You have a 54-year-old black patient with gout, diabetes mellitus, hypertension, and chronic kidney disease (CKD). He has an acute gout flare involving his right knee. In the past year he has had four attacks of gout in the ankles and knees, which you treated with intra-articular glucocorticoid injections. He has been on allopurinol (Zyloprim) 200 mg daily, but his last serum urate level was 9.4 mg/dL (reference range 3.0–8.0). His creatinine clearance is 45 mL/minute (reference range 85–125).
In view of his kidney disease, you are concerned about increasing his dose of allopurinol, but also about the need to treat his frequent attacks. How should you manage this patient?
GOUT IS CHALLENGING TO TREAT IN PATIENTS WITH KIDNEY DISEASE
A major challenge in treating patients with gout is to avoid therapeutic interactions with common comorbidities, including hypertension, insulin resistance, coronary artery disease, heart failure, and especially CKD.1
In this paper, we discuss approaches to and controversies in the management of gout and hyperuricemia in patients with CKD. Unfortunately, the evidence from clinical trials to guide treatment decisions is limited; therefore, decisions must often be based on experience and pathophysiologic principles.
GENERAL GOALS OF GOUT THERAPY
Depending on the patient and the stage of the disease, the goals in treating patients with gout are to:
Terminate acute attacks as promptly and safely as possible
Prevent recurrences of acute gout attacks
Prevent or reverse complications resulting from deposition of monosodium urate in the joints, in the kidneys, or at other sites.
These goals are more difficult to achieve in patients with CKD because of the potential complications from many of the available drugs.
TERMINATING ACUTE GOUT FLARES
In patients with acute gout, treatment is aimed at quickly resolving pain and inflammation.
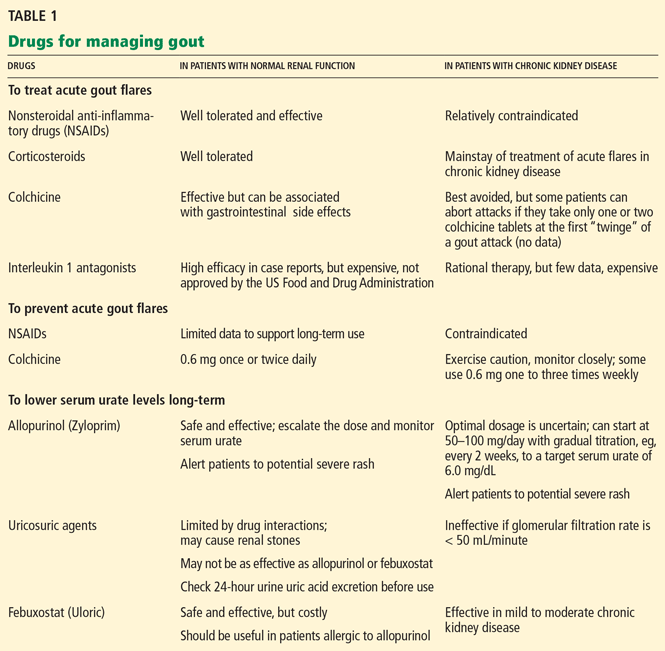
Several types of drugs can terminate acute gout flares. The choice in most situations is colchicine (Colcrys); a nonsteroidal anti-inflammatory drug (NSAID); a corticosteroid; or corticotropin (ACTH).
However, in patients with CKD, there are concerns about using colchicine or NSAIDs, and corticotropin is very expensive; thus, corticosteroids are often used.
Colchicine’s clearance is reduced in CKD
Colchicine is somewhat effective in treating acute gout attacks and probably more effective in preventing attacks.
Due to concerns about inappropriate dosing and reported deaths,2 the intravenous formulation is not available in many countries, including the United States.
After oral administration, colchicine is rapidly absorbed, with a bioavailability of up to 50%. It undergoes metabolism by the liver, and its metabolites are excreted by renal and biliary-intestinal routes. Up to 20% of the active drug is excreted by the kidneys.3
Colchicine’s clearance is significantly reduced in patients with renal or hepatic insufficiency, and the drug may accumulate in cells, with resultant toxicity.4 Colchicine-induced toxicity has been observed when the drug was used for acute treatment, as well as for chronic prophylaxis of gout in patients with CKD; thus, alternative agents for treating acute attacks should be considered.5,6 With prolonged use, reversible colchicine-induced axonal neuropathy, neutropenia, and vacuolar myopathy can develop in patients with CKD.7
In a trial in patients with normal renal function, nearly 100% who received an initial dose of 1 mg followed by 0.5 mg every 2 hours developed diarrhea at a median time of 24 hours.8 Emesis may also occur.
A lower dose of 1.8 mg (two 0.6-mg pills followed by one pill an hour later) was well tolerated but only moderately effective in treating acute gout, causing at least a 50% reduction in pain at 24 hours in only 38% of patients.9 This study does not clarify the dosage to use to completely resolve attacks. Using additional colchicine likely will increase the response rate, but will also increase side effects. Patients with CKD were not included.
Some patients, as shown in the above trial, can abort attacks by taking only one or two colchicine tablets when they feel the first “twinge” of an attack. This approach is likely to be safe in CKD, but it may be of value to only a few patients.
Nonsteroidal anti-inflammatory drugs can worsen chronic kidney disease
NSAIDs in high doses can effectively treat the pain and inflammation of acute gout. Indomethacin (Indocin) 50 mg three times daily has been standard NSAID therapy.
Other nonselective NSAIDs and NSAIDs that selectively inhibit cyclooxygenase 2 (COX-2) are effective, but all can cause acute renal toxicity or worsen CKD.10 Renal side effects include salt and water retention, acute tubular necrosis, acute interstitial nephritis, proteinuria, hypertension, hyperkalemia, and chronic renal injury.11
Even short-term use of high-dose NSAIDs should generally be avoided in patients with preexisting CKD, for whom there is no established safe threshold dose. When NSAIDs (including selective COX-2 inhibitors) are used, renal function should be monitored closely and the duration limited as much as possible.
Corticosteroids are often used to treat acute attacks
Due to the concerns about NSAIDs or colchicine to treat acute gout attacks in patients with CKD, corticosteroids are often used in this setting.
Intra-articular steroid injections are useful in treating acute gout limited to a single joint or bursa.12 However, one should first make sure that the joint is not infected: septic arthritis should ideally be excluded by arthrocentesis, particularly in immunosuppressed patients13 or those with end-stage renal disease, who are predisposed to bacteremia.
Oral, intramuscular, or intravenous steroids can provide complete relief from acute gout, although high doses (eg, prednisone 30–60 mg/day or the equivalent) are often needed. Common errors resulting in inefficacy include using too low a dose or not treating for a sufficient time before tapering or stopping. Groff and colleagues14 described 13 patients who received oral or intravenous steroids for acute gout. Nine patients received an initial single dose of prednisone ranging from 20 to 50 mg, with tapering over a mean of 10 days. Twelve of the 13 patients had improvement within 48 hours, and the signs and symptoms of acute gout resolved completely within 7 to 10 days.
We often give prednisone 40 mg daily until a day after the acute attack resolves and then taper over another 7 to 10 days. There are no data to guide steroid dosing in an evidence-based way, but we believe too short a course of therapy may result in return of symptoms.
Corticotropin and other agents: Effective but costly
Corticotropin shares the same indications as systemic corticosteroids, being used to treat flares when NSAIDs, intra-articular steroids, and colchicine are contraindicated. However, corticotropin is far more expensive than generic corticosteroids, costing nearly $2,000 for a single 80-IU dose, which may need to be repeated.
Corticotropin is available for subcutaneous or intramuscular injection. A single intramuscular injection of corticotropin gel (H.P. Acthar, 25–80 IU) may terminate an acute gout attack.15 However, many patients need another injection after 24 to 72 hours, which would require another visit to the physician. This treatment has been touted by some as being more effective than corticosteroid therapy, possibly because of a unique peripheral mechanism of action in addition to stimulating cortisol release.16
We rarely use corticotropin, in view of its cost as well as concerns about excessive sodium and water retention due to the release of multiple hormones from the adrenal gland. This may be especially deleterious in patients with CKD or congestive heart failure.17
Parenteral anti-tumor necrosis factor agents or interleukin 1 antagonists can be dramatically effective but are also expensive.18,19 For example, anakinra (Kineret) 100 mg costs about $73, and multiple daily doses may be necessary.
Under unique conditions in which they can be safely used (eg, patients with CKD, diabetes mellitus, liver disease), they may be cost-effective if they can shorten the stay of a hospitalized patient with acute gout.
PROPHYLACTIC ANTI-INFLAMMATORY THERAPY FOR PATIENTS WITH GOUT
Between attacks, the goal is to prevent new attacks through prophylactic management, which may include anti-inflammatory and hypouricemic therapy along with dietary instruction (such as avoiding excessive beer, liquor, and fructose ingestion).
Colchicine can be used as prophylaxis, with caution and monitoring
Although colchicine is not 100% effective, it markedly reduces the flare rate when started in low doses at the time hypouricemic therapy is initiated.20,21 (Hypouricemic therapy is discussed below.) We generally try to continue this prophylactic therapy, if the patient tolerates it, for at least 6 months—longer if tophi are still present or if attacks continue to occur.
If renal function is intact, colchicine can be prescribed at a dosage of 0.6 mg orally once or twice daily.21 In CKD, since the clearance of colchicine is reduced,4 the dosage should be reduced. Patients on colchicine for prophylaxis must be carefully monitored if the glomerular filtration rate is less than 50 mL/minute, or colchicine should be avoided altogether.6 Laboratory testing for colchicine levels is not routinely available and may be of limited value in predicting adverse effects; thus, recommendations about dose adjustments in CKD are empiric.
Wallace et al22 recommended a dose of 0.6 mg once daily if the creatinine clearance is 35 to 49 mL/minute and 0.6 mg every 2 to 3 days if it is 10 to 34 mL/minute, but there are no published long-term safety or efficacy data validating these reasonable (based on available information) dosing regimens.
Even with dose adjustment, caution is needed. Low-dose daily colchicine may be associated with reversible neuromyopathy and bone marrow suppression.7,23 Patients with neuromyopathy may complain of myalgias, proximal muscle weakness, and numbness and may have areflexia and decreased sensation. Laboratory findings include elevated creatine kinase and aminotransferase levels. We regularly check for leukopenia or elevated creatine kinase and aspartate aminotransferase levels in patients with CKD who are receiving colchicine in any dose.
Prolonged colchicine therapy should probably be avoided in patients on hemodialysis, as this drug is not removed by dialysis or by exchange transfusion, and the risk of toxicity under these circumstances may be high.22 When there is no viable alternative and the drug is given, patients should be closely monitored for signs of toxicity.
Concurrent (even short-term) treatment with most macrolide antibiotics, particularly clarithromycin (Biaxin), most statin drugs, ketoconazole (Nizoral), cyclosporine, and likely other drugs predisposes to colchicine toxicity by altering its distribution and elimination, and can in rare cases cause morbidity or death.24–26
NSAIDs are not optimal as prophylaxis in patients with chronic kidney disease
Little information has been published about using NSAIDs chronically to prevent flares, but they are not the optimal drugs to use in patients with CKD, as discussed above. In patients with end-stage renal disease, there are also concerns about NSAID-induced gastric and intestinal bleeding.
Low-dose steroids may not be effective as prophylaxis
Lower doses of steroids may not be effective as prophylaxis against gout flares, consistent with the common observation that gout flares still occur in organ transplant recipients who are taking maintenance doses of prednisone.13
PREVENTING FLARES BY LOWERING SERUM URATE LEVELS
If tophi are present, if radiography shows evidence of damage, if attacks are frequent or disabling, or if there are relative contraindications to the drugs that would be needed to treat acute attacks, then hypouricemic therapy should be strongly considered to reduce the burden of urate in the body, resorb tophi, and ultimately reduce the frequency of gout flares.20
Although intermittent therapy for attacks or prolonged prophylactic use of colchicine may prevent recurrent episodes of gouty arthritis and may be reasonable for many patients, this approach does not prevent continued urate deposition, with the potential development of bony erosions, tophaceous deposits, and chronic arthritis.
The definitive therapy for gouty arthritis is to deplete the periarticular deposits of urate by maintaining a low serum urate level. Urate-lowering therapy, when indicated, is almost always lifelong.
Four strategies for lowering serum urate
The serum urate concentration can be lowered in four ways:
Increasing renal uric acid excretion
Altering the diet
Decreasing urate synthesis
Converting urate to a more soluble metabolite.
Increasing uric acid excretion is rarely effective if renal function is impaired
Probenecid, sulfinpyrazone (Anturane), and losartan (Cozaar) modestly increase uric acid secretion and reduce serum urate levels, but they are rarely effective if the creatinine clearance rate is less than 60 mL/minute, and they require significant fluid intake for maximal efficacy.
Uricosuric drugs probably should be avoided in patients who excrete more than 1,000 mg of uric acid per day on a normal diet, since urinary uric acid stones may form. In practice, however, patients are given losartan to treat hypertension without attention to uric acid excretion.
More-potent urocosuric drugs are being tested in clinical trials.
Altering the diet: Traditional advice confirmed
The Health Professionals Follow-up Study27,28 prospectively examined the relation between diet and gout over 12 years in 47,150 men. The study confirmed some long-standing beliefs, such as that consuming meat, seafood, beer, and liquor increases the risk. Other risk factors were consumption of sugar-sweetened soft drinks and fructose, adiposity, weight gain, hypertension, and diuretic use. On the other hand, protein, wine, and purine-rich vegetables were not associated with gout flares. Low-fat dairy products may have a protective effect. Weight loss was found to be protective.
Low-purine diets are not very palatable, are difficult to adhere to, and are at best only minimally effective, lowering serum urate by 1 to 2 mg/dL. Low-protein diets designed to slow progression of CKD will likely also have only a slight effect on serum urate. Dietary change alone is not likely to dramatically lower serum urate levels.
Metabolizing urate with exogenous uricase
Rasburicase (Elitek) effectively converts urate to allantoin, which is more soluble, but rasburicase is fraught with allergic reactions and cannot be used as chronic therapy.
A pegylated intravenous uricase29 has just been approved by the US Food and Drug Administration (FDA); the retail cost is not yet known. It is dramatically effective in those patients able to use it chronically, but it has not been fully evaluated in patients with CKD.
Decreasing urate synthesis with allopurinol
Allopurinol acts by competitively inhibiting xanthine oxidase, the enzyme that converts hypoxanthine to xanthine and xanthine to uric acid. The drug, a structural analogue of hypoxanthine, is converted by xanthine oxidase to oxypurinol, which is an even more effective inhibitor of xanthine oxidase than allopurinol.
Allopurinol is metabolized in the liver and has a half-life of 1 to 3 hours, but oxypurinol, which is excreted in the urine, has a half-life of 12 to 17 hours. Because of these pharmacokinetic properties, allopurinol can usually be given once daily, and the dosage required to reduce serum urate levels should in theory be lower in patients with lower glomerular filtration rates.
Allopurinol (100- and 300-mg tablets) is approved by the FDA in doses of up to 800 mg/day to treat hyperuricemia in patients with gout,30 while guidelines from the British Society of Rheumatology advocate a maximum dose of 900 mg/day.31 These maximum doses are based on the limited amount of data with higher doses, not on documented toxicity.
Practice survey data in the United States indicate that most physicians prescribe no greater than 300 mg daily, although this dosage is likely to reduce the serum urate to less than 6 mg/dL—the goal level—in fewer than 50% of patients.20,32 Patients with normal renal function occasionally require more than 1,000 mg daily to reduce the serum urate level to less than 6 mg/dL.
How low should the serum urate level be?
Ideally, therapy should keep the serum urate level significantly below 6.7 mg/dL, the approximate saturation point of urate in physiologic fluids.
Lowering the serum urate level from 10 mg/dL to 7 mg/dL may seem encouraging, and the urate level may be in the laboratory “normal” range; however, urate may continue to precipitate in tissues if the concentration is greater than 6.7 mg/dL. A target of 6 mg/dL, used in clinical studies, is far enough below the saturation level to provide some margin for fluctuations in serum levels. A serum level of 6.0 mg/dL has thus been arbitrarily proposed as a reasonable therapeutic target.
The lower the serum urate level achieved during hypouricemic therapy, the faster the reduction in tophaceous deposits. With adequate urate lowering, tophi can be visibly reduced in less than a year of hypouricemic therapy.33,34
We have as yet no convincing evidence that lowering the serum urate level to less than 6.0 mg/dL is harmful, despite theoretical concerns that urate is a beneficial circulating antioxidant and epidemiologic observations that urate levels have been inversely correlated with progression of Parkinson disease.
Start low, go slow to avoid a flare
Rapid reduction of the serum urate level in a patient with chronic hyperuricemia and gout is likely to induce an acute flare.20 We have traditionally used a “start low and increase slowly” approach to escalating hypouricemic therapy in hopes of reducing the likelihood of causing a gout flare.
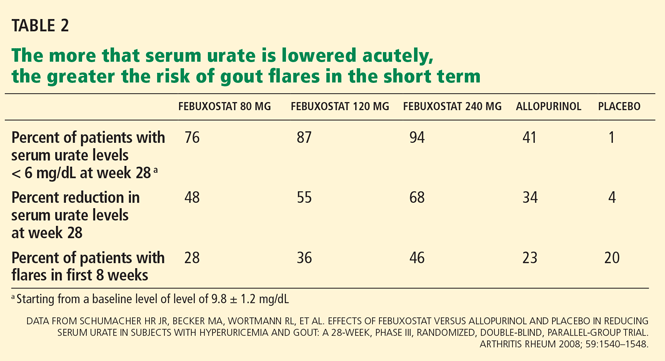
Without anti-inflammatory prophylaxis, acute flares associated with urate-lowering are extremely likely. In a 28-week trial of allopurinol, febuxostat, and placebo by Schumacher et al,33 during the first 8 weeks, when prophylaxis against gout flare was provided with either colchicine 0.6 mg once daily or naproxen (Naprosyn) 250 mg twice daily, the proportion of patients requiring treatment of gout flares was still 23% to 46%. When prophylaxis was stopped, the flare rate increased further.33
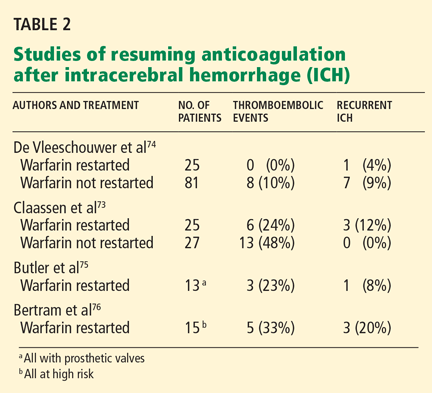
The more we acutely lower serum urate levels, the more likely flares are to occur. In the study by Schumacher et al,33 the percentage of patients needing treatment for gout flares during the first 8 weeks of the study, despite gout flare prophylaxis, was related to the percent reduction in serum urate by week 28 of the trial (Table 2).
IS IT NECESSARY TO ADJUST THE ALLOPURINOL DOSE IN CHRONIC KIDNEY DISEASE?
In 1984, Hande et al35 proposed that allopurinol doses be lower in patients with renal insufficiency, with a dosage scale based on creatinine clearance.
Their thoughtful proposal was based on data from six of their own patients and 72 others with severe allopurinol toxicity, mainly allopurinol hypersensitivity syndrome, reported in the literature.
Perez-Ruiz et al36 noted that patients who had experienced adverse effects from allopurinol in their series were likely to have had received “higher” doses of allopurinol, if the dosage was corrected for reduced oxypurinol elimination based on their estimated creatinine clearance.
However, most of these reactions occurred soon after initiating therapy, a temporal pattern more typical of non-dose-dependent allergic reactions. Additionally, allopurinol hypersensitivity has been linked to T-cell-mediated immune reactions to oxypurinol,37 a mechanism not likely linked to drug levels.
Arguments against dose adjustment
Despite the compelling information that allopurinol reactions are more common in CKD, adjusting the dosage of allopurinol has not been clearly shown to reduce the frequency of these reactions.
In a small retrospective analysis, Vázquez-Mellado et al38 reported that adjusting the allopurinol dosage according to creatinine clearance did not decrease the incidence of allopurinol hypersensitivity.
In a study in 250 patients, Dalbeth et al39 showed that the overall incidence of hypersensitivity reaction was 1.6%, and the incidence of allergic reactions did not decrease when allopurinol was given according to the dosing guidelines proposed by Hande et al.35 However, it is worth noting that, of the patients who received the recommended lower doses, only 19% achieved the target serum urate level of 6 mg/dL.39
Silverberg et al40 found that of 15 patients who developed hypersensitivity reactions to allopurinol, 10 had received doses that were low or appropriate according to the guidelines of Hande et al.35
More recently, Stamp et al41 found that gradually increasing the allopurinol dose above the proposed creatinine clearance-based dose was safe and effective. Thirty-one (89%) of the 35 patients who completed the study achieved the target serum urate level of 6 mg/dL, while only 3 of 45 who started the study developed rashes, which were not serious.
The small number of patients in these studies limits any strong conclusion, but at present there is no interventional study showing that allopurinol dosing adjustment based on glomerular filtration rate is effective or safer than dosing based on the serum urate level.
Our view on allopurinol dosing adjustment
We believe the initial observations of Hande et al35 and the subsequent meticulous data from Perez-Ruiz et al36 suggest a relationship between CKD and the occurrence of severe allopurinol reactions. However, these observations do not prove that dose adjustment will prevent these reactions.
In patients with normal kidney function, the FDA30 and the European League Against Rheumatism (EULAR)42 recommend slow upward titration, starting with 100 to 200 mg/day, which we agree should decrease the frequency of acute gout flares. The dose is increased by increments of 100 mg/day at intervals of 1 week (FDA recommendation) or 2 to 4 weeks (EULAR recommendation) until the serum urate level is lower than 6 mg/dL.
We believe the optimal approach to allopurinol dosing in patients with CKD remains uncertain. We generally escalate the dose slowly, with ongoing frequent laboratory and clinical monitoring, and we do not limit the maximal dose as suggested by Hande et al.35
An alternative strategy is to use the newer, far more expensive xanthine oxidase inhibitor febuxostat in patients with CKD, since it is not excreted by the kidney. We usually first try escalating doses of allopurinol.
FEBUXOSTAT, AN ALTERNATIVE TO ALLOPURINOL
Febuxostat is an oral nonpurine inhibitor of xanthine oxidase.43 Approved by the FDA in 2009, it is available in 40- and 80-mg tablets.
Unlike allopurinol, febuxostat is metabolized primarily by hepatic glucuronide formation and oxidation and then excreted in stool and urine,44 making it in theory an attractive agent in patients with renal insufficiency, bypassing the controversial dose-adjustment issue with allopurinol.
In the Febuxostat Versus Allopurinol Controlled Trial (FACT),20 a 52-week randomized, double-blind study in hyperuricemic patients with gout, serum urate levels were reduced to less than 6.0 mg/dL in over 50% of patients receiving febuxostat 80 mg or 120 mg once daily, while only 21% of patients receiving 300 mg of allopurinol achieved this goal. This does not imply that allopurinol at higher doses, as should be used in clinical practice,45 would not be equally effective. Patients with CKD were not included in this trial.
In the study by Schumacher et al,33 febuxostat 80, 120, or 240 mg once daily reduced serum urate. A small subset (35 patients) had mild to moderate renal insufficiency (serum creatinine 1.5–2 mg/dL).33 The number of patients with renal insufficiency who achieved the primary end point of a serum urate level lower than 6 mg/dL was 4 (44%) of 9 in the febuxostat 80-mg group, 5 (46%) of 11 in the 120-mg group, and 3 (60%) of 5 in the 240-mg group, while none of the 10 patients in the dose-adjusted allopurinol group achieved the primary end point (P < .05). Of note, 41% of the patients with normal renal function who received allopurinol achieved the primary end point.33 As proposed above, if the allopurinol dose had been slowly increased in the patients with renal insufficiency, it might have been equally effective.
Febuxostat has not been thoroughly evaluated in patients with severe CKD or in patients on hemodialysis.
A presumed niche indication of febuxostat is in patients allergic to allopurinol, since the drugs are not similar in chemical structure. However, at present, experience with this use is limited. Allopurinol-allergic patients were excluded from the clinical trials; thus, if there is any allergic overlap, it would not likely have been recognized in those studies. The FDA has received reports of patients who were allergic to allopurinol also having reactions to febuxostat, and it is currently evaluating these reports (personal communication).
Concern was raised over cardiovascular adverse events in patients treated with febuxostat during clinical trials. In the FACT trial, two patients died of cardiac causes.20 In the study by Schumacher et al,33 11 of 670 patients experienced cardiac adverse events in the febuxostat group vs 3 of 268 in the allopurinol group. Events included atrial fibrillation, chest pain, coronary artery disease, and myocardial infarction. However, this difference was not statistically significant.
Febuxostat costs much more than allopurinol. Currently, patients pay $153.88 for 1 month of febuxostat 40 or 80 mg from Cleveland Clinic pharmacy; 1 month of allopurinol costs $17.45 (300 mg) or $14.00 (100 mg). We believe febuxostat should be reserved for patients with documented intolerance to allopurinol in effective doses.
Monitoring serum urate levels is important in all patients on hypouricemic therapy so that dosage adjustments can be made until the target serum urate concentration is reached. In patients failing to meet target serum urate levels, patient adherence with the prescribed dosing should be specifically addressed because as many as 50% of patients do not adhere to their prescribed regimen.
DOES URATE-LOWERING THERAPY HAVE BENEFITS BEYOND GOUT?
Despite experimental animal data and a strong epidemiologic association between hyperuri-cemia and hypertension,46 metabolic syndrome, and rates of cardiovascular and all-cause mortality,47 the evidence from interventional trials so far does not support the routine use of hypo-uricemic therapy to prevent these outcomes.
Similarly, hyperuricemia has long been associated with renal disease, and there has been debate as to whether hyperuricemia is a result of kidney dysfunction or a contributing factor.46,48–51 A few studies have documented improvement of renal function after initiation of hypouricemic therapy.52 However, treating asymptomatic hyperuricemia to preserve kidney function remains controversial.
A recent study indicates that lowering the serum urate level with allopurinol can lower the blood pressure in hyperuricemic adolescents who have newly diagnosed primary hypertension.53 This does not indicate, however, that initiating hypouricemic therapy in patients with preexisting, long-standing hypertension will be successful.
RECOMMENDED FOR OUR PATIENT
As for our diabetic patient with an acute gout flare and creatinine clearance rate of 45 mL/minute, we would recommend:
Aspirating the knee, sending the fluid for bacterial culture, and then treating it with a local glucocorticoid injection
Starting colchicine 0.6 mg every day, with frequent monitoring for signs of toxicity (muscle pain, weakness, leukopenia, and elevations of creatine kinase and aspartate aminotransferase)
Increasing his allopurinol dose by 100 mg every 2 to 4 weeks until the target serum urate level of less than 6.0 mg/dL is reached
If he cannot tolerate allopurinol or if the target serum urate level is not achieved despite adequate doses of allopurinol (about 800 mg), we would switch to febuxostat 40 mg and increase the dose as needed to achieve the desired urate level.
References
Vázquez-Mellado J, García CG, Vázquez SG, et al. Metabolic syndrome and ischemic heart disease in gout. J Clin Rheumatol2004; 10:105–109.
Bonnel RA, Villalba ML, Karwoski CB, Beitz J. Deaths associated with inappropriate intravenous colchicine administration. J Emerg Med2002; 22:385–387.
Achtert G, Scherrmann JM, Christen MO. Pharmacokinetics/bioavailability of colchicine in healthy male volunteers. Eur J Drug Metab Pharmacokinet1989; 14:317–322.
Ben-Chetrit E, Scherrmann JM, Zylber-Katz E, Levy M. Colchicine disposition in patients with familial Mediterranean fever with renal impairment. J Rheumatol1994; 21:710–713.
Putterman C, Ben-Chetrit E, Caraco Y, Levy M. Colchicine intoxication: clinical pharmacology, risk factors, features, and management. Semin Arthritis Rheum1991; 21:143–155.
Aronoff G, Brater DC, Schrier R, Bennett WM. Use of drugs in patients with renal insufficiency. Workshop report. Blood Purif1994; 12:14–19.
Kuncl RW, Duncan G, Watson D, Alderson K, Rogawski MA, Peper M. Colchicine myopathy and neuropathy. N Engl J Med1987; 316:1562–1568.
Ahern MJ, Reid C, Gordon TP, McCredie M, Brooks PM, Jones M. Does colchicine work? The results of the first controlled study in acute gout. Aust N Z J Med1987; 17:301–304.
Terkeltaub R, Furst D, Bennett K, Kook K, Crockett RS, Davis WM. High versus low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum2010, 62:1060–1068.
Whelton A. Nephrotoxicity of nonsteroidal anti-inflammatory drugs: physiologic foundations and clinical implications. Am J Med1999; 106:13S–24S.
Wali RK, Henrich WL. Recent developments in toxic nephropathy. Curr Opin Nephrol Hypertens2002; 11:155–163.
Fernández C, Noguera R, González JA, Pascual E. Treatment of acute attacks of gout with a small dose of intraarticular triamcinolone acetonide. J Rheumatol1999; 26:2285–2286.
Clive DM. Renal transplant-associated hyperuricemia and gout. J Am Soc Nephrol2000; 11:974–979.
Groff GD, Franck WA, Raddatz DA. Systemic steroid therapy for acute gout: a clinical trial and review of the literature. Semin Arthritis Rheum1990; 19:329–336.
Ritter J, Kerr LD, Valeriano-Marcet J, Spiera H. ACTH revisited: effective treatment for acute crystal induced synovitis in patients with multiple medical problems. J Rheumatol1994; 21:696–699.
Getting SJ, Christian HC, Flower RJ, Perretti M. Activation of melanocortin type 3 receptor as a molecular mechanism for adrenocorticotropic hormone efficacy in gouty arthritis. Arthritis Rheum2002; 46:2765–2775.
Connell JM, Whitworth JA, Davies DL, Lever AF, Richards AM, Fraser R. Effects of ACTH and cortisol administration on blood pressure, electrolyte metabolism, atrial natriuretic peptide and renal function in normal man. J Hypertens1987; 5:425–433.
Tausche AK, Richter K, Grässler A, Hänsel S, Roch B, Schröder HE. Severe gouty arthritis refractory to anti-inflammatory drugs: treatment with anti-tumour necrosis factor alpha as a new therapeutic option. Ann Rheum Dis2004; 63:1351–1352.
So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther2007; 9:R28.
Becker MA, Schumacher HR, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med2005; 353:2450–2461.
Borstad GC, Bryant LR, Abel MP, Scroggie DA, Harris MD, Alloway JA. Colchicine for prophylaxis of acute flares when initiating allopurinol for chronic gouty arthritis. J Rheumatol2004; 31:2429–2432.
Wallace SL, Singer JZ, Duncan GJ, Wigley FM, Kuncl RW. Renal function predicts colchicine toxicity: guidelines for the prophylactic use of colchicine in gout. J Rheumatol1991; 18:264–269.
Wilbur K, Makowsky M. Colchicine myotoxicity: case reports and literature review. Pharmacotherapy2004; 24:1784–1792.
Hung IF, Wu AK, Cheng VC, et al. Fatal interaction between clarithromycin and colchicine in patients with renal insufficiency: a retrospective study. Clin Infect Dis2005; 41:291–300.
Alayli G, Cengiz K, Cantürk F, Durmus D, Akyol Y, Menekse EB. Acute myopathy in a patient with concomitant use of pravastatin and colchicine. Ann Pharmacother2005; 39:1358–1361.
Ducloux D, Schuller V, Bresson-Vautrin C, Chalopin JM. Colchicine myopathy in renal transplant recipients on cyclosporin. Nephrol Dial Transplant1997; 12:2389–2392.
Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med2004; 350:1093–1103.
Choi HK, Curhan G. Soft drinks, fructose consumption, and the risk of gout in men: prospective cohort study. BMJ2008; 336:309–312.
Sundy JS, Becker MA, Baraf HS, et al; Pegloticase Phase 2 Study Investigators. Reduction of plasma urate levels following treatment with multiple doses of pegloticase (polyethylene glycol-conjugated uricase) in patients with treatment-failure gout: results of a phase II randomized study. Arthritis Rheum2008; 58:2882–2891.
Jordan KM, Cameron JS, Snaith M, et al; British Society for Rheumatology and British Health Professionals in Rheumatology Standards, Guidelines and Audit Working Group (SGAWG). British Society for Rheumatology and British Health Professionals in Rheumatology guideline for the management of gout. Rheumatology (Oxford)2007; 46:1372–1374.
Perez-Ruiz F, Alonso-Ruiz A, Calabozo M, Herrero-Beites A, García-Erauskin G, Ruiz-Lucea E. Efficacy of allopurinol and benzbromarone for the control of hyperuricaemia. A pathogenic approach to the treatment of primary chronic gout. Ann Rheum Dis1998; 57:545–549.
Schumacher HR, Becker MA, Wortmann RL, et al. Effects of febuxostat versus allopurinol and placebo in reducing serum urate in subjects with hyperuricemia and gout: a 28-week, phase III, randomized, double-blind, parallel-group trial. Arthritis Rheum2008; 59:1540–1548.
Perez-Ruiz F, Calabozo M, Pijoan JI, Herrero-Beites AM, Ruibal A. Effect of urate-lowering therapy on the velocity of size reduction of tophi in chronic gout. Arthritis Rheum2002; 47:356–360.
Hande KR, Noone RM, Stone WJ. Severe allopurinol toxicity. Description and guidelines for prevention in patients with renal insufficiency. Am J Med1984; 76:47–56.
Perez-Ruiz F, Hernando I, Villar I, Nolla JM. Correction of allopurinol dosing should be based on clearance of creatinine, but not plasma creatinine levels: another insight to allopurinol-related toxicity. J Clin Rheumatol2005; 11:129–133.
Hung SI, Chung WH, Liou LB, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A2005; 102:4134–4139.
Vázquez-Mellado J, Morales EM, Pacheco-Tena C, Burgos-Vargas R. Relation between adverse events associated with allopurinol and renal function in patients with gout. Ann Rheum Dis2001; 60:981–983.
Dalbeth N, Kumar S, Stamp L, Gow P. Dose adjustment of allopurinol according to creatinine clearance does not provide adequate control of hyperuricemia in patients with gout. J Rheumatol2006; 33:1646–1650.
Silverberg MS, Mallela R, Lesse AJ, Bonner MR, Baer AN, Li C. Allopurinol hypersensitivity reactions: a case-control study of the role of renal dosing (abstract). Arthritis Rheum2009; 60(suppl 10):1106.
Stamp LK, O'Donnell JL, Zhang M, et al. Using allopurinol above the dose based on creatinine clearance is effective and safe in chronic gout, including in those with renal impairment. Arthritis Rhem2010; doi:10.1002/art.30119. E-pub ahead of print. Accessed 10/29/2010.
Zhang W, Doherty M, Bardin T, et al; EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis2006; 65:1312–1324.
Okamoto K, Eger BT, Nishino T, Kondo S, Pai EF, Nishino T. An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J Biol Chem2003; 278:1848–1855.
Khosravan R, Grabowski BA, Wu JT, Joseph-Ridge N, Vernillet L. Pharmacokinetics, pharmacodynamics and safety of febuxostat, a non-purine selective inhibitor of xanthine oxidase, in a dose escalation study in healthy subjects. Clin Pharmacokinet2006; 45:821–841.
Reinders MK, Haagsma C, Jansen TL, et al. A randomised controlled trial on the efficacy and tolerability with dose escalation of allopurinol 300–600 mg/day versus benzbromarone 100–200 mg/day in patients with gout. Ann Rheum Dis2009; 68:892–897.
Mazzali M, Hughes J, Kim YG, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension2001; 38:1101–1106.
Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med2008; 359:1811–1821.
Beck LH. Requiem for gouty nephropathy. Kidney Int1986; 30:280–287.
Tomita M, Mizuno S, Yamanaka H, et al. Does hyperuricemia affect mortality? A prospective cohort study of Japanese male workers. J Epidemiol2000; 10:403–409.
Kang DH, Nakagawa T. Uric acid and chronic renal disease: possible implication of hyperuricemia on progression of renal disease. Semin Nephrol2005; 25:43–49.
Iseki K, Oshiro S, Tozawa M, Iseki C, Ikemiya Y, Takishita S. Significance of hyperuricemia on the early detection of renal failure in a cohort of screened subjects. Hypertens Res2001; 24:691–697.
Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med1987; 82:421–426.
Siu YP, Leung KT, Tong MK, Kwan TH. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis2006; 47:51–59.
Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA2008; 300:924–932.
Hossam El-Zawawy, MD, MS Assistant Professor, Charles E. Schmidt College of Medicine, Florida Atlantic University, Boca Raton; Associate Staff, Department of Rheumatic and Immunologic Diseases, Cleveland Clinic, Weston, FL
Brian F. Mandell, MD, PhD Chairman, Department of Medicine, Education Institute, Cleveland Clinic; Professor of Medicine, Lerner College of Medicine of Case Western Reserve University, Cleveland, OH; Department of Rheumatic and Immunologic Diseases, Cleveland Clinic; and Editor-in-Chief, Cleveland Clinic Journal of Medicine
Address: Brian F. Mandell, MD, PhD, Education Institute, NA10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
Dr. Mandell has disclosed that he has been a consultant for Takeda Pharmaceutical Company and has conducted a clinical trial for Savient Pharmaceuticals.
Hossam El-Zawawy, MD, MS Assistant Professor, Charles E. Schmidt College of Medicine, Florida Atlantic University, Boca Raton; Associate Staff, Department of Rheumatic and Immunologic Diseases, Cleveland Clinic, Weston, FL
Brian F. Mandell, MD, PhD Chairman, Department of Medicine, Education Institute, Cleveland Clinic; Professor of Medicine, Lerner College of Medicine of Case Western Reserve University, Cleveland, OH; Department of Rheumatic and Immunologic Diseases, Cleveland Clinic; and Editor-in-Chief, Cleveland Clinic Journal of Medicine
Address: Brian F. Mandell, MD, PhD, Education Institute, NA10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
Dr. Mandell has disclosed that he has been a consultant for Takeda Pharmaceutical Company and has conducted a clinical trial for Savient Pharmaceuticals.
Author and Disclosure Information
Hossam El-Zawawy, MD, MS Assistant Professor, Charles E. Schmidt College of Medicine, Florida Atlantic University, Boca Raton; Associate Staff, Department of Rheumatic and Immunologic Diseases, Cleveland Clinic, Weston, FL
Brian F. Mandell, MD, PhD Chairman, Department of Medicine, Education Institute, Cleveland Clinic; Professor of Medicine, Lerner College of Medicine of Case Western Reserve University, Cleveland, OH; Department of Rheumatic and Immunologic Diseases, Cleveland Clinic; and Editor-in-Chief, Cleveland Clinic Journal of Medicine
Address: Brian F. Mandell, MD, PhD, Education Institute, NA10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
Dr. Mandell has disclosed that he has been a consultant for Takeda Pharmaceutical Company and has conducted a clinical trial for Savient Pharmaceuticals.
You have a 54-year-old black patient with gout, diabetes mellitus, hypertension, and chronic kidney disease (CKD). He has an acute gout flare involving his right knee. In the past year he has had four attacks of gout in the ankles and knees, which you treated with intra-articular glucocorticoid injections. He has been on allopurinol (Zyloprim) 200 mg daily, but his last serum urate level was 9.4 mg/dL (reference range 3.0–8.0). His creatinine clearance is 45 mL/minute (reference range 85–125).
In view of his kidney disease, you are concerned about increasing his dose of allopurinol, but also about the need to treat his frequent attacks. How should you manage this patient?
GOUT IS CHALLENGING TO TREAT IN PATIENTS WITH KIDNEY DISEASE
A major challenge in treating patients with gout is to avoid therapeutic interactions with common comorbidities, including hypertension, insulin resistance, coronary artery disease, heart failure, and especially CKD.1
In this paper, we discuss approaches to and controversies in the management of gout and hyperuricemia in patients with CKD. Unfortunately, the evidence from clinical trials to guide treatment decisions is limited; therefore, decisions must often be based on experience and pathophysiologic principles.
GENERAL GOALS OF GOUT THERAPY
Depending on the patient and the stage of the disease, the goals in treating patients with gout are to:
Terminate acute attacks as promptly and safely as possible
Prevent recurrences of acute gout attacks
Prevent or reverse complications resulting from deposition of monosodium urate in the joints, in the kidneys, or at other sites.
These goals are more difficult to achieve in patients with CKD because of the potential complications from many of the available drugs.
TERMINATING ACUTE GOUT FLARES
In patients with acute gout, treatment is aimed at quickly resolving pain and inflammation.
Several types of drugs can terminate acute gout flares. The choice in most situations is colchicine (Colcrys); a nonsteroidal anti-inflammatory drug (NSAID); a corticosteroid; or corticotropin (ACTH).
However, in patients with CKD, there are concerns about using colchicine or NSAIDs, and corticotropin is very expensive; thus, corticosteroids are often used.
Colchicine’s clearance is reduced in CKD
Colchicine is somewhat effective in treating acute gout attacks and probably more effective in preventing attacks.
Due to concerns about inappropriate dosing and reported deaths,2 the intravenous formulation is not available in many countries, including the United States.
After oral administration, colchicine is rapidly absorbed, with a bioavailability of up to 50%. It undergoes metabolism by the liver, and its metabolites are excreted by renal and biliary-intestinal routes. Up to 20% of the active drug is excreted by the kidneys.3
Colchicine’s clearance is significantly reduced in patients with renal or hepatic insufficiency, and the drug may accumulate in cells, with resultant toxicity.4 Colchicine-induced toxicity has been observed when the drug was used for acute treatment, as well as for chronic prophylaxis of gout in patients with CKD; thus, alternative agents for treating acute attacks should be considered.5,6 With prolonged use, reversible colchicine-induced axonal neuropathy, neutropenia, and vacuolar myopathy can develop in patients with CKD.7
In a trial in patients with normal renal function, nearly 100% who received an initial dose of 1 mg followed by 0.5 mg every 2 hours developed diarrhea at a median time of 24 hours.8 Emesis may also occur.
A lower dose of 1.8 mg (two 0.6-mg pills followed by one pill an hour later) was well tolerated but only moderately effective in treating acute gout, causing at least a 50% reduction in pain at 24 hours in only 38% of patients.9 This study does not clarify the dosage to use to completely resolve attacks. Using additional colchicine likely will increase the response rate, but will also increase side effects. Patients with CKD were not included.
Some patients, as shown in the above trial, can abort attacks by taking only one or two colchicine tablets when they feel the first “twinge” of an attack. This approach is likely to be safe in CKD, but it may be of value to only a few patients.
Nonsteroidal anti-inflammatory drugs can worsen chronic kidney disease
NSAIDs in high doses can effectively treat the pain and inflammation of acute gout. Indomethacin (Indocin) 50 mg three times daily has been standard NSAID therapy.
Other nonselective NSAIDs and NSAIDs that selectively inhibit cyclooxygenase 2 (COX-2) are effective, but all can cause acute renal toxicity or worsen CKD.10 Renal side effects include salt and water retention, acute tubular necrosis, acute interstitial nephritis, proteinuria, hypertension, hyperkalemia, and chronic renal injury.11
Even short-term use of high-dose NSAIDs should generally be avoided in patients with preexisting CKD, for whom there is no established safe threshold dose. When NSAIDs (including selective COX-2 inhibitors) are used, renal function should be monitored closely and the duration limited as much as possible.
Corticosteroids are often used to treat acute attacks
Due to the concerns about NSAIDs or colchicine to treat acute gout attacks in patients with CKD, corticosteroids are often used in this setting.
Intra-articular steroid injections are useful in treating acute gout limited to a single joint or bursa.12 However, one should first make sure that the joint is not infected: septic arthritis should ideally be excluded by arthrocentesis, particularly in immunosuppressed patients13 or those with end-stage renal disease, who are predisposed to bacteremia.
Oral, intramuscular, or intravenous steroids can provide complete relief from acute gout, although high doses (eg, prednisone 30–60 mg/day or the equivalent) are often needed. Common errors resulting in inefficacy include using too low a dose or not treating for a sufficient time before tapering or stopping. Groff and colleagues14 described 13 patients who received oral or intravenous steroids for acute gout. Nine patients received an initial single dose of prednisone ranging from 20 to 50 mg, with tapering over a mean of 10 days. Twelve of the 13 patients had improvement within 48 hours, and the signs and symptoms of acute gout resolved completely within 7 to 10 days.
We often give prednisone 40 mg daily until a day after the acute attack resolves and then taper over another 7 to 10 days. There are no data to guide steroid dosing in an evidence-based way, but we believe too short a course of therapy may result in return of symptoms.
Corticotropin and other agents: Effective but costly
Corticotropin shares the same indications as systemic corticosteroids, being used to treat flares when NSAIDs, intra-articular steroids, and colchicine are contraindicated. However, corticotropin is far more expensive than generic corticosteroids, costing nearly $2,000 for a single 80-IU dose, which may need to be repeated.
Corticotropin is available for subcutaneous or intramuscular injection. A single intramuscular injection of corticotropin gel (H.P. Acthar, 25–80 IU) may terminate an acute gout attack.15 However, many patients need another injection after 24 to 72 hours, which would require another visit to the physician. This treatment has been touted by some as being more effective than corticosteroid therapy, possibly because of a unique peripheral mechanism of action in addition to stimulating cortisol release.16
We rarely use corticotropin, in view of its cost as well as concerns about excessive sodium and water retention due to the release of multiple hormones from the adrenal gland. This may be especially deleterious in patients with CKD or congestive heart failure.17
Parenteral anti-tumor necrosis factor agents or interleukin 1 antagonists can be dramatically effective but are also expensive.18,19 For example, anakinra (Kineret) 100 mg costs about $73, and multiple daily doses may be necessary.
Under unique conditions in which they can be safely used (eg, patients with CKD, diabetes mellitus, liver disease), they may be cost-effective if they can shorten the stay of a hospitalized patient with acute gout.
PROPHYLACTIC ANTI-INFLAMMATORY THERAPY FOR PATIENTS WITH GOUT
Between attacks, the goal is to prevent new attacks through prophylactic management, which may include anti-inflammatory and hypouricemic therapy along with dietary instruction (such as avoiding excessive beer, liquor, and fructose ingestion).
Colchicine can be used as prophylaxis, with caution and monitoring
Although colchicine is not 100% effective, it markedly reduces the flare rate when started in low doses at the time hypouricemic therapy is initiated.20,21 (Hypouricemic therapy is discussed below.) We generally try to continue this prophylactic therapy, if the patient tolerates it, for at least 6 months—longer if tophi are still present or if attacks continue to occur.
If renal function is intact, colchicine can be prescribed at a dosage of 0.6 mg orally once or twice daily.21 In CKD, since the clearance of colchicine is reduced,4 the dosage should be reduced. Patients on colchicine for prophylaxis must be carefully monitored if the glomerular filtration rate is less than 50 mL/minute, or colchicine should be avoided altogether.6 Laboratory testing for colchicine levels is not routinely available and may be of limited value in predicting adverse effects; thus, recommendations about dose adjustments in CKD are empiric.
Wallace et al22 recommended a dose of 0.6 mg once daily if the creatinine clearance is 35 to 49 mL/minute and 0.6 mg every 2 to 3 days if it is 10 to 34 mL/minute, but there are no published long-term safety or efficacy data validating these reasonable (based on available information) dosing regimens.
Even with dose adjustment, caution is needed. Low-dose daily colchicine may be associated with reversible neuromyopathy and bone marrow suppression.7,23 Patients with neuromyopathy may complain of myalgias, proximal muscle weakness, and numbness and may have areflexia and decreased sensation. Laboratory findings include elevated creatine kinase and aminotransferase levels. We regularly check for leukopenia or elevated creatine kinase and aspartate aminotransferase levels in patients with CKD who are receiving colchicine in any dose.
Prolonged colchicine therapy should probably be avoided in patients on hemodialysis, as this drug is not removed by dialysis or by exchange transfusion, and the risk of toxicity under these circumstances may be high.22 When there is no viable alternative and the drug is given, patients should be closely monitored for signs of toxicity.
Concurrent (even short-term) treatment with most macrolide antibiotics, particularly clarithromycin (Biaxin), most statin drugs, ketoconazole (Nizoral), cyclosporine, and likely other drugs predisposes to colchicine toxicity by altering its distribution and elimination, and can in rare cases cause morbidity or death.24–26
NSAIDs are not optimal as prophylaxis in patients with chronic kidney disease
Little information has been published about using NSAIDs chronically to prevent flares, but they are not the optimal drugs to use in patients with CKD, as discussed above. In patients with end-stage renal disease, there are also concerns about NSAID-induced gastric and intestinal bleeding.
Low-dose steroids may not be effective as prophylaxis
Lower doses of steroids may not be effective as prophylaxis against gout flares, consistent with the common observation that gout flares still occur in organ transplant recipients who are taking maintenance doses of prednisone.13
PREVENTING FLARES BY LOWERING SERUM URATE LEVELS
If tophi are present, if radiography shows evidence of damage, if attacks are frequent or disabling, or if there are relative contraindications to the drugs that would be needed to treat acute attacks, then hypouricemic therapy should be strongly considered to reduce the burden of urate in the body, resorb tophi, and ultimately reduce the frequency of gout flares.20
Although intermittent therapy for attacks or prolonged prophylactic use of colchicine may prevent recurrent episodes of gouty arthritis and may be reasonable for many patients, this approach does not prevent continued urate deposition, with the potential development of bony erosions, tophaceous deposits, and chronic arthritis.
The definitive therapy for gouty arthritis is to deplete the periarticular deposits of urate by maintaining a low serum urate level. Urate-lowering therapy, when indicated, is almost always lifelong.
Four strategies for lowering serum urate
The serum urate concentration can be lowered in four ways:
Increasing renal uric acid excretion
Altering the diet
Decreasing urate synthesis
Converting urate to a more soluble metabolite.
Increasing uric acid excretion is rarely effective if renal function is impaired
Probenecid, sulfinpyrazone (Anturane), and losartan (Cozaar) modestly increase uric acid secretion and reduce serum urate levels, but they are rarely effective if the creatinine clearance rate is less than 60 mL/minute, and they require significant fluid intake for maximal efficacy.
Uricosuric drugs probably should be avoided in patients who excrete more than 1,000 mg of uric acid per day on a normal diet, since urinary uric acid stones may form. In practice, however, patients are given losartan to treat hypertension without attention to uric acid excretion.
More-potent urocosuric drugs are being tested in clinical trials.
Altering the diet: Traditional advice confirmed
The Health Professionals Follow-up Study27,28 prospectively examined the relation between diet and gout over 12 years in 47,150 men. The study confirmed some long-standing beliefs, such as that consuming meat, seafood, beer, and liquor increases the risk. Other risk factors were consumption of sugar-sweetened soft drinks and fructose, adiposity, weight gain, hypertension, and diuretic use. On the other hand, protein, wine, and purine-rich vegetables were not associated with gout flares. Low-fat dairy products may have a protective effect. Weight loss was found to be protective.
Low-purine diets are not very palatable, are difficult to adhere to, and are at best only minimally effective, lowering serum urate by 1 to 2 mg/dL. Low-protein diets designed to slow progression of CKD will likely also have only a slight effect on serum urate. Dietary change alone is not likely to dramatically lower serum urate levels.
Metabolizing urate with exogenous uricase
Rasburicase (Elitek) effectively converts urate to allantoin, which is more soluble, but rasburicase is fraught with allergic reactions and cannot be used as chronic therapy.
A pegylated intravenous uricase29 has just been approved by the US Food and Drug Administration (FDA); the retail cost is not yet known. It is dramatically effective in those patients able to use it chronically, but it has not been fully evaluated in patients with CKD.
Decreasing urate synthesis with allopurinol
Allopurinol acts by competitively inhibiting xanthine oxidase, the enzyme that converts hypoxanthine to xanthine and xanthine to uric acid. The drug, a structural analogue of hypoxanthine, is converted by xanthine oxidase to oxypurinol, which is an even more effective inhibitor of xanthine oxidase than allopurinol.
Allopurinol is metabolized in the liver and has a half-life of 1 to 3 hours, but oxypurinol, which is excreted in the urine, has a half-life of 12 to 17 hours. Because of these pharmacokinetic properties, allopurinol can usually be given once daily, and the dosage required to reduce serum urate levels should in theory be lower in patients with lower glomerular filtration rates.
Allopurinol (100- and 300-mg tablets) is approved by the FDA in doses of up to 800 mg/day to treat hyperuricemia in patients with gout,30 while guidelines from the British Society of Rheumatology advocate a maximum dose of 900 mg/day.31 These maximum doses are based on the limited amount of data with higher doses, not on documented toxicity.
Practice survey data in the United States indicate that most physicians prescribe no greater than 300 mg daily, although this dosage is likely to reduce the serum urate to less than 6 mg/dL—the goal level—in fewer than 50% of patients.20,32 Patients with normal renal function occasionally require more than 1,000 mg daily to reduce the serum urate level to less than 6 mg/dL.
How low should the serum urate level be?
Ideally, therapy should keep the serum urate level significantly below 6.7 mg/dL, the approximate saturation point of urate in physiologic fluids.
Lowering the serum urate level from 10 mg/dL to 7 mg/dL may seem encouraging, and the urate level may be in the laboratory “normal” range; however, urate may continue to precipitate in tissues if the concentration is greater than 6.7 mg/dL. A target of 6 mg/dL, used in clinical studies, is far enough below the saturation level to provide some margin for fluctuations in serum levels. A serum level of 6.0 mg/dL has thus been arbitrarily proposed as a reasonable therapeutic target.
The lower the serum urate level achieved during hypouricemic therapy, the faster the reduction in tophaceous deposits. With adequate urate lowering, tophi can be visibly reduced in less than a year of hypouricemic therapy.33,34
We have as yet no convincing evidence that lowering the serum urate level to less than 6.0 mg/dL is harmful, despite theoretical concerns that urate is a beneficial circulating antioxidant and epidemiologic observations that urate levels have been inversely correlated with progression of Parkinson disease.
Start low, go slow to avoid a flare
Rapid reduction of the serum urate level in a patient with chronic hyperuricemia and gout is likely to induce an acute flare.20 We have traditionally used a “start low and increase slowly” approach to escalating hypouricemic therapy in hopes of reducing the likelihood of causing a gout flare.
Without anti-inflammatory prophylaxis, acute flares associated with urate-lowering are extremely likely. In a 28-week trial of allopurinol, febuxostat, and placebo by Schumacher et al,33 during the first 8 weeks, when prophylaxis against gout flare was provided with either colchicine 0.6 mg once daily or naproxen (Naprosyn) 250 mg twice daily, the proportion of patients requiring treatment of gout flares was still 23% to 46%. When prophylaxis was stopped, the flare rate increased further.33
The more we acutely lower serum urate levels, the more likely flares are to occur. In the study by Schumacher et al,33 the percentage of patients needing treatment for gout flares during the first 8 weeks of the study, despite gout flare prophylaxis, was related to the percent reduction in serum urate by week 28 of the trial (Table 2).
IS IT NECESSARY TO ADJUST THE ALLOPURINOL DOSE IN CHRONIC KIDNEY DISEASE?
In 1984, Hande et al35 proposed that allopurinol doses be lower in patients with renal insufficiency, with a dosage scale based on creatinine clearance.
Their thoughtful proposal was based on data from six of their own patients and 72 others with severe allopurinol toxicity, mainly allopurinol hypersensitivity syndrome, reported in the literature.
Perez-Ruiz et al36 noted that patients who had experienced adverse effects from allopurinol in their series were likely to have had received “higher” doses of allopurinol, if the dosage was corrected for reduced oxypurinol elimination based on their estimated creatinine clearance.
However, most of these reactions occurred soon after initiating therapy, a temporal pattern more typical of non-dose-dependent allergic reactions. Additionally, allopurinol hypersensitivity has been linked to T-cell-mediated immune reactions to oxypurinol,37 a mechanism not likely linked to drug levels.
Arguments against dose adjustment
Despite the compelling information that allopurinol reactions are more common in CKD, adjusting the dosage of allopurinol has not been clearly shown to reduce the frequency of these reactions.
In a small retrospective analysis, Vázquez-Mellado et al38 reported that adjusting the allopurinol dosage according to creatinine clearance did not decrease the incidence of allopurinol hypersensitivity.
In a study in 250 patients, Dalbeth et al39 showed that the overall incidence of hypersensitivity reaction was 1.6%, and the incidence of allergic reactions did not decrease when allopurinol was given according to the dosing guidelines proposed by Hande et al.35 However, it is worth noting that, of the patients who received the recommended lower doses, only 19% achieved the target serum urate level of 6 mg/dL.39
Silverberg et al40 found that of 15 patients who developed hypersensitivity reactions to allopurinol, 10 had received doses that were low or appropriate according to the guidelines of Hande et al.35
More recently, Stamp et al41 found that gradually increasing the allopurinol dose above the proposed creatinine clearance-based dose was safe and effective. Thirty-one (89%) of the 35 patients who completed the study achieved the target serum urate level of 6 mg/dL, while only 3 of 45 who started the study developed rashes, which were not serious.
The small number of patients in these studies limits any strong conclusion, but at present there is no interventional study showing that allopurinol dosing adjustment based on glomerular filtration rate is effective or safer than dosing based on the serum urate level.
Our view on allopurinol dosing adjustment
We believe the initial observations of Hande et al35 and the subsequent meticulous data from Perez-Ruiz et al36 suggest a relationship between CKD and the occurrence of severe allopurinol reactions. However, these observations do not prove that dose adjustment will prevent these reactions.
In patients with normal kidney function, the FDA30 and the European League Against Rheumatism (EULAR)42 recommend slow upward titration, starting with 100 to 200 mg/day, which we agree should decrease the frequency of acute gout flares. The dose is increased by increments of 100 mg/day at intervals of 1 week (FDA recommendation) or 2 to 4 weeks (EULAR recommendation) until the serum urate level is lower than 6 mg/dL.
We believe the optimal approach to allopurinol dosing in patients with CKD remains uncertain. We generally escalate the dose slowly, with ongoing frequent laboratory and clinical monitoring, and we do not limit the maximal dose as suggested by Hande et al.35
An alternative strategy is to use the newer, far more expensive xanthine oxidase inhibitor febuxostat in patients with CKD, since it is not excreted by the kidney. We usually first try escalating doses of allopurinol.
FEBUXOSTAT, AN ALTERNATIVE TO ALLOPURINOL
Febuxostat is an oral nonpurine inhibitor of xanthine oxidase.43 Approved by the FDA in 2009, it is available in 40- and 80-mg tablets.
Unlike allopurinol, febuxostat is metabolized primarily by hepatic glucuronide formation and oxidation and then excreted in stool and urine,44 making it in theory an attractive agent in patients with renal insufficiency, bypassing the controversial dose-adjustment issue with allopurinol.
In the Febuxostat Versus Allopurinol Controlled Trial (FACT),20 a 52-week randomized, double-blind study in hyperuricemic patients with gout, serum urate levels were reduced to less than 6.0 mg/dL in over 50% of patients receiving febuxostat 80 mg or 120 mg once daily, while only 21% of patients receiving 300 mg of allopurinol achieved this goal. This does not imply that allopurinol at higher doses, as should be used in clinical practice,45 would not be equally effective. Patients with CKD were not included in this trial.
In the study by Schumacher et al,33 febuxostat 80, 120, or 240 mg once daily reduced serum urate. A small subset (35 patients) had mild to moderate renal insufficiency (serum creatinine 1.5–2 mg/dL).33 The number of patients with renal insufficiency who achieved the primary end point of a serum urate level lower than 6 mg/dL was 4 (44%) of 9 in the febuxostat 80-mg group, 5 (46%) of 11 in the 120-mg group, and 3 (60%) of 5 in the 240-mg group, while none of the 10 patients in the dose-adjusted allopurinol group achieved the primary end point (P < .05). Of note, 41% of the patients with normal renal function who received allopurinol achieved the primary end point.33 As proposed above, if the allopurinol dose had been slowly increased in the patients with renal insufficiency, it might have been equally effective.
Febuxostat has not been thoroughly evaluated in patients with severe CKD or in patients on hemodialysis.
A presumed niche indication of febuxostat is in patients allergic to allopurinol, since the drugs are not similar in chemical structure. However, at present, experience with this use is limited. Allopurinol-allergic patients were excluded from the clinical trials; thus, if there is any allergic overlap, it would not likely have been recognized in those studies. The FDA has received reports of patients who were allergic to allopurinol also having reactions to febuxostat, and it is currently evaluating these reports (personal communication).
Concern was raised over cardiovascular adverse events in patients treated with febuxostat during clinical trials. In the FACT trial, two patients died of cardiac causes.20 In the study by Schumacher et al,33 11 of 670 patients experienced cardiac adverse events in the febuxostat group vs 3 of 268 in the allopurinol group. Events included atrial fibrillation, chest pain, coronary artery disease, and myocardial infarction. However, this difference was not statistically significant.
Febuxostat costs much more than allopurinol. Currently, patients pay $153.88 for 1 month of febuxostat 40 or 80 mg from Cleveland Clinic pharmacy; 1 month of allopurinol costs $17.45 (300 mg) or $14.00 (100 mg). We believe febuxostat should be reserved for patients with documented intolerance to allopurinol in effective doses.
Monitoring serum urate levels is important in all patients on hypouricemic therapy so that dosage adjustments can be made until the target serum urate concentration is reached. In patients failing to meet target serum urate levels, patient adherence with the prescribed dosing should be specifically addressed because as many as 50% of patients do not adhere to their prescribed regimen.
DOES URATE-LOWERING THERAPY HAVE BENEFITS BEYOND GOUT?
Despite experimental animal data and a strong epidemiologic association between hyperuri-cemia and hypertension,46 metabolic syndrome, and rates of cardiovascular and all-cause mortality,47 the evidence from interventional trials so far does not support the routine use of hypo-uricemic therapy to prevent these outcomes.
Similarly, hyperuricemia has long been associated with renal disease, and there has been debate as to whether hyperuricemia is a result of kidney dysfunction or a contributing factor.46,48–51 A few studies have documented improvement of renal function after initiation of hypouricemic therapy.52 However, treating asymptomatic hyperuricemia to preserve kidney function remains controversial.
A recent study indicates that lowering the serum urate level with allopurinol can lower the blood pressure in hyperuricemic adolescents who have newly diagnosed primary hypertension.53 This does not indicate, however, that initiating hypouricemic therapy in patients with preexisting, long-standing hypertension will be successful.
RECOMMENDED FOR OUR PATIENT
As for our diabetic patient with an acute gout flare and creatinine clearance rate of 45 mL/minute, we would recommend:
Aspirating the knee, sending the fluid for bacterial culture, and then treating it with a local glucocorticoid injection
Starting colchicine 0.6 mg every day, with frequent monitoring for signs of toxicity (muscle pain, weakness, leukopenia, and elevations of creatine kinase and aspartate aminotransferase)
Increasing his allopurinol dose by 100 mg every 2 to 4 weeks until the target serum urate level of less than 6.0 mg/dL is reached
If he cannot tolerate allopurinol or if the target serum urate level is not achieved despite adequate doses of allopurinol (about 800 mg), we would switch to febuxostat 40 mg and increase the dose as needed to achieve the desired urate level.
You have a 54-year-old black patient with gout, diabetes mellitus, hypertension, and chronic kidney disease (CKD). He has an acute gout flare involving his right knee. In the past year he has had four attacks of gout in the ankles and knees, which you treated with intra-articular glucocorticoid injections. He has been on allopurinol (Zyloprim) 200 mg daily, but his last serum urate level was 9.4 mg/dL (reference range 3.0–8.0). His creatinine clearance is 45 mL/minute (reference range 85–125).
In view of his kidney disease, you are concerned about increasing his dose of allopurinol, but also about the need to treat his frequent attacks. How should you manage this patient?
GOUT IS CHALLENGING TO TREAT IN PATIENTS WITH KIDNEY DISEASE
A major challenge in treating patients with gout is to avoid therapeutic interactions with common comorbidities, including hypertension, insulin resistance, coronary artery disease, heart failure, and especially CKD.1
In this paper, we discuss approaches to and controversies in the management of gout and hyperuricemia in patients with CKD. Unfortunately, the evidence from clinical trials to guide treatment decisions is limited; therefore, decisions must often be based on experience and pathophysiologic principles.
GENERAL GOALS OF GOUT THERAPY
Depending on the patient and the stage of the disease, the goals in treating patients with gout are to:
Terminate acute attacks as promptly and safely as possible
Prevent recurrences of acute gout attacks
Prevent or reverse complications resulting from deposition of monosodium urate in the joints, in the kidneys, or at other sites.
These goals are more difficult to achieve in patients with CKD because of the potential complications from many of the available drugs.
TERMINATING ACUTE GOUT FLARES
In patients with acute gout, treatment is aimed at quickly resolving pain and inflammation.
Several types of drugs can terminate acute gout flares. The choice in most situations is colchicine (Colcrys); a nonsteroidal anti-inflammatory drug (NSAID); a corticosteroid; or corticotropin (ACTH).
However, in patients with CKD, there are concerns about using colchicine or NSAIDs, and corticotropin is very expensive; thus, corticosteroids are often used.
Colchicine’s clearance is reduced in CKD
Colchicine is somewhat effective in treating acute gout attacks and probably more effective in preventing attacks.
Due to concerns about inappropriate dosing and reported deaths,2 the intravenous formulation is not available in many countries, including the United States.
After oral administration, colchicine is rapidly absorbed, with a bioavailability of up to 50%. It undergoes metabolism by the liver, and its metabolites are excreted by renal and biliary-intestinal routes. Up to 20% of the active drug is excreted by the kidneys.3
Colchicine’s clearance is significantly reduced in patients with renal or hepatic insufficiency, and the drug may accumulate in cells, with resultant toxicity.4 Colchicine-induced toxicity has been observed when the drug was used for acute treatment, as well as for chronic prophylaxis of gout in patients with CKD; thus, alternative agents for treating acute attacks should be considered.5,6 With prolonged use, reversible colchicine-induced axonal neuropathy, neutropenia, and vacuolar myopathy can develop in patients with CKD.7
In a trial in patients with normal renal function, nearly 100% who received an initial dose of 1 mg followed by 0.5 mg every 2 hours developed diarrhea at a median time of 24 hours.8 Emesis may also occur.
A lower dose of 1.8 mg (two 0.6-mg pills followed by one pill an hour later) was well tolerated but only moderately effective in treating acute gout, causing at least a 50% reduction in pain at 24 hours in only 38% of patients.9 This study does not clarify the dosage to use to completely resolve attacks. Using additional colchicine likely will increase the response rate, but will also increase side effects. Patients with CKD were not included.
Some patients, as shown in the above trial, can abort attacks by taking only one or two colchicine tablets when they feel the first “twinge” of an attack. This approach is likely to be safe in CKD, but it may be of value to only a few patients.
Nonsteroidal anti-inflammatory drugs can worsen chronic kidney disease
NSAIDs in high doses can effectively treat the pain and inflammation of acute gout. Indomethacin (Indocin) 50 mg three times daily has been standard NSAID therapy.
Other nonselective NSAIDs and NSAIDs that selectively inhibit cyclooxygenase 2 (COX-2) are effective, but all can cause acute renal toxicity or worsen CKD.10 Renal side effects include salt and water retention, acute tubular necrosis, acute interstitial nephritis, proteinuria, hypertension, hyperkalemia, and chronic renal injury.11
Even short-term use of high-dose NSAIDs should generally be avoided in patients with preexisting CKD, for whom there is no established safe threshold dose. When NSAIDs (including selective COX-2 inhibitors) are used, renal function should be monitored closely and the duration limited as much as possible.
Corticosteroids are often used to treat acute attacks
Due to the concerns about NSAIDs or colchicine to treat acute gout attacks in patients with CKD, corticosteroids are often used in this setting.
Intra-articular steroid injections are useful in treating acute gout limited to a single joint or bursa.12 However, one should first make sure that the joint is not infected: septic arthritis should ideally be excluded by arthrocentesis, particularly in immunosuppressed patients13 or those with end-stage renal disease, who are predisposed to bacteremia.
Oral, intramuscular, or intravenous steroids can provide complete relief from acute gout, although high doses (eg, prednisone 30–60 mg/day or the equivalent) are often needed. Common errors resulting in inefficacy include using too low a dose or not treating for a sufficient time before tapering or stopping. Groff and colleagues14 described 13 patients who received oral or intravenous steroids for acute gout. Nine patients received an initial single dose of prednisone ranging from 20 to 50 mg, with tapering over a mean of 10 days. Twelve of the 13 patients had improvement within 48 hours, and the signs and symptoms of acute gout resolved completely within 7 to 10 days.
We often give prednisone 40 mg daily until a day after the acute attack resolves and then taper over another 7 to 10 days. There are no data to guide steroid dosing in an evidence-based way, but we believe too short a course of therapy may result in return of symptoms.
Corticotropin and other agents: Effective but costly
Corticotropin shares the same indications as systemic corticosteroids, being used to treat flares when NSAIDs, intra-articular steroids, and colchicine are contraindicated. However, corticotropin is far more expensive than generic corticosteroids, costing nearly $2,000 for a single 80-IU dose, which may need to be repeated.
Corticotropin is available for subcutaneous or intramuscular injection. A single intramuscular injection of corticotropin gel (H.P. Acthar, 25–80 IU) may terminate an acute gout attack.15 However, many patients need another injection after 24 to 72 hours, which would require another visit to the physician. This treatment has been touted by some as being more effective than corticosteroid therapy, possibly because of a unique peripheral mechanism of action in addition to stimulating cortisol release.16
We rarely use corticotropin, in view of its cost as well as concerns about excessive sodium and water retention due to the release of multiple hormones from the adrenal gland. This may be especially deleterious in patients with CKD or congestive heart failure.17
Parenteral anti-tumor necrosis factor agents or interleukin 1 antagonists can be dramatically effective but are also expensive.18,19 For example, anakinra (Kineret) 100 mg costs about $73, and multiple daily doses may be necessary.
Under unique conditions in which they can be safely used (eg, patients with CKD, diabetes mellitus, liver disease), they may be cost-effective if they can shorten the stay of a hospitalized patient with acute gout.
PROPHYLACTIC ANTI-INFLAMMATORY THERAPY FOR PATIENTS WITH GOUT
Between attacks, the goal is to prevent new attacks through prophylactic management, which may include anti-inflammatory and hypouricemic therapy along with dietary instruction (such as avoiding excessive beer, liquor, and fructose ingestion).
Colchicine can be used as prophylaxis, with caution and monitoring
Although colchicine is not 100% effective, it markedly reduces the flare rate when started in low doses at the time hypouricemic therapy is initiated.20,21 (Hypouricemic therapy is discussed below.) We generally try to continue this prophylactic therapy, if the patient tolerates it, for at least 6 months—longer if tophi are still present or if attacks continue to occur.
If renal function is intact, colchicine can be prescribed at a dosage of 0.6 mg orally once or twice daily.21 In CKD, since the clearance of colchicine is reduced,4 the dosage should be reduced. Patients on colchicine for prophylaxis must be carefully monitored if the glomerular filtration rate is less than 50 mL/minute, or colchicine should be avoided altogether.6 Laboratory testing for colchicine levels is not routinely available and may be of limited value in predicting adverse effects; thus, recommendations about dose adjustments in CKD are empiric.
Wallace et al22 recommended a dose of 0.6 mg once daily if the creatinine clearance is 35 to 49 mL/minute and 0.6 mg every 2 to 3 days if it is 10 to 34 mL/minute, but there are no published long-term safety or efficacy data validating these reasonable (based on available information) dosing regimens.
Even with dose adjustment, caution is needed. Low-dose daily colchicine may be associated with reversible neuromyopathy and bone marrow suppression.7,23 Patients with neuromyopathy may complain of myalgias, proximal muscle weakness, and numbness and may have areflexia and decreased sensation. Laboratory findings include elevated creatine kinase and aminotransferase levels. We regularly check for leukopenia or elevated creatine kinase and aspartate aminotransferase levels in patients with CKD who are receiving colchicine in any dose.
Prolonged colchicine therapy should probably be avoided in patients on hemodialysis, as this drug is not removed by dialysis or by exchange transfusion, and the risk of toxicity under these circumstances may be high.22 When there is no viable alternative and the drug is given, patients should be closely monitored for signs of toxicity.
Concurrent (even short-term) treatment with most macrolide antibiotics, particularly clarithromycin (Biaxin), most statin drugs, ketoconazole (Nizoral), cyclosporine, and likely other drugs predisposes to colchicine toxicity by altering its distribution and elimination, and can in rare cases cause morbidity or death.24–26
NSAIDs are not optimal as prophylaxis in patients with chronic kidney disease
Little information has been published about using NSAIDs chronically to prevent flares, but they are not the optimal drugs to use in patients with CKD, as discussed above. In patients with end-stage renal disease, there are also concerns about NSAID-induced gastric and intestinal bleeding.
Low-dose steroids may not be effective as prophylaxis
Lower doses of steroids may not be effective as prophylaxis against gout flares, consistent with the common observation that gout flares still occur in organ transplant recipients who are taking maintenance doses of prednisone.13
PREVENTING FLARES BY LOWERING SERUM URATE LEVELS
If tophi are present, if radiography shows evidence of damage, if attacks are frequent or disabling, or if there are relative contraindications to the drugs that would be needed to treat acute attacks, then hypouricemic therapy should be strongly considered to reduce the burden of urate in the body, resorb tophi, and ultimately reduce the frequency of gout flares.20
Although intermittent therapy for attacks or prolonged prophylactic use of colchicine may prevent recurrent episodes of gouty arthritis and may be reasonable for many patients, this approach does not prevent continued urate deposition, with the potential development of bony erosions, tophaceous deposits, and chronic arthritis.
The definitive therapy for gouty arthritis is to deplete the periarticular deposits of urate by maintaining a low serum urate level. Urate-lowering therapy, when indicated, is almost always lifelong.
Four strategies for lowering serum urate
The serum urate concentration can be lowered in four ways:
Increasing renal uric acid excretion
Altering the diet
Decreasing urate synthesis
Converting urate to a more soluble metabolite.
Increasing uric acid excretion is rarely effective if renal function is impaired
Probenecid, sulfinpyrazone (Anturane), and losartan (Cozaar) modestly increase uric acid secretion and reduce serum urate levels, but they are rarely effective if the creatinine clearance rate is less than 60 mL/minute, and they require significant fluid intake for maximal efficacy.
Uricosuric drugs probably should be avoided in patients who excrete more than 1,000 mg of uric acid per day on a normal diet, since urinary uric acid stones may form. In practice, however, patients are given losartan to treat hypertension without attention to uric acid excretion.
More-potent urocosuric drugs are being tested in clinical trials.
Altering the diet: Traditional advice confirmed
The Health Professionals Follow-up Study27,28 prospectively examined the relation between diet and gout over 12 years in 47,150 men. The study confirmed some long-standing beliefs, such as that consuming meat, seafood, beer, and liquor increases the risk. Other risk factors were consumption of sugar-sweetened soft drinks and fructose, adiposity, weight gain, hypertension, and diuretic use. On the other hand, protein, wine, and purine-rich vegetables were not associated with gout flares. Low-fat dairy products may have a protective effect. Weight loss was found to be protective.
Low-purine diets are not very palatable, are difficult to adhere to, and are at best only minimally effective, lowering serum urate by 1 to 2 mg/dL. Low-protein diets designed to slow progression of CKD will likely also have only a slight effect on serum urate. Dietary change alone is not likely to dramatically lower serum urate levels.
Metabolizing urate with exogenous uricase
Rasburicase (Elitek) effectively converts urate to allantoin, which is more soluble, but rasburicase is fraught with allergic reactions and cannot be used as chronic therapy.
A pegylated intravenous uricase29 has just been approved by the US Food and Drug Administration (FDA); the retail cost is not yet known. It is dramatically effective in those patients able to use it chronically, but it has not been fully evaluated in patients with CKD.
Decreasing urate synthesis with allopurinol
Allopurinol acts by competitively inhibiting xanthine oxidase, the enzyme that converts hypoxanthine to xanthine and xanthine to uric acid. The drug, a structural analogue of hypoxanthine, is converted by xanthine oxidase to oxypurinol, which is an even more effective inhibitor of xanthine oxidase than allopurinol.
Allopurinol is metabolized in the liver and has a half-life of 1 to 3 hours, but oxypurinol, which is excreted in the urine, has a half-life of 12 to 17 hours. Because of these pharmacokinetic properties, allopurinol can usually be given once daily, and the dosage required to reduce serum urate levels should in theory be lower in patients with lower glomerular filtration rates.
Allopurinol (100- and 300-mg tablets) is approved by the FDA in doses of up to 800 mg/day to treat hyperuricemia in patients with gout,30 while guidelines from the British Society of Rheumatology advocate a maximum dose of 900 mg/day.31 These maximum doses are based on the limited amount of data with higher doses, not on documented toxicity.
Practice survey data in the United States indicate that most physicians prescribe no greater than 300 mg daily, although this dosage is likely to reduce the serum urate to less than 6 mg/dL—the goal level—in fewer than 50% of patients.20,32 Patients with normal renal function occasionally require more than 1,000 mg daily to reduce the serum urate level to less than 6 mg/dL.
How low should the serum urate level be?
Ideally, therapy should keep the serum urate level significantly below 6.7 mg/dL, the approximate saturation point of urate in physiologic fluids.
Lowering the serum urate level from 10 mg/dL to 7 mg/dL may seem encouraging, and the urate level may be in the laboratory “normal” range; however, urate may continue to precipitate in tissues if the concentration is greater than 6.7 mg/dL. A target of 6 mg/dL, used in clinical studies, is far enough below the saturation level to provide some margin for fluctuations in serum levels. A serum level of 6.0 mg/dL has thus been arbitrarily proposed as a reasonable therapeutic target.
The lower the serum urate level achieved during hypouricemic therapy, the faster the reduction in tophaceous deposits. With adequate urate lowering, tophi can be visibly reduced in less than a year of hypouricemic therapy.33,34
We have as yet no convincing evidence that lowering the serum urate level to less than 6.0 mg/dL is harmful, despite theoretical concerns that urate is a beneficial circulating antioxidant and epidemiologic observations that urate levels have been inversely correlated with progression of Parkinson disease.
Start low, go slow to avoid a flare
Rapid reduction of the serum urate level in a patient with chronic hyperuricemia and gout is likely to induce an acute flare.20 We have traditionally used a “start low and increase slowly” approach to escalating hypouricemic therapy in hopes of reducing the likelihood of causing a gout flare.
Without anti-inflammatory prophylaxis, acute flares associated with urate-lowering are extremely likely. In a 28-week trial of allopurinol, febuxostat, and placebo by Schumacher et al,33 during the first 8 weeks, when prophylaxis against gout flare was provided with either colchicine 0.6 mg once daily or naproxen (Naprosyn) 250 mg twice daily, the proportion of patients requiring treatment of gout flares was still 23% to 46%. When prophylaxis was stopped, the flare rate increased further.33
The more we acutely lower serum urate levels, the more likely flares are to occur. In the study by Schumacher et al,33 the percentage of patients needing treatment for gout flares during the first 8 weeks of the study, despite gout flare prophylaxis, was related to the percent reduction in serum urate by week 28 of the trial (Table 2).
IS IT NECESSARY TO ADJUST THE ALLOPURINOL DOSE IN CHRONIC KIDNEY DISEASE?
In 1984, Hande et al35 proposed that allopurinol doses be lower in patients with renal insufficiency, with a dosage scale based on creatinine clearance.
Their thoughtful proposal was based on data from six of their own patients and 72 others with severe allopurinol toxicity, mainly allopurinol hypersensitivity syndrome, reported in the literature.
