User login
Prasugrel for acute coronary syndromes: Faster, more potent, but higher bleeding risk
Prasugrel (Effient) is more potent and consistent in its effects than clopidogrel (Plavix), thus preventing more thrombotic events—but at a price of more bleeding. Therefore, the drugs must be appropriately selected for the individual patient.
Over the last 9 years, the thienopyridines—ticlopidine (Ticlid), clopidogrel, and now prasugrel—have become essential tools for treating acute coronary syndromes.
The usual underlying mechanism of acute coronary syndromes is thrombosis, caused by rupture of atherosclerotic plaque.1 Accordingly, antithrombotic agents—aspirin, heparin, lowmolecular-weight heparin, glycoprotein IIb/IIIa inhibitors, the direct thrombin inhibitor bivalirudin (Angiomax), and thienopyridines—have all been shown to reduce the risk of major adverse cardiac outcomes in this setting.
In this article, we review the pharmacology and evidence of effectiveness of the thienopyridine drugs, focusing on prasugrel, the latest thienopyridine to be approved by the US Food and Drug Administration (FDA).
THIENOPYRIDINES INHIBIT PLATELET ACTIVATION AND AGGREGATION
Thienopyridines are prodrugs that require conversion by hepatic cytochrome P450 enzymes. The active metabolites bind irreversibly to platelet P2Y12 receptors. Consequently, they permanently block signalling mediated by platelet adenosine diphosphate-P2Y12 receptors, thereby inhibiting glycoprotein IIb/IIIa receptor activation and platelet aggregation.
Aspirin, in contrast, inhibits platelets by blocking the thromboxane-mediated pathway. Therefore, the combination of aspirin plus a thienopyridine has an additive effect.2
The effect of thienopyridines on platelets is irreversible. Therefore, although the half-life of prasugrel’s active metabolite is 3.7 hours, its inhibitory effects last for 96 hours, essentially the time for half the body’s circulating platelets to be replaced.
TICLOPIDINE, THE FIRST THIENOPYRIDINE
Ticlopidine was the first thienopyridine to be approved by the FDA. Its initial studies in unstable angina were small, their designs did not call for patients to concurrently receive aspirin, and all they showed was that ticlopidine was about as beneficial as aspirin. Consequently, the studies had little impact on clinical practice.3
In a pivotal trial,4 patients who received coronary stents were randomized to afterward receive either the combination of ticlopidine plus aspirin or anticoagulation therapy with heparin, phenprocoumon (a coumarin derivative available in Europe), and aspirin. At 30 days, an ischemic complication (death, myocardial infarction [MI], repeat intervention) had occurred in 6.2% of the anticoagulation therapy group vs 1.6% of the ticlopidine group, a risk reduction of 75%. Rates of stent occlusion, MI, and revascularization were 80% to 85% lower in the ticlodipine group. This study paved the way for widespread use of thienopyridines.
Ticlopidine’s use was limited, however, by a 2.4% incidence of serious granulocytopenia and rare cases of thrombocytopenic purpura.
BENEFIT OF CLOPIDOGREL
Although prasugrel is the focus of this review, the trials of prasugrel all compared its efficacy with that of clopidogrel. Furthermore, many patients should still receive clopidogrel and not prasugrel, so it is important to be familiar with the evidence of clopidogrel’s benefit.
Once approved for clinical use, clopidogrel was substituted for ticlopidine in patients undergoing coronary stenting on the basis of studies showing it to be at least as effective as ticlopidine and more tolerable. A series of trials of clopidogrel were done in patients across a spectrum of risk groups, from those at high risk of coronary heart disease to those presenting with ST-elevation MI. The time of pretreatment in the studies ranged from 3 hours to 6 days before percutaneous coronary intervention, and the duration of treatment following intervention ranged from 30 days to 1 year.
Clopidogrel in non-ST-elevation acute coronary syndromes
The CURE trial2 (Clopidogrel in Unstable Angina to Prevent Recurrent Events), published in 2001, established clopidogrel as a therapy for unstable ischemic syndromes, whether treated medically or with revascularization. In that trial, 12,562 patients with acute coronary syndromes without ST elevation (ie, unstable angina or non-ST-elevation MI), as defined by electrocardiographic changes or positive cardiac markers, were randomized to receive clopidogrel (a 300-mg loading dose followed by 75-mg maintenance doses) or placebo for a mean duration of 9 months. All patients also received aspirin 75 mg to 325 mg daily.
The composite outcome of death from cardiovascular causes, nonfatal MI, or stroke occurred in 20% fewer patients treated with clopidogrel than with placebo (9.3% vs 11.4%). The benefit was similar in patients undergoing revascularization compared with those treated medically.
Although there were significantly more cases of major bleeding in the clopidogrel group than in the placebo group (3.7% vs 2.7%), the number of episodes of life-threatening bleeding or hemorrhagic strokes was the same.
PCI-CURE5 was a substudy of the CURE trial in patients who underwent a percutaneous coronary intervention. Patients were pretreated with clopidogrel or placebo for a mean of 6 days before the procedure. Afterward, they all received clopidogrel plus aspirin in an unblinded fashion for 2 to 4 weeks, and then the randomized study drug was resumed for a mean of 8 months.
Significantly fewer adverse events occurred in the clopidogrel group as tallied at the time of the intervention, 1 month later, and 8 months later.
Clopidogrel in ST-elevation acute MI
The CLARITY-TIMI 28 trial6 (Clopidogrel as Adjunctive Reperfusion Therapy—Thrombolysis in Myocardial Infarction 28) showed that adding clopidogrel (a 300-mg loading dose, then 75 mg daily) to aspirin benefitted patients with ST-elevation MI receiving fibrinolytic therapy. At 30 days, cardiovascular death, recurrent MI, or urgent revascularization had occurred in 11.6% of the clopidogrel group vs 14.1% of the placebo group, a statistically significant difference. The rates of major or minor bleeding were no higher in the clopidogrel group than in the placebo group, an especially remarkable finding in patients receiving thrombolytic therapy.
PCI-CLARITY.7 About half of the patients in the CLARITY trial ultimately underwent a percutaneous coronary intervention after fibrinolytic therapy, with results reported as the PCI-CLARITY substudy. Like those in PCI-CURE, these patients were randomized to receive pretreatment with either clopidogrel or placebo before the procedure, in this study for a median of 3 days. Both groups received clopidogrel afterward. At 30 days from randomization, the outcome of cardiovascular death, MI, or stroke had occurred in 7.5% of the clopidogrel group compared with 12.0% of the placebo group, which was statistically significant, without any significant excess in the rates of major or minor bleeding.
COMMIT8 (the Clopidogrel and Metoprolol in Myocardial Infarction Trial) also showed clopidogrel to be beneficial in patients with acute MI. This trial included more than 45,000 patients in China with acute MI, 93% of whom had ST-segment elevation. In contrast to CLARITY, in COMMIT barely more than half of the patients received fibrinolysis, fewer than 5% proceeded to percutaneous interventions, and no loading dose was given: patients in the clopidogrel group received 75 mg/day from the outset.
At 15 days, the incidence of death, reinfarction, or stroke was 9.2% with clopidogrel compared with 10.1% with placebo, a small but statistically significant difference. Again, the rate of major bleeding was not significantly higher, either overall or in patients over age 70.
Of note, patients over age 75 were excluded from CLARITY, and as mentioned, no loading dose was used in COMMIT. Thus, for patients receiving fibrinolysis who are over age 75, there is no evidence to support the safety of a loading dose, and clopidogrel should be started at 75 mg daily.
Clopidogrel in elective percutaneous coronary intervention
The CREDO trial9 (Clopidogrel for the Reduction of Events During Observation) was in patients referred for elective percutaneous coronary intervention. Three to 24 hours before the procedure, the patients received either a 300-mg loading dose of clopidogrel or placebo; afterward, all patients received clopidogrel 75 mg/day for 28 days. All patients also received aspirin.
A clopidogrel loading dose 3 to 24 hours before the intervention did not produce a statistically significant reduction in ischemic events, although a post hoc subgroup analysis suggested that patients who received the loading dose between 6 and 24 hours before did benefit, with a relative risk reduction of 38.6% in the composite end point (P = .051).
After 28 days, the patients who had received the clopidogrel loading dose were continued on clopidogrel, while those in the placebo group were switched back to placebo. At 1 year, the investigators found a significantly lower rate of the composite end point with the prolonged course of clopidogrel (8.5% vs 11.5%).
In summary, these studies found clopidogrel to be beneficial in a broad spectrum of coronary diseases. Subgroup analyses suggest that pretreatment before percutaneous coronary intervention provides additional benefit, particularly if clopidogrel is given at least 6 hours in advance (the time necessary for clopidogrel to cause substantial platelet inhibition).
SOME PATIENTS RESPOND LESS TO CLOPIDOGREL
The level of platelet inhibition induced by clopidogrel varies. In different studies, the frequency of clopidogrel “nonresponsiveness” ranged from 5% to 56% of patients, depending on which test and which cutoff values were used. The distribution of responses to clopidogrel is wide and fits a normal gaussian curve.10
A large fraction of the population carries a gene that may account for some of the interpatient variation in platelet inhibition with clopidogrel. Carriers of a reduced-function CYP2C19 allele—approximately 30% of people in one study—have significantly lower levels of the active metabolite of clopidogrel, less platelet inhibition from clopidogrel therapy, and a 53% higher rate of death from cardiovascular causes, MI, or stroke.11
PRASUGREL, THE NEWEST THIENOPYRIDINE
Prasugrel, FDA-approved in July 2009 for the treatment of acute coronary syndromes, is given in an oral loading dose of 60 mg followed by an oral maintenance dose of 10 mg daily.
Pharmacology of prasugrel vs clopidogrel
As noted previously, the thienopyridines are prodrugs that require hepatic conversion to exert antiplatelet effects.
Metabolism. Prasugrel’s hepatic activation involves a single step, in contrast to the multiple-step process required for activation of clopidogrel. Clopidogrel is primarily hydrolyzed by intestinal and plasma esterases to an inactive terminal metabolite, with the residual unhydrolized drug undergoing a two-step metabolism that depends on cytochrome P450 enzymes. Prasugrel is also extensively hydrolyzed by these esterases, but the intermediate product is then metabolized in a single step to the active sulfhydryl compound, mainly by CYP3A4 and CYP2B6.
Thus, about 80% of an orally absorbed dose of prasugrel is converted to active drug, compared with only 10% to 20% of absorbed clopidogrel.
Time to peak effect. With clopidogrel, maximal inhibition of platelet aggregation occurs 3 to 5 days after starting therapy with 75 mg daily without a loading dose, but within 4 to 6 hours if a loading dose of 300 to 600 mg is given. In contrast, a prasugrel loading dose produces more than 80% of its platelet inhibitory effects by 30 minutes, and peak activity is observed within 4 hours.12 The platelet inhibition induced by prasugrel at 30 minutes after administration is comparable to the peak effect of clopidogrel at 6 hours.13
Dose-response. Prasugrel’s inhibition of platelet aggregation is dose-related.
Prasugrel is about 10 times more potent than clopidogrel and 100 times more potent than ticlopidine. Thus, treatment with 5 mg of prasugrel results in inhibition of platelet activity (distributed in a gaussian curve) very similar to that produced by 75 mg of clopidogrel. On the other hand, even a maintenance dose of 150 mg of clopidogrel inhibits platelet activity to a lesser degree than 10 mg of prasugrel (46% vs 61%),14 so clopidogrel appears to reach a plateau of platelet inhibition that prasugrel can overcome.
At the approved dose of prasugrel, inhibition of platelet aggregation is significantly greater and there are fewer “nonresponders” than with clopidogrel.
Interactions. Drugs that inhibit CYP3A4 do not inhibit the efficacy of prasugrel, but they can inhibit that of clopidogrel. Some commonly used drugs that have this effect are the statins (eg, atorvastain [Lipitor]) and the macrolide antibiotics (eg, erythromycin). Furthermore, whereas proton pump inhibitors have been shown to diminish the effect of clopidogrel by reducing the formation of its active metabolite, no such effect has been noted with prasugrel.
Prasugrel in phase 2 trials: Finding the optimal dosage
A phase 2 trial compared three prasugrel regimens (loading dose/daily maintenance dose of 40 mg/7.5 mg, 60 mg/10 mg, and 60 mg/15 mg) and standard clopidogrel therapy (300 mg/75 mg) in patients undergoing elective or urgent percutaneous coronary intervention.15 No significant difference in outcomes was seen in the groups receiving the three prasugrel regimens. However, more “minimal bleeding events” (defined by the criteria of the TIMI trial16) occurred with high-dose prasugrel than with lower-dose prasugrel or with clopidogrel, leading to use of the intermediate-dose prasugrel regimen (60-mg loading dose, 10-mg daily maintenance) for later trials.
Another phase 2 trial randomized 201 patients undergoing elective percutaneous coronary intervention to receive prasugrel 60 mg/10 mg or clopidogrel 600 mg/150 mg.14 In all patients, the loading dose was given about 1 hour before cardiac catheterization. As soon as 30 minutes after the loading dose, platelet inhibition was superior with prasugrel (31% vs 5% inhibition of platelet aggregation), and it remained significantly higher at 6 hours (75% vs 32%) and during the maintenance phase (61% vs 46%).
Phase 3 trial of prasugrel vs clopidogrel: TRITON-TIMI 38
Only one large phase 3 trial of prasugrel has been completed: TRITON-TIMI 38 (the Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel—Thrombolysis in Myocardial Infarction),17 which enrolled adults with moderate-risk to high-risk acute coronary syndromes scheduled to undergo a percutaneous coronary intervention. In this trial, 10,074 patients were enrolled who had moderate-to high-risk unstable angina or non-ST-elevation MI, and 3,534 patients were enrolled who had ST-elevation MI.
Patients were randomized to receive prasugrel (a 60-mg loading dose, then 10 mg daily) or clopidogrel (a 300-mg loading dose, then 75 mg daily) and were treated for 6 to 15 months. All patients also received aspirin.
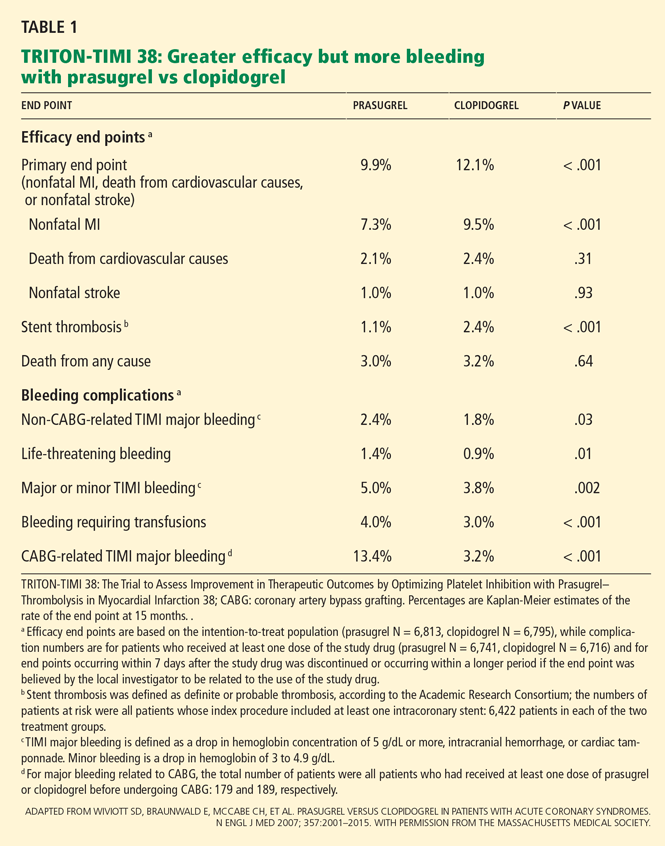
These benefits came at a price of more bleeding. Of those patients who did not undergo coronary artery bypass grafting, more experienced bleeding in the prasugrel group than in the clopidogrel group (2.4% vs 1.8%, P = .03), including a higher rate of life-threatening bleeding (1.4% vs 0.89%, P = .01) and fatal bleeding (0.4% vs 0.1%, P = .002). More patients discontinued prasugrel because of hemorrhage (2.5% vs 1.4%, P < .001). In patients who proceeded to coronary artery bypass grafting, the rate of major bleeding was more than four times higher in those who received prasugrel than in those who received clopidogrel (13.4% vs 3.2%, P < .001).
A higher rate of adverse events related to colon cancer was also noted in patients treated with prasugrel, although the authors suggest this may have resulted from the stronger antiplatelet effects of prasugrel bringing more tumors to medical attention due to bleeding.
Overall death rates did not differ significantly between the treatment groups.
In a post hoc analysis,18 prasugrel was superior to clopidogrel in preventing ischemic events both during the first 3 days following randomization (the “loading phase”) and for the remainder of the trial (the “maintenance phase”). Whereas bleeding risk was similar with the two drugs during the loading phase, prasugrel was subsequently associated with more bleeding during the maintenance phase.
Certain patient subgroups had no net benefit or even suffered harm from prasugrel compared with clopidogrel.17 Patients with previous stroke or transient ischemic attack had net harm from prasugrel (hazard ratio 1.54, P = .04) and showed a strong trend toward a greater rate of major bleeding (P = .06). Patients age 75 and older and those weighing less than 60 kg had no net benefit from prasugrel.
Cost of prasugrel
Prasugrel is currently priced at 18% more than clopidogrel, with average wholesale prices per pill of $6.65 for prasugrel 10 mg compared with $5.63 for clopidogrel 75 mg. (Prasugrel 10-mg pills cost $6.33 at drugstore.com or $7.60 at CVS; clopidogrel 75-mg pills cost $5.33 at drugstore.com or $6.43 at CVS.) The patent on clopidogrel expires in November 2011, after which the price differential is expected to become significantly greater.
TICAGRELOR, A REVERSIBLE ORAL AGENT
Ticagrelor, the first reversible oral P2Y12 receptor antagonist, is an alternative to thienopyridine therapy for acute coronary syndromes.
Ticagrelor is quickly absorbed, does not require metabolic activation, and has a rapid antiplatelet effect and offset of effect, which closely follow drug-exposure levels. In a large randomized controlled trial in patients with acute coronary syndromes with or without STsegment elevation, treatment with ticagrelor compared with clopidogrel resulted in a significant reduction in death from vascular causes, MI, or stroke (9.8% vs 11.7%).19
Given its reversible effect on platelet inhibition, ticagrelor may be preferred in patients whose coronary anatomy is unknown and for whom coronary artery bypass grafting is deemed probable. It is still undergoing trials and is not yet approved.
TAKE-HOME POINTS
Prasugrel is more potent, more rapid in onset, and more consistent in inhibiting platelet aggregation than clopidogrel. A large clinical trial17 found prasugrel to be superior to clopidogrel for patients with moderate-to high-risk acute coronary syndromes with high probability of undergoing a percutaneous coronary intervention.
Who should receive prasugrel, and how?
Prasugrel should be given after angiography to patients with non-ST-elevation acute coronary syndromes or at presentation to patients with ST-elevation MI. When used for planned percutaneous coronary intervention, prasugrel should be given at least 30 minutes before the intervention, as was done in phase 2 trials (although its routine use in this situation is not recommended—see below).
It is given in a one-time loading dose of 60 mg by mouth and then maintained with 10 mg by mouth once daily for at least 1 year. (At least 9 months of treatment with a thienopyridine is indicated for patients with acute coronary syndromes who are medically treated, and at least 1 year is indicated following urgent or elective percutaneous coronary intervention, including balloon angioplasty and placement of a bare-metal or drug-eluting stent.)
Who should not receive prasugrel?
For now, prasugrel should be avoided in favor of clopidogrel in patients at higher risk of bleeding. It is clearly contraindicated in patients with prior transient ischemic attack or stroke, for whom the risk of serious bleeding seems to be prohibitive. It should generally be avoided in patients age 75 and older, although it might be considered in those at particularly high risk of stent thrombosis, such as those with diabetes or prior MI. In patients weighing less than 60 kg, the package insert advises a reduced dose (5 mg), although clinical evidence for this practice is lacking.
As yet, we have no data assuring that prasugrel is safe to use in combination with fibrinolytic agents, so patients on thrombolytic therapy for acute MI should continue to receive clopidogrel starting immediately after lysis. Furthermore, in patients who proceeded to coronary artery bypass grafting, the rate of major bleeding was more than four times higher in the prasugrel group than in the clopidogrel group in the TRITON-TIMI 38 trial.17 No thienopyridine should be given to patients likely to proceed to coronary artery bypass grafting.
Only clopidogrel has evidence supporting its use as an alternative to aspirin for patients with atherosclerotic disease who cannot tolerate aspirin. Neither drug has evidence for use for primary prevention.
Other areas of uncertainty
Prior to angiography. Indications for prasugrel are currently limited by the narrow scope of the trial data. TRITON-TIMI 38,17 the only large trial completed to date, randomized patients to receive prasugrel only after their coronary anatomy was known, except for ST-elevation MI patients. It is unknown whether the benefits of prasugrel will outweigh the higher risk of bleeding in patients with acute coronary syndromes who do not proceed to percutaneous coronary interventions.
A clinical trial is currently under way comparing prasugrel with clopidogrel in 10,000 patients with acute coronary syndromes who will be medically managed without planned revascularization: A Comparison of Prasugrel and Clopidogrel in Acute Coronary Syndrome Subjects (TRILOGY ACS), ClinicalTrials.gov Identifier: NCT00699998. The trial has an estimated completion date of March 2011.
In cases of non-ST-elevation acute coronary syndrome, it is reasonable to wait to give a thienopyridine until after the coronary anatomy has been defined, if angiography will be completed soon after presentation. For example, a 1-hour delay before giving prasugrel still delivers antiplatelet therapy more quickly than giving clopidogrel on presentation. If longer delays are expected before angiography, however, the patient should be given a loading dose of clopidogrel “up front,” in accordance with guidelines published by the American College of Cardiology, American Heart Association, and European Society of Cardiology,20 which recommend starting a thienopyridine early during hospitalization based on trial data with clopidogrel.
Patients undergoing elective percutaneous coronary intervention are at lower risk of stent thrombosis and other ischemic complications, so it is possible that the benefits of prasugrel would not outweigh the risks in these patients. Thus, prasugrel cannot yet be recommended for routine elective percutaneous coronary intervention except in individual cases in which the interventionalist feels that the patient may be at higher risk of thrombosis.
- Yeghiazarians Y, Braunstein JB, Askari A, Stone PH. Unstable angina pectoris. N Engl J Med 2000; 342:101–114.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Balsano F, Rizzon P, Violi F, et al. Antiplatelet treatment with ticlopidine in unstable angina. A controlled multicenter clinical trial. The Studio della Ticlopidina nell'Angina Instabile Group. Circulation 1990; 82:17–26.
- Schömig A, Neumann FJ, Kastrati A, et al. A randomized comparison of antiplatelet and anticoagulant therapy after the placement of coronary-artery stents. N Engl J Med 1996; 334:1084–1089.
- Mehta SR, Yusuf S, Peters RJG, et al; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- Sabatine MS, Cannon CP, Gibson CM, et al; CLA RITY-TIMI 28 Investigators. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with STsegment elevation. N Engl J Med 2005; 352:1179–1189.
- Sabatine MS, Cannon CP, Gibson CM, et al; Clopidogrel as Adjunctive Reperfusion Therapy (CLARITY)-Thrombolysis in Myocardial Infarction (TIMI) 28 Investigators. Effect of clopidogrel pretreatment before percutaneous coronary intervention in patients with ST-elevation myocardial infarction treated with fibrinolytics: the PCI-CLARITY study. JAMA 2005: 294:1224–1232.
- Chen ZM, Jiang LX, Chen YP, et al; COMMIT (ClOpidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Steinhubl SR, Berger PB, Mann JT, et al; CREDO Investigators. Clopidogrel for the reduction of events during observation. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 288:2411–2420.
- Serebruany VL, Steinhubl SR, Berger PB, Malinin AI, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45:246–251.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome P-450 polymorphisms and response to clopidogrel. N Engl J Med 2009; 360:354–362.
- Helft G, Osende JI, Worthley SG, et al. Acute antithrombotic effect of a front-loaded regimen of clopidogrel in patients with atherosclerosis on aspirin. Arterioscler Thromb Vasc Biol 2000; 20:2316–2321.
- Weerakkody GJ, Jakubowski JA, Brandt JT, et al. Comparison of speed of onset of platelet inhibition after loading doses of clopidogrel versus prasugrel in healthy volunteers and correlation with responder status. Am J Cardiol 2007; 100:331–336.
- Wiviott SD, Trenk D, Frelinger AL, et al; PRINCIPLETIMI 44 Investigators. Prasugrel compared with high loading-and maintenance-dose clopidogrel in patients with planned percutaneous coronary intervention: the Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation-Thrombolysis in Myocardial Infarction 44 trial. Circulation 2007; 116:2923–2932.
- Wiviott SD, Antman EM, Winters KJ, et al; JUMBO-TIMI 26 Investigators. Randomized comparison of prasugrel (CS-747, LY640315), a novel thienopyridine P2Y12 antagonist, with clopidogrel in percutaneous coronary intervention: results of the Joint Utilization of Medications to Block Platelets Optimally (JUMBO)-TIMI 26 Trial. Circulation 2005; 111:3366–3373.
- Bovill EG, Terrin ML, Stump DC, et al. Hemorrhagic events during therapy with recombinant tissue-type plasminogen activator, heparin, and aspirin for acute myocardial infarction. Results of the Thrombolysis in Myocardial Infarction (TIMI) Phase II Trial. Ann Intern Med 1991; 115:256–265.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITONTIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Antman EM, Wiviott SD, Murphy SA, et al. Early and late benefits of prasugrel in patients with acute coronary syndromes undergoing percutaneous coronary intervention: a TRITON-TIMI 38 (TRial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet InhibitioN with Prasugrel-Thrombolysis In Myocardial Infarction) analysis. J Am Coll Cardiol 2008; 51:2028–2033.
- Wallentin L, Becker RC, Budaj A, Freij A, Thorsén M, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Braunwald E, Antman EM, Beasley JW, et al. ACC/AHA 2002 guideline update for the management of patients with unstable angina and non–ST-segment elevation myocardial infarction—summary article*1: A report of the American College of Cardiology/American Heart Association task force on practice guidelines (Committee on the Management of Patients With Unstable Angina). J Am Coll Cardiol 2002; 40:1366–1374.
Prasugrel (Effient) is more potent and consistent in its effects than clopidogrel (Plavix), thus preventing more thrombotic events—but at a price of more bleeding. Therefore, the drugs must be appropriately selected for the individual patient.
Over the last 9 years, the thienopyridines—ticlopidine (Ticlid), clopidogrel, and now prasugrel—have become essential tools for treating acute coronary syndromes.
The usual underlying mechanism of acute coronary syndromes is thrombosis, caused by rupture of atherosclerotic plaque.1 Accordingly, antithrombotic agents—aspirin, heparin, lowmolecular-weight heparin, glycoprotein IIb/IIIa inhibitors, the direct thrombin inhibitor bivalirudin (Angiomax), and thienopyridines—have all been shown to reduce the risk of major adverse cardiac outcomes in this setting.
In this article, we review the pharmacology and evidence of effectiveness of the thienopyridine drugs, focusing on prasugrel, the latest thienopyridine to be approved by the US Food and Drug Administration (FDA).
THIENOPYRIDINES INHIBIT PLATELET ACTIVATION AND AGGREGATION
Thienopyridines are prodrugs that require conversion by hepatic cytochrome P450 enzymes. The active metabolites bind irreversibly to platelet P2Y12 receptors. Consequently, they permanently block signalling mediated by platelet adenosine diphosphate-P2Y12 receptors, thereby inhibiting glycoprotein IIb/IIIa receptor activation and platelet aggregation.
Aspirin, in contrast, inhibits platelets by blocking the thromboxane-mediated pathway. Therefore, the combination of aspirin plus a thienopyridine has an additive effect.2
The effect of thienopyridines on platelets is irreversible. Therefore, although the half-life of prasugrel’s active metabolite is 3.7 hours, its inhibitory effects last for 96 hours, essentially the time for half the body’s circulating platelets to be replaced.
TICLOPIDINE, THE FIRST THIENOPYRIDINE
Ticlopidine was the first thienopyridine to be approved by the FDA. Its initial studies in unstable angina were small, their designs did not call for patients to concurrently receive aspirin, and all they showed was that ticlopidine was about as beneficial as aspirin. Consequently, the studies had little impact on clinical practice.3
In a pivotal trial,4 patients who received coronary stents were randomized to afterward receive either the combination of ticlopidine plus aspirin or anticoagulation therapy with heparin, phenprocoumon (a coumarin derivative available in Europe), and aspirin. At 30 days, an ischemic complication (death, myocardial infarction [MI], repeat intervention) had occurred in 6.2% of the anticoagulation therapy group vs 1.6% of the ticlopidine group, a risk reduction of 75%. Rates of stent occlusion, MI, and revascularization were 80% to 85% lower in the ticlodipine group. This study paved the way for widespread use of thienopyridines.
Ticlopidine’s use was limited, however, by a 2.4% incidence of serious granulocytopenia and rare cases of thrombocytopenic purpura.
BENEFIT OF CLOPIDOGREL
Although prasugrel is the focus of this review, the trials of prasugrel all compared its efficacy with that of clopidogrel. Furthermore, many patients should still receive clopidogrel and not prasugrel, so it is important to be familiar with the evidence of clopidogrel’s benefit.
Once approved for clinical use, clopidogrel was substituted for ticlopidine in patients undergoing coronary stenting on the basis of studies showing it to be at least as effective as ticlopidine and more tolerable. A series of trials of clopidogrel were done in patients across a spectrum of risk groups, from those at high risk of coronary heart disease to those presenting with ST-elevation MI. The time of pretreatment in the studies ranged from 3 hours to 6 days before percutaneous coronary intervention, and the duration of treatment following intervention ranged from 30 days to 1 year.
Clopidogrel in non-ST-elevation acute coronary syndromes
The CURE trial2 (Clopidogrel in Unstable Angina to Prevent Recurrent Events), published in 2001, established clopidogrel as a therapy for unstable ischemic syndromes, whether treated medically or with revascularization. In that trial, 12,562 patients with acute coronary syndromes without ST elevation (ie, unstable angina or non-ST-elevation MI), as defined by electrocardiographic changes or positive cardiac markers, were randomized to receive clopidogrel (a 300-mg loading dose followed by 75-mg maintenance doses) or placebo for a mean duration of 9 months. All patients also received aspirin 75 mg to 325 mg daily.
The composite outcome of death from cardiovascular causes, nonfatal MI, or stroke occurred in 20% fewer patients treated with clopidogrel than with placebo (9.3% vs 11.4%). The benefit was similar in patients undergoing revascularization compared with those treated medically.
Although there were significantly more cases of major bleeding in the clopidogrel group than in the placebo group (3.7% vs 2.7%), the number of episodes of life-threatening bleeding or hemorrhagic strokes was the same.
PCI-CURE5 was a substudy of the CURE trial in patients who underwent a percutaneous coronary intervention. Patients were pretreated with clopidogrel or placebo for a mean of 6 days before the procedure. Afterward, they all received clopidogrel plus aspirin in an unblinded fashion for 2 to 4 weeks, and then the randomized study drug was resumed for a mean of 8 months.
Significantly fewer adverse events occurred in the clopidogrel group as tallied at the time of the intervention, 1 month later, and 8 months later.
Clopidogrel in ST-elevation acute MI
The CLARITY-TIMI 28 trial6 (Clopidogrel as Adjunctive Reperfusion Therapy—Thrombolysis in Myocardial Infarction 28) showed that adding clopidogrel (a 300-mg loading dose, then 75 mg daily) to aspirin benefitted patients with ST-elevation MI receiving fibrinolytic therapy. At 30 days, cardiovascular death, recurrent MI, or urgent revascularization had occurred in 11.6% of the clopidogrel group vs 14.1% of the placebo group, a statistically significant difference. The rates of major or minor bleeding were no higher in the clopidogrel group than in the placebo group, an especially remarkable finding in patients receiving thrombolytic therapy.
PCI-CLARITY.7 About half of the patients in the CLARITY trial ultimately underwent a percutaneous coronary intervention after fibrinolytic therapy, with results reported as the PCI-CLARITY substudy. Like those in PCI-CURE, these patients were randomized to receive pretreatment with either clopidogrel or placebo before the procedure, in this study for a median of 3 days. Both groups received clopidogrel afterward. At 30 days from randomization, the outcome of cardiovascular death, MI, or stroke had occurred in 7.5% of the clopidogrel group compared with 12.0% of the placebo group, which was statistically significant, without any significant excess in the rates of major or minor bleeding.
COMMIT8 (the Clopidogrel and Metoprolol in Myocardial Infarction Trial) also showed clopidogrel to be beneficial in patients with acute MI. This trial included more than 45,000 patients in China with acute MI, 93% of whom had ST-segment elevation. In contrast to CLARITY, in COMMIT barely more than half of the patients received fibrinolysis, fewer than 5% proceeded to percutaneous interventions, and no loading dose was given: patients in the clopidogrel group received 75 mg/day from the outset.
At 15 days, the incidence of death, reinfarction, or stroke was 9.2% with clopidogrel compared with 10.1% with placebo, a small but statistically significant difference. Again, the rate of major bleeding was not significantly higher, either overall or in patients over age 70.
Of note, patients over age 75 were excluded from CLARITY, and as mentioned, no loading dose was used in COMMIT. Thus, for patients receiving fibrinolysis who are over age 75, there is no evidence to support the safety of a loading dose, and clopidogrel should be started at 75 mg daily.
Clopidogrel in elective percutaneous coronary intervention
The CREDO trial9 (Clopidogrel for the Reduction of Events During Observation) was in patients referred for elective percutaneous coronary intervention. Three to 24 hours before the procedure, the patients received either a 300-mg loading dose of clopidogrel or placebo; afterward, all patients received clopidogrel 75 mg/day for 28 days. All patients also received aspirin.
A clopidogrel loading dose 3 to 24 hours before the intervention did not produce a statistically significant reduction in ischemic events, although a post hoc subgroup analysis suggested that patients who received the loading dose between 6 and 24 hours before did benefit, with a relative risk reduction of 38.6% in the composite end point (P = .051).
After 28 days, the patients who had received the clopidogrel loading dose were continued on clopidogrel, while those in the placebo group were switched back to placebo. At 1 year, the investigators found a significantly lower rate of the composite end point with the prolonged course of clopidogrel (8.5% vs 11.5%).
In summary, these studies found clopidogrel to be beneficial in a broad spectrum of coronary diseases. Subgroup analyses suggest that pretreatment before percutaneous coronary intervention provides additional benefit, particularly if clopidogrel is given at least 6 hours in advance (the time necessary for clopidogrel to cause substantial platelet inhibition).
SOME PATIENTS RESPOND LESS TO CLOPIDOGREL
The level of platelet inhibition induced by clopidogrel varies. In different studies, the frequency of clopidogrel “nonresponsiveness” ranged from 5% to 56% of patients, depending on which test and which cutoff values were used. The distribution of responses to clopidogrel is wide and fits a normal gaussian curve.10
A large fraction of the population carries a gene that may account for some of the interpatient variation in platelet inhibition with clopidogrel. Carriers of a reduced-function CYP2C19 allele—approximately 30% of people in one study—have significantly lower levels of the active metabolite of clopidogrel, less platelet inhibition from clopidogrel therapy, and a 53% higher rate of death from cardiovascular causes, MI, or stroke.11
PRASUGREL, THE NEWEST THIENOPYRIDINE
Prasugrel, FDA-approved in July 2009 for the treatment of acute coronary syndromes, is given in an oral loading dose of 60 mg followed by an oral maintenance dose of 10 mg daily.
Pharmacology of prasugrel vs clopidogrel
As noted previously, the thienopyridines are prodrugs that require hepatic conversion to exert antiplatelet effects.
Metabolism. Prasugrel’s hepatic activation involves a single step, in contrast to the multiple-step process required for activation of clopidogrel. Clopidogrel is primarily hydrolyzed by intestinal and plasma esterases to an inactive terminal metabolite, with the residual unhydrolized drug undergoing a two-step metabolism that depends on cytochrome P450 enzymes. Prasugrel is also extensively hydrolyzed by these esterases, but the intermediate product is then metabolized in a single step to the active sulfhydryl compound, mainly by CYP3A4 and CYP2B6.
Thus, about 80% of an orally absorbed dose of prasugrel is converted to active drug, compared with only 10% to 20% of absorbed clopidogrel.
Time to peak effect. With clopidogrel, maximal inhibition of platelet aggregation occurs 3 to 5 days after starting therapy with 75 mg daily without a loading dose, but within 4 to 6 hours if a loading dose of 300 to 600 mg is given. In contrast, a prasugrel loading dose produces more than 80% of its platelet inhibitory effects by 30 minutes, and peak activity is observed within 4 hours.12 The platelet inhibition induced by prasugrel at 30 minutes after administration is comparable to the peak effect of clopidogrel at 6 hours.13
Dose-response. Prasugrel’s inhibition of platelet aggregation is dose-related.
Prasugrel is about 10 times more potent than clopidogrel and 100 times more potent than ticlopidine. Thus, treatment with 5 mg of prasugrel results in inhibition of platelet activity (distributed in a gaussian curve) very similar to that produced by 75 mg of clopidogrel. On the other hand, even a maintenance dose of 150 mg of clopidogrel inhibits platelet activity to a lesser degree than 10 mg of prasugrel (46% vs 61%),14 so clopidogrel appears to reach a plateau of platelet inhibition that prasugrel can overcome.
At the approved dose of prasugrel, inhibition of platelet aggregation is significantly greater and there are fewer “nonresponders” than with clopidogrel.
Interactions. Drugs that inhibit CYP3A4 do not inhibit the efficacy of prasugrel, but they can inhibit that of clopidogrel. Some commonly used drugs that have this effect are the statins (eg, atorvastain [Lipitor]) and the macrolide antibiotics (eg, erythromycin). Furthermore, whereas proton pump inhibitors have been shown to diminish the effect of clopidogrel by reducing the formation of its active metabolite, no such effect has been noted with prasugrel.
Prasugrel in phase 2 trials: Finding the optimal dosage
A phase 2 trial compared three prasugrel regimens (loading dose/daily maintenance dose of 40 mg/7.5 mg, 60 mg/10 mg, and 60 mg/15 mg) and standard clopidogrel therapy (300 mg/75 mg) in patients undergoing elective or urgent percutaneous coronary intervention.15 No significant difference in outcomes was seen in the groups receiving the three prasugrel regimens. However, more “minimal bleeding events” (defined by the criteria of the TIMI trial16) occurred with high-dose prasugrel than with lower-dose prasugrel or with clopidogrel, leading to use of the intermediate-dose prasugrel regimen (60-mg loading dose, 10-mg daily maintenance) for later trials.
Another phase 2 trial randomized 201 patients undergoing elective percutaneous coronary intervention to receive prasugrel 60 mg/10 mg or clopidogrel 600 mg/150 mg.14 In all patients, the loading dose was given about 1 hour before cardiac catheterization. As soon as 30 minutes after the loading dose, platelet inhibition was superior with prasugrel (31% vs 5% inhibition of platelet aggregation), and it remained significantly higher at 6 hours (75% vs 32%) and during the maintenance phase (61% vs 46%).
Phase 3 trial of prasugrel vs clopidogrel: TRITON-TIMI 38
Only one large phase 3 trial of prasugrel has been completed: TRITON-TIMI 38 (the Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel—Thrombolysis in Myocardial Infarction),17 which enrolled adults with moderate-risk to high-risk acute coronary syndromes scheduled to undergo a percutaneous coronary intervention. In this trial, 10,074 patients were enrolled who had moderate-to high-risk unstable angina or non-ST-elevation MI, and 3,534 patients were enrolled who had ST-elevation MI.
Patients were randomized to receive prasugrel (a 60-mg loading dose, then 10 mg daily) or clopidogrel (a 300-mg loading dose, then 75 mg daily) and were treated for 6 to 15 months. All patients also received aspirin.
These benefits came at a price of more bleeding. Of those patients who did not undergo coronary artery bypass grafting, more experienced bleeding in the prasugrel group than in the clopidogrel group (2.4% vs 1.8%, P = .03), including a higher rate of life-threatening bleeding (1.4% vs 0.89%, P = .01) and fatal bleeding (0.4% vs 0.1%, P = .002). More patients discontinued prasugrel because of hemorrhage (2.5% vs 1.4%, P < .001). In patients who proceeded to coronary artery bypass grafting, the rate of major bleeding was more than four times higher in those who received prasugrel than in those who received clopidogrel (13.4% vs 3.2%, P < .001).
A higher rate of adverse events related to colon cancer was also noted in patients treated with prasugrel, although the authors suggest this may have resulted from the stronger antiplatelet effects of prasugrel bringing more tumors to medical attention due to bleeding.
Overall death rates did not differ significantly between the treatment groups.
In a post hoc analysis,18 prasugrel was superior to clopidogrel in preventing ischemic events both during the first 3 days following randomization (the “loading phase”) and for the remainder of the trial (the “maintenance phase”). Whereas bleeding risk was similar with the two drugs during the loading phase, prasugrel was subsequently associated with more bleeding during the maintenance phase.
Certain patient subgroups had no net benefit or even suffered harm from prasugrel compared with clopidogrel.17 Patients with previous stroke or transient ischemic attack had net harm from prasugrel (hazard ratio 1.54, P = .04) and showed a strong trend toward a greater rate of major bleeding (P = .06). Patients age 75 and older and those weighing less than 60 kg had no net benefit from prasugrel.
Cost of prasugrel
Prasugrel is currently priced at 18% more than clopidogrel, with average wholesale prices per pill of $6.65 for prasugrel 10 mg compared with $5.63 for clopidogrel 75 mg. (Prasugrel 10-mg pills cost $6.33 at drugstore.com or $7.60 at CVS; clopidogrel 75-mg pills cost $5.33 at drugstore.com or $6.43 at CVS.) The patent on clopidogrel expires in November 2011, after which the price differential is expected to become significantly greater.
TICAGRELOR, A REVERSIBLE ORAL AGENT
Ticagrelor, the first reversible oral P2Y12 receptor antagonist, is an alternative to thienopyridine therapy for acute coronary syndromes.
Ticagrelor is quickly absorbed, does not require metabolic activation, and has a rapid antiplatelet effect and offset of effect, which closely follow drug-exposure levels. In a large randomized controlled trial in patients with acute coronary syndromes with or without STsegment elevation, treatment with ticagrelor compared with clopidogrel resulted in a significant reduction in death from vascular causes, MI, or stroke (9.8% vs 11.7%).19
Given its reversible effect on platelet inhibition, ticagrelor may be preferred in patients whose coronary anatomy is unknown and for whom coronary artery bypass grafting is deemed probable. It is still undergoing trials and is not yet approved.
TAKE-HOME POINTS
Prasugrel is more potent, more rapid in onset, and more consistent in inhibiting platelet aggregation than clopidogrel. A large clinical trial17 found prasugrel to be superior to clopidogrel for patients with moderate-to high-risk acute coronary syndromes with high probability of undergoing a percutaneous coronary intervention.
Who should receive prasugrel, and how?
Prasugrel should be given after angiography to patients with non-ST-elevation acute coronary syndromes or at presentation to patients with ST-elevation MI. When used for planned percutaneous coronary intervention, prasugrel should be given at least 30 minutes before the intervention, as was done in phase 2 trials (although its routine use in this situation is not recommended—see below).
It is given in a one-time loading dose of 60 mg by mouth and then maintained with 10 mg by mouth once daily for at least 1 year. (At least 9 months of treatment with a thienopyridine is indicated for patients with acute coronary syndromes who are medically treated, and at least 1 year is indicated following urgent or elective percutaneous coronary intervention, including balloon angioplasty and placement of a bare-metal or drug-eluting stent.)
Who should not receive prasugrel?
For now, prasugrel should be avoided in favor of clopidogrel in patients at higher risk of bleeding. It is clearly contraindicated in patients with prior transient ischemic attack or stroke, for whom the risk of serious bleeding seems to be prohibitive. It should generally be avoided in patients age 75 and older, although it might be considered in those at particularly high risk of stent thrombosis, such as those with diabetes or prior MI. In patients weighing less than 60 kg, the package insert advises a reduced dose (5 mg), although clinical evidence for this practice is lacking.
As yet, we have no data assuring that prasugrel is safe to use in combination with fibrinolytic agents, so patients on thrombolytic therapy for acute MI should continue to receive clopidogrel starting immediately after lysis. Furthermore, in patients who proceeded to coronary artery bypass grafting, the rate of major bleeding was more than four times higher in the prasugrel group than in the clopidogrel group in the TRITON-TIMI 38 trial.17 No thienopyridine should be given to patients likely to proceed to coronary artery bypass grafting.
Only clopidogrel has evidence supporting its use as an alternative to aspirin for patients with atherosclerotic disease who cannot tolerate aspirin. Neither drug has evidence for use for primary prevention.
Other areas of uncertainty
Prior to angiography. Indications for prasugrel are currently limited by the narrow scope of the trial data. TRITON-TIMI 38,17 the only large trial completed to date, randomized patients to receive prasugrel only after their coronary anatomy was known, except for ST-elevation MI patients. It is unknown whether the benefits of prasugrel will outweigh the higher risk of bleeding in patients with acute coronary syndromes who do not proceed to percutaneous coronary interventions.
A clinical trial is currently under way comparing prasugrel with clopidogrel in 10,000 patients with acute coronary syndromes who will be medically managed without planned revascularization: A Comparison of Prasugrel and Clopidogrel in Acute Coronary Syndrome Subjects (TRILOGY ACS), ClinicalTrials.gov Identifier: NCT00699998. The trial has an estimated completion date of March 2011.
In cases of non-ST-elevation acute coronary syndrome, it is reasonable to wait to give a thienopyridine until after the coronary anatomy has been defined, if angiography will be completed soon after presentation. For example, a 1-hour delay before giving prasugrel still delivers antiplatelet therapy more quickly than giving clopidogrel on presentation. If longer delays are expected before angiography, however, the patient should be given a loading dose of clopidogrel “up front,” in accordance with guidelines published by the American College of Cardiology, American Heart Association, and European Society of Cardiology,20 which recommend starting a thienopyridine early during hospitalization based on trial data with clopidogrel.
Patients undergoing elective percutaneous coronary intervention are at lower risk of stent thrombosis and other ischemic complications, so it is possible that the benefits of prasugrel would not outweigh the risks in these patients. Thus, prasugrel cannot yet be recommended for routine elective percutaneous coronary intervention except in individual cases in which the interventionalist feels that the patient may be at higher risk of thrombosis.
Prasugrel (Effient) is more potent and consistent in its effects than clopidogrel (Plavix), thus preventing more thrombotic events—but at a price of more bleeding. Therefore, the drugs must be appropriately selected for the individual patient.
Over the last 9 years, the thienopyridines—ticlopidine (Ticlid), clopidogrel, and now prasugrel—have become essential tools for treating acute coronary syndromes.
The usual underlying mechanism of acute coronary syndromes is thrombosis, caused by rupture of atherosclerotic plaque.1 Accordingly, antithrombotic agents—aspirin, heparin, lowmolecular-weight heparin, glycoprotein IIb/IIIa inhibitors, the direct thrombin inhibitor bivalirudin (Angiomax), and thienopyridines—have all been shown to reduce the risk of major adverse cardiac outcomes in this setting.
In this article, we review the pharmacology and evidence of effectiveness of the thienopyridine drugs, focusing on prasugrel, the latest thienopyridine to be approved by the US Food and Drug Administration (FDA).
THIENOPYRIDINES INHIBIT PLATELET ACTIVATION AND AGGREGATION
Thienopyridines are prodrugs that require conversion by hepatic cytochrome P450 enzymes. The active metabolites bind irreversibly to platelet P2Y12 receptors. Consequently, they permanently block signalling mediated by platelet adenosine diphosphate-P2Y12 receptors, thereby inhibiting glycoprotein IIb/IIIa receptor activation and platelet aggregation.
Aspirin, in contrast, inhibits platelets by blocking the thromboxane-mediated pathway. Therefore, the combination of aspirin plus a thienopyridine has an additive effect.2
The effect of thienopyridines on platelets is irreversible. Therefore, although the half-life of prasugrel’s active metabolite is 3.7 hours, its inhibitory effects last for 96 hours, essentially the time for half the body’s circulating platelets to be replaced.
TICLOPIDINE, THE FIRST THIENOPYRIDINE
Ticlopidine was the first thienopyridine to be approved by the FDA. Its initial studies in unstable angina were small, their designs did not call for patients to concurrently receive aspirin, and all they showed was that ticlopidine was about as beneficial as aspirin. Consequently, the studies had little impact on clinical practice.3
In a pivotal trial,4 patients who received coronary stents were randomized to afterward receive either the combination of ticlopidine plus aspirin or anticoagulation therapy with heparin, phenprocoumon (a coumarin derivative available in Europe), and aspirin. At 30 days, an ischemic complication (death, myocardial infarction [MI], repeat intervention) had occurred in 6.2% of the anticoagulation therapy group vs 1.6% of the ticlopidine group, a risk reduction of 75%. Rates of stent occlusion, MI, and revascularization were 80% to 85% lower in the ticlodipine group. This study paved the way for widespread use of thienopyridines.
Ticlopidine’s use was limited, however, by a 2.4% incidence of serious granulocytopenia and rare cases of thrombocytopenic purpura.
BENEFIT OF CLOPIDOGREL
Although prasugrel is the focus of this review, the trials of prasugrel all compared its efficacy with that of clopidogrel. Furthermore, many patients should still receive clopidogrel and not prasugrel, so it is important to be familiar with the evidence of clopidogrel’s benefit.
Once approved for clinical use, clopidogrel was substituted for ticlopidine in patients undergoing coronary stenting on the basis of studies showing it to be at least as effective as ticlopidine and more tolerable. A series of trials of clopidogrel were done in patients across a spectrum of risk groups, from those at high risk of coronary heart disease to those presenting with ST-elevation MI. The time of pretreatment in the studies ranged from 3 hours to 6 days before percutaneous coronary intervention, and the duration of treatment following intervention ranged from 30 days to 1 year.
Clopidogrel in non-ST-elevation acute coronary syndromes
The CURE trial2 (Clopidogrel in Unstable Angina to Prevent Recurrent Events), published in 2001, established clopidogrel as a therapy for unstable ischemic syndromes, whether treated medically or with revascularization. In that trial, 12,562 patients with acute coronary syndromes without ST elevation (ie, unstable angina or non-ST-elevation MI), as defined by electrocardiographic changes or positive cardiac markers, were randomized to receive clopidogrel (a 300-mg loading dose followed by 75-mg maintenance doses) or placebo for a mean duration of 9 months. All patients also received aspirin 75 mg to 325 mg daily.
The composite outcome of death from cardiovascular causes, nonfatal MI, or stroke occurred in 20% fewer patients treated with clopidogrel than with placebo (9.3% vs 11.4%). The benefit was similar in patients undergoing revascularization compared with those treated medically.
Although there were significantly more cases of major bleeding in the clopidogrel group than in the placebo group (3.7% vs 2.7%), the number of episodes of life-threatening bleeding or hemorrhagic strokes was the same.
PCI-CURE5 was a substudy of the CURE trial in patients who underwent a percutaneous coronary intervention. Patients were pretreated with clopidogrel or placebo for a mean of 6 days before the procedure. Afterward, they all received clopidogrel plus aspirin in an unblinded fashion for 2 to 4 weeks, and then the randomized study drug was resumed for a mean of 8 months.
Significantly fewer adverse events occurred in the clopidogrel group as tallied at the time of the intervention, 1 month later, and 8 months later.
Clopidogrel in ST-elevation acute MI
The CLARITY-TIMI 28 trial6 (Clopidogrel as Adjunctive Reperfusion Therapy—Thrombolysis in Myocardial Infarction 28) showed that adding clopidogrel (a 300-mg loading dose, then 75 mg daily) to aspirin benefitted patients with ST-elevation MI receiving fibrinolytic therapy. At 30 days, cardiovascular death, recurrent MI, or urgent revascularization had occurred in 11.6% of the clopidogrel group vs 14.1% of the placebo group, a statistically significant difference. The rates of major or minor bleeding were no higher in the clopidogrel group than in the placebo group, an especially remarkable finding in patients receiving thrombolytic therapy.
PCI-CLARITY.7 About half of the patients in the CLARITY trial ultimately underwent a percutaneous coronary intervention after fibrinolytic therapy, with results reported as the PCI-CLARITY substudy. Like those in PCI-CURE, these patients were randomized to receive pretreatment with either clopidogrel or placebo before the procedure, in this study for a median of 3 days. Both groups received clopidogrel afterward. At 30 days from randomization, the outcome of cardiovascular death, MI, or stroke had occurred in 7.5% of the clopidogrel group compared with 12.0% of the placebo group, which was statistically significant, without any significant excess in the rates of major or minor bleeding.
COMMIT8 (the Clopidogrel and Metoprolol in Myocardial Infarction Trial) also showed clopidogrel to be beneficial in patients with acute MI. This trial included more than 45,000 patients in China with acute MI, 93% of whom had ST-segment elevation. In contrast to CLARITY, in COMMIT barely more than half of the patients received fibrinolysis, fewer than 5% proceeded to percutaneous interventions, and no loading dose was given: patients in the clopidogrel group received 75 mg/day from the outset.
At 15 days, the incidence of death, reinfarction, or stroke was 9.2% with clopidogrel compared with 10.1% with placebo, a small but statistically significant difference. Again, the rate of major bleeding was not significantly higher, either overall or in patients over age 70.
Of note, patients over age 75 were excluded from CLARITY, and as mentioned, no loading dose was used in COMMIT. Thus, for patients receiving fibrinolysis who are over age 75, there is no evidence to support the safety of a loading dose, and clopidogrel should be started at 75 mg daily.
Clopidogrel in elective percutaneous coronary intervention
The CREDO trial9 (Clopidogrel for the Reduction of Events During Observation) was in patients referred for elective percutaneous coronary intervention. Three to 24 hours before the procedure, the patients received either a 300-mg loading dose of clopidogrel or placebo; afterward, all patients received clopidogrel 75 mg/day for 28 days. All patients also received aspirin.
A clopidogrel loading dose 3 to 24 hours before the intervention did not produce a statistically significant reduction in ischemic events, although a post hoc subgroup analysis suggested that patients who received the loading dose between 6 and 24 hours before did benefit, with a relative risk reduction of 38.6% in the composite end point (P = .051).
After 28 days, the patients who had received the clopidogrel loading dose were continued on clopidogrel, while those in the placebo group were switched back to placebo. At 1 year, the investigators found a significantly lower rate of the composite end point with the prolonged course of clopidogrel (8.5% vs 11.5%).
In summary, these studies found clopidogrel to be beneficial in a broad spectrum of coronary diseases. Subgroup analyses suggest that pretreatment before percutaneous coronary intervention provides additional benefit, particularly if clopidogrel is given at least 6 hours in advance (the time necessary for clopidogrel to cause substantial platelet inhibition).
