, ie, full-dose tenecteplase (TNKase) with immediate transfer for PCI or standard care, ie, tenecteplase with transfer for rescue PCI if the patient had persistent ST-segment elevation, chest pain, or hemodynamic instability.15 The goal was to perform PCI within 6 hours of thrombolysis, and the median time to PCI was 3.9 hours (range 2–6 hours). In the standard-care group, 35% of patients needed to be transferred for rescue PCI. Unlike in the ASSENT-4 trial, over 80% of patients received aggressive antiplatelet therapy with both 300 mg of clopidogrel and glycoprotein IIb/IIIa inhibitors.
The rate of cardiovascular events at 30 days was significantly lower with pharmacoinvasive therapy than with standard care and rescue PCI (11% vs 17%, P = .004). This difference was driven by lower rates of recurrent ischemia, reinfarction, and heart failure.
The CARESS-in-AMI study
The CARESS-in-AMI study16 found a similar improvement in ischemic outcomes in 600 patients with high-risk ST-elevation MI arriving at non-PCI centers if they had received pharmacoinvasive therapy. Patients received half-dose reteplase and abciximab and were randomized either to be immediately transferred for PCI (median time to PCI 2.25 hours) or to be transferred only if they had persistent ST-segment elevation or clinical deterioration.16 The event rate was low with pharmacoinvasive therapy, comparable to that achieved in primary PCI trials.
Interestingly, no significant increase was seen in the risk of major and minor bleeding in these two trials despite the use of a femoral approach for PCI in over 80% of the cases; this is probably due to the delays between thrombolytic administration and PCI and to the use of a highly fibrin-specific thrombolytic agent and adjusted-dose heparin.
Meta-analysis of pharmacoinvasive trials
A meta-analysis29 of studies of systematic early PCI (mainly with stenting) within 24 hours of thrombolysis showed a reduction in the rates of mortality and reinfarction with this strategy, without an increase in the risk of major or intracranial bleeding.30 In contrast to the results of the trials of facilitated PCI, a pharmacoinvasive strategy improved outcomes in these trials because the delay between thrombolysis and PCI was more than 2 hours, ie, long enough to prevent bleeding complications, and because most patients randomized in these trials presented within 2 to 3 hours of symptom onset, when the time to reperfusion is critical. After 3 hours, the PCI-mediated myocardial salvage is less time-dependent. Moreover, trials of pharmacoinvasive strategy used aggressive antiplatelet therapy with clopidogrel and glycoprotein IIb/IIIa inhibitors.
Comment: Pharmacoinvasive strategy in the guidelines
These results and those of the subgroup analysis from the FINESSE trial suggest that patients with high-risk ST-elevation MI treated at non-PCI hospitals have better outcomes without an increase in major bleeding events when given thrombolysis and then immediately transferred for routine PCI, rather than being transferred only if reperfusion fails.
Hence, the 2009 update of the ACC/AHA guidelines31 gives a class IIa recommendation for transferring patients with anterior ST-elevation MI or high-risk inferior ST-elevation MI treated with thrombolysis to a PCI-capable facility where PCI is performed as part of a pharmacoinvasive or rescue strategy soon after thrombolysis.
This strategy has been particularly studied in patients younger than 75 years presenting with high-risk types of ST-elevation MI early (< 3 hours) after symptom onset. If not at high risk, the patient may be transferred to a PCI facility after receiving thrombolysis or observed in the initial facility (class IIb recommendation). Consideration should be given to starting anticoagulant and antiplatelet therapy before and during transfer—ie, 300 mg of clopidogrel before transfer for PCI and glycoprotein IIb/IIIa inhibitor therapy during PCI.
The European Society of Cardiology (ESC) guidelines32 recommend early routine angiography 3 to 24 hours after successful thrombolysis. This time window was selected to avoid PCI during the prothrombotic period in the first few hours after thrombolysis and to minimize the risk of reocclusion with PCI delays of more than 24 hours (class IIa recommendation).
Larger randomized trials are still needed to establish whether the pharmacoinvasive strategy confers a survival benefit, to determine its usefulness in low-risk inferior or lateral ST-elevation MI, and to further refine the time window when PCI is both safe and beneficial after thrombolysis.33
RESCUE PCI REDUCES MORTALITY RATES
Rescue PCI is the most accepted form of thrombolysis-PCI combination.
The REACT trial
The REACT trial20 showed that rescue PCI performed at a mean of 4.5 hours after failed thrombolysis reduces the rate of adverse cardiovascular events by more than 50% at 6 to 12 months and reduces the 5-year mortality rate by more than 50% compared with conservative management.20 As in the pharmacoinvasive strategy, aggressive antiplatelet regimens were used in the REACT trial.
A meta-analysis of rescue PCI trials
A meta-analysis of rescue PCI trials34 confirmed these results, showing a reduction in heart failure and reinfarction and a trend toward a lower mortality rate with rescue PCI.34 After thrombolysis, 40% of patients do not achieve grade 3 TIMI flow, which explains why in modern clinical trials 30% of patients treated with thrombolysis require rescue PCI.5,15,16,35
For patients with high-risk ST-elevation MI, current ACC/AHA guidelines assign a class IIa recommendation to rescue PCI.31
WHEN PATIENTS WITH ST-ELEVATION MI PRESENT TO A NON-PCI HOSPITAL
Transfer for primary PCI vs thrombolysis at the non-PCI hospital
The DANAMI-2 trial36 found that immediate transfer for PCI was superior to onsite thrombolytic therapy, as measured by a reduction in the rate of ischemic events (composite of death, myocardial infarction, or stroke at 30 days): 8.5% vs 14.2% (P < .001). There were no deaths during transfer.3
The PRAGUE-2 trial4 showed similar results for patients presenting 3 to 12 hours after symptom onset (30-day mortality rate 6% with immediate transfer vs 15.3% with on-site thrombolysis, P < .002), whereas patients presenting within 3 hours of symptom onset had a similar mortality rate with either therapy.4
Comment. These trials showed that transfer for primary PCI is superior to thrombolytic therapy when performed in a timely fashion. However, they were done in countries with established transfer networks and short distances between community hospitals and PCI centers, with a PCI-related delay of only 44 minutes and a door-to-balloon time of 90 minutes despite transfer. The large-scale application of this prompt transfer policy is not practical in most regions in the United States. Thus, a strategy of local thrombolysis followed by routine early transfer for routine or rescue PCI seems warranted when the door-to-balloon time or the PCI-related delay time is expected to be too long.
Experiences with community-based systems of care and prehospital thrombolysis
In Minnesota, Henry et al37 developed a PCI-based treatment system and an integrated transfer program for ST-elevation MI involving 30 hospitals within 210 miles of the Minneapolis Heart Institute. Participating hospitals were divided into two zones: zone 1 hospitals were within 60 miles, and zone 2 facilities were between 60 and 210 miles from the Heart Institute. Zone 2 patients received half-dose tenecteplase (if thrombolytic therapy was not contraindicated) in anticipation of a lengthy transfer time.
The median door-to-balloon time for zone 1 patients was 95 minutes (interquartile range 82 and 116 minutes) and for zone 2 patients 120 minutes (interquartile range 100 and 145 minutes). The diagnosis of ST-elevation MI was made by the emergency department physician, who activated the system with a phone call. The patient was then directly transferred to the catheterization laboratory, most often by helicopter.
The in-hospital death rate for patients who presented to the PCI center and for patients in zones 1 and 2 was similarly low (about 5%).37
In France, the FAST-MI registry,17 which collected outcome data for different reperfusion strategies, found that thrombolysis yielded in-hospital and midterm results that were comparable to those of primary PCI. Of note, thrombolysis was started early after symptom onset (about 2 hours), and was started in the ambulance in two-thirds of cases. Nearly all patients underwent a pharmacoinvasive strategy that combined thrombolysis with coronary angiography and PCI within 24 hours of symptom onset. These findings suggest that timely thrombolysis followed by semiurgent transfer for PCI is an alternative to primary PCI for patients presenting to hospitals with no PCI capability, and that this alternative offers similar benefit to that of primary PCI.
Five centers in the United States have reported their experience with half-dose thrombolysis in the prehospital setting (in the field or during transfer) or at a non-PCI hospital, followed by prompt transfer to a PCI facility. In this registry of almost 3,000 patients,38 patients treated with thrombolysis had better outcomes than patients directly transferred for primary PCI, with a significantly lower 30-day mortality rate (3.8% vs from 6.4%), and no increase in bleeding.38,39 The mean door-to-balloon time was long (168 minutes in the primary PCI group and 196 minutes in the thrombolysis-PCI group), which might explain the benefit achieved with prompt thrombolysis.
CARDIOGENIC SHOCK
Patients presenting with left ventricular cardiogenic shock derive a large mortality benefit from revascularization, whether they are transferred or directly admitted to a PCI center. 40 Moreover, in the SHOCK registry, patients with predominant right ventricular cardiogenic shock had an in-hospital mortality rate similar to that of patients with predominant left ventricular cardiogenic shock, and revascularization (PCI or surgical revascularization) was associated with a strikingly lower mortality rate in both groups.41
Thus, all patients with left or right cardiogenic shock should be revascularized on an emergency basis, either surgically or percutaneously.
While trials of pharmacoinvasive therapy excluded patients with cardiogenic shock,15,16 thrombolytic therapy was associated with improved outcomes in the drug-therapy group of the SHOCK trial and in hypotensive patients randomized in the early thrombolysis trials.13 Thus, the ACC/AHA guidelines recommend thrombolytic therapy before transfer if a patient presents in shock within 3 to 6 hours of onset of the MI and delays in transport and intervention are anticipated.8
PUTTING IT ALL TOGETHER: MANAGEMENT STRATEGIES
If an effective transfer system is in place, primary PCI not preceded by thrombolytic therapy or glycoprotein IIb/IIIa inhibitor therapy is the preferred approach, according to ACC/AHA and ESC guidelines.31,32 Giving thrombolytics immediately before PCI is harmful and thus should be avoided when the expected door-to-balloon time is 90 minutes or less.
All hospitals (whether or not they offer PCI) and regional emergency medical services should participate in a community-based system of care for ST-elevation MI, with protocols for expeditious transfer as defined and coordinated by the American Heart Association initiative “Mission: Lifeline.” In addition, a system of field triage and direct transport to the catheterization laboratory of a PCI facility after field activation significantly reduces door-to-balloon times and improves outcomes.42
If such a system is not in place, then a pharmacoinvasive strategy seems best: ie, local full-dose thrombolysis (if not contraindicated) followed by transfer to a PCI facility and routine performance of PCI 2 to 6 hours after thrombolysis—in conjunction with aggressive early dual oral antiplatelet therapy and “downstream” glycoprotein IIb/IIIa inhibition. This approach is associated with outcomes similar to those of primary PCI.15–17,37
Prehospital thrombolysis delivered by paramedics and followed by early transfer to a PCI facility has been associated with further reduction in mortality rates compared with in-hospital thrombolysis (as in the Swedish registry43), and a reduction in death rate comparable to that of primary PCI in patients presenting early. This is an adequate strategy in regions where such a system can be established.5,17,38,43,44
Patients presenting more than 3 to 4 hours after symptom onset, older patients, and patients with lower-risk MI or a higher risk of bleeding may still be suited for primary PCI even when the door-to-balloon time is 90 to 120 minutes, as stated by the European guidelines,32 or when the PCI-related delay time is as long as 100 minutes. 10 On the other hand, while the ACC/AHA guidelines recognize that in these patients the mortality advantage of primary PCI vs thrombolytic therapy is maintained with more prolonged door-to-balloon times, they nevertheless state that the focus should be on developing systems of care to increase the number of patients with access to primary PCI in less than 90 minutes rather than extending the acceptable window for door-to-balloon time.
In conclusion, for patients presenting with ST-elevation MI who cannot undergo timely primary PCI, the best approach seems to be prehospital thrombolysis delivered by paramedics or local thrombolysis at the non-PCI hospital followed by transferring the patient and performing PCI within a few hours. This is especially important in patients with high-risk ST-elevation MI who present early after symptom onset, when the extent of myocardial necrosis associated with delayed primary PCI is largest.
In addition, every community should develop a coordinated transfer strategy between non-PCI and PCI hospitals.
- Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet 2003; 361:13–20.
- Waters RE, Singh KP, Roe MT, et al. Rationale and strategies for implementing community-based transfer protocols for primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. J Am Coll Cardiol 2004; 43:2153–2159.
- Chakrabarti A, Krumholz HM, Wang Y, Rumsfeld JS, Nallamothu BK; National Cardiovascular Data Registry. Time-to-reperfusion in patients undergoing interhospital transfer for primary percutaneous coronary intervention in the U.S: an analysis of 2005 and 2006 data from the National Cardiovascular Data Registry. J Am Coll Cardiol 2008; 51:2442–2443.
- Widimský P, Budesínský T, Vorác D, et al; ‘PRAGUE’ Study Group Investigators. Long distance transport for primary angioplasty vs immediate thrombolysis in acute myocardial infarction. Final results of the randomized national multicentre trial—PRAGUE-2. Eur Heart J 2003; 24:94–104.
- Steg PG, Bonnefoy E, Chabaud S, et al; Comparison of Angioplasty and Prehospital Thrombolysis in Acute Myocardial infarction (CAPTIM) Investigators. Impact of time to treatment on mortality after prehospital fibrinolysis or primary angioplasty: data from the CAPTIM randomized clinical trial. Circulation 2003; 108:2851–2856.
- Kalla K, Christ G, Karnik R, et al; Vienna STEMI Registry Group. Implementation of guidelines improves the standard of care: the Viennese registry on reperfusion strategies in ST-elevation myocardial infarction (Vienna STEMI registry). Circulation 2006; 113:2398–2405.
- Boersma E, Maas AC, Deckers JW, Simoons ML. Early thrombolytic treatment in acute myocardial infarction: reappraisal of the golden hour. Lancet 1996; 348:771–775.
- Antman EM, Anbe DT, Armstrong PW, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines; Canadian Cardiovascular Society. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of Patients with Acute Myocardial Infarction). Circulation 2004; 110:e82–e292.
- Nallamothu BK, Bates ER. Percutaneous coronary intervention versus fibrinolytic therapy in acute myocardial infarction: is timing (almost) everything? Am J Cardiol 2003; 92:824–826.
- Pinto DS, Kirtane AJ, Nallamothu BK, et al. Hospital delays in reperfusion for ST-elevation myocardial infarction: implications when selecting a reperfusion strategy. Circulation 2006; 114:2019–2025.
- Boersma E; Primary Coronary Angioplasty vs Thrombolysis Group. Does time matter? A pooled analysis of randomized clinical trials comparing primary percutaneous coronary intervention and in-hospital fibrinolysis in acute myocardial infarction patients. Eur Heart J 2006; 27:779–788.
- Stenestrand U, Wallentin L; Register of Information and Knowledge About Swedish Heart Intensive Care Admissions (RIKS-HIA). Fibrinolytic therapy in patients 75 years and older with ST-segment-elevation myocardial infarction: one-year follow-up of a large prospective cohort. Arch Intern Med 2003; 163:965–971.
- Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group. Lancet 1994; 343:311–322.
- Grines CL, Browne KF, Marco J, et al. A comparison of immediate angioplasty with thrombolytic therapy for acute myocardial infarction. The Primary Angioplasty in Myocardial Infarction Study Group. N Engl J Med 1993; 328:673–679.
- Cantor WJ, Fitchett D, Borgundvaag B, et al; TRANSFER-AMI Trial Investigators. Routine early angioplasty after fibrinolysis for acute myocardial infarction. N Engl J Med 2009; 360:2705–2718.
- Di Mario C, Dudek D, Piscione F, et al; CARESS-in-AMI (Combined Abciximab RE-teplase Stent Study in Acute Myocardial Infarction) Investigators. Immediate angioplasty versus standard therapy with rescue angioplasty after thrombolysis in the Combined Abciximab REteplase Stent Study in Acute Myocardial Infarction (CARESS-in-AMI): an open, prospective, randomised, multicentre trial. Lancet 2008; 371:559–568.
- Danchin N, Coste P, Ferrières J, et al; FAST-MI Investigators. Comparison of thrombolysis followed by broad use of percutaneous coronary intervention with primary percutaneous coronary intervention for ST-segment-elevation acute myocardial infarction: data from the French registry on Acute ST-elevation Myocardial Infarction (FAST-MI). Circulation 2008; 118:268–276.
- Assessment of the Safety and Efficacy of a New Treatment Strategy with Percutaneous Coronary Intervention (ASSENT-4 PCI) investigators. Primary versus tenecteplase-facilitated percutaneous coronary intervention in patients with ST-segment elevation acute myocardial infarction (ASSENT-4 PCI): randomised trial. Lancet 2006; 367:569–578.
- Ellis SG, Tendera M, de Belder MA, et al; FINESSE Investigators. Facilitated PCI in patients with ST-elevation myocardial infarction. N Engl J Med 2008; 358:2205–2217.
- Carver A, Rafelt S, Gershlick AH, Fairbrother KL, Hughes S, Wilcox R; REACT Investigators. Longer-term follow-up of patients recruited to the REACT (Rescue Angioplasty Versus Conservative Treatment or Repeat Thrombolysis) trial. J Am Coll Cardiol 2009; 54:118–126.
- Rasmanis G, Vesterqvist O, Gréen K, Edhag O, Henriksson P. Evidence of increased platelet activation after thrombolysis in patients with acute myocardial infarction. Br Heart J 1992; 68:374–376.
- Gurbel PA, Serebruany VL, Shustov AR, et al. Effects of reteplase and alteplase on platelet aggregation and major receptor expression during the first 24 hours of acute myocardial infarction treatment. GUSTO-III Investigators. Global Use of Strategies to Open Occluded Coronary Arteries. J Am Coll Cardiol 1998; 31:1466–1473.
- Coulter SA, Cannon CP, Ault KA, et al. High levels of platelet inhibition with abciximab despite heightened platelet activation and aggregation during thrombolysis for acute myocardial infarction: results from TIMI (thrombolysis in myocardial infarction) 14. Circulation 2000; 101:2690–2695.
- Ross AM, Huber K, Zeymer U, et al. The impact of place of enrollment and delay to reperfusion on 90-day post-infarction mortality in the ASSENT-4 PCI trial: assessment of the safety and efficacy of a new treatment strategy with percutaneous coronary intervention. JACC Cardiovasc Interv 2009; 2:925–930.
- Herrmann HC, Lu J, Brodie BR, et al; FINESSE Investigators. Benefit of facilitated percutaneous coronary intervention in high-risk ST-segment elevation myocardial infarction patients presenting to nonpercutaneous coronary intervention hospitals. JACC Cardiovasc Interv 2009; 2:917–924.
- Scheller B, Hennen B, Hammer B, et al; SIAM III Study Group. Beneficial effects of immediate stenting after thrombolysis in acute myocardial infarction. J Am Coll Cardiol 2003; 42:634–641.
- Fernandez-Avilés F, Alonso JJ, Castro-Beiras A, et al; GRACIA (Grupo de Análisis de la Cardiopatía Isquémica Aguda) Group. Routine invasive strategy within 24 hours of thrombolysis versus ischaemiaguided conservative approach for acute myocardial infarction with ST-segment elevation (GRACIA-1): a randomised controlled trial. Lancet 2004; 364:1045–1053.
- Le May MR, Wells GA, Labinaz M, et al. Combined angioplasty and pharmacological intervention versus thrombolysis alone in acute myocardial infarction (CAPITAL AMI study). J Am Coll Cardiol 2005; 46:417–424.
- Bøhmer E, Hoffmann P, Abdelnoor M, Arnesen H, Halvorsen S. Efficacy and safety of immediate angioplasty versus ischemia-guided management after thrombolysis in acute myocardial infarction in areas with very long transfer distances results of the NORDISTEMI (NORwegian study on DIstrict treatment of ST-elevation myocardial infarction). J Am Coll Cardiol 2010; 55:102–110.
- Wijeysundera HC, You JJ, Nallamothu BK, Krumholz HM, Cantor WJ, Ko DT. An early invasive strategy versus ischemia-guided management after fibrinolytic therapy for ST-segment elevation myocardial infarction: a meta-analysis of contemporary randomized controlled trials. Am Heart J 2008; 156:564–572,572.e1–e2.
- Kushner FG, Hand M, Smith SC, et al. 2009 focused updates: ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction (updating the 2004 guideline and 2007 focused update) and ACC/AHA/SCAI guidelines on percutaneous coronary intervention (updating the 2005 guideline and 2007 focused update) a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2009; 54:2205–2241.
- Van de Werf F, Bax J, Betriu A, et al. Management of acute myocardial infarction in patients presenting with persistent ST-segment elevation: the Task Force on the Management of ST-Segment Elevation Acute Myocardial Infarction of the European Society of Cardiology. Eur Heart J 2008; 29:2909–2945.
- Mukherjee D, Moliterno DJ. The timely coupling of mechanical revascularization following thrombolysis for myocardial infarction. Cardiology 2007; 107:337–339.
- Wijeysundera HC, Vijayaraghavan R, Nallamothu BK, et al. Rescue angioplasty or repeat fibrinolysis after failed fibrinolytic therapy for ST-segment myocardial infarction: a meta-analysis of randomized trials. J Am Coll Cardiol 2007; 49:422–430.
- The GUSTO Angiographic Investigators. The effects of tissue plasminogen activator, streptokinase, or both on coronary-artery patency, ventricular function, and survival after acute myocardial infarction. N Engl J Med 1993; 329:1615–1622.
- Andersen HR, Nielsen TT, Rasmussen K, et al; DANAMI-2 Investigators. A comparison of coronary angioplasty with fibrinolytic therapy in acute myocardial infarction. N Engl J Med 2003; 349:733–742.
- Henry TD, Sharkey SW, Burke MN, et al. A regional system to provide timely access to percutaneous coronary intervention for ST-elevation myocardial infarction. Circulation 2007; 116:721–728.
- Denktas AE, Athar H, Henry TD, et al. Reduced-dose fibrinolytic acceleration of ST-segment elevation myocardial infarction treatment coupled with urgent percutaneous coronary intervention compared to primary percutaneous coronary intervention alone results of the AMICO (Alliance for Myocardial Infarction Care Optimization) Registry. JACC Cardiovasc Interv 2008; 1:504–510.
- Smalling RW. Ischemic time: the new gold standard for ST-segment elevation myocardial infarction care. J Am Coll Cardiol 2009; 54:2154–2156.
- Hochman JS, Sleeper LA, White HD, et al; SHOCK Investigators. Should We Emergently Revascularize Occluded Coronaries for Cardiogenic Shock. One-year survival following early revascularization for cardiogenic shock. JAMA 2001; 285:190–192.
- Jacobs AK, Leopold JA, Bates E, et al. Cardiogenic shock caused by right ventricular infarction: a report from the SHOCK registry. J Am Coll Cardiol 2003; 41:1273–1279.
- Pedersen SH, Galatius S, Hansen PR, et al. Field triage reduces treatment delay and improves long-term clinical outcome in patients with acute ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. J Am Coll Cardiol 2009; 54:2296–2302.
- Björklund E, Stenestrand U, Lindbäck J, Svensson L, Wallentin L, Lindahl B. Pre-hospital thrombolysis delivered by paramedics is associated with reduced time delay and mortality in ambulance-transported real-life patients with ST-elevation myocardial infarction. Eur Heart J 2006; 27:1146–1152.
- The European Myocardial Infarction Project Group. Prehospital thrombolytic therapy in patients with suspected acute myocardial infarction. N Engl J Med 1993; 329:383–389.
- Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet 2003; 361:13–20.
- Waters RE, Singh KP, Roe MT, et al. Rationale and strategies for implementing community-based transfer protocols for primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. J Am Coll Cardiol 2004; 43:2153–2159.
- Chakrabarti A, Krumholz HM, Wang Y, Rumsfeld JS, Nallamothu BK; National Cardiovascular Data Registry. Time-to-reperfusion in patients undergoing interhospital transfer for primary percutaneous coronary intervention in the U.S: an analysis of 2005 and 2006 data from the National Cardiovascular Data Registry. J Am Coll Cardiol 2008; 51:2442–2443.
- Widimský P, Budesínský T, Vorác D, et al; ‘PRAGUE’ Study Group Investigators. Long distance transport for primary angioplasty vs immediate thrombolysis in acute myocardial infarction. Final results of the randomized national multicentre trial—PRAGUE-2. Eur Heart J 2003; 24:94–104.
- Steg PG, Bonnefoy E, Chabaud S, et al; Comparison of Angioplasty and Prehospital Thrombolysis in Acute Myocardial infarction (CAPTIM) Investigators. Impact of time to treatment on mortality after prehospital fibrinolysis or primary angioplasty: data from the CAPTIM randomized clinical trial. Circulation 2003; 108:2851–2856.
- Kalla K, Christ G, Karnik R, et al; Vienna STEMI Registry Group. Implementation of guidelines improves the standard of care: the Viennese registry on reperfusion strategies in ST-elevation myocardial infarction (Vienna STEMI registry). Circulation 2006; 113:2398–2405.
- Boersma E, Maas AC, Deckers JW, Simoons ML. Early thrombolytic treatment in acute myocardial infarction: reappraisal of the golden hour. Lancet 1996; 348:771–775.
- Antman EM, Anbe DT, Armstrong PW, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines; Canadian Cardiovascular Society. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of Patients with Acute Myocardial Infarction). Circulation 2004; 110:e82–e292.
- Nallamothu BK, Bates ER. Percutaneous coronary intervention versus fibrinolytic therapy in acute myocardial infarction: is timing (almost) everything? Am J Cardiol 2003; 92:824–826.
- Pinto DS, Kirtane AJ, Nallamothu BK, et al. Hospital delays in reperfusion for ST-elevation myocardial infarction: implications when selecting a reperfusion strategy. Circulation 2006; 114:2019–2025.
- Boersma E; Primary Coronary Angioplasty vs Thrombolysis Group. Does time matter? A pooled analysis of randomized clinical trials comparing primary percutaneous coronary intervention and in-hospital fibrinolysis in acute myocardial infarction patients. Eur Heart J 2006; 27:779–788.
- Stenestrand U, Wallentin L; Register of Information and Knowledge About Swedish Heart Intensive Care Admissions (RIKS-HIA). Fibrinolytic therapy in patients 75 years and older with ST-segment-elevation myocardial infarction: one-year follow-up of a large prospective cohort. Arch Intern Med 2003; 163:965–971.
- Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group. Lancet 1994; 343:311–322.
- Grines CL, Browne KF, Marco J, et al. A comparison of immediate angioplasty with thrombolytic therapy for acute myocardial infarction. The Primary Angioplasty in Myocardial Infarction Study Group. N Engl J Med 1993; 328:673–679.
- Cantor WJ, Fitchett D, Borgundvaag B, et al; TRANSFER-AMI Trial Investigators. Routine early angioplasty after fibrinolysis for acute myocardial infarction. N Engl J Med 2009; 360:2705–2718.
- Di Mario C, Dudek D, Piscione F, et al; CARESS-in-AMI (Combined Abciximab RE-teplase Stent Study in Acute Myocardial Infarction) Investigators. Immediate angioplasty versus standard therapy with rescue angioplasty after thrombolysis in the Combined Abciximab REteplase Stent Study in Acute Myocardial Infarction (CARESS-in-AMI): an open, prospective, randomised, multicentre trial. Lancet 2008; 371:559–568.
- Danchin N, Coste P, Ferrières J, et al; FAST-MI Investigators. Comparison of thrombolysis followed by broad use of percutaneous coronary intervention with primary percutaneous coronary intervention for ST-segment-elevation acute myocardial infarction: data from the French registry on Acute ST-elevation Myocardial Infarction (FAST-MI). Circulation 2008; 118:268–276.
- Assessment of the Safety and Efficacy of a New Treatment Strategy with Percutaneous Coronary Intervention (ASSENT-4 PCI) investigators. Primary versus tenecteplase-facilitated percutaneous coronary intervention in patients with ST-segment elevation acute myocardial infarction (ASSENT-4 PCI): randomised trial. Lancet 2006; 367:569–578.
- Ellis SG, Tendera M, de Belder MA, et al; FINESSE Investigators. Facilitated PCI in patients with ST-elevation myocardial infarction. N Engl J Med 2008; 358:2205–2217.
- Carver A, Rafelt S, Gershlick AH, Fairbrother KL, Hughes S, Wilcox R; REACT Investigators. Longer-term follow-up of patients recruited to the REACT (Rescue Angioplasty Versus Conservative Treatment or Repeat Thrombolysis) trial. J Am Coll Cardiol 2009; 54:118–126.
- Rasmanis G, Vesterqvist O, Gréen K, Edhag O, Henriksson P. Evidence of increased platelet activation after thrombolysis in patients with acute myocardial infarction. Br Heart J 1992; 68:374–376.
- Gurbel PA, Serebruany VL, Shustov AR, et al. Effects of reteplase and alteplase on platelet aggregation and major receptor expression during the first 24 hours of acute myocardial infarction treatment. GUSTO-III Investigators. Global Use of Strategies to Open Occluded Coronary Arteries. J Am Coll Cardiol 1998; 31:1466–1473.
- Coulter SA, Cannon CP, Ault KA, et al. High levels of platelet inhibition with abciximab despite heightened platelet activation and aggregation during thrombolysis for acute myocardial infarction: results from TIMI (thrombolysis in myocardial infarction) 14. Circulation 2000; 101:2690–2695.
- Ross AM, Huber K, Zeymer U, et al. The impact of place of enrollment and delay to reperfusion on 90-day post-infarction mortality in the ASSENT-4 PCI trial: assessment of the safety and efficacy of a new treatment strategy with percutaneous coronary intervention. JACC Cardiovasc Interv 2009; 2:925–930.
- Herrmann HC, Lu J, Brodie BR, et al; FINESSE Investigators. Benefit of facilitated percutaneous coronary intervention in high-risk ST-segment elevation myocardial infarction patients presenting to nonpercutaneous coronary intervention hospitals. JACC Cardiovasc Interv 2009; 2:917–924.
- Scheller B, Hennen B, Hammer B, et al; SIAM III Study Group. Beneficial effects of immediate stenting after thrombolysis in acute myocardial infarction. J Am Coll Cardiol 2003; 42:634–641.
- Fernandez-Avilés F, Alonso JJ, Castro-Beiras A, et al; GRACIA (Grupo de Análisis de la Cardiopatía Isquémica Aguda) Group. Routine invasive strategy within 24 hours of thrombolysis versus ischaemiaguided conservative approach for acute myocardial infarction with ST-segment elevation (GRACIA-1): a randomised controlled trial. Lancet 2004; 364:1045–1053.
- Le May MR, Wells GA, Labinaz M, et al. Combined angioplasty and pharmacological intervention versus thrombolysis alone in acute myocardial infarction (CAPITAL AMI study). J Am Coll Cardiol 2005; 46:417–424.
- Bøhmer E, Hoffmann P, Abdelnoor M, Arnesen H, Halvorsen S. Efficacy and safety of immediate angioplasty versus ischemia-guided management after thrombolysis in acute myocardial infarction in areas with very long transfer distances results of the NORDISTEMI (NORwegian study on DIstrict treatment of ST-elevation myocardial infarction). J Am Coll Cardiol 2010; 55:102–110.
- Wijeysundera HC, You JJ, Nallamothu BK, Krumholz HM, Cantor WJ, Ko DT. An early invasive strategy versus ischemia-guided management after fibrinolytic therapy for ST-segment elevation myocardial infarction: a meta-analysis of contemporary randomized controlled trials. Am Heart J 2008; 156:564–572,572.e1–e2.
- Kushner FG, Hand M, Smith SC, et al. 2009 focused updates: ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction (updating the 2004 guideline and 2007 focused update) and ACC/AHA/SCAI guidelines on percutaneous coronary intervention (updating the 2005 guideline and 2007 focused update) a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2009; 54:2205–2241.
- Van de Werf F, Bax J, Betriu A, et al. Management of acute myocardial infarction in patients presenting with persistent ST-segment elevation: the Task Force on the Management of ST-Segment Elevation Acute Myocardial Infarction of the European Society of Cardiology. Eur Heart J 2008; 29:2909–2945.
- Mukherjee D, Moliterno DJ. The timely coupling of mechanical revascularization following thrombolysis for myocardial infarction. Cardiology 2007; 107:337–339.
- Wijeysundera HC, Vijayaraghavan R, Nallamothu BK, et al. Rescue angioplasty or repeat fibrinolysis after failed fibrinolytic therapy for ST-segment myocardial infarction: a meta-analysis of randomized trials. J Am Coll Cardiol 2007; 49:422–430.
- The GUSTO Angiographic Investigators. The effects of tissue plasminogen activator, streptokinase, or both on coronary-artery patency, ventricular function, and survival after acute myocardial infarction. N Engl J Med 1993; 329:1615–1622.
- Andersen HR, Nielsen TT, Rasmussen K, et al; DANAMI-2 Investigators. A comparison of coronary angioplasty with fibrinolytic therapy in acute myocardial infarction. N Engl J Med 2003; 349:733–742.
- Henry TD, Sharkey SW, Burke MN, et al. A regional system to provide timely access to percutaneous coronary intervention for ST-elevation myocardial infarction. Circulation 2007; 116:721–728.
- Denktas AE, Athar H, Henry TD, et al. Reduced-dose fibrinolytic acceleration of ST-segment elevation myocardial infarction treatment coupled with urgent percutaneous coronary intervention compared to primary percutaneous coronary intervention alone results of the AMICO (Alliance for Myocardial Infarction Care Optimization) Registry. JACC Cardiovasc Interv 2008; 1:504–510.
- Smalling RW. Ischemic time: the new gold standard for ST-segment elevation myocardial infarction care. J Am Coll Cardiol 2009; 54:2154–2156.
- Hochman JS, Sleeper LA, White HD, et al; SHOCK Investigators. Should We Emergently Revascularize Occluded Coronaries for Cardiogenic Shock. One-year survival following early revascularization for cardiogenic shock. JAMA 2001; 285:190–192.
- Jacobs AK, Leopold JA, Bates E, et al. Cardiogenic shock caused by right ventricular infarction: a report from the SHOCK registry. J Am Coll Cardiol 2003; 41:1273–1279.
- Pedersen SH, Galatius S, Hansen PR, et al. Field triage reduces treatment delay and improves long-term clinical outcome in patients with acute ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. J Am Coll Cardiol 2009; 54:2296–2302.
- Björklund E, Stenestrand U, Lindbäck J, Svensson L, Wallentin L, Lindahl B. Pre-hospital thrombolysis delivered by paramedics is associated with reduced time delay and mortality in ambulance-transported real-life patients with ST-elevation myocardial infarction. Eur Heart J 2006; 27:1146–1152.
- The European Myocardial Infarction Project Group. Prehospital thrombolytic therapy in patients with suspected acute myocardial infarction. N Engl J Med 1993; 329:383–389.
KEY POINTS
- When the expected door-to-balloon time is less than 90 minutes and the door-to-balloon time minus the door-to-needle time is less than 60 minutes, the preferred approach is PCI not preceded by thrombolysis.
- Evidence suggests that routine early (but not immediate) PCI—ie, 2 to 6 hours after thrombolysis—is beneficial, particularly in patients with high-risk ST-elevation MI.
- Hospitals and emergency services should participate in community-based and regional systems of care, with standardized protocols to ensure expeditious transfer and prompt reperfusion.
- Prehospital thrombolysis followed by early transfer to a PCI facility as part of a community-based system of care may further improve outcomes of patients with very long transfer times.
Do incretin drugs for type 2 diabetes increase the risk of acute pancreatitis?
Probably not. Although cases of acute pancreatitis have occurred in patients taking these drugs, cases have been reported in patients taking other drugs as well. Furthermore, the incidence of acute pancreatitis is higher in patients with type 2 diabetes (for which incretin-type drugs are indicated) than in the general population, regardless of treatment.
INCRETINS, A NEW CLASS OF DRUGS FOR TYPE 2 DIABETES
Incretins are hormones secreted by the small intestine in response to glucose in food. Glucagon-like peptide 1 (GLP-1) is an endogenous incretin that stimulates insulin secretion, suppresses glucagon secretion, and delays gastric emptying.
Current incretin-based therapies for type 2 diabetes include two types of agents. First are drugs that mimic the action of native GLP-1, such as the injectable GLP-1 analogues exenatide (Byetta) and liraglutide (Victoza). Second are agents that interfere with the metabolism of native GLP-1, mainly by inhibiting the endogenous enzyme dipeptidyl peptidase 4 (DPP-4), thus extending the life of native GLP-1. Two DPP-4 inhibitors pertinent to this discussion are saxagliptin (Onglyza) and sitagliptin (Januvia), both of which are taken orally.
The question has been raised whether incretin-based therapy causes pancreatitis. The package inserts for exenatide and sitagliptin have been updated to reflect this possibility, thus causing concern to practitioners. Is this concern warranted?
MANY DRUGS ARE ASSOCIATED WITH ACUTE PANCREATIS
In a review published in 2005, Trivedi and Pitchumoni1 reported that, of the top 100 prescribed drugs in the United States, 44 had been associated with acute pancreatitis. These agents included over-the-counter drugs such as acetaminophen (Tylenol), common antibiotics such as trimethoprim-sulfamethoxazole (Bactrim) and erythromycin, and drugs used to treat acquired immunodeficiency syndrome and cancer. No clear pathophysiologic basis connects these agents.
In 2002, Blomgren et al2 suggested that glyburide (Micronase) use might be a risk factor for acute pancreatitis, and that the risk of pancreatitis is higher if the body mass index is 30 kg/m2 or more. In 2008, more concern was raised with a report of hemorrhagic or necrotizing pancreatitis in six patients taking exenatide, two of whom died.3 And more recently, reports of 88 cases of acute pancreatitis (including 2 cases of hemorrhagic or necrotizing pancreatitis) from October 2006 to February 2009 in patients taking sitagliptin or the sitagliptin-metformin combination Janumet4 prompted a revision of the package inserts.
Do these cases represent unexpected toxicities not appreciated in premarket clinical trials, or are they to be expected in the population treated with these agents as greater numbers are exposed?
TYPE 2 DIABETES ALSO POSES A RISK OF PANCREATITIS
A number of comorbidities associated with type 2 diabetes predispose to pancreatitis, particularly hypertriglyceridemia and gallbladder disease.5–7 People with diabetes can also be exposed to alcohol or other drugs reported to be associated with pancreatitis.
What is the risk of pancreatitis in patients with type 2 diabetes? Is there evidence of a greater risk when incretin-based drugs are used to control hyperglycemia rather than other agents?
Pancreatitis appears to be increasingly prevalent in the general population in western countries. Some 60% to 80% of cases are attributed to alcohol or gallstones, but 20% do not have a clear cause.
In 2009, a new cause of acute pancreatitis was introduced when Frulloni et al8 reported that a novel antibody that recognizes epitopes shared with Helicobacter pylori was associated with autoimmune pancreatitis. H pylori is a common gastrointestinal organism, found in diabetic and nondiabetic patients, and it may well account for what has up to now been considered idiopathic pancreatitis.
Type 2 diabetes is associated with obesity and hyperlipidemia, each of which has been considered a putative risk factor for pancreatits.5–7
Noel et al9 examined the risk of pancreatitis in patients with type 2 diabetes in a large insurance database (29,332,477 covered lives). They identified people with type 2 diabetes and those without diabetes eligible for coverage by the plan, using medical and pharmacy claims from January 1, 1999, to December 31, 2005. The authors also used medical claims to identify episodes of acute pancreatitis and gallbladder disease. They found that the risk of acute pancreatitis was 2.8 times higher in the overall diabetic cohort than in the nondiabetic cohort, and five times higher in the youngest diabetic cohort (ages 18 to 44) than in nondiabetic people of the same age. The risk was three times higher in diabetic men than in nondiabetic men, and 2.6 times higher in diabetic women than in nondiabetic women.
The time period examined in this study is fortuitous, since exenatide was approved in June 2005 and had very little market penetration during its first 6 months, corresponding to the last 6 months of the study period. Sitagliptin, the first DPP-4 inhibitor, had not yet reached the market.
Noel et al9 also found that the risk of biliary disease in patients with diabetes was 1.9 times higher than in those without diabetes. The relative risk of gallbladder disease was proportionally greater in a younger population with diabetes than in the population without diabetes, in whom the risk of gallbladder disease increases with age. Cholelithiasis was believed to be the underlying cause in at least 50% of the cases of pancreatitis.
PANCREATITIS AND INCRETIN-BASED THERAPIES
The estimated risk of acute pancreatitis in the population at large is reported as 0.33 to 0.44 events per 1,000 adults per year10; 15% to 20% of cases are considered severe, and 2% to 4% result in death.5,10 A relatively small number (1%–2%) are believed to be drug-induced.10
Exenatide. In the exenatide development program, six cases of acute pancreatitis were observed in about 3,489 subject-years of exposure (1.7 per 1,000 subject-years), compared with one case in about 336 subject-years with placebo (3.0 per 1,000 subject-years) and one case in about 497 subject-years (2.0 per 1,000 subject-years) with insulin.11
Sitagliptin. Dore et al12 examined claims from another database for the period of June 2005 through June 2008 to look specifically at the risk with incretin-based therapies. This database included 27,996 people starting exenatide and 16,276 people starting sitagliptin, matched with people with type 2 diabetes taking metformin (Glucophage) or glyburide. Over a period of 1 year, 0.13% of exenatide users and 0.12% of sitagliptin users suffered acute pancreatitis. The risk of pancreatitis was comparable in each group:
- For exenatide, relative risk (RR) 1.0, 95% confidence interval (CI) 0.6 to 1.7, compared with metformin or glyburide
- For sitagliptin, RR 1.0, 95% CI 0.5 to 2.0.
Saxagliptin. In clinical trials of saxagliptin, the incidence of pancreatitis was 0.2% in 3,422 patients receiving saxagliptin and 0.2% in 1,066 controls,13 similar to the rates for sitagliptin and exenatide.
Liraglutide appeared to be associated with a risk of acute pancreatitis, with seven cases in 3,900 patients receiving liraglutide vs one case in a patient taking another diabetes drug.14 This rate is similar to that reported in exenatide clinical trials, suggesting that pancreatitis has been underreported in the comparator subjects. We need more experience to see if this agent really poses more risk than other antidiabetic therapies.
As new antidiabetic agents enter the market and their use becomes common, it would not be surprising to see rates of pancreatitis similar to those reported by Blomgren et al2 in 2002, when glyburide was becoming a mainstay of therapy for type 2 diabetes.
- Trivedi CD, Pitchumoni CS. Drug-induced pancreatitis: an update. J Clin Gastroenterol 2005; 39:709–716.
- Blomgren KB, Sundström A, Steineck G, Wiholm BE. Obesity and treatment of diabetes with glyburide may both be risk factors for acute pancreatitis. Diabetes Care 2002; 25:298–302.
- US Food and Drug Administration. Information for healthcare professionals: exenatide (marketed as Byetta)—8/2008 update. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm124713.htm. Accessed July 1, 2010.
- US Food and Drug Administration. Information for healthcare professionals—acute pancreatitis and sitagliptin (marketed as Januvia and Janumet). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm183764.htm. Accessed July 1, 2010.
- Forsmark CE, Baillie J; AGA Institute Clinical Practice and Economics Committee. AGA Institute technical review on acute pancreatitis. Gastroenterology 2007; 132:2022–2044.
- Pagliarulo M, Fornari F, Fraquelli M, et al. Gallstone disease and related risk factors in a large cohort of diabetic patients. Dig Liver Dis 2004; 36:130–134.
