User login
Breaking the cycle of medication overuse headache
Some migraine patients fall into a trap by overusing the medications they take when they get their headaches, ending in a downward spiral of daily or near-daily headaches for which their medications become less and less effective.
This condition, called medication overuse headache, makes for a poor quality of life. It is often associated with nonrestorative sleep, neck pain, and vasomotor instability. Comorbid depression and anxiety are common and may complicate treatment. (Depression and anxiety, however, do not cause daily headaches.) Patients can also suffer from the physiologic and psychological consequences of the overused medications.
Fortunately, we can break the cycle.1,2 Treatment involves completely weaning the patient from the overused medications and educating her or him to follow a new regimen of prophylaxis and acute treatment with clear limits on frequency of use. Nondrug treatments such as relaxation therapy, biofeedback, and cognitive behavioral therapy can be useful adjuncts.
CROSSING THE LINE: 15 HEADACHE DAYS A MONTH
Chronic daily headache
We define chronic daily headache as occurring on at least 15 days per month for at least 3 months in a row and lasting at least 4 hours if untreated.
Most patients start with episodic migraine, and many of them remember the period of transformation. Crossing the 15-day-per-month threshold changes the clinical presentation, prognosis, and treatment, all for the worse.
In a large population-based study,3 2.5% of patients who began with episodic migraine (headaches on fewer than 15 days per month) had “transformed migraine” (headaches on 15 or more days per month) 1 year later. The prevalence of chronic daily headache is almost 5% of the general population and may account for up to 70% of the initial diagnoses seen in headache centers.
The closer a patient is to having 15 headaches per month, the more likely she or he will cross the line.4,5 Katsarava and colleagues5 followed patients for 1 year in a neurology clinic in Germany and found that those starting the year with 6 to 9 headache days per month were 6.2 times more likely to develop chronic daily headache in the next year than those who began the year with 0 to 4 per month—and those starting with 10 to 14 headaches per month were 20 times more likely.
Medication overuse headache
Medication overuse headache is a subset of chronic daily headache, also occurring on 15 or more days per month but with the added criterion of medication overuse, ie, regular overuse for more than 3 months of at least one acute treatment drug:
- Ergotamine, triptans, opioids, or combination analgesic medications on 10 or more days per month on a regular basis for more than 3 months, or
- Simple analgesics or any combination of ergotamine, triptans, analgesics, or opioids on 15 or more days per month on a regular basis for more than 3 months without overuse of any single class alone.
Another criterion is that the patient’s headaches must worsen in some way (usually frequency) as the use of acute medications becomes more frequent.6,7
Medication overuse headache is the most common form of secondary chronic daily headache seen in headache practice,8–10 and probably accounts for about half of cases of chronic daily headache.11–13
Different terminology confuses the issue
Many terms have been used to describe medication overuse headache in the past, such as analgesic-rebound headache (or just rebound headache), transformed migraine with medication overuse, and even chronic migraine. The lack of uniformity in terminology makes for confusion in the literature and difficulty in communicating with patients and colleagues. Some authors mean medication overuse headache when they say chronic daily headache.
Complicating this diagnostic confusion is a debate as to whether chronic daily headache in general should be treated as a primary or secondary headache disorder. Some European headache specialists insist on a strict division between primary and secondary daily headaches, and medication overuse headache is one of the latter. Many American specialists believe that chronic daily headache is a collective description or phenotype rather than a diagnostic category, and that it is usually associated with and exacerbated by medication overuse.14,15 The International Classification of Headache Disorders uses the term “chronic migraine” for primary daily headache, and “medication overuse headache” for secondary daily headache or rebound.
Many American specialists approach the disorder clinically, treating chronic daily headache in the same way regardless of whether there is medication overuse. They cite randomized controlled trials of topiramate (Topamax) and onabotulinum toxin type A (Botox) that reported comparable benefit with these medications in patients with chronic daily headache with or without medication overuse.16–18
MORE IS LESS: THE PARADOX OF TREATING ACUTE HEADACHE
The clinical paradox and dilemma of treating acute episodic migraine is that more is less: the more days of acute treatment, the less well the migraines are controlled. And thus, the patient is likely to veer out of control.3
The compassion that motivates us to prescribe medications for acute episodic migraine must be tempered by the realization that too much of a good thing will result in its malignant transformation to medication overuse headache. Once this develops, preventive and migrainespecific acute medications are less effective, and patients need far more complex interventions.
Complicating the dilemma, acute migraine-specific medications such as triptans and dihydroergotamine (Migranal) work better when taken early in migraine attacks, before central sensitization and allodynia develop with attendant photophonophobia and sensitivity to other stimuli. On the other hand, overuse will lead to medication overuse headache.
SYMPTOMS VARY
The symptoms of medication overuse headache vary in frequency, severity, location, quality, and associated features, both among patients and in the same patient. This is because the disease itself varies and also because of differences in the type and frequency of medication intake. Still, some features help to define this problem, and failing to recognize them may account for a widely held clinical feeling that these patients are “difficult.”
History of episodic migraine. Generally, medication overuse headache does not occur in nonmigraineurs.
Headache on most days of the month. Whenever a migraineur starts having headaches on more days than not, the diagnosis of medication overuse should be considered.
Overuse of acute medications. The criteria (see above) allow for combining days of acute medication use. For example, if a patient takes a combination analgesic on 5 days and a triptan on 5 different days, that would still be enough days of acute treatment to trigger medication overuse headache.
Variable pain location is a particular characteristic of medication overuse headache. Although the location may differ from day to day (front or back, rostral or caudal, unilateral or bilateral), it is the quantity not the quality or location of the headaches that suggests the diagnosis.
A drug-dependent rhythm. Predictably, the headaches come on in the early morning or awaken the patient from sleep. This may be due to variable drug withdrawal.
Neck pain. Medication overuse headache frequently involves the neck, and patients often seek and receive treatments such as muscle relaxants or injections to the neck. When patients are weaned from their acute migraine medications, neck pain generally dissipates. The neck pain, however, can recur episodically with their remaining, now-episodic acute migraines. Neck pain associated with medication overuse headache is not usually a sign of a primary neck disorder; rather, it is a symptom of medication overuse headache itself.
Concomitant depression and anxiety are comorbid with episodic migraine, but appear to be more common with medication overuse headache. Treating the depression or anxiety does not restore an episodic pattern of migraine; weaning from the overused medications remains the most important intervention. A frequent clinical error is to diagnose and treat the psychiatric issues without recognizing medication overuse as the primary problem.
Nonrestorative sleep is almost always reported by patients with medication overuse headache. This is often due to the caffeine contained in combination analgesics or to excessive dietary caffeine intake, but it may also be part of the daily acute drug withdrawal syndrome. The sleep problems are also associated with the concomitant depression. Sleep often improves after weaning from the offending substance or substances. As with neck pain, patients do not have a primary sleep disorder—the sleep disturbance is a symptom of medication overuse headache.
Vasomotor instability. Autonomic features are commonly associated with medication overuse headache. Rhinorrhea, nasal stuffiness, and lacrimation are features of medication withdrawal, especially from opioids, and are frequently attributed to sinus disease or “sinus headaches.” Many patients undergo unnecessary sinus procedures or are given antibiotics, decongestants, and other wrong medications for incorrect diagnoses. Decongestants can cause and exacerbate medication overuse headache, so they need to be withdrawn. The sinus features generally remit when the overused migraine medications are eliminated.
Preventive medications are less effective or ineffective until the acute medications are withdrawn. Thus, prescribing prevention without weaning is usually futile, and the patients are often dismissed as having a refractory problem. At the same time, migraine-specific acute treatments, ie, triptans and ergots, are usually also less effective. When patients complain that “nothing works,” either preventively or acutely, medication overuse headache should spring to mind.
Weaning from overused medications can restore the efficacy of previously ineffective treatments at the same time that a patient is restored to an episodic headache pattern. Thus, complete weaning is the pivotal clinical intervention. Clinically, there is no spontaneous remission from rebound without absolute detoxification, maintained for months.9,19–22
Other diagnoses entertained. The more diagnoses suggested for daily headache, and the more treatments tried unsuccessfully, the more likely the diagnosis is actually medication overuse headache. Because this condition is protean, patients and caregivers alike make more and more fanciful diagnoses such as allergies, cervicogenic headache, temperomandibular disorder, occipital neuralgia, chronic Lyme disease, and systemic candidiasis. A useful strategy is to assume that daily headache is likely due to medication overuse. And since medication overuse headache is generally treatable, patients labeled as having refractory headaches often are dramatically improved by appropriate intervention.
WHY ARE MIGRAINEURS SO SUSCEPTIBLE?
Medication overuse headache occurs primarily in people with a history of episodic migraine, but the unique susceptibility of migraineurs is not fully understood.
Structural changes in the brain?
Episodic migraine attacks appear to be generated in the upper brainstem. This region in turn activates a set of peripheral pain mechanisms, ie, meningeal inflammation and vasodilation. The peripheral pain processes turn on afferent circuits that carry the pain signals to the lower brainstem, where these signals are integrated. Finally, the central signals ascend the brainstem, stimulating autonomic nuclei that account for nausea and other vasomotor changes, proceed through the thalamus, and terminate in the cortex where pain is perceived. Thus, migraine without aura consists of three steps—a central generator, a set of peripheral pain mechanisms, and a series of steps culminating in central integration. (Aura involves other steps, not outlined here.)
A possible explanation of why migraine becomes chronic is that a yo-yo effect of repeated migrainous pain processes, followed by repeated medication, results in structural changes. These propagate central sensitization with a lowered threshold for activation of all of the central processing of head pain.
This set of disturbances may occur due to undertreatment of migraine pain. With inadequate pain control, headaches recur, and the process repeats until damage occurs. Evidence for this is seen in up-regulation of excitatory serotonin receptors when analgesics are repetitively given to laboratory animals.23
A pure withdrawal phenomenon?
Also possible is that medication overuse headache is just a complex dependence-and-withdrawal phenomenon. Thus, the cyclical use of various medications results in withdrawal headaches and a set of symptoms, including disturbed sleep, morning headache, and vasomotor signs of withdrawal. Arguing against its being a pure withdrawal phenomenon is that daily use of analgesics or opioids generally does not cause daily headache in nonmigraineurs.24
HOW MUCH MEDICATION USE IS TOO MUCH?
For an episodic migraine condition to transform into a chronic one, medications need to be taken on only a modest number of days per month: 5 to 10, depending on the type of medication.
A pivotal study3 found that butalbital combinations were most likely to cause medication overuse headache, needing to be taken on merely 5 or more days per month to cause it in migraineurs. Opioids caused it if taken 8 or more days per month, and triptans if taken 10 or more days per month. Nonsteroidal anti-inflammatory drugs (NSAIDs) actually protected against transformation to daily headache if used 5 or fewer days per month, but caused medication overuse headache if used 10 or more days per month.
Thus, there was a hierarchy of risk, with butalbital being the worst, opioids in the middle, and NSAIDs and triptans the least risky. None of the agents had to be taken daily to trigger medication overuse headache.
PREVENTION IS THE BEST TREATMENT
The best approach to medication overuse headache is to prevent it while the patient still has episodic migraine.
Outcomes are better with triptans or ergots
Undertreatment of migraine leads quickly to overuse of symptomatic medications, and from there to medication overuse headache.
Outcomes of episodic migraine are better when triptans or ergots (which are migrainespecific) are used first-line in patients with disabling migraine and no vascular contraindications. Patients who start with nonspecific treatment and step up to a more specific treatment when lower-level medications fail have less favorable outcomes in terms of migraine relief and disability time than those treated with triptans from the beginning.25
To put this in perspective, if a patient takes an acute medication, gets only partial relief (not a pain-free response) at 2 hours and then takes another pill, or gets a recurrence and takes another pill, the likelihood of prolonging an attack and using more medications goes up. If a patient takes a triptan and gets a sustained pain-free response, the attack is truncated and the medication usage reduced. Therefore, migraine-specific acute treatments are more likely to not be overused.
Daily preventive medication, if necessary
As noted above, if the number of headache days exceeds 10 per month, the likelihood of developing daily headache escalates steeply. Thus, patients with 10 or more days of headache per month should be prescribed preventive medications to be taken daily to reduce the frequency, severity, and duration of attacks. Preventive treatment may also increase the efficacy of the acute treatments.
The drugs used for preventive treatment are different than those used for acute treatment and are not likely to cause medication rebound headache. However, they are not very effective. Those that have the best evidence of efficacy are beta-blockers, tricyclic antidepressants, and anticonvulsants; calcium channel blockers and NSAIDs are also popular. This topic has been reviewed in detail elsewhere.26,27
REVERSING MEDICATION OVERUSE HEADACHE
If a patient already has medication overuse headache, the clinician is faced with the problem of weaning her or him from the overused medication while establishing a reasonable regimen of prophylaxis and acute medications with limits.
For the most part, these tasks can be accomplished in a series of clinic visits. However, some patients have such severe comorbid medical and psychiatric illnesses that outpatient treatment is impossible. For them, a day hospital or inpatient program with infusion capabilities is often useful.
Outpatient treatment of medication overuse headache
Outpatient treatment of medication overuse headache involves:
Educating patients about the genesis of the problem and reassuring them that you are not accusing them of being an addict. Most patients who develop medication overuse headache are habituated inadvertently, and this needs to be made clear, along with the overall plan and the likely prognosis.

Establishing daily preventive medications. The prophylactic regimen can be established either before or during the weaning.
Providing acute medications, with limits. At a certain point in the weaning, advise the patient not to treat low-level headaches, and provide a triptan or dihydroergotamine to use for severe attacks, no more than twice weekly and less than 10 days per month. If the patient is in triptan rebound, dihydroergotamine would be the choice.
Instructing the patient to keep a headache diary to follow adherence and outcomes.
Psychology consultation can be very helpful to teach patients behavioral techniques to deal with anticipatory anxiety during the weaning.
Multidisciplinary programs with infusion capability
Some patients need a more intensive approach to restore an episodic migraine pattern. Examples: those on very high doses of narcotics or barbiturates, those with comorbid medical illnesses that limit both acute and preventive treatments, and those with severe and complicating comorbid psychiatric illnesses.
Multidisciplinary programs are available, with specialists in neurology, primary care, psychology, and physical and occupational therapy providing treatment. Patients check into the hospital or a “day hospital,” where they can also receive intravenous infusions to get through the weaning. The goal is to shift the locus of control back to patients as they revert from daily headache to episodic migraine. Patient education is crucial.
OUTCOMES ARE GOOD
There is much good news about medication overuse headache.
It can be prevented with careful monitoring of acute medication outcomes and number of headache days. Prophylaxis should be used when treating high-frequency or very disabling migraine.
Most patients improve when weaned and treated with preventive medications. “Recovery” means at least 3 months off the overused medications. In studies, more than half of patients who underwent treatment for medication overuse headache remained better and had an episodic pattern of headache 5 years later.26
Unfortunately, the initial improvement often seen with patients after weaning and being given preventive medication (72%–85% of patients improve) in the first year is often followed by preventable relapse, so it is very important to follow up with patients regularly. 28–32
Helping restore a patient’s quality of life is an outcome rewarding to primary care provider and specialist alike.
- Schwartz BS, Stewart WF, Lipton RB. Lost workdays and decreased work effectiveness associated with headache in the workplace. J Occup Environ Med 1997; 39:320–327.
- Meletiche DM, Lofland JH, Young WB. Quality-of-life differences between patients with episodic and transformed migraine. Headache 2001; 41:573–578.
- Bigal ME, Serrano D, Buse D, Scher A, Stewart WF, Lipton RB. Acute migraine medications and evolution from episodic to chronic migraine: a longitudinal populationbased study. Headache 2008; 48:1157–1168.
- Scher AI, Stewart WF, Ricci JA, Lipton RB. Factors associated with the onset and remission of chronic daily headache in a population-based study. Pain 2003; 106:81–89.
- Katsarava Z, Schneeweiss S, Kurth T, et al Incidence and predictors for chronicity of headache in patients with episodic migraine. Neurology 2004; 62:788–790.
- Olesen J, Bousser MG, Diener HC, et al., Headache Classification Committee New appendix criteria open for a broader concept of chronic migraine. Cephalalgia 2006; 26:742–746.
- Bigal M, Rapoport A, Sheftell F, Tepper S, Lipton R. The international classification of headache disorders revised criteria for chronic migraine—field testing in a headache specialty clinic. Cephalalgia 2007; 27:230–234.
- Rapoport A, Stang P, Gutterman DL, et al Analgesic rebound headache in clinical practice: data from a physician survey. Headache 1996; 36:14–19.
- Mathew NT. Transformed migraine, analgesic rebound, and other chronic daily headaches. Neurol Clin 1997; 15:167–186.
- Bigal ME, Sheftell FD, Rapoport AM, Tepper SJ, Lipton RB. Chronic daily headache: identification of factors associated with induction and transformation. Headache 2002; 42:575–581.
- Silberstein SD. Tension-type and chronic daily headache. Neurology 1993; 43:1644–1649.
- Castillo J, Muñoz P, Guitera V, Pascual J. Kaplan Award 1998. Epidemiology of chronic daily headache in the general population. Headache 1999; 39:190–196.
- Scher AI, Stewart WF, Liberman J, Lipton RB. Prevalence of frequent headache in a population sample. Headache 1998; 38:497–506.
- Headache Classification Subcommittee of the International Headache Society. The international classification of headache disorders: 2nd edition. Cephalalgia 2004; 24( suppl 1):9–160.
- Silberstein SD, Lipton RB, Sliwinski M. Classification of daily and near-daily headaches: field trial of revised IHS criteria. Neurology 1996; 47:871–875.
- Diener HC, Bussone G, Van Oene JC, Lahaye M, Schwalen S, Goadsby PJTOPMAT-MIG-201 (TOP-CHROME) Study Group. Topiramate reduces headache days in chronic migraine: a randomized, double-blind, placebo-controlled study. Cephalalgia 2007; 27:814–823.
- Silberstein SD, Lipton RB, Dodick DW, et al., Topiramate Chronic Migraine Study Group. Efficacy and safety of topiramate for the treatment of chronic migraine: a randomized, double-blind, placebo-controlled trial. Headache 2007; 47:170–180.
- Silberstein SD, Blumenfeld AM, Cady RK, et al Botulinum neurotoxin type A for the treatment of chronic migraine: analysis of the PREEMPT chronic migraine subgroup with baseline acute headache medication overuse. Cephalalgia 2009; 29( suppl 1):1–176.
- Mathew NT. Medication misuse headache. Cephalalgia 1998; 18( suppl 21):34–36.
- Diener HC, Katasarva Z. Analgesic/abortive overuse and misuse in chronic daily headache. Curr Pain Headache Rep 2001; 5:545–550.
- Tepper SJ, Rapoport AM, Sheftell FD, Bigal ME. Chronic daily headache: an update. Headache Care 2004; 1:233–245.
- Dodick DW. Clinical practice. Chronic daily headache. N Engl J Med 2006; 354:158–165.
- Srikiatkhachorn A, Tarasub N, Govitrapong P. Effect of chronic analgesic exposure on the central serotonin system: a possible mechanism of analgesic abuse headache. Headache 2000; 40:343–350.
- Wilkinson SM, Becker WJ, Heine JA. Opiate use to control bowel motility may induce chronic daily headache in patients with migraine. Headache 2001; 41:303–309.
- Lipton RB, Stewart WF, Stone AM, Láinez MJ, Sawyer JPDisability in Strategies of Care Study group. Stratified care vs step care strategies for migraine: the Disability in Strategies of Care (DISC) Study: a randomized trial. JAMA 2000; 284:2599–605.
- Bamford CC, Tepper SJ. Daily pharmacologic prophylaxis of episodic migraine. Tech Regional Anesthesia Pain Manage 2009; 13:20–37.
- Tepper SJ. Preventive pharmacologic treatment of migraine and tension-type headache. In:Levin M, editor. Comprehensive Review of Headache Medicine. Oxford: Oxford University Press, 2008:231–234.
- Zed PJ, Loewen PS, Robinson G. Medication-induced headache: overview and systematic review of therapeutic approaches. Ann Pharmacother 1999; 33:61–72.
- Fritsche G, Eberl A, Katsarava Z, Limmroth V, Diener HC. Drug-induced headache: long-term follow-up of withdrawal therapy and persistence of drug misuse. Eur Neurol 2001; 45:229–235.
- Pini LA, Cicero AF, Sandrini M. Long-term follow-up of patients treated for chronic headache with analgesic overuse. Cephalalgia 2001; 21:878–883.
- Katsarava Z, Limmroth V, Finke M, Diener HC, Fritsche G. Rates and predictors for relapse in medication overuse headache: a 1-year prospective study. Neurology 2003; 60:1682–1683.
- Zidverc-Trajkovic J, Pekmezovic T, Jovanovic Z, et al Medication overuse headache: clinical features predicting treatment outcome at 1-year follow-up. Cephalalgia 2007; 27:1219–1225.
Some migraine patients fall into a trap by overusing the medications they take when they get their headaches, ending in a downward spiral of daily or near-daily headaches for which their medications become less and less effective.
This condition, called medication overuse headache, makes for a poor quality of life. It is often associated with nonrestorative sleep, neck pain, and vasomotor instability. Comorbid depression and anxiety are common and may complicate treatment. (Depression and anxiety, however, do not cause daily headaches.) Patients can also suffer from the physiologic and psychological consequences of the overused medications.
Fortunately, we can break the cycle.1,2 Treatment involves completely weaning the patient from the overused medications and educating her or him to follow a new regimen of prophylaxis and acute treatment with clear limits on frequency of use. Nondrug treatments such as relaxation therapy, biofeedback, and cognitive behavioral therapy can be useful adjuncts.
CROSSING THE LINE: 15 HEADACHE DAYS A MONTH
Chronic daily headache
We define chronic daily headache as occurring on at least 15 days per month for at least 3 months in a row and lasting at least 4 hours if untreated.
Most patients start with episodic migraine, and many of them remember the period of transformation. Crossing the 15-day-per-month threshold changes the clinical presentation, prognosis, and treatment, all for the worse.
In a large population-based study,3 2.5% of patients who began with episodic migraine (headaches on fewer than 15 days per month) had “transformed migraine” (headaches on 15 or more days per month) 1 year later. The prevalence of chronic daily headache is almost 5% of the general population and may account for up to 70% of the initial diagnoses seen in headache centers.
The closer a patient is to having 15 headaches per month, the more likely she or he will cross the line.4,5 Katsarava and colleagues5 followed patients for 1 year in a neurology clinic in Germany and found that those starting the year with 6 to 9 headache days per month were 6.2 times more likely to develop chronic daily headache in the next year than those who began the year with 0 to 4 per month—and those starting with 10 to 14 headaches per month were 20 times more likely.
Medication overuse headache
Medication overuse headache is a subset of chronic daily headache, also occurring on 15 or more days per month but with the added criterion of medication overuse, ie, regular overuse for more than 3 months of at least one acute treatment drug:
- Ergotamine, triptans, opioids, or combination analgesic medications on 10 or more days per month on a regular basis for more than 3 months, or
- Simple analgesics or any combination of ergotamine, triptans, analgesics, or opioids on 15 or more days per month on a regular basis for more than 3 months without overuse of any single class alone.
Another criterion is that the patient’s headaches must worsen in some way (usually frequency) as the use of acute medications becomes more frequent.6,7
Medication overuse headache is the most common form of secondary chronic daily headache seen in headache practice,8–10 and probably accounts for about half of cases of chronic daily headache.11–13
Different terminology confuses the issue
Many terms have been used to describe medication overuse headache in the past, such as analgesic-rebound headache (or just rebound headache), transformed migraine with medication overuse, and even chronic migraine. The lack of uniformity in terminology makes for confusion in the literature and difficulty in communicating with patients and colleagues. Some authors mean medication overuse headache when they say chronic daily headache.
Complicating this diagnostic confusion is a debate as to whether chronic daily headache in general should be treated as a primary or secondary headache disorder. Some European headache specialists insist on a strict division between primary and secondary daily headaches, and medication overuse headache is one of the latter. Many American specialists believe that chronic daily headache is a collective description or phenotype rather than a diagnostic category, and that it is usually associated with and exacerbated by medication overuse.14,15 The International Classification of Headache Disorders uses the term “chronic migraine” for primary daily headache, and “medication overuse headache” for secondary daily headache or rebound.
Many American specialists approach the disorder clinically, treating chronic daily headache in the same way regardless of whether there is medication overuse. They cite randomized controlled trials of topiramate (Topamax) and onabotulinum toxin type A (Botox) that reported comparable benefit with these medications in patients with chronic daily headache with or without medication overuse.16–18
MORE IS LESS: THE PARADOX OF TREATING ACUTE HEADACHE
The clinical paradox and dilemma of treating acute episodic migraine is that more is less: the more days of acute treatment, the less well the migraines are controlled. And thus, the patient is likely to veer out of control.3
The compassion that motivates us to prescribe medications for acute episodic migraine must be tempered by the realization that too much of a good thing will result in its malignant transformation to medication overuse headache. Once this develops, preventive and migrainespecific acute medications are less effective, and patients need far more complex interventions.
Complicating the dilemma, acute migraine-specific medications such as triptans and dihydroergotamine (Migranal) work better when taken early in migraine attacks, before central sensitization and allodynia develop with attendant photophonophobia and sensitivity to other stimuli. On the other hand, overuse will lead to medication overuse headache.
SYMPTOMS VARY
The symptoms of medication overuse headache vary in frequency, severity, location, quality, and associated features, both among patients and in the same patient. This is because the disease itself varies and also because of differences in the type and frequency of medication intake. Still, some features help to define this problem, and failing to recognize them may account for a widely held clinical feeling that these patients are “difficult.”
History of episodic migraine. Generally, medication overuse headache does not occur in nonmigraineurs.
Headache on most days of the month. Whenever a migraineur starts having headaches on more days than not, the diagnosis of medication overuse should be considered.
Overuse of acute medications. The criteria (see above) allow for combining days of acute medication use. For example, if a patient takes a combination analgesic on 5 days and a triptan on 5 different days, that would still be enough days of acute treatment to trigger medication overuse headache.
Variable pain location is a particular characteristic of medication overuse headache. Although the location may differ from day to day (front or back, rostral or caudal, unilateral or bilateral), it is the quantity not the quality or location of the headaches that suggests the diagnosis.
A drug-dependent rhythm. Predictably, the headaches come on in the early morning or awaken the patient from sleep. This may be due to variable drug withdrawal.
Neck pain. Medication overuse headache frequently involves the neck, and patients often seek and receive treatments such as muscle relaxants or injections to the neck. When patients are weaned from their acute migraine medications, neck pain generally dissipates. The neck pain, however, can recur episodically with their remaining, now-episodic acute migraines. Neck pain associated with medication overuse headache is not usually a sign of a primary neck disorder; rather, it is a symptom of medication overuse headache itself.
Concomitant depression and anxiety are comorbid with episodic migraine, but appear to be more common with medication overuse headache. Treating the depression or anxiety does not restore an episodic pattern of migraine; weaning from the overused medications remains the most important intervention. A frequent clinical error is to diagnose and treat the psychiatric issues without recognizing medication overuse as the primary problem.
Nonrestorative sleep is almost always reported by patients with medication overuse headache. This is often due to the caffeine contained in combination analgesics or to excessive dietary caffeine intake, but it may also be part of the daily acute drug withdrawal syndrome. The sleep problems are also associated with the concomitant depression. Sleep often improves after weaning from the offending substance or substances. As with neck pain, patients do not have a primary sleep disorder—the sleep disturbance is a symptom of medication overuse headache.
Vasomotor instability. Autonomic features are commonly associated with medication overuse headache. Rhinorrhea, nasal stuffiness, and lacrimation are features of medication withdrawal, especially from opioids, and are frequently attributed to sinus disease or “sinus headaches.” Many patients undergo unnecessary sinus procedures or are given antibiotics, decongestants, and other wrong medications for incorrect diagnoses. Decongestants can cause and exacerbate medication overuse headache, so they need to be withdrawn. The sinus features generally remit when the overused migraine medications are eliminated.
Preventive medications are less effective or ineffective until the acute medications are withdrawn. Thus, prescribing prevention without weaning is usually futile, and the patients are often dismissed as having a refractory problem. At the same time, migraine-specific acute treatments, ie, triptans and ergots, are usually also less effective. When patients complain that “nothing works,” either preventively or acutely, medication overuse headache should spring to mind.
Weaning from overused medications can restore the efficacy of previously ineffective treatments at the same time that a patient is restored to an episodic headache pattern. Thus, complete weaning is the pivotal clinical intervention. Clinically, there is no spontaneous remission from rebound without absolute detoxification, maintained for months.9,19–22
Other diagnoses entertained. The more diagnoses suggested for daily headache, and the more treatments tried unsuccessfully, the more likely the diagnosis is actually medication overuse headache. Because this condition is protean, patients and caregivers alike make more and more fanciful diagnoses such as allergies, cervicogenic headache, temperomandibular disorder, occipital neuralgia, chronic Lyme disease, and systemic candidiasis. A useful strategy is to assume that daily headache is likely due to medication overuse. And since medication overuse headache is generally treatable, patients labeled as having refractory headaches often are dramatically improved by appropriate intervention.
WHY ARE MIGRAINEURS SO SUSCEPTIBLE?
Medication overuse headache occurs primarily in people with a history of episodic migraine, but the unique susceptibility of migraineurs is not fully understood.
Structural changes in the brain?
Episodic migraine attacks appear to be generated in the upper brainstem. This region in turn activates a set of peripheral pain mechanisms, ie, meningeal inflammation and vasodilation. The peripheral pain processes turn on afferent circuits that carry the pain signals to the lower brainstem, where these signals are integrated. Finally, the central signals ascend the brainstem, stimulating autonomic nuclei that account for nausea and other vasomotor changes, proceed through the thalamus, and terminate in the cortex where pain is perceived. Thus, migraine without aura consists of three steps—a central generator, a set of peripheral pain mechanisms, and a series of steps culminating in central integration. (Aura involves other steps, not outlined here.)
A possible explanation of why migraine becomes chronic is that a yo-yo effect of repeated migrainous pain processes, followed by repeated medication, results in structural changes. These propagate central sensitization with a lowered threshold for activation of all of the central processing of head pain.
This set of disturbances may occur due to undertreatment of migraine pain. With inadequate pain control, headaches recur, and the process repeats until damage occurs. Evidence for this is seen in up-regulation of excitatory serotonin receptors when analgesics are repetitively given to laboratory animals.23
A pure withdrawal phenomenon?
Also possible is that medication overuse headache is just a complex dependence-and-withdrawal phenomenon. Thus, the cyclical use of various medications results in withdrawal headaches and a set of symptoms, including disturbed sleep, morning headache, and vasomotor signs of withdrawal. Arguing against its being a pure withdrawal phenomenon is that daily use of analgesics or opioids generally does not cause daily headache in nonmigraineurs.24
HOW MUCH MEDICATION USE IS TOO MUCH?
For an episodic migraine condition to transform into a chronic one, medications need to be taken on only a modest number of days per month: 5 to 10, depending on the type of medication.
A pivotal study3 found that butalbital combinations were most likely to cause medication overuse headache, needing to be taken on merely 5 or more days per month to cause it in migraineurs. Opioids caused it if taken 8 or more days per month, and triptans if taken 10 or more days per month. Nonsteroidal anti-inflammatory drugs (NSAIDs) actually protected against transformation to daily headache if used 5 or fewer days per month, but caused medication overuse headache if used 10 or more days per month.
Thus, there was a hierarchy of risk, with butalbital being the worst, opioids in the middle, and NSAIDs and triptans the least risky. None of the agents had to be taken daily to trigger medication overuse headache.
PREVENTION IS THE BEST TREATMENT
The best approach to medication overuse headache is to prevent it while the patient still has episodic migraine.
Outcomes are better with triptans or ergots
Undertreatment of migraine leads quickly to overuse of symptomatic medications, and from there to medication overuse headache.
Outcomes of episodic migraine are better when triptans or ergots (which are migrainespecific) are used first-line in patients with disabling migraine and no vascular contraindications. Patients who start with nonspecific treatment and step up to a more specific treatment when lower-level medications fail have less favorable outcomes in terms of migraine relief and disability time than those treated with triptans from the beginning.25
To put this in perspective, if a patient takes an acute medication, gets only partial relief (not a pain-free response) at 2 hours and then takes another pill, or gets a recurrence and takes another pill, the likelihood of prolonging an attack and using more medications goes up. If a patient takes a triptan and gets a sustained pain-free response, the attack is truncated and the medication usage reduced. Therefore, migraine-specific acute treatments are more likely to not be overused.
Daily preventive medication, if necessary
As noted above, if the number of headache days exceeds 10 per month, the likelihood of developing daily headache escalates steeply. Thus, patients with 10 or more days of headache per month should be prescribed preventive medications to be taken daily to reduce the frequency, severity, and duration of attacks. Preventive treatment may also increase the efficacy of the acute treatments.
The drugs used for preventive treatment are different than those used for acute treatment and are not likely to cause medication rebound headache. However, they are not very effective. Those that have the best evidence of efficacy are beta-blockers, tricyclic antidepressants, and anticonvulsants; calcium channel blockers and NSAIDs are also popular. This topic has been reviewed in detail elsewhere.26,27
REVERSING MEDICATION OVERUSE HEADACHE
If a patient already has medication overuse headache, the clinician is faced with the problem of weaning her or him from the overused medication while establishing a reasonable regimen of prophylaxis and acute medications with limits.
For the most part, these tasks can be accomplished in a series of clinic visits. However, some patients have such severe comorbid medical and psychiatric illnesses that outpatient treatment is impossible. For them, a day hospital or inpatient program with infusion capabilities is often useful.
Outpatient treatment of medication overuse headache
Outpatient treatment of medication overuse headache involves:
Educating patients about the genesis of the problem and reassuring them that you are not accusing them of being an addict. Most patients who develop medication overuse headache are habituated inadvertently, and this needs to be made clear, along with the overall plan and the likely prognosis.
Establishing daily preventive medications. The prophylactic regimen can be established either before or during the weaning.
Providing acute medications, with limits. At a certain point in the weaning, advise the patient not to treat low-level headaches, and provide a triptan or dihydroergotamine to use for severe attacks, no more than twice weekly and less than 10 days per month. If the patient is in triptan rebound, dihydroergotamine would be the choice.
Instructing the patient to keep a headache diary to follow adherence and outcomes.
Psychology consultation can be very helpful to teach patients behavioral techniques to deal with anticipatory anxiety during the weaning.
Multidisciplinary programs with infusion capability
Some patients need a more intensive approach to restore an episodic migraine pattern. Examples: those on very high doses of narcotics or barbiturates, those with comorbid medical illnesses that limit both acute and preventive treatments, and those with severe and complicating comorbid psychiatric illnesses.
Multidisciplinary programs are available, with specialists in neurology, primary care, psychology, and physical and occupational therapy providing treatment. Patients check into the hospital or a “day hospital,” where they can also receive intravenous infusions to get through the weaning. The goal is to shift the locus of control back to patients as they revert from daily headache to episodic migraine. Patient education is crucial.
OUTCOMES ARE GOOD
There is much good news about medication overuse headache.
It can be prevented with careful monitoring of acute medication outcomes and number of headache days. Prophylaxis should be used when treating high-frequency or very disabling migraine.
Most patients improve when weaned and treated with preventive medications. “Recovery” means at least 3 months off the overused medications. In studies, more than half of patients who underwent treatment for medication overuse headache remained better and had an episodic pattern of headache 5 years later.26
Unfortunately, the initial improvement often seen with patients after weaning and being given preventive medication (72%–85% of patients improve) in the first year is often followed by preventable relapse, so it is very important to follow up with patients regularly. 28–32
Helping restore a patient’s quality of life is an outcome rewarding to primary care provider and specialist alike.
Some migraine patients fall into a trap by overusing the medications they take when they get their headaches, ending in a downward spiral of daily or near-daily headaches for which their medications become less and less effective.
This condition, called medication overuse headache, makes for a poor quality of life. It is often associated with nonrestorative sleep, neck pain, and vasomotor instability. Comorbid depression and anxiety are common and may complicate treatment. (Depression and anxiety, however, do not cause daily headaches.) Patients can also suffer from the physiologic and psychological consequences of the overused medications.
Fortunately, we can break the cycle.1,2 Treatment involves completely weaning the patient from the overused medications and educating her or him to follow a new regimen of prophylaxis and acute treatment with clear limits on frequency of use. Nondrug treatments such as relaxation therapy, biofeedback, and cognitive behavioral therapy can be useful adjuncts.
CROSSING THE LINE: 15 HEADACHE DAYS A MONTH
Chronic daily headache
We define chronic daily headache as occurring on at least 15 days per month for at least 3 months in a row and lasting at least 4 hours if untreated.
Most patients start with episodic migraine, and many of them remember the period of transformation. Crossing the 15-day-per-month threshold changes the clinical presentation, prognosis, and treatment, all for the worse.
In a large population-based study,3 2.5% of patients who began with episodic migraine (headaches on fewer than 15 days per month) had “transformed migraine” (headaches on 15 or more days per month) 1 year later. The prevalence of chronic daily headache is almost 5% of the general population and may account for up to 70% of the initial diagnoses seen in headache centers.
The closer a patient is to having 15 headaches per month, the more likely she or he will cross the line.4,5 Katsarava and colleagues5 followed patients for 1 year in a neurology clinic in Germany and found that those starting the year with 6 to 9 headache days per month were 6.2 times more likely to develop chronic daily headache in the next year than those who began the year with 0 to 4 per month—and those starting with 10 to 14 headaches per month were 20 times more likely.
Medication overuse headache
Medication overuse headache is a subset of chronic daily headache, also occurring on 15 or more days per month but with the added criterion of medication overuse, ie, regular overuse for more than 3 months of at least one acute treatment drug:
- Ergotamine, triptans, opioids, or combination analgesic medications on 10 or more days per month on a regular basis for more than 3 months, or
- Simple analgesics or any combination of ergotamine, triptans, analgesics, or opioids on 15 or more days per month on a regular basis for more than 3 months without overuse of any single class alone.
Another criterion is that the patient’s headaches must worsen in some way (usually frequency) as the use of acute medications becomes more frequent.6,7
Medication overuse headache is the most common form of secondary chronic daily headache seen in headache practice,8–10 and probably accounts for about half of cases of chronic daily headache.11–13
Different terminology confuses the issue
Many terms have been used to describe medication overuse headache in the past, such as analgesic-rebound headache (or just rebound headache), transformed migraine with medication overuse, and even chronic migraine. The lack of uniformity in terminology makes for confusion in the literature and difficulty in communicating with patients and colleagues. Some authors mean medication overuse headache when they say chronic daily headache.
Complicating this diagnostic confusion is a debate as to whether chronic daily headache in general should be treated as a primary or secondary headache disorder. Some European headache specialists insist on a strict division between primary and secondary daily headaches, and medication overuse headache is one of the latter. Many American specialists believe that chronic daily headache is a collective description or phenotype rather than a diagnostic category, and that it is usually associated with and exacerbated by medication overuse.14,15 The International Classification of Headache Disorders uses the term “chronic migraine” for primary daily headache, and “medication overuse headache” for secondary daily headache or rebound.
Many American specialists approach the disorder clinically, treating chronic daily headache in the same way regardless of whether there is medication overuse. They cite randomized controlled trials of topiramate (Topamax) and onabotulinum toxin type A (Botox) that reported comparable benefit with these medications in patients with chronic daily headache with or without medication overuse.16–18
MORE IS LESS: THE PARADOX OF TREATING ACUTE HEADACHE
The clinical paradox and dilemma of treating acute episodic migraine is that more is less: the more days of acute treatment, the less well the migraines are controlled. And thus, the patient is likely to veer out of control.3
The compassion that motivates us to prescribe medications for acute episodic migraine must be tempered by the realization that too much of a good thing will result in its malignant transformation to medication overuse headache. Once this develops, preventive and migrainespecific acute medications are less effective, and patients need far more complex interventions.
Complicating the dilemma, acute migraine-specific medications such as triptans and dihydroergotamine (Migranal) work better when taken early in migraine attacks, before central sensitization and allodynia develop with attendant photophonophobia and sensitivity to other stimuli. On the other hand, overuse will lead to medication overuse headache.
SYMPTOMS VARY
The symptoms of medication overuse headache vary in frequency, severity, location, quality, and associated features, both among patients and in the same patient. This is because the disease itself varies and also because of differences in the type and frequency of medication intake. Still, some features help to define this problem, and failing to recognize them may account for a widely held clinical feeling that these patients are “difficult.”
History of episodic migraine. Generally, medication overuse headache does not occur in nonmigraineurs.
Headache on most days of the month. Whenever a migraineur starts having headaches on more days than not, the diagnosis of medication overuse should be considered.
Overuse of acute medications. The criteria (see above) allow for combining days of acute medication use. For example, if a patient takes a combination analgesic on 5 days and a triptan on 5 different days, that would still be enough days of acute treatment to trigger medication overuse headache.
Variable pain location is a particular characteristic of medication overuse headache. Although the location may differ from day to day (front or back, rostral or caudal, unilateral or bilateral), it is the quantity not the quality or location of the headaches that suggests the diagnosis.
A drug-dependent rhythm. Predictably, the headaches come on in the early morning or awaken the patient from sleep. This may be due to variable drug withdrawal.
Neck pain. Medication overuse headache frequently involves the neck, and patients often seek and receive treatments such as muscle relaxants or injections to the neck. When patients are weaned from their acute migraine medications, neck pain generally dissipates. The neck pain, however, can recur episodically with their remaining, now-episodic acute migraines. Neck pain associated with medication overuse headache is not usually a sign of a primary neck disorder; rather, it is a symptom of medication overuse headache itself.
Concomitant depression and anxiety are comorbid with episodic migraine, but appear to be more common with medication overuse headache. Treating the depression or anxiety does not restore an episodic pattern of migraine; weaning from the overused medications remains the most important intervention. A frequent clinical error is to diagnose and treat the psychiatric issues without recognizing medication overuse as the primary problem.
Nonrestorative sleep is almost always reported by patients with medication overuse headache. This is often due to the caffeine contained in combination analgesics or to excessive dietary caffeine intake, but it may also be part of the daily acute drug withdrawal syndrome. The sleep problems are also associated with the concomitant depression. Sleep often improves after weaning from the offending substance or substances. As with neck pain, patients do not have a primary sleep disorder—the sleep disturbance is a symptom of medication overuse headache.
Vasomotor instability. Autonomic features are commonly associated with medication overuse headache. Rhinorrhea, nasal stuffiness, and lacrimation are features of medication withdrawal, especially from opioids, and are frequently attributed to sinus disease or “sinus headaches.” Many patients undergo unnecessary sinus procedures or are given antibiotics, decongestants, and other wrong medications for incorrect diagnoses. Decongestants can cause and exacerbate medication overuse headache, so they need to be withdrawn. The sinus features generally remit when the overused migraine medications are eliminated.
Preventive medications are less effective or ineffective until the acute medications are withdrawn. Thus, prescribing prevention without weaning is usually futile, and the patients are often dismissed as having a refractory problem. At the same time, migraine-specific acute treatments, ie, triptans and ergots, are usually also less effective. When patients complain that “nothing works,” either preventively or acutely, medication overuse headache should spring to mind.
Weaning from overused medications can restore the efficacy of previously ineffective treatments at the same time that a patient is restored to an episodic headache pattern. Thus, complete weaning is the pivotal clinical intervention. Clinically, there is no spontaneous remission from rebound without absolute detoxification, maintained for months.9,19–22
Other diagnoses entertained. The more diagnoses suggested for daily headache, and the more treatments tried unsuccessfully, the more likely the diagnosis is actually medication overuse headache. Because this condition is protean, patients and caregivers alike make more and more fanciful diagnoses such as allergies, cervicogenic headache, temperomandibular disorder, occipital neuralgia, chronic Lyme disease, and systemic candidiasis. A useful strategy is to assume that daily headache is likely due to medication overuse. And since medication overuse headache is generally treatable, patients labeled as having refractory headaches often are dramatically improved by appropriate intervention.
WHY ARE MIGRAINEURS SO SUSCEPTIBLE?
Medication overuse headache occurs primarily in people with a history of episodic migraine, but the unique susceptibility of migraineurs is not fully understood.
Structural changes in the brain?
Episodic migraine attacks appear to be generated in the upper brainstem. This region in turn activates a set of peripheral pain mechanisms, ie, meningeal inflammation and vasodilation. The peripheral pain processes turn on afferent circuits that carry the pain signals to the lower brainstem, where these signals are integrated. Finally, the central signals ascend the brainstem, stimulating autonomic nuclei that account for nausea and other vasomotor changes, proceed through the thalamus, and terminate in the cortex where pain is perceived. Thus, migraine without aura consists of three steps—a central generator, a set of peripheral pain mechanisms, and a series of steps culminating in central integration. (Aura involves other steps, not outlined here.)
A possible explanation of why migraine becomes chronic is that a yo-yo effect of repeated migrainous pain processes, followed by repeated medication, results in structural changes. These propagate central sensitization with a lowered threshold for activation of all of the central processing of head pain.
This set of disturbances may occur due to undertreatment of migraine pain. With inadequate pain control, headaches recur, and the process repeats until damage occurs. Evidence for this is seen in up-regulation of excitatory serotonin receptors when analgesics are repetitively given to laboratory animals.23
A pure withdrawal phenomenon?
Also possible is that medication overuse headache is just a complex dependence-and-withdrawal phenomenon. Thus, the cyclical use of various medications results in withdrawal headaches and a set of symptoms, including disturbed sleep, morning headache, and vasomotor signs of withdrawal. Arguing against its being a pure withdrawal phenomenon is that daily use of analgesics or opioids generally does not cause daily headache in nonmigraineurs.24
HOW MUCH MEDICATION USE IS TOO MUCH?
For an episodic migraine condition to transform into a chronic one, medications need to be taken on only a modest number of days per month: 5 to 10, depending on the type of medication.
A pivotal study3 found that butalbital combinations were most likely to cause medication overuse headache, needing to be taken on merely 5 or more days per month to cause it in migraineurs. Opioids caused it if taken 8 or more days per month, and triptans if taken 10 or more days per month. Nonsteroidal anti-inflammatory drugs (NSAIDs) actually protected against transformation to daily headache if used 5 or fewer days per month, but caused medication overuse headache if used 10 or more days per month.
Thus, there was a hierarchy of risk, with butalbital being the worst, opioids in the middle, and NSAIDs and triptans the least risky. None of the agents had to be taken daily to trigger medication overuse headache.
PREVENTION IS THE BEST TREATMENT
The best approach to medication overuse headache is to prevent it while the patient still has episodic migraine.
Outcomes are better with triptans or ergots
Undertreatment of migraine leads quickly to overuse of symptomatic medications, and from there to medication overuse headache.
Outcomes of episodic migraine are better when triptans or ergots (which are migrainespecific) are used first-line in patients with disabling migraine and no vascular contraindications. Patients who start with nonspecific treatment and step up to a more specific treatment when lower-level medications fail have less favorable outcomes in terms of migraine relief and disability time than those treated with triptans from the beginning.25
To put this in perspective, if a patient takes an acute medication, gets only partial relief (not a pain-free response) at 2 hours and then takes another pill, or gets a recurrence and takes another pill, the likelihood of prolonging an attack and using more medications goes up. If a patient takes a triptan and gets a sustained pain-free response, the attack is truncated and the medication usage reduced. Therefore, migraine-specific acute treatments are more likely to not be overused.
Daily preventive medication, if necessary
As noted above, if the number of headache days exceeds 10 per month, the likelihood of developing daily headache escalates steeply. Thus, patients with 10 or more days of headache per month should be prescribed preventive medications to be taken daily to reduce the frequency, severity, and duration of attacks. Preventive treatment may also increase the efficacy of the acute treatments.
The drugs used for preventive treatment are different than those used for acute treatment and are not likely to cause medication rebound headache. However, they are not very effective. Those that have the best evidence of efficacy are beta-blockers, tricyclic antidepressants, and anticonvulsants; calcium channel blockers and NSAIDs are also popular. This topic has been reviewed in detail elsewhere.26,27
REVERSING MEDICATION OVERUSE HEADACHE
If a patient already has medication overuse headache, the clinician is faced with the problem of weaning her or him from the overused medication while establishing a reasonable regimen of prophylaxis and acute medications with limits.
For the most part, these tasks can be accomplished in a series of clinic visits. However, some patients have such severe comorbid medical and psychiatric illnesses that outpatient treatment is impossible. For them, a day hospital or inpatient program with infusion capabilities is often useful.
Outpatient treatment of medication overuse headache
Outpatient treatment of medication overuse headache involves:
Educating patients about the genesis of the problem and reassuring them that you are not accusing them of being an addict. Most patients who develop medication overuse headache are habituated inadvertently, and this needs to be made clear, along with the overall plan and the likely prognosis.
Establishing daily preventive medications. The prophylactic regimen can be established either before or during the weaning.
Providing acute medications, with limits. At a certain point in the weaning, advise the patient not to treat low-level headaches, and provide a triptan or dihydroergotamine to use for severe attacks, no more than twice weekly and less than 10 days per month. If the patient is in triptan rebound, dihydroergotamine would be the choice.
Instructing the patient to keep a headache diary to follow adherence and outcomes.
Psychology consultation can be very helpful to teach patients behavioral techniques to deal with anticipatory anxiety during the weaning.
Multidisciplinary programs with infusion capability
Some patients need a more intensive approach to restore an episodic migraine pattern. Examples: those on very high doses of narcotics or barbiturates, those with comorbid medical illnesses that limit both acute and preventive treatments, and those with severe and complicating comorbid psychiatric illnesses.
Multidisciplinary programs are available, with specialists in neurology, primary care, psychology, and physical and occupational therapy providing treatment. Patients check into the hospital or a “day hospital,” where they can also receive intravenous infusions to get through the weaning. The goal is to shift the locus of control back to patients as they revert from daily headache to episodic migraine. Patient education is crucial.
OUTCOMES ARE GOOD
There is much good news about medication overuse headache.
It can be prevented with careful monitoring of acute medication outcomes and number of headache days. Prophylaxis should be used when treating high-frequency or very disabling migraine.
Most patients improve when weaned and treated with preventive medications. “Recovery” means at least 3 months off the overused medications. In studies, more than half of patients who underwent treatment for medication overuse headache remained better and had an episodic pattern of headache 5 years later.26
Unfortunately, the initial improvement often seen with patients after weaning and being given preventive medication (72%–85% of patients improve) in the first year is often followed by preventable relapse, so it is very important to follow up with patients regularly. 28–32
Helping restore a patient’s quality of life is an outcome rewarding to primary care provider and specialist alike.
- Schwartz BS, Stewart WF, Lipton RB. Lost workdays and decreased work effectiveness associated with headache in the workplace. J Occup Environ Med 1997; 39:320–327.
- Meletiche DM, Lofland JH, Young WB. Quality-of-life differences between patients with episodic and transformed migraine. Headache 2001; 41:573–578.
- Bigal ME, Serrano D, Buse D, Scher A, Stewart WF, Lipton RB. Acute migraine medications and evolution from episodic to chronic migraine: a longitudinal populationbased study. Headache 2008; 48:1157–1168.
- Scher AI, Stewart WF, Ricci JA, Lipton RB. Factors associated with the onset and remission of chronic daily headache in a population-based study. Pain 2003; 106:81–89.
- Katsarava Z, Schneeweiss S, Kurth T, et al Incidence and predictors for chronicity of headache in patients with episodic migraine. Neurology 2004; 62:788–790.
- Olesen J, Bousser MG, Diener HC, et al., Headache Classification Committee New appendix criteria open for a broader concept of chronic migraine. Cephalalgia 2006; 26:742–746.
- Bigal M, Rapoport A, Sheftell F, Tepper S, Lipton R. The international classification of headache disorders revised criteria for chronic migraine—field testing in a headache specialty clinic. Cephalalgia 2007; 27:230–234.
- Rapoport A, Stang P, Gutterman DL, et al Analgesic rebound headache in clinical practice: data from a physician survey. Headache 1996; 36:14–19.
- Mathew NT. Transformed migraine, analgesic rebound, and other chronic daily headaches. Neurol Clin 1997; 15:167–186.
- Bigal ME, Sheftell FD, Rapoport AM, Tepper SJ, Lipton RB. Chronic daily headache: identification of factors associated with induction and transformation. Headache 2002; 42:575–581.
- Silberstein SD. Tension-type and chronic daily headache. Neurology 1993; 43:1644–1649.
- Castillo J, Muñoz P, Guitera V, Pascual J. Kaplan Award 1998. Epidemiology of chronic daily headache in the general population. Headache 1999; 39:190–196.
- Scher AI, Stewart WF, Liberman J, Lipton RB. Prevalence of frequent headache in a population sample. Headache 1998; 38:497–506.
- Headache Classification Subcommittee of the International Headache Society. The international classification of headache disorders: 2nd edition. Cephalalgia 2004; 24( suppl 1):9–160.
- Silberstein SD, Lipton RB, Sliwinski M. Classification of daily and near-daily headaches: field trial of revised IHS criteria. Neurology 1996; 47:871–875.
- Diener HC, Bussone G, Van Oene JC, Lahaye M, Schwalen S, Goadsby PJTOPMAT-MIG-201 (TOP-CHROME) Study Group. Topiramate reduces headache days in chronic migraine: a randomized, double-blind, placebo-controlled study. Cephalalgia 2007; 27:814–823.
- Silberstein SD, Lipton RB, Dodick DW, et al., Topiramate Chronic Migraine Study Group. Efficacy and safety of topiramate for the treatment of chronic migraine: a randomized, double-blind, placebo-controlled trial. Headache 2007; 47:170–180.
- Silberstein SD, Blumenfeld AM, Cady RK, et al Botulinum neurotoxin type A for the treatment of chronic migraine: analysis of the PREEMPT chronic migraine subgroup with baseline acute headache medication overuse. Cephalalgia 2009; 29( suppl 1):1–176.
- Mathew NT. Medication misuse headache. Cephalalgia 1998; 18( suppl 21):34–36.
- Diener HC, Katasarva Z. Analgesic/abortive overuse and misuse in chronic daily headache. Curr Pain Headache Rep 2001; 5:545–550.
- Tepper SJ, Rapoport AM, Sheftell FD, Bigal ME. Chronic daily headache: an update. Headache Care 2004; 1:233–245.
- Dodick DW. Clinical practice. Chronic daily headache. N Engl J Med 2006; 354:158–165.
- Srikiatkhachorn A, Tarasub N, Govitrapong P. Effect of chronic analgesic exposure on the central serotonin system: a possible mechanism of analgesic abuse headache. Headache 2000; 40:343–350.
- Wilkinson SM, Becker WJ, Heine JA. Opiate use to control bowel motility may induce chronic daily headache in patients with migraine. Headache 2001; 41:303–309.
- Lipton RB, Stewart WF, Stone AM, Láinez MJ, Sawyer JPDisability in Strategies of Care Study group. Stratified care vs step care strategies for migraine: the Disability in Strategies of Care (DISC) Study: a randomized trial. JAMA 2000; 284:2599–605.
- Bamford CC, Tepper SJ. Daily pharmacologic prophylaxis of episodic migraine. Tech Regional Anesthesia Pain Manage 2009; 13:20–37.
- Tepper SJ. Preventive pharmacologic treatment of migraine and tension-type headache. In:Levin M, editor. Comprehensive Review of Headache Medicine. Oxford: Oxford University Press, 2008:231–234.
- Zed PJ, Loewen PS, Robinson G. Medication-induced headache: overview and systematic review of therapeutic approaches. Ann Pharmacother 1999; 33:61–72.
- Fritsche G, Eberl A, Katsarava Z, Limmroth V, Diener HC. Drug-induced headache: long-term follow-up of withdrawal therapy and persistence of drug misuse. Eur Neurol 2001; 45:229–235.
- Pini LA, Cicero AF, Sandrini M. Long-term follow-up of patients treated for chronic headache with analgesic overuse. Cephalalgia 2001; 21:878–883.
- Katsarava Z, Limmroth V, Finke M, Diener HC, Fritsche G. Rates and predictors for relapse in medication overuse headache: a 1-year prospective study. Neurology 2003; 60:1682–1683.
- Zidverc-Trajkovic J, Pekmezovic T, Jovanovic Z, et al Medication overuse headache: clinical features predicting treatment outcome at 1-year follow-up. Cephalalgia 2007; 27:1219–1225.
- Schwartz BS, Stewart WF, Lipton RB. Lost workdays and decreased work effectiveness associated with headache in the workplace. J Occup Environ Med 1997; 39:320–327.
- Meletiche DM, Lofland JH, Young WB. Quality-of-life differences between patients with episodic and transformed migraine. Headache 2001; 41:573–578.
- Bigal ME, Serrano D, Buse D, Scher A, Stewart WF, Lipton RB. Acute migraine medications and evolution from episodic to chronic migraine: a longitudinal populationbased study. Headache 2008; 48:1157–1168.
- Scher AI, Stewart WF, Ricci JA, Lipton RB. Factors associated with the onset and remission of chronic daily headache in a population-based study. Pain 2003; 106:81–89.
- Katsarava Z, Schneeweiss S, Kurth T, et al Incidence and predictors for chronicity of headache in patients with episodic migraine. Neurology 2004; 62:788–790.
- Olesen J, Bousser MG, Diener HC, et al., Headache Classification Committee New appendix criteria open for a broader concept of chronic migraine. Cephalalgia 2006; 26:742–746.
- Bigal M, Rapoport A, Sheftell F, Tepper S, Lipton R. The international classification of headache disorders revised criteria for chronic migraine—field testing in a headache specialty clinic. Cephalalgia 2007; 27:230–234.
- Rapoport A, Stang P, Gutterman DL, et al Analgesic rebound headache in clinical practice: data from a physician survey. Headache 1996; 36:14–19.
- Mathew NT. Transformed migraine, analgesic rebound, and other chronic daily headaches. Neurol Clin 1997; 15:167–186.
- Bigal ME, Sheftell FD, Rapoport AM, Tepper SJ, Lipton RB. Chronic daily headache: identification of factors associated with induction and transformation. Headache 2002; 42:575–581.
- Silberstein SD. Tension-type and chronic daily headache. Neurology 1993; 43:1644–1649.
- Castillo J, Muñoz P, Guitera V, Pascual J. Kaplan Award 1998. Epidemiology of chronic daily headache in the general population. Headache 1999; 39:190–196.
- Scher AI, Stewart WF, Liberman J, Lipton RB. Prevalence of frequent headache in a population sample. Headache 1998; 38:497–506.
- Headache Classification Subcommittee of the International Headache Society. The international classification of headache disorders: 2nd edition. Cephalalgia 2004; 24( suppl 1):9–160.
- Silberstein SD, Lipton RB, Sliwinski M. Classification of daily and near-daily headaches: field trial of revised IHS criteria. Neurology 1996; 47:871–875.
- Diener HC, Bussone G, Van Oene JC, Lahaye M, Schwalen S, Goadsby PJTOPMAT-MIG-201 (TOP-CHROME) Study Group. Topiramate reduces headache days in chronic migraine: a randomized, double-blind, placebo-controlled study. Cephalalgia 2007; 27:814–823.
- Silberstein SD, Lipton RB, Dodick DW, et al., Topiramate Chronic Migraine Study Group. Efficacy and safety of topiramate for the treatment of chronic migraine: a randomized, double-blind, placebo-controlled trial. Headache 2007; 47:170–180.
- Silberstein SD, Blumenfeld AM, Cady RK, et al Botulinum neurotoxin type A for the treatment of chronic migraine: analysis of the PREEMPT chronic migraine subgroup with baseline acute headache medication overuse. Cephalalgia 2009; 29( suppl 1):1–176.
- Mathew NT. Medication misuse headache. Cephalalgia 1998; 18( suppl 21):34–36.
- Diener HC, Katasarva Z. Analgesic/abortive overuse and misuse in chronic daily headache. Curr Pain Headache Rep 2001; 5:545–550.
- Tepper SJ, Rapoport AM, Sheftell FD, Bigal ME. Chronic daily headache: an update. Headache Care 2004; 1:233–245.
- Dodick DW. Clinical practice. Chronic daily headache. N Engl J Med 2006; 354:158–165.
- Srikiatkhachorn A, Tarasub N, Govitrapong P. Effect of chronic analgesic exposure on the central serotonin system: a possible mechanism of analgesic abuse headache. Headache 2000; 40:343–350.
- Wilkinson SM, Becker WJ, Heine JA. Opiate use to control bowel motility may induce chronic daily headache in patients with migraine. Headache 2001; 41:303–309.
- Lipton RB, Stewart WF, Stone AM, Láinez MJ, Sawyer JPDisability in Strategies of Care Study group. Stratified care vs step care strategies for migraine: the Disability in Strategies of Care (DISC) Study: a randomized trial. JAMA 2000; 284:2599–605.
- Bamford CC, Tepper SJ. Daily pharmacologic prophylaxis of episodic migraine. Tech Regional Anesthesia Pain Manage 2009; 13:20–37.
- Tepper SJ. Preventive pharmacologic treatment of migraine and tension-type headache. In:Levin M, editor. Comprehensive Review of Headache Medicine. Oxford: Oxford University Press, 2008:231–234.
- Zed PJ, Loewen PS, Robinson G. Medication-induced headache: overview and systematic review of therapeutic approaches. Ann Pharmacother 1999; 33:61–72.
- Fritsche G, Eberl A, Katsarava Z, Limmroth V, Diener HC. Drug-induced headache: long-term follow-up of withdrawal therapy and persistence of drug misuse. Eur Neurol 2001; 45:229–235.
- Pini LA, Cicero AF, Sandrini M. Long-term follow-up of patients treated for chronic headache with analgesic overuse. Cephalalgia 2001; 21:878–883.
- Katsarava Z, Limmroth V, Finke M, Diener HC, Fritsche G. Rates and predictors for relapse in medication overuse headache: a 1-year prospective study. Neurology 2003; 60:1682–1683.
- Zidverc-Trajkovic J, Pekmezovic T, Jovanovic Z, et al Medication overuse headache: clinical features predicting treatment outcome at 1-year follow-up. Cephalalgia 2007; 27:1219–1225.
KEY POINTS
- Medication overuse headache can occur with as few as 5 days per month of treatment with butalbital or 8 days per month with opioids.
- The features vary, but the most important is headache on 15 or more days per month, lasting at least 4 hours if untreated, for at least 3 consecutive months. Other common symptoms are morning headaches, neck pain, nonrestorative sleep, and vasomotor instability, all of which tend to improve with weaning from the overused medications.
- Daily preventive treatment is indicated when patients have 10 or more headaches per month or severe disability from their attacks.
- With treatment, the prognosis for medication overuse headache is good. However, patients need close followup to prevent recidivism.
Interpreting The JUPITER Trial: Statins can prevent VTE, but more study is needed
A major placebo-controlled trial has found that a statin can reduce the risk of venous thromboembolism (VTE).1
We do not recommend prescribing this class of drugs for this purpose until much more research has been done, and we certainly do not recommend substituting a statin for anticoagulant therapy in a patient at risk of VTE.
Nevertheless, we are excited by the latest findings, and we find comfort in knowing that if a patient is taking a statin for an approved indication, ie, reducing the risk of cardiovascular disease in a patient with hyperlipidemia or a previous cardiovascular event, the drug will also reduce the risk of VTE.
In the pages that follow, we describe and comment on what is known about the effect of statins on the risk of VTE.
ARTERIAL AND VENOUS THROMBOSIS: HOW ARE THEY LINKED?
The causes of arterial thrombosis may not be entirely distinct from those of deep vein thrombosis and pulmonary embolism, collectively referred to as VTE. Some studies have found that risk factors for arterial thrombosis overlap with those for VTE.2–4 However, other studies have shown no association between venous and arterial events.5–10
Hyperlipidemia, in particular, has been evaluated to see if it is a risk factor for VTE. As with other risk factors for arterial thrombosis, the data have been mixed, with some reports favoring an association with VTE and others not.4,5,11 Even so, preventive strategies targeting arterial risk factors have shown promise in reducing VTE events.12
Although commonly used to treat hyperlipidemia, statins (3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors) are believed to reduce the incidence of thrombosis by a number of mechanisms13:
- Decreasing platelet aggregation
- Inhibiting expression of tissue factor and plasminogen activator inhibitor 1
- Increasing expression of tissue plasminogen activator
- Increasing expression of thrombomodulin, which can activate protein C and prevent thrombin-induced platelet and factor V activation and fibrinogen clotting.
STATINS AND VTE IN OBSERVATIONAL AND CASE-CONTROL STUDIES
In view of the multiple effects of statins, several studies have looked at whether these drugs reduce the occurrence of both arterial thrombosis and VTE.14–19
Two prospective observational studies and four case-control studies found that statins reduced the risk of VTE by 20% to 60%.14–19 Interestingly, two of the case-control studies found that antiplatelet therapy did not reduce the risk of VTE.18,19
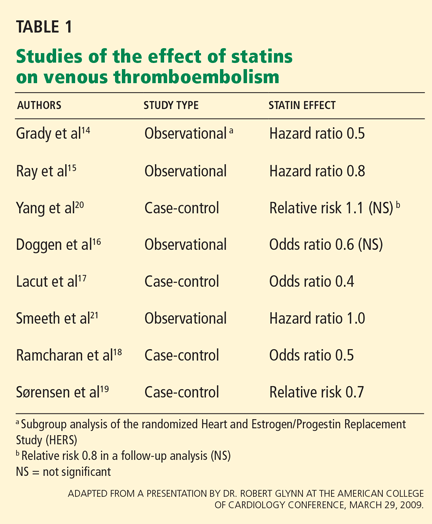
THE JUPITER STUDY
The Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) study primarily sought to determine if rosuvastatin (Crestor) 20 mg/day, compared with placebo, would reduce the rate of first major cardiovascular events.22 A prespecified secondary end point of the trial was VTE, making JUPITER the first randomized, placebo-controlled trial to specifically test whether statins prevent VTE.1
Inclusion criteria: Normal LDL, high CRP
The study included men age 50 and older and women age 60 and older with no history of cardiovascular disease. In addition, their lowdensity lipoprotein (LDL) cholesterol levels had to be lower than 130 mg/dL (3.4 mmol/L), their triglyceride levels had to be lower than 500 mg/dL (5.6 mmol/L), and their highsensitivity C-reactive protein (hs-CRP) levels had to be 2.0 mg/L or higher.
Since high levels of hs-CRP, a marker of inflammation, predict cardiovascular events and since statins lower hs-CRP levels, the investigators hypothesized that people with elevated hs-CRP but without hyperlipidemia might benefit from statin treatment.21
Patients were excluded if they had received lipid-lowering therapy within 6 weeks of the trial screening, had diabetes mellitus or uncontrolled hypertension, were currently using postmenopausal hormone-replacement therapy, or had had cancer within the previous 5 years, except for certain skin cancers.
Candidates who complied well during a 4-week placebo run-in phase were randomly assigned to receive either rosuvastatin 20 mg daily (an intermediate dose) or a matching placebo. In all, 17,802 people were randomized. The two assigned groups appeared to be well matched.