SOME PATIENTS RESPOND LESS TO CLOPIDOGREL
The level of platelet inhibition induced by clopidogrel varies. In different studies, the frequency of clopidogrel “nonresponsiveness” ranged from 5% to 56% of patients, depending on which test and which cutoff values were used. The distribution of responses to clopidogrel is wide and fits a normal gaussian curve.10
A large fraction of the population carries a gene that may account for some of the interpatient variation in platelet inhibition with clopidogrel. Carriers of a reduced-function CYP2C19 allele—approximately 30% of people in one study—have significantly lower levels of the active metabolite of clopidogrel, less platelet inhibition from clopidogrel therapy, and a 53% higher rate of death from cardiovascular causes, MI, or stroke.11
PRASUGREL, THE NEWEST THIENOPYRIDINE
Prasugrel, FDA-approved in July 2009 for the treatment of acute coronary syndromes, is given in an oral loading dose of 60 mg followed by an oral maintenance dose of 10 mg daily.
Pharmacology of prasugrel vs clopidogrel
As noted previously, the thienopyridines are prodrugs that require hepatic conversion to exert antiplatelet effects.
Metabolism. Prasugrel’s hepatic activation involves a single step, in contrast to the multiple-step process required for activation of clopidogrel. Clopidogrel is primarily hydrolyzed by intestinal and plasma esterases to an inactive terminal metabolite, with the residual unhydrolized drug undergoing a two-step metabolism that depends on cytochrome P450 enzymes. Prasugrel is also extensively hydrolyzed by these esterases, but the intermediate product is then metabolized in a single step to the active sulfhydryl compound, mainly by CYP3A4 and CYP2B6.
Thus, about 80% of an orally absorbed dose of prasugrel is converted to active drug, compared with only 10% to 20% of absorbed clopidogrel.
Time to peak effect. With clopidogrel, maximal inhibition of platelet aggregation occurs 3 to 5 days after starting therapy with 75 mg daily without a loading dose, but within 4 to 6 hours if a loading dose of 300 to 600 mg is given. In contrast, a prasugrel loading dose produces more than 80% of its platelet inhibitory effects by 30 minutes, and peak activity is observed within 4 hours.12 The platelet inhibition induced by prasugrel at 30 minutes after administration is comparable to the peak effect of clopidogrel at 6 hours.13
Dose-response. Prasugrel’s inhibition of platelet aggregation is dose-related.
Prasugrel is about 10 times more potent than clopidogrel and 100 times more potent than ticlopidine. Thus, treatment with 5 mg of prasugrel results in inhibition of platelet activity (distributed in a gaussian curve) very similar to that produced by 75 mg of clopidogrel. On the other hand, even a maintenance dose of 150 mg of clopidogrel inhibits platelet activity to a lesser degree than 10 mg of prasugrel (46% vs 61%),14 so clopidogrel appears to reach a plateau of platelet inhibition that prasugrel can overcome.
At the approved dose of prasugrel, inhibition of platelet aggregation is significantly greater and there are fewer “nonresponders” than with clopidogrel.
Interactions. Drugs that inhibit CYP3A4 do not inhibit the efficacy of prasugrel, but they can inhibit that of clopidogrel. Some commonly used drugs that have this effect are the statins (eg, atorvastain [Lipitor]) and the macrolide antibiotics (eg, erythromycin). Furthermore, whereas proton pump inhibitors have been shown to diminish the effect of clopidogrel by reducing the formation of its active metabolite, no such effect has been noted with prasugrel.
Prasugrel in phase 2 trials: Finding the optimal dosage
A phase 2 trial compared three prasugrel regimens (loading dose/daily maintenance dose of 40 mg/7.5 mg, 60 mg/10 mg, and 60 mg/15 mg) and standard clopidogrel therapy (300 mg/75 mg) in patients undergoing elective or urgent percutaneous coronary intervention.15 No significant difference in outcomes was seen in the groups receiving the three prasugrel regimens. However, more “minimal bleeding events” (defined by the criteria of the TIMI trial16) occurred with high-dose prasugrel than with lower-dose prasugrel or with clopidogrel, leading to use of the intermediate-dose prasugrel regimen (60-mg loading dose, 10-mg daily maintenance) for later trials.
Another phase 2 trial randomized 201 patients undergoing elective percutaneous coronary intervention to receive prasugrel 60 mg/10 mg or clopidogrel 600 mg/150 mg.14 In all patients, the loading dose was given about 1 hour before cardiac catheterization. As soon as 30 minutes after the loading dose, platelet inhibition was superior with prasugrel (31% vs 5% inhibition of platelet aggregation), and it remained significantly higher at 6 hours (75% vs 32%) and during the maintenance phase (61% vs 46%).
Phase 3 trial of prasugrel vs clopidogrel: TRITON-TIMI 38
Only one large phase 3 trial of prasugrel has been completed: TRITON-TIMI 38 (the Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel—Thrombolysis in Myocardial Infarction),17 which enrolled adults with moderate-risk to high-risk acute coronary syndromes scheduled to undergo a percutaneous coronary intervention. In this trial, 10,074 patients were enrolled who had moderate-to high-risk unstable angina or non-ST-elevation MI, and 3,534 patients were enrolled who had ST-elevation MI.
Patients were randomized to receive prasugrel (a 60-mg loading dose, then 10 mg daily) or clopidogrel (a 300-mg loading dose, then 75 mg daily) and were treated for 6 to 15 months. All patients also received aspirin.
These benefits came at a price of more bleeding. Of those patients who did not undergo coronary artery bypass grafting, more experienced bleeding in the prasugrel group than in the clopidogrel group (2.4% vs 1.8%, P = .03), including a higher rate of life-threatening bleeding (1.4% vs 0.89%, P = .01) and fatal bleeding (0.4% vs 0.1%, P = .002). More patients discontinued prasugrel because of hemorrhage (2.5% vs 1.4%, P < .001). In patients who proceeded to coronary artery bypass grafting, the rate of major bleeding was more than four times higher in those who received prasugrel than in those who received clopidogrel (13.4% vs 3.2%, P < .001).
A higher rate of adverse events related to colon cancer was also noted in patients treated with prasugrel, although the authors suggest this may have resulted from the stronger antiplatelet effects of prasugrel bringing more tumors to medical attention due to bleeding.
Overall death rates did not differ significantly between the treatment groups.
In a post hoc analysis,18 prasugrel was superior to clopidogrel in preventing ischemic events both during the first 3 days following randomization (the “loading phase”) and for the remainder of the trial (the “maintenance phase”). Whereas bleeding risk was similar with the two drugs during the loading phase, prasugrel was subsequently associated with more bleeding during the maintenance phase.
Certain patient subgroups had no net benefit or even suffered harm from prasugrel compared with clopidogrel.17 Patients with previous stroke or transient ischemic attack had net harm from prasugrel (hazard ratio 1.54, P = .04) and showed a strong trend toward a greater rate of major bleeding (P = .06). Patients age 75 and older and those weighing less than 60 kg had no net benefit from prasugrel.
Cost of prasugrel
Prasugrel is currently priced at 18% more than clopidogrel, with average wholesale prices per pill of $6.65 for prasugrel 10 mg compared with $5.63 for clopidogrel 75 mg. (Prasugrel 10-mg pills cost $6.33 at drugstore.com or $7.60 at CVS; clopidogrel 75-mg pills cost $5.33 at drugstore.com or $6.43 at CVS.) The patent on clopidogrel expires in November 2011, after which the price differential is expected to become significantly greater.
TICAGRELOR, A REVERSIBLE ORAL AGENT
Ticagrelor, the first reversible oral P2Y12 receptor antagonist, is an alternative to thienopyridine therapy for acute coronary syndromes.
Ticagrelor is quickly absorbed, does not require metabolic activation, and has a rapid antiplatelet effect and offset of effect, which closely follow drug-exposure levels. In a large randomized controlled trial in patients with acute coronary syndromes with or without STsegment elevation, treatment with ticagrelor compared with clopidogrel resulted in a significant reduction in death from vascular causes, MI, or stroke (9.8% vs 11.7%).19
Given its reversible effect on platelet inhibition, ticagrelor may be preferred in patients whose coronary anatomy is unknown and for whom coronary artery bypass grafting is deemed probable. It is still undergoing trials and is not yet approved.
TAKE-HOME POINTS
Prasugrel is more potent, more rapid in onset, and more consistent in inhibiting platelet aggregation than clopidogrel. A large clinical trial17 found prasugrel to be superior to clopidogrel for patients with moderate-to high-risk acute coronary syndromes with high probability of undergoing a percutaneous coronary intervention.
Who should receive prasugrel, and how?
Prasugrel should be given after angiography to patients with non-ST-elevation acute coronary syndromes or at presentation to patients with ST-elevation MI. When used for planned percutaneous coronary intervention, prasugrel should be given at least 30 minutes before the intervention, as was done in phase 2 trials (although its routine use in this situation is not recommended—see below).
It is given in a one-time loading dose of 60 mg by mouth and then maintained with 10 mg by mouth once daily for at least 1 year. (At least 9 months of treatment with a thienopyridine is indicated for patients with acute coronary syndromes who are medically treated, and at least 1 year is indicated following urgent or elective percutaneous coronary intervention, including balloon angioplasty and placement of a bare-metal or drug-eluting stent.)
Who should not receive prasugrel?
For now, prasugrel should be avoided in favor of clopidogrel in patients at higher risk of bleeding. It is clearly contraindicated in patients with prior transient ischemic attack or stroke, for whom the risk of serious bleeding seems to be prohibitive. It should generally be avoided in patients age 75 and older, although it might be considered in those at particularly high risk of stent thrombosis, such as those with diabetes or prior MI. In patients weighing less than 60 kg, the package insert advises a reduced dose (5 mg), although clinical evidence for this practice is lacking.
As yet, we have no data assuring that prasugrel is safe to use in combination with fibrinolytic agents, so patients on thrombolytic therapy for acute MI should continue to receive clopidogrel starting immediately after lysis. Furthermore, in patients who proceeded to coronary artery bypass grafting, the rate of major bleeding was more than four times higher in the prasugrel group than in the clopidogrel group in the TRITON-TIMI 38 trial.17 No thienopyridine should be given to patients likely to proceed to coronary artery bypass grafting.
Only clopidogrel has evidence supporting its use as an alternative to aspirin for patients with atherosclerotic disease who cannot tolerate aspirin. Neither drug has evidence for use for primary prevention.
Other areas of uncertainty
Prior to angiography. Indications for prasugrel are currently limited by the narrow scope of the trial data. TRITON-TIMI 38,17 the only large trial completed to date, randomized patients to receive prasugrel only after their coronary anatomy was known, except for ST-elevation MI patients. It is unknown whether the benefits of prasugrel will outweigh the higher risk of bleeding in patients with acute coronary syndromes who do not proceed to percutaneous coronary interventions.
A clinical trial is currently under way comparing prasugrel with clopidogrel in 10,000 patients with acute coronary syndromes who will be medically managed without planned revascularization: A Comparison of Prasugrel and Clopidogrel in Acute Coronary Syndrome Subjects (TRILOGY ACS), ClinicalTrials.gov Identifier: NCT00699998. The trial has an estimated completion date of March 2011.
In cases of non-ST-elevation acute coronary syndrome, it is reasonable to wait to give a thienopyridine until after the coronary anatomy has been defined, if angiography will be completed soon after presentation. For example, a 1-hour delay before giving prasugrel still delivers antiplatelet therapy more quickly than giving clopidogrel on presentation. If longer delays are expected before angiography, however, the patient should be given a loading dose of clopidogrel “up front,” in accordance with guidelines published by the American College of Cardiology, American Heart Association, and European Society of Cardiology,20 which recommend starting a thienopyridine early during hospitalization based on trial data with clopidogrel.
Patients undergoing elective percutaneous coronary intervention are at lower risk of stent thrombosis and other ischemic complications, so it is possible that the benefits of prasugrel would not outweigh the risks in these patients. Thus, prasugrel cannot yet be recommended for routine elective percutaneous coronary intervention except in individual cases in which the interventionalist feels that the patient may be at higher risk of thrombosis.
- Yeghiazarians Y, Braunstein JB, Askari A, Stone PH. Unstable angina pectoris. N Engl J Med 2000; 342:101–114.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Balsano F, Rizzon P, Violi F, et al. Antiplatelet treatment with ticlopidine in unstable angina. A controlled multicenter clinical trial. The Studio della Ticlopidina nell'Angina Instabile Group. Circulation 1990; 82:17–26.
- Schömig A, Neumann FJ, Kastrati A, et al. A randomized comparison of antiplatelet and anticoagulant therapy after the placement of coronary-artery stents. N Engl J Med 1996; 334:1084–1089.
- Mehta SR, Yusuf S, Peters RJG, et al; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- Sabatine MS, Cannon CP, Gibson CM, et al; CLA RITY-TIMI 28 Investigators. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with STsegment elevation. N Engl J Med 2005; 352:1179–1189.
- Sabatine MS, Cannon CP, Gibson CM, et al; Clopidogrel as Adjunctive Reperfusion Therapy (CLARITY)-Thrombolysis in Myocardial Infarction (TIMI) 28 Investigators. Effect of clopidogrel pretreatment before percutaneous coronary intervention in patients with ST-elevation myocardial infarction treated with fibrinolytics: the PCI-CLARITY study. JAMA 2005: 294:1224–1232.
- Chen ZM, Jiang LX, Chen YP, et al; COMMIT (ClOpidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Steinhubl SR, Berger PB, Mann JT, et al; CREDO Investigators. Clopidogrel for the reduction of events during observation. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 288:2411–2420.
- Serebruany VL, Steinhubl SR, Berger PB, Malinin AI, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45:246–251.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome P-450 polymorphisms and response to clopidogrel. N Engl J Med 2009; 360:354–362.
- Helft G, Osende JI, Worthley SG, et al. Acute antithrombotic effect of a front-loaded regimen of clopidogrel in patients with atherosclerosis on aspirin. Arterioscler Thromb Vasc Biol 2000; 20:2316–2321.
- Weerakkody GJ, Jakubowski JA, Brandt JT, et al. Comparison of speed of onset of platelet inhibition after loading doses of clopidogrel versus prasugrel in healthy volunteers and correlation with responder status. Am J Cardiol 2007; 100:331–336.
- Wiviott SD, Trenk D, Frelinger AL, et al; PRINCIPLETIMI 44 Investigators. Prasugrel compared with high loading-and maintenance-dose clopidogrel in patients with planned percutaneous coronary intervention: the Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation-Thrombolysis in Myocardial Infarction 44 trial. Circulation 2007; 116:2923–2932.
- Wiviott SD, Antman EM, Winters KJ, et al; JUMBO-TIMI 26 Investigators. Randomized comparison of prasugrel (CS-747, LY640315), a novel thienopyridine P2Y12 antagonist, with clopidogrel in percutaneous coronary intervention: results of the Joint Utilization of Medications to Block Platelets Optimally (JUMBO)-TIMI 26 Trial. Circulation 2005; 111:3366–3373.
- Bovill EG, Terrin ML, Stump DC, et al. Hemorrhagic events during therapy with recombinant tissue-type plasminogen activator, heparin, and aspirin for acute myocardial infarction. Results of the Thrombolysis in Myocardial Infarction (TIMI) Phase II Trial. Ann Intern Med 1991; 115:256–265.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITONTIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Antman EM, Wiviott SD, Murphy SA, et al. Early and late benefits of prasugrel in patients with acute coronary syndromes undergoing percutaneous coronary intervention: a TRITON-TIMI 38 (TRial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet InhibitioN with Prasugrel-Thrombolysis In Myocardial Infarction) analysis. J Am Coll Cardiol 2008; 51:2028–2033.
- Wallentin L, Becker RC, Budaj A, Freij A, Thorsén M, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Braunwald E, Antman EM, Beasley JW, et al. ACC/AHA 2002 guideline update for the management of patients with unstable angina and non–ST-segment elevation myocardial infarction—summary article*1: A report of the American College of Cardiology/American Heart Association task force on practice guidelines (Committee on the Management of Patients With Unstable Angina). J Am Coll Cardiol 2002; 40:1366–1374.
- Yeghiazarians Y, Braunstein JB, Askari A, Stone PH. Unstable angina pectoris. N Engl J Med 2000; 342:101–114.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Balsano F, Rizzon P, Violi F, et al. Antiplatelet treatment with ticlopidine in unstable angina. A controlled multicenter clinical trial. The Studio della Ticlopidina nell'Angina Instabile Group. Circulation 1990; 82:17–26.
- Schömig A, Neumann FJ, Kastrati A, et al. A randomized comparison of antiplatelet and anticoagulant therapy after the placement of coronary-artery stents. N Engl J Med 1996; 334:1084–1089.
- Mehta SR, Yusuf S, Peters RJG, et al; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- Sabatine MS, Cannon CP, Gibson CM, et al; CLA RITY-TIMI 28 Investigators. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with STsegment elevation. N Engl J Med 2005; 352:1179–1189.
- Sabatine MS, Cannon CP, Gibson CM, et al; Clopidogrel as Adjunctive Reperfusion Therapy (CLARITY)-Thrombolysis in Myocardial Infarction (TIMI) 28 Investigators. Effect of clopidogrel pretreatment before percutaneous coronary intervention in patients with ST-elevation myocardial infarction treated with fibrinolytics: the PCI-CLARITY study. JAMA 2005: 294:1224–1232.
- Chen ZM, Jiang LX, Chen YP, et al; COMMIT (ClOpidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Steinhubl SR, Berger PB, Mann JT, et al; CREDO Investigators. Clopidogrel for the reduction of events during observation. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 288:2411–2420.
- Serebruany VL, Steinhubl SR, Berger PB, Malinin AI, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45:246–251.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome P-450 polymorphisms and response to clopidogrel. N Engl J Med 2009; 360:354–362.
- Helft G, Osende JI, Worthley SG, et al. Acute antithrombotic effect of a front-loaded regimen of clopidogrel in patients with atherosclerosis on aspirin. Arterioscler Thromb Vasc Biol 2000; 20:2316–2321.
- Weerakkody GJ, Jakubowski JA, Brandt JT, et al. Comparison of speed of onset of platelet inhibition after loading doses of clopidogrel versus prasugrel in healthy volunteers and correlation with responder status. Am J Cardiol 2007; 100:331–336.
- Wiviott SD, Trenk D, Frelinger AL, et al; PRINCIPLETIMI 44 Investigators. Prasugrel compared with high loading-and maintenance-dose clopidogrel in patients with planned percutaneous coronary intervention: the Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation-Thrombolysis in Myocardial Infarction 44 trial. Circulation 2007; 116:2923–2932.
- Wiviott SD, Antman EM, Winters KJ, et al; JUMBO-TIMI 26 Investigators. Randomized comparison of prasugrel (CS-747, LY640315), a novel thienopyridine P2Y12 antagonist, with clopidogrel in percutaneous coronary intervention: results of the Joint Utilization of Medications to Block Platelets Optimally (JUMBO)-TIMI 26 Trial. Circulation 2005; 111:3366–3373.
- Bovill EG, Terrin ML, Stump DC, et al. Hemorrhagic events during therapy with recombinant tissue-type plasminogen activator, heparin, and aspirin for acute myocardial infarction. Results of the Thrombolysis in Myocardial Infarction (TIMI) Phase II Trial. Ann Intern Med 1991; 115:256–265.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITONTIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Antman EM, Wiviott SD, Murphy SA, et al. Early and late benefits of prasugrel in patients with acute coronary syndromes undergoing percutaneous coronary intervention: a TRITON-TIMI 38 (TRial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet InhibitioN with Prasugrel-Thrombolysis In Myocardial Infarction) analysis. J Am Coll Cardiol 2008; 51:2028–2033.
- Wallentin L, Becker RC, Budaj A, Freij A, Thorsén M, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Braunwald E, Antman EM, Beasley JW, et al. ACC/AHA 2002 guideline update for the management of patients with unstable angina and non–ST-segment elevation myocardial infarction—summary article*1: A report of the American College of Cardiology/American Heart Association task force on practice guidelines (Committee on the Management of Patients With Unstable Angina). J Am Coll Cardiol 2002; 40:1366–1374.
KEY POINTS
- The thienopyridines—ticlopidine (Ticlid), clopidogrel (Plavix), and now prasugrel—reduce the risk of death from and serious complications of acute coronary syndromes by inhibiting platelet aggregation.
- Compared with clopidogrel, prasugrel is more potent, faster in onset, and more consistent in inhibiting platelets.
- Prasugrel should be avoided in patients at higher risk of bleeding, including those with a history of stroke or transient ischemic attack, those age 75 or older, or those who weigh less than 60 kg.
Is an ACE inhibitor plus an ARB more effective than either drug alone?
No. Although randomized, controlled trials have shown convincingly that angiotensin-converting enzyme (ACE) inhibitors reduce the rates of death, myocardial infarction, stroke, and heart failure in patients with known coronary artery disease or left ventricular dysfunction,1 and that angiotensin receptor blockers (ARBs) are “noninferior” to and better tolerated than ACE inhibitors, causing less angioedema and cough but costing more,2 dual renin-angiotensin system (RAS) blockade—an ACE inhibitor plus an ARB—has never been shown to reduce the rates of morbidity or death from any cause.
In fact, the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET)3,4 found that dual RAS blockade was no more beneficial than monotherapy with an ACE inhibitor or an ARB in preventing serious outcomes in patients with known vascular disease or diabetes with end-organ damage. Furthermore, patients on dual RAS blockade had higher rates of renal insufficiency, hyperkalemia, and hypotension.
THE RATIONALE FOR DUAL RAS BLOCKADE
Dual RAS blockade was first proposed in the early 1990s as a way to avoid the “escape phenomenon” (incomplete suppression of angiotensin II) with ACE inhibitor monotherapy.5 Indeed, studies in rats showed a synergistic effect on blood pressure with an ACE inhibitor combined with an ARB,6 and these results were encouraging enough for the medical community to make a remarkably quick transition to adopting dual RAS blockade in clinical practice.
The concept of dual RAS blockade was so appealing that effects on surrogate end points—lower blood pressure, less protein in the urine, and improved endothelial function—were accepted as free passes, obviating the need for evidence of an effect on hard end points such as lower rates of cardiovascular death or kidney failure. Currently, in the United States, about 1.5% of all patients on RAS blockers are currently receiving both an ACE inhibitor and an ARB.
CONDITIONS IN WHICH DUAL RAS BLOCKADE WAS THOUGHT BENEFICIAL
Hypertension
The European Society of Cardiology’s 2007 clinical practice guidelines7 say that treatment with an ACE inhibitor plus an ARB is preferred for hypertensive patients with metabolic syndrome and its major components (eg, abdominal obesity, insulin resistance, frank diabetes).
Dulton et al, in a meta-analysis,8 calculated that the combination of an ACE inhibitor and an ARB lowered 24-hour blood pressure by 4/3 mm Hg more than monotherapy did. However, most of the studies were of short duration (6 to 8 weeks) and used submaximal doses or once-daily doses of a short-acting ACE inhibitor. Interestingly, studies that used a long-acting ACE inhibitor or a larger dose of a short-acting ACE inhibitor generally showed no additive effect on blood pressure when an ARB was added.
Hence, more evidence from larger randomized and appropriately designed studies is needed before we can conclude that dual RAS blockade is safe and significantly superior to monotherapy in blood pressure control.
Proteinuria
Proteinuria is a surrogate end point for cardiovascular death and is a marker as well as a cause of progressive renal insufficiency. It therefore seemed rational that modifying the degree of proteinuria would translate into robust clinical benefits. Several studies9 showed better renal outcomes, such as fewer patients needing dialysis with combination therapy than with an ACE inhibitor or ARB alone. However, this has never been proven in an adequately powered trial.
ONTARGET was a perfect opportunity to convert what seemed like reliable mechanistic information into solid outcome data.3 The trial enrolled 25,620 patients with established atherosclerotic disease or with diabetes and evidence of end-organ damage. At baseline, 13.1% had microalbuminuria and 4.0% had macroalbuminuria.3 The amount of protein in the urine increased by a significantly lesser amount in the ARB group and in the dualtherapy group than in the group taking only an ACE inhibitor, but in the dual-therapy group this apparent advantage came at the expense of hard end points: more patients reached the primary composite end point of needing dialysis, doubling of their serum creatinine level, or death.
Reducing proteinuria could be an important benefit, but it certainly does not outweigh the risk of increased rates of renal failure and death.
Atherosclerosis and acute coronary syndrome
The road to myocardial infarction begins with inflammation in the “shoulders” of atherosclerotic plaques, which subsequently rupture. Tissue ACE activity and expression of the angiotensin II type 1 receptor are significantly increased in patients with acute coronary syndrome and primarily co-localized to the shoulder regions of the plaque.10 Giving an ACE inhibitor or an ARB to patients who have unstable angina or who have had a myocardial infarction may decrease the rate of reinfarction and lessens the inflammatory process in the atherosclerotic plaque.
Large randomized clinical trials such as HOPE (Heart Outcomes Prevention Evaluation)11 and EUROPA (European Trial on Reduction of Cardiac Events With Perindopril in Stable Coronary Artery Disease)12 showed a lower rate of cardiovascular death in patients with established coronary artery disease and normal left ventricular function if they received an ACE inhibitor. In the HOPE trial, the rate of cardiovascular death was 25% lower in patients treated with ramipril (Altace) vs placebo.11 (The year after HOPE was published, the number of prescriptions for ramipril went up 400%). Interestingly, studies of ARBs for secondary prevention failed to show any lowering of the rate of cardiovascular death or myocardial infarction.13
In ONTARGET,4 although the combination of telmisartan (Micardis) and ramipril had a greater effect on blood pressure, it was not significantly better than ramipril alone in terms of the primary outcome of death from cardiovascular causes, myocardial infarction, stroke, or hospitalization for heart failure (relative risk 0.99).
Heart failure
The bulk of data on dual RAS blockade in heart failure patients comes from three large randomized trials: CHARM-Added (Candesartan in Heart Failure: Assessment of Reduction in Mortality and Morbidity),14 VALIANT (Valsartan in Acute Myocardial Infarction Trial),15 and VAL-HeFT (Valsartan Heart Failure Trial).16
CHARM-Added14 was the only trial that showed a reduction in cardiovascular deaths with dual RAS therapy (absolute risk reduction 3.6%). It also showed a lower rate of hospitalization for heart failure (absolute risk reduction 4%). However, the rate of allcause mortality was not different between the groups. Of note, more patients receiving dual RAS blockade had to stop taking the study drug because of adverse effects.
Val-HeFT16 showed, in a post hoc analysis, higher rates of morbidity (cardiac arrest, hospitalization for heart failure, or receipt of intravenous inotropic or vasodilator therapy for at least 4 hours) and death when the ARB valsartan (Diovan) was added to the combination of an ACE inhibitor plus a beta-blocker.
A recent meta-analysis17 of safety and tolerability of dual RAS blockade compared with an ACE inhibitor alone found a higher risk of discontinuation because of adverse effects such as hyperkalemia, renal dysfunction, and hypotension in patients on dual RAS blockade. The authors concluded that, given the adverse effects and the lack of consistent survival benefit, the available data do not support the routine addition of an ARB to ACE inhibitor therapy in heart failure patients.
WHAT ABOUT DIRECT RENIN INHIBITORS?
Another class of RAS blockers is available: direct renin inhibitors. Therefore, dual RAS blockade can be achieved by combining an ACE inhibitor with an ARB, an ACE inhibitor with a direct renin inhibitor, or an ARB with a direct renin inhibitor.
We have some outcome data on the combination of an ACE inhibitor plus an ARB,3,4,17 but none for the other two possible dual RAS combinations. Thus far, we know that dual RAS blockade with an ARB and an ACE inhibitor is not beneficial in patients like those in ONTARGET, and that it has questionable benefit in heart failure. However, little is known about combining a direct renin inhibitor with either an ACE inhibitor or an ARB.
Since ARBs and ACE inhibitors both increase plasma renin activity and only partially block the RAS, the argument has been put forward that the addition of a drug such as a direct renin inhibitor, which really decreases plasma renin activity, has the potential to be more beneficial than blockade with either an ACE inhibitor or an ARB. In theory, this is an attractive concept and certainly deserves scrutiny in outcome studies such as ALTITUDE (Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints).18
SURROGATE END POINTS: A CAVEAT
As defined by Temple,19 a surrogate end point of a clinical trial is a laboratory measurement or a physical sign used as a substitute for a clinically meaningful end point that measures directly how patients feel or function, or if they survive. Effects on surrogate end points often fail to predict the true clinical effects of an intervention, as the ONTARGET data demonstrated. Among several explanations for this failure is that interventions may affect the clinical outcome by unintended, unanticipated, and unrecognized mechanisms that operate independently of the disease process.20 Nonetheless, surrogate end point cosmetics remains attractive for many clinicians.
The ONTARGET findings indicate that there is no clinically important benefit in adding an ARB for patients with hypertension, proteinuria, heart failure, or coronary artery disease if they are already being treated with an ACE inhibitor. This would indicate that dual RAS blockade should be avoided in clinical practice until we are provided with better evidence.
- Father MD, Yusuf S, Kober L, et al. Long-term ACE-inhibitor therapy in patients with heart failure or left ventricular dysfunction: a systemic overview of data from individual patients. ACEInhibitor Myocardial Infarction Collaborative Group. Lancet 2000; 355:1575–1581.
- Pitt B, Poole-Wilson PA, Segal R, et al. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: randomized trial—the Losartan Heart Failure Survival Study ELITE II. Lancet 2000; 355:1582–1587.
- Mann JF, Schmiede RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomized, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Yusuf S, Teo KK, Pogue J, et al; ONTARGET Investigators Telmisartan, ramipril, or both in patients at high risk for vascular events, N Engl J Med 2008; 358:1547–1559.
- van den Meiracker AH, Man in ‘t Veld AJ, Admiraal PJ, et al. Partial escape of angiotensin converting enzyme (ACE) inhibition during prolonged ACE inhibitor treatment: dose it exist and does it affect the antihypertensive response? J Hypertens 1992; 10:803–812.
- Menard J, Campbell DJ, Azizi M, Gonzales MF. Synergistic effects of ACE inhibition and Ang II antagonism on blood pressure, cardiac weight, and renin in spontaneously hypertensive rats. Circulation 1997; 96:3072–3078.
- Mancia G, De Backer G, Dominiczak A, et al; Task Force for the Management of Arterial Hypertension of the European Society of Hypertension. 2007 guidelines for the management of arterial hypertension. J Hypertens 2007; 25:1105–1187.
- Dulton TW, He FJ, MacGregor GA. Systematic review of combined angiotensin-converting enzyme inhibition and angiotensin receptor blockade in hypertension. Hypertension 2005; 45:880–886.
- Kunz R, Fredrich C, Wolbers M, Mann JF. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med 2008; 148:30–48.
- Schieffer B, Schieffer E, Hilfiker-Kleiner D, et al. Expression of angiotensin II and interleukin 6 in human coronary atherosclerotic plaques: potential implications for inflammation and plaque instability. Circulation 2000; 101:1372–1378.
- Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med 2000; 342:145–153.
- Fox KM; European trial On reduction of cardiac events with Perindopril in stable coronary Artery disease Investigators. Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: randomized, double-blind, placebo-controlled, multicentre trial (the EUROPA study). Lancet 2003; 362:782–788.
- Dahlöf B, Devereux RB, Kjeldsen SE, et al; LIFE Study Group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomized trial against atenolol. Lancet 2002; 359:995–1003.
- McMurray JJ, Ostergren J, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced leftventricular systolic function taking angiotensin converting enzyme inhibitors: the CHARM-Added trial. Lancet 2003; 362:767–771.
- Pfeffer MA, McMurray JJ, Velzquez E, et al; Valsartan in Acute Myocardial Infarction Trial Investigators. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 2003; 349:1893–1906.
- Cohn JN, Tognoni GValsartan Heart Failure Trial Investigators. A randomized trial of the angiotensin receptor blocker valsartan in chronic heart failure. N Engl J Med 2001: 345:1667–1675.
- Lakhdar R, Al-Mallah MH, Lanfear DE. Safety and tolerability of angiotensin-converting enzyme inhibitor versus the combination of angiotensin-converting enzyme inhibitor and angiotensin receptor blocker in patients with left ventricular dysfunction: a systematic review and meta-analysis of randomized controlled trials. J Card Fail 2008; 14:181–188.
- Parving H-H, Brenner BM, McMurray JJV, et al. Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints (ALTITUDE): rationale and study design [published online ahead of print January 14, 2009]. Nephrol Dial Transplant. doi:10.1093/ndt/gfn721.
- Temple RJ. A regulatory authority’s opinion about surrogate endpoints. In:Nimmo WS, Tucker GT, eds. Clinical Measurement in Drug Evaluation. J Wiley: New York, 1995.
- Messerli FH. The sudden demise of dual renin-angiotensin system blockade or the soft science of the surrogate end point. J Am Coll Cardiol 2009; 53:468–470.
No. Although randomized, controlled trials have shown convincingly that angiotensin-converting enzyme (ACE) inhibitors reduce the rates of death, myocardial infarction, stroke, and heart failure in patients with known coronary artery disease or left ventricular dysfunction,1 and that angiotensin receptor blockers (ARBs) are “noninferior” to and better tolerated than ACE inhibitors, causing less angioedema and cough but costing more,2 dual renin-angiotensin system (RAS) blockade—an ACE inhibitor plus an ARB—has never been shown to reduce the rates of morbidity or death from any cause.
In fact, the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET)3,4 found that dual RAS blockade was no more beneficial than monotherapy with an ACE inhibitor or an ARB in preventing serious outcomes in patients with known vascular disease or diabetes with end-organ damage. Furthermore, patients on dual RAS blockade had higher rates of renal insufficiency, hyperkalemia, and hypotension.
THE RATIONALE FOR DUAL RAS BLOCKADE
Dual RAS blockade was first proposed in the early 1990s as a way to avoid the “escape phenomenon” (incomplete suppression of angiotensin II) with ACE inhibitor monotherapy.5 Indeed, studies in rats showed a synergistic effect on blood pressure with an ACE inhibitor combined with an ARB,6 and these results were encouraging enough for the medical community to make a remarkably quick transition to adopting dual RAS blockade in clinical practice.
The concept of dual RAS blockade was so appealing that effects on surrogate end points—lower blood pressure, less protein in the urine, and improved endothelial function—were accepted as free passes, obviating the need for evidence of an effect on hard end points such as lower rates of cardiovascular death or kidney failure. Currently, in the United States, about 1.5% of all patients on RAS blockers are currently receiving both an ACE inhibitor and an ARB.
CONDITIONS IN WHICH DUAL RAS BLOCKADE WAS THOUGHT BENEFICIAL
Hypertension
The European Society of Cardiology’s 2007 clinical practice guidelines7 say that treatment with an ACE inhibitor plus an ARB is preferred for hypertensive patients with metabolic syndrome and its major components (eg, abdominal obesity, insulin resistance, frank diabetes).
Dulton et al, in a meta-analysis,8 calculated that the combination of an ACE inhibitor and an ARB lowered 24-hour blood pressure by 4/3 mm Hg more than monotherapy did. However, most of the studies were of short duration (6 to 8 weeks) and used submaximal doses or once-daily doses of a short-acting ACE inhibitor. Interestingly, studies that used a long-acting ACE inhibitor or a larger dose of a short-acting ACE inhibitor generally showed no additive effect on blood pressure when an ARB was added.
Hence, more evidence from larger randomized and appropriately designed studies is needed before we can conclude that dual RAS blockade is safe and significantly superior to monotherapy in blood pressure control.
Proteinuria
Proteinuria is a surrogate end point for cardiovascular death and is a marker as well as a cause of progressive renal insufficiency. It therefore seemed rational that modifying the degree of proteinuria would translate into robust clinical benefits. Several studies9 showed better renal outcomes, such as fewer patients needing dialysis with combination therapy than with an ACE inhibitor or ARB alone. However, this has never been proven in an adequately powered trial.
ONTARGET was a perfect opportunity to convert what seemed like reliable mechanistic information into solid outcome data.3 The trial enrolled 25,620 patients with established atherosclerotic disease or with diabetes and evidence of end-organ damage. At baseline, 13.1% had microalbuminuria and 4.0% had macroalbuminuria.3 The amount of protein in the urine increased by a significantly lesser amount in the ARB group and in the dualtherapy group than in the group taking only an ACE inhibitor, but in the dual-therapy group this apparent advantage came at the expense of hard end points: more patients reached the primary composite end point of needing dialysis, doubling of their serum creatinine level, or death.
Reducing proteinuria could be an important benefit, but it certainly does not outweigh the risk of increased rates of renal failure and death.
Atherosclerosis and acute coronary syndrome
The road to myocardial infarction begins with inflammation in the “shoulders” of atherosclerotic plaques, which subsequently rupture. Tissue ACE activity and expression of the angiotensin II type 1 receptor are significantly increased in patients with acute coronary syndrome and primarily co-localized to the shoulder regions of the plaque.10 Giving an ACE inhibitor or an ARB to patients who have unstable angina or who have had a myocardial infarction may decrease the rate of reinfarction and lessens the inflammatory process in the atherosclerotic plaque.
Large randomized clinical trials such as HOPE (Heart Outcomes Prevention Evaluation)11 and EUROPA (European Trial on Reduction of Cardiac Events With Perindopril in Stable Coronary Artery Disease)12 showed a lower rate of cardiovascular death in patients with established coronary artery disease and normal left ventricular function if they received an ACE inhibitor. In the HOPE trial, the rate of cardiovascular death was 25% lower in patients treated with ramipril (Altace) vs placebo.11 (The year after HOPE was published, the number of prescriptions for ramipril went up 400%). Interestingly, studies of ARBs for secondary prevention failed to show any lowering of the rate of cardiovascular death or myocardial infarction.13
In ONTARGET,4 although the combination of telmisartan (Micardis) and ramipril had a greater effect on blood pressure, it was not significantly better than ramipril alone in terms of the primary outcome of death from cardiovascular causes, myocardial infarction, stroke, or hospitalization for heart failure (relative risk 0.99).
Heart failure
The bulk of data on dual RAS blockade in heart failure patients comes from three large randomized trials: CHARM-Added (Candesartan in Heart Failure: Assessment of Reduction in Mortality and Morbidity),14 VALIANT (Valsartan in Acute Myocardial Infarction Trial),15 and VAL-HeFT (Valsartan Heart Failure Trial).16
CHARM-Added14 was the only trial that showed a reduction in cardiovascular deaths with dual RAS therapy (absolute risk reduction 3.6%). It also showed a lower rate of hospitalization for heart failure (absolute risk reduction 4%). However, the rate of allcause mortality was not different between the groups. Of note, more patients receiving dual RAS blockade had to stop taking the study drug because of adverse effects.
Val-HeFT16 showed, in a post hoc analysis, higher rates of morbidity (cardiac arrest, hospitalization for heart failure, or receipt of intravenous inotropic or vasodilator therapy for at least 4 hours) and death when the ARB valsartan (Diovan) was added to the combination of an ACE inhibitor plus a beta-blocker.
A recent meta-analysis17 of safety and tolerability of dual RAS blockade compared with an ACE inhibitor alone found a higher risk of discontinuation because of adverse effects such as hyperkalemia, renal dysfunction, and hypotension in patients on dual RAS blockade. The authors concluded that, given the adverse effects and the lack of consistent survival benefit, the available data do not support the routine addition of an ARB to ACE inhibitor therapy in heart failure patients.
WHAT ABOUT DIRECT RENIN INHIBITORS?
Another class of RAS blockers is available: direct renin inhibitors. Therefore, dual RAS blockade can be achieved by combining an ACE inhibitor with an ARB, an ACE inhibitor with a direct renin inhibitor, or an ARB with a direct renin inhibitor.
We have some outcome data on the combination of an ACE inhibitor plus an ARB,3,4,17 but none for the other two possible dual RAS combinations. Thus far, we know that dual RAS blockade with an ARB and an ACE inhibitor is not beneficial in patients like those in ONTARGET, and that it has questionable benefit in heart failure. However, little is known about combining a direct renin inhibitor with either an ACE inhibitor or an ARB.
Since ARBs and ACE inhibitors both increase plasma renin activity and only partially block the RAS, the argument has been put forward that the addition of a drug such as a direct renin inhibitor, which really decreases plasma renin activity, has the potential to be more beneficial than blockade with either an ACE inhibitor or an ARB. In theory, this is an attractive concept and certainly deserves scrutiny in outcome studies such as ALTITUDE (Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints).18
SURROGATE END POINTS: A CAVEAT
As defined by Temple,19 a surrogate end point of a clinical trial is a laboratory measurement or a physical sign used as a substitute for a clinically meaningful end point that measures directly how patients feel or function, or if they survive. Effects on surrogate end points often fail to predict the true clinical effects of an intervention, as the ONTARGET data demonstrated. Among several explanations for this failure is that interventions may affect the clinical outcome by unintended, unanticipated, and unrecognized mechanisms that operate independently of the disease process.20 Nonetheless, surrogate end point cosmetics remains attractive for many clinicians.
The ONTARGET findings indicate that there is no clinically important benefit in adding an ARB for patients with hypertension, proteinuria, heart failure, or coronary artery disease if they are already being treated with an ACE inhibitor. This would indicate that dual RAS blockade should be avoided in clinical practice until we are provided with better evidence.
No. Although randomized, controlled trials have shown convincingly that angiotensin-converting enzyme (ACE) inhibitors reduce the rates of death, myocardial infarction, stroke, and heart failure in patients with known coronary artery disease or left ventricular dysfunction,1 and that angiotensin receptor blockers (ARBs) are “noninferior” to and better tolerated than ACE inhibitors, causing less angioedema and cough but costing more,2 dual renin-angiotensin system (RAS) blockade—an ACE inhibitor plus an ARB—has never been shown to reduce the rates of morbidity or death from any cause.
In fact, the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET)3,4 found that dual RAS blockade was no more beneficial than monotherapy with an ACE inhibitor or an ARB in preventing serious outcomes in patients with known vascular disease or diabetes with end-organ damage. Furthermore, patients on dual RAS blockade had higher rates of renal insufficiency, hyperkalemia, and hypotension.
THE RATIONALE FOR DUAL RAS BLOCKADE
Dual RAS blockade was first proposed in the early 1990s as a way to avoid the “escape phenomenon” (incomplete suppression of angiotensin II) with ACE inhibitor monotherapy.5 Indeed, studies in rats showed a synergistic effect on blood pressure with an ACE inhibitor combined with an ARB,6 and these results were encouraging enough for the medical community to make a remarkably quick transition to adopting dual RAS blockade in clinical practice.
The concept of dual RAS blockade was so appealing that effects on surrogate end points—lower blood pressure, less protein in the urine, and improved endothelial function—were accepted as free passes, obviating the need for evidence of an effect on hard end points such as lower rates of cardiovascular death or kidney failure. Currently, in the United States, about 1.5% of all patients on RAS blockers are currently receiving both an ACE inhibitor and an ARB.
CONDITIONS IN WHICH DUAL RAS BLOCKADE WAS THOUGHT BENEFICIAL
Hypertension
The European Society of Cardiology’s 2007 clinical practice guidelines7 say that treatment with an ACE inhibitor plus an ARB is preferred for hypertensive patients with metabolic syndrome and its major components (eg, abdominal obesity, insulin resistance, frank diabetes).
Dulton et al, in a meta-analysis,8 calculated that the combination of an ACE inhibitor and an ARB lowered 24-hour blood pressure by 4/3 mm Hg more than monotherapy did. However, most of the studies were of short duration (6 to 8 weeks) and used submaximal doses or once-daily doses of a short-acting ACE inhibitor. Interestingly, studies that used a long-acting ACE inhibitor or a larger dose of a short-acting ACE inhibitor generally showed no additive effect on blood pressure when an ARB was added.
Hence, more evidence from larger randomized and appropriately designed studies is needed before we can conclude that dual RAS blockade is safe and significantly superior to monotherapy in blood pressure control.
Proteinuria
Proteinuria is a surrogate end point for cardiovascular death and is a marker as well as a cause of progressive renal insufficiency. It therefore seemed rational that modifying the degree of proteinuria would translate into robust clinical benefits. Several studies9 showed better renal outcomes, such as fewer patients needing dialysis with combination therapy than with an ACE inhibitor or ARB alone. However, this has never been proven in an adequately powered trial.
ONTARGET was a perfect opportunity to convert what seemed like reliable mechanistic information into solid outcome data.3 The trial enrolled 25,620 patients with established atherosclerotic disease or with diabetes and evidence of end-organ damage. At baseline, 13.1% had microalbuminuria and 4.0% had macroalbuminuria.3 The amount of protein in the urine increased by a significantly lesser amount in the ARB group and in the dualtherapy group than in the group taking only an ACE inhibitor, but in the dual-therapy group this apparent advantage came at the expense of hard end points: more patients reached the primary composite end point of needing dialysis, doubling of their serum creatinine level, or death.
Reducing proteinuria could be an important benefit, but it certainly does not outweigh the risk of increased rates of renal failure and death.
Atherosclerosis and acute coronary syndrome
The road to myocardial infarction begins with inflammation in the “shoulders” of atherosclerotic plaques, which subsequently rupture. Tissue ACE activity and expression of the angiotensin II type 1 receptor are significantly increased in patients with acute coronary syndrome and primarily co-localized to the shoulder regions of the plaque.10 Giving an ACE inhibitor or an ARB to patients who have unstable angina or who have had a myocardial infarction may decrease the rate of reinfarction and lessens the inflammatory process in the atherosclerotic plaque.
Large randomized clinical trials such as HOPE (Heart Outcomes Prevention Evaluation)11 and EUROPA (European Trial on Reduction of Cardiac Events With Perindopril in Stable Coronary Artery Disease)12 showed a lower rate of cardiovascular death in patients with established coronary artery disease and normal left ventricular function if they received an ACE inhibitor. In the HOPE trial, the rate of cardiovascular death was 25% lower in patients treated with ramipril (Altace) vs placebo.11 (The year after HOPE was published, the number of prescriptions for ramipril went up 400%). Interestingly, studies of ARBs for secondary prevention failed to show any lowering of the rate of cardiovascular death or myocardial infarction.13
In ONTARGET,4 although the combination of telmisartan (Micardis) and ramipril had a greater effect on blood pressure, it was not significantly better than ramipril alone in terms of the primary outcome of death from cardiovascular causes, myocardial infarction, stroke, or hospitalization for heart failure (relative risk 0.99).
Heart failure
The bulk of data on dual RAS blockade in heart failure patients comes from three large randomized trials: CHARM-Added (Candesartan in Heart Failure: Assessment of Reduction in Mortality and Morbidity),14 VALIANT (Valsartan in Acute Myocardial Infarction Trial),15 and VAL-HeFT (Valsartan Heart Failure Trial).16
CHARM-Added14 was the only trial that showed a reduction in cardiovascular deaths with dual RAS therapy (absolute risk reduction 3.6%). It also showed a lower rate of hospitalization for heart failure (absolute risk reduction 4%). However, the rate of allcause mortality was not different between the groups. Of note, more patients receiving dual RAS blockade had to stop taking the study drug because of adverse effects.
Val-HeFT16 showed, in a post hoc analysis, higher rates of morbidity (cardiac arrest, hospitalization for heart failure, or receipt of intravenous inotropic or vasodilator therapy for at least 4 hours) and death when the ARB valsartan (Diovan) was added to the combination of an ACE inhibitor plus a beta-blocker.
A recent meta-analysis17 of safety and tolerability of dual RAS blockade compared with an ACE inhibitor alone found a higher risk of discontinuation because of adverse effects such as hyperkalemia, renal dysfunction, and hypotension in patients on dual RAS blockade. The authors concluded that, given the adverse effects and the lack of consistent survival benefit, the available data do not support the routine addition of an ARB to ACE inhibitor therapy in heart failure patients.
WHAT ABOUT DIRECT RENIN INHIBITORS?
Another class of RAS blockers is available: direct renin inhibitors. Therefore, dual RAS blockade can be achieved by combining an ACE inhibitor with an ARB, an ACE inhibitor with a direct renin inhibitor, or an ARB with a direct renin inhibitor.
We have some outcome data on the combination of an ACE inhibitor plus an ARB,3,4,17 but none for the other two possible dual RAS combinations. Thus far, we know that dual RAS blockade with an ARB and an ACE inhibitor is not beneficial in patients like those in ONTARGET, and that it has questionable benefit in heart failure. However, little is known about combining a direct renin inhibitor with either an ACE inhibitor or an ARB.
Since ARBs and ACE inhibitors both increase plasma renin activity and only partially block the RAS, the argument has been put forward that the addition of a drug such as a direct renin inhibitor, which really decreases plasma renin activity, has the potential to be more beneficial than blockade with either an ACE inhibitor or an ARB. In theory, this is an attractive concept and certainly deserves scrutiny in outcome studies such as ALTITUDE (Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints).18
SURROGATE END POINTS: A CAVEAT
As defined by Temple,19 a surrogate end point of a clinical trial is a laboratory measurement or a physical sign used as a substitute for a clinically meaningful end point that measures directly how patients feel or function, or if they survive. Effects on surrogate end points often fail to predict the true clinical effects of an intervention, as the ONTARGET data demonstrated. Among several explanations for this failure is that interventions may affect the clinical outcome by unintended, unanticipated, and unrecognized mechanisms that operate independently of the disease process.20 Nonetheless, surrogate end point cosmetics remains attractive for many clinicians.
The ONTARGET findings indicate that there is no clinically important benefit in adding an ARB for patients with hypertension, proteinuria, heart failure, or coronary artery disease if they are already being treated with an ACE inhibitor. This would indicate that dual RAS blockade should be avoided in clinical practice until we are provided with better evidence.
- Father MD, Yusuf S, Kober L, et al. Long-term ACE-inhibitor therapy in patients with heart failure or left ventricular dysfunction: a systemic overview of data from individual patients. ACEInhibitor Myocardial Infarction Collaborative Group. Lancet 2000; 355:1575–1581.
- Pitt B, Poole-Wilson PA, Segal R, et al. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: randomized trial—the Losartan Heart Failure Survival Study ELITE II. Lancet 2000; 355:1582–1587.
- Mann JF, Schmiede RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomized, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Yusuf S, Teo KK, Pogue J, et al; ONTARGET Investigators Telmisartan, ramipril, or both in patients at high risk for vascular events, N Engl J Med 2008; 358:1547–1559.
- van den Meiracker AH, Man in ‘t Veld AJ, Admiraal PJ, et al. Partial escape of angiotensin converting enzyme (ACE) inhibition during prolonged ACE inhibitor treatment: dose it exist and does it affect the antihypertensive response? J Hypertens 1992; 10:803–812.
- Menard J, Campbell DJ, Azizi M, Gonzales MF. Synergistic effects of ACE inhibition and Ang II antagonism on blood pressure, cardiac weight, and renin in spontaneously hypertensive rats. Circulation 1997; 96:3072–3078.
- Mancia G, De Backer G, Dominiczak A, et al; Task Force for the Management of Arterial Hypertension of the European Society of Hypertension. 2007 guidelines for the management of arterial hypertension. J Hypertens 2007; 25:1105–1187.
- Dulton TW, He FJ, MacGregor GA. Systematic review of combined angiotensin-converting enzyme inhibition and angiotensin receptor blockade in hypertension. Hypertension 2005; 45:880–886.
- Kunz R, Fredrich C, Wolbers M, Mann JF. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med 2008; 148:30–48.
- Schieffer B, Schieffer E, Hilfiker-Kleiner D, et al. Expression of angiotensin II and interleukin 6 in human coronary atherosclerotic plaques: potential implications for inflammation and plaque instability. Circulation 2000; 101:1372–1378.
- Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med 2000; 342:145–153.
- Fox KM; European trial On reduction of cardiac events with Perindopril in stable coronary Artery disease Investigators. Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: randomized, double-blind, placebo-controlled, multicentre trial (the EUROPA study). Lancet 2003; 362:782–788.
- Dahlöf B, Devereux RB, Kjeldsen SE, et al; LIFE Study Group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomized trial against atenolol. Lancet 2002; 359:995–1003.
- McMurray JJ, Ostergren J, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced leftventricular systolic function taking angiotensin converting enzyme inhibitors: the CHARM-Added trial. Lancet 2003; 362:767–771.
- Pfeffer MA, McMurray JJ, Velzquez E, et al; Valsartan in Acute Myocardial Infarction Trial Investigators. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 2003; 349:1893–1906.
- Cohn JN, Tognoni GValsartan Heart Failure Trial Investigators. A randomized trial of the angiotensin receptor blocker valsartan in chronic heart failure. N Engl J Med 2001: 345:1667–1675.
- Lakhdar R, Al-Mallah MH, Lanfear DE. Safety and tolerability of angiotensin-converting enzyme inhibitor versus the combination of angiotensin-converting enzyme inhibitor and angiotensin receptor blocker in patients with left ventricular dysfunction: a systematic review and meta-analysis of randomized controlled trials. J Card Fail 2008; 14:181–188.
- Parving H-H, Brenner BM, McMurray JJV, et al. Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints (ALTITUDE): rationale and study design [published online ahead of print January 14, 2009]. Nephrol Dial Transplant. doi:10.1093/ndt/gfn721.
- Temple RJ. A regulatory authority’s opinion about surrogate endpoints. In:Nimmo WS, Tucker GT, eds. Clinical Measurement in Drug Evaluation. J Wiley: New York, 1995.
- Messerli FH. The sudden demise of dual renin-angiotensin system blockade or the soft science of the surrogate end point. J Am Coll Cardiol 2009; 53:468–470.
- Father MD, Yusuf S, Kober L, et al. Long-term ACE-inhibitor therapy in patients with heart failure or left ventricular dysfunction: a systemic overview of data from individual patients. ACEInhibitor Myocardial Infarction Collaborative Group. Lancet 2000; 355:1575–1581.
- Pitt B, Poole-Wilson PA, Segal R, et al. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: randomized trial—the Losartan Heart Failure Survival Study ELITE II. Lancet 2000; 355:1582–1587.
- Mann JF, Schmiede RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomized, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Yusuf S, Teo KK, Pogue J, et al; ONTARGET Investigators Telmisartan, ramipril, or both in patients at high risk for vascular events, N Engl J Med 2008; 358:1547–1559.
- van den Meiracker AH, Man in ‘t Veld AJ, Admiraal PJ, et al. Partial escape of angiotensin converting enzyme (ACE) inhibition during prolonged ACE inhibitor treatment: dose it exist and does it affect the antihypertensive response? J Hypertens 1992; 10:803–812.
- Menard J, Campbell DJ, Azizi M, Gonzales MF. Synergistic effects of ACE inhibition and Ang II antagonism on blood pressure, cardiac weight, and renin in spontaneously hypertensive rats. Circulation 1997; 96:3072–3078.
- Mancia G, De Backer G, Dominiczak A, et al; Task Force for the Management of Arterial Hypertension of the European Society of Hypertension. 2007 guidelines for the management of arterial hypertension. J Hypertens 2007; 25:1105–1187.
- Dulton TW, He FJ, MacGregor GA. Systematic review of combined angiotensin-converting enzyme inhibition and angiotensin receptor blockade in hypertension. Hypertension 2005; 45:880–886.