Perez-Ruiz et al36 noted that patients who had experienced adverse effects from allopurinol in their series were likely to have had received “higher” doses of allopurinol, if the dosage was corrected for reduced oxypurinol elimination based on their estimated creatinine clearance.
However, most of these reactions occurred soon after initiating therapy, a temporal pattern more typical of non-dose-dependent allergic reactions. Additionally, allopurinol hypersensitivity has been linked to T-cell-mediated immune reactions to oxypurinol,37 a mechanism not likely linked to drug levels.
Arguments against dose adjustment
Despite the compelling information that allopurinol reactions are more common in CKD, adjusting the dosage of allopurinol has not been clearly shown to reduce the frequency of these reactions.
In a small retrospective analysis, Vázquez-Mellado et al38 reported that adjusting the allopurinol dosage according to creatinine clearance did not decrease the incidence of allopurinol hypersensitivity.
In a study in 250 patients, Dalbeth et al39 showed that the overall incidence of hypersensitivity reaction was 1.6%, and the incidence of allergic reactions did not decrease when allopurinol was given according to the dosing guidelines proposed by Hande et al.35 However, it is worth noting that, of the patients who received the recommended lower doses, only 19% achieved the target serum urate level of 6 mg/dL.39
Silverberg et al40 found that of 15 patients who developed hypersensitivity reactions to allopurinol, 10 had received doses that were low or appropriate according to the guidelines of Hande et al.35
More recently, Stamp et al41 found that gradually increasing the allopurinol dose above the proposed creatinine clearance-based dose was safe and effective. Thirty-one (89%) of the 35 patients who completed the study achieved the target serum urate level of 6 mg/dL, while only 3 of 45 who started the study developed rashes, which were not serious.
The small number of patients in these studies limits any strong conclusion, but at present there is no interventional study showing that allopurinol dosing adjustment based on glomerular filtration rate is effective or safer than dosing based on the serum urate level.
Our view on allopurinol dosing adjustment
We believe the initial observations of Hande et al35 and the subsequent meticulous data from Perez-Ruiz et al36 suggest a relationship between CKD and the occurrence of severe allopurinol reactions. However, these observations do not prove that dose adjustment will prevent these reactions.
In patients with normal kidney function, the FDA30 and the European League Against Rheumatism (EULAR)42 recommend slow upward titration, starting with 100 to 200 mg/day, which we agree should decrease the frequency of acute gout flares. The dose is increased by increments of 100 mg/day at intervals of 1 week (FDA recommendation) or 2 to 4 weeks (EULAR recommendation) until the serum urate level is lower than 6 mg/dL.
We believe the optimal approach to allopurinol dosing in patients with CKD remains uncertain. We generally escalate the dose slowly, with ongoing frequent laboratory and clinical monitoring, and we do not limit the maximal dose as suggested by Hande et al.35
An alternative strategy is to use the newer, far more expensive xanthine oxidase inhibitor febuxostat in patients with CKD, since it is not excreted by the kidney. We usually first try escalating doses of allopurinol.
FEBUXOSTAT, AN ALTERNATIVE TO ALLOPURINOL
Febuxostat is an oral nonpurine inhibitor of xanthine oxidase.43 Approved by the FDA in 2009, it is available in 40- and 80-mg tablets.
Unlike allopurinol, febuxostat is metabolized primarily by hepatic glucuronide formation and oxidation and then excreted in stool and urine,44 making it in theory an attractive agent in patients with renal insufficiency, bypassing the controversial dose-adjustment issue with allopurinol.
In the Febuxostat Versus Allopurinol Controlled Trial (FACT),20 a 52-week randomized, double-blind study in hyperuricemic patients with gout, serum urate levels were reduced to less than 6.0 mg/dL in over 50% of patients receiving febuxostat 80 mg or 120 mg once daily, while only 21% of patients receiving 300 mg of allopurinol achieved this goal. This does not imply that allopurinol at higher doses, as should be used in clinical practice,45 would not be equally effective. Patients with CKD were not included in this trial.
In the study by Schumacher et al,33 febuxostat 80, 120, or 240 mg once daily reduced serum urate. A small subset (35 patients) had mild to moderate renal insufficiency (serum creatinine 1.5–2 mg/dL).33 The number of patients with renal insufficiency who achieved the primary end point of a serum urate level lower than 6 mg/dL was 4 (44%) of 9 in the febuxostat 80-mg group, 5 (46%) of 11 in the 120-mg group, and 3 (60%) of 5 in the 240-mg group, while none of the 10 patients in the dose-adjusted allopurinol group achieved the primary end point (P < .05). Of note, 41% of the patients with normal renal function who received allopurinol achieved the primary end point.33 As proposed above, if the allopurinol dose had been slowly increased in the patients with renal insufficiency, it might have been equally effective.
Febuxostat has not been thoroughly evaluated in patients with severe CKD or in patients on hemodialysis.
A presumed niche indication of febuxostat is in patients allergic to allopurinol, since the drugs are not similar in chemical structure. However, at present, experience with this use is limited. Allopurinol-allergic patients were excluded from the clinical trials; thus, if there is any allergic overlap, it would not likely have been recognized in those studies. The FDA has received reports of patients who were allergic to allopurinol also having reactions to febuxostat, and it is currently evaluating these reports (personal communication).
Concern was raised over cardiovascular adverse events in patients treated with febuxostat during clinical trials. In the FACT trial, two patients died of cardiac causes.20 In the study by Schumacher et al,33 11 of 670 patients experienced cardiac adverse events in the febuxostat group vs 3 of 268 in the allopurinol group. Events included atrial fibrillation, chest pain, coronary artery disease, and myocardial infarction. However, this difference was not statistically significant.
Febuxostat costs much more than allopurinol. Currently, patients pay $153.88 for 1 month of febuxostat 40 or 80 mg from Cleveland Clinic pharmacy; 1 month of allopurinol costs $17.45 (300 mg) or $14.00 (100 mg). We believe febuxostat should be reserved for patients with documented intolerance to allopurinol in effective doses.
Monitoring serum urate levels is important in all patients on hypouricemic therapy so that dosage adjustments can be made until the target serum urate concentration is reached. In patients failing to meet target serum urate levels, patient adherence with the prescribed dosing should be specifically addressed because as many as 50% of patients do not adhere to their prescribed regimen.
DOES URATE-LOWERING THERAPY HAVE BENEFITS BEYOND GOUT?
Despite experimental animal data and a strong epidemiologic association between hyperuri-cemia and hypertension,46 metabolic syndrome, and rates of cardiovascular and all-cause mortality,47 the evidence from interventional trials so far does not support the routine use of hypo-uricemic therapy to prevent these outcomes.
Similarly, hyperuricemia has long been associated with renal disease, and there has been debate as to whether hyperuricemia is a result of kidney dysfunction or a contributing factor.46,48–51 A few studies have documented improvement of renal function after initiation of hypouricemic therapy.52 However, treating asymptomatic hyperuricemia to preserve kidney function remains controversial.
A recent study indicates that lowering the serum urate level with allopurinol can lower the blood pressure in hyperuricemic adolescents who have newly diagnosed primary hypertension.53 This does not indicate, however, that initiating hypouricemic therapy in patients with preexisting, long-standing hypertension will be successful.
RECOMMENDED FOR OUR PATIENT
As for our diabetic patient with an acute gout flare and creatinine clearance rate of 45 mL/minute, we would recommend:
Aspirating the knee, sending the fluid for bacterial culture, and then treating it with a local glucocorticoid injection
Starting colchicine 0.6 mg every day, with frequent monitoring for signs of toxicity (muscle pain, weakness, leukopenia, and elevations of creatine kinase and aspartate aminotransferase)
Increasing his allopurinol dose by 100 mg every 2 to 4 weeks until the target serum urate level of less than 6.0 mg/dL is reached
If he cannot tolerate allopurinol or if the target serum urate level is not achieved despite adequate doses of allopurinol (about 800 mg), we would switch to febuxostat 40 mg and increase the dose as needed to achieve the desired urate level.
References
Vázquez-Mellado J, García CG, Vázquez SG, et al. Metabolic syndrome and ischemic heart disease in gout. J Clin Rheumatol2004; 10:105–109.
Bonnel RA, Villalba ML, Karwoski CB, Beitz J. Deaths associated with inappropriate intravenous colchicine administration. J Emerg Med2002; 22:385–387.
Achtert G, Scherrmann JM, Christen MO. Pharmacokinetics/bioavailability of colchicine in healthy male volunteers. Eur J Drug Metab Pharmacokinet1989; 14:317–322.
Ben-Chetrit E, Scherrmann JM, Zylber-Katz E, Levy M. Colchicine disposition in patients with familial Mediterranean fever with renal impairment. J Rheumatol1994; 21:710–713.
Putterman C, Ben-Chetrit E, Caraco Y, Levy M. Colchicine intoxication: clinical pharmacology, risk factors, features, and management. Semin Arthritis Rheum1991; 21:143–155.
Aronoff G, Brater DC, Schrier R, Bennett WM. Use of drugs in patients with renal insufficiency. Workshop report. Blood Purif1994; 12:14–19.
Kuncl RW, Duncan G, Watson D, Alderson K, Rogawski MA, Peper M. Colchicine myopathy and neuropathy. N Engl J Med1987; 316:1562–1568.
Ahern MJ, Reid C, Gordon TP, McCredie M, Brooks PM, Jones M. Does colchicine work? The results of the first controlled study in acute gout. Aust N Z J Med1987; 17:301–304.
Terkeltaub R, Furst D, Bennett K, Kook K, Crockett RS, Davis WM. High versus low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum2010, 62:1060–1068.
Whelton A. Nephrotoxicity of nonsteroidal anti-inflammatory drugs: physiologic foundations and clinical implications. Am J Med1999; 106:13S–24S.
Wali RK, Henrich WL. Recent developments in toxic nephropathy. Curr Opin Nephrol Hypertens2002; 11:155–163.
Fernández C, Noguera R, González JA, Pascual E. Treatment of acute attacks of gout with a small dose of intraarticular triamcinolone acetonide. J Rheumatol1999; 26:2285–2286.
Clive DM. Renal transplant-associated hyperuricemia and gout. J Am Soc Nephrol2000; 11:974–979.
Groff GD, Franck WA, Raddatz DA. Systemic steroid therapy for acute gout: a clinical trial and review of the literature. Semin Arthritis Rheum1990; 19:329–336.
Ritter J, Kerr LD, Valeriano-Marcet J, Spiera H. ACTH revisited: effective treatment for acute crystal induced synovitis in patients with multiple medical problems. J Rheumatol1994; 21:696–699.
Getting SJ, Christian HC, Flower RJ, Perretti M. Activation of melanocortin type 3 receptor as a molecular mechanism for adrenocorticotropic hormone efficacy in gouty arthritis. Arthritis Rheum2002; 46:2765–2775.
Connell JM, Whitworth JA, Davies DL, Lever AF, Richards AM, Fraser R. Effects of ACTH and cortisol administration on blood pressure, electrolyte metabolism, atrial natriuretic peptide and renal function in normal man. J Hypertens1987; 5:425–433.
Tausche AK, Richter K, Grässler A, Hänsel S, Roch B, Schröder HE. Severe gouty arthritis refractory to anti-inflammatory drugs: treatment with anti-tumour necrosis factor alpha as a new therapeutic option. Ann Rheum Dis2004; 63:1351–1352.
So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther2007; 9:R28.
Becker MA, Schumacher HR, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med2005; 353:2450–2461.
Borstad GC, Bryant LR, Abel MP, Scroggie DA, Harris MD, Alloway JA. Colchicine for prophylaxis of acute flares when initiating allopurinol for chronic gouty arthritis. J Rheumatol2004; 31:2429–2432.
Wallace SL, Singer JZ, Duncan GJ, Wigley FM, Kuncl RW. Renal function predicts colchicine toxicity: guidelines for the prophylactic use of colchicine in gout. J Rheumatol1991; 18:264–269.
Wilbur K, Makowsky M. Colchicine myotoxicity: case reports and literature review. Pharmacotherapy2004; 24:1784–1792.
Hung IF, Wu AK, Cheng VC, et al. Fatal interaction between clarithromycin and colchicine in patients with renal insufficiency: a retrospective study. Clin Infect Dis2005; 41:291–300.
Alayli G, Cengiz K, Cantürk F, Durmus D, Akyol Y, Menekse EB. Acute myopathy in a patient with concomitant use of pravastatin and colchicine. Ann Pharmacother2005; 39:1358–1361.
Ducloux D, Schuller V, Bresson-Vautrin C, Chalopin JM. Colchicine myopathy in renal transplant recipients on cyclosporin. Nephrol Dial Transplant1997; 12:2389–2392.
Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med2004; 350:1093–1103.
Choi HK, Curhan G. Soft drinks, fructose consumption, and the risk of gout in men: prospective cohort study. BMJ2008; 336:309–312.
Sundy JS, Becker MA, Baraf HS, et al; Pegloticase Phase 2 Study Investigators. Reduction of plasma urate levels following treatment with multiple doses of pegloticase (polyethylene glycol-conjugated uricase) in patients with treatment-failure gout: results of a phase II randomized study. Arthritis Rheum2008; 58:2882–2891.
Jordan KM, Cameron JS, Snaith M, et al; British Society for Rheumatology and British Health Professionals in Rheumatology Standards, Guidelines and Audit Working Group (SGAWG). British Society for Rheumatology and British Health Professionals in Rheumatology guideline for the management of gout. Rheumatology (Oxford)2007; 46:1372–1374.
Perez-Ruiz F, Alonso-Ruiz A, Calabozo M, Herrero-Beites A, García-Erauskin G, Ruiz-Lucea E. Efficacy of allopurinol and benzbromarone for the control of hyperuricaemia. A pathogenic approach to the treatment of primary chronic gout. Ann Rheum Dis1998; 57:545–549.
Schumacher HR, Becker MA, Wortmann RL, et al. Effects of febuxostat versus allopurinol and placebo in reducing serum urate in subjects with hyperuricemia and gout: a 28-week, phase III, randomized, double-blind, parallel-group trial. Arthritis Rheum2008; 59:1540–1548.
Perez-Ruiz F, Calabozo M, Pijoan JI, Herrero-Beites AM, Ruibal A. Effect of urate-lowering therapy on the velocity of size reduction of tophi in chronic gout. Arthritis Rheum2002; 47:356–360.
Hande KR, Noone RM, Stone WJ. Severe allopurinol toxicity. Description and guidelines for prevention in patients with renal insufficiency. Am J Med1984; 76:47–56.
Perez-Ruiz F, Hernando I, Villar I, Nolla JM. Correction of allopurinol dosing should be based on clearance of creatinine, but not plasma creatinine levels: another insight to allopurinol-related toxicity. J Clin Rheumatol2005; 11:129–133.
Hung SI, Chung WH, Liou LB, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A2005; 102:4134–4139.
Vázquez-Mellado J, Morales EM, Pacheco-Tena C, Burgos-Vargas R. Relation between adverse events associated with allopurinol and renal function in patients with gout. Ann Rheum Dis2001; 60:981–983.
Dalbeth N, Kumar S, Stamp L, Gow P. Dose adjustment of allopurinol according to creatinine clearance does not provide adequate control of hyperuricemia in patients with gout. J Rheumatol2006; 33:1646–1650.
Silverberg MS, Mallela R, Lesse AJ, Bonner MR, Baer AN, Li C. Allopurinol hypersensitivity reactions: a case-control study of the role of renal dosing (abstract). Arthritis Rheum2009; 60(suppl 10):1106.
Stamp LK, O'Donnell JL, Zhang M, et al. Using allopurinol above the dose based on creatinine clearance is effective and safe in chronic gout, including in those with renal impairment. Arthritis Rhem2010; doi:10.1002/art.30119. E-pub ahead of print. Accessed 10/29/2010.
Zhang W, Doherty M, Bardin T, et al; EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis2006; 65:1312–1324.
Okamoto K, Eger BT, Nishino T, Kondo S, Pai EF, Nishino T. An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J Biol Chem2003; 278:1848–1855.
Khosravan R, Grabowski BA, Wu JT, Joseph-Ridge N, Vernillet L. Pharmacokinetics, pharmacodynamics and safety of febuxostat, a non-purine selective inhibitor of xanthine oxidase, in a dose escalation study in healthy subjects. Clin Pharmacokinet2006; 45:821–841.
Reinders MK, Haagsma C, Jansen TL, et al. A randomised controlled trial on the efficacy and tolerability with dose escalation of allopurinol 300–600 mg/day versus benzbromarone 100–200 mg/day in patients with gout. Ann Rheum Dis2009; 68:892–897.
Mazzali M, Hughes J, Kim YG, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension2001; 38:1101–1106.
Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med2008; 359:1811–1821.
Beck LH. Requiem for gouty nephropathy. Kidney Int1986; 30:280–287.
Tomita M, Mizuno S, Yamanaka H, et al. Does hyperuricemia affect mortality? A prospective cohort study of Japanese male workers. J Epidemiol2000; 10:403–409.
Kang DH, Nakagawa T. Uric acid and chronic renal disease: possible implication of hyperuricemia on progression of renal disease. Semin Nephrol2005; 25:43–49.
Iseki K, Oshiro S, Tozawa M, Iseki C, Ikemiya Y, Takishita S. Significance of hyperuricemia on the early detection of renal failure in a cohort of screened subjects. Hypertens Res2001; 24:691–697.
Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med1987; 82:421–426.
Siu YP, Leung KT, Tong MK, Kwan TH. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis2006; 47:51–59.
Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA2008; 300:924–932.
References
Vázquez-Mellado J, García CG, Vázquez SG, et al. Metabolic syndrome and ischemic heart disease in gout. J Clin Rheumatol2004; 10:105–109.
Bonnel RA, Villalba ML, Karwoski CB, Beitz J. Deaths associated with inappropriate intravenous colchicine administration. J Emerg Med2002; 22:385–387.
Achtert G, Scherrmann JM, Christen MO. Pharmacokinetics/bioavailability of colchicine in healthy male volunteers. Eur J Drug Metab Pharmacokinet1989; 14:317–322.
Ben-Chetrit E, Scherrmann JM, Zylber-Katz E, Levy M. Colchicine disposition in patients with familial Mediterranean fever with renal impairment. J Rheumatol1994; 21:710–713.
Putterman C, Ben-Chetrit E, Caraco Y, Levy M. Colchicine intoxication: clinical pharmacology, risk factors, features, and management. Semin Arthritis Rheum1991; 21:143–155.
Aronoff G, Brater DC, Schrier R, Bennett WM. Use of drugs in patients with renal insufficiency. Workshop report. Blood Purif1994; 12:14–19.
Kuncl RW, Duncan G, Watson D, Alderson K, Rogawski MA, Peper M. Colchicine myopathy and neuropathy. N Engl J Med1987; 316:1562–1568.
Ahern MJ, Reid C, Gordon TP, McCredie M, Brooks PM, Jones M. Does colchicine work? The results of the first controlled study in acute gout. Aust N Z J Med1987; 17:301–304.
Terkeltaub R, Furst D, Bennett K, Kook K, Crockett RS, Davis WM. High versus low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum2010, 62:1060–1068.
Whelton A. Nephrotoxicity of nonsteroidal anti-inflammatory drugs: physiologic foundations and clinical implications. Am J Med1999; 106:13S–24S.
Wali RK, Henrich WL. Recent developments in toxic nephropathy. Curr Opin Nephrol Hypertens2002; 11:155–163.
Fernández C, Noguera R, González JA, Pascual E. Treatment of acute attacks of gout with a small dose of intraarticular triamcinolone acetonide. J Rheumatol1999; 26:2285–2286.
Clive DM. Renal transplant-associated hyperuricemia and gout. J Am Soc Nephrol2000; 11:974–979.
Groff GD, Franck WA, Raddatz DA. Systemic steroid therapy for acute gout: a clinical trial and review of the literature. Semin Arthritis Rheum1990; 19:329–336.
Ritter J, Kerr LD, Valeriano-Marcet J, Spiera H. ACTH revisited: effective treatment for acute crystal induced synovitis in patients with multiple medical problems. J Rheumatol1994; 21:696–699.
Getting SJ, Christian HC, Flower RJ, Perretti M. Activation of melanocortin type 3 receptor as a molecular mechanism for adrenocorticotropic hormone efficacy in gouty arthritis. Arthritis Rheum2002; 46:2765–2775.
Connell JM, Whitworth JA, Davies DL, Lever AF, Richards AM, Fraser R. Effects of ACTH and cortisol administration on blood pressure, electrolyte metabolism, atrial natriuretic peptide and renal function in normal man. J Hypertens1987; 5:425–433.
Tausche AK, Richter K, Grässler A, Hänsel S, Roch B, Schröder HE. Severe gouty arthritis refractory to anti-inflammatory drugs: treatment with anti-tumour necrosis factor alpha as a new therapeutic option. Ann Rheum Dis2004; 63:1351–1352.
So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther2007; 9:R28.
Becker MA, Schumacher HR, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med2005; 353:2450–2461.
Borstad GC, Bryant LR, Abel MP, Scroggie DA, Harris MD, Alloway JA. Colchicine for prophylaxis of acute flares when initiating allopurinol for chronic gouty arthritis. J Rheumatol2004; 31:2429–2432.
Wallace SL, Singer JZ, Duncan GJ, Wigley FM, Kuncl RW. Renal function predicts colchicine toxicity: guidelines for the prophylactic use of colchicine in gout. J Rheumatol1991; 18:264–269.
Wilbur K, Makowsky M. Colchicine myotoxicity: case reports and literature review. Pharmacotherapy2004; 24:1784–1792.
Hung IF, Wu AK, Cheng VC, et al. Fatal interaction between clarithromycin and colchicine in patients with renal insufficiency: a retrospective study. Clin Infect Dis2005; 41:291–300.
Alayli G, Cengiz K, Cantürk F, Durmus D, Akyol Y, Menekse EB. Acute myopathy in a patient with concomitant use of pravastatin and colchicine. Ann Pharmacother2005; 39:1358–1361.
Ducloux D, Schuller V, Bresson-Vautrin C, Chalopin JM. Colchicine myopathy in renal transplant recipients on cyclosporin. Nephrol Dial Transplant1997; 12:2389–2392.
Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med2004; 350:1093–1103.
Choi HK, Curhan G. Soft drinks, fructose consumption, and the risk of gout in men: prospective cohort study. BMJ2008; 336:309–312.
Sundy JS, Becker MA, Baraf HS, et al; Pegloticase Phase 2 Study Investigators. Reduction of plasma urate levels following treatment with multiple doses of pegloticase (polyethylene glycol-conjugated uricase) in patients with treatment-failure gout: results of a phase II randomized study. Arthritis Rheum2008; 58:2882–2891.
Jordan KM, Cameron JS, Snaith M, et al; British Society for Rheumatology and British Health Professionals in Rheumatology Standards, Guidelines and Audit Working Group (SGAWG). British Society for Rheumatology and British Health Professionals in Rheumatology guideline for the management of gout. Rheumatology (Oxford)2007; 46:1372–1374.
Perez-Ruiz F, Alonso-Ruiz A, Calabozo M, Herrero-Beites A, García-Erauskin G, Ruiz-Lucea E. Efficacy of allopurinol and benzbromarone for the control of hyperuricaemia. A pathogenic approach to the treatment of primary chronic gout. Ann Rheum Dis1998; 57:545–549.
Schumacher HR, Becker MA, Wortmann RL, et al. Effects of febuxostat versus allopurinol and placebo in reducing serum urate in subjects with hyperuricemia and gout: a 28-week, phase III, randomized, double-blind, parallel-group trial. Arthritis Rheum2008; 59:1540–1548.
Perez-Ruiz F, Calabozo M, Pijoan JI, Herrero-Beites AM, Ruibal A. Effect of urate-lowering therapy on the velocity of size reduction of tophi in chronic gout. Arthritis Rheum2002; 47:356–360.
Hande KR, Noone RM, Stone WJ. Severe allopurinol toxicity. Description and guidelines for prevention in patients with renal insufficiency. Am J Med1984; 76:47–56.
Perez-Ruiz F, Hernando I, Villar I, Nolla JM. Correction of allopurinol dosing should be based on clearance of creatinine, but not plasma creatinine levels: another insight to allopurinol-related toxicity. J Clin Rheumatol2005; 11:129–133.
Hung SI, Chung WH, Liou LB, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A2005; 102:4134–4139.
Vázquez-Mellado J, Morales EM, Pacheco-Tena C, Burgos-Vargas R. Relation between adverse events associated with allopurinol and renal function in patients with gout. Ann Rheum Dis2001; 60:981–983.
Dalbeth N, Kumar S, Stamp L, Gow P. Dose adjustment of allopurinol according to creatinine clearance does not provide adequate control of hyperuricemia in patients with gout. J Rheumatol2006; 33:1646–1650.
Silverberg MS, Mallela R, Lesse AJ, Bonner MR, Baer AN, Li C. Allopurinol hypersensitivity reactions: a case-control study of the role of renal dosing (abstract). Arthritis Rheum2009; 60(suppl 10):1106.
Stamp LK, O'Donnell JL, Zhang M, et al. Using allopurinol above the dose based on creatinine clearance is effective and safe in chronic gout, including in those with renal impairment. Arthritis Rhem2010; doi:10.1002/art.30119. E-pub ahead of print. Accessed 10/29/2010.
Zhang W, Doherty M, Bardin T, et al; EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis2006; 65:1312–1324.
Okamoto K, Eger BT, Nishino T, Kondo S, Pai EF, Nishino T. An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J Biol Chem2003; 278:1848–1855.
Khosravan R, Grabowski BA, Wu JT, Joseph-Ridge N, Vernillet L. Pharmacokinetics, pharmacodynamics and safety of febuxostat, a non-purine selective inhibitor of xanthine oxidase, in a dose escalation study in healthy subjects. Clin Pharmacokinet2006; 45:821–841.
Reinders MK, Haagsma C, Jansen TL, et al. A randomised controlled trial on the efficacy and tolerability with dose escalation of allopurinol 300–600 mg/day versus benzbromarone 100–200 mg/day in patients with gout. Ann Rheum Dis2009; 68:892–897.
Mazzali M, Hughes J, Kim YG, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension2001; 38:1101–1106.
Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med2008; 359:1811–1821.
Beck LH. Requiem for gouty nephropathy. Kidney Int1986; 30:280–287.
Tomita M, Mizuno S, Yamanaka H, et al. Does hyperuricemia affect mortality? A prospective cohort study of Japanese male workers. J Epidemiol2000; 10:403–409.
Kang DH, Nakagawa T. Uric acid and chronic renal disease: possible implication of hyperuricemia on progression of renal disease. Semin Nephrol2005; 25:43–49.
Iseki K, Oshiro S, Tozawa M, Iseki C, Ikemiya Y, Takishita S. Significance of hyperuricemia on the early detection of renal failure in a cohort of screened subjects. Hypertens Res2001; 24:691–697.
Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med1987; 82:421–426.
Siu YP, Leung KT, Tong MK, Kwan TH. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis2006; 47:51–59.
Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA2008; 300:924–932.
Owing to concerns about using colchicine and nonsteroidal anti-inflammatory drugs (NSAIDs) in patients with CKD, glucocorticoids (local injections or systemic therapy) are often used to treat acute attacks. Corticotropin (Acthar), anti-tumor necrosis factor agents, and interleukin 1 antagonists are effective but expensive.
Colchicine can be used in low doses as prophylaxis, with caution and appropriate monitoring. NSAIDs should be avoided, and glucocorticoids may not be effective for this purpose.
Whether the dosage of allopurinol should be lower in patients with CKD remains controversial. We start with a low dose and slowly increase it, with a goal serum urate level of less than 6.0 mg/dL.
Febuxostat (Uloric), like allopurinol, is a xanthine oxidase inhibitor, but the elimination of the active drug is not by the kidney. Nevertheless, we try allopurinol in escalating doses first, due to major cost differences.
Vitamin D is viewed as a promising supplement by the medical, public health, and lay communities, potentially offering many health benefits. But enthusiasm for a new intervention too often gets far ahead of the evidence, as was the case with beta-carotene, selenium, folic acid, and vitamins C and E.
Despite the enthusiasm for vitamin D, there have been no large-scale primary prevention trials that have had either cardiovascular disease or cancer as a prespecified primary outcome. Previous randomized trials of vitamin D have focused primarily on osteoporosis, fracture, falls, and physical function. Although the investigators often reported their findings on vitamin D and cardiovascular disease or cancer, these outcomes were generally secondary or tertiary end points that were not prespecified. These studies should be viewed as hypothesis-generating rather than hypothesis-testing. The increasing prevalence of use of vitamin D supplements underscores the need for rigorous and conclusive evidence from randomized clinical trials that have cardiovascular disease and cancer as primary outcomes.
This article will explain the rationale for a large-scale, randomized clinical trial to evaluate the role of vitamin D in the prevention of cardiovascular disease and cancer. It will also describe the biological mechanisms and currently available evidence relating vitamin D to potential health benefits. Finally, the design, dosage considerations, and logistics of the Vitamin D and Omega-3 Trial (VITAL) will be presented.
EVIDENCE IS MOUNTING FOR VITAMIN D’S BIOLOGICAL IMPORTANCE
Vitamin D is undoubtedly important to health: not only is it produced endogenously, but at least 500 genes have been identified with vitamin D response elements. The vitamin D receptor is found in nearly all cells in the body, and the 1-alpha-hydroxylase enzyme is present in many tissues. Some studies suggest that almost 10% of the human genome may be at least partially regulated by vitamin D.
From Hajjar V, et al. Does vitamin D deficiency play a role in the pathogenesis of chronic heart failure? Do supplements improve survival? Cleve Clin J Med 2010; 77:290–293.
Figure 1.
Vitamin D is a prohormone, and people obtain it both endogenously and exogenously (Figure 1). With exposure to ultraviolet B light, 7-dehydrocholesterol in the skin converts to vitamin D3. We also obtain it through diet or supplements. The plant form (vitamin D2) and the animal form (vitamin D3) undergo 25-hydroxylation in the liver. Then, 1-alpha-hydroxylase converts the 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3, primarily in the kidney. Increasing evidence shows that 1-alpha-hydroxylase is present in many other cells and tissues, and that 1,25-dihydroxyvitamin D3 may be locally produced and possibly even have autocrine effects (acting on surface receptors of the same cell it is secreted by) and paracrine effects (acting on adjacent cells).
Although we know vitamin D is important, what our optimal intake and our blood level of 25-hydroxyvitamin D3 should be are key unknowns.
RECOMMENDATIONS FOR VITAMIN D INTAKE
During winter, late fall, and early spring, people who live above the 37th parallel (geographically, about one-half of the contiguous United States) do not get enough ultraviolet B energy from the sun to make all the vitamin D they need, even if they spend several hours outside every day. In addition, dark skin pigmentation serves as a sun block, as do sunscreens.
The Institute of Medicine (IOM) provided guidelines for vitamin D intake in 1997 and, most recently, in 2010. However, these guidelines are based on the amount of vitamin D required for bone health and do not address the amount that may be of benefit for prevention of cancer and cardiovascular disease. The latter outcomes are not addressed because the IOM committee believed that evidence was insufficient to determine the role of vitamin D in the prevention of cardiovascular disease, cancer, and other chronic diseases. Thus, current IOM guidelines, which generally recommend less than 1,000 IU of vitamin D daily, are relevant to bone health but not necessarily to other health outcomes. More research is needed to understand whether the guidelines should be modified for the prevention of other chronic diseases.
Moreover, whether or not everyone should be screened for 25-hydroxyvitamin D3 blood levels is controversial. Most experts agree that a level less than 20 ng/mL is deficient or insufficient. Conversely, potentially harmful are levels 150 ng/mL or more (> 375 nmol/L), which entail the risk of hypercalcemia, vascular soft tissue calcification, and hyperphosphatemia.
People do not reach toxic levels with ultraviolet light exposure because the amount of 25-hydroxyvitamin D3 synthesis is well regulated. Dietary supplements, however, can bring about toxic levels, and patients taking high doses need to be monitored carefully. The level that should be considered optimal is controversial and requires further study.
RISK FACTORS FOR LOW VITAMIN D LEVELS
Risk factors for low vitamin D levels include older age, living in northern latitudes, sun avoidance, dark skin pigmentation, obesity, low dietary intake, and various medical conditions, especially malabsorption syndromes. Some of these are also risk factors for cardiovascular disease, cancer, and other chronic diseases, and potentially confound outcomes in many studies. Older age, which is usually adjusted for in multivariate models, is important to recognize as a major risk factor for vitamin D deficiency, owing to reduced absorption and synthesis, less time outdoors, and low dietary intake.
Wearing sunscreen decreases the synthesis of vitamin D in the skin, but because ultra-violet light has been clearly classified as a carcinogen, it is a not advisable to increase sun exposure for the sake of increasing vitamin D levels. That is a poor trade-off, given the high incidence rate of skin cancer and the adverse effects of solar radiation on skin aging.
Obesity is a risk factor for vitamin D deficiency because vitamin D is fat-soluble and becomes sequestered in fat tissue. Vitamin D may also play a role in the differentiation of adipocytes and may affect their function. In observational studies, it is very important for researchers to adjust for body mass index, physical activity (which may be correlated with more time outdoors), and other potential confounders in their analyses.
HOW VITAMIN D MAY LOWER CANCER RISK
Because of the important effect of vitamin D in regulating cell differentiation and cell growth, there are multiple ways that it may affect cancer risk. Laboratory, cell culture, and animal studies suggest that vitamin D may lower cancer risk by inhibiting cell proliferation, angiogenesis, metastasis, and inflammation and inducing apoptosis and cellular differentiation. Several of these mechanisms are also relevant to atherosclerosis and cardiovascular disease. Although VITAL is addressing the role of vitamin D in preventing both cancer and cardiovascular disease, the remainder of this article will focus on cardiovascular outcomes.
HOW VITAMIN D MAY REDUCE CARDIOVASCULAR RISK
Vitamin D may lower cardiovascular risk via several mechanisms:
Inhibiting inflammation. Vitamin D has a powerful immunomodulatory effect: laboratory studies show that it inhibits prostaglandin and cyclooxygenase 2 pathways, reduces matrix metalloproteinase 9 and several proinflammatory cytokines, and increases interleukin 10, all of which result in suppressed inflammation.1
Inhibiting vascular muscle proliferation and vascular calcification. Animal studies indicate that in moderate doses vitamin D decreases calcium cellular influx and increases matrix Gla protein, which inhibits vascular smooth muscle proliferation and vascular calcification. These protective effects contrast with the hypercalcemia associated with a high intake of vitamin D, especially in the context of renal failure or other risk factors, which may lead to increased vascular calcification.1
Regulates blood pressure. Vitamin D decreases renin gene expression and the synthesis of renin, which reduces activity of the renin-angiotensin-aldosterone system, leading to a reduction of blood pressure and a favorable effect on volume homeostasis.1
Regulates glucose metabolism. Limited evidence shows that vitamin D may increase insulin sensitivity and regulate glucose metabolism.1
Vitamin D and cardiac hypertrophy
The vitamin D receptor is present in virtually all tissues, including cardiac myocytes and endothelial cells. Animals with vitamin D deficiency have higher blood pressures, and animals genetically altered to have no vitamin D receptors (knock-out models) develop left ventricular hypertrophy and heart failure.
Animals genetically altered to have no 1-alpha-hydroxylase (so that the most active form of vitamin D is not made) also develop left ventricular hypertrophy. They can be rescued by the administration of 1,25-dihydroxy vitamin D3.1
These findings are consistent with what is observed in patients with end-stage renal disease, who produce very little 1,25-dihydroxyvitamin D3: they often develop left ventricular hypertrophy, diastolic heart failure, atherosclerosis, and vascular calcification.
EVIDENCE FOR CARDIOVASCULAR DISEASE REDUCTION
Wang et al1 recently reviewed available prospective cohort and randomized clinical trials from 1966 to 2009 that examined vitamin D or calcium supplementation and cardiovascular disease. Comparing people with the lowest to the highest levels of serum 25-hydroxyvitamin D3 indicated that a low level is a risk factor for coronary artery disease and cardiovascular death. Unfortunately, most studies were not designed to assess primary effects on cardiovascular outcomes, and so have many potential confounders.
Prospective observational studies
Observational studies suggest that vitamin D deficiency is associated with an increased risk of cardiovascular disease. Some examples:
The Framingham Offspring Study2 followed 1,739 men and women with a mean age of 59 for 5.4 years. The study compared the incidence of cardiovascular events in those with a serum 25-dihydroxyvitamin D level of at least 37.5 nmol/L vs those with lower levels. The risk of cardiovascular disease was 1.62 times higher in those with the lowest levels of vitamin D, a statistically significant difference. However, a threshold effect was apparent (discussed below).
The Health Professionals Follow-up Study3 prospectively evaluated more than 18,000 men ages 40 to 75 for 10 years. The study compared men with a low serum level of vitamin D (< 37.5 nmol/L) to those with a more optimal level (> 75 nmol/L). The incidence of cardiovascular events was 2.09 times higher in men with low levels of vitamin D, a difference that was statistically significant.
The Third National Health and Nutrition Examination Survey (NHANES III) included data for more than 13,300 men and women age 20 years and older. Using a cohort that was followed for 8.7 years, Melamed et al4 compared the quartile with the lowest serum vitamin D level (< 44.4 nmol/L) against the quartile with the highest level (> 80.1 nmol/L). The associations were modest: those with low levels had a 1.20-times higher rate of death from cardiovascular disease and a statistically significant 1.26-times higher rate of death from all causes.
Randomized clinical trials
A meta-analysis of 18 randomized trials5 of vitamin D supplementation (300–2,000 IU/day, mean 528 IU/day vs placebo), including 57,311 participants, evaluated the rate of death from all causes and found a modest but significant reduction in risk (relative risk 0.93, 95% confidence interval [CI] 0.87–0.99). These were generally trials looking at fracture rates or physical performance, and a dose-response relationship was not evident. A recent systematic review of randomized controlled trials of vitamin D1 that included cardiovascular disease as a secondary outcome found a pooled relative risk for cardiovascular disease of 0.90 (95% CI 0.77–1.05) for vitamin D supplementation compared with placebo and 1.04 (95% CI 0.92–1.18) for combination vitamin D plus calcium supplementation vs placebo.1 Two individual trials are discussed below.
Trivedi et al6 randomized 2,686 British men and women to vitamin D3 100,000 IU given every 4 months over 5 years (equivalent to 800 IU/day) or placebo. The relative risk of cardiovascular events was 0.90 (95% CI 0.77–1.06) and of cardiovascular deaths 0.84 (95% CI 0.65–1.10). Although the results were promising, the trial was designed to assess fracture risk and was not large enough for the differences in cardiovascular outcomes to reach statistical significance.
The Women’s Health Initiative,7,8 which included 36,282 postmenopausal women aged 50 to 79, tested combined vitamin D3 (400 IU/day) with calcium (1,000 mg/day) vs placebo. No benefit was seen for preventing coronary events or stroke, which may be due to the low dosage of vitamin D. The hazard ratio for coronary disease was 1.04 (0.92–1.18). Regarding mortality, the hazard ratio for cardiovascular death was 0.92, for cerebrovascular death 0.89, for cancer death 0.89, and for other deaths 0.95. None of these hazard ratios reached statistical significance.
MORE MAY NOT BE BETTER
As is probably true for everything in biological systems, there apparently is an optimal level of intake to meet vitamin D needs.
The Framingham Offspring Study,2 which found a higher risk with vitamin D deficiency, also found a suggestion of a threshold. Participants who had levels of 50 to 65 nmol/L had the lowest risk. Higher levels did not confer lower risk and even suggested a slight upturn.
Evidence from the Women’s Health Initiative8 also indicates that high dosages may not be better than moderate dosages. The meta-analysis of vitamin D and all-cause mortality5 found a relative risk of 0.93, but one of the largest studies in that meta-analysis tested only 400 IU a day and found a similar relative risk of 0.91 (95% confidence interval, 0.83–1.01).
Moreover, the NHANES study found that with increasing serum 25-hydroxyvitamin D3 levels, the risk of all-cause mortality fell until about 100 nmol/L, but then plateaued and even increased with higher serum levels.4
VITAL: STUDY DESIGN AND LOGISTICS
In VITAL, the investigators aim to recruit 20,000 healthy men (age 60 and older) and women (65 and older) who are representative of the US population (www.vitalstudy.org). Because it is a primary prevention trial, people with a known history of cardiovascular disease or cancer will be excluded. Participants will be randomized to receive either 2,000 IU of vitamin D3 per day or placebo. Each group will be further randomized to receive either 1 g per day of fish oil (combined eicosapentaenoic acid [EPA] and docosahexaenoic acid [DHA]) or placebo. The mean treatment period will be 5 years. Recruitment began in early 2010.
Blood will be collected in about 80% (ideally 100%) of participants, with follow-up blood collection in at least 2,000.
Primary aims of the trial are to test whether vitamin D3 and the omega-3 fatty acids reduce the risk of total cancer and major cardiovascular events (a composite of myocardial infarction, stroke, and death due to cardiovascular events).
Secondary aims are to test whether these agents lower the risk of:
Site-specific cancer, including colorectal, breast, and prostate cancer, and the total cancer mortality rate
An expanded composite outcome including myocardial infarction, stroke, cardiovascular death, coronary artery bypass grafting, percutaneous coronary intervention, and its individual components.
Tertiary aims are to explore whether vitamin D3 and omega-3 fatty acids have additive effects on the primary and secondary end points. The trial will also explore whether the effects of vitamin D3 and omega-3 fatty acids on cancer and cardiovascular disease vary by baseline blood levels of these nutrients, and whether race, skin pigmentation, or body mass index modify the effects of vitamin D3.
Ancillary studies will assess the effect of the interventions on risk of diabetes, hypertension, cognitive decline, depression, fracture, infections, respiratory disorders, and autoimmune diseases. The primary sponsor of this trial is the National Cancer Institute, and the secondary sponsor is the National Heart, Lung and Blood Institute. Other institutes and agencies also are cosponsors of the study.
The timing of VITAL is optimal
There is a limited window of opportunity for conducting a randomized clinical trial: the evidence must be strong enough to justify mounting a very large trial with enough power to look at cardiovascular events and cancer, but the evidence must not be so strong that it would be unethical to have a placebo group. Thus, there must be a state of equipoise. Our trial allows the study population to have a background intake of vitamin D that is currently recommended by national guidelines. Therefore, even the placebo group should have adequate intake of vitamin D.
The growing use of vitamin D supplementation by the public underscores the need for conclusive evidence of its benefits and risks. No previous large-scale randomized clinical trial has tested moderate to high doses of vitamin D for the primary prevention of cancer and cardiovascular disease.
Setting the dosage
VITAL set the vitamin D3 dosage at 2,000 IU per day (50 μg/day), which is designed to provide the best balance of efficacy and safety. As a general rule, each microgram of vitamin D3 is expected to raise the serum 25-hydroxyvitamin D3 level about 1 nmol/L, although the response is not linear: if baseline levels are lower, the increase is greater. In the United States, people commonly have a baseline level of about 40 nmol/L, so we expect that levels of people treated in the study will reach about 90 nmol/L (range 75–100 nmol/L), about 35 to 50 nmol/L higher than in the placebo group.
The target range of 75 to 100 nmol/L is the level at which greatest efficacy has been suggested in observational studies. Previous randomized trials of vitamin D have not tested high enough doses to achieve this level of 25-hydroxyvitamin D3. VITAL will test whether reaching this serum level lowers the risk of cardiovascular disease, cancer, and other chronic diseases. This level may be associated with benefit and has minimal risk of hypercalcemia. Risk of hypercalcemia may be present in participants with an occult chronic granulomatous condition such as sarcoidosis or Wegener granulomatosis, in which activated macrophages synthesize 1,25-dihydroxyvitamin D3. These conditions are very rare, however, and the risk of hypercalcemia in the trial is exceedingly low.
VITAL participants will also be randomized to take placebo or 1 g per day of combined EPA and DHA, about 5 to 10 times more than most Americans consume.
Nationwide recruitment among senior citizens
We aim to recruit 20,000 people (10,000 men and 10,000 women) nationwide who are willing, eligible, and compliant (ie, who take more than two-thirds of study pills during a 3-month placebo “run-in” phase of the trial). The trial aims to enroll 40,000 in the run-in period, and 20,000 will be randomized. To get this many participants, we will send invitational mailings and screening questionnaires to at least 2.5 million people around the United States, with mailing lists selected by age—ie, members of the American Association of Retired Persons, health professionals, teachers, and subscription lists for selected magazines. A pilot study in 5,000 people has indicated that recruiting and randomizing 20,000 participants via large mailings should be possible.
The trial is expected to be extremely cost-effective because it will be conducted largely by mail. Medication will be mailed in calendar blister packs. Participants report outcomes, which are then confirmed by medical record review. The Centers for Medicare and Medicaid Services and the National Death Index will also be used to ascertain outcomes.
We hope to recruit a more racially diverse study population than is typically seen in US trials: 63% (12,620) whites, 25% (5,000) African Americans, 7% (1,400) Hispanics, 2.5% (500) Asians, 2% (400) American Indians and Alaska natives, and 0.4% (80) native Hawaiian and Pacific Islanders.
Eligibility criteria ensure primary prevention is tested
To enter the study, men must be at least 60 years old and women at least 65. At a minimum, a high school education is required due to the detailed forms and questionnaires to be completed. Because this is a primary prevention trial, anyone with a history of cancer (except nonmelanoma skin cancer) or cardiovascular disease (including myocardial infarction, stroke, or coronary revascularization) will be excluded, as will anyone with a history of kidney stones, renal failure or dialysis, hypercalcemia, hypoparathyroidism or hyperparathyroidism, severe liver disease (eg, cirrhosis), sarcoidosis, tuberculosis, or other granulomatous disease. People with an allergy to fish will also be excluded.
We do not expect that those in the placebo group will develop vitamin D deficiency due to their participation in the study. The trial will allow a background intake in the study population of up to 800 IU of vitamin D and 1,200 mg of calcium per day in supplements. Assuming they also get about 200 IU of vitamin D in the diet, the background intake in the placebo group may be close to 1,000 IU of vitamin D. Assuming that the active treatment group has a similar background intake, their total intake will be about 3,000 IU per day (about 1,000 IU/day from background intake plus 2,000 IU/day from the intervention).
Cohort power sufficient to see effect in 5 years
The trial is expected to have sufficient power to evaluate cardiovascular disease and cancer end points as primary outcomes during 5 years of follow-up. The trial is designed to have a power of 91% to 92% to detect a relative risk of 0.85 for the primary cancer end point of total cancer incidence and 0.80 for the cardiovascular disease end point of myocardial infarction, stroke, and cardiovascular mortality. Power will be even greater for the expanded composite outcome for cardiovascular disease.
Ancillary studies
Ancillary studies include evaluating the interventions’ role in preventing diabetes and glucose intolerance, hypertension, heart failure, atrial fibrillation, cognitive decline, mood disorders, osteoporosis and fractures, asthma and respiratory diseases, infections, macular degeneration, rheumatoid arthritis, systemic lupus erythematosus, and a composite of autoimmune diseases. Imaging studies also are planned, including dual energy x-ray absorptiometry, mammographic density, and non-invasive vascular imaging (carotid intima medial thickness, coronary calcium measurements, and two-dimensional echocardiography to assess cardiac function).
Several biomarker and genetic studies will also be carried out. We intend to perform genetic studies on most of the study population to evaluate gene variants in the vitamin D receptor, vitamin D binding protein, and other vitamin-D-related genes that may contribute to lower baseline levels of 25-hydroxyvitamin D3 or different responses to the interventions.
Clinical and Translational Science Center visits are planned to provide more detailed assessments of 1,000 participants, including blood pressure measurements, height, weight, waist circumference, other anthropometric measurements, a 2-hour glucose tolerance test, a fasting blood collection, hemoglobin A1c measurements, spirometry, and assessment of physical performance, strength, frailty, cognitive function, mood, and depression. Dual-energy x-ray absorptiometry and noninvasive vascular imaging studies are also planned for those visits.
References
Wang L, Manson JE, Song Y, Sesso HD. Systematic review: vitamin D and calcium supplementation in prevention of cardiovascular events. Ann Intern Med2010; 152:315–323.
Wang TJ, Pencina MJ, Booth SL, et al. Vitamin D deficiency and risk of cardiovascular disease. Circulation2008; 117:503–511.
Giovannucci E, Liu Y, Hollis BW, Rimm EB. 25-Hydroxyvitamin D and risk of myocardial infarction in men: a prospective study. Arch Intern Med2008; 168:1174–1180.
Melamed ML, Michos ED, Post W, Astor B. 25-Hydroxyvitamin D levels and the risk of mortality in the general population. Arch Intern Med2008; 168:1629–1637.
Autier P, Gandini S. Vitamin D supplementation and total mortality: a meta-analysis of randomized controlled trials. Arch Intern Med2007; 167:1730–1737.
Trivedi DP, Doll R, Khaw KT. Effect of four monthly oral vitamin D3 (cholecalciferol) supplementation on fractures and mortality in men and women living in the community: randomised double blind controlled trial. BMJ2003; 326:469.
Hsia J, Heiss G, Ren H, et al; Women’s Health Initiative Investigators. Calcium/vitamin D supplementation and cardiovascular events. Circulation2007; 115:846–854.
LaCroix AZ, Kotchen J, Anderson G, et al. Calcium plus vitamin D supplementation and mortality in postmenopausal women: the Women’s Health Initiative calcium-vitamin D randomized controlled trial. J Gerontol A Biol Sci Med Sci2009; 64:559–567.
Joann E. Manson, MD, DrPH, FAHA Chief, Division of Preventive Medicine; Co-Director, Connors Center for Women’s Health and Gender Biology, Brigham and Women’s Hospital; Professor of Medicine, Harvard Medical School, Boston, MA; Principal Investigator, Vitamin D and Omega-3 Trial (VITAL)
Address: JoAnn E. Manson, MD, DrPH, Division of Preventive Medicine, Brigham and Women’s Hospital, Harvard Medical School, 900 Commonwealth Avenue, Boston, MA 02215; email [email protected]
Dr. Manson has received funding from the National Institutes of Health to conduct a large-scale randomized trial of vitamin D and omega-3 fatty acids for the prevention of cardiovascular disease and cancer.
Joann E. Manson, MD, DrPH, FAHA Chief, Division of Preventive Medicine; Co-Director, Connors Center for Women’s Health and Gender Biology, Brigham and Women’s Hospital; Professor of Medicine, Harvard Medical School, Boston, MA; Principal Investigator, Vitamin D and Omega-3 Trial (VITAL)
Address: JoAnn E. Manson, MD, DrPH, Division of Preventive Medicine, Brigham and Women’s Hospital, Harvard Medical School, 900 Commonwealth Avenue, Boston, MA 02215; email [email protected]
Dr. Manson has received funding from the National Institutes of Health to conduct a large-scale randomized trial of vitamin D and omega-3 fatty acids for the prevention of cardiovascular disease and cancer.
Author and Disclosure Information
Joann E. Manson, MD, DrPH, FAHA Chief, Division of Preventive Medicine; Co-Director, Connors Center for Women’s Health and Gender Biology, Brigham and Women’s Hospital; Professor of Medicine, Harvard Medical School, Boston, MA; Principal Investigator, Vitamin D and Omega-3 Trial (VITAL)
Address: JoAnn E. Manson, MD, DrPH, Division of Preventive Medicine, Brigham and Women’s Hospital, Harvard Medical School, 900 Commonwealth Avenue, Boston, MA 02215; email [email protected]
Dr. Manson has received funding from the National Institutes of Health to conduct a large-scale randomized trial of vitamin D and omega-3 fatty acids for the prevention of cardiovascular disease and cancer.
Vitamin D is viewed as a promising supplement by the medical, public health, and lay communities, potentially offering many health benefits. But enthusiasm for a new intervention too often gets far ahead of the evidence, as was the case with beta-carotene, selenium, folic acid, and vitamins C and E.
Despite the enthusiasm for vitamin D, there have been no large-scale primary prevention trials that have had either cardiovascular disease or cancer as a prespecified primary outcome. Previous randomized trials of vitamin D have focused primarily on osteoporosis, fracture, falls, and physical function. Although the investigators often reported their findings on vitamin D and cardiovascular disease or cancer, these outcomes were generally secondary or tertiary end points that were not prespecified. These studies should be viewed as hypothesis-generating rather than hypothesis-testing. The increasing prevalence of use of vitamin D supplements underscores the need for rigorous and conclusive evidence from randomized clinical trials that have cardiovascular disease and cancer as primary outcomes.
This article will explain the rationale for a large-scale, randomized clinical trial to evaluate the role of vitamin D in the prevention of cardiovascular disease and cancer. It will also describe the biological mechanisms and currently available evidence relating vitamin D to potential health benefits. Finally, the design, dosage considerations, and logistics of the Vitamin D and Omega-3 Trial (VITAL) will be presented.
EVIDENCE IS MOUNTING FOR VITAMIN D’S BIOLOGICAL IMPORTANCE
Vitamin D is undoubtedly important to health: not only is it produced endogenously, but at least 500 genes have been identified with vitamin D response elements. The vitamin D receptor is found in nearly all cells in the body, and the 1-alpha-hydroxylase enzyme is present in many tissues. Some studies suggest that almost 10% of the human genome may be at least partially regulated by vitamin D.
From Hajjar V, et al. Does vitamin D deficiency play a role in the pathogenesis of chronic heart failure? Do supplements improve survival? Cleve Clin J Med 2010; 77:290–293.
Figure 1.
Vitamin D is a prohormone, and people obtain it both endogenously and exogenously (Figure 1). With exposure to ultraviolet B light, 7-dehydrocholesterol in the skin converts to vitamin D3. We also obtain it through diet or supplements. The plant form (vitamin D2) and the animal form (vitamin D3) undergo 25-hydroxylation in the liver. Then, 1-alpha-hydroxylase converts the 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3, primarily in the kidney. Increasing evidence shows that 1-alpha-hydroxylase is present in many other cells and tissues, and that 1,25-dihydroxyvitamin D3 may be locally produced and possibly even have autocrine effects (acting on surface receptors of the same cell it is secreted by) and paracrine effects (acting on adjacent cells).
Although we know vitamin D is important, what our optimal intake and our blood level of 25-hydroxyvitamin D3 should be are key unknowns.
RECOMMENDATIONS FOR VITAMIN D INTAKE
During winter, late fall, and early spring, people who live above the 37th parallel (geographically, about one-half of the contiguous United States) do not get enough ultraviolet B energy from the sun to make all the vitamin D they need, even if they spend several hours outside every day. In addition, dark skin pigmentation serves as a sun block, as do sunscreens.
The Institute of Medicine (IOM) provided guidelines for vitamin D intake in 1997 and, most recently, in 2010. However, these guidelines are based on the amount of vitamin D required for bone health and do not address the amount that may be of benefit for prevention of cancer and cardiovascular disease. The latter outcomes are not addressed because the IOM committee believed that evidence was insufficient to determine the role of vitamin D in the prevention of cardiovascular disease, cancer, and other chronic diseases. Thus, current IOM guidelines, which generally recommend less than 1,000 IU of vitamin D daily, are relevant to bone health but not necessarily to other health outcomes. More research is needed to understand whether the guidelines should be modified for the prevention of other chronic diseases.
Moreover, whether or not everyone should be screened for 25-hydroxyvitamin D3 blood levels is controversial. Most experts agree that a level less than 20 ng/mL is deficient or insufficient. Conversely, potentially harmful are levels 150 ng/mL or more (> 375 nmol/L), which entail the risk of hypercalcemia, vascular soft tissue calcification, and hyperphosphatemia.
People do not reach toxic levels with ultraviolet light exposure because the amount of 25-hydroxyvitamin D3 synthesis is well regulated. Dietary supplements, however, can bring about toxic levels, and patients taking high doses need to be monitored carefully. The level that should be considered optimal is controversial and requires further study.
RISK FACTORS FOR LOW VITAMIN D LEVELS
Risk factors for low vitamin D levels include older age, living in northern latitudes, sun avoidance, dark skin pigmentation, obesity, low dietary intake, and various medical conditions, especially malabsorption syndromes. Some of these are also risk factors for cardiovascular disease, cancer, and other chronic diseases, and potentially confound outcomes in many studies. Older age, which is usually adjusted for in multivariate models, is important to recognize as a major risk factor for vitamin D deficiency, owing to reduced absorption and synthesis, less time outdoors, and low dietary intake.
Wearing sunscreen decreases the synthesis of vitamin D in the skin, but because ultra-violet light has been clearly classified as a carcinogen, it is a not advisable to increase sun exposure for the sake of increasing vitamin D levels. That is a poor trade-off, given the high incidence rate of skin cancer and the adverse effects of solar radiation on skin aging.
Obesity is a risk factor for vitamin D deficiency because vitamin D is fat-soluble and becomes sequestered in fat tissue. Vitamin D may also play a role in the differentiation of adipocytes and may affect their function. In observational studies, it is very important for researchers to adjust for body mass index, physical activity (which may be correlated with more time outdoors), and other potential confounders in their analyses.
HOW VITAMIN D MAY LOWER CANCER RISK
Because of the important effect of vitamin D in regulating cell differentiation and cell growth, there are multiple ways that it may affect cancer risk. Laboratory, cell culture, and animal studies suggest that vitamin D may lower cancer risk by inhibiting cell proliferation, angiogenesis, metastasis, and inflammation and inducing apoptosis and cellular differentiation. Several of these mechanisms are also relevant to atherosclerosis and cardiovascular disease. Although VITAL is addressing the role of vitamin D in preventing both cancer and cardiovascular disease, the remainder of this article will focus on cardiovascular outcomes.
HOW VITAMIN D MAY REDUCE CARDIOVASCULAR RISK
Vitamin D may lower cardiovascular risk via several mechanisms:
Inhibiting inflammation. Vitamin D has a powerful immunomodulatory effect: laboratory studies show that it inhibits prostaglandin and cyclooxygenase 2 pathways, reduces matrix metalloproteinase 9 and several proinflammatory cytokines, and increases interleukin 10, all of which result in suppressed inflammation.1
Inhibiting vascular muscle proliferation and vascular calcification. Animal studies indicate that in moderate doses vitamin D decreases calcium cellular influx and increases matrix Gla protein, which inhibits vascular smooth muscle proliferation and vascular calcification. These protective effects contrast with the hypercalcemia associated with a high intake of vitamin D, especially in the context of renal failure or other risk factors, which may lead to increased vascular calcification.1
Regulates blood pressure. Vitamin D decreases renin gene expression and the synthesis of renin, which reduces activity of the renin-angiotensin-aldosterone system, leading to a reduction of blood pressure and a favorable effect on volume homeostasis.1
Regulates glucose metabolism. Limited evidence shows that vitamin D may increase insulin sensitivity and regulate glucose metabolism.1
Vitamin D and cardiac hypertrophy
The vitamin D receptor is present in virtually all tissues, including cardiac myocytes and endothelial cells. Animals with vitamin D deficiency have higher blood pressures, and animals genetically altered to have no vitamin D receptors (knock-out models) develop left ventricular hypertrophy and heart failure.
Animals genetically altered to have no 1-alpha-hydroxylase (so that the most active form of vitamin D is not made) also develop left ventricular hypertrophy. They can be rescued by the administration of 1,25-dihydroxy vitamin D3.1
These findings are consistent with what is observed in patients with end-stage renal disease, who produce very little 1,25-dihydroxyvitamin D3: they often develop left ventricular hypertrophy, diastolic heart failure, atherosclerosis, and vascular calcification.
EVIDENCE FOR CARDIOVASCULAR DISEASE REDUCTION
Wang et al1 recently reviewed available prospective cohort and randomized clinical trials from 1966 to 2009 that examined vitamin D or calcium supplementation and cardiovascular disease. Comparing people with the lowest to the highest levels of serum 25-hydroxyvitamin D3 indicated that a low level is a risk factor for coronary artery disease and cardiovascular death. Unfortunately, most studies were not designed to assess primary effects on cardiovascular outcomes, and so have many potential confounders.
Prospective observational studies
Observational studies suggest that vitamin D deficiency is associated with an increased risk of cardiovascular disease. Some examples:
The Framingham Offspring Study2 followed 1,739 men and women with a mean age of 59 for 5.4 years. The study compared the incidence of cardiovascular events in those with a serum 25-dihydroxyvitamin D level of at least 37.5 nmol/L vs those with lower levels. The risk of cardiovascular disease was 1.62 times higher in those with the lowest levels of vitamin D, a statistically significant difference. However, a threshold effect was apparent (discussed below).
The Health Professionals Follow-up Study3 prospectively evaluated more than 18,000 men ages 40 to 75 for 10 years. The study compared men with a low serum level of vitamin D (< 37.5 nmol/L) to those with a more optimal level (> 75 nmol/L). The incidence of cardiovascular events was 2.09 times higher in men with low levels of vitamin D, a difference that was statistically significant.
The Third National Health and Nutrition Examination Survey (NHANES III) included data for more than 13,300 men and women age 20 years and older. Using a cohort that was followed for 8.7 years, Melamed et al4 compared the quartile with the lowest serum vitamin D level (< 44.4 nmol/L) against the quartile with the highest level (> 80.1 nmol/L). The associations were modest: those with low levels had a 1.20-times higher rate of death from cardiovascular disease and a statistically significant 1.26-times higher rate of death from all causes.
Randomized clinical trials
A meta-analysis of 18 randomized trials5 of vitamin D supplementation (300–2,000 IU/day, mean 528 IU/day vs placebo), including 57,311 participants, evaluated the rate of death from all causes and found a modest but significant reduction in risk (relative risk 0.93, 95% confidence interval [CI] 0.87–0.99). These were generally trials looking at fracture rates or physical performance, and a dose-response relationship was not evident. A recent systematic review of randomized controlled trials of vitamin D1 that included cardiovascular disease as a secondary outcome found a pooled relative risk for cardiovascular disease of 0.90 (95% CI 0.77–1.05) for vitamin D supplementation compared with placebo and 1.04 (95% CI 0.92–1.18) for combination vitamin D plus calcium supplementation vs placebo.1 Two individual trials are discussed below.
Trivedi et al6 randomized 2,686 British men and women to vitamin D3 100,000 IU given every 4 months over 5 years (equivalent to 800 IU/day) or placebo. The relative risk of cardiovascular events was 0.90 (95% CI 0.77–1.06) and of cardiovascular deaths 0.84 (95% CI 0.65–1.10). Although the results were promising, the trial was designed to assess fracture risk and was not large enough for the differences in cardiovascular outcomes to reach statistical significance.
The Women’s Health Initiative,7,8 which included 36,282 postmenopausal women aged 50 to 79, tested combined vitamin D3 (400 IU/day) with calcium (1,000 mg/day) vs placebo. No benefit was seen for preventing coronary events or stroke, which may be due to the low dosage of vitamin D. The hazard ratio for coronary disease was 1.04 (0.92–1.18). Regarding mortality, the hazard ratio for cardiovascular death was 0.92, for cerebrovascular death 0.89, for cancer death 0.89, and for other deaths 0.95. None of these hazard ratios reached statistical significance.
MORE MAY NOT BE BETTER
As is probably true for everything in biological systems, there apparently is an optimal level of intake to meet vitamin D needs.
The Framingham Offspring Study,2 which found a higher risk with vitamin D deficiency, also found a suggestion of a threshold. Participants who had levels of 50 to 65 nmol/L had the lowest risk. Higher levels did not confer lower risk and even suggested a slight upturn.
Evidence from the Women’s Health Initiative8 also indicates that high dosages may not be better than moderate dosages. The meta-analysis of vitamin D and all-cause mortality5 found a relative risk of 0.93, but one of the largest studies in that meta-analysis tested only 400 IU a day and found a similar relative risk of 0.91 (95% confidence interval, 0.83–1.01).
Moreover, the NHANES study found that with increasing serum 25-hydroxyvitamin D3 levels, the risk of all-cause mortality fell until about 100 nmol/L, but then plateaued and even increased with higher serum levels.4
VITAL: STUDY DESIGN AND LOGISTICS
In VITAL, the investigators aim to recruit 20,000 healthy men (age 60 and older) and women (65 and older) who are representative of the US population (www.vitalstudy.org). Because it is a primary prevention trial, people with a known history of cardiovascular disease or cancer will be excluded. Participants will be randomized to receive either 2,000 IU of vitamin D3 per day or placebo. Each group will be further randomized to receive either 1 g per day of fish oil (combined eicosapentaenoic acid [EPA] and docosahexaenoic acid [DHA]) or placebo. The mean treatment period will be 5 years. Recruitment began in early 2010.
Blood will be collected in about 80% (ideally 100%) of participants, with follow-up blood collection in at least 2,000.
Primary aims of the trial are to test whether vitamin D3 and the omega-3 fatty acids reduce the risk of total cancer and major cardiovascular events (a composite of myocardial infarction, stroke, and death due to cardiovascular events).
Secondary aims are to test whether these agents lower the risk of:
Site-specific cancer, including colorectal, breast, and prostate cancer, and the total cancer mortality rate
An expanded composite outcome including myocardial infarction, stroke, cardiovascular death, coronary artery bypass grafting, percutaneous coronary intervention, and its individual components.
Tertiary aims are to explore whether vitamin D3 and omega-3 fatty acids have additive effects on the primary and secondary end points. The trial will also explore whether the effects of vitamin D3 and omega-3 fatty acids on cancer and cardiovascular disease vary by baseline blood levels of these nutrients, and whether race, skin pigmentation, or body mass index modify the effects of vitamin D3.
Ancillary studies will assess the effect of the interventions on risk of diabetes, hypertension, cognitive decline, depression, fracture, infections, respiratory disorders, and autoimmune diseases. The primary sponsor of this trial is the National Cancer Institute, and the secondary sponsor is the National Heart, Lung and Blood Institute. Other institutes and agencies also are cosponsors of the study.
The timing of VITAL is optimal
There is a limited window of opportunity for conducting a randomized clinical trial: the evidence must be strong enough to justify mounting a very large trial with enough power to look at cardiovascular events and cancer, but the evidence must not be so strong that it would be unethical to have a placebo group. Thus, there must be a state of equipoise. Our trial allows the study population to have a background intake of vitamin D that is currently recommended by national guidelines. Therefore, even the placebo group should have adequate intake of vitamin D.
The growing use of vitamin D supplementation by the public underscores the need for conclusive evidence of its benefits and risks. No previous large-scale randomized clinical trial has tested moderate to high doses of vitamin D for the primary prevention of cancer and cardiovascular disease.
Setting the dosage
VITAL set the vitamin D3 dosage at 2,000 IU per day (50 μg/day), which is designed to provide the best balance of efficacy and safety. As a general rule, each microgram of vitamin D3 is expected to raise the serum 25-hydroxyvitamin D3 level about 1 nmol/L, although the response is not linear: if baseline levels are lower, the increase is greater. In the United States, people commonly have a baseline level of about 40 nmol/L, so we expect that levels of people treated in the study will reach about 90 nmol/L (range 75–100 nmol/L), about 35 to 50 nmol/L higher than in the placebo group.
The target range of 75 to 100 nmol/L is the level at which greatest efficacy has been suggested in observational studies. Previous randomized trials of vitamin D have not tested high enough doses to achieve this level of 25-hydroxyvitamin D3. VITAL will test whether reaching this serum level lowers the risk of cardiovascular disease, cancer, and other chronic diseases. This level may be associated with benefit and has minimal risk of hypercalcemia. Risk of hypercalcemia may be present in participants with an occult chronic granulomatous condition such as sarcoidosis or Wegener granulomatosis, in which activated macrophages synthesize 1,25-dihydroxyvitamin D3. These conditions are very rare, however, and the risk of hypercalcemia in the trial is exceedingly low.
VITAL participants will also be randomized to take placebo or 1 g per day of combined EPA and DHA, about 5 to 10 times more than most Americans consume.
Nationwide recruitment among senior citizens
We aim to recruit 20,000 people (10,000 men and 10,000 women) nationwide who are willing, eligible, and compliant (ie, who take more than two-thirds of study pills during a 3-month placebo “run-in” phase of the trial). The trial aims to enroll 40,000 in the run-in period, and 20,000 will be randomized. To get this many participants, we will send invitational mailings and screening questionnaires to at least 2.5 million people around the United States, with mailing lists selected by age—ie, members of the American Association of Retired Persons, health professionals, teachers, and subscription lists for selected magazines. A pilot study in 5,000 people has indicated that recruiting and randomizing 20,000 participants via large mailings should be possible.
The trial is expected to be extremely cost-effective because it will be conducted largely by mail. Medication will be mailed in calendar blister packs. Participants report outcomes, which are then confirmed by medical record review. The Centers for Medicare and Medicaid Services and the National Death Index will also be used to ascertain outcomes.
We hope to recruit a more racially diverse study population than is typically seen in US trials: 63% (12,620) whites, 25% (5,000) African Americans, 7% (1,400) Hispanics, 2.5% (500) Asians, 2% (400) American Indians and Alaska natives, and 0.4% (80) native Hawaiian and Pacific Islanders.
Eligibility criteria ensure primary prevention is tested
To enter the study, men must be at least 60 years old and women at least 65. At a minimum, a high school education is required due to the detailed forms and questionnaires to be completed. Because this is a primary prevention trial, anyone with a history of cancer (except nonmelanoma skin cancer) or cardiovascular disease (including myocardial infarction, stroke, or coronary revascularization) will be excluded, as will anyone with a history of kidney stones, renal failure or dialysis, hypercalcemia, hypoparathyroidism or hyperparathyroidism, severe liver disease (eg, cirrhosis), sarcoidosis, tuberculosis, or other granulomatous disease. People with an allergy to fish will also be excluded.
We do not expect that those in the placebo group will develop vitamin D deficiency due to their participation in the study. The trial will allow a background intake in the study population of up to 800 IU of vitamin D and 1,200 mg of calcium per day in supplements. Assuming they also get about 200 IU of vitamin D in the diet, the background intake in the placebo group may be close to 1,000 IU of vitamin D. Assuming that the active treatment group has a similar background intake, their total intake will be about 3,000 IU per day (about 1,000 IU/day from background intake plus 2,000 IU/day from the intervention).
Cohort power sufficient to see effect in 5 years
The trial is expected to have sufficient power to evaluate cardiovascular disease and cancer end points as primary outcomes during 5 years of follow-up. The trial is designed to have a power of 91% to 92% to detect a relative risk of 0.85 for the primary cancer end point of total cancer incidence and 0.80 for the cardiovascular disease end point of myocardial infarction, stroke, and cardiovascular mortality. Power will be even greater for the expanded composite outcome for cardiovascular disease.
Ancillary studies
Ancillary studies include evaluating the interventions’ role in preventing diabetes and glucose intolerance, hypertension, heart failure, atrial fibrillation, cognitive decline, mood disorders, osteoporosis and fractures, asthma and respiratory diseases, infections, macular degeneration, rheumatoid arthritis, systemic lupus erythematosus, and a composite of autoimmune diseases. Imaging studies also are planned, including dual energy x-ray absorptiometry, mammographic density, and non-invasive vascular imaging (carotid intima medial thickness, coronary calcium measurements, and two-dimensional echocardiography to assess cardiac function).
Several biomarker and genetic studies will also be carried out. We intend to perform genetic studies on most of the study population to evaluate gene variants in the vitamin D receptor, vitamin D binding protein, and other vitamin-D-related genes that may contribute to lower baseline levels of 25-hydroxyvitamin D3 or different responses to the interventions.
Clinical and Translational Science Center visits are planned to provide more detailed assessments of 1,000 participants, including blood pressure measurements, height, weight, waist circumference, other anthropometric measurements, a 2-hour glucose tolerance test, a fasting blood collection, hemoglobin A1c measurements, spirometry, and assessment of physical performance, strength, frailty, cognitive function, mood, and depression. Dual-energy x-ray absorptiometry and noninvasive vascular imaging studies are also planned for those visits.
Vitamin D is viewed as a promising supplement by the medical, public health, and lay communities, potentially offering many health benefits. But enthusiasm for a new intervention too often gets far ahead of the evidence, as was the case with beta-carotene, selenium, folic acid, and vitamins C and E.
Despite the enthusiasm for vitamin D, there have been no large-scale primary prevention trials that have had either cardiovascular disease or cancer as a prespecified primary outcome. Previous randomized trials of vitamin D have focused primarily on osteoporosis, fracture, falls, and physical function. Although the investigators often reported their findings on vitamin D and cardiovascular disease or cancer, these outcomes were generally secondary or tertiary end points that were not prespecified. These studies should be viewed as hypothesis-generating rather than hypothesis-testing. The increasing prevalence of use of vitamin D supplements underscores the need for rigorous and conclusive evidence from randomized clinical trials that have cardiovascular disease and cancer as primary outcomes.
This article will explain the rationale for a large-scale, randomized clinical trial to evaluate the role of vitamin D in the prevention of cardiovascular disease and cancer. It will also describe the biological mechanisms and currently available evidence relating vitamin D to potential health benefits. Finally, the design, dosage considerations, and logistics of the Vitamin D and Omega-3 Trial (VITAL) will be presented.
EVIDENCE IS MOUNTING FOR VITAMIN D’S BIOLOGICAL IMPORTANCE
Vitamin D is undoubtedly important to health: not only is it produced endogenously, but at least 500 genes have been identified with vitamin D response elements. The vitamin D receptor is found in nearly all cells in the body, and the 1-alpha-hydroxylase enzyme is present in many tissues. Some studies suggest that almost 10% of the human genome may be at least partially regulated by vitamin D.
From Hajjar V, et al. Does vitamin D deficiency play a role in the pathogenesis of chronic heart failure? Do supplements improve survival? Cleve Clin J Med 2010; 77:290–293.
Figure 1.
Vitamin D is a prohormone, and people obtain it both endogenously and exogenously (Figure 1). With exposure to ultraviolet B light, 7-dehydrocholesterol in the skin converts to vitamin D3. We also obtain it through diet or supplements. The plant form (vitamin D2) and the animal form (vitamin D3) undergo 25-hydroxylation in the liver. Then, 1-alpha-hydroxylase converts the 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3, primarily in the kidney. Increasing evidence shows that 1-alpha-hydroxylase is present in many other cells and tissues, and that 1,25-dihydroxyvitamin D3 may be locally produced and possibly even have autocrine effects (acting on surface receptors of the same cell it is secreted by) and paracrine effects (acting on adjacent cells).
Although we know vitamin D is important, what our optimal intake and our blood level of 25-hydroxyvitamin D3 should be are key unknowns.
RECOMMENDATIONS FOR VITAMIN D INTAKE
During winter, late fall, and early spring, people who live above the 37th parallel (geographically, about one-half of the contiguous United States) do not get enough ultraviolet B energy from the sun to make all the vitamin D they need, even if they spend several hours outside every day. In addition, dark skin pigmentation serves as a sun block, as do sunscreens.
The Institute of Medicine (IOM) provided guidelines for vitamin D intake in 1997 and, most recently, in 2010. However, these guidelines are based on the amount of vitamin D required for bone health and do not address the amount that may be of benefit for prevention of cancer and cardiovascular disease. The latter outcomes are not addressed because the IOM committee believed that evidence was insufficient to determine the role of vitamin D in the prevention of cardiovascular disease, cancer, and other chronic diseases. Thus, current IOM guidelines, which generally recommend less than 1,000 IU of vitamin D daily, are relevant to bone health but not necessarily to other health outcomes. More research is needed to understand whether the guidelines should be modified for the prevention of other chronic diseases.
Moreover, whether or not everyone should be screened for 25-hydroxyvitamin D3 blood levels is controversial. Most experts agree that a level less than 20 ng/mL is deficient or insufficient. Conversely, potentially harmful are levels 150 ng/mL or more (> 375 nmol/L), which entail the risk of hypercalcemia, vascular soft tissue calcification, and hyperphosphatemia.
People do not reach toxic levels with ultraviolet light exposure because the amount of 25-hydroxyvitamin D3 synthesis is well regulated. Dietary supplements, however, can bring about toxic levels, and patients taking high doses need to be monitored carefully. The level that should be considered optimal is controversial and requires further study.
RISK FACTORS FOR LOW VITAMIN D LEVELS
Risk factors for low vitamin D levels include older age, living in northern latitudes, sun avoidance, dark skin pigmentation, obesity, low dietary intake, and various medical conditions, especially malabsorption syndromes. Some of these are also risk factors for cardiovascular disease, cancer, and other chronic diseases, and potentially confound outcomes in many studies. Older age, which is usually adjusted for in multivariate models, is important to recognize as a major risk factor for vitamin D deficiency, owing to reduced absorption and synthesis, less time outdoors, and low dietary intake.
Wearing sunscreen decreases the synthesis of vitamin D in the skin, but because ultra-violet light has been clearly classified as a carcinogen, it is a not advisable to increase sun exposure for the sake of increasing vitamin D levels. That is a poor trade-off, given the high incidence rate of skin cancer and the adverse effects of solar radiation on skin aging.
Obesity is a risk factor for vitamin D deficiency because vitamin D is fat-soluble and becomes sequestered in fat tissue. Vitamin D may also play a role in the differentiation of adipocytes and may affect their function. In observational studies, it is very important for researchers to adjust for body mass index, physical activity (which may be correlated with more time outdoors), and other potential confounders in their analyses.
HOW VITAMIN D MAY LOWER CANCER RISK
Because of the important effect of vitamin D in regulating cell differentiation and cell growth, there are multiple ways that it may affect cancer risk. Laboratory, cell culture, and animal studies suggest that vitamin D may lower cancer risk by inhibiting cell proliferation, angiogenesis, metastasis, and inflammation and inducing apoptosis and cellular differentiation. Several of these mechanisms are also relevant to atherosclerosis and cardiovascular disease. Although VITAL is addressing the role of vitamin D in preventing both cancer and cardiovascular disease, the remainder of this article will focus on cardiovascular outcomes.
HOW VITAMIN D MAY REDUCE CARDIOVASCULAR RISK
Vitamin D may lower cardiovascular risk via several mechanisms:
Inhibiting inflammation. Vitamin D has a powerful immunomodulatory effect: laboratory studies show that it inhibits prostaglandin and cyclooxygenase 2 pathways, reduces matrix metalloproteinase 9 and several proinflammatory cytokines, and increases interleukin 10, all of which result in suppressed inflammation.1
Inhibiting vascular muscle proliferation and vascular calcification. Animal studies indicate that in moderate doses vitamin D decreases calcium cellular influx and increases matrix Gla protein, which inhibits vascular smooth muscle proliferation and vascular calcification. These protective effects contrast with the hypercalcemia associated with a high intake of vitamin D, especially in the context of renal failure or other risk factors, which may lead to increased vascular calcification.1
Regulates blood pressure. Vitamin D decreases renin gene expression and the synthesis of renin, which reduces activity of the renin-angiotensin-aldosterone system, leading to a reduction of blood pressure and a favorable effect on volume homeostasis.1
Regulates glucose metabolism. Limited evidence shows that vitamin D may increase insulin sensitivity and regulate glucose metabolism.1
Vitamin D and cardiac hypertrophy
The vitamin D receptor is present in virtually all tissues, including cardiac myocytes and endothelial cells. Animals with vitamin D deficiency have higher blood pressures, and animals genetically altered to have no vitamin D receptors (knock-out models) develop left ventricular hypertrophy and heart failure.
Animals genetically altered to have no 1-alpha-hydroxylase (so that the most active form of vitamin D is not made) also develop left ventricular hypertrophy. They can be rescued by the administration of 1,25-dihydroxy vitamin D3.1
These findings are consistent with what is observed in patients with end-stage renal disease, who produce very little 1,25-dihydroxyvitamin D3: they often develop left ventricular hypertrophy, diastolic heart failure, atherosclerosis, and vascular calcification.
