User login
Gene-based, rational drug-dosing: An evolving, complex opportunity

For some drugs there is a key step in metabolism, often in a rate-limiting pathway, with an enzyme that has known and detectable polymorphisms that differ dramatically in their ability to affect the drug’s degradation. In theory, by determining the patient’s specific genotype ahead of time, the initial dose of the drug can be determined more rationally. In this issue of the Journal, Kitzmiller et al describe several drugs for which this may be true.
However, for this approach to be practical and cost-effective, several conditions should be met. The drug must be one that needs to be dosed to its therapeutic level rapidly: if there is time to titrate slowly, then there is little need for the extra expense associated with genotyping in order to titrate it more rapidly. Also, it should be proven that dosing based on advance knowledge of the genotype of the target actually results in safer or more efficacious dosing.
For carbamazepine (Tegretol, Equetro) and allopurinol (Zyloprim), specific human leukocyte antigen haplotypes are associated with a strikingly increased frequency of serious hypersensitivity reactions. In some patients, these should be checked before giving the drug.
But the concept of pharmacogenomics is broad, and it may yet explain many vagaries of drug-responsiveness in individual patients. Polymorphisms in renal anion transporters may dictate the level of anionic drugs. Drug-receptor polymorphisms may determine the affinity of a drug for its target and, hence, its efficacy. Cell-membrane transporters, which may have functionally different stable alleles or polymorphisms, may regulate intracellular drug levels by pumping the drug into or out of cells with different efficiencies.
As the entire human genome is dissected and analyzed, and as more and more genes (with their polymorphisms) are linked to specific functions readily detectable in specific patients, we will have more opportunities to match the right drug and dose to the right patient. We are not there yet, but that day is coming.
For some drugs there is a key step in metabolism, often in a rate-limiting pathway, with an enzyme that has known and detectable polymorphisms that differ dramatically in their ability to affect the drug’s degradation. In theory, by determining the patient’s specific genotype ahead of time, the initial dose of the drug can be determined more rationally. In this issue of the Journal, Kitzmiller et al describe several drugs for which this may be true.
However, for this approach to be practical and cost-effective, several conditions should be met. The drug must be one that needs to be dosed to its therapeutic level rapidly: if there is time to titrate slowly, then there is little need for the extra expense associated with genotyping in order to titrate it more rapidly. Also, it should be proven that dosing based on advance knowledge of the genotype of the target actually results in safer or more efficacious dosing.
For carbamazepine (Tegretol, Equetro) and allopurinol (Zyloprim), specific human leukocyte antigen haplotypes are associated with a strikingly increased frequency of serious hypersensitivity reactions. In some patients, these should be checked before giving the drug.
But the concept of pharmacogenomics is broad, and it may yet explain many vagaries of drug-responsiveness in individual patients. Polymorphisms in renal anion transporters may dictate the level of anionic drugs. Drug-receptor polymorphisms may determine the affinity of a drug for its target and, hence, its efficacy. Cell-membrane transporters, which may have functionally different stable alleles or polymorphisms, may regulate intracellular drug levels by pumping the drug into or out of cells with different efficiencies.
As the entire human genome is dissected and analyzed, and as more and more genes (with their polymorphisms) are linked to specific functions readily detectable in specific patients, we will have more opportunities to match the right drug and dose to the right patient. We are not there yet, but that day is coming.
For some drugs there is a key step in metabolism, often in a rate-limiting pathway, with an enzyme that has known and detectable polymorphisms that differ dramatically in their ability to affect the drug’s degradation. In theory, by determining the patient’s specific genotype ahead of time, the initial dose of the drug can be determined more rationally. In this issue of the Journal, Kitzmiller et al describe several drugs for which this may be true.
However, for this approach to be practical and cost-effective, several conditions should be met. The drug must be one that needs to be dosed to its therapeutic level rapidly: if there is time to titrate slowly, then there is little need for the extra expense associated with genotyping in order to titrate it more rapidly. Also, it should be proven that dosing based on advance knowledge of the genotype of the target actually results in safer or more efficacious dosing.
For carbamazepine (Tegretol, Equetro) and allopurinol (Zyloprim), specific human leukocyte antigen haplotypes are associated with a strikingly increased frequency of serious hypersensitivity reactions. In some patients, these should be checked before giving the drug.
But the concept of pharmacogenomics is broad, and it may yet explain many vagaries of drug-responsiveness in individual patients. Polymorphisms in renal anion transporters may dictate the level of anionic drugs. Drug-receptor polymorphisms may determine the affinity of a drug for its target and, hence, its efficacy. Cell-membrane transporters, which may have functionally different stable alleles or polymorphisms, may regulate intracellular drug levels by pumping the drug into or out of cells with different efficiencies.
As the entire human genome is dissected and analyzed, and as more and more genes (with their polymorphisms) are linked to specific functions readily detectable in specific patients, we will have more opportunities to match the right drug and dose to the right patient. We are not there yet, but that day is coming.
Pharmacogenomics for the primary care provider: Why should we care?
Since the human genome was sequenced in 2000, the American public has continued to hold hope that our growing understanding of genetics will revolutionize the practice of medicine.
One way genetics promises to improve the quality and value of health care is in personalized medicine, by helping us tailor treatment to a person’s individual genetic makeup. One such approach is called pharmacogenomics.
Pharmacogenomics uses knowledge of a person’s genetics to understand how a particular drug will work, or not work, in his or her body. For instance, some people might carry genes that make them more sensitive than average to a drug, and therefore they would require a lower dose. Others might have genes that make them resistant to the drug, meaning the drug is ineffective unless they receive a higher dose.
Adverse drug reactions are a leading cause of death in hospitalized patients in the United States and are responsible for billions of dollars in health care costs.1,2 Our current practice of prescribing for adult patients is largely trial-and-error, with the same dose given to all patients, in many cases with little regard even to sex, height, or weight.
Pharmacogenomics promises to change this way of prescribing to a customized approach that uses genetic information to predict an individual’s response to medications. It is one piece of an overall initiative to personalize patient care based on the patient’s individual characteristics and preferences.
OVERCOMING BARRIERS TO USING PHARMACOGENOMICS IN PRACTICE
If personalized medicine has promised to improve the quality and value of health care for our patients, why have we been so slow to adopt this information in clinical practice?
The usual barriers to clinical adoption certainly exist. We need further studies to determine whether genetic-based prescribing is truly valid, and for which patient populations. We need to determine whether this approach is cost-effective and better than the current standard of care. We need to work on payment options.
However, one of the largest barriers for busy primary care physicians is the lack of time to keep up with new information. Many practicing physicians were taught little about formal genetics in medical school. The body of scientific literature on pharmacogenomics is fragmented, and it crosses disease states and specialties, making it difficult to unite. Given the breadth of diseases treated and drugs prescribed by primary care physicians, it is unrealistic for most to keep track of the vast body of literature of pharmacogenomic testing and to decipher how to apply this to clinical practice.
In this issue of the Journal, Kitzmiller et al3 provide one solution to this problem, giving an overview of pharmacogenomic applications that might be pertinent to practicing physicians. However, as we try to make pharmacogenomics accessible to busy physicians, we need other solutions to integrate pharmacogenomic information efficiently into the clinical work flow. One approach might be to build pharmacogenomics into the electronic medical record. We can also store the integrated information in research databases and provide clinical recommendations on Internet sites such as www.pharmgkb.org, and we can develop applications to run on cell phones and iPads.
QUESTIONS REMAIN
Kitzmiller et al discuss an important step in this process, highlighting several key questions:
Should we seek genetics-based information to personalize drug selection? Based on the information presented in the literature and in the Kitzmiller paper, there may be circumstances when it is appropriate to consider doing so. While the evidence is not yet compelling to order these tests on a regular basis in clinical practice, this information might be helpful in some situations, such as for patients who have had adverse effects from minimal doses of antidepressants.
For now, clinicians should not abandon their current practice of personalizing patient care on the basis of personal, cultural, and economic preferences. Rather, they should consider pharmacogenomic information an additional piece of information when selecting drug therapy. We should also encourage health care systems and interested providers to be early adopters and to study how their outcomes compare with the standard of care.
Once we have this information, what is our obligation to use it? An increasing number of patients already have genetic information in their health record, either ordered by or provided to their physicians. However, there is little in the scientific literature to guide us in this arena.
Yet most of us would agree that if we have information (genetic or otherwise) that can help to select a drug type or dose or reduce adverse events or costs, we should consider this information in our decision-making. Several circumstances are documented in this paper and in the literature in which prior knowledge about drug metabolism can help in selecting a dose of medication. One example would be the 50% recommended reduction in tricyclic antidepressant dose if the patient is a CYP2D6 poor metabolizer.4
MOVING FORWARD AS A TEAM
In summary, Kitzmiller et al bring to light the promise and the uncertainties that currently exist in the field of pharmacogenomics. While it is unclear if we should incorporate pharmacogenomic tests into standard medical practice at this time, it is clear that this information is becoming more readily available, whether or not we have requested it. Some would argue that, once we have the information, we have an obligation to use it, just as we use other information in our clinical decision-making. This means we need to develop tools and resources to help practitioners evaluate pharmacogenomic data and incorporate it into clinical care in an efficient manner.
The authors also highlight the need for more education about drug metabolism in general, and they cite several instances in which knowledge of drug interactions and metabolism can clearly influence decision-making. An example is paroxetine (Paxil) inhibition of tamoxifen (Nolvadex).5
Lastly, regardless of our personal feelings about the clinical usefulness of genetic testing in large populations, we need to work together to determine clinical utility and validity and to develop efficient ways to put into practice findings that could affect patient care. As we move forward, we need to work as a team, utilizing our clinical partners—pharmacists, pharmacologists, metabolism and health information technology experts, and medical geneticists. Working as a team, pooling our resources and tools, we move closer to providing world-class personalized health care.
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998; 279:1200–1205.
- Field TS, Gilman BH, Subramanian S, Fuller JC, Bates DW, Gurwitz JH. The costs associated with adverse drug events among older adults in the ambulatory setting. Med Care 2005; 43:1171–1176.
- Kitzmiller JP, Groen DK, Phelps MA, Sadee W. Pharmacogenomic testing: relevance in medical practice. Why drugs work in some patients but not others. Cleve Clin J Med 2011; 78:243–257.
- Thuerauf N, Lunkenheimer J. The impact of the CYP2D6-polymorphism on dose recommendations for current antidepressants. Eur Arch Psychiatry Clin Neurosci 2006; 256:287–293.
- Schwarz EB, McNamara M, Miller RG, Walsh JM. Update in women’s health for the general internist. J Gen Intern Med201; 26:207–213.
Since the human genome was sequenced in 2000, the American public has continued to hold hope that our growing understanding of genetics will revolutionize the practice of medicine.
One way genetics promises to improve the quality and value of health care is in personalized medicine, by helping us tailor treatment to a person’s individual genetic makeup. One such approach is called pharmacogenomics.
Pharmacogenomics uses knowledge of a person’s genetics to understand how a particular drug will work, or not work, in his or her body. For instance, some people might carry genes that make them more sensitive than average to a drug, and therefore they would require a lower dose. Others might have genes that make them resistant to the drug, meaning the drug is ineffective unless they receive a higher dose.
Adverse drug reactions are a leading cause of death in hospitalized patients in the United States and are responsible for billions of dollars in health care costs.1,2 Our current practice of prescribing for adult patients is largely trial-and-error, with the same dose given to all patients, in many cases with little regard even to sex, height, or weight.
Pharmacogenomics promises to change this way of prescribing to a customized approach that uses genetic information to predict an individual’s response to medications. It is one piece of an overall initiative to personalize patient care based on the patient’s individual characteristics and preferences.
OVERCOMING BARRIERS TO USING PHARMACOGENOMICS IN PRACTICE
If personalized medicine has promised to improve the quality and value of health care for our patients, why have we been so slow to adopt this information in clinical practice?
The usual barriers to clinical adoption certainly exist. We need further studies to determine whether genetic-based prescribing is truly valid, and for which patient populations. We need to determine whether this approach is cost-effective and better than the current standard of care. We need to work on payment options.
However, one of the largest barriers for busy primary care physicians is the lack of time to keep up with new information. Many practicing physicians were taught little about formal genetics in medical school. The body of scientific literature on pharmacogenomics is fragmented, and it crosses disease states and specialties, making it difficult to unite. Given the breadth of diseases treated and drugs prescribed by primary care physicians, it is unrealistic for most to keep track of the vast body of literature of pharmacogenomic testing and to decipher how to apply this to clinical practice.
In this issue of the Journal, Kitzmiller et al3 provide one solution to this problem, giving an overview of pharmacogenomic applications that might be pertinent to practicing physicians. However, as we try to make pharmacogenomics accessible to busy physicians, we need other solutions to integrate pharmacogenomic information efficiently into the clinical work flow. One approach might be to build pharmacogenomics into the electronic medical record. We can also store the integrated information in research databases and provide clinical recommendations on Internet sites such as www.pharmgkb.org, and we can develop applications to run on cell phones and iPads.
QUESTIONS REMAIN
Kitzmiller et al discuss an important step in this process, highlighting several key questions:
Should we seek genetics-based information to personalize drug selection? Based on the information presented in the literature and in the Kitzmiller paper, there may be circumstances when it is appropriate to consider doing so. While the evidence is not yet compelling to order these tests on a regular basis in clinical practice, this information might be helpful in some situations, such as for patients who have had adverse effects from minimal doses of antidepressants.
For now, clinicians should not abandon their current practice of personalizing patient care on the basis of personal, cultural, and economic preferences. Rather, they should consider pharmacogenomic information an additional piece of information when selecting drug therapy. We should also encourage health care systems and interested providers to be early adopters and to study how their outcomes compare with the standard of care.
Once we have this information, what is our obligation to use it? An increasing number of patients already have genetic information in their health record, either ordered by or provided to their physicians. However, there is little in the scientific literature to guide us in this arena.
Yet most of us would agree that if we have information (genetic or otherwise) that can help to select a drug type or dose or reduce adverse events or costs, we should consider this information in our decision-making. Several circumstances are documented in this paper and in the literature in which prior knowledge about drug metabolism can help in selecting a dose of medication. One example would be the 50% recommended reduction in tricyclic antidepressant dose if the patient is a CYP2D6 poor metabolizer.4
MOVING FORWARD AS A TEAM
In summary, Kitzmiller et al bring to light the promise and the uncertainties that currently exist in the field of pharmacogenomics. While it is unclear if we should incorporate pharmacogenomic tests into standard medical practice at this time, it is clear that this information is becoming more readily available, whether or not we have requested it. Some would argue that, once we have the information, we have an obligation to use it, just as we use other information in our clinical decision-making. This means we need to develop tools and resources to help practitioners evaluate pharmacogenomic data and incorporate it into clinical care in an efficient manner.
The authors also highlight the need for more education about drug metabolism in general, and they cite several instances in which knowledge of drug interactions and metabolism can clearly influence decision-making. An example is paroxetine (Paxil) inhibition of tamoxifen (Nolvadex).5
Lastly, regardless of our personal feelings about the clinical usefulness of genetic testing in large populations, we need to work together to determine clinical utility and validity and to develop efficient ways to put into practice findings that could affect patient care. As we move forward, we need to work as a team, utilizing our clinical partners—pharmacists, pharmacologists, metabolism and health information technology experts, and medical geneticists. Working as a team, pooling our resources and tools, we move closer to providing world-class personalized health care.
Since the human genome was sequenced in 2000, the American public has continued to hold hope that our growing understanding of genetics will revolutionize the practice of medicine.
One way genetics promises to improve the quality and value of health care is in personalized medicine, by helping us tailor treatment to a person’s individual genetic makeup. One such approach is called pharmacogenomics.
Pharmacogenomics uses knowledge of a person’s genetics to understand how a particular drug will work, or not work, in his or her body. For instance, some people might carry genes that make them more sensitive than average to a drug, and therefore they would require a lower dose. Others might have genes that make them resistant to the drug, meaning the drug is ineffective unless they receive a higher dose.
Adverse drug reactions are a leading cause of death in hospitalized patients in the United States and are responsible for billions of dollars in health care costs.1,2 Our current practice of prescribing for adult patients is largely trial-and-error, with the same dose given to all patients, in many cases with little regard even to sex, height, or weight.
Pharmacogenomics promises to change this way of prescribing to a customized approach that uses genetic information to predict an individual’s response to medications. It is one piece of an overall initiative to personalize patient care based on the patient’s individual characteristics and preferences.
OVERCOMING BARRIERS TO USING PHARMACOGENOMICS IN PRACTICE
If personalized medicine has promised to improve the quality and value of health care for our patients, why have we been so slow to adopt this information in clinical practice?
The usual barriers to clinical adoption certainly exist. We need further studies to determine whether genetic-based prescribing is truly valid, and for which patient populations. We need to determine whether this approach is cost-effective and better than the current standard of care. We need to work on payment options.
However, one of the largest barriers for busy primary care physicians is the lack of time to keep up with new information. Many practicing physicians were taught little about formal genetics in medical school. The body of scientific literature on pharmacogenomics is fragmented, and it crosses disease states and specialties, making it difficult to unite. Given the breadth of diseases treated and drugs prescribed by primary care physicians, it is unrealistic for most to keep track of the vast body of literature of pharmacogenomic testing and to decipher how to apply this to clinical practice.
In this issue of the Journal, Kitzmiller et al3 provide one solution to this problem, giving an overview of pharmacogenomic applications that might be pertinent to practicing physicians. However, as we try to make pharmacogenomics accessible to busy physicians, we need other solutions to integrate pharmacogenomic information efficiently into the clinical work flow. One approach might be to build pharmacogenomics into the electronic medical record. We can also store the integrated information in research databases and provide clinical recommendations on Internet sites such as www.pharmgkb.org, and we can develop applications to run on cell phones and iPads.
QUESTIONS REMAIN
Kitzmiller et al discuss an important step in this process, highlighting several key questions:
Should we seek genetics-based information to personalize drug selection? Based on the information presented in the literature and in the Kitzmiller paper, there may be circumstances when it is appropriate to consider doing so. While the evidence is not yet compelling to order these tests on a regular basis in clinical practice, this information might be helpful in some situations, such as for patients who have had adverse effects from minimal doses of antidepressants.
For now, clinicians should not abandon their current practice of personalizing patient care on the basis of personal, cultural, and economic preferences. Rather, they should consider pharmacogenomic information an additional piece of information when selecting drug therapy. We should also encourage health care systems and interested providers to be early adopters and to study how their outcomes compare with the standard of care.
Once we have this information, what is our obligation to use it? An increasing number of patients already have genetic information in their health record, either ordered by or provided to their physicians. However, there is little in the scientific literature to guide us in this arena.
Yet most of us would agree that if we have information (genetic or otherwise) that can help to select a drug type or dose or reduce adverse events or costs, we should consider this information in our decision-making. Several circumstances are documented in this paper and in the literature in which prior knowledge about drug metabolism can help in selecting a dose of medication. One example would be the 50% recommended reduction in tricyclic antidepressant dose if the patient is a CYP2D6 poor metabolizer.4
MOVING FORWARD AS A TEAM
In summary, Kitzmiller et al bring to light the promise and the uncertainties that currently exist in the field of pharmacogenomics. While it is unclear if we should incorporate pharmacogenomic tests into standard medical practice at this time, it is clear that this information is becoming more readily available, whether or not we have requested it. Some would argue that, once we have the information, we have an obligation to use it, just as we use other information in our clinical decision-making. This means we need to develop tools and resources to help practitioners evaluate pharmacogenomic data and incorporate it into clinical care in an efficient manner.
The authors also highlight the need for more education about drug metabolism in general, and they cite several instances in which knowledge of drug interactions and metabolism can clearly influence decision-making. An example is paroxetine (Paxil) inhibition of tamoxifen (Nolvadex).5
Lastly, regardless of our personal feelings about the clinical usefulness of genetic testing in large populations, we need to work together to determine clinical utility and validity and to develop efficient ways to put into practice findings that could affect patient care. As we move forward, we need to work as a team, utilizing our clinical partners—pharmacists, pharmacologists, metabolism and health information technology experts, and medical geneticists. Working as a team, pooling our resources and tools, we move closer to providing world-class personalized health care.
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998; 279:1200–1205.
- Field TS, Gilman BH, Subramanian S, Fuller JC, Bates DW, Gurwitz JH. The costs associated with adverse drug events among older adults in the ambulatory setting. Med Care 2005; 43:1171–1176.
- Kitzmiller JP, Groen DK, Phelps MA, Sadee W. Pharmacogenomic testing: relevance in medical practice. Why drugs work in some patients but not others. Cleve Clin J Med 2011; 78:243–257.
- Thuerauf N, Lunkenheimer J. The impact of the CYP2D6-polymorphism on dose recommendations for current antidepressants. Eur Arch Psychiatry Clin Neurosci 2006; 256:287–293.
- Schwarz EB, McNamara M, Miller RG, Walsh JM. Update in women’s health for the general internist. J Gen Intern Med201; 26:207–213.
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998; 279:1200–1205.
- Field TS, Gilman BH, Subramanian S, Fuller JC, Bates DW, Gurwitz JH. The costs associated with adverse drug events among older adults in the ambulatory setting. Med Care 2005; 43:1171–1176.
- Kitzmiller JP, Groen DK, Phelps MA, Sadee W. Pharmacogenomic testing: relevance in medical practice. Why drugs work in some patients but not others. Cleve Clin J Med 2011; 78:243–257.
- Thuerauf N, Lunkenheimer J. The impact of the CYP2D6-polymorphism on dose recommendations for current antidepressants. Eur Arch Psychiatry Clin Neurosci 2006; 256:287–293.
- Schwarz EB, McNamara M, Miller RG, Walsh JM. Update in women’s health for the general internist. J Gen Intern Med201; 26:207–213.
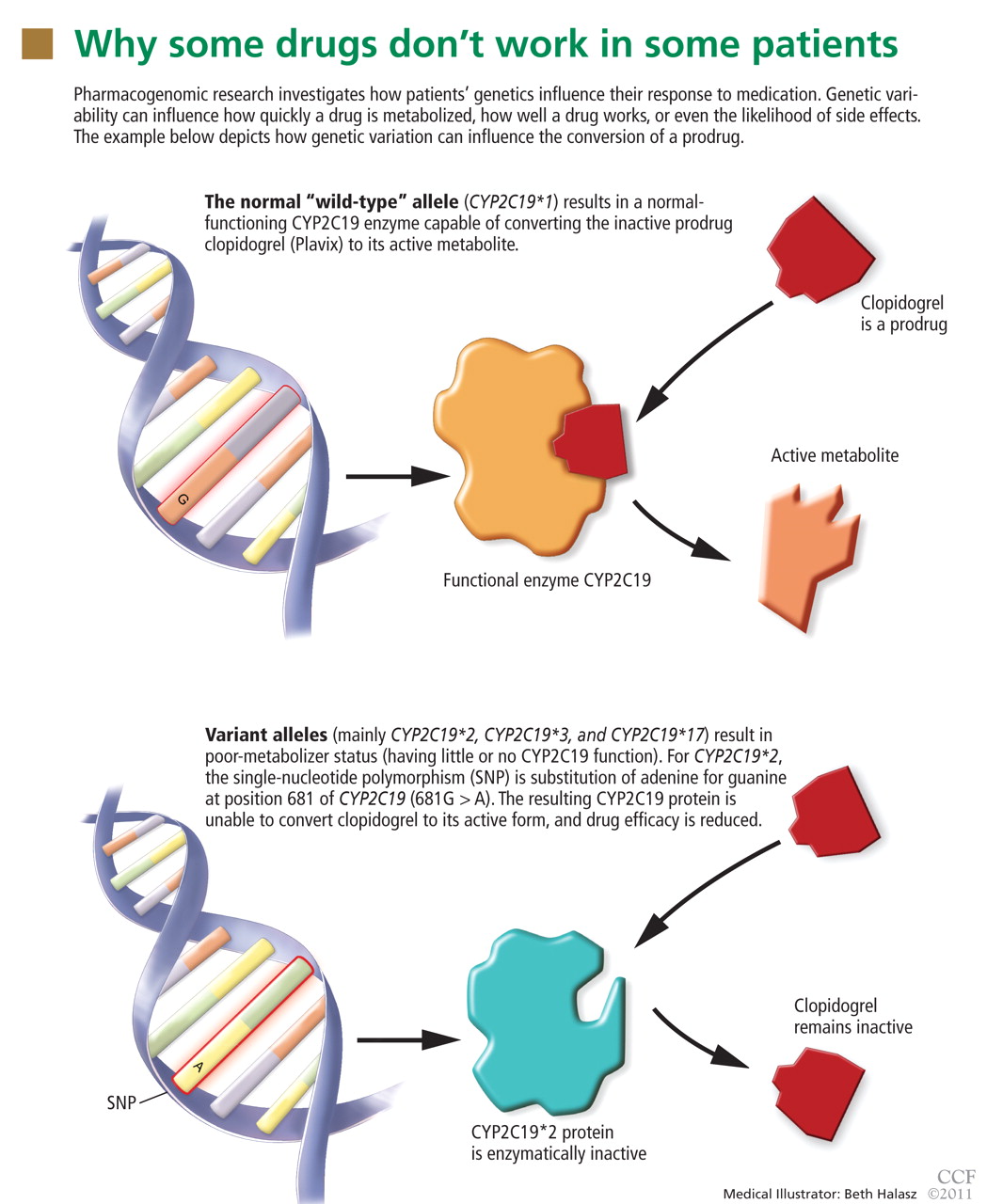
Pharmacogenomic testing: Relevance in medical practice
In many patients, certain drugs do not work as well as expected, whereas in other patients they cause toxic effects, even at lower doses. For some patients, the reason may be genetic.
Sizeable minorities of the population carry genetic variants—polymorphisms— that affect their response to various drugs. Thanks to genetic research, our understanding of the variability of drug response has advanced markedly in the last decade. Many relevant polymorphisms have been identified, and tests for some of them are available.
Armed with the knowledge of their patients’ genetic status, physicians could predict their response to certain drugs, leading to better efficacy, fewer adverse drug reactions, and a better cost-benefit ratio.
The possible impact is substantial, since more than half of the drugs most commonly involved in adverse drug reactions are metabolized by polymorphic enzymes.1 Adverse drug reactions remain a significant detriment to public health, having a substantial impact on rates of morbidity and death and on healthcare costs.2–8 In the United States, adverse drug reactions are a leading cause of death in hospitalized patients4 and are annually responsible for hundreds of thousands of deaths and hundreds of billions of dollars in added costs.2,4,6–8

In the meantime, physicians can educate their patients and promote efforts to incorporate genomic information into standard clinical decision-making.
This article offers an overview of pharmacogenomic testing, discussing implications and limitations of a few validated tests. Specifically, we will discuss testing that is relevant when using warfarin (Coumadin), clopidogrel (Plavix), statins, tamoxifen (Nolvadex), codeine, and psychotropic medications, as well as the future role of pharmacogenomic testing in medicine.
WHAT IS PHARMACOGENOMICS?
Pharmacogenomics is the study of how genetic factors relate to interindividual variability of drug response.
Many clinicians may not be familiar with the background and terminology used in the pharmacogenomic literature. Below, a brief review of the terminology is followed by a schematic describing the various stages of research involved in pharmacogenomics and the advancement of a test into standard practice.
The review and schematic may be helpful for evaluating the clinical significance of pharmacogenomics-related articles.
From genotype to phenotype
Genotype refers to the coding sequence of DNA base pairs for a particular gene, and phenotype (eg, disease or drug response) refers to a trait resulting from the protein product encoded by the gene. The name of a gene often refers to its protein product and is italicized (eg, the CYP3A4 gene encodes for the CYP3A4 enzyme).
Two alleles per autosomal gene (one paternal and one maternal) form the genotype. Heterozygotes possess two different alleles, and homozygotes possess two of the same alleles. The most common allele in a population is referred to as the wild type, and allele frequencies can vary greatly in different populations.9
Most sequence variations are single nucleotide polymorphisms (SNPs, pronounced “snips”), a single DNA base pair substitution that may result in a different gene product. SNPs can be classified as structural RNA polymorphisms (srSNPs), regulatory polymorphisms (rSNPs), or polymorphisms in coding regions (cSNPs)10: srSNPs alter mRNA processing and translation, rSNPs alter transcription, and cSNPs alter protein sequence and function.
Recently, genetic associations with a phenotype have been done on a large scale, with millions of SNPs measured in each of many subjects. This approach, called a genomewide association study or GWAS, has revealed countless candidate genes for clinical traits, but only a few have resulted in a practical clinical application. SNPs may by themselves exert a pharmacokinetic effect (ie, how the body processes the drug), a pharmacodynamic effect (ie, how the drug affects the body), or both, or they may act in concert with other genetic factors. Pharmacodynamic effects can result from a pharmacokinetic effect or can result from variations in a pharmacologic target.
Establishing a genotype-phenotype association can involve clinical studies, animal transgenic studies, or molecular and cellular functional assays.
Clinical applications are emerging

WARFARIN: IMPORTANCE OF CYP2C9, VKORC1
Warfarin is used for the long-term treatment and prevention of thromboembolic events.
This drug has a narrow therapeutic window and shows substantial interpatient dose variability. The start of warfarin therapy is associated with one of the highest rates of adverse events and emergency room visits of any single drug.12 More than 2 million patients start warfarin each year in the United States alone,13 and about 20% of them are hospitalized within the first 6 months because of bleeding due to overanticoagulation.14
The findings from a recent study suggest that pharmacogenomic testing may eventually allow more patients to safely benefit from warfarin therapy. In this large, nationwide, prospective study, hospitalization rates were 30% lower when pharmacogenomic testing was used.14 However, no reduction was seen in the time needed to reach the target international normalized ratio (INR) or in the need for INR checks at 6 months. Furthermore, this study used historical control data, and some or all of the reduction in hospitalization rates may be attributed to more frequent INR checks in the patients who underwent testing than in the historical control group.
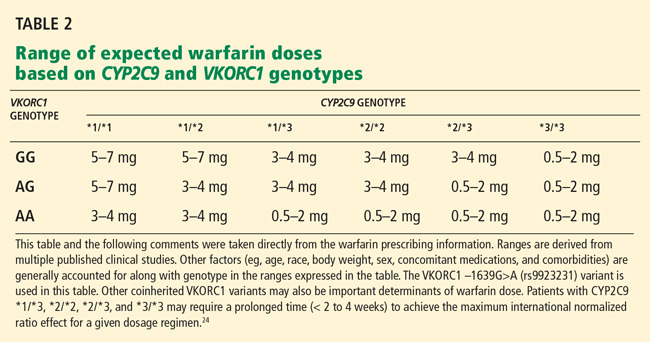
A relationship between warfarin dose requirements and the genetic status of CYP2C9, which encodes a major drug-metabolizing enzyme, has been demonstrated in retrospective and prospective studies.15–17
S-warfarin is metabolized by CYP2C9, which is polymorphic
Warfarin contains equal amounts of two isomers, designated S and R. S-warfarin, which is more potent, is metabolized principally by CYP2C9, while R-warfarin is metabolized by CYP1A2, CYP2C19, and CYP3A4.
People who possess two copies of the wild type CYP2C9 gene CYP2C9*1 metabolize warfarin very well and so are called “extensive warfarin metabolizers.” Carriers of the allelic variants CYP2C9*2 and CYP2C9*3 (which have point mutations in exons 3 and 7 of CYP2C9, respectively), have less capacity. Compared with those who are homozygous for the wild-type gene, homozygous carriers of CYP2C9* 3 clear S-warfarin at a rate that is 90% lower, and those with the CYP2C9*1/*3, CYP2C9* 1/*2, CYP2C9*2/*2, or CYP2C9*2/*3 genotypes clear it at a rate 50% to 75% lower. A meta-analysis of 12 studies found that the CYP2C9 genotype accounted for 12% of the interindividual variability of warfarin dose requirements.18
About 8% of whites carry at least one copy of CYP2C9*2, as do 1% of African Americans; the allele is rare in Asian populations. The frequency of CYP2C9*3 is 6% in whites, 1% in African Americans, and 3% in Asians.19,20 People with CYP2C9*4 or CYP2C9*5 have a diminished capacity to clear warfarin; however, these variants occur so infrequently that their clinical relevance may be minimal.
Warfarin’s target, VKOR, is also polymorphic
Genetic variation in warfarin’s pharmacologic target, vitamin K 2,3-epoxide reductase (VKOR), also influences dose requirements. Warfarin decreases the synthesis of vitamin-K-dependent clotting factors by inhibiting VKOR. This inhibition depends on the patient’s C1 subunit gene, VKORC1. Patients with a guanine-to-adenine SNP 1,639 bases upstream of VKORC1 (−1639G>A) need lower warfarin doses—an average of 25% lower in those with the GA genotype (ie, one allele has guanine in the −1639 position and the other allele has adenine in that position) and 50% lower in those with the AA genotype compared with the wild-type genotype GG.21 This promoter SNP, positioned upstream (ie, before the gene-coding region), greatly influences VKORC1 expression.
A meta-analysis of 10 studies found that the VKORC1 polymorphism accounts for 25% of the interindividual variation in warfarin dose.18 In one study, the frequency of the AA genotype in a white population was 14%, AG 47%, and GG 39%; in a Chinese population the frequency of AA was 82%, AG 18%, and GG 0.35%.22
CYP4F2 and GGCX also affect warfarin’s dose requirements
Genetic variations in the enzymes CYP4F2 and gamma-glutamyl carboxylase (GGCX) also influence warfarin dose requirements. Although the data are limited and the effects are smaller than those of CYP2C9 and VKORC1, people with a SNP in CYP4F2 need 8% higher doses of warfarin, while those with a SNP in GGCX need 6% lower doses.23
CYP2C9 and VKORC1 testing is available
Currently, the clinical pharmacogenetic tests relevant for warfarin use are for CYP2C9 and VKORC1.10
The FDA has approved four warfarin pharmacogenetic test kits, but most third-party payers are reluctant to reimburse for testing because it is not currently considered a standard of care. Testing typically costs a few hundred dollars, but it should become less expensive as it becomes more commonplace. The current FDA-approved product label for warfarin does not recommend routine pharmacogenomic testing for determining initial or maintenance doses, but it does acknowledge that dose requirements are influenced by CYP2C9 and VKORC1 and states that genotype information, when available, can assist in selecting the starting dose.24
Clinical trials of warfarin pharmacogenomic testing are under way
Although genetic status can greatly influence an individual patient’s warfarin dosing requirement, routine prospective pharmacogenomic testing is not endorsed by the FDA or by other expert panels26 because there is currently insufficient evidence to recommend for or against it.
Several large prospective trials are under way. For example, the National Heart, Lung, and Blood Institute began a prospective trial in about 1,200 patients to evaluate the use of clinical plus genetic information to guide the initiation of warfarin therapy and to improve anticoagulation control for patients.27 The results, expected in September 2011, and those of other large prospective trials should provide adequate evidence for making recommendations about the clinical utility of routine pharmacogenetic testing for guiding warfarin therapy.
Several recent cost-utility and cost-effectiveness studies have attempted to quantify the value of pharmacogenomic testing for warfarin therapy,28–30 but their analyses are largely limited because the benefit (clinical utility) is yet to be sufficiently characterized.
The relevance of such analyses may soon be drastically diminished, as several non-vitamin-K-dependent blood thinners such as rivaroxaban (Xarelto), dabigatran (Pradaxa), and apixaban are poised to enter clinical practice.31
CLOPIDOGREL IS ACTIVATED BY CYP2C19
Clopidogrel, taken by about 40 million patients worldwide, is used to prevent atherothrombotic events and cardiac stent thrombosis when given along with aspirin.
Studies of clopidogrel pharmacogenomics
A recent genome-wide association study conducted in a cohort of 429 healthy Amish persons revealed a SNP in CYP2C19 to be associated with a diminished response to clopidogrel and to account for 12% of the variation in drug response.33 Traditional factors (the patient’s age, body-mass index, and cholesterol level) combined accounted for less than 10% of the variation.
Findings were similar in a subsequent investigation in 227 cardiac patients receiving clopidogrel: 21% of those with the variant had a cardiovascular ischemic event or died during a 1-year follow-up period compared with 10% of those without the variant (hazard ratio 2.42, P = .02).33
A 12-year prospective study investigating clopidogrel efficacy in 300 cardiac patients under the age of 45 used cardiovascular death, nonfatal myocardial infarction, and urgent coronary revascularization as end points. It concluded that the only independent predictor of these events was the patient’s CYP2C19 status.34
A study in 2,200 patients with recent myocardial infarction examined whether any of the known allelic variations that modulate clopidogrel’s absorption (ABCB1), metabolic activation (CYP3A4/5 and CYP2C19), or biologic activity (P2RY12 and ITGB3) was associated with a higher rate of the combined end point of all-cause mortality, nonfatal myocardial infarction, or stroke. None of the SNPs in CYP3A4/5, P2RY12, or ITGB3 that were evaluated was associated with a higher risk at 1 year. However, the allelic variations modulating clopidogrel’s absorption (ABCB1) and metabolism (CYP2C19) were associated with higher event rates. Patients with two variant ABCB1 alleles had a higher adjusted hazard ratio (95% confidence interval [CI] 1.2–2.47) than those with the wild-type allele. Patients who had one or two CYP2C19 loss-of-function alleles had a higher event rate than those with two wild-type alleles (95% CI 1.10–3.58 and 1.71–7.51, respectively).35
Conversely, researchers from the Population Health Research Institute found no association between poor-metabolizer status and treatment outcomes when CYP2C19 analysis was retrospectively added to the findings of two large clinical trials (combined N > 5,000). However, patients with acute coronary syndrome benefited more from clopidogrel treatment if they were ultra-rapid metabolizers (possessing the gain-of-function allele CYP2C19*17).36
Current status of clopidogrel testing: Uncertain
A current FDA boxed warning states that poor CYP2C19 metabolizers may not benefit from clopidogrel and recommends that prescribers consider alternative treatment for patients in this category.37 However, routine CYP2C19 testing is not recommended, and no firm recommendations have been established regarding dose adjustments for CYP2C19 status.

In 2010, the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the American Heart Association collectively pronounced the current evidence base insufficient for recommending routine pharmacogenomic testing.39
Needed are large-scale studies examining the cost-effectiveness and clinical utility of genotype-guided clopidogrel therapy compared with other therapy options such as prasugrel (Effient), an analogue not metabolized by CYP2C19. One such study, sponsored by Medco Health Solutions, plans to enroll 14,600 cardiac patients and has an estimated completion date in June 2011.40 The expectation that clopidogrel will be available in generic form in 2012 adds to the uncertainty regarding the cost-effectiveness of CYP2C19 testing for clopidogrel therapy.
STATINS: SLC01B1*5 INCREASES MYOPATHY RISK
Statins lower the concentration of low-density lipoprotein cholesterol (LDL-C), resulting in a relative-risk reduction of about 20% for each 1 mmol/L (39 mg/dL) decrement in LDL-C.41 They are one of the most commonly prescribed classes of drugs, but their side effects can limit their appeal: statin-induced myopathy occurs in about 1:1,000 to 1:10,000 patients and is difficult to predict.
SLC01B1. The Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH), a genome-wide association study, recently found a SNP (SLCO1B1* 5) in the SLC01B1 gene to be associated with a higher risk of statin-induced myopathy in cardiac patients receiving simvastatin (Zocor) 40 or 80 mg daily.42 The SLC01B1 gene, located on chromosome 12, influences the extent of the drug’s hepatic uptake and its serum concentration. Only the SLC01B1*5 SNP emerged as a predictor of statin-induced myopathy across the entire genome.42
The authors believe the findings are likely to apply to other statins. The mechanisms leading to statin-induced myopathy and the impact of statin pharmacogenomics are still unclear.43
CYP3A4. Other genetic variants may play a vital role in determining response to statin therapy. Carriers of a newly identified CYP3A4 polymorphism (intron 6 SNP, rs35599367, C>T) required significantly lower statin doses (0.2–0.6 times less) for optimal lipid control. The analyses included atorvastatin (Lipitor), simvastatin, and lovastatin (Mevacor), and the association was robust (P = .019).44
Statin pharmacogenomic testing is not routinely recommended
Routine pharmacogenomic testing for statin therapy is not recommended. Additional studies are needed to determine the clinical utility and cost-effectiveness of pharmacogenomic testing (involving a combination of several polymorphisms) in various patient populations delineated by type of statin, dose, and concomitant use of other drugs.
TAMOXIFEN IS ACTIVATED BY CYP2D6
Tamoxifen is prescribed to prevent the recurrence of estrogen-receptor-positive breast cancer, to treat metastatic breast cancer, to prevent cancer in high-risk populations, and to treat ductal carcinoma in situ.
Tamoxifen is metabolized to form endoxifen, which has much higher potency and higher systemic levels than tamoxifen.45 Both CYP2D6 and CYP3A4/5 are required to produce endoxifen via two intermediates, but CYP2D6 catalyzes the critical step leading to metabolic activation.
The CYP2D6 gene is highly polymorphic, with more than 75 allelic variants identified. Extensive literature is available describing the influence of CYP2D6 polymorphisms on tamoxifen metabolism and therapy outcomes.46–52 Several CYP2D6 variants result in reduced or no enzyme activity, and people who have more than two normally functioning alleles have exaggerated enzyme activity (gene amplification).
Classification of CYP2D6 status
Several systems have been developed to categorize the phenotypic activity of CYP2D6 based on genotype.
A genetic basis for the observed diversity in the metabolism of cytochrome P450 substrates was recognized more than 30 years ago. People were categorized as either extensive or poor metabolizers, reflecting normal vs impaired ability to metabolize the CYP2D6 substrates sparteine and debrisoquine. Later work expanded this system to include categories for intermediate (between poor and extensive) and ultra-rapid (better than extensive) metabolizers.
The genetic basis for these categories includes homozygosity for dysfunctional variants (the poor-metabolizer group) or extra copies of normal functioning variants (the ultra-rapid-metabolizer group).
Newer systems have been described for characterizing the CYP2D6 activity phenotype whereby CYP2D6 variants are assigned activity scores.53–56 The various scoring systems have been reviewed by Kirchheiner.57

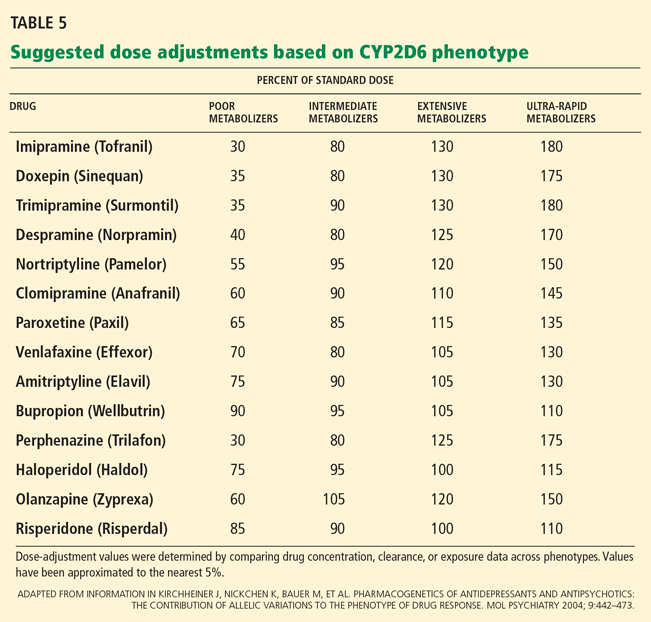
A recent version of the activity scoring system also takes into consideration the many drugs that inhibit CYP2D6, such as amiodarone (Cordarone) and fluoxetine (Prozac) that can reduce the action of tamoxifen if given with it (Table 4).58 For example, the tamoxifen exposure (as predicted by the CYP2D6-activity score) experienced by a CYP2D6 extensive metabolizer taking a CYP2D6-inhibiting drug may be similar to the exposure experienced by a CYP2D6 poor metabolizer receiving the same tamoxifen dose but not taking a CYP2D6-inhibiting drug.
Likewise, the activity score of a CYP2D6 intermediate metabolizer taking a CYP2D6-inducing drug may be similar to that of a CYP2D6 ultra-rapid metabolizer not taking a CYP2D6-inducing drug. Examples of CYP2D6 inducers are dexamethasone, rifampin, and hyperforin (St. John’s wort).
While the newer systems are reported to provide better correlations between genotype and phenotype scores, the older scoring systems and the categorical names are still widely used (eg, in the FDA-approved AmpliChip CYP450 test from Roche,59 which includes genotype data for CYP2D6 and CYP2C19).
No firm recommendations for CYP2D6 testing in tamoxifen users
The different genotypes and phenotypes vary in prevalence in different ethnic groups, and significantly different activity levels for endoxifen formation are observed. Tamoxifen lacks efficacy in those who are poor CYP2D6 metabolizers—ie, about 7% of the white population.
However, the FDA has not made firm recommendations about CYP2D6 testing for prescribing tamoxifen because the evidence of benefit, although suggestive, has been considered insufficient.
Clinicians should be aware that tamoxifen’s efficacy is greatly reduced by concomitant therapy with CYP2D6-inhibiting drugs (Table 4).
Other genes affecting tamoxifen: CYP3A4/5, SULT1A1, and UGT2B15
Some investigators propose that polymorphisms in additional genes encoding enzymes in the tamoxifen metabolic and elimination pathways (eg, CYP3A4/5, SULT1A1, and UGT2B15) also need to be considered to account adequately for interindividual variation in drug response.
For example, CYP3A4 and CYP3A5 are also polymorphic, and large interindividual variation exists in their enzyme activities. These enzymes have overlapping substrate specificities, represent the most abundant drug-metabolizing enzymes in the human liver, and are involved in the biotransformation of a broad range of endogenous substrates and most drugs.60
Clinical studies evaluating the impact of CYP3A4/5 polymorphisms have been inconsistent in their conclusions, which is generally attributed to the relatively low functional impact or the low prevalence of the SNPs evaluated. Many of the nearly 100 CYP3A4/5 polymorphisms identified have not yet been characterized regarding their functional impact on enzyme expression or activity. CYP-3A4/5 enzyme activity is highly variable between individuals and warrants further study of its role in outcomes of tamoxifen therapy. Ongoing and future prospective clinical trials evaluating CYP2D6, CYP3A4/5, and other relevant polymorphisms are necessary to define their clinical relevance before routine genetic testing for tamoxifen can be justified.
CODEINE IS ALSO ACTIVATED BY CYP2D6
Codeine also depends on the CYP2D6 gene, as it must be activated to its more potent opioid metabolites, including morphine. Poor CYP2D6 metabolizers do not benefit from codeine therapy.
The pharmacogenomics of codeine has become a hot topic, especially regarding breast-feeding mothers. The debate was ignited with the publication in 2006 of a case report of an infant’s death, apparently the result of metabolic polymorphisms.61 The evolution of this debate and the outcome of the case may be noteworthy to clinicians, as they illustrate the gravity of public and patient interest in pharmacogenomic testing. In this case, the breast-feeding mother had taken codeine regularly for about 14 days when her 13-day-old infant died from toxic levels of morphine. Unknown to her and the prescriber, both the mother and infant were ultra-rapid CYP2D6 metabolizers, resulting in a more rapid and extensive conversion of codeine to morphine.
A logical strategy for preventing similar deaths would be routine CYP2D6 genotyping when prescribing codeine to breast-feeding mothers. However, after several investigations examined the metabolic and excretion pathways of codeine in their entirety, the FDA did not recommend routine CYP2D6 testing when prescribing codeine to breastfeeding mothers because several other factors, including rare genetic variations of other enzymes, proved necessary for reaching the opioid toxicity leading to the infant’s death.62
PHARMACOGENOMICS OF PSYCHOTROPIC DRUGS
Pharmacogenomic testing has clinical utility for some psychotropic drugs.
HLA-B and carbamazepine
Considered a standard of care, HLA-B genotyping is appropriate before prescribing carbamazepine (Tegretol, Equetro) to patients in populations in which HLAB*1502 is likely to be present, such as Asians. Carriers of HLAB* 1502 are at higher risk of life-threatening skin reactions such as Stevens-Johnson syndrome.11
Several other pharmacogenomic applications for psychotropic medications have been suggested, but routine testing has not been recommended by the FDA or endorsed by any expert panel because sufficient clinical utility and cost-effectiveness have not been demonstrated. A brief summary of study findings and a few practical suggestions follow.
Polymorphisms in metabolizing enzymes have been investigated in patients receiving psychotropic drugs.
CYP2D6 and antidepressants
Many antidepressants show significant differences in plasma drug levels with CYP2D6 polymorphisms (in descending order of influence)55:
- Imipramine (Tofranil)
- Doxepin (Adapin, Silenor, Sinequan)
- Maprotiline (Deprilept, Ludiomil, Psymion)
- Trimipramine (Surmontil)
- Desipramine (Noraprim)
- Nortriptyline (Aventyl, Pamelor)
- Clomipramine (Anafranil)
- Paroxetine (Paxil)
- Venlafaxine (Effexor)
- Amitriptyline (Elavil)
- Mianserin
- Trazadone (Desyrel)
- Bupropion (Wellbutrin)
- Nefazodone (Serzone)
- Citalopram (Celexa)
- Sertraline (Zoloft).
CYP2D6 and antipsychotics
Several antipsychotics are also influenced by CYP2D6 polymorphisms (also in descending order of influence)55:
- Perphenazine (Trilafon)
- Thioridazine (Mellaril)
- Olanzapine (Zyprexa)
- Zuclopenthixol (Cisordinol, Clopixol, Acuphase)
- Aripiprazole (Abilify)
- Flupentixol (Depixol, Fluanxol)
- Haloperidol (Haldol)
- Perazine (Taxilan)
- Risperidone (Risperdal)
- Pimozide (Orap).
CYP2C19 and antidepressants
CYP2C19 polymorphisms are likewise associated with differences in drug metabolism for many antidepressants, such as (in descending order of CYP2C19-mediated influence)55:
- Trimipramine
- Doxepin
- Amitriptyline
- Imipramine
- Citalopram (Celexa)
- Clomipramine
- Moclobemide (Aurorix, Manerix)
- Sertraline
- Fluvoxamine (Luvox).