- Kunz R, Fredrich C, Wolbers M, Mann JF. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med 2008; 148:30–48.
- Schieffer B, Schieffer E, Hilfiker-Kleiner D, et al. Expression of angiotensin II and interleukin 6 in human coronary atherosclerotic plaques: potential implications for inflammation and plaque instability. Circulation 2000; 101:1372–1378.
- Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med 2000; 342:145–153.
- Fox KM; European trial On reduction of cardiac events with Perindopril in stable coronary Artery disease Investigators. Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: randomized, double-blind, placebo-controlled, multicentre trial (the EUROPA study). Lancet 2003; 362:782–788.
- Dahlöf B, Devereux RB, Kjeldsen SE, et al; LIFE Study Group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomized trial against atenolol. Lancet 2002; 359:995–1003.
- McMurray JJ, Ostergren J, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced leftventricular systolic function taking angiotensin converting enzyme inhibitors: the CHARM-Added trial. Lancet 2003; 362:767–771.
- Pfeffer MA, McMurray JJ, Velzquez E, et al; Valsartan in Acute Myocardial Infarction Trial Investigators. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 2003; 349:1893–1906.
- Cohn JN, Tognoni GValsartan Heart Failure Trial Investigators. A randomized trial of the angiotensin receptor blocker valsartan in chronic heart failure. N Engl J Med 2001: 345:1667–1675.
- Lakhdar R, Al-Mallah MH, Lanfear DE. Safety and tolerability of angiotensin-converting enzyme inhibitor versus the combination of angiotensin-converting enzyme inhibitor and angiotensin receptor blocker in patients with left ventricular dysfunction: a systematic review and meta-analysis of randomized controlled trials. J Card Fail 2008; 14:181–188.
- Parving H-H, Brenner BM, McMurray JJV, et al. Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints (ALTITUDE): rationale and study design [published online ahead of print January 14, 2009]. Nephrol Dial Transplant. doi:10.1093/ndt/gfn721.
- Temple RJ. A regulatory authority’s opinion about surrogate endpoints. In:Nimmo WS, Tucker GT, eds. Clinical Measurement in Drug Evaluation. J Wiley: New York, 1995.
- Messerli FH. The sudden demise of dual renin-angiotensin system blockade or the soft science of the surrogate end point. J Am Coll Cardiol 2009; 53:468–470.
Dual antiplatelet therapy in coronary artery disease: A case-based approach
Plaque rupture and thrombosis play central roles in the genesis of acute coronary syndrome. Aspirin has long been the preventive agent of choice. But dual antiplatelet therapy with aspirin plus clopidogrel (Plavix) is warranted in many patients to further reduce their risk of future cardiovascular events.
Although dual antiplatelet therapy is usually started by a subspecialist, the primary care physician is often the one who ensures that the patient remains compliant with it in the long term. A review of the seminal published data is helpful in understanding the rationale behind dual antiplatelet therapy and its risks and benefits.
In the mid-1990s, the thienopyridine ticlopidine (Ticlid) was found to significantly decrease the number of deaths, target-lesion revascularizations, and myocardial infarctions (MIs) in the 30 days following stent placement. 1 However, 2% to 3% of patients experienced neutropenia2 and thrombotic thrombocytopenic purpura with this drug,3 leading to the use of clopidogrel, another agent in the same class. Over the past decade, a large body of evidence has established the usefulness of clopidogrel in a number of clinical settings.
In this paper we review the current use of clopidogrel in ST-elevation MI, non-ST-elevation acute coronary syndromes, and percutaneous coronary intervention, and discuss the landmark trials that are the basis for the treatment guidelines published jointly by the American College of Cardiology (ACC) and the American Heart Association (AHA).4–6 We also briefly discuss the use of prasugrel (Effient), the newest antiplatelet agent to gain approval from the US Food and Drug Administration (FDA).
CLOPIDOGREL AS AN ALTERNATIVE TO ASPIRIN
Clopidogrel, a prodrug, is converted into its active form in the liver.7 It then irreversibly binds to the platelet P2Y12 receptor and inhibits adenosine diphosphate-induced platelet aggregation.
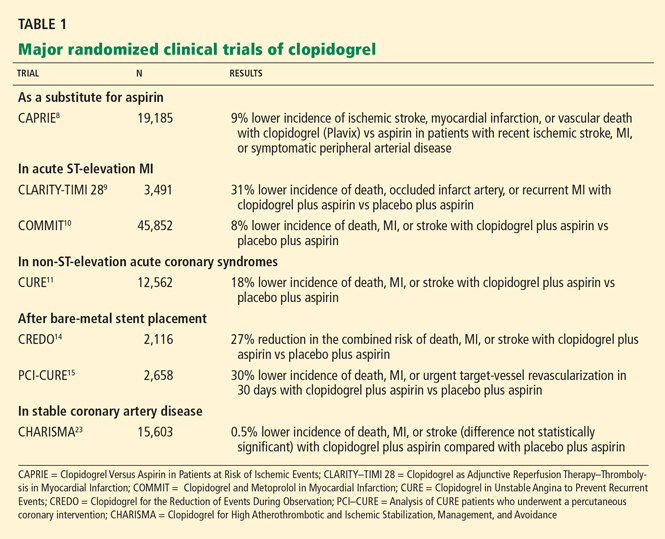
Those treated with clopidogrel had an annual risk of ischemic stroke, MI, or vascular death of 5.32%, compared with 5.83% in the aspirin group, for a statistically significant 8.7% relative risk reduction (P = .043). The observed frequency of neutropenia (neutrophils < 1.2 × 109/L) was 0.10% with clopidogrel vs 0.17% with aspirin. This study showed clopidogrel to be an effective alternative in patients who cannot tolerate aspirin.
CASE 1: ST-ELEVATION MI
A 57-year-old farmer in rural Ohio with a history of hypertension and hyperlipidemia presents to the local emergency department 45 minutes after the onset, while he was chopping wood, of dull, aching, substernal chest pain that radiates to his jaw. Electrocardiography reveals 2-mm ST-segment elevation in leads V1 through V6. He is treated with aspirin 162 mg, low-molecular-weight heparin, and tenecteplase.
What would be the value of starting dual antiplatelet therapy with clopidogrel in this patient?
Clopidogrel, aspirin, and fibrinolysis in ST-elevation MI

The CLARITY-TIMI 28 trial (Clopidogrel as Adjunctive Reperfusion Therapy–Thrombolysis in Myocardial Infarction)9 included 3,491 patients (ages 18 to 75) from 319 international sites. All patients received a fibrinolytic agent, aspirin (162 mg to 325 mg on the first day and 75 mg to 162 mg thereafter), and heparin as part of standard care for acute ST-elevation MI (Table 1). Patients were randomized to receive a 300-mg loading dose of clopidogrel followed by 75 mg daily or placebo within 12 hours of onset of ST-elevation MI. The status of the infarct-related artery was ascertained by protocol-mandated coronary angiography 48 to 192 hours after starting the study medication. The primary end point was the composite of an occluded infarct-related artery on angiography, death from any cause prior to angiography, or recurrent MI prior to angiography.
Significantly fewer patients had an end point event in the clopidogrel group than in the placebo group, 15% vs 21.7% (P < .001), for a relative risk reduction of 31%. There was no significant increase in major or minor bleeding events.
Of note, the CLARITY-TIMI 28 patients were relatively young (average age 57 years) and at low cardiovascular risk (30-day mortality risk < 5%).
The COMMIT trial (Clopidogrel and Metoprolol in Myocardial Infarction)10 consisted of 45,852 patients with suspected acute MI admitted to 1,250 hospitals in China. Each patient received aspirin 162 mg daily plus either clopidogrel 75 mg daily (n = 22,961) or placebo (n = 22,891) for the duration of hospitalization (average 16 days) or 28 days, whichever came first.
The incidence of the primary composite end point of death, reinfarction, or stroke was significantly lower with clopidogrel than with placebo (9.2% vs 10.1%, P = .002). This was regardless of age (the average age was 61, and 26% of patients were older than 70), sex, time to presentation (67% presented within 12 hours), or reperfusion strategy (49% underwent fibrinolysis). The clopidogrel group did not have a significantly higher incidence of bleeding, but patients in this trial did not receive a loading dose of clopidogrel.
Comment. In view of the results of these trials, our 57-year-old patient should start clopidogrel early.
CASE 2: NON-ST-ELEVATION ACUTE CORONARY SYNDROME
A 65-year-old woman living independently with no significant medical history presents to the emergency room with 2 hours of waxing and waning substernal chest pain. Her blood pressure is 145/90 mm Hg, her heart rate is 95 beats per minute, and the results of her physical examination are unremarkable. Resting electrocardiography reveals 1.5-mm ST-segment depression in the inferior leads, and her troponin T level on admission is two times the upper limit of normal. She is given aspirin and is started on low-molecular-weight heparin and intravenous nitroglycerin.
What would be the value of starting clopidogrel in this patient?
Clopidogrel in non-ST-elevation acute coronary syndromes
The ACC/AHA guidelines strongly support starting clopidogrel in patients with non-ST-elevation acute coronary syndromes (Table 2).5
The CURE trial (Clopidogrel in Unstable Angina to Prevent Recurrent Events)11 provided the evidence for this recommendation. In this trial, 12,562 patients from 482 centers in 28 countries who presented within 24 hours of coronary symptoms, without ST elevation, were randomized to receive either clopidogrel (a 300-mg loading dose, followed by 75 mg daily) or placebo for 3 to 12 months (mean 9 months).
Significantly fewer patients in the clopidogrel group reached one of the end points of the composite primary outcome (cardiovascular death, nonfatal MI, or stroke): 9.3% vs 11.4%, 95% confidence interval (CI) 0.72–0.90, P < .001. Significantly fewer of them also suffered one of the secondary outcomes, ie, severe ischemia, heart failure, or need for revascularization.
Of concern was a higher rate of major bleeding in the clopidogrel group (3.7%) than in the placebo group (2.7%) without an excess of fatal bleeding. For every 1,000 patients treated with clopidogrel, 6 required a blood transfusion. Nevertheless, CURE proved that patients with non-ST-elevation acute coronary syndromes benefited from clopidogrel, regardless of whether they underwent percutaneous coronary intervention.
Comment. Our patient should receive clopidogrel and, if she has no significant bleeding, she should continue to take it for at least 12 months after discharge. It is important for the primary care physician to ensure compliance with this agent and not discontinue it on routine clinical follow-up.
CASE 3: BARE-METAL STENT PLACEMENT
A 62-year-old man with a history of hypertension, diabetes, and hyperlipidemia presents to his primary care physician’s office with stable-effort angina that is not responding to an excellent anti-ischemic regimen and is affecting his quality of life. He is referred for coronary angiography, which reveals 80% stenosis of the proximal left circumflex artery. He undergoes a percutaneous coronary intervention with placement of a bare-metal stent.
How long should he be on clopidogrel? And what if a drug-eluting stent had been placed instead of a bare-metal stent?
Dual therapy after bare-metal stent placement
Dual antiplatelet therapy with clopidogrel and aspirin is recommended in all patients receiving a stent (Table 2). The better safety and efficacy of clopidogrel compared with ticlopidine has been established in patients receiving a coronary artery stent,12,13 and clopidogrel’s favorable safety profile soon made it the thienopyridine of choice.
The CREDO trial (Clopidogrel for the Reduction of Events During Observation)14 randomized 2,116 patients undergoing an elective percutaneous coronary intervention (bare-metal stent placement only) to receive a 300-mg loading dose of clopidogrel 3 to 24 hours before the procedure, or placebo. All patients received 325 mg of aspirin. After the intervention, all patients received clopidogrel 75 mg daily and aspirin 325 mg daily through day 28. For day 29 through 12 months, those who had received the 300-mg preprocedural loading dose of clopidogrel continued with 75 mg daily, and those who had not received clopidogrel before the procedure received placebo.
No significant difference was seen in the primary outcome for those who received pretreatment with clopidogrel; however, in a subgroup analysis, those who received clopidogrel at least 6 hours before the percutaneous coronary intervention had a 38.6% relative risk reduction (Table 1). Long-term use of clopidogrel (ie, for 12 months) was associated with an overall relative reduction of 26.9% in the combined risk of death, MI, or stroke.
PCI-CURE, an analysis of 2,658 patients in the CURE trial with non-ST-elevation acute coronary syndrome who underwent PCI,15 yielded results similar to those of CREDO, with a 31% reduction in the rate of cardiovascular death or MI at 30 days and at 9 months. Of note, however, clopidogrel was given for a median of 6 days prior to the procedure.
Comment. The minimum suggested duration of clopidogrel treatment after placement of a bare-metal stent is 1 month. However, these trial results indicate that patients who are not at high risk of bleeding should take clopidogrel for at least 12 months.
Dual antiplatelet therapy with drug-eluting stents
Although rates of in-stent restenosis are clearly lower with drug-eluting stents than with bare-metal stents, the antiproliferative effect of drug-eluting stents may delay complete endothelialization of every strut. This may contribute to late (> 1 month after placement) or very late (> 1 year) thrombosis of the stent after clopidogrel is discontinued.16–18
In 2006, the FDA indicated that dual antiplatelet therapy was needed for 6 months with paclitaxel-eluting (Taxus) stents and 3 months with sirolimus-eluting (Cipher) stents. As reports of very late stent thrombosis began to appear in 2007, concern arose over the need to extend the duration of clopidogrel treatment.
Bavry et al19 quantified the incidence of late and very late stent thrombosis in a meta-analysis of 14 clinical trials that randomized patients to receive either a drug-eluting stent (paclitaxel or sirolimus) or a bare-metal stent.19 The incidence of stent thrombosis within 30 days in this analysis was similar for both groups—4.4 per 1,000 patients vs 5 per 1,000 (relative risk 0.89; 95% CI 0.46–1.75; P = .74). However, the rate of very late stent thrombosis was significantly higher in those receiving a drug-eluting stent vs a bare-metal stent—5 per 1,000 patients treated (relative risk 5.02, 95% CI 1.29–19.52; P = .02).
The results of this and other studies led the ACC and AHA to revise their joint guidelines to recommend thienopyridine treatment for at least 1 year for patients who receive a drug-eluting stent.6,20–22 In fact, many cardiologists consider indefinite dual antiplatelet therapy in patients with a drug-eluting stent to avoid very late in-stent thrombosis, especially in patients undergoing high-risk interventions such as placement of multiple stents, bifurcation lesions, and unprotected left main trunk interventions.
Thus, when faced with a patient with a recent coronary stent implantation, the primary care physician should be aware of the type of stent and the duration of therapy recommended by the interventional cardiologist. Also, in the absence of a pressing indication, elective surgery should be deferred for 1 year after placement of a drug-eluting stent, as this would necessitate stopping clopidogrel and would increase the risk of perioperative stent thrombosis, which is associated with high rates of morbidity and death.
CASE 4: HIGH-RISK CORONARY ARTERY DISEASE
A 67-year-old woman presents to your office to establish care. She has a history of diabetes and established coronary artery disease with two bare-metal stents placed 2 years ago. She is taking aspirin 81 mg.
What would be the value of adding clopidogrel to her regimen?
No indication for clopidogrel in chronic coronary artery disease
The CHARISMA trial (Clopidogrel for High Atherothrombotic and Ischemic Stabilization, Management, and Avoidance)23 randomized 15,603 patients with stable cardiovascular disease or multiple risk factors to receive either clopidogrel plus low-dose aspirin or placebo plus low-dose aspirin and followed them for a median of 28 months (Table 1).
The primary end point (a composite of MI, stroke, or death) was 6.8% with clopidogrel plus aspirin and 7.3% with aspirin alone, indicating no significant benefit with clopidogrel plus aspirin compared with aspirin alone in reducing the rate of MI, stroke, or cardiovascular death in patients with high-risk but stable atherothrombotic disease. A marginal statistical benefit with dual antiplatelet therapy was noted in the subgroup of patients with previously documented coronary, cerebrovascular, or peripheral vascular disease—6.9% with aspirin plus clopidogrel vs 7.9% with aspirin alone (relative risk 0.88; 95% CI 0.77–0.998; P = .046).
Consequently, there is no compelling reason to start clopidogrel in this patient.
PRASUGREL, THE NEWEST THIENOPYRIDINE
Prasugrel was recently approved by the FDA as antiplatelet treatment for patients with acute coronary syndromes planning to undergo a percutaneous coronary intervention.24 It has been shown to inhibit adenosine-diphosphate-induced platelet activation in a more consistent and effective manner than clopidogrel.25,26
Although both clopidogrel and prasugrel are prodrugs, 80% of absorbed clopidogrel is metabolized by esterases into inactive metabolites, and the availability of active metabolite can vary, as it is significantly influenced by polymorphisms in the cytochrome P450 system. 27 In contrast, prasugrel is not degraded by esterases, and its conversion to active metabolite by the cytochrome P450 system is not influenced by common genetic polymorphisms, particularly CYP2C19*2.
TRITON-TIMI 38 (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel—Thrombolysis in Myocardial Infarction) provided most of the evidence for the approval of prasugrel for clinical use.28,29 In this trial, a 60-mg loading dose of prasugrel followed by a daily maintenance dose of 10 mg was significantly superior to the current clopidogrel regimen in preventing death from cardiovascular causes, nonfatal MI, or nonfatal stroke during a study period of 15 months.28 Also observed was a 24% lower rate of MI, a 34% lower rate of urgent target-vessel revascularization, and a 52% lower rate of stent thrombosis.
These benefits, however, came at the cost of a significantly higher risk of major bleeding, including the potential for three excess fatal bleeding events for every 1,000 patients treated. Patients at highest risk at the dosages evaluated included the elderly (age 75 and older), patients who weigh less than 60 kg, and patients with a history of stroke or transient ischemic attack. Based on these results, we recommend caution with the use of prasugrel in these patient subsets.
Clinical use of prasugrel is likely to be highest in patients presenting with ST-elevation MI who are undergoing a primary percutaneous coronary intervention. There is currently no evidence from any randomized clinical trial to support the safety of prasugrel given in the emergency room or “upstream” in the setting of non-ST-elevation acute coronary syndromes.
Of note, patients with non-ST-elevation acute coronary syndromes in the TRITON trial were randomized only after angiographic definition. As a result, only 179 patients exposed to prasugrel were referred for coronary artery bypass surgery, but the rate of surgery-related major bleeding in this group was 13.4% (vs 3.2% in the clopidogrel group). Based on these data, prasugrel should be withheld for at least 1 week prior to any surgery.
- Leon MB, Baim DS, Popma JJ, et al. A clinical trial comparing three antithrombotic-drug regimens after coronary-artery stenting. Stent Anticoagulation Restenosis Study Investigators. N Engl J Med 1998; 339:1665–1671.
- Yeh SP, Hsueh EJ, Wu H, Wang YC. Ticlopidine-associated aplastic anemia. A case report and review of literature, Ann Hematol 1998; 76:87–90.
- Page Y, Tardy B, Zeni F, Comtet C, Terrana R, Bertrand JC. Thrombotic thrombocytopenic purpura related to ticlopidine. Lancet 1991; 337:774–776.
- Canadian Cardiovascular Society; Antman EM, Hand M, Armstrong PW, et al. 2007 focused update of the ACC/AHA 2004 guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2008; 51:210–247.
- Anderson JL, Adams CD, Antman EM, et al; American College of Cardiology, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol 2007; 50:e1–e157.
- King SB, Smith SC, Hirshfeld JW, et al. 2007 focused update of the ACC/AHA/SCAI 2005 guideline update for percutaneous coronary intervention: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2008; 51:172–209.
- Kam PC, Nethery CM. The thienopyridine derivatives (platelet adenosine diphosphate receptor antagonists), pharmacology and clinical developments. Anaesthesia 2003; 58:28–35.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet 1996; 348:1329–1339.
- Sabatine MS, Cannon CP, Gibson M, et al; CLARITY-TIMI 28 Investigators. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation, N Engl J Med 2005; 352:1179–1189.
- Chen ZM, Jiang LX, Chen YP, et al; COMMIT (ClOpidogrel and Metoprolol in Myocardial Infarction Trial) Collaborative Group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: a randomized placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Bhatt DL, Bertrand ME, Berger PB, et al. Meta-analysis of randomized and registry comparisons of ticlopidine with clopidogrel after stenting. J Am Coll Cardiol 2002; 39:9–14.
- Bertrand ME, Rupprecht HJ, Urban P, Gershlick AH; CLASSICS Investigators. Double-blind study of the safety of clopidogrel with and without a loading dose in combination with aspirin compared with ticlopidine in combination with aspirin after coronary stenting: The Clopidogrel Aspirin Stent International Cooperative Study (CLASSICS). Circulation 2000; 102:624–629.
- Steinhubl SR, Berger PB, Mann JT, et al; CREDO Investigators. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 288:2411–2420.
- Mehta SR, Yusuf S, Peters RJ, et a; Clopidogrel in Unstable Angina to Prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCICURE study. Lancet 2001; 358:527–533.
- Lüscher TF, Steffel J, Eberli FR, et al. Drug-eluting stent and coronary thrombosis: biological mechanisms and clinical implications. Circulation 2007; 115:1051–1058.
- Kotani J, Awata M, Nanto S, et al. Incomplete neointimal coverage of sirolimus-eluting stents: angioscopic findings. J Am Coll Cardiol 2006; 47:2108–2111.
- Pfisterer M, Brunner-La Rocca HP, Buser PT, et al; BASKETLATE Investigators. Late clinical events after clopidogrel discontinuation may limit the benefit of drug-eluting stents: an observational study of drug-eluting versus bare-metal stents. J Am Coll Cardiol 2006; 48:2584–2591.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Stone GW, Moses JW, Ellis SG, et al. Safety and efficacy of sirolimus- and paclitaxel-eluting coronary stents. N Engl J Med 2007; 356:998–1008.
- Mauri L, Hsieh WH, Massaro JM, Ho KK, D’Agostino R, Cutlip DE. Stent thrombosis in randomized clinical trials of drug-eluting stents. N Engl J Med 2007; 356:1020–1029.
- Kastrati A, Mehilli J, Pache J, et al. Analysis of 14 trials comparing sirolimus-eluting stents with bare-metal stents. N Engl J Med 2007; 356:1030–1039.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- US Food and Drug Administration. FDA Approves Effient to Reduce the Risk of Heart Attack in Angioplasty Patients. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm171497.htm. Accessed October 2, 2009.
- Jernber T, Payne CD, Winters KJ, et al. Prasugrel achieves greater inhibition of platelet aggregation and a lower rate of non-responders compared with clopidogrel in aspirin-treated patients with stable coronary artery disease. Eur Heart J 2006; 27:1166–1173.
- Wiviott SD, Trenk D, Frelinger AL, et al; PRINCIPLE-TIMI 44 Investigators. Prasugrel compared with high loading and maintenance-dose clopidogrel in patients with planned percutaneous coronary intervention: the Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation-Thrombolysis in Myocardial Infarction 44 trial. Circulation 2007; 116:2923–2932.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med 2009; 360:354–362.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Antman EM, Wiviott SD, Murphy SA, et al. Early and late benefits of prasugrel in patients with acute coronary syndromes undergoing percutaneous coronary intervention: A TRITON-TIMI (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel-Thrombolysis In Myocardial Infarction) analysis. J Am Coll Card 2008; 51:2028–2033.
Plaque rupture and thrombosis play central roles in the genesis of acute coronary syndrome. Aspirin has long been the preventive agent of choice. But dual antiplatelet therapy with aspirin plus clopidogrel (Plavix) is warranted in many patients to further reduce their risk of future cardiovascular events.
Although dual antiplatelet therapy is usually started by a subspecialist, the primary care physician is often the one who ensures that the patient remains compliant with it in the long term. A review of the seminal published data is helpful in understanding the rationale behind dual antiplatelet therapy and its risks and benefits.
In the mid-1990s, the thienopyridine ticlopidine (Ticlid) was found to significantly decrease the number of deaths, target-lesion revascularizations, and myocardial infarctions (MIs) in the 30 days following stent placement. 1 However, 2% to 3% of patients experienced neutropenia2 and thrombotic thrombocytopenic purpura with this drug,3 leading to the use of clopidogrel, another agent in the same class. Over the past decade, a large body of evidence has established the usefulness of clopidogrel in a number of clinical settings.
In this paper we review the current use of clopidogrel in ST-elevation MI, non-ST-elevation acute coronary syndromes, and percutaneous coronary intervention, and discuss the landmark trials that are the basis for the treatment guidelines published jointly by the American College of Cardiology (ACC) and the American Heart Association (AHA).4–6 We also briefly discuss the use of prasugrel (Effient), the newest antiplatelet agent to gain approval from the US Food and Drug Administration (FDA).
CLOPIDOGREL AS AN ALTERNATIVE TO ASPIRIN
Clopidogrel, a prodrug, is converted into its active form in the liver.7 It then irreversibly binds to the platelet P2Y12 receptor and inhibits adenosine diphosphate-induced platelet aggregation.
Those treated with clopidogrel had an annual risk of ischemic stroke, MI, or vascular death of 5.32%, compared with 5.83% in the aspirin group, for a statistically significant 8.7% relative risk reduction (P = .043). The observed frequency of neutropenia (neutrophils < 1.2 × 109/L) was 0.10% with clopidogrel vs 0.17% with aspirin. This study showed clopidogrel to be an effective alternative in patients who cannot tolerate aspirin.
CASE 1: ST-ELEVATION MI
A 57-year-old farmer in rural Ohio with a history of hypertension and hyperlipidemia presents to the local emergency department 45 minutes after the onset, while he was chopping wood, of dull, aching, substernal chest pain that radiates to his jaw. Electrocardiography reveals 2-mm ST-segment elevation in leads V1 through V6. He is treated with aspirin 162 mg, low-molecular-weight heparin, and tenecteplase.
What would be the value of starting dual antiplatelet therapy with clopidogrel in this patient?
Clopidogrel, aspirin, and fibrinolysis in ST-elevation MI
The CLARITY-TIMI 28 trial (Clopidogrel as Adjunctive Reperfusion Therapy–Thrombolysis in Myocardial Infarction)9 included 3,491 patients (ages 18 to 75) from 319 international sites. All patients received a fibrinolytic agent, aspirin (162 mg to 325 mg on the first day and 75 mg to 162 mg thereafter), and heparin as part of standard care for acute ST-elevation MI (Table 1). Patients were randomized to receive a 300-mg loading dose of clopidogrel followed by 75 mg daily or placebo within 12 hours of onset of ST-elevation MI. The status of the infarct-related artery was ascertained by protocol-mandated coronary angiography 48 to 192 hours after starting the study medication. The primary end point was the composite of an occluded infarct-related artery on angiography, death from any cause prior to angiography, or recurrent MI prior to angiography.
Significantly fewer patients had an end point event in the clopidogrel group than in the placebo group, 15% vs 21.7% (P < .001), for a relative risk reduction of 31%. There was no significant increase in major or minor bleeding events.
Of note, the CLARITY-TIMI 28 patients were relatively young (average age 57 years) and at low cardiovascular risk (30-day mortality risk < 5%).
The COMMIT trial (Clopidogrel and Metoprolol in Myocardial Infarction)10 consisted of 45,852 patients with suspected acute MI admitted to 1,250 hospitals in China. Each patient received aspirin 162 mg daily plus either clopidogrel 75 mg daily (n = 22,961) or placebo (n = 22,891) for the duration of hospitalization (average 16 days) or 28 days, whichever came first.
The incidence of the primary composite end point of death, reinfarction, or stroke was significantly lower with clopidogrel than with placebo (9.2% vs 10.1%, P = .002). This was regardless of age (the average age was 61, and 26% of patients were older than 70), sex, time to presentation (67% presented within 12 hours), or reperfusion strategy (49% underwent fibrinolysis). The clopidogrel group did not have a significantly higher incidence of bleeding, but patients in this trial did not receive a loading dose of clopidogrel.
Comment. In view of the results of these trials, our 57-year-old patient should start clopidogrel early.
CASE 2: NON-ST-ELEVATION ACUTE CORONARY SYNDROME
A 65-year-old woman living independently with no significant medical history presents to the emergency room with 2 hours of waxing and waning substernal chest pain. Her blood pressure is 145/90 mm Hg, her heart rate is 95 beats per minute, and the results of her physical examination are unremarkable. Resting electrocardiography reveals 1.5-mm ST-segment depression in the inferior leads, and her troponin T level on admission is two times the upper limit of normal. She is given aspirin and is started on low-molecular-weight heparin and intravenous nitroglycerin.
What would be the value of starting clopidogrel in this patient?
Clopidogrel in non-ST-elevation acute coronary syndromes
The ACC/AHA guidelines strongly support starting clopidogrel in patients with non-ST-elevation acute coronary syndromes (Table 2).5
The CURE trial (Clopidogrel in Unstable Angina to Prevent Recurrent Events)11 provided the evidence for this recommendation. In this trial, 12,562 patients from 482 centers in 28 countries who presented within 24 hours of coronary symptoms, without ST elevation, were randomized to receive either clopidogrel (a 300-mg loading dose, followed by 75 mg daily) or placebo for 3 to 12 months (mean 9 months).
Significantly fewer patients in the clopidogrel group reached one of the end points of the composite primary outcome (cardiovascular death, nonfatal MI, or stroke): 9.3% vs 11.4%, 95% confidence interval (CI) 0.72–0.90, P < .001. Significantly fewer of them also suffered one of the secondary outcomes, ie, severe ischemia, heart failure, or need for revascularization.
Of concern was a higher rate of major bleeding in the clopidogrel group (3.7%) than in the placebo group (2.7%) without an excess of fatal bleeding. For every 1,000 patients treated with clopidogrel, 6 required a blood transfusion. Nevertheless, CURE proved that patients with non-ST-elevation acute coronary syndromes benefited from clopidogrel, regardless of whether they underwent percutaneous coronary intervention.
Comment. Our patient should receive clopidogrel and, if she has no significant bleeding, she should continue to take it for at least 12 months after discharge. It is important for the primary care physician to ensure compliance with this agent and not discontinue it on routine clinical follow-up.
CASE 3: BARE-METAL STENT PLACEMENT
A 62-year-old man with a history of hypertension, diabetes, and hyperlipidemia presents to his primary care physician’s office with stable-effort angina that is not responding to an excellent anti-ischemic regimen and is affecting his quality of life. He is referred for coronary angiography, which reveals 80% stenosis of the proximal left circumflex artery. He undergoes a percutaneous coronary intervention with placement of a bare-metal stent.
How long should he be on clopidogrel? And what if a drug-eluting stent had been placed instead of a bare-metal stent?
Dual therapy after bare-metal stent placement
Dual antiplatelet therapy with clopidogrel and aspirin is recommended in all patients receiving a stent (Table 2). The better safety and efficacy of clopidogrel compared with ticlopidine has been established in patients receiving a coronary artery stent,12,13 and clopidogrel’s favorable safety profile soon made it the thienopyridine of choice.
The CREDO trial (Clopidogrel for the Reduction of Events During Observation)14 randomized 2,116 patients undergoing an elective percutaneous coronary intervention (bare-metal stent placement only) to receive a 300-mg loading dose of clopidogrel 3 to 24 hours before the procedure, or placebo. All patients received 325 mg of aspirin. After the intervention, all patients received clopidogrel 75 mg daily and aspirin 325 mg daily through day 28. For day 29 through 12 months, those who had received the 300-mg preprocedural loading dose of clopidogrel continued with 75 mg daily, and those who had not received clopidogrel before the procedure received placebo.
No significant difference was seen in the primary outcome for those who received pretreatment with clopidogrel; however, in a subgroup analysis, those who received clopidogrel at least 6 hours before the percutaneous coronary intervention had a 38.6% relative risk reduction (Table 1). Long-term use of clopidogrel (ie, for 12 months) was associated with an overall relative reduction of 26.9% in the combined risk of death, MI, or stroke.
PCI-CURE, an analysis of 2,658 patients in the CURE trial with non-ST-elevation acute coronary syndrome who underwent PCI,15 yielded results similar to those of CREDO, with a 31% reduction in the rate of cardiovascular death or MI at 30 days and at 9 months. Of note, however, clopidogrel was given for a median of 6 days prior to the procedure.
Comment. The minimum suggested duration of clopidogrel treatment after placement of a bare-metal stent is 1 month. However, these trial results indicate that patients who are not at high risk of bleeding should take clopidogrel for at least 12 months.
Dual antiplatelet therapy with drug-eluting stents
Although rates of in-stent restenosis are clearly lower with drug-eluting stents than with bare-metal stents, the antiproliferative effect of drug-eluting stents may delay complete endothelialization of every strut. This may contribute to late (> 1 month after placement) or very late (> 1 year) thrombosis of the stent after clopidogrel is discontinued.16–18
In 2006, the FDA indicated that dual antiplatelet therapy was needed for 6 months with paclitaxel-eluting (Taxus) stents and 3 months with sirolimus-eluting (Cipher) stents. As reports of very late stent thrombosis began to appear in 2007, concern arose over the need to extend the duration of clopidogrel treatment.
Bavry et al19 quantified the incidence of late and very late stent thrombosis in a meta-analysis of 14 clinical trials that randomized patients to receive either a drug-eluting stent (paclitaxel or sirolimus) or a bare-metal stent.19 The incidence of stent thrombosis within 30 days in this analysis was similar for both groups—4.4 per 1,000 patients vs 5 per 1,000 (relative risk 0.89; 95% CI 0.46–1.75; P = .74). However, the rate of very late stent thrombosis was significantly higher in those receiving a drug-eluting stent vs a bare-metal stent—5 per 1,000 patients treated (relative risk 5.02, 95% CI 1.29–19.52; P = .02).
The results of this and other studies led the ACC and AHA to revise their joint guidelines to recommend thienopyridine treatment for at least 1 year for patients who receive a drug-eluting stent.6,20–22 In fact, many cardiologists consider indefinite dual antiplatelet therapy in patients with a drug-eluting stent to avoid very late in-stent thrombosis, especially in patients undergoing high-risk interventions such as placement of multiple stents, bifurcation lesions, and unprotected left main trunk interventions.
Thus, when faced with a patient with a recent coronary stent implantation, the primary care physician should be aware of the type of stent and the duration of therapy recommended by the interventional cardiologist. Also, in the absence of a pressing indication, elective surgery should be deferred for 1 year after placement of a drug-eluting stent, as this would necessitate stopping clopidogrel and would increase the risk of perioperative stent thrombosis, which is associated with high rates of morbidity and death.
CASE 4: HIGH-RISK CORONARY ARTERY DISEASE
A 67-year-old woman presents to your office to establish care. She has a history of diabetes and established coronary artery disease with two bare-metal stents placed 2 years ago. She is taking aspirin 81 mg.
What would be the value of adding clopidogrel to her regimen?
No indication for clopidogrel in chronic coronary artery disease
The CHARISMA trial (Clopidogrel for High Atherothrombotic and Ischemic Stabilization, Management, and Avoidance)23 randomized 15,603 patients with stable cardiovascular disease or multiple risk factors to receive either clopidogrel plus low-dose aspirin or placebo plus low-dose aspirin and followed them for a median of 28 months (Table 1).
The primary end point (a composite of MI, stroke, or death) was 6.8% with clopidogrel plus aspirin and 7.3% with aspirin alone, indicating no significant benefit with clopidogrel plus aspirin compared with aspirin alone in reducing the rate of MI, stroke, or cardiovascular death in patients with high-risk but stable atherothrombotic disease. A marginal statistical benefit with dual antiplatelet therapy was noted in the subgroup of patients with previously documented coronary, cerebrovascular, or peripheral vascular disease—6.9% with aspirin plus clopidogrel vs 7.9% with aspirin alone (relative risk 0.88; 95% CI 0.77–0.998; P = .046).
Consequently, there is no compelling reason to start clopidogrel in this patient.
PRASUGREL, THE NEWEST THIENOPYRIDINE
Prasugrel was recently approved by the FDA as antiplatelet treatment for patients with acute coronary syndromes planning to undergo a percutaneous coronary intervention.24 It has been shown to inhibit adenosine-diphosphate-induced platelet activation in a more consistent and effective manner than clopidogrel.25,26
Although both clopidogrel and prasugrel are prodrugs, 80% of absorbed clopidogrel is metabolized by esterases into inactive metabolites, and the availability of active metabolite can vary, as it is significantly influenced by polymorphisms in the cytochrome P450 system. 27 In contrast, prasugrel is not degraded by esterases, and its conversion to active metabolite by the cytochrome P450 system is not influenced by common genetic polymorphisms, particularly CYP2C19*2.
TRITON-TIMI 38 (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel—Thrombolysis in Myocardial Infarction) provided most of the evidence for the approval of prasugrel for clinical use.28,29 In this trial, a 60-mg loading dose of prasugrel followed by a daily maintenance dose of 10 mg was significantly superior to the current clopidogrel regimen in preventing death from cardiovascular causes, nonfatal MI, or nonfatal stroke during a study period of 15 months.28 Also observed was a 24% lower rate of MI, a 34% lower rate of urgent target-vessel revascularization, and a 52% lower rate of stent thrombosis.
These benefits, however, came at the cost of a significantly higher risk of major bleeding, including the potential for three excess fatal bleeding events for every 1,000 patients treated. Patients at highest risk at the dosages evaluated included the elderly (age 75 and older), patients who weigh less than 60 kg, and patients with a history of stroke or transient ischemic attack. Based on these results, we recommend caution with the use of prasugrel in these patient subsets.
Clinical use of prasugrel is likely to be highest in patients presenting with ST-elevation MI who are undergoing a primary percutaneous coronary intervention. There is currently no evidence from any randomized clinical trial to support the safety of prasugrel given in the emergency room or “upstream” in the setting of non-ST-elevation acute coronary syndromes.
Of note, patients with non-ST-elevation acute coronary syndromes in the TRITON trial were randomized only after angiographic definition. As a result, only 179 patients exposed to prasugrel were referred for coronary artery bypass surgery, but the rate of surgery-related major bleeding in this group was 13.4% (vs 3.2% in the clopidogrel group). Based on these data, prasugrel should be withheld for at least 1 week prior to any surgery.
Plaque rupture and thrombosis play central roles in the genesis of acute coronary syndrome. Aspirin has long been the preventive agent of choice. But dual antiplatelet therapy with aspirin plus clopidogrel (Plavix) is warranted in many patients to further reduce their risk of future cardiovascular events.
Although dual antiplatelet therapy is usually started by a subspecialist, the primary care physician is often the one who ensures that the patient remains compliant with it in the long term. A review of the seminal published data is helpful in understanding the rationale behind dual antiplatelet therapy and its risks and benefits.
In the mid-1990s, the thienopyridine ticlopidine (Ticlid) was found to significantly decrease the number of deaths, target-lesion revascularizations, and myocardial infarctions (MIs) in the 30 days following stent placement. 1 However, 2% to 3% of patients experienced neutropenia2 and thrombotic thrombocytopenic purpura with this drug,3 leading to the use of clopidogrel, another agent in the same class. Over the past decade, a large body of evidence has established the usefulness of clopidogrel in a number of clinical settings.
In this paper we review the current use of clopidogrel in ST-elevation MI, non-ST-elevation acute coronary syndromes, and percutaneous coronary intervention, and discuss the landmark trials that are the basis for the treatment guidelines published jointly by the American College of Cardiology (ACC) and the American Heart Association (AHA).4–6 We also briefly discuss the use of prasugrel (Effient), the newest antiplatelet agent to gain approval from the US Food and Drug Administration (FDA).
CLOPIDOGREL AS AN ALTERNATIVE TO ASPIRIN
Clopidogrel, a prodrug, is converted into its active form in the liver.7 It then irreversibly binds to the platelet P2Y12 receptor and inhibits adenosine diphosphate-induced platelet aggregation.
Those treated with clopidogrel had an annual risk of ischemic stroke, MI, or vascular death of 5.32%, compared with 5.83% in the aspirin group, for a statistically significant 8.7% relative risk reduction (P = .043). The observed frequency of neutropenia (neutrophils < 1.2 × 109/L) was 0.10% with clopidogrel vs 0.17% with aspirin. This study showed clopidogrel to be an effective alternative in patients who cannot tolerate aspirin.
CASE 1: ST-ELEVATION MI
A 57-year-old farmer in rural Ohio with a history of hypertension and hyperlipidemia presents to the local emergency department 45 minutes after the onset, while he was chopping wood, of dull, aching, substernal chest pain that radiates to his jaw. Electrocardiography reveals 2-mm ST-segment elevation in leads V1 through V6. He is treated with aspirin 162 mg, low-molecular-weight heparin, and tenecteplase.
What would be the value of starting dual antiplatelet therapy with clopidogrel in this patient?
Clopidogrel, aspirin, and fibrinolysis in ST-elevation MI
The CLARITY-TIMI 28 trial (Clopidogrel as Adjunctive Reperfusion Therapy–Thrombolysis in Myocardial Infarction)9 included 3,491 patients (ages 18 to 75) from 319 international sites. All patients received a fibrinolytic agent, aspirin (162 mg to 325 mg on the first day and 75 mg to 162 mg thereafter), and heparin as part of standard care for acute ST-elevation MI (Table 1). Patients were randomized to receive a 300-mg loading dose of clopidogrel followed by 75 mg daily or placebo within 12 hours of onset of ST-elevation MI. The status of the infarct-related artery was ascertained by protocol-mandated coronary angiography 48 to 192 hours after starting the study medication. The primary end point was the composite of an occluded infarct-related artery on angiography, death from any cause prior to angiography, or recurrent MI prior to angiography.
Significantly fewer patients had an end point event in the clopidogrel group than in the placebo group, 15% vs 21.7% (P < .001), for a relative risk reduction of 31%. There was no significant increase in major or minor bleeding events.
Of note, the CLARITY-TIMI 28 patients were relatively young (average age 57 years) and at low cardiovascular risk (30-day mortality risk < 5%).
The COMMIT trial (Clopidogrel and Metoprolol in Myocardial Infarction)10 consisted of 45,852 patients with suspected acute MI admitted to 1,250 hospitals in China. Each patient received aspirin 162 mg daily plus either clopidogrel 75 mg daily (n = 22,961) or placebo (n = 22,891) for the duration of hospitalization (average 16 days) or 28 days, whichever came first.
The incidence of the primary composite end point of death, reinfarction, or stroke was significantly lower with clopidogrel than with placebo (9.2% vs 10.1%, P = .002). This was regardless of age (the average age was 61, and 26% of patients were older than 70), sex, time to presentation (67% presented within 12 hours), or reperfusion strategy (49% underwent fibrinolysis). The clopidogrel group did not have a significantly higher incidence of bleeding, but patients in this trial did not receive a loading dose of clopidogrel.
Comment. In view of the results of these trials, our 57-year-old patient should start clopidogrel early.
CASE 2: NON-ST-ELEVATION ACUTE CORONARY SYNDROME
A 65-year-old woman living independently with no significant medical history presents to the emergency room with 2 hours of waxing and waning substernal chest pain. Her blood pressure is 145/90 mm Hg, her heart rate is 95 beats per minute, and the results of her physical examination are unremarkable. Resting electrocardiography reveals 1.5-mm ST-segment depression in the inferior leads, and her troponin T level on admission is two times the upper limit of normal. She is given aspirin and is started on low-molecular-weight heparin and intravenous nitroglycerin.
What would be the value of starting clopidogrel in this patient?
Clopidogrel in non-ST-elevation acute coronary syndromes
The ACC/AHA guidelines strongly support starting clopidogrel in patients with non-ST-elevation acute coronary syndromes (Table 2).5
The CURE trial (Clopidogrel in Unstable Angina to Prevent Recurrent Events)11 provided the evidence for this recommendation. In this trial, 12,562 patients from 482 centers in 28 countries who presented within 24 hours of coronary symptoms, without ST elevation, were randomized to receive either clopidogrel (a 300-mg loading dose, followed by 75 mg daily) or placebo for 3 to 12 months (mean 9 months).
Significantly fewer patients in the clopidogrel group reached one of the end points of the composite primary outcome (cardiovascular death, nonfatal MI, or stroke): 9.3% vs 11.4%, 95% confidence interval (CI) 0.72–0.90, P < .001. Significantly fewer of them also suffered one of the secondary outcomes, ie, severe ischemia, heart failure, or need for revascularization.
Of concern was a higher rate of major bleeding in the clopidogrel group (3.7%) than in the placebo group (2.7%) without an excess of fatal bleeding. For every 1,000 patients treated with clopidogrel, 6 required a blood transfusion. Nevertheless, CURE proved that patients with non-ST-elevation acute coronary syndromes benefited from clopidogrel, regardless of whether they underwent percutaneous coronary intervention.
Comment. Our patient should receive clopidogrel and, if she has no significant bleeding, she should continue to take it for at least 12 months after discharge. It is important for the primary care physician to ensure compliance with this agent and not discontinue it on routine clinical follow-up.
CASE 3: BARE-METAL STENT PLACEMENT
A 62-year-old man with a history of hypertension, diabetes, and hyperlipidemia presents to his primary care physician’s office with stable-effort angina that is not responding to an excellent anti-ischemic regimen and is affecting his quality of life. He is referred for coronary angiography, which reveals 80% stenosis of the proximal left circumflex artery. He undergoes a percutaneous coronary intervention with placement of a bare-metal stent.
How long should he be on clopidogrel? And what if a drug-eluting stent had been placed instead of a bare-metal stent?
Dual therapy after bare-metal stent placement
Dual antiplatelet therapy with clopidogrel and aspirin is recommended in all patients receiving a stent (Table 2). The better safety and efficacy of clopidogrel compared with ticlopidine has been established in patients receiving a coronary artery stent,12,13 and clopidogrel’s favorable safety profile soon made it the thienopyridine of choice.
The CREDO trial (Clopidogrel for the Reduction of Events During Observation)14 randomized 2,116 patients undergoing an elective percutaneous coronary intervention (bare-metal stent placement only) to receive a 300-mg loading dose of clopidogrel 3 to 24 hours before the procedure, or placebo. All patients received 325 mg of aspirin. After the intervention, all patients received clopidogrel 75 mg daily and aspirin 325 mg daily through day 28. For day 29 through 12 months, those who had received the 300-mg preprocedural loading dose of clopidogrel continued with 75 mg daily, and those who had not received clopidogrel before the procedure received placebo.
No significant difference was seen in the primary outcome for those who received pretreatment with clopidogrel; however, in a subgroup analysis, those who received clopidogrel at least 6 hours before the percutaneous coronary intervention had a 38.6% relative risk reduction (Table 1). Long-term use of clopidogrel (ie, for 12 months) was associated with an overall relative reduction of 26.9% in the combined risk of death, MI, or stroke.
PCI-CURE, an analysis of 2,658 patients in the CURE trial with non-ST-elevation acute coronary syndrome who underwent PCI,15 yielded results similar to those of CREDO, with a 31% reduction in the rate of cardiovascular death or MI at 30 days and at 9 months. Of note, however, clopidogrel was given for a median of 6 days prior to the procedure.
Comment. The minimum suggested duration of clopidogrel treatment after placement of a bare-metal stent is 1 month. However, these trial results indicate that patients who are not at high risk of bleeding should take clopidogrel for at least 12 months.
Dual antiplatelet therapy with drug-eluting stents
Although rates of in-stent restenosis are clearly lower with drug-eluting stents than with bare-metal stents, the antiproliferative effect of drug-eluting stents may delay complete endothelialization of every strut. This may contribute to late (> 1 month after placement) or very late (> 1 year) thrombosis of the stent after clopidogrel is discontinued.16–18
In 2006, the FDA indicated that dual antiplatelet therapy was needed for 6 months with paclitaxel-eluting (Taxus) stents and 3 months with sirolimus-eluting (Cipher) stents. As reports of very late stent thrombosis began to appear in 2007, concern arose over the need to extend the duration of clopidogrel treatment.
Bavry et al19 quantified the incidence of late and very late stent thrombosis in a meta-analysis of 14 clinical trials that randomized patients to receive either a drug-eluting stent (paclitaxel or sirolimus) or a bare-metal stent.19 The incidence of stent thrombosis within 30 days in this analysis was similar for both groups—4.4 per 1,000 patients vs 5 per 1,000 (relative risk 0.89; 95% CI 0.46–1.75; P = .74). However, the rate of very late stent thrombosis was significantly higher in those receiving a drug-eluting stent vs a bare-metal stent—5 per 1,000 patients treated (relative risk 5.02, 95% CI 1.29–19.52; P = .02).
The results of this and other studies led the ACC and AHA to revise their joint guidelines to recommend thienopyridine treatment for at least 1 year for patients who receive a drug-eluting stent.6,20–22 In fact, many cardiologists consider indefinite dual antiplatelet therapy in patients with a drug-eluting stent to avoid very late in-stent thrombosis, especially in patients undergoing high-risk interventions such as placement of multiple stents, bifurcation lesions, and unprotected left main trunk interventions.
Thus, when faced with a patient with a recent coronary stent implantation, the primary care physician should be aware of the type of stent and the duration of therapy recommended by the interventional cardiologist. Also, in the absence of a pressing indication, elective surgery should be deferred for 1 year after placement of a drug-eluting stent, as this would necessitate stopping clopidogrel and would increase the risk of perioperative stent thrombosis, which is associated with high rates of morbidity and death.
CASE 4: HIGH-RISK CORONARY ARTERY DISEASE
A 67-year-old woman presents to your office to establish care. She has a history of diabetes and established coronary artery disease with two bare-metal stents placed 2 years ago. She is taking aspirin 81 mg.
What would be the value of adding clopidogrel to her regimen?
No indication for clopidogrel in chronic coronary artery disease
The CHARISMA trial (Clopidogrel for High Atherothrombotic and Ischemic Stabilization, Management, and Avoidance)23 randomized 15,603 patients with stable cardiovascular disease or multiple risk factors to receive either clopidogrel plus low-dose aspirin or placebo plus low-dose aspirin and followed them for a median of 28 months (Table 1).
The primary end point (a composite of MI, stroke, or death) was 6.8% with clopidogrel plus aspirin and 7.3% with aspirin alone, indicating no significant benefit with clopidogrel plus aspirin compared with aspirin alone in reducing the rate of MI, stroke, or cardiovascular death in patients with high-risk but stable atherothrombotic disease. A marginal statistical benefit with dual antiplatelet therapy was noted in the subgroup of patients with previously documented coronary, cerebrovascular, or peripheral vascular disease—6.9% with aspirin plus clopidogrel vs 7.9% with aspirin alone (relative risk 0.88; 95% CI 0.77–0.998; P = .046).
Consequently, there is no compelling reason to start clopidogrel in this patient.
PRASUGREL, THE NEWEST THIENOPYRIDINE
Prasugrel was recently approved by the FDA as antiplatelet treatment for patients with acute coronary syndromes planning to undergo a percutaneous coronary intervention.24 It has been shown to inhibit adenosine-diphosphate-induced platelet activation in a more consistent and effective manner than clopidogrel.25,26
Although both clopidogrel and prasugrel are prodrugs, 80% of absorbed clopidogrel is metabolized by esterases into inactive metabolites, and the availability of active metabolite can vary, as it is significantly influenced by polymorphisms in the cytochrome P450 system. 27 In contrast, prasugrel is not degraded by esterases, and its conversion to active metabolite by the cytochrome P450 system is not influenced by common genetic polymorphisms, particularly CYP2C19*2.
TRITON-TIMI 38 (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel—Thrombolysis in Myocardial Infarction) provided most of the evidence for the approval of prasugrel for clinical use.28,29 In this trial, a 60-mg loading dose of prasugrel followed by a daily maintenance dose of 10 mg was significantly superior to the current clopidogrel regimen in preventing death from cardiovascular causes, nonfatal MI, or nonfatal stroke during a study period of 15 months.28 Also observed was a 24% lower rate of MI, a 34% lower rate of urgent target-vessel revascularization, and a 52% lower rate of stent thrombosis.
These benefits, however, came at the cost of a significantly higher risk of major bleeding, including the potential for three excess fatal bleeding events for every 1,000 patients treated. Patients at highest risk at the dosages evaluated included the elderly (age 75 and older), patients who weigh less than 60 kg, and patients with a history of stroke or transient ischemic attack. Based on these results, we recommend caution with the use of prasugrel in these patient subsets.
Clinical use of prasugrel is likely to be highest in patients presenting with ST-elevation MI who are undergoing a primary percutaneous coronary intervention. There is currently no evidence from any randomized clinical trial to support the safety of prasugrel given in the emergency room or “upstream” in the setting of non-ST-elevation acute coronary syndromes.
Of note, patients with non-ST-elevation acute coronary syndromes in the TRITON trial were randomized only after angiographic definition. As a result, only 179 patients exposed to prasugrel were referred for coronary artery bypass surgery, but the rate of surgery-related major bleeding in this group was 13.4% (vs 3.2% in the clopidogrel group). Based on these data, prasugrel should be withheld for at least 1 week prior to any surgery.
- Leon MB, Baim DS, Popma JJ, et al. A clinical trial comparing three antithrombotic-drug regimens after coronary-artery stenting. Stent Anticoagulation Restenosis Study Investigators. N Engl J Med 1998; 339:1665–1671.
- Yeh SP, Hsueh EJ, Wu H, Wang YC. Ticlopidine-associated aplastic anemia. A case report and review of literature, Ann Hematol 1998; 76:87–90.
- Page Y, Tardy B, Zeni F, Comtet C, Terrana R, Bertrand JC. Thrombotic thrombocytopenic purpura related to ticlopidine. Lancet 1991; 337:774–776.
- Canadian Cardiovascular Society; Antman EM, Hand M, Armstrong PW, et al. 2007 focused update of the ACC/AHA 2004 guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2008; 51:210–247.
- Anderson JL, Adams CD, Antman EM, et al; American College of Cardiology, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol 2007; 50:e1–e157.
- King SB, Smith SC, Hirshfeld JW, et al. 2007 focused update of the ACC/AHA/SCAI 2005 guideline update for percutaneous coronary intervention: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2008; 51:172–209.
- Kam PC, Nethery CM. The thienopyridine derivatives (platelet adenosine diphosphate receptor antagonists), pharmacology and clinical developments. Anaesthesia 2003; 58:28–35.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet 1996; 348:1329–1339.
- Sabatine MS, Cannon CP, Gibson M, et al; CLARITY-TIMI 28 Investigators. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation, N Engl J Med 2005; 352:1179–1189.
- Chen ZM, Jiang LX, Chen YP, et al; COMMIT (ClOpidogrel and Metoprolol in Myocardial Infarction Trial) Collaborative Group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: a randomized placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Bhatt DL, Bertrand ME, Berger PB, et al. Meta-analysis of randomized and registry comparisons of ticlopidine with clopidogrel after stenting. J Am Coll Cardiol 2002; 39:9–14.
- Bertrand ME, Rupprecht HJ, Urban P, Gershlick AH; CLASSICS Investigators. Double-blind study of the safety of clopidogrel with and without a loading dose in combination with aspirin compared with ticlopidine in combination with aspirin after coronary stenting: The Clopidogrel Aspirin Stent International Cooperative Study (CLASSICS). Circulation 2000; 102:624–629.
- Steinhubl SR, Berger PB, Mann JT, et al; CREDO Investigators. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 288:2411–2420.