EVIDENCE FOR CARDIOVASCULAR DISEASE REDUCTION
Wang et al1 recently reviewed available prospective cohort and randomized clinical trials from 1966 to 2009 that examined vitamin D or calcium supplementation and cardiovascular disease. Comparing people with the lowest to the highest levels of serum 25-hydroxyvitamin D3 indicated that a low level is a risk factor for coronary artery disease and cardiovascular death. Unfortunately, most studies were not designed to assess primary effects on cardiovascular outcomes, and so have many potential confounders.
Prospective observational studies
Observational studies suggest that vitamin D deficiency is associated with an increased risk of cardiovascular disease. Some examples:
The Framingham Offspring Study2 followed 1,739 men and women with a mean age of 59 for 5.4 years. The study compared the incidence of cardiovascular events in those with a serum 25-dihydroxyvitamin D level of at least 37.5 nmol/L vs those with lower levels. The risk of cardiovascular disease was 1.62 times higher in those with the lowest levels of vitamin D, a statistically significant difference. However, a threshold effect was apparent (discussed below).
The Health Professionals Follow-up Study3 prospectively evaluated more than 18,000 men ages 40 to 75 for 10 years. The study compared men with a low serum level of vitamin D (< 37.5 nmol/L) to those with a more optimal level (> 75 nmol/L). The incidence of cardiovascular events was 2.09 times higher in men with low levels of vitamin D, a difference that was statistically significant.
The Third National Health and Nutrition Examination Survey (NHANES III) included data for more than 13,300 men and women age 20 years and older. Using a cohort that was followed for 8.7 years, Melamed et al4 compared the quartile with the lowest serum vitamin D level (< 44.4 nmol/L) against the quartile with the highest level (> 80.1 nmol/L). The associations were modest: those with low levels had a 1.20-times higher rate of death from cardiovascular disease and a statistically significant 1.26-times higher rate of death from all causes.
Randomized clinical trials
A meta-analysis of 18 randomized trials5 of vitamin D supplementation (300–2,000 IU/day, mean 528 IU/day vs placebo), including 57,311 participants, evaluated the rate of death from all causes and found a modest but significant reduction in risk (relative risk 0.93, 95% confidence interval [CI] 0.87–0.99). These were generally trials looking at fracture rates or physical performance, and a dose-response relationship was not evident. A recent systematic review of randomized controlled trials of vitamin D1 that included cardiovascular disease as a secondary outcome found a pooled relative risk for cardiovascular disease of 0.90 (95% CI 0.77–1.05) for vitamin D supplementation compared with placebo and 1.04 (95% CI 0.92–1.18) for combination vitamin D plus calcium supplementation vs placebo.1 Two individual trials are discussed below.
Trivedi et al6 randomized 2,686 British men and women to vitamin D3 100,000 IU given every 4 months over 5 years (equivalent to 800 IU/day) or placebo. The relative risk of cardiovascular events was 0.90 (95% CI 0.77–1.06) and of cardiovascular deaths 0.84 (95% CI 0.65–1.10). Although the results were promising, the trial was designed to assess fracture risk and was not large enough for the differences in cardiovascular outcomes to reach statistical significance.
The Women’s Health Initiative,7,8 which included 36,282 postmenopausal women aged 50 to 79, tested combined vitamin D3 (400 IU/day) with calcium (1,000 mg/day) vs placebo. No benefit was seen for preventing coronary events or stroke, which may be due to the low dosage of vitamin D. The hazard ratio for coronary disease was 1.04 (0.92–1.18). Regarding mortality, the hazard ratio for cardiovascular death was 0.92, for cerebrovascular death 0.89, for cancer death 0.89, and for other deaths 0.95. None of these hazard ratios reached statistical significance.
MORE MAY NOT BE BETTER
As is probably true for everything in biological systems, there apparently is an optimal level of intake to meet vitamin D needs.
The Framingham Offspring Study,2 which found a higher risk with vitamin D deficiency, also found a suggestion of a threshold. Participants who had levels of 50 to 65 nmol/L had the lowest risk. Higher levels did not confer lower risk and even suggested a slight upturn.
Evidence from the Women’s Health Initiative8 also indicates that high dosages may not be better than moderate dosages. The meta-analysis of vitamin D and all-cause mortality5 found a relative risk of 0.93, but one of the largest studies in that meta-analysis tested only 400 IU a day and found a similar relative risk of 0.91 (95% confidence interval, 0.83–1.01).
Moreover, the NHANES study found that with increasing serum 25-hydroxyvitamin D3 levels, the risk of all-cause mortality fell until about 100 nmol/L, but then plateaued and even increased with higher serum levels.4
VITAL: STUDY DESIGN AND LOGISTICS
In VITAL, the investigators aim to recruit 20,000 healthy men (age 60 and older) and women (65 and older) who are representative of the US population (www.vitalstudy.org). Because it is a primary prevention trial, people with a known history of cardiovascular disease or cancer will be excluded. Participants will be randomized to receive either 2,000 IU of vitamin D3 per day or placebo. Each group will be further randomized to receive either 1 g per day of fish oil (combined eicosapentaenoic acid [EPA] and docosahexaenoic acid [DHA]) or placebo. The mean treatment period will be 5 years. Recruitment began in early 2010.
Blood will be collected in about 80% (ideally 100%) of participants, with follow-up blood collection in at least 2,000.
Primary aims of the trial are to test whether vitamin D3 and the omega-3 fatty acids reduce the risk of total cancer and major cardiovascular events (a composite of myocardial infarction, stroke, and death due to cardiovascular events).
Secondary aims are to test whether these agents lower the risk of:
Site-specific cancer, including colorectal, breast, and prostate cancer, and the total cancer mortality rate
An expanded composite outcome including myocardial infarction, stroke, cardiovascular death, coronary artery bypass grafting, percutaneous coronary intervention, and its individual components.
Tertiary aims are to explore whether vitamin D3 and omega-3 fatty acids have additive effects on the primary and secondary end points. The trial will also explore whether the effects of vitamin D3 and omega-3 fatty acids on cancer and cardiovascular disease vary by baseline blood levels of these nutrients, and whether race, skin pigmentation, or body mass index modify the effects of vitamin D3.
Ancillary studies will assess the effect of the interventions on risk of diabetes, hypertension, cognitive decline, depression, fracture, infections, respiratory disorders, and autoimmune diseases. The primary sponsor of this trial is the National Cancer Institute, and the secondary sponsor is the National Heart, Lung and Blood Institute. Other institutes and agencies also are cosponsors of the study.
The timing of VITAL is optimal
There is a limited window of opportunity for conducting a randomized clinical trial: the evidence must be strong enough to justify mounting a very large trial with enough power to look at cardiovascular events and cancer, but the evidence must not be so strong that it would be unethical to have a placebo group. Thus, there must be a state of equipoise. Our trial allows the study population to have a background intake of vitamin D that is currently recommended by national guidelines. Therefore, even the placebo group should have adequate intake of vitamin D.
The growing use of vitamin D supplementation by the public underscores the need for conclusive evidence of its benefits and risks. No previous large-scale randomized clinical trial has tested moderate to high doses of vitamin D for the primary prevention of cancer and cardiovascular disease.
Setting the dosage
VITAL set the vitamin D3 dosage at 2,000 IU per day (50 μg/day), which is designed to provide the best balance of efficacy and safety. As a general rule, each microgram of vitamin D3 is expected to raise the serum 25-hydroxyvitamin D3 level about 1 nmol/L, although the response is not linear: if baseline levels are lower, the increase is greater. In the United States, people commonly have a baseline level of about 40 nmol/L, so we expect that levels of people treated in the study will reach about 90 nmol/L (range 75–100 nmol/L), about 35 to 50 nmol/L higher than in the placebo group.
The target range of 75 to 100 nmol/L is the level at which greatest efficacy has been suggested in observational studies. Previous randomized trials of vitamin D have not tested high enough doses to achieve this level of 25-hydroxyvitamin D3. VITAL will test whether reaching this serum level lowers the risk of cardiovascular disease, cancer, and other chronic diseases. This level may be associated with benefit and has minimal risk of hypercalcemia. Risk of hypercalcemia may be present in participants with an occult chronic granulomatous condition such as sarcoidosis or Wegener granulomatosis, in which activated macrophages synthesize 1,25-dihydroxyvitamin D3. These conditions are very rare, however, and the risk of hypercalcemia in the trial is exceedingly low.
VITAL participants will also be randomized to take placebo or 1 g per day of combined EPA and DHA, about 5 to 10 times more than most Americans consume.
Nationwide recruitment among senior citizens
We aim to recruit 20,000 people (10,000 men and 10,000 women) nationwide who are willing, eligible, and compliant (ie, who take more than two-thirds of study pills during a 3-month placebo “run-in” phase of the trial). The trial aims to enroll 40,000 in the run-in period, and 20,000 will be randomized. To get this many participants, we will send invitational mailings and screening questionnaires to at least 2.5 million people around the United States, with mailing lists selected by age—ie, members of the American Association of Retired Persons, health professionals, teachers, and subscription lists for selected magazines. A pilot study in 5,000 people has indicated that recruiting and randomizing 20,000 participants via large mailings should be possible.
The trial is expected to be extremely cost-effective because it will be conducted largely by mail. Medication will be mailed in calendar blister packs. Participants report outcomes, which are then confirmed by medical record review. The Centers for Medicare and Medicaid Services and the National Death Index will also be used to ascertain outcomes.
We hope to recruit a more racially diverse study population than is typically seen in US trials: 63% (12,620) whites, 25% (5,000) African Americans, 7% (1,400) Hispanics, 2.5% (500) Asians, 2% (400) American Indians and Alaska natives, and 0.4% (80) native Hawaiian and Pacific Islanders.
Eligibility criteria ensure primary prevention is tested
To enter the study, men must be at least 60 years old and women at least 65. At a minimum, a high school education is required due to the detailed forms and questionnaires to be completed. Because this is a primary prevention trial, anyone with a history of cancer (except nonmelanoma skin cancer) or cardiovascular disease (including myocardial infarction, stroke, or coronary revascularization) will be excluded, as will anyone with a history of kidney stones, renal failure or dialysis, hypercalcemia, hypoparathyroidism or hyperparathyroidism, severe liver disease (eg, cirrhosis), sarcoidosis, tuberculosis, or other granulomatous disease. People with an allergy to fish will also be excluded.
We do not expect that those in the placebo group will develop vitamin D deficiency due to their participation in the study. The trial will allow a background intake in the study population of up to 800 IU of vitamin D and 1,200 mg of calcium per day in supplements. Assuming they also get about 200 IU of vitamin D in the diet, the background intake in the placebo group may be close to 1,000 IU of vitamin D. Assuming that the active treatment group has a similar background intake, their total intake will be about 3,000 IU per day (about 1,000 IU/day from background intake plus 2,000 IU/day from the intervention).
Cohort power sufficient to see effect in 5 years
The trial is expected to have sufficient power to evaluate cardiovascular disease and cancer end points as primary outcomes during 5 years of follow-up. The trial is designed to have a power of 91% to 92% to detect a relative risk of 0.85 for the primary cancer end point of total cancer incidence and 0.80 for the cardiovascular disease end point of myocardial infarction, stroke, and cardiovascular mortality. Power will be even greater for the expanded composite outcome for cardiovascular disease.
Ancillary studies
Ancillary studies include evaluating the interventions’ role in preventing diabetes and glucose intolerance, hypertension, heart failure, atrial fibrillation, cognitive decline, mood disorders, osteoporosis and fractures, asthma and respiratory diseases, infections, macular degeneration, rheumatoid arthritis, systemic lupus erythematosus, and a composite of autoimmune diseases. Imaging studies also are planned, including dual energy x-ray absorptiometry, mammographic density, and non-invasive vascular imaging (carotid intima medial thickness, coronary calcium measurements, and two-dimensional echocardiography to assess cardiac function).
Several biomarker and genetic studies will also be carried out. We intend to perform genetic studies on most of the study population to evaluate gene variants in the vitamin D receptor, vitamin D binding protein, and other vitamin-D-related genes that may contribute to lower baseline levels of 25-hydroxyvitamin D3 or different responses to the interventions.
Clinical and Translational Science Center visits are planned to provide more detailed assessments of 1,000 participants, including blood pressure measurements, height, weight, waist circumference, other anthropometric measurements, a 2-hour glucose tolerance test, a fasting blood collection, hemoglobin A1c measurements, spirometry, and assessment of physical performance, strength, frailty, cognitive function, mood, and depression. Dual-energy x-ray absorptiometry and noninvasive vascular imaging studies are also planned for those visits.
References
Wang L, Manson JE, Song Y, Sesso HD. Systematic review: vitamin D and calcium supplementation in prevention of cardiovascular events. Ann Intern Med2010; 152:315–323.
Wang TJ, Pencina MJ, Booth SL, et al. Vitamin D deficiency and risk of cardiovascular disease. Circulation2008; 117:503–511.
Giovannucci E, Liu Y, Hollis BW, Rimm EB. 25-Hydroxyvitamin D and risk of myocardial infarction in men: a prospective study. Arch Intern Med2008; 168:1174–1180.
Melamed ML, Michos ED, Post W, Astor B. 25-Hydroxyvitamin D levels and the risk of mortality in the general population. Arch Intern Med2008; 168:1629–1637.
Autier P, Gandini S. Vitamin D supplementation and total mortality: a meta-analysis of randomized controlled trials. Arch Intern Med2007; 167:1730–1737.
Trivedi DP, Doll R, Khaw KT. Effect of four monthly oral vitamin D3 (cholecalciferol) supplementation on fractures and mortality in men and women living in the community: randomised double blind controlled trial. BMJ2003; 326:469.
Hsia J, Heiss G, Ren H, et al; Women’s Health Initiative Investigators. Calcium/vitamin D supplementation and cardiovascular events. Circulation2007; 115:846–854.
LaCroix AZ, Kotchen J, Anderson G, et al. Calcium plus vitamin D supplementation and mortality in postmenopausal women: the Women’s Health Initiative calcium-vitamin D randomized controlled trial. J Gerontol A Biol Sci Med Sci2009; 64:559–567.
References
Wang L, Manson JE, Song Y, Sesso HD. Systematic review: vitamin D and calcium supplementation in prevention of cardiovascular events. Ann Intern Med2010; 152:315–323.
Wang TJ, Pencina MJ, Booth SL, et al. Vitamin D deficiency and risk of cardiovascular disease. Circulation2008; 117:503–511.
Giovannucci E, Liu Y, Hollis BW, Rimm EB. 25-Hydroxyvitamin D and risk of myocardial infarction in men: a prospective study. Arch Intern Med2008; 168:1174–1180.
Melamed ML, Michos ED, Post W, Astor B. 25-Hydroxyvitamin D levels and the risk of mortality in the general population. Arch Intern Med2008; 168:1629–1637.
Autier P, Gandini S. Vitamin D supplementation and total mortality: a meta-analysis of randomized controlled trials. Arch Intern Med2007; 167:1730–1737.
Trivedi DP, Doll R, Khaw KT. Effect of four monthly oral vitamin D3 (cholecalciferol) supplementation on fractures and mortality in men and women living in the community: randomised double blind controlled trial. BMJ2003; 326:469.
Hsia J, Heiss G, Ren H, et al; Women’s Health Initiative Investigators. Calcium/vitamin D supplementation and cardiovascular events. Circulation2007; 115:846–854.
LaCroix AZ, Kotchen J, Anderson G, et al. Calcium plus vitamin D supplementation and mortality in postmenopausal women: the Women’s Health Initiative calcium-vitamin D randomized controlled trial. J Gerontol A Biol Sci Med Sci2009; 64:559–567.
Laboratory evidence suggests that vitamin D may lower cancer risk by inhibiting cell proliferation, angiogenesis, metastasis, and inflammation.
Vitamin D may also reduce cardiovascular risk by inhibiting vascular smooth muscle proliferation, regulating blood pressure and glucose metabolism, and reducing inflammation.
Some observational studies indicate there may be a threshold for vitamin D intake above which there is no increase in benefit and which may increase risk.
The VITAL trial is currently randomizing 20,000 healthy older men and women throughout the United States to receive either 2,000 IU of vitamin D3 (cholecalciferol) per day or placebo, as well as 1 g of marine omega-3 fatty acids per day or placebo, for 5 years.
Alpha-blockers should not be used as first-line therapy for hypertension. However, an alpha-blocker can be considered as a second-line or third-line add-on in a patient whose blood pressure is not under control despite treatment with other drugs.
In addition, alpha-blockers are useful in relieving lower urinary tract symptoms in patients with benign prostatic hypertrophy. However, even in a patient who has both hypertension and benign prostatic hypertrophy, we advise physicians to use alpha-blockers primarily to relieve the urinary symptoms, and we recommend lowering the blood pressure with a drug of a class shown to reduce rates of illness and death.
NOT FIRST-LINE THERAPY
All antihypertensive drugs, including alpha-blockers, lower blood pressure. Alpha-blockers have been approved by the US Food and Drug Administration for treating high blood pressure, and they are just as effective as other antihypertensive drugs—if efficacy is defined as a decrease in millimeters of mercury.
However, lowering the blood pressure is not the main goal of antihypertensive therapy. What we want to achieve when prescribing antihypertensive drugs is to reduce the rates of heart attacks, strokes, and other adverse cardiovascular adverse outcomes, including death.
Unfortunately, alpha-blockers fall short in this regard. In the Antihypertensive and Lipid Lowering Treatment to Prevent Heart Attack (ALLHAT) trial,1,2 doxazosin (Cardura) was found to carry a higher risk of combined cardiovascular disease (relative risk 1.19, P = .04), mostly stroke. Alarmingly, the incidence of symptomatic heart failure in patients on doxazosin was twice that in patients on chlorthalidone (relative risk 2.04, P < .001). Doxazosin was minimally more effective in lowering blood pressure than chlorthalidone, but the small difference in blood pressure was unlikely to have accounted for the significant difference in the risk of heart failure.3
This experience with doxazosin illustrates a key drawback to surrogate end points: a treatment may produce a favorable outcome in the surrogate end point (blood pressure) but produce little or no benefit in terms of the real end point (stroke, myocardial infarction, and heart failure).4
Based on the ALLHAT data as well as on a Veterans Administration study in patients with chronic heart failure in which survival with prazosin (Minipress) was no better than with placebo,5 it seems reasonable to no longer use alpha-blockers as initial therapy for hypertension. This view is reflected by current European6 and American7 guidelines.
ALPHA-BLOCKERS AS PART OF COMBINATION THERAPY
In several clinical trials, alpha-blockers were allowed8 or were specified9,10 as add-on therapy if other drugs failed to control the blood pressure, but they were not used in a randomized fashion. Thus, we cannot judge their effect on cardiovascular outcomes such as heart attack and stroke.
The choice of drugs for combination therapy very often is still empirical and based on personal preference. Doxazosin as add-on therapy, in general, has been shown to be safe and well tolerated.11 But even if it is acceptable, it is not a preferred combination.
In the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT),9 patients received extended-release doxazosin as a third drug if they did not reach their goal blood pressure with either the combination of amlodipine (Norvasc) plus perindopril (Aceon) or atenolol (Tenormin) plus bendroflumethiazide. Extended-release doxazosin was an effective add-on, and there was no apparent excess rate of heart failure in doxazosin users.
In other studies, in patients with uncontrolled hypertension, adding doxazosin as a second- or third-line agent to a gold-standard drug—calcium channel blocker, diuretic, beta-blocker, angiotensin-converting enzyme inhibitor, angiotensin receptor blocker, or combinations of these—allowed significantly more participants to achieve their blood pressure goal.11
Personally, we consider doxazosin in patients whose blood pressure is not controlled with triple therapy with a renin-angiotensin system blocker, a diuretic, and a calcium channel antagonist in full doses. In patients with stage 3 or stage 4 kidney disease who can no longer tolerate renin-angiotensin system blockers, doxazosin may also be a useful adjunct. Whether the metabolic effects of alpha-blockers, such as a reduction in insulin resistance and a decrease in total and low-density lipoprotein cholesterol, will result in lower rates of morbidity and death has not been conclusively determined.
A point of view somewhat more favorable to the use of alpha-blockers has recently been put forward by Chapman et al.12
ALPHA-BLOCKERS ALLEVIATE SYMPTOMS OF BENIGN PROSTATIC HYPERTROPHY
Doxazosin and other alpha-blockers are commonly used to alleviate lower urinary tract symptoms in patients with benign prostatic hypertrophy.
Both high blood pressure and benign prostatic hypertrophy become more common with advancing age, and it has been estimated that both are present in more than 25% of men over age 60.13 Indeed, two trials documented that a significant reduction in symptoms of benign prostatic hypertrophy and in systolic and diastolic blood pressure can be achieved with an alpha-blocker.13,14
This raises the question whether such a “twofer” (treating two disease states with one drug) should be used in clinical practice. We have to consider that the principle of the twofer has never been tested and agree with Davis et al,3 who, in a further analysis of the ALLHAT data, stated that, “In older men with benign prostatic hypertrophy in whom an [alpha]-adrenergic blocker seems like the best treatment for the uropathy, coexisting hypertension should be treated with another antihypertensive drug as well.”3
Again, this would clearly relegate doxazosin to second-line or third-line status, even in patients with benign prostatic hypertrophy, in whom it has been shown to be indicated.
ADVERSE EFFECTS OF ALPHA-BLOCKERS
Dizziness, fatigue, and somnolence are occasionally reported but appear to be well tolerated. Postural hypotension is much less common with proper titration of standard doxazosin or with the use of controlled-release formulations.9–15 However, in patients with impaired autonomic function, even long-acting alpha-blockers can cause postural hypotension and syncope.
Patients using phosphodiesterase type 5 inhibitors—sildenafil (Viagra), vardenafil (Levitra), or tadalafil (Cialis)—for erectile dysfunction should avoid alpha-blockers because the blood-pressure-lowering effects of the two drug classes may be additive.
References
Messerli FH. Implications of discontinuation of doxazosin arm of ALLHAT. Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (commentary). Lancet2000; 355:863–864.
Major cardiovascular events in hypertensive patients randomized to doxazosin vs chlorthalidone: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). ALLHAT Collaborative Research Group. JAMA2000; 283:1967–1975.
Davis BR, Cutler JA, Furberg CD, et al; ALLHAT Collaborative Research Group. Relationship of antihypertensive treatment regimens and change in blood pressure to risk for heart failure in hypertensive patients randomly assigned to doxazosin or chlorthalidone: further analyses from the Antihypertensive and Lipid-Lowering treatment to prevent Heart Attack Trial. Ann Intern Med2002; 137:313–320.
Messerli FH. Doxazosin and congestive heart failure (viewpoint). J Am Coll Cardiol2001; 38:1295–1296.
Cohn JN, Archibald DG, Ziesche S, et al. Effect of vasodilator therapy on mortality in chronic congestive heart failure. Results of a Veterans Administration Cooperative Study. N Engl J Med1986; 314:1547–1552.
Mancia G, De Backer G, Dominiczak A, et al; ESH-ESC Task Force on the Management of Arterial Hypertension. 2007 ESH-ESC practice guidelines for the management of arterial hypertension: ESH-ESC Task Force on the Management of Arterial Hypertension. J Hypertens2007; 25:1751–1762.
Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. JAMA2003; 289:2560–2572.
Jamerson K, Weber MA, Bakris GL, et al; for the ACCOMPLISH Trial Investigators. Benazepril plus amlodipine or hydrochlorothiazide for hypertension in high-risk patients. N Engl J Med2008; 359:2417–2428.
Chapman N, Chang CL, Dahlöf B, Sever PS, Wedel H, Poulter NR; ASCOT Investigators. Effect of doxazosin gastrointestinal therapeutic system as third-line antihypertensive therapy on blood pressure and lipids in the Anglo-Scandinavian Cardiac Outcomes Trial. Circulation2008; 118:42–48.
de Alvaro F, Hernandez-Presa MAASOCIA Study. Effect of doxazosin gastrointestinal therapeutic system on patients with uncontrolled hypertension: the ASOCIA Study. J Cardiovasc Pharmacol2006; 47:271–276.
Black HR. Doxazosin as combination therapy for patients with stage 1 and stage 2 hypertension. J Cardiovasc Pharmacol2003; 41:866–869.
Chapman N, Chen C-Y, Fujita T, et al. Time to re-appraise the role of alpha-1 adrenoceptor antagonists in the management of hypertension?J Hypertens2010; 28:1796–1803.
Steers WD, Kirby RS. Clinical ease of using doxazosin in BPH patients with and without hypertension. Prostate Cancer Prostatic Dis2005; 8:152–157.
Guthrie RM, Siegel RL. A multicenter, community-based study of doxazosin in the treatment of concomitant hypertension and symptomatic benign prostatic hyperplasia: the Hypertension and BPH Intervention Trial (HABIT). Clin Ther1999; 21:1732–1748.
MacDonald R, Wilt TJ, Howe RW. Doxazosin for treating lower urinary tract symptoms compatible with benign prostatic obstruction: a systematic review of efficacy and adverse effects. BJU Int2004; 94:1263–1270.
Giacomo Rossitto, MD Department of Clinical and Experimental Medicine, University of Padua School of Medicine, Padua, Italy
Ganesh Kamath, MD Department of Medicine, St. Luke’s-Roosevelt Hospital Center, New York, NY
Franz H. Messerli, MD, FACC, FACP Director, Hypertension Program, Professor of Clinical Medicine, Columbia University College of Physicians and Surgeons; Division of Cardiology, St. Luke’s-Roosevelt Hospital Center, New York, NY
Address: Franz H. Messerli, MD, Hypertension Program, Division of Cardiology, St. Luke’s-Roosevelt Hospital Center, 1000 Tenth Avenue, New York, NY 10019; e-mail [email protected]
Giacomo Rossitto, MD Department of Clinical and Experimental Medicine, University of Padua School of Medicine, Padua, Italy
Ganesh Kamath, MD Department of Medicine, St. Luke’s-Roosevelt Hospital Center, New York, NY
Franz H. Messerli, MD, FACC, FACP Director, Hypertension Program, Professor of Clinical Medicine, Columbia University College of Physicians and Surgeons; Division of Cardiology, St. Luke’s-Roosevelt Hospital Center, New York, NY
Address: Franz H. Messerli, MD, Hypertension Program, Division of Cardiology, St. Luke’s-Roosevelt Hospital Center, 1000 Tenth Avenue, New York, NY 10019; e-mail [email protected]
Author and Disclosure Information
Giacomo Rossitto, MD Department of Clinical and Experimental Medicine, University of Padua School of Medicine, Padua, Italy
Ganesh Kamath, MD Department of Medicine, St. Luke’s-Roosevelt Hospital Center, New York, NY
Franz H. Messerli, MD, FACC, FACP Director, Hypertension Program, Professor of Clinical Medicine, Columbia University College of Physicians and Surgeons; Division of Cardiology, St. Luke’s-Roosevelt Hospital Center, New York, NY
Address: Franz H. Messerli, MD, Hypertension Program, Division of Cardiology, St. Luke’s-Roosevelt Hospital Center, 1000 Tenth Avenue, New York, NY 10019; e-mail [email protected]
Alpha-blockers should not be used as first-line therapy for hypertension. However, an alpha-blocker can be considered as a second-line or third-line add-on in a patient whose blood pressure is not under control despite treatment with other drugs.
In addition, alpha-blockers are useful in relieving lower urinary tract symptoms in patients with benign prostatic hypertrophy. However, even in a patient who has both hypertension and benign prostatic hypertrophy, we advise physicians to use alpha-blockers primarily to relieve the urinary symptoms, and we recommend lowering the blood pressure with a drug of a class shown to reduce rates of illness and death.
NOT FIRST-LINE THERAPY
All antihypertensive drugs, including alpha-blockers, lower blood pressure. Alpha-blockers have been approved by the US Food and Drug Administration for treating high blood pressure, and they are just as effective as other antihypertensive drugs—if efficacy is defined as a decrease in millimeters of mercury.
However, lowering the blood pressure is not the main goal of antihypertensive therapy. What we want to achieve when prescribing antihypertensive drugs is to reduce the rates of heart attacks, strokes, and other adverse cardiovascular adverse outcomes, including death.
Unfortunately, alpha-blockers fall short in this regard. In the Antihypertensive and Lipid Lowering Treatment to Prevent Heart Attack (ALLHAT) trial,1,2 doxazosin (Cardura) was found to carry a higher risk of combined cardiovascular disease (relative risk 1.19, P = .04), mostly stroke. Alarmingly, the incidence of symptomatic heart failure in patients on doxazosin was twice that in patients on chlorthalidone (relative risk 2.04, P < .001). Doxazosin was minimally more effective in lowering blood pressure than chlorthalidone, but the small difference in blood pressure was unlikely to have accounted for the significant difference in the risk of heart failure.3
This experience with doxazosin illustrates a key drawback to surrogate end points: a treatment may produce a favorable outcome in the surrogate end point (blood pressure) but produce little or no benefit in terms of the real end point (stroke, myocardial infarction, and heart failure).4
Based on the ALLHAT data as well as on a Veterans Administration study in patients with chronic heart failure in which survival with prazosin (Minipress) was no better than with placebo,5 it seems reasonable to no longer use alpha-blockers as initial therapy for hypertension. This view is reflected by current European6 and American7 guidelines.
ALPHA-BLOCKERS AS PART OF COMBINATION THERAPY
In several clinical trials, alpha-blockers were allowed8 or were specified9,10 as add-on therapy if other drugs failed to control the blood pressure, but they were not used in a randomized fashion. Thus, we cannot judge their effect on cardiovascular outcomes such as heart attack and stroke.
The choice of drugs for combination therapy very often is still empirical and based on personal preference. Doxazosin as add-on therapy, in general, has been shown to be safe and well tolerated.11 But even if it is acceptable, it is not a preferred combination.
In the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT),9 patients received extended-release doxazosin as a third drug if they did not reach their goal blood pressure with either the combination of amlodipine (Norvasc) plus perindopril (Aceon) or atenolol (Tenormin) plus bendroflumethiazide. Extended-release doxazosin was an effective add-on, and there was no apparent excess rate of heart failure in doxazosin users.
In other studies, in patients with uncontrolled hypertension, adding doxazosin as a second- or third-line agent to a gold-standard drug—calcium channel blocker, diuretic, beta-blocker, angiotensin-converting enzyme inhibitor, angiotensin receptor blocker, or combinations of these—allowed significantly more participants to achieve their blood pressure goal.11
Personally, we consider doxazosin in patients whose blood pressure is not controlled with triple therapy with a renin-angiotensin system blocker, a diuretic, and a calcium channel antagonist in full doses. In patients with stage 3 or stage 4 kidney disease who can no longer tolerate renin-angiotensin system blockers, doxazosin may also be a useful adjunct. Whether the metabolic effects of alpha-blockers, such as a reduction in insulin resistance and a decrease in total and low-density lipoprotein cholesterol, will result in lower rates of morbidity and death has not been conclusively determined.
A point of view somewhat more favorable to the use of alpha-blockers has recently been put forward by Chapman et al.12
ALPHA-BLOCKERS ALLEVIATE SYMPTOMS OF BENIGN PROSTATIC HYPERTROPHY
Doxazosin and other alpha-blockers are commonly used to alleviate lower urinary tract symptoms in patients with benign prostatic hypertrophy.
Both high blood pressure and benign prostatic hypertrophy become more common with advancing age, and it has been estimated that both are present in more than 25% of men over age 60.13 Indeed, two trials documented that a significant reduction in symptoms of benign prostatic hypertrophy and in systolic and diastolic blood pressure can be achieved with an alpha-blocker.13,14
This raises the question whether such a “twofer” (treating two disease states with one drug) should be used in clinical practice. We have to consider that the principle of the twofer has never been tested and agree with Davis et al,3 who, in a further analysis of the ALLHAT data, stated that, “In older men with benign prostatic hypertrophy in whom an [alpha]-adrenergic blocker seems like the best treatment for the uropathy, coexisting hypertension should be treated with another antihypertensive drug as well.”3
Again, this would clearly relegate doxazosin to second-line or third-line status, even in patients with benign prostatic hypertrophy, in whom it has been shown to be indicated.
ADVERSE EFFECTS OF ALPHA-BLOCKERS
Dizziness, fatigue, and somnolence are occasionally reported but appear to be well tolerated. Postural hypotension is much less common with proper titration of standard doxazosin or with the use of controlled-release formulations.9–15 However, in patients with impaired autonomic function, even long-acting alpha-blockers can cause postural hypotension and syncope.
Patients using phosphodiesterase type 5 inhibitors—sildenafil (Viagra), vardenafil (Levitra), or tadalafil (Cialis)—for erectile dysfunction should avoid alpha-blockers because the blood-pressure-lowering effects of the two drug classes may be additive.
Alpha-blockers should not be used as first-line therapy for hypertension. However, an alpha-blocker can be considered as a second-line or third-line add-on in a patient whose blood pressure is not under control despite treatment with other drugs.
In addition, alpha-blockers are useful in relieving lower urinary tract symptoms in patients with benign prostatic hypertrophy. However, even in a patient who has both hypertension and benign prostatic hypertrophy, we advise physicians to use alpha-blockers primarily to relieve the urinary symptoms, and we recommend lowering the blood pressure with a drug of a class shown to reduce rates of illness and death.
NOT FIRST-LINE THERAPY
All antihypertensive drugs, including alpha-blockers, lower blood pressure. Alpha-blockers have been approved by the US Food and Drug Administration for treating high blood pressure, and they are just as effective as other antihypertensive drugs—if efficacy is defined as a decrease in millimeters of mercury.
However, lowering the blood pressure is not the main goal of antihypertensive therapy. What we want to achieve when prescribing antihypertensive drugs is to reduce the rates of heart attacks, strokes, and other adverse cardiovascular adverse outcomes, including death.
Unfortunately, alpha-blockers fall short in this regard. In the Antihypertensive and Lipid Lowering Treatment to Prevent Heart Attack (ALLHAT) trial,1,2 doxazosin (Cardura) was found to carry a higher risk of combined cardiovascular disease (relative risk 1.19, P = .04), mostly stroke. Alarmingly, the incidence of symptomatic heart failure in patients on doxazosin was twice that in patients on chlorthalidone (relative risk 2.04, P < .001). Doxazosin was minimally more effective in lowering blood pressure than chlorthalidone, but the small difference in blood pressure was unlikely to have accounted for the significant difference in the risk of heart failure.3
This experience with doxazosin illustrates a key drawback to surrogate end points: a treatment may produce a favorable outcome in the surrogate end point (blood pressure) but produce little or no benefit in terms of the real end point (stroke, myocardial infarction, and heart failure).4
Based on the ALLHAT data as well as on a Veterans Administration study in patients with chronic heart failure in which survival with prazosin (Minipress) was no better than with placebo,5 it seems reasonable to no longer use alpha-blockers as initial therapy for hypertension. This view is reflected by current European6 and American7 guidelines.
ALPHA-BLOCKERS AS PART OF COMBINATION THERAPY
In several clinical trials, alpha-blockers were allowed8 or were specified9,10 as add-on therapy if other drugs failed to control the blood pressure, but they were not used in a randomized fashion. Thus, we cannot judge their effect on cardiovascular outcomes such as heart attack and stroke.
The choice of drugs for combination therapy very often is still empirical and based on personal preference. Doxazosin as add-on therapy, in general, has been shown to be safe and well tolerated.11 But even if it is acceptable, it is not a preferred combination.
In the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT),9 patients received extended-release doxazosin as a third drug if they did not reach their goal blood pressure with either the combination of amlodipine (Norvasc) plus perindopril (Aceon) or atenolol (Tenormin) plus bendroflumethiazide. Extended-release doxazosin was an effective add-on, and there was no apparent excess rate of heart failure in doxazosin users.
In other studies, in patients with uncontrolled hypertension, adding doxazosin as a second- or third-line agent to a gold-standard drug—calcium channel blocker, diuretic, beta-blocker, angiotensin-converting enzyme inhibitor, angiotensin receptor blocker, or combinations of these—allowed significantly more participants to achieve their blood pressure goal.11
Personally, we consider doxazosin in patients whose blood pressure is not controlled with triple therapy with a renin-angiotensin system blocker, a diuretic, and a calcium channel antagonist in full doses. In patients with stage 3 or stage 4 kidney disease who can no longer tolerate renin-angiotensin system blockers, doxazosin may also be a useful adjunct. Whether the metabolic effects of alpha-blockers, such as a reduction in insulin resistance and a decrease in total and low-density lipoprotein cholesterol, will result in lower rates of morbidity and death has not been conclusively determined.
A point of view somewhat more favorable to the use of alpha-blockers has recently been put forward by Chapman et al.12
ALPHA-BLOCKERS ALLEVIATE SYMPTOMS OF BENIGN PROSTATIC HYPERTROPHY
Doxazosin and other alpha-blockers are commonly used to alleviate lower urinary tract symptoms in patients with benign prostatic hypertrophy.
Both high blood pressure and benign prostatic hypertrophy become more common with advancing age, and it has been estimated that both are present in more than 25% of men over age 60.13 Indeed, two trials documented that a significant reduction in symptoms of benign prostatic hypertrophy and in systolic and diastolic blood pressure can be achieved with an alpha-blocker.13,14
This raises the question whether such a “twofer” (treating two disease states with one drug) should be used in clinical practice. We have to consider that the principle of the twofer has never been tested and agree with Davis et al,3 who, in a further analysis of the ALLHAT data, stated that, “In older men with benign prostatic hypertrophy in whom an [alpha]-adrenergic blocker seems like the best treatment for the uropathy, coexisting hypertension should be treated with another antihypertensive drug as well.”3
Again, this would clearly relegate doxazosin to second-line or third-line status, even in patients with benign prostatic hypertrophy, in whom it has been shown to be indicated.
ADVERSE EFFECTS OF ALPHA-BLOCKERS
Dizziness, fatigue, and somnolence are occasionally reported but appear to be well tolerated. Postural hypotension is much less common with proper titration of standard doxazosin or with the use of controlled-release formulations.9–15 However, in patients with impaired autonomic function, even long-acting alpha-blockers can cause postural hypotension and syncope.
Patients using phosphodiesterase type 5 inhibitors—sildenafil (Viagra), vardenafil (Levitra), or tadalafil (Cialis)—for erectile dysfunction should avoid alpha-blockers because the blood-pressure-lowering effects of the two drug classes may be additive.
References
Messerli FH. Implications of discontinuation of doxazosin arm of ALLHAT. Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (commentary). Lancet2000; 355:863–864.
Major cardiovascular events in hypertensive patients randomized to doxazosin vs chlorthalidone: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). ALLHAT Collaborative Research Group. JAMA2000; 283:1967–1975.
Davis BR, Cutler JA, Furberg CD, et al; ALLHAT Collaborative Research Group. Relationship of antihypertensive treatment regimens and change in blood pressure to risk for heart failure in hypertensive patients randomly assigned to doxazosin or chlorthalidone: further analyses from the Antihypertensive and Lipid-Lowering treatment to prevent Heart Attack Trial. Ann Intern Med2002; 137:313–320.
Messerli FH. Doxazosin and congestive heart failure (viewpoint). J Am Coll Cardiol2001; 38:1295–1296.
Cohn JN, Archibald DG, Ziesche S, et al. Effect of vasodilator therapy on mortality in chronic congestive heart failure. Results of a Veterans Administration Cooperative Study. N Engl J Med1986; 314:1547–1552.
Mancia G, De Backer G, Dominiczak A, et al; ESH-ESC Task Force on the Management of Arterial Hypertension. 2007 ESH-ESC practice guidelines for the management of arterial hypertension: ESH-ESC Task Force on the Management of Arterial Hypertension. J Hypertens2007; 25:1751–1762.
Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. JAMA2003; 289:2560–2572.
Jamerson K, Weber MA, Bakris GL, et al; for the ACCOMPLISH Trial Investigators. Benazepril plus amlodipine or hydrochlorothiazide for hypertension in high-risk patients. N Engl J Med2008; 359:2417–2428.
Chapman N, Chang CL, Dahlöf B, Sever PS, Wedel H, Poulter NR; ASCOT Investigators. Effect of doxazosin gastrointestinal therapeutic system as third-line antihypertensive therapy on blood pressure and lipids in the Anglo-Scandinavian Cardiac Outcomes Trial. Circulation2008; 118:42–48.
de Alvaro F, Hernandez-Presa MAASOCIA Study. Effect of doxazosin gastrointestinal therapeutic system on patients with uncontrolled hypertension: the ASOCIA Study. J Cardiovasc Pharmacol2006; 47:271–276.
Black HR. Doxazosin as combination therapy for patients with stage 1 and stage 2 hypertension. J Cardiovasc Pharmacol2003; 41:866–869.
Chapman N, Chen C-Y, Fujita T, et al. Time to re-appraise the role of alpha-1 adrenoceptor antagonists in the management of hypertension?J Hypertens2010; 28:1796–1803.
Steers WD, Kirby RS. Clinical ease of using doxazosin in BPH patients with and without hypertension. Prostate Cancer Prostatic Dis2005; 8:152–157.
Guthrie RM, Siegel RL. A multicenter, community-based study of doxazosin in the treatment of concomitant hypertension and symptomatic benign prostatic hyperplasia: the Hypertension and BPH Intervention Trial (HABIT). Clin Ther1999; 21:1732–1748.
MacDonald R, Wilt TJ, Howe RW. Doxazosin for treating lower urinary tract symptoms compatible with benign prostatic obstruction: a systematic review of efficacy and adverse effects. BJU Int2004; 94:1263–1270.
References
Messerli FH. Implications of discontinuation of doxazosin arm of ALLHAT. Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (commentary). Lancet2000; 355:863–864.
Major cardiovascular events in hypertensive patients randomized to doxazosin vs chlorthalidone: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). ALLHAT Collaborative Research Group. JAMA2000; 283:1967–1975.
Davis BR, Cutler JA, Furberg CD, et al; ALLHAT Collaborative Research Group. Relationship of antihypertensive treatment regimens and change in blood pressure to risk for heart failure in hypertensive patients randomly assigned to doxazosin or chlorthalidone: further analyses from the Antihypertensive and Lipid-Lowering treatment to prevent Heart Attack Trial. Ann Intern Med2002; 137:313–320.
Messerli FH. Doxazosin and congestive heart failure (viewpoint). J Am Coll Cardiol2001; 38:1295–1296.
Cohn JN, Archibald DG, Ziesche S, et al. Effect of vasodilator therapy on mortality in chronic congestive heart failure. Results of a Veterans Administration Cooperative Study. N Engl J Med1986; 314:1547–1552.
Mancia G, De Backer G, Dominiczak A, et al; ESH-ESC Task Force on the Management of Arterial Hypertension. 2007 ESH-ESC practice guidelines for the management of arterial hypertension: ESH-ESC Task Force on the Management of Arterial Hypertension. J Hypertens2007; 25:1751–1762.
Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. JAMA2003; 289:2560–2572.
Jamerson K, Weber MA, Bakris GL, et al; for the ACCOMPLISH Trial Investigators. Benazepril plus amlodipine or hydrochlorothiazide for hypertension in high-risk patients. N Engl J Med2008; 359:2417–2428.
Chapman N, Chang CL, Dahlöf B, Sever PS, Wedel H, Poulter NR; ASCOT Investigators. Effect of doxazosin gastrointestinal therapeutic system as third-line antihypertensive therapy on blood pressure and lipids in the Anglo-Scandinavian Cardiac Outcomes Trial. Circulation2008; 118:42–48.
de Alvaro F, Hernandez-Presa MAASOCIA Study. Effect of doxazosin gastrointestinal therapeutic system on patients with uncontrolled hypertension: the ASOCIA Study. J Cardiovasc Pharmacol2006; 47:271–276.
Black HR. Doxazosin as combination therapy for patients with stage 1 and stage 2 hypertension. J Cardiovasc Pharmacol2003; 41:866–869.
Chapman N, Chen C-Y, Fujita T, et al. Time to re-appraise the role of alpha-1 adrenoceptor antagonists in the management of hypertension?J Hypertens2010; 28:1796–1803.
Steers WD, Kirby RS. Clinical ease of using doxazosin in BPH patients with and without hypertension. Prostate Cancer Prostatic Dis2005; 8:152–157.
Guthrie RM, Siegel RL. A multicenter, community-based study of doxazosin in the treatment of concomitant hypertension and symptomatic benign prostatic hyperplasia: the Hypertension and BPH Intervention Trial (HABIT). Clin Ther1999; 21:1732–1748.
MacDonald R, Wilt TJ, Howe RW. Doxazosin for treating lower urinary tract symptoms compatible with benign prostatic obstruction: a systematic review of efficacy and adverse effects. BJU Int2004; 94:1263–1270.
Monoamine oxidase (MAO) inhibitors were the first drugs for treating depression. Introduced in the 1950s, they were used extensively for the next two decades. Their use declined substantially since then because of their reported side effects, their food and drug interactions, and the introduction of new classes of antidepressants.
This trend may be changing. These drugs can be effective in major depressive disorder, and particularly in major depressive disorder with atypical features and in treatment-resistant depression.
New, selective MAO inhibitors are being developed. Moreover, the selegiline transdermal system (Emsam),1,2 introduced in 2006, offers the potential advantage of eliminating the need for burdensome dietary restrictions and has renewed interest in this group of drugs.
In this article, we discuss the history, pharmacology, safety and tolerability of MAO inhibitors, and we summarize recent MAO inhibitor research. Our goal is to familiarize physicians with this class of drugs, including recent updates regarding their safety profile and liberalized dietary recommendations.
DEPRESSION IS COMMON, DIFFICULT
Depression affects 121 million people worldwide.3 According to a study that compared two surveys of 40,000 people each, the prevalence of major depressive disorder in the United States more than doubled (from 3.3% to 7.0%) from 1992 to 2002.4 Another survey, in 2002 and 2003, revealed the lifetime prevalence of major depressive disorder to be 16.6%.5
Treatments for depression have expanded over the past 20 years, with new classes of drugs such as selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs). However, depression has remained a difficult condition to treat. In the National Institute of Mental Health’s Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study,6 the remission rate in patients treated with the SSRI citalopram (Celexa) for up to 14 weeks was 28% using one measure and 33% using another. Diversifying and understanding existing and emerging therapeutic options is important to the effective treatment of this disease.
THE RISE AND FALL OF MAO INHIBITORS
The first antidepressant introduced was an MAO inhibitor, iproniazid, followed shortly thereafter by a tricyclic antidepressant, imipramine (Tofranil). When iproniazid, originally an antituberculosis agent, was promoted for its antidepressant properties in the 1950s, very little was known about its side effects. It was later removed from the market because of hepatotoxicity, but several other MAO inhibitors had surfaced for the treatment of depression—eg, phenelzine (Nardil), isocarboxazid (Marplan), and tranylcypromine (Parnate).
Currently, MAO inhibitors are typically reserved for third- or fourth-line treatment. As a result, even psychiatrists have little experience with these agents. In a 1999 survey of the Michigan Psychiatric Association,7 12% of practicing psychiatrists said they had never prescribed an MAO inhibitor, another 27% had not prescribed one in the prior 3 years, and only 2% said they prescribed them frequently. A decade earlier, about 25% had said they prescribed them often.8
The prescription rate of MAO inhibitors has remained low during the past 10 years. In a Canadian population-based study9 conducted among older adults in a large health care database from January 1997 to April 2007, the yearly incidence of MAO inhibitor prescriptions decreased from a rate of 3.1 per 100,000 to 1.4 per 100,000. Drug interactions, side effects, preference for other treatments, and dietary restrictions were the reasons most often cited for not prescribing these drugs.7
The side effects of MAO inhibitors were recognized by the mid-1960s, when more than 40 cases of tyramine-induced hypertensive crisis were reported (particularly with tranylcypromine).10,11 Many of the reported events happened after the patient ate tyramine-rich foods such as aged cheese (hence, “the cheese reaction”—more on this below) or drank draft beer.10,11 The US Food and Drug Administration (FDA) consequently established dietary restrictions for patients taking MAO inhibitors, but people found the guidelines cumbersome and often switched to newer drugs that did not require a restrictive diet, such as tricyclics and, much later (in the 1980s), SSRIs.
MAO HAS TWO SUBTYPES
MAO is a flavin-containing enzyme critical for regulating neurotransmitter levels by catabolizing endogenous monoamines (eg, norepinephrine, serotonin, and dopamine) and exogenous amines (eg, dietary tyramine). It is found throughout the body but is more highly concentrated in the liver, kidneys, intestinal wall, and brain.
MAO has two subtypes, isoenzyme A (MAO-A) and isoenzyme B (MAO-B), which vary in their distribution. MAO-A is found primarily in the intestinal tract, liver, and peripheral adrenergic neurons (adrenal glands, arterial vessels, and sympathetic nerves) and preferentially metabolizes serotonin and norepinephrine. MAO-B is found mostly in the brain and liver. However, both isotypes are found in all of the areas mentioned. Since 80% of intestinal MAO is MAO-A, this isoenzyme is primarily responsible for degradation of tyramine, and thus inhibition of MAO-A is associated with the cheese reaction.10,11
TYPES OF MAO INHIBITORS
MAO inhibitors can be classified on the basis of whether they are nonselective or selective for either MAO-A or MAO-B, and whether their effect is reversible.
Nonselective MAO inhibitors are phenelzine, isocarboxazid, and tranylcypromine.
Selective MAO inhibitors. Selegiline is selective for MAO-B. Clorgyline is selective for MAO-A, but it is not available in the United States.
A reversible MAO inhibitor is moclobemide (not available in the United States).
Do selectivity and reversibility matter?
Classic MAO inhibitors such as tranylcypromine and phenelzine are neither reversible (binding to the enzyme for the extent of its lifetime of 14–28 days) nor selective for the subtypes. These drugs were used extensively several decades ago to treat atypical depression, anxiety, and phobias. The only selective MAO inhibitor now available in the United States is selegiline, which inhibits MAO-B at low doses but loses its selectivity at dosages greater than 20 mg/day.
Experimental studies suggest that inhibition of more than 70% of MAO-A activity is necessary for the antidepressant effect of selegiline.12 At oral doses that selectively inhibit MAO-B (5–10 mg/day), selegiline does not seem to have potent antidepressant activity, although it does show success as an adjunctive treatment for Parkinson disease and does not necessitate any dietary restriction. Only at higher oral doses (20–60 mg/day), at which MAO-B selectivity is lost, is the antidepressant effect seen. But the higher doses necessitate dietary restrictions. Therefore, patients who are taking the oral selective MAO inhibitor selegiline have to follow the same dietary restrictions as patients taking the nonselective ones.
Reversible inhibitors of MAO-A have the distinction of being easily displaced by ingested tyramine in the gut and thus do not cause the cheese reaction. However, the only reversible agent available in the world market is moclobemide. It is not available in the United States, and appears to be less effective than older, nonselective MAO inhibitors.13
SELEGILINE TRANSDERMAL SYSTEM
The selegiline transdermal system (Emsam) is the first FDA-approved transdermal patch for treatment of major depression. Patients who are using Emsam at its lowest effective dose of 6 mg/24 hours do not need to follow the dietary restrictions that are needed for all oral MAO inhibitors.
Pharmacokinetics of the selegiline patch
With the transdermal patch, selegiline is extensively absorbed through the skin. Plasma levels are maintained over a 24-hour period, allowing once-daily application. Patches are available that deliver 6, 9, or 12 mg per 24 hours. Steady-state plasma levels are reached after about 5 days.
The bioavailability of selegiline is about 75% with the transdermal delivery system vs 4.4% after oral administration, the lower number being due to first-pass metabolism.1 About 90% of selegiline is bound to plasma proteins and quickly penetrates the central nervous system.
This drug is metabolized by cytochrome P450 isoenzymes, including CYP2C9, CYP2B6, and CYP3A4. Its metabolites are l-methamphetamine and n-desmethylselegiline.
Clinical research showed that dosage adjustments were not necessary in specific populations studied, including patients with various stages of renal or hepatic failure.1 Clearance of selegiline was independent of dose, age, sex, renal function, body weight, or concomitant medications.1
Advantages of the patch system
Since selegiline delivered via the patch is not absorbed through the gut, it has little effect on gut MAO-A and therefore is unlikely to lead to tyramine-induced hypertensive crisis. Studies of the selegiline patch show that inhibition of more than 80% of gut MAO-A is necessary to impair metabolism of tyramine in the gut.1 Therefore, the 6-mg patch will not significantly impair tyramine degradation in the gut. In phase III testing of the selegiline patch, no hypertensive crises were reported among 2,656 outpatients without dietary restrictions. However, it is still recommended that patients on the 9-mg and 12-mg patches follow a tyramine-free diet.1
Although there are no data available to suggest that higher dosages are more effective, it is recommended that the dose be titrated in 3-mg increments at intervals of at least 2 weeks until the maximum recommended dosage of 12 mg/24 hours is reached.2
Disadvantage of the selegiline patch: Cost
The selegiline patch is expensive: $692.99 for 1 month’s supply at a dose of 6 mg/24 hours and $638.99 for 1 month’s supply at a dose of 9 or 12 mg/24 hours (verified with a national pharmacy chain at the time of this writing). Insurance coverage for the patch varies, and documentation may be required from the physician. Oral MAO inhibitors are much less expensive.
SAFETY, TOLERABILITY OF MAO INHIBITORS
Side effects of oral agents
Orthostatic hypotension, dizziness, drowsiness, insomnia, and nausea are the most frequently reported side effects of oral MAO inhibitors.14,15 These side effects can generally be managed symptomatically by slowing the titration, dividing the doses, changing the time it is taken, or, in the case of orthostatic hypotension, increasing fluid intake.14 Phenelzine has the strongest association with sedation.14
Weight gain, edema, muscle pain, myoclonus, paresthesias, sexual dysfunction, and, rarely, hepatotoxicity are late side effects.15–18 Paresthesias, an infrequent side effect, are often treated with pyridoxine supplementation.15
Transient hypertensive episodes within 2 hours after ingestion of MAO inhibitors, which were independent of dietary or drug interactions, have been reported.19 The hypertensive episodes are usually self-limited but in rare cases result in hypertensive crisis.19–21
Serotonin syndrome has been reported with MAO inhibitor monotherapy in rare cases.22 Serotonin syndrome is characterized by mental status changes, restlessness, myoclonus, hyperreflexia, diaphoresis, or evidence of autonomic hyperactivity.23 The syndrome is potentially fatal and is treated symptomatically by removing the offending drugs and giving intravenous rehydration.23
Side effects of the selegiline patch
The most common adverse events with the selegiline patch include application-site reaction (24% vs 12% with placebo), headache (18% vs 17%), insomnia, diarrhea, dry mouth, and dyspepsia.24,25 Dose-related orthostatic hypotension was reported (occurring in 9.8% vs 6.7% with placebo) and was most likely to occur in elderly patients.25 It is suggested that insomnia may be lessened by removing the patch before bedtime. Also, rotating the patch application sites and prompt topical treatment of irritation may lessen local effects.24
Observe a washout period when switching between serotonergic drugs
Most MAO inhibitors irreversibly inhibit MAO for the life of the enzyme, and thus the physiologic effects of phenelzine, isocarboxazid, and tranylcypromine last for up to 2 to 3 weeks.26 Although the elimination half-life of typical MAO inhibitors is short (1.5–4 hours),27,28 their physiologic effects are long-lasting.14
Switching from a MAO inhibitor to another serotonergic agent. Concomitant use of MAO inhibitors and other serotonergic drugs is associated with the risk of serotonin syndrome. After stopping an MAO inhibitor, a 14-day washout period is recommended before starting another serotonergic agent.29 Patients should continue to be monitored closely after the washout period, as cases of serotonin syndrome have been reported later.30 A 14-day washout period is also recommended when switching between MAO inhibitors, although more rapid switches have been made safely.31
Switching from another serotonergic agent to an oral MAO inhibitor. Similarly, a 14-day washout period (or five half-lives) is necessary after stopping most of the serotonergic agents mentioned above before beginning treatment with an oral MAO inhibitor. Fluoxetine (Prozac) has a longer half-life and therefore requires a longer washout period, ie, 5 weeks.
Switching from another serotonergic agent to the selegiline patch. When switching to the selegiline patch from another serotonergic drug, the washout period is 1 week after stopping most drugs or 5 weeks after stopping fluoxetine. One must wait 2 weeks after stopping the selegiline patch before starting therapy with any of the other serotonergic drugs.
Drugs to avoid due to interactions
In view of the risk of severe of drug-drug interactions, particularly the risk of serotonin syndrome, the following serotonin-enhancing compounds are contraindicated in patients taking a MAO inhibitor: SSRIs, SNRIs, tricyclic antidepressants, other MAO inhibitors, mirtazapine, and St. John’s wort. Other pharmaceuticals to be avoided include bupropion, meperidine, tramadol, methadone, propoxyphene, pentazocine, dextromethorphan, and cyclobenzaprine (Table 1). Also, there have been numerous reports of serotonin syndrome with the use of the broad-spectrum, MAO-based antibiotic linezolid (Zyvox), by itself or in conjunction with other serotonergic agents.32–35
Several studies suggested a hazardous combination of nonsubcutaneous sumatriptans (5-HT1B/1D agonists) and MAO-B inhibitors, while subcutaneous sumatriptan migraine-abortive treatment and MAO-B inhibitors appear to be safe.36,37
Also, amphetamines, cough-and-cold preparations, and weight-reducing preparations that contain vasoconstrictors (eg, pseudoephedrine, phenylephrine, phenylpropanolamine, and ephedrine) should be avoided, as the risk of hypertensive crisis increases with these products.
Patients on MAO inhibitors should wear a medical alert bracelet in case they need to undergo emergency surgery and are unable to verbally communicate their drug history. They should be instructed to alert all health care providers about their MAO inhibitor use.14
Beware of worsening depression
Physicians, patients, and family members should be advised to observe for worsening depression or “suicidality” during the course of treatment with MAO inhibitors, as with all antidepressants.
Diet can be more lenient than in the past
The dietary restrictions classically advised for patients taking oral MAO inhibitors were established to prevent hypertensive crises associated with tyramine ingestion. However, some of these restrictions were unsubstantiated,38 and evidence from more recent studies suggests that they are unnecessarily strict39 and may lead to resistance by the physician, the patient, or both to using this potentially beneficial therapy.14 There is also a risk that patients will inadvertently discover that a food that was in the “restricted” list caused them no harm upon ingestion and thus will become cavalier about dietary adherence.39
To prevent dietary noncompliance, physicians should conduct ongoing diet surveys and encourage adherence to evidence-based dietary recommendations.40
The FDA and drug-package inserts for oral MAO inhibitors continue to recommend stringent dietary restrictions, including no aged cheeses or meats, soy sauce, soy beans, soy paste, miso soup, Italian green beans (fava beans), snow peas, broad bean pods, sauerkraut, kimchee, concentrated yeast extracts (Marmite), wine, beer (including alcohol-free beer), and many other foods. However, several studies have measured the tyramine content of food and determined that less than 6 mg per serving is generally safe.39,41 The results of these investigations have led to more lenient dietary guidelines.39
Absolute dietary restrictions include39:
Aged cheeses and meats
Banana peels
Broad bean (fava) pods
Spoiled meats
Marmite
Sauerkraut
Soybean products
Draft beers.
Among the many foods determined to be unnecessarily restricted are avocados; bananas; beef or chicken bouillon; chocolate; fresh and mild cheeses, eg, ricotta, cottage cheese, cream cheese, processed cheese slices; fresh meat, poultry, or fish; meat gravy (fresh); monosodium glutamate; peanuts; properly stored pickled or smoked fish (eg, herring); raspberries; and yeast extracts (except Marmite).39
Dietary restrictions should continue for 2 weeks after stopping an MAO inhibitor.
Dietary restrictions for the selegiline patch
Tyramine-containing foods pose less risk with the selegiline patch than with oral MAO inhibitors, and studies42 show that the 6-mg patch does not necessitate dietary restrictions. The accumulating data suggest that the risk of a tyramine-induced event is extremely low with the patch even in doses above 6 mg. But in the meantime, the recommendations for the 9-mg and 12-mg patches remain the same as for the classic oral MAO inhibitors, and tyramine-containing food should be restricted.
EFFICACY OF MAO INHIBITORS IN CLINICAL PRACTICE
Data from numerous studies suggest MAO inhibitors are effective in managing major depressive disorder, and specifically atypical depression,43–48 treatment-resistant major depressive disorder,49,50 and bipolar depression.51,52 Guidelines from the American Psychiatric Association and the British Association for Psychopharmacology suggest that MAO inhibitors be recommended for treatment of major depressive disorder in patients with atypical features and when other antidepressants have failed.53
MAO inhibitors have also been used in the treatment of Parkinson disease, bulimia, anxiety disorders, anorexia nervosa, and body dysmorphic disorder.54
Major depressive disorder
In controlled trials in outpatients with depression who were treated with therapeutic doses of MAO inhibitors, the response rate was 50% to 70%.55 When tranylcypromine was used in severely depressed inpatients, its efficacy was comparable to that of electroconvulsive therapy, imipramine, and amitriptyline.56 Thase et al,57 in a meta-analysis, found that the MAO inhibitors tranylcypromine, phenelzine, and isocarboxazid were equally effective in treating depression.
Atypical depression
Atypical depression is one of the most common subtypes of major depressive disorder. Diagnostic criteria for major depressive disorder with atypical features include mood reactivity and two of the following: weight gain or hyperphagia, hypersomnolence, leaden paralysis, or an enduring pattern of rejection sensitivity.58 An estimated 30% of outpatients with unipolar depression meet these criteria.59
Multiple randomized controlled trials showed that MAO inhibitors were superior to tricyclic antidepressants in treating atypical depression. One study, involving more than 400 patients, determined that atypical depression responded better to phenelzine than to imipramine.43 Another study evaluating 153 critically depressed patients showed significantly greater response with phenelzine than with imipramine or placebo.49 Furthermore, in another double-blind controlled crossover study, 89 mood-reactive, nonmelancholic, chronically depressed outpatients were found to have a striking response to phenelzine after being unresponsive to imipramine.50 Another report48 indicated that in a double-blind, randomized, placebo-controlled trial among 119 patients with atypical depression treated for 6 weeks, the overall response rates were 78% with phenelzine, 50% with imipramine, and 28% with placebo.
A recent meta-analysis of treatment trials in atypical depression revealed a large mean effect size of 0.45 for the superiority of MAO inhibitors over placebo and a medium mean effect size of 0.27 for the superiority of MAO inhibitors over tricyclic antidepressants.60 Additionally, in a randomized, double-blind placebo-controlled trial, patients with comorbid atypical depression and bulimia showed significant improvement in both bulimic and depressive symptoms when given phenelzine vs imipramine or placebo.61
The current data comparing SSRIs and MAO inhibitors in the treatment of atypical depression are limited. The above-mentioned meta-analysis of three such trials (when moclobemide was used in two out of three trials) revealed no significant difference in efficacy.60 However, the authors themselves warned about the limitations of the studies, including low power to detect differences. Parker and Crawford59 compared self-rating of effectiveness of the various previous treatments in patients with depression with and without atypical features using an online survey. The analysis of the responses of 1,934 patients showed no overall difference in treatment response to both drug and non-drug therapies between respondents with and without atypical features, except with SSRIs. The “atypical” group had a significantly lower mean effectiveness score for SSRIs overall, and a lower mean effectiveness rating for two of six SSRIs examined. The authors speculated that even though there was no differential outcome detected in individuals with atypical depression treated with MAO inhibitors, this negative finding may simply have reflected the low prevalence of sample respondents who received MAO inhibitors (which was 4% in the “atypical depression” group of 338).59
Treatment-resistant depression
The ultimate goal in treating major depressive disorder is to achieve complete remission. If complete remission is not achieved, the risk of relapse is high,62,63 as is the risk of more severe future depressive episodes63 and death from any cause.64 Therefore, the ability of clinicians to make appropriate and evidence-based changes in treatment strategy is of high importance.
The use of MAO inhibitors as a third-line or fourth-line choice for treatment-resistant depression is supported by a number of studies.49,50,65–67 MAO inhibitors appear to be especially effective in the subgroup of patients who have treatment-resistant depression with atypical or anergic bipolar features.
Bipolar depression
Anergic bipolar depression is defined as a condition associated with fatigue, psychomotor retardation, and at least one reversed neurovegetative symptom in a patient with bipolar disorder meeting the criteria for a major depressive episode. According to several trials,51,52,68 MAO inhibitors may be more effective than a tricyclic antidepressant in the treatment of anergic bipolar depression. However, more studies are required to determine the role of antidepressants in general and MAO inhibitors in particular in the management of bipolar depression.
Efficacy of the selegiline patch
The efficacy of the selegiline patch in the treatment of depression was examined in four double-blind placebo-controlled studies.69–72 There were three short-term studies (a 6-week study of 177 patients,69 an 8-week study of 265 patients,72 and an 8-week study of 289 patients70) and a fixed-dose 1-year relapse prevention study of 322 patients.71 The inclusion criterion for the short-term studies was diagnosis of a first or a recurrent episode of major depressive disorder in patients with a Hamilton Depression Rating Scale (HDRS) score higher than 20. The HDRS score was used to assess improvement in depressive symptoms. In all studies, patients on active patch had significant improvement in depressive symptoms on the HDRS compared with placebo. In the relapse prevention study,71 patients with major depressive disorder that responded to transdermal selegiline 6 mg within the first 10 weeks were stratified either to continue receiving the selegiline 6-mg patch or to receive placebo. Those continually receiving selegiline experienced a significantly longer time to relapse. At 12 months, the relapse rate was 16.8% with the selegiline patch vs 30.7% with placebo. The patch was reported to be well tolerated, with the most common side effect being application site reaction. The adherence to the treatment was high—84.2% in the active-patch group and 89.6% in the placebo group.71
DO MAO INHIBITORS HAVE A PLACE IN PRIMARY CARE?
MAO inhibitors have secured their place in the history of psychiatry as the first antidepressants. Overall, MAO inhibitors remain underused. However, with the introduction of new and selective MAO inhibitors including the selegiline patch, and with data suggesting efficacy in the management of certain subtypes of depression, we expect that interest in this class of drugs will grow among psychiatrists. Based on the current guidelines for MAO inhibitors to be used as a third- or fourth-line treatment, as well as on research data, it is premature to recommend their more extensive use in a primary care setting. Whether this will change in the future depends on both the research advances and new, safer formulations of MAO inhibitors.
Compton WM, Conway KP, Stinson FS, Grant BF. Changes in the prevalence of major depression and comorbid substance use disorders in the United States between 1991–1992 and 2001–2002. Am J Psychiatry2006; 163:2141–2147.
Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry2005; 62:593–602.
Trivedi MH, Rush AJ, Wisniewski SR, et al; STAR*D Study Team. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry2006; 163:28–40.
Balon R, Mufti R, Arfken CL. A survey of prescribing practices for monoamine oxidase inhibitors. Psychiatr Serv1999; 50:945–947.
Clary C, Mandos LA, Schweizer E. Results of a brief survey on the prescribing practices for monoamine oxidase inhibitor antidepressants. J Clin Psychiatry1990; 51:226–231.
Shulman KI, Fischer HD, Herrmann N, Huo CY, Anderson GM, Rochon PA. Current prescription patterns and safety profile of irreversible monoamine oxidase inhibitors: a population-based cohort study of older adults. J Clin Psychiatry2009; 70:1681–1686.
Horwitz D, Lovenberg W, Engelman K, Sjoerdsma A. Monoamine oxidase inhibitors, tyramine, and cheese. JAMA1964; 188:1108–1110.
Asatoor AM, Levi AJ, Milne MD. Tranylcypromine and cheese. Lancet1963; 2:733–734.
Gordon MN, Muller CD, Sherman KA, Morgan DG, Azzaro AJ, Wecker L. Oral versus transdermal selegiline: antidepressant-like activity in rats. Pharmacol Biochem Behav1999; 63:501–506.
Lotufu-Neto F, Trivedi M, Thase ME. Meta-analysis of the reversible inhibitors of monoamine oxidase type A moclobemide and brofaromine for the treatment of depression. Neuropsychopharmacology1999; 20:226–247.
Fiedorowicz JG, Swartz KL. The role of monoamine oxidase inhibitors in current psychiatric practice. J Psychiatr Pract2004; 10:239–248.
Evans DL, Davidson J, Raft D. Early and late side effects of phenelzine. J Clin Psychopharmacol1982; 2:208–210.
Fava M. Weight gain and antidepressants. J Clin Psychiatry2000; 61(suppl 11):37–41.
Rabkin J, Quitkin F, Harrison W, Tricamo E, McGrath P. Adverse reactions to monoamine oxidase inhibitors. Part I. A comparative study. J Clin Psychopharmacol1984; 4:270–278.
Gomez-Gil E, Salmeron JM, Mas A. Phenelzine-induced fulminant hepatic failure. Ann Intern Med1996; 124:692–693.
Lavin MR, Mendelwitz A, Kronig MH. Spontaneous hypertensive reactions with monoamine oxidase inhibitors. Biol Psychiatry1993; 34:146–151.
Fisher P. Serotonin syndrome in the elderly after antidepressive monotherapy. J Clin Psychopharmacol1995; 15:440–442.
Sternback H. The serotonin syndrome. Am J Psychiatry1991; 148:705–713.
Thase M. Novel transdermal delivery formulation of the monoamine oxidase inhibitor selegiline nearing release for treatment of depression. J Clin Psychiatry2006; 67:671–672.
Lee KC, Chen JJ. Transdermal selegiline for the treatment of major depressive disorder. Neuropsychiatr Dis Treat2007; 3:527–537.
Cooper AJ. Tyramine and irreversible monoamine oxidase inhibitors in clinical practice. Br J Psychiatry Suppl1989; Oct(6):38–45.
Mallinger AG, Smith E. Pharmacokinetics of monoamine oxidase inhibitors. Psychopharmacol Bull1991; 27:493–502.
Fulton B, Benfield P. Moclobemide. An update of its pharmacological properties and therapeutic use. Drugs1996; 52:450–474.
Marangell LB. Switching antidepressants for treatment-resistant major depression. J Clin Psychiatry2001; 62(suppl 18):12–17.
Gitlin MJ. Venlafaxine, monoamine oxidase inhibitors, and the serotonin syndrome. J Clin Psychopharmacol1997; 17:66–67.
Szuba MP, Hornig-Rohan M, Amsterdam JD. Rapid conversion from one monoamine oxidase inhibitor to another. J Clin Psychiatry1997; 58:307–310.
Lorenz RA, Vandenberg AM, Canepa EA. Serotonergic antidepressants and linezolid: a retrospective chart review and presentation of cases. Int J Psychiatry Med2008; 38:81–90.
Miller DG, Lovell EO. Antibiotic-induced serotonin syndrome. J Emerg Med2008, May1(Epub ahead of print).
Das PK, Wakentin DI, Hewko R, Forrest DL. Serotonin syndrome after concomitant treatment with linezolid and meperidine. Clin Infect Dis2008; 46:264–265.
Packer S, Berman SA. Serotonin syndrome precipitated by the monoamine oxidase inhibitor linezolid. Am J Psychiatry2007; 164:346–347.
Diamond S. The use of sumatriptan in patients on monoamine oxidase inhibitors. Neurology1995; 45:1039–1040.
Fox AW. Subcutaneous sumatriptan pharmacokinetics: delimiting the monoamine oxidase inhibitor effect. Headache2010; 50:249–255.
Gardner DM, Shulman KI, Walker SE, Tailor SA. The making of a user friendly MAOI diet. J Clin Psychiatry1996; 57:99–104.
Sweet RA, Brown EJ, Heimberg RG, et al. Monoamine oxidase inhibitor dietary restrictions: what are we asking patients to give up?J Clin Psychiatry1995; 56:196–201.
Shulman KI, Walker SE. Refining the MAOI diet: tyramine content of pizzas and soy products. J Clin Psychiatry1999; 60:191–193.
Azzaro AJ, Vandenberg CM, Blob LF, et al. Tyramine pressor sensitivity during treatment with the selegiline transdermal system 6 mg/24 h in healthy subjects. J Clin Pharmacol2006; 46:933–944.
Quitkin FM, Stewart JW, McGrath PJ, et al. Columbia atypical depression. A subgroup of depressives with better response to MAOI than to tricyclic antidepressants or placebo. Br J Psychiatry Suppl1993; Sep(21):30–34.
Davidson J, Pelton S. Forms of atypical depression and their response to antidepressant drugs. Psychiatry Res1986; 17:87–95.
Liebowitz MR, Quitkin FM, Stewart JW, et al. Antidepressant specificity in atypical depression. Arch Gen Psychiatry1988; 45:129–137.
Rapaport MH, Thase ME. Translating the evidence on atypical depression into clinical practice. J Clin Psychiatry2007; 68:e11.
Liebowitz MR. Depression with anxiety and atypical depression. J Clin Psychiatry1993; 54(suppl):10–14.
Stewart JW, McGrath PJ, Quitkin FM, et al. Chronic depression: response to placebo, imipramine, and phenelzine. J Clin Psychopharmacol1993; 13:391–396.
McGrath PJ, Stewart JW, Nunes EV, et al. A double-blind crossover trial of imipramine and phenelzine for outpatients with treatment-refractory depression. Am J Psychiatry1993; 150:118–123.
Zarate CA, Tohen M, Baraibar G, Kando JC, Mirin J. Prescribing trends of antidepressants in bipolar depression. J Clin Psychiatry1995; 56:260–264.
Mallinger AG, Frank E, Thase ME, Barwell MM, Diazgranados N, Luckenbaugh DA, Kupfer DJ. Revisiting the effectiveness of standard antidepressants in bipolar disorder: are monoamine oxidase inhibitors superior?Psychopharmacol Bull2009; 42:64–74.
Anderson IM, Nutt DJ, Deakin JF. Evidence-based guidelines for treating depressive disorders with antidepressants: a revision of the 1993 British Association for Psychopharmacology guidelines. British Association for Psychopharmacology. J Psychopharmacol2000; 14:3–20.
Liebowitz MR, Hollander E, Schneier F, et al. Reversible and irreversible monoamine oxidase inhibitors in other psychiatric disorders. Acta Psychiatr Scand Suppl1990; 360:29–34.
Davidson JR, Giller EL, Zisook S, Overall JE. An efficacy study of isocarboxazid and placebo in depression, and its relationship to depressive nosology. Arch Gen Psychiatry1988; 45:120–127.
Razani J, White KL, White J, et al. The safety and efficacy of combined amitriptyline and tranylcypromine antidepressant treatment: a controlled trial. Arch Gen Psychiatry1983; 40:657–661.
Thase ME, Triverdi MH, Rush AJ. MAOIs in the contemporary treatment of depression. Neuropsychopharmacology1995; 12:185–219.
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision. Washington, DC, American Psychiatric Association, 2000.
Parker G, Crawford J. Atypical depression: retrospective self-reporting of treatment effectiveness. Acta Psychiatr Scand2009; 120:213–221.
Henkel V, Mergl R, Algaier AK, Kohnen R, Moller HJ, Hergerl U. Treatment of depression with atypical features: a meta-analytic approach. Psychiatry Res2006; 141:89–101.
Rothschild R, Quitkin HM, Quitkin FM, et al. A double-blind placebo-controlled comparison of phenelzine and imipramine in the treatment of bulimia in atypical depressives. Int J Eat Disord1994; 15:1–9.
Judd LL, Paulus MJ, Schettler PJ, et al. Does incomplete recovery from first life-time major depressive episode herald a chronic course of illness?Am J Psychiatry2000; 157:1501–1504.
Murphy JM, Monson RR, Olivier DC, Sobol AM, Leighton AH. Affective disorders and mortality: a general population study. Arch Gen Psychiatry1987; 44:473–480.
Thase ME, Frank E, Mallinger AG, Hamer T, Kupfer DJ. Treatment of imipramine-resistant recurrent depression, III: efficacy of monoamine oxidase inhibitors. J Clin Psychiatry1992; 53:5–11.
Thase ME, Mallinger AG, McKnight D, Himmelhoch JM. Treatment of imipramine-resistant recurrent depression, IV: a double blind crossover study of tranylcypromine for anergic bipolar depression. Am J Psychiatry1992; 149:195–198.
Quitkin FM, McGrath PJ, Stewart JW, et al. Atypical depression, panic attacks, and response to imipramine and phenelzine: a replication. Arch Gen Psychiatry1990; 47:935–941.
Himmelhoch JM, Thase ME, Mallinger AG, Houck P. Tranylcypromine versus imipramine in anergic bipolar depression. Am J Psychiatry1991; 148:910–916.
Bodkin JA, Amsterdam JD. Transdermal selegiline in major depression: a double-blind, placebo-controlled, parallel-group study in outpatients. Am J Psychiatry2002; 159:1869–1875.
Amsterdam JD. A double-blind, placebo-controlled trial of the safety and efficacy of selegiline transdermal system without dietary restrictions in patients with major depressive disorder. J Clin Psychiatry2003; 64:208–214.
Amsterdam JD, Bodkin JA. Selegiline transdermal system in the prevention of relapse of major depressive disorder: a 52-week, double-blind, placebo-substitution, parallel-group clinical trial. J Clin Psychopharmacol2006; 26:579–586.
Feiger AD, Rickels K, Rynn MA, et al. Selegiline transdermal system for the treatment of major depressive disorder: an 8-week, double-blind, placebo-controlled, flexible-dose titration trial. J Clin Psychiatry2006; 67:1354–1361.
Monoamine oxidase (MAO) inhibitors were the first drugs for treating depression. Introduced in the 1950s, they were used extensively for the next two decades. Their use declined substantially since then because of their reported side effects, their food and drug interactions, and the introduction of new classes of antidepressants.
This trend may be changing. These drugs can be effective in major depressive disorder, and particularly in major depressive disorder with atypical features and in treatment-resistant depression.
New, selective MAO inhibitors are being developed. Moreover, the selegiline transdermal system (Emsam),1,2 introduced in 2006, offers the potential advantage of eliminating the need for burdensome dietary restrictions and has renewed interest in this group of drugs.
In this article, we discuss the history, pharmacology, safety and tolerability of MAO inhibitors, and we summarize recent MAO inhibitor research. Our goal is to familiarize physicians with this class of drugs, including recent updates regarding their safety profile and liberalized dietary recommendations.
DEPRESSION IS COMMON, DIFFICULT
Depression affects 121 million people worldwide.3 According to a study that compared two surveys of 40,000 people each, the prevalence of major depressive disorder in the United States more than doubled (from 3.3% to 7.0%) from 1992 to 2002.4 Another survey, in 2002 and 2003, revealed the lifetime prevalence of major depressive disorder to be 16.6%.5
Treatments for depression have expanded over the past 20 years, with new classes of drugs such as selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs). However, depression has remained a difficult condition to treat. In the National Institute of Mental Health’s Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study,6 the remission rate in patients treated with the SSRI citalopram (Celexa) for up to 14 weeks was 28% using one measure and 33% using another. Diversifying and understanding existing and emerging therapeutic options is important to the effective treatment of this disease.
THE RISE AND FALL OF MAO INHIBITORS
The first antidepressant introduced was an MAO inhibitor, iproniazid, followed shortly thereafter by a tricyclic antidepressant, imipramine (Tofranil). When iproniazid, originally an antituberculosis agent, was promoted for its antidepressant properties in the 1950s, very little was known about its side effects. It was later removed from the market because of hepatotoxicity, but several other MAO inhibitors had surfaced for the treatment of depression—eg, phenelzine (Nardil), isocarboxazid (Marplan), and tranylcypromine (Parnate).