Clinical relevance of CYP2D6 and CYP2C19
Several studies have demonstrated that poor and intermediate CYP2D6 metabolizers have a higher incidence of adverse effects when taking CYP2D6-dependent antidepressants63–68; however, an almost equal number of studies did not find statistically significant associations.69–72 Likewise, several studies have found an association between ultra-rapid CYP2D6 metabolizer status and diminished response to antidepressants,65,73,74 but no association was found in a larger retrospective study.75
Routine CYP2D6 and CYP2C19 screening is not recommended when prescribing psychotropic drugs. However, reviews of the pharmacokinetic data have suggested a few practical applications when genetic status is already known. In general, clinicians can consider reducing the dose of tricyclic antidepressants by about 50% when prescribing to CYP2D6-poor-metabolizers.55,76–78
Genes that affect serotonin metabolism
Several genes in the serotonin pathway have been investigated to determine whether they influence patients’ susceptibility to depression and adverse effects and response to psychotropic medications.
SLC6A4. Polymorphisms in the promoter region of the serotonin transporter gene SLC6A4 appear to influence the treatment response and side-effect profiles of selective serotonin reuptake inhibitors (SSRIs). Carriers of the SLC6A4 5-HTTLPR L alleles have fewer side effects79 and better response to SSRI treatment, and carriers of the S allele have a higher incidence of antidepressant-induced mania80 and poorer response to SSRI treatment.81
5-HT. Polymorphisms in serotonin receptors (2A and 2C subtypes) appear to influence SSRI response and side effects. Carriers of 5-HT 2A C alleles had more severe adverse effects from paroxetine,71 but another 5-HT 2A polymorphism common to Asians is associated with better response to antidepressant therapy.82 A 5-HT 2C polymorphism was associated with a lower incidence of antipsychotic-induced weight gain.83
Although the understanding of these relationships is incomplete and routine pharmacogenomic testing is not currently recommended, reviews of the pharmacodynamic data have suggested a few practical applications when a patient’s genetic status is already known. One should consider:
- Selecting treatments other than SSRIs for depressed patients known to possess the SLC6A4 variant
- Selecting citalopram for depressed patients known to carry the 5-HT 2A polymorphism
- Avoiding treatment with antipsychotic drugs for patients known to possess the 5-HT 2C polymorphism.
THE FUTURE OF PHARMACOGENOMIC TESTING
The examples discussed in this article provide some insight about how pharmacogenomic testing is maturing and slowly being integrated into the practice of medicine. They also illustrate the complexity of the multiple stages of research that pharmacogenomic applications must go through in order to be adopted as standard practice.
In the future, pharmacogenomic data will continue to accumulate, and the clinical utility of many other pharmacogenomic tests may be uncovered. The FDA provides information on emerging pharmacogenomic tests at its Web site, www.fda.gov.11 Its up-to-date “Table of Valid Genomic Biomarkers in the Context of Approved Drug Labels” includes boxed warnings, recommendations, research outcomes, and relevant population genetics.
If the FDA continues its current policy, prospective randomized trials that show improvement in patient outcomes will remain the gold standard for determining the clinical significance of a pharmacogenomic test. Furthermore, cost-benefit analyses are likely to continue dictating policy regarding pharmacogenomic testing, and cost-benefit profiles should improve as technology advances and as information gathered from a single test becomes applicable to multiple medications and clinical scenarios.
In the meantime, physicians should become familiar with the terms used in medical genetics and pharmacogenomics and begin to understand genetic contributions to the outcomes of drug therapy. For example, understanding the consequences of metabolizer status and the frequency of variants in a given population can be tremendously helpful when advising our patients about anticipating potential problems when taking specific medications and about making informed decisions about pharmacogenomic testing.
This exchange of information alone may go a long way in improving therapy outcomes even when prospective pharmacogenomic testing is not routinely performed. Furthermore, an increasing number of patients will already have genotyping information available when they come to us, and clinicians need to be aware of the many pharmacogenomic applications recommended by the FDA when genetic status is known.10
- King HC, Sinha AA. Gene expression profile analysis by DNA micro-arrays: promise and pitfalls. JAMA 2001; 286:2280–2288.
- Nuckols TK, Paddock SM, Bower AG, et al. Costs of intravenous adverse drug events in academic and nonacademic intensive care units. Med Care 2008; 46:17–24.
- Gurwitz JH, Field TS, Judge J, et al. The incidence of adverse events in two long-term care facilities. Am J Med 2005; 118:251–258.
- Vargas E, Terleira A, Hernando F, et al. Effects of adverse drug reactions on length of stay in surgical intensive care units. Crit Care Med 2003; 31:694–698.
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998; 279:1200–1205.
- Field TS, Gilman BH, Subramanian S, Fuller JC, Bates DW, Gurwitz JH. The costs associated with adverse drug events in older adults in the ambulatory setting. Med Care 2005; 43:1171–1176.
- Classen DC, Pestotnik SL, Evans RS, Lloyd JF, Burke JP. Adverse drug events in hospitalized patients. Excess length of stay, extra costs, and attributable mortality. JAMA 1997; 277:301–306.
- Ernst FR, Grizzle AJ. Drug-related morbidity and mortality: updating the cost-of-illness model. J Am Pharm Assoc (Wash) 2001; 41:192–199.
- Bradford LD. CYP2D6 allele frequency in European Caucasians, Asians, Africans and their descendants. Pharmacogenomics 2002; 3:229–243.
- Sadee W. Measuring cis-acting regulatory variants genome-wide: new insights into expression genetics and disease susceptibility. Genome Med 2009; 1:116.
- US Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labels. http://www.fda.gov/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/ucm083378.htm. Accessed 1/18/2011.
- Budnitz DS, Shehab N, Kegler SR, Richards CL. Medication use leading to emergency department visits for adverse drug events in older adults. Ann Intern Med 2007; 147:755–765.
- Elias DJ, Topol EJ. Warfarin pharmacogenomics: a big step forward for individualized medicine: enlightened dosing of warfarin. Eur J Hum Genet 2008; 16:532–534.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study). J Am Coll Cardiol 2010; 55:2804–2812.
- Rettie AE, Wienkers LC, Gonzalez FJ, Trager WF, Korzekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allelic variant of CYP2C9. Pharmacogenetics 1994; 4:39–42.
- Higashi MK, Veenstra DL, Kondo LM, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 2002; 287:1690–1698.
- Sanderson S, Emery J, Higgins J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: a HuGEnet systematic review and meta-analysis. Genet Med 2005; 7:97–104.
- Au N, Rettie AE. Pharmacogenomics of 4-hydroxycoumarin anticoagulants. Drug Metab Rev 2008; 40:355–375.
- García-Martín E, Martínez C, Ladero JM, Agúndez JA. Interethnic and intraethnic variability of CYP2C8 and CYP2C9 polymorphisms in healthy individuals. Mol Diagn Ther 2006; 10:29–40.
- Sullivan-Klose TH, Ghanayem BI, Bell DA, et al. The role of the CYP2C9-Leu359 allelic variant in the tolbutamide polymorphism. Pharmacogenetics 1996; 6:341–349.
- Wen MS, Lee M, Chen JJ, et al. Prospective study of warfarin dosage requirements based on CYP2C9 and VKORC1 genotypes. Clin Pharmacol Ther 2008; 84:83–89.
- Larramendy-Gozalo C, Yang JQ, Verstuyft C, et al. Genetic polymorphism of vitamin K epoxide reductase (VKORC1) 1173C>T in a Chinese and a Caucasian population. Basic Clin Pharmacol Toxicol 2006; 98:611–613.
- Caldwell MD, Awad T, Johnson JA, et al. CYP4F2 genetic variant alters required warfarin dose. Blood 2008; 111:4106–4112.
- Bristol-Myers Squibb. Coumadin (warfarin sodium) Prescribing Information. January 2010.
- Barnes-Jewish Hospital at Washington University Medical Center. Warfarin dosing. http://warfarindosing.orgAccessed 1/20/2011.
- Flockhart DA, O’Kane D, Williams MS, et al. Pharmacogenetic testing of CYP2C9 and VKORC1 alleles for warfarin. Genet Med 2008; 10:139–150.
- National Institutes of Health. ClinicalTrials.gov. http://clinicaltrials.gov. Accessed January 20, 2011.
- Eckman MH, Rosand J, Greenberg SM, Gage BF. Cost-effectiveness of using pharmacogenetic information in warfarin dosing for patients with nonvalvular atrial fibrillation. Ann Intern Med 2009; 150:73–83.
- Meckley LM, Gudgeon JM, Anderson JL, Williams MS, Veenstra DL. A policy model to evaluate the benefits, risks, and costs of warfarin pharmacogenomic testing. Pharmacoeconomics 2010; 28:61–74.
- Patrick AR, Avron J, Choudhry NK. Cost-effectiveness of genotype-guided warfarin dosing for patients with atrial fibrillation. Circ Cardiovasc Qual Outcomes 2009; 2:429–436.
- Haas S. New oral Xa and IIa inhibitors: updates on clinical trial results. J Thromb Thrombolysis 2008; 25:52–60.
- Bhatt DL. Tailoring antiplatelet therapy based on pharmacogenomics: how well do the data fit? JAMA 2009; 302:896–897.
- Shuldiner AR, O’Connell JR, Bliden KP, et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 2009; 302:849–857.
- Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 2009; 373:309–317.
- Simon T, Verstuyft C, Mary-Krause M, et al; French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360:363–375.
- Paré G, Mehta SR, Yusuf S, et al. Effects of CYP2C19 genotype on outcomes of clopidogrel treatment. N Engl J Med 2010; 363:1704–1714.
- Bristol-Myers Squibb/Sanofi Pharmaceutical Partnership. Plavix (clopidogrel bisulfate) prescribing information. August 2010.
- P450 Drug Interaction Table. Indiana University School of Medicine. http://medicine.iupui.edu/clinpharm/ddis/table.asp. Accessed 1/21/2011.
- Society for Cardiovascular Angiography and Interventions; Holmes DR, Dehmer GJ, Kaul S, Leifer D, O’Gara PT, Stein CM. ACCF/AHA clopidogrel clinical alert: approaches to the FDA “boxed warning”: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the American Heart Association. Circulation 2010; 122:537–557.
- National Institutes of Health. Genotype Guided Comparison of Clopidogrel and Prasugrel Outcomes Study. http://clinicaltrialsfeeds.org/clinical-trials/show/NCT00995514. Accessed 1/20/2011.
- Amarenco P, Labreuche J. Lipid management in the prevention of stroke: Review and updated meta-analysis of statins for stroke prevention. Lancet Neurol 2009; 8:453–463.
- SEARCH Collaborative Group, Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Romaine SP, Bailey KM, Hall AS, Balmforth AJ. The influence of SLC01B1 (OATP1B1) gene polymorphisms on response to statin therapy. Pharmacogenomics J 2010; 10:1–11.
- Wang D, Guo Y, Wrighton SA, Cooke GE, Sadee W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J 2010; Apr 13 [Epub ahead of print].
- Goetz MP, Rae JM, Suman VJ, et al. Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J Clin Oncol 2005; 23:9312–9318.
- Beverage JN, Sissung TM, Sion AM, Danesi R, Figg WD. CYP2D6 polymorphisms and the impact on tamoxifen therapy. J Pharm Sci 2007; 96:2224–2231.
- Flockhart D. CYP2D6 genotyping and the pharmacogenetics of tamoxifen. Clin Adv Hematol Oncol 2008; 6:493–494.
- Goetz MP, Kamal A, Ames MM. Tamoxifen pharmacogenomics: the role of CYP2D6 as a predictor of drug response. Clin Pharmacol Ther 2008; 83:160–166.
- Stearns V, Rae JM. Pharmacogenetics and breast cancer endocrine therapy: CYP2D6 as a predictive factor for tamoxifen metabolism and drug response? Expert Rev Mol Med 2008; 10:e34.
- Dezentjé VO, Guchelaar HJ, Nortier JW, van del Velde CJ, Gelderblom H. Clinical implications of CYP2D6 genotyping in tamoxifen treatment for breast cancer. Clin Cancer Res 2009; 15:15–21.
- Higgins MJ, Rae JM, Flockhart DA, Hayes DF, Stearns V. Pharmacogenetics of tamoxifen: who should undergo CYP2D6 genetic testing? J Natl Compr Canc Netw 2009; 7:203–213.
- Hoskins JM, Carey LA, McLeod HL. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat Rev Cancer 2009; 9:576–586.
- Steimer W, Zöpf K, von Amelunxen S, et al. Allele-specific change of concentration and functional gene dose for the prediction of steady-state serum concentrations of amitriptyline and nortriptyline in CYP2C19 and CYP2D6 extensive and intermediate metabolizers. Clinical Cancer 2004; 50:1623–1633.
- Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharm Ther 2008; 83:234–242.
- Kirchheiner J, Nickchen K, Bauer M, et al. Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol Psychiatry 2004; 9:442–473.
- Kirchheiner J, Schmidt H, Tzvetkov M, et al. Pharmacokinetics of codeine and its metabolite morphine in ultra-rapid metabolizers due to CYP2D6 duplication. Pharmacogenomics J 2007; 7:257–265.
- Kirchheiner J. CYP2D6 phenotype prediction from genotype: which system is the best? Clin Pharmacol Ther 2008; 83:225–227.
- Borges S, Desta Z, Jin Y, et al. Composite functional genetic and comedication CYP2D6 activity score in predicting tamoxifen drug exposure among breast cancer patients. J Clin Pharmacol 2010; 50:450–458.
- Hoffmann-La Roche Ltd. AmpliChip CYP450 Test. http://www.roche.com/assays/Pages/AmpliChipCYP450Test.aspx. Accessed 1/21/2011.
- Anzenbacher P, Anzenbacherová E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci 2001; 58:737–747.
- Koren G, Cairns J, Chitayat D, Gaedigk A, Leeder SJ. Pharmacogenetics of morphine poisoning in a breastfed neonate of a codeine-prescribed mother. Lancet 2006; 368:704.
- Willmann S, Edginton AN, Coboeken K, Ahr G, Lippert J. Risk to the breast-fed neonate from codeine treatment to the mother: a quantitative mechanistic modeling study. Clin Pharm Ther 2009; 86:634–643.
- Grzesiak M, Beszłej A, Lebioda A, Jonkisz A, Dobosz T, Kienja A. [Retrospective assessment of the antidepressants tolerance in the group of patients with diagnosis of depression and different CYP2D6 genotype.] [In Polish] Psychiatr Pol 2003; 37:433–444.
- Laika B, Leucht S, Heres S, Steimer W. Intermediate metabolizer: increased side effects in psychoactive drug therapy. The key to cost-effectiveness of pretreatment CYP2D6 screening? Pharmacogenomics J 2009; 9:395–403.
- Rau T, Wohlleben G, Wuttke H, et al. CYP2D6 genotype: Impact on adverse effects and nonresponse during treatment with antidepressants—a pilot study. Clin Pharm Ther 2004; 75:386–393.
- McAlpine DE, O’Kane DJ, Black JL, Mrazek DA. Cytochrome P450 2D6 genotype variation and venlafaxine dosage. Mayo Clin Proc 2007; 82:1065–1068.
- Chen S, Chou WH, Blouin RA, et al. The cytochrome P450 2D6 (CYP2D6) enzyme polymorphism: screening costs and influence on clinical outcomes in psychiatry. Clin Pharmacol Ther 1996; 60:522–534.
- Shams ME, Arneth B, Hiemke C, et al. CYP2D6 polymorphism and clinical effect of the antidepressant venlafaxine. J Clin Pharm Ther 2006; 31:493–502.
- Whyte EM, Romkes M, Mulsant BH, et al. CYP2D6 genotype and venlafaxine-XR concentrations in depressed elderly. Int J Geriatr Psychiatry 2006; 21:542–549.
- Roberts RL, Mulder RT, Joyce PR, Luty SE, Kennedy MA. No evidence of increased adverse drug reactions in cytochrome P450 CYP2D6 poor metabolizers treated with fluoxetine or nortriptyline. Hum Psychopharmacol 2004; 19:17–23.
- Murphy GM, Kremer C, Rodrigues HE, Schatzberg AF. Pharmacogenetics of antidepressant medication intolerance. Am J Psychiatry 2003; 160:1830–1835.
- Gillman PK. Re: no evidence of increased adverse drug reactions in cytochrome P450 CYP2D6 poor metabolizers treated with fluoxetine or nortriptyline. Hum Psychopharmacol 2005; 20:61–62.
- Gex-Fabry M, Eap CB, Oneda B, et al. CYP2D6 and ABCB1 genetic variability: influence on paroxetine plasma level and therapeutic response. Ther Drug Monit 2008; 30:474–482.
- Kawanishi C, Lundgren S, Agren H, Bertilsson L. Increased incidence of CYP2D6 gene duplication in patients with persistent mood disorders: ultrarapid metabolism of antidepressants as a cause of nonresponse. A pilot study. Eur J Clin Pharmacol 2004; 59:803–807.
- Serretti A, Calati R, Massat I, et al. Cytochrome P450 CYP1A2, CYP2C9, CYP2C19 and CYP2D6 genes are not associated with response and remission in a sample of depressive patients. Int Clin Psychopharmacol 2009; 24:250–256.
- de Leon J, Armstrong SC, Cozza KL. Clinical guidelines for psychiatrists for the use of pharmacogenetic testing for CYP450 2D6 and CYP450 2C19. Psychosomatics 2006; 47:75–85.
- de Leon J, Susce MT, Johnson M, et al. DNA microarray technology in the clinical environment: the AmpliChip CYP450 test for CYP2D6 and CYP2C19 genotyping. CNS Spectr 2009; 14:19–34.
- Thuerauf N, Lunkenheimer J. The impact of the CYP2D6-polymorphism on dose recommendations for current antidepressants. Eur Arch Psychiatry Clin Neurosci 2006; 256:287–293.
- Horstmann S, Binder EB. Pharmacogenomics of antidepressant drugs. Pharmacol Ther 2009; 124:57–73.
- Ferreira Ade A, Neves FS, da Rocha FF, et al. The role of 5-HTTLPR polymorphism in antidepressant-associated mania in bipolar disorder. J Affect Disord 2009; 112:267–272.
- Serretti A, Kato M, De Ronchi D, Kinoshita T. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with selective serotonin reuptake inhibitor efficacy in depressed patients. Mol Psychiatry 2007; 12:247–257.
- Kato M, Serretti A. Review and meta-analysis of antidepressant pharmacogenetic findings in major depressive disorder. Mol Psychiatry 2010; 15:473–500.
- Reynolds GP, Zhang Z, Zhang X. Polymorphism of the promoter region of the serotonin 5-HT(2C) receptor gene and clozapine-induced weight gain. Am J Psychiatry 2003; 160:677–679.
In many patients, certain drugs do not work as well as expected, whereas in other patients they cause toxic effects, even at lower doses. For some patients, the reason may be genetic.
Sizeable minorities of the population carry genetic variants—polymorphisms— that affect their response to various drugs. Thanks to genetic research, our understanding of the variability of drug response has advanced markedly in the last decade. Many relevant polymorphisms have been identified, and tests for some of them are available.
Armed with the knowledge of their patients’ genetic status, physicians could predict their response to certain drugs, leading to better efficacy, fewer adverse drug reactions, and a better cost-benefit ratio.
The possible impact is substantial, since more than half of the drugs most commonly involved in adverse drug reactions are metabolized by polymorphic enzymes.1 Adverse drug reactions remain a significant detriment to public health, having a substantial impact on rates of morbidity and death and on healthcare costs.2–8 In the United States, adverse drug reactions are a leading cause of death in hospitalized patients4 and are annually responsible for hundreds of thousands of deaths and hundreds of billions of dollars in added costs.2,4,6–8
In the meantime, physicians can educate their patients and promote efforts to incorporate genomic information into standard clinical decision-making.
This article offers an overview of pharmacogenomic testing, discussing implications and limitations of a few validated tests. Specifically, we will discuss testing that is relevant when using warfarin (Coumadin), clopidogrel (Plavix), statins, tamoxifen (Nolvadex), codeine, and psychotropic medications, as well as the future role of pharmacogenomic testing in medicine.
WHAT IS PHARMACOGENOMICS?
Pharmacogenomics is the study of how genetic factors relate to interindividual variability of drug response.
Many clinicians may not be familiar with the background and terminology used in the pharmacogenomic literature. Below, a brief review of the terminology is followed by a schematic describing the various stages of research involved in pharmacogenomics and the advancement of a test into standard practice.
The review and schematic may be helpful for evaluating the clinical significance of pharmacogenomics-related articles.
From genotype to phenotype
Genotype refers to the coding sequence of DNA base pairs for a particular gene, and phenotype (eg, disease or drug response) refers to a trait resulting from the protein product encoded by the gene. The name of a gene often refers to its protein product and is italicized (eg, the CYP3A4 gene encodes for the CYP3A4 enzyme).
Two alleles per autosomal gene (one paternal and one maternal) form the genotype. Heterozygotes possess two different alleles, and homozygotes possess two of the same alleles. The most common allele in a population is referred to as the wild type, and allele frequencies can vary greatly in different populations.9
Most sequence variations are single nucleotide polymorphisms (SNPs, pronounced “snips”), a single DNA base pair substitution that may result in a different gene product. SNPs can be classified as structural RNA polymorphisms (srSNPs), regulatory polymorphisms (rSNPs), or polymorphisms in coding regions (cSNPs)10: srSNPs alter mRNA processing and translation, rSNPs alter transcription, and cSNPs alter protein sequence and function.
Recently, genetic associations with a phenotype have been done on a large scale, with millions of SNPs measured in each of many subjects. This approach, called a genomewide association study or GWAS, has revealed countless candidate genes for clinical traits, but only a few have resulted in a practical clinical application. SNPs may by themselves exert a pharmacokinetic effect (ie, how the body processes the drug), a pharmacodynamic effect (ie, how the drug affects the body), or both, or they may act in concert with other genetic factors. Pharmacodynamic effects can result from a pharmacokinetic effect or can result from variations in a pharmacologic target.
Establishing a genotype-phenotype association can involve clinical studies, animal transgenic studies, or molecular and cellular functional assays.
Clinical applications are emerging
WARFARIN: IMPORTANCE OF CYP2C9, VKORC1
Warfarin is used for the long-term treatment and prevention of thromboembolic events.
This drug has a narrow therapeutic window and shows substantial interpatient dose variability. The start of warfarin therapy is associated with one of the highest rates of adverse events and emergency room visits of any single drug.12 More than 2 million patients start warfarin each year in the United States alone,13 and about 20% of them are hospitalized within the first 6 months because of bleeding due to overanticoagulation.14
The findings from a recent study suggest that pharmacogenomic testing may eventually allow more patients to safely benefit from warfarin therapy. In this large, nationwide, prospective study, hospitalization rates were 30% lower when pharmacogenomic testing was used.14 However, no reduction was seen in the time needed to reach the target international normalized ratio (INR) or in the need for INR checks at 6 months. Furthermore, this study used historical control data, and some or all of the reduction in hospitalization rates may be attributed to more frequent INR checks in the patients who underwent testing than in the historical control group.
A relationship between warfarin dose requirements and the genetic status of CYP2C9, which encodes a major drug-metabolizing enzyme, has been demonstrated in retrospective and prospective studies.15–17
S-warfarin is metabolized by CYP2C9, which is polymorphic
Warfarin contains equal amounts of two isomers, designated S and R. S-warfarin, which is more potent, is metabolized principally by CYP2C9, while R-warfarin is metabolized by CYP1A2, CYP2C19, and CYP3A4.
People who possess two copies of the wild type CYP2C9 gene CYP2C9*1 metabolize warfarin very well and so are called “extensive warfarin metabolizers.” Carriers of the allelic variants CYP2C9*2 and CYP2C9*3 (which have point mutations in exons 3 and 7 of CYP2C9, respectively), have less capacity. Compared with those who are homozygous for the wild-type gene, homozygous carriers of CYP2C9* 3 clear S-warfarin at a rate that is 90% lower, and those with the CYP2C9*1/*3, CYP2C9* 1/*2, CYP2C9*2/*2, or CYP2C9*2/*3 genotypes clear it at a rate 50% to 75% lower. A meta-analysis of 12 studies found that the CYP2C9 genotype accounted for 12% of the interindividual variability of warfarin dose requirements.18
About 8% of whites carry at least one copy of CYP2C9*2, as do 1% of African Americans; the allele is rare in Asian populations. The frequency of CYP2C9*3 is 6% in whites, 1% in African Americans, and 3% in Asians.19,20 People with CYP2C9*4 or CYP2C9*5 have a diminished capacity to clear warfarin; however, these variants occur so infrequently that their clinical relevance may be minimal.
Warfarin’s target, VKOR, is also polymorphic
Genetic variation in warfarin’s pharmacologic target, vitamin K 2,3-epoxide reductase (VKOR), also influences dose requirements. Warfarin decreases the synthesis of vitamin-K-dependent clotting factors by inhibiting VKOR. This inhibition depends on the patient’s C1 subunit gene, VKORC1. Patients with a guanine-to-adenine SNP 1,639 bases upstream of VKORC1 (−1639G>A) need lower warfarin doses—an average of 25% lower in those with the GA genotype (ie, one allele has guanine in the −1639 position and the other allele has adenine in that position) and 50% lower in those with the AA genotype compared with the wild-type genotype GG.21 This promoter SNP, positioned upstream (ie, before the gene-coding region), greatly influences VKORC1 expression.
A meta-analysis of 10 studies found that the VKORC1 polymorphism accounts for 25% of the interindividual variation in warfarin dose.18 In one study, the frequency of the AA genotype in a white population was 14%, AG 47%, and GG 39%; in a Chinese population the frequency of AA was 82%, AG 18%, and GG 0.35%.22
CYP4F2 and GGCX also affect warfarin’s dose requirements
Genetic variations in the enzymes CYP4F2 and gamma-glutamyl carboxylase (GGCX) also influence warfarin dose requirements. Although the data are limited and the effects are smaller than those of CYP2C9 and VKORC1, people with a SNP in CYP4F2 need 8% higher doses of warfarin, while those with a SNP in GGCX need 6% lower doses.23
CYP2C9 and VKORC1 testing is available
Currently, the clinical pharmacogenetic tests relevant for warfarin use are for CYP2C9 and VKORC1.10
The FDA has approved four warfarin pharmacogenetic test kits, but most third-party payers are reluctant to reimburse for testing because it is not currently considered a standard of care. Testing typically costs a few hundred dollars, but it should become less expensive as it becomes more commonplace. The current FDA-approved product label for warfarin does not recommend routine pharmacogenomic testing for determining initial or maintenance doses, but it does acknowledge that dose requirements are influenced by CYP2C9 and VKORC1 and states that genotype information, when available, can assist in selecting the starting dose.24
Clinical trials of warfarin pharmacogenomic testing are under way
Although genetic status can greatly influence an individual patient’s warfarin dosing requirement, routine prospective pharmacogenomic testing is not endorsed by the FDA or by other expert panels26 because there is currently insufficient evidence to recommend for or against it.
Several large prospective trials are under way. For example, the National Heart, Lung, and Blood Institute began a prospective trial in about 1,200 patients to evaluate the use of clinical plus genetic information to guide the initiation of warfarin therapy and to improve anticoagulation control for patients.27 The results, expected in September 2011, and those of other large prospective trials should provide adequate evidence for making recommendations about the clinical utility of routine pharmacogenetic testing for guiding warfarin therapy.
Several recent cost-utility and cost-effectiveness studies have attempted to quantify the value of pharmacogenomic testing for warfarin therapy,28–30 but their analyses are largely limited because the benefit (clinical utility) is yet to be sufficiently characterized.
The relevance of such analyses may soon be drastically diminished, as several non-vitamin-K-dependent blood thinners such as rivaroxaban (Xarelto), dabigatran (Pradaxa), and apixaban are poised to enter clinical practice.31
CLOPIDOGREL IS ACTIVATED BY CYP2C19
Clopidogrel, taken by about 40 million patients worldwide, is used to prevent atherothrombotic events and cardiac stent thrombosis when given along with aspirin.
Studies of clopidogrel pharmacogenomics
A recent genome-wide association study conducted in a cohort of 429 healthy Amish persons revealed a SNP in CYP2C19 to be associated with a diminished response to clopidogrel and to account for 12% of the variation in drug response.33 Traditional factors (the patient’s age, body-mass index, and cholesterol level) combined accounted for less than 10% of the variation.
Findings were similar in a subsequent investigation in 227 cardiac patients receiving clopidogrel: 21% of those with the variant had a cardiovascular ischemic event or died during a 1-year follow-up period compared with 10% of those without the variant (hazard ratio 2.42, P = .02).33
A 12-year prospective study investigating clopidogrel efficacy in 300 cardiac patients under the age of 45 used cardiovascular death, nonfatal myocardial infarction, and urgent coronary revascularization as end points. It concluded that the only independent predictor of these events was the patient’s CYP2C19 status.34
A study in 2,200 patients with recent myocardial infarction examined whether any of the known allelic variations that modulate clopidogrel’s absorption (ABCB1), metabolic activation (CYP3A4/5 and CYP2C19), or biologic activity (P2RY12 and ITGB3) was associated with a higher rate of the combined end point of all-cause mortality, nonfatal myocardial infarction, or stroke. None of the SNPs in CYP3A4/5, P2RY12, or ITGB3 that were evaluated was associated with a higher risk at 1 year. However, the allelic variations modulating clopidogrel’s absorption (ABCB1) and metabolism (CYP2C19) were associated with higher event rates. Patients with two variant ABCB1 alleles had a higher adjusted hazard ratio (95% confidence interval [CI] 1.2–2.47) than those with the wild-type allele. Patients who had one or two CYP2C19 loss-of-function alleles had a higher event rate than those with two wild-type alleles (95% CI 1.10–3.58 and 1.71–7.51, respectively).35
Conversely, researchers from the Population Health Research Institute found no association between poor-metabolizer status and treatment outcomes when CYP2C19 analysis was retrospectively added to the findings of two large clinical trials (combined N > 5,000). However, patients with acute coronary syndrome benefited more from clopidogrel treatment if they were ultra-rapid metabolizers (possessing the gain-of-function allele CYP2C19*17).36
Current status of clopidogrel testing: Uncertain
A current FDA boxed warning states that poor CYP2C19 metabolizers may not benefit from clopidogrel and recommends that prescribers consider alternative treatment for patients in this category.37 However, routine CYP2C19 testing is not recommended, and no firm recommendations have been established regarding dose adjustments for CYP2C19 status.
In 2010, the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the American Heart Association collectively pronounced the current evidence base insufficient for recommending routine pharmacogenomic testing.39
Needed are large-scale studies examining the cost-effectiveness and clinical utility of genotype-guided clopidogrel therapy compared with other therapy options such as prasugrel (Effient), an analogue not metabolized by CYP2C19. One such study, sponsored by Medco Health Solutions, plans to enroll 14,600 cardiac patients and has an estimated completion date in June 2011.40 The expectation that clopidogrel will be available in generic form in 2012 adds to the uncertainty regarding the cost-effectiveness of CYP2C19 testing for clopidogrel therapy.
STATINS: SLC01B1*5 INCREASES MYOPATHY RISK
Statins lower the concentration of low-density lipoprotein cholesterol (LDL-C), resulting in a relative-risk reduction of about 20% for each 1 mmol/L (39 mg/dL) decrement in LDL-C.41 They are one of the most commonly prescribed classes of drugs, but their side effects can limit their appeal: statin-induced myopathy occurs in about 1:1,000 to 1:10,000 patients and is difficult to predict.
SLC01B1. The Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH), a genome-wide association study, recently found a SNP (SLCO1B1* 5) in the SLC01B1 gene to be associated with a higher risk of statin-induced myopathy in cardiac patients receiving simvastatin (Zocor) 40 or 80 mg daily.42 The SLC01B1 gene, located on chromosome 12, influences the extent of the drug’s hepatic uptake and its serum concentration. Only the SLC01B1*5 SNP emerged as a predictor of statin-induced myopathy across the entire genome.42
The authors believe the findings are likely to apply to other statins. The mechanisms leading to statin-induced myopathy and the impact of statin pharmacogenomics are still unclear.43
CYP3A4. Other genetic variants may play a vital role in determining response to statin therapy. Carriers of a newly identified CYP3A4 polymorphism (intron 6 SNP, rs35599367, C>T) required significantly lower statin doses (0.2–0.6 times less) for optimal lipid control. The analyses included atorvastatin (Lipitor), simvastatin, and lovastatin (Mevacor), and the association was robust (P = .019).44
Statin pharmacogenomic testing is not routinely recommended
Routine pharmacogenomic testing for statin therapy is not recommended. Additional studies are needed to determine the clinical utility and cost-effectiveness of pharmacogenomic testing (involving a combination of several polymorphisms) in various patient populations delineated by type of statin, dose, and concomitant use of other drugs.
TAMOXIFEN IS ACTIVATED BY CYP2D6
Tamoxifen is prescribed to prevent the recurrence of estrogen-receptor-positive breast cancer, to treat metastatic breast cancer, to prevent cancer in high-risk populations, and to treat ductal carcinoma in situ.
Tamoxifen is metabolized to form endoxifen, which has much higher potency and higher systemic levels than tamoxifen.45 Both CYP2D6 and CYP3A4/5 are required to produce endoxifen via two intermediates, but CYP2D6 catalyzes the critical step leading to metabolic activation.
The CYP2D6 gene is highly polymorphic, with more than 75 allelic variants identified. Extensive literature is available describing the influence of CYP2D6 polymorphisms on tamoxifen metabolism and therapy outcomes.46–52 Several CYP2D6 variants result in reduced or no enzyme activity, and people who have more than two normally functioning alleles have exaggerated enzyme activity (gene amplification).
Classification of CYP2D6 status
Several systems have been developed to categorize the phenotypic activity of CYP2D6 based on genotype.
A genetic basis for the observed diversity in the metabolism of cytochrome P450 substrates was recognized more than 30 years ago. People were categorized as either extensive or poor metabolizers, reflecting normal vs impaired ability to metabolize the CYP2D6 substrates sparteine and debrisoquine. Later work expanded this system to include categories for intermediate (between poor and extensive) and ultra-rapid (better than extensive) metabolizers.
The genetic basis for these categories includes homozygosity for dysfunctional variants (the poor-metabolizer group) or extra copies of normal functioning variants (the ultra-rapid-metabolizer group).
Newer systems have been described for characterizing the CYP2D6 activity phenotype whereby CYP2D6 variants are assigned activity scores.53–56 The various scoring systems have been reviewed by Kirchheiner.57
A recent version of the activity scoring system also takes into consideration the many drugs that inhibit CYP2D6, such as amiodarone (Cordarone) and fluoxetine (Prozac) that can reduce the action of tamoxifen if given with it (Table 4).58 For example, the tamoxifen exposure (as predicted by the CYP2D6-activity score) experienced by a CYP2D6 extensive metabolizer taking a CYP2D6-inhibiting drug may be similar to the exposure experienced by a CYP2D6 poor metabolizer receiving the same tamoxifen dose but not taking a CYP2D6-inhibiting drug.
Likewise, the activity score of a CYP2D6 intermediate metabolizer taking a CYP2D6-inducing drug may be similar to that of a CYP2D6 ultra-rapid metabolizer not taking a CYP2D6-inducing drug. Examples of CYP2D6 inducers are dexamethasone, rifampin, and hyperforin (St. John’s wort).
While the newer systems are reported to provide better correlations between genotype and phenotype scores, the older scoring systems and the categorical names are still widely used (eg, in the FDA-approved AmpliChip CYP450 test from Roche,59 which includes genotype data for CYP2D6 and CYP2C19).
No firm recommendations for CYP2D6 testing in tamoxifen users
The different genotypes and phenotypes vary in prevalence in different ethnic groups, and significantly different activity levels for endoxifen formation are observed. Tamoxifen lacks efficacy in those who are poor CYP2D6 metabolizers—ie, about 7% of the white population.
However, the FDA has not made firm recommendations about CYP2D6 testing for prescribing tamoxifen because the evidence of benefit, although suggestive, has been considered insufficient.
Clinicians should be aware that tamoxifen’s efficacy is greatly reduced by concomitant therapy with CYP2D6-inhibiting drugs (Table 4).
Other genes affecting tamoxifen: CYP3A4/5, SULT1A1, and UGT2B15
Some investigators propose that polymorphisms in additional genes encoding enzymes in the tamoxifen metabolic and elimination pathways (eg, CYP3A4/5, SULT1A1, and UGT2B15) also need to be considered to account adequately for interindividual variation in drug response.
For example, CYP3A4 and CYP3A5 are also polymorphic, and large interindividual variation exists in their enzyme activities. These enzymes have overlapping substrate specificities, represent the most abundant drug-metabolizing enzymes in the human liver, and are involved in the biotransformation of a broad range of endogenous substrates and most drugs.60
Clinical studies evaluating the impact of CYP3A4/5 polymorphisms have been inconsistent in their conclusions, which is generally attributed to the relatively low functional impact or the low prevalence of the SNPs evaluated. Many of the nearly 100 CYP3A4/5 polymorphisms identified have not yet been characterized regarding their functional impact on enzyme expression or activity. CYP-3A4/5 enzyme activity is highly variable between individuals and warrants further study of its role in outcomes of tamoxifen therapy. Ongoing and future prospective clinical trials evaluating CYP2D6, CYP3A4/5, and other relevant polymorphisms are necessary to define their clinical relevance before routine genetic testing for tamoxifen can be justified.
CODEINE IS ALSO ACTIVATED BY CYP2D6
Codeine also depends on the CYP2D6 gene, as it must be activated to its more potent opioid metabolites, including morphine. Poor CYP2D6 metabolizers do not benefit from codeine therapy.
The pharmacogenomics of codeine has become a hot topic, especially regarding breast-feeding mothers. The debate was ignited with the publication in 2006 of a case report of an infant’s death, apparently the result of metabolic polymorphisms.61 The evolution of this debate and the outcome of the case may be noteworthy to clinicians, as they illustrate the gravity of public and patient interest in pharmacogenomic testing. In this case, the breast-feeding mother had taken codeine regularly for about 14 days when her 13-day-old infant died from toxic levels of morphine. Unknown to her and the prescriber, both the mother and infant were ultra-rapid CYP2D6 metabolizers, resulting in a more rapid and extensive conversion of codeine to morphine.
A logical strategy for preventing similar deaths would be routine CYP2D6 genotyping when prescribing codeine to breast-feeding mothers. However, after several investigations examined the metabolic and excretion pathways of codeine in their entirety, the FDA did not recommend routine CYP2D6 testing when prescribing codeine to breastfeeding mothers because several other factors, including rare genetic variations of other enzymes, proved necessary for reaching the opioid toxicity leading to the infant’s death.62
PHARMACOGENOMICS OF PSYCHOTROPIC DRUGS
Pharmacogenomic testing has clinical utility for some psychotropic drugs.
HLA-B and carbamazepine
Considered a standard of care, HLA-B genotyping is appropriate before prescribing carbamazepine (Tegretol, Equetro) to patients in populations in which HLAB*1502 is likely to be present, such as Asians. Carriers of HLAB* 1502 are at higher risk of life-threatening skin reactions such as Stevens-Johnson syndrome.11
Several other pharmacogenomic applications for psychotropic medications have been suggested, but routine testing has not been recommended by the FDA or endorsed by any expert panel because sufficient clinical utility and cost-effectiveness have not been demonstrated. A brief summary of study findings and a few practical suggestions follow.
Polymorphisms in metabolizing enzymes have been investigated in patients receiving psychotropic drugs.
CYP2D6 and antidepressants
Many antidepressants show significant differences in plasma drug levels with CYP2D6 polymorphisms (in descending order of influence)55:
- Imipramine (Tofranil)
- Doxepin (Adapin, Silenor, Sinequan)
- Maprotiline (Deprilept, Ludiomil, Psymion)
- Trimipramine (Surmontil)
- Desipramine (Noraprim)
- Nortriptyline (Aventyl, Pamelor)
- Clomipramine (Anafranil)
- Paroxetine (Paxil)
- Venlafaxine (Effexor)
- Amitriptyline (Elavil)
- Mianserin
- Trazadone (Desyrel)
- Bupropion (Wellbutrin)
- Nefazodone (Serzone)
- Citalopram (Celexa)
- Sertraline (Zoloft).
CYP2D6 and antipsychotics
Several antipsychotics are also influenced by CYP2D6 polymorphisms (also in descending order of influence)55:
- Perphenazine (Trilafon)
- Thioridazine (Mellaril)
- Olanzapine (Zyprexa)
- Zuclopenthixol (Cisordinol, Clopixol, Acuphase)
- Aripiprazole (Abilify)
- Flupentixol (Depixol, Fluanxol)
- Haloperidol (Haldol)
- Perazine (Taxilan)
- Risperidone (Risperdal)
- Pimozide (Orap).
CYP2C19 and antidepressants
CYP2C19 polymorphisms are likewise associated with differences in drug metabolism for many antidepressants, such as (in descending order of CYP2C19-mediated influence)55:
- Trimipramine
- Doxepin
- Amitriptyline
- Imipramine
- Citalopram (Celexa)
- Clomipramine
- Moclobemide (Aurorix, Manerix)
- Sertraline
- Fluvoxamine (Luvox).
Clinical relevance of CYP2D6 and CYP2C19
Several studies have demonstrated that poor and intermediate CYP2D6 metabolizers have a higher incidence of adverse effects when taking CYP2D6-dependent antidepressants63–68; however, an almost equal number of studies did not find statistically significant associations.69–72 Likewise, several studies have found an association between ultra-rapid CYP2D6 metabolizer status and diminished response to antidepressants,65,73,74 but no association was found in a larger retrospective study.75
Routine CYP2D6 and CYP2C19 screening is not recommended when prescribing psychotropic drugs. However, reviews of the pharmacokinetic data have suggested a few practical applications when genetic status is already known. In general, clinicians can consider reducing the dose of tricyclic antidepressants by about 50% when prescribing to CYP2D6-poor-metabolizers.55,76–78
Genes that affect serotonin metabolism
Several genes in the serotonin pathway have been investigated to determine whether they influence patients’ susceptibility to depression and adverse effects and response to psychotropic medications.
SLC6A4. Polymorphisms in the promoter region of the serotonin transporter gene SLC6A4 appear to influence the treatment response and side-effect profiles of selective serotonin reuptake inhibitors (SSRIs). Carriers of the SLC6A4 5-HTTLPR L alleles have fewer side effects79 and better response to SSRI treatment, and carriers of the S allele have a higher incidence of antidepressant-induced mania80 and poorer response to SSRI treatment.81
5-HT. Polymorphisms in serotonin receptors (2A and 2C subtypes) appear to influence SSRI response and side effects. Carriers of 5-HT 2A C alleles had more severe adverse effects from paroxetine,71 but another 5-HT 2A polymorphism common to Asians is associated with better response to antidepressant therapy.82 A 5-HT 2C polymorphism was associated with a lower incidence of antipsychotic-induced weight gain.83
Although the understanding of these relationships is incomplete and routine pharmacogenomic testing is not currently recommended, reviews of the pharmacodynamic data have suggested a few practical applications when a patient’s genetic status is already known. One should consider:
- Selecting treatments other than SSRIs for depressed patients known to possess the SLC6A4 variant
- Selecting citalopram for depressed patients known to carry the 5-HT 2A polymorphism
- Avoiding treatment with antipsychotic drugs for patients known to possess the 5-HT 2C polymorphism.
THE FUTURE OF PHARMACOGENOMIC TESTING
The examples discussed in this article provide some insight about how pharmacogenomic testing is maturing and slowly being integrated into the practice of medicine. They also illustrate the complexity of the multiple stages of research that pharmacogenomic applications must go through in order to be adopted as standard practice.
In the future, pharmacogenomic data will continue to accumulate, and the clinical utility of many other pharmacogenomic tests may be uncovered. The FDA provides information on emerging pharmacogenomic tests at its Web site, www.fda.gov.11 Its up-to-date “Table of Valid Genomic Biomarkers in the Context of Approved Drug Labels” includes boxed warnings, recommendations, research outcomes, and relevant population genetics.
If the FDA continues its current policy, prospective randomized trials that show improvement in patient outcomes will remain the gold standard for determining the clinical significance of a pharmacogenomic test. Furthermore, cost-benefit analyses are likely to continue dictating policy regarding pharmacogenomic testing, and cost-benefit profiles should improve as technology advances and as information gathered from a single test becomes applicable to multiple medications and clinical scenarios.
In the meantime, physicians should become familiar with the terms used in medical genetics and pharmacogenomics and begin to understand genetic contributions to the outcomes of drug therapy. For example, understanding the consequences of metabolizer status and the frequency of variants in a given population can be tremendously helpful when advising our patients about anticipating potential problems when taking specific medications and about making informed decisions about pharmacogenomic testing.
This exchange of information alone may go a long way in improving therapy outcomes even when prospective pharmacogenomic testing is not routinely performed. Furthermore, an increasing number of patients will already have genotyping information available when they come to us, and clinicians need to be aware of the many pharmacogenomic applications recommended by the FDA when genetic status is known.10
In many patients, certain drugs do not work as well as expected, whereas in other patients they cause toxic effects, even at lower doses. For some patients, the reason may be genetic.
Sizeable minorities of the population carry genetic variants—polymorphisms— that affect their response to various drugs. Thanks to genetic research, our understanding of the variability of drug response has advanced markedly in the last decade. Many relevant polymorphisms have been identified, and tests for some of them are available.
Armed with the knowledge of their patients’ genetic status, physicians could predict their response to certain drugs, leading to better efficacy, fewer adverse drug reactions, and a better cost-benefit ratio.
The possible impact is substantial, since more than half of the drugs most commonly involved in adverse drug reactions are metabolized by polymorphic enzymes.1 Adverse drug reactions remain a significant detriment to public health, having a substantial impact on rates of morbidity and death and on healthcare costs.2–8 In the United States, adverse drug reactions are a leading cause of death in hospitalized patients4 and are annually responsible for hundreds of thousands of deaths and hundreds of billions of dollars in added costs.2,4,6–8
In the meantime, physicians can educate their patients and promote efforts to incorporate genomic information into standard clinical decision-making.
This article offers an overview of pharmacogenomic testing, discussing implications and limitations of a few validated tests. Specifically, we will discuss testing that is relevant when using warfarin (Coumadin), clopidogrel (Plavix), statins, tamoxifen (Nolvadex), codeine, and psychotropic medications, as well as the future role of pharmacogenomic testing in medicine.
WHAT IS PHARMACOGENOMICS?
Pharmacogenomics is the study of how genetic factors relate to interindividual variability of drug response.
Many clinicians may not be familiar with the background and terminology used in the pharmacogenomic literature. Below, a brief review of the terminology is followed by a schematic describing the various stages of research involved in pharmacogenomics and the advancement of a test into standard practice.
The review and schematic may be helpful for evaluating the clinical significance of pharmacogenomics-related articles.
From genotype to phenotype
Genotype refers to the coding sequence of DNA base pairs for a particular gene, and phenotype (eg, disease or drug response) refers to a trait resulting from the protein product encoded by the gene. The name of a gene often refers to its protein product and is italicized (eg, the CYP3A4 gene encodes for the CYP3A4 enzyme).
Two alleles per autosomal gene (one paternal and one maternal) form the genotype. Heterozygotes possess two different alleles, and homozygotes possess two of the same alleles. The most common allele in a population is referred to as the wild type, and allele frequencies can vary greatly in different populations.9
Most sequence variations are single nucleotide polymorphisms (SNPs, pronounced “snips”), a single DNA base pair substitution that may result in a different gene product. SNPs can be classified as structural RNA polymorphisms (srSNPs), regulatory polymorphisms (rSNPs), or polymorphisms in coding regions (cSNPs)10: srSNPs alter mRNA processing and translation, rSNPs alter transcription, and cSNPs alter protein sequence and function.
Recently, genetic associations with a phenotype have been done on a large scale, with millions of SNPs measured in each of many subjects. This approach, called a genomewide association study or GWAS, has revealed countless candidate genes for clinical traits, but only a few have resulted in a practical clinical application. SNPs may by themselves exert a pharmacokinetic effect (ie, how the body processes the drug), a pharmacodynamic effect (ie, how the drug affects the body), or both, or they may act in concert with other genetic factors. Pharmacodynamic effects can result from a pharmacokinetic effect or can result from variations in a pharmacologic target.
Establishing a genotype-phenotype association can involve clinical studies, animal transgenic studies, or molecular and cellular functional assays.
Clinical applications are emerging
WARFARIN: IMPORTANCE OF CYP2C9, VKORC1
Warfarin is used for the long-term treatment and prevention of thromboembolic events.
This drug has a narrow therapeutic window and shows substantial interpatient dose variability. The start of warfarin therapy is associated with one of the highest rates of adverse events and emergency room visits of any single drug.12 More than 2 million patients start warfarin each year in the United States alone,13 and about 20% of them are hospitalized within the first 6 months because of bleeding due to overanticoagulation.14
The findings from a recent study suggest that pharmacogenomic testing may eventually allow more patients to safely benefit from warfarin therapy. In this large, nationwide, prospective study, hospitalization rates were 30% lower when pharmacogenomic testing was used.14 However, no reduction was seen in the time needed to reach the target international normalized ratio (INR) or in the need for INR checks at 6 months. Furthermore, this study used historical control data, and some or all of the reduction in hospitalization rates may be attributed to more frequent INR checks in the patients who underwent testing than in the historical control group.
A relationship between warfarin dose requirements and the genetic status of CYP2C9, which encodes a major drug-metabolizing enzyme, has been demonstrated in retrospective and prospective studies.15–17
S-warfarin is metabolized by CYP2C9, which is polymorphic
Warfarin contains equal amounts of two isomers, designated S and R. S-warfarin, which is more potent, is metabolized principally by CYP2C9, while R-warfarin is metabolized by CYP1A2, CYP2C19, and CYP3A4.