- Mehta SR, Yusuf S, Peters RJ, et a; Clopidogrel in Unstable Angina to Prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCICURE study. Lancet 2001; 358:527–533.
- Lüscher TF, Steffel J, Eberli FR, et al. Drug-eluting stent and coronary thrombosis: biological mechanisms and clinical implications. Circulation 2007; 115:1051–1058.
- Kotani J, Awata M, Nanto S, et al. Incomplete neointimal coverage of sirolimus-eluting stents: angioscopic findings. J Am Coll Cardiol 2006; 47:2108–2111.
- Pfisterer M, Brunner-La Rocca HP, Buser PT, et al; BASKETLATE Investigators. Late clinical events after clopidogrel discontinuation may limit the benefit of drug-eluting stents: an observational study of drug-eluting versus bare-metal stents. J Am Coll Cardiol 2006; 48:2584–2591.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Stone GW, Moses JW, Ellis SG, et al. Safety and efficacy of sirolimus- and paclitaxel-eluting coronary stents. N Engl J Med 2007; 356:998–1008.
- Mauri L, Hsieh WH, Massaro JM, Ho KK, D’Agostino R, Cutlip DE. Stent thrombosis in randomized clinical trials of drug-eluting stents. N Engl J Med 2007; 356:1020–1029.
- Kastrati A, Mehilli J, Pache J, et al. Analysis of 14 trials comparing sirolimus-eluting stents with bare-metal stents. N Engl J Med 2007; 356:1030–1039.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- US Food and Drug Administration. FDA Approves Effient to Reduce the Risk of Heart Attack in Angioplasty Patients. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm171497.htm. Accessed October 2, 2009.
- Jernber T, Payne CD, Winters KJ, et al. Prasugrel achieves greater inhibition of platelet aggregation and a lower rate of non-responders compared with clopidogrel in aspirin-treated patients with stable coronary artery disease. Eur Heart J 2006; 27:1166–1173.
- Wiviott SD, Trenk D, Frelinger AL, et al; PRINCIPLE-TIMI 44 Investigators. Prasugrel compared with high loading and maintenance-dose clopidogrel in patients with planned percutaneous coronary intervention: the Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation-Thrombolysis in Myocardial Infarction 44 trial. Circulation 2007; 116:2923–2932.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med 2009; 360:354–362.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Antman EM, Wiviott SD, Murphy SA, et al. Early and late benefits of prasugrel in patients with acute coronary syndromes undergoing percutaneous coronary intervention: A TRITON-TIMI (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel-Thrombolysis In Myocardial Infarction) analysis. J Am Coll Card 2008; 51:2028–2033.
- Leon MB, Baim DS, Popma JJ, et al. A clinical trial comparing three antithrombotic-drug regimens after coronary-artery stenting. Stent Anticoagulation Restenosis Study Investigators. N Engl J Med 1998; 339:1665–1671.
- Yeh SP, Hsueh EJ, Wu H, Wang YC. Ticlopidine-associated aplastic anemia. A case report and review of literature, Ann Hematol 1998; 76:87–90.
- Page Y, Tardy B, Zeni F, Comtet C, Terrana R, Bertrand JC. Thrombotic thrombocytopenic purpura related to ticlopidine. Lancet 1991; 337:774–776.
- Canadian Cardiovascular Society; Antman EM, Hand M, Armstrong PW, et al. 2007 focused update of the ACC/AHA 2004 guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2008; 51:210–247.
- Anderson JL, Adams CD, Antman EM, et al; American College of Cardiology, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol 2007; 50:e1–e157.
- King SB, Smith SC, Hirshfeld JW, et al. 2007 focused update of the ACC/AHA/SCAI 2005 guideline update for percutaneous coronary intervention: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2008; 51:172–209.
- Kam PC, Nethery CM. The thienopyridine derivatives (platelet adenosine diphosphate receptor antagonists), pharmacology and clinical developments. Anaesthesia 2003; 58:28–35.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet 1996; 348:1329–1339.
- Sabatine MS, Cannon CP, Gibson M, et al; CLARITY-TIMI 28 Investigators. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation, N Engl J Med 2005; 352:1179–1189.
- Chen ZM, Jiang LX, Chen YP, et al; COMMIT (ClOpidogrel and Metoprolol in Myocardial Infarction Trial) Collaborative Group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: a randomized placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Bhatt DL, Bertrand ME, Berger PB, et al. Meta-analysis of randomized and registry comparisons of ticlopidine with clopidogrel after stenting. J Am Coll Cardiol 2002; 39:9–14.
- Bertrand ME, Rupprecht HJ, Urban P, Gershlick AH; CLASSICS Investigators. Double-blind study of the safety of clopidogrel with and without a loading dose in combination with aspirin compared with ticlopidine in combination with aspirin after coronary stenting: The Clopidogrel Aspirin Stent International Cooperative Study (CLASSICS). Circulation 2000; 102:624–629.
- Steinhubl SR, Berger PB, Mann JT, et al; CREDO Investigators. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 288:2411–2420.
- Mehta SR, Yusuf S, Peters RJ, et a; Clopidogrel in Unstable Angina to Prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCICURE study. Lancet 2001; 358:527–533.
- Lüscher TF, Steffel J, Eberli FR, et al. Drug-eluting stent and coronary thrombosis: biological mechanisms and clinical implications. Circulation 2007; 115:1051–1058.
- Kotani J, Awata M, Nanto S, et al. Incomplete neointimal coverage of sirolimus-eluting stents: angioscopic findings. J Am Coll Cardiol 2006; 47:2108–2111.
- Pfisterer M, Brunner-La Rocca HP, Buser PT, et al; BASKETLATE Investigators. Late clinical events after clopidogrel discontinuation may limit the benefit of drug-eluting stents: an observational study of drug-eluting versus bare-metal stents. J Am Coll Cardiol 2006; 48:2584–2591.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Stone GW, Moses JW, Ellis SG, et al. Safety and efficacy of sirolimus- and paclitaxel-eluting coronary stents. N Engl J Med 2007; 356:998–1008.
- Mauri L, Hsieh WH, Massaro JM, Ho KK, D’Agostino R, Cutlip DE. Stent thrombosis in randomized clinical trials of drug-eluting stents. N Engl J Med 2007; 356:1020–1029.
- Kastrati A, Mehilli J, Pache J, et al. Analysis of 14 trials comparing sirolimus-eluting stents with bare-metal stents. N Engl J Med 2007; 356:1030–1039.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- US Food and Drug Administration. FDA Approves Effient to Reduce the Risk of Heart Attack in Angioplasty Patients. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm171497.htm. Accessed October 2, 2009.
- Jernber T, Payne CD, Winters KJ, et al. Prasugrel achieves greater inhibition of platelet aggregation and a lower rate of non-responders compared with clopidogrel in aspirin-treated patients with stable coronary artery disease. Eur Heart J 2006; 27:1166–1173.
- Wiviott SD, Trenk D, Frelinger AL, et al; PRINCIPLE-TIMI 44 Investigators. Prasugrel compared with high loading and maintenance-dose clopidogrel in patients with planned percutaneous coronary intervention: the Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation-Thrombolysis in Myocardial Infarction 44 trial. Circulation 2007; 116:2923–2932.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med 2009; 360:354–362.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Antman EM, Wiviott SD, Murphy SA, et al. Early and late benefits of prasugrel in patients with acute coronary syndromes undergoing percutaneous coronary intervention: A TRITON-TIMI (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel-Thrombolysis In Myocardial Infarction) analysis. J Am Coll Card 2008; 51:2028–2033.
KEY POINTS
- Dual antiplatelet therapy is recommended after ST-elevation MI or non-ST-elevation acute coronary syndromes, with aspirin indefinitely and clopidogrel for up to 1 year.
- Dual antiplatelet therapy is recommended for at least 1 month after placement of a bare-metal stent and for at least 1 year (or possibly indefinitely) after placement of a drug-eluting stent.
- There is no compelling indication for clopidogrel in patients with chronic coronary artery disease.
- Compared with clopidogrel, prasugrel (Effient) is associated with lower rates of MI, urgent target-vessel revascularization, and in-stent thrombosis, but at the cost of a higher risk of major bleeding.
Beta-blockers for hypertension: Are they going out of style?
In recent years the role of beta-blockers as a primary tool to treat hypertension has come under question. These drugs have shown disappointing results when used as antihypertensive therapy in patients without heart disease, ie, when used as primary prevention. At the same time, beta-blockers clearly reduce the risk of future cardiovascular events in patients who already have heart disease, eg, who already have had a myocardial infarction or who have congestive heart failure.
Several meta-analyses and a few clinical trials have shown that beta-blockers may have no advantage over other antihypertensive drugs, and in fact may not reduce the risk of stroke as effectively as other classes of blood pressure medications.
Why should this be? Is it that the patients in the antihypertensive trials were mostly older, and that beta-blockers do not work as well in older patients as in younger ones? Or does it have to do with the fact that atenolol (Tenormin) was the drug most often used in the trials? Would newer, different beta-blockers be better?
Hypertension experts currently disagree on how to interpret the available data, and this has led to conflict and confusion among clinicians as to the role of beta-blockers in managing hypertension. Current evidence suggests that older beta-blockers may not be the preferred first-line antihypertensive drugs for hypertensive patients who have no compelling indications for them (eg, heart failure, myocardial infarction, diabetes, high risk of coronary heart disease). However, newer beta-blockers with vasodilatory properties should be considered in cases of uncontrolled or resistant hypertension, especially in younger patients.
Further, while controversy and debate continue over the benefits and adverse effects of one class of antihypertensive drugs vs another, it is indisputable that controlling arterial blood pressure to the recommended goal offers major protection against cardiovascular and renal events in patients with hypertension.1,2
MECHANISM OF ACTION OF BETA-BLOCKERS
Beta-blockers effectively reduce blood pressure in both systolic-diastolic hypertension and isolated systolic hypertension.3–5 Exactly how is not known, but it has been proposed that they may do so by:
Reducing the heart rate and cardiac output. When catecholamines activate beta-1 receptors in the heart, the heart rate and myocardial contractility increase. By blocking beta-1 receptors, beta-blockers reduce the heart rate and myocardial contractility, thus lowering cardiac output and arterial blood pressure.6
Inhibiting renin release. Activation of the renin-angiotensin system is another major pathway that can lead to elevated arterial blood pressure. Renin release is mediated through the sympathetic nervous system via beta-1 receptors on the juxtaglomerular cells of the kidney. Beta-blockers can therefore lower blood pressure by inhibiting renin release.7
Inhibiting central nervous sympathetic outflow, thereby inducing presynaptic blockade, which in turn reduces the release of catecholamines.
Reducing venous return and plasma volume.
Generating nitric oxide, thus reducing peripheral vascular resistance (some agents).8
Reducing vasomotor tone.
Reducing vascular tone.
Improving vascular compliance.
Resetting baroreceptor levels.
Attenuating the pressor response to catecholamines with exercise and stress.
HETEROGENEITY OF BETA-BLOCKERS
Selectivity
Beta-blockers are not all the same. They can be classified into three categories.
Nonselective beta-blockers block both beta-1 and beta-2 adrenergic receptors. It is generally accepted that beta-blockers exert their primary antihypertensive effect by blocking beta-1 adrenergic receptors.6 Of interest, nonselective beta-blockers inhibit beta-2 receptors on arteries and thus cause an unopposed alpha-adrenergic effect, leading to increased peripheral vascular resistance.9 Examples of this category:
- Nadolol (Corgard)
- Pindolol (Visken)
- Propranolol (Inderal)
- Timolol (Blocadren).
Selective beta-blockers specifically block beta-1 receptors alone, although they are known to be nonselective at higher doses. Examples:
- Atenolol (Tenormin)
- Betaxolol (Kerlone)
- Bisoprolol (Zebeta)
- Esmolol (Brevibloc)
- Metoprolol (Lopressor, Toprol).
Beta-blockers with peripheral vasodilatatory effects act either via antagonism of the alpha-1 receptor, as with labetolol (Normodyne) and carvedilol (Coreg),10 or via enhanced release of nitric oxide, as with nebivolol (Bystolic).8
Lipid and water solubility
The lipid solubility and water solubility of each beta-blocker determine its bioavailability and side-effect profile.
Lipid solubility determines the degree to which a beta-blocker penetrates the blood-brain barrier and thereby leads to central nervous system side effects such as lethargy, nightmares, confusion, and depression. Propranolol is highly lipid-soluble; metoprolol and labetalol are moderately so.
Water-soluble beta-blockers such as atenolol have less tissue permeation, have a longer half-life, and cause fewer central nervous system effects and symptoms.11
Routes of elimination
Beta-blockers also differ in their route of elimination.
Atenolol and nadolol are eliminated by the kidney and require dose adjustment in patients with impaired renal function.12,13
On the other hand, propranolol, metoprolol, labetalol, carvedilol, and nebivolol are excreted primarily via hepatic metabolism.13
BETA-BLOCKERS IN THE MANAGEMENT OF HYPERTENSION
Beta-blockers were initially used to treat arrhythmias, but by the early 1970s they were also widely accepted for managing hypertension. 14 Their initial acceptance as one of the first-line classes of drugs for hypertension was based on their better side-effect profile compared with other antihypertensive drugs available at that time.
In the 1980s and 1990s, beta-blockers were listed as preferred first-line antihypertensive drugs along with diuretics in national hypertension guidelines.15 Subsequent updates of the guidelines favored diuretics as initial therapy and relegated all other classes of antihypertensive medications to be alternatives to diuretics.16 Although beta-blockers remain alternative first-line drugs in the latest guidelines (published in 2003; see reference 66), they are the preferred antihypertensive agents for patients with cardiac disease.
The current recommendations reflect the findings from hypertension trials in which patients with myocardial infarction and congestive heart failure had better cardiovascular outcomes if they received these drugs,17–19 including a lower risk of death.20,21 It was widely assumed that beta-blockers would also prevent first episodes of cardiovascular events.
However, to date, there is no evidence that beta-blockers are effective as primary prevention. Several large randomized controlled trials showed no benefit with beta-blockers compared with other antihypertensive drugs—in fact, there were more cardiovascular events with beta-blockers (see below).
Beta-blockers are well tolerated in clinical practice, although they can have side effects that include fatigue, depression, impaired exercise tolerance, sexual dysfunction, and asthma attacks.
Wiysonge et al22 analyzed how many patients withdrew from randomized trials of antihypertensive treatment because of drug-related adverse events. There was no significant difference in the incidence of fatigue, depressive symptoms, or sexual dysfunction with beta-blockers compared with placebo, and trial participants on a beta-blocker were not statistically significantly more likely to discontinue treatment than those receiving a placebo in three trials with 22,729 participants (relative risk [RR] 2.34, 95% confidence interval [CI] 0.84–6.52).
THE CONTROVERSY: WHAT THE TRIALS SHOWED
Messerli et al23 performed a meta-analysis published in 1998 that suggested that beta-blockers may not be as effective as diuretics in preventing cardiovascular events when used as first-line antihypertensive therapy in elderly patients. In 10 randomized controlled trials in 16,164 patients who were treated with either a diuretic or a beta-blocker (atenolol), blood pressure was normalized in two-thirds of diuretic-treated patients but only one-third of patients treated with atenolol as monotherapy. Diuretic therapy was superior with regard to all end points, and beta-blockers were found to be ineffective except in reducing cerebrovascular events.
The LIFE study (Losartan Intervention for Endpoint Reduction in Hypertension)24 compared the angiotensin-receptor blocker losartan (Cozaar) and atenolol in 9,193 patients with hypertension and left ventricular hypertrophy. At 4 years of follow-up, the rate of primary cardiovascular events (death, myocardial infarction, or stroke) was lower in the losartan group than in the atenolol group. The difference was mainly due to a 25% lower incidence of stroke, which was statistically significant. The rates of myocardial infarction and death from cardiovascular causes were not significantly different between the two treatment groups. The systolic blood pressure was 1 mm Hg lower in the losartan group than in the atenolol group, which was statistically significant.
Carlberg et al25 performed another important meta-analysis that questioned whether atenolol reduces rates of cardiovascular morbidity and death in hypertensive patients. The results were surprising: eight randomized controlled trials including more than 6,000 patients and comparing atenolol with placebo or no treatment showed no differences between the treatment groups with regard to the outcomes of all-cause mortality (RR 1.01, 95% CI 0.89–1.15), cardiovascular mortality (RR 0.99, 95% CI 0.83–1.18), or myocardial infarction (RR 0.99, 95% CI 0.83–1.19).
In addition, when atenolol was compared with other antihypertensives in five other randomized controlled trials that included more than 14,000 patients, those treated with atenolol had a higher risk of stroke (RR 1.30, 95% CI 1.12–1.50) and death (RR 1.13, 95% CI 1.02–1.25).
The ASCOT-BPLA trial (Anglo-Scandinavian Cardiac Outcomes Trial—Blood Pressure Lowering Arm)26 had similar results. This trial compared the combination of atenolol plus the diuretic bendroflumethiazide against the combination of the calcium channel blocker amlodipine (Norvasc) plus the angiotensin-converting enzyme (ACE) inhibitor perindopril (Aceon). Although no significant difference was seen in the primary outcome of nonfatal myocardial infarction or fatal coronary heart disease (unadjusted hazard ratio [HR] with amlodipine-perindopril 0.90, 95% CI 0.79–1.02, P = .1052), the amlodipine-plus-perindopril group had significantly fewer strokes (327 vs 422, HR 0.77, 95% CI 0.66–0.89, P = .0003), fewer total cardiovascular events (1,362 vs 1,602, HR 0.84, 95% CI 0.78–0.90, P = .0001), and fewer deaths from any cause (738 vs 820; HR 0.89, 95% CI 0.81–0.99, P = .025).
Lindholm et al27 performed a meta-analysis that included studies of selective beta-blockers (including atenolol) and nonselective beta-blockers, with a follow-up time of more than 2 years. Compared with placebo or no treatment, beta-blockers reduced the risk of stroke by 19% but had no effect on myocardial infarction or all-cause mortality. Compared with other antihypertensive drugs, beta-blockers were less than optimum, and the relative risk of stroke was 16% higher. Atenolol was the beta-blocker used in most of the randomized clinical trials included in this meta-analysis.
The Cochrane group22 found beta-blockers to be inferior to all other antihypertensive drugs with respect to the ability to lower the risk of stroke.
WHY WERE THE RESULTS SO DISAPPOINTING?
Problems with atenolol
Most of the trials in the meta-analyses discussed above used atenolol and other beta-blockers that had no vasodilatory properties.
Further, in most of the trials atenolol was used in a once-daily dosage, whereas ideally it needs to be taken more frequently, based on its pharmacokinetic and pharmacodynamic properties (a half-life of 6–9 hours).3 Neutel et al28 confirmed that atenolol, when taken once daily, leaves the patient unprotected in the last 6 hours of a 24-hour period, as demonstrated by 24-hour ambulatory blood pressure monitoring. It is possible that this short duration of action of atenolol may have contributed to the results observed in clinical trials that used atenolol to treat hypertension.
Differences between older and younger patients
Another possible reason for the disappointing results is that the trials included many elderly patients, in whom beta-blockers may not be as effective. The pathophysiology of hypertension in younger people is different from that in older patients.29 Hemodynamic characteristics of younger hypertensive patients include a high cardiac output and hyperdynamic circulation with a low pulse pressure, while older patients have lower arterial compliance with an elevated vascular resistance.
The notion of choosing antihypertensive medications on the basis of age and age-related pathophysiology is supported by several clinical studies. Randomized controlled trials appear to show that beta-blockers are effective in younger hypertensive patients.30
Conversely, the CAFE (Conduit Artery Function Evaluation) trial,31 a substudy of the main ASCOT trial,26 indicated that betablocker-based therapy was less effective in reducing central aortic pressure than were regimens based on an ACE inhibitor or a calcium channel blocker.
The CAFE researchers recruited 2,073 patients from five ASCOT centers and used radial artery applanation tonometry and pulse-wave analysis to derive central aortic pressures and hemodynamic indices during study visits up to a period of 4 years. Although the two treatment groups achieved similar brachial systolic blood pressures, the central aortic systolic pressure was 4.3 mm Hg lower in the amlodipine group (95% CI 3.3–5.4; P < .0001), and the central aortic pulse pressure was 3.0 mm Hg lower (95% CI 2.1–3.9; P < .0001).
Pulse-wave dyssynchrony
Bangalore et al47 offer an interesting hypothesis to explain the probable adverse effect of beta-blockers. Their theory concerns the effect of these drugs on the arterial pulse wave.
Normally, with each contraction of the left ventricle during systole, an arterial pulse wave is generated and propagated forward to the peripheral arteries. This wave is then reflected back to the heart from the branching points of peripheral arteries. The final form of the pressure wave at the aortic root is a synchronized summation of the forward-traveling wave and the backward-reflected wave.
In healthy people with normal arteries, the reflected wave merges with the forward-traveling wave in diastole and augments coronary blood flow. In patients whose arteries are stiff due to aging or vascular comorbidities, the reflected wave returns faster and merges with the incident wave in systole, resulting in higher left ventricular afterload and less coronary perfusion.48
Bangalore et al47 propose that artificially reducing the heart rate with beta-blockers may further dyssynchronize the pulse wave, adversely affecting coronary perfusion and leading to an increased risk of cardiovascular events and death.
Metabolic side effects
Older beta-blockers, and especially atenolol, have well-known metabolic adverse effects, particularly impairment of glycemic control. This adverse effect appears to occur only with beta-blockers that do not possess vasodilatory properties and thus increase peripheral vascular resistance, which results in lower glucose availability and reduced uptake by skeletal muscles.49
Bangalore et al50 evaluated the effect of beta-blockers in a meta-analysis of 12 studies in 94,492 patients followed up for more than 1 year. Beta-blocker therapy resulted in a 22% higher risk of new-onset diabetes mellitus (RR 1.22, 95% CI 1.12–1.33) than with other nondiuretic antihypertensive agents.
Of note, however, the meta-analysis did not show a significantly higher risk of the onset of diabetes with propranolol or metoprolol than with other nondiuretic antihypertensives when studies of these beta-blockers were separated from atenolol-based studies.
Further, the United Kingdom Prospective Diabetes Study40 found that cardiovascular outcomes in patients with good blood pressure control were similar when atenolol-based therapy was compared with therapy with the ACE inhibitor captopril (Capoten).
A meta-analysis conducted by Balamuthusamy et al51 in 2009 found no higher risk of stroke in patients with hypertension and diabetes mellitus who received beta-blockers than in those who received other antihypertensive medications. However, beta-blockers were associated with a higher risk of death from cardiovascular causes (RR 1.39, 95% CI 1.07–1.804; P < .01) compared with reninangiotensin blockade.
NEWER BETA-BLOCKERS MAY BE BETTER
In the United States, more than 40 million prescriptions for atenolol are written every year, making it by far the most commonly used beta-blocker for the treatment of hypertension. 52 It is clear, however, that atenolol is not an ideal representative of this class of antihypertensive medications.
Preliminary data from studies of newer beta-blockers that possess beneficial vasodilatory properties are encouraging. Animal studies and preliminary human studies find that these new-generation beta-blockers cause fewer adverse metabolic effects and improve endothelial function, measures of arterial stiffness, and cardiovascular outcomes.
Carvedilol
Carvedilol is a nonselective beta-blocker with vasodilatory effects that are thought to be due to its ability to concurrently block alpha-1 receptors in addition to beta receptors. 53 In experiments in vitro and in trials in patients with diabetes and hypertension, carvedilol increased endothelial vasodilation and reduced inflammation and platelet aggregation. These effects may be achieved though antioxidant actions, thereby preserving nitric oxide bioactivity.54,55
In the Glycemic Effects in Diabetes Mellitus: Carvedilol-Metoprolol Comparison in Hypertensives (GEMINI) trial,56 carvedilol was associated with better maintenance of glycemic control in diabetic hypertensive patients than was metoprolol. Insulin sensitivity improved with carvedilol but not with metoprolol, and fewer patients on carvedilol progressed to microalbuminuria.
Nebivolol
Nebivolol is a novel selective beta-blocker with a much higher affinity for beta-1 adrenergic receptors than for beta-2 adrenergic receptors. Among all the beta-blockers in clinical use today, nebivolol has the highest selectivity for beta-1 receptors.8
Nebivolol causes vasodilation through activation of the l-arginine/nitric oxide pathway.57–59 Blockade of synthesis of nitric oxide leads to local arterial stiffness. Endothelial dysfunction is characterized by decreased bioavailability of nitric oxide and has been shown to be a strong predictor of cardiovascular outcomes. By generating nitric oxide, nebivolol reduces peripheral vascular resistance, overcoming a significant side effect of earlier beta-blockers that lowered blood pressure but ultimately increased peripheral vascular tone and resistance.8
In an experiment in a bovine model,60 nebivolol significantly reduced the pulse-wave velocity (a measure of arterial stiffness), while atenolol had no effect. Moreover, evidence for the role of the l-arginine/nitric oxide pathway in the vasodilatory effect of nebivolol was demonstrated by co-infusion of NG-monomethyl-L-arginine, a specific endothelial nitric oxide synthetase inhibitor that attenuated the reduction of pulse-wave velocity by nebivolol.
In studies in hypertensive patients, nebivolol was associated with a better metabolic profile than atenolol, with none of the adverse effects on insulin sensitivity that atenolol had.61 In the Study of Effects of Nebivolol Interventions on Outcomes and Rehospitalization in Seniors With Heart Failure (SENIORS) trial, significantly fewer patients receiving nebivolol died or were admitted to the hospital for cardiovascular reasons compared with those receiving placebo.62
Although these findings are encouraging, we do not yet know if these effects will translate into a significant reduction in cardiovascular outcomes in clinical trials. Large, prospective hypertension outcome trials, particularly to evaluate primary prevention of cardiovascular outcomes, are needed for an evidence-based approach to using the newer beta-blockers as preferred first-line therapy for hypertension.
WHAT RECENT GUIDELINES SAY ABOUT BETA-BLOCKERS
The British National Institute for Health and Clinical Excellence and the British Hypertension Society, in their 2004 guidelines, recommended beta-blockers as one of several first-line antihypertensive medications in young, nonblack patients.63 On the other hand, they advised clinicians to be aware of the reported increase in onset of diabetes mellitus in patients treated with these medications. After the LIFE24 and ASCOT26 study results were published, these guidelines were amended to exclude beta-blockers as preferred routine initial therapy for hypertension.64
More recently, the 2007 European Society of Hypertension and European Society of Cardiology reconsidered the role of beta-blockers, recommending them as an option in both initial and subsequent antihypertensive treatment strategies.65
The current guidelines from the National Heart, Lung, and Blood Institute,66 which were published in 2003, were highly influenced by the results of the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT),2 and favor diuretics as the first-line therapy. However, they indicate that beta-blockers are a suitable alternative, particularly when a compelling cardiac indication is present.53 We hope that the next update, expected late in 2009, will re-address this issue in the light of more recent data.
- Staessen JA, Wang JG, Thijs L. Cardiovascular protection and blood pressure reduction: a meta-analysis. Lancet 2001; 358:1305–1315.
- ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002; 288:2981–2997.
- Neutel JM, Smith DH, Ram CV, et al. Application of ambulatory blood pressure monitoring in differentiating between antihypertensive agents. Am J Med 1993; 94:181–187.
- Materson BJ, Reda DJ, Cushman WC, et al. Single-drug therapy for hypertension in men. A comparison of six antihypertensive agents with placebo. The Department of Veterans Affairs Cooperative Study Group on Antihypertensive Agents. N Engl J Med 1993; 328:914–921.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- Frishman W, Silverman R. Clinical pharmacology of the new beta-adrenergic blocking drugs. Part 2. Physiologic and metabolic effects. Am Heart J 1979; 97:797–807.
- Garrett BN, Kaplan NM. Plasma renin activity suppression: duration after withdrawal from beta-adrenergic blockade. Arch Intern Med 1980; 140:1316–1318.
- Pedersen ME, Cockcroft JR. The latest generation of beta-blockers: new pharmacologic properties. Curr Hypertens Rep 2006; 8:279–286.
- Man in’t Veld AJ, Van den Meiracker AH, Schalekamp MA. Do beta-blockers really increase peripheral vascular resistance? Review of the literature and new observations under basal conditions. Am J Hypertens 1988; 1:91–96.
- Pearce CJ, Wallin JD. Labetalol and other agents that block both alpha- and beta-adrenergic receptors. Cleve Clin J Med 1994; 61:59–69.
- Dimsdale JE, Newton RP, Joist T. Neuropsychological side effects of beta-blockers. Arch Intern Med 1989; 149:514–525.
- Agarwal R. Supervised atenolol therapy in the management of hemodialysis hypertension. Kidney Int 1999; 55:1528–1535.
- Sica DA, Black HR. Pharmacologic considerations in the positioning of beta-blockers in antihypertensive therapy. Curr Hypertens Rep 2008; 10:330–335.
- Prichard BN, Gillam GP. Use of propranolol (Inderal) in treatment of hypertension. Br Med J 1964; 19; 2:725–727.
- The fifth report of the Joint National Committee on Detection, Evaluation, and Treatment of High Blood Pressure (JNC V). Arch Intern Med 1993; 153:154–183.
- Moser M. Evolution of the treatment of hypertension from the 1940s to JNC V. Am J Hypertens 1997; 10:2S–8S.
- Houghton T, Freemantle N, Cleland JG. Are beta-blockers effective in patients who develop heart failure soon after myocardial infarction? A meta-regression analysis of randomised trials. Eur J Heart Fail 2000; 2:333–340.
- The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet 1999; 353:9–13.
- Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 1999; 353:2001–2007.
- Yusuf S, Peto R, Lewis J, Collins R, Sleight P. Beta blockade during and after myocardial infarction: an overview of the randomized trials. Prog Cardiovasc Dis 1985; 27:335–371.
- Brophy JM, Joseph L, Rouleau JL. Beta-blockers in congestive heart failure. A Bayesian meta-analysis. Ann Intern Med 2001; 134:550–560.
- Wiysonge CS, Bradley H, Mayosi BM, et al. Beta-blockers for hypertension. Cochrane Database Syst Rev 2007;CD002003.
- Messerli FH, Grossman E, Goldbourt U. Are beta-blockers efficacious as first-line therapy for hypertension in the elderly? A systematic review. JAMA 1998; 279:1903–1907.
- Dahlöf B, Devereux RB, Kjeldsen SE, et al; for the LIFE study group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint Reduction in Hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002; 359:995–1003.
- Carlberg B, Samuelsson O, Lindholm LH. Atenolol in hypertension: is it a wise choice? Lancet 2004; 364:1684–1689.
- Dahlöf B, Sever PS, Poulter NR, et al; ASCOT Investigators. Prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA): a multicentre randomised controlled trial. Lancet 2005; 366:895–906.
- Lindholm LH, Carlberg B, Samuelsson O. Should beta-blockers remain first choice in the treatment of primary hypertension? A meta-analysis. Lancet 2005; 366:1545–1553.
- Neutel JM, Schnaper H, Cheung DG, Graettinger WF, Weber MA. Antihypertensive effects of beta-blockers administered once daily: 24-hour measurements. Am Heart J 1990; 120:166–171.
- Franklin SS, Gustin W, Wong ND, et al. Hemodynamic patterns of age-related changes in blood pressure. The Framingham Heart Study. Circulation 1997; 96:308–315.
- The IPPPSH Collaborative Group. Cardiovascular risk and risk factors in a randomized trial of treatment based on the beta-blocker oxprenolol: the International Prospective Primary Prevention Study in Hypertension (IPPPSH). J Hypertens 1985; 3:379–392.
- Williams B, Lacy PS, Thom SM, et al; CAFE Investigators. Differential impact of blood pressure-lowering drugs on central aortic pressure and clinical outcomes: principal results of the Conduit Artery Function Evaluation (CAFE) study. Circulation 2006; 113:1213–1225.
- Khan N, McAlister FA. Re-examining the efficacy of beta-blockers for the treatment of hypertension: a meta-analysis. CMAJ 2006; 174:1737–1742.
- Medical Research Council Working Party. MRC trial of treatment of mild hypertension: principal results. BMJ 1985; 291:97–104.
- Coope J, Warrender TS. Randomised trial of treatment of hypertension in elderly patients in primary care. BMJ 1986; 293:1145–1151.
- Dahlöf B, Lindholm LH, Hansson L, et al. Morbidity and mortality in the Swedish Trial in Old Patients with Hypertension (STOP-Hypertension). Lancet 1991; 338:1281–1285.
- MRC Working Party. Medical Research Council trial of treatment of hypertension in older adults: principal results. BMJ 1992; 304:405–412.
- The Dutch TIA Study Group. Trial of secondary prevention with atenolol after transient ischemic attack or nondisabling ischemic stroke. Stroke 1993; 24:543–548.
- Eriksson S, Olofsson B-O, Wester P-O; for the TEST Study Group. Atenolol in secondary prevention after stroke. Cerebrovasc Dis 1995; 5:21–25.
- Wilhelmsen L, Berglund G, Elmfeldt D, et al. Beta-blockers versus diuretics in hypertensive men: main results from the HAPPHY trial. J Hypertens 1987; 5:561–572.
- UK Prospective Diabetes Study Group. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. BMJ 1998; 317:713–720.
- Hansson L, Lindholm LH, Niskanen L, et al. Effect of angiotensin-converting enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPP) randomised trial. Lancet 1999; 353:611–616.
- Lanchetti A, Bond MG, Henning M, et al. Calcium antagonist lacidipine slow down progression of asymptomatic carotid atherosclerosis. Principal results of the European Lacidipine Study on Atherosclerosis (ELSA), a randomized, double-blind, long-term trial. Circulation 2002; 106:2422–2427.
- Hansson L, Lindholm LH, Ekbom T, et al. Randomised trial of old and new antihypertensive drugs in elderly patients: cardiovascular mortality and morbidity in the Swedish Trial in Old Patients with Hypertension-2 study. Lancet 1999; 354:1751–1756.
- Hansson L, Hedner T, Lund-Johansen P, et al. Randomised trial of effects of calcium antagonists compared with diuretics and ß blockers on cardiovascular morbidity and mortality in hypertension: the Nordic Diltiazem (NORDIL) study. Lancet 2000; 356:359–365.
- Pepine CJ, Handsberg EM, Cooper-DeHoff RM, et al. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003; 290:2805–2816.
- Black HR, Elliott WJ, Grandits G, et al. Principal results of the Controlled Onset Verapamil Investigation of Cardiovascular End Points (CONVINCE) Trial. JAMA 2003; 289:2073–2082.
- Bangalore S, Sawhney S, Messerli FH. Relation of beta-blocker-induced heart rate lowering and cardioprotection in hypertension. J Am Coll Cardiol 2008; 52:1482–1489.
- Boutouyrie P, Vermersch S, Laurent S, Briet M. Cardiovascular risk assessment through target organ damage: role of carotid to femoral pulse wave velocity. Clin Exp Pharmacol Physiol 2008; 35:530–533.
- Kveiborg B, Christiansen B, Major-Petersen A, Torp-Pedersen C. Metabolic effects of beta-adrenoceptor antagonists with special emphasis on carvedilol. Am J Cardiovasc Drugs 2006; 6:209–217.
- Bangalore S, Parkar S, Grossman E, Messerli FH. A meta-analysis of 94,492 patients with hypertension treated with beta-blockers to determine the risk of new-onset diabetes mellitus. Am J Cardiol 2007; 100:1254–1262.
- Balamuthusamy S, Molnar J, Adigopula S, Arora R. Comparative analysis of beta-blockers with other antihypertensive agents on cardiovascular outcomes in hypertensive patients with diabetes mellitus: a systematic review and meta-analysis. Am J Ther 2009; 16:133–142.
- Berenson A. Big drug makers see sales decline with their image. New York Times 2005 Nov 14.
- Bristow MR. Beta-adrenergic receptor blockade in chronic heart failure. Circulation 2000; 101:558–569.
- Giugliano D, Marfella R, Acampora R, Giunta R, Coppola L, D’Onofrio F. Effects of perindopril and carvedilol on endothelium-dependent vascular functions in patients with diabetes and hypertension. Diabetes Care 1998; 21:631–636.
- Lopez BL, Christopher TA, Yue TL, Ruffolo R, Feuerstein GZ, Ma XL. Carvedilol, a new beta-adrenoreceptor blocker antihypertensive drug, protects against free-radical-induced endothelial dysfunction. Pharmacology 1995; 51:165–173.
- Bakris GL, Fonseca V, Katholi RE, et al; GEMINI Investigators. Metabolic effects of carvedilol vs metoprolol in patients with type 2 diabetes mellitus and hypertension: a randomized controlled trial. JAMA 2004; 292:2227–2236.
- Georgescu A, Pluteanu F, Flonta ML, Badila E, Dorobantu M, Popov D. The cellular mechanisms involved in the vasodilator effect of nebivolol on the renal artery. Eur J Pharmacol 2005; 508:159–166.
- Kalinowski L, Dobrucki LW, Szczepanska-Konkel M, et al. Third-generation beta-blockers stimulate nitric oxide release from endothelial cells through ATP efflux: a novel mechanism for antihypertensive action. Circulation 2003; 107:2747–2752.
- Cockcroft JR, Chowienczyk PJ, Brett SE, et al. Nebivolol vasodilates human forearm vasculature: evidence for an L-arginine/NO-dependent mechanism. J Pharmacol Exp Ther 1995; 274:1067–1071.
- McEniery CM, Schmitt M, Qasem A, et al. Nebivolol increases arterial distensibility in vivo. Hypertension 2004; 44:305–310.
- Poirier L, Cleroux J, Nadeau A, Lacourciere Y. Effects of nebivolol and atenolol on insulin sensitivity and haemodynamics in hypertensive patients. J Hypertens 2001; 19:1429–1435.
- Flather MD, Shibata MC, Coats AJ, et al; SENIORS Investigators. Randomized trial to determine the effect of nebivolol on mortality and cardiovascular hospital admission in elderly patients with heart failure (SENIORS). Eur Heart J 2005; 26:215–225.
- Williams B, Poulter NR, Brown MJ, et al; BHS guidelines working party, for the British Hypertension Society. British Hypertension Society guidelines for hypertension management 2004 (BHS-IV): summary. BMJ 2004; 328:634–640.
- Sever P. New hypertension guidelines from the National Institute for Health and Clinical Excellence and the British Hypertension Society. J Renin Angiotensin Aldosterone Syst 2006; 7:61–63.
- Mancia G, De Backer G, Dominiczak A, et al; Management of Arterial Hypertension of the European Society of Hypertension. 2007 Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens 2007; 25:1105–1187.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289:2560–2572.
In recent years the role of beta-blockers as a primary tool to treat hypertension has come under question. These drugs have shown disappointing results when used as antihypertensive therapy in patients without heart disease, ie, when used as primary prevention. At the same time, beta-blockers clearly reduce the risk of future cardiovascular events in patients who already have heart disease, eg, who already have had a myocardial infarction or who have congestive heart failure.
Several meta-analyses and a few clinical trials have shown that beta-blockers may have no advantage over other antihypertensive drugs, and in fact may not reduce the risk of stroke as effectively as other classes of blood pressure medications.
Why should this be? Is it that the patients in the antihypertensive trials were mostly older, and that beta-blockers do not work as well in older patients as in younger ones? Or does it have to do with the fact that atenolol (Tenormin) was the drug most often used in the trials? Would newer, different beta-blockers be better?
Hypertension experts currently disagree on how to interpret the available data, and this has led to conflict and confusion among clinicians as to the role of beta-blockers in managing hypertension. Current evidence suggests that older beta-blockers may not be the preferred first-line antihypertensive drugs for hypertensive patients who have no compelling indications for them (eg, heart failure, myocardial infarction, diabetes, high risk of coronary heart disease). However, newer beta-blockers with vasodilatory properties should be considered in cases of uncontrolled or resistant hypertension, especially in younger patients.
Further, while controversy and debate continue over the benefits and adverse effects of one class of antihypertensive drugs vs another, it is indisputable that controlling arterial blood pressure to the recommended goal offers major protection against cardiovascular and renal events in patients with hypertension.1,2
MECHANISM OF ACTION OF BETA-BLOCKERS
Beta-blockers effectively reduce blood pressure in both systolic-diastolic hypertension and isolated systolic hypertension.3–5 Exactly how is not known, but it has been proposed that they may do so by:
Reducing the heart rate and cardiac output. When catecholamines activate beta-1 receptors in the heart, the heart rate and myocardial contractility increase. By blocking beta-1 receptors, beta-blockers reduce the heart rate and myocardial contractility, thus lowering cardiac output and arterial blood pressure.6
Inhibiting renin release. Activation of the renin-angiotensin system is another major pathway that can lead to elevated arterial blood pressure. Renin release is mediated through the sympathetic nervous system via beta-1 receptors on the juxtaglomerular cells of the kidney. Beta-blockers can therefore lower blood pressure by inhibiting renin release.7
Inhibiting central nervous sympathetic outflow, thereby inducing presynaptic blockade, which in turn reduces the release of catecholamines.
Reducing venous return and plasma volume.
Generating nitric oxide, thus reducing peripheral vascular resistance (some agents).8
Reducing vasomotor tone.
Reducing vascular tone.
Improving vascular compliance.
Resetting baroreceptor levels.
Attenuating the pressor response to catecholamines with exercise and stress.
HETEROGENEITY OF BETA-BLOCKERS
Selectivity
Beta-blockers are not all the same. They can be classified into three categories.
Nonselective beta-blockers block both beta-1 and beta-2 adrenergic receptors. It is generally accepted that beta-blockers exert their primary antihypertensive effect by blocking beta-1 adrenergic receptors.6 Of interest, nonselective beta-blockers inhibit beta-2 receptors on arteries and thus cause an unopposed alpha-adrenergic effect, leading to increased peripheral vascular resistance.9 Examples of this category:
- Nadolol (Corgard)
- Pindolol (Visken)
- Propranolol (Inderal)
- Timolol (Blocadren).
Selective beta-blockers specifically block beta-1 receptors alone, although they are known to be nonselective at higher doses. Examples:
- Atenolol (Tenormin)
- Betaxolol (Kerlone)
- Bisoprolol (Zebeta)
- Esmolol (Brevibloc)
- Metoprolol (Lopressor, Toprol).
Beta-blockers with peripheral vasodilatatory effects act either via antagonism of the alpha-1 receptor, as with labetolol (Normodyne) and carvedilol (Coreg),10 or via enhanced release of nitric oxide, as with nebivolol (Bystolic).8
Lipid and water solubility
The lipid solubility and water solubility of each beta-blocker determine its bioavailability and side-effect profile.
Lipid solubility determines the degree to which a beta-blocker penetrates the blood-brain barrier and thereby leads to central nervous system side effects such as lethargy, nightmares, confusion, and depression. Propranolol is highly lipid-soluble; metoprolol and labetalol are moderately so.
Water-soluble beta-blockers such as atenolol have less tissue permeation, have a longer half-life, and cause fewer central nervous system effects and symptoms.11
Routes of elimination
Beta-blockers also differ in their route of elimination.
Atenolol and nadolol are eliminated by the kidney and require dose adjustment in patients with impaired renal function.12,13
On the other hand, propranolol, metoprolol, labetalol, carvedilol, and nebivolol are excreted primarily via hepatic metabolism.13
BETA-BLOCKERS IN THE MANAGEMENT OF HYPERTENSION
Beta-blockers were initially used to treat arrhythmias, but by the early 1970s they were also widely accepted for managing hypertension. 14 Their initial acceptance as one of the first-line classes of drugs for hypertension was based on their better side-effect profile compared with other antihypertensive drugs available at that time.
In the 1980s and 1990s, beta-blockers were listed as preferred first-line antihypertensive drugs along with diuretics in national hypertension guidelines.15 Subsequent updates of the guidelines favored diuretics as initial therapy and relegated all other classes of antihypertensive medications to be alternatives to diuretics.16 Although beta-blockers remain alternative first-line drugs in the latest guidelines (published in 2003; see reference 66), they are the preferred antihypertensive agents for patients with cardiac disease.
The current recommendations reflect the findings from hypertension trials in which patients with myocardial infarction and congestive heart failure had better cardiovascular outcomes if they received these drugs,17–19 including a lower risk of death.20,21 It was widely assumed that beta-blockers would also prevent first episodes of cardiovascular events.
However, to date, there is no evidence that beta-blockers are effective as primary prevention. Several large randomized controlled trials showed no benefit with beta-blockers compared with other antihypertensive drugs—in fact, there were more cardiovascular events with beta-blockers (see below).
Beta-blockers are well tolerated in clinical practice, although they can have side effects that include fatigue, depression, impaired exercise tolerance, sexual dysfunction, and asthma attacks.
Wiysonge et al22 analyzed how many patients withdrew from randomized trials of antihypertensive treatment because of drug-related adverse events. There was no significant difference in the incidence of fatigue, depressive symptoms, or sexual dysfunction with beta-blockers compared with placebo, and trial participants on a beta-blocker were not statistically significantly more likely to discontinue treatment than those receiving a placebo in three trials with 22,729 participants (relative risk [RR] 2.34, 95% confidence interval [CI] 0.84–6.52).
THE CONTROVERSY: WHAT THE TRIALS SHOWED
Messerli et al23 performed a meta-analysis published in 1998 that suggested that beta-blockers may not be as effective as diuretics in preventing cardiovascular events when used as first-line antihypertensive therapy in elderly patients. In 10 randomized controlled trials in 16,164 patients who were treated with either a diuretic or a beta-blocker (atenolol), blood pressure was normalized in two-thirds of diuretic-treated patients but only one-third of patients treated with atenolol as monotherapy. Diuretic therapy was superior with regard to all end points, and beta-blockers were found to be ineffective except in reducing cerebrovascular events.
The LIFE study (Losartan Intervention for Endpoint Reduction in Hypertension)24 compared the angiotensin-receptor blocker losartan (Cozaar) and atenolol in 9,193 patients with hypertension and left ventricular hypertrophy. At 4 years of follow-up, the rate of primary cardiovascular events (death, myocardial infarction, or stroke) was lower in the losartan group than in the atenolol group. The difference was mainly due to a 25% lower incidence of stroke, which was statistically significant. The rates of myocardial infarction and death from cardiovascular causes were not significantly different between the two treatment groups. The systolic blood pressure was 1 mm Hg lower in the losartan group than in the atenolol group, which was statistically significant.
Carlberg et al25 performed another important meta-analysis that questioned whether atenolol reduces rates of cardiovascular morbidity and death in hypertensive patients. The results were surprising: eight randomized controlled trials including more than 6,000 patients and comparing atenolol with placebo or no treatment showed no differences between the treatment groups with regard to the outcomes of all-cause mortality (RR 1.01, 95% CI 0.89–1.15), cardiovascular mortality (RR 0.99, 95% CI 0.83–1.18), or myocardial infarction (RR 0.99, 95% CI 0.83–1.19).
In addition, when atenolol was compared with other antihypertensives in five other randomized controlled trials that included more than 14,000 patients, those treated with atenolol had a higher risk of stroke (RR 1.30, 95% CI 1.12–1.50) and death (RR 1.13, 95% CI 1.02–1.25).
The ASCOT-BPLA trial (Anglo-Scandinavian Cardiac Outcomes Trial—Blood Pressure Lowering Arm)26 had similar results. This trial compared the combination of atenolol plus the diuretic bendroflumethiazide against the combination of the calcium channel blocker amlodipine (Norvasc) plus the angiotensin-converting enzyme (ACE) inhibitor perindopril (Aceon). Although no significant difference was seen in the primary outcome of nonfatal myocardial infarction or fatal coronary heart disease (unadjusted hazard ratio [HR] with amlodipine-perindopril 0.90, 95% CI 0.79–1.02, P = .1052), the amlodipine-plus-perindopril group had significantly fewer strokes (327 vs 422, HR 0.77, 95% CI 0.66–0.89, P = .0003), fewer total cardiovascular events (1,362 vs 1,602, HR 0.84, 95% CI 0.78–0.90, P = .0001), and fewer deaths from any cause (738 vs 820; HR 0.89, 95% CI 0.81–0.99, P = .025).
Lindholm et al27 performed a meta-analysis that included studies of selective beta-blockers (including atenolol) and nonselective beta-blockers, with a follow-up time of more than 2 years. Compared with placebo or no treatment, beta-blockers reduced the risk of stroke by 19% but had no effect on myocardial infarction or all-cause mortality. Compared with other antihypertensive drugs, beta-blockers were less than optimum, and the relative risk of stroke was 16% higher. Atenolol was the beta-blocker used in most of the randomized clinical trials included in this meta-analysis.
The Cochrane group22 found beta-blockers to be inferior to all other antihypertensive drugs with respect to the ability to lower the risk of stroke.
WHY WERE THE RESULTS SO DISAPPOINTING?
Problems with atenolol
Most of the trials in the meta-analyses discussed above used atenolol and other beta-blockers that had no vasodilatory properties.
Further, in most of the trials atenolol was used in a once-daily dosage, whereas ideally it needs to be taken more frequently, based on its pharmacokinetic and pharmacodynamic properties (a half-life of 6–9 hours).3 Neutel et al28 confirmed that atenolol, when taken once daily, leaves the patient unprotected in the last 6 hours of a 24-hour period, as demonstrated by 24-hour ambulatory blood pressure monitoring. It is possible that this short duration of action of atenolol may have contributed to the results observed in clinical trials that used atenolol to treat hypertension.
Differences between older and younger patients
Another possible reason for the disappointing results is that the trials included many elderly patients, in whom beta-blockers may not be as effective. The pathophysiology of hypertension in younger people is different from that in older patients.29 Hemodynamic characteristics of younger hypertensive patients include a high cardiac output and hyperdynamic circulation with a low pulse pressure, while older patients have lower arterial compliance with an elevated vascular resistance.
The notion of choosing antihypertensive medications on the basis of age and age-related pathophysiology is supported by several clinical studies. Randomized controlled trials appear to show that beta-blockers are effective in younger hypertensive patients.30
Conversely, the CAFE (Conduit Artery Function Evaluation) trial,31 a substudy of the main ASCOT trial,26 indicated that betablocker-based therapy was less effective in reducing central aortic pressure than were regimens based on an ACE inhibitor or a calcium channel blocker.
The CAFE researchers recruited 2,073 patients from five ASCOT centers and used radial artery applanation tonometry and pulse-wave analysis to derive central aortic pressures and hemodynamic indices during study visits up to a period of 4 years. Although the two treatment groups achieved similar brachial systolic blood pressures, the central aortic systolic pressure was 4.3 mm Hg lower in the amlodipine group (95% CI 3.3–5.4; P < .0001), and the central aortic pulse pressure was 3.0 mm Hg lower (95% CI 2.1–3.9; P < .0001).
Pulse-wave dyssynchrony
Bangalore et al47 offer an interesting hypothesis to explain the probable adverse effect of beta-blockers. Their theory concerns the effect of these drugs on the arterial pulse wave.
Normally, with each contraction of the left ventricle during systole, an arterial pulse wave is generated and propagated forward to the peripheral arteries. This wave is then reflected back to the heart from the branching points of peripheral arteries. The final form of the pressure wave at the aortic root is a synchronized summation of the forward-traveling wave and the backward-reflected wave.
In healthy people with normal arteries, the reflected wave merges with the forward-traveling wave in diastole and augments coronary blood flow. In patients whose arteries are stiff due to aging or vascular comorbidities, the reflected wave returns faster and merges with the incident wave in systole, resulting in higher left ventricular afterload and less coronary perfusion.48
Bangalore et al47 propose that artificially reducing the heart rate with beta-blockers may further dyssynchronize the pulse wave, adversely affecting coronary perfusion and leading to an increased risk of cardiovascular events and death.
Metabolic side effects
Older beta-blockers, and especially atenolol, have well-known metabolic adverse effects, particularly impairment of glycemic control. This adverse effect appears to occur only with beta-blockers that do not possess vasodilatory properties and thus increase peripheral vascular resistance, which results in lower glucose availability and reduced uptake by skeletal muscles.49
Bangalore et al50 evaluated the effect of beta-blockers in a meta-analysis of 12 studies in 94,492 patients followed up for more than 1 year. Beta-blocker therapy resulted in a 22% higher risk of new-onset diabetes mellitus (RR 1.22, 95% CI 1.12–1.33) than with other nondiuretic antihypertensive agents.
Of note, however, the meta-analysis did not show a significantly higher risk of the onset of diabetes with propranolol or metoprolol than with other nondiuretic antihypertensives when studies of these beta-blockers were separated from atenolol-based studies.
Further, the United Kingdom Prospective Diabetes Study40 found that cardiovascular outcomes in patients with good blood pressure control were similar when atenolol-based therapy was compared with therapy with the ACE inhibitor captopril (Capoten).
A meta-analysis conducted by Balamuthusamy et al51 in 2009 found no higher risk of stroke in patients with hypertension and diabetes mellitus who received beta-blockers than in those who received other antihypertensive medications. However, beta-blockers were associated with a higher risk of death from cardiovascular causes (RR 1.39, 95% CI 1.07–1.804; P < .01) compared with reninangiotensin blockade.
NEWER BETA-BLOCKERS MAY BE BETTER
In the United States, more than 40 million prescriptions for atenolol are written every year, making it by far the most commonly used beta-blocker for the treatment of hypertension. 52 It is clear, however, that atenolol is not an ideal representative of this class of antihypertensive medications.
Preliminary data from studies of newer beta-blockers that possess beneficial vasodilatory properties are encouraging. Animal studies and preliminary human studies find that these new-generation beta-blockers cause fewer adverse metabolic effects and improve endothelial function, measures of arterial stiffness, and cardiovascular outcomes.
Carvedilol
Carvedilol is a nonselective beta-blocker with vasodilatory effects that are thought to be due to its ability to concurrently block alpha-1 receptors in addition to beta receptors. 53 In experiments in vitro and in trials in patients with diabetes and hypertension, carvedilol increased endothelial vasodilation and reduced inflammation and platelet aggregation. These effects may be achieved though antioxidant actions, thereby preserving nitric oxide bioactivity.54,55
In the Glycemic Effects in Diabetes Mellitus: Carvedilol-Metoprolol Comparison in Hypertensives (GEMINI) trial,56 carvedilol was associated with better maintenance of glycemic control in diabetic hypertensive patients than was metoprolol. Insulin sensitivity improved with carvedilol but not with metoprolol, and fewer patients on carvedilol progressed to microalbuminuria.
Nebivolol
Nebivolol is a novel selective beta-blocker with a much higher affinity for beta-1 adrenergic receptors than for beta-2 adrenergic receptors. Among all the beta-blockers in clinical use today, nebivolol has the highest selectivity for beta-1 receptors.8
Nebivolol causes vasodilation through activation of the l-arginine/nitric oxide pathway.57–59 Blockade of synthesis of nitric oxide leads to local arterial stiffness. Endothelial dysfunction is characterized by decreased bioavailability of nitric oxide and has been shown to be a strong predictor of cardiovascular outcomes. By generating nitric oxide, nebivolol reduces peripheral vascular resistance, overcoming a significant side effect of earlier beta-blockers that lowered blood pressure but ultimately increased peripheral vascular tone and resistance.8
In an experiment in a bovine model,60 nebivolol significantly reduced the pulse-wave velocity (a measure of arterial stiffness), while atenolol had no effect. Moreover, evidence for the role of the l-arginine/nitric oxide pathway in the vasodilatory effect of nebivolol was demonstrated by co-infusion of NG-monomethyl-L-arginine, a specific endothelial nitric oxide synthetase inhibitor that attenuated the reduction of pulse-wave velocity by nebivolol.