- Field AE, Coakley EH, Must A, et al. Impact of overweight on the risk of developing common chronic diseases during a 10-year period. Arch Intern Med 2001; 161:1581–1586.
- Frulloni L, Lunardi C, Simone R, et al. Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med 2009; 361:2135–2142.
- Noel RA, Braun DK, Patterson RE, Bloomgren GL. Increased risk of acute pancreatitis and biliary disease observed in patients with type 2 diabetes: a retrospective cohort study. Diabetes Care 2009; 32:834–838.
- Whitcomb DC. Clinical practice. Acute pancreatitis. N Engl J Med 2006; 354:2142–2150.
- Data on file, Amylin Pharmaceuticals, Inc. and Eli Lilly.
- Dore DD, Seeger JD, Arnold Chan K. Use of a claims-based active drug safety surveillance system to assess the risk of acute pancreatitis with exenatide or sitagliptin compared to metformin or glyburide. Curr Med Res Opin 2009; 25:1019–1027.
- US Food and Drug Administration. Controlled Phase 2b/3 Pooled Population—Day 120 Update. http://www.fda.gov/downloads/AdvisoryCommittees/Committees-MeetingMaterials/Drugs/EndocrinologicandMetabolic-DrugsAdvisoryCommittee/UCM149589. Accessed July 4, 2010.
- US Food and Drug Administration. Questions and answers—safety requirements for Victoza (liraglutide). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrug-SafetyInformationforPatientsandProviders/ucm198543.htm. Accessed July 4, 2010.
Probably not. Although cases of acute pancreatitis have occurred in patients taking these drugs, cases have been reported in patients taking other drugs as well. Furthermore, the incidence of acute pancreatitis is higher in patients with type 2 diabetes (for which incretin-type drugs are indicated) than in the general population, regardless of treatment.
INCRETINS, A NEW CLASS OF DRUGS FOR TYPE 2 DIABETES
Incretins are hormones secreted by the small intestine in response to glucose in food. Glucagon-like peptide 1 (GLP-1) is an endogenous incretin that stimulates insulin secretion, suppresses glucagon secretion, and delays gastric emptying.
Current incretin-based therapies for type 2 diabetes include two types of agents. First are drugs that mimic the action of native GLP-1, such as the injectable GLP-1 analogues exenatide (Byetta) and liraglutide (Victoza). Second are agents that interfere with the metabolism of native GLP-1, mainly by inhibiting the endogenous enzyme dipeptidyl peptidase 4 (DPP-4), thus extending the life of native GLP-1. Two DPP-4 inhibitors pertinent to this discussion are saxagliptin (Onglyza) and sitagliptin (Januvia), both of which are taken orally.
The question has been raised whether incretin-based therapy causes pancreatitis. The package inserts for exenatide and sitagliptin have been updated to reflect this possibility, thus causing concern to practitioners. Is this concern warranted?
MANY DRUGS ARE ASSOCIATED WITH ACUTE PANCREATIS
In a review published in 2005, Trivedi and Pitchumoni1 reported that, of the top 100 prescribed drugs in the United States, 44 had been associated with acute pancreatitis. These agents included over-the-counter drugs such as acetaminophen (Tylenol), common antibiotics such as trimethoprim-sulfamethoxazole (Bactrim) and erythromycin, and drugs used to treat acquired immunodeficiency syndrome and cancer. No clear pathophysiologic basis connects these agents.
In 2002, Blomgren et al2 suggested that glyburide (Micronase) use might be a risk factor for acute pancreatitis, and that the risk of pancreatitis is higher if the body mass index is 30 kg/m2 or more. In 2008, more concern was raised with a report of hemorrhagic or necrotizing pancreatitis in six patients taking exenatide, two of whom died.3 And more recently, reports of 88 cases of acute pancreatitis (including 2 cases of hemorrhagic or necrotizing pancreatitis) from October 2006 to February 2009 in patients taking sitagliptin or the sitagliptin-metformin combination Janumet4 prompted a revision of the package inserts.
Do these cases represent unexpected toxicities not appreciated in premarket clinical trials, or are they to be expected in the population treated with these agents as greater numbers are exposed?
TYPE 2 DIABETES ALSO POSES A RISK OF PANCREATITIS
A number of comorbidities associated with type 2 diabetes predispose to pancreatitis, particularly hypertriglyceridemia and gallbladder disease.5–7 People with diabetes can also be exposed to alcohol or other drugs reported to be associated with pancreatitis.
What is the risk of pancreatitis in patients with type 2 diabetes? Is there evidence of a greater risk when incretin-based drugs are used to control hyperglycemia rather than other agents?
Pancreatitis appears to be increasingly prevalent in the general population in western countries. Some 60% to 80% of cases are attributed to alcohol or gallstones, but 20% do not have a clear cause.
In 2009, a new cause of acute pancreatitis was introduced when Frulloni et al8 reported that a novel antibody that recognizes epitopes shared with Helicobacter pylori was associated with autoimmune pancreatitis. H pylori is a common gastrointestinal organism, found in diabetic and nondiabetic patients, and it may well account for what has up to now been considered idiopathic pancreatitis.
Type 2 diabetes is associated with obesity and hyperlipidemia, each of which has been considered a putative risk factor for pancreatits.5–7
Noel et al9 examined the risk of pancreatitis in patients with type 2 diabetes in a large insurance database (29,332,477 covered lives). They identified people with type 2 diabetes and those without diabetes eligible for coverage by the plan, using medical and pharmacy claims from January 1, 1999, to December 31, 2005. The authors also used medical claims to identify episodes of acute pancreatitis and gallbladder disease. They found that the risk of acute pancreatitis was 2.8 times higher in the overall diabetic cohort than in the nondiabetic cohort, and five times higher in the youngest diabetic cohort (ages 18 to 44) than in nondiabetic people of the same age. The risk was three times higher in diabetic men than in nondiabetic men, and 2.6 times higher in diabetic women than in nondiabetic women.
The time period examined in this study is fortuitous, since exenatide was approved in June 2005 and had very little market penetration during its first 6 months, corresponding to the last 6 months of the study period. Sitagliptin, the first DPP-4 inhibitor, had not yet reached the market.
Noel et al9 also found that the risk of biliary disease in patients with diabetes was 1.9 times higher than in those without diabetes. The relative risk of gallbladder disease was proportionally greater in a younger population with diabetes than in the population without diabetes, in whom the risk of gallbladder disease increases with age. Cholelithiasis was believed to be the underlying cause in at least 50% of the cases of pancreatitis.
PANCREATITIS AND INCRETIN-BASED THERAPIES
The estimated risk of acute pancreatitis in the population at large is reported as 0.33 to 0.44 events per 1,000 adults per year10; 15% to 20% of cases are considered severe, and 2% to 4% result in death.5,10 A relatively small number (1%–2%) are believed to be drug-induced.10
Exenatide. In the exenatide development program, six cases of acute pancreatitis were observed in about 3,489 subject-years of exposure (1.7 per 1,000 subject-years), compared with one case in about 336 subject-years with placebo (3.0 per 1,000 subject-years) and one case in about 497 subject-years (2.0 per 1,000 subject-years) with insulin.11
Sitagliptin. Dore et al12 examined claims from another database for the period of June 2005 through June 2008 to look specifically at the risk with incretin-based therapies. This database included 27,996 people starting exenatide and 16,276 people starting sitagliptin, matched with people with type 2 diabetes taking metformin (Glucophage) or glyburide. Over a period of 1 year, 0.13% of exenatide users and 0.12% of sitagliptin users suffered acute pancreatitis. The risk of pancreatitis was comparable in each group:
- For exenatide, relative risk (RR) 1.0, 95% confidence interval (CI) 0.6 to 1.7, compared with metformin or glyburide
- For sitagliptin, RR 1.0, 95% CI 0.5 to 2.0.
Saxagliptin. In clinical trials of saxagliptin, the incidence of pancreatitis was 0.2% in 3,422 patients receiving saxagliptin and 0.2% in 1,066 controls,13 similar to the rates for sitagliptin and exenatide.
Liraglutide appeared to be associated with a risk of acute pancreatitis, with seven cases in 3,900 patients receiving liraglutide vs one case in a patient taking another diabetes drug.14 This rate is similar to that reported in exenatide clinical trials, suggesting that pancreatitis has been underreported in the comparator subjects. We need more experience to see if this agent really poses more risk than other antidiabetic therapies.
As new antidiabetic agents enter the market and their use becomes common, it would not be surprising to see rates of pancreatitis similar to those reported by Blomgren et al2 in 2002, when glyburide was becoming a mainstay of therapy for type 2 diabetes.
Probably not. Although cases of acute pancreatitis have occurred in patients taking these drugs, cases have been reported in patients taking other drugs as well. Furthermore, the incidence of acute pancreatitis is higher in patients with type 2 diabetes (for which incretin-type drugs are indicated) than in the general population, regardless of treatment.
INCRETINS, A NEW CLASS OF DRUGS FOR TYPE 2 DIABETES
Incretins are hormones secreted by the small intestine in response to glucose in food. Glucagon-like peptide 1 (GLP-1) is an endogenous incretin that stimulates insulin secretion, suppresses glucagon secretion, and delays gastric emptying.
Current incretin-based therapies for type 2 diabetes include two types of agents. First are drugs that mimic the action of native GLP-1, such as the injectable GLP-1 analogues exenatide (Byetta) and liraglutide (Victoza). Second are agents that interfere with the metabolism of native GLP-1, mainly by inhibiting the endogenous enzyme dipeptidyl peptidase 4 (DPP-4), thus extending the life of native GLP-1. Two DPP-4 inhibitors pertinent to this discussion are saxagliptin (Onglyza) and sitagliptin (Januvia), both of which are taken orally.
The question has been raised whether incretin-based therapy causes pancreatitis. The package inserts for exenatide and sitagliptin have been updated to reflect this possibility, thus causing concern to practitioners. Is this concern warranted?
MANY DRUGS ARE ASSOCIATED WITH ACUTE PANCREATIS
In a review published in 2005, Trivedi and Pitchumoni1 reported that, of the top 100 prescribed drugs in the United States, 44 had been associated with acute pancreatitis. These agents included over-the-counter drugs such as acetaminophen (Tylenol), common antibiotics such as trimethoprim-sulfamethoxazole (Bactrim) and erythromycin, and drugs used to treat acquired immunodeficiency syndrome and cancer. No clear pathophysiologic basis connects these agents.
In 2002, Blomgren et al2 suggested that glyburide (Micronase) use might be a risk factor for acute pancreatitis, and that the risk of pancreatitis is higher if the body mass index is 30 kg/m2 or more. In 2008, more concern was raised with a report of hemorrhagic or necrotizing pancreatitis in six patients taking exenatide, two of whom died.3 And more recently, reports of 88 cases of acute pancreatitis (including 2 cases of hemorrhagic or necrotizing pancreatitis) from October 2006 to February 2009 in patients taking sitagliptin or the sitagliptin-metformin combination Janumet4 prompted a revision of the package inserts.
Do these cases represent unexpected toxicities not appreciated in premarket clinical trials, or are they to be expected in the population treated with these agents as greater numbers are exposed?
TYPE 2 DIABETES ALSO POSES A RISK OF PANCREATITIS
A number of comorbidities associated with type 2 diabetes predispose to pancreatitis, particularly hypertriglyceridemia and gallbladder disease.5–7 People with diabetes can also be exposed to alcohol or other drugs reported to be associated with pancreatitis.
What is the risk of pancreatitis in patients with type 2 diabetes? Is there evidence of a greater risk when incretin-based drugs are used to control hyperglycemia rather than other agents?
Pancreatitis appears to be increasingly prevalent in the general population in western countries. Some 60% to 80% of cases are attributed to alcohol or gallstones, but 20% do not have a clear cause.
In 2009, a new cause of acute pancreatitis was introduced when Frulloni et al8 reported that a novel antibody that recognizes epitopes shared with Helicobacter pylori was associated with autoimmune pancreatitis. H pylori is a common gastrointestinal organism, found in diabetic and nondiabetic patients, and it may well account for what has up to now been considered idiopathic pancreatitis.
Type 2 diabetes is associated with obesity and hyperlipidemia, each of which has been considered a putative risk factor for pancreatits.5–7
Noel et al9 examined the risk of pancreatitis in patients with type 2 diabetes in a large insurance database (29,332,477 covered lives). They identified people with type 2 diabetes and those without diabetes eligible for coverage by the plan, using medical and pharmacy claims from January 1, 1999, to December 31, 2005. The authors also used medical claims to identify episodes of acute pancreatitis and gallbladder disease. They found that the risk of acute pancreatitis was 2.8 times higher in the overall diabetic cohort than in the nondiabetic cohort, and five times higher in the youngest diabetic cohort (ages 18 to 44) than in nondiabetic people of the same age. The risk was three times higher in diabetic men than in nondiabetic men, and 2.6 times higher in diabetic women than in nondiabetic women.
The time period examined in this study is fortuitous, since exenatide was approved in June 2005 and had very little market penetration during its first 6 months, corresponding to the last 6 months of the study period. Sitagliptin, the first DPP-4 inhibitor, had not yet reached the market.
Noel et al9 also found that the risk of biliary disease in patients with diabetes was 1.9 times higher than in those without diabetes. The relative risk of gallbladder disease was proportionally greater in a younger population with diabetes than in the population without diabetes, in whom the risk of gallbladder disease increases with age. Cholelithiasis was believed to be the underlying cause in at least 50% of the cases of pancreatitis.
PANCREATITIS AND INCRETIN-BASED THERAPIES
The estimated risk of acute pancreatitis in the population at large is reported as 0.33 to 0.44 events per 1,000 adults per year10; 15% to 20% of cases are considered severe, and 2% to 4% result in death.5,10 A relatively small number (1%–2%) are believed to be drug-induced.10
Exenatide. In the exenatide development program, six cases of acute pancreatitis were observed in about 3,489 subject-years of exposure (1.7 per 1,000 subject-years), compared with one case in about 336 subject-years with placebo (3.0 per 1,000 subject-years) and one case in about 497 subject-years (2.0 per 1,000 subject-years) with insulin.11
Sitagliptin. Dore et al12 examined claims from another database for the period of June 2005 through June 2008 to look specifically at the risk with incretin-based therapies. This database included 27,996 people starting exenatide and 16,276 people starting sitagliptin, matched with people with type 2 diabetes taking metformin (Glucophage) or glyburide. Over a period of 1 year, 0.13% of exenatide users and 0.12% of sitagliptin users suffered acute pancreatitis. The risk of pancreatitis was comparable in each group:
- For exenatide, relative risk (RR) 1.0, 95% confidence interval (CI) 0.6 to 1.7, compared with metformin or glyburide
- For sitagliptin, RR 1.0, 95% CI 0.5 to 2.0.
Saxagliptin. In clinical trials of saxagliptin, the incidence of pancreatitis was 0.2% in 3,422 patients receiving saxagliptin and 0.2% in 1,066 controls,13 similar to the rates for sitagliptin and exenatide.
Liraglutide appeared to be associated with a risk of acute pancreatitis, with seven cases in 3,900 patients receiving liraglutide vs one case in a patient taking another diabetes drug.14 This rate is similar to that reported in exenatide clinical trials, suggesting that pancreatitis has been underreported in the comparator subjects. We need more experience to see if this agent really poses more risk than other antidiabetic therapies.
As new antidiabetic agents enter the market and their use becomes common, it would not be surprising to see rates of pancreatitis similar to those reported by Blomgren et al2 in 2002, when glyburide was becoming a mainstay of therapy for type 2 diabetes.
- Trivedi CD, Pitchumoni CS. Drug-induced pancreatitis: an update. J Clin Gastroenterol 2005; 39:709–716.
- Blomgren KB, Sundström A, Steineck G, Wiholm BE. Obesity and treatment of diabetes with glyburide may both be risk factors for acute pancreatitis. Diabetes Care 2002; 25:298–302.
- US Food and Drug Administration. Information for healthcare professionals: exenatide (marketed as Byetta)—8/2008 update. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm124713.htm. Accessed July 1, 2010.
- US Food and Drug Administration. Information for healthcare professionals—acute pancreatitis and sitagliptin (marketed as Januvia and Janumet). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm183764.htm. Accessed July 1, 2010.
- Forsmark CE, Baillie J; AGA Institute Clinical Practice and Economics Committee. AGA Institute technical review on acute pancreatitis. Gastroenterology 2007; 132:2022–2044.
- Pagliarulo M, Fornari F, Fraquelli M, et al. Gallstone disease and related risk factors in a large cohort of diabetic patients. Dig Liver Dis 2004; 36:130–134.
- Field AE, Coakley EH, Must A, et al. Impact of overweight on the risk of developing common chronic diseases during a 10-year period. Arch Intern Med 2001; 161:1581–1586.
- Frulloni L, Lunardi C, Simone R, et al. Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med 2009; 361:2135–2142.
- Noel RA, Braun DK, Patterson RE, Bloomgren GL. Increased risk of acute pancreatitis and biliary disease observed in patients with type 2 diabetes: a retrospective cohort study. Diabetes Care 2009; 32:834–838.
- Whitcomb DC. Clinical practice. Acute pancreatitis. N Engl J Med 2006; 354:2142–2150.
- Data on file, Amylin Pharmaceuticals, Inc. and Eli Lilly.
- Dore DD, Seeger JD, Arnold Chan K. Use of a claims-based active drug safety surveillance system to assess the risk of acute pancreatitis with exenatide or sitagliptin compared to metformin or glyburide. Curr Med Res Opin 2009; 25:1019–1027.
- US Food and Drug Administration. Controlled Phase 2b/3 Pooled Population—Day 120 Update. http://www.fda.gov/downloads/AdvisoryCommittees/Committees-MeetingMaterials/Drugs/EndocrinologicandMetabolic-DrugsAdvisoryCommittee/UCM149589. Accessed July 4, 2010.
- US Food and Drug Administration. Questions and answers—safety requirements for Victoza (liraglutide). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrug-SafetyInformationforPatientsandProviders/ucm198543.htm. Accessed July 4, 2010.
- Trivedi CD, Pitchumoni CS. Drug-induced pancreatitis: an update. J Clin Gastroenterol 2005; 39:709–716.
- Blomgren KB, Sundström A, Steineck G, Wiholm BE. Obesity and treatment of diabetes with glyburide may both be risk factors for acute pancreatitis. Diabetes Care 2002; 25:298–302.
- US Food and Drug Administration. Information for healthcare professionals: exenatide (marketed as Byetta)—8/2008 update. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm124713.htm. Accessed July 1, 2010.
- US Food and Drug Administration. Information for healthcare professionals—acute pancreatitis and sitagliptin (marketed as Januvia and Janumet). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm183764.htm. Accessed July 1, 2010.
- Forsmark CE, Baillie J; AGA Institute Clinical Practice and Economics Committee. AGA Institute technical review on acute pancreatitis. Gastroenterology 2007; 132:2022–2044.
- Pagliarulo M, Fornari F, Fraquelli M, et al. Gallstone disease and related risk factors in a large cohort of diabetic patients. Dig Liver Dis 2004; 36:130–134.
- Field AE, Coakley EH, Must A, et al. Impact of overweight on the risk of developing common chronic diseases during a 10-year period. Arch Intern Med 2001; 161:1581–1586.
- Frulloni L, Lunardi C, Simone R, et al. Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med 2009; 361:2135–2142.
- Noel RA, Braun DK, Patterson RE, Bloomgren GL. Increased risk of acute pancreatitis and biliary disease observed in patients with type 2 diabetes: a retrospective cohort study. Diabetes Care 2009; 32:834–838.
- Whitcomb DC. Clinical practice. Acute pancreatitis. N Engl J Med 2006; 354:2142–2150.
- Data on file, Amylin Pharmaceuticals, Inc. and Eli Lilly.
- Dore DD, Seeger JD, Arnold Chan K. Use of a claims-based active drug safety surveillance system to assess the risk of acute pancreatitis with exenatide or sitagliptin compared to metformin or glyburide. Curr Med Res Opin 2009; 25:1019–1027.
- US Food and Drug Administration. Controlled Phase 2b/3 Pooled Population—Day 120 Update. http://www.fda.gov/downloads/AdvisoryCommittees/Committees-MeetingMaterials/Drugs/EndocrinologicandMetabolic-DrugsAdvisoryCommittee/UCM149589. Accessed July 4, 2010.
- US Food and Drug Administration. Questions and answers—safety requirements for Victoza (liraglutide). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrug-SafetyInformationforPatientsandProviders/ucm198543.htm. Accessed July 4, 2010.
HIV: Just another chronic disease

In subsequent years we learned about HIV—the retrovirus, and the immune system that it cleverly and efficiently disables. For the most part, we matured professionally and moved past the social stigmas of the disease, although that was painful. We developed systems to keep acutely ill patients out of the hospital while providing them with “long-term” (weeks or months of) intravenous antibiotics and humane palliative care.
We learned about AZT and argued about when to use it. But mainly, we watched many, many young men (and some women) die in corner hospital rooms. For me, from the ′80s, there remain heartrending personal images, notes, and cassette tapes voicing thanks for my concern and time spent, but no notes of thanks like those I’ve received from my patients with chronic rheumatoid arthritis who, after years of care, are able to hold their nieces or grandchildren.
A few long-term survivors have raised the hope that immune systems could recover and exist in symbiosis with the virus, and that maybe a drug cocktail or vaccine could provide a cure or remission. Magic Johnson, known to be infected since at least 1991, is likely the most public example of a long-term survivor on highly active antiviral therapy—a hope in the flesh.
But did we ever expect a time when HIV would be viewed as a chronic disease, with patients warranting screening for coronary artery disease in order to decrease long-term coronary complications? Did we ever expect a time that we would be offering organ transplants to HIV-infected patients?
In this issue of the Journal, Drs. Malvestutto and Aberg discuss coronary issues that need to be recognized and managed in HIV-infected patients. This further complicates the management of these patients, and draws cardiologists and primary care providers back into management plans.
I can’t think of a management “complication” of a chronic illness that is more welcome—or more surprising.
In subsequent years we learned about HIV—the retrovirus, and the immune system that it cleverly and efficiently disables. For the most part, we matured professionally and moved past the social stigmas of the disease, although that was painful. We developed systems to keep acutely ill patients out of the hospital while providing them with “long-term” (weeks or months of) intravenous antibiotics and humane palliative care.
We learned about AZT and argued about when to use it. But mainly, we watched many, many young men (and some women) die in corner hospital rooms. For me, from the ′80s, there remain heartrending personal images, notes, and cassette tapes voicing thanks for my concern and time spent, but no notes of thanks like those I’ve received from my patients with chronic rheumatoid arthritis who, after years of care, are able to hold their nieces or grandchildren.
A few long-term survivors have raised the hope that immune systems could recover and exist in symbiosis with the virus, and that maybe a drug cocktail or vaccine could provide a cure or remission. Magic Johnson, known to be infected since at least 1991, is likely the most public example of a long-term survivor on highly active antiviral therapy—a hope in the flesh.
But did we ever expect a time when HIV would be viewed as a chronic disease, with patients warranting screening for coronary artery disease in order to decrease long-term coronary complications? Did we ever expect a time that we would be offering organ transplants to HIV-infected patients?
In this issue of the Journal, Drs. Malvestutto and Aberg discuss coronary issues that need to be recognized and managed in HIV-infected patients. This further complicates the management of these patients, and draws cardiologists and primary care providers back into management plans.
I can’t think of a management “complication” of a chronic illness that is more welcome—or more surprising.
In subsequent years we learned about HIV—the retrovirus, and the immune system that it cleverly and efficiently disables. For the most part, we matured professionally and moved past the social stigmas of the disease, although that was painful. We developed systems to keep acutely ill patients out of the hospital while providing them with “long-term” (weeks or months of) intravenous antibiotics and humane palliative care.
We learned about AZT and argued about when to use it. But mainly, we watched many, many young men (and some women) die in corner hospital rooms. For me, from the ′80s, there remain heartrending personal images, notes, and cassette tapes voicing thanks for my concern and time spent, but no notes of thanks like those I’ve received from my patients with chronic rheumatoid arthritis who, after years of care, are able to hold their nieces or grandchildren.
A few long-term survivors have raised the hope that immune systems could recover and exist in symbiosis with the virus, and that maybe a drug cocktail or vaccine could provide a cure or remission. Magic Johnson, known to be infected since at least 1991, is likely the most public example of a long-term survivor on highly active antiviral therapy—a hope in the flesh.
But did we ever expect a time when HIV would be viewed as a chronic disease, with patients warranting screening for coronary artery disease in order to decrease long-term coronary complications? Did we ever expect a time that we would be offering organ transplants to HIV-infected patients?
In this issue of the Journal, Drs. Malvestutto and Aberg discuss coronary issues that need to be recognized and managed in HIV-infected patients. This further complicates the management of these patients, and draws cardiologists and primary care providers back into management plans.
I can’t think of a management “complication” of a chronic illness that is more welcome—or more surprising.
Coronary heart disease in people infected with HIV
Widespread use of antiretroviral therapy has caused a remarkable decline in rates of morbidity and death related to acquired immunodeficiency syndrome (AIDS) and has effectively made human immunodeficiency virus (HIV) infection a manageable—although not yet curable— chronic condition. And as the HIV-infected population on antiretroviral therapy ages, the prevalence of chronic conditions (eg, cardiovascular disease, hepatic disease, pulmonary disease, non-AIDS cancers) and deaths attributable to these conditions have also increased.1
Many of the traditional risk factors for cardiovascular disease in the general population, including smoking, dyslipidemia, and diabetes, are common in HIV-infected patients, and HIV infection itself independently increases the risk of coronary heart disease. In addition, different antiretroviral combinations can contribute, in varying degrees, to changes in lipid levels and insulin resistance, further increasing coronary risk.
Ultimately, however, the immunologic benefits of antiretroviral therapy for individual patients far exceed the modest increase in cardiovascular risk associated with certain regimens. In most cases, careful selection of the initial antiretroviral regimen and the addition of lipid-lowering or glucose-controlling medications (with close attention to drug interactions) can effectively manage the metabolic changes associated with antiretroviral therapy and obviate any premature modification of virologically suppressive regimens.
TRADITIONAL CARDIAC RISK FACTORS IN HIV PATIENTS
The risk of coronary heart disease in HIV patients is influenced mostly by traditional factors such as age, smoking, diabetes, and dyslipidemia, including high levels of total cholesterol and low-density lipoprotein cholesterol (LDL-C) and low levels of high-density lipoprotein cholesterol (HDL-C).2
In various large cohorts, HIV-infected men had a higher prevalence of smoking,3 a lower mean HDL-C level,3,4 and a higher mean triglyceride level3,4 than men without HIV infection, placing them at greater risk of coronary heart disease. However, even after adjusting for traditional risk factors, rates of atherosclerosis are still higher in people who are infected with HIV than in those who are not.5
EFFECT OF HIV INFECTION ON CORONARY RISK
HIV infection has been shown to increase coronary risk.
In the Kaiser Permanente database,6 HIV-positive patients had a significantly higher rate of hospitalizations for coronary heart disease than did people who were not infected.
Similarly, in a cohort study of almost 4,000 HIV-infected patients and more than 1 million controls, the risk of acute myocardial infarction was 75% higher for HIV-positive patients than for HIV-negative patients, even after adjusting for sex, race, hypertension, diabetes, and dyslipidemia.5
The Fat Redistribution and Metabolism (FRAM) cross-sectional study7 showed that HIV infection was associated with greater carotid intima media thickness, an established marker of atherosclerosis, independently of traditional risk factors and to virtually the same degree as smoking and male sex.
Other studies of subclinical atherosclerosis in HIV patients have yielded disparate results, likely because of differences in study design, methods of measuring carotid thickness, and characteristics of the study populations (eg, prevalence of cardiovascular risk factors and stage of HIV disease). However, a meta-analysis of six prospective cohort studies, three case-control studies, and four cross-sectional studies confirmed that HIV patients had slightly but statistically significantly greater carotid intima media thickness than HIV-negative people.8
MECHANISMS BY WHICH HIV MAY PROMOTE CORONARY HEART DISEASE
The pathogenesis of coronary heart disease in HIV infection has not been fully elucidated, but the virus appears to contribute directly to the accelerated development of atherosclerosis. It may do so through direct effects on cholesterol processing and transport, attraction of monocytes to the intimal wall, and activation of monocytes to induce an inflammatory response and endothelial proliferation.
Effects on lipids
In early HIV infection, levels of total cholesterol and HDL-C are lower. In more advanced infection, lower CD4+ lymphocyte counts have been associated with lower levels of apolipoprotein B and with smaller LDL-C particles, suggesting that HIV affects lipid processing and delivery to vessel walls.9 HIV infection is also associated with reduced clearance of LDL-C.10 HIV appears to specifically inhibit the compensatory efflux of excess cholesterol from macrophages, thus promoting the formation of foam cells in atherosclerotic plaque.11
Attraction of monocytes to the vessel wall
In vitro studies also suggest that HIV enhances migration of monocytes into the vascular intima during atherosclerotic plaque development by promoting secretion of the chemokine monocyte chemoattractant protein 112 and the expression of endothelial cell adhesion molecules such as intercellular adhesion molecule 1, vascular cell adhesion molecule 1 (VCAM-1), and E-selectin.13
Inflammation
A recent study suggests that chronic inflammation may be a key contributor to the accelerated development of atherosclerosis in HIV patients. Hsue et al14 compared carotid intima media thickness and levels of C-reactive protein (a marker of systemic inflammation) in HIV-positive and HIV-negative patients. The carotid intima media thickness was greater in all groups of HIV patients, irrespective of level of viremia or exposure to antiretroviral therapy, than in healthy controls. In addition, C-reactive protein levels remained elevated in HIV-infected participants regardless of their level of viremia.
These findings suggest not only that HIV-associated atherosclerosis is determined by advanced immunodeficiency, high-level viremia, and exposure to antiretroviral drugs, but also that persistent inflammation due to HIV infection may play an important role in accelerated atherosclerosis.
EFFECT OF ANTIRETROVIRAL THERAPY ON CORONARY RISK
Antiretroviral therapy is associated with a small but significant increase in coronary risk.
Medi-Cal,15 a retrospective study of 28,513 patients, found antiretroviral therapy to be associated with coronary heart disease among patients 18 to 33 years of age (relative risk 2.06, P < .001).
The Data Collection on Adverse Events of Anti-HIV Drugs study16 prospectively followed 23,437 patients for 94,469 person-years. Adjusted for exposure to nonnucleoside reverse transcriptase inhibitors and for hypertension and diabetes, the relative risk of myocardial infarction per year of protease inhibitor exposure was 1.16 (95% confidence interval [CI] 1.10–1.23). The relative risk was lower after adjusting for serum lipid levels but remained significant at 1.10 (95% CI 1.04–1.18).
Reports have been mixed regarding a possible association between myocardial infarction and the nucleoside reverse transcriptase inhibitor abacavir (Ziagen): several studies found a statistically significant association,17–20 and others did not.21–23 Differences in study design (observational cohort studies vs prospective randomized clinical trials), populations studied (differing in age, cardiovascular risk factor prevalence, and whether the patients had already been exposed to treatment), and outcome definition probably contributed to the different conclusions.
On the other hand, several studies have shown that suppression of HIV with antiretroviral therapy actually improves some of the surrogate markers of cardiovascular disease. For example:
- Markers of endothelial function such as flow-mediated vasodilation improve significantly within 4 weeks of a patient’s starting antiretroviral therapy, regardless of the class of antiretroviral drug used.24
- After viral suppression is achieved, levels of the markers of endothelial activation VCAM-1 and P-selectin decline significantly, as do levels of the adipocyte activation marker leptin and the coagulation marker D-dimer.25,26
- Levels of the anti-inflammatory markers adiponectin and interleukin 10 increase. 25,26
Interrupting antiretroviral therapy may increase coronary risk
Not only is uncontrolled viral replication in untreated HIV infection associated with cardiovascular disease, but interrupting antiretroviral therapy may result in a supplementary increase in coronary risk.
In the 5,472-patient Strategies for Management of Antiretroviral Therapy (SMART) trial, the rate of cardiovascular disease events was higher if treatment was interrupted than with continuous treatment, with a hazard ratio of 1.57 (95% CI 1.0–2.46, P = .05).27
This association between treatment interruption and coronary events does not appear to be related to the level of viremia.28 Rather, development of cardiovascular disease in HIV-infected patients who interrupt antiretroviral therapy may be mediated, to a large extent, by chronic inflammation in the setting of viral replication. In the treatment-interruption group, levels of the inflammatory cytokine interleukin 6 (IL-6) and the coagulation marker D-dimer were significantly elevated 1 month after randomization, and these differences were strongly associated with death (odds ratio [OR] 12.6, P < .0001 for IL-6; OR 13.1, P < .0001 for D-dimer). Elevated IL-6 levels were also significantly associated with the development of cardiovascular disease (OR 2.8, P = .03).29
METABOLIC COMPLICATIONS OF ANTIRETROVIRAL THERAPY
Persons with HIV infection may experience metabolic complications that are due to HIV itself or to its treatment.
Cross-sectional studies that included HIV-negative patients as controls have demonstrated changes in lipid processing that are known to promote atherosclerosis. For example, persons with HIV infection have smaller LDL-C particles30 and higher levels of circulating oxidized LDL-C.31
In the Multicenter AIDS Cohort Study (MACS), after HIV seroconversion, nonfasting total cholesterol, LDL-C, and HDL-C levels declined, which is consistent with a chronic inflammatory state. After antiretroviral therapy was started, lipid levels returned to baseline levels or slightly higher except for HDL-C, which remained low.9 These changes may be due to a general “return to health,” or they may be direct medication effects.
Similar patterns were seen in the SMART study.28 Participants randomized to receive intermittent antiretroviral therapy had overall decreases in all lipid levels, with a marked reduction in HDL-C, while those randomized to receive continuous therapy had increased levels of all lipids, including HDL-C, at 12 months. Overall, the ratio of total cholesterol to HDL-C actually increased for participants on episodic therapy, while it decreased in the continuous-treatment group. Along with continued vascular inflammation, the low HDL-C may have contributed to the worse cardiovascular outcomes in patients who received intermittent antiretroviral therapy.
Some lipid changes associated with antiretroviral therapy may actually be beneficial. For example, nonnucleoside reverse transcriptase inhibitors may raise HDL-C levels. However, such increases alone do not necessarily offset the other lipid changes or translate to an observed improvement in coronary risk.32
The degree of dyslipidemia and specific lipid changes differ among the different classes of antiretroviral drugs and even among the individual drugs within each class. Furthermore, the magnitude of the observed lipid changes varies widely among patients on the same antiretroviral regimen, reflecting the likely important role of host genomics.
While the protease inhibitors and nonnucleoside reverse transcriptase inhibitors have well-described effects on lipids (described in greater detail in the following sections), there have been no reported significant changes in lipid profiles or cardiovascular risk associated with the newest classes, ie, fusion inhibitors such as enfuvirtide (Fuzeon), CC chemokine receptor type 5 (CCR5) receptor inhibitors such as maraviroc (Selzentry), or integrase inhibitors such as raltegravir (Isentress).
Impact of protease inhibitors on lipids
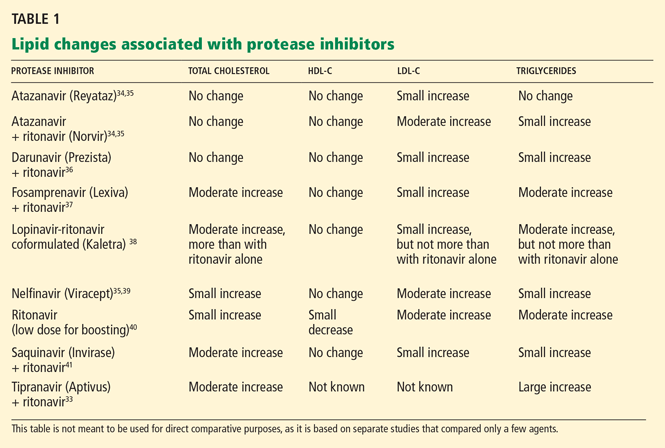
Ritonavir (Norvir) and ritonavir-boosted protease inhibitor combinations cause the most significant increases in lipids. Currently, ritonavir is used in low doses to boost the levels of most other protease inhibitors as the standard of care in protease inhibitor-based regimens. However, in most patients, giving ritonavir with protease inhibitors raises lipid levels, particularly triglycerides.
Most boosted protease inhibitor regimens have similar effects on lipid levels, with some exceptions.
Tipranavir (Aptivus) plus ritonavir, for example, markedly raises total cholesterol and triglyceride levels and would not be recommended for patients with dyslipidemia at baseline.33
Atazanavir (Reyataz)34,35 plus ritonavir and darunavir (Prezista)36 plus ritonavir cause more modest lipid changes. Unboosted atazanavir raises lipid levels only minimally, if at all,34,35 but it is no longer a preferred regimen according to US Department of Health and Human Services guidelines.42
Impact of nonnucleoside reverse transcriptase inhibitors on lipids

Efavirenz (Sustiva), a nonnucleoside reverse transcriptase inhibitor, when added to a regimen of two or three nucleoside reverse transcriptase inhibitors, resulted in modest increases in all lipids, including HDL-C (a potentially beneficial change) at 96 weeks compared with a regimen of three nucleoside reverse transcriptase inhibitors only.43
Nevirapine (Viramune), compared with efavirenz, results in a more favorable lipid profile in previously untreated patients, as shown by larger increases in HDL-C and smaller increases in triglycerides at 48 weeks.44
Etravirine (Intelence), the newest nonnucleoside reverse transcriptase inhibitor, does not appear to cause any further increase in lipids when added to a regimen containing darunavir-ritonavir and nucleoside agents.45
Impact of nucleoside reverse transcriptase inhibitors on lipids
As a class, nucleoside reverse transcriptase inhibitors have been associated with mitochondrial toxicity and insulin resistance,46 but the lipid changes associated with them are generally less significant than those caused by protease inhibitors or nonnucleoside reverse transcriptase inhibitors. Nevertheless, within the class, there is considerable variability in lipid changes associated with specific agents.
Stavudine (Zerit), for example, is associated with hypertriglyceridemia.
Tenofovir (Viread), for another example, in combination with emtricitabine (Emtriva) and the nonnucleoside reverse transcriptase inhibitor efavirenz (the three drugs are contained in a formulation called Atripla) was associated with a smaller increase in fasting total cholesterol than with zidovudine-lamivudine and efavirenz at 96 weeks.47
A recent placebo-controlled, crossover, pilot study of 17 HIV-infected patients suggested that tenofovir may actually have independent lipid-lowering properties.48
Abacavir, as discussed above, has been reported to be associated with a higher risk of myocardial infarction, but this is debatable.
MANAGING CORONARY RISK FACTORS IN HIV-INFECTED PATIENTS
Cardiovascular risk assessment
In HIV patients, cardiovascular risk can be assessed using models derived from large epidemiologic studies such as the Framingham Heart Study.49
Current guidelines from the Infectious Diseases Society of America and the AIDS Clinical Trials Group (ACTG) for evaluating and managing dyslipidemia in HIV-infected adults are based on the National Cholesterol Education Program Adult Treatment Panel III.50 They recommend obtaining a fasting lipid profile before starting antiretroviral therapy and within 3 to 6 months after starting a new regimen.
The guidelines also recommend stratifying risk by counting the number of cardiovascular risk factors, as is done for the general population. If the patient has more than two factors, the Framingham equation should be used to calculate the 10-year risk of myocardial infarction or cardiac death. Interventions should be offered for modifiable cardiovascular risk factors such as smoking, hypertension, physical inactivity, and diabetes mellitus. LDL-C goals should be determined, and lipid-lowering drugs should be initiated accordingly. If triglyceride levels are 200 to 500 mg/dL and levels of “non-HDL-C” (total cholesterol minus the HDL-C level) are high, a statin is recommended. If the triglyceride level is higher than 500 mg/dL, a fibrate should be started.51
Dyslipidemia management
In HIV patients, statin and fibrate therapy must be considered cautiously, given the important drug interactions with protease inhibitors and especially ritonavir. Many statins are metabolized by cytochrome P3A4, which protease inhibitors inhibit.
Statins generally considered safe to use with most protease inhibitors:
- Pravastatin (Pravachol)
- Rosuvastatin (Crestor)
- Atorvastatin (Lipitor).
Exceptions and caveats:
- Pravastatin should not be prescribed with boosted darunavir.
- Data for fluvastatin (Lescol) in HIV-infected patients on antiretroviral therapy are limited.
- Lovastatin (Mevacor) and simvastatin (Zocor) are contraindicated with protease inhibitor therapy.52
- In contrast to the increase in statin levels seen with protease inhibitors, efavirenz lowers levels of simvastatin, pravastatin, and atorvastatin.53,54
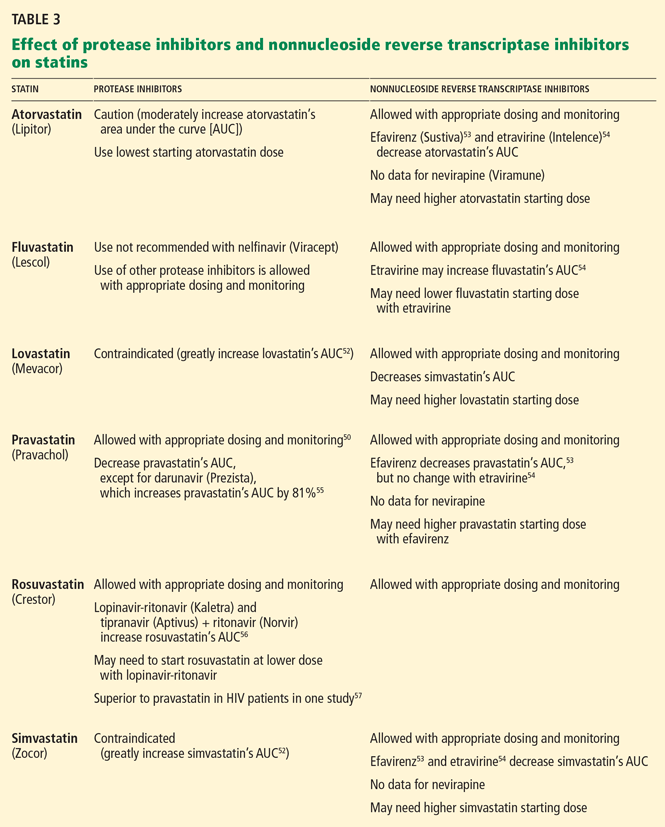
Ezetimibe (Zetia), which is metabolized independently of the cytochrome P450 system, has been shown to be safe and effective when given to HIV-infected patients on antiretroviral therapy.58
Fenofibrate (Lofibra) is recommended by current guidelines for patients with elevated triglyceride levels (> 500 mg/dL).51 In the ACTG 5087 study, a combination of fenofibrate plus pravastatin was found to be safe and effective in improving lipid profiles.59
Long-acting niacin resulted in significant improvements in triglycerides, total cholesterol, HDL-C, and LDL-C after 48 weeks of use, although insulin sensitivity worsened.60
Fish oil has been shown to be an effective alternative to fibrates, or it can be used in combination with them.61
Switching antiretroviral agents vs adding lipid-lowering agents. In some patients with significant dyslipidemia, switching antiretro viral agents may lower lipid levels without compromising virologic control.62 However, due to the multifactorial nature of dyslipidemia in HIV patients on antiretroviral therapy, switching the HIV therapy alone may not result in sufficient improvement in the lipid profile45 and may be associated with virologic failure, particularly among patients who have underlying treatment-resistant HIV.63
In many cases, adding lipid-lowering agents may be more beneficial than switching the antiretroviral therapy. For example, a randomized trial in HIV-infected patients with hyperlipidemia found that adding a lipid-lowering agent such as pravastatin or bezafibrate to the unchanged antiretroviral regimen resulted in greater improvement in total cholesterol, LDL-C, and triglyceride levels than switching from a protease inhibitor to either nevirapine or efavirenz.64
Given the complexity of prescribing lipid-lowering therapies to patients on antiretroviral therapy, we recommend that providers check with a pharmacist or refer to package inserts and other medical literature if they are unfamiliar with these drug interactions and responses to lipid-lowering therapies.