Patients were to come in for visits twice a year for 60 months after randomization to be assessed for symptomatic deep venous thrombosis and pulmonary embolism. New cases of VTE were confirmed by imaging studies, by the initiation of anticoagulation therapy, or by death ascribed to pulmonary embolism.
Idiopathic VTE was classified as unprovoked if it occurred in the absence of trauma, hospitalization, or surgery within 3 months before the event, and in the absence of any diagnosed cancer within 3 months before and after the event. Provoked VTE events were those that occurred in a participant with cancer or when a precipitating event was associated with trauma, hospitalization, or surgery.
Rosuvastatin prevents heart attack, stroke
On the recommendation of the trial’s independent data and safety monitoring board, JUPITER was stopped early because the trial drug showed evidence of efficacy in preventing the combined primary end point of a first major cardiovascular event—ie, nonfatal myocardial infarction, nonfatal stroke, hospitalization for unstable angina, an arterial revascularization procedure, or confirmed death from a cardiovascular cause.22 (The cardiovascular outcomes of the JUPITER study were reviewed by Shishehbor and Hazen23 in the January 2009 issue of the Cleveland Clinic Journal of Medicine; see doi:10.3949/ccjm.75a.08105).
Formal follow-up for the trial's primary and secondary efficacy end points ended then, but data on VTE continued to be collected until each patient’s closeout visit as part of a safety monitoring protocol. The last closeout visit occurred on August 20, 2008. The primary analysis focused on events occurring up to March 30, 2008, the date the study was stopped.
Secondary end point results: Rosuvastatin prevents VTE
At a median follow-up of 1.9 years, an episode of VTE had occurred in 94 (0.53%) of the 17,802 patients—34 in the rosuvastatin group and 60 in the placebo group.1 This translates to 0.18 and 0.32 events per 100 person-years of follow-up in the rosuvastatin and placebo groups, respectively (hazard ratio for the rosuvastatin group 0.57, 95% confidence interval [CI] 0.37–0.86, P = .007).
Forty-four cases of VTE were classified as provoked and 50 cases were categorized as unprovoked. The risk reduction was statistically significant for provoked cases (hazard ratio 0.52, 95% CI 0.28–0.96, P = .03), but not for unprovoked events (hazard ratio 0.61, 95% CI 0.35–1.09, P = .09).
Subgroup analysis revealed no significant association between patient characteristics and the impact of rosuvastatin on the risk of a VTE event, but, as expected, more benefit was associated with higher baseline lipid levels.
STILL TOO SOON TO ADVISE ROUTINE STATIN USE TO PREVENT VTE
While the JUPITER trial data show an apparent benefit of statin use on the rate of VTE events, advising routine use of statins to prevent VTE is premature, for three main reasons.
Many must be treated to prevent one case of VTE. The number needed to treat (NNT) with rosuvastatin for 5 years to prevent either a case of VTE or a cardiovascular event was 21, and the NNT to prevent one cardiovascular event was 25. In a review of the two most recent case-control studies investigating the effects of statins on VTE,18,19 Cushman24 calculated that the NNT to prevent one VTE event each year was 333 for those age 75 and older. Though the Jupiter data did not provide the specific incidence of VTE at 1 year, except graphically, we can estimate that the NNT to prevent one VTE event at 1 year in the study is also very high.
Practically speaking, the perceived benefits of VTE prevention require large numbers to be treated, and the net clinical gain is still largely in preventing arterial events such as heart attack and stroke rather than VTE.
Statins, though safe, can still have adverse effects. The JUPITER study found a trend (albeit nonsignificant) toward more muscle complaints and elevations on liver function testing in apparently healthy persons taking a statin.22 Although severe complications of statin therapy such as rhabdomyolysis and elevations of creatine phosphokinase are rare, patients taking a statin have a 39% higher risk of an adverse event, most commonly myalgias or abnormalities on liver function testing.25 Were statins to be given routinely to even more people than they are now, more adverse outcomes would be likely.
More study is needed. The JUPITER study did not address a high risk of VTE. In fact, the investigators provided no information as to the VTE history of those enrolled.
Clearly, statins should not be substituted for proven prophylaxis and anticoagulation without further investigation, especially for patients with recurrent deep venous thrombosis, hospitalized patients, postoperative patients, and other patients prone to VTE.
OUR VIEW
The JUPITER study is an important leap forward in adding to our knowledge of how to prevent VTE. For people with another indication for taking a statin (eg, a previous cardiovascular event, hyperlipidemia), it is helpful to know that their risk of VTE may be reduced without exposure to the risks of other kinds of conventional thromboprophylaxis.
We look forward to additional studies to elaborate on the benefits of statins in both the prevention and treatment of VTE for averagerisk and VTE-prone populations.
- Glynn RJ, Danielson E, Fonseca FA, et al. A randomized trial of rosuvastatin in the prevention of venous thromboembolism. N Engl J Med 2009; 360:1851–1861.
- Prandoni P, Bilora F, Marchiori A, et al. An association between atherosclerosis and venous thrombosis. N Engl J Med 2003; 348:1435–1441.
- Prandoni P, Ghirarduzzi A, Prins MH, et al. Venous thromboembolism and the risk of subsequent symptomatic atherosclerosis. J Thromb Haemost 2006; 4:1891–1896.
- Braekkan SK, Mathiesen EB, Njolstad I, Wilsgaard T, Stormer J, Hansen JB. Family history of myocardial infarction is an independent risk factor for venous thromboembolism: the Tromso study. J Thromb Haemost 2008; 6:1851–1857.
- Tsai AW, Cushman M, Rosamond WD, Heckbert SR, Polak JF, Folsom AR. Cardiovascular risk factors and venous thromboembolism incidence: the longitudinal investigation of thromboembolism etiology. Arch Intern Med 2002; 162:1182–1189.
- van der Hagen PB, Folsom AR, Jenny NS, et al. Subclinical atherosclerosis and the risk of future venous thrombosis in the Cardiovascular Health Study. J Thromb Haemost 2006; 4:1903–1908.
- Reich LM, Folsom AR, Key NS, et al. Prospective study of subclinical atherosclerosis as a risk factor for venous thromboembolism. J Thromb Haemost 2006; 4:1909–1913.
- Huerta C, Johansson S, Wallander MA, Rodriguez LA. Risk of myocardial infarction and overall mortality in survivors of venous thromboembolism. Thromb J 2008; 6:10.
- Linnemann B, Schindewolf M, Zgouras D, Erbe M, Jarosch-Preusche M, Lindhoff-Last E. Are patients with thrombophilia and previous venous thromboembolism at higher risk to arterial thrombosis? Thromb Res 2008; 121:743–750.
- Schwaiger J, Kiechl S, Stockner H, et al. Burden of atherosclerosis and risk of venous thromboembolism in patients with migraine. Neurology 2008; 71:937–943.
- Linnemann B, Zgouras D, Schindewolf M, Schwonberg J, Jarosch-Preusche M, Lindhoff-Last E. Impact of sex and traditional cardiovascular risk factors on the risk of recurrent venous thromboembolism: results from the German MAISTHRO Registry. Blood Coagul Fibrinolysis 2008; 19:159–165.
- Steffen LM, Folsom AR, Cushman M, Jacobs DR, Rosamond WD. Greater fish, fruit, and vegetable intakes are related to lower incidence of venous thromboembolism: the Longitudinal Investigation of Thromboembolism Etiology. Circulation 2007; 115:188–195.
- Arslan F, Pasterkamp G, de Kleijn DP. Unraveling pleiotropic effects of statins: bit by bit, a slow case with perspective. Circ Res 2008; 103:334–336.
- Grady D, Wenger NK, Herrington D, et al. Postmenopausal hormone therapy increases risk for venous thromboembolic disease. The Heart and Estrogen/progestin Replacement Study. Ann Intern Med 2000; 132:689–696.
- Ray JG, Mamdani M, Tsuyuki RT, Anderson DR, Yeo EL, Laupacis A. Use of statins and the subsequent development of deep vein thrombosis. Arch Intern Med 2001; 161:1405–1410.
- Doggen CJ, Lemaitre RN, Smith NL, Heckbert SR, Psaty BM. HMG CoA reductase inhibitors and the risk of venous thrombosis among postmenopausal women. J Thromb Haemost 2004; 2:700–701.
- Lacut K, Oger E, Le Gal G, et al. Statins but not fibrates are associated with a reduced risk of venous thromboembolism: a hospitalbased case-control study. Fundam Clin Pharmacol 2004; 18:477–482.
- Ramcharan AS, Van Stralen KJ, Snoep JD, Mantel-Teeuwisse AK, Rosendaal FR, Doggen CJ. HMG-CoA reductase inhibitors, other lipid-lowering medication, antiplatelet therapy, and the risk of venous thrombosis. J Thromb Haemost 2009; 7:514–520.
- Sørensen HT, Horvath-Puho E, Sogaard KK, et al. Arterial cardiovascular events, statins, low-dose aspirin and subsequent risk of venous thromboembolism: a population-based case-control study. J Thromb Haemost 2009; 7:521–528.
- Yang CC, Jick SS, Jick H. Statins and the risk of idiopathic venous thromboembolism. Br J Clin Pharmacol 2002; 53:101–105.
- Smeeth L, Douglas I, Hall AJ, Hubbard R, Evans S. Effect of statins on a wide range of health outcomes: a cohort study validated by comparison with randomized trials. Br J Clin Pharmacol 2009; 67:99–109.
- Ridker PM, Danielson E, Fonseca FA, et al; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Shishehbor MH, Hazen SL. JUPITER to Earth: A statin helps peole with normal LDL-C and high hs-CRP, but what does it mean? Cleve Clin J Med 2009; 76:37–44.
- Cushman M. A new indication for statins to prevent venous thromboembolism? Not yet. J Thromb Haemost 2009; 7:511–513.
- Silva MA, Swanson AC, Gandhi PJ, Tataronis GR. Statin-related adverse events: a meta-analysis. Clin Ther 2006; 28:26–35.
A major placebo-controlled trial has found that a statin can reduce the risk of venous thromboembolism (VTE).1
We do not recommend prescribing this class of drugs for this purpose until much more research has been done, and we certainly do not recommend substituting a statin for anticoagulant therapy in a patient at risk of VTE.
Nevertheless, we are excited by the latest findings, and we find comfort in knowing that if a patient is taking a statin for an approved indication, ie, reducing the risk of cardiovascular disease in a patient with hyperlipidemia or a previous cardiovascular event, the drug will also reduce the risk of VTE.
In the pages that follow, we describe and comment on what is known about the effect of statins on the risk of VTE.
ARTERIAL AND VENOUS THROMBOSIS: HOW ARE THEY LINKED?
The causes of arterial thrombosis may not be entirely distinct from those of deep vein thrombosis and pulmonary embolism, collectively referred to as VTE. Some studies have found that risk factors for arterial thrombosis overlap with those for VTE.2–4 However, other studies have shown no association between venous and arterial events.5–10
Hyperlipidemia, in particular, has been evaluated to see if it is a risk factor for VTE. As with other risk factors for arterial thrombosis, the data have been mixed, with some reports favoring an association with VTE and others not.4,5,11 Even so, preventive strategies targeting arterial risk factors have shown promise in reducing VTE events.12
Although commonly used to treat hyperlipidemia, statins (3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors) are believed to reduce the incidence of thrombosis by a number of mechanisms13:
- Decreasing platelet aggregation
- Inhibiting expression of tissue factor and plasminogen activator inhibitor 1
- Increasing expression of tissue plasminogen activator
- Increasing expression of thrombomodulin, which can activate protein C and prevent thrombin-induced platelet and factor V activation and fibrinogen clotting.
STATINS AND VTE IN OBSERVATIONAL AND CASE-CONTROL STUDIES
In view of the multiple effects of statins, several studies have looked at whether these drugs reduce the occurrence of both arterial thrombosis and VTE.14–19
Two prospective observational studies and four case-control studies found that statins reduced the risk of VTE by 20% to 60%.14–19 Interestingly, two of the case-control studies found that antiplatelet therapy did not reduce the risk of VTE.18,19
THE JUPITER STUDY
The Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) study primarily sought to determine if rosuvastatin (Crestor) 20 mg/day, compared with placebo, would reduce the rate of first major cardiovascular events.22 A prespecified secondary end point of the trial was VTE, making JUPITER the first randomized, placebo-controlled trial to specifically test whether statins prevent VTE.1
Inclusion criteria: Normal LDL, high CRP
The study included men age 50 and older and women age 60 and older with no history of cardiovascular disease. In addition, their lowdensity lipoprotein (LDL) cholesterol levels had to be lower than 130 mg/dL (3.4 mmol/L), their triglyceride levels had to be lower than 500 mg/dL (5.6 mmol/L), and their highsensitivity C-reactive protein (hs-CRP) levels had to be 2.0 mg/L or higher.
Since high levels of hs-CRP, a marker of inflammation, predict cardiovascular events and since statins lower hs-CRP levels, the investigators hypothesized that people with elevated hs-CRP but without hyperlipidemia might benefit from statin treatment.21
Patients were excluded if they had received lipid-lowering therapy within 6 weeks of the trial screening, had diabetes mellitus or uncontrolled hypertension, were currently using postmenopausal hormone-replacement therapy, or had had cancer within the previous 5 years, except for certain skin cancers.
Candidates who complied well during a 4-week placebo run-in phase were randomly assigned to receive either rosuvastatin 20 mg daily (an intermediate dose) or a matching placebo. In all, 17,802 people were randomized. The two assigned groups appeared to be well matched.
Patients were to come in for visits twice a year for 60 months after randomization to be assessed for symptomatic deep venous thrombosis and pulmonary embolism. New cases of VTE were confirmed by imaging studies, by the initiation of anticoagulation therapy, or by death ascribed to pulmonary embolism.
Idiopathic VTE was classified as unprovoked if it occurred in the absence of trauma, hospitalization, or surgery within 3 months before the event, and in the absence of any diagnosed cancer within 3 months before and after the event. Provoked VTE events were those that occurred in a participant with cancer or when a precipitating event was associated with trauma, hospitalization, or surgery.
Rosuvastatin prevents heart attack, stroke
On the recommendation of the trial’s independent data and safety monitoring board, JUPITER was stopped early because the trial drug showed evidence of efficacy in preventing the combined primary end point of a first major cardiovascular event—ie, nonfatal myocardial infarction, nonfatal stroke, hospitalization for unstable angina, an arterial revascularization procedure, or confirmed death from a cardiovascular cause.22 (The cardiovascular outcomes of the JUPITER study were reviewed by Shishehbor and Hazen23 in the January 2009 issue of the Cleveland Clinic Journal of Medicine; see doi:10.3949/ccjm.75a.08105).
Formal follow-up for the trial's primary and secondary efficacy end points ended then, but data on VTE continued to be collected until each patient’s closeout visit as part of a safety monitoring protocol. The last closeout visit occurred on August 20, 2008. The primary analysis focused on events occurring up to March 30, 2008, the date the study was stopped.
Secondary end point results: Rosuvastatin prevents VTE
At a median follow-up of 1.9 years, an episode of VTE had occurred in 94 (0.53%) of the 17,802 patients—34 in the rosuvastatin group and 60 in the placebo group.1 This translates to 0.18 and 0.32 events per 100 person-years of follow-up in the rosuvastatin and placebo groups, respectively (hazard ratio for the rosuvastatin group 0.57, 95% confidence interval [CI] 0.37–0.86, P = .007).
Forty-four cases of VTE were classified as provoked and 50 cases were categorized as unprovoked. The risk reduction was statistically significant for provoked cases (hazard ratio 0.52, 95% CI 0.28–0.96, P = .03), but not for unprovoked events (hazard ratio 0.61, 95% CI 0.35–1.09, P = .09).
Subgroup analysis revealed no significant association between patient characteristics and the impact of rosuvastatin on the risk of a VTE event, but, as expected, more benefit was associated with higher baseline lipid levels.
STILL TOO SOON TO ADVISE ROUTINE STATIN USE TO PREVENT VTE
While the JUPITER trial data show an apparent benefit of statin use on the rate of VTE events, advising routine use of statins to prevent VTE is premature, for three main reasons.
Many must be treated to prevent one case of VTE. The number needed to treat (NNT) with rosuvastatin for 5 years to prevent either a case of VTE or a cardiovascular event was 21, and the NNT to prevent one cardiovascular event was 25. In a review of the two most recent case-control studies investigating the effects of statins on VTE,18,19 Cushman24 calculated that the NNT to prevent one VTE event each year was 333 for those age 75 and older. Though the Jupiter data did not provide the specific incidence of VTE at 1 year, except graphically, we can estimate that the NNT to prevent one VTE event at 1 year in the study is also very high.
Practically speaking, the perceived benefits of VTE prevention require large numbers to be treated, and the net clinical gain is still largely in preventing arterial events such as heart attack and stroke rather than VTE.
Statins, though safe, can still have adverse effects. The JUPITER study found a trend (albeit nonsignificant) toward more muscle complaints and elevations on liver function testing in apparently healthy persons taking a statin.22 Although severe complications of statin therapy such as rhabdomyolysis and elevations of creatine phosphokinase are rare, patients taking a statin have a 39% higher risk of an adverse event, most commonly myalgias or abnormalities on liver function testing.25 Were statins to be given routinely to even more people than they are now, more adverse outcomes would be likely.
More study is needed. The JUPITER study did not address a high risk of VTE. In fact, the investigators provided no information as to the VTE history of those enrolled.
Clearly, statins should not be substituted for proven prophylaxis and anticoagulation without further investigation, especially for patients with recurrent deep venous thrombosis, hospitalized patients, postoperative patients, and other patients prone to VTE.
OUR VIEW
The JUPITER study is an important leap forward in adding to our knowledge of how to prevent VTE. For people with another indication for taking a statin (eg, a previous cardiovascular event, hyperlipidemia), it is helpful to know that their risk of VTE may be reduced without exposure to the risks of other kinds of conventional thromboprophylaxis.
We look forward to additional studies to elaborate on the benefits of statins in both the prevention and treatment of VTE for averagerisk and VTE-prone populations.
A major placebo-controlled trial has found that a statin can reduce the risk of venous thromboembolism (VTE).1
We do not recommend prescribing this class of drugs for this purpose until much more research has been done, and we certainly do not recommend substituting a statin for anticoagulant therapy in a patient at risk of VTE.
Nevertheless, we are excited by the latest findings, and we find comfort in knowing that if a patient is taking a statin for an approved indication, ie, reducing the risk of cardiovascular disease in a patient with hyperlipidemia or a previous cardiovascular event, the drug will also reduce the risk of VTE.
In the pages that follow, we describe and comment on what is known about the effect of statins on the risk of VTE.
ARTERIAL AND VENOUS THROMBOSIS: HOW ARE THEY LINKED?
The causes of arterial thrombosis may not be entirely distinct from those of deep vein thrombosis and pulmonary embolism, collectively referred to as VTE. Some studies have found that risk factors for arterial thrombosis overlap with those for VTE.2–4 However, other studies have shown no association between venous and arterial events.5–10
Hyperlipidemia, in particular, has been evaluated to see if it is a risk factor for VTE. As with other risk factors for arterial thrombosis, the data have been mixed, with some reports favoring an association with VTE and others not.4,5,11 Even so, preventive strategies targeting arterial risk factors have shown promise in reducing VTE events.12
Although commonly used to treat hyperlipidemia, statins (3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors) are believed to reduce the incidence of thrombosis by a number of mechanisms13:
- Decreasing platelet aggregation
- Inhibiting expression of tissue factor and plasminogen activator inhibitor 1
- Increasing expression of tissue plasminogen activator
- Increasing expression of thrombomodulin, which can activate protein C and prevent thrombin-induced platelet and factor V activation and fibrinogen clotting.
STATINS AND VTE IN OBSERVATIONAL AND CASE-CONTROL STUDIES
In view of the multiple effects of statins, several studies have looked at whether these drugs reduce the occurrence of both arterial thrombosis and VTE.14–19
Two prospective observational studies and four case-control studies found that statins reduced the risk of VTE by 20% to 60%.14–19 Interestingly, two of the case-control studies found that antiplatelet therapy did not reduce the risk of VTE.18,19
THE JUPITER STUDY
The Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) study primarily sought to determine if rosuvastatin (Crestor) 20 mg/day, compared with placebo, would reduce the rate of first major cardiovascular events.22 A prespecified secondary end point of the trial was VTE, making JUPITER the first randomized, placebo-controlled trial to specifically test whether statins prevent VTE.1
Inclusion criteria: Normal LDL, high CRP
The study included men age 50 and older and women age 60 and older with no history of cardiovascular disease. In addition, their lowdensity lipoprotein (LDL) cholesterol levels had to be lower than 130 mg/dL (3.4 mmol/L), their triglyceride levels had to be lower than 500 mg/dL (5.6 mmol/L), and their highsensitivity C-reactive protein (hs-CRP) levels had to be 2.0 mg/L or higher.
Since high levels of hs-CRP, a marker of inflammation, predict cardiovascular events and since statins lower hs-CRP levels, the investigators hypothesized that people with elevated hs-CRP but without hyperlipidemia might benefit from statin treatment.21
Patients were excluded if they had received lipid-lowering therapy within 6 weeks of the trial screening, had diabetes mellitus or uncontrolled hypertension, were currently using postmenopausal hormone-replacement therapy, or had had cancer within the previous 5 years, except for certain skin cancers.
Candidates who complied well during a 4-week placebo run-in phase were randomly assigned to receive either rosuvastatin 20 mg daily (an intermediate dose) or a matching placebo. In all, 17,802 people were randomized. The two assigned groups appeared to be well matched.
Patients were to come in for visits twice a year for 60 months after randomization to be assessed for symptomatic deep venous thrombosis and pulmonary embolism. New cases of VTE were confirmed by imaging studies, by the initiation of anticoagulation therapy, or by death ascribed to pulmonary embolism.
Idiopathic VTE was classified as unprovoked if it occurred in the absence of trauma, hospitalization, or surgery within 3 months before the event, and in the absence of any diagnosed cancer within 3 months before and after the event. Provoked VTE events were those that occurred in a participant with cancer or when a precipitating event was associated with trauma, hospitalization, or surgery.
Rosuvastatin prevents heart attack, stroke
On the recommendation of the trial’s independent data and safety monitoring board, JUPITER was stopped early because the trial drug showed evidence of efficacy in preventing the combined primary end point of a first major cardiovascular event—ie, nonfatal myocardial infarction, nonfatal stroke, hospitalization for unstable angina, an arterial revascularization procedure, or confirmed death from a cardiovascular cause.22 (The cardiovascular outcomes of the JUPITER study were reviewed by Shishehbor and Hazen23 in the January 2009 issue of the Cleveland Clinic Journal of Medicine; see doi:10.3949/ccjm.75a.08105).
Formal follow-up for the trial's primary and secondary efficacy end points ended then, but data on VTE continued to be collected until each patient’s closeout visit as part of a safety monitoring protocol. The last closeout visit occurred on August 20, 2008. The primary analysis focused on events occurring up to March 30, 2008, the date the study was stopped.
Secondary end point results: Rosuvastatin prevents VTE
At a median follow-up of 1.9 years, an episode of VTE had occurred in 94 (0.53%) of the 17,802 patients—34 in the rosuvastatin group and 60 in the placebo group.1 This translates to 0.18 and 0.32 events per 100 person-years of follow-up in the rosuvastatin and placebo groups, respectively (hazard ratio for the rosuvastatin group 0.57, 95% confidence interval [CI] 0.37–0.86, P = .007).
Forty-four cases of VTE were classified as provoked and 50 cases were categorized as unprovoked. The risk reduction was statistically significant for provoked cases (hazard ratio 0.52, 95% CI 0.28–0.96, P = .03), but not for unprovoked events (hazard ratio 0.61, 95% CI 0.35–1.09, P = .09).
Subgroup analysis revealed no significant association between patient characteristics and the impact of rosuvastatin on the risk of a VTE event, but, as expected, more benefit was associated with higher baseline lipid levels.
STILL TOO SOON TO ADVISE ROUTINE STATIN USE TO PREVENT VTE
While the JUPITER trial data show an apparent benefit of statin use on the rate of VTE events, advising routine use of statins to prevent VTE is premature, for three main reasons.
Many must be treated to prevent one case of VTE. The number needed to treat (NNT) with rosuvastatin for 5 years to prevent either a case of VTE or a cardiovascular event was 21, and the NNT to prevent one cardiovascular event was 25. In a review of the two most recent case-control studies investigating the effects of statins on VTE,18,19 Cushman24 calculated that the NNT to prevent one VTE event each year was 333 for those age 75 and older. Though the Jupiter data did not provide the specific incidence of VTE at 1 year, except graphically, we can estimate that the NNT to prevent one VTE event at 1 year in the study is also very high.
Practically speaking, the perceived benefits of VTE prevention require large numbers to be treated, and the net clinical gain is still largely in preventing arterial events such as heart attack and stroke rather than VTE.
Statins, though safe, can still have adverse effects. The JUPITER study found a trend (albeit nonsignificant) toward more muscle complaints and elevations on liver function testing in apparently healthy persons taking a statin.22 Although severe complications of statin therapy such as rhabdomyolysis and elevations of creatine phosphokinase are rare, patients taking a statin have a 39% higher risk of an adverse event, most commonly myalgias or abnormalities on liver function testing.25 Were statins to be given routinely to even more people than they are now, more adverse outcomes would be likely.
More study is needed. The JUPITER study did not address a high risk of VTE. In fact, the investigators provided no information as to the VTE history of those enrolled.
Clearly, statins should not be substituted for proven prophylaxis and anticoagulation without further investigation, especially for patients with recurrent deep venous thrombosis, hospitalized patients, postoperative patients, and other patients prone to VTE.
OUR VIEW
The JUPITER study is an important leap forward in adding to our knowledge of how to prevent VTE. For people with another indication for taking a statin (eg, a previous cardiovascular event, hyperlipidemia), it is helpful to know that their risk of VTE may be reduced without exposure to the risks of other kinds of conventional thromboprophylaxis.
We look forward to additional studies to elaborate on the benefits of statins in both the prevention and treatment of VTE for averagerisk and VTE-prone populations.
- Glynn RJ, Danielson E, Fonseca FA, et al. A randomized trial of rosuvastatin in the prevention of venous thromboembolism. N Engl J Med 2009; 360:1851–1861.
- Prandoni P, Bilora F, Marchiori A, et al. An association between atherosclerosis and venous thrombosis. N Engl J Med 2003; 348:1435–1441.
- Prandoni P, Ghirarduzzi A, Prins MH, et al. Venous thromboembolism and the risk of subsequent symptomatic atherosclerosis. J Thromb Haemost 2006; 4:1891–1896.
- Braekkan SK, Mathiesen EB, Njolstad I, Wilsgaard T, Stormer J, Hansen JB. Family history of myocardial infarction is an independent risk factor for venous thromboembolism: the Tromso study. J Thromb Haemost 2008; 6:1851–1857.
- Tsai AW, Cushman M, Rosamond WD, Heckbert SR, Polak JF, Folsom AR. Cardiovascular risk factors and venous thromboembolism incidence: the longitudinal investigation of thromboembolism etiology. Arch Intern Med 2002; 162:1182–1189.
- van der Hagen PB, Folsom AR, Jenny NS, et al. Subclinical atherosclerosis and the risk of future venous thrombosis in the Cardiovascular Health Study. J Thromb Haemost 2006; 4:1903–1908.
- Reich LM, Folsom AR, Key NS, et al. Prospective study of subclinical atherosclerosis as a risk factor for venous thromboembolism. J Thromb Haemost 2006; 4:1909–1913.
- Huerta C, Johansson S, Wallander MA, Rodriguez LA. Risk of myocardial infarction and overall mortality in survivors of venous thromboembolism. Thromb J 2008; 6:10.
- Linnemann B, Schindewolf M, Zgouras D, Erbe M, Jarosch-Preusche M, Lindhoff-Last E. Are patients with thrombophilia and previous venous thromboembolism at higher risk to arterial thrombosis? Thromb Res 2008; 121:743–750.
- Schwaiger J, Kiechl S, Stockner H, et al. Burden of atherosclerosis and risk of venous thromboembolism in patients with migraine. Neurology 2008; 71:937–943.
- Linnemann B, Zgouras D, Schindewolf M, Schwonberg J, Jarosch-Preusche M, Lindhoff-Last E. Impact of sex and traditional cardiovascular risk factors on the risk of recurrent venous thromboembolism: results from the German MAISTHRO Registry. Blood Coagul Fibrinolysis 2008; 19:159–165.
- Steffen LM, Folsom AR, Cushman M, Jacobs DR, Rosamond WD. Greater fish, fruit, and vegetable intakes are related to lower incidence of venous thromboembolism: the Longitudinal Investigation of Thromboembolism Etiology. Circulation 2007; 115:188–195.
- Arslan F, Pasterkamp G, de Kleijn DP. Unraveling pleiotropic effects of statins: bit by bit, a slow case with perspective. Circ Res 2008; 103:334–336.
- Grady D, Wenger NK, Herrington D, et al. Postmenopausal hormone therapy increases risk for venous thromboembolic disease. The Heart and Estrogen/progestin Replacement Study. Ann Intern Med 2000; 132:689–696.
- Ray JG, Mamdani M, Tsuyuki RT, Anderson DR, Yeo EL, Laupacis A. Use of statins and the subsequent development of deep vein thrombosis. Arch Intern Med 2001; 161:1405–1410.
- Doggen CJ, Lemaitre RN, Smith NL, Heckbert SR, Psaty BM. HMG CoA reductase inhibitors and the risk of venous thrombosis among postmenopausal women. J Thromb Haemost 2004; 2:700–701.
- Lacut K, Oger E, Le Gal G, et al. Statins but not fibrates are associated with a reduced risk of venous thromboembolism: a hospitalbased case-control study. Fundam Clin Pharmacol 2004; 18:477–482.
- Ramcharan AS, Van Stralen KJ, Snoep JD, Mantel-Teeuwisse AK, Rosendaal FR, Doggen CJ. HMG-CoA reductase inhibitors, other lipid-lowering medication, antiplatelet therapy, and the risk of venous thrombosis. J Thromb Haemost 2009; 7:514–520.
- Sørensen HT, Horvath-Puho E, Sogaard KK, et al. Arterial cardiovascular events, statins, low-dose aspirin and subsequent risk of venous thromboembolism: a population-based case-control study. J Thromb Haemost 2009; 7:521–528.
- Yang CC, Jick SS, Jick H. Statins and the risk of idiopathic venous thromboembolism. Br J Clin Pharmacol 2002; 53:101–105.
- Smeeth L, Douglas I, Hall AJ, Hubbard R, Evans S. Effect of statins on a wide range of health outcomes: a cohort study validated by comparison with randomized trials. Br J Clin Pharmacol 2009; 67:99–109.
- Ridker PM, Danielson E, Fonseca FA, et al; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Shishehbor MH, Hazen SL. JUPITER to Earth: A statin helps peole with normal LDL-C and high hs-CRP, but what does it mean? Cleve Clin J Med 2009; 76:37–44.
- Cushman M. A new indication for statins to prevent venous thromboembolism? Not yet. J Thromb Haemost 2009; 7:511–513.
- Silva MA, Swanson AC, Gandhi PJ, Tataronis GR. Statin-related adverse events: a meta-analysis. Clin Ther 2006; 28:26–35.
- Glynn RJ, Danielson E, Fonseca FA, et al. A randomized trial of rosuvastatin in the prevention of venous thromboembolism. N Engl J Med 2009; 360:1851–1861.
- Prandoni P, Bilora F, Marchiori A, et al. An association between atherosclerosis and venous thrombosis. N Engl J Med 2003; 348:1435–1441.
- Prandoni P, Ghirarduzzi A, Prins MH, et al. Venous thromboembolism and the risk of subsequent symptomatic atherosclerosis. J Thromb Haemost 2006; 4:1891–1896.
- Braekkan SK, Mathiesen EB, Njolstad I, Wilsgaard T, Stormer J, Hansen JB. Family history of myocardial infarction is an independent risk factor for venous thromboembolism: the Tromso study. J Thromb Haemost 2008; 6:1851–1857.
- Tsai AW, Cushman M, Rosamond WD, Heckbert SR, Polak JF, Folsom AR. Cardiovascular risk factors and venous thromboembolism incidence: the longitudinal investigation of thromboembolism etiology. Arch Intern Med 2002; 162:1182–1189.
- van der Hagen PB, Folsom AR, Jenny NS, et al. Subclinical atherosclerosis and the risk of future venous thrombosis in the Cardiovascular Health Study. J Thromb Haemost 2006; 4:1903–1908.
- Reich LM, Folsom AR, Key NS, et al. Prospective study of subclinical atherosclerosis as a risk factor for venous thromboembolism. J Thromb Haemost 2006; 4:1909–1913.
- Huerta C, Johansson S, Wallander MA, Rodriguez LA. Risk of myocardial infarction and overall mortality in survivors of venous thromboembolism. Thromb J 2008; 6:10.
- Linnemann B, Schindewolf M, Zgouras D, Erbe M, Jarosch-Preusche M, Lindhoff-Last E. Are patients with thrombophilia and previous venous thromboembolism at higher risk to arterial thrombosis? Thromb Res 2008; 121:743–750.
- Schwaiger J, Kiechl S, Stockner H, et al. Burden of atherosclerosis and risk of venous thromboembolism in patients with migraine. Neurology 2008; 71:937–943.
- Linnemann B, Zgouras D, Schindewolf M, Schwonberg J, Jarosch-Preusche M, Lindhoff-Last E. Impact of sex and traditional cardiovascular risk factors on the risk of recurrent venous thromboembolism: results from the German MAISTHRO Registry. Blood Coagul Fibrinolysis 2008; 19:159–165.
- Steffen LM, Folsom AR, Cushman M, Jacobs DR, Rosamond WD. Greater fish, fruit, and vegetable intakes are related to lower incidence of venous thromboembolism: the Longitudinal Investigation of Thromboembolism Etiology. Circulation 2007; 115:188–195.
- Arslan F, Pasterkamp G, de Kleijn DP. Unraveling pleiotropic effects of statins: bit by bit, a slow case with perspective. Circ Res 2008; 103:334–336.
- Grady D, Wenger NK, Herrington D, et al. Postmenopausal hormone therapy increases risk for venous thromboembolic disease. The Heart and Estrogen/progestin Replacement Study. Ann Intern Med 2000; 132:689–696.
- Ray JG, Mamdani M, Tsuyuki RT, Anderson DR, Yeo EL, Laupacis A. Use of statins and the subsequent development of deep vein thrombosis. Arch Intern Med 2001; 161:1405–1410.
- Doggen CJ, Lemaitre RN, Smith NL, Heckbert SR, Psaty BM. HMG CoA reductase inhibitors and the risk of venous thrombosis among postmenopausal women. J Thromb Haemost 2004; 2:700–701.
- Lacut K, Oger E, Le Gal G, et al. Statins but not fibrates are associated with a reduced risk of venous thromboembolism: a hospitalbased case-control study. Fundam Clin Pharmacol 2004; 18:477–482.
- Ramcharan AS, Van Stralen KJ, Snoep JD, Mantel-Teeuwisse AK, Rosendaal FR, Doggen CJ. HMG-CoA reductase inhibitors, other lipid-lowering medication, antiplatelet therapy, and the risk of venous thrombosis. J Thromb Haemost 2009; 7:514–520.
- Sørensen HT, Horvath-Puho E, Sogaard KK, et al. Arterial cardiovascular events, statins, low-dose aspirin and subsequent risk of venous thromboembolism: a population-based case-control study. J Thromb Haemost 2009; 7:521–528.
- Yang CC, Jick SS, Jick H. Statins and the risk of idiopathic venous thromboembolism. Br J Clin Pharmacol 2002; 53:101–105.
- Smeeth L, Douglas I, Hall AJ, Hubbard R, Evans S. Effect of statins on a wide range of health outcomes: a cohort study validated by comparison with randomized trials. Br J Clin Pharmacol 2009; 67:99–109.
- Ridker PM, Danielson E, Fonseca FA, et al; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Shishehbor MH, Hazen SL. JUPITER to Earth: A statin helps peole with normal LDL-C and high hs-CRP, but what does it mean? Cleve Clin J Med 2009; 76:37–44.
- Cushman M. A new indication for statins to prevent venous thromboembolism? Not yet. J Thromb Haemost 2009; 7:511–513.
- Silva MA, Swanson AC, Gandhi PJ, Tataronis GR. Statin-related adverse events: a meta-analysis. Clin Ther 2006; 28:26–35.
KEY POINTS
- Risk factors for VTE overlap with those for arterial thrombosis, although the data are mixed.
- The statin drugs have a number of effects on factors other than lipid levels, notably on markers of inflammation and on clotting factors.
- In the JUPITER trial, the incidence of VTE in people taking rosuvastatin (Crestor) 20 mg/day was about half that in people taking placebo. This was a relatively healthy population, and the incidence in both groups was low.
- Further study is needed in patients at risk of VTE.
Kidney stones
To the Editor: Thanks for the excellent review articles on nephrolithiasis in your October 2009 issue.1,2
Dr. Hall1 cites studies in which patients given the alpha blocker tamsulosin (Flomax) or the calcium channel blocker nifedipine (Procardia) had improved rates of kidney stone passage compared with placebo. As a primary care physician, I am often confronted with the challenge of managing a patient who is waiting for a kidney stone to pass while taking tamsulosin. Is Dr. Hall aware of any clinical studies, or at least theoretical reasons, which would support adding nifedipine in such cases?
Secondly, Dr. Hall cites studies that demonstrated that a higher intake of dietary calcium is actually associated with fewer calcium stone events in both men and women. An unanswered question is whether patients taking calcium supplements for osteoporosis or osteopenia can safely continue to do so after a calcium stone event, or indeed, whether calcium supplementation might actually be helpful in preventing a recurrent calcum stone.
If there is an absence of randomized studies to answer these questions, Dr. Hall’s recommendations based on his expert experience would be most welcome.
- Hall PM. Nephrolithiasis: treatment, causes, and prevention. Cleve Clin J Med 2009; 76:583–591.
- Samplaski MK, Irwin BH, Desai M. Less-invasive ways to remove stones from the kidneys and ureters. Cleve Clin J Med 2009; 76:592–598.
To the Editor: Thanks for the excellent review articles on nephrolithiasis in your October 2009 issue.1,2
Dr. Hall1 cites studies in which patients given the alpha blocker tamsulosin (Flomax) or the calcium channel blocker nifedipine (Procardia) had improved rates of kidney stone passage compared with placebo. As a primary care physician, I am often confronted with the challenge of managing a patient who is waiting for a kidney stone to pass while taking tamsulosin. Is Dr. Hall aware of any clinical studies, or at least theoretical reasons, which would support adding nifedipine in such cases?
Secondly, Dr. Hall cites studies that demonstrated that a higher intake of dietary calcium is actually associated with fewer calcium stone events in both men and women. An unanswered question is whether patients taking calcium supplements for osteoporosis or osteopenia can safely continue to do so after a calcium stone event, or indeed, whether calcium supplementation might actually be helpful in preventing a recurrent calcum stone.
If there is an absence of randomized studies to answer these questions, Dr. Hall’s recommendations based on his expert experience would be most welcome.
To the Editor: Thanks for the excellent review articles on nephrolithiasis in your October 2009 issue.1,2
Dr. Hall1 cites studies in which patients given the alpha blocker tamsulosin (Flomax) or the calcium channel blocker nifedipine (Procardia) had improved rates of kidney stone passage compared with placebo. As a primary care physician, I am often confronted with the challenge of managing a patient who is waiting for a kidney stone to pass while taking tamsulosin. Is Dr. Hall aware of any clinical studies, or at least theoretical reasons, which would support adding nifedipine in such cases?
Secondly, Dr. Hall cites studies that demonstrated that a higher intake of dietary calcium is actually associated with fewer calcium stone events in both men and women. An unanswered question is whether patients taking calcium supplements for osteoporosis or osteopenia can safely continue to do so after a calcium stone event, or indeed, whether calcium supplementation might actually be helpful in preventing a recurrent calcum stone.
If there is an absence of randomized studies to answer these questions, Dr. Hall’s recommendations based on his expert experience would be most welcome.
- Hall PM. Nephrolithiasis: treatment, causes, and prevention. Cleve Clin J Med 2009; 76:583–591.
- Samplaski MK, Irwin BH, Desai M. Less-invasive ways to remove stones from the kidneys and ureters. Cleve Clin J Med 2009; 76:592–598.
- Hall PM. Nephrolithiasis: treatment, causes, and prevention. Cleve Clin J Med 2009; 76:583–591.
- Samplaski MK, Irwin BH, Desai M. Less-invasive ways to remove stones from the kidneys and ureters. Cleve Clin J Med 2009; 76:592–598.
In reply: Kidney stones
In Reply: I thank Dr. Keller for his kind letter.
With respect to expulsive therapy, Dellabella et al1 randomly assigned 210 patients to receive nifedipine, tamsulosin, or phloroglucinol. All the patients also received a corticosteroid. The most effective therapy was tamsulosin, though this was not a placebo-controlled study. In a separate study, Borghi et al2 compared methylprednisolone plus nifedipine and methylprednisolone plus placebo. The nifedipine-methylpednisolone combination seemed to result in more prompt stone passage.
With respect to calcium supplements in calcium kidney stone disease, Curhan et al3 prospectively examined stone risk associated with dietary calcium as well as calcium supplements. This seemed to show that with calcium supplements there was no increased risk, and there may have even been some benefit. In another study by Borghi et al,4 normal dietary calcium intake was shown to be associated with lower stone risk than a low calcium intake. Further, the study by Curhan et al3 seemed to indicate the same.
- Dellabella M, Milanese G, Muzzonigro G. Randomized trial of the efficacy of tamsulosin, nifedipine and phloroglucinol in medical expulsive therapy for distal ureteral calculi. J Urol 2005; 174:167–172.
- Borghi L, Meschi T, Amato F, et al. Nifedipine and methylprednisolone
in facilitating ureteral stone passage: a randomized, double blind, placebo-controlled study. J Urol 1994; 152:1095–1098. - Curhan GC, Willett WC, Knight EL, Stampfer MJ. Dietary factors and the risk of incident kidney stones in younger women: Nurses’ Health Study II. Arch Intern Med 2004; 164:885–891.
- Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med 2002; 346:77–84.
In Reply: I thank Dr. Keller for his kind letter.
With respect to expulsive therapy, Dellabella et al1 randomly assigned 210 patients to receive nifedipine, tamsulosin, or phloroglucinol. All the patients also received a corticosteroid. The most effective therapy was tamsulosin, though this was not a placebo-controlled study. In a separate study, Borghi et al2 compared methylprednisolone plus nifedipine and methylprednisolone plus placebo. The nifedipine-methylpednisolone combination seemed to result in more prompt stone passage.
With respect to calcium supplements in calcium kidney stone disease, Curhan et al3 prospectively examined stone risk associated with dietary calcium as well as calcium supplements. This seemed to show that with calcium supplements there was no increased risk, and there may have even been some benefit. In another study by Borghi et al,4 normal dietary calcium intake was shown to be associated with lower stone risk than a low calcium intake. Further, the study by Curhan et al3 seemed to indicate the same.
In Reply: I thank Dr. Keller for his kind letter.
With respect to expulsive therapy, Dellabella et al1 randomly assigned 210 patients to receive nifedipine, tamsulosin, or phloroglucinol. All the patients also received a corticosteroid. The most effective therapy was tamsulosin, though this was not a placebo-controlled study. In a separate study, Borghi et al2 compared methylprednisolone plus nifedipine and methylprednisolone plus placebo. The nifedipine-methylpednisolone combination seemed to result in more prompt stone passage.
With respect to calcium supplements in calcium kidney stone disease, Curhan et al3 prospectively examined stone risk associated with dietary calcium as well as calcium supplements. This seemed to show that with calcium supplements there was no increased risk, and there may have even been some benefit. In another study by Borghi et al,4 normal dietary calcium intake was shown to be associated with lower stone risk than a low calcium intake. Further, the study by Curhan et al3 seemed to indicate the same.
- Dellabella M, Milanese G, Muzzonigro G. Randomized trial of the efficacy of tamsulosin, nifedipine and phloroglucinol in medical expulsive therapy for distal ureteral calculi. J Urol 2005; 174:167–172.
- Borghi L, Meschi T, Amato F, et al. Nifedipine and methylprednisolone
in facilitating ureteral stone passage: a randomized, double blind, placebo-controlled study. J Urol 1994; 152:1095–1098. - Curhan GC, Willett WC, Knight EL, Stampfer MJ. Dietary factors and the risk of incident kidney stones in younger women: Nurses’ Health Study II. Arch Intern Med 2004; 164:885–891.
- Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med 2002; 346:77–84.
- Dellabella M, Milanese G, Muzzonigro G. Randomized trial of the efficacy of tamsulosin, nifedipine and phloroglucinol in medical expulsive therapy for distal ureteral calculi. J Urol 2005; 174:167–172.
- Borghi L, Meschi T, Amato F, et al. Nifedipine and methylprednisolone
in facilitating ureteral stone passage: a randomized, double blind, placebo-controlled study. J Urol 1994; 152:1095–1098. - Curhan GC, Willett WC, Knight EL, Stampfer MJ. Dietary factors and the risk of incident kidney stones in younger women: Nurses’ Health Study II. Arch Intern Med 2004; 164:885–891.
- Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med 2002; 346:77–84.
Managing acute upper GI bleeding, preventing recurrences
Upper gastrointestinal (GI) bleeding is common, costly, and potentially life-threatening. It must be managed promptly and appropriately to prevent adverse outcomes.
More people are admitted to the hospital for upper GI bleeding than for congestive heart failure or deep vein thrombosis. In the United States, the annual rate of hospitalization for upper GI bleeding is estimated to be 165 per 100,000—more than 300,000 hospitalizations per year, at a cost of $2.5 billion.1,2
Furthermore, despite advances in therapy, the case-fatality rate has remained unchanged at 7% to 10%.3 This may be because today’s patients are older and have more comorbidities than those in the past.4
CAUSES OF UPPER GI BLEEDING
Peptic ulcers account for about 60% of severe cases of upper GI bleeding,5 and they are the focus of this paper. Fortunately, up to 80% of bleeding ulcers stop bleeding spontaneously without any intervention.6
Gastroduodenal erosions account for about 12%.3
Varices due to cirrhosis are less common but more dangerous. Variceal bleeding accounts for a relatively small percentage (6%) of upper GI bleeding, but the mortality rate from a single episode of variceal bleeding is 30%, and 60% to 70% of patients die within 1 year, mostly of underlying liver disease.
Less frequent causes include Mallory-Weiss tears, erosive duodenitis, Dieulafoy ulcer (a type of vascular malformation), other vascular lesions, neoplasms, aortoenteric fistula, gastric antral vascular ectasia, and prolapse gastropathy.5
HEMATEMESIS AND MELENA
The most common presenting signs of acute upper GI bleeding are hematemesis (vomiting of blood), “coffee grounds” emesis, and melena (tarry black stools). About 30% of patients with bleeding ulcers present with hematemesis, 20% with melena, and 50% with both.7
Hematochezia (red blood in the stool) usually suggests a lower GI source of bleeding, since blood from an upper source turns black and tarry as it passes through the gut, producing melena. However, up to 5% of patients with bleeding ulcers have hematochezia,7 and it indicates heavy bleeding: bleeding of approximately 1,000 mL into the upper GI tract is needed to cause hematochezia, whereas only 50 to 100 mL is needed to cause melena.8,9 Hematochezia with signs and symptoms of hemodynamic compromise such as syncope, postural hypotension, tachycardia, and shock should therefore direct one’s attention to an upper GI source of bleeding.
Nonspecific features include nausea, vomiting, epigastric pain, vasovagal phenomena, and syncope.
WHAT IS THE PATIENT’S RISK?
An assessment of clinical severity is the first critical task, as it helps in planning treatment. Advanced age, multiple comorbidities, and hemodynamic instability call for aggressive treatment. Apart from this simple clinical rule, scoring systems have been developed.
The Rockall scoring system, the most widely used, gives estimates of the risks of recurrent bleeding and death. It is based on the three clinical factors mentioned above and on two endoscopic ones, awarding points for:
- Age—0 points if less than 60; 1 point if 60 to 79; or 2 points if 80 years or older
- Shock—1 point if the pulse is more than 100; 2 points if the systolic blood pressure is less than 100 mm Hg
- Comorbid illness—2 points for ischemic heart disease, congestive heart failure, or other major comorbidity; 3 points for renal failure, hepatic failure, or metastatic disease
- Endoscopic diagnosis—0 points if no lesion found or a Mallory-Weiss tear; 1 point for peptic ulcer, esophagitis, or erosive disease; 2 points for GI malignancy
- Endoscopic stigmata or recent hemorrhage—0 points for a clean-based ulcer or flat pigmented spot; 2 points for blood in the upper GI tract, active bleeding, a nonbleeding visible vessel, or adherent clot.
The Rockall score can thus range from 0 to 11 points, with an overall score of 0, 1, or 2 associated with an excellent prognosis.10

Other systems that are used less often include the Baylor severity scale and the Acute Physiology and Chronic Health Evaluation (APACHE) II score.
Does the patient have varices?
All variceal bleeding should be considered severe, since the 1-year death rate is so high (up to 70%). Clues pointing to variceal bleeding include previous variceal bleeding, thrombocytopenia, history of liver disease, and signs of liver disease on clinical examination.
All patients suspected of having bleeding varices should be admitted to the intensive care unit for close monitoring and should be given the highest priority, even if they are hemodynamically stable.
Is the patient hemodynamically stable?
Appropriate hemodynamic assessment includes monitoring of heart rate, blood pressure, and mental status. Tachycardia at rest, hypotension, and orthostatic changes in vital signs indicate a considerable loss of blood volume. Low urine output, dry mucous membranes, and sunken neck veins are also useful signs. (Tachycardia may be blunted if the patient is taking a beta-blocker.)
If these signs of hypovolemia are present, the initial management focuses on treating shock and on improving oxygen delivery to the vital organs. This involves repletion of the intravascular volume with intravenous infusions or blood transfusions. Supplemental oxygen also is useful, especially in elderly patients with heart disease.12
Inspection of nasogastric aspirate
In the initial assessment, it is useful to insert a nasogastric tube and inspect the aspirate. If it contains bright red blood, the patient needs an urgent endoscopic evaluation and an intensive level of care13,14; if it contains coffee-grounds material, the patient needs to be admitted to the hospital and to undergo endoscopic evaluation within 24 hours.
However, a normal aspirate does not rule out upper GI bleeding. Aljebreen et al15 found that 15% of patients with upper GI bleeding and normal nasogastric aspirate still had high-risk lesions (ie, visible bleeding or nonbleeding visible vessels) on endoscopy.
ACID-SUPPRESSION HELPS ULCERS HEAL
Acid and pepsin interfere with the healing of ulcers and other nonvariceal upper GI lesions. Further, an acidic environment promotes platelet disaggregation and fibrinolysis and impairs clot formation.16 This suggests that inhibiting gastric acid secretion and raising the gastric pH to 6 or higher may stabilize clots. Moreover, pepsinogen in the stomach is converted to its active form (pepsin) if the pH is less than 4. Therefore, keeping the pH above 4 keeps pepsinogen in an inactive form.
Histamine-2 receptor antagonists
Histamine-2 receptor antagonists were the first drugs to inhibit acid secretion, reversibly blocking histamine-2 receptors on the basolateral membrane of parietal cells. However, these drugs did not prove very useful in managing upper GI bleeding in clinical trials.17,18 In their intravenous form, they often fail to keep the gastric pH at 6 or higher, due to tachyphylaxis.19 The use of this class of drugs has declined in favor of proton pump inhibitors.
Proton pump inhibitors
Proton pump inhibitors reduce both basal and stimulated acid secretion by inhibiting hydrogen-potassium adenosine triphosphatase, the proton pump of the parietal cell.
Multiple studies have shown that proton pump inhibitors raise the gastric pH and keep it high. For example, an infusion of omeprazole (Prilosec) can keep the gastric pH above 6 for 72 hours without inducing tachyphylaxis.20,21
Started after endoscopy. Randomized controlled trials have found proton pump inhibitors to be effective when given in high doses intravenously for 72 hours after successful endoscopic treatment of bleeding ulcers with high-risk endoscopic signs, such as active bleeding or nonbleeding visible vessels.22,23
A meta-analysis indicated that these drugs decrease the incidence of recurrent peptic ulcer bleeding, the need for blood transfusions, the need for surgery, and the duration of hospitalization, but not the mortality rate.24,25 These studies also illustrate the benefit of following up endoscopic treatment to stop the bleeding with an intravenous infusion of a proton pump inhibitor.
The recommended dose of omeprazole for patients with high-risk findings on endoscopy is an 80-mg bolus followed by an 8-mg/hour infusion for 72 hours. After the patient’s condition stabilizes, oral therapy can be substituted for intravenous therapy. In patients with low-risk endoscopic findings (a clean-based ulcer or flat spot), oral proton pump inhibitors in high doses are recommended.
In either case, after the initial bleeding is treated endoscopically and hemostasis is achieved, a proton pump inhibitor is recommended for 6 to 8 weeks, or longer if the patient is also positive for Helicobacter pylori or is on daily treatment with aspirin or a nonsteroidal anti-inflammatory drug (NSAID) that is not selective for cyclo-oxygenase 2 (see below).
Started before endoscopy, these drugs reduced the frequency of actively bleeding ulcers, the duration of hospitalization, and the need for endoscopic therapy in a randomized controlled trial.26 A meta-analysis found that significantly fewer patients had signs of recent bleeding on endoscopy if they received a proton pump inhibitor 24 to 48 hours before the procedure, but it did not find any significant difference in important clinical outcomes such as death, recurrent bleeding, or surgery.27 Nevertheless, we believe that intravenous proton pump inhibitor therapy should be started before endoscopy in patients with upper GI bleeding.
Somatostatin analogues
Octreotide (Sandostatin), an analogue of the hormone somatostatin, decreases splanchnic blood flow, decreases secretion of gastric acid and pepsin, and stimulates mucus production. Although it is beneficial in treating upper GI bleeding due to varices, its benefit has not been confirmed in patients with nonvariceal upper GI bleeding.
A meta-analysis revealed that outcomes were better with high-dose intravenous proton pump inhibitor therapy than with octreotide when these drugs were started after endoscopic treatment of acute peptic ulcer bleeding.28 Nevertheless, octreotide may be useful in patients with uncontrolled nonvariceal bleeding who are awaiting endoscopy, since it is relatively safe to use.
ALL PATIENTS NEED ENDOSCOPY
All patients with upper GI bleeding need an upper endoscopic examination to diagnose and assess the risk posed by the bleeding lesion and to treat the lesion, reducing the risk of recurrent bleeding.
How urgently does endoscopy need to be done?
Endoscopy within the first 24 hours of upper GI bleeding is considered the standard of care. Patients with uncontrolled or recurrent bleeding should undergo endoscopy on an urgent basis to control the bleeding and reduce the risk of death.
However, how urgently endoscopy needs to be done is often debated. A multicenter randomized controlled trial compared outcomes in patients who underwent endoscopy within 6 hours of coming to the emergency department vs within 24 hours after the initial evaluation. The study found no significant difference in outcomes between the two groups; however, the group that underwent endoscopy sooner needed fewer transfusions.29
For a better view of the stomach
Gastric lavage improves the view of the gastric fundus but has not been proven to improve outcome.30
Promotility agents such as erythromycin and metoclopramide (Reglan) are also used to empty the stomach for better visualization.31–35 Erythromycin has been shown to improve visualization, shorten the procedure time, and prevent the need for additional endoscopy attempts in two randomized controlled studies.33,34 Furthermore, a cost-effectiveness study confirmed that giving intravenous erythromycin before endoscopy for acute upper GI bleeding saved money and resulted in an increase in quality-adjusted life-years.35
Endoscopy to diagnose bleeding and assess risk
Upper endoscopy is 90% to 95% diagnostic for acute upper GI bleeding.36
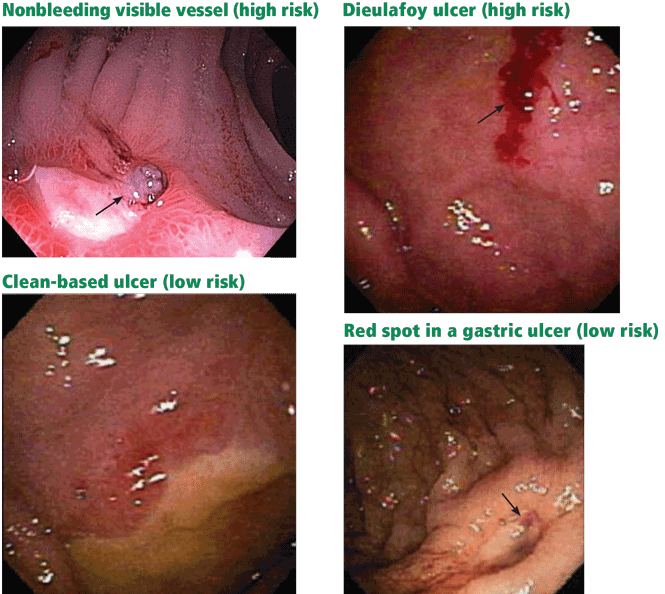
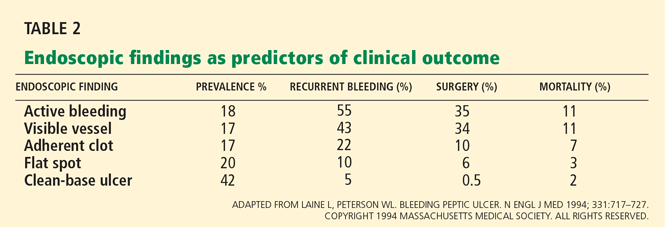
Patients at high risk (ie, older than 60 years, with severe comorbidity, or hemodynamically compromised) who have active bleeding (ie, witnessed hematemesis, red blood per nasogastric tube, or fresh blood per rectum) or a nonbleeding visible vessel should be admitted to a monitored bed or intensive care unit. Observation in a regular medical ward is appropriate for high-risk patients found to have an adherent clot. Patients with low-risk findings (eg, a clean ulcer base) are at low risk of recurrent bleeding and may be considered for early hospital discharge with appropriate outpatient follow-up.
Endoscopy to treat bleeding
About 25% of endoscopic procedures performed for upper GI bleeding include some type of treatment,39 such as injections of epinephrine, normal saline, or sclerosants; thermal cautery; argon plasma coagulation; electrocautery; or application of clips or bands. They are all equally effective, and combinations of these therapies are more effective than when they are used individually. A recent meta-analysis found dual therapy to be superior to epinephrine monotherapy in preventing recurrent bleeding, need for surgery, and death.40

How to manage adherent clots is controversial, but recent studies have revealed a significant benefit from removing them and treating the underlying lesions compared with drug therapy alone.43,45
Flat, pigmented spots and nonbleeding ulcers with a clean base do not require endoscopic treatment because the risk of recurrent bleeding is low.
Endoscopic therapy stops the bleeding in more than 90% of patients, but bleeding recurs after endoscopic therapy in 10% to 25%.46 Reversal of any severe coagulopathy with transfusions of platelets or fresh frozen plasma is essential for endoscopic hemostasis. However, coagulopathy at the time of initial bleeding and endoscopy does not appear to be associated with higher rates of recurrent bleeding following endoscopic therapy for nonvariceal upper GI bleeding.47
Patients with refractory bleeding are candidates for angiography or surgery. However, even when endoscopic hemostasis fails, endoscopy is important before angiography or surgery to pinpoint the site of bleeding and diagnose the cause.
A second endoscopic procedure is generally not recommended within 24 hours after the initial procedure.48 However, it is appropriate in cases in which clinical signs indicate recurrent bleeding or if hemostasis during the initial procedure is questionable. A meta-analysis found that routinely repeating endoscopy reduces the rate of recurrent bleeding but not the need for surgery or the risk of death.49
ALL PATIENTS SHOULD BE ADMITTED
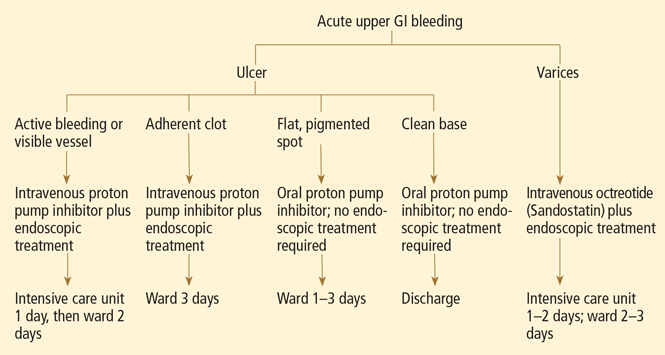
VARICEAL BLEEDING
Variceal bleeding, a severe outcome of portal hypertension secondary to cirrhosis, carries a 6-week mortality rate of 10% to 20%.50 In view of the risk, primary prevention is indicated in patients with high-risk varices.
The mainstays of primary and secondary prevention are the nonselective beta-blockers such as nadolol (Corgard) and propranolol (Inderal). Several randomized controlled trials have shown lower rates of recurrent bleeding and death with propranolol or nadolol than with placebo.51 In doses that decrease the heart rate by 25%, beta-blockers have been shown to delay and decrease variceal hemorrhage. However, most patients require prophylactic endoscopic variceal ligation because they cannot tolerate beta-blocker therapy.
In suspected acute variceal bleeding, a somatostatin analogue should be started to decrease the portal pressure, and antibiotics should be started to reduce the risks of infection and death. Vasoactive drugs, ie, somatostatin analogues, should be started before endoscopy and continued for 5 days to reduce the chances of recurrent bleeding.52,53
Terlipressin is the only drug proven to improve the odds of survival in acute variceal bleeding. Although widely used in Europe, it has not been approved for use in the United States.
Octreotide, another option, improves hemostasis to the same extent, although it does not increase the survival rate.54,55 The recommended dose of octreotide for patients with variceal bleeding is a 50-μg intravenous bolus, followed by a 50-μg/hour infusion for 5 days.
Combining endoscopic and drug therapy improves the chances of stopping the bleeding and reduces the risk of recurrent bleeding compared with endoscopic therapy alone.56
Transjugular intrahepatic portosystemic shunting is indicated in recurrent variceal hemorrhage or in those with initial bleeding that is refractory to standard medical and endoscopic therapy. It is not the primary therapy because it doubles the risk of encephalopathy and has a high stent occlusion rate (up to 60%, lower with covered stents).
GI BLEEDING CAN CAUSE ACUTE MYOCARDIAL INFARCTION
The simultaneous presentation of acute myocardial infarction (MI) and GI hemorrhage is very serious and unfortunately common.
An acute MI occurring simultaneously with or after GI bleeding is usually precipitated by massive bleeding causing hypovolemia, hemodynamic compromise, and hypoperfusion. Conversely, the anticoagulant, antiplatelet, or thrombolytic drugs given to treat MI can precipitate GI bleeding (see below).
This distinction is important because the two scenarios have different clinical courses and prognoses. GI bleeding that precipitates an acute MI tends to be massive, whereas GI bleeding after treatment of acute MI tends to be self-limited and often resolves with reversal of underlying coagulopathy.57
Endoscopy carries a higher than average risk in patients with recent acute MI, with all-cause mortality rates as high as 1%.58 (The usual rate is 0.0004%.59) Nevertheless, endoscopy can be safely performed early on in patients with acute MI if it is done under strict monitoring in a coronary care unit.
Several studies have shown that MI patients who present with upper GI bleeding as the inciting event or patients with acute MI who are vomiting blood or who are hemodynamically unstable due to GI bleeding are significantly more likely to have a high-risk lesion and so have the greatest need for endoscopic therapy. Therefore, endoscopic intervention may be offered to MI patients at high risk who have been started on antiplatelet agents.
WARFARIN CAN PRECIPITATE BLEEDING
Acute upper GI bleeding can be a severe complication of long-term oral anticoagulation, not because the drugs cause ulcers, but rather because they exacerbate ulcers that are already present.60 Therefore, when starting warfarin (Coumadin), patients should be evaluated to determine if they have other risk factors for GI bleeding, such as ulcers.
The number of people presenting with upper GI bleeding while on warfarin therapy is increasing because of the expanding indications for long-term anticoagulation therapy, such as atrial fibrillation and deep venous thrombosis.
The risk of GI bleeding in patients who use oral anticoagulants is estimated to be 2.3 to 4.9 times higher than in nonusers.61
The goal international normalized ratio (INR) for patients on warfarin therapy is usually 2.0 to 3.0. Recent studies found that endoscopy can be safely performed in patients with acute GI bleeding whose INR is between 2.0 and 3.0.62,63 Some suggest that both the length of warfarin therapy and the INR affect the risk of bleeding.64,65
Managing patients with an INR higher than 3.0 who have an episode of GI bleeding is always a challenge. It is not uncommon to find pathologic lesions causing GI bleeding in patients who are on warfarin with a supratherapeutic INR, and thus, endoscopy is indicated. However, before endoscopy, reversal of anticoagulation should be considered.
BLEEDING IN PATIENTS ON ANTIPLATELET DRUGS
Aspirin
Aspirin decreases production of prostaglandins in the GI tract, thereby decreasing the protective and restorative properties of the gastric and duodenal mucosa and predisposing to ulcers and bleeding.
The higher the aspirin dose, the higher the risk. Aspirin doubles the risk of upper GI bleeding at daily doses of 75 mg and quadruples it at doses of 300 mg.66 Even doses as low as 10 mg can decrease gastric mucosal prostaglandin production.67 Thus, it appears that there is no risk-free dose of aspirin, and enteric-coated or buffered formulations do not appear to reduce the risk.68–70
The most important risk factor for upper GI bleeding in patients taking aspirin is a history of peptic ulcer bleeding. Approximately 15% of aspirin users who have bleeding from ulcers have recurrent bleeding within 1 year.71
As aspirin-induced GI bleeding becomes more common, health care providers often feel caught between the GI risk and the cardiovascular benefit. When considering whether to discontinue antiplatelet therapy, a cardiologist should be consulted along with a gastroenterologist to weigh the risks of GI bleeding vs thrombosis. To date, there have been no clinical trials published to suggest when antiplatelet therapy should be stopped to optimize GI and cardiovascular outcomes. An alternative is to replace aspirin with another antiplatelet drug that does not induce ulcers.
Clopidogrel
Clopidogrel (Plavix) is recommended for hospitalized patients with acute coronary syndrome who cannot tolerate the GI side effects of aspirin, according to the joint guidelines of the American College of Cardiology and the American Heart Association, with the highest level of evidence.72 This recommendation was largely based on the safety data from the CAPRIE (Clopidogrel Versus Aspirin in Patients at Risk of Ischemic Events) trial, in which the incidence of major GI bleeding was lower in the clopidogrel group (0.52%) than in the aspirin group (0.72%; P < .05).73
Aspirin plus a proton pump inhibitor
Patients who have had an episode of upper GI bleeding and who need long-term aspirin therapy should also receive a proton pump inhibitor indefinitely to prevent ulcer recurrence.