People who possess two copies of the wild type CYP2C9 gene CYP2C9*1 metabolize warfarin very well and so are called “extensive warfarin metabolizers.” Carriers of the allelic variants CYP2C9*2 and CYP2C9*3 (which have point mutations in exons 3 and 7 of CYP2C9, respectively), have less capacity. Compared with those who are homozygous for the wild-type gene, homozygous carriers of CYP2C9* 3 clear S-warfarin at a rate that is 90% lower, and those with the CYP2C9*1/*3, CYP2C9* 1/*2, CYP2C9*2/*2, or CYP2C9*2/*3 genotypes clear it at a rate 50% to 75% lower. A meta-analysis of 12 studies found that the CYP2C9 genotype accounted for 12% of the interindividual variability of warfarin dose requirements.18
About 8% of whites carry at least one copy of CYP2C9*2, as do 1% of African Americans; the allele is rare in Asian populations. The frequency of CYP2C9*3 is 6% in whites, 1% in African Americans, and 3% in Asians.19,20 People with CYP2C9*4 or CYP2C9*5 have a diminished capacity to clear warfarin; however, these variants occur so infrequently that their clinical relevance may be minimal.
Warfarin’s target, VKOR, is also polymorphic
Genetic variation in warfarin’s pharmacologic target, vitamin K 2,3-epoxide reductase (VKOR), also influences dose requirements. Warfarin decreases the synthesis of vitamin-K-dependent clotting factors by inhibiting VKOR. This inhibition depends on the patient’s C1 subunit gene, VKORC1. Patients with a guanine-to-adenine SNP 1,639 bases upstream of VKORC1 (−1639G>A) need lower warfarin doses—an average of 25% lower in those with the GA genotype (ie, one allele has guanine in the −1639 position and the other allele has adenine in that position) and 50% lower in those with the AA genotype compared with the wild-type genotype GG.21 This promoter SNP, positioned upstream (ie, before the gene-coding region), greatly influences VKORC1 expression.
A meta-analysis of 10 studies found that the VKORC1 polymorphism accounts for 25% of the interindividual variation in warfarin dose.18 In one study, the frequency of the AA genotype in a white population was 14%, AG 47%, and GG 39%; in a Chinese population the frequency of AA was 82%, AG 18%, and GG 0.35%.22
CYP4F2 and GGCX also affect warfarin’s dose requirements
Genetic variations in the enzymes CYP4F2 and gamma-glutamyl carboxylase (GGCX) also influence warfarin dose requirements. Although the data are limited and the effects are smaller than those of CYP2C9 and VKORC1, people with a SNP in CYP4F2 need 8% higher doses of warfarin, while those with a SNP in GGCX need 6% lower doses.23
CYP2C9 and VKORC1 testing is available
Currently, the clinical pharmacogenetic tests relevant for warfarin use are for CYP2C9 and VKORC1.10
The FDA has approved four warfarin pharmacogenetic test kits, but most third-party payers are reluctant to reimburse for testing because it is not currently considered a standard of care. Testing typically costs a few hundred dollars, but it should become less expensive as it becomes more commonplace. The current FDA-approved product label for warfarin does not recommend routine pharmacogenomic testing for determining initial or maintenance doses, but it does acknowledge that dose requirements are influenced by CYP2C9 and VKORC1 and states that genotype information, when available, can assist in selecting the starting dose.24
Clinical trials of warfarin pharmacogenomic testing are under way
Although genetic status can greatly influence an individual patient’s warfarin dosing requirement, routine prospective pharmacogenomic testing is not endorsed by the FDA or by other expert panels26 because there is currently insufficient evidence to recommend for or against it.
Several large prospective trials are under way. For example, the National Heart, Lung, and Blood Institute began a prospective trial in about 1,200 patients to evaluate the use of clinical plus genetic information to guide the initiation of warfarin therapy and to improve anticoagulation control for patients.27 The results, expected in September 2011, and those of other large prospective trials should provide adequate evidence for making recommendations about the clinical utility of routine pharmacogenetic testing for guiding warfarin therapy.
Several recent cost-utility and cost-effectiveness studies have attempted to quantify the value of pharmacogenomic testing for warfarin therapy,28–30 but their analyses are largely limited because the benefit (clinical utility) is yet to be sufficiently characterized.
The relevance of such analyses may soon be drastically diminished, as several non-vitamin-K-dependent blood thinners such as rivaroxaban (Xarelto), dabigatran (Pradaxa), and apixaban are poised to enter clinical practice.31
CLOPIDOGREL IS ACTIVATED BY CYP2C19
Clopidogrel, taken by about 40 million patients worldwide, is used to prevent atherothrombotic events and cardiac stent thrombosis when given along with aspirin.
Studies of clopidogrel pharmacogenomics
A recent genome-wide association study conducted in a cohort of 429 healthy Amish persons revealed a SNP in CYP2C19 to be associated with a diminished response to clopidogrel and to account for 12% of the variation in drug response.33 Traditional factors (the patient’s age, body-mass index, and cholesterol level) combined accounted for less than 10% of the variation.
Findings were similar in a subsequent investigation in 227 cardiac patients receiving clopidogrel: 21% of those with the variant had a cardiovascular ischemic event or died during a 1-year follow-up period compared with 10% of those without the variant (hazard ratio 2.42, P = .02).33
A 12-year prospective study investigating clopidogrel efficacy in 300 cardiac patients under the age of 45 used cardiovascular death, nonfatal myocardial infarction, and urgent coronary revascularization as end points. It concluded that the only independent predictor of these events was the patient’s CYP2C19 status.34
A study in 2,200 patients with recent myocardial infarction examined whether any of the known allelic variations that modulate clopidogrel’s absorption (ABCB1), metabolic activation (CYP3A4/5 and CYP2C19), or biologic activity (P2RY12 and ITGB3) was associated with a higher rate of the combined end point of all-cause mortality, nonfatal myocardial infarction, or stroke. None of the SNPs in CYP3A4/5, P2RY12, or ITGB3 that were evaluated was associated with a higher risk at 1 year. However, the allelic variations modulating clopidogrel’s absorption (ABCB1) and metabolism (CYP2C19) were associated with higher event rates. Patients with two variant ABCB1 alleles had a higher adjusted hazard ratio (95% confidence interval [CI] 1.2–2.47) than those with the wild-type allele. Patients who had one or two CYP2C19 loss-of-function alleles had a higher event rate than those with two wild-type alleles (95% CI 1.10–3.58 and 1.71–7.51, respectively).35
Conversely, researchers from the Population Health Research Institute found no association between poor-metabolizer status and treatment outcomes when CYP2C19 analysis was retrospectively added to the findings of two large clinical trials (combined N > 5,000). However, patients with acute coronary syndrome benefited more from clopidogrel treatment if they were ultra-rapid metabolizers (possessing the gain-of-function allele CYP2C19*17).36
Current status of clopidogrel testing: Uncertain
A current FDA boxed warning states that poor CYP2C19 metabolizers may not benefit from clopidogrel and recommends that prescribers consider alternative treatment for patients in this category.37 However, routine CYP2C19 testing is not recommended, and no firm recommendations have been established regarding dose adjustments for CYP2C19 status.
In 2010, the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the American Heart Association collectively pronounced the current evidence base insufficient for recommending routine pharmacogenomic testing.39
Needed are large-scale studies examining the cost-effectiveness and clinical utility of genotype-guided clopidogrel therapy compared with other therapy options such as prasugrel (Effient), an analogue not metabolized by CYP2C19. One such study, sponsored by Medco Health Solutions, plans to enroll 14,600 cardiac patients and has an estimated completion date in June 2011.40 The expectation that clopidogrel will be available in generic form in 2012 adds to the uncertainty regarding the cost-effectiveness of CYP2C19 testing for clopidogrel therapy.
STATINS: SLC01B1*5 INCREASES MYOPATHY RISK
Statins lower the concentration of low-density lipoprotein cholesterol (LDL-C), resulting in a relative-risk reduction of about 20% for each 1 mmol/L (39 mg/dL) decrement in LDL-C.41 They are one of the most commonly prescribed classes of drugs, but their side effects can limit their appeal: statin-induced myopathy occurs in about 1:1,000 to 1:10,000 patients and is difficult to predict.
SLC01B1. The Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH), a genome-wide association study, recently found a SNP (SLCO1B1* 5) in the SLC01B1 gene to be associated with a higher risk of statin-induced myopathy in cardiac patients receiving simvastatin (Zocor) 40 or 80 mg daily.42 The SLC01B1 gene, located on chromosome 12, influences the extent of the drug’s hepatic uptake and its serum concentration. Only the SLC01B1*5 SNP emerged as a predictor of statin-induced myopathy across the entire genome.42
The authors believe the findings are likely to apply to other statins. The mechanisms leading to statin-induced myopathy and the impact of statin pharmacogenomics are still unclear.43
CYP3A4. Other genetic variants may play a vital role in determining response to statin therapy. Carriers of a newly identified CYP3A4 polymorphism (intron 6 SNP, rs35599367, C>T) required significantly lower statin doses (0.2–0.6 times less) for optimal lipid control. The analyses included atorvastatin (Lipitor), simvastatin, and lovastatin (Mevacor), and the association was robust (P = .019).44
Statin pharmacogenomic testing is not routinely recommended
Routine pharmacogenomic testing for statin therapy is not recommended. Additional studies are needed to determine the clinical utility and cost-effectiveness of pharmacogenomic testing (involving a combination of several polymorphisms) in various patient populations delineated by type of statin, dose, and concomitant use of other drugs.
TAMOXIFEN IS ACTIVATED BY CYP2D6
Tamoxifen is prescribed to prevent the recurrence of estrogen-receptor-positive breast cancer, to treat metastatic breast cancer, to prevent cancer in high-risk populations, and to treat ductal carcinoma in situ.
Tamoxifen is metabolized to form endoxifen, which has much higher potency and higher systemic levels than tamoxifen.45 Both CYP2D6 and CYP3A4/5 are required to produce endoxifen via two intermediates, but CYP2D6 catalyzes the critical step leading to metabolic activation.
The CYP2D6 gene is highly polymorphic, with more than 75 allelic variants identified. Extensive literature is available describing the influence of CYP2D6 polymorphisms on tamoxifen metabolism and therapy outcomes.46–52 Several CYP2D6 variants result in reduced or no enzyme activity, and people who have more than two normally functioning alleles have exaggerated enzyme activity (gene amplification).
Classification of CYP2D6 status
Several systems have been developed to categorize the phenotypic activity of CYP2D6 based on genotype.
A genetic basis for the observed diversity in the metabolism of cytochrome P450 substrates was recognized more than 30 years ago. People were categorized as either extensive or poor metabolizers, reflecting normal vs impaired ability to metabolize the CYP2D6 substrates sparteine and debrisoquine. Later work expanded this system to include categories for intermediate (between poor and extensive) and ultra-rapid (better than extensive) metabolizers.
The genetic basis for these categories includes homozygosity for dysfunctional variants (the poor-metabolizer group) or extra copies of normal functioning variants (the ultra-rapid-metabolizer group).
Newer systems have been described for characterizing the CYP2D6 activity phenotype whereby CYP2D6 variants are assigned activity scores.53–56 The various scoring systems have been reviewed by Kirchheiner.57
A recent version of the activity scoring system also takes into consideration the many drugs that inhibit CYP2D6, such as amiodarone (Cordarone) and fluoxetine (Prozac) that can reduce the action of tamoxifen if given with it (Table 4).58 For example, the tamoxifen exposure (as predicted by the CYP2D6-activity score) experienced by a CYP2D6 extensive metabolizer taking a CYP2D6-inhibiting drug may be similar to the exposure experienced by a CYP2D6 poor metabolizer receiving the same tamoxifen dose but not taking a CYP2D6-inhibiting drug.
Likewise, the activity score of a CYP2D6 intermediate metabolizer taking a CYP2D6-inducing drug may be similar to that of a CYP2D6 ultra-rapid metabolizer not taking a CYP2D6-inducing drug. Examples of CYP2D6 inducers are dexamethasone, rifampin, and hyperforin (St. John’s wort).
While the newer systems are reported to provide better correlations between genotype and phenotype scores, the older scoring systems and the categorical names are still widely used (eg, in the FDA-approved AmpliChip CYP450 test from Roche,59 which includes genotype data for CYP2D6 and CYP2C19).
No firm recommendations for CYP2D6 testing in tamoxifen users
The different genotypes and phenotypes vary in prevalence in different ethnic groups, and significantly different activity levels for endoxifen formation are observed. Tamoxifen lacks efficacy in those who are poor CYP2D6 metabolizers—ie, about 7% of the white population.
However, the FDA has not made firm recommendations about CYP2D6 testing for prescribing tamoxifen because the evidence of benefit, although suggestive, has been considered insufficient.
Clinicians should be aware that tamoxifen’s efficacy is greatly reduced by concomitant therapy with CYP2D6-inhibiting drugs (Table 4).
Other genes affecting tamoxifen: CYP3A4/5, SULT1A1, and UGT2B15
Some investigators propose that polymorphisms in additional genes encoding enzymes in the tamoxifen metabolic and elimination pathways (eg, CYP3A4/5, SULT1A1, and UGT2B15) also need to be considered to account adequately for interindividual variation in drug response.
For example, CYP3A4 and CYP3A5 are also polymorphic, and large interindividual variation exists in their enzyme activities. These enzymes have overlapping substrate specificities, represent the most abundant drug-metabolizing enzymes in the human liver, and are involved in the biotransformation of a broad range of endogenous substrates and most drugs.60
Clinical studies evaluating the impact of CYP3A4/5 polymorphisms have been inconsistent in their conclusions, which is generally attributed to the relatively low functional impact or the low prevalence of the SNPs evaluated. Many of the nearly 100 CYP3A4/5 polymorphisms identified have not yet been characterized regarding their functional impact on enzyme expression or activity. CYP-3A4/5 enzyme activity is highly variable between individuals and warrants further study of its role in outcomes of tamoxifen therapy. Ongoing and future prospective clinical trials evaluating CYP2D6, CYP3A4/5, and other relevant polymorphisms are necessary to define their clinical relevance before routine genetic testing for tamoxifen can be justified.
CODEINE IS ALSO ACTIVATED BY CYP2D6
Codeine also depends on the CYP2D6 gene, as it must be activated to its more potent opioid metabolites, including morphine. Poor CYP2D6 metabolizers do not benefit from codeine therapy.
The pharmacogenomics of codeine has become a hot topic, especially regarding breast-feeding mothers. The debate was ignited with the publication in 2006 of a case report of an infant’s death, apparently the result of metabolic polymorphisms.61 The evolution of this debate and the outcome of the case may be noteworthy to clinicians, as they illustrate the gravity of public and patient interest in pharmacogenomic testing. In this case, the breast-feeding mother had taken codeine regularly for about 14 days when her 13-day-old infant died from toxic levels of morphine. Unknown to her and the prescriber, both the mother and infant were ultra-rapid CYP2D6 metabolizers, resulting in a more rapid and extensive conversion of codeine to morphine.
A logical strategy for preventing similar deaths would be routine CYP2D6 genotyping when prescribing codeine to breast-feeding mothers. However, after several investigations examined the metabolic and excretion pathways of codeine in their entirety, the FDA did not recommend routine CYP2D6 testing when prescribing codeine to breastfeeding mothers because several other factors, including rare genetic variations of other enzymes, proved necessary for reaching the opioid toxicity leading to the infant’s death.62
PHARMACOGENOMICS OF PSYCHOTROPIC DRUGS
Pharmacogenomic testing has clinical utility for some psychotropic drugs.
HLA-B and carbamazepine
Considered a standard of care, HLA-B genotyping is appropriate before prescribing carbamazepine (Tegretol, Equetro) to patients in populations in which HLAB*1502 is likely to be present, such as Asians. Carriers of HLAB* 1502 are at higher risk of life-threatening skin reactions such as Stevens-Johnson syndrome.11
Several other pharmacogenomic applications for psychotropic medications have been suggested, but routine testing has not been recommended by the FDA or endorsed by any expert panel because sufficient clinical utility and cost-effectiveness have not been demonstrated. A brief summary of study findings and a few practical suggestions follow.
Polymorphisms in metabolizing enzymes have been investigated in patients receiving psychotropic drugs.
CYP2D6 and antidepressants
Many antidepressants show significant differences in plasma drug levels with CYP2D6 polymorphisms (in descending order of influence)55:
- Imipramine (Tofranil)
- Doxepin (Adapin, Silenor, Sinequan)
- Maprotiline (Deprilept, Ludiomil, Psymion)
- Trimipramine (Surmontil)
- Desipramine (Noraprim)
- Nortriptyline (Aventyl, Pamelor)
- Clomipramine (Anafranil)
- Paroxetine (Paxil)
- Venlafaxine (Effexor)
- Amitriptyline (Elavil)
- Mianserin
- Trazadone (Desyrel)
- Bupropion (Wellbutrin)
- Nefazodone (Serzone)
- Citalopram (Celexa)
- Sertraline (Zoloft).
CYP2D6 and antipsychotics
Several antipsychotics are also influenced by CYP2D6 polymorphisms (also in descending order of influence)55:
- Perphenazine (Trilafon)
- Thioridazine (Mellaril)
- Olanzapine (Zyprexa)
- Zuclopenthixol (Cisordinol, Clopixol, Acuphase)
- Aripiprazole (Abilify)
- Flupentixol (Depixol, Fluanxol)
- Haloperidol (Haldol)
- Perazine (Taxilan)
- Risperidone (Risperdal)
- Pimozide (Orap).
CYP2C19 and antidepressants
CYP2C19 polymorphisms are likewise associated with differences in drug metabolism for many antidepressants, such as (in descending order of CYP2C19-mediated influence)55:
- Trimipramine
- Doxepin
- Amitriptyline
- Imipramine
- Citalopram (Celexa)
- Clomipramine
- Moclobemide (Aurorix, Manerix)
- Sertraline
- Fluvoxamine (Luvox).
Clinical relevance of CYP2D6 and CYP2C19
Several studies have demonstrated that poor and intermediate CYP2D6 metabolizers have a higher incidence of adverse effects when taking CYP2D6-dependent antidepressants63–68; however, an almost equal number of studies did not find statistically significant associations.69–72 Likewise, several studies have found an association between ultra-rapid CYP2D6 metabolizer status and diminished response to antidepressants,65,73,74 but no association was found in a larger retrospective study.75
Routine CYP2D6 and CYP2C19 screening is not recommended when prescribing psychotropic drugs. However, reviews of the pharmacokinetic data have suggested a few practical applications when genetic status is already known. In general, clinicians can consider reducing the dose of tricyclic antidepressants by about 50% when prescribing to CYP2D6-poor-metabolizers.55,76–78
Genes that affect serotonin metabolism
Several genes in the serotonin pathway have been investigated to determine whether they influence patients’ susceptibility to depression and adverse effects and response to psychotropic medications.
SLC6A4. Polymorphisms in the promoter region of the serotonin transporter gene SLC6A4 appear to influence the treatment response and side-effect profiles of selective serotonin reuptake inhibitors (SSRIs). Carriers of the SLC6A4 5-HTTLPR L alleles have fewer side effects79 and better response to SSRI treatment, and carriers of the S allele have a higher incidence of antidepressant-induced mania80 and poorer response to SSRI treatment.81
5-HT. Polymorphisms in serotonin receptors (2A and 2C subtypes) appear to influence SSRI response and side effects. Carriers of 5-HT 2A C alleles had more severe adverse effects from paroxetine,71 but another 5-HT 2A polymorphism common to Asians is associated with better response to antidepressant therapy.82 A 5-HT 2C polymorphism was associated with a lower incidence of antipsychotic-induced weight gain.83
Although the understanding of these relationships is incomplete and routine pharmacogenomic testing is not currently recommended, reviews of the pharmacodynamic data have suggested a few practical applications when a patient’s genetic status is already known. One should consider:
- Selecting treatments other than SSRIs for depressed patients known to possess the SLC6A4 variant
- Selecting citalopram for depressed patients known to carry the 5-HT 2A polymorphism
- Avoiding treatment with antipsychotic drugs for patients known to possess the 5-HT 2C polymorphism.
THE FUTURE OF PHARMACOGENOMIC TESTING
The examples discussed in this article provide some insight about how pharmacogenomic testing is maturing and slowly being integrated into the practice of medicine. They also illustrate the complexity of the multiple stages of research that pharmacogenomic applications must go through in order to be adopted as standard practice.
In the future, pharmacogenomic data will continue to accumulate, and the clinical utility of many other pharmacogenomic tests may be uncovered. The FDA provides information on emerging pharmacogenomic tests at its Web site, www.fda.gov.11 Its up-to-date “Table of Valid Genomic Biomarkers in the Context of Approved Drug Labels” includes boxed warnings, recommendations, research outcomes, and relevant population genetics.
If the FDA continues its current policy, prospective randomized trials that show improvement in patient outcomes will remain the gold standard for determining the clinical significance of a pharmacogenomic test. Furthermore, cost-benefit analyses are likely to continue dictating policy regarding pharmacogenomic testing, and cost-benefit profiles should improve as technology advances and as information gathered from a single test becomes applicable to multiple medications and clinical scenarios.
In the meantime, physicians should become familiar with the terms used in medical genetics and pharmacogenomics and begin to understand genetic contributions to the outcomes of drug therapy. For example, understanding the consequences of metabolizer status and the frequency of variants in a given population can be tremendously helpful when advising our patients about anticipating potential problems when taking specific medications and about making informed decisions about pharmacogenomic testing.
This exchange of information alone may go a long way in improving therapy outcomes even when prospective pharmacogenomic testing is not routinely performed. Furthermore, an increasing number of patients will already have genotyping information available when they come to us, and clinicians need to be aware of the many pharmacogenomic applications recommended by the FDA when genetic status is known.10
- King HC, Sinha AA. Gene expression profile analysis by DNA micro-arrays: promise and pitfalls. JAMA 2001; 286:2280–2288.
- Nuckols TK, Paddock SM, Bower AG, et al. Costs of intravenous adverse drug events in academic and nonacademic intensive care units. Med Care 2008; 46:17–24.
- Gurwitz JH, Field TS, Judge J, et al. The incidence of adverse events in two long-term care facilities. Am J Med 2005; 118:251–258.
- Vargas E, Terleira A, Hernando F, et al. Effects of adverse drug reactions on length of stay in surgical intensive care units. Crit Care Med 2003; 31:694–698.
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998; 279:1200–1205.
- Field TS, Gilman BH, Subramanian S, Fuller JC, Bates DW, Gurwitz JH. The costs associated with adverse drug events in older adults in the ambulatory setting. Med Care 2005; 43:1171–1176.
- Classen DC, Pestotnik SL, Evans RS, Lloyd JF, Burke JP. Adverse drug events in hospitalized patients. Excess length of stay, extra costs, and attributable mortality. JAMA 1997; 277:301–306.
- Ernst FR, Grizzle AJ. Drug-related morbidity and mortality: updating the cost-of-illness model. J Am Pharm Assoc (Wash) 2001; 41:192–199.
- Bradford LD. CYP2D6 allele frequency in European Caucasians, Asians, Africans and their descendants. Pharmacogenomics 2002; 3:229–243.
- Sadee W. Measuring cis-acting regulatory variants genome-wide: new insights into expression genetics and disease susceptibility. Genome Med 2009; 1:116.
- US Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labels. http://www.fda.gov/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/ucm083378.htm. Accessed 1/18/2011.
- Budnitz DS, Shehab N, Kegler SR, Richards CL. Medication use leading to emergency department visits for adverse drug events in older adults. Ann Intern Med 2007; 147:755–765.
- Elias DJ, Topol EJ. Warfarin pharmacogenomics: a big step forward for individualized medicine: enlightened dosing of warfarin. Eur J Hum Genet 2008; 16:532–534.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study). J Am Coll Cardiol 2010; 55:2804–2812.
- Rettie AE, Wienkers LC, Gonzalez FJ, Trager WF, Korzekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allelic variant of CYP2C9. Pharmacogenetics 1994; 4:39–42.
- Higashi MK, Veenstra DL, Kondo LM, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 2002; 287:1690–1698.
- Sanderson S, Emery J, Higgins J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: a HuGEnet systematic review and meta-analysis. Genet Med 2005; 7:97–104.
- Au N, Rettie AE. Pharmacogenomics of 4-hydroxycoumarin anticoagulants. Drug Metab Rev 2008; 40:355–375.
- García-Martín E, Martínez C, Ladero JM, Agúndez JA. Interethnic and intraethnic variability of CYP2C8 and CYP2C9 polymorphisms in healthy individuals. Mol Diagn Ther 2006; 10:29–40.
- Sullivan-Klose TH, Ghanayem BI, Bell DA, et al. The role of the CYP2C9-Leu359 allelic variant in the tolbutamide polymorphism. Pharmacogenetics 1996; 6:341–349.
- Wen MS, Lee M, Chen JJ, et al. Prospective study of warfarin dosage requirements based on CYP2C9 and VKORC1 genotypes. Clin Pharmacol Ther 2008; 84:83–89.
- Larramendy-Gozalo C, Yang JQ, Verstuyft C, et al. Genetic polymorphism of vitamin K epoxide reductase (VKORC1) 1173C>T in a Chinese and a Caucasian population. Basic Clin Pharmacol Toxicol 2006; 98:611–613.
- Caldwell MD, Awad T, Johnson JA, et al. CYP4F2 genetic variant alters required warfarin dose. Blood 2008; 111:4106–4112.
- Bristol-Myers Squibb. Coumadin (warfarin sodium) Prescribing Information. January 2010.
- Barnes-Jewish Hospital at Washington University Medical Center. Warfarin dosing. http://warfarindosing.orgAccessed 1/20/2011.
- Flockhart DA, O’Kane D, Williams MS, et al. Pharmacogenetic testing of CYP2C9 and VKORC1 alleles for warfarin. Genet Med 2008; 10:139–150.
- National Institutes of Health. ClinicalTrials.gov. http://clinicaltrials.gov. Accessed January 20, 2011.
- Eckman MH, Rosand J, Greenberg SM, Gage BF. Cost-effectiveness of using pharmacogenetic information in warfarin dosing for patients with nonvalvular atrial fibrillation. Ann Intern Med 2009; 150:73–83.
- Meckley LM, Gudgeon JM, Anderson JL, Williams MS, Veenstra DL. A policy model to evaluate the benefits, risks, and costs of warfarin pharmacogenomic testing. Pharmacoeconomics 2010; 28:61–74.
- Patrick AR, Avron J, Choudhry NK. Cost-effectiveness of genotype-guided warfarin dosing for patients with atrial fibrillation. Circ Cardiovasc Qual Outcomes 2009; 2:429–436.
- Haas S. New oral Xa and IIa inhibitors: updates on clinical trial results. J Thromb Thrombolysis 2008; 25:52–60.
- Bhatt DL. Tailoring antiplatelet therapy based on pharmacogenomics: how well do the data fit? JAMA 2009; 302:896–897.
- Shuldiner AR, O’Connell JR, Bliden KP, et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 2009; 302:849–857.
- Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 2009; 373:309–317.
- Simon T, Verstuyft C, Mary-Krause M, et al; French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360:363–375.
- Paré G, Mehta SR, Yusuf S, et al. Effects of CYP2C19 genotype on outcomes of clopidogrel treatment. N Engl J Med 2010; 363:1704–1714.
- Bristol-Myers Squibb/Sanofi Pharmaceutical Partnership. Plavix (clopidogrel bisulfate) prescribing information. August 2010.
- P450 Drug Interaction Table. Indiana University School of Medicine. http://medicine.iupui.edu/clinpharm/ddis/table.asp. Accessed 1/21/2011.
- Society for Cardiovascular Angiography and Interventions; Holmes DR, Dehmer GJ, Kaul S, Leifer D, O’Gara PT, Stein CM. ACCF/AHA clopidogrel clinical alert: approaches to the FDA “boxed warning”: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the American Heart Association. Circulation 2010; 122:537–557.
- National Institutes of Health. Genotype Guided Comparison of Clopidogrel and Prasugrel Outcomes Study. http://clinicaltrialsfeeds.org/clinical-trials/show/NCT00995514. Accessed 1/20/2011.
- Amarenco P, Labreuche J. Lipid management in the prevention of stroke: Review and updated meta-analysis of statins for stroke prevention. Lancet Neurol 2009; 8:453–463.
- SEARCH Collaborative Group, Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Romaine SP, Bailey KM, Hall AS, Balmforth AJ. The influence of SLC01B1 (OATP1B1) gene polymorphisms on response to statin therapy. Pharmacogenomics J 2010; 10:1–11.
- Wang D, Guo Y, Wrighton SA, Cooke GE, Sadee W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J 2010; Apr 13 [Epub ahead of print].
- Goetz MP, Rae JM, Suman VJ, et al. Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J Clin Oncol 2005; 23:9312–9318.
- Beverage JN, Sissung TM, Sion AM, Danesi R, Figg WD. CYP2D6 polymorphisms and the impact on tamoxifen therapy. J Pharm Sci 2007; 96:2224–2231.
- Flockhart D. CYP2D6 genotyping and the pharmacogenetics of tamoxifen. Clin Adv Hematol Oncol 2008; 6:493–494.
- Goetz MP, Kamal A, Ames MM. Tamoxifen pharmacogenomics: the role of CYP2D6 as a predictor of drug response. Clin Pharmacol Ther 2008; 83:160–166.
- Stearns V, Rae JM. Pharmacogenetics and breast cancer endocrine therapy: CYP2D6 as a predictive factor for tamoxifen metabolism and drug response? Expert Rev Mol Med 2008; 10:e34.
- Dezentjé VO, Guchelaar HJ, Nortier JW, van del Velde CJ, Gelderblom H. Clinical implications of CYP2D6 genotyping in tamoxifen treatment for breast cancer. Clin Cancer Res 2009; 15:15–21.
- Higgins MJ, Rae JM, Flockhart DA, Hayes DF, Stearns V. Pharmacogenetics of tamoxifen: who should undergo CYP2D6 genetic testing? J Natl Compr Canc Netw 2009; 7:203–213.
- Hoskins JM, Carey LA, McLeod HL. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat Rev Cancer 2009; 9:576–586.
- Steimer W, Zöpf K, von Amelunxen S, et al. Allele-specific change of concentration and functional gene dose for the prediction of steady-state serum concentrations of amitriptyline and nortriptyline in CYP2C19 and CYP2D6 extensive and intermediate metabolizers. Clinical Cancer 2004; 50:1623–1633.
- Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharm Ther 2008; 83:234–242.
- Kirchheiner J, Nickchen K, Bauer M, et al. Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol Psychiatry 2004; 9:442–473.
- Kirchheiner J, Schmidt H, Tzvetkov M, et al. Pharmacokinetics of codeine and its metabolite morphine in ultra-rapid metabolizers due to CYP2D6 duplication. Pharmacogenomics J 2007; 7:257–265.
- Kirchheiner J. CYP2D6 phenotype prediction from genotype: which system is the best? Clin Pharmacol Ther 2008; 83:225–227.
- Borges S, Desta Z, Jin Y, et al. Composite functional genetic and comedication CYP2D6 activity score in predicting tamoxifen drug exposure among breast cancer patients. J Clin Pharmacol 2010; 50:450–458.
- Hoffmann-La Roche Ltd. AmpliChip CYP450 Test. http://www.roche.com/assays/Pages/AmpliChipCYP450Test.aspx. Accessed 1/21/2011.
- Anzenbacher P, Anzenbacherová E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci 2001; 58:737–747.
- Koren G, Cairns J, Chitayat D, Gaedigk A, Leeder SJ. Pharmacogenetics of morphine poisoning in a breastfed neonate of a codeine-prescribed mother. Lancet 2006; 368:704.
- Willmann S, Edginton AN, Coboeken K, Ahr G, Lippert J. Risk to the breast-fed neonate from codeine treatment to the mother: a quantitative mechanistic modeling study. Clin Pharm Ther 2009; 86:634–643.
- Grzesiak M, Beszłej A, Lebioda A, Jonkisz A, Dobosz T, Kienja A. [Retrospective assessment of the antidepressants tolerance in the group of patients with diagnosis of depression and different CYP2D6 genotype.] [In Polish] Psychiatr Pol 2003; 37:433–444.
- Laika B, Leucht S, Heres S, Steimer W. Intermediate metabolizer: increased side effects in psychoactive drug therapy. The key to cost-effectiveness of pretreatment CYP2D6 screening? Pharmacogenomics J 2009; 9:395–403.
- Rau T, Wohlleben G, Wuttke H, et al. CYP2D6 genotype: Impact on adverse effects and nonresponse during treatment with antidepressants—a pilot study. Clin Pharm Ther 2004; 75:386–393.
- McAlpine DE, O’Kane DJ, Black JL, Mrazek DA. Cytochrome P450 2D6 genotype variation and venlafaxine dosage. Mayo Clin Proc 2007; 82:1065–1068.
- Chen S, Chou WH, Blouin RA, et al. The cytochrome P450 2D6 (CYP2D6) enzyme polymorphism: screening costs and influence on clinical outcomes in psychiatry. Clin Pharmacol Ther 1996; 60:522–534.
- Shams ME, Arneth B, Hiemke C, et al. CYP2D6 polymorphism and clinical effect of the antidepressant venlafaxine. J Clin Pharm Ther 2006; 31:493–502.
- Whyte EM, Romkes M, Mulsant BH, et al. CYP2D6 genotype and venlafaxine-XR concentrations in depressed elderly. Int J Geriatr Psychiatry 2006; 21:542–549.
- Roberts RL, Mulder RT, Joyce PR, Luty SE, Kennedy MA. No evidence of increased adverse drug reactions in cytochrome P450 CYP2D6 poor metabolizers treated with fluoxetine or nortriptyline. Hum Psychopharmacol 2004; 19:17–23.
- Murphy GM, Kremer C, Rodrigues HE, Schatzberg AF. Pharmacogenetics of antidepressant medication intolerance. Am J Psychiatry 2003; 160:1830–1835.
- Gillman PK. Re: no evidence of increased adverse drug reactions in cytochrome P450 CYP2D6 poor metabolizers treated with fluoxetine or nortriptyline. Hum Psychopharmacol 2005; 20:61–62.
- Gex-Fabry M, Eap CB, Oneda B, et al. CYP2D6 and ABCB1 genetic variability: influence on paroxetine plasma level and therapeutic response. Ther Drug Monit 2008; 30:474–482.
- Kawanishi C, Lundgren S, Agren H, Bertilsson L. Increased incidence of CYP2D6 gene duplication in patients with persistent mood disorders: ultrarapid metabolism of antidepressants as a cause of nonresponse. A pilot study. Eur J Clin Pharmacol 2004; 59:803–807.
- Serretti A, Calati R, Massat I, et al. Cytochrome P450 CYP1A2, CYP2C9, CYP2C19 and CYP2D6 genes are not associated with response and remission in a sample of depressive patients. Int Clin Psychopharmacol 2009; 24:250–256.
- de Leon J, Armstrong SC, Cozza KL. Clinical guidelines for psychiatrists for the use of pharmacogenetic testing for CYP450 2D6 and CYP450 2C19. Psychosomatics 2006; 47:75–85.
- de Leon J, Susce MT, Johnson M, et al. DNA microarray technology in the clinical environment: the AmpliChip CYP450 test for CYP2D6 and CYP2C19 genotyping. CNS Spectr 2009; 14:19–34.
- Thuerauf N, Lunkenheimer J. The impact of the CYP2D6-polymorphism on dose recommendations for current antidepressants. Eur Arch Psychiatry Clin Neurosci 2006; 256:287–293.
- Horstmann S, Binder EB. Pharmacogenomics of antidepressant drugs. Pharmacol Ther 2009; 124:57–73.
- Ferreira Ade A, Neves FS, da Rocha FF, et al. The role of 5-HTTLPR polymorphism in antidepressant-associated mania in bipolar disorder. J Affect Disord 2009; 112:267–272.
- Serretti A, Kato M, De Ronchi D, Kinoshita T. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with selective serotonin reuptake inhibitor efficacy in depressed patients. Mol Psychiatry 2007; 12:247–257.
- Kato M, Serretti A. Review and meta-analysis of antidepressant pharmacogenetic findings in major depressive disorder. Mol Psychiatry 2010; 15:473–500.
- Reynolds GP, Zhang Z, Zhang X. Polymorphism of the promoter region of the serotonin 5-HT(2C) receptor gene and clozapine-induced weight gain. Am J Psychiatry 2003; 160:677–679.
- King HC, Sinha AA. Gene expression profile analysis by DNA micro-arrays: promise and pitfalls. JAMA 2001; 286:2280–2288.
- Nuckols TK, Paddock SM, Bower AG, et al. Costs of intravenous adverse drug events in academic and nonacademic intensive care units. Med Care 2008; 46:17–24.
- Gurwitz JH, Field TS, Judge J, et al. The incidence of adverse events in two long-term care facilities. Am J Med 2005; 118:251–258.
- Vargas E, Terleira A, Hernando F, et al. Effects of adverse drug reactions on length of stay in surgical intensive care units. Crit Care Med 2003; 31:694–698.
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998; 279:1200–1205.
- Field TS, Gilman BH, Subramanian S, Fuller JC, Bates DW, Gurwitz JH. The costs associated with adverse drug events in older adults in the ambulatory setting. Med Care 2005; 43:1171–1176.
- Classen DC, Pestotnik SL, Evans RS, Lloyd JF, Burke JP. Adverse drug events in hospitalized patients. Excess length of stay, extra costs, and attributable mortality. JAMA 1997; 277:301–306.
- Ernst FR, Grizzle AJ. Drug-related morbidity and mortality: updating the cost-of-illness model. J Am Pharm Assoc (Wash) 2001; 41:192–199.
- Bradford LD. CYP2D6 allele frequency in European Caucasians, Asians, Africans and their descendants. Pharmacogenomics 2002; 3:229–243.
- Sadee W. Measuring cis-acting regulatory variants genome-wide: new insights into expression genetics and disease susceptibility. Genome Med 2009; 1:116.
- US Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labels. http://www.fda.gov/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/ucm083378.htm. Accessed 1/18/2011.
- Budnitz DS, Shehab N, Kegler SR, Richards CL. Medication use leading to emergency department visits for adverse drug events in older adults. Ann Intern Med 2007; 147:755–765.
- Elias DJ, Topol EJ. Warfarin pharmacogenomics: a big step forward for individualized medicine: enlightened dosing of warfarin. Eur J Hum Genet 2008; 16:532–534.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study). J Am Coll Cardiol 2010; 55:2804–2812.
- Rettie AE, Wienkers LC, Gonzalez FJ, Trager WF, Korzekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allelic variant of CYP2C9. Pharmacogenetics 1994; 4:39–42.
- Higashi MK, Veenstra DL, Kondo LM, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 2002; 287:1690–1698.
- Sanderson S, Emery J, Higgins J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: a HuGEnet systematic review and meta-analysis. Genet Med 2005; 7:97–104.
- Au N, Rettie AE. Pharmacogenomics of 4-hydroxycoumarin anticoagulants. Drug Metab Rev 2008; 40:355–375.
- García-Martín E, Martínez C, Ladero JM, Agúndez JA. Interethnic and intraethnic variability of CYP2C8 and CYP2C9 polymorphisms in healthy individuals. Mol Diagn Ther 2006; 10:29–40.
- Sullivan-Klose TH, Ghanayem BI, Bell DA, et al. The role of the CYP2C9-Leu359 allelic variant in the tolbutamide polymorphism. Pharmacogenetics 1996; 6:341–349.
- Wen MS, Lee M, Chen JJ, et al. Prospective study of warfarin dosage requirements based on CYP2C9 and VKORC1 genotypes. Clin Pharmacol Ther 2008; 84:83–89.
- Larramendy-Gozalo C, Yang JQ, Verstuyft C, et al. Genetic polymorphism of vitamin K epoxide reductase (VKORC1) 1173C>T in a Chinese and a Caucasian population. Basic Clin Pharmacol Toxicol 2006; 98:611–613.
- Caldwell MD, Awad T, Johnson JA, et al. CYP4F2 genetic variant alters required warfarin dose. Blood 2008; 111:4106–4112.
- Bristol-Myers Squibb. Coumadin (warfarin sodium) Prescribing Information. January 2010.
- Barnes-Jewish Hospital at Washington University Medical Center. Warfarin dosing. http://warfarindosing.orgAccessed 1/20/2011.
- Flockhart DA, O’Kane D, Williams MS, et al. Pharmacogenetic testing of CYP2C9 and VKORC1 alleles for warfarin. Genet Med 2008; 10:139–150.
- National Institutes of Health. ClinicalTrials.gov. http://clinicaltrials.gov. Accessed January 20, 2011.
- Eckman MH, Rosand J, Greenberg SM, Gage BF. Cost-effectiveness of using pharmacogenetic information in warfarin dosing for patients with nonvalvular atrial fibrillation. Ann Intern Med 2009; 150:73–83.
- Meckley LM, Gudgeon JM, Anderson JL, Williams MS, Veenstra DL. A policy model to evaluate the benefits, risks, and costs of warfarin pharmacogenomic testing. Pharmacoeconomics 2010; 28:61–74.
- Patrick AR, Avron J, Choudhry NK. Cost-effectiveness of genotype-guided warfarin dosing for patients with atrial fibrillation. Circ Cardiovasc Qual Outcomes 2009; 2:429–436.
- Haas S. New oral Xa and IIa inhibitors: updates on clinical trial results. J Thromb Thrombolysis 2008; 25:52–60.
- Bhatt DL. Tailoring antiplatelet therapy based on pharmacogenomics: how well do the data fit? JAMA 2009; 302:896–897.
- Shuldiner AR, O’Connell JR, Bliden KP, et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 2009; 302:849–857.
- Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 2009; 373:309–317.
- Simon T, Verstuyft C, Mary-Krause M, et al; French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360:363–375.
- Paré G, Mehta SR, Yusuf S, et al. Effects of CYP2C19 genotype on outcomes of clopidogrel treatment. N Engl J Med 2010; 363:1704–1714.
- Bristol-Myers Squibb/Sanofi Pharmaceutical Partnership. Plavix (clopidogrel bisulfate) prescribing information. August 2010.
- P450 Drug Interaction Table. Indiana University School of Medicine. http://medicine.iupui.edu/clinpharm/ddis/table.asp. Accessed 1/21/2011.
- Society for Cardiovascular Angiography and Interventions; Holmes DR, Dehmer GJ, Kaul S, Leifer D, O’Gara PT, Stein CM. ACCF/AHA clopidogrel clinical alert: approaches to the FDA “boxed warning”: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the American Heart Association. Circulation 2010; 122:537–557.
- National Institutes of Health. Genotype Guided Comparison of Clopidogrel and Prasugrel Outcomes Study. http://clinicaltrialsfeeds.org/clinical-trials/show/NCT00995514. Accessed 1/20/2011.
- Amarenco P, Labreuche J. Lipid management in the prevention of stroke: Review and updated meta-analysis of statins for stroke prevention. Lancet Neurol 2009; 8:453–463.
- SEARCH Collaborative Group, Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Romaine SP, Bailey KM, Hall AS, Balmforth AJ. The influence of SLC01B1 (OATP1B1) gene polymorphisms on response to statin therapy. Pharmacogenomics J 2010; 10:1–11.
- Wang D, Guo Y, Wrighton SA, Cooke GE, Sadee W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J 2010; Apr 13 [Epub ahead of print].
- Goetz MP, Rae JM, Suman VJ, et al. Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J Clin Oncol 2005; 23:9312–9318.
- Beverage JN, Sissung TM, Sion AM, Danesi R, Figg WD. CYP2D6 polymorphisms and the impact on tamoxifen therapy. J Pharm Sci 2007; 96:2224–2231.
- Flockhart D. CYP2D6 genotyping and the pharmacogenetics of tamoxifen. Clin Adv Hematol Oncol 2008; 6:493–494.
- Goetz MP, Kamal A, Ames MM. Tamoxifen pharmacogenomics: the role of CYP2D6 as a predictor of drug response. Clin Pharmacol Ther 2008; 83:160–166.
- Stearns V, Rae JM. Pharmacogenetics and breast cancer endocrine therapy: CYP2D6 as a predictive factor for tamoxifen metabolism and drug response? Expert Rev Mol Med 2008; 10:e34.
- Dezentjé VO, Guchelaar HJ, Nortier JW, van del Velde CJ, Gelderblom H. Clinical implications of CYP2D6 genotyping in tamoxifen treatment for breast cancer. Clin Cancer Res 2009; 15:15–21.
- Higgins MJ, Rae JM, Flockhart DA, Hayes DF, Stearns V. Pharmacogenetics of tamoxifen: who should undergo CYP2D6 genetic testing? J Natl Compr Canc Netw 2009; 7:203–213.
- Hoskins JM, Carey LA, McLeod HL. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat Rev Cancer 2009; 9:576–586.
- Steimer W, Zöpf K, von Amelunxen S, et al. Allele-specific change of concentration and functional gene dose for the prediction of steady-state serum concentrations of amitriptyline and nortriptyline in CYP2C19 and CYP2D6 extensive and intermediate metabolizers. Clinical Cancer 2004; 50:1623–1633.
- Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharm Ther 2008; 83:234–242.
- Kirchheiner J, Nickchen K, Bauer M, et al. Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol Psychiatry 2004; 9:442–473.
- Kirchheiner J, Schmidt H, Tzvetkov M, et al. Pharmacokinetics of codeine and its metabolite morphine in ultra-rapid metabolizers due to CYP2D6 duplication. Pharmacogenomics J 2007; 7:257–265.
- Kirchheiner J. CYP2D6 phenotype prediction from genotype: which system is the best? Clin Pharmacol Ther 2008; 83:225–227.
- Borges S, Desta Z, Jin Y, et al. Composite functional genetic and comedication CYP2D6 activity score in predicting tamoxifen drug exposure among breast cancer patients. J Clin Pharmacol 2010; 50:450–458.
- Hoffmann-La Roche Ltd. AmpliChip CYP450 Test. http://www.roche.com/assays/Pages/AmpliChipCYP450Test.aspx. Accessed 1/21/2011.
- Anzenbacher P, Anzenbacherová E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci 2001; 58:737–747.
- Koren G, Cairns J, Chitayat D, Gaedigk A, Leeder SJ. Pharmacogenetics of morphine poisoning in a breastfed neonate of a codeine-prescribed mother. Lancet 2006; 368:704.
- Willmann S, Edginton AN, Coboeken K, Ahr G, Lippert J. Risk to the breast-fed neonate from codeine treatment to the mother: a quantitative mechanistic modeling study. Clin Pharm Ther 2009; 86:634–643.
- Grzesiak M, Beszłej A, Lebioda A, Jonkisz A, Dobosz T, Kienja A. [Retrospective assessment of the antidepressants tolerance in the group of patients with diagnosis of depression and different CYP2D6 genotype.] [In Polish] Psychiatr Pol 2003; 37:433–444.
- Laika B, Leucht S, Heres S, Steimer W. Intermediate metabolizer: increased side effects in psychoactive drug therapy. The key to cost-effectiveness of pretreatment CYP2D6 screening? Pharmacogenomics J 2009; 9:395–403.
- Rau T, Wohlleben G, Wuttke H, et al. CYP2D6 genotype: Impact on adverse effects and nonresponse during treatment with antidepressants—a pilot study. Clin Pharm Ther 2004; 75:386–393.
- McAlpine DE, O’Kane DJ, Black JL, Mrazek DA. Cytochrome P450 2D6 genotype variation and venlafaxine dosage. Mayo Clin Proc 2007; 82:1065–1068.
- Chen S, Chou WH, Blouin RA, et al. The cytochrome P450 2D6 (CYP2D6) enzyme polymorphism: screening costs and influence on clinical outcomes in psychiatry. Clin Pharmacol Ther 1996; 60:522–534.
- Shams ME, Arneth B, Hiemke C, et al. CYP2D6 polymorphism and clinical effect of the antidepressant venlafaxine. J Clin Pharm Ther 2006; 31:493–502.
- Whyte EM, Romkes M, Mulsant BH, et al. CYP2D6 genotype and venlafaxine-XR concentrations in depressed elderly. Int J Geriatr Psychiatry 2006; 21:542–549.
- Roberts RL, Mulder RT, Joyce PR, Luty SE, Kennedy MA. No evidence of increased adverse drug reactions in cytochrome P450 CYP2D6 poor metabolizers treated with fluoxetine or nortriptyline. Hum Psychopharmacol 2004; 19:17–23.
- Murphy GM, Kremer C, Rodrigues HE, Schatzberg AF. Pharmacogenetics of antidepressant medication intolerance. Am J Psychiatry 2003; 160:1830–1835.
- Gillman PK. Re: no evidence of increased adverse drug reactions in cytochrome P450 CYP2D6 poor metabolizers treated with fluoxetine or nortriptyline. Hum Psychopharmacol 2005; 20:61–62.
- Gex-Fabry M, Eap CB, Oneda B, et al. CYP2D6 and ABCB1 genetic variability: influence on paroxetine plasma level and therapeutic response. Ther Drug Monit 2008; 30:474–482.
- Kawanishi C, Lundgren S, Agren H, Bertilsson L. Increased incidence of CYP2D6 gene duplication in patients with persistent mood disorders: ultrarapid metabolism of antidepressants as a cause of nonresponse. A pilot study. Eur J Clin Pharmacol 2004; 59:803–807.
- Serretti A, Calati R, Massat I, et al. Cytochrome P450 CYP1A2, CYP2C9, CYP2C19 and CYP2D6 genes are not associated with response and remission in a sample of depressive patients. Int Clin Psychopharmacol 2009; 24:250–256.
- de Leon J, Armstrong SC, Cozza KL. Clinical guidelines for psychiatrists for the use of pharmacogenetic testing for CYP450 2D6 and CYP450 2C19. Psychosomatics 2006; 47:75–85.
- de Leon J, Susce MT, Johnson M, et al. DNA microarray technology in the clinical environment: the AmpliChip CYP450 test for CYP2D6 and CYP2C19 genotyping. CNS Spectr 2009; 14:19–34.
- Thuerauf N, Lunkenheimer J. The impact of the CYP2D6-polymorphism on dose recommendations for current antidepressants. Eur Arch Psychiatry Clin Neurosci 2006; 256:287–293.
- Horstmann S, Binder EB. Pharmacogenomics of antidepressant drugs. Pharmacol Ther 2009; 124:57–73.
- Ferreira Ade A, Neves FS, da Rocha FF, et al. The role of 5-HTTLPR polymorphism in antidepressant-associated mania in bipolar disorder. J Affect Disord 2009; 112:267–272.
- Serretti A, Kato M, De Ronchi D, Kinoshita T. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with selective serotonin reuptake inhibitor efficacy in depressed patients. Mol Psychiatry 2007; 12:247–257.
- Kato M, Serretti A. Review and meta-analysis of antidepressant pharmacogenetic findings in major depressive disorder. Mol Psychiatry 2010; 15:473–500.