Managing insulin resistance
Diabetes mellitus is a well-known risk factor for coronary heart disease. The Data Collection on Adverse Events of Anti-HIV Drugs study found a higher incidence of coronary heart disease in HIV-infected patients, with higher rates in those with longer duration of diabetes.65 The prevalence of diabetes in HIV-infected populations varies, depending on demographic characteristics,65,66 prevalence of coinfection with hepatitis C virus,66 and prevalence of exposure to antiretroviral drugs67 in the study population.
Drugs that lessen insulin resistance include the thiazolidinedione rosiglitazone (Avandia) and the biguanide metformin (Glucophage). In a randomized trial, both drugs, alone or in combination, improved insulin sensitivity in HIV-infected patients, but neither lessened the amount of visceral or subcutaneous fat.68
Smoking cessation
Smoking is another well-known modifiable risk factor for coronary heart disease.
The prevalence of smoking is usually higher in HIV patients than in HIV-negative people. For example, a French cohort study reported smoking prevalence rates of 56.6% in HIV-infected men vs 32.7% in HIV-negative men; in women, the rates were 58% vs 28.1%. The 5-year relative risk of coronary heart disease in HIV-infected vs HIV-negative persons was 1.20 for men and 1.59 for women. The estimated attributable risk due to smoking was 65% for men and 29% for women.3
Therefore, smoking cessation should be a top priority in managing cardiovascular risk in HIV-infected patients. In fact, control of modifiable risk factors through lifestyle changes such as smoking cessation, dietary changes, and exercise is likely to have a significant impact on cardiovascular risk in this population.
- Palella FJ, Baker RK, Moorman AC, et al; HIV Outpatient Study Investigators. Mortality in the highly active antiretroviral therapy era: changing causes of death and disease in the HIV outpatient study. J Acquir Immune Defic Syndr 2006; 43:27–34.
- Lichtenstein KA, Armon C, Buchacz K, Moorman AC, Wood KC, Brooks JT; HOPS investigators. Analysis of cardiovascular risk factors in the HIV Outpatient Study (HOPS) cohort. Presented at the 13th Conference on Retroviruses and Opportunistic Infections; Denver, CO; 2006.
- Savès M, Chêne G, Ducimetière P, et al; French WHO MONICA Project and the APROCO (ANRS EP11) Study Group. Risk factors for coronary heart disease in patients treated for human immunodeficiency virus infection compared with the general population. Clin Infect Dis 2003; 37:292–298.
- Kaplan RC, Kingsley LA, Sharrett AR, et al. Ten-year predicted coronary heart disease risk in HIV-infected men and women. Clin Infect Dis 2007; 45:1074–1081.
- Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab 2007; 92:2506–2512.
- Klein D, Hurley LB, Quesenberry CP, Sidney S. Do protease inhibitors increase the risk for coronary heart disease in patients with HIV-1 infection? J Acquir Immune Defic Syndr 2002; 30:471–477.
- Grunfeld C, Delaney JA, Wanke C, et al. Preclinical atherosclerosis due to HIV infection: carotid intima-medial thickness measurements from the FRAM study. AIDS 2009; 23:1841–1849.
- Hulten E, Mitchell J, Scally J, Gibbs B, Villines TC. HIV positivity, protease inhibitor exposure and subclinical atherosclerosis: a systematic review and meta-analysis of observational studies. Heart 2009; 95:1826–1835.
- Riddler SA, Smit E, Cole SR, et al. Impact of HIV infection and HAART on serum lipids in men. JAMA 2003; 289:2978–2982.
- Shahmanesh M, Das S, Stolinski M, et al. Antiretroviral treatment reduces very-low-density lipoprotein and intermediate-density lipoprotein apolipoprotein B fractional catabolic rate in human immunodeficiency virus-infected patients with mild dyslipidemia. J Clin Endocrinol Metab 2005; 90:755–760.
- Mujawar Z, Rose H, Morrow MP, et al. Human immunodeficiency virus impairs reverse cholesterol transport from macrophages. PLoS Biol 2006; 4:e365.
- Park IW, Wang JF, Groopman JE. HIV-1 Tat promotes monocyte chemoattractant protein-1 secretion followed by transmigration of monocytes. Blood 2001; 97:352–358.
- Fisher SD, Miller TL, Lipshultz SE. Impact of HIV and highly active antiretroviral therapy on leukocyte adhesion molecules, arterial inflammation, dyslipidemia, and atherosclerosis. Atherosclerosis 2006; 185:1–11.
- Hsue PY, Hunt PW, Schnell A, et al. Role of viral replication, antiretroviral therapy, and immunodeficiency in HIV-associated atherosclerosis. AIDS 2009; 23:1059–1067.
- Currier JS, Taylor A, Boyd F, et al. Coronary heart disease in HIV-infected individuals. J Acquir Immune Defic Syndr 2003; 33:506–512.
- DAD Study Group; Friis-Møller N, Reiss P, Sabin CA, et al. Class of antiretroviral drugs and the risk of myocardial infarction. N Engl J Med 2007: 356:1723–1735.
- DAD Study Group; Sabin CA, Worm SW, Weber R, et al. Use of nucleoside reverse transcriptase inhibitors and risk of myocardial infarction in HIV-infected patients enrolled in the D:A:D study: a multi-cohort collaboration. Lancet 2008; 371:1417–1426.
- Durand M, Sheehy O, Baril JG, Lelorier J, Tremblay C; GRUCHUM Research Center (Groupe de Recherche de l’UHRESS du Centre Hospitalier Universitaire de Montréal). Relation between use of nucleoside reverse transcriptase inhibitors (NRTI) and risk of myocardial infarction (MI): a nested case control study using Quebec’s public health insurance database (QPHID). Presented at the 5th IAS Conference on HIV Pathogenesis, Treatment and Prevention in Cape Town, South Africa, July 17–22, 2009.
- Lang S, Mary-Krause M, Cotte L, et al; the Clinical Epi Group of the French Hospital Database on HIV. Impact of specific NRTI and PI exposure on the risk of myocardial infarction: a case-control study nested within FHDH ANRS CO4. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Strategies for Management of Anti-Retroviral Therapy/INSIGHT. Use of nucleoside reverse transcriptase inhibitors and risk of myocardial infarction in HIV-infected patients. AIDS 2008; 22:F17–F24.
- Bedimo R, Westfall A, Drechsler H, Tebas P. Abacavir use and risk of acute myocardial infarction and cerebrovascular disease in the HAART era. Presented at the 5th IAS Conference on HIV Pathogenesis, Treatment and Prevention in Cape Town, South Africa, July 19–22, 2009.
- Brothers CH, Hernandez JE, Cutrell AG, et al. Risk of myocardial infarction and abacavir therapy: no increased risk across 52 GlaxoSmithKline-sponsored clinical trials in adult subjects. J Acquir Immune Defic Syndr 2009; 51:20–28.
- Benson C, Ribaudo H, Zheng E, et al; the ACTG A5001/ALLRT Protocol Team. No Association of Abacavir Use with Risk of Myocardial Infarction or Severe Cardiovascular Disease Events: Results from ACTG A5001. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Torriani FJ, Komarow L, Parker RA, et al; ACTG 5152s Study Team. Endothelial function in human immunodeficiency virus-infected antiretroviral-naive subjects before and after starting potent antiretroviral therapy: The ACTG (AIDS Clinical Trials Group) Study 5152s. J Am Coll Cardiol 2008; 52:569–576.
- Calmy A, Gayet-Ageron A, Montecucco F, et al; STACCATO Study Group. HIV increases markers of cardiovascular risk: results from a randomized, treatment interruption trial. AIDS 2009; 23:929–939.
- van Vonderen MG, Hassink EA, van Agtmael MA, et al. Increase in carotid artery intima-media thickness and arterial stiffness but improvement in several markers of endothelial function after initiation of antiretroviral therapy. J Infect Dis 2009; 199:1186–1194.
- Strategies for Management of Antiretroviral Therapy (SMART) Study Group; El-Sadr WM, Lundgren JD, Neaton JD, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med 2006; 355:2283–2296.
- Phillips AN, Carr A, Neuhaus J, et al. Interruption of antiretroviral therapy and risk of cardiovascular disease in persons with HIV-1 infection: exploratory analyses from the SMART trial. Antivir Ther 2008; 13:177–187.
- Kuller LH, Tracy R, Belloso WINSIGHT SMART Study Group. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med 2008; 5:e203.
- Badiou S, De Boever CM, Dupuy AM, Baillat V, Cristol JP, Reynes J. Small dense LDL and atherogenic lipid profile in HIV-positive adults: influence of lopinavir/ritonavir-containing regimen. AIDS 2003; 17:772–774.
- Duong M, Petit JM, Martha B, et al. Concentration of circulating oxidized LDL in HIV-infected patients treated with antiretroviral agents: relation to HIV-related lipodystrophy. HIV Clin Trials 2006; 7:41–47.
- Fisac C, Fumero E, Crespo M, et al. Metabolic benefits 24 months after replacing a protease inhibitor with abacavir, efavirenz or nevirapine. AIDS 2005; 19:917–925.
- Hicks CB, Cahn P, Cooper DA, et al; RESIST investigator group. Durable efficacy of tipranavir-ritonavir in combination with an optimised background regimen of antiretroviral drugs for treatmentexperienced HIV-1-infected patients at 48 weeks in the Randomized Evaluation of Strategic Intervention in multi-drug reSistant patients with Tipranavir (RESIST) studies: an analysis of combined data from two randomised open-label trials. Lancet 2006; 368:466–475.
- Malan DR, Krantz E, David N, Wirtz V, Hammond J, McGrath D; 089 Study Group. Efficacy and safety of atazanavir, with or without ritonavir, as part of once-daily highly active antiretroviral therapy regimens in antiretroviral-naive patients. J Acquir Immune Defic Syndr 2008; 47:161–167.
- Anastos K, Lu D, Shi Q, et al. Association of serum lipid levels with HIV serostatus, specific antiretroviral agents, and treatment regimens. J Acquir Immune Defic Syndr 2007; 45:34–42.
- Tomaka F, Lefebvre E, Sekar V, et al. Effects of ritonavir-boosted darunavir vs ritonavir-boosted atazanavir on lipid and glucose parameters in HIV-negative, healthy volunteers. HIV Med 2009; 10:318–327.
- Eron J, Yeni P, Gathe J, et al; KLEAN study team. The KLEAN study of fosamprenavir-ritonavir versus lopinavir-ritonavir, each in combination with abacavir-lamivudine, for initial treatment of HIV infection over 48 weeks: a randomised non-inferiority trial. Lancet 2006; 368:476–482.
- Shafran SD, Mashinter LD, Roberts SE. The effect of low-dose ritonavir monotherapy on fasting serum lipid concentrations. HIV Med 2005; 6:421–425.
- Kumar PN, Rodriguez-French A, Thompson MA, et al; ESS40002 Study Team. A prospective, 96-week study of the impact of trizivir, combivir/nelfinavir, and lamivudine/stavudine/nelfinavir on lipids, metabolic parameters and efficacy in antiretroviral-naive patients: effect of sex and ethnicity. HIV Med 2006; 7:85–98.
- Shafran SD, Mashinter LD, Roberts SE. The effect of low-dose ritonavir monotherapy on fasting serum lipid concentrations. HIV Med 2005; 6:421–425.
- Walmsley S, Avihingsanon A, Slim J, et al. Gemini: a noninferiority study of saquinavir/ritonavir versus lopinavir/ritonavir as initial HIV-1 therapy in adults. J Acquir Immune Defic Syndr 2009; 50:367–374.
- DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents— A Working Group of the Office of AIDS Research Advisory Council (OARAC). Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. December 1, 1009. http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Accessed June 29, 2010.
- Shikuma CM, Yang Y, Glesby MJ, et al. Metabolic effects of protease inhibitor-sparing antiretroviral regimens given as initial treatment of HIV-1 Infection (AIDS Clinical Trials Group Study A5095). J Acquir Immune Defic Syndr 2007; 44:540–550.
- van Leth F, Phanuphak P, Stroes E, et al. Nevirapine and efavirenz elicit different changes in lipid profiles in antiretroviral-therapynaive patients infected with HIV-1. PLoS Med 2004; 1:e19.
- Katlama C, Haubrich R, Lalezari J, et al; DUET-1, DUET-2 study groups. Efficacy and safety of etravirine in treatment-experienced, HIV-1 patients: pooled 48 week analysis of two randomized, controlled trials. AIDS 2009; 23:2289–2300.
- Hammond E, Nolan D, James I, Metcalf C, Mallal S. Reduction of mitochondrial DNA content and respiratory chain activity occurs in adipocytes within 6–12 months of commencing nucleoside reverse transcriptase inhibitor therapy. AIDS 2004; 18:815–817.
- Pozniak AL, Gallant JE, DeJesus E, et al. Tenofovir disoproxil fumarate, emtricitabine, and efavirenz versus fixed-dose zidovudine/lamivudine and efavirenz in antiretroviral-naive patients: virologic, immunologic, and morphologic changes—a 96-week analysis. J Acquir Immune Defic Syndr 2006; 43:535–540.
- Tungsiripat M, Kitch D, Glesby M, et al. A pilot study to determine the effect on dyslipidemia of the addition of tenofovir to stable background ART in HIV-infected subjects: results from the A5206 Study Team. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Law MG, Friis-Møller N, El-Sadr WM, et al; D:A:D Study Group. The use of the Framingham equation to predict myocardial infarctions in HIV-infected patients: comparison with observed events in the D:A:D Study. HIV Med 2006; 7:218–230.
- Aberg JA. Cardiovascular complications in HIV management: past, present, and future. J Acquir Immune Defic Syndr 2009; 50:54–64.
- Dubé MP, Stein JH, Aberg JA, et al; Adult AIDS Clinical Trials Group Cardiovascular Subcommittee. Guidelines for the evaluation and management of dyslipidemia in human immunodeficiency virus (HIV)-infected adults receiving antiretroviral therapy: recommendations of the HIV Medical Association of the Infectious Disease Society of America and the Adult AIDS Clinical Trials Group. Clin Infect Dis 2003; 37:613–627.
- Fichtenbaum CJ. Metabolic abnormalities associated with HIV infection and antiretroviral therapy. Curr Infect Dis Rep 2009; 11:84–92.
- Gerber JG, Rosenkranz SL, Fichtenbaum CJ, et al; AIDS Clinical Trials Group A5108 Team. Effect of efavirenz on the pharmacokinetics of simvastatin, atorvastatin, and pravastatin: results of AIDS Clinical Trials Group 5108 Study. J Acquir Immune Defic Syndr 2005; 39:307–312.
- Grennan T, Walmsley S. Etravirine for HIV-I: addressing the limitations of the nonnucleoside reverse transcriptase inhibitor class. J Int Assoc Physicians AIDS Care (Chic Ill) 2009; 8:354–363.
- Sekar V S-GS, Marien K. Pharmacokinetic drug-drug interaction between the new HIV protease inhibitor darunavir (TMC114) and the lipid-lowering agent pravastatin. Presented at the 8th International Workshop on Pharmacology of HIV Therapy; Budapest, Hungary, April 16–18, 2007.
- Kiser JJ, Gerber JG, Predhomme JA, Wolfe P, Flynn DM, Hoody DW. Drug/drug interaction between lopinavir/ritonavir and rosuvastatin in healthy volunteers. J Acquir Immune Defic Syndr 2008; 47:570–578.
- Aslangul E, Assoumou L, Bittar R, et al. Rosuvastatin versus pravastatin in dyslipidemic HIV-1-infected patients receiving protease inhibitors: a randomized trial. AIDS 2010; 24:77–83.
- Chow D, Chen H, Glesby MJ, et al. Short-term ezetimibe is well tolerated and effective in combination with statin therapy to treat elevated LDL cholesterol in HIV-infected patients. AIDS 2009; 23:2133–2141.
- Aberg JA, Zackin RA, Brobst SW, et al; ACTG 5087 Study Team. A randomized trial of the efficacy and safety of fenofibrate versus pravastatin in HIV-infected subjects with lipid abnormalities: AIDS Clinical Trials Group Study 5087. AIDS Res Hum Retroviruses 2005; 21:757–767.
- Dubé MP, Wu JW, Aberg JA, et al; AIDS Clinical Trials Group A5148 Study Team. Safety and efficacy of extended-release niacin for the treatment of dyslipidaemia in patients with HIV infection: AIDS Clinical Trials Group Study A5148. Antivir Ther 2006; 11:1081–1089.
- Gerber JG, Kitch DW, Fichtenbaum CJ, et al. Fish oil and fenofibrate for the treatment of hypertriglyceridemia in HIV-infected subjects on antiretroviral therapy: results of ACTG A5186. J Acquir Immune Defic Syndr 2008; 47:459–466.
- Mallolas J, Podzamczer D, Milinkovic A, et al; ATAZIP Study Group. Efficacy and safety of switching from boosted lopinavir to boosted atazanavir in patients with virological suppression receiving a LPV/rcontaining HAART: the ATAZIP study. J Acquir Immune Defic Syndr 2009; 51:29–36.
- Eron J, Andrade J, Zajdenverg R, et al. Switching from stable lopinavir/ritonavir-based to raltegravir-based combination ART resulted in a superior lipid profile at week 12 but did not demonstrate noninferior virologic efficacy at week 24. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Calza L, Manfredi R, Colangeli V, et al. Substitution of nevirapine or efavirenz for protease inhibitor versus lipid-lowering therapy for the management of dyslipidaemia. AIDS 2005; 19:1051–1058.
- Worm SW, De Wit S, Weber R, et al. Diabetes mellitus, preexisting coronary heart disease, and the risk of subsequent coronary heart disease events in patients infected with human immunodeficiency virus: the Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D Study). Circulation 2009; 119:805–811.
- Brown TT, Cole SR, Li X, et al. Antiretroviral therapy and the prevalence and incidence of diabetes mellitus in the multicenter AIDS cohort study. Arch Intern Med 2005; 165:1179–1184.
- Butt AA, McGinnis K, Rodriguez-Barradas MC, et al; Veterans Aging Cohort Study. HIV infection and the risk of diabetes mellitus. AIDS 2009; 23:1227–1234.
- Mulligan K, Yang Y, Wininger DA, et al. Effects of metformin and rosiglitazone in HIV-infected patients with hyperinsulinemia and elevated waist/hip ratio. AIDS 2007; 21:47–57.
Widespread use of antiretroviral therapy has caused a remarkable decline in rates of morbidity and death related to acquired immunodeficiency syndrome (AIDS) and has effectively made human immunodeficiency virus (HIV) infection a manageable—although not yet curable— chronic condition. And as the HIV-infected population on antiretroviral therapy ages, the prevalence of chronic conditions (eg, cardiovascular disease, hepatic disease, pulmonary disease, non-AIDS cancers) and deaths attributable to these conditions have also increased.1
Many of the traditional risk factors for cardiovascular disease in the general population, including smoking, dyslipidemia, and diabetes, are common in HIV-infected patients, and HIV infection itself independently increases the risk of coronary heart disease. In addition, different antiretroviral combinations can contribute, in varying degrees, to changes in lipid levels and insulin resistance, further increasing coronary risk.
Ultimately, however, the immunologic benefits of antiretroviral therapy for individual patients far exceed the modest increase in cardiovascular risk associated with certain regimens. In most cases, careful selection of the initial antiretroviral regimen and the addition of lipid-lowering or glucose-controlling medications (with close attention to drug interactions) can effectively manage the metabolic changes associated with antiretroviral therapy and obviate any premature modification of virologically suppressive regimens.
TRADITIONAL CARDIAC RISK FACTORS IN HIV PATIENTS
The risk of coronary heart disease in HIV patients is influenced mostly by traditional factors such as age, smoking, diabetes, and dyslipidemia, including high levels of total cholesterol and low-density lipoprotein cholesterol (LDL-C) and low levels of high-density lipoprotein cholesterol (HDL-C).2
In various large cohorts, HIV-infected men had a higher prevalence of smoking,3 a lower mean HDL-C level,3,4 and a higher mean triglyceride level3,4 than men without HIV infection, placing them at greater risk of coronary heart disease. However, even after adjusting for traditional risk factors, rates of atherosclerosis are still higher in people who are infected with HIV than in those who are not.5
EFFECT OF HIV INFECTION ON CORONARY RISK
HIV infection has been shown to increase coronary risk.
In the Kaiser Permanente database,6 HIV-positive patients had a significantly higher rate of hospitalizations for coronary heart disease than did people who were not infected.
Similarly, in a cohort study of almost 4,000 HIV-infected patients and more than 1 million controls, the risk of acute myocardial infarction was 75% higher for HIV-positive patients than for HIV-negative patients, even after adjusting for sex, race, hypertension, diabetes, and dyslipidemia.5
The Fat Redistribution and Metabolism (FRAM) cross-sectional study7 showed that HIV infection was associated with greater carotid intima media thickness, an established marker of atherosclerosis, independently of traditional risk factors and to virtually the same degree as smoking and male sex.
Other studies of subclinical atherosclerosis in HIV patients have yielded disparate results, likely because of differences in study design, methods of measuring carotid thickness, and characteristics of the study populations (eg, prevalence of cardiovascular risk factors and stage of HIV disease). However, a meta-analysis of six prospective cohort studies, three case-control studies, and four cross-sectional studies confirmed that HIV patients had slightly but statistically significantly greater carotid intima media thickness than HIV-negative people.8
MECHANISMS BY WHICH HIV MAY PROMOTE CORONARY HEART DISEASE
The pathogenesis of coronary heart disease in HIV infection has not been fully elucidated, but the virus appears to contribute directly to the accelerated development of atherosclerosis. It may do so through direct effects on cholesterol processing and transport, attraction of monocytes to the intimal wall, and activation of monocytes to induce an inflammatory response and endothelial proliferation.
Effects on lipids
In early HIV infection, levels of total cholesterol and HDL-C are lower. In more advanced infection, lower CD4+ lymphocyte counts have been associated with lower levels of apolipoprotein B and with smaller LDL-C particles, suggesting that HIV affects lipid processing and delivery to vessel walls.9 HIV infection is also associated with reduced clearance of LDL-C.10 HIV appears to specifically inhibit the compensatory efflux of excess cholesterol from macrophages, thus promoting the formation of foam cells in atherosclerotic plaque.11
Attraction of monocytes to the vessel wall
In vitro studies also suggest that HIV enhances migration of monocytes into the vascular intima during atherosclerotic plaque development by promoting secretion of the chemokine monocyte chemoattractant protein 112 and the expression of endothelial cell adhesion molecules such as intercellular adhesion molecule 1, vascular cell adhesion molecule 1 (VCAM-1), and E-selectin.13
Inflammation
A recent study suggests that chronic inflammation may be a key contributor to the accelerated development of atherosclerosis in HIV patients. Hsue et al14 compared carotid intima media thickness and levels of C-reactive protein (a marker of systemic inflammation) in HIV-positive and HIV-negative patients. The carotid intima media thickness was greater in all groups of HIV patients, irrespective of level of viremia or exposure to antiretroviral therapy, than in healthy controls. In addition, C-reactive protein levels remained elevated in HIV-infected participants regardless of their level of viremia.
These findings suggest not only that HIV-associated atherosclerosis is determined by advanced immunodeficiency, high-level viremia, and exposure to antiretroviral drugs, but also that persistent inflammation due to HIV infection may play an important role in accelerated atherosclerosis.
EFFECT OF ANTIRETROVIRAL THERAPY ON CORONARY RISK
Antiretroviral therapy is associated with a small but significant increase in coronary risk.
Medi-Cal,15 a retrospective study of 28,513 patients, found antiretroviral therapy to be associated with coronary heart disease among patients 18 to 33 years of age (relative risk 2.06, P < .001).
The Data Collection on Adverse Events of Anti-HIV Drugs study16 prospectively followed 23,437 patients for 94,469 person-years. Adjusted for exposure to nonnucleoside reverse transcriptase inhibitors and for hypertension and diabetes, the relative risk of myocardial infarction per year of protease inhibitor exposure was 1.16 (95% confidence interval [CI] 1.10–1.23). The relative risk was lower after adjusting for serum lipid levels but remained significant at 1.10 (95% CI 1.04–1.18).
Reports have been mixed regarding a possible association between myocardial infarction and the nucleoside reverse transcriptase inhibitor abacavir (Ziagen): several studies found a statistically significant association,17–20 and others did not.21–23 Differences in study design (observational cohort studies vs prospective randomized clinical trials), populations studied (differing in age, cardiovascular risk factor prevalence, and whether the patients had already been exposed to treatment), and outcome definition probably contributed to the different conclusions.
On the other hand, several studies have shown that suppression of HIV with antiretroviral therapy actually improves some of the surrogate markers of cardiovascular disease. For example:
- Markers of endothelial function such as flow-mediated vasodilation improve significantly within 4 weeks of a patient’s starting antiretroviral therapy, regardless of the class of antiretroviral drug used.24
- After viral suppression is achieved, levels of the markers of endothelial activation VCAM-1 and P-selectin decline significantly, as do levels of the adipocyte activation marker leptin and the coagulation marker D-dimer.25,26
- Levels of the anti-inflammatory markers adiponectin and interleukin 10 increase. 25,26
Interrupting antiretroviral therapy may increase coronary risk
Not only is uncontrolled viral replication in untreated HIV infection associated with cardiovascular disease, but interrupting antiretroviral therapy may result in a supplementary increase in coronary risk.
In the 5,472-patient Strategies for Management of Antiretroviral Therapy (SMART) trial, the rate of cardiovascular disease events was higher if treatment was interrupted than with continuous treatment, with a hazard ratio of 1.57 (95% CI 1.0–2.46, P = .05).27
This association between treatment interruption and coronary events does not appear to be related to the level of viremia.28 Rather, development of cardiovascular disease in HIV-infected patients who interrupt antiretroviral therapy may be mediated, to a large extent, by chronic inflammation in the setting of viral replication. In the treatment-interruption group, levels of the inflammatory cytokine interleukin 6 (IL-6) and the coagulation marker D-dimer were significantly elevated 1 month after randomization, and these differences were strongly associated with death (odds ratio [OR] 12.6, P < .0001 for IL-6; OR 13.1, P < .0001 for D-dimer). Elevated IL-6 levels were also significantly associated with the development of cardiovascular disease (OR 2.8, P = .03).29
METABOLIC COMPLICATIONS OF ANTIRETROVIRAL THERAPY
Persons with HIV infection may experience metabolic complications that are due to HIV itself or to its treatment.
Cross-sectional studies that included HIV-negative patients as controls have demonstrated changes in lipid processing that are known to promote atherosclerosis. For example, persons with HIV infection have smaller LDL-C particles30 and higher levels of circulating oxidized LDL-C.31
In the Multicenter AIDS Cohort Study (MACS), after HIV seroconversion, nonfasting total cholesterol, LDL-C, and HDL-C levels declined, which is consistent with a chronic inflammatory state. After antiretroviral therapy was started, lipid levels returned to baseline levels or slightly higher except for HDL-C, which remained low.9 These changes may be due to a general “return to health,” or they may be direct medication effects.
Similar patterns were seen in the SMART study.28 Participants randomized to receive intermittent antiretroviral therapy had overall decreases in all lipid levels, with a marked reduction in HDL-C, while those randomized to receive continuous therapy had increased levels of all lipids, including HDL-C, at 12 months. Overall, the ratio of total cholesterol to HDL-C actually increased for participants on episodic therapy, while it decreased in the continuous-treatment group. Along with continued vascular inflammation, the low HDL-C may have contributed to the worse cardiovascular outcomes in patients who received intermittent antiretroviral therapy.
Some lipid changes associated with antiretroviral therapy may actually be beneficial. For example, nonnucleoside reverse transcriptase inhibitors may raise HDL-C levels. However, such increases alone do not necessarily offset the other lipid changes or translate to an observed improvement in coronary risk.32
The degree of dyslipidemia and specific lipid changes differ among the different classes of antiretroviral drugs and even among the individual drugs within each class. Furthermore, the magnitude of the observed lipid changes varies widely among patients on the same antiretroviral regimen, reflecting the likely important role of host genomics.
While the protease inhibitors and nonnucleoside reverse transcriptase inhibitors have well-described effects on lipids (described in greater detail in the following sections), there have been no reported significant changes in lipid profiles or cardiovascular risk associated with the newest classes, ie, fusion inhibitors such as enfuvirtide (Fuzeon), CC chemokine receptor type 5 (CCR5) receptor inhibitors such as maraviroc (Selzentry), or integrase inhibitors such as raltegravir (Isentress).
Impact of protease inhibitors on lipids
Ritonavir (Norvir) and ritonavir-boosted protease inhibitor combinations cause the most significant increases in lipids. Currently, ritonavir is used in low doses to boost the levels of most other protease inhibitors as the standard of care in protease inhibitor-based regimens. However, in most patients, giving ritonavir with protease inhibitors raises lipid levels, particularly triglycerides.
Most boosted protease inhibitor regimens have similar effects on lipid levels, with some exceptions.
Tipranavir (Aptivus) plus ritonavir, for example, markedly raises total cholesterol and triglyceride levels and would not be recommended for patients with dyslipidemia at baseline.33
Atazanavir (Reyataz)34,35 plus ritonavir and darunavir (Prezista)36 plus ritonavir cause more modest lipid changes. Unboosted atazanavir raises lipid levels only minimally, if at all,34,35 but it is no longer a preferred regimen according to US Department of Health and Human Services guidelines.42
Impact of nonnucleoside reverse transcriptase inhibitors on lipids
Efavirenz (Sustiva), a nonnucleoside reverse transcriptase inhibitor, when added to a regimen of two or three nucleoside reverse transcriptase inhibitors, resulted in modest increases in all lipids, including HDL-C (a potentially beneficial change) at 96 weeks compared with a regimen of three nucleoside reverse transcriptase inhibitors only.43
Nevirapine (Viramune), compared with efavirenz, results in a more favorable lipid profile in previously untreated patients, as shown by larger increases in HDL-C and smaller increases in triglycerides at 48 weeks.44
Etravirine (Intelence), the newest nonnucleoside reverse transcriptase inhibitor, does not appear to cause any further increase in lipids when added to a regimen containing darunavir-ritonavir and nucleoside agents.45
Impact of nucleoside reverse transcriptase inhibitors on lipids
As a class, nucleoside reverse transcriptase inhibitors have been associated with mitochondrial toxicity and insulin resistance,46 but the lipid changes associated with them are generally less significant than those caused by protease inhibitors or nonnucleoside reverse transcriptase inhibitors. Nevertheless, within the class, there is considerable variability in lipid changes associated with specific agents.
Stavudine (Zerit), for example, is associated with hypertriglyceridemia.
Tenofovir (Viread), for another example, in combination with emtricitabine (Emtriva) and the nonnucleoside reverse transcriptase inhibitor efavirenz (the three drugs are contained in a formulation called Atripla) was associated with a smaller increase in fasting total cholesterol than with zidovudine-lamivudine and efavirenz at 96 weeks.47
A recent placebo-controlled, crossover, pilot study of 17 HIV-infected patients suggested that tenofovir may actually have independent lipid-lowering properties.48
Abacavir, as discussed above, has been reported to be associated with a higher risk of myocardial infarction, but this is debatable.
MANAGING CORONARY RISK FACTORS IN HIV-INFECTED PATIENTS
Cardiovascular risk assessment
In HIV patients, cardiovascular risk can be assessed using models derived from large epidemiologic studies such as the Framingham Heart Study.49
Current guidelines from the Infectious Diseases Society of America and the AIDS Clinical Trials Group (ACTG) for evaluating and managing dyslipidemia in HIV-infected adults are based on the National Cholesterol Education Program Adult Treatment Panel III.50 They recommend obtaining a fasting lipid profile before starting antiretroviral therapy and within 3 to 6 months after starting a new regimen.
The guidelines also recommend stratifying risk by counting the number of cardiovascular risk factors, as is done for the general population. If the patient has more than two factors, the Framingham equation should be used to calculate the 10-year risk of myocardial infarction or cardiac death. Interventions should be offered for modifiable cardiovascular risk factors such as smoking, hypertension, physical inactivity, and diabetes mellitus. LDL-C goals should be determined, and lipid-lowering drugs should be initiated accordingly. If triglyceride levels are 200 to 500 mg/dL and levels of “non-HDL-C” (total cholesterol minus the HDL-C level) are high, a statin is recommended. If the triglyceride level is higher than 500 mg/dL, a fibrate should be started.51
Dyslipidemia management
In HIV patients, statin and fibrate therapy must be considered cautiously, given the important drug interactions with protease inhibitors and especially ritonavir. Many statins are metabolized by cytochrome P3A4, which protease inhibitors inhibit.
Statins generally considered safe to use with most protease inhibitors:
- Pravastatin (Pravachol)
- Rosuvastatin (Crestor)
- Atorvastatin (Lipitor).
Exceptions and caveats:
- Pravastatin should not be prescribed with boosted darunavir.
- Data for fluvastatin (Lescol) in HIV-infected patients on antiretroviral therapy are limited.
- Lovastatin (Mevacor) and simvastatin (Zocor) are contraindicated with protease inhibitor therapy.52
- In contrast to the increase in statin levels seen with protease inhibitors, efavirenz lowers levels of simvastatin, pravastatin, and atorvastatin.53,54
Ezetimibe (Zetia), which is metabolized independently of the cytochrome P450 system, has been shown to be safe and effective when given to HIV-infected patients on antiretroviral therapy.58
Fenofibrate (Lofibra) is recommended by current guidelines for patients with elevated triglyceride levels (> 500 mg/dL).51 In the ACTG 5087 study, a combination of fenofibrate plus pravastatin was found to be safe and effective in improving lipid profiles.59
Long-acting niacin resulted in significant improvements in triglycerides, total cholesterol, HDL-C, and LDL-C after 48 weeks of use, although insulin sensitivity worsened.60
Fish oil has been shown to be an effective alternative to fibrates, or it can be used in combination with them.61
Switching antiretroviral agents vs adding lipid-lowering agents. In some patients with significant dyslipidemia, switching antiretro viral agents may lower lipid levels without compromising virologic control.62 However, due to the multifactorial nature of dyslipidemia in HIV patients on antiretroviral therapy, switching the HIV therapy alone may not result in sufficient improvement in the lipid profile45 and may be associated with virologic failure, particularly among patients who have underlying treatment-resistant HIV.63
In many cases, adding lipid-lowering agents may be more beneficial than switching the antiretroviral therapy. For example, a randomized trial in HIV-infected patients with hyperlipidemia found that adding a lipid-lowering agent such as pravastatin or bezafibrate to the unchanged antiretroviral regimen resulted in greater improvement in total cholesterol, LDL-C, and triglyceride levels than switching from a protease inhibitor to either nevirapine or efavirenz.64
Given the complexity of prescribing lipid-lowering therapies to patients on antiretroviral therapy, we recommend that providers check with a pharmacist or refer to package inserts and other medical literature if they are unfamiliar with these drug interactions and responses to lipid-lowering therapies.
Managing insulin resistance
Diabetes mellitus is a well-known risk factor for coronary heart disease. The Data Collection on Adverse Events of Anti-HIV Drugs study found a higher incidence of coronary heart disease in HIV-infected patients, with higher rates in those with longer duration of diabetes.65 The prevalence of diabetes in HIV-infected populations varies, depending on demographic characteristics,65,66 prevalence of coinfection with hepatitis C virus,66 and prevalence of exposure to antiretroviral drugs67 in the study population.
Drugs that lessen insulin resistance include the thiazolidinedione rosiglitazone (Avandia) and the biguanide metformin (Glucophage). In a randomized trial, both drugs, alone or in combination, improved insulin sensitivity in HIV-infected patients, but neither lessened the amount of visceral or subcutaneous fat.68
Smoking cessation
Smoking is another well-known modifiable risk factor for coronary heart disease.
The prevalence of smoking is usually higher in HIV patients than in HIV-negative people. For example, a French cohort study reported smoking prevalence rates of 56.6% in HIV-infected men vs 32.7% in HIV-negative men; in women, the rates were 58% vs 28.1%. The 5-year relative risk of coronary heart disease in HIV-infected vs HIV-negative persons was 1.20 for men and 1.59 for women. The estimated attributable risk due to smoking was 65% for men and 29% for women.3
Therefore, smoking cessation should be a top priority in managing cardiovascular risk in HIV-infected patients. In fact, control of modifiable risk factors through lifestyle changes such as smoking cessation, dietary changes, and exercise is likely to have a significant impact on cardiovascular risk in this population.
Widespread use of antiretroviral therapy has caused a remarkable decline in rates of morbidity and death related to acquired immunodeficiency syndrome (AIDS) and has effectively made human immunodeficiency virus (HIV) infection a manageable—although not yet curable— chronic condition. And as the HIV-infected population on antiretroviral therapy ages, the prevalence of chronic conditions (eg, cardiovascular disease, hepatic disease, pulmonary disease, non-AIDS cancers) and deaths attributable to these conditions have also increased.1
Many of the traditional risk factors for cardiovascular disease in the general population, including smoking, dyslipidemia, and diabetes, are common in HIV-infected patients, and HIV infection itself independently increases the risk of coronary heart disease. In addition, different antiretroviral combinations can contribute, in varying degrees, to changes in lipid levels and insulin resistance, further increasing coronary risk.
Ultimately, however, the immunologic benefits of antiretroviral therapy for individual patients far exceed the modest increase in cardiovascular risk associated with certain regimens. In most cases, careful selection of the initial antiretroviral regimen and the addition of lipid-lowering or glucose-controlling medications (with close attention to drug interactions) can effectively manage the metabolic changes associated with antiretroviral therapy and obviate any premature modification of virologically suppressive regimens.
TRADITIONAL CARDIAC RISK FACTORS IN HIV PATIENTS
The risk of coronary heart disease in HIV patients is influenced mostly by traditional factors such as age, smoking, diabetes, and dyslipidemia, including high levels of total cholesterol and low-density lipoprotein cholesterol (LDL-C) and low levels of high-density lipoprotein cholesterol (HDL-C).2
In various large cohorts, HIV-infected men had a higher prevalence of smoking,3 a lower mean HDL-C level,3,4 and a higher mean triglyceride level3,4 than men without HIV infection, placing them at greater risk of coronary heart disease. However, even after adjusting for traditional risk factors, rates of atherosclerosis are still higher in people who are infected with HIV than in those who are not.5
EFFECT OF HIV INFECTION ON CORONARY RISK
HIV infection has been shown to increase coronary risk.
In the Kaiser Permanente database,6 HIV-positive patients had a significantly higher rate of hospitalizations for coronary heart disease than did people who were not infected.
Similarly, in a cohort study of almost 4,000 HIV-infected patients and more than 1 million controls, the risk of acute myocardial infarction was 75% higher for HIV-positive patients than for HIV-negative patients, even after adjusting for sex, race, hypertension, diabetes, and dyslipidemia.5
The Fat Redistribution and Metabolism (FRAM) cross-sectional study7 showed that HIV infection was associated with greater carotid intima media thickness, an established marker of atherosclerosis, independently of traditional risk factors and to virtually the same degree as smoking and male sex.
Other studies of subclinical atherosclerosis in HIV patients have yielded disparate results, likely because of differences in study design, methods of measuring carotid thickness, and characteristics of the study populations (eg, prevalence of cardiovascular risk factors and stage of HIV disease). However, a meta-analysis of six prospective cohort studies, three case-control studies, and four cross-sectional studies confirmed that HIV patients had slightly but statistically significantly greater carotid intima media thickness than HIV-negative people.8
MECHANISMS BY WHICH HIV MAY PROMOTE CORONARY HEART DISEASE
The pathogenesis of coronary heart disease in HIV infection has not been fully elucidated, but the virus appears to contribute directly to the accelerated development of atherosclerosis. It may do so through direct effects on cholesterol processing and transport, attraction of monocytes to the intimal wall, and activation of monocytes to induce an inflammatory response and endothelial proliferation.
Effects on lipids
In early HIV infection, levels of total cholesterol and HDL-C are lower. In more advanced infection, lower CD4+ lymphocyte counts have been associated with lower levels of apolipoprotein B and with smaller LDL-C particles, suggesting that HIV affects lipid processing and delivery to vessel walls.9 HIV infection is also associated with reduced clearance of LDL-C.10 HIV appears to specifically inhibit the compensatory efflux of excess cholesterol from macrophages, thus promoting the formation of foam cells in atherosclerotic plaque.11
Attraction of monocytes to the vessel wall
In vitro studies also suggest that HIV enhances migration of monocytes into the vascular intima during atherosclerotic plaque development by promoting secretion of the chemokine monocyte chemoattractant protein 112 and the expression of endothelial cell adhesion molecules such as intercellular adhesion molecule 1, vascular cell adhesion molecule 1 (VCAM-1), and E-selectin.13
Inflammation
A recent study suggests that chronic inflammation may be a key contributor to the accelerated development of atherosclerosis in HIV patients. Hsue et al14 compared carotid intima media thickness and levels of C-reactive protein (a marker of systemic inflammation) in HIV-positive and HIV-negative patients. The carotid intima media thickness was greater in all groups of HIV patients, irrespective of level of viremia or exposure to antiretroviral therapy, than in healthy controls. In addition, C-reactive protein levels remained elevated in HIV-infected participants regardless of their level of viremia.
These findings suggest not only that HIV-associated atherosclerosis is determined by advanced immunodeficiency, high-level viremia, and exposure to antiretroviral drugs, but also that persistent inflammation due to HIV infection may play an important role in accelerated atherosclerosis.
EFFECT OF ANTIRETROVIRAL THERAPY ON CORONARY RISK
Antiretroviral therapy is associated with a small but significant increase in coronary risk.
Medi-Cal,15 a retrospective study of 28,513 patients, found antiretroviral therapy to be associated with coronary heart disease among patients 18 to 33 years of age (relative risk 2.06, P < .001).
The Data Collection on Adverse Events of Anti-HIV Drugs study16 prospectively followed 23,437 patients for 94,469 person-years. Adjusted for exposure to nonnucleoside reverse transcriptase inhibitors and for hypertension and diabetes, the relative risk of myocardial infarction per year of protease inhibitor exposure was 1.16 (95% confidence interval [CI] 1.10–1.23). The relative risk was lower after adjusting for serum lipid levels but remained significant at 1.10 (95% CI 1.04–1.18).
Reports have been mixed regarding a possible association between myocardial infarction and the nucleoside reverse transcriptase inhibitor abacavir (Ziagen): several studies found a statistically significant association,17–20 and others did not.21–23 Differences in study design (observational cohort studies vs prospective randomized clinical trials), populations studied (differing in age, cardiovascular risk factor prevalence, and whether the patients had already been exposed to treatment), and outcome definition probably contributed to the different conclusions.
On the other hand, several studies have shown that suppression of HIV with antiretroviral therapy actually improves some of the surrogate markers of cardiovascular disease. For example:
- Markers of endothelial function such as flow-mediated vasodilation improve significantly within 4 weeks of a patient’s starting antiretroviral therapy, regardless of the class of antiretroviral drug used.24
- After viral suppression is achieved, levels of the markers of endothelial activation VCAM-1 and P-selectin decline significantly, as do levels of the adipocyte activation marker leptin and the coagulation marker D-dimer.25,26
- Levels of the anti-inflammatory markers adiponectin and interleukin 10 increase. 25,26
Interrupting antiretroviral therapy may increase coronary risk
Not only is uncontrolled viral replication in untreated HIV infection associated with cardiovascular disease, but interrupting antiretroviral therapy may result in a supplementary increase in coronary risk.