In a recent double-blind randomized controlled trial in patients with a history of aspirin-induced bleeding, the combination of low-dose aspirin plus esomeprazole (Nexium) twice a day was superior to clopidogrel by itself in terms of the rate of recurrent bleeding (0.7% vs 8.6%; P < .05).74 A similar trial showed nearly identical results: 0% upper GI bleeding in the group receiving aspirin plus esomeprazole 20 mg daily, vs 13.6% in the clopidogrel group (P = .0019).75 These studies suggest that a once-daily proton pump inhibitor combined with aspirin is a safer alternative than clopidogrel alone.
Clopidogrel plus a proton pump inhibitor
Interestingly, recent studies have shown that omeprazole decreases the antiplatelet effect of clopidogrel, possibly by inhibiting the CYP2C19 enzyme.76 However, concomitant use of pantoprazole (Protonix), lansoprazole (Prevacid), and esomeprazole did not have this effect, suggesting that although all proton pump inhibitors are metabolized to a varying degree by CYP2C19, the interaction between proton pump inhibitors and clopidogrel is not a class effect.77–79 Therefore, pantoprazole, lansoprazole, and esomeprazole may be the appropriate proton pump inhibitors to use with clopidogrel in patients who have a clear indication for the medication, consistent with current guideline recommendations.
Helicobacter pylori infection in antiplatelet drug users
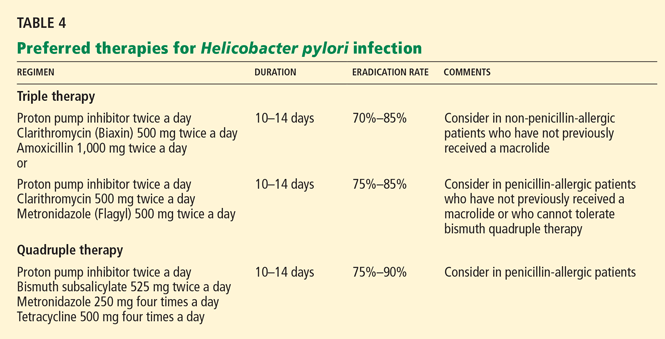
TREATMENT AND PREVENTION OF NSAID-RELATED GI INJURY
About 1 in 20 users of NSAIDs develop GI complications and ulcers of varying degrees of severity, as do one in seven NSAID users over the age of 65. In fact, NSAID use accounts for 30% of hospitalizations for upper GI bleeding and deaths from this cause.82–85 In addition, approximately 15% to 30% of NSAID users have clinically silent but endoscopically evident peptic ulcers.86
NSAIDs contribute to ulcer development by depleting prostaglandins. Thus, misoprostol (Cytotec), a synthetic prostaglandin, has been used to reduce this side effect.
In a clinical trial, misoprostol reduced the incidence of NSAID-associated GI complications by 40%.87 Furthermore, it has been shown to be better than placebo in preventing recurrent gastric ulcers in patients with a history of gastric ulcer who were receiving low-dose aspirin.88
However, misoprostol is rarely used because it can cause diarrhea and abdominal cramping. Rather, the preferred drugs for preventing and treating NSAID- and aspirin-related GI lesions are proton pump inhibitors.
Numerous clinical trials using endoscopic end points showed that proton pump inhibitors in standard doses significantly reduce the incidence of ulcers associated with the use of NSAIDs.89 Proton pump inhibitor therapy has achieved a significant reduction in relative risk of upper GI bleeding in patients who received low-dose aspirin therapy, as confirmed by epidemiologic studies.90,91 The number of NSAID-related ulcers found on endoscopy could be reduced by an estimated 90% simply by using proton pump inhibitors.92
- Lewis JD, Bilker WB, Brensinger C, Farrar JT, Strom BL. Hospitalization and mortality rates from peptic ulcer disease and GI bleeding in the 1990s: relationship to sales of non-steroidal anti-inflammatory drugs and acid suppression medications. Am J Gastroenterol 2002; 97:2540–2549.
- Viviane A, Alan BN. Estimates of costs of hospital stays for variceal and nonvariceal upper gastrointestinal bleeding in the United States. Value Health 2008; 11:1–3.
- Yavorski RT, Wong RK, Maydonovitch C, Battin LS, Furnia A, Amundson DE. Analysis of 3,294 cases of upper gastrointestinal bleeding in military medical facilities. Am J Gastroenterol 1995; 90:568–573.
- Kaplan RC, Heckbert SR, Koepsell TD, et al. Risk factors for hospitalized gastrointestinal bleeding among older persons. Cardiovascular Health Study Investigators. J Am Geriatr Soc 2001; 49:126–133.
- Longstreth GF. Epidemiology of hospitalization for acute upper gastrointestinal hemorrhage: a population-based study. Am J Gastroenterol 1995; 90:206–210.
- Laine L, Peterson WL. Bleeding peptic ulcer. N Engl J Med 1994; 331:717–727.
- Wara P, Stodkilde H. Bleeding pattern before admission as guideline for emergency endoscopy. Scand J Gastroenterol 1985; 20:72–78.
- Jensen DM, Machicado GA. Diagnosis and treatment of severe hematochezia. The role of urgent colonoscopy after purge. Gastroenterology 1988; 95:1569–1574.
- Daniel WA, Egan S. The quantity of blood required to produce a tarry stool. J Am Med Assoc 1939; 113:2232.
- Rockall TA, Logan RF, Devlin HB, Northfield TC. Risk assessment after acute upper gastrointestinal hemorrhage. Gut 1996; 38:316–321.
- Blatchford O, Murray WR, Blatchford M. A risk score to predict need for treatment for upper-gastrointestinal hemorrhage. Lancet 2000; 356:1318–1321.
- Gralnek IM, Barkun AN, Bardou M. Management of acute bleeding from a peptic ulcer. N Engl J Med 2008; 359:928–937.
- Silverstein FE, Gilbert DA, Tedesco FJ, Buenger NK, Persing J. The national ASGE survey on upper gastrointestinal bleeding II. Clinical prognostic factors. Gastrointest Endosc 1981; 27:80–93.
- Corley DA, Stefan AM, Wolf M, Cook EF, Lee TH. Early indicators of prognosis in upper gastrointestinal hemorrhage. Am J Gastroenterol 1998; 93:336–340.
- Aljebreen AM, Fallone CA, Barkun AN. Nasogastric aspirate predicts high-risk endoscopic lesions in patients with acute upper-GI bleeding. Gastrointest Endosc 2004; 59:172–178.
- Barkun AN, Cockeram AW, Plourde V, Fedorak RN. Review article: acid suppression in non-variceal acute upper gastrointestinal bleeding. Aliment Pharmacol Ther 1999; 13:1565–1584.
- Levine JE, Leontiadis JI, Sharma VK, Howden CW. Meta-analysis: the efficacy of intravenous H2-receptor antagonists in bleeding peptic ulcer. Aliment Pharmacol Ther 2002; 16:1137–1142.
- Walt RP, Cottrell J, Mann SG, Freemantle NP, Langman MJ. Continuous intravenous famotidine for hemorrhage from peptic ulcer. Lancet 1992; 340:1058–1062.
- Labenz J, Peitz U, Leusing C, Tillenburg B, Blum AL, Börsch G. Efficacy of primed infusion with high dose ranitidine and omeprazole to maintain high intragastric pH in patients with peptic ulcer bleeding: a prospective randomized controlled study. Gut 1997; 40:36–41.
- Merki HS, Wilder-Smith CH. Do continuous infusions of omeprazole and ranitidine retain their effect with prolonged dosing? Gastroenterology 1994; 106:60–64.
- Netzer P, Gaia C, Sandoz M, et al. Effect of repeated injection and continuous infusion of omeprazole and ranitidine on intragastric pH over 72 hours. Am J Gastroenterol 1999; 94:351–357.
- Lin HJ, Lo WC, Cheng YC, Perng CL. Role of intravenous omeprazole in patients with high-risk peptic ulcer bleeding after successful endoscopic epinephrine injection: a prospective randomized comparative trial. Am J Gastroenterol 2006; 101:500–505.
- Lau JY, Sung JJ, Lee KK, et al. Effects of intravenous omeprazole on recurrent bleeding after endoscopic treatment of bleeding peptic ulcers. N Engl J Med 2000; 343:310–316.
- Leontiadis GI, Sharma VK, Howden CW. Proton pump inhibitor treatment for acute peptic ulcer bleeding. Cochrane Database Syst Rev 2006;CD002094.
- Andriulli A, Annese V, Caruso N, et al. Proton-pump inhibitors and outcome of endoscopic hemostasis in bleeding peptic ulcers: a series of meta-analyses. Am J Gastroenterol 2005; 100:207–219.
- Lau JY, Leung WK, Wu JC, et al. Omeprazole before endoscopy in patients with gastrointestinal bleeding. N Engl J Med 2007; 356:1631–1640.
- Dorward S, Sreedharan A, Leontiadis GI, Howden CW, Moayyedi P, Forman D. Proton pump inhibitor treatment initiated prior to endoscopic diagnosis in upper gastrointestinal bleeding. Cochrane Database Syst Rev 2006;CD005415.
- Bardou M, Toubouti Y, Benhaberou-Brun D, Rahme E, Barkun AN. Meta-analysis: proton-pump inhibition in high-risk patients with acute peptic ulcer bleeding. Aliment Pharmacol Ther 2005; 21:677–686.
- Bjorkman DJ, Zaman A, Fennerty MB, Lieberman D, Disario JA, Guest-Warnick G. Urgent vs elective endoscopy for acute non-variceal upper-GI bleeding: an effectiveness study. Gastointest Endosc 2004; 60:1–8.
- Lee SD, Kearney DJ. A randomized controlled trial of gastric lavage prior to endoscopy for acute upper gastrointestinal bleeding. J Clin Gastroenterol 2004; 38:861–865.
- Tack J, Janssens J, Vantrappen G, et al. Effect of erythromycin on gastric motility in controls and in diabetic gastroparesis. Gastroenterology 1992; 103:72–79.
- Xynos E, Mantides A, Papageorgiou A, Fountos A, Pechlivanides G, Vassilakis JS. Erythromycin accelerates delayed gastric emptying of solids in patients after truncal vagotomy and pyloroplasty. Eur J Surg 1992; 158:407–411.
- Coffin B, Pocard M, Panis Y, et al; Groupe des endoscopistes de garde á l’AP-HP. Erythromycin improves the quality of EGD in patients with acute upper GI bleeding: a randomized controlled study. Gastrointest Endosc 2002; 56:174–179.
- Frossard JL, Spahr L, Queneau PE, et al. Erythromycin intravenous bolus infusion in acute upper gastrointestinal bleeding: a randomized, controlled, double-blind trial. Gastroenterology 2002; 123:17–23.
- Winstead NS, Wilcox CM. Erythromycin prior to endoscopy for acute upper gastrointestinal hemorrhage: a cost-effectiveness analysis. Aliment Pharmacol Ther 2007; 26:1371–1377.
- Chak A, Cooper GS, Lloyd LE, Kolz CS, Barnhart BA, Wong RC. Effectiveness of endoscopy in patients admitted to the intensive care unit with upper GI hemorrhage. Gastrointest Endosc 2001; 53:6–13.
- Lau JY, Chung SC, Leung JW, Lo KK, Yung MY, Li AK. The evolution of stigmata of hemorrhage in bleeding peptic ulcers: a sequential endoscopic study. Endoscopy 1998; 30:513–518.
- Chung IK, Kim EJ, Lee MS, et al. Endoscopic factors predisposing to rebleeding following endoscopic hemostasis in bleeding peptic ulcers. Endoscopy 2001; 33:969–975.
- Elta GH. Acute nonvariceal upper gastrointestinal hemorrhage. Curr Treat Options Gastroenterol 2002; 5:147–152.
- Marmo R, Rotondano G, Piscopo R, Bianco MA, D’Angella R, Cipolletta L. Dual therapy versus monotherapy in the endoscopic treatment of high-risk bleeding ulcers: a meta-analysis of controlled trials. Am J Gastroenterol 2007; 102:279–289.
- Kovacs TO, Jensen DM. Recent advances in the endoscopic diagnosis and therapy of upper gastrointestinal, small intestinal, and colonic bleeding. Med Clin North Am 2002; 86:1319–1356.
- Kovacs TO, Jensen DM. Endoscopic treatment of ulcer bleeding. Curr Treat Options Gastroenterol 2007; 10:143–148.
- Jensen DM, Kovacs TO, Jutabha R, et al. Randomized trial of medical or endoscopic therapy to prevent recurrent ulcer hemorrhage in patients with adherent clots. Gastroenterology 2002; 123:407–413.
- Jensen DM, Machicado GA. Endoscopic hemostasis of ulcer hemorrhage with injection, thermal, and combination methods. Techniques Gastrointest Endosc 2005; 7:124–131.
- Bleau BL, Gostout CJ, Sherman KE, et al. Recurrent bleeding from peptic ulcer associated with adherent clot: a randomized study comparing endoscopic treatment with medical therapy. Gastrointest Endosc 2002; 56:1–6.
- Lau JY, Sung JJ, Lam YH, et al. Endoscopic retreatment compared with surgery in patients with recurrent bleeding after initial endoscopic control of bleeding ulcers. N Engl J Med 1999; 340:751–756.
- Wolf AT, Wasan SK, Saltzman JR. Impact of anticoagulation on rebleeding following endoscopic therapy for nonvariceal upper gastrointestinal hemorrhage. Am J Gastroenterol 2007; 102:290–296.
- Barkun A, Bardou M, Marshall JK; Nonvariceal Upper GI Bleeding Consensus Conference Group. Consensus recommendations for managing patients with nonvariceal upper gastrointestinal bleeding. Ann Intern Med 2003; 139:843–857.
- Marmo R, Rotondano G, Bianco MA, Piscopo R, Prisco A, Cipolletta L. Outcome of endoscopic treatment for peptic ulcer bleeding: is a second look necessary? A meta-analysis. Gastrointest Endosc 2003; 57:62–67.
- Dell’Era A, deFrancis R, Iannuzzi F. Acute variceal bleeding: pharmacological treatment and primary/secondary prophylaxis. Best Pract Res Clin Gastroenterol 2008; 22:279–294.
- Jalan R, Hayes PC. UK guidelines on the management of variceal hemorrhage in cirrhotic patients. British Society of Gastroenterology. Gut 2000; 46( suppl 3–4):III1–III15.
- Bernard B, Lebrec D, Mathurin P, Opolon P, Poynard T. Beta-adrenergic antagonists in the prevention of gastrointestinal rebleeding in patients with cirrhosis: a meta-analysis. Hepatology 1997; 25:63–70.
- De Franchis R. Evolving consensus in portal hypertension. Report of the Baveno IV consensus workshop on methodology of diagnosis and therapy in portal hypertension. J Hepatol 2005; 43:167–176.
- Levacher S, Letoumelin P, Pateron D, Blaise M, Lapandry C, Pourriat JL. Early administration of terlipressin plus glyceryl trinitrate to control active upper gastrointestinal bleeding in cirrhotic patients. Lancet 1995; 346:865–868.
- Abraldes JG, Bosch J. Somatostatin and analogues in portal hypertension. Hepatology 2002; 35:1305–1312.
- Banares R, Albillos A, Rincon D, et al. Endoscopic treatment versus endoscopic plus pharmacological treatment for acute variceal bleeding: a meta analysis. Hepatology 2002; 35:609–615.
- Cappell M. Gastrointenstinal bleeding associated with myocardial infarction. Gastroenterol Clin North Am 2000; 29:423–444.
- Lin S, Konstance R, Jollis J, Fisher DA. The utility of upper endoscopy in patients with concomitant upper gastrointestinal bleeding and acute myocardial infarction. Dig Dis Sci 2006; 51:2377–2383.
- Silvis SE, Nebel O, Rogers G, Sugawa C, Mandelstam P. Endoscopic complications. Results of the 1974 American Society for Gastrointestinal Endoscopy Survey. JAMA 1976; 235:928–930.
- Lanas A, Garcia-Rodriguez LA, Arroyo MT, et al; Investigators of the Asociación Española de Gastroenterología (AEG). Effect of antisecretory drugs and nitrates on the risk of ulcer bleeding associated with nonsteroidal anti-inflammatory drugs, antiplatelet agents, and anticoagulants. Am J Gastroenterol 2007; 102:507–515.
- Shorr RI, Ray WA, Daugherty JR, Griffin MR. Concurrent use of nonsteroidal anti-inflammatory drugs and oral anticoagulants places elderly persons at high risk for hemorrhagic peptic ulcer disease. Arch Intern Med 1993; 153:1665–1670.
- Tabibian N. Acute gastrointestinal bleeding in anticoagulated patients: a prospective evaluation. Am J Gastroenterol 1989; 84:10–12.
- Choudari CP, Rajgopal C, Palmer KR. Acute gastrointestinal hemorrhage in anticoagulated patients: diagnoses and response to endoscopic treatment. Gut 1994; 35:464–466.
- Petty GW, Brown RD, Whisnant JP, Sicks JD, O’Fallon WM, Wiebers DO. Frequency of major complications of aspirin, warfarin, and intravenous heparin for secondary stroke prevention: a population-based study. Ann Intern Med 1999; 130:14–22.
- Landefeld CS, Rosenblatt MW, Goldman L. Bleeding in outpatients treated with warfarin: relation to the prothrombin time and important remediable lesions. Am J Med 1989; 87:153–159.
- Weil J, Colin-Jones D, Langman M, et al. Prophylactic aspirin and risk of peptic ulcer bleeding. BMJ 1995; 310:827–830.
- Cryer B, Feldman M. Effects of very low dose daily, long-term aspirin therapy on gastric, duodenal, and rectal prostaglandin levels and on mucosal injury in healthy humans. Gastroenterology 1999; 117:17–25.
- De Abajo FJ, Garcia Rodriguez LA. Risk of upper gastrointestinal bleeding and perforation associated with low-dose aspirin as plain and enteric-coated formulations. BMC Clin Pharmacol 2001; 1:1.
- Kelly JP, Kaufman DW, Jurgelon JM, Sheehan J, Koff RS, Shapiro S. Risk of aspirin-associated major upper gastrointestinal bleeding with enteric coated or buffered product. Lancet 1996; 348:1413–1416.
- Garcia Rodriguez LA, Hernandez-Diaz S, De Abajo FJ. Association between aspirin and upper gastrointestinal complications: systematic review of epidemiological studies. Br J Clin Pharmacol 2001; 52:563–571.
- Wilcox CM, Ladabaum U. A patient with high risk of gastrointestinal bleeding requiring nonsteroidal anti-inflammatory drugs. Clin Gastroenterol Hepatol 2006; 4:1090–1093.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet 1996; 348:1329–1339.
- Chan FK, Ching JY, Hung LC, et al. Clopidogrel versus aspirin and esomeprazole to prevent recurrent ulcer bleeding. N Engl J Med 2005; 352:238–244.
- Lai KC, Chu KM, Hui WM, et al. Esomeprazole with aspirin versus clopidogrel for prevention of recurrent gastrointestinal ulcer complications. Clin Gastroenterol Hepatol 2006; 4:860–865.
- Ho MP, Maddox TM, Wang L, et al. Risk of adverse outcomes associated with concomitant use of clopidogrel and proton pump inhibitors following acute coronary syndrome. JAMA 2009; 301:937–944.
- Siller-Matula JM, Spiel AO, Lang IM, Kreiner G, Christ G, Jilma B. Effects of pantoprazole and esomeprazole on platelet inhibition by clopidogrel. Am Heart J 2009; 157:148.e1–e5.
- Small DS, Farid NA, Payne CD, et al. Effects of proton pump inhibitor lansoprazole on the pharmacokinetics and pharmacodynamics of prasugel and clopidogrel. J Clin Pharmacol 2008; 48:475–484.
- Ishizaki T, Horai Y. Review article: cytochrome P450 and the metabolism of proton pump inhibitors—emphasis on rabeprazole. Aliment Pharacol Ther 1999; 13 (suppl 3):27–36.
- Lanas A, Fuentes J, Benito R, Serrano P, Bajador E, Sainz R. Helicobacter pylori increases the risk of upper gastrointestinal bleeding in patients taking low-dose aspirin. Aliment Pharmacol Ther 2002; 16:779–786.
- Chan FK. NSAID-Induced peptic ulcers and Helicobacter pylori infection: implications for patient management. Drug Saf 2005; 28:287–300.
- Bombardier V, Laine L, Reicin A, et al; VIGOR Study Group. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis: VIGOR Study Group. N Eng J Med 2000; 343:1520–1528.
- Griffin MR, Ray WA, Schaffner W. Nonsteroidal anti-inflammatory drug use and death from peptic ulcer in elderly persons. Ann Intern Med 1988; 109:359–363.
- Griffin MR, Piper JM, Daugherty JR, Snowden M, Ray WA. Nonsteroidal anti-inflammatory drug use and increased risk for peptic ulcer disease in elderly persons. Ann Intern Med 1991; 114:257–263.
- Smalley WE, Ray WA, Daugherty JR, Griffin MR. Nonsteroidal anti-inflammatory drugs and the incidence of hospitalizations for peptic ulcer disease in elderly persons. Am J Epidemiol 1995; 141:539–545.
- Laine L. Approaches to nonsteroidal anti-inflammatory drug use in the high-risk patient. Gastroenterology 2001; 120:594–606.
- Silverstein FE, Graham DY, Senior JR, et al. Misoprostol reduces serious gastrointestinal complications in patients with rheumatoid arthritis receiving nonsteroidal anti-inflammatory drugs. A randomized, double blind, placebo controlled trial. Ann Intern Med 1995; 123:241–249.
- Goldstein JL, Huang B, Amer F, Christopoulos NG. Ulcer recurrence in high-risk patients receiving nonsteroidal anti-inflammatory drugs plus low dose aspirin: results of a post hoc subanalysis. Clin Ther 2004; 26:1637–1643.
- Berger JS, Stebbins A, Granger CB, et al. Initial aspirin dose and outcome among ST-elevation myocardial infarction patients treated with fibrinolytic therapy. Circulation 2008; 117:192–199.
- Lanas A, Garcia-Rodriguez LA, Arroyo MT, et al; Investigators of the Asociación Española de Gastroenterología (AEG). Effect of antisecretory drugs and nitrates on the risk of ulcer bleeding associated with nonsteroidal anti-inflammatory drugs, antiplatelet agents, and anticoagulants. Am J Gastroenterol 2007; 102:507–515.
- Chin MW, Yong G, Bulsara MK, Rankin J, Forbes GM. Predictive and protective factors associated with upper gastrointestinal bleeding after percutaneous coronary intervention: a case-control study. Am J Gastroenterol 2007; 102:2411–2416.
- Hunt RH, Bazzoli F. Should NSAID/low dose aspirin takers be tested routinely for H. Pylori infection and treated if positive? Implications for primary risk of ulcer and ulcer relapse after initial healing. Aliment Pharmacol Ther 2004; 19 (suppl 1):9–16.
Upper gastrointestinal (GI) bleeding is common, costly, and potentially life-threatening. It must be managed promptly and appropriately to prevent adverse outcomes.
More people are admitted to the hospital for upper GI bleeding than for congestive heart failure or deep vein thrombosis. In the United States, the annual rate of hospitalization for upper GI bleeding is estimated to be 165 per 100,000—more than 300,000 hospitalizations per year, at a cost of $2.5 billion.1,2
Furthermore, despite advances in therapy, the case-fatality rate has remained unchanged at 7% to 10%.3 This may be because today’s patients are older and have more comorbidities than those in the past.4
CAUSES OF UPPER GI BLEEDING
Peptic ulcers account for about 60% of severe cases of upper GI bleeding,5 and they are the focus of this paper. Fortunately, up to 80% of bleeding ulcers stop bleeding spontaneously without any intervention.6
Gastroduodenal erosions account for about 12%.3
Varices due to cirrhosis are less common but more dangerous. Variceal bleeding accounts for a relatively small percentage (6%) of upper GI bleeding, but the mortality rate from a single episode of variceal bleeding is 30%, and 60% to 70% of patients die within 1 year, mostly of underlying liver disease.
Less frequent causes include Mallory-Weiss tears, erosive duodenitis, Dieulafoy ulcer (a type of vascular malformation), other vascular lesions, neoplasms, aortoenteric fistula, gastric antral vascular ectasia, and prolapse gastropathy.5
HEMATEMESIS AND MELENA
The most common presenting signs of acute upper GI bleeding are hematemesis (vomiting of blood), “coffee grounds” emesis, and melena (tarry black stools). About 30% of patients with bleeding ulcers present with hematemesis, 20% with melena, and 50% with both.7
Hematochezia (red blood in the stool) usually suggests a lower GI source of bleeding, since blood from an upper source turns black and tarry as it passes through the gut, producing melena. However, up to 5% of patients with bleeding ulcers have hematochezia,7 and it indicates heavy bleeding: bleeding of approximately 1,000 mL into the upper GI tract is needed to cause hematochezia, whereas only 50 to 100 mL is needed to cause melena.8,9 Hematochezia with signs and symptoms of hemodynamic compromise such as syncope, postural hypotension, tachycardia, and shock should therefore direct one’s attention to an upper GI source of bleeding.
Nonspecific features include nausea, vomiting, epigastric pain, vasovagal phenomena, and syncope.
WHAT IS THE PATIENT’S RISK?
An assessment of clinical severity is the first critical task, as it helps in planning treatment. Advanced age, multiple comorbidities, and hemodynamic instability call for aggressive treatment. Apart from this simple clinical rule, scoring systems have been developed.
The Rockall scoring system, the most widely used, gives estimates of the risks of recurrent bleeding and death. It is based on the three clinical factors mentioned above and on two endoscopic ones, awarding points for:
- Age—0 points if less than 60; 1 point if 60 to 79; or 2 points if 80 years or older
- Shock—1 point if the pulse is more than 100; 2 points if the systolic blood pressure is less than 100 mm Hg
- Comorbid illness—2 points for ischemic heart disease, congestive heart failure, or other major comorbidity; 3 points for renal failure, hepatic failure, or metastatic disease
- Endoscopic diagnosis—0 points if no lesion found or a Mallory-Weiss tear; 1 point for peptic ulcer, esophagitis, or erosive disease; 2 points for GI malignancy
- Endoscopic stigmata or recent hemorrhage—0 points for a clean-based ulcer or flat pigmented spot; 2 points for blood in the upper GI tract, active bleeding, a nonbleeding visible vessel, or adherent clot.
The Rockall score can thus range from 0 to 11 points, with an overall score of 0, 1, or 2 associated with an excellent prognosis.10
Other systems that are used less often include the Baylor severity scale and the Acute Physiology and Chronic Health Evaluation (APACHE) II score.
Does the patient have varices?
All variceal bleeding should be considered severe, since the 1-year death rate is so high (up to 70%). Clues pointing to variceal bleeding include previous variceal bleeding, thrombocytopenia, history of liver disease, and signs of liver disease on clinical examination.
All patients suspected of having bleeding varices should be admitted to the intensive care unit for close monitoring and should be given the highest priority, even if they are hemodynamically stable.
Is the patient hemodynamically stable?
Appropriate hemodynamic assessment includes monitoring of heart rate, blood pressure, and mental status. Tachycardia at rest, hypotension, and orthostatic changes in vital signs indicate a considerable loss of blood volume. Low urine output, dry mucous membranes, and sunken neck veins are also useful signs. (Tachycardia may be blunted if the patient is taking a beta-blocker.)
If these signs of hypovolemia are present, the initial management focuses on treating shock and on improving oxygen delivery to the vital organs. This involves repletion of the intravascular volume with intravenous infusions or blood transfusions. Supplemental oxygen also is useful, especially in elderly patients with heart disease.12
Inspection of nasogastric aspirate
In the initial assessment, it is useful to insert a nasogastric tube and inspect the aspirate. If it contains bright red blood, the patient needs an urgent endoscopic evaluation and an intensive level of care13,14; if it contains coffee-grounds material, the patient needs to be admitted to the hospital and to undergo endoscopic evaluation within 24 hours.
However, a normal aspirate does not rule out upper GI bleeding. Aljebreen et al15 found that 15% of patients with upper GI bleeding and normal nasogastric aspirate still had high-risk lesions (ie, visible bleeding or nonbleeding visible vessels) on endoscopy.
ACID-SUPPRESSION HELPS ULCERS HEAL
Acid and pepsin interfere with the healing of ulcers and other nonvariceal upper GI lesions. Further, an acidic environment promotes platelet disaggregation and fibrinolysis and impairs clot formation.16 This suggests that inhibiting gastric acid secretion and raising the gastric pH to 6 or higher may stabilize clots. Moreover, pepsinogen in the stomach is converted to its active form (pepsin) if the pH is less than 4. Therefore, keeping the pH above 4 keeps pepsinogen in an inactive form.
Histamine-2 receptor antagonists
Histamine-2 receptor antagonists were the first drugs to inhibit acid secretion, reversibly blocking histamine-2 receptors on the basolateral membrane of parietal cells. However, these drugs did not prove very useful in managing upper GI bleeding in clinical trials.17,18 In their intravenous form, they often fail to keep the gastric pH at 6 or higher, due to tachyphylaxis.19 The use of this class of drugs has declined in favor of proton pump inhibitors.
Proton pump inhibitors
Proton pump inhibitors reduce both basal and stimulated acid secretion by inhibiting hydrogen-potassium adenosine triphosphatase, the proton pump of the parietal cell.
Multiple studies have shown that proton pump inhibitors raise the gastric pH and keep it high. For example, an infusion of omeprazole (Prilosec) can keep the gastric pH above 6 for 72 hours without inducing tachyphylaxis.20,21
Started after endoscopy. Randomized controlled trials have found proton pump inhibitors to be effective when given in high doses intravenously for 72 hours after successful endoscopic treatment of bleeding ulcers with high-risk endoscopic signs, such as active bleeding or nonbleeding visible vessels.22,23
A meta-analysis indicated that these drugs decrease the incidence of recurrent peptic ulcer bleeding, the need for blood transfusions, the need for surgery, and the duration of hospitalization, but not the mortality rate.24,25 These studies also illustrate the benefit of following up endoscopic treatment to stop the bleeding with an intravenous infusion of a proton pump inhibitor.
The recommended dose of omeprazole for patients with high-risk findings on endoscopy is an 80-mg bolus followed by an 8-mg/hour infusion for 72 hours. After the patient’s condition stabilizes, oral therapy can be substituted for intravenous therapy. In patients with low-risk endoscopic findings (a clean-based ulcer or flat spot), oral proton pump inhibitors in high doses are recommended.
In either case, after the initial bleeding is treated endoscopically and hemostasis is achieved, a proton pump inhibitor is recommended for 6 to 8 weeks, or longer if the patient is also positive for Helicobacter pylori or is on daily treatment with aspirin or a nonsteroidal anti-inflammatory drug (NSAID) that is not selective for cyclo-oxygenase 2 (see below).
Started before endoscopy, these drugs reduced the frequency of actively bleeding ulcers, the duration of hospitalization, and the need for endoscopic therapy in a randomized controlled trial.26 A meta-analysis found that significantly fewer patients had signs of recent bleeding on endoscopy if they received a proton pump inhibitor 24 to 48 hours before the procedure, but it did not find any significant difference in important clinical outcomes such as death, recurrent bleeding, or surgery.27 Nevertheless, we believe that intravenous proton pump inhibitor therapy should be started before endoscopy in patients with upper GI bleeding.
Somatostatin analogues
Octreotide (Sandostatin), an analogue of the hormone somatostatin, decreases splanchnic blood flow, decreases secretion of gastric acid and pepsin, and stimulates mucus production. Although it is beneficial in treating upper GI bleeding due to varices, its benefit has not been confirmed in patients with nonvariceal upper GI bleeding.
A meta-analysis revealed that outcomes were better with high-dose intravenous proton pump inhibitor therapy than with octreotide when these drugs were started after endoscopic treatment of acute peptic ulcer bleeding.28 Nevertheless, octreotide may be useful in patients with uncontrolled nonvariceal bleeding who are awaiting endoscopy, since it is relatively safe to use.
ALL PATIENTS NEED ENDOSCOPY
All patients with upper GI bleeding need an upper endoscopic examination to diagnose and assess the risk posed by the bleeding lesion and to treat the lesion, reducing the risk of recurrent bleeding.
How urgently does endoscopy need to be done?
Endoscopy within the first 24 hours of upper GI bleeding is considered the standard of care. Patients with uncontrolled or recurrent bleeding should undergo endoscopy on an urgent basis to control the bleeding and reduce the risk of death.
However, how urgently endoscopy needs to be done is often debated. A multicenter randomized controlled trial compared outcomes in patients who underwent endoscopy within 6 hours of coming to the emergency department vs within 24 hours after the initial evaluation. The study found no significant difference in outcomes between the two groups; however, the group that underwent endoscopy sooner needed fewer transfusions.29
For a better view of the stomach
Gastric lavage improves the view of the gastric fundus but has not been proven to improve outcome.30
Promotility agents such as erythromycin and metoclopramide (Reglan) are also used to empty the stomach for better visualization.31–35 Erythromycin has been shown to improve visualization, shorten the procedure time, and prevent the need for additional endoscopy attempts in two randomized controlled studies.33,34 Furthermore, a cost-effectiveness study confirmed that giving intravenous erythromycin before endoscopy for acute upper GI bleeding saved money and resulted in an increase in quality-adjusted life-years.35
Endoscopy to diagnose bleeding and assess risk
Upper endoscopy is 90% to 95% diagnostic for acute upper GI bleeding.36
Patients at high risk (ie, older than 60 years, with severe comorbidity, or hemodynamically compromised) who have active bleeding (ie, witnessed hematemesis, red blood per nasogastric tube, or fresh blood per rectum) or a nonbleeding visible vessel should be admitted to a monitored bed or intensive care unit. Observation in a regular medical ward is appropriate for high-risk patients found to have an adherent clot. Patients with low-risk findings (eg, a clean ulcer base) are at low risk of recurrent bleeding and may be considered for early hospital discharge with appropriate outpatient follow-up.
Endoscopy to treat bleeding
About 25% of endoscopic procedures performed for upper GI bleeding include some type of treatment,39 such as injections of epinephrine, normal saline, or sclerosants; thermal cautery; argon plasma coagulation; electrocautery; or application of clips or bands. They are all equally effective, and combinations of these therapies are more effective than when they are used individually. A recent meta-analysis found dual therapy to be superior to epinephrine monotherapy in preventing recurrent bleeding, need for surgery, and death.40
How to manage adherent clots is controversial, but recent studies have revealed a significant benefit from removing them and treating the underlying lesions compared with drug therapy alone.43,45
Flat, pigmented spots and nonbleeding ulcers with a clean base do not require endoscopic treatment because the risk of recurrent bleeding is low.
Endoscopic therapy stops the bleeding in more than 90% of patients, but bleeding recurs after endoscopic therapy in 10% to 25%.46 Reversal of any severe coagulopathy with transfusions of platelets or fresh frozen plasma is essential for endoscopic hemostasis. However, coagulopathy at the time of initial bleeding and endoscopy does not appear to be associated with higher rates of recurrent bleeding following endoscopic therapy for nonvariceal upper GI bleeding.47
Patients with refractory bleeding are candidates for angiography or surgery. However, even when endoscopic hemostasis fails, endoscopy is important before angiography or surgery to pinpoint the site of bleeding and diagnose the cause.
A second endoscopic procedure is generally not recommended within 24 hours after the initial procedure.48 However, it is appropriate in cases in which clinical signs indicate recurrent bleeding or if hemostasis during the initial procedure is questionable. A meta-analysis found that routinely repeating endoscopy reduces the rate of recurrent bleeding but not the need for surgery or the risk of death.49
ALL PATIENTS SHOULD BE ADMITTED
VARICEAL BLEEDING
Variceal bleeding, a severe outcome of portal hypertension secondary to cirrhosis, carries a 6-week mortality rate of 10% to 20%.50 In view of the risk, primary prevention is indicated in patients with high-risk varices.
The mainstays of primary and secondary prevention are the nonselective beta-blockers such as nadolol (Corgard) and propranolol (Inderal). Several randomized controlled trials have shown lower rates of recurrent bleeding and death with propranolol or nadolol than with placebo.51 In doses that decrease the heart rate by 25%, beta-blockers have been shown to delay and decrease variceal hemorrhage. However, most patients require prophylactic endoscopic variceal ligation because they cannot tolerate beta-blocker therapy.
In suspected acute variceal bleeding, a somatostatin analogue should be started to decrease the portal pressure, and antibiotics should be started to reduce the risks of infection and death. Vasoactive drugs, ie, somatostatin analogues, should be started before endoscopy and continued for 5 days to reduce the chances of recurrent bleeding.52,53
Terlipressin is the only drug proven to improve the odds of survival in acute variceal bleeding. Although widely used in Europe, it has not been approved for use in the United States.
Octreotide, another option, improves hemostasis to the same extent, although it does not increase the survival rate.54,55 The recommended dose of octreotide for patients with variceal bleeding is a 50-μg intravenous bolus, followed by a 50-μg/hour infusion for 5 days.
Combining endoscopic and drug therapy improves the chances of stopping the bleeding and reduces the risk of recurrent bleeding compared with endoscopic therapy alone.56
Transjugular intrahepatic portosystemic shunting is indicated in recurrent variceal hemorrhage or in those with initial bleeding that is refractory to standard medical and endoscopic therapy. It is not the primary therapy because it doubles the risk of encephalopathy and has a high stent occlusion rate (up to 60%, lower with covered stents).
GI BLEEDING CAN CAUSE ACUTE MYOCARDIAL INFARCTION
The simultaneous presentation of acute myocardial infarction (MI) and GI hemorrhage is very serious and unfortunately common.
An acute MI occurring simultaneously with or after GI bleeding is usually precipitated by massive bleeding causing hypovolemia, hemodynamic compromise, and hypoperfusion. Conversely, the anticoagulant, antiplatelet, or thrombolytic drugs given to treat MI can precipitate GI bleeding (see below).
This distinction is important because the two scenarios have different clinical courses and prognoses. GI bleeding that precipitates an acute MI tends to be massive, whereas GI bleeding after treatment of acute MI tends to be self-limited and often resolves with reversal of underlying coagulopathy.57
Endoscopy carries a higher than average risk in patients with recent acute MI, with all-cause mortality rates as high as 1%.58 (The usual rate is 0.0004%.59) Nevertheless, endoscopy can be safely performed early on in patients with acute MI if it is done under strict monitoring in a coronary care unit.
Several studies have shown that MI patients who present with upper GI bleeding as the inciting event or patients with acute MI who are vomiting blood or who are hemodynamically unstable due to GI bleeding are significantly more likely to have a high-risk lesion and so have the greatest need for endoscopic therapy. Therefore, endoscopic intervention may be offered to MI patients at high risk who have been started on antiplatelet agents.
WARFARIN CAN PRECIPITATE BLEEDING
Acute upper GI bleeding can be a severe complication of long-term oral anticoagulation, not because the drugs cause ulcers, but rather because they exacerbate ulcers that are already present.60 Therefore, when starting warfarin (Coumadin), patients should be evaluated to determine if they have other risk factors for GI bleeding, such as ulcers.
The number of people presenting with upper GI bleeding while on warfarin therapy is increasing because of the expanding indications for long-term anticoagulation therapy, such as atrial fibrillation and deep venous thrombosis.
The risk of GI bleeding in patients who use oral anticoagulants is estimated to be 2.3 to 4.9 times higher than in nonusers.61
The goal international normalized ratio (INR) for patients on warfarin therapy is usually 2.0 to 3.0. Recent studies found that endoscopy can be safely performed in patients with acute GI bleeding whose INR is between 2.0 and 3.0.62,63 Some suggest that both the length of warfarin therapy and the INR affect the risk of bleeding.64,65
Managing patients with an INR higher than 3.0 who have an episode of GI bleeding is always a challenge. It is not uncommon to find pathologic lesions causing GI bleeding in patients who are on warfarin with a supratherapeutic INR, and thus, endoscopy is indicated. However, before endoscopy, reversal of anticoagulation should be considered.
BLEEDING IN PATIENTS ON ANTIPLATELET DRUGS
Aspirin
Aspirin decreases production of prostaglandins in the GI tract, thereby decreasing the protective and restorative properties of the gastric and duodenal mucosa and predisposing to ulcers and bleeding.
The higher the aspirin dose, the higher the risk. Aspirin doubles the risk of upper GI bleeding at daily doses of 75 mg and quadruples it at doses of 300 mg.66 Even doses as low as 10 mg can decrease gastric mucosal prostaglandin production.67 Thus, it appears that there is no risk-free dose of aspirin, and enteric-coated or buffered formulations do not appear to reduce the risk.68–70
The most important risk factor for upper GI bleeding in patients taking aspirin is a history of peptic ulcer bleeding. Approximately 15% of aspirin users who have bleeding from ulcers have recurrent bleeding within 1 year.71
As aspirin-induced GI bleeding becomes more common, health care providers often feel caught between the GI risk and the cardiovascular benefit. When considering whether to discontinue antiplatelet therapy, a cardiologist should be consulted along with a gastroenterologist to weigh the risks of GI bleeding vs thrombosis. To date, there have been no clinical trials published to suggest when antiplatelet therapy should be stopped to optimize GI and cardiovascular outcomes. An alternative is to replace aspirin with another antiplatelet drug that does not induce ulcers.
Clopidogrel
Clopidogrel (Plavix) is recommended for hospitalized patients with acute coronary syndrome who cannot tolerate the GI side effects of aspirin, according to the joint guidelines of the American College of Cardiology and the American Heart Association, with the highest level of evidence.72 This recommendation was largely based on the safety data from the CAPRIE (Clopidogrel Versus Aspirin in Patients at Risk of Ischemic Events) trial, in which the incidence of major GI bleeding was lower in the clopidogrel group (0.52%) than in the aspirin group (0.72%; P < .05).73
Aspirin plus a proton pump inhibitor
Patients who have had an episode of upper GI bleeding and who need long-term aspirin therapy should also receive a proton pump inhibitor indefinitely to prevent ulcer recurrence.
In a recent double-blind randomized controlled trial in patients with a history of aspirin-induced bleeding, the combination of low-dose aspirin plus esomeprazole (Nexium) twice a day was superior to clopidogrel by itself in terms of the rate of recurrent bleeding (0.7% vs 8.6%; P < .05).74 A similar trial showed nearly identical results: 0% upper GI bleeding in the group receiving aspirin plus esomeprazole 20 mg daily, vs 13.6% in the clopidogrel group (P = .0019).75 These studies suggest that a once-daily proton pump inhibitor combined with aspirin is a safer alternative than clopidogrel alone.
Clopidogrel plus a proton pump inhibitor
Interestingly, recent studies have shown that omeprazole decreases the antiplatelet effect of clopidogrel, possibly by inhibiting the CYP2C19 enzyme.76 However, concomitant use of pantoprazole (Protonix), lansoprazole (Prevacid), and esomeprazole did not have this effect, suggesting that although all proton pump inhibitors are metabolized to a varying degree by CYP2C19, the interaction between proton pump inhibitors and clopidogrel is not a class effect.77–79 Therefore, pantoprazole, lansoprazole, and esomeprazole may be the appropriate proton pump inhibitors to use with clopidogrel in patients who have a clear indication for the medication, consistent with current guideline recommendations.
Helicobacter pylori infection in antiplatelet drug users
TREATMENT AND PREVENTION OF NSAID-RELATED GI INJURY
About 1 in 20 users of NSAIDs develop GI complications and ulcers of varying degrees of severity, as do one in seven NSAID users over the age of 65. In fact, NSAID use accounts for 30% of hospitalizations for upper GI bleeding and deaths from this cause.82–85 In addition, approximately 15% to 30% of NSAID users have clinically silent but endoscopically evident peptic ulcers.86
NSAIDs contribute to ulcer development by depleting prostaglandins. Thus, misoprostol (Cytotec), a synthetic prostaglandin, has been used to reduce this side effect.
In a clinical trial, misoprostol reduced the incidence of NSAID-associated GI complications by 40%.87 Furthermore, it has been shown to be better than placebo in preventing recurrent gastric ulcers in patients with a history of gastric ulcer who were receiving low-dose aspirin.88
However, misoprostol is rarely used because it can cause diarrhea and abdominal cramping. Rather, the preferred drugs for preventing and treating NSAID- and aspirin-related GI lesions are proton pump inhibitors.
Numerous clinical trials using endoscopic end points showed that proton pump inhibitors in standard doses significantly reduce the incidence of ulcers associated with the use of NSAIDs.89 Proton pump inhibitor therapy has achieved a significant reduction in relative risk of upper GI bleeding in patients who received low-dose aspirin therapy, as confirmed by epidemiologic studies.90,91 The number of NSAID-related ulcers found on endoscopy could be reduced by an estimated 90% simply by using proton pump inhibitors.92
Upper gastrointestinal (GI) bleeding is common, costly, and potentially life-threatening. It must be managed promptly and appropriately to prevent adverse outcomes.
More people are admitted to the hospital for upper GI bleeding than for congestive heart failure or deep vein thrombosis. In the United States, the annual rate of hospitalization for upper GI bleeding is estimated to be 165 per 100,000—more than 300,000 hospitalizations per year, at a cost of $2.5 billion.1,2
Furthermore, despite advances in therapy, the case-fatality rate has remained unchanged at 7% to 10%.3 This may be because today’s patients are older and have more comorbidities than those in the past.4
CAUSES OF UPPER GI BLEEDING
Peptic ulcers account for about 60% of severe cases of upper GI bleeding,5 and they are the focus of this paper. Fortunately, up to 80% of bleeding ulcers stop bleeding spontaneously without any intervention.6
Gastroduodenal erosions account for about 12%.3
Varices due to cirrhosis are less common but more dangerous. Variceal bleeding accounts for a relatively small percentage (6%) of upper GI bleeding, but the mortality rate from a single episode of variceal bleeding is 30%, and 60% to 70% of patients die within 1 year, mostly of underlying liver disease.
Less frequent causes include Mallory-Weiss tears, erosive duodenitis, Dieulafoy ulcer (a type of vascular malformation), other vascular lesions, neoplasms, aortoenteric fistula, gastric antral vascular ectasia, and prolapse gastropathy.5
HEMATEMESIS AND MELENA
The most common presenting signs of acute upper GI bleeding are hematemesis (vomiting of blood), “coffee grounds” emesis, and melena (tarry black stools). About 30% of patients with bleeding ulcers present with hematemesis, 20% with melena, and 50% with both.7
Hematochezia (red blood in the stool) usually suggests a lower GI source of bleeding, since blood from an upper source turns black and tarry as it passes through the gut, producing melena. However, up to 5% of patients with bleeding ulcers have hematochezia,7 and it indicates heavy bleeding: bleeding of approximately 1,000 mL into the upper GI tract is needed to cause hematochezia, whereas only 50 to 100 mL is needed to cause melena.8,9 Hematochezia with signs and symptoms of hemodynamic compromise such as syncope, postural hypotension, tachycardia, and shock should therefore direct one’s attention to an upper GI source of bleeding.
Nonspecific features include nausea, vomiting, epigastric pain, vasovagal phenomena, and syncope.
WHAT IS THE PATIENT’S RISK?
An assessment of clinical severity is the first critical task, as it helps in planning treatment. Advanced age, multiple comorbidities, and hemodynamic instability call for aggressive treatment. Apart from this simple clinical rule, scoring systems have been developed.
The Rockall scoring system, the most widely used, gives estimates of the risks of recurrent bleeding and death. It is based on the three clinical factors mentioned above and on two endoscopic ones, awarding points for:
- Age—0 points if less than 60; 1 point if 60 to 79; or 2 points if 80 years or older
- Shock—1 point if the pulse is more than 100; 2 points if the systolic blood pressure is less than 100 mm Hg
- Comorbid illness—2 points for ischemic heart disease, congestive heart failure, or other major comorbidity; 3 points for renal failure, hepatic failure, or metastatic disease
- Endoscopic diagnosis—0 points if no lesion found or a Mallory-Weiss tear; 1 point for peptic ulcer, esophagitis, or erosive disease; 2 points for GI malignancy
- Endoscopic stigmata or recent hemorrhage—0 points for a clean-based ulcer or flat pigmented spot; 2 points for blood in the upper GI tract, active bleeding, a nonbleeding visible vessel, or adherent clot.
The Rockall score can thus range from 0 to 11 points, with an overall score of 0, 1, or 2 associated with an excellent prognosis.10
Other systems that are used less often include the Baylor severity scale and the Acute Physiology and Chronic Health Evaluation (APACHE) II score.
Does the patient have varices?
All variceal bleeding should be considered severe, since the 1-year death rate is so high (up to 70%). Clues pointing to variceal bleeding include previous variceal bleeding, thrombocytopenia, history of liver disease, and signs of liver disease on clinical examination.
All patients suspected of having bleeding varices should be admitted to the intensive care unit for close monitoring and should be given the highest priority, even if they are hemodynamically stable.
Is the patient hemodynamically stable?
Appropriate hemodynamic assessment includes monitoring of heart rate, blood pressure, and mental status. Tachycardia at rest, hypotension, and orthostatic changes in vital signs indicate a considerable loss of blood volume. Low urine output, dry mucous membranes, and sunken neck veins are also useful signs. (Tachycardia may be blunted if the patient is taking a beta-blocker.)
If these signs of hypovolemia are present, the initial management focuses on treating shock and on improving oxygen delivery to the vital organs. This involves repletion of the intravascular volume with intravenous infusions or blood transfusions. Supplemental oxygen also is useful, especially in elderly patients with heart disease.12
Inspection of nasogastric aspirate
In the initial assessment, it is useful to insert a nasogastric tube and inspect the aspirate. If it contains bright red blood, the patient needs an urgent endoscopic evaluation and an intensive level of care13,14; if it contains coffee-grounds material, the patient needs to be admitted to the hospital and to undergo endoscopic evaluation within 24 hours.
However, a normal aspirate does not rule out upper GI bleeding. Aljebreen et al15 found that 15% of patients with upper GI bleeding and normal nasogastric aspirate still had high-risk lesions (ie, visible bleeding or nonbleeding visible vessels) on endoscopy.
ACID-SUPPRESSION HELPS ULCERS HEAL
Acid and pepsin interfere with the healing of ulcers and other nonvariceal upper GI lesions. Further, an acidic environment promotes platelet disaggregation and fibrinolysis and impairs clot formation.16 This suggests that inhibiting gastric acid secretion and raising the gastric pH to 6 or higher may stabilize clots. Moreover, pepsinogen in the stomach is converted to its active form (pepsin) if the pH is less than 4. Therefore, keeping the pH above 4 keeps pepsinogen in an inactive form.
Histamine-2 receptor antagonists
Histamine-2 receptor antagonists were the first drugs to inhibit acid secretion, reversibly blocking histamine-2 receptors on the basolateral membrane of parietal cells. However, these drugs did not prove very useful in managing upper GI bleeding in clinical trials.17,18 In their intravenous form, they often fail to keep the gastric pH at 6 or higher, due to tachyphylaxis.19 The use of this class of drugs has declined in favor of proton pump inhibitors.
Proton pump inhibitors
Proton pump inhibitors reduce both basal and stimulated acid secretion by inhibiting hydrogen-potassium adenosine triphosphatase, the proton pump of the parietal cell.
Multiple studies have shown that proton pump inhibitors raise the gastric pH and keep it high. For example, an infusion of omeprazole (Prilosec) can keep the gastric pH above 6 for 72 hours without inducing tachyphylaxis.20,21
Started after endoscopy. Randomized controlled trials have found proton pump inhibitors to be effective when given in high doses intravenously for 72 hours after successful endoscopic treatment of bleeding ulcers with high-risk endoscopic signs, such as active bleeding or nonbleeding visible vessels.22,23
A meta-analysis indicated that these drugs decrease the incidence of recurrent peptic ulcer bleeding, the need for blood transfusions, the need for surgery, and the duration of hospitalization, but not the mortality rate.24,25 These studies also illustrate the benefit of following up endoscopic treatment to stop the bleeding with an intravenous infusion of a proton pump inhibitor.
The recommended dose of omeprazole for patients with high-risk findings on endoscopy is an 80-mg bolus followed by an 8-mg/hour infusion for 72 hours. After the patient’s condition stabilizes, oral therapy can be substituted for intravenous therapy. In patients with low-risk endoscopic findings (a clean-based ulcer or flat spot), oral proton pump inhibitors in high doses are recommended.
In either case, after the initial bleeding is treated endoscopically and hemostasis is achieved, a proton pump inhibitor is recommended for 6 to 8 weeks, or longer if the patient is also positive for Helicobacter pylori or is on daily treatment with aspirin or a nonsteroidal anti-inflammatory drug (NSAID) that is not selective for cyclo-oxygenase 2 (see below).
Started before endoscopy, these drugs reduced the frequency of actively bleeding ulcers, the duration of hospitalization, and the need for endoscopic therapy in a randomized controlled trial.26 A meta-analysis found that significantly fewer patients had signs of recent bleeding on endoscopy if they received a proton pump inhibitor 24 to 48 hours before the procedure, but it did not find any significant difference in important clinical outcomes such as death, recurrent bleeding, or surgery.27 Nevertheless, we believe that intravenous proton pump inhibitor therapy should be started before endoscopy in patients with upper GI bleeding.
Somatostatin analogues
Octreotide (Sandostatin), an analogue of the hormone somatostatin, decreases splanchnic blood flow, decreases secretion of gastric acid and pepsin, and stimulates mucus production. Although it is beneficial in treating upper GI bleeding due to varices, its benefit has not been confirmed in patients with nonvariceal upper GI bleeding.
A meta-analysis revealed that outcomes were better with high-dose intravenous proton pump inhibitor therapy than with octreotide when these drugs were started after endoscopic treatment of acute peptic ulcer bleeding.28 Nevertheless, octreotide may be useful in patients with uncontrolled nonvariceal bleeding who are awaiting endoscopy, since it is relatively safe to use.
ALL PATIENTS NEED ENDOSCOPY
All patients with upper GI bleeding need an upper endoscopic examination to diagnose and assess the risk posed by the bleeding lesion and to treat the lesion, reducing the risk of recurrent bleeding.
How urgently does endoscopy need to be done?
Endoscopy within the first 24 hours of upper GI bleeding is considered the standard of care. Patients with uncontrolled or recurrent bleeding should undergo endoscopy on an urgent basis to control the bleeding and reduce the risk of death.
However, how urgently endoscopy needs to be done is often debated. A multicenter randomized controlled trial compared outcomes in patients who underwent endoscopy within 6 hours of coming to the emergency department vs within 24 hours after the initial evaluation. The study found no significant difference in outcomes between the two groups; however, the group that underwent endoscopy sooner needed fewer transfusions.29
For a better view of the stomach
Gastric lavage improves the view of the gastric fundus but has not been proven to improve outcome.30
Promotility agents such as erythromycin and metoclopramide (Reglan) are also used to empty the stomach for better visualization.31–35 Erythromycin has been shown to improve visualization, shorten the procedure time, and prevent the need for additional endoscopy attempts in two randomized controlled studies.33,34 Furthermore, a cost-effectiveness study confirmed that giving intravenous erythromycin before endoscopy for acute upper GI bleeding saved money and resulted in an increase in quality-adjusted life-years.35
Endoscopy to diagnose bleeding and assess risk
Upper endoscopy is 90% to 95% diagnostic for acute upper GI bleeding.36
Patients at high risk (ie, older than 60 years, with severe comorbidity, or hemodynamically compromised) who have active bleeding (ie, witnessed hematemesis, red blood per nasogastric tube, or fresh blood per rectum) or a nonbleeding visible vessel should be admitted to a monitored bed or intensive care unit. Observation in a regular medical ward is appropriate for high-risk patients found to have an adherent clot. Patients with low-risk findings (eg, a clean ulcer base) are at low risk of recurrent bleeding and may be considered for early hospital discharge with appropriate outpatient follow-up.
Endoscopy to treat bleeding
About 25% of endoscopic procedures performed for upper GI bleeding include some type of treatment,39 such as injections of epinephrine, normal saline, or sclerosants; thermal cautery; argon plasma coagulation; electrocautery; or application of clips or bands. They are all equally effective, and combinations of these therapies are more effective than when they are used individually. A recent meta-analysis found dual therapy to be superior to epinephrine monotherapy in preventing recurrent bleeding, need for surgery, and death.40
How to manage adherent clots is controversial, but recent studies have revealed a significant benefit from removing them and treating the underlying lesions compared with drug therapy alone.43,45
Flat, pigmented spots and nonbleeding ulcers with a clean base do not require endoscopic treatment because the risk of recurrent bleeding is low.
Endoscopic therapy stops the bleeding in more than 90% of patients, but bleeding recurs after endoscopic therapy in 10% to 25%.46 Reversal of any severe coagulopathy with transfusions of platelets or fresh frozen plasma is essential for endoscopic hemostasis. However, coagulopathy at the time of initial bleeding and endoscopy does not appear to be associated with higher rates of recurrent bleeding following endoscopic therapy for nonvariceal upper GI bleeding.47
Patients with refractory bleeding are candidates for angiography or surgery. However, even when endoscopic hemostasis fails, endoscopy is important before angiography or surgery to pinpoint the site of bleeding and diagnose the cause.
A second endoscopic procedure is generally not recommended within 24 hours after the initial procedure.48 However, it is appropriate in cases in which clinical signs indicate recurrent bleeding or if hemostasis during the initial procedure is questionable. A meta-analysis found that routinely repeating endoscopy reduces the rate of recurrent bleeding but not the need for surgery or the risk of death.49
ALL PATIENTS SHOULD BE ADMITTED
VARICEAL BLEEDING
Variceal bleeding, a severe outcome of portal hypertension secondary to cirrhosis, carries a 6-week mortality rate of 10% to 20%.50 In view of the risk, primary prevention is indicated in patients with high-risk varices.
The mainstays of primary and secondary prevention are the nonselective beta-blockers such as nadolol (Corgard) and propranolol (Inderal). Several randomized controlled trials have shown lower rates of recurrent bleeding and death with propranolol or nadolol than with placebo.51 In doses that decrease the heart rate by 25%, beta-blockers have been shown to delay and decrease variceal hemorrhage. However, most patients require prophylactic endoscopic variceal ligation because they cannot tolerate beta-blocker therapy.
In suspected acute variceal bleeding, a somatostatin analogue should be started to decrease the portal pressure, and antibiotics should be started to reduce the risks of infection and death. Vasoactive drugs, ie, somatostatin analogues, should be started before endoscopy and continued for 5 days to reduce the chances of recurrent bleeding.52,53
Terlipressin is the only drug proven to improve the odds of survival in acute variceal bleeding. Although widely used in Europe, it has not been approved for use in the United States.
Octreotide, another option, improves hemostasis to the same extent, although it does not increase the survival rate.54,55 The recommended dose of octreotide for patients with variceal bleeding is a 50-μg intravenous bolus, followed by a 50-μg/hour infusion for 5 days.
Combining endoscopic and drug therapy improves the chances of stopping the bleeding and reduces the risk of recurrent bleeding compared with endoscopic therapy alone.56
Transjugular intrahepatic portosystemic shunting is indicated in recurrent variceal hemorrhage or in those with initial bleeding that is refractory to standard medical and endoscopic therapy. It is not the primary therapy because it doubles the risk of encephalopathy and has a high stent occlusion rate (up to 60%, lower with covered stents).
GI BLEEDING CAN CAUSE ACUTE MYOCARDIAL INFARCTION
The simultaneous presentation of acute myocardial infarction (MI) and GI hemorrhage is very serious and unfortunately common.
An acute MI occurring simultaneously with or after GI bleeding is usually precipitated by massive bleeding causing hypovolemia, hemodynamic compromise, and hypoperfusion. Conversely, the anticoagulant, antiplatelet, or thrombolytic drugs given to treat MI can precipitate GI bleeding (see below).
This distinction is important because the two scenarios have different clinical courses and prognoses. GI bleeding that precipitates an acute MI tends to be massive, whereas GI bleeding after treatment of acute MI tends to be self-limited and often resolves with reversal of underlying coagulopathy.57
Endoscopy carries a higher than average risk in patients with recent acute MI, with all-cause mortality rates as high as 1%.58 (The usual rate is 0.0004%.59) Nevertheless, endoscopy can be safely performed early on in patients with acute MI if it is done under strict monitoring in a coronary care unit.
Several studies have shown that MI patients who present with upper GI bleeding as the inciting event or patients with acute MI who are vomiting blood or who are hemodynamically unstable due to GI bleeding are significantly more likely to have a high-risk lesion and so have the greatest need for endoscopic therapy. Therefore, endoscopic intervention may be offered to MI patients at high risk who have been started on antiplatelet agents.
WARFARIN CAN PRECIPITATE BLEEDING
Acute upper GI bleeding can be a severe complication of long-term oral anticoagulation, not because the drugs cause ulcers, but rather because they exacerbate ulcers that are already present.60 Therefore, when starting warfarin (Coumadin), patients should be evaluated to determine if they have other risk factors for GI bleeding, such as ulcers.
The number of people presenting with upper GI bleeding while on warfarin therapy is increasing because of the expanding indications for long-term anticoagulation therapy, such as atrial fibrillation and deep venous thrombosis.
The risk of GI bleeding in patients who use oral anticoagulants is estimated to be 2.3 to 4.9 times higher than in nonusers.61
The goal international normalized ratio (INR) for patients on warfarin therapy is usually 2.0 to 3.0. Recent studies found that endoscopy can be safely performed in patients with acute GI bleeding whose INR is between 2.0 and 3.0.62,63 Some suggest that both the length of warfarin therapy and the INR affect the risk of bleeding.64,65
Managing patients with an INR higher than 3.0 who have an episode of GI bleeding is always a challenge. It is not uncommon to find pathologic lesions causing GI bleeding in patients who are on warfarin with a supratherapeutic INR, and thus, endoscopy is indicated. However, before endoscopy, reversal of anticoagulation should be considered.
BLEEDING IN PATIENTS ON ANTIPLATELET DRUGS
Aspirin
Aspirin decreases production of prostaglandins in the GI tract, thereby decreasing the protective and restorative properties of the gastric and duodenal mucosa and predisposing to ulcers and bleeding.
The higher the aspirin dose, the higher the risk. Aspirin doubles the risk of upper GI bleeding at daily doses of 75 mg and quadruples it at doses of 300 mg.66 Even doses as low as 10 mg can decrease gastric mucosal prostaglandin production.67 Thus, it appears that there is no risk-free dose of aspirin, and enteric-coated or buffered formulations do not appear to reduce the risk.68–70
The most important risk factor for upper GI bleeding in patients taking aspirin is a history of peptic ulcer bleeding. Approximately 15% of aspirin users who have bleeding from ulcers have recurrent bleeding within 1 year.71
As aspirin-induced GI bleeding becomes more common, health care providers often feel caught between the GI risk and the cardiovascular benefit. When considering whether to discontinue antiplatelet therapy, a cardiologist should be consulted along with a gastroenterologist to weigh the risks of GI bleeding vs thrombosis. To date, there have been no clinical trials published to suggest when antiplatelet therapy should be stopped to optimize GI and cardiovascular outcomes. An alternative is to replace aspirin with another antiplatelet drug that does not induce ulcers.
Clopidogrel
Clopidogrel (Plavix) is recommended for hospitalized patients with acute coronary syndrome who cannot tolerate the GI side effects of aspirin, according to the joint guidelines of the American College of Cardiology and the American Heart Association, with the highest level of evidence.72 This recommendation was largely based on the safety data from the CAPRIE (Clopidogrel Versus Aspirin in Patients at Risk of Ischemic Events) trial, in which the incidence of major GI bleeding was lower in the clopidogrel group (0.52%) than in the aspirin group (0.72%; P < .05).73
Aspirin plus a proton pump inhibitor
Patients who have had an episode of upper GI bleeding and who need long-term aspirin therapy should also receive a proton pump inhibitor indefinitely to prevent ulcer recurrence.
In a recent double-blind randomized controlled trial in patients with a history of aspirin-induced bleeding, the combination of low-dose aspirin plus esomeprazole (Nexium) twice a day was superior to clopidogrel by itself in terms of the rate of recurrent bleeding (0.7% vs 8.6%; P < .05).74 A similar trial showed nearly identical results: 0% upper GI bleeding in the group receiving aspirin plus esomeprazole 20 mg daily, vs 13.6% in the clopidogrel group (P = .0019).75 These studies suggest that a once-daily proton pump inhibitor combined with aspirin is a safer alternative than clopidogrel alone.
Clopidogrel plus a proton pump inhibitor
Interestingly, recent studies have shown that omeprazole decreases the antiplatelet effect of clopidogrel, possibly by inhibiting the CYP2C19 enzyme.76 However, concomitant use of pantoprazole (Protonix), lansoprazole (Prevacid), and esomeprazole did not have this effect, suggesting that although all proton pump inhibitors are metabolized to a varying degree by CYP2C19, the interaction between proton pump inhibitors and clopidogrel is not a class effect.77–79 Therefore, pantoprazole, lansoprazole, and esomeprazole may be the appropriate proton pump inhibitors to use with clopidogrel in patients who have a clear indication for the medication, consistent with current guideline recommendations.
Helicobacter pylori infection in antiplatelet drug users
TREATMENT AND PREVENTION OF NSAID-RELATED GI INJURY
About 1 in 20 users of NSAIDs develop GI complications and ulcers of varying degrees of severity, as do one in seven NSAID users over the age of 65. In fact, NSAID use accounts for 30% of hospitalizations for upper GI bleeding and deaths from this cause.82–85 In addition, approximately 15% to 30% of NSAID users have clinically silent but endoscopically evident peptic ulcers.86
NSAIDs contribute to ulcer development by depleting prostaglandins. Thus, misoprostol (Cytotec), a synthetic prostaglandin, has been used to reduce this side effect.
In a clinical trial, misoprostol reduced the incidence of NSAID-associated GI complications by 40%.87 Furthermore, it has been shown to be better than placebo in preventing recurrent gastric ulcers in patients with a history of gastric ulcer who were receiving low-dose aspirin.88
However, misoprostol is rarely used because it can cause diarrhea and abdominal cramping. Rather, the preferred drugs for preventing and treating NSAID- and aspirin-related GI lesions are proton pump inhibitors.
Numerous clinical trials using endoscopic end points showed that proton pump inhibitors in standard doses significantly reduce the incidence of ulcers associated with the use of NSAIDs.89 Proton pump inhibitor therapy has achieved a significant reduction in relative risk of upper GI bleeding in patients who received low-dose aspirin therapy, as confirmed by epidemiologic studies.90,91 The number of NSAID-related ulcers found on endoscopy could be reduced by an estimated 90% simply by using proton pump inhibitors.92
- Lewis JD, Bilker WB, Brensinger C, Farrar JT, Strom BL. Hospitalization and mortality rates from peptic ulcer disease and GI bleeding in the 1990s: relationship to sales of non-steroidal anti-inflammatory drugs and acid suppression medications. Am J Gastroenterol 2002; 97:2540–2549.
- Viviane A, Alan BN. Estimates of costs of hospital stays for variceal and nonvariceal upper gastrointestinal bleeding in the United States. Value Health 2008; 11:1–3.
