User login
What is the role of probiotics in the treatment of acute Clostridium difficile-associated diarrhea?
Overall, the evidence does not support using probiotics to treat Clostridium difficile-associated diarrhea (CDAD). More studies are needed to determine if they are helpful and, if so, which ones and at what dosages.
WHAT ARE PROBIOTICS?
Probiotics are live bacteria or fungi that carry health benefits when ingested. There is great interest in using these agents to treat and prevent gastrointestinal disorders, as they have been said to inhibit the growth or invasion of pathogenic bacteria, enhance the intestinal barrier, and augment the immune system by regulating cytokines. Their proposed use in treating and preventing CDAD is based on their presumed mechanisms of action and effectiveness in other disorders of the gastrointestinal tract. Given that these readily available bacteria and fungi appear to be safe and well tolerated, their potential use in CDAD is of substantial interest.
LIMITED STUDIES AVAILABLE
Few clinical trials have tested probiotics in CDAD. Two recent systematic reviews did not find a clear benefit to adding probiotics to antibiotics to treat CDAD.1,2 Six trials of various probiotics were included in a 2006 meta-analysis.3 Overall, the analysis did find a benefit, but this was mostly derived from two trials of Saccharomyces boulardii.4,5 This yeast has a mechanism other probiotics do not have: a protease that it produces can degrade the exotoxins produced by C difficile.6
McFarland et al4 gave either S boulardii or placebo to 124 patients who were having either a first episode or a recurrence of CDAD. All patients also received either vancomycin (Vancocin) or metronidazole (Flagyl) in doses chosen by their physician. Patients taking S boulardii were more likely to have their diarrhea resolve and not recur, though post hoc analysis found that this benefit was limited to those with recurrent CDAD.4
Surawicz et al5 gave either S boulardii or placebo to 168 patients with recurrent CDAD who were also participating in a trial comparing vancomycin in a high dose, vancomycin in a low dose, and metronidazole. The probiotic was beneficial, but only in patients on high-dose vancomycin (2 g/day). These patients tended to have a more severe form of CDAD with colitis.
YOGURT, OVER-THE-COUNTER PRODUCTS MAY NOT CONTAIN ACTIVE BACTERIA
The efficacy of over-the-counter probiotic preparations and probiotic-containing foods, such as yogurt, is difficult to determine. For example, in the case of yogurts with “live and active cultures,” the inocula must remain stable from the factory to the grocery store shelf to the table and then through the gastrointestinal tract to the colon. The number of bacteria that survive this long journey is variable.
Another issue is whether probiotic products contain the species and quantities of organisms listed on their labels. In studies that have attempted to examine this issue, many of the products contained species not listed on the label. Most products that did contain viable cells of the stated therapeutic agent did so at a lower number than listed.7,8 The contents and dosages of these over-the-counter products are not regulated and may vary even within the same brand.
The US Food and Drug Administration (FDA) classifies these products as dietary supplements and therefore does not test them for efficacy or safety, though it does have the ability to remove them from the market if they are proven harmful.
FEW ADVERSE EFFECTS
Adverse effects seem to be uncommon with probiotics. Untoward symptoms include flatulence, bloating, and thirst. There are reports of invasive disease, including Lactobacillus bacteremia and Saccharomyces fungemia, occurring after these probiotics were given to patients with severe comorbidities.9,10,11
BENIGN STRAINS OF C DIFFICILE MAY PROTECT AGAINST CDAD
Interestingly, C difficile itself may serve as a probiotic, preventing future episodes of CDAD. Several studies in hamsters showed that colonization with nontoxigenic strains of C difficile can prevent infection with toxigenic strains. In these studies, hamsters receiving clindamycin (Cleocin) or cefoxitin (Mefoxin) were given nontoxigenic strains of C difficile that were either susceptible or resistant to the antibiotic, followed by a toxigenic strain. Those given resistant nontoxigenic strains were significantly less likely to develop CDAD. One study, for example, found that 100% of hamsters given a clindamycin-resistant, nontoxigenic strain of C difficile were protected from CDAD.12
INFECTION CONTROL IS KEY
Novel treatments for CDAD and ways to prevent it are constantly being sought as C difficile has reemerged in hospitals across North America and Europe. However, CDAD is fundamentally a hospital-acquired infection transmitted from patient to patient via the hands of health care workers. The most common predisposing factor is antibiotic use. While new therapeutic advances would be welcome, hand hygiene, basic infection control practice, and judicious use of antimicrobials are essential to decreasing the incidence of this disease.
- Dendukuri N, Costa V, McGregor M, Brophy JM. Probiotic therapy for the prevention and treatment of Clostridium difficile-associated diarrhea: a systematic review. CMAJ 2005; 173:167–170.
- Pillai A, Nelson R. Probiotics for treatment of Clostridium difficile-associated colitis in adults. Cochrane Database Syst Rev 2008;CD004611.
- McFarland LV. Meta-analysis of probiotics for the prevention of antibiotic associated diarrhea and the treatment of Clostridium difficile disease. Am J Gastroenterol 2006; 101:812–822.
- McFarland LV, Surawicz CM, Greenberg RN, et al. A randomized placebo-controlled trial of Saccharomyces boulardii in combination with standard antibiotics for Clostridium difficile disease. JAMA 1994; 271:1913–1918.
- Surawicz CM, McFarland LV, Greenberg RN, et al. The search for a better treatment for recurrent Clostridium difficile disease: use of high-dose vancomycin combined with Saccharomyces boulardii. Clin Infect Dis 2000; 31:1012–1017.
- Castagliuolo I, Riegler MF, Valenick L, LaMont JT, Pothoulakis C. Saccharomyces boulardii protease inhibits the effects of Clostridium difficile toxins A and B in human colonic mucosa. Infect Immun 1999; 67:302–307.
- Huff BA. Caveat emptor. “Probiotics” might not be what they seem. Can Fam Physician 2004; 50:583–587.
- Coeuret V, Gueguen M, Vernoux J. Numbers and strains of lactobacilli in some probiotic products. Int J Food Microbiol 2004; 97:147–156.
- Land MH, Rouster-Stevens K, Woods CR, Cannon ML, Cnota J, Shetty AK. Lactobacillus sepsis associated with probiotic therapy. Pediatrics 2005; 115;178–181.
- Lherm T, Monet C, Nougière B, et al. Seven cases of fungemia with Saccharomyces boulardii in critically ill patients. Intensive Care Med 2002; 28:797–801.
- Salminen MK, Rautelin H, Tynkkynen S, et al. Lactobacillus bacteremia, clinical significance, and patient outcome, with special focus on probiotic L. rhamnosus GG. Clin Infect Dis 2004; 38:62–69.
- Merrigan MM, Sambol SP, Johnson S, Gerding DN. Prevention of fatal Clostridium difficile-associated disease during continuous administration of clindamycin in hamsters. J Infect Dis 2003; 188:1922–1927.
Overall, the evidence does not support using probiotics to treat Clostridium difficile-associated diarrhea (CDAD). More studies are needed to determine if they are helpful and, if so, which ones and at what dosages.
WHAT ARE PROBIOTICS?
Probiotics are live bacteria or fungi that carry health benefits when ingested. There is great interest in using these agents to treat and prevent gastrointestinal disorders, as they have been said to inhibit the growth or invasion of pathogenic bacteria, enhance the intestinal barrier, and augment the immune system by regulating cytokines. Their proposed use in treating and preventing CDAD is based on their presumed mechanisms of action and effectiveness in other disorders of the gastrointestinal tract. Given that these readily available bacteria and fungi appear to be safe and well tolerated, their potential use in CDAD is of substantial interest.
LIMITED STUDIES AVAILABLE
Few clinical trials have tested probiotics in CDAD. Two recent systematic reviews did not find a clear benefit to adding probiotics to antibiotics to treat CDAD.1,2 Six trials of various probiotics were included in a 2006 meta-analysis.3 Overall, the analysis did find a benefit, but this was mostly derived from two trials of Saccharomyces boulardii.4,5 This yeast has a mechanism other probiotics do not have: a protease that it produces can degrade the exotoxins produced by C difficile.6
McFarland et al4 gave either S boulardii or placebo to 124 patients who were having either a first episode or a recurrence of CDAD. All patients also received either vancomycin (Vancocin) or metronidazole (Flagyl) in doses chosen by their physician. Patients taking S boulardii were more likely to have their diarrhea resolve and not recur, though post hoc analysis found that this benefit was limited to those with recurrent CDAD.4
Surawicz et al5 gave either S boulardii or placebo to 168 patients with recurrent CDAD who were also participating in a trial comparing vancomycin in a high dose, vancomycin in a low dose, and metronidazole. The probiotic was beneficial, but only in patients on high-dose vancomycin (2 g/day). These patients tended to have a more severe form of CDAD with colitis.
YOGURT, OVER-THE-COUNTER PRODUCTS MAY NOT CONTAIN ACTIVE BACTERIA
The efficacy of over-the-counter probiotic preparations and probiotic-containing foods, such as yogurt, is difficult to determine. For example, in the case of yogurts with “live and active cultures,” the inocula must remain stable from the factory to the grocery store shelf to the table and then through the gastrointestinal tract to the colon. The number of bacteria that survive this long journey is variable.
Another issue is whether probiotic products contain the species and quantities of organisms listed on their labels. In studies that have attempted to examine this issue, many of the products contained species not listed on the label. Most products that did contain viable cells of the stated therapeutic agent did so at a lower number than listed.7,8 The contents and dosages of these over-the-counter products are not regulated and may vary even within the same brand.
The US Food and Drug Administration (FDA) classifies these products as dietary supplements and therefore does not test them for efficacy or safety, though it does have the ability to remove them from the market if they are proven harmful.
FEW ADVERSE EFFECTS
Adverse effects seem to be uncommon with probiotics. Untoward symptoms include flatulence, bloating, and thirst. There are reports of invasive disease, including Lactobacillus bacteremia and Saccharomyces fungemia, occurring after these probiotics were given to patients with severe comorbidities.9,10,11
BENIGN STRAINS OF C DIFFICILE MAY PROTECT AGAINST CDAD
Interestingly, C difficile itself may serve as a probiotic, preventing future episodes of CDAD. Several studies in hamsters showed that colonization with nontoxigenic strains of C difficile can prevent infection with toxigenic strains. In these studies, hamsters receiving clindamycin (Cleocin) or cefoxitin (Mefoxin) were given nontoxigenic strains of C difficile that were either susceptible or resistant to the antibiotic, followed by a toxigenic strain. Those given resistant nontoxigenic strains were significantly less likely to develop CDAD. One study, for example, found that 100% of hamsters given a clindamycin-resistant, nontoxigenic strain of C difficile were protected from CDAD.12
INFECTION CONTROL IS KEY
Novel treatments for CDAD and ways to prevent it are constantly being sought as C difficile has reemerged in hospitals across North America and Europe. However, CDAD is fundamentally a hospital-acquired infection transmitted from patient to patient via the hands of health care workers. The most common predisposing factor is antibiotic use. While new therapeutic advances would be welcome, hand hygiene, basic infection control practice, and judicious use of antimicrobials are essential to decreasing the incidence of this disease.
Overall, the evidence does not support using probiotics to treat Clostridium difficile-associated diarrhea (CDAD). More studies are needed to determine if they are helpful and, if so, which ones and at what dosages.
WHAT ARE PROBIOTICS?
Probiotics are live bacteria or fungi that carry health benefits when ingested. There is great interest in using these agents to treat and prevent gastrointestinal disorders, as they have been said to inhibit the growth or invasion of pathogenic bacteria, enhance the intestinal barrier, and augment the immune system by regulating cytokines. Their proposed use in treating and preventing CDAD is based on their presumed mechanisms of action and effectiveness in other disorders of the gastrointestinal tract. Given that these readily available bacteria and fungi appear to be safe and well tolerated, their potential use in CDAD is of substantial interest.
LIMITED STUDIES AVAILABLE
Few clinical trials have tested probiotics in CDAD. Two recent systematic reviews did not find a clear benefit to adding probiotics to antibiotics to treat CDAD.1,2 Six trials of various probiotics were included in a 2006 meta-analysis.3 Overall, the analysis did find a benefit, but this was mostly derived from two trials of Saccharomyces boulardii.4,5 This yeast has a mechanism other probiotics do not have: a protease that it produces can degrade the exotoxins produced by C difficile.6
McFarland et al4 gave either S boulardii or placebo to 124 patients who were having either a first episode or a recurrence of CDAD. All patients also received either vancomycin (Vancocin) or metronidazole (Flagyl) in doses chosen by their physician. Patients taking S boulardii were more likely to have their diarrhea resolve and not recur, though post hoc analysis found that this benefit was limited to those with recurrent CDAD.4
Surawicz et al5 gave either S boulardii or placebo to 168 patients with recurrent CDAD who were also participating in a trial comparing vancomycin in a high dose, vancomycin in a low dose, and metronidazole. The probiotic was beneficial, but only in patients on high-dose vancomycin (2 g/day). These patients tended to have a more severe form of CDAD with colitis.
YOGURT, OVER-THE-COUNTER PRODUCTS MAY NOT CONTAIN ACTIVE BACTERIA
The efficacy of over-the-counter probiotic preparations and probiotic-containing foods, such as yogurt, is difficult to determine. For example, in the case of yogurts with “live and active cultures,” the inocula must remain stable from the factory to the grocery store shelf to the table and then through the gastrointestinal tract to the colon. The number of bacteria that survive this long journey is variable.
Another issue is whether probiotic products contain the species and quantities of organisms listed on their labels. In studies that have attempted to examine this issue, many of the products contained species not listed on the label. Most products that did contain viable cells of the stated therapeutic agent did so at a lower number than listed.7,8 The contents and dosages of these over-the-counter products are not regulated and may vary even within the same brand.
The US Food and Drug Administration (FDA) classifies these products as dietary supplements and therefore does not test them for efficacy or safety, though it does have the ability to remove them from the market if they are proven harmful.
FEW ADVERSE EFFECTS
Adverse effects seem to be uncommon with probiotics. Untoward symptoms include flatulence, bloating, and thirst. There are reports of invasive disease, including Lactobacillus bacteremia and Saccharomyces fungemia, occurring after these probiotics were given to patients with severe comorbidities.9,10,11
BENIGN STRAINS OF C DIFFICILE MAY PROTECT AGAINST CDAD
Interestingly, C difficile itself may serve as a probiotic, preventing future episodes of CDAD. Several studies in hamsters showed that colonization with nontoxigenic strains of C difficile can prevent infection with toxigenic strains. In these studies, hamsters receiving clindamycin (Cleocin) or cefoxitin (Mefoxin) were given nontoxigenic strains of C difficile that were either susceptible or resistant to the antibiotic, followed by a toxigenic strain. Those given resistant nontoxigenic strains were significantly less likely to develop CDAD. One study, for example, found that 100% of hamsters given a clindamycin-resistant, nontoxigenic strain of C difficile were protected from CDAD.12
INFECTION CONTROL IS KEY
Novel treatments for CDAD and ways to prevent it are constantly being sought as C difficile has reemerged in hospitals across North America and Europe. However, CDAD is fundamentally a hospital-acquired infection transmitted from patient to patient via the hands of health care workers. The most common predisposing factor is antibiotic use. While new therapeutic advances would be welcome, hand hygiene, basic infection control practice, and judicious use of antimicrobials are essential to decreasing the incidence of this disease.
- Dendukuri N, Costa V, McGregor M, Brophy JM. Probiotic therapy for the prevention and treatment of Clostridium difficile-associated diarrhea: a systematic review. CMAJ 2005; 173:167–170.
- Pillai A, Nelson R. Probiotics for treatment of Clostridium difficile-associated colitis in adults. Cochrane Database Syst Rev 2008;CD004611.
- McFarland LV. Meta-analysis of probiotics for the prevention of antibiotic associated diarrhea and the treatment of Clostridium difficile disease. Am J Gastroenterol 2006; 101:812–822.
- McFarland LV, Surawicz CM, Greenberg RN, et al. A randomized placebo-controlled trial of Saccharomyces boulardii in combination with standard antibiotics for Clostridium difficile disease. JAMA 1994; 271:1913–1918.
- Surawicz CM, McFarland LV, Greenberg RN, et al. The search for a better treatment for recurrent Clostridium difficile disease: use of high-dose vancomycin combined with Saccharomyces boulardii. Clin Infect Dis 2000; 31:1012–1017.
- Castagliuolo I, Riegler MF, Valenick L, LaMont JT, Pothoulakis C. Saccharomyces boulardii protease inhibits the effects of Clostridium difficile toxins A and B in human colonic mucosa. Infect Immun 1999; 67:302–307.
- Huff BA. Caveat emptor. “Probiotics” might not be what they seem. Can Fam Physician 2004; 50:583–587.
- Coeuret V, Gueguen M, Vernoux J. Numbers and strains of lactobacilli in some probiotic products. Int J Food Microbiol 2004; 97:147–156.
- Land MH, Rouster-Stevens K, Woods CR, Cannon ML, Cnota J, Shetty AK. Lactobacillus sepsis associated with probiotic therapy. Pediatrics 2005; 115;178–181.
- Lherm T, Monet C, Nougière B, et al. Seven cases of fungemia with Saccharomyces boulardii in critically ill patients. Intensive Care Med 2002; 28:797–801.
- Salminen MK, Rautelin H, Tynkkynen S, et al. Lactobacillus bacteremia, clinical significance, and patient outcome, with special focus on probiotic L. rhamnosus GG. Clin Infect Dis 2004; 38:62–69.
- Merrigan MM, Sambol SP, Johnson S, Gerding DN. Prevention of fatal Clostridium difficile-associated disease during continuous administration of clindamycin in hamsters. J Infect Dis 2003; 188:1922–1927.
- Dendukuri N, Costa V, McGregor M, Brophy JM. Probiotic therapy for the prevention and treatment of Clostridium difficile-associated diarrhea: a systematic review. CMAJ 2005; 173:167–170.
- Pillai A, Nelson R. Probiotics for treatment of Clostridium difficile-associated colitis in adults. Cochrane Database Syst Rev 2008;CD004611.
- McFarland LV. Meta-analysis of probiotics for the prevention of antibiotic associated diarrhea and the treatment of Clostridium difficile disease. Am J Gastroenterol 2006; 101:812–822.
- McFarland LV, Surawicz CM, Greenberg RN, et al. A randomized placebo-controlled trial of Saccharomyces boulardii in combination with standard antibiotics for Clostridium difficile disease. JAMA 1994; 271:1913–1918.
- Surawicz CM, McFarland LV, Greenberg RN, et al. The search for a better treatment for recurrent Clostridium difficile disease: use of high-dose vancomycin combined with Saccharomyces boulardii. Clin Infect Dis 2000; 31:1012–1017.
- Castagliuolo I, Riegler MF, Valenick L, LaMont JT, Pothoulakis C. Saccharomyces boulardii protease inhibits the effects of Clostridium difficile toxins A and B in human colonic mucosa. Infect Immun 1999; 67:302–307.
- Huff BA. Caveat emptor. “Probiotics” might not be what they seem. Can Fam Physician 2004; 50:583–587.
- Coeuret V, Gueguen M, Vernoux J. Numbers and strains of lactobacilli in some probiotic products. Int J Food Microbiol 2004; 97:147–156.
- Land MH, Rouster-Stevens K, Woods CR, Cannon ML, Cnota J, Shetty AK. Lactobacillus sepsis associated with probiotic therapy. Pediatrics 2005; 115;178–181.
- Lherm T, Monet C, Nougière B, et al. Seven cases of fungemia with Saccharomyces boulardii in critically ill patients. Intensive Care Med 2002; 28:797–801.
- Salminen MK, Rautelin H, Tynkkynen S, et al. Lactobacillus bacteremia, clinical significance, and patient outcome, with special focus on probiotic L. rhamnosus GG. Clin Infect Dis 2004; 38:62–69.
- Merrigan MM, Sambol SP, Johnson S, Gerding DN. Prevention of fatal Clostridium difficile-associated disease during continuous administration of clindamycin in hamsters. J Infect Dis 2003; 188:1922–1927.
Soft tissue atrophy after corticosteroid injection
A 27-year-old woman presents with pain and tenderness over her right radial styloid. Examination reveals tenderness to palpation and a positive Finkelstein test, and her condition is diagnosed as de Quervain tenosynovitis. She is referred for occupational therapy and for a corticosteroid injection.
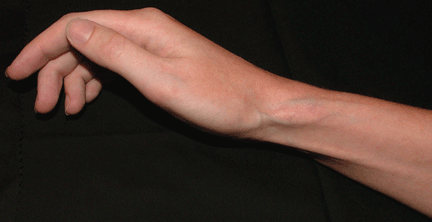
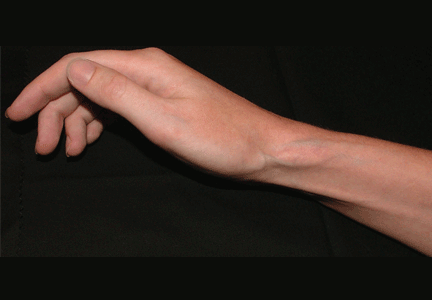
Q: On the basis of the skin findings, which corticosteroid injection was most likely used?
- Triamcinolone hexacetonide (Aristospan)
- Dexamethasone sodium phosphate (Decadron)
- Betamethasone sodium phosphate and betamethasone acetate (Celestone Soluspan)
- Triamcinolone acetonide (Kenalog-40)
A: Both triamcinolone hexacetonide and triamcinolone acetonide are correct, as they are the least soluble of the agents listed.
ADVERSE EFFECTS OF STEROID INJECTIONS
Soft tissue atrophy and local depigmentation are possible adverse effects of any steroid injection, particularly when given at a superficial site.1,2 Although these are rare, with an estimated risk of less than 1%, patients still need to be told about these potential side effects.3 In addition, these adverse effects of injection may be prevented by applying pressure with gauze over the injection site as the needle is withdrawn to prevent leakage of corticosteroid along the needle track.3
Soft tissue atrophy generally appears in 1 to 4 months and resolves 6 to 30 months later. 4 Patients with darker skin are at greater risk of depigmentation.