In the 5,472-patient Strategies for Management of Antiretroviral Therapy (SMART) trial, the rate of cardiovascular disease events was higher if treatment was interrupted than with continuous treatment, with a hazard ratio of 1.57 (95% CI 1.0–2.46, P = .05).27
This association between treatment interruption and coronary events does not appear to be related to the level of viremia.28 Rather, development of cardiovascular disease in HIV-infected patients who interrupt antiretroviral therapy may be mediated, to a large extent, by chronic inflammation in the setting of viral replication. In the treatment-interruption group, levels of the inflammatory cytokine interleukin 6 (IL-6) and the coagulation marker D-dimer were significantly elevated 1 month after randomization, and these differences were strongly associated with death (odds ratio [OR] 12.6, P < .0001 for IL-6; OR 13.1, P < .0001 for D-dimer). Elevated IL-6 levels were also significantly associated with the development of cardiovascular disease (OR 2.8, P = .03).29
METABOLIC COMPLICATIONS OF ANTIRETROVIRAL THERAPY
Persons with HIV infection may experience metabolic complications that are due to HIV itself or to its treatment.
Cross-sectional studies that included HIV-negative patients as controls have demonstrated changes in lipid processing that are known to promote atherosclerosis. For example, persons with HIV infection have smaller LDL-C particles30 and higher levels of circulating oxidized LDL-C.31
In the Multicenter AIDS Cohort Study (MACS), after HIV seroconversion, nonfasting total cholesterol, LDL-C, and HDL-C levels declined, which is consistent with a chronic inflammatory state. After antiretroviral therapy was started, lipid levels returned to baseline levels or slightly higher except for HDL-C, which remained low.9 These changes may be due to a general “return to health,” or they may be direct medication effects.
Similar patterns were seen in the SMART study.28 Participants randomized to receive intermittent antiretroviral therapy had overall decreases in all lipid levels, with a marked reduction in HDL-C, while those randomized to receive continuous therapy had increased levels of all lipids, including HDL-C, at 12 months. Overall, the ratio of total cholesterol to HDL-C actually increased for participants on episodic therapy, while it decreased in the continuous-treatment group. Along with continued vascular inflammation, the low HDL-C may have contributed to the worse cardiovascular outcomes in patients who received intermittent antiretroviral therapy.
Some lipid changes associated with antiretroviral therapy may actually be beneficial. For example, nonnucleoside reverse transcriptase inhibitors may raise HDL-C levels. However, such increases alone do not necessarily offset the other lipid changes or translate to an observed improvement in coronary risk.32
The degree of dyslipidemia and specific lipid changes differ among the different classes of antiretroviral drugs and even among the individual drugs within each class. Furthermore, the magnitude of the observed lipid changes varies widely among patients on the same antiretroviral regimen, reflecting the likely important role of host genomics.
While the protease inhibitors and nonnucleoside reverse transcriptase inhibitors have well-described effects on lipids (described in greater detail in the following sections), there have been no reported significant changes in lipid profiles or cardiovascular risk associated with the newest classes, ie, fusion inhibitors such as enfuvirtide (Fuzeon), CC chemokine receptor type 5 (CCR5) receptor inhibitors such as maraviroc (Selzentry), or integrase inhibitors such as raltegravir (Isentress).
Impact of protease inhibitors on lipids
Ritonavir (Norvir) and ritonavir-boosted protease inhibitor combinations cause the most significant increases in lipids. Currently, ritonavir is used in low doses to boost the levels of most other protease inhibitors as the standard of care in protease inhibitor-based regimens. However, in most patients, giving ritonavir with protease inhibitors raises lipid levels, particularly triglycerides.
Most boosted protease inhibitor regimens have similar effects on lipid levels, with some exceptions.
Tipranavir (Aptivus) plus ritonavir, for example, markedly raises total cholesterol and triglyceride levels and would not be recommended for patients with dyslipidemia at baseline.33
Atazanavir (Reyataz)34,35 plus ritonavir and darunavir (Prezista)36 plus ritonavir cause more modest lipid changes. Unboosted atazanavir raises lipid levels only minimally, if at all,34,35 but it is no longer a preferred regimen according to US Department of Health and Human Services guidelines.42
Impact of nonnucleoside reverse transcriptase inhibitors on lipids
Efavirenz (Sustiva), a nonnucleoside reverse transcriptase inhibitor, when added to a regimen of two or three nucleoside reverse transcriptase inhibitors, resulted in modest increases in all lipids, including HDL-C (a potentially beneficial change) at 96 weeks compared with a regimen of three nucleoside reverse transcriptase inhibitors only.43
Nevirapine (Viramune), compared with efavirenz, results in a more favorable lipid profile in previously untreated patients, as shown by larger increases in HDL-C and smaller increases in triglycerides at 48 weeks.44
Etravirine (Intelence), the newest nonnucleoside reverse transcriptase inhibitor, does not appear to cause any further increase in lipids when added to a regimen containing darunavir-ritonavir and nucleoside agents.45
Impact of nucleoside reverse transcriptase inhibitors on lipids
As a class, nucleoside reverse transcriptase inhibitors have been associated with mitochondrial toxicity and insulin resistance,46 but the lipid changes associated with them are generally less significant than those caused by protease inhibitors or nonnucleoside reverse transcriptase inhibitors. Nevertheless, within the class, there is considerable variability in lipid changes associated with specific agents.
Stavudine (Zerit), for example, is associated with hypertriglyceridemia.
Tenofovir (Viread), for another example, in combination with emtricitabine (Emtriva) and the nonnucleoside reverse transcriptase inhibitor efavirenz (the three drugs are contained in a formulation called Atripla) was associated with a smaller increase in fasting total cholesterol than with zidovudine-lamivudine and efavirenz at 96 weeks.47
A recent placebo-controlled, crossover, pilot study of 17 HIV-infected patients suggested that tenofovir may actually have independent lipid-lowering properties.48
Abacavir, as discussed above, has been reported to be associated with a higher risk of myocardial infarction, but this is debatable.
MANAGING CORONARY RISK FACTORS IN HIV-INFECTED PATIENTS
Cardiovascular risk assessment
In HIV patients, cardiovascular risk can be assessed using models derived from large epidemiologic studies such as the Framingham Heart Study.49
Current guidelines from the Infectious Diseases Society of America and the AIDS Clinical Trials Group (ACTG) for evaluating and managing dyslipidemia in HIV-infected adults are based on the National Cholesterol Education Program Adult Treatment Panel III.50 They recommend obtaining a fasting lipid profile before starting antiretroviral therapy and within 3 to 6 months after starting a new regimen.
The guidelines also recommend stratifying risk by counting the number of cardiovascular risk factors, as is done for the general population. If the patient has more than two factors, the Framingham equation should be used to calculate the 10-year risk of myocardial infarction or cardiac death. Interventions should be offered for modifiable cardiovascular risk factors such as smoking, hypertension, physical inactivity, and diabetes mellitus. LDL-C goals should be determined, and lipid-lowering drugs should be initiated accordingly. If triglyceride levels are 200 to 500 mg/dL and levels of “non-HDL-C” (total cholesterol minus the HDL-C level) are high, a statin is recommended. If the triglyceride level is higher than 500 mg/dL, a fibrate should be started.51
Dyslipidemia management
In HIV patients, statin and fibrate therapy must be considered cautiously, given the important drug interactions with protease inhibitors and especially ritonavir. Many statins are metabolized by cytochrome P3A4, which protease inhibitors inhibit.
Statins generally considered safe to use with most protease inhibitors:
- Pravastatin (Pravachol)
- Rosuvastatin (Crestor)
- Atorvastatin (Lipitor).
Exceptions and caveats:
- Pravastatin should not be prescribed with boosted darunavir.
- Data for fluvastatin (Lescol) in HIV-infected patients on antiretroviral therapy are limited.
- Lovastatin (Mevacor) and simvastatin (Zocor) are contraindicated with protease inhibitor therapy.52
- In contrast to the increase in statin levels seen with protease inhibitors, efavirenz lowers levels of simvastatin, pravastatin, and atorvastatin.53,54
Ezetimibe (Zetia), which is metabolized independently of the cytochrome P450 system, has been shown to be safe and effective when given to HIV-infected patients on antiretroviral therapy.58
Fenofibrate (Lofibra) is recommended by current guidelines for patients with elevated triglyceride levels (> 500 mg/dL).51 In the ACTG 5087 study, a combination of fenofibrate plus pravastatin was found to be safe and effective in improving lipid profiles.59
Long-acting niacin resulted in significant improvements in triglycerides, total cholesterol, HDL-C, and LDL-C after 48 weeks of use, although insulin sensitivity worsened.60
Fish oil has been shown to be an effective alternative to fibrates, or it can be used in combination with them.61
Switching antiretroviral agents vs adding lipid-lowering agents. In some patients with significant dyslipidemia, switching antiretro viral agents may lower lipid levels without compromising virologic control.62 However, due to the multifactorial nature of dyslipidemia in HIV patients on antiretroviral therapy, switching the HIV therapy alone may not result in sufficient improvement in the lipid profile45 and may be associated with virologic failure, particularly among patients who have underlying treatment-resistant HIV.63
In many cases, adding lipid-lowering agents may be more beneficial than switching the antiretroviral therapy. For example, a randomized trial in HIV-infected patients with hyperlipidemia found that adding a lipid-lowering agent such as pravastatin or bezafibrate to the unchanged antiretroviral regimen resulted in greater improvement in total cholesterol, LDL-C, and triglyceride levels than switching from a protease inhibitor to either nevirapine or efavirenz.64
Given the complexity of prescribing lipid-lowering therapies to patients on antiretroviral therapy, we recommend that providers check with a pharmacist or refer to package inserts and other medical literature if they are unfamiliar with these drug interactions and responses to lipid-lowering therapies.
Managing insulin resistance
Diabetes mellitus is a well-known risk factor for coronary heart disease. The Data Collection on Adverse Events of Anti-HIV Drugs study found a higher incidence of coronary heart disease in HIV-infected patients, with higher rates in those with longer duration of diabetes.65 The prevalence of diabetes in HIV-infected populations varies, depending on demographic characteristics,65,66 prevalence of coinfection with hepatitis C virus,66 and prevalence of exposure to antiretroviral drugs67 in the study population.
Drugs that lessen insulin resistance include the thiazolidinedione rosiglitazone (Avandia) and the biguanide metformin (Glucophage). In a randomized trial, both drugs, alone or in combination, improved insulin sensitivity in HIV-infected patients, but neither lessened the amount of visceral or subcutaneous fat.68
Smoking cessation
Smoking is another well-known modifiable risk factor for coronary heart disease.
The prevalence of smoking is usually higher in HIV patients than in HIV-negative people. For example, a French cohort study reported smoking prevalence rates of 56.6% in HIV-infected men vs 32.7% in HIV-negative men; in women, the rates were 58% vs 28.1%. The 5-year relative risk of coronary heart disease in HIV-infected vs HIV-negative persons was 1.20 for men and 1.59 for women. The estimated attributable risk due to smoking was 65% for men and 29% for women.3
Therefore, smoking cessation should be a top priority in managing cardiovascular risk in HIV-infected patients. In fact, control of modifiable risk factors through lifestyle changes such as smoking cessation, dietary changes, and exercise is likely to have a significant impact on cardiovascular risk in this population.
- Palella FJ, Baker RK, Moorman AC, et al; HIV Outpatient Study Investigators. Mortality in the highly active antiretroviral therapy era: changing causes of death and disease in the HIV outpatient study. J Acquir Immune Defic Syndr 2006; 43:27–34.
- Lichtenstein KA, Armon C, Buchacz K, Moorman AC, Wood KC, Brooks JT; HOPS investigators. Analysis of cardiovascular risk factors in the HIV Outpatient Study (HOPS) cohort. Presented at the 13th Conference on Retroviruses and Opportunistic Infections; Denver, CO; 2006.
- Savès M, Chêne G, Ducimetière P, et al; French WHO MONICA Project and the APROCO (ANRS EP11) Study Group. Risk factors for coronary heart disease in patients treated for human immunodeficiency virus infection compared with the general population. Clin Infect Dis 2003; 37:292–298.
- Kaplan RC, Kingsley LA, Sharrett AR, et al. Ten-year predicted coronary heart disease risk in HIV-infected men and women. Clin Infect Dis 2007; 45:1074–1081.
- Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab 2007; 92:2506–2512.
- Klein D, Hurley LB, Quesenberry CP, Sidney S. Do protease inhibitors increase the risk for coronary heart disease in patients with HIV-1 infection? J Acquir Immune Defic Syndr 2002; 30:471–477.
- Grunfeld C, Delaney JA, Wanke C, et al. Preclinical atherosclerosis due to HIV infection: carotid intima-medial thickness measurements from the FRAM study. AIDS 2009; 23:1841–1849.
- Hulten E, Mitchell J, Scally J, Gibbs B, Villines TC. HIV positivity, protease inhibitor exposure and subclinical atherosclerosis: a systematic review and meta-analysis of observational studies. Heart 2009; 95:1826–1835.
- Riddler SA, Smit E, Cole SR, et al. Impact of HIV infection and HAART on serum lipids in men. JAMA 2003; 289:2978–2982.
- Shahmanesh M, Das S, Stolinski M, et al. Antiretroviral treatment reduces very-low-density lipoprotein and intermediate-density lipoprotein apolipoprotein B fractional catabolic rate in human immunodeficiency virus-infected patients with mild dyslipidemia. J Clin Endocrinol Metab 2005; 90:755–760.
- Mujawar Z, Rose H, Morrow MP, et al. Human immunodeficiency virus impairs reverse cholesterol transport from macrophages. PLoS Biol 2006; 4:e365.
- Park IW, Wang JF, Groopman JE. HIV-1 Tat promotes monocyte chemoattractant protein-1 secretion followed by transmigration of monocytes. Blood 2001; 97:352–358.
- Fisher SD, Miller TL, Lipshultz SE. Impact of HIV and highly active antiretroviral therapy on leukocyte adhesion molecules, arterial inflammation, dyslipidemia, and atherosclerosis. Atherosclerosis 2006; 185:1–11.
- Hsue PY, Hunt PW, Schnell A, et al. Role of viral replication, antiretroviral therapy, and immunodeficiency in HIV-associated atherosclerosis. AIDS 2009; 23:1059–1067.
- Currier JS, Taylor A, Boyd F, et al. Coronary heart disease in HIV-infected individuals. J Acquir Immune Defic Syndr 2003; 33:506–512.
- DAD Study Group; Friis-Møller N, Reiss P, Sabin CA, et al. Class of antiretroviral drugs and the risk of myocardial infarction. N Engl J Med 2007: 356:1723–1735.
- DAD Study Group; Sabin CA, Worm SW, Weber R, et al. Use of nucleoside reverse transcriptase inhibitors and risk of myocardial infarction in HIV-infected patients enrolled in the D:A:D study: a multi-cohort collaboration. Lancet 2008; 371:1417–1426.
- Durand M, Sheehy O, Baril JG, Lelorier J, Tremblay C; GRUCHUM Research Center (Groupe de Recherche de l’UHRESS du Centre Hospitalier Universitaire de Montréal). Relation between use of nucleoside reverse transcriptase inhibitors (NRTI) and risk of myocardial infarction (MI): a nested case control study using Quebec’s public health insurance database (QPHID). Presented at the 5th IAS Conference on HIV Pathogenesis, Treatment and Prevention in Cape Town, South Africa, July 17–22, 2009.
- Lang S, Mary-Krause M, Cotte L, et al; the Clinical Epi Group of the French Hospital Database on HIV. Impact of specific NRTI and PI exposure on the risk of myocardial infarction: a case-control study nested within FHDH ANRS CO4. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Strategies for Management of Anti-Retroviral Therapy/INSIGHT. Use of nucleoside reverse transcriptase inhibitors and risk of myocardial infarction in HIV-infected patients. AIDS 2008; 22:F17–F24.
- Bedimo R, Westfall A, Drechsler H, Tebas P. Abacavir use and risk of acute myocardial infarction and cerebrovascular disease in the HAART era. Presented at the 5th IAS Conference on HIV Pathogenesis, Treatment and Prevention in Cape Town, South Africa, July 19–22, 2009.
- Brothers CH, Hernandez JE, Cutrell AG, et al. Risk of myocardial infarction and abacavir therapy: no increased risk across 52 GlaxoSmithKline-sponsored clinical trials in adult subjects. J Acquir Immune Defic Syndr 2009; 51:20–28.
- Benson C, Ribaudo H, Zheng E, et al; the ACTG A5001/ALLRT Protocol Team. No Association of Abacavir Use with Risk of Myocardial Infarction or Severe Cardiovascular Disease Events: Results from ACTG A5001. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Torriani FJ, Komarow L, Parker RA, et al; ACTG 5152s Study Team. Endothelial function in human immunodeficiency virus-infected antiretroviral-naive subjects before and after starting potent antiretroviral therapy: The ACTG (AIDS Clinical Trials Group) Study 5152s. J Am Coll Cardiol 2008; 52:569–576.
- Calmy A, Gayet-Ageron A, Montecucco F, et al; STACCATO Study Group. HIV increases markers of cardiovascular risk: results from a randomized, treatment interruption trial. AIDS 2009; 23:929–939.
- van Vonderen MG, Hassink EA, van Agtmael MA, et al. Increase in carotid artery intima-media thickness and arterial stiffness but improvement in several markers of endothelial function after initiation of antiretroviral therapy. J Infect Dis 2009; 199:1186–1194.
- Strategies for Management of Antiretroviral Therapy (SMART) Study Group; El-Sadr WM, Lundgren JD, Neaton JD, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med 2006; 355:2283–2296.
- Phillips AN, Carr A, Neuhaus J, et al. Interruption of antiretroviral therapy and risk of cardiovascular disease in persons with HIV-1 infection: exploratory analyses from the SMART trial. Antivir Ther 2008; 13:177–187.
- Kuller LH, Tracy R, Belloso WINSIGHT SMART Study Group. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med 2008; 5:e203.
- Badiou S, De Boever CM, Dupuy AM, Baillat V, Cristol JP, Reynes J. Small dense LDL and atherogenic lipid profile in HIV-positive adults: influence of lopinavir/ritonavir-containing regimen. AIDS 2003; 17:772–774.
- Duong M, Petit JM, Martha B, et al. Concentration of circulating oxidized LDL in HIV-infected patients treated with antiretroviral agents: relation to HIV-related lipodystrophy. HIV Clin Trials 2006; 7:41–47.
- Fisac C, Fumero E, Crespo M, et al. Metabolic benefits 24 months after replacing a protease inhibitor with abacavir, efavirenz or nevirapine. AIDS 2005; 19:917–925.
- Hicks CB, Cahn P, Cooper DA, et al; RESIST investigator group. Durable efficacy of tipranavir-ritonavir in combination with an optimised background regimen of antiretroviral drugs for treatmentexperienced HIV-1-infected patients at 48 weeks in the Randomized Evaluation of Strategic Intervention in multi-drug reSistant patients with Tipranavir (RESIST) studies: an analysis of combined data from two randomised open-label trials. Lancet 2006; 368:466–475.
- Malan DR, Krantz E, David N, Wirtz V, Hammond J, McGrath D; 089 Study Group. Efficacy and safety of atazanavir, with or without ritonavir, as part of once-daily highly active antiretroviral therapy regimens in antiretroviral-naive patients. J Acquir Immune Defic Syndr 2008; 47:161–167.
- Anastos K, Lu D, Shi Q, et al. Association of serum lipid levels with HIV serostatus, specific antiretroviral agents, and treatment regimens. J Acquir Immune Defic Syndr 2007; 45:34–42.
- Tomaka F, Lefebvre E, Sekar V, et al. Effects of ritonavir-boosted darunavir vs ritonavir-boosted atazanavir on lipid and glucose parameters in HIV-negative, healthy volunteers. HIV Med 2009; 10:318–327.
- Eron J, Yeni P, Gathe J, et al; KLEAN study team. The KLEAN study of fosamprenavir-ritonavir versus lopinavir-ritonavir, each in combination with abacavir-lamivudine, for initial treatment of HIV infection over 48 weeks: a randomised non-inferiority trial. Lancet 2006; 368:476–482.
- Shafran SD, Mashinter LD, Roberts SE. The effect of low-dose ritonavir monotherapy on fasting serum lipid concentrations. HIV Med 2005; 6:421–425.
- Kumar PN, Rodriguez-French A, Thompson MA, et al; ESS40002 Study Team. A prospective, 96-week study of the impact of trizivir, combivir/nelfinavir, and lamivudine/stavudine/nelfinavir on lipids, metabolic parameters and efficacy in antiretroviral-naive patients: effect of sex and ethnicity. HIV Med 2006; 7:85–98.
- Shafran SD, Mashinter LD, Roberts SE. The effect of low-dose ritonavir monotherapy on fasting serum lipid concentrations. HIV Med 2005; 6:421–425.
- Walmsley S, Avihingsanon A, Slim J, et al. Gemini: a noninferiority study of saquinavir/ritonavir versus lopinavir/ritonavir as initial HIV-1 therapy in adults. J Acquir Immune Defic Syndr 2009; 50:367–374.
- DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents— A Working Group of the Office of AIDS Research Advisory Council (OARAC). Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. December 1, 1009. http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Accessed June 29, 2010.
- Shikuma CM, Yang Y, Glesby MJ, et al. Metabolic effects of protease inhibitor-sparing antiretroviral regimens given as initial treatment of HIV-1 Infection (AIDS Clinical Trials Group Study A5095). J Acquir Immune Defic Syndr 2007; 44:540–550.
- van Leth F, Phanuphak P, Stroes E, et al. Nevirapine and efavirenz elicit different changes in lipid profiles in antiretroviral-therapynaive patients infected with HIV-1. PLoS Med 2004; 1:e19.
- Katlama C, Haubrich R, Lalezari J, et al; DUET-1, DUET-2 study groups. Efficacy and safety of etravirine in treatment-experienced, HIV-1 patients: pooled 48 week analysis of two randomized, controlled trials. AIDS 2009; 23:2289–2300.
- Hammond E, Nolan D, James I, Metcalf C, Mallal S. Reduction of mitochondrial DNA content and respiratory chain activity occurs in adipocytes within 6–12 months of commencing nucleoside reverse transcriptase inhibitor therapy. AIDS 2004; 18:815–817.
- Pozniak AL, Gallant JE, DeJesus E, et al. Tenofovir disoproxil fumarate, emtricitabine, and efavirenz versus fixed-dose zidovudine/lamivudine and efavirenz in antiretroviral-naive patients: virologic, immunologic, and morphologic changes—a 96-week analysis. J Acquir Immune Defic Syndr 2006; 43:535–540.
- Tungsiripat M, Kitch D, Glesby M, et al. A pilot study to determine the effect on dyslipidemia of the addition of tenofovir to stable background ART in HIV-infected subjects: results from the A5206 Study Team. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Law MG, Friis-Møller N, El-Sadr WM, et al; D:A:D Study Group. The use of the Framingham equation to predict myocardial infarctions in HIV-infected patients: comparison with observed events in the D:A:D Study. HIV Med 2006; 7:218–230.
- Aberg JA. Cardiovascular complications in HIV management: past, present, and future. J Acquir Immune Defic Syndr 2009; 50:54–64.
- Dubé MP, Stein JH, Aberg JA, et al; Adult AIDS Clinical Trials Group Cardiovascular Subcommittee. Guidelines for the evaluation and management of dyslipidemia in human immunodeficiency virus (HIV)-infected adults receiving antiretroviral therapy: recommendations of the HIV Medical Association of the Infectious Disease Society of America and the Adult AIDS Clinical Trials Group. Clin Infect Dis 2003; 37:613–627.
- Fichtenbaum CJ. Metabolic abnormalities associated with HIV infection and antiretroviral therapy. Curr Infect Dis Rep 2009; 11:84–92.
- Gerber JG, Rosenkranz SL, Fichtenbaum CJ, et al; AIDS Clinical Trials Group A5108 Team. Effect of efavirenz on the pharmacokinetics of simvastatin, atorvastatin, and pravastatin: results of AIDS Clinical Trials Group 5108 Study. J Acquir Immune Defic Syndr 2005; 39:307–312.
- Grennan T, Walmsley S. Etravirine for HIV-I: addressing the limitations of the nonnucleoside reverse transcriptase inhibitor class. J Int Assoc Physicians AIDS Care (Chic Ill) 2009; 8:354–363.
- Sekar V S-GS, Marien K. Pharmacokinetic drug-drug interaction between the new HIV protease inhibitor darunavir (TMC114) and the lipid-lowering agent pravastatin. Presented at the 8th International Workshop on Pharmacology of HIV Therapy; Budapest, Hungary, April 16–18, 2007.
- Kiser JJ, Gerber JG, Predhomme JA, Wolfe P, Flynn DM, Hoody DW. Drug/drug interaction between lopinavir/ritonavir and rosuvastatin in healthy volunteers. J Acquir Immune Defic Syndr 2008; 47:570–578.
- Aslangul E, Assoumou L, Bittar R, et al. Rosuvastatin versus pravastatin in dyslipidemic HIV-1-infected patients receiving protease inhibitors: a randomized trial. AIDS 2010; 24:77–83.
- Chow D, Chen H, Glesby MJ, et al. Short-term ezetimibe is well tolerated and effective in combination with statin therapy to treat elevated LDL cholesterol in HIV-infected patients. AIDS 2009; 23:2133–2141.
- Aberg JA, Zackin RA, Brobst SW, et al; ACTG 5087 Study Team. A randomized trial of the efficacy and safety of fenofibrate versus pravastatin in HIV-infected subjects with lipid abnormalities: AIDS Clinical Trials Group Study 5087. AIDS Res Hum Retroviruses 2005; 21:757–767.
- Dubé MP, Wu JW, Aberg JA, et al; AIDS Clinical Trials Group A5148 Study Team. Safety and efficacy of extended-release niacin for the treatment of dyslipidaemia in patients with HIV infection: AIDS Clinical Trials Group Study A5148. Antivir Ther 2006; 11:1081–1089.
- Gerber JG, Kitch DW, Fichtenbaum CJ, et al. Fish oil and fenofibrate for the treatment of hypertriglyceridemia in HIV-infected subjects on antiretroviral therapy: results of ACTG A5186. J Acquir Immune Defic Syndr 2008; 47:459–466.
- Mallolas J, Podzamczer D, Milinkovic A, et al; ATAZIP Study Group. Efficacy and safety of switching from boosted lopinavir to boosted atazanavir in patients with virological suppression receiving a LPV/rcontaining HAART: the ATAZIP study. J Acquir Immune Defic Syndr 2009; 51:29–36.
- Eron J, Andrade J, Zajdenverg R, et al. Switching from stable lopinavir/ritonavir-based to raltegravir-based combination ART resulted in a superior lipid profile at week 12 but did not demonstrate noninferior virologic efficacy at week 24. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Calza L, Manfredi R, Colangeli V, et al. Substitution of nevirapine or efavirenz for protease inhibitor versus lipid-lowering therapy for the management of dyslipidaemia. AIDS 2005; 19:1051–1058.
- Worm SW, De Wit S, Weber R, et al. Diabetes mellitus, preexisting coronary heart disease, and the risk of subsequent coronary heart disease events in patients infected with human immunodeficiency virus: the Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D Study). Circulation 2009; 119:805–811.
- Brown TT, Cole SR, Li X, et al. Antiretroviral therapy and the prevalence and incidence of diabetes mellitus in the multicenter AIDS cohort study. Arch Intern Med 2005; 165:1179–1184.
- Butt AA, McGinnis K, Rodriguez-Barradas MC, et al; Veterans Aging Cohort Study. HIV infection and the risk of diabetes mellitus. AIDS 2009; 23:1227–1234.
- Mulligan K, Yang Y, Wininger DA, et al. Effects of metformin and rosiglitazone in HIV-infected patients with hyperinsulinemia and elevated waist/hip ratio. AIDS 2007; 21:47–57.
- Palella FJ, Baker RK, Moorman AC, et al; HIV Outpatient Study Investigators. Mortality in the highly active antiretroviral therapy era: changing causes of death and disease in the HIV outpatient study. J Acquir Immune Defic Syndr 2006; 43:27–34.
- Lichtenstein KA, Armon C, Buchacz K, Moorman AC, Wood KC, Brooks JT; HOPS investigators. Analysis of cardiovascular risk factors in the HIV Outpatient Study (HOPS) cohort. Presented at the 13th Conference on Retroviruses and Opportunistic Infections; Denver, CO; 2006.
- Savès M, Chêne G, Ducimetière P, et al; French WHO MONICA Project and the APROCO (ANRS EP11) Study Group. Risk factors for coronary heart disease in patients treated for human immunodeficiency virus infection compared with the general population. Clin Infect Dis 2003; 37:292–298.
- Kaplan RC, Kingsley LA, Sharrett AR, et al. Ten-year predicted coronary heart disease risk in HIV-infected men and women. Clin Infect Dis 2007; 45:1074–1081.
- Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab 2007; 92:2506–2512.
- Klein D, Hurley LB, Quesenberry CP, Sidney S. Do protease inhibitors increase the risk for coronary heart disease in patients with HIV-1 infection? J Acquir Immune Defic Syndr 2002; 30:471–477.
- Grunfeld C, Delaney JA, Wanke C, et al. Preclinical atherosclerosis due to HIV infection: carotid intima-medial thickness measurements from the FRAM study. AIDS 2009; 23:1841–1849.
- Hulten E, Mitchell J, Scally J, Gibbs B, Villines TC. HIV positivity, protease inhibitor exposure and subclinical atherosclerosis: a systematic review and meta-analysis of observational studies. Heart 2009; 95:1826–1835.
- Riddler SA, Smit E, Cole SR, et al. Impact of HIV infection and HAART on serum lipids in men. JAMA 2003; 289:2978–2982.
- Shahmanesh M, Das S, Stolinski M, et al. Antiretroviral treatment reduces very-low-density lipoprotein and intermediate-density lipoprotein apolipoprotein B fractional catabolic rate in human immunodeficiency virus-infected patients with mild dyslipidemia. J Clin Endocrinol Metab 2005; 90:755–760.
- Mujawar Z, Rose H, Morrow MP, et al. Human immunodeficiency virus impairs reverse cholesterol transport from macrophages. PLoS Biol 2006; 4:e365.
- Park IW, Wang JF, Groopman JE. HIV-1 Tat promotes monocyte chemoattractant protein-1 secretion followed by transmigration of monocytes. Blood 2001; 97:352–358.
- Fisher SD, Miller TL, Lipshultz SE. Impact of HIV and highly active antiretroviral therapy on leukocyte adhesion molecules, arterial inflammation, dyslipidemia, and atherosclerosis. Atherosclerosis 2006; 185:1–11.
- Hsue PY, Hunt PW, Schnell A, et al. Role of viral replication, antiretroviral therapy, and immunodeficiency in HIV-associated atherosclerosis. AIDS 2009; 23:1059–1067.
- Currier JS, Taylor A, Boyd F, et al. Coronary heart disease in HIV-infected individuals. J Acquir Immune Defic Syndr 2003; 33:506–512.
- DAD Study Group; Friis-Møller N, Reiss P, Sabin CA, et al. Class of antiretroviral drugs and the risk of myocardial infarction. N Engl J Med 2007: 356:1723–1735.
- DAD Study Group; Sabin CA, Worm SW, Weber R, et al. Use of nucleoside reverse transcriptase inhibitors and risk of myocardial infarction in HIV-infected patients enrolled in the D:A:D study: a multi-cohort collaboration. Lancet 2008; 371:1417–1426.
- Durand M, Sheehy O, Baril JG, Lelorier J, Tremblay C; GRUCHUM Research Center (Groupe de Recherche de l’UHRESS du Centre Hospitalier Universitaire de Montréal). Relation between use of nucleoside reverse transcriptase inhibitors (NRTI) and risk of myocardial infarction (MI): a nested case control study using Quebec’s public health insurance database (QPHID). Presented at the 5th IAS Conference on HIV Pathogenesis, Treatment and Prevention in Cape Town, South Africa, July 17–22, 2009.
- Lang S, Mary-Krause M, Cotte L, et al; the Clinical Epi Group of the French Hospital Database on HIV. Impact of specific NRTI and PI exposure on the risk of myocardial infarction: a case-control study nested within FHDH ANRS CO4. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Strategies for Management of Anti-Retroviral Therapy/INSIGHT. Use of nucleoside reverse transcriptase inhibitors and risk of myocardial infarction in HIV-infected patients. AIDS 2008; 22:F17–F24.
- Bedimo R, Westfall A, Drechsler H, Tebas P. Abacavir use and risk of acute myocardial infarction and cerebrovascular disease in the HAART era. Presented at the 5th IAS Conference on HIV Pathogenesis, Treatment and Prevention in Cape Town, South Africa, July 19–22, 2009.
- Brothers CH, Hernandez JE, Cutrell AG, et al. Risk of myocardial infarction and abacavir therapy: no increased risk across 52 GlaxoSmithKline-sponsored clinical trials in adult subjects. J Acquir Immune Defic Syndr 2009; 51:20–28.
- Benson C, Ribaudo H, Zheng E, et al; the ACTG A5001/ALLRT Protocol Team. No Association of Abacavir Use with Risk of Myocardial Infarction or Severe Cardiovascular Disease Events: Results from ACTG A5001. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Torriani FJ, Komarow L, Parker RA, et al; ACTG 5152s Study Team. Endothelial function in human immunodeficiency virus-infected antiretroviral-naive subjects before and after starting potent antiretroviral therapy: The ACTG (AIDS Clinical Trials Group) Study 5152s. J Am Coll Cardiol 2008; 52:569–576.
- Calmy A, Gayet-Ageron A, Montecucco F, et al; STACCATO Study Group. HIV increases markers of cardiovascular risk: results from a randomized, treatment interruption trial. AIDS 2009; 23:929–939.
- van Vonderen MG, Hassink EA, van Agtmael MA, et al. Increase in carotid artery intima-media thickness and arterial stiffness but improvement in several markers of endothelial function after initiation of antiretroviral therapy. J Infect Dis 2009; 199:1186–1194.
- Strategies for Management of Antiretroviral Therapy (SMART) Study Group; El-Sadr WM, Lundgren JD, Neaton JD, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med 2006; 355:2283–2296.
- Phillips AN, Carr A, Neuhaus J, et al. Interruption of antiretroviral therapy and risk of cardiovascular disease in persons with HIV-1 infection: exploratory analyses from the SMART trial. Antivir Ther 2008; 13:177–187.
- Kuller LH, Tracy R, Belloso WINSIGHT SMART Study Group. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med 2008; 5:e203.
- Badiou S, De Boever CM, Dupuy AM, Baillat V, Cristol JP, Reynes J. Small dense LDL and atherogenic lipid profile in HIV-positive adults: influence of lopinavir/ritonavir-containing regimen. AIDS 2003; 17:772–774.
- Duong M, Petit JM, Martha B, et al. Concentration of circulating oxidized LDL in HIV-infected patients treated with antiretroviral agents: relation to HIV-related lipodystrophy. HIV Clin Trials 2006; 7:41–47.
- Fisac C, Fumero E, Crespo M, et al. Metabolic benefits 24 months after replacing a protease inhibitor with abacavir, efavirenz or nevirapine. AIDS 2005; 19:917–925.
- Hicks CB, Cahn P, Cooper DA, et al; RESIST investigator group. Durable efficacy of tipranavir-ritonavir in combination with an optimised background regimen of antiretroviral drugs for treatmentexperienced HIV-1-infected patients at 48 weeks in the Randomized Evaluation of Strategic Intervention in multi-drug reSistant patients with Tipranavir (RESIST) studies: an analysis of combined data from two randomised open-label trials. Lancet 2006; 368:466–475.
- Malan DR, Krantz E, David N, Wirtz V, Hammond J, McGrath D; 089 Study Group. Efficacy and safety of atazanavir, with or without ritonavir, as part of once-daily highly active antiretroviral therapy regimens in antiretroviral-naive patients. J Acquir Immune Defic Syndr 2008; 47:161–167.
- Anastos K, Lu D, Shi Q, et al. Association of serum lipid levels with HIV serostatus, specific antiretroviral agents, and treatment regimens. J Acquir Immune Defic Syndr 2007; 45:34–42.
- Tomaka F, Lefebvre E, Sekar V, et al. Effects of ritonavir-boosted darunavir vs ritonavir-boosted atazanavir on lipid and glucose parameters in HIV-negative, healthy volunteers. HIV Med 2009; 10:318–327.
- Eron J, Yeni P, Gathe J, et al; KLEAN study team. The KLEAN study of fosamprenavir-ritonavir versus lopinavir-ritonavir, each in combination with abacavir-lamivudine, for initial treatment of HIV infection over 48 weeks: a randomised non-inferiority trial. Lancet 2006; 368:476–482.
- Shafran SD, Mashinter LD, Roberts SE. The effect of low-dose ritonavir monotherapy on fasting serum lipid concentrations. HIV Med 2005; 6:421–425.
- Kumar PN, Rodriguez-French A, Thompson MA, et al; ESS40002 Study Team. A prospective, 96-week study of the impact of trizivir, combivir/nelfinavir, and lamivudine/stavudine/nelfinavir on lipids, metabolic parameters and efficacy in antiretroviral-naive patients: effect of sex and ethnicity. HIV Med 2006; 7:85–98.
- Shafran SD, Mashinter LD, Roberts SE. The effect of low-dose ritonavir monotherapy on fasting serum lipid concentrations. HIV Med 2005; 6:421–425.
- Walmsley S, Avihingsanon A, Slim J, et al. Gemini: a noninferiority study of saquinavir/ritonavir versus lopinavir/ritonavir as initial HIV-1 therapy in adults. J Acquir Immune Defic Syndr 2009; 50:367–374.
- DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents— A Working Group of the Office of AIDS Research Advisory Council (OARAC). Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. December 1, 1009. http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Accessed June 29, 2010.
- Shikuma CM, Yang Y, Glesby MJ, et al. Metabolic effects of protease inhibitor-sparing antiretroviral regimens given as initial treatment of HIV-1 Infection (AIDS Clinical Trials Group Study A5095). J Acquir Immune Defic Syndr 2007; 44:540–550.
- van Leth F, Phanuphak P, Stroes E, et al. Nevirapine and efavirenz elicit different changes in lipid profiles in antiretroviral-therapynaive patients infected with HIV-1. PLoS Med 2004; 1:e19.
- Katlama C, Haubrich R, Lalezari J, et al; DUET-1, DUET-2 study groups. Efficacy and safety of etravirine in treatment-experienced, HIV-1 patients: pooled 48 week analysis of two randomized, controlled trials. AIDS 2009; 23:2289–2300.
- Hammond E, Nolan D, James I, Metcalf C, Mallal S. Reduction of mitochondrial DNA content and respiratory chain activity occurs in adipocytes within 6–12 months of commencing nucleoside reverse transcriptase inhibitor therapy. AIDS 2004; 18:815–817.
- Pozniak AL, Gallant JE, DeJesus E, et al. Tenofovir disoproxil fumarate, emtricitabine, and efavirenz versus fixed-dose zidovudine/lamivudine and efavirenz in antiretroviral-naive patients: virologic, immunologic, and morphologic changes—a 96-week analysis. J Acquir Immune Defic Syndr 2006; 43:535–540.
- Tungsiripat M, Kitch D, Glesby M, et al. A pilot study to determine the effect on dyslipidemia of the addition of tenofovir to stable background ART in HIV-infected subjects: results from the A5206 Study Team. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Law MG, Friis-Møller N, El-Sadr WM, et al; D:A:D Study Group. The use of the Framingham equation to predict myocardial infarctions in HIV-infected patients: comparison with observed events in the D:A:D Study. HIV Med 2006; 7:218–230.
- Aberg JA. Cardiovascular complications in HIV management: past, present, and future. J Acquir Immune Defic Syndr 2009; 50:54–64.
- Dubé MP, Stein JH, Aberg JA, et al; Adult AIDS Clinical Trials Group Cardiovascular Subcommittee. Guidelines for the evaluation and management of dyslipidemia in human immunodeficiency virus (HIV)-infected adults receiving antiretroviral therapy: recommendations of the HIV Medical Association of the Infectious Disease Society of America and the Adult AIDS Clinical Trials Group. Clin Infect Dis 2003; 37:613–627.
- Fichtenbaum CJ. Metabolic abnormalities associated with HIV infection and antiretroviral therapy. Curr Infect Dis Rep 2009; 11:84–92.
- Gerber JG, Rosenkranz SL, Fichtenbaum CJ, et al; AIDS Clinical Trials Group A5108 Team. Effect of efavirenz on the pharmacokinetics of simvastatin, atorvastatin, and pravastatin: results of AIDS Clinical Trials Group 5108 Study. J Acquir Immune Defic Syndr 2005; 39:307–312.
- Grennan T, Walmsley S. Etravirine for HIV-I: addressing the limitations of the nonnucleoside reverse transcriptase inhibitor class. J Int Assoc Physicians AIDS Care (Chic Ill) 2009; 8:354–363.
- Sekar V S-GS, Marien K. Pharmacokinetic drug-drug interaction between the new HIV protease inhibitor darunavir (TMC114) and the lipid-lowering agent pravastatin. Presented at the 8th International Workshop on Pharmacology of HIV Therapy; Budapest, Hungary, April 16–18, 2007.
- Kiser JJ, Gerber JG, Predhomme JA, Wolfe P, Flynn DM, Hoody DW. Drug/drug interaction between lopinavir/ritonavir and rosuvastatin in healthy volunteers. J Acquir Immune Defic Syndr 2008; 47:570–578.
- Aslangul E, Assoumou L, Bittar R, et al. Rosuvastatin versus pravastatin in dyslipidemic HIV-1-infected patients receiving protease inhibitors: a randomized trial. AIDS 2010; 24:77–83.
- Chow D, Chen H, Glesby MJ, et al. Short-term ezetimibe is well tolerated and effective in combination with statin therapy to treat elevated LDL cholesterol in HIV-infected patients. AIDS 2009; 23:2133–2141.
- Aberg JA, Zackin RA, Brobst SW, et al; ACTG 5087 Study Team. A randomized trial of the efficacy and safety of fenofibrate versus pravastatin in HIV-infected subjects with lipid abnormalities: AIDS Clinical Trials Group Study 5087. AIDS Res Hum Retroviruses 2005; 21:757–767.
- Dubé MP, Wu JW, Aberg JA, et al; AIDS Clinical Trials Group A5148 Study Team. Safety and efficacy of extended-release niacin for the treatment of dyslipidaemia in patients with HIV infection: AIDS Clinical Trials Group Study A5148. Antivir Ther 2006; 11:1081–1089.
- Gerber JG, Kitch DW, Fichtenbaum CJ, et al. Fish oil and fenofibrate for the treatment of hypertriglyceridemia in HIV-infected subjects on antiretroviral therapy: results of ACTG A5186. J Acquir Immune Defic Syndr 2008; 47:459–466.
- Mallolas J, Podzamczer D, Milinkovic A, et al; ATAZIP Study Group. Efficacy and safety of switching from boosted lopinavir to boosted atazanavir in patients with virological suppression receiving a LPV/rcontaining HAART: the ATAZIP study. J Acquir Immune Defic Syndr 2009; 51:29–36.