- Yavorski RT, Wong RK, Maydonovitch C, Battin LS, Furnia A, Amundson DE. Analysis of 3,294 cases of upper gastrointestinal bleeding in military medical facilities. Am J Gastroenterol 1995; 90:568–573.
- Kaplan RC, Heckbert SR, Koepsell TD, et al. Risk factors for hospitalized gastrointestinal bleeding among older persons. Cardiovascular Health Study Investigators. J Am Geriatr Soc 2001; 49:126–133.
- Longstreth GF. Epidemiology of hospitalization for acute upper gastrointestinal hemorrhage: a population-based study. Am J Gastroenterol 1995; 90:206–210.
- Laine L, Peterson WL. Bleeding peptic ulcer. N Engl J Med 1994; 331:717–727.
- Wara P, Stodkilde H. Bleeding pattern before admission as guideline for emergency endoscopy. Scand J Gastroenterol 1985; 20:72–78.
- Jensen DM, Machicado GA. Diagnosis and treatment of severe hematochezia. The role of urgent colonoscopy after purge. Gastroenterology 1988; 95:1569–1574.
- Daniel WA, Egan S. The quantity of blood required to produce a tarry stool. J Am Med Assoc 1939; 113:2232.
- Rockall TA, Logan RF, Devlin HB, Northfield TC. Risk assessment after acute upper gastrointestinal hemorrhage. Gut 1996; 38:316–321.
- Blatchford O, Murray WR, Blatchford M. A risk score to predict need for treatment for upper-gastrointestinal hemorrhage. Lancet 2000; 356:1318–1321.
- Gralnek IM, Barkun AN, Bardou M. Management of acute bleeding from a peptic ulcer. N Engl J Med 2008; 359:928–937.
- Silverstein FE, Gilbert DA, Tedesco FJ, Buenger NK, Persing J. The national ASGE survey on upper gastrointestinal bleeding II. Clinical prognostic factors. Gastrointest Endosc 1981; 27:80–93.
- Corley DA, Stefan AM, Wolf M, Cook EF, Lee TH. Early indicators of prognosis in upper gastrointestinal hemorrhage. Am J Gastroenterol 1998; 93:336–340.
- Aljebreen AM, Fallone CA, Barkun AN. Nasogastric aspirate predicts high-risk endoscopic lesions in patients with acute upper-GI bleeding. Gastrointest Endosc 2004; 59:172–178.
- Barkun AN, Cockeram AW, Plourde V, Fedorak RN. Review article: acid suppression in non-variceal acute upper gastrointestinal bleeding. Aliment Pharmacol Ther 1999; 13:1565–1584.
- Levine JE, Leontiadis JI, Sharma VK, Howden CW. Meta-analysis: the efficacy of intravenous H2-receptor antagonists in bleeding peptic ulcer. Aliment Pharmacol Ther 2002; 16:1137–1142.
- Walt RP, Cottrell J, Mann SG, Freemantle NP, Langman MJ. Continuous intravenous famotidine for hemorrhage from peptic ulcer. Lancet 1992; 340:1058–1062.
- Labenz J, Peitz U, Leusing C, Tillenburg B, Blum AL, Börsch G. Efficacy of primed infusion with high dose ranitidine and omeprazole to maintain high intragastric pH in patients with peptic ulcer bleeding: a prospective randomized controlled study. Gut 1997; 40:36–41.
- Merki HS, Wilder-Smith CH. Do continuous infusions of omeprazole and ranitidine retain their effect with prolonged dosing? Gastroenterology 1994; 106:60–64.
- Netzer P, Gaia C, Sandoz M, et al. Effect of repeated injection and continuous infusion of omeprazole and ranitidine on intragastric pH over 72 hours. Am J Gastroenterol 1999; 94:351–357.
- Lin HJ, Lo WC, Cheng YC, Perng CL. Role of intravenous omeprazole in patients with high-risk peptic ulcer bleeding after successful endoscopic epinephrine injection: a prospective randomized comparative trial. Am J Gastroenterol 2006; 101:500–505.
- Lau JY, Sung JJ, Lee KK, et al. Effects of intravenous omeprazole on recurrent bleeding after endoscopic treatment of bleeding peptic ulcers. N Engl J Med 2000; 343:310–316.
- Leontiadis GI, Sharma VK, Howden CW. Proton pump inhibitor treatment for acute peptic ulcer bleeding. Cochrane Database Syst Rev 2006;CD002094.
- Andriulli A, Annese V, Caruso N, et al. Proton-pump inhibitors and outcome of endoscopic hemostasis in bleeding peptic ulcers: a series of meta-analyses. Am J Gastroenterol 2005; 100:207–219.
- Lau JY, Leung WK, Wu JC, et al. Omeprazole before endoscopy in patients with gastrointestinal bleeding. N Engl J Med 2007; 356:1631–1640.
- Dorward S, Sreedharan A, Leontiadis GI, Howden CW, Moayyedi P, Forman D. Proton pump inhibitor treatment initiated prior to endoscopic diagnosis in upper gastrointestinal bleeding. Cochrane Database Syst Rev 2006;CD005415.
- Bardou M, Toubouti Y, Benhaberou-Brun D, Rahme E, Barkun AN. Meta-analysis: proton-pump inhibition in high-risk patients with acute peptic ulcer bleeding. Aliment Pharmacol Ther 2005; 21:677–686.
- Bjorkman DJ, Zaman A, Fennerty MB, Lieberman D, Disario JA, Guest-Warnick G. Urgent vs elective endoscopy for acute non-variceal upper-GI bleeding: an effectiveness study. Gastointest Endosc 2004; 60:1–8.
- Lee SD, Kearney DJ. A randomized controlled trial of gastric lavage prior to endoscopy for acute upper gastrointestinal bleeding. J Clin Gastroenterol 2004; 38:861–865.
- Tack J, Janssens J, Vantrappen G, et al. Effect of erythromycin on gastric motility in controls and in diabetic gastroparesis. Gastroenterology 1992; 103:72–79.
- Xynos E, Mantides A, Papageorgiou A, Fountos A, Pechlivanides G, Vassilakis JS. Erythromycin accelerates delayed gastric emptying of solids in patients after truncal vagotomy and pyloroplasty. Eur J Surg 1992; 158:407–411.
- Coffin B, Pocard M, Panis Y, et al; Groupe des endoscopistes de garde á l’AP-HP. Erythromycin improves the quality of EGD in patients with acute upper GI bleeding: a randomized controlled study. Gastrointest Endosc 2002; 56:174–179.
- Frossard JL, Spahr L, Queneau PE, et al. Erythromycin intravenous bolus infusion in acute upper gastrointestinal bleeding: a randomized, controlled, double-blind trial. Gastroenterology 2002; 123:17–23.
- Winstead NS, Wilcox CM. Erythromycin prior to endoscopy for acute upper gastrointestinal hemorrhage: a cost-effectiveness analysis. Aliment Pharmacol Ther 2007; 26:1371–1377.
- Chak A, Cooper GS, Lloyd LE, Kolz CS, Barnhart BA, Wong RC. Effectiveness of endoscopy in patients admitted to the intensive care unit with upper GI hemorrhage. Gastrointest Endosc 2001; 53:6–13.
- Lau JY, Chung SC, Leung JW, Lo KK, Yung MY, Li AK. The evolution of stigmata of hemorrhage in bleeding peptic ulcers: a sequential endoscopic study. Endoscopy 1998; 30:513–518.
- Chung IK, Kim EJ, Lee MS, et al. Endoscopic factors predisposing to rebleeding following endoscopic hemostasis in bleeding peptic ulcers. Endoscopy 2001; 33:969–975.
- Elta GH. Acute nonvariceal upper gastrointestinal hemorrhage. Curr Treat Options Gastroenterol 2002; 5:147–152.
- Marmo R, Rotondano G, Piscopo R, Bianco MA, D’Angella R, Cipolletta L. Dual therapy versus monotherapy in the endoscopic treatment of high-risk bleeding ulcers: a meta-analysis of controlled trials. Am J Gastroenterol 2007; 102:279–289.
- Kovacs TO, Jensen DM. Recent advances in the endoscopic diagnosis and therapy of upper gastrointestinal, small intestinal, and colonic bleeding. Med Clin North Am 2002; 86:1319–1356.
- Kovacs TO, Jensen DM. Endoscopic treatment of ulcer bleeding. Curr Treat Options Gastroenterol 2007; 10:143–148.
- Jensen DM, Kovacs TO, Jutabha R, et al. Randomized trial of medical or endoscopic therapy to prevent recurrent ulcer hemorrhage in patients with adherent clots. Gastroenterology 2002; 123:407–413.
- Jensen DM, Machicado GA. Endoscopic hemostasis of ulcer hemorrhage with injection, thermal, and combination methods. Techniques Gastrointest Endosc 2005; 7:124–131.
- Bleau BL, Gostout CJ, Sherman KE, et al. Recurrent bleeding from peptic ulcer associated with adherent clot: a randomized study comparing endoscopic treatment with medical therapy. Gastrointest Endosc 2002; 56:1–6.
- Lau JY, Sung JJ, Lam YH, et al. Endoscopic retreatment compared with surgery in patients with recurrent bleeding after initial endoscopic control of bleeding ulcers. N Engl J Med 1999; 340:751–756.
- Wolf AT, Wasan SK, Saltzman JR. Impact of anticoagulation on rebleeding following endoscopic therapy for nonvariceal upper gastrointestinal hemorrhage. Am J Gastroenterol 2007; 102:290–296.
- Barkun A, Bardou M, Marshall JK; Nonvariceal Upper GI Bleeding Consensus Conference Group. Consensus recommendations for managing patients with nonvariceal upper gastrointestinal bleeding. Ann Intern Med 2003; 139:843–857.
- Marmo R, Rotondano G, Bianco MA, Piscopo R, Prisco A, Cipolletta L. Outcome of endoscopic treatment for peptic ulcer bleeding: is a second look necessary? A meta-analysis. Gastrointest Endosc 2003; 57:62–67.
- Dell’Era A, deFrancis R, Iannuzzi F. Acute variceal bleeding: pharmacological treatment and primary/secondary prophylaxis. Best Pract Res Clin Gastroenterol 2008; 22:279–294.
- Jalan R, Hayes PC. UK guidelines on the management of variceal hemorrhage in cirrhotic patients. British Society of Gastroenterology. Gut 2000; 46( suppl 3–4):III1–III15.
- Bernard B, Lebrec D, Mathurin P, Opolon P, Poynard T. Beta-adrenergic antagonists in the prevention of gastrointestinal rebleeding in patients with cirrhosis: a meta-analysis. Hepatology 1997; 25:63–70.
- De Franchis R. Evolving consensus in portal hypertension. Report of the Baveno IV consensus workshop on methodology of diagnosis and therapy in portal hypertension. J Hepatol 2005; 43:167–176.
- Levacher S, Letoumelin P, Pateron D, Blaise M, Lapandry C, Pourriat JL. Early administration of terlipressin plus glyceryl trinitrate to control active upper gastrointestinal bleeding in cirrhotic patients. Lancet 1995; 346:865–868.
- Abraldes JG, Bosch J. Somatostatin and analogues in portal hypertension. Hepatology 2002; 35:1305–1312.
- Banares R, Albillos A, Rincon D, et al. Endoscopic treatment versus endoscopic plus pharmacological treatment for acute variceal bleeding: a meta analysis. Hepatology 2002; 35:609–615.
- Cappell M. Gastrointenstinal bleeding associated with myocardial infarction. Gastroenterol Clin North Am 2000; 29:423–444.
- Lin S, Konstance R, Jollis J, Fisher DA. The utility of upper endoscopy in patients with concomitant upper gastrointestinal bleeding and acute myocardial infarction. Dig Dis Sci 2006; 51:2377–2383.
- Silvis SE, Nebel O, Rogers G, Sugawa C, Mandelstam P. Endoscopic complications. Results of the 1974 American Society for Gastrointestinal Endoscopy Survey. JAMA 1976; 235:928–930.
- Lanas A, Garcia-Rodriguez LA, Arroyo MT, et al; Investigators of the Asociación Española de Gastroenterología (AEG). Effect of antisecretory drugs and nitrates on the risk of ulcer bleeding associated with nonsteroidal anti-inflammatory drugs, antiplatelet agents, and anticoagulants. Am J Gastroenterol 2007; 102:507–515.
- Shorr RI, Ray WA, Daugherty JR, Griffin MR. Concurrent use of nonsteroidal anti-inflammatory drugs and oral anticoagulants places elderly persons at high risk for hemorrhagic peptic ulcer disease. Arch Intern Med 1993; 153:1665–1670.
- Tabibian N. Acute gastrointestinal bleeding in anticoagulated patients: a prospective evaluation. Am J Gastroenterol 1989; 84:10–12.
- Choudari CP, Rajgopal C, Palmer KR. Acute gastrointestinal hemorrhage in anticoagulated patients: diagnoses and response to endoscopic treatment. Gut 1994; 35:464–466.
- Petty GW, Brown RD, Whisnant JP, Sicks JD, O’Fallon WM, Wiebers DO. Frequency of major complications of aspirin, warfarin, and intravenous heparin for secondary stroke prevention: a population-based study. Ann Intern Med 1999; 130:14–22.
- Landefeld CS, Rosenblatt MW, Goldman L. Bleeding in outpatients treated with warfarin: relation to the prothrombin time and important remediable lesions. Am J Med 1989; 87:153–159.
- Weil J, Colin-Jones D, Langman M, et al. Prophylactic aspirin and risk of peptic ulcer bleeding. BMJ 1995; 310:827–830.
- Cryer B, Feldman M. Effects of very low dose daily, long-term aspirin therapy on gastric, duodenal, and rectal prostaglandin levels and on mucosal injury in healthy humans. Gastroenterology 1999; 117:17–25.
- De Abajo FJ, Garcia Rodriguez LA. Risk of upper gastrointestinal bleeding and perforation associated with low-dose aspirin as plain and enteric-coated formulations. BMC Clin Pharmacol 2001; 1:1.
- Kelly JP, Kaufman DW, Jurgelon JM, Sheehan J, Koff RS, Shapiro S. Risk of aspirin-associated major upper gastrointestinal bleeding with enteric coated or buffered product. Lancet 1996; 348:1413–1416.
- Garcia Rodriguez LA, Hernandez-Diaz S, De Abajo FJ. Association between aspirin and upper gastrointestinal complications: systematic review of epidemiological studies. Br J Clin Pharmacol 2001; 52:563–571.
- Wilcox CM, Ladabaum U. A patient with high risk of gastrointestinal bleeding requiring nonsteroidal anti-inflammatory drugs. Clin Gastroenterol Hepatol 2006; 4:1090–1093.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet 1996; 348:1329–1339.
- Chan FK, Ching JY, Hung LC, et al. Clopidogrel versus aspirin and esomeprazole to prevent recurrent ulcer bleeding. N Engl J Med 2005; 352:238–244.
- Lai KC, Chu KM, Hui WM, et al. Esomeprazole with aspirin versus clopidogrel for prevention of recurrent gastrointestinal ulcer complications. Clin Gastroenterol Hepatol 2006; 4:860–865.
- Ho MP, Maddox TM, Wang L, et al. Risk of adverse outcomes associated with concomitant use of clopidogrel and proton pump inhibitors following acute coronary syndrome. JAMA 2009; 301:937–944.
- Siller-Matula JM, Spiel AO, Lang IM, Kreiner G, Christ G, Jilma B. Effects of pantoprazole and esomeprazole on platelet inhibition by clopidogrel. Am Heart J 2009; 157:148.e1–e5.
- Small DS, Farid NA, Payne CD, et al. Effects of proton pump inhibitor lansoprazole on the pharmacokinetics and pharmacodynamics of prasugel and clopidogrel. J Clin Pharmacol 2008; 48:475–484.
- Ishizaki T, Horai Y. Review article: cytochrome P450 and the metabolism of proton pump inhibitors—emphasis on rabeprazole. Aliment Pharacol Ther 1999; 13 (suppl 3):27–36.
- Lanas A, Fuentes J, Benito R, Serrano P, Bajador E, Sainz R. Helicobacter pylori increases the risk of upper gastrointestinal bleeding in patients taking low-dose aspirin. Aliment Pharmacol Ther 2002; 16:779–786.
- Chan FK. NSAID-Induced peptic ulcers and Helicobacter pylori infection: implications for patient management. Drug Saf 2005; 28:287–300.
- Bombardier V, Laine L, Reicin A, et al; VIGOR Study Group. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis: VIGOR Study Group. N Eng J Med 2000; 343:1520–1528.
- Griffin MR, Ray WA, Schaffner W. Nonsteroidal anti-inflammatory drug use and death from peptic ulcer in elderly persons. Ann Intern Med 1988; 109:359–363.
- Griffin MR, Piper JM, Daugherty JR, Snowden M, Ray WA. Nonsteroidal anti-inflammatory drug use and increased risk for peptic ulcer disease in elderly persons. Ann Intern Med 1991; 114:257–263.
- Smalley WE, Ray WA, Daugherty JR, Griffin MR. Nonsteroidal anti-inflammatory drugs and the incidence of hospitalizations for peptic ulcer disease in elderly persons. Am J Epidemiol 1995; 141:539–545.
- Laine L. Approaches to nonsteroidal anti-inflammatory drug use in the high-risk patient. Gastroenterology 2001; 120:594–606.
- Silverstein FE, Graham DY, Senior JR, et al. Misoprostol reduces serious gastrointestinal complications in patients with rheumatoid arthritis receiving nonsteroidal anti-inflammatory drugs. A randomized, double blind, placebo controlled trial. Ann Intern Med 1995; 123:241–249.
- Goldstein JL, Huang B, Amer F, Christopoulos NG. Ulcer recurrence in high-risk patients receiving nonsteroidal anti-inflammatory drugs plus low dose aspirin: results of a post hoc subanalysis. Clin Ther 2004; 26:1637–1643.
- Berger JS, Stebbins A, Granger CB, et al. Initial aspirin dose and outcome among ST-elevation myocardial infarction patients treated with fibrinolytic therapy. Circulation 2008; 117:192–199.
- Lanas A, Garcia-Rodriguez LA, Arroyo MT, et al; Investigators of the Asociación Española de Gastroenterología (AEG). Effect of antisecretory drugs and nitrates on the risk of ulcer bleeding associated with nonsteroidal anti-inflammatory drugs, antiplatelet agents, and anticoagulants. Am J Gastroenterol 2007; 102:507–515.
- Chin MW, Yong G, Bulsara MK, Rankin J, Forbes GM. Predictive and protective factors associated with upper gastrointestinal bleeding after percutaneous coronary intervention: a case-control study. Am J Gastroenterol 2007; 102:2411–2416.
- Hunt RH, Bazzoli F. Should NSAID/low dose aspirin takers be tested routinely for H. Pylori infection and treated if positive? Implications for primary risk of ulcer and ulcer relapse after initial healing. Aliment Pharmacol Ther 2004; 19 (suppl 1):9–16.
- Lewis JD, Bilker WB, Brensinger C, Farrar JT, Strom BL. Hospitalization and mortality rates from peptic ulcer disease and GI bleeding in the 1990s: relationship to sales of non-steroidal anti-inflammatory drugs and acid suppression medications. Am J Gastroenterol 2002; 97:2540–2549.
- Viviane A, Alan BN. Estimates of costs of hospital stays for variceal and nonvariceal upper gastrointestinal bleeding in the United States. Value Health 2008; 11:1–3.
- Yavorski RT, Wong RK, Maydonovitch C, Battin LS, Furnia A, Amundson DE. Analysis of 3,294 cases of upper gastrointestinal bleeding in military medical facilities. Am J Gastroenterol 1995; 90:568–573.
- Kaplan RC, Heckbert SR, Koepsell TD, et al. Risk factors for hospitalized gastrointestinal bleeding among older persons. Cardiovascular Health Study Investigators. J Am Geriatr Soc 2001; 49:126–133.
- Longstreth GF. Epidemiology of hospitalization for acute upper gastrointestinal hemorrhage: a population-based study. Am J Gastroenterol 1995; 90:206–210.
- Laine L, Peterson WL. Bleeding peptic ulcer. N Engl J Med 1994; 331:717–727.
- Wara P, Stodkilde H. Bleeding pattern before admission as guideline for emergency endoscopy. Scand J Gastroenterol 1985; 20:72–78.
- Jensen DM, Machicado GA. Diagnosis and treatment of severe hematochezia. The role of urgent colonoscopy after purge. Gastroenterology 1988; 95:1569–1574.
- Daniel WA, Egan S. The quantity of blood required to produce a tarry stool. J Am Med Assoc 1939; 113:2232.
- Rockall TA, Logan RF, Devlin HB, Northfield TC. Risk assessment after acute upper gastrointestinal hemorrhage. Gut 1996; 38:316–321.
- Blatchford O, Murray WR, Blatchford M. A risk score to predict need for treatment for upper-gastrointestinal hemorrhage. Lancet 2000; 356:1318–1321.
- Gralnek IM, Barkun AN, Bardou M. Management of acute bleeding from a peptic ulcer. N Engl J Med 2008; 359:928–937.
- Silverstein FE, Gilbert DA, Tedesco FJ, Buenger NK, Persing J. The national ASGE survey on upper gastrointestinal bleeding II. Clinical prognostic factors. Gastrointest Endosc 1981; 27:80–93.
- Corley DA, Stefan AM, Wolf M, Cook EF, Lee TH. Early indicators of prognosis in upper gastrointestinal hemorrhage. Am J Gastroenterol 1998; 93:336–340.
- Aljebreen AM, Fallone CA, Barkun AN. Nasogastric aspirate predicts high-risk endoscopic lesions in patients with acute upper-GI bleeding. Gastrointest Endosc 2004; 59:172–178.
- Barkun AN, Cockeram AW, Plourde V, Fedorak RN. Review article: acid suppression in non-variceal acute upper gastrointestinal bleeding. Aliment Pharmacol Ther 1999; 13:1565–1584.
- Levine JE, Leontiadis JI, Sharma VK, Howden CW. Meta-analysis: the efficacy of intravenous H2-receptor antagonists in bleeding peptic ulcer. Aliment Pharmacol Ther 2002; 16:1137–1142.
- Walt RP, Cottrell J, Mann SG, Freemantle NP, Langman MJ. Continuous intravenous famotidine for hemorrhage from peptic ulcer. Lancet 1992; 340:1058–1062.
- Labenz J, Peitz U, Leusing C, Tillenburg B, Blum AL, Börsch G. Efficacy of primed infusion with high dose ranitidine and omeprazole to maintain high intragastric pH in patients with peptic ulcer bleeding: a prospective randomized controlled study. Gut 1997; 40:36–41.
- Merki HS, Wilder-Smith CH. Do continuous infusions of omeprazole and ranitidine retain their effect with prolonged dosing? Gastroenterology 1994; 106:60–64.
- Netzer P, Gaia C, Sandoz M, et al. Effect of repeated injection and continuous infusion of omeprazole and ranitidine on intragastric pH over 72 hours. Am J Gastroenterol 1999; 94:351–357.
- Lin HJ, Lo WC, Cheng YC, Perng CL. Role of intravenous omeprazole in patients with high-risk peptic ulcer bleeding after successful endoscopic epinephrine injection: a prospective randomized comparative trial. Am J Gastroenterol 2006; 101:500–505.
- Lau JY, Sung JJ, Lee KK, et al. Effects of intravenous omeprazole on recurrent bleeding after endoscopic treatment of bleeding peptic ulcers. N Engl J Med 2000; 343:310–316.
- Leontiadis GI, Sharma VK, Howden CW. Proton pump inhibitor treatment for acute peptic ulcer bleeding. Cochrane Database Syst Rev 2006;CD002094.
- Andriulli A, Annese V, Caruso N, et al. Proton-pump inhibitors and outcome of endoscopic hemostasis in bleeding peptic ulcers: a series of meta-analyses. Am J Gastroenterol 2005; 100:207–219.
- Lau JY, Leung WK, Wu JC, et al. Omeprazole before endoscopy in patients with gastrointestinal bleeding. N Engl J Med 2007; 356:1631–1640.
- Dorward S, Sreedharan A, Leontiadis GI, Howden CW, Moayyedi P, Forman D. Proton pump inhibitor treatment initiated prior to endoscopic diagnosis in upper gastrointestinal bleeding. Cochrane Database Syst Rev 2006;CD005415.
- Bardou M, Toubouti Y, Benhaberou-Brun D, Rahme E, Barkun AN. Meta-analysis: proton-pump inhibition in high-risk patients with acute peptic ulcer bleeding. Aliment Pharmacol Ther 2005; 21:677–686.
- Bjorkman DJ, Zaman A, Fennerty MB, Lieberman D, Disario JA, Guest-Warnick G. Urgent vs elective endoscopy for acute non-variceal upper-GI bleeding: an effectiveness study. Gastointest Endosc 2004; 60:1–8.
- Lee SD, Kearney DJ. A randomized controlled trial of gastric lavage prior to endoscopy for acute upper gastrointestinal bleeding. J Clin Gastroenterol 2004; 38:861–865.
- Tack J, Janssens J, Vantrappen G, et al. Effect of erythromycin on gastric motility in controls and in diabetic gastroparesis. Gastroenterology 1992; 103:72–79.
- Xynos E, Mantides A, Papageorgiou A, Fountos A, Pechlivanides G, Vassilakis JS. Erythromycin accelerates delayed gastric emptying of solids in patients after truncal vagotomy and pyloroplasty. Eur J Surg 1992; 158:407–411.
- Coffin B, Pocard M, Panis Y, et al; Groupe des endoscopistes de garde á l’AP-HP. Erythromycin improves the quality of EGD in patients with acute upper GI bleeding: a randomized controlled study. Gastrointest Endosc 2002; 56:174–179.
- Frossard JL, Spahr L, Queneau PE, et al. Erythromycin intravenous bolus infusion in acute upper gastrointestinal bleeding: a randomized, controlled, double-blind trial. Gastroenterology 2002; 123:17–23.
- Winstead NS, Wilcox CM. Erythromycin prior to endoscopy for acute upper gastrointestinal hemorrhage: a cost-effectiveness analysis. Aliment Pharmacol Ther 2007; 26:1371–1377.
- Chak A, Cooper GS, Lloyd LE, Kolz CS, Barnhart BA, Wong RC. Effectiveness of endoscopy in patients admitted to the intensive care unit with upper GI hemorrhage. Gastrointest Endosc 2001; 53:6–13.
- Lau JY, Chung SC, Leung JW, Lo KK, Yung MY, Li AK. The evolution of stigmata of hemorrhage in bleeding peptic ulcers: a sequential endoscopic study. Endoscopy 1998; 30:513–518.
- Chung IK, Kim EJ, Lee MS, et al. Endoscopic factors predisposing to rebleeding following endoscopic hemostasis in bleeding peptic ulcers. Endoscopy 2001; 33:969–975.
- Elta GH. Acute nonvariceal upper gastrointestinal hemorrhage. Curr Treat Options Gastroenterol 2002; 5:147–152.
- Marmo R, Rotondano G, Piscopo R, Bianco MA, D’Angella R, Cipolletta L. Dual therapy versus monotherapy in the endoscopic treatment of high-risk bleeding ulcers: a meta-analysis of controlled trials. Am J Gastroenterol 2007; 102:279–289.
- Kovacs TO, Jensen DM. Recent advances in the endoscopic diagnosis and therapy of upper gastrointestinal, small intestinal, and colonic bleeding. Med Clin North Am 2002; 86:1319–1356.
- Kovacs TO, Jensen DM. Endoscopic treatment of ulcer bleeding. Curr Treat Options Gastroenterol 2007; 10:143–148.
- Jensen DM, Kovacs TO, Jutabha R, et al. Randomized trial of medical or endoscopic therapy to prevent recurrent ulcer hemorrhage in patients with adherent clots. Gastroenterology 2002; 123:407–413.
- Jensen DM, Machicado GA. Endoscopic hemostasis of ulcer hemorrhage with injection, thermal, and combination methods. Techniques Gastrointest Endosc 2005; 7:124–131.
- Bleau BL, Gostout CJ, Sherman KE, et al. Recurrent bleeding from peptic ulcer associated with adherent clot: a randomized study comparing endoscopic treatment with medical therapy. Gastrointest Endosc 2002; 56:1–6.
- Lau JY, Sung JJ, Lam YH, et al. Endoscopic retreatment compared with surgery in patients with recurrent bleeding after initial endoscopic control of bleeding ulcers. N Engl J Med 1999; 340:751–756.
- Wolf AT, Wasan SK, Saltzman JR. Impact of anticoagulation on rebleeding following endoscopic therapy for nonvariceal upper gastrointestinal hemorrhage. Am J Gastroenterol 2007; 102:290–296.
- Barkun A, Bardou M, Marshall JK; Nonvariceal Upper GI Bleeding Consensus Conference Group. Consensus recommendations for managing patients with nonvariceal upper gastrointestinal bleeding. Ann Intern Med 2003; 139:843–857.
- Marmo R, Rotondano G, Bianco MA, Piscopo R, Prisco A, Cipolletta L. Outcome of endoscopic treatment for peptic ulcer bleeding: is a second look necessary? A meta-analysis. Gastrointest Endosc 2003; 57:62–67.
- Dell’Era A, deFrancis R, Iannuzzi F. Acute variceal bleeding: pharmacological treatment and primary/secondary prophylaxis. Best Pract Res Clin Gastroenterol 2008; 22:279–294.
- Jalan R, Hayes PC. UK guidelines on the management of variceal hemorrhage in cirrhotic patients. British Society of Gastroenterology. Gut 2000; 46( suppl 3–4):III1–III15.
- Bernard B, Lebrec D, Mathurin P, Opolon P, Poynard T. Beta-adrenergic antagonists in the prevention of gastrointestinal rebleeding in patients with cirrhosis: a meta-analysis. Hepatology 1997; 25:63–70.
- De Franchis R. Evolving consensus in portal hypertension. Report of the Baveno IV consensus workshop on methodology of diagnosis and therapy in portal hypertension. J Hepatol 2005; 43:167–176.
- Levacher S, Letoumelin P, Pateron D, Blaise M, Lapandry C, Pourriat JL. Early administration of terlipressin plus glyceryl trinitrate to control active upper gastrointestinal bleeding in cirrhotic patients. Lancet 1995; 346:865–868.
- Abraldes JG, Bosch J. Somatostatin and analogues in portal hypertension. Hepatology 2002; 35:1305–1312.
- Banares R, Albillos A, Rincon D, et al. Endoscopic treatment versus endoscopic plus pharmacological treatment for acute variceal bleeding: a meta analysis. Hepatology 2002; 35:609–615.
- Cappell M. Gastrointenstinal bleeding associated with myocardial infarction. Gastroenterol Clin North Am 2000; 29:423–444.
- Lin S, Konstance R, Jollis J, Fisher DA. The utility of upper endoscopy in patients with concomitant upper gastrointestinal bleeding and acute myocardial infarction. Dig Dis Sci 2006; 51:2377–2383.
- Silvis SE, Nebel O, Rogers G, Sugawa C, Mandelstam P. Endoscopic complications. Results of the 1974 American Society for Gastrointestinal Endoscopy Survey. JAMA 1976; 235:928–930.
- Lanas A, Garcia-Rodriguez LA, Arroyo MT, et al; Investigators of the Asociación Española de Gastroenterología (AEG). Effect of antisecretory drugs and nitrates on the risk of ulcer bleeding associated with nonsteroidal anti-inflammatory drugs, antiplatelet agents, and anticoagulants. Am J Gastroenterol 2007; 102:507–515.
- Shorr RI, Ray WA, Daugherty JR, Griffin MR. Concurrent use of nonsteroidal anti-inflammatory drugs and oral anticoagulants places elderly persons at high risk for hemorrhagic peptic ulcer disease. Arch Intern Med 1993; 153:1665–1670.
- Tabibian N. Acute gastrointestinal bleeding in anticoagulated patients: a prospective evaluation. Am J Gastroenterol 1989; 84:10–12.
- Choudari CP, Rajgopal C, Palmer KR. Acute gastrointestinal hemorrhage in anticoagulated patients: diagnoses and response to endoscopic treatment. Gut 1994; 35:464–466.
- Petty GW, Brown RD, Whisnant JP, Sicks JD, O’Fallon WM, Wiebers DO. Frequency of major complications of aspirin, warfarin, and intravenous heparin for secondary stroke prevention: a population-based study. Ann Intern Med 1999; 130:14–22.
- Landefeld CS, Rosenblatt MW, Goldman L. Bleeding in outpatients treated with warfarin: relation to the prothrombin time and important remediable lesions. Am J Med 1989; 87:153–159.
- Weil J, Colin-Jones D, Langman M, et al. Prophylactic aspirin and risk of peptic ulcer bleeding. BMJ 1995; 310:827–830.
- Cryer B, Feldman M. Effects of very low dose daily, long-term aspirin therapy on gastric, duodenal, and rectal prostaglandin levels and on mucosal injury in healthy humans. Gastroenterology 1999; 117:17–25.
- De Abajo FJ, Garcia Rodriguez LA. Risk of upper gastrointestinal bleeding and perforation associated with low-dose aspirin as plain and enteric-coated formulations. BMC Clin Pharmacol 2001; 1:1.
- Kelly JP, Kaufman DW, Jurgelon JM, Sheehan J, Koff RS, Shapiro S. Risk of aspirin-associated major upper gastrointestinal bleeding with enteric coated or buffered product. Lancet 1996; 348:1413–1416.
- Garcia Rodriguez LA, Hernandez-Diaz S, De Abajo FJ. Association between aspirin and upper gastrointestinal complications: systematic review of epidemiological studies. Br J Clin Pharmacol 2001; 52:563–571.
- Wilcox CM, Ladabaum U. A patient with high risk of gastrointestinal bleeding requiring nonsteroidal anti-inflammatory drugs. Clin Gastroenterol Hepatol 2006; 4:1090–1093.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet 1996; 348:1329–1339.
- Chan FK, Ching JY, Hung LC, et al. Clopidogrel versus aspirin and esomeprazole to prevent recurrent ulcer bleeding. N Engl J Med 2005; 352:238–244.
- Lai KC, Chu KM, Hui WM, et al. Esomeprazole with aspirin versus clopidogrel for prevention of recurrent gastrointestinal ulcer complications. Clin Gastroenterol Hepatol 2006; 4:860–865.
- Ho MP, Maddox TM, Wang L, et al. Risk of adverse outcomes associated with concomitant use of clopidogrel and proton pump inhibitors following acute coronary syndrome. JAMA 2009; 301:937–944.
- Siller-Matula JM, Spiel AO, Lang IM, Kreiner G, Christ G, Jilma B. Effects of pantoprazole and esomeprazole on platelet inhibition by clopidogrel. Am Heart J 2009; 157:148.e1–e5.
- Small DS, Farid NA, Payne CD, et al. Effects of proton pump inhibitor lansoprazole on the pharmacokinetics and pharmacodynamics of prasugel and clopidogrel. J Clin Pharmacol 2008; 48:475–484.
- Ishizaki T, Horai Y. Review article: cytochrome P450 and the metabolism of proton pump inhibitors—emphasis on rabeprazole. Aliment Pharacol Ther 1999; 13 (suppl 3):27–36.
- Lanas A, Fuentes J, Benito R, Serrano P, Bajador E, Sainz R. Helicobacter pylori increases the risk of upper gastrointestinal bleeding in patients taking low-dose aspirin. Aliment Pharmacol Ther 2002; 16:779–786.
- Chan FK. NSAID-Induced peptic ulcers and Helicobacter pylori infection: implications for patient management. Drug Saf 2005; 28:287–300.
- Bombardier V, Laine L, Reicin A, et al; VIGOR Study Group. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis: VIGOR Study Group. N Eng J Med 2000; 343:1520–1528.
- Griffin MR, Ray WA, Schaffner W. Nonsteroidal anti-inflammatory drug use and death from peptic ulcer in elderly persons. Ann Intern Med 1988; 109:359–363.
- Griffin MR, Piper JM, Daugherty JR, Snowden M, Ray WA. Nonsteroidal anti-inflammatory drug use and increased risk for peptic ulcer disease in elderly persons. Ann Intern Med 1991; 114:257–263.
- Smalley WE, Ray WA, Daugherty JR, Griffin MR. Nonsteroidal anti-inflammatory drugs and the incidence of hospitalizations for peptic ulcer disease in elderly persons. Am J Epidemiol 1995; 141:539–545.
- Laine L. Approaches to nonsteroidal anti-inflammatory drug use in the high-risk patient. Gastroenterology 2001; 120:594–606.
- Silverstein FE, Graham DY, Senior JR, et al. Misoprostol reduces serious gastrointestinal complications in patients with rheumatoid arthritis receiving nonsteroidal anti-inflammatory drugs. A randomized, double blind, placebo controlled trial. Ann Intern Med 1995; 123:241–249.
- Goldstein JL, Huang B, Amer F, Christopoulos NG. Ulcer recurrence in high-risk patients receiving nonsteroidal anti-inflammatory drugs plus low dose aspirin: results of a post hoc subanalysis. Clin Ther 2004; 26:1637–1643.
- Berger JS, Stebbins A, Granger CB, et al. Initial aspirin dose and outcome among ST-elevation myocardial infarction patients treated with fibrinolytic therapy. Circulation 2008; 117:192–199.
- Lanas A, Garcia-Rodriguez LA, Arroyo MT, et al; Investigators of the Asociación Española de Gastroenterología (AEG). Effect of antisecretory drugs and nitrates on the risk of ulcer bleeding associated with nonsteroidal anti-inflammatory drugs, antiplatelet agents, and anticoagulants. Am J Gastroenterol 2007; 102:507–515.
- Chin MW, Yong G, Bulsara MK, Rankin J, Forbes GM. Predictive and protective factors associated with upper gastrointestinal bleeding after percutaneous coronary intervention: a case-control study. Am J Gastroenterol 2007; 102:2411–2416.
- Hunt RH, Bazzoli F. Should NSAID/low dose aspirin takers be tested routinely for H. Pylori infection and treated if positive? Implications for primary risk of ulcer and ulcer relapse after initial healing. Aliment Pharmacol Ther 2004; 19 (suppl 1):9–16.
KEY POINTS
- The first priority is to ensure that the patient is hemodynamically stable, which often requires admission to the intensive care unit for monitoring and fluid resuscitation.
- Peptic ulcers account for most cases of upper GI bleeding, but bleeding from varices has a much higher case-fatality rate and always demands aggressive treatment.
- Patients with ulcer disease should be tested and treated for Helicobacter pylori infection.
- Patients with a history of bleeding ulcers who need long-term treatment with aspirin or a nonsteroidal anti-inflammatory drug should also be prescribed a proton pump inhibitor.
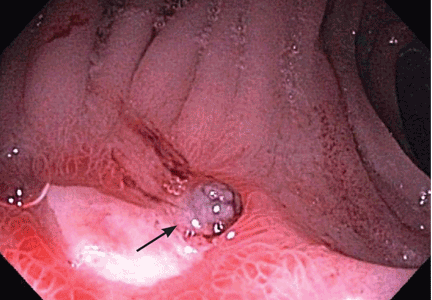
Preventing venous thromboembolism in long-term care residents: Cautious advice based on limited data
Randomized trials that included more than 20,000 medical patients have shown that anticoagulant therapy is safe and effective in preventing venous thromboembolism (VTE), ie, deep vein thrombosis and pulmonary embolism.
However, these trials were done in hospitalized patients, who typically had an acute medical illness and who, if eligible, received a short (7- to 10-day) course of anticoagulant prophylaxis.
Little attention has been given to VTE prophylaxis in residents of long-term care facilities. These patients have risk profiles similar to those of hospitalized medical patients. Some of them may have been transferred from an acute care hospital. In addition, most are elderly, and many have reduced mobility and are at risk for illnesses such as stroke and cardiorespiratory insufficiency, which increase the risk of VTE.
VTE in residents of long-term care facilities is a growing concern. By some estimates, by the year 2030 more than 20% of the US population (70.2 million people) will be over 65 years of age.1 Of those who reached age 65 in 1990, an estimated 43% will enter a nursing home at least once before they die—32% for 3 months, 24% for at least a year, and 9% for at least 5 years.2
Against this background, the objectives of this review are to consider:
- The scope of the problem of VTE in long-term care residents
- Why VTE prophylaxis is often overlooked in medical patients
- Evidence—or lack of evidence—for the safety and efficacy of VTE prophylaxis in long-term care residents and other medical patients
- Available options for VTE prophylaxis
- Which long-term care residents should or should not be considered for prophylaxis.
THE TRUE SCOPE OF THE PROBLEM IS UNKNOWN
The incidence of acute VTE among nursing home residents is reported to be 1.3 events per 100 person-years.3 About 8% of cases of pulmonary embolism and 10% of cases of deep venous thrombosis in the elderly are in nursing home residents.4
However, only 20% of patients with VTE have typical symptoms such as leg pain and swelling or acute dyspnea and chest pain, while 80% have no symptoms.5
Furthermore, deep venous thrombosis is more likely to be clinically silent in patients whose mobility is impaired, such as nursing home residents, as the symptoms arising from obstruction of venous flow are more pronounced with walking.
Pulmonary embolism is also underdiagnosed in this group. An autopsy study of 234 nursing home residents found undiagnosed pulmonary embolism to be the cause of death in 8%, and 40% of cases of pulmonary embolism were not suspected before the patient died.6 Yet pulmonary embolism has a higher case-fatality rate in the elderly than in younger patients, particularly when elderly patients have comorbidities.7
A reason why the diagnosis is so often missed is that pulmonary embolism can present atypically in the elderly, with syncope being more common and tachycardia being less common than in younger patients.8
Since so many cases of VTE are clinically silent and most long-term care residents who die do not undergo autopsy, the true scope of VTE as a clinical problem in these patients is unknown. Consequently, the best way to diagnose, prevent, and treat VTE is also unclear.
WHY IS VTE PREVENTION SO OFTEN OVERLOOKED IN MEDICAL PATIENTS?
In general, nonsurgical patients receive suboptimal thromboprophylaxis. National and international chart audits and cross-sectional studies show that only 16% to 33% of hospitalized medical patients at risk for VTE receive appropriate anticoagulant prophylaxis.9 Though no audits in long-term care facilities have been published, the rate of appropriate prophylaxis is likely comparable to or possibly less than that in medical patients in the hospital. In contrast, in surgical patients the rate is much higher—up to 90%.10,11
Why is VTE prophylaxis so underused in medical patients?
One reason is that we do not really know the baseline risk of VTE in medical patients, particularly in those with chronic illness who require long-term care.12 This is relevant because, in the absence of data about patients’ baseline risk, anticoagulant prophylaxis should be ordered selectively, as it poses known risks of bleeding. The risk is greater in elderly people with comorbidities, as are the associated costs.
In addition, relatively few studies have assessed thromboprophylaxis in medical patients, especially in residents of long-term care facilities.
Another reason is that we lack practice guidelines for patients who need long-term care. The well-accepted guidelines from the American College of Chest Physicians (ACCP) cite advanced age and immobility as risk factors for VTE and strongly recommend prophylaxis in acutely ill medical patients who have limited mobility and an additional risk factor such as infection or cancer.13 Though elderly residents of long-term care facilities may share some of these risk factors, the ACCP guidelines make no specific recommendations for this group.
The attitudes of health care professionals may also pose a barrier. Lloyd et al (unpublished data, 2009) surveyed 1,601 health care professionals in Ontario, Canada, in 2007, to assess potential barriers to anticoagulant prophylaxis in hospitalized medical patients. Respondents cited concerns about the risk of bleeding from anticoagulants, lack of clear indications and contraindications for anticoagulant prophylaxis, and lack of time to consider VTE prophylaxis in every patient. (They did not, however, cite disagreement with guidelines or patient discomfort from subcutaneous anticoagulant injections as barriers.) It is reasonable to assume that these attitudes may also pose a problem in long-term care residents.
Finally, no randomized trials have evaluated the efficacy and safety of anticoagulant drugs or mechanical methods of prophylaxis in long-term care residents. Studies have shown that a short course (7–10 days) of an anticoagulant drug effectively prevents VTE in acutely ill patients, but the efficacy of an extended course in patients with chronic illness who require long-term care is not clear. Therefore, recommendations about thromboprophylaxis in long-term care residents should be made with the caveat that they are based on indirect evidence from other patient groups. This is a considerable limitation.
OPTIONS FOR THROMBOPROPHYLAXIS IN LONG-TERM CARE RESIDENTS
Options for thromboprophylaxis fall into two broad categories: anticoagulant drugs and mechanical devices.
Anticoagulant prophylactic drugs

These agents have been assessed in randomized trials in surgical or acutely ill medical patients, although fondaparinux and tinzaparin are not approved for use in medical patients. Furthermore, none of them has been evaluated in residents of long-term care facilities.
The choice of anticoagulant for prophylaxis is determined largely by clinical factors.
Low-molecular-weight heparins are popular both in and out of the hospital because they have predictable pharmacokinetic properties, they come in convenient prefilled syringes, and they can be given once daily. However, some of them may bioaccumulate in patients with impaired renal function, as they are cleared primarily by the kidney.
Unfractionated heparin is likely to be safer in patients with severe renal insufficiency (creatinine clearance < 30 mL/min), as it is cleared via nonrenal mechanisms.
However, a recent single-arm trial of dalteparin 5,000 IU once daily in critically ill patients with severe renal insufficiency found no evidence of an excessive anticoagulant effect or of drug bioaccumulation.15 Dalteparin may thus be an alternative to unfractionated heparin in medical patients with impaired renal function.
Fondaparinux, a newer anticoagulant, is also given once daily. It is the anticoagulant of choice in patients who have had heparin-induced thrombocytopenia because it is not derived from heparin and likely does not cross-react with heparin-induced thrombocytopenia antibodies.16,17
Limited data on benefit of prophylactic anticoagulant drugs
As mentioned, the trials that confirmed the efficacy and safety of anticoagulant prophylaxis were in surgical patients and hospitalized medical patients, not elderly long-term care residents. The poor evidence for anticoagulant prophylaxis in these patients may be strengthened if extended-duration, out-of-hospital prophylaxis were shown to be effective in medical patients. Long-term care residents could more reasonably be compared with medical patients discharged home with a chronic or resolving illness than with those who are hospitalized.
There is some evidence, although with caveats, that extended anticoagulant prophylaxis, started after an acute illness has resolved, confers a benefit. A recent randomized trial compared extended-duration and short-duration prophylaxis (5 weeks vs 10 days) with enoxaparin 40 mg once daily in 4,726 medical patients with impaired mobility.18 The risk of any VTE event was 44% lower with extended-duration prophylaxis (2.8% vs 4.9%; P = .001) and the risk of symptomatic VTE was 73% lower (0.3% vs 1.1%; P = .004), and this benefit persisted 2 months after treatment was stopped (3.0% vs 5.2%; P = .0015). However, extended treatment conferred a fourfold higher risk of major bleeding (0.6% vs 0.15%; P = .019).
These findings should also be considered in terms of absolute benefit and harm. Treating 1,000 patients for 5 weeks instead of 10 days would prevent eight episodes of symptomatic VTE (absolute risk reduction = 0.8%, number needed to treat = 125) at the cost of four to five episodes of major bleeding (absolute risk increase = 0.45%, number needed to harm = 222). This is a modest net therapeutic benefit.
The therapeutic benefit would be greater if we consider all episodes of VTE, both symptomatic and asymptomatic. Treating 1,000 patients for 5 weeks would prevent 20 episodes of symptomatic or asymptomatic VTE (absolute risk reduction = 2.1%, number needed to treat = 48). However, the clinical importance of asymptomatic VTE is questionable.
Given these considerations, if extended-duration anticoagulant prophylaxis is considered, it should be for patients at highest risk to optimize both its net therapeutic benefits and its cost-effectiveness.
Mechanical prophylaxis
Mechanical thromboprophylactic devices—graduated or elastic compression stockings and intermittent pneumatic compression devices—are effective when used by themselves in surgical patients.13 However, in a randomized controlled trial in patients with ischemic stroke, the rate of VTE was 10.0% with graduated compression stockings in addition to “usual care VTE prophylaxis” vs 10.5% with usual care alone, and patients in the stocking group had a fourfold higher risk of developing skin breaks, ulcers, blisters, or necrosis (5% vs 1%; odds ratio 4.18; 95% CI 2.4–7.3).19 Furthermore, improperly fitted stockings, especially those that are thigh-length, can be uncomfortable to wear and difficult to apply.
Overall, the role of mechanical thromboprophylaxis in long-term care facilities is not clear. If it is considered, there should be a compelling reason to use it—for example, for patients at high risk in whom anticoagulants are contraindicated because of ongoing bleeding or a higher risk of bleeding (eg, recent gastrointestinal bleeding, hemorrhagic stroke, coagulopathy, or thrombocytopenia). Furthermore, if stockings are used, they should be properly fitted and routinely monitored for adverse effects, since elderly patients are likely to be most susceptible to skin breakdown.
WHICH LONG-TERM CARE RESIDENTS SHOULD RECEIVE VTE PROPHYLAXIS?

Old age and immobility are not the only risk factors
The current ACCP guidelines suggest considering thromboprophylaxis for hospitalized medical patients over age 75 who cannot walk without assistance.13 However, we lack evidence to suggest a similar strategy in long-term care residents.
The ACCP guidelines are based on data on risk. Nearly 25% of elderly patients with confirmed pulmonary embolism had been immobile prior to their diagnosis.8 In addition, prolonged bed rest (> 14 days) has been reported to be the strongest independent risk factor for symptomatic deep venous thrombosis, increasing the risk more than fivefold.20 Advanced age is also considered a risk factor for VTE, as risk starts to increase at age 40 and doubles each decade of life thereafter.18
No study has assessed the impact of these factors on the risk of VTE in long-term care residents. Since most of such patients are elderly and have impaired mobility, we believe a more selective approach should be used in assigning VTE risk status, one that does not use advanced age and immobility as the only criteria for starting thromboprophylaxis.
Residents of long-term care facilities may be immobile because of underlying illness or disability, such as cognitive impairment, sensory impairment (eg, poor access to corrective lenses and hearing aids), or poor access to assist devices (eg, walkers, canes). In addition, iatrogenic factors that decrease mobility such as indwelling bladder catheters and physical restraints are also common in such patients.
Efforts to improve mobility should be encouraged. However, we recommend that thromboprophylaxis be considered only in patients who have both impaired mobility and an intercurrent acute medical illness such as an acute infection or acute inflammatory disease.13
A related issue is the difference between long-term care residents with a chronic but stable disease and those with acute disease. Patients with acute exacerbations of congestive heart failure or chronic obstructive lung disease may be considered for thromboprophylaxis, as they become more comparable to acutely ill medical patients in whom clinical trials have shown the effectiveness of anticoagulant prophylaxis. On the other hand, patients with these diseases who remain stable may not need prophylaxis.
This approach avoids giving long-term anticoagulant prophylaxis to patients who have irreversible diseases and limits the use of these drugs and devices to higher-risk periods.
Consider thromboprophylaxis if…

- An acute exacerbation of congestive heart failure or chronic obstructive pulmonary disease
- Acute infection (eg, urosepsis, pneumonia, cellulitis, infectious diarrhea)
- An acute exacerbation of an inflammatory disease (eg, rheumatoid arthritis)
- Active cancer (eg, patient receiving radiation therapy or chemotherapy)
- Immobility and prior VTE.
Do not routinely consider prophylaxis if…
We also believe patients should not be routinely considered for anticoagulant VTE prophylaxis if they have:
- Chronic but stable cardiorespiratory disease
- Chronic but stable infectious or inflammatory disease
- Terminal cancer with very limited life expectancy
- Any contraindication to anticoagulants (eg, active bleeding, recent bleeding, coagulopathy, thrombocytopenia).
ANTICOAGULANT PROPHYLAXIS POSES RISKS IN LONG-TERM CARE RESIDENTS
Bleeding is the principal risk

The incidence of clinically important bleeding associated with anticoagulant prophylaxis is 0.2% to 5.6%, and the risk of fatal bleeding is 0.02% to 0.5%.21–24
As no randomized trial has examined anticoagulant prophylaxis in elderly long-term care residents, their bleeding risk with this therapy is unclear. However, older patients are likely to be at higher risk than younger patients because they have more comorbidities, take more drugs that could interact with heparin and potentiate bleeding, and have fragile skin, predisposing to injury from subcutaneous injections.
Also, renal function tends to decline with age. In a retrospective study of 854 outpatients over age 65, 29% had moderate renal insufficiency (creatinine clearance 30–50 mL/min), and 6% had severe renal insufficiency (creatinine clearance < 30 mL/min).25 Recent evidence suggests that some low-molecular-weight heparins (dalteparin and tinzaparin) do not bioaccumulate in patients with impaired renal function. However, enoxaparin and fondaparinux should be used with caution in patients with moderate to severe renal impairment.
Though much attention has recently been paid to increasing anticoagulant doses if the patient is obese, residents of long-term care facilities are more likely to be underweight. Dose adjustment should be considered when a low-molecular-weight heparin or fondaparinux is given to patients weighing less than 50 kg.
Heparin-induced thrombocytopenia
The other major risk of anticoagulant prophylaxis is heparin-induced thrombocytopenia, an infrequent but life-threatening complication caused by the formation of antibodies to the heparin-derived anticoagulant and a platelet surface antigen. It is associated with moderate thrombocytopenia and an incidence of venous or arterial thrombosis that is over 50%.26
No study has assessed the incidence of heparin-induced thrombocytopenia in long-term care residents. A meta-analysis reported that the risk with anticoagulant prophylaxis was 1.6% with unfractionated heparin (95% confidence interval [CI] 1.2%–2.1%) and 0.6% with low-molecular-weight heparin (95% CI 0.4%–0.9%), and that this risk increased with the duration of prophylaxis.27 If anticoagulant prophylaxis were given to all long-term care residents for extended durations (eg, for the duration of reduced mobility), the incidence and prevalence of heparin-induced thrombocytopenia would likely become a major concern.
Whenever anticoagulant prophylaxis is considered, the risks of both thrombosis and bleeding should be considered. Patients who are receiving anticoagulant prophylaxis should also be monitored for bleeding and heparin-induced thrombocytopenia. This is particularly true in long-term care residents, in whom the risks and benefits of anticoagulant prophylaxis are extrapolated from data from other populations.
MORE RESEARCH IS NEEDED
To date, we lack audits of thromboprophylaxis, clinical practice guidelines, and clear indications and contraindications for anticoagulant prophylaxis in long-term care residents. In the absence of such data, extrapolating the efficacy and safety of thromboprophylaxis from hospitalized patients to long-term care residents is difficult.
Clearly, additional research is needed to identify which long-term care residents would benefit most from thromboprophylaxis. In the meantime, a selective approach to identifying patients who should be considered for thromboprophylaxis should be adopted.
- Cornman JM. Questions for societies with “third age” populations. The Extension-of-Life Working Group, The Gerontological Society of America. Acad Med 1997; 72:856–862.
- Kemper P, Murtaugh CM. Lifetime use of nursing home care. N Engl J Med 1991; 324:595–600.
- Gomes JP, Shaheen WH, Truong SV, Brown EF, Beasley BW, Gajewski BJ. Incidence of venous thromboembolic events among nursing home residents. J Gen Intern Med 2003; 18:934–936.
- Kniffin WD, Baron JA, Barrett J, Birkmeyer JD, Anderson FA. The epidemiology of diagnosed pulmonary embolism and deep venous thrombosis in the elderly. Arch Intern Med 1994; 154:861–866.
- Bounameaux H. Integrating pharmacologic and mechanical prophylaxis of venous thromboembolism. Thromb Haemost 1999; 82:931–937.
- Gross JS, Neufeld RR, Libow LS, Gerber I, Rodstein M. Autopsy study of the elderly institutionalized patient. Review of 234 autopsies. Arch Intern Med 1988; 148:173–176.
- Spyropoulos AC, Merli G. Management of venous thromboembolism in the elderly. Drugs Aging 2006; 23:651–671.
- Punukollu H, Khan IA, Punukollu G, Gowda RM, Mendoza C, Sacchi TJ. Acute pulmonary embolism in elderly: clinical characteristics and outcome. Int J Cardiol 2005; 99:213–216.
- Douketis JD. Prevention of venous thromboembolism in hospitalized medical patients: addressing some practical questions. Curr Opin Pulm Med 2008; 14:381–388.
- Cohen AT, Tapson VF, Bergmann JF, et al; ENDORSE Investigators. Venous thromboembolism risk and prophylaxis in the acute hospital care setting (ENDORSE study): a multinational cross-sectional study. Lancet 2008; 371:387–394.
- Kahn SR, Panju A, Geerts W, et al; CURVE study investigators. Multicenter evaluation of the use of venous thromboembolism prophylaxis in acutely ill medical patients in Canada. Thromb Res 2007; 119:145–155.
- Haas S, Spyropoulos AC. Primary prevention of venous thromboembolism in long-term care: identifying and managing the risk. Clin Appl Thromb Hemost 2008; 14:149–158.
- Geerts WH, Bergqvist D, Pineo GF, et al; American College of Chest Physicians. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133( suppl 6):381S–453S.
- Francis CW. Clinical practice. Prophylaxis for thromboembolism in hospitalized medical patients. N Engl J Med 2007; 356:1438–1444.
- Douketis J, Cook D, Meade M, et al; Canadian Critical Care Trials Group. Prophylaxis against deep vein thrombosis in critically ill patients with severe renal insufficiency with the low-molecular-weight heparin dalteparin: an assessment of safety and pharmacodynamics: the DIRECT study. Arch Intern Med 2008; 168:1805–1812.
- Lobo B, Finch C, Howard A, Minhas S. Fondaparinux for the treatment of patients with acute heparin-induced thrombocytopenia. Thromb Haemost 2008; 99:208–214.
- Spinler SA. New concepts in heparin-induced thrombocytopenia: diagnosis and management. J Thromb Thrombolysis 2006; 21:17–21.
- Hull RD, Schellong SM, Tapson VF, et al. Extended-duration thromboprophylaxis in acutely ill medical patients with recent reduced mobility: methodology for the EXCLAIM study. J Thromb Thrombolysis 2006; 22:31–38.
- Dennis M, Sandercock PA, Reid J, et al; CLOTS Trials Collaboration Effectiveness of thigh-length graduated compression stockings to reduce the risk of deep vein thrombosis after stroke (CLOTS trial 1): a multicentre, randomised controlled trial. Lancet 2009; 373:1958–1965.
- Weill-Engerer S, Meaume S, Lahlou A, et al. Risk factors for deep vein thrombosis in inpatients aged 65 and older: a case-control multicenter study. J Am Geriatr Soc 2004; 52:1299–1304.
- Dentali F, Douketis JD, Gianni M, Lim W, Crowther MA. Meta-analysis: anticoagulant prophylaxis to prevent symptomatic venous thromboembolism in hospitalized medical patients. Ann Intern Med 2007; 146:278–288.
- Douketis JD, Arneklev K, Goldhaber SZ, Spandorfer J, Halperin F, Horrow J. Comparison of bleeding in patients with nonvalvular atrial fibrillation treated with ximelagatran or warfarin: assessment of incidence, case-fatality rate, time course and sites of bleeding, and risk factors for bleeding. Arch Intern Med 2006; 166:853–859.
- Linkins LA, Choi PT, Douketis JD. Clinical impact of bleeding in patients taking oral anticoagulant therapy for venous thromboembolism: a meta-analysis. Ann Intern Med 2003; 139:893–900.
- Lloyd NS, Douketis JD, Moinuddin I, Lim W, Crowther MA. Anticoagulant prophylaxis to prevent asymptomatic deep vein thrombosis in hospitalized medical patients: a systematic review and meta-analysis. J Thromb Haemost 2008; 6:405–414.
- Swedko PJ, Clark HD, Paramsothy K, Akbari A. Serum creatinine is an inadequate screening test for renal failure in elderly patients. Arch Intern Med 2003; 163:356–360.
- Martel N, Lee J, Wells PS. Risk for heparin-induced thrombocytopenia with unfractionated and low-molecular-weight heparin thromboprophylaxis: a meta-analysis. Blood 2005; 106:2710–2715.
- Stein PD, Hull RD, Matta F, Yaekoub AY, Liang J. Incidence of thrombocytopenia in hospitalized patients with venous thromboembolism. Am J Med 2009; 122:919–930.
Randomized trials that included more than 20,000 medical patients have shown that anticoagulant therapy is safe and effective in preventing venous thromboembolism (VTE), ie, deep vein thrombosis and pulmonary embolism.
However, these trials were done in hospitalized patients, who typically had an acute medical illness and who, if eligible, received a short (7- to 10-day) course of anticoagulant prophylaxis.
Little attention has been given to VTE prophylaxis in residents of long-term care facilities. These patients have risk profiles similar to those of hospitalized medical patients. Some of them may have been transferred from an acute care hospital. In addition, most are elderly, and many have reduced mobility and are at risk for illnesses such as stroke and cardiorespiratory insufficiency, which increase the risk of VTE.
VTE in residents of long-term care facilities is a growing concern. By some estimates, by the year 2030 more than 20% of the US population (70.2 million people) will be over 65 years of age.1 Of those who reached age 65 in 1990, an estimated 43% will enter a nursing home at least once before they die—32% for 3 months, 24% for at least a year, and 9% for at least 5 years.2
Against this background, the objectives of this review are to consider:
- The scope of the problem of VTE in long-term care residents
- Why VTE prophylaxis is often overlooked in medical patients
- Evidence—or lack of evidence—for the safety and efficacy of VTE prophylaxis in long-term care residents and other medical patients
- Available options for VTE prophylaxis
- Which long-term care residents should or should not be considered for prophylaxis.
THE TRUE SCOPE OF THE PROBLEM IS UNKNOWN
The incidence of acute VTE among nursing home residents is reported to be 1.3 events per 100 person-years.3 About 8% of cases of pulmonary embolism and 10% of cases of deep venous thrombosis in the elderly are in nursing home residents.4
However, only 20% of patients with VTE have typical symptoms such as leg pain and swelling or acute dyspnea and chest pain, while 80% have no symptoms.5
Furthermore, deep venous thrombosis is more likely to be clinically silent in patients whose mobility is impaired, such as nursing home residents, as the symptoms arising from obstruction of venous flow are more pronounced with walking.
Pulmonary embolism is also underdiagnosed in this group. An autopsy study of 234 nursing home residents found undiagnosed pulmonary embolism to be the cause of death in 8%, and 40% of cases of pulmonary embolism were not suspected before the patient died.6 Yet pulmonary embolism has a higher case-fatality rate in the elderly than in younger patients, particularly when elderly patients have comorbidities.7
A reason why the diagnosis is so often missed is that pulmonary embolism can present atypically in the elderly, with syncope being more common and tachycardia being less common than in younger patients.8
Since so many cases of VTE are clinically silent and most long-term care residents who die do not undergo autopsy, the true scope of VTE as a clinical problem in these patients is unknown. Consequently, the best way to diagnose, prevent, and treat VTE is also unclear.
WHY IS VTE PREVENTION SO OFTEN OVERLOOKED IN MEDICAL PATIENTS?
In general, nonsurgical patients receive suboptimal thromboprophylaxis. National and international chart audits and cross-sectional studies show that only 16% to 33% of hospitalized medical patients at risk for VTE receive appropriate anticoagulant prophylaxis.9 Though no audits in long-term care facilities have been published, the rate of appropriate prophylaxis is likely comparable to or possibly less than that in medical patients in the hospital. In contrast, in surgical patients the rate is much higher—up to 90%.10,11
Why is VTE prophylaxis so underused in medical patients?
One reason is that we do not really know the baseline risk of VTE in medical patients, particularly in those with chronic illness who require long-term care.12 This is relevant because, in the absence of data about patients’ baseline risk, anticoagulant prophylaxis should be ordered selectively, as it poses known risks of bleeding. The risk is greater in elderly people with comorbidities, as are the associated costs.
In addition, relatively few studies have assessed thromboprophylaxis in medical patients, especially in residents of long-term care facilities.
Another reason is that we lack practice guidelines for patients who need long-term care. The well-accepted guidelines from the American College of Chest Physicians (ACCP) cite advanced age and immobility as risk factors for VTE and strongly recommend prophylaxis in acutely ill medical patients who have limited mobility and an additional risk factor such as infection or cancer.13 Though elderly residents of long-term care facilities may share some of these risk factors, the ACCP guidelines make no specific recommendations for this group.
The attitudes of health care professionals may also pose a barrier. Lloyd et al (unpublished data, 2009) surveyed 1,601 health care professionals in Ontario, Canada, in 2007, to assess potential barriers to anticoagulant prophylaxis in hospitalized medical patients. Respondents cited concerns about the risk of bleeding from anticoagulants, lack of clear indications and contraindications for anticoagulant prophylaxis, and lack of time to consider VTE prophylaxis in every patient. (They did not, however, cite disagreement with guidelines or patient discomfort from subcutaneous anticoagulant injections as barriers.) It is reasonable to assume that these attitudes may also pose a problem in long-term care residents.
Finally, no randomized trials have evaluated the efficacy and safety of anticoagulant drugs or mechanical methods of prophylaxis in long-term care residents. Studies have shown that a short course (7–10 days) of an anticoagulant drug effectively prevents VTE in acutely ill patients, but the efficacy of an extended course in patients with chronic illness who require long-term care is not clear. Therefore, recommendations about thromboprophylaxis in long-term care residents should be made with the caveat that they are based on indirect evidence from other patient groups. This is a considerable limitation.
OPTIONS FOR THROMBOPROPHYLAXIS IN LONG-TERM CARE RESIDENTS
Options for thromboprophylaxis fall into two broad categories: anticoagulant drugs and mechanical devices.
Anticoagulant prophylactic drugs
These agents have been assessed in randomized trials in surgical or acutely ill medical patients, although fondaparinux and tinzaparin are not approved for use in medical patients. Furthermore, none of them has been evaluated in residents of long-term care facilities.
The choice of anticoagulant for prophylaxis is determined largely by clinical factors.
Low-molecular-weight heparins are popular both in and out of the hospital because they have predictable pharmacokinetic properties, they come in convenient prefilled syringes, and they can be given once daily. However, some of them may bioaccumulate in patients with impaired renal function, as they are cleared primarily by the kidney.
Unfractionated heparin is likely to be safer in patients with severe renal insufficiency (creatinine clearance < 30 mL/min), as it is cleared via nonrenal mechanisms.
However, a recent single-arm trial of dalteparin 5,000 IU once daily in critically ill patients with severe renal insufficiency found no evidence of an excessive anticoagulant effect or of drug bioaccumulation.15 Dalteparin may thus be an alternative to unfractionated heparin in medical patients with impaired renal function.
Fondaparinux, a newer anticoagulant, is also given once daily. It is the anticoagulant of choice in patients who have had heparin-induced thrombocytopenia because it is not derived from heparin and likely does not cross-react with heparin-induced thrombocytopenia antibodies.16,17
Limited data on benefit of prophylactic anticoagulant drugs
As mentioned, the trials that confirmed the efficacy and safety of anticoagulant prophylaxis were in surgical patients and hospitalized medical patients, not elderly long-term care residents. The poor evidence for anticoagulant prophylaxis in these patients may be strengthened if extended-duration, out-of-hospital prophylaxis were shown to be effective in medical patients. Long-term care residents could more reasonably be compared with medical patients discharged home with a chronic or resolving illness than with those who are hospitalized.