The cause of the pigment changes is not fully understood but may be related either to the steroid or to the constituents of the vehicle in which the steroid is suspended.5
CHOOSING THE APPROPRIATE STEROID PREPARATION
Although soft tissue (fat) atrophy and local depigmentation are possible with any steroid preparation injected into soft tissue, the risk can be modulated by using a corticosteroid agent with appropriate solubility. A less soluble agent such as triamcinolone acetonide or hexacetonide is preferred for intra-articular injections of deep structures, such as the knee, elbow, or shoulder. A more soluble agent, such as betamethasone sodium phosphate and acetate or dexamethasone sodium phosphate, is preferred for soft tissue injections of bursae, tendon sheaths, metacarpophalangeal joints, proximal phalangeal joints, and the carpal tunnel.
OTHER POSSIBLE COMPLICATIONS
Other potential complications of corticosteroid injection include pain, bleeding, infection (risk 1 in 40,000), flushing, post-injection flare (< 1%), nerve damage, tendon weakening, and rarely, tendon rupture. In cases of tendonitis, it is very important to ensure that the drug is injected into the tendon sheath and not the tendon. A general rule for tendon sheath injection is to not inject if resistance is met.
PATIENT UNWILLING TO RECEIVE MORE INJECTIONS
This patient’s symptoms persist, with a painful right wrist, perhaps due to refractory de Quervain tenosynovitis, nerve damage, or tendinosis. In time, the tenosynovitis and atrophy may improve, but she is reluctant to receive any more injections, as she was not forewarned about the possibility of atrophy.
- Saunders S, Longworth S. Injection Techniques in Orthopaedics and Sports Medicine. 3rd ed. London: Elsevier; 2006.
- Cardone DA, Tallia AF. Joint and soft tissue injection. Am Fam Physician 2002; 66:283–288.
- Gray RG, Gottlieb NL. Intra-articular corticosteroids: an updated assessment. Clin Orthop Relat Res 1983; 177:235–263.
- Cassidy JT, Bole GG. Cutaneous atrophy secondary to intra-articular corticosteroid administration. Ann Intern Med 1966; 65:1008–1018.
- Newman RJ. Local skin depigmentation due to corticosteroid injection. Br Med J (Clin Res Ed) 1984; 288:1725–1726.
A 27-year-old woman presents with pain and tenderness over her right radial styloid. Examination reveals tenderness to palpation and a positive Finkelstein test, and her condition is diagnosed as de Quervain tenosynovitis. She is referred for occupational therapy and for a corticosteroid injection.
Q: On the basis of the skin findings, which corticosteroid injection was most likely used?
- Triamcinolone hexacetonide (Aristospan)
- Dexamethasone sodium phosphate (Decadron)
- Betamethasone sodium phosphate and betamethasone acetate (Celestone Soluspan)
- Triamcinolone acetonide (Kenalog-40)
A: Both triamcinolone hexacetonide and triamcinolone acetonide are correct, as they are the least soluble of the agents listed.
ADVERSE EFFECTS OF STEROID INJECTIONS
Soft tissue atrophy and local depigmentation are possible adverse effects of any steroid injection, particularly when given at a superficial site.1,2 Although these are rare, with an estimated risk of less than 1%, patients still need to be told about these potential side effects.3 In addition, these adverse effects of injection may be prevented by applying pressure with gauze over the injection site as the needle is withdrawn to prevent leakage of corticosteroid along the needle track.3
Soft tissue atrophy generally appears in 1 to 4 months and resolves 6 to 30 months later. 4 Patients with darker skin are at greater risk of depigmentation.
The cause of the pigment changes is not fully understood but may be related either to the steroid or to the constituents of the vehicle in which the steroid is suspended.5
CHOOSING THE APPROPRIATE STEROID PREPARATION
Although soft tissue (fat) atrophy and local depigmentation are possible with any steroid preparation injected into soft tissue, the risk can be modulated by using a corticosteroid agent with appropriate solubility. A less soluble agent such as triamcinolone acetonide or hexacetonide is preferred for intra-articular injections of deep structures, such as the knee, elbow, or shoulder. A more soluble agent, such as betamethasone sodium phosphate and acetate or dexamethasone sodium phosphate, is preferred for soft tissue injections of bursae, tendon sheaths, metacarpophalangeal joints, proximal phalangeal joints, and the carpal tunnel.
OTHER POSSIBLE COMPLICATIONS
Other potential complications of corticosteroid injection include pain, bleeding, infection (risk 1 in 40,000), flushing, post-injection flare (< 1%), nerve damage, tendon weakening, and rarely, tendon rupture. In cases of tendonitis, it is very important to ensure that the drug is injected into the tendon sheath and not the tendon. A general rule for tendon sheath injection is to not inject if resistance is met.
PATIENT UNWILLING TO RECEIVE MORE INJECTIONS
This patient’s symptoms persist, with a painful right wrist, perhaps due to refractory de Quervain tenosynovitis, nerve damage, or tendinosis. In time, the tenosynovitis and atrophy may improve, but she is reluctant to receive any more injections, as she was not forewarned about the possibility of atrophy.
A 27-year-old woman presents with pain and tenderness over her right radial styloid. Examination reveals tenderness to palpation and a positive Finkelstein test, and her condition is diagnosed as de Quervain tenosynovitis. She is referred for occupational therapy and for a corticosteroid injection.
Q: On the basis of the skin findings, which corticosteroid injection was most likely used?
- Triamcinolone hexacetonide (Aristospan)
- Dexamethasone sodium phosphate (Decadron)
- Betamethasone sodium phosphate and betamethasone acetate (Celestone Soluspan)
- Triamcinolone acetonide (Kenalog-40)
A: Both triamcinolone hexacetonide and triamcinolone acetonide are correct, as they are the least soluble of the agents listed.
ADVERSE EFFECTS OF STEROID INJECTIONS
Soft tissue atrophy and local depigmentation are possible adverse effects of any steroid injection, particularly when given at a superficial site.1,2 Although these are rare, with an estimated risk of less than 1%, patients still need to be told about these potential side effects.3 In addition, these adverse effects of injection may be prevented by applying pressure with gauze over the injection site as the needle is withdrawn to prevent leakage of corticosteroid along the needle track.3
Soft tissue atrophy generally appears in 1 to 4 months and resolves 6 to 30 months later. 4 Patients with darker skin are at greater risk of depigmentation.
The cause of the pigment changes is not fully understood but may be related either to the steroid or to the constituents of the vehicle in which the steroid is suspended.5
CHOOSING THE APPROPRIATE STEROID PREPARATION
Although soft tissue (fat) atrophy and local depigmentation are possible with any steroid preparation injected into soft tissue, the risk can be modulated by using a corticosteroid agent with appropriate solubility. A less soluble agent such as triamcinolone acetonide or hexacetonide is preferred for intra-articular injections of deep structures, such as the knee, elbow, or shoulder. A more soluble agent, such as betamethasone sodium phosphate and acetate or dexamethasone sodium phosphate, is preferred for soft tissue injections of bursae, tendon sheaths, metacarpophalangeal joints, proximal phalangeal joints, and the carpal tunnel.
OTHER POSSIBLE COMPLICATIONS
Other potential complications of corticosteroid injection include pain, bleeding, infection (risk 1 in 40,000), flushing, post-injection flare (< 1%), nerve damage, tendon weakening, and rarely, tendon rupture. In cases of tendonitis, it is very important to ensure that the drug is injected into the tendon sheath and not the tendon. A general rule for tendon sheath injection is to not inject if resistance is met.
PATIENT UNWILLING TO RECEIVE MORE INJECTIONS
This patient’s symptoms persist, with a painful right wrist, perhaps due to refractory de Quervain tenosynovitis, nerve damage, or tendinosis. In time, the tenosynovitis and atrophy may improve, but she is reluctant to receive any more injections, as she was not forewarned about the possibility of atrophy.
- Saunders S, Longworth S. Injection Techniques in Orthopaedics and Sports Medicine. 3rd ed. London: Elsevier; 2006.
- Cardone DA, Tallia AF. Joint and soft tissue injection. Am Fam Physician 2002; 66:283–288.
- Gray RG, Gottlieb NL. Intra-articular corticosteroids: an updated assessment. Clin Orthop Relat Res 1983; 177:235–263.
- Cassidy JT, Bole GG. Cutaneous atrophy secondary to intra-articular corticosteroid administration. Ann Intern Med 1966; 65:1008–1018.
- Newman RJ. Local skin depigmentation due to corticosteroid injection. Br Med J (Clin Res Ed) 1984; 288:1725–1726.
- Saunders S, Longworth S. Injection Techniques in Orthopaedics and Sports Medicine. 3rd ed. London: Elsevier; 2006.
- Cardone DA, Tallia AF. Joint and soft tissue injection. Am Fam Physician 2002; 66:283–288.
- Gray RG, Gottlieb NL. Intra-articular corticosteroids: an updated assessment. Clin Orthop Relat Res 1983; 177:235–263.
- Cassidy JT, Bole GG. Cutaneous atrophy secondary to intra-articular corticosteroid administration. Ann Intern Med 1966; 65:1008–1018.
- Newman RJ. Local skin depigmentation due to corticosteroid injection. Br Med J (Clin Res Ed) 1984; 288:1725–1726.
Diffuse hair loss: Its triggers and management
Diffuse hair shedding is distressing. In many cases, the patient notes an increase in hair on the pillow, or when brushing, or in the shower drain.1 It is usually recognized more readily by women than men.1,2 However, diffuse hair loss can affect both sexes at any age.
In this article, we review the triggers of diffuse hair loss and outline an approach to diagnosis and management.
THE NORMAL HAIR CYCLE
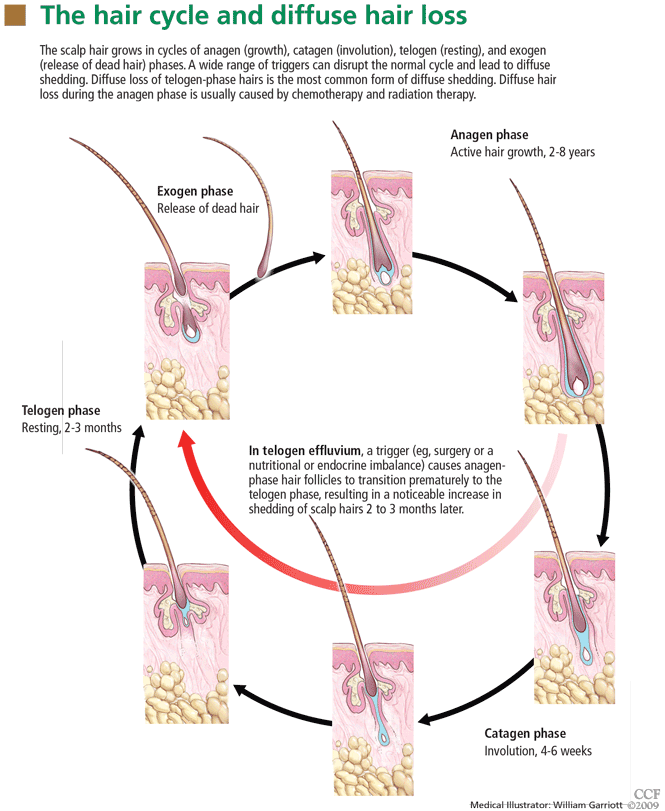
Normally, each hair follicle cycles independently, so that while some hairs are growing, others are resting and others are shedding. Thus, the density of the scalp hair and the total number of scalp hairs remain stable. Most people have about 100,000 scalp hairs, and normally 10% to 15% of these are in the telogen phase.6 Shedding of 100 to 150 telogen hairs per day is normal.5 Anagen hair loss is never normal.
The most common type of diffuse shedding is telogen effluvium, in which anagen-phase hair follicles prematurely transition to the telogen phase, resulting in a noticeable increase in hair shedding at the end of the telogen phase 2 to 3 months later.2,4 Telogen effluvium is a sign of an underlying condition and, thus, is not itself a complete diagnosis.
THE DIFFERENTIAL DIAGNOSIS OF DIFFUSE HAIR LOSS
Telogen Hair Loss
Telogen effluvium has many triggers, and these determine the characteristics of the telogen hair loss.
Telogen effluvium can be acute (lasting < 6 months), chronic (6 months or more), or chronic-repetitive.1,7 If a trigger is acute and short-lived, the telogen effluvium will likely be acute and will resolve. If a trigger is ongoing, if repeated or sequential triggers occur, or if a trigger is not reversed, then the telogen hair shedding can be ongoing.7
Rule out androgenetic alopecia. Important in the differential diagnosis of telogen hair loss is early androgenetic alopecia (pattern hair loss). Early androgenetic alopecia can present as episodic telogen hair shedding before the distinctive pattern of hair loss is seen.8 Androgenetic alopecia is a distinct condition, but the signs of telogen hair shedding can be noted.
Anagen hair loss
Anagen hair shedding is due to the premature termination of anagen hair growth or anagen arrest, after an acute, severe metabolic insult.9 It is most often iatrogenic, caused by treatment with cytotoxic drugs9,10 or radiation.9
Rule out alopecia areata. Important in the differential diagnosis of anagen hair loss is alopecia areata. A detailed history and physical examination to identify the temporal association of possible triggers and any underlying systemic disease should be done in patients with a history of hair shedding. In some cases, further workup is required.
TRIGGERS OF DIFFUSE TELOGEN HAIR LOSS
Triggers of telogen effluvium are numerous.2,4,11–13
Physiologic stress
Physiologic stress such as surgical trauma,4 high fever,4 chronic systemic illness,4 and hemorrhage11 are well known to cause telogen effluvium 2 to 3 months after the insult. Telogen hair shedding can be experienced 2 to 4 months after childbirth (telogen gravidarum).4
Emotional stress
The relationship between emotional stress and hair loss is difficult to ascertain, and hair loss itself is stressful to the patient.14 Historically, acute reversible hair loss occurring with great stress has been reported.11 However, the relationship between chronic diffuse hair loss and psychological stress is controversial.11,14 Evidence for this association appears to be weak, as everyday stresses are likely not enough to trigger hair loss.3,14
Medical conditions
Both hypothyroidism and hyperthyroidism can cause diffuse telogen hair loss that is usually reversible once the euthyroid state is restored.9,11 Chronic systemic disorders such as systemic amyloidosis,14 hepatic failure,4 chronic renal failure,4 inflammatory bowel disease,4,14 and lymphoproliferative disorders2 can cause telogen hair shedding. Telogen hair loss has also been reported in autoimmune diseases such as systemic lupus erythematosus and dermatomyositis,14 as well as in chronic infections such as human immunodeficiency virus type 19 and secondary syphilis.11 Inflammatory disorders such as psoriasis, seborrheic dermatitis, and allergic contact dermatitis can all cause diffuse telogen hair loss.7,15
Dietary triggers
Nutritional causes of diffuse telogen hair loss are zinc deficiency and iron deficiency.11,14 Severe protein, fatty acid and caloric restriction with chronic starvation2,11,14 and crash dieting12 can also induce diffuse telogen hair loss. Malabsorption syndromes and pancreatic disease can precipitate telogen hair shedding.11 Essential fatty acid deficiency can also be associated with diffuse telogen hair shedding usually 2 to 4 months after inadequate intake.11 Vitamin D is an essential vitamin in cell growth, and vitamin D deficiency may be associated with diffuse hair loss.1,7 Biotin deficiency can result in alopecia, but this is a very rare cause of hair loss.14
Drugs that cause hair loss
Drugs can cause telogen hair loss that starts about 12 weeks after starting the drug and continues while on the drug.10 Dosing changes can also precipitate hair shedding.7 Any medication or over-the-counter product the patient is taking should be suspected in hair loss.
Drugs known to cause telogen effluvium are oral contraceptive pills, androgens, retinoids, beta-blockers, angiotensin-converting enzyme inhibitors, anticonvulsants, antidepressants, and the anticoagulants heparin and warfarin (Coumadin).10,14 Changing or stopping any oral contraceptive can precipitate telogen hair shedding.10,14 Oral contraceptives containing an androgenic progestin and hormonal replacement therapy with high-dose progesterone can cause telogen hair shedding with or without patterned alopecia.7,11,14
IDENTIFYING THE TRIGGERS
Normal hair shedding usually goes unnoticed. However, at the onset of telogen effluvium, hair shedding increases by 25%.7
To determine the true trigger of telogen hair loss, the relationship between the trigger and the hair loss must be reproducible, with improvement of the hair shedding following correction of or removal of the trigger, and deterioration on rechallenge.3
In acute telogen effluvium, ie, the acute onset of telogen hair loss 2 to 3 months after an acute, short-lived triggering event,4 a detailed history is important to determine an accurate timeline. No trigger can be identified in some cases.2 Regrowth is not visible for 4 to 6 months.7 If the trigger is identified and removed, recovery can be expected to be complete.4,7
In chronic diffuse telogen hair loss, ie, telogen hair loss lasting more than 6 months,3,14 a range of triggers can precipitate the hair loss. It can be due to idiopathic chronic telogen effluvium. It can also be secondary to prolonged, sequential, or repeated triggers, such as a nutritional deficiency or underlying systemic disorder, and shedding can be less pronounced than in acute telogen effluvium.7
Chronic telogen effluvium is an idiopathic condition with telogen hair shedding lasting longer than 6 months, and with a fluctuating chronic course over many years without an identifiable trigger.16,17 These patients can present with a full head of hair or with bitemporal recession and no widening of the midline part.16,17 Histologic study shows no miniaturization of the hair follicles.17 The diagnosis of chronic telogen effluvium is made by the exclusion of causes of diffuse telogen hair loss, including androgenetic alopecia.
Androgenetic alopecia typically presents as well-defined, patterned scalp hair loss in patients with a family history of androgenetic alopecia. Diffuse hair loss over the vertex and widening of the central part in women, with or without frontal accentuation (“Christmastree” pattern), is characteristic.14,18
The functional mechanism of patterned hair loss is related to a shortening of the anagen phase and a progressive miniaturization of the hair follicles.18 In some instances, androgenetic alopecia may present as diffuse scalp hair loss with episodic increases in telogen hair shedding.8,14 This presentation can be mistaken for other causes of diffuse telogen hair loss.14
Although, most women with patterned hair loss have normal androgen levels,14 androgen excess disorders such as polycystic ovarian syndrome can cause diffuse scalp hair loss or patterned hair loss.7,18 Laboratory testing can exclude other causes of telogen hair loss, and an androgen screen should be performed in women who present with signs of androgen excess, such as irregular menstrual periods, hirsutism, or acne.18 Scalp biopsy can confirm the diagnosis of androgenetic alopecia. 14
ANAGEN HAIR LOSS: KEY FEATURES
Anagen hair loss, the result of interruption of the anagen hair cycle, presents as abrupt anagen hair shedding with a severe diffuse scalp alopecia.9 A serious insult to the hair follicles can cause up to an 80% loss of scalp hair.7 The time course for anagen effluvium is usually rapid compared with telogen effluvium, occurring within days to weeks of the insult to the hair follicles.9 The hair-pull test (see below) is positive for dystrophic anagen hairs with tapered ends.9 If the insult ceases, hair growth restarts again within weeks.
Causes of anagen effluvium include cancer therapies and alopecia areata
Antimitotic chemotherapeutic agents induce arrest of the anagen phase and present a toxic insult to the rapidly dividing hair matrix.9 Hair loss usually begins 1 to 2 weeks after chemotherapy is started and is most noticeable by 1 to 2 months.19 The scalp hair is usually most affected, but all body hair including eyelashes and eyebrows can be affected.10
Other triggers of anagen hair loss include radiation,9 heavy-metal poisoning, and boric acid poisoning.19 Radiation has also been known to cause telogen hair loss and permanent hair loss.9,10
Alopecia areata is another cause of anagen hair shedding.9 This autoimmune condition of the hair20 can cause patchy hair loss, complete hair loss of the scalp (alopecia totalis), or complete loss of scalp and body hair (alopecia universalis).
THE IMPORTANCE OF THE HISTORY IN IDENTIFYING TRIGGERS

The history should concentrate especially on events in the 3 months before the start of the hair loss in the case of telogen hair loss. A history of recent illness or surgery should be recorded. A dietary history is also helpful.21 A detailed drug history including new medications or over-the-counter supplements should be recorded, as should any change in dosages.
As mentioned above, other important factors include recent chemotherapy or radiation therapy, a family history of pattern hair loss such as androgenetic alopecia, oral contraceptive use, and hormone replacement therapy.
PHYSICAL EXAMINATION
Given the complexity of the diagnosis of diffuse hair loss, the clinical examination is of great importance. The scalp should be examined for degree and pattern of hair loss. The hair shafts should be assessed for length, diameter, and breakage.21 The scalp should be examined for inflammation, erythema, and scaling.21
The hair-pull test should be done in all patients with hair loss.22 This involves gentle traction from the base to the tips of a group of 25 to 50 hairs. Normally, only 1 or 2 hairs are dislodged.1 However, in shedding conditions, 10 to 15 hairs can be dislodged.1 Light-microscopy helps differentiate the pulled hairs into telogen hairs or dystrophic anagen hairs.1 Hair shaft microscopy can also indicate nutritional deficiencies.11
A daily count of shed hair can sometimes be useful,22 as can a hair collection.7 A hair collection is done by the patient at home over 2 weeks.7 The shed hair is collected daily at one specific time, usually in the morning, and is placed in dated envelopes. It is important to note the dates of shampooing.7 Daily hair collections of more than 100 hairs per day suggest effluvium.7 Hairs can then be examined and identified as telogen hairs or anagen hairs.
LABORATORY EVALUATION AND SCALP BIOPSY
A laboratory workup can identify triggers or causes of diffuse telogen hair loss. This should include the following:
- A complete blood count and serum ferritin level to look for anemia and iron deficiency
- A thyroid-stimulating hormone and thyroxine (T4) level to detect thyroid disease
- A serum zinc level to detect zinc deficiency
- A comprehensive metabolic panel to exclude chronic renal or liver disease.
If the history and physical examination suggest lupus erythematosus or syphilis, serologic testing can be ordered. Also, an androgen screen should be performed if signs of hyperandrogenism are present18 or if a hormonal cause for the telogen hair loss is suspected.