Currently, MAO inhibitors are typically reserved for third- or fourth-line treatment. As a result, even psychiatrists have little experience with these agents. In a 1999 survey of the Michigan Psychiatric Association,7 12% of practicing psychiatrists said they had never prescribed an MAO inhibitor, another 27% had not prescribed one in the prior 3 years, and only 2% said they prescribed them frequently. A decade earlier, about 25% had said they prescribed them often.8
The prescription rate of MAO inhibitors has remained low during the past 10 years. In a Canadian population-based study9 conducted among older adults in a large health care database from January 1997 to April 2007, the yearly incidence of MAO inhibitor prescriptions decreased from a rate of 3.1 per 100,000 to 1.4 per 100,000. Drug interactions, side effects, preference for other treatments, and dietary restrictions were the reasons most often cited for not prescribing these drugs.7
The side effects of MAO inhibitors were recognized by the mid-1960s, when more than 40 cases of tyramine-induced hypertensive crisis were reported (particularly with tranylcypromine).10,11 Many of the reported events happened after the patient ate tyramine-rich foods such as aged cheese (hence, “the cheese reaction”—more on this below) or drank draft beer.10,11 The US Food and Drug Administration (FDA) consequently established dietary restrictions for patients taking MAO inhibitors, but people found the guidelines cumbersome and often switched to newer drugs that did not require a restrictive diet, such as tricyclics and, much later (in the 1980s), SSRIs.
MAO HAS TWO SUBTYPES
MAO is a flavin-containing enzyme critical for regulating neurotransmitter levels by catabolizing endogenous monoamines (eg, norepinephrine, serotonin, and dopamine) and exogenous amines (eg, dietary tyramine). It is found throughout the body but is more highly concentrated in the liver, kidneys, intestinal wall, and brain.
MAO has two subtypes, isoenzyme A (MAO-A) and isoenzyme B (MAO-B), which vary in their distribution. MAO-A is found primarily in the intestinal tract, liver, and peripheral adrenergic neurons (adrenal glands, arterial vessels, and sympathetic nerves) and preferentially metabolizes serotonin and norepinephrine. MAO-B is found mostly in the brain and liver. However, both isotypes are found in all of the areas mentioned. Since 80% of intestinal MAO is MAO-A, this isoenzyme is primarily responsible for degradation of tyramine, and thus inhibition of MAO-A is associated with the cheese reaction.10,11
TYPES OF MAO INHIBITORS
MAO inhibitors can be classified on the basis of whether they are nonselective or selective for either MAO-A or MAO-B, and whether their effect is reversible.
Nonselective MAO inhibitors are phenelzine, isocarboxazid, and tranylcypromine.
Selective MAO inhibitors. Selegiline is selective for MAO-B. Clorgyline is selective for MAO-A, but it is not available in the United States.
A reversible MAO inhibitor is moclobemide (not available in the United States).
Do selectivity and reversibility matter?
Classic MAO inhibitors such as tranylcypromine and phenelzine are neither reversible (binding to the enzyme for the extent of its lifetime of 14–28 days) nor selective for the subtypes. These drugs were used extensively several decades ago to treat atypical depression, anxiety, and phobias. The only selective MAO inhibitor now available in the United States is selegiline, which inhibits MAO-B at low doses but loses its selectivity at dosages greater than 20 mg/day.
Experimental studies suggest that inhibition of more than 70% of MAO-A activity is necessary for the antidepressant effect of selegiline.12 At oral doses that selectively inhibit MAO-B (5–10 mg/day), selegiline does not seem to have potent antidepressant activity, although it does show success as an adjunctive treatment for Parkinson disease and does not necessitate any dietary restriction. Only at higher oral doses (20–60 mg/day), at which MAO-B selectivity is lost, is the antidepressant effect seen. But the higher doses necessitate dietary restrictions. Therefore, patients who are taking the oral selective MAO inhibitor selegiline have to follow the same dietary restrictions as patients taking the nonselective ones.
Reversible inhibitors of MAO-A have the distinction of being easily displaced by ingested tyramine in the gut and thus do not cause the cheese reaction. However, the only reversible agent available in the world market is moclobemide. It is not available in the United States, and appears to be less effective than older, nonselective MAO inhibitors.13
SELEGILINE TRANSDERMAL SYSTEM
The selegiline transdermal system (Emsam) is the first FDA-approved transdermal patch for treatment of major depression. Patients who are using Emsam at its lowest effective dose of 6 mg/24 hours do not need to follow the dietary restrictions that are needed for all oral MAO inhibitors.
Pharmacokinetics of the selegiline patch
With the transdermal patch, selegiline is extensively absorbed through the skin. Plasma levels are maintained over a 24-hour period, allowing once-daily application. Patches are available that deliver 6, 9, or 12 mg per 24 hours. Steady-state plasma levels are reached after about 5 days.
The bioavailability of selegiline is about 75% with the transdermal delivery system vs 4.4% after oral administration, the lower number being due to first-pass metabolism.1 About 90% of selegiline is bound to plasma proteins and quickly penetrates the central nervous system.
This drug is metabolized by cytochrome P450 isoenzymes, including CYP2C9, CYP2B6, and CYP3A4. Its metabolites are l-methamphetamine and n-desmethylselegiline.
Clinical research showed that dosage adjustments were not necessary in specific populations studied, including patients with various stages of renal or hepatic failure.1 Clearance of selegiline was independent of dose, age, sex, renal function, body weight, or concomitant medications.1
Advantages of the patch system
Since selegiline delivered via the patch is not absorbed through the gut, it has little effect on gut MAO-A and therefore is unlikely to lead to tyramine-induced hypertensive crisis. Studies of the selegiline patch show that inhibition of more than 80% of gut MAO-A is necessary to impair metabolism of tyramine in the gut.1 Therefore, the 6-mg patch will not significantly impair tyramine degradation in the gut. In phase III testing of the selegiline patch, no hypertensive crises were reported among 2,656 outpatients without dietary restrictions. However, it is still recommended that patients on the 9-mg and 12-mg patches follow a tyramine-free diet.1
Although there are no data available to suggest that higher dosages are more effective, it is recommended that the dose be titrated in 3-mg increments at intervals of at least 2 weeks until the maximum recommended dosage of 12 mg/24 hours is reached.2
Disadvantage of the selegiline patch: Cost
The selegiline patch is expensive: $692.99 for 1 month’s supply at a dose of 6 mg/24 hours and $638.99 for 1 month’s supply at a dose of 9 or 12 mg/24 hours (verified with a national pharmacy chain at the time of this writing). Insurance coverage for the patch varies, and documentation may be required from the physician. Oral MAO inhibitors are much less expensive.
SAFETY, TOLERABILITY OF MAO INHIBITORS
Side effects of oral agents
Orthostatic hypotension, dizziness, drowsiness, insomnia, and nausea are the most frequently reported side effects of oral MAO inhibitors.14,15 These side effects can generally be managed symptomatically by slowing the titration, dividing the doses, changing the time it is taken, or, in the case of orthostatic hypotension, increasing fluid intake.14 Phenelzine has the strongest association with sedation.14
Weight gain, edema, muscle pain, myoclonus, paresthesias, sexual dysfunction, and, rarely, hepatotoxicity are late side effects.15–18 Paresthesias, an infrequent side effect, are often treated with pyridoxine supplementation.15
Transient hypertensive episodes within 2 hours after ingestion of MAO inhibitors, which were independent of dietary or drug interactions, have been reported.19 The hypertensive episodes are usually self-limited but in rare cases result in hypertensive crisis.19–21
Serotonin syndrome has been reported with MAO inhibitor monotherapy in rare cases.22 Serotonin syndrome is characterized by mental status changes, restlessness, myoclonus, hyperreflexia, diaphoresis, or evidence of autonomic hyperactivity.23 The syndrome is potentially fatal and is treated symptomatically by removing the offending drugs and giving intravenous rehydration.23
Side effects of the selegiline patch
The most common adverse events with the selegiline patch include application-site reaction (24% vs 12% with placebo), headache (18% vs 17%), insomnia, diarrhea, dry mouth, and dyspepsia.24,25 Dose-related orthostatic hypotension was reported (occurring in 9.8% vs 6.7% with placebo) and was most likely to occur in elderly patients.25 It is suggested that insomnia may be lessened by removing the patch before bedtime. Also, rotating the patch application sites and prompt topical treatment of irritation may lessen local effects.24
Observe a washout period when switching between serotonergic drugs
Most MAO inhibitors irreversibly inhibit MAO for the life of the enzyme, and thus the physiologic effects of phenelzine, isocarboxazid, and tranylcypromine last for up to 2 to 3 weeks.26 Although the elimination half-life of typical MAO inhibitors is short (1.5–4 hours),27,28 their physiologic effects are long-lasting.14
Switching from a MAO inhibitor to another serotonergic agent. Concomitant use of MAO inhibitors and other serotonergic drugs is associated with the risk of serotonin syndrome. After stopping an MAO inhibitor, a 14-day washout period is recommended before starting another serotonergic agent.29 Patients should continue to be monitored closely after the washout period, as cases of serotonin syndrome have been reported later.30 A 14-day washout period is also recommended when switching between MAO inhibitors, although more rapid switches have been made safely.31
Switching from another serotonergic agent to an oral MAO inhibitor. Similarly, a 14-day washout period (or five half-lives) is necessary after stopping most of the serotonergic agents mentioned above before beginning treatment with an oral MAO inhibitor. Fluoxetine (Prozac) has a longer half-life and therefore requires a longer washout period, ie, 5 weeks.
Switching from another serotonergic agent to the selegiline patch. When switching to the selegiline patch from another serotonergic drug, the washout period is 1 week after stopping most drugs or 5 weeks after stopping fluoxetine. One must wait 2 weeks after stopping the selegiline patch before starting therapy with any of the other serotonergic drugs.
Drugs to avoid due to interactions
In view of the risk of severe of drug-drug interactions, particularly the risk of serotonin syndrome, the following serotonin-enhancing compounds are contraindicated in patients taking a MAO inhibitor: SSRIs, SNRIs, tricyclic antidepressants, other MAO inhibitors, mirtazapine, and St. John’s wort. Other pharmaceuticals to be avoided include bupropion, meperidine, tramadol, methadone, propoxyphene, pentazocine, dextromethorphan, and cyclobenzaprine (Table 1). Also, there have been numerous reports of serotonin syndrome with the use of the broad-spectrum, MAO-based antibiotic linezolid (Zyvox), by itself or in conjunction with other serotonergic agents.32–35
Several studies suggested a hazardous combination of nonsubcutaneous sumatriptans (5-HT1B/1D agonists) and MAO-B inhibitors, while subcutaneous sumatriptan migraine-abortive treatment and MAO-B inhibitors appear to be safe.36,37
Also, amphetamines, cough-and-cold preparations, and weight-reducing preparations that contain vasoconstrictors (eg, pseudoephedrine, phenylephrine, phenylpropanolamine, and ephedrine) should be avoided, as the risk of hypertensive crisis increases with these products.
Patients on MAO inhibitors should wear a medical alert bracelet in case they need to undergo emergency surgery and are unable to verbally communicate their drug history. They should be instructed to alert all health care providers about their MAO inhibitor use.14
Beware of worsening depression
Physicians, patients, and family members should be advised to observe for worsening depression or “suicidality” during the course of treatment with MAO inhibitors, as with all antidepressants.
Diet can be more lenient than in the past
The dietary restrictions classically advised for patients taking oral MAO inhibitors were established to prevent hypertensive crises associated with tyramine ingestion. However, some of these restrictions were unsubstantiated,38 and evidence from more recent studies suggests that they are unnecessarily strict39 and may lead to resistance by the physician, the patient, or both to using this potentially beneficial therapy.14 There is also a risk that patients will inadvertently discover that a food that was in the “restricted” list caused them no harm upon ingestion and thus will become cavalier about dietary adherence.39
To prevent dietary noncompliance, physicians should conduct ongoing diet surveys and encourage adherence to evidence-based dietary recommendations.40
The FDA and drug-package inserts for oral MAO inhibitors continue to recommend stringent dietary restrictions, including no aged cheeses or meats, soy sauce, soy beans, soy paste, miso soup, Italian green beans (fava beans), snow peas, broad bean pods, sauerkraut, kimchee, concentrated yeast extracts (Marmite), wine, beer (including alcohol-free beer), and many other foods. However, several studies have measured the tyramine content of food and determined that less than 6 mg per serving is generally safe.39,41 The results of these investigations have led to more lenient dietary guidelines.39
Absolute dietary restrictions include39:
Aged cheeses and meats
Banana peels
Broad bean (fava) pods
Spoiled meats
Marmite
Sauerkraut
Soybean products
Draft beers.
Among the many foods determined to be unnecessarily restricted are avocados; bananas; beef or chicken bouillon; chocolate; fresh and mild cheeses, eg, ricotta, cottage cheese, cream cheese, processed cheese slices; fresh meat, poultry, or fish; meat gravy (fresh); monosodium glutamate; peanuts; properly stored pickled or smoked fish (eg, herring); raspberries; and yeast extracts (except Marmite).39
Dietary restrictions should continue for 2 weeks after stopping an MAO inhibitor.
Dietary restrictions for the selegiline patch
Tyramine-containing foods pose less risk with the selegiline patch than with oral MAO inhibitors, and studies42 show that the 6-mg patch does not necessitate dietary restrictions. The accumulating data suggest that the risk of a tyramine-induced event is extremely low with the patch even in doses above 6 mg. But in the meantime, the recommendations for the 9-mg and 12-mg patches remain the same as for the classic oral MAO inhibitors, and tyramine-containing food should be restricted.
EFFICACY OF MAO INHIBITORS IN CLINICAL PRACTICE
Data from numerous studies suggest MAO inhibitors are effective in managing major depressive disorder, and specifically atypical depression,43–48 treatment-resistant major depressive disorder,49,50 and bipolar depression.51,52 Guidelines from the American Psychiatric Association and the British Association for Psychopharmacology suggest that MAO inhibitors be recommended for treatment of major depressive disorder in patients with atypical features and when other antidepressants have failed.53
MAO inhibitors have also been used in the treatment of Parkinson disease, bulimia, anxiety disorders, anorexia nervosa, and body dysmorphic disorder.54
Major depressive disorder
In controlled trials in outpatients with depression who were treated with therapeutic doses of MAO inhibitors, the response rate was 50% to 70%.55 When tranylcypromine was used in severely depressed inpatients, its efficacy was comparable to that of electroconvulsive therapy, imipramine, and amitriptyline.56 Thase et al,57 in a meta-analysis, found that the MAO inhibitors tranylcypromine, phenelzine, and isocarboxazid were equally effective in treating depression.
Atypical depression
Atypical depression is one of the most common subtypes of major depressive disorder. Diagnostic criteria for major depressive disorder with atypical features include mood reactivity and two of the following: weight gain or hyperphagia, hypersomnolence, leaden paralysis, or an enduring pattern of rejection sensitivity.58 An estimated 30% of outpatients with unipolar depression meet these criteria.59
Multiple randomized controlled trials showed that MAO inhibitors were superior to tricyclic antidepressants in treating atypical depression. One study, involving more than 400 patients, determined that atypical depression responded better to phenelzine than to imipramine.43 Another study evaluating 153 critically depressed patients showed significantly greater response with phenelzine than with imipramine or placebo.49 Furthermore, in another double-blind controlled crossover study, 89 mood-reactive, nonmelancholic, chronically depressed outpatients were found to have a striking response to phenelzine after being unresponsive to imipramine.50 Another report48 indicated that in a double-blind, randomized, placebo-controlled trial among 119 patients with atypical depression treated for 6 weeks, the overall response rates were 78% with phenelzine, 50% with imipramine, and 28% with placebo.
A recent meta-analysis of treatment trials in atypical depression revealed a large mean effect size of 0.45 for the superiority of MAO inhibitors over placebo and a medium mean effect size of 0.27 for the superiority of MAO inhibitors over tricyclic antidepressants.60 Additionally, in a randomized, double-blind placebo-controlled trial, patients with comorbid atypical depression and bulimia showed significant improvement in both bulimic and depressive symptoms when given phenelzine vs imipramine or placebo.61
The current data comparing SSRIs and MAO inhibitors in the treatment of atypical depression are limited. The above-mentioned meta-analysis of three such trials (when moclobemide was used in two out of three trials) revealed no significant difference in efficacy.60 However, the authors themselves warned about the limitations of the studies, including low power to detect differences. Parker and Crawford59 compared self-rating of effectiveness of the various previous treatments in patients with depression with and without atypical features using an online survey. The analysis of the responses of 1,934 patients showed no overall difference in treatment response to both drug and non-drug therapies between respondents with and without atypical features, except with SSRIs. The “atypical” group had a significantly lower mean effectiveness score for SSRIs overall, and a lower mean effectiveness rating for two of six SSRIs examined. The authors speculated that even though there was no differential outcome detected in individuals with atypical depression treated with MAO inhibitors, this negative finding may simply have reflected the low prevalence of sample respondents who received MAO inhibitors (which was 4% in the “atypical depression” group of 338).59
Treatment-resistant depression
The ultimate goal in treating major depressive disorder is to achieve complete remission. If complete remission is not achieved, the risk of relapse is high,62,63 as is the risk of more severe future depressive episodes63 and death from any cause.64 Therefore, the ability of clinicians to make appropriate and evidence-based changes in treatment strategy is of high importance.
The use of MAO inhibitors as a third-line or fourth-line choice for treatment-resistant depression is supported by a number of studies.49,50,65–67 MAO inhibitors appear to be especially effective in the subgroup of patients who have treatment-resistant depression with atypical or anergic bipolar features.
Bipolar depression
Anergic bipolar depression is defined as a condition associated with fatigue, psychomotor retardation, and at least one reversed neurovegetative symptom in a patient with bipolar disorder meeting the criteria for a major depressive episode. According to several trials,51,52,68 MAO inhibitors may be more effective than a tricyclic antidepressant in the treatment of anergic bipolar depression. However, more studies are required to determine the role of antidepressants in general and MAO inhibitors in particular in the management of bipolar depression.
Efficacy of the selegiline patch
The efficacy of the selegiline patch in the treatment of depression was examined in four double-blind placebo-controlled studies.69–72 There were three short-term studies (a 6-week study of 177 patients,69 an 8-week study of 265 patients,72 and an 8-week study of 289 patients70) and a fixed-dose 1-year relapse prevention study of 322 patients.71 The inclusion criterion for the short-term studies was diagnosis of a first or a recurrent episode of major depressive disorder in patients with a Hamilton Depression Rating Scale (HDRS) score higher than 20. The HDRS score was used to assess improvement in depressive symptoms. In all studies, patients on active patch had significant improvement in depressive symptoms on the HDRS compared with placebo. In the relapse prevention study,71 patients with major depressive disorder that responded to transdermal selegiline 6 mg within the first 10 weeks were stratified either to continue receiving the selegiline 6-mg patch or to receive placebo. Those continually receiving selegiline experienced a significantly longer time to relapse. At 12 months, the relapse rate was 16.8% with the selegiline patch vs 30.7% with placebo. The patch was reported to be well tolerated, with the most common side effect being application site reaction. The adherence to the treatment was high—84.2% in the active-patch group and 89.6% in the placebo group.71
DO MAO INHIBITORS HAVE A PLACE IN PRIMARY CARE?
MAO inhibitors have secured their place in the history of psychiatry as the first antidepressants. Overall, MAO inhibitors remain underused. However, with the introduction of new and selective MAO inhibitors including the selegiline patch, and with data suggesting efficacy in the management of certain subtypes of depression, we expect that interest in this class of drugs will grow among psychiatrists. Based on the current guidelines for MAO inhibitors to be used as a third- or fourth-line treatment, as well as on research data, it is premature to recommend their more extensive use in a primary care setting. Whether this will change in the future depends on both the research advances and new, safer formulations of MAO inhibitors.
Monoamine oxidase (MAO) inhibitors were the first drugs for treating depression. Introduced in the 1950s, they were used extensively for the next two decades. Their use declined substantially since then because of their reported side effects, their food and drug interactions, and the introduction of new classes of antidepressants.
This trend may be changing. These drugs can be effective in major depressive disorder, and particularly in major depressive disorder with atypical features and in treatment-resistant depression.
New, selective MAO inhibitors are being developed. Moreover, the selegiline transdermal system (Emsam),1,2 introduced in 2006, offers the potential advantage of eliminating the need for burdensome dietary restrictions and has renewed interest in this group of drugs.
In this article, we discuss the history, pharmacology, safety and tolerability of MAO inhibitors, and we summarize recent MAO inhibitor research. Our goal is to familiarize physicians with this class of drugs, including recent updates regarding their safety profile and liberalized dietary recommendations.
DEPRESSION IS COMMON, DIFFICULT
Depression affects 121 million people worldwide.3 According to a study that compared two surveys of 40,000 people each, the prevalence of major depressive disorder in the United States more than doubled (from 3.3% to 7.0%) from 1992 to 2002.4 Another survey, in 2002 and 2003, revealed the lifetime prevalence of major depressive disorder to be 16.6%.5
Treatments for depression have expanded over the past 20 years, with new classes of drugs such as selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs). However, depression has remained a difficult condition to treat. In the National Institute of Mental Health’s Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study,6 the remission rate in patients treated with the SSRI citalopram (Celexa) for up to 14 weeks was 28% using one measure and 33% using another. Diversifying and understanding existing and emerging therapeutic options is important to the effective treatment of this disease.
THE RISE AND FALL OF MAO INHIBITORS
The first antidepressant introduced was an MAO inhibitor, iproniazid, followed shortly thereafter by a tricyclic antidepressant, imipramine (Tofranil). When iproniazid, originally an antituberculosis agent, was promoted for its antidepressant properties in the 1950s, very little was known about its side effects. It was later removed from the market because of hepatotoxicity, but several other MAO inhibitors had surfaced for the treatment of depression—eg, phenelzine (Nardil), isocarboxazid (Marplan), and tranylcypromine (Parnate).
Currently, MAO inhibitors are typically reserved for third- or fourth-line treatment. As a result, even psychiatrists have little experience with these agents. In a 1999 survey of the Michigan Psychiatric Association,7 12% of practicing psychiatrists said they had never prescribed an MAO inhibitor, another 27% had not prescribed one in the prior 3 years, and only 2% said they prescribed them frequently. A decade earlier, about 25% had said they prescribed them often.8
The prescription rate of MAO inhibitors has remained low during the past 10 years. In a Canadian population-based study9 conducted among older adults in a large health care database from January 1997 to April 2007, the yearly incidence of MAO inhibitor prescriptions decreased from a rate of 3.1 per 100,000 to 1.4 per 100,000. Drug interactions, side effects, preference for other treatments, and dietary restrictions were the reasons most often cited for not prescribing these drugs.7
The side effects of MAO inhibitors were recognized by the mid-1960s, when more than 40 cases of tyramine-induced hypertensive crisis were reported (particularly with tranylcypromine).10,11 Many of the reported events happened after the patient ate tyramine-rich foods such as aged cheese (hence, “the cheese reaction”—more on this below) or drank draft beer.10,11 The US Food and Drug Administration (FDA) consequently established dietary restrictions for patients taking MAO inhibitors, but people found the guidelines cumbersome and often switched to newer drugs that did not require a restrictive diet, such as tricyclics and, much later (in the 1980s), SSRIs.
MAO HAS TWO SUBTYPES
MAO is a flavin-containing enzyme critical for regulating neurotransmitter levels by catabolizing endogenous monoamines (eg, norepinephrine, serotonin, and dopamine) and exogenous amines (eg, dietary tyramine). It is found throughout the body but is more highly concentrated in the liver, kidneys, intestinal wall, and brain.
MAO has two subtypes, isoenzyme A (MAO-A) and isoenzyme B (MAO-B), which vary in their distribution. MAO-A is found primarily in the intestinal tract, liver, and peripheral adrenergic neurons (adrenal glands, arterial vessels, and sympathetic nerves) and preferentially metabolizes serotonin and norepinephrine. MAO-B is found mostly in the brain and liver. However, both isotypes are found in all of the areas mentioned. Since 80% of intestinal MAO is MAO-A, this isoenzyme is primarily responsible for degradation of tyramine, and thus inhibition of MAO-A is associated with the cheese reaction.10,11
TYPES OF MAO INHIBITORS
MAO inhibitors can be classified on the basis of whether they are nonselective or selective for either MAO-A or MAO-B, and whether their effect is reversible.
Nonselective MAO inhibitors are phenelzine, isocarboxazid, and tranylcypromine.
Selective MAO inhibitors. Selegiline is selective for MAO-B. Clorgyline is selective for MAO-A, but it is not available in the United States.
A reversible MAO inhibitor is moclobemide (not available in the United States).
Do selectivity and reversibility matter?
Classic MAO inhibitors such as tranylcypromine and phenelzine are neither reversible (binding to the enzyme for the extent of its lifetime of 14–28 days) nor selective for the subtypes. These drugs were used extensively several decades ago to treat atypical depression, anxiety, and phobias. The only selective MAO inhibitor now available in the United States is selegiline, which inhibits MAO-B at low doses but loses its selectivity at dosages greater than 20 mg/day.
Experimental studies suggest that inhibition of more than 70% of MAO-A activity is necessary for the antidepressant effect of selegiline.12 At oral doses that selectively inhibit MAO-B (5–10 mg/day), selegiline does not seem to have potent antidepressant activity, although it does show success as an adjunctive treatment for Parkinson disease and does not necessitate any dietary restriction. Only at higher oral doses (20–60 mg/day), at which MAO-B selectivity is lost, is the antidepressant effect seen. But the higher doses necessitate dietary restrictions. Therefore, patients who are taking the oral selective MAO inhibitor selegiline have to follow the same dietary restrictions as patients taking the nonselective ones.
Reversible inhibitors of MAO-A have the distinction of being easily displaced by ingested tyramine in the gut and thus do not cause the cheese reaction. However, the only reversible agent available in the world market is moclobemide. It is not available in the United States, and appears to be less effective than older, nonselective MAO inhibitors.13
SELEGILINE TRANSDERMAL SYSTEM
The selegiline transdermal system (Emsam) is the first FDA-approved transdermal patch for treatment of major depression. Patients who are using Emsam at its lowest effective dose of 6 mg/24 hours do not need to follow the dietary restrictions that are needed for all oral MAO inhibitors.
Pharmacokinetics of the selegiline patch
With the transdermal patch, selegiline is extensively absorbed through the skin. Plasma levels are maintained over a 24-hour period, allowing once-daily application. Patches are available that deliver 6, 9, or 12 mg per 24 hours. Steady-state plasma levels are reached after about 5 days.
The bioavailability of selegiline is about 75% with the transdermal delivery system vs 4.4% after oral administration, the lower number being due to first-pass metabolism.1 About 90% of selegiline is bound to plasma proteins and quickly penetrates the central nervous system.
This drug is metabolized by cytochrome P450 isoenzymes, including CYP2C9, CYP2B6, and CYP3A4. Its metabolites are l-methamphetamine and n-desmethylselegiline.
Clinical research showed that dosage adjustments were not necessary in specific populations studied, including patients with various stages of renal or hepatic failure.1 Clearance of selegiline was independent of dose, age, sex, renal function, body weight, or concomitant medications.1
Advantages of the patch system
Since selegiline delivered via the patch is not absorbed through the gut, it has little effect on gut MAO-A and therefore is unlikely to lead to tyramine-induced hypertensive crisis. Studies of the selegiline patch show that inhibition of more than 80% of gut MAO-A is necessary to impair metabolism of tyramine in the gut.1 Therefore, the 6-mg patch will not significantly impair tyramine degradation in the gut. In phase III testing of the selegiline patch, no hypertensive crises were reported among 2,656 outpatients without dietary restrictions. However, it is still recommended that patients on the 9-mg and 12-mg patches follow a tyramine-free diet.1
Although there are no data available to suggest that higher dosages are more effective, it is recommended that the dose be titrated in 3-mg increments at intervals of at least 2 weeks until the maximum recommended dosage of 12 mg/24 hours is reached.2
Disadvantage of the selegiline patch: Cost
The selegiline patch is expensive: $692.99 for 1 month’s supply at a dose of 6 mg/24 hours and $638.99 for 1 month’s supply at a dose of 9 or 12 mg/24 hours (verified with a national pharmacy chain at the time of this writing). Insurance coverage for the patch varies, and documentation may be required from the physician. Oral MAO inhibitors are much less expensive.
SAFETY, TOLERABILITY OF MAO INHIBITORS
Side effects of oral agents
Orthostatic hypotension, dizziness, drowsiness, insomnia, and nausea are the most frequently reported side effects of oral MAO inhibitors.14,15 These side effects can generally be managed symptomatically by slowing the titration, dividing the doses, changing the time it is taken, or, in the case of orthostatic hypotension, increasing fluid intake.14 Phenelzine has the strongest association with sedation.14
Weight gain, edema, muscle pain, myoclonus, paresthesias, sexual dysfunction, and, rarely, hepatotoxicity are late side effects.15–18 Paresthesias, an infrequent side effect, are often treated with pyridoxine supplementation.15
Transient hypertensive episodes within 2 hours after ingestion of MAO inhibitors, which were independent of dietary or drug interactions, have been reported.19 The hypertensive episodes are usually self-limited but in rare cases result in hypertensive crisis.19–21
Serotonin syndrome has been reported with MAO inhibitor monotherapy in rare cases.22 Serotonin syndrome is characterized by mental status changes, restlessness, myoclonus, hyperreflexia, diaphoresis, or evidence of autonomic hyperactivity.23 The syndrome is potentially fatal and is treated symptomatically by removing the offending drugs and giving intravenous rehydration.23
Side effects of the selegiline patch
The most common adverse events with the selegiline patch include application-site reaction (24% vs 12% with placebo), headache (18% vs 17%), insomnia, diarrhea, dry mouth, and dyspepsia.24,25 Dose-related orthostatic hypotension was reported (occurring in 9.8% vs 6.7% with placebo) and was most likely to occur in elderly patients.25 It is suggested that insomnia may be lessened by removing the patch before bedtime. Also, rotating the patch application sites and prompt topical treatment of irritation may lessen local effects.24
Observe a washout period when switching between serotonergic drugs
Most MAO inhibitors irreversibly inhibit MAO for the life of the enzyme, and thus the physiologic effects of phenelzine, isocarboxazid, and tranylcypromine last for up to 2 to 3 weeks.26 Although the elimination half-life of typical MAO inhibitors is short (1.5–4 hours),27,28 their physiologic effects are long-lasting.14
Switching from a MAO inhibitor to another serotonergic agent. Concomitant use of MAO inhibitors and other serotonergic drugs is associated with the risk of serotonin syndrome. After stopping an MAO inhibitor, a 14-day washout period is recommended before starting another serotonergic agent.29 Patients should continue to be monitored closely after the washout period, as cases of serotonin syndrome have been reported later.30 A 14-day washout period is also recommended when switching between MAO inhibitors, although more rapid switches have been made safely.31
Switching from another serotonergic agent to an oral MAO inhibitor. Similarly, a 14-day washout period (or five half-lives) is necessary after stopping most of the serotonergic agents mentioned above before beginning treatment with an oral MAO inhibitor. Fluoxetine (Prozac) has a longer half-life and therefore requires a longer washout period, ie, 5 weeks.
Switching from another serotonergic agent to the selegiline patch. When switching to the selegiline patch from another serotonergic drug, the washout period is 1 week after stopping most drugs or 5 weeks after stopping fluoxetine. One must wait 2 weeks after stopping the selegiline patch before starting therapy with any of the other serotonergic drugs.
Drugs to avoid due to interactions
In view of the risk of severe of drug-drug interactions, particularly the risk of serotonin syndrome, the following serotonin-enhancing compounds are contraindicated in patients taking a MAO inhibitor: SSRIs, SNRIs, tricyclic antidepressants, other MAO inhibitors, mirtazapine, and St. John’s wort. Other pharmaceuticals to be avoided include bupropion, meperidine, tramadol, methadone, propoxyphene, pentazocine, dextromethorphan, and cyclobenzaprine (Table 1). Also, there have been numerous reports of serotonin syndrome with the use of the broad-spectrum, MAO-based antibiotic linezolid (Zyvox), by itself or in conjunction with other serotonergic agents.32–35
Several studies suggested a hazardous combination of nonsubcutaneous sumatriptans (5-HT1B/1D agonists) and MAO-B inhibitors, while subcutaneous sumatriptan migraine-abortive treatment and MAO-B inhibitors appear to be safe.36,37
Also, amphetamines, cough-and-cold preparations, and weight-reducing preparations that contain vasoconstrictors (eg, pseudoephedrine, phenylephrine, phenylpropanolamine, and ephedrine) should be avoided, as the risk of hypertensive crisis increases with these products.
Patients on MAO inhibitors should wear a medical alert bracelet in case they need to undergo emergency surgery and are unable to verbally communicate their drug history. They should be instructed to alert all health care providers about their MAO inhibitor use.14
Beware of worsening depression
Physicians, patients, and family members should be advised to observe for worsening depression or “suicidality” during the course of treatment with MAO inhibitors, as with all antidepressants.
Diet can be more lenient than in the past
The dietary restrictions classically advised for patients taking oral MAO inhibitors were established to prevent hypertensive crises associated with tyramine ingestion. However, some of these restrictions were unsubstantiated,38 and evidence from more recent studies suggests that they are unnecessarily strict39 and may lead to resistance by the physician, the patient, or both to using this potentially beneficial therapy.14 There is also a risk that patients will inadvertently discover that a food that was in the “restricted” list caused them no harm upon ingestion and thus will become cavalier about dietary adherence.39
To prevent dietary noncompliance, physicians should conduct ongoing diet surveys and encourage adherence to evidence-based dietary recommendations.40
The FDA and drug-package inserts for oral MAO inhibitors continue to recommend stringent dietary restrictions, including no aged cheeses or meats, soy sauce, soy beans, soy paste, miso soup, Italian green beans (fava beans), snow peas, broad bean pods, sauerkraut, kimchee, concentrated yeast extracts (Marmite), wine, beer (including alcohol-free beer), and many other foods. However, several studies have measured the tyramine content of food and determined that less than 6 mg per serving is generally safe.39,41 The results of these investigations have led to more lenient dietary guidelines.39
Absolute dietary restrictions include39:
Aged cheeses and meats
Banana peels
Broad bean (fava) pods
Spoiled meats
Marmite
Sauerkraut
Soybean products
Draft beers.
Among the many foods determined to be unnecessarily restricted are avocados; bananas; beef or chicken bouillon; chocolate; fresh and mild cheeses, eg, ricotta, cottage cheese, cream cheese, processed cheese slices; fresh meat, poultry, or fish; meat gravy (fresh); monosodium glutamate; peanuts; properly stored pickled or smoked fish (eg, herring); raspberries; and yeast extracts (except Marmite).39
Dietary restrictions should continue for 2 weeks after stopping an MAO inhibitor.
Dietary restrictions for the selegiline patch
Tyramine-containing foods pose less risk with the selegiline patch than with oral MAO inhibitors, and studies42 show that the 6-mg patch does not necessitate dietary restrictions. The accumulating data suggest that the risk of a tyramine-induced event is extremely low with the patch even in doses above 6 mg. But in the meantime, the recommendations for the 9-mg and 12-mg patches remain the same as for the classic oral MAO inhibitors, and tyramine-containing food should be restricted.
EFFICACY OF MAO INHIBITORS IN CLINICAL PRACTICE
Data from numerous studies suggest MAO inhibitors are effective in managing major depressive disorder, and specifically atypical depression,43–48 treatment-resistant major depressive disorder,49,50 and bipolar depression.51,52 Guidelines from the American Psychiatric Association and the British Association for Psychopharmacology suggest that MAO inhibitors be recommended for treatment of major depressive disorder in patients with atypical features and when other antidepressants have failed.53
MAO inhibitors have also been used in the treatment of Parkinson disease, bulimia, anxiety disorders, anorexia nervosa, and body dysmorphic disorder.54
Major depressive disorder
In controlled trials in outpatients with depression who were treated with therapeutic doses of MAO inhibitors, the response rate was 50% to 70%.55 When tranylcypromine was used in severely depressed inpatients, its efficacy was comparable to that of electroconvulsive therapy, imipramine, and amitriptyline.56 Thase et al,57 in a meta-analysis, found that the MAO inhibitors tranylcypromine, phenelzine, and isocarboxazid were equally effective in treating depression.
Atypical depression
Atypical depression is one of the most common subtypes of major depressive disorder. Diagnostic criteria for major depressive disorder with atypical features include mood reactivity and two of the following: weight gain or hyperphagia, hypersomnolence, leaden paralysis, or an enduring pattern of rejection sensitivity.58 An estimated 30% of outpatients with unipolar depression meet these criteria.59
Multiple randomized controlled trials showed that MAO inhibitors were superior to tricyclic antidepressants in treating atypical depression. One study, involving more than 400 patients, determined that atypical depression responded better to phenelzine than to imipramine.43 Another study evaluating 153 critically depressed patients showed significantly greater response with phenelzine than with imipramine or placebo.49 Furthermore, in another double-blind controlled crossover study, 89 mood-reactive, nonmelancholic, chronically depressed outpatients were found to have a striking response to phenelzine after being unresponsive to imipramine.50 Another report48 indicated that in a double-blind, randomized, placebo-controlled trial among 119 patients with atypical depression treated for 6 weeks, the overall response rates were 78% with phenelzine, 50% with imipramine, and 28% with placebo.
A recent meta-analysis of treatment trials in atypical depression revealed a large mean effect size of 0.45 for the superiority of MAO inhibitors over placebo and a medium mean effect size of 0.27 for the superiority of MAO inhibitors over tricyclic antidepressants.60 Additionally, in a randomized, double-blind placebo-controlled trial, patients with comorbid atypical depression and bulimia showed significant improvement in both bulimic and depressive symptoms when given phenelzine vs imipramine or placebo.61
The current data comparing SSRIs and MAO inhibitors in the treatment of atypical depression are limited. The above-mentioned meta-analysis of three such trials (when moclobemide was used in two out of three trials) revealed no significant difference in efficacy.60 However, the authors themselves warned about the limitations of the studies, including low power to detect differences. Parker and Crawford59 compared self-rating of effectiveness of the various previous treatments in patients with depression with and without atypical features using an online survey. The analysis of the responses of 1,934 patients showed no overall difference in treatment response to both drug and non-drug therapies between respondents with and without atypical features, except with SSRIs. The “atypical” group had a significantly lower mean effectiveness score for SSRIs overall, and a lower mean effectiveness rating for two of six SSRIs examined. The authors speculated that even though there was no differential outcome detected in individuals with atypical depression treated with MAO inhibitors, this negative finding may simply have reflected the low prevalence of sample respondents who received MAO inhibitors (which was 4% in the “atypical depression” group of 338).59
Treatment-resistant depression
The ultimate goal in treating major depressive disorder is to achieve complete remission. If complete remission is not achieved, the risk of relapse is high,62,63 as is the risk of more severe future depressive episodes63 and death from any cause.64 Therefore, the ability of clinicians to make appropriate and evidence-based changes in treatment strategy is of high importance.
The use of MAO inhibitors as a third-line or fourth-line choice for treatment-resistant depression is supported by a number of studies.49,50,65–67 MAO inhibitors appear to be especially effective in the subgroup of patients who have treatment-resistant depression with atypical or anergic bipolar features.
Bipolar depression
Anergic bipolar depression is defined as a condition associated with fatigue, psychomotor retardation, and at least one reversed neurovegetative symptom in a patient with bipolar disorder meeting the criteria for a major depressive episode. According to several trials,51,52,68 MAO inhibitors may be more effective than a tricyclic antidepressant in the treatment of anergic bipolar depression. However, more studies are required to determine the role of antidepressants in general and MAO inhibitors in particular in the management of bipolar depression.
Efficacy of the selegiline patch
The efficacy of the selegiline patch in the treatment of depression was examined in four double-blind placebo-controlled studies.69–72 There were three short-term studies (a 6-week study of 177 patients,69 an 8-week study of 265 patients,72 and an 8-week study of 289 patients70) and a fixed-dose 1-year relapse prevention study of 322 patients.71 The inclusion criterion for the short-term studies was diagnosis of a first or a recurrent episode of major depressive disorder in patients with a Hamilton Depression Rating Scale (HDRS) score higher than 20. The HDRS score was used to assess improvement in depressive symptoms. In all studies, patients on active patch had significant improvement in depressive symptoms on the HDRS compared with placebo. In the relapse prevention study,71 patients with major depressive disorder that responded to transdermal selegiline 6 mg within the first 10 weeks were stratified either to continue receiving the selegiline 6-mg patch or to receive placebo. Those continually receiving selegiline experienced a significantly longer time to relapse. At 12 months, the relapse rate was 16.8% with the selegiline patch vs 30.7% with placebo. The patch was reported to be well tolerated, with the most common side effect being application site reaction. The adherence to the treatment was high—84.2% in the active-patch group and 89.6% in the placebo group.71
DO MAO INHIBITORS HAVE A PLACE IN PRIMARY CARE?
MAO inhibitors have secured their place in the history of psychiatry as the first antidepressants. Overall, MAO inhibitors remain underused. However, with the introduction of new and selective MAO inhibitors including the selegiline patch, and with data suggesting efficacy in the management of certain subtypes of depression, we expect that interest in this class of drugs will grow among psychiatrists. Based on the current guidelines for MAO inhibitors to be used as a third- or fourth-line treatment, as well as on research data, it is premature to recommend their more extensive use in a primary care setting. Whether this will change in the future depends on both the research advances and new, safer formulations of MAO inhibitors.
Compton WM, Conway KP, Stinson FS, Grant BF. Changes in the prevalence of major depression and comorbid substance use disorders in the United States between 1991–1992 and 2001–2002. Am J Psychiatry2006; 163:2141–2147.
Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry2005; 62:593–602.
Trivedi MH, Rush AJ, Wisniewski SR, et al; STAR*D Study Team. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry2006; 163:28–40.
Balon R, Mufti R, Arfken CL. A survey of prescribing practices for monoamine oxidase inhibitors. Psychiatr Serv1999; 50:945–947.
Clary C, Mandos LA, Schweizer E. Results of a brief survey on the prescribing practices for monoamine oxidase inhibitor antidepressants. J Clin Psychiatry1990; 51:226–231.
Shulman KI, Fischer HD, Herrmann N, Huo CY, Anderson GM, Rochon PA. Current prescription patterns and safety profile of irreversible monoamine oxidase inhibitors: a population-based cohort study of older adults. J Clin Psychiatry2009; 70:1681–1686.
Horwitz D, Lovenberg W, Engelman K, Sjoerdsma A. Monoamine oxidase inhibitors, tyramine, and cheese. JAMA1964; 188:1108–1110.
Asatoor AM, Levi AJ, Milne MD. Tranylcypromine and cheese. Lancet1963; 2:733–734.
Gordon MN, Muller CD, Sherman KA, Morgan DG, Azzaro AJ, Wecker L. Oral versus transdermal selegiline: antidepressant-like activity in rats. Pharmacol Biochem Behav1999; 63:501–506.
Lotufu-Neto F, Trivedi M, Thase ME. Meta-analysis of the reversible inhibitors of monoamine oxidase type A moclobemide and brofaromine for the treatment of depression. Neuropsychopharmacology1999; 20:226–247.
Fiedorowicz JG, Swartz KL. The role of monoamine oxidase inhibitors in current psychiatric practice. J Psychiatr Pract2004; 10:239–248.
Evans DL, Davidson J, Raft D. Early and late side effects of phenelzine. J Clin Psychopharmacol1982; 2:208–210.
Fava M. Weight gain and antidepressants. J Clin Psychiatry2000; 61(suppl 11):37–41.
Rabkin J, Quitkin F, Harrison W, Tricamo E, McGrath P. Adverse reactions to monoamine oxidase inhibitors. Part I. A comparative study. J Clin Psychopharmacol1984; 4:270–278.
Gomez-Gil E, Salmeron JM, Mas A. Phenelzine-induced fulminant hepatic failure. Ann Intern Med1996; 124:692–693.
Lavin MR, Mendelwitz A, Kronig MH. Spontaneous hypertensive reactions with monoamine oxidase inhibitors. Biol Psychiatry1993; 34:146–151.
Fisher P. Serotonin syndrome in the elderly after antidepressive monotherapy. J Clin Psychopharmacol1995; 15:440–442.
Sternback H. The serotonin syndrome. Am J Psychiatry1991; 148:705–713.
Thase M. Novel transdermal delivery formulation of the monoamine oxidase inhibitor selegiline nearing release for treatment of depression. J Clin Psychiatry2006; 67:671–672.
Lee KC, Chen JJ. Transdermal selegiline for the treatment of major depressive disorder. Neuropsychiatr Dis Treat2007; 3:527–537.
Cooper AJ. Tyramine and irreversible monoamine oxidase inhibitors in clinical practice. Br J Psychiatry Suppl1989; Oct(6):38–45.
Mallinger AG, Smith E. Pharmacokinetics of monoamine oxidase inhibitors. Psychopharmacol Bull1991; 27:493–502.
Fulton B, Benfield P. Moclobemide. An update of its pharmacological properties and therapeutic use. Drugs1996; 52:450–474.
Marangell LB. Switching antidepressants for treatment-resistant major depression. J Clin Psychiatry2001; 62(suppl 18):12–17.
Gitlin MJ. Venlafaxine, monoamine oxidase inhibitors, and the serotonin syndrome. J Clin Psychopharmacol1997; 17:66–67.
Szuba MP, Hornig-Rohan M, Amsterdam JD. Rapid conversion from one monoamine oxidase inhibitor to another. J Clin Psychiatry1997; 58:307–310.
Lorenz RA, Vandenberg AM, Canepa EA. Serotonergic antidepressants and linezolid: a retrospective chart review and presentation of cases. Int J Psychiatry Med2008; 38:81–90.
Miller DG, Lovell EO. Antibiotic-induced serotonin syndrome. J Emerg Med2008, May1(Epub ahead of print).
Das PK, Wakentin DI, Hewko R, Forrest DL. Serotonin syndrome after concomitant treatment with linezolid and meperidine. Clin Infect Dis2008; 46:264–265.
Packer S, Berman SA. Serotonin syndrome precipitated by the monoamine oxidase inhibitor linezolid. Am J Psychiatry2007; 164:346–347.
Diamond S. The use of sumatriptan in patients on monoamine oxidase inhibitors. Neurology1995; 45:1039–1040.
Fox AW. Subcutaneous sumatriptan pharmacokinetics: delimiting the monoamine oxidase inhibitor effect. Headache2010; 50:249–255.
Gardner DM, Shulman KI, Walker SE, Tailor SA. The making of a user friendly MAOI diet. J Clin Psychiatry1996; 57:99–104.
Sweet RA, Brown EJ, Heimberg RG, et al. Monoamine oxidase inhibitor dietary restrictions: what are we asking patients to give up?J Clin Psychiatry1995; 56:196–201.
Shulman KI, Walker SE. Refining the MAOI diet: tyramine content of pizzas and soy products. J Clin Psychiatry1999; 60:191–193.
Azzaro AJ, Vandenberg CM, Blob LF, et al. Tyramine pressor sensitivity during treatment with the selegiline transdermal system 6 mg/24 h in healthy subjects. J Clin Pharmacol2006; 46:933–944.
Quitkin FM, Stewart JW, McGrath PJ, et al. Columbia atypical depression. A subgroup of depressives with better response to MAOI than to tricyclic antidepressants or placebo. Br J Psychiatry Suppl1993; Sep(21):30–34.
Davidson J, Pelton S. Forms of atypical depression and their response to antidepressant drugs. Psychiatry Res1986; 17:87–95.
Liebowitz MR, Quitkin FM, Stewart JW, et al. Antidepressant specificity in atypical depression. Arch Gen Psychiatry1988; 45:129–137.
Rapaport MH, Thase ME. Translating the evidence on atypical depression into clinical practice. J Clin Psychiatry2007; 68:e11.
Liebowitz MR. Depression with anxiety and atypical depression. J Clin Psychiatry1993; 54(suppl):10–14.
Stewart JW, McGrath PJ, Quitkin FM, et al. Chronic depression: response to placebo, imipramine, and phenelzine. J Clin Psychopharmacol1993; 13:391–396.
McGrath PJ, Stewart JW, Nunes EV, et al. A double-blind crossover trial of imipramine and phenelzine for outpatients with treatment-refractory depression. Am J Psychiatry1993; 150:118–123.
Zarate CA, Tohen M, Baraibar G, Kando JC, Mirin J. Prescribing trends of antidepressants in bipolar depression. J Clin Psychiatry1995; 56:260–264.
Mallinger AG, Frank E, Thase ME, Barwell MM, Diazgranados N, Luckenbaugh DA, Kupfer DJ. Revisiting the effectiveness of standard antidepressants in bipolar disorder: are monoamine oxidase inhibitors superior?Psychopharmacol Bull2009; 42:64–74.
Anderson IM, Nutt DJ, Deakin JF. Evidence-based guidelines for treating depressive disorders with antidepressants: a revision of the 1993 British Association for Psychopharmacology guidelines. British Association for Psychopharmacology. J Psychopharmacol2000; 14:3–20.
Liebowitz MR, Hollander E, Schneier F, et al. Reversible and irreversible monoamine oxidase inhibitors in other psychiatric disorders. Acta Psychiatr Scand Suppl1990; 360:29–34.
Davidson JR, Giller EL, Zisook S, Overall JE. An efficacy study of isocarboxazid and placebo in depression, and its relationship to depressive nosology. Arch Gen Psychiatry1988; 45:120–127.
Razani J, White KL, White J, et al. The safety and efficacy of combined amitriptyline and tranylcypromine antidepressant treatment: a controlled trial. Arch Gen Psychiatry1983; 40:657–661.
Thase ME, Triverdi MH, Rush AJ. MAOIs in the contemporary treatment of depression. Neuropsychopharmacology1995; 12:185–219.
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision. Washington, DC, American Psychiatric Association, 2000.
Parker G, Crawford J. Atypical depression: retrospective self-reporting of treatment effectiveness. Acta Psychiatr Scand2009; 120:213–221.
Henkel V, Mergl R, Algaier AK, Kohnen R, Moller HJ, Hergerl U. Treatment of depression with atypical features: a meta-analytic approach. Psychiatry Res2006; 141:89–101.
Rothschild R, Quitkin HM, Quitkin FM, et al. A double-blind placebo-controlled comparison of phenelzine and imipramine in the treatment of bulimia in atypical depressives. Int J Eat Disord1994; 15:1–9.
Judd LL, Paulus MJ, Schettler PJ, et al. Does incomplete recovery from first life-time major depressive episode herald a chronic course of illness?Am J Psychiatry2000; 157:1501–1504.
Murphy JM, Monson RR, Olivier DC, Sobol AM, Leighton AH. Affective disorders and mortality: a general population study. Arch Gen Psychiatry1987; 44:473–480.
Thase ME, Frank E, Mallinger AG, Hamer T, Kupfer DJ. Treatment of imipramine-resistant recurrent depression, III: efficacy of monoamine oxidase inhibitors. J Clin Psychiatry1992; 53:5–11.
Thase ME, Mallinger AG, McKnight D, Himmelhoch JM. Treatment of imipramine-resistant recurrent depression, IV: a double blind crossover study of tranylcypromine for anergic bipolar depression. Am J Psychiatry1992; 149:195–198.
Quitkin FM, McGrath PJ, Stewart JW, et al. Atypical depression, panic attacks, and response to imipramine and phenelzine: a replication. Arch Gen Psychiatry1990; 47:935–941.
Himmelhoch JM, Thase ME, Mallinger AG, Houck P. Tranylcypromine versus imipramine in anergic bipolar depression. Am J Psychiatry1991; 148:910–916.
Bodkin JA, Amsterdam JD. Transdermal selegiline in major depression: a double-blind, placebo-controlled, parallel-group study in outpatients. Am J Psychiatry2002; 159:1869–1875.
Amsterdam JD. A double-blind, placebo-controlled trial of the safety and efficacy of selegiline transdermal system without dietary restrictions in patients with major depressive disorder. J Clin Psychiatry2003; 64:208–214.
Amsterdam JD, Bodkin JA. Selegiline transdermal system in the prevention of relapse of major depressive disorder: a 52-week, double-blind, placebo-substitution, parallel-group clinical trial. J Clin Psychopharmacol2006; 26:579–586.
Feiger AD, Rickels K, Rynn MA, et al. Selegiline transdermal system for the treatment of major depressive disorder: an 8-week, double-blind, placebo-controlled, flexible-dose titration trial. J Clin Psychiatry2006; 67:1354–1361.
Compton WM, Conway KP, Stinson FS, Grant BF. Changes in the prevalence of major depression and comorbid substance use disorders in the United States between 1991–1992 and 2001–2002. Am J Psychiatry2006; 163:2141–2147.
Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry2005; 62:593–602.
Trivedi MH, Rush AJ, Wisniewski SR, et al; STAR*D Study Team. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry2006; 163:28–40.
Balon R, Mufti R, Arfken CL. A survey of prescribing practices for monoamine oxidase inhibitors. Psychiatr Serv1999; 50:945–947.
Clary C, Mandos LA, Schweizer E. Results of a brief survey on the prescribing practices for monoamine oxidase inhibitor antidepressants. J Clin Psychiatry1990; 51:226–231.
Shulman KI, Fischer HD, Herrmann N, Huo CY, Anderson GM, Rochon PA. Current prescription patterns and safety profile of irreversible monoamine oxidase inhibitors: a population-based cohort study of older adults. J Clin Psychiatry2009; 70:1681–1686.
Horwitz D, Lovenberg W, Engelman K, Sjoerdsma A. Monoamine oxidase inhibitors, tyramine, and cheese. JAMA1964; 188:1108–1110.
Asatoor AM, Levi AJ, Milne MD. Tranylcypromine and cheese. Lancet1963; 2:733–734.
Gordon MN, Muller CD, Sherman KA, Morgan DG, Azzaro AJ, Wecker L. Oral versus transdermal selegiline: antidepressant-like activity in rats. Pharmacol Biochem Behav1999; 63:501–506.
Lotufu-Neto F, Trivedi M, Thase ME. Meta-analysis of the reversible inhibitors of monoamine oxidase type A moclobemide and brofaromine for the treatment of depression. Neuropsychopharmacology1999; 20:226–247.
Fiedorowicz JG, Swartz KL. The role of monoamine oxidase inhibitors in current psychiatric practice. J Psychiatr Pract2004; 10:239–248.
Evans DL, Davidson J, Raft D. Early and late side effects of phenelzine. J Clin Psychopharmacol1982; 2:208–210.
Fava M. Weight gain and antidepressants. J Clin Psychiatry2000; 61(suppl 11):37–41.
Rabkin J, Quitkin F, Harrison W, Tricamo E, McGrath P. Adverse reactions to monoamine oxidase inhibitors. Part I. A comparative study. J Clin Psychopharmacol1984; 4:270–278.
Gomez-Gil E, Salmeron JM, Mas A. Phenelzine-induced fulminant hepatic failure. Ann Intern Med1996; 124:692–693.
Lavin MR, Mendelwitz A, Kronig MH. Spontaneous hypertensive reactions with monoamine oxidase inhibitors. Biol Psychiatry1993; 34:146–151.
Fisher P. Serotonin syndrome in the elderly after antidepressive monotherapy. J Clin Psychopharmacol1995; 15:440–442.
Sternback H. The serotonin syndrome. Am J Psychiatry1991; 148:705–713.
Thase M. Novel transdermal delivery formulation of the monoamine oxidase inhibitor selegiline nearing release for treatment of depression. J Clin Psychiatry2006; 67:671–672.
Lee KC, Chen JJ. Transdermal selegiline for the treatment of major depressive disorder. Neuropsychiatr Dis Treat2007; 3:527–537.
Cooper AJ. Tyramine and irreversible monoamine oxidase inhibitors in clinical practice. Br J Psychiatry Suppl1989; Oct(6):38–45.
Mallinger AG, Smith E. Pharmacokinetics of monoamine oxidase inhibitors. Psychopharmacol Bull1991; 27:493–502.
Fulton B, Benfield P. Moclobemide. An update of its pharmacological properties and therapeutic use. Drugs1996; 52:450–474.
Marangell LB. Switching antidepressants for treatment-resistant major depression. J Clin Psychiatry2001; 62(suppl 18):12–17.
Gitlin MJ. Venlafaxine, monoamine oxidase inhibitors, and the serotonin syndrome. J Clin Psychopharmacol1997; 17:66–67.
Szuba MP, Hornig-Rohan M, Amsterdam JD. Rapid conversion from one monoamine oxidase inhibitor to another. J Clin Psychiatry1997; 58:307–310.
Lorenz RA, Vandenberg AM, Canepa EA. Serotonergic antidepressants and linezolid: a retrospective chart review and presentation of cases. Int J Psychiatry Med2008; 38:81–90.
Miller DG, Lovell EO. Antibiotic-induced serotonin syndrome. J Emerg Med2008, May1(Epub ahead of print).
Das PK, Wakentin DI, Hewko R, Forrest DL. Serotonin syndrome after concomitant treatment with linezolid and meperidine. Clin Infect Dis2008; 46:264–265.
Packer S, Berman SA. Serotonin syndrome precipitated by the monoamine oxidase inhibitor linezolid. Am J Psychiatry2007; 164:346–347.
Diamond S. The use of sumatriptan in patients on monoamine oxidase inhibitors. Neurology1995; 45:1039–1040.
Fox AW. Subcutaneous sumatriptan pharmacokinetics: delimiting the monoamine oxidase inhibitor effect. Headache2010; 50:249–255.
Gardner DM, Shulman KI, Walker SE, Tailor SA. The making of a user friendly MAOI diet. J Clin Psychiatry1996; 57:99–104.
Sweet RA, Brown EJ, Heimberg RG, et al. Monoamine oxidase inhibitor dietary restrictions: what are we asking patients to give up?J Clin Psychiatry1995; 56:196–201.
Shulman KI, Walker SE. Refining the MAOI diet: tyramine content of pizzas and soy products. J Clin Psychiatry1999; 60:191–193.
Azzaro AJ, Vandenberg CM, Blob LF, et al. Tyramine pressor sensitivity during treatment with the selegiline transdermal system 6 mg/24 h in healthy subjects. J Clin Pharmacol2006; 46:933–944.
Quitkin FM, Stewart JW, McGrath PJ, et al. Columbia atypical depression. A subgroup of depressives with better response to MAOI than to tricyclic antidepressants or placebo. Br J Psychiatry Suppl1993; Sep(21):30–34.
Davidson J, Pelton S. Forms of atypical depression and their response to antidepressant drugs. Psychiatry Res1986; 17:87–95.
Liebowitz MR, Quitkin FM, Stewart JW, et al. Antidepressant specificity in atypical depression. Arch Gen Psychiatry1988; 45:129–137.
Rapaport MH, Thase ME. Translating the evidence on atypical depression into clinical practice. J Clin Psychiatry2007; 68:e11.
Liebowitz MR. Depression with anxiety and atypical depression. J Clin Psychiatry1993; 54(suppl):10–14.
Stewart JW, McGrath PJ, Quitkin FM, et al. Chronic depression: response to placebo, imipramine, and phenelzine. J Clin Psychopharmacol1993; 13:391–396.
McGrath PJ, Stewart JW, Nunes EV, et al. A double-blind crossover trial of imipramine and phenelzine for outpatients with treatment-refractory depression. Am J Psychiatry1993; 150:118–123.
Zarate CA, Tohen M, Baraibar G, Kando JC, Mirin J. Prescribing trends of antidepressants in bipolar depression. J Clin Psychiatry1995; 56:260–264.
Mallinger AG, Frank E, Thase ME, Barwell MM, Diazgranados N, Luckenbaugh DA, Kupfer DJ. Revisiting the effectiveness of standard antidepressants in bipolar disorder: are monoamine oxidase inhibitors superior?Psychopharmacol Bull2009; 42:64–74.
Anderson IM, Nutt DJ, Deakin JF. Evidence-based guidelines for treating depressive disorders with antidepressants: a revision of the 1993 British Association for Psychopharmacology guidelines. British Association for Psychopharmacology. J Psychopharmacol2000; 14:3–20.
Liebowitz MR, Hollander E, Schneier F, et al. Reversible and irreversible monoamine oxidase inhibitors in other psychiatric disorders. Acta Psychiatr Scand Suppl1990; 360:29–34.
Davidson JR, Giller EL, Zisook S, Overall JE. An efficacy study of isocarboxazid and placebo in depression, and its relationship to depressive nosology. Arch Gen Psychiatry1988; 45:120–127.
Razani J, White KL, White J, et al. The safety and efficacy of combined amitriptyline and tranylcypromine antidepressant treatment: a controlled trial. Arch Gen Psychiatry1983; 40:657–661.
Thase ME, Triverdi MH, Rush AJ. MAOIs in the contemporary treatment of depression. Neuropsychopharmacology1995; 12:185–219.
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision. Washington, DC, American Psychiatric Association, 2000.
Parker G, Crawford J. Atypical depression: retrospective self-reporting of treatment effectiveness. Acta Psychiatr Scand2009; 120:213–221.
Henkel V, Mergl R, Algaier AK, Kohnen R, Moller HJ, Hergerl U. Treatment of depression with atypical features: a meta-analytic approach. Psychiatry Res2006; 141:89–101.
Rothschild R, Quitkin HM, Quitkin FM, et al. A double-blind placebo-controlled comparison of phenelzine and imipramine in the treatment of bulimia in atypical depressives. Int J Eat Disord1994; 15:1–9.
Judd LL, Paulus MJ, Schettler PJ, et al. Does incomplete recovery from first life-time major depressive episode herald a chronic course of illness?Am J Psychiatry2000; 157:1501–1504.
Murphy JM, Monson RR, Olivier DC, Sobol AM, Leighton AH. Affective disorders and mortality: a general population study. Arch Gen Psychiatry1987; 44:473–480.
Thase ME, Frank E, Mallinger AG, Hamer T, Kupfer DJ. Treatment of imipramine-resistant recurrent depression, III: efficacy of monoamine oxidase inhibitors. J Clin Psychiatry1992; 53:5–11.
Thase ME, Mallinger AG, McKnight D, Himmelhoch JM. Treatment of imipramine-resistant recurrent depression, IV: a double blind crossover study of tranylcypromine for anergic bipolar depression. Am J Psychiatry1992; 149:195–198.
Quitkin FM, McGrath PJ, Stewart JW, et al. Atypical depression, panic attacks, and response to imipramine and phenelzine: a replication. Arch Gen Psychiatry1990; 47:935–941.
Himmelhoch JM, Thase ME, Mallinger AG, Houck P. Tranylcypromine versus imipramine in anergic bipolar depression. Am J Psychiatry1991; 148:910–916.
Bodkin JA, Amsterdam JD. Transdermal selegiline in major depression: a double-blind, placebo-controlled, parallel-group study in outpatients. Am J Psychiatry2002; 159:1869–1875.
Amsterdam JD. A double-blind, placebo-controlled trial of the safety and efficacy of selegiline transdermal system without dietary restrictions in patients with major depressive disorder. J Clin Psychiatry2003; 64:208–214.
Amsterdam JD, Bodkin JA. Selegiline transdermal system in the prevention of relapse of major depressive disorder: a 52-week, double-blind, placebo-substitution, parallel-group clinical trial. J Clin Psychopharmacol2006; 26:579–586.
Feiger AD, Rickels K, Rynn MA, et al. Selegiline transdermal system for the treatment of major depressive disorder: an 8-week, double-blind, placebo-controlled, flexible-dose titration trial. J Clin Psychiatry2006; 67:1354–1361.
Data from multiple studies suggest the efficacy of MAO inhibitors in the management of major depressive disorder and, in particular, major depressive disorder with atypical features and in treatment-resistant depression.
When using oral MAO inhibitors, patients must follow a low-tyramine diet to avoid the “cheese reaction,” ie, tyramine-induced hypertensive crisis. However, recent studies suggest that traditional dietary advice may be unnecessarily restrictive.
The selegiline transdermal system (Emsam) is the first approved transdermal patch for treatment of major depression. Unlike oral MAO inhibitors, the patch can be used without the dietary restrictions at its lowest effective dose of 6 mg/24 hours. Because of its transdermal delivery, it has the advantage of not inhibiting the metabolism of dietary tyramine by MAO subtype A in the gut, while providing antidepressant effect in the brain. The patch may be a promising alternative to existing strategies for the management of major depressive disorder.
New clinical trials and observational studies are shedding light on ways to improve the health of elderly patients. Here is a brief summary of these trials and how they might influence your clinical practice.
EXERCISE HAS NEWLY DISCOVERED BENEFITS
According to government data,1 exercise has a dose-dependent effect on rates of all-cause mortality: the more hours one exercises per week, the lower the risk of death. The difference in risk is most pronounced as one goes from no exercise to about 3 hours of exercise per week; above 3 hours per week, the curve flattens out but continues to decline. Hence, we advise patients to engage in about 30 minutes of moderate-intensity exercise every day.
Lately, physical exercise has been found to have other, unexpected benefits.
Exercise helps cognition
ERICKSON KI, PRAKASH RS, VOSS MW, ET AL. AEROBIC FITNESS IS ASSOCIATED WITH HIPPOCAMPAL VOLUME IN ELDERLY HUMANS. HIPPOCAMPUS 2009; 19:1030–1039.
ETGEN T, SANDER D, HUNTGEBURTH U, POPPERT H, FÖRSTL H, BICKEL H. PHYSICAL ACTIVITY AND INCIDENT COGNITIVE IMPAIRMENT IN ELDERLY PERSONS: THE INVADE STUDY. ARCH INTERN MED 2010; 170:186–193.
The hippocampus is a structure deep in the brain that is involved in short-term memory. It atrophies with age, more so with dementia. Erickson2 found a correlation between aerobic fitness (as measured by maximum oxygen consumption), hippocampal volume, and spatial memory performance.
Etgen and colleagues3 studied nearly 4,000 older adults in Bavaria for 2 years. Among those reporting no physical activity, 21.4% had cognitive impairment at baseline, compared with 7.3% of those with high activity at baseline. Following those without cognitive impairment over a 2-year period, they found the incidence of new cognitive impairment was 13.9% in those with no physical activity at baseline, 6.7% in those with moderate activity, and 5.1% in those with high activity.
Exercise boosts the effect of influenza vaccine
WOODS JA, KEYLOCK KT, LOWDER T, ET AL. CARDIOVASCULAR EXERCISE TRAINING EXTENDS INFLUENZA VACCINE SEROPROTECTION IN SEDENTARY OLDER ADULTS: THE IMMUNE FUNCTION INTERVENTION TRIAL. J AM GERIATR SOC 2009; 57:2183–2191.
In a study in 144 sedentary but healthy older adults (ages 60 to 83), Woods et al4 randomized the participants to undergo either flexibility or cardiovascular training for 10 months, starting 4 months before their annual influenza shot. Exercise extended the duration of antibody protection, with more participants in the cardiovascular group than in the flexibility group showing protection at 24 weeks against all three strains covered by the vaccine: H1N1, H3N2, and influenza B.
PREVENTING FRACTURES
Each year, about 30% of people age 65 or older fall, sustaining serious injuries in 5% to 10% of cases. Unintentional falls are the main cause of hip fractures, which number 300,000 per year. They are also a common cause of death.
Vitamin D prevents fractures, but can there be too much of a good thing?
BISCHOFF-FERRARI HA, WILLETT WC, WONG JB, ET AL. PREVENTION OF NONVERTEBRAL FRACTURES WITH ORAL VITAMIN D AND DOSE DEPENDENCY: A META-ANALYSIS OF RANDOMIZED CONTROLLED TRIALS. ARCH INTERN MED 2009; 169:551–561.
SANDERS KM, STUART AL, WILLIAMSON EJ, ET AL. ANNUAL HIGH-DOSE ORAL VITAMIN D AND FALLS AND FRACTURES IN OLDER WOMEN: A RANDOMIZED CONTROLLED TRIAL. JAMA 2010; 303:1815–1822.
Bischoff-Ferrari5 performed a meta-analysis of 12 randomized controlled trials of oral supplemental vitamin D3 for preventing nonvertebral fractures in people age 65 and older, and eight trials for preventing hip fractures in the same age group. They found that the higher the daily dose of vitamin D, the lower the relative risk of hip fracture. The threshold dose at which supplementation significantly reduced the risk of falling was about 400 units per day. Higher doses of vitamin D reduced both falls and hip fractures by about 20%. The maximal effect was seen with studies using the maximum daily doses, ie, 770 to 800 units per day—not megadoses, but more than most Americans are taking. The threshold serum level of vitamin D of significance was 60 nmol/L (24 ng/mL).
Of interest, the effect on fractures was independent of calcium supplementation. This is important because calcium supplementation over and above ordinary dietary intake may increase the risk of cardiovascular events.6,7
Despite the benefits of vitamin D, too much may be too much of a good thing. Sanders et al8 performed a double-blind, placebo-controlled trial in 2,256 community-dwelling women, age 70 or older, who were considered to be at high risk for fractures. Half received a large oral dose (500,000 units) once a year for 3 to 5 years, and half got placebo. Their initial serum vitamin D level was 49 nmol/L; the level 30 days after a dose in the treatment group was 120 nmol/L.
Contrary to expectations, the incidence of falls was 15% higher in the vitamin D group than in the placebo group (P = .03), and the incidence of fractures was 26% higher (P = .047). The falls and fractures tended to cluster in the first 3 months after the dose in the active treatment group, when serum vitamin D levels were highest.
Comments. Unless future studies suggest a benefit to megadoses of vitamin D or prove calcium supplementation greater than 1,000 mg is safe, the optimal daily intake of vitamin D is likely 1,000 units, with approximately 200 units from diet and 800 units from supplements. A diet rich in low-fat dairy products may not require calcium supplementation. In those consuming a low-calcium diet, supplements of 500 to 1,000 mg/day are likely adequate.
Denosumab, a new drug for preventing fractures
CUMMINGS SR, SAN MARTIN J, MCCLUNG MR, ET AL; FREEDOM TRIAL. DENOSUMAB FOR PREVENTION OF FRACTURES IN POSTMENOPAUSAL WOMEN WITH OSTEOPOROSIS. N ENGL J MED 2009; 361:756–765.
SMITH MR, EGERDIE B, HERNÁNDEZ TORIZ N, ET AL; DENOSUMAB HALT PROSTATE CANCER STUDY GROUP. DENOSUMAB IN MEN RECEIVING ANDROGEN-DEPRIVATION THERAPY FOR PROSTATE CANCER. N ENGL J MED 2009; 361:745–755.
Denosumab (Prolia) is the first of a new class of drugs for the treatment of osteoporosis. It is a monoclonal antibody and member of the tumor necrosis factor superfamily that binds to the receptor activator nuclear factor kappa B (RANK) ligand. It has an antiresorptive effect, preventing osteoclast differentiation and activation. It is given by subcutaneous injection of 60 mg every 6 months; it is cleared by a nonrenal mechanism.
In a randomized controlled trial in 7,868 women between the ages of 60 and 90 who had osteoporosis, Cummings et al9 reported that denosumab reduced the 3-year incidence of vertebral fractures by 68% (P < .001), reduced the incidence of hip fractures by 40% (P = .01), and reduced the incidence of nonvertebral fractures by 20% (P = .01). In a trial in men receiving androgen deprivation therapy for prostate cancer, Smith et al10 reported that denosumab reduced the incidence of vertebral fracture by 62% (P = .006).
Comment. Denosumab was approved by the US Food and Drug Administration (FDA) on June 1, 2010, and is emerging in specialty clinics at the time of this publication. Its potential impact on clinical care is not yet known. It is costly—about $825 (average wholesale price) per injection—but since it is given by injection it may be easier than a yearly infusion of zoledronic acid (Reclast). It has the potential to suppress immune function, although this was not reported in the clinical trials. It may ultimately have a role in treating osteoporosis in men and women, prostate cancer following androgen deprivation, metastatic prostate cancer, metastatic breast cancer, osteoporosis with renal impairment, and other diseases.
DIALYSIS IN THE ELDERLY: A BLEAK STORY
KURELLA TAMURA M, COVINSKY KE, CHERTOW GM, YAFFE K, LANDEFELD CS, MCCOLLOCH CE. FUNCTIONAL STATUS OF ELDERLY ADULTS BEFORE AND AFTER INITIATION OF DIALYSIS. N ENGL J MED 2009; 361:1539–1547.
JASSAL SV, CHIU E, HLADUNEWITH M. LOSS OF INDEPENDENCE IN PATIENTS STARTING DIALYSIS AT 80 YEARS OF AGE OR OLDER (LETTER). N ENGL J MED 2009; 361:1612–1613.
Nursing home residents account for 4% of all patients in end-stage renal disease. However, the benefits of dialysis in older patients are uncertain. The mortality rate during the first year of dialysis is 35% in patients 70 years of age and older and 50% in patients 80 years and older.
Is dialysis helpful in the elderly, ie, does it improve survival and function?
Kurella Tamura et al11 retrospectively identified 3,702 nursing home residents starting dialysis in whom functional assessments had been done. The numbers told a bleak story. Initiation of dialysis was associated with a sharp decline in functional status, as reflected in an increase of 2.8 points on the 28-point Minimum Data Set–Activities of Daily Living (MDS-ADL) scale (the higher the score, the worse the function). MDS-ADL scores stabilized at a plateau for about 6 months and then continued to decline. Moreover, at 12 months, 58% of the patients had died.
The MDS-ADL score is based on seven components: eating, bed mobility, locomotion, transferring, toileting, hygiene, and dressing; function declined in all of these areas when patients started dialysis.
Patients were more likely to decline in activities of daily living after starting dialysis if they were older, were white, had cerebrovascular disease, had a diagnosis of dementia, were hospitalized at the start of dialysis, or had a serum albumin level lower than 3.5 g/dL.
The same thing happens to elders living in the community when they start dialysis. Jassal and colleagues12 reported that, of 97 community-dwelling patients (mean age 85), 46 (47%) were dead 2 years after starting dialysis. Although 76 (78%) had been living independently at the start of dialysis, only 11 (11%) were still doing so at 2 years.
Comment. These findings indicate that we do not know if hemodialysis improves survival. Hemodialysis may buy about 3 months of stable function, but it clearly does not restore function.
Is this the best we can do? Standard hemodialysis may have flaws, and nocturnal dialysis and peritoneal dialysis are used more in other countries. These dialysis techniques require more study in our older population. The lesson from these two publications on dialysis is that we should attend more carefully to slowing the decline in renal function before patients reach end-stage renal disease.
DABIGATRAN: AN ALTERNATIVE TO WARFARIN FOR ATRIAL FIBRILLATION
CONNOLLY SJ, EZEKOWITZ MD, YUSUF S, ET AL; RE-LY STEERING COMMITTEE AND INVESTIGATORS. DABIGATRAN VERSUS WARFARIN IN PATIENTS WITH ATRIAL FIBRILLATION. N ENGL J MED 2009; 361:1139–1151.
Atrial fibrillation is common, affecting 2.2 million adults. The median age of people who have atrial fibrillation is 75 years, and it is the most common arrhythmia in the elderly. Some 20% of ischemic strokes are attributed to it.13–15
Warfarin (Coumadin) is still the mainstay of treatment to prevent stroke in patients with atrial fibrillation. In an analysis of pooled data from five clinical trials,16 the relative risk reduction with warfarin was about 68% in the overall population (number needed to treat 32), 51% in people older than 75 years with no other risk factors (number needed to treat 56), and 85% in people older than 75 years with one or more risk factors (number needed to treat 15).
But warfarin carries a risk of bleeding, and its dose must be periodically adjusted on the basis of the international normalized ratio (INR) of the prothrombin time, so it carries a burden of laboratory monitoring. It is less safe in people who eat erratically, resulting in wide fluctuations in the INR.
Dabigatran (Pradaxa), a direct thrombin inhibitor, is expected to become an alternative to warfarin. It has been approved in Europe but not yet in the United States.
Connolly et al,17 in a randomized, double-blind trial, assigned 18,113 patients who had atrial fibrillation to receive either dabigatran 110 or 150 mg twice daily or adjusted-dose warfarin in an unblinded fashion. At 2 years, the rates of stroke and systemic embolism were about the same with dabigatran 110 mg as with warfarin but were lower with dabigatran 150 mg (relative risk 0.66, 95% confidence interval [CI] 0.53–0.82, P < .001). The rate of major bleeding was lower with dabigatran 110 mg than with warfarin (2.71% per year vs 3.36% per year, P = .003), but it was similar with dabigatran 150 mg (3.11% per year). Rates of life-threatening bleeding were 1.80% with warfarin, 1.22% with dabigatran 110 mg (P < .05), and 1.45% with dabigatran 150 mg (P < .05).
Comment. I suspect that warfarin’s days are numbered. Dabigatran 110 or 150 mg was as safe and as effective as warfarin in clinical trials, and probably will be more effective than warfarin in clinical practice. It will also probably be safer than warfarin in clinical practice, particularly in challenging settings such as long-term care. On the other hand, it will likely be much more expensive than warfarin.
DEMENTIA
Adverse effects of cholinesterase inhibitors
GILL SS, ANDERSON GM, FISCHER HD, ET AL. SYNCOPE AND ITS CONSEQUENCES IN PATIENTS WITH DEMENTIA RECEIVING CHOLINESTERASE INHIBITORS: A POPULATION-BASED COHORT STUDY. ARCH INTERN MED 2009; 169:867–873.
Cholinesterase inhibitors, eg, donepezil (Aricept), galantamine (Razadyne), and rivastigmine (Exelon), are commonly used to treat Alzheimer disease. However, these drugs carry risks of serious adverse effects.
Gill et al18 retrospectively reviewed a database from Ontario, Canada, and identified about 20,000 community-dwelling elderly persons admitted to the hospital who had been prescribed cholinesterase inhibitors and about three times as many matched controls.
Several adverse events were more frequent in people receiving cholinesterase inhibitors. Findings (events per 1,000 person-years):
Hospital visits for syncope: 31.5 vs 18.6, adjusted hazard ratio (HR) 1.76, 95% CI 1.57–1.98
Hip fractures: 22.4 vs 19.8, HR 1.18, 85% CI 1.04–1.34
Hospital visits for bradycardia: 6.9 vs 4.4, HR 1.69, 95% CI 1.32–2.15
Permanent pacemaker insertion: 4.7 vs 3.3, HR 1.49, 95% CI 1.12–2.00.
Comment. This study adds to the concerns that cholinesterase inhibitors, which have only modest cognitive benefits, may increase the risk of falls, injury, and need for pacemaker placement in demented patients. A low threshold to stop medications in this class should be considered when a patient on a cholinesterase inhibitor presents with bradycardia, falls, and syncope.
The importance of ‘staging’ dementia
IVERSON DJ, GRONSETH GS, REGER MA, ET AL; STANDARDS SUBCOMMITTEE OF THE AMERICAN ACADEMY OF NEUROLOGY. PRACTICE PARAMETER UPDATE: EVALUATION AND MANAGEMENT OF DRIVING RISK IN DEMENTIA: REPORT OF THE QUALITY STANDARDS SUBCOMMITTEE OF THE AMERICAN ACADEMY OF NEUROLOGY. NEUROLOGY 2010; 74:1316–1324.
The Clinical Dementia Rating (CDR) is a simple scale that should be applied by clinicians to describe stage of dementia in patients with Alzheimer disease. This scale can be useful in a variety of settings, from prescribing antidementia drugs to determining whether a patient should still drive. Although research protocols utilize a survey or semistructured interview to derive the stage, the clinician can estimate the stage easily in the office, particularly if there is an informant who can comment on performance outside the office.
There are four stages to the CDR19:
0: No dementia
0.5: Mild memory deficit but intact function
1.0: Moderate memory loss with mild functional impairment
2.0: Severe memory loss, moderate functional impairment
3.0: Severe memory loss, no significant function outside of the house.
Comment. The first stage (0.5, mild memory deficit but intact function) corresponds to “mild cognitive impairment.” In the clinic, these patients tend to take more notes. They come to the appointment with a little book and they write everything down so they don’t forget. They do arrive at their appointments on time; they are not crashing the car; they are paying their bills.
Patients with CDR stage 1.0 dementia (moderate memory loss with mild functional impairment) may miss appointments, they may confuse their medications, and they may have problems driving. They are still taking care of their basic needs, and they show up for appointments acceptably washed and dressed. However, they are likely having trouble shopping and managing their finances.