In studies in hypertensive patients, nebivolol was associated with a better metabolic profile than atenolol, with none of the adverse effects on insulin sensitivity that atenolol had.61 In the Study of Effects of Nebivolol Interventions on Outcomes and Rehospitalization in Seniors With Heart Failure (SENIORS) trial, significantly fewer patients receiving nebivolol died or were admitted to the hospital for cardiovascular reasons compared with those receiving placebo.62
Although these findings are encouraging, we do not yet know if these effects will translate into a significant reduction in cardiovascular outcomes in clinical trials. Large, prospective hypertension outcome trials, particularly to evaluate primary prevention of cardiovascular outcomes, are needed for an evidence-based approach to using the newer beta-blockers as preferred first-line therapy for hypertension.
WHAT RECENT GUIDELINES SAY ABOUT BETA-BLOCKERS
The British National Institute for Health and Clinical Excellence and the British Hypertension Society, in their 2004 guidelines, recommended beta-blockers as one of several first-line antihypertensive medications in young, nonblack patients.63 On the other hand, they advised clinicians to be aware of the reported increase in onset of diabetes mellitus in patients treated with these medications. After the LIFE24 and ASCOT26 study results were published, these guidelines were amended to exclude beta-blockers as preferred routine initial therapy for hypertension.64
More recently, the 2007 European Society of Hypertension and European Society of Cardiology reconsidered the role of beta-blockers, recommending them as an option in both initial and subsequent antihypertensive treatment strategies.65
The current guidelines from the National Heart, Lung, and Blood Institute,66 which were published in 2003, were highly influenced by the results of the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT),2 and favor diuretics as the first-line therapy. However, they indicate that beta-blockers are a suitable alternative, particularly when a compelling cardiac indication is present.53 We hope that the next update, expected late in 2009, will re-address this issue in the light of more recent data.
In recent years the role of beta-blockers as a primary tool to treat hypertension has come under question. These drugs have shown disappointing results when used as antihypertensive therapy in patients without heart disease, ie, when used as primary prevention. At the same time, beta-blockers clearly reduce the risk of future cardiovascular events in patients who already have heart disease, eg, who already have had a myocardial infarction or who have congestive heart failure.
Several meta-analyses and a few clinical trials have shown that beta-blockers may have no advantage over other antihypertensive drugs, and in fact may not reduce the risk of stroke as effectively as other classes of blood pressure medications.
Why should this be? Is it that the patients in the antihypertensive trials were mostly older, and that beta-blockers do not work as well in older patients as in younger ones? Or does it have to do with the fact that atenolol (Tenormin) was the drug most often used in the trials? Would newer, different beta-blockers be better?
Hypertension experts currently disagree on how to interpret the available data, and this has led to conflict and confusion among clinicians as to the role of beta-blockers in managing hypertension. Current evidence suggests that older beta-blockers may not be the preferred first-line antihypertensive drugs for hypertensive patients who have no compelling indications for them (eg, heart failure, myocardial infarction, diabetes, high risk of coronary heart disease). However, newer beta-blockers with vasodilatory properties should be considered in cases of uncontrolled or resistant hypertension, especially in younger patients.
Further, while controversy and debate continue over the benefits and adverse effects of one class of antihypertensive drugs vs another, it is indisputable that controlling arterial blood pressure to the recommended goal offers major protection against cardiovascular and renal events in patients with hypertension.1,2
MECHANISM OF ACTION OF BETA-BLOCKERS
Beta-blockers effectively reduce blood pressure in both systolic-diastolic hypertension and isolated systolic hypertension.3–5 Exactly how is not known, but it has been proposed that they may do so by:
Reducing the heart rate and cardiac output. When catecholamines activate beta-1 receptors in the heart, the heart rate and myocardial contractility increase. By blocking beta-1 receptors, beta-blockers reduce the heart rate and myocardial contractility, thus lowering cardiac output and arterial blood pressure.6
Inhibiting renin release. Activation of the renin-angiotensin system is another major pathway that can lead to elevated arterial blood pressure. Renin release is mediated through the sympathetic nervous system via beta-1 receptors on the juxtaglomerular cells of the kidney. Beta-blockers can therefore lower blood pressure by inhibiting renin release.7
Inhibiting central nervous sympathetic outflow, thereby inducing presynaptic blockade, which in turn reduces the release of catecholamines.
Reducing venous return and plasma volume.
Generating nitric oxide, thus reducing peripheral vascular resistance (some agents).8
Reducing vasomotor tone.
Reducing vascular tone.
Improving vascular compliance.
Resetting baroreceptor levels.
Attenuating the pressor response to catecholamines with exercise and stress.
HETEROGENEITY OF BETA-BLOCKERS
Selectivity
Beta-blockers are not all the same. They can be classified into three categories.
Nonselective beta-blockers block both beta-1 and beta-2 adrenergic receptors. It is generally accepted that beta-blockers exert their primary antihypertensive effect by blocking beta-1 adrenergic receptors.6 Of interest, nonselective beta-blockers inhibit beta-2 receptors on arteries and thus cause an unopposed alpha-adrenergic effect, leading to increased peripheral vascular resistance.9 Examples of this category:
- Nadolol (Corgard)
- Pindolol (Visken)
- Propranolol (Inderal)
- Timolol (Blocadren).
Selective beta-blockers specifically block beta-1 receptors alone, although they are known to be nonselective at higher doses. Examples:
- Atenolol (Tenormin)
- Betaxolol (Kerlone)
- Bisoprolol (Zebeta)
- Esmolol (Brevibloc)
- Metoprolol (Lopressor, Toprol).
Beta-blockers with peripheral vasodilatatory effects act either via antagonism of the alpha-1 receptor, as with labetolol (Normodyne) and carvedilol (Coreg),10 or via enhanced release of nitric oxide, as with nebivolol (Bystolic).8
Lipid and water solubility
The lipid solubility and water solubility of each beta-blocker determine its bioavailability and side-effect profile.
Lipid solubility determines the degree to which a beta-blocker penetrates the blood-brain barrier and thereby leads to central nervous system side effects such as lethargy, nightmares, confusion, and depression. Propranolol is highly lipid-soluble; metoprolol and labetalol are moderately so.
Water-soluble beta-blockers such as atenolol have less tissue permeation, have a longer half-life, and cause fewer central nervous system effects and symptoms.11
Routes of elimination
Beta-blockers also differ in their route of elimination.
Atenolol and nadolol are eliminated by the kidney and require dose adjustment in patients with impaired renal function.12,13
On the other hand, propranolol, metoprolol, labetalol, carvedilol, and nebivolol are excreted primarily via hepatic metabolism.13
BETA-BLOCKERS IN THE MANAGEMENT OF HYPERTENSION
Beta-blockers were initially used to treat arrhythmias, but by the early 1970s they were also widely accepted for managing hypertension. 14 Their initial acceptance as one of the first-line classes of drugs for hypertension was based on their better side-effect profile compared with other antihypertensive drugs available at that time.
In the 1980s and 1990s, beta-blockers were listed as preferred first-line antihypertensive drugs along with diuretics in national hypertension guidelines.15 Subsequent updates of the guidelines favored diuretics as initial therapy and relegated all other classes of antihypertensive medications to be alternatives to diuretics.16 Although beta-blockers remain alternative first-line drugs in the latest guidelines (published in 2003; see reference 66), they are the preferred antihypertensive agents for patients with cardiac disease.
The current recommendations reflect the findings from hypertension trials in which patients with myocardial infarction and congestive heart failure had better cardiovascular outcomes if they received these drugs,17–19 including a lower risk of death.20,21 It was widely assumed that beta-blockers would also prevent first episodes of cardiovascular events.
However, to date, there is no evidence that beta-blockers are effective as primary prevention. Several large randomized controlled trials showed no benefit with beta-blockers compared with other antihypertensive drugs—in fact, there were more cardiovascular events with beta-blockers (see below).
Beta-blockers are well tolerated in clinical practice, although they can have side effects that include fatigue, depression, impaired exercise tolerance, sexual dysfunction, and asthma attacks.
Wiysonge et al22 analyzed how many patients withdrew from randomized trials of antihypertensive treatment because of drug-related adverse events. There was no significant difference in the incidence of fatigue, depressive symptoms, or sexual dysfunction with beta-blockers compared with placebo, and trial participants on a beta-blocker were not statistically significantly more likely to discontinue treatment than those receiving a placebo in three trials with 22,729 participants (relative risk [RR] 2.34, 95% confidence interval [CI] 0.84–6.52).
THE CONTROVERSY: WHAT THE TRIALS SHOWED
Messerli et al23 performed a meta-analysis published in 1998 that suggested that beta-blockers may not be as effective as diuretics in preventing cardiovascular events when used as first-line antihypertensive therapy in elderly patients. In 10 randomized controlled trials in 16,164 patients who were treated with either a diuretic or a beta-blocker (atenolol), blood pressure was normalized in two-thirds of diuretic-treated patients but only one-third of patients treated with atenolol as monotherapy. Diuretic therapy was superior with regard to all end points, and beta-blockers were found to be ineffective except in reducing cerebrovascular events.
The LIFE study (Losartan Intervention for Endpoint Reduction in Hypertension)24 compared the angiotensin-receptor blocker losartan (Cozaar) and atenolol in 9,193 patients with hypertension and left ventricular hypertrophy. At 4 years of follow-up, the rate of primary cardiovascular events (death, myocardial infarction, or stroke) was lower in the losartan group than in the atenolol group. The difference was mainly due to a 25% lower incidence of stroke, which was statistically significant. The rates of myocardial infarction and death from cardiovascular causes were not significantly different between the two treatment groups. The systolic blood pressure was 1 mm Hg lower in the losartan group than in the atenolol group, which was statistically significant.
Carlberg et al25 performed another important meta-analysis that questioned whether atenolol reduces rates of cardiovascular morbidity and death in hypertensive patients. The results were surprising: eight randomized controlled trials including more than 6,000 patients and comparing atenolol with placebo or no treatment showed no differences between the treatment groups with regard to the outcomes of all-cause mortality (RR 1.01, 95% CI 0.89–1.15), cardiovascular mortality (RR 0.99, 95% CI 0.83–1.18), or myocardial infarction (RR 0.99, 95% CI 0.83–1.19).
In addition, when atenolol was compared with other antihypertensives in five other randomized controlled trials that included more than 14,000 patients, those treated with atenolol had a higher risk of stroke (RR 1.30, 95% CI 1.12–1.50) and death (RR 1.13, 95% CI 1.02–1.25).
The ASCOT-BPLA trial (Anglo-Scandinavian Cardiac Outcomes Trial—Blood Pressure Lowering Arm)26 had similar results. This trial compared the combination of atenolol plus the diuretic bendroflumethiazide against the combination of the calcium channel blocker amlodipine (Norvasc) plus the angiotensin-converting enzyme (ACE) inhibitor perindopril (Aceon). Although no significant difference was seen in the primary outcome of nonfatal myocardial infarction or fatal coronary heart disease (unadjusted hazard ratio [HR] with amlodipine-perindopril 0.90, 95% CI 0.79–1.02, P = .1052), the amlodipine-plus-perindopril group had significantly fewer strokes (327 vs 422, HR 0.77, 95% CI 0.66–0.89, P = .0003), fewer total cardiovascular events (1,362 vs 1,602, HR 0.84, 95% CI 0.78–0.90, P = .0001), and fewer deaths from any cause (738 vs 820; HR 0.89, 95% CI 0.81–0.99, P = .025).
Lindholm et al27 performed a meta-analysis that included studies of selective beta-blockers (including atenolol) and nonselective beta-blockers, with a follow-up time of more than 2 years. Compared with placebo or no treatment, beta-blockers reduced the risk of stroke by 19% but had no effect on myocardial infarction or all-cause mortality. Compared with other antihypertensive drugs, beta-blockers were less than optimum, and the relative risk of stroke was 16% higher. Atenolol was the beta-blocker used in most of the randomized clinical trials included in this meta-analysis.
The Cochrane group22 found beta-blockers to be inferior to all other antihypertensive drugs with respect to the ability to lower the risk of stroke.
WHY WERE THE RESULTS SO DISAPPOINTING?
Problems with atenolol
Most of the trials in the meta-analyses discussed above used atenolol and other beta-blockers that had no vasodilatory properties.
Further, in most of the trials atenolol was used in a once-daily dosage, whereas ideally it needs to be taken more frequently, based on its pharmacokinetic and pharmacodynamic properties (a half-life of 6–9 hours).3 Neutel et al28 confirmed that atenolol, when taken once daily, leaves the patient unprotected in the last 6 hours of a 24-hour period, as demonstrated by 24-hour ambulatory blood pressure monitoring. It is possible that this short duration of action of atenolol may have contributed to the results observed in clinical trials that used atenolol to treat hypertension.
Differences between older and younger patients
Another possible reason for the disappointing results is that the trials included many elderly patients, in whom beta-blockers may not be as effective. The pathophysiology of hypertension in younger people is different from that in older patients.29 Hemodynamic characteristics of younger hypertensive patients include a high cardiac output and hyperdynamic circulation with a low pulse pressure, while older patients have lower arterial compliance with an elevated vascular resistance.
The notion of choosing antihypertensive medications on the basis of age and age-related pathophysiology is supported by several clinical studies. Randomized controlled trials appear to show that beta-blockers are effective in younger hypertensive patients.30
Conversely, the CAFE (Conduit Artery Function Evaluation) trial,31 a substudy of the main ASCOT trial,26 indicated that betablocker-based therapy was less effective in reducing central aortic pressure than were regimens based on an ACE inhibitor or a calcium channel blocker.
The CAFE researchers recruited 2,073 patients from five ASCOT centers and used radial artery applanation tonometry and pulse-wave analysis to derive central aortic pressures and hemodynamic indices during study visits up to a period of 4 years. Although the two treatment groups achieved similar brachial systolic blood pressures, the central aortic systolic pressure was 4.3 mm Hg lower in the amlodipine group (95% CI 3.3–5.4; P < .0001), and the central aortic pulse pressure was 3.0 mm Hg lower (95% CI 2.1–3.9; P < .0001).
Pulse-wave dyssynchrony
Bangalore et al47 offer an interesting hypothesis to explain the probable adverse effect of beta-blockers. Their theory concerns the effect of these drugs on the arterial pulse wave.
Normally, with each contraction of the left ventricle during systole, an arterial pulse wave is generated and propagated forward to the peripheral arteries. This wave is then reflected back to the heart from the branching points of peripheral arteries. The final form of the pressure wave at the aortic root is a synchronized summation of the forward-traveling wave and the backward-reflected wave.
In healthy people with normal arteries, the reflected wave merges with the forward-traveling wave in diastole and augments coronary blood flow. In patients whose arteries are stiff due to aging or vascular comorbidities, the reflected wave returns faster and merges with the incident wave in systole, resulting in higher left ventricular afterload and less coronary perfusion.48
Bangalore et al47 propose that artificially reducing the heart rate with beta-blockers may further dyssynchronize the pulse wave, adversely affecting coronary perfusion and leading to an increased risk of cardiovascular events and death.
Metabolic side effects
Older beta-blockers, and especially atenolol, have well-known metabolic adverse effects, particularly impairment of glycemic control. This adverse effect appears to occur only with beta-blockers that do not possess vasodilatory properties and thus increase peripheral vascular resistance, which results in lower glucose availability and reduced uptake by skeletal muscles.49
Bangalore et al50 evaluated the effect of beta-blockers in a meta-analysis of 12 studies in 94,492 patients followed up for more than 1 year. Beta-blocker therapy resulted in a 22% higher risk of new-onset diabetes mellitus (RR 1.22, 95% CI 1.12–1.33) than with other nondiuretic antihypertensive agents.
Of note, however, the meta-analysis did not show a significantly higher risk of the onset of diabetes with propranolol or metoprolol than with other nondiuretic antihypertensives when studies of these beta-blockers were separated from atenolol-based studies.
Further, the United Kingdom Prospective Diabetes Study40 found that cardiovascular outcomes in patients with good blood pressure control were similar when atenolol-based therapy was compared with therapy with the ACE inhibitor captopril (Capoten).
A meta-analysis conducted by Balamuthusamy et al51 in 2009 found no higher risk of stroke in patients with hypertension and diabetes mellitus who received beta-blockers than in those who received other antihypertensive medications. However, beta-blockers were associated with a higher risk of death from cardiovascular causes (RR 1.39, 95% CI 1.07–1.804; P < .01) compared with reninangiotensin blockade.
NEWER BETA-BLOCKERS MAY BE BETTER
In the United States, more than 40 million prescriptions for atenolol are written every year, making it by far the most commonly used beta-blocker for the treatment of hypertension. 52 It is clear, however, that atenolol is not an ideal representative of this class of antihypertensive medications.
Preliminary data from studies of newer beta-blockers that possess beneficial vasodilatory properties are encouraging. Animal studies and preliminary human studies find that these new-generation beta-blockers cause fewer adverse metabolic effects and improve endothelial function, measures of arterial stiffness, and cardiovascular outcomes.
Carvedilol
Carvedilol is a nonselective beta-blocker with vasodilatory effects that are thought to be due to its ability to concurrently block alpha-1 receptors in addition to beta receptors. 53 In experiments in vitro and in trials in patients with diabetes and hypertension, carvedilol increased endothelial vasodilation and reduced inflammation and platelet aggregation. These effects may be achieved though antioxidant actions, thereby preserving nitric oxide bioactivity.54,55
In the Glycemic Effects in Diabetes Mellitus: Carvedilol-Metoprolol Comparison in Hypertensives (GEMINI) trial,56 carvedilol was associated with better maintenance of glycemic control in diabetic hypertensive patients than was metoprolol. Insulin sensitivity improved with carvedilol but not with metoprolol, and fewer patients on carvedilol progressed to microalbuminuria.
Nebivolol
Nebivolol is a novel selective beta-blocker with a much higher affinity for beta-1 adrenergic receptors than for beta-2 adrenergic receptors. Among all the beta-blockers in clinical use today, nebivolol has the highest selectivity for beta-1 receptors.8
Nebivolol causes vasodilation through activation of the l-arginine/nitric oxide pathway.57–59 Blockade of synthesis of nitric oxide leads to local arterial stiffness. Endothelial dysfunction is characterized by decreased bioavailability of nitric oxide and has been shown to be a strong predictor of cardiovascular outcomes. By generating nitric oxide, nebivolol reduces peripheral vascular resistance, overcoming a significant side effect of earlier beta-blockers that lowered blood pressure but ultimately increased peripheral vascular tone and resistance.8
In an experiment in a bovine model,60 nebivolol significantly reduced the pulse-wave velocity (a measure of arterial stiffness), while atenolol had no effect. Moreover, evidence for the role of the l-arginine/nitric oxide pathway in the vasodilatory effect of nebivolol was demonstrated by co-infusion of NG-monomethyl-L-arginine, a specific endothelial nitric oxide synthetase inhibitor that attenuated the reduction of pulse-wave velocity by nebivolol.
In studies in hypertensive patients, nebivolol was associated with a better metabolic profile than atenolol, with none of the adverse effects on insulin sensitivity that atenolol had.61 In the Study of Effects of Nebivolol Interventions on Outcomes and Rehospitalization in Seniors With Heart Failure (SENIORS) trial, significantly fewer patients receiving nebivolol died or were admitted to the hospital for cardiovascular reasons compared with those receiving placebo.62
Although these findings are encouraging, we do not yet know if these effects will translate into a significant reduction in cardiovascular outcomes in clinical trials. Large, prospective hypertension outcome trials, particularly to evaluate primary prevention of cardiovascular outcomes, are needed for an evidence-based approach to using the newer beta-blockers as preferred first-line therapy for hypertension.
WHAT RECENT GUIDELINES SAY ABOUT BETA-BLOCKERS
The British National Institute for Health and Clinical Excellence and the British Hypertension Society, in their 2004 guidelines, recommended beta-blockers as one of several first-line antihypertensive medications in young, nonblack patients.63 On the other hand, they advised clinicians to be aware of the reported increase in onset of diabetes mellitus in patients treated with these medications. After the LIFE24 and ASCOT26 study results were published, these guidelines were amended to exclude beta-blockers as preferred routine initial therapy for hypertension.64
More recently, the 2007 European Society of Hypertension and European Society of Cardiology reconsidered the role of beta-blockers, recommending them as an option in both initial and subsequent antihypertensive treatment strategies.65
The current guidelines from the National Heart, Lung, and Blood Institute,66 which were published in 2003, were highly influenced by the results of the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT),2 and favor diuretics as the first-line therapy. However, they indicate that beta-blockers are a suitable alternative, particularly when a compelling cardiac indication is present.53 We hope that the next update, expected late in 2009, will re-address this issue in the light of more recent data.
- Staessen JA, Wang JG, Thijs L. Cardiovascular protection and blood pressure reduction: a meta-analysis. Lancet 2001; 358:1305–1315.
- ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002; 288:2981–2997.
- Neutel JM, Smith DH, Ram CV, et al. Application of ambulatory blood pressure monitoring in differentiating between antihypertensive agents. Am J Med 1993; 94:181–187.
- Materson BJ, Reda DJ, Cushman WC, et al. Single-drug therapy for hypertension in men. A comparison of six antihypertensive agents with placebo. The Department of Veterans Affairs Cooperative Study Group on Antihypertensive Agents. N Engl J Med 1993; 328:914–921.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- Frishman W, Silverman R. Clinical pharmacology of the new beta-adrenergic blocking drugs. Part 2. Physiologic and metabolic effects. Am Heart J 1979; 97:797–807.
- Garrett BN, Kaplan NM. Plasma renin activity suppression: duration after withdrawal from beta-adrenergic blockade. Arch Intern Med 1980; 140:1316–1318.
- Pedersen ME, Cockcroft JR. The latest generation of beta-blockers: new pharmacologic properties. Curr Hypertens Rep 2006; 8:279–286.
- Man in’t Veld AJ, Van den Meiracker AH, Schalekamp MA. Do beta-blockers really increase peripheral vascular resistance? Review of the literature and new observations under basal conditions. Am J Hypertens 1988; 1:91–96.
- Pearce CJ, Wallin JD. Labetalol and other agents that block both alpha- and beta-adrenergic receptors. Cleve Clin J Med 1994; 61:59–69.
- Dimsdale JE, Newton RP, Joist T. Neuropsychological side effects of beta-blockers. Arch Intern Med 1989; 149:514–525.
- Agarwal R. Supervised atenolol therapy in the management of hemodialysis hypertension. Kidney Int 1999; 55:1528–1535.
- Sica DA, Black HR. Pharmacologic considerations in the positioning of beta-blockers in antihypertensive therapy. Curr Hypertens Rep 2008; 10:330–335.
- Prichard BN, Gillam GP. Use of propranolol (Inderal) in treatment of hypertension. Br Med J 1964; 19; 2:725–727.
- The fifth report of the Joint National Committee on Detection, Evaluation, and Treatment of High Blood Pressure (JNC V). Arch Intern Med 1993; 153:154–183.
- Moser M. Evolution of the treatment of hypertension from the 1940s to JNC V. Am J Hypertens 1997; 10:2S–8S.
- Houghton T, Freemantle N, Cleland JG. Are beta-blockers effective in patients who develop heart failure soon after myocardial infarction? A meta-regression analysis of randomised trials. Eur J Heart Fail 2000; 2:333–340.
- The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet 1999; 353:9–13.
- Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 1999; 353:2001–2007.
- Yusuf S, Peto R, Lewis J, Collins R, Sleight P. Beta blockade during and after myocardial infarction: an overview of the randomized trials. Prog Cardiovasc Dis 1985; 27:335–371.
- Brophy JM, Joseph L, Rouleau JL. Beta-blockers in congestive heart failure. A Bayesian meta-analysis. Ann Intern Med 2001; 134:550–560.
- Wiysonge CS, Bradley H, Mayosi BM, et al. Beta-blockers for hypertension. Cochrane Database Syst Rev 2007;CD002003.
- Messerli FH, Grossman E, Goldbourt U. Are beta-blockers efficacious as first-line therapy for hypertension in the elderly? A systematic review. JAMA 1998; 279:1903–1907.
- Dahlöf B, Devereux RB, Kjeldsen SE, et al; for the LIFE study group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint Reduction in Hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002; 359:995–1003.
- Carlberg B, Samuelsson O, Lindholm LH. Atenolol in hypertension: is it a wise choice? Lancet 2004; 364:1684–1689.
- Dahlöf B, Sever PS, Poulter NR, et al; ASCOT Investigators. Prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA): a multicentre randomised controlled trial. Lancet 2005; 366:895–906.
- Lindholm LH, Carlberg B, Samuelsson O. Should beta-blockers remain first choice in the treatment of primary hypertension? A meta-analysis. Lancet 2005; 366:1545–1553.
- Neutel JM, Schnaper H, Cheung DG, Graettinger WF, Weber MA. Antihypertensive effects of beta-blockers administered once daily: 24-hour measurements. Am Heart J 1990; 120:166–171.
- Franklin SS, Gustin W, Wong ND, et al. Hemodynamic patterns of age-related changes in blood pressure. The Framingham Heart Study. Circulation 1997; 96:308–315.
- The IPPPSH Collaborative Group. Cardiovascular risk and risk factors in a randomized trial of treatment based on the beta-blocker oxprenolol: the International Prospective Primary Prevention Study in Hypertension (IPPPSH). J Hypertens 1985; 3:379–392.
- Williams B, Lacy PS, Thom SM, et al; CAFE Investigators. Differential impact of blood pressure-lowering drugs on central aortic pressure and clinical outcomes: principal results of the Conduit Artery Function Evaluation (CAFE) study. Circulation 2006; 113:1213–1225.
- Khan N, McAlister FA. Re-examining the efficacy of beta-blockers for the treatment of hypertension: a meta-analysis. CMAJ 2006; 174:1737–1742.
- Medical Research Council Working Party. MRC trial of treatment of mild hypertension: principal results. BMJ 1985; 291:97–104.
- Coope J, Warrender TS. Randomised trial of treatment of hypertension in elderly patients in primary care. BMJ 1986; 293:1145–1151.
- Dahlöf B, Lindholm LH, Hansson L, et al. Morbidity and mortality in the Swedish Trial in Old Patients with Hypertension (STOP-Hypertension). Lancet 1991; 338:1281–1285.
- MRC Working Party. Medical Research Council trial of treatment of hypertension in older adults: principal results. BMJ 1992; 304:405–412.
- The Dutch TIA Study Group. Trial of secondary prevention with atenolol after transient ischemic attack or nondisabling ischemic stroke. Stroke 1993; 24:543–548.
- Eriksson S, Olofsson B-O, Wester P-O; for the TEST Study Group. Atenolol in secondary prevention after stroke. Cerebrovasc Dis 1995; 5:21–25.
- Wilhelmsen L, Berglund G, Elmfeldt D, et al. Beta-blockers versus diuretics in hypertensive men: main results from the HAPPHY trial. J Hypertens 1987; 5:561–572.
- UK Prospective Diabetes Study Group. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. BMJ 1998; 317:713–720.
- Hansson L, Lindholm LH, Niskanen L, et al. Effect of angiotensin-converting enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPP) randomised trial. Lancet 1999; 353:611–616.
- Lanchetti A, Bond MG, Henning M, et al. Calcium antagonist lacidipine slow down progression of asymptomatic carotid atherosclerosis. Principal results of the European Lacidipine Study on Atherosclerosis (ELSA), a randomized, double-blind, long-term trial. Circulation 2002; 106:2422–2427.
- Hansson L, Lindholm LH, Ekbom T, et al. Randomised trial of old and new antihypertensive drugs in elderly patients: cardiovascular mortality and morbidity in the Swedish Trial in Old Patients with Hypertension-2 study. Lancet 1999; 354:1751–1756.
- Hansson L, Hedner T, Lund-Johansen P, et al. Randomised trial of effects of calcium antagonists compared with diuretics and ß blockers on cardiovascular morbidity and mortality in hypertension: the Nordic Diltiazem (NORDIL) study. Lancet 2000; 356:359–365.
- Pepine CJ, Handsberg EM, Cooper-DeHoff RM, et al. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003; 290:2805–2816.
- Black HR, Elliott WJ, Grandits G, et al. Principal results of the Controlled Onset Verapamil Investigation of Cardiovascular End Points (CONVINCE) Trial. JAMA 2003; 289:2073–2082.
- Bangalore S, Sawhney S, Messerli FH. Relation of beta-blocker-induced heart rate lowering and cardioprotection in hypertension. J Am Coll Cardiol 2008; 52:1482–1489.
- Boutouyrie P, Vermersch S, Laurent S, Briet M. Cardiovascular risk assessment through target organ damage: role of carotid to femoral pulse wave velocity. Clin Exp Pharmacol Physiol 2008; 35:530–533.
- Kveiborg B, Christiansen B, Major-Petersen A, Torp-Pedersen C. Metabolic effects of beta-adrenoceptor antagonists with special emphasis on carvedilol. Am J Cardiovasc Drugs 2006; 6:209–217.
- Bangalore S, Parkar S, Grossman E, Messerli FH. A meta-analysis of 94,492 patients with hypertension treated with beta-blockers to determine the risk of new-onset diabetes mellitus. Am J Cardiol 2007; 100:1254–1262.
- Balamuthusamy S, Molnar J, Adigopula S, Arora R. Comparative analysis of beta-blockers with other antihypertensive agents on cardiovascular outcomes in hypertensive patients with diabetes mellitus: a systematic review and meta-analysis. Am J Ther 2009; 16:133–142.
- Berenson A. Big drug makers see sales decline with their image. New York Times 2005 Nov 14.
- Bristow MR. Beta-adrenergic receptor blockade in chronic heart failure. Circulation 2000; 101:558–569.
- Giugliano D, Marfella R, Acampora R, Giunta R, Coppola L, D’Onofrio F. Effects of perindopril and carvedilol on endothelium-dependent vascular functions in patients with diabetes and hypertension. Diabetes Care 1998; 21:631–636.
- Lopez BL, Christopher TA, Yue TL, Ruffolo R, Feuerstein GZ, Ma XL. Carvedilol, a new beta-adrenoreceptor blocker antihypertensive drug, protects against free-radical-induced endothelial dysfunction. Pharmacology 1995; 51:165–173.
- Bakris GL, Fonseca V, Katholi RE, et al; GEMINI Investigators. Metabolic effects of carvedilol vs metoprolol in patients with type 2 diabetes mellitus and hypertension: a randomized controlled trial. JAMA 2004; 292:2227–2236.
- Georgescu A, Pluteanu F, Flonta ML, Badila E, Dorobantu M, Popov D. The cellular mechanisms involved in the vasodilator effect of nebivolol on the renal artery. Eur J Pharmacol 2005; 508:159–166.
- Kalinowski L, Dobrucki LW, Szczepanska-Konkel M, et al. Third-generation beta-blockers stimulate nitric oxide release from endothelial cells through ATP efflux: a novel mechanism for antihypertensive action. Circulation 2003; 107:2747–2752.
- Cockcroft JR, Chowienczyk PJ, Brett SE, et al. Nebivolol vasodilates human forearm vasculature: evidence for an L-arginine/NO-dependent mechanism. J Pharmacol Exp Ther 1995; 274:1067–1071.
- McEniery CM, Schmitt M, Qasem A, et al. Nebivolol increases arterial distensibility in vivo. Hypertension 2004; 44:305–310.
- Poirier L, Cleroux J, Nadeau A, Lacourciere Y. Effects of nebivolol and atenolol on insulin sensitivity and haemodynamics in hypertensive patients. J Hypertens 2001; 19:1429–1435.
- Flather MD, Shibata MC, Coats AJ, et al; SENIORS Investigators. Randomized trial to determine the effect of nebivolol on mortality and cardiovascular hospital admission in elderly patients with heart failure (SENIORS). Eur Heart J 2005; 26:215–225.
- Williams B, Poulter NR, Brown MJ, et al; BHS guidelines working party, for the British Hypertension Society. British Hypertension Society guidelines for hypertension management 2004 (BHS-IV): summary. BMJ 2004; 328:634–640.
- Sever P. New hypertension guidelines from the National Institute for Health and Clinical Excellence and the British Hypertension Society. J Renin Angiotensin Aldosterone Syst 2006; 7:61–63.
- Mancia G, De Backer G, Dominiczak A, et al; Management of Arterial Hypertension of the European Society of Hypertension. 2007 Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens 2007; 25:1105–1187.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289:2560–2572.
- Staessen JA, Wang JG, Thijs L. Cardiovascular protection and blood pressure reduction: a meta-analysis. Lancet 2001; 358:1305–1315.
- ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002; 288:2981–2997.
- Neutel JM, Smith DH, Ram CV, et al. Application of ambulatory blood pressure monitoring in differentiating between antihypertensive agents. Am J Med 1993; 94:181–187.
- Materson BJ, Reda DJ, Cushman WC, et al. Single-drug therapy for hypertension in men. A comparison of six antihypertensive agents with placebo. The Department of Veterans Affairs Cooperative Study Group on Antihypertensive Agents. N Engl J Med 1993; 328:914–921.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- Frishman W, Silverman R. Clinical pharmacology of the new beta-adrenergic blocking drugs. Part 2. Physiologic and metabolic effects. Am Heart J 1979; 97:797–807.
- Garrett BN, Kaplan NM. Plasma renin activity suppression: duration after withdrawal from beta-adrenergic blockade. Arch Intern Med 1980; 140:1316–1318.
- Pedersen ME, Cockcroft JR. The latest generation of beta-blockers: new pharmacologic properties. Curr Hypertens Rep 2006; 8:279–286.
- Man in’t Veld AJ, Van den Meiracker AH, Schalekamp MA. Do beta-blockers really increase peripheral vascular resistance? Review of the literature and new observations under basal conditions. Am J Hypertens 1988; 1:91–96.
- Pearce CJ, Wallin JD. Labetalol and other agents that block both alpha- and beta-adrenergic receptors. Cleve Clin J Med 1994; 61:59–69.
- Dimsdale JE, Newton RP, Joist T. Neuropsychological side effects of beta-blockers. Arch Intern Med 1989; 149:514–525.
- Agarwal R. Supervised atenolol therapy in the management of hemodialysis hypertension. Kidney Int 1999; 55:1528–1535.
- Sica DA, Black HR. Pharmacologic considerations in the positioning of beta-blockers in antihypertensive therapy. Curr Hypertens Rep 2008; 10:330–335.
- Prichard BN, Gillam GP. Use of propranolol (Inderal) in treatment of hypertension. Br Med J 1964; 19; 2:725–727.
- The fifth report of the Joint National Committee on Detection, Evaluation, and Treatment of High Blood Pressure (JNC V). Arch Intern Med 1993; 153:154–183.
- Moser M. Evolution of the treatment of hypertension from the 1940s to JNC V. Am J Hypertens 1997; 10:2S–8S.
- Houghton T, Freemantle N, Cleland JG. Are beta-blockers effective in patients who develop heart failure soon after myocardial infarction? A meta-regression analysis of randomised trials. Eur J Heart Fail 2000; 2:333–340.
- The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet 1999; 353:9–13.
- Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 1999; 353:2001–2007.
- Yusuf S, Peto R, Lewis J, Collins R, Sleight P. Beta blockade during and after myocardial infarction: an overview of the randomized trials. Prog Cardiovasc Dis 1985; 27:335–371.
- Brophy JM, Joseph L, Rouleau JL. Beta-blockers in congestive heart failure. A Bayesian meta-analysis. Ann Intern Med 2001; 134:550–560.
- Wiysonge CS, Bradley H, Mayosi BM, et al. Beta-blockers for hypertension. Cochrane Database Syst Rev 2007;CD002003.
- Messerli FH, Grossman E, Goldbourt U. Are beta-blockers efficacious as first-line therapy for hypertension in the elderly? A systematic review. JAMA 1998; 279:1903–1907.
- Dahlöf B, Devereux RB, Kjeldsen SE, et al; for the LIFE study group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint Reduction in Hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002; 359:995–1003.
- Carlberg B, Samuelsson O, Lindholm LH. Atenolol in hypertension: is it a wise choice? Lancet 2004; 364:1684–1689.
- Dahlöf B, Sever PS, Poulter NR, et al; ASCOT Investigators. Prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA): a multicentre randomised controlled trial. Lancet 2005; 366:895–906.
- Lindholm LH, Carlberg B, Samuelsson O. Should beta-blockers remain first choice in the treatment of primary hypertension? A meta-analysis. Lancet 2005; 366:1545–1553.
- Neutel JM, Schnaper H, Cheung DG, Graettinger WF, Weber MA. Antihypertensive effects of beta-blockers administered once daily: 24-hour measurements. Am Heart J 1990; 120:166–171.
- Franklin SS, Gustin W, Wong ND, et al. Hemodynamic patterns of age-related changes in blood pressure. The Framingham Heart Study. Circulation 1997; 96:308–315.
- The IPPPSH Collaborative Group. Cardiovascular risk and risk factors in a randomized trial of treatment based on the beta-blocker oxprenolol: the International Prospective Primary Prevention Study in Hypertension (IPPPSH). J Hypertens 1985; 3:379–392.
- Williams B, Lacy PS, Thom SM, et al; CAFE Investigators. Differential impact of blood pressure-lowering drugs on central aortic pressure and clinical outcomes: principal results of the Conduit Artery Function Evaluation (CAFE) study. Circulation 2006; 113:1213–1225.
- Khan N, McAlister FA. Re-examining the efficacy of beta-blockers for the treatment of hypertension: a meta-analysis. CMAJ 2006; 174:1737–1742.
- Medical Research Council Working Party. MRC trial of treatment of mild hypertension: principal results. BMJ 1985; 291:97–104.
- Coope J, Warrender TS. Randomised trial of treatment of hypertension in elderly patients in primary care. BMJ 1986; 293:1145–1151.
- Dahlöf B, Lindholm LH, Hansson L, et al. Morbidity and mortality in the Swedish Trial in Old Patients with Hypertension (STOP-Hypertension). Lancet 1991; 338:1281–1285.
- MRC Working Party. Medical Research Council trial of treatment of hypertension in older adults: principal results. BMJ 1992; 304:405–412.
- The Dutch TIA Study Group. Trial of secondary prevention with atenolol after transient ischemic attack or nondisabling ischemic stroke. Stroke 1993; 24:543–548.
- Eriksson S, Olofsson B-O, Wester P-O; for the TEST Study Group. Atenolol in secondary prevention after stroke. Cerebrovasc Dis 1995; 5:21–25.
- Wilhelmsen L, Berglund G, Elmfeldt D, et al. Beta-blockers versus diuretics in hypertensive men: main results from the HAPPHY trial. J Hypertens 1987; 5:561–572.
- UK Prospective Diabetes Study Group. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. BMJ 1998; 317:713–720.
- Hansson L, Lindholm LH, Niskanen L, et al. Effect of angiotensin-converting enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPP) randomised trial. Lancet 1999; 353:611–616.
- Lanchetti A, Bond MG, Henning M, et al. Calcium antagonist lacidipine slow down progression of asymptomatic carotid atherosclerosis. Principal results of the European Lacidipine Study on Atherosclerosis (ELSA), a randomized, double-blind, long-term trial. Circulation 2002; 106:2422–2427.
- Hansson L, Lindholm LH, Ekbom T, et al. Randomised trial of old and new antihypertensive drugs in elderly patients: cardiovascular mortality and morbidity in the Swedish Trial in Old Patients with Hypertension-2 study. Lancet 1999; 354:1751–1756.
- Hansson L, Hedner T, Lund-Johansen P, et al. Randomised trial of effects of calcium antagonists compared with diuretics and ß blockers on cardiovascular morbidity and mortality in hypertension: the Nordic Diltiazem (NORDIL) study. Lancet 2000; 356:359–365.
- Pepine CJ, Handsberg EM, Cooper-DeHoff RM, et al. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003; 290:2805–2816.
- Black HR, Elliott WJ, Grandits G, et al. Principal results of the Controlled Onset Verapamil Investigation of Cardiovascular End Points (CONVINCE) Trial. JAMA 2003; 289:2073–2082.
- Bangalore S, Sawhney S, Messerli FH. Relation of beta-blocker-induced heart rate lowering and cardioprotection in hypertension. J Am Coll Cardiol 2008; 52:1482–1489.
- Boutouyrie P, Vermersch S, Laurent S, Briet M. Cardiovascular risk assessment through target organ damage: role of carotid to femoral pulse wave velocity. Clin Exp Pharmacol Physiol 2008; 35:530–533.
- Kveiborg B, Christiansen B, Major-Petersen A, Torp-Pedersen C. Metabolic effects of beta-adrenoceptor antagonists with special emphasis on carvedilol. Am J Cardiovasc Drugs 2006; 6:209–217.
- Bangalore S, Parkar S, Grossman E, Messerli FH. A meta-analysis of 94,492 patients with hypertension treated with beta-blockers to determine the risk of new-onset diabetes mellitus. Am J Cardiol 2007; 100:1254–1262.
- Balamuthusamy S, Molnar J, Adigopula S, Arora R. Comparative analysis of beta-blockers with other antihypertensive agents on cardiovascular outcomes in hypertensive patients with diabetes mellitus: a systematic review and meta-analysis. Am J Ther 2009; 16:133–142.
- Berenson A. Big drug makers see sales decline with their image. New York Times 2005 Nov 14.
- Bristow MR. Beta-adrenergic receptor blockade in chronic heart failure. Circulation 2000; 101:558–569.
- Giugliano D, Marfella R, Acampora R, Giunta R, Coppola L, D’Onofrio F. Effects of perindopril and carvedilol on endothelium-dependent vascular functions in patients with diabetes and hypertension. Diabetes Care 1998; 21:631–636.
- Lopez BL, Christopher TA, Yue TL, Ruffolo R, Feuerstein GZ, Ma XL. Carvedilol, a new beta-adrenoreceptor blocker antihypertensive drug, protects against free-radical-induced endothelial dysfunction. Pharmacology 1995; 51:165–173.
- Bakris GL, Fonseca V, Katholi RE, et al; GEMINI Investigators. Metabolic effects of carvedilol vs metoprolol in patients with type 2 diabetes mellitus and hypertension: a randomized controlled trial. JAMA 2004; 292:2227–2236.
- Georgescu A, Pluteanu F, Flonta ML, Badila E, Dorobantu M, Popov D. The cellular mechanisms involved in the vasodilator effect of nebivolol on the renal artery. Eur J Pharmacol 2005; 508:159–166.
- Kalinowski L, Dobrucki LW, Szczepanska-Konkel M, et al. Third-generation beta-blockers stimulate nitric oxide release from endothelial cells through ATP efflux: a novel mechanism for antihypertensive action. Circulation 2003; 107:2747–2752.
- Cockcroft JR, Chowienczyk PJ, Brett SE, et al. Nebivolol vasodilates human forearm vasculature: evidence for an L-arginine/NO-dependent mechanism. J Pharmacol Exp Ther 1995; 274:1067–1071.
- McEniery CM, Schmitt M, Qasem A, et al. Nebivolol increases arterial distensibility in vivo. Hypertension 2004; 44:305–310.
- Poirier L, Cleroux J, Nadeau A, Lacourciere Y. Effects of nebivolol and atenolol on insulin sensitivity and haemodynamics in hypertensive patients. J Hypertens 2001; 19:1429–1435.
- Flather MD, Shibata MC, Coats AJ, et al; SENIORS Investigators. Randomized trial to determine the effect of nebivolol on mortality and cardiovascular hospital admission in elderly patients with heart failure (SENIORS). Eur Heart J 2005; 26:215–225.
- Williams B, Poulter NR, Brown MJ, et al; BHS guidelines working party, for the British Hypertension Society. British Hypertension Society guidelines for hypertension management 2004 (BHS-IV): summary. BMJ 2004; 328:634–640.
- Sever P. New hypertension guidelines from the National Institute for Health and Clinical Excellence and the British Hypertension Society. J Renin Angiotensin Aldosterone Syst 2006; 7:61–63.
- Mancia G, De Backer G, Dominiczak A, et al; Management of Arterial Hypertension of the European Society of Hypertension. 2007 Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens 2007; 25:1105–1187.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289:2560–2572.
KEY POINTS
- No evidence exists that beta-blockers prevent first episodes of cardiovascular events in patients with hypertension, and in some trials, outcomes were worse with beta-blockers than with antihypertensive drugs of other classes.
- Younger hypertensive patients have hemodynamic characteristics that would seem to be amenable to beta-blocker therapy. However, most clinical trials of beta blockers did not stratify patients by age.
- Most trials of the antihypertensive effects of beta-blockers used atenolol (Tenormin), which is not an ideal representative of this class of drugs.
- Newer beta-blockers with vasodilatory properties may overcome the adverse effect of increased peripheral vascular resistance that occurs with older agents such as atenolol.
Should catheter ablation be the first line of treatment for atrial fibrillation?
Catheter ablation for atrial fibrillation has evolved since it was introduced a decade ago. It will continue to improve as we gain experience with the procedure, better understand the pathophysiology of atrial fibrillation, and develop new technologies for imaging, catheter navigation, and more effective ablation of atrial tissue. The topic is reviewed by Chowdhury et al1 in this issue of the Cleveland Clinic Journal of Medicine.
An important question is whether catheter ablation should replace antiarrhythmic drugs as the first line of therapy. The answer will be determined by the procedure’s success rate, complication rate, cost, and long-term outcomes compared with drug therapy.
RELATIVELY FEW RANDOMIZED TRIALS, BUT ENCOURAGING RESULTS
Relatively few randomized trials have compared catheter ablation and medical therapy.
In patients with paroxysmal atrial fibrillation, three important randomized trials2–4 have shown catheter ablation to be superior to antiarrhythmic drug therapy. In these trials, freedom from atrial fibrillation or atrial flutter was achieved in 63% to 93% of patients who underwent ablation compared with 17% to 35% of those assigned to drug therapy. However, more than one ablation procedure may be required to achieve success rates in the higher range. Further, these studies were done at “high-volume” centers, and they excluded patients with major comorbidities.
Persistent or long-standing atrial fibrillation is more complex than paroxysmal atrial fibrillation. It is more often accompanied by significant comorbidities, and comparative trials have generally excluded patients with these attributes. Fewer of such patients obtain complete success (ie, cure), and more of them need a second ablation procedure.
Oral et al5 randomly assigned patients with long-standing atrial fibrillation to be treated with amiodarone (Cordarone) or catheter ablation. The analysis of this study was complicated by a high rate of crossover from the drug therapy group to the ablation group. Twenty-five (32%) of the 77 patients assigned to undergo ablation needed a second procedure, but at 12 months 74% were in sinus rhythm without amiodarone, compared with only 4% treated with amiodarone without ablation.
These results indicate that ablation is more effective than medical therapy for paroxysmal atrial fibrillation, and it appears to be more effective than drugs alone for long-standing persistent atrial fibrillation. In addition, quality of life was better after ablation, and complications were relatively few.2–4
The limitations are that the trials were done at hospitals in which the ablation teams had a lot of experience, did many ablation procedures per year, and tracked their outcomes carefully: other hospitals may not be able to achieve the same results. Moreover, many patients referred for ablation have heart failure, significant valvular disease, or left atrial enlargement, which would have excluded them from the published trials.
MORE STUDIES UNDER WAY
Two other initiatives may help define the role of ablation for atrial fibrillation.
The Cardiac Ablation vs Antiarrhythmic Drug Therapy for Atrial Fibrillation (the CABANA) trial is a multicenter randomized longitudinal study designed to determine whether ablation is more effective than drug therapy. Target enrollment is 3,000 patients.
The National Cardiovascular Data Registry is exploring the possibility of establishing a registry for ablation of atrial fibrillation. This database could be used by physicians, hospitals, the Centers for Medicare & Medicaid Services, and the US Food and Drug Administration to track overall outcomes of these complex procedures.
FOR NOW, DRUGS ARE STILL THE FIRST-LINE TREATMENT
For now, I believe that antiarrhythmic drugs should remain the first line of treatment for atrial fibrillation until cumulative evidence from additional randomized multicenter trials proves otherwise. However, the threshold for deciding to do an ablation procedure is getting lower, and it is reasonable for patients to make an informed decision to move directly to ablation as an alternative to drug therapy if that is their preference.
To make these decisions, patients need accurate information about success rates and the risk of complications at the center where the procedure is to be performed. At Cleveland Clinic, where more than 4,300 ablation procedures have been performed for atrial fibrillation, substantial resources are devoted to tracking outcomes. As the government and insurance companies focus on pay for performance and as ablation procedures for atrial fibrillation become more widespread and new technologies are introduced, it will be especially important for hospitals to track their own costs and outcomes.
The cumulative experience from well-designed clinical trials will provide guidance, but unless hospitals verify that they achieve results equivalent to those in the trials, physicians should be cautious about recommending ablation as the first-line therapy.
- Chowdhury P, Lewis R, Schweikert R, Cummings JE. Catheter ablation for the treatment of atrial fibrillation. Cleve Clin J Med 2009; 76:543–550.
- Pappone C, Augello G, Sala S, et al. A randomized trial of circumferential pulmonary vein ablation versus antiarrhythmic drug therapy in paroxysmal atrial fibrillation. the APAF Study. J Am Coll Cardiol 2006; 48:2340–2347.
- Jais P, Cauchemez , Macle L, et al. Catheter ablation versus antiarrhythmic drugs for atrial fibrillation: the A4 Study. Circulation 2008; 118:2498–2505.
- Wilber DJ, Pappone C, Neuzil P, et al; The Thermocool AF Investigators. Recurrent atrial arrhythmias and quality-of-life in patients with paroxysmal atrial fibrillation treated by radiofrequency catheter ablation compared to antiarrhythmic drug therapy: final results of the Thermocool AF trial. Presented at the Heart Rhythm Society Annual Scientific Sessions, May 13–19 2009, Boston, MA.
- Oral H, Pappone C, Chugh A, et al. Circumferential pulmonary-vein ablation for chronic atrial fibrillation. N Engl J Med 2006; 354:934–941.
Catheter ablation for atrial fibrillation has evolved since it was introduced a decade ago. It will continue to improve as we gain experience with the procedure, better understand the pathophysiology of atrial fibrillation, and develop new technologies for imaging, catheter navigation, and more effective ablation of atrial tissue. The topic is reviewed by Chowdhury et al1 in this issue of the Cleveland Clinic Journal of Medicine.
An important question is whether catheter ablation should replace antiarrhythmic drugs as the first line of therapy. The answer will be determined by the procedure’s success rate, complication rate, cost, and long-term outcomes compared with drug therapy.
RELATIVELY FEW RANDOMIZED TRIALS, BUT ENCOURAGING RESULTS
Relatively few randomized trials have compared catheter ablation and medical therapy.
In patients with paroxysmal atrial fibrillation, three important randomized trials2–4 have shown catheter ablation to be superior to antiarrhythmic drug therapy. In these trials, freedom from atrial fibrillation or atrial flutter was achieved in 63% to 93% of patients who underwent ablation compared with 17% to 35% of those assigned to drug therapy. However, more than one ablation procedure may be required to achieve success rates in the higher range. Further, these studies were done at “high-volume” centers, and they excluded patients with major comorbidities.
Persistent or long-standing atrial fibrillation is more complex than paroxysmal atrial fibrillation. It is more often accompanied by significant comorbidities, and comparative trials have generally excluded patients with these attributes. Fewer of such patients obtain complete success (ie, cure), and more of them need a second ablation procedure.
Oral et al5 randomly assigned patients with long-standing atrial fibrillation to be treated with amiodarone (Cordarone) or catheter ablation. The analysis of this study was complicated by a high rate of crossover from the drug therapy group to the ablation group. Twenty-five (32%) of the 77 patients assigned to undergo ablation needed a second procedure, but at 12 months 74% were in sinus rhythm without amiodarone, compared with only 4% treated with amiodarone without ablation.
These results indicate that ablation is more effective than medical therapy for paroxysmal atrial fibrillation, and it appears to be more effective than drugs alone for long-standing persistent atrial fibrillation. In addition, quality of life was better after ablation, and complications were relatively few.2–4
The limitations are that the trials were done at hospitals in which the ablation teams had a lot of experience, did many ablation procedures per year, and tracked their outcomes carefully: other hospitals may not be able to achieve the same results. Moreover, many patients referred for ablation have heart failure, significant valvular disease, or left atrial enlargement, which would have excluded them from the published trials.
MORE STUDIES UNDER WAY
Two other initiatives may help define the role of ablation for atrial fibrillation.
The Cardiac Ablation vs Antiarrhythmic Drug Therapy for Atrial Fibrillation (the CABANA) trial is a multicenter randomized longitudinal study designed to determine whether ablation is more effective than drug therapy. Target enrollment is 3,000 patients.
The National Cardiovascular Data Registry is exploring the possibility of establishing a registry for ablation of atrial fibrillation. This database could be used by physicians, hospitals, the Centers for Medicare & Medicaid Services, and the US Food and Drug Administration to track overall outcomes of these complex procedures.
FOR NOW, DRUGS ARE STILL THE FIRST-LINE TREATMENT
For now, I believe that antiarrhythmic drugs should remain the first line of treatment for atrial fibrillation until cumulative evidence from additional randomized multicenter trials proves otherwise. However, the threshold for deciding to do an ablation procedure is getting lower, and it is reasonable for patients to make an informed decision to move directly to ablation as an alternative to drug therapy if that is their preference.
To make these decisions, patients need accurate information about success rates and the risk of complications at the center where the procedure is to be performed. At Cleveland Clinic, where more than 4,300 ablation procedures have been performed for atrial fibrillation, substantial resources are devoted to tracking outcomes. As the government and insurance companies focus on pay for performance and as ablation procedures for atrial fibrillation become more widespread and new technologies are introduced, it will be especially important for hospitals to track their own costs and outcomes.
The cumulative experience from well-designed clinical trials will provide guidance, but unless hospitals verify that they achieve results equivalent to those in the trials, physicians should be cautious about recommending ablation as the first-line therapy.
Catheter ablation for atrial fibrillation has evolved since it was introduced a decade ago. It will continue to improve as we gain experience with the procedure, better understand the pathophysiology of atrial fibrillation, and develop new technologies for imaging, catheter navigation, and more effective ablation of atrial tissue. The topic is reviewed by Chowdhury et al1 in this issue of the Cleveland Clinic Journal of Medicine.
An important question is whether catheter ablation should replace antiarrhythmic drugs as the first line of therapy. The answer will be determined by the procedure’s success rate, complication rate, cost, and long-term outcomes compared with drug therapy.
RELATIVELY FEW RANDOMIZED TRIALS, BUT ENCOURAGING RESULTS
Relatively few randomized trials have compared catheter ablation and medical therapy.
In patients with paroxysmal atrial fibrillation, three important randomized trials2–4 have shown catheter ablation to be superior to antiarrhythmic drug therapy. In these trials, freedom from atrial fibrillation or atrial flutter was achieved in 63% to 93% of patients who underwent ablation compared with 17% to 35% of those assigned to drug therapy. However, more than one ablation procedure may be required to achieve success rates in the higher range. Further, these studies were done at “high-volume” centers, and they excluded patients with major comorbidities.