- Reynolds GP, Zhang Z, Zhang X. Polymorphism of the promoter region of the serotonin 5-HT(2C) receptor gene and clozapine-induced weight gain. Am J Psychiatry 2003; 160:677–679.
KEY POINTS
- Polymorphisms that affect the pharmacokinetics and pharmacodynamics of specific drugs are common.
- Testing for certain polymorphisms before prescribing certain drugs could help avoid adverse drug effects and improve efficacy.
- Pharmacogenomic testing has only recently begun to enter clinical practice, and routine testing is currently limited to a few clinical scenarios. However, additional applications may be just around the corner.
- Many pharmacogenomic tests are available, but testing has not yet been recommended for most drugs. Needed are large-scale trials to show that routine testing improves patient outcomes.
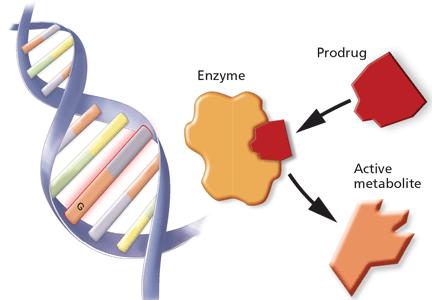
Managing newly diagnosed atrial fibrillation: Rate, rhythm, and risk
Three general concerns dictate the management of atrial fibrillation:
- Controlling the heart rate
- Controlling symptoms
- Preventing thromboembolic events, including stroke.
When seeing a patient with newly diagnosed atrial fibrillation, these same three concerns should be kept in mind, but several additional issues must be addressed:
- Reversible causes of atrial fibrillation must be ruled out
- The true time of onset of the atrial fibrillation and the frequency of the episodes should be ascertained, if possible
- A careful estimation of the patient’s symptom burden should be made.
Atrial fibrillation is common and has a huge impact in terms of the morbidity, death, and costs associated with it. It affects more than 2.2 million Americans.1 Approximately 1 in 10 people over the age of 80 has atrial fibrillation, and for those over the age of 40, the lifetime risk of developing it is one in four.2 Framingham data suggest that the risk of death is approximately twice as high for patients with atrial fibrillation compared with a similar cohort without.3–5
IMPORTANT QUESTIONS DURING THE INITIAL WORKUP
Does the patient have a reversible cause of atrial fibrillation?
Atrial fibrillation is thought to be due to triggers that initiate it and to a myocardial substrate that supports it. While it may develop in the absence of other heart disease, it is often associated with hypertension, diabetes, obesity, structural heart disease (including congenital heart disease), obstructive sleep apnea, advanced age, and alcohol abuse.
Therefore, once atrial fibrillation has been diagnosed, the history, examination, and diagnostic workup should be directed toward looking for potentially reversible causes and for frequently associated comorbidities. Common reversible causes include:
Hyperthyroidism. The laboratory evaluation should include a thyrotropin (thyroid-stimulating hormone, or TSH) level.
Alcohol use, especially binge drinking.
Obstructive sleep apnea, if suspected on the basis of the history or the body habitus.
Structural heart disease such as valvular heart disease or congenital heart defects may also predispose to atrial fibrillation. Therefore, listen carefully to the heart and obtain a transthoracic echocardiogram if one has not already been done or if you suspect a change in valvular disease or systolic function since the most recent study.
How long has the patient been in atrial fibrillation?
The duration of the atrial fibrillation often affects the treatment strategy. Therefore, when the diagnosis has been made, it is important to try to estimate how long the patient has been in atrial fibrillation.
Often, we must settle for an estimate, as the patient’s recollection may be vague. However, in some cases, the symptoms are pronounced or electrocardiographic or telemetric data are available, allowing the time of onset to be clearly defined.
In addition, it is helpful to know if the patient has had prior episodes that were never brought to medical attention. To this end, elicit the patient’s spectrum of symptoms and encourage him or her to think back months or years and try to recall other times when similar symptoms might have occurred.
How do the symptoms affect the patient’s quality of life?
The clinician must also estimate the extent to which the symptoms affect the patient’s quality of life. This is best done when the heart rate is under control. If the patient still has significant symptoms despite adequate rate control, then a rhythm control strategy should probably be pursued.
MANAGING NEWLY DIAGNOSED ATRIAL FIBRILLATION
Control the heart rate with a beta-blocker, a calcium channel blocker, or digoxin
Many patients present during their first episode of atrial fibrillation with a rapid ventricular rate, especially if they are not already taking a drug to slow conduction through the atrioventricular node. If the symptoms are particularly profound, one should try to get the heart rate under control quickly.
Oral agents take time to be absorbed and are not always easy to titrate. Intravenous beta-blockers such as metoprolol (Lopressor) and labetalol (Normodyne, Trandate) or intravenous diltiazem (Cardizem) can slow the heart rate quickly and can be titrated. Once the heart rate is controlled, the oral form can be started, to allow weaning from the intravenous agent. In acute management, we seek a heart rate of less than about 100 to 110 beats per minute.
If the patient’s blood pressure is marginal, loading with intravenous digoxin may be considered. The dosage is 0.5 mg intravenously, then 0.25 mg intravenously in the first 6 hours and another 0.25 mg intravenously in another 6 hours. In patients with renal insufficiency the dosage should be less, or digoxin should be avoided altogether. Often, the blood pressure will improve once the heart rate is decreased, allowing other agents to be initiated. However, if the patient is frankly hypotensive with chest pain, shortness of breath, or a diminished level of consciousness, then emergency electrical cardioversion is indicated even if anticoagulation has not yet been started (more about anticoagulation below).
Oral forms of these same agents are the workhorses for heart rate control in the outpatient setting. Oral beta-blockers and nondihydropyridine calcium channel blockers (ie, diltiazem or verapamil [Calan, Verelan]) are the first-line agents, because when digoxin is used alone, it is relatively poor at controlling the heart rate, especially when the patient is not at rest.
The choice between these agents should be dictated by whether the patient has comorbidities such as coronary artery disease, heart failure, or reactive airway disease. Nondihydropyridine calcium channel blockers are relatively contraindicated in patients with heart failure, while beta-blockers can exacerbate reactive airway disease.6
It is also important to document that the heart rate is adequately controlled outside the hospital or outpatient clinic, where the patient is typically sitting or supine. This can be done with a 6-minute walk, exercise test, or Holter monitor once rate-controlling agents have been titrated.7
When to try to restore sinus rhythm
When atrial fibrillation is first diagnosed, it may not be possible to determine if it is paroxysmal (ie, self-terminating) or persistent. If the episode does not quickly end on its own, consideration may be given to restoring sinus rhythm.
Although experts debate the merits of a rate control approach vs a rhythm control approach for managing atrial fibrillation in the long term, many, including ourselves, recommend trying to restore sinus rhythm at least once when atrial fibrillation is first discovered. It is not always clear if atrial fibrillation is truly asymptomatic. Symptoms such as fatigue or decreased exercise tolerance can be subtle. Additionally, these symptoms may be attributed to other factors such as deconditioning, obesity, or advancing age. Thus, in many cases, only restoring normal sinus rhythm for a time allows the patient and clinician to fully assess the symptoms attributable to atrial fibrillation.
Therefore, in patients with newly diagnosed atrial fibrillation, an attempt to restore sinus rhythm is often warranted. Exceptions are in select patients who have no apparent symptoms and who are very old or are deemed too frail to tolerate cardioversion.
Direct-current cardioversion is typically the treatment of choice when attempting to restore sinus rhythm. The procedure can be done without anticoagulation within 48 hours of the onset of atrial fibrillation, if the time of onset is clear.7 However, clinicians must be careful in defining the onset of atrial fibrillation for this purpose.
Symptoms such as fatigue or shortness of breath can be vague in terms of the exact time of onset and often cannot be relied upon for the purpose of deciding whether cardioversion can be done without anticoagulation. When in doubt, it is best to err on the side of safety and assume that the atrial fibrillation has been going on for more than 48 hours.
If the time of onset is unclear or if more than 48 hours have passed, there are two general strategies for proceeding to electrical cardioversion.
One is to order transesophageal echocardiography and begin anticoagulation therapy at the same time. If there is no thrombus in the left atrium, then cardioversion can be done.8 Therapeutic anticoagulation with heparin, low-molecular-weight heparin, or warfarin (Coumadin) should be achieved within 24 to 48 hours of transesophageal echocardiography and cardioversion to minimize the risk of thromboembolic events, which can occur even after sinus rhythm has been restored.
At our institution, we typically strive to achieve therapeutic anticoagulation with either heparin or low-molecular-weight heparin before cardioversion in this scenario so as to avoid situations in which a patient may undergo cardioversion but then fail to achieve therapeutic anticoagulation for some time due to unforeseen factors.
The other approach is to start warfarin and maintain a goal international normalized ratio (INR) of 2 to 3 for 3 weeks, at which time cardioversion can be performed safely without transesophageal echocardiography.8
Regardless of which strategy is used, anticoagulation should be continued for at least 4 weeks after cardioversion,8 as atrial dysfunction and the risk of stroke may persist for days to weeks after normal sinus rhythm is restored.9
Role of antiarrhythmic drugs
Antiarrhythmic drugs can be used for chemical cardioversion or, more often, to help maintain sinus rhythm after direct-current cardioversion.
Because most of these drugs have at least a small chance of restoring normal sinus rhythm, we need to follow the same rules when starting them as when performing direct-current cardioversion. Patients should not be started on an antiarrhythmic medication until they have had adequate anticoagulation for at least 3 weeks or adequate anticoagulation and a transesophageal echocardiogram confirming that there is no thrombus in the left atrium.
Antiarrhythmic drugs should be started in select patients with newly diagnosed atrial fibrillation in whom a rhythm control strategy will be pursued. For patients whose history suggests a single episode, or episodes that previously self-terminated, an antiarrhythmic may not be necessary. For those with frequent episodes or whose history suggests ongoing atrial fibrillation for a long period, an antiarrhythmic will likely be required to provide a reasonable chance of achieving freedom from atrial fibrillation.
The choice of antiarrhythmic drug should be tailored to the specific patient.
Propafenone (Rythmol) and flecainide (Tambocor) are first-line drugs7 but are contraindicated in patients with coronary artery disease and significant structural heart disease.10
Sotalol (Betapace) and dofetilide (Tikosyn) can be used in patients with coronary artery disease. However, sotalol is contraindicated in patients with congestive heart failure, and dofetilide carries a long list of drug interactions. Both must be used with extreme caution in patients with renal insufficiency, and hospital admission is required for initiation or upward titration of the dose.
Amiodarone (Cordarone) is effective, and in the short term it is typically very well tolerated. However, it has a long half-life, and its potential for long-term toxicity often makes it a poor choice for long-term treatment. The toxicity of amiodarone increases with the cumulative dose. Therefore, this drug should be avoided for long-term therapy of atrial fibrillation in younger patients.
The ‘pill-in-the-pocket’ strategy
The “pill-in-the-pocket” strategy, in which patients are instructed to take their medication only when they have a bout of atrial fibrillation, is a reasonable option for patients with symptomatic recurrences of paroxysmal atrial fibrillation. This strategy is mainly reserved for patients who have relatively infrequent recurrences. Those who have frequent recurrences are usually more effectively treated with daily dosing of an antiarrhythmic. Flecainide and propafenone are the agents of choice for this approach because of their safety profile and efficacy in chemical cardioversion.
While this strategy may be started on an outpatient basis in patients with lone, paroxysmal atrial fibrillation, those with structural heart disease or conduction abnormalities should be observed in the hospital during initiation of therapy to observe for excessive PR prolongation or development of dangerous or worrisome arrhythmias.11–13
Additionally, these agents can decrease the refractory period of the atrioventricular node, thereby increasing the ventricular rate. In the case of atrial flutter, patients may be converted from variable or 2:1 response to a 1:1 conduction. Thus, one should consider also using a beta-blocker with this strategy.
Since the goal of this approach is to convert the patient to sinus rhythm within a few hours of the onset of atrial fibrillation, it may be implemented without the use of warfarin. Patients are instructed that if they do not convert to normal sinus rhythm within a few hours, they should notify the physician so they can undergo electrical cardioversion within the 48-hour window from the onset of atrial fibrillation.
Dronedarone, a new antiarrhythmic drug
Dronedarone (Multaq) is now available and has been shown to be effective in treating atrial fibrillation.14 It has a long half-life and a mechanism of action similar to that of amiodarone. However, it may be inferior to amiodarone in terms of efficacy.15 It is metabolized by CYP3A4. No dosage adjustment is needed for patients with renal insufficiency.
Because dronedarone lacks the iodine moiety found in amiodarone, it should not carry the same toxicity profile. No pulmonary or thyroid toxicity was reported in early trials.16
Nevertheless, dronedarone has several important limitations. It carries a black box warning stating it is contraindicated in patients with severe or recently decompensated heart failure, as the mortality rate was twice as high when this drug was used in such patients in initial studies.17 Additionally, there have been reports of hepatotoxicity requiring liver transplantation in a small number of patients. The extent of this problem and strategies for avoiding it are not yet defined as of the writing of this paper. As with any new medication, patients who are started on dronedarone should be observed closely for any side effects, and these should be reported to assist in the development of the drug’s safety profile.
Pulmonary vein isolation
In a procedure that can potentially cure atrial fibrillation, catheters are inserted into the left atrium and rings of scar tissue are created around the ostia of the pulmonary veins using radiofrequency energy, electrically isolating them from the rest of the left atrium.
Some debate exists as to whether this procedure may be reasonable as a first-line therapy for some patients with atrial fibrillation.18,19 It may be considered as an early treatment strategy in a small subset of patients, specifically young patients with symptomatic, recurrent atrial fibrillation, especially if they are averse to long-term antiarrhythmic therapy.
Because patients may still be more prone to atrial arrhythmias for several weeks to months after the procedure, they must be able to tolerate anticoagulation with warfarin for at least several months.
Rate control vs rhythm control
The choice between a rate control strategy or a rhythm control strategy in the long term is not always straightforward. While atrial fibrillation is clearly associated with higher morbidity and mortality rates, there are few data to date showing that restoring and maintaining sinus rhythm in patients with atrial fibrillation reduce the incidence of morbid complications or the likelihood of death.
Thus, current guidelines recommend a rate control strategy in patients who have no symptoms, and a rhythm control strategy if rate control cannot be achieved or if symptoms persist despite adequate control of the heart rate.7 The circumstances and preferences of the individual patient should carry weight in this decision.
Trials are under way that may shed more light on the relative benefits of rhythm control with ablation or antiarrhythmics and rate control.
PREVENTING THROMBOEMBOLIC EVENTS
Warfarin
In the short term, warfarin therapy may be dictated by plans to restore sinus rhythm. Patients need warfarin for at least 4 weeks after cardioversion unless it is performed within 48 hours of the onset of atrial fibrillation.
The CHADS2score (1 point each for congestive heart failure, hypertension, age 75 or older, and diabetes mellitus; 2 points for prior stroke or transient ischemic attack) is useful when deciding whether to give long-term anticoagulation.
For patients with a score of 0, the risk of stroke is lower than the risk of a major bleeding complication while on therapeutic warfarin.20,21 For these patients, aspirin 81 to 325 mg daily is recommended for stroke prophylaxis.
For those with a score of 2 or greater, the risk of stroke without warfarin is greater than the risk of a major bleeding complication with warfarin. These patients should receive warfarin with a goal INR of 2.0 to 3.0.7
Patients with a CHADS2 score of 1 present a dilemma, as their risk of stroke without warfarin is about the same as their risk of a major bleeding complication with warfarin. They can be managed with either warfarin or aspirin, according to the physician’s judgment.7 In these cases, factors such as hobbies or professions that might increase the risk of bleeding, perceived frequency of atrial fibrillation episodes, and even patient preconceptions about warfarin are often used when deciding between aspirin and warfarin.
Patients with a CHADS2 score of 2 or greater with a single episode of atrial fibrillation and a likely reversible cause may also pose a dilemma when deciding whether to start warfarin. These patients have demonstrated they at least have the substrate to maintain atrial fibrillation. This situation again calls for physician judgment. Bear in mind that asymptomatic recurrences are common in patients with atrial fibrillation.22,23 A higher CHADS2 score denotes a greater risk of stroke and may influence this decision. It is usually beneficial to enlist the patient in this decision-making process, as patients often have very strong opinions about tolerance of the risk of stroke or of warfarin therapy itself.
Another strategy is to start anticoagulation with warfarin and aggressively monitor for recurrences. If the patient has no recurrences of atrial fibrillation after 6 to 12 months and the reversible cause is resolved, one can then revisit the need for warfarin.
Role of aspirin and clopidogrel
Aspirin, alone or in conjunction with clopidogrel (Plavix), may provide an alternative for stroke prophylaxis in patients in whom warfarin is contraindicated. While inferior to warfarin, the combination of aspirin and clopidogrel has been shown to decrease the incidence of major thromboembolic events, especially stroke.24 However, the risk of a major bleeding complication was also significantly increased.
This combination may be a reasonable strategy in select patients with a CHADS2 score of 2 or greater in whom warfarin cannot be used for reasons such as personal aversion to the medication, side effects, or nonbleeding complications or in patients whose INR is exceedingly difficult to keep within the therapeutic range.
Dabigatran, a new anticoagulant
The newest option for anticoagulation in patients with atrial fibrillation is a direct thrombin inhibitor, dabigatran (Pradaxa).
In the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) trial,25 dabigatran was studied head-to-head with warfarin. The doses of dabigatran studied were 110 mg and 150 mg twice a day. At 150 mg twice a day, patients on dabigatran had a lower rate of stroke than with warfarin (1.11% vs 1.69%, P < .001), as well as a lower rate of central nervous system bleeding (0.10% vs 0.38% with warfarin, P < .001). The rates of major bleeding were comparable in the patients receiving warfarin or dabigatran 150 mg twice a day, but the rate of gastrointestinal bleeding was higher in the dabigatran group (1.51% vs 1.02% with warfarin, P < .001).25
Dabigatran was recently approved by the US Food and Drug Administration for use in patients with atrial fibrillation. Doses of 150 mg and 75 mg are available.
Dabigatran is renally excreted, and the 150 mg twice-a-day dosing is intended for patients with a creatinine clearance greater than 30 mL/min. The 75-mg twice-a-day dosing is intended for patients with a creatinine clearance of 15 to 30 mL/min. However, it should be noted that currently there are no data to support the 75-mg twice-a-day dosing.
Dabigatran does have several advantages over warfarin. Patients do not need to avoid foods containing vitamin K, and routine serial monitoring does not appear to be needed. As with any new medication, patients who are started on dabigatran should be observed closely for any side effects, and these should be reported to assist in the development of the drug’s safety profile.
SPECIAL CIRCUMSTANCES
After cardiac or noncardiac surgery
Atrial fibrillation is common after open heart surgery, occurring in approximately 25% to 50% of patients.26–28
When this happens, at least one or two attempts are made to restore sinus rhythm. Especially in the early postoperative period, anticoagulation with heparin or warfarin may be contraindicated, so careful attention must be paid to the patient’s heart rhythm so that atrial fibrillation can be recognized quickly and cardioversion performed within a 48-hour window of onset. Beta-blockers, diltiazem, and verapamil are typically used for rate control.
When atrial fibrillation recurs in patients who have undergone open heart surgery, antiarrhythmics are started early to help prevent further recurrences. At our institution, we usually use amiodarone, as it is highly effective and well tolerated in the short term. When started on amiodarone for postoperative atrial fibrillation, patients are informed that the drug will be stopped after about 2 to 3 months. For patients who continue to have bouts of atrial fibrillation, the need for antiarrhythmic medications can be reassessed, and, if needed, the optimal antiarrhythmic medication for long-term therapy for the patient can be chosen.
Atrial fibrillation in severe, acute illness
Atrial fibrillation is common in the setting of extreme systemic stressors such as shock and sepsis and when the patient is being supported with inotropic agents. In this setting, patients may be in a high-catecholamine state, and both the heart rate and the heart rhythm may be very difficult to control.
Beta-blockers and nondihydropyridine calcium channel blockers should not be used when patients are on medications to support blood pressure, and in this setting, when the patient’s hemodynamic status permits the use of these agents, their effect may be minimal.
Amiodarone or perhaps digoxin may slow the heart rate somewhat without too much effect on the blood pressure. However, with amiodarone, one may have to accept a risk of chemical cardioversion.
Electrical cardioversion with or without the assistance of an antiarrhythmic drug may control the heart rate by restoring sinus rhythm. However, atrial fibrillation often recurs, and if it recurs quickly one may have to accept elevated heart rates until the underlying process is addressed.
- Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the Anticoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001; 285:2370–2375.
- Lloyd-Jones DM, Wang TJ, Leip EP, et al. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation 2004; 110:1042–1046.
- Benjamin EJ, Wolf PA, D’Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation 1998; 98:946–952.
- Kannel WB, Wolf PA, Benjamin EJ, Levy D. Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: population-based estimates. Am J Cardiol 1998; 82( 8A):2N–9N.
- Kannel WB, Abbott RD, Savage DD, McNamara PM. Epidemiologic features of chronic atrial fibrillation: the Framingham study. N Engl J Med 1982; 306:1018–1022.
- The Multicenter Diltiazem Postinfarction Trial Research Group. The effect of diltiazem on mortality and reinfarction after myocardial infarction. N Engl J Med 1988; 319:385–392.
- European heart Rhythm Association; Heart Rhythm society, Fuster V, Rydén LE, Cannom DS, et al. ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation—executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 Guidelines for the Management of Patients With Atrial Fibrillation). J Am Coll Cardiol 2006; 48:854–906.
- Klein AL, Grimm RA, Murray RD, et al; Assessment of Cardioversion Using Transesophageal Echocardiography Investigators. Use of transesophageal echocardiography to guide cardioversion in patients with atrial fibrillation. N Engl J Med 2001; 344:1411–1120.
- Grimm RA, Leung DY, Black IW, Stewart WJ, Thomas JD, Klein AL. Left atrial appendage “stunning” after spontaneous conversion of atrial fibrillation demonstrated by transesophageal Doppler echocardiography. Am Heart J 1995; 130:174–176.
- The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med 1989; 321:406–412.
- Alboni P, Tomasi C, Menozzi C, et al. Efficacy and safety of out-of-hospital self-administered single-dose oral drug treatment in the management of infrequent, well-tolerated paroxysmal supraventricular tachycardia. J Am Coll Cardiol 2001; 37:548–553.
- Capucci A, Villani GQ, Piepoli MF. Reproducible efficacy of loading oral propafenone in restoring sinus rhythm in patients with paroxysmal atrial fibrillation. Am J Cardiol 2003; 92:1345–1347.
- Khan IA. Single oral loading dose of propafenone for pharmacological cardioversion of recent-onset atrial fibrillation. J Am Coll Cardiol 2001; 37:542–547.
- Singh BN, Connolly SJ, Crijns HJ, et al; EURIDIS and ADONIS Investigators. Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N Engl J Med 2007; 357:987–999.
- Le Heuzey J, De Ferrari GM, Radzik D, Santini M, Zhu J, Davy JM. A short-term, randomized, double-blind, parallel-group study to evaluate the efficacy and safety of dronedarone versus amiodarone in patients with persistent atrial fibrillation: the DIONYSOS study. J Cardiovasc Electrophysiol 2010; 21:597–605.
- Hohnloser SH, Crijns HJ, van Eickels M, et al. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med 2009; 360:668–678.
- Køber L, Torp-Pedersen C, McMurray JJ, et al; Dronedarone Study Group. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med 2008; 358:2678–2687.
- Pappone C, Rosanio S, Augello G, et al. Mortality, morbidity, and quality of life after circumferential pulmonary vein ablation for atrial fibrillation: outcomes from a controlled nonrandomized long-term study. J Am Coll Cardiol 2003; 42:185–197.
- Wazni OM, Marrouche NF, Martin DO, et al. Radiofrequency ablation vs antiarrhythmic drugs as first-line treatment of symptomatic atrial fibrillation: a randomized trial. JAMA 2005; 293:2634–2640.
- van Walraven C, Hart RG, Singer DE, et al. Oral anticoagulants vs aspirin in nonvalvular atrial fibrillation: an individual patient metaanalysis. JAMA 2002; 288:2441–2448.
- Hart RG, Benavente O, McBride R, Pearce LA. Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: a metaanalysis. Ann Intern Med 1999; 131:492–501.
- Page RL, Wilkinson WE, Clair WK, McCarthy EA, Pritchett EL. Asymptomatic arrhythmias in patients with symptomatic paroxysmal atrial fibrillation and paroxysmal supraventricular tachycardia. Circulation 1994; 89:224–227.
- Savelieva I, Camm AJ. Clinical relevance of silent atrial fibrillation: prevalence, prognosis, quality of life, and management. J Intervent Card Electrophysiol 2000; 4:369–382.
- ACTIVE Investigators, Connolly SJ, Pogue J, Hart RG, et al. Effect of clopidogrel added to aspirin in patients with atrial fibrillation. N Engl J Med 2009; 360:2066–2078.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151. Erratum in: N Engl J Med 2010; 363:1877.
- Almassi GH, Schowalter T, Nicolosi AC, et al. Atrial fibrillation after cardiac surgery: a major morbid event? Ann Surg 1997; 226:501–511.
- Creswell LL, Schuessler RB, Rosenbloom M, Cox JL. Hazards of postoperative atrial arrhythmias. Ann Thorac Surg 1993; 56:539–549.
- Mathew JP, Fontes ML, Tudor IC, et al; Investigators of the Ischemia Research and Education Foundation; Multicenter Study of Perioperative Ischemia Research Group. A multicenter risk index for atrial fibrillation after cardiac surgery. JAMA 2004; 291:1720–1729.
Three general concerns dictate the management of atrial fibrillation:
- Controlling the heart rate
- Controlling symptoms
- Preventing thromboembolic events, including stroke.
When seeing a patient with newly diagnosed atrial fibrillation, these same three concerns should be kept in mind, but several additional issues must be addressed:
- Reversible causes of atrial fibrillation must be ruled out
- The true time of onset of the atrial fibrillation and the frequency of the episodes should be ascertained, if possible
- A careful estimation of the patient’s symptom burden should be made.
Atrial fibrillation is common and has a huge impact in terms of the morbidity, death, and costs associated with it. It affects more than 2.2 million Americans.1 Approximately 1 in 10 people over the age of 80 has atrial fibrillation, and for those over the age of 40, the lifetime risk of developing it is one in four.2 Framingham data suggest that the risk of death is approximately twice as high for patients with atrial fibrillation compared with a similar cohort without.3–5
IMPORTANT QUESTIONS DURING THE INITIAL WORKUP
Does the patient have a reversible cause of atrial fibrillation?
Atrial fibrillation is thought to be due to triggers that initiate it and to a myocardial substrate that supports it. While it may develop in the absence of other heart disease, it is often associated with hypertension, diabetes, obesity, structural heart disease (including congenital heart disease), obstructive sleep apnea, advanced age, and alcohol abuse.
Therefore, once atrial fibrillation has been diagnosed, the history, examination, and diagnostic workup should be directed toward looking for potentially reversible causes and for frequently associated comorbidities. Common reversible causes include:
Hyperthyroidism. The laboratory evaluation should include a thyrotropin (thyroid-stimulating hormone, or TSH) level.
Alcohol use, especially binge drinking.
Obstructive sleep apnea, if suspected on the basis of the history or the body habitus.
Structural heart disease such as valvular heart disease or congenital heart defects may also predispose to atrial fibrillation. Therefore, listen carefully to the heart and obtain a transthoracic echocardiogram if one has not already been done or if you suspect a change in valvular disease or systolic function since the most recent study.
How long has the patient been in atrial fibrillation?
The duration of the atrial fibrillation often affects the treatment strategy. Therefore, when the diagnosis has been made, it is important to try to estimate how long the patient has been in atrial fibrillation.
Often, we must settle for an estimate, as the patient’s recollection may be vague. However, in some cases, the symptoms are pronounced or electrocardiographic or telemetric data are available, allowing the time of onset to be clearly defined.
In addition, it is helpful to know if the patient has had prior episodes that were never brought to medical attention. To this end, elicit the patient’s spectrum of symptoms and encourage him or her to think back months or years and try to recall other times when similar symptoms might have occurred.
How do the symptoms affect the patient’s quality of life?
The clinician must also estimate the extent to which the symptoms affect the patient’s quality of life. This is best done when the heart rate is under control. If the patient still has significant symptoms despite adequate rate control, then a rhythm control strategy should probably be pursued.
MANAGING NEWLY DIAGNOSED ATRIAL FIBRILLATION
Control the heart rate with a beta-blocker, a calcium channel blocker, or digoxin
Many patients present during their first episode of atrial fibrillation with a rapid ventricular rate, especially if they are not already taking a drug to slow conduction through the atrioventricular node. If the symptoms are particularly profound, one should try to get the heart rate under control quickly.
Oral agents take time to be absorbed and are not always easy to titrate. Intravenous beta-blockers such as metoprolol (Lopressor) and labetalol (Normodyne, Trandate) or intravenous diltiazem (Cardizem) can slow the heart rate quickly and can be titrated. Once the heart rate is controlled, the oral form can be started, to allow weaning from the intravenous agent. In acute management, we seek a heart rate of less than about 100 to 110 beats per minute.
If the patient’s blood pressure is marginal, loading with intravenous digoxin may be considered. The dosage is 0.5 mg intravenously, then 0.25 mg intravenously in the first 6 hours and another 0.25 mg intravenously in another 6 hours. In patients with renal insufficiency the dosage should be less, or digoxin should be avoided altogether. Often, the blood pressure will improve once the heart rate is decreased, allowing other agents to be initiated. However, if the patient is frankly hypotensive with chest pain, shortness of breath, or a diminished level of consciousness, then emergency electrical cardioversion is indicated even if anticoagulation has not yet been started (more about anticoagulation below).
Oral forms of these same agents are the workhorses for heart rate control in the outpatient setting. Oral beta-blockers and nondihydropyridine calcium channel blockers (ie, diltiazem or verapamil [Calan, Verelan]) are the first-line agents, because when digoxin is used alone, it is relatively poor at controlling the heart rate, especially when the patient is not at rest.
The choice between these agents should be dictated by whether the patient has comorbidities such as coronary artery disease, heart failure, or reactive airway disease. Nondihydropyridine calcium channel blockers are relatively contraindicated in patients with heart failure, while beta-blockers can exacerbate reactive airway disease.6
It is also important to document that the heart rate is adequately controlled outside the hospital or outpatient clinic, where the patient is typically sitting or supine. This can be done with a 6-minute walk, exercise test, or Holter monitor once rate-controlling agents have been titrated.7
When to try to restore sinus rhythm
When atrial fibrillation is first diagnosed, it may not be possible to determine if it is paroxysmal (ie, self-terminating) or persistent. If the episode does not quickly end on its own, consideration may be given to restoring sinus rhythm.
Although experts debate the merits of a rate control approach vs a rhythm control approach for managing atrial fibrillation in the long term, many, including ourselves, recommend trying to restore sinus rhythm at least once when atrial fibrillation is first discovered. It is not always clear if atrial fibrillation is truly asymptomatic. Symptoms such as fatigue or decreased exercise tolerance can be subtle. Additionally, these symptoms may be attributed to other factors such as deconditioning, obesity, or advancing age. Thus, in many cases, only restoring normal sinus rhythm for a time allows the patient and clinician to fully assess the symptoms attributable to atrial fibrillation.
Therefore, in patients with newly diagnosed atrial fibrillation, an attempt to restore sinus rhythm is often warranted. Exceptions are in select patients who have no apparent symptoms and who are very old or are deemed too frail to tolerate cardioversion.
Direct-current cardioversion is typically the treatment of choice when attempting to restore sinus rhythm. The procedure can be done without anticoagulation within 48 hours of the onset of atrial fibrillation, if the time of onset is clear.7 However, clinicians must be careful in defining the onset of atrial fibrillation for this purpose.
Symptoms such as fatigue or shortness of breath can be vague in terms of the exact time of onset and often cannot be relied upon for the purpose of deciding whether cardioversion can be done without anticoagulation. When in doubt, it is best to err on the side of safety and assume that the atrial fibrillation has been going on for more than 48 hours.
If the time of onset is unclear or if more than 48 hours have passed, there are two general strategies for proceeding to electrical cardioversion.
One is to order transesophageal echocardiography and begin anticoagulation therapy at the same time. If there is no thrombus in the left atrium, then cardioversion can be done.8 Therapeutic anticoagulation with heparin, low-molecular-weight heparin, or warfarin (Coumadin) should be achieved within 24 to 48 hours of transesophageal echocardiography and cardioversion to minimize the risk of thromboembolic events, which can occur even after sinus rhythm has been restored.
At our institution, we typically strive to achieve therapeutic anticoagulation with either heparin or low-molecular-weight heparin before cardioversion in this scenario so as to avoid situations in which a patient may undergo cardioversion but then fail to achieve therapeutic anticoagulation for some time due to unforeseen factors.
The other approach is to start warfarin and maintain a goal international normalized ratio (INR) of 2 to 3 for 3 weeks, at which time cardioversion can be performed safely without transesophageal echocardiography.8
Regardless of which strategy is used, anticoagulation should be continued for at least 4 weeks after cardioversion,8 as atrial dysfunction and the risk of stroke may persist for days to weeks after normal sinus rhythm is restored.9
Role of antiarrhythmic drugs
Antiarrhythmic drugs can be used for chemical cardioversion or, more often, to help maintain sinus rhythm after direct-current cardioversion.
Because most of these drugs have at least a small chance of restoring normal sinus rhythm, we need to follow the same rules when starting them as when performing direct-current cardioversion. Patients should not be started on an antiarrhythmic medication until they have had adequate anticoagulation for at least 3 weeks or adequate anticoagulation and a transesophageal echocardiogram confirming that there is no thrombus in the left atrium.
Antiarrhythmic drugs should be started in select patients with newly diagnosed atrial fibrillation in whom a rhythm control strategy will be pursued. For patients whose history suggests a single episode, or episodes that previously self-terminated, an antiarrhythmic may not be necessary. For those with frequent episodes or whose history suggests ongoing atrial fibrillation for a long period, an antiarrhythmic will likely be required to provide a reasonable chance of achieving freedom from atrial fibrillation.
The choice of antiarrhythmic drug should be tailored to the specific patient.
Propafenone (Rythmol) and flecainide (Tambocor) are first-line drugs7 but are contraindicated in patients with coronary artery disease and significant structural heart disease.10
Sotalol (Betapace) and dofetilide (Tikosyn) can be used in patients with coronary artery disease. However, sotalol is contraindicated in patients with congestive heart failure, and dofetilide carries a long list of drug interactions. Both must be used with extreme caution in patients with renal insufficiency, and hospital admission is required for initiation or upward titration of the dose.
Amiodarone (Cordarone) is effective, and in the short term it is typically very well tolerated. However, it has a long half-life, and its potential for long-term toxicity often makes it a poor choice for long-term treatment. The toxicity of amiodarone increases with the cumulative dose. Therefore, this drug should be avoided for long-term therapy of atrial fibrillation in younger patients.
The ‘pill-in-the-pocket’ strategy
The “pill-in-the-pocket” strategy, in which patients are instructed to take their medication only when they have a bout of atrial fibrillation, is a reasonable option for patients with symptomatic recurrences of paroxysmal atrial fibrillation. This strategy is mainly reserved for patients who have relatively infrequent recurrences. Those who have frequent recurrences are usually more effectively treated with daily dosing of an antiarrhythmic. Flecainide and propafenone are the agents of choice for this approach because of their safety profile and efficacy in chemical cardioversion.
While this strategy may be started on an outpatient basis in patients with lone, paroxysmal atrial fibrillation, those with structural heart disease or conduction abnormalities should be observed in the hospital during initiation of therapy to observe for excessive PR prolongation or development of dangerous or worrisome arrhythmias.11–13
Additionally, these agents can decrease the refractory period of the atrioventricular node, thereby increasing the ventricular rate. In the case of atrial flutter, patients may be converted from variable or 2:1 response to a 1:1 conduction. Thus, one should consider also using a beta-blocker with this strategy.
Since the goal of this approach is to convert the patient to sinus rhythm within a few hours of the onset of atrial fibrillation, it may be implemented without the use of warfarin. Patients are instructed that if they do not convert to normal sinus rhythm within a few hours, they should notify the physician so they can undergo electrical cardioversion within the 48-hour window from the onset of atrial fibrillation.
Dronedarone, a new antiarrhythmic drug
Dronedarone (Multaq) is now available and has been shown to be effective in treating atrial fibrillation.14 It has a long half-life and a mechanism of action similar to that of amiodarone. However, it may be inferior to amiodarone in terms of efficacy.15 It is metabolized by CYP3A4. No dosage adjustment is needed for patients with renal insufficiency.
Because dronedarone lacks the iodine moiety found in amiodarone, it should not carry the same toxicity profile. No pulmonary or thyroid toxicity was reported in early trials.16
Nevertheless, dronedarone has several important limitations. It carries a black box warning stating it is contraindicated in patients with severe or recently decompensated heart failure, as the mortality rate was twice as high when this drug was used in such patients in initial studies.17 Additionally, there have been reports of hepatotoxicity requiring liver transplantation in a small number of patients. The extent of this problem and strategies for avoiding it are not yet defined as of the writing of this paper. As with any new medication, patients who are started on dronedarone should be observed closely for any side effects, and these should be reported to assist in the development of the drug’s safety profile.
Pulmonary vein isolation
In a procedure that can potentially cure atrial fibrillation, catheters are inserted into the left atrium and rings of scar tissue are created around the ostia of the pulmonary veins using radiofrequency energy, electrically isolating them from the rest of the left atrium.
Some debate exists as to whether this procedure may be reasonable as a first-line therapy for some patients with atrial fibrillation.18,19 It may be considered as an early treatment strategy in a small subset of patients, specifically young patients with symptomatic, recurrent atrial fibrillation, especially if they are averse to long-term antiarrhythmic therapy.
Because patients may still be more prone to atrial arrhythmias for several weeks to months after the procedure, they must be able to tolerate anticoagulation with warfarin for at least several months.
Rate control vs rhythm control
The choice between a rate control strategy or a rhythm control strategy in the long term is not always straightforward. While atrial fibrillation is clearly associated with higher morbidity and mortality rates, there are few data to date showing that restoring and maintaining sinus rhythm in patients with atrial fibrillation reduce the incidence of morbid complications or the likelihood of death.
Thus, current guidelines recommend a rate control strategy in patients who have no symptoms, and a rhythm control strategy if rate control cannot be achieved or if symptoms persist despite adequate control of the heart rate.7 The circumstances and preferences of the individual patient should carry weight in this decision.
Trials are under way that may shed more light on the relative benefits of rhythm control with ablation or antiarrhythmics and rate control.
PREVENTING THROMBOEMBOLIC EVENTS
Warfarin
In the short term, warfarin therapy may be dictated by plans to restore sinus rhythm. Patients need warfarin for at least 4 weeks after cardioversion unless it is performed within 48 hours of the onset of atrial fibrillation.
The CHADS2score (1 point each for congestive heart failure, hypertension, age 75 or older, and diabetes mellitus; 2 points for prior stroke or transient ischemic attack) is useful when deciding whether to give long-term anticoagulation.
For patients with a score of 0, the risk of stroke is lower than the risk of a major bleeding complication while on therapeutic warfarin.20,21 For these patients, aspirin 81 to 325 mg daily is recommended for stroke prophylaxis.
For those with a score of 2 or greater, the risk of stroke without warfarin is greater than the risk of a major bleeding complication with warfarin. These patients should receive warfarin with a goal INR of 2.0 to 3.0.7
Patients with a CHADS2 score of 1 present a dilemma, as their risk of stroke without warfarin is about the same as their risk of a major bleeding complication with warfarin. They can be managed with either warfarin or aspirin, according to the physician’s judgment.7 In these cases, factors such as hobbies or professions that might increase the risk of bleeding, perceived frequency of atrial fibrillation episodes, and even patient preconceptions about warfarin are often used when deciding between aspirin and warfarin.
Patients with a CHADS2 score of 2 or greater with a single episode of atrial fibrillation and a likely reversible cause may also pose a dilemma when deciding whether to start warfarin. These patients have demonstrated they at least have the substrate to maintain atrial fibrillation. This situation again calls for physician judgment. Bear in mind that asymptomatic recurrences are common in patients with atrial fibrillation.22,23 A higher CHADS2 score denotes a greater risk of stroke and may influence this decision. It is usually beneficial to enlist the patient in this decision-making process, as patients often have very strong opinions about tolerance of the risk of stroke or of warfarin therapy itself.
Another strategy is to start anticoagulation with warfarin and aggressively monitor for recurrences. If the patient has no recurrences of atrial fibrillation after 6 to 12 months and the reversible cause is resolved, one can then revisit the need for warfarin.
Role of aspirin and clopidogrel
Aspirin, alone or in conjunction with clopidogrel (Plavix), may provide an alternative for stroke prophylaxis in patients in whom warfarin is contraindicated. While inferior to warfarin, the combination of aspirin and clopidogrel has been shown to decrease the incidence of major thromboembolic events, especially stroke.24 However, the risk of a major bleeding complication was also significantly increased.
This combination may be a reasonable strategy in select patients with a CHADS2 score of 2 or greater in whom warfarin cannot be used for reasons such as personal aversion to the medication, side effects, or nonbleeding complications or in patients whose INR is exceedingly difficult to keep within the therapeutic range.
Dabigatran, a new anticoagulant
The newest option for anticoagulation in patients with atrial fibrillation is a direct thrombin inhibitor, dabigatran (Pradaxa).
In the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) trial,25 dabigatran was studied head-to-head with warfarin. The doses of dabigatran studied were 110 mg and 150 mg twice a day. At 150 mg twice a day, patients on dabigatran had a lower rate of stroke than with warfarin (1.11% vs 1.69%, P < .001), as well as a lower rate of central nervous system bleeding (0.10% vs 0.38% with warfarin, P < .001). The rates of major bleeding were comparable in the patients receiving warfarin or dabigatran 150 mg twice a day, but the rate of gastrointestinal bleeding was higher in the dabigatran group (1.51% vs 1.02% with warfarin, P < .001).25
Dabigatran was recently approved by the US Food and Drug Administration for use in patients with atrial fibrillation. Doses of 150 mg and 75 mg are available.
Dabigatran is renally excreted, and the 150 mg twice-a-day dosing is intended for patients with a creatinine clearance greater than 30 mL/min. The 75-mg twice-a-day dosing is intended for patients with a creatinine clearance of 15 to 30 mL/min. However, it should be noted that currently there are no data to support the 75-mg twice-a-day dosing.
Dabigatran does have several advantages over warfarin. Patients do not need to avoid foods containing vitamin K, and routine serial monitoring does not appear to be needed. As with any new medication, patients who are started on dabigatran should be observed closely for any side effects, and these should be reported to assist in the development of the drug’s safety profile.
SPECIAL CIRCUMSTANCES
After cardiac or noncardiac surgery
Atrial fibrillation is common after open heart surgery, occurring in approximately 25% to 50% of patients.26–28
When this happens, at least one or two attempts are made to restore sinus rhythm. Especially in the early postoperative period, anticoagulation with heparin or warfarin may be contraindicated, so careful attention must be paid to the patient’s heart rhythm so that atrial fibrillation can be recognized quickly and cardioversion performed within a 48-hour window of onset. Beta-blockers, diltiazem, and verapamil are typically used for rate control.
When atrial fibrillation recurs in patients who have undergone open heart surgery, antiarrhythmics are started early to help prevent further recurrences. At our institution, we usually use amiodarone, as it is highly effective and well tolerated in the short term. When started on amiodarone for postoperative atrial fibrillation, patients are informed that the drug will be stopped after about 2 to 3 months. For patients who continue to have bouts of atrial fibrillation, the need for antiarrhythmic medications can be reassessed, and, if needed, the optimal antiarrhythmic medication for long-term therapy for the patient can be chosen.
Atrial fibrillation in severe, acute illness
Atrial fibrillation is common in the setting of extreme systemic stressors such as shock and sepsis and when the patient is being supported with inotropic agents. In this setting, patients may be in a high-catecholamine state, and both the heart rate and the heart rhythm may be very difficult to control.
Beta-blockers and nondihydropyridine calcium channel blockers should not be used when patients are on medications to support blood pressure, and in this setting, when the patient’s hemodynamic status permits the use of these agents, their effect may be minimal.
Amiodarone or perhaps digoxin may slow the heart rate somewhat without too much effect on the blood pressure. However, with amiodarone, one may have to accept a risk of chemical cardioversion.
Electrical cardioversion with or without the assistance of an antiarrhythmic drug may control the heart rate by restoring sinus rhythm. However, atrial fibrillation often recurs, and if it recurs quickly one may have to accept elevated heart rates until the underlying process is addressed.
Three general concerns dictate the management of atrial fibrillation:
- Controlling the heart rate
- Controlling symptoms
- Preventing thromboembolic events, including stroke.
When seeing a patient with newly diagnosed atrial fibrillation, these same three concerns should be kept in mind, but several additional issues must be addressed:
- Reversible causes of atrial fibrillation must be ruled out
- The true time of onset of the atrial fibrillation and the frequency of the episodes should be ascertained, if possible
- A careful estimation of the patient’s symptom burden should be made.
Atrial fibrillation is common and has a huge impact in terms of the morbidity, death, and costs associated with it. It affects more than 2.2 million Americans.1 Approximately 1 in 10 people over the age of 80 has atrial fibrillation, and for those over the age of 40, the lifetime risk of developing it is one in four.2 Framingham data suggest that the risk of death is approximately twice as high for patients with atrial fibrillation compared with a similar cohort without.3–5
IMPORTANT QUESTIONS DURING THE INITIAL WORKUP
Does the patient have a reversible cause of atrial fibrillation?
Atrial fibrillation is thought to be due to triggers that initiate it and to a myocardial substrate that supports it. While it may develop in the absence of other heart disease, it is often associated with hypertension, diabetes, obesity, structural heart disease (including congenital heart disease), obstructive sleep apnea, advanced age, and alcohol abuse.
Therefore, once atrial fibrillation has been diagnosed, the history, examination, and diagnostic workup should be directed toward looking for potentially reversible causes and for frequently associated comorbidities. Common reversible causes include:
Hyperthyroidism. The laboratory evaluation should include a thyrotropin (thyroid-stimulating hormone, or TSH) level.
Alcohol use, especially binge drinking.
Obstructive sleep apnea, if suspected on the basis of the history or the body habitus.
Structural heart disease such as valvular heart disease or congenital heart defects may also predispose to atrial fibrillation. Therefore, listen carefully to the heart and obtain a transthoracic echocardiogram if one has not already been done or if you suspect a change in valvular disease or systolic function since the most recent study.
How long has the patient been in atrial fibrillation?
The duration of the atrial fibrillation often affects the treatment strategy. Therefore, when the diagnosis has been made, it is important to try to estimate how long the patient has been in atrial fibrillation.
Often, we must settle for an estimate, as the patient’s recollection may be vague. However, in some cases, the symptoms are pronounced or electrocardiographic or telemetric data are available, allowing the time of onset to be clearly defined.
In addition, it is helpful to know if the patient has had prior episodes that were never brought to medical attention. To this end, elicit the patient’s spectrum of symptoms and encourage him or her to think back months or years and try to recall other times when similar symptoms might have occurred.
How do the symptoms affect the patient’s quality of life?
The clinician must also estimate the extent to which the symptoms affect the patient’s quality of life. This is best done when the heart rate is under control. If the patient still has significant symptoms despite adequate rate control, then a rhythm control strategy should probably be pursued.
MANAGING NEWLY DIAGNOSED ATRIAL FIBRILLATION
Control the heart rate with a beta-blocker, a calcium channel blocker, or digoxin
Many patients present during their first episode of atrial fibrillation with a rapid ventricular rate, especially if they are not already taking a drug to slow conduction through the atrioventricular node. If the symptoms are particularly profound, one should try to get the heart rate under control quickly.
Oral agents take time to be absorbed and are not always easy to titrate. Intravenous beta-blockers such as metoprolol (Lopressor) and labetalol (Normodyne, Trandate) or intravenous diltiazem (Cardizem) can slow the heart rate quickly and can be titrated. Once the heart rate is controlled, the oral form can be started, to allow weaning from the intravenous agent. In acute management, we seek a heart rate of less than about 100 to 110 beats per minute.
If the patient’s blood pressure is marginal, loading with intravenous digoxin may be considered. The dosage is 0.5 mg intravenously, then 0.25 mg intravenously in the first 6 hours and another 0.25 mg intravenously in another 6 hours. In patients with renal insufficiency the dosage should be less, or digoxin should be avoided altogether. Often, the blood pressure will improve once the heart rate is decreased, allowing other agents to be initiated. However, if the patient is frankly hypotensive with chest pain, shortness of breath, or a diminished level of consciousness, then emergency electrical cardioversion is indicated even if anticoagulation has not yet been started (more about anticoagulation below).
Oral forms of these same agents are the workhorses for heart rate control in the outpatient setting. Oral beta-blockers and nondihydropyridine calcium channel blockers (ie, diltiazem or verapamil [Calan, Verelan]) are the first-line agents, because when digoxin is used alone, it is relatively poor at controlling the heart rate, especially when the patient is not at rest.
The choice between these agents should be dictated by whether the patient has comorbidities such as coronary artery disease, heart failure, or reactive airway disease. Nondihydropyridine calcium channel blockers are relatively contraindicated in patients with heart failure, while beta-blockers can exacerbate reactive airway disease.6
It is also important to document that the heart rate is adequately controlled outside the hospital or outpatient clinic, where the patient is typically sitting or supine. This can be done with a 6-minute walk, exercise test, or Holter monitor once rate-controlling agents have been titrated.7
When to try to restore sinus rhythm
When atrial fibrillation is first diagnosed, it may not be possible to determine if it is paroxysmal (ie, self-terminating) or persistent. If the episode does not quickly end on its own, consideration may be given to restoring sinus rhythm.