Patients with severe memory loss and moderate functional impairment (CDR stage 2.0) may not realize they haven’t bathed for a week or have worn the same clothes repeatedly. They are having trouble with basic activities of daily living, such as bathing and toilet hygiene. However, if you were to encounter them socially and didn’t talk to them for too long, you might think they were normal.
Those with severe memory loss and no significant function outside the house (CDR stage 3.0) are the most severely disabled. Dementia in these individuals is recognizable at a glance, from across the room.
Alzheimer patients progress through the stages, from CDR stage 0.5 at about 1 year to stage 1 by about 2 years, to stage 2 by 5 years, and to stage 3 at 8 or 9 years.20
In prescribing antidementia medications. The CDR can help with prescribing antidementia drugs. No medications are approved by the FDA for stage 0 or 0.5. Cholinesterase inhibitors are approved for stages 1, 2, and 3; memantine (Namenda) is approved for stages 2 and 3.
Advising about driving. The CDR is the only risk predictor with a quality-of-evidence rating of A. More than half of people with stage 0.5 memory impairment are safe drivers; fewer than half of those with stage 1.0 are still safe drivers; and patients with stage 2.0 dementia should not be driving at all.21 An adverse rating by a caregiver carries a quality-of-evidence rating of B. Predictors of driving risk with a quality-of-evidence rating of C are decreased mileage due to self-restriction, agitation, or aggression; a crash in the past 1 to 5 years; a citation in the past 2 to 3 years; and a Folstein Mini-Mental State Examination score of 24 or less. Studies also show that a memory-impaired person’s self-rating of safe driving ability or of assurance that he or she avoids unsafe situations is not reliable.21
DELIRIUM
Delirium goes by a number of synonyms, eg, “sundowning,” acute confusional state, acute change in mental status, metabolic encephalopathy, toxic encephalopathy (psychosis), acute brain syndrome, and acute toxic psychosis.
Delirium is common in hospitalized elderly patients, occurring in 11% to 42% of elderly hospitalized patients overall, up to 53% of elderly surgical patients on regular hospital floors, 80% of elderly surgical patients in intensive care, and about half of elderly patients after undergoing coronary artery bypass grafting. Unfortunately, it is undiagnosed in 30% to 60% of cases.22–24
Many pathways can lead to delirium, including hypoxemia, metabolic derangement, drug effects, systemic inflammation, and infection.25
Outcomes can vary from full recovery to death. After 1 year, 50% of those who leave the hospital with some evidence of delirium have not regained their baseline function. Delirium also increases the cost of care and the risk of institutionalization.
Delirium can accelerate dementia
FONG TG, JONES RN, SHI P, ET AL. DELIRIUM ACCELERATES COGNITIVE DECLINE IN ALZHEIMER DISEASE. NEUROLOGY 2009; 72:1570–1575.
Delirium accelerates the course of dementia in patients who had some evidence of dementia before they entered the hospital. Often, the change is noticeable by the family.26
Preventing delirium
INOUYE SK BOGARDUS ST JR, CHARPENTIER PA, ET AL. A MULTICOMPONENT INTERVENTION TO PREVENT DELIRIUM IN HOSPITALIZED OLDER PATIENTS. N ENGL J MED 1999; 340:669–676.
LUNDSTRÖM M, OLOFSSON B, STENVALL M, ET AL. POSTOPERATIVE DELIRIUM IN OLD PATIENTS WITH FEMORAL NECK FRACTURE: A RANDOMIZED INTERVENTION STUDY. AGING CLIN EXP RES 2007; 19:178–186.
Delirium can often be prevented. In a report published in 1999, Inouye et al27 described the outcomes of a program to prevent delirium in hospitalized medically ill elderly patients. Interventions were aimed at optimizing cognitive function, preventing sleep deprivation, avoiding immobility, improving vision and hearing, and treating dehydration. The incidence of delirium was 9.9% in the intervention group vs 15% in the control group, a 40% reduction (P < .05).
Lundström et al28 implemented a similar program for elderly patients with hip fractures. Interventions included staff education and teamwork; active prevention, detection, and treatment of delirium; transfusions if hemoglobin levels were less than 10 g/dL; prompt removal of indwelling urinary catheters, with screening for urinary retention; active prevention and treatment of constipation; and protein-enriched meals. The incidence of delirium was 55% in the intervention group vs 75% in the control group, a 27% reduction.
Comment. Although we have long known that the risk of delirium in medical and surgical patients can be reduced, most hospitals do not have systematic programs to detect delirium and reduce its incidence. Hopefully, reduction in delirium risk will also reduce its adverse consequences, including worsening of dementia and increased mortality.
Erickson KI, Prakash RS, Voss MW, et al. Aerobic fitness is associated with hippocampal volume in elderly humans. Hippocampus2009; 19:1030–1039.
Etgen T, Sander D, Huntgeburth U, Poppert H, Förstl H, Bickel H. Physical activity and incident cognitive impairment in elderly persons: the INVADE study. Arch Intern Med2010; 170:186–193.
Woods JA, Keylock KT, Lowder T, et al. Cardiovascular exercise training extends influenza vaccine seroprotection in sedentary older adults: the immune function intervention trial. J Am Geriatr Soc2009; 57:2183–2191.
Bischoff-Ferrari HA, Willett WC, Wong JB, et al. Prevention of nonvertebral fractures with oral vitamin D and dose dependency: a meta-analysis of randomized controlled trials. Arch Intern Med2009; 169:551–561.
Bolland MJ, Avenell A, Baron JA, et al. Effect of calcium supplements on risk of myocardial infarction and cardiovascular events: meta-analysis. BMJ2010; 341:c3691. doi:10.1136/bmj.c3691.
Bolland MJ, Barber PA, Doughty RN, et al. Vascular events in healthy older women receiving calcium supplementation: randomised controlled trial. BMJ2008; 336:262–266.
Sanders KM, Stuart AL, Williamson EJ, et al. Annual high-dose oral vitamin D and falls and fractures in older women: a randomized controlled trial. JAMA2010; 303:1815–1822.
Cummings SR, San Martin J, McClung MR, et al; FREEDOM Trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med2009; 361:756–765.
Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med2009; 361:745–755.
Kurella Tamura M, Covinsky KE, Chertow GM, Yaffe K, Landefeld CS, McColloch CE. Functional status of elderly adults before and after initiation of dialysis. N Engl J Med2009; 361:1539–1547.
Jassal SV, Chiu E, Hladunewich M. Loss of independence in patients starting dialysis at 80 years of age or older (letter). N Engl J Med2009; 361:1612–1613.
Feinberg WM, Blackshear JL, Laupacis A, Kronmal R, Hart RG. Prevalence, age distribution and gender of patients with atrial fibrillation. Analysis and implications. Arch Intern Med1995; 155:469–473.
Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation: a major contributor to stroke in the elderly. The Framingham Study. Arch Intern Med1987; 147:1561–1564.
Lin HJ, Wolf PA, Kelly-Hayes M, et al. Stroke severity in atrial fibrillation. The Framingham Study. Stroke1996; 27:1760–1764.
Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis of pooled data from five randomized controlled trials. Arch Intern Med1994; 154:1449–1457.
Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med2009; 361:1139–1151.
Gill SS, Anderson GM, Fischer HD, et al. Syncope and its consequences in patients with dementia receiving cholinesterase inhibitors: a population-based cohort study. Arch Intern Med2009; 169:867–873.
Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology1993; 43:2412–2414.
Sloane PD. Advances in the treatment of Alzheimer’s disease. Am Fam Physician1998; 58:1577–1586.
Iverson DJ, Gronseth GS, Reger MA, et al; Standards Subcommittee of the American Academy of Neurology. Practice parameter update: evaluation and management of driving risk in dementia: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology2010; 74:1316–1324.
Demeure MJ, Fain MJ. The elderly surgical patient and postoperative delirium. J Am Coll Surg2006; 203:752–757.
Siddiqi N, House AO, Holmes JD. Occurrence and outcome of delirium in medical in-patients: a systematic literature review. Age Ageing2006; 35:350–364.
Rudolph JL, Jones RN, Levkoff SE, et al. Derivation and validation of a preoperative prediction rule for delirium after cardiac surgery. Circulation2009; 119:229–236.
Fong TG, Tulebaev SR, Inouye SK. Delirium in elderly adults: diagnosis, prevention and treatment. Nat Rev Neurol2009; 5:210–220.
Fong TG, Jones RN, Shi P, et al. Delirium accelerates cognitive decline in Alzheimer disease. Neurology2009; 72:1570–1575.
Inouye SK, Bogardus ST, Charpentier PA, et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med1999; 340:669–676.
Lundström M, Olofsson B, Stenvall M, et al. Postoperative delirium in old patients with femoral neck fracture: a randomized intervention study. Aging Clin Exp Res2007; 19:178–186.
Barbara Messinger-Rapport, MD, PhD, CMD Director, Center for Geriatric Medicine, Medicine Institute, Cleveland Clinic
Address: Barbara M. Messinger-Rapport, MD, PhD, Center for Geriatric Medicine, G10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
Medical Grand Rounds articles are based on edited transcripts from Medicine Grand Rounds presentations at Cleveland Clinic. They are approved by the author but are not peer-reviewed.
Barbara Messinger-Rapport, MD, PhD, CMD Director, Center for Geriatric Medicine, Medicine Institute, Cleveland Clinic
Address: Barbara M. Messinger-Rapport, MD, PhD, Center for Geriatric Medicine, G10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
Medical Grand Rounds articles are based on edited transcripts from Medicine Grand Rounds presentations at Cleveland Clinic. They are approved by the author but are not peer-reviewed.
Author and Disclosure Information
Barbara Messinger-Rapport, MD, PhD, CMD Director, Center for Geriatric Medicine, Medicine Institute, Cleveland Clinic
Address: Barbara M. Messinger-Rapport, MD, PhD, Center for Geriatric Medicine, G10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
Medical Grand Rounds articles are based on edited transcripts from Medicine Grand Rounds presentations at Cleveland Clinic. They are approved by the author but are not peer-reviewed.
New clinical trials and observational studies are shedding light on ways to improve the health of elderly patients. Here is a brief summary of these trials and how they might influence your clinical practice.
EXERCISE HAS NEWLY DISCOVERED BENEFITS
According to government data,1 exercise has a dose-dependent effect on rates of all-cause mortality: the more hours one exercises per week, the lower the risk of death. The difference in risk is most pronounced as one goes from no exercise to about 3 hours of exercise per week; above 3 hours per week, the curve flattens out but continues to decline. Hence, we advise patients to engage in about 30 minutes of moderate-intensity exercise every day.
Lately, physical exercise has been found to have other, unexpected benefits.
Exercise helps cognition
ERICKSON KI, PRAKASH RS, VOSS MW, ET AL. AEROBIC FITNESS IS ASSOCIATED WITH HIPPOCAMPAL VOLUME IN ELDERLY HUMANS. HIPPOCAMPUS 2009; 19:1030–1039.
ETGEN T, SANDER D, HUNTGEBURTH U, POPPERT H, FÖRSTL H, BICKEL H. PHYSICAL ACTIVITY AND INCIDENT COGNITIVE IMPAIRMENT IN ELDERLY PERSONS: THE INVADE STUDY. ARCH INTERN MED 2010; 170:186–193.
The hippocampus is a structure deep in the brain that is involved in short-term memory. It atrophies with age, more so with dementia. Erickson2 found a correlation between aerobic fitness (as measured by maximum oxygen consumption), hippocampal volume, and spatial memory performance.
Etgen and colleagues3 studied nearly 4,000 older adults in Bavaria for 2 years. Among those reporting no physical activity, 21.4% had cognitive impairment at baseline, compared with 7.3% of those with high activity at baseline. Following those without cognitive impairment over a 2-year period, they found the incidence of new cognitive impairment was 13.9% in those with no physical activity at baseline, 6.7% in those with moderate activity, and 5.1% in those with high activity.
Exercise boosts the effect of influenza vaccine
WOODS JA, KEYLOCK KT, LOWDER T, ET AL. CARDIOVASCULAR EXERCISE TRAINING EXTENDS INFLUENZA VACCINE SEROPROTECTION IN SEDENTARY OLDER ADULTS: THE IMMUNE FUNCTION INTERVENTION TRIAL. J AM GERIATR SOC 2009; 57:2183–2191.
In a study in 144 sedentary but healthy older adults (ages 60 to 83), Woods et al4 randomized the participants to undergo either flexibility or cardiovascular training for 10 months, starting 4 months before their annual influenza shot. Exercise extended the duration of antibody protection, with more participants in the cardiovascular group than in the flexibility group showing protection at 24 weeks against all three strains covered by the vaccine: H1N1, H3N2, and influenza B.
PREVENTING FRACTURES
Each year, about 30% of people age 65 or older fall, sustaining serious injuries in 5% to 10% of cases. Unintentional falls are the main cause of hip fractures, which number 300,000 per year. They are also a common cause of death.
Vitamin D prevents fractures, but can there be too much of a good thing?
BISCHOFF-FERRARI HA, WILLETT WC, WONG JB, ET AL. PREVENTION OF NONVERTEBRAL FRACTURES WITH ORAL VITAMIN D AND DOSE DEPENDENCY: A META-ANALYSIS OF RANDOMIZED CONTROLLED TRIALS. ARCH INTERN MED 2009; 169:551–561.
SANDERS KM, STUART AL, WILLIAMSON EJ, ET AL. ANNUAL HIGH-DOSE ORAL VITAMIN D AND FALLS AND FRACTURES IN OLDER WOMEN: A RANDOMIZED CONTROLLED TRIAL. JAMA 2010; 303:1815–1822.
Bischoff-Ferrari5 performed a meta-analysis of 12 randomized controlled trials of oral supplemental vitamin D3 for preventing nonvertebral fractures in people age 65 and older, and eight trials for preventing hip fractures in the same age group. They found that the higher the daily dose of vitamin D, the lower the relative risk of hip fracture. The threshold dose at which supplementation significantly reduced the risk of falling was about 400 units per day. Higher doses of vitamin D reduced both falls and hip fractures by about 20%. The maximal effect was seen with studies using the maximum daily doses, ie, 770 to 800 units per day—not megadoses, but more than most Americans are taking. The threshold serum level of vitamin D of significance was 60 nmol/L (24 ng/mL).
Of interest, the effect on fractures was independent of calcium supplementation. This is important because calcium supplementation over and above ordinary dietary intake may increase the risk of cardiovascular events.6,7
Despite the benefits of vitamin D, too much may be too much of a good thing. Sanders et al8 performed a double-blind, placebo-controlled trial in 2,256 community-dwelling women, age 70 or older, who were considered to be at high risk for fractures. Half received a large oral dose (500,000 units) once a year for 3 to 5 years, and half got placebo. Their initial serum vitamin D level was 49 nmol/L; the level 30 days after a dose in the treatment group was 120 nmol/L.
Contrary to expectations, the incidence of falls was 15% higher in the vitamin D group than in the placebo group (P = .03), and the incidence of fractures was 26% higher (P = .047). The falls and fractures tended to cluster in the first 3 months after the dose in the active treatment group, when serum vitamin D levels were highest.
Comments. Unless future studies suggest a benefit to megadoses of vitamin D or prove calcium supplementation greater than 1,000 mg is safe, the optimal daily intake of vitamin D is likely 1,000 units, with approximately 200 units from diet and 800 units from supplements. A diet rich in low-fat dairy products may not require calcium supplementation. In those consuming a low-calcium diet, supplements of 500 to 1,000 mg/day are likely adequate.
Denosumab, a new drug for preventing fractures
CUMMINGS SR, SAN MARTIN J, MCCLUNG MR, ET AL; FREEDOM TRIAL. DENOSUMAB FOR PREVENTION OF FRACTURES IN POSTMENOPAUSAL WOMEN WITH OSTEOPOROSIS. N ENGL J MED 2009; 361:756–765.
SMITH MR, EGERDIE B, HERNÁNDEZ TORIZ N, ET AL; DENOSUMAB HALT PROSTATE CANCER STUDY GROUP. DENOSUMAB IN MEN RECEIVING ANDROGEN-DEPRIVATION THERAPY FOR PROSTATE CANCER. N ENGL J MED 2009; 361:745–755.
Denosumab (Prolia) is the first of a new class of drugs for the treatment of osteoporosis. It is a monoclonal antibody and member of the tumor necrosis factor superfamily that binds to the receptor activator nuclear factor kappa B (RANK) ligand. It has an antiresorptive effect, preventing osteoclast differentiation and activation. It is given by subcutaneous injection of 60 mg every 6 months; it is cleared by a nonrenal mechanism.
In a randomized controlled trial in 7,868 women between the ages of 60 and 90 who had osteoporosis, Cummings et al9 reported that denosumab reduced the 3-year incidence of vertebral fractures by 68% (P < .001), reduced the incidence of hip fractures by 40% (P = .01), and reduced the incidence of nonvertebral fractures by 20% (P = .01). In a trial in men receiving androgen deprivation therapy for prostate cancer, Smith et al10 reported that denosumab reduced the incidence of vertebral fracture by 62% (P = .006).
Comment. Denosumab was approved by the US Food and Drug Administration (FDA) on June 1, 2010, and is emerging in specialty clinics at the time of this publication. Its potential impact on clinical care is not yet known. It is costly—about $825 (average wholesale price) per injection—but since it is given by injection it may be easier than a yearly infusion of zoledronic acid (Reclast). It has the potential to suppress immune function, although this was not reported in the clinical trials. It may ultimately have a role in treating osteoporosis in men and women, prostate cancer following androgen deprivation, metastatic prostate cancer, metastatic breast cancer, osteoporosis with renal impairment, and other diseases.
DIALYSIS IN THE ELDERLY: A BLEAK STORY
KURELLA TAMURA M, COVINSKY KE, CHERTOW GM, YAFFE K, LANDEFELD CS, MCCOLLOCH CE. FUNCTIONAL STATUS OF ELDERLY ADULTS BEFORE AND AFTER INITIATION OF DIALYSIS. N ENGL J MED 2009; 361:1539–1547.
JASSAL SV, CHIU E, HLADUNEWITH M. LOSS OF INDEPENDENCE IN PATIENTS STARTING DIALYSIS AT 80 YEARS OF AGE OR OLDER (LETTER). N ENGL J MED 2009; 361:1612–1613.
Nursing home residents account for 4% of all patients in end-stage renal disease. However, the benefits of dialysis in older patients are uncertain. The mortality rate during the first year of dialysis is 35% in patients 70 years of age and older and 50% in patients 80 years and older.
Is dialysis helpful in the elderly, ie, does it improve survival and function?
Kurella Tamura et al11 retrospectively identified 3,702 nursing home residents starting dialysis in whom functional assessments had been done. The numbers told a bleak story. Initiation of dialysis was associated with a sharp decline in functional status, as reflected in an increase of 2.8 points on the 28-point Minimum Data Set–Activities of Daily Living (MDS-ADL) scale (the higher the score, the worse the function). MDS-ADL scores stabilized at a plateau for about 6 months and then continued to decline. Moreover, at 12 months, 58% of the patients had died.
The MDS-ADL score is based on seven components: eating, bed mobility, locomotion, transferring, toileting, hygiene, and dressing; function declined in all of these areas when patients started dialysis.
Patients were more likely to decline in activities of daily living after starting dialysis if they were older, were white, had cerebrovascular disease, had a diagnosis of dementia, were hospitalized at the start of dialysis, or had a serum albumin level lower than 3.5 g/dL.
The same thing happens to elders living in the community when they start dialysis. Jassal and colleagues12 reported that, of 97 community-dwelling patients (mean age 85), 46 (47%) were dead 2 years after starting dialysis. Although 76 (78%) had been living independently at the start of dialysis, only 11 (11%) were still doing so at 2 years.
Comment. These findings indicate that we do not know if hemodialysis improves survival. Hemodialysis may buy about 3 months of stable function, but it clearly does not restore function.
Is this the best we can do? Standard hemodialysis may have flaws, and nocturnal dialysis and peritoneal dialysis are used more in other countries. These dialysis techniques require more study in our older population. The lesson from these two publications on dialysis is that we should attend more carefully to slowing the decline in renal function before patients reach end-stage renal disease.
DABIGATRAN: AN ALTERNATIVE TO WARFARIN FOR ATRIAL FIBRILLATION
CONNOLLY SJ, EZEKOWITZ MD, YUSUF S, ET AL; RE-LY STEERING COMMITTEE AND INVESTIGATORS. DABIGATRAN VERSUS WARFARIN IN PATIENTS WITH ATRIAL FIBRILLATION. N ENGL J MED 2009; 361:1139–1151.
Atrial fibrillation is common, affecting 2.2 million adults. The median age of people who have atrial fibrillation is 75 years, and it is the most common arrhythmia in the elderly. Some 20% of ischemic strokes are attributed to it.13–15
Warfarin (Coumadin) is still the mainstay of treatment to prevent stroke in patients with atrial fibrillation. In an analysis of pooled data from five clinical trials,16 the relative risk reduction with warfarin was about 68% in the overall population (number needed to treat 32), 51% in people older than 75 years with no other risk factors (number needed to treat 56), and 85% in people older than 75 years with one or more risk factors (number needed to treat 15).
But warfarin carries a risk of bleeding, and its dose must be periodically adjusted on the basis of the international normalized ratio (INR) of the prothrombin time, so it carries a burden of laboratory monitoring. It is less safe in people who eat erratically, resulting in wide fluctuations in the INR.
Dabigatran (Pradaxa), a direct thrombin inhibitor, is expected to become an alternative to warfarin. It has been approved in Europe but not yet in the United States.
Connolly et al,17 in a randomized, double-blind trial, assigned 18,113 patients who had atrial fibrillation to receive either dabigatran 110 or 150 mg twice daily or adjusted-dose warfarin in an unblinded fashion. At 2 years, the rates of stroke and systemic embolism were about the same with dabigatran 110 mg as with warfarin but were lower with dabigatran 150 mg (relative risk 0.66, 95% confidence interval [CI] 0.53–0.82, P < .001). The rate of major bleeding was lower with dabigatran 110 mg than with warfarin (2.71% per year vs 3.36% per year, P = .003), but it was similar with dabigatran 150 mg (3.11% per year). Rates of life-threatening bleeding were 1.80% with warfarin, 1.22% with dabigatran 110 mg (P < .05), and 1.45% with dabigatran 150 mg (P < .05).
Comment. I suspect that warfarin’s days are numbered. Dabigatran 110 or 150 mg was as safe and as effective as warfarin in clinical trials, and probably will be more effective than warfarin in clinical practice. It will also probably be safer than warfarin in clinical practice, particularly in challenging settings such as long-term care. On the other hand, it will likely be much more expensive than warfarin.
DEMENTIA
Adverse effects of cholinesterase inhibitors
GILL SS, ANDERSON GM, FISCHER HD, ET AL. SYNCOPE AND ITS CONSEQUENCES IN PATIENTS WITH DEMENTIA RECEIVING CHOLINESTERASE INHIBITORS: A POPULATION-BASED COHORT STUDY. ARCH INTERN MED 2009; 169:867–873.
Cholinesterase inhibitors, eg, donepezil (Aricept), galantamine (Razadyne), and rivastigmine (Exelon), are commonly used to treat Alzheimer disease. However, these drugs carry risks of serious adverse effects.
Gill et al18 retrospectively reviewed a database from Ontario, Canada, and identified about 20,000 community-dwelling elderly persons admitted to the hospital who had been prescribed cholinesterase inhibitors and about three times as many matched controls.
Several adverse events were more frequent in people receiving cholinesterase inhibitors. Findings (events per 1,000 person-years):
Hospital visits for syncope: 31.5 vs 18.6, adjusted hazard ratio (HR) 1.76, 95% CI 1.57–1.98
Hip fractures: 22.4 vs 19.8, HR 1.18, 85% CI 1.04–1.34
Hospital visits for bradycardia: 6.9 vs 4.4, HR 1.69, 95% CI 1.32–2.15
Permanent pacemaker insertion: 4.7 vs 3.3, HR 1.49, 95% CI 1.12–2.00.
Comment. This study adds to the concerns that cholinesterase inhibitors, which have only modest cognitive benefits, may increase the risk of falls, injury, and need for pacemaker placement in demented patients. A low threshold to stop medications in this class should be considered when a patient on a cholinesterase inhibitor presents with bradycardia, falls, and syncope.
The importance of ‘staging’ dementia
IVERSON DJ, GRONSETH GS, REGER MA, ET AL; STANDARDS SUBCOMMITTEE OF THE AMERICAN ACADEMY OF NEUROLOGY. PRACTICE PARAMETER UPDATE: EVALUATION AND MANAGEMENT OF DRIVING RISK IN DEMENTIA: REPORT OF THE QUALITY STANDARDS SUBCOMMITTEE OF THE AMERICAN ACADEMY OF NEUROLOGY. NEUROLOGY 2010; 74:1316–1324.
The Clinical Dementia Rating (CDR) is a simple scale that should be applied by clinicians to describe stage of dementia in patients with Alzheimer disease. This scale can be useful in a variety of settings, from prescribing antidementia drugs to determining whether a patient should still drive. Although research protocols utilize a survey or semistructured interview to derive the stage, the clinician can estimate the stage easily in the office, particularly if there is an informant who can comment on performance outside the office.
There are four stages to the CDR19:
0: No dementia
0.5: Mild memory deficit but intact function
1.0: Moderate memory loss with mild functional impairment
2.0: Severe memory loss, moderate functional impairment
3.0: Severe memory loss, no significant function outside of the house.
Comment. The first stage (0.5, mild memory deficit but intact function) corresponds to “mild cognitive impairment.” In the clinic, these patients tend to take more notes. They come to the appointment with a little book and they write everything down so they don’t forget. They do arrive at their appointments on time; they are not crashing the car; they are paying their bills.
Patients with CDR stage 1.0 dementia (moderate memory loss with mild functional impairment) may miss appointments, they may confuse their medications, and they may have problems driving. They are still taking care of their basic needs, and they show up for appointments acceptably washed and dressed. However, they are likely having trouble shopping and managing their finances.
Patients with severe memory loss and moderate functional impairment (CDR stage 2.0) may not realize they haven’t bathed for a week or have worn the same clothes repeatedly. They are having trouble with basic activities of daily living, such as bathing and toilet hygiene. However, if you were to encounter them socially and didn’t talk to them for too long, you might think they were normal.
Those with severe memory loss and no significant function outside the house (CDR stage 3.0) are the most severely disabled. Dementia in these individuals is recognizable at a glance, from across the room.
Alzheimer patients progress through the stages, from CDR stage 0.5 at about 1 year to stage 1 by about 2 years, to stage 2 by 5 years, and to stage 3 at 8 or 9 years.20
In prescribing antidementia medications. The CDR can help with prescribing antidementia drugs. No medications are approved by the FDA for stage 0 or 0.5. Cholinesterase inhibitors are approved for stages 1, 2, and 3; memantine (Namenda) is approved for stages 2 and 3.
Advising about driving. The CDR is the only risk predictor with a quality-of-evidence rating of A. More than half of people with stage 0.5 memory impairment are safe drivers; fewer than half of those with stage 1.0 are still safe drivers; and patients with stage 2.0 dementia should not be driving at all.21 An adverse rating by a caregiver carries a quality-of-evidence rating of B. Predictors of driving risk with a quality-of-evidence rating of C are decreased mileage due to self-restriction, agitation, or aggression; a crash in the past 1 to 5 years; a citation in the past 2 to 3 years; and a Folstein Mini-Mental State Examination score of 24 or less. Studies also show that a memory-impaired person’s self-rating of safe driving ability or of assurance that he or she avoids unsafe situations is not reliable.21
DELIRIUM
Delirium goes by a number of synonyms, eg, “sundowning,” acute confusional state, acute change in mental status, metabolic encephalopathy, toxic encephalopathy (psychosis), acute brain syndrome, and acute toxic psychosis.
Delirium is common in hospitalized elderly patients, occurring in 11% to 42% of elderly hospitalized patients overall, up to 53% of elderly surgical patients on regular hospital floors, 80% of elderly surgical patients in intensive care, and about half of elderly patients after undergoing coronary artery bypass grafting. Unfortunately, it is undiagnosed in 30% to 60% of cases.22–24
Many pathways can lead to delirium, including hypoxemia, metabolic derangement, drug effects, systemic inflammation, and infection.25
Outcomes can vary from full recovery to death. After 1 year, 50% of those who leave the hospital with some evidence of delirium have not regained their baseline function. Delirium also increases the cost of care and the risk of institutionalization.
Delirium can accelerate dementia
FONG TG, JONES RN, SHI P, ET AL. DELIRIUM ACCELERATES COGNITIVE DECLINE IN ALZHEIMER DISEASE. NEUROLOGY 2009; 72:1570–1575.
Delirium accelerates the course of dementia in patients who had some evidence of dementia before they entered the hospital. Often, the change is noticeable by the family.26
Preventing delirium
INOUYE SK BOGARDUS ST JR, CHARPENTIER PA, ET AL. A MULTICOMPONENT INTERVENTION TO PREVENT DELIRIUM IN HOSPITALIZED OLDER PATIENTS. N ENGL J MED 1999; 340:669–676.
LUNDSTRÖM M, OLOFSSON B, STENVALL M, ET AL. POSTOPERATIVE DELIRIUM IN OLD PATIENTS WITH FEMORAL NECK FRACTURE: A RANDOMIZED INTERVENTION STUDY. AGING CLIN EXP RES 2007; 19:178–186.
Delirium can often be prevented. In a report published in 1999, Inouye et al27 described the outcomes of a program to prevent delirium in hospitalized medically ill elderly patients. Interventions were aimed at optimizing cognitive function, preventing sleep deprivation, avoiding immobility, improving vision and hearing, and treating dehydration. The incidence of delirium was 9.9% in the intervention group vs 15% in the control group, a 40% reduction (P < .05).
Lundström et al28 implemented a similar program for elderly patients with hip fractures. Interventions included staff education and teamwork; active prevention, detection, and treatment of delirium; transfusions if hemoglobin levels were less than 10 g/dL; prompt removal of indwelling urinary catheters, with screening for urinary retention; active prevention and treatment of constipation; and protein-enriched meals. The incidence of delirium was 55% in the intervention group vs 75% in the control group, a 27% reduction.
Comment. Although we have long known that the risk of delirium in medical and surgical patients can be reduced, most hospitals do not have systematic programs to detect delirium and reduce its incidence. Hopefully, reduction in delirium risk will also reduce its adverse consequences, including worsening of dementia and increased mortality.
New clinical trials and observational studies are shedding light on ways to improve the health of elderly patients. Here is a brief summary of these trials and how they might influence your clinical practice.
EXERCISE HAS NEWLY DISCOVERED BENEFITS
According to government data,1 exercise has a dose-dependent effect on rates of all-cause mortality: the more hours one exercises per week, the lower the risk of death. The difference in risk is most pronounced as one goes from no exercise to about 3 hours of exercise per week; above 3 hours per week, the curve flattens out but continues to decline. Hence, we advise patients to engage in about 30 minutes of moderate-intensity exercise every day.
Lately, physical exercise has been found to have other, unexpected benefits.
Exercise helps cognition
ERICKSON KI, PRAKASH RS, VOSS MW, ET AL. AEROBIC FITNESS IS ASSOCIATED WITH HIPPOCAMPAL VOLUME IN ELDERLY HUMANS. HIPPOCAMPUS 2009; 19:1030–1039.
ETGEN T, SANDER D, HUNTGEBURTH U, POPPERT H, FÖRSTL H, BICKEL H. PHYSICAL ACTIVITY AND INCIDENT COGNITIVE IMPAIRMENT IN ELDERLY PERSONS: THE INVADE STUDY. ARCH INTERN MED 2010; 170:186–193.
The hippocampus is a structure deep in the brain that is involved in short-term memory. It atrophies with age, more so with dementia. Erickson2 found a correlation between aerobic fitness (as measured by maximum oxygen consumption), hippocampal volume, and spatial memory performance.
Etgen and colleagues3 studied nearly 4,000 older adults in Bavaria for 2 years. Among those reporting no physical activity, 21.4% had cognitive impairment at baseline, compared with 7.3% of those with high activity at baseline. Following those without cognitive impairment over a 2-year period, they found the incidence of new cognitive impairment was 13.9% in those with no physical activity at baseline, 6.7% in those with moderate activity, and 5.1% in those with high activity.
Exercise boosts the effect of influenza vaccine
WOODS JA, KEYLOCK KT, LOWDER T, ET AL. CARDIOVASCULAR EXERCISE TRAINING EXTENDS INFLUENZA VACCINE SEROPROTECTION IN SEDENTARY OLDER ADULTS: THE IMMUNE FUNCTION INTERVENTION TRIAL. J AM GERIATR SOC 2009; 57:2183–2191.
In a study in 144 sedentary but healthy older adults (ages 60 to 83), Woods et al4 randomized the participants to undergo either flexibility or cardiovascular training for 10 months, starting 4 months before their annual influenza shot. Exercise extended the duration of antibody protection, with more participants in the cardiovascular group than in the flexibility group showing protection at 24 weeks against all three strains covered by the vaccine: H1N1, H3N2, and influenza B.
PREVENTING FRACTURES
Each year, about 30% of people age 65 or older fall, sustaining serious injuries in 5% to 10% of cases. Unintentional falls are the main cause of hip fractures, which number 300,000 per year. They are also a common cause of death.
Vitamin D prevents fractures, but can there be too much of a good thing?
BISCHOFF-FERRARI HA, WILLETT WC, WONG JB, ET AL. PREVENTION OF NONVERTEBRAL FRACTURES WITH ORAL VITAMIN D AND DOSE DEPENDENCY: A META-ANALYSIS OF RANDOMIZED CONTROLLED TRIALS. ARCH INTERN MED 2009; 169:551–561.
SANDERS KM, STUART AL, WILLIAMSON EJ, ET AL. ANNUAL HIGH-DOSE ORAL VITAMIN D AND FALLS AND FRACTURES IN OLDER WOMEN: A RANDOMIZED CONTROLLED TRIAL. JAMA 2010; 303:1815–1822.
Bischoff-Ferrari5 performed a meta-analysis of 12 randomized controlled trials of oral supplemental vitamin D3 for preventing nonvertebral fractures in people age 65 and older, and eight trials for preventing hip fractures in the same age group. They found that the higher the daily dose of vitamin D, the lower the relative risk of hip fracture. The threshold dose at which supplementation significantly reduced the risk of falling was about 400 units per day. Higher doses of vitamin D reduced both falls and hip fractures by about 20%. The maximal effect was seen with studies using the maximum daily doses, ie, 770 to 800 units per day—not megadoses, but more than most Americans are taking. The threshold serum level of vitamin D of significance was 60 nmol/L (24 ng/mL).
Of interest, the effect on fractures was independent of calcium supplementation. This is important because calcium supplementation over and above ordinary dietary intake may increase the risk of cardiovascular events.6,7
Despite the benefits of vitamin D, too much may be too much of a good thing. Sanders et al8 performed a double-blind, placebo-controlled trial in 2,256 community-dwelling women, age 70 or older, who were considered to be at high risk for fractures. Half received a large oral dose (500,000 units) once a year for 3 to 5 years, and half got placebo. Their initial serum vitamin D level was 49 nmol/L; the level 30 days after a dose in the treatment group was 120 nmol/L.
Contrary to expectations, the incidence of falls was 15% higher in the vitamin D group than in the placebo group (P = .03), and the incidence of fractures was 26% higher (P = .047). The falls and fractures tended to cluster in the first 3 months after the dose in the active treatment group, when serum vitamin D levels were highest.
Comments. Unless future studies suggest a benefit to megadoses of vitamin D or prove calcium supplementation greater than 1,000 mg is safe, the optimal daily intake of vitamin D is likely 1,000 units, with approximately 200 units from diet and 800 units from supplements. A diet rich in low-fat dairy products may not require calcium supplementation. In those consuming a low-calcium diet, supplements of 500 to 1,000 mg/day are likely adequate.
Denosumab, a new drug for preventing fractures
CUMMINGS SR, SAN MARTIN J, MCCLUNG MR, ET AL; FREEDOM TRIAL. DENOSUMAB FOR PREVENTION OF FRACTURES IN POSTMENOPAUSAL WOMEN WITH OSTEOPOROSIS. N ENGL J MED 2009; 361:756–765.
SMITH MR, EGERDIE B, HERNÁNDEZ TORIZ N, ET AL; DENOSUMAB HALT PROSTATE CANCER STUDY GROUP. DENOSUMAB IN MEN RECEIVING ANDROGEN-DEPRIVATION THERAPY FOR PROSTATE CANCER. N ENGL J MED 2009; 361:745–755.
Denosumab (Prolia) is the first of a new class of drugs for the treatment of osteoporosis. It is a monoclonal antibody and member of the tumor necrosis factor superfamily that binds to the receptor activator nuclear factor kappa B (RANK) ligand. It has an antiresorptive effect, preventing osteoclast differentiation and activation. It is given by subcutaneous injection of 60 mg every 6 months; it is cleared by a nonrenal mechanism.
In a randomized controlled trial in 7,868 women between the ages of 60 and 90 who had osteoporosis, Cummings et al9 reported that denosumab reduced the 3-year incidence of vertebral fractures by 68% (P < .001), reduced the incidence of hip fractures by 40% (P = .01), and reduced the incidence of nonvertebral fractures by 20% (P = .01). In a trial in men receiving androgen deprivation therapy for prostate cancer, Smith et al10 reported that denosumab reduced the incidence of vertebral fracture by 62% (P = .006).
Comment. Denosumab was approved by the US Food and Drug Administration (FDA) on June 1, 2010, and is emerging in specialty clinics at the time of this publication. Its potential impact on clinical care is not yet known. It is costly—about $825 (average wholesale price) per injection—but since it is given by injection it may be easier than a yearly infusion of zoledronic acid (Reclast). It has the potential to suppress immune function, although this was not reported in the clinical trials. It may ultimately have a role in treating osteoporosis in men and women, prostate cancer following androgen deprivation, metastatic prostate cancer, metastatic breast cancer, osteoporosis with renal impairment, and other diseases.
DIALYSIS IN THE ELDERLY: A BLEAK STORY
KURELLA TAMURA M, COVINSKY KE, CHERTOW GM, YAFFE K, LANDEFELD CS, MCCOLLOCH CE. FUNCTIONAL STATUS OF ELDERLY ADULTS BEFORE AND AFTER INITIATION OF DIALYSIS. N ENGL J MED 2009; 361:1539–1547.
JASSAL SV, CHIU E, HLADUNEWITH M. LOSS OF INDEPENDENCE IN PATIENTS STARTING DIALYSIS AT 80 YEARS OF AGE OR OLDER (LETTER). N ENGL J MED 2009; 361:1612–1613.
Nursing home residents account for 4% of all patients in end-stage renal disease. However, the benefits of dialysis in older patients are uncertain. The mortality rate during the first year of dialysis is 35% in patients 70 years of age and older and 50% in patients 80 years and older.
Is dialysis helpful in the elderly, ie, does it improve survival and function?
Kurella Tamura et al11 retrospectively identified 3,702 nursing home residents starting dialysis in whom functional assessments had been done. The numbers told a bleak story. Initiation of dialysis was associated with a sharp decline in functional status, as reflected in an increase of 2.8 points on the 28-point Minimum Data Set–Activities of Daily Living (MDS-ADL) scale (the higher the score, the worse the function). MDS-ADL scores stabilized at a plateau for about 6 months and then continued to decline. Moreover, at 12 months, 58% of the patients had died.
The MDS-ADL score is based on seven components: eating, bed mobility, locomotion, transferring, toileting, hygiene, and dressing; function declined in all of these areas when patients started dialysis.
Patients were more likely to decline in activities of daily living after starting dialysis if they were older, were white, had cerebrovascular disease, had a diagnosis of dementia, were hospitalized at the start of dialysis, or had a serum albumin level lower than 3.5 g/dL.
The same thing happens to elders living in the community when they start dialysis. Jassal and colleagues12 reported that, of 97 community-dwelling patients (mean age 85), 46 (47%) were dead 2 years after starting dialysis. Although 76 (78%) had been living independently at the start of dialysis, only 11 (11%) were still doing so at 2 years.
Comment. These findings indicate that we do not know if hemodialysis improves survival. Hemodialysis may buy about 3 months of stable function, but it clearly does not restore function.
Is this the best we can do? Standard hemodialysis may have flaws, and nocturnal dialysis and peritoneal dialysis are used more in other countries. These dialysis techniques require more study in our older population. The lesson from these two publications on dialysis is that we should attend more carefully to slowing the decline in renal function before patients reach end-stage renal disease.
DABIGATRAN: AN ALTERNATIVE TO WARFARIN FOR ATRIAL FIBRILLATION
CONNOLLY SJ, EZEKOWITZ MD, YUSUF S, ET AL; RE-LY STEERING COMMITTEE AND INVESTIGATORS. DABIGATRAN VERSUS WARFARIN IN PATIENTS WITH ATRIAL FIBRILLATION. N ENGL J MED 2009; 361:1139–1151.
Atrial fibrillation is common, affecting 2.2 million adults. The median age of people who have atrial fibrillation is 75 years, and it is the most common arrhythmia in the elderly. Some 20% of ischemic strokes are attributed to it.13–15
Warfarin (Coumadin) is still the mainstay of treatment to prevent stroke in patients with atrial fibrillation. In an analysis of pooled data from five clinical trials,16 the relative risk reduction with warfarin was about 68% in the overall population (number needed to treat 32), 51% in people older than 75 years with no other risk factors (number needed to treat 56), and 85% in people older than 75 years with one or more risk factors (number needed to treat 15).
But warfarin carries a risk of bleeding, and its dose must be periodically adjusted on the basis of the international normalized ratio (INR) of the prothrombin time, so it carries a burden of laboratory monitoring. It is less safe in people who eat erratically, resulting in wide fluctuations in the INR.
Dabigatran (Pradaxa), a direct thrombin inhibitor, is expected to become an alternative to warfarin. It has been approved in Europe but not yet in the United States.
Connolly et al,17 in a randomized, double-blind trial, assigned 18,113 patients who had atrial fibrillation to receive either dabigatran 110 or 150 mg twice daily or adjusted-dose warfarin in an unblinded fashion. At 2 years, the rates of stroke and systemic embolism were about the same with dabigatran 110 mg as with warfarin but were lower with dabigatran 150 mg (relative risk 0.66, 95% confidence interval [CI] 0.53–0.82, P < .001). The rate of major bleeding was lower with dabigatran 110 mg than with warfarin (2.71% per year vs 3.36% per year, P = .003), but it was similar with dabigatran 150 mg (3.11% per year). Rates of life-threatening bleeding were 1.80% with warfarin, 1.22% with dabigatran 110 mg (P < .05), and 1.45% with dabigatran 150 mg (P < .05).
Comment. I suspect that warfarin’s days are numbered. Dabigatran 110 or 150 mg was as safe and as effective as warfarin in clinical trials, and probably will be more effective than warfarin in clinical practice. It will also probably be safer than warfarin in clinical practice, particularly in challenging settings such as long-term care. On the other hand, it will likely be much more expensive than warfarin.
DEMENTIA
Adverse effects of cholinesterase inhibitors
GILL SS, ANDERSON GM, FISCHER HD, ET AL. SYNCOPE AND ITS CONSEQUENCES IN PATIENTS WITH DEMENTIA RECEIVING CHOLINESTERASE INHIBITORS: A POPULATION-BASED COHORT STUDY. ARCH INTERN MED 2009; 169:867–873.
Cholinesterase inhibitors, eg, donepezil (Aricept), galantamine (Razadyne), and rivastigmine (Exelon), are commonly used to treat Alzheimer disease. However, these drugs carry risks of serious adverse effects.
Gill et al18 retrospectively reviewed a database from Ontario, Canada, and identified about 20,000 community-dwelling elderly persons admitted to the hospital who had been prescribed cholinesterase inhibitors and about three times as many matched controls.
Several adverse events were more frequent in people receiving cholinesterase inhibitors. Findings (events per 1,000 person-years):
Hospital visits for syncope: 31.5 vs 18.6, adjusted hazard ratio (HR) 1.76, 95% CI 1.57–1.98
Hip fractures: 22.4 vs 19.8, HR 1.18, 85% CI 1.04–1.34
Hospital visits for bradycardia: 6.9 vs 4.4, HR 1.69, 95% CI 1.32–2.15
Permanent pacemaker insertion: 4.7 vs 3.3, HR 1.49, 95% CI 1.12–2.00.
Comment. This study adds to the concerns that cholinesterase inhibitors, which have only modest cognitive benefits, may increase the risk of falls, injury, and need for pacemaker placement in demented patients. A low threshold to stop medications in this class should be considered when a patient on a cholinesterase inhibitor presents with bradycardia, falls, and syncope.
The importance of ‘staging’ dementia
IVERSON DJ, GRONSETH GS, REGER MA, ET AL; STANDARDS SUBCOMMITTEE OF THE AMERICAN ACADEMY OF NEUROLOGY. PRACTICE PARAMETER UPDATE: EVALUATION AND MANAGEMENT OF DRIVING RISK IN DEMENTIA: REPORT OF THE QUALITY STANDARDS SUBCOMMITTEE OF THE AMERICAN ACADEMY OF NEUROLOGY. NEUROLOGY 2010; 74:1316–1324.
The Clinical Dementia Rating (CDR) is a simple scale that should be applied by clinicians to describe stage of dementia in patients with Alzheimer disease. This scale can be useful in a variety of settings, from prescribing antidementia drugs to determining whether a patient should still drive. Although research protocols utilize a survey or semistructured interview to derive the stage, the clinician can estimate the stage easily in the office, particularly if there is an informant who can comment on performance outside the office.
There are four stages to the CDR19:
0: No dementia
0.5: Mild memory deficit but intact function
1.0: Moderate memory loss with mild functional impairment
2.0: Severe memory loss, moderate functional impairment
3.0: Severe memory loss, no significant function outside of the house.
Comment. The first stage (0.5, mild memory deficit but intact function) corresponds to “mild cognitive impairment.” In the clinic, these patients tend to take more notes. They come to the appointment with a little book and they write everything down so they don’t forget. They do arrive at their appointments on time; they are not crashing the car; they are paying their bills.
Patients with CDR stage 1.0 dementia (moderate memory loss with mild functional impairment) may miss appointments, they may confuse their medications, and they may have problems driving. They are still taking care of their basic needs, and they show up for appointments acceptably washed and dressed. However, they are likely having trouble shopping and managing their finances.
Patients with severe memory loss and moderate functional impairment (CDR stage 2.0) may not realize they haven’t bathed for a week or have worn the same clothes repeatedly. They are having trouble with basic activities of daily living, such as bathing and toilet hygiene. However, if you were to encounter them socially and didn’t talk to them for too long, you might think they were normal.
Those with severe memory loss and no significant function outside the house (CDR stage 3.0) are the most severely disabled. Dementia in these individuals is recognizable at a glance, from across the room.
Alzheimer patients progress through the stages, from CDR stage 0.5 at about 1 year to stage 1 by about 2 years, to stage 2 by 5 years, and to stage 3 at 8 or 9 years.20
In prescribing antidementia medications. The CDR can help with prescribing antidementia drugs. No medications are approved by the FDA for stage 0 or 0.5. Cholinesterase inhibitors are approved for stages 1, 2, and 3; memantine (Namenda) is approved for stages 2 and 3.
Advising about driving. The CDR is the only risk predictor with a quality-of-evidence rating of A. More than half of people with stage 0.5 memory impairment are safe drivers; fewer than half of those with stage 1.0 are still safe drivers; and patients with stage 2.0 dementia should not be driving at all.21 An adverse rating by a caregiver carries a quality-of-evidence rating of B. Predictors of driving risk with a quality-of-evidence rating of C are decreased mileage due to self-restriction, agitation, or aggression; a crash in the past 1 to 5 years; a citation in the past 2 to 3 years; and a Folstein Mini-Mental State Examination score of 24 or less. Studies also show that a memory-impaired person’s self-rating of safe driving ability or of assurance that he or she avoids unsafe situations is not reliable.21
DELIRIUM
Delirium goes by a number of synonyms, eg, “sundowning,” acute confusional state, acute change in mental status, metabolic encephalopathy, toxic encephalopathy (psychosis), acute brain syndrome, and acute toxic psychosis.
Delirium is common in hospitalized elderly patients, occurring in 11% to 42% of elderly hospitalized patients overall, up to 53% of elderly surgical patients on regular hospital floors, 80% of elderly surgical patients in intensive care, and about half of elderly patients after undergoing coronary artery bypass grafting. Unfortunately, it is undiagnosed in 30% to 60% of cases.22–24
Many pathways can lead to delirium, including hypoxemia, metabolic derangement, drug effects, systemic inflammation, and infection.25
Outcomes can vary from full recovery to death. After 1 year, 50% of those who leave the hospital with some evidence of delirium have not regained their baseline function. Delirium also increases the cost of care and the risk of institutionalization.
Delirium can accelerate dementia
FONG TG, JONES RN, SHI P, ET AL. DELIRIUM ACCELERATES COGNITIVE DECLINE IN ALZHEIMER DISEASE. NEUROLOGY 2009; 72:1570–1575.
Delirium accelerates the course of dementia in patients who had some evidence of dementia before they entered the hospital. Often, the change is noticeable by the family.26
Preventing delirium
INOUYE SK BOGARDUS ST JR, CHARPENTIER PA, ET AL. A MULTICOMPONENT INTERVENTION TO PREVENT DELIRIUM IN HOSPITALIZED OLDER PATIENTS. N ENGL J MED 1999; 340:669–676.
LUNDSTRÖM M, OLOFSSON B, STENVALL M, ET AL. POSTOPERATIVE DELIRIUM IN OLD PATIENTS WITH FEMORAL NECK FRACTURE: A RANDOMIZED INTERVENTION STUDY. AGING CLIN EXP RES 2007; 19:178–186.
Delirium can often be prevented. In a report published in 1999, Inouye et al27 described the outcomes of a program to prevent delirium in hospitalized medically ill elderly patients. Interventions were aimed at optimizing cognitive function, preventing sleep deprivation, avoiding immobility, improving vision and hearing, and treating dehydration. The incidence of delirium was 9.9% in the intervention group vs 15% in the control group, a 40% reduction (P < .05).
Lundström et al28 implemented a similar program for elderly patients with hip fractures. Interventions included staff education and teamwork; active prevention, detection, and treatment of delirium; transfusions if hemoglobin levels were less than 10 g/dL; prompt removal of indwelling urinary catheters, with screening for urinary retention; active prevention and treatment of constipation; and protein-enriched meals. The incidence of delirium was 55% in the intervention group vs 75% in the control group, a 27% reduction.
Comment. Although we have long known that the risk of delirium in medical and surgical patients can be reduced, most hospitals do not have systematic programs to detect delirium and reduce its incidence. Hopefully, reduction in delirium risk will also reduce its adverse consequences, including worsening of dementia and increased mortality.
Erickson KI, Prakash RS, Voss MW, et al. Aerobic fitness is associated with hippocampal volume in elderly humans. Hippocampus2009; 19:1030–1039.
Etgen T, Sander D, Huntgeburth U, Poppert H, Förstl H, Bickel H. Physical activity and incident cognitive impairment in elderly persons: the INVADE study. Arch Intern Med2010; 170:186–193.
Woods JA, Keylock KT, Lowder T, et al. Cardiovascular exercise training extends influenza vaccine seroprotection in sedentary older adults: the immune function intervention trial. J Am Geriatr Soc2009; 57:2183–2191.
Bischoff-Ferrari HA, Willett WC, Wong JB, et al. Prevention of nonvertebral fractures with oral vitamin D and dose dependency: a meta-analysis of randomized controlled trials. Arch Intern Med2009; 169:551–561.
Bolland MJ, Avenell A, Baron JA, et al. Effect of calcium supplements on risk of myocardial infarction and cardiovascular events: meta-analysis. BMJ2010; 341:c3691. doi:10.1136/bmj.c3691.
Bolland MJ, Barber PA, Doughty RN, et al. Vascular events in healthy older women receiving calcium supplementation: randomised controlled trial. BMJ2008; 336:262–266.
Sanders KM, Stuart AL, Williamson EJ, et al. Annual high-dose oral vitamin D and falls and fractures in older women: a randomized controlled trial. JAMA2010; 303:1815–1822.
Cummings SR, San Martin J, McClung MR, et al; FREEDOM Trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med2009; 361:756–765.
Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med2009; 361:745–755.
Kurella Tamura M, Covinsky KE, Chertow GM, Yaffe K, Landefeld CS, McColloch CE. Functional status of elderly adults before and after initiation of dialysis. N Engl J Med2009; 361:1539–1547.
Jassal SV, Chiu E, Hladunewich M. Loss of independence in patients starting dialysis at 80 years of age or older (letter). N Engl J Med2009; 361:1612–1613.
Feinberg WM, Blackshear JL, Laupacis A, Kronmal R, Hart RG. Prevalence, age distribution and gender of patients with atrial fibrillation. Analysis and implications. Arch Intern Med1995; 155:469–473.
Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation: a major contributor to stroke in the elderly. The Framingham Study. Arch Intern Med1987; 147:1561–1564.
Lin HJ, Wolf PA, Kelly-Hayes M, et al. Stroke severity in atrial fibrillation. The Framingham Study. Stroke1996; 27:1760–1764.
Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis of pooled data from five randomized controlled trials. Arch Intern Med1994; 154:1449–1457.
Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med2009; 361:1139–1151.
Gill SS, Anderson GM, Fischer HD, et al. Syncope and its consequences in patients with dementia receiving cholinesterase inhibitors: a population-based cohort study. Arch Intern Med2009; 169:867–873.
Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology1993; 43:2412–2414.
Sloane PD. Advances in the treatment of Alzheimer’s disease. Am Fam Physician1998; 58:1577–1586.
Iverson DJ, Gronseth GS, Reger MA, et al; Standards Subcommittee of the American Academy of Neurology. Practice parameter update: evaluation and management of driving risk in dementia: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology2010; 74:1316–1324.
Demeure MJ, Fain MJ. The elderly surgical patient and postoperative delirium. J Am Coll Surg2006; 203:752–757.
Siddiqi N, House AO, Holmes JD. Occurrence and outcome of delirium in medical in-patients: a systematic literature review. Age Ageing2006; 35:350–364.
Rudolph JL, Jones RN, Levkoff SE, et al. Derivation and validation of a preoperative prediction rule for delirium after cardiac surgery. Circulation2009; 119:229–236.
Fong TG, Tulebaev SR, Inouye SK. Delirium in elderly adults: diagnosis, prevention and treatment. Nat Rev Neurol2009; 5:210–220.
Fong TG, Jones RN, Shi P, et al. Delirium accelerates cognitive decline in Alzheimer disease. Neurology2009; 72:1570–1575.
Inouye SK, Bogardus ST, Charpentier PA, et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med1999; 340:669–676.
Lundström M, Olofsson B, Stenvall M, et al. Postoperative delirium in old patients with femoral neck fracture: a randomized intervention study. Aging Clin Exp Res2007; 19:178–186.
Erickson KI, Prakash RS, Voss MW, et al. Aerobic fitness is associated with hippocampal volume in elderly humans. Hippocampus2009; 19:1030–1039.
Etgen T, Sander D, Huntgeburth U, Poppert H, Förstl H, Bickel H. Physical activity and incident cognitive impairment in elderly persons: the INVADE study. Arch Intern Med2010; 170:186–193.
Woods JA, Keylock KT, Lowder T, et al. Cardiovascular exercise training extends influenza vaccine seroprotection in sedentary older adults: the immune function intervention trial. J Am Geriatr Soc2009; 57:2183–2191.
Bischoff-Ferrari HA, Willett WC, Wong JB, et al. Prevention of nonvertebral fractures with oral vitamin D and dose dependency: a meta-analysis of randomized controlled trials. Arch Intern Med2009; 169:551–561.
Bolland MJ, Avenell A, Baron JA, et al. Effect of calcium supplements on risk of myocardial infarction and cardiovascular events: meta-analysis. BMJ2010; 341:c3691. doi:10.1136/bmj.c3691.
Bolland MJ, Barber PA, Doughty RN, et al. Vascular events in healthy older women receiving calcium supplementation: randomised controlled trial. BMJ2008; 336:262–266.
Sanders KM, Stuart AL, Williamson EJ, et al. Annual high-dose oral vitamin D and falls and fractures in older women: a randomized controlled trial. JAMA2010; 303:1815–1822.
Cummings SR, San Martin J, McClung MR, et al; FREEDOM Trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med2009; 361:756–765.
Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med2009; 361:745–755.
Kurella Tamura M, Covinsky KE, Chertow GM, Yaffe K, Landefeld CS, McColloch CE. Functional status of elderly adults before and after initiation of dialysis. N Engl J Med2009; 361:1539–1547.
Jassal SV, Chiu E, Hladunewich M. Loss of independence in patients starting dialysis at 80 years of age or older (letter). N Engl J Med2009; 361:1612–1613.
Feinberg WM, Blackshear JL, Laupacis A, Kronmal R, Hart RG. Prevalence, age distribution and gender of patients with atrial fibrillation. Analysis and implications. Arch Intern Med1995; 155:469–473.
Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation: a major contributor to stroke in the elderly. The Framingham Study. Arch Intern Med1987; 147:1561–1564.
Lin HJ, Wolf PA, Kelly-Hayes M, et al. Stroke severity in atrial fibrillation. The Framingham Study. Stroke1996; 27:1760–1764.
Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis of pooled data from five randomized controlled trials. Arch Intern Med1994; 154:1449–1457.
Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med2009; 361:1139–1151.
Gill SS, Anderson GM, Fischer HD, et al. Syncope and its consequences in patients with dementia receiving cholinesterase inhibitors: a population-based cohort study. Arch Intern Med2009; 169:867–873.
Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology1993; 43:2412–2414.
Sloane PD. Advances in the treatment of Alzheimer’s disease. Am Fam Physician1998; 58:1577–1586.
Iverson DJ, Gronseth GS, Reger MA, et al; Standards Subcommittee of the American Academy of Neurology. Practice parameter update: evaluation and management of driving risk in dementia: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology2010; 74:1316–1324.
Demeure MJ, Fain MJ. The elderly surgical patient and postoperative delirium. J Am Coll Surg2006; 203:752–757.
Siddiqi N, House AO, Holmes JD. Occurrence and outcome of delirium in medical in-patients: a systematic literature review. Age Ageing2006; 35:350–364.
Rudolph JL, Jones RN, Levkoff SE, et al. Derivation and validation of a preoperative prediction rule for delirium after cardiac surgery. Circulation2009; 119:229–236.
Fong TG, Tulebaev SR, Inouye SK. Delirium in elderly adults: diagnosis, prevention and treatment. Nat Rev Neurol2009; 5:210–220.
Fong TG, Jones RN, Shi P, et al. Delirium accelerates cognitive decline in Alzheimer disease. Neurology2009; 72:1570–1575.
Inouye SK, Bogardus ST, Charpentier PA, et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med1999; 340:669–676.
Lundström M, Olofsson B, Stenvall M, et al. Postoperative delirium in old patients with femoral neck fracture: a randomized intervention study. Aging Clin Exp Res2007; 19:178–186.
Exercise has newly discovered benefits, such as preserving cognition and boosting the response to vaccination.
Vitamin D supplementation has been found to prevent fractures, but yearly megadoses had the opposite effect.
Denosumab (Prolia) has been approved for preventing fractures. It acts by inhibiting the receptor activator of nuclear factor kappa B (RANK) ligand.
The outlook for elderly patients starting hemodialysis is bleak, with loss of function and a high risk of death.
Dabigatran (Pradaxa), a direct thrombin inhibitor, may prove to be a safer alternative to warfarin (Coumadin).
Cholinesterase inhibitors for Alzheimer disease are associated with higher risks of hospitalization for syncope, hip fractures, bradycardia, and pacemaker insertion.
The Clinical Dementia Rating should be estimated when prescribing a cognitive enhancer and when advising a patient with memory impairment on driving safety.
Delirium often accelerates dementia; interventions for hospitalized elderly patients may reduce its incidence.
Patients with an osteoporotic hip fracture suffer from profound morbidity and are at a heightened risk of death. It is therefore essential that they receive treatment with a bisphosphonate known to modify the subsequent risk of fracture at any site—eg, alendronate (Fosamax), risedronate (Actonel), or zoledronic acid (Reclast).
However, there is concern that starting a bisphosphonate too soon after surgery could disrupt bone remodeling and delay fracture repair.
Only one clinical study addressed the timing of bisphosphonate therapy after hip fracture repair. In this study, Eriksen et al1 performed a post hoc analysis of data from the Health Outcomes and Reduced Incidence With Zoledronic Acid Once Yearly Recurrent Fracture Trial (HORIZON-RFT)2 and concluded that the optimal time to give intravenous zoledronic acid is 2 to 12 weeks after surgical repair of the fracture.
In a frail, elderly patient with comorbidities, a single intravenous 5-mg dose of zoledronic acid guarantees adequate treatment, obviating issues of poor compliance and oral absorption and loss to follow-up. Sufficient levels of vitamin D and calcium should be ensured.
THE EVIDENCE
The original HORIZON-RFT study,2 published in 2007, compared intravenous zoledronic acid against placebo in elderly patients with osteoporotic hip fracture. Most of the patients were white women; their mean age was 74; 1,065 received intravenous zoledronic acid, and 1,062 received placebo. All received vitamin D and calcium.
The trial showed a clear reduction in the rate of recurrent fractures at other sites (a primary end point) and a reduction in the rate of all-cause mortality in patients treated within 90 days of fracture. A total of 424 fractures occurred in 231 patients. The risk of any new clinical fracture was 35% lower with treatment than with placebo (occurring in 8.6% vs 13.9% of patients, P = .001), and the number of deaths due to any cause was 28% lower with treatment than with placebo (occurring in 101 vs 141, P = .01).2
The mean time to fracture was 39.8 months in the treated group vs 36.4 in the placebo group. The fracture risk reduction began to be apparent by 12 months, and the reduction in mortality rate by 16 months.2
In a post hoc analysis of the trial, Eriksen et al1 attempted to ascertain the optimal time for therapy in terms of fracture risk and mortality reduction. Analyzing the data by 2-week intervals beginning after the surgical repair of the fracture, the authors found that only 56 patients (5.3%) had received zoledronic acid within 2 weeks of surgery and only 47 had received placebo, and they saw no advantage to intravenous zoledronic acid compared with placebo in these first 2 weeks with respect to bone mineral density, fracture risk, or risk of death. However, excluding this small subset, antifracture efficacy and reduction in mortality rate were present when patients were treated with zoledronic acid in the 2 to 12 weeks after hip fracture repair, and improvement in bone mineral density at the hip was noted at 12 months in all cohorts.
Colón-Emeric et al3 performed another post hoc analysis, attempting to explain the lower mortality rate seen in patients treated with zoledronic acid. It had been an unexpected finding, and determinants of mortality rate reduction were hampered by a limited knowledge of the true cause of death or the circumstances of care after fracture. The authors concluded that only 8% of the reduction in mortality rate evident early in the second year of treatment with zoledronic acid could be attributed to a reduction in fractures.3 Other mechanisms by which the mortality rate reduction occurred remained unclear.
Curiously, in another large randomized controlled trial of zoledronic acid, in women with postmenopausal osteoporosis, Black et al4 reported that more patients died in the treated group (130 of 3,862) than in the placebo group (112 of 3,852). This difference was not statistically significant, but neither was it explained.
A meta-analysis by Bolland et al5 examined the effect of other osteoporosis treatments on mortality rate, using randomized controlled trials that lasted more than 12 months and that reported more than 10 deaths. The authors concluded the following:
In the trials in which bisphosphonates reduced the mortality rate, the mortality rate in the placebo group was higher than 10 per 1,000 patient-years
The effect of osteoporosis treatment on the mortality rate in a frail, elderly population is evident using agents with proven efficacy in reducing vertebral and nonvertebral fractures, eg, alendronate, risedronate, and zoledronic acid.5
THE SCIENCE
Osteoporotic fractures occur with minimal trauma, with the failure of bone attributed to impaired integrity of bone microarchitecture. The ultimate goal of fracture repair is to restore bone size, shape, and tissue properties. The issue of when to treat with a bisphosphonate after hip fracture arises because bisphosphonates are known to disrupt bone remodeling and so delay fracture repair.
After fracture, both anabolic and catabolic phases occur.6 The final outcome depends on the following:
The type of intervention to stabilize the fracture site (eg, surgical repair)
The inflammatory cytokines and growth factors released by the cellular elements in bloody and disrupted tissue.
Oxygen tension, angiogenesis, and osteoblasts are critical to primary bone formation, and osteoclasts are essential in remodeling this initial bone deposition. These late phases of fracture repair are most vulnerable to the bisphosphonates, through suppression of osteoclast resorption and possibly through decreased angiogenesis.6 Callus formation is sustained, but bone remodeling is delayed.
Amanat et al7 examined the timing of a single dose of zoledronic acid after fracture repair in a rat model of diaphyseal fracture and found that the callus was larger and stronger if the bisphosphonate dose had been delayed 1 or 2 weeks. The animals treated with zoledronic acid showed a remarkable trabecular network of bone between the original femoral cortex and the new cortical bone that was not present in the control group, perhaps contributing to the enhanced mechanical properties of the callus. Other studies suggest single dosing rather than continuous dosing may be advantageous in fracture healing.8
THE REALITY
Healthy dogs or growing rats with linear diaphyseal fractures are imperfect models for elderly osteoporotic patients with hip fracture, as Dr. Herbert Fleisch noted in his editorial, “Can bisphosphonates be given to patients with fractures?”9 Still, if retained primary bone can be used in the process of fracture repair to gain an early mechanical advantage, then perhaps delayed remodeling will permit early mobilization and further fracture prevention in humans.
How soon after hip fracture surgery should a patient start a bisphosphonate? The only data we have are from a single randomized controlled trial designed to measure fracture risk reduction in osteoporotic patients with hip fracture using intravenous zoledronic acid 5 mg compared with placebo.2 A post hoc analysis of this study1 generated the limited clinical data we have on the optimal timing of the treatment. Linking these study data with the laboratory data, one would intuit that delaying the infusion of zoledronic acid for at least 2 weeks after hip fracture repair would offer a clinical reduction in fracture risk and improvement (or stabilization) in bone mineral density by 12 months, and a reduction in the rate of all-cause mortality beginning at 16 months.
References
Eriksen EF, Lyles KW, Colón-Emeric CS, et al. Antifracture efficacy and reduction of mortality in relation to timing of the first dose of zoledronic acid after hip fracture. J Bone Miner Res2009; 24:1308–1313.
Lyles KW, Colón-Emeric CS, Magaziner JS, et al; for the HORIZON Recurrent Fracture Trial. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med2007; 357:1799–1809.
Colón-Emeric CS, Mesenbrink P, Lyles KW, et al. Potential mediators of the mortality reduction with zoledronic acid after hip fracture. J Bone Miner Res2010; 25:91–97.
Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med2007; 356:1809–1822.
Bolland MJ, Grey AB, Gamble GD, Reid IR. Effect of osteoporosis treatment on mortality: a meta-analysis. J Clin Endocrinol Metab2010; 95:1174–1181.
Schindeler A, McDonald MM, Bokko P, Little DG. Bone remodeling during fracture repair: the cellular picture. Semin Cell Dev Biol2008; 19:459–466.
Amanat N, McDonald M, Godfrey C, Bilston L, Little D. Optimal timing of a single dose of zoledronic acid to increase strength in rat fracture repair. J Bone Miner Res2007; 22:867–876.
Li J, Mori S, Kaji Y, Mashiba T, Kawanishi J, Norimatsu H. Effect of bisphosphonate (incadronate) on fracture healing of long bones in rats. J Bone Miner Res1999; 14:969–979.
Fleisch H. Can bisphosphonates be given to patients with fractures?J Bone Miner Res2001; 16:437–440.
Margaret Seton, MD Assistant Professor of Medicine, Harvard Medical School; Director, Rheumatology Fellowship Program, Division of Rheumatology, Allergy, and Immunology, Massachusetts General Hospital, Boston, MA
Address: Margaret Seton, MD, Rheumatology, Allergy, and Immunology, Massachusetts General Hospital, Bulfinch 165, 55 Fruit Street, Boston, MA 02114; e-mail [email protected]
Margaret Seton, MD Assistant Professor of Medicine, Harvard Medical School; Director, Rheumatology Fellowship Program, Division of Rheumatology, Allergy, and Immunology, Massachusetts General Hospital, Boston, MA
Address: Margaret Seton, MD, Rheumatology, Allergy, and Immunology, Massachusetts General Hospital, Bulfinch 165, 55 Fruit Street, Boston, MA 02114; e-mail [email protected]
Author and Disclosure Information
Margaret Seton, MD Assistant Professor of Medicine, Harvard Medical School; Director, Rheumatology Fellowship Program, Division of Rheumatology, Allergy, and Immunology, Massachusetts General Hospital, Boston, MA
Address: Margaret Seton, MD, Rheumatology, Allergy, and Immunology, Massachusetts General Hospital, Bulfinch 165, 55 Fruit Street, Boston, MA 02114; e-mail [email protected]
Patients with an osteoporotic hip fracture suffer from profound morbidity and are at a heightened risk of death. It is therefore essential that they receive treatment with a bisphosphonate known to modify the subsequent risk of fracture at any site—eg, alendronate (Fosamax), risedronate (Actonel), or zoledronic acid (Reclast).
However, there is concern that starting a bisphosphonate too soon after surgery could disrupt bone remodeling and delay fracture repair.
Only one clinical study addressed the timing of bisphosphonate therapy after hip fracture repair. In this study, Eriksen et al1 performed a post hoc analysis of data from the Health Outcomes and Reduced Incidence With Zoledronic Acid Once Yearly Recurrent Fracture Trial (HORIZON-RFT)2 and concluded that the optimal time to give intravenous zoledronic acid is 2 to 12 weeks after surgical repair of the fracture.
In a frail, elderly patient with comorbidities, a single intravenous 5-mg dose of zoledronic acid guarantees adequate treatment, obviating issues of poor compliance and oral absorption and loss to follow-up. Sufficient levels of vitamin D and calcium should be ensured.
THE EVIDENCE
The original HORIZON-RFT study,2 published in 2007, compared intravenous zoledronic acid against placebo in elderly patients with osteoporotic hip fracture. Most of the patients were white women; their mean age was 74; 1,065 received intravenous zoledronic acid, and 1,062 received placebo. All received vitamin D and calcium.
The trial showed a clear reduction in the rate of recurrent fractures at other sites (a primary end point) and a reduction in the rate of all-cause mortality in patients treated within 90 days of fracture. A total of 424 fractures occurred in 231 patients. The risk of any new clinical fracture was 35% lower with treatment than with placebo (occurring in 8.6% vs 13.9% of patients, P = .001), and the number of deaths due to any cause was 28% lower with treatment than with placebo (occurring in 101 vs 141, P = .01).2
The mean time to fracture was 39.8 months in the treated group vs 36.4 in the placebo group. The fracture risk reduction began to be apparent by 12 months, and the reduction in mortality rate by 16 months.2
In a post hoc analysis of the trial, Eriksen et al1 attempted to ascertain the optimal time for therapy in terms of fracture risk and mortality reduction. Analyzing the data by 2-week intervals beginning after the surgical repair of the fracture, the authors found that only 56 patients (5.3%) had received zoledronic acid within 2 weeks of surgery and only 47 had received placebo, and they saw no advantage to intravenous zoledronic acid compared with placebo in these first 2 weeks with respect to bone mineral density, fracture risk, or risk of death. However, excluding this small subset, antifracture efficacy and reduction in mortality rate were present when patients were treated with zoledronic acid in the 2 to 12 weeks after hip fracture repair, and improvement in bone mineral density at the hip was noted at 12 months in all cohorts.
Colón-Emeric et al3 performed another post hoc analysis, attempting to explain the lower mortality rate seen in patients treated with zoledronic acid. It had been an unexpected finding, and determinants of mortality rate reduction were hampered by a limited knowledge of the true cause of death or the circumstances of care after fracture. The authors concluded that only 8% of the reduction in mortality rate evident early in the second year of treatment with zoledronic acid could be attributed to a reduction in fractures.3 Other mechanisms by which the mortality rate reduction occurred remained unclear.
Curiously, in another large randomized controlled trial of zoledronic acid, in women with postmenopausal osteoporosis, Black et al4 reported that more patients died in the treated group (130 of 3,862) than in the placebo group (112 of 3,852). This difference was not statistically significant, but neither was it explained.
A meta-analysis by Bolland et al5 examined the effect of other osteoporosis treatments on mortality rate, using randomized controlled trials that lasted more than 12 months and that reported more than 10 deaths. The authors concluded the following:
In the trials in which bisphosphonates reduced the mortality rate, the mortality rate in the placebo group was higher than 10 per 1,000 patient-years
The effect of osteoporosis treatment on the mortality rate in a frail, elderly population is evident using agents with proven efficacy in reducing vertebral and nonvertebral fractures, eg, alendronate, risedronate, and zoledronic acid.5
THE SCIENCE
Osteoporotic fractures occur with minimal trauma, with the failure of bone attributed to impaired integrity of bone microarchitecture. The ultimate goal of fracture repair is to restore bone size, shape, and tissue properties. The issue of when to treat with a bisphosphonate after hip fracture arises because bisphosphonates are known to disrupt bone remodeling and so delay fracture repair.
After fracture, both anabolic and catabolic phases occur.6 The final outcome depends on the following:
The type of intervention to stabilize the fracture site (eg, surgical repair)
The inflammatory cytokines and growth factors released by the cellular elements in bloody and disrupted tissue.
Oxygen tension, angiogenesis, and osteoblasts are critical to primary bone formation, and osteoclasts are essential in remodeling this initial bone deposition. These late phases of fracture repair are most vulnerable to the bisphosphonates, through suppression of osteoclast resorption and possibly through decreased angiogenesis.6 Callus formation is sustained, but bone remodeling is delayed.
Amanat et al7 examined the timing of a single dose of zoledronic acid after fracture repair in a rat model of diaphyseal fracture and found that the callus was larger and stronger if the bisphosphonate dose had been delayed 1 or 2 weeks. The animals treated with zoledronic acid showed a remarkable trabecular network of bone between the original femoral cortex and the new cortical bone that was not present in the control group, perhaps contributing to the enhanced mechanical properties of the callus. Other studies suggest single dosing rather than continuous dosing may be advantageous in fracture healing.8
THE REALITY
Healthy dogs or growing rats with linear diaphyseal fractures are imperfect models for elderly osteoporotic patients with hip fracture, as Dr. Herbert Fleisch noted in his editorial, “Can bisphosphonates be given to patients with fractures?”9 Still, if retained primary bone can be used in the process of fracture repair to gain an early mechanical advantage, then perhaps delayed remodeling will permit early mobilization and further fracture prevention in humans.
How soon after hip fracture surgery should a patient start a bisphosphonate? The only data we have are from a single randomized controlled trial designed to measure fracture risk reduction in osteoporotic patients with hip fracture using intravenous zoledronic acid 5 mg compared with placebo.2 A post hoc analysis of this study1 generated the limited clinical data we have on the optimal timing of the treatment. Linking these study data with the laboratory data, one would intuit that delaying the infusion of zoledronic acid for at least 2 weeks after hip fracture repair would offer a clinical reduction in fracture risk and improvement (or stabilization) in bone mineral density by 12 months, and a reduction in the rate of all-cause mortality beginning at 16 months.
Patients with an osteoporotic hip fracture suffer from profound morbidity and are at a heightened risk of death. It is therefore essential that they receive treatment with a bisphosphonate known to modify the subsequent risk of fracture at any site—eg, alendronate (Fosamax), risedronate (Actonel), or zoledronic acid (Reclast).
However, there is concern that starting a bisphosphonate too soon after surgery could disrupt bone remodeling and delay fracture repair.
Only one clinical study addressed the timing of bisphosphonate therapy after hip fracture repair. In this study, Eriksen et al1 performed a post hoc analysis of data from the Health Outcomes and Reduced Incidence With Zoledronic Acid Once Yearly Recurrent Fracture Trial (HORIZON-RFT)2 and concluded that the optimal time to give intravenous zoledronic acid is 2 to 12 weeks after surgical repair of the fracture.
In a frail, elderly patient with comorbidities, a single intravenous 5-mg dose of zoledronic acid guarantees adequate treatment, obviating issues of poor compliance and oral absorption and loss to follow-up. Sufficient levels of vitamin D and calcium should be ensured.
THE EVIDENCE
The original HORIZON-RFT study,2 published in 2007, compared intravenous zoledronic acid against placebo in elderly patients with osteoporotic hip fracture. Most of the patients were white women; their mean age was 74; 1,065 received intravenous zoledronic acid, and 1,062 received placebo. All received vitamin D and calcium.
The trial showed a clear reduction in the rate of recurrent fractures at other sites (a primary end point) and a reduction in the rate of all-cause mortality in patients treated within 90 days of fracture. A total of 424 fractures occurred in 231 patients. The risk of any new clinical fracture was 35% lower with treatment than with placebo (occurring in 8.6% vs 13.9% of patients, P = .001), and the number of deaths due to any cause was 28% lower with treatment than with placebo (occurring in 101 vs 141, P = .01).2
The mean time to fracture was 39.8 months in the treated group vs 36.4 in the placebo group. The fracture risk reduction began to be apparent by 12 months, and the reduction in mortality rate by 16 months.2
In a post hoc analysis of the trial, Eriksen et al1 attempted to ascertain the optimal time for therapy in terms of fracture risk and mortality reduction. Analyzing the data by 2-week intervals beginning after the surgical repair of the fracture, the authors found that only 56 patients (5.3%) had received zoledronic acid within 2 weeks of surgery and only 47 had received placebo, and they saw no advantage to intravenous zoledronic acid compared with placebo in these first 2 weeks with respect to bone mineral density, fracture risk, or risk of death. However, excluding this small subset, antifracture efficacy and reduction in mortality rate were present when patients were treated with zoledronic acid in the 2 to 12 weeks after hip fracture repair, and improvement in bone mineral density at the hip was noted at 12 months in all cohorts.
Colón-Emeric et al3 performed another post hoc analysis, attempting to explain the lower mortality rate seen in patients treated with zoledronic acid. It had been an unexpected finding, and determinants of mortality rate reduction were hampered by a limited knowledge of the true cause of death or the circumstances of care after fracture. The authors concluded that only 8% of the reduction in mortality rate evident early in the second year of treatment with zoledronic acid could be attributed to a reduction in fractures.3 Other mechanisms by which the mortality rate reduction occurred remained unclear.
Curiously, in another large randomized controlled trial of zoledronic acid, in women with postmenopausal osteoporosis, Black et al4 reported that more patients died in the treated group (130 of 3,862) than in the placebo group (112 of 3,852). This difference was not statistically significant, but neither was it explained.
A meta-analysis by Bolland et al5 examined the effect of other osteoporosis treatments on mortality rate, using randomized controlled trials that lasted more than 12 months and that reported more than 10 deaths. The authors concluded the following:
In the trials in which bisphosphonates reduced the mortality rate, the mortality rate in the placebo group was higher than 10 per 1,000 patient-years
The effect of osteoporosis treatment on the mortality rate in a frail, elderly population is evident using agents with proven efficacy in reducing vertebral and nonvertebral fractures, eg, alendronate, risedronate, and zoledronic acid.5
THE SCIENCE
Osteoporotic fractures occur with minimal trauma, with the failure of bone attributed to impaired integrity of bone microarchitecture. The ultimate goal of fracture repair is to restore bone size, shape, and tissue properties. The issue of when to treat with a bisphosphonate after hip fracture arises because bisphosphonates are known to disrupt bone remodeling and so delay fracture repair.
After fracture, both anabolic and catabolic phases occur.6 The final outcome depends on the following:
The type of intervention to stabilize the fracture site (eg, surgical repair)
The inflammatory cytokines and growth factors released by the cellular elements in bloody and disrupted tissue.