Scalp biopsy is helpful in most cases of hair loss.21 Lack of identifiable triggers, chronic hair loss, miniaturized hair shafts, and failure to exclude alopecia areata are all indications for scalp biopsy.1,2
Two 4-mm biopsy specimens are recommended to provide for adequate horizontal and vertical sectioning.7 Terminal and vellus hair counts can be done, and the anagen-to-telogen hair ratio can be calculated. In acute telogen effluvium, a reversal of the normal anagen-to-telogen ratio can be seen.23 Miniaturization of the hair shafts and low terminalto-vellus hair counts are seen in androgenetic alopecia.23 Characteristic peribulbar lymphocytic inflammation can be seen in alopecia areata.20
MANAGEMENT: THE IMPORTANCE OF PATIENT EDUCATION
The most important aspect in the management of telogen effluvium is educating the patient about the natural history of the condition. The normal hair cycle should be explained, as well as the relationship between triggers and the timing of hair loss. For example, in telogen effluvium, shedding usually is noted 2 to 3 months after a trigger, although it can in rare cases begin as soon as 2 weeks after a trigger.7
To help identify triggers, a health diary or calendar can be useful. The patient should be instructed to record any stresses, hospital admissions, surgical procedures, new medications, dosage changes, or other potential triggers of hair loss.1,7
The patient should understand that, once the trigger is identified and removed or treated, the shedding settles but can continue for up to 6 months.1 Regrowth can be noted 3 to 6 months after the trigger has been removed, but cosmetically significant regrowth can take 12 to 18 months.1,7
In acute telogen effluvium, if the trigger can be identified and removed, the shedding is short-lived and no further treatment is required.1,4 Patients can be reassured that they are unlikely to go bald.
Adequate nutrition is essential. If a drug is suspected, it should be ceased or changed for at least 3 months to determine whether it is a contributing factor.3 Any underlying scalp inflammation (for example, seborrheic dermatitis or psoriasis) should be treated with an anti-dandruff shampoo and a topical corticosteroid. 1,7
Chronic diffuse telogen hair loss is more complex because multiple sequential or repetitive triggers can be involved.7 Nutritional deficiencies, thyroid disease, systemic illnesses, and infections should be treated.
For acute telogen effluvium, chronic diffuse telogen hair loss, and chronic-repetitive telogen effluvium, biotin and zinc replacement can support hair regrowth.1,7
No specific medical treatment exists for telogen effluvium, but applying the topical hair-growth promoter minoxidil (Rogaine) 2% and 5% to the scalp once a day can be useful in chronic diffuse telogen hair loss and chronic telogen effluvium7 (W. F. Bergfeld, personal communication, November 12, 2008).
In men, medical treatment of androgenetic alopecia includes topical minoxidil 2% or 5% and oral finasteride (Propecia).18 Women can also use topical minoxidil; however, only the 2% solution is approved by the US Food and Drug Administration for female androgenetic alopecia.18 Antiandrogens such as spironolactone (Aldactone) are used off-label for females with androgenetic alopecia. Antiandrogens cause feminization of the male fetus; hence, all women of childbearing years should be on a reliable form of contraceptive.18 Small studies show spironolactone combined with an oral contraceptive can be useful in the treatment of androgenetic alopecia in women.18,24
Anagen hair loss is usually managed with observation and support, as the cause will be obvious from the history. If no iatrogenic cause can be found for anagen hair loss, then other causes such as alopecia areata and heavy-metal poisoning should be investigated and the underlying condition treated.
- Bergfeld WF, Mulinari-Brenner F. Shedding: how to manage a common cause of hair loss. Cleve Clin J Med 2001; 68:256–261.
- Headington JT. Telogen effluvium: new concepts and review. Arch Dermatol 1993; 129:356–363.
- Harrison S, Sinclair R. Telogen effluvium. Clin Exp Dermatol 2002; 27:389–395.
- Kligman AM. Pathologic dynamics of human hair loss. I. Telogen effluvium. Arch Dermatol 1961; 83:175–198.
- Paus R, Cotsarelis G. The biology of hair follicles. N Engl J Med 1999; 341:491–497.
- Rook A, Dawber R. Chapter 1. The comparative physiology, embryology and physiology of human hair. In: Rook A, Dawber R, eds. Diseases of the Hair and Scalp. Oxford, UK: Blackwell Science Publications; 1982:1–17.
- Bergfeld WF. Chapter 9. Telogen effluvium. In: McMichael J, Hordin MK, eds. Hair and Scalp Diseases: Medical, Surgical, and Cosmetic Treatments. London, UK: Informa Health Care; 2008:119–136.
- Sinclair RD, Dawber RP. Androgenetic alopecia in men and women. Clin Dermatol 2001; 19:167–178.
- Sperling LC. Hair and systemic disease. Dermatol Clin 2001; 19:711–726.
- Tosti A, Pazzaglia M. Drug reactions affecting hair: diagnosis. Dermatol Clin 2007; 25:223–231.
- Rook A, Dawber R. Chapter 5. Diffuse alopecia: endocrine, metabolic and chemical influences on the follicular cycle. In: Rook A, Dawber R, eds. Diseases of the Hair and Scalp. Oxford, UK: Blackwell Science Publications; 1982:115–145.
- Goette DK, Odum RB. Alopecia in crash dieters. JAMA 1976; 235:2622–2623.
- Pillans PI, Woods DJ. Drug-induced alopecia. Int J Dermatol 1995; 34:149–158.
- Fiedler VC, Gray AC. Chapter 10. Diffuse alopecia: telogen hair loss. In: Olsen EA, ed. Disorders of Hair Growth: Diagnosis and Treatment. 2nd ed. New York, NY: McGraw-Hill Publishing; 2003:303–320.
- Apache PG. Eczematous dermatitis of the scalp. In: Zviak C, ed. The Science of Hair Care. New York, NY: Marcel Dekker, 1986:513–521.
- Whiting DA. Chronic telogen effluvium. Dermatol Clin 1996; 14:723–731.
- Whiting DA. Chronic telogen effluvium: increased scalp hair shedding in middle-aged women. J Am Acad Dermatol 1996; 35:899–906.
- Olsen EA, Messenger AG, Shapiro J, et al. Evaluation and treatment of male and female pattern hair loss. J Am Acad Dermatol 2005; 52:301–311.
- Sinclair R, Grossman KL, Kvedar JC. Chapter 9: Anagen hair loss. In: Olsen EA, ed. Disorders of Hair Growth: Diagnosis and Treatment. 2nd ed. New York, NY: McGraw-Hill Publishing; 2003:275–302.
- Madani S, Shapiro J. Alopecia areata update. J Am Acad Dermatol 2000; 42:549–566.
- Shapiro J. Clinical practice. Hair loss in women. N Engl J Med 2007; 357:1620–1630.
- Piérard GE, Piérard-Franchimont C, Marks R, Elsner PEEMCO group (European Expert Group on Efficacy Measurement of Cosmetics and other Topical Products). EEMCO guidance for the assessment of hair shedding and alopecia. Skin Pharmacol Physiol 2004; 17:98–110.
- Sellheyer K, Bergfeld WF. Histopathologic evaluation of alopecias. Am J Dermatopathol 2006; 28:236–259.
- Burke BM, Cunliffe WJ. Oral spironolactone therapy for female patients with acne, hirsutism, and androgenetic alopecia. Br J Dermatol 1985; 112:124–125.
Diffuse hair shedding is distressing. In many cases, the patient notes an increase in hair on the pillow, or when brushing, or in the shower drain.1 It is usually recognized more readily by women than men.1,2 However, diffuse hair loss can affect both sexes at any age.
In this article, we review the triggers of diffuse hair loss and outline an approach to diagnosis and management.
THE NORMAL HAIR CYCLE
Normally, each hair follicle cycles independently, so that while some hairs are growing, others are resting and others are shedding. Thus, the density of the scalp hair and the total number of scalp hairs remain stable. Most people have about 100,000 scalp hairs, and normally 10% to 15% of these are in the telogen phase.6 Shedding of 100 to 150 telogen hairs per day is normal.5 Anagen hair loss is never normal.
The most common type of diffuse shedding is telogen effluvium, in which anagen-phase hair follicles prematurely transition to the telogen phase, resulting in a noticeable increase in hair shedding at the end of the telogen phase 2 to 3 months later.2,4 Telogen effluvium is a sign of an underlying condition and, thus, is not itself a complete diagnosis.
THE DIFFERENTIAL DIAGNOSIS OF DIFFUSE HAIR LOSS
Telogen Hair Loss
Telogen effluvium has many triggers, and these determine the characteristics of the telogen hair loss.
Telogen effluvium can be acute (lasting < 6 months), chronic (6 months or more), or chronic-repetitive.1,7 If a trigger is acute and short-lived, the telogen effluvium will likely be acute and will resolve. If a trigger is ongoing, if repeated or sequential triggers occur, or if a trigger is not reversed, then the telogen hair shedding can be ongoing.7
Rule out androgenetic alopecia. Important in the differential diagnosis of telogen hair loss is early androgenetic alopecia (pattern hair loss). Early androgenetic alopecia can present as episodic telogen hair shedding before the distinctive pattern of hair loss is seen.8 Androgenetic alopecia is a distinct condition, but the signs of telogen hair shedding can be noted.
Anagen hair loss
Anagen hair shedding is due to the premature termination of anagen hair growth or anagen arrest, after an acute, severe metabolic insult.9 It is most often iatrogenic, caused by treatment with cytotoxic drugs9,10 or radiation.9
Rule out alopecia areata. Important in the differential diagnosis of anagen hair loss is alopecia areata. A detailed history and physical examination to identify the temporal association of possible triggers and any underlying systemic disease should be done in patients with a history of hair shedding. In some cases, further workup is required.
TRIGGERS OF DIFFUSE TELOGEN HAIR LOSS
Triggers of telogen effluvium are numerous.2,4,11–13
Physiologic stress
Physiologic stress such as surgical trauma,4 high fever,4 chronic systemic illness,4 and hemorrhage11 are well known to cause telogen effluvium 2 to 3 months after the insult. Telogen hair shedding can be experienced 2 to 4 months after childbirth (telogen gravidarum).4
Emotional stress
The relationship between emotional stress and hair loss is difficult to ascertain, and hair loss itself is stressful to the patient.14 Historically, acute reversible hair loss occurring with great stress has been reported.11 However, the relationship between chronic diffuse hair loss and psychological stress is controversial.11,14 Evidence for this association appears to be weak, as everyday stresses are likely not enough to trigger hair loss.3,14
Medical conditions
Both hypothyroidism and hyperthyroidism can cause diffuse telogen hair loss that is usually reversible once the euthyroid state is restored.9,11 Chronic systemic disorders such as systemic amyloidosis,14 hepatic failure,4 chronic renal failure,4 inflammatory bowel disease,4,14 and lymphoproliferative disorders2 can cause telogen hair shedding. Telogen hair loss has also been reported in autoimmune diseases such as systemic lupus erythematosus and dermatomyositis,14 as well as in chronic infections such as human immunodeficiency virus type 19 and secondary syphilis.11 Inflammatory disorders such as psoriasis, seborrheic dermatitis, and allergic contact dermatitis can all cause diffuse telogen hair loss.7,15
Dietary triggers
Nutritional causes of diffuse telogen hair loss are zinc deficiency and iron deficiency.11,14 Severe protein, fatty acid and caloric restriction with chronic starvation2,11,14 and crash dieting12 can also induce diffuse telogen hair loss. Malabsorption syndromes and pancreatic disease can precipitate telogen hair shedding.11 Essential fatty acid deficiency can also be associated with diffuse telogen hair shedding usually 2 to 4 months after inadequate intake.11 Vitamin D is an essential vitamin in cell growth, and vitamin D deficiency may be associated with diffuse hair loss.1,7 Biotin deficiency can result in alopecia, but this is a very rare cause of hair loss.14
Drugs that cause hair loss
Drugs can cause telogen hair loss that starts about 12 weeks after starting the drug and continues while on the drug.10 Dosing changes can also precipitate hair shedding.7 Any medication or over-the-counter product the patient is taking should be suspected in hair loss.
Drugs known to cause telogen effluvium are oral contraceptive pills, androgens, retinoids, beta-blockers, angiotensin-converting enzyme inhibitors, anticonvulsants, antidepressants, and the anticoagulants heparin and warfarin (Coumadin).10,14 Changing or stopping any oral contraceptive can precipitate telogen hair shedding.10,14 Oral contraceptives containing an androgenic progestin and hormonal replacement therapy with high-dose progesterone can cause telogen hair shedding with or without patterned alopecia.7,11,14
IDENTIFYING THE TRIGGERS
Normal hair shedding usually goes unnoticed. However, at the onset of telogen effluvium, hair shedding increases by 25%.7
To determine the true trigger of telogen hair loss, the relationship between the trigger and the hair loss must be reproducible, with improvement of the hair shedding following correction of or removal of the trigger, and deterioration on rechallenge.3
In acute telogen effluvium, ie, the acute onset of telogen hair loss 2 to 3 months after an acute, short-lived triggering event,4 a detailed history is important to determine an accurate timeline. No trigger can be identified in some cases.2 Regrowth is not visible for 4 to 6 months.7 If the trigger is identified and removed, recovery can be expected to be complete.4,7
In chronic diffuse telogen hair loss, ie, telogen hair loss lasting more than 6 months,3,14 a range of triggers can precipitate the hair loss. It can be due to idiopathic chronic telogen effluvium. It can also be secondary to prolonged, sequential, or repeated triggers, such as a nutritional deficiency or underlying systemic disorder, and shedding can be less pronounced than in acute telogen effluvium.7
Chronic telogen effluvium is an idiopathic condition with telogen hair shedding lasting longer than 6 months, and with a fluctuating chronic course over many years without an identifiable trigger.16,17 These patients can present with a full head of hair or with bitemporal recession and no widening of the midline part.16,17 Histologic study shows no miniaturization of the hair follicles.17 The diagnosis of chronic telogen effluvium is made by the exclusion of causes of diffuse telogen hair loss, including androgenetic alopecia.
Androgenetic alopecia typically presents as well-defined, patterned scalp hair loss in patients with a family history of androgenetic alopecia. Diffuse hair loss over the vertex and widening of the central part in women, with or without frontal accentuation (“Christmastree” pattern), is characteristic.14,18
The functional mechanism of patterned hair loss is related to a shortening of the anagen phase and a progressive miniaturization of the hair follicles.18 In some instances, androgenetic alopecia may present as diffuse scalp hair loss with episodic increases in telogen hair shedding.8,14 This presentation can be mistaken for other causes of diffuse telogen hair loss.14
Although, most women with patterned hair loss have normal androgen levels,14 androgen excess disorders such as polycystic ovarian syndrome can cause diffuse scalp hair loss or patterned hair loss.7,18 Laboratory testing can exclude other causes of telogen hair loss, and an androgen screen should be performed in women who present with signs of androgen excess, such as irregular menstrual periods, hirsutism, or acne.18 Scalp biopsy can confirm the diagnosis of androgenetic alopecia. 14
ANAGEN HAIR LOSS: KEY FEATURES
Anagen hair loss, the result of interruption of the anagen hair cycle, presents as abrupt anagen hair shedding with a severe diffuse scalp alopecia.9 A serious insult to the hair follicles can cause up to an 80% loss of scalp hair.7 The time course for anagen effluvium is usually rapid compared with telogen effluvium, occurring within days to weeks of the insult to the hair follicles.9 The hair-pull test (see below) is positive for dystrophic anagen hairs with tapered ends.9 If the insult ceases, hair growth restarts again within weeks.
Causes of anagen effluvium include cancer therapies and alopecia areata
Antimitotic chemotherapeutic agents induce arrest of the anagen phase and present a toxic insult to the rapidly dividing hair matrix.9 Hair loss usually begins 1 to 2 weeks after chemotherapy is started and is most noticeable by 1 to 2 months.19 The scalp hair is usually most affected, but all body hair including eyelashes and eyebrows can be affected.10
Other triggers of anagen hair loss include radiation,9 heavy-metal poisoning, and boric acid poisoning.19 Radiation has also been known to cause telogen hair loss and permanent hair loss.9,10
Alopecia areata is another cause of anagen hair shedding.9 This autoimmune condition of the hair20 can cause patchy hair loss, complete hair loss of the scalp (alopecia totalis), or complete loss of scalp and body hair (alopecia universalis).
THE IMPORTANCE OF THE HISTORY IN IDENTIFYING TRIGGERS
The history should concentrate especially on events in the 3 months before the start of the hair loss in the case of telogen hair loss. A history of recent illness or surgery should be recorded. A dietary history is also helpful.21 A detailed drug history including new medications or over-the-counter supplements should be recorded, as should any change in dosages.
As mentioned above, other important factors include recent chemotherapy or radiation therapy, a family history of pattern hair loss such as androgenetic alopecia, oral contraceptive use, and hormone replacement therapy.
PHYSICAL EXAMINATION
Given the complexity of the diagnosis of diffuse hair loss, the clinical examination is of great importance. The scalp should be examined for degree and pattern of hair loss. The hair shafts should be assessed for length, diameter, and breakage.21 The scalp should be examined for inflammation, erythema, and scaling.21
The hair-pull test should be done in all patients with hair loss.22 This involves gentle traction from the base to the tips of a group of 25 to 50 hairs. Normally, only 1 or 2 hairs are dislodged.1 However, in shedding conditions, 10 to 15 hairs can be dislodged.1 Light-microscopy helps differentiate the pulled hairs into telogen hairs or dystrophic anagen hairs.1 Hair shaft microscopy can also indicate nutritional deficiencies.11
A daily count of shed hair can sometimes be useful,22 as can a hair collection.7 A hair collection is done by the patient at home over 2 weeks.7 The shed hair is collected daily at one specific time, usually in the morning, and is placed in dated envelopes. It is important to note the dates of shampooing.7 Daily hair collections of more than 100 hairs per day suggest effluvium.7 Hairs can then be examined and identified as telogen hairs or anagen hairs.
LABORATORY EVALUATION AND SCALP BIOPSY
A laboratory workup can identify triggers or causes of diffuse telogen hair loss. This should include the following:
- A complete blood count and serum ferritin level to look for anemia and iron deficiency
- A thyroid-stimulating hormone and thyroxine (T4) level to detect thyroid disease
- A serum zinc level to detect zinc deficiency
- A comprehensive metabolic panel to exclude chronic renal or liver disease.
If the history and physical examination suggest lupus erythematosus or syphilis, serologic testing can be ordered. Also, an androgen screen should be performed if signs of hyperandrogenism are present18 or if a hormonal cause for the telogen hair loss is suspected.
Scalp biopsy is helpful in most cases of hair loss.21 Lack of identifiable triggers, chronic hair loss, miniaturized hair shafts, and failure to exclude alopecia areata are all indications for scalp biopsy.1,2
Two 4-mm biopsy specimens are recommended to provide for adequate horizontal and vertical sectioning.7 Terminal and vellus hair counts can be done, and the anagen-to-telogen hair ratio can be calculated. In acute telogen effluvium, a reversal of the normal anagen-to-telogen ratio can be seen.23 Miniaturization of the hair shafts and low terminalto-vellus hair counts are seen in androgenetic alopecia.23 Characteristic peribulbar lymphocytic inflammation can be seen in alopecia areata.20
MANAGEMENT: THE IMPORTANCE OF PATIENT EDUCATION
The most important aspect in the management of telogen effluvium is educating the patient about the natural history of the condition. The normal hair cycle should be explained, as well as the relationship between triggers and the timing of hair loss. For example, in telogen effluvium, shedding usually is noted 2 to 3 months after a trigger, although it can in rare cases begin as soon as 2 weeks after a trigger.7
To help identify triggers, a health diary or calendar can be useful. The patient should be instructed to record any stresses, hospital admissions, surgical procedures, new medications, dosage changes, or other potential triggers of hair loss.1,7
The patient should understand that, once the trigger is identified and removed or treated, the shedding settles but can continue for up to 6 months.1 Regrowth can be noted 3 to 6 months after the trigger has been removed, but cosmetically significant regrowth can take 12 to 18 months.1,7
In acute telogen effluvium, if the trigger can be identified and removed, the shedding is short-lived and no further treatment is required.1,4 Patients can be reassured that they are unlikely to go bald.
Adequate nutrition is essential. If a drug is suspected, it should be ceased or changed for at least 3 months to determine whether it is a contributing factor.3 Any underlying scalp inflammation (for example, seborrheic dermatitis or psoriasis) should be treated with an anti-dandruff shampoo and a topical corticosteroid. 1,7
Chronic diffuse telogen hair loss is more complex because multiple sequential or repetitive triggers can be involved.7 Nutritional deficiencies, thyroid disease, systemic illnesses, and infections should be treated.
For acute telogen effluvium, chronic diffuse telogen hair loss, and chronic-repetitive telogen effluvium, biotin and zinc replacement can support hair regrowth.1,7
No specific medical treatment exists for telogen effluvium, but applying the topical hair-growth promoter minoxidil (Rogaine) 2% and 5% to the scalp once a day can be useful in chronic diffuse telogen hair loss and chronic telogen effluvium7 (W. F. Bergfeld, personal communication, November 12, 2008).
In men, medical treatment of androgenetic alopecia includes topical minoxidil 2% or 5% and oral finasteride (Propecia).18 Women can also use topical minoxidil; however, only the 2% solution is approved by the US Food and Drug Administration for female androgenetic alopecia.18 Antiandrogens such as spironolactone (Aldactone) are used off-label for females with androgenetic alopecia. Antiandrogens cause feminization of the male fetus; hence, all women of childbearing years should be on a reliable form of contraceptive.18 Small studies show spironolactone combined with an oral contraceptive can be useful in the treatment of androgenetic alopecia in women.18,24
Anagen hair loss is usually managed with observation and support, as the cause will be obvious from the history. If no iatrogenic cause can be found for anagen hair loss, then other causes such as alopecia areata and heavy-metal poisoning should be investigated and the underlying condition treated.
Diffuse hair shedding is distressing. In many cases, the patient notes an increase in hair on the pillow, or when brushing, or in the shower drain.1 It is usually recognized more readily by women than men.1,2 However, diffuse hair loss can affect both sexes at any age.
In this article, we review the triggers of diffuse hair loss and outline an approach to diagnosis and management.
THE NORMAL HAIR CYCLE
Normally, each hair follicle cycles independently, so that while some hairs are growing, others are resting and others are shedding. Thus, the density of the scalp hair and the total number of scalp hairs remain stable. Most people have about 100,000 scalp hairs, and normally 10% to 15% of these are in the telogen phase.6 Shedding of 100 to 150 telogen hairs per day is normal.5 Anagen hair loss is never normal.
The most common type of diffuse shedding is telogen effluvium, in which anagen-phase hair follicles prematurely transition to the telogen phase, resulting in a noticeable increase in hair shedding at the end of the telogen phase 2 to 3 months later.2,4 Telogen effluvium is a sign of an underlying condition and, thus, is not itself a complete diagnosis.
THE DIFFERENTIAL DIAGNOSIS OF DIFFUSE HAIR LOSS
Telogen Hair Loss
Telogen effluvium has many triggers, and these determine the characteristics of the telogen hair loss.