There is some evidence, although with caveats, that extended anticoagulant prophylaxis, started after an acute illness has resolved, confers a benefit. A recent randomized trial compared extended-duration and short-duration prophylaxis (5 weeks vs 10 days) with enoxaparin 40 mg once daily in 4,726 medical patients with impaired mobility.18 The risk of any VTE event was 44% lower with extended-duration prophylaxis (2.8% vs 4.9%; P = .001) and the risk of symptomatic VTE was 73% lower (0.3% vs 1.1%; P = .004), and this benefit persisted 2 months after treatment was stopped (3.0% vs 5.2%; P = .0015). However, extended treatment conferred a fourfold higher risk of major bleeding (0.6% vs 0.15%; P = .019).
These findings should also be considered in terms of absolute benefit and harm. Treating 1,000 patients for 5 weeks instead of 10 days would prevent eight episodes of symptomatic VTE (absolute risk reduction = 0.8%, number needed to treat = 125) at the cost of four to five episodes of major bleeding (absolute risk increase = 0.45%, number needed to harm = 222). This is a modest net therapeutic benefit.
The therapeutic benefit would be greater if we consider all episodes of VTE, both symptomatic and asymptomatic. Treating 1,000 patients for 5 weeks would prevent 20 episodes of symptomatic or asymptomatic VTE (absolute risk reduction = 2.1%, number needed to treat = 48). However, the clinical importance of asymptomatic VTE is questionable.
Given these considerations, if extended-duration anticoagulant prophylaxis is considered, it should be for patients at highest risk to optimize both its net therapeutic benefits and its cost-effectiveness.
Mechanical prophylaxis
Mechanical thromboprophylactic devices—graduated or elastic compression stockings and intermittent pneumatic compression devices—are effective when used by themselves in surgical patients.13 However, in a randomized controlled trial in patients with ischemic stroke, the rate of VTE was 10.0% with graduated compression stockings in addition to “usual care VTE prophylaxis” vs 10.5% with usual care alone, and patients in the stocking group had a fourfold higher risk of developing skin breaks, ulcers, blisters, or necrosis (5% vs 1%; odds ratio 4.18; 95% CI 2.4–7.3).19 Furthermore, improperly fitted stockings, especially those that are thigh-length, can be uncomfortable to wear and difficult to apply.
Overall, the role of mechanical thromboprophylaxis in long-term care facilities is not clear. If it is considered, there should be a compelling reason to use it—for example, for patients at high risk in whom anticoagulants are contraindicated because of ongoing bleeding or a higher risk of bleeding (eg, recent gastrointestinal bleeding, hemorrhagic stroke, coagulopathy, or thrombocytopenia). Furthermore, if stockings are used, they should be properly fitted and routinely monitored for adverse effects, since elderly patients are likely to be most susceptible to skin breakdown.
WHICH LONG-TERM CARE RESIDENTS SHOULD RECEIVE VTE PROPHYLAXIS?
Old age and immobility are not the only risk factors
The current ACCP guidelines suggest considering thromboprophylaxis for hospitalized medical patients over age 75 who cannot walk without assistance.13 However, we lack evidence to suggest a similar strategy in long-term care residents.
The ACCP guidelines are based on data on risk. Nearly 25% of elderly patients with confirmed pulmonary embolism had been immobile prior to their diagnosis.8 In addition, prolonged bed rest (> 14 days) has been reported to be the strongest independent risk factor for symptomatic deep venous thrombosis, increasing the risk more than fivefold.20 Advanced age is also considered a risk factor for VTE, as risk starts to increase at age 40 and doubles each decade of life thereafter.18
No study has assessed the impact of these factors on the risk of VTE in long-term care residents. Since most of such patients are elderly and have impaired mobility, we believe a more selective approach should be used in assigning VTE risk status, one that does not use advanced age and immobility as the only criteria for starting thromboprophylaxis.
Residents of long-term care facilities may be immobile because of underlying illness or disability, such as cognitive impairment, sensory impairment (eg, poor access to corrective lenses and hearing aids), or poor access to assist devices (eg, walkers, canes). In addition, iatrogenic factors that decrease mobility such as indwelling bladder catheters and physical restraints are also common in such patients.
Efforts to improve mobility should be encouraged. However, we recommend that thromboprophylaxis be considered only in patients who have both impaired mobility and an intercurrent acute medical illness such as an acute infection or acute inflammatory disease.13
A related issue is the difference between long-term care residents with a chronic but stable disease and those with acute disease. Patients with acute exacerbations of congestive heart failure or chronic obstructive lung disease may be considered for thromboprophylaxis, as they become more comparable to acutely ill medical patients in whom clinical trials have shown the effectiveness of anticoagulant prophylaxis. On the other hand, patients with these diseases who remain stable may not need prophylaxis.
This approach avoids giving long-term anticoagulant prophylaxis to patients who have irreversible diseases and limits the use of these drugs and devices to higher-risk periods.
Consider thromboprophylaxis if…
- An acute exacerbation of congestive heart failure or chronic obstructive pulmonary disease
- Acute infection (eg, urosepsis, pneumonia, cellulitis, infectious diarrhea)
- An acute exacerbation of an inflammatory disease (eg, rheumatoid arthritis)
- Active cancer (eg, patient receiving radiation therapy or chemotherapy)
- Immobility and prior VTE.
Do not routinely consider prophylaxis if…
We also believe patients should not be routinely considered for anticoagulant VTE prophylaxis if they have:
- Chronic but stable cardiorespiratory disease
- Chronic but stable infectious or inflammatory disease
- Terminal cancer with very limited life expectancy
- Any contraindication to anticoagulants (eg, active bleeding, recent bleeding, coagulopathy, thrombocytopenia).
ANTICOAGULANT PROPHYLAXIS POSES RISKS IN LONG-TERM CARE RESIDENTS
Bleeding is the principal risk
The incidence of clinically important bleeding associated with anticoagulant prophylaxis is 0.2% to 5.6%, and the risk of fatal bleeding is 0.02% to 0.5%.21–24
As no randomized trial has examined anticoagulant prophylaxis in elderly long-term care residents, their bleeding risk with this therapy is unclear. However, older patients are likely to be at higher risk than younger patients because they have more comorbidities, take more drugs that could interact with heparin and potentiate bleeding, and have fragile skin, predisposing to injury from subcutaneous injections.
Also, renal function tends to decline with age. In a retrospective study of 854 outpatients over age 65, 29% had moderate renal insufficiency (creatinine clearance 30–50 mL/min), and 6% had severe renal insufficiency (creatinine clearance < 30 mL/min).25 Recent evidence suggests that some low-molecular-weight heparins (dalteparin and tinzaparin) do not bioaccumulate in patients with impaired renal function. However, enoxaparin and fondaparinux should be used with caution in patients with moderate to severe renal impairment.
Though much attention has recently been paid to increasing anticoagulant doses if the patient is obese, residents of long-term care facilities are more likely to be underweight. Dose adjustment should be considered when a low-molecular-weight heparin or fondaparinux is given to patients weighing less than 50 kg.
Heparin-induced thrombocytopenia
The other major risk of anticoagulant prophylaxis is heparin-induced thrombocytopenia, an infrequent but life-threatening complication caused by the formation of antibodies to the heparin-derived anticoagulant and a platelet surface antigen. It is associated with moderate thrombocytopenia and an incidence of venous or arterial thrombosis that is over 50%.26
No study has assessed the incidence of heparin-induced thrombocytopenia in long-term care residents. A meta-analysis reported that the risk with anticoagulant prophylaxis was 1.6% with unfractionated heparin (95% confidence interval [CI] 1.2%–2.1%) and 0.6% with low-molecular-weight heparin (95% CI 0.4%–0.9%), and that this risk increased with the duration of prophylaxis.27 If anticoagulant prophylaxis were given to all long-term care residents for extended durations (eg, for the duration of reduced mobility), the incidence and prevalence of heparin-induced thrombocytopenia would likely become a major concern.
Whenever anticoagulant prophylaxis is considered, the risks of both thrombosis and bleeding should be considered. Patients who are receiving anticoagulant prophylaxis should also be monitored for bleeding and heparin-induced thrombocytopenia. This is particularly true in long-term care residents, in whom the risks and benefits of anticoagulant prophylaxis are extrapolated from data from other populations.
MORE RESEARCH IS NEEDED
To date, we lack audits of thromboprophylaxis, clinical practice guidelines, and clear indications and contraindications for anticoagulant prophylaxis in long-term care residents. In the absence of such data, extrapolating the efficacy and safety of thromboprophylaxis from hospitalized patients to long-term care residents is difficult.
Clearly, additional research is needed to identify which long-term care residents would benefit most from thromboprophylaxis. In the meantime, a selective approach to identifying patients who should be considered for thromboprophylaxis should be adopted.
Randomized trials that included more than 20,000 medical patients have shown that anticoagulant therapy is safe and effective in preventing venous thromboembolism (VTE), ie, deep vein thrombosis and pulmonary embolism.
However, these trials were done in hospitalized patients, who typically had an acute medical illness and who, if eligible, received a short (7- to 10-day) course of anticoagulant prophylaxis.
Little attention has been given to VTE prophylaxis in residents of long-term care facilities. These patients have risk profiles similar to those of hospitalized medical patients. Some of them may have been transferred from an acute care hospital. In addition, most are elderly, and many have reduced mobility and are at risk for illnesses such as stroke and cardiorespiratory insufficiency, which increase the risk of VTE.
VTE in residents of long-term care facilities is a growing concern. By some estimates, by the year 2030 more than 20% of the US population (70.2 million people) will be over 65 years of age.1 Of those who reached age 65 in 1990, an estimated 43% will enter a nursing home at least once before they die—32% for 3 months, 24% for at least a year, and 9% for at least 5 years.2
Against this background, the objectives of this review are to consider:
- The scope of the problem of VTE in long-term care residents
- Why VTE prophylaxis is often overlooked in medical patients
- Evidence—or lack of evidence—for the safety and efficacy of VTE prophylaxis in long-term care residents and other medical patients
- Available options for VTE prophylaxis
- Which long-term care residents should or should not be considered for prophylaxis.
THE TRUE SCOPE OF THE PROBLEM IS UNKNOWN
The incidence of acute VTE among nursing home residents is reported to be 1.3 events per 100 person-years.3 About 8% of cases of pulmonary embolism and 10% of cases of deep venous thrombosis in the elderly are in nursing home residents.4
However, only 20% of patients with VTE have typical symptoms such as leg pain and swelling or acute dyspnea and chest pain, while 80% have no symptoms.5
Furthermore, deep venous thrombosis is more likely to be clinically silent in patients whose mobility is impaired, such as nursing home residents, as the symptoms arising from obstruction of venous flow are more pronounced with walking.
Pulmonary embolism is also underdiagnosed in this group. An autopsy study of 234 nursing home residents found undiagnosed pulmonary embolism to be the cause of death in 8%, and 40% of cases of pulmonary embolism were not suspected before the patient died.6 Yet pulmonary embolism has a higher case-fatality rate in the elderly than in younger patients, particularly when elderly patients have comorbidities.7
A reason why the diagnosis is so often missed is that pulmonary embolism can present atypically in the elderly, with syncope being more common and tachycardia being less common than in younger patients.8
Since so many cases of VTE are clinically silent and most long-term care residents who die do not undergo autopsy, the true scope of VTE as a clinical problem in these patients is unknown. Consequently, the best way to diagnose, prevent, and treat VTE is also unclear.
WHY IS VTE PREVENTION SO OFTEN OVERLOOKED IN MEDICAL PATIENTS?
In general, nonsurgical patients receive suboptimal thromboprophylaxis. National and international chart audits and cross-sectional studies show that only 16% to 33% of hospitalized medical patients at risk for VTE receive appropriate anticoagulant prophylaxis.9 Though no audits in long-term care facilities have been published, the rate of appropriate prophylaxis is likely comparable to or possibly less than that in medical patients in the hospital. In contrast, in surgical patients the rate is much higher—up to 90%.10,11
Why is VTE prophylaxis so underused in medical patients?
One reason is that we do not really know the baseline risk of VTE in medical patients, particularly in those with chronic illness who require long-term care.12 This is relevant because, in the absence of data about patients’ baseline risk, anticoagulant prophylaxis should be ordered selectively, as it poses known risks of bleeding. The risk is greater in elderly people with comorbidities, as are the associated costs.
In addition, relatively few studies have assessed thromboprophylaxis in medical patients, especially in residents of long-term care facilities.
Another reason is that we lack practice guidelines for patients who need long-term care. The well-accepted guidelines from the American College of Chest Physicians (ACCP) cite advanced age and immobility as risk factors for VTE and strongly recommend prophylaxis in acutely ill medical patients who have limited mobility and an additional risk factor such as infection or cancer.13 Though elderly residents of long-term care facilities may share some of these risk factors, the ACCP guidelines make no specific recommendations for this group.
The attitudes of health care professionals may also pose a barrier. Lloyd et al (unpublished data, 2009) surveyed 1,601 health care professionals in Ontario, Canada, in 2007, to assess potential barriers to anticoagulant prophylaxis in hospitalized medical patients. Respondents cited concerns about the risk of bleeding from anticoagulants, lack of clear indications and contraindications for anticoagulant prophylaxis, and lack of time to consider VTE prophylaxis in every patient. (They did not, however, cite disagreement with guidelines or patient discomfort from subcutaneous anticoagulant injections as barriers.) It is reasonable to assume that these attitudes may also pose a problem in long-term care residents.
Finally, no randomized trials have evaluated the efficacy and safety of anticoagulant drugs or mechanical methods of prophylaxis in long-term care residents. Studies have shown that a short course (7–10 days) of an anticoagulant drug effectively prevents VTE in acutely ill patients, but the efficacy of an extended course in patients with chronic illness who require long-term care is not clear. Therefore, recommendations about thromboprophylaxis in long-term care residents should be made with the caveat that they are based on indirect evidence from other patient groups. This is a considerable limitation.
OPTIONS FOR THROMBOPROPHYLAXIS IN LONG-TERM CARE RESIDENTS
Options for thromboprophylaxis fall into two broad categories: anticoagulant drugs and mechanical devices.
Anticoagulant prophylactic drugs
These agents have been assessed in randomized trials in surgical or acutely ill medical patients, although fondaparinux and tinzaparin are not approved for use in medical patients. Furthermore, none of them has been evaluated in residents of long-term care facilities.
The choice of anticoagulant for prophylaxis is determined largely by clinical factors.
Low-molecular-weight heparins are popular both in and out of the hospital because they have predictable pharmacokinetic properties, they come in convenient prefilled syringes, and they can be given once daily. However, some of them may bioaccumulate in patients with impaired renal function, as they are cleared primarily by the kidney.
Unfractionated heparin is likely to be safer in patients with severe renal insufficiency (creatinine clearance < 30 mL/min), as it is cleared via nonrenal mechanisms.
However, a recent single-arm trial of dalteparin 5,000 IU once daily in critically ill patients with severe renal insufficiency found no evidence of an excessive anticoagulant effect or of drug bioaccumulation.15 Dalteparin may thus be an alternative to unfractionated heparin in medical patients with impaired renal function.
Fondaparinux, a newer anticoagulant, is also given once daily. It is the anticoagulant of choice in patients who have had heparin-induced thrombocytopenia because it is not derived from heparin and likely does not cross-react with heparin-induced thrombocytopenia antibodies.16,17
Limited data on benefit of prophylactic anticoagulant drugs
As mentioned, the trials that confirmed the efficacy and safety of anticoagulant prophylaxis were in surgical patients and hospitalized medical patients, not elderly long-term care residents. The poor evidence for anticoagulant prophylaxis in these patients may be strengthened if extended-duration, out-of-hospital prophylaxis were shown to be effective in medical patients. Long-term care residents could more reasonably be compared with medical patients discharged home with a chronic or resolving illness than with those who are hospitalized.
There is some evidence, although with caveats, that extended anticoagulant prophylaxis, started after an acute illness has resolved, confers a benefit. A recent randomized trial compared extended-duration and short-duration prophylaxis (5 weeks vs 10 days) with enoxaparin 40 mg once daily in 4,726 medical patients with impaired mobility.18 The risk of any VTE event was 44% lower with extended-duration prophylaxis (2.8% vs 4.9%; P = .001) and the risk of symptomatic VTE was 73% lower (0.3% vs 1.1%; P = .004), and this benefit persisted 2 months after treatment was stopped (3.0% vs 5.2%; P = .0015). However, extended treatment conferred a fourfold higher risk of major bleeding (0.6% vs 0.15%; P = .019).
These findings should also be considered in terms of absolute benefit and harm. Treating 1,000 patients for 5 weeks instead of 10 days would prevent eight episodes of symptomatic VTE (absolute risk reduction = 0.8%, number needed to treat = 125) at the cost of four to five episodes of major bleeding (absolute risk increase = 0.45%, number needed to harm = 222). This is a modest net therapeutic benefit.
The therapeutic benefit would be greater if we consider all episodes of VTE, both symptomatic and asymptomatic. Treating 1,000 patients for 5 weeks would prevent 20 episodes of symptomatic or asymptomatic VTE (absolute risk reduction = 2.1%, number needed to treat = 48). However, the clinical importance of asymptomatic VTE is questionable.
Given these considerations, if extended-duration anticoagulant prophylaxis is considered, it should be for patients at highest risk to optimize both its net therapeutic benefits and its cost-effectiveness.
Mechanical prophylaxis
Mechanical thromboprophylactic devices—graduated or elastic compression stockings and intermittent pneumatic compression devices—are effective when used by themselves in surgical patients.13 However, in a randomized controlled trial in patients with ischemic stroke, the rate of VTE was 10.0% with graduated compression stockings in addition to “usual care VTE prophylaxis” vs 10.5% with usual care alone, and patients in the stocking group had a fourfold higher risk of developing skin breaks, ulcers, blisters, or necrosis (5% vs 1%; odds ratio 4.18; 95% CI 2.4–7.3).19 Furthermore, improperly fitted stockings, especially those that are thigh-length, can be uncomfortable to wear and difficult to apply.
Overall, the role of mechanical thromboprophylaxis in long-term care facilities is not clear. If it is considered, there should be a compelling reason to use it—for example, for patients at high risk in whom anticoagulants are contraindicated because of ongoing bleeding or a higher risk of bleeding (eg, recent gastrointestinal bleeding, hemorrhagic stroke, coagulopathy, or thrombocytopenia). Furthermore, if stockings are used, they should be properly fitted and routinely monitored for adverse effects, since elderly patients are likely to be most susceptible to skin breakdown.
WHICH LONG-TERM CARE RESIDENTS SHOULD RECEIVE VTE PROPHYLAXIS?
Old age and immobility are not the only risk factors
The current ACCP guidelines suggest considering thromboprophylaxis for hospitalized medical patients over age 75 who cannot walk without assistance.13 However, we lack evidence to suggest a similar strategy in long-term care residents.
The ACCP guidelines are based on data on risk. Nearly 25% of elderly patients with confirmed pulmonary embolism had been immobile prior to their diagnosis.8 In addition, prolonged bed rest (> 14 days) has been reported to be the strongest independent risk factor for symptomatic deep venous thrombosis, increasing the risk more than fivefold.20 Advanced age is also considered a risk factor for VTE, as risk starts to increase at age 40 and doubles each decade of life thereafter.18
No study has assessed the impact of these factors on the risk of VTE in long-term care residents. Since most of such patients are elderly and have impaired mobility, we believe a more selective approach should be used in assigning VTE risk status, one that does not use advanced age and immobility as the only criteria for starting thromboprophylaxis.
Residents of long-term care facilities may be immobile because of underlying illness or disability, such as cognitive impairment, sensory impairment (eg, poor access to corrective lenses and hearing aids), or poor access to assist devices (eg, walkers, canes). In addition, iatrogenic factors that decrease mobility such as indwelling bladder catheters and physical restraints are also common in such patients.
Efforts to improve mobility should be encouraged. However, we recommend that thromboprophylaxis be considered only in patients who have both impaired mobility and an intercurrent acute medical illness such as an acute infection or acute inflammatory disease.13
A related issue is the difference between long-term care residents with a chronic but stable disease and those with acute disease. Patients with acute exacerbations of congestive heart failure or chronic obstructive lung disease may be considered for thromboprophylaxis, as they become more comparable to acutely ill medical patients in whom clinical trials have shown the effectiveness of anticoagulant prophylaxis. On the other hand, patients with these diseases who remain stable may not need prophylaxis.
This approach avoids giving long-term anticoagulant prophylaxis to patients who have irreversible diseases and limits the use of these drugs and devices to higher-risk periods.
Consider thromboprophylaxis if…
- An acute exacerbation of congestive heart failure or chronic obstructive pulmonary disease
- Acute infection (eg, urosepsis, pneumonia, cellulitis, infectious diarrhea)
- An acute exacerbation of an inflammatory disease (eg, rheumatoid arthritis)
- Active cancer (eg, patient receiving radiation therapy or chemotherapy)
- Immobility and prior VTE.
Do not routinely consider prophylaxis if…
We also believe patients should not be routinely considered for anticoagulant VTE prophylaxis if they have:
- Chronic but stable cardiorespiratory disease
- Chronic but stable infectious or inflammatory disease
- Terminal cancer with very limited life expectancy
- Any contraindication to anticoagulants (eg, active bleeding, recent bleeding, coagulopathy, thrombocytopenia).
ANTICOAGULANT PROPHYLAXIS POSES RISKS IN LONG-TERM CARE RESIDENTS
Bleeding is the principal risk
The incidence of clinically important bleeding associated with anticoagulant prophylaxis is 0.2% to 5.6%, and the risk of fatal bleeding is 0.02% to 0.5%.21–24
As no randomized trial has examined anticoagulant prophylaxis in elderly long-term care residents, their bleeding risk with this therapy is unclear. However, older patients are likely to be at higher risk than younger patients because they have more comorbidities, take more drugs that could interact with heparin and potentiate bleeding, and have fragile skin, predisposing to injury from subcutaneous injections.
Also, renal function tends to decline with age. In a retrospective study of 854 outpatients over age 65, 29% had moderate renal insufficiency (creatinine clearance 30–50 mL/min), and 6% had severe renal insufficiency (creatinine clearance < 30 mL/min).25 Recent evidence suggests that some low-molecular-weight heparins (dalteparin and tinzaparin) do not bioaccumulate in patients with impaired renal function. However, enoxaparin and fondaparinux should be used with caution in patients with moderate to severe renal impairment.
Though much attention has recently been paid to increasing anticoagulant doses if the patient is obese, residents of long-term care facilities are more likely to be underweight. Dose adjustment should be considered when a low-molecular-weight heparin or fondaparinux is given to patients weighing less than 50 kg.
Heparin-induced thrombocytopenia
The other major risk of anticoagulant prophylaxis is heparin-induced thrombocytopenia, an infrequent but life-threatening complication caused by the formation of antibodies to the heparin-derived anticoagulant and a platelet surface antigen. It is associated with moderate thrombocytopenia and an incidence of venous or arterial thrombosis that is over 50%.26
No study has assessed the incidence of heparin-induced thrombocytopenia in long-term care residents. A meta-analysis reported that the risk with anticoagulant prophylaxis was 1.6% with unfractionated heparin (95% confidence interval [CI] 1.2%–2.1%) and 0.6% with low-molecular-weight heparin (95% CI 0.4%–0.9%), and that this risk increased with the duration of prophylaxis.27 If anticoagulant prophylaxis were given to all long-term care residents for extended durations (eg, for the duration of reduced mobility), the incidence and prevalence of heparin-induced thrombocytopenia would likely become a major concern.
Whenever anticoagulant prophylaxis is considered, the risks of both thrombosis and bleeding should be considered. Patients who are receiving anticoagulant prophylaxis should also be monitored for bleeding and heparin-induced thrombocytopenia. This is particularly true in long-term care residents, in whom the risks and benefits of anticoagulant prophylaxis are extrapolated from data from other populations.
MORE RESEARCH IS NEEDED
To date, we lack audits of thromboprophylaxis, clinical practice guidelines, and clear indications and contraindications for anticoagulant prophylaxis in long-term care residents. In the absence of such data, extrapolating the efficacy and safety of thromboprophylaxis from hospitalized patients to long-term care residents is difficult.
Clearly, additional research is needed to identify which long-term care residents would benefit most from thromboprophylaxis. In the meantime, a selective approach to identifying patients who should be considered for thromboprophylaxis should be adopted.
- Cornman JM. Questions for societies with “third age” populations. The Extension-of-Life Working Group, The Gerontological Society of America. Acad Med 1997; 72:856–862.
- Kemper P, Murtaugh CM. Lifetime use of nursing home care. N Engl J Med 1991; 324:595–600.
- Gomes JP, Shaheen WH, Truong SV, Brown EF, Beasley BW, Gajewski BJ. Incidence of venous thromboembolic events among nursing home residents. J Gen Intern Med 2003; 18:934–936.
- Kniffin WD, Baron JA, Barrett J, Birkmeyer JD, Anderson FA. The epidemiology of diagnosed pulmonary embolism and deep venous thrombosis in the elderly. Arch Intern Med 1994; 154:861–866.
- Bounameaux H. Integrating pharmacologic and mechanical prophylaxis of venous thromboembolism. Thromb Haemost 1999; 82:931–937.
- Gross JS, Neufeld RR, Libow LS, Gerber I, Rodstein M. Autopsy study of the elderly institutionalized patient. Review of 234 autopsies. Arch Intern Med 1988; 148:173–176.
- Spyropoulos AC, Merli G. Management of venous thromboembolism in the elderly. Drugs Aging 2006; 23:651–671.
- Punukollu H, Khan IA, Punukollu G, Gowda RM, Mendoza C, Sacchi TJ. Acute pulmonary embolism in elderly: clinical characteristics and outcome. Int J Cardiol 2005; 99:213–216.
- Douketis JD. Prevention of venous thromboembolism in hospitalized medical patients: addressing some practical questions. Curr Opin Pulm Med 2008; 14:381–388.
- Cohen AT, Tapson VF, Bergmann JF, et al; ENDORSE Investigators. Venous thromboembolism risk and prophylaxis in the acute hospital care setting (ENDORSE study): a multinational cross-sectional study. Lancet 2008; 371:387–394.
- Kahn SR, Panju A, Geerts W, et al; CURVE study investigators. Multicenter evaluation of the use of venous thromboembolism prophylaxis in acutely ill medical patients in Canada. Thromb Res 2007; 119:145–155.
- Haas S, Spyropoulos AC. Primary prevention of venous thromboembolism in long-term care: identifying and managing the risk. Clin Appl Thromb Hemost 2008; 14:149–158.
- Geerts WH, Bergqvist D, Pineo GF, et al; American College of Chest Physicians. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133( suppl 6):381S–453S.
- Francis CW. Clinical practice. Prophylaxis for thromboembolism in hospitalized medical patients. N Engl J Med 2007; 356:1438–1444.
- Douketis J, Cook D, Meade M, et al; Canadian Critical Care Trials Group. Prophylaxis against deep vein thrombosis in critically ill patients with severe renal insufficiency with the low-molecular-weight heparin dalteparin: an assessment of safety and pharmacodynamics: the DIRECT study. Arch Intern Med 2008; 168:1805–1812.
- Lobo B, Finch C, Howard A, Minhas S. Fondaparinux for the treatment of patients with acute heparin-induced thrombocytopenia. Thromb Haemost 2008; 99:208–214.
- Spinler SA. New concepts in heparin-induced thrombocytopenia: diagnosis and management. J Thromb Thrombolysis 2006; 21:17–21.
- Hull RD, Schellong SM, Tapson VF, et al. Extended-duration thromboprophylaxis in acutely ill medical patients with recent reduced mobility: methodology for the EXCLAIM study. J Thromb Thrombolysis 2006; 22:31–38.
- Dennis M, Sandercock PA, Reid J, et al; CLOTS Trials Collaboration Effectiveness of thigh-length graduated compression stockings to reduce the risk of deep vein thrombosis after stroke (CLOTS trial 1): a multicentre, randomised controlled trial. Lancet 2009; 373:1958–1965.
- Weill-Engerer S, Meaume S, Lahlou A, et al. Risk factors for deep vein thrombosis in inpatients aged 65 and older: a case-control multicenter study. J Am Geriatr Soc 2004; 52:1299–1304.
- Dentali F, Douketis JD, Gianni M, Lim W, Crowther MA. Meta-analysis: anticoagulant prophylaxis to prevent symptomatic venous thromboembolism in hospitalized medical patients. Ann Intern Med 2007; 146:278–288.
- Douketis JD, Arneklev K, Goldhaber SZ, Spandorfer J, Halperin F, Horrow J. Comparison of bleeding in patients with nonvalvular atrial fibrillation treated with ximelagatran or warfarin: assessment of incidence, case-fatality rate, time course and sites of bleeding, and risk factors for bleeding. Arch Intern Med 2006; 166:853–859.
- Linkins LA, Choi PT, Douketis JD. Clinical impact of bleeding in patients taking oral anticoagulant therapy for venous thromboembolism: a meta-analysis. Ann Intern Med 2003; 139:893–900.
- Lloyd NS, Douketis JD, Moinuddin I, Lim W, Crowther MA. Anticoagulant prophylaxis to prevent asymptomatic deep vein thrombosis in hospitalized medical patients: a systematic review and meta-analysis. J Thromb Haemost 2008; 6:405–414.
- Swedko PJ, Clark HD, Paramsothy K, Akbari A. Serum creatinine is an inadequate screening test for renal failure in elderly patients. Arch Intern Med 2003; 163:356–360.
- Martel N, Lee J, Wells PS. Risk for heparin-induced thrombocytopenia with unfractionated and low-molecular-weight heparin thromboprophylaxis: a meta-analysis. Blood 2005; 106:2710–2715.
- Stein PD, Hull RD, Matta F, Yaekoub AY, Liang J. Incidence of thrombocytopenia in hospitalized patients with venous thromboembolism. Am J Med 2009; 122:919–930.
- Cornman JM. Questions for societies with “third age” populations. The Extension-of-Life Working Group, The Gerontological Society of America. Acad Med 1997; 72:856–862.
- Kemper P, Murtaugh CM. Lifetime use of nursing home care. N Engl J Med 1991; 324:595–600.
- Gomes JP, Shaheen WH, Truong SV, Brown EF, Beasley BW, Gajewski BJ. Incidence of venous thromboembolic events among nursing home residents. J Gen Intern Med 2003; 18:934–936.
- Kniffin WD, Baron JA, Barrett J, Birkmeyer JD, Anderson FA. The epidemiology of diagnosed pulmonary embolism and deep venous thrombosis in the elderly. Arch Intern Med 1994; 154:861–866.
- Bounameaux H. Integrating pharmacologic and mechanical prophylaxis of venous thromboembolism. Thromb Haemost 1999; 82:931–937.
- Gross JS, Neufeld RR, Libow LS, Gerber I, Rodstein M. Autopsy study of the elderly institutionalized patient. Review of 234 autopsies. Arch Intern Med 1988; 148:173–176.
- Spyropoulos AC, Merli G. Management of venous thromboembolism in the elderly. Drugs Aging 2006; 23:651–671.
- Punukollu H, Khan IA, Punukollu G, Gowda RM, Mendoza C, Sacchi TJ. Acute pulmonary embolism in elderly: clinical characteristics and outcome. Int J Cardiol 2005; 99:213–216.
- Douketis JD. Prevention of venous thromboembolism in hospitalized medical patients: addressing some practical questions. Curr Opin Pulm Med 2008; 14:381–388.
- Cohen AT, Tapson VF, Bergmann JF, et al; ENDORSE Investigators. Venous thromboembolism risk and prophylaxis in the acute hospital care setting (ENDORSE study): a multinational cross-sectional study. Lancet 2008; 371:387–394.
- Kahn SR, Panju A, Geerts W, et al; CURVE study investigators. Multicenter evaluation of the use of venous thromboembolism prophylaxis in acutely ill medical patients in Canada. Thromb Res 2007; 119:145–155.
- Haas S, Spyropoulos AC. Primary prevention of venous thromboembolism in long-term care: identifying and managing the risk. Clin Appl Thromb Hemost 2008; 14:149–158.
- Geerts WH, Bergqvist D, Pineo GF, et al; American College of Chest Physicians. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133( suppl 6):381S–453S.
- Francis CW. Clinical practice. Prophylaxis for thromboembolism in hospitalized medical patients. N Engl J Med 2007; 356:1438–1444.
- Douketis J, Cook D, Meade M, et al; Canadian Critical Care Trials Group. Prophylaxis against deep vein thrombosis in critically ill patients with severe renal insufficiency with the low-molecular-weight heparin dalteparin: an assessment of safety and pharmacodynamics: the DIRECT study. Arch Intern Med 2008; 168:1805–1812.
- Lobo B, Finch C, Howard A, Minhas S. Fondaparinux for the treatment of patients with acute heparin-induced thrombocytopenia. Thromb Haemost 2008; 99:208–214.
- Spinler SA. New concepts in heparin-induced thrombocytopenia: diagnosis and management. J Thromb Thrombolysis 2006; 21:17–21.
- Hull RD, Schellong SM, Tapson VF, et al. Extended-duration thromboprophylaxis in acutely ill medical patients with recent reduced mobility: methodology for the EXCLAIM study. J Thromb Thrombolysis 2006; 22:31–38.
- Dennis M, Sandercock PA, Reid J, et al; CLOTS Trials Collaboration Effectiveness of thigh-length graduated compression stockings to reduce the risk of deep vein thrombosis after stroke (CLOTS trial 1): a multicentre, randomised controlled trial. Lancet 2009; 373:1958–1965.
- Weill-Engerer S, Meaume S, Lahlou A, et al. Risk factors for deep vein thrombosis in inpatients aged 65 and older: a case-control multicenter study. J Am Geriatr Soc 2004; 52:1299–1304.
- Dentali F, Douketis JD, Gianni M, Lim W, Crowther MA. Meta-analysis: anticoagulant prophylaxis to prevent symptomatic venous thromboembolism in hospitalized medical patients. Ann Intern Med 2007; 146:278–288.
- Douketis JD, Arneklev K, Goldhaber SZ, Spandorfer J, Halperin F, Horrow J. Comparison of bleeding in patients with nonvalvular atrial fibrillation treated with ximelagatran or warfarin: assessment of incidence, case-fatality rate, time course and sites of bleeding, and risk factors for bleeding. Arch Intern Med 2006; 166:853–859.
- Linkins LA, Choi PT, Douketis JD. Clinical impact of bleeding in patients taking oral anticoagulant therapy for venous thromboembolism: a meta-analysis. Ann Intern Med 2003; 139:893–900.
- Lloyd NS, Douketis JD, Moinuddin I, Lim W, Crowther MA. Anticoagulant prophylaxis to prevent asymptomatic deep vein thrombosis in hospitalized medical patients: a systematic review and meta-analysis. J Thromb Haemost 2008; 6:405–414.
- Swedko PJ, Clark HD, Paramsothy K, Akbari A. Serum creatinine is an inadequate screening test for renal failure in elderly patients. Arch Intern Med 2003; 163:356–360.
- Martel N, Lee J, Wells PS. Risk for heparin-induced thrombocytopenia with unfractionated and low-molecular-weight heparin thromboprophylaxis: a meta-analysis. Blood 2005; 106:2710–2715.
- Stein PD, Hull RD, Matta F, Yaekoub AY, Liang J. Incidence of thrombocytopenia in hospitalized patients with venous thromboembolism. Am J Med 2009; 122:919–930.
KEY POINTS
- Assessment of VTE risk and consideration of need for anticoagulant prophylaxis in long-term care residents are based on indirect data, derived primarily from studies of acutely ill hospitalized medical patients.
- Drugs and devices for thromboprophylaxis have been studied in medical and surgical populations, but issues of efficacy and safety are likely to also pertain to long-term care residents.
- Thromboprophylaxis should be considered for long-term care residents if they are definitely at increased risk of VTE—ie, if they have an acute exacerbation of congestive heart failure or chronic obstructive pulmonary disease; acute inflammatory disease; acute infection; active cancer; or immobility and prior VTE.
Omeprazole and clopidogrel: Should clinicians be worried?
Many clinicians are concerned about a possible interaction between the proton pump inhibitor omeprazole (Prilosec) and the antiplatelet drug clopidogrel (Plavix), which is often given to patients as part of dual antiplatelet therapy after an acute coronary syndrome or a percutaneous coronary intervention. Indeed, the US Food and Drug Administration (FDA) has warned that omeprazole reduces the antiplatelet effect of clopidogrel.
Although we should not take such warnings lightly, we also should not be alarmed. The data on which the FDA warning was based came mostly from laboratory assays of platelet function. Preliminary results from a randomized, controlled clinical trial with hard end points show that, for the time being, we should not change the way we manage patients.
PROTON PUMP INHIBITORS DECREASE GASTROINTESTINAL BLEEDING
Dual antiplatelet therapy with aspirin and clopidogrel decreases the risk of major adverse cardiac events after an acute coronary syndrome or a percutaneous coronary intervention compared with aspirin alone.1 However, it also increases the risk of gastrointestinal bleeding. A recent analysis determined that dual antiplatelet therapy was the most significant risk factor associated with serious or fatal gastrointestinal bleeding in high-risk survivors of myocardial infarction.2
Given the risks of significant morbidity and death in patients on dual antiplatelet therapy who develop gastrointestinal bleeding, an expert consensus panel recommended the use of proton pump inhibitors in patients on dual antiplatelet therapy who have risk factors for gastrointestinal bleeding.3 Accordingly, these drugs are commonly used for gastrointestinal protection in patients requiring dual antiplatelet therapy.
A POSSIBLE CYP450 INTERACTION
Clopidogrel is metabolized from a prodrug to its active metabolite by the cytochrome P450 (CYP450) system. Proton pump inhibitors also are metabolized by the CYP450 system.4 Proton pump inhibitors are thought to diminish the activity of clopidogrel via inhibition of the CYP2C19 isoenzyme. However, the clinical significance of this inhibition is not clear. Different drugs of this class inhibit the CYP450 system to varying degrees.
The potential interaction between proton pump inhibitors and clopidogrel is worrisome for many physicians, since adverse cardiovascular outcomes are more common in patients in whom the antiplatelet response to clopidogrel is impaired.1 This interaction led to the publication of numerous articles, and prompted the FDA to carefully analyze the potential clinical implications.
In several randomized trials, omeprazole diminished the response to clopidogrel (measured via platelet function assays).5,6 It is unclear if this is a class effect, as proton pump inhibitors other than omeprazole have not consistently been shown to have this effect.6,7 Observational studies of the effect of co-administration of a proton pump inhibitor and clopidogrel on cardiovascular outcomes following acute coronary syndromes have had conflicting findings.8–11
THE FDA ISSUES AN ADVISORY
Given the reports of an impaired platelet response to clopidogrel with omeprazole, the FDA asked the manufacturer for data on this potential interaction. The data showed diminished platelet inhibition when clopidogrel was co-administered with omeprazole or when the two were taken 12 hours apart.
On November 17, 2009, the FDA issued a patient advisory and updated the patient safety information on the package insert for clopidogrel about this drug interaction.12 Specifically, the FDA warns that omeprazole reduces the antiplatelet effect of clopidogrel by about 50%. The FDA warning sparked debate in the medical community, as the decision was based in part on ex vivo data.
POST HOC ANALYSES FROM RANDOMIZED CONTROLLED TRIALS
Several post hoc analyses of large randomized clinical trials have studied the potential interaction between proton pump inhibitors and clopidogrel.
In the Clopidogrel for the Reduction of Events During Observation (CREDO) trial, clopidogrel reduced the incidence of death, myocardial infarction, or stroke to a similar extent regardless of baseline use of a proton pump inhibitor.13
In patients undergoing percutaneous coronary intervention, the Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation—Thrombolysis in Myocardial Infarction 44 (PRINCIPLE-TIMI 44) trial found that those taking a proton pump inhibitor had significantly less platelet inhibition with clopidogrel compared with those not on one.14 However, patients taking prasugrel (Effient) and a proton pump inhibitor only had a slight trend towards diminished platelet inhibition.14
The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel—Thrombolysis in Myocardial Infarction 38 (TRITON-TIMI 38) found that proton pump inhibitors did not influence the long-term outcome of cardiovascular death, myocardial infarction, or stroke for patients on clopidogrel or prasugrel after an acute coronary syndrome.14 A subanalysis did not reveal any differences between omeprazole or other drugs of this class as to an effect on the primary outcome.
Though informative, the results of these post hoc analyses need to be validated with data from randomized clinical trials.
‘COGENT’ TRIAL HALTED EARLY, BUT PRELIMINARY RESULTS AVAILABLE
The Clopidogrel and the Optimization of Gastrointestinal Events (COGENT) trial was the first randomized clinical study of the effect of the interaction between clopidogrel and omeprazole on cardiovascular and gastrointestinal outcomes.15 In a double-blind fashion, patients with acute coronary syndromes or undergoing percutaneous coronary interventions were randomized to receive a fixed-dose combination pill containing either clopidogrel and delayed-release omeprazole or clopidogrel alone. All patients also received aspirin.
Unfortunately, the trial was stopped early because the sponsor declared bankruptcy. However, preliminary results revealed no significant difference in cardiovascular outcomes for patients on clopidogrel and omeprazole compared with clopidogrel alone.15 Furthermore, adverse gastrointestinal events were significantly fewer in patients on clopidogrel and omeprazole.
Thus, omeprazole appears to be safe and may offer gastrointestinal protection to patients on dual antiplatelet therapy, though we need to await publication of the full results.
‘SPICE ’ TRIAL TO EVALUATE POSSIBLE MECHANISMS OF INTERACTION
The Evaluation of the Influence of Statins and Proton Pump Inhibitors on Clopidogrel Antiplatelet Effects (SPICE) trial is a mechanistic study that will evaluate platelet function and genetic polymorphisms in patients on clopidogrel and aspirin after a percutaneous coronary intervention. They will be randomized to statin therapy plus different proton pump inhibitors.16 Prior concerns about an ex vivo interaction between clopidogrel and certain statins were not validated by clinical data.17
OUR RECOMMENDATIONS
Based on the current evidence, patients on aspirin and clopidogrel who have an indication for a proton pump inhibitor or who are at risk of gastrointestinal bleeding can continue or start taking a proton pump inhibitor, including omeprazole.
Switching to another proton pump inhibitor is not currently supported by any randomized clinical trial, nor is changing to a histamine H2-receptor antagonist. The effect of proton pump inhibitors other than omeprazole on clopidogrel is unclear, and it is not known if the interaction with clopidogrel is a class effect or specific to certain drugs of this class.18 On the other hand, we still have no compelling evidence of any major clinical interaction between alternative proton pump inhibitors and clopidogrel.18
Also, separating the dosing times of clopidogrel and omeprazole by 12 hours is not supported by any randomized clinical trial, and runs contrary to at least some ex vivo data.
It is important that all physicians assess the need for a proton pump inhibitor in their patients, as overuse of these drugs has been documented in certain settings.19
Clopidogrel and omeprazole share a common metabolic link via CYP2C19. Omeprazole, along with some other proton pump inhibitors, interacts with clopidogrel at the level of the CYP450 system. Platelet function studies show that platelet inhibition by clopidogrel is impaired, though the astute clinician should be aware of the wide variability associated with platelet function assays and clopidogrel.1,20 However, what may appear to be an interaction at the enzymatic level does not necessarily translate into worse clinical outcomes. Additionally, reliance on nonrandomized studies rather than on randomized clinical trials can be misleading.
- Depta JP, Bhatt DL. Aspirin and platelet adenosine diphosphate receptor antagonists in acute coronary syndromes and percutaneous coronary intervention: role in therapy and strategies to overcome resistance. Am J Cardiovasc Drugs 2008; 8:91–112.
- Moukarbel GV, Signorovitch JE, Pfeffer MA, et al. Gastrointestinal bleeding in high risk survivors of myocardial infarction: the VALIANT trial. Eur Heart J 2009; 30:2226–2232.
- Bhatt DL, Scheiman J, Abraham NS, et al; American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. J Am Coll Cardiol 2008; 52:1502–1517.
- Li XQ, Andersson TB, Ahlström M, Weidolf L. Comparison of inhibitory effects of the proton pump-inhibiting drugs omeprazole, esomeprazole, lansoprazole, pantoprazole, and rabeprazole on human cytochrome P450 activities. Drug Metab Dispos 2004; 32:821–827.
- Gilard M, Arnaud B, Cornily JC, et al. Influence of omeprazole on the antiplatelet action of clopidogrel associated with aspirin: the randomized, double-blind OCLA (Omeprazole CLopidogrel Aspirin) study. J Am Coll Cardiol 2008; 51:256–260.
- Cuisset T, Frere C, Quilici J, et al. Comparison of omeprazole and pantoprazole influence on a high 150-mg clopidogrel maintenance dose. The PACA (Proton Pump Inhibitors and Clopidogrel Association) prospective randomized study. J Am Coll Cardiol 2009; 54:1149–1153.
- Siller-Matula JM, Spiel AO, Lang IM, Kreiner G, Christ G, Jilma B. Effects of pantoprazole and esomeprazole on platelet inhibition by clopidogrel. Am Heart J 2009; 157:148.e1–148.e5.
- Aubert RE, Epstein RS, Teagarden JR, et al. Proton pump inhibitors effect on clopidogrel effectiveness: the Clopidogrel Medco Outcomes Study [abstract]. Circulation 2008; 118( suppl):S28–10–2008.
- Ho PM, Maddox TM, Wang L, et al. Risk of adverse outcomes associated with concomitant use of clopidogrel and proton pump inhibitors following acute coronary syndrome. JAMA 2009; 301:937–944.
- Juurlink DN, Gomes T, Ko DT, et al. A population-based study of the drug interaction between proton pump inhibitors and clopidogrel. CMAJ 2009; 180:713–718.
- Rassen JA, Choudhry NK, Avorn J, Schneeweiss S. Cardiovascular outcomes and mortality in patients using clopidogrel with proton pump inhibitors after percutaneous coronary intervention or acute coronary syndrome. Circulation 2009; 120:2322–2329.
- US Food and Drug Administration. Public health advisory: updated safety information about a drug interaction between clopidogrel bisulfate (marketed as Plavix) and omeprazole (marketed as Prilosec and Prilosec OTC). www.fda.gov/Drugs/DrugSafety/PublicHealthAdvisories/ucm190825.htm. Accessed 1/6/2010.
- Dunn SP, Macaulay TE, Brennan DM, et al. Baseline proton pump inhibitor use associated with increased cardiovascular events with and without the use of clopidogrel in the CREDO trial. Circulation 2008; 118:S_815. Abstract 3999.
- O‘Donoghue ML, Braunwald E, Antman EM, et al. Pharmacodynamic effect and clinical efficacy of clopidogrel and prasugrel with or without a proton-pump inhibitor: an analysis of two randomised trials. Lancet 2009; 374:989–997.
- Bhatt DL. COGENT: A prospective, randomized, placebo-controlled trial of omeprazole in patients receiving aspirin and clopidogrel. Presented at Transcatheter Cardiovascular Therapeutics; September 24, 2009; San Francisco, Calif.
- National Institutes of Health. Evaluation of the Influence of Statins and Proton Pump Inhibitors on Clopidogrel Antiplatelet Effects (SPICE). http://clinicaltrials.gov/ct2/show/NCT00930670. Accessed 1/6/2010.
- Saw J, Brennan DM, Steinhubl SR, et al. Lack of evidence of clopidogrel-statin interaction in the CHARISMA trial. J Am Coll Cardiol 2007; 50:291–295.
- Laine L, Hennekens C. Proton pump inhibitor and clopidogrel interaction: fact or fiction? Am J Gastroenterol 2009 Nov 10. [Epub ahead of print].
- Forgacs I, Loganayagam A. Overprescribing proton pump inhibitors. BMJ 2008; 336:2–3.
- Serebruany VL, Steinhubl SR, Berger PB, Malinin AI, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45:246–251.
Many clinicians are concerned about a possible interaction between the proton pump inhibitor omeprazole (Prilosec) and the antiplatelet drug clopidogrel (Plavix), which is often given to patients as part of dual antiplatelet therapy after an acute coronary syndrome or a percutaneous coronary intervention. Indeed, the US Food and Drug Administration (FDA) has warned that omeprazole reduces the antiplatelet effect of clopidogrel.
Although we should not take such warnings lightly, we also should not be alarmed. The data on which the FDA warning was based came mostly from laboratory assays of platelet function. Preliminary results from a randomized, controlled clinical trial with hard end points show that, for the time being, we should not change the way we manage patients.
PROTON PUMP INHIBITORS DECREASE GASTROINTESTINAL BLEEDING
Dual antiplatelet therapy with aspirin and clopidogrel decreases the risk of major adverse cardiac events after an acute coronary syndrome or a percutaneous coronary intervention compared with aspirin alone.1 However, it also increases the risk of gastrointestinal bleeding. A recent analysis determined that dual antiplatelet therapy was the most significant risk factor associated with serious or fatal gastrointestinal bleeding in high-risk survivors of myocardial infarction.2
Given the risks of significant morbidity and death in patients on dual antiplatelet therapy who develop gastrointestinal bleeding, an expert consensus panel recommended the use of proton pump inhibitors in patients on dual antiplatelet therapy who have risk factors for gastrointestinal bleeding.3 Accordingly, these drugs are commonly used for gastrointestinal protection in patients requiring dual antiplatelet therapy.
A POSSIBLE CYP450 INTERACTION
Clopidogrel is metabolized from a prodrug to its active metabolite by the cytochrome P450 (CYP450) system. Proton pump inhibitors also are metabolized by the CYP450 system.4 Proton pump inhibitors are thought to diminish the activity of clopidogrel via inhibition of the CYP2C19 isoenzyme. However, the clinical significance of this inhibition is not clear. Different drugs of this class inhibit the CYP450 system to varying degrees.
The potential interaction between proton pump inhibitors and clopidogrel is worrisome for many physicians, since adverse cardiovascular outcomes are more common in patients in whom the antiplatelet response to clopidogrel is impaired.1 This interaction led to the publication of numerous articles, and prompted the FDA to carefully analyze the potential clinical implications.
In several randomized trials, omeprazole diminished the response to clopidogrel (measured via platelet function assays).5,6 It is unclear if this is a class effect, as proton pump inhibitors other than omeprazole have not consistently been shown to have this effect.6,7 Observational studies of the effect of co-administration of a proton pump inhibitor and clopidogrel on cardiovascular outcomes following acute coronary syndromes have had conflicting findings.8–11
THE FDA ISSUES AN ADVISORY
Given the reports of an impaired platelet response to clopidogrel with omeprazole, the FDA asked the manufacturer for data on this potential interaction. The data showed diminished platelet inhibition when clopidogrel was co-administered with omeprazole or when the two were taken 12 hours apart.
On November 17, 2009, the FDA issued a patient advisory and updated the patient safety information on the package insert for clopidogrel about this drug interaction.12 Specifically, the FDA warns that omeprazole reduces the antiplatelet effect of clopidogrel by about 50%. The FDA warning sparked debate in the medical community, as the decision was based in part on ex vivo data.
POST HOC ANALYSES FROM RANDOMIZED CONTROLLED TRIALS
Several post hoc analyses of large randomized clinical trials have studied the potential interaction between proton pump inhibitors and clopidogrel.
In the Clopidogrel for the Reduction of Events During Observation (CREDO) trial, clopidogrel reduced the incidence of death, myocardial infarction, or stroke to a similar extent regardless of baseline use of a proton pump inhibitor.13
In patients undergoing percutaneous coronary intervention, the Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation—Thrombolysis in Myocardial Infarction 44 (PRINCIPLE-TIMI 44) trial found that those taking a proton pump inhibitor had significantly less platelet inhibition with clopidogrel compared with those not on one.14 However, patients taking prasugrel (Effient) and a proton pump inhibitor only had a slight trend towards diminished platelet inhibition.14
The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel—Thrombolysis in Myocardial Infarction 38 (TRITON-TIMI 38) found that proton pump inhibitors did not influence the long-term outcome of cardiovascular death, myocardial infarction, or stroke for patients on clopidogrel or prasugrel after an acute coronary syndrome.14 A subanalysis did not reveal any differences between omeprazole or other drugs of this class as to an effect on the primary outcome.
Though informative, the results of these post hoc analyses need to be validated with data from randomized clinical trials.
‘COGENT’ TRIAL HALTED EARLY, BUT PRELIMINARY RESULTS AVAILABLE
The Clopidogrel and the Optimization of Gastrointestinal Events (COGENT) trial was the first randomized clinical study of the effect of the interaction between clopidogrel and omeprazole on cardiovascular and gastrointestinal outcomes.15 In a double-blind fashion, patients with acute coronary syndromes or undergoing percutaneous coronary interventions were randomized to receive a fixed-dose combination pill containing either clopidogrel and delayed-release omeprazole or clopidogrel alone. All patients also received aspirin.
Unfortunately, the trial was stopped early because the sponsor declared bankruptcy. However, preliminary results revealed no significant difference in cardiovascular outcomes for patients on clopidogrel and omeprazole compared with clopidogrel alone.15 Furthermore, adverse gastrointestinal events were significantly fewer in patients on clopidogrel and omeprazole.
Thus, omeprazole appears to be safe and may offer gastrointestinal protection to patients on dual antiplatelet therapy, though we need to await publication of the full results.
‘SPICE ’ TRIAL TO EVALUATE POSSIBLE MECHANISMS OF INTERACTION
The Evaluation of the Influence of Statins and Proton Pump Inhibitors on Clopidogrel Antiplatelet Effects (SPICE) trial is a mechanistic study that will evaluate platelet function and genetic polymorphisms in patients on clopidogrel and aspirin after a percutaneous coronary intervention. They will be randomized to statin therapy plus different proton pump inhibitors.16 Prior concerns about an ex vivo interaction between clopidogrel and certain statins were not validated by clinical data.17
OUR RECOMMENDATIONS
Based on the current evidence, patients on aspirin and clopidogrel who have an indication for a proton pump inhibitor or who are at risk of gastrointestinal bleeding can continue or start taking a proton pump inhibitor, including omeprazole.
Switching to another proton pump inhibitor is not currently supported by any randomized clinical trial, nor is changing to a histamine H2-receptor antagonist. The effect of proton pump inhibitors other than omeprazole on clopidogrel is unclear, and it is not known if the interaction with clopidogrel is a class effect or specific to certain drugs of this class.18 On the other hand, we still have no compelling evidence of any major clinical interaction between alternative proton pump inhibitors and clopidogrel.18
Also, separating the dosing times of clopidogrel and omeprazole by 12 hours is not supported by any randomized clinical trial, and runs contrary to at least some ex vivo data.
It is important that all physicians assess the need for a proton pump inhibitor in their patients, as overuse of these drugs has been documented in certain settings.19
Clopidogrel and omeprazole share a common metabolic link via CYP2C19. Omeprazole, along with some other proton pump inhibitors, interacts with clopidogrel at the level of the CYP450 system. Platelet function studies show that platelet inhibition by clopidogrel is impaired, though the astute clinician should be aware of the wide variability associated with platelet function assays and clopidogrel.1,20 However, what may appear to be an interaction at the enzymatic level does not necessarily translate into worse clinical outcomes. Additionally, reliance on nonrandomized studies rather than on randomized clinical trials can be misleading.
Many clinicians are concerned about a possible interaction between the proton pump inhibitor omeprazole (Prilosec) and the antiplatelet drug clopidogrel (Plavix), which is often given to patients as part of dual antiplatelet therapy after an acute coronary syndrome or a percutaneous coronary intervention. Indeed, the US Food and Drug Administration (FDA) has warned that omeprazole reduces the antiplatelet effect of clopidogrel.
Although we should not take such warnings lightly, we also should not be alarmed. The data on which the FDA warning was based came mostly from laboratory assays of platelet function. Preliminary results from a randomized, controlled clinical trial with hard end points show that, for the time being, we should not change the way we manage patients.
PROTON PUMP INHIBITORS DECREASE GASTROINTESTINAL BLEEDING
Dual antiplatelet therapy with aspirin and clopidogrel decreases the risk of major adverse cardiac events after an acute coronary syndrome or a percutaneous coronary intervention compared with aspirin alone.1 However, it also increases the risk of gastrointestinal bleeding. A recent analysis determined that dual antiplatelet therapy was the most significant risk factor associated with serious or fatal gastrointestinal bleeding in high-risk survivors of myocardial infarction.2
Given the risks of significant morbidity and death in patients on dual antiplatelet therapy who develop gastrointestinal bleeding, an expert consensus panel recommended the use of proton pump inhibitors in patients on dual antiplatelet therapy who have risk factors for gastrointestinal bleeding.3 Accordingly, these drugs are commonly used for gastrointestinal protection in patients requiring dual antiplatelet therapy.
A POSSIBLE CYP450 INTERACTION
Clopidogrel is metabolized from a prodrug to its active metabolite by the cytochrome P450 (CYP450) system. Proton pump inhibitors also are metabolized by the CYP450 system.4 Proton pump inhibitors are thought to diminish the activity of clopidogrel via inhibition of the CYP2C19 isoenzyme. However, the clinical significance of this inhibition is not clear. Different drugs of this class inhibit the CYP450 system to varying degrees.
The potential interaction between proton pump inhibitors and clopidogrel is worrisome for many physicians, since adverse cardiovascular outcomes are more common in patients in whom the antiplatelet response to clopidogrel is impaired.1 This interaction led to the publication of numerous articles, and prompted the FDA to carefully analyze the potential clinical implications.
In several randomized trials, omeprazole diminished the response to clopidogrel (measured via platelet function assays).5,6 It is unclear if this is a class effect, as proton pump inhibitors other than omeprazole have not consistently been shown to have this effect.6,7 Observational studies of the effect of co-administration of a proton pump inhibitor and clopidogrel on cardiovascular outcomes following acute coronary syndromes have had conflicting findings.8–11
THE FDA ISSUES AN ADVISORY
Given the reports of an impaired platelet response to clopidogrel with omeprazole, the FDA asked the manufacturer for data on this potential interaction. The data showed diminished platelet inhibition when clopidogrel was co-administered with omeprazole or when the two were taken 12 hours apart.
On November 17, 2009, the FDA issued a patient advisory and updated the patient safety information on the package insert for clopidogrel about this drug interaction.12 Specifically, the FDA warns that omeprazole reduces the antiplatelet effect of clopidogrel by about 50%. The FDA warning sparked debate in the medical community, as the decision was based in part on ex vivo data.
POST HOC ANALYSES FROM RANDOMIZED CONTROLLED TRIALS
Several post hoc analyses of large randomized clinical trials have studied the potential interaction between proton pump inhibitors and clopidogrel.
In the Clopidogrel for the Reduction of Events During Observation (CREDO) trial, clopidogrel reduced the incidence of death, myocardial infarction, or stroke to a similar extent regardless of baseline use of a proton pump inhibitor.13
In patients undergoing percutaneous coronary intervention, the Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation—Thrombolysis in Myocardial Infarction 44 (PRINCIPLE-TIMI 44) trial found that those taking a proton pump inhibitor had significantly less platelet inhibition with clopidogrel compared with those not on one.14 However, patients taking prasugrel (Effient) and a proton pump inhibitor only had a slight trend towards diminished platelet inhibition.14
The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel—Thrombolysis in Myocardial Infarction 38 (TRITON-TIMI 38) found that proton pump inhibitors did not influence the long-term outcome of cardiovascular death, myocardial infarction, or stroke for patients on clopidogrel or prasugrel after an acute coronary syndrome.14 A subanalysis did not reveal any differences between omeprazole or other drugs of this class as to an effect on the primary outcome.
Though informative, the results of these post hoc analyses need to be validated with data from randomized clinical trials.
‘COGENT’ TRIAL HALTED EARLY, BUT PRELIMINARY RESULTS AVAILABLE
The Clopidogrel and the Optimization of Gastrointestinal Events (COGENT) trial was the first randomized clinical study of the effect of the interaction between clopidogrel and omeprazole on cardiovascular and gastrointestinal outcomes.15 In a double-blind fashion, patients with acute coronary syndromes or undergoing percutaneous coronary interventions were randomized to receive a fixed-dose combination pill containing either clopidogrel and delayed-release omeprazole or clopidogrel alone. All patients also received aspirin.
Unfortunately, the trial was stopped early because the sponsor declared bankruptcy. However, preliminary results revealed no significant difference in cardiovascular outcomes for patients on clopidogrel and omeprazole compared with clopidogrel alone.15 Furthermore, adverse gastrointestinal events were significantly fewer in patients on clopidogrel and omeprazole.
Thus, omeprazole appears to be safe and may offer gastrointestinal protection to patients on dual antiplatelet therapy, though we need to await publication of the full results.
‘SPICE ’ TRIAL TO EVALUATE POSSIBLE MECHANISMS OF INTERACTION
The Evaluation of the Influence of Statins and Proton Pump Inhibitors on Clopidogrel Antiplatelet Effects (SPICE) trial is a mechanistic study that will evaluate platelet function and genetic polymorphisms in patients on clopidogrel and aspirin after a percutaneous coronary intervention. They will be randomized to statin therapy plus different proton pump inhibitors.16 Prior concerns about an ex vivo interaction between clopidogrel and certain statins were not validated by clinical data.17
OUR RECOMMENDATIONS
Based on the current evidence, patients on aspirin and clopidogrel who have an indication for a proton pump inhibitor or who are at risk of gastrointestinal bleeding can continue or start taking a proton pump inhibitor, including omeprazole.
Switching to another proton pump inhibitor is not currently supported by any randomized clinical trial, nor is changing to a histamine H2-receptor antagonist. The effect of proton pump inhibitors other than omeprazole on clopidogrel is unclear, and it is not known if the interaction with clopidogrel is a class effect or specific to certain drugs of this class.18 On the other hand, we still have no compelling evidence of any major clinical interaction between alternative proton pump inhibitors and clopidogrel.18
Also, separating the dosing times of clopidogrel and omeprazole by 12 hours is not supported by any randomized clinical trial, and runs contrary to at least some ex vivo data.
It is important that all physicians assess the need for a proton pump inhibitor in their patients, as overuse of these drugs has been documented in certain settings.19
Clopidogrel and omeprazole share a common metabolic link via CYP2C19. Omeprazole, along with some other proton pump inhibitors, interacts with clopidogrel at the level of the CYP450 system. Platelet function studies show that platelet inhibition by clopidogrel is impaired, though the astute clinician should be aware of the wide variability associated with platelet function assays and clopidogrel.1,20 However, what may appear to be an interaction at the enzymatic level does not necessarily translate into worse clinical outcomes. Additionally, reliance on nonrandomized studies rather than on randomized clinical trials can be misleading.
- Depta JP, Bhatt DL. Aspirin and platelet adenosine diphosphate receptor antagonists in acute coronary syndromes and percutaneous coronary intervention: role in therapy and strategies to overcome resistance. Am J Cardiovasc Drugs 2008; 8:91–112.
- Moukarbel GV, Signorovitch JE, Pfeffer MA, et al. Gastrointestinal bleeding in high risk survivors of myocardial infarction: the VALIANT trial. Eur Heart J 2009; 30:2226–2232.
- Bhatt DL, Scheiman J, Abraham NS, et al; American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. J Am Coll Cardiol 2008; 52:1502–1517.
- Li XQ, Andersson TB, Ahlström M, Weidolf L. Comparison of inhibitory effects of the proton pump-inhibiting drugs omeprazole, esomeprazole, lansoprazole, pantoprazole, and rabeprazole on human cytochrome P450 activities. Drug Metab Dispos 2004; 32:821–827.
- Gilard M, Arnaud B, Cornily JC, et al. Influence of omeprazole on the antiplatelet action of clopidogrel associated with aspirin: the randomized, double-blind OCLA (Omeprazole CLopidogrel Aspirin) study. J Am Coll Cardiol 2008; 51:256–260.
- Cuisset T, Frere C, Quilici J, et al. Comparison of omeprazole and pantoprazole influence on a high 150-mg clopidogrel maintenance dose. The PACA (Proton Pump Inhibitors and Clopidogrel Association) prospective randomized study. J Am Coll Cardiol 2009; 54:1149–1153.
- Siller-Matula JM, Spiel AO, Lang IM, Kreiner G, Christ G, Jilma B. Effects of pantoprazole and esomeprazole on platelet inhibition by clopidogrel. Am Heart J 2009; 157:148.e1–148.e5.
- Aubert RE, Epstein RS, Teagarden JR, et al. Proton pump inhibitors effect on clopidogrel effectiveness: the Clopidogrel Medco Outcomes Study [abstract]. Circulation 2008; 118( suppl):S28–10–2008.
- Ho PM, Maddox TM, Wang L, et al. Risk of adverse outcomes associated with concomitant use of clopidogrel and proton pump inhibitors following acute coronary syndrome. JAMA 2009; 301:937–944.
- Juurlink DN, Gomes T, Ko DT, et al. A population-based study of the drug interaction between proton pump inhibitors and clopidogrel. CMAJ 2009; 180:713–718.
- Rassen JA, Choudhry NK, Avorn J, Schneeweiss S. Cardiovascular outcomes and mortality in patients using clopidogrel with proton pump inhibitors after percutaneous coronary intervention or acute coronary syndrome. Circulation 2009; 120:2322–2329.
- US Food and Drug Administration. Public health advisory: updated safety information about a drug interaction between clopidogrel bisulfate (marketed as Plavix) and omeprazole (marketed as Prilosec and Prilosec OTC). www.fda.gov/Drugs/DrugSafety/PublicHealthAdvisories/ucm190825.htm. Accessed 1/6/2010.
- Dunn SP, Macaulay TE, Brennan DM, et al. Baseline proton pump inhibitor use associated with increased cardiovascular events with and without the use of clopidogrel in the CREDO trial. Circulation 2008; 118:S_815. Abstract 3999.
- O‘Donoghue ML, Braunwald E, Antman EM, et al. Pharmacodynamic effect and clinical efficacy of clopidogrel and prasugrel with or without a proton-pump inhibitor: an analysis of two randomised trials. Lancet 2009; 374:989–997.
- Bhatt DL. COGENT: A prospective, randomized, placebo-controlled trial of omeprazole in patients receiving aspirin and clopidogrel. Presented at Transcatheter Cardiovascular Therapeutics; September 24, 2009; San Francisco, Calif.
- National Institutes of Health. Evaluation of the Influence of Statins and Proton Pump Inhibitors on Clopidogrel Antiplatelet Effects (SPICE). http://clinicaltrials.gov/ct2/show/NCT00930670. Accessed 1/6/2010.
- Saw J, Brennan DM, Steinhubl SR, et al. Lack of evidence of clopidogrel-statin interaction in the CHARISMA trial. J Am Coll Cardiol 2007; 50:291–295.
- Laine L, Hennekens C. Proton pump inhibitor and clopidogrel interaction: fact or fiction? Am J Gastroenterol 2009 Nov 10. [Epub ahead of print].
- Forgacs I, Loganayagam A. Overprescribing proton pump inhibitors. BMJ 2008; 336:2–3.
- Serebruany VL, Steinhubl SR, Berger PB, Malinin AI, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45:246–251.