Oxygen tension, angiogenesis, and osteoblasts are critical to primary bone formation, and osteoclasts are essential in remodeling this initial bone deposition. These late phases of fracture repair are most vulnerable to the bisphosphonates, through suppression of osteoclast resorption and possibly through decreased angiogenesis.6 Callus formation is sustained, but bone remodeling is delayed.
Amanat et al7 examined the timing of a single dose of zoledronic acid after fracture repair in a rat model of diaphyseal fracture and found that the callus was larger and stronger if the bisphosphonate dose had been delayed 1 or 2 weeks. The animals treated with zoledronic acid showed a remarkable trabecular network of bone between the original femoral cortex and the new cortical bone that was not present in the control group, perhaps contributing to the enhanced mechanical properties of the callus. Other studies suggest single dosing rather than continuous dosing may be advantageous in fracture healing.8
THE REALITY
Healthy dogs or growing rats with linear diaphyseal fractures are imperfect models for elderly osteoporotic patients with hip fracture, as Dr. Herbert Fleisch noted in his editorial, “Can bisphosphonates be given to patients with fractures?”9 Still, if retained primary bone can be used in the process of fracture repair to gain an early mechanical advantage, then perhaps delayed remodeling will permit early mobilization and further fracture prevention in humans.
How soon after hip fracture surgery should a patient start a bisphosphonate? The only data we have are from a single randomized controlled trial designed to measure fracture risk reduction in osteoporotic patients with hip fracture using intravenous zoledronic acid 5 mg compared with placebo.2 A post hoc analysis of this study1 generated the limited clinical data we have on the optimal timing of the treatment. Linking these study data with the laboratory data, one would intuit that delaying the infusion of zoledronic acid for at least 2 weeks after hip fracture repair would offer a clinical reduction in fracture risk and improvement (or stabilization) in bone mineral density by 12 months, and a reduction in the rate of all-cause mortality beginning at 16 months.
References
Eriksen EF, Lyles KW, Colón-Emeric CS, et al. Antifracture efficacy and reduction of mortality in relation to timing of the first dose of zoledronic acid after hip fracture. J Bone Miner Res2009; 24:1308–1313.
Lyles KW, Colón-Emeric CS, Magaziner JS, et al; for the HORIZON Recurrent Fracture Trial. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med2007; 357:1799–1809.
Colón-Emeric CS, Mesenbrink P, Lyles KW, et al. Potential mediators of the mortality reduction with zoledronic acid after hip fracture. J Bone Miner Res2010; 25:91–97.
Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med2007; 356:1809–1822.
Bolland MJ, Grey AB, Gamble GD, Reid IR. Effect of osteoporosis treatment on mortality: a meta-analysis. J Clin Endocrinol Metab2010; 95:1174–1181.
Schindeler A, McDonald MM, Bokko P, Little DG. Bone remodeling during fracture repair: the cellular picture. Semin Cell Dev Biol2008; 19:459–466.
Amanat N, McDonald M, Godfrey C, Bilston L, Little D. Optimal timing of a single dose of zoledronic acid to increase strength in rat fracture repair. J Bone Miner Res2007; 22:867–876.
Li J, Mori S, Kaji Y, Mashiba T, Kawanishi J, Norimatsu H. Effect of bisphosphonate (incadronate) on fracture healing of long bones in rats. J Bone Miner Res1999; 14:969–979.
Fleisch H. Can bisphosphonates be given to patients with fractures?J Bone Miner Res2001; 16:437–440.
References
Eriksen EF, Lyles KW, Colón-Emeric CS, et al. Antifracture efficacy and reduction of mortality in relation to timing of the first dose of zoledronic acid after hip fracture. J Bone Miner Res2009; 24:1308–1313.
Lyles KW, Colón-Emeric CS, Magaziner JS, et al; for the HORIZON Recurrent Fracture Trial. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med2007; 357:1799–1809.
Colón-Emeric CS, Mesenbrink P, Lyles KW, et al. Potential mediators of the mortality reduction with zoledronic acid after hip fracture. J Bone Miner Res2010; 25:91–97.
Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med2007; 356:1809–1822.
Bolland MJ, Grey AB, Gamble GD, Reid IR. Effect of osteoporosis treatment on mortality: a meta-analysis. J Clin Endocrinol Metab2010; 95:1174–1181.
Schindeler A, McDonald MM, Bokko P, Little DG. Bone remodeling during fracture repair: the cellular picture. Semin Cell Dev Biol2008; 19:459–466.
Amanat N, McDonald M, Godfrey C, Bilston L, Little D. Optimal timing of a single dose of zoledronic acid to increase strength in rat fracture repair. J Bone Miner Res2007; 22:867–876.
Li J, Mori S, Kaji Y, Mashiba T, Kawanishi J, Norimatsu H. Effect of bisphosphonate (incadronate) on fracture healing of long bones in rats. J Bone Miner Res1999; 14:969–979.
Fleisch H. Can bisphosphonates be given to patients with fractures?J Bone Miner Res2001; 16:437–440.
Although we often approach anticoagulation therapy with a confidence born of familiarity, it is not for the faint of heart. We start chronic anticoagulation in several clinical settings, such as to prevent a recurrence after a thromboembolic event. But this decision requires weighing the increased risk of bleeding from the anticoagulant therapy against the risk of another thromboembolic event.
Along with massive pulmonary embolism, the most feared thromboembolic event is the clot that migrates to the brain, resulting in life-altering stroke. We assess this risk in a semiquantitative manner in patients with atrial fibrillation using the CHADS2 score, hoping to maximize the benefits of anticoagulation while reducing the risks. We recognize that patients at the greatest risk of stroke in this setting are those with a history of a prior stroke. Also, patients bedridden with a recent cerebrovascular accident (CVA) seem to be hypercoagulable, potentially adding risk to recent injury. Thus, we try to start anticoagulation as soon as feasible after the diagnosis of a possible thrombotic event.
But the decision to start or resume anticoagulation is especially agonizing in a patient who has suffered an intracerebral hemorrhage. In this issue of the Journal, Drs. Joshua Goldstein and Steven Greenberg and Dr. Franklin Michota provide a thoughtful discussion of the issues we need to consider in these patients.
While not contributing to the prevention of additional CVAs or other arterial thrombotic events, a modality often underused in the prevention of thrombotic disease is the application (not just the ordering) of compressive leg stockings to bedridden hospitalized patients who cannot, for any reason, be provided pharmacologic anticoagulation therapy. I just completed a stint of hospital consultation, and I was pleased to see the widespread integration of prophylactic anticoagulation therapy, but somewhat dismayed by the number of compressive stockings I watched pumping with vigor, but to no one’s benefit, as they were draped over a bed rail.
As we struggle with complex clinical decisions, we need to also be attentive to the simple and the seemingly mundane: using the foam dispenser at the door, offering the verbal greeting and patient touch at the bedside, and rewrapping the pneumatic stockings that have somehow migrated between mattress and footboard.
Although we often approach anticoagulation therapy with a confidence born of familiarity, it is not for the faint of heart. We start chronic anticoagulation in several clinical settings, such as to prevent a recurrence after a thromboembolic event. But this decision requires weighing the increased risk of bleeding from the anticoagulant therapy against the risk of another thromboembolic event.
Along with massive pulmonary embolism, the most feared thromboembolic event is the clot that migrates to the brain, resulting in life-altering stroke. We assess this risk in a semiquantitative manner in patients with atrial fibrillation using the CHADS2 score, hoping to maximize the benefits of anticoagulation while reducing the risks. We recognize that patients at the greatest risk of stroke in this setting are those with a history of a prior stroke. Also, patients bedridden with a recent cerebrovascular accident (CVA) seem to be hypercoagulable, potentially adding risk to recent injury. Thus, we try to start anticoagulation as soon as feasible after the diagnosis of a possible thrombotic event.
But the decision to start or resume anticoagulation is especially agonizing in a patient who has suffered an intracerebral hemorrhage. In this issue of the Journal, Drs. Joshua Goldstein and Steven Greenberg and Dr. Franklin Michota provide a thoughtful discussion of the issues we need to consider in these patients.
While not contributing to the prevention of additional CVAs or other arterial thrombotic events, a modality often underused in the prevention of thrombotic disease is the application (not just the ordering) of compressive leg stockings to bedridden hospitalized patients who cannot, for any reason, be provided pharmacologic anticoagulation therapy. I just completed a stint of hospital consultation, and I was pleased to see the widespread integration of prophylactic anticoagulation therapy, but somewhat dismayed by the number of compressive stockings I watched pumping with vigor, but to no one’s benefit, as they were draped over a bed rail.
As we struggle with complex clinical decisions, we need to also be attentive to the simple and the seemingly mundane: using the foam dispenser at the door, offering the verbal greeting and patient touch at the bedside, and rewrapping the pneumatic stockings that have somehow migrated between mattress and footboard.
Although we often approach anticoagulation therapy with a confidence born of familiarity, it is not for the faint of heart. We start chronic anticoagulation in several clinical settings, such as to prevent a recurrence after a thromboembolic event. But this decision requires weighing the increased risk of bleeding from the anticoagulant therapy against the risk of another thromboembolic event.
Along with massive pulmonary embolism, the most feared thromboembolic event is the clot that migrates to the brain, resulting in life-altering stroke. We assess this risk in a semiquantitative manner in patients with atrial fibrillation using the CHADS2 score, hoping to maximize the benefits of anticoagulation while reducing the risks. We recognize that patients at the greatest risk of stroke in this setting are those with a history of a prior stroke. Also, patients bedridden with a recent cerebrovascular accident (CVA) seem to be hypercoagulable, potentially adding risk to recent injury. Thus, we try to start anticoagulation as soon as feasible after the diagnosis of a possible thrombotic event.
But the decision to start or resume anticoagulation is especially agonizing in a patient who has suffered an intracerebral hemorrhage. In this issue of the Journal, Drs. Joshua Goldstein and Steven Greenberg and Dr. Franklin Michota provide a thoughtful discussion of the issues we need to consider in these patients.
While not contributing to the prevention of additional CVAs or other arterial thrombotic events, a modality often underused in the prevention of thrombotic disease is the application (not just the ordering) of compressive leg stockings to bedridden hospitalized patients who cannot, for any reason, be provided pharmacologic anticoagulation therapy. I just completed a stint of hospital consultation, and I was pleased to see the widespread integration of prophylactic anticoagulation therapy, but somewhat dismayed by the number of compressive stockings I watched pumping with vigor, but to no one’s benefit, as they were draped over a bed rail.
As we struggle with complex clinical decisions, we need to also be attentive to the simple and the seemingly mundane: using the foam dispenser at the door, offering the verbal greeting and patient touch at the bedside, and rewrapping the pneumatic stockings that have somehow migrated between mattress and footboard.
Anticoagulants have been helping patients at risk of thrombosis since the late 1930s.1,2 Although the indications for these agents are many, the development of anticoagulants beyond oral vitamin K antagonists and parenteral heparin has been slow. In the United States, the vitamin K antagonist warfarin (Coumadin) is still the only oral anticoagulant available.
The major complication of anticoagulant therapy is bleeding, and vitamin K antagonists have proven challenging to use in clinical practice.1,3 They have a narrow therapeutic window, they vary considerably in dose-response from patient to patient, and they are subject to significant interactions with other drugs and with foods. For these reasons, therapy must be monitored with laboratory testing, and good patient compliance and patient education are essential. Yet even with these measures, life-threatening hemorrhage still can occur.
In this issue of the Cleveland Clinic Journal of Medicine, Goldstein and Greenberg4 review warfarin-related intracerebral hemorrhage (ICH) and provide a framework for considering whether to resume anticoagulant therapy.
WHAT TO DO IN THE ACUTE PHASE
Goldstein and Greenberg divide the difficult clinical question of what to do after ICH into the acute phase and the chronic phase.
What to do in the acute phase appears straightforward, as the risk of hematoma expansion in the hours immediately after warfarin-related ICH outweighs the risk of arterial or venous thromboembolism. Anticoagulant reversal should be the primary consideration in the first 24 hours, and, assuming the patient does not have acute (< 4-week-old) deep vein thrombosis, intermittent pneumatic compression should be applied to the lower extremities to reduce the risk of venous thromboembolism associated with ICH.5
Prophylactic anticoagulation with subcutaneous fixed-dose heparin or low-molecular-weight heparin is recommended starting 72 hours after ICH is diagnosed, provided the patient is not underweight (< 50 kg), has relatively normal renal function (creatinine clearance > 30 mL/minute/1.73 m2) and normal platelet function, and does not have coagulopathy. 6 If any one of these criteria is not met, the risk of bleeding can be higher, even with only prophylactic doses of anticoagulant drugs. Prophylactic anticoagulation should be continued until hospital and rehabilitation discharge, typically 1 to 2 weeks after ICH, depending on the severity of the patient’s neurologic impairment.
If a patient with warfarin-related ICH has concomitant acute proximal deep vein thrombosis or pulmonary embolism (ie, < 4 weeks old), then caval interruption therapy would be indicated.7 Although retrievable inferior vena cava filters are increasingly preferred over permanent filters, it is important to recognize the relative lack of both longitudinal and prospective data on retrievable devices. Given that provoked venous thromboembolism requires a minimum of 3 months of anticoagulation, and retrievable filters generally need to be removed before 3 months, a retrievable filter should be chosen only if the clinician has already decided that oral anticoagulation will be restarted in the next 3 to 4 weeks after filter removal.
WHAT TO DO IN THE CHRONIC PHASE
A more difficult question in patients with warfarin-related ICH arises in the chronic phase: should oral anticoagulation be resumed at all?
Goldstein and Greenberg outline important considerations. Under the principle of primum non nocere, patients who have suffered a warfarin-related ICH should first be evaluated for their risk of thrombosis in light of their original indication for oral anticoagulant therapy. As the authors point out, oral anticoagulation for primary prevention of thrombosis after warfarin-related ICH must be viewed differently than oral anticoagulation for secondary prevention of thrombosis. In addition, Douketis et al8 have described a method of stratifying a patient’s risk of thrombosis as low, moderate, or high (Table 1), which is the basis for decisions about perioperative anticoagulation. Based on Goldstein and Greenberg’s review, we can similarly categorize these patients as being at low, moderate, or high risk of ICH recurrence (Table 2). Patients at low risk of thrombosis should probably not resume taking a vitamin K antagonist, regardless of their ICH risk (Table 3). It would be reasonable, however, for patients at moderate or high risk of thrombosis and at low risk of ICH to resume taking their vitamin K antagonist.
Uncertainty remains for patients with a moderate or high risk of thrombosis and a moderate or high risk of ICH. For patients with these combinations of risk, individualized approaches need to be explored. All attempts should be made to widen the margin of safety of vitamin K antagonist therapy; these include referring the patient to an anticoagulation management service, frequent laboratory monitoring, and ongoing patient education.1
Since the risk of ICH is related to the intensity of anticoagulation, a lower target international normalized ratio may be the best compromise, depending on the patient. Alternatively, antiplatelet therapy alone may offer some benefit with less risk of ICH.
THE NEWER ORAL ANTICOAGULANTS
As Goldstein and Greenberg mention, the ongoing development of new and potentially safer oral anticoagulants may affect how we approach these risk-benefit equations.
Three new oral anticoagulants—dabigatran (Pradaxa), apixaban, and rivaroxaban (Xarelto)—are being tested for various anticoagulant indications, and several phase III studies have recently closed or are nearing completion.
Dabigatran is an oral direct thrombin inhibitor currently available in Europe and Canada.
In the Randomized Evaluation of Long-term Anticoagulant Therapy (RE-LY) trial, the efficacy and safety of two different doses of dabigatran (110 mg twice daily or 150 mg twice daily) relative to warfarin were studied in more than 18,000 patients with atrial fibrillation. 9 The primary outcome measure was stroke or systemic embolism. Dabigatran 110 mg was not inferior to warfarin in terms of the primary outcome, while dabigatran 150 mg was superior. The rate of major bleeding was 3.36% per year in the warfarin group vs 2.71% in the 110-mg group (P = .003) and 3.11% in the 150-mg group (P not significant).
Additional safety data on this drug are available from the 2,500-patient RE-COVER trial.10 Dabigatran was not inferior to warfarin in the treatment of acute venous thromboembolism, with a similar rate of major bleeding and a lower rate of combined major plus nonmajor bleeding.
Apixaban, an oral direct factor Xa inhibitor, is in a phase III trial in patients with atrial fibrillation—Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE)11—comparing apixaban vs warfarin. Another phase III trial, AVERROES,12 was stopped early after a predefined interim analysis by the independent data-monitoring committee found clear evidence of benefit in the apixaban group.13 The AVERROES results were presented at the 2010 European Society of Cardiology Congress, August 28–September 1, Stockholm, Sweden.14
Rivaroxaban, another promising oral direct factor Xa inhibitor, is currently available in Europe and Canada for the prevention of thrombosis in orthopedic surgery patients. Rivaroxaban is also in large phase III trials for the treatment of acute venous thromboembolism15–17 and for the prevention of stroke in atrial fibrillation.18
Newer agents have drawbacks, too
These new agents need no laboratory monitoring, and they do not appear to be subject to the dose variability and the interactions with drugs and foods seen with vitamin K antagonists. As a result, they may pose less risk of anticoagulant-related ICH.
The decision to resume anticoagulation after anticoagulant-associated intracranial hemorrhage should be based on the risk of rebleeding vs the risk of thrombosis. Patients determined to be at high risk of thrombosis and low risk of rebleeding are the best candidates for resuming anticoagulation.
Still, for patients who suffer an anticoagulant- or warfarin-related ICH, these new anticoagulants are not likely to simplify the issue of restarting anticoagulant therapy. Unlike vitamin K antagonists, dabigatran and the direct factor Xa inhibitors have no known antidote for their anticoagulant effects. Animal data suggest that factor Xa concentrates may help,19 but for patients at risk of a second anticoagulant-related ICH, this does not provide much reassurance.
As with all clinical decisions in medicine, the potential benefits of any therapy should outweigh the risks. In the case of warfarin-related ICH, resuming anticoagulant therapy requires careful consideration of many factors, including patient preferences and tolerance of different levels of risk. As new and perhaps safer anticoagulants become available, clinicians may face such difficult questions less and less. But in the meantime, doctors and their patients are left to pick their poison.
References
Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G; American College of Chest Physicians. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):160S–198S.
Hirsh J, Bauer KA, Donati MB, Gould M, Samama MM, Weitz JI; American College of Chest Physicians. Parenteral anticoagulants: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):141S–159S.
Schulman S, Beyth RJ, Kearon C, Levine MN; American College of Chest Physicians. Hemorrhagic complications of anticoagulant and thrombolytic treatment: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th edition). Chest2008; 133(suppl 6):257S–298S.
Goldstein JN, Greenberg SM. Should anticoagulation be resumed after intracerebral hemorrhage?Cleve Clin J Med2010; 77:791–799.
Geerts WH, Bergqvist D, Pineo GF, et al; American College of Chest Physicians. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):381S–453S.
Michota F, Merli G. Anticoagulation in special patient populations: are special dosing considerations required?Cleve Clin J Med2005; 72(suppl 1):S37–S42.
Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ; American College of Chest Physicians. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):454S–545S.
Douketis JD, Berger PB, Dunn AS, et al; American College of Chest Physicians. The perioperative management of antithrombotic therapy: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):299S–339S.
Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med2009; 361:1139–1151.
Schulman S, Kearon C, Kakkar AK, et al; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med2009; 361:2342–2352.
Lopes RD, Alexander JH, Al-Khatib SM; ARISTOTLE Investigators. Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial: design and rationale. Am Heart J2010; 159:331–339.
Eikelboom JW, O’Donnell M, Yusuf S, et al. Rationale and design of AVERROES: apixaban versus acetylsalicylic acid to prevent stroke in atrial fibrillation patients who have failed or are unsuitable for vitamin K antagonist treatment. Am Heart J2010; 159:348–353.
Pfizer/Bristol-Myers Squibb. AVERROES study of investigational agent apixaban closes early due to clear evidence of efficacy, June 9, 2010. www.theheart.org/article/1087291.do. Accessed September 26, 2010.
Once-daily oral direct factor Xa inhibitor rivaroxaban in the long-term prevention of recurrent symptomatic venous thromboembolism in patients with symptomatic deep-vein thrombosis or pulmonary embolism. The Einstein-Extension Study. http://clinicaltrials.gov/ct2/show/NCT00439725. Accessed September 26, 2010.
Oral direct factor Xa inhibitor rivaroxaban in patients with acute symptomatic deep-vein thrombosis without symptomatic pulmonary embolism: Einstein-DVT Evaluation. http://clinicaltrials.gov/ct2/show/NCT00440193. Accessed September 26, 2010.
Oral direct factor Xa inhibitor rivaroxaban in patients with acute symptomatic pulmonary embolism with or without symptomatic deep-vein thrombosis: Einstein-PE Evaluation. http://clinicaltrials.gov/ct2/show/NCT00439777. Accessed September 26, 2010.
ROCKET AF Study Investigators. Rivaroxaban once-daily, oral, direct factor Xa inhibition compared with vitamin K antagonism for prevention of stroke and embolism trial in atrial fibrillation: rationale and design of the ROCKET AF study. Am Heart J2010; 159:340–347.
Weitz JI, Hirsh J, Samama MM; American College of Chest Physicians. New antithrombotic drugs: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):234S–256S.
Franklin Michota, MD, FHM Associate Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH, and Director of Academic Affairs, Department of Hospital Medicine, Cleveland Clinic
Address: Franklin Michota, MD, Department of Hospital Medicine, M2 Anx, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
Franklin Michota, MD, FHM Associate Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH, and Director of Academic Affairs, Department of Hospital Medicine, Cleveland Clinic
Address: Franklin Michota, MD, Department of Hospital Medicine, M2 Anx, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
Author and Disclosure Information
Franklin Michota, MD, FHM Associate Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH, and Director of Academic Affairs, Department of Hospital Medicine, Cleveland Clinic
Address: Franklin Michota, MD, Department of Hospital Medicine, M2 Anx, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
Anticoagulants have been helping patients at risk of thrombosis since the late 1930s.1,2 Although the indications for these agents are many, the development of anticoagulants beyond oral vitamin K antagonists and parenteral heparin has been slow. In the United States, the vitamin K antagonist warfarin (Coumadin) is still the only oral anticoagulant available.
The major complication of anticoagulant therapy is bleeding, and vitamin K antagonists have proven challenging to use in clinical practice.1,3 They have a narrow therapeutic window, they vary considerably in dose-response from patient to patient, and they are subject to significant interactions with other drugs and with foods. For these reasons, therapy must be monitored with laboratory testing, and good patient compliance and patient education are essential. Yet even with these measures, life-threatening hemorrhage still can occur.
In this issue of the Cleveland Clinic Journal of Medicine, Goldstein and Greenberg4 review warfarin-related intracerebral hemorrhage (ICH) and provide a framework for considering whether to resume anticoagulant therapy.
WHAT TO DO IN THE ACUTE PHASE
Goldstein and Greenberg divide the difficult clinical question of what to do after ICH into the acute phase and the chronic phase.
What to do in the acute phase appears straightforward, as the risk of hematoma expansion in the hours immediately after warfarin-related ICH outweighs the risk of arterial or venous thromboembolism. Anticoagulant reversal should be the primary consideration in the first 24 hours, and, assuming the patient does not have acute (< 4-week-old) deep vein thrombosis, intermittent pneumatic compression should be applied to the lower extremities to reduce the risk of venous thromboembolism associated with ICH.5
Prophylactic anticoagulation with subcutaneous fixed-dose heparin or low-molecular-weight heparin is recommended starting 72 hours after ICH is diagnosed, provided the patient is not underweight (< 50 kg), has relatively normal renal function (creatinine clearance > 30 mL/minute/1.73 m2) and normal platelet function, and does not have coagulopathy. 6 If any one of these criteria is not met, the risk of bleeding can be higher, even with only prophylactic doses of anticoagulant drugs. Prophylactic anticoagulation should be continued until hospital and rehabilitation discharge, typically 1 to 2 weeks after ICH, depending on the severity of the patient’s neurologic impairment.
If a patient with warfarin-related ICH has concomitant acute proximal deep vein thrombosis or pulmonary embolism (ie, < 4 weeks old), then caval interruption therapy would be indicated.7 Although retrievable inferior vena cava filters are increasingly preferred over permanent filters, it is important to recognize the relative lack of both longitudinal and prospective data on retrievable devices. Given that provoked venous thromboembolism requires a minimum of 3 months of anticoagulation, and retrievable filters generally need to be removed before 3 months, a retrievable filter should be chosen only if the clinician has already decided that oral anticoagulation will be restarted in the next 3 to 4 weeks after filter removal.
WHAT TO DO IN THE CHRONIC PHASE
A more difficult question in patients with warfarin-related ICH arises in the chronic phase: should oral anticoagulation be resumed at all?
Goldstein and Greenberg outline important considerations. Under the principle of primum non nocere, patients who have suffered a warfarin-related ICH should first be evaluated for their risk of thrombosis in light of their original indication for oral anticoagulant therapy. As the authors point out, oral anticoagulation for primary prevention of thrombosis after warfarin-related ICH must be viewed differently than oral anticoagulation for secondary prevention of thrombosis. In addition, Douketis et al8 have described a method of stratifying a patient’s risk of thrombosis as low, moderate, or high (Table 1), which is the basis for decisions about perioperative anticoagulation. Based on Goldstein and Greenberg’s review, we can similarly categorize these patients as being at low, moderate, or high risk of ICH recurrence (Table 2). Patients at low risk of thrombosis should probably not resume taking a vitamin K antagonist, regardless of their ICH risk (Table 3). It would be reasonable, however, for patients at moderate or high risk of thrombosis and at low risk of ICH to resume taking their vitamin K antagonist.
Uncertainty remains for patients with a moderate or high risk of thrombosis and a moderate or high risk of ICH. For patients with these combinations of risk, individualized approaches need to be explored. All attempts should be made to widen the margin of safety of vitamin K antagonist therapy; these include referring the patient to an anticoagulation management service, frequent laboratory monitoring, and ongoing patient education.1
Since the risk of ICH is related to the intensity of anticoagulation, a lower target international normalized ratio may be the best compromise, depending on the patient. Alternatively, antiplatelet therapy alone may offer some benefit with less risk of ICH.
THE NEWER ORAL ANTICOAGULANTS
As Goldstein and Greenberg mention, the ongoing development of new and potentially safer oral anticoagulants may affect how we approach these risk-benefit equations.
Three new oral anticoagulants—dabigatran (Pradaxa), apixaban, and rivaroxaban (Xarelto)—are being tested for various anticoagulant indications, and several phase III studies have recently closed or are nearing completion.
Dabigatran is an oral direct thrombin inhibitor currently available in Europe and Canada.
In the Randomized Evaluation of Long-term Anticoagulant Therapy (RE-LY) trial, the efficacy and safety of two different doses of dabigatran (110 mg twice daily or 150 mg twice daily) relative to warfarin were studied in more than 18,000 patients with atrial fibrillation. 9 The primary outcome measure was stroke or systemic embolism. Dabigatran 110 mg was not inferior to warfarin in terms of the primary outcome, while dabigatran 150 mg was superior. The rate of major bleeding was 3.36% per year in the warfarin group vs 2.71% in the 110-mg group (P = .003) and 3.11% in the 150-mg group (P not significant).
Additional safety data on this drug are available from the 2,500-patient RE-COVER trial.10 Dabigatran was not inferior to warfarin in the treatment of acute venous thromboembolism, with a similar rate of major bleeding and a lower rate of combined major plus nonmajor bleeding.
Apixaban, an oral direct factor Xa inhibitor, is in a phase III trial in patients with atrial fibrillation—Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE)11—comparing apixaban vs warfarin. Another phase III trial, AVERROES,12 was stopped early after a predefined interim analysis by the independent data-monitoring committee found clear evidence of benefit in the apixaban group.13 The AVERROES results were presented at the 2010 European Society of Cardiology Congress, August 28–September 1, Stockholm, Sweden.14
Rivaroxaban, another promising oral direct factor Xa inhibitor, is currently available in Europe and Canada for the prevention of thrombosis in orthopedic surgery patients. Rivaroxaban is also in large phase III trials for the treatment of acute venous thromboembolism15–17 and for the prevention of stroke in atrial fibrillation.18
Newer agents have drawbacks, too
These new agents need no laboratory monitoring, and they do not appear to be subject to the dose variability and the interactions with drugs and foods seen with vitamin K antagonists. As a result, they may pose less risk of anticoagulant-related ICH.
The decision to resume anticoagulation after anticoagulant-associated intracranial hemorrhage should be based on the risk of rebleeding vs the risk of thrombosis. Patients determined to be at high risk of thrombosis and low risk of rebleeding are the best candidates for resuming anticoagulation.
Still, for patients who suffer an anticoagulant- or warfarin-related ICH, these new anticoagulants are not likely to simplify the issue of restarting anticoagulant therapy. Unlike vitamin K antagonists, dabigatran and the direct factor Xa inhibitors have no known antidote for their anticoagulant effects. Animal data suggest that factor Xa concentrates may help,19 but for patients at risk of a second anticoagulant-related ICH, this does not provide much reassurance.
As with all clinical decisions in medicine, the potential benefits of any therapy should outweigh the risks. In the case of warfarin-related ICH, resuming anticoagulant therapy requires careful consideration of many factors, including patient preferences and tolerance of different levels of risk. As new and perhaps safer anticoagulants become available, clinicians may face such difficult questions less and less. But in the meantime, doctors and their patients are left to pick their poison.
Anticoagulants have been helping patients at risk of thrombosis since the late 1930s.1,2 Although the indications for these agents are many, the development of anticoagulants beyond oral vitamin K antagonists and parenteral heparin has been slow. In the United States, the vitamin K antagonist warfarin (Coumadin) is still the only oral anticoagulant available.
The major complication of anticoagulant therapy is bleeding, and vitamin K antagonists have proven challenging to use in clinical practice.1,3 They have a narrow therapeutic window, they vary considerably in dose-response from patient to patient, and they are subject to significant interactions with other drugs and with foods. For these reasons, therapy must be monitored with laboratory testing, and good patient compliance and patient education are essential. Yet even with these measures, life-threatening hemorrhage still can occur.
In this issue of the Cleveland Clinic Journal of Medicine, Goldstein and Greenberg4 review warfarin-related intracerebral hemorrhage (ICH) and provide a framework for considering whether to resume anticoagulant therapy.
WHAT TO DO IN THE ACUTE PHASE
Goldstein and Greenberg divide the difficult clinical question of what to do after ICH into the acute phase and the chronic phase.
What to do in the acute phase appears straightforward, as the risk of hematoma expansion in the hours immediately after warfarin-related ICH outweighs the risk of arterial or venous thromboembolism. Anticoagulant reversal should be the primary consideration in the first 24 hours, and, assuming the patient does not have acute (< 4-week-old) deep vein thrombosis, intermittent pneumatic compression should be applied to the lower extremities to reduce the risk of venous thromboembolism associated with ICH.5
Prophylactic anticoagulation with subcutaneous fixed-dose heparin or low-molecular-weight heparin is recommended starting 72 hours after ICH is diagnosed, provided the patient is not underweight (< 50 kg), has relatively normal renal function (creatinine clearance > 30 mL/minute/1.73 m2) and normal platelet function, and does not have coagulopathy. 6 If any one of these criteria is not met, the risk of bleeding can be higher, even with only prophylactic doses of anticoagulant drugs. Prophylactic anticoagulation should be continued until hospital and rehabilitation discharge, typically 1 to 2 weeks after ICH, depending on the severity of the patient’s neurologic impairment.
If a patient with warfarin-related ICH has concomitant acute proximal deep vein thrombosis or pulmonary embolism (ie, < 4 weeks old), then caval interruption therapy would be indicated.7 Although retrievable inferior vena cava filters are increasingly preferred over permanent filters, it is important to recognize the relative lack of both longitudinal and prospective data on retrievable devices. Given that provoked venous thromboembolism requires a minimum of 3 months of anticoagulation, and retrievable filters generally need to be removed before 3 months, a retrievable filter should be chosen only if the clinician has already decided that oral anticoagulation will be restarted in the next 3 to 4 weeks after filter removal.
WHAT TO DO IN THE CHRONIC PHASE
A more difficult question in patients with warfarin-related ICH arises in the chronic phase: should oral anticoagulation be resumed at all?
Goldstein and Greenberg outline important considerations. Under the principle of primum non nocere, patients who have suffered a warfarin-related ICH should first be evaluated for their risk of thrombosis in light of their original indication for oral anticoagulant therapy. As the authors point out, oral anticoagulation for primary prevention of thrombosis after warfarin-related ICH must be viewed differently than oral anticoagulation for secondary prevention of thrombosis. In addition, Douketis et al8 have described a method of stratifying a patient’s risk of thrombosis as low, moderate, or high (Table 1), which is the basis for decisions about perioperative anticoagulation. Based on Goldstein and Greenberg’s review, we can similarly categorize these patients as being at low, moderate, or high risk of ICH recurrence (Table 2). Patients at low risk of thrombosis should probably not resume taking a vitamin K antagonist, regardless of their ICH risk (Table 3). It would be reasonable, however, for patients at moderate or high risk of thrombosis and at low risk of ICH to resume taking their vitamin K antagonist.
Uncertainty remains for patients with a moderate or high risk of thrombosis and a moderate or high risk of ICH. For patients with these combinations of risk, individualized approaches need to be explored. All attempts should be made to widen the margin of safety of vitamin K antagonist therapy; these include referring the patient to an anticoagulation management service, frequent laboratory monitoring, and ongoing patient education.1
Since the risk of ICH is related to the intensity of anticoagulation, a lower target international normalized ratio may be the best compromise, depending on the patient. Alternatively, antiplatelet therapy alone may offer some benefit with less risk of ICH.
THE NEWER ORAL ANTICOAGULANTS
As Goldstein and Greenberg mention, the ongoing development of new and potentially safer oral anticoagulants may affect how we approach these risk-benefit equations.
Three new oral anticoagulants—dabigatran (Pradaxa), apixaban, and rivaroxaban (Xarelto)—are being tested for various anticoagulant indications, and several phase III studies have recently closed or are nearing completion.
Dabigatran is an oral direct thrombin inhibitor currently available in Europe and Canada.
In the Randomized Evaluation of Long-term Anticoagulant Therapy (RE-LY) trial, the efficacy and safety of two different doses of dabigatran (110 mg twice daily or 150 mg twice daily) relative to warfarin were studied in more than 18,000 patients with atrial fibrillation. 9 The primary outcome measure was stroke or systemic embolism. Dabigatran 110 mg was not inferior to warfarin in terms of the primary outcome, while dabigatran 150 mg was superior. The rate of major bleeding was 3.36% per year in the warfarin group vs 2.71% in the 110-mg group (P = .003) and 3.11% in the 150-mg group (P not significant).
Additional safety data on this drug are available from the 2,500-patient RE-COVER trial.10 Dabigatran was not inferior to warfarin in the treatment of acute venous thromboembolism, with a similar rate of major bleeding and a lower rate of combined major plus nonmajor bleeding.
Apixaban, an oral direct factor Xa inhibitor, is in a phase III trial in patients with atrial fibrillation—Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE)11—comparing apixaban vs warfarin. Another phase III trial, AVERROES,12 was stopped early after a predefined interim analysis by the independent data-monitoring committee found clear evidence of benefit in the apixaban group.13 The AVERROES results were presented at the 2010 European Society of Cardiology Congress, August 28–September 1, Stockholm, Sweden.14
Rivaroxaban, another promising oral direct factor Xa inhibitor, is currently available in Europe and Canada for the prevention of thrombosis in orthopedic surgery patients. Rivaroxaban is also in large phase III trials for the treatment of acute venous thromboembolism15–17 and for the prevention of stroke in atrial fibrillation.18
Newer agents have drawbacks, too
These new agents need no laboratory monitoring, and they do not appear to be subject to the dose variability and the interactions with drugs and foods seen with vitamin K antagonists. As a result, they may pose less risk of anticoagulant-related ICH.
The decision to resume anticoagulation after anticoagulant-associated intracranial hemorrhage should be based on the risk of rebleeding vs the risk of thrombosis. Patients determined to be at high risk of thrombosis and low risk of rebleeding are the best candidates for resuming anticoagulation.
Still, for patients who suffer an anticoagulant- or warfarin-related ICH, these new anticoagulants are not likely to simplify the issue of restarting anticoagulant therapy. Unlike vitamin K antagonists, dabigatran and the direct factor Xa inhibitors have no known antidote for their anticoagulant effects. Animal data suggest that factor Xa concentrates may help,19 but for patients at risk of a second anticoagulant-related ICH, this does not provide much reassurance.
As with all clinical decisions in medicine, the potential benefits of any therapy should outweigh the risks. In the case of warfarin-related ICH, resuming anticoagulant therapy requires careful consideration of many factors, including patient preferences and tolerance of different levels of risk. As new and perhaps safer anticoagulants become available, clinicians may face such difficult questions less and less. But in the meantime, doctors and their patients are left to pick their poison.
References
Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G; American College of Chest Physicians. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):160S–198S.
Hirsh J, Bauer KA, Donati MB, Gould M, Samama MM, Weitz JI; American College of Chest Physicians. Parenteral anticoagulants: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):141S–159S.
Schulman S, Beyth RJ, Kearon C, Levine MN; American College of Chest Physicians. Hemorrhagic complications of anticoagulant and thrombolytic treatment: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th edition). Chest2008; 133(suppl 6):257S–298S.
Goldstein JN, Greenberg SM. Should anticoagulation be resumed after intracerebral hemorrhage?Cleve Clin J Med2010; 77:791–799.
Geerts WH, Bergqvist D, Pineo GF, et al; American College of Chest Physicians. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):381S–453S.
Michota F, Merli G. Anticoagulation in special patient populations: are special dosing considerations required?Cleve Clin J Med2005; 72(suppl 1):S37–S42.
Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ; American College of Chest Physicians. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):454S–545S.
Douketis JD, Berger PB, Dunn AS, et al; American College of Chest Physicians. The perioperative management of antithrombotic therapy: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):299S–339S.
Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med2009; 361:1139–1151.
Schulman S, Kearon C, Kakkar AK, et al; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med2009; 361:2342–2352.
Lopes RD, Alexander JH, Al-Khatib SM; ARISTOTLE Investigators. Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial: design and rationale. Am Heart J2010; 159:331–339.
Eikelboom JW, O’Donnell M, Yusuf S, et al. Rationale and design of AVERROES: apixaban versus acetylsalicylic acid to prevent stroke in atrial fibrillation patients who have failed or are unsuitable for vitamin K antagonist treatment. Am Heart J2010; 159:348–353.
Pfizer/Bristol-Myers Squibb. AVERROES study of investigational agent apixaban closes early due to clear evidence of efficacy, June 9, 2010. www.theheart.org/article/1087291.do. Accessed September 26, 2010.
Once-daily oral direct factor Xa inhibitor rivaroxaban in the long-term prevention of recurrent symptomatic venous thromboembolism in patients with symptomatic deep-vein thrombosis or pulmonary embolism. The Einstein-Extension Study. http://clinicaltrials.gov/ct2/show/NCT00439725. Accessed September 26, 2010.
Oral direct factor Xa inhibitor rivaroxaban in patients with acute symptomatic deep-vein thrombosis without symptomatic pulmonary embolism: Einstein-DVT Evaluation. http://clinicaltrials.gov/ct2/show/NCT00440193. Accessed September 26, 2010.
Oral direct factor Xa inhibitor rivaroxaban in patients with acute symptomatic pulmonary embolism with or without symptomatic deep-vein thrombosis: Einstein-PE Evaluation. http://clinicaltrials.gov/ct2/show/NCT00439777. Accessed September 26, 2010.
ROCKET AF Study Investigators. Rivaroxaban once-daily, oral, direct factor Xa inhibition compared with vitamin K antagonism for prevention of stroke and embolism trial in atrial fibrillation: rationale and design of the ROCKET AF study. Am Heart J2010; 159:340–347.
Weitz JI, Hirsh J, Samama MM; American College of Chest Physicians. New antithrombotic drugs: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):234S–256S.
References
Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G; American College of Chest Physicians. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):160S–198S.
Hirsh J, Bauer KA, Donati MB, Gould M, Samama MM, Weitz JI; American College of Chest Physicians. Parenteral anticoagulants: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):141S–159S.
Schulman S, Beyth RJ, Kearon C, Levine MN; American College of Chest Physicians. Hemorrhagic complications of anticoagulant and thrombolytic treatment: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th edition). Chest2008; 133(suppl 6):257S–298S.
Goldstein JN, Greenberg SM. Should anticoagulation be resumed after intracerebral hemorrhage?Cleve Clin J Med2010; 77:791–799.
Geerts WH, Bergqvist D, Pineo GF, et al; American College of Chest Physicians. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):381S–453S.
Michota F, Merli G. Anticoagulation in special patient populations: are special dosing considerations required?Cleve Clin J Med2005; 72(suppl 1):S37–S42.
Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ; American College of Chest Physicians. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):454S–545S.
Douketis JD, Berger PB, Dunn AS, et al; American College of Chest Physicians. The perioperative management of antithrombotic therapy: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):299S–339S.
Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med2009; 361:1139–1151.
Schulman S, Kearon C, Kakkar AK, et al; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med2009; 361:2342–2352.
Lopes RD, Alexander JH, Al-Khatib SM; ARISTOTLE Investigators. Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial: design and rationale. Am Heart J2010; 159:331–339.
Eikelboom JW, O’Donnell M, Yusuf S, et al. Rationale and design of AVERROES: apixaban versus acetylsalicylic acid to prevent stroke in atrial fibrillation patients who have failed or are unsuitable for vitamin K antagonist treatment. Am Heart J2010; 159:348–353.
Pfizer/Bristol-Myers Squibb. AVERROES study of investigational agent apixaban closes early due to clear evidence of efficacy, June 9, 2010. www.theheart.org/article/1087291.do. Accessed September 26, 2010.
Once-daily oral direct factor Xa inhibitor rivaroxaban in the long-term prevention of recurrent symptomatic venous thromboembolism in patients with symptomatic deep-vein thrombosis or pulmonary embolism. The Einstein-Extension Study. http://clinicaltrials.gov/ct2/show/NCT00439725. Accessed September 26, 2010.
Oral direct factor Xa inhibitor rivaroxaban in patients with acute symptomatic deep-vein thrombosis without symptomatic pulmonary embolism: Einstein-DVT Evaluation. http://clinicaltrials.gov/ct2/show/NCT00440193. Accessed September 26, 2010.
Oral direct factor Xa inhibitor rivaroxaban in patients with acute symptomatic pulmonary embolism with or without symptomatic deep-vein thrombosis: Einstein-PE Evaluation. http://clinicaltrials.gov/ct2/show/NCT00439777. Accessed September 26, 2010.
ROCKET AF Study Investigators. Rivaroxaban once-daily, oral, direct factor Xa inhibition compared with vitamin K antagonism for prevention of stroke and embolism trial in atrial fibrillation: rationale and design of the ROCKET AF study. Am Heart J2010; 159:340–347.
Weitz JI, Hirsh J, Samama MM; American College of Chest Physicians. New antithrombotic drugs: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):234S–256S.
If a patient taking warfarin (Coumadin) or other anticoagulant drug suffers an intracerebral hemorrhage (ICH) and survives, the physician faces the dilemma of whether to resume the anticoagulant. On one hand, the drug was prescribed because the patient was at risk of a thromboembolic event such as stroke or pulmonary embolism. On the other hand, warfarin use may increase the risk of another ICH.
Unfortunately, we have little evidence from clinical trials on which to base the decision. Nevertheless, we believe that in selected patients the potential benefit of resuming anticoagulation outweighs the considerable risk.
In the pages that follow, we summarize when and how anticoagulation therapy should be resumed after ICH.
A DEADLY COMPLICATION OF ANTICOAGULANT THERAPY
Intracranial bleeding is the most feared and the most deadly complication of oral anticoagulant therapy.1 The substantial risks associated with oral anticoagulants likely account for these drugs being underprescribed in patients who have indications for them.2–4
While bleeding is the major risk, not all bleeding events are equally damaging. Extracranial bleeding (eg, gastrointestinal bleeding, hematuria, epistaxis) leads to death or disability in only 3% of cases, whereas intracranial bleeding such as ICH leads to death or disability 76% of cases.5
Even without anticoagulation, ICH is the deadliest form of stroke,6–9 and if the patient has been taking warfarin, the risk of disability and death is substantially higher.6,10 Warfarin has a striking effect on the incidence and outcomes of ICH. While the overall incidence of ICH in the general population is approximately 25 per 100,000 person-years, the incidence in patients on warfarin is exponentially higher, at 2 to 3 per 100 per year, and appears to be increasing.11,12 In addition, once ICH occurs, the risk of death is up to twice as high in those on warfarin.6 The bulk of this effect is likely due to a higher risk of ongoing bleeding after the event.10,13–16
Major risk factors for ICH in patients taking oral anticoagulants include a higher international normalized ratio (INR) and older age.11,17
TWO KEY QUESTIONS
Once a patient is diagnosed with warfarin-related ICH, clinicians typically take urgent measures to restore normal coagulation, hoping to limit ongoing bleeding and improve outcome.18,19
The higher the INR at presentation, the greater the risk of death.6 In addition, in retrospective studies, some authors have noted that earlier correction of the INR is associated with better outcome.14,16
While emergency reversal of warfarin is widely considered standard treatment in the acute phase,20–24 concern persists about its safety in patients at high risk of thromboembolism.
Until the results of clinical trials are available, decisions about whether to reverse and when to resume anticoagulation hinge on two questions:
In the acute phase, how does the risk of further bleeding (hematoma expansion) compare with the short-term risk of thromboembolism?
In the chronic phase, how does the risk of recurrent hemorrhage compare with the excess risk of thromboembolism if the patient does not resume anticoagulation therapy?
ACUTELY, THE RISK OF BLEEDING OUTWEIGHS THAT OF CLOTTING
High risk of hematoma expansion after ICH
Unfortunately, continued bleeding is common after ICH. In patients who present within 3 hours of symptom onset, 26% of hematomas expand more than 33% over the first hour, and another 12% expand this amount over the next 20 hours.6 In warfarin-associated ICH, up to 50% of patients develop this level of hematoma expansion, but it appears to take place over a more prolonged period of time.10,13–16 Over 70% of patients presenting acutely develop at least some amount of expansion within 24 hours.25 Therefore, the risk of hematoma expansion in the first 24 hours is likely so high that patients cannot safely receive anticoagulants during this time frame.
But not all patients are at equal risk of hematoma expansion. Several features are associated with higher risk (Table 1)10,26–40:
A large hematoma volume on presentation is a significant predictor of expansion, possibly reflecting a more severe underlying insult.26,27
Early presentation, especially within 3 hours of symptom onset, also appears to mark those at higher risk, presumably because such patients undergo computed tomography (CT) while still bleeding.26,27
Figure 1. The “spot sign” (arrow), contrast extravasation after contrast-enhanced computed tomography, is associated with a high risk of hematoma expansion.
For those on warfarin, a higher INR is a significant predictor, not just of higher risk, but also of a more delayed expansion.10,28
Certain radiographic findings indicate higher risk. One is the “spot sign,” ie, contrast extravasation after contrast-enhanced CT27,29–31 (Figure 1). Apparently, the more spots present, and the denser the contrast, the greater the risk, an observation that has led to a proposed “spot-sign score” that may predict both expansion and poor outcome. 32,41
Given the high risk of hematoma expansion in the early phase, and given our inability to predict hematoma expansion, most authorities recommend immediate reversal of anticoagulation after diagnosis.42–44 Reversal of anticoagulation typically includes intravenous vitamin K, which begins to act within several hours, and repletion of coagulation factors, which act within minutes (prothrombin complex concentrates and recombinant factor VIIa [NovoSeven]) or a few hours (fresh frozen plasma).1
Dosages:
Vitamin K 5 to 10 mg intravenously
Prothrombin complex concentrates 10 to 50 U/kg
Recombinant factor VIIa 40 to 80 μg/kg
Fresh frozen plasma 10 to 50 U/kg.
Risk of thromboembolism after ICH: Ongoing and cumulative
Thromboembolism after ICH is a major concern, for two main reasons.
First, patients on oral anticoagulation typically have a preexisting risk factor and are thus at higher risk of a thromboembolic event, particularly while they are off anticoagulation. Patients with atrial fibrillation or a mechanical valve are at risk of arterial events such as ischemic stroke, whereas patients with a known venous thromboembolic condition such as deep venous thrombosis or pulmonary embolism are at risk of extension of the thrombosis or recurrence of a venous thrombotic event.
Second, ICH itself increases the risk of arterial and venous thromboembolic events. Including patients not previously on anticoagulation, this risk is as high as 7% during the initial hospitalization and 9% during the first 90 days.45,46 Worth noting is that patients who previously received anticoagulant drugs (and who are off this therapy in the acute phase) are at no higher risk of thromboembolism compared with those who never received anticoagulants.45
However, while the risk of hematoma expansion is highest at presentation and then decreases with time, the risk of thromboembolism (particularly venous thromboembolism) is ongoing and cumulative. Arterial thromboembolism is more likely to occur early, within the first week, whereas venous thromboembolism can occur later.45
Overall, studies have estimated the short-term risk of pulmonary embolism to be 1% to 2%, deep venous thrombosis 1% to 4%, myocardial ischemia about 2%, and cerebral ischemia 2% to 3%.45,46 However, when patients are actively screened, the incidence of asymptomatic deep venous thrombosis is found to be as high as 16% in the first 10 days,47 and evidence of myocardial ischemia can be detected in up to 27% of patients.48
Therefore, the risk of hematoma expansion appears to be high and the risk of thromboembolism appears to be low during the first day after ICH. Over the next days, as the risk of hematoma expansion recedes, this ratio shifts.
Studies of in-hospital anticoagulation after ICH
The data on restarting oral anticoagulation in the acute phase are sparse. In practice, clinicians typically start heparinoids in low subcutaneous doses to prevent deep venous thrombosis and, after the first few days or a week, consider increasing to a full anticoagulation dose or starting an oral anticoagulant and subsequently discontinuing the heparin when the INR is in the therapeutic range (see discussion below).
ICH patients in general may benefit from starting prophylactic-dose heparin therapy early. One randomized trial found that starting heparin in a low subcutaneous dose the day after an ICH decreased the risk of thromboembolism without increasing the risk of rebleeding.49 Another study also found no increased risk of rebleeding with early prophylactic-dose subcutaneous heparin.50
As the benefit appears to outweigh the risk, national guidelines suggest starting subcutaneous heparin early in all ICH patients, including those not previously on warfarin.42,43
Commonly used heparinoid regimens include unfractionated heparin 5,000 units subcutaneously twice a day; enoxaparin (Lovenox) 40 mg once a day; and dalteparin (Fragmin) 5,000 units once a day.51 In addition, all patients should receive optimal mechanical thromboprophylaxis, including graduated compression stockings or intermittent pneumatic compression stockings, or both.
LONG-TERM MANAGEMENT: ICH RECURRENCE VS THROMBOEMBOLISM
Risk of ICH recurrence on warfarin is not precisely known
Overall, the risk of ICH recurrence is about 1% at 3 months, and warfarin likely increases this risk.42,52 Unfortunately, the risk of ICH recurrence in patients on anticoagulation therapy after a first ICH is not clear, and no population-based study has clarified this risk. Therefore, the best we can do at present is to try to estimate the risk of recurrent warfarin-related ICH by separately examining two issues:
The risk of ICH recurrence in general
The risk of major bleeding (including ICH) in the general population of patients on warfarin.
The risk of ICH recurrence in general is about 2% to 4% per patient-year.52–54 However, this risk appears to be a function of the underlying vasculopathy. ICH location is often used as a surrogate for underlying cause. Most ICHs in deep hemispheric (basal ganglia, thalamus) or brainstem territories are likely caused by hypertensive vasculopathy, whereas lobar ICH is often associated with cerebral amyloid angiopathy.52–54 Presumably because of this distinction, ICH in a deep location recurs in about 2% of cases per year, compared with 4% for lobar ICH.53 The presence and number of microbleeds on T2-weighted gradient-echo magnetic resonance imaging appear to predict ICH recurrence; microbleeds likely are markers of more severe or widespread underlying vasculopathy.55–57
A genetic risk factor for the recurrence of lobar ICH is apolipoprotein E genotype58; future studies may highlight genetic variations that specifically modify the risk of warfarin-related ICH.59 Unfortunately, there is currently no way to modify the risk of ICH associated with cerebral amyloid angiopathy. On the other hand, in patients with hypertensive hemorrhage, antihypertensive therapy likely reduces the risk of recurrent ICH. One randomized controlled trial showed that such therapy decreased the risk of ICH by more than half.60
The risk of major bleeding in the general population of patients on warfarin may be 2% to 3% per year and is likely higher in the first month.11 The risk is higher in older patients and if the INR rises above 4.0.11,17 For some patients, it is possible to estimate the likelihood of major bleeding using validated decision-support tools that include factors such as age, sex, and medical history.11,61–64
Given the lack of data specifically addressing the risk of ICH recurrence on warfarin, the clinician is left to try to extrapolate this risk from available data using specific patient characteristics that modify the presumed risk. For example, one can combine factors such as ICH location (or better yet, the underlying cause) with decision-support tools that predict the risk of major bleeding. Close control of both blood pressure and the INR appears especially critical for patients receiving anticoagulation after ICH.11,60,65 Still, the risk does not disappear with good INR control, and most patients with anticoagulation-related ICH present with INRs within the therapeutic range.5,10,65
Long-term risk of thromboembolism depends on underlying condition
In the long term, the risk of thromboembolism depends on the reason for which the patient was originally given anticoagulation. In addition, many patients with ICH suffer decreased mobility and are therefore at higher risk of venous thromboembolism than before their event.
Nonvalvular atrial fibrillation is the most common indication for anticoagulation. For these patients, the risk of ischemic stroke is 2% to 5% per year.66,67 The system usually used to stratify this risk is CHADS2, an acronym for five key risk factors:
Congestive heart failure (1 point)
Hypertension (1 point)
Age over 75 (1 point)
Diabetes mellitus (1 point)
Prior stroke or transient ischemic attack (2 points).
The annual risk of stroke ranges from 1.9% (score of 0) to 18.2% (score of 6).68,69 In patients with nonvalvular atrial fibrillation, the excess risk of ischemic stroke without anticoagulation must be weighed against the risk of ICH recurrence.
A mechanical heart valve, another common indication, carries a risk of ischemic stroke of about 4% per year.70 A mechanical valve is traditionally considered an absolute indication for anticoagulation. However, patients with lobar ICH face a risk of recurrence that is greater than 4% per year, and so the risks of resuming anticoagulation may well outweigh the benefits.
Heart failure may be associated with a risk of ischemic stroke of 1% to 4% per year, and this is likely a function of disease severity.71
Venous thromboembolism. The risk of recurrent venous thromboembolism in patients with deep venous thrombosis or pulmonary embolism is around 4% per year.72 Given that ICH itself confers a 2% to 3% risk of these conditions, the rate of recurrence of deep venous thrombosis may well be much higher in those ICH patients who have also already had deep venous thrombosis.
Data on resuming oral anticoagulation after ICH
Several studies have examined the outcomes when oral anticoagulants were resumed after ICH (Table 2),73–76 but experts differ on when these drugs should be resumed (eg, between 1 and 10 days after onset), or even whether they should be resumed at all.19
Notably, an analysis of 52 patients found a high risk of ICH recurrence (and gastrointestinal bleeding) in patients who restarted warfarin, and a high risk of myocardial infarction and ischemic stroke in those who did not restart, with neither strategy demonstrating a clear benefit in the rate of death or disability.73 All patients with a thromboembolic event were being treated for a previous event, suggesting that secondary prevention is a stronger indication for anticoagulation than primary prevention in this population.73
IF AND WHEN TO RESTART
Two major questions to consider are whether the benefits of restarting anticoagulation outweigh the risk, and if so, when and how should anticoagulation be restarted?
Whether to restart anticoagulation
As for the risk-benefit ratio, many think that anticoagulation should be restarted only with extreme caution and possibly only in those with deep ICH or a documented history of thromboembolism.19
In one decision analysis examining whether to restart anticoagulation after ICH in patients with atrial fibrillation, the risk of thromboembolism would need to exceed 7% per year to justify restarting anticoagulation after deep ICH,67 and no risk level was high enough to justify restarting anticoagulation after lobar ICH.
For patients at sufficiently high risk of ICH recurrence, antiplatelet treatment is probably safer, as antiplatelet agents carry a substantially lower risk of bleeding.54,77–79 The American Heart Association comments that for nonvalvular atrial fibrillation, long-term anticoagulation should be avoided after spontaneous lobar ICH, but that antiplatelet agents may be considered. 42 They note that anticoagulation after nonlobar ICH may be considered depending on the indication.42
The decision to restart anticoagulation may also be a function of whether the underlying risk factor is a temporary one. For example, atrial fibrillation or a mechanical heart valve confers a long-term, ongoing risk of arterial thromboembolism, and such patients would not normally be considered for a short course of warfarin therapy. However, isolated deep vein thrombosis may only require anticoagulation for a limited time, such as 3 to 6 months.80 Perhaps for such patients the long-term outcome is maximized with a narrowly defined, temporary course of anticoagulation.
When to restart anticoagulation
As for when to restart, it is not certain how long after symptom onset the risk of ongoing bleeding continues. Clearly, the risk is high on the first day, but small after the first few days.
The European Stroke Initiative recommends that patients with a strong indication for anticoagulation, such as a history of embolic stroke with atrial fibrillation, should be restarted on warfarin after 10 to 14 days, depending on the risk of thromboembolism and ICH recurrence.43
The American Heart Association suggests that, in patients with a very high risk of thromboembolism for whom restarting warfarin is considered, warfarin may be restarted 7 to 10 days after ICH onset.42
The American College of Chest Physicians recommends starting prophylactic-dose heparin the day after an ICH, with no clear guidance on restarting warfarin.81
ALTERNATIVES TO WARFARIN
Alternatives to warfarin that show promise in reducing bleeding risk include factor Xa and direct thrombin inhibitors, which may reduce the risk of thromboembolism to an extent similar to that of warfarin, but with fewer bleeding complications.82
In patients with atrial fibrillation, the direct thrombin inhibitor dabigatran (Pradaxa) was shown to prevent ischemic stroke to a similar or greater degree than warfarin, with fewer bleeding complications.83 Further patient follow-up is under way to ensure that this drug does not cause liver problems, as did a similarly designed predecessor.84
The availability of this and other agents in various stages of development82 will probably not make warfarin extinct. Rather, they may change the “tipping point,” the threshold at which the risk of thromboembolism is high enough to justify the risks associated with restarting warfarin therapy. In addition, clinical decision tools clarifying the individual patient’s risk of thromboembolism vs the risk of ICH recurrence will help physicians tailor the therapy to the patient.
For the moment, in situations in which the decision is difficult, maximizing the use of antiplatelet agents offers the best hope.85
RECOMMENDATIONS IN LIEU OF GUIDELINES
No guideline can broadly cover every clinical scenario. Many factors go into assessing a patient’s risk of hematoma expansion or recurrent hemorrhage (Table 3) and the extent to which anticoagulation can reduce the risk of thromboembolism.
In the short term, most patients with ICH will likely benefit from acute reversal of anticoagulation, followed by gradual reinstitution of prophylactic-dose anticoagulation after the first 24 to 72 hours.
In the long term, many patients with lobar hemorrhage, cerebral amyloid angiopathy, or other risk factors may remain at higher risk of anticoagulant-related ICH recurrence than of fatal or disabling thromboembolic events and would therefore be best managed without anticoagulants. Conversely, those with deep hemispheric ICH, hypertension that can be well controlled, and a high risk of disabling thromboembolism may receive a net benefit from restarting anticoagulation.
Wysowski DK, Nourjah P, Swartz L. Bleeding complications with warfarin use: a prevalent adverse effect resulting in regulatory action. Arch Intern Med2007; 167:1414–1419.
Choudhry NK, Anderson GM, Laupacis A, Ross-Degnan D, Normand SL, Soumerai SB. Impact of adverse events on prescribing warfarin in patients with atrial fibrillation: matched pair analysis. BMJ2006; 332:141–145.
Choudhry NK, Soumerai SB, Normand SL, Ross-Degnan D, Laupacis A, Anderson GM. Warfarin prescribing in atrial fibrillation: the impact of physician, patient, and hospital characteristics. Am J Med2006; 119:607–615.
Fang MC, Go AS, Chang Y, et al. Death and disability from warfarin-associated intracranial and extracranial hemorrhages. Am J Med2007; 120:700–705.
Rosand J, Eckman MH, Knudsen KA, Singer DE, Greenberg SM. The effect of warfarin and intensity of anticoagulation on outcome of intracerebral hemorrhage. Arch Intern Med2004; 164:880–884.
Dixon AA, Holness RO, Howes WJ, Garner JB. Spontaneous intracerebral haemorrhage: an analysis of factors affecting prognosis. Can J Neurol Sci1985; 12:267–271.
Broderick JP, Adams HP, Barsan W, et al. Guidelines for the management of spontaneous intracerebral hemorrhage: a statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Stroke1999; 30:905–915.
Flibotte JJ, Hagan N, O’Donnell J, Greenberg SM, Rosand J. Warfarin, hematoma expansion, and outcome of intracerebral hemorrhage. Neurology2004; 63:1059–1064.
Schulman S, Beyth RJ, Kearon C, Levine MN; American College of Chest Physicians. Hemorrhagic complications of anticoagulant and thrombolytic treatment: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):257S–298S.
Flaherty ML, Kissela B, Woo D, et al. The increasing incidence of anticoagulant-associated intracerebral hemorrhage. Neurology2007; 68:116–121.
Lee SB, Manno EM, Layton KF, Wijdicks EF. Progression of warfarin-associated intracerebral hemorrhage after INR normalization with FFP. Neurology2006; 67:1272–1274.
Huttner HB, Schellinger PD, Hartmann M, et al. Hematoma growth and outcome in treated neurocritical care patients with intracerebral hemorrhage related to oral anticoagulant therapy: comparison of acute treatment strategies using vitamin K, fresh frozen plasma, and prothrombin complex concentrates. Stroke2006; 37:1465–1470.
Sjöblom L, Hårdemark HG, Lindgren A, et al. Management and prognostic features of intracerebral hemorrhage during anticoagulant therapy: a Swedish multicenter study. Stroke2001; 32:2567–2574.
Yasaka M, Minematsu K, Naritomi H, Sakata T, Yamaguchi T. Predisposing factors for enlargement of intracerebral hemorrhage in patients treated with warfarin. Thromb Haemost2003; 89:278–283.
Hylek EM, Evans-Molina C, Shea C, Henault LE, Regan S. Major hemorrhage and tolerability of warfarin in the first year of therapy among elderly patients with atrial fibrillation. Circulation2007; 115:2689–2696.
Goldstein JN, Thomas SH, Frontiero V, et al. Timing of fresh frozen plasma administration and rapid correction of coagulopathy in warfarin-related intracerebral hemorrhage. Stroke2006; 37:151–155.
Aguilar MI, Hart RG, Kase CS, et al. Treatment of warfarin-associated intracerebral hemorrhage: literature review and expert opinion. Mayo Clin Proc2007; 82:82–92.
Guidelines on oral anticoagulation: third edition. Br J Haematol1998; 101:374–387.
Baglin TP, Keeling DM, Watson HG; British Committee for Standards in Haematology. Guidelines on oral anticoagulation (warfarin): third edition—2005 update. Br J Haematol2006; 132:277–285.
O’Shaughnessy DF, Atterbury C, Bolton Maggs P, et al; British Committee for Standards in Haematology, Blood Transfusion Task Force. Guidelines for the use of fresh-frozen plasma, cryoprecipitate and cryosupernatant. Br J Haematol2004; 126:11–28.
Baker RI, Coughlin PB, Gallus AS, Harper PL, Salem HH, Wood EM; Warfarin Reversal Consensus Group. Warfarin reversal: consensus guidelines, on behalf of the Australasian Society of Thrombosis and Haemostasis. Med J Aust2004; 181:492–497.
Davis SM, Broderick J, Hennerici M, et al; Recombinant Activated Factor VII Intracerebral Hemorrhage Trial Investigators. Hematoma growth is a determinant of mortality and poor outcome after intracerebral hemorrhage. Neurology2006; 66:1175–1181.
Broderick JP, Diringer MN, Hill MD, et al; Recombinant Activated Factor VII Intracerebral Hemorrhage Trial Investigators. Determinants of intracerebral hemorrhage growth: an exploratory analysis. Stroke2007; 38:1072–1075.
Goldstein JN, Fazen LE, Snider R, et al. Contrast extravasation on CT angiography predicts hematoma expansion in intracerebral hemorrhage. Neurology2007; 68:889–894.
Kazui S, Naritomi H, Yamamoto H, Sawada T, Yamaguchi T. Enlargement of spontaneous intracerebral hemorrhage. Incidence and time course. Stroke1996; 27:1783–1787.
Kim J, Smith A, Hemphill JC, et al. Contrast extravasation on CT predicts mortality in primary intracerebral hemorrhage. AJNR Am J Neuroradiol2008; 29:520–525.
Wada R, Aviv RI, Fox AJ, et al. CT angiography “spot sign” predicts hematoma expansion in acute intracerebral hemorrhage. Stroke2007; 38:1257–1262.
Barras CD, Tress BM, Christensen S, et al; Recombinant Activated Factor VII Intracerebral Hemorrhage Trial Investigators. Density and shape as CT predictors of intracerebral hemorrhage growth. Stroke2009; 40:1325–1331.
Delgado Almandoz JE, Yoo AJ, Stone MJ, et al. Systematic characterization of the computed tomography angiography spot sign in primary intracerebral hemorrhage identifies patients at highest risk for hematoma expansion: the spot sign score. Stroke2009; 40:2994–3000.
Fujii Y, Takeuchi S, Sasaki O, Minakawa T, Tanaka R. Multivariate analysis of predictors of hematoma enlargement in spontaneous intracerebral hemorrhage. Stroke1998; 29:1160–1166.
Ederies A, Demchuk A, Chia T, et al. Postcontrast CT extravasation is associated with hematoma expansion in CTA spot negative patients. Stroke2009; 40:1672–1676.
Delgado P, Alvarez-Sabín J, Abilleira S, et al. Plasma d-dimer predicts poor outcome after acute intracerebral hemorrhage. Neurology2006; 67:94–98.
Naidech AM, Jovanovic B, Liebling S, et al. Reduced platelet activity is associated with early clot growth and worse 3-month outcome after intracerebral hemorrhage. Stroke2009; 40:2398–2401.
Silva Y, Leira R, Tejada J, Lainez JM, Castillo J, Dávalos A; Stroke Project, Cerebrovascular Diseases Group of the Spanish Neurological Society. Molecular signatures of vascular injury are associated with early growth of intracerebral hemorrhage. Stroke2005; 36:86–91.
Sansing LH, Messe SR, Cucchiara BL, Cohen SN, Lyden PD, Kasner SE; CHANT Investigators. Prior antiplatelet use does not affect hemorrhage growth or outcome after ICH. Neurology2009; 72:1397–1402.
Moussouttas M, Malhotra R, Fernandez L, et al. Role of antiplatelet agents in hematoma expansion during the acute period of intracerebral hemorrhage. Neurocrit Care2010; 12:24–29.
Delgado Almandoz JE, Yoo AJ, Stone MJ, et al. The spot sign score in primary intracerebral hemorrhage identifies patients at highest risk of in-hospital mortality and poor outcome among survivors. Stroke2010; 41:54–60.
Broderick J, Connolly S, Feldmann E, et al; American Heart Association; American Stroke Association Stroke Council; High Blood Pressure Research Council; Quality of Care and Outcomes in Research Interdisciplinary Working Group. Guidelines for the management of spontaneous intracerebral hemorrhage in adults: 2007 update: a guideline from the American Heart Association/American Stroke Association Stroke Council, High Blood Pressure Research Council, and the Quality of Care and Outcomes in Research Interdisciplinary Working Group. Stroke2007; 38:2001–2023.
Steiner T, Kaste M, Forsting M, et al. Recommendations for the management of intracranial haemorrhage - part I: spontaneous intracerebral haemorrhage. The European Stroke Initiative Writing Committee and the Writing Committee for the EUSI Executive Committee. Cerebrovasc Dis2006; 22:294–316.
Steiner T, Rosand J, Diringer M. Intracerebral hemorrhage associated with oral anticoagulant therapy: current practices and unresolved questions. Stroke2006; 37:256–262.
Goldstein JN, Fazen LE, Wendell L, et al. Risk of thromboembolism following acute intracerebral hemorrhage. Neurocrit Care2009; 10:28–34.
Christensen MC, Dawson J, Vincent C. Risk of thromboembolic complications after intracerebral hemorrhage according to ethnicity. Adv Ther2008; 25:831–841.
Lacut K, Bressollette L, Le Gal G, et al; VICTORIAh (Venous Intermittent Compression and Thrombosis Occurrence Related to Intracerebral Acute hemorrhage) Investigators. Prevention of venous thrombosis in patients with acute intracerebral hemorrhage. Neurology2005; 65:865–869.
Diringer MN, Skolnick BE, Mayer SA, et al. Thromboembolic events with recombinant activated factor VII in spontaneous intracerebral hemorrhage: results from the Factor Seven for Acute Hemorrhagic Stroke (FAST) trial. Stroke2010; 41:48–53.
Boeer A, Voth E, Henze T, Prange HW. Early heparin therapy in patients with spontaneous intracerebral haemorrhage. J Neurol Neurosurg Psychiatry1991; 54:466–467.
Dickmann U, Voth E, Schicha H, Henze T, Prange H, Emrich D. Heparin therapy, deep-vein thrombosis and pulmonary embolism after intracerebral hemorrhage. Klin Wochenschr1988; 66:1182–1183.
Geerts WH, Bergqvist D, Pineo GF, et al; American College of Chest Physicians. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):381S–453S.
Vermeer SE, Algra A, Franke CL, Koudstaal PJ, Rinkel GJ. Long-term prognosis after recovery from primary intracerebral hemorrhage. Neurology2002; 59:205–209.
Bailey RD, Hart RG, Benavente O, Pearce LA. Recurrent brain hemorrhage is more frequent than ischemic stroke after intracranial hemorrhage. Neurology2001; 56:773–777.
Viswanathan A, Rakich SM, Engel C, et al. Antiplatelet use after intracerebral hemorrhage. Neurology2006; 66:206–209.
Greenberg SM, Eng JA, Ning M, Smith EE, Rosand J. Hemorrhage burden predicts recurrent intracerebral hemorrhage after lobar hemorrhage. Stroke2004; 35:1415–1420.
Lee SH, Ryu WS, Roh JK. Cerebral microbleeds are a risk factor for warfarin-related intracerebral hemorrhage. Neurology2009; 72:171–176.
Ueno H, Naka H, Ohshita T, et al. Association between cerebral microbleeds on T2*-weighted MR images and recurrent hemorrhagic stroke in patients treated with warfarin following ischemic stroke. AJNR Am J Neuroradiol2008; 29:1483–1486.
O’Donnell HC, Rosand J, Knudsen KA, et al. Apolipoprotein E genotype and the risk of recurrent lobar intracerebral hemorrhage. N Engl J Med2000; 342:240–245.
Genes for Cerebral Hemorrhage on Anticoagulation (GOCHA) Collaborative Group. Exploiting common genetic variation to make anticoagulation safer. Stroke2009; 40(suppl 3):S64–S66.
Tzourio C, Arima H, Harrap S, et al. APOE genotype, ethnicity, and the risk of cerebral hemorrhage. Neurology2008; 70:1322–1328.
Beyth RJ, Quinn LM, Landefeld CS. Prospective evaluation of an index for predicting the risk of major bleeding in outpatients treated with warfarin. Am J Med1998; 105:91–99.
Kuijer PM, Hutten BA, Prins MH, Büller HR. Prediction of the risk of bleeding during anticoagulant treatment for venous thromboembolism. Arch Intern Med1999; 159:457–460.
Wess ML, Schauer DP, Johnston JA, et al. Application of a decision support tool for anticoagulation in patients with non-valvular atrial fibrillation. J Gen Intern Med2008; 23:411–417.
Singer DE, Chang Y, Fang MC, et al. Should patient characteristics influence target anticoagulation intensity for stroke prevention in nonvalvular atrial fibrillation? The ATRIA study. Circ Cardiovasc Qual Outcomes2009; 2:297–304.
Fang MC, Chang Y, Hylek EM, et al. Advanced age, anticoagulation intensity, and risk for intracranial hemorrhage among patients taking warfarin for atrial fibrillation. Ann Intern Med2004; 141:745–752.
Singer DE, Chang Y, Fang MC, et al. The net clinical benefit of warfarin anticoagulation in atrial fibrillation. Ann Intern Med2009; 151:297–305.
Eckman MH, Rosand J, Knudsen KA, Singer DE, Greenberg SM. Can patients be anticoagulated after intracerebral hemorrhage? A decision analysis. Stroke2003; 34:1710–1716.
Baruch L, Gage BF, Horrow J, et al. Can patients at elevated risk of stroke treated with anticoagulants be further risk stratified?Stroke2007; 38:2459–2463.
Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA2001; 285:2864–2870.
Cannegieter SC, Rosendaal FR, Briët E. Thromboembolic and bleeding complications in patients with mechanical heart valve prostheses. Circulation1994; 89:635–641.
Freudenberger RS, Hellkamp AS, Halperin JL, et al; SCD-HeFT Investigators. Risk of thromboembolism in heart failure: an analysis from the Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT). Circulation2007; 115:2637–2641.
Prandoni P, Lensing AW, Cogo A, et al. The long-term clinical course of acute deep venous thrombosis. Ann Intern Med1996; 125:1–7.
Claassen DO, Kazemi N, Zubkov AY, Wijdicks EF, Rabinstein AA. Restarting anticoagulation therapy after warfarin-associated intracerebral hemorrhage. Arch Neurol2008; 65:1313–1318.
De Vleeschouwer S, Van Calenbergh F, van Loon J, Nuttin B, Goffin J, Plets C. Risk analysis of thrombo-embolic and recurrent bleeding events in the management of intracranial haemorrhage due to oral anticoagulation. Acta Chir Belg2005; 105:268–274.
Butler AC, Tait RC. Restarting anticoagulation in prosthetic heart valve patients after intracranial haemorrhage: a 2-year follow-up. Br J Haematol1998; 103:1064–1066.
Bertram M, Bonsanto M, Hacke W, Schwab S. Managing the therapeutic dilemma: patients with spontaneous intracerebral hemorrhage and urgent need for anticoagulation. J Neurol2000; 247:209–214.
Taylor FC, Cohen H, Ebrahim S. Systematic review of long term anticoagulation or antiplatelet treatment in patients with nonrheumatic atrial fibrillation. BMJ2001; 322:321–326.
Flaherty ML, Tao H, Haverbusch M, et al. Warfarin use leads to larger intracerebral hematomas. Neurology2008; 71:1084–1089.
Sansing LH, Messe SR, Cucchiara BL, Cohen SN, Lyden PD, Kasner SE; CHANT Investigators. Prior antiplatelet use does not affect hemorrhage growth or outcome after ICH. Neurology2009; 72:1397–1402.
Snow V, Qaseem A, Barry P, et al; American College of Physicians; American Academy of Family Physicians Panel on Deep Venous Thrombosis/Pulmonary Embolism. Management of venous thromboembolism: a clinical practice guideline from the American College of Physicians and the American Academy of Family Physicians. Ann Intern Med2007; 146:204–210.
Albers GW, Amarenco P, Easton JD, Sacco RL, Teal PA; merican College of Chest Physicians. Antithrombotic and thrombolytic therapy for ischemic stroke: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):630S–639S.
Hankey GJ, Eikelboom JW. Antithrombotic drugs for patients with ischaemic stroke and transient ischaemic attack to prevent recurrent major vascular events. Lancet Neurol2010; 9:273–284.
Schulman S, Kearon C, Kakkar AK, et al; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med2009; 361:2342–2352.
Schulman S, Wåhlander K, Lundström T, Clason SB, Eriksson H; THRIVE III Investigators. Secondary prevention of venous thromboembolism with the oral direct thrombin inhibitor ximelagatran. N Engl J Med2003; 349:1713–1721.
Singer DE, Albers GW, Dalen JE, et al; American College of Chest Physicians. Antithrombotic therapy in atrial fibrillation: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):546S–592S.
Joshua N. Goldstein, MD, PhD Assistant Professor, Department of Emergency Medicine, Massachusetts General Hospital, Boston, MA
Steven M. Greenberg, MD, PhD Director, Hemorrhagic Stroke Research Program, Department of Neurology, and the Vascular Center, Massachusetts General Hospital, Boston, MA
Address: Joshua N. Goldstein, MD, PhD, Department of Emergency Medicine, Massachusetts General Hospital, Zero Emerson Place, Suite 3B, Boston, MA 02114; e-mail [email protected]
Dr. Goldstein has acted as a consultant and a member of advisory committees or review panels for CSL Behring.
Joshua N. Goldstein, MD, PhD Assistant Professor, Department of Emergency Medicine, Massachusetts General Hospital, Boston, MA
Steven M. Greenberg, MD, PhD Director, Hemorrhagic Stroke Research Program, Department of Neurology, and the Vascular Center, Massachusetts General Hospital, Boston, MA
Address: Joshua N. Goldstein, MD, PhD, Department of Emergency Medicine, Massachusetts General Hospital, Zero Emerson Place, Suite 3B, Boston, MA 02114; e-mail [email protected]
Dr. Goldstein has acted as a consultant and a member of advisory committees or review panels for CSL Behring.
Author and Disclosure Information
Joshua N. Goldstein, MD, PhD Assistant Professor, Department of Emergency Medicine, Massachusetts General Hospital, Boston, MA
Steven M. Greenberg, MD, PhD Director, Hemorrhagic Stroke Research Program, Department of Neurology, and the Vascular Center, Massachusetts General Hospital, Boston, MA
Address: Joshua N. Goldstein, MD, PhD, Department of Emergency Medicine, Massachusetts General Hospital, Zero Emerson Place, Suite 3B, Boston, MA 02114; e-mail [email protected]
Dr. Goldstein has acted as a consultant and a member of advisory committees or review panels for CSL Behring.
If a patient taking warfarin (Coumadin) or other anticoagulant drug suffers an intracerebral hemorrhage (ICH) and survives, the physician faces the dilemma of whether to resume the anticoagulant. On one hand, the drug was prescribed because the patient was at risk of a thromboembolic event such as stroke or pulmonary embolism. On the other hand, warfarin use may increase the risk of another ICH.
Unfortunately, we have little evidence from clinical trials on which to base the decision. Nevertheless, we believe that in selected patients the potential benefit of resuming anticoagulation outweighs the considerable risk.
In the pages that follow, we summarize when and how anticoagulation therapy should be resumed after ICH.
A DEADLY COMPLICATION OF ANTICOAGULANT THERAPY
Intracranial bleeding is the most feared and the most deadly complication of oral anticoagulant therapy.1 The substantial risks associated with oral anticoagulants likely account for these drugs being underprescribed in patients who have indications for them.2–4
While bleeding is the major risk, not all bleeding events are equally damaging. Extracranial bleeding (eg, gastrointestinal bleeding, hematuria, epistaxis) leads to death or disability in only 3% of cases, whereas intracranial bleeding such as ICH leads to death or disability 76% of cases.5
Even without anticoagulation, ICH is the deadliest form of stroke,6–9 and if the patient has been taking warfarin, the risk of disability and death is substantially higher.6,10 Warfarin has a striking effect on the incidence and outcomes of ICH. While the overall incidence of ICH in the general population is approximately 25 per 100,000 person-years, the incidence in patients on warfarin is exponentially higher, at 2 to 3 per 100 per year, and appears to be increasing.11,12 In addition, once ICH occurs, the risk of death is up to twice as high in those on warfarin.6 The bulk of this effect is likely due to a higher risk of ongoing bleeding after the event.10,13–16
Major risk factors for ICH in patients taking oral anticoagulants include a higher international normalized ratio (INR) and older age.11,17
TWO KEY QUESTIONS
Once a patient is diagnosed with warfarin-related ICH, clinicians typically take urgent measures to restore normal coagulation, hoping to limit ongoing bleeding and improve outcome.18,19
The higher the INR at presentation, the greater the risk of death.6 In addition, in retrospective studies, some authors have noted that earlier correction of the INR is associated with better outcome.14,16
While emergency reversal of warfarin is widely considered standard treatment in the acute phase,20–24 concern persists about its safety in patients at high risk of thromboembolism.