Telogen effluvium can be acute (lasting < 6 months), chronic (6 months or more), or chronic-repetitive.1,7 If a trigger is acute and short-lived, the telogen effluvium will likely be acute and will resolve. If a trigger is ongoing, if repeated or sequential triggers occur, or if a trigger is not reversed, then the telogen hair shedding can be ongoing.7
Rule out androgenetic alopecia. Important in the differential diagnosis of telogen hair loss is early androgenetic alopecia (pattern hair loss). Early androgenetic alopecia can present as episodic telogen hair shedding before the distinctive pattern of hair loss is seen.8 Androgenetic alopecia is a distinct condition, but the signs of telogen hair shedding can be noted.
Anagen hair loss
Anagen hair shedding is due to the premature termination of anagen hair growth or anagen arrest, after an acute, severe metabolic insult.9 It is most often iatrogenic, caused by treatment with cytotoxic drugs9,10 or radiation.9
Rule out alopecia areata. Important in the differential diagnosis of anagen hair loss is alopecia areata. A detailed history and physical examination to identify the temporal association of possible triggers and any underlying systemic disease should be done in patients with a history of hair shedding. In some cases, further workup is required.
TRIGGERS OF DIFFUSE TELOGEN HAIR LOSS
Triggers of telogen effluvium are numerous.2,4,11–13
Physiologic stress
Physiologic stress such as surgical trauma,4 high fever,4 chronic systemic illness,4 and hemorrhage11 are well known to cause telogen effluvium 2 to 3 months after the insult. Telogen hair shedding can be experienced 2 to 4 months after childbirth (telogen gravidarum).4
Emotional stress
The relationship between emotional stress and hair loss is difficult to ascertain, and hair loss itself is stressful to the patient.14 Historically, acute reversible hair loss occurring with great stress has been reported.11 However, the relationship between chronic diffuse hair loss and psychological stress is controversial.11,14 Evidence for this association appears to be weak, as everyday stresses are likely not enough to trigger hair loss.3,14
Medical conditions
Both hypothyroidism and hyperthyroidism can cause diffuse telogen hair loss that is usually reversible once the euthyroid state is restored.9,11 Chronic systemic disorders such as systemic amyloidosis,14 hepatic failure,4 chronic renal failure,4 inflammatory bowel disease,4,14 and lymphoproliferative disorders2 can cause telogen hair shedding. Telogen hair loss has also been reported in autoimmune diseases such as systemic lupus erythematosus and dermatomyositis,14 as well as in chronic infections such as human immunodeficiency virus type 19 and secondary syphilis.11 Inflammatory disorders such as psoriasis, seborrheic dermatitis, and allergic contact dermatitis can all cause diffuse telogen hair loss.7,15
Dietary triggers
Nutritional causes of diffuse telogen hair loss are zinc deficiency and iron deficiency.11,14 Severe protein, fatty acid and caloric restriction with chronic starvation2,11,14 and crash dieting12 can also induce diffuse telogen hair loss. Malabsorption syndromes and pancreatic disease can precipitate telogen hair shedding.11 Essential fatty acid deficiency can also be associated with diffuse telogen hair shedding usually 2 to 4 months after inadequate intake.11 Vitamin D is an essential vitamin in cell growth, and vitamin D deficiency may be associated with diffuse hair loss.1,7 Biotin deficiency can result in alopecia, but this is a very rare cause of hair loss.14
Drugs that cause hair loss
Drugs can cause telogen hair loss that starts about 12 weeks after starting the drug and continues while on the drug.10 Dosing changes can also precipitate hair shedding.7 Any medication or over-the-counter product the patient is taking should be suspected in hair loss.
Drugs known to cause telogen effluvium are oral contraceptive pills, androgens, retinoids, beta-blockers, angiotensin-converting enzyme inhibitors, anticonvulsants, antidepressants, and the anticoagulants heparin and warfarin (Coumadin).10,14 Changing or stopping any oral contraceptive can precipitate telogen hair shedding.10,14 Oral contraceptives containing an androgenic progestin and hormonal replacement therapy with high-dose progesterone can cause telogen hair shedding with or without patterned alopecia.7,11,14
IDENTIFYING THE TRIGGERS
Normal hair shedding usually goes unnoticed. However, at the onset of telogen effluvium, hair shedding increases by 25%.7
To determine the true trigger of telogen hair loss, the relationship between the trigger and the hair loss must be reproducible, with improvement of the hair shedding following correction of or removal of the trigger, and deterioration on rechallenge.3
In acute telogen effluvium, ie, the acute onset of telogen hair loss 2 to 3 months after an acute, short-lived triggering event,4 a detailed history is important to determine an accurate timeline. No trigger can be identified in some cases.2 Regrowth is not visible for 4 to 6 months.7 If the trigger is identified and removed, recovery can be expected to be complete.4,7
In chronic diffuse telogen hair loss, ie, telogen hair loss lasting more than 6 months,3,14 a range of triggers can precipitate the hair loss. It can be due to idiopathic chronic telogen effluvium. It can also be secondary to prolonged, sequential, or repeated triggers, such as a nutritional deficiency or underlying systemic disorder, and shedding can be less pronounced than in acute telogen effluvium.7
Chronic telogen effluvium is an idiopathic condition with telogen hair shedding lasting longer than 6 months, and with a fluctuating chronic course over many years without an identifiable trigger.16,17 These patients can present with a full head of hair or with bitemporal recession and no widening of the midline part.16,17 Histologic study shows no miniaturization of the hair follicles.17 The diagnosis of chronic telogen effluvium is made by the exclusion of causes of diffuse telogen hair loss, including androgenetic alopecia.
Androgenetic alopecia typically presents as well-defined, patterned scalp hair loss in patients with a family history of androgenetic alopecia. Diffuse hair loss over the vertex and widening of the central part in women, with or without frontal accentuation (“Christmastree” pattern), is characteristic.14,18
The functional mechanism of patterned hair loss is related to a shortening of the anagen phase and a progressive miniaturization of the hair follicles.18 In some instances, androgenetic alopecia may present as diffuse scalp hair loss with episodic increases in telogen hair shedding.8,14 This presentation can be mistaken for other causes of diffuse telogen hair loss.14
Although, most women with patterned hair loss have normal androgen levels,14 androgen excess disorders such as polycystic ovarian syndrome can cause diffuse scalp hair loss or patterned hair loss.7,18 Laboratory testing can exclude other causes of telogen hair loss, and an androgen screen should be performed in women who present with signs of androgen excess, such as irregular menstrual periods, hirsutism, or acne.18 Scalp biopsy can confirm the diagnosis of androgenetic alopecia. 14
ANAGEN HAIR LOSS: KEY FEATURES
Anagen hair loss, the result of interruption of the anagen hair cycle, presents as abrupt anagen hair shedding with a severe diffuse scalp alopecia.9 A serious insult to the hair follicles can cause up to an 80% loss of scalp hair.7 The time course for anagen effluvium is usually rapid compared with telogen effluvium, occurring within days to weeks of the insult to the hair follicles.9 The hair-pull test (see below) is positive for dystrophic anagen hairs with tapered ends.9 If the insult ceases, hair growth restarts again within weeks.
Causes of anagen effluvium include cancer therapies and alopecia areata
Antimitotic chemotherapeutic agents induce arrest of the anagen phase and present a toxic insult to the rapidly dividing hair matrix.9 Hair loss usually begins 1 to 2 weeks after chemotherapy is started and is most noticeable by 1 to 2 months.19 The scalp hair is usually most affected, but all body hair including eyelashes and eyebrows can be affected.10
Other triggers of anagen hair loss include radiation,9 heavy-metal poisoning, and boric acid poisoning.19 Radiation has also been known to cause telogen hair loss and permanent hair loss.9,10
Alopecia areata is another cause of anagen hair shedding.9 This autoimmune condition of the hair20 can cause patchy hair loss, complete hair loss of the scalp (alopecia totalis), or complete loss of scalp and body hair (alopecia universalis).
THE IMPORTANCE OF THE HISTORY IN IDENTIFYING TRIGGERS
The history should concentrate especially on events in the 3 months before the start of the hair loss in the case of telogen hair loss. A history of recent illness or surgery should be recorded. A dietary history is also helpful.21 A detailed drug history including new medications or over-the-counter supplements should be recorded, as should any change in dosages.
As mentioned above, other important factors include recent chemotherapy or radiation therapy, a family history of pattern hair loss such as androgenetic alopecia, oral contraceptive use, and hormone replacement therapy.
PHYSICAL EXAMINATION
Given the complexity of the diagnosis of diffuse hair loss, the clinical examination is of great importance. The scalp should be examined for degree and pattern of hair loss. The hair shafts should be assessed for length, diameter, and breakage.21 The scalp should be examined for inflammation, erythema, and scaling.21
The hair-pull test should be done in all patients with hair loss.22 This involves gentle traction from the base to the tips of a group of 25 to 50 hairs. Normally, only 1 or 2 hairs are dislodged.1 However, in shedding conditions, 10 to 15 hairs can be dislodged.1 Light-microscopy helps differentiate the pulled hairs into telogen hairs or dystrophic anagen hairs.1 Hair shaft microscopy can also indicate nutritional deficiencies.11
A daily count of shed hair can sometimes be useful,22 as can a hair collection.7 A hair collection is done by the patient at home over 2 weeks.7 The shed hair is collected daily at one specific time, usually in the morning, and is placed in dated envelopes. It is important to note the dates of shampooing.7 Daily hair collections of more than 100 hairs per day suggest effluvium.7 Hairs can then be examined and identified as telogen hairs or anagen hairs.
LABORATORY EVALUATION AND SCALP BIOPSY
A laboratory workup can identify triggers or causes of diffuse telogen hair loss. This should include the following:
- A complete blood count and serum ferritin level to look for anemia and iron deficiency
- A thyroid-stimulating hormone and thyroxine (T4) level to detect thyroid disease
- A serum zinc level to detect zinc deficiency
- A comprehensive metabolic panel to exclude chronic renal or liver disease.
If the history and physical examination suggest lupus erythematosus or syphilis, serologic testing can be ordered. Also, an androgen screen should be performed if signs of hyperandrogenism are present18 or if a hormonal cause for the telogen hair loss is suspected.
Scalp biopsy is helpful in most cases of hair loss.21 Lack of identifiable triggers, chronic hair loss, miniaturized hair shafts, and failure to exclude alopecia areata are all indications for scalp biopsy.1,2
Two 4-mm biopsy specimens are recommended to provide for adequate horizontal and vertical sectioning.7 Terminal and vellus hair counts can be done, and the anagen-to-telogen hair ratio can be calculated. In acute telogen effluvium, a reversal of the normal anagen-to-telogen ratio can be seen.23 Miniaturization of the hair shafts and low terminalto-vellus hair counts are seen in androgenetic alopecia.23 Characteristic peribulbar lymphocytic inflammation can be seen in alopecia areata.20
MANAGEMENT: THE IMPORTANCE OF PATIENT EDUCATION
The most important aspect in the management of telogen effluvium is educating the patient about the natural history of the condition. The normal hair cycle should be explained, as well as the relationship between triggers and the timing of hair loss. For example, in telogen effluvium, shedding usually is noted 2 to 3 months after a trigger, although it can in rare cases begin as soon as 2 weeks after a trigger.7
To help identify triggers, a health diary or calendar can be useful. The patient should be instructed to record any stresses, hospital admissions, surgical procedures, new medications, dosage changes, or other potential triggers of hair loss.1,7
The patient should understand that, once the trigger is identified and removed or treated, the shedding settles but can continue for up to 6 months.1 Regrowth can be noted 3 to 6 months after the trigger has been removed, but cosmetically significant regrowth can take 12 to 18 months.1,7
In acute telogen effluvium, if the trigger can be identified and removed, the shedding is short-lived and no further treatment is required.1,4 Patients can be reassured that they are unlikely to go bald.
Adequate nutrition is essential. If a drug is suspected, it should be ceased or changed for at least 3 months to determine whether it is a contributing factor.3 Any underlying scalp inflammation (for example, seborrheic dermatitis or psoriasis) should be treated with an anti-dandruff shampoo and a topical corticosteroid. 1,7
Chronic diffuse telogen hair loss is more complex because multiple sequential or repetitive triggers can be involved.7 Nutritional deficiencies, thyroid disease, systemic illnesses, and infections should be treated.
For acute telogen effluvium, chronic diffuse telogen hair loss, and chronic-repetitive telogen effluvium, biotin and zinc replacement can support hair regrowth.1,7
No specific medical treatment exists for telogen effluvium, but applying the topical hair-growth promoter minoxidil (Rogaine) 2% and 5% to the scalp once a day can be useful in chronic diffuse telogen hair loss and chronic telogen effluvium7 (W. F. Bergfeld, personal communication, November 12, 2008).
In men, medical treatment of androgenetic alopecia includes topical minoxidil 2% or 5% and oral finasteride (Propecia).18 Women can also use topical minoxidil; however, only the 2% solution is approved by the US Food and Drug Administration for female androgenetic alopecia.18 Antiandrogens such as spironolactone (Aldactone) are used off-label for females with androgenetic alopecia. Antiandrogens cause feminization of the male fetus; hence, all women of childbearing years should be on a reliable form of contraceptive.18 Small studies show spironolactone combined with an oral contraceptive can be useful in the treatment of androgenetic alopecia in women.18,24
Anagen hair loss is usually managed with observation and support, as the cause will be obvious from the history. If no iatrogenic cause can be found for anagen hair loss, then other causes such as alopecia areata and heavy-metal poisoning should be investigated and the underlying condition treated.
- Bergfeld WF, Mulinari-Brenner F. Shedding: how to manage a common cause of hair loss. Cleve Clin J Med 2001; 68:256–261.
- Headington JT. Telogen effluvium: new concepts and review. Arch Dermatol 1993; 129:356–363.
- Harrison S, Sinclair R. Telogen effluvium. Clin Exp Dermatol 2002; 27:389–395.
- Kligman AM. Pathologic dynamics of human hair loss. I. Telogen effluvium. Arch Dermatol 1961; 83:175–198.
- Paus R, Cotsarelis G. The biology of hair follicles. N Engl J Med 1999; 341:491–497.
- Rook A, Dawber R. Chapter 1. The comparative physiology, embryology and physiology of human hair. In: Rook A, Dawber R, eds. Diseases of the Hair and Scalp. Oxford, UK: Blackwell Science Publications; 1982:1–17.
- Bergfeld WF. Chapter 9. Telogen effluvium. In: McMichael J, Hordin MK, eds. Hair and Scalp Diseases: Medical, Surgical, and Cosmetic Treatments. London, UK: Informa Health Care; 2008:119–136.
- Sinclair RD, Dawber RP. Androgenetic alopecia in men and women. Clin Dermatol 2001; 19:167–178.
- Sperling LC. Hair and systemic disease. Dermatol Clin 2001; 19:711–726.
- Tosti A, Pazzaglia M. Drug reactions affecting hair: diagnosis. Dermatol Clin 2007; 25:223–231.
- Rook A, Dawber R. Chapter 5. Diffuse alopecia: endocrine, metabolic and chemical influences on the follicular cycle. In: Rook A, Dawber R, eds. Diseases of the Hair and Scalp. Oxford, UK: Blackwell Science Publications; 1982:115–145.
- Goette DK, Odum RB. Alopecia in crash dieters. JAMA 1976; 235:2622–2623.
- Pillans PI, Woods DJ. Drug-induced alopecia. Int J Dermatol 1995; 34:149–158.
- Fiedler VC, Gray AC. Chapter 10. Diffuse alopecia: telogen hair loss. In: Olsen EA, ed. Disorders of Hair Growth: Diagnosis and Treatment. 2nd ed. New York, NY: McGraw-Hill Publishing; 2003:303–320.
- Apache PG. Eczematous dermatitis of the scalp. In: Zviak C, ed. The Science of Hair Care. New York, NY: Marcel Dekker, 1986:513–521.
- Whiting DA. Chronic telogen effluvium. Dermatol Clin 1996; 14:723–731.
- Whiting DA. Chronic telogen effluvium: increased scalp hair shedding in middle-aged women. J Am Acad Dermatol 1996; 35:899–906.
- Olsen EA, Messenger AG, Shapiro J, et al. Evaluation and treatment of male and female pattern hair loss. J Am Acad Dermatol 2005; 52:301–311.
- Sinclair R, Grossman KL, Kvedar JC. Chapter 9: Anagen hair loss. In: Olsen EA, ed. Disorders of Hair Growth: Diagnosis and Treatment. 2nd ed. New York, NY: McGraw-Hill Publishing; 2003:275–302.
- Madani S, Shapiro J. Alopecia areata update. J Am Acad Dermatol 2000; 42:549–566.
- Shapiro J. Clinical practice. Hair loss in women. N Engl J Med 2007; 357:1620–1630.
- Piérard GE, Piérard-Franchimont C, Marks R, Elsner PEEMCO group (European Expert Group on Efficacy Measurement of Cosmetics and other Topical Products). EEMCO guidance for the assessment of hair shedding and alopecia. Skin Pharmacol Physiol 2004; 17:98–110.
- Sellheyer K, Bergfeld WF. Histopathologic evaluation of alopecias. Am J Dermatopathol 2006; 28:236–259.
- Burke BM, Cunliffe WJ. Oral spironolactone therapy for female patients with acne, hirsutism, and androgenetic alopecia. Br J Dermatol 1985; 112:124–125.
- Bergfeld WF, Mulinari-Brenner F. Shedding: how to manage a common cause of hair loss. Cleve Clin J Med 2001; 68:256–261.
- Headington JT. Telogen effluvium: new concepts and review. Arch Dermatol 1993; 129:356–363.
- Harrison S, Sinclair R. Telogen effluvium. Clin Exp Dermatol 2002; 27:389–395.
- Kligman AM. Pathologic dynamics of human hair loss. I. Telogen effluvium. Arch Dermatol 1961; 83:175–198.
- Paus R, Cotsarelis G. The biology of hair follicles. N Engl J Med 1999; 341:491–497.
- Rook A, Dawber R. Chapter 1. The comparative physiology, embryology and physiology of human hair. In: Rook A, Dawber R, eds. Diseases of the Hair and Scalp. Oxford, UK: Blackwell Science Publications; 1982:1–17.
- Bergfeld WF. Chapter 9. Telogen effluvium. In: McMichael J, Hordin MK, eds. Hair and Scalp Diseases: Medical, Surgical, and Cosmetic Treatments. London, UK: Informa Health Care; 2008:119–136.
- Sinclair RD, Dawber RP. Androgenetic alopecia in men and women. Clin Dermatol 2001; 19:167–178.
- Sperling LC. Hair and systemic disease. Dermatol Clin 2001; 19:711–726.
- Tosti A, Pazzaglia M. Drug reactions affecting hair: diagnosis. Dermatol Clin 2007; 25:223–231.
- Rook A, Dawber R. Chapter 5. Diffuse alopecia: endocrine, metabolic and chemical influences on the follicular cycle. In: Rook A, Dawber R, eds. Diseases of the Hair and Scalp. Oxford, UK: Blackwell Science Publications; 1982:115–145.
- Goette DK, Odum RB. Alopecia in crash dieters. JAMA 1976; 235:2622–2623.
- Pillans PI, Woods DJ. Drug-induced alopecia. Int J Dermatol 1995; 34:149–158.
- Fiedler VC, Gray AC. Chapter 10. Diffuse alopecia: telogen hair loss. In: Olsen EA, ed. Disorders of Hair Growth: Diagnosis and Treatment. 2nd ed. New York, NY: McGraw-Hill Publishing; 2003:303–320.
- Apache PG. Eczematous dermatitis of the scalp. In: Zviak C, ed. The Science of Hair Care. New York, NY: Marcel Dekker, 1986:513–521.
- Whiting DA. Chronic telogen effluvium. Dermatol Clin 1996; 14:723–731.
- Whiting DA. Chronic telogen effluvium: increased scalp hair shedding in middle-aged women. J Am Acad Dermatol 1996; 35:899–906.
- Olsen EA, Messenger AG, Shapiro J, et al. Evaluation and treatment of male and female pattern hair loss. J Am Acad Dermatol 2005; 52:301–311.
- Sinclair R, Grossman KL, Kvedar JC. Chapter 9: Anagen hair loss. In: Olsen EA, ed. Disorders of Hair Growth: Diagnosis and Treatment. 2nd ed. New York, NY: McGraw-Hill Publishing; 2003:275–302.
- Madani S, Shapiro J. Alopecia areata update. J Am Acad Dermatol 2000; 42:549–566.
- Shapiro J. Clinical practice. Hair loss in women. N Engl J Med 2007; 357:1620–1630.
- Piérard GE, Piérard-Franchimont C, Marks R, Elsner PEEMCO group (European Expert Group on Efficacy Measurement of Cosmetics and other Topical Products). EEMCO guidance for the assessment of hair shedding and alopecia. Skin Pharmacol Physiol 2004; 17:98–110.
- Sellheyer K, Bergfeld WF. Histopathologic evaluation of alopecias. Am J Dermatopathol 2006; 28:236–259.
- Burke BM, Cunliffe WJ. Oral spironolactone therapy for female patients with acne, hirsutism, and androgenetic alopecia. Br J Dermatol 1985; 112:124–125.
KEY POINTS
- Early androgenetic alopecia can present as episodic telogen hair shedding, before the distinctive pattern of hair loss is seen.
- Telogen effluvium is a sign of an underlying condition and, thus, is not itself a complete diagnosis.
- Androgenetic alopecia should not be overlooked as an important cause of diffuse telogen hair shedding.
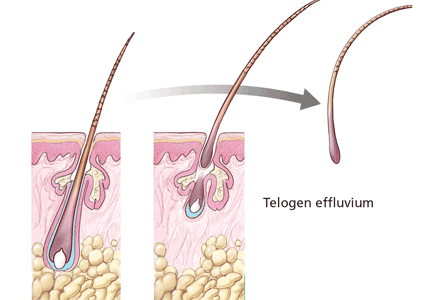
Small fiber neuropathy: A burning problem
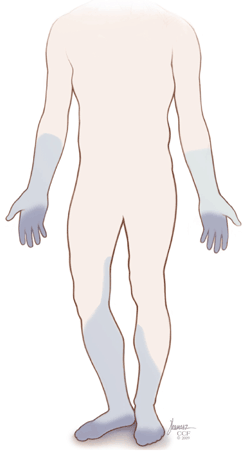
In many of these patients, the findings on neurologic examination, nerve conduction studies, and electromyography are normal, although some may show signs of mild distal sensory loss on physical examination. The lack of objective findings on routine nerve conduction studies and electromyography may lead many physicians to attribute the symptoms to other disorders such as plantar fasciitis, vascular insufficiency, or degenerative lumbosacral spine disease.