Although experts debate the merits of a rate control approach vs a rhythm control approach for managing atrial fibrillation in the long term, many, including ourselves, recommend trying to restore sinus rhythm at least once when atrial fibrillation is first discovered. It is not always clear if atrial fibrillation is truly asymptomatic. Symptoms such as fatigue or decreased exercise tolerance can be subtle. Additionally, these symptoms may be attributed to other factors such as deconditioning, obesity, or advancing age. Thus, in many cases, only restoring normal sinus rhythm for a time allows the patient and clinician to fully assess the symptoms attributable to atrial fibrillation.
Therefore, in patients with newly diagnosed atrial fibrillation, an attempt to restore sinus rhythm is often warranted. Exceptions are in select patients who have no apparent symptoms and who are very old or are deemed too frail to tolerate cardioversion.
Direct-current cardioversion is typically the treatment of choice when attempting to restore sinus rhythm. The procedure can be done without anticoagulation within 48 hours of the onset of atrial fibrillation, if the time of onset is clear.7 However, clinicians must be careful in defining the onset of atrial fibrillation for this purpose.
Symptoms such as fatigue or shortness of breath can be vague in terms of the exact time of onset and often cannot be relied upon for the purpose of deciding whether cardioversion can be done without anticoagulation. When in doubt, it is best to err on the side of safety and assume that the atrial fibrillation has been going on for more than 48 hours.
If the time of onset is unclear or if more than 48 hours have passed, there are two general strategies for proceeding to electrical cardioversion.
One is to order transesophageal echocardiography and begin anticoagulation therapy at the same time. If there is no thrombus in the left atrium, then cardioversion can be done.8 Therapeutic anticoagulation with heparin, low-molecular-weight heparin, or warfarin (Coumadin) should be achieved within 24 to 48 hours of transesophageal echocardiography and cardioversion to minimize the risk of thromboembolic events, which can occur even after sinus rhythm has been restored.
At our institution, we typically strive to achieve therapeutic anticoagulation with either heparin or low-molecular-weight heparin before cardioversion in this scenario so as to avoid situations in which a patient may undergo cardioversion but then fail to achieve therapeutic anticoagulation for some time due to unforeseen factors.
The other approach is to start warfarin and maintain a goal international normalized ratio (INR) of 2 to 3 for 3 weeks, at which time cardioversion can be performed safely without transesophageal echocardiography.8
Regardless of which strategy is used, anticoagulation should be continued for at least 4 weeks after cardioversion,8 as atrial dysfunction and the risk of stroke may persist for days to weeks after normal sinus rhythm is restored.9
Role of antiarrhythmic drugs
Antiarrhythmic drugs can be used for chemical cardioversion or, more often, to help maintain sinus rhythm after direct-current cardioversion.
Because most of these drugs have at least a small chance of restoring normal sinus rhythm, we need to follow the same rules when starting them as when performing direct-current cardioversion. Patients should not be started on an antiarrhythmic medication until they have had adequate anticoagulation for at least 3 weeks or adequate anticoagulation and a transesophageal echocardiogram confirming that there is no thrombus in the left atrium.
Antiarrhythmic drugs should be started in select patients with newly diagnosed atrial fibrillation in whom a rhythm control strategy will be pursued. For patients whose history suggests a single episode, or episodes that previously self-terminated, an antiarrhythmic may not be necessary. For those with frequent episodes or whose history suggests ongoing atrial fibrillation for a long period, an antiarrhythmic will likely be required to provide a reasonable chance of achieving freedom from atrial fibrillation.
The choice of antiarrhythmic drug should be tailored to the specific patient.
Propafenone (Rythmol) and flecainide (Tambocor) are first-line drugs7 but are contraindicated in patients with coronary artery disease and significant structural heart disease.10
Sotalol (Betapace) and dofetilide (Tikosyn) can be used in patients with coronary artery disease. However, sotalol is contraindicated in patients with congestive heart failure, and dofetilide carries a long list of drug interactions. Both must be used with extreme caution in patients with renal insufficiency, and hospital admission is required for initiation or upward titration of the dose.
Amiodarone (Cordarone) is effective, and in the short term it is typically very well tolerated. However, it has a long half-life, and its potential for long-term toxicity often makes it a poor choice for long-term treatment. The toxicity of amiodarone increases with the cumulative dose. Therefore, this drug should be avoided for long-term therapy of atrial fibrillation in younger patients.
The ‘pill-in-the-pocket’ strategy
The “pill-in-the-pocket” strategy, in which patients are instructed to take their medication only when they have a bout of atrial fibrillation, is a reasonable option for patients with symptomatic recurrences of paroxysmal atrial fibrillation. This strategy is mainly reserved for patients who have relatively infrequent recurrences. Those who have frequent recurrences are usually more effectively treated with daily dosing of an antiarrhythmic. Flecainide and propafenone are the agents of choice for this approach because of their safety profile and efficacy in chemical cardioversion.
While this strategy may be started on an outpatient basis in patients with lone, paroxysmal atrial fibrillation, those with structural heart disease or conduction abnormalities should be observed in the hospital during initiation of therapy to observe for excessive PR prolongation or development of dangerous or worrisome arrhythmias.11–13
Additionally, these agents can decrease the refractory period of the atrioventricular node, thereby increasing the ventricular rate. In the case of atrial flutter, patients may be converted from variable or 2:1 response to a 1:1 conduction. Thus, one should consider also using a beta-blocker with this strategy.
Since the goal of this approach is to convert the patient to sinus rhythm within a few hours of the onset of atrial fibrillation, it may be implemented without the use of warfarin. Patients are instructed that if they do not convert to normal sinus rhythm within a few hours, they should notify the physician so they can undergo electrical cardioversion within the 48-hour window from the onset of atrial fibrillation.
Dronedarone, a new antiarrhythmic drug
Dronedarone (Multaq) is now available and has been shown to be effective in treating atrial fibrillation.14 It has a long half-life and a mechanism of action similar to that of amiodarone. However, it may be inferior to amiodarone in terms of efficacy.15 It is metabolized by CYP3A4. No dosage adjustment is needed for patients with renal insufficiency.
Because dronedarone lacks the iodine moiety found in amiodarone, it should not carry the same toxicity profile. No pulmonary or thyroid toxicity was reported in early trials.16
Nevertheless, dronedarone has several important limitations. It carries a black box warning stating it is contraindicated in patients with severe or recently decompensated heart failure, as the mortality rate was twice as high when this drug was used in such patients in initial studies.17 Additionally, there have been reports of hepatotoxicity requiring liver transplantation in a small number of patients. The extent of this problem and strategies for avoiding it are not yet defined as of the writing of this paper. As with any new medication, patients who are started on dronedarone should be observed closely for any side effects, and these should be reported to assist in the development of the drug’s safety profile.
Pulmonary vein isolation
In a procedure that can potentially cure atrial fibrillation, catheters are inserted into the left atrium and rings of scar tissue are created around the ostia of the pulmonary veins using radiofrequency energy, electrically isolating them from the rest of the left atrium.
Some debate exists as to whether this procedure may be reasonable as a first-line therapy for some patients with atrial fibrillation.18,19 It may be considered as an early treatment strategy in a small subset of patients, specifically young patients with symptomatic, recurrent atrial fibrillation, especially if they are averse to long-term antiarrhythmic therapy.
Because patients may still be more prone to atrial arrhythmias for several weeks to months after the procedure, they must be able to tolerate anticoagulation with warfarin for at least several months.
Rate control vs rhythm control
The choice between a rate control strategy or a rhythm control strategy in the long term is not always straightforward. While atrial fibrillation is clearly associated with higher morbidity and mortality rates, there are few data to date showing that restoring and maintaining sinus rhythm in patients with atrial fibrillation reduce the incidence of morbid complications or the likelihood of death.
Thus, current guidelines recommend a rate control strategy in patients who have no symptoms, and a rhythm control strategy if rate control cannot be achieved or if symptoms persist despite adequate control of the heart rate.7 The circumstances and preferences of the individual patient should carry weight in this decision.
Trials are under way that may shed more light on the relative benefits of rhythm control with ablation or antiarrhythmics and rate control.
PREVENTING THROMBOEMBOLIC EVENTS
Warfarin
In the short term, warfarin therapy may be dictated by plans to restore sinus rhythm. Patients need warfarin for at least 4 weeks after cardioversion unless it is performed within 48 hours of the onset of atrial fibrillation.
The CHADS2score (1 point each for congestive heart failure, hypertension, age 75 or older, and diabetes mellitus; 2 points for prior stroke or transient ischemic attack) is useful when deciding whether to give long-term anticoagulation.
For patients with a score of 0, the risk of stroke is lower than the risk of a major bleeding complication while on therapeutic warfarin.20,21 For these patients, aspirin 81 to 325 mg daily is recommended for stroke prophylaxis.
For those with a score of 2 or greater, the risk of stroke without warfarin is greater than the risk of a major bleeding complication with warfarin. These patients should receive warfarin with a goal INR of 2.0 to 3.0.7
Patients with a CHADS2 score of 1 present a dilemma, as their risk of stroke without warfarin is about the same as their risk of a major bleeding complication with warfarin. They can be managed with either warfarin or aspirin, according to the physician’s judgment.7 In these cases, factors such as hobbies or professions that might increase the risk of bleeding, perceived frequency of atrial fibrillation episodes, and even patient preconceptions about warfarin are often used when deciding between aspirin and warfarin.
Patients with a CHADS2 score of 2 or greater with a single episode of atrial fibrillation and a likely reversible cause may also pose a dilemma when deciding whether to start warfarin. These patients have demonstrated they at least have the substrate to maintain atrial fibrillation. This situation again calls for physician judgment. Bear in mind that asymptomatic recurrences are common in patients with atrial fibrillation.22,23 A higher CHADS2 score denotes a greater risk of stroke and may influence this decision. It is usually beneficial to enlist the patient in this decision-making process, as patients often have very strong opinions about tolerance of the risk of stroke or of warfarin therapy itself.
Another strategy is to start anticoagulation with warfarin and aggressively monitor for recurrences. If the patient has no recurrences of atrial fibrillation after 6 to 12 months and the reversible cause is resolved, one can then revisit the need for warfarin.
Role of aspirin and clopidogrel
Aspirin, alone or in conjunction with clopidogrel (Plavix), may provide an alternative for stroke prophylaxis in patients in whom warfarin is contraindicated. While inferior to warfarin, the combination of aspirin and clopidogrel has been shown to decrease the incidence of major thromboembolic events, especially stroke.24 However, the risk of a major bleeding complication was also significantly increased.
This combination may be a reasonable strategy in select patients with a CHADS2 score of 2 or greater in whom warfarin cannot be used for reasons such as personal aversion to the medication, side effects, or nonbleeding complications or in patients whose INR is exceedingly difficult to keep within the therapeutic range.
Dabigatran, a new anticoagulant
The newest option for anticoagulation in patients with atrial fibrillation is a direct thrombin inhibitor, dabigatran (Pradaxa).
In the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) trial,25 dabigatran was studied head-to-head with warfarin. The doses of dabigatran studied were 110 mg and 150 mg twice a day. At 150 mg twice a day, patients on dabigatran had a lower rate of stroke than with warfarin (1.11% vs 1.69%, P < .001), as well as a lower rate of central nervous system bleeding (0.10% vs 0.38% with warfarin, P < .001). The rates of major bleeding were comparable in the patients receiving warfarin or dabigatran 150 mg twice a day, but the rate of gastrointestinal bleeding was higher in the dabigatran group (1.51% vs 1.02% with warfarin, P < .001).25
Dabigatran was recently approved by the US Food and Drug Administration for use in patients with atrial fibrillation. Doses of 150 mg and 75 mg are available.
Dabigatran is renally excreted, and the 150 mg twice-a-day dosing is intended for patients with a creatinine clearance greater than 30 mL/min. The 75-mg twice-a-day dosing is intended for patients with a creatinine clearance of 15 to 30 mL/min. However, it should be noted that currently there are no data to support the 75-mg twice-a-day dosing.
Dabigatran does have several advantages over warfarin. Patients do not need to avoid foods containing vitamin K, and routine serial monitoring does not appear to be needed. As with any new medication, patients who are started on dabigatran should be observed closely for any side effects, and these should be reported to assist in the development of the drug’s safety profile.
SPECIAL CIRCUMSTANCES
After cardiac or noncardiac surgery
Atrial fibrillation is common after open heart surgery, occurring in approximately 25% to 50% of patients.26–28
When this happens, at least one or two attempts are made to restore sinus rhythm. Especially in the early postoperative period, anticoagulation with heparin or warfarin may be contraindicated, so careful attention must be paid to the patient’s heart rhythm so that atrial fibrillation can be recognized quickly and cardioversion performed within a 48-hour window of onset. Beta-blockers, diltiazem, and verapamil are typically used for rate control.
When atrial fibrillation recurs in patients who have undergone open heart surgery, antiarrhythmics are started early to help prevent further recurrences. At our institution, we usually use amiodarone, as it is highly effective and well tolerated in the short term. When started on amiodarone for postoperative atrial fibrillation, patients are informed that the drug will be stopped after about 2 to 3 months. For patients who continue to have bouts of atrial fibrillation, the need for antiarrhythmic medications can be reassessed, and, if needed, the optimal antiarrhythmic medication for long-term therapy for the patient can be chosen.
Atrial fibrillation in severe, acute illness
Atrial fibrillation is common in the setting of extreme systemic stressors such as shock and sepsis and when the patient is being supported with inotropic agents. In this setting, patients may be in a high-catecholamine state, and both the heart rate and the heart rhythm may be very difficult to control.
Beta-blockers and nondihydropyridine calcium channel blockers should not be used when patients are on medications to support blood pressure, and in this setting, when the patient’s hemodynamic status permits the use of these agents, their effect may be minimal.
Amiodarone or perhaps digoxin may slow the heart rate somewhat without too much effect on the blood pressure. However, with amiodarone, one may have to accept a risk of chemical cardioversion.
Electrical cardioversion with or without the assistance of an antiarrhythmic drug may control the heart rate by restoring sinus rhythm. However, atrial fibrillation often recurs, and if it recurs quickly one may have to accept elevated heart rates until the underlying process is addressed.
- Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the Anticoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001; 285:2370–2375.
- Lloyd-Jones DM, Wang TJ, Leip EP, et al. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation 2004; 110:1042–1046.
- Benjamin EJ, Wolf PA, D’Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation 1998; 98:946–952.
- Kannel WB, Wolf PA, Benjamin EJ, Levy D. Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: population-based estimates. Am J Cardiol 1998; 82( 8A):2N–9N.
- Kannel WB, Abbott RD, Savage DD, McNamara PM. Epidemiologic features of chronic atrial fibrillation: the Framingham study. N Engl J Med 1982; 306:1018–1022.
- The Multicenter Diltiazem Postinfarction Trial Research Group. The effect of diltiazem on mortality and reinfarction after myocardial infarction. N Engl J Med 1988; 319:385–392.
- European heart Rhythm Association; Heart Rhythm society, Fuster V, Rydén LE, Cannom DS, et al. ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation—executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 Guidelines for the Management of Patients With Atrial Fibrillation). J Am Coll Cardiol 2006; 48:854–906.
- Klein AL, Grimm RA, Murray RD, et al; Assessment of Cardioversion Using Transesophageal Echocardiography Investigators. Use of transesophageal echocardiography to guide cardioversion in patients with atrial fibrillation. N Engl J Med 2001; 344:1411–1120.
- Grimm RA, Leung DY, Black IW, Stewart WJ, Thomas JD, Klein AL. Left atrial appendage “stunning” after spontaneous conversion of atrial fibrillation demonstrated by transesophageal Doppler echocardiography. Am Heart J 1995; 130:174–176.
- The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med 1989; 321:406–412.
- Alboni P, Tomasi C, Menozzi C, et al. Efficacy and safety of out-of-hospital self-administered single-dose oral drug treatment in the management of infrequent, well-tolerated paroxysmal supraventricular tachycardia. J Am Coll Cardiol 2001; 37:548–553.
- Capucci A, Villani GQ, Piepoli MF. Reproducible efficacy of loading oral propafenone in restoring sinus rhythm in patients with paroxysmal atrial fibrillation. Am J Cardiol 2003; 92:1345–1347.
- Khan IA. Single oral loading dose of propafenone for pharmacological cardioversion of recent-onset atrial fibrillation. J Am Coll Cardiol 2001; 37:542–547.
- Singh BN, Connolly SJ, Crijns HJ, et al; EURIDIS and ADONIS Investigators. Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N Engl J Med 2007; 357:987–999.
- Le Heuzey J, De Ferrari GM, Radzik D, Santini M, Zhu J, Davy JM. A short-term, randomized, double-blind, parallel-group study to evaluate the efficacy and safety of dronedarone versus amiodarone in patients with persistent atrial fibrillation: the DIONYSOS study. J Cardiovasc Electrophysiol 2010; 21:597–605.
- Hohnloser SH, Crijns HJ, van Eickels M, et al. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med 2009; 360:668–678.
- Køber L, Torp-Pedersen C, McMurray JJ, et al; Dronedarone Study Group. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med 2008; 358:2678–2687.
- Pappone C, Rosanio S, Augello G, et al. Mortality, morbidity, and quality of life after circumferential pulmonary vein ablation for atrial fibrillation: outcomes from a controlled nonrandomized long-term study. J Am Coll Cardiol 2003; 42:185–197.
- Wazni OM, Marrouche NF, Martin DO, et al. Radiofrequency ablation vs antiarrhythmic drugs as first-line treatment of symptomatic atrial fibrillation: a randomized trial. JAMA 2005; 293:2634–2640.
- van Walraven C, Hart RG, Singer DE, et al. Oral anticoagulants vs aspirin in nonvalvular atrial fibrillation: an individual patient metaanalysis. JAMA 2002; 288:2441–2448.
- Hart RG, Benavente O, McBride R, Pearce LA. Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: a metaanalysis. Ann Intern Med 1999; 131:492–501.
- Page RL, Wilkinson WE, Clair WK, McCarthy EA, Pritchett EL. Asymptomatic arrhythmias in patients with symptomatic paroxysmal atrial fibrillation and paroxysmal supraventricular tachycardia. Circulation 1994; 89:224–227.
- Savelieva I, Camm AJ. Clinical relevance of silent atrial fibrillation: prevalence, prognosis, quality of life, and management. J Intervent Card Electrophysiol 2000; 4:369–382.
- ACTIVE Investigators, Connolly SJ, Pogue J, Hart RG, et al. Effect of clopidogrel added to aspirin in patients with atrial fibrillation. N Engl J Med 2009; 360:2066–2078.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151. Erratum in: N Engl J Med 2010; 363:1877.
- Almassi GH, Schowalter T, Nicolosi AC, et al. Atrial fibrillation after cardiac surgery: a major morbid event? Ann Surg 1997; 226:501–511.
- Creswell LL, Schuessler RB, Rosenbloom M, Cox JL. Hazards of postoperative atrial arrhythmias. Ann Thorac Surg 1993; 56:539–549.
- Mathew JP, Fontes ML, Tudor IC, et al; Investigators of the Ischemia Research and Education Foundation; Multicenter Study of Perioperative Ischemia Research Group. A multicenter risk index for atrial fibrillation after cardiac surgery. JAMA 2004; 291:1720–1729.
- Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the Anticoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001; 285:2370–2375.
- Lloyd-Jones DM, Wang TJ, Leip EP, et al. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation 2004; 110:1042–1046.
- Benjamin EJ, Wolf PA, D’Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation 1998; 98:946–952.
- Kannel WB, Wolf PA, Benjamin EJ, Levy D. Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: population-based estimates. Am J Cardiol 1998; 82( 8A):2N–9N.
- Kannel WB, Abbott RD, Savage DD, McNamara PM. Epidemiologic features of chronic atrial fibrillation: the Framingham study. N Engl J Med 1982; 306:1018–1022.
- The Multicenter Diltiazem Postinfarction Trial Research Group. The effect of diltiazem on mortality and reinfarction after myocardial infarction. N Engl J Med 1988; 319:385–392.
- European heart Rhythm Association; Heart Rhythm society, Fuster V, Rydén LE, Cannom DS, et al. ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation—executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 Guidelines for the Management of Patients With Atrial Fibrillation). J Am Coll Cardiol 2006; 48:854–906.
- Klein AL, Grimm RA, Murray RD, et al; Assessment of Cardioversion Using Transesophageal Echocardiography Investigators. Use of transesophageal echocardiography to guide cardioversion in patients with atrial fibrillation. N Engl J Med 2001; 344:1411–1120.
- Grimm RA, Leung DY, Black IW, Stewart WJ, Thomas JD, Klein AL. Left atrial appendage “stunning” after spontaneous conversion of atrial fibrillation demonstrated by transesophageal Doppler echocardiography. Am Heart J 1995; 130:174–176.
- The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med 1989; 321:406–412.
- Alboni P, Tomasi C, Menozzi C, et al. Efficacy and safety of out-of-hospital self-administered single-dose oral drug treatment in the management of infrequent, well-tolerated paroxysmal supraventricular tachycardia. J Am Coll Cardiol 2001; 37:548–553.
- Capucci A, Villani GQ, Piepoli MF. Reproducible efficacy of loading oral propafenone in restoring sinus rhythm in patients with paroxysmal atrial fibrillation. Am J Cardiol 2003; 92:1345–1347.
- Khan IA. Single oral loading dose of propafenone for pharmacological cardioversion of recent-onset atrial fibrillation. J Am Coll Cardiol 2001; 37:542–547.
- Singh BN, Connolly SJ, Crijns HJ, et al; EURIDIS and ADONIS Investigators. Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N Engl J Med 2007; 357:987–999.
- Le Heuzey J, De Ferrari GM, Radzik D, Santini M, Zhu J, Davy JM. A short-term, randomized, double-blind, parallel-group study to evaluate the efficacy and safety of dronedarone versus amiodarone in patients with persistent atrial fibrillation: the DIONYSOS study. J Cardiovasc Electrophysiol 2010; 21:597–605.
- Hohnloser SH, Crijns HJ, van Eickels M, et al. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med 2009; 360:668–678.
- Køber L, Torp-Pedersen C, McMurray JJ, et al; Dronedarone Study Group. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med 2008; 358:2678–2687.
- Pappone C, Rosanio S, Augello G, et al. Mortality, morbidity, and quality of life after circumferential pulmonary vein ablation for atrial fibrillation: outcomes from a controlled nonrandomized long-term study. J Am Coll Cardiol 2003; 42:185–197.
- Wazni OM, Marrouche NF, Martin DO, et al. Radiofrequency ablation vs antiarrhythmic drugs as first-line treatment of symptomatic atrial fibrillation: a randomized trial. JAMA 2005; 293:2634–2640.
- van Walraven C, Hart RG, Singer DE, et al. Oral anticoagulants vs aspirin in nonvalvular atrial fibrillation: an individual patient metaanalysis. JAMA 2002; 288:2441–2448.
- Hart RG, Benavente O, McBride R, Pearce LA. Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: a metaanalysis. Ann Intern Med 1999; 131:492–501.
- Page RL, Wilkinson WE, Clair WK, McCarthy EA, Pritchett EL. Asymptomatic arrhythmias in patients with symptomatic paroxysmal atrial fibrillation and paroxysmal supraventricular tachycardia. Circulation 1994; 89:224–227.
- Savelieva I, Camm AJ. Clinical relevance of silent atrial fibrillation: prevalence, prognosis, quality of life, and management. J Intervent Card Electrophysiol 2000; 4:369–382.
- ACTIVE Investigators, Connolly SJ, Pogue J, Hart RG, et al. Effect of clopidogrel added to aspirin in patients with atrial fibrillation. N Engl J Med 2009; 360:2066–2078.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151. Erratum in: N Engl J Med 2010; 363:1877.
- Almassi GH, Schowalter T, Nicolosi AC, et al. Atrial fibrillation after cardiac surgery: a major morbid event? Ann Surg 1997; 226:501–511.
- Creswell LL, Schuessler RB, Rosenbloom M, Cox JL. Hazards of postoperative atrial arrhythmias. Ann Thorac Surg 1993; 56:539–549.
- Mathew JP, Fontes ML, Tudor IC, et al; Investigators of the Ischemia Research and Education Foundation; Multicenter Study of Perioperative Ischemia Research Group. A multicenter risk index for atrial fibrillation after cardiac surgery. JAMA 2004; 291:1720–1729.
KEY POINTS
- When atrial fibrillation is newly diagnosed, reversible causes and commonly associated processes should be sought.
- Agents to control the heart rate, eg, beta-blockers or nondihydropyridine calcium channel blockers, are often started and titrated intravenously and then changed to oral dosing.
- The benefit of rhythm control has not been firmly established. Although we try cardioversion at least once when atrial fibrillation is first diagnosed, rhythm control is generally reserved for patients whose symptoms persist despite rate control, or for patients in whom the heart rate cannot be controlled.
- The need for short-term or long-term anticoagulation must be estimated.
Dronedarone for atrial fibrillation: How does it compare with amiodarone?
Dronedarone (Multaq), approved by the US Food and Drug Administration in July 2009, is a congener of the antiarrhythmic drug amiodarone (Cordarone). Designed in the hope that it would be safer than amiodarone, its official indication is to lower the risk of hospitalization in patients with paroxysmal or persistent atrial fibrillation or atrial flutter. However, its precise role in the management of atrial fibrillation is yet to be defined. If dronedarone remains well tolerated, it may permit clinicians to pursue a rhythm control strategy more often. In this article, we present a progress report on this new agent.
BETTER ANTIARRHYTHMIC DRUGS ARE NEEDED
Atrial fibrillation increases the risk of stroke fivefold and accounts for 15% to 20% of all strokes.1 It also increases the risk of heart failure. Drugs are the mainstay of therapy, but many antiarrhythmic drugs are not very effective and cause cardiac and extracardiac toxicity. Thus, the need for safe and effective new drugs.2
Much effort is going into the development of drugs that target specific ion channels or proteins expressed predominantly in atrial myocardium. The rationale is to avoid the unwanted effects of ionic currents on the ventricle and thus avoid ventricular proarrhythmic effects. At the same time, alternatives to the multiple channel blocker amiodarone, the mainstay of heart rhythm control therapy in atrial fibrillation, are being developed to retain the electrophysiologic efficacy of the mother compound but avoid its extracardiac toxicity.
RATE CONTROL VS RHYTHM CONTROL
In the acute care setting, heart rate control with atrioventricular nodal agents (beta-blockers, calcium channel blockers, and digitalis) is the preferred initial strategy in most hemodynamically stable patients presenting with new-onset atrial fibrillation.3
Since we lack an effective method for maintaining sinus rhythm without incurring significant adverse effects, rate control is also often chosen for chronic management of atrial fibrillation. This is particularly true for patients who have no symptoms or only minimal symptoms and in whom adequate rate control is easily attained. Indeed, results of large clinical trials suggest that rate control is satisfactory for many patients.
The main purpose of rate control is to control symptoms as opposed to merely lowering the ventricular rate. Effective rate control often prevents hemodynamic instability in patients with underlying heart disease who present acutely with atrial fibrillation. In patients with permanent atrial fibrillation, the RACE II study4 (Rate Control Efficacy in Permanent Atrial Fibrillation: a Comparison between Lenient Versus Strict Rate Control II), during a 3-year follow-up, showed that lenient rate control (resting heart rate < 110 beats per minute) is not inferior to strict rate control (resting heart rate < 80 beats per minute) in preventing major cardiovascular events (heart failure, stroke) or arrhythmic events such as syncope and sustained ventricular tachycardia.4
As a long-term strategy, rate control also prevents tachycardia-induced cardiomyopathy, reduces the risk of worsening of underlying heart failure, and can improve symptoms and quality of life.
Although maintenance of sinus rhythm is most likely associated with a survival benefit, heart rhythm control with antiarrhythmic drugs has not shown an advantage over rate control in overall or cardiovascular death rates, thromboembolic complications, or impact on heart failure. Indeed, a rhythm control strategy has been associated only with better exercise tolerance and, although less clear, with better quality of life.5
One possible explanation as to why a rhythm control strategy has not been shown to be superior to a rate control strategy is the side effects of the presently available drugs for rhythm control.
In a subgroup analysis of the Atrial Fibrillation Follow-up Investigation of Rhythm Management (AFFIRM) trial,6 antiarrhythmic therapy was associated with a 49% increase in the mortality rate that offset the benefits of conversion and maintenance of sinus rhythm, which was associated with a 53% reduction in mortality rates.
The hope is that newer drugs with less toxicity may produce better outcomes for patients treated with rhythm control.
AN ANALOGUE OF AMIODARONE, WITHOUT THE IODINE
Dronedarone is a structurally modified version of amiodarone, the antiarrhythmic drug that has shown the greatest efficacy at maintaining sinus rhythm in patients with paroxysmal atrial fibrillation. Although historically amiodarone has been effective in maintaining sinus rhythm and has been used safely in patients with advanced heart failure, its use has been limited by cumulative and often irreversible extracardiac organ toxicity.
Dronedarone was designed to match amiodarone’s efficacy but with a better safety profile. An iodine radical makes up more than one-third of amiodarone’s molecular weight. The omission of iodine in dronedarone was intended to reduce the likelihood of toxic side effects.
Dronedarone is a benzofuran derivative pharmacologically related to amiodarone, with the addition of a methylsulfonamide group. This reduces lipophilicity and the propensity to cross the blood-brain barrier; over a 2-year period this drug has not been shown to have neurotoxic effects.7
Dronedarone has proved efficacious without toxic or proarrhythmic effects and has minimal side effects, but concerns remain regarding its use in advanced heart failure. To date, its adverse-event profile appears comparable to that of placebo. However, whether its efficacy and incidence of adverse effects are comparable to what has been reported in the literature may take time to assess.
DRONEDARONE’S PHARMACOLOGY
Dronedarone, like amiodarone, blocks multiple sodium and potassium ion channels. It also exerts an antiadrenergic effect by noncompetitive binding to beta-adrenergic receptors as well as by inhibiting an agonist-induced increase in adenylate cyclase activity.8 Compared with amiodarone, dronedarone is a more potent blocker of peak sodium current.
Dronedarone is largely metabolized by the hepatic enzyme cytochrome P450 3A4 isoform (CYP3A4). Only 6% of dronedarone is excreted renally; however, no trial has yet assessed dronedarone’s safety in patients with marked kidney dysfunction.89
Dronedarone’s steady-state terminal elimination half-life is approximately 30 hours. When taken twice a day, it achieves steady-state concentrations in 5 to 7 days.
Dronedarone is available only for oral administration at 400 mg twice daily. Dose adjustment or titration is not recommended.
CLINICAL TRIALS OF DRONEDARONE
Dronedarone vs placebo
ATHENA (A Placebo-Controlled, Double-Blind, Parallel Arm Trial to Assess the Efficacy of Dronedarone 400 mg bid for the Prevention of Cardiovascular Hospitalization or Death From any Cause in Patients With Atrial Fibrillation/Atrial Flutter)10 was a prospective, double-blind study to assess morbidity and death rates in 4,628 patients with atrial fibrillation or atrial flutter and at least one other cardiovascular risk factor.
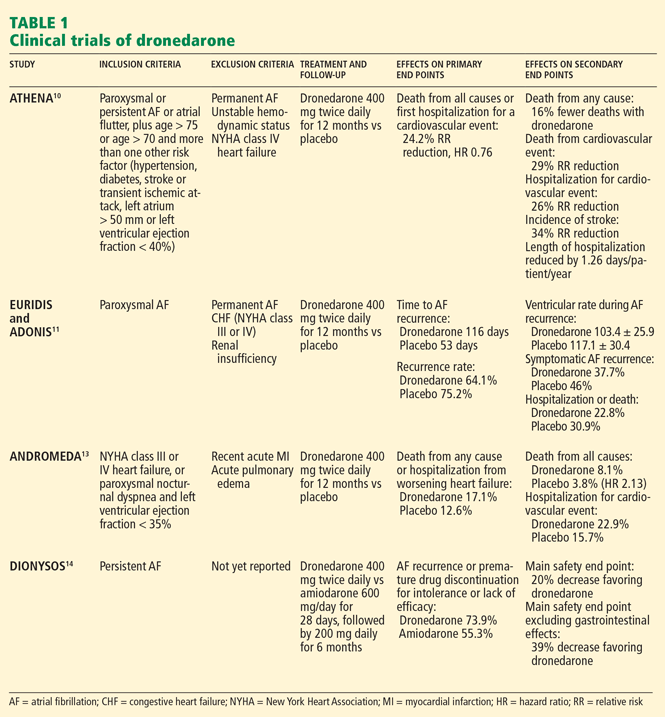
EURIDIS and ADONIS. Two trials,11 EURIDIS (European Trial in Atrial Fibrillation or Flutter Patients Receiving Dronedarone for the Maintenance of Sinus Rhythm) and ADONIS (American-Australian Trial With Dronedarone in Atrial Fibrillation or Flutter Patients for the Maintenance of Sinus Rhythm), enrolled a total of more than 1,200 patients and showed that dronedarone 400 mg twice a day produced a significantly lower rate of recurrence of atrial fibrillation after electrical cardioversion compared with placebo.
Overall, treatment with dronedarone significantly reduced the risk of a first recurrence of atrial fibrillation by 22% (ADONIS) and 27.5% (EURIDIS) (Table 1).
ERATO (Efficacy and Safety of Dronedarone for the Control of Ventricular Rate During Atrial Fibrillation),12 an additional phase III study, showed that dronedarone controlled the heart rate in patients with persistently accelerated ventricular rates despite concomitant standard therapy with a beta-blocker, digitalis, or a calcium-channel blocker. Dronedarone reduced the mean 24-hour heart rate by 11.7 beats per minute and the maximal exercise ventricular rate by 24.5 beats per minute at the 14th day.
ANDROMEDA (Anti-arrhythmic Trial With Dronedarone in Moderate to Severe CHF Evaluating Morbidity Decrease)13 was a study not of patients with atrial fibrillation but rather of patients with symptomatic congestive heart failure, a left ventricular ejection fraction of 35% or less, and recent hospitalization with new or worsening heart failure. The study was terminated early because of a higher rate of death with dronedarone13 (Table 1).
Dronedarone vs amiodarone
DIONYSOS (Efficacy and Safety of Dronedarone Versus Amiodarone for the Maintenance of Sinus Rhythm in Patients With Atrial Fibrillation)14 was a randomized double-blind trial. It evaluated the efficacy and safety of dronedarone (400 mg twice daily) or amiodarone (600 mg daily for 28 days, then 200 mg daily thereafter) for at least 6 months for the maintenance of sinus rhythm in patients with atrial fibrillation. It enrolled 504 patients with persistent atrial fibrillation; patients had not previously taken amiodarone. Dronedarone was less effective than amiodarone in maintaining sinus rhythm: the rate of recurrent atrial fibrillation was 63% with dronedarone and 42% with amiodarone. But dronedarone was associated with fewer adverse effects and less need for premature discontinuation of drug treatment at a mean follow-up of 7 months (Table 1).
WHERE DOES DRONEDARONE FIT IN ATRIAL FIBRILLATION MANAGEMENT?
Dronedarone is indicated in persistent or paroxysmal atrial fibrillation, based on the observed reduction of the rate of hospitalization. It is indicated for the maintenance of sinus rhythm and may be used in patients with persistent or paroxysmal atrial fibrillation and flutter who are in sinus rhythm or will be undergoing cardioversion soon after starting the drug. Dronedarone has no role in the acute management of atrial fibrillation, such as in cardioversion to sinus rhythm in the emergency department.
We do not have substantial evidence of the efficacy of dronedarone in patients with resistant atrial fibrillation, in whom multiple antiarrhythmics have failed to maintain sinus rhythm, and no published trial has used the inclusion criterion of treatment failure with other antiarrhythmic drugs.
The role of dronedarone in heart failure with preserved systolic function is unclear. Patients taking dronedarone are twice as likely as those taking amiodarone to have a recurrence of atrial fibrillation.
The main advantage of dronedarone is its lower adverse effect profile. However, this statement is based on only a few years of observation. If the patient has developed adverse effects with amiodarone, or if the clinician is concerned about the risk of serious adverse effects, dronedarone presents an alternative for those patients without heart failure or significant left ventricular dysfunction. One such group may be younger patients, because of concerns about the cumulative effects of amiodarone taken over a lifetime.
Dronedarone may represent an acceptable alternative to many of the current antiarrhythmic drugs. Based on the results of the Cardiac Arrhythmia Suppression Trial (CAST),15 class IC antiarrhythmics such as flecainide (Tambocor) are generally avoided in patients with prior myocardial infarction or with known or even suspected coronary artery disease. Similarly, sotalol (Betapace) is generally avoided in patients with marked left ventricular hypertrophy because of adverse effects.16 Dofetilide (Tikosyn) and often sotalol require hospitalization with telemetric monitoring for QTc prolongation and the risk of proarrhythmia with torsades de pointes. Dronedarone, however, generally can be safely started in the outpatient setting.
As when considering prescribing any antiarrhythmic, the clinician must assess the patient’s thromboembolic risk, since this risk persists with a rhythm control strategy.
There is substantial evidence from the ATHENA trial,10 in which 30% of the patients had coronary artery disease, that dronedarone is safe and effective in patients with coronary artery disease. Its use in patients who have undergone coronary artery bypass surgery remains to be defined.
WHEN SHOULD WE SWITCH PATIENTS TO DRONEDARONE?
Preliminary experience suggests that dronedarone, unlike most antiarrhythmic drugs, can be safely started about 48 hours after amiodarone is discontinued. Cumulative toxicity has not been noted with dronedarone. Caution should be exercised when switching if the patient has baseline bradycardia or QT interval prolongation. No algorithm has been developed for switching from other antiarrhythmic drugs to dronedarone.
CONTRAINDICATIONS TO DRONEDARONE
Dronedarone is contraindicated in:
- Patients with New York Heart Association (NYHA) class IV heart failure or NYHA class II or III heart failure with recent decompensation requiring hospitalization or referral to a specialized heart failure clinic
- Patients with second- or third-degree atrioventricular block or sick sinus syndrome (except when used in conjunction with a functioning pacemaker) or bradycardia (a heart rate < 50 beats per minute)
- Patients with a QTc interval of 500 ms or longer
- Patients with severe hepatic impairment
- Women who are pregnant, are attempting to become pregnant, or are breast-feeding
- Patients taking potent CYP3A inhibitors—antifungals like ketoconazole (Nizoral), itraconazole (Sporanox), or voriconazole (VFEND); macrolide antibiotics like telithromycin (Ketek) or clarithromycin (Biaxin); protease inhibitors; or other drugs that prolong the QT interval.
In patients with new or worsening heart failure, one should consider suspending or stopping dronedarone therapy.
DRONEDARONE’S ADVERSE EFFECTS
In trials to date, dronedarone has not shown evidence of proarrhythmia (tachyarrhythmia or bradyarrythmia), torsades de pointes, or amiodarone-like organ toxicity affecting the thyroid or the lungs. Recently, rare cases of severe hepatic injury were associated with dronedarone; therefore, periodic liver function testing is advised for patients taking dronedarone, especially during the first 6 months of therapy.
Dronedarone has been associated with higher rates of diarrhea, nausea, bradycardia, QT interval prolongation, and cutaneous rash compared with placebo. In DAFNE (Dronedarone Atrial Fibrillation Study After Electrical Cardioversion),17 10.8% of patients taking dronedarone had to stop taking it because of adverse events. With 800 mg daily, the discontinuation rate was only 3.9%. The most common cause of drug discontinuation was gastrointestinal effects. Anecdotal reports suggest that the gastrointestinal side effects may be self-limited and may not always require discontinuation of the drug.
Serum creatinine levels increase by about 0.1 mg/dL after the start of treatment. This elevation occurs after 1 to 2 days, reaches a plateau after 7 days, and is reversible. The mechanism is thought to be that dronedarone partially inhibits tubular organic cation transporters, which in turn reduces renal creatinine clearance by about 18%, but with no evidence of an effect on glomerular filtration, renal plasma flow, or electrolyte exchanges.18 A limited increase in serum creatinine is, therefore, expected with dronedarone treatment, but this does not mean there is a decline in renal function.
DRONEDARONE AND POTENTIAL DRUG INTERACTIONS
Warfarin. Dronedarone does not increase the international normalized ratio when used with warfarin (Coumadin).
Verapamil, diltiazem. Dose reduction is required to avoid bradyarrhythmias with co-administration of moderate CYP3A4 inhibitors such as verapamil (Calan, Verelan) and diltiazem (Cardizem).
Simvastatin. Dronedarone increases levels of simvastatin (Zocor), a CYP3A4 substrate, two to four times, thus increasing the risk of statin-induced myopathy.
Digoxin. Dronedarone increases the serum digoxin concentration about 2.5 times, and this necessitates monitoring the digoxin level and possibly reducing the digoxin dose.13
Diuretics. Hypokalemia and hypomagnesemia may occur with concomitant administration of potassium-depleting diuretics. Potassium levels should be maintained in the normal range before and during administration of dronedarone.
Tacrolimus, sirolimus. Dronedarone may increase levels of tacrolimus (Prograf) or sirolimus (Rapamune) in posttransplantation patients. This requires dose monitoring and adjustment in concomitant therapy with these agents.
COST VARIES
The cost of dronedarone varies based on factors that include location. Dronedarone’s retail cost ranges from $3.20 to $4.00 per pill (approximately $7.20 per day). It is not available in generic form. It is presently covered by many health plans as a tier 2 drug, representing a $15 to $40 monthly copay.
MORE DATA NEEDED
Dronedarone represents the first in what may well be a number of new antiarrhythmic drugs for the treatment of patients with paroxysmal atrial fibrillation. Although less efficacious then amiodarone, dronedarone appears to be better tolerated and have less serious side effects. It is contraindicated in patients with severe systolic dysfunction and in those with recent heart failure decompensation. It appears safe in coronary artery disease and marked left ventricular hypertrophy, unlike flecainide, propafenone (Rythmol), and sotalol.
To further understand how dronedarone will fare against other antiarrhythmic drugs, more studies with longer follow-up are needed. These studies need to demonstrate superior tolerability of dronedarone, acceptable quality of life without unacceptable loss of efficacy, or a decrease in morbidity or mortality rates compared with amiodarone.
Dronedarone can be safely started in most patients on an outpatient basis. The risk of proarrhythmia with dronedarone appears to be very low.
- Mathew ST, Patel J, Joseph S. Atrial fibrillation: mechanistic insights and treatment options. Eur J Intern Med 2009; 20:672–681.
- Schmitt J, Ehrlich JR, Hohnloser SH. New antiarrhythmic drugs for the treatment of atrial fibrillation. Herz 2008; 33:562–567.
- Wyse DG, Waldo AL, DiMarco JP, et al; Atrial Fibrillation Follow-up Investigation of Rhythm Management (AFFIRM) Investigators. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med 2002; 347:1825–1833.
- Van Gelder IC, Groenveld HF, Crijns HJ, et al; RACE II Investigators. Lenient versus strict rate control in patients with atrial fibrillation. N Engl J Med 2010; 362:1363–1373.
- Singh BN, Singh SN, Reda DJ, et al; Sotalol Amiodarone Atrial Fibrillation Efficacy Trial (SAFE-T) Investigators. Amiodarone versus sotalol for atrial fibrillation. N Engl J Med 2005; 352:1861–1872.
- The AFFIRM Investigators. Relationships between sinus rhythm, treatment, and survival in the Atrial Fibrillation Follow-Up Investigation of Rhythm Management (AFFIRM) study. Circulation 2004; 109:1509–1513.
- Van Beeren HC, Jong WM, Kaptein E, Visser TJ, Bakker O, Wiersinga WM. Dronerarone [sic] acts as a selective inhibitor of 3,5,3′-triiodothyronine binding to thyroid hormone receptor-alpha1: in vitro and in vivo evidence. Endocrinology 2003; 144:552–558.
- Patel C, Yan GX, Kowey PR. Dronedarone. Circulation 2009; 120:636–644.
- Dale KM, White CM. Dronedarone: an amiodarone analog for the treatment of atrial fibrillation and atrial flutter. Ann Pharmacother 2007; 41:599–605.
- Hohnloser SH, Crijns HJ, van Eickels M; ATHENA Investigators. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med 2009; 360:668–678.
- Singh BN, Connolly SJ, Crijns HJ, et al; EURIDIS and ADONIS Investigators. Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N Engl J Med 2007; 357:987–999.
- Davy JM, Herold M, Hoglund CERATO Study Investigators. Dronedarone for the control of ventricular rate in permanent atrial fibrillation: the Efficacy and safety of dRonedArone for the cOntrol of ventricular rate during atrial fibrillation (ERATO) study. Am Heart J 2008; 156:527.e1–e9.
- Køber L, Torp-Pedersen C, McMurray JJ; Dronedarone Study Group. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med 2008; 358:2678–2687.
- Le Heuzey JY, De Ferrari GM, Radzik D, Santini M, Zhu J, Davy JM. A short-term, randomized, double-blind, parallel-group study to evaluate the efficacy and safety of dronedarone versus amiodarone in patients with persistent atrial fibrillation: the DIONYSOS study. J Cardiovasc Electrophysiol 2010; 21:597–605.
- The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med 1989; 321:406–412.
- Pratt CM. Clinical implications of the Survival With Oral D-sotalol (SWORD) trial: an investigation of patients with left ventricular dysfunction after myocardial infarction. Card Electrophysiol Rev 1998; 2:28–29.
- Touboul P, Brugada J, Capucci A, Crijns HJ, Edvardsson N, Hohnloser SH. Dronedarone for prevention of atrial fibrillation: a dose-ranging study. Eur Heart J 2003; 24:1481–1487.
- Tschuppert Y, Buclin T, Rothuizen LE, et al. Effect of dronedarone on renal function in healthy subjects. Br J Clin Pharmacol 2007; 64:785–791.
Dronedarone (Multaq), approved by the US Food and Drug Administration in July 2009, is a congener of the antiarrhythmic drug amiodarone (Cordarone). Designed in the hope that it would be safer than amiodarone, its official indication is to lower the risk of hospitalization in patients with paroxysmal or persistent atrial fibrillation or atrial flutter. However, its precise role in the management of atrial fibrillation is yet to be defined. If dronedarone remains well tolerated, it may permit clinicians to pursue a rhythm control strategy more often. In this article, we present a progress report on this new agent.
BETTER ANTIARRHYTHMIC DRUGS ARE NEEDED
Atrial fibrillation increases the risk of stroke fivefold and accounts for 15% to 20% of all strokes.1 It also increases the risk of heart failure. Drugs are the mainstay of therapy, but many antiarrhythmic drugs are not very effective and cause cardiac and extracardiac toxicity. Thus, the need for safe and effective new drugs.2
Much effort is going into the development of drugs that target specific ion channels or proteins expressed predominantly in atrial myocardium. The rationale is to avoid the unwanted effects of ionic currents on the ventricle and thus avoid ventricular proarrhythmic effects. At the same time, alternatives to the multiple channel blocker amiodarone, the mainstay of heart rhythm control therapy in atrial fibrillation, are being developed to retain the electrophysiologic efficacy of the mother compound but avoid its extracardiac toxicity.
RATE CONTROL VS RHYTHM CONTROL
In the acute care setting, heart rate control with atrioventricular nodal agents (beta-blockers, calcium channel blockers, and digitalis) is the preferred initial strategy in most hemodynamically stable patients presenting with new-onset atrial fibrillation.3
Since we lack an effective method for maintaining sinus rhythm without incurring significant adverse effects, rate control is also often chosen for chronic management of atrial fibrillation. This is particularly true for patients who have no symptoms or only minimal symptoms and in whom adequate rate control is easily attained. Indeed, results of large clinical trials suggest that rate control is satisfactory for many patients.
The main purpose of rate control is to control symptoms as opposed to merely lowering the ventricular rate. Effective rate control often prevents hemodynamic instability in patients with underlying heart disease who present acutely with atrial fibrillation. In patients with permanent atrial fibrillation, the RACE II study4 (Rate Control Efficacy in Permanent Atrial Fibrillation: a Comparison between Lenient Versus Strict Rate Control II), during a 3-year follow-up, showed that lenient rate control (resting heart rate < 110 beats per minute) is not inferior to strict rate control (resting heart rate < 80 beats per minute) in preventing major cardiovascular events (heart failure, stroke) or arrhythmic events such as syncope and sustained ventricular tachycardia.4
As a long-term strategy, rate control also prevents tachycardia-induced cardiomyopathy, reduces the risk of worsening of underlying heart failure, and can improve symptoms and quality of life.
Although maintenance of sinus rhythm is most likely associated with a survival benefit, heart rhythm control with antiarrhythmic drugs has not shown an advantage over rate control in overall or cardiovascular death rates, thromboembolic complications, or impact on heart failure. Indeed, a rhythm control strategy has been associated only with better exercise tolerance and, although less clear, with better quality of life.5
One possible explanation as to why a rhythm control strategy has not been shown to be superior to a rate control strategy is the side effects of the presently available drugs for rhythm control.
In a subgroup analysis of the Atrial Fibrillation Follow-up Investigation of Rhythm Management (AFFIRM) trial,6 antiarrhythmic therapy was associated with a 49% increase in the mortality rate that offset the benefits of conversion and maintenance of sinus rhythm, which was associated with a 53% reduction in mortality rates.
The hope is that newer drugs with less toxicity may produce better outcomes for patients treated with rhythm control.
AN ANALOGUE OF AMIODARONE, WITHOUT THE IODINE
Dronedarone is a structurally modified version of amiodarone, the antiarrhythmic drug that has shown the greatest efficacy at maintaining sinus rhythm in patients with paroxysmal atrial fibrillation. Although historically amiodarone has been effective in maintaining sinus rhythm and has been used safely in patients with advanced heart failure, its use has been limited by cumulative and often irreversible extracardiac organ toxicity.
Dronedarone was designed to match amiodarone’s efficacy but with a better safety profile. An iodine radical makes up more than one-third of amiodarone’s molecular weight. The omission of iodine in dronedarone was intended to reduce the likelihood of toxic side effects.
Dronedarone is a benzofuran derivative pharmacologically related to amiodarone, with the addition of a methylsulfonamide group. This reduces lipophilicity and the propensity to cross the blood-brain barrier; over a 2-year period this drug has not been shown to have neurotoxic effects.7
Dronedarone has proved efficacious without toxic or proarrhythmic effects and has minimal side effects, but concerns remain regarding its use in advanced heart failure. To date, its adverse-event profile appears comparable to that of placebo. However, whether its efficacy and incidence of adverse effects are comparable to what has been reported in the literature may take time to assess.