- Depta JP, Bhatt DL. Aspirin and platelet adenosine diphosphate receptor antagonists in acute coronary syndromes and percutaneous coronary intervention: role in therapy and strategies to overcome resistance. Am J Cardiovasc Drugs 2008; 8:91–112.
- Moukarbel GV, Signorovitch JE, Pfeffer MA, et al. Gastrointestinal bleeding in high risk survivors of myocardial infarction: the VALIANT trial. Eur Heart J 2009; 30:2226–2232.
- Bhatt DL, Scheiman J, Abraham NS, et al; American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. J Am Coll Cardiol 2008; 52:1502–1517.
- Li XQ, Andersson TB, Ahlström M, Weidolf L. Comparison of inhibitory effects of the proton pump-inhibiting drugs omeprazole, esomeprazole, lansoprazole, pantoprazole, and rabeprazole on human cytochrome P450 activities. Drug Metab Dispos 2004; 32:821–827.
- Gilard M, Arnaud B, Cornily JC, et al. Influence of omeprazole on the antiplatelet action of clopidogrel associated with aspirin: the randomized, double-blind OCLA (Omeprazole CLopidogrel Aspirin) study. J Am Coll Cardiol 2008; 51:256–260.
- Cuisset T, Frere C, Quilici J, et al. Comparison of omeprazole and pantoprazole influence on a high 150-mg clopidogrel maintenance dose. The PACA (Proton Pump Inhibitors and Clopidogrel Association) prospective randomized study. J Am Coll Cardiol 2009; 54:1149–1153.
- Siller-Matula JM, Spiel AO, Lang IM, Kreiner G, Christ G, Jilma B. Effects of pantoprazole and esomeprazole on platelet inhibition by clopidogrel. Am Heart J 2009; 157:148.e1–148.e5.
- Aubert RE, Epstein RS, Teagarden JR, et al. Proton pump inhibitors effect on clopidogrel effectiveness: the Clopidogrel Medco Outcomes Study [abstract]. Circulation 2008; 118( suppl):S28–10–2008.
- Ho PM, Maddox TM, Wang L, et al. Risk of adverse outcomes associated with concomitant use of clopidogrel and proton pump inhibitors following acute coronary syndrome. JAMA 2009; 301:937–944.
- Juurlink DN, Gomes T, Ko DT, et al. A population-based study of the drug interaction between proton pump inhibitors and clopidogrel. CMAJ 2009; 180:713–718.
- Rassen JA, Choudhry NK, Avorn J, Schneeweiss S. Cardiovascular outcomes and mortality in patients using clopidogrel with proton pump inhibitors after percutaneous coronary intervention or acute coronary syndrome. Circulation 2009; 120:2322–2329.
- US Food and Drug Administration. Public health advisory: updated safety information about a drug interaction between clopidogrel bisulfate (marketed as Plavix) and omeprazole (marketed as Prilosec and Prilosec OTC). www.fda.gov/Drugs/DrugSafety/PublicHealthAdvisories/ucm190825.htm. Accessed 1/6/2010.
- Dunn SP, Macaulay TE, Brennan DM, et al. Baseline proton pump inhibitor use associated with increased cardiovascular events with and without the use of clopidogrel in the CREDO trial. Circulation 2008; 118:S_815. Abstract 3999.
- O‘Donoghue ML, Braunwald E, Antman EM, et al. Pharmacodynamic effect and clinical efficacy of clopidogrel and prasugrel with or without a proton-pump inhibitor: an analysis of two randomised trials. Lancet 2009; 374:989–997.
- Bhatt DL. COGENT: A prospective, randomized, placebo-controlled trial of omeprazole in patients receiving aspirin and clopidogrel. Presented at Transcatheter Cardiovascular Therapeutics; September 24, 2009; San Francisco, Calif.
- National Institutes of Health. Evaluation of the Influence of Statins and Proton Pump Inhibitors on Clopidogrel Antiplatelet Effects (SPICE). http://clinicaltrials.gov/ct2/show/NCT00930670. Accessed 1/6/2010.
- Saw J, Brennan DM, Steinhubl SR, et al. Lack of evidence of clopidogrel-statin interaction in the CHARISMA trial. J Am Coll Cardiol 2007; 50:291–295.
- Laine L, Hennekens C. Proton pump inhibitor and clopidogrel interaction: fact or fiction? Am J Gastroenterol 2009 Nov 10. [Epub ahead of print].
- Forgacs I, Loganayagam A. Overprescribing proton pump inhibitors. BMJ 2008; 336:2–3.
- Serebruany VL, Steinhubl SR, Berger PB, Malinin AI, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45:246–251.
KEY POINTS
- Proton pump inhibitors such as omeprazole reduce the risk of gastrointestinal bleeding in patients on antiplatelet therapy after an acute coronary syndrome or percutaneous coronary intervention.
- Omeprazole diminishes the antiplatelet activity of clopidogrel by inhibiting the CYP2C19 isoenzyme.
- Although the interaction between omeprazole and clopidogrel can be demonstrated on platelet function studies, the clinical significance of this interaction is not clear.
Controversies in non-ST-elevation acute coronary syndromes and percutaneous coronary interventions
Despite all the attention paid to ST-segment-elevation myocardial infarction (MI), in terms of sheer numbers, non-ST-elevation MI and unstable angina are where the action is. Acute coronary syndromes account for 2.43 million hospital discharges per year. Of these, 0.46 million are for ST-elevation MI and 1.97 million are for non-ST-elevation MI and unstable angina.1,2
A number of recent studies have begun to answer some of the pressing questions about treating these types of acute coronary syndromes. In this article, I update the reader on these studies, along with recent findings regarding stenting and antiplatelet agents. As you will see, they are all interconnected.
TO CATHETERIZE IS BETTER THAN NOT TO CATHETERIZE
In the 1990s, a topic of debate was whether patients presenting with unstable angina or non-ST-elevation MI should routinely undergo catheterization or whether they would do just as well with a conservative approach, ie, undergoing catheterization only if they developed recurrent, spontaneous, or stress-induced ischemia. Now, the data are reasonably clear and favor an aggressive strategy.3
Mehta et al4 performed a meta-analysis of seven randomized controlled trials (N = 9,212 patients) of aggressive vs conservative angiography and revascularization for non-ST-elevation MI or unstable angina. The results favored the aggressive strategy. At 17 months of follow-up, death or MI had occurred in 7.4% of patients who received the aggressive therapy compared with 11.0% of those who received the conservative therapy, for an odds ratio of 0.82 (P = .001).
The CRUSADE (Can Rapid Risk Stratification of Unstable Angina Patients Suppress Adverse Outcomes With Early Implemention of the ACC/AHA Guidelines?) Quality Improvement Initiative5 analyzed data from a registry of 17,926 patients with non-ST-elevation acute coronary syndrome who were at high risk because of positive cardiac markers or ischemic electrocardiographic changes. Overall, 2.0% of patients who received early invasive care (catheterization within the first 48 hours) died in the hospital compared with 6.2% of those who got no early invasive care, for an adjusted odds ratio of 0.63 (95% confidence interval [CI] 0.52–0.77).
The investigators also stratified the patients into those at low, medium, and high risk, using the criteria of the PURSUIT (Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin [eptifibatide] Therapy) risk score. There were fewer deaths with early invasive therapy in each risk group, and the risk reduction was greatest in the high-risk group.5
Bavry et al6 performed an updated meta-analysis of randomized trials. At a mean follow-up of 24 months, the relative risk of death from any cause was 0.75 in patients who received early invasive therapy.
In another meta-analysis, O’Donoghue et al7 found that the odds ratio of death, MI, or rehospitalization with acute coronary syndromes was 0.73 (95% CI 0.55–0.98) in men who received invasive vs conservative therapy; in women it was 0.81 (95% CI 0.65–1.01). In women, the benefit was statistically significant in those who had elevations of creatine kinase MB or troponin but not in those who did not, though the benefit in men appeared to be less dependent on the presence of biomarker abnormalities.
MUST ANGIOGRAPHY BE DONE IN THE FIRST 24 HOURS?
Although a number of trials showed that a routine invasive strategy leads to better outcomes than a conservative strategy, until recently we had no information as to whether the catheterization needed to be done early (eg, within the first 24 hours) or if it could be delayed a day or two while the patient received medical therapy.
Mehta et al8 conducted a trial to find out: the Timing of Intervention in Acute Coronary Syndrome (TIMACS) trial. Patients were included if they had unstable angina or non-ST-elevation MI, presented to a hospital within 24 hours of the onset of symptoms, and had two of three high-risk features: age 60 years or older, elevated cardiac biomarkers, or electrocardiographic findings compatible with ischemia. All received standard medical therapy, and 3,031 were randomly assigned to undergo angiography either within 24 hours after randomization or 36 or more hours after randomization.
At 6 months, the primary outcome of death, new MI, or stroke had occurred in 9.6% of the patients in the early-intervention group and in 11.3% of those in the delayed-intervention group, but the difference was not statistically significant. However, the difference in the rate of a secondary end point, death, MI, or refractory ischemia, was statistically significant: 9.5% vs 12.9%, P = .003, owing mainly to less refractory ischemia with early intervention.
The patients were also stratified into two groups by baseline risk. The rate of the primary outcome was significantly lower with early intervention in high-risk patients, but not in those at intermediate or low risk. Thus, early intervention may be beneficial in patients at high risk, such as those with ongoing chest pain, but not necessarily in those at low risk.
LEAVE NO LESION BEHIND?
Coronary artery disease often affects more than one segment. Until recently, it was not known whether we should stent all stenotic segments in patients presenting with non-ST-elevation MI or unstable angina, or only the “culprit lesion.”
Shishehbor et al9 examined data from a Cleveland Clinic registry of 1,240 patients with acute coronary syndrome and multivessel coronary artery disease who underwent bare-metal stenting. The median follow-up was 2.3 years. Using a propensity model to match patients in the two groups with similar baseline characteristics, they found that the rate of repeat revascularization was less with multivessel intervention than with culprit-only stenting, as was the rate of the combined end point of death, MI, or revascularization, but not that of all-cause mortality or the composite of death or MI.
BARE-METAL VS DRUG-ELUTING STENTS: BALANCING THE RISKS AND BENEFITS
After a patient receives a stent, two bad things can happen: the artery can close up again either gradually, in a process called restenosis, or suddenly, via thrombosis.
Drug-eluting stents were invented to solve the problem of restenosis, and they work very well. Stone et al10 pooled the data from four double-blind trials of sirolimus (Rapamune) stents and five double-blind trials of paclitaxel (Taxol) stents and found that, at 4 years, the rates of target-lesion revascularization (for restenosis) were 7.8% with sirolimus stents vs 23.6% with bare-metal stents (P < .001), and 10.1% with paclitaxel stents vs 20.0% with bare-metal stents (P < .001).
Thrombosis was much less common in these studies, occurring in 1.2% of the sirolimus stent groups vs 0.6% of the bare-metal stent groups (P = .20), and in 1.3% of the paclitaxel stent groups vs 0.9% of the bare-metal stent groups (P = .30).10
However, drug-eluting stents appear to increase the risk of thrombosis later on, ie, after 1 year. Bavry et al,11 in a meta-analysis, calculated that when stent thrombosis occurred, the median time after implantation was 15.5 months with sirolimus stents vs 4 months with bare-metal stents (P = .0052), and 18 months with paclitaxel stents vs 3.5 months with bare-metal stents (P = .04). The absolute risk of very late stent thrombosis after 1 year was very low, with five events per 1,000 patients with drug-eluting stents vs no events with bare-metal stents (P = .02). Nevertheless, this finding has practical implications. How long must patients continue dual antiplatelet therapy? And what if a patient needs surgery a year later?
Restenosis is not always so gradual
Although stent thrombosis is serious and often fatal, bare-metal stent restenosis is not always benign either, despite the classic view that stent restenosis is a gradual process that results in exertional angina. Reviewing 1,186 cases of bare-metal stent restenosis in 984 patients at Cleveland Clinic, Chen et al12 reported that 9.5% of cases presented as acute MI (2.2% as ST-elevation MI and 7.3% as non-ST-elevation MI), and 26.4% as unstable angina requiring hospitalization.
A Mayo Clinic study13 corroborated these findings. The 10-year incidence of clinical bare-metal stent restenosis was 18.1%, and the incidence of MI was 2.1%. The 10-year rate of bare-metal stent thrombosis was 2%. Off-label use, primarily in saphenous vein grafts, increased the incidence; other correlates were prior MI, peripheral arterial disease, and ulcerated lesions.
Furthermore, bare-metal stent thrombosis can also occur later. We saw a case that occurred 13 years after the procedure, 3 days after the patient stopped taking aspirin because he was experiencing flu-like symptoms, ran out of aspirin, and felt too sick to go out and buy more. The presentation was with ST-elevation MI. The patient recovered after treatment with intracoronary abciximab (ReoPro), percutaneous thrombectomy, balloon angioplasty, and, eventually, bypass surgery.14
No difference in risk of death with drug-eluting vs bare-metal stents
Even though drug-eluting stents pose a slightly higher risk of thrombosis than bare-metal stents, the risk of death is no higher.15
I believe the reason is that there are competing risks, and that the higher risk of thrombosis with first-generation drug-eluting stents and the higher risk of restenosis with bare-metal stents essentially cancel each other out. For most patients, there is an absolute benefit with drug-eluting stents, which reduce the need for revascularization with no effect in terms of either increasing or decreasing the risk of MI or death. Second-generation drug-eluting stents may have advantages in reducing rates of death or MI compared with first-generation drug-eluting stents, though this remains to be proven conclusively.
The right revascularization for the right patient
Bavry and I16 developed an algorithm for deciding on revascularization, posing a series of questions:
- Does the patient need any form of revascularization?
- Is he or she at higher risk of both stent thrombosis and restenosis, as in patients with diabetes, diffuse multivessel disease with bifurcation lesions, or chronic total occlusions? If so, coronary artery bypass grafting remains an excellent option.
- Does he or she have a low risk of restenosis, as in patients without diabetes with focal lesions in large vessels? If so, one could consider a bare-metal stent, which would probably be more cost-effective than a drug-eluting stent in this situation.
- Does the patient have relative contraindications to drug-eluting stents? Examples are a history of noncompliance with medical therapy, financial issues such as lack of insurance that would make buying clopidogrel (Plavix) a problem, long-term anticoagulation, or anticipated need for surgery in the next few years.
If a drug-eluting stent is used, certain measures can help ensure that it is used optimally. It should often be placed under high pressure with a noncompliant balloon so that it achieves contact with the artery wall all around. One should consider intravascular ultrasonographic guidance to make sure the stent is well opposed if it is in a very calcified lesion. Dual antiplatelet therapy with clopidogrel and aspirin should be given for at least 1 year, and if there is no bleeding, perhaps longer, pending further data.16
LEAVE NO PLATELET ACTIVATED?
Platelets have several types of receptors that, when bound by their respective ligands, lead to platelet activation and aggregation and, ultimately, thrombus formation. Antagonists to some of these receptors are available or are being developed.17
For long-term therapy, blocking the process “upstream,” ie, preventing platelet activation, is better than blocking it “downstream,” ie, preventing aggregation. For example, clopidogrel, ticlopipine (Ticlid), and prasugrel (Effient) have active metabolites that bind to a subtype of the adenosine diphosphate receptor and prevent platelet activation, whereas the glycoprotein IIb/IIIa inhibitors such as abciximab work downstream, binding to a different receptor and preventing aggregation.18
Dual therapy for 1 year is the standard of care after acute coronary syndromes
The evidence for using dual antiplatelet therapy (ie, aspirin plus clopidogrel) in patients with acute coronary syndromes without ST-elevation is very well established.
The Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) trial,19 published in 2001, found a 20% relative risk reduction and a 2% absolute risk reduction in the incidence of MI, stroke, or cardiovascular death in patients randomly assigned to receive clopidogrel plus aspirin for 1 year vs aspirin alone for 1 year (P < .001). In the subgroup of patients who underwent percutaneous coronary intervention, the relative risk reduction in the incidence of MI or cardiovascular death at 1 year of follow-up was 31% (P = .002).20
As a result of these findings, the cardiology society guidelines21 recommend a year of dual antiplatelet therapy after acute coronary syndromes, regardless of whether the patient is treated medically, percutaneously, or surgically.
But what happens after clopidogrel is withdrawn? Ho et al22 retrospectively analyzed data from Veterans Affairs hospitals and found a spike in the incidence of death or MI in the first 90 days after stopping clopidogrel treatment. This was true in medically treated patients as well as in those treated with percutaneous coronary interventions, in those with or without diabetes mellitus, in those who received a drug-eluting stent or a bare-metal stent, and in those treated longer than 9 months.
The investigators concluded that there might be a “clopidogrel rebound effect.” However, I believe that a true rebound effect, such as after withdrawal of heparin or warfarin, is biologically unlikely with clopidogrel, since clopidogrel irreversibly binds to its receptor for the 7- to 10-day life span of the platelet. Rather, I believe the phenomenon must be due to withdrawal of protection in patients at risk.
In stable patients, dual therapy is not as beneficial
Would dual antiplatelet therapy with clopidogrel and aspirin also benefit patients at risk of atherothrombotic events but without acute coronary syndromes?
The Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial23 included 15,603 patients with either clinically evident but stable cardiovascular disease or multiple risk factors for athero-thrombosis. They were randomly assigned to receive either clopidogrel 75 mg/day plus aspirin 75 to 162 mg/day or placebo plus aspirin. At a median of 28 months, the groups did not differ significantly in the rate of MI, stroke, or death from cardiovascular causes.
However, the subgroup of patients who had documented prior MI, ischemic stroke, or symptomatic peripheral arterial disease did appear to derive significant benefit from dual therapy.24 In this subgroup, the rate of MI, stroke, or cardiovascular death at a median follow-up of 27.6 months was 8.8% with placebo plus aspirin compared with 7.3% with clopidogrel plus aspirin, for a hazard ratio of 0.83 (95% CI 0.72–0.96, P = .01). Unstented patients with stable coronary artery disease but without prior MI derived no benefit.
Bleeding and thrombosis: The Scylla and Charybdis of antiplatelet therapy
However, with dual antiplatelet therapy, we steer between the Scylla of bleeding and the Charybdis of thrombosis.25
In the CHARISMA subgroup who had prior MI, ischemic stroke, or symptomatic peripheral arterial disease, the incidence of moderate or severe bleeding was higher with dual therapy than with aspirin alone, but the rates converged after about 1 year of treatment.24 Further, there was no difference in fatal bleeding or intracranial bleeding, although the rate of moderate bleeding (defined as the need for transfusion) was higher with dual therapy (2.0% vs 1.3%, P = .004).
I believe the data indicate that if a patient can tolerate dual antiplatelet therapy for 9 to 12 months without any bleeding issues, he or she is unlikely to have a major bleeding episode if dual therapy is continued beyond this time.
About half of bleeding events in patients on chronic antiplatelet therapy are gastrointestinal. To address this risk, in 2008 an expert committee from the American College of Cardiology, American College of Gastroenterology, and American Heart Association issued a consensus document26 in which they recommended assessing gastrointestinal risk factors in patients on antiplatelet therapy, such as history of ulcers (and testing for and treating Helicobacter pylori infection if present), history of gastrointestinal bleeding, concomitant anticoagulant therapy, and dual antiplatelet therapy. If any of these were present, the committee recommended considering a proton pump inhibitor. The committee also recommended a proton pump inhibitor for patients on antiplatelet therapy who have more than one of the following: age 60 years or more, corticosteroid use, or dyspepsia or gastroesophageal reflux symptoms.
Some ex vivo platelet studies and observational analyses have suggested that there might be an adverse interaction between clopidogrel and proton pump inhibitors due to a blunting of clopidogrel’s antiplatelet effect. A large randomized clinical trial was designed and launched to determine if a single-pill combination of the proton pump inhibitor omeprazole (Prilosec) and clopidogrel would be safer than clopidogrel alone when added to aspirin. Called COGENT-1 (Clopidogrel and the Optimization of GI Events Trial), it was halted early in 2009 when it lost its funding. However, preliminary data did not show an adverse interaction between clopidogrel and omeprazole.
What is the right dose of aspirin?
Steinhubl et al27 performed a post hoc observational analysis of data from the CHARISMA trial. Their findings suggested that higher doses of aspirin are not more effective than lower doses for chronic therapy. Furthermore, in the group receiving clopidogrel plus aspirin, the incidence of severe or life-threatening bleeding was significantly greater with aspirin doses higher than 100 mg than with doses lower than 100 mg, 2.6% vs 1.7%, P = .040.
A randomized, controlled trial called Clopidogrel Optimal Loading Dose Usage to Reduce Recurrent Events/Optimal Antiplatelet Strategy for Interventions (CURRENT/OASIS 7)28 recently reported that higher-dose aspirin (ie, 325 mg) may be better than lower dose aspirin (ie, 81 mg) in patients with acute coronary syndromes undergoing percutaneous coronary intervention and receiving clopidogrel. During this 30-day study, there was no increase in overall bleeding with the higher dose of aspirin, though gastrointestinal bleeding was slightly increased.29 In a factorial design, the second part of this trial found that a higher-dose clopidogrel regimen reduced stent thrombosis.29
Should nonresponders get higher doses of clopidogrel?
In vitro, response to clopidogrel shows a normal bell-shaped distribution.30 In theory, therefore, patients who are hyperresponders may be at higher risk of bleeding, and those who are hyporesponders may be at risk of ischemic events.
A clinical trial is under way to examine whether hyporesponders should get higher doses. Called GRAVITAS (Gauging Responsiveness With a VerifyNow Assay Impact on Thrombosis and Safety), it will use a point-of-care platelet assay and then allocate patients to receive either standard therapy or double the dose of clopidogrel. The primary end point will be the rate of cardiovascular death, nonfatal MI, or stent thrombosis at 6 months.
Is prasugrel better than clopidogrel?
Prasugrel (Effient) is a new drug of the same class as clopidogrel, ie, a thienopyridine, with its active metabolite binding to the same platelet receptor as clopidogrel and inhibiting platelet aggregation more rapidly, more consistently, and to a greater extent than clopidogrel. Prasugrel was recently approved by the Food and Drug Administration. But is it better?31
The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel–Thrombolysis in Myocardial Infarction (TRITON-TIMI 38) compared prasugrel and clopidogrel in 13,608 patients with moderate- to high-risk acute coronary syndromes who were scheduled to undergo percutaneous coronary intervention.32
Overall, prasugrel was better. At 15 months, the incidence of the primary end point (death from cardiovascular causes, nonfatal MI, or nonfatal stroke) was significantly lower with prasugrel therapy than with clopidogrel in the entire cohort (9.9% vs 12.1%, hazard ratio 0.81, 95% CI 0.73–0.90, P < .001), in the subgroup with ST-segment elevation MI, and in the subgroup with unstable angina or non-ST-elevation MI.
However, there was a price to pay. The rate of major bleeding was higher with prasugrel (2.4% vs 1.8%, hazard ratio 1.32, 95% CI 1.03–1.68, P = .03). Assessing the balance between the risk and the benefit, the investigators identified three subgroups who did not derive a net clinical benefit from prasugrel: patients who had had a previous stroke or transient ischemic attack (this group actually had a net harm from prasugrel), patients 75 years of age or older, and patients weighing less than 60 kg (132 pounds).
More work is needed to determine which patients are best served by standard-dose clopidogrel, higher doses of clopidogrel, platelet-assay-guided dosing of clopidogrel, or prasugrel.24
Short-acting, potent intravenous platelet blockade with an agent such as cangrelor is theoretically appealing, but further research is necessary.33,34 Ticagrelor, a reversible adenosine diphosphate receptor antagonist, provides yet another potential option in antiplatelet therapy for acute coronary syndromes. In the recent PLATO trial (Study of Platelet Inhibition and Patient Outcomes), compared with clopidogrel, ticagrelor reduced the risk of ischemic events, including death.35,36 Here, too, there was more major bleeding (unrelated to coronary artery bypass grafting) with ticagrelor.
Thus, clinical assessment of an individual patient’s ischemic and bleeding risks will continue to be critical as therapeutic strategies evolve.
- Wiviott SD, Morrow DA, Giugliano RP, et al. Performance of the Thrombolysis In Myocardial Infarction risk index for early acute coronary syndrome in the National Registry of Myocardial Infarction: a simple risk index predicts mortality in both ST and non-ST elevation myocardial infarction [abstract]. J Am Coll Cardiol 2003; 43( suppl 2):365A–366A.
- Thom T, Haase N, Rosamond W, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2006; 113:e85–e151. Errata in Circulation 2006; 113:e696 and Circulation 2006 114:e630.
- Bhatt DL. To cath or not to cath. That is no longer the question. JAMA 2005; 293:2935–2937.
- Mehta SR, Cannon CP, Fox KA, et al. Routine vs selective invasive strategies in patients with acute coronary syndromes: a collaborative meta-analysis of randomized trials. JAMA 2005; 293:2908–2917.
- Bhatt DL, Roe MT, Peterson ED, et al; for the CRUSADE Investigators. Utilization of early invasive management strategies for high-risk patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE Quality Improvement Initiative. JAMA 2004; 292:2096–2104.
- Bavry AA, Kumbhani DJ, Rassi AN, Bhatt DL, Askari AT. Benefit of early invasive therapy in acute coronary syndromes: a meta-analysis of contemporary randomized clinical trials. J Am Coll Cardiol 2006; 48:1319–1325.
- O’Donoghue MO, Boden WE, Braunwald E, et al. Early invasive vs conservative treatment strategies in women and men with unstable angina and non-ST segment elevation myocardial infarction: a meta-analysis. JAMA 2008; 300:71–80.
- Mehta SR, Granger CB, Boden WE, et al; TIMACS Investigators. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med 2009; 360:2165–2175.
- Shishehbor MH, Lauer MS, Singh IM, et al. In unstable angina or non-ST-segment acute coronary syndrome, should patients with multivessel coronary artery disease undergo multivessel or culpritonly stenting? J Am Coll Cardiol 2007; 49:849–854.
- Stone GW, Moses JW, Ellis SG, et al. Safety and efficacy of sirolimus- and paclitaxel-eluting coronary stents. N Engl J Med 2007; 356:998–1008.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Chen MS, John JM, Chew DP, Lee DS, Ellis SG, Bhatt DL. Bare metal stent restenosis is not a benign clinical entity. Am Heart J 2006; 151:1260–1264.
- Doyle B, Rihal CS, O’Sullivan CJ, et al. Outcomes of stent thrombosis and restenosis during extended follow-up of patients treated with bare-metal coronary stents. Circulation 2007; 116:2391–2398.
- Sarkees ML, Bavry AA, Galla JM, Bhatt DL. Bare metal stent thrombosis 13 years after implantation. Cardiovasc Revasc Med 2009; 10:58–91.
- Bavry AA, Bhatt DL. Appropriate use of drug-eluting stents: balancing the reduction in restenosis with the concern of late thrombosis. Lancet 2008; 371:2134–2143.
- Bavry AA, Bhatt DL. Drug-eluting stents: dual antiplatelet therapy for every survivor? Circulation 2007; 116:696–699.
- Meadows TA, Bhatt DL. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 2007; 100:1261–1275.
- Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov 2003; 2:15–28.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502. Errata in N Engl J Med 2001; 345:1506 and N Engl J Med 2001; 345:1716.
- Mehta SR, Yusuf S, Peters RJ, et al; Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- Anderson JL, Adams CD, Antman EM, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction); american College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons; American Association of Cardiovascular and Pulmonary Rehabilitation; Society for Academic Emergency Medicine. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol 2007; 50:e1–e157.
- Ho PM, Peterson ED, Wang L, et al. Incidence of death and acute myocardial infarction associated with stopping clopidogrel after acute coronary syndrome. JAMA 2008; 299:532–539. Erratum in JAMA 2008; 299:2390.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bhatt DL. Intensifying platelet inhibition—navigating between Scylla and Charybdis. N Engl J Med 2007; 357:2078–2081.
- Bhatt DL, Scheiman J, Abraham NS, et al; American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation 2008; 118:1894–1909.
- Steinhubl SR, Bhatt DL, Brennan DM, et al; CHARISMA Investigators. Aspirin to prevent cardiovascular disease: the association of aspirin dose and clopidogrel with thrombosis and bleeding. Ann Intern Med 2009; 150:379–386.
- Mehta SR, Bassand JP, Chrolavicius S, et al; CURRENT-OASIS 7 Steering Committee. Design and rationale of CURRENT-OASIS 7: a randomized, 2 x 2 factorial trial evaluating optimal dosing strategies for clopidogrel and aspirin in patients with ST and non-ST-elevation acute coronary syndromes managed with an early invasive strategy. Am Heart J 2008; 156:1080–1088.
- Mehta SR, Van de Werf F. A randomized comparison of a clopidogrel high loading and maintenance dose regimen versus standard dose and high versus low dose aspirin in 25,000 patients with acute coronary syndromes: results of the CURRENT OASIS 7 trial. Paper presented at the European Society of Cardiology Congress; August 30, 2009; Barcelona, Spain. Also available online at www.Escardio.org/congresses/esc-2009/congress-reports. Accessed December 12, 2009.
- Serebruany VL, Steinhubl SR, Berger PB, Malinin AT, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45:246–251.
- Bhatt DL. Prasugrel in clinical practice [perspective]. N Engl J Med 2009; 361:940–942.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Bhatt DL, Lincoff AM, Gibson CM, et al; for the CHAMPION PLATFORM Investigators. Intravenous platelet blockade with cangrelor during PCI. N Engl J Med 2009 Nov 15(epub ahead of print).
- Harrington RA, Stone GW, McNulty S, et al. Platelet inhibition with cangrelor in patient sundergoing PCI. N Engl J Med 2009 Nov 17(epub ahead of print).
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Bhatt DL. Ticagrelor in ACS—what does PLATO teach us? Nat Rev Cardiol 2009; 6:737–738.
Despite all the attention paid to ST-segment-elevation myocardial infarction (MI), in terms of sheer numbers, non-ST-elevation MI and unstable angina are where the action is. Acute coronary syndromes account for 2.43 million hospital discharges per year. Of these, 0.46 million are for ST-elevation MI and 1.97 million are for non-ST-elevation MI and unstable angina.1,2
A number of recent studies have begun to answer some of the pressing questions about treating these types of acute coronary syndromes. In this article, I update the reader on these studies, along with recent findings regarding stenting and antiplatelet agents. As you will see, they are all interconnected.
TO CATHETERIZE IS BETTER THAN NOT TO CATHETERIZE
In the 1990s, a topic of debate was whether patients presenting with unstable angina or non-ST-elevation MI should routinely undergo catheterization or whether they would do just as well with a conservative approach, ie, undergoing catheterization only if they developed recurrent, spontaneous, or stress-induced ischemia. Now, the data are reasonably clear and favor an aggressive strategy.3
Mehta et al4 performed a meta-analysis of seven randomized controlled trials (N = 9,212 patients) of aggressive vs conservative angiography and revascularization for non-ST-elevation MI or unstable angina. The results favored the aggressive strategy. At 17 months of follow-up, death or MI had occurred in 7.4% of patients who received the aggressive therapy compared with 11.0% of those who received the conservative therapy, for an odds ratio of 0.82 (P = .001).
The CRUSADE (Can Rapid Risk Stratification of Unstable Angina Patients Suppress Adverse Outcomes With Early Implemention of the ACC/AHA Guidelines?) Quality Improvement Initiative5 analyzed data from a registry of 17,926 patients with non-ST-elevation acute coronary syndrome who were at high risk because of positive cardiac markers or ischemic electrocardiographic changes. Overall, 2.0% of patients who received early invasive care (catheterization within the first 48 hours) died in the hospital compared with 6.2% of those who got no early invasive care, for an adjusted odds ratio of 0.63 (95% confidence interval [CI] 0.52–0.77).
The investigators also stratified the patients into those at low, medium, and high risk, using the criteria of the PURSUIT (Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin [eptifibatide] Therapy) risk score. There were fewer deaths with early invasive therapy in each risk group, and the risk reduction was greatest in the high-risk group.5
Bavry et al6 performed an updated meta-analysis of randomized trials. At a mean follow-up of 24 months, the relative risk of death from any cause was 0.75 in patients who received early invasive therapy.
In another meta-analysis, O’Donoghue et al7 found that the odds ratio of death, MI, or rehospitalization with acute coronary syndromes was 0.73 (95% CI 0.55–0.98) in men who received invasive vs conservative therapy; in women it was 0.81 (95% CI 0.65–1.01). In women, the benefit was statistically significant in those who had elevations of creatine kinase MB or troponin but not in those who did not, though the benefit in men appeared to be less dependent on the presence of biomarker abnormalities.
MUST ANGIOGRAPHY BE DONE IN THE FIRST 24 HOURS?
Although a number of trials showed that a routine invasive strategy leads to better outcomes than a conservative strategy, until recently we had no information as to whether the catheterization needed to be done early (eg, within the first 24 hours) or if it could be delayed a day or two while the patient received medical therapy.
Mehta et al8 conducted a trial to find out: the Timing of Intervention in Acute Coronary Syndrome (TIMACS) trial. Patients were included if they had unstable angina or non-ST-elevation MI, presented to a hospital within 24 hours of the onset of symptoms, and had two of three high-risk features: age 60 years or older, elevated cardiac biomarkers, or electrocardiographic findings compatible with ischemia. All received standard medical therapy, and 3,031 were randomly assigned to undergo angiography either within 24 hours after randomization or 36 or more hours after randomization.
At 6 months, the primary outcome of death, new MI, or stroke had occurred in 9.6% of the patients in the early-intervention group and in 11.3% of those in the delayed-intervention group, but the difference was not statistically significant. However, the difference in the rate of a secondary end point, death, MI, or refractory ischemia, was statistically significant: 9.5% vs 12.9%, P = .003, owing mainly to less refractory ischemia with early intervention.
The patients were also stratified into two groups by baseline risk. The rate of the primary outcome was significantly lower with early intervention in high-risk patients, but not in those at intermediate or low risk. Thus, early intervention may be beneficial in patients at high risk, such as those with ongoing chest pain, but not necessarily in those at low risk.
LEAVE NO LESION BEHIND?
Coronary artery disease often affects more than one segment. Until recently, it was not known whether we should stent all stenotic segments in patients presenting with non-ST-elevation MI or unstable angina, or only the “culprit lesion.”
Shishehbor et al9 examined data from a Cleveland Clinic registry of 1,240 patients with acute coronary syndrome and multivessel coronary artery disease who underwent bare-metal stenting. The median follow-up was 2.3 years. Using a propensity model to match patients in the two groups with similar baseline characteristics, they found that the rate of repeat revascularization was less with multivessel intervention than with culprit-only stenting, as was the rate of the combined end point of death, MI, or revascularization, but not that of all-cause mortality or the composite of death or MI.
BARE-METAL VS DRUG-ELUTING STENTS: BALANCING THE RISKS AND BENEFITS
After a patient receives a stent, two bad things can happen: the artery can close up again either gradually, in a process called restenosis, or suddenly, via thrombosis.
Drug-eluting stents were invented to solve the problem of restenosis, and they work very well. Stone et al10 pooled the data from four double-blind trials of sirolimus (Rapamune) stents and five double-blind trials of paclitaxel (Taxol) stents and found that, at 4 years, the rates of target-lesion revascularization (for restenosis) were 7.8% with sirolimus stents vs 23.6% with bare-metal stents (P < .001), and 10.1% with paclitaxel stents vs 20.0% with bare-metal stents (P < .001).
Thrombosis was much less common in these studies, occurring in 1.2% of the sirolimus stent groups vs 0.6% of the bare-metal stent groups (P = .20), and in 1.3% of the paclitaxel stent groups vs 0.9% of the bare-metal stent groups (P = .30).10
However, drug-eluting stents appear to increase the risk of thrombosis later on, ie, after 1 year. Bavry et al,11 in a meta-analysis, calculated that when stent thrombosis occurred, the median time after implantation was 15.5 months with sirolimus stents vs 4 months with bare-metal stents (P = .0052), and 18 months with paclitaxel stents vs 3.5 months with bare-metal stents (P = .04). The absolute risk of very late stent thrombosis after 1 year was very low, with five events per 1,000 patients with drug-eluting stents vs no events with bare-metal stents (P = .02). Nevertheless, this finding has practical implications. How long must patients continue dual antiplatelet therapy? And what if a patient needs surgery a year later?
Restenosis is not always so gradual
Although stent thrombosis is serious and often fatal, bare-metal stent restenosis is not always benign either, despite the classic view that stent restenosis is a gradual process that results in exertional angina. Reviewing 1,186 cases of bare-metal stent restenosis in 984 patients at Cleveland Clinic, Chen et al12 reported that 9.5% of cases presented as acute MI (2.2% as ST-elevation MI and 7.3% as non-ST-elevation MI), and 26.4% as unstable angina requiring hospitalization.
A Mayo Clinic study13 corroborated these findings. The 10-year incidence of clinical bare-metal stent restenosis was 18.1%, and the incidence of MI was 2.1%. The 10-year rate of bare-metal stent thrombosis was 2%. Off-label use, primarily in saphenous vein grafts, increased the incidence; other correlates were prior MI, peripheral arterial disease, and ulcerated lesions.
Furthermore, bare-metal stent thrombosis can also occur later. We saw a case that occurred 13 years after the procedure, 3 days after the patient stopped taking aspirin because he was experiencing flu-like symptoms, ran out of aspirin, and felt too sick to go out and buy more. The presentation was with ST-elevation MI. The patient recovered after treatment with intracoronary abciximab (ReoPro), percutaneous thrombectomy, balloon angioplasty, and, eventually, bypass surgery.14
No difference in risk of death with drug-eluting vs bare-metal stents
Even though drug-eluting stents pose a slightly higher risk of thrombosis than bare-metal stents, the risk of death is no higher.15
I believe the reason is that there are competing risks, and that the higher risk of thrombosis with first-generation drug-eluting stents and the higher risk of restenosis with bare-metal stents essentially cancel each other out. For most patients, there is an absolute benefit with drug-eluting stents, which reduce the need for revascularization with no effect in terms of either increasing or decreasing the risk of MI or death. Second-generation drug-eluting stents may have advantages in reducing rates of death or MI compared with first-generation drug-eluting stents, though this remains to be proven conclusively.
The right revascularization for the right patient
Bavry and I16 developed an algorithm for deciding on revascularization, posing a series of questions:
- Does the patient need any form of revascularization?
- Is he or she at higher risk of both stent thrombosis and restenosis, as in patients with diabetes, diffuse multivessel disease with bifurcation lesions, or chronic total occlusions? If so, coronary artery bypass grafting remains an excellent option.
- Does he or she have a low risk of restenosis, as in patients without diabetes with focal lesions in large vessels? If so, one could consider a bare-metal stent, which would probably be more cost-effective than a drug-eluting stent in this situation.
- Does the patient have relative contraindications to drug-eluting stents? Examples are a history of noncompliance with medical therapy, financial issues such as lack of insurance that would make buying clopidogrel (Plavix) a problem, long-term anticoagulation, or anticipated need for surgery in the next few years.
If a drug-eluting stent is used, certain measures can help ensure that it is used optimally. It should often be placed under high pressure with a noncompliant balloon so that it achieves contact with the artery wall all around. One should consider intravascular ultrasonographic guidance to make sure the stent is well opposed if it is in a very calcified lesion. Dual antiplatelet therapy with clopidogrel and aspirin should be given for at least 1 year, and if there is no bleeding, perhaps longer, pending further data.16
LEAVE NO PLATELET ACTIVATED?
Platelets have several types of receptors that, when bound by their respective ligands, lead to platelet activation and aggregation and, ultimately, thrombus formation. Antagonists to some of these receptors are available or are being developed.17
For long-term therapy, blocking the process “upstream,” ie, preventing platelet activation, is better than blocking it “downstream,” ie, preventing aggregation. For example, clopidogrel, ticlopipine (Ticlid), and prasugrel (Effient) have active metabolites that bind to a subtype of the adenosine diphosphate receptor and prevent platelet activation, whereas the glycoprotein IIb/IIIa inhibitors such as abciximab work downstream, binding to a different receptor and preventing aggregation.18
Dual therapy for 1 year is the standard of care after acute coronary syndromes
The evidence for using dual antiplatelet therapy (ie, aspirin plus clopidogrel) in patients with acute coronary syndromes without ST-elevation is very well established.
The Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) trial,19 published in 2001, found a 20% relative risk reduction and a 2% absolute risk reduction in the incidence of MI, stroke, or cardiovascular death in patients randomly assigned to receive clopidogrel plus aspirin for 1 year vs aspirin alone for 1 year (P < .001). In the subgroup of patients who underwent percutaneous coronary intervention, the relative risk reduction in the incidence of MI or cardiovascular death at 1 year of follow-up was 31% (P = .002).20
As a result of these findings, the cardiology society guidelines21 recommend a year of dual antiplatelet therapy after acute coronary syndromes, regardless of whether the patient is treated medically, percutaneously, or surgically.
But what happens after clopidogrel is withdrawn? Ho et al22 retrospectively analyzed data from Veterans Affairs hospitals and found a spike in the incidence of death or MI in the first 90 days after stopping clopidogrel treatment. This was true in medically treated patients as well as in those treated with percutaneous coronary interventions, in those with or without diabetes mellitus, in those who received a drug-eluting stent or a bare-metal stent, and in those treated longer than 9 months.
The investigators concluded that there might be a “clopidogrel rebound effect.” However, I believe that a true rebound effect, such as after withdrawal of heparin or warfarin, is biologically unlikely with clopidogrel, since clopidogrel irreversibly binds to its receptor for the 7- to 10-day life span of the platelet. Rather, I believe the phenomenon must be due to withdrawal of protection in patients at risk.
In stable patients, dual therapy is not as beneficial
Would dual antiplatelet therapy with clopidogrel and aspirin also benefit patients at risk of atherothrombotic events but without acute coronary syndromes?
The Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial23 included 15,603 patients with either clinically evident but stable cardiovascular disease or multiple risk factors for athero-thrombosis. They were randomly assigned to receive either clopidogrel 75 mg/day plus aspirin 75 to 162 mg/day or placebo plus aspirin. At a median of 28 months, the groups did not differ significantly in the rate of MI, stroke, or death from cardiovascular causes.
However, the subgroup of patients who had documented prior MI, ischemic stroke, or symptomatic peripheral arterial disease did appear to derive significant benefit from dual therapy.24 In this subgroup, the rate of MI, stroke, or cardiovascular death at a median follow-up of 27.6 months was 8.8% with placebo plus aspirin compared with 7.3% with clopidogrel plus aspirin, for a hazard ratio of 0.83 (95% CI 0.72–0.96, P = .01). Unstented patients with stable coronary artery disease but without prior MI derived no benefit.
Bleeding and thrombosis: The Scylla and Charybdis of antiplatelet therapy
However, with dual antiplatelet therapy, we steer between the Scylla of bleeding and the Charybdis of thrombosis.25
In the CHARISMA subgroup who had prior MI, ischemic stroke, or symptomatic peripheral arterial disease, the incidence of moderate or severe bleeding was higher with dual therapy than with aspirin alone, but the rates converged after about 1 year of treatment.24 Further, there was no difference in fatal bleeding or intracranial bleeding, although the rate of moderate bleeding (defined as the need for transfusion) was higher with dual therapy (2.0% vs 1.3%, P = .004).
I believe the data indicate that if a patient can tolerate dual antiplatelet therapy for 9 to 12 months without any bleeding issues, he or she is unlikely to have a major bleeding episode if dual therapy is continued beyond this time.
About half of bleeding events in patients on chronic antiplatelet therapy are gastrointestinal. To address this risk, in 2008 an expert committee from the American College of Cardiology, American College of Gastroenterology, and American Heart Association issued a consensus document26 in which they recommended assessing gastrointestinal risk factors in patients on antiplatelet therapy, such as history of ulcers (and testing for and treating Helicobacter pylori infection if present), history of gastrointestinal bleeding, concomitant anticoagulant therapy, and dual antiplatelet therapy. If any of these were present, the committee recommended considering a proton pump inhibitor. The committee also recommended a proton pump inhibitor for patients on antiplatelet therapy who have more than one of the following: age 60 years or more, corticosteroid use, or dyspepsia or gastroesophageal reflux symptoms.
Some ex vivo platelet studies and observational analyses have suggested that there might be an adverse interaction between clopidogrel and proton pump inhibitors due to a blunting of clopidogrel’s antiplatelet effect. A large randomized clinical trial was designed and launched to determine if a single-pill combination of the proton pump inhibitor omeprazole (Prilosec) and clopidogrel would be safer than clopidogrel alone when added to aspirin. Called COGENT-1 (Clopidogrel and the Optimization of GI Events Trial), it was halted early in 2009 when it lost its funding. However, preliminary data did not show an adverse interaction between clopidogrel and omeprazole.
What is the right dose of aspirin?
Steinhubl et al27 performed a post hoc observational analysis of data from the CHARISMA trial. Their findings suggested that higher doses of aspirin are not more effective than lower doses for chronic therapy. Furthermore, in the group receiving clopidogrel plus aspirin, the incidence of severe or life-threatening bleeding was significantly greater with aspirin doses higher than 100 mg than with doses lower than 100 mg, 2.6% vs 1.7%, P = .040.
A randomized, controlled trial called Clopidogrel Optimal Loading Dose Usage to Reduce Recurrent Events/Optimal Antiplatelet Strategy for Interventions (CURRENT/OASIS 7)28 recently reported that higher-dose aspirin (ie, 325 mg) may be better than lower dose aspirin (ie, 81 mg) in patients with acute coronary syndromes undergoing percutaneous coronary intervention and receiving clopidogrel. During this 30-day study, there was no increase in overall bleeding with the higher dose of aspirin, though gastrointestinal bleeding was slightly increased.29 In a factorial design, the second part of this trial found that a higher-dose clopidogrel regimen reduced stent thrombosis.29
Should nonresponders get higher doses of clopidogrel?
In vitro, response to clopidogrel shows a normal bell-shaped distribution.30 In theory, therefore, patients who are hyperresponders may be at higher risk of bleeding, and those who are hyporesponders may be at risk of ischemic events.
A clinical trial is under way to examine whether hyporesponders should get higher doses. Called GRAVITAS (Gauging Responsiveness With a VerifyNow Assay Impact on Thrombosis and Safety), it will use a point-of-care platelet assay and then allocate patients to receive either standard therapy or double the dose of clopidogrel. The primary end point will be the rate of cardiovascular death, nonfatal MI, or stent thrombosis at 6 months.
Is prasugrel better than clopidogrel?
Prasugrel (Effient) is a new drug of the same class as clopidogrel, ie, a thienopyridine, with its active metabolite binding to the same platelet receptor as clopidogrel and inhibiting platelet aggregation more rapidly, more consistently, and to a greater extent than clopidogrel. Prasugrel was recently approved by the Food and Drug Administration. But is it better?31
The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel–Thrombolysis in Myocardial Infarction (TRITON-TIMI 38) compared prasugrel and clopidogrel in 13,608 patients with moderate- to high-risk acute coronary syndromes who were scheduled to undergo percutaneous coronary intervention.32
Overall, prasugrel was better. At 15 months, the incidence of the primary end point (death from cardiovascular causes, nonfatal MI, or nonfatal stroke) was significantly lower with prasugrel therapy than with clopidogrel in the entire cohort (9.9% vs 12.1%, hazard ratio 0.81, 95% CI 0.73–0.90, P < .001), in the subgroup with ST-segment elevation MI, and in the subgroup with unstable angina or non-ST-elevation MI.
However, there was a price to pay. The rate of major bleeding was higher with prasugrel (2.4% vs 1.8%, hazard ratio 1.32, 95% CI 1.03–1.68, P = .03). Assessing the balance between the risk and the benefit, the investigators identified three subgroups who did not derive a net clinical benefit from prasugrel: patients who had had a previous stroke or transient ischemic attack (this group actually had a net harm from prasugrel), patients 75 years of age or older, and patients weighing less than 60 kg (132 pounds).
More work is needed to determine which patients are best served by standard-dose clopidogrel, higher doses of clopidogrel, platelet-assay-guided dosing of clopidogrel, or prasugrel.24
Short-acting, potent intravenous platelet blockade with an agent such as cangrelor is theoretically appealing, but further research is necessary.33,34 Ticagrelor, a reversible adenosine diphosphate receptor antagonist, provides yet another potential option in antiplatelet therapy for acute coronary syndromes. In the recent PLATO trial (Study of Platelet Inhibition and Patient Outcomes), compared with clopidogrel, ticagrelor reduced the risk of ischemic events, including death.35,36 Here, too, there was more major bleeding (unrelated to coronary artery bypass grafting) with ticagrelor.
Thus, clinical assessment of an individual patient’s ischemic and bleeding risks will continue to be critical as therapeutic strategies evolve.
Despite all the attention paid to ST-segment-elevation myocardial infarction (MI), in terms of sheer numbers, non-ST-elevation MI and unstable angina are where the action is. Acute coronary syndromes account for 2.43 million hospital discharges per year. Of these, 0.46 million are for ST-elevation MI and 1.97 million are for non-ST-elevation MI and unstable angina.1,2
A number of recent studies have begun to answer some of the pressing questions about treating these types of acute coronary syndromes. In this article, I update the reader on these studies, along with recent findings regarding stenting and antiplatelet agents. As you will see, they are all interconnected.
TO CATHETERIZE IS BETTER THAN NOT TO CATHETERIZE
In the 1990s, a topic of debate was whether patients presenting with unstable angina or non-ST-elevation MI should routinely undergo catheterization or whether they would do just as well with a conservative approach, ie, undergoing catheterization only if they developed recurrent, spontaneous, or stress-induced ischemia. Now, the data are reasonably clear and favor an aggressive strategy.3
Mehta et al4 performed a meta-analysis of seven randomized controlled trials (N = 9,212 patients) of aggressive vs conservative angiography and revascularization for non-ST-elevation MI or unstable angina. The results favored the aggressive strategy. At 17 months of follow-up, death or MI had occurred in 7.4% of patients who received the aggressive therapy compared with 11.0% of those who received the conservative therapy, for an odds ratio of 0.82 (P = .001).
The CRUSADE (Can Rapid Risk Stratification of Unstable Angina Patients Suppress Adverse Outcomes With Early Implemention of the ACC/AHA Guidelines?) Quality Improvement Initiative5 analyzed data from a registry of 17,926 patients with non-ST-elevation acute coronary syndrome who were at high risk because of positive cardiac markers or ischemic electrocardiographic changes. Overall, 2.0% of patients who received early invasive care (catheterization within the first 48 hours) died in the hospital compared with 6.2% of those who got no early invasive care, for an adjusted odds ratio of 0.63 (95% confidence interval [CI] 0.52–0.77).
The investigators also stratified the patients into those at low, medium, and high risk, using the criteria of the PURSUIT (Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin [eptifibatide] Therapy) risk score. There were fewer deaths with early invasive therapy in each risk group, and the risk reduction was greatest in the high-risk group.5
Bavry et al6 performed an updated meta-analysis of randomized trials. At a mean follow-up of 24 months, the relative risk of death from any cause was 0.75 in patients who received early invasive therapy.
In another meta-analysis, O’Donoghue et al7 found that the odds ratio of death, MI, or rehospitalization with acute coronary syndromes was 0.73 (95% CI 0.55–0.98) in men who received invasive vs conservative therapy; in women it was 0.81 (95% CI 0.65–1.01). In women, the benefit was statistically significant in those who had elevations of creatine kinase MB or troponin but not in those who did not, though the benefit in men appeared to be less dependent on the presence of biomarker abnormalities.
MUST ANGIOGRAPHY BE DONE IN THE FIRST 24 HOURS?
Although a number of trials showed that a routine invasive strategy leads to better outcomes than a conservative strategy, until recently we had no information as to whether the catheterization needed to be done early (eg, within the first 24 hours) or if it could be delayed a day or two while the patient received medical therapy.
Mehta et al8 conducted a trial to find out: the Timing of Intervention in Acute Coronary Syndrome (TIMACS) trial. Patients were included if they had unstable angina or non-ST-elevation MI, presented to a hospital within 24 hours of the onset of symptoms, and had two of three high-risk features: age 60 years or older, elevated cardiac biomarkers, or electrocardiographic findings compatible with ischemia. All received standard medical therapy, and 3,031 were randomly assigned to undergo angiography either within 24 hours after randomization or 36 or more hours after randomization.
At 6 months, the primary outcome of death, new MI, or stroke had occurred in 9.6% of the patients in the early-intervention group and in 11.3% of those in the delayed-intervention group, but the difference was not statistically significant. However, the difference in the rate of a secondary end point, death, MI, or refractory ischemia, was statistically significant: 9.5% vs 12.9%, P = .003, owing mainly to less refractory ischemia with early intervention.
The patients were also stratified into two groups by baseline risk. The rate of the primary outcome was significantly lower with early intervention in high-risk patients, but not in those at intermediate or low risk. Thus, early intervention may be beneficial in patients at high risk, such as those with ongoing chest pain, but not necessarily in those at low risk.
LEAVE NO LESION BEHIND?
Coronary artery disease often affects more than one segment. Until recently, it was not known whether we should stent all stenotic segments in patients presenting with non-ST-elevation MI or unstable angina, or only the “culprit lesion.”
Shishehbor et al9 examined data from a Cleveland Clinic registry of 1,240 patients with acute coronary syndrome and multivessel coronary artery disease who underwent bare-metal stenting. The median follow-up was 2.3 years. Using a propensity model to match patients in the two groups with similar baseline characteristics, they found that the rate of repeat revascularization was less with multivessel intervention than with culprit-only stenting, as was the rate of the combined end point of death, MI, or revascularization, but not that of all-cause mortality or the composite of death or MI.
BARE-METAL VS DRUG-ELUTING STENTS: BALANCING THE RISKS AND BENEFITS
After a patient receives a stent, two bad things can happen: the artery can close up again either gradually, in a process called restenosis, or suddenly, via thrombosis.
Drug-eluting stents were invented to solve the problem of restenosis, and they work very well. Stone et al10 pooled the data from four double-blind trials of sirolimus (Rapamune) stents and five double-blind trials of paclitaxel (Taxol) stents and found that, at 4 years, the rates of target-lesion revascularization (for restenosis) were 7.8% with sirolimus stents vs 23.6% with bare-metal stents (P < .001), and 10.1% with paclitaxel stents vs 20.0% with bare-metal stents (P < .001).
Thrombosis was much less common in these studies, occurring in 1.2% of the sirolimus stent groups vs 0.6% of the bare-metal stent groups (P = .20), and in 1.3% of the paclitaxel stent groups vs 0.9% of the bare-metal stent groups (P = .30).10
However, drug-eluting stents appear to increase the risk of thrombosis later on, ie, after 1 year. Bavry et al,11 in a meta-analysis, calculated that when stent thrombosis occurred, the median time after implantation was 15.5 months with sirolimus stents vs 4 months with bare-metal stents (P = .0052), and 18 months with paclitaxel stents vs 3.5 months with bare-metal stents (P = .04). The absolute risk of very late stent thrombosis after 1 year was very low, with five events per 1,000 patients with drug-eluting stents vs no events with bare-metal stents (P = .02). Nevertheless, this finding has practical implications. How long must patients continue dual antiplatelet therapy? And what if a patient needs surgery a year later?
Restenosis is not always so gradual
Although stent thrombosis is serious and often fatal, bare-metal stent restenosis is not always benign either, despite the classic view that stent restenosis is a gradual process that results in exertional angina. Reviewing 1,186 cases of bare-metal stent restenosis in 984 patients at Cleveland Clinic, Chen et al12 reported that 9.5% of cases presented as acute MI (2.2% as ST-elevation MI and 7.3% as non-ST-elevation MI), and 26.4% as unstable angina requiring hospitalization.
A Mayo Clinic study13 corroborated these findings. The 10-year incidence of clinical bare-metal stent restenosis was 18.1%, and the incidence of MI was 2.1%. The 10-year rate of bare-metal stent thrombosis was 2%. Off-label use, primarily in saphenous vein grafts, increased the incidence; other correlates were prior MI, peripheral arterial disease, and ulcerated lesions.
Furthermore, bare-metal stent thrombosis can also occur later. We saw a case that occurred 13 years after the procedure, 3 days after the patient stopped taking aspirin because he was experiencing flu-like symptoms, ran out of aspirin, and felt too sick to go out and buy more. The presentation was with ST-elevation MI. The patient recovered after treatment with intracoronary abciximab (ReoPro), percutaneous thrombectomy, balloon angioplasty, and, eventually, bypass surgery.14
No difference in risk of death with drug-eluting vs bare-metal stents
Even though drug-eluting stents pose a slightly higher risk of thrombosis than bare-metal stents, the risk of death is no higher.15
I believe the reason is that there are competing risks, and that the higher risk of thrombosis with first-generation drug-eluting stents and the higher risk of restenosis with bare-metal stents essentially cancel each other out. For most patients, there is an absolute benefit with drug-eluting stents, which reduce the need for revascularization with no effect in terms of either increasing or decreasing the risk of MI or death. Second-generation drug-eluting stents may have advantages in reducing rates of death or MI compared with first-generation drug-eluting stents, though this remains to be proven conclusively.
The right revascularization for the right patient
Bavry and I16 developed an algorithm for deciding on revascularization, posing a series of questions:
- Does the patient need any form of revascularization?
- Is he or she at higher risk of both stent thrombosis and restenosis, as in patients with diabetes, diffuse multivessel disease with bifurcation lesions, or chronic total occlusions? If so, coronary artery bypass grafting remains an excellent option.
- Does he or she have a low risk of restenosis, as in patients without diabetes with focal lesions in large vessels? If so, one could consider a bare-metal stent, which would probably be more cost-effective than a drug-eluting stent in this situation.
- Does the patient have relative contraindications to drug-eluting stents? Examples are a history of noncompliance with medical therapy, financial issues such as lack of insurance that would make buying clopidogrel (Plavix) a problem, long-term anticoagulation, or anticipated need for surgery in the next few years.
If a drug-eluting stent is used, certain measures can help ensure that it is used optimally. It should often be placed under high pressure with a noncompliant balloon so that it achieves contact with the artery wall all around. One should consider intravascular ultrasonographic guidance to make sure the stent is well opposed if it is in a very calcified lesion. Dual antiplatelet therapy with clopidogrel and aspirin should be given for at least 1 year, and if there is no bleeding, perhaps longer, pending further data.16
LEAVE NO PLATELET ACTIVATED?
Platelets have several types of receptors that, when bound by their respective ligands, lead to platelet activation and aggregation and, ultimately, thrombus formation. Antagonists to some of these receptors are available or are being developed.17
For long-term therapy, blocking the process “upstream,” ie, preventing platelet activation, is better than blocking it “downstream,” ie, preventing aggregation. For example, clopidogrel, ticlopipine (Ticlid), and prasugrel (Effient) have active metabolites that bind to a subtype of the adenosine diphosphate receptor and prevent platelet activation, whereas the glycoprotein IIb/IIIa inhibitors such as abciximab work downstream, binding to a different receptor and preventing aggregation.18
Dual therapy for 1 year is the standard of care after acute coronary syndromes
The evidence for using dual antiplatelet therapy (ie, aspirin plus clopidogrel) in patients with acute coronary syndromes without ST-elevation is very well established.
The Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) trial,19 published in 2001, found a 20% relative risk reduction and a 2% absolute risk reduction in the incidence of MI, stroke, or cardiovascular death in patients randomly assigned to receive clopidogrel plus aspirin for 1 year vs aspirin alone for 1 year (P < .001). In the subgroup of patients who underwent percutaneous coronary intervention, the relative risk reduction in the incidence of MI or cardiovascular death at 1 year of follow-up was 31% (P = .002).20
As a result of these findings, the cardiology society guidelines21 recommend a year of dual antiplatelet therapy after acute coronary syndromes, regardless of whether the patient is treated medically, percutaneously, or surgically.
But what happens after clopidogrel is withdrawn? Ho et al22 retrospectively analyzed data from Veterans Affairs hospitals and found a spike in the incidence of death or MI in the first 90 days after stopping clopidogrel treatment. This was true in medically treated patients as well as in those treated with percutaneous coronary interventions, in those with or without diabetes mellitus, in those who received a drug-eluting stent or a bare-metal stent, and in those treated longer than 9 months.
The investigators concluded that there might be a “clopidogrel rebound effect.” However, I believe that a true rebound effect, such as after withdrawal of heparin or warfarin, is biologically unlikely with clopidogrel, since clopidogrel irreversibly binds to its receptor for the 7- to 10-day life span of the platelet. Rather, I believe the phenomenon must be due to withdrawal of protection in patients at risk.
In stable patients, dual therapy is not as beneficial
Would dual antiplatelet therapy with clopidogrel and aspirin also benefit patients at risk of atherothrombotic events but without acute coronary syndromes?
The Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial23 included 15,603 patients with either clinically evident but stable cardiovascular disease or multiple risk factors for athero-thrombosis. They were randomly assigned to receive either clopidogrel 75 mg/day plus aspirin 75 to 162 mg/day or placebo plus aspirin. At a median of 28 months, the groups did not differ significantly in the rate of MI, stroke, or death from cardiovascular causes.
However, the subgroup of patients who had documented prior MI, ischemic stroke, or symptomatic peripheral arterial disease did appear to derive significant benefit from dual therapy.24 In this subgroup, the rate of MI, stroke, or cardiovascular death at a median follow-up of 27.6 months was 8.8% with placebo plus aspirin compared with 7.3% with clopidogrel plus aspirin, for a hazard ratio of 0.83 (95% CI 0.72–0.96, P = .01). Unstented patients with stable coronary artery disease but without prior MI derived no benefit.
Bleeding and thrombosis: The Scylla and Charybdis of antiplatelet therapy
However, with dual antiplatelet therapy, we steer between the Scylla of bleeding and the Charybdis of thrombosis.25
In the CHARISMA subgroup who had prior MI, ischemic stroke, or symptomatic peripheral arterial disease, the incidence of moderate or severe bleeding was higher with dual therapy than with aspirin alone, but the rates converged after about 1 year of treatment.24 Further, there was no difference in fatal bleeding or intracranial bleeding, although the rate of moderate bleeding (defined as the need for transfusion) was higher with dual therapy (2.0% vs 1.3%, P = .004).
I believe the data indicate that if a patient can tolerate dual antiplatelet therapy for 9 to 12 months without any bleeding issues, he or she is unlikely to have a major bleeding episode if dual therapy is continued beyond this time.
About half of bleeding events in patients on chronic antiplatelet therapy are gastrointestinal. To address this risk, in 2008 an expert committee from the American College of Cardiology, American College of Gastroenterology, and American Heart Association issued a consensus document26 in which they recommended assessing gastrointestinal risk factors in patients on antiplatelet therapy, such as history of ulcers (and testing for and treating Helicobacter pylori infection if present), history of gastrointestinal bleeding, concomitant anticoagulant therapy, and dual antiplatelet therapy. If any of these were present, the committee recommended considering a proton pump inhibitor. The committee also recommended a proton pump inhibitor for patients on antiplatelet therapy who have more than one of the following: age 60 years or more, corticosteroid use, or dyspepsia or gastroesophageal reflux symptoms.
Some ex vivo platelet studies and observational analyses have suggested that there might be an adverse interaction between clopidogrel and proton pump inhibitors due to a blunting of clopidogrel’s antiplatelet effect. A large randomized clinical trial was designed and launched to determine if a single-pill combination of the proton pump inhibitor omeprazole (Prilosec) and clopidogrel would be safer than clopidogrel alone when added to aspirin. Called COGENT-1 (Clopidogrel and the Optimization of GI Events Trial), it was halted early in 2009 when it lost its funding. However, preliminary data did not show an adverse interaction between clopidogrel and omeprazole.
What is the right dose of aspirin?
Steinhubl et al27 performed a post hoc observational analysis of data from the CHARISMA trial. Their findings suggested that higher doses of aspirin are not more effective than lower doses for chronic therapy. Furthermore, in the group receiving clopidogrel plus aspirin, the incidence of severe or life-threatening bleeding was significantly greater with aspirin doses higher than 100 mg than with doses lower than 100 mg, 2.6% vs 1.7%, P = .040.
A randomized, controlled trial called Clopidogrel Optimal Loading Dose Usage to Reduce Recurrent Events/Optimal Antiplatelet Strategy for Interventions (CURRENT/OASIS 7)28 recently reported that higher-dose aspirin (ie, 325 mg) may be better than lower dose aspirin (ie, 81 mg) in patients with acute coronary syndromes undergoing percutaneous coronary intervention and receiving clopidogrel. During this 30-day study, there was no increase in overall bleeding with the higher dose of aspirin, though gastrointestinal bleeding was slightly increased.29 In a factorial design, the second part of this trial found that a higher-dose clopidogrel regimen reduced stent thrombosis.29
Should nonresponders get higher doses of clopidogrel?
In vitro, response to clopidogrel shows a normal bell-shaped distribution.30 In theory, therefore, patients who are hyperresponders may be at higher risk of bleeding, and those who are hyporesponders may be at risk of ischemic events.
A clinical trial is under way to examine whether hyporesponders should get higher doses. Called GRAVITAS (Gauging Responsiveness With a VerifyNow Assay Impact on Thrombosis and Safety), it will use a point-of-care platelet assay and then allocate patients to receive either standard therapy or double the dose of clopidogrel. The primary end point will be the rate of cardiovascular death, nonfatal MI, or stent thrombosis at 6 months.
Is prasugrel better than clopidogrel?
Prasugrel (Effient) is a new drug of the same class as clopidogrel, ie, a thienopyridine, with its active metabolite binding to the same platelet receptor as clopidogrel and inhibiting platelet aggregation more rapidly, more consistently, and to a greater extent than clopidogrel. Prasugrel was recently approved by the Food and Drug Administration. But is it better?31
The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel–Thrombolysis in Myocardial Infarction (TRITON-TIMI 38) compared prasugrel and clopidogrel in 13,608 patients with moderate- to high-risk acute coronary syndromes who were scheduled to undergo percutaneous coronary intervention.32
Overall, prasugrel was better. At 15 months, the incidence of the primary end point (death from cardiovascular causes, nonfatal MI, or nonfatal stroke) was significantly lower with prasugrel therapy than with clopidogrel in the entire cohort (9.9% vs 12.1%, hazard ratio 0.81, 95% CI 0.73–0.90, P < .001), in the subgroup with ST-segment elevation MI, and in the subgroup with unstable angina or non-ST-elevation MI.
However, there was a price to pay. The rate of major bleeding was higher with prasugrel (2.4% vs 1.8%, hazard ratio 1.32, 95% CI 1.03–1.68, P = .03). Assessing the balance between the risk and the benefit, the investigators identified three subgroups who did not derive a net clinical benefit from prasugrel: patients who had had a previous stroke or transient ischemic attack (this group actually had a net harm from prasugrel), patients 75 years of age or older, and patients weighing less than 60 kg (132 pounds).