The past 2 decades have seen the development of specialized tests that have greatly facilitated the diagnosis of small fiber neuropathy; these include skin biopsy to evaluate the density of nerve fibers in the epidermis and studies of autonomic nerve function. Common etiologies have been identified for small fiber neuropathy and can be specifically treated, which is critical for controlling progression of the disease. Pain management is becoming easier with more available options but is still quite challenging.
WHAT IS SMALL FIBER NEUROPATHY?
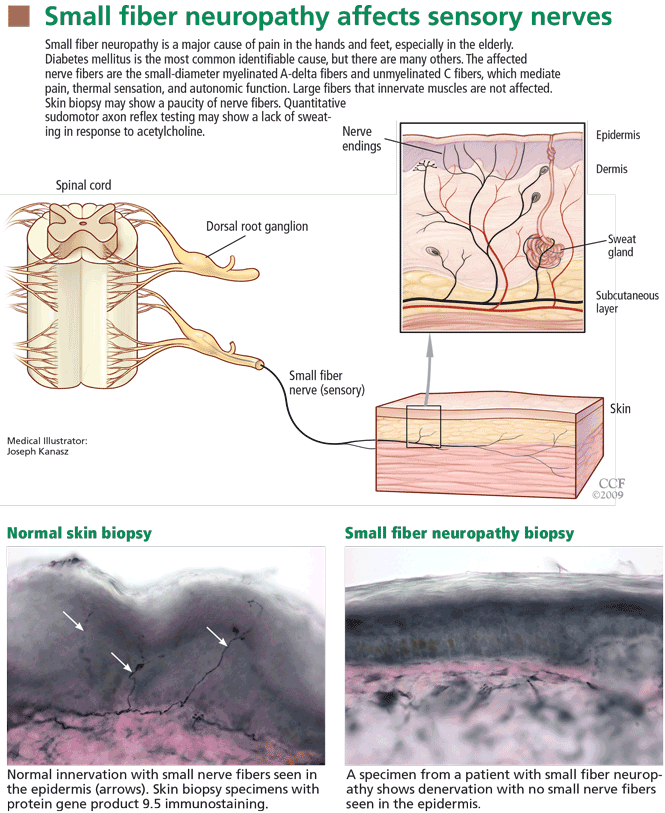
Peripheral nerve fibers can be classified according to size, which correlates with the degree of myelination.
- Large nerve fibers are heavily myelinated and include A-alpha fibers, which mediate motor strength, and A-beta fibers, which mediate vibratory and touch sensation.
- Medium-sized fibers, known as A-gamma fibers, are also myelinated and carry information to muscle spindles.
- Small fibers include myelinated A-delta fibers and unmyelinated C fibers, which innervate skin (somatic fibers) and involuntary muscles, including cardiac and smooth muscles (autonomic fibers). Together, they mediate pain, thermal sensation, and autonomic function.
Small fiber neuropathy results from selective impairment of small myelinated A-delta and unmyelinated C fibers.
Sensory symptoms: Pain, burning, tingling, numbness
Damage to or loss of small somatic nerve fibers results in pain, burning, tingling, or numbness that typically affects the limbs in a distal-to-proximal gradient. In rare cases, small fiber neuropathy follows a non-length-dependent distribution in which symptoms may be manifested predominantly in the arms, face, or trunk.
Symptoms may be mild initially, with some patients complaining of vague discomfort in one or both feet similar to the sensation of a sock gathering at the end of a shoe. Others report a wooden quality in their feet, numbness in their toes, or a feeling as if they are walking on pebbles, sand, or golf balls. The most bothersome and fairly typical symptom is burning pain in the feet that extends proximally in a stocking-glove distribution and is often accompanied by stabbing or aching pains, electric shock-like or pins-and-needles sensations, or cramping of the feet and calves.
Symptoms are usually worse at night and often affect sleep. Some patients say that their feet have become so exquisitely tender that they cannot bear having the bed sheets touch them, and so they sleep with their feet uncovered. A small number of patients do not have pain but report a feeling of tightness and swelling in their feet (even though the feet appear normal).
Examination often reveals allodynia (perception of nonpainful stimuli as being painful), hyperalgesia (perception of painful stimuli as being more painful than expected), or reduced pinprick and thermal sensation in the affected area. Vibratory sensation can be mildly reduced at the toes. Motor strength, tendon reflexes, and proprioception, however, are preserved because they are functions of large nerve fibers.
Autonomic symptoms
When autonomic fibers are affected, patients may experience dry eyes, dry mouth, orthostatic dizziness, constipation, bladder incontinence, sexual dysfunction, trouble sweating, or red or white skin discoloration.2 Examination may show orthostatic hypotension and skin changes. The skin over the affected area may appear atrophic, dry, shiny, discolored, or mildly edematous as the result of sudomotor and vasomotor abnormalities.
WHAT CAUSES SMALL FIBER NEUROPATHY?
Small fiber neuropathy has been associated with many medical conditions, including glucose dysmetabolism,3 connective tissue disease,4,5 dysthyroidism,6 vitamin B12 deficiency, paraproteinemia, human immunodeficiency virus (HIV) infection,7 hepatitis C virus infection, celiac disease,8 restless legs syndrome,9 neurotoxic drug exposure, hereditary diseases, and paraneoplastic syndrome. While most of these conditions cause a length-dependent small fiber neuropathy, others (Sjögren disease, celiac disease, and paraneoplastic syndrome) can cause a form of small fiber neuropathy that is not length-dependent.4,8,10
Diabetes and prediabetes
Glucose dysmetabolism, including diabetes and prediabetes with impaired oral glucose tolerance (a glucose level 140–199 mg/dL 2 hours after a 75-g oral dextrose load), is the most common identifiable associated condition, present in about one-third of patients with painful sensory neuropathy11 and in nearly half of those with otherwise idiopathic small fiber neuropathy.12–14
Research findings strongly suggest that even prediabetes is a risk factor for small fiber neuropathy, and that so-called “impaired glucose tolerance neuropathy” may represent the earliest stage of diabetic neuropathy. Several recent studies have found a high prevalence of impaired glucose tolerance in patients with sensory peripheral neuropathy,12–14 with a rate of up to 42% in cases initially thought to be idiopathic14 compared with 14% in the general population.15 Also, a dose-response relationship between the severity of hyperglycemia and the degree of neuropathy was demonstrated in one study, in which patients with impaired glucose tolerance more often had small fiber neuropathy, whereas those with diabetes more often had polyneuropathy involving both small and large fibers.14 And studies in animals and cell cultures have shown that intermittent hyperglycemia, which can be seen in patients with impaired glucose tolerance, caused sensory neuron and nerve fiber damage and increased spontaneous C-fiber firing, resulting in neuropathic pain.8,16,17
Metabolic syndrome
Insulin resistance with prediabetes and diabetes is a part of the metabolic syndrome, which also consists of hypertension, hyperlipidemia, and obesity. The individual components of the metabolic syndrome have been implicated as risk factors not only for cardiovascular and cerebrovascular disease but also for small fiber neuropathy.
One study in 548 patients with type 2 diabetes showed that those with the metabolic syndrome were twice as likely to have neuropathy as those without.18 Another study showed that in 1,200 patients with type 1 diabetes without neuropathy at baseline, hypertension, hyperlipidemia, and increased body mass index were each independently associated with a higher risk of developing neuropathy.19
A recent study of 219 patients with idiopathic distal symmetrical peripheral neuropathy and 175 diabetic patients without neuropathy found a higher prevalence of metabolic syndrome in patients with neuropathy than in normal populations. The prevalence of dyslipidemia (high levels of total and low-density lipoprotein cholesterol and triglycerides and low levels of high-density lipoprotein cholesterol), but not hypertension or obesity, was higher in patients with neuropathy than in patients with diabetes but no neuropathy.20 The findings linked dyslipidemia to neuropathy and showed the need for further studies of the potential pathogenic role of dyslipidemia in neuropathy.
Hereditary causes
Hereditary causes of small fiber neuropathy are rare and include Fabry disease, Tangier disease, hereditary sensory autonomic neuropathy, and hereditary amyloidosis.
HOW DO YOU EVALUATE PATIENTS WITH SUSPECTED SMALL FIBER NEUROPATHY?
A thorough history should be taken to obtain details regarding onset and features of neuropathy symptoms, exacerbating factors, and progression. It is also important to ascertain whether the patient has any associated conditions as mentioned above, a family history of neuropathy, risk factors for HIV or hepatitis C virus infection, or a history of neurotoxic drug exposure.
Clinical suspicion of small fiber neuropathy should be high if a patient presents with predominant small fiber symptoms and signs with preserved large fiber functions.
Nerve conduction studies and electromyography
For diagnostic testing, routine nerve conduction studies and electromyography assess the function of large nerve fibers only and are thus normal in small fiber neuropathy. These tests should still be ordered to rule out subclinical involvement of large fibers, which may affect the diagnostic evaluation, prognosis, and treatment plan. However, if the results of these tests are normal, specialized studies are needed to evaluate small fibers.
Although several tests are available to evaluate somatic and autonomic small fibers, the two that have the highest diagnostic efficiency for small fiber neuropathy and that are used most often are skin biopsy, to evaluate intraepidermal nerve fiber density, and quantitative sudomotor axon reflex testing (QSART), to assess sudomotor autonomic function.21–23
Skin biopsy
Skin biopsy is a minimally invasive procedure in which 3-mm-diameter punch biopsy specimens are taken from the distal leg, distal thigh, and proximal thigh of one lower limb. The procedure takes only 10 to 15 minutes.
Biopsy specimens are immunostained using an antibody against protein gene product 9.5, which is a panaxonal marker. Small nerve fibers in the epidermis are counted under a microscope, and intraepithelial nerve fiber densities are calculated and compared with established normative values. The diagnosis of small fiber neuropathy can be established if the intraepidermal nerve fiber density is lower than normal (Figure 1). Nerve fiber density may be normal in the early stage of small fiber neuropathy, but in this setting skin biopsy often shows abnormal morphologic changes in the small fibers, especially large swellings,24 and repeat biopsy in 6 to 12 months may be considered.
The diagnostic efficiency of skin biopsy is about 88%.21,23 For diagnosing small fiber neuropathy, it is more sensitive than quantitative sensory testing21,25 and more sensitive and less invasive than sural nerve biopsy.26 Intraepidermal nerve fiber density also correlates well with a variety of measures of severity of HIV distal sensory neuropathy and thus may be used to measure the severity and treatment response of small fiber neuropathy.27
Quantitative sudomotor axon reflex testing
QSART is an autonomic study that measures sweat output in response to acetylcholine, which reflects the function of postganglionic sympathetic unmyelinated sudomotor nerve fibers. Electrodes are placed on the arms and legs to record the volume of sweat produced by acetylcholine iontophoresis, in which a mild electrical stimulation on the skin allows acetylcholine to stimulate the sweat glands. The output is compared with normative values.
One prospective study showed that 67 (72.8%) of 92 patients with painful feet had abnormal results on QSART, ie, low sweat output.28 A retrospective study found that 77 (62%) of 125 patients with clinical features of distal small fiber neuropathy had a length-dependent pattern of QSART abnormalities.22 QSART abnormalities were detected in some patients without autonomic symptoms.
If these tests are not available
Skin biopsy and QSART are objective, reproducible, sensitive, and complementary in diagnosing small fiber neuropathy. One or both can be ordered, depending on whether the patient has somatic symptoms, autonomic symptoms, or both. However, these two tests are not widely available. Only a few laboratories in the country can process skin biopsy specimens to evaluate intraepidermal nerve fiber density. Nevertheless, it is easy to learn the skin punch biopsy procedure, and primary care physicians and neurologists can perform it after appropriate training. (A concern is avoiding damage to the epidermis.) They can then send specimens to one of the cutaneous nerve laboratories (but not to a routine reference laboratory).
A special technique, including unique fixative and cryoprotectant, is used to fix and process the biopsy specimens, because routine techniques for processing dermatologic punch biopsy specimens often result in lower intraepidermal nerve fiber densities. Therefore, it is very important to contact the laboratory regarding fixative and processing before performing a biopsy.
QSART requires specialized equipment and must be performed on site. In addition, the test is very sensitive to drugs that can affect sweating, such as antihistamines and antidepressants, and such drugs must be discontinued 48 hours before the study.
Basic laboratory tests to find the cause
Once the diagnosis of small fiber neuropathy is established, the next important step is to order a battery of laboratory tests to search for an underlying cause. The tests should include the following:
- Complete blood cell count
- Comprehensive metabolic panel
- Lipid panel
- Erythrocyte sedimentation rate
- Thyroid-stimulating hormone level
- Free thyroxine (T4) level
- Antinuclear antibody
- Extractable nuclear antigens
- Angiotensin-converting enzyme (ACE) level
- Serum and urine immunofixation tests
- Vitamin B12 level
- 2-hour oral glucose tolerance test.
Oral glucose tolerance testing is much more sensitive than measuring the hemoglobin A1c and fasting glucose levels in detecting diabetes and prediabetes. These two conditions were detected by oral glucose tolerance testing in more than 50% of patients with otherwise idiopathic sensory-predominant peripheral neuropathy and normal hemoglobin A1c and fasting glucose levels.13,14 Therefore, every patient with small fiber neuropathy without a known history of diabetes or prediabetes should have an oral glucose tolerance test.
Special laboratory tests in special cases
- If there is a history of gastrointestinal symptoms or herpetiform-like rash, then testing for gliadin antibody and tissue transglutaminase antibodies as well as small-bowel biopsy may be pursued to evaluate for celiac sprue.
- Serologic tests for HIV or hepatitis C should be ordered if the patient has risk factors.
- If there is a significant family history, further genetic testing should be considered.
- Lip biopsy or bone marrow biopsy should be considered if clinical suspicion is high for Sjögren disease, seronegative sicca syndrome, or amyloidosis.
- The serum ACE level has a low sensitivity and specificity; therefore, if sarcoid is suspected clinically, additional confirmatory testing, such as computed tomography of the chest, should be ordered despite a normal ACE value.
HOW DO YOU TREAT SMALL FIBER NEUROPATHY?
Treatment of small fiber neuropathy should target the underlying cause and neuropathic pain. Cause-specific treatment is a key in preventing small fiber neuropathy or slowing its progression.
Glucose control, weight control, and regular exercise
As glucose dysmetabolism is the condition most often associated with small fiber neuropathy (and since individual components of the metabolic syndrome are potential risk factors for it), tight glycemic control and lifestyle modification with diet control, weight control, and regular exercise are of paramount importance in patients with these conditions.
The Diabetic Prevention Program,29 a study in 3,234 people with prediabetes, found that diet and exercise were more effective than metformin (Glucophage) in preventing full-blown diabetes. At an average of 2.8 years of follow-up, the incidence of diabetes was 11.0 cases per 100 patient-years in a group assigned to receive placebo, compared with 7.8 in those assigned to receive metformin (31% lower), and 4.8 (58% lower) in those who were assigned to undergo a lifestyle intervention that included at least 150 minutes of physical activity per week with a weight-loss goal of 7%. Put another way, to prevent one case of diabetes over 3 years, 6.9 patients would have to undergo the lifestyle intervention program, or 13.9 would have to receive metformin. Since impaired glucose tolerance neuropathy may represent the earliest stage of diabetic neuropathy, the neuropathy at this stage may be reversible with lifestyle intervention and improvement of impaired glucose tolerance.
This concept is supported by a 3-year study in 31 people, which showed that lifestyle intervention significantly improved impaired glucose tolerance, reduced the body mass index, and lowered total serum cholesterol levels.30 Changes in these metabolic variables were accompanied by significant improvement of neuropathy as evidenced by significantly increased intraepidermal nerve fiber density, increased foot sweat volume, and decreased neuropathic pain.30
Treatment of other diseases
It has also been reported that treatment of sarcoidosis, autoimmune diseases, and celiac disease improved the symptoms of small fiber neuropathy resulting from these conditions.8,31 Therefore, it is important to identify the cause and treat it to prevent and slow the progression of small fiber neuropathy, and doing so may improve the disease in some mild cases.
Pain management
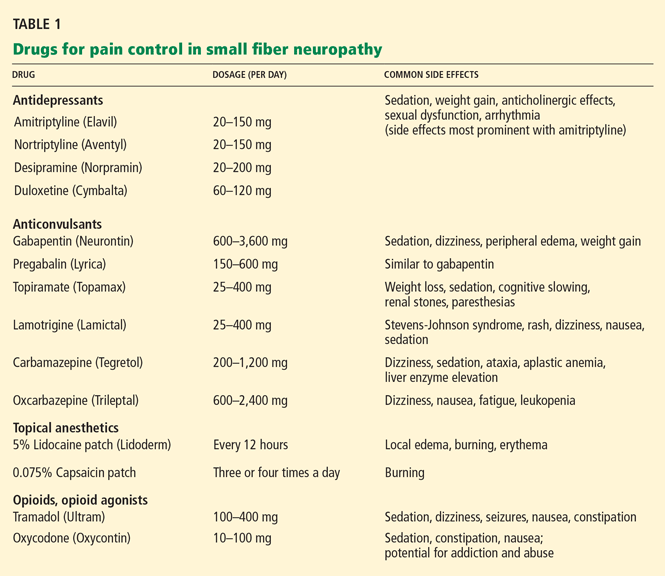
First-line choices of pain medications are the anticonvulsants gabapentin (Neurontin) and pregabalin (Lyrica), the tricyclic antidepressants amitriptyline (Elavil) and nortriptyline (Aventyl), a 5% lidocaine patch (Lidoderm), and the semisynthetic opioid analgesic tramadol (Ultram). These can be used alone or in combination.
Gabapentin is relatively well tolerated, but drowsiness can occur, especially with high starting doses. We usually start with 300 mg daily and increase it by 300 mg every week up to 1,200 mg three times a day as tolerated. Most patients need 600 to 900 mg three times a day.
Pregabalin is a newer antiepileptic drug, similar to gabapentin but less sedating. It can be started at 75 mg twice a day and gradually increased to 300 mg twice a day as needed. Weight gain and, rarely, swelling of the lower extremities may limit the use of both of these drugs.
Tricyclic antidepressants, such as amitriptyline, nortriptyline, and desipramine (Norpramin), are proven effective in controlling neuropathic pain, although no response with amitriptyline was seen in patients with painful HIV distal sensory neuropathy.34
Lidocaine patch is preferred if the painful area is small. Patients should be instructed to use the patch to cover the painful area 12 hours on and 12 hours off. If it does not provide relief within 1 week, it should be discontinued.
Tramadol is also helpful in treating neuropathic pain. It can be started at 50 mg two to four times a day as needed.
Nonsteroidal anti-inflammatory drugs and selective serotonin reuptake inhibitors are typically less effective than the other drugs mentioned.
Opioids should be reserved for refractory cases, given the potential for addiction, but they are sometimes necessary in patients with disabling pain that does not respond to other drugs.
TENS may be of benefit. The patient controls a pocket-size device that sends electrical signals to leads placed on affected areas.
Alternative therapies for small fiber neuropathy, such as meditation, yoga, and acupuncture, have yet to be studied.
It is also important to explain to patients that the typical course of small fiber neuropathy is relatively benign, as many patients worry about developing weakness and eventually not being able to walk. These concerns and fears can aggravate pain and depression, which can make treatment difficult.
WHAT IS THE PROGNOSIS OF SMALL FIBER NEUROPATHY?
Most patients with small fiber neuropathy experience a slowly progressive course, with symptoms and signs spreading proximally over time.
In one study, only 13% of 124 patients with small fiber neuropathy showed evidence of large-fiber involvement over a 2-year period. 21 None went on to develop Charcot joints, foot ulcers, weakness, or sensory ataxia, as is often seen in patients with long-standing or severe large fiber neuropathy. Neuropathic pain worsened in 30% and resolved spontaneously in 11%.21
Most patients with small fiber neuropathy require chronic pain management. Again, treatment of the underlying cause is important and can improve the prognosis.
We believe that the overall progression of small fiber neuropathy is slow. A longitudinal study with a follow-up longer than 2 years would be useful to confirm this.
TAKE-HOME POINTS
As the population continues to age and as more patients develop diabetes and the metabolic syndrome, the prevalence of small fiber neuropathy will rise. Patients who present to their primary care physicians with painful, burning feet require a thorough diagnostic evaluation, which may include referral for specialized neurodiagnostic testing. Aggressive cause-specific treatment, lifestyle modification, and pain control are key elements of a team approach to managing small fiber neuropathy.
- Gregg EW, Gu Q, Williams D, et al. Prevalence of lower extremity diseases associated with normal glucose levels, impaired fasting glucose, and diabetes among U.S. adults aged 40 or older. Diabetes Res Clin Pract 2007; 77:485–488.
- Lacomis D. Small fiber neuropathy. Muscle Nerve 2002; 26:173–188.
- Smith AG, Singleton JR. Impaired glucose tolerance and neuropathy. Neurologist 2008; 14:23–29.
- Chai J, Herrmann DN, Stanton M, Barbano RL, Logigian EL. Painful small-fiber neuropathy in Sjogren syndrome. Neurology 2005; 65:925–927.
- Goransson LG, Tjensvoll AB, Herigstad A, Mellgren SI, Omdal R. Small-diameter nerve fiber neuropathy in systemic lupus erythematosus. Arch Neurol 2006; 63:401–404.
- Orstavik K, Norheim I, Jorum E. Pain and small-fiber neuropathy in patients with hypothyroidism. Neurology 2006; 67:786–791.
- McArthur JC, Brew BJ, Nath A. Neurological complications of HIV infection. Lancet Neurol 2005; 4:543–555.
- Brannagan TH, Hays AP, Chin SS, et al. Small-fiber neuropathy/neuronopathy associated with celiac disease: skin biopsy findings. Arch Neurol 2005; 62:1574–1578.
- Polydefkis M, Allen RP, Hauer P, Earley CJ, Griffin JW, McArthur JC. Subclinical sensory neuropathy in late-onset restless legs syndrome. Neurology 2000; 55:1115–1121.
- Gorson KC, Herrmann DN, Thiagarajan R, et al. Non-length dependent small fibre neuropathy/ganglionopathy. J Neurol Neurosurg Psychiatry 2008; 79:163–169.