- Eron J, Andrade J, Zajdenverg R, et al. Switching from stable lopinavir/ritonavir-based to raltegravir-based combination ART resulted in a superior lipid profile at week 12 but did not demonstrate noninferior virologic efficacy at week 24. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Calza L, Manfredi R, Colangeli V, et al. Substitution of nevirapine or efavirenz for protease inhibitor versus lipid-lowering therapy for the management of dyslipidaemia. AIDS 2005; 19:1051–1058.
- Worm SW, De Wit S, Weber R, et al. Diabetes mellitus, preexisting coronary heart disease, and the risk of subsequent coronary heart disease events in patients infected with human immunodeficiency virus: the Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D Study). Circulation 2009; 119:805–811.
- Brown TT, Cole SR, Li X, et al. Antiretroviral therapy and the prevalence and incidence of diabetes mellitus in the multicenter AIDS cohort study. Arch Intern Med 2005; 165:1179–1184.
- Butt AA, McGinnis K, Rodriguez-Barradas MC, et al; Veterans Aging Cohort Study. HIV infection and the risk of diabetes mellitus. AIDS 2009; 23:1227–1234.
- Mulligan K, Yang Y, Wininger DA, et al. Effects of metformin and rosiglitazone in HIV-infected patients with hyperinsulinemia and elevated waist/hip ratio. AIDS 2007; 21:47–57.
KEY POINTS
- Traditional risk factors are the main contributors to cardiovascular disease in this population, although HIV infection is independently associated with increased cardiovascular risk.
- Antiretroviral therapy contributes modestly to the risk of coronary heart disease. Antiretroviral combinations that include protease inhibitors cause the most substantial deleterious changes in lipid levels.
- Most changes in lipids and insulin resistance can be managed by adding lipid-lowering and antiglycemic agents and may not require changes to the antiretroviral regimen.
- Close attention to drug interactions is important when selecting lipid-lowering medications for patients on antiretroviral therapy to avoid dangerous increases in the levels of certain statins.
- Addressing modifiable risk factors such as smoking, obesity, and sedentary lifestyle can have a far greater impact on cardiovascular risk than changes in antiretroviral therapy.
How to prevent glucocorticoid-induced osteoporosis
Although glucocorticoid drugs such as prednisone, methylprednisolone, and dexamethasone have many benefits, they are the number-one cause of secondary osteoporosis. 1 When prescribing them for long-term therapy, physicians should take steps to prevent bone loss and fractures.
Being inexpensive and potent anti-inflammatory drugs, glucocorticoids are widely used to treat many diseases affecting millions of Americans, such as dermatologic conditions, inflammatory bowel disease, pulmonary diseases (eg, asthma, chronic obstructive pulmonary disease, interstitial lung disease), renal diseases (eg, glomerulonephritis), rheumatologic disorders (eg, rheumatoid arthritis, lupus, vasculitis, polymyalgia rheumatica), and transplant rejection.
This article discusses the mechanisms of glucocorticoid-induced bone loss and guidelines for preventing and treating it.
GLUCOCORTICOIDS PROMOTE BONE LOSS DIRECTLY AND INDIRECTLY
The pathophysiology of glucocorticoid-induced osteoporosis is much more complicated than was previously thought.
The older view was that these drugs mostly affect bone indirectly by inhibiting calcium absorption, causing secondary hyperparathyroidism. Indeed, they do inhibit calcium absorption from the gastrointestinal tract and induce renal calcium loss. However, most patients do not have elevated levels of parathyroid hormone.
Now, reduced bone formation rather than increased bone resorption is thought to be the predominant effect of glucocorticoids on bone turnover, as these drugs suppress the number and the activity of osteoblasts.
Direct effects on bone
Glucocorticoids directly affect bone cells in a number of ways—eg, by stimulating osteoclastogenesis, decreasing osteoblast function and life span, increasing osteoblast apoptosis, and impairing preosteoblast formation.2
Glucocorticoids also increase osteocyte apoptosis.3 Osteocytes, the most numerous bone cells, are thought to be an integral part of the “nervous system” of bone, directing bone-remodeling units to locations where repair of bone microfractures or removal of bone is needed. Osteocyte apoptosis caused by glucocorticoids may disrupt the signaling process, resulting in increased osteoclast activity in an area of apoptotic osteocytes and the inability to directly repair bone, thus impairing the bone’s ability to preserve its strength and architecture. Such disruption may affect bone quality and increase the risk of fracture independent of any decrease in bone mineral density. 4
Direct molecular effects
Glucocorticoids have been found to:
- Block the stimulatory effect of insulin-like growth factor 1 on bone formation5
- Oppose Wnt/beta-catenin signaling, resulting in decreased bone formation6
- Affect stromal cell differentiation, shunting cell formation towards more adipocyte formation so that fewer osteoblasts and chondrocytes are formed, resulting in less bone formation
- Increase levels of receptor activator of nuclear factor kappa (RANK) ligand and macrophage colony-stimulating factor and decrease levels of osteoprotegerin, resulting in increased osteoclastogenesis and increased bone resorption7
- Decrease estrogen, testosterone, and adrenal androgen levels, which also have adverse effects on bone cells.8
Inflammatory diseases also affect bone
Furthermore, many patients taking glucocorticoids are already at risk of osteoporosis because many of the diseases that require these drugs for treatment are associated with bone loss due to their inflammatory nature. In rheumatoid arthritis, RANK ligand, one of the cytokines involved in inflammation, causes bony erosions and also causes localized osteopenia. The malabsorption of calcium and vitamin D in inflammatory bowel disease is a cause of secondary osteoporosis.
Trabecular bone is affected first
The degree of bone loss in patients receiving glucocorticoids can vary markedly, depending on the skeletal site. Initially, these drugs affect trabecular bone because of its higher metabolic activity, but with prolonged use cortical bone is also affected.2 Greater trabecular thinning is seen in glucocorticoid-induced osteoporosis than in postmenopausal osteoporosis, in which more trabecular perforations are seen.9
Bone loss occurs rapidly during the first few months of glucocorticoid therapy, followed by a slower but continued loss with ongoing use.
FRACTURE RISK INCREASES RAPIDLY
With this decrease in bone mass comes a rapid increase in fracture risk, which correlates with the dose of glucocorticoids and the duration of use.10 Vertebral fractures resulting from prolonged cortisone use were first described in 1954.11
A dosage of 5 mg or more of prednisolone or its equivalent per day decreases bone mineral density and rapidly increases the risk of fracture over 3 to 6 months. The relative risks12:
- Any fracture—1.33 to 1.91
- Hip fracture—1.61 to 2.01
- Vertebral fracture—2.60 to 2.86
- Forearm fracture—1.09 to 1.13.
These risks are independent of age, sex, and underlying disease.12
Patients receiving glucocorticoids may suffer vertebral and hip fractures at higher bone mineral density values than patients with postmenopausal osteoporosis. In 2003, van Staa et al13 reported that, at any given bone mineral density, the incidence of new vertebral fracture in postmenopausal women receiving glucocorticoids was higher than in nonusers. This suggests that glucocorticoids have both a qualitative and a quantitative effect on bone.
Glucocorticoids also cause a form of myopathy, which increases the propensity to fall, further increasing the risk of fractures.
Fracture risk declines after oral glucocorticoids are stopped, reaching a relative risk of 1 approximately 2 years later.12 However, keep in mind that the underlying conditions being treated by the glucocorticoids also increase the patient’s fracture risk. Therefore, the patient’s risk of fracture needs to be evaluated even after stopping the glucocorticoid.
INHALED STEROIDS IN HIGH DOSES MAY ALSO INCREASE RISK
Although inhaled glucocorticoids are generally believed not to affect bone, some evidence suggests that in high doses (> 2,000 μg/day) they may result in significant osteoporosis over several years.14,15
In a retrospective cohort study, van Staa et al15 compared the risk of fracture in 171,000 patients taking the inhaled glucocorticoids fluticasone (Flovent), budesonide (Pulmicort), or beclomethasone (Beconase); 109,000 patients taking inhaled nonglucocorticoid bronchodilators; and 171,000 controls not using inhalers. They found no differences between the inhaled glucocorticoid and nonglucocorticoid bronchodilator groups in the risk of nonvertebral fracture. Users of inhaled glucocorticoids had a higher risk of fracture, particularly of the hip and spine, than did controls, but this may have been related more to the severity of the underlying respiratory disease than to the inhaled glucocorticoids.
Weldon et al16 suggested preventive measures to prevent glucocorticoid-induced effects on bone metabolism when prescribing inhaled glucocorticoids to children. They stated that prophylaxis against osteoporosis requires suspicion, assessment of bone density, supplemental calcium and vitamin D, and, if indicated, bisphosphonates to prevent bone fractures that could compromise the patient’s quality of life.
PREVENTING AND TREATING BONE LOSS DUE TO GLUCOCORTICOIDS
Effective options are available to prevent the deleterious effects of glucocorticoids on bone.
A plethora of guidelines offer direction on how to reduce fracture risk—ie, how to maintain bone mineral density while preventing additional bone loss, alleviating pain associated with existing fractures, maintaining and increasing muscle strength, and initiating lifestyle changes as needed.17,18 Guidelines from the American College of Rheumatology (ACR),17 published in 2001, are being updated. United Kingdom (UK) guidelines,18 published in December 2002, differ slightly from those of the ACR.
Limit exposure to glucocorticoids
Oral glucocorticoids should be given in the lowest effective dose for the shortest possible time. However, there is no safe oral glucocorticoid dose with respect to bone. Alternate-day dosing suppresses the adrenal axis less but has the same effect as daily dosing with regard to bone.
Recommend lifestyle measures from day 1
All guidelines recommend that as soon as a patient is prescribed a glucocorticoid, the clinician should prescribe certain preventive measures, including:
- Smoking cessation
- Weight-bearing and strength-building exercises
- Calcium intake of 1,000 to 1,500 mg per day
- Vitamin D 800 to 1,000 IU per day.
Calcium and vitamin D for all
The Cochrane Database of Systematic Reviews19 evaluated the data supporting the recommendation to use calcium and vitamin D as preventive therapy in patients receiving glucocorticoids. Five trials with 274 patients were included in the meta-analysis. At 2 years after starting calcium and vitamin D, there was a significant weighted mean difference of 2.6% (95% confidence interval [CI] 0.7–4.5) between the treatment and control groups in lumbar spine bone mineral density.
The authors concluded that because calcium and vitamin D have low toxicity and are inexpensive, all patients starting glucocorticoids should also take a calcium and a vitamin D supplement prophylactically.
Bisphosphonates are effective and recommended
The ACR17 and UK18 guidelines said that bisphosphonates are effective for preventing and treating bone loss in patients receiving glucocorticoids.
More recently, Stoch et al20 evaluated the efficacy and safety of alendronate (Fosamax) 70 mg weekly for preventing and treating bone loss in patients on glucocorticoid therapy. At 12 months, bone mineral density in the lumbar spine, trochanter, and total hip had increased from baseline in the alendronate group and was significantly higher than in the placebo group. At the same time, levels of biochemical markers of bone remodeling were significantly lower than at baseline in the alendronate group.
For premenopausal women, postmenopausal women on estrogen replacement therapy, and men, the ACR17 recommends risedronate (Actonel) 5 mg per day or alendronate 5 mg per day; for postmenopausal women not on estrogen, risedronate 5 mg per day or alendronate 10 mg per day is recommended.
Who should receive a bisphosphonate?
In men and postmenopausal women, the ACR17 recommends a bisphosphonate for patients starting long-term glucocorticoid treatment (ie, expected to last 3 months or more) in doses of 5 mg or more per day of prednisone or its equivalent, irrespective of bone mineral density values.
In patients already taking glucocorticoids, a bisphosphonate should be started if the bone mineral density is below a certain threshold. The rationale for using bone mineral thresholds instead of giving bisphosphonates to all is that these drugs have potentially significant side effects and so should not be prescribed if not needed. The appropriate threshold at which intervention should be considered in glucocorticoid-treated patients is a matter of controversy. Based on evidence that fractures occur at a higher bone mineral density in glucocorticoid-treated patients than in postmenopausal women, the UK guidelines18 recommend starting a bisphosphonate if the T score is less than −1.5 at the spine or hip, but the ACR17 guidelines propose a T-score cutoff of −1.0. Whichever cutoff is chosen, its significance in terms of absolute fracture risk will differ according to the age of the patient. Therefore, use of T scores as an intervention threshold is not advisable.
The ACR and the UK guidelines both recommend measuring the bone mineral density by dual-energy x-ray absorptiometry at baseline (even though preventive therapy is not based on this value) and repeating it 6 months later and then yearly.
In premenopausal women, bisphosphonates should be used with caution, as they cross the placenta and are teratogenic in animals. Nevertheless, the ACR guidelines17 state they can be given after appropriate counseling and instruction about contraception.
The UK guidelines18 note that in the large clinical trials of alendronate and risedronate, the incidence of vertebral fractures was low in premenopausal women, indicating a very low fracture risk. Therefore, the UK guidelines state that bone-active drugs should be reserved for premenopausal women who have very low bone mineral density or who suffer fragility fractures or who have other strong risk factors for fracture.
In children and adolescents, the data are insufficient to produce evidence-based guidelines for the prevention and treatment of glucocorticoid-induced osteoporosis. General measures include using the lowest effective dose of glucocorticoids for the shortest period of time, and considering alternate therapies, calcium and vitamin D supplementation, weight-bearing exercise, and proper nutrition.
Bisphosphonates are recommended when bone mineral density is falling despite these general measures and when “high-dose” glucocorticoids are likely to be used for a “prolonged” time, or in patients who have already had a fracture.21
Weekly doses may improve compliance
Risedronate is approved by the US Food and Drug Administration (FDA) for the prevention of glucocorticoid-induced osteoporosis, and both risedronate and alendronate are approved for its treatment.
The ACR guidelines recommend the FDA-approved (ie, daily) doses of alendronate and risedronate for glucocorticoid-induced osteoporosis. Most patients, however, are pre-scribed weekly doses of these two agents, as compliance is much greater with this schedule of administration.
Estrogen is being used more selectively
The 2001 ACR guidelines said that, although there were no randomized controlled trials of hormone replacement (or testosterone) therapy to prevent glucocorticoid-induced bone loss, patients receiving long-term glucocorticoid therapy who are hypogonadal should be offered hormone replacement therapy.17
In 2002, the principal results of the Women’s Health Initiative22 showed that hormone replacement therapy with estrogen and progesterone was associated with a higher risk of breast cancer. Since then, the consensus has been that hormone replacement therapy should be restricted to women with menopausal symptoms or to older women who cannot tolerate other therapies or who express a strong preference for hormone replacement therapy despite being informed about potential adverse events.23
A role for testosterone?
Since a daily dose of more than 5 to 7.5 mg of prednisone increases the risk of gonadotropin and testosterone suppression,24 testosterone replacement therapy has been used to treat glucocorticoid-induced osteoporosis in men.
In two placebo-controlled trials in men receiving glucocorticoid therapy for bronchial asthma or chronic obstructive pulmonary disease, testosterone therapy was associated with a significant 4% increase (95% CI 2–7) in bone mineral density in the lumbar spine.25,26
While these studies cannot be considered conclusive in view of their small size and the lack of fracture data, the Endocrine Society currently recommends that men with chronic obstructive pulmonary disease who are receiving glucocorticoids, are hypogonadal, and have no contraindications to androgen replacement therapy (eg, prostate cancer) be offered testosterone therapy to preserve lean body mass and bone mineral density.27
Calcitonin is not a first-line therapy
Neither the ACR nor the UK guidelines recommended calcitonin as first-line therapy.
A Cochrane systematic review28 evaluated the data on the use of calcitonin to prevent and treat glucocorticoid-induced osteoporosis. Nine trials met the inclusion criteria, and included 221 patients randomized to receive calcitonin and 220 patients who received placebo. Calcitonin was more effective than placebo in preserving bone density in the lumbar spine, with a weighted mean difference of 2.8% (95% CI 1.4–4.3) at 6 months and 3.2% (95% CI 0.3–6.1) at 12 months. However, at 24 months, the lumbar spine bone mineral density was not statistically different between groups, nor was the relative risk of fractures. Calcitonin was given subcutaneously in one trial, in which it showed a substantially greater degree of prevention of bone loss than in the other trials, in which it was given nasally.
NEWLY APPROVED AND INVESTIGATIONAL AGENTS
Zoledronic acid once a year
Zoledronic acid (Reclast), a bisphosphonate given intravenously once a year, was approved for glucocorticoid-induced osteoporosis after the ACR and UK guidelines were published.
Zoledronic acid underwent a randomized multicenter, double-blind, active control trial29 in 833 men and women, age range 18 to 85 years, who had glucocorticoid-induced osteoporosis (they had been treated with 7.5 mg per day or more of prednisone or its equivalent). Of these patients, 416 received a single infusion of 5 mg of zoledronic acid and daily oral placebo, and 417 received a single placebo infusion and daily oral risedronate 5 mg as an active control. All patients also received 1,000 mg of calcium and 400 to 1,000 IU of vitamin D per day. The study duration was 1 year.
Of those who had received a glucocorticoid for more than 3 months, those who received zoledronic acid had a significantly greater mean increase in lumbar spine bone mineral density compared with those in the oral risedronate group: 4.1% vs 2.7%, an absolute difference of 1.4% (P < .0001).
In those who had received a glucocorticoid for 3 months or less, those who received zoledronic acid also had a significantly greater mean increase in lumbar spine bone mineral density compared with those in the risedronate group at 1 year: 2.6% vs 0.6%, a treatment difference of 2% (P < .0001).
Bone biopsy specimens were obtained from 23 patients, 12 in the zoledronic acid group and 11 in the risedronate group.30 Qualitative assessment showed normal bone architecture and quality without mineralization defects. Apparent reductions in activation frequency and remodeling rates were seen when compared with the histomorphometric results in the zoledronic acid postmenopausal osteoporosis population.31 The long-term consequences of this degree of suppression of bone remodeling in the glucocorticoid-treated patients are unknown.
The overall safety and tolerability of zoledronic acid in the glucocorticoid-induced osteoporosis population was similar to that in the postmenopausal osteoporosis clinical trial.29,31 Adverse reactions reported in at least 2% of patients that were either not reported in the postmenopausal osteoporosis trial or were reported more frequently in the glucocorticoid-induced trial included the following: abdominal pain, musculoskeletal pain, nausea, and dyspepsia. The incidence of serious adverse events was similar in the zoledronic acid and the active control groups. In the zoledronic acid group, 2.2% of the patients withdrew from the study due to adverse events vs 1.4% in the active control group.
Teriparatide, a parathyroid hormone drug
Teriparatide (Forteo) consists of a fragment of the human parathyroid hormone molecule. It is given once daily by subcutaneous injection. It was also approved for treating glucocorticoid-induced osteoporosis after the current guidelines were written.
Teriparatide was compared with alendronate in a randomized, double-blind trial in patients with glucocorticoid-induced osteoporosis. 32 Entry criteria were treatment with at least 5 mg of prednisone per day for at least 3 months before screening and a T score of −2.0 or less in the lumbar spine, total hip, or femoral neck, or −1.0 or less plus one or more fragility fractures.
Eighty-three men and 345 women ages 21 or older were enrolled and randomized to receive injectable teriparatide 20 μg per day plus oral placebo or oral alendronate 10 mg per day plus injectable placebo. All of them also received calcium 1,000 mg per day and vitamin D 800 IU per day.
At 18 months, the bone mineral density had increased significantly more in the teriparatide group than in the alendronate group in the lumbar spine (P < .001) and in the total hip (P < .01). As expected, markers of bone turnover were suppressed in the alendronate group but were increased in the teriparatide group.
New vertebral fractures were found on radiography in 10 of 165 patients in the alendronate group vs 1 of 171 patients in the teriparatide group (P = .004). Clinical vertebral fractures occurred in 3 of 165 patients treated with alendronate but in none of the teriparatide-treated patients (P = .07). Nonvertebral fractures occurred in 8 of 214 patients treated with alendronate and 12 of 214 patients treated with teriparatide (P = .362). Three of 214 patients treated with alendronate suffered nonvertebral fragility fractures, compared with 5 of 214 patients treated with teriparatide (P = .455).
Denosumab, an antibody to RANK ligand
Denosumab (Prolia) is a fully human monoclonal antibody to RANK ligand. (Recall that glucocorticoids are associated with increases in RANK ligand and decreases in osteoprotegerin.) Denosumab is given subcutaneously in a dosage of 60 mg every 6 months. It was recently approved for the treatment of postmenopausal osteoporosis.
In a phase 2 study of denosumab33 in men and women with rheumatoid arthritis (an independent risk factor for bone loss), the bone mineral density of the lumbar spine increased irrespective of whether the patients were treated with bisphosphonates and glucocorticoids.
ADHERENCE TO GUIDELINES IS POOR
Unfortunately, prevention and treatment in actual clinical practice still lag behind what is recommended in the current guidelines, even though multiple therapies are available.
In 2005, Blalock et al34 expressed concerns about patients’ knowledge, beliefs, and behavior and the prevention and treatment of glucocorticoid-induced osteoporosis. They found that most patients taking oral glucocorticoids are not adequately educated about the prevention of osteoporosis, stating that “patients either are not being counseled or they are being counseled in a manner that is not sufficient to promote subsequent recall and behavior change.”34 They concluded that research is needed to develop effective ways to educate patients about how to prevent glucocorticoid-induced osteoporosis.
Also in 2005, Curtis et al35 reviewed the records of managed-care patients taking glucocorticoids, comparing the prescription of antiresorptive therapy and the use of over-the-counter calcium or vitamin D or both in the periods 2001 to 2003 vs 1995 to 1998. The frequency of bone mineral density measurement in 2001 to 2003 had increased threefold compared with 1995 to 1998, and the use of a prescription antiresorptive drug had increased approximately twofold. However, only 42% of the patients underwent bone mineral density testing or were prescribed bone-protective medicine. The rates were lowest for men, at 25%.
A CALL TO ACTION
Evidenced-based guidelines exist to guide the clinician in an attempt to prevent the deleterious effects of glucocorticoids on bone. Physicians, physician assistants, nurse practitioners, and pharmacists need to coordinate their effects to ensure that adherence to these guidelines improves. Only then will the bone health of patients treated with glucocorticoids improve.
- Bouvard B, Legrand E, Audran M, Chappard D. Glucocorticoid-induced osteoporosis: a review. Clin Rev Bone Miner Metab 2010; 8:15–26.
- Yao W, Cheng Z, Busse C, Pham A, Nakamura MC, Lane NE. Glucocorticoid excess in mice results in early activation of osteoclastogenesis and adipogenesis and prolonged suppression of osteogenesis: a longitudinal study of gene expression in bone tissue from glucocorticoid-treated mice. Arthritis Rheum 2008; 58:1674–1686.
- Manolagas SC. Corticosteroids and fractures: a close encounter of the third cell kind. J Bone Miner Res 2000; 15:1001–1005.
- Manolagas SC, Weinstein RS. New developments in the pathogenesis and treatment of steroid-induced osteoporosis. J Bone Miner Res 1999; 14:1061–1066.
- Canalis E, Bilezikian JP, Angeli A, Giustina A. Perspectives on glucocorticoid-induced osteoporosis. Bone 2004; 34:593–598.
- Ohnaka K, Tanabe M, Kawate H, Nawata H, Takayanagi R. Glucocorticoid suppresses the canonical Wnt signal in cultured human osteoblasts. Biochem Biophys Res Commun 2005; 329:177–181.
- Deal C. Potential new drug targets for osteoporosis. Nat Clin Pract Rheumatol 2009; 5:20–27.
- Lane NE, Lukert B. The science and therapy of glucocorticoid-induced bone loss. Endocrinol Metab Clin North Am 1998; 27:465–483.
- Dalle Carbonare L, Arlot ME, Chavassieux PM, Roux JP, Portero NR, Meunier PJ. Comparison of trabecular bone microarchitecture and remodeling in glucocorticoid-induced and postmenopausal osteoporosis. J Bone Miner Res 2001; 16:97–103.
- van Staa TP, Leufkens HG, Abenhaim L, Begaud B, Zhang B, Cooper C. Use of oral corticosteroids in the United Kingdom. QJM 2000; 93:105–111.
- Curtiss PH, Clark WS, Herndon CH. Vertebral fractures resulting from prolonged cortisone and corticotropin therapy. J Am Med Assoc 1954; 156:467–469.
- van Staa TP, Leufkens HG, Cooper C. The epidemiology of corticosteroid-induced osteoporosis: a meta-analysis. Osteoporos Int 2002; 13:777–787.
- van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Wong CA, Walsh LJ, Smith CJ, et al. Inhaled corticosteroid use and bone-mineral density in patients with asthma. Lancet 2000; 355:1399–1403.
- van Staa TP, Leufkens HG, Cooper C. Use of inhaled corticosteroids and risk of fractures. J Bone Miner Res 2001; 16:581–588.
- Weldon D. The effects of corticosteroids on bone growth and bone density. Ann Allergy Asthma Immunol 2009; 103:3–11.
- American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. Arthritis Rheum 2001; 44:1496–1503.
- Compston J, Barlow D, Brown P, et al. Glucocorticoid-induced osteoporosis. Guidelines for prevention and treatment. London: Royal College of Physicians; 2002. http://www.rcplondon.ac.uk/pubs/books/glucocorticoid/Glucocorticoid.pdf. Accessed 5/20/2010.
- Homik J, Suarez-Almazor ME, Shea B, Cranney A, Wells G, Tugwell P. Calcium and vitamin D for corticosteroid-induced osteoporosis. Cochrane Database Syst Rev 2000; ( 2):CD000952.
- Stoch SA, Saag KG, Greenwald M, et al. Once-weekly oral alendronate 70 mg in patients with glucocorticoid-induced bone loss: a 12-month randomized, placebocontrolled clinical trial. J Rheumatol 2009; 36:1705–1714.
- Bianchi ML. Glucorticoids and bone: some general remarks and some special observations in pediatric patients. Calcif Tissue Int 2002; 70:384–390.
- Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women. Principal results from the Women’s Health Initiative Randomized Controlled Trial. JAMA 2002; 288:321–333.
- Compston JE. The risks and benefits of HRT. J Musculoskelet Neuronal Interact 2004; 4:187–190.
- Reid IR, Ibbertson HK, France JT, Pybus J. Plasma testosterone concentrations in asthmatic men treated with glucocorticoids. Br Med J (Clin Res Ed) 1985; 291:574.
- Reid IR, Wattie DJ, Evans MC, Stapleton JP. Testosterone therapy in glucocorticoid-treated men. Arch Intern Med 1996; 156:1173–1177.
- Crawford BA, Liu PY, Kean MT, Bleasel JF, Handelsman DJ. Randomized placebo-controlled trial of androgen effects on muscle and bone in men requiring long-term systemic glucocorticoid treatment. J Clin Endocrinol Metab 2003; 88:3167–3176.
- Bhasin S, Cunningham GR, Hayes FJ, et al. Testosterone therapy in adult men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2006; 91:1995–2010.
- Cranney A, Welch V, Adachi J, et al. Calcitonin for the treatment and prevention of corticosteroid-induced osteoporosis. Cochrane Database Syst Rev 2000; ( 2):CD0019830.
- Reid DM, Devogelaer JP, Saag K, et al; HORIZON investigators. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 2009; 373:1253–1263.
- Recker RR, Delmas PD, Halse J, et al. Effects of intravenous zoledronic acid once yearly on bone remodeling and bone structure. J Bone Miner Res 2008; 23:6–16.
- Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med 2007; 356:1809–1822.
- Saag KG, Shane E, Boonen S, et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med 2007; 357:2028–2039.
- Dore RK, Cohen SB, Lane NE, et al; Denosumab RA Study Group. Effects of denosumab on bone mineral density and bone turnover in patients with rheumatoid arthritis receiving concurrent glucocorticoids or bisphosphonates. Ann Rheum Dis 2010; 69:872–875.
- Blalock SJ, Norton LL, Patel RA, Dooley MA. Patient knowledge, beliefs, and behavior concerning the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Rheum 2005; 53:732–739.
- Curtis JR, Westfall AO, Allison JJ, et al. Longitudinal patterns in the prevention of osteoporosis in glucocorticoid-treated patients. Arthritis Rheum 2005; 52:2485–2494.
Although glucocorticoid drugs such as prednisone, methylprednisolone, and dexamethasone have many benefits, they are the number-one cause of secondary osteoporosis. 1 When prescribing them for long-term therapy, physicians should take steps to prevent bone loss and fractures.
Being inexpensive and potent anti-inflammatory drugs, glucocorticoids are widely used to treat many diseases affecting millions of Americans, such as dermatologic conditions, inflammatory bowel disease, pulmonary diseases (eg, asthma, chronic obstructive pulmonary disease, interstitial lung disease), renal diseases (eg, glomerulonephritis), rheumatologic disorders (eg, rheumatoid arthritis, lupus, vasculitis, polymyalgia rheumatica), and transplant rejection.
This article discusses the mechanisms of glucocorticoid-induced bone loss and guidelines for preventing and treating it.
GLUCOCORTICOIDS PROMOTE BONE LOSS DIRECTLY AND INDIRECTLY
The pathophysiology of glucocorticoid-induced osteoporosis is much more complicated than was previously thought.
The older view was that these drugs mostly affect bone indirectly by inhibiting calcium absorption, causing secondary hyperparathyroidism. Indeed, they do inhibit calcium absorption from the gastrointestinal tract and induce renal calcium loss. However, most patients do not have elevated levels of parathyroid hormone.
Now, reduced bone formation rather than increased bone resorption is thought to be the predominant effect of glucocorticoids on bone turnover, as these drugs suppress the number and the activity of osteoblasts.
Direct effects on bone
Glucocorticoids directly affect bone cells in a number of ways—eg, by stimulating osteoclastogenesis, decreasing osteoblast function and life span, increasing osteoblast apoptosis, and impairing preosteoblast formation.2
Glucocorticoids also increase osteocyte apoptosis.3 Osteocytes, the most numerous bone cells, are thought to be an integral part of the “nervous system” of bone, directing bone-remodeling units to locations where repair of bone microfractures or removal of bone is needed. Osteocyte apoptosis caused by glucocorticoids may disrupt the signaling process, resulting in increased osteoclast activity in an area of apoptotic osteocytes and the inability to directly repair bone, thus impairing the bone’s ability to preserve its strength and architecture. Such disruption may affect bone quality and increase the risk of fracture independent of any decrease in bone mineral density. 4
Direct molecular effects
Glucocorticoids have been found to:
- Block the stimulatory effect of insulin-like growth factor 1 on bone formation5
- Oppose Wnt/beta-catenin signaling, resulting in decreased bone formation6
- Affect stromal cell differentiation, shunting cell formation towards more adipocyte formation so that fewer osteoblasts and chondrocytes are formed, resulting in less bone formation
- Increase levels of receptor activator of nuclear factor kappa (RANK) ligand and macrophage colony-stimulating factor and decrease levels of osteoprotegerin, resulting in increased osteoclastogenesis and increased bone resorption7
- Decrease estrogen, testosterone, and adrenal androgen levels, which also have adverse effects on bone cells.8
Inflammatory diseases also affect bone
Furthermore, many patients taking glucocorticoids are already at risk of osteoporosis because many of the diseases that require these drugs for treatment are associated with bone loss due to their inflammatory nature. In rheumatoid arthritis, RANK ligand, one of the cytokines involved in inflammation, causes bony erosions and also causes localized osteopenia. The malabsorption of calcium and vitamin D in inflammatory bowel disease is a cause of secondary osteoporosis.
Trabecular bone is affected first
The degree of bone loss in patients receiving glucocorticoids can vary markedly, depending on the skeletal site. Initially, these drugs affect trabecular bone because of its higher metabolic activity, but with prolonged use cortical bone is also affected.2 Greater trabecular thinning is seen in glucocorticoid-induced osteoporosis than in postmenopausal osteoporosis, in which more trabecular perforations are seen.9
Bone loss occurs rapidly during the first few months of glucocorticoid therapy, followed by a slower but continued loss with ongoing use.
FRACTURE RISK INCREASES RAPIDLY
With this decrease in bone mass comes a rapid increase in fracture risk, which correlates with the dose of glucocorticoids and the duration of use.10 Vertebral fractures resulting from prolonged cortisone use were first described in 1954.11
A dosage of 5 mg or more of prednisolone or its equivalent per day decreases bone mineral density and rapidly increases the risk of fracture over 3 to 6 months. The relative risks12:
- Any fracture—1.33 to 1.91
- Hip fracture—1.61 to 2.01
- Vertebral fracture—2.60 to 2.86
- Forearm fracture—1.09 to 1.13.
These risks are independent of age, sex, and underlying disease.12
Patients receiving glucocorticoids may suffer vertebral and hip fractures at higher bone mineral density values than patients with postmenopausal osteoporosis. In 2003, van Staa et al13 reported that, at any given bone mineral density, the incidence of new vertebral fracture in postmenopausal women receiving glucocorticoids was higher than in nonusers. This suggests that glucocorticoids have both a qualitative and a quantitative effect on bone.
Glucocorticoids also cause a form of myopathy, which increases the propensity to fall, further increasing the risk of fractures.
Fracture risk declines after oral glucocorticoids are stopped, reaching a relative risk of 1 approximately 2 years later.12 However, keep in mind that the underlying conditions being treated by the glucocorticoids also increase the patient’s fracture risk. Therefore, the patient’s risk of fracture needs to be evaluated even after stopping the glucocorticoid.
INHALED STEROIDS IN HIGH DOSES MAY ALSO INCREASE RISK
Although inhaled glucocorticoids are generally believed not to affect bone, some evidence suggests that in high doses (> 2,000 μg/day) they may result in significant osteoporosis over several years.14,15
In a retrospective cohort study, van Staa et al15 compared the risk of fracture in 171,000 patients taking the inhaled glucocorticoids fluticasone (Flovent), budesonide (Pulmicort), or beclomethasone (Beconase); 109,000 patients taking inhaled nonglucocorticoid bronchodilators; and 171,000 controls not using inhalers. They found no differences between the inhaled glucocorticoid and nonglucocorticoid bronchodilator groups in the risk of nonvertebral fracture. Users of inhaled glucocorticoids had a higher risk of fracture, particularly of the hip and spine, than did controls, but this may have been related more to the severity of the underlying respiratory disease than to the inhaled glucocorticoids.
Weldon et al16 suggested preventive measures to prevent glucocorticoid-induced effects on bone metabolism when prescribing inhaled glucocorticoids to children. They stated that prophylaxis against osteoporosis requires suspicion, assessment of bone density, supplemental calcium and vitamin D, and, if indicated, bisphosphonates to prevent bone fractures that could compromise the patient’s quality of life.
PREVENTING AND TREATING BONE LOSS DUE TO GLUCOCORTICOIDS
Effective options are available to prevent the deleterious effects of glucocorticoids on bone.
A plethora of guidelines offer direction on how to reduce fracture risk—ie, how to maintain bone mineral density while preventing additional bone loss, alleviating pain associated with existing fractures, maintaining and increasing muscle strength, and initiating lifestyle changes as needed.17,18 Guidelines from the American College of Rheumatology (ACR),17 published in 2001, are being updated. United Kingdom (UK) guidelines,18 published in December 2002, differ slightly from those of the ACR.
Limit exposure to glucocorticoids
Oral glucocorticoids should be given in the lowest effective dose for the shortest possible time. However, there is no safe oral glucocorticoid dose with respect to bone. Alternate-day dosing suppresses the adrenal axis less but has the same effect as daily dosing with regard to bone.
Recommend lifestyle measures from day 1
All guidelines recommend that as soon as a patient is prescribed a glucocorticoid, the clinician should prescribe certain preventive measures, including:
- Smoking cessation
- Weight-bearing and strength-building exercises
- Calcium intake of 1,000 to 1,500 mg per day
- Vitamin D 800 to 1,000 IU per day.
Calcium and vitamin D for all
The Cochrane Database of Systematic Reviews19 evaluated the data supporting the recommendation to use calcium and vitamin D as preventive therapy in patients receiving glucocorticoids. Five trials with 274 patients were included in the meta-analysis. At 2 years after starting calcium and vitamin D, there was a significant weighted mean difference of 2.6% (95% confidence interval [CI] 0.7–4.5) between the treatment and control groups in lumbar spine bone mineral density.
The authors concluded that because calcium and vitamin D have low toxicity and are inexpensive, all patients starting glucocorticoids should also take a calcium and a vitamin D supplement prophylactically.
Bisphosphonates are effective and recommended
The ACR17 and UK18 guidelines said that bisphosphonates are effective for preventing and treating bone loss in patients receiving glucocorticoids.
More recently, Stoch et al20 evaluated the efficacy and safety of alendronate (Fosamax) 70 mg weekly for preventing and treating bone loss in patients on glucocorticoid therapy. At 12 months, bone mineral density in the lumbar spine, trochanter, and total hip had increased from baseline in the alendronate group and was significantly higher than in the placebo group. At the same time, levels of biochemical markers of bone remodeling were significantly lower than at baseline in the alendronate group.
For premenopausal women, postmenopausal women on estrogen replacement therapy, and men, the ACR17 recommends risedronate (Actonel) 5 mg per day or alendronate 5 mg per day; for postmenopausal women not on estrogen, risedronate 5 mg per day or alendronate 10 mg per day is recommended.
Who should receive a bisphosphonate?
In men and postmenopausal women, the ACR17 recommends a bisphosphonate for patients starting long-term glucocorticoid treatment (ie, expected to last 3 months or more) in doses of 5 mg or more per day of prednisone or its equivalent, irrespective of bone mineral density values.
In patients already taking glucocorticoids, a bisphosphonate should be started if the bone mineral density is below a certain threshold. The rationale for using bone mineral thresholds instead of giving bisphosphonates to all is that these drugs have potentially significant side effects and so should not be prescribed if not needed. The appropriate threshold at which intervention should be considered in glucocorticoid-treated patients is a matter of controversy. Based on evidence that fractures occur at a higher bone mineral density in glucocorticoid-treated patients than in postmenopausal women, the UK guidelines18 recommend starting a bisphosphonate if the T score is less than −1.5 at the spine or hip, but the ACR17 guidelines propose a T-score cutoff of −1.0. Whichever cutoff is chosen, its significance in terms of absolute fracture risk will differ according to the age of the patient. Therefore, use of T scores as an intervention threshold is not advisable.
The ACR and the UK guidelines both recommend measuring the bone mineral density by dual-energy x-ray absorptiometry at baseline (even though preventive therapy is not based on this value) and repeating it 6 months later and then yearly.
In premenopausal women, bisphosphonates should be used with caution, as they cross the placenta and are teratogenic in animals. Nevertheless, the ACR guidelines17 state they can be given after appropriate counseling and instruction about contraception.
The UK guidelines18 note that in the large clinical trials of alendronate and risedronate, the incidence of vertebral fractures was low in premenopausal women, indicating a very low fracture risk. Therefore, the UK guidelines state that bone-active drugs should be reserved for premenopausal women who have very low bone mineral density or who suffer fragility fractures or who have other strong risk factors for fracture.
In children and adolescents, the data are insufficient to produce evidence-based guidelines for the prevention and treatment of glucocorticoid-induced osteoporosis. General measures include using the lowest effective dose of glucocorticoids for the shortest period of time, and considering alternate therapies, calcium and vitamin D supplementation, weight-bearing exercise, and proper nutrition.
Bisphosphonates are recommended when bone mineral density is falling despite these general measures and when “high-dose” glucocorticoids are likely to be used for a “prolonged” time, or in patients who have already had a fracture.21
Weekly doses may improve compliance
Risedronate is approved by the US Food and Drug Administration (FDA) for the prevention of glucocorticoid-induced osteoporosis, and both risedronate and alendronate are approved for its treatment.
The ACR guidelines recommend the FDA-approved (ie, daily) doses of alendronate and risedronate for glucocorticoid-induced osteoporosis. Most patients, however, are pre-scribed weekly doses of these two agents, as compliance is much greater with this schedule of administration.
Estrogen is being used more selectively
The 2001 ACR guidelines said that, although there were no randomized controlled trials of hormone replacement (or testosterone) therapy to prevent glucocorticoid-induced bone loss, patients receiving long-term glucocorticoid therapy who are hypogonadal should be offered hormone replacement therapy.17
In 2002, the principal results of the Women’s Health Initiative22 showed that hormone replacement therapy with estrogen and progesterone was associated with a higher risk of breast cancer. Since then, the consensus has been that hormone replacement therapy should be restricted to women with menopausal symptoms or to older women who cannot tolerate other therapies or who express a strong preference for hormone replacement therapy despite being informed about potential adverse events.23
A role for testosterone?
Since a daily dose of more than 5 to 7.5 mg of prednisone increases the risk of gonadotropin and testosterone suppression,24 testosterone replacement therapy has been used to treat glucocorticoid-induced osteoporosis in men.
In two placebo-controlled trials in men receiving glucocorticoid therapy for bronchial asthma or chronic obstructive pulmonary disease, testosterone therapy was associated with a significant 4% increase (95% CI 2–7) in bone mineral density in the lumbar spine.25,26
While these studies cannot be considered conclusive in view of their small size and the lack of fracture data, the Endocrine Society currently recommends that men with chronic obstructive pulmonary disease who are receiving glucocorticoids, are hypogonadal, and have no contraindications to androgen replacement therapy (eg, prostate cancer) be offered testosterone therapy to preserve lean body mass and bone mineral density.27
Calcitonin is not a first-line therapy
Neither the ACR nor the UK guidelines recommended calcitonin as first-line therapy.
A Cochrane systematic review28 evaluated the data on the use of calcitonin to prevent and treat glucocorticoid-induced osteoporosis. Nine trials met the inclusion criteria, and included 221 patients randomized to receive calcitonin and 220 patients who received placebo. Calcitonin was more effective than placebo in preserving bone density in the lumbar spine, with a weighted mean difference of 2.8% (95% CI 1.4–4.3) at 6 months and 3.2% (95% CI 0.3–6.1) at 12 months. However, at 24 months, the lumbar spine bone mineral density was not statistically different between groups, nor was the relative risk of fractures. Calcitonin was given subcutaneously in one trial, in which it showed a substantially greater degree of prevention of bone loss than in the other trials, in which it was given nasally.
NEWLY APPROVED AND INVESTIGATIONAL AGENTS
Zoledronic acid once a year
Zoledronic acid (Reclast), a bisphosphonate given intravenously once a year, was approved for glucocorticoid-induced osteoporosis after the ACR and UK guidelines were published.
Zoledronic acid underwent a randomized multicenter, double-blind, active control trial29 in 833 men and women, age range 18 to 85 years, who had glucocorticoid-induced osteoporosis (they had been treated with 7.5 mg per day or more of prednisone or its equivalent). Of these patients, 416 received a single infusion of 5 mg of zoledronic acid and daily oral placebo, and 417 received a single placebo infusion and daily oral risedronate 5 mg as an active control. All patients also received 1,000 mg of calcium and 400 to 1,000 IU of vitamin D per day. The study duration was 1 year.
Of those who had received a glucocorticoid for more than 3 months, those who received zoledronic acid had a significantly greater mean increase in lumbar spine bone mineral density compared with those in the oral risedronate group: 4.1% vs 2.7%, an absolute difference of 1.4% (P < .0001).
In those who had received a glucocorticoid for 3 months or less, those who received zoledronic acid also had a significantly greater mean increase in lumbar spine bone mineral density compared with those in the risedronate group at 1 year: 2.6% vs 0.6%, a treatment difference of 2% (P < .0001).