More work is needed to determine which patients are best served by standard-dose clopidogrel, higher doses of clopidogrel, platelet-assay-guided dosing of clopidogrel, or prasugrel.24
Short-acting, potent intravenous platelet blockade with an agent such as cangrelor is theoretically appealing, but further research is necessary.33,34 Ticagrelor, a reversible adenosine diphosphate receptor antagonist, provides yet another potential option in antiplatelet therapy for acute coronary syndromes. In the recent PLATO trial (Study of Platelet Inhibition and Patient Outcomes), compared with clopidogrel, ticagrelor reduced the risk of ischemic events, including death.35,36 Here, too, there was more major bleeding (unrelated to coronary artery bypass grafting) with ticagrelor.
Thus, clinical assessment of an individual patient’s ischemic and bleeding risks will continue to be critical as therapeutic strategies evolve.
- Wiviott SD, Morrow DA, Giugliano RP, et al. Performance of the Thrombolysis In Myocardial Infarction risk index for early acute coronary syndrome in the National Registry of Myocardial Infarction: a simple risk index predicts mortality in both ST and non-ST elevation myocardial infarction [abstract]. J Am Coll Cardiol 2003; 43( suppl 2):365A–366A.
- Thom T, Haase N, Rosamond W, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2006; 113:e85–e151. Errata in Circulation 2006; 113:e696 and Circulation 2006 114:e630.
- Bhatt DL. To cath or not to cath. That is no longer the question. JAMA 2005; 293:2935–2937.
- Mehta SR, Cannon CP, Fox KA, et al. Routine vs selective invasive strategies in patients with acute coronary syndromes: a collaborative meta-analysis of randomized trials. JAMA 2005; 293:2908–2917.
- Bhatt DL, Roe MT, Peterson ED, et al; for the CRUSADE Investigators. Utilization of early invasive management strategies for high-risk patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE Quality Improvement Initiative. JAMA 2004; 292:2096–2104.
- Bavry AA, Kumbhani DJ, Rassi AN, Bhatt DL, Askari AT. Benefit of early invasive therapy in acute coronary syndromes: a meta-analysis of contemporary randomized clinical trials. J Am Coll Cardiol 2006; 48:1319–1325.
- O’Donoghue MO, Boden WE, Braunwald E, et al. Early invasive vs conservative treatment strategies in women and men with unstable angina and non-ST segment elevation myocardial infarction: a meta-analysis. JAMA 2008; 300:71–80.
- Mehta SR, Granger CB, Boden WE, et al; TIMACS Investigators. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med 2009; 360:2165–2175.
- Shishehbor MH, Lauer MS, Singh IM, et al. In unstable angina or non-ST-segment acute coronary syndrome, should patients with multivessel coronary artery disease undergo multivessel or culpritonly stenting? J Am Coll Cardiol 2007; 49:849–854.
- Stone GW, Moses JW, Ellis SG, et al. Safety and efficacy of sirolimus- and paclitaxel-eluting coronary stents. N Engl J Med 2007; 356:998–1008.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Chen MS, John JM, Chew DP, Lee DS, Ellis SG, Bhatt DL. Bare metal stent restenosis is not a benign clinical entity. Am Heart J 2006; 151:1260–1264.
- Doyle B, Rihal CS, O’Sullivan CJ, et al. Outcomes of stent thrombosis and restenosis during extended follow-up of patients treated with bare-metal coronary stents. Circulation 2007; 116:2391–2398.
- Sarkees ML, Bavry AA, Galla JM, Bhatt DL. Bare metal stent thrombosis 13 years after implantation. Cardiovasc Revasc Med 2009; 10:58–91.
- Bavry AA, Bhatt DL. Appropriate use of drug-eluting stents: balancing the reduction in restenosis with the concern of late thrombosis. Lancet 2008; 371:2134–2143.
- Bavry AA, Bhatt DL. Drug-eluting stents: dual antiplatelet therapy for every survivor? Circulation 2007; 116:696–699.
- Meadows TA, Bhatt DL. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 2007; 100:1261–1275.
- Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov 2003; 2:15–28.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502. Errata in N Engl J Med 2001; 345:1506 and N Engl J Med 2001; 345:1716.
- Mehta SR, Yusuf S, Peters RJ, et al; Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- Anderson JL, Adams CD, Antman EM, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction); american College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons; American Association of Cardiovascular and Pulmonary Rehabilitation; Society for Academic Emergency Medicine. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol 2007; 50:e1–e157.
- Ho PM, Peterson ED, Wang L, et al. Incidence of death and acute myocardial infarction associated with stopping clopidogrel after acute coronary syndrome. JAMA 2008; 299:532–539. Erratum in JAMA 2008; 299:2390.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bhatt DL. Intensifying platelet inhibition—navigating between Scylla and Charybdis. N Engl J Med 2007; 357:2078–2081.
- Bhatt DL, Scheiman J, Abraham NS, et al; American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation 2008; 118:1894–1909.
- Steinhubl SR, Bhatt DL, Brennan DM, et al; CHARISMA Investigators. Aspirin to prevent cardiovascular disease: the association of aspirin dose and clopidogrel with thrombosis and bleeding. Ann Intern Med 2009; 150:379–386.
- Mehta SR, Bassand JP, Chrolavicius S, et al; CURRENT-OASIS 7 Steering Committee. Design and rationale of CURRENT-OASIS 7: a randomized, 2 x 2 factorial trial evaluating optimal dosing strategies for clopidogrel and aspirin in patients with ST and non-ST-elevation acute coronary syndromes managed with an early invasive strategy. Am Heart J 2008; 156:1080–1088.
- Mehta SR, Van de Werf F. A randomized comparison of a clopidogrel high loading and maintenance dose regimen versus standard dose and high versus low dose aspirin in 25,000 patients with acute coronary syndromes: results of the CURRENT OASIS 7 trial. Paper presented at the European Society of Cardiology Congress; August 30, 2009; Barcelona, Spain. Also available online at www.Escardio.org/congresses/esc-2009/congress-reports. Accessed December 12, 2009.
- Serebruany VL, Steinhubl SR, Berger PB, Malinin AT, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45:246–251.
- Bhatt DL. Prasugrel in clinical practice [perspective]. N Engl J Med 2009; 361:940–942.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Bhatt DL, Lincoff AM, Gibson CM, et al; for the CHAMPION PLATFORM Investigators. Intravenous platelet blockade with cangrelor during PCI. N Engl J Med 2009 Nov 15(epub ahead of print).
- Harrington RA, Stone GW, McNulty S, et al. Platelet inhibition with cangrelor in patient sundergoing PCI. N Engl J Med 2009 Nov 17(epub ahead of print).
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Bhatt DL. Ticagrelor in ACS—what does PLATO teach us? Nat Rev Cardiol 2009; 6:737–738.
- Wiviott SD, Morrow DA, Giugliano RP, et al. Performance of the Thrombolysis In Myocardial Infarction risk index for early acute coronary syndrome in the National Registry of Myocardial Infarction: a simple risk index predicts mortality in both ST and non-ST elevation myocardial infarction [abstract]. J Am Coll Cardiol 2003; 43( suppl 2):365A–366A.
- Thom T, Haase N, Rosamond W, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2006; 113:e85–e151. Errata in Circulation 2006; 113:e696 and Circulation 2006 114:e630.
- Bhatt DL. To cath or not to cath. That is no longer the question. JAMA 2005; 293:2935–2937.
- Mehta SR, Cannon CP, Fox KA, et al. Routine vs selective invasive strategies in patients with acute coronary syndromes: a collaborative meta-analysis of randomized trials. JAMA 2005; 293:2908–2917.
- Bhatt DL, Roe MT, Peterson ED, et al; for the CRUSADE Investigators. Utilization of early invasive management strategies for high-risk patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE Quality Improvement Initiative. JAMA 2004; 292:2096–2104.
- Bavry AA, Kumbhani DJ, Rassi AN, Bhatt DL, Askari AT. Benefit of early invasive therapy in acute coronary syndromes: a meta-analysis of contemporary randomized clinical trials. J Am Coll Cardiol 2006; 48:1319–1325.
- O’Donoghue MO, Boden WE, Braunwald E, et al. Early invasive vs conservative treatment strategies in women and men with unstable angina and non-ST segment elevation myocardial infarction: a meta-analysis. JAMA 2008; 300:71–80.
- Mehta SR, Granger CB, Boden WE, et al; TIMACS Investigators. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med 2009; 360:2165–2175.
- Shishehbor MH, Lauer MS, Singh IM, et al. In unstable angina or non-ST-segment acute coronary syndrome, should patients with multivessel coronary artery disease undergo multivessel or culpritonly stenting? J Am Coll Cardiol 2007; 49:849–854.
- Stone GW, Moses JW, Ellis SG, et al. Safety and efficacy of sirolimus- and paclitaxel-eluting coronary stents. N Engl J Med 2007; 356:998–1008.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Chen MS, John JM, Chew DP, Lee DS, Ellis SG, Bhatt DL. Bare metal stent restenosis is not a benign clinical entity. Am Heart J 2006; 151:1260–1264.
- Doyle B, Rihal CS, O’Sullivan CJ, et al. Outcomes of stent thrombosis and restenosis during extended follow-up of patients treated with bare-metal coronary stents. Circulation 2007; 116:2391–2398.
- Sarkees ML, Bavry AA, Galla JM, Bhatt DL. Bare metal stent thrombosis 13 years after implantation. Cardiovasc Revasc Med 2009; 10:58–91.
- Bavry AA, Bhatt DL. Appropriate use of drug-eluting stents: balancing the reduction in restenosis with the concern of late thrombosis. Lancet 2008; 371:2134–2143.
- Bavry AA, Bhatt DL. Drug-eluting stents: dual antiplatelet therapy for every survivor? Circulation 2007; 116:696–699.
- Meadows TA, Bhatt DL. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 2007; 100:1261–1275.
- Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov 2003; 2:15–28.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502. Errata in N Engl J Med 2001; 345:1506 and N Engl J Med 2001; 345:1716.
- Mehta SR, Yusuf S, Peters RJ, et al; Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- Anderson JL, Adams CD, Antman EM, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction); american College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons; American Association of Cardiovascular and Pulmonary Rehabilitation; Society for Academic Emergency Medicine. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol 2007; 50:e1–e157.
- Ho PM, Peterson ED, Wang L, et al. Incidence of death and acute myocardial infarction associated with stopping clopidogrel after acute coronary syndrome. JAMA 2008; 299:532–539. Erratum in JAMA 2008; 299:2390.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bhatt DL. Intensifying platelet inhibition—navigating between Scylla and Charybdis. N Engl J Med 2007; 357:2078–2081.
- Bhatt DL, Scheiman J, Abraham NS, et al; American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation 2008; 118:1894–1909.
- Steinhubl SR, Bhatt DL, Brennan DM, et al; CHARISMA Investigators. Aspirin to prevent cardiovascular disease: the association of aspirin dose and clopidogrel with thrombosis and bleeding. Ann Intern Med 2009; 150:379–386.
- Mehta SR, Bassand JP, Chrolavicius S, et al; CURRENT-OASIS 7 Steering Committee. Design and rationale of CURRENT-OASIS 7: a randomized, 2 x 2 factorial trial evaluating optimal dosing strategies for clopidogrel and aspirin in patients with ST and non-ST-elevation acute coronary syndromes managed with an early invasive strategy. Am Heart J 2008; 156:1080–1088.
- Mehta SR, Van de Werf F. A randomized comparison of a clopidogrel high loading and maintenance dose regimen versus standard dose and high versus low dose aspirin in 25,000 patients with acute coronary syndromes: results of the CURRENT OASIS 7 trial. Paper presented at the European Society of Cardiology Congress; August 30, 2009; Barcelona, Spain. Also available online at www.Escardio.org/congresses/esc-2009/congress-reports. Accessed December 12, 2009.
- Serebruany VL, Steinhubl SR, Berger PB, Malinin AT, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45:246–251.
- Bhatt DL. Prasugrel in clinical practice [perspective]. N Engl J Med 2009; 361:940–942.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Bhatt DL, Lincoff AM, Gibson CM, et al; for the CHAMPION PLATFORM Investigators. Intravenous platelet blockade with cangrelor during PCI. N Engl J Med 2009 Nov 15(epub ahead of print).
- Harrington RA, Stone GW, McNulty S, et al. Platelet inhibition with cangrelor in patient sundergoing PCI. N Engl J Med 2009 Nov 17(epub ahead of print).
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Bhatt DL. Ticagrelor in ACS—what does PLATO teach us? Nat Rev Cardiol 2009; 6:737–738.
KEY POINTS
- The data favor an aggressive strategy of routine catheterization, rather than a conservative strategy of catheterization only if a patient develops recurrent, spontaneous, or stress-induced ischemia.
- Early percutaneous intervention (within 24 hours) may be beneficial in patients at higher risk, but not necessarily in those at lower risk.
- Drug-eluting stents appear safe, assuming dual antiplatelet therapy is used. It is unclear how long this therapy needs to be continued.
- The choice of revascularization strategy—bypass surgery, bare-metal stent, or drug-eluting stent—should be individualized based on the risk of restenosis, thrombosis, and other factors.
Acetaminophen: Old drug, new warnings
Editor’s note: Portions of this article are based on an article previously published in an internal Cleveland Clinic publication, Pharmacotherapy Update. The version here has been revised, updated, and peer-reviewed.
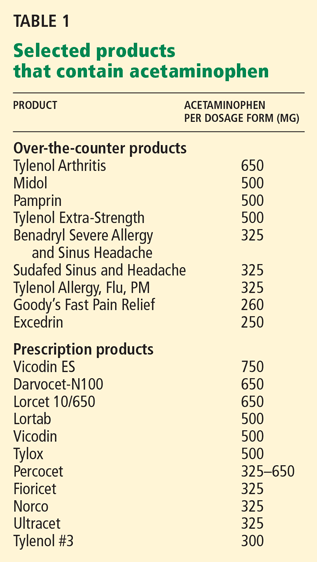
This drug is generally considered safe, but high doses can be toxic. The number of overdoses is worrisome. In 2006 alone, the American Association of Poison Control Centers implicated acetaminophen in nearly 140,000 poisoning cases, in which more than 100 patients died.2 It is responsible for more emergency room visits than any other drug on the market.
According to a position statement from the American Association for the Study of Liver Diseases (AASLD),3 the incidence of acetaminophen-related liver toxicity has been steadily increasing over the past decade, and this drug is now the most common cause of acute liver failure.
MANY OVERDOSES ARE UNINTENTIONAL
Cases of acetaminophen-related liver toxicity can be categorized as either intentional (ie, due to a suicide attempt) or unintentional (ie, due to multiple therapeutic but excessive doses over a period of time, usually more than 3 days).
Up to 50% of cases are unintentional. Bower et al4 reviewed cases of acute liver failure that occurred in the Atlanta, GA, area between November 2000 and October 2004. Acetaminophen was the most common cause in adult patients. Of greater concern is that 61% of the acetaminophen-related cases were due to unintentional overdose. According to the Institute for Safe Medication Practices,5 one hospital (not named) reported that an average of one patient per day was given more than the recommended maximum daily acetaminophen dose of 4 g while in the hospital.
Many patients take more than one acetaminophen product
Unintentional overdoses or “therapeutic misadventures” are most often due to taking multiple products that contain acetaminophen, taking acetaminophen-narcotic combinations, and impulsive behavior involving a lack of understanding of possible injury in consuming multiple acetaminophen-containing products.3
In the US Food and Drug Administration (FDA) Medwatch Database, in 307 cases of unintentional acetaminophen overdose between 1998 and 2001, 25% of patients had been taking more than one acetaminophencontaining product.5
Larson et al6 found that one-third of patients who had had an unintentional acetaminophen overdose were taking an acetaminophen-narcotic combination in addition to another acetaminophen-containing product.
Many consumers don’t know they are taking acetaminophen
Many consumers don’t know that some of the drugs they take contain acetaminophen. This may be because many drug labels contain abbreviations for acetaminophen such as “APAP” or have inconsistent formatting that makes it difficult to determine if the product contains acetaminophen.
Others may not be aware of the total maximum recommended daily dose or may not be able to calculate the total daily intake from the information on the label. The problem is not only with over-the-counter products. For example, if a physician prescribes one or two tablets of hydrocodone/acetaminophen (Vicodin) 5 mg/500 mg every 4 to 6 hours, a patient could easily exceed the recommended maximum daily dose of 4 g of acetaminophen.
Toxicity can occur even at therapeutic doses
Acetaminophen hepatotoxicity can also occur even with therapeutic doses in certain conditions. Risk factors:
- Chronic alcohol use (ie, more than three drinks per day)
- Malnutrition
- Concurrent use of drugs that induce cytochrome P450 (CYP450) enzymes (more on this below).6
FIRST USED IN 1893
Acetaminophen was first used in medicine in 1893, and it became widely used after 1949, when it was found to be a less-toxic metabolite of two parent compounds, acetanilide and phenacetin.1
Acetaminophen is an effective antipyretic and analgesic, but its anti-inflammatory properties are minimal, especially compared with nonsteroidal anti-inflammatory drugs (NSAIDs). Nevertheless, acetaminophen is preferred over NSAIDs in some patients because it carries a lower risk of gastrointestinal toxicity (eg, ulceration, bleeding) and so may be better tolerated.1
INDICATIONS AND DOSAGE
Acetaminophen is indicated for mild to moderate pain or fever, including the pain of osteoarthritis. It is not recommended for chronic inflammatory conditions such as rheumatoid arthritis, since it lacks anti-inflammatory properties.
In adults and in children over age 12, the usual dosage is 325 to 650 mg orally or rectally every 4 to 6 hours, or 1,000 mg three to four times daily.
The current package label recommends that the total daily dose not exceed 4 g in most adults. Lower maximum daily doses (eg, 2 g) are recommended in patients who may be at higher risk of hepatotoxicity, such as those who drink heavily, are malnourished, or take enzyme-inducing drugs. Tylenol products currently include an alcohol warning, advising those who consume three or more alcoholic drinks a day to ask their doctor if they should take acetaminophen.
In children up to 12 years of age, the recommended dosage is 10 to 15 mg/kg orally or rectally every 4 to 6 hours. The maximum dosing for children in this age group should not exceed five doses (or 50 to 75 mg/kg) in 24 hours.7 In children under age 2 or weighing less than 11 kg, acetaminophen should only be used under the direction of a physician.
MOST ACETAMINOPHEN IS CONJUGATED AND THEN EXCRETED IN THE URINE
Acetaminophen usually has excellent bioavailability (up to 98%), but the exact amount absorbed varies, depending on the dosage form and concomitant use of other drugs.8
At therapeutic doses, the elimination halflife is about 2 hours. Peak plasma concentrations are reached 30 to 60 minutes after the dose is taken,1 but taking acetaminophen with opioids, anticholinergic drugs, or even food may delay the time to peak concentration by delaying gastric emptying.8
NAPQI is a toxic metabolite

Under normal circumstances, a small amount of acetaminophen undergoes hepatic metabolism by a different pathway, ie, by CYP450 enzymes, primarily CYP2E1 and to a lesser extent CYP1A2, CYP2A6, and CYP3A4, forming a toxic metabolite, N-acetyl-p-benzoquinone imine (NAPQI). Then, the sulfhydryl groups of glutathione convert NAPQI into harmless metabolites that are excreted in the urine.1,7
Well tolerated and relatively safe
At recommended doses, acetaminophen is well tolerated, and it is considered relatively safe when used according to labeling instructions.
Rarely, patients experience an erythematous or urticarial rash or other allergic complications.1
However, acetaminophen is a dose-dependent hepatotoxin, and excessive doses (intentional or unintentional) may lead to acute liver failure. In addition, even in therapeutic doses, acetaminophen may still cause transient liver enzyme elevations and possibly hepatotoxicity, particularly in people who are malnourished or alcoholic or are taking certain CYP450-inducing drugs.3,6,10
HOW ACETAMINOPHEN CAN INJURE THE LIVER
Glucuronidation and sulfation, the major metabolic pathways, become saturated after an acetaminophen overdose.7 When this happens, more of the toxic metabolite NAPQI is formed by CYP450-mediated N-hydroxylation. When glutathione is depleted after large doses of acetaminophen or in malnourished people, the toxic metabolite accumulates, resulting in liver damage (Figure 1).6
The liver is damaged by two mechanisms. In one, NAPQI binds to hepatic cell macromolecules, causing dysfunction of the enzymatic systems, structural and metabolic disarray, and eventually necrotic cell death. The other mechanism is oxidative stress due to depletion of glutathione.
In children, single acetaminophen doses of 120 to 150 mg/kg of body weight have been associated with hepatotoxicity,11 as have single doses of more than 150 mg/kg or a total dose of greater than 7.5 g in adults. However, the minimal dose associated with liver injury has ranged from 4 to 10 g, and in healthy volunteers even therapeutic doses of 1 g orally every 6 hours resulted in mild liver injury.12
Patients who are malnourished or fasting are thought to be at greater risk of acetaminophen hepatotoxicity because they may be deficient in glutathione at baseline. In addition, even at lower-than-therapeutic doses, induction of CYP450 enzymes by drugs or chronic alcohol consumption may lead to an increase in the formation of NAPQI, increasing the risk of hepatotoxicity. Examples of drugs that induce CYP450 enzymes to produce more NAPQI include the anticonvuslants phenytoin (Dilantin) and phenobarbital.
CLINICAL PRESENTATION OF ACETAMINOPHEN OVERDOSE
The diagnosis of acetaminophen overdose is often established by a thorough history. The pertinent information may be difficult to obtain, however, because the patient may be confused or stuporous at presentation, may not know that the overthe-counter products he or she has been taking contain acetaminophen, or may be embarrassed about taking too much acetaminophen.13
In acute intentional overdose, signs may not be apparent immediately
The symptoms of toxicity may not be apparent immediately after ingestion of an acute overdose of acetaminophen, but early recognition and treatment can prevent more severe liver damage, decreasing morbidity and the risk of death.9
Phase 1 of an acetaminophen overdose begins shortly after ingestion and can last for 12 to 24 hours. Patients may have signs of gastrointestinal upset, nausea, vomiting, anorexia, diaphoresis, and pallor. Although the signs and symptoms show a consistent pattern and are more pronounced after larger acute overdoses, they are not diagnostic or specific.
Phase 2 (up to 48 hours after ingestion). Patients may begin to feel better during this phase. However, the hepatic enzyme levels, the prothrombin time (PT), and the international normalized ratio (INR) may continue to rise, and right upper quadrant pain may develop. Additionally, other laboratory results may be abnormal, and renal insufficiency can occur due to acetaminophen-induced renal tubular necrosis.6 Most patients receive the antidote, acetylcysteine (Mycomyst, Acetadote) before or during this phase, and consequently, liver function gradually returns to normal.
Phase 3, if reached, may be marked by severe hepatic necrosis, typically 3 to 5 days after ingestion. Symptoms during this phase range from less severe (eg, nausea and general malaise) to more severe (eg, confusion and stupor). Also, at this time, liver enzyme levels can be as high as 10,000 IU/L or even higher, and lactic acidosis and coagulopathy may worsen. If death should occur, it is most likely from complications associated with fulminant hepatic failure, including cerebral edema, multiorgan-system failure, or sepsis.6
Phase 4. Patients who recover generally have complete recovery of liver function with no long-term sequelae..
In unintentional overdoses, patients may have low drug levels
Many patients who present with unintentional acetaminophen toxicity have been taking the drug or products that contain the drug over several days to treat an acute or chronic medical condition. They often have low or undetectable serum acetaminophen levels after 2 to 3 days of nonspecific symptoms.12
MANAGING ACETAMINOPHENHEN OVERDOSE
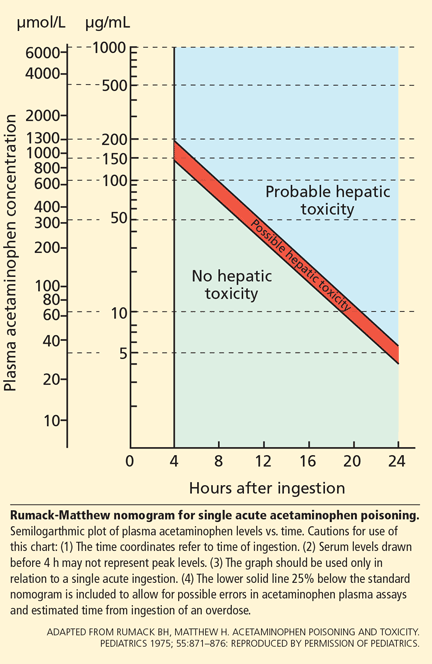
Unintentional overdoses occur over a more prolonged period, and therefore the nomogram is not useful in this situation.
Acetylcysteine is the antidote
Acetylcysteine is the antidote for acetaminophen toxicity and should be given within 8 hours of ingestion for maximal protection against hepatic injury in patients whose serum acetaminophen levels are above the “possible” toxicity line on the nomogram.15 If acetaminophen overdose is suspected but the time elapsed since ingestion cannot be determined, acetylcysteine should be given immediately regardless of the quantity of acetaminophen ingested. 16 In cases of unintentional overdose, it is often given at the discretion of the physician.
Acetylcysteine limits the toxicity of acetaminophen by increasing glutathione stores, binding with NAPQI as a substitute for glutathione, and enhancing sulfate conjugation.6 It may further limit acetaminophen toxicity by nonspecific mechanisms including anti-inflammatory, antioxidant, inotropic, and vasodilating effects.6 In addition, it may prevent further hepatic damage in any patient thought to have acetaminophen-related liver toxicity even beyond the first 12 hours of an overdose.1
Acetylcysteine is available in an oral (Mucomyst) and an intravenous (Acetadote) formulation, which are similar in efficacy. Because many patients find the taste of the oral solution unpleasant and difficult to tolerate, it should be diluted in a 1:3 ratio with cola, orange juice, or other drink to mask its flavor. This mixture should be used within 1 hour of preparation.
Anaphylactoid reactions have occurred in patients receiving intravenous acetylcysteine for acetaminophen overdose soon after the infusion was started, most commonly during the loading dose. The frequency of infusion-related reactions has been reported to be 0.2% to 20.8%.15
The recommended dosage for intravenous acetylcysteine is a loading dose of 150 mg/kg given over 60 minutes, followed by a second dose of 50 mg/kg given over 4 hours, and finally a third dose of 100 mg/kg given over 16 hours.15 For oral acetylcysteine, the loading dose is 140 mg/kg followed by 70 mg/kg every 4 hours for 17 additional doses.17
Other measures
Activated charcoal can be used if the patient presents within 1 to 2 hours after taking acetaminophen. However, the rapid gastrointestinal absorption of acetaminophen makes this treatment ineffective in most cases.9
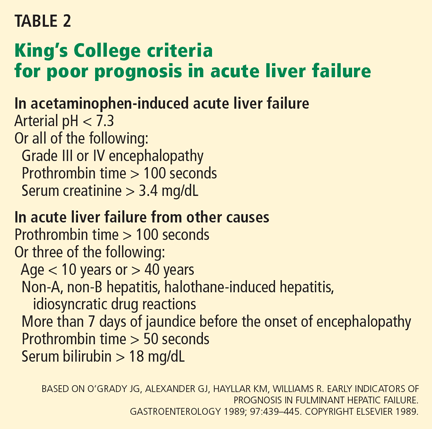
FINDINGS FROM LARGE REGISTRIES
Findings in adults
Ostapowicz et al,19 as part of the Acute Liver Failure Study Group, in 2002 prospectively characterized the short-term outcomes of acute liver failure in a large number of patients at 17 tertiary care centers in the United States over approximately 41 months. All centers except one performed liver transplants. Eligible patients had to meet criteria for acute liver failure, including an INR higher than 1.5, evidence of hepatic encephalopathy, and presentation within 26 weeks of illness onset without apparent chronic liver disease.
Of the 308 cases of acute liver failure, 120 (39%) were from acetaminophen overdose, making this drug the most common cause of acute liver failure. Forty-four (37%) of the patients with acetaminophen-related acute liver failure were trying to commit suicide, 57% of cases were accidental, and the remaining reasons for acetaminophen overdose were unknown. The median amount ingested was 13.2 g/day (range 2.6–75 g), and 99 (83%) of the 120 patients took more than 4 g/day.
The patients with acetaminophen-related acute liver toxicity differed from those with other causes of acute liver failure such as idiosyncratic drug reactions or indeterminate causes. The acetaminophen group had a shorter duration of disease and higher serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and serum creatinine than those with acute liver failure from other causes. They also had lower bilirubin levels and a lower arterial pH.
Rates of liver transplantation were 6% in the acetaminophen group, 53% in the group with other drug-induced liver toxicity, 51% in the indeterminate group, and 36% in the remaining groups. Of the acetaminophen group, 47% met the criteria for transplantation, but only 57% of those eligible were listed for transplantation. Those excluded from the transplant list had medical contraindications or were excluded for psychosocial reasons. The short-term transplant-free survival rate was 68% in the acetaminophen group.
Overall, 11% of the patients died, including 28% of the acetaminophen group. The authors concluded that most cases of liver injury in the United States are due to medications and may be preventable.
Larson et al,20 as part of the Acute Liver Failure Study Group, examined the incidence, risk factors, and outcomes of acetaminopheninduced acute liver failure at 22 tertiary care centers in the United States over a 6-year period. Of the 662 patients in the study, 302 had acetaminophen-related toxicity and 275 were included in the final analysis; some of them had been included in the study by Ostapowicz et al.19 During the study period, the number of cases of acute liver toxicity related to acetaminophen increased from 28% to 51%.
Of the patients enrolled in the study, 56% met the criteria for potentially toxic acetaminophen ingestion. Of those who met the criteria, 77% had detectable acetaminophen levels in their serum, and 91% had ALT levels higher than 1,000 IU/L. The time between ingestion and symptom onset ranged from 1 to 32 days, and the median dose was 24 g (range 1.2–180 g).
Of the overdose cases, 48% were unintentional, 44% were intentional, and the remaining 8% had no definable reason. Of the patients in the unintentional-overdose group, 38% were using more than one acetaminophen-containing product and 63% were taking combination products containing narcotics. Overeall, 44% of patients reported using narcotic-acetaminophen combination products. The unintentional-overdose group had lower serum acetaminophen levels than the intentional-overdose group, but they were more likely to present with severe hepatic encephalopathy.
The number of patients with an unintentional overdose was worrisome. Furthermore, one-third of the patients who were receiving an acetaminophen-narcotic combination product were taking an additional acetaminophencontaining product. The authors concluded that unintentional overdose is the leading cause of acetaminophen-related hepatotoxicity, and efforts to limit the over-the-counter package size and to restrict prescriptions of acetaminophen-narcotic combinations may be necessary to decrease the incidence of this preventable cause of acute liver failure.
Findings in children
Squires et al21 and the Pediatric Acute Liver Failure Study Group examined the pathogenesis, treatment, and outcome of acute liver failure in children (any age from birth to 18 years) with no previous evidence of chronic liver disease, evidence of acute liver injury, or hepatic-based coagulopathy. From December 1999 to December 2004, 348 patients were enrolled.
Fourteen percent of the cases were due to acetaminophen. The median dose of acetaminophen ingested was 183 mg/kg. Most of these patients were white and female, and 96% were over age 3. Hepatic encephalopathy was more common in the non-acetaminophen groups than in the acetaminophen group, although this is often difficult to assess in infants and children. Children with acetaminophen toxicity had the highest rate of spontaneous recovery: 45 (94%) of 48 recovered.
Although there are fewer acetaminophenrelated cases of acute liver failure in children than in adults, the use of acetaminophen in children is still worrisome. The authors concluded that if they do not have hepatic encephalopathy, children with acetaminopheninduced liver toxicity have an excellent prognosis.
THE FDA LOOKS AT THE PROBLEM
Why overdoses occur
A recent FDA report22 cited the following reasons for acetaminophen overdose:
- Some patients may experience liver injury at doses only slightly above the recommended 4-g daily limit.
- Some patients may be more prone to liver injury from acetaminophen.
- The symptoms associated with liver injury due to acetaminophen can be nonspecific and tend to evolve over several days.
- Many acetaminophen-containing products are available, including over-the-counter and prescription drugs with many different strengths and indications.
- Consumers are unaware of the risk of liver toxicity with acetaminophen.
- Prescription products are not always clearly labeled with acetaminophen as an ingredient.
- Pediatric dosage forms are available in many different concentrations.
New package labeling

Recommendations from an advisory panel
In addition, an FDA advisory panel recommended decreasing the maximum recommended daily dose (possibly to 3,250 mg/day in adults, although this is not final) to help prevent overdoses, and reducing the maximum amount in a single nonprescription dose of the drug to 650 mg.23 The panel also voted (by a narrow margin) to ban all acetaminophen-narcotic combination products.
As of this writing, the FDA has not adopted these recommendations. (Although the FDA is not obliged to follow the advice of its advisory panels, in most cases it does.) The recommendations about acetaminophen could take years to implement fully.
THE UNITED KINGDOM ACTED IN 1998
In response to a rising number of analgesicrelated deaths, the United Kingdom enacted legislation in 1998 to limit the package size available to consumers.24 Packages sold in general stores can contain no more than 16 capsules, while those sold in pharmacies can contain 24. Blister packs were also introduced to make it harder for people to impulsively take handfuls of tablets.25
Two studies since then both found that the number of deaths related to paracetamol (as acetaminophen is known in the United Kingdom) had fallen since the legislation was implemented.24,26 Greene et al27 found that patients who ingested potentially toxic doses of paracetamol had obtained the drug “in a manner contravening the 1998 legislation.”27 In other words, shops in London were not obeying the law.
SUMMARY
The new labeling and the proposed changes are sensible and draw needed attention to the problem of acetaminophen toxicity. To prevent unintentional acetaminophen overdoses, education of patients and health care professionals is urgently needed so that the dangers of consuming excess acetaminophen daily are understood.
- Burke A, Smyth EM, Fitzgerald GA. Analgesic-antipyretic agents: pharmacotherapy of gout. In:Brunton LL, Lazo JS, Parker K, editors. Goodman and Gilman’s the Pharmacological Basis of Therapeutics, 11th ed. New York: McGraw-Hill, 2006:671–716.
- Bronstein AC, Spyker DA, Cantilena LR, Green J, Rumack BH, Heard SE. 2006 Annual Report of the American Association of Poison Control Centers National Poison Data System (NPDS). Clin Toxicol (Phila) 2007; 45:815–917.
- Schwartz J, Stravitz T, Lee WM; American Association for the Study of Liver Disease Study Group. AASLD position on acetaminophen. www.aasld.org/about/publicpolicy/Documents/Public%2520Policy%2520Documents/AcetaminophenPosition.pdf.
- Bower WA, Johns M, Margolis HS, Williams IT, Bell BP. Populationbased surveillance for acute liver failure. Am J Gastroenterol 2007; 102:2459–2463.
- Institute for Safe Medication Practices. How are you preventing acetaminophen overdoses? www.ismp.org/newsletters/acutecare/articles/20030808.asp. Accessed 11/17/2009.
- Larson AM. Acetaminophen hepatotoxicity. Clin Liver Dis 2007; 11:525–548.
- Tylenol package insert. Fort Washington, PA: McNeil-PPC Inc.; 1999.
- Bizovi KE, Hendrickson RG. Chapter 34. Acetaminophen. In:Hoffman RS, Nelson LS, Howland MA, Lewin NA, Flomenbaum NE, Goldfrank LR, editors. Goldfrank’s Manual of Toxicologic Emergencies. 3rd ed. McGraw-Hill: New York, 2007. www.accessemergencymedicine.com/content.aspx?aID=88781. Accessed 11/7/2009.
- Hung OL, Nelson LS. Chapter 171. Acetaminophen. In:Tintinalli JE, Kelen GD, Stapcynski S, editors. Tintinalli's Emergency Medicine: A Comprehensive Study Guide. 6th ed. McGraw-Hill: New York, 2004. www.accessmedicine.com/content.aspx?aID=602606. Accessed 11/17/2009.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- American Academy of Pediatrics Committee on Drugs. Acetaminophen toxicity in children. Pediatrics 2001; 108:1020–1024.
- Fontana RJ. Acute liver failure including acetaminophen overdose. Med Clin North Am 2008; 92:761–794.
- Heard KJ. Acetylcysteine for acetaminophen poisoning. N Engl J Med 2008; 359:285–292.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Acetadote package insert. Nashville, TN: Cumberland Pharmaceuticals; 2008Dec.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- Product Information: acetylcysteine inhalation solution, acetylcysteine inhalation solution. Hospira,Inc, Lake Forest, IL, 2004.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Ostapowicz G, Fontana RJ, Schiødt FV, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Larson AM, Polson J, Fontana RJ, et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Squires RH, Shneider BL, Bucuvalas J, et al. Acute liver failure in children: the first 348 patients in the Pediatric Acute Liver Failure Study Group. J Pediatr 2006; 148:652–658.
- US Food and Drug Administration. Questions and answers on final rule for labeling changes to over-the-counter pain relievers. www.fda.gov/Drugs/NewsEvents/ucm144068.htm. Accessed 10/30/2009.
- Perrone M. FDA panel recommends smaller doses of painkillers. Associated Press. Adelphi, MD. June 30, 2009.
- Hawton K, Simkin S, Deeks J, et al. UK legislation on analgesic packs: before and after study of long term effect on poisonings. BMJ 2004; 329:1076.
- Hughes B, Durran A, Langford NJ, Mutimer D. Paracetamol poisoning—impact of pack size restrictions. J Clin Pharmacol Ther 2003; 28:307–310.
- Wilkinson S, Taylor G, Templeton L, Mistral W, Salter E, Bennett P. Admissions to hospital for deliberate self-harm in England 1995–2000: an analysis of hospital episode statistics. J Public Health Med 2002; 24:179–183.
- Greene SL, Dargan PI, Leman P, Jones AL. Paracetamol availability and recent changes in paracetamol poisoning: is the 1998 legislation limiting availability of paracetamol being followed? Postgrad Med J 2006; 82:520–523.
Editor’s note: Portions of this article are based on an article previously published in an internal Cleveland Clinic publication, Pharmacotherapy Update. The version here has been revised, updated, and peer-reviewed.
This drug is generally considered safe, but high doses can be toxic. The number of overdoses is worrisome. In 2006 alone, the American Association of Poison Control Centers implicated acetaminophen in nearly 140,000 poisoning cases, in which more than 100 patients died.2 It is responsible for more emergency room visits than any other drug on the market.
According to a position statement from the American Association for the Study of Liver Diseases (AASLD),3 the incidence of acetaminophen-related liver toxicity has been steadily increasing over the past decade, and this drug is now the most common cause of acute liver failure.
MANY OVERDOSES ARE UNINTENTIONAL
Cases of acetaminophen-related liver toxicity can be categorized as either intentional (ie, due to a suicide attempt) or unintentional (ie, due to multiple therapeutic but excessive doses over a period of time, usually more than 3 days).
Up to 50% of cases are unintentional. Bower et al4 reviewed cases of acute liver failure that occurred in the Atlanta, GA, area between November 2000 and October 2004. Acetaminophen was the most common cause in adult patients. Of greater concern is that 61% of the acetaminophen-related cases were due to unintentional overdose. According to the Institute for Safe Medication Practices,5 one hospital (not named) reported that an average of one patient per day was given more than the recommended maximum daily acetaminophen dose of 4 g while in the hospital.
Many patients take more than one acetaminophen product
Unintentional overdoses or “therapeutic misadventures” are most often due to taking multiple products that contain acetaminophen, taking acetaminophen-narcotic combinations, and impulsive behavior involving a lack of understanding of possible injury in consuming multiple acetaminophen-containing products.3
In the US Food and Drug Administration (FDA) Medwatch Database, in 307 cases of unintentional acetaminophen overdose between 1998 and 2001, 25% of patients had been taking more than one acetaminophencontaining product.5
Larson et al6 found that one-third of patients who had had an unintentional acetaminophen overdose were taking an acetaminophen-narcotic combination in addition to another acetaminophen-containing product.
Many consumers don’t know they are taking acetaminophen
Many consumers don’t know that some of the drugs they take contain acetaminophen. This may be because many drug labels contain abbreviations for acetaminophen such as “APAP” or have inconsistent formatting that makes it difficult to determine if the product contains acetaminophen.
Others may not be aware of the total maximum recommended daily dose or may not be able to calculate the total daily intake from the information on the label. The problem is not only with over-the-counter products. For example, if a physician prescribes one or two tablets of hydrocodone/acetaminophen (Vicodin) 5 mg/500 mg every 4 to 6 hours, a patient could easily exceed the recommended maximum daily dose of 4 g of acetaminophen.
Toxicity can occur even at therapeutic doses
Acetaminophen hepatotoxicity can also occur even with therapeutic doses in certain conditions. Risk factors:
- Chronic alcohol use (ie, more than three drinks per day)
- Malnutrition
- Concurrent use of drugs that induce cytochrome P450 (CYP450) enzymes (more on this below).6
FIRST USED IN 1893
Acetaminophen was first used in medicine in 1893, and it became widely used after 1949, when it was found to be a less-toxic metabolite of two parent compounds, acetanilide and phenacetin.1
Acetaminophen is an effective antipyretic and analgesic, but its anti-inflammatory properties are minimal, especially compared with nonsteroidal anti-inflammatory drugs (NSAIDs). Nevertheless, acetaminophen is preferred over NSAIDs in some patients because it carries a lower risk of gastrointestinal toxicity (eg, ulceration, bleeding) and so may be better tolerated.1
INDICATIONS AND DOSAGE
Acetaminophen is indicated for mild to moderate pain or fever, including the pain of osteoarthritis. It is not recommended for chronic inflammatory conditions such as rheumatoid arthritis, since it lacks anti-inflammatory properties.
In adults and in children over age 12, the usual dosage is 325 to 650 mg orally or rectally every 4 to 6 hours, or 1,000 mg three to four times daily.
The current package label recommends that the total daily dose not exceed 4 g in most adults. Lower maximum daily doses (eg, 2 g) are recommended in patients who may be at higher risk of hepatotoxicity, such as those who drink heavily, are malnourished, or take enzyme-inducing drugs. Tylenol products currently include an alcohol warning, advising those who consume three or more alcoholic drinks a day to ask their doctor if they should take acetaminophen.
In children up to 12 years of age, the recommended dosage is 10 to 15 mg/kg orally or rectally every 4 to 6 hours. The maximum dosing for children in this age group should not exceed five doses (or 50 to 75 mg/kg) in 24 hours.7 In children under age 2 or weighing less than 11 kg, acetaminophen should only be used under the direction of a physician.
MOST ACETAMINOPHEN IS CONJUGATED AND THEN EXCRETED IN THE URINE
Acetaminophen usually has excellent bioavailability (up to 98%), but the exact amount absorbed varies, depending on the dosage form and concomitant use of other drugs.8
At therapeutic doses, the elimination halflife is about 2 hours. Peak plasma concentrations are reached 30 to 60 minutes after the dose is taken,1 but taking acetaminophen with opioids, anticholinergic drugs, or even food may delay the time to peak concentration by delaying gastric emptying.8
NAPQI is a toxic metabolite
Under normal circumstances, a small amount of acetaminophen undergoes hepatic metabolism by a different pathway, ie, by CYP450 enzymes, primarily CYP2E1 and to a lesser extent CYP1A2, CYP2A6, and CYP3A4, forming a toxic metabolite, N-acetyl-p-benzoquinone imine (NAPQI). Then, the sulfhydryl groups of glutathione convert NAPQI into harmless metabolites that are excreted in the urine.1,7
Well tolerated and relatively safe
At recommended doses, acetaminophen is well tolerated, and it is considered relatively safe when used according to labeling instructions.
Rarely, patients experience an erythematous or urticarial rash or other allergic complications.1
However, acetaminophen is a dose-dependent hepatotoxin, and excessive doses (intentional or unintentional) may lead to acute liver failure. In addition, even in therapeutic doses, acetaminophen may still cause transient liver enzyme elevations and possibly hepatotoxicity, particularly in people who are malnourished or alcoholic or are taking certain CYP450-inducing drugs.3,6,10
HOW ACETAMINOPHEN CAN INJURE THE LIVER
Glucuronidation and sulfation, the major metabolic pathways, become saturated after an acetaminophen overdose.7 When this happens, more of the toxic metabolite NAPQI is formed by CYP450-mediated N-hydroxylation. When glutathione is depleted after large doses of acetaminophen or in malnourished people, the toxic metabolite accumulates, resulting in liver damage (Figure 1).6
The liver is damaged by two mechanisms. In one, NAPQI binds to hepatic cell macromolecules, causing dysfunction of the enzymatic systems, structural and metabolic disarray, and eventually necrotic cell death. The other mechanism is oxidative stress due to depletion of glutathione.
In children, single acetaminophen doses of 120 to 150 mg/kg of body weight have been associated with hepatotoxicity,11 as have single doses of more than 150 mg/kg or a total dose of greater than 7.5 g in adults. However, the minimal dose associated with liver injury has ranged from 4 to 10 g, and in healthy volunteers even therapeutic doses of 1 g orally every 6 hours resulted in mild liver injury.12
Patients who are malnourished or fasting are thought to be at greater risk of acetaminophen hepatotoxicity because they may be deficient in glutathione at baseline. In addition, even at lower-than-therapeutic doses, induction of CYP450 enzymes by drugs or chronic alcohol consumption may lead to an increase in the formation of NAPQI, increasing the risk of hepatotoxicity. Examples of drugs that induce CYP450 enzymes to produce more NAPQI include the anticonvuslants phenytoin (Dilantin) and phenobarbital.
CLINICAL PRESENTATION OF ACETAMINOPHEN OVERDOSE
The diagnosis of acetaminophen overdose is often established by a thorough history. The pertinent information may be difficult to obtain, however, because the patient may be confused or stuporous at presentation, may not know that the overthe-counter products he or she has been taking contain acetaminophen, or may be embarrassed about taking too much acetaminophen.13
In acute intentional overdose, signs may not be apparent immediately
The symptoms of toxicity may not be apparent immediately after ingestion of an acute overdose of acetaminophen, but early recognition and treatment can prevent more severe liver damage, decreasing morbidity and the risk of death.9
Phase 1 of an acetaminophen overdose begins shortly after ingestion and can last for 12 to 24 hours. Patients may have signs of gastrointestinal upset, nausea, vomiting, anorexia, diaphoresis, and pallor. Although the signs and symptoms show a consistent pattern and are more pronounced after larger acute overdoses, they are not diagnostic or specific.
Phase 2 (up to 48 hours after ingestion). Patients may begin to feel better during this phase. However, the hepatic enzyme levels, the prothrombin time (PT), and the international normalized ratio (INR) may continue to rise, and right upper quadrant pain may develop. Additionally, other laboratory results may be abnormal, and renal insufficiency can occur due to acetaminophen-induced renal tubular necrosis.6 Most patients receive the antidote, acetylcysteine (Mycomyst, Acetadote) before or during this phase, and consequently, liver function gradually returns to normal.
Phase 3, if reached, may be marked by severe hepatic necrosis, typically 3 to 5 days after ingestion. Symptoms during this phase range from less severe (eg, nausea and general malaise) to more severe (eg, confusion and stupor). Also, at this time, liver enzyme levels can be as high as 10,000 IU/L or even higher, and lactic acidosis and coagulopathy may worsen. If death should occur, it is most likely from complications associated with fulminant hepatic failure, including cerebral edema, multiorgan-system failure, or sepsis.6
Phase 4. Patients who recover generally have complete recovery of liver function with no long-term sequelae..
In unintentional overdoses, patients may have low drug levels
Many patients who present with unintentional acetaminophen toxicity have been taking the drug or products that contain the drug over several days to treat an acute or chronic medical condition. They often have low or undetectable serum acetaminophen levels after 2 to 3 days of nonspecific symptoms.12
MANAGING ACETAMINOPHENHEN OVERDOSE
Unintentional overdoses occur over a more prolonged period, and therefore the nomogram is not useful in this situation.
Acetylcysteine is the antidote
Acetylcysteine is the antidote for acetaminophen toxicity and should be given within 8 hours of ingestion for maximal protection against hepatic injury in patients whose serum acetaminophen levels are above the “possible” toxicity line on the nomogram.15 If acetaminophen overdose is suspected but the time elapsed since ingestion cannot be determined, acetylcysteine should be given immediately regardless of the quantity of acetaminophen ingested. 16 In cases of unintentional overdose, it is often given at the discretion of the physician.
Acetylcysteine limits the toxicity of acetaminophen by increasing glutathione stores, binding with NAPQI as a substitute for glutathione, and enhancing sulfate conjugation.6 It may further limit acetaminophen toxicity by nonspecific mechanisms including anti-inflammatory, antioxidant, inotropic, and vasodilating effects.6 In addition, it may prevent further hepatic damage in any patient thought to have acetaminophen-related liver toxicity even beyond the first 12 hours of an overdose.1
Acetylcysteine is available in an oral (Mucomyst) and an intravenous (Acetadote) formulation, which are similar in efficacy. Because many patients find the taste of the oral solution unpleasant and difficult to tolerate, it should be diluted in a 1:3 ratio with cola, orange juice, or other drink to mask its flavor. This mixture should be used within 1 hour of preparation.
Anaphylactoid reactions have occurred in patients receiving intravenous acetylcysteine for acetaminophen overdose soon after the infusion was started, most commonly during the loading dose. The frequency of infusion-related reactions has been reported to be 0.2% to 20.8%.15
The recommended dosage for intravenous acetylcysteine is a loading dose of 150 mg/kg given over 60 minutes, followed by a second dose of 50 mg/kg given over 4 hours, and finally a third dose of 100 mg/kg given over 16 hours.15 For oral acetylcysteine, the loading dose is 140 mg/kg followed by 70 mg/kg every 4 hours for 17 additional doses.17
Other measures
Activated charcoal can be used if the patient presents within 1 to 2 hours after taking acetaminophen. However, the rapid gastrointestinal absorption of acetaminophen makes this treatment ineffective in most cases.9
FINDINGS FROM LARGE REGISTRIES
Findings in adults
Ostapowicz et al,19 as part of the Acute Liver Failure Study Group, in 2002 prospectively characterized the short-term outcomes of acute liver failure in a large number of patients at 17 tertiary care centers in the United States over approximately 41 months. All centers except one performed liver transplants. Eligible patients had to meet criteria for acute liver failure, including an INR higher than 1.5, evidence of hepatic encephalopathy, and presentation within 26 weeks of illness onset without apparent chronic liver disease.
Of the 308 cases of acute liver failure, 120 (39%) were from acetaminophen overdose, making this drug the most common cause of acute liver failure. Forty-four (37%) of the patients with acetaminophen-related acute liver failure were trying to commit suicide, 57% of cases were accidental, and the remaining reasons for acetaminophen overdose were unknown. The median amount ingested was 13.2 g/day (range 2.6–75 g), and 99 (83%) of the 120 patients took more than 4 g/day.
The patients with acetaminophen-related acute liver toxicity differed from those with other causes of acute liver failure such as idiosyncratic drug reactions or indeterminate causes. The acetaminophen group had a shorter duration of disease and higher serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and serum creatinine than those with acute liver failure from other causes. They also had lower bilirubin levels and a lower arterial pH.
Rates of liver transplantation were 6% in the acetaminophen group, 53% in the group with other drug-induced liver toxicity, 51% in the indeterminate group, and 36% in the remaining groups. Of the acetaminophen group, 47% met the criteria for transplantation, but only 57% of those eligible were listed for transplantation. Those excluded from the transplant list had medical contraindications or were excluded for psychosocial reasons. The short-term transplant-free survival rate was 68% in the acetaminophen group.
Overall, 11% of the patients died, including 28% of the acetaminophen group. The authors concluded that most cases of liver injury in the United States are due to medications and may be preventable.
Larson et al,20 as part of the Acute Liver Failure Study Group, examined the incidence, risk factors, and outcomes of acetaminopheninduced acute liver failure at 22 tertiary care centers in the United States over a 6-year period. Of the 662 patients in the study, 302 had acetaminophen-related toxicity and 275 were included in the final analysis; some of them had been included in the study by Ostapowicz et al.19 During the study period, the number of cases of acute liver toxicity related to acetaminophen increased from 28% to 51%.
Of the patients enrolled in the study, 56% met the criteria for potentially toxic acetaminophen ingestion. Of those who met the criteria, 77% had detectable acetaminophen levels in their serum, and 91% had ALT levels higher than 1,000 IU/L. The time between ingestion and symptom onset ranged from 1 to 32 days, and the median dose was 24 g (range 1.2–180 g).
Of the overdose cases, 48% were unintentional, 44% were intentional, and the remaining 8% had no definable reason. Of the patients in the unintentional-overdose group, 38% were using more than one acetaminophen-containing product and 63% were taking combination products containing narcotics. Overeall, 44% of patients reported using narcotic-acetaminophen combination products. The unintentional-overdose group had lower serum acetaminophen levels than the intentional-overdose group, but they were more likely to present with severe hepatic encephalopathy.
The number of patients with an unintentional overdose was worrisome. Furthermore, one-third of the patients who were receiving an acetaminophen-narcotic combination product were taking an additional acetaminophencontaining product. The authors concluded that unintentional overdose is the leading cause of acetaminophen-related hepatotoxicity, and efforts to limit the over-the-counter package size and to restrict prescriptions of acetaminophen-narcotic combinations may be necessary to decrease the incidence of this preventable cause of acute liver failure.
Findings in children
Squires et al21 and the Pediatric Acute Liver Failure Study Group examined the pathogenesis, treatment, and outcome of acute liver failure in children (any age from birth to 18 years) with no previous evidence of chronic liver disease, evidence of acute liver injury, or hepatic-based coagulopathy. From December 1999 to December 2004, 348 patients were enrolled.
Fourteen percent of the cases were due to acetaminophen. The median dose of acetaminophen ingested was 183 mg/kg. Most of these patients were white and female, and 96% were over age 3. Hepatic encephalopathy was more common in the non-acetaminophen groups than in the acetaminophen group, although this is often difficult to assess in infants and children. Children with acetaminophen toxicity had the highest rate of spontaneous recovery: 45 (94%) of 48 recovered.
Although there are fewer acetaminophenrelated cases of acute liver failure in children than in adults, the use of acetaminophen in children is still worrisome. The authors concluded that if they do not have hepatic encephalopathy, children with acetaminopheninduced liver toxicity have an excellent prognosis.
THE FDA LOOKS AT THE PROBLEM
Why overdoses occur
A recent FDA report22 cited the following reasons for acetaminophen overdose:
- Some patients may experience liver injury at doses only slightly above the recommended 4-g daily limit.
- Some patients may be more prone to liver injury from acetaminophen.
- The symptoms associated with liver injury due to acetaminophen can be nonspecific and tend to evolve over several days.
- Many acetaminophen-containing products are available, including over-the-counter and prescription drugs with many different strengths and indications.
- Consumers are unaware of the risk of liver toxicity with acetaminophen.
- Prescription products are not always clearly labeled with acetaminophen as an ingredient.
- Pediatric dosage forms are available in many different concentrations.
New package labeling
Recommendations from an advisory panel
In addition, an FDA advisory panel recommended decreasing the maximum recommended daily dose (possibly to 3,250 mg/day in adults, although this is not final) to help prevent overdoses, and reducing the maximum amount in a single nonprescription dose of the drug to 650 mg.23 The panel also voted (by a narrow margin) to ban all acetaminophen-narcotic combination products.
As of this writing, the FDA has not adopted these recommendations. (Although the FDA is not obliged to follow the advice of its advisory panels, in most cases it does.) The recommendations about acetaminophen could take years to implement fully.
THE UNITED KINGDOM ACTED IN 1998
In response to a rising number of analgesicrelated deaths, the United Kingdom enacted legislation in 1998 to limit the package size available to consumers.24 Packages sold in general stores can contain no more than 16 capsules, while those sold in pharmacies can contain 24. Blister packs were also introduced to make it harder for people to impulsively take handfuls of tablets.25
Two studies since then both found that the number of deaths related to paracetamol (as acetaminophen is known in the United Kingdom) had fallen since the legislation was implemented.24,26 Greene et al27 found that patients who ingested potentially toxic doses of paracetamol had obtained the drug “in a manner contravening the 1998 legislation.”27 In other words, shops in London were not obeying the law.
SUMMARY
The new labeling and the proposed changes are sensible and draw needed attention to the problem of acetaminophen toxicity. To prevent unintentional acetaminophen overdoses, education of patients and health care professionals is urgently needed so that the dangers of consuming excess acetaminophen daily are understood.
Editor’s note: Portions of this article are based on an article previously published in an internal Cleveland Clinic publication, Pharmacotherapy Update. The version here has been revised, updated, and peer-reviewed.
This drug is generally considered safe, but high doses can be toxic. The number of overdoses is worrisome. In 2006 alone, the American Association of Poison Control Centers implicated acetaminophen in nearly 140,000 poisoning cases, in which more than 100 patients died.2 It is responsible for more emergency room visits than any other drug on the market.
According to a position statement from the American Association for the Study of Liver Diseases (AASLD),3 the incidence of acetaminophen-related liver toxicity has been steadily increasing over the past decade, and this drug is now the most common cause of acute liver failure.
MANY OVERDOSES ARE UNINTENTIONAL
Cases of acetaminophen-related liver toxicity can be categorized as either intentional (ie, due to a suicide attempt) or unintentional (ie, due to multiple therapeutic but excessive doses over a period of time, usually more than 3 days).
Up to 50% of cases are unintentional. Bower et al4 reviewed cases of acute liver failure that occurred in the Atlanta, GA, area between November 2000 and October 2004. Acetaminophen was the most common cause in adult patients. Of greater concern is that 61% of the acetaminophen-related cases were due to unintentional overdose. According to the Institute for Safe Medication Practices,5 one hospital (not named) reported that an average of one patient per day was given more than the recommended maximum daily acetaminophen dose of 4 g while in the hospital.
Many patients take more than one acetaminophen product
Unintentional overdoses or “therapeutic misadventures” are most often due to taking multiple products that contain acetaminophen, taking acetaminophen-narcotic combinations, and impulsive behavior involving a lack of understanding of possible injury in consuming multiple acetaminophen-containing products.3
In the US Food and Drug Administration (FDA) Medwatch Database, in 307 cases of unintentional acetaminophen overdose between 1998 and 2001, 25% of patients had been taking more than one acetaminophencontaining product.5
Larson et al6 found that one-third of patients who had had an unintentional acetaminophen overdose were taking an acetaminophen-narcotic combination in addition to another acetaminophen-containing product.
Many consumers don’t know they are taking acetaminophen
Many consumers don’t know that some of the drugs they take contain acetaminophen. This may be because many drug labels contain abbreviations for acetaminophen such as “APAP” or have inconsistent formatting that makes it difficult to determine if the product contains acetaminophen.
Others may not be aware of the total maximum recommended daily dose or may not be able to calculate the total daily intake from the information on the label. The problem is not only with over-the-counter products. For example, if a physician prescribes one or two tablets of hydrocodone/acetaminophen (Vicodin) 5 mg/500 mg every 4 to 6 hours, a patient could easily exceed the recommended maximum daily dose of 4 g of acetaminophen.
Toxicity can occur even at therapeutic doses
Acetaminophen hepatotoxicity can also occur even with therapeutic doses in certain conditions. Risk factors:
- Chronic alcohol use (ie, more than three drinks per day)
- Malnutrition
- Concurrent use of drugs that induce cytochrome P450 (CYP450) enzymes (more on this below).6
FIRST USED IN 1893
Acetaminophen was first used in medicine in 1893, and it became widely used after 1949, when it was found to be a less-toxic metabolite of two parent compounds, acetanilide and phenacetin.1
Acetaminophen is an effective antipyretic and analgesic, but its anti-inflammatory properties are minimal, especially compared with nonsteroidal anti-inflammatory drugs (NSAIDs). Nevertheless, acetaminophen is preferred over NSAIDs in some patients because it carries a lower risk of gastrointestinal toxicity (eg, ulceration, bleeding) and so may be better tolerated.1
INDICATIONS AND DOSAGE
Acetaminophen is indicated for mild to moderate pain or fever, including the pain of osteoarthritis. It is not recommended for chronic inflammatory conditions such as rheumatoid arthritis, since it lacks anti-inflammatory properties.
In adults and in children over age 12, the usual dosage is 325 to 650 mg orally or rectally every 4 to 6 hours, or 1,000 mg three to four times daily.
The current package label recommends that the total daily dose not exceed 4 g in most adults. Lower maximum daily doses (eg, 2 g) are recommended in patients who may be at higher risk of hepatotoxicity, such as those who drink heavily, are malnourished, or take enzyme-inducing drugs. Tylenol products currently include an alcohol warning, advising those who consume three or more alcoholic drinks a day to ask their doctor if they should take acetaminophen.
In children up to 12 years of age, the recommended dosage is 10 to 15 mg/kg orally or rectally every 4 to 6 hours. The maximum dosing for children in this age group should not exceed five doses (or 50 to 75 mg/kg) in 24 hours.7 In children under age 2 or weighing less than 11 kg, acetaminophen should only be used under the direction of a physician.
MOST ACETAMINOPHEN IS CONJUGATED AND THEN EXCRETED IN THE URINE
Acetaminophen usually has excellent bioavailability (up to 98%), but the exact amount absorbed varies, depending on the dosage form and concomitant use of other drugs.8
At therapeutic doses, the elimination halflife is about 2 hours. Peak plasma concentrations are reached 30 to 60 minutes after the dose is taken,1 but taking acetaminophen with opioids, anticholinergic drugs, or even food may delay the time to peak concentration by delaying gastric emptying.8
NAPQI is a toxic metabolite
Under normal circumstances, a small amount of acetaminophen undergoes hepatic metabolism by a different pathway, ie, by CYP450 enzymes, primarily CYP2E1 and to a lesser extent CYP1A2, CYP2A6, and CYP3A4, forming a toxic metabolite, N-acetyl-p-benzoquinone imine (NAPQI). Then, the sulfhydryl groups of glutathione convert NAPQI into harmless metabolites that are excreted in the urine.1,7
Well tolerated and relatively safe
At recommended doses, acetaminophen is well tolerated, and it is considered relatively safe when used according to labeling instructions.
Rarely, patients experience an erythematous or urticarial rash or other allergic complications.1
However, acetaminophen is a dose-dependent hepatotoxin, and excessive doses (intentional or unintentional) may lead to acute liver failure. In addition, even in therapeutic doses, acetaminophen may still cause transient liver enzyme elevations and possibly hepatotoxicity, particularly in people who are malnourished or alcoholic or are taking certain CYP450-inducing drugs.3,6,10
HOW ACETAMINOPHEN CAN INJURE THE LIVER
Glucuronidation and sulfation, the major metabolic pathways, become saturated after an acetaminophen overdose.7 When this happens, more of the toxic metabolite NAPQI is formed by CYP450-mediated N-hydroxylation. When glutathione is depleted after large doses of acetaminophen or in malnourished people, the toxic metabolite accumulates, resulting in liver damage (Figure 1).6
The liver is damaged by two mechanisms. In one, NAPQI binds to hepatic cell macromolecules, causing dysfunction of the enzymatic systems, structural and metabolic disarray, and eventually necrotic cell death. The other mechanism is oxidative stress due to depletion of glutathione.
In children, single acetaminophen doses of 120 to 150 mg/kg of body weight have been associated with hepatotoxicity,11 as have single doses of more than 150 mg/kg or a total dose of greater than 7.5 g in adults. However, the minimal dose associated with liver injury has ranged from 4 to 10 g, and in healthy volunteers even therapeutic doses of 1 g orally every 6 hours resulted in mild liver injury.12
Patients who are malnourished or fasting are thought to be at greater risk of acetaminophen hepatotoxicity because they may be deficient in glutathione at baseline. In addition, even at lower-than-therapeutic doses, induction of CYP450 enzymes by drugs or chronic alcohol consumption may lead to an increase in the formation of NAPQI, increasing the risk of hepatotoxicity. Examples of drugs that induce CYP450 enzymes to produce more NAPQI include the anticonvuslants phenytoin (Dilantin) and phenobarbital.
CLINICAL PRESENTATION OF ACETAMINOPHEN OVERDOSE
The diagnosis of acetaminophen overdose is often established by a thorough history. The pertinent information may be difficult to obtain, however, because the patient may be confused or stuporous at presentation, may not know that the overthe-counter products he or she has been taking contain acetaminophen, or may be embarrassed about taking too much acetaminophen.13
In acute intentional overdose, signs may not be apparent immediately
The symptoms of toxicity may not be apparent immediately after ingestion of an acute overdose of acetaminophen, but early recognition and treatment can prevent more severe liver damage, decreasing morbidity and the risk of death.9
Phase 1 of an acetaminophen overdose begins shortly after ingestion and can last for 12 to 24 hours. Patients may have signs of gastrointestinal upset, nausea, vomiting, anorexia, diaphoresis, and pallor. Although the signs and symptoms show a consistent pattern and are more pronounced after larger acute overdoses, they are not diagnostic or specific.
Phase 2 (up to 48 hours after ingestion). Patients may begin to feel better during this phase. However, the hepatic enzyme levels, the prothrombin time (PT), and the international normalized ratio (INR) may continue to rise, and right upper quadrant pain may develop. Additionally, other laboratory results may be abnormal, and renal insufficiency can occur due to acetaminophen-induced renal tubular necrosis.6 Most patients receive the antidote, acetylcysteine (Mycomyst, Acetadote) before or during this phase, and consequently, liver function gradually returns to normal.
Phase 3, if reached, may be marked by severe hepatic necrosis, typically 3 to 5 days after ingestion. Symptoms during this phase range from less severe (eg, nausea and general malaise) to more severe (eg, confusion and stupor). Also, at this time, liver enzyme levels can be as high as 10,000 IU/L or even higher, and lactic acidosis and coagulopathy may worsen. If death should occur, it is most likely from complications associated with fulminant hepatic failure, including cerebral edema, multiorgan-system failure, or sepsis.6
Phase 4. Patients who recover generally have complete recovery of liver function with no long-term sequelae..
In unintentional overdoses, patients may have low drug levels
Many patients who present with unintentional acetaminophen toxicity have been taking the drug or products that contain the drug over several days to treat an acute or chronic medical condition. They often have low or undetectable serum acetaminophen levels after 2 to 3 days of nonspecific symptoms.12
MANAGING ACETAMINOPHENHEN OVERDOSE
Unintentional overdoses occur over a more prolonged period, and therefore the nomogram is not useful in this situation.
Acetylcysteine is the antidote
Acetylcysteine is the antidote for acetaminophen toxicity and should be given within 8 hours of ingestion for maximal protection against hepatic injury in patients whose serum acetaminophen levels are above the “possible” toxicity line on the nomogram.15 If acetaminophen overdose is suspected but the time elapsed since ingestion cannot be determined, acetylcysteine should be given immediately regardless of the quantity of acetaminophen ingested. 16 In cases of unintentional overdose, it is often given at the discretion of the physician.
Acetylcysteine limits the toxicity of acetaminophen by increasing glutathione stores, binding with NAPQI as a substitute for glutathione, and enhancing sulfate conjugation.6 It may further limit acetaminophen toxicity by nonspecific mechanisms including anti-inflammatory, antioxidant, inotropic, and vasodilating effects.6 In addition, it may prevent further hepatic damage in any patient thought to have acetaminophen-related liver toxicity even beyond the first 12 hours of an overdose.1
Acetylcysteine is available in an oral (Mucomyst) and an intravenous (Acetadote) formulation, which are similar in efficacy. Because many patients find the taste of the oral solution unpleasant and difficult to tolerate, it should be diluted in a 1:3 ratio with cola, orange juice, or other drink to mask its flavor. This mixture should be used within 1 hour of preparation.