- Singleton JR, Smith AG, Bromberg MB. Increased prevalence of impaired glucose tolerance in patients with painful sensory neuropathy. Diabetes Care 2001; 24:1448–1453.
- Novella SP, Inzucchi SE, Goldstein JM. The frequency of undiagnosed diabetes and impaired glucose tolerance in patients with idiopathic sensory neuropathy. Muscle Nerve 2001; 24:1229–1231.
- Smith AG, Singleton JR. The diagnostic yield of a standardized approach to idiopathic sensory-predominant neuropathy. Arch Intern Med 2004; 164:1021–1025.
- Sumner CJ, Sheth S, Griffin JW, Cornblath DR, Polydefkis M. The spectrum of neuropathy in diabetes and impaired glucose tolerance. Neurology 2003; 60:108–111.
- Gregg EW, Sorlie P, Paulose-Ram R, et al. Prevalence of lower-extremity disease in the US adult population >=40 years of age with and without diabetes: 1999–2000 National Health and Nutrition Examination Survey. Diabetes Care 2004; 27:1591–1597.
- Boulton A. What causes neuropathic pain? J Diabetes Complications 1992; 6:58–63.
- Russell JW, Sullivan KA, Windebank AJ, Herrmann DN, Feldman EL. Neurons undergo apoptosis in animal and cell culture models of diabetes. Neurobiol Dis 1999; 6:347–363.
- Costa LA, Canani LH, Lisboa HR, Tres GS, Gross JL. Aggregation of features of the metabolic syndrome is associated with increased prevalence of chronic complications in type 2 diabetes. Diabet Med 2004; 21:252–255.
- Tesfaye S, Chaturvedi N, Eaton SE, et al. Vascular risk factors and diabetic neuropathy. N Engl J Med 2005; 352:341–350.
- Smith A, Rose K, Singleton J. Idiopathic neuropathy patients are at high risk for metabolic syndrome. J Neurol Sci 2008; 273:25–28.
- Devigili G, Tugnoli V, Penza P, et al. The diagnostic criteria for small fibre neuropathy: from symptoms to neuropathology. Brain 2008; 131:1912– 1925.
- Low VA, Sandroni P, Fealey RD, Low PA. Detection of small-fiber neuropathy by sudomotor testing. Muscle Nerve 2006; 34:57–61.
- McArthur JC, Stocks EA, Hauer P, Cornblath DR, Griffin JW. Epidermal nerve fiber density: normative reference range and diagnostic efficiency. Arch Neurol 1998; 55:1513–1520.
- Gibbons CH, Griffin JW, Polydefkis M, et al. The utility of skin biopsy for prediction of progression in suspected small fiber neuropathy. Neurology 2006; 66:256–258.
- Polydefkis M, Yiannoutsos CT, Cohen BA, et al. Reduced intraepidermal nerve fiber density in HIV-associated sensory neuropathy. Neurology 2002; 58:115–119.
- Herrmann DN, Griffin JW, Hauer P, Cornblath DR, McArthur JC. Epidermal nerve fiber density and sural nerve morphometry in peripheral neuropathies. Neurology 1999; 53:1634–1640.
- Zhou L, Kitch DW, Evans SR, et al. Correlates of epidermal nerve fiber densities in HIV-associated distal sensory polyneuropathy. Neurology 2007; 68:2113–2119.
- Novak V, Freimer ML, Kissel JT, et al. Autonomic impairment in painful neuropathy. Neurology 2001; 56:861–868.
- Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346:393–403.
- Smith AG, Russell J, Feldman EL, et al. Lifestyle intervention for prediabetic neuropathy. Diabetes Care 2006; 29:1294–1299.
- Hoitsma E, Faber CG, van Santen-Hoeufft M, De Vries J, Reulen JP, Drent M. Improvement of small fiber neuropathy in a sarcoidosis patient after treatment with infliximab. Sarcoidosis Vasc Diffuse Lung Dis 2006; 23:73–77.
- Chen H, Lamer TJ, Rho RH, et al. Contemporary management of neuropathic pain for the primary care physician. Mayo Clin Proc 2004; 79:1533–1545.
- Galluzzi KE. Managing neuropathic pain. J Am Osteopath Assoc 2007; 107( suppl 6):ES39–ES48.
- Kieburtz K, Simpson D, Yiannoutsos C, et al. A randomized trial of amitriptyline and mexiletine for painful neuropathy in HIV infection. AIDS Clinical Trial Group 242 Protocol Team. Neurology 1998; 51:1682–1688.
In many of these patients, the findings on neurologic examination, nerve conduction studies, and electromyography are normal, although some may show signs of mild distal sensory loss on physical examination. The lack of objective findings on routine nerve conduction studies and electromyography may lead many physicians to attribute the symptoms to other disorders such as plantar fasciitis, vascular insufficiency, or degenerative lumbosacral spine disease.
The past 2 decades have seen the development of specialized tests that have greatly facilitated the diagnosis of small fiber neuropathy; these include skin biopsy to evaluate the density of nerve fibers in the epidermis and studies of autonomic nerve function. Common etiologies have been identified for small fiber neuropathy and can be specifically treated, which is critical for controlling progression of the disease. Pain management is becoming easier with more available options but is still quite challenging.
WHAT IS SMALL FIBER NEUROPATHY?
Peripheral nerve fibers can be classified according to size, which correlates with the degree of myelination.
- Large nerve fibers are heavily myelinated and include A-alpha fibers, which mediate motor strength, and A-beta fibers, which mediate vibratory and touch sensation.
- Medium-sized fibers, known as A-gamma fibers, are also myelinated and carry information to muscle spindles.
- Small fibers include myelinated A-delta fibers and unmyelinated C fibers, which innervate skin (somatic fibers) and involuntary muscles, including cardiac and smooth muscles (autonomic fibers). Together, they mediate pain, thermal sensation, and autonomic function.
Small fiber neuropathy results from selective impairment of small myelinated A-delta and unmyelinated C fibers.
Sensory symptoms: Pain, burning, tingling, numbness
Damage to or loss of small somatic nerve fibers results in pain, burning, tingling, or numbness that typically affects the limbs in a distal-to-proximal gradient. In rare cases, small fiber neuropathy follows a non-length-dependent distribution in which symptoms may be manifested predominantly in the arms, face, or trunk.
Symptoms may be mild initially, with some patients complaining of vague discomfort in one or both feet similar to the sensation of a sock gathering at the end of a shoe. Others report a wooden quality in their feet, numbness in their toes, or a feeling as if they are walking on pebbles, sand, or golf balls. The most bothersome and fairly typical symptom is burning pain in the feet that extends proximally in a stocking-glove distribution and is often accompanied by stabbing or aching pains, electric shock-like or pins-and-needles sensations, or cramping of the feet and calves.
Symptoms are usually worse at night and often affect sleep. Some patients say that their feet have become so exquisitely tender that they cannot bear having the bed sheets touch them, and so they sleep with their feet uncovered. A small number of patients do not have pain but report a feeling of tightness and swelling in their feet (even though the feet appear normal).
Examination often reveals allodynia (perception of nonpainful stimuli as being painful), hyperalgesia (perception of painful stimuli as being more painful than expected), or reduced pinprick and thermal sensation in the affected area. Vibratory sensation can be mildly reduced at the toes. Motor strength, tendon reflexes, and proprioception, however, are preserved because they are functions of large nerve fibers.
Autonomic symptoms
When autonomic fibers are affected, patients may experience dry eyes, dry mouth, orthostatic dizziness, constipation, bladder incontinence, sexual dysfunction, trouble sweating, or red or white skin discoloration.2 Examination may show orthostatic hypotension and skin changes. The skin over the affected area may appear atrophic, dry, shiny, discolored, or mildly edematous as the result of sudomotor and vasomotor abnormalities.
WHAT CAUSES SMALL FIBER NEUROPATHY?
Small fiber neuropathy has been associated with many medical conditions, including glucose dysmetabolism,3 connective tissue disease,4,5 dysthyroidism,6 vitamin B12 deficiency, paraproteinemia, human immunodeficiency virus (HIV) infection,7 hepatitis C virus infection, celiac disease,8 restless legs syndrome,9 neurotoxic drug exposure, hereditary diseases, and paraneoplastic syndrome. While most of these conditions cause a length-dependent small fiber neuropathy, others (Sjögren disease, celiac disease, and paraneoplastic syndrome) can cause a form of small fiber neuropathy that is not length-dependent.4,8,10
Diabetes and prediabetes
Glucose dysmetabolism, including diabetes and prediabetes with impaired oral glucose tolerance (a glucose level 140–199 mg/dL 2 hours after a 75-g oral dextrose load), is the most common identifiable associated condition, present in about one-third of patients with painful sensory neuropathy11 and in nearly half of those with otherwise idiopathic small fiber neuropathy.12–14
Research findings strongly suggest that even prediabetes is a risk factor for small fiber neuropathy, and that so-called “impaired glucose tolerance neuropathy” may represent the earliest stage of diabetic neuropathy. Several recent studies have found a high prevalence of impaired glucose tolerance in patients with sensory peripheral neuropathy,12–14 with a rate of up to 42% in cases initially thought to be idiopathic14 compared with 14% in the general population.15 Also, a dose-response relationship between the severity of hyperglycemia and the degree of neuropathy was demonstrated in one study, in which patients with impaired glucose tolerance more often had small fiber neuropathy, whereas those with diabetes more often had polyneuropathy involving both small and large fibers.14 And studies in animals and cell cultures have shown that intermittent hyperglycemia, which can be seen in patients with impaired glucose tolerance, caused sensory neuron and nerve fiber damage and increased spontaneous C-fiber firing, resulting in neuropathic pain.8,16,17
Metabolic syndrome
Insulin resistance with prediabetes and diabetes is a part of the metabolic syndrome, which also consists of hypertension, hyperlipidemia, and obesity. The individual components of the metabolic syndrome have been implicated as risk factors not only for cardiovascular and cerebrovascular disease but also for small fiber neuropathy.
One study in 548 patients with type 2 diabetes showed that those with the metabolic syndrome were twice as likely to have neuropathy as those without.18 Another study showed that in 1,200 patients with type 1 diabetes without neuropathy at baseline, hypertension, hyperlipidemia, and increased body mass index were each independently associated with a higher risk of developing neuropathy.19
A recent study of 219 patients with idiopathic distal symmetrical peripheral neuropathy and 175 diabetic patients without neuropathy found a higher prevalence of metabolic syndrome in patients with neuropathy than in normal populations. The prevalence of dyslipidemia (high levels of total and low-density lipoprotein cholesterol and triglycerides and low levels of high-density lipoprotein cholesterol), but not hypertension or obesity, was higher in patients with neuropathy than in patients with diabetes but no neuropathy.20 The findings linked dyslipidemia to neuropathy and showed the need for further studies of the potential pathogenic role of dyslipidemia in neuropathy.
Hereditary causes
Hereditary causes of small fiber neuropathy are rare and include Fabry disease, Tangier disease, hereditary sensory autonomic neuropathy, and hereditary amyloidosis.
HOW DO YOU EVALUATE PATIENTS WITH SUSPECTED SMALL FIBER NEUROPATHY?
A thorough history should be taken to obtain details regarding onset and features of neuropathy symptoms, exacerbating factors, and progression. It is also important to ascertain whether the patient has any associated conditions as mentioned above, a family history of neuropathy, risk factors for HIV or hepatitis C virus infection, or a history of neurotoxic drug exposure.
Clinical suspicion of small fiber neuropathy should be high if a patient presents with predominant small fiber symptoms and signs with preserved large fiber functions.
Nerve conduction studies and electromyography
For diagnostic testing, routine nerve conduction studies and electromyography assess the function of large nerve fibers only and are thus normal in small fiber neuropathy. These tests should still be ordered to rule out subclinical involvement of large fibers, which may affect the diagnostic evaluation, prognosis, and treatment plan. However, if the results of these tests are normal, specialized studies are needed to evaluate small fibers.
Although several tests are available to evaluate somatic and autonomic small fibers, the two that have the highest diagnostic efficiency for small fiber neuropathy and that are used most often are skin biopsy, to evaluate intraepidermal nerve fiber density, and quantitative sudomotor axon reflex testing (QSART), to assess sudomotor autonomic function.21–23
Skin biopsy
Skin biopsy is a minimally invasive procedure in which 3-mm-diameter punch biopsy specimens are taken from the distal leg, distal thigh, and proximal thigh of one lower limb. The procedure takes only 10 to 15 minutes.
Biopsy specimens are immunostained using an antibody against protein gene product 9.5, which is a panaxonal marker. Small nerve fibers in the epidermis are counted under a microscope, and intraepithelial nerve fiber densities are calculated and compared with established normative values. The diagnosis of small fiber neuropathy can be established if the intraepidermal nerve fiber density is lower than normal (Figure 1). Nerve fiber density may be normal in the early stage of small fiber neuropathy, but in this setting skin biopsy often shows abnormal morphologic changes in the small fibers, especially large swellings,24 and repeat biopsy in 6 to 12 months may be considered.
The diagnostic efficiency of skin biopsy is about 88%.21,23 For diagnosing small fiber neuropathy, it is more sensitive than quantitative sensory testing21,25 and more sensitive and less invasive than sural nerve biopsy.26 Intraepidermal nerve fiber density also correlates well with a variety of measures of severity of HIV distal sensory neuropathy and thus may be used to measure the severity and treatment response of small fiber neuropathy.27
Quantitative sudomotor axon reflex testing
QSART is an autonomic study that measures sweat output in response to acetylcholine, which reflects the function of postganglionic sympathetic unmyelinated sudomotor nerve fibers. Electrodes are placed on the arms and legs to record the volume of sweat produced by acetylcholine iontophoresis, in which a mild electrical stimulation on the skin allows acetylcholine to stimulate the sweat glands. The output is compared with normative values.
One prospective study showed that 67 (72.8%) of 92 patients with painful feet had abnormal results on QSART, ie, low sweat output.28 A retrospective study found that 77 (62%) of 125 patients with clinical features of distal small fiber neuropathy had a length-dependent pattern of QSART abnormalities.22 QSART abnormalities were detected in some patients without autonomic symptoms.
If these tests are not available
Skin biopsy and QSART are objective, reproducible, sensitive, and complementary in diagnosing small fiber neuropathy. One or both can be ordered, depending on whether the patient has somatic symptoms, autonomic symptoms, or both. However, these two tests are not widely available. Only a few laboratories in the country can process skin biopsy specimens to evaluate intraepidermal nerve fiber density. Nevertheless, it is easy to learn the skin punch biopsy procedure, and primary care physicians and neurologists can perform it after appropriate training. (A concern is avoiding damage to the epidermis.) They can then send specimens to one of the cutaneous nerve laboratories (but not to a routine reference laboratory).
A special technique, including unique fixative and cryoprotectant, is used to fix and process the biopsy specimens, because routine techniques for processing dermatologic punch biopsy specimens often result in lower intraepidermal nerve fiber densities. Therefore, it is very important to contact the laboratory regarding fixative and processing before performing a biopsy.
QSART requires specialized equipment and must be performed on site. In addition, the test is very sensitive to drugs that can affect sweating, such as antihistamines and antidepressants, and such drugs must be discontinued 48 hours before the study.
Basic laboratory tests to find the cause
Once the diagnosis of small fiber neuropathy is established, the next important step is to order a battery of laboratory tests to search for an underlying cause. The tests should include the following:
- Complete blood cell count
- Comprehensive metabolic panel
- Lipid panel
- Erythrocyte sedimentation rate
- Thyroid-stimulating hormone level
- Free thyroxine (T4) level
- Antinuclear antibody
- Extractable nuclear antigens
- Angiotensin-converting enzyme (ACE) level
- Serum and urine immunofixation tests
- Vitamin B12 level
- 2-hour oral glucose tolerance test.
Oral glucose tolerance testing is much more sensitive than measuring the hemoglobin A1c and fasting glucose levels in detecting diabetes and prediabetes. These two conditions were detected by oral glucose tolerance testing in more than 50% of patients with otherwise idiopathic sensory-predominant peripheral neuropathy and normal hemoglobin A1c and fasting glucose levels.13,14 Therefore, every patient with small fiber neuropathy without a known history of diabetes or prediabetes should have an oral glucose tolerance test.
Special laboratory tests in special cases
- If there is a history of gastrointestinal symptoms or herpetiform-like rash, then testing for gliadin antibody and tissue transglutaminase antibodies as well as small-bowel biopsy may be pursued to evaluate for celiac sprue.
- Serologic tests for HIV or hepatitis C should be ordered if the patient has risk factors.
- If there is a significant family history, further genetic testing should be considered.
- Lip biopsy or bone marrow biopsy should be considered if clinical suspicion is high for Sjögren disease, seronegative sicca syndrome, or amyloidosis.
- The serum ACE level has a low sensitivity and specificity; therefore, if sarcoid is suspected clinically, additional confirmatory testing, such as computed tomography of the chest, should be ordered despite a normal ACE value.
HOW DO YOU TREAT SMALL FIBER NEUROPATHY?
Treatment of small fiber neuropathy should target the underlying cause and neuropathic pain. Cause-specific treatment is a key in preventing small fiber neuropathy or slowing its progression.
Glucose control, weight control, and regular exercise
As glucose dysmetabolism is the condition most often associated with small fiber neuropathy (and since individual components of the metabolic syndrome are potential risk factors for it), tight glycemic control and lifestyle modification with diet control, weight control, and regular exercise are of paramount importance in patients with these conditions.
The Diabetic Prevention Program,29 a study in 3,234 people with prediabetes, found that diet and exercise were more effective than metformin (Glucophage) in preventing full-blown diabetes. At an average of 2.8 years of follow-up, the incidence of diabetes was 11.0 cases per 100 patient-years in a group assigned to receive placebo, compared with 7.8 in those assigned to receive metformin (31% lower), and 4.8 (58% lower) in those who were assigned to undergo a lifestyle intervention that included at least 150 minutes of physical activity per week with a weight-loss goal of 7%. Put another way, to prevent one case of diabetes over 3 years, 6.9 patients would have to undergo the lifestyle intervention program, or 13.9 would have to receive metformin. Since impaired glucose tolerance neuropathy may represent the earliest stage of diabetic neuropathy, the neuropathy at this stage may be reversible with lifestyle intervention and improvement of impaired glucose tolerance.
This concept is supported by a 3-year study in 31 people, which showed that lifestyle intervention significantly improved impaired glucose tolerance, reduced the body mass index, and lowered total serum cholesterol levels.30 Changes in these metabolic variables were accompanied by significant improvement of neuropathy as evidenced by significantly increased intraepidermal nerve fiber density, increased foot sweat volume, and decreased neuropathic pain.30
Treatment of other diseases
It has also been reported that treatment of sarcoidosis, autoimmune diseases, and celiac disease improved the symptoms of small fiber neuropathy resulting from these conditions.8,31 Therefore, it is important to identify the cause and treat it to prevent and slow the progression of small fiber neuropathy, and doing so may improve the disease in some mild cases.
Pain management
First-line choices of pain medications are the anticonvulsants gabapentin (Neurontin) and pregabalin (Lyrica), the tricyclic antidepressants amitriptyline (Elavil) and nortriptyline (Aventyl), a 5% lidocaine patch (Lidoderm), and the semisynthetic opioid analgesic tramadol (Ultram). These can be used alone or in combination.
Gabapentin is relatively well tolerated, but drowsiness can occur, especially with high starting doses. We usually start with 300 mg daily and increase it by 300 mg every week up to 1,200 mg three times a day as tolerated. Most patients need 600 to 900 mg three times a day.
Pregabalin is a newer antiepileptic drug, similar to gabapentin but less sedating. It can be started at 75 mg twice a day and gradually increased to 300 mg twice a day as needed. Weight gain and, rarely, swelling of the lower extremities may limit the use of both of these drugs.
Tricyclic antidepressants, such as amitriptyline, nortriptyline, and desipramine (Norpramin), are proven effective in controlling neuropathic pain, although no response with amitriptyline was seen in patients with painful HIV distal sensory neuropathy.34
Lidocaine patch is preferred if the painful area is small. Patients should be instructed to use the patch to cover the painful area 12 hours on and 12 hours off. If it does not provide relief within 1 week, it should be discontinued.
Tramadol is also helpful in treating neuropathic pain. It can be started at 50 mg two to four times a day as needed.
Nonsteroidal anti-inflammatory drugs and selective serotonin reuptake inhibitors are typically less effective than the other drugs mentioned.
Opioids should be reserved for refractory cases, given the potential for addiction, but they are sometimes necessary in patients with disabling pain that does not respond to other drugs.
TENS may be of benefit. The patient controls a pocket-size device that sends electrical signals to leads placed on affected areas.
Alternative therapies for small fiber neuropathy, such as meditation, yoga, and acupuncture, have yet to be studied.
It is also important to explain to patients that the typical course of small fiber neuropathy is relatively benign, as many patients worry about developing weakness and eventually not being able to walk. These concerns and fears can aggravate pain and depression, which can make treatment difficult.
WHAT IS THE PROGNOSIS OF SMALL FIBER NEUROPATHY?
Most patients with small fiber neuropathy experience a slowly progressive course, with symptoms and signs spreading proximally over time.
In one study, only 13% of 124 patients with small fiber neuropathy showed evidence of large-fiber involvement over a 2-year period. 21 None went on to develop Charcot joints, foot ulcers, weakness, or sensory ataxia, as is often seen in patients with long-standing or severe large fiber neuropathy. Neuropathic pain worsened in 30% and resolved spontaneously in 11%.21
Most patients with small fiber neuropathy require chronic pain management. Again, treatment of the underlying cause is important and can improve the prognosis.
We believe that the overall progression of small fiber neuropathy is slow. A longitudinal study with a follow-up longer than 2 years would be useful to confirm this.
TAKE-HOME POINTS
As the population continues to age and as more patients develop diabetes and the metabolic syndrome, the prevalence of small fiber neuropathy will rise. Patients who present to their primary care physicians with painful, burning feet require a thorough diagnostic evaluation, which may include referral for specialized neurodiagnostic testing. Aggressive cause-specific treatment, lifestyle modification, and pain control are key elements of a team approach to managing small fiber neuropathy.
In many of these patients, the findings on neurologic examination, nerve conduction studies, and electromyography are normal, although some may show signs of mild distal sensory loss on physical examination. The lack of objective findings on routine nerve conduction studies and electromyography may lead many physicians to attribute the symptoms to other disorders such as plantar fasciitis, vascular insufficiency, or degenerative lumbosacral spine disease.