Bone biopsy specimens were obtained from 23 patients, 12 in the zoledronic acid group and 11 in the risedronate group.30 Qualitative assessment showed normal bone architecture and quality without mineralization defects. Apparent reductions in activation frequency and remodeling rates were seen when compared with the histomorphometric results in the zoledronic acid postmenopausal osteoporosis population.31 The long-term consequences of this degree of suppression of bone remodeling in the glucocorticoid-treated patients are unknown.
The overall safety and tolerability of zoledronic acid in the glucocorticoid-induced osteoporosis population was similar to that in the postmenopausal osteoporosis clinical trial.29,31 Adverse reactions reported in at least 2% of patients that were either not reported in the postmenopausal osteoporosis trial or were reported more frequently in the glucocorticoid-induced trial included the following: abdominal pain, musculoskeletal pain, nausea, and dyspepsia. The incidence of serious adverse events was similar in the zoledronic acid and the active control groups. In the zoledronic acid group, 2.2% of the patients withdrew from the study due to adverse events vs 1.4% in the active control group.
Teriparatide, a parathyroid hormone drug
Teriparatide (Forteo) consists of a fragment of the human parathyroid hormone molecule. It is given once daily by subcutaneous injection. It was also approved for treating glucocorticoid-induced osteoporosis after the current guidelines were written.
Teriparatide was compared with alendronate in a randomized, double-blind trial in patients with glucocorticoid-induced osteoporosis. 32 Entry criteria were treatment with at least 5 mg of prednisone per day for at least 3 months before screening and a T score of −2.0 or less in the lumbar spine, total hip, or femoral neck, or −1.0 or less plus one or more fragility fractures.
Eighty-three men and 345 women ages 21 or older were enrolled and randomized to receive injectable teriparatide 20 μg per day plus oral placebo or oral alendronate 10 mg per day plus injectable placebo. All of them also received calcium 1,000 mg per day and vitamin D 800 IU per day.
At 18 months, the bone mineral density had increased significantly more in the teriparatide group than in the alendronate group in the lumbar spine (P < .001) and in the total hip (P < .01). As expected, markers of bone turnover were suppressed in the alendronate group but were increased in the teriparatide group.
New vertebral fractures were found on radiography in 10 of 165 patients in the alendronate group vs 1 of 171 patients in the teriparatide group (P = .004). Clinical vertebral fractures occurred in 3 of 165 patients treated with alendronate but in none of the teriparatide-treated patients (P = .07). Nonvertebral fractures occurred in 8 of 214 patients treated with alendronate and 12 of 214 patients treated with teriparatide (P = .362). Three of 214 patients treated with alendronate suffered nonvertebral fragility fractures, compared with 5 of 214 patients treated with teriparatide (P = .455).
Denosumab, an antibody to RANK ligand
Denosumab (Prolia) is a fully human monoclonal antibody to RANK ligand. (Recall that glucocorticoids are associated with increases in RANK ligand and decreases in osteoprotegerin.) Denosumab is given subcutaneously in a dosage of 60 mg every 6 months. It was recently approved for the treatment of postmenopausal osteoporosis.
In a phase 2 study of denosumab33 in men and women with rheumatoid arthritis (an independent risk factor for bone loss), the bone mineral density of the lumbar spine increased irrespective of whether the patients were treated with bisphosphonates and glucocorticoids.
ADHERENCE TO GUIDELINES IS POOR
Unfortunately, prevention and treatment in actual clinical practice still lag behind what is recommended in the current guidelines, even though multiple therapies are available.
In 2005, Blalock et al34 expressed concerns about patients’ knowledge, beliefs, and behavior and the prevention and treatment of glucocorticoid-induced osteoporosis. They found that most patients taking oral glucocorticoids are not adequately educated about the prevention of osteoporosis, stating that “patients either are not being counseled or they are being counseled in a manner that is not sufficient to promote subsequent recall and behavior change.”34 They concluded that research is needed to develop effective ways to educate patients about how to prevent glucocorticoid-induced osteoporosis.
Also in 2005, Curtis et al35 reviewed the records of managed-care patients taking glucocorticoids, comparing the prescription of antiresorptive therapy and the use of over-the-counter calcium or vitamin D or both in the periods 2001 to 2003 vs 1995 to 1998. The frequency of bone mineral density measurement in 2001 to 2003 had increased threefold compared with 1995 to 1998, and the use of a prescription antiresorptive drug had increased approximately twofold. However, only 42% of the patients underwent bone mineral density testing or were prescribed bone-protective medicine. The rates were lowest for men, at 25%.
A CALL TO ACTION
Evidenced-based guidelines exist to guide the clinician in an attempt to prevent the deleterious effects of glucocorticoids on bone. Physicians, physician assistants, nurse practitioners, and pharmacists need to coordinate their effects to ensure that adherence to these guidelines improves. Only then will the bone health of patients treated with glucocorticoids improve.
Although glucocorticoid drugs such as prednisone, methylprednisolone, and dexamethasone have many benefits, they are the number-one cause of secondary osteoporosis. 1 When prescribing them for long-term therapy, physicians should take steps to prevent bone loss and fractures.
Being inexpensive and potent anti-inflammatory drugs, glucocorticoids are widely used to treat many diseases affecting millions of Americans, such as dermatologic conditions, inflammatory bowel disease, pulmonary diseases (eg, asthma, chronic obstructive pulmonary disease, interstitial lung disease), renal diseases (eg, glomerulonephritis), rheumatologic disorders (eg, rheumatoid arthritis, lupus, vasculitis, polymyalgia rheumatica), and transplant rejection.
This article discusses the mechanisms of glucocorticoid-induced bone loss and guidelines for preventing and treating it.
GLUCOCORTICOIDS PROMOTE BONE LOSS DIRECTLY AND INDIRECTLY
The pathophysiology of glucocorticoid-induced osteoporosis is much more complicated than was previously thought.
The older view was that these drugs mostly affect bone indirectly by inhibiting calcium absorption, causing secondary hyperparathyroidism. Indeed, they do inhibit calcium absorption from the gastrointestinal tract and induce renal calcium loss. However, most patients do not have elevated levels of parathyroid hormone.
Now, reduced bone formation rather than increased bone resorption is thought to be the predominant effect of glucocorticoids on bone turnover, as these drugs suppress the number and the activity of osteoblasts.
Direct effects on bone
Glucocorticoids directly affect bone cells in a number of ways—eg, by stimulating osteoclastogenesis, decreasing osteoblast function and life span, increasing osteoblast apoptosis, and impairing preosteoblast formation.2
Glucocorticoids also increase osteocyte apoptosis.3 Osteocytes, the most numerous bone cells, are thought to be an integral part of the “nervous system” of bone, directing bone-remodeling units to locations where repair of bone microfractures or removal of bone is needed. Osteocyte apoptosis caused by glucocorticoids may disrupt the signaling process, resulting in increased osteoclast activity in an area of apoptotic osteocytes and the inability to directly repair bone, thus impairing the bone’s ability to preserve its strength and architecture. Such disruption may affect bone quality and increase the risk of fracture independent of any decrease in bone mineral density. 4
Direct molecular effects
Glucocorticoids have been found to:
- Block the stimulatory effect of insulin-like growth factor 1 on bone formation5
- Oppose Wnt/beta-catenin signaling, resulting in decreased bone formation6
- Affect stromal cell differentiation, shunting cell formation towards more adipocyte formation so that fewer osteoblasts and chondrocytes are formed, resulting in less bone formation
- Increase levels of receptor activator of nuclear factor kappa (RANK) ligand and macrophage colony-stimulating factor and decrease levels of osteoprotegerin, resulting in increased osteoclastogenesis and increased bone resorption7
- Decrease estrogen, testosterone, and adrenal androgen levels, which also have adverse effects on bone cells.8
Inflammatory diseases also affect bone
Furthermore, many patients taking glucocorticoids are already at risk of osteoporosis because many of the diseases that require these drugs for treatment are associated with bone loss due to their inflammatory nature. In rheumatoid arthritis, RANK ligand, one of the cytokines involved in inflammation, causes bony erosions and also causes localized osteopenia. The malabsorption of calcium and vitamin D in inflammatory bowel disease is a cause of secondary osteoporosis.
Trabecular bone is affected first
The degree of bone loss in patients receiving glucocorticoids can vary markedly, depending on the skeletal site. Initially, these drugs affect trabecular bone because of its higher metabolic activity, but with prolonged use cortical bone is also affected.2 Greater trabecular thinning is seen in glucocorticoid-induced osteoporosis than in postmenopausal osteoporosis, in which more trabecular perforations are seen.9
Bone loss occurs rapidly during the first few months of glucocorticoid therapy, followed by a slower but continued loss with ongoing use.
FRACTURE RISK INCREASES RAPIDLY
With this decrease in bone mass comes a rapid increase in fracture risk, which correlates with the dose of glucocorticoids and the duration of use.10 Vertebral fractures resulting from prolonged cortisone use were first described in 1954.11
A dosage of 5 mg or more of prednisolone or its equivalent per day decreases bone mineral density and rapidly increases the risk of fracture over 3 to 6 months. The relative risks12:
- Any fracture—1.33 to 1.91
- Hip fracture—1.61 to 2.01
- Vertebral fracture—2.60 to 2.86
- Forearm fracture—1.09 to 1.13.
These risks are independent of age, sex, and underlying disease.12
Patients receiving glucocorticoids may suffer vertebral and hip fractures at higher bone mineral density values than patients with postmenopausal osteoporosis. In 2003, van Staa et al13 reported that, at any given bone mineral density, the incidence of new vertebral fracture in postmenopausal women receiving glucocorticoids was higher than in nonusers. This suggests that glucocorticoids have both a qualitative and a quantitative effect on bone.
Glucocorticoids also cause a form of myopathy, which increases the propensity to fall, further increasing the risk of fractures.
Fracture risk declines after oral glucocorticoids are stopped, reaching a relative risk of 1 approximately 2 years later.12 However, keep in mind that the underlying conditions being treated by the glucocorticoids also increase the patient’s fracture risk. Therefore, the patient’s risk of fracture needs to be evaluated even after stopping the glucocorticoid.
INHALED STEROIDS IN HIGH DOSES MAY ALSO INCREASE RISK
Although inhaled glucocorticoids are generally believed not to affect bone, some evidence suggests that in high doses (> 2,000 μg/day) they may result in significant osteoporosis over several years.14,15
In a retrospective cohort study, van Staa et al15 compared the risk of fracture in 171,000 patients taking the inhaled glucocorticoids fluticasone (Flovent), budesonide (Pulmicort), or beclomethasone (Beconase); 109,000 patients taking inhaled nonglucocorticoid bronchodilators; and 171,000 controls not using inhalers. They found no differences between the inhaled glucocorticoid and nonglucocorticoid bronchodilator groups in the risk of nonvertebral fracture. Users of inhaled glucocorticoids had a higher risk of fracture, particularly of the hip and spine, than did controls, but this may have been related more to the severity of the underlying respiratory disease than to the inhaled glucocorticoids.
Weldon et al16 suggested preventive measures to prevent glucocorticoid-induced effects on bone metabolism when prescribing inhaled glucocorticoids to children. They stated that prophylaxis against osteoporosis requires suspicion, assessment of bone density, supplemental calcium and vitamin D, and, if indicated, bisphosphonates to prevent bone fractures that could compromise the patient’s quality of life.
PREVENTING AND TREATING BONE LOSS DUE TO GLUCOCORTICOIDS
Effective options are available to prevent the deleterious effects of glucocorticoids on bone.
A plethora of guidelines offer direction on how to reduce fracture risk—ie, how to maintain bone mineral density while preventing additional bone loss, alleviating pain associated with existing fractures, maintaining and increasing muscle strength, and initiating lifestyle changes as needed.17,18 Guidelines from the American College of Rheumatology (ACR),17 published in 2001, are being updated. United Kingdom (UK) guidelines,18 published in December 2002, differ slightly from those of the ACR.
Limit exposure to glucocorticoids
Oral glucocorticoids should be given in the lowest effective dose for the shortest possible time. However, there is no safe oral glucocorticoid dose with respect to bone. Alternate-day dosing suppresses the adrenal axis less but has the same effect as daily dosing with regard to bone.
Recommend lifestyle measures from day 1
All guidelines recommend that as soon as a patient is prescribed a glucocorticoid, the clinician should prescribe certain preventive measures, including:
- Smoking cessation
- Weight-bearing and strength-building exercises
- Calcium intake of 1,000 to 1,500 mg per day
- Vitamin D 800 to 1,000 IU per day.
Calcium and vitamin D for all
The Cochrane Database of Systematic Reviews19 evaluated the data supporting the recommendation to use calcium and vitamin D as preventive therapy in patients receiving glucocorticoids. Five trials with 274 patients were included in the meta-analysis. At 2 years after starting calcium and vitamin D, there was a significant weighted mean difference of 2.6% (95% confidence interval [CI] 0.7–4.5) between the treatment and control groups in lumbar spine bone mineral density.
The authors concluded that because calcium and vitamin D have low toxicity and are inexpensive, all patients starting glucocorticoids should also take a calcium and a vitamin D supplement prophylactically.
Bisphosphonates are effective and recommended
The ACR17 and UK18 guidelines said that bisphosphonates are effective for preventing and treating bone loss in patients receiving glucocorticoids.
More recently, Stoch et al20 evaluated the efficacy and safety of alendronate (Fosamax) 70 mg weekly for preventing and treating bone loss in patients on glucocorticoid therapy. At 12 months, bone mineral density in the lumbar spine, trochanter, and total hip had increased from baseline in the alendronate group and was significantly higher than in the placebo group. At the same time, levels of biochemical markers of bone remodeling were significantly lower than at baseline in the alendronate group.
For premenopausal women, postmenopausal women on estrogen replacement therapy, and men, the ACR17 recommends risedronate (Actonel) 5 mg per day or alendronate 5 mg per day; for postmenopausal women not on estrogen, risedronate 5 mg per day or alendronate 10 mg per day is recommended.
Who should receive a bisphosphonate?
In men and postmenopausal women, the ACR17 recommends a bisphosphonate for patients starting long-term glucocorticoid treatment (ie, expected to last 3 months or more) in doses of 5 mg or more per day of prednisone or its equivalent, irrespective of bone mineral density values.
In patients already taking glucocorticoids, a bisphosphonate should be started if the bone mineral density is below a certain threshold. The rationale for using bone mineral thresholds instead of giving bisphosphonates to all is that these drugs have potentially significant side effects and so should not be prescribed if not needed. The appropriate threshold at which intervention should be considered in glucocorticoid-treated patients is a matter of controversy. Based on evidence that fractures occur at a higher bone mineral density in glucocorticoid-treated patients than in postmenopausal women, the UK guidelines18 recommend starting a bisphosphonate if the T score is less than −1.5 at the spine or hip, but the ACR17 guidelines propose a T-score cutoff of −1.0. Whichever cutoff is chosen, its significance in terms of absolute fracture risk will differ according to the age of the patient. Therefore, use of T scores as an intervention threshold is not advisable.
The ACR and the UK guidelines both recommend measuring the bone mineral density by dual-energy x-ray absorptiometry at baseline (even though preventive therapy is not based on this value) and repeating it 6 months later and then yearly.
In premenopausal women, bisphosphonates should be used with caution, as they cross the placenta and are teratogenic in animals. Nevertheless, the ACR guidelines17 state they can be given after appropriate counseling and instruction about contraception.
The UK guidelines18 note that in the large clinical trials of alendronate and risedronate, the incidence of vertebral fractures was low in premenopausal women, indicating a very low fracture risk. Therefore, the UK guidelines state that bone-active drugs should be reserved for premenopausal women who have very low bone mineral density or who suffer fragility fractures or who have other strong risk factors for fracture.
In children and adolescents, the data are insufficient to produce evidence-based guidelines for the prevention and treatment of glucocorticoid-induced osteoporosis. General measures include using the lowest effective dose of glucocorticoids for the shortest period of time, and considering alternate therapies, calcium and vitamin D supplementation, weight-bearing exercise, and proper nutrition.
Bisphosphonates are recommended when bone mineral density is falling despite these general measures and when “high-dose” glucocorticoids are likely to be used for a “prolonged” time, or in patients who have already had a fracture.21
Weekly doses may improve compliance
Risedronate is approved by the US Food and Drug Administration (FDA) for the prevention of glucocorticoid-induced osteoporosis, and both risedronate and alendronate are approved for its treatment.
The ACR guidelines recommend the FDA-approved (ie, daily) doses of alendronate and risedronate for glucocorticoid-induced osteoporosis. Most patients, however, are pre-scribed weekly doses of these two agents, as compliance is much greater with this schedule of administration.
Estrogen is being used more selectively
The 2001 ACR guidelines said that, although there were no randomized controlled trials of hormone replacement (or testosterone) therapy to prevent glucocorticoid-induced bone loss, patients receiving long-term glucocorticoid therapy who are hypogonadal should be offered hormone replacement therapy.17
In 2002, the principal results of the Women’s Health Initiative22 showed that hormone replacement therapy with estrogen and progesterone was associated with a higher risk of breast cancer. Since then, the consensus has been that hormone replacement therapy should be restricted to women with menopausal symptoms or to older women who cannot tolerate other therapies or who express a strong preference for hormone replacement therapy despite being informed about potential adverse events.23
A role for testosterone?
Since a daily dose of more than 5 to 7.5 mg of prednisone increases the risk of gonadotropin and testosterone suppression,24 testosterone replacement therapy has been used to treat glucocorticoid-induced osteoporosis in men.
In two placebo-controlled trials in men receiving glucocorticoid therapy for bronchial asthma or chronic obstructive pulmonary disease, testosterone therapy was associated with a significant 4% increase (95% CI 2–7) in bone mineral density in the lumbar spine.25,26
While these studies cannot be considered conclusive in view of their small size and the lack of fracture data, the Endocrine Society currently recommends that men with chronic obstructive pulmonary disease who are receiving glucocorticoids, are hypogonadal, and have no contraindications to androgen replacement therapy (eg, prostate cancer) be offered testosterone therapy to preserve lean body mass and bone mineral density.27
Calcitonin is not a first-line therapy
Neither the ACR nor the UK guidelines recommended calcitonin as first-line therapy.
A Cochrane systematic review28 evaluated the data on the use of calcitonin to prevent and treat glucocorticoid-induced osteoporosis. Nine trials met the inclusion criteria, and included 221 patients randomized to receive calcitonin and 220 patients who received placebo. Calcitonin was more effective than placebo in preserving bone density in the lumbar spine, with a weighted mean difference of 2.8% (95% CI 1.4–4.3) at 6 months and 3.2% (95% CI 0.3–6.1) at 12 months. However, at 24 months, the lumbar spine bone mineral density was not statistically different between groups, nor was the relative risk of fractures. Calcitonin was given subcutaneously in one trial, in which it showed a substantially greater degree of prevention of bone loss than in the other trials, in which it was given nasally.
NEWLY APPROVED AND INVESTIGATIONAL AGENTS
Zoledronic acid once a year
Zoledronic acid (Reclast), a bisphosphonate given intravenously once a year, was approved for glucocorticoid-induced osteoporosis after the ACR and UK guidelines were published.
Zoledronic acid underwent a randomized multicenter, double-blind, active control trial29 in 833 men and women, age range 18 to 85 years, who had glucocorticoid-induced osteoporosis (they had been treated with 7.5 mg per day or more of prednisone or its equivalent). Of these patients, 416 received a single infusion of 5 mg of zoledronic acid and daily oral placebo, and 417 received a single placebo infusion and daily oral risedronate 5 mg as an active control. All patients also received 1,000 mg of calcium and 400 to 1,000 IU of vitamin D per day. The study duration was 1 year.
Of those who had received a glucocorticoid for more than 3 months, those who received zoledronic acid had a significantly greater mean increase in lumbar spine bone mineral density compared with those in the oral risedronate group: 4.1% vs 2.7%, an absolute difference of 1.4% (P < .0001).
In those who had received a glucocorticoid for 3 months or less, those who received zoledronic acid also had a significantly greater mean increase in lumbar spine bone mineral density compared with those in the risedronate group at 1 year: 2.6% vs 0.6%, a treatment difference of 2% (P < .0001).
Bone biopsy specimens were obtained from 23 patients, 12 in the zoledronic acid group and 11 in the risedronate group.30 Qualitative assessment showed normal bone architecture and quality without mineralization defects. Apparent reductions in activation frequency and remodeling rates were seen when compared with the histomorphometric results in the zoledronic acid postmenopausal osteoporosis population.31 The long-term consequences of this degree of suppression of bone remodeling in the glucocorticoid-treated patients are unknown.
The overall safety and tolerability of zoledronic acid in the glucocorticoid-induced osteoporosis population was similar to that in the postmenopausal osteoporosis clinical trial.29,31 Adverse reactions reported in at least 2% of patients that were either not reported in the postmenopausal osteoporosis trial or were reported more frequently in the glucocorticoid-induced trial included the following: abdominal pain, musculoskeletal pain, nausea, and dyspepsia. The incidence of serious adverse events was similar in the zoledronic acid and the active control groups. In the zoledronic acid group, 2.2% of the patients withdrew from the study due to adverse events vs 1.4% in the active control group.
Teriparatide, a parathyroid hormone drug
Teriparatide (Forteo) consists of a fragment of the human parathyroid hormone molecule. It is given once daily by subcutaneous injection. It was also approved for treating glucocorticoid-induced osteoporosis after the current guidelines were written.
Teriparatide was compared with alendronate in a randomized, double-blind trial in patients with glucocorticoid-induced osteoporosis. 32 Entry criteria were treatment with at least 5 mg of prednisone per day for at least 3 months before screening and a T score of −2.0 or less in the lumbar spine, total hip, or femoral neck, or −1.0 or less plus one or more fragility fractures.
Eighty-three men and 345 women ages 21 or older were enrolled and randomized to receive injectable teriparatide 20 μg per day plus oral placebo or oral alendronate 10 mg per day plus injectable placebo. All of them also received calcium 1,000 mg per day and vitamin D 800 IU per day.
At 18 months, the bone mineral density had increased significantly more in the teriparatide group than in the alendronate group in the lumbar spine (P < .001) and in the total hip (P < .01). As expected, markers of bone turnover were suppressed in the alendronate group but were increased in the teriparatide group.
New vertebral fractures were found on radiography in 10 of 165 patients in the alendronate group vs 1 of 171 patients in the teriparatide group (P = .004). Clinical vertebral fractures occurred in 3 of 165 patients treated with alendronate but in none of the teriparatide-treated patients (P = .07). Nonvertebral fractures occurred in 8 of 214 patients treated with alendronate and 12 of 214 patients treated with teriparatide (P = .362). Three of 214 patients treated with alendronate suffered nonvertebral fragility fractures, compared with 5 of 214 patients treated with teriparatide (P = .455).
Denosumab, an antibody to RANK ligand
Denosumab (Prolia) is a fully human monoclonal antibody to RANK ligand. (Recall that glucocorticoids are associated with increases in RANK ligand and decreases in osteoprotegerin.) Denosumab is given subcutaneously in a dosage of 60 mg every 6 months. It was recently approved for the treatment of postmenopausal osteoporosis.
In a phase 2 study of denosumab33 in men and women with rheumatoid arthritis (an independent risk factor for bone loss), the bone mineral density of the lumbar spine increased irrespective of whether the patients were treated with bisphosphonates and glucocorticoids.
ADHERENCE TO GUIDELINES IS POOR
Unfortunately, prevention and treatment in actual clinical practice still lag behind what is recommended in the current guidelines, even though multiple therapies are available.
In 2005, Blalock et al34 expressed concerns about patients’ knowledge, beliefs, and behavior and the prevention and treatment of glucocorticoid-induced osteoporosis. They found that most patients taking oral glucocorticoids are not adequately educated about the prevention of osteoporosis, stating that “patients either are not being counseled or they are being counseled in a manner that is not sufficient to promote subsequent recall and behavior change.”34 They concluded that research is needed to develop effective ways to educate patients about how to prevent glucocorticoid-induced osteoporosis.
Also in 2005, Curtis et al35 reviewed the records of managed-care patients taking glucocorticoids, comparing the prescription of antiresorptive therapy and the use of over-the-counter calcium or vitamin D or both in the periods 2001 to 2003 vs 1995 to 1998. The frequency of bone mineral density measurement in 2001 to 2003 had increased threefold compared with 1995 to 1998, and the use of a prescription antiresorptive drug had increased approximately twofold. However, only 42% of the patients underwent bone mineral density testing or were prescribed bone-protective medicine. The rates were lowest for men, at 25%.
A CALL TO ACTION
Evidenced-based guidelines exist to guide the clinician in an attempt to prevent the deleterious effects of glucocorticoids on bone. Physicians, physician assistants, nurse practitioners, and pharmacists need to coordinate their effects to ensure that adherence to these guidelines improves. Only then will the bone health of patients treated with glucocorticoids improve.
- Bouvard B, Legrand E, Audran M, Chappard D. Glucocorticoid-induced osteoporosis: a review. Clin Rev Bone Miner Metab 2010; 8:15–26.
- Yao W, Cheng Z, Busse C, Pham A, Nakamura MC, Lane NE. Glucocorticoid excess in mice results in early activation of osteoclastogenesis and adipogenesis and prolonged suppression of osteogenesis: a longitudinal study of gene expression in bone tissue from glucocorticoid-treated mice. Arthritis Rheum 2008; 58:1674–1686.
- Manolagas SC. Corticosteroids and fractures: a close encounter of the third cell kind. J Bone Miner Res 2000; 15:1001–1005.
- Manolagas SC, Weinstein RS. New developments in the pathogenesis and treatment of steroid-induced osteoporosis. J Bone Miner Res 1999; 14:1061–1066.
- Canalis E, Bilezikian JP, Angeli A, Giustina A. Perspectives on glucocorticoid-induced osteoporosis. Bone 2004; 34:593–598.
- Ohnaka K, Tanabe M, Kawate H, Nawata H, Takayanagi R. Glucocorticoid suppresses the canonical Wnt signal in cultured human osteoblasts. Biochem Biophys Res Commun 2005; 329:177–181.
- Deal C. Potential new drug targets for osteoporosis. Nat Clin Pract Rheumatol 2009; 5:20–27.
- Lane NE, Lukert B. The science and therapy of glucocorticoid-induced bone loss. Endocrinol Metab Clin North Am 1998; 27:465–483.
- Dalle Carbonare L, Arlot ME, Chavassieux PM, Roux JP, Portero NR, Meunier PJ. Comparison of trabecular bone microarchitecture and remodeling in glucocorticoid-induced and postmenopausal osteoporosis. J Bone Miner Res 2001; 16:97–103.
- van Staa TP, Leufkens HG, Abenhaim L, Begaud B, Zhang B, Cooper C. Use of oral corticosteroids in the United Kingdom. QJM 2000; 93:105–111.
- Curtiss PH, Clark WS, Herndon CH. Vertebral fractures resulting from prolonged cortisone and corticotropin therapy. J Am Med Assoc 1954; 156:467–469.
- van Staa TP, Leufkens HG, Cooper C. The epidemiology of corticosteroid-induced osteoporosis: a meta-analysis. Osteoporos Int 2002; 13:777–787.
- van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Wong CA, Walsh LJ, Smith CJ, et al. Inhaled corticosteroid use and bone-mineral density in patients with asthma. Lancet 2000; 355:1399–1403.
- van Staa TP, Leufkens HG, Cooper C. Use of inhaled corticosteroids and risk of fractures. J Bone Miner Res 2001; 16:581–588.
- Weldon D. The effects of corticosteroids on bone growth and bone density. Ann Allergy Asthma Immunol 2009; 103:3–11.
- American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. Arthritis Rheum 2001; 44:1496–1503.
- Compston J, Barlow D, Brown P, et al. Glucocorticoid-induced osteoporosis. Guidelines for prevention and treatment. London: Royal College of Physicians; 2002. http://www.rcplondon.ac.uk/pubs/books/glucocorticoid/Glucocorticoid.pdf. Accessed 5/20/2010.
- Homik J, Suarez-Almazor ME, Shea B, Cranney A, Wells G, Tugwell P. Calcium and vitamin D for corticosteroid-induced osteoporosis. Cochrane Database Syst Rev 2000; ( 2):CD000952.
- Stoch SA, Saag KG, Greenwald M, et al. Once-weekly oral alendronate 70 mg in patients with glucocorticoid-induced bone loss: a 12-month randomized, placebocontrolled clinical trial. J Rheumatol 2009; 36:1705–1714.
- Bianchi ML. Glucorticoids and bone: some general remarks and some special observations in pediatric patients. Calcif Tissue Int 2002; 70:384–390.
- Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women. Principal results from the Women’s Health Initiative Randomized Controlled Trial. JAMA 2002; 288:321–333.
- Compston JE. The risks and benefits of HRT. J Musculoskelet Neuronal Interact 2004; 4:187–190.
- Reid IR, Ibbertson HK, France JT, Pybus J. Plasma testosterone concentrations in asthmatic men treated with glucocorticoids. Br Med J (Clin Res Ed) 1985; 291:574.
- Reid IR, Wattie DJ, Evans MC, Stapleton JP. Testosterone therapy in glucocorticoid-treated men. Arch Intern Med 1996; 156:1173–1177.
- Crawford BA, Liu PY, Kean MT, Bleasel JF, Handelsman DJ. Randomized placebo-controlled trial of androgen effects on muscle and bone in men requiring long-term systemic glucocorticoid treatment. J Clin Endocrinol Metab 2003; 88:3167–3176.
- Bhasin S, Cunningham GR, Hayes FJ, et al. Testosterone therapy in adult men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2006; 91:1995–2010.
- Cranney A, Welch V, Adachi J, et al. Calcitonin for the treatment and prevention of corticosteroid-induced osteoporosis. Cochrane Database Syst Rev 2000; ( 2):CD0019830.
- Reid DM, Devogelaer JP, Saag K, et al; HORIZON investigators. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 2009; 373:1253–1263.
- Recker RR, Delmas PD, Halse J, et al. Effects of intravenous zoledronic acid once yearly on bone remodeling and bone structure. J Bone Miner Res 2008; 23:6–16.
- Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med 2007; 356:1809–1822.
- Saag KG, Shane E, Boonen S, et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med 2007; 357:2028–2039.
- Dore RK, Cohen SB, Lane NE, et al; Denosumab RA Study Group. Effects of denosumab on bone mineral density and bone turnover in patients with rheumatoid arthritis receiving concurrent glucocorticoids or bisphosphonates. Ann Rheum Dis 2010; 69:872–875.
- Blalock SJ, Norton LL, Patel RA, Dooley MA. Patient knowledge, beliefs, and behavior concerning the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Rheum 2005; 53:732–739.
- Curtis JR, Westfall AO, Allison JJ, et al. Longitudinal patterns in the prevention of osteoporosis in glucocorticoid-treated patients. Arthritis Rheum 2005; 52:2485–2494.
- Bouvard B, Legrand E, Audran M, Chappard D. Glucocorticoid-induced osteoporosis: a review. Clin Rev Bone Miner Metab 2010; 8:15–26.
- Yao W, Cheng Z, Busse C, Pham A, Nakamura MC, Lane NE. Glucocorticoid excess in mice results in early activation of osteoclastogenesis and adipogenesis and prolonged suppression of osteogenesis: a longitudinal study of gene expression in bone tissue from glucocorticoid-treated mice. Arthritis Rheum 2008; 58:1674–1686.
- Manolagas SC. Corticosteroids and fractures: a close encounter of the third cell kind. J Bone Miner Res 2000; 15:1001–1005.
- Manolagas SC, Weinstein RS. New developments in the pathogenesis and treatment of steroid-induced osteoporosis. J Bone Miner Res 1999; 14:1061–1066.
- Canalis E, Bilezikian JP, Angeli A, Giustina A. Perspectives on glucocorticoid-induced osteoporosis. Bone 2004; 34:593–598.
- Ohnaka K, Tanabe M, Kawate H, Nawata H, Takayanagi R. Glucocorticoid suppresses the canonical Wnt signal in cultured human osteoblasts. Biochem Biophys Res Commun 2005; 329:177–181.
- Deal C. Potential new drug targets for osteoporosis. Nat Clin Pract Rheumatol 2009; 5:20–27.
- Lane NE, Lukert B. The science and therapy of glucocorticoid-induced bone loss. Endocrinol Metab Clin North Am 1998; 27:465–483.
- Dalle Carbonare L, Arlot ME, Chavassieux PM, Roux JP, Portero NR, Meunier PJ. Comparison of trabecular bone microarchitecture and remodeling in glucocorticoid-induced and postmenopausal osteoporosis. J Bone Miner Res 2001; 16:97–103.
- van Staa TP, Leufkens HG, Abenhaim L, Begaud B, Zhang B, Cooper C. Use of oral corticosteroids in the United Kingdom. QJM 2000; 93:105–111.
- Curtiss PH, Clark WS, Herndon CH. Vertebral fractures resulting from prolonged cortisone and corticotropin therapy. J Am Med Assoc 1954; 156:467–469.
- van Staa TP, Leufkens HG, Cooper C. The epidemiology of corticosteroid-induced osteoporosis: a meta-analysis. Osteoporos Int 2002; 13:777–787.
- van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Wong CA, Walsh LJ, Smith CJ, et al. Inhaled corticosteroid use and bone-mineral density in patients with asthma. Lancet 2000; 355:1399–1403.
- van Staa TP, Leufkens HG, Cooper C. Use of inhaled corticosteroids and risk of fractures. J Bone Miner Res 2001; 16:581–588.
- Weldon D. The effects of corticosteroids on bone growth and bone density. Ann Allergy Asthma Immunol 2009; 103:3–11.
- American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. Arthritis Rheum 2001; 44:1496–1503.
- Compston J, Barlow D, Brown P, et al. Glucocorticoid-induced osteoporosis. Guidelines for prevention and treatment. London: Royal College of Physicians; 2002. http://www.rcplondon.ac.uk/pubs/books/glucocorticoid/Glucocorticoid.pdf. Accessed 5/20/2010.
- Homik J, Suarez-Almazor ME, Shea B, Cranney A, Wells G, Tugwell P. Calcium and vitamin D for corticosteroid-induced osteoporosis. Cochrane Database Syst Rev 2000; ( 2):CD000952.
- Stoch SA, Saag KG, Greenwald M, et al. Once-weekly oral alendronate 70 mg in patients with glucocorticoid-induced bone loss: a 12-month randomized, placebocontrolled clinical trial. J Rheumatol 2009; 36:1705–1714.
- Bianchi ML. Glucorticoids and bone: some general remarks and some special observations in pediatric patients. Calcif Tissue Int 2002; 70:384–390.
- Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women. Principal results from the Women’s Health Initiative Randomized Controlled Trial. JAMA 2002; 288:321–333.
- Compston JE. The risks and benefits of HRT. J Musculoskelet Neuronal Interact 2004; 4:187–190.
- Reid IR, Ibbertson HK, France JT, Pybus J. Plasma testosterone concentrations in asthmatic men treated with glucocorticoids. Br Med J (Clin Res Ed) 1985; 291:574.
- Reid IR, Wattie DJ, Evans MC, Stapleton JP. Testosterone therapy in glucocorticoid-treated men. Arch Intern Med 1996; 156:1173–1177.
- Crawford BA, Liu PY, Kean MT, Bleasel JF, Handelsman DJ. Randomized placebo-controlled trial of androgen effects on muscle and bone in men requiring long-term systemic glucocorticoid treatment. J Clin Endocrinol Metab 2003; 88:3167–3176.
- Bhasin S, Cunningham GR, Hayes FJ, et al. Testosterone therapy in adult men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2006; 91:1995–2010.
- Cranney A, Welch V, Adachi J, et al. Calcitonin for the treatment and prevention of corticosteroid-induced osteoporosis. Cochrane Database Syst Rev 2000; ( 2):CD0019830.
- Reid DM, Devogelaer JP, Saag K, et al; HORIZON investigators. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 2009; 373:1253–1263.
- Recker RR, Delmas PD, Halse J, et al. Effects of intravenous zoledronic acid once yearly on bone remodeling and bone structure. J Bone Miner Res 2008; 23:6–16.
- Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med 2007; 356:1809–1822.
- Saag KG, Shane E, Boonen S, et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med 2007; 357:2028–2039.
- Dore RK, Cohen SB, Lane NE, et al; Denosumab RA Study Group. Effects of denosumab on bone mineral density and bone turnover in patients with rheumatoid arthritis receiving concurrent glucocorticoids or bisphosphonates. Ann Rheum Dis 2010; 69:872–875.
- Blalock SJ, Norton LL, Patel RA, Dooley MA. Patient knowledge, beliefs, and behavior concerning the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Rheum 2005; 53:732–739.
- Curtis JR, Westfall AO, Allison JJ, et al. Longitudinal patterns in the prevention of osteoporosis in glucocorticoid-treated patients. Arthritis Rheum 2005; 52:2485–2494.
KEY POINTS
- Glucocorticoids have both direct and indirect effects on bone cells, and they both suppress bone formation and promote resorption.
- Patients who need glucocorticoids should receive the lowest effective dose for the shortest possible time. They should also be advised to undertake general health measures, including stopping smoking, reducing alcohol intake, exercising daily, and taking in adequate amounts of calcium and vitamin D.
- Bisphosphonates and teriparatide (Forteo) are approved for treating glucocorticoid-induced osteoporosis, but adherence to guidelines for managing this condition is far from optimal.
Pharmacoresistant epilepsy: From pathogenesis to current and emerging therapies
Although more than 10 new antiepileptic drugs have been developed in the past decade, epilepsy remains resistant to drug therapy in about one-third of patients. Approximately 20% of patients with primary generalized epilepsy and up to 60% of patients who have focal epilepsy develop drug resistance during the course of their condition, which for many is lifelong.1
Those who get no response or only a partial response to drugs continue to have incapacitating seizures that lead to significant neuropsychiatric and social impairment, lower quality of life, greater morbidity, and a higher risk of death.
Managing these patients is a challenge and requires a structured multidisciplinary approach in specialized clinics. Newer research, particularly in pharmacogenomics, holds promise of therapy that more closely suits an individual’s profile and type of epilepsy.
This article reviews recent developments in the pathogenesis and treatment of pharmacoresistant epilepsy, placing these topics in clinical context to facilitate and enhance the physician’s ability to manage it.
THE COSTS OF RESISTANT EPILEPSY
A US study in the early 1990s estimated that the annual cost of refractory epilepsy in adults exceeds $11,745 per person2; the cost would be considerably higher today. Another study found that costs correlate with severity of illness and that patients who have intractable seizures incur a cost eight times greater than in those whose epilepsy is controlled.3
Higher risk of death
In any given interval of time, people with pharmacoresistant epilepsy are about two to 10 times more likely to die compared with the general population.4 The risk is inversely linked to seizure control.
“Sudden unexpected death in epilepsy”is the most frequent type of death in patients with pharmacoresistant epilepsy. This category excludes deaths from trauma or drowning. The death can be witnessed or unwitnessed and with or without evidence of a seizure (but not documented status epilepticus). Postmortem examination does not reveal a toxic or anatomic cause of death, and the underlying mechanisms remain unknown. However, the risk is closely associated with drug resistance (which manifests with uncontrolled convulsive seizures and need for polytherapy with antiepileptic drugs).5
Case-control studies have shown that the risk of sudden unexpected death is closely and inversely associated with seizure control; the rate is significantly higher in patients who have a higher frequency of convulsive seizures.6 In addition, freedom from seizures, achieved after successful epilepsy surgery, reduces the risk of death from all causes.7
Other causes of death in patients with epilepsy may be directly related to seizures (accidental trauma, drowning, burns) or to the underlying condition causing the seizures. Furthermore, people with epilepsy are at higher risk of suicide than the general population.
CONCEPT OF PHARMACORESISTANCE AS IT PERTAINS TO EPILEPSY
There is no uniformly accepted definition of pharmacoresistant epilepsy. Most studies defined it according to the number of antiepileptic drugs the patient had tried without success, the frequency of seizures, the duration of illness, and the period of remission. Its true definition awaits a better understanding of underlying mechanisms.
Nevertheless, a useful operational definition at present is failure to control seizures despite a trial of two or three drugs that are suitable for the type of epilepsy and have been appropriately prescribed at maximum tolerated doses. This is because the chances of controlling epilepsy decline sharply after failure of the second or third antiepileptic drug trial. In fact, some clinicians would argue against trying another antiepileptic drug in these patients, who may be candidates for surgical procedures that have high rates of success.8
Common causes of treatment failure, such as poor compliance or inappropriate selection of first-line antiepileptic drugs, should be addressed early on by the treating physician. Nonadherence to the prescribed regimen is a very common cause of uncontrolled seizures, so it is critical to maintain a good rapport with the patient and to inquire about reasons for noncompliance.
Factors that have been associated with treatment-resistant epilepsy include:
- Early onset of seizures
- Long history of poor seizure control
- Having more than one type of seizure
- Remote symptomatic etiology (eg, patients with a history of brain infection or head trauma)
- Certain structural abnormalities (eg, cortical dysplasia)
- Certain abnormalities on electroencephalography (EEG)
- Cognitive disability
- History of status epilepticus.
When to consider referral
A topic of debate is how long a patient must have active epilepsy before he or she is considered to have pharmacoresistance and should be referred to a specialist center.
Both the rate of remission and the time needed to achieve remission depend on multiple factors such as the type and etiology of the epilepsy and the definition of sustained intractability.
Importantly, the prognosis for most patients with newly diagnosed epilepsy, whether good or bad, becomes apparent within a few years of starting treatment. Although pharmacoresistant epilepsy will become refractory within 8 years in some patients, in others a second drug may not fail for more than 1 or 2 decades after diagnosis. Nonetheless, a history of a lack of a sustained seizure-free period for 12 consecutive months, in spite of two or three suitable and tolerated antiepileptic drugs, is a definite red flag for clinicians and should prompt referral to a specialist center.9,10 The National Association of Epilepsy Centers recommends referral to a specialized epilepsy center if seizure control is not achieved by a general neurologist within a period of 9 months.11

AN APPROACH TO PHARMACORESISTANT EPILEPSY FOR THE NONSPECIALIST
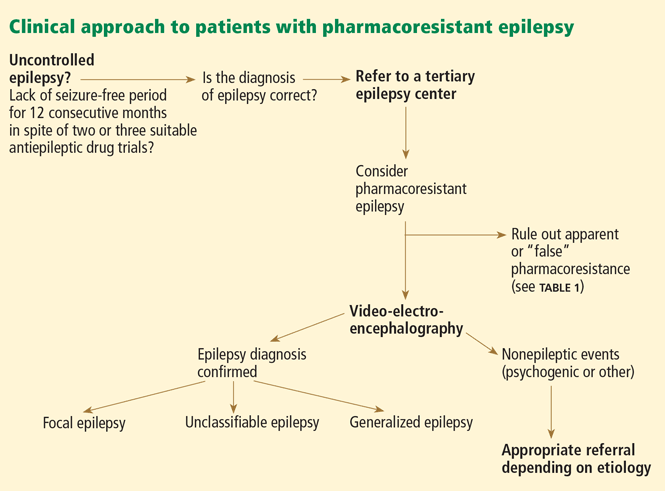
Review and confirm the diagnosis of epilepsy with the help of a careful history, video-EEG, and imaging.
When seizures cannot be controlled with drugs, it is important to verify that the events in question are indeed epileptic. Continuous video-EEG monitoring may be necessary to capture and characterize the clinical manifestations and corresponding EEG changes.14 When a typical spell is not associated with an EEG change, one can often make the diagnosis of nonepileptic events, which commonly are psychogenic nonepileptic seizures. Of note, however: scalp EEG may fail to pick up ictal EEG changes in focal seizures arising from a small or deeply situated focus: for example, as in focal sensorimotor seizures from restricted perirolandic cortex.15,16
Identify the cause, type of seizure or seizures, and syndromic classification, if any.