DRONEDARONE’S PHARMACOLOGY
Dronedarone, like amiodarone, blocks multiple sodium and potassium ion channels. It also exerts an antiadrenergic effect by noncompetitive binding to beta-adrenergic receptors as well as by inhibiting an agonist-induced increase in adenylate cyclase activity.8 Compared with amiodarone, dronedarone is a more potent blocker of peak sodium current.
Dronedarone is largely metabolized by the hepatic enzyme cytochrome P450 3A4 isoform (CYP3A4). Only 6% of dronedarone is excreted renally; however, no trial has yet assessed dronedarone’s safety in patients with marked kidney dysfunction.89
Dronedarone’s steady-state terminal elimination half-life is approximately 30 hours. When taken twice a day, it achieves steady-state concentrations in 5 to 7 days.
Dronedarone is available only for oral administration at 400 mg twice daily. Dose adjustment or titration is not recommended.
CLINICAL TRIALS OF DRONEDARONE
Dronedarone vs placebo
ATHENA (A Placebo-Controlled, Double-Blind, Parallel Arm Trial to Assess the Efficacy of Dronedarone 400 mg bid for the Prevention of Cardiovascular Hospitalization or Death From any Cause in Patients With Atrial Fibrillation/Atrial Flutter)10 was a prospective, double-blind study to assess morbidity and death rates in 4,628 patients with atrial fibrillation or atrial flutter and at least one other cardiovascular risk factor.
EURIDIS and ADONIS. Two trials,11 EURIDIS (European Trial in Atrial Fibrillation or Flutter Patients Receiving Dronedarone for the Maintenance of Sinus Rhythm) and ADONIS (American-Australian Trial With Dronedarone in Atrial Fibrillation or Flutter Patients for the Maintenance of Sinus Rhythm), enrolled a total of more than 1,200 patients and showed that dronedarone 400 mg twice a day produced a significantly lower rate of recurrence of atrial fibrillation after electrical cardioversion compared with placebo.
Overall, treatment with dronedarone significantly reduced the risk of a first recurrence of atrial fibrillation by 22% (ADONIS) and 27.5% (EURIDIS) (Table 1).
ERATO (Efficacy and Safety of Dronedarone for the Control of Ventricular Rate During Atrial Fibrillation),12 an additional phase III study, showed that dronedarone controlled the heart rate in patients with persistently accelerated ventricular rates despite concomitant standard therapy with a beta-blocker, digitalis, or a calcium-channel blocker. Dronedarone reduced the mean 24-hour heart rate by 11.7 beats per minute and the maximal exercise ventricular rate by 24.5 beats per minute at the 14th day.
ANDROMEDA (Anti-arrhythmic Trial With Dronedarone in Moderate to Severe CHF Evaluating Morbidity Decrease)13 was a study not of patients with atrial fibrillation but rather of patients with symptomatic congestive heart failure, a left ventricular ejection fraction of 35% or less, and recent hospitalization with new or worsening heart failure. The study was terminated early because of a higher rate of death with dronedarone13 (Table 1).
Dronedarone vs amiodarone
DIONYSOS (Efficacy and Safety of Dronedarone Versus Amiodarone for the Maintenance of Sinus Rhythm in Patients With Atrial Fibrillation)14 was a randomized double-blind trial. It evaluated the efficacy and safety of dronedarone (400 mg twice daily) or amiodarone (600 mg daily for 28 days, then 200 mg daily thereafter) for at least 6 months for the maintenance of sinus rhythm in patients with atrial fibrillation. It enrolled 504 patients with persistent atrial fibrillation; patients had not previously taken amiodarone. Dronedarone was less effective than amiodarone in maintaining sinus rhythm: the rate of recurrent atrial fibrillation was 63% with dronedarone and 42% with amiodarone. But dronedarone was associated with fewer adverse effects and less need for premature discontinuation of drug treatment at a mean follow-up of 7 months (Table 1).
WHERE DOES DRONEDARONE FIT IN ATRIAL FIBRILLATION MANAGEMENT?
Dronedarone is indicated in persistent or paroxysmal atrial fibrillation, based on the observed reduction of the rate of hospitalization. It is indicated for the maintenance of sinus rhythm and may be used in patients with persistent or paroxysmal atrial fibrillation and flutter who are in sinus rhythm or will be undergoing cardioversion soon after starting the drug. Dronedarone has no role in the acute management of atrial fibrillation, such as in cardioversion to sinus rhythm in the emergency department.
We do not have substantial evidence of the efficacy of dronedarone in patients with resistant atrial fibrillation, in whom multiple antiarrhythmics have failed to maintain sinus rhythm, and no published trial has used the inclusion criterion of treatment failure with other antiarrhythmic drugs.
The role of dronedarone in heart failure with preserved systolic function is unclear. Patients taking dronedarone are twice as likely as those taking amiodarone to have a recurrence of atrial fibrillation.
The main advantage of dronedarone is its lower adverse effect profile. However, this statement is based on only a few years of observation. If the patient has developed adverse effects with amiodarone, or if the clinician is concerned about the risk of serious adverse effects, dronedarone presents an alternative for those patients without heart failure or significant left ventricular dysfunction. One such group may be younger patients, because of concerns about the cumulative effects of amiodarone taken over a lifetime.
Dronedarone may represent an acceptable alternative to many of the current antiarrhythmic drugs. Based on the results of the Cardiac Arrhythmia Suppression Trial (CAST),15 class IC antiarrhythmics such as flecainide (Tambocor) are generally avoided in patients with prior myocardial infarction or with known or even suspected coronary artery disease. Similarly, sotalol (Betapace) is generally avoided in patients with marked left ventricular hypertrophy because of adverse effects.16 Dofetilide (Tikosyn) and often sotalol require hospitalization with telemetric monitoring for QTc prolongation and the risk of proarrhythmia with torsades de pointes. Dronedarone, however, generally can be safely started in the outpatient setting.
As when considering prescribing any antiarrhythmic, the clinician must assess the patient’s thromboembolic risk, since this risk persists with a rhythm control strategy.
There is substantial evidence from the ATHENA trial,10 in which 30% of the patients had coronary artery disease, that dronedarone is safe and effective in patients with coronary artery disease. Its use in patients who have undergone coronary artery bypass surgery remains to be defined.
WHEN SHOULD WE SWITCH PATIENTS TO DRONEDARONE?
Preliminary experience suggests that dronedarone, unlike most antiarrhythmic drugs, can be safely started about 48 hours after amiodarone is discontinued. Cumulative toxicity has not been noted with dronedarone. Caution should be exercised when switching if the patient has baseline bradycardia or QT interval prolongation. No algorithm has been developed for switching from other antiarrhythmic drugs to dronedarone.
CONTRAINDICATIONS TO DRONEDARONE
Dronedarone is contraindicated in:
- Patients with New York Heart Association (NYHA) class IV heart failure or NYHA class II or III heart failure with recent decompensation requiring hospitalization or referral to a specialized heart failure clinic
- Patients with second- or third-degree atrioventricular block or sick sinus syndrome (except when used in conjunction with a functioning pacemaker) or bradycardia (a heart rate < 50 beats per minute)
- Patients with a QTc interval of 500 ms or longer
- Patients with severe hepatic impairment
- Women who are pregnant, are attempting to become pregnant, or are breast-feeding
- Patients taking potent CYP3A inhibitors—antifungals like ketoconazole (Nizoral), itraconazole (Sporanox), or voriconazole (VFEND); macrolide antibiotics like telithromycin (Ketek) or clarithromycin (Biaxin); protease inhibitors; or other drugs that prolong the QT interval.
In patients with new or worsening heart failure, one should consider suspending or stopping dronedarone therapy.
DRONEDARONE’S ADVERSE EFFECTS
In trials to date, dronedarone has not shown evidence of proarrhythmia (tachyarrhythmia or bradyarrythmia), torsades de pointes, or amiodarone-like organ toxicity affecting the thyroid or the lungs. Recently, rare cases of severe hepatic injury were associated with dronedarone; therefore, periodic liver function testing is advised for patients taking dronedarone, especially during the first 6 months of therapy.
Dronedarone has been associated with higher rates of diarrhea, nausea, bradycardia, QT interval prolongation, and cutaneous rash compared with placebo. In DAFNE (Dronedarone Atrial Fibrillation Study After Electrical Cardioversion),17 10.8% of patients taking dronedarone had to stop taking it because of adverse events. With 800 mg daily, the discontinuation rate was only 3.9%. The most common cause of drug discontinuation was gastrointestinal effects. Anecdotal reports suggest that the gastrointestinal side effects may be self-limited and may not always require discontinuation of the drug.
Serum creatinine levels increase by about 0.1 mg/dL after the start of treatment. This elevation occurs after 1 to 2 days, reaches a plateau after 7 days, and is reversible. The mechanism is thought to be that dronedarone partially inhibits tubular organic cation transporters, which in turn reduces renal creatinine clearance by about 18%, but with no evidence of an effect on glomerular filtration, renal plasma flow, or electrolyte exchanges.18 A limited increase in serum creatinine is, therefore, expected with dronedarone treatment, but this does not mean there is a decline in renal function.
DRONEDARONE AND POTENTIAL DRUG INTERACTIONS
Warfarin. Dronedarone does not increase the international normalized ratio when used with warfarin (Coumadin).
Verapamil, diltiazem. Dose reduction is required to avoid bradyarrhythmias with co-administration of moderate CYP3A4 inhibitors such as verapamil (Calan, Verelan) and diltiazem (Cardizem).
Simvastatin. Dronedarone increases levels of simvastatin (Zocor), a CYP3A4 substrate, two to four times, thus increasing the risk of statin-induced myopathy.
Digoxin. Dronedarone increases the serum digoxin concentration about 2.5 times, and this necessitates monitoring the digoxin level and possibly reducing the digoxin dose.13
Diuretics. Hypokalemia and hypomagnesemia may occur with concomitant administration of potassium-depleting diuretics. Potassium levels should be maintained in the normal range before and during administration of dronedarone.
Tacrolimus, sirolimus. Dronedarone may increase levels of tacrolimus (Prograf) or sirolimus (Rapamune) in posttransplantation patients. This requires dose monitoring and adjustment in concomitant therapy with these agents.
COST VARIES
The cost of dronedarone varies based on factors that include location. Dronedarone’s retail cost ranges from $3.20 to $4.00 per pill (approximately $7.20 per day). It is not available in generic form. It is presently covered by many health plans as a tier 2 drug, representing a $15 to $40 monthly copay.
MORE DATA NEEDED
Dronedarone represents the first in what may well be a number of new antiarrhythmic drugs for the treatment of patients with paroxysmal atrial fibrillation. Although less efficacious then amiodarone, dronedarone appears to be better tolerated and have less serious side effects. It is contraindicated in patients with severe systolic dysfunction and in those with recent heart failure decompensation. It appears safe in coronary artery disease and marked left ventricular hypertrophy, unlike flecainide, propafenone (Rythmol), and sotalol.
To further understand how dronedarone will fare against other antiarrhythmic drugs, more studies with longer follow-up are needed. These studies need to demonstrate superior tolerability of dronedarone, acceptable quality of life without unacceptable loss of efficacy, or a decrease in morbidity or mortality rates compared with amiodarone.
Dronedarone can be safely started in most patients on an outpatient basis. The risk of proarrhythmia with dronedarone appears to be very low.
Dronedarone (Multaq), approved by the US Food and Drug Administration in July 2009, is a congener of the antiarrhythmic drug amiodarone (Cordarone). Designed in the hope that it would be safer than amiodarone, its official indication is to lower the risk of hospitalization in patients with paroxysmal or persistent atrial fibrillation or atrial flutter. However, its precise role in the management of atrial fibrillation is yet to be defined. If dronedarone remains well tolerated, it may permit clinicians to pursue a rhythm control strategy more often. In this article, we present a progress report on this new agent.
BETTER ANTIARRHYTHMIC DRUGS ARE NEEDED
Atrial fibrillation increases the risk of stroke fivefold and accounts for 15% to 20% of all strokes.1 It also increases the risk of heart failure. Drugs are the mainstay of therapy, but many antiarrhythmic drugs are not very effective and cause cardiac and extracardiac toxicity. Thus, the need for safe and effective new drugs.2
Much effort is going into the development of drugs that target specific ion channels or proteins expressed predominantly in atrial myocardium. The rationale is to avoid the unwanted effects of ionic currents on the ventricle and thus avoid ventricular proarrhythmic effects. At the same time, alternatives to the multiple channel blocker amiodarone, the mainstay of heart rhythm control therapy in atrial fibrillation, are being developed to retain the electrophysiologic efficacy of the mother compound but avoid its extracardiac toxicity.
RATE CONTROL VS RHYTHM CONTROL
In the acute care setting, heart rate control with atrioventricular nodal agents (beta-blockers, calcium channel blockers, and digitalis) is the preferred initial strategy in most hemodynamically stable patients presenting with new-onset atrial fibrillation.3
Since we lack an effective method for maintaining sinus rhythm without incurring significant adverse effects, rate control is also often chosen for chronic management of atrial fibrillation. This is particularly true for patients who have no symptoms or only minimal symptoms and in whom adequate rate control is easily attained. Indeed, results of large clinical trials suggest that rate control is satisfactory for many patients.
The main purpose of rate control is to control symptoms as opposed to merely lowering the ventricular rate. Effective rate control often prevents hemodynamic instability in patients with underlying heart disease who present acutely with atrial fibrillation. In patients with permanent atrial fibrillation, the RACE II study4 (Rate Control Efficacy in Permanent Atrial Fibrillation: a Comparison between Lenient Versus Strict Rate Control II), during a 3-year follow-up, showed that lenient rate control (resting heart rate < 110 beats per minute) is not inferior to strict rate control (resting heart rate < 80 beats per minute) in preventing major cardiovascular events (heart failure, stroke) or arrhythmic events such as syncope and sustained ventricular tachycardia.4
As a long-term strategy, rate control also prevents tachycardia-induced cardiomyopathy, reduces the risk of worsening of underlying heart failure, and can improve symptoms and quality of life.
Although maintenance of sinus rhythm is most likely associated with a survival benefit, heart rhythm control with antiarrhythmic drugs has not shown an advantage over rate control in overall or cardiovascular death rates, thromboembolic complications, or impact on heart failure. Indeed, a rhythm control strategy has been associated only with better exercise tolerance and, although less clear, with better quality of life.5
One possible explanation as to why a rhythm control strategy has not been shown to be superior to a rate control strategy is the side effects of the presently available drugs for rhythm control.
In a subgroup analysis of the Atrial Fibrillation Follow-up Investigation of Rhythm Management (AFFIRM) trial,6 antiarrhythmic therapy was associated with a 49% increase in the mortality rate that offset the benefits of conversion and maintenance of sinus rhythm, which was associated with a 53% reduction in mortality rates.
The hope is that newer drugs with less toxicity may produce better outcomes for patients treated with rhythm control.
AN ANALOGUE OF AMIODARONE, WITHOUT THE IODINE
Dronedarone is a structurally modified version of amiodarone, the antiarrhythmic drug that has shown the greatest efficacy at maintaining sinus rhythm in patients with paroxysmal atrial fibrillation. Although historically amiodarone has been effective in maintaining sinus rhythm and has been used safely in patients with advanced heart failure, its use has been limited by cumulative and often irreversible extracardiac organ toxicity.
Dronedarone was designed to match amiodarone’s efficacy but with a better safety profile. An iodine radical makes up more than one-third of amiodarone’s molecular weight. The omission of iodine in dronedarone was intended to reduce the likelihood of toxic side effects.
Dronedarone is a benzofuran derivative pharmacologically related to amiodarone, with the addition of a methylsulfonamide group. This reduces lipophilicity and the propensity to cross the blood-brain barrier; over a 2-year period this drug has not been shown to have neurotoxic effects.7
Dronedarone has proved efficacious without toxic or proarrhythmic effects and has minimal side effects, but concerns remain regarding its use in advanced heart failure. To date, its adverse-event profile appears comparable to that of placebo. However, whether its efficacy and incidence of adverse effects are comparable to what has been reported in the literature may take time to assess.
DRONEDARONE’S PHARMACOLOGY
Dronedarone, like amiodarone, blocks multiple sodium and potassium ion channels. It also exerts an antiadrenergic effect by noncompetitive binding to beta-adrenergic receptors as well as by inhibiting an agonist-induced increase in adenylate cyclase activity.8 Compared with amiodarone, dronedarone is a more potent blocker of peak sodium current.
Dronedarone is largely metabolized by the hepatic enzyme cytochrome P450 3A4 isoform (CYP3A4). Only 6% of dronedarone is excreted renally; however, no trial has yet assessed dronedarone’s safety in patients with marked kidney dysfunction.89
Dronedarone’s steady-state terminal elimination half-life is approximately 30 hours. When taken twice a day, it achieves steady-state concentrations in 5 to 7 days.
Dronedarone is available only for oral administration at 400 mg twice daily. Dose adjustment or titration is not recommended.
CLINICAL TRIALS OF DRONEDARONE
Dronedarone vs placebo
ATHENA (A Placebo-Controlled, Double-Blind, Parallel Arm Trial to Assess the Efficacy of Dronedarone 400 mg bid for the Prevention of Cardiovascular Hospitalization or Death From any Cause in Patients With Atrial Fibrillation/Atrial Flutter)10 was a prospective, double-blind study to assess morbidity and death rates in 4,628 patients with atrial fibrillation or atrial flutter and at least one other cardiovascular risk factor.
EURIDIS and ADONIS. Two trials,11 EURIDIS (European Trial in Atrial Fibrillation or Flutter Patients Receiving Dronedarone for the Maintenance of Sinus Rhythm) and ADONIS (American-Australian Trial With Dronedarone in Atrial Fibrillation or Flutter Patients for the Maintenance of Sinus Rhythm), enrolled a total of more than 1,200 patients and showed that dronedarone 400 mg twice a day produced a significantly lower rate of recurrence of atrial fibrillation after electrical cardioversion compared with placebo.
Overall, treatment with dronedarone significantly reduced the risk of a first recurrence of atrial fibrillation by 22% (ADONIS) and 27.5% (EURIDIS) (Table 1).
ERATO (Efficacy and Safety of Dronedarone for the Control of Ventricular Rate During Atrial Fibrillation),12 an additional phase III study, showed that dronedarone controlled the heart rate in patients with persistently accelerated ventricular rates despite concomitant standard therapy with a beta-blocker, digitalis, or a calcium-channel blocker. Dronedarone reduced the mean 24-hour heart rate by 11.7 beats per minute and the maximal exercise ventricular rate by 24.5 beats per minute at the 14th day.
ANDROMEDA (Anti-arrhythmic Trial With Dronedarone in Moderate to Severe CHF Evaluating Morbidity Decrease)13 was a study not of patients with atrial fibrillation but rather of patients with symptomatic congestive heart failure, a left ventricular ejection fraction of 35% or less, and recent hospitalization with new or worsening heart failure. The study was terminated early because of a higher rate of death with dronedarone13 (Table 1).
Dronedarone vs amiodarone
DIONYSOS (Efficacy and Safety of Dronedarone Versus Amiodarone for the Maintenance of Sinus Rhythm in Patients With Atrial Fibrillation)14 was a randomized double-blind trial. It evaluated the efficacy and safety of dronedarone (400 mg twice daily) or amiodarone (600 mg daily for 28 days, then 200 mg daily thereafter) for at least 6 months for the maintenance of sinus rhythm in patients with atrial fibrillation. It enrolled 504 patients with persistent atrial fibrillation; patients had not previously taken amiodarone. Dronedarone was less effective than amiodarone in maintaining sinus rhythm: the rate of recurrent atrial fibrillation was 63% with dronedarone and 42% with amiodarone. But dronedarone was associated with fewer adverse effects and less need for premature discontinuation of drug treatment at a mean follow-up of 7 months (Table 1).
WHERE DOES DRONEDARONE FIT IN ATRIAL FIBRILLATION MANAGEMENT?
Dronedarone is indicated in persistent or paroxysmal atrial fibrillation, based on the observed reduction of the rate of hospitalization. It is indicated for the maintenance of sinus rhythm and may be used in patients with persistent or paroxysmal atrial fibrillation and flutter who are in sinus rhythm or will be undergoing cardioversion soon after starting the drug. Dronedarone has no role in the acute management of atrial fibrillation, such as in cardioversion to sinus rhythm in the emergency department.
We do not have substantial evidence of the efficacy of dronedarone in patients with resistant atrial fibrillation, in whom multiple antiarrhythmics have failed to maintain sinus rhythm, and no published trial has used the inclusion criterion of treatment failure with other antiarrhythmic drugs.
The role of dronedarone in heart failure with preserved systolic function is unclear. Patients taking dronedarone are twice as likely as those taking amiodarone to have a recurrence of atrial fibrillation.
The main advantage of dronedarone is its lower adverse effect profile. However, this statement is based on only a few years of observation. If the patient has developed adverse effects with amiodarone, or if the clinician is concerned about the risk of serious adverse effects, dronedarone presents an alternative for those patients without heart failure or significant left ventricular dysfunction. One such group may be younger patients, because of concerns about the cumulative effects of amiodarone taken over a lifetime.
Dronedarone may represent an acceptable alternative to many of the current antiarrhythmic drugs. Based on the results of the Cardiac Arrhythmia Suppression Trial (CAST),15 class IC antiarrhythmics such as flecainide (Tambocor) are generally avoided in patients with prior myocardial infarction or with known or even suspected coronary artery disease. Similarly, sotalol (Betapace) is generally avoided in patients with marked left ventricular hypertrophy because of adverse effects.16 Dofetilide (Tikosyn) and often sotalol require hospitalization with telemetric monitoring for QTc prolongation and the risk of proarrhythmia with torsades de pointes. Dronedarone, however, generally can be safely started in the outpatient setting.
As when considering prescribing any antiarrhythmic, the clinician must assess the patient’s thromboembolic risk, since this risk persists with a rhythm control strategy.
There is substantial evidence from the ATHENA trial,10 in which 30% of the patients had coronary artery disease, that dronedarone is safe and effective in patients with coronary artery disease. Its use in patients who have undergone coronary artery bypass surgery remains to be defined.
WHEN SHOULD WE SWITCH PATIENTS TO DRONEDARONE?
Preliminary experience suggests that dronedarone, unlike most antiarrhythmic drugs, can be safely started about 48 hours after amiodarone is discontinued. Cumulative toxicity has not been noted with dronedarone. Caution should be exercised when switching if the patient has baseline bradycardia or QT interval prolongation. No algorithm has been developed for switching from other antiarrhythmic drugs to dronedarone.
CONTRAINDICATIONS TO DRONEDARONE
Dronedarone is contraindicated in:
- Patients with New York Heart Association (NYHA) class IV heart failure or NYHA class II or III heart failure with recent decompensation requiring hospitalization or referral to a specialized heart failure clinic
- Patients with second- or third-degree atrioventricular block or sick sinus syndrome (except when used in conjunction with a functioning pacemaker) or bradycardia (a heart rate < 50 beats per minute)
- Patients with a QTc interval of 500 ms or longer
- Patients with severe hepatic impairment
- Women who are pregnant, are attempting to become pregnant, or are breast-feeding
- Patients taking potent CYP3A inhibitors—antifungals like ketoconazole (Nizoral), itraconazole (Sporanox), or voriconazole (VFEND); macrolide antibiotics like telithromycin (Ketek) or clarithromycin (Biaxin); protease inhibitors; or other drugs that prolong the QT interval.
In patients with new or worsening heart failure, one should consider suspending or stopping dronedarone therapy.
DRONEDARONE’S ADVERSE EFFECTS
In trials to date, dronedarone has not shown evidence of proarrhythmia (tachyarrhythmia or bradyarrythmia), torsades de pointes, or amiodarone-like organ toxicity affecting the thyroid or the lungs. Recently, rare cases of severe hepatic injury were associated with dronedarone; therefore, periodic liver function testing is advised for patients taking dronedarone, especially during the first 6 months of therapy.
Dronedarone has been associated with higher rates of diarrhea, nausea, bradycardia, QT interval prolongation, and cutaneous rash compared with placebo. In DAFNE (Dronedarone Atrial Fibrillation Study After Electrical Cardioversion),17 10.8% of patients taking dronedarone had to stop taking it because of adverse events. With 800 mg daily, the discontinuation rate was only 3.9%. The most common cause of drug discontinuation was gastrointestinal effects. Anecdotal reports suggest that the gastrointestinal side effects may be self-limited and may not always require discontinuation of the drug.
Serum creatinine levels increase by about 0.1 mg/dL after the start of treatment. This elevation occurs after 1 to 2 days, reaches a plateau after 7 days, and is reversible. The mechanism is thought to be that dronedarone partially inhibits tubular organic cation transporters, which in turn reduces renal creatinine clearance by about 18%, but with no evidence of an effect on glomerular filtration, renal plasma flow, or electrolyte exchanges.18 A limited increase in serum creatinine is, therefore, expected with dronedarone treatment, but this does not mean there is a decline in renal function.
DRONEDARONE AND POTENTIAL DRUG INTERACTIONS
Warfarin. Dronedarone does not increase the international normalized ratio when used with warfarin (Coumadin).
Verapamil, diltiazem. Dose reduction is required to avoid bradyarrhythmias with co-administration of moderate CYP3A4 inhibitors such as verapamil (Calan, Verelan) and diltiazem (Cardizem).
Simvastatin. Dronedarone increases levels of simvastatin (Zocor), a CYP3A4 substrate, two to four times, thus increasing the risk of statin-induced myopathy.
Digoxin. Dronedarone increases the serum digoxin concentration about 2.5 times, and this necessitates monitoring the digoxin level and possibly reducing the digoxin dose.13
Diuretics. Hypokalemia and hypomagnesemia may occur with concomitant administration of potassium-depleting diuretics. Potassium levels should be maintained in the normal range before and during administration of dronedarone.
Tacrolimus, sirolimus. Dronedarone may increase levels of tacrolimus (Prograf) or sirolimus (Rapamune) in posttransplantation patients. This requires dose monitoring and adjustment in concomitant therapy with these agents.
COST VARIES
The cost of dronedarone varies based on factors that include location. Dronedarone’s retail cost ranges from $3.20 to $4.00 per pill (approximately $7.20 per day). It is not available in generic form. It is presently covered by many health plans as a tier 2 drug, representing a $15 to $40 monthly copay.
MORE DATA NEEDED
Dronedarone represents the first in what may well be a number of new antiarrhythmic drugs for the treatment of patients with paroxysmal atrial fibrillation. Although less efficacious then amiodarone, dronedarone appears to be better tolerated and have less serious side effects. It is contraindicated in patients with severe systolic dysfunction and in those with recent heart failure decompensation. It appears safe in coronary artery disease and marked left ventricular hypertrophy, unlike flecainide, propafenone (Rythmol), and sotalol.
To further understand how dronedarone will fare against other antiarrhythmic drugs, more studies with longer follow-up are needed. These studies need to demonstrate superior tolerability of dronedarone, acceptable quality of life without unacceptable loss of efficacy, or a decrease in morbidity or mortality rates compared with amiodarone.
Dronedarone can be safely started in most patients on an outpatient basis. The risk of proarrhythmia with dronedarone appears to be very low.
- Mathew ST, Patel J, Joseph S. Atrial fibrillation: mechanistic insights and treatment options. Eur J Intern Med 2009; 20:672–681.
- Schmitt J, Ehrlich JR, Hohnloser SH. New antiarrhythmic drugs for the treatment of atrial fibrillation. Herz 2008; 33:562–567.
- Wyse DG, Waldo AL, DiMarco JP, et al; Atrial Fibrillation Follow-up Investigation of Rhythm Management (AFFIRM) Investigators. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med 2002; 347:1825–1833.
- Van Gelder IC, Groenveld HF, Crijns HJ, et al; RACE II Investigators. Lenient versus strict rate control in patients with atrial fibrillation. N Engl J Med 2010; 362:1363–1373.
- Singh BN, Singh SN, Reda DJ, et al; Sotalol Amiodarone Atrial Fibrillation Efficacy Trial (SAFE-T) Investigators. Amiodarone versus sotalol for atrial fibrillation. N Engl J Med 2005; 352:1861–1872.
- The AFFIRM Investigators. Relationships between sinus rhythm, treatment, and survival in the Atrial Fibrillation Follow-Up Investigation of Rhythm Management (AFFIRM) study. Circulation 2004; 109:1509–1513.
- Van Beeren HC, Jong WM, Kaptein E, Visser TJ, Bakker O, Wiersinga WM. Dronerarone [sic] acts as a selective inhibitor of 3,5,3′-triiodothyronine binding to thyroid hormone receptor-alpha1: in vitro and in vivo evidence. Endocrinology 2003; 144:552–558.
- Patel C, Yan GX, Kowey PR. Dronedarone. Circulation 2009; 120:636–644.
- Dale KM, White CM. Dronedarone: an amiodarone analog for the treatment of atrial fibrillation and atrial flutter. Ann Pharmacother 2007; 41:599–605.
- Hohnloser SH, Crijns HJ, van Eickels M; ATHENA Investigators. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med 2009; 360:668–678.
- Singh BN, Connolly SJ, Crijns HJ, et al; EURIDIS and ADONIS Investigators. Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N Engl J Med 2007; 357:987–999.
- Davy JM, Herold M, Hoglund CERATO Study Investigators. Dronedarone for the control of ventricular rate in permanent atrial fibrillation: the Efficacy and safety of dRonedArone for the cOntrol of ventricular rate during atrial fibrillation (ERATO) study. Am Heart J 2008; 156:527.e1–e9.
- Køber L, Torp-Pedersen C, McMurray JJ; Dronedarone Study Group. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med 2008; 358:2678–2687.
- Le Heuzey JY, De Ferrari GM, Radzik D, Santini M, Zhu J, Davy JM. A short-term, randomized, double-blind, parallel-group study to evaluate the efficacy and safety of dronedarone versus amiodarone in patients with persistent atrial fibrillation: the DIONYSOS study. J Cardiovasc Electrophysiol 2010; 21:597–605.
- The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med 1989; 321:406–412.
- Pratt CM. Clinical implications of the Survival With Oral D-sotalol (SWORD) trial: an investigation of patients with left ventricular dysfunction after myocardial infarction. Card Electrophysiol Rev 1998; 2:28–29.
- Touboul P, Brugada J, Capucci A, Crijns HJ, Edvardsson N, Hohnloser SH. Dronedarone for prevention of atrial fibrillation: a dose-ranging study. Eur Heart J 2003; 24:1481–1487.
- Tschuppert Y, Buclin T, Rothuizen LE, et al. Effect of dronedarone on renal function in healthy subjects. Br J Clin Pharmacol 2007; 64:785–791.
- Mathew ST, Patel J, Joseph S. Atrial fibrillation: mechanistic insights and treatment options. Eur J Intern Med 2009; 20:672–681.
- Schmitt J, Ehrlich JR, Hohnloser SH. New antiarrhythmic drugs for the treatment of atrial fibrillation. Herz 2008; 33:562–567.
- Wyse DG, Waldo AL, DiMarco JP, et al; Atrial Fibrillation Follow-up Investigation of Rhythm Management (AFFIRM) Investigators. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med 2002; 347:1825–1833.
- Van Gelder IC, Groenveld HF, Crijns HJ, et al; RACE II Investigators. Lenient versus strict rate control in patients with atrial fibrillation. N Engl J Med 2010; 362:1363–1373.
- Singh BN, Singh SN, Reda DJ, et al; Sotalol Amiodarone Atrial Fibrillation Efficacy Trial (SAFE-T) Investigators. Amiodarone versus sotalol for atrial fibrillation. N Engl J Med 2005; 352:1861–1872.
- The AFFIRM Investigators. Relationships between sinus rhythm, treatment, and survival in the Atrial Fibrillation Follow-Up Investigation of Rhythm Management (AFFIRM) study. Circulation 2004; 109:1509–1513.
- Van Beeren HC, Jong WM, Kaptein E, Visser TJ, Bakker O, Wiersinga WM. Dronerarone [sic] acts as a selective inhibitor of 3,5,3′-triiodothyronine binding to thyroid hormone receptor-alpha1: in vitro and in vivo evidence. Endocrinology 2003; 144:552–558.
- Patel C, Yan GX, Kowey PR. Dronedarone. Circulation 2009; 120:636–644.
- Dale KM, White CM. Dronedarone: an amiodarone analog for the treatment of atrial fibrillation and atrial flutter. Ann Pharmacother 2007; 41:599–605.
- Hohnloser SH, Crijns HJ, van Eickels M; ATHENA Investigators. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med 2009; 360:668–678.
- Singh BN, Connolly SJ, Crijns HJ, et al; EURIDIS and ADONIS Investigators. Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N Engl J Med 2007; 357:987–999.
- Davy JM, Herold M, Hoglund CERATO Study Investigators. Dronedarone for the control of ventricular rate in permanent atrial fibrillation: the Efficacy and safety of dRonedArone for the cOntrol of ventricular rate during atrial fibrillation (ERATO) study. Am Heart J 2008; 156:527.e1–e9.
- Køber L, Torp-Pedersen C, McMurray JJ; Dronedarone Study Group. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med 2008; 358:2678–2687.
- Le Heuzey JY, De Ferrari GM, Radzik D, Santini M, Zhu J, Davy JM. A short-term, randomized, double-blind, parallel-group study to evaluate the efficacy and safety of dronedarone versus amiodarone in patients with persistent atrial fibrillation: the DIONYSOS study. J Cardiovasc Electrophysiol 2010; 21:597–605.
- The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med 1989; 321:406–412.
- Pratt CM. Clinical implications of the Survival With Oral D-sotalol (SWORD) trial: an investigation of patients with left ventricular dysfunction after myocardial infarction. Card Electrophysiol Rev 1998; 2:28–29.
- Touboul P, Brugada J, Capucci A, Crijns HJ, Edvardsson N, Hohnloser SH. Dronedarone for prevention of atrial fibrillation: a dose-ranging study. Eur Heart J 2003; 24:1481–1487.
- Tschuppert Y, Buclin T, Rothuizen LE, et al. Effect of dronedarone on renal function in healthy subjects. Br J Clin Pharmacol 2007; 64:785–791.
KEY POINTS
- Patients with persistent or paroxysmal atrial fibrillation are candidates for dronedarone therapy if they are in sinus rhythm or will be cardioverted soon after starting. This drug is not indicated for the acute management of atrial fibrillation, for example, in the emergency department.
- Dronedarone is an option if a patient cannot tolerate amiodarone or has an underlying condition such as pulmonary or thyroid disease that is a contraindication to amiodarone.
- Dronedarone is contraindicated in patients with significant left ventricular dysfunction or heart failure with recent decompensation.
- The ultimate role for dronedarone is yet to be defined. Little evidence exists as to whether it will succeed when other drugs have failed.
In reply: Menstrual manipulation
In Reply: Thank you for reading our article. Although the focus was geared more toward a comparison of different means of menstrual manipulation, we appreciate your comments on oral contraceptives and the link to premenopausal breast cancer.
As you noted, oral contraceptives have been linked to an increased risk of breast cancer, both in your meta-analysis1 and again more recently in a prospective study of 116,608 female nurses from 25 to 42 years of age.2 Interestingly, data from the latter study suggested that different formulations of oral contraceptives may pose different risks, and specifically that the use of triphasic preparations with levonorgestrel as the progestin had the highest risk. However, there is otherwise a paucity of data regarding the risk of specific formulations. There is currently no evidence of an association between oral contraceptive use and death from breast cancer, nor is there evidence that longer use of an oral contraceptive increases one’s risk of death from breast cancer.3
Oral contraceptives have also been associated with a reduced risk of ovarian cancer,4 and they appear to protect against death from ovarian cancer and uterine cancer.3 Therefore, the clinician must consider the individual patient before making treatment recommendations, taking into account personal risk factors and other health concerns. (For a full list of contraindications to oral contraceptives, please refer to Table 2 in our original article.) Further guidelines may also be obtained from the “US Medical Eligibility Criteria for Contraceptive Use 2010,” issued by the US Centers for Disease Control and Prevention in May 2010,5 which delineates the eligibility criteria for initiating and continuing specific contraceptive methods, including oral contraceptives.
Thank you again for sharing your concerns. We appreciate the opportunity to clarify this important point.
- Kahlenborn C, Modugno F, Potter DM, et al. Oral contraceptive use as a risk factor for premenopausal breast cancer: a meta-analysis. Mayo Clin Proc 2006; 81:1290–1302.
- Hunter DJ, Colditz GA, Hankinson SE, et al. Oral contraceptive use and breast cancer: a prospective study of young women. Cancer Epidemiol Biomarkers Prev 2010; 19:2496–2502.
- Vessey M, Yeates D, Flynn S. Factors affecting mortality in a large cohort study with special reference to oral contraceptive use. Contraception 2010; 82:221–229.
- Lurie G, Thompson P, McDuffie KE, et al. Association of estrogen and progestin potency of oral contraceptives with ovarian carcinoma risk. Obstet Gynecol 2007; 109:597–607.
- Division of Reproductive Health, National Center for Chronic Disease Prevention and Health Promotion, US Centers for Disease Control and Prevention (CDC), Farr S, et al. US medical eligibility criteria for contraceptive use, 2010: adapted from the World Health Organization medical eligibility criteria for contraceptive use, 4th edition. MMWR Recomm Rep 2010; 59:1–86.
In Reply: Thank you for reading our article. Although the focus was geared more toward a comparison of different means of menstrual manipulation, we appreciate your comments on oral contraceptives and the link to premenopausal breast cancer.
As you noted, oral contraceptives have been linked to an increased risk of breast cancer, both in your meta-analysis1 and again more recently in a prospective study of 116,608 female nurses from 25 to 42 years of age.2 Interestingly, data from the latter study suggested that different formulations of oral contraceptives may pose different risks, and specifically that the use of triphasic preparations with levonorgestrel as the progestin had the highest risk. However, there is otherwise a paucity of data regarding the risk of specific formulations. There is currently no evidence of an association between oral contraceptive use and death from breast cancer, nor is there evidence that longer use of an oral contraceptive increases one’s risk of death from breast cancer.3
Oral contraceptives have also been associated with a reduced risk of ovarian cancer,4 and they appear to protect against death from ovarian cancer and uterine cancer.3 Therefore, the clinician must consider the individual patient before making treatment recommendations, taking into account personal risk factors and other health concerns. (For a full list of contraindications to oral contraceptives, please refer to Table 2 in our original article.) Further guidelines may also be obtained from the “US Medical Eligibility Criteria for Contraceptive Use 2010,” issued by the US Centers for Disease Control and Prevention in May 2010,5 which delineates the eligibility criteria for initiating and continuing specific contraceptive methods, including oral contraceptives.
Thank you again for sharing your concerns. We appreciate the opportunity to clarify this important point.
In Reply: Thank you for reading our article. Although the focus was geared more toward a comparison of different means of menstrual manipulation, we appreciate your comments on oral contraceptives and the link to premenopausal breast cancer.
As you noted, oral contraceptives have been linked to an increased risk of breast cancer, both in your meta-analysis1 and again more recently in a prospective study of 116,608 female nurses from 25 to 42 years of age.2 Interestingly, data from the latter study suggested that different formulations of oral contraceptives may pose different risks, and specifically that the use of triphasic preparations with levonorgestrel as the progestin had the highest risk. However, there is otherwise a paucity of data regarding the risk of specific formulations. There is currently no evidence of an association between oral contraceptive use and death from breast cancer, nor is there evidence that longer use of an oral contraceptive increases one’s risk of death from breast cancer.3
Oral contraceptives have also been associated with a reduced risk of ovarian cancer,4 and they appear to protect against death from ovarian cancer and uterine cancer.3 Therefore, the clinician must consider the individual patient before making treatment recommendations, taking into account personal risk factors and other health concerns. (For a full list of contraindications to oral contraceptives, please refer to Table 2 in our original article.) Further guidelines may also be obtained from the “US Medical Eligibility Criteria for Contraceptive Use 2010,” issued by the US Centers for Disease Control and Prevention in May 2010,5 which delineates the eligibility criteria for initiating and continuing specific contraceptive methods, including oral contraceptives.
Thank you again for sharing your concerns. We appreciate the opportunity to clarify this important point.
- Kahlenborn C, Modugno F, Potter DM, et al. Oral contraceptive use as a risk factor for premenopausal breast cancer: a meta-analysis. Mayo Clin Proc 2006; 81:1290–1302.
- Hunter DJ, Colditz GA, Hankinson SE, et al. Oral contraceptive use and breast cancer: a prospective study of young women. Cancer Epidemiol Biomarkers Prev 2010; 19:2496–2502.
- Vessey M, Yeates D, Flynn S. Factors affecting mortality in a large cohort study with special reference to oral contraceptive use. Contraception 2010; 82:221–229.
- Lurie G, Thompson P, McDuffie KE, et al. Association of estrogen and progestin potency of oral contraceptives with ovarian carcinoma risk. Obstet Gynecol 2007; 109:597–607.
- Division of Reproductive Health, National Center for Chronic Disease Prevention and Health Promotion, US Centers for Disease Control and Prevention (CDC), Farr S, et al. US medical eligibility criteria for contraceptive use, 2010: adapted from the World Health Organization medical eligibility criteria for contraceptive use, 4th edition. MMWR Recomm Rep 2010; 59:1–86.
- Kahlenborn C, Modugno F, Potter DM, et al. Oral contraceptive use as a risk factor for premenopausal breast cancer: a meta-analysis. Mayo Clin Proc 2006; 81:1290–1302.
- Hunter DJ, Colditz GA, Hankinson SE, et al. Oral contraceptive use and breast cancer: a prospective study of young women. Cancer Epidemiol Biomarkers Prev 2010; 19:2496–2502.
- Vessey M, Yeates D, Flynn S. Factors affecting mortality in a large cohort study with special reference to oral contraceptive use. Contraception 2010; 82:221–229.
- Lurie G, Thompson P, McDuffie KE, et al. Association of estrogen and progestin potency of oral contraceptives with ovarian carcinoma risk. Obstet Gynecol 2007; 109:597–607.
- Division of Reproductive Health, National Center for Chronic Disease Prevention and Health Promotion, US Centers for Disease Control and Prevention (CDC), Farr S, et al. US medical eligibility criteria for contraceptive use, 2010: adapted from the World Health Organization medical eligibility criteria for contraceptive use, 4th edition. MMWR Recomm Rep 2010; 59:1–86.
Menstrual manipulation
To the Editor: In the article, “Menstrual manipulation: Options for suppressing the cycle,”1 the authors described advantages and disadvantages of various hormone-based methods of menstrual manipulation, including prolonged use of oral contraceptives. We believe the authors underemphasized the risks associated with oral contraceptives. Blood clots, stroke, and death are often included in print and television ads by law firms recruiting patients harmed by these drugs. In addition, the authors failed to mention the risk of premenopausal breast cancer due to oral contraceptives, which are now classified as group 1 carcinogens by the World Health Organization.2
In October 2006, we published the most current meta-analysis to date regarding oral contraceptive use and the risk of premenopausal breast cancer.3 We found that 21 out of 23 studies showed a positive trend or positive risk for premenopausal breast cancer with oral contraceptive use prior to first-term pregnancy. This resulted in a highly statistically significant cumulative risk of 44% (ie, odds ratio 1.44, 95% confidence interval 1.24–1.68). Our meta-analysis remains the most recent study in this area and updates the Oxford pooled analysis,4 which relied on older studies with older women (two-thirds of whom were over age 45).
A more recent collaborative study coauthored by investigators from the National Cancer Institute, the Hutchinson Cancer Research Center, and the University of Washington includes oral contraceptives in the list of risk factors for breast cancer in younger women.5 We ask your readers to consider that patients are entitled to know about this important risk factor before making a decision regarding hormonal menstrual manipulation.
- Hicks CW, Rome ES. Menstrual manipulation: options for suppressing the cycle. Clev Clin J Med 2010; 77:445–453.
- Cogliano V, Grosse Y, Baan R, et al; WHO International Agency for Research on Cancer. Carcinogenicity of combined oestrogen-progestagen contraceptives and menopausal treatment. Lancet Oncol 2005; 6:552–553.
- Kahlenborn C, Modugno F, Potter DM, Severs WB. Oral contraceptive use as a risk factor for premenopausal breast cancer: a meta-analysis. Mayo Clin Proc 2006; 81:1290–1302.
- Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and hormonal contraceptives: further results. Contraception 1996; 54(3 suppl):1S–106S.
- Dolle JM, Daling JR, White E, et al. Risk factors of triple-negative breast cancer in women under the age of 45 years. Cancer Epidemiol Biomarkers Prev 2009; 18:1157–1166.
To the Editor: In the article, “Menstrual manipulation: Options for suppressing the cycle,”1 the authors described advantages and disadvantages of various hormone-based methods of menstrual manipulation, including prolonged use of oral contraceptives. We believe the authors underemphasized the risks associated with oral contraceptives. Blood clots, stroke, and death are often included in print and television ads by law firms recruiting patients harmed by these drugs. In addition, the authors failed to mention the risk of premenopausal breast cancer due to oral contraceptives, which are now classified as group 1 carcinogens by the World Health Organization.2
In October 2006, we published the most current meta-analysis to date regarding oral contraceptive use and the risk of premenopausal breast cancer.3 We found that 21 out of 23 studies showed a positive trend or positive risk for premenopausal breast cancer with oral contraceptive use prior to first-term pregnancy. This resulted in a highly statistically significant cumulative risk of 44% (ie, odds ratio 1.44, 95% confidence interval 1.24–1.68). Our meta-analysis remains the most recent study in this area and updates the Oxford pooled analysis,4 which relied on older studies with older women (two-thirds of whom were over age 45).
A more recent collaborative study coauthored by investigators from the National Cancer Institute, the Hutchinson Cancer Research Center, and the University of Washington includes oral contraceptives in the list of risk factors for breast cancer in younger women.5 We ask your readers to consider that patients are entitled to know about this important risk factor before making a decision regarding hormonal menstrual manipulation.
To the Editor: In the article, “Menstrual manipulation: Options for suppressing the cycle,”1 the authors described advantages and disadvantages of various hormone-based methods of menstrual manipulation, including prolonged use of oral contraceptives. We believe the authors underemphasized the risks associated with oral contraceptives. Blood clots, stroke, and death are often included in print and television ads by law firms recruiting patients harmed by these drugs. In addition, the authors failed to mention the risk of premenopausal breast cancer due to oral contraceptives, which are now classified as group 1 carcinogens by the World Health Organization.2
In October 2006, we published the most current meta-analysis to date regarding oral contraceptive use and the risk of premenopausal breast cancer.3 We found that 21 out of 23 studies showed a positive trend or positive risk for premenopausal breast cancer with oral contraceptive use prior to first-term pregnancy. This resulted in a highly statistically significant cumulative risk of 44% (ie, odds ratio 1.44, 95% confidence interval 1.24–1.68). Our meta-analysis remains the most recent study in this area and updates the Oxford pooled analysis,4 which relied on older studies with older women (two-thirds of whom were over age 45).
A more recent collaborative study coauthored by investigators from the National Cancer Institute, the Hutchinson Cancer Research Center, and the University of Washington includes oral contraceptives in the list of risk factors for breast cancer in younger women.5 We ask your readers to consider that patients are entitled to know about this important risk factor before making a decision regarding hormonal menstrual manipulation.
- Hicks CW, Rome ES. Menstrual manipulation: options for suppressing the cycle. Clev Clin J Med 2010; 77:445–453.
- Cogliano V, Grosse Y, Baan R, et al; WHO International Agency for Research on Cancer. Carcinogenicity of combined oestrogen-progestagen contraceptives and menopausal treatment. Lancet Oncol 2005; 6:552–553.
- Kahlenborn C, Modugno F, Potter DM, Severs WB. Oral contraceptive use as a risk factor for premenopausal breast cancer: a meta-analysis. Mayo Clin Proc 2006; 81:1290–1302.
- Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and hormonal contraceptives: further results. Contraception 1996; 54(3 suppl):1S–106S.
- Dolle JM, Daling JR, White E, et al. Risk factors of triple-negative breast cancer in women under the age of 45 years. Cancer Epidemiol Biomarkers Prev 2009; 18:1157–1166.
- Hicks CW, Rome ES. Menstrual manipulation: options for suppressing the cycle. Clev Clin J Med 2010; 77:445–453.
- Cogliano V, Grosse Y, Baan R, et al; WHO International Agency for Research on Cancer. Carcinogenicity of combined oestrogen-progestagen contraceptives and menopausal treatment. Lancet Oncol 2005; 6:552–553.
- Kahlenborn C, Modugno F, Potter DM, Severs WB. Oral contraceptive use as a risk factor for premenopausal breast cancer: a meta-analysis. Mayo Clin Proc 2006; 81:1290–1302.
- Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and hormonal contraceptives: further results. Contraception 1996; 54(3 suppl):1S–106S.
- Dolle JM, Daling JR, White E, et al. Risk factors of triple-negative breast cancer in women under the age of 45 years. Cancer Epidemiol Biomarkers Prev 2009; 18:1157–1166.
Proton pump inhibitor side effects and drug interactions: Much ado about nothing?
The development and introduction of the first proton pump inhibitor (PPI), omeprazole (Prilosec), for the management of acid-peptic disorders marks one of the great success stories in gastroenterology. Until the latter part of the 20th century, complications of acid-peptic disease were among the most common problems faced in gastroenterology. Severe peptic strictures were once a highly prevalent cause of dysphagia, and operations for peptic ulcer disease were routinely learned by surgical trainees.

The success of these drugs, with sales total-ling $13.6 billion worldwide in 2009,1 is not just a result of their potency and effectiveness in improving symptoms and complications of acid-peptic disease. Their safety among pharmacologic agents has been unparalleled. When the drugs were first introduced, their use was limited to short courses out of concern that gastric carcinoids could develop, but decades of use have not shown this issue to be of clinical relevance. Serious, acute adverse effects are also exceedingly uncommon.
However, recent reports have questioned the long-term safety of PPIs. Furthermore, these drugs are too often used in patients who have no valid indication for them,2,3 exposing these patients to unnecessary risks.
The goals of this review are to analyze the recent literature about the risks of PPIs and to provide a rational approach for managing patients on PPI therapy in light of these concerns.
DO PPIs REDUCE THE EFFECT OF CLOPIDOGREL?