The past 2 decades have seen the development of specialized tests that have greatly facilitated the diagnosis of small fiber neuropathy; these include skin biopsy to evaluate the density of nerve fibers in the epidermis and studies of autonomic nerve function. Common etiologies have been identified for small fiber neuropathy and can be specifically treated, which is critical for controlling progression of the disease. Pain management is becoming easier with more available options but is still quite challenging.
WHAT IS SMALL FIBER NEUROPATHY?
Peripheral nerve fibers can be classified according to size, which correlates with the degree of myelination.
- Large nerve fibers are heavily myelinated and include A-alpha fibers, which mediate motor strength, and A-beta fibers, which mediate vibratory and touch sensation.
- Medium-sized fibers, known as A-gamma fibers, are also myelinated and carry information to muscle spindles.
- Small fibers include myelinated A-delta fibers and unmyelinated C fibers, which innervate skin (somatic fibers) and involuntary muscles, including cardiac and smooth muscles (autonomic fibers). Together, they mediate pain, thermal sensation, and autonomic function.
Small fiber neuropathy results from selective impairment of small myelinated A-delta and unmyelinated C fibers.
Sensory symptoms: Pain, burning, tingling, numbness
Damage to or loss of small somatic nerve fibers results in pain, burning, tingling, or numbness that typically affects the limbs in a distal-to-proximal gradient. In rare cases, small fiber neuropathy follows a non-length-dependent distribution in which symptoms may be manifested predominantly in the arms, face, or trunk.
Symptoms may be mild initially, with some patients complaining of vague discomfort in one or both feet similar to the sensation of a sock gathering at the end of a shoe. Others report a wooden quality in their feet, numbness in their toes, or a feeling as if they are walking on pebbles, sand, or golf balls. The most bothersome and fairly typical symptom is burning pain in the feet that extends proximally in a stocking-glove distribution and is often accompanied by stabbing or aching pains, electric shock-like or pins-and-needles sensations, or cramping of the feet and calves.
Symptoms are usually worse at night and often affect sleep. Some patients say that their feet have become so exquisitely tender that they cannot bear having the bed sheets touch them, and so they sleep with their feet uncovered. A small number of patients do not have pain but report a feeling of tightness and swelling in their feet (even though the feet appear normal).
Examination often reveals allodynia (perception of nonpainful stimuli as being painful), hyperalgesia (perception of painful stimuli as being more painful than expected), or reduced pinprick and thermal sensation in the affected area. Vibratory sensation can be mildly reduced at the toes. Motor strength, tendon reflexes, and proprioception, however, are preserved because they are functions of large nerve fibers.
Autonomic symptoms
When autonomic fibers are affected, patients may experience dry eyes, dry mouth, orthostatic dizziness, constipation, bladder incontinence, sexual dysfunction, trouble sweating, or red or white skin discoloration.2 Examination may show orthostatic hypotension and skin changes. The skin over the affected area may appear atrophic, dry, shiny, discolored, or mildly edematous as the result of sudomotor and vasomotor abnormalities.
WHAT CAUSES SMALL FIBER NEUROPATHY?
Small fiber neuropathy has been associated with many medical conditions, including glucose dysmetabolism,3 connective tissue disease,4,5 dysthyroidism,6 vitamin B12 deficiency, paraproteinemia, human immunodeficiency virus (HIV) infection,7 hepatitis C virus infection, celiac disease,8 restless legs syndrome,9 neurotoxic drug exposure, hereditary diseases, and paraneoplastic syndrome. While most of these conditions cause a length-dependent small fiber neuropathy, others (Sjögren disease, celiac disease, and paraneoplastic syndrome) can cause a form of small fiber neuropathy that is not length-dependent.4,8,10
Diabetes and prediabetes
Glucose dysmetabolism, including diabetes and prediabetes with impaired oral glucose tolerance (a glucose level 140–199 mg/dL 2 hours after a 75-g oral dextrose load), is the most common identifiable associated condition, present in about one-third of patients with painful sensory neuropathy11 and in nearly half of those with otherwise idiopathic small fiber neuropathy.12–14
Research findings strongly suggest that even prediabetes is a risk factor for small fiber neuropathy, and that so-called “impaired glucose tolerance neuropathy” may represent the earliest stage of diabetic neuropathy. Several recent studies have found a high prevalence of impaired glucose tolerance in patients with sensory peripheral neuropathy,12–14 with a rate of up to 42% in cases initially thought to be idiopathic14 compared with 14% in the general population.15 Also, a dose-response relationship between the severity of hyperglycemia and the degree of neuropathy was demonstrated in one study, in which patients with impaired glucose tolerance more often had small fiber neuropathy, whereas those with diabetes more often had polyneuropathy involving both small and large fibers.14 And studies in animals and cell cultures have shown that intermittent hyperglycemia, which can be seen in patients with impaired glucose tolerance, caused sensory neuron and nerve fiber damage and increased spontaneous C-fiber firing, resulting in neuropathic pain.8,16,17
Metabolic syndrome
Insulin resistance with prediabetes and diabetes is a part of the metabolic syndrome, which also consists of hypertension, hyperlipidemia, and obesity. The individual components of the metabolic syndrome have been implicated as risk factors not only for cardiovascular and cerebrovascular disease but also for small fiber neuropathy.
One study in 548 patients with type 2 diabetes showed that those with the metabolic syndrome were twice as likely to have neuropathy as those without.18 Another study showed that in 1,200 patients with type 1 diabetes without neuropathy at baseline, hypertension, hyperlipidemia, and increased body mass index were each independently associated with a higher risk of developing neuropathy.19
A recent study of 219 patients with idiopathic distal symmetrical peripheral neuropathy and 175 diabetic patients without neuropathy found a higher prevalence of metabolic syndrome in patients with neuropathy than in normal populations. The prevalence of dyslipidemia (high levels of total and low-density lipoprotein cholesterol and triglycerides and low levels of high-density lipoprotein cholesterol), but not hypertension or obesity, was higher in patients with neuropathy than in patients with diabetes but no neuropathy.20 The findings linked dyslipidemia to neuropathy and showed the need for further studies of the potential pathogenic role of dyslipidemia in neuropathy.
Hereditary causes
Hereditary causes of small fiber neuropathy are rare and include Fabry disease, Tangier disease, hereditary sensory autonomic neuropathy, and hereditary amyloidosis.
HOW DO YOU EVALUATE PATIENTS WITH SUSPECTED SMALL FIBER NEUROPATHY?
A thorough history should be taken to obtain details regarding onset and features of neuropathy symptoms, exacerbating factors, and progression. It is also important to ascertain whether the patient has any associated conditions as mentioned above, a family history of neuropathy, risk factors for HIV or hepatitis C virus infection, or a history of neurotoxic drug exposure.
Clinical suspicion of small fiber neuropathy should be high if a patient presents with predominant small fiber symptoms and signs with preserved large fiber functions.
Nerve conduction studies and electromyography
For diagnostic testing, routine nerve conduction studies and electromyography assess the function of large nerve fibers only and are thus normal in small fiber neuropathy. These tests should still be ordered to rule out subclinical involvement of large fibers, which may affect the diagnostic evaluation, prognosis, and treatment plan. However, if the results of these tests are normal, specialized studies are needed to evaluate small fibers.
Although several tests are available to evaluate somatic and autonomic small fibers, the two that have the highest diagnostic efficiency for small fiber neuropathy and that are used most often are skin biopsy, to evaluate intraepidermal nerve fiber density, and quantitative sudomotor axon reflex testing (QSART), to assess sudomotor autonomic function.21–23
Skin biopsy
Skin biopsy is a minimally invasive procedure in which 3-mm-diameter punch biopsy specimens are taken from the distal leg, distal thigh, and proximal thigh of one lower limb. The procedure takes only 10 to 15 minutes.
Biopsy specimens are immunostained using an antibody against protein gene product 9.5, which is a panaxonal marker. Small nerve fibers in the epidermis are counted under a microscope, and intraepithelial nerve fiber densities are calculated and compared with established normative values. The diagnosis of small fiber neuropathy can be established if the intraepidermal nerve fiber density is lower than normal (Figure 1). Nerve fiber density may be normal in the early stage of small fiber neuropathy, but in this setting skin biopsy often shows abnormal morphologic changes in the small fibers, especially large swellings,24 and repeat biopsy in 6 to 12 months may be considered.
The diagnostic efficiency of skin biopsy is about 88%.21,23 For diagnosing small fiber neuropathy, it is more sensitive than quantitative sensory testing21,25 and more sensitive and less invasive than sural nerve biopsy.26 Intraepidermal nerve fiber density also correlates well with a variety of measures of severity of HIV distal sensory neuropathy and thus may be used to measure the severity and treatment response of small fiber neuropathy.27
Quantitative sudomotor axon reflex testing
QSART is an autonomic study that measures sweat output in response to acetylcholine, which reflects the function of postganglionic sympathetic unmyelinated sudomotor nerve fibers. Electrodes are placed on the arms and legs to record the volume of sweat produced by acetylcholine iontophoresis, in which a mild electrical stimulation on the skin allows acetylcholine to stimulate the sweat glands. The output is compared with normative values.
One prospective study showed that 67 (72.8%) of 92 patients with painful feet had abnormal results on QSART, ie, low sweat output.28 A retrospective study found that 77 (62%) of 125 patients with clinical features of distal small fiber neuropathy had a length-dependent pattern of QSART abnormalities.22 QSART abnormalities were detected in some patients without autonomic symptoms.
If these tests are not available
Skin biopsy and QSART are objective, reproducible, sensitive, and complementary in diagnosing small fiber neuropathy. One or both can be ordered, depending on whether the patient has somatic symptoms, autonomic symptoms, or both. However, these two tests are not widely available. Only a few laboratories in the country can process skin biopsy specimens to evaluate intraepidermal nerve fiber density. Nevertheless, it is easy to learn the skin punch biopsy procedure, and primary care physicians and neurologists can perform it after appropriate training. (A concern is avoiding damage to the epidermis.) They can then send specimens to one of the cutaneous nerve laboratories (but not to a routine reference laboratory).
A special technique, including unique fixative and cryoprotectant, is used to fix and process the biopsy specimens, because routine techniques for processing dermatologic punch biopsy specimens often result in lower intraepidermal nerve fiber densities. Therefore, it is very important to contact the laboratory regarding fixative and processing before performing a biopsy.
QSART requires specialized equipment and must be performed on site. In addition, the test is very sensitive to drugs that can affect sweating, such as antihistamines and antidepressants, and such drugs must be discontinued 48 hours before the study.
Basic laboratory tests to find the cause
Once the diagnosis of small fiber neuropathy is established, the next important step is to order a battery of laboratory tests to search for an underlying cause. The tests should include the following:
- Complete blood cell count
- Comprehensive metabolic panel
- Lipid panel
- Erythrocyte sedimentation rate
- Thyroid-stimulating hormone level
- Free thyroxine (T4) level
- Antinuclear antibody
- Extractable nuclear antigens
- Angiotensin-converting enzyme (ACE) level
- Serum and urine immunofixation tests
- Vitamin B12 level
- 2-hour oral glucose tolerance test.
Oral glucose tolerance testing is much more sensitive than measuring the hemoglobin A1c and fasting glucose levels in detecting diabetes and prediabetes. These two conditions were detected by oral glucose tolerance testing in more than 50% of patients with otherwise idiopathic sensory-predominant peripheral neuropathy and normal hemoglobin A1c and fasting glucose levels.13,14 Therefore, every patient with small fiber neuropathy without a known history of diabetes or prediabetes should have an oral glucose tolerance test.
Special laboratory tests in special cases
- If there is a history of gastrointestinal symptoms or herpetiform-like rash, then testing for gliadin antibody and tissue transglutaminase antibodies as well as small-bowel biopsy may be pursued to evaluate for celiac sprue.
- Serologic tests for HIV or hepatitis C should be ordered if the patient has risk factors.
- If there is a significant family history, further genetic testing should be considered.
- Lip biopsy or bone marrow biopsy should be considered if clinical suspicion is high for Sjögren disease, seronegative sicca syndrome, or amyloidosis.
- The serum ACE level has a low sensitivity and specificity; therefore, if sarcoid is suspected clinically, additional confirmatory testing, such as computed tomography of the chest, should be ordered despite a normal ACE value.
HOW DO YOU TREAT SMALL FIBER NEUROPATHY?
Treatment of small fiber neuropathy should target the underlying cause and neuropathic pain. Cause-specific treatment is a key in preventing small fiber neuropathy or slowing its progression.
Glucose control, weight control, and regular exercise
As glucose dysmetabolism is the condition most often associated with small fiber neuropathy (and since individual components of the metabolic syndrome are potential risk factors for it), tight glycemic control and lifestyle modification with diet control, weight control, and regular exercise are of paramount importance in patients with these conditions.
The Diabetic Prevention Program,29 a study in 3,234 people with prediabetes, found that diet and exercise were more effective than metformin (Glucophage) in preventing full-blown diabetes. At an average of 2.8 years of follow-up, the incidence of diabetes was 11.0 cases per 100 patient-years in a group assigned to receive placebo, compared with 7.8 in those assigned to receive metformin (31% lower), and 4.8 (58% lower) in those who were assigned to undergo a lifestyle intervention that included at least 150 minutes of physical activity per week with a weight-loss goal of 7%. Put another way, to prevent one case of diabetes over 3 years, 6.9 patients would have to undergo the lifestyle intervention program, or 13.9 would have to receive metformin. Since impaired glucose tolerance neuropathy may represent the earliest stage of diabetic neuropathy, the neuropathy at this stage may be reversible with lifestyle intervention and improvement of impaired glucose tolerance.
This concept is supported by a 3-year study in 31 people, which showed that lifestyle intervention significantly improved impaired glucose tolerance, reduced the body mass index, and lowered total serum cholesterol levels.30 Changes in these metabolic variables were accompanied by significant improvement of neuropathy as evidenced by significantly increased intraepidermal nerve fiber density, increased foot sweat volume, and decreased neuropathic pain.30
Treatment of other diseases
It has also been reported that treatment of sarcoidosis, autoimmune diseases, and celiac disease improved the symptoms of small fiber neuropathy resulting from these conditions.8,31 Therefore, it is important to identify the cause and treat it to prevent and slow the progression of small fiber neuropathy, and doing so may improve the disease in some mild cases.
Pain management
First-line choices of pain medications are the anticonvulsants gabapentin (Neurontin) and pregabalin (Lyrica), the tricyclic antidepressants amitriptyline (Elavil) and nortriptyline (Aventyl), a 5% lidocaine patch (Lidoderm), and the semisynthetic opioid analgesic tramadol (Ultram). These can be used alone or in combination.
Gabapentin is relatively well tolerated, but drowsiness can occur, especially with high starting doses. We usually start with 300 mg daily and increase it by 300 mg every week up to 1,200 mg three times a day as tolerated. Most patients need 600 to 900 mg three times a day.
Pregabalin is a newer antiepileptic drug, similar to gabapentin but less sedating. It can be started at 75 mg twice a day and gradually increased to 300 mg twice a day as needed. Weight gain and, rarely, swelling of the lower extremities may limit the use of both of these drugs.
Tricyclic antidepressants, such as amitriptyline, nortriptyline, and desipramine (Norpramin), are proven effective in controlling neuropathic pain, although no response with amitriptyline was seen in patients with painful HIV distal sensory neuropathy.34
Lidocaine patch is preferred if the painful area is small. Patients should be instructed to use the patch to cover the painful area 12 hours on and 12 hours off. If it does not provide relief within 1 week, it should be discontinued.
Tramadol is also helpful in treating neuropathic pain. It can be started at 50 mg two to four times a day as needed.
Nonsteroidal anti-inflammatory drugs and selective serotonin reuptake inhibitors are typically less effective than the other drugs mentioned.
Opioids should be reserved for refractory cases, given the potential for addiction, but they are sometimes necessary in patients with disabling pain that does not respond to other drugs.
TENS may be of benefit. The patient controls a pocket-size device that sends electrical signals to leads placed on affected areas.
Alternative therapies for small fiber neuropathy, such as meditation, yoga, and acupuncture, have yet to be studied.
It is also important to explain to patients that the typical course of small fiber neuropathy is relatively benign, as many patients worry about developing weakness and eventually not being able to walk. These concerns and fears can aggravate pain and depression, which can make treatment difficult.
WHAT IS THE PROGNOSIS OF SMALL FIBER NEUROPATHY?
Most patients with small fiber neuropathy experience a slowly progressive course, with symptoms and signs spreading proximally over time.
In one study, only 13% of 124 patients with small fiber neuropathy showed evidence of large-fiber involvement over a 2-year period. 21 None went on to develop Charcot joints, foot ulcers, weakness, or sensory ataxia, as is often seen in patients with long-standing or severe large fiber neuropathy. Neuropathic pain worsened in 30% and resolved spontaneously in 11%.21
Most patients with small fiber neuropathy require chronic pain management. Again, treatment of the underlying cause is important and can improve the prognosis.
We believe that the overall progression of small fiber neuropathy is slow. A longitudinal study with a follow-up longer than 2 years would be useful to confirm this.
TAKE-HOME POINTS
As the population continues to age and as more patients develop diabetes and the metabolic syndrome, the prevalence of small fiber neuropathy will rise. Patients who present to their primary care physicians with painful, burning feet require a thorough diagnostic evaluation, which may include referral for specialized neurodiagnostic testing. Aggressive cause-specific treatment, lifestyle modification, and pain control are key elements of a team approach to managing small fiber neuropathy.
- Gregg EW, Gu Q, Williams D, et al. Prevalence of lower extremity diseases associated with normal glucose levels, impaired fasting glucose, and diabetes among U.S. adults aged 40 or older. Diabetes Res Clin Pract 2007; 77:485–488.
- Lacomis D. Small fiber neuropathy. Muscle Nerve 2002; 26:173–188.
- Smith AG, Singleton JR. Impaired glucose tolerance and neuropathy. Neurologist 2008; 14:23–29.
- Chai J, Herrmann DN, Stanton M, Barbano RL, Logigian EL. Painful small-fiber neuropathy in Sjogren syndrome. Neurology 2005; 65:925–927.
- Goransson LG, Tjensvoll AB, Herigstad A, Mellgren SI, Omdal R. Small-diameter nerve fiber neuropathy in systemic lupus erythematosus. Arch Neurol 2006; 63:401–404.
- Orstavik K, Norheim I, Jorum E. Pain and small-fiber neuropathy in patients with hypothyroidism. Neurology 2006; 67:786–791.
- McArthur JC, Brew BJ, Nath A. Neurological complications of HIV infection. Lancet Neurol 2005; 4:543–555.
- Brannagan TH, Hays AP, Chin SS, et al. Small-fiber neuropathy/neuronopathy associated with celiac disease: skin biopsy findings. Arch Neurol 2005; 62:1574–1578.
- Polydefkis M, Allen RP, Hauer P, Earley CJ, Griffin JW, McArthur JC. Subclinical sensory neuropathy in late-onset restless legs syndrome. Neurology 2000; 55:1115–1121.
- Gorson KC, Herrmann DN, Thiagarajan R, et al. Non-length dependent small fibre neuropathy/ganglionopathy. J Neurol Neurosurg Psychiatry 2008; 79:163–169.
- Singleton JR, Smith AG, Bromberg MB. Increased prevalence of impaired glucose tolerance in patients with painful sensory neuropathy. Diabetes Care 2001; 24:1448–1453.
- Novella SP, Inzucchi SE, Goldstein JM. The frequency of undiagnosed diabetes and impaired glucose tolerance in patients with idiopathic sensory neuropathy. Muscle Nerve 2001; 24:1229–1231.
- Smith AG, Singleton JR. The diagnostic yield of a standardized approach to idiopathic sensory-predominant neuropathy. Arch Intern Med 2004; 164:1021–1025.
- Sumner CJ, Sheth S, Griffin JW, Cornblath DR, Polydefkis M. The spectrum of neuropathy in diabetes and impaired glucose tolerance. Neurology 2003; 60:108–111.
- Gregg EW, Sorlie P, Paulose-Ram R, et al. Prevalence of lower-extremity disease in the US adult population >=40 years of age with and without diabetes: 1999–2000 National Health and Nutrition Examination Survey. Diabetes Care 2004; 27:1591–1597.
- Boulton A. What causes neuropathic pain? J Diabetes Complications 1992; 6:58–63.
- Russell JW, Sullivan KA, Windebank AJ, Herrmann DN, Feldman EL. Neurons undergo apoptosis in animal and cell culture models of diabetes. Neurobiol Dis 1999; 6:347–363.
- Costa LA, Canani LH, Lisboa HR, Tres GS, Gross JL. Aggregation of features of the metabolic syndrome is associated with increased prevalence of chronic complications in type 2 diabetes. Diabet Med 2004; 21:252–255.
- Tesfaye S, Chaturvedi N, Eaton SE, et al. Vascular risk factors and diabetic neuropathy. N Engl J Med 2005; 352:341–350.
- Smith A, Rose K, Singleton J. Idiopathic neuropathy patients are at high risk for metabolic syndrome. J Neurol Sci 2008; 273:25–28.
- Devigili G, Tugnoli V, Penza P, et al. The diagnostic criteria for small fibre neuropathy: from symptoms to neuropathology. Brain 2008; 131:1912– 1925.
- Low VA, Sandroni P, Fealey RD, Low PA. Detection of small-fiber neuropathy by sudomotor testing. Muscle Nerve 2006; 34:57–61.
- McArthur JC, Stocks EA, Hauer P, Cornblath DR, Griffin JW. Epidermal nerve fiber density: normative reference range and diagnostic efficiency. Arch Neurol 1998; 55:1513–1520.
- Gibbons CH, Griffin JW, Polydefkis M, et al. The utility of skin biopsy for prediction of progression in suspected small fiber neuropathy. Neurology 2006; 66:256–258.
- Polydefkis M, Yiannoutsos CT, Cohen BA, et al. Reduced intraepidermal nerve fiber density in HIV-associated sensory neuropathy. Neurology 2002; 58:115–119.
- Herrmann DN, Griffin JW, Hauer P, Cornblath DR, McArthur JC. Epidermal nerve fiber density and sural nerve morphometry in peripheral neuropathies. Neurology 1999; 53:1634–1640.
- Zhou L, Kitch DW, Evans SR, et al. Correlates of epidermal nerve fiber densities in HIV-associated distal sensory polyneuropathy. Neurology 2007; 68:2113–2119.