Until the results of clinical trials are available, decisions about whether to reverse and when to resume anticoagulation hinge on two questions:
In the acute phase, how does the risk of further bleeding (hematoma expansion) compare with the short-term risk of thromboembolism?
In the chronic phase, how does the risk of recurrent hemorrhage compare with the excess risk of thromboembolism if the patient does not resume anticoagulation therapy?
ACUTELY, THE RISK OF BLEEDING OUTWEIGHS THAT OF CLOTTING
High risk of hematoma expansion after ICH
Unfortunately, continued bleeding is common after ICH. In patients who present within 3 hours of symptom onset, 26% of hematomas expand more than 33% over the first hour, and another 12% expand this amount over the next 20 hours.6 In warfarin-associated ICH, up to 50% of patients develop this level of hematoma expansion, but it appears to take place over a more prolonged period of time.10,13–16 Over 70% of patients presenting acutely develop at least some amount of expansion within 24 hours.25 Therefore, the risk of hematoma expansion in the first 24 hours is likely so high that patients cannot safely receive anticoagulants during this time frame.
But not all patients are at equal risk of hematoma expansion. Several features are associated with higher risk (Table 1)10,26–40:
A large hematoma volume on presentation is a significant predictor of expansion, possibly reflecting a more severe underlying insult.26,27
Early presentation, especially within 3 hours of symptom onset, also appears to mark those at higher risk, presumably because such patients undergo computed tomography (CT) while still bleeding.26,27
Figure 1. The “spot sign” (arrow), contrast extravasation after contrast-enhanced computed tomography, is associated with a high risk of hematoma expansion.
For those on warfarin, a higher INR is a significant predictor, not just of higher risk, but also of a more delayed expansion.10,28
Certain radiographic findings indicate higher risk. One is the “spot sign,” ie, contrast extravasation after contrast-enhanced CT27,29–31 (Figure 1). Apparently, the more spots present, and the denser the contrast, the greater the risk, an observation that has led to a proposed “spot-sign score” that may predict both expansion and poor outcome. 32,41
Given the high risk of hematoma expansion in the early phase, and given our inability to predict hematoma expansion, most authorities recommend immediate reversal of anticoagulation after diagnosis.42–44 Reversal of anticoagulation typically includes intravenous vitamin K, which begins to act within several hours, and repletion of coagulation factors, which act within minutes (prothrombin complex concentrates and recombinant factor VIIa [NovoSeven]) or a few hours (fresh frozen plasma).1
Dosages:
Vitamin K 5 to 10 mg intravenously
Prothrombin complex concentrates 10 to 50 U/kg
Recombinant factor VIIa 40 to 80 μg/kg
Fresh frozen plasma 10 to 50 U/kg.
Risk of thromboembolism after ICH: Ongoing and cumulative
Thromboembolism after ICH is a major concern, for two main reasons.
First, patients on oral anticoagulation typically have a preexisting risk factor and are thus at higher risk of a thromboembolic event, particularly while they are off anticoagulation. Patients with atrial fibrillation or a mechanical valve are at risk of arterial events such as ischemic stroke, whereas patients with a known venous thromboembolic condition such as deep venous thrombosis or pulmonary embolism are at risk of extension of the thrombosis or recurrence of a venous thrombotic event.
Second, ICH itself increases the risk of arterial and venous thromboembolic events. Including patients not previously on anticoagulation, this risk is as high as 7% during the initial hospitalization and 9% during the first 90 days.45,46 Worth noting is that patients who previously received anticoagulant drugs (and who are off this therapy in the acute phase) are at no higher risk of thromboembolism compared with those who never received anticoagulants.45
However, while the risk of hematoma expansion is highest at presentation and then decreases with time, the risk of thromboembolism (particularly venous thromboembolism) is ongoing and cumulative. Arterial thromboembolism is more likely to occur early, within the first week, whereas venous thromboembolism can occur later.45
Overall, studies have estimated the short-term risk of pulmonary embolism to be 1% to 2%, deep venous thrombosis 1% to 4%, myocardial ischemia about 2%, and cerebral ischemia 2% to 3%.45,46 However, when patients are actively screened, the incidence of asymptomatic deep venous thrombosis is found to be as high as 16% in the first 10 days,47 and evidence of myocardial ischemia can be detected in up to 27% of patients.48
Therefore, the risk of hematoma expansion appears to be high and the risk of thromboembolism appears to be low during the first day after ICH. Over the next days, as the risk of hematoma expansion recedes, this ratio shifts.
Studies of in-hospital anticoagulation after ICH
The data on restarting oral anticoagulation in the acute phase are sparse. In practice, clinicians typically start heparinoids in low subcutaneous doses to prevent deep venous thrombosis and, after the first few days or a week, consider increasing to a full anticoagulation dose or starting an oral anticoagulant and subsequently discontinuing the heparin when the INR is in the therapeutic range (see discussion below).
ICH patients in general may benefit from starting prophylactic-dose heparin therapy early. One randomized trial found that starting heparin in a low subcutaneous dose the day after an ICH decreased the risk of thromboembolism without increasing the risk of rebleeding.49 Another study also found no increased risk of rebleeding with early prophylactic-dose subcutaneous heparin.50
As the benefit appears to outweigh the risk, national guidelines suggest starting subcutaneous heparin early in all ICH patients, including those not previously on warfarin.42,43
Commonly used heparinoid regimens include unfractionated heparin 5,000 units subcutaneously twice a day; enoxaparin (Lovenox) 40 mg once a day; and dalteparin (Fragmin) 5,000 units once a day.51 In addition, all patients should receive optimal mechanical thromboprophylaxis, including graduated compression stockings or intermittent pneumatic compression stockings, or both.
LONG-TERM MANAGEMENT: ICH RECURRENCE VS THROMBOEMBOLISM
Risk of ICH recurrence on warfarin is not precisely known
Overall, the risk of ICH recurrence is about 1% at 3 months, and warfarin likely increases this risk.42,52 Unfortunately, the risk of ICH recurrence in patients on anticoagulation therapy after a first ICH is not clear, and no population-based study has clarified this risk. Therefore, the best we can do at present is to try to estimate the risk of recurrent warfarin-related ICH by separately examining two issues:
The risk of ICH recurrence in general
The risk of major bleeding (including ICH) in the general population of patients on warfarin.
The risk of ICH recurrence in general is about 2% to 4% per patient-year.52–54 However, this risk appears to be a function of the underlying vasculopathy. ICH location is often used as a surrogate for underlying cause. Most ICHs in deep hemispheric (basal ganglia, thalamus) or brainstem territories are likely caused by hypertensive vasculopathy, whereas lobar ICH is often associated with cerebral amyloid angiopathy.52–54 Presumably because of this distinction, ICH in a deep location recurs in about 2% of cases per year, compared with 4% for lobar ICH.53 The presence and number of microbleeds on T2-weighted gradient-echo magnetic resonance imaging appear to predict ICH recurrence; microbleeds likely are markers of more severe or widespread underlying vasculopathy.55–57
A genetic risk factor for the recurrence of lobar ICH is apolipoprotein E genotype58; future studies may highlight genetic variations that specifically modify the risk of warfarin-related ICH.59 Unfortunately, there is currently no way to modify the risk of ICH associated with cerebral amyloid angiopathy. On the other hand, in patients with hypertensive hemorrhage, antihypertensive therapy likely reduces the risk of recurrent ICH. One randomized controlled trial showed that such therapy decreased the risk of ICH by more than half.60
The risk of major bleeding in the general population of patients on warfarin may be 2% to 3% per year and is likely higher in the first month.11 The risk is higher in older patients and if the INR rises above 4.0.11,17 For some patients, it is possible to estimate the likelihood of major bleeding using validated decision-support tools that include factors such as age, sex, and medical history.11,61–64
Given the lack of data specifically addressing the risk of ICH recurrence on warfarin, the clinician is left to try to extrapolate this risk from available data using specific patient characteristics that modify the presumed risk. For example, one can combine factors such as ICH location (or better yet, the underlying cause) with decision-support tools that predict the risk of major bleeding. Close control of both blood pressure and the INR appears especially critical for patients receiving anticoagulation after ICH.11,60,65 Still, the risk does not disappear with good INR control, and most patients with anticoagulation-related ICH present with INRs within the therapeutic range.5,10,65
Long-term risk of thromboembolism depends on underlying condition
In the long term, the risk of thromboembolism depends on the reason for which the patient was originally given anticoagulation. In addition, many patients with ICH suffer decreased mobility and are therefore at higher risk of venous thromboembolism than before their event.
Nonvalvular atrial fibrillation is the most common indication for anticoagulation. For these patients, the risk of ischemic stroke is 2% to 5% per year.66,67 The system usually used to stratify this risk is CHADS2, an acronym for five key risk factors:
Congestive heart failure (1 point)
Hypertension (1 point)
Age over 75 (1 point)
Diabetes mellitus (1 point)
Prior stroke or transient ischemic attack (2 points).
The annual risk of stroke ranges from 1.9% (score of 0) to 18.2% (score of 6).68,69 In patients with nonvalvular atrial fibrillation, the excess risk of ischemic stroke without anticoagulation must be weighed against the risk of ICH recurrence.
A mechanical heart valve, another common indication, carries a risk of ischemic stroke of about 4% per year.70 A mechanical valve is traditionally considered an absolute indication for anticoagulation. However, patients with lobar ICH face a risk of recurrence that is greater than 4% per year, and so the risks of resuming anticoagulation may well outweigh the benefits.
Heart failure may be associated with a risk of ischemic stroke of 1% to 4% per year, and this is likely a function of disease severity.71
Venous thromboembolism. The risk of recurrent venous thromboembolism in patients with deep venous thrombosis or pulmonary embolism is around 4% per year.72 Given that ICH itself confers a 2% to 3% risk of these conditions, the rate of recurrence of deep venous thrombosis may well be much higher in those ICH patients who have also already had deep venous thrombosis.
Data on resuming oral anticoagulation after ICH
Several studies have examined the outcomes when oral anticoagulants were resumed after ICH (Table 2),73–76 but experts differ on when these drugs should be resumed (eg, between 1 and 10 days after onset), or even whether they should be resumed at all.19
Notably, an analysis of 52 patients found a high risk of ICH recurrence (and gastrointestinal bleeding) in patients who restarted warfarin, and a high risk of myocardial infarction and ischemic stroke in those who did not restart, with neither strategy demonstrating a clear benefit in the rate of death or disability.73 All patients with a thromboembolic event were being treated for a previous event, suggesting that secondary prevention is a stronger indication for anticoagulation than primary prevention in this population.73
IF AND WHEN TO RESTART
Two major questions to consider are whether the benefits of restarting anticoagulation outweigh the risk, and if so, when and how should anticoagulation be restarted?
Whether to restart anticoagulation
As for the risk-benefit ratio, many think that anticoagulation should be restarted only with extreme caution and possibly only in those with deep ICH or a documented history of thromboembolism.19
In one decision analysis examining whether to restart anticoagulation after ICH in patients with atrial fibrillation, the risk of thromboembolism would need to exceed 7% per year to justify restarting anticoagulation after deep ICH,67 and no risk level was high enough to justify restarting anticoagulation after lobar ICH.
For patients at sufficiently high risk of ICH recurrence, antiplatelet treatment is probably safer, as antiplatelet agents carry a substantially lower risk of bleeding.54,77–79 The American Heart Association comments that for nonvalvular atrial fibrillation, long-term anticoagulation should be avoided after spontaneous lobar ICH, but that antiplatelet agents may be considered. 42 They note that anticoagulation after nonlobar ICH may be considered depending on the indication.42
The decision to restart anticoagulation may also be a function of whether the underlying risk factor is a temporary one. For example, atrial fibrillation or a mechanical heart valve confers a long-term, ongoing risk of arterial thromboembolism, and such patients would not normally be considered for a short course of warfarin therapy. However, isolated deep vein thrombosis may only require anticoagulation for a limited time, such as 3 to 6 months.80 Perhaps for such patients the long-term outcome is maximized with a narrowly defined, temporary course of anticoagulation.
When to restart anticoagulation
As for when to restart, it is not certain how long after symptom onset the risk of ongoing bleeding continues. Clearly, the risk is high on the first day, but small after the first few days.
The European Stroke Initiative recommends that patients with a strong indication for anticoagulation, such as a history of embolic stroke with atrial fibrillation, should be restarted on warfarin after 10 to 14 days, depending on the risk of thromboembolism and ICH recurrence.43
The American Heart Association suggests that, in patients with a very high risk of thromboembolism for whom restarting warfarin is considered, warfarin may be restarted 7 to 10 days after ICH onset.42
The American College of Chest Physicians recommends starting prophylactic-dose heparin the day after an ICH, with no clear guidance on restarting warfarin.81
ALTERNATIVES TO WARFARIN
Alternatives to warfarin that show promise in reducing bleeding risk include factor Xa and direct thrombin inhibitors, which may reduce the risk of thromboembolism to an extent similar to that of warfarin, but with fewer bleeding complications.82
In patients with atrial fibrillation, the direct thrombin inhibitor dabigatran (Pradaxa) was shown to prevent ischemic stroke to a similar or greater degree than warfarin, with fewer bleeding complications.83 Further patient follow-up is under way to ensure that this drug does not cause liver problems, as did a similarly designed predecessor.84
The availability of this and other agents in various stages of development82 will probably not make warfarin extinct. Rather, they may change the “tipping point,” the threshold at which the risk of thromboembolism is high enough to justify the risks associated with restarting warfarin therapy. In addition, clinical decision tools clarifying the individual patient’s risk of thromboembolism vs the risk of ICH recurrence will help physicians tailor the therapy to the patient.
For the moment, in situations in which the decision is difficult, maximizing the use of antiplatelet agents offers the best hope.85
RECOMMENDATIONS IN LIEU OF GUIDELINES
No guideline can broadly cover every clinical scenario. Many factors go into assessing a patient’s risk of hematoma expansion or recurrent hemorrhage (Table 3) and the extent to which anticoagulation can reduce the risk of thromboembolism.
In the short term, most patients with ICH will likely benefit from acute reversal of anticoagulation, followed by gradual reinstitution of prophylactic-dose anticoagulation after the first 24 to 72 hours.
In the long term, many patients with lobar hemorrhage, cerebral amyloid angiopathy, or other risk factors may remain at higher risk of anticoagulant-related ICH recurrence than of fatal or disabling thromboembolic events and would therefore be best managed without anticoagulants. Conversely, those with deep hemispheric ICH, hypertension that can be well controlled, and a high risk of disabling thromboembolism may receive a net benefit from restarting anticoagulation.
If a patient taking warfarin (Coumadin) or other anticoagulant drug suffers an intracerebral hemorrhage (ICH) and survives, the physician faces the dilemma of whether to resume the anticoagulant. On one hand, the drug was prescribed because the patient was at risk of a thromboembolic event such as stroke or pulmonary embolism. On the other hand, warfarin use may increase the risk of another ICH.
Unfortunately, we have little evidence from clinical trials on which to base the decision. Nevertheless, we believe that in selected patients the potential benefit of resuming anticoagulation outweighs the considerable risk.
In the pages that follow, we summarize when and how anticoagulation therapy should be resumed after ICH.
A DEADLY COMPLICATION OF ANTICOAGULANT THERAPY
Intracranial bleeding is the most feared and the most deadly complication of oral anticoagulant therapy.1 The substantial risks associated with oral anticoagulants likely account for these drugs being underprescribed in patients who have indications for them.2–4
While bleeding is the major risk, not all bleeding events are equally damaging. Extracranial bleeding (eg, gastrointestinal bleeding, hematuria, epistaxis) leads to death or disability in only 3% of cases, whereas intracranial bleeding such as ICH leads to death or disability 76% of cases.5
Even without anticoagulation, ICH is the deadliest form of stroke,6–9 and if the patient has been taking warfarin, the risk of disability and death is substantially higher.6,10 Warfarin has a striking effect on the incidence and outcomes of ICH. While the overall incidence of ICH in the general population is approximately 25 per 100,000 person-years, the incidence in patients on warfarin is exponentially higher, at 2 to 3 per 100 per year, and appears to be increasing.11,12 In addition, once ICH occurs, the risk of death is up to twice as high in those on warfarin.6 The bulk of this effect is likely due to a higher risk of ongoing bleeding after the event.10,13–16
Major risk factors for ICH in patients taking oral anticoagulants include a higher international normalized ratio (INR) and older age.11,17
TWO KEY QUESTIONS
Once a patient is diagnosed with warfarin-related ICH, clinicians typically take urgent measures to restore normal coagulation, hoping to limit ongoing bleeding and improve outcome.18,19
The higher the INR at presentation, the greater the risk of death.6 In addition, in retrospective studies, some authors have noted that earlier correction of the INR is associated with better outcome.14,16
While emergency reversal of warfarin is widely considered standard treatment in the acute phase,20–24 concern persists about its safety in patients at high risk of thromboembolism.
Until the results of clinical trials are available, decisions about whether to reverse and when to resume anticoagulation hinge on two questions:
In the acute phase, how does the risk of further bleeding (hematoma expansion) compare with the short-term risk of thromboembolism?
In the chronic phase, how does the risk of recurrent hemorrhage compare with the excess risk of thromboembolism if the patient does not resume anticoagulation therapy?
ACUTELY, THE RISK OF BLEEDING OUTWEIGHS THAT OF CLOTTING
High risk of hematoma expansion after ICH
Unfortunately, continued bleeding is common after ICH. In patients who present within 3 hours of symptom onset, 26% of hematomas expand more than 33% over the first hour, and another 12% expand this amount over the next 20 hours.6 In warfarin-associated ICH, up to 50% of patients develop this level of hematoma expansion, but it appears to take place over a more prolonged period of time.10,13–16 Over 70% of patients presenting acutely develop at least some amount of expansion within 24 hours.25 Therefore, the risk of hematoma expansion in the first 24 hours is likely so high that patients cannot safely receive anticoagulants during this time frame.
But not all patients are at equal risk of hematoma expansion. Several features are associated with higher risk (Table 1)10,26–40:
A large hematoma volume on presentation is a significant predictor of expansion, possibly reflecting a more severe underlying insult.26,27
Early presentation, especially within 3 hours of symptom onset, also appears to mark those at higher risk, presumably because such patients undergo computed tomography (CT) while still bleeding.26,27
Figure 1. The “spot sign” (arrow), contrast extravasation after contrast-enhanced computed tomography, is associated with a high risk of hematoma expansion.
For those on warfarin, a higher INR is a significant predictor, not just of higher risk, but also of a more delayed expansion.10,28
Certain radiographic findings indicate higher risk. One is the “spot sign,” ie, contrast extravasation after contrast-enhanced CT27,29–31 (Figure 1). Apparently, the more spots present, and the denser the contrast, the greater the risk, an observation that has led to a proposed “spot-sign score” that may predict both expansion and poor outcome. 32,41
Given the high risk of hematoma expansion in the early phase, and given our inability to predict hematoma expansion, most authorities recommend immediate reversal of anticoagulation after diagnosis.42–44 Reversal of anticoagulation typically includes intravenous vitamin K, which begins to act within several hours, and repletion of coagulation factors, which act within minutes (prothrombin complex concentrates and recombinant factor VIIa [NovoSeven]) or a few hours (fresh frozen plasma).1
Dosages:
Vitamin K 5 to 10 mg intravenously
Prothrombin complex concentrates 10 to 50 U/kg
Recombinant factor VIIa 40 to 80 μg/kg
Fresh frozen plasma 10 to 50 U/kg.
Risk of thromboembolism after ICH: Ongoing and cumulative
Thromboembolism after ICH is a major concern, for two main reasons.
First, patients on oral anticoagulation typically have a preexisting risk factor and are thus at higher risk of a thromboembolic event, particularly while they are off anticoagulation. Patients with atrial fibrillation or a mechanical valve are at risk of arterial events such as ischemic stroke, whereas patients with a known venous thromboembolic condition such as deep venous thrombosis or pulmonary embolism are at risk of extension of the thrombosis or recurrence of a venous thrombotic event.
Second, ICH itself increases the risk of arterial and venous thromboembolic events. Including patients not previously on anticoagulation, this risk is as high as 7% during the initial hospitalization and 9% during the first 90 days.45,46 Worth noting is that patients who previously received anticoagulant drugs (and who are off this therapy in the acute phase) are at no higher risk of thromboembolism compared with those who never received anticoagulants.45
However, while the risk of hematoma expansion is highest at presentation and then decreases with time, the risk of thromboembolism (particularly venous thromboembolism) is ongoing and cumulative. Arterial thromboembolism is more likely to occur early, within the first week, whereas venous thromboembolism can occur later.45
Overall, studies have estimated the short-term risk of pulmonary embolism to be 1% to 2%, deep venous thrombosis 1% to 4%, myocardial ischemia about 2%, and cerebral ischemia 2% to 3%.45,46 However, when patients are actively screened, the incidence of asymptomatic deep venous thrombosis is found to be as high as 16% in the first 10 days,47 and evidence of myocardial ischemia can be detected in up to 27% of patients.48
Therefore, the risk of hematoma expansion appears to be high and the risk of thromboembolism appears to be low during the first day after ICH. Over the next days, as the risk of hematoma expansion recedes, this ratio shifts.
Studies of in-hospital anticoagulation after ICH
The data on restarting oral anticoagulation in the acute phase are sparse. In practice, clinicians typically start heparinoids in low subcutaneous doses to prevent deep venous thrombosis and, after the first few days or a week, consider increasing to a full anticoagulation dose or starting an oral anticoagulant and subsequently discontinuing the heparin when the INR is in the therapeutic range (see discussion below).
ICH patients in general may benefit from starting prophylactic-dose heparin therapy early. One randomized trial found that starting heparin in a low subcutaneous dose the day after an ICH decreased the risk of thromboembolism without increasing the risk of rebleeding.49 Another study also found no increased risk of rebleeding with early prophylactic-dose subcutaneous heparin.50
As the benefit appears to outweigh the risk, national guidelines suggest starting subcutaneous heparin early in all ICH patients, including those not previously on warfarin.42,43
Commonly used heparinoid regimens include unfractionated heparin 5,000 units subcutaneously twice a day; enoxaparin (Lovenox) 40 mg once a day; and dalteparin (Fragmin) 5,000 units once a day.51 In addition, all patients should receive optimal mechanical thromboprophylaxis, including graduated compression stockings or intermittent pneumatic compression stockings, or both.
LONG-TERM MANAGEMENT: ICH RECURRENCE VS THROMBOEMBOLISM
Risk of ICH recurrence on warfarin is not precisely known
Overall, the risk of ICH recurrence is about 1% at 3 months, and warfarin likely increases this risk.42,52 Unfortunately, the risk of ICH recurrence in patients on anticoagulation therapy after a first ICH is not clear, and no population-based study has clarified this risk. Therefore, the best we can do at present is to try to estimate the risk of recurrent warfarin-related ICH by separately examining two issues:
The risk of ICH recurrence in general
The risk of major bleeding (including ICH) in the general population of patients on warfarin.
The risk of ICH recurrence in general is about 2% to 4% per patient-year.52–54 However, this risk appears to be a function of the underlying vasculopathy. ICH location is often used as a surrogate for underlying cause. Most ICHs in deep hemispheric (basal ganglia, thalamus) or brainstem territories are likely caused by hypertensive vasculopathy, whereas lobar ICH is often associated with cerebral amyloid angiopathy.52–54 Presumably because of this distinction, ICH in a deep location recurs in about 2% of cases per year, compared with 4% for lobar ICH.53 The presence and number of microbleeds on T2-weighted gradient-echo magnetic resonance imaging appear to predict ICH recurrence; microbleeds likely are markers of more severe or widespread underlying vasculopathy.55–57
A genetic risk factor for the recurrence of lobar ICH is apolipoprotein E genotype58; future studies may highlight genetic variations that specifically modify the risk of warfarin-related ICH.59 Unfortunately, there is currently no way to modify the risk of ICH associated with cerebral amyloid angiopathy. On the other hand, in patients with hypertensive hemorrhage, antihypertensive therapy likely reduces the risk of recurrent ICH. One randomized controlled trial showed that such therapy decreased the risk of ICH by more than half.60
The risk of major bleeding in the general population of patients on warfarin may be 2% to 3% per year and is likely higher in the first month.11 The risk is higher in older patients and if the INR rises above 4.0.11,17 For some patients, it is possible to estimate the likelihood of major bleeding using validated decision-support tools that include factors such as age, sex, and medical history.11,61–64
Given the lack of data specifically addressing the risk of ICH recurrence on warfarin, the clinician is left to try to extrapolate this risk from available data using specific patient characteristics that modify the presumed risk. For example, one can combine factors such as ICH location (or better yet, the underlying cause) with decision-support tools that predict the risk of major bleeding. Close control of both blood pressure and the INR appears especially critical for patients receiving anticoagulation after ICH.11,60,65 Still, the risk does not disappear with good INR control, and most patients with anticoagulation-related ICH present with INRs within the therapeutic range.5,10,65
Long-term risk of thromboembolism depends on underlying condition
In the long term, the risk of thromboembolism depends on the reason for which the patient was originally given anticoagulation. In addition, many patients with ICH suffer decreased mobility and are therefore at higher risk of venous thromboembolism than before their event.
Nonvalvular atrial fibrillation is the most common indication for anticoagulation. For these patients, the risk of ischemic stroke is 2% to 5% per year.66,67 The system usually used to stratify this risk is CHADS2, an acronym for five key risk factors:
Congestive heart failure (1 point)
Hypertension (1 point)
Age over 75 (1 point)
Diabetes mellitus (1 point)
Prior stroke or transient ischemic attack (2 points).
The annual risk of stroke ranges from 1.9% (score of 0) to 18.2% (score of 6).68,69 In patients with nonvalvular atrial fibrillation, the excess risk of ischemic stroke without anticoagulation must be weighed against the risk of ICH recurrence.
A mechanical heart valve, another common indication, carries a risk of ischemic stroke of about 4% per year.70 A mechanical valve is traditionally considered an absolute indication for anticoagulation. However, patients with lobar ICH face a risk of recurrence that is greater than 4% per year, and so the risks of resuming anticoagulation may well outweigh the benefits.
Heart failure may be associated with a risk of ischemic stroke of 1% to 4% per year, and this is likely a function of disease severity.71
Venous thromboembolism. The risk of recurrent venous thromboembolism in patients with deep venous thrombosis or pulmonary embolism is around 4% per year.72 Given that ICH itself confers a 2% to 3% risk of these conditions, the rate of recurrence of deep venous thrombosis may well be much higher in those ICH patients who have also already had deep venous thrombosis.
Data on resuming oral anticoagulation after ICH
Several studies have examined the outcomes when oral anticoagulants were resumed after ICH (Table 2),73–76 but experts differ on when these drugs should be resumed (eg, between 1 and 10 days after onset), or even whether they should be resumed at all.19
Notably, an analysis of 52 patients found a high risk of ICH recurrence (and gastrointestinal bleeding) in patients who restarted warfarin, and a high risk of myocardial infarction and ischemic stroke in those who did not restart, with neither strategy demonstrating a clear benefit in the rate of death or disability.73 All patients with a thromboembolic event were being treated for a previous event, suggesting that secondary prevention is a stronger indication for anticoagulation than primary prevention in this population.73
IF AND WHEN TO RESTART
Two major questions to consider are whether the benefits of restarting anticoagulation outweigh the risk, and if so, when and how should anticoagulation be restarted?
Whether to restart anticoagulation
As for the risk-benefit ratio, many think that anticoagulation should be restarted only with extreme caution and possibly only in those with deep ICH or a documented history of thromboembolism.19
In one decision analysis examining whether to restart anticoagulation after ICH in patients with atrial fibrillation, the risk of thromboembolism would need to exceed 7% per year to justify restarting anticoagulation after deep ICH,67 and no risk level was high enough to justify restarting anticoagulation after lobar ICH.
For patients at sufficiently high risk of ICH recurrence, antiplatelet treatment is probably safer, as antiplatelet agents carry a substantially lower risk of bleeding.54,77–79 The American Heart Association comments that for nonvalvular atrial fibrillation, long-term anticoagulation should be avoided after spontaneous lobar ICH, but that antiplatelet agents may be considered. 42 They note that anticoagulation after nonlobar ICH may be considered depending on the indication.42
The decision to restart anticoagulation may also be a function of whether the underlying risk factor is a temporary one. For example, atrial fibrillation or a mechanical heart valve confers a long-term, ongoing risk of arterial thromboembolism, and such patients would not normally be considered for a short course of warfarin therapy. However, isolated deep vein thrombosis may only require anticoagulation for a limited time, such as 3 to 6 months.80 Perhaps for such patients the long-term outcome is maximized with a narrowly defined, temporary course of anticoagulation.
When to restart anticoagulation
As for when to restart, it is not certain how long after symptom onset the risk of ongoing bleeding continues. Clearly, the risk is high on the first day, but small after the first few days.
The European Stroke Initiative recommends that patients with a strong indication for anticoagulation, such as a history of embolic stroke with atrial fibrillation, should be restarted on warfarin after 10 to 14 days, depending on the risk of thromboembolism and ICH recurrence.43
The American Heart Association suggests that, in patients with a very high risk of thromboembolism for whom restarting warfarin is considered, warfarin may be restarted 7 to 10 days after ICH onset.42
The American College of Chest Physicians recommends starting prophylactic-dose heparin the day after an ICH, with no clear guidance on restarting warfarin.81
ALTERNATIVES TO WARFARIN
Alternatives to warfarin that show promise in reducing bleeding risk include factor Xa and direct thrombin inhibitors, which may reduce the risk of thromboembolism to an extent similar to that of warfarin, but with fewer bleeding complications.82
In patients with atrial fibrillation, the direct thrombin inhibitor dabigatran (Pradaxa) was shown to prevent ischemic stroke to a similar or greater degree than warfarin, with fewer bleeding complications.83 Further patient follow-up is under way to ensure that this drug does not cause liver problems, as did a similarly designed predecessor.84
The availability of this and other agents in various stages of development82 will probably not make warfarin extinct. Rather, they may change the “tipping point,” the threshold at which the risk of thromboembolism is high enough to justify the risks associated with restarting warfarin therapy. In addition, clinical decision tools clarifying the individual patient’s risk of thromboembolism vs the risk of ICH recurrence will help physicians tailor the therapy to the patient.
For the moment, in situations in which the decision is difficult, maximizing the use of antiplatelet agents offers the best hope.85
RECOMMENDATIONS IN LIEU OF GUIDELINES
No guideline can broadly cover every clinical scenario. Many factors go into assessing a patient’s risk of hematoma expansion or recurrent hemorrhage (Table 3) and the extent to which anticoagulation can reduce the risk of thromboembolism.
In the short term, most patients with ICH will likely benefit from acute reversal of anticoagulation, followed by gradual reinstitution of prophylactic-dose anticoagulation after the first 24 to 72 hours.
In the long term, many patients with lobar hemorrhage, cerebral amyloid angiopathy, or other risk factors may remain at higher risk of anticoagulant-related ICH recurrence than of fatal or disabling thromboembolic events and would therefore be best managed without anticoagulants. Conversely, those with deep hemispheric ICH, hypertension that can be well controlled, and a high risk of disabling thromboembolism may receive a net benefit from restarting anticoagulation.
Wysowski DK, Nourjah P, Swartz L. Bleeding complications with warfarin use: a prevalent adverse effect resulting in regulatory action. Arch Intern Med2007; 167:1414–1419.
Choudhry NK, Anderson GM, Laupacis A, Ross-Degnan D, Normand SL, Soumerai SB. Impact of adverse events on prescribing warfarin in patients with atrial fibrillation: matched pair analysis. BMJ2006; 332:141–145.
Choudhry NK, Soumerai SB, Normand SL, Ross-Degnan D, Laupacis A, Anderson GM. Warfarin prescribing in atrial fibrillation: the impact of physician, patient, and hospital characteristics. Am J Med2006; 119:607–615.
Fang MC, Go AS, Chang Y, et al. Death and disability from warfarin-associated intracranial and extracranial hemorrhages. Am J Med2007; 120:700–705.
Rosand J, Eckman MH, Knudsen KA, Singer DE, Greenberg SM. The effect of warfarin and intensity of anticoagulation on outcome of intracerebral hemorrhage. Arch Intern Med2004; 164:880–884.
Dixon AA, Holness RO, Howes WJ, Garner JB. Spontaneous intracerebral haemorrhage: an analysis of factors affecting prognosis. Can J Neurol Sci1985; 12:267–271.
Broderick JP, Adams HP, Barsan W, et al. Guidelines for the management of spontaneous intracerebral hemorrhage: a statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Stroke1999; 30:905–915.
Flibotte JJ, Hagan N, O’Donnell J, Greenberg SM, Rosand J. Warfarin, hematoma expansion, and outcome of intracerebral hemorrhage. Neurology2004; 63:1059–1064.
Schulman S, Beyth RJ, Kearon C, Levine MN; American College of Chest Physicians. Hemorrhagic complications of anticoagulant and thrombolytic treatment: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):257S–298S.
Flaherty ML, Kissela B, Woo D, et al. The increasing incidence of anticoagulant-associated intracerebral hemorrhage. Neurology2007; 68:116–121.
Lee SB, Manno EM, Layton KF, Wijdicks EF. Progression of warfarin-associated intracerebral hemorrhage after INR normalization with FFP. Neurology2006; 67:1272–1274.
Huttner HB, Schellinger PD, Hartmann M, et al. Hematoma growth and outcome in treated neurocritical care patients with intracerebral hemorrhage related to oral anticoagulant therapy: comparison of acute treatment strategies using vitamin K, fresh frozen plasma, and prothrombin complex concentrates. Stroke2006; 37:1465–1470.
Sjöblom L, Hårdemark HG, Lindgren A, et al. Management and prognostic features of intracerebral hemorrhage during anticoagulant therapy: a Swedish multicenter study. Stroke2001; 32:2567–2574.
Yasaka M, Minematsu K, Naritomi H, Sakata T, Yamaguchi T. Predisposing factors for enlargement of intracerebral hemorrhage in patients treated with warfarin. Thromb Haemost2003; 89:278–283.
Hylek EM, Evans-Molina C, Shea C, Henault LE, Regan S. Major hemorrhage and tolerability of warfarin in the first year of therapy among elderly patients with atrial fibrillation. Circulation2007; 115:2689–2696.
Goldstein JN, Thomas SH, Frontiero V, et al. Timing of fresh frozen plasma administration and rapid correction of coagulopathy in warfarin-related intracerebral hemorrhage. Stroke2006; 37:151–155.
Aguilar MI, Hart RG, Kase CS, et al. Treatment of warfarin-associated intracerebral hemorrhage: literature review and expert opinion. Mayo Clin Proc2007; 82:82–92.
Guidelines on oral anticoagulation: third edition. Br J Haematol1998; 101:374–387.
Baglin TP, Keeling DM, Watson HG; British Committee for Standards in Haematology. Guidelines on oral anticoagulation (warfarin): third edition—2005 update. Br J Haematol2006; 132:277–285.
O’Shaughnessy DF, Atterbury C, Bolton Maggs P, et al; British Committee for Standards in Haematology, Blood Transfusion Task Force. Guidelines for the use of fresh-frozen plasma, cryoprecipitate and cryosupernatant. Br J Haematol2004; 126:11–28.
Baker RI, Coughlin PB, Gallus AS, Harper PL, Salem HH, Wood EM; Warfarin Reversal Consensus Group. Warfarin reversal: consensus guidelines, on behalf of the Australasian Society of Thrombosis and Haemostasis. Med J Aust2004; 181:492–497.
Davis SM, Broderick J, Hennerici M, et al; Recombinant Activated Factor VII Intracerebral Hemorrhage Trial Investigators. Hematoma growth is a determinant of mortality and poor outcome after intracerebral hemorrhage. Neurology2006; 66:1175–1181.
Broderick JP, Diringer MN, Hill MD, et al; Recombinant Activated Factor VII Intracerebral Hemorrhage Trial Investigators. Determinants of intracerebral hemorrhage growth: an exploratory analysis. Stroke2007; 38:1072–1075.
Goldstein JN, Fazen LE, Snider R, et al. Contrast extravasation on CT angiography predicts hematoma expansion in intracerebral hemorrhage. Neurology2007; 68:889–894.
Kazui S, Naritomi H, Yamamoto H, Sawada T, Yamaguchi T. Enlargement of spontaneous intracerebral hemorrhage. Incidence and time course. Stroke1996; 27:1783–1787.
Kim J, Smith A, Hemphill JC, et al. Contrast extravasation on CT predicts mortality in primary intracerebral hemorrhage. AJNR Am J Neuroradiol2008; 29:520–525.
Wada R, Aviv RI, Fox AJ, et al. CT angiography “spot sign” predicts hematoma expansion in acute intracerebral hemorrhage. Stroke2007; 38:1257–1262.
Barras CD, Tress BM, Christensen S, et al; Recombinant Activated Factor VII Intracerebral Hemorrhage Trial Investigators. Density and shape as CT predictors of intracerebral hemorrhage growth. Stroke2009; 40:1325–1331.
Delgado Almandoz JE, Yoo AJ, Stone MJ, et al. Systematic characterization of the computed tomography angiography spot sign in primary intracerebral hemorrhage identifies patients at highest risk for hematoma expansion: the spot sign score. Stroke2009; 40:2994–3000.
Fujii Y, Takeuchi S, Sasaki O, Minakawa T, Tanaka R. Multivariate analysis of predictors of hematoma enlargement in spontaneous intracerebral hemorrhage. Stroke1998; 29:1160–1166.
Ederies A, Demchuk A, Chia T, et al. Postcontrast CT extravasation is associated with hematoma expansion in CTA spot negative patients. Stroke2009; 40:1672–1676.
Delgado P, Alvarez-Sabín J, Abilleira S, et al. Plasma d-dimer predicts poor outcome after acute intracerebral hemorrhage. Neurology2006; 67:94–98.
Naidech AM, Jovanovic B, Liebling S, et al. Reduced platelet activity is associated with early clot growth and worse 3-month outcome after intracerebral hemorrhage. Stroke2009; 40:2398–2401.
Silva Y, Leira R, Tejada J, Lainez JM, Castillo J, Dávalos A; Stroke Project, Cerebrovascular Diseases Group of the Spanish Neurological Society. Molecular signatures of vascular injury are associated with early growth of intracerebral hemorrhage. Stroke2005; 36:86–91.
Sansing LH, Messe SR, Cucchiara BL, Cohen SN, Lyden PD, Kasner SE; CHANT Investigators. Prior antiplatelet use does not affect hemorrhage growth or outcome after ICH. Neurology2009; 72:1397–1402.
Moussouttas M, Malhotra R, Fernandez L, et al. Role of antiplatelet agents in hematoma expansion during the acute period of intracerebral hemorrhage. Neurocrit Care2010; 12:24–29.
Delgado Almandoz JE, Yoo AJ, Stone MJ, et al. The spot sign score in primary intracerebral hemorrhage identifies patients at highest risk of in-hospital mortality and poor outcome among survivors. Stroke2010; 41:54–60.
Broderick J, Connolly S, Feldmann E, et al; American Heart Association; American Stroke Association Stroke Council; High Blood Pressure Research Council; Quality of Care and Outcomes in Research Interdisciplinary Working Group. Guidelines for the management of spontaneous intracerebral hemorrhage in adults: 2007 update: a guideline from the American Heart Association/American Stroke Association Stroke Council, High Blood Pressure Research Council, and the Quality of Care and Outcomes in Research Interdisciplinary Working Group. Stroke2007; 38:2001–2023.
Steiner T, Kaste M, Forsting M, et al. Recommendations for the management of intracranial haemorrhage - part I: spontaneous intracerebral haemorrhage. The European Stroke Initiative Writing Committee and the Writing Committee for the EUSI Executive Committee. Cerebrovasc Dis2006; 22:294–316.
Steiner T, Rosand J, Diringer M. Intracerebral hemorrhage associated with oral anticoagulant therapy: current practices and unresolved questions. Stroke2006; 37:256–262.
Goldstein JN, Fazen LE, Wendell L, et al. Risk of thromboembolism following acute intracerebral hemorrhage. Neurocrit Care2009; 10:28–34.
Christensen MC, Dawson J, Vincent C. Risk of thromboembolic complications after intracerebral hemorrhage according to ethnicity. Adv Ther2008; 25:831–841.
Lacut K, Bressollette L, Le Gal G, et al; VICTORIAh (Venous Intermittent Compression and Thrombosis Occurrence Related to Intracerebral Acute hemorrhage) Investigators. Prevention of venous thrombosis in patients with acute intracerebral hemorrhage. Neurology2005; 65:865–869.
Diringer MN, Skolnick BE, Mayer SA, et al. Thromboembolic events with recombinant activated factor VII in spontaneous intracerebral hemorrhage: results from the Factor Seven for Acute Hemorrhagic Stroke (FAST) trial. Stroke2010; 41:48–53.
Boeer A, Voth E, Henze T, Prange HW. Early heparin therapy in patients with spontaneous intracerebral haemorrhage. J Neurol Neurosurg Psychiatry1991; 54:466–467.
Dickmann U, Voth E, Schicha H, Henze T, Prange H, Emrich D. Heparin therapy, deep-vein thrombosis and pulmonary embolism after intracerebral hemorrhage. Klin Wochenschr1988; 66:1182–1183.
Geerts WH, Bergqvist D, Pineo GF, et al; American College of Chest Physicians. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):381S–453S.
Vermeer SE, Algra A, Franke CL, Koudstaal PJ, Rinkel GJ. Long-term prognosis after recovery from primary intracerebral hemorrhage. Neurology2002; 59:205–209.
Bailey RD, Hart RG, Benavente O, Pearce LA. Recurrent brain hemorrhage is more frequent than ischemic stroke after intracranial hemorrhage. Neurology2001; 56:773–777.
Viswanathan A, Rakich SM, Engel C, et al. Antiplatelet use after intracerebral hemorrhage. Neurology2006; 66:206–209.
Greenberg SM, Eng JA, Ning M, Smith EE, Rosand J. Hemorrhage burden predicts recurrent intracerebral hemorrhage after lobar hemorrhage. Stroke2004; 35:1415–1420.
Lee SH, Ryu WS, Roh JK. Cerebral microbleeds are a risk factor for warfarin-related intracerebral hemorrhage. Neurology2009; 72:171–176.
Ueno H, Naka H, Ohshita T, et al. Association between cerebral microbleeds on T2*-weighted MR images and recurrent hemorrhagic stroke in patients treated with warfarin following ischemic stroke. AJNR Am J Neuroradiol2008; 29:1483–1486.
O’Donnell HC, Rosand J, Knudsen KA, et al. Apolipoprotein E genotype and the risk of recurrent lobar intracerebral hemorrhage. N Engl J Med2000; 342:240–245.
Genes for Cerebral Hemorrhage on Anticoagulation (GOCHA) Collaborative Group. Exploiting common genetic variation to make anticoagulation safer. Stroke2009; 40(suppl 3):S64–S66.
Tzourio C, Arima H, Harrap S, et al. APOE genotype, ethnicity, and the risk of cerebral hemorrhage. Neurology2008; 70:1322–1328.
Beyth RJ, Quinn LM, Landefeld CS. Prospective evaluation of an index for predicting the risk of major bleeding in outpatients treated with warfarin. Am J Med1998; 105:91–99.
Kuijer PM, Hutten BA, Prins MH, Büller HR. Prediction of the risk of bleeding during anticoagulant treatment for venous thromboembolism. Arch Intern Med1999; 159:457–460.
Wess ML, Schauer DP, Johnston JA, et al. Application of a decision support tool for anticoagulation in patients with non-valvular atrial fibrillation. J Gen Intern Med2008; 23:411–417.
Singer DE, Chang Y, Fang MC, et al. Should patient characteristics influence target anticoagulation intensity for stroke prevention in nonvalvular atrial fibrillation? The ATRIA study. Circ Cardiovasc Qual Outcomes2009; 2:297–304.
Fang MC, Chang Y, Hylek EM, et al. Advanced age, anticoagulation intensity, and risk for intracranial hemorrhage among patients taking warfarin for atrial fibrillation. Ann Intern Med2004; 141:745–752.
Singer DE, Chang Y, Fang MC, et al. The net clinical benefit of warfarin anticoagulation in atrial fibrillation. Ann Intern Med2009; 151:297–305.
Eckman MH, Rosand J, Knudsen KA, Singer DE, Greenberg SM. Can patients be anticoagulated after intracerebral hemorrhage? A decision analysis. Stroke2003; 34:1710–1716.
Baruch L, Gage BF, Horrow J, et al. Can patients at elevated risk of stroke treated with anticoagulants be further risk stratified?Stroke2007; 38:2459–2463.
Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA2001; 285:2864–2870.
Cannegieter SC, Rosendaal FR, Briët E. Thromboembolic and bleeding complications in patients with mechanical heart valve prostheses. Circulation1994; 89:635–641.
Freudenberger RS, Hellkamp AS, Halperin JL, et al; SCD-HeFT Investigators. Risk of thromboembolism in heart failure: an analysis from the Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT). Circulation2007; 115:2637–2641.
Prandoni P, Lensing AW, Cogo A, et al. The long-term clinical course of acute deep venous thrombosis. Ann Intern Med1996; 125:1–7.
Claassen DO, Kazemi N, Zubkov AY, Wijdicks EF, Rabinstein AA. Restarting anticoagulation therapy after warfarin-associated intracerebral hemorrhage. Arch Neurol2008; 65:1313–1318.
De Vleeschouwer S, Van Calenbergh F, van Loon J, Nuttin B, Goffin J, Plets C. Risk analysis of thrombo-embolic and recurrent bleeding events in the management of intracranial haemorrhage due to oral anticoagulation. Acta Chir Belg2005; 105:268–274.
Butler AC, Tait RC. Restarting anticoagulation in prosthetic heart valve patients after intracranial haemorrhage: a 2-year follow-up. Br J Haematol1998; 103:1064–1066.
Bertram M, Bonsanto M, Hacke W, Schwab S. Managing the therapeutic dilemma: patients with spontaneous intracerebral hemorrhage and urgent need for anticoagulation. J Neurol2000; 247:209–214.
Taylor FC, Cohen H, Ebrahim S. Systematic review of long term anticoagulation or antiplatelet treatment in patients with nonrheumatic atrial fibrillation. BMJ2001; 322:321–326.
Flaherty ML, Tao H, Haverbusch M, et al. Warfarin use leads to larger intracerebral hematomas. Neurology2008; 71:1084–1089.
Sansing LH, Messe SR, Cucchiara BL, Cohen SN, Lyden PD, Kasner SE; CHANT Investigators. Prior antiplatelet use does not affect hemorrhage growth or outcome after ICH. Neurology2009; 72:1397–1402.
Snow V, Qaseem A, Barry P, et al; American College of Physicians; American Academy of Family Physicians Panel on Deep Venous Thrombosis/Pulmonary Embolism. Management of venous thromboembolism: a clinical practice guideline from the American College of Physicians and the American Academy of Family Physicians. Ann Intern Med2007; 146:204–210.
Albers GW, Amarenco P, Easton JD, Sacco RL, Teal PA; merican College of Chest Physicians. Antithrombotic and thrombolytic therapy for ischemic stroke: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):630S–639S.
Hankey GJ, Eikelboom JW. Antithrombotic drugs for patients with ischaemic stroke and transient ischaemic attack to prevent recurrent major vascular events. Lancet Neurol2010; 9:273–284.
Schulman S, Kearon C, Kakkar AK, et al; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med2009; 361:2342–2352.
Schulman S, Wåhlander K, Lundström T, Clason SB, Eriksson H; THRIVE III Investigators. Secondary prevention of venous thromboembolism with the oral direct thrombin inhibitor ximelagatran. N Engl J Med2003; 349:1713–1721.
Singer DE, Albers GW, Dalen JE, et al; American College of Chest Physicians. Antithrombotic therapy in atrial fibrillation: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):546S–592S.
Wysowski DK, Nourjah P, Swartz L. Bleeding complications with warfarin use: a prevalent adverse effect resulting in regulatory action. Arch Intern Med2007; 167:1414–1419.
Choudhry NK, Anderson GM, Laupacis A, Ross-Degnan D, Normand SL, Soumerai SB. Impact of adverse events on prescribing warfarin in patients with atrial fibrillation: matched pair analysis. BMJ2006; 332:141–145.
Choudhry NK, Soumerai SB, Normand SL, Ross-Degnan D, Laupacis A, Anderson GM. Warfarin prescribing in atrial fibrillation: the impact of physician, patient, and hospital characteristics. Am J Med2006; 119:607–615.
Fang MC, Go AS, Chang Y, et al. Death and disability from warfarin-associated intracranial and extracranial hemorrhages. Am J Med2007; 120:700–705.
Rosand J, Eckman MH, Knudsen KA, Singer DE, Greenberg SM. The effect of warfarin and intensity of anticoagulation on outcome of intracerebral hemorrhage. Arch Intern Med2004; 164:880–884.
Dixon AA, Holness RO, Howes WJ, Garner JB. Spontaneous intracerebral haemorrhage: an analysis of factors affecting prognosis. Can J Neurol Sci1985; 12:267–271.
Broderick JP, Adams HP, Barsan W, et al. Guidelines for the management of spontaneous intracerebral hemorrhage: a statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Stroke1999; 30:905–915.
Flibotte JJ, Hagan N, O’Donnell J, Greenberg SM, Rosand J. Warfarin, hematoma expansion, and outcome of intracerebral hemorrhage. Neurology2004; 63:1059–1064.
Schulman S, Beyth RJ, Kearon C, Levine MN; American College of Chest Physicians. Hemorrhagic complications of anticoagulant and thrombolytic treatment: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):257S–298S.
Flaherty ML, Kissela B, Woo D, et al. The increasing incidence of anticoagulant-associated intracerebral hemorrhage. Neurology2007; 68:116–121.
Lee SB, Manno EM, Layton KF, Wijdicks EF. Progression of warfarin-associated intracerebral hemorrhage after INR normalization with FFP. Neurology2006; 67:1272–1274.
Huttner HB, Schellinger PD, Hartmann M, et al. Hematoma growth and outcome in treated neurocritical care patients with intracerebral hemorrhage related to oral anticoagulant therapy: comparison of acute treatment strategies using vitamin K, fresh frozen plasma, and prothrombin complex concentrates. Stroke2006; 37:1465–1470.
Sjöblom L, Hårdemark HG, Lindgren A, et al. Management and prognostic features of intracerebral hemorrhage during anticoagulant therapy: a Swedish multicenter study. Stroke2001; 32:2567–2574.
Yasaka M, Minematsu K, Naritomi H, Sakata T, Yamaguchi T. Predisposing factors for enlargement of intracerebral hemorrhage in patients treated with warfarin. Thromb Haemost2003; 89:278–283.
Hylek EM, Evans-Molina C, Shea C, Henault LE, Regan S. Major hemorrhage and tolerability of warfarin in the first year of therapy among elderly patients with atrial fibrillation. Circulation2007; 115:2689–2696.
Goldstein JN, Thomas SH, Frontiero V, et al. Timing of fresh frozen plasma administration and rapid correction of coagulopathy in warfarin-related intracerebral hemorrhage. Stroke2006; 37:151–155.
Aguilar MI, Hart RG, Kase CS, et al. Treatment of warfarin-associated intracerebral hemorrhage: literature review and expert opinion. Mayo Clin Proc2007; 82:82–92.
Guidelines on oral anticoagulation: third edition. Br J Haematol1998; 101:374–387.
Baglin TP, Keeling DM, Watson HG; British Committee for Standards in Haematology. Guidelines on oral anticoagulation (warfarin): third edition—2005 update. Br J Haematol2006; 132:277–285.
O’Shaughnessy DF, Atterbury C, Bolton Maggs P, et al; British Committee for Standards in Haematology, Blood Transfusion Task Force. Guidelines for the use of fresh-frozen plasma, cryoprecipitate and cryosupernatant. Br J Haematol2004; 126:11–28.
Baker RI, Coughlin PB, Gallus AS, Harper PL, Salem HH, Wood EM; Warfarin Reversal Consensus Group. Warfarin reversal: consensus guidelines, on behalf of the Australasian Society of Thrombosis and Haemostasis. Med J Aust2004; 181:492–497.
Davis SM, Broderick J, Hennerici M, et al; Recombinant Activated Factor VII Intracerebral Hemorrhage Trial Investigators. Hematoma growth is a determinant of mortality and poor outcome after intracerebral hemorrhage. Neurology2006; 66:1175–1181.
Broderick JP, Diringer MN, Hill MD, et al; Recombinant Activated Factor VII Intracerebral Hemorrhage Trial Investigators. Determinants of intracerebral hemorrhage growth: an exploratory analysis. Stroke2007; 38:1072–1075.
Goldstein JN, Fazen LE, Snider R, et al. Contrast extravasation on CT angiography predicts hematoma expansion in intracerebral hemorrhage. Neurology2007; 68:889–894.
Kazui S, Naritomi H, Yamamoto H, Sawada T, Yamaguchi T. Enlargement of spontaneous intracerebral hemorrhage. Incidence and time course. Stroke1996; 27:1783–1787.
Kim J, Smith A, Hemphill JC, et al. Contrast extravasation on CT predicts mortality in primary intracerebral hemorrhage. AJNR Am J Neuroradiol2008; 29:520–525.
Wada R, Aviv RI, Fox AJ, et al. CT angiography “spot sign” predicts hematoma expansion in acute intracerebral hemorrhage. Stroke2007; 38:1257–1262.
Barras CD, Tress BM, Christensen S, et al; Recombinant Activated Factor VII Intracerebral Hemorrhage Trial Investigators. Density and shape as CT predictors of intracerebral hemorrhage growth. Stroke2009; 40:1325–1331.
Delgado Almandoz JE, Yoo AJ, Stone MJ, et al. Systematic characterization of the computed tomography angiography spot sign in primary intracerebral hemorrhage identifies patients at highest risk for hematoma expansion: the spot sign score. Stroke2009; 40:2994–3000.
Fujii Y, Takeuchi S, Sasaki O, Minakawa T, Tanaka R. Multivariate analysis of predictors of hematoma enlargement in spontaneous intracerebral hemorrhage. Stroke1998; 29:1160–1166.
Ederies A, Demchuk A, Chia T, et al. Postcontrast CT extravasation is associated with hematoma expansion in CTA spot negative patients. Stroke2009; 40:1672–1676.
Delgado P, Alvarez-Sabín J, Abilleira S, et al. Plasma d-dimer predicts poor outcome after acute intracerebral hemorrhage. Neurology2006; 67:94–98.
Naidech AM, Jovanovic B, Liebling S, et al. Reduced platelet activity is associated with early clot growth and worse 3-month outcome after intracerebral hemorrhage. Stroke2009; 40:2398–2401.
Silva Y, Leira R, Tejada J, Lainez JM, Castillo J, Dávalos A; Stroke Project, Cerebrovascular Diseases Group of the Spanish Neurological Society. Molecular signatures of vascular injury are associated with early growth of intracerebral hemorrhage. Stroke2005; 36:86–91.
Sansing LH, Messe SR, Cucchiara BL, Cohen SN, Lyden PD, Kasner SE; CHANT Investigators. Prior antiplatelet use does not affect hemorrhage growth or outcome after ICH. Neurology2009; 72:1397–1402.
Moussouttas M, Malhotra R, Fernandez L, et al. Role of antiplatelet agents in hematoma expansion during the acute period of intracerebral hemorrhage. Neurocrit Care2010; 12:24–29.
Delgado Almandoz JE, Yoo AJ, Stone MJ, et al. The spot sign score in primary intracerebral hemorrhage identifies patients at highest risk of in-hospital mortality and poor outcome among survivors. Stroke2010; 41:54–60.
Broderick J, Connolly S, Feldmann E, et al; American Heart Association; American Stroke Association Stroke Council; High Blood Pressure Research Council; Quality of Care and Outcomes in Research Interdisciplinary Working Group. Guidelines for the management of spontaneous intracerebral hemorrhage in adults: 2007 update: a guideline from the American Heart Association/American Stroke Association Stroke Council, High Blood Pressure Research Council, and the Quality of Care and Outcomes in Research Interdisciplinary Working Group. Stroke2007; 38:2001–2023.
Steiner T, Kaste M, Forsting M, et al. Recommendations for the management of intracranial haemorrhage - part I: spontaneous intracerebral haemorrhage. The European Stroke Initiative Writing Committee and the Writing Committee for the EUSI Executive Committee. Cerebrovasc Dis2006; 22:294–316.
Steiner T, Rosand J, Diringer M. Intracerebral hemorrhage associated with oral anticoagulant therapy: current practices and unresolved questions. Stroke2006; 37:256–262.
Goldstein JN, Fazen LE, Wendell L, et al. Risk of thromboembolism following acute intracerebral hemorrhage. Neurocrit Care2009; 10:28–34.
Christensen MC, Dawson J, Vincent C. Risk of thromboembolic complications after intracerebral hemorrhage according to ethnicity. Adv Ther2008; 25:831–841.
Lacut K, Bressollette L, Le Gal G, et al; VICTORIAh (Venous Intermittent Compression and Thrombosis Occurrence Related to Intracerebral Acute hemorrhage) Investigators. Prevention of venous thrombosis in patients with acute intracerebral hemorrhage. Neurology2005; 65:865–869.
Diringer MN, Skolnick BE, Mayer SA, et al. Thromboembolic events with recombinant activated factor VII in spontaneous intracerebral hemorrhage: results from the Factor Seven for Acute Hemorrhagic Stroke (FAST) trial. Stroke2010; 41:48–53.
Boeer A, Voth E, Henze T, Prange HW. Early heparin therapy in patients with spontaneous intracerebral haemorrhage. J Neurol Neurosurg Psychiatry1991; 54:466–467.
Dickmann U, Voth E, Schicha H, Henze T, Prange H, Emrich D. Heparin therapy, deep-vein thrombosis and pulmonary embolism after intracerebral hemorrhage. Klin Wochenschr1988; 66:1182–1183.
Geerts WH, Bergqvist D, Pineo GF, et al; American College of Chest Physicians. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):381S–453S.
Vermeer SE, Algra A, Franke CL, Koudstaal PJ, Rinkel GJ. Long-term prognosis after recovery from primary intracerebral hemorrhage. Neurology2002; 59:205–209.
Bailey RD, Hart RG, Benavente O, Pearce LA. Recurrent brain hemorrhage is more frequent than ischemic stroke after intracranial hemorrhage. Neurology2001; 56:773–777.
Viswanathan A, Rakich SM, Engel C, et al. Antiplatelet use after intracerebral hemorrhage. Neurology2006; 66:206–209.
Greenberg SM, Eng JA, Ning M, Smith EE, Rosand J. Hemorrhage burden predicts recurrent intracerebral hemorrhage after lobar hemorrhage. Stroke2004; 35:1415–1420.
Lee SH, Ryu WS, Roh JK. Cerebral microbleeds are a risk factor for warfarin-related intracerebral hemorrhage. Neurology2009; 72:171–176.
Ueno H, Naka H, Ohshita T, et al. Association between cerebral microbleeds on T2*-weighted MR images and recurrent hemorrhagic stroke in patients treated with warfarin following ischemic stroke. AJNR Am J Neuroradiol2008; 29:1483–1486.
O’Donnell HC, Rosand J, Knudsen KA, et al. Apolipoprotein E genotype and the risk of recurrent lobar intracerebral hemorrhage. N Engl J Med2000; 342:240–245.
Genes for Cerebral Hemorrhage on Anticoagulation (GOCHA) Collaborative Group. Exploiting common genetic variation to make anticoagulation safer. Stroke2009; 40(suppl 3):S64–S66.
Tzourio C, Arima H, Harrap S, et al. APOE genotype, ethnicity, and the risk of cerebral hemorrhage. Neurology2008; 70:1322–1328.
Beyth RJ, Quinn LM, Landefeld CS. Prospective evaluation of an index for predicting the risk of major bleeding in outpatients treated with warfarin. Am J Med1998; 105:91–99.
Kuijer PM, Hutten BA, Prins MH, Büller HR. Prediction of the risk of bleeding during anticoagulant treatment for venous thromboembolism. Arch Intern Med1999; 159:457–460.
Wess ML, Schauer DP, Johnston JA, et al. Application of a decision support tool for anticoagulation in patients with non-valvular atrial fibrillation. J Gen Intern Med2008; 23:411–417.
Singer DE, Chang Y, Fang MC, et al. Should patient characteristics influence target anticoagulation intensity for stroke prevention in nonvalvular atrial fibrillation? The ATRIA study. Circ Cardiovasc Qual Outcomes2009; 2:297–304.
Fang MC, Chang Y, Hylek EM, et al. Advanced age, anticoagulation intensity, and risk for intracranial hemorrhage among patients taking warfarin for atrial fibrillation. Ann Intern Med2004; 141:745–752.
Singer DE, Chang Y, Fang MC, et al. The net clinical benefit of warfarin anticoagulation in atrial fibrillation. Ann Intern Med2009; 151:297–305.
Eckman MH, Rosand J, Knudsen KA, Singer DE, Greenberg SM. Can patients be anticoagulated after intracerebral hemorrhage? A decision analysis. Stroke2003; 34:1710–1716.
Baruch L, Gage BF, Horrow J, et al. Can patients at elevated risk of stroke treated with anticoagulants be further risk stratified?Stroke2007; 38:2459–2463.
Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA2001; 285:2864–2870.
Cannegieter SC, Rosendaal FR, Briët E. Thromboembolic and bleeding complications in patients with mechanical heart valve prostheses. Circulation1994; 89:635–641.
Freudenberger RS, Hellkamp AS, Halperin JL, et al; SCD-HeFT Investigators. Risk of thromboembolism in heart failure: an analysis from the Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT). Circulation2007; 115:2637–2641.
Prandoni P, Lensing AW, Cogo A, et al. The long-term clinical course of acute deep venous thrombosis. Ann Intern Med1996; 125:1–7.
Claassen DO, Kazemi N, Zubkov AY, Wijdicks EF, Rabinstein AA. Restarting anticoagulation therapy after warfarin-associated intracerebral hemorrhage. Arch Neurol2008; 65:1313–1318.
De Vleeschouwer S, Van Calenbergh F, van Loon J, Nuttin B, Goffin J, Plets C. Risk analysis of thrombo-embolic and recurrent bleeding events in the management of intracranial haemorrhage due to oral anticoagulation. Acta Chir Belg2005; 105:268–274.
Butler AC, Tait RC. Restarting anticoagulation in prosthetic heart valve patients after intracranial haemorrhage: a 2-year follow-up. Br J Haematol1998; 103:1064–1066.
Bertram M, Bonsanto M, Hacke W, Schwab S. Managing the therapeutic dilemma: patients with spontaneous intracerebral hemorrhage and urgent need for anticoagulation. J Neurol2000; 247:209–214.
Taylor FC, Cohen H, Ebrahim S. Systematic review of long term anticoagulation or antiplatelet treatment in patients with nonrheumatic atrial fibrillation. BMJ2001; 322:321–326.
Flaherty ML, Tao H, Haverbusch M, et al. Warfarin use leads to larger intracerebral hematomas. Neurology2008; 71:1084–1089.
Sansing LH, Messe SR, Cucchiara BL, Cohen SN, Lyden PD, Kasner SE; CHANT Investigators. Prior antiplatelet use does not affect hemorrhage growth or outcome after ICH. Neurology2009; 72:1397–1402.
Snow V, Qaseem A, Barry P, et al; American College of Physicians; American Academy of Family Physicians Panel on Deep Venous Thrombosis/Pulmonary Embolism. Management of venous thromboembolism: a clinical practice guideline from the American College of Physicians and the American Academy of Family Physicians. Ann Intern Med2007; 146:204–210.
Albers GW, Amarenco P, Easton JD, Sacco RL, Teal PA; merican College of Chest Physicians. Antithrombotic and thrombolytic therapy for ischemic stroke: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):630S–639S.
Hankey GJ, Eikelboom JW. Antithrombotic drugs for patients with ischaemic stroke and transient ischaemic attack to prevent recurrent major vascular events. Lancet Neurol2010; 9:273–284.
Schulman S, Kearon C, Kakkar AK, et al; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med2009; 361:2342–2352.
Schulman S, Wåhlander K, Lundström T, Clason SB, Eriksson H; THRIVE III Investigators. Secondary prevention of venous thromboembolism with the oral direct thrombin inhibitor ximelagatran. N Engl J Med2003; 349:1713–1721.
Singer DE, Albers GW, Dalen JE, et al; American College of Chest Physicians. Antithrombotic therapy in atrial fibrillation: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest2008; 133(suppl 6):546S–592S.
Given the high risk of hematoma expansion in the early phase of acute ICH, most experts recommend reversing anticoagulation immediately.
Many clinicians start subcutaneous heparinoids in low doses 24 to 72 hours after ICH to prevent deep vein thrombosis, and after the first few days or a week, consider either increasing the dose to a full anticoagulation dose or making a transition to oral anticoagulants.
Many patients with lobar hemorrhage or cerebral amyloid angiopathy may remain at higher risk of anticoagulant-related ICH recurrence than thromboembolic events and would therefore be best managed without anticoagulants.
Those with deep hemispheric ICH, hypertension that can be well controlled, and a high risk of disabling thromboembolism may receive net benefit from restarting anticoagulation.
Hyponatremia, defined as a serum sodium concentration below 135 mmol/L, is one of the most frequently encountered electrolyte disorders. In 1981, Flear et al1 reported that 15% of their hospitalized patients had plasma sodium concentrations lower than 134 mmol/L, the cutoff they were using at that time.
Hyponatremia is sometimes merely a laboratory artifact or a result of improper blood collection. If real, it can be due to excessive water intake or, most often, the inability of the kidney to excrete water coupled with continued water intake. Patients with significant underlying cardiac, hepatic, or renal dysfunction are at greatest risk of developing hyponatremia, secondary to the nonosmotic release of antidiuretic hormone (ADH). Others at risk include postoperative patients (especially menstruating women), older patients on thiazide diuretics, patients with malignant or psychiatric illness, and endurance athletes.
In this article, we review the treatment of acute and chronic hyponatremia, emphasizing the importance of basing the therapy on the severity of symptoms and taking care not to raise the serum sodium level too rapidly, which can cause neurologic dysfunction.
Guidelines for managing hyponatremia2 are based mostly on retrospective studies and expert opinion, since few prospective studies have been done. Despite the paucity of evidence-based recommendations, we will attempt to incorporate findings from important human and animal studies and consensus guidelines from expert panels. We will focus initially on the critical diagnostic considerations necessary to initiate treatment.
SYMPTOMATIC VS ASYMPTOMATIC
Subsequent sections will address therapeutic approaches in two clinical settings:
Symptomatic hyponatremia, ie, with severe signs or symptoms of cerebral edema—a medical emergency; and
Asymptomatic hyponatremia, ie, without serious signs or symptoms of cerebral edema.
KEY DIAGNOSTIC STEPS WHEN STARTING TREATMENT
The treatment of hyponatremia begins by confirming a truly hypo-osmolar state, assessing its clinical significance, and determining its cause (Table 1).
The clinical and laboratory evaluations form the foundation of a proper approach to any patient with hyponatremia. The rationale behind making several important diagnostic distinctions will be discussed here briefly and then expanded on in the remaining text. The reader is referred to another review on the diagnostic evaluation of hyponatremia.3
Confirm that the patient truly has hypo-osmolar hyponatremia
The serum osmolality should be measured to confirm that it is low (ie, < 275 mOsm/kg). In addition, the arterial serum sodium concentration can be measured using a blood gas device if pseudohyponatremia (see below) is suspected. This method uses direct potentiometry and bypasses the dilutional step in the processing of venous samples.4
Rationale. The clinical consequences of hyponatremia are due to water moving from hypo-osmolar extracellular fluid into the relatively hyperosmolar interior of the cell. This water movement can cause progressive cerebral edema, resulting in a spectrum of signs and symptoms from headache and ataxia to seizures and coma. But significant fluid shifts and cerebral edema occur only if the extracellular fluid is hypo-osmolar relative to the intracellular fluid.
In fact, hyponatremia can occur in several situations in which the extracellular fluid is not hypo-osmolar. An increase in effective plasma osmoles (substances in the extracellular fluid that do not readily move across the plasma membrane) can cause water to move out of cells, resulting in translocational hyponatremia. This may be seen in hyperglycemia or when mannitol or contrast dye has been given. In these situations, the plasma is either isotonic or even hypertonic to the intracellular fluid, resulting in no movement of water into the cells and therefore no clinical consequences relating to the hyponatremia. Importantly, no therapy is required for the hyponatremia.
Other situations in which hyponatremia is present but not associated with true hypotonicity include states of excess protein or lipid in the blood (pseudohyponatremia). Also, if an infusion of hypotonic fluid is running, clinicians must be sure that blood samples are not drawn proximally in the same vein.
Are there significant signs or symptoms of cerebral edema?
Hyponatremia can cause brain swelling within the confined space of the skull as water shifts from the extracellular fluid into the cells. Depending on underlying risk factors (Table 2)5 and the severity and duration of hyponatremia (see below), this may result in signs or symptoms of cerebral edema, including visual changes, focal neurologic changes, encephalopathy, respiratory depression, and seizures. Ultimately, brain herniation can occur.
Patients need to be assessed quickly because those with serious neurologic signs or symptoms thought to be related to hyponatremia require urgent treatment with hypertonic saline to increase the serum sodium concentration, regardless of the underlying volume status, the cause of hyponatremia, or the time of onset.
Determine the duration of hyponatremia
One should try to ascertain when the hyponatremia started, as its duration is important in determining the proper pace of correction.
Figure 1.
The brain begins to adapt to hyponatremia within minutes, and the cerebral adaptation is maximal within 2 to 3 days (Figure 1).6
At the onset of hyponatremia, water moves from the extracellular fluid into cells, pulled in by osmosis. The brain can decrease the net amount of water entering into the neurons (and thus regulate its volume) by increasing the flow of water from the interstitium into the cerebrospinal fluid via increased interstitial hydraulic pressure.7
Over the next several days, inorganic solutes (eg, potassium and sodium salts) and various organic solutes are transported out of the cells. In patients in whom this process has had time to occur, treatment of hyponatremia with hypertonic fluids raises the plasma osmolality faster than the cells can recapture the previously transported osmoles. In this situation, overly rapid correction can cause excessive loss of intracellular water, resulting in cell shrinkage and osmotic demyelination syndrome. Osmotic demyelination usually presents during treatment of hyponatremia after an initial improvement in mental status, with worsening neurologic function and various neurologic signs, including paresis and ultimately even death.6
In patients with acute-onset hyponatremia (ie, with onset within the past 48 hours), in whom the above cerebral adaptations have not had time to occur completely, rapid correction is unlikely to result in osmotic demyelination.
In view of the serious risk of osmotic demyelination, if the timing of development of hyponatremia cannot be determined, one should assume it is chronic (> 48 hours) and avoid rapid overcorrection (see discussion below on the rate of correction).
On the other hand, patients who have severe neurologic signs or symptoms initially need their serum sodium increased urgently to safer levels, regardless of the timing of onset (see below for suggested approach). Subsequent treatment of hyponatremia—after the serum sodium level has been raised enough to reverse neurologic symptoms—will be influenced by the duration of the hyponatremia, with careful avoidance of overly rapid correction, especially in patients with chronic hyponatremia.
Assess the patient’s volume status to determine the proper initial treatment
In patients with hypo-osmolar hyponatremia who do not need urgent therapy with hypertonic saline, the initial treatment is based on clinical and laboratory assessment of extracellular fluid volume status, including spot urine sodium measurement (Table 3).3 This will be discussed further below.
Check urine osmolality to assess for hyponatremic states in which urinary dilution is intact
Measuring urine osmolality is useful in ascertaining whether hyponatremic patients are making appropriately dilute urine (< 100 mOsm/kg). If they are, the cause of the hyponatremia may be excessive water intake, a reset osmostat, or low solute intake. In addition, patients with hypovolemic hyponatremia may have appropriately dilute urine soon after treatment with isotonic intravenous fluids.
The serum sodium concentration often returns to normal if the underlying cause is eliminated (eg, if excessive fluid intake is stopped). If there are no serious signs or symptoms, this can usually be accomplished without additional therapy with intravenous fluids or medications, thereby avoiding the risk of overcorrection.
Search for causes of rapidly correctable hyponatremia
If hyponatremia is due to one of several important underlying causes, it may reverse rapidly once the underlying cause has been eliminated (Table 4). Examples: restricting water intake in patients with psychogenic polydipsia, discontinuing thiazide diuretics, replenishing depleted fluid volume, stopping desmopressin (DDAVP), and giving glucocorticoid replacement to those who are glucocorticoid-deficient.
TREATING HYPONATREMIC PATIENTS WITH SERIOUS SIGNS OR SYMPTOMS
Patients with hypo-osmolar hyponatremia and serious signs or symptoms of cerebral edema (lethargy, respiratory depression, seizures) need rapid initial correction of the serum sodium level, as this is a true medical emergency.
Certain patients are at greater risk of developing cerebral edema from hyponatremia (Table 2).
On the other hand, patients with chronic hyponatremia are very unlikely to present with signs or symptoms of cerebral edema. In fact, in a patient with chronic hyponatremia, care must be taken to avoid overcorrection beyond that needed to reverse severe signs and symptoms. In the rare case in which a patient with chronic hyponatremia presents with signs or symptoms of cerebral edema, the hypertonic saline infusion must be stopped as soon as the signs or symptoms have resolved. Further rapid changes in serum sodium must be avoided.
During correction of hyponatremia, some patients are at particularly high risk of osmotic demyelination syndrome secondary to underlying abnormalities in cerebral osmotic regulation. These include patients with alcoholism, malnutrition, hypokalemia, and burns, and elderly women on thiazide diuretics.8 These patients should be monitored vigilantly for overly rapid correction during treatment.
INITIAL TREATMENT: REVERSE CEREBRAL EDEMA WITH 3% SALINE
The goal of the initial, rapid phase of correction is to reverse cerebral edema.
Patients with serious signs or symptoms should receive hypertonic (3%) saline at a rate of about 1 mL/kg/hour for the first several hours.8 Those with concomitant hypervolemia (as in congestive heart failure) or underlying cardiovascular disease should also receive a loop diuretic to aid in free-water excretion and to prevent volume overload from the saline infusion. This regimen usually raises the serum sodium concentration enough (usually by about 1 mmol/L/hour) to decrease cerebral edema and improve symptoms.
In patients having active seizures or showing signs of brain herniation, 3% saline can be given initially at a higher rate of about 2 to 3 mL/kg/hour over the first few hours. An alternative approach is an initial 50-mL bolus of 3% saline and an additional 200 mL given over the subsequent 4 to 6 hours.9
No study has compared the efficacy and safety of these approaches, and clinicians should always monitor extracellular fluid volume status, neurologic status, and serum sodium levels closely in any patient treated with hypertonic saline.
After severe signs and symptoms have resolved, 3% saline is promptly discontinued and appropriate therapy is initiated based on the patient’s volume status and underlying cause of hyponatremia (see discussion below).
NEXT, FIND THE APPROPRIATE RATE OF CORRECTION
After the initial serious signs or symptoms have been addressed with hypertonic saline, management should focus on limiting the rate of correction in patients with chronic hyponatremia or hyponatremia of unknown duration.
Animal studies and retrospective human studies have suggested certain guidelines on the appropriate pace and magnitude of correction during treatment of hyponatremia to avoid osmotic demyelination syndrome.2
Clinicians must not attempt to correct the serum sodium to “normal” values. Although patients with acute hyponatremia may tolerate complete correction, there is little evidence that raising the serum sodium concentration acutely by more than 5 to 8 mmol/L is advantageous. Therefore, correction should be judicious in all patients.
Appropriate rates of correction
A recent expert consensus panel suggested that the serum sodium level be raised by no more than 10 to 12 mmol/L during the first 24 hours of treatment, and by less than 18 mmol/L over 48 hours.2
Patients with chronic hyponatremia and signs or symptoms of cerebral edema should have their sodium level raised at an even slower rate—some recommend less than 10 mmol/L in the first 24 hours.10 Aggressive initial correction at the rate of 1.5 to 2 mmol/hour for the first 3 to 4 hours with 3% saline is indicated until serious symptoms (seizure, obtundation) resolve, but correction beyond 10 to 12 mmol/L in the first 24 hours should be avoided. Hypertonic saline therapy should usually be discontinued well before the serum sodium level has risen this much, to avoid a continuing rise in the sodium level after the infusion has stopped.
While hypertonic saline is being infused, serum sodium levels should be checked every 1 to 2 hours. In a study in 56 patients with severe hyponatremia (serum sodium ≤ 105 mmol/L),11 no neurologic complications were observed in patients with chronic hyponatremia whose serum sodium was corrected by less than 12 mmol/L in 24 hours or by less than 18 mmol/L in 48 hours or in whom the average rate of correction to a serum sodium of 120 mmol/L was less than or equal to 0.55 mmol/L per hour.
If the serum sodium concentration has been overcorrected
Desmopressin is effective in preventing and reversing inadvertent overcorrection of hyponatremia. 12 In one study, desmopressin lowered the sodium concentration by 2 to 9 mmol/L in 14 of 20 patients. None of the patients developed any serious adverse consequences.
In addition, intravenous water (dextrose 5%) can be given alone or in combination with desmopressin to prevent or reverse an excessive increase in serum sodium.13 Such therapy may be considered in patients who continue to excrete hypotonic urine and have already reached a serum sodium concentration that meets or exceeds the recommended rate or magnitude of change.
Formulas for estimating the rate of correction
Various formulas have been devised for estimating the change in serum sodium concentration during treatment of hyponatremia.14
The Adrogué-Madias formula, one of the most commonly used, gives an estimate of how much the serum sodium concentration will rise when 1 L of various intravenous fluids is given (Table 5).15 This formula also accounts for the increase in serum sodium that takes place during concomitant correction of hypokalemia with potassium. Recently, however, a retrospective study16 found that this formula underestimated the change in serum sodium in 23 (74.2%) of 31 patients with hyponatremia treated with hypertonic saline.
An alternative is the Barsoum-Levine equation, which takes into account ongoing urinary losses. Although it is more cumbersome to calculate, it may be more precise.17
Alternatively, in patients without hypovolemia, the clinician can calculate the amount of urinary excretion of free water required to achieve a specific target serum sodium and then measure hourly urinary water excretion during aquaresis induced by furosemide (Lasix).8 Although more physiologic, this method can be clinically cumbersome, requiring timely handling of urine specimens, accurate recording of urine output, and rapid reporting of laboratory results.
Ultimately, these methods serve only as estimates of the change in serum sodium and do not replace careful monitoring of electrolytes (every 1 to 2 hours during acute therapy) and fastidious assessment for clinical signs or symptoms of osmotic demyelination syndrome.
PATIENTS WITH HYPONATREMIA AND NO SERIOUS SIGNS OR SYMPTOMS
General approach
Hyponatremic patients without serious signs or symptoms of cerebral edema do not require urgent therapy to raise the serum sodium.