Anaphylactoid reactions have occurred in patients receiving intravenous acetylcysteine for acetaminophen overdose soon after the infusion was started, most commonly during the loading dose. The frequency of infusion-related reactions has been reported to be 0.2% to 20.8%.15
The recommended dosage for intravenous acetylcysteine is a loading dose of 150 mg/kg given over 60 minutes, followed by a second dose of 50 mg/kg given over 4 hours, and finally a third dose of 100 mg/kg given over 16 hours.15 For oral acetylcysteine, the loading dose is 140 mg/kg followed by 70 mg/kg every 4 hours for 17 additional doses.17
Other measures
Activated charcoal can be used if the patient presents within 1 to 2 hours after taking acetaminophen. However, the rapid gastrointestinal absorption of acetaminophen makes this treatment ineffective in most cases.9
FINDINGS FROM LARGE REGISTRIES
Findings in adults
Ostapowicz et al,19 as part of the Acute Liver Failure Study Group, in 2002 prospectively characterized the short-term outcomes of acute liver failure in a large number of patients at 17 tertiary care centers in the United States over approximately 41 months. All centers except one performed liver transplants. Eligible patients had to meet criteria for acute liver failure, including an INR higher than 1.5, evidence of hepatic encephalopathy, and presentation within 26 weeks of illness onset without apparent chronic liver disease.
Of the 308 cases of acute liver failure, 120 (39%) were from acetaminophen overdose, making this drug the most common cause of acute liver failure. Forty-four (37%) of the patients with acetaminophen-related acute liver failure were trying to commit suicide, 57% of cases were accidental, and the remaining reasons for acetaminophen overdose were unknown. The median amount ingested was 13.2 g/day (range 2.6–75 g), and 99 (83%) of the 120 patients took more than 4 g/day.
The patients with acetaminophen-related acute liver toxicity differed from those with other causes of acute liver failure such as idiosyncratic drug reactions or indeterminate causes. The acetaminophen group had a shorter duration of disease and higher serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and serum creatinine than those with acute liver failure from other causes. They also had lower bilirubin levels and a lower arterial pH.
Rates of liver transplantation were 6% in the acetaminophen group, 53% in the group with other drug-induced liver toxicity, 51% in the indeterminate group, and 36% in the remaining groups. Of the acetaminophen group, 47% met the criteria for transplantation, but only 57% of those eligible were listed for transplantation. Those excluded from the transplant list had medical contraindications or were excluded for psychosocial reasons. The short-term transplant-free survival rate was 68% in the acetaminophen group.
Overall, 11% of the patients died, including 28% of the acetaminophen group. The authors concluded that most cases of liver injury in the United States are due to medications and may be preventable.
Larson et al,20 as part of the Acute Liver Failure Study Group, examined the incidence, risk factors, and outcomes of acetaminopheninduced acute liver failure at 22 tertiary care centers in the United States over a 6-year period. Of the 662 patients in the study, 302 had acetaminophen-related toxicity and 275 were included in the final analysis; some of them had been included in the study by Ostapowicz et al.19 During the study period, the number of cases of acute liver toxicity related to acetaminophen increased from 28% to 51%.
Of the patients enrolled in the study, 56% met the criteria for potentially toxic acetaminophen ingestion. Of those who met the criteria, 77% had detectable acetaminophen levels in their serum, and 91% had ALT levels higher than 1,000 IU/L. The time between ingestion and symptom onset ranged from 1 to 32 days, and the median dose was 24 g (range 1.2–180 g).
Of the overdose cases, 48% were unintentional, 44% were intentional, and the remaining 8% had no definable reason. Of the patients in the unintentional-overdose group, 38% were using more than one acetaminophen-containing product and 63% were taking combination products containing narcotics. Overeall, 44% of patients reported using narcotic-acetaminophen combination products. The unintentional-overdose group had lower serum acetaminophen levels than the intentional-overdose group, but they were more likely to present with severe hepatic encephalopathy.
The number of patients with an unintentional overdose was worrisome. Furthermore, one-third of the patients who were receiving an acetaminophen-narcotic combination product were taking an additional acetaminophencontaining product. The authors concluded that unintentional overdose is the leading cause of acetaminophen-related hepatotoxicity, and efforts to limit the over-the-counter package size and to restrict prescriptions of acetaminophen-narcotic combinations may be necessary to decrease the incidence of this preventable cause of acute liver failure.
Findings in children
Squires et al21 and the Pediatric Acute Liver Failure Study Group examined the pathogenesis, treatment, and outcome of acute liver failure in children (any age from birth to 18 years) with no previous evidence of chronic liver disease, evidence of acute liver injury, or hepatic-based coagulopathy. From December 1999 to December 2004, 348 patients were enrolled.
Fourteen percent of the cases were due to acetaminophen. The median dose of acetaminophen ingested was 183 mg/kg. Most of these patients were white and female, and 96% were over age 3. Hepatic encephalopathy was more common in the non-acetaminophen groups than in the acetaminophen group, although this is often difficult to assess in infants and children. Children with acetaminophen toxicity had the highest rate of spontaneous recovery: 45 (94%) of 48 recovered.
Although there are fewer acetaminophenrelated cases of acute liver failure in children than in adults, the use of acetaminophen in children is still worrisome. The authors concluded that if they do not have hepatic encephalopathy, children with acetaminopheninduced liver toxicity have an excellent prognosis.
THE FDA LOOKS AT THE PROBLEM
Why overdoses occur
A recent FDA report22 cited the following reasons for acetaminophen overdose:
- Some patients may experience liver injury at doses only slightly above the recommended 4-g daily limit.
- Some patients may be more prone to liver injury from acetaminophen.
- The symptoms associated with liver injury due to acetaminophen can be nonspecific and tend to evolve over several days.
- Many acetaminophen-containing products are available, including over-the-counter and prescription drugs with many different strengths and indications.
- Consumers are unaware of the risk of liver toxicity with acetaminophen.
- Prescription products are not always clearly labeled with acetaminophen as an ingredient.
- Pediatric dosage forms are available in many different concentrations.
New package labeling
Recommendations from an advisory panel
In addition, an FDA advisory panel recommended decreasing the maximum recommended daily dose (possibly to 3,250 mg/day in adults, although this is not final) to help prevent overdoses, and reducing the maximum amount in a single nonprescription dose of the drug to 650 mg.23 The panel also voted (by a narrow margin) to ban all acetaminophen-narcotic combination products.
As of this writing, the FDA has not adopted these recommendations. (Although the FDA is not obliged to follow the advice of its advisory panels, in most cases it does.) The recommendations about acetaminophen could take years to implement fully.
THE UNITED KINGDOM ACTED IN 1998
In response to a rising number of analgesicrelated deaths, the United Kingdom enacted legislation in 1998 to limit the package size available to consumers.24 Packages sold in general stores can contain no more than 16 capsules, while those sold in pharmacies can contain 24. Blister packs were also introduced to make it harder for people to impulsively take handfuls of tablets.25
Two studies since then both found that the number of deaths related to paracetamol (as acetaminophen is known in the United Kingdom) had fallen since the legislation was implemented.24,26 Greene et al27 found that patients who ingested potentially toxic doses of paracetamol had obtained the drug “in a manner contravening the 1998 legislation.”27 In other words, shops in London were not obeying the law.
SUMMARY
The new labeling and the proposed changes are sensible and draw needed attention to the problem of acetaminophen toxicity. To prevent unintentional acetaminophen overdoses, education of patients and health care professionals is urgently needed so that the dangers of consuming excess acetaminophen daily are understood.
- Burke A, Smyth EM, Fitzgerald GA. Analgesic-antipyretic agents: pharmacotherapy of gout. In:Brunton LL, Lazo JS, Parker K, editors. Goodman and Gilman’s the Pharmacological Basis of Therapeutics, 11th ed. New York: McGraw-Hill, 2006:671–716.
- Bronstein AC, Spyker DA, Cantilena LR, Green J, Rumack BH, Heard SE. 2006 Annual Report of the American Association of Poison Control Centers National Poison Data System (NPDS). Clin Toxicol (Phila) 2007; 45:815–917.
- Schwartz J, Stravitz T, Lee WM; American Association for the Study of Liver Disease Study Group. AASLD position on acetaminophen. www.aasld.org/about/publicpolicy/Documents/Public%2520Policy%2520Documents/AcetaminophenPosition.pdf.
- Bower WA, Johns M, Margolis HS, Williams IT, Bell BP. Populationbased surveillance for acute liver failure. Am J Gastroenterol 2007; 102:2459–2463.
- Institute for Safe Medication Practices. How are you preventing acetaminophen overdoses? www.ismp.org/newsletters/acutecare/articles/20030808.asp. Accessed 11/17/2009.
- Larson AM. Acetaminophen hepatotoxicity. Clin Liver Dis 2007; 11:525–548.
- Tylenol package insert. Fort Washington, PA: McNeil-PPC Inc.; 1999.
- Bizovi KE, Hendrickson RG. Chapter 34. Acetaminophen. In:Hoffman RS, Nelson LS, Howland MA, Lewin NA, Flomenbaum NE, Goldfrank LR, editors. Goldfrank’s Manual of Toxicologic Emergencies. 3rd ed. McGraw-Hill: New York, 2007. www.accessemergencymedicine.com/content.aspx?aID=88781. Accessed 11/7/2009.
- Hung OL, Nelson LS. Chapter 171. Acetaminophen. In:Tintinalli JE, Kelen GD, Stapcynski S, editors. Tintinalli's Emergency Medicine: A Comprehensive Study Guide. 6th ed. McGraw-Hill: New York, 2004. www.accessmedicine.com/content.aspx?aID=602606. Accessed 11/17/2009.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- American Academy of Pediatrics Committee on Drugs. Acetaminophen toxicity in children. Pediatrics 2001; 108:1020–1024.
- Fontana RJ. Acute liver failure including acetaminophen overdose. Med Clin North Am 2008; 92:761–794.
- Heard KJ. Acetylcysteine for acetaminophen poisoning. N Engl J Med 2008; 359:285–292.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Acetadote package insert. Nashville, TN: Cumberland Pharmaceuticals; 2008Dec.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- Product Information: acetylcysteine inhalation solution, acetylcysteine inhalation solution. Hospira,Inc, Lake Forest, IL, 2004.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Ostapowicz G, Fontana RJ, Schiødt FV, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Larson AM, Polson J, Fontana RJ, et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Squires RH, Shneider BL, Bucuvalas J, et al. Acute liver failure in children: the first 348 patients in the Pediatric Acute Liver Failure Study Group. J Pediatr 2006; 148:652–658.
- US Food and Drug Administration. Questions and answers on final rule for labeling changes to over-the-counter pain relievers. www.fda.gov/Drugs/NewsEvents/ucm144068.htm. Accessed 10/30/2009.
- Perrone M. FDA panel recommends smaller doses of painkillers. Associated Press. Adelphi, MD. June 30, 2009.
- Hawton K, Simkin S, Deeks J, et al. UK legislation on analgesic packs: before and after study of long term effect on poisonings. BMJ 2004; 329:1076.
- Hughes B, Durran A, Langford NJ, Mutimer D. Paracetamol poisoning—impact of pack size restrictions. J Clin Pharmacol Ther 2003; 28:307–310.
- Wilkinson S, Taylor G, Templeton L, Mistral W, Salter E, Bennett P. Admissions to hospital for deliberate self-harm in England 1995–2000: an analysis of hospital episode statistics. J Public Health Med 2002; 24:179–183.
- Greene SL, Dargan PI, Leman P, Jones AL. Paracetamol availability and recent changes in paracetamol poisoning: is the 1998 legislation limiting availability of paracetamol being followed? Postgrad Med J 2006; 82:520–523.
- Burke A, Smyth EM, Fitzgerald GA. Analgesic-antipyretic agents: pharmacotherapy of gout. In:Brunton LL, Lazo JS, Parker K, editors. Goodman and Gilman’s the Pharmacological Basis of Therapeutics, 11th ed. New York: McGraw-Hill, 2006:671–716.
- Bronstein AC, Spyker DA, Cantilena LR, Green J, Rumack BH, Heard SE. 2006 Annual Report of the American Association of Poison Control Centers National Poison Data System (NPDS). Clin Toxicol (Phila) 2007; 45:815–917.
- Schwartz J, Stravitz T, Lee WM; American Association for the Study of Liver Disease Study Group. AASLD position on acetaminophen. www.aasld.org/about/publicpolicy/Documents/Public%2520Policy%2520Documents/AcetaminophenPosition.pdf.
- Bower WA, Johns M, Margolis HS, Williams IT, Bell BP. Populationbased surveillance for acute liver failure. Am J Gastroenterol 2007; 102:2459–2463.
- Institute for Safe Medication Practices. How are you preventing acetaminophen overdoses? www.ismp.org/newsletters/acutecare/articles/20030808.asp. Accessed 11/17/2009.
- Larson AM. Acetaminophen hepatotoxicity. Clin Liver Dis 2007; 11:525–548.
- Tylenol package insert. Fort Washington, PA: McNeil-PPC Inc.; 1999.
- Bizovi KE, Hendrickson RG. Chapter 34. Acetaminophen. In:Hoffman RS, Nelson LS, Howland MA, Lewin NA, Flomenbaum NE, Goldfrank LR, editors. Goldfrank’s Manual of Toxicologic Emergencies. 3rd ed. McGraw-Hill: New York, 2007. www.accessemergencymedicine.com/content.aspx?aID=88781. Accessed 11/7/2009.
- Hung OL, Nelson LS. Chapter 171. Acetaminophen. In:Tintinalli JE, Kelen GD, Stapcynski S, editors. Tintinalli's Emergency Medicine: A Comprehensive Study Guide. 6th ed. McGraw-Hill: New York, 2004. www.accessmedicine.com/content.aspx?aID=602606. Accessed 11/17/2009.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- American Academy of Pediatrics Committee on Drugs. Acetaminophen toxicity in children. Pediatrics 2001; 108:1020–1024.
- Fontana RJ. Acute liver failure including acetaminophen overdose. Med Clin North Am 2008; 92:761–794.
- Heard KJ. Acetylcysteine for acetaminophen poisoning. N Engl J Med 2008; 359:285–292.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Acetadote package insert. Nashville, TN: Cumberland Pharmaceuticals; 2008Dec.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- Product Information: acetylcysteine inhalation solution, acetylcysteine inhalation solution. Hospira,Inc, Lake Forest, IL, 2004.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Ostapowicz G, Fontana RJ, Schiødt FV, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Larson AM, Polson J, Fontana RJ, et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Squires RH, Shneider BL, Bucuvalas J, et al. Acute liver failure in children: the first 348 patients in the Pediatric Acute Liver Failure Study Group. J Pediatr 2006; 148:652–658.
- US Food and Drug Administration. Questions and answers on final rule for labeling changes to over-the-counter pain relievers. www.fda.gov/Drugs/NewsEvents/ucm144068.htm. Accessed 10/30/2009.
- Perrone M. FDA panel recommends smaller doses of painkillers. Associated Press. Adelphi, MD. June 30, 2009.
- Hawton K, Simkin S, Deeks J, et al. UK legislation on analgesic packs: before and after study of long term effect on poisonings. BMJ 2004; 329:1076.
- Hughes B, Durran A, Langford NJ, Mutimer D. Paracetamol poisoning—impact of pack size restrictions. J Clin Pharmacol Ther 2003; 28:307–310.
- Wilkinson S, Taylor G, Templeton L, Mistral W, Salter E, Bennett P. Admissions to hospital for deliberate self-harm in England 1995–2000: an analysis of hospital episode statistics. J Public Health Med 2002; 24:179–183.
- Greene SL, Dargan PI, Leman P, Jones AL. Paracetamol availability and recent changes in paracetamol poisoning: is the 1998 legislation limiting availability of paracetamol being followed? Postgrad Med J 2006; 82:520–523.
KEY POINTS
- Acetaminophen is the leading cause of acute liver failure in the United States, and nearly half of acetaminophenassociated cases are due to unintentional overdose.
- In many cases of unintentional overdose, patients took more than one acetaminophen-containing product and did not know that both products contained this drug.
- Prescribers need to inform all patients, especially vulnerable ones (eg, those taking enzyme-inducing drugs, those who chronically use alcohol, and those who are malnourished) of the risks associated with acetaminophen.
- Although no consensus has been reached on what is a safe dose in patients with liver disease, 4 g/day is too much: a total daily dose of no more than 2 g is recommended to decrease the risk of toxicity in these patients.
An algorithm for managing warfarin resistance
Warfarin (coumadin) differs from most other drugs in that the dosage required to achieve a desired therapeutic effect varies greatly among individuals. This variability can lead to therapeutic failure, potentially resulting in new thrombosis, or, at the other extreme, to life-threatening bleeding.
Further, there is no reliable means to identify patients who require unusually high doses of warfarin, although genetic testing may become available in the future.
See related patient information
Warfarin, a coumarin derivative first synthesized in 1948, is still the only oral anticoagulant available for long-term use in the United States. Indications for its use include the treatment and, to a lesser extent, the prevention of arterial and venous thromboembolism. It is also used for long-term anticoagulation in patients with atrial arrhythmias (atrial fibrillation and atrial flutter) and mechanical heart valves.
In the paragraphs that follow, we review the causes of warfarin resistance and how to recognize and manage it.
WHAT IS WARFARIN RESISTANCE?
Resistance to warfarin has been described as the inability to prolong the prothrombin time or raise the international normalized ratio (INR) into the therapeutic range when the drug is given at normally prescribed doses.1
However, a higher warfarin requirement does not itself establish the diagnosis of warfarin resistance. The prevalence of warfarin resistance varies by patient population and is difficult to determine. The difficulty lies largely in accounting for dietary factors and in defining normal metabolic variations among individuals.
The range of normally recommended daily or weekly warfarin doses to maintain a therapeutic prothrombin time or INR depends on the study population. Nevertheless, patients who need more than 105 mg per week (15 mg/day) should be considered warfarin-resistant. These patients are likely to be in the top 5% for warfarin doses within an anticoagulated cohort.
Warfarin resistance is different than warfarin failure, which is defined as a new thrombotic event despite a therapeutic prothrombin time and INR. This situation is commonly seen in patients with malignant diseases.
An important characteristic of warfarin resistance is that patients need much smaller doses of vitamin K to reverse the effect of warfarin.2 Thijssen3 showed that, in warfarin-resistant rats, warfarin did not irreversibly inhibit vitamin K1 2,3-epoxide reductase (VKORC1) activity. This is consistent with the vitamin K hypersensitivity observed in warfarin-resistant people.2,3

WHAT CAUSES WARFARIN RESISTANCE?
Warfarin resistance can be classified in practical terms as acquired vs hereditary, or in mechanistic terms as pharmacokinetic vs pharmacodynamic.
Acquired vs hereditary resistance
Hulse4 categorizes warfarin resistance as either acquired or hereditary.
Acquired resistance to warfarin may result from:
- Poor patient compliance (the most common cause)
- High consumption of vitamin K

- Increased clearance (see Warfarin is metabolized by P450 enzymes5–11)
- Drug interactions (Table 1).12,13

Hereditary resistance has been postulated to be caused by genetic factors that result either in faster metabolism of the drug (a form of pharmacokinetic resistance) or in lower activity of the drug (pharmacodynamic resistance). Polymorphisms may play a role, as some VKORC1 and CYP2C9 variant alleles are known to be associated with increased sensitivity to warfarin.14
However, the genetic mechanisms of warfarin resistance are not clearly understood, despite several case reports of hereditary resistance confirmed by similar patterns of resistance in immediate family members.15–19 More than one mechanism is likely. There is ample room for further insight into genetic polymorphisms underlying hereditary warfarin resistance. More on this topic is included in the sections below.
Pharmacokinetic resistance
Pharmacokinetic resistance can result from diminished absorption or increased elimination of the drug. Causes of diminished absorption include emesis, diarrhea, and malabsorption syndrome.
The mechanism of increased warfarin clearance has not been delineated, although the following have been implicated.
Genetic factors. Duplication or multiplication of cytochrome P450 enzyme genes has been described as contributing to a phenotype of ultrarapid metabolism. Some people may carry multiple copies of the CYP2C9 gene, as has already been reported for cytochrome P450 CYP2D6 and CYP2A6.7,8 It is also plausible that rare allelic variants of CYP2C9 exist that are associated with higher-than-normal activity, given that there are alleles known to predispose to warfarin sensitivity.
Hypoalbuminemia may increase the free fraction of warfarin, leading to enhanced rates of clearance and a shorter plasma half-life.15
Hyperalbuminemia may paradoxically also contribute to warfarin resistance via drug binding.
Hyperlipidemia. Several observers have found that lowering serum lipids, primarily triglycerides, increases the sensitivity to warfarin irrespective of the means used to achieve this decrease.20 This most likely results in a decreased pool of vitamin K, some of which is bound to triglycerides.21 Conversely, patients receiving intravenous lipids with total parenteral nutrition have also been diagnosed clinically with warfarin resistance,22 and rat models have shown an association between a lipidrich diet and increased vitamin K-dependent factor activity.23
Diuretics may decrease the response to warfarin by reducing the plasma volume, with a subsequent increase in clotting factor activity.24
Pharmacodynamic resistance
Potential mechanisms of pharmacodynamic warfarin resistance described in rats and in people include:
- Increased affinity of vitamin K1, 2,3-epoxide reductase complex (VKOR) for vitamin K25,26 (see How warfarin works2,10,11,27–30)
- Prolongation of normal clotting factor activity16
- Production of clotting factors that is not dependent on vitamin K16
- Decreased VKOR sensitivity to warfarin.26
In rats, these mechanisms are manifested by relatively high doses of warfarin being required to achieve poisoning. In humans, they result in high doses being needed to achieve a therapeutic effect in the setting of normal warfarin pharmacokinetics, normal warfarin concentration, and normal half-lives of blood clotting proteins.
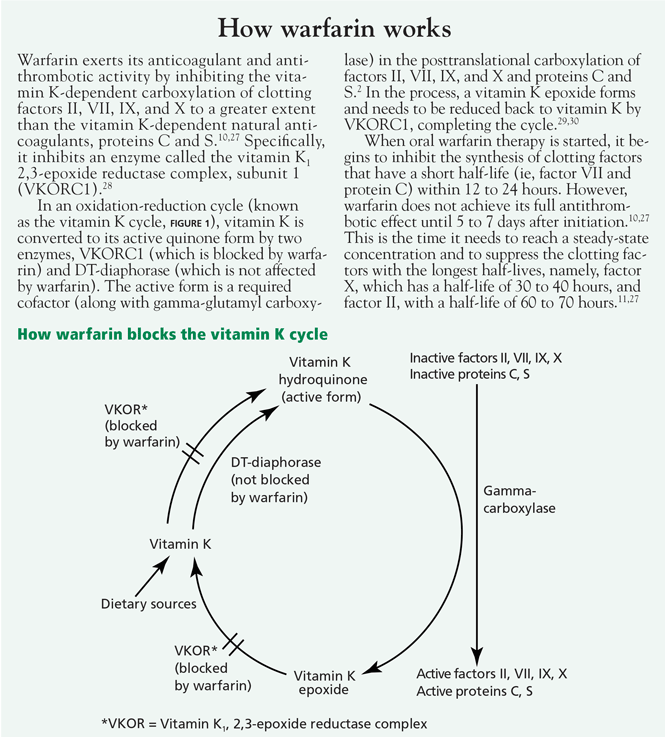
Genetics of pharmacodynamic resistance. Pharmacodynamic warfarin resistance has also been described with inheritance of a monogenetic dominant trait. An early study by O’Reilly24 traced anticoagulation resistance to a genetically linked abnormality of interaction between warfarin and a putative vitamin K receptor.
In one patient with hereditary resistance and high warfarin requirements, a heterozygous point mutation in the VKORC1 gene was identified.31 This results in a substitution that lies in a conserved (normally constant or unchanging DNA sequence in a genome) region of VKORC1 that contains three of four previously identified amino acid substitutions associated with warfarin resistance (Val29Leu, Val45Ala, and Arg58Gly). Further investigation is required to fully characterize the structure-function relationship for VKORC1 and to determine the relationship between the VKORC1 genotype and other pharmacogenetic determinants of warfarin dose-response.
Separately, Loebstein et al32 reported a new mutation, Asp36Tyr, which was common in Jewish ethnic groups of Ethiopian descent (in whom the prevalence is 5%) and Ashkenazi descent (prevalence 4%). In that study, Asp36Tyr carriers needed doses of more than 70 mg per week, placing them towards the high end of the usual warfarin dosing range.
Daly and Aithal7 discovered that warfarinresistant rats overexpressed a protein known as calumenin. This protein is situated in the endoplasmic reticulum and appears to interact with VKOR, decreasing the binding of warfarin. In mice, the calumenin gene is located on chromosome 7, where the gene for VKORC1 is also located.
DIAGNOSIS BY HISTORY AND LABORATORY STUDIES
A full drug and diet history is invaluable in diagnosing potential causes of warfarin resistance (Table 1).
Plasma warfarin levels that are subtherapeutic should raise suspicion of intestinal malabsorption or poor compliance. Poor compliance might be more appropriately seen as a mimic of warfarin resistance. Studies in humans suggest that a therapeutic total plasma warfarin level lies between 0.5 μg/mL and 3.0 μg/mL,10 though the range may vary among laboratories and patient populations.
Warfarin absorption and clearance can be evaluated by analyzing plasma levels at specific intervals after administration, eg, every 60 to 180 minutes. The drug’s half-life can be determined on the basis of its concentrations in different time samples. Normally, the S-enantiomer of warfarin is cleared at twice the rate of the R-enantiomer (5.2 vs 2.5 mL/min/70 kg).8 A normal clearance rate confirms that resistance to warfarin is not due to enhanced elimination.
Clotting assays of factors II, VII, IX, and X may be a more precise way to assess the pharmacodynamics of warfarin,10 although there is no strong evidence to support routine use of such assays. Some studies suggest targeting factor II and factor X activity levels of 10% to 30% of normal biologic activity for a therapeutic warfarin effect in patients with an unreliable or prolonged baseline prothrombin time and INR, such as those with lupus anticoagulant.

TREAT THE CAUSE
Once the type of warfarin resistance has been determined, treatment should be oriented toward the cause.
Educate the patient
The importance of compliance should be reinforced. Educating the patient about diet and other medications that may interact with warfarin is also important. (See an example of patient education material.)
Increase the warfarin dose
If the patient truly has hereditary resistance, there are two approaches to treatment.
The first is to increase the warfarin dose until the prothrombin time and INR are in the therapeutic ranges. When indicated, the warfarin dose can be safely titrated upward to more than 100 mg per day in patients who are monitored regularly—as all patients on chronic warfarin therapy should be—and whose other medications are otherwise stable. One such example is reported in a warfarinresistant patient who needed 145 mg/day to maintain a therapeutic prothrombin time.22
Try other anticoagulants?
The second approach is to change to another type of anticoagulant. However, there is no strong evidence in favor of this approach over prescribing larger dosages of warfarin.
Other anticoagulant drugs currently available in the United States include subcutaneous heparins (unfractionated and low-molecular-weight heparins) and the subcutaneous factor Xa inhibitor fondaparinux (Arixtra).
Agents not available in the United States include the following.
Dabigatran, an oral direct thrombin inhibitor, is undergoing phase 3 studies of its use for long-term anticoagulation.
Rivaroxaban (a direct factor Xa inhibitor) and dabigatran have been approved in Canada and the European Union to prevent venous thromboembolism after knee and hip arthroplasty, based on prospective comparisons with enoxaparin (Lovenox).34–37
Vitamin K antagonists other than warfarin that are not available in the United States include bishydroxycoumarin (which has limitations including slow absorption and high frequency of gastrointestinal side effects), phenprocoumon, and acenocoumarol. Another is phenindione, which has been associated with serious hypersensitivity reactions, some of which proved fatal and occurred within a few weeks of initiating therapy.
- Lefrere JJ, Horellou MH, Conard J, Samama M. Proposed classification of resistance to oral anticoagulant therapy. J Clin Pathol 1987; 40:242.
- Linder MW. Genetic mechanisms for hypersensitivity and resistance to the anticoagulant warfarin. Clin Chim Acta 2001; 308:9–15.
- Thijssen HH. Warfarin resistance. Vitamin K epoxide reductase of Scottish resistance gene is not irreversibly blocked by warfarin. Biochem Pharmacol 1987; 36:2753–2757.
- Hulse ML. Warfarin resistance: diagnosis and therapeutic alternative. Pharmacotherapy 1996; 16:1009–1017.
- Hirsh J, Dalen JE, Deykin D, Poller L, Bussey H. Oral anticoagulants. Mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 1995; 108( suppl 4):231S–234S.
- Daly AK, King BP. Pharmacogenetics of oral anticoagulants. Pharmacogenetics 2003; 13:247–252.
- Daly AK, Aithal GP. Genetic regulation of warfarin metabolism and response. Semin Vasc Med 2003; 3:231–238.
- Takahashi H, Echizen H. Pharmacogenetics of warfarin elimination and its clinical implications. Clin Pharmacokinet 2001; 40:587–603.
- Retti AE, Wienkers LC, Gonzalez FJ, Trager WF, Korezekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allele variant of CYP2C9. Pharmacogenetics 1994; 4:39–42.
- Porter RS, Sawyer WR. Warfarin. In:Evans WE, Shentag JJ, Jusko WJ, editors. Applied Pharmacokinetics. Principles of Therapeutics Drug Monitoring, 3rd ed. Washington, DC: Applied Therapeutics, 1992: 31.1–31.46.
- Warrell DA, Cox TM, Firth JD. Oxford Textbook of Medicine, 4th ed. Oxford University Press, 2003:734.
- Holbrook AM, Pereira JA, Labiris R, et al. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med 2005; 165:1095–1106.
- Medical Economics Staff. Physicians’ Desk Reference, 55th Ed. Medical Economics, 2001:1139–1140.
- Schwarz UI, Ritchie MD, Bradford Y, et al. Genetic determinants of response to warfarin during initial anticoagulation. N Engl J Med 2008; 358:999–1008.
- Diab F, Feffer S. Hereditary warfarin resistance. South Med J 1994; 87:407–409.
- O’Reilly RA. The second reported kindred with hereditary resistance to oral anticoagulant drugs. N Engl J Med 1970; 282:1448–1451.
- O’Reilly RA, Aggeler PM, Hoag MS, Leong LS, Kropatkin ML. Hereditary transmission of exceptional resistance to coumarin anticoagulant drugs. The first reported kindred. N Engl J Med 1964; 271:809–815.
- Alving BM, Strickler MP, Knight RD, Barr CF, Berenberg JL, Peek CC. Hereditary warfarin resistance. Investigation of rare phenomenon. Arch Intern Med 1985; 145:499–501.
- Warrier L, Brennan CA, Lusher JM. Familial warfarin resistance in a black child. Am J Pediatr Hematol Oncol 1986; 8:346–347.
- Nikkila EA, Pelkonen R. Serum lipid-reducing agents and anticoagulant requirement. Lancet 1963; 1:332.
- Robinson A, Liau FO, Routledge PA, Backhouse G, Spragg BP, Bentley DP. Lipids and warfarin requirements. Thromb Haemost 1990; 63:148–149.
- MacLaren R, Wachsman BA, Swift DK, Kuhl DA. Warfarin resistance associated with intravenous lipid administration: discussion of propofol and review of the literature. Pharmacotherapy 1997; 17:1331–1337.
- DeCurtis A, D’Adamo MC, Amore C, et al. Experimental arterial thrombosis in genetically or diet induced hyperlipidemia in rats—role of vitamin K-dependent clotting factors and prevention by low-intensity oral anticoagulation. Thromb Haemost 2001; 86:1440–1448.
- O’Reilly RA. Drug interaction involving oral anticoagulation. In:Melmon KL, editor. Cardiovascular Drug Therapy, Philadelphia; FA Davis, 1975:23–41.
- O’ Reilly RA, Pool JG, Aggeler PM. Hereditary resistance to coumarin anticoagulation drugs in man and rat. Ann N Y Acad Sci 1968; 151:913–931.
- Cain D, Hutson SM, Wallin R. Warfarin resistance is associated with a protein component of the vitamin K 2,3-epoxide reductase enzyme complex in rat liver. Thromb Haemost 1998; 80:128–133.
- Rodvold KA, Quandt CM, Friedenberg WR. Thromboembolic disorders. In:DiPiro JT, Talbert RL, editors. Pharmacotherapy. A Pathophysiologic Approach, 2nd ed. New York: Elsevier, 1992:312–335.
- Park BK. Warfarin: metabolism and mode of action. Biochem Pharmacol 1988; 37:19–27.
- Cain D, Hutson SM, Wallin R. Assembly of the warfarin-sensitive vitamin K 2,3-epoxide reductase enzyme complex in the endoplasmic reticulum membrane. J Biol Chem 1997; 272:29068–29075.
- Gallop PM, Lian JB, Hauschka PV. Carboxylated calcium binding proteins and vitamin K. N Engl J Med 1980; 302:1460–1466.
- Rost S, Fregin A, Ivaskevicius V, et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 2004; 427:537–541.
- Loebstein R, Dovskin I, Halkin H, et al. A coding VKORC1 Asp36-Tyr polymorphism predisposes to warfarin resistance. Blood 2007; 109:2477–2480.
- Bentley DP, Backhouse G, Hutchings A, Haddon RL, Spragg B, Routledge PA. Investigation of patients with abnormal response to warfarin. Br J Clin Pharmacol 1986; 22:37–41.
- Eriksson BI, Borris LC, Friedman RJ, et al. RECORD1 Study Group. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med 2008; 358:2765–2775.
- Kakkar AK, Brenner B, Dahl OE, et al; RECORD2 Investigators. Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial. Lancet 2008; 372:31–39.
- Lassen MR, Ageno W, Borris LC, et al; RECORD3 Investigators. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med 2008; 358:2776–2786.
- Wolowacz SE, Roskell NS, Plumb JM, Caprini JA, Eriksson BI. Efficacy and safety of dabigatran etexilate for the prevention of venous thromboembolism following total hip or knee arthroplasty. A meta-analysis. Thromb Haemost 2009; 101:77–85.
Warfarin (coumadin) differs from most other drugs in that the dosage required to achieve a desired therapeutic effect varies greatly among individuals. This variability can lead to therapeutic failure, potentially resulting in new thrombosis, or, at the other extreme, to life-threatening bleeding.
Further, there is no reliable means to identify patients who require unusually high doses of warfarin, although genetic testing may become available in the future.
See related patient information
Warfarin, a coumarin derivative first synthesized in 1948, is still the only oral anticoagulant available for long-term use in the United States. Indications for its use include the treatment and, to a lesser extent, the prevention of arterial and venous thromboembolism. It is also used for long-term anticoagulation in patients with atrial arrhythmias (atrial fibrillation and atrial flutter) and mechanical heart valves.
In the paragraphs that follow, we review the causes of warfarin resistance and how to recognize and manage it.
WHAT IS WARFARIN RESISTANCE?
Resistance to warfarin has been described as the inability to prolong the prothrombin time or raise the international normalized ratio (INR) into the therapeutic range when the drug is given at normally prescribed doses.1
However, a higher warfarin requirement does not itself establish the diagnosis of warfarin resistance. The prevalence of warfarin resistance varies by patient population and is difficult to determine. The difficulty lies largely in accounting for dietary factors and in defining normal metabolic variations among individuals.
The range of normally recommended daily or weekly warfarin doses to maintain a therapeutic prothrombin time or INR depends on the study population. Nevertheless, patients who need more than 105 mg per week (15 mg/day) should be considered warfarin-resistant. These patients are likely to be in the top 5% for warfarin doses within an anticoagulated cohort.
Warfarin resistance is different than warfarin failure, which is defined as a new thrombotic event despite a therapeutic prothrombin time and INR. This situation is commonly seen in patients with malignant diseases.
An important characteristic of warfarin resistance is that patients need much smaller doses of vitamin K to reverse the effect of warfarin.2 Thijssen3 showed that, in warfarin-resistant rats, warfarin did not irreversibly inhibit vitamin K1 2,3-epoxide reductase (VKORC1) activity. This is consistent with the vitamin K hypersensitivity observed in warfarin-resistant people.2,3
WHAT CAUSES WARFARIN RESISTANCE?
Warfarin resistance can be classified in practical terms as acquired vs hereditary, or in mechanistic terms as pharmacokinetic vs pharmacodynamic.
Acquired vs hereditary resistance
Hulse4 categorizes warfarin resistance as either acquired or hereditary.
Acquired resistance to warfarin may result from:
- Poor patient compliance (the most common cause)
- High consumption of vitamin K
- Increased clearance (see Warfarin is metabolized by P450 enzymes5–11)
- Drug interactions (Table 1).12,13
Hereditary resistance has been postulated to be caused by genetic factors that result either in faster metabolism of the drug (a form of pharmacokinetic resistance) or in lower activity of the drug (pharmacodynamic resistance). Polymorphisms may play a role, as some VKORC1 and CYP2C9 variant alleles are known to be associated with increased sensitivity to warfarin.14
However, the genetic mechanisms of warfarin resistance are not clearly understood, despite several case reports of hereditary resistance confirmed by similar patterns of resistance in immediate family members.15–19 More than one mechanism is likely. There is ample room for further insight into genetic polymorphisms underlying hereditary warfarin resistance. More on this topic is included in the sections below.
Pharmacokinetic resistance
Pharmacokinetic resistance can result from diminished absorption or increased elimination of the drug. Causes of diminished absorption include emesis, diarrhea, and malabsorption syndrome.
The mechanism of increased warfarin clearance has not been delineated, although the following have been implicated.
Genetic factors. Duplication or multiplication of cytochrome P450 enzyme genes has been described as contributing to a phenotype of ultrarapid metabolism. Some people may carry multiple copies of the CYP2C9 gene, as has already been reported for cytochrome P450 CYP2D6 and CYP2A6.7,8 It is also plausible that rare allelic variants of CYP2C9 exist that are associated with higher-than-normal activity, given that there are alleles known to predispose to warfarin sensitivity.
Hypoalbuminemia may increase the free fraction of warfarin, leading to enhanced rates of clearance and a shorter plasma half-life.15
Hyperalbuminemia may paradoxically also contribute to warfarin resistance via drug binding.
Hyperlipidemia. Several observers have found that lowering serum lipids, primarily triglycerides, increases the sensitivity to warfarin irrespective of the means used to achieve this decrease.20 This most likely results in a decreased pool of vitamin K, some of which is bound to triglycerides.21 Conversely, patients receiving intravenous lipids with total parenteral nutrition have also been diagnosed clinically with warfarin resistance,22 and rat models have shown an association between a lipidrich diet and increased vitamin K-dependent factor activity.23
Diuretics may decrease the response to warfarin by reducing the plasma volume, with a subsequent increase in clotting factor activity.24
Pharmacodynamic resistance
Potential mechanisms of pharmacodynamic warfarin resistance described in rats and in people include:
- Increased affinity of vitamin K1, 2,3-epoxide reductase complex (VKOR) for vitamin K25,26 (see How warfarin works2,10,11,27–30)
- Prolongation of normal clotting factor activity16
- Production of clotting factors that is not dependent on vitamin K16
- Decreased VKOR sensitivity to warfarin.26
In rats, these mechanisms are manifested by relatively high doses of warfarin being required to achieve poisoning. In humans, they result in high doses being needed to achieve a therapeutic effect in the setting of normal warfarin pharmacokinetics, normal warfarin concentration, and normal half-lives of blood clotting proteins.
Genetics of pharmacodynamic resistance. Pharmacodynamic warfarin resistance has also been described with inheritance of a monogenetic dominant trait. An early study by O’Reilly24 traced anticoagulation resistance to a genetically linked abnormality of interaction between warfarin and a putative vitamin K receptor.
In one patient with hereditary resistance and high warfarin requirements, a heterozygous point mutation in the VKORC1 gene was identified.31 This results in a substitution that lies in a conserved (normally constant or unchanging DNA sequence in a genome) region of VKORC1 that contains three of four previously identified amino acid substitutions associated with warfarin resistance (Val29Leu, Val45Ala, and Arg58Gly). Further investigation is required to fully characterize the structure-function relationship for VKORC1 and to determine the relationship between the VKORC1 genotype and other pharmacogenetic determinants of warfarin dose-response.
Separately, Loebstein et al32 reported a new mutation, Asp36Tyr, which was common in Jewish ethnic groups of Ethiopian descent (in whom the prevalence is 5%) and Ashkenazi descent (prevalence 4%). In that study, Asp36Tyr carriers needed doses of more than 70 mg per week, placing them towards the high end of the usual warfarin dosing range.
Daly and Aithal7 discovered that warfarinresistant rats overexpressed a protein known as calumenin. This protein is situated in the endoplasmic reticulum and appears to interact with VKOR, decreasing the binding of warfarin. In mice, the calumenin gene is located on chromosome 7, where the gene for VKORC1 is also located.
DIAGNOSIS BY HISTORY AND LABORATORY STUDIES
A full drug and diet history is invaluable in diagnosing potential causes of warfarin resistance (Table 1).
Plasma warfarin levels that are subtherapeutic should raise suspicion of intestinal malabsorption or poor compliance. Poor compliance might be more appropriately seen as a mimic of warfarin resistance. Studies in humans suggest that a therapeutic total plasma warfarin level lies between 0.5 μg/mL and 3.0 μg/mL,10 though the range may vary among laboratories and patient populations.
Warfarin absorption and clearance can be evaluated by analyzing plasma levels at specific intervals after administration, eg, every 60 to 180 minutes. The drug’s half-life can be determined on the basis of its concentrations in different time samples. Normally, the S-enantiomer of warfarin is cleared at twice the rate of the R-enantiomer (5.2 vs 2.5 mL/min/70 kg).8 A normal clearance rate confirms that resistance to warfarin is not due to enhanced elimination.
Clotting assays of factors II, VII, IX, and X may be a more precise way to assess the pharmacodynamics of warfarin,10 although there is no strong evidence to support routine use of such assays. Some studies suggest targeting factor II and factor X activity levels of 10% to 30% of normal biologic activity for a therapeutic warfarin effect in patients with an unreliable or prolonged baseline prothrombin time and INR, such as those with lupus anticoagulant.
TREAT THE CAUSE
Once the type of warfarin resistance has been determined, treatment should be oriented toward the cause.
Educate the patient
The importance of compliance should be reinforced. Educating the patient about diet and other medications that may interact with warfarin is also important. (See an example of patient education material.)
Increase the warfarin dose
If the patient truly has hereditary resistance, there are two approaches to treatment.
The first is to increase the warfarin dose until the prothrombin time and INR are in the therapeutic ranges. When indicated, the warfarin dose can be safely titrated upward to more than 100 mg per day in patients who are monitored regularly—as all patients on chronic warfarin therapy should be—and whose other medications are otherwise stable. One such example is reported in a warfarinresistant patient who needed 145 mg/day to maintain a therapeutic prothrombin time.22
Try other anticoagulants?
The second approach is to change to another type of anticoagulant. However, there is no strong evidence in favor of this approach over prescribing larger dosages of warfarin.
Other anticoagulant drugs currently available in the United States include subcutaneous heparins (unfractionated and low-molecular-weight heparins) and the subcutaneous factor Xa inhibitor fondaparinux (Arixtra).
Agents not available in the United States include the following.
Dabigatran, an oral direct thrombin inhibitor, is undergoing phase 3 studies of its use for long-term anticoagulation.
Rivaroxaban (a direct factor Xa inhibitor) and dabigatran have been approved in Canada and the European Union to prevent venous thromboembolism after knee and hip arthroplasty, based on prospective comparisons with enoxaparin (Lovenox).34–37
Vitamin K antagonists other than warfarin that are not available in the United States include bishydroxycoumarin (which has limitations including slow absorption and high frequency of gastrointestinal side effects), phenprocoumon, and acenocoumarol. Another is phenindione, which has been associated with serious hypersensitivity reactions, some of which proved fatal and occurred within a few weeks of initiating therapy.
Warfarin (coumadin) differs from most other drugs in that the dosage required to achieve a desired therapeutic effect varies greatly among individuals. This variability can lead to therapeutic failure, potentially resulting in new thrombosis, or, at the other extreme, to life-threatening bleeding.
Further, there is no reliable means to identify patients who require unusually high doses of warfarin, although genetic testing may become available in the future.
See related patient information
Warfarin, a coumarin derivative first synthesized in 1948, is still the only oral anticoagulant available for long-term use in the United States. Indications for its use include the treatment and, to a lesser extent, the prevention of arterial and venous thromboembolism. It is also used for long-term anticoagulation in patients with atrial arrhythmias (atrial fibrillation and atrial flutter) and mechanical heart valves.
In the paragraphs that follow, we review the causes of warfarin resistance and how to recognize and manage it.
WHAT IS WARFARIN RESISTANCE?
Resistance to warfarin has been described as the inability to prolong the prothrombin time or raise the international normalized ratio (INR) into the therapeutic range when the drug is given at normally prescribed doses.1
However, a higher warfarin requirement does not itself establish the diagnosis of warfarin resistance. The prevalence of warfarin resistance varies by patient population and is difficult to determine. The difficulty lies largely in accounting for dietary factors and in defining normal metabolic variations among individuals.
The range of normally recommended daily or weekly warfarin doses to maintain a therapeutic prothrombin time or INR depends on the study population. Nevertheless, patients who need more than 105 mg per week (15 mg/day) should be considered warfarin-resistant. These patients are likely to be in the top 5% for warfarin doses within an anticoagulated cohort.
Warfarin resistance is different than warfarin failure, which is defined as a new thrombotic event despite a therapeutic prothrombin time and INR. This situation is commonly seen in patients with malignant diseases.
An important characteristic of warfarin resistance is that patients need much smaller doses of vitamin K to reverse the effect of warfarin.2 Thijssen3 showed that, in warfarin-resistant rats, warfarin did not irreversibly inhibit vitamin K1 2,3-epoxide reductase (VKORC1) activity. This is consistent with the vitamin K hypersensitivity observed in warfarin-resistant people.2,3
WHAT CAUSES WARFARIN RESISTANCE?
Warfarin resistance can be classified in practical terms as acquired vs hereditary, or in mechanistic terms as pharmacokinetic vs pharmacodynamic.
Acquired vs hereditary resistance
Hulse4 categorizes warfarin resistance as either acquired or hereditary.
Acquired resistance to warfarin may result from:
- Poor patient compliance (the most common cause)
- High consumption of vitamin K
- Increased clearance (see Warfarin is metabolized by P450 enzymes5–11)
- Drug interactions (Table 1).12,13
Hereditary resistance has been postulated to be caused by genetic factors that result either in faster metabolism of the drug (a form of pharmacokinetic resistance) or in lower activity of the drug (pharmacodynamic resistance). Polymorphisms may play a role, as some VKORC1 and CYP2C9 variant alleles are known to be associated with increased sensitivity to warfarin.14
However, the genetic mechanisms of warfarin resistance are not clearly understood, despite several case reports of hereditary resistance confirmed by similar patterns of resistance in immediate family members.15–19 More than one mechanism is likely. There is ample room for further insight into genetic polymorphisms underlying hereditary warfarin resistance. More on this topic is included in the sections below.
Pharmacokinetic resistance
Pharmacokinetic resistance can result from diminished absorption or increased elimination of the drug. Causes of diminished absorption include emesis, diarrhea, and malabsorption syndrome.
The mechanism of increased warfarin clearance has not been delineated, although the following have been implicated.
Genetic factors. Duplication or multiplication of cytochrome P450 enzyme genes has been described as contributing to a phenotype of ultrarapid metabolism. Some people may carry multiple copies of the CYP2C9 gene, as has already been reported for cytochrome P450 CYP2D6 and CYP2A6.7,8 It is also plausible that rare allelic variants of CYP2C9 exist that are associated with higher-than-normal activity, given that there are alleles known to predispose to warfarin sensitivity.
Hypoalbuminemia may increase the free fraction of warfarin, leading to enhanced rates of clearance and a shorter plasma half-life.15
Hyperalbuminemia may paradoxically also contribute to warfarin resistance via drug binding.
Hyperlipidemia. Several observers have found that lowering serum lipids, primarily triglycerides, increases the sensitivity to warfarin irrespective of the means used to achieve this decrease.20 This most likely results in a decreased pool of vitamin K, some of which is bound to triglycerides.21 Conversely, patients receiving intravenous lipids with total parenteral nutrition have also been diagnosed clinically with warfarin resistance,22 and rat models have shown an association between a lipidrich diet and increased vitamin K-dependent factor activity.23
Diuretics may decrease the response to warfarin by reducing the plasma volume, with a subsequent increase in clotting factor activity.24
Pharmacodynamic resistance
Potential mechanisms of pharmacodynamic warfarin resistance described in rats and in people include:
- Increased affinity of vitamin K1, 2,3-epoxide reductase complex (VKOR) for vitamin K25,26 (see How warfarin works2,10,11,27–30)
- Prolongation of normal clotting factor activity16
- Production of clotting factors that is not dependent on vitamin K16
- Decreased VKOR sensitivity to warfarin.26
In rats, these mechanisms are manifested by relatively high doses of warfarin being required to achieve poisoning. In humans, they result in high doses being needed to achieve a therapeutic effect in the setting of normal warfarin pharmacokinetics, normal warfarin concentration, and normal half-lives of blood clotting proteins.
Genetics of pharmacodynamic resistance. Pharmacodynamic warfarin resistance has also been described with inheritance of a monogenetic dominant trait. An early study by O’Reilly24 traced anticoagulation resistance to a genetically linked abnormality of interaction between warfarin and a putative vitamin K receptor.
In one patient with hereditary resistance and high warfarin requirements, a heterozygous point mutation in the VKORC1 gene was identified.31 This results in a substitution that lies in a conserved (normally constant or unchanging DNA sequence in a genome) region of VKORC1 that contains three of four previously identified amino acid substitutions associated with warfarin resistance (Val29Leu, Val45Ala, and Arg58Gly). Further investigation is required to fully characterize the structure-function relationship for VKORC1 and to determine the relationship between the VKORC1 genotype and other pharmacogenetic determinants of warfarin dose-response.
Separately, Loebstein et al32 reported a new mutation, Asp36Tyr, which was common in Jewish ethnic groups of Ethiopian descent (in whom the prevalence is 5%) and Ashkenazi descent (prevalence 4%). In that study, Asp36Tyr carriers needed doses of more than 70 mg per week, placing them towards the high end of the usual warfarin dosing range.
Daly and Aithal7 discovered that warfarinresistant rats overexpressed a protein known as calumenin. This protein is situated in the endoplasmic reticulum and appears to interact with VKOR, decreasing the binding of warfarin. In mice, the calumenin gene is located on chromosome 7, where the gene for VKORC1 is also located.
DIAGNOSIS BY HISTORY AND LABORATORY STUDIES
A full drug and diet history is invaluable in diagnosing potential causes of warfarin resistance (Table 1).
Plasma warfarin levels that are subtherapeutic should raise suspicion of intestinal malabsorption or poor compliance. Poor compliance might be more appropriately seen as a mimic of warfarin resistance. Studies in humans suggest that a therapeutic total plasma warfarin level lies between 0.5 μg/mL and 3.0 μg/mL,10 though the range may vary among laboratories and patient populations.
Warfarin absorption and clearance can be evaluated by analyzing plasma levels at specific intervals after administration, eg, every 60 to 180 minutes. The drug’s half-life can be determined on the basis of its concentrations in different time samples. Normally, the S-enantiomer of warfarin is cleared at twice the rate of the R-enantiomer (5.2 vs 2.5 mL/min/70 kg).8 A normal clearance rate confirms that resistance to warfarin is not due to enhanced elimination.
Clotting assays of factors II, VII, IX, and X may be a more precise way to assess the pharmacodynamics of warfarin,10 although there is no strong evidence to support routine use of such assays. Some studies suggest targeting factor II and factor X activity levels of 10% to 30% of normal biologic activity for a therapeutic warfarin effect in patients with an unreliable or prolonged baseline prothrombin time and INR, such as those with lupus anticoagulant.
TREAT THE CAUSE
Once the type of warfarin resistance has been determined, treatment should be oriented toward the cause.
Educate the patient
The importance of compliance should be reinforced. Educating the patient about diet and other medications that may interact with warfarin is also important. (See an example of patient education material.)
Increase the warfarin dose
If the patient truly has hereditary resistance, there are two approaches to treatment.
The first is to increase the warfarin dose until the prothrombin time and INR are in the therapeutic ranges. When indicated, the warfarin dose can be safely titrated upward to more than 100 mg per day in patients who are monitored regularly—as all patients on chronic warfarin therapy should be—and whose other medications are otherwise stable. One such example is reported in a warfarinresistant patient who needed 145 mg/day to maintain a therapeutic prothrombin time.22
Try other anticoagulants?
The second approach is to change to another type of anticoagulant. However, there is no strong evidence in favor of this approach over prescribing larger dosages of warfarin.
Other anticoagulant drugs currently available in the United States include subcutaneous heparins (unfractionated and low-molecular-weight heparins) and the subcutaneous factor Xa inhibitor fondaparinux (Arixtra).
Agents not available in the United States include the following.
Dabigatran, an oral direct thrombin inhibitor, is undergoing phase 3 studies of its use for long-term anticoagulation.
Rivaroxaban (a direct factor Xa inhibitor) and dabigatran have been approved in Canada and the European Union to prevent venous thromboembolism after knee and hip arthroplasty, based on prospective comparisons with enoxaparin (Lovenox).34–37
Vitamin K antagonists other than warfarin that are not available in the United States include bishydroxycoumarin (which has limitations including slow absorption and high frequency of gastrointestinal side effects), phenprocoumon, and acenocoumarol. Another is phenindione, which has been associated with serious hypersensitivity reactions, some of which proved fatal and occurred within a few weeks of initiating therapy.
- Lefrere JJ, Horellou MH, Conard J, Samama M. Proposed classification of resistance to oral anticoagulant therapy. J Clin Pathol 1987; 40:242.
- Linder MW. Genetic mechanisms for hypersensitivity and resistance to the anticoagulant warfarin. Clin Chim Acta 2001; 308:9–15.
- Thijssen HH. Warfarin resistance. Vitamin K epoxide reductase of Scottish resistance gene is not irreversibly blocked by warfarin. Biochem Pharmacol 1987; 36:2753–2757.
- Hulse ML. Warfarin resistance: diagnosis and therapeutic alternative. Pharmacotherapy 1996; 16:1009–1017.
- Hirsh J, Dalen JE, Deykin D, Poller L, Bussey H. Oral anticoagulants. Mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 1995; 108( suppl 4):231S–234S.
- Daly AK, King BP. Pharmacogenetics of oral anticoagulants. Pharmacogenetics 2003; 13:247–252.
- Daly AK, Aithal GP. Genetic regulation of warfarin metabolism and response. Semin Vasc Med 2003; 3:231–238.
- Takahashi H, Echizen H. Pharmacogenetics of warfarin elimination and its clinical implications. Clin Pharmacokinet 2001; 40:587–603.
- Retti AE, Wienkers LC, Gonzalez FJ, Trager WF, Korezekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allele variant of CYP2C9. Pharmacogenetics 1994; 4:39–42.
- Porter RS, Sawyer WR. Warfarin. In:Evans WE, Shentag JJ, Jusko WJ, editors. Applied Pharmacokinetics. Principles of Therapeutics Drug Monitoring, 3rd ed. Washington, DC: Applied Therapeutics, 1992: 31.1–31.46.
- Warrell DA, Cox TM, Firth JD. Oxford Textbook of Medicine, 4th ed. Oxford University Press, 2003:734.
- Holbrook AM, Pereira JA, Labiris R, et al. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med 2005; 165:1095–1106.
- Medical Economics Staff. Physicians’ Desk Reference, 55th Ed. Medical Economics, 2001:1139–1140.
- Schwarz UI, Ritchie MD, Bradford Y, et al. Genetic determinants of response to warfarin during initial anticoagulation. N Engl J Med 2008; 358:999–1008.
- Diab F, Feffer S. Hereditary warfarin resistance. South Med J 1994; 87:407–409.
- O’Reilly RA. The second reported kindred with hereditary resistance to oral anticoagulant drugs. N Engl J Med 1970; 282:1448–1451.
- O’Reilly RA, Aggeler PM, Hoag MS, Leong LS, Kropatkin ML. Hereditary transmission of exceptional resistance to coumarin anticoagulant drugs. The first reported kindred. N Engl J Med 1964; 271:809–815.
- Alving BM, Strickler MP, Knight RD, Barr CF, Berenberg JL, Peek CC. Hereditary warfarin resistance. Investigation of rare phenomenon. Arch Intern Med 1985; 145:499–501.
- Warrier L, Brennan CA, Lusher JM. Familial warfarin resistance in a black child. Am J Pediatr Hematol Oncol 1986; 8:346–347.
- Nikkila EA, Pelkonen R. Serum lipid-reducing agents and anticoagulant requirement. Lancet 1963; 1:332.
- Robinson A, Liau FO, Routledge PA, Backhouse G, Spragg BP, Bentley DP. Lipids and warfarin requirements. Thromb Haemost 1990; 63:148–149.
- MacLaren R, Wachsman BA, Swift DK, Kuhl DA. Warfarin resistance associated with intravenous lipid administration: discussion of propofol and review of the literature. Pharmacotherapy 1997; 17:1331–1337.
- DeCurtis A, D’Adamo MC, Amore C, et al. Experimental arterial thrombosis in genetically or diet induced hyperlipidemia in rats—role of vitamin K-dependent clotting factors and prevention by low-intensity oral anticoagulation. Thromb Haemost 2001; 86:1440–1448.
- O’Reilly RA. Drug interaction involving oral anticoagulation. In:Melmon KL, editor. Cardiovascular Drug Therapy, Philadelphia; FA Davis, 1975:23–41.
- O’ Reilly RA, Pool JG, Aggeler PM. Hereditary resistance to coumarin anticoagulation drugs in man and rat. Ann N Y Acad Sci 1968; 151:913–931.
- Cain D, Hutson SM, Wallin R. Warfarin resistance is associated with a protein component of the vitamin K 2,3-epoxide reductase enzyme complex in rat liver. Thromb Haemost 1998; 80:128–133.
- Rodvold KA, Quandt CM, Friedenberg WR. Thromboembolic disorders. In:DiPiro JT, Talbert RL, editors. Pharmacotherapy. A Pathophysiologic Approach, 2nd ed. New York: Elsevier, 1992:312–335.
- Park BK. Warfarin: metabolism and mode of action. Biochem Pharmacol 1988; 37:19–27.
- Cain D, Hutson SM, Wallin R. Assembly of the warfarin-sensitive vitamin K 2,3-epoxide reductase enzyme complex in the endoplasmic reticulum membrane. J Biol Chem 1997; 272:29068–29075.
- Gallop PM, Lian JB, Hauschka PV. Carboxylated calcium binding proteins and vitamin K. N Engl J Med 1980; 302:1460–1466.
- Rost S, Fregin A, Ivaskevicius V, et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 2004; 427:537–541.
- Loebstein R, Dovskin I, Halkin H, et al. A coding VKORC1 Asp36-Tyr polymorphism predisposes to warfarin resistance. Blood 2007; 109:2477–2480.
- Bentley DP, Backhouse G, Hutchings A, Haddon RL, Spragg B, Routledge PA. Investigation of patients with abnormal response to warfarin. Br J Clin Pharmacol 1986; 22:37–41.
- Eriksson BI, Borris LC, Friedman RJ, et al. RECORD1 Study Group. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med 2008; 358:2765–2775.
- Kakkar AK, Brenner B, Dahl OE, et al; RECORD2 Investigators. Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial. Lancet 2008; 372:31–39.
- Lassen MR, Ageno W, Borris LC, et al; RECORD3 Investigators. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med 2008; 358:2776–2786.
- Wolowacz SE, Roskell NS, Plumb JM, Caprini JA, Eriksson BI. Efficacy and safety of dabigatran etexilate for the prevention of venous thromboembolism following total hip or knee arthroplasty. A meta-analysis. Thromb Haemost 2009; 101:77–85.
- Lefrere JJ, Horellou MH, Conard J, Samama M. Proposed classification of resistance to oral anticoagulant therapy. J Clin Pathol 1987; 40:242.
- Linder MW. Genetic mechanisms for hypersensitivity and resistance to the anticoagulant warfarin. Clin Chim Acta 2001; 308:9–15.
- Thijssen HH. Warfarin resistance. Vitamin K epoxide reductase of Scottish resistance gene is not irreversibly blocked by warfarin. Biochem Pharmacol 1987; 36:2753–2757.
- Hulse ML. Warfarin resistance: diagnosis and therapeutic alternative. Pharmacotherapy 1996; 16:1009–1017.
- Hirsh J, Dalen JE, Deykin D, Poller L, Bussey H. Oral anticoagulants. Mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 1995; 108( suppl 4):231S–234S.
- Daly AK, King BP. Pharmacogenetics of oral anticoagulants. Pharmacogenetics 2003; 13:247–252.
- Daly AK, Aithal GP. Genetic regulation of warfarin metabolism and response. Semin Vasc Med 2003; 3:231–238.
- Takahashi H, Echizen H. Pharmacogenetics of warfarin elimination and its clinical implications. Clin Pharmacokinet 2001; 40:587–603.
- Retti AE, Wienkers LC, Gonzalez FJ, Trager WF, Korezekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allele variant of CYP2C9. Pharmacogenetics 1994; 4:39–42.
- Porter RS, Sawyer WR. Warfarin. In:Evans WE, Shentag JJ, Jusko WJ, editors. Applied Pharmacokinetics. Principles of Therapeutics Drug Monitoring, 3rd ed. Washington, DC: Applied Therapeutics, 1992: 31.1–31.46.
- Warrell DA, Cox TM, Firth JD. Oxford Textbook of Medicine, 4th ed. Oxford University Press, 2003:734.
- Holbrook AM, Pereira JA, Labiris R, et al. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med 2005; 165:1095–1106.
- Medical Economics Staff. Physicians’ Desk Reference, 55th Ed. Medical Economics, 2001:1139–1140.
- Schwarz UI, Ritchie MD, Bradford Y, et al. Genetic determinants of response to warfarin during initial anticoagulation. N Engl J Med 2008; 358:999–1008.
- Diab F, Feffer S. Hereditary warfarin resistance. South Med J 1994; 87:407–409.
- O’Reilly RA. The second reported kindred with hereditary resistance to oral anticoagulant drugs. N Engl J Med 1970; 282:1448–1451.
- O’Reilly RA, Aggeler PM, Hoag MS, Leong LS, Kropatkin ML. Hereditary transmission of exceptional resistance to coumarin anticoagulant drugs. The first reported kindred. N Engl J Med 1964; 271:809–815.
- Alving BM, Strickler MP, Knight RD, Barr CF, Berenberg JL, Peek CC. Hereditary warfarin resistance. Investigation of rare phenomenon. Arch Intern Med 1985; 145:499–501.
- Warrier L, Brennan CA, Lusher JM. Familial warfarin resistance in a black child. Am J Pediatr Hematol Oncol 1986; 8:346–347.
- Nikkila EA, Pelkonen R. Serum lipid-reducing agents and anticoagulant requirement. Lancet 1963; 1:332.
- Robinson A, Liau FO, Routledge PA, Backhouse G, Spragg BP, Bentley DP. Lipids and warfarin requirements. Thromb Haemost 1990; 63:148–149.
- MacLaren R, Wachsman BA, Swift DK, Kuhl DA. Warfarin resistance associated with intravenous lipid administration: discussion of propofol and review of the literature. Pharmacotherapy 1997; 17:1331–1337.
- DeCurtis A, D’Adamo MC, Amore C, et al. Experimental arterial thrombosis in genetically or diet induced hyperlipidemia in rats—role of vitamin K-dependent clotting factors and prevention by low-intensity oral anticoagulation. Thromb Haemost 2001; 86:1440–1448.
- O’Reilly RA. Drug interaction involving oral anticoagulation. In:Melmon KL, editor. Cardiovascular Drug Therapy, Philadelphia; FA Davis, 1975:23–41.
- O’ Reilly RA, Pool JG, Aggeler PM. Hereditary resistance to coumarin anticoagulation drugs in man and rat. Ann N Y Acad Sci 1968; 151:913–931.
- Cain D, Hutson SM, Wallin R. Warfarin resistance is associated with a protein component of the vitamin K 2,3-epoxide reductase enzyme complex in rat liver. Thromb Haemost 1998; 80:128–133.
- Rodvold KA, Quandt CM, Friedenberg WR. Thromboembolic disorders. In:DiPiro JT, Talbert RL, editors. Pharmacotherapy. A Pathophysiologic Approach, 2nd ed. New York: Elsevier, 1992:312–335.
- Park BK. Warfarin: metabolism and mode of action. Biochem Pharmacol 1988; 37:19–27.
- Cain D, Hutson SM, Wallin R. Assembly of the warfarin-sensitive vitamin K 2,3-epoxide reductase enzyme complex in the endoplasmic reticulum membrane. J Biol Chem 1997; 272:29068–29075.
- Gallop PM, Lian JB, Hauschka PV. Carboxylated calcium binding proteins and vitamin K. N Engl J Med 1980; 302:1460–1466.
- Rost S, Fregin A, Ivaskevicius V, et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 2004; 427:537–541.
- Loebstein R, Dovskin I, Halkin H, et al. A coding VKORC1 Asp36-Tyr polymorphism predisposes to warfarin resistance. Blood 2007; 109:2477–2480.
- Bentley DP, Backhouse G, Hutchings A, Haddon RL, Spragg B, Routledge PA. Investigation of patients with abnormal response to warfarin. Br J Clin Pharmacol 1986; 22:37–41.
- Eriksson BI, Borris LC, Friedman RJ, et al. RECORD1 Study Group. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med 2008; 358:2765–2775.
- Kakkar AK, Brenner B, Dahl OE, et al; RECORD2 Investigators. Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial. Lancet 2008; 372:31–39.
- Lassen MR, Ageno W, Borris LC, et al; RECORD3 Investigators. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med 2008; 358:2776–2786.
- Wolowacz SE, Roskell NS, Plumb JM, Caprini JA, Eriksson BI. Efficacy and safety of dabigatran etexilate for the prevention of venous thromboembolism following total hip or knee arthroplasty. A meta-analysis. Thromb Haemost 2009; 101:77–85.
KEY POINTS
- The most common cause of warfarin resistance is noncompliance. Others include poor absorption, high vitamin K intake, hypersensitivity to vitamin K, and rapid drug deactivation.
- Patient education is necessary to improve compliance and to mitigate adverse effects of warfarin therapy, regardless of the dose.
- In time, it may be possible to individualize anticoagulant dosing on the basis of genetic testing for patients with warfarin resistance, although currently such tests are not routinely advocated and are usually done only in specialized laboratories.
- In true hereditary warfarin resistance, there are two approaches to treatment: increase the warfarin dosage (perhaps to as high as 100 mg/day or more), or switch to another anticoagulant.