- Novak V, Freimer ML, Kissel JT, et al. Autonomic impairment in painful neuropathy. Neurology 2001; 56:861–868.
- Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346:393–403.
- Smith AG, Russell J, Feldman EL, et al. Lifestyle intervention for prediabetic neuropathy. Diabetes Care 2006; 29:1294–1299.
- Hoitsma E, Faber CG, van Santen-Hoeufft M, De Vries J, Reulen JP, Drent M. Improvement of small fiber neuropathy in a sarcoidosis patient after treatment with infliximab. Sarcoidosis Vasc Diffuse Lung Dis 2006; 23:73–77.
- Chen H, Lamer TJ, Rho RH, et al. Contemporary management of neuropathic pain for the primary care physician. Mayo Clin Proc 2004; 79:1533–1545.
- Galluzzi KE. Managing neuropathic pain. J Am Osteopath Assoc 2007; 107( suppl 6):ES39–ES48.
- Kieburtz K, Simpson D, Yiannoutsos C, et al. A randomized trial of amitriptyline and mexiletine for painful neuropathy in HIV infection. AIDS Clinical Trial Group 242 Protocol Team. Neurology 1998; 51:1682–1688.
- Gregg EW, Gu Q, Williams D, et al. Prevalence of lower extremity diseases associated with normal glucose levels, impaired fasting glucose, and diabetes among U.S. adults aged 40 or older. Diabetes Res Clin Pract 2007; 77:485–488.
- Lacomis D. Small fiber neuropathy. Muscle Nerve 2002; 26:173–188.
- Smith AG, Singleton JR. Impaired glucose tolerance and neuropathy. Neurologist 2008; 14:23–29.
- Chai J, Herrmann DN, Stanton M, Barbano RL, Logigian EL. Painful small-fiber neuropathy in Sjogren syndrome. Neurology 2005; 65:925–927.
- Goransson LG, Tjensvoll AB, Herigstad A, Mellgren SI, Omdal R. Small-diameter nerve fiber neuropathy in systemic lupus erythematosus. Arch Neurol 2006; 63:401–404.
- Orstavik K, Norheim I, Jorum E. Pain and small-fiber neuropathy in patients with hypothyroidism. Neurology 2006; 67:786–791.
- McArthur JC, Brew BJ, Nath A. Neurological complications of HIV infection. Lancet Neurol 2005; 4:543–555.
- Brannagan TH, Hays AP, Chin SS, et al. Small-fiber neuropathy/neuronopathy associated with celiac disease: skin biopsy findings. Arch Neurol 2005; 62:1574–1578.
- Polydefkis M, Allen RP, Hauer P, Earley CJ, Griffin JW, McArthur JC. Subclinical sensory neuropathy in late-onset restless legs syndrome. Neurology 2000; 55:1115–1121.
- Gorson KC, Herrmann DN, Thiagarajan R, et al. Non-length dependent small fibre neuropathy/ganglionopathy. J Neurol Neurosurg Psychiatry 2008; 79:163–169.
- Singleton JR, Smith AG, Bromberg MB. Increased prevalence of impaired glucose tolerance in patients with painful sensory neuropathy. Diabetes Care 2001; 24:1448–1453.
- Novella SP, Inzucchi SE, Goldstein JM. The frequency of undiagnosed diabetes and impaired glucose tolerance in patients with idiopathic sensory neuropathy. Muscle Nerve 2001; 24:1229–1231.
- Smith AG, Singleton JR. The diagnostic yield of a standardized approach to idiopathic sensory-predominant neuropathy. Arch Intern Med 2004; 164:1021–1025.
- Sumner CJ, Sheth S, Griffin JW, Cornblath DR, Polydefkis M. The spectrum of neuropathy in diabetes and impaired glucose tolerance. Neurology 2003; 60:108–111.
- Gregg EW, Sorlie P, Paulose-Ram R, et al. Prevalence of lower-extremity disease in the US adult population >=40 years of age with and without diabetes: 1999–2000 National Health and Nutrition Examination Survey. Diabetes Care 2004; 27:1591–1597.
- Boulton A. What causes neuropathic pain? J Diabetes Complications 1992; 6:58–63.
- Russell JW, Sullivan KA, Windebank AJ, Herrmann DN, Feldman EL. Neurons undergo apoptosis in animal and cell culture models of diabetes. Neurobiol Dis 1999; 6:347–363.
- Costa LA, Canani LH, Lisboa HR, Tres GS, Gross JL. Aggregation of features of the metabolic syndrome is associated with increased prevalence of chronic complications in type 2 diabetes. Diabet Med 2004; 21:252–255.
- Tesfaye S, Chaturvedi N, Eaton SE, et al. Vascular risk factors and diabetic neuropathy. N Engl J Med 2005; 352:341–350.
- Smith A, Rose K, Singleton J. Idiopathic neuropathy patients are at high risk for metabolic syndrome. J Neurol Sci 2008; 273:25–28.
- Devigili G, Tugnoli V, Penza P, et al. The diagnostic criteria for small fibre neuropathy: from symptoms to neuropathology. Brain 2008; 131:1912– 1925.
- Low VA, Sandroni P, Fealey RD, Low PA. Detection of small-fiber neuropathy by sudomotor testing. Muscle Nerve 2006; 34:57–61.
- McArthur JC, Stocks EA, Hauer P, Cornblath DR, Griffin JW. Epidermal nerve fiber density: normative reference range and diagnostic efficiency. Arch Neurol 1998; 55:1513–1520.
- Gibbons CH, Griffin JW, Polydefkis M, et al. The utility of skin biopsy for prediction of progression in suspected small fiber neuropathy. Neurology 2006; 66:256–258.
- Polydefkis M, Yiannoutsos CT, Cohen BA, et al. Reduced intraepidermal nerve fiber density in HIV-associated sensory neuropathy. Neurology 2002; 58:115–119.
- Herrmann DN, Griffin JW, Hauer P, Cornblath DR, McArthur JC. Epidermal nerve fiber density and sural nerve morphometry in peripheral neuropathies. Neurology 1999; 53:1634–1640.
- Zhou L, Kitch DW, Evans SR, et al. Correlates of epidermal nerve fiber densities in HIV-associated distal sensory polyneuropathy. Neurology 2007; 68:2113–2119.
- Novak V, Freimer ML, Kissel JT, et al. Autonomic impairment in painful neuropathy. Neurology 2001; 56:861–868.
- Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346:393–403.
- Smith AG, Russell J, Feldman EL, et al. Lifestyle intervention for prediabetic neuropathy. Diabetes Care 2006; 29:1294–1299.
- Hoitsma E, Faber CG, van Santen-Hoeufft M, De Vries J, Reulen JP, Drent M. Improvement of small fiber neuropathy in a sarcoidosis patient after treatment with infliximab. Sarcoidosis Vasc Diffuse Lung Dis 2006; 23:73–77.
- Chen H, Lamer TJ, Rho RH, et al. Contemporary management of neuropathic pain for the primary care physician. Mayo Clin Proc 2004; 79:1533–1545.
- Galluzzi KE. Managing neuropathic pain. J Am Osteopath Assoc 2007; 107( suppl 6):ES39–ES48.
- Kieburtz K, Simpson D, Yiannoutsos C, et al. A randomized trial of amitriptyline and mexiletine for painful neuropathy in HIV infection. AIDS Clinical Trial Group 242 Protocol Team. Neurology 1998; 51:1682–1688.
KEY POINTS
- Symptoms of small fiber neuropathy typically start with burning feet and numb toes.
- Causes and associated conditions can be found in over 50% of cases. These include glucose dysmetabolism, connective tissue diseases, sarcoidosis, dysthyroidism, vitamin B12 deficiency, paraproteinemia, human immunodeficiency virus infection, celiac disease, neurotoxic drug exposure, and paraneoplastic syndrome.
- Findings on routine nerve conduction studies and electromyography are typically normal in this disease.
- Management includes aggressively identifying and treating the underlying cause, advising lifestyle modifications, and alleviating pain.
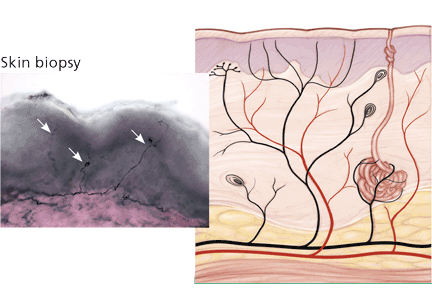
Pregabalin for fibromyalgia: Some relief but no cure
Pregabalin (Lyrica) is a novel analogue of the neurotransmitter gamma aminobutyric acid (GABA) with analgesic, anticonvulsant, and anxiolytic activity. Its approval by the US Food and Drug Administration (FDA) in 2007 for the treatment of fibromyalgia made it the first drug approved for this indication. Until then, management of fibromyalgia entailed drugs to treat pain, sleep, fatigue, and psychological disorders, and a strong emphasis on exercise and physical therapy.
Those who still question the validity of fibromyalgia as a diagnosis object to drug companies “benefiting” from the sale of such drugs.1 But many hail pregabalin as an important advance in our understanding of the pathogenesis of fibromyalgia and how to treat it. A key question remains: How will pregabalin fit into the treatment of this often-challenging disease?
FROM FIBROSITIS TO FIBROMYALGIA
Fibromyalgia is a syndrome characterized by widespread pain. Chronic muscular pain is a common problem, but fibromyalgia is distinguished from other pain disorders by additional findings, such as consistent areas of tenderness (tender points), nonrestorative sleep, severe fatigue, and frequent psychological comorbidities such as depression and anxiety.
Fibromyalgia was originally termed “fibrositis” in 1904 by Sir William Gowers, who described it as a painful condition of the fibrous tissue, which he believed was due to inflammation in the muscles.2,3 For several decades, research was dedicated to looking for pathology in the muscle tissue, which was thought to be the major source of pain for most patients with fibromyalgia.
In the mid-1970s, Dr. H. Moldofsky, a noted sleep researcher, reported on abnormalities of the alpha-delta component of nonrapid-eye-movement sleep in these patients. He subsequently collaborated with Dr. Hugh A. Smythe, who helped define the fibromyalgia tender points. Fibrositis was subsequently renamed fibromyalgia syndrome, since it was agreed that there was no true inflammation in muscles or fibrous tissue.
In 1990, the American College of Rheumatology published “classification criteria” for the disease.4 The criteria include two main features:
- A history of widespread pain (“widespread” being defined as in the axial distribution, in both the left and right sides of the body, and above and below the waist), which must be present for 3 months or more, and
- Tenderness in at least 11 of 18 specified points that is elicited when a pressure of 4 kg (the amount of pressure required to blanch a thumbnail) is applied in steady increments starting at 1 kg.
Although pain is subjective and therefore difficult to assess, the classification criteria did make it easier to study the disease in a uniform way and led to an explosion of research in this field.
FUNCTIONAL ABNORMALITIES NI THE CENTRAL NERVOUS SYSTEM
Research to date points to the pain in fibromyalgia as being mediated by changes in the central nervous system rather than in the musculoskeletal system, as was initially thought.
In the dorsal horn of the spinal cord, nociceptive (pain-sensing) neurons from the periphery synapse with the second-order neurons that carry the pain signal to the brain. In fibromyalgia, several processes seem to amplify the signal.
Central sensitization is defined as enhanced excitability of neurons in the dorsal horn. Its features include augmented spontaneous neuronal activity, enlarged receptive field areas, and enhanced responses generated by large- and small-caliber primary afferent fibers. It can result from prolonged or strong activity in the dorsal horn neurons, and it leads to the spread of hyperactivity across multiple spinal segments.5–7
While much of the evidence for central sensitization in fibromyalgia is from animal studies, the phenomenon has also been studied in humans. Desmeules et al8 found that, compared with people without fibromyalgia, those with fibromyalgia had significantly lower thresholds of pain as assessed subjectively and measured objectively using the nociceptive flexion R-III reflex, which the authors described as “a specific physiologic correlate for the objective evaluation of central nociceptive pathways.”
Wind-up. Prolonged stimulation of C fibers in the dorsal horn can result in the phenomenon of wind-up, which refers to the temporal summation of second pain.
A painful stimulus evokes two pain signals. The first signal is brief and travels rapidly to the spinal cord via myelinated fibers (A fibers). The second signal, which is related to chronic pain and is described as dull, aching, or burning, travels more slowly to the dorsal horn via unmyelinated fibers (C fibers), the synapses of which use the neurotransmitter glutamate. Temporal summation is a phenomenon observed in experiments in which a series of painful stimuli are applied at regular intervals of about 2 seconds; although each stimulus is identical in intensity, subjects perceive them as increasing in intensity. The reason: during this repetitive stimulation, N-methyl-d-aspartate (NMDA) receptors become activated, leading to the removal of a magnesium block within the receptor. This results in an influx of calcium into the neuron and activation of protein kinase C, nitric oxide synthase, and cyclooxygenase. Ultimately, the firing rates of the nociceptive neurons are increased and the peripheral pain signal is strongly amplified.6
Wind-up has been shown to lead to characteristics of central sensitization related to C-fiber activity in animals.7 Staud et al9 studied wind-up in patients with and without fibromyalgia using series of repetitive thermal stimulation to produce temporal summation. Though wind-up was evoked in both groups, differences were observed both in the magnitude of sensory response to the first stimulus within a series and in the amount of temporal summation within a series.
Elevated excitatory neurotransmitters. In 1994, Russell et al10 showed that the concentration of substance P, an excitatory neurotransmitter, was three times higher in the cerebrospinal fluid of people with fibromyalgia than in normal controls.
Harris and colleagues11 reported that glutamate, another excitatory neurotransmitter, is elevated within the brain in people with fibromyalgia. They further showed that the levels of glutamate within the insula of the brain are directly associated with the levels of both experimental pressure-evoked pain thresholds and clinical pain ratings in fibromyalgia patients.
Evidence from imaging studies. Other objective evidence of central sensitization in fibromyalgia patients comes from studies using novel imaging.
Gracely et al12 performed functional magnetic resonance imaging (MRI) in people with and without fibromyalgia while applying pressure to their thumbs with a thumbscrew-type device. At equal levels of pressure, the people with fibromyalgia said the pressure hurt more, and specific areas of their brains lit up more on functional MRI. When the experimenters increased the pressure in the people without fibromyalgia until this group subjectively rated the pain as high as the fibromyalgia patients rated the lower level of pressure, their brains lit up to a similar degree in the same areas. These findings provide objective evidence of significantly lower pain thresholds in patients with fibromyalgia than in healthy controls, and they support the theory of central augmentation of pain sensitivity in fibromyalgia.
Staud et al13 also used functional MRI and found greater brain activity associated with temporal summation in fibromyalgia patients compared with controls. (In this experiment, the painful stimulus consisted of heat pulses to the foot.)
Drugs other than pregabalin that modulate the dorsal horn activity of the pain pathway include opioids, tramadol (Ultram), gabapentin (Neurontin), GABA agonists such as baclofen (Lioresal), antidepressants, alpha-2 adrenergic agonists (phenylephrine), and 5-HT3 antagonists such as ondansetron (Zofran), but none has been consistently effective for fibromyalgia.5,14
PREGABALIN
Pregabalin is an alpha-2-delta ligand similar to GABA, but it does not act on GABA receptors. Rather, it binds with high affinity to the alpha-2-delta subunit of voltage-gated presynaptic calcium channels, resulting in reduction of calcium flow through the channels, which subsequently inhibits the release of neurotransmitters including glutamate, norepinephrine, and substance P.15–17 Animal studies suggest that the decrease in the levels of these excitatory neurotransmitters is the mechanism of action of pregabalin, resulting in its analgesic, anticonvulsant, and anxiolytic benefit.15 Another potential mechanism of pregabalin is enhancement of slow-wave sleep, demonstrated in one study in healthy human subjects.18
Besides fibromyalgia, pregabalin is also approved for the treatment of diabetic peripheral neuropathy, postherpetic neuralgia, generalized anxiety disorder, and social anxiety disorder, and as adjunctive therapy for partial-onset seizure in adults.
Pharmacokinetics
Pregabalin is quickly absorbed, primarily in the proximal colon (bioavailability > 90%), and has highly predictable and linear pharmacokinetics.15 Food consumption does not affect its absorption or elimination but can delay its peak plasma concentration, which occurs at 1.5 hours. Its elimination half-life is approximately 6 hours.15 Because it does not bind to plasma proteins, it freely crosses the blood-brain barrier. The drug reaches its steady-state concentration within 2 days of starting therapy.
Its clearance is not affected by the sex or race of the patient, but its total clearance may be lower in the elderly because of age-related loss of renal function. Patients on hemodialysis may require a supplemental dose after dialysis because hemodialysis removes the pregabalin.
The drug is not metabolized by the P450 system in the liver, so it interacts only minimally with drugs that do use the P450 system. However, its clearance may be decreased when it is used concomitantly with drugs that can reduce the glomerular filtration rate, such as nonsteroidal anti-inflammatory drugs, aminoglycosides, and cyclosporine.15
Efficacy
The efficacy of pregabalin in fibromyalgia was evaluated in several recent trials.19
Crofford et al16 assessed pregabalin’s effects on pain, sleep, fatigue, and health-related quality of life. Some 529 patients with fibromyalgia were randomized in a double-blind fashion to four treatment groups: placebo, and pregabalin 150 mg/day, 300 mg/day, and 450 mg/day. The baseline mean pain scores (a 0-to-10 scale derived from daily diary ratings) were 6.9 in the placebo group, 6.9 for the pregabalin 150 mg/day group, 7.3 for the pregabalin 300 mg/day group, and 7.0 for the pregabalin 450 mg/day group.
The pain scores declined in all groups, but at 8 weeks, the mean score had declined 0.93 points more in the group receiving pregabalin 450 mg/day than in the placebo group (P ≤ .001). The scores in the groups taking pregabalin 150 mg/day and 300 mg/day were not significantly different from those in the placebo group. Significantly more patients in the 450-mg/day group (29%, vs 13% in the placebo group) had at least 50% improvement in pain at the end of the study. Patients in both the 300-mg/day group and the 450-mg/day had statistically significant improvement in their quality of sleep, in fatigue, and on the Patient Global Impression of Change (PGIC) scale.
Arnold et al20 conducted a trial with 750 patients in which three doses of pregabalin were compared with placebo: 300 mg/day, 450 mg/day, and 600 mg/day. The primary end point was also the change in pain score from baseline (using the 0-to-10 scale derived from a daily pain diary). The mean baseline pain score was 6.7.
At 14 weeks, the mean pain score was lower than at baseline in all the groups, but it had declined 0.71 more in the pregabalin 300-mg/day group than in the placebo group, 0.98 points more in the 450-mg/day group, and 1.0 points more in the 600-mg/day group. All three pregabalin groups also showed significant improvement on the PGIC scale, and patients in the 450-mg/day and 600-mg/day groups showed statistically significant improvement in the Fibromyalgia Impact Questionnaire (FIQ) score. All three pregabalin treatment groups also had significantly better patient-reported sleep outcomes than in the placebo group, both in measures of overall sleep and quality of sleep. With the exception of a significant improvement of anxiety on 600 mg/day, there was no significant difference between the treatment and placebo groups in the secondary outcomes of depression and anxiety symptoms and fatigue.
Duan et al21 presented a pooled analysis of this and a similarly designed double-blind, placebo-controlled trial (the results of which were not available individually) at the 71st annual meeting of the American College of Rheumatology in November 2007. The analysis included 1,493 patients with a mean baseline pain score of 6.9. Compared with the mean pain score in the placebo group, those in the pregabalin groups had declined more by the end of the study: 0.55 points more with 300 mg/day, 0.71 points more with 450 mg/day, and 0.82 points more with 600 mg/day. This pooled analysis also showed significant improvement in PGIC score with all pregabalin doses and in the FIQ score with 450 mg/day and 600 mg/day.
The FREEDOM trial22 (Fibromyalgia Relapse Evaluation and Efficacy for Durability of Meaningful Relief) evaluated the durability of effect of pregabalin in reducing pain and symptoms associated with fibromyalgia in 1,051 patients who initially responded to the drug.
The patients received 6 weeks of open-label treatment with pregabalin and then 26 weeks of double-blind treatment (dose adjustment was allowed based on efficacy and tolerability for the first 3 weeks). The time to loss of therapeutic response was significantly longer with pregabalin than with placebo. Loss of therapeutic response was defined as worsening of pain for two consecutive visits or worsening of fibromyalgia symptoms requiring alternative therapy.
By the end of the double-blind phase, 61% of those in the placebo group had loss of therapeutic response compared with only 32% in the pregabalin group. The time to worsening of the FIQ score was also significantly longer in the pregabalin group than in the placebo group.
Adverse effects: Dizziness, sleepiness, weight gain
Dizziness and sleepiness were the most common adverse events in these studies.
In the 8-week study by Crofford et al,16 dizziness was dose-related, occurring in 10.7% of those receiving placebo (one patient withdrew because of dizziness), 22.7% of those receiving 150 mg/day (two patients withdrew), 31.3% of those receiving 300 mg/day (four patients withdrew), and 49.2% of those receiving 450 mg/day (five patients withdrew). Somnolence was also dose-related, occurring in 4.6% in the placebo group, 15.9% in the 150-mg/day group (two patients withdrew due to somnolence), 27.6% in the 300-mg/day group (three withdrew), and 28.0% in the 450-mg/day group (five withdrew).
The 14-week study by Arnold et al20 also showed higher frequencies of adverse events with higher doses. The rates of dizziness were 7.6% with placebo, 27.9% with pregabalin 300 mg/day, 37.4% with 450 mg/day, and 42.0% with 600 mg/day. The rates of somnolence were 3.8% with placebo, 12.6% with 300 mg/day of pregabalin, 19.5% with 450 mg/day, and 21.8% with 600 mg/day. Dizziness and somnolence were also the most common adverse effects that led to discontinuation of pregabalin, with rates of 4% and 3%, respectively.
The open-label phase of the FREEDOM trial showed rates of 36% for dizziness and 22% for somnolence among pregabalin-treated patients.
Weight gain and peripheral edema were also common adverse effects in these studies.22 Definitions of weight gain varied, and edema was not accompanied by evidence of cardiac or renal dysfunction.
Less common side effects seen more frequently in the treated groups included dry mouth, blurred vision, and difficulty with concentration and attention. The package insert also warns of angioedema, hypersensitivity reaction, mild asymptomatic creatine kinase elevation, decreased platelet count (without bleeding), and prolongation of the PR interval on electrocardiography.