Patients with chronic asymptomatic hyponatremia are commonly encountered in clinical practice. As a result of cerebral adaptation, they can appear to have no symptoms despite serum sodium levels as low as 115 to 120 mmol/L. However, even if they have no serious signs or symptoms of cerebral edema, some patients may complain of fatigue, lethargy, nausea, gait abnormalities, and muscle cramps and have evidence of milder forms of neurocognitive impairment.18
In a recent case-control study,18 elderly patients with chronic hyponatremia (mean serum sodium concentration 126 ± 5 mmol/L) were more likely to present to the hospital with falls compared with age-matched controls. Further analysis suggested these patients had marked impairments in gait and attention, which improved in some as the serum sodium increased.
Another recent study19 reported that mild hyponatremia (mean serum sodium concentration 132 mmol/L) was independently associated with the risk of fracture, even after adjustment for known osteoporotic risk factors.
Even when there is no need for acute therapy to raise the serum sodium level, the clinician should scrutinize the medical regimen and available clinical data to rule out reversible causes of water excess. These may include ongoing administration of hypotonic fluids (eg, parenteral nutrition or dextrose 5% to “keep the vein open”) or of medications that cause inappropriate release of ADH (eg, selective serotonin reuptake inhibitors) or that impair water excretion (eg, nonsteroidal anti-inflammatory drugs). The clinician should also search for an underlying diagnosis that predisposes to water retention, such as hypothyroidism, adrenal insufficiency, congestive heart failure, or hepatic or renal failure. If hyponatremia is due to endocrine disease, correction of hypothyroidism or adrenal insufficiency should result in water excretion and improvement in the serum sodium.
If the cause of the hyponatremia is not immediately apparent, treatment can be started on the basis of assessment of the patient’s extracellular fluid volume status using clinical examination and supplementary laboratory data such as the serum uric acid concentration and urinary sodium concentration.3Table 3 outlines general treatment options for hypoosmolar hyponatremia according to extracellular fluid volume status.
Of note, physical examination alone has poor sensitivity and specificity in assessing extracellular fluid volume status in patients with hyponatremia.20,21 This highlights the importance of spot measurements of urine sodium and serum uric acid and, when appropriate, isotonic intravenous saline challenge to detect occult hypovolemia.
In general, patients with euvolemia are treated with fluid restriction, and patients with hypovolemia are given isotonic saline. Patients with hypervolemia can be difficult to treat, but in general they are prescribed both sodium and fluid restriction. Loop diuretics can be given to promote excretion of water and sodium. Thiazide diuretics are avoided, as they impair urinary dilution and worsen hyponatremia. Attempts should be made to optimize the treatment of the underlying hypervolemic disorder (congestive heart failure, cirrhosis, advanced renal failure). Vasopressin receptor antagonists can also be used in selected cases of hypervolemic or euvolemic hyponatremia (see discussion below).
How to prescribe fluid restriction rationally
Ideally, patients should not ingest any more fluid than they can excrete in urine and insensible losses—otherwise, the serum sodium can continue to decrease.
Water excretion can be estimated from solute intake and urine osmolarity. In theory, a 70-kg person with a typical daily solute intake of about 10 mOsm/kg and intact urinary dilution to a urine osmolarity of 50 mOsm/L can excrete up to 14 L of urine (700 mOsm/50 mOsm/L) per day. However, a patient with the syndrome of inappropriate ADH secretion (SIADH) and a fixed urine osmolality of 700 mOsm/kg would excrete a similar solute load in only 1 L of urine. Thus, any fluid intake in excess of this volume could worsen hyponatremia.
To excrete free water, urinary sodium plus urinary potassium must be less than the serum sodium concentration. In this regard, the necessary degree of fluid restriction can also be estimated made on the basis of the patient’s urinary electrolytes.22
Increased solute intake to augment water excretion
In patients without hypervolemia, solute intake can be increased to augment water excretion. 22 This can be achieved with salt tablets or oral urea. Although urea can be effective, it is not commonly used because it is not available the United States and it has poor gastrointestinal tolerability. In patients whose nutritional intake is limited and who continue to ingest fluids (such as, for example, an elderly patient subsisting on tea and toast) every effort should be made to increase solute intake with high-protein foods or supplements.
DRUGS TO INHIBIT VASOPRESSIN
Unfortunately, patients often do not adhere to these strategies, as fluid restriction and unpalatable salt tablets or urea can become too burdensome. In such instances, pharmacologic inhibition of vasopressin-mediated water reabsorption can be considered using the following agents.
Demeclocycline (Declomycin) and lithium inhibit the kidney’s response to vasopressin. Because lithium may be nephrotoxic and has unwanted effects on the central nervous system, demeclocycline has become the preferred agent. Given in doses of 300 to 600 mg twice daily, demeclocycline promotes free water excretion, but often takes 1 to 2 weeks of therapy to begin working.
Renal failure due to demeclocycline has been reported in patients with concomitant liver disease.23 Demeclocycline can also cause photosensitivity and is contraindicated in children and pregnant women due to abnormalities in bone and enamel formation. In addition, it can be expensive and may not be covered fully by some prescription plans.
Vasopressin receptor antagonists (‘vaptans’)
ADH, also called vasopressin, interacts with various receptor subtypes, including V1a (causing vasoconstriction, platelet aggregation, inotropic stimulation, myocardial protein synthesis), V1b (causing secretion of adrenocorticotropic hormone), and V2 (causing water reabsorption and release of von Willebrand factor and factor VIII).
Drugs that block V2 receptors in the renal tubule increase water excretion, making them attractive as therapy for some hyponatremic states (Table 6).24,25 These drugs exert their aquaretic effect by causing a decrease in transcription and insertion of aquaporin-2 channels (“water pores”) into the apical collecting duct membrane. As a result, the water permeability of the collecting duct is decreased even in the presence of circulating ADH.
Conivaptan (Vaprisol) is a combined V1a-V2 antagonist that has been approved for the treatment of euvolemic and hypervolemic hyponatremia. Conivaptan inhibits the cytochrome P450 3A4 system and thus may interact with other drugs; therefore, its use has been limited to no more than 4 days of intravenous administration in the hospital setting. The recommended dosage is an initial 20-mg infusion over 30 minutes, followed by continuous infusions of 20 to 40 mg/day. Dosing adjustments in renal and hepatic impairment have not been well defined.
Tolvaptan (Samsca) is an oral selective V2 antagonist that has been studied in patients with euvolemic and hypervolemic hyponatremia. 26 Studies have included patients with congestive heart failure, cirrhosis, and SIADH. Although tolvaptan has not been shown to reduce rates of rehospitalization or death in congestive heart failure, it improves serum sodium, overall fluid balance, and congestive symptoms.27 Tolvaptan has recently been approved for the treatment of euvolemic and hypervolemic hyponatremia.
A recent study has confirmed the longterm efficacy of tolvaptan in 111 patients over a mean duration of treatment greater than 700 days.28 While the clinical benefits of chronic tolvaptan therapy have yet to be clearly demonstrated, this study shows that tolvaptan therapy can result in a sustained improvement in serum sodium concentration without an unacceptable increase in adverse events.29
Lixivaptan (VPA-985), another oral selective V2 receptor antagonist, is being studied in patients with euvolemic and hypervolemic hyponatremia.
Current role of vasopressin antagonists
Current studies of vasopressin antagonists in the treatment of hyponatremia are promising, though definite recommendations are needed to ensure slow, careful correction of hyponatremia. Most studies suggest that these agents provide slow, reliable increases in serum sodium. In one large study of patients with congestive heart failure, serum sodium rose by more than 12 mmol/L in 24 hours in fewer than 2% of patients.26
Notably, no cases of osmotic demyelination syndrome have been reported in these studies. However, it should be noted that therapy was started in the hospital with close monitoring of serum sodium levels and discontinuation of fluid restriction; the incidence of overly rapid correction of sodium may be higher outside of carefully done clinical studies. Clinicians should adopt monitoring strategies similar to those used in these rigorous studies.
At present, there is little experience with vasopressin antagonists in hyponatremic patients with serious signs or symptoms of cerebral edema, and most clinicians still view 3% saline as the gold standard for these patients.
Vasopressin antagonists should not be used in patients with hypovolemic hyponatremia, due to concerns about V1a blockade causing hypotension and about V2 blockade producing water excretion and a worsening of the volume-depleted state.
Recent clinical trials have reported that patients often experience increased thirst while taking these agents. This highlights the need to monitor serum sodium during treatment.
These agents are expensive. Tolvaptan costs about $250 per tablet; conivaptan, which is administered intravenously, may cost a little more per treatment course.
THERAPY IN SPECIFIC DISEASE STATES
Patients with hyponatremia and cirrhosis
The focus of treatment remains water and salt restriction and judicious use of loop diuretics and aldosterone antagonists such as spironolactone (Aldactone).
Tolvaptan has been effective at raising the serum sodium level in patients with cirrhosis, 26 while conivaptan should be avoided at present because of vasodilation from V1a receptor antagonism and its potential effects on systemic hemodynamics and risk of variceal bleeding.30
As the severity of cirrhosis increases, the only effective treatment of hyponatremia is liver transplantation.
Patients with SIADH
In most cases, water restriction is the mainstay of therapy. Adequate nutritional intake should also be stressed so that enough solute is available for ongoing water excretion. Although fluid restriction is usually effective, many patients cannot adhere to the level of restriction required.
In cases in which fluid restriction is not effective on its own, demeclocyline can be used to antagonize ADH action and increase water excretion. Sodium tablets and loop diuretics can also be used, taking care to avoid hypovolemia from diuretic-induced sodium losses. The use of tolvaptan in patients with SIADH has resulted in short-term increases in serum sodium.26 A recent study has suggested that this effect can be sustained with longer-term treatment,28 but further studies are needed to show a complementary clinical benefit (eg, improved neurocognition) to guide the use of these costly agents in clinical practice.
Patients with diuretic-induced hyponatremia
Thiazide diuretics should be discontinued and hypovolemia and hypokalemia should be corrected with isotonic saline and potassium supplementation. As the hypokalemia is corrected and the diuretic effect and hypovolemic stimulus to ADH dissipates, water excretion can increase rapidly, resulting in a brisk change in serum sodium.
Serum sodium levels should be closely monitored during therapy to avoid overcorrection. For this reason, use of hypertonic saline should generally be avoided. Hypotonic fluid— eg, half-normal (0.45%) or quarter-normal (0.22%) saline or even desmopressin—may become necessary in the later stages of therapy to avoid overly rapid correction.
Patients with exercise-associated hyponatremia
Patients at highest risk of exercise-associated hyponatremia include those who drink too much fluid during a long-distance race, who have low body weight, who are female, who exercise longer than 4 hours, and who use nonsteroidal anti-inflammatory drugs.31 The cause of hyponatremia is likely multifactorial, with excessive water intake coupled with sodium losses and impaired renal excretion of water due to ADH action and impaired renal dilution. To prevent exercise-associated hyponatremia, fluid intake should be limited to 400 to 800 mL/hour, with the higher end recommended for larger athletes and hotter climates.
Consensus recommendations suggest that most patients with mild hyponatremia (serum sodium 130 to 135 mmol/L) should be treated with fluid restriction and clinical observation, as spontaneous water diuresis leads to improvement in the serum sodium level. Importantly, the reflex to provide isotonic saline infusions should be avoided unless clear signs of volume depletion are present. Intravenous saline has the potential to worsen hyponatremia in the presence of ADH. In addition, some athletes will have retained water in the gastrointestinal tract that may be mobilized after the race, resulting in worsening of hyponatremia.32
In athletes with severe hyponatremia (serum sodium < 120 mmol/L) or symptomatic exercise-associated hyponatremia (lethargy, respiratory depression, seizures), hypertonic saline is the treatment of choice. One protocol suggests giving 100 mL of 3% saline over 10 minutes in the field, followed by prompt transportation to hospital.33
SUMMARY POINTS
Hyponatremia is a common electrolyte disorder that in its most severe form requires urgent therapy with hypertonic saline to correct cerebral edema.
In patients without serious signs or symptoms of cerebral edema, recent observations suggest there may be clinically important symptomatology relating to mild neurocognitive dysfunction and an association with risk of bone fracture.
Multiple treatment strategies are available according to the underlying extracellular fluid volume status and cause of hyponatremia. These include fluid and sodium restriction and augmentation of urinary water excretion with various nutritional and pharmacologic strategies. The most novel therapy includes antagonism of the vasopressin V2 receptor with a class of aquaretic agents known as vaptans.
There can be serious neurologic injury associated with overly rapid correction of chronic hyponatremia or undercorrection of acute symptomatic hyponatremia.
Clinicians must be familiar with the details of each of the treatments and have an appreciation of the importance of careful monitoring during treatment.
References
Flear CTG, Gill GV, Burn J. Hyponatremia: mechanisms and management. Lancet1981; 2:26–31.
Freda BJ, Davidson MB, Hall PM. Evaluation of hyponatremia: a little physiology goes a long way. Cleve Clin J Med2004; 71:639–650.
Weisberg LS. Pseudohyponatremia: a reappraisal. Am J Med1989; 86:315–318.
Moritz L, Ayus JC. The pathophysiology and treatment of hyponatraemic encephalopathy: an update. Nephrol Dial Transplant2003; 18:2486–2491.
Widdess-Walsh P, Sabharwal V, Demirjian S, DeGeorgia M. Neurologic effects of hyponatremia and its treatment. Cleve Clin J Med2007; 74:377–383.
Melton JE, Patlak CS, Pettigrew KD, Cserr HF. Volume regulatory loss of Na, Cl, and K from rat brain during acute hyponatremia. Am J Physiol1987; 252:F661–F669.
Lauriat SM, Berl T. The hyponatremic patient: practical focus on therapy. J Am Soc Nephrol1997; 8:1599–1607.
Kokko JP. Symptomatic hyponatremia with hypoxia is a medical emergency. Kidney Int2006; 69:1291–1293.
Ellis SJ. Severe hyponatraemia: complications and treatment. QJM1995; 88:905–909.
Sterns RH, Cappuccio JD, Silver SM, Cohen EP. Neurologic sequelae after treatment of severe hyponatremia: a multicenter perspective. J Am Soc Nephrol1994; 4:1522–1530.
Perianayagam A, Sterns RH, Silver SM, et al. DDAVP is effective in preventing and reversing inadvertent overcorrection of hyponatremia. Clin J Am Soc Nephrol2008; 3:331–336.
Sterns RH, Hix JK. Overcorrection of hyponatremia is a medical emergency. Kidney Int2009; 76:587–589.
Nguyen MK, Kurtz I. Analysis of current formulas used for treatment of the dysnatremias. Clin Exp Nephrol2004; 8:12–16.
Adrogué HJ, Madias NE. Hyponatremia. N Engl J Med2000; 342:1581–1589.
Mohmand HK, Issa D, Ahmad Z, Cappuccio JD, Kouides RW, Sterns RH. Hypertonic saline for hyponatremia: risk of inadvertent overcorrection. Clin J Am Soc Nephrol2007; 2:1110–1117.
Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med2007; 356:2064–2072.
Renneboog B, Musch W, Vandemergel X, Manto MU, Decaux G. Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits. Am J Med2006; 119:71.e1–e8.
Kinsella S, Moran S, Sullivan MO, Molloy MG, Eustace JA. Hyponatremia independent of osteoporosis is associated with fracture occurrence. Clin J Am Soc Nephrol2010; 5:275–280.
Chung HM, Kluge R, Schrier RW, Anderson RJ. Clinical assessment of extracellular fluid volume in hyponatremia. Am J Med1987; 83:905–988.
Hoorn EJ, Halperin ML, Zietse R. Diagnostic approach to a patient with hyponatraemia: traditional versus physiology-based options. QJM2005; 98:529–540.
Berl T. Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol2008; 19:1076–1078.
Carrilho F, Bosch J, Arroyo V, Mas A, Viver J, Rodes J. Renal failure associated with demeclocycline in cirrhosis. Ann Intern Med1977; 87:195–197.
Lehrich RW, Greenberg A. When is it appropriate to use vasopressin receptor antagonists?J Am Soc Nephrol2008; 19:1054–1058.
Decaux G, Soupart A, Vassart G. Non-peptide arginine-vasopressin antagonists: the vaptans. Lancet2008; 371:1624–1632.
Schrier RW, Gross P, Gheorghiade M, et al; SALT Investigators. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med2006; 355:2099–2112.
Konstam MA, Gheorghiade M, Burnett JC, et al; Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA2007; 297:1319–1331.
Berl T, Quittnat-Pelletier F, Verbalis JG, et al; SALTWATER Investigators. Oral tolvaptan is safe and effective in chronic hyponatremia. J Am Soc Nephrol2010; 21:705–712.
Greenberg A, Lehrich RW. Treatment of chronic hyponatremia: now we know how, but do we know when or if?J Am Soc Nephrol2010; 21:552–555.
Greenberg A, Verbalis JG. Vasopressin receptor antagonists. Kidney Int2006; 69:2124–2130.
Rosner MH, Kirven J. Exercise-associated hyponatremia. Clin J Am Soc Nephrol2007; 2:151–161.
Halperin ML, Kamel KS, Sterns R. Hyponatremia in marathon runners. N Engl J Med2005; 353:427–428.
Hew-Butler T, Almond C, Ayus JC, et al; Exercise-Associated Hyponatremia (EAH) Consensus Panel. Consensus statement of the 1st International Exercise-Associated Hyponatremia Consensus Development Conference, Cape Town, South Africa 2005. Clin J Sport Med2005; 15:208–213.
Hyponatremia, defined as a serum sodium concentration below 135 mmol/L, is one of the most frequently encountered electrolyte disorders. In 1981, Flear et al1 reported that 15% of their hospitalized patients had plasma sodium concentrations lower than 134 mmol/L, the cutoff they were using at that time.
Hyponatremia is sometimes merely a laboratory artifact or a result of improper blood collection. If real, it can be due to excessive water intake or, most often, the inability of the kidney to excrete water coupled with continued water intake. Patients with significant underlying cardiac, hepatic, or renal dysfunction are at greatest risk of developing hyponatremia, secondary to the nonosmotic release of antidiuretic hormone (ADH). Others at risk include postoperative patients (especially menstruating women), older patients on thiazide diuretics, patients with malignant or psychiatric illness, and endurance athletes.
In this article, we review the treatment of acute and chronic hyponatremia, emphasizing the importance of basing the therapy on the severity of symptoms and taking care not to raise the serum sodium level too rapidly, which can cause neurologic dysfunction.
Guidelines for managing hyponatremia2 are based mostly on retrospective studies and expert opinion, since few prospective studies have been done. Despite the paucity of evidence-based recommendations, we will attempt to incorporate findings from important human and animal studies and consensus guidelines from expert panels. We will focus initially on the critical diagnostic considerations necessary to initiate treatment.
SYMPTOMATIC VS ASYMPTOMATIC
Subsequent sections will address therapeutic approaches in two clinical settings:
Symptomatic hyponatremia, ie, with severe signs or symptoms of cerebral edema—a medical emergency; and
Asymptomatic hyponatremia, ie, without serious signs or symptoms of cerebral edema.
KEY DIAGNOSTIC STEPS WHEN STARTING TREATMENT
The treatment of hyponatremia begins by confirming a truly hypo-osmolar state, assessing its clinical significance, and determining its cause (Table 1).
The clinical and laboratory evaluations form the foundation of a proper approach to any patient with hyponatremia. The rationale behind making several important diagnostic distinctions will be discussed here briefly and then expanded on in the remaining text. The reader is referred to another review on the diagnostic evaluation of hyponatremia.3
Confirm that the patient truly has hypo-osmolar hyponatremia
The serum osmolality should be measured to confirm that it is low (ie, < 275 mOsm/kg). In addition, the arterial serum sodium concentration can be measured using a blood gas device if pseudohyponatremia (see below) is suspected. This method uses direct potentiometry and bypasses the dilutional step in the processing of venous samples.4
Rationale. The clinical consequences of hyponatremia are due to water moving from hypo-osmolar extracellular fluid into the relatively hyperosmolar interior of the cell. This water movement can cause progressive cerebral edema, resulting in a spectrum of signs and symptoms from headache and ataxia to seizures and coma. But significant fluid shifts and cerebral edema occur only if the extracellular fluid is hypo-osmolar relative to the intracellular fluid.
In fact, hyponatremia can occur in several situations in which the extracellular fluid is not hypo-osmolar. An increase in effective plasma osmoles (substances in the extracellular fluid that do not readily move across the plasma membrane) can cause water to move out of cells, resulting in translocational hyponatremia. This may be seen in hyperglycemia or when mannitol or contrast dye has been given. In these situations, the plasma is either isotonic or even hypertonic to the intracellular fluid, resulting in no movement of water into the cells and therefore no clinical consequences relating to the hyponatremia. Importantly, no therapy is required for the hyponatremia.
Other situations in which hyponatremia is present but not associated with true hypotonicity include states of excess protein or lipid in the blood (pseudohyponatremia). Also, if an infusion of hypotonic fluid is running, clinicians must be sure that blood samples are not drawn proximally in the same vein.
Are there significant signs or symptoms of cerebral edema?
Hyponatremia can cause brain swelling within the confined space of the skull as water shifts from the extracellular fluid into the cells. Depending on underlying risk factors (Table 2)5 and the severity and duration of hyponatremia (see below), this may result in signs or symptoms of cerebral edema, including visual changes, focal neurologic changes, encephalopathy, respiratory depression, and seizures. Ultimately, brain herniation can occur.
Patients need to be assessed quickly because those with serious neurologic signs or symptoms thought to be related to hyponatremia require urgent treatment with hypertonic saline to increase the serum sodium concentration, regardless of the underlying volume status, the cause of hyponatremia, or the time of onset.
Determine the duration of hyponatremia
One should try to ascertain when the hyponatremia started, as its duration is important in determining the proper pace of correction.
Figure 1.
The brain begins to adapt to hyponatremia within minutes, and the cerebral adaptation is maximal within 2 to 3 days (Figure 1).6
At the onset of hyponatremia, water moves from the extracellular fluid into cells, pulled in by osmosis. The brain can decrease the net amount of water entering into the neurons (and thus regulate its volume) by increasing the flow of water from the interstitium into the cerebrospinal fluid via increased interstitial hydraulic pressure.7
Over the next several days, inorganic solutes (eg, potassium and sodium salts) and various organic solutes are transported out of the cells. In patients in whom this process has had time to occur, treatment of hyponatremia with hypertonic fluids raises the plasma osmolality faster than the cells can recapture the previously transported osmoles. In this situation, overly rapid correction can cause excessive loss of intracellular water, resulting in cell shrinkage and osmotic demyelination syndrome. Osmotic demyelination usually presents during treatment of hyponatremia after an initial improvement in mental status, with worsening neurologic function and various neurologic signs, including paresis and ultimately even death.6
In patients with acute-onset hyponatremia (ie, with onset within the past 48 hours), in whom the above cerebral adaptations have not had time to occur completely, rapid correction is unlikely to result in osmotic demyelination.
In view of the serious risk of osmotic demyelination, if the timing of development of hyponatremia cannot be determined, one should assume it is chronic (> 48 hours) and avoid rapid overcorrection (see discussion below on the rate of correction).
On the other hand, patients who have severe neurologic signs or symptoms initially need their serum sodium increased urgently to safer levels, regardless of the timing of onset (see below for suggested approach). Subsequent treatment of hyponatremia—after the serum sodium level has been raised enough to reverse neurologic symptoms—will be influenced by the duration of the hyponatremia, with careful avoidance of overly rapid correction, especially in patients with chronic hyponatremia.
Assess the patient’s volume status to determine the proper initial treatment
In patients with hypo-osmolar hyponatremia who do not need urgent therapy with hypertonic saline, the initial treatment is based on clinical and laboratory assessment of extracellular fluid volume status, including spot urine sodium measurement (Table 3).3 This will be discussed further below.
Check urine osmolality to assess for hyponatremic states in which urinary dilution is intact
Measuring urine osmolality is useful in ascertaining whether hyponatremic patients are making appropriately dilute urine (< 100 mOsm/kg). If they are, the cause of the hyponatremia may be excessive water intake, a reset osmostat, or low solute intake. In addition, patients with hypovolemic hyponatremia may have appropriately dilute urine soon after treatment with isotonic intravenous fluids.
The serum sodium concentration often returns to normal if the underlying cause is eliminated (eg, if excessive fluid intake is stopped). If there are no serious signs or symptoms, this can usually be accomplished without additional therapy with intravenous fluids or medications, thereby avoiding the risk of overcorrection.
Search for causes of rapidly correctable hyponatremia
If hyponatremia is due to one of several important underlying causes, it may reverse rapidly once the underlying cause has been eliminated (Table 4). Examples: restricting water intake in patients with psychogenic polydipsia, discontinuing thiazide diuretics, replenishing depleted fluid volume, stopping desmopressin (DDAVP), and giving glucocorticoid replacement to those who are glucocorticoid-deficient.
TREATING HYPONATREMIC PATIENTS WITH SERIOUS SIGNS OR SYMPTOMS
Patients with hypo-osmolar hyponatremia and serious signs or symptoms of cerebral edema (lethargy, respiratory depression, seizures) need rapid initial correction of the serum sodium level, as this is a true medical emergency.
Certain patients are at greater risk of developing cerebral edema from hyponatremia (Table 2).
On the other hand, patients with chronic hyponatremia are very unlikely to present with signs or symptoms of cerebral edema. In fact, in a patient with chronic hyponatremia, care must be taken to avoid overcorrection beyond that needed to reverse severe signs and symptoms. In the rare case in which a patient with chronic hyponatremia presents with signs or symptoms of cerebral edema, the hypertonic saline infusion must be stopped as soon as the signs or symptoms have resolved. Further rapid changes in serum sodium must be avoided.
During correction of hyponatremia, some patients are at particularly high risk of osmotic demyelination syndrome secondary to underlying abnormalities in cerebral osmotic regulation. These include patients with alcoholism, malnutrition, hypokalemia, and burns, and elderly women on thiazide diuretics.8 These patients should be monitored vigilantly for overly rapid correction during treatment.
INITIAL TREATMENT: REVERSE CEREBRAL EDEMA WITH 3% SALINE
The goal of the initial, rapid phase of correction is to reverse cerebral edema.
Patients with serious signs or symptoms should receive hypertonic (3%) saline at a rate of about 1 mL/kg/hour for the first several hours.8 Those with concomitant hypervolemia (as in congestive heart failure) or underlying cardiovascular disease should also receive a loop diuretic to aid in free-water excretion and to prevent volume overload from the saline infusion. This regimen usually raises the serum sodium concentration enough (usually by about 1 mmol/L/hour) to decrease cerebral edema and improve symptoms.
In patients having active seizures or showing signs of brain herniation, 3% saline can be given initially at a higher rate of about 2 to 3 mL/kg/hour over the first few hours. An alternative approach is an initial 50-mL bolus of 3% saline and an additional 200 mL given over the subsequent 4 to 6 hours.9
No study has compared the efficacy and safety of these approaches, and clinicians should always monitor extracellular fluid volume status, neurologic status, and serum sodium levels closely in any patient treated with hypertonic saline.
After severe signs and symptoms have resolved, 3% saline is promptly discontinued and appropriate therapy is initiated based on the patient’s volume status and underlying cause of hyponatremia (see discussion below).
NEXT, FIND THE APPROPRIATE RATE OF CORRECTION
After the initial serious signs or symptoms have been addressed with hypertonic saline, management should focus on limiting the rate of correction in patients with chronic hyponatremia or hyponatremia of unknown duration.
Animal studies and retrospective human studies have suggested certain guidelines on the appropriate pace and magnitude of correction during treatment of hyponatremia to avoid osmotic demyelination syndrome.2
Clinicians must not attempt to correct the serum sodium to “normal” values. Although patients with acute hyponatremia may tolerate complete correction, there is little evidence that raising the serum sodium concentration acutely by more than 5 to 8 mmol/L is advantageous. Therefore, correction should be judicious in all patients.
Appropriate rates of correction
A recent expert consensus panel suggested that the serum sodium level be raised by no more than 10 to 12 mmol/L during the first 24 hours of treatment, and by less than 18 mmol/L over 48 hours.2
Patients with chronic hyponatremia and signs or symptoms of cerebral edema should have their sodium level raised at an even slower rate—some recommend less than 10 mmol/L in the first 24 hours.10 Aggressive initial correction at the rate of 1.5 to 2 mmol/hour for the first 3 to 4 hours with 3% saline is indicated until serious symptoms (seizure, obtundation) resolve, but correction beyond 10 to 12 mmol/L in the first 24 hours should be avoided. Hypertonic saline therapy should usually be discontinued well before the serum sodium level has risen this much, to avoid a continuing rise in the sodium level after the infusion has stopped.
While hypertonic saline is being infused, serum sodium levels should be checked every 1 to 2 hours. In a study in 56 patients with severe hyponatremia (serum sodium ≤ 105 mmol/L),11 no neurologic complications were observed in patients with chronic hyponatremia whose serum sodium was corrected by less than 12 mmol/L in 24 hours or by less than 18 mmol/L in 48 hours or in whom the average rate of correction to a serum sodium of 120 mmol/L was less than or equal to 0.55 mmol/L per hour.
If the serum sodium concentration has been overcorrected
Desmopressin is effective in preventing and reversing inadvertent overcorrection of hyponatremia. 12 In one study, desmopressin lowered the sodium concentration by 2 to 9 mmol/L in 14 of 20 patients. None of the patients developed any serious adverse consequences.
In addition, intravenous water (dextrose 5%) can be given alone or in combination with desmopressin to prevent or reverse an excessive increase in serum sodium.13 Such therapy may be considered in patients who continue to excrete hypotonic urine and have already reached a serum sodium concentration that meets or exceeds the recommended rate or magnitude of change.
Formulas for estimating the rate of correction
Various formulas have been devised for estimating the change in serum sodium concentration during treatment of hyponatremia.14
The Adrogué-Madias formula, one of the most commonly used, gives an estimate of how much the serum sodium concentration will rise when 1 L of various intravenous fluids is given (Table 5).15 This formula also accounts for the increase in serum sodium that takes place during concomitant correction of hypokalemia with potassium. Recently, however, a retrospective study16 found that this formula underestimated the change in serum sodium in 23 (74.2%) of 31 patients with hyponatremia treated with hypertonic saline.
An alternative is the Barsoum-Levine equation, which takes into account ongoing urinary losses. Although it is more cumbersome to calculate, it may be more precise.17
Alternatively, in patients without hypovolemia, the clinician can calculate the amount of urinary excretion of free water required to achieve a specific target serum sodium and then measure hourly urinary water excretion during aquaresis induced by furosemide (Lasix).8 Although more physiologic, this method can be clinically cumbersome, requiring timely handling of urine specimens, accurate recording of urine output, and rapid reporting of laboratory results.
Ultimately, these methods serve only as estimates of the change in serum sodium and do not replace careful monitoring of electrolytes (every 1 to 2 hours during acute therapy) and fastidious assessment for clinical signs or symptoms of osmotic demyelination syndrome.
PATIENTS WITH HYPONATREMIA AND NO SERIOUS SIGNS OR SYMPTOMS
General approach
Hyponatremic patients without serious signs or symptoms of cerebral edema do not require urgent therapy to raise the serum sodium.
Patients with chronic asymptomatic hyponatremia are commonly encountered in clinical practice. As a result of cerebral adaptation, they can appear to have no symptoms despite serum sodium levels as low as 115 to 120 mmol/L. However, even if they have no serious signs or symptoms of cerebral edema, some patients may complain of fatigue, lethargy, nausea, gait abnormalities, and muscle cramps and have evidence of milder forms of neurocognitive impairment.18
In a recent case-control study,18 elderly patients with chronic hyponatremia (mean serum sodium concentration 126 ± 5 mmol/L) were more likely to present to the hospital with falls compared with age-matched controls. Further analysis suggested these patients had marked impairments in gait and attention, which improved in some as the serum sodium increased.
Another recent study19 reported that mild hyponatremia (mean serum sodium concentration 132 mmol/L) was independently associated with the risk of fracture, even after adjustment for known osteoporotic risk factors.
Even when there is no need for acute therapy to raise the serum sodium level, the clinician should scrutinize the medical regimen and available clinical data to rule out reversible causes of water excess. These may include ongoing administration of hypotonic fluids (eg, parenteral nutrition or dextrose 5% to “keep the vein open”) or of medications that cause inappropriate release of ADH (eg, selective serotonin reuptake inhibitors) or that impair water excretion (eg, nonsteroidal anti-inflammatory drugs). The clinician should also search for an underlying diagnosis that predisposes to water retention, such as hypothyroidism, adrenal insufficiency, congestive heart failure, or hepatic or renal failure. If hyponatremia is due to endocrine disease, correction of hypothyroidism or adrenal insufficiency should result in water excretion and improvement in the serum sodium.
If the cause of the hyponatremia is not immediately apparent, treatment can be started on the basis of assessment of the patient’s extracellular fluid volume status using clinical examination and supplementary laboratory data such as the serum uric acid concentration and urinary sodium concentration.3Table 3 outlines general treatment options for hypoosmolar hyponatremia according to extracellular fluid volume status.
Of note, physical examination alone has poor sensitivity and specificity in assessing extracellular fluid volume status in patients with hyponatremia.20,21 This highlights the importance of spot measurements of urine sodium and serum uric acid and, when appropriate, isotonic intravenous saline challenge to detect occult hypovolemia.
In general, patients with euvolemia are treated with fluid restriction, and patients with hypovolemia are given isotonic saline. Patients with hypervolemia can be difficult to treat, but in general they are prescribed both sodium and fluid restriction. Loop diuretics can be given to promote excretion of water and sodium. Thiazide diuretics are avoided, as they impair urinary dilution and worsen hyponatremia. Attempts should be made to optimize the treatment of the underlying hypervolemic disorder (congestive heart failure, cirrhosis, advanced renal failure). Vasopressin receptor antagonists can also be used in selected cases of hypervolemic or euvolemic hyponatremia (see discussion below).
How to prescribe fluid restriction rationally
Ideally, patients should not ingest any more fluid than they can excrete in urine and insensible losses—otherwise, the serum sodium can continue to decrease.
Water excretion can be estimated from solute intake and urine osmolarity. In theory, a 70-kg person with a typical daily solute intake of about 10 mOsm/kg and intact urinary dilution to a urine osmolarity of 50 mOsm/L can excrete up to 14 L of urine (700 mOsm/50 mOsm/L) per day. However, a patient with the syndrome of inappropriate ADH secretion (SIADH) and a fixed urine osmolality of 700 mOsm/kg would excrete a similar solute load in only 1 L of urine. Thus, any fluid intake in excess of this volume could worsen hyponatremia.
To excrete free water, urinary sodium plus urinary potassium must be less than the serum sodium concentration. In this regard, the necessary degree of fluid restriction can also be estimated made on the basis of the patient’s urinary electrolytes.22
Increased solute intake to augment water excretion
In patients without hypervolemia, solute intake can be increased to augment water excretion. 22 This can be achieved with salt tablets or oral urea. Although urea can be effective, it is not commonly used because it is not available the United States and it has poor gastrointestinal tolerability. In patients whose nutritional intake is limited and who continue to ingest fluids (such as, for example, an elderly patient subsisting on tea and toast) every effort should be made to increase solute intake with high-protein foods or supplements.
DRUGS TO INHIBIT VASOPRESSIN
Unfortunately, patients often do not adhere to these strategies, as fluid restriction and unpalatable salt tablets or urea can become too burdensome. In such instances, pharmacologic inhibition of vasopressin-mediated water reabsorption can be considered using the following agents.
Demeclocycline (Declomycin) and lithium inhibit the kidney’s response to vasopressin. Because lithium may be nephrotoxic and has unwanted effects on the central nervous system, demeclocycline has become the preferred agent. Given in doses of 300 to 600 mg twice daily, demeclocycline promotes free water excretion, but often takes 1 to 2 weeks of therapy to begin working.
Renal failure due to demeclocycline has been reported in patients with concomitant liver disease.23 Demeclocycline can also cause photosensitivity and is contraindicated in children and pregnant women due to abnormalities in bone and enamel formation. In addition, it can be expensive and may not be covered fully by some prescription plans.
Vasopressin receptor antagonists (‘vaptans’)
ADH, also called vasopressin, interacts with various receptor subtypes, including V1a (causing vasoconstriction, platelet aggregation, inotropic stimulation, myocardial protein synthesis), V1b (causing secretion of adrenocorticotropic hormone), and V2 (causing water reabsorption and release of von Willebrand factor and factor VIII).
Drugs that block V2 receptors in the renal tubule increase water excretion, making them attractive as therapy for some hyponatremic states (Table 6).24,25 These drugs exert their aquaretic effect by causing a decrease in transcription and insertion of aquaporin-2 channels (“water pores”) into the apical collecting duct membrane. As a result, the water permeability of the collecting duct is decreased even in the presence of circulating ADH.
Conivaptan (Vaprisol) is a combined V1a-V2 antagonist that has been approved for the treatment of euvolemic and hypervolemic hyponatremia. Conivaptan inhibits the cytochrome P450 3A4 system and thus may interact with other drugs; therefore, its use has been limited to no more than 4 days of intravenous administration in the hospital setting. The recommended dosage is an initial 20-mg infusion over 30 minutes, followed by continuous infusions of 20 to 40 mg/day. Dosing adjustments in renal and hepatic impairment have not been well defined.
Tolvaptan (Samsca) is an oral selective V2 antagonist that has been studied in patients with euvolemic and hypervolemic hyponatremia. 26 Studies have included patients with congestive heart failure, cirrhosis, and SIADH. Although tolvaptan has not been shown to reduce rates of rehospitalization or death in congestive heart failure, it improves serum sodium, overall fluid balance, and congestive symptoms.27 Tolvaptan has recently been approved for the treatment of euvolemic and hypervolemic hyponatremia.
A recent study has confirmed the longterm efficacy of tolvaptan in 111 patients over a mean duration of treatment greater than 700 days.28 While the clinical benefits of chronic tolvaptan therapy have yet to be clearly demonstrated, this study shows that tolvaptan therapy can result in a sustained improvement in serum sodium concentration without an unacceptable increase in adverse events.29
Lixivaptan (VPA-985), another oral selective V2 receptor antagonist, is being studied in patients with euvolemic and hypervolemic hyponatremia.
Current role of vasopressin antagonists
Current studies of vasopressin antagonists in the treatment of hyponatremia are promising, though definite recommendations are needed to ensure slow, careful correction of hyponatremia. Most studies suggest that these agents provide slow, reliable increases in serum sodium. In one large study of patients with congestive heart failure, serum sodium rose by more than 12 mmol/L in 24 hours in fewer than 2% of patients.26
Notably, no cases of osmotic demyelination syndrome have been reported in these studies. However, it should be noted that therapy was started in the hospital with close monitoring of serum sodium levels and discontinuation of fluid restriction; the incidence of overly rapid correction of sodium may be higher outside of carefully done clinical studies. Clinicians should adopt monitoring strategies similar to those used in these rigorous studies.
At present, there is little experience with vasopressin antagonists in hyponatremic patients with serious signs or symptoms of cerebral edema, and most clinicians still view 3% saline as the gold standard for these patients.
Vasopressin antagonists should not be used in patients with hypovolemic hyponatremia, due to concerns about V1a blockade causing hypotension and about V2 blockade producing water excretion and a worsening of the volume-depleted state.
Recent clinical trials have reported that patients often experience increased thirst while taking these agents. This highlights the need to monitor serum sodium during treatment.
These agents are expensive. Tolvaptan costs about $250 per tablet; conivaptan, which is administered intravenously, may cost a little more per treatment course.
THERAPY IN SPECIFIC DISEASE STATES
Patients with hyponatremia and cirrhosis
The focus of treatment remains water and salt restriction and judicious use of loop diuretics and aldosterone antagonists such as spironolactone (Aldactone).
Tolvaptan has been effective at raising the serum sodium level in patients with cirrhosis, 26 while conivaptan should be avoided at present because of vasodilation from V1a receptor antagonism and its potential effects on systemic hemodynamics and risk of variceal bleeding.30
As the severity of cirrhosis increases, the only effective treatment of hyponatremia is liver transplantation.
Patients with SIADH
In most cases, water restriction is the mainstay of therapy. Adequate nutritional intake should also be stressed so that enough solute is available for ongoing water excretion. Although fluid restriction is usually effective, many patients cannot adhere to the level of restriction required.
In cases in which fluid restriction is not effective on its own, demeclocyline can be used to antagonize ADH action and increase water excretion. Sodium tablets and loop diuretics can also be used, taking care to avoid hypovolemia from diuretic-induced sodium losses. The use of tolvaptan in patients with SIADH has resulted in short-term increases in serum sodium.26 A recent study has suggested that this effect can be sustained with longer-term treatment,28 but further studies are needed to show a complementary clinical benefit (eg, improved neurocognition) to guide the use of these costly agents in clinical practice.
Patients with diuretic-induced hyponatremia
Thiazide diuretics should be discontinued and hypovolemia and hypokalemia should be corrected with isotonic saline and potassium supplementation. As the hypokalemia is corrected and the diuretic effect and hypovolemic stimulus to ADH dissipates, water excretion can increase rapidly, resulting in a brisk change in serum sodium.
Serum sodium levels should be closely monitored during therapy to avoid overcorrection. For this reason, use of hypertonic saline should generally be avoided. Hypotonic fluid— eg, half-normal (0.45%) or quarter-normal (0.22%) saline or even desmopressin—may become necessary in the later stages of therapy to avoid overly rapid correction.
Patients with exercise-associated hyponatremia
Patients at highest risk of exercise-associated hyponatremia include those who drink too much fluid during a long-distance race, who have low body weight, who are female, who exercise longer than 4 hours, and who use nonsteroidal anti-inflammatory drugs.31 The cause of hyponatremia is likely multifactorial, with excessive water intake coupled with sodium losses and impaired renal excretion of water due to ADH action and impaired renal dilution. To prevent exercise-associated hyponatremia, fluid intake should be limited to 400 to 800 mL/hour, with the higher end recommended for larger athletes and hotter climates.
Consensus recommendations suggest that most patients with mild hyponatremia (serum sodium 130 to 135 mmol/L) should be treated with fluid restriction and clinical observation, as spontaneous water diuresis leads to improvement in the serum sodium level. Importantly, the reflex to provide isotonic saline infusions should be avoided unless clear signs of volume depletion are present. Intravenous saline has the potential to worsen hyponatremia in the presence of ADH. In addition, some athletes will have retained water in the gastrointestinal tract that may be mobilized after the race, resulting in worsening of hyponatremia.32
In athletes with severe hyponatremia (serum sodium < 120 mmol/L) or symptomatic exercise-associated hyponatremia (lethargy, respiratory depression, seizures), hypertonic saline is the treatment of choice. One protocol suggests giving 100 mL of 3% saline over 10 minutes in the field, followed by prompt transportation to hospital.33
SUMMARY POINTS
Hyponatremia is a common electrolyte disorder that in its most severe form requires urgent therapy with hypertonic saline to correct cerebral edema.
In patients without serious signs or symptoms of cerebral edema, recent observations suggest there may be clinically important symptomatology relating to mild neurocognitive dysfunction and an association with risk of bone fracture.
Multiple treatment strategies are available according to the underlying extracellular fluid volume status and cause of hyponatremia. These include fluid and sodium restriction and augmentation of urinary water excretion with various nutritional and pharmacologic strategies. The most novel therapy includes antagonism of the vasopressin V2 receptor with a class of aquaretic agents known as vaptans.
There can be serious neurologic injury associated with overly rapid correction of chronic hyponatremia or undercorrection of acute symptomatic hyponatremia.
Clinicians must be familiar with the details of each of the treatments and have an appreciation of the importance of careful monitoring during treatment.
Hyponatremia, defined as a serum sodium concentration below 135 mmol/L, is one of the most frequently encountered electrolyte disorders. In 1981, Flear et al1 reported that 15% of their hospitalized patients had plasma sodium concentrations lower than 134 mmol/L, the cutoff they were using at that time.
Hyponatremia is sometimes merely a laboratory artifact or a result of improper blood collection. If real, it can be due to excessive water intake or, most often, the inability of the kidney to excrete water coupled with continued water intake. Patients with significant underlying cardiac, hepatic, or renal dysfunction are at greatest risk of developing hyponatremia, secondary to the nonosmotic release of antidiuretic hormone (ADH). Others at risk include postoperative patients (especially menstruating women), older patients on thiazide diuretics, patients with malignant or psychiatric illness, and endurance athletes.
In this article, we review the treatment of acute and chronic hyponatremia, emphasizing the importance of basing the therapy on the severity of symptoms and taking care not to raise the serum sodium level too rapidly, which can cause neurologic dysfunction.
Guidelines for managing hyponatremia2 are based mostly on retrospective studies and expert opinion, since few prospective studies have been done. Despite the paucity of evidence-based recommendations, we will attempt to incorporate findings from important human and animal studies and consensus guidelines from expert panels. We will focus initially on the critical diagnostic considerations necessary to initiate treatment.
SYMPTOMATIC VS ASYMPTOMATIC
Subsequent sections will address therapeutic approaches in two clinical settings:
Symptomatic hyponatremia, ie, with severe signs or symptoms of cerebral edema—a medical emergency; and
Asymptomatic hyponatremia, ie, without serious signs or symptoms of cerebral edema.
KEY DIAGNOSTIC STEPS WHEN STARTING TREATMENT
The treatment of hyponatremia begins by confirming a truly hypo-osmolar state, assessing its clinical significance, and determining its cause (Table 1).
The clinical and laboratory evaluations form the foundation of a proper approach to any patient with hyponatremia. The rationale behind making several important diagnostic distinctions will be discussed here briefly and then expanded on in the remaining text. The reader is referred to another review on the diagnostic evaluation of hyponatremia.3
Confirm that the patient truly has hypo-osmolar hyponatremia
The serum osmolality should be measured to confirm that it is low (ie, < 275 mOsm/kg). In addition, the arterial serum sodium concentration can be measured using a blood gas device if pseudohyponatremia (see below) is suspected. This method uses direct potentiometry and bypasses the dilutional step in the processing of venous samples.4
Rationale. The clinical consequences of hyponatremia are due to water moving from hypo-osmolar extracellular fluid into the relatively hyperosmolar interior of the cell. This water movement can cause progressive cerebral edema, resulting in a spectrum of signs and symptoms from headache and ataxia to seizures and coma. But significant fluid shifts and cerebral edema occur only if the extracellular fluid is hypo-osmolar relative to the intracellular fluid.
In fact, hyponatremia can occur in several situations in which the extracellular fluid is not hypo-osmolar. An increase in effective plasma osmoles (substances in the extracellular fluid that do not readily move across the plasma membrane) can cause water to move out of cells, resulting in translocational hyponatremia. This may be seen in hyperglycemia or when mannitol or contrast dye has been given. In these situations, the plasma is either isotonic or even hypertonic to the intracellular fluid, resulting in no movement of water into the cells and therefore no clinical consequences relating to the hyponatremia. Importantly, no therapy is required for the hyponatremia.
Other situations in which hyponatremia is present but not associated with true hypotonicity include states of excess protein or lipid in the blood (pseudohyponatremia). Also, if an infusion of hypotonic fluid is running, clinicians must be sure that blood samples are not drawn proximally in the same vein.
Are there significant signs or symptoms of cerebral edema?
Hyponatremia can cause brain swelling within the confined space of the skull as water shifts from the extracellular fluid into the cells. Depending on underlying risk factors (Table 2)5 and the severity and duration of hyponatremia (see below), this may result in signs or symptoms of cerebral edema, including visual changes, focal neurologic changes, encephalopathy, respiratory depression, and seizures. Ultimately, brain herniation can occur.
Patients need to be assessed quickly because those with serious neurologic signs or symptoms thought to be related to hyponatremia require urgent treatment with hypertonic saline to increase the serum sodium concentration, regardless of the underlying volume status, the cause of hyponatremia, or the time of onset.
Determine the duration of hyponatremia
One should try to ascertain when the hyponatremia started, as its duration is important in determining the proper pace of correction.
Figure 1.
The brain begins to adapt to hyponatremia within minutes, and the cerebral adaptation is maximal within 2 to 3 days (Figure 1).6
At the onset of hyponatremia, water moves from the extracellular fluid into cells, pulled in by osmosis. The brain can decrease the net amount of water entering into the neurons (and thus regulate its volume) by increasing the flow of water from the interstitium into the cerebrospinal fluid via increased interstitial hydraulic pressure.7
Over the next several days, inorganic solutes (eg, potassium and sodium salts) and various organic solutes are transported out of the cells. In patients in whom this process has had time to occur, treatment of hyponatremia with hypertonic fluids raises the plasma osmolality faster than the cells can recapture the previously transported osmoles. In this situation, overly rapid correction can cause excessive loss of intracellular water, resulting in cell shrinkage and osmotic demyelination syndrome. Osmotic demyelination usually presents during treatment of hyponatremia after an initial improvement in mental status, with worsening neurologic function and various neurologic signs, including paresis and ultimately even death.6
In patients with acute-onset hyponatremia (ie, with onset within the past 48 hours), in whom the above cerebral adaptations have not had time to occur completely, rapid correction is unlikely to result in osmotic demyelination.
In view of the serious risk of osmotic demyelination, if the timing of development of hyponatremia cannot be determined, one should assume it is chronic (> 48 hours) and avoid rapid overcorrection (see discussion below on the rate of correction).
On the other hand, patients who have severe neurologic signs or symptoms initially need their serum sodium increased urgently to safer levels, regardless of the timing of onset (see below for suggested approach). Subsequent treatment of hyponatremia—after the serum sodium level has been raised enough to reverse neurologic symptoms—will be influenced by the duration of the hyponatremia, with careful avoidance of overly rapid correction, especially in patients with chronic hyponatremia.
Assess the patient’s volume status to determine the proper initial treatment
In patients with hypo-osmolar hyponatremia who do not need urgent therapy with hypertonic saline, the initial treatment is based on clinical and laboratory assessment of extracellular fluid volume status, including spot urine sodium measurement (Table 3).3 This will be discussed further below.
Check urine osmolality to assess for hyponatremic states in which urinary dilution is intact
Measuring urine osmolality is useful in ascertaining whether hyponatremic patients are making appropriately dilute urine (< 100 mOsm/kg). If they are, the cause of the hyponatremia may be excessive water intake, a reset osmostat, or low solute intake. In addition, patients with hypovolemic hyponatremia may have appropriately dilute urine soon after treatment with isotonic intravenous fluids.
The serum sodium concentration often returns to normal if the underlying cause is eliminated (eg, if excessive fluid intake is stopped). If there are no serious signs or symptoms, this can usually be accomplished without additional therapy with intravenous fluids or medications, thereby avoiding the risk of overcorrection.
Search for causes of rapidly correctable hyponatremia
If hyponatremia is due to one of several important underlying causes, it may reverse rapidly once the underlying cause has been eliminated (Table 4). Examples: restricting water intake in patients with psychogenic polydipsia, discontinuing thiazide diuretics, replenishing depleted fluid volume, stopping desmopressin (DDAVP), and giving glucocorticoid replacement to those who are glucocorticoid-deficient.
TREATING HYPONATREMIC PATIENTS WITH SERIOUS SIGNS OR SYMPTOMS
Patients with hypo-osmolar hyponatremia and serious signs or symptoms of cerebral edema (lethargy, respiratory depression, seizures) need rapid initial correction of the serum sodium level, as this is a true medical emergency.
Certain patients are at greater risk of developing cerebral edema from hyponatremia (Table 2).
On the other hand, patients with chronic hyponatremia are very unlikely to present with signs or symptoms of cerebral edema. In fact, in a patient with chronic hyponatremia, care must be taken to avoid overcorrection beyond that needed to reverse severe signs and symptoms. In the rare case in which a patient with chronic hyponatremia presents with signs or symptoms of cerebral edema, the hypertonic saline infusion must be stopped as soon as the signs or symptoms have resolved. Further rapid changes in serum sodium must be avoided.
During correction of hyponatremia, some patients are at particularly high risk of osmotic demyelination syndrome secondary to underlying abnormalities in cerebral osmotic regulation. These include patients with alcoholism, malnutrition, hypokalemia, and burns, and elderly women on thiazide diuretics.8 These patients should be monitored vigilantly for overly rapid correction during treatment.
INITIAL TREATMENT: REVERSE CEREBRAL EDEMA WITH 3% SALINE
The goal of the initial, rapid phase of correction is to reverse cerebral edema.
Patients with serious signs or symptoms should receive hypertonic (3%) saline at a rate of about 1 mL/kg/hour for the first several hours.8 Those with concomitant hypervolemia (as in congestive heart failure) or underlying cardiovascular disease should also receive a loop diuretic to aid in free-water excretion and to prevent volume overload from the saline infusion. This regimen usually raises the serum sodium concentration enough (usually by about 1 mmol/L/hour) to decrease cerebral edema and improve symptoms.
In patients having active seizures or showing signs of brain herniation, 3% saline can be given initially at a higher rate of about 2 to 3 mL/kg/hour over the first few hours. An alternative approach is an initial 50-mL bolus of 3% saline and an additional 200 mL given over the subsequent 4 to 6 hours.9
No study has compared the efficacy and safety of these approaches, and clinicians should always monitor extracellular fluid volume status, neurologic status, and serum sodium levels closely in any patient treated with hypertonic saline.
After severe signs and symptoms have resolved, 3% saline is promptly discontinued and appropriate therapy is initiated based on the patient’s volume status and underlying cause of hyponatremia (see discussion below).
NEXT, FIND THE APPROPRIATE RATE OF CORRECTION
After the initial serious signs or symptoms have been addressed with hypertonic saline, management should focus on limiting the rate of correction in patients with chronic hyponatremia or hyponatremia of unknown duration.
Animal studies and retrospective human studies have suggested certain guidelines on the appropriate pace and magnitude of correction during treatment of hyponatremia to avoid osmotic demyelination syndrome.2
Clinicians must not attempt to correct the serum sodium to “normal” values. Although patients with acute hyponatremia may tolerate complete correction, there is little evidence that raising the serum sodium concentration acutely by more than 5 to 8 mmol/L is advantageous. Therefore, correction should be judicious in all patients.
Appropriate rates of correction
A recent expert consensus panel suggested that the serum sodium level be raised by no more than 10 to 12 mmol/L during the first 24 hours of treatment, and by less than 18 mmol/L over 48 hours.2
Patients with chronic hyponatremia and signs or symptoms of cerebral edema should have their sodium level raised at an even slower rate—some recommend less than 10 mmol/L in the first 24 hours.10 Aggressive initial correction at the rate of 1.5 to 2 mmol/hour for the first 3 to 4 hours with 3% saline is indicated until serious symptoms (seizure, obtundation) resolve, but correction beyond 10 to 12 mmol/L in the first 24 hours should be avoided. Hypertonic saline therapy should usually be discontinued well before the serum sodium level has risen this much, to avoid a continuing rise in the sodium level after the infusion has stopped.
While hypertonic saline is being infused, serum sodium levels should be checked every 1 to 2 hours. In a study in 56 patients with severe hyponatremia (serum sodium ≤ 105 mmol/L),11 no neurologic complications were observed in patients with chronic hyponatremia whose serum sodium was corrected by less than 12 mmol/L in 24 hours or by less than 18 mmol/L in 48 hours or in whom the average rate of correction to a serum sodium of 120 mmol/L was less than or equal to 0.55 mmol/L per hour.
If the serum sodium concentration has been overcorrected
Desmopressin is effective in preventing and reversing inadvertent overcorrection of hyponatremia. 12 In one study, desmopressin lowered the sodium concentration by 2 to 9 mmol/L in 14 of 20 patients. None of the patients developed any serious adverse consequences.
In addition, intravenous water (dextrose 5%) can be given alone or in combination with desmopressin to prevent or reverse an excessive increase in serum sodium.13 Such therapy may be considered in patients who continue to excrete hypotonic urine and have already reached a serum sodium concentration that meets or exceeds the recommended rate or magnitude of change.
Formulas for estimating the rate of correction
Various formulas have been devised for estimating the change in serum sodium concentration during treatment of hyponatremia.14
The Adrogué-Madias formula, one of the most commonly used, gives an estimate of how much the serum sodium concentration will rise when 1 L of various intravenous fluids is given (Table 5).15 This formula also accounts for the increase in serum sodium that takes place during concomitant correction of hypokalemia with potassium. Recently, however, a retrospective study16 found that this formula underestimated the change in serum sodium in 23 (74.2%) of 31 patients with hyponatremia treated with hypertonic saline.
An alternative is the Barsoum-Levine equation, which takes into account ongoing urinary losses. Although it is more cumbersome to calculate, it may be more precise.17
Alternatively, in patients without hypovolemia, the clinician can calculate the amount of urinary excretion of free water required to achieve a specific target serum sodium and then measure hourly urinary water excretion during aquaresis induced by furosemide (Lasix).8 Although more physiologic, this method can be clinically cumbersome, requiring timely handling of urine specimens, accurate recording of urine output, and rapid reporting of laboratory results.
Ultimately, these methods serve only as estimates of the change in serum sodium and do not replace careful monitoring of electrolytes (every 1 to 2 hours during acute therapy) and fastidious assessment for clinical signs or symptoms of osmotic demyelination syndrome.
PATIENTS WITH HYPONATREMIA AND NO SERIOUS SIGNS OR SYMPTOMS
General approach
Hyponatremic patients without serious signs or symptoms of cerebral edema do not require urgent therapy to raise the serum sodium.
Patients with chronic asymptomatic hyponatremia are commonly encountered in clinical practice. As a result of cerebral adaptation, they can appear to have no symptoms despite serum sodium levels as low as 115 to 120 mmol/L. However, even if they have no serious signs or symptoms of cerebral edema, some patients may complain of fatigue, lethargy, nausea, gait abnormalities, and muscle cramps and have evidence of milder forms of neurocognitive impairment.18
In a recent case-control study,18 elderly patients with chronic hyponatremia (mean serum sodium concentration 126 ± 5 mmol/L) were more likely to present to the hospital with falls compared with age-matched controls. Further analysis suggested these patients had marked impairments in gait and attention, which improved in some as the serum sodium increased.
Another recent study19 reported that mild hyponatremia (mean serum sodium concentration 132 mmol/L) was independently associated with the risk of fracture, even after adjustment for known osteoporotic risk factors.
Even when there is no need for acute therapy to raise the serum sodium level, the clinician should scrutinize the medical regimen and available clinical data to rule out reversible causes of water excess. These may include ongoing administration of hypotonic fluids (eg, parenteral nutrition or dextrose 5% to “keep the vein open”) or of medications that cause inappropriate release of ADH (eg, selective serotonin reuptake inhibitors) or that impair water excretion (eg, nonsteroidal anti-inflammatory drugs). The clinician should also search for an underlying diagnosis that predisposes to water retention, such as hypothyroidism, adrenal insufficiency, congestive heart failure, or hepatic or renal failure. If hyponatremia is due to endocrine disease, correction of hypothyroidism or adrenal insufficiency should result in water excretion and improvement in the serum sodium.
If the cause of the hyponatremia is not immediately apparent, treatment can be started on the basis of assessment of the patient’s extracellular fluid volume status using clinical examination and supplementary laboratory data such as the serum uric acid concentration and urinary sodium concentration.3Table 3 outlines general treatment options for hypoosmolar hyponatremia according to extracellular fluid volume status.
Of note, physical examination alone has poor sensitivity and specificity in assessing extracellular fluid volume status in patients with hyponatremia.20,21 This highlights the importance of spot measurements of urine sodium and serum uric acid and, when appropriate, isotonic intravenous saline challenge to detect occult hypovolemia.
In general, patients with euvolemia are treated with fluid restriction, and patients with hypovolemia are given isotonic saline. Patients with hypervolemia can be difficult to treat, but in general they are prescribed both sodium and fluid restriction. Loop diuretics can be given to promote excretion of water and sodium. Thiazide diuretics are avoided, as they impair urinary dilution and worsen hyponatremia. Attempts should be made to optimize the treatment of the underlying hypervolemic disorder (congestive heart failure, cirrhosis, advanced renal failure). Vasopressin receptor antagonists can also be used in selected cases of hypervolemic or euvolemic hyponatremia (see discussion below).
How to prescribe fluid restriction rationally
Ideally, patients should not ingest any more fluid than they can excrete in urine and insensible losses—otherwise, the serum sodium can continue to decrease.
Water excretion can be estimated from solute intake and urine osmolarity. In theory, a 70-kg person with a typical daily solute intake of about 10 mOsm/kg and intact urinary dilution to a urine osmolarity of 50 mOsm/L can excrete up to 14 L of urine (700 mOsm/50 mOsm/L) per day. However, a patient with the syndrome of inappropriate ADH secretion (SIADH) and a fixed urine osmolality of 700 mOsm/kg would excrete a similar solute load in only 1 L of urine. Thus, any fluid intake in excess of this volume could worsen hyponatremia.
To excrete free water, urinary sodium plus urinary potassium must be less than the serum sodium concentration. In this regard, the necessary degree of fluid restriction can also be estimated made on the basis of the patient’s urinary electrolytes.22
Increased solute intake to augment water excretion
In patients without hypervolemia, solute intake can be increased to augment water excretion. 22 This can be achieved with salt tablets or oral urea. Although urea can be effective, it is not commonly used because it is not available the United States and it has poor gastrointestinal tolerability. In patients whose nutritional intake is limited and who continue to ingest fluids (such as, for example, an elderly patient subsisting on tea and toast) every effort should be made to increase solute intake with high-protein foods or supplements.
DRUGS TO INHIBIT VASOPRESSIN
Unfortunately, patients often do not adhere to these strategies, as fluid restriction and unpalatable salt tablets or urea can become too burdensome. In such instances, pharmacologic inhibition of vasopressin-mediated water reabsorption can be considered using the following agents.
Demeclocycline (Declomycin) and lithium inhibit the kidney’s response to vasopressin. Because lithium may be nephrotoxic and has unwanted effects on the central nervous system, demeclocycline has become the preferred agent. Given in doses of 300 to 600 mg twice daily, demeclocycline promotes free water excretion, but often takes 1 to 2 weeks of therapy to begin working.
Renal failure due to demeclocycline has been reported in patients with concomitant liver disease.23 Demeclocycline can also cause photosensitivity and is contraindicated in children and pregnant women due to abnormalities in bone and enamel formation. In addition, it can be expensive and may not be covered fully by some prescription plans.
Vasopressin receptor antagonists (‘vaptans’)
ADH, also called vasopressin, interacts with various receptor subtypes, including V1a (causing vasoconstriction, platelet aggregation, inotropic stimulation, myocardial protein synthesis), V1b (causing secretion of adrenocorticotropic hormone), and V2 (causing water reabsorption and release of von Willebrand factor and factor VIII).
Drugs that block V2 receptors in the renal tubule increase water excretion, making them attractive as therapy for some hyponatremic states (Table 6).24,25 These drugs exert their aquaretic effect by causing a decrease in transcription and insertion of aquaporin-2 channels (“water pores”) into the apical collecting duct membrane. As a result, the water permeability of the collecting duct is decreased even in the presence of circulating ADH.
Conivaptan (Vaprisol) is a combined V1a-V2 antagonist that has been approved for the treatment of euvolemic and hypervolemic hyponatremia. Conivaptan inhibits the cytochrome P450 3A4 system and thus may interact with other drugs; therefore, its use has been limited to no more than 4 days of intravenous administration in the hospital setting. The recommended dosage is an initial 20-mg infusion over 30 minutes, followed by continuous infusions of 20 to 40 mg/day. Dosing adjustments in renal and hepatic impairment have not been well defined.
Tolvaptan (Samsca) is an oral selective V2 antagonist that has been studied in patients with euvolemic and hypervolemic hyponatremia. 26 Studies have included patients with congestive heart failure, cirrhosis, and SIADH. Although tolvaptan has not been shown to reduce rates of rehospitalization or death in congestive heart failure, it improves serum sodium, overall fluid balance, and congestive symptoms.27 Tolvaptan has recently been approved for the treatment of euvolemic and hypervolemic hyponatremia.
A recent study has confirmed the longterm efficacy of tolvaptan in 111 patients over a mean duration of treatment greater than 700 days.28 While the clinical benefits of chronic tolvaptan therapy have yet to be clearly demonstrated, this study shows that tolvaptan therapy can result in a sustained improvement in serum sodium concentration without an unacceptable increase in adverse events.29
Lixivaptan (VPA-985), another oral selective V2 receptor antagonist, is being studied in patients with euvolemic and hypervolemic hyponatremia.
Current role of vasopressin antagonists
Current studies of vasopressin antagonists in the treatment of hyponatremia are promising, though definite recommendations are needed to ensure slow, careful correction of hyponatremia. Most studies suggest that these agents provide slow, reliable increases in serum sodium. In one large study of patients with congestive heart failure, serum sodium rose by more than 12 mmol/L in 24 hours in fewer than 2% of patients.26
Notably, no cases of osmotic demyelination syndrome have been reported in these studies. However, it should be noted that therapy was started in the hospital with close monitoring of serum sodium levels and discontinuation of fluid restriction; the incidence of overly rapid correction of sodium may be higher outside of carefully done clinical studies. Clinicians should adopt monitoring strategies similar to those used in these rigorous studies.
At present, there is little experience with vasopressin antagonists in hyponatremic patients with serious signs or symptoms of cerebral edema, and most clinicians still view 3% saline as the gold standard for these patients.
Vasopressin antagonists should not be used in patients with hypovolemic hyponatremia, due to concerns about V1a blockade causing hypotension and about V2 blockade producing water excretion and a worsening of the volume-depleted state.
Recent clinical trials have reported that patients often experience increased thirst while taking these agents. This highlights the need to monitor serum sodium during treatment.
These agents are expensive. Tolvaptan costs about $250 per tablet; conivaptan, which is administered intravenously, may cost a little more per treatment course.
THERAPY IN SPECIFIC DISEASE STATES
Patients with hyponatremia and cirrhosis
The focus of treatment remains water and salt restriction and judicious use of loop diuretics and aldosterone antagonists such as spironolactone (Aldactone).
Tolvaptan has been effective at raising the serum sodium level in patients with cirrhosis, 26 while conivaptan should be avoided at present because of vasodilation from V1a receptor antagonism and its potential effects on systemic hemodynamics and risk of variceal bleeding.30
As the severity of cirrhosis increases, the only effective treatment of hyponatremia is liver transplantation.
Patients with SIADH
In most cases, water restriction is the mainstay of therapy. Adequate nutritional intake should also be stressed so that enough solute is available for ongoing water excretion. Although fluid restriction is usually effective, many patients cannot adhere to the level of restriction required.
In cases in which fluid restriction is not effective on its own, demeclocyline can be used to antagonize ADH action and increase water excretion. Sodium tablets and loop diuretics can also be used, taking care to avoid hypovolemia from diuretic-induced sodium losses. The use of tolvaptan in patients with SIADH has resulted in short-term increases in serum sodium.26 A recent study has suggested that this effect can be sustained with longer-term treatment,28 but further studies are needed to show a complementary clinical benefit (eg, improved neurocognition) to guide the use of these costly agents in clinical practice.
Patients with diuretic-induced hyponatremia
Thiazide diuretics should be discontinued and hypovolemia and hypokalemia should be corrected with isotonic saline and potassium supplementation. As the hypokalemia is corrected and the diuretic effect and hypovolemic stimulus to ADH dissipates, water excretion can increase rapidly, resulting in a brisk change in serum sodium.
Serum sodium levels should be closely monitored during therapy to avoid overcorrection. For this reason, use of hypertonic saline should generally be avoided. Hypotonic fluid— eg, half-normal (0.45%) or quarter-normal (0.22%) saline or even desmopressin—may become necessary in the later stages of therapy to avoid overly rapid correction.
Patients with exercise-associated hyponatremia
Patients at highest risk of exercise-associated hyponatremia include those who drink too much fluid during a long-distance race, who have low body weight, who are female, who exercise longer than 4 hours, and who use nonsteroidal anti-inflammatory drugs.31 The cause of hyponatremia is likely multifactorial, with excessive water intake coupled with sodium losses and impaired renal excretion of water due to ADH action and impaired renal dilution. To prevent exercise-associated hyponatremia, fluid intake should be limited to 400 to 800 mL/hour, with the higher end recommended for larger athletes and hotter climates.
Consensus recommendations suggest that most patients with mild hyponatremia (serum sodium 130 to 135 mmol/L) should be treated with fluid restriction and clinical observation, as spontaneous water diuresis leads to improvement in the serum sodium level. Importantly, the reflex to provide isotonic saline infusions should be avoided unless clear signs of volume depletion are present. Intravenous saline has the potential to worsen hyponatremia in the presence of ADH. In addition, some athletes will have retained water in the gastrointestinal tract that may be mobilized after the race, resulting in worsening of hyponatremia.32
In athletes with severe hyponatremia (serum sodium < 120 mmol/L) or symptomatic exercise-associated hyponatremia (lethargy, respiratory depression, seizures), hypertonic saline is the treatment of choice. One protocol suggests giving 100 mL of 3% saline over 10 minutes in the field, followed by prompt transportation to hospital.33
SUMMARY POINTS
Hyponatremia is a common electrolyte disorder that in its most severe form requires urgent therapy with hypertonic saline to correct cerebral edema.
In patients without serious signs or symptoms of cerebral edema, recent observations suggest there may be clinically important symptomatology relating to mild neurocognitive dysfunction and an association with risk of bone fracture.
Multiple treatment strategies are available according to the underlying extracellular fluid volume status and cause of hyponatremia. These include fluid and sodium restriction and augmentation of urinary water excretion with various nutritional and pharmacologic strategies. The most novel therapy includes antagonism of the vasopressin V2 receptor with a class of aquaretic agents known as vaptans.
There can be serious neurologic injury associated with overly rapid correction of chronic hyponatremia or undercorrection of acute symptomatic hyponatremia.
Clinicians must be familiar with the details of each of the treatments and have an appreciation of the importance of careful monitoring during treatment.
References
Flear CTG, Gill GV, Burn J. Hyponatremia: mechanisms and management. Lancet1981; 2:26–31.
Freda BJ, Davidson MB, Hall PM. Evaluation of hyponatremia: a little physiology goes a long way. Cleve Clin J Med2004; 71:639–650.
Weisberg LS. Pseudohyponatremia: a reappraisal. Am J Med1989; 86:315–318.
Moritz L, Ayus JC. The pathophysiology and treatment of hyponatraemic encephalopathy: an update. Nephrol Dial Transplant2003; 18:2486–2491.
Widdess-Walsh P, Sabharwal V, Demirjian S, DeGeorgia M. Neurologic effects of hyponatremia and its treatment. Cleve Clin J Med2007; 74:377–383.
Melton JE, Patlak CS, Pettigrew KD, Cserr HF. Volume regulatory loss of Na, Cl, and K from rat brain during acute hyponatremia. Am J Physiol1987; 252:F661–F669.
Lauriat SM, Berl T. The hyponatremic patient: practical focus on therapy. J Am Soc Nephrol1997; 8:1599–1607.
Kokko JP. Symptomatic hyponatremia with hypoxia is a medical emergency. Kidney Int2006; 69:1291–1293.
Ellis SJ. Severe hyponatraemia: complications and treatment. QJM1995; 88:905–909.
Sterns RH, Cappuccio JD, Silver SM, Cohen EP. Neurologic sequelae after treatment of severe hyponatremia: a multicenter perspective. J Am Soc Nephrol1994; 4:1522–1530.
Perianayagam A, Sterns RH, Silver SM, et al. DDAVP is effective in preventing and reversing inadvertent overcorrection of hyponatremia. Clin J Am Soc Nephrol2008; 3:331–336.
Sterns RH, Hix JK. Overcorrection of hyponatremia is a medical emergency. Kidney Int2009; 76:587–589.
Nguyen MK, Kurtz I. Analysis of current formulas used for treatment of the dysnatremias. Clin Exp Nephrol2004; 8:12–16.
Adrogué HJ, Madias NE. Hyponatremia. N Engl J Med2000; 342:1581–1589.
Mohmand HK, Issa D, Ahmad Z, Cappuccio JD, Kouides RW, Sterns RH. Hypertonic saline for hyponatremia: risk of inadvertent overcorrection. Clin J Am Soc Nephrol2007; 2:1110–1117.
Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med2007; 356:2064–2072.
Renneboog B, Musch W, Vandemergel X, Manto MU, Decaux G. Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits. Am J Med2006; 119:71.e1–e8.
Kinsella S, Moran S, Sullivan MO, Molloy MG, Eustace JA. Hyponatremia independent of osteoporosis is associated with fracture occurrence. Clin J Am Soc Nephrol2010; 5:275–280.
Chung HM, Kluge R, Schrier RW, Anderson RJ. Clinical assessment of extracellular fluid volume in hyponatremia. Am J Med1987; 83:905–988.
Hoorn EJ, Halperin ML, Zietse R. Diagnostic approach to a patient with hyponatraemia: traditional versus physiology-based options. QJM2005; 98:529–540.
Berl T. Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol2008; 19:1076–1078.
Carrilho F, Bosch J, Arroyo V, Mas A, Viver J, Rodes J. Renal failure associated with demeclocycline in cirrhosis. Ann Intern Med1977; 87:195–197.
Lehrich RW, Greenberg A. When is it appropriate to use vasopressin receptor antagonists?J Am Soc Nephrol2008; 19:1054–1058.
Decaux G, Soupart A, Vassart G. Non-peptide arginine-vasopressin antagonists: the vaptans. Lancet2008; 371:1624–1632.
Schrier RW, Gross P, Gheorghiade M, et al; SALT Investigators. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med2006; 355:2099–2112.
Konstam MA, Gheorghiade M, Burnett JC, et al; Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA2007; 297:1319–1331.
Berl T, Quittnat-Pelletier F, Verbalis JG, et al; SALTWATER Investigators. Oral tolvaptan is safe and effective in chronic hyponatremia. J Am Soc Nephrol2010; 21:705–712.
Greenberg A, Lehrich RW. Treatment of chronic hyponatremia: now we know how, but do we know when or if?J Am Soc Nephrol2010; 21:552–555.
Greenberg A, Verbalis JG. Vasopressin receptor antagonists. Kidney Int2006; 69:2124–2130.
Rosner MH, Kirven J. Exercise-associated hyponatremia. Clin J Am Soc Nephrol2007; 2:151–161.
Halperin ML, Kamel KS, Sterns R. Hyponatremia in marathon runners. N Engl J Med2005; 353:427–428.
Hew-Butler T, Almond C, Ayus JC, et al; Exercise-Associated Hyponatremia (EAH) Consensus Panel. Consensus statement of the 1st International Exercise-Associated Hyponatremia Consensus Development Conference, Cape Town, South Africa 2005. Clin J Sport Med2005; 15:208–213.
References
Flear CTG, Gill GV, Burn J. Hyponatremia: mechanisms and management. Lancet1981; 2:26–31.
Freda BJ, Davidson MB, Hall PM. Evaluation of hyponatremia: a little physiology goes a long way. Cleve Clin J Med2004; 71:639–650.
Weisberg LS. Pseudohyponatremia: a reappraisal. Am J Med1989; 86:315–318.
Moritz L, Ayus JC. The pathophysiology and treatment of hyponatraemic encephalopathy: an update. Nephrol Dial Transplant2003; 18:2486–2491.
Widdess-Walsh P, Sabharwal V, Demirjian S, DeGeorgia M. Neurologic effects of hyponatremia and its treatment. Cleve Clin J Med2007; 74:377–383.
Melton JE, Patlak CS, Pettigrew KD, Cserr HF. Volume regulatory loss of Na, Cl, and K from rat brain during acute hyponatremia. Am J Physiol1987; 252:F661–F669.
Lauriat SM, Berl T. The hyponatremic patient: practical focus on therapy. J Am Soc Nephrol1997; 8:1599–1607.
Kokko JP. Symptomatic hyponatremia with hypoxia is a medical emergency. Kidney Int2006; 69:1291–1293.
Ellis SJ. Severe hyponatraemia: complications and treatment. QJM1995; 88:905–909.
Sterns RH, Cappuccio JD, Silver SM, Cohen EP. Neurologic sequelae after treatment of severe hyponatremia: a multicenter perspective. J Am Soc Nephrol1994; 4:1522–1530.
Perianayagam A, Sterns RH, Silver SM, et al. DDAVP is effective in preventing and reversing inadvertent overcorrection of hyponatremia. Clin J Am Soc Nephrol2008; 3:331–336.
Sterns RH, Hix JK. Overcorrection of hyponatremia is a medical emergency. Kidney Int2009; 76:587–589.
Nguyen MK, Kurtz I. Analysis of current formulas used for treatment of the dysnatremias. Clin Exp Nephrol2004; 8:12–16.
Adrogué HJ, Madias NE. Hyponatremia. N Engl J Med2000; 342:1581–1589.
Mohmand HK, Issa D, Ahmad Z, Cappuccio JD, Kouides RW, Sterns RH. Hypertonic saline for hyponatremia: risk of inadvertent overcorrection. Clin J Am Soc Nephrol2007; 2:1110–1117.
Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med2007; 356:2064–2072.
Renneboog B, Musch W, Vandemergel X, Manto MU, Decaux G. Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits. Am J Med2006; 119:71.e1–e8.
Kinsella S, Moran S, Sullivan MO, Molloy MG, Eustace JA. Hyponatremia independent of osteoporosis is associated with fracture occurrence. Clin J Am Soc Nephrol2010; 5:275–280.
Chung HM, Kluge R, Schrier RW, Anderson RJ. Clinical assessment of extracellular fluid volume in hyponatremia. Am J Med1987; 83:905–988.
Hoorn EJ, Halperin ML, Zietse R. Diagnostic approach to a patient with hyponatraemia: traditional versus physiology-based options. QJM2005; 98:529–540.
Berl T. Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol2008; 19:1076–1078.
Carrilho F, Bosch J, Arroyo V, Mas A, Viver J, Rodes J. Renal failure associated with demeclocycline in cirrhosis. Ann Intern Med1977; 87:195–197.
Lehrich RW, Greenberg A. When is it appropriate to use vasopressin receptor antagonists?J Am Soc Nephrol2008; 19:1054–1058.
Decaux G, Soupart A, Vassart G. Non-peptide arginine-vasopressin antagonists: the vaptans. Lancet2008; 371:1624–1632.
Schrier RW, Gross P, Gheorghiade M, et al; SALT Investigators. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med2006; 355:2099–2112.
Konstam MA, Gheorghiade M, Burnett JC, et al; Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA2007; 297:1319–1331.
Berl T, Quittnat-Pelletier F, Verbalis JG, et al; SALTWATER Investigators. Oral tolvaptan is safe and effective in chronic hyponatremia. J Am Soc Nephrol2010; 21:705–712.
Greenberg A, Lehrich RW. Treatment of chronic hyponatremia: now we know how, but do we know when or if?J Am Soc Nephrol2010; 21:552–555.
Greenberg A, Verbalis JG. Vasopressin receptor antagonists. Kidney Int2006; 69:2124–2130.
Rosner MH, Kirven J. Exercise-associated hyponatremia. Clin J Am Soc Nephrol2007; 2:151–161.
Halperin ML, Kamel KS, Sterns R. Hyponatremia in marathon runners. N Engl J Med2005; 353:427–428.
Hew-Butler T, Almond C, Ayus JC, et al; Exercise-Associated Hyponatremia (EAH) Consensus Panel. Consensus statement of the 1st International Exercise-Associated Hyponatremia Consensus Development Conference, Cape Town, South Africa 2005. Clin J Sport Med2005; 15:208–213.
Some hyponatremic patients present with acute, life-threatening cerebral edema due to severe hyponatremia. In others, the hyponatremia may be chronic and less severe, causing relatively few symptoms, but representing an important, independent marker of poor prognosis due to an underlying disease (eg, heart failure).
Even patients with chronic, less severe hyponatremia may have subtle symptoms of neurocognitive dysfunction and a higher risk of bone fractures.
Overly rapid correction of chronic hyponatremia or undercorrection of acute symptomatic hyponatremia can lead to serious neurologic injury.
Treatment strategies vary depending on the extracellular fluid volume status and the cause of hyponatremia.
Vasopressin antagonists (“vaptans”), a new class of aquaretic agents, specifically target the mechanism driving hyponatremia in some patients.