Review past and present medications, doses, efficacy, and side effects. Consider the possibility of drug interactions.
Different drugs may have different pharmacokinetic and dose-efficacy curves. With most of the newer-generation antiepileptic compounds such as lamotrigine (Lamictal), levetiracetam (Keppra), pregabalin (Lyrica), and topiramate (Topamax), efficacy may continue to increase in some patients as the dose is increased without reaching toxicity. An important exception is phenytoin (Dilantin): due to its nonlinear and saturable pharmacokinetics, even minor dose increases may lead to a large increase in phenytoin concentration (the drug accumulates as elimination becomes saturated). A high degree of individual variability exists, which is determined by factors that include the patient’s age, genetic and enzymatic profile, comorbidities, and concurrent medications.17 Understanding these relationships in individual patients facilitates dose selection and titration and enhances compliance. Testing serum drug levels may help when compliance is questionable.
Choose antiepileptic drugs primarily on the basis of the type of seizures and the individual clinical scenario: Which drug is likely to be most efficacious with the fewest side effects, and which one is appropriate for the patient’s comorbidities and concomitant medications?
When changing the dosage, withdrawing a drug, or adding a second antiepileptic drug, always do it in a systematic way, one step at a time. Reassess the response to the change before moving to the next step.
Discuss issues such as seizure precautions, lifestyle modifications, psychosocial dysfunction, and sudden unexpected death. Offer access to epilepsy-specialist nurses and epilepsy support groups such as those offered by the Epilepsy Foundation of America, the agency dedicated to the welfare of individuals with epilepsy in the United States (http://epilepsyfoundation.ning.com/).
PATTERNS OF DRUG RESISTANCE
Epidemiologic studies suggest three different patterns of drug resistance in epilepsy: de novo, progressive, and waxing-and-waning.
De novo drug resistance
In some patients, resistance is present from the time of onset of the very first seizure, before any antiepileptic drug is even started. One landmark study showed that patients with newly diagnosed epilepsy for whom the first drug was ineffective had only an 11% probability of future success, compared with 41% to 55% in patients who had had to stop taking the drug because of intolerable side effects or idiosyncratic reactions.10 Most patients for whom the first drug fails will be resistant to most and often all antiepileptic drugs.18 These results suggest that seizures in newly diagnosed patients are either easy to control or difficult to control right from the start.
Progressive drug resistance
In some patients, epilepsy is initially controlled but then gradually becomes refractory. This pattern may be seen, for example, in childhood epilepsies or in patients with hippocampal sclerosis.19,20
Waxing and waning resistance
In some patients, epilepsy has a waxing-and-waning pattern: ie, it alternates between a remitting (pharmacoresponsive) and relapsing (pharmacoresistant) course. Patients thought to have drug-resistant epilepsy may become seizure-free when other drugs are tried. Changes in drug bioavailability, local concentration of the drug in the brain, receptor changes, the development of tolerance, and interactions with new medications may be implicated, though the exact mechanism is not understood.21
BIOLOGIC BASIS OF PHARMACORESISTANT EPILEPSY
Pharmacoresistance is not unique to epilepsy: it is now recognized in diverse brain disorders, including depression and schizophrenia,22 and in other diseases affecting the brain, such as human immunodeficiency virus infection and many forms of cancer.23
Multiple drug resistance is characterized by insensitivity to a broad spectrum of drugs that presumably act on different receptors and by different mechanisms.24
Conceptually, the variable response to antiepileptic drugs can be attributed to factors related to the disease, the patient, and the drugs, or to other unknown factors. These factors are not mutually exclusive and may be either constitutive or acquired during the course of the disease.
Factors related to the disease (independent of the host)
These factors include etiology, epilepsy progression resulting in persistent changes of the epileptogenic network, and alterations of drug targets (ie, the “target” or pharmacodynamic hypothesis that reduced sensitivity to antiepileptic drugs is due to seizure-related alterations of specific drug targets25,26) or drug uptake into the brain27 (the “transporter” or pharmacokinetic hypothesis that the drugs are ineffective due to intrinsic or acquired overexpression of multidrug transporter proteins that hamper local drug delivery to target sites).28
Factors related to the drugs
Several drug-related factors have been implicated, such as the development of tolerance, lack of antiepileptogenic (disease-modifying) actions to interrupt the ongoing process of epileptogenesis rather than only suppressing seizures, and paucity of drugs with specific mechanisms of action tailored to difficult-tocontrol epilepsies.29
Patient characteristics
Variability in response (efficacy and adverse effects) to each antiepileptic drug can be due to interindividual differences in any of four interrelated fundamental factors: DNA, RNA, proteins, or metabolites. The field of study that aims to assess the effect of DNA variations (genotype) on a patient’s clinical response to a drug (phenotype) is known as pharmacogenetics. Age-related changes in pharmacokinetic and pharmacodynamic variables may contribute to age-dependent pharmacoresistance. Least studied are environmental factors that may play a role in the development or expression of pharmacoresistance.
NONPHARMACOLOGIC TREATMENTS
Ketogenic diet
The ketogenic diet, an important nonpharmacologic alternative, is usually reserved for young patients with difficult-to-control seizures. Originally developed almost a century ago, the diet mimics the biochemical changes associated with starvation. It is a strict regimen, high in fat and low in carbohydrate and protein (typically in a ratio of 4:1 or 3:1 in adolescents and very young children).
Such a strict regimen is difficult to implement and maintain and requires close supervision by a dietician and physician. In addition to the practical complexities, concerns also exist about the long-term effects of the diet on the child’s growth and overall health. For these reasons, the ketogenic diet is restricted to a small group of young patients with pharmacoresistant epilepsy and is not usually used long-term. There are few data indicating when it is appropriate to terminate the diet in patients who have a favorable response, but most clinicians wean the patient after 2 to 3 years.
Reports on the use of the ketogenic diet in adults are scarce, although benefit was seen in a small series.30 No long-term follow-up data exist for adults, especially regarding the risk of atherosclerosis.
Vagus nerve stimulation
Vagus nerve stimulation is a nonpharmacologic alternative for adults and for adolescents over age 12 years who have intractable focal seizures and who are not favorable surgical candidates. 31 Its effectiveness in younger patients and in those who have intractable generalized seizures is less clear, although published uncontrolled series have reported benefit (fewer seizures and better quality of life).32–34
A device consisting of a pulse generator is implanted subcutaneously in the precordium, and a lead wire is tunneled under the skin and attached to the left vagus nerve. The generator is programmed using a telemetry wand held over the device, with settings for current intensity (typically 1–2 mA), pulse width (250–300 μsec), frequency (30 Hz), and “duty cycle” (typically 30 seconds on stimulation, followed by 3 to 5 minutes off, cycling 24 hours/day). Hence, it provides “open-loop stimulation,” ie, continuous stimulation that is not modified in response to the patient’s EEG seizure activity. Patients or caregivers can also activate the device manually (“on demand”) at the first sign or warning of an impending seizure by swiping a handheld magnet.
Common side effects such as cough, voice alteration, and hoarseness are usually stimulation-dependent and tend to diminish with time. Notably, vagus nerve stimulation has none of the cognitive side effects often encountered with increasing doses of antiepileptic drugs. As with other implantable stimulators, some safety concerns exist in patients undergoing magnetic resonance imaging (MRI).
At least one-third of patients who receive this treatment show a sustained response, defined as a 50% or greater reduction in seizures. However, few achieve freedom from seizures, and therefore this therapy is considered palliative and is reserved for patients who are not candidates for surgery or for whom surgery has failed.
Unfortunately, it has not been possible to predict which patients will benefit from vagus nerve stimulation. The American Academy of Neurology recommends that this treatment be considered only after a thorough evaluation by a subspecialist to rule out nonepileptic conditions, false pharmacoresistance, and surgically treatable types of epilepsy.31
IS THE PATIENT A CANDIDATE FOR EPILEPSY SURGERY?
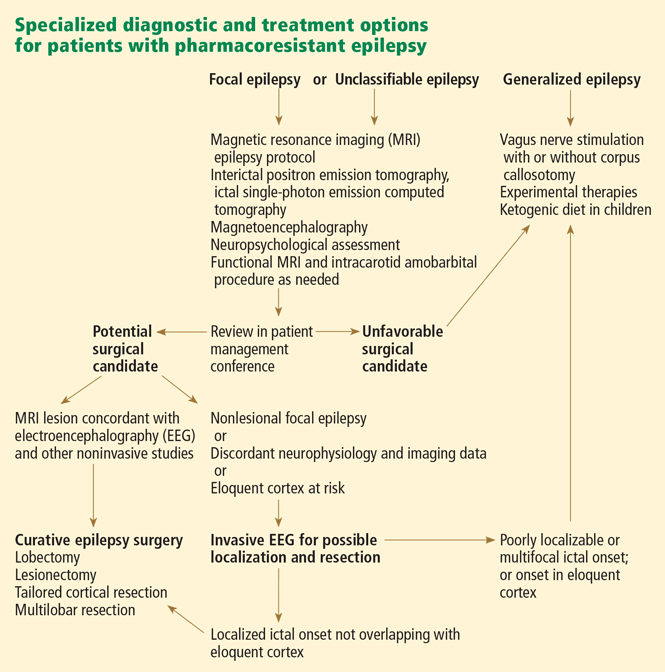
- Is the epilepsy diagnosis correct?
- Is the epilepsy focal? Have the following possibilities been excluded: generalized or multifocal epilepsy, situational or provoked seizures, or an epilepsy syndrome with spontaneous remission?
- Do seizures remain poorly controlled despite adequate pharmacologic trials?
- If so, do the seizures or medication side effects significantly affect the patient’s quality of life?
- Can an epileptogenic lesion be seen on MRI, and what is the suspected etiology?
- Is there converging evidence for a single epileptogenic focus?
- Are there abnormalities elsewhere in the brain?
- What are the chances of a good outcome in terms of seizure control and improvement in quality of life?
- What are the risks of surgery, and how do these compare with the risks of not having surgery?
- What are the patient’s perceptions and attitudes toward epilepsy surgery?
Surgical recommendations should be made after a thorough discussion of all preoperative data in a multidisciplinary patient management conference (Figure 1, Figure 2), in which epileptologists, neurosurgeons, neuroradiologists, neuropsychologists, and psychiatrists all actively participate.
Preoperative counseling is essential for the patient and his or her family, addressing the goals, risks, and benefits of the surgery. Treatment decisions should take into account the possible impact of surgery on the patient’s medical and psychosocial circumstances (risks of ongoing seizures vs surgical intervention; impact on the patient’s independence, employment status, emotional well-being, and psychiatric and other comorbidities).
EPILEPSY SURGERY: CURATIVE OR PALLIATIVE
Epilepsy surgery can be classified as curative or palliative, depending on the goal.
Curative procedures
Curative procedures include lobectomy, lesionectomy, and multilobar or hemispheric surgery (hemispherectomy).
Anterior temporal lobectomy and hippocampectomy are used for temporal lobe epilepsy and have several variations.
“Mesial temporal lobe epilepsy associated with hippocampal sclerosis,” a recognizable syndrome with complex partial seizures, stereotyped electroclinical features, and fairly typical natural history, is the most common of the focal epilepsies in adults.36 Its prognosis is poor when treated medically, but it responds well to surgery.37,38 In a landmark prospective trial, 58% of 40 patients randomized to surgical treatment were free of disabling seizures after 1 year, compared with only 8% of 40 patients treated medically.39
Lesionectomy and lobectomy are resective approaches targeting seizure foci outside the temporal lobe (most often in the frontal lobe, less commonly in the parietal or occipital lobes) or within the temporal lobe but outside the hippocampus (neocortical temporal lobe epilepsies).
The nature of the underlying substrate plays a significant role in determining the natural history,40 surgical strategy, and prognosis for freedom from seizures postoperatively. Patients with seizures due to structural lesions that are visible on MRI (“lesional epilepsies,” eg, cavernous angiomas or circumscribed lowgrade tumors) may become seizure-free after limited resections targeting the lesion itself (lesionectomy) or extending to involve part of a lobe or an entire lobe (lobectomy).
A favorable surgical outcome is much more likely if the lesion can be completely removed.41 At times, however, the lesion cannot be resected in its entirety, for example if it is located within inaccessible or essential (eloquent) cortex.
On the other hand, identifying the epileptogenic focus in patients with no visible structural abnormality on MRI (“nonlesional epilepsies”) can be challenging and usually requires intracranial investigations. In this instance, the aim of surgery is to resect regions that are electrographically abnormal. In general, the postoperative outcome is less favorable in nonlesional focal epilepsies than in lesional epilepsies.42
Multilobar resections and hemispherectomy are indicated when seizures arise from extensive, diffuse, or multiple regions of a single hemisphere.
If the neurologic function supported by the abnormal hemisphere is intact, a tailored multilobar resection aims at eliminating the epileptogenic focus without creating new deficits.
If, however, the underlying hemispheric abnormality is associated with significant contralateral hemiparesis, hemiplegia, or visual field deficits, the need to preserve function does not limit surgery, and hemispherectomy can be considered. Hemispherectomy can be the procedure of choice for selected infants and young children with catastrophic epilepsies and unilateral brain damage.43,44 The goal is to control seizures by completely disconnecting the abnormal epileptogenic hemisphere from the opposite, “good” hemisphere. A second major goal is to improve psychosocial and cognitive development by eliminating the child’s uncontrolled seizures.
Palliative procedures
Palliative procedures, in contrast to curative ones, rarely eliminate seizures entirely. It is important to determine that patients are not candidates for a more definitive, potentially curative resective procedure before considering palliative surgical options such as corpus callosotomy, multiple subpial transections, or vagus nerve stimulation.
Corpus callosotomy (transection of the corpus callosum) is performed in a small number of patients, ie, those who have disabling seizures that rapidly become generalized or injurious drop attacks and are not candidates for focal resection. By disconnecting the two hemispheres, this procedure aims to hinder the fast interhemispheric spread of seizure discharges.
Callosotomy may be complete or involve only a portion of the corpus callosum. The extent of resection has been correlated with favorable outcome.45
Some investigators report a 50% or greater reduction in seizure frequency, with drop attacks and generalized tonic-clonic seizures showing the most consistent improvement. In addition, behavior and quality of life may also improve.46
Multiple subpial transections are reserved for seizures arising from eloquent cortex (ie, from areas that cannot be removed without incurring unacceptable neurologic deficits). Therefore, the surgeon only transects the epileptogenic cortex in a vertical manner, so as to interrupt the horizontal cortical connections without resection. This approach is thought to disrupt the synchrony of seizure propagation while preserving physiologic function.
A meta-analysis of small case series suggests some decrease in seizure frequency with no or minimal neurologic compromise in up to 60% of patients.47
COMPLICATIONS OF EPILEPSY SURGERY
Resective surgery is not without risk, but often the risk is much less than that posed by uncontrolled epilepsy in the long term. Operative mortality rates vary from almost zero for temporal lobe surgery to 2.5% for hemispherectomy. The reported risk of permanent surgical morbidity varies by type of surgery from 1.1% for temporal lobe resection to about 5% for frontal lobe resection.4
NOVEL EPILEPSY THERAPIES
The failure of available antiepileptic drugs to control seizures in a substantial number of patients underscores the need to develop novel therapies such as electrical stimulation, local drug delivery, cell transplantation, and genebased therapies.48 Future targeted therapies could be coupled to seizure-forecasting systems to create “smart” implantable devices that predict, detect, and preemptively treat the seizures in a “closed-loop” fashion.
Targeted electrical stimulation
To modulate abnormal cortical hyperexcitability, electrical stimulation can be applied to the peripheral nervous system (eg, vagus nerve stimulation) or central nervous system. Central nervous system stimulation can be broadly divided into two approaches:
Direct stimulation targets presumed epileptogenic brain tissue such as the neocortex or hippocampus.
Indirect stimulation targets presumed seizure-gating networks such as in the cerebellum and various deep brain nuclei in the basal ganglia or thalamus (deep brain stimulation), which are believed to play a central role in modulating the synchronization and propagation of seizure activity.
Systematic, well-designed studies are currently under way. The budding field of electrical stimulation faces a number of challenges, which include optimizing stimulation variables and target sites, selecting favorable candidates, validating long-term safety and efficacy, evaluating long-term effects on tissue reorganization, plasticity, and epileptogenicity, and developing reliable algorithms for seizure detection and prediction.
Local drug delivery
Direct delivery of drugs into the epileptogenic brain tissue holds promise, particularly for patients whose foci cannot be surgically removed. By bypassing the systemic circulation, this approach has the potential to avoid systemic and even whole-brain side effects.
However, only a few proof-of-principle experiments have been conducted in animals, and to date no clinical study has explored the utility of intraparenchymal or intraventricular antiepileptic drug delivery in humans.
Cell and gene therapies
The emerging field of experimental cell- and gene-based neuropharmacology holds promise for location-specific therapeutic strategies. In ex vivo gene therapy, bioengineered cells capable of delivering anticonvulsant compounds might be transplanted into specific areas of the brain. On the other hand, in vivo gene therapy would involve delivering genes by viral vectors to induce the localized production of antiepileptic compounds in situ.
Endogenous anticonvulsants such as gamma-aminobutyric acid (GABA) and adenosine have been tried in various animal experiments. 49 Before they can be applied clinically, significant questions need to be addressed, including the potential for toxicity or maladaptive plasticity and long-term therapeutic safety and efficacy.
Cell transplantation is aimed at restoring the physiologic balance of neurotransmitters, and is currently being investigated for the treatment of several neurologic disorders such as Parkinson disease and Huntington disease.50 Unlike delivery of exogenous compounds, cell transplantation (heterologous fetal cell grafts or embryonic or adult stem cells) has the potential to form restorative synaptic connections and assimilate within existing cells and networks in the host tissue. An essential limitation to xenotransplantation in humans is the risk of immunologic rejection.
The future
We hope that continued progress in genomics will lead to targeted development of disease-modifying drugs that can impede or reverse the process of epileptogenesis. Advances in informatics and genetics may be harnessed to predict which patients are likely to develop pharmacoresistance, to cure certain genetic epilepsies, and to individualize antiepileptic drug selection on the basis of each person’s genetic profile.
- Siegel AM. Presurgical evaluation and surgical treatment of medically refractory epilepsy. Neurosurg Rev 2004; 27:1–18.
- Murray MI, Halpern MT, Leppik IE. Cost of refractory epilepsy in adults in the USA. Epilepsy Res 1996; 23:139–148.
- Jacoby A, Buck D, Baker G, McNamee P, Graham-Jones S, Chadwick D. Uptake and costs of care for epilepsy: findings from a U.K. regional study. Epilepsia 1998; 39:776–786.
- Chapell R, Reston J, Snyder D, Treadwell J, Treager S, Turkelson C. Management of treatment-resistant epilepsy. Evid Rep Technol Assess (Summ) 2003 Apr; 77:1–8.
- Nei M, Bagla R. Seizure-related injury and death. Curr Neurol Neurosci Rep 2007; 7:335–341.
- Langan Y, Nashef L, Sander JW. Case-control study of SUDEP. Neurology 2005; 64:1131–1133.
- Sperling MR, Feldman H, Kinman J, Liporace JD, O’Connor MJ. Seizure control and mortality in epilepsy. Ann Neurol 1999; 46:45–50.
- Berg AT. Understanding the delay before epilepsy surgery: who develops intractable focal epilepsy and when? CNS Spectr 2004; 9:136–144.
- Perucca E. Pharmacoresistance in epilepsy: how should it be defined? CNS Drugs 1998; 10:171–179.
- Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med 2000; 342:314–319.
- Gumnit RJ, Walczak TS; National Association of Epilepsy Centers. Guidelines for essential services, personnel, and facilities in specialized epilepsy centers in the United States. Epilepsia 2001; 42:804–814.
- Smith D, Defalla BA, Chadwick DW. The misdiagnosis of epilepsy and the management of refractory epilepsy in a specialist clinic. QJM 1999; 92:15–23.
- Schuele SU, Lüders HO. Intractable epilepsy: management and therapeutic alternatives. Lancet Neurol 2008; 7:514–524.
- Engel J, Burchfiel J, Ebersole J, et al. Long-term monitoring for epilepsy. Report of an IFCN committee. Electroencephalogr Clin Neurophysiol 1993; 87:437–458.
- Kanner AM, Morris HH, Lüders H, et al. Supplementary motor seizures mimicking pseudoseizures: some clinical differences. Neurology 1990; 40:1404–1407.
- Alexopoulos AV, Dinner DS. Focal motor seizures, epilepsia partialis continua, and supplementary sensorimotor seizures. In:Wyllie E, Gupta A, Lachhwani DK, editors. The Treatment of Epilepsy: Principles & Practice. Philadelphia, PA: Lippincott Williams & Wilkins, 2006:257–277.
- French JA. Refractory epilepsy: clinical overview. Epilepsia 2007; 48(suppl 1):3–7.
- Regesta G, Tanganelli P. Clinical aspects and biological bases of drug-resistant epilepsies. Epilepsy Res 1999; 34:109–122.
- Berg AT, Langfitt J, Shinnar S, et al. How long does it take for partial epilepsy to become intractable? Neurology 2003; 60:186–190.
- Berg AT, Vickrey BG, Testa FM, et al. How long does it take for epilepsy to become intractable? A prospective investigation. Ann Neurol 2006; 60:73–79.
- Löscher W, Schmidt D. Experimental and clinical evidence for loss of effect (tolerance) during prolonged treatment with antiepileptic drugs. Epilepsia 2006; 47:1253–1284.
- Löscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci 2005; 6:591–602.
- Siddiqui A, Kerb R, Weale ME, et al. Association of multidrug resistance in epilepsy with a polymorphism in the drug-transporter gene ABCB1. N Engl J Med 2003; 348:1442–1448.
- Granata T, Marchi N, Carlton E, et al. Management of the patient with medically refractory epilepsy. Expert Rev Neurother 2009; 9:1791–1802.
- Marchi N, Hallene KL, Kight KM, et al. Significance of MDR1 and multiple drug resistance in refractory human epileptic brain. BMC Med 2004; 2:37.
- Oby E, Janigro D. The blood-brain barrier and epilepsy. Epilepsia 2006; 47:1761–1774.
- Schmidt D, Löscher W. New developments in antiepileptic drug resistance: an integrative view. Epilepsy Curr 2009; 9:47–52.
- Remy S, Beck H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain 2006; 129:18–35.
- Löscher W, Schmidt D. New horizons in the development of antiepileptic drugs. Epilepsy Res 2002; 50:3–16.
- Sirven J, Whedon B, Caplan D, et al. The ketogenic diet for intractable epilepsy in adults: preliminary results. Epilepsia 1999; 40:1721–1726.
- Fisher RS, Handforth A. Reassessment: vagus nerve stimulation for epilepsy: a report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 1999; 53:666–669.
- Hallböök T, Lundgren J, Stjernqvist K, Blennow G, Strömblad LG, Rosén I. Vagus nerve stimulation in 15 children with therapy resistant epilepsy; its impact on cognition, quality of life, behaviour and mood. Seizure 2005; 14:504–513.
- Holmes MD, Silbergeld DL, Drouhard D, Wilensky AJ, Ojemann LM. Effect of vagus nerve stimulation on adults with pharmacoresistant generalized epilepsy syndromes. Seizure 2004; 13:340–345.
- Murphy JV, Torkelson R, Dowler I, Simon S, Hudson S. Vagal nerve stimulation in refractory epilepsy: the first 100 patients receiving vagal nerve stimulation at a pediatric epilepsy center. Arch Pediatr Adolesc Med 2003; 157:560–564.
- Alexopoulos AV, Najm IM. Neurosurgical management of focal epilepsies in adults. In:Panayiotopoulos CP, et al, editors. Focal Epilepsies: Seizures, Syndromes and Management. Oxford, UK: Medicinae; 2009:204–220.
- Wieser HGILAE Commission on Neurosurgery of Epilepsy. ILAE Commission Report. Mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia 2004; 45:695–714.
- Engel J. Surgery for seizures. N Engl J Med 1996; 334:647–652.
- Engel J, Wiebe S, French J, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: temporal lobe and localized neocortical resections for epilepsy: report of the Quality Standards Subcommittee of the American Academy of Neurology, in association with the American Epilepsy Society and the American Association of Neurological Surgeons. Neurology 2003; 60:538–547.
- Wiebe S, Blume WT, Girvin JP, Eliasziw M; Effectiveness and Efficiency of Surgery for Temporal Lobe Epilepsy Study Group. A randomized, controlled trial of surgery for temporal-lobe epilepsy. N Engl J Med 2001; 345:311–318.
- Semah F, Picot MC, Adam C, et al. Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology 1998; 51:1256–1262.
- Awad IA, Rosenfeld J, Ahl J, Hahn JF, Lüders H. Intractable epilepsy and structural lesions of the brain: mapping, resection strategies, and seizure outcome. Epilepsia 1991; 32:179–186.
- Cascino GD. Surgical treatment for extratemporal epilepsy. Curr Treat Options Neurol 2004; 6:257–262.
- González-Martínez JA, Gupta A, Kotagal P, et al. Hemispherectomy for catastrophic epilepsy in infants. Epilepsia 2005; 46:1518–1525.
- Wyllie E. Surgery for catastrophic localization-related epilepsy in infants. Epilepsia 1996; 37(suppl 1):S22–S25.
- Tanriverdi T, Olivier A, Poulin N, Andermann F, Dubeau F. Long-term seizure outcome after corpus callosotomy: a retrospective analysis of 95 patients. J Neurosurg 2009; 110:332–342.
- Asadi-Pooya AA, Sharan A, Nei M, Sperling MR. Corpus callosotomy. Epilepsy Behav 2008; 13:271–278.
- Spencer SS, Schramm J, Wyler A, et al. Multiple subpial transection for intractable partial epilepsy: an international meta-analysis. Epilepsia 2002; 43:141–145.
- Alexopoulos AV, Gonugunta V, Yang J, Boulis NM. Electrical stimulation and gene-based neuromodulation for control of medically-refractory epilepsy. Acta Neurochir Suppl 2007; 97:293–309.
- Detlev B. Cell and gene therapies for refractory epilepsy. Curr Neuropharmacol 2007; 5:115–125.
- Fisher RS, Ho J. Potential new methods for antiepileptic drug delivery. CNS Drugs 2002; 16:579–593.
Although more than 10 new antiepileptic drugs have been developed in the past decade, epilepsy remains resistant to drug therapy in about one-third of patients. Approximately 20% of patients with primary generalized epilepsy and up to 60% of patients who have focal epilepsy develop drug resistance during the course of their condition, which for many is lifelong.1
Those who get no response or only a partial response to drugs continue to have incapacitating seizures that lead to significant neuropsychiatric and social impairment, lower quality of life, greater morbidity, and a higher risk of death.
Managing these patients is a challenge and requires a structured multidisciplinary approach in specialized clinics. Newer research, particularly in pharmacogenomics, holds promise of therapy that more closely suits an individual’s profile and type of epilepsy.
This article reviews recent developments in the pathogenesis and treatment of pharmacoresistant epilepsy, placing these topics in clinical context to facilitate and enhance the physician’s ability to manage it.
THE COSTS OF RESISTANT EPILEPSY
A US study in the early 1990s estimated that the annual cost of refractory epilepsy in adults exceeds $11,745 per person2; the cost would be considerably higher today. Another study found that costs correlate with severity of illness and that patients who have intractable seizures incur a cost eight times greater than in those whose epilepsy is controlled.3
Higher risk of death
In any given interval of time, people with pharmacoresistant epilepsy are about two to 10 times more likely to die compared with the general population.4 The risk is inversely linked to seizure control.
“Sudden unexpected death in epilepsy”is the most frequent type of death in patients with pharmacoresistant epilepsy. This category excludes deaths from trauma or drowning. The death can be witnessed or unwitnessed and with or without evidence of a seizure (but not documented status epilepticus). Postmortem examination does not reveal a toxic or anatomic cause of death, and the underlying mechanisms remain unknown. However, the risk is closely associated with drug resistance (which manifests with uncontrolled convulsive seizures and need for polytherapy with antiepileptic drugs).5
Case-control studies have shown that the risk of sudden unexpected death is closely and inversely associated with seizure control; the rate is significantly higher in patients who have a higher frequency of convulsive seizures.6 In addition, freedom from seizures, achieved after successful epilepsy surgery, reduces the risk of death from all causes.7
Other causes of death in patients with epilepsy may be directly related to seizures (accidental trauma, drowning, burns) or to the underlying condition causing the seizures. Furthermore, people with epilepsy are at higher risk of suicide than the general population.
CONCEPT OF PHARMACORESISTANCE AS IT PERTAINS TO EPILEPSY
There is no uniformly accepted definition of pharmacoresistant epilepsy. Most studies defined it according to the number of antiepileptic drugs the patient had tried without success, the frequency of seizures, the duration of illness, and the period of remission. Its true definition awaits a better understanding of underlying mechanisms.
Nevertheless, a useful operational definition at present is failure to control seizures despite a trial of two or three drugs that are suitable for the type of epilepsy and have been appropriately prescribed at maximum tolerated doses. This is because the chances of controlling epilepsy decline sharply after failure of the second or third antiepileptic drug trial. In fact, some clinicians would argue against trying another antiepileptic drug in these patients, who may be candidates for surgical procedures that have high rates of success.8
Common causes of treatment failure, such as poor compliance or inappropriate selection of first-line antiepileptic drugs, should be addressed early on by the treating physician. Nonadherence to the prescribed regimen is a very common cause of uncontrolled seizures, so it is critical to maintain a good rapport with the patient and to inquire about reasons for noncompliance.
Factors that have been associated with treatment-resistant epilepsy include:
- Early onset of seizures
- Long history of poor seizure control
- Having more than one type of seizure
- Remote symptomatic etiology (eg, patients with a history of brain infection or head trauma)
- Certain structural abnormalities (eg, cortical dysplasia)
- Certain abnormalities on electroencephalography (EEG)
- Cognitive disability
- History of status epilepticus.
When to consider referral
A topic of debate is how long a patient must have active epilepsy before he or she is considered to have pharmacoresistance and should be referred to a specialist center.
Both the rate of remission and the time needed to achieve remission depend on multiple factors such as the type and etiology of the epilepsy and the definition of sustained intractability.
Importantly, the prognosis for most patients with newly diagnosed epilepsy, whether good or bad, becomes apparent within a few years of starting treatment. Although pharmacoresistant epilepsy will become refractory within 8 years in some patients, in others a second drug may not fail for more than 1 or 2 decades after diagnosis. Nonetheless, a history of a lack of a sustained seizure-free period for 12 consecutive months, in spite of two or three suitable and tolerated antiepileptic drugs, is a definite red flag for clinicians and should prompt referral to a specialist center.9,10 The National Association of Epilepsy Centers recommends referral to a specialized epilepsy center if seizure control is not achieved by a general neurologist within a period of 9 months.11
AN APPROACH TO PHARMACORESISTANT EPILEPSY FOR THE NONSPECIALIST
Review and confirm the diagnosis of epilepsy with the help of a careful history, video-EEG, and imaging.
When seizures cannot be controlled with drugs, it is important to verify that the events in question are indeed epileptic. Continuous video-EEG monitoring may be necessary to capture and characterize the clinical manifestations and corresponding EEG changes.14 When a typical spell is not associated with an EEG change, one can often make the diagnosis of nonepileptic events, which commonly are psychogenic nonepileptic seizures. Of note, however: scalp EEG may fail to pick up ictal EEG changes in focal seizures arising from a small or deeply situated focus: for example, as in focal sensorimotor seizures from restricted perirolandic cortex.15,16
Identify the cause, type of seizure or seizures, and syndromic classification, if any.
Review past and present medications, doses, efficacy, and side effects. Consider the possibility of drug interactions.
Different drugs may have different pharmacokinetic and dose-efficacy curves. With most of the newer-generation antiepileptic compounds such as lamotrigine (Lamictal), levetiracetam (Keppra), pregabalin (Lyrica), and topiramate (Topamax), efficacy may continue to increase in some patients as the dose is increased without reaching toxicity. An important exception is phenytoin (Dilantin): due to its nonlinear and saturable pharmacokinetics, even minor dose increases may lead to a large increase in phenytoin concentration (the drug accumulates as elimination becomes saturated). A high degree of individual variability exists, which is determined by factors that include the patient’s age, genetic and enzymatic profile, comorbidities, and concurrent medications.17 Understanding these relationships in individual patients facilitates dose selection and titration and enhances compliance. Testing serum drug levels may help when compliance is questionable.
Choose antiepileptic drugs primarily on the basis of the type of seizures and the individual clinical scenario: Which drug is likely to be most efficacious with the fewest side effects, and which one is appropriate for the patient’s comorbidities and concomitant medications?
When changing the dosage, withdrawing a drug, or adding a second antiepileptic drug, always do it in a systematic way, one step at a time. Reassess the response to the change before moving to the next step.
Discuss issues such as seizure precautions, lifestyle modifications, psychosocial dysfunction, and sudden unexpected death. Offer access to epilepsy-specialist nurses and epilepsy support groups such as those offered by the Epilepsy Foundation of America, the agency dedicated to the welfare of individuals with epilepsy in the United States (http://epilepsyfoundation.ning.com/).
PATTERNS OF DRUG RESISTANCE
Epidemiologic studies suggest three different patterns of drug resistance in epilepsy: de novo, progressive, and waxing-and-waning.
De novo drug resistance
In some patients, resistance is present from the time of onset of the very first seizure, before any antiepileptic drug is even started. One landmark study showed that patients with newly diagnosed epilepsy for whom the first drug was ineffective had only an 11% probability of future success, compared with 41% to 55% in patients who had had to stop taking the drug because of intolerable side effects or idiosyncratic reactions.10 Most patients for whom the first drug fails will be resistant to most and often all antiepileptic drugs.18 These results suggest that seizures in newly diagnosed patients are either easy to control or difficult to control right from the start.
Progressive drug resistance
In some patients, epilepsy is initially controlled but then gradually becomes refractory. This pattern may be seen, for example, in childhood epilepsies or in patients with hippocampal sclerosis.19,20
Waxing and waning resistance
In some patients, epilepsy has a waxing-and-waning pattern: ie, it alternates between a remitting (pharmacoresponsive) and relapsing (pharmacoresistant) course. Patients thought to have drug-resistant epilepsy may become seizure-free when other drugs are tried. Changes in drug bioavailability, local concentration of the drug in the brain, receptor changes, the development of tolerance, and interactions with new medications may be implicated, though the exact mechanism is not understood.21
BIOLOGIC BASIS OF PHARMACORESISTANT EPILEPSY
Pharmacoresistance is not unique to epilepsy: it is now recognized in diverse brain disorders, including depression and schizophrenia,22 and in other diseases affecting the brain, such as human immunodeficiency virus infection and many forms of cancer.23
Multiple drug resistance is characterized by insensitivity to a broad spectrum of drugs that presumably act on different receptors and by different mechanisms.24
Conceptually, the variable response to antiepileptic drugs can be attributed to factors related to the disease, the patient, and the drugs, or to other unknown factors. These factors are not mutually exclusive and may be either constitutive or acquired during the course of the disease.
Factors related to the disease (independent of the host)
These factors include etiology, epilepsy progression resulting in persistent changes of the epileptogenic network, and alterations of drug targets (ie, the “target” or pharmacodynamic hypothesis that reduced sensitivity to antiepileptic drugs is due to seizure-related alterations of specific drug targets25,26) or drug uptake into the brain27 (the “transporter” or pharmacokinetic hypothesis that the drugs are ineffective due to intrinsic or acquired overexpression of multidrug transporter proteins that hamper local drug delivery to target sites).28
Factors related to the drugs
Several drug-related factors have been implicated, such as the development of tolerance, lack of antiepileptogenic (disease-modifying) actions to interrupt the ongoing process of epileptogenesis rather than only suppressing seizures, and paucity of drugs with specific mechanisms of action tailored to difficult-tocontrol epilepsies.29
Patient characteristics
Variability in response (efficacy and adverse effects) to each antiepileptic drug can be due to interindividual differences in any of four interrelated fundamental factors: DNA, RNA, proteins, or metabolites. The field of study that aims to assess the effect of DNA variations (genotype) on a patient’s clinical response to a drug (phenotype) is known as pharmacogenetics. Age-related changes in pharmacokinetic and pharmacodynamic variables may contribute to age-dependent pharmacoresistance. Least studied are environmental factors that may play a role in the development or expression of pharmacoresistance.
NONPHARMACOLOGIC TREATMENTS
Ketogenic diet
The ketogenic diet, an important nonpharmacologic alternative, is usually reserved for young patients with difficult-to-control seizures. Originally developed almost a century ago, the diet mimics the biochemical changes associated with starvation. It is a strict regimen, high in fat and low in carbohydrate and protein (typically in a ratio of 4:1 or 3:1 in adolescents and very young children).
Such a strict regimen is difficult to implement and maintain and requires close supervision by a dietician and physician. In addition to the practical complexities, concerns also exist about the long-term effects of the diet on the child’s growth and overall health. For these reasons, the ketogenic diet is restricted to a small group of young patients with pharmacoresistant epilepsy and is not usually used long-term. There are few data indicating when it is appropriate to terminate the diet in patients who have a favorable response, but most clinicians wean the patient after 2 to 3 years.
Reports on the use of the ketogenic diet in adults are scarce, although benefit was seen in a small series.30 No long-term follow-up data exist for adults, especially regarding the risk of atherosclerosis.
Vagus nerve stimulation
Vagus nerve stimulation is a nonpharmacologic alternative for adults and for adolescents over age 12 years who have intractable focal seizures and who are not favorable surgical candidates. 31 Its effectiveness in younger patients and in those who have intractable generalized seizures is less clear, although published uncontrolled series have reported benefit (fewer seizures and better quality of life).32–34
A device consisting of a pulse generator is implanted subcutaneously in the precordium, and a lead wire is tunneled under the skin and attached to the left vagus nerve. The generator is programmed using a telemetry wand held over the device, with settings for current intensity (typically 1–2 mA), pulse width (250–300 μsec), frequency (30 Hz), and “duty cycle” (typically 30 seconds on stimulation, followed by 3 to 5 minutes off, cycling 24 hours/day). Hence, it provides “open-loop stimulation,” ie, continuous stimulation that is not modified in response to the patient’s EEG seizure activity. Patients or caregivers can also activate the device manually (“on demand”) at the first sign or warning of an impending seizure by swiping a handheld magnet.
Common side effects such as cough, voice alteration, and hoarseness are usually stimulation-dependent and tend to diminish with time. Notably, vagus nerve stimulation has none of the cognitive side effects often encountered with increasing doses of antiepileptic drugs. As with other implantable stimulators, some safety concerns exist in patients undergoing magnetic resonance imaging (MRI).
At least one-third of patients who receive this treatment show a sustained response, defined as a 50% or greater reduction in seizures. However, few achieve freedom from seizures, and therefore this therapy is considered palliative and is reserved for patients who are not candidates for surgery or for whom surgery has failed.
Unfortunately, it has not been possible to predict which patients will benefit from vagus nerve stimulation. The American Academy of Neurology recommends that this treatment be considered only after a thorough evaluation by a subspecialist to rule out nonepileptic conditions, false pharmacoresistance, and surgically treatable types of epilepsy.31
IS THE PATIENT A CANDIDATE FOR EPILEPSY SURGERY?
- Is the epilepsy diagnosis correct?
- Is the epilepsy focal? Have the following possibilities been excluded: generalized or multifocal epilepsy, situational or provoked seizures, or an epilepsy syndrome with spontaneous remission?
- Do seizures remain poorly controlled despite adequate pharmacologic trials?
- If so, do the seizures or medication side effects significantly affect the patient’s quality of life?
- Can an epileptogenic lesion be seen on MRI, and what is the suspected etiology?
- Is there converging evidence for a single epileptogenic focus?
- Are there abnormalities elsewhere in the brain?
- What are the chances of a good outcome in terms of seizure control and improvement in quality of life?
- What are the risks of surgery, and how do these compare with the risks of not having surgery?
- What are the patient’s perceptions and attitudes toward epilepsy surgery?
Surgical recommendations should be made after a thorough discussion of all preoperative data in a multidisciplinary patient management conference (Figure 1, Figure 2), in which epileptologists, neurosurgeons, neuroradiologists, neuropsychologists, and psychiatrists all actively participate.
Preoperative counseling is essential for the patient and his or her family, addressing the goals, risks, and benefits of the surgery. Treatment decisions should take into account the possible impact of surgery on the patient’s medical and psychosocial circumstances (risks of ongoing seizures vs surgical intervention; impact on the patient’s independence, employment status, emotional well-being, and psychiatric and other comorbidities).
EPILEPSY SURGERY: CURATIVE OR PALLIATIVE
Epilepsy surgery can be classified as curative or palliative, depending on the goal.
Curative procedures
Curative procedures include lobectomy, lesionectomy, and multilobar or hemispheric surgery (hemispherectomy).
Anterior temporal lobectomy and hippocampectomy are used for temporal lobe epilepsy and have several variations.
“Mesial temporal lobe epilepsy associated with hippocampal sclerosis,” a recognizable syndrome with complex partial seizures, stereotyped electroclinical features, and fairly typical natural history, is the most common of the focal epilepsies in adults.36 Its prognosis is poor when treated medically, but it responds well to surgery.37,38 In a landmark prospective trial, 58% of 40 patients randomized to surgical treatment were free of disabling seizures after 1 year, compared with only 8% of 40 patients treated medically.39
Lesionectomy and lobectomy are resective approaches targeting seizure foci outside the temporal lobe (most often in the frontal lobe, less commonly in the parietal or occipital lobes) or within the temporal lobe but outside the hippocampus (neocortical temporal lobe epilepsies).
The nature of the underlying substrate plays a significant role in determining the natural history,40 surgical strategy, and prognosis for freedom from seizures postoperatively. Patients with seizures due to structural lesions that are visible on MRI (“lesional epilepsies,” eg, cavernous angiomas or circumscribed lowgrade tumors) may become seizure-free after limited resections targeting the lesion itself (lesionectomy) or extending to involve part of a lobe or an entire lobe (lobectomy).
A favorable surgical outcome is much more likely if the lesion can be completely removed.41 At times, however, the lesion cannot be resected in its entirety, for example if it is located within inaccessible or essential (eloquent) cortex.
On the other hand, identifying the epileptogenic focus in patients with no visible structural abnormality on MRI (“nonlesional epilepsies”) can be challenging and usually requires intracranial investigations. In this instance, the aim of surgery is to resect regions that are electrographically abnormal. In general, the postoperative outcome is less favorable in nonlesional focal epilepsies than in lesional epilepsies.42
Multilobar resections and hemispherectomy are indicated when seizures arise from extensive, diffuse, or multiple regions of a single hemisphere.
If the neurologic function supported by the abnormal hemisphere is intact, a tailored multilobar resection aims at eliminating the epileptogenic focus without creating new deficits.
If, however, the underlying hemispheric abnormality is associated with significant contralateral hemiparesis, hemiplegia, or visual field deficits, the need to preserve function does not limit surgery, and hemispherectomy can be considered. Hemispherectomy can be the procedure of choice for selected infants and young children with catastrophic epilepsies and unilateral brain damage.43,44 The goal is to control seizures by completely disconnecting the abnormal epileptogenic hemisphere from the opposite, “good” hemisphere. A second major goal is to improve psychosocial and cognitive development by eliminating the child’s uncontrolled seizures.