Clopidogrel (Plavix) is a potent antiplatelet agent commonly used in patients with atherosclerotic cardiac or cerebrovascular disease, sometimes in combination with aspirin. Because of the risk of significant gastrointestinal bleeding, a 2008 multisociety task force recommended prescribing a PPI when both clopidogrel and aspirin are used as dual antiplatelet therapy.4
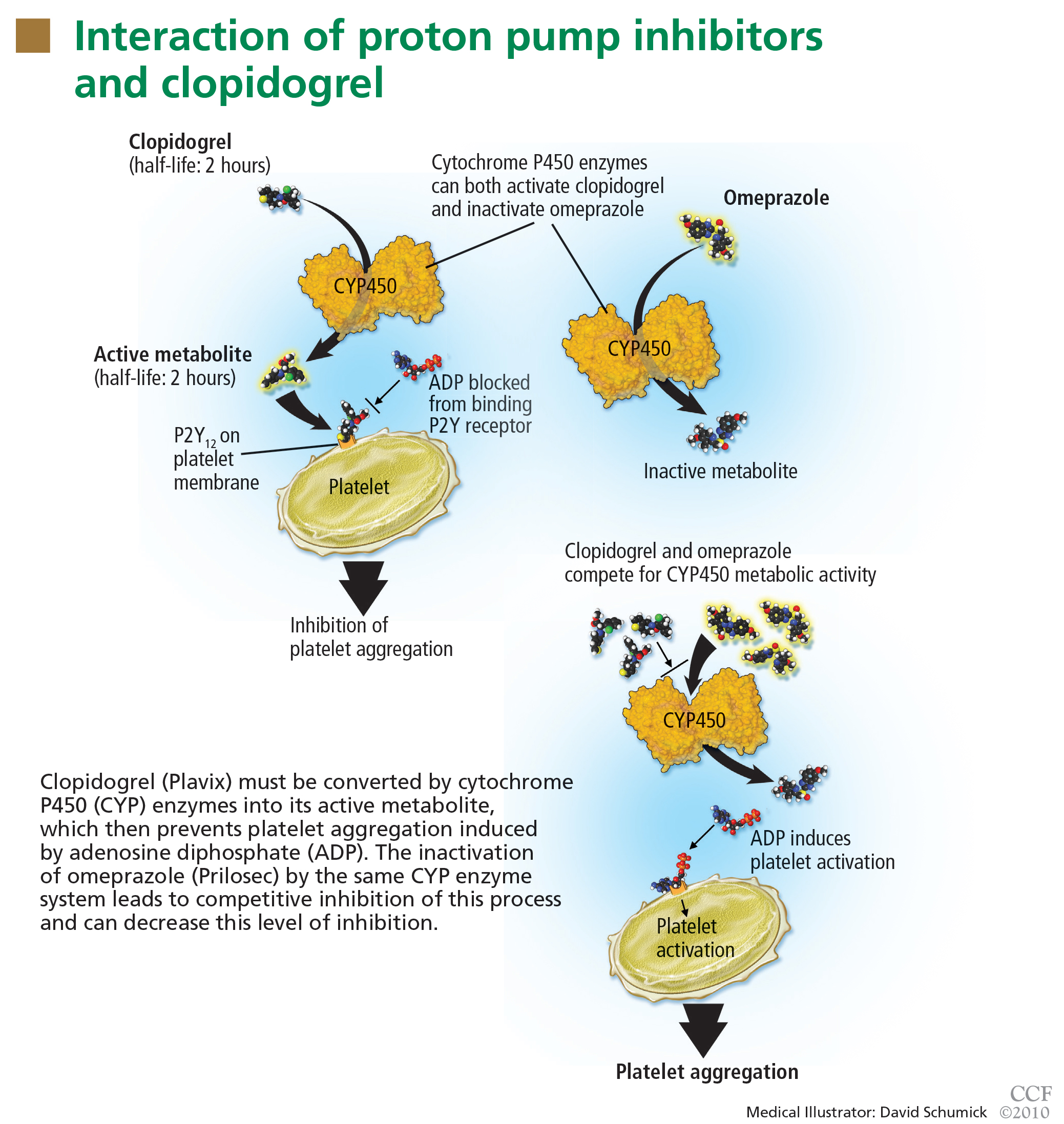
Studies of clopidogrel plus PPIs: Discrepant results
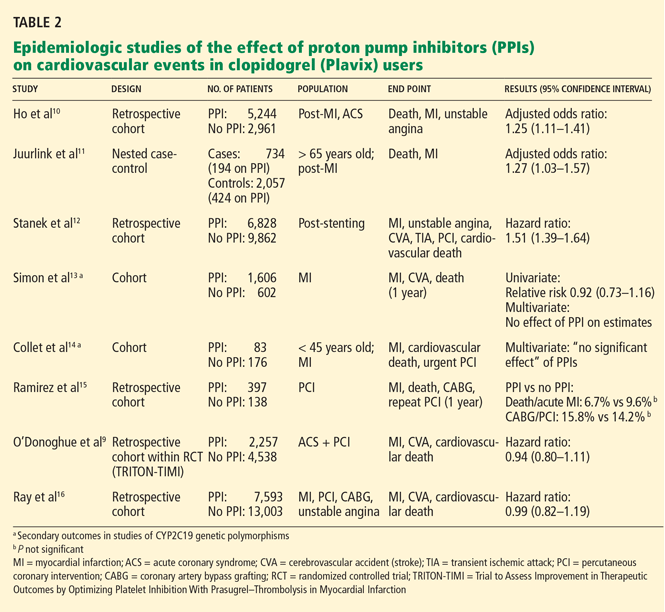
To date, only one prospective randomized controlled trial has specifically investigated the effect of PPIs on cardiovascular outcomes in patients using clopidogrel. In this trial, patients on dual antiplatelet therapy with clopidogrel and aspirin were randomized to receive either omeprazole 20 mg or placebo. Analysis of the data revealed no significant increase in the composite end point of cardiovascular events (hazard ratio [HR] 0.99, 95% confidence interval [CI] 0.68–1.44, P = .96), but a statistically significant decrease in composite gastrointestinal events (HR 0.34, 95% CI 0.18–0.63, P < .001).17
Unfortunately, this trial had to be terminated before the prespecified sample size and duration of follow-up were reached because the study sponsor declared bankruptcy.
One additional recent retrospective cohort study16 likewise found no significant risk of serious cardiovascular disease related to PPI use in clopidogrel users. It also found that the adjusted incidence of hospitalization for upper gastrointestinal bleeding was 50% lower in patients who used PPIs than in those who did not (HR 0.50, 95% CI 0.39–0.65).
Do factors other than PPIs account for the higher risk in some of the studies?
The discrepant results of these studies suggest that the higher risk of cardiovascular events may be due, either completely or in part, to a factor other than the pharmacologic interaction of PPIs and clopidogrel. It is difficult to infer causality from the available data. In situations in which no randomized controlled trials exist, one looks to observational (case-control or cohort) studies to try to obtain the best estimate of the actual risk. With PPIs and clopidogrel, a randomized controlled trial was performed but terminated before patient enrollment was complete.
The increased risk found in some of these studies may be real, may be due to chance, or may even represent an increased risk from PPIs alone (although data do not support this possibility).18 However, the major concern in observational studies is the inability to account for unmeasured confounders, a problem virtually eliminated by randomization strategies in prospective studies.
In the studies that found a higher risk with the combination of omeprazole plus clopidogrel, the principal concern is confounding by indication, in which distortions of the risk estimates arise from an imbalance in prognostic factors between compared treatment groups that remains unmeasured.19 Stated another way, physicians who believed some patients to be “sicker” or to have a higher risk of serious events may have treated them with a PPI on the basis of factors that remained unaccounted for in the epidemiologic investigation.
This possibility has been reinforced by findings from a nonrandomized subgroup analysis of a randomized controlled trial in which patients who had been receiving a PPI had a higher rate of cardiovascular events whether they received clopidogrel or placebo.20
FDA alert: Avoid using omeprazole or esomeprazole with clopidogrel
Nonetheless, on November 17, 2009, the US Food and Drug Administration (FDA) issued an alert to health care professionals and the public about the potential interaction between clopidogrel and omeprazole.21 In this alert, the FDA stated that the use of omeprazole or esomeprazole (Nexium) with clopidogrel should be avoided.
An algorithm to use when considering clopidogrel plus a PPI
Physicians are now left in a bind between the minimal, if any, pooled risk seen in the available data and the FDA recommendation. What is the best action to take?
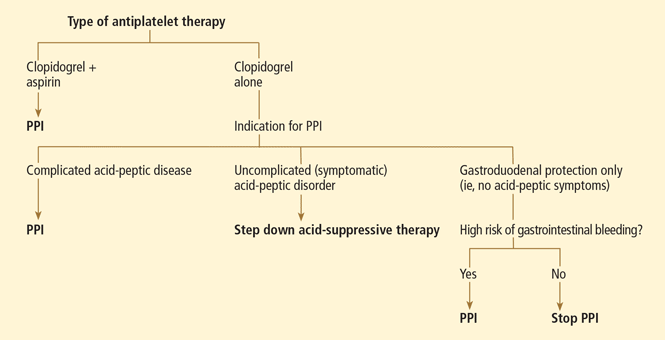
First, assess the need for dual antiplatelet therapy. If dual antiplatelet therapy (clopidogrel plus aspirin) is required, then a PPI is warranted for gastric protection because the risk of life-threatening bleeding outweighs any increased risk of cardiovascular events.4
If antiplatelet monotherapy (clopidogrel alone) is required, then assess the reason for antisecretory therapy.
For complicated disease, such as gastroesophageal reflux disease with Barrett esophagus or peptic strictures, PPI therapy is warranted to prevent progression or recurrence of complications. If the antisecretory therapy is being provided for noncomplicated symptomatic disorders such as nonerosive gastroesophageal reflux disease or dyspepsia, then one should try to “step down” the therapy by lowering the PPI dose as much as possible while still controlling symptoms to the patient’s tolerance, then possibly stepping further by substituting a histamine-2-receptor antagonist, an antacid, or “on-demand” use of PPIs.22,23
However, if the rationale for antisecretory therapy is simply for gastrointestinal protection, then further risk stratification for gastro intestinal bleeding should be undertaken.4 For patients with a high risk of future gastrointestinal bleeding, such as those with prior episodes of bleeding or concurrent use of nonsteroidal anti-inflammatory drugs, antisecretory therapy is still recommended. Therefore, if a patient is on monotherapy with clopidogrel, has no complicated or symptomatic gastrointestinal disorder, and does not have a high risk of gastrointestinal bleeding, then therapy with a PPI should be reconsidered.
There are no strong data to indicate that one particular PPI should be used or avoided if one of the above criteria indicates the concurrent need for clopidogrel and a PPI. In their health alert about the potential interaction, the FDA did not issue the same warning for PPIs other than omeprazole and esomeprazole, but fell short of recommending a change to another PPI because of a lack of data to support or refute a similar interaction.
Because the half-lives of clopidogrel and PPIs are short, separating their administration could in theory decrease or eliminate the risk of competitive inhibition. The PPI could be given in the morning before breakfast and the clopidogrel could be given at night, or the clopidogrel could be given at lunchtime and the PPI before dinner. Although the FDA does not believe this strategy will reduce this interaction,21 one expert in the field has suggested it.18
DO PPIs CAUSE OSTEOPOROSIS, FRACTURES?
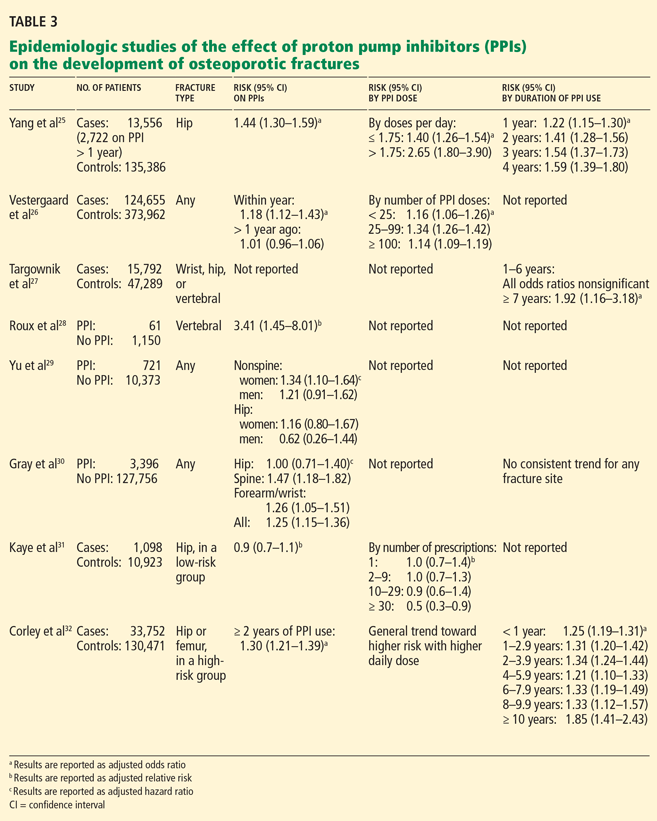
In a widely publicized paper published in 2006, Yang and colleagues25 reported the results of a large nested case-control study in the United Kingdom. The risk of hip fracture was significantly greater in patients who had been using PPIs for at least 1 year than in those who had not. The risk appeared to increase with longer use and higher doses of PPIs.
A similar risk of hip fracture was seen in a larger Danish case-control study published the same year.26 This study also found an increased odds ratio for PPI use in patients with spine fractures as well as in patients with any type of fracture. Interestingly, this study found a lower risk of fracture in patients using a histamine-2-receptor antagonist instead of a PPI.
Targownik et al27 found that the risk of hip fracture was not significantly higher until after 5 years of PPI exposure, with an even stronger risk after 7 years.
However, the data on both association and causal relationship are not uniform.
The Women’s Health Initiative,30 with more than 1 million person-years of followup, found no association between PPI use and hip fracture, but a modest association between PPI use and spine, arm, and wrist fractures, as well as total fractures.
A study in the United Kingdom found that patients without any major risk factors for hip fracture (defined by a risk ratio > 2) accounted for only 25% of cases but 53% of controls. When only these two average-risk groups were compared, the risk of hip fracture was similar in cases and controls.31
Corley et al32 also found that the risk of fracture associated with PPI use was only significant in the presence of another risk factor. These findings suggest that residual confounding may be to blame, at least in part, for the estimates of increased risk in the prior studies.
Another way to interpret these data is that PPIs increase the risk in patients at high risk to begin with, but not in those at average risk. This is an example of interaction (or effect modification) in which the risk is unequally distributed across groups with different characteristics.
A recently published study refutes the theory that impaired calcium absorption is responsible for the increase in fractures.33 In this study, investigators queried the Manitoba Bone Mineral Density Database to determine the relationship between antisecretory therapy with PPIs and osteoporosis or loss of bone mineral density—and they found none. This study may support the theory that residual confounding is the reason for the finding of an increased risk, but it also leaves open the possibility that PPIs induce other changes in bone microstructure that could increase the risk of fracture.
FDA labeling: Possible risk of fracture with PPIs
Based on the data so far, it appears possible that there is a small, albeit statistically significant, association between PPI use and fracture risk. The association is indeed biologically plausible, but it remains to be seen if this association is clinically significant, as the risk is relatively low. Even though the studies had methodologic limitations, on May 25, 2010, the FDA announced a change in the required labeling information for PPIs to indicate a possible risk of fracture with these drugs.34
Reassess the need for chronic PPI therapy
Although patients may worry that they will develop osteoporosis and fractures if they take PPIs, the data do not support a strong risk. Nevertheless, when faced with a patient on chronic PPI therapy, especially with a high dose, providers should use the opportunity to reassess the indication for the PPI to decide if chronic therapy is required, in a matter similar to the algorithm provided for PPI-clopidogrel cotherapy (FIGURE 2). Providers should educate patients about the data, and limit new and recurring PPI prescriptions to patients who require a PPI for appropriate indications, at the lowest dose, and for the shortest time possible.
DO PPIs INCREASE THE RISK OF PNEUMONIA?
Several recent studies have also raised concern about an association between PPI use and pneumonia.
Normally, the stomach remains free of bacteria (except for Helicobacter pylori) because its acidic milieu destroys nearly all bacteria swallowed. If the stomach becomes less acidic, it loses this protective mechanism, and ingested organisms can survive and proliferate.35 In theory, when gastroesophageal reflux occurs, these bacteria could be carried up to the hypopharynx where microaspiration into the lower airways could lead to pneumonia, especially in patients with compromised oropharyngeal protective reflexes (eg, patients on mechanical ventilation).
This possible association came to the attention of the general medical community when a Dutch study,36 in which 5,551 cases of community-acquired pneumonia developed in 364,683 people, found that the incidence of pneumonia was about 4.5 times higher in people exposed to acid-suppressive drugs (both PPIs and histamine-2-receptor antagonists) than in unexposed individuals. Patients who developed pneumonia also had higher odds of significant comorbid conditions, including heart failure and chronic obstructive pulmonary disease. The authors calculated that about one case of pneumonia per 226 patients treated with a PPI would be attributable to the PPI. A major limitation of this study, however, was that only 18% of the patients diagnosed with pneumonia actually had radiologic or microbiologic confirmation of pneumonia.
Other studies later examined the relationship between PPIs and community-acquired pneumonia,37–41 and most have revealed a modestly higher risk of community-acquired pneumonia in patients exposed to PPIs.
This risk was confirmed in a recent metaanalysis, which found a higher risk of community-acquired pneumonia with PPI use (odds ratio 1.36, 95% CI 1.12–1.65).42 However, the authors refrained from drawing definitive conclusions from these data because of significant heterogeneity between the studies. One study37 found that recent onset of use (within 7 days) had a much stronger association with community-acquired pneumonia than longer-term use, which is contradictory to a causal association, since longer-term use should lead to more cases of pneumonia.
Another study investigated the association between acid-suppressive drugs and hospital-acquired pneumonia in nonventilated patients.43 In a 4-year period, there were 63,878 admissions in 42,093 unique patients. Acid-suppressive drugs were prescribed in 32,922 admissions (52%); the drugs included PPIs in 83% of these. Hospital-acquired pneumonia occurred in 2,219 admissions (3.5%), with a higher incidence in patients exposed to acid-suppressive drugs than in the unexposed group (4.6% vs 2.0%). The adjusted odds ratio for pneumonia was 1.3 (95% CI 1.1–1.4) in the exposed group. Subgroup analysis revealed that the association remained significant for PPIs but not for histamine-2-receptor antagonists.
Adequate studies of mechanically ventilated patients in the current era of intravenous PPI use are lacking. Older studies in this group of patients may not be generalizable to current practice because of the reduction in gastric volume with intravenous PPIs that may offset the theoretical risk of aspiration.35
Although the data supporting the association are not exceedingly strong, the relationship is biologically plausible. If there is a risk, it seems to be greatest in the sickest patients, who can least afford to develop pneumonia. Therefore, prudent prescribing should be the rule for both inpatients and outpatients, especially in patients with comorbidities, in whom pneumonia could have serious consequences.
PPIs AND ENTERIC INFECTIONS
Traditionally, gastric acid was not believed to be important in protecting against Clostridium difficile infection because acid-resistant spores were presumed to be the principal vector of transmission.44 Recently, this thought has been challenged, as several studies have found a higher risk of C difficile infection in PPI users. In theory, PPIs may increase the risk of C difficile infection by increasing the ability of the spore to convert to the vegetative form and to survive intraluminally.
A recent meta-analysis of 11 papers, including nearly 127,000 patients, found a significant relationship between PPI use and C difficile infection, with an odds ratio of 2.05 (95% CI 1.47–2.85).45 Further supporting the hypothesis of a direct causative association, a recent study found a significant dose-response, with more aggressive acid-suppression associated with higher odds ratios.46 In view of this association, patients using PPIs who develop diarrhea should be evaluated for C difficile, perhaps even in the absence of other risk factors.
Other enteric infections have been found to be associated with PPIs.44,45 Small intestinal bacterial overgrowth, a condition that is associated with bloating, diarrhea, and malabsorption, has recently been associated with PPI use, although the significance of the association is uncertain.47
Based on a change in the intestinal flora, recent reports have additionally implied that there is a relationship between PPI use and the development of spontaneous bacterial peritonitis in hospitalized cirrhotic patients with ascites. One study found a strong association (odds ratio 4.3, 95% CI 1.3–11.7) between PPIs and spontaneous bacterial pneumonitis,48 whereas another study found no significant association (odds ratio 1.0, 95% CI 0.4–2.6).49
Both studies were small case-control studies of hospitalized patients. No firm conclusion can be drawn about the relevance of this association from these investigations at this point.
PPIs AND ACUTE INTERSTITIAL NEPHRITIS
Several case reports have implicated PPIs as a cause of acute interstitial nephritis.
A systematic review from 2007 found 64 cases documented in the literature, 12 of which were considered certainly associated, and 9 of which were probably associated.50 Initial symptoms were nonspecific and included nausea, malaise, and fever. With such extensive use worldwide as the denominator, the authors concluded that acute interstitial nephritis was a rare, idiosyncratic occurrence related to PPI use, but did not find enough evidence to support a causative relationship. Despite the rarity of the syndrome, they recommended maintaining a high level of clinical suspicion to detect acute interstitial nephritis early in its course, especially soon after the initiation of PPI therapy.
POSSIBLE ASSOCIATIONS WITH IRON AND B12 DEFICIENCIES
Long-term PPI therapy has been thought to be associated with micronutrient deficiencies, especially of iron and vitamin B12. Hydrochloric acid in the stomach assists in the dissociation of iron salts from food and the reduction of ferric iron to the more soluble ferrous iron.51 Gastric acid also facilitates the release of vitamin B12 bound to proteins within ingested foodstuffs to permit binding to R-proteins for eventual absorption in the terminal ileum.51,52
Despite the biologic plausibility of these deficiencies, there is currently little evidence to support a clinically relevant association to recommend a change in current practice.
NO THERAPY IS COMPLETELY WITHOUT RISK
Although concerns have been raised about the long-term safety of PPIs, the preponderance of the evidence does not strongly support the apprehensions publicized over the last few years. When translating these studies into the routine management of patients, it is important to recall some very basic tenets of good patient care.
No therapy is completely without risk—whether pharmacologic, surgical, or psychological, and no matter how benign or straightforward. Consequently, no drug, procedure, or treatment plan should be ordered without a valid indication. Even with an indication, the risk-benefit ratio of the therapy prescribed should always be considered. If the indication for the PPI is weak or uncertain, then even a slight risk tips the balance away from the drug, and the drug should be discontinued.
When seeing patients in long-term care, the indication and necessity for all drugs, including PPIs, should be reviewed. The algorithm proposed in Figure 2 can be adapted for virtually any of the possible associations.
Consider the indication for the PPI. Was the PPI started during a hospitalization and then routinely continued after discharge? This is one situation in which the use of a PPI could potentially be discontinued.2
For complicated acid-peptic disease, dose reduction or cessation of PPI therapy may not be possible.
If the PPI was prescribed only for symptom relief, as in cases of dyspepsia or nonerosive gastroesophageal reflux disease, reduce the dose of PPI to as low as possible to maintain symptom control. Should chronic therapy still be required, no specific monitoring is recommended, apart from routine monitoring that takes place in the course of patient care.
Lastly, because of the media attention that several of these concerns have garnered, patients may still harbor significant concerns about PPIs, even their short-term use. In such cases, the prescriber should take the opportunity to communicate the reason for the decision to prescribe the PPI, as well as the best available data about the risks PPIs may pose. None of these outcomes is very common in the absence of PPIs, with the possible exception of recurrent cardiovascular events, and the risks provided in all of these studies are relative to the baseline risk. Even if the risk of a particular outcome doubles with long-term PPI use, twice a small risk remains a small risk.
- Gatyas G. IMS Health reports U.S. prescription sales grew 5.1 percent in 2009, to $300.3 Billion. IMS Health. http://www.imshealth.com/portal/site/imshealth/menuitem.a46c6d4df3db4b3d88f611019418c22a/?vgnextoid=d690a27e9d5b7210VgnVCM100000ed152ca2RCRD&vgnextfmt=default. Accessed 10/7/2010.
- Zink DA, Pohlman M, Barnes M, Cannon ME. Long-term use of acid suppression started inappropriately during hospitalization. Aliment Pharmacol Ther 2005; 21:1203–1209.
- Heidelbaugh JJ, Inadomi JM. Magnitude and economic impact of inappropriate use of stress ulcer prophylaxis in non-ICU hospitalized patients. Am J Gastroenterol 2006; 101:2200–2205.
- Bhatt DL, Scheiman J, Abraham NS, et al; American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation 2008; 118:1894–1909.
- Klotz U, Schwab M, Treiber G. CYP2C19 polymorphism and proton pump inhibitors. Basic Clin Pharmacol Toxicol 2004; 95:2–8.
- Gilard M, Arnaud B, Cornily JC, et al. Influence of omeprazole on the antiplatelet action of clopidogrel associated with aspirin: the randomized, double-blind OCLA (Omeprazole CLopidogrel Aspirin) study. J Am Coll Cardiol 2008; 51:256–260.
- Small DS, Farid NA, Payne CD, et al. Effects of the proton pump inhibitor lansoprazole on the pharmacokinetics and pharmacodynamics of prasugrel and clopidogrel. J Clin Pharmacol 2008; 48:475–484.
- Sibbing D, Morath T, Stegherr J, et al. Impact of proton pump inhibitors on the antiplatelet effects of clopidogrel. Thromb Haemost 2009; 101:714–719.
- O’Donoghue ML, Braunwald E, Antman EM, et al. Pharmacodynamic effect and clinical efficacy of clopidogrel and prasugrel with or without a proton-pump inhibitor: an analysis of two randomised trials. Lancet 2009; 374:989–997.
- Ho PM, Maddox TM, Wang L, et al. Risk of adverse outcomes associated with concomitant use of clopidogrel and proton pump inhibitors following acute coronary syndrome. JAMA 2009; 301:937–944.
- Juurlink DN, Gomes T, Ko DT, et al. A population-based study of the drug interaction between proton pump inhibitors and clopidogrel. CMAJ 2009; 180:713–718.
- Stanek EJ, Aubert RE, Flockhart DA, et al. A national study of the effect of individual proton pump inhibitors on cardiovascular outcomes in patients treated with clopidogrel following coronary stenting: the Clopidogrel Medco Outcomes Study. Program and abstracts of the 32nd Annual SCAI Scientific Sessions May 6, 2009; Las Vegas, Nevada.
- Simon T, Verstuyft C, Mary-Krause M, et al; French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360:363–375.
- Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 2009; 373:309–317.
- Ramirez JF, Selzer F, Chakaprani R, et al. Proton pump inhibitor and clopidogrel combination is not associated with adverse clinical outcomes after PCI: the NHLBI dynamic registry (abstract). J Am Coll Cardiol 2009; 53(suppl 1):A27.
- Ray WA, Murray KT, Griffin MR, et al. Outcomes with concurrent use of clopidogrel and proton-pump inhibitors: a cohort study. Ann Intern Med 2010; 152:337–345.
- Bhatt DL, Cryer B, Contant CF, et al. Clopidogrel with or without omeprazole in coronary artery disease. N Engl J Med 2010; 363:1909–1917.
- Laine L, Hennekens C. Proton pump inhibitor and clopidogrel interaction: fact or fiction? Am J Gastroenterol 2010; 105:34–41.
- Walker AM. Confounding by indication. Epidemiology 1996; 7:335–336.
- Dunn SP, Macaulay TE, Brennan DM, et al. Baseline proton pump inhibitor use is associated with increased cardiovascular events with and without the use of clopidogrel in the CREDO trial (abstract). Circulation 2008; 118:S815.
- US Food and Drug Administration. Information for healthcare professionals: update to the labeling of clopidogrel bisulfate (marketed as Plavix) to alert healthcare professionals about a drug interaction with omeprazole (marketed as Prilosec and Prilosec OTC). U.S. Department of Health and Human Services, 11/17/2009. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm190787.htm. Accessed 9/23/2010.
- Inadomi JM, Jamal R, Murata GH, et al. Step-down management of gastroesophageal reflux disease. Gastroenterology 2001; 121:1095–1100.
- Inadomi JM, McIntyre L, Bernard L, Fendrick AM. Step-down from multiple- to single-dose proton pump inhibitors (PPIs): a prospective study of patients with heartburn or acid regurgitation completely relieved with PPIs. Am J Gastroenterol 2003; 98:1940–1944.
- O’Connell MB, Madden DM, Murray AM, Heaney RP, Kerzner LJ. Effects of proton pump inhibitors on calcium carbonate absorption in women: a randomized crossover trial. Am J Med 2005; 118:778–781.
- Yang YX, Lewis JD, Epstein S, Metz DC. Long-term proton pump inhibitor therapy and risk of hip fracture. JAMA 2006; 296:2947–2953.
- Vestergaard P, Rejnmark L, Mosekilde L. Proton pump inhibitors, histamine H2 receptor antagonists, and other antacid medications and the risk of fracture. Calcif Tissue Int 2006; 79:76–83.
- Targownik LE, Lix LM, Metge CJ, Prior HJ, Leung S, Leslie WD. Use of proton pump inhibitors and risk of osteoporosis-related fractures. CMAJ 2008; 179:319–326.
- Roux C, Briot K, Gossec L, et al. Increase in vertebral fracture risk in postmenopausal women using omeprazole. Calcif Tissue Int 2009; 84:13–19.
- Yu EW, Blackwell T, Ensrud KE, et al. Acid-suppressive medications and risk of bone loss and fracture in older adults. Calcif Tissue Int 2008; 83:251–259.
- Gray SL, LaCroix AZ, Larson J, et al. Proton pump inhibitor use, hip fracture, and change in bone mineral density in postmenopausal women: results from the Women’s Health Initiative. Arch Intern Med 2010; 170:765–771.
- Kaye JA, Jick H. Proton pump inhibitor use and risk of hip fractures in patients without major risk factors. Pharmacotherapy 2008; 28:951–959.
- Corley DA, Kubo A, Zhao W, Quesenberry C. Proton pump inhibitors and histamine-2 receptor antagonists are associated with hip fractures among at-risk patients. Gastroenterology 2010; 139:93–101.
- Targownik LE, Lix LM, Leung S, Leslie WD. Proton-pump inhibitor use is not associated with osteoporosis or accelerated bone mineral density loss. Gastroenterology 2010; 138:896–904.
- US Food and Drug Administration. FDA Drug Safety Communication: possible increased risk of fractures of the hip, wrist, and spine with the use of proton pump inhibitors. U.S. Department of Health and Human Services, 5/25/2010. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm213206.htm. Accessed 12/7/2010.
- Vakil N. Acid inhibition and infections outside the gastrointestinal tract. Am J Gastroenterol 2009; 104(suppl 2):S17–S20.
- Laheij RJ, Sturkenboom MC, Hassing RJ, Dieleman J, Stricker BH, Jansen JB. Risk of community-acquired pneumonia and use of gastric acid-suppressive drugs. JAMA 2004; 292:1955–1960.
- Gulmez SE, Holm A, Frederiksen H, Jensen TG, Pedersen C, Hallas J. Use of proton pump inhibitors and the risk of community-acquired pneumonia: a population-based case-control study. Arch Intern Med 2007; 167:950–955.
- Sarkar M, Hennessy S, Yang YX. Proton-pump inhibitor use and the risk for community-acquired pneumonia. Ann Intern Med 2008; 149:391–398.
- Myles PR, Hubbard RB, McKeever TM, Pogson Z, Smith CJ, Gibson JE. Risk of community-acquired pneumonia and the use of statins, ACE inhibitors and gastric acid suppressants: a population-based case-control study. Pharmacoepidemiol Drug Saf 2009; 18:269–275.
- Rodríguez LA, Ruigómez A, Wallander MA, Johansson S. Acid-suppressive drugs and community-acquired pneumonia. Epidemiology 2009; 20:800–806.
- Eurich DT, Sadowski CA, Simpson SH, Marrie TJ, Majumdar SR. Recurrent community-acquired pneumonia in patients starting acid-suppressing drugs. Am J Med 2010; 123:47–53.
- Johnstone J, Nerenberg K, Loeb M. Meta-analysis: proton pump inhibitor use and the risk of community-acquired pneumonia. Aliment Pharmacol Ther 2010; 31:1165–1177.
- Herzig SJ, Howell MD, Ngo LH, Marcantonio ER. Acid-suppressive medication use and the risk for hospital-acquired pneumonia. JAMA 2009; 301:2120–2128.
- Dial MS. Proton pump inhibitor use and enteric infections. Am J Gastroenterol 2009; 104(suppl 2):S10–S16.
- Leonard J, Marshall JK, Moayyedi P. Systematic review of the risk of enteric infection in patients taking acid suppression. Am J Gastroenterol 2007; 102:2047–2056.
- Howell MD, Novack V, Grgurich P, et al. Iatrogenic gastric acid suppression and the risk of nosocomial Clostridium difficile infection. Arch Intern Med 2010; 170:784–790.
- Lombardo L, Foti M, Ruggia O, Chiecchio A. Increased incidence of small intestinal bacterial overgrowth during proton pump inhibitor therapy. Clin Gastroenterol Hepatol 2010; 8:504–508.
- Bajaj JS, Zadvornova Y, Heuman DM, et al. Association of proton pump inhibitor therapy with spontaneous bacterial peritonitis in cirrhotic patients with ascites. Am J Gastroenterol 2009; 104:1130–1134.
- Campbell MS, Obstein K, Reddy KR, Yang YX. Association between proton pump inhibitor use and spontaneous bacterial peritonitis. Dig Dis Sci 2008; 53:394–398.
- Sierra F, Suarez M, Rey M, Vela MF. Systematic review: proton pump inhibitor-associated acute interstitial nephritis. Aliment Pharmacol Ther 2007; 26:545–553.
- McColl KE. Effect of proton pump inhibitors on vitamins and iron. Am J Gastroenterol 2009; 104(suppl 2):S5–S9.
- Ali T, Roberts DN, Tierney WM. Long-term safety concerns with proton pump inhibitors. Am J Med 2009; 122:896–903.
The development and introduction of the first proton pump inhibitor (PPI), omeprazole (Prilosec), for the management of acid-peptic disorders marks one of the great success stories in gastroenterology. Until the latter part of the 20th century, complications of acid-peptic disease were among the most common problems faced in gastroenterology. Severe peptic strictures were once a highly prevalent cause of dysphagia, and operations for peptic ulcer disease were routinely learned by surgical trainees.
The success of these drugs, with sales total-ling $13.6 billion worldwide in 2009,1 is not just a result of their potency and effectiveness in improving symptoms and complications of acid-peptic disease. Their safety among pharmacologic agents has been unparalleled. When the drugs were first introduced, their use was limited to short courses out of concern that gastric carcinoids could develop, but decades of use have not shown this issue to be of clinical relevance. Serious, acute adverse effects are also exceedingly uncommon.
However, recent reports have questioned the long-term safety of PPIs. Furthermore, these drugs are too often used in patients who have no valid indication for them,2,3 exposing these patients to unnecessary risks.
The goals of this review are to analyze the recent literature about the risks of PPIs and to provide a rational approach for managing patients on PPI therapy in light of these concerns.
DO PPIs REDUCE THE EFFECT OF CLOPIDOGREL?
Clopidogrel (Plavix) is a potent antiplatelet agent commonly used in patients with atherosclerotic cardiac or cerebrovascular disease, sometimes in combination with aspirin. Because of the risk of significant gastrointestinal bleeding, a 2008 multisociety task force recommended prescribing a PPI when both clopidogrel and aspirin are used as dual antiplatelet therapy.4
Studies of clopidogrel plus PPIs: Discrepant results
To date, only one prospective randomized controlled trial has specifically investigated the effect of PPIs on cardiovascular outcomes in patients using clopidogrel. In this trial, patients on dual antiplatelet therapy with clopidogrel and aspirin were randomized to receive either omeprazole 20 mg or placebo. Analysis of the data revealed no significant increase in the composite end point of cardiovascular events (hazard ratio [HR] 0.99, 95% confidence interval [CI] 0.68–1.44, P = .96), but a statistically significant decrease in composite gastrointestinal events (HR 0.34, 95% CI 0.18–0.63, P < .001).17
Unfortunately, this trial had to be terminated before the prespecified sample size and duration of follow-up were reached because the study sponsor declared bankruptcy.
One additional recent retrospective cohort study16 likewise found no significant risk of serious cardiovascular disease related to PPI use in clopidogrel users. It also found that the adjusted incidence of hospitalization for upper gastrointestinal bleeding was 50% lower in patients who used PPIs than in those who did not (HR 0.50, 95% CI 0.39–0.65).
Do factors other than PPIs account for the higher risk in some of the studies?
The discrepant results of these studies suggest that the higher risk of cardiovascular events may be due, either completely or in part, to a factor other than the pharmacologic interaction of PPIs and clopidogrel. It is difficult to infer causality from the available data. In situations in which no randomized controlled trials exist, one looks to observational (case-control or cohort) studies to try to obtain the best estimate of the actual risk. With PPIs and clopidogrel, a randomized controlled trial was performed but terminated before patient enrollment was complete.
The increased risk found in some of these studies may be real, may be due to chance, or may even represent an increased risk from PPIs alone (although data do not support this possibility).18 However, the major concern in observational studies is the inability to account for unmeasured confounders, a problem virtually eliminated by randomization strategies in prospective studies.
In the studies that found a higher risk with the combination of omeprazole plus clopidogrel, the principal concern is confounding by indication, in which distortions of the risk estimates arise from an imbalance in prognostic factors between compared treatment groups that remains unmeasured.19 Stated another way, physicians who believed some patients to be “sicker” or to have a higher risk of serious events may have treated them with a PPI on the basis of factors that remained unaccounted for in the epidemiologic investigation.
This possibility has been reinforced by findings from a nonrandomized subgroup analysis of a randomized controlled trial in which patients who had been receiving a PPI had a higher rate of cardiovascular events whether they received clopidogrel or placebo.20
FDA alert: Avoid using omeprazole or esomeprazole with clopidogrel
Nonetheless, on November 17, 2009, the US Food and Drug Administration (FDA) issued an alert to health care professionals and the public about the potential interaction between clopidogrel and omeprazole.21 In this alert, the FDA stated that the use of omeprazole or esomeprazole (Nexium) with clopidogrel should be avoided.
An algorithm to use when considering clopidogrel plus a PPI
Physicians are now left in a bind between the minimal, if any, pooled risk seen in the available data and the FDA recommendation. What is the best action to take?
First, assess the need for dual antiplatelet therapy. If dual antiplatelet therapy (clopidogrel plus aspirin) is required, then a PPI is warranted for gastric protection because the risk of life-threatening bleeding outweighs any increased risk of cardiovascular events.4
If antiplatelet monotherapy (clopidogrel alone) is required, then assess the reason for antisecretory therapy.
For complicated disease, such as gastroesophageal reflux disease with Barrett esophagus or peptic strictures, PPI therapy is warranted to prevent progression or recurrence of complications. If the antisecretory therapy is being provided for noncomplicated symptomatic disorders such as nonerosive gastroesophageal reflux disease or dyspepsia, then one should try to “step down” the therapy by lowering the PPI dose as much as possible while still controlling symptoms to the patient’s tolerance, then possibly stepping further by substituting a histamine-2-receptor antagonist, an antacid, or “on-demand” use of PPIs.22,23
However, if the rationale for antisecretory therapy is simply for gastrointestinal protection, then further risk stratification for gastro intestinal bleeding should be undertaken.4 For patients with a high risk of future gastrointestinal bleeding, such as those with prior episodes of bleeding or concurrent use of nonsteroidal anti-inflammatory drugs, antisecretory therapy is still recommended. Therefore, if a patient is on monotherapy with clopidogrel, has no complicated or symptomatic gastrointestinal disorder, and does not have a high risk of gastrointestinal bleeding, then therapy with a PPI should be reconsidered.
There are no strong data to indicate that one particular PPI should be used or avoided if one of the above criteria indicates the concurrent need for clopidogrel and a PPI. In their health alert about the potential interaction, the FDA did not issue the same warning for PPIs other than omeprazole and esomeprazole, but fell short of recommending a change to another PPI because of a lack of data to support or refute a similar interaction.
Because the half-lives of clopidogrel and PPIs are short, separating their administration could in theory decrease or eliminate the risk of competitive inhibition. The PPI could be given in the morning before breakfast and the clopidogrel could be given at night, or the clopidogrel could be given at lunchtime and the PPI before dinner. Although the FDA does not believe this strategy will reduce this interaction,21 one expert in the field has suggested it.18
DO PPIs CAUSE OSTEOPOROSIS, FRACTURES?
In a widely publicized paper published in 2006, Yang and colleagues25 reported the results of a large nested case-control study in the United Kingdom. The risk of hip fracture was significantly greater in patients who had been using PPIs for at least 1 year than in those who had not. The risk appeared to increase with longer use and higher doses of PPIs.
A similar risk of hip fracture was seen in a larger Danish case-control study published the same year.26 This study also found an increased odds ratio for PPI use in patients with spine fractures as well as in patients with any type of fracture. Interestingly, this study found a lower risk of fracture in patients using a histamine-2-receptor antagonist instead of a PPI.
Targownik et al27 found that the risk of hip fracture was not significantly higher until after 5 years of PPI exposure, with an even stronger risk after 7 years.
However, the data on both association and causal relationship are not uniform.
The Women’s Health Initiative,30 with more than 1 million person-years of followup, found no association between PPI use and hip fracture, but a modest association between PPI use and spine, arm, and wrist fractures, as well as total fractures.
A study in the United Kingdom found that patients without any major risk factors for hip fracture (defined by a risk ratio > 2) accounted for only 25% of cases but 53% of controls. When only these two average-risk groups were compared, the risk of hip fracture was similar in cases and controls.31
Corley et al32 also found that the risk of fracture associated with PPI use was only significant in the presence of another risk factor. These findings suggest that residual confounding may be to blame, at least in part, for the estimates of increased risk in the prior studies.
Another way to interpret these data is that PPIs increase the risk in patients at high risk to begin with, but not in those at average risk. This is an example of interaction (or effect modification) in which the risk is unequally distributed across groups with different characteristics.
A recently published study refutes the theory that impaired calcium absorption is responsible for the increase in fractures.33 In this study, investigators queried the Manitoba Bone Mineral Density Database to determine the relationship between antisecretory therapy with PPIs and osteoporosis or loss of bone mineral density—and they found none. This study may support the theory that residual confounding is the reason for the finding of an increased risk, but it also leaves open the possibility that PPIs induce other changes in bone microstructure that could increase the risk of fracture.
FDA labeling: Possible risk of fracture with PPIs
Based on the data so far, it appears possible that there is a small, albeit statistically significant, association between PPI use and fracture risk. The association is indeed biologically plausible, but it remains to be seen if this association is clinically significant, as the risk is relatively low. Even though the studies had methodologic limitations, on May 25, 2010, the FDA announced a change in the required labeling information for PPIs to indicate a possible risk of fracture with these drugs.34
Reassess the need for chronic PPI therapy
Although patients may worry that they will develop osteoporosis and fractures if they take PPIs, the data do not support a strong risk. Nevertheless, when faced with a patient on chronic PPI therapy, especially with a high dose, providers should use the opportunity to reassess the indication for the PPI to decide if chronic therapy is required, in a matter similar to the algorithm provided for PPI-clopidogrel cotherapy (FIGURE 2). Providers should educate patients about the data, and limit new and recurring PPI prescriptions to patients who require a PPI for appropriate indications, at the lowest dose, and for the shortest time possible.
DO PPIs INCREASE THE RISK OF PNEUMONIA?
Several recent studies have also raised concern about an association between PPI use and pneumonia.
Normally, the stomach remains free of bacteria (except for Helicobacter pylori) because its acidic milieu destroys nearly all bacteria swallowed. If the stomach becomes less acidic, it loses this protective mechanism, and ingested organisms can survive and proliferate.35 In theory, when gastroesophageal reflux occurs, these bacteria could be carried up to the hypopharynx where microaspiration into the lower airways could lead to pneumonia, especially in patients with compromised oropharyngeal protective reflexes (eg, patients on mechanical ventilation).
This possible association came to the attention of the general medical community when a Dutch study,36 in which 5,551 cases of community-acquired pneumonia developed in 364,683 people, found that the incidence of pneumonia was about 4.5 times higher in people exposed to acid-suppressive drugs (both PPIs and histamine-2-receptor antagonists) than in unexposed individuals. Patients who developed pneumonia also had higher odds of significant comorbid conditions, including heart failure and chronic obstructive pulmonary disease. The authors calculated that about one case of pneumonia per 226 patients treated with a PPI would be attributable to the PPI. A major limitation of this study, however, was that only 18% of the patients diagnosed with pneumonia actually had radiologic or microbiologic confirmation of pneumonia.
Other studies later examined the relationship between PPIs and community-acquired pneumonia,37–41 and most have revealed a modestly higher risk of community-acquired pneumonia in patients exposed to PPIs.
This risk was confirmed in a recent metaanalysis, which found a higher risk of community-acquired pneumonia with PPI use (odds ratio 1.36, 95% CI 1.12–1.65).42 However, the authors refrained from drawing definitive conclusions from these data because of significant heterogeneity between the studies. One study37 found that recent onset of use (within 7 days) had a much stronger association with community-acquired pneumonia than longer-term use, which is contradictory to a causal association, since longer-term use should lead to more cases of pneumonia.
Another study investigated the association between acid-suppressive drugs and hospital-acquired pneumonia in nonventilated patients.43 In a 4-year period, there were 63,878 admissions in 42,093 unique patients. Acid-suppressive drugs were prescribed in 32,922 admissions (52%); the drugs included PPIs in 83% of these. Hospital-acquired pneumonia occurred in 2,219 admissions (3.5%), with a higher incidence in patients exposed to acid-suppressive drugs than in the unexposed group (4.6% vs 2.0%). The adjusted odds ratio for pneumonia was 1.3 (95% CI 1.1–1.4) in the exposed group. Subgroup analysis revealed that the association remained significant for PPIs but not for histamine-2-receptor antagonists.
Adequate studies of mechanically ventilated patients in the current era of intravenous PPI use are lacking. Older studies in this group of patients may not be generalizable to current practice because of the reduction in gastric volume with intravenous PPIs that may offset the theoretical risk of aspiration.35
Although the data supporting the association are not exceedingly strong, the relationship is biologically plausible. If there is a risk, it seems to be greatest in the sickest patients, who can least afford to develop pneumonia. Therefore, prudent prescribing should be the rule for both inpatients and outpatients, especially in patients with comorbidities, in whom pneumonia could have serious consequences.
PPIs AND ENTERIC INFECTIONS
Traditionally, gastric acid was not believed to be important in protecting against Clostridium difficile infection because acid-resistant spores were presumed to be the principal vector of transmission.44 Recently, this thought has been challenged, as several studies have found a higher risk of C difficile infection in PPI users. In theory, PPIs may increase the risk of C difficile infection by increasing the ability of the spore to convert to the vegetative form and to survive intraluminally.
A recent meta-analysis of 11 papers, including nearly 127,000 patients, found a significant relationship between PPI use and C difficile infection, with an odds ratio of 2.05 (95% CI 1.47–2.85).45 Further supporting the hypothesis of a direct causative association, a recent study found a significant dose-response, with more aggressive acid-suppression associated with higher odds ratios.46 In view of this association, patients using PPIs who develop diarrhea should be evaluated for C difficile, perhaps even in the absence of other risk factors.
Other enteric infections have been found to be associated with PPIs.44,45 Small intestinal bacterial overgrowth, a condition that is associated with bloating, diarrhea, and malabsorption, has recently been associated with PPI use, although the significance of the association is uncertain.47
Based on a change in the intestinal flora, recent reports have additionally implied that there is a relationship between PPI use and the development of spontaneous bacterial peritonitis in hospitalized cirrhotic patients with ascites. One study found a strong association (odds ratio 4.3, 95% CI 1.3–11.7) between PPIs and spontaneous bacterial pneumonitis,48 whereas another study found no significant association (odds ratio 1.0, 95% CI 0.4–2.6).49
Both studies were small case-control studies of hospitalized patients. No firm conclusion can be drawn about the relevance of this association from these investigations at this point.
PPIs AND ACUTE INTERSTITIAL NEPHRITIS
Several case reports have implicated PPIs as a cause of acute interstitial nephritis.
A systematic review from 2007 found 64 cases documented in the literature, 12 of which were considered certainly associated, and 9 of which were probably associated.50 Initial symptoms were nonspecific and included nausea, malaise, and fever. With such extensive use worldwide as the denominator, the authors concluded that acute interstitial nephritis was a rare, idiosyncratic occurrence related to PPI use, but did not find enough evidence to support a causative relationship. Despite the rarity of the syndrome, they recommended maintaining a high level of clinical suspicion to detect acute interstitial nephritis early in its course, especially soon after the initiation of PPI therapy.
POSSIBLE ASSOCIATIONS WITH IRON AND B12 DEFICIENCIES
Long-term PPI therapy has been thought to be associated with micronutrient deficiencies, especially of iron and vitamin B12. Hydrochloric acid in the stomach assists in the dissociation of iron salts from food and the reduction of ferric iron to the more soluble ferrous iron.51 Gastric acid also facilitates the release of vitamin B12 bound to proteins within ingested foodstuffs to permit binding to R-proteins for eventual absorption in the terminal ileum.51,52
Despite the biologic plausibility of these deficiencies, there is currently little evidence to support a clinically relevant association to recommend a change in current practice.
NO THERAPY IS COMPLETELY WITHOUT RISK
Although concerns have been raised about the long-term safety of PPIs, the preponderance of the evidence does not strongly support the apprehensions publicized over the last few years. When translating these studies into the routine management of patients, it is important to recall some very basic tenets of good patient care.
No therapy is completely without risk—whether pharmacologic, surgical, or psychological, and no matter how benign or straightforward. Consequently, no drug, procedure, or treatment plan should be ordered without a valid indication. Even with an indication, the risk-benefit ratio of the therapy prescribed should always be considered. If the indication for the PPI is weak or uncertain, then even a slight risk tips the balance away from the drug, and the drug should be discontinued.
When seeing patients in long-term care, the indication and necessity for all drugs, including PPIs, should be reviewed. The algorithm proposed in Figure 2 can be adapted for virtually any of the possible associations.