Persistent or long-standing atrial fibrillation is more complex than paroxysmal atrial fibrillation. It is more often accompanied by significant comorbidities, and comparative trials have generally excluded patients with these attributes. Fewer of such patients obtain complete success (ie, cure), and more of them need a second ablation procedure.
Oral et al5 randomly assigned patients with long-standing atrial fibrillation to be treated with amiodarone (Cordarone) or catheter ablation. The analysis of this study was complicated by a high rate of crossover from the drug therapy group to the ablation group. Twenty-five (32%) of the 77 patients assigned to undergo ablation needed a second procedure, but at 12 months 74% were in sinus rhythm without amiodarone, compared with only 4% treated with amiodarone without ablation.
These results indicate that ablation is more effective than medical therapy for paroxysmal atrial fibrillation, and it appears to be more effective than drugs alone for long-standing persistent atrial fibrillation. In addition, quality of life was better after ablation, and complications were relatively few.2–4
The limitations are that the trials were done at hospitals in which the ablation teams had a lot of experience, did many ablation procedures per year, and tracked their outcomes carefully: other hospitals may not be able to achieve the same results. Moreover, many patients referred for ablation have heart failure, significant valvular disease, or left atrial enlargement, which would have excluded them from the published trials.
MORE STUDIES UNDER WAY
Two other initiatives may help define the role of ablation for atrial fibrillation.
The Cardiac Ablation vs Antiarrhythmic Drug Therapy for Atrial Fibrillation (the CABANA) trial is a multicenter randomized longitudinal study designed to determine whether ablation is more effective than drug therapy. Target enrollment is 3,000 patients.
The National Cardiovascular Data Registry is exploring the possibility of establishing a registry for ablation of atrial fibrillation. This database could be used by physicians, hospitals, the Centers for Medicare & Medicaid Services, and the US Food and Drug Administration to track overall outcomes of these complex procedures.
FOR NOW, DRUGS ARE STILL THE FIRST-LINE TREATMENT
For now, I believe that antiarrhythmic drugs should remain the first line of treatment for atrial fibrillation until cumulative evidence from additional randomized multicenter trials proves otherwise. However, the threshold for deciding to do an ablation procedure is getting lower, and it is reasonable for patients to make an informed decision to move directly to ablation as an alternative to drug therapy if that is their preference.
To make these decisions, patients need accurate information about success rates and the risk of complications at the center where the procedure is to be performed. At Cleveland Clinic, where more than 4,300 ablation procedures have been performed for atrial fibrillation, substantial resources are devoted to tracking outcomes. As the government and insurance companies focus on pay for performance and as ablation procedures for atrial fibrillation become more widespread and new technologies are introduced, it will be especially important for hospitals to track their own costs and outcomes.
The cumulative experience from well-designed clinical trials will provide guidance, but unless hospitals verify that they achieve results equivalent to those in the trials, physicians should be cautious about recommending ablation as the first-line therapy.
- Chowdhury P, Lewis R, Schweikert R, Cummings JE. Catheter ablation for the treatment of atrial fibrillation. Cleve Clin J Med 2009; 76:543–550.
- Pappone C, Augello G, Sala S, et al. A randomized trial of circumferential pulmonary vein ablation versus antiarrhythmic drug therapy in paroxysmal atrial fibrillation. the APAF Study. J Am Coll Cardiol 2006; 48:2340–2347.
- Jais P, Cauchemez , Macle L, et al. Catheter ablation versus antiarrhythmic drugs for atrial fibrillation: the A4 Study. Circulation 2008; 118:2498–2505.
- Wilber DJ, Pappone C, Neuzil P, et al; The Thermocool AF Investigators. Recurrent atrial arrhythmias and quality-of-life in patients with paroxysmal atrial fibrillation treated by radiofrequency catheter ablation compared to antiarrhythmic drug therapy: final results of the Thermocool AF trial. Presented at the Heart Rhythm Society Annual Scientific Sessions, May 13–19 2009, Boston, MA.
- Oral H, Pappone C, Chugh A, et al. Circumferential pulmonary-vein ablation for chronic atrial fibrillation. N Engl J Med 2006; 354:934–941.
- Chowdhury P, Lewis R, Schweikert R, Cummings JE. Catheter ablation for the treatment of atrial fibrillation. Cleve Clin J Med 2009; 76:543–550.
- Pappone C, Augello G, Sala S, et al. A randomized trial of circumferential pulmonary vein ablation versus antiarrhythmic drug therapy in paroxysmal atrial fibrillation. the APAF Study. J Am Coll Cardiol 2006; 48:2340–2347.
- Jais P, Cauchemez , Macle L, et al. Catheter ablation versus antiarrhythmic drugs for atrial fibrillation: the A4 Study. Circulation 2008; 118:2498–2505.
- Wilber DJ, Pappone C, Neuzil P, et al; The Thermocool AF Investigators. Recurrent atrial arrhythmias and quality-of-life in patients with paroxysmal atrial fibrillation treated by radiofrequency catheter ablation compared to antiarrhythmic drug therapy: final results of the Thermocool AF trial. Presented at the Heart Rhythm Society Annual Scientific Sessions, May 13–19 2009, Boston, MA.
- Oral H, Pappone C, Chugh A, et al. Circumferential pulmonary-vein ablation for chronic atrial fibrillation. N Engl J Med 2006; 354:934–941.
Pregabalin for fibromyalgia
To the Editor: The article by Kim et al regarding the use of pregabalin (Lyrica) in fibromyalgia is interesting and timely.1 We would like make some additional comments.
They claim that “many hail pregabalin as an important advance in our understanding of the pathogenesis of fibromyalgia and how to treat it,” but they fail to cite who those “many” are. We contend that aside from the pharmaceutical company’s representatives, physicians on its speaker’s bureau, and those participating in paid drug studies, it would be difficult to substantiate this statement.
The authors’ historical overview discusses Gowers’ description of fibrositis but misinterprets his discussion. Gowers did not believe that “inflammation of muscles” was a problem, but that fibrous tissue itself was inflamed (thus the term “fibrositis,” not “myositis”) and could thus produce pain such as pharyngitis and sciatica, as well as “muscular rheumatism.”2
The authors review functional abnormalities in central nervous system processing as an etiology of pain. Russell et al are cited as elucidating the role of substance P in the process.3 Although they showed that substance P was three times higher in the cerebrospinal fluid of fibromyalgia patients compared with normal controls, the cited paper also notes that there was an inverse relationship between substance P levels and tenderness. Substance P also did not correlate with the Visual Analogue Scale self-assessment of pain severity or “with any other clinical variable.”3
A question of whether appropriate controls were chosen for the study has to be raised as well, since 25 of 32 fibromyalgia patients and only 3 of 30 controls had “possible depression.”3 The discussion of other therapies has limitations. The authors rely on two meta-analyses and a review to suggest that the efficacy of tricyclics is supported by a variety of studies. We don’t disagree, but as we previously noted, long-term studies don’t support prolonged efficacy of these drugs, which raises questions of treatment of a chronic illness.4 Duloxetine (Cymbalta) and milnacipran (Savella) are briefly mentioned. The authors should have used at least a sentence for each drug to indicate that the drugs have their own substantial shortcomings.
Finally, the authors conclude their article by asking, “What role for pregabalin?” A careful reading of that section does not appear to provide an answer. We recently presented a study suggesting that pregabalin and standard therapy were equally effective (or equally not effective), suggesting that pregabalin neither represents a major pharmaceutical advance in therapy nor is likely to “advance our understanding of the pathogenesis of fibromyalgia.”5 We do agree with the authors that medications are only part of a comprehensive program of therapy, and further point out that fibromyalgia undoubtedly represents the end point of a variety of etiologic insults and is unlikely to be one specific syndrome.
- Kim L, Lipton S, Deodhar A. Pregabalin for fibromyalgia: some relief but no cure. Cleve Clin J Med 2009; 76:255–261.
- Abeles M. Fibromyalgia syndrome. In:Manu P, Editor: Functional Somatic Syndromes. New York: Cambridge University Press, 1998:32–57.
- Russell IJ, Orr MD, Littman B, et al. Elevated cerebrospinal fluid levels of substance P in patients with fibromyalgia syndrome. Arthritis Rheum 1994; 37:1593–1601.
- Abeles M, Abeles SR, Abeles AM. Fibromyalgia remains a controversial medical enigma [letter]. Am Fam Physician 2008; 77:1220.
- Abeles M, Abeles AM. Is pregabalin better than conventional therapy in fibromyalgia? [abstract] Arthritis Rheum 2008; 58( suppl):S396.
To the Editor: The article by Kim et al regarding the use of pregabalin (Lyrica) in fibromyalgia is interesting and timely.1 We would like make some additional comments.
They claim that “many hail pregabalin as an important advance in our understanding of the pathogenesis of fibromyalgia and how to treat it,” but they fail to cite who those “many” are. We contend that aside from the pharmaceutical company’s representatives, physicians on its speaker’s bureau, and those participating in paid drug studies, it would be difficult to substantiate this statement.
The authors’ historical overview discusses Gowers’ description of fibrositis but misinterprets his discussion. Gowers did not believe that “inflammation of muscles” was a problem, but that fibrous tissue itself was inflamed (thus the term “fibrositis,” not “myositis”) and could thus produce pain such as pharyngitis and sciatica, as well as “muscular rheumatism.”2
The authors review functional abnormalities in central nervous system processing as an etiology of pain. Russell et al are cited as elucidating the role of substance P in the process.3 Although they showed that substance P was three times higher in the cerebrospinal fluid of fibromyalgia patients compared with normal controls, the cited paper also notes that there was an inverse relationship between substance P levels and tenderness. Substance P also did not correlate with the Visual Analogue Scale self-assessment of pain severity or “with any other clinical variable.”3
A question of whether appropriate controls were chosen for the study has to be raised as well, since 25 of 32 fibromyalgia patients and only 3 of 30 controls had “possible depression.”3 The discussion of other therapies has limitations. The authors rely on two meta-analyses and a review to suggest that the efficacy of tricyclics is supported by a variety of studies. We don’t disagree, but as we previously noted, long-term studies don’t support prolonged efficacy of these drugs, which raises questions of treatment of a chronic illness.4 Duloxetine (Cymbalta) and milnacipran (Savella) are briefly mentioned. The authors should have used at least a sentence for each drug to indicate that the drugs have their own substantial shortcomings.
Finally, the authors conclude their article by asking, “What role for pregabalin?” A careful reading of that section does not appear to provide an answer. We recently presented a study suggesting that pregabalin and standard therapy were equally effective (or equally not effective), suggesting that pregabalin neither represents a major pharmaceutical advance in therapy nor is likely to “advance our understanding of the pathogenesis of fibromyalgia.”5 We do agree with the authors that medications are only part of a comprehensive program of therapy, and further point out that fibromyalgia undoubtedly represents the end point of a variety of etiologic insults and is unlikely to be one specific syndrome.
To the Editor: The article by Kim et al regarding the use of pregabalin (Lyrica) in fibromyalgia is interesting and timely.1 We would like make some additional comments.
They claim that “many hail pregabalin as an important advance in our understanding of the pathogenesis of fibromyalgia and how to treat it,” but they fail to cite who those “many” are. We contend that aside from the pharmaceutical company’s representatives, physicians on its speaker’s bureau, and those participating in paid drug studies, it would be difficult to substantiate this statement.
The authors’ historical overview discusses Gowers’ description of fibrositis but misinterprets his discussion. Gowers did not believe that “inflammation of muscles” was a problem, but that fibrous tissue itself was inflamed (thus the term “fibrositis,” not “myositis”) and could thus produce pain such as pharyngitis and sciatica, as well as “muscular rheumatism.”2
The authors review functional abnormalities in central nervous system processing as an etiology of pain. Russell et al are cited as elucidating the role of substance P in the process.3 Although they showed that substance P was three times higher in the cerebrospinal fluid of fibromyalgia patients compared with normal controls, the cited paper also notes that there was an inverse relationship between substance P levels and tenderness. Substance P also did not correlate with the Visual Analogue Scale self-assessment of pain severity or “with any other clinical variable.”3
A question of whether appropriate controls were chosen for the study has to be raised as well, since 25 of 32 fibromyalgia patients and only 3 of 30 controls had “possible depression.”3 The discussion of other therapies has limitations. The authors rely on two meta-analyses and a review to suggest that the efficacy of tricyclics is supported by a variety of studies. We don’t disagree, but as we previously noted, long-term studies don’t support prolonged efficacy of these drugs, which raises questions of treatment of a chronic illness.4 Duloxetine (Cymbalta) and milnacipran (Savella) are briefly mentioned. The authors should have used at least a sentence for each drug to indicate that the drugs have their own substantial shortcomings.
Finally, the authors conclude their article by asking, “What role for pregabalin?” A careful reading of that section does not appear to provide an answer. We recently presented a study suggesting that pregabalin and standard therapy were equally effective (or equally not effective), suggesting that pregabalin neither represents a major pharmaceutical advance in therapy nor is likely to “advance our understanding of the pathogenesis of fibromyalgia.”5 We do agree with the authors that medications are only part of a comprehensive program of therapy, and further point out that fibromyalgia undoubtedly represents the end point of a variety of etiologic insults and is unlikely to be one specific syndrome.
- Kim L, Lipton S, Deodhar A. Pregabalin for fibromyalgia: some relief but no cure. Cleve Clin J Med 2009; 76:255–261.
- Abeles M. Fibromyalgia syndrome. In:Manu P, Editor: Functional Somatic Syndromes. New York: Cambridge University Press, 1998:32–57.
- Russell IJ, Orr MD, Littman B, et al. Elevated cerebrospinal fluid levels of substance P in patients with fibromyalgia syndrome. Arthritis Rheum 1994; 37:1593–1601.
- Abeles M, Abeles SR, Abeles AM. Fibromyalgia remains a controversial medical enigma [letter]. Am Fam Physician 2008; 77:1220.
- Abeles M, Abeles AM. Is pregabalin better than conventional therapy in fibromyalgia? [abstract] Arthritis Rheum 2008; 58( suppl):S396.
- Kim L, Lipton S, Deodhar A. Pregabalin for fibromyalgia: some relief but no cure. Cleve Clin J Med 2009; 76:255–261.
- Abeles M. Fibromyalgia syndrome. In:Manu P, Editor: Functional Somatic Syndromes. New York: Cambridge University Press, 1998:32–57.
- Russell IJ, Orr MD, Littman B, et al. Elevated cerebrospinal fluid levels of substance P in patients with fibromyalgia syndrome. Arthritis Rheum 1994; 37:1593–1601.
- Abeles M, Abeles SR, Abeles AM. Fibromyalgia remains a controversial medical enigma [letter]. Am Fam Physician 2008; 77:1220.
- Abeles M, Abeles AM. Is pregabalin better than conventional therapy in fibromyalgia? [abstract] Arthritis Rheum 2008; 58( suppl):S396.
In reply: Pregabalin for fibromyalgia
In Reply: We would like to thank the Drs. Abeles for reading our paper1 and providing valuable input. We are, however, surprised by their question as to who the “many” people are who believe that pregabalin is an important advance in the treatment of fibromyalgia. Anyone who is involved in taking care of fibromyalgia patients would know that several patients regularly report being helped by this medication to a varying degree. These patients rightly believe that this drug—the first drug approved by the US Food and Drug Administration for their oft-misunderstood condition—has started a much-needed dialogue in the medical community, and that in itself is a major advance.
We accept that Gowers, in his original paper on “fibrositis,” believed that fibrous tissue and not muscle was the source of inflammation in this condition.
We do believe that the paper by Russell et al2 was one of the many investigations that helped establish the role of central sensitization or abnormalities in pain processing in the central nervous system as the root cause of fibromyalgia pain. However, we do not believe our paper on pregabalin was the right place to discuss the merits or shortcomings of that paper in any more detail.
As we mentioned in our paper, therapies for fibromyalgia have limitations, and duloxetine and milnacipran are no exceptions. However, both these drugs were approved after our review was completed. We believe that the role of pregabalin in the treatment of fibromyalgia is going to be limited simply because medications overall form a small part of the comprehensive program of therapy for this condition.
- Kim L, Lipton S, Deodhar A. Pregabalin for fibromyalgia: some relief but no cure. Cleve Clin J Med 2009; 76:255–261.
- Russell IJ, Orr MD, Littman B, et al. Elevated cerebrospinal fluid levels of substance P in patients with fibromyalgia syndrome. Arthritis Rheum 1994; 37:1593–1601.
In Reply: We would like to thank the Drs. Abeles for reading our paper1 and providing valuable input. We are, however, surprised by their question as to who the “many” people are who believe that pregabalin is an important advance in the treatment of fibromyalgia. Anyone who is involved in taking care of fibromyalgia patients would know that several patients regularly report being helped by this medication to a varying degree. These patients rightly believe that this drug—the first drug approved by the US Food and Drug Administration for their oft-misunderstood condition—has started a much-needed dialogue in the medical community, and that in itself is a major advance.
We accept that Gowers, in his original paper on “fibrositis,” believed that fibrous tissue and not muscle was the source of inflammation in this condition.
We do believe that the paper by Russell et al2 was one of the many investigations that helped establish the role of central sensitization or abnormalities in pain processing in the central nervous system as the root cause of fibromyalgia pain. However, we do not believe our paper on pregabalin was the right place to discuss the merits or shortcomings of that paper in any more detail.
As we mentioned in our paper, therapies for fibromyalgia have limitations, and duloxetine and milnacipran are no exceptions. However, both these drugs were approved after our review was completed. We believe that the role of pregabalin in the treatment of fibromyalgia is going to be limited simply because medications overall form a small part of the comprehensive program of therapy for this condition.
In Reply: We would like to thank the Drs. Abeles for reading our paper1 and providing valuable input. We are, however, surprised by their question as to who the “many” people are who believe that pregabalin is an important advance in the treatment of fibromyalgia. Anyone who is involved in taking care of fibromyalgia patients would know that several patients regularly report being helped by this medication to a varying degree. These patients rightly believe that this drug—the first drug approved by the US Food and Drug Administration for their oft-misunderstood condition—has started a much-needed dialogue in the medical community, and that in itself is a major advance.
We accept that Gowers, in his original paper on “fibrositis,” believed that fibrous tissue and not muscle was the source of inflammation in this condition.
We do believe that the paper by Russell et al2 was one of the many investigations that helped establish the role of central sensitization or abnormalities in pain processing in the central nervous system as the root cause of fibromyalgia pain. However, we do not believe our paper on pregabalin was the right place to discuss the merits or shortcomings of that paper in any more detail.
As we mentioned in our paper, therapies for fibromyalgia have limitations, and duloxetine and milnacipran are no exceptions. However, both these drugs were approved after our review was completed. We believe that the role of pregabalin in the treatment of fibromyalgia is going to be limited simply because medications overall form a small part of the comprehensive program of therapy for this condition.
- Kim L, Lipton S, Deodhar A. Pregabalin for fibromyalgia: some relief but no cure. Cleve Clin J Med 2009; 76:255–261.
- Russell IJ, Orr MD, Littman B, et al. Elevated cerebrospinal fluid levels of substance P in patients with fibromyalgia syndrome. Arthritis Rheum 1994; 37:1593–1601.
- Kim L, Lipton S, Deodhar A. Pregabalin for fibromyalgia: some relief but no cure. Cleve Clin J Med 2009; 76:255–261.
- Russell IJ, Orr MD, Littman B, et al. Elevated cerebrospinal fluid levels of substance P in patients with fibromyalgia syndrome. Arthritis Rheum 1994; 37:1593–1601.
How menopause affects oral health, and what we can do about it
Menopause can bring oral health problems that physicians ought to keep in mind. The same processes that lead to loss of bone in the spine and hips can also lead to loss of the alveolar bone of the jaws, resulting in periodontal disease, loose teeth, and tooth loss. Although the mouth is traditionally the dentist’s responsibility, patients may need encouragement from their physicians to practice good oral hygiene and to see their dentists, and should be referred to a periodontist at the first sign of periodontal disease.
Moreover, bisphosphonates, the class of drugs most often prescribed for osteoporosis, have been linked by case reports (unfairly, we believe) to osteonecrosis of the jaw. This low-evidence-level information, its far-reaching interpretation, and misinformation in the lay media about hormonal changes associated with menopause have led to confusion among women; for clarification and reliable information, they are driven to ask their physicians challenging questions related to oral health.
This article reviews the published studies of the association between menopause and periodontal disease, specifically, the effects of hormonal changes, osteoporosis, and bisphosphonate use on the periodontal status of postmenopausal women. We will highlight the interrelationship of dental health and postmenopausal health and underscore the need for cross-communication and patient referral between physicians and dentists.
GINGIVITIS CAN PROGRESS TO PERIODONTITIS
AS ESTROGEN DECLINES, SO DO THE BONES AND, MAYBE, THE TEETH
The rate of bone loss in postmenopausal women predicts tooth loss—for every 1%-per-year decrease in whole-body bone mineral density, the risk of tooth loss increases more than four times.2 In fact, Kribbs3 found that women with severe osteoporosis were three times more likely than healthy, age-matched controls to be edentulous (ie, to have fewer teeth).
Although a number of studies have found that the density of the alveolar bone in the mandible correlated with the density of the bone in the rest of the skeleton and that generalized bone loss may render the jaw susceptible to accelerated alveolar bone resorption,3–11 these findings are not universal. In a longitudinal study, Famili et al12 found no association between systemic bone loss, periodontal disease, and edentulism. This shows that the relationship between alveolar bone loss and systemic bone loss is multifactorial and not yet fully understood.13
Nevertheless, the American Academy of Periodontology considers osteoporosis to be a risk factor for periodontal disease.10 In fact, alveolar bone loss has been related not only to osteoporosis but also to osteopenia.14
Bone mineral density has also been studied in relation to the loss of periodontal ligament—the collagenous attachment of tooth to bone. Klemetti et al15 found that healthy postmenopausal women with high bone mineral density seemed to retain teeth more readily than those with low bone density or those with osteoporosis, even if they had deep periodontal pockets (a sign of periodontal disease). These findings were reiterated when osteoporotic women were found to have significantly greater loss of attachment compared with nonosteoporotic women.7
However, Hildebolt16 reported that loss of tooth attachment correlated with tooth loss but not with the density of the vertebrae or the proximal femur. This study called into question the findings of the previous studies and provoked debate.
Tezal et al17 found that low bone mineral density was related to the loss of interproximal alveolar bone (the alveolar bone between adjacent teeth) and, to a lesser extent, ligamentous attachment loss. These data implicated osteoporosis as a possible risk indicator for periodontal disease in white women. (This study was limited to white women because of different demographics in the incidence of osteoporosis.)
Another study showed only a weak correlation between changes in alveolar bone height (in periodontal disease, bone height decreases) and attachment levels. Although a correlation might be present, the relationship was complex and required further examination. The authors found no clear association between clinical attachment levels and bone mineral density in the lumbar spine, but they recognized that attachment loss often precedes the loss of alveolar bone by a significant time period.13
Several studies have found a possible relationship between the bone density in the jaw and the density in the rest of the skeleton. It appears that loss of bone mineral density in the hip, wrist, and lumbar areas is correlated with low density in the mandible. Taguchi et al18 reported that the density in the lumbar spine correlated with the density of the mandibular cortex in early menopause, and with the density of both the cortex and cancellous bone in later menopause.
But whatever the statistical measurement, the susceptibility to progressive periodontitis increases after menopause, and the primary cause is bacterial plaque. The best hedge against this increased susceptibility is regular dental care to remove bacterial plaque biofilm under the gum-line.
HORMONE THERAPY PRESERVES BONE IN THE JAW
Hormones have long been recognized as having some role in periodontal disease.
Payne et al19 reported that postmenopausal women who were estrogen-deficient had a higher frequency of sites with a net loss of alveolar bone density at follow-up. Furthermore, estrogen-deficient women undergoing supportive periodontal therapy following treatment of moderate to severe periodontitis had three times as many sites losing more than 0.4 mm of interproximal alveolar bone height. Patients who had sufficient estrogen levels did not lose bone during 1 year of follow-up.20
Estrogen replacement improves bone density in postmenopausal women. In a 3-year randomized trial in postmenopausal women with moderate or advanced periodontal disease, estrogen therapy significantly increased alveolar bone mass compared with placebo (P = .04), and it increased bone density in the femur but not the lumbar spine.21 Furthermore, women receiving hormonal therapy had significantly less gingival inflammation, lower plaque scores, and less loss of attachment.
On the other hand, a report by Albandar and Kingman22 suggested that women who comply with hormonal therapy also comply with oral hygiene instructions. This compliance could explain the lower gingival inflammation scores, lower plaque scores, and lesser loss of attachment.
Norderyd et al,23 in a cross-sectional study, found less periodontal disease in postmenopausal women who were on estrogen therapy than in those who were not, although the difference was not statistically significant.
In a 5-year longitudinal study of 69 postmenopausal women receiving estrogen, a moderate but significant relationship was found between bone mineral density of the lumbar spine and the mandible, and estrogen replacement therapy had a positive effect on the mandibular bone mass.24
In a longitudinal study of 24 postmenopausal women, estrogen-deficient women had a mean net loss of alveolar bone density over time, while estrogen-sufficient women had a mean net gain, suggesting that estrogen deficiency may be a risk factor for alveolar bone loss.20 More-recent studies had similar findings. A cross-sectional study by Meisel et al25 found that hormone therapy significantly reduced the extent of clinical attachment loss and, hence, periodontal disease.
The findings of these studies are generally consistent, suggesting that estrogen builds up mandibular bone mass and attenuates the severity of periodontal disease in postmenopausal women.26
DOES ESTROGEN THERAPY PROTECT THE TEETH?
Studies of the Leisure World,27 Framingham,28 and Nurses Health Study29 cohorts suggest that hormone therapy protects against tooth loss in postmenopausal women.
On the other hand, Taguchi et al30 evaluated more than 300 postmenopausal Japanese women and found no significant difference in the total number of teeth between estrogen users and nonusers. The population in this study was younger than in the other studies mentioned above,27–29 which may explain the negative finding. However, the duration of estrogen use was significantly associated with the total number of teeth remaining, independent of age.30 Meisel et al25 reported that women receiving hormonal therapy had more teeth, though the difference was not significant.
CYTOKINES, PERIODONTITIS, AND SKELETAL BONE LOSS
Studies suggest that low estrogen production after menopause is associated with increased production of interleukin 1 (IL-1), IL-6, IL-8, IL-10, tumor necrosis factor alpha, granulocyte colony-stimulating factor, and granulocyte-macrophage colony-stimulating factor, which stimulate mature osteoclasts, modulate bone cell proliferation, and induce resorption of both skeletal and alveolar bone.31–34
In this regard, osteoporosis and periodontitis appear to be mediated by common cytokines. Managing osteoporosis, removing bacterial plaque biofilm with good oral hygiene, and regular dental visits are important in avoiding periodontitis in susceptible women.
BISPHOSPHONATES PROTECT BONE
In the skeleton
Bisphosphonates, the most commonly prescribed therapy for osteoporosis, inhibit systemic bone resorption and reduce the incidence of vertebral and nonvertebral fractures. Among the bisphosphonates, alendronate (Fosamax), risedronate (Actonel), and intravenous zoledronic acid (Reclast) have been shown to reduce the risk of both hip and vertebral fractures, whereas ibandronate (Boniva) has only been shown to decrease the risk of vertebral fracture.36 Specific findings:
- In the Fracture Intervention Trial,37 alendronate reduced the risk of vertebral fracture by 47% and hip fracture by 51% in women with low bone mineral density and previous vertebral fractures.
- In the Hip Intervention Program,38 risedronate decreased the risk of hip fracture by 40% in postmenopausal women 70 to 79 years old with osteoporosis, but not in those 80 years and older, who are at high risk of falls. Risedronate also reduced vertebral fracture risk by 49% after 3 years of treatment.39
- In the Health Outcomes and Reduced Incidence With Zoledronic Acid Once Yearly Recurrent Fracture Trial,40 annual infusion of zoledronic acid after a hip fracture reduced the rates of new clinical vertebral and nonvertebral fractures and death from all causes.
In the jaw
Not surprisingly, recent studies suggest that bisphosphonates slow the resorption of alveolar bone of the maxilla and mandible as well. Alendronate and risedronate, in particular, have been noted to improve periodontal status.41–43 Findings:
- In a cross-sectional study by Palomo et al,41 postmenopausal women with low bone density using risedronate for at least 3 months showed significantly less plaque accumulation, less gingival inflammation, lower probing-depth measurments, less periodontal attachment loss, and greater alveolar bone levels.
- In a double-blind, controlled, prospective study by Rocha et al,42 6 months of alendronate therapy significantly improved periodontal disease as assessed radiographically and clinically in 40 postmenopausal women with established periodontal disease.
- Jeffcoat et al43 reported that 2 years of alendronate treatment significantly reduced alveolar bone loss relative to placebo in patients with low mandibular bone mineral density at baseline but not in those with normal baseline mandibular bone mineral density.
DO BISPHOSPHONATES CAUSE OSTEONECROSIS OF THE JAW?
The intravenous bisphosphonates most commonly used to treat hypercalcemia of malignancy, multiple myeloma, or metastatic bone disease are47:
- Pamidronate (Aredia) 90 mg infused over 2 to 24 hours every 3 to 4 weeks
- Zoledronic acid (Zometa) 4 mg infused over 15 minutes monthly.
The doses of bisphosphonates indicated for the treatment of osteoporosis are much lower,1 eg:
- Alendronate 70 mg by mouth once a week
- Risedronate 35 mg by mouth once a week or 150 mg once a month
- Ibandronate 150 mg by mouth once a month
- Ibandronate 3 mg intravenously every 3 months
- Zoledronic acid 5 mg intravenously once a year.
Moreover, less than 1% of an oral dose is absorbed by the gastrointestinal tract,49 whereas more than 50% of the dose of bisphosphonates given intravenously is bioavailable,50 which may account for the lower incidence of jaw ostenonecrosis with oral agents.
Osteonecrosis of the jaw can occur spontaneously but is more often associated with dental procedures that traumatize bone, such as tooth extraction.51 In a systematic review,45 patients with multiple myeloma and metastatic cancer to the bone who were receiving intravenous bisphosphonates accounted for 94% of published cases. Sixty percent of cases were preceded by dental surgical procedures, and in 39% of cases that occurred spontaneously the lesions were located on bony exostoses, a possible source of trauma. Of 63 cases reported by Ruggiero et al,47 56 patients were receiving intravenous bisphosphonates and 7 were receiving oral bisphosphonates. Older age (> 65 years), chronic systemic steroid use, periodontitis, and prolonged use of bisphosphonates have also been associated with a higher risk of osteonecrosis of the jaw.51
The risk of developing osteonecrosis of the jaw in people taking bisphosphonates in doses recommended by the US Food and Drug Administration for treating osteoporosis is very low (the incidence is calculated at 0.7 per 100,000 person-years of exposure to alendronate).51,52 In a 3-year prospective study in more than 7,000 women with post-menopausal osteoporosis, the incidence of osteonecrosis of the jaw was no different in those treated with zoledronic acid 5 mg intravenously than in those receiving placebo.53 In a randomized, placebo-controlled study of the effect of 2 years of alendronate treatment on alveolar bone loss involving 335 patients with periodontal disease, no cases of osteonecrosis of the jaw were reported.43
The American Dental Association (ADA) released a statement noting that osteonecrosis of the jaw can occur with or without bisphosphonate use.51 To date, a true cause-and-effect relationship between osteonecrosis of the jaw and bisphosphonate use has not been established. Further studies are needed to fully explore this relationship. Our group is currently exploring novel periodontal assessments comparing the oral health of postmenopausal women with osteoporosis who are on no bone therapy vs postmenopausal women with osteoporosis treated with bisphosphonates for 2 or more years.

Discussion of treatment for bisphosphonate-associated osteonecrosis of the jaw is beyond the scope of this article.
REGULAR DENTAL CARE IS ESSENTIAL
Regardless of whether the patient is receiving a bisphosphonate drug, physicians caring for postmenopausal women should be vigilant and encourage their patients to seek regular dental evaluation for prevention and early management of oral disorders. Conversely, dentists should be aware of the potential effects of menopause and its treatments on bone and dental health.
Questions from postmenopausal women can be managed, in part, by returning to the basics suggested by the ADA:
- Regular dental examinations; regular professional cleaning to remove bacterial plaque biofilm under the gum-line where a toothbrush will not reach
- Daily oral hygiene practices to remove biofilm at and above the gum-line including brushing twice daily with an ADA-accepted toothpaste
- Replacing the toothbrush every 3 to 4 months (or sooner if the bristles begin to look frayed)
- Cleaning interproximally (between teeth) with floss or interdental cleaner
- Maintaining a balanced diet
- No smoking.
- North American Menopause Society. Menopause Practice: A Clinician’s Guide. 3rd ed; 2007.
- Krall EA, Garcia RI, Dawson-Hughes B. Increased risk of tooth loss is related to bone loss at the whole body, hip and spine. Calcif Tissue Int 1996; 59:433–437.
- Kribbs PJ. Comparison of mandibular bone in normal and osteoporotic women. J Prosthet Dent 1990; 63:218–222.
- Kribbs PJ, Chesnut CH, Ott SM, Kilcoyne RF. Relationship between mandibular and skeletal bone in an osteoporotic population. J Prosthet Dent 1989; 62:703–707.
- Kribbs PJ, Chestnut CH, Ott SM, Kilcyne RE. Relationship between mandibular and skeletal bone in a population of normal women. J Prosthet Dent 1990; 63:86–89.
- Kribbs PJ, Smith DE, Chestnut CH. Oral findings in osteoporosis. Part II: relationship between residual ridge and alveolar bone resorption and generalized skeletal osteopenia. J Prosthet Dent 1983; 50:719–724.
- von Wowern N, Klausen B, Kollerup G. Osteoporosis: a risk factor in periodontal disease. J Periodontol 1994; 65:1134–1138.
- Wactawski-Wende J, Grossi SG, Trevisan M, et al. The role of osteopenia in oral bone loss and periodontal disease. J Peridontol 1996; 67(suppl 10):1076–1084.
- Ronderos M, Jacobs DR, Himes JH, Pihlstrom BL. Associations of periodontal disease with femoral bone mineral density and estrogen replacement therapy: cross-sectional evaluation of US adults from the NHANES III. J Clin Periodontol 2000; 27:778–86.
- American Dental Association Council on Access, Prevention and Interprofessional Relations. Women’s Oral Health Issues. November 2006.
- Jeffcoat MK, Lewis CE, Reddy MS, Wang CY, Redford M. Post-menopausal bone loss and its relationship to oral bone loss. Periodontol 2000 2000; 23:94–102.
- Famili P, Cauley J, Suzuki JB, Weyant R. Longitudinal study of periodontal disease and edentulism with rates of bone loss in older women. J Periodontol 2005; 76:11–15.
- Pilgram TK, Hildebolt CF, Yokoyama N, et al. Relationships between longitudinal changes in radiographic alveolar bone height and probing depth measurements: data from postmenopausal women. J Periodontol 1999; 70:829–833.
- Jeffcoat MK, Lewis CE, Reddy MS, et al. Oral bone loss and systemic osteopenia, osteoporosis. InMarcus R, Feldman D, Kelsey J, editors. Osteoporosis. New York Academic Press 1996:969–990.
- Klemetti E, Collin HL, Forss H, Markkanen H, Lassila V. Mineral status of skeletal and advanced periodontal disease. J Clin Periodontol 1994; 21:184–188.
- Hildebolt CF. Osteoporosis and oral bone loss. Dentomaxillofac Radiol 1997; 26:3–15.
- Tezal M, Wactawski-Wende J, Grossi SG, Ho AW, Dunford R, Genco RJ. The relationship between bone mineral density and periodontitis in postmenopausal women. J Periodontol 2000; 71:1492–1498.
- Taguchi A, Tanimoto K, Suei Y, Ohama K, Wada T. Relationship between the mandibular and lumbar vertebral bone mineral density at different postmenopausal stages. Dentomaxillofac Radiol 1996; 25:130–135.
- Payne JB, Reinhardt RA, Nummikoski PV, Patil KD. Longitudinal alveolar bone loss in postmenopausal osteoporotic/osteopenic women. Osteoporos Int 1999; 10:34–40.
- Payne JB, Zachs NR, Reinhardt RA, Nummikoski PV, Patil K. The association between estrogen status and alveolar bone density changes in postmenopausal women with a history of periodontitis. J Periodontol 1997; 68:24–31.
- Civitelli R, Pilgram TK, Dotson M, et al. Alveolar and postcranial bone density in postmenopausal women receiving hormone/estrogen replacement: a randomized, double blind, placebo-controlled trial. Arch Intern Med 2002; 162:1409–1415.
- Albandar JM, Kingman A. Gingival recession, gingival bleeding, and dental calculus in adults 30 years of age and older in the United States, 1988–1994. J Periodontol 1999; 70:30–43.
- Norderyd OM, Grossi SG, Machtel EE, et al. Periodontal status of women taking postmenopausal estrogen supplementation. J Periodontol 1993; 64:957–962.
- Jacobs R, Ghyselen J, Koninckx P, van Steenberghe D. Long-term bone mass evaluation of mandible and lumbar spine in a group of women receiving hormone replacement therapy. Eur J Oral Sci 1996; 104:10–16.
- Meisel P, Reifenberger J, Haase R, Nauck M, Bandt C, Kocher T. Women are periodontally healthier than men, but why don’t they have more teeth than men? Menopause 2008; 15:270–275.
- Genco RJ, Grossi SG. Is estrogen deficiency a risk factor for periodontal disease? Compend Contin Educ Dent Suppl 1998; 22:S23–S29.
- Paganini-Hill A. The benefits of estrogen replacement therapy on oral health. The Leisure World cohort. Arch Intern Med 1995; 155:2325–2329.
- Krall EA, Dawson-Hughes B, Hannan MT, Wilson PW, Kiel DP. Post-menopausal estrogen replacement and tooth retention. Am J Med 1997; 102:536–542.
- Grodstein F, Colditz GA, Stampfer MJ. Postmenopausal hormone use and tooth loss: a prospective study. J Am Dent Assoc 1996; 127:370–377.
- Taguchi A, Sanada M, Suei Y, et al. Effect of estrogen use on tooth retention, oral bone height, and oral bone porosity in Japanese postmenopausal women. Menopause 2004; 11:556–562.
- Pacifici R. Estrogen, cytokines and pathogenesis of postmenopausal osteoporosis. J Bone Miner Res 1996; 11:1043–1051.
- Pacifici R. Is there a causal role for IL-1 in postmenopausal bone loss? Calcif Tissue Int 1992; 50:295–299.
- Girasole G, Jilka RL, Passeri G, et al. 17 beta-estradiol inhibits interleukin-6 production by bone marrow-derived stromal cells and osteoblasts in vitro: a potential mechanism for the antiosteoporotic effect of estrogens. J Clin Invest 1992; 89:883–891.
- Pacifici R, Brown C, Pusheck E, et al. Effect of surgical menopause and estrogen replacement on cytokine release from human blood mononuclear cells. Proc Natl Acad Sci USA 1991; 88:5134–5138.
- Brennan RM, Genco RJ, Wilding GE, Hovey KM, Trevisan M, Wactawski-Wende J. Bacterial species in subgingival plaque and oral bone loss in postmenopausal women. J Periodontol 2007; 78:1051–1061.
- Chestnut CH, Skag A, Christiansen C, et al. Effects of oral ibandronate administered daily or intermittently on fracture risk in post-menopausal osteoporosis. J Bone Miner Res 2004; 19:1241–1249.
- Black DM, Cummings SR, Karpf DB, et al. Randomized trial of effect of alendronate on risk of fracture in women with existing vertebral fractures: Fracture Intervention Trial Research Group. Lancet 1996; 348:1535–1541.
- McClung MR, Geusen P, Miller PD, et al. Effect of risedronate on the risk of hip fracture in elderly women. Hip Intervention Program Study Group. N Engl J Med 2001; 344:333–340.
- Reginster JY, Minne HW, Sorensen OH, et al. Randomized trial of effects of risedronate on vertebral fractures in women with established postmenopausal osteoporosis. Vertebral Efficacy with Risedronate Therapy (VERT) Study Group. Osteoporos Int 2000; 11:83–91.
- Lyles KW, Colon-Emeric CS, Magaziner JS, et al. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med 2007; 357:1799–1809.
- Palomo L, Bissada N, Liu J. Periodontal assessment of postmenopausal women receiving risedronate. Menopause 2005; 12:685–690.
- Rocha ML, Malacara JM, Sánchez-Marin FJ, Vazquez de la Torre CJ, Fajardo ME. Effect of alendronate on periodontal disease in postmenopausal women: a randomized placebo-controlled trial. J Periodontol 2004; 75:1579–1585.
- Jeffcoat MK, Cizza G, Shih WJ, Genco R, Lombardi A. Efficacy of bisphosphonates for the control of alveolar bone loss in periodontitis. J Int Acad Periodontol 2007; 9:70–76.
- Carey JJ, Palomo L. Bisphosphonates and osteonecrosis of the jaw: innocent association or significant risk? Cleve Clin J Med 2008; 75:871–879.
- Woo SB, Hellstein JW, Kalamare JR. Narrative [corrected] review: bisphosphonates and osteonecrosis of the jaws. Ann Intern Med 2006; 144:753–761.
- Dodson TB, Raje NS, Caruso PA, Rosenberg AE. Case records of the Massachusetts General Hospital. Case 9–2008. A 65-year-old woman with a nonhealing ulcer of the jaw. N Engl J Med 2008; 358:1283–1291.
- Ruggiero SL, Mehrotra B, Rosenberg TJ, Engroff SL. Osteonecrosis of the jaws associated with the use of bisphosphonates: a review of 63 cases. J Oral Maxillofacial Surg 2004; 62:527–534.
- Palomo L, Liu J, Bissada NF. Skeletal bone diseases impact the periodontium: a review of bisphosphonate therapy. Expert Opin Pharmacother 2007; 8:309–315.
- Ezra A, Golomb G. Administration routes and delivery systems of bisphosphonates for the treatment of bone resorption. Adv Drug Deliv Rev 2000; 42:175–195.
- Berenson JR, Rosen L, Vescio R, et al. Pharmacokinetics of pamidronate disodium in patients with cancer with normal or impaired renal function. J Clin Pharmacol 1997; 37:285–290.
- American Dental Association Council on Scientific Affairs. Dental management of patients receiving oral bisphosphonate therapy: expert panel recommendations. J Am Dent Assoc 2006; 137:1144–1150.
- Advisory Task Force on Bisphosphonate-Related Ostenonecrosis of the Jaws. American Association of Oral and Maxillofacial Surgeons position paper on bisphosphonate-related osteonecrosis of the jaws. J Oral Maxillofacial Surg 2007; 65:369–376.
- Grbic JT, Landesberg R, Lin SQ, et al; Health Outcomes and Reduced Incidence with Zoledronic Acid Once Yearly Pivotal Fracture Trial Research Group. Incidence of osteonecrosis of the jaw in women with postmenopausal osteoporosis in the Health Outcomes and Reduced Incidence with Zoledronic Acid Once Yearly Pivotal Fracture Trial. J Am Dent Assoc 2008; 139:32–40.
Menopause can bring oral health problems that physicians ought to keep in mind. The same processes that lead to loss of bone in the spine and hips can also lead to loss of the alveolar bone of the jaws, resulting in periodontal disease, loose teeth, and tooth loss. Although the mouth is traditionally the dentist’s responsibility, patients may need encouragement from their physicians to practice good oral hygiene and to see their dentists, and should be referred to a periodontist at the first sign of periodontal disease.
Moreover, bisphosphonates, the class of drugs most often prescribed for osteoporosis, have been linked by case reports (unfairly, we believe) to osteonecrosis of the jaw. This low-evidence-level information, its far-reaching interpretation, and misinformation in the lay media about hormonal changes associated with menopause have led to confusion among women; for clarification and reliable information, they are driven to ask their physicians challenging questions related to oral health.
This article reviews the published studies of the association between menopause and periodontal disease, specifically, the effects of hormonal changes, osteoporosis, and bisphosphonate use on the periodontal status of postmenopausal women. We will highlight the interrelationship of dental health and postmenopausal health and underscore the need for cross-communication and patient referral between physicians and dentists.
GINGIVITIS CAN PROGRESS TO PERIODONTITIS
AS ESTROGEN DECLINES, SO DO THE BONES AND, MAYBE, THE TEETH
The rate of bone loss in postmenopausal women predicts tooth loss—for every 1%-per-year decrease in whole-body bone mineral density, the risk of tooth loss increases more than four times.2 In fact, Kribbs3 found that women with severe osteoporosis were three times more likely than healthy, age-matched controls to be edentulous (ie, to have fewer teeth).
Although a number of studies have found that the density of the alveolar bone in the mandible correlated with the density of the bone in the rest of the skeleton and that generalized bone loss may render the jaw susceptible to accelerated alveolar bone resorption,3–11 these findings are not universal. In a longitudinal study, Famili et al12 found no association between systemic bone loss, periodontal disease, and edentulism. This shows that the relationship between alveolar bone loss and systemic bone loss is multifactorial and not yet fully understood.13
Nevertheless, the American Academy of Periodontology considers osteoporosis to be a risk factor for periodontal disease.10 In fact, alveolar bone loss has been related not only to osteoporosis but also to osteopenia.14
Bone mineral density has also been studied in relation to the loss of periodontal ligament—the collagenous attachment of tooth to bone. Klemetti et al15 found that healthy postmenopausal women with high bone mineral density seemed to retain teeth more readily than those with low bone density or those with osteoporosis, even if they had deep periodontal pockets (a sign of periodontal disease). These findings were reiterated when osteoporotic women were found to have significantly greater loss of attachment compared with nonosteoporotic women.7
However, Hildebolt16 reported that loss of tooth attachment correlated with tooth loss but not with the density of the vertebrae or the proximal femur. This study called into question the findings of the previous studies and provoked debate.
Tezal et al17 found that low bone mineral density was related to the loss of interproximal alveolar bone (the alveolar bone between adjacent teeth) and, to a lesser extent, ligamentous attachment loss. These data implicated osteoporosis as a possible risk indicator for periodontal disease in white women. (This study was limited to white women because of different demographics in the incidence of osteoporosis.)
Another study showed only a weak correlation between changes in alveolar bone height (in periodontal disease, bone height decreases) and attachment levels. Although a correlation might be present, the relationship was complex and required further examination. The authors found no clear association between clinical attachment levels and bone mineral density in the lumbar spine, but they recognized that attachment loss often precedes the loss of alveolar bone by a significant time period.13
Several studies have found a possible relationship between the bone density in the jaw and the density in the rest of the skeleton. It appears that loss of bone mineral density in the hip, wrist, and lumbar areas is correlated with low density in the mandible. Taguchi et al18 reported that the density in the lumbar spine correlated with the density of the mandibular cortex in early menopause, and with the density of both the cortex and cancellous bone in later menopause.
But whatever the statistical measurement, the susceptibility to progressive periodontitis increases after menopause, and the primary cause is bacterial plaque. The best hedge against this increased susceptibility is regular dental care to remove bacterial plaque biofilm under the gum-line.
HORMONE THERAPY PRESERVES BONE IN THE JAW
Hormones have long been recognized as having some role in periodontal disease.
Payne et al19 reported that postmenopausal women who were estrogen-deficient had a higher frequency of sites with a net loss of alveolar bone density at follow-up. Furthermore, estrogen-deficient women undergoing supportive periodontal therapy following treatment of moderate to severe periodontitis had three times as many sites losing more than 0.4 mm of interproximal alveolar bone height. Patients who had sufficient estrogen levels did not lose bone during 1 year of follow-up.20
Estrogen replacement improves bone density in postmenopausal women. In a 3-year randomized trial in postmenopausal women with moderate or advanced periodontal disease, estrogen therapy significantly increased alveolar bone mass compared with placebo (P = .04), and it increased bone density in the femur but not the lumbar spine.21 Furthermore, women receiving hormonal therapy had significantly less gingival inflammation, lower plaque scores, and less loss of attachment.
On the other hand, a report by Albandar and Kingman22 suggested that women who comply with hormonal therapy also comply with oral hygiene instructions. This compliance could explain the lower gingival inflammation scores, lower plaque scores, and lesser loss of attachment.
Norderyd et al,23 in a cross-sectional study, found less periodontal disease in postmenopausal women who were on estrogen therapy than in those who were not, although the difference was not statistically significant.
In a 5-year longitudinal study of 69 postmenopausal women receiving estrogen, a moderate but significant relationship was found between bone mineral density of the lumbar spine and the mandible, and estrogen replacement therapy had a positive effect on the mandibular bone mass.24
In a longitudinal study of 24 postmenopausal women, estrogen-deficient women had a mean net loss of alveolar bone density over time, while estrogen-sufficient women had a mean net gain, suggesting that estrogen deficiency may be a risk factor for alveolar bone loss.20 More-recent studies had similar findings. A cross-sectional study by Meisel et al25 found that hormone therapy significantly reduced the extent of clinical attachment loss and, hence, periodontal disease.
The findings of these studies are generally consistent, suggesting that estrogen builds up mandibular bone mass and attenuates the severity of periodontal disease in postmenopausal women.26
DOES ESTROGEN THERAPY PROTECT THE TEETH?
Studies of the Leisure World,27 Framingham,28 and Nurses Health Study29 cohorts suggest that hormone therapy protects against tooth loss in postmenopausal women.
On the other hand, Taguchi et al30 evaluated more than 300 postmenopausal Japanese women and found no significant difference in the total number of teeth between estrogen users and nonusers. The population in this study was younger than in the other studies mentioned above,27–29 which may explain the negative finding. However, the duration of estrogen use was significantly associated with the total number of teeth remaining, independent of age.30 Meisel et al25 reported that women receiving hormonal therapy had more teeth, though the difference was not significant.
CYTOKINES, PERIODONTITIS, AND SKELETAL BONE LOSS
Studies suggest that low estrogen production after menopause is associated with increased production of interleukin 1 (IL-1), IL-6, IL-8, IL-10, tumor necrosis factor alpha, granulocyte colony-stimulating factor, and granulocyte-macrophage colony-stimulating factor, which stimulate mature osteoclasts, modulate bone cell proliferation, and induce resorption of both skeletal and alveolar bone.31–34
In this regard, osteoporosis and periodontitis appear to be mediated by common cytokines. Managing osteoporosis, removing bacterial plaque biofilm with good oral hygiene, and regular dental visits are important in avoiding periodontitis in susceptible women.
BISPHOSPHONATES PROTECT BONE
In the skeleton
Bisphosphonates, the most commonly prescribed therapy for osteoporosis, inhibit systemic bone resorption and reduce the incidence of vertebral and nonvertebral fractures. Among the bisphosphonates, alendronate (Fosamax), risedronate (Actonel), and intravenous zoledronic acid (Reclast) have been shown to reduce the risk of both hip and vertebral fractures, whereas ibandronate (Boniva) has only been shown to decrease the risk of vertebral fracture.36 Specific findings:
- In the Fracture Intervention Trial,37 alendronate reduced the risk of vertebral fracture by 47% and hip fracture by 51% in women with low bone mineral density and previous vertebral fractures.
- In the Hip Intervention Program,38 risedronate decreased the risk of hip fracture by 40% in postmenopausal women 70 to 79 years old with osteoporosis, but not in those 80 years and older, who are at high risk of falls. Risedronate also reduced vertebral fracture risk by 49% after 3 years of treatment.39
- In the Health Outcomes and Reduced Incidence With Zoledronic Acid Once Yearly Recurrent Fracture Trial,40 annual infusion of zoledronic acid after a hip fracture reduced the rates of new clinical vertebral and nonvertebral fractures and death from all causes.
In the jaw
Not surprisingly, recent studies suggest that bisphosphonates slow the resorption of alveolar bone of the maxilla and mandible as well. Alendronate and risedronate, in particular, have been noted to improve periodontal status.41–43 Findings:
- In a cross-sectional study by Palomo et al,41 postmenopausal women with low bone density using risedronate for at least 3 months showed significantly less plaque accumulation, less gingival inflammation, lower probing-depth measurments, less periodontal attachment loss, and greater alveolar bone levels.
- In a double-blind, controlled, prospective study by Rocha et al,42 6 months of alendronate therapy significantly improved periodontal disease as assessed radiographically and clinically in 40 postmenopausal women with established periodontal disease.
- Jeffcoat et al43 reported that 2 years of alendronate treatment significantly reduced alveolar bone loss relative to placebo in patients with low mandibular bone mineral density at baseline but not in those with normal baseline mandibular bone mineral density.
DO BISPHOSPHONATES CAUSE OSTEONECROSIS OF THE JAW?
The intravenous bisphosphonates most commonly used to treat hypercalcemia of malignancy, multiple myeloma, or metastatic bone disease are47:
- Pamidronate (Aredia) 90 mg infused over 2 to 24 hours every 3 to 4 weeks
- Zoledronic acid (Zometa) 4 mg infused over 15 minutes monthly.
The doses of bisphosphonates indicated for the treatment of osteoporosis are much lower,1 eg:
- Alendronate 70 mg by mouth once a week
- Risedronate 35 mg by mouth once a week or 150 mg once a month
- Ibandronate 150 mg by mouth once a month
- Ibandronate 3 mg intravenously every 3 months
- Zoledronic acid 5 mg intravenously once a year.
Moreover, less than 1% of an oral dose is absorbed by the gastrointestinal tract,49 whereas more than 50% of the dose of bisphosphonates given intravenously is bioavailable,50 which may account for the lower incidence of jaw ostenonecrosis with oral agents.
Osteonecrosis of the jaw can occur spontaneously but is more often associated with dental procedures that traumatize bone, such as tooth extraction.51 In a systematic review,45 patients with multiple myeloma and metastatic cancer to the bone who were receiving intravenous bisphosphonates accounted for 94% of published cases. Sixty percent of cases were preceded by dental surgical procedures, and in 39% of cases that occurred spontaneously the lesions were located on bony exostoses, a possible source of trauma. Of 63 cases reported by Ruggiero et al,47 56 patients were receiving intravenous bisphosphonates and 7 were receiving oral bisphosphonates. Older age (> 65 years), chronic systemic steroid use, periodontitis, and prolonged use of bisphosphonates have also been associated with a higher risk of osteonecrosis of the jaw.51
The risk of developing osteonecrosis of the jaw in people taking bisphosphonates in doses recommended by the US Food and Drug Administration for treating osteoporosis is very low (the incidence is calculated at 0.7 per 100,000 person-years of exposure to alendronate).51,52 In a 3-year prospective study in more than 7,000 women with post-menopausal osteoporosis, the incidence of osteonecrosis of the jaw was no different in those treated with zoledronic acid 5 mg intravenously than in those receiving placebo.53 In a randomized, placebo-controlled study of the effect of 2 years of alendronate treatment on alveolar bone loss involving 335 patients with periodontal disease, no cases of osteonecrosis of the jaw were reported.43
The American Dental Association (ADA) released a statement noting that osteonecrosis of the jaw can occur with or without bisphosphonate use.51 To date, a true cause-and-effect relationship between osteonecrosis of the jaw and bisphosphonate use has not been established. Further studies are needed to fully explore this relationship. Our group is currently exploring novel periodontal assessments comparing the oral health of postmenopausal women with osteoporosis who are on no bone therapy vs postmenopausal women with osteoporosis treated with bisphosphonates for 2 or more years.