Palliative procedures
Palliative procedures, in contrast to curative ones, rarely eliminate seizures entirely. It is important to determine that patients are not candidates for a more definitive, potentially curative resective procedure before considering palliative surgical options such as corpus callosotomy, multiple subpial transections, or vagus nerve stimulation.
Corpus callosotomy (transection of the corpus callosum) is performed in a small number of patients, ie, those who have disabling seizures that rapidly become generalized or injurious drop attacks and are not candidates for focal resection. By disconnecting the two hemispheres, this procedure aims to hinder the fast interhemispheric spread of seizure discharges.
Callosotomy may be complete or involve only a portion of the corpus callosum. The extent of resection has been correlated with favorable outcome.45
Some investigators report a 50% or greater reduction in seizure frequency, with drop attacks and generalized tonic-clonic seizures showing the most consistent improvement. In addition, behavior and quality of life may also improve.46
Multiple subpial transections are reserved for seizures arising from eloquent cortex (ie, from areas that cannot be removed without incurring unacceptable neurologic deficits). Therefore, the surgeon only transects the epileptogenic cortex in a vertical manner, so as to interrupt the horizontal cortical connections without resection. This approach is thought to disrupt the synchrony of seizure propagation while preserving physiologic function.
A meta-analysis of small case series suggests some decrease in seizure frequency with no or minimal neurologic compromise in up to 60% of patients.47
COMPLICATIONS OF EPILEPSY SURGERY
Resective surgery is not without risk, but often the risk is much less than that posed by uncontrolled epilepsy in the long term. Operative mortality rates vary from almost zero for temporal lobe surgery to 2.5% for hemispherectomy. The reported risk of permanent surgical morbidity varies by type of surgery from 1.1% for temporal lobe resection to about 5% for frontal lobe resection.4
NOVEL EPILEPSY THERAPIES
The failure of available antiepileptic drugs to control seizures in a substantial number of patients underscores the need to develop novel therapies such as electrical stimulation, local drug delivery, cell transplantation, and genebased therapies.48 Future targeted therapies could be coupled to seizure-forecasting systems to create “smart” implantable devices that predict, detect, and preemptively treat the seizures in a “closed-loop” fashion.
Targeted electrical stimulation
To modulate abnormal cortical hyperexcitability, electrical stimulation can be applied to the peripheral nervous system (eg, vagus nerve stimulation) or central nervous system. Central nervous system stimulation can be broadly divided into two approaches:
Direct stimulation targets presumed epileptogenic brain tissue such as the neocortex or hippocampus.
Indirect stimulation targets presumed seizure-gating networks such as in the cerebellum and various deep brain nuclei in the basal ganglia or thalamus (deep brain stimulation), which are believed to play a central role in modulating the synchronization and propagation of seizure activity.
Systematic, well-designed studies are currently under way. The budding field of electrical stimulation faces a number of challenges, which include optimizing stimulation variables and target sites, selecting favorable candidates, validating long-term safety and efficacy, evaluating long-term effects on tissue reorganization, plasticity, and epileptogenicity, and developing reliable algorithms for seizure detection and prediction.
Local drug delivery
Direct delivery of drugs into the epileptogenic brain tissue holds promise, particularly for patients whose foci cannot be surgically removed. By bypassing the systemic circulation, this approach has the potential to avoid systemic and even whole-brain side effects.
However, only a few proof-of-principle experiments have been conducted in animals, and to date no clinical study has explored the utility of intraparenchymal or intraventricular antiepileptic drug delivery in humans.
Cell and gene therapies
The emerging field of experimental cell- and gene-based neuropharmacology holds promise for location-specific therapeutic strategies. In ex vivo gene therapy, bioengineered cells capable of delivering anticonvulsant compounds might be transplanted into specific areas of the brain. On the other hand, in vivo gene therapy would involve delivering genes by viral vectors to induce the localized production of antiepileptic compounds in situ.
Endogenous anticonvulsants such as gamma-aminobutyric acid (GABA) and adenosine have been tried in various animal experiments. 49 Before they can be applied clinically, significant questions need to be addressed, including the potential for toxicity or maladaptive plasticity and long-term therapeutic safety and efficacy.
Cell transplantation is aimed at restoring the physiologic balance of neurotransmitters, and is currently being investigated for the treatment of several neurologic disorders such as Parkinson disease and Huntington disease.50 Unlike delivery of exogenous compounds, cell transplantation (heterologous fetal cell grafts or embryonic or adult stem cells) has the potential to form restorative synaptic connections and assimilate within existing cells and networks in the host tissue. An essential limitation to xenotransplantation in humans is the risk of immunologic rejection.
The future
We hope that continued progress in genomics will lead to targeted development of disease-modifying drugs that can impede or reverse the process of epileptogenesis. Advances in informatics and genetics may be harnessed to predict which patients are likely to develop pharmacoresistance, to cure certain genetic epilepsies, and to individualize antiepileptic drug selection on the basis of each person’s genetic profile.
Although more than 10 new antiepileptic drugs have been developed in the past decade, epilepsy remains resistant to drug therapy in about one-third of patients. Approximately 20% of patients with primary generalized epilepsy and up to 60% of patients who have focal epilepsy develop drug resistance during the course of their condition, which for many is lifelong.1
Those who get no response or only a partial response to drugs continue to have incapacitating seizures that lead to significant neuropsychiatric and social impairment, lower quality of life, greater morbidity, and a higher risk of death.
Managing these patients is a challenge and requires a structured multidisciplinary approach in specialized clinics. Newer research, particularly in pharmacogenomics, holds promise of therapy that more closely suits an individual’s profile and type of epilepsy.
This article reviews recent developments in the pathogenesis and treatment of pharmacoresistant epilepsy, placing these topics in clinical context to facilitate and enhance the physician’s ability to manage it.
THE COSTS OF RESISTANT EPILEPSY
A US study in the early 1990s estimated that the annual cost of refractory epilepsy in adults exceeds $11,745 per person2; the cost would be considerably higher today. Another study found that costs correlate with severity of illness and that patients who have intractable seizures incur a cost eight times greater than in those whose epilepsy is controlled.3
Higher risk of death
In any given interval of time, people with pharmacoresistant epilepsy are about two to 10 times more likely to die compared with the general population.4 The risk is inversely linked to seizure control.
“Sudden unexpected death in epilepsy”is the most frequent type of death in patients with pharmacoresistant epilepsy. This category excludes deaths from trauma or drowning. The death can be witnessed or unwitnessed and with or without evidence of a seizure (but not documented status epilepticus). Postmortem examination does not reveal a toxic or anatomic cause of death, and the underlying mechanisms remain unknown. However, the risk is closely associated with drug resistance (which manifests with uncontrolled convulsive seizures and need for polytherapy with antiepileptic drugs).5
Case-control studies have shown that the risk of sudden unexpected death is closely and inversely associated with seizure control; the rate is significantly higher in patients who have a higher frequency of convulsive seizures.6 In addition, freedom from seizures, achieved after successful epilepsy surgery, reduces the risk of death from all causes.7
Other causes of death in patients with epilepsy may be directly related to seizures (accidental trauma, drowning, burns) or to the underlying condition causing the seizures. Furthermore, people with epilepsy are at higher risk of suicide than the general population.
CONCEPT OF PHARMACORESISTANCE AS IT PERTAINS TO EPILEPSY
There is no uniformly accepted definition of pharmacoresistant epilepsy. Most studies defined it according to the number of antiepileptic drugs the patient had tried without success, the frequency of seizures, the duration of illness, and the period of remission. Its true definition awaits a better understanding of underlying mechanisms.
Nevertheless, a useful operational definition at present is failure to control seizures despite a trial of two or three drugs that are suitable for the type of epilepsy and have been appropriately prescribed at maximum tolerated doses. This is because the chances of controlling epilepsy decline sharply after failure of the second or third antiepileptic drug trial. In fact, some clinicians would argue against trying another antiepileptic drug in these patients, who may be candidates for surgical procedures that have high rates of success.8
Common causes of treatment failure, such as poor compliance or inappropriate selection of first-line antiepileptic drugs, should be addressed early on by the treating physician. Nonadherence to the prescribed regimen is a very common cause of uncontrolled seizures, so it is critical to maintain a good rapport with the patient and to inquire about reasons for noncompliance.
Factors that have been associated with treatment-resistant epilepsy include:
- Early onset of seizures
- Long history of poor seizure control
- Having more than one type of seizure
- Remote symptomatic etiology (eg, patients with a history of brain infection or head trauma)
- Certain structural abnormalities (eg, cortical dysplasia)
- Certain abnormalities on electroencephalography (EEG)
- Cognitive disability
- History of status epilepticus.
When to consider referral
A topic of debate is how long a patient must have active epilepsy before he or she is considered to have pharmacoresistance and should be referred to a specialist center.
Both the rate of remission and the time needed to achieve remission depend on multiple factors such as the type and etiology of the epilepsy and the definition of sustained intractability.
Importantly, the prognosis for most patients with newly diagnosed epilepsy, whether good or bad, becomes apparent within a few years of starting treatment. Although pharmacoresistant epilepsy will become refractory within 8 years in some patients, in others a second drug may not fail for more than 1 or 2 decades after diagnosis. Nonetheless, a history of a lack of a sustained seizure-free period for 12 consecutive months, in spite of two or three suitable and tolerated antiepileptic drugs, is a definite red flag for clinicians and should prompt referral to a specialist center.9,10 The National Association of Epilepsy Centers recommends referral to a specialized epilepsy center if seizure control is not achieved by a general neurologist within a period of 9 months.11
AN APPROACH TO PHARMACORESISTANT EPILEPSY FOR THE NONSPECIALIST
Review and confirm the diagnosis of epilepsy with the help of a careful history, video-EEG, and imaging.
When seizures cannot be controlled with drugs, it is important to verify that the events in question are indeed epileptic. Continuous video-EEG monitoring may be necessary to capture and characterize the clinical manifestations and corresponding EEG changes.14 When a typical spell is not associated with an EEG change, one can often make the diagnosis of nonepileptic events, which commonly are psychogenic nonepileptic seizures. Of note, however: scalp EEG may fail to pick up ictal EEG changes in focal seizures arising from a small or deeply situated focus: for example, as in focal sensorimotor seizures from restricted perirolandic cortex.15,16
Identify the cause, type of seizure or seizures, and syndromic classification, if any.
Review past and present medications, doses, efficacy, and side effects. Consider the possibility of drug interactions.
Different drugs may have different pharmacokinetic and dose-efficacy curves. With most of the newer-generation antiepileptic compounds such as lamotrigine (Lamictal), levetiracetam (Keppra), pregabalin (Lyrica), and topiramate (Topamax), efficacy may continue to increase in some patients as the dose is increased without reaching toxicity. An important exception is phenytoin (Dilantin): due to its nonlinear and saturable pharmacokinetics, even minor dose increases may lead to a large increase in phenytoin concentration (the drug accumulates as elimination becomes saturated). A high degree of individual variability exists, which is determined by factors that include the patient’s age, genetic and enzymatic profile, comorbidities, and concurrent medications.17 Understanding these relationships in individual patients facilitates dose selection and titration and enhances compliance. Testing serum drug levels may help when compliance is questionable.
Choose antiepileptic drugs primarily on the basis of the type of seizures and the individual clinical scenario: Which drug is likely to be most efficacious with the fewest side effects, and which one is appropriate for the patient’s comorbidities and concomitant medications?
When changing the dosage, withdrawing a drug, or adding a second antiepileptic drug, always do it in a systematic way, one step at a time. Reassess the response to the change before moving to the next step.
Discuss issues such as seizure precautions, lifestyle modifications, psychosocial dysfunction, and sudden unexpected death. Offer access to epilepsy-specialist nurses and epilepsy support groups such as those offered by the Epilepsy Foundation of America, the agency dedicated to the welfare of individuals with epilepsy in the United States (http://epilepsyfoundation.ning.com/).
PATTERNS OF DRUG RESISTANCE
Epidemiologic studies suggest three different patterns of drug resistance in epilepsy: de novo, progressive, and waxing-and-waning.
De novo drug resistance
In some patients, resistance is present from the time of onset of the very first seizure, before any antiepileptic drug is even started. One landmark study showed that patients with newly diagnosed epilepsy for whom the first drug was ineffective had only an 11% probability of future success, compared with 41% to 55% in patients who had had to stop taking the drug because of intolerable side effects or idiosyncratic reactions.10 Most patients for whom the first drug fails will be resistant to most and often all antiepileptic drugs.18 These results suggest that seizures in newly diagnosed patients are either easy to control or difficult to control right from the start.
Progressive drug resistance
In some patients, epilepsy is initially controlled but then gradually becomes refractory. This pattern may be seen, for example, in childhood epilepsies or in patients with hippocampal sclerosis.19,20
Waxing and waning resistance
In some patients, epilepsy has a waxing-and-waning pattern: ie, it alternates between a remitting (pharmacoresponsive) and relapsing (pharmacoresistant) course. Patients thought to have drug-resistant epilepsy may become seizure-free when other drugs are tried. Changes in drug bioavailability, local concentration of the drug in the brain, receptor changes, the development of tolerance, and interactions with new medications may be implicated, though the exact mechanism is not understood.21
BIOLOGIC BASIS OF PHARMACORESISTANT EPILEPSY
Pharmacoresistance is not unique to epilepsy: it is now recognized in diverse brain disorders, including depression and schizophrenia,22 and in other diseases affecting the brain, such as human immunodeficiency virus infection and many forms of cancer.23
Multiple drug resistance is characterized by insensitivity to a broad spectrum of drugs that presumably act on different receptors and by different mechanisms.24
Conceptually, the variable response to antiepileptic drugs can be attributed to factors related to the disease, the patient, and the drugs, or to other unknown factors. These factors are not mutually exclusive and may be either constitutive or acquired during the course of the disease.
Factors related to the disease (independent of the host)
These factors include etiology, epilepsy progression resulting in persistent changes of the epileptogenic network, and alterations of drug targets (ie, the “target” or pharmacodynamic hypothesis that reduced sensitivity to antiepileptic drugs is due to seizure-related alterations of specific drug targets25,26) or drug uptake into the brain27 (the “transporter” or pharmacokinetic hypothesis that the drugs are ineffective due to intrinsic or acquired overexpression of multidrug transporter proteins that hamper local drug delivery to target sites).28
Factors related to the drugs
Several drug-related factors have been implicated, such as the development of tolerance, lack of antiepileptogenic (disease-modifying) actions to interrupt the ongoing process of epileptogenesis rather than only suppressing seizures, and paucity of drugs with specific mechanisms of action tailored to difficult-tocontrol epilepsies.29
Patient characteristics
Variability in response (efficacy and adverse effects) to each antiepileptic drug can be due to interindividual differences in any of four interrelated fundamental factors: DNA, RNA, proteins, or metabolites. The field of study that aims to assess the effect of DNA variations (genotype) on a patient’s clinical response to a drug (phenotype) is known as pharmacogenetics. Age-related changes in pharmacokinetic and pharmacodynamic variables may contribute to age-dependent pharmacoresistance. Least studied are environmental factors that may play a role in the development or expression of pharmacoresistance.
NONPHARMACOLOGIC TREATMENTS
Ketogenic diet
The ketogenic diet, an important nonpharmacologic alternative, is usually reserved for young patients with difficult-to-control seizures. Originally developed almost a century ago, the diet mimics the biochemical changes associated with starvation. It is a strict regimen, high in fat and low in carbohydrate and protein (typically in a ratio of 4:1 or 3:1 in adolescents and very young children).
Such a strict regimen is difficult to implement and maintain and requires close supervision by a dietician and physician. In addition to the practical complexities, concerns also exist about the long-term effects of the diet on the child’s growth and overall health. For these reasons, the ketogenic diet is restricted to a small group of young patients with pharmacoresistant epilepsy and is not usually used long-term. There are few data indicating when it is appropriate to terminate the diet in patients who have a favorable response, but most clinicians wean the patient after 2 to 3 years.
Reports on the use of the ketogenic diet in adults are scarce, although benefit was seen in a small series.30 No long-term follow-up data exist for adults, especially regarding the risk of atherosclerosis.
Vagus nerve stimulation
Vagus nerve stimulation is a nonpharmacologic alternative for adults and for adolescents over age 12 years who have intractable focal seizures and who are not favorable surgical candidates. 31 Its effectiveness in younger patients and in those who have intractable generalized seizures is less clear, although published uncontrolled series have reported benefit (fewer seizures and better quality of life).32–34
A device consisting of a pulse generator is implanted subcutaneously in the precordium, and a lead wire is tunneled under the skin and attached to the left vagus nerve. The generator is programmed using a telemetry wand held over the device, with settings for current intensity (typically 1–2 mA), pulse width (250–300 μsec), frequency (30 Hz), and “duty cycle” (typically 30 seconds on stimulation, followed by 3 to 5 minutes off, cycling 24 hours/day). Hence, it provides “open-loop stimulation,” ie, continuous stimulation that is not modified in response to the patient’s EEG seizure activity. Patients or caregivers can also activate the device manually (“on demand”) at the first sign or warning of an impending seizure by swiping a handheld magnet.
Common side effects such as cough, voice alteration, and hoarseness are usually stimulation-dependent and tend to diminish with time. Notably, vagus nerve stimulation has none of the cognitive side effects often encountered with increasing doses of antiepileptic drugs. As with other implantable stimulators, some safety concerns exist in patients undergoing magnetic resonance imaging (MRI).
At least one-third of patients who receive this treatment show a sustained response, defined as a 50% or greater reduction in seizures. However, few achieve freedom from seizures, and therefore this therapy is considered palliative and is reserved for patients who are not candidates for surgery or for whom surgery has failed.
Unfortunately, it has not been possible to predict which patients will benefit from vagus nerve stimulation. The American Academy of Neurology recommends that this treatment be considered only after a thorough evaluation by a subspecialist to rule out nonepileptic conditions, false pharmacoresistance, and surgically treatable types of epilepsy.31
IS THE PATIENT A CANDIDATE FOR EPILEPSY SURGERY?
- Is the epilepsy diagnosis correct?
- Is the epilepsy focal? Have the following possibilities been excluded: generalized or multifocal epilepsy, situational or provoked seizures, or an epilepsy syndrome with spontaneous remission?
- Do seizures remain poorly controlled despite adequate pharmacologic trials?
- If so, do the seizures or medication side effects significantly affect the patient’s quality of life?
- Can an epileptogenic lesion be seen on MRI, and what is the suspected etiology?
- Is there converging evidence for a single epileptogenic focus?
- Are there abnormalities elsewhere in the brain?
- What are the chances of a good outcome in terms of seizure control and improvement in quality of life?
- What are the risks of surgery, and how do these compare with the risks of not having surgery?
- What are the patient’s perceptions and attitudes toward epilepsy surgery?
Surgical recommendations should be made after a thorough discussion of all preoperative data in a multidisciplinary patient management conference (Figure 1, Figure 2), in which epileptologists, neurosurgeons, neuroradiologists, neuropsychologists, and psychiatrists all actively participate.
Preoperative counseling is essential for the patient and his or her family, addressing the goals, risks, and benefits of the surgery. Treatment decisions should take into account the possible impact of surgery on the patient’s medical and psychosocial circumstances (risks of ongoing seizures vs surgical intervention; impact on the patient’s independence, employment status, emotional well-being, and psychiatric and other comorbidities).
EPILEPSY SURGERY: CURATIVE OR PALLIATIVE
Epilepsy surgery can be classified as curative or palliative, depending on the goal.
Curative procedures
Curative procedures include lobectomy, lesionectomy, and multilobar or hemispheric surgery (hemispherectomy).
Anterior temporal lobectomy and hippocampectomy are used for temporal lobe epilepsy and have several variations.
“Mesial temporal lobe epilepsy associated with hippocampal sclerosis,” a recognizable syndrome with complex partial seizures, stereotyped electroclinical features, and fairly typical natural history, is the most common of the focal epilepsies in adults.36 Its prognosis is poor when treated medically, but it responds well to surgery.37,38 In a landmark prospective trial, 58% of 40 patients randomized to surgical treatment were free of disabling seizures after 1 year, compared with only 8% of 40 patients treated medically.39
Lesionectomy and lobectomy are resective approaches targeting seizure foci outside the temporal lobe (most often in the frontal lobe, less commonly in the parietal or occipital lobes) or within the temporal lobe but outside the hippocampus (neocortical temporal lobe epilepsies).
The nature of the underlying substrate plays a significant role in determining the natural history,40 surgical strategy, and prognosis for freedom from seizures postoperatively. Patients with seizures due to structural lesions that are visible on MRI (“lesional epilepsies,” eg, cavernous angiomas or circumscribed lowgrade tumors) may become seizure-free after limited resections targeting the lesion itself (lesionectomy) or extending to involve part of a lobe or an entire lobe (lobectomy).
A favorable surgical outcome is much more likely if the lesion can be completely removed.41 At times, however, the lesion cannot be resected in its entirety, for example if it is located within inaccessible or essential (eloquent) cortex.
On the other hand, identifying the epileptogenic focus in patients with no visible structural abnormality on MRI (“nonlesional epilepsies”) can be challenging and usually requires intracranial investigations. In this instance, the aim of surgery is to resect regions that are electrographically abnormal. In general, the postoperative outcome is less favorable in nonlesional focal epilepsies than in lesional epilepsies.42
Multilobar resections and hemispherectomy are indicated when seizures arise from extensive, diffuse, or multiple regions of a single hemisphere.
If the neurologic function supported by the abnormal hemisphere is intact, a tailored multilobar resection aims at eliminating the epileptogenic focus without creating new deficits.
If, however, the underlying hemispheric abnormality is associated with significant contralateral hemiparesis, hemiplegia, or visual field deficits, the need to preserve function does not limit surgery, and hemispherectomy can be considered. Hemispherectomy can be the procedure of choice for selected infants and young children with catastrophic epilepsies and unilateral brain damage.43,44 The goal is to control seizures by completely disconnecting the abnormal epileptogenic hemisphere from the opposite, “good” hemisphere. A second major goal is to improve psychosocial and cognitive development by eliminating the child’s uncontrolled seizures.
Palliative procedures
Palliative procedures, in contrast to curative ones, rarely eliminate seizures entirely. It is important to determine that patients are not candidates for a more definitive, potentially curative resective procedure before considering palliative surgical options such as corpus callosotomy, multiple subpial transections, or vagus nerve stimulation.
Corpus callosotomy (transection of the corpus callosum) is performed in a small number of patients, ie, those who have disabling seizures that rapidly become generalized or injurious drop attacks and are not candidates for focal resection. By disconnecting the two hemispheres, this procedure aims to hinder the fast interhemispheric spread of seizure discharges.
Callosotomy may be complete or involve only a portion of the corpus callosum. The extent of resection has been correlated with favorable outcome.45
Some investigators report a 50% or greater reduction in seizure frequency, with drop attacks and generalized tonic-clonic seizures showing the most consistent improvement. In addition, behavior and quality of life may also improve.46
Multiple subpial transections are reserved for seizures arising from eloquent cortex (ie, from areas that cannot be removed without incurring unacceptable neurologic deficits). Therefore, the surgeon only transects the epileptogenic cortex in a vertical manner, so as to interrupt the horizontal cortical connections without resection. This approach is thought to disrupt the synchrony of seizure propagation while preserving physiologic function.
A meta-analysis of small case series suggests some decrease in seizure frequency with no or minimal neurologic compromise in up to 60% of patients.47
COMPLICATIONS OF EPILEPSY SURGERY
Resective surgery is not without risk, but often the risk is much less than that posed by uncontrolled epilepsy in the long term. Operative mortality rates vary from almost zero for temporal lobe surgery to 2.5% for hemispherectomy. The reported risk of permanent surgical morbidity varies by type of surgery from 1.1% for temporal lobe resection to about 5% for frontal lobe resection.4
NOVEL EPILEPSY THERAPIES
The failure of available antiepileptic drugs to control seizures in a substantial number of patients underscores the need to develop novel therapies such as electrical stimulation, local drug delivery, cell transplantation, and genebased therapies.48 Future targeted therapies could be coupled to seizure-forecasting systems to create “smart” implantable devices that predict, detect, and preemptively treat the seizures in a “closed-loop” fashion.
Targeted electrical stimulation
To modulate abnormal cortical hyperexcitability, electrical stimulation can be applied to the peripheral nervous system (eg, vagus nerve stimulation) or central nervous system. Central nervous system stimulation can be broadly divided into two approaches:
Direct stimulation targets presumed epileptogenic brain tissue such as the neocortex or hippocampus.
Indirect stimulation targets presumed seizure-gating networks such as in the cerebellum and various deep brain nuclei in the basal ganglia or thalamus (deep brain stimulation), which are believed to play a central role in modulating the synchronization and propagation of seizure activity.
Systematic, well-designed studies are currently under way. The budding field of electrical stimulation faces a number of challenges, which include optimizing stimulation variables and target sites, selecting favorable candidates, validating long-term safety and efficacy, evaluating long-term effects on tissue reorganization, plasticity, and epileptogenicity, and developing reliable algorithms for seizure detection and prediction.
Local drug delivery
Direct delivery of drugs into the epileptogenic brain tissue holds promise, particularly for patients whose foci cannot be surgically removed. By bypassing the systemic circulation, this approach has the potential to avoid systemic and even whole-brain side effects.
However, only a few proof-of-principle experiments have been conducted in animals, and to date no clinical study has explored the utility of intraparenchymal or intraventricular antiepileptic drug delivery in humans.
Cell and gene therapies
The emerging field of experimental cell- and gene-based neuropharmacology holds promise for location-specific therapeutic strategies. In ex vivo gene therapy, bioengineered cells capable of delivering anticonvulsant compounds might be transplanted into specific areas of the brain. On the other hand, in vivo gene therapy would involve delivering genes by viral vectors to induce the localized production of antiepileptic compounds in situ.
Endogenous anticonvulsants such as gamma-aminobutyric acid (GABA) and adenosine have been tried in various animal experiments. 49 Before they can be applied clinically, significant questions need to be addressed, including the potential for toxicity or maladaptive plasticity and long-term therapeutic safety and efficacy.
Cell transplantation is aimed at restoring the physiologic balance of neurotransmitters, and is currently being investigated for the treatment of several neurologic disorders such as Parkinson disease and Huntington disease.50 Unlike delivery of exogenous compounds, cell transplantation (heterologous fetal cell grafts or embryonic or adult stem cells) has the potential to form restorative synaptic connections and assimilate within existing cells and networks in the host tissue. An essential limitation to xenotransplantation in humans is the risk of immunologic rejection.
The future
We hope that continued progress in genomics will lead to targeted development of disease-modifying drugs that can impede or reverse the process of epileptogenesis. Advances in informatics and genetics may be harnessed to predict which patients are likely to develop pharmacoresistance, to cure certain genetic epilepsies, and to individualize antiepileptic drug selection on the basis of each person’s genetic profile.
- Siegel AM. Presurgical evaluation and surgical treatment of medically refractory epilepsy. Neurosurg Rev 2004; 27:1–18.
- Murray MI, Halpern MT, Leppik IE. Cost of refractory epilepsy in adults in the USA. Epilepsy Res 1996; 23:139–148.
- Jacoby A, Buck D, Baker G, McNamee P, Graham-Jones S, Chadwick D. Uptake and costs of care for epilepsy: findings from a U.K. regional study. Epilepsia 1998; 39:776–786.
- Chapell R, Reston J, Snyder D, Treadwell J, Treager S, Turkelson C. Management of treatment-resistant epilepsy. Evid Rep Technol Assess (Summ) 2003 Apr; 77:1–8.
- Nei M, Bagla R. Seizure-related injury and death. Curr Neurol Neurosci Rep 2007; 7:335–341.
- Langan Y, Nashef L, Sander JW. Case-control study of SUDEP. Neurology 2005; 64:1131–1133.
- Sperling MR, Feldman H, Kinman J, Liporace JD, O’Connor MJ. Seizure control and mortality in epilepsy. Ann Neurol 1999; 46:45–50.
- Berg AT. Understanding the delay before epilepsy surgery: who develops intractable focal epilepsy and when? CNS Spectr 2004; 9:136–144.
- Perucca E. Pharmacoresistance in epilepsy: how should it be defined? CNS Drugs 1998; 10:171–179.
- Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med 2000; 342:314–319.
- Gumnit RJ, Walczak TS; National Association of Epilepsy Centers. Guidelines for essential services, personnel, and facilities in specialized epilepsy centers in the United States. Epilepsia 2001; 42:804–814.
- Smith D, Defalla BA, Chadwick DW. The misdiagnosis of epilepsy and the management of refractory epilepsy in a specialist clinic. QJM 1999; 92:15–23.
- Schuele SU, Lüders HO. Intractable epilepsy: management and therapeutic alternatives. Lancet Neurol 2008; 7:514–524.
- Engel J, Burchfiel J, Ebersole J, et al. Long-term monitoring for epilepsy. Report of an IFCN committee. Electroencephalogr Clin Neurophysiol 1993; 87:437–458.
- Kanner AM, Morris HH, Lüders H, et al. Supplementary motor seizures mimicking pseudoseizures: some clinical differences. Neurology 1990; 40:1404–1407.
- Alexopoulos AV, Dinner DS. Focal motor seizures, epilepsia partialis continua, and supplementary sensorimotor seizures. In:Wyllie E, Gupta A, Lachhwani DK, editors. The Treatment of Epilepsy: Principles & Practice. Philadelphia, PA: Lippincott Williams & Wilkins, 2006:257–277.
- French JA. Refractory epilepsy: clinical overview. Epilepsia 2007; 48(suppl 1):3–7.
- Regesta G, Tanganelli P. Clinical aspects and biological bases of drug-resistant epilepsies. Epilepsy Res 1999; 34:109–122.
- Berg AT, Langfitt J, Shinnar S, et al. How long does it take for partial epilepsy to become intractable? Neurology 2003; 60:186–190.
- Berg AT, Vickrey BG, Testa FM, et al. How long does it take for epilepsy to become intractable? A prospective investigation. Ann Neurol 2006; 60:73–79.
- Löscher W, Schmidt D. Experimental and clinical evidence for loss of effect (tolerance) during prolonged treatment with antiepileptic drugs. Epilepsia 2006; 47:1253–1284.
- Löscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci 2005; 6:591–602.
- Siddiqui A, Kerb R, Weale ME, et al. Association of multidrug resistance in epilepsy with a polymorphism in the drug-transporter gene ABCB1. N Engl J Med 2003; 348:1442–1448.
- Granata T, Marchi N, Carlton E, et al. Management of the patient with medically refractory epilepsy. Expert Rev Neurother 2009; 9:1791–1802.
- Marchi N, Hallene KL, Kight KM, et al. Significance of MDR1 and multiple drug resistance in refractory human epileptic brain. BMC Med 2004; 2:37.
- Oby E, Janigro D. The blood-brain barrier and epilepsy. Epilepsia 2006; 47:1761–1774.
- Schmidt D, Löscher W. New developments in antiepileptic drug resistance: an integrative view. Epilepsy Curr 2009; 9:47–52.
- Remy S, Beck H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain 2006; 129:18–35.
- Löscher W, Schmidt D. New horizons in the development of antiepileptic drugs. Epilepsy Res 2002; 50:3–16.
- Sirven J, Whedon B, Caplan D, et al. The ketogenic diet for intractable epilepsy in adults: preliminary results. Epilepsia 1999; 40:1721–1726.
- Fisher RS, Handforth A. Reassessment: vagus nerve stimulation for epilepsy: a report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 1999; 53:666–669.
- Hallböök T, Lundgren J, Stjernqvist K, Blennow G, Strömblad LG, Rosén I. Vagus nerve stimulation in 15 children with therapy resistant epilepsy; its impact on cognition, quality of life, behaviour and mood. Seizure 2005; 14:504–513.
- Holmes MD, Silbergeld DL, Drouhard D, Wilensky AJ, Ojemann LM. Effect of vagus nerve stimulation on adults with pharmacoresistant generalized epilepsy syndromes. Seizure 2004; 13:340–345.
- Murphy JV, Torkelson R, Dowler I, Simon S, Hudson S. Vagal nerve stimulation in refractory epilepsy: the first 100 patients receiving vagal nerve stimulation at a pediatric epilepsy center. Arch Pediatr Adolesc Med 2003; 157:560–564.
- Alexopoulos AV, Najm IM. Neurosurgical management of focal epilepsies in adults. In:Panayiotopoulos CP, et al, editors. Focal Epilepsies: Seizures, Syndromes and Management. Oxford, UK: Medicinae; 2009:204–220.
- Wieser HGILAE Commission on Neurosurgery of Epilepsy. ILAE Commission Report. Mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia 2004; 45:695–714.
- Engel J. Surgery for seizures. N Engl J Med 1996; 334:647–652.
- Engel J, Wiebe S, French J, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: temporal lobe and localized neocortical resections for epilepsy: report of the Quality Standards Subcommittee of the American Academy of Neurology, in association with the American Epilepsy Society and the American Association of Neurological Surgeons. Neurology 2003; 60:538–547.
- Wiebe S, Blume WT, Girvin JP, Eliasziw M; Effectiveness and Efficiency of Surgery for Temporal Lobe Epilepsy Study Group. A randomized, controlled trial of surgery for temporal-lobe epilepsy. N Engl J Med 2001; 345:311–318.
- Semah F, Picot MC, Adam C, et al. Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology 1998; 51:1256–1262.
- Awad IA, Rosenfeld J, Ahl J, Hahn JF, Lüders H. Intractable epilepsy and structural lesions of the brain: mapping, resection strategies, and seizure outcome. Epilepsia 1991; 32:179–186.
- Cascino GD. Surgical treatment for extratemporal epilepsy. Curr Treat Options Neurol 2004; 6:257–262.
- González-Martínez JA, Gupta A, Kotagal P, et al. Hemispherectomy for catastrophic epilepsy in infants. Epilepsia 2005; 46:1518–1525.
- Wyllie E. Surgery for catastrophic localization-related epilepsy in infants. Epilepsia 1996; 37(suppl 1):S22–S25.
- Tanriverdi T, Olivier A, Poulin N, Andermann F, Dubeau F. Long-term seizure outcome after corpus callosotomy: a retrospective analysis of 95 patients. J Neurosurg 2009; 110:332–342.
- Asadi-Pooya AA, Sharan A, Nei M, Sperling MR. Corpus callosotomy. Epilepsy Behav 2008; 13:271–278.
- Spencer SS, Schramm J, Wyler A, et al. Multiple subpial transection for intractable partial epilepsy: an international meta-analysis. Epilepsia 2002; 43:141–145.
- Alexopoulos AV, Gonugunta V, Yang J, Boulis NM. Electrical stimulation and gene-based neuromodulation for control of medically-refractory epilepsy. Acta Neurochir Suppl 2007; 97:293–309.
- Detlev B. Cell and gene therapies for refractory epilepsy. Curr Neuropharmacol 2007; 5:115–125.
- Fisher RS, Ho J. Potential new methods for antiepileptic drug delivery. CNS Drugs 2002; 16:579–593.
- Siegel AM. Presurgical evaluation and surgical treatment of medically refractory epilepsy. Neurosurg Rev 2004; 27:1–18.
- Murray MI, Halpern MT, Leppik IE. Cost of refractory epilepsy in adults in the USA. Epilepsy Res 1996; 23:139–148.
- Jacoby A, Buck D, Baker G, McNamee P, Graham-Jones S, Chadwick D. Uptake and costs of care for epilepsy: findings from a U.K. regional study. Epilepsia 1998; 39:776–786.
- Chapell R, Reston J, Snyder D, Treadwell J, Treager S, Turkelson C. Management of treatment-resistant epilepsy. Evid Rep Technol Assess (Summ) 2003 Apr; 77:1–8.
- Nei M, Bagla R. Seizure-related injury and death. Curr Neurol Neurosci Rep 2007; 7:335–341.
- Langan Y, Nashef L, Sander JW. Case-control study of SUDEP. Neurology 2005; 64:1131–1133.
- Sperling MR, Feldman H, Kinman J, Liporace JD, O’Connor MJ. Seizure control and mortality in epilepsy. Ann Neurol 1999; 46:45–50.
- Berg AT. Understanding the delay before epilepsy surgery: who develops intractable focal epilepsy and when? CNS Spectr 2004; 9:136–144.
- Perucca E. Pharmacoresistance in epilepsy: how should it be defined? CNS Drugs 1998; 10:171–179.
- Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med 2000; 342:314–319.
- Gumnit RJ, Walczak TS; National Association of Epilepsy Centers. Guidelines for essential services, personnel, and facilities in specialized epilepsy centers in the United States. Epilepsia 2001; 42:804–814.
- Smith D, Defalla BA, Chadwick DW. The misdiagnosis of epilepsy and the management of refractory epilepsy in a specialist clinic. QJM 1999; 92:15–23.
- Schuele SU, Lüders HO. Intractable epilepsy: management and therapeutic alternatives. Lancet Neurol 2008; 7:514–524.
- Engel J, Burchfiel J, Ebersole J, et al. Long-term monitoring for epilepsy. Report of an IFCN committee. Electroencephalogr Clin Neurophysiol 1993; 87:437–458.
- Kanner AM, Morris HH, Lüders H, et al. Supplementary motor seizures mimicking pseudoseizures: some clinical differences. Neurology 1990; 40:1404–1407.
- Alexopoulos AV, Dinner DS. Focal motor seizures, epilepsia partialis continua, and supplementary sensorimotor seizures. In:Wyllie E, Gupta A, Lachhwani DK, editors. The Treatment of Epilepsy: Principles & Practice. Philadelphia, PA: Lippincott Williams & Wilkins, 2006:257–277.
- French JA. Refractory epilepsy: clinical overview. Epilepsia 2007; 48(suppl 1):3–7.
- Regesta G, Tanganelli P. Clinical aspects and biological bases of drug-resistant epilepsies. Epilepsy Res 1999; 34:109–122.
- Berg AT, Langfitt J, Shinnar S, et al. How long does it take for partial epilepsy to become intractable? Neurology 2003; 60:186–190.
- Berg AT, Vickrey BG, Testa FM, et al. How long does it take for epilepsy to become intractable? A prospective investigation. Ann Neurol 2006; 60:73–79.
- Löscher W, Schmidt D. Experimental and clinical evidence for loss of effect (tolerance) during prolonged treatment with antiepileptic drugs. Epilepsia 2006; 47:1253–1284.
- Löscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci 2005; 6:591–602.
- Siddiqui A, Kerb R, Weale ME, et al. Association of multidrug resistance in epilepsy with a polymorphism in the drug-transporter gene ABCB1. N Engl J Med 2003; 348:1442–1448.
- Granata T, Marchi N, Carlton E, et al. Management of the patient with medically refractory epilepsy. Expert Rev Neurother 2009; 9:1791–1802.
- Marchi N, Hallene KL, Kight KM, et al. Significance of MDR1 and multiple drug resistance in refractory human epileptic brain. BMC Med 2004; 2:37.
- Oby E, Janigro D. The blood-brain barrier and epilepsy. Epilepsia 2006; 47:1761–1774.
- Schmidt D, Löscher W. New developments in antiepileptic drug resistance: an integrative view. Epilepsy Curr 2009; 9:47–52.
- Remy S, Beck H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain 2006; 129:18–35.
- Löscher W, Schmidt D. New horizons in the development of antiepileptic drugs. Epilepsy Res 2002; 50:3–16.
- Sirven J, Whedon B, Caplan D, et al. The ketogenic diet for intractable epilepsy in adults: preliminary results. Epilepsia 1999; 40:1721–1726.
- Fisher RS, Handforth A. Reassessment: vagus nerve stimulation for epilepsy: a report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 1999; 53:666–669.
- Hallböök T, Lundgren J, Stjernqvist K, Blennow G, Strömblad LG, Rosén I. Vagus nerve stimulation in 15 children with therapy resistant epilepsy; its impact on cognition, quality of life, behaviour and mood. Seizure 2005; 14:504–513.
- Holmes MD, Silbergeld DL, Drouhard D, Wilensky AJ, Ojemann LM. Effect of vagus nerve stimulation on adults with pharmacoresistant generalized epilepsy syndromes. Seizure 2004; 13:340–345.
- Murphy JV, Torkelson R, Dowler I, Simon S, Hudson S. Vagal nerve stimulation in refractory epilepsy: the first 100 patients receiving vagal nerve stimulation at a pediatric epilepsy center. Arch Pediatr Adolesc Med 2003; 157:560–564.
- Alexopoulos AV, Najm IM. Neurosurgical management of focal epilepsies in adults. In:Panayiotopoulos CP, et al, editors. Focal Epilepsies: Seizures, Syndromes and Management. Oxford, UK: Medicinae; 2009:204–220.
- Wieser HGILAE Commission on Neurosurgery of Epilepsy. ILAE Commission Report. Mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia 2004; 45:695–714.
- Engel J. Surgery for seizures. N Engl J Med 1996; 334:647–652.
- Engel J, Wiebe S, French J, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: temporal lobe and localized neocortical resections for epilepsy: report of the Quality Standards Subcommittee of the American Academy of Neurology, in association with the American Epilepsy Society and the American Association of Neurological Surgeons. Neurology 2003; 60:538–547.
- Wiebe S, Blume WT, Girvin JP, Eliasziw M; Effectiveness and Efficiency of Surgery for Temporal Lobe Epilepsy Study Group. A randomized, controlled trial of surgery for temporal-lobe epilepsy. N Engl J Med 2001; 345:311–318.
- Semah F, Picot MC, Adam C, et al. Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology 1998; 51:1256–1262.
- Awad IA, Rosenfeld J, Ahl J, Hahn JF, Lüders H. Intractable epilepsy and structural lesions of the brain: mapping, resection strategies, and seizure outcome. Epilepsia 1991; 32:179–186.
- Cascino GD. Surgical treatment for extratemporal epilepsy. Curr Treat Options Neurol 2004; 6:257–262.
- González-Martínez JA, Gupta A, Kotagal P, et al. Hemispherectomy for catastrophic epilepsy in infants. Epilepsia 2005; 46:1518–1525.
- Wyllie E. Surgery for catastrophic localization-related epilepsy in infants. Epilepsia 1996; 37(suppl 1):S22–S25.
- Tanriverdi T, Olivier A, Poulin N, Andermann F, Dubeau F. Long-term seizure outcome after corpus callosotomy: a retrospective analysis of 95 patients. J Neurosurg 2009; 110:332–342.
- Asadi-Pooya AA, Sharan A, Nei M, Sperling MR. Corpus callosotomy. Epilepsy Behav 2008; 13:271–278.
- Spencer SS, Schramm J, Wyler A, et al. Multiple subpial transection for intractable partial epilepsy: an international meta-analysis. Epilepsia 2002; 43:141–145.
- Alexopoulos AV, Gonugunta V, Yang J, Boulis NM. Electrical stimulation and gene-based neuromodulation for control of medically-refractory epilepsy. Acta Neurochir Suppl 2007; 97:293–309.
- Detlev B. Cell and gene therapies for refractory epilepsy. Curr Neuropharmacol 2007; 5:115–125.
- Fisher RS, Ho J. Potential new methods for antiepileptic drug delivery. CNS Drugs 2002; 16:579–593.
KEY POINTS
- When seizures have failed to respond to two or three appropriate antiepileptic drugs, the chance of significant benefit from other drugs is 10% or less.
- The biologic basis of pharmacoresistance is multifactorial and varies from one patient to another.
- Social and lifestyle factors, including alcohol misuse and nonadherence to prescribed antiepileptic drugs, can contribute to or masquerade as pharmacoresistance.
- Current options for patients with pharmacoresistant epilepsy are surgery (the best option when feasible), vagus nerve stimulation, investigational drugs or devices, and aggressive combination treatment with available antiepileptic drugs.