Consider the indication for the PPI. Was the PPI started during a hospitalization and then routinely continued after discharge? This is one situation in which the use of a PPI could potentially be discontinued.2
For complicated acid-peptic disease, dose reduction or cessation of PPI therapy may not be possible.
If the PPI was prescribed only for symptom relief, as in cases of dyspepsia or nonerosive gastroesophageal reflux disease, reduce the dose of PPI to as low as possible to maintain symptom control. Should chronic therapy still be required, no specific monitoring is recommended, apart from routine monitoring that takes place in the course of patient care.
Lastly, because of the media attention that several of these concerns have garnered, patients may still harbor significant concerns about PPIs, even their short-term use. In such cases, the prescriber should take the opportunity to communicate the reason for the decision to prescribe the PPI, as well as the best available data about the risks PPIs may pose. None of these outcomes is very common in the absence of PPIs, with the possible exception of recurrent cardiovascular events, and the risks provided in all of these studies are relative to the baseline risk. Even if the risk of a particular outcome doubles with long-term PPI use, twice a small risk remains a small risk.
The development and introduction of the first proton pump inhibitor (PPI), omeprazole (Prilosec), for the management of acid-peptic disorders marks one of the great success stories in gastroenterology. Until the latter part of the 20th century, complications of acid-peptic disease were among the most common problems faced in gastroenterology. Severe peptic strictures were once a highly prevalent cause of dysphagia, and operations for peptic ulcer disease were routinely learned by surgical trainees.
The success of these drugs, with sales total-ling $13.6 billion worldwide in 2009,1 is not just a result of their potency and effectiveness in improving symptoms and complications of acid-peptic disease. Their safety among pharmacologic agents has been unparalleled. When the drugs were first introduced, their use was limited to short courses out of concern that gastric carcinoids could develop, but decades of use have not shown this issue to be of clinical relevance. Serious, acute adverse effects are also exceedingly uncommon.
However, recent reports have questioned the long-term safety of PPIs. Furthermore, these drugs are too often used in patients who have no valid indication for them,2,3 exposing these patients to unnecessary risks.
The goals of this review are to analyze the recent literature about the risks of PPIs and to provide a rational approach for managing patients on PPI therapy in light of these concerns.
DO PPIs REDUCE THE EFFECT OF CLOPIDOGREL?
Clopidogrel (Plavix) is a potent antiplatelet agent commonly used in patients with atherosclerotic cardiac or cerebrovascular disease, sometimes in combination with aspirin. Because of the risk of significant gastrointestinal bleeding, a 2008 multisociety task force recommended prescribing a PPI when both clopidogrel and aspirin are used as dual antiplatelet therapy.4
Studies of clopidogrel plus PPIs: Discrepant results
To date, only one prospective randomized controlled trial has specifically investigated the effect of PPIs on cardiovascular outcomes in patients using clopidogrel. In this trial, patients on dual antiplatelet therapy with clopidogrel and aspirin were randomized to receive either omeprazole 20 mg or placebo. Analysis of the data revealed no significant increase in the composite end point of cardiovascular events (hazard ratio [HR] 0.99, 95% confidence interval [CI] 0.68–1.44, P = .96), but a statistically significant decrease in composite gastrointestinal events (HR 0.34, 95% CI 0.18–0.63, P < .001).17
Unfortunately, this trial had to be terminated before the prespecified sample size and duration of follow-up were reached because the study sponsor declared bankruptcy.
One additional recent retrospective cohort study16 likewise found no significant risk of serious cardiovascular disease related to PPI use in clopidogrel users. It also found that the adjusted incidence of hospitalization for upper gastrointestinal bleeding was 50% lower in patients who used PPIs than in those who did not (HR 0.50, 95% CI 0.39–0.65).
Do factors other than PPIs account for the higher risk in some of the studies?
The discrepant results of these studies suggest that the higher risk of cardiovascular events may be due, either completely or in part, to a factor other than the pharmacologic interaction of PPIs and clopidogrel. It is difficult to infer causality from the available data. In situations in which no randomized controlled trials exist, one looks to observational (case-control or cohort) studies to try to obtain the best estimate of the actual risk. With PPIs and clopidogrel, a randomized controlled trial was performed but terminated before patient enrollment was complete.
The increased risk found in some of these studies may be real, may be due to chance, or may even represent an increased risk from PPIs alone (although data do not support this possibility).18 However, the major concern in observational studies is the inability to account for unmeasured confounders, a problem virtually eliminated by randomization strategies in prospective studies.
In the studies that found a higher risk with the combination of omeprazole plus clopidogrel, the principal concern is confounding by indication, in which distortions of the risk estimates arise from an imbalance in prognostic factors between compared treatment groups that remains unmeasured.19 Stated another way, physicians who believed some patients to be “sicker” or to have a higher risk of serious events may have treated them with a PPI on the basis of factors that remained unaccounted for in the epidemiologic investigation.
This possibility has been reinforced by findings from a nonrandomized subgroup analysis of a randomized controlled trial in which patients who had been receiving a PPI had a higher rate of cardiovascular events whether they received clopidogrel or placebo.20
FDA alert: Avoid using omeprazole or esomeprazole with clopidogrel
Nonetheless, on November 17, 2009, the US Food and Drug Administration (FDA) issued an alert to health care professionals and the public about the potential interaction between clopidogrel and omeprazole.21 In this alert, the FDA stated that the use of omeprazole or esomeprazole (Nexium) with clopidogrel should be avoided.
An algorithm to use when considering clopidogrel plus a PPI
Physicians are now left in a bind between the minimal, if any, pooled risk seen in the available data and the FDA recommendation. What is the best action to take?
First, assess the need for dual antiplatelet therapy. If dual antiplatelet therapy (clopidogrel plus aspirin) is required, then a PPI is warranted for gastric protection because the risk of life-threatening bleeding outweighs any increased risk of cardiovascular events.4
If antiplatelet monotherapy (clopidogrel alone) is required, then assess the reason for antisecretory therapy.
For complicated disease, such as gastroesophageal reflux disease with Barrett esophagus or peptic strictures, PPI therapy is warranted to prevent progression or recurrence of complications. If the antisecretory therapy is being provided for noncomplicated symptomatic disorders such as nonerosive gastroesophageal reflux disease or dyspepsia, then one should try to “step down” the therapy by lowering the PPI dose as much as possible while still controlling symptoms to the patient’s tolerance, then possibly stepping further by substituting a histamine-2-receptor antagonist, an antacid, or “on-demand” use of PPIs.22,23
However, if the rationale for antisecretory therapy is simply for gastrointestinal protection, then further risk stratification for gastro intestinal bleeding should be undertaken.4 For patients with a high risk of future gastrointestinal bleeding, such as those with prior episodes of bleeding or concurrent use of nonsteroidal anti-inflammatory drugs, antisecretory therapy is still recommended. Therefore, if a patient is on monotherapy with clopidogrel, has no complicated or symptomatic gastrointestinal disorder, and does not have a high risk of gastrointestinal bleeding, then therapy with a PPI should be reconsidered.
There are no strong data to indicate that one particular PPI should be used or avoided if one of the above criteria indicates the concurrent need for clopidogrel and a PPI. In their health alert about the potential interaction, the FDA did not issue the same warning for PPIs other than omeprazole and esomeprazole, but fell short of recommending a change to another PPI because of a lack of data to support or refute a similar interaction.
Because the half-lives of clopidogrel and PPIs are short, separating their administration could in theory decrease or eliminate the risk of competitive inhibition. The PPI could be given in the morning before breakfast and the clopidogrel could be given at night, or the clopidogrel could be given at lunchtime and the PPI before dinner. Although the FDA does not believe this strategy will reduce this interaction,21 one expert in the field has suggested it.18
DO PPIs CAUSE OSTEOPOROSIS, FRACTURES?
In a widely publicized paper published in 2006, Yang and colleagues25 reported the results of a large nested case-control study in the United Kingdom. The risk of hip fracture was significantly greater in patients who had been using PPIs for at least 1 year than in those who had not. The risk appeared to increase with longer use and higher doses of PPIs.
A similar risk of hip fracture was seen in a larger Danish case-control study published the same year.26 This study also found an increased odds ratio for PPI use in patients with spine fractures as well as in patients with any type of fracture. Interestingly, this study found a lower risk of fracture in patients using a histamine-2-receptor antagonist instead of a PPI.
Targownik et al27 found that the risk of hip fracture was not significantly higher until after 5 years of PPI exposure, with an even stronger risk after 7 years.
However, the data on both association and causal relationship are not uniform.
The Women’s Health Initiative,30 with more than 1 million person-years of followup, found no association between PPI use and hip fracture, but a modest association between PPI use and spine, arm, and wrist fractures, as well as total fractures.
A study in the United Kingdom found that patients without any major risk factors for hip fracture (defined by a risk ratio > 2) accounted for only 25% of cases but 53% of controls. When only these two average-risk groups were compared, the risk of hip fracture was similar in cases and controls.31
Corley et al32 also found that the risk of fracture associated with PPI use was only significant in the presence of another risk factor. These findings suggest that residual confounding may be to blame, at least in part, for the estimates of increased risk in the prior studies.
Another way to interpret these data is that PPIs increase the risk in patients at high risk to begin with, but not in those at average risk. This is an example of interaction (or effect modification) in which the risk is unequally distributed across groups with different characteristics.
A recently published study refutes the theory that impaired calcium absorption is responsible for the increase in fractures.33 In this study, investigators queried the Manitoba Bone Mineral Density Database to determine the relationship between antisecretory therapy with PPIs and osteoporosis or loss of bone mineral density—and they found none. This study may support the theory that residual confounding is the reason for the finding of an increased risk, but it also leaves open the possibility that PPIs induce other changes in bone microstructure that could increase the risk of fracture.
FDA labeling: Possible risk of fracture with PPIs
Based on the data so far, it appears possible that there is a small, albeit statistically significant, association between PPI use and fracture risk. The association is indeed biologically plausible, but it remains to be seen if this association is clinically significant, as the risk is relatively low. Even though the studies had methodologic limitations, on May 25, 2010, the FDA announced a change in the required labeling information for PPIs to indicate a possible risk of fracture with these drugs.34
Reassess the need for chronic PPI therapy
Although patients may worry that they will develop osteoporosis and fractures if they take PPIs, the data do not support a strong risk. Nevertheless, when faced with a patient on chronic PPI therapy, especially with a high dose, providers should use the opportunity to reassess the indication for the PPI to decide if chronic therapy is required, in a matter similar to the algorithm provided for PPI-clopidogrel cotherapy (FIGURE 2). Providers should educate patients about the data, and limit new and recurring PPI prescriptions to patients who require a PPI for appropriate indications, at the lowest dose, and for the shortest time possible.
DO PPIs INCREASE THE RISK OF PNEUMONIA?
Several recent studies have also raised concern about an association between PPI use and pneumonia.
Normally, the stomach remains free of bacteria (except for Helicobacter pylori) because its acidic milieu destroys nearly all bacteria swallowed. If the stomach becomes less acidic, it loses this protective mechanism, and ingested organisms can survive and proliferate.35 In theory, when gastroesophageal reflux occurs, these bacteria could be carried up to the hypopharynx where microaspiration into the lower airways could lead to pneumonia, especially in patients with compromised oropharyngeal protective reflexes (eg, patients on mechanical ventilation).
This possible association came to the attention of the general medical community when a Dutch study,36 in which 5,551 cases of community-acquired pneumonia developed in 364,683 people, found that the incidence of pneumonia was about 4.5 times higher in people exposed to acid-suppressive drugs (both PPIs and histamine-2-receptor antagonists) than in unexposed individuals. Patients who developed pneumonia also had higher odds of significant comorbid conditions, including heart failure and chronic obstructive pulmonary disease. The authors calculated that about one case of pneumonia per 226 patients treated with a PPI would be attributable to the PPI. A major limitation of this study, however, was that only 18% of the patients diagnosed with pneumonia actually had radiologic or microbiologic confirmation of pneumonia.
Other studies later examined the relationship between PPIs and community-acquired pneumonia,37–41 and most have revealed a modestly higher risk of community-acquired pneumonia in patients exposed to PPIs.
This risk was confirmed in a recent metaanalysis, which found a higher risk of community-acquired pneumonia with PPI use (odds ratio 1.36, 95% CI 1.12–1.65).42 However, the authors refrained from drawing definitive conclusions from these data because of significant heterogeneity between the studies. One study37 found that recent onset of use (within 7 days) had a much stronger association with community-acquired pneumonia than longer-term use, which is contradictory to a causal association, since longer-term use should lead to more cases of pneumonia.
Another study investigated the association between acid-suppressive drugs and hospital-acquired pneumonia in nonventilated patients.43 In a 4-year period, there were 63,878 admissions in 42,093 unique patients. Acid-suppressive drugs were prescribed in 32,922 admissions (52%); the drugs included PPIs in 83% of these. Hospital-acquired pneumonia occurred in 2,219 admissions (3.5%), with a higher incidence in patients exposed to acid-suppressive drugs than in the unexposed group (4.6% vs 2.0%). The adjusted odds ratio for pneumonia was 1.3 (95% CI 1.1–1.4) in the exposed group. Subgroup analysis revealed that the association remained significant for PPIs but not for histamine-2-receptor antagonists.
Adequate studies of mechanically ventilated patients in the current era of intravenous PPI use are lacking. Older studies in this group of patients may not be generalizable to current practice because of the reduction in gastric volume with intravenous PPIs that may offset the theoretical risk of aspiration.35
Although the data supporting the association are not exceedingly strong, the relationship is biologically plausible. If there is a risk, it seems to be greatest in the sickest patients, who can least afford to develop pneumonia. Therefore, prudent prescribing should be the rule for both inpatients and outpatients, especially in patients with comorbidities, in whom pneumonia could have serious consequences.
PPIs AND ENTERIC INFECTIONS
Traditionally, gastric acid was not believed to be important in protecting against Clostridium difficile infection because acid-resistant spores were presumed to be the principal vector of transmission.44 Recently, this thought has been challenged, as several studies have found a higher risk of C difficile infection in PPI users. In theory, PPIs may increase the risk of C difficile infection by increasing the ability of the spore to convert to the vegetative form and to survive intraluminally.
A recent meta-analysis of 11 papers, including nearly 127,000 patients, found a significant relationship between PPI use and C difficile infection, with an odds ratio of 2.05 (95% CI 1.47–2.85).45 Further supporting the hypothesis of a direct causative association, a recent study found a significant dose-response, with more aggressive acid-suppression associated with higher odds ratios.46 In view of this association, patients using PPIs who develop diarrhea should be evaluated for C difficile, perhaps even in the absence of other risk factors.
Other enteric infections have been found to be associated with PPIs.44,45 Small intestinal bacterial overgrowth, a condition that is associated with bloating, diarrhea, and malabsorption, has recently been associated with PPI use, although the significance of the association is uncertain.47
Based on a change in the intestinal flora, recent reports have additionally implied that there is a relationship between PPI use and the development of spontaneous bacterial peritonitis in hospitalized cirrhotic patients with ascites. One study found a strong association (odds ratio 4.3, 95% CI 1.3–11.7) between PPIs and spontaneous bacterial pneumonitis,48 whereas another study found no significant association (odds ratio 1.0, 95% CI 0.4–2.6).49
Both studies were small case-control studies of hospitalized patients. No firm conclusion can be drawn about the relevance of this association from these investigations at this point.
PPIs AND ACUTE INTERSTITIAL NEPHRITIS
Several case reports have implicated PPIs as a cause of acute interstitial nephritis.
A systematic review from 2007 found 64 cases documented in the literature, 12 of which were considered certainly associated, and 9 of which were probably associated.50 Initial symptoms were nonspecific and included nausea, malaise, and fever. With such extensive use worldwide as the denominator, the authors concluded that acute interstitial nephritis was a rare, idiosyncratic occurrence related to PPI use, but did not find enough evidence to support a causative relationship. Despite the rarity of the syndrome, they recommended maintaining a high level of clinical suspicion to detect acute interstitial nephritis early in its course, especially soon after the initiation of PPI therapy.
POSSIBLE ASSOCIATIONS WITH IRON AND B12 DEFICIENCIES
Long-term PPI therapy has been thought to be associated with micronutrient deficiencies, especially of iron and vitamin B12. Hydrochloric acid in the stomach assists in the dissociation of iron salts from food and the reduction of ferric iron to the more soluble ferrous iron.51 Gastric acid also facilitates the release of vitamin B12 bound to proteins within ingested foodstuffs to permit binding to R-proteins for eventual absorption in the terminal ileum.51,52
Despite the biologic plausibility of these deficiencies, there is currently little evidence to support a clinically relevant association to recommend a change in current practice.
NO THERAPY IS COMPLETELY WITHOUT RISK
Although concerns have been raised about the long-term safety of PPIs, the preponderance of the evidence does not strongly support the apprehensions publicized over the last few years. When translating these studies into the routine management of patients, it is important to recall some very basic tenets of good patient care.
No therapy is completely without risk—whether pharmacologic, surgical, or psychological, and no matter how benign or straightforward. Consequently, no drug, procedure, or treatment plan should be ordered without a valid indication. Even with an indication, the risk-benefit ratio of the therapy prescribed should always be considered. If the indication for the PPI is weak or uncertain, then even a slight risk tips the balance away from the drug, and the drug should be discontinued.
When seeing patients in long-term care, the indication and necessity for all drugs, including PPIs, should be reviewed. The algorithm proposed in Figure 2 can be adapted for virtually any of the possible associations.
Consider the indication for the PPI. Was the PPI started during a hospitalization and then routinely continued after discharge? This is one situation in which the use of a PPI could potentially be discontinued.2
For complicated acid-peptic disease, dose reduction or cessation of PPI therapy may not be possible.
If the PPI was prescribed only for symptom relief, as in cases of dyspepsia or nonerosive gastroesophageal reflux disease, reduce the dose of PPI to as low as possible to maintain symptom control. Should chronic therapy still be required, no specific monitoring is recommended, apart from routine monitoring that takes place in the course of patient care.
Lastly, because of the media attention that several of these concerns have garnered, patients may still harbor significant concerns about PPIs, even their short-term use. In such cases, the prescriber should take the opportunity to communicate the reason for the decision to prescribe the PPI, as well as the best available data about the risks PPIs may pose. None of these outcomes is very common in the absence of PPIs, with the possible exception of recurrent cardiovascular events, and the risks provided in all of these studies are relative to the baseline risk. Even if the risk of a particular outcome doubles with long-term PPI use, twice a small risk remains a small risk.
- Gatyas G. IMS Health reports U.S. prescription sales grew 5.1 percent in 2009, to $300.3 Billion. IMS Health. http://www.imshealth.com/portal/site/imshealth/menuitem.a46c6d4df3db4b3d88f611019418c22a/?vgnextoid=d690a27e9d5b7210VgnVCM100000ed152ca2RCRD&vgnextfmt=default. Accessed 10/7/2010.
- Zink DA, Pohlman M, Barnes M, Cannon ME. Long-term use of acid suppression started inappropriately during hospitalization. Aliment Pharmacol Ther 2005; 21:1203–1209.
- Heidelbaugh JJ, Inadomi JM. Magnitude and economic impact of inappropriate use of stress ulcer prophylaxis in non-ICU hospitalized patients. Am J Gastroenterol 2006; 101:2200–2205.
- Bhatt DL, Scheiman J, Abraham NS, et al; American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation 2008; 118:1894–1909.
- Klotz U, Schwab M, Treiber G. CYP2C19 polymorphism and proton pump inhibitors. Basic Clin Pharmacol Toxicol 2004; 95:2–8.
- Gilard M, Arnaud B, Cornily JC, et al. Influence of omeprazole on the antiplatelet action of clopidogrel associated with aspirin: the randomized, double-blind OCLA (Omeprazole CLopidogrel Aspirin) study. J Am Coll Cardiol 2008; 51:256–260.
- Small DS, Farid NA, Payne CD, et al. Effects of the proton pump inhibitor lansoprazole on the pharmacokinetics and pharmacodynamics of prasugrel and clopidogrel. J Clin Pharmacol 2008; 48:475–484.
- Sibbing D, Morath T, Stegherr J, et al. Impact of proton pump inhibitors on the antiplatelet effects of clopidogrel. Thromb Haemost 2009; 101:714–719.
- O’Donoghue ML, Braunwald E, Antman EM, et al. Pharmacodynamic effect and clinical efficacy of clopidogrel and prasugrel with or without a proton-pump inhibitor: an analysis of two randomised trials. Lancet 2009; 374:989–997.
- Ho PM, Maddox TM, Wang L, et al. Risk of adverse outcomes associated with concomitant use of clopidogrel and proton pump inhibitors following acute coronary syndrome. JAMA 2009; 301:937–944.
- Juurlink DN, Gomes T, Ko DT, et al. A population-based study of the drug interaction between proton pump inhibitors and clopidogrel. CMAJ 2009; 180:713–718.
- Stanek EJ, Aubert RE, Flockhart DA, et al. A national study of the effect of individual proton pump inhibitors on cardiovascular outcomes in patients treated with clopidogrel following coronary stenting: the Clopidogrel Medco Outcomes Study. Program and abstracts of the 32nd Annual SCAI Scientific Sessions May 6, 2009; Las Vegas, Nevada.
- Simon T, Verstuyft C, Mary-Krause M, et al; French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360:363–375.
- Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 2009; 373:309–317.
- Ramirez JF, Selzer F, Chakaprani R, et al. Proton pump inhibitor and clopidogrel combination is not associated with adverse clinical outcomes after PCI: the NHLBI dynamic registry (abstract). J Am Coll Cardiol 2009; 53(suppl 1):A27.
- Ray WA, Murray KT, Griffin MR, et al. Outcomes with concurrent use of clopidogrel and proton-pump inhibitors: a cohort study. Ann Intern Med 2010; 152:337–345.
- Bhatt DL, Cryer B, Contant CF, et al. Clopidogrel with or without omeprazole in coronary artery disease. N Engl J Med 2010; 363:1909–1917.
- Laine L, Hennekens C. Proton pump inhibitor and clopidogrel interaction: fact or fiction? Am J Gastroenterol 2010; 105:34–41.
- Walker AM. Confounding by indication. Epidemiology 1996; 7:335–336.
- Dunn SP, Macaulay TE, Brennan DM, et al. Baseline proton pump inhibitor use is associated with increased cardiovascular events with and without the use of clopidogrel in the CREDO trial (abstract). Circulation 2008; 118:S815.
- US Food and Drug Administration. Information for healthcare professionals: update to the labeling of clopidogrel bisulfate (marketed as Plavix) to alert healthcare professionals about a drug interaction with omeprazole (marketed as Prilosec and Prilosec OTC). U.S. Department of Health and Human Services, 11/17/2009. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm190787.htm. Accessed 9/23/2010.
- Inadomi JM, Jamal R, Murata GH, et al. Step-down management of gastroesophageal reflux disease. Gastroenterology 2001; 121:1095–1100.
- Inadomi JM, McIntyre L, Bernard L, Fendrick AM. Step-down from multiple- to single-dose proton pump inhibitors (PPIs): a prospective study of patients with heartburn or acid regurgitation completely relieved with PPIs. Am J Gastroenterol 2003; 98:1940–1944.
- O’Connell MB, Madden DM, Murray AM, Heaney RP, Kerzner LJ. Effects of proton pump inhibitors on calcium carbonate absorption in women: a randomized crossover trial. Am J Med 2005; 118:778–781.
- Yang YX, Lewis JD, Epstein S, Metz DC. Long-term proton pump inhibitor therapy and risk of hip fracture. JAMA 2006; 296:2947–2953.
- Vestergaard P, Rejnmark L, Mosekilde L. Proton pump inhibitors, histamine H2 receptor antagonists, and other antacid medications and the risk of fracture. Calcif Tissue Int 2006; 79:76–83.
- Targownik LE, Lix LM, Metge CJ, Prior HJ, Leung S, Leslie WD. Use of proton pump inhibitors and risk of osteoporosis-related fractures. CMAJ 2008; 179:319–326.
- Roux C, Briot K, Gossec L, et al. Increase in vertebral fracture risk in postmenopausal women using omeprazole. Calcif Tissue Int 2009; 84:13–19.
- Yu EW, Blackwell T, Ensrud KE, et al. Acid-suppressive medications and risk of bone loss and fracture in older adults. Calcif Tissue Int 2008; 83:251–259.
- Gray SL, LaCroix AZ, Larson J, et al. Proton pump inhibitor use, hip fracture, and change in bone mineral density in postmenopausal women: results from the Women’s Health Initiative. Arch Intern Med 2010; 170:765–771.
- Kaye JA, Jick H. Proton pump inhibitor use and risk of hip fractures in patients without major risk factors. Pharmacotherapy 2008; 28:951–959.
- Corley DA, Kubo A, Zhao W, Quesenberry C. Proton pump inhibitors and histamine-2 receptor antagonists are associated with hip fractures among at-risk patients. Gastroenterology 2010; 139:93–101.
- Targownik LE, Lix LM, Leung S, Leslie WD. Proton-pump inhibitor use is not associated with osteoporosis or accelerated bone mineral density loss. Gastroenterology 2010; 138:896–904.
- US Food and Drug Administration. FDA Drug Safety Communication: possible increased risk of fractures of the hip, wrist, and spine with the use of proton pump inhibitors. U.S. Department of Health and Human Services, 5/25/2010. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm213206.htm. Accessed 12/7/2010.
- Vakil N. Acid inhibition and infections outside the gastrointestinal tract. Am J Gastroenterol 2009; 104(suppl 2):S17–S20.
- Laheij RJ, Sturkenboom MC, Hassing RJ, Dieleman J, Stricker BH, Jansen JB. Risk of community-acquired pneumonia and use of gastric acid-suppressive drugs. JAMA 2004; 292:1955–1960.
- Gulmez SE, Holm A, Frederiksen H, Jensen TG, Pedersen C, Hallas J. Use of proton pump inhibitors and the risk of community-acquired pneumonia: a population-based case-control study. Arch Intern Med 2007; 167:950–955.
- Sarkar M, Hennessy S, Yang YX. Proton-pump inhibitor use and the risk for community-acquired pneumonia. Ann Intern Med 2008; 149:391–398.
- Myles PR, Hubbard RB, McKeever TM, Pogson Z, Smith CJ, Gibson JE. Risk of community-acquired pneumonia and the use of statins, ACE inhibitors and gastric acid suppressants: a population-based case-control study. Pharmacoepidemiol Drug Saf 2009; 18:269–275.
- Rodríguez LA, Ruigómez A, Wallander MA, Johansson S. Acid-suppressive drugs and community-acquired pneumonia. Epidemiology 2009; 20:800–806.
- Eurich DT, Sadowski CA, Simpson SH, Marrie TJ, Majumdar SR. Recurrent community-acquired pneumonia in patients starting acid-suppressing drugs. Am J Med 2010; 123:47–53.
- Johnstone J, Nerenberg K, Loeb M. Meta-analysis: proton pump inhibitor use and the risk of community-acquired pneumonia. Aliment Pharmacol Ther 2010; 31:1165–1177.
- Herzig SJ, Howell MD, Ngo LH, Marcantonio ER. Acid-suppressive medication use and the risk for hospital-acquired pneumonia. JAMA 2009; 301:2120–2128.
- Dial MS. Proton pump inhibitor use and enteric infections. Am J Gastroenterol 2009; 104(suppl 2):S10–S16.
- Leonard J, Marshall JK, Moayyedi P. Systematic review of the risk of enteric infection in patients taking acid suppression. Am J Gastroenterol 2007; 102:2047–2056.
- Howell MD, Novack V, Grgurich P, et al. Iatrogenic gastric acid suppression and the risk of nosocomial Clostridium difficile infection. Arch Intern Med 2010; 170:784–790.
- Lombardo L, Foti M, Ruggia O, Chiecchio A. Increased incidence of small intestinal bacterial overgrowth during proton pump inhibitor therapy. Clin Gastroenterol Hepatol 2010; 8:504–508.
- Bajaj JS, Zadvornova Y, Heuman DM, et al. Association of proton pump inhibitor therapy with spontaneous bacterial peritonitis in cirrhotic patients with ascites. Am J Gastroenterol 2009; 104:1130–1134.
- Campbell MS, Obstein K, Reddy KR, Yang YX. Association between proton pump inhibitor use and spontaneous bacterial peritonitis. Dig Dis Sci 2008; 53:394–398.
- Sierra F, Suarez M, Rey M, Vela MF. Systematic review: proton pump inhibitor-associated acute interstitial nephritis. Aliment Pharmacol Ther 2007; 26:545–553.
- McColl KE. Effect of proton pump inhibitors on vitamins and iron. Am J Gastroenterol 2009; 104(suppl 2):S5–S9.
- Ali T, Roberts DN, Tierney WM. Long-term safety concerns with proton pump inhibitors. Am J Med 2009; 122:896–903.
- Gatyas G. IMS Health reports U.S. prescription sales grew 5.1 percent in 2009, to $300.3 Billion. IMS Health. http://www.imshealth.com/portal/site/imshealth/menuitem.a46c6d4df3db4b3d88f611019418c22a/?vgnextoid=d690a27e9d5b7210VgnVCM100000ed152ca2RCRD&vgnextfmt=default. Accessed 10/7/2010.
- Zink DA, Pohlman M, Barnes M, Cannon ME. Long-term use of acid suppression started inappropriately during hospitalization. Aliment Pharmacol Ther 2005; 21:1203–1209.
- Heidelbaugh JJ, Inadomi JM. Magnitude and economic impact of inappropriate use of stress ulcer prophylaxis in non-ICU hospitalized patients. Am J Gastroenterol 2006; 101:2200–2205.
- Bhatt DL, Scheiman J, Abraham NS, et al; American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation 2008; 118:1894–1909.
- Klotz U, Schwab M, Treiber G. CYP2C19 polymorphism and proton pump inhibitors. Basic Clin Pharmacol Toxicol 2004; 95:2–8.
- Gilard M, Arnaud B, Cornily JC, et al. Influence of omeprazole on the antiplatelet action of clopidogrel associated with aspirin: the randomized, double-blind OCLA (Omeprazole CLopidogrel Aspirin) study. J Am Coll Cardiol 2008; 51:256–260.
- Small DS, Farid NA, Payne CD, et al. Effects of the proton pump inhibitor lansoprazole on the pharmacokinetics and pharmacodynamics of prasugrel and clopidogrel. J Clin Pharmacol 2008; 48:475–484.
- Sibbing D, Morath T, Stegherr J, et al. Impact of proton pump inhibitors on the antiplatelet effects of clopidogrel. Thromb Haemost 2009; 101:714–719.
- O’Donoghue ML, Braunwald E, Antman EM, et al. Pharmacodynamic effect and clinical efficacy of clopidogrel and prasugrel with or without a proton-pump inhibitor: an analysis of two randomised trials. Lancet 2009; 374:989–997.
- Ho PM, Maddox TM, Wang L, et al. Risk of adverse outcomes associated with concomitant use of clopidogrel and proton pump inhibitors following acute coronary syndrome. JAMA 2009; 301:937–944.
- Juurlink DN, Gomes T, Ko DT, et al. A population-based study of the drug interaction between proton pump inhibitors and clopidogrel. CMAJ 2009; 180:713–718.
- Stanek EJ, Aubert RE, Flockhart DA, et al. A national study of the effect of individual proton pump inhibitors on cardiovascular outcomes in patients treated with clopidogrel following coronary stenting: the Clopidogrel Medco Outcomes Study. Program and abstracts of the 32nd Annual SCAI Scientific Sessions May 6, 2009; Las Vegas, Nevada.
- Simon T, Verstuyft C, Mary-Krause M, et al; French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360:363–375.
- Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 2009; 373:309–317.
- Ramirez JF, Selzer F, Chakaprani R, et al. Proton pump inhibitor and clopidogrel combination is not associated with adverse clinical outcomes after PCI: the NHLBI dynamic registry (abstract). J Am Coll Cardiol 2009; 53(suppl 1):A27.
- Ray WA, Murray KT, Griffin MR, et al. Outcomes with concurrent use of clopidogrel and proton-pump inhibitors: a cohort study. Ann Intern Med 2010; 152:337–345.
- Bhatt DL, Cryer B, Contant CF, et al. Clopidogrel with or without omeprazole in coronary artery disease. N Engl J Med 2010; 363:1909–1917.
- Laine L, Hennekens C. Proton pump inhibitor and clopidogrel interaction: fact or fiction? Am J Gastroenterol 2010; 105:34–41.
- Walker AM. Confounding by indication. Epidemiology 1996; 7:335–336.
- Dunn SP, Macaulay TE, Brennan DM, et al. Baseline proton pump inhibitor use is associated with increased cardiovascular events with and without the use of clopidogrel in the CREDO trial (abstract). Circulation 2008; 118:S815.
- US Food and Drug Administration. Information for healthcare professionals: update to the labeling of clopidogrel bisulfate (marketed as Plavix) to alert healthcare professionals about a drug interaction with omeprazole (marketed as Prilosec and Prilosec OTC). U.S. Department of Health and Human Services, 11/17/2009. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm190787.htm. Accessed 9/23/2010.
- Inadomi JM, Jamal R, Murata GH, et al. Step-down management of gastroesophageal reflux disease. Gastroenterology 2001; 121:1095–1100.
- Inadomi JM, McIntyre L, Bernard L, Fendrick AM. Step-down from multiple- to single-dose proton pump inhibitors (PPIs): a prospective study of patients with heartburn or acid regurgitation completely relieved with PPIs. Am J Gastroenterol 2003; 98:1940–1944.
- O’Connell MB, Madden DM, Murray AM, Heaney RP, Kerzner LJ. Effects of proton pump inhibitors on calcium carbonate absorption in women: a randomized crossover trial. Am J Med 2005; 118:778–781.
- Yang YX, Lewis JD, Epstein S, Metz DC. Long-term proton pump inhibitor therapy and risk of hip fracture. JAMA 2006; 296:2947–2953.
- Vestergaard P, Rejnmark L, Mosekilde L. Proton pump inhibitors, histamine H2 receptor antagonists, and other antacid medications and the risk of fracture. Calcif Tissue Int 2006; 79:76–83.
- Targownik LE, Lix LM, Metge CJ, Prior HJ, Leung S, Leslie WD. Use of proton pump inhibitors and risk of osteoporosis-related fractures. CMAJ 2008; 179:319–326.
- Roux C, Briot K, Gossec L, et al. Increase in vertebral fracture risk in postmenopausal women using omeprazole. Calcif Tissue Int 2009; 84:13–19.
- Yu EW, Blackwell T, Ensrud KE, et al. Acid-suppressive medications and risk of bone loss and fracture in older adults. Calcif Tissue Int 2008; 83:251–259.
- Gray SL, LaCroix AZ, Larson J, et al. Proton pump inhibitor use, hip fracture, and change in bone mineral density in postmenopausal women: results from the Women’s Health Initiative. Arch Intern Med 2010; 170:765–771.
- Kaye JA, Jick H. Proton pump inhibitor use and risk of hip fractures in patients without major risk factors. Pharmacotherapy 2008; 28:951–959.
- Corley DA, Kubo A, Zhao W, Quesenberry C. Proton pump inhibitors and histamine-2 receptor antagonists are associated with hip fractures among at-risk patients. Gastroenterology 2010; 139:93–101.
- Targownik LE, Lix LM, Leung S, Leslie WD. Proton-pump inhibitor use is not associated with osteoporosis or accelerated bone mineral density loss. Gastroenterology 2010; 138:896–904.
- US Food and Drug Administration. FDA Drug Safety Communication: possible increased risk of fractures of the hip, wrist, and spine with the use of proton pump inhibitors. U.S. Department of Health and Human Services, 5/25/2010. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm213206.htm. Accessed 12/7/2010.
- Vakil N. Acid inhibition and infections outside the gastrointestinal tract. Am J Gastroenterol 2009; 104(suppl 2):S17–S20.
- Laheij RJ, Sturkenboom MC, Hassing RJ, Dieleman J, Stricker BH, Jansen JB. Risk of community-acquired pneumonia and use of gastric acid-suppressive drugs. JAMA 2004; 292:1955–1960.
- Gulmez SE, Holm A, Frederiksen H, Jensen TG, Pedersen C, Hallas J. Use of proton pump inhibitors and the risk of community-acquired pneumonia: a population-based case-control study. Arch Intern Med 2007; 167:950–955.
- Sarkar M, Hennessy S, Yang YX. Proton-pump inhibitor use and the risk for community-acquired pneumonia. Ann Intern Med 2008; 149:391–398.
- Myles PR, Hubbard RB, McKeever TM, Pogson Z, Smith CJ, Gibson JE. Risk of community-acquired pneumonia and the use of statins, ACE inhibitors and gastric acid suppressants: a population-based case-control study. Pharmacoepidemiol Drug Saf 2009; 18:269–275.
- Rodríguez LA, Ruigómez A, Wallander MA, Johansson S. Acid-suppressive drugs and community-acquired pneumonia. Epidemiology 2009; 20:800–806.
- Eurich DT, Sadowski CA, Simpson SH, Marrie TJ, Majumdar SR. Recurrent community-acquired pneumonia in patients starting acid-suppressing drugs. Am J Med 2010; 123:47–53.
- Johnstone J, Nerenberg K, Loeb M. Meta-analysis: proton pump inhibitor use and the risk of community-acquired pneumonia. Aliment Pharmacol Ther 2010; 31:1165–1177.
- Herzig SJ, Howell MD, Ngo LH, Marcantonio ER. Acid-suppressive medication use and the risk for hospital-acquired pneumonia. JAMA 2009; 301:2120–2128.
- Dial MS. Proton pump inhibitor use and enteric infections. Am J Gastroenterol 2009; 104(suppl 2):S10–S16.
- Leonard J, Marshall JK, Moayyedi P. Systematic review of the risk of enteric infection in patients taking acid suppression. Am J Gastroenterol 2007; 102:2047–2056.
- Howell MD, Novack V, Grgurich P, et al. Iatrogenic gastric acid suppression and the risk of nosocomial Clostridium difficile infection. Arch Intern Med 2010; 170:784–790.
- Lombardo L, Foti M, Ruggia O, Chiecchio A. Increased incidence of small intestinal bacterial overgrowth during proton pump inhibitor therapy. Clin Gastroenterol Hepatol 2010; 8:504–508.
- Bajaj JS, Zadvornova Y, Heuman DM, et al. Association of proton pump inhibitor therapy with spontaneous bacterial peritonitis in cirrhotic patients with ascites. Am J Gastroenterol 2009; 104:1130–1134.
- Campbell MS, Obstein K, Reddy KR, Yang YX. Association between proton pump inhibitor use and spontaneous bacterial peritonitis. Dig Dis Sci 2008; 53:394–398.
- Sierra F, Suarez M, Rey M, Vela MF. Systematic review: proton pump inhibitor-associated acute interstitial nephritis. Aliment Pharmacol Ther 2007; 26:545–553.
- McColl KE. Effect of proton pump inhibitors on vitamins and iron. Am J Gastroenterol 2009; 104(suppl 2):S5–S9.
- Ali T, Roberts DN, Tierney WM. Long-term safety concerns with proton pump inhibitors. Am J Med 2009; 122:896–903.
KEY POINTS
- The US Food and Drug Administration has issued alerts that PPIs may increase the rate of osteoporosis-related fractures and may decrease the effectiveness of clopidogrel (Plavix) for preventing serious cardiovascular events.
- Other concerns include increased rates of pneumonia, Clostridium difficile infection, and other infections.
- A prudent approach to managing these concerns in day-to-day practice is required: PPIs, like any other drugs, should be prescribed only if indicated.
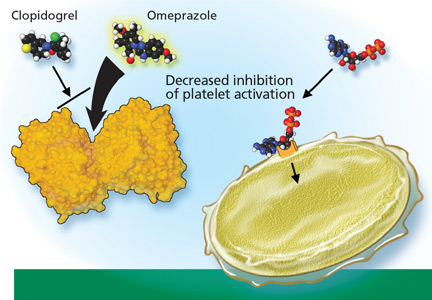
Glucocorticoid-induced osteoporosis
To the Editor: I have to say I am disappointed, but not surprised, at Dr. Dore’s article, “How to prevent glucocorticoid-induced osteoporosis” in your August issue.1 The section “Estrogen is being used more selectively” was shorter and had older and out of date references compared with the section “A role for testosterone?” and it was actually blatantly sexist: the comment in the estrogen section is that “…the consensus…that hormone replacement therapy should be restricted to women with menopausal symptoms or to older women who cannot tolerate other therapies or who express a strong preference for hormone replacement therapy despite being informed about potential adverse events” [my italics],1 while the comment in the testosterone section is that males who “… are hypogonadal, and have no contraindications to androgen replacement therapy (eg, prostate cancer) be offered testosterone therapy to preserve lean body mass and bone mineral density” [my italics].1
While I am not arguing that menopausal hormone therapy should be used first-line for the prevention or treatment of glucocorticoid-induced osteoporosis, I would like to note the following:
First, the referenced 2002 Women’s Health Initiative study2 was a prevention trial, not a therapeutic menopausal trial, and to reference it as a position statement on the use of hormone therapy is ridiculous and perpetuates misinformation about the role of menopausal hormone therapy.
Next, there has been updated information from the Women’s Health Initiative, as well as updated position statements on the use of hormone therapy—the 2010 position statement on the use of estrogen and progestogen in menopausal women3 as well as the 2008 American Association of Clinical Endocrinologists position statement4 noting that the benefits of hormone therapy outweigh the risks for most women under age 60. So Dr. Dore’s reference citation from 20045 is hopelessly outdated.
And lastly, females, unlike males, routinely become hypogonadal at midlife. When faced with a medical condition that requires glucocorticoids that further intensifies the hypogonadal state by suppressing adrenal adrenogens, females may face a “triple whammy” on the bone.
The Women’s Health Initiative actually showed fracture reduction in postmenopausal women who did not even carry the diagnosis of osteoporosis, while the referenced studies in Dr. Dore’s article related to males admittedly “cannot be considered conclusive in view of their small size and the lack of fracture data…”1
So what is bad (actually potentially good) for the goose is apparently just fine for the gander.
- Dore RK. How to prevent glucocorticoid-induced osteoporosis. Cleve Clin J Med 2010; 77:529–536.
- Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women. Principal results from the Women’s Health Initiative Randomized Controlled Trial. JAMA 2002; 288:321–333.
- Estrogen and progestogen use in postmenopausal women: 2010 position statement of The North American Menopause Society. Menopause 2010. www.menopause.org. Accessed October 28, 2010.
- American Association of Clinical Endocrinologists. Position statement on hormone replacement therapy and cardiovascular risk. www.aace.com/pub/pdf/guidelines/HRTCVRISKposition_statement.pdf. Accessed October 28, 2010.
- Compston JE. The risks and benefits of HRT. J Musculo-skelet Neuronal Interact 2004; 4:187–190.
To the Editor: I have to say I am disappointed, but not surprised, at Dr. Dore’s article, “How to prevent glucocorticoid-induced osteoporosis” in your August issue.1 The section “Estrogen is being used more selectively” was shorter and had older and out of date references compared with the section “A role for testosterone?” and it was actually blatantly sexist: the comment in the estrogen section is that “…the consensus…that hormone replacement therapy should be restricted to women with menopausal symptoms or to older women who cannot tolerate other therapies or who express a strong preference for hormone replacement therapy despite being informed about potential adverse events” [my italics],1 while the comment in the testosterone section is that males who “… are hypogonadal, and have no contraindications to androgen replacement therapy (eg, prostate cancer) be offered testosterone therapy to preserve lean body mass and bone mineral density” [my italics].1
While I am not arguing that menopausal hormone therapy should be used first-line for the prevention or treatment of glucocorticoid-induced osteoporosis, I would like to note the following:
First, the referenced 2002 Women’s Health Initiative study2 was a prevention trial, not a therapeutic menopausal trial, and to reference it as a position statement on the use of hormone therapy is ridiculous and perpetuates misinformation about the role of menopausal hormone therapy.
Next, there has been updated information from the Women’s Health Initiative, as well as updated position statements on the use of hormone therapy—the 2010 position statement on the use of estrogen and progestogen in menopausal women3 as well as the 2008 American Association of Clinical Endocrinologists position statement4 noting that the benefits of hormone therapy outweigh the risks for most women under age 60. So Dr. Dore’s reference citation from 20045 is hopelessly outdated.
And lastly, females, unlike males, routinely become hypogonadal at midlife. When faced with a medical condition that requires glucocorticoids that further intensifies the hypogonadal state by suppressing adrenal adrenogens, females may face a “triple whammy” on the bone.
The Women’s Health Initiative actually showed fracture reduction in postmenopausal women who did not even carry the diagnosis of osteoporosis, while the referenced studies in Dr. Dore’s article related to males admittedly “cannot be considered conclusive in view of their small size and the lack of fracture data…”1
So what is bad (actually potentially good) for the goose is apparently just fine for the gander.
To the Editor: I have to say I am disappointed, but not surprised, at Dr. Dore’s article, “How to prevent glucocorticoid-induced osteoporosis” in your August issue.1 The section “Estrogen is being used more selectively” was shorter and had older and out of date references compared with the section “A role for testosterone?” and it was actually blatantly sexist: the comment in the estrogen section is that “…the consensus…that hormone replacement therapy should be restricted to women with menopausal symptoms or to older women who cannot tolerate other therapies or who express a strong preference for hormone replacement therapy despite being informed about potential adverse events” [my italics],1 while the comment in the testosterone section is that males who “… are hypogonadal, and have no contraindications to androgen replacement therapy (eg, prostate cancer) be offered testosterone therapy to preserve lean body mass and bone mineral density” [my italics].1
While I am not arguing that menopausal hormone therapy should be used first-line for the prevention or treatment of glucocorticoid-induced osteoporosis, I would like to note the following:
First, the referenced 2002 Women’s Health Initiative study2 was a prevention trial, not a therapeutic menopausal trial, and to reference it as a position statement on the use of hormone therapy is ridiculous and perpetuates misinformation about the role of menopausal hormone therapy.
Next, there has been updated information from the Women’s Health Initiative, as well as updated position statements on the use of hormone therapy—the 2010 position statement on the use of estrogen and progestogen in menopausal women3 as well as the 2008 American Association of Clinical Endocrinologists position statement4 noting that the benefits of hormone therapy outweigh the risks for most women under age 60. So Dr. Dore’s reference citation from 20045 is hopelessly outdated.
And lastly, females, unlike males, routinely become hypogonadal at midlife. When faced with a medical condition that requires glucocorticoids that further intensifies the hypogonadal state by suppressing adrenal adrenogens, females may face a “triple whammy” on the bone.
The Women’s Health Initiative actually showed fracture reduction in postmenopausal women who did not even carry the diagnosis of osteoporosis, while the referenced studies in Dr. Dore’s article related to males admittedly “cannot be considered conclusive in view of their small size and the lack of fracture data…”1
So what is bad (actually potentially good) for the goose is apparently just fine for the gander.
- Dore RK. How to prevent glucocorticoid-induced osteoporosis. Cleve Clin J Med 2010; 77:529–536.
- Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women. Principal results from the Women’s Health Initiative Randomized Controlled Trial. JAMA 2002; 288:321–333.
- Estrogen and progestogen use in postmenopausal women: 2010 position statement of The North American Menopause Society. Menopause 2010. www.menopause.org. Accessed October 28, 2010.
- American Association of Clinical Endocrinologists. Position statement on hormone replacement therapy and cardiovascular risk. www.aace.com/pub/pdf/guidelines/HRTCVRISKposition_statement.pdf. Accessed October 28, 2010.
- Compston JE. The risks and benefits of HRT. J Musculo-skelet Neuronal Interact 2004; 4:187–190.
- Dore RK. How to prevent glucocorticoid-induced osteoporosis. Cleve Clin J Med 2010; 77:529–536.
- Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women. Principal results from the Women’s Health Initiative Randomized Controlled Trial. JAMA 2002; 288:321–333.
- Estrogen and progestogen use in postmenopausal women: 2010 position statement of The North American Menopause Society. Menopause 2010. www.menopause.org. Accessed October 28, 2010.
- American Association of Clinical Endocrinologists. Position statement on hormone replacement therapy and cardiovascular risk. www.aace.com/pub/pdf/guidelines/HRTCVRISKposition_statement.pdf. Accessed October 28, 2010.
- Compston JE. The risks and benefits of HRT. J Musculo-skelet Neuronal Interact 2004; 4:187–190.
In reply: Glucocorticoid-induced osteoporosis
In Reply: I could find references for the use of testosterone in glucocorticoid-induced osteoporosis and could not find any references for the use of estrogen in this condition, except for the outdated American College of Rheumatology guidelines from the 1990s, which included Dr. Nancy Lane’s work. So perhaps it is the research that is gender-biased rather than my article. I agree that in osteoporosis that is not glucocorticoid-induced, estrogen has great fracture efficacy even in those without osteoporosis, as you stated, but I tried to keep my article evidence-based and on-topic regarding glucocorticoid-induced osteoporosis. As usual, topics that involve estrogen are highly volatile, and I did not mean to fuel the fire.
In Reply: I could find references for the use of testosterone in glucocorticoid-induced osteoporosis and could not find any references for the use of estrogen in this condition, except for the outdated American College of Rheumatology guidelines from the 1990s, which included Dr. Nancy Lane’s work. So perhaps it is the research that is gender-biased rather than my article. I agree that in osteoporosis that is not glucocorticoid-induced, estrogen has great fracture efficacy even in those without osteoporosis, as you stated, but I tried to keep my article evidence-based and on-topic regarding glucocorticoid-induced osteoporosis. As usual, topics that involve estrogen are highly volatile, and I did not mean to fuel the fire.
In Reply: I could find references for the use of testosterone in glucocorticoid-induced osteoporosis and could not find any references for the use of estrogen in this condition, except for the outdated American College of Rheumatology guidelines from the 1990s, which included Dr. Nancy Lane’s work. So perhaps it is the research that is gender-biased rather than my article. I agree that in osteoporosis that is not glucocorticoid-induced, estrogen has great fracture efficacy even in those without osteoporosis, as you stated, but I tried to keep my article evidence-based and on-topic regarding glucocorticoid-induced osteoporosis. As usual, topics that involve estrogen are highly volatile, and I did not mean to fuel the fire.