Discussion of treatment for bisphosphonate-associated osteonecrosis of the jaw is beyond the scope of this article.
REGULAR DENTAL CARE IS ESSENTIAL
Regardless of whether the patient is receiving a bisphosphonate drug, physicians caring for postmenopausal women should be vigilant and encourage their patients to seek regular dental evaluation for prevention and early management of oral disorders. Conversely, dentists should be aware of the potential effects of menopause and its treatments on bone and dental health.
Questions from postmenopausal women can be managed, in part, by returning to the basics suggested by the ADA:
- Regular dental examinations; regular professional cleaning to remove bacterial plaque biofilm under the gum-line where a toothbrush will not reach
- Daily oral hygiene practices to remove biofilm at and above the gum-line including brushing twice daily with an ADA-accepted toothpaste
- Replacing the toothbrush every 3 to 4 months (or sooner if the bristles begin to look frayed)
- Cleaning interproximally (between teeth) with floss or interdental cleaner
- Maintaining a balanced diet
- No smoking.
Menopause can bring oral health problems that physicians ought to keep in mind. The same processes that lead to loss of bone in the spine and hips can also lead to loss of the alveolar bone of the jaws, resulting in periodontal disease, loose teeth, and tooth loss. Although the mouth is traditionally the dentist’s responsibility, patients may need encouragement from their physicians to practice good oral hygiene and to see their dentists, and should be referred to a periodontist at the first sign of periodontal disease.
Moreover, bisphosphonates, the class of drugs most often prescribed for osteoporosis, have been linked by case reports (unfairly, we believe) to osteonecrosis of the jaw. This low-evidence-level information, its far-reaching interpretation, and misinformation in the lay media about hormonal changes associated with menopause have led to confusion among women; for clarification and reliable information, they are driven to ask their physicians challenging questions related to oral health.
This article reviews the published studies of the association between menopause and periodontal disease, specifically, the effects of hormonal changes, osteoporosis, and bisphosphonate use on the periodontal status of postmenopausal women. We will highlight the interrelationship of dental health and postmenopausal health and underscore the need for cross-communication and patient referral between physicians and dentists.
GINGIVITIS CAN PROGRESS TO PERIODONTITIS
AS ESTROGEN DECLINES, SO DO THE BONES AND, MAYBE, THE TEETH
The rate of bone loss in postmenopausal women predicts tooth loss—for every 1%-per-year decrease in whole-body bone mineral density, the risk of tooth loss increases more than four times.2 In fact, Kribbs3 found that women with severe osteoporosis were three times more likely than healthy, age-matched controls to be edentulous (ie, to have fewer teeth).
Although a number of studies have found that the density of the alveolar bone in the mandible correlated with the density of the bone in the rest of the skeleton and that generalized bone loss may render the jaw susceptible to accelerated alveolar bone resorption,3–11 these findings are not universal. In a longitudinal study, Famili et al12 found no association between systemic bone loss, periodontal disease, and edentulism. This shows that the relationship between alveolar bone loss and systemic bone loss is multifactorial and not yet fully understood.13
Nevertheless, the American Academy of Periodontology considers osteoporosis to be a risk factor for periodontal disease.10 In fact, alveolar bone loss has been related not only to osteoporosis but also to osteopenia.14
Bone mineral density has also been studied in relation to the loss of periodontal ligament—the collagenous attachment of tooth to bone. Klemetti et al15 found that healthy postmenopausal women with high bone mineral density seemed to retain teeth more readily than those with low bone density or those with osteoporosis, even if they had deep periodontal pockets (a sign of periodontal disease). These findings were reiterated when osteoporotic women were found to have significantly greater loss of attachment compared with nonosteoporotic women.7
However, Hildebolt16 reported that loss of tooth attachment correlated with tooth loss but not with the density of the vertebrae or the proximal femur. This study called into question the findings of the previous studies and provoked debate.
Tezal et al17 found that low bone mineral density was related to the loss of interproximal alveolar bone (the alveolar bone between adjacent teeth) and, to a lesser extent, ligamentous attachment loss. These data implicated osteoporosis as a possible risk indicator for periodontal disease in white women. (This study was limited to white women because of different demographics in the incidence of osteoporosis.)
Another study showed only a weak correlation between changes in alveolar bone height (in periodontal disease, bone height decreases) and attachment levels. Although a correlation might be present, the relationship was complex and required further examination. The authors found no clear association between clinical attachment levels and bone mineral density in the lumbar spine, but they recognized that attachment loss often precedes the loss of alveolar bone by a significant time period.13
Several studies have found a possible relationship between the bone density in the jaw and the density in the rest of the skeleton. It appears that loss of bone mineral density in the hip, wrist, and lumbar areas is correlated with low density in the mandible. Taguchi et al18 reported that the density in the lumbar spine correlated with the density of the mandibular cortex in early menopause, and with the density of both the cortex and cancellous bone in later menopause.
But whatever the statistical measurement, the susceptibility to progressive periodontitis increases after menopause, and the primary cause is bacterial plaque. The best hedge against this increased susceptibility is regular dental care to remove bacterial plaque biofilm under the gum-line.
HORMONE THERAPY PRESERVES BONE IN THE JAW
Hormones have long been recognized as having some role in periodontal disease.
Payne et al19 reported that postmenopausal women who were estrogen-deficient had a higher frequency of sites with a net loss of alveolar bone density at follow-up. Furthermore, estrogen-deficient women undergoing supportive periodontal therapy following treatment of moderate to severe periodontitis had three times as many sites losing more than 0.4 mm of interproximal alveolar bone height. Patients who had sufficient estrogen levels did not lose bone during 1 year of follow-up.20
Estrogen replacement improves bone density in postmenopausal women. In a 3-year randomized trial in postmenopausal women with moderate or advanced periodontal disease, estrogen therapy significantly increased alveolar bone mass compared with placebo (P = .04), and it increased bone density in the femur but not the lumbar spine.21 Furthermore, women receiving hormonal therapy had significantly less gingival inflammation, lower plaque scores, and less loss of attachment.
On the other hand, a report by Albandar and Kingman22 suggested that women who comply with hormonal therapy also comply with oral hygiene instructions. This compliance could explain the lower gingival inflammation scores, lower plaque scores, and lesser loss of attachment.
Norderyd et al,23 in a cross-sectional study, found less periodontal disease in postmenopausal women who were on estrogen therapy than in those who were not, although the difference was not statistically significant.
In a 5-year longitudinal study of 69 postmenopausal women receiving estrogen, a moderate but significant relationship was found between bone mineral density of the lumbar spine and the mandible, and estrogen replacement therapy had a positive effect on the mandibular bone mass.24
In a longitudinal study of 24 postmenopausal women, estrogen-deficient women had a mean net loss of alveolar bone density over time, while estrogen-sufficient women had a mean net gain, suggesting that estrogen deficiency may be a risk factor for alveolar bone loss.20 More-recent studies had similar findings. A cross-sectional study by Meisel et al25 found that hormone therapy significantly reduced the extent of clinical attachment loss and, hence, periodontal disease.
The findings of these studies are generally consistent, suggesting that estrogen builds up mandibular bone mass and attenuates the severity of periodontal disease in postmenopausal women.26
DOES ESTROGEN THERAPY PROTECT THE TEETH?
Studies of the Leisure World,27 Framingham,28 and Nurses Health Study29 cohorts suggest that hormone therapy protects against tooth loss in postmenopausal women.
On the other hand, Taguchi et al30 evaluated more than 300 postmenopausal Japanese women and found no significant difference in the total number of teeth between estrogen users and nonusers. The population in this study was younger than in the other studies mentioned above,27–29 which may explain the negative finding. However, the duration of estrogen use was significantly associated with the total number of teeth remaining, independent of age.30 Meisel et al25 reported that women receiving hormonal therapy had more teeth, though the difference was not significant.
CYTOKINES, PERIODONTITIS, AND SKELETAL BONE LOSS
Studies suggest that low estrogen production after menopause is associated with increased production of interleukin 1 (IL-1), IL-6, IL-8, IL-10, tumor necrosis factor alpha, granulocyte colony-stimulating factor, and granulocyte-macrophage colony-stimulating factor, which stimulate mature osteoclasts, modulate bone cell proliferation, and induce resorption of both skeletal and alveolar bone.31–34
In this regard, osteoporosis and periodontitis appear to be mediated by common cytokines. Managing osteoporosis, removing bacterial plaque biofilm with good oral hygiene, and regular dental visits are important in avoiding periodontitis in susceptible women.
BISPHOSPHONATES PROTECT BONE
In the skeleton
Bisphosphonates, the most commonly prescribed therapy for osteoporosis, inhibit systemic bone resorption and reduce the incidence of vertebral and nonvertebral fractures. Among the bisphosphonates, alendronate (Fosamax), risedronate (Actonel), and intravenous zoledronic acid (Reclast) have been shown to reduce the risk of both hip and vertebral fractures, whereas ibandronate (Boniva) has only been shown to decrease the risk of vertebral fracture.36 Specific findings:
- In the Fracture Intervention Trial,37 alendronate reduced the risk of vertebral fracture by 47% and hip fracture by 51% in women with low bone mineral density and previous vertebral fractures.
- In the Hip Intervention Program,38 risedronate decreased the risk of hip fracture by 40% in postmenopausal women 70 to 79 years old with osteoporosis, but not in those 80 years and older, who are at high risk of falls. Risedronate also reduced vertebral fracture risk by 49% after 3 years of treatment.39
- In the Health Outcomes and Reduced Incidence With Zoledronic Acid Once Yearly Recurrent Fracture Trial,40 annual infusion of zoledronic acid after a hip fracture reduced the rates of new clinical vertebral and nonvertebral fractures and death from all causes.
In the jaw
Not surprisingly, recent studies suggest that bisphosphonates slow the resorption of alveolar bone of the maxilla and mandible as well. Alendronate and risedronate, in particular, have been noted to improve periodontal status.41–43 Findings:
- In a cross-sectional study by Palomo et al,41 postmenopausal women with low bone density using risedronate for at least 3 months showed significantly less plaque accumulation, less gingival inflammation, lower probing-depth measurments, less periodontal attachment loss, and greater alveolar bone levels.
- In a double-blind, controlled, prospective study by Rocha et al,42 6 months of alendronate therapy significantly improved periodontal disease as assessed radiographically and clinically in 40 postmenopausal women with established periodontal disease.
- Jeffcoat et al43 reported that 2 years of alendronate treatment significantly reduced alveolar bone loss relative to placebo in patients with low mandibular bone mineral density at baseline but not in those with normal baseline mandibular bone mineral density.
DO BISPHOSPHONATES CAUSE OSTEONECROSIS OF THE JAW?
The intravenous bisphosphonates most commonly used to treat hypercalcemia of malignancy, multiple myeloma, or metastatic bone disease are47:
- Pamidronate (Aredia) 90 mg infused over 2 to 24 hours every 3 to 4 weeks
- Zoledronic acid (Zometa) 4 mg infused over 15 minutes monthly.
The doses of bisphosphonates indicated for the treatment of osteoporosis are much lower,1 eg:
- Alendronate 70 mg by mouth once a week
- Risedronate 35 mg by mouth once a week or 150 mg once a month
- Ibandronate 150 mg by mouth once a month
- Ibandronate 3 mg intravenously every 3 months
- Zoledronic acid 5 mg intravenously once a year.
Moreover, less than 1% of an oral dose is absorbed by the gastrointestinal tract,49 whereas more than 50% of the dose of bisphosphonates given intravenously is bioavailable,50 which may account for the lower incidence of jaw ostenonecrosis with oral agents.
Osteonecrosis of the jaw can occur spontaneously but is more often associated with dental procedures that traumatize bone, such as tooth extraction.51 In a systematic review,45 patients with multiple myeloma and metastatic cancer to the bone who were receiving intravenous bisphosphonates accounted for 94% of published cases. Sixty percent of cases were preceded by dental surgical procedures, and in 39% of cases that occurred spontaneously the lesions were located on bony exostoses, a possible source of trauma. Of 63 cases reported by Ruggiero et al,47 56 patients were receiving intravenous bisphosphonates and 7 were receiving oral bisphosphonates. Older age (> 65 years), chronic systemic steroid use, periodontitis, and prolonged use of bisphosphonates have also been associated with a higher risk of osteonecrosis of the jaw.51
The risk of developing osteonecrosis of the jaw in people taking bisphosphonates in doses recommended by the US Food and Drug Administration for treating osteoporosis is very low (the incidence is calculated at 0.7 per 100,000 person-years of exposure to alendronate).51,52 In a 3-year prospective study in more than 7,000 women with post-menopausal osteoporosis, the incidence of osteonecrosis of the jaw was no different in those treated with zoledronic acid 5 mg intravenously than in those receiving placebo.53 In a randomized, placebo-controlled study of the effect of 2 years of alendronate treatment on alveolar bone loss involving 335 patients with periodontal disease, no cases of osteonecrosis of the jaw were reported.43
The American Dental Association (ADA) released a statement noting that osteonecrosis of the jaw can occur with or without bisphosphonate use.51 To date, a true cause-and-effect relationship between osteonecrosis of the jaw and bisphosphonate use has not been established. Further studies are needed to fully explore this relationship. Our group is currently exploring novel periodontal assessments comparing the oral health of postmenopausal women with osteoporosis who are on no bone therapy vs postmenopausal women with osteoporosis treated with bisphosphonates for 2 or more years.
Discussion of treatment for bisphosphonate-associated osteonecrosis of the jaw is beyond the scope of this article.
REGULAR DENTAL CARE IS ESSENTIAL
Regardless of whether the patient is receiving a bisphosphonate drug, physicians caring for postmenopausal women should be vigilant and encourage their patients to seek regular dental evaluation for prevention and early management of oral disorders. Conversely, dentists should be aware of the potential effects of menopause and its treatments on bone and dental health.
Questions from postmenopausal women can be managed, in part, by returning to the basics suggested by the ADA:
- Regular dental examinations; regular professional cleaning to remove bacterial plaque biofilm under the gum-line where a toothbrush will not reach
- Daily oral hygiene practices to remove biofilm at and above the gum-line including brushing twice daily with an ADA-accepted toothpaste
- Replacing the toothbrush every 3 to 4 months (or sooner if the bristles begin to look frayed)
- Cleaning interproximally (between teeth) with floss or interdental cleaner
- Maintaining a balanced diet
- No smoking.
- North American Menopause Society. Menopause Practice: A Clinician’s Guide. 3rd ed; 2007.
- Krall EA, Garcia RI, Dawson-Hughes B. Increased risk of tooth loss is related to bone loss at the whole body, hip and spine. Calcif Tissue Int 1996; 59:433–437.
- Kribbs PJ. Comparison of mandibular bone in normal and osteoporotic women. J Prosthet Dent 1990; 63:218–222.
- Kribbs PJ, Chesnut CH, Ott SM, Kilcoyne RF. Relationship between mandibular and skeletal bone in an osteoporotic population. J Prosthet Dent 1989; 62:703–707.
- Kribbs PJ, Chestnut CH, Ott SM, Kilcyne RE. Relationship between mandibular and skeletal bone in a population of normal women. J Prosthet Dent 1990; 63:86–89.
- Kribbs PJ, Smith DE, Chestnut CH. Oral findings in osteoporosis. Part II: relationship between residual ridge and alveolar bone resorption and generalized skeletal osteopenia. J Prosthet Dent 1983; 50:719–724.
- von Wowern N, Klausen B, Kollerup G. Osteoporosis: a risk factor in periodontal disease. J Periodontol 1994; 65:1134–1138.
- Wactawski-Wende J, Grossi SG, Trevisan M, et al. The role of osteopenia in oral bone loss and periodontal disease. J Peridontol 1996; 67(suppl 10):1076–1084.
- Ronderos M, Jacobs DR, Himes JH, Pihlstrom BL. Associations of periodontal disease with femoral bone mineral density and estrogen replacement therapy: cross-sectional evaluation of US adults from the NHANES III. J Clin Periodontol 2000; 27:778–86.
- American Dental Association Council on Access, Prevention and Interprofessional Relations. Women’s Oral Health Issues. November 2006.
- Jeffcoat MK, Lewis CE, Reddy MS, Wang CY, Redford M. Post-menopausal bone loss and its relationship to oral bone loss. Periodontol 2000 2000; 23:94–102.
- Famili P, Cauley J, Suzuki JB, Weyant R. Longitudinal study of periodontal disease and edentulism with rates of bone loss in older women. J Periodontol 2005; 76:11–15.
- Pilgram TK, Hildebolt CF, Yokoyama N, et al. Relationships between longitudinal changes in radiographic alveolar bone height and probing depth measurements: data from postmenopausal women. J Periodontol 1999; 70:829–833.
- Jeffcoat MK, Lewis CE, Reddy MS, et al. Oral bone loss and systemic osteopenia, osteoporosis. InMarcus R, Feldman D, Kelsey J, editors. Osteoporosis. New York Academic Press 1996:969–990.
- Klemetti E, Collin HL, Forss H, Markkanen H, Lassila V. Mineral status of skeletal and advanced periodontal disease. J Clin Periodontol 1994; 21:184–188.
- Hildebolt CF. Osteoporosis and oral bone loss. Dentomaxillofac Radiol 1997; 26:3–15.
- Tezal M, Wactawski-Wende J, Grossi SG, Ho AW, Dunford R, Genco RJ. The relationship between bone mineral density and periodontitis in postmenopausal women. J Periodontol 2000; 71:1492–1498.
- Taguchi A, Tanimoto K, Suei Y, Ohama K, Wada T. Relationship between the mandibular and lumbar vertebral bone mineral density at different postmenopausal stages. Dentomaxillofac Radiol 1996; 25:130–135.
- Payne JB, Reinhardt RA, Nummikoski PV, Patil KD. Longitudinal alveolar bone loss in postmenopausal osteoporotic/osteopenic women. Osteoporos Int 1999; 10:34–40.
- Payne JB, Zachs NR, Reinhardt RA, Nummikoski PV, Patil K. The association between estrogen status and alveolar bone density changes in postmenopausal women with a history of periodontitis. J Periodontol 1997; 68:24–31.
- Civitelli R, Pilgram TK, Dotson M, et al. Alveolar and postcranial bone density in postmenopausal women receiving hormone/estrogen replacement: a randomized, double blind, placebo-controlled trial. Arch Intern Med 2002; 162:1409–1415.
- Albandar JM, Kingman A. Gingival recession, gingival bleeding, and dental calculus in adults 30 years of age and older in the United States, 1988–1994. J Periodontol 1999; 70:30–43.
- Norderyd OM, Grossi SG, Machtel EE, et al. Periodontal status of women taking postmenopausal estrogen supplementation. J Periodontol 1993; 64:957–962.
- Jacobs R, Ghyselen J, Koninckx P, van Steenberghe D. Long-term bone mass evaluation of mandible and lumbar spine in a group of women receiving hormone replacement therapy. Eur J Oral Sci 1996; 104:10–16.
- Meisel P, Reifenberger J, Haase R, Nauck M, Bandt C, Kocher T. Women are periodontally healthier than men, but why don’t they have more teeth than men? Menopause 2008; 15:270–275.
- Genco RJ, Grossi SG. Is estrogen deficiency a risk factor for periodontal disease? Compend Contin Educ Dent Suppl 1998; 22:S23–S29.
- Paganini-Hill A. The benefits of estrogen replacement therapy on oral health. The Leisure World cohort. Arch Intern Med 1995; 155:2325–2329.
- Krall EA, Dawson-Hughes B, Hannan MT, Wilson PW, Kiel DP. Post-menopausal estrogen replacement and tooth retention. Am J Med 1997; 102:536–542.
- Grodstein F, Colditz GA, Stampfer MJ. Postmenopausal hormone use and tooth loss: a prospective study. J Am Dent Assoc 1996; 127:370–377.
- Taguchi A, Sanada M, Suei Y, et al. Effect of estrogen use on tooth retention, oral bone height, and oral bone porosity in Japanese postmenopausal women. Menopause 2004; 11:556–562.
- Pacifici R. Estrogen, cytokines and pathogenesis of postmenopausal osteoporosis. J Bone Miner Res 1996; 11:1043–1051.
- Pacifici R. Is there a causal role for IL-1 in postmenopausal bone loss? Calcif Tissue Int 1992; 50:295–299.
- Girasole G, Jilka RL, Passeri G, et al. 17 beta-estradiol inhibits interleukin-6 production by bone marrow-derived stromal cells and osteoblasts in vitro: a potential mechanism for the antiosteoporotic effect of estrogens. J Clin Invest 1992; 89:883–891.
- Pacifici R, Brown C, Pusheck E, et al. Effect of surgical menopause and estrogen replacement on cytokine release from human blood mononuclear cells. Proc Natl Acad Sci USA 1991; 88:5134–5138.
- Brennan RM, Genco RJ, Wilding GE, Hovey KM, Trevisan M, Wactawski-Wende J. Bacterial species in subgingival plaque and oral bone loss in postmenopausal women. J Periodontol 2007; 78:1051–1061.
- Chestnut CH, Skag A, Christiansen C, et al. Effects of oral ibandronate administered daily or intermittently on fracture risk in post-menopausal osteoporosis. J Bone Miner Res 2004; 19:1241–1249.
- Black DM, Cummings SR, Karpf DB, et al. Randomized trial of effect of alendronate on risk of fracture in women with existing vertebral fractures: Fracture Intervention Trial Research Group. Lancet 1996; 348:1535–1541.
- McClung MR, Geusen P, Miller PD, et al. Effect of risedronate on the risk of hip fracture in elderly women. Hip Intervention Program Study Group. N Engl J Med 2001; 344:333–340.
- Reginster JY, Minne HW, Sorensen OH, et al. Randomized trial of effects of risedronate on vertebral fractures in women with established postmenopausal osteoporosis. Vertebral Efficacy with Risedronate Therapy (VERT) Study Group. Osteoporos Int 2000; 11:83–91.
- Lyles KW, Colon-Emeric CS, Magaziner JS, et al. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med 2007; 357:1799–1809.
- Palomo L, Bissada N, Liu J. Periodontal assessment of postmenopausal women receiving risedronate. Menopause 2005; 12:685–690.
- Rocha ML, Malacara JM, Sánchez-Marin FJ, Vazquez de la Torre CJ, Fajardo ME. Effect of alendronate on periodontal disease in postmenopausal women: a randomized placebo-controlled trial. J Periodontol 2004; 75:1579–1585.
- Jeffcoat MK, Cizza G, Shih WJ, Genco R, Lombardi A. Efficacy of bisphosphonates for the control of alveolar bone loss in periodontitis. J Int Acad Periodontol 2007; 9:70–76.
- Carey JJ, Palomo L. Bisphosphonates and osteonecrosis of the jaw: innocent association or significant risk? Cleve Clin J Med 2008; 75:871–879.
- Woo SB, Hellstein JW, Kalamare JR. Narrative [corrected] review: bisphosphonates and osteonecrosis of the jaws. Ann Intern Med 2006; 144:753–761.
- Dodson TB, Raje NS, Caruso PA, Rosenberg AE. Case records of the Massachusetts General Hospital. Case 9–2008. A 65-year-old woman with a nonhealing ulcer of the jaw. N Engl J Med 2008; 358:1283–1291.
- Ruggiero SL, Mehrotra B, Rosenberg TJ, Engroff SL. Osteonecrosis of the jaws associated with the use of bisphosphonates: a review of 63 cases. J Oral Maxillofacial Surg 2004; 62:527–534.
- Palomo L, Liu J, Bissada NF. Skeletal bone diseases impact the periodontium: a review of bisphosphonate therapy. Expert Opin Pharmacother 2007; 8:309–315.
- Ezra A, Golomb G. Administration routes and delivery systems of bisphosphonates for the treatment of bone resorption. Adv Drug Deliv Rev 2000; 42:175–195.
- Berenson JR, Rosen L, Vescio R, et al. Pharmacokinetics of pamidronate disodium in patients with cancer with normal or impaired renal function. J Clin Pharmacol 1997; 37:285–290.
- American Dental Association Council on Scientific Affairs. Dental management of patients receiving oral bisphosphonate therapy: expert panel recommendations. J Am Dent Assoc 2006; 137:1144–1150.
- Advisory Task Force on Bisphosphonate-Related Ostenonecrosis of the Jaws. American Association of Oral and Maxillofacial Surgeons position paper on bisphosphonate-related osteonecrosis of the jaws. J Oral Maxillofacial Surg 2007; 65:369–376.
- Grbic JT, Landesberg R, Lin SQ, et al; Health Outcomes and Reduced Incidence with Zoledronic Acid Once Yearly Pivotal Fracture Trial Research Group. Incidence of osteonecrosis of the jaw in women with postmenopausal osteoporosis in the Health Outcomes and Reduced Incidence with Zoledronic Acid Once Yearly Pivotal Fracture Trial. J Am Dent Assoc 2008; 139:32–40.
- North American Menopause Society. Menopause Practice: A Clinician’s Guide. 3rd ed; 2007.
- Krall EA, Garcia RI, Dawson-Hughes B. Increased risk of tooth loss is related to bone loss at the whole body, hip and spine. Calcif Tissue Int 1996; 59:433–437.
- Kribbs PJ. Comparison of mandibular bone in normal and osteoporotic women. J Prosthet Dent 1990; 63:218–222.
- Kribbs PJ, Chesnut CH, Ott SM, Kilcoyne RF. Relationship between mandibular and skeletal bone in an osteoporotic population. J Prosthet Dent 1989; 62:703–707.
- Kribbs PJ, Chestnut CH, Ott SM, Kilcyne RE. Relationship between mandibular and skeletal bone in a population of normal women. J Prosthet Dent 1990; 63:86–89.
- Kribbs PJ, Smith DE, Chestnut CH. Oral findings in osteoporosis. Part II: relationship between residual ridge and alveolar bone resorption and generalized skeletal osteopenia. J Prosthet Dent 1983; 50:719–724.
- von Wowern N, Klausen B, Kollerup G. Osteoporosis: a risk factor in periodontal disease. J Periodontol 1994; 65:1134–1138.
- Wactawski-Wende J, Grossi SG, Trevisan M, et al. The role of osteopenia in oral bone loss and periodontal disease. J Peridontol 1996; 67(suppl 10):1076–1084.
- Ronderos M, Jacobs DR, Himes JH, Pihlstrom BL. Associations of periodontal disease with femoral bone mineral density and estrogen replacement therapy: cross-sectional evaluation of US adults from the NHANES III. J Clin Periodontol 2000; 27:778–86.
- American Dental Association Council on Access, Prevention and Interprofessional Relations. Women’s Oral Health Issues. November 2006.
- Jeffcoat MK, Lewis CE, Reddy MS, Wang CY, Redford M. Post-menopausal bone loss and its relationship to oral bone loss. Periodontol 2000 2000; 23:94–102.
- Famili P, Cauley J, Suzuki JB, Weyant R. Longitudinal study of periodontal disease and edentulism with rates of bone loss in older women. J Periodontol 2005; 76:11–15.
- Pilgram TK, Hildebolt CF, Yokoyama N, et al. Relationships between longitudinal changes in radiographic alveolar bone height and probing depth measurements: data from postmenopausal women. J Periodontol 1999; 70:829–833.
- Jeffcoat MK, Lewis CE, Reddy MS, et al. Oral bone loss and systemic osteopenia, osteoporosis. InMarcus R, Feldman D, Kelsey J, editors. Osteoporosis. New York Academic Press 1996:969–990.
- Klemetti E, Collin HL, Forss H, Markkanen H, Lassila V. Mineral status of skeletal and advanced periodontal disease. J Clin Periodontol 1994; 21:184–188.
- Hildebolt CF. Osteoporosis and oral bone loss. Dentomaxillofac Radiol 1997; 26:3–15.
- Tezal M, Wactawski-Wende J, Grossi SG, Ho AW, Dunford R, Genco RJ. The relationship between bone mineral density and periodontitis in postmenopausal women. J Periodontol 2000; 71:1492–1498.
- Taguchi A, Tanimoto K, Suei Y, Ohama K, Wada T. Relationship between the mandibular and lumbar vertebral bone mineral density at different postmenopausal stages. Dentomaxillofac Radiol 1996; 25:130–135.
- Payne JB, Reinhardt RA, Nummikoski PV, Patil KD. Longitudinal alveolar bone loss in postmenopausal osteoporotic/osteopenic women. Osteoporos Int 1999; 10:34–40.
- Payne JB, Zachs NR, Reinhardt RA, Nummikoski PV, Patil K. The association between estrogen status and alveolar bone density changes in postmenopausal women with a history of periodontitis. J Periodontol 1997; 68:24–31.
- Civitelli R, Pilgram TK, Dotson M, et al. Alveolar and postcranial bone density in postmenopausal women receiving hormone/estrogen replacement: a randomized, double blind, placebo-controlled trial. Arch Intern Med 2002; 162:1409–1415.
- Albandar JM, Kingman A. Gingival recession, gingival bleeding, and dental calculus in adults 30 years of age and older in the United States, 1988–1994. J Periodontol 1999; 70:30–43.
- Norderyd OM, Grossi SG, Machtel EE, et al. Periodontal status of women taking postmenopausal estrogen supplementation. J Periodontol 1993; 64:957–962.
- Jacobs R, Ghyselen J, Koninckx P, van Steenberghe D. Long-term bone mass evaluation of mandible and lumbar spine in a group of women receiving hormone replacement therapy. Eur J Oral Sci 1996; 104:10–16.
- Meisel P, Reifenberger J, Haase R, Nauck M, Bandt C, Kocher T. Women are periodontally healthier than men, but why don’t they have more teeth than men? Menopause 2008; 15:270–275.
- Genco RJ, Grossi SG. Is estrogen deficiency a risk factor for periodontal disease? Compend Contin Educ Dent Suppl 1998; 22:S23–S29.
- Paganini-Hill A. The benefits of estrogen replacement therapy on oral health. The Leisure World cohort. Arch Intern Med 1995; 155:2325–2329.
- Krall EA, Dawson-Hughes B, Hannan MT, Wilson PW, Kiel DP. Post-menopausal estrogen replacement and tooth retention. Am J Med 1997; 102:536–542.
- Grodstein F, Colditz GA, Stampfer MJ. Postmenopausal hormone use and tooth loss: a prospective study. J Am Dent Assoc 1996; 127:370–377.
- Taguchi A, Sanada M, Suei Y, et al. Effect of estrogen use on tooth retention, oral bone height, and oral bone porosity in Japanese postmenopausal women. Menopause 2004; 11:556–562.
- Pacifici R. Estrogen, cytokines and pathogenesis of postmenopausal osteoporosis. J Bone Miner Res 1996; 11:1043–1051.
- Pacifici R. Is there a causal role for IL-1 in postmenopausal bone loss? Calcif Tissue Int 1992; 50:295–299.
- Girasole G, Jilka RL, Passeri G, et al. 17 beta-estradiol inhibits interleukin-6 production by bone marrow-derived stromal cells and osteoblasts in vitro: a potential mechanism for the antiosteoporotic effect of estrogens. J Clin Invest 1992; 89:883–891.
- Pacifici R, Brown C, Pusheck E, et al. Effect of surgical menopause and estrogen replacement on cytokine release from human blood mononuclear cells. Proc Natl Acad Sci USA 1991; 88:5134–5138.
- Brennan RM, Genco RJ, Wilding GE, Hovey KM, Trevisan M, Wactawski-Wende J. Bacterial species in subgingival plaque and oral bone loss in postmenopausal women. J Periodontol 2007; 78:1051–1061.
- Chestnut CH, Skag A, Christiansen C, et al. Effects of oral ibandronate administered daily or intermittently on fracture risk in post-menopausal osteoporosis. J Bone Miner Res 2004; 19:1241–1249.
- Black DM, Cummings SR, Karpf DB, et al. Randomized trial of effect of alendronate on risk of fracture in women with existing vertebral fractures: Fracture Intervention Trial Research Group. Lancet 1996; 348:1535–1541.
- McClung MR, Geusen P, Miller PD, et al. Effect of risedronate on the risk of hip fracture in elderly women. Hip Intervention Program Study Group. N Engl J Med 2001; 344:333–340.
- Reginster JY, Minne HW, Sorensen OH, et al. Randomized trial of effects of risedronate on vertebral fractures in women with established postmenopausal osteoporosis. Vertebral Efficacy with Risedronate Therapy (VERT) Study Group. Osteoporos Int 2000; 11:83–91.
- Lyles KW, Colon-Emeric CS, Magaziner JS, et al. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med 2007; 357:1799–1809.
- Palomo L, Bissada N, Liu J. Periodontal assessment of postmenopausal women receiving risedronate. Menopause 2005; 12:685–690.
- Rocha ML, Malacara JM, Sánchez-Marin FJ, Vazquez de la Torre CJ, Fajardo ME. Effect of alendronate on periodontal disease in postmenopausal women: a randomized placebo-controlled trial. J Periodontol 2004; 75:1579–1585.
- Jeffcoat MK, Cizza G, Shih WJ, Genco R, Lombardi A. Efficacy of bisphosphonates for the control of alveolar bone loss in periodontitis. J Int Acad Periodontol 2007; 9:70–76.
- Carey JJ, Palomo L. Bisphosphonates and osteonecrosis of the jaw: innocent association or significant risk? Cleve Clin J Med 2008; 75:871–879.
- Woo SB, Hellstein JW, Kalamare JR. Narrative [corrected] review: bisphosphonates and osteonecrosis of the jaws. Ann Intern Med 2006; 144:753–761.
- Dodson TB, Raje NS, Caruso PA, Rosenberg AE. Case records of the Massachusetts General Hospital. Case 9–2008. A 65-year-old woman with a nonhealing ulcer of the jaw. N Engl J Med 2008; 358:1283–1291.
- Ruggiero SL, Mehrotra B, Rosenberg TJ, Engroff SL. Osteonecrosis of the jaws associated with the use of bisphosphonates: a review of 63 cases. J Oral Maxillofacial Surg 2004; 62:527–534.
- Palomo L, Liu J, Bissada NF. Skeletal bone diseases impact the periodontium: a review of bisphosphonate therapy. Expert Opin Pharmacother 2007; 8:309–315.
- Ezra A, Golomb G. Administration routes and delivery systems of bisphosphonates for the treatment of bone resorption. Adv Drug Deliv Rev 2000; 42:175–195.
- Berenson JR, Rosen L, Vescio R, et al. Pharmacokinetics of pamidronate disodium in patients with cancer with normal or impaired renal function. J Clin Pharmacol 1997; 37:285–290.
- American Dental Association Council on Scientific Affairs. Dental management of patients receiving oral bisphosphonate therapy: expert panel recommendations. J Am Dent Assoc 2006; 137:1144–1150.
- Advisory Task Force on Bisphosphonate-Related Ostenonecrosis of the Jaws. American Association of Oral and Maxillofacial Surgeons position paper on bisphosphonate-related osteonecrosis of the jaws. J Oral Maxillofacial Surg 2007; 65:369–376.
- Grbic JT, Landesberg R, Lin SQ, et al; Health Outcomes and Reduced Incidence with Zoledronic Acid Once Yearly Pivotal Fracture Trial Research Group. Incidence of osteonecrosis of the jaw in women with postmenopausal osteoporosis in the Health Outcomes and Reduced Incidence with Zoledronic Acid Once Yearly Pivotal Fracture Trial. J Am Dent Assoc 2008; 139:32–40.
KEY POINTS
- Physicians should be vigilant for dental problems and should encourage their patients to practice good oral hygiene and to seek regular dental care.
- Available information suggests that hormone therapy and bisphosphonate drugs may be developed to protect against alveolar bone loss and perhaps slow the progression of periodontal disease.
- Bisphosphonate-associated osteonecrosis of the jaw is rare, and most of the reported cases have been in cancer patients who received high doses of bisphosphonates intravenously and who had other risk factors for it.
Painful eye with a facial rash
A 75-year-old man presents 4 days after painful cutaneous lesions appeared on the left side of his face, associated with severe ocular pain. Two days before the eruption, he had had an intense headache, which was diagnosed as a tension headache and was treated with oral acetaminophen (Tylenol), but with no improvement.
The remainder of his physical examination is normal. Laboratory tests, including red and white blood cell counts, hemoglobin, and basic metabolic and coagulation tests reveal no abnormalities.
Q: What is your diagnosis?
- Allergic contact dermatitis
- Herpes simplex
- Varicella
- Ramsay-Hunt syndrome
- Herpes zoster ophthalmicus and herpetic keratitis
A: Herpes zoster ophthalmicus is the correct diagnosis. It represents a reactivation of the varicella zoster virus.1
Varicella zoster virus, like others of the herpes family, has developed a complex control of virus-host interactions to ensure its survival in humans. It lies dormant in the sensory ganglia and, when reactivated, moves down the neurons and satellite cells along the sensory axons to the skin.1 The reactivation is related to diminished cell-mediated immunity, which occurs as a physiologic part of aging, which is why the elderly tend to be the most often affected. 2 The incidence of herpes zoster varies from 2.2 to 3.4 per 1,000 people per year.3 Its incidence in people over age 80 is about 10 per 1,000 people per year.3
CLINICAL PRESENTATION
Herpes zoster typically presents as a dermatome-grouped vesicular eruption over an erythematous base, accompanied or preceded by local pain. It has two main complications, postherpetic neuralgia and ocular involvement. Postherpetic neuralgia is neuropathic pain that persists or develops after the dermatomal rash has healed.4 Independent predictors of postherpetic neuralgia are older age, severe acute pain, severe rash, a shorter duration of rash before consultation, and ocular involvement.5 It occurs in 36.6% of patients over age 60, and in 47.5% over 70.6 Persistent postherpetic neuralgia has been linked to suicide in patients over 70.7
Ocular infection occurs with involvement of the ophthalmic division of the fifth cranial nerve. Before the antiviral era, it was seen in as many as 50% of patients.8 Hutchinson’s sign is skin lesions on the tip, side, or root of the nose and is an important predictor of ocular involvement.1 Lesions may include folliculopapillar conjunctivitis, episcleritis, scleritis, keratitis (dendritic, pseudodendritic, and interstitial), uveitis, and necrotizing retinitis.
DIAGNOSIS
The diagnosis of herpes zoster is usually based on clinical observation of the characteristic rash, although viral culture and molecular techniques are available when definitive diagnosis is required. When ophthalmic division is affected and Hutchinson’s sign, unexplained ocular redness with pain, or complaints of visual problems are present, the patient should be referred promptly to an ophthalmologist, because serious visual impairment can occur. The fluorescein dye may show no staining or the typical dendritic keratitis (Figure 2).
TREATMENT
Oral antiviral drugs have made the treatment of zoster possible when, effectively, no treatment existed before. Ideally, an antiviral should be given within 72 hours of symptom onset. Starting treatment as early as possible—especially within 72 hours of onset—has been shown to be effective in alleviating acute pain and in preventing or limiting the duration and severity of postherpetic neuralgia.3
Acyclovir (Zovirax) 800 mg five times a day for 7 days or one of its derivatives—eg, famciclovir (Famvir), penciclovir (Denavir), or valacyclovir (Valtrex)—has been shown to be safe and effective in the treatment of active disease, as well as in preventing or shortening the duration of postherpetic neuralgia.3 It has also been shown to reduce the rate of eye involvement from 50% to 20% or 30%.9 This is why all patients with this dermatomal involvement must be treated.
Second-generation antivirals
Valacyclovir 1,000 mg three times a day and famciclovir 500 mg three times a day seem to be as effective as acyclovir in reducing zoster-associated pain, but their efficacy in reducing eye involvement has not been studied. In clinical practice, however, these second-generation antivirals may be more effective than acyclovir because patients are more likely to comply with the treatment regimen of three rather than five daily doses.
Other considerations
In patients with kidney failure, the non-nephrotoxic antiviral brivudine is preferred, but it is not available in the United States. Therefore, one must use acyclovir or one of the other drugs, carefully adjusting the dose according to the creatinine clearance and making sure the patient is well hydrated.
The efficacy of antiviral treatment that is started more than 72 hours after the onset of skin rash has never been confirmed.
Although the additional effectiveness of acyclovir eye ointment has never been established, topical acyclovir can be considered in cases of dendritic or pseudodendritic keratitis.
- Liesegang TJ. Herpes zoster ophthalmicus: natural history, risk factors, clinical presentation, and morbidity. Ophthalmology 2008; 115( suppl 2):S3–S12.
- Opstelten W, Eekhof J, Neven AK, Verheij T. Treatment of herpes zoster. Can Fam Physician 2008; 54:373–377.
- Opstelten W, Zaal MJ. Managing ophthalmic herpes zoster in primary care. BMJ 2005; 331:147–151.
- Donahue JG, Choo PW, Manson JE, Platt R. The incidence of herpes zoster. Arch Intern Med 1995; 155:1605–1609.
- Opstelten W, Zuithoff NP, van Essen GA, et al. Predicting postherpetic neuralgia in elderly primary care patients with herpes zoster: prospective prognostic study. Pain 2007; 132( suppl 1):S52–S59.
- De Morgas JM, Kierland RR. The outcome of patients with herpes zoster. AMA Arch Derm 1957; 75:193–196.
- Hess TM, Lutz LJ, Nauss LA, Lamer TJ. Treatment of acute herpetic neuralgia. A case report and review of the literature. Minn Med 1990; 73:37–40.
- Harding SP, Lipton JR, Wells JC. Natural history of herpes zoster ophthalmicus: predictors of postherpetic neuralgia and ocular involvement. Br J Ophthalmol 1987; 71:353–358.
- Cobo LM, Foulks GN, Liesegang T, et al. Oral acyclovir in the treatment of acute herpes zoster ophthalmicus. Ophthalmology 1986; 93:763–770.
A 75-year-old man presents 4 days after painful cutaneous lesions appeared on the left side of his face, associated with severe ocular pain. Two days before the eruption, he had had an intense headache, which was diagnosed as a tension headache and was treated with oral acetaminophen (Tylenol), but with no improvement.
The remainder of his physical examination is normal. Laboratory tests, including red and white blood cell counts, hemoglobin, and basic metabolic and coagulation tests reveal no abnormalities.
Q: What is your diagnosis?
- Allergic contact dermatitis
- Herpes simplex
- Varicella
- Ramsay-Hunt syndrome
- Herpes zoster ophthalmicus and herpetic keratitis
A: Herpes zoster ophthalmicus is the correct diagnosis. It represents a reactivation of the varicella zoster virus.1
Varicella zoster virus, like others of the herpes family, has developed a complex control of virus-host interactions to ensure its survival in humans. It lies dormant in the sensory ganglia and, when reactivated, moves down the neurons and satellite cells along the sensory axons to the skin.1 The reactivation is related to diminished cell-mediated immunity, which occurs as a physiologic part of aging, which is why the elderly tend to be the most often affected. 2 The incidence of herpes zoster varies from 2.2 to 3.4 per 1,000 people per year.3 Its incidence in people over age 80 is about 10 per 1,000 people per year.3
CLINICAL PRESENTATION
Herpes zoster typically presents as a dermatome-grouped vesicular eruption over an erythematous base, accompanied or preceded by local pain. It has two main complications, postherpetic neuralgia and ocular involvement. Postherpetic neuralgia is neuropathic pain that persists or develops after the dermatomal rash has healed.4 Independent predictors of postherpetic neuralgia are older age, severe acute pain, severe rash, a shorter duration of rash before consultation, and ocular involvement.5 It occurs in 36.6% of patients over age 60, and in 47.5% over 70.6 Persistent postherpetic neuralgia has been linked to suicide in patients over 70.7
Ocular infection occurs with involvement of the ophthalmic division of the fifth cranial nerve. Before the antiviral era, it was seen in as many as 50% of patients.8 Hutchinson’s sign is skin lesions on the tip, side, or root of the nose and is an important predictor of ocular involvement.1 Lesions may include folliculopapillar conjunctivitis, episcleritis, scleritis, keratitis (dendritic, pseudodendritic, and interstitial), uveitis, and necrotizing retinitis.
DIAGNOSIS
The diagnosis of herpes zoster is usually based on clinical observation of the characteristic rash, although viral culture and molecular techniques are available when definitive diagnosis is required. When ophthalmic division is affected and Hutchinson’s sign, unexplained ocular redness with pain, or complaints of visual problems are present, the patient should be referred promptly to an ophthalmologist, because serious visual impairment can occur. The fluorescein dye may show no staining or the typical dendritic keratitis (Figure 2).
TREATMENT
Oral antiviral drugs have made the treatment of zoster possible when, effectively, no treatment existed before. Ideally, an antiviral should be given within 72 hours of symptom onset. Starting treatment as early as possible—especially within 72 hours of onset—has been shown to be effective in alleviating acute pain and in preventing or limiting the duration and severity of postherpetic neuralgia.3
Acyclovir (Zovirax) 800 mg five times a day for 7 days or one of its derivatives—eg, famciclovir (Famvir), penciclovir (Denavir), or valacyclovir (Valtrex)—has been shown to be safe and effective in the treatment of active disease, as well as in preventing or shortening the duration of postherpetic neuralgia.3 It has also been shown to reduce the rate of eye involvement from 50% to 20% or 30%.9 This is why all patients with this dermatomal involvement must be treated.
Second-generation antivirals
Valacyclovir 1,000 mg three times a day and famciclovir 500 mg three times a day seem to be as effective as acyclovir in reducing zoster-associated pain, but their efficacy in reducing eye involvement has not been studied. In clinical practice, however, these second-generation antivirals may be more effective than acyclovir because patients are more likely to comply with the treatment regimen of three rather than five daily doses.
Other considerations
In patients with kidney failure, the non-nephrotoxic antiviral brivudine is preferred, but it is not available in the United States. Therefore, one must use acyclovir or one of the other drugs, carefully adjusting the dose according to the creatinine clearance and making sure the patient is well hydrated.
The efficacy of antiviral treatment that is started more than 72 hours after the onset of skin rash has never been confirmed.
Although the additional effectiveness of acyclovir eye ointment has never been established, topical acyclovir can be considered in cases of dendritic or pseudodendritic keratitis.
A 75-year-old man presents 4 days after painful cutaneous lesions appeared on the left side of his face, associated with severe ocular pain. Two days before the eruption, he had had an intense headache, which was diagnosed as a tension headache and was treated with oral acetaminophen (Tylenol), but with no improvement.
The remainder of his physical examination is normal. Laboratory tests, including red and white blood cell counts, hemoglobin, and basic metabolic and coagulation tests reveal no abnormalities.
Q: What is your diagnosis?
- Allergic contact dermatitis
- Herpes simplex
- Varicella
- Ramsay-Hunt syndrome
- Herpes zoster ophthalmicus and herpetic keratitis
A: Herpes zoster ophthalmicus is the correct diagnosis. It represents a reactivation of the varicella zoster virus.1
Varicella zoster virus, like others of the herpes family, has developed a complex control of virus-host interactions to ensure its survival in humans. It lies dormant in the sensory ganglia and, when reactivated, moves down the neurons and satellite cells along the sensory axons to the skin.1 The reactivation is related to diminished cell-mediated immunity, which occurs as a physiologic part of aging, which is why the elderly tend to be the most often affected. 2 The incidence of herpes zoster varies from 2.2 to 3.4 per 1,000 people per year.3 Its incidence in people over age 80 is about 10 per 1,000 people per year.3
CLINICAL PRESENTATION
Herpes zoster typically presents as a dermatome-grouped vesicular eruption over an erythematous base, accompanied or preceded by local pain. It has two main complications, postherpetic neuralgia and ocular involvement. Postherpetic neuralgia is neuropathic pain that persists or develops after the dermatomal rash has healed.4 Independent predictors of postherpetic neuralgia are older age, severe acute pain, severe rash, a shorter duration of rash before consultation, and ocular involvement.5 It occurs in 36.6% of patients over age 60, and in 47.5% over 70.6 Persistent postherpetic neuralgia has been linked to suicide in patients over 70.7
Ocular infection occurs with involvement of the ophthalmic division of the fifth cranial nerve. Before the antiviral era, it was seen in as many as 50% of patients.8 Hutchinson’s sign is skin lesions on the tip, side, or root of the nose and is an important predictor of ocular involvement.1 Lesions may include folliculopapillar conjunctivitis, episcleritis, scleritis, keratitis (dendritic, pseudodendritic, and interstitial), uveitis, and necrotizing retinitis.
DIAGNOSIS
The diagnosis of herpes zoster is usually based on clinical observation of the characteristic rash, although viral culture and molecular techniques are available when definitive diagnosis is required. When ophthalmic division is affected and Hutchinson’s sign, unexplained ocular redness with pain, or complaints of visual problems are present, the patient should be referred promptly to an ophthalmologist, because serious visual impairment can occur. The fluorescein dye may show no staining or the typical dendritic keratitis (Figure 2).
TREATMENT
Oral antiviral drugs have made the treatment of zoster possible when, effectively, no treatment existed before. Ideally, an antiviral should be given within 72 hours of symptom onset. Starting treatment as early as possible—especially within 72 hours of onset—has been shown to be effective in alleviating acute pain and in preventing or limiting the duration and severity of postherpetic neuralgia.3
Acyclovir (Zovirax) 800 mg five times a day for 7 days or one of its derivatives—eg, famciclovir (Famvir), penciclovir (Denavir), or valacyclovir (Valtrex)—has been shown to be safe and effective in the treatment of active disease, as well as in preventing or shortening the duration of postherpetic neuralgia.3 It has also been shown to reduce the rate of eye involvement from 50% to 20% or 30%.9 This is why all patients with this dermatomal involvement must be treated.
Second-generation antivirals
Valacyclovir 1,000 mg three times a day and famciclovir 500 mg three times a day seem to be as effective as acyclovir in reducing zoster-associated pain, but their efficacy in reducing eye involvement has not been studied. In clinical practice, however, these second-generation antivirals may be more effective than acyclovir because patients are more likely to comply with the treatment regimen of three rather than five daily doses.
Other considerations
In patients with kidney failure, the non-nephrotoxic antiviral brivudine is preferred, but it is not available in the United States. Therefore, one must use acyclovir or one of the other drugs, carefully adjusting the dose according to the creatinine clearance and making sure the patient is well hydrated.
The efficacy of antiviral treatment that is started more than 72 hours after the onset of skin rash has never been confirmed.
Although the additional effectiveness of acyclovir eye ointment has never been established, topical acyclovir can be considered in cases of dendritic or pseudodendritic keratitis.
- Liesegang TJ. Herpes zoster ophthalmicus: natural history, risk factors, clinical presentation, and morbidity. Ophthalmology 2008; 115( suppl 2):S3–S12.
- Opstelten W, Eekhof J, Neven AK, Verheij T. Treatment of herpes zoster. Can Fam Physician 2008; 54:373–377.
- Opstelten W, Zaal MJ. Managing ophthalmic herpes zoster in primary care. BMJ 2005; 331:147–151.
- Donahue JG, Choo PW, Manson JE, Platt R. The incidence of herpes zoster. Arch Intern Med 1995; 155:1605–1609.
- Opstelten W, Zuithoff NP, van Essen GA, et al. Predicting postherpetic neuralgia in elderly primary care patients with herpes zoster: prospective prognostic study. Pain 2007; 132( suppl 1):S52–S59.
- De Morgas JM, Kierland RR. The outcome of patients with herpes zoster. AMA Arch Derm 1957; 75:193–196.
- Hess TM, Lutz LJ, Nauss LA, Lamer TJ. Treatment of acute herpetic neuralgia. A case report and review of the literature. Minn Med 1990; 73:37–40.
- Harding SP, Lipton JR, Wells JC. Natural history of herpes zoster ophthalmicus: predictors of postherpetic neuralgia and ocular involvement. Br J Ophthalmol 1987; 71:353–358.
- Cobo LM, Foulks GN, Liesegang T, et al. Oral acyclovir in the treatment of acute herpes zoster ophthalmicus. Ophthalmology 1986; 93:763–770.
- Liesegang TJ. Herpes zoster ophthalmicus: natural history, risk factors, clinical presentation, and morbidity. Ophthalmology 2008; 115( suppl 2):S3–S12.
- Opstelten W, Eekhof J, Neven AK, Verheij T. Treatment of herpes zoster. Can Fam Physician 2008; 54:373–377.
- Opstelten W, Zaal MJ. Managing ophthalmic herpes zoster in primary care. BMJ 2005; 331:147–151.
- Donahue JG, Choo PW, Manson JE, Platt R. The incidence of herpes zoster. Arch Intern Med 1995; 155:1605–1609.
- Opstelten W, Zuithoff NP, van Essen GA, et al. Predicting postherpetic neuralgia in elderly primary care patients with herpes zoster: prospective prognostic study. Pain 2007; 132( suppl 1):S52–S59.
- De Morgas JM, Kierland RR. The outcome of patients with herpes zoster. AMA Arch Derm 1957; 75:193–196.
- Hess TM, Lutz LJ, Nauss LA, Lamer TJ. Treatment of acute herpetic neuralgia. A case report and review of the literature. Minn Med 1990; 73:37–40.
- Harding SP, Lipton JR, Wells JC. Natural history of herpes zoster ophthalmicus: predictors of postherpetic neuralgia and ocular involvement. Br J Ophthalmol 1987; 71:353–358.
- Cobo LM, Foulks GN, Liesegang T, et al. Oral acyclovir in the treatment of acute herpes zoster ophthalmicus. Ophthalmology 1986; 93:763–770.
Back pain made simple: An approach based on principles and evidence
Low back pain should be understood as a remittent, intermittent predicament of life. Its cause is indeterminate, but its course is predictable. Its link to work-related injury is tenuous and confounded by psychosocial issues, including workers’ compensation. It challenges function, compromises performance, and calls for empathy and understanding.1
In this brief paper, we offer a simple approach to one of the most common human afflictions, based on principles and evidence.
WHY IS BACK PAIN IMPORTANT?
Low back pain is common and affects people of all ages. It is second only to the common cold as the most common affliction of mankind, and it is among the leading complaints bringing patients to physicians’ offices. Its lifetime prevalence exceeds 70% in most industrialized countries, with an annual incidence of 15% to 20% in the United States.
Its social and economic impact is substantial. It is the most frequent cause of disability for people under age 45. In 2005, the mean age- and sex-adjusted medical expenditure among respondents with spine problems was $6,096 vs $3,516 in those without spine problems, and it had increased by 65% (adjusted for inflation) from 1997 to 2005.2
WHAT ARE THE GOALS AND PRINCIPLES OF MANAGING LOW BACK PAIN?
The goals of management for patients with low back pain are to:
- Decrease the pain
- Restore mobility
- Hasten recovery so the patient can resume normal daily activities as soon as possible
- Prevent development of a chronic recurrent condition: low back pain is considered acute when it persists for less than 6 weeks, subacute between 6 weeks and 3 months, and chronic when it lasts longer than 3 months
- Restore and preserve physical and financial independence and comfort.
Principles of management
- Most back pain has no recognizable cause and is therefore termed “mechanical” or “musculoskeletal.”
- Underlying systemic disease is rare.
- Most episodes of back pain are unpreventable.
- Confounding psychosocial issues are often contributory, important, and relevant.
- A careful, informed history and physical examination are invaluable; diagnostic studies, however technologically sophisticated, are never a substitute.
- Defer diagnostic studies for specific indications.
- Refer patients only if they have underlying disease or progressive neurologic dysfunction, or if they do not respond to conservative management.
- Encouragement of activity is benign and perhaps salutary for back pain and is desirable for general physical and mental health; there is only scant evidence to support bed rest.3
- Few if any treatments have been proven effective for low back pain.
- Talking to the patient and explaining the issues involved are critical to successful management.4
INITIAL CONSIDERATIONS WHEN EVALUATING A PATIENT
When encountering a patient with back pain, the initial consideration is whether the symptoms are regional—ie, local, mechanical, and musculoskeletal—or if they reflect a systemic disease. It is also important to look for evidence of social or psychological distress that may amplify, prolong, or confound the pain or the patient’s perception of it.
What are the clues to a systemic process?
Does the patient have a regional low back syndrome?
Regional low back syndromes account for 90% of the causes of low back pain. They are usually mechanical in origin.
Regional back pain is due to overuse of a normal mechanical structure (muscle strain, “lumbago”) or is secondary to trauma, deformity, or degeneration of an anatomical structure (herniated nucleus pulposus, fracture, and spondyloarthropathy, including facet joint arthritis). Chronic regional back syndromes include osteoarthritis of the spine (ie, spondylosis), spinal stenosis, and facet joint arthropathy.
Characteristically, mechanical disorders are exacerbated by certain physical activities, such as lifting, and are relieved by others, such as assuming a supine position.
Does the patient have sciatica or another nerve root compression syndrome?
The obvious manifestation of nerve root irritation is usually sciatica, a sharp or burning pain radiating down the posterior or lateral aspect of the leg usually to the foot or the ankle and often associated with numbness or paresthesias. The pain is sometimes aggravated by coughing, sneezing, or the Valsalva maneuver. It is most commonly seen in lumbar disk herniation, cauda equina syndrome, and spinal stenosis.
Might the patient have spinal stenosis?
More than 20% of people over age 60 have radiographic evidence of lumbar spinal canal stenosis, even if they have no symptoms.5 For this reason, the diagnosis of spinal stenosis as a cause of low back pain must be based on the history and physical examination.
The classic history of spinal stenosis is that of neurogenic claudication (“pseudoclaudication”), which is pain that occurs in the legs after walking or prolonged standing and is relieved with sitting. It may sometimes be associated with a varying and transient neurologic deficit. Lumbar flexion increases and lumbar extension decreases the cross-sectional area of the spinal canal—hence, the relief of symptoms of spinal stenosis on stooping or bending forward. Pain is commonly perceived in the back, buttock, or thigh and is elicited by prolonged lumbar extension.
On neurologic examination, about 50% of patients with spinal stenosis have a deficit in vibratory sensibility, temperature sensitivity, or muscle strength. The nerve root involved is most commonly L5, followed by S1 and L4.
Many patients have balance disturbance (wide-based gait or Romberg sign), particularly later in the course of the disorder, with normal cerebellar signs (“pseudocerebellar” presentation).
Patients with bilateral hip osteoarthritis may present with similar symptoms of buttock or thigh pain, which can be distinguished with the above clinical examination. Rotation of the hip is painful in osteoarthritis but not in spinal stenosis. If both conditions overlap, injection of a steroid or lidocaine in the painful hip should decrease the pain associated with hip osteoarthritis.
Does the patient have evidence of neurologic compromise?

Muscle strength is tested by examining the:
- L2 nerve root (which supplies the iliopsoas muscle and is tested by hip flexion)
- L3 nerve root (quadriceps, tested by knee extension)
- L4 nerve root (tibialis anterior, assessed by evaluating ankle dorsiflexion and inversion at the subtalar joint)
- L5 nerve root (extensor hallucis longus and extensor digitorum longus, tested by asking the patient to dorsiflex the great toe, then the other toes)
- S1 nerve root (flexor hallucis longus, flexor digitorum longus, and tendoachilles, tested by asking the patient to plantar-flex the great toe, then the other toes, and then the ankle).
The patient is also asked to walk a few steps on the toes and then on the heels. Inability to toe-walk indicates S1 nerve root involvement; inability to heel-walk may indicate L4 or L5 involvement. If the patient cannot heelwalk, ask him or her to squat; inability to do so indicates L4 problems.6
Radiculopathy. Detecting and locating the cause of radiculopathy may be helpful. In L3–L4 disk herniation, there is pain and paresthesia with numbness and hypalgesia in the anteromedial thigh and the knee. In L4–L5 disk herniation, there is usually involvement of the exiting L5 nerve root, which presents as numbness or paresthesias in the anterolateral calf, great toe, first web space, and medial foot. In L5-S1 disk herniation, the S1 nerve root is involved, presenting as numbness and hypalgesia in the fifth toe, lateral aspect of the foot, sole, and posterolateral calf and thigh.
Reflexes. Exaggerated or decreased reflexes do not always indicate a neurologic abnormality, but reflex asymmetry is significant. The knee-jerk reflex is diminished in L3–L4 nerve root involvement, and the ankle-jerk reflex is diminished with S1 nerve root involvement. The Babinski sign indicates pyramidal tract involvement.
Gait. Observe the patient’s gait as he or she rises and moves to the examining table, to determine whether it is shortened, asymmetrical, or antalgic.7 Also note any foot drop, which may indicate a potentially serious problem (L5 radiculopathy).
What is an adequate examination of the back?
A good back examination can elicit important information about the cause and the extent of back pain. It includes inspection, palpation, and range of movement of the spine along with a detailed neurologic examination.
Inspect it for any deformities, scoliosis, asymmetry, paraspinal muscle spasm, unusual hair growth, listing to one side, decrease or increase in lumbar lordosis, or muscle atrophy or fasciculation.
Palpate it for paraspinal muscle spasm, warmth, and localized bone pain.
Move it. The normal ranges of motion of the lumbar spine are 15 degrees of extension, 40 degrees of flexion, 30 degrees of lateral bending, and 40 degrees of lateral rotation to each side.
Assess it. This includes estimating the tone and nutrition of the muscles, testing their strength (Table 2), examining vibratory or proprioception and pinprick sensation in each dermatome (see below), testing the Achilles and patellar reflexes, and looking for the Babinski sign and clonus. In addition, perform the straight-leg-raising and the cross-straightleg-raising tests, which are positive in most patients with lower lumbar disk herniations.
The femoral stretch test is usually positive in upper lumbar disk herniations (L2–L3, L3– L4). It is performed with the patient in the prone position, with the knee being gradually flexed from full extension. Pain radiating along the anterior aspect of the thigh indicates a positive test.
The examination of the spine must be supplemented with examination of the hip and sacroiliac joints, since back pain may be a referred symptom from any pathology affecting these joints.
When should patients be referred to a specialist?
Patients should be referred to a neurologist, neurosurgeon, orthopedist, or other specialist if they have cauda equina syndrome; severe or progressive neurologic deficits; infections, tumors, or fractures compressing the spinal cord; or, perhaps, no response to conservative therapy for 4 to 6 weeks for patients with a herniated lumbar disk or 8 to 12 weeks for those with spinal stenosis.
If there is profound motor involvement at the time of the initial evaluation, patients must be promptly given systemic corticosteroids such as methylprednisolone (Medrol) or dexamethasone (Decadron) to decrease spinal cord edema.
Are there signs of psychological distress?
Psychosocial factors can significantly affect pain and functional disability in patients who have low back pain.8,9 These are known as “yellow flags” and are better predictors of treatment outcome than physical factors. 10 Anatomically inappropriate signs may be helpful in identifying psychological distress as a result of or as an amplifier of low back symptoms.
Waddell et al11 proposed five categories of these nonorganic signs. These are:
- Inappropriate tenderness that is superficial or widespread
- Pain on simulated axial loading by pressing on the top of the head or simulated spine rotation
- Distraction signs such as inconsistent performance between straight-leg-raising in the seated position vs the supine position
- Regional disturbances in strength and sensation that do not correspond with nerve root innervation patterns
- Overreaction during the physical examination.
The occurrence of any one of the signs is of limited value, but positive findings in three of the five categories suggest psychological distress.11
Which diagnostic studies are useful, cost-effective, and supported by evidence?
Since most abnormalities found on imaging studies are nonspecific, such studies are not necessary during the initial evaluation of acute low back pain unless there are red flags that suggest a more ominous source of pain.
Routine plain lumbosacral spine radiographs with anteroposterior and lateral views may be appropriate initially if the patient has risk factors for vertebral fractures (Table 1), or if the patient does not improve after a course of conservative treatment (usually 4–6 weeks).
Magnetic resonance imaging (MRI) is the preferred test if one suspects a tumor, infection, disk pathology, or spinal stenosis.
Computed tomography (CT) shows bony details better than MRI does. Hence, it is preferred when one needs to evaluate bony details (fractures, scoliosis) and when there are contraindications to MRI, as in patients with metal implant devices and those who are claustrophobic (although now there are “open system” MRI machines, in which the feeling of claustrophobia is much less).
MRI and CT should not be ordered routinely, but only for specific indications to answer specific questions, when specific findings would indicate specific treatment.
In most cases, contrast is not needed for CT or MRI to rule out common causes of low back pain, except in cases of suspected intraspinal tumor. Patients with compromised renal function who need contrast for CT need to be hydrated before the scan to lower the risk of contrast-induced nephropathy. These patients are also at higher risk of nephrogenic fibrosing dermopathy when they receive gadolinium contrast for MRI.
Bone scans can be used to look for infections or fractures not noted on plain radiography. However, MRI provides similar or better diagnostic accuracy without radiation.
Electrodiagnostic studies may be used in patients with radiculopathy when clinical examination suggests multilevel root lesions, when symptoms do not match imaging studies, and when patients have breakaway weakness (fluctuating levels of strength in one or more muscle groups).
Other useful diagnostic and laboratory studies may include the erythrocyte sedimentation rate to screen for malignancy and infection when these are suspected, blood culture for osteomyelitis, and bone aspiration and biopsy for histopathologic diagnosis of infection, malignancy, or other lesions.
WHICH TREATMENTS ARE SUPPORTED BY ROBUST EVIDENCE?
The primary treatment of low back pain should be conservative care, reassurance, and education, allowing patients to improve on their own and helping them cope with their predicament.
Limited bed rest. While 2 or 3 days of limited bed rest may help improve symptoms in patients who have acute radiculopathy, several studies have shown that long periods of bed rest are not beneficial for acute or subacute low back pain.12 Encouraging activity modification allows patients with nonspecific back pain or radicular symptoms to remain active while avoiding activities that may aggravate pain and is shown to lead to a more rapid recovery than bed rest.13,14 The most common situations to avoid are prolonged sitting or standing.15 Low-stress aerobic activities, especially walking, are the best early activities.15
Exercise is one of the only evidence-based, effective treatments for chronic low back pain.16 The most commonly prescribed exercises are aimed at retraining the multifidus (a back muscle) and transversus abdominis (a deep abdominal muscle), supplemented with exercises for the pelvic floor and breathing control.
Nonsteroidal anti-inflammatory drugs (NSAIDs) and acetaminophen (Tylenol) are the drugs of choice for pain control in acute back pain17,18 and are as effective as muscle relaxants or opioids.
Muscle relaxants and opioids offer few advantages over NSAIDs and acetaminophen, except when there is severe muscle spasm associated with the back pain or if acetaminophen or NSAIDs do not relieve the pain. Muscle relaxants and opioids are both associated with more severe adverse effects. If prescribed, they should be used for a short, clearly defined period (1 to 2 weeks).19
Epidural corticosteroids, when used for sciatica, give mild to moderate short-term improvement in leg pain and sensory deficit but no significant long-term functional benefit or reduction in the need for surgery.20
Surgery may be considered in cases of cauda equina syndrome, which is a surgical emergency; severe or progressive neurologic deficit; infections, tumors, and fractures compressing the spinal cord; mechanical instability of the back; and, perhaps, intractable pain (leg pain equal to or greater than back pain) with a positive straight-leg-raising test and no response to conservative therapy.
The term “instability” implies an abnormal motion under physiologic loads. Lumbar instability is defined as translation of more than 4 mm or 10 degrees of angular motion between flexion and extension on an upright lateral radiograph.
Although Weinstein et al21 showed that patients with spinal stenosis who underwent surgery showed significantly more improvement in all primary outcomes than did patients treated nonsurgically, many patients can be effectively treated without surgery.
WHAT SHOULD BE REMEMBERED ABOUT LOW BACK PAIN?
Low back pain is a common and costly medical condition with only a weak correlation between symptoms and pathologic changes, resulting in a lack of objective clinical findings on which a definitive diagnosis can be based.22 Most back pain has no recognizable cause and is usually regional and musculoskeletal. Back pain as a result of an underlying systemic disease is rare and needs to be excluded by a good history and physical examination. Diagnostic studies are best reserved for specific indications.
Referral to a specialist is warranted when the patient is not responding to conservative treatment, when a progressive neurologic deficit or cauda equina syndrome is noted or suspected, or when the patient has an underlying malignancy, infection, fracture, or spinal instability.
Bed rest is best avoided, and activity within the limits of pain is encouraged. NSAIDs and acetaminophen are usually the drugs of choice for controlling acute low back pain.
Ultimately, the goal for clinicians is to identify serious conditions and to prevent the back pain from becoming chronic pain by promptly identifying the various risk factors.
- Hadler NM. Low back pain. In:Koopman WJ, editor. Arthritis and Allied Conditions. 14th ed. Philadelphia, PA: Lipincott Williams and Wilkins; 2001:2026–2041.
- Martin BI, Deyo RA, Mirza SK, et al. Expenditures and health status among adults with back and neck problems. JAMA 2008; 299:656–664.
- NASS Task Force on clinical guidelines. Phase III clinical guidelines for multidisciplinary spine care specialists. Unremitting low back pain. 1st ed. Burr Ridge, IL: North American Spine Society; 2000.
- Cailliet R. Low back pain. In: Soft Tissue Pain and Disability. 3rd ed. Philadelphia, PA: FA Davis; 1996:101–170.
- Jensen MC, Brant-Zawadzki MN, Obuchowski N, Modic MT, Malkasian D, Ross JS. Magnetic resonance imaging of the lumbar spine in people without back pain. N Engl J Med 1994; 331:69–73.
- A gency for Health Care Policy and Research. Acute low back pain problems in adults: assessment and treatment. http://www.chirobase.org/07Strategy/AHCPR/ahcprclinician.html. Accessed March 2009.
- Cohen R, Chopra P, Upshur C. Primary care work-up of acute and chronic symptoms. Geriatrics 2001; 56:26–37.
- Pincus T, Burton AK, Vogel S, Field AP. A systematic review of psychological factors as predictors of chronicity/disability in prospective cohorts of low back pain. Spine 2002; 27:E109–E120.
- Carragee EJ. Clinical practice: persistent low back pain. N Engl J Med 2005; 352:1891–1898.
- Pincus T, Vlaeyen JW, Kendall NA, Von Korff MR, Kalauokalani DA, Reis S. Cognitive-behavioral therapy and psychosocial factors in low back pain: directions for the future. Spine 2002; 27:E133–E138.
- Waddell G, McCulloch JA, Kummel E, Venner RM. Nonorganic physical signs in low back pain. Spine 1980; 5:117–125.
- Hagen KB, Hilde G, Jamtvedt G, Winnem M. Bed rest for acute low-back pain and sciatica. Cochrane Database Syst Rev 2004; 4:CD001254.
- Patel AT, Ogle AA. Diagnosis and management of acute low back pain. Am Fam Physician 2000; 61:1779–1790.
- Grotle M, Brox JI, Glomsrød B, Lønn JH, Vøllestad NK. Prognostic factors in first-time care seekers due to acute low back pain. Eur J Pain 2007; 11:290–298.
- Atlas SJ, Deyo RA. Evaluating and managing acute low back pain in the primary care setting. J Gen Intern Med 2001; 16:120–131.
- Maher CG. Effective physical treatment for low back pain. Orthop Clin North Am 2004; 35:57–64.
- Chou R, Qaseem A, Snow V, et al. Diagnosis and treatment of low back pain: a joint clinical practice guideline from the American College of Physicians and the American Pain Society. Ann Intern Med 2007; 147:478–491.
- Chou R, Huffman LHAmerican Pain Society. Medications for acute and chronic low back pain: a review of the evidence for an American Pain Society/American College of Physicians clinical practice guideline. Ann Intern Med 2007; 147:505–514.
- Cherkin DC, Wheeler KJ, Barlow W, Deyo RA. Medication use for low back pain in primary care. Spine 1998; 23:607–614.
- Carette S, Leclaire R, Marcoux S, et al. Epidural corticosteroid injections for sciatica due to herniated nucleus pulposus. N Engl J Med 1997; 336:1634–1640.
- Weinstein JN, Tosteson TD, Lurie JD, et al. Surgical versus non-surgical therapy for lumbar spinal stenosis. N Engl J Med 2008; 358:794–810.
- Deyo RA, Rainville J, Kent DL. What can the history and physical examination tell us about low back pain? JAMA 1992; 268:760–765.
Low back pain should be understood as a remittent, intermittent predicament of life. Its cause is indeterminate, but its course is predictable. Its link to work-related injury is tenuous and confounded by psychosocial issues, including workers’ compensation. It challenges function, compromises performance, and calls for empathy and understanding.1
In this brief paper, we offer a simple approach to one of the most common human afflictions, based on principles and evidence.
WHY IS BACK PAIN IMPORTANT?
Low back pain is common and affects people of all ages. It is second only to the common cold as the most common affliction of mankind, and it is among the leading complaints bringing patients to physicians’ offices. Its lifetime prevalence exceeds 70% in most industrialized countries, with an annual incidence of 15% to 20% in the United States.
Its social and economic impact is substantial. It is the most frequent cause of disability for people under age 45. In 2005, the mean age- and sex-adjusted medical expenditure among respondents with spine problems was $6,096 vs $3,516 in those without spine problems, and it had increased by 65% (adjusted for inflation) from 1997 to 2005.2
WHAT ARE THE GOALS AND PRINCIPLES OF MANAGING LOW BACK PAIN?
The goals of management for patients with low back pain are to:
- Decrease the pain
- Restore mobility
- Hasten recovery so the patient can resume normal daily activities as soon as possible
- Prevent development of a chronic recurrent condition: low back pain is considered acute when it persists for less than 6 weeks, subacute between 6 weeks and 3 months, and chronic when it lasts longer than 3 months
- Restore and preserve physical and financial independence and comfort.
Principles of management
- Most back pain has no recognizable cause and is therefore termed “mechanical” or “musculoskeletal.”
- Underlying systemic disease is rare.
- Most episodes of back pain are unpreventable.
- Confounding psychosocial issues are often contributory, important, and relevant.
- A careful, informed history and physical examination are invaluable; diagnostic studies, however technologically sophisticated, are never a substitute.
- Defer diagnostic studies for specific indications.
- Refer patients only if they have underlying disease or progressive neurologic dysfunction, or if they do not respond to conservative management.
- Encouragement of activity is benign and perhaps salutary for back pain and is desirable for general physical and mental health; there is only scant evidence to support bed rest.3
- Few if any treatments have been proven effective for low back pain.
- Talking to the patient and explaining the issues involved are critical to successful management.4
INITIAL CONSIDERATIONS WHEN EVALUATING A PATIENT
When encountering a patient with back pain, the initial consideration is whether the symptoms are regional—ie, local, mechanical, and musculoskeletal—or if they reflect a systemic disease. It is also important to look for evidence of social or psychological distress that may amplify, prolong, or confound the pain or the patient’s perception of it.
What are the clues to a systemic process?
Does the patient have a regional low back syndrome?
Regional low back syndromes account for 90% of the causes of low back pain. They are usually mechanical in origin.
Regional back pain is due to overuse of a normal mechanical structure (muscle strain, “lumbago”) or is secondary to trauma, deformity, or degeneration of an anatomical structure (herniated nucleus pulposus, fracture, and spondyloarthropathy, including facet joint arthritis). Chronic regional back syndromes include osteoarthritis of the spine (ie, spondylosis), spinal stenosis, and facet joint arthropathy.
Characteristically, mechanical disorders are exacerbated by certain physical activities, such as lifting, and are relieved by others, such as assuming a supine position.
Does the patient have sciatica or another nerve root compression syndrome?
The obvious manifestation of nerve root irritation is usually sciatica, a sharp or burning pain radiating down the posterior or lateral aspect of the leg usually to the foot or the ankle and often associated with numbness or paresthesias. The pain is sometimes aggravated by coughing, sneezing, or the Valsalva maneuver. It is most commonly seen in lumbar disk herniation, cauda equina syndrome, and spinal stenosis.
Might the patient have spinal stenosis?
More than 20% of people over age 60 have radiographic evidence of lumbar spinal canal stenosis, even if they have no symptoms.5 For this reason, the diagnosis of spinal stenosis as a cause of low back pain must be based on the history and physical examination.
The classic history of spinal stenosis is that of neurogenic claudication (“pseudoclaudication”), which is pain that occurs in the legs after walking or prolonged standing and is relieved with sitting. It may sometimes be associated with a varying and transient neurologic deficit. Lumbar flexion increases and lumbar extension decreases the cross-sectional area of the spinal canal—hence, the relief of symptoms of spinal stenosis on stooping or bending forward. Pain is commonly perceived in the back, buttock, or thigh and is elicited by prolonged lumbar extension.
On neurologic examination, about 50% of patients with spinal stenosis have a deficit in vibratory sensibility, temperature sensitivity, or muscle strength. The nerve root involved is most commonly L5, followed by S1 and L4.
Many patients have balance disturbance (wide-based gait or Romberg sign), particularly later in the course of the disorder, with normal cerebellar signs (“pseudocerebellar” presentation).
Patients with bilateral hip osteoarthritis may present with similar symptoms of buttock or thigh pain, which can be distinguished with the above clinical examination. Rotation of the hip is painful in osteoarthritis but not in spinal stenosis. If both conditions overlap, injection of a steroid or lidocaine in the painful hip should decrease the pain associated with hip osteoarthritis.
Does the patient have evidence of neurologic compromise?
Muscle strength is tested by examining the:
- L2 nerve root (which supplies the iliopsoas muscle and is tested by hip flexion)
- L3 nerve root (quadriceps, tested by knee extension)
- L4 nerve root (tibialis anterior, assessed by evaluating ankle dorsiflexion and inversion at the subtalar joint)
- L5 nerve root (extensor hallucis longus and extensor digitorum longus, tested by asking the patient to dorsiflex the great toe, then the other toes)
- S1 nerve root (flexor hallucis longus, flexor digitorum longus, and tendoachilles, tested by asking the patient to plantar-flex the great toe, then the other toes, and then the ankle).
The patient is also asked to walk a few steps on the toes and then on the heels. Inability to toe-walk indicates S1 nerve root involvement; inability to heel-walk may indicate L4 or L5 involvement. If the patient cannot heelwalk, ask him or her to squat; inability to do so indicates L4 problems.6
Radiculopathy. Detecting and locating the cause of radiculopathy may be helpful. In L3–L4 disk herniation, there is pain and paresthesia with numbness and hypalgesia in the anteromedial thigh and the knee. In L4–L5 disk herniation, there is usually involvement of the exiting L5 nerve root, which presents as numbness or paresthesias in the anterolateral calf, great toe, first web space, and medial foot. In L5-S1 disk herniation, the S1 nerve root is involved, presenting as numbness and hypalgesia in the fifth toe, lateral aspect of the foot, sole, and posterolateral calf and thigh.
Reflexes. Exaggerated or decreased reflexes do not always indicate a neurologic abnormality, but reflex asymmetry is significant. The knee-jerk reflex is diminished in L3–L4 nerve root involvement, and the ankle-jerk reflex is diminished with S1 nerve root involvement. The Babinski sign indicates pyramidal tract involvement.
Gait. Observe the patient’s gait as he or she rises and moves to the examining table, to determine whether it is shortened, asymmetrical, or antalgic.7 Also note any foot drop, which may indicate a potentially serious problem (L5 radiculopathy).
What is an adequate examination of the back?
A good back examination can elicit important information about the cause and the extent of back pain. It includes inspection, palpation, and range of movement of the spine along with a detailed neurologic examination.
Inspect it for any deformities, scoliosis, asymmetry, paraspinal muscle spasm, unusual hair growth, listing to one side, decrease or increase in lumbar lordosis, or muscle atrophy or fasciculation.
Palpate it for paraspinal muscle spasm, warmth, and localized bone pain.
Move it. The normal ranges of motion of the lumbar spine are 15 degrees of extension, 40 degrees of flexion, 30 degrees of lateral bending, and 40 degrees of lateral rotation to each side.
Assess it. This includes estimating the tone and nutrition of the muscles, testing their strength (Table 2), examining vibratory or proprioception and pinprick sensation in each dermatome (see below), testing the Achilles and patellar reflexes, and looking for the Babinski sign and clonus. In addition, perform the straight-leg-raising and the cross-straightleg-raising tests, which are positive in most patients with lower lumbar disk herniations.
The femoral stretch test is usually positive in upper lumbar disk herniations (L2–L3, L3– L4). It is performed with the patient in the prone position, with the knee being gradually flexed from full extension. Pain radiating along the anterior aspect of the thigh indicates a positive test.
The examination of the spine must be supplemented with examination of the hip and sacroiliac joints, since back pain may be a referred symptom from any pathology affecting these joints.
When should patients be referred to a specialist?
Patients should be referred to a neurologist, neurosurgeon, orthopedist, or other specialist if they have cauda equina syndrome; severe or progressive neurologic deficits; infections, tumors, or fractures compressing the spinal cord; or, perhaps, no response to conservative therapy for 4 to 6 weeks for patients with a herniated lumbar disk or 8 to 12 weeks for those with spinal stenosis.
If there is profound motor involvement at the time of the initial evaluation, patients must be promptly given systemic corticosteroids such as methylprednisolone (Medrol) or dexamethasone (Decadron) to decrease spinal cord edema.
Are there signs of psychological distress?
Psychosocial factors can significantly affect pain and functional disability in patients who have low back pain.8,9 These are known as “yellow flags” and are better predictors of treatment outcome than physical factors. 10 Anatomically inappropriate signs may be helpful in identifying psychological distress as a result of or as an amplifier of low back symptoms.
Waddell et al11 proposed five categories of these nonorganic signs. These are:
- Inappropriate tenderness that is superficial or widespread
- Pain on simulated axial loading by pressing on the top of the head or simulated spine rotation
- Distraction signs such as inconsistent performance between straight-leg-raising in the seated position vs the supine position
- Regional disturbances in strength and sensation that do not correspond with nerve root innervation patterns
- Overreaction during the physical examination.
The occurrence of any one of the signs is of limited value, but positive findings in three of the five categories suggest psychological distress.11
Which diagnostic studies are useful, cost-effective, and supported by evidence?
Since most abnormalities found on imaging studies are nonspecific, such studies are not necessary during the initial evaluation of acute low back pain unless there are red flags that suggest a more ominous source of pain.
Routine plain lumbosacral spine radiographs with anteroposterior and lateral views may be appropriate initially if the patient has risk factors for vertebral fractures (Table 1), or if the patient does not improve after a course of conservative treatment (usually 4–6 weeks).
Magnetic resonance imaging (MRI) is the preferred test if one suspects a tumor, infection, disk pathology, or spinal stenosis.
Computed tomography (CT) shows bony details better than MRI does. Hence, it is preferred when one needs to evaluate bony details (fractures, scoliosis) and when there are contraindications to MRI, as in patients with metal implant devices and those who are claustrophobic (although now there are “open system” MRI machines, in which the feeling of claustrophobia is much less).
MRI and CT should not be ordered routinely, but only for specific indications to answer specific questions, when specific findings would indicate specific treatment.
In most cases, contrast is not needed for CT or MRI to rule out common causes of low back pain, except in cases of suspected intraspinal tumor. Patients with compromised renal function who need contrast for CT need to be hydrated before the scan to lower the risk of contrast-induced nephropathy. These patients are also at higher risk of nephrogenic fibrosing dermopathy when they receive gadolinium contrast for MRI.
Bone scans can be used to look for infections or fractures not noted on plain radiography. However, MRI provides similar or better diagnostic accuracy without radiation.
Electrodiagnostic studies may be used in patients with radiculopathy when clinical examination suggests multilevel root lesions, when symptoms do not match imaging studies, and when patients have breakaway weakness (fluctuating levels of strength in one or more muscle groups).
Other useful diagnostic and laboratory studies may include the erythrocyte sedimentation rate to screen for malignancy and infection when these are suspected, blood culture for osteomyelitis, and bone aspiration and biopsy for histopathologic diagnosis of infection, malignancy, or other lesions.
WHICH TREATMENTS ARE SUPPORTED BY ROBUST EVIDENCE?
The primary treatment of low back pain should be conservative care, reassurance, and education, allowing patients to improve on their own and helping them cope with their predicament.
Limited bed rest. While 2 or 3 days of limited bed rest may help improve symptoms in patients who have acute radiculopathy, several studies have shown that long periods of bed rest are not beneficial for acute or subacute low back pain.12 Encouraging activity modification allows patients with nonspecific back pain or radicular symptoms to remain active while avoiding activities that may aggravate pain and is shown to lead to a more rapid recovery than bed rest.13,14 The most common situations to avoid are prolonged sitting or standing.15 Low-stress aerobic activities, especially walking, are the best early activities.15
Exercise is one of the only evidence-based, effective treatments for chronic low back pain.16 The most commonly prescribed exercises are aimed at retraining the multifidus (a back muscle) and transversus abdominis (a deep abdominal muscle), supplemented with exercises for the pelvic floor and breathing control.
Nonsteroidal anti-inflammatory drugs (NSAIDs) and acetaminophen (Tylenol) are the drugs of choice for pain control in acute back pain17,18 and are as effective as muscle relaxants or opioids.
Muscle relaxants and opioids offer few advantages over NSAIDs and acetaminophen, except when there is severe muscle spasm associated with the back pain or if acetaminophen or NSAIDs do not relieve the pain. Muscle relaxants and opioids are both associated with more severe adverse effects. If prescribed, they should be used for a short, clearly defined period (1 to 2 weeks).19
Epidural corticosteroids, when used for sciatica, give mild to moderate short-term improvement in leg pain and sensory deficit but no significant long-term functional benefit or reduction in the need for surgery.20
Surgery may be considered in cases of cauda equina syndrome, which is a surgical emergency; severe or progressive neurologic deficit; infections, tumors, and fractures compressing the spinal cord; mechanical instability of the back; and, perhaps, intractable pain (leg pain equal to or greater than back pain) with a positive straight-leg-raising test and no response to conservative therapy.
The term “instability” implies an abnormal motion under physiologic loads. Lumbar instability is defined as translation of more than 4 mm or 10 degrees of angular motion between flexion and extension on an upright lateral radiograph.
Although Weinstein et al21 showed that patients with spinal stenosis who underwent surgery showed significantly more improvement in all primary outcomes than did patients treated nonsurgically, many patients can be effectively treated without surgery.
WHAT SHOULD BE REMEMBERED ABOUT LOW BACK PAIN?
Low back pain is a common and costly medical condition with only a weak correlation between symptoms and pathologic changes, resulting in a lack of objective clinical findings on which a definitive diagnosis can be based.22 Most back pain has no recognizable cause and is usually regional and musculoskeletal. Back pain as a result of an underlying systemic disease is rare and needs to be excluded by a good history and physical examination. Diagnostic studies are best reserved for specific indications.
Referral to a specialist is warranted when the patient is not responding to conservative treatment, when a progressive neurologic deficit or cauda equina syndrome is noted or suspected, or when the patient has an underlying malignancy, infection, fracture, or spinal instability.
Bed rest is best avoided, and activity within the limits of pain is encouraged. NSAIDs and acetaminophen are usually the drugs of choice for controlling acute low back pain.
Ultimately, the goal for clinicians is to identify serious conditions and to prevent the back pain from becoming chronic pain by promptly identifying the various risk factors.
Low back pain should be understood as a remittent, intermittent predicament of life. Its cause is indeterminate, but its course is predictable. Its link to work-related injury is tenuous and confounded by psychosocial issues, including workers’ compensation. It challenges function, compromises performance, and calls for empathy and understanding.1
In this brief paper, we offer a simple approach to one of the most common human afflictions, based on principles and evidence.
WHY IS BACK PAIN IMPORTANT?
Low back pain is common and affects people of all ages. It is second only to the common cold as the most common affliction of mankind, and it is among the leading complaints bringing patients to physicians’ offices. Its lifetime prevalence exceeds 70% in most industrialized countries, with an annual incidence of 15% to 20% in the United States.
Its social and economic impact is substantial. It is the most frequent cause of disability for people under age 45. In 2005, the mean age- and sex-adjusted medical expenditure among respondents with spine problems was $6,096 vs $3,516 in those without spine problems, and it had increased by 65% (adjusted for inflation) from 1997 to 2005.2
WHAT ARE THE GOALS AND PRINCIPLES OF MANAGING LOW BACK PAIN?
The goals of management for patients with low back pain are to:
- Decrease the pain
- Restore mobility
- Hasten recovery so the patient can resume normal daily activities as soon as possible
- Prevent development of a chronic recurrent condition: low back pain is considered acute when it persists for less than 6 weeks, subacute between 6 weeks and 3 months, and chronic when it lasts longer than 3 months
- Restore and preserve physical and financial independence and comfort.
Principles of management
- Most back pain has no recognizable cause and is therefore termed “mechanical” or “musculoskeletal.”
- Underlying systemic disease is rare.
- Most episodes of back pain are unpreventable.
- Confounding psychosocial issues are often contributory, important, and relevant.
- A careful, informed history and physical examination are invaluable; diagnostic studies, however technologically sophisticated, are never a substitute.
- Defer diagnostic studies for specific indications.
- Refer patients only if they have underlying disease or progressive neurologic dysfunction, or if they do not respond to conservative management.
- Encouragement of activity is benign and perhaps salutary for back pain and is desirable for general physical and mental health; there is only scant evidence to support bed rest.3
- Few if any treatments have been proven effective for low back pain.
- Talking to the patient and explaining the issues involved are critical to successful management.4
INITIAL CONSIDERATIONS WHEN EVALUATING A PATIENT
When encountering a patient with back pain, the initial consideration is whether the symptoms are regional—ie, local, mechanical, and musculoskeletal—or if they reflect a systemic disease. It is also important to look for evidence of social or psychological distress that may amplify, prolong, or confound the pain or the patient’s perception of it.
What are the clues to a systemic process?
Does the patient have a regional low back syndrome?
Regional low back syndromes account for 90% of the causes of low back pain. They are usually mechanical in origin.
Regional back pain is due to overuse of a normal mechanical structure (muscle strain, “lumbago”) or is secondary to trauma, deformity, or degeneration of an anatomical structure (herniated nucleus pulposus, fracture, and spondyloarthropathy, including facet joint arthritis). Chronic regional back syndromes include osteoarthritis of the spine (ie, spondylosis), spinal stenosis, and facet joint arthropathy.
Characteristically, mechanical disorders are exacerbated by certain physical activities, such as lifting, and are relieved by others, such as assuming a supine position.
Does the patient have sciatica or another nerve root compression syndrome?
The obvious manifestation of nerve root irritation is usually sciatica, a sharp or burning pain radiating down the posterior or lateral aspect of the leg usually to the foot or the ankle and often associated with numbness or paresthesias. The pain is sometimes aggravated by coughing, sneezing, or the Valsalva maneuver. It is most commonly seen in lumbar disk herniation, cauda equina syndrome, and spinal stenosis.
Might the patient have spinal stenosis?
More than 20% of people over age 60 have radiographic evidence of lumbar spinal canal stenosis, even if they have no symptoms.5 For this reason, the diagnosis of spinal stenosis as a cause of low back pain must be based on the history and physical examination.
The classic history of spinal stenosis is that of neurogenic claudication (“pseudoclaudication”), which is pain that occurs in the legs after walking or prolonged standing and is relieved with sitting. It may sometimes be associated with a varying and transient neurologic deficit. Lumbar flexion increases and lumbar extension decreases the cross-sectional area of the spinal canal—hence, the relief of symptoms of spinal stenosis on stooping or bending forward. Pain is commonly perceived in the back, buttock, or thigh and is elicited by prolonged lumbar extension.
On neurologic examination, about 50% of patients with spinal stenosis have a deficit in vibratory sensibility, temperature sensitivity, or muscle strength. The nerve root involved is most commonly L5, followed by S1 and L4.
Many patients have balance disturbance (wide-based gait or Romberg sign), particularly later in the course of the disorder, with normal cerebellar signs (“pseudocerebellar” presentation).
Patients with bilateral hip osteoarthritis may present with similar symptoms of buttock or thigh pain, which can be distinguished with the above clinical examination. Rotation of the hip is painful in osteoarthritis but not in spinal stenosis. If both conditions overlap, injection of a steroid or lidocaine in the painful hip should decrease the pain associated with hip osteoarthritis.
Does the patient have evidence of neurologic compromise?
Muscle strength is tested by examining the:
- L2 nerve root (which supplies the iliopsoas muscle and is tested by hip flexion)
- L3 nerve root (quadriceps, tested by knee extension)
- L4 nerve root (tibialis anterior, assessed by evaluating ankle dorsiflexion and inversion at the subtalar joint)
- L5 nerve root (extensor hallucis longus and extensor digitorum longus, tested by asking the patient to dorsiflex the great toe, then the other toes)
- S1 nerve root (flexor hallucis longus, flexor digitorum longus, and tendoachilles, tested by asking the patient to plantar-flex the great toe, then the other toes, and then the ankle).
The patient is also asked to walk a few steps on the toes and then on the heels. Inability to toe-walk indicates S1 nerve root involvement; inability to heel-walk may indicate L4 or L5 involvement. If the patient cannot heelwalk, ask him or her to squat; inability to do so indicates L4 problems.6
Radiculopathy. Detecting and locating the cause of radiculopathy may be helpful. In L3–L4 disk herniation, there is pain and paresthesia with numbness and hypalgesia in the anteromedial thigh and the knee. In L4–L5 disk herniation, there is usually involvement of the exiting L5 nerve root, which presents as numbness or paresthesias in the anterolateral calf, great toe, first web space, and medial foot. In L5-S1 disk herniation, the S1 nerve root is involved, presenting as numbness and hypalgesia in the fifth toe, lateral aspect of the foot, sole, and posterolateral calf and thigh.
Reflexes. Exaggerated or decreased reflexes do not always indicate a neurologic abnormality, but reflex asymmetry is significant. The knee-jerk reflex is diminished in L3–L4 nerve root involvement, and the ankle-jerk reflex is diminished with S1 nerve root involvement. The Babinski sign indicates pyramidal tract involvement.
Gait. Observe the patient’s gait as he or she rises and moves to the examining table, to determine whether it is shortened, asymmetrical, or antalgic.7 Also note any foot drop, which may indicate a potentially serious problem (L5 radiculopathy).
What is an adequate examination of the back?
A good back examination can elicit important information about the cause and the extent of back pain. It includes inspection, palpation, and range of movement of the spine along with a detailed neurologic examination.
Inspect it for any deformities, scoliosis, asymmetry, paraspinal muscle spasm, unusual hair growth, listing to one side, decrease or increase in lumbar lordosis, or muscle atrophy or fasciculation.
Palpate it for paraspinal muscle spasm, warmth, and localized bone pain.
Move it. The normal ranges of motion of the lumbar spine are 15 degrees of extension, 40 degrees of flexion, 30 degrees of lateral bending, and 40 degrees of lateral rotation to each side.
Assess it. This includes estimating the tone and nutrition of the muscles, testing their strength (Table 2), examining vibratory or proprioception and pinprick sensation in each dermatome (see below), testing the Achilles and patellar reflexes, and looking for the Babinski sign and clonus. In addition, perform the straight-leg-raising and the cross-straightleg-raising tests, which are positive in most patients with lower lumbar disk herniations.
The femoral stretch test is usually positive in upper lumbar disk herniations (L2–L3, L3– L4). It is performed with the patient in the prone position, with the knee being gradually flexed from full extension. Pain radiating along the anterior aspect of the thigh indicates a positive test.
The examination of the spine must be supplemented with examination of the hip and sacroiliac joints, since back pain may be a referred symptom from any pathology affecting these joints.
When should patients be referred to a specialist?
Patients should be referred to a neurologist, neurosurgeon, orthopedist, or other specialist if they have cauda equina syndrome; severe or progressive neurologic deficits; infections, tumors, or fractures compressing the spinal cord; or, perhaps, no response to conservative therapy for 4 to 6 weeks for patients with a herniated lumbar disk or 8 to 12 weeks for those with spinal stenosis.
If there is profound motor involvement at the time of the initial evaluation, patients must be promptly given systemic corticosteroids such as methylprednisolone (Medrol) or dexamethasone (Decadron) to decrease spinal cord edema.
Are there signs of psychological distress?
Psychosocial factors can significantly affect pain and functional disability in patients who have low back pain.8,9 These are known as “yellow flags” and are better predictors of treatment outcome than physical factors. 10 Anatomically inappropriate signs may be helpful in identifying psychological distress as a result of or as an amplifier of low back symptoms.
Waddell et al11 proposed five categories of these nonorganic signs. These are:
- Inappropriate tenderness that is superficial or widespread
- Pain on simulated axial loading by pressing on the top of the head or simulated spine rotation
- Distraction signs such as inconsistent performance between straight-leg-raising in the seated position vs the supine position
- Regional disturbances in strength and sensation that do not correspond with nerve root innervation patterns
- Overreaction during the physical examination.
The occurrence of any one of the signs is of limited value, but positive findings in three of the five categories suggest psychological distress.11
Which diagnostic studies are useful, cost-effective, and supported by evidence?
Since most abnormalities found on imaging studies are nonspecific, such studies are not necessary during the initial evaluation of acute low back pain unless there are red flags that suggest a more ominous source of pain.
Routine plain lumbosacral spine radiographs with anteroposterior and lateral views may be appropriate initially if the patient has risk factors for vertebral fractures (Table 1), or if the patient does not improve after a course of conservative treatment (usually 4–6 weeks).
Magnetic resonance imaging (MRI) is the preferred test if one suspects a tumor, infection, disk pathology, or spinal stenosis.
Computed tomography (CT) shows bony details better than MRI does. Hence, it is preferred when one needs to evaluate bony details (fractures, scoliosis) and when there are contraindications to MRI, as in patients with metal implant devices and those who are claustrophobic (although now there are “open system” MRI machines, in which the feeling of claustrophobia is much less).
MRI and CT should not be ordered routinely, but only for specific indications to answer specific questions, when specific findings would indicate specific treatment.
In most cases, contrast is not needed for CT or MRI to rule out common causes of low back pain, except in cases of suspected intraspinal tumor. Patients with compromised renal function who need contrast for CT need to be hydrated before the scan to lower the risk of contrast-induced nephropathy. These patients are also at higher risk of nephrogenic fibrosing dermopathy when they receive gadolinium contrast for MRI.
Bone scans can be used to look for infections or fractures not noted on plain radiography. However, MRI provides similar or better diagnostic accuracy without radiation.
Electrodiagnostic studies may be used in patients with radiculopathy when clinical examination suggests multilevel root lesions, when symptoms do not match imaging studies, and when patients have breakaway weakness (fluctuating levels of strength in one or more muscle groups).
Other useful diagnostic and laboratory studies may include the erythrocyte sedimentation rate to screen for malignancy and infection when these are suspected, blood culture for osteomyelitis, and bone aspiration and biopsy for histopathologic diagnosis of infection, malignancy, or other lesions.
WHICH TREATMENTS ARE SUPPORTED BY ROBUST EVIDENCE?
The primary treatment of low back pain should be conservative care, reassurance, and education, allowing patients to improve on their own and helping them cope with their predicament.
Limited bed rest. While 2 or 3 days of limited bed rest may help improve symptoms in patients who have acute radiculopathy, several studies have shown that long periods of bed rest are not beneficial for acute or subacute low back pain.12 Encouraging activity modification allows patients with nonspecific back pain or radicular symptoms to remain active while avoiding activities that may aggravate pain and is shown to lead to a more rapid recovery than bed rest.13,14 The most common situations to avoid are prolonged sitting or standing.15 Low-stress aerobic activities, especially walking, are the best early activities.15
Exercise is one of the only evidence-based, effective treatments for chronic low back pain.16 The most commonly prescribed exercises are aimed at retraining the multifidus (a back muscle) and transversus abdominis (a deep abdominal muscle), supplemented with exercises for the pelvic floor and breathing control.
Nonsteroidal anti-inflammatory drugs (NSAIDs) and acetaminophen (Tylenol) are the drugs of choice for pain control in acute back pain17,18 and are as effective as muscle relaxants or opioids.
Muscle relaxants and opioids offer few advantages over NSAIDs and acetaminophen, except when there is severe muscle spasm associated with the back pain or if acetaminophen or NSAIDs do not relieve the pain. Muscle relaxants and opioids are both associated with more severe adverse effects. If prescribed, they should be used for a short, clearly defined period (1 to 2 weeks).19
Epidural corticosteroids, when used for sciatica, give mild to moderate short-term improvement in leg pain and sensory deficit but no significant long-term functional benefit or reduction in the need for surgery.20
Surgery may be considered in cases of cauda equina syndrome, which is a surgical emergency; severe or progressive neurologic deficit; infections, tumors, and fractures compressing the spinal cord; mechanical instability of the back; and, perhaps, intractable pain (leg pain equal to or greater than back pain) with a positive straight-leg-raising test and no response to conservative therapy.
The term “instability” implies an abnormal motion under physiologic loads. Lumbar instability is defined as translation of more than 4 mm or 10 degrees of angular motion between flexion and extension on an upright lateral radiograph.
Although Weinstein et al21 showed that patients with spinal stenosis who underwent surgery showed significantly more improvement in all primary outcomes than did patients treated nonsurgically, many patients can be effectively treated without surgery.
WHAT SHOULD BE REMEMBERED ABOUT LOW BACK PAIN?
Low back pain is a common and costly medical condition with only a weak correlation between symptoms and pathologic changes, resulting in a lack of objective clinical findings on which a definitive diagnosis can be based.22 Most back pain has no recognizable cause and is usually regional and musculoskeletal. Back pain as a result of an underlying systemic disease is rare and needs to be excluded by a good history and physical examination. Diagnostic studies are best reserved for specific indications.
Referral to a specialist is warranted when the patient is not responding to conservative treatment, when a progressive neurologic deficit or cauda equina syndrome is noted or suspected, or when the patient has an underlying malignancy, infection, fracture, or spinal instability.
Bed rest is best avoided, and activity within the limits of pain is encouraged. NSAIDs and acetaminophen are usually the drugs of choice for controlling acute low back pain.
Ultimately, the goal for clinicians is to identify serious conditions and to prevent the back pain from becoming chronic pain by promptly identifying the various risk factors.
- Hadler NM. Low back pain. In:Koopman WJ, editor. Arthritis and Allied Conditions. 14th ed. Philadelphia, PA: Lipincott Williams and Wilkins; 2001:2026–2041.
- Martin BI, Deyo RA, Mirza SK, et al. Expenditures and health status among adults with back and neck problems. JAMA 2008; 299:656–664.
- NASS Task Force on clinical guidelines. Phase III clinical guidelines for multidisciplinary spine care specialists. Unremitting low back pain. 1st ed. Burr Ridge, IL: North American Spine Society; 2000.
- Cailliet R. Low back pain. In: Soft Tissue Pain and Disability. 3rd ed. Philadelphia, PA: FA Davis; 1996:101–170.
- Jensen MC, Brant-Zawadzki MN, Obuchowski N, Modic MT, Malkasian D, Ross JS. Magnetic resonance imaging of the lumbar spine in people without back pain. N Engl J Med 1994; 331:69–73.
- A gency for Health Care Policy and Research. Acute low back pain problems in adults: assessment and treatment. http://www.chirobase.org/07Strategy/AHCPR/ahcprclinician.html. Accessed March 2009.
- Cohen R, Chopra P, Upshur C. Primary care work-up of acute and chronic symptoms. Geriatrics 2001; 56:26–37.
- Pincus T, Burton AK, Vogel S, Field AP. A systematic review of psychological factors as predictors of chronicity/disability in prospective cohorts of low back pain. Spine 2002; 27:E109–E120.
- Carragee EJ. Clinical practice: persistent low back pain. N Engl J Med 2005; 352:1891–1898.
- Pincus T, Vlaeyen JW, Kendall NA, Von Korff MR, Kalauokalani DA, Reis S. Cognitive-behavioral therapy and psychosocial factors in low back pain: directions for the future. Spine 2002; 27:E133–E138.
- Waddell G, McCulloch JA, Kummel E, Venner RM. Nonorganic physical signs in low back pain. Spine 1980; 5:117–125.
- Hagen KB, Hilde G, Jamtvedt G, Winnem M. Bed rest for acute low-back pain and sciatica. Cochrane Database Syst Rev 2004; 4:CD001254.
- Patel AT, Ogle AA. Diagnosis and management of acute low back pain. Am Fam Physician 2000; 61:1779–1790.
- Grotle M, Brox JI, Glomsrød B, Lønn JH, Vøllestad NK. Prognostic factors in first-time care seekers due to acute low back pain. Eur J Pain 2007; 11:290–298.
- Atlas SJ, Deyo RA. Evaluating and managing acute low back pain in the primary care setting. J Gen Intern Med 2001; 16:120–131.
- Maher CG. Effective physical treatment for low back pain. Orthop Clin North Am 2004; 35:57–64.
- Chou R, Qaseem A, Snow V, et al. Diagnosis and treatment of low back pain: a joint clinical practice guideline from the American College of Physicians and the American Pain Society. Ann Intern Med 2007; 147:478–491.
- Chou R, Huffman LHAmerican Pain Society. Medications for acute and chronic low back pain: a review of the evidence for an American Pain Society/American College of Physicians clinical practice guideline. Ann Intern Med 2007; 147:505–514.
- Cherkin DC, Wheeler KJ, Barlow W, Deyo RA. Medication use for low back pain in primary care. Spine 1998; 23:607–614.
- Carette S, Leclaire R, Marcoux S, et al. Epidural corticosteroid injections for sciatica due to herniated nucleus pulposus. N Engl J Med 1997; 336:1634–1640.
- Weinstein JN, Tosteson TD, Lurie JD, et al. Surgical versus non-surgical therapy for lumbar spinal stenosis. N Engl J Med 2008; 358:794–810.
- Deyo RA, Rainville J, Kent DL. What can the history and physical examination tell us about low back pain? JAMA 1992; 268:760–765.
- Hadler NM. Low back pain. In:Koopman WJ, editor. Arthritis and Allied Conditions. 14th ed. Philadelphia, PA: Lipincott Williams and Wilkins; 2001:2026–2041.
- Martin BI, Deyo RA, Mirza SK, et al. Expenditures and health status among adults with back and neck problems. JAMA 2008; 299:656–664.
- NASS Task Force on clinical guidelines. Phase III clinical guidelines for multidisciplinary spine care specialists. Unremitting low back pain. 1st ed. Burr Ridge, IL: North American Spine Society; 2000.
- Cailliet R. Low back pain. In: Soft Tissue Pain and Disability. 3rd ed. Philadelphia, PA: FA Davis; 1996:101–170.
- Jensen MC, Brant-Zawadzki MN, Obuchowski N, Modic MT, Malkasian D, Ross JS. Magnetic resonance imaging of the lumbar spine in people without back pain. N Engl J Med 1994; 331:69–73.
- A gency for Health Care Policy and Research. Acute low back pain problems in adults: assessment and treatment. http://www.chirobase.org/07Strategy/AHCPR/ahcprclinician.html. Accessed March 2009.
- Cohen R, Chopra P, Upshur C. Primary care work-up of acute and chronic symptoms. Geriatrics 2001; 56:26–37.
- Pincus T, Burton AK, Vogel S, Field AP. A systematic review of psychological factors as predictors of chronicity/disability in prospective cohorts of low back pain. Spine 2002; 27:E109–E120.
- Carragee EJ. Clinical practice: persistent low back pain. N Engl J Med 2005; 352:1891–1898.
- Pincus T, Vlaeyen JW, Kendall NA, Von Korff MR, Kalauokalani DA, Reis S. Cognitive-behavioral therapy and psychosocial factors in low back pain: directions for the future. Spine 2002; 27:E133–E138.
- Waddell G, McCulloch JA, Kummel E, Venner RM. Nonorganic physical signs in low back pain. Spine 1980; 5:117–125.
- Hagen KB, Hilde G, Jamtvedt G, Winnem M. Bed rest for acute low-back pain and sciatica. Cochrane Database Syst Rev 2004; 4:CD001254.
- Patel AT, Ogle AA. Diagnosis and management of acute low back pain. Am Fam Physician 2000; 61:1779–1790.
- Grotle M, Brox JI, Glomsrød B, Lønn JH, Vøllestad NK. Prognostic factors in first-time care seekers due to acute low back pain. Eur J Pain 2007; 11:290–298.
- Atlas SJ, Deyo RA. Evaluating and managing acute low back pain in the primary care setting. J Gen Intern Med 2001; 16:120–131.
- Maher CG. Effective physical treatment for low back pain. Orthop Clin North Am 2004; 35:57–64.
- Chou R, Qaseem A, Snow V, et al. Diagnosis and treatment of low back pain: a joint clinical practice guideline from the American College of Physicians and the American Pain Society. Ann Intern Med 2007; 147:478–491.
- Chou R, Huffman LHAmerican Pain Society. Medications for acute and chronic low back pain: a review of the evidence for an American Pain Society/American College of Physicians clinical practice guideline. Ann Intern Med 2007; 147:505–514.
- Cherkin DC, Wheeler KJ, Barlow W, Deyo RA. Medication use for low back pain in primary care. Spine 1998; 23:607–614.
- Carette S, Leclaire R, Marcoux S, et al. Epidural corticosteroid injections for sciatica due to herniated nucleus pulposus. N Engl J Med 1997; 336:1634–1640.
- Weinstein JN, Tosteson TD, Lurie JD, et al. Surgical versus non-surgical therapy for lumbar spinal stenosis. N Engl J Med 2008; 358:794–810.
- Deyo RA, Rainville J, Kent DL. What can the history and physical examination tell us about low back pain? JAMA 1992; 268:760–765.
KEY POINTS
- Most back pain has no recognizable cause and is therefore termed “mechanical” or “musculoskeletal.” Underlying systemic disease is rare.
- Most episodes of back pain are not preventable.
- Confounding psychosocial issues are common.
- A careful, informed history and physical examination are invaluable; diagnostic studies, however sophisticated, are never a substitute. Defer them for specific indications.
- Refer patients only if they have underlying disease or progressive neurologic dysfunction or do not respond to conservative management.
- Encouragement of activity is benign and perhaps salutary for back pain and is desirable for general physical and mental health. Evidence to support bed rest is scant.
- Few if any treatments have been proven effective for low back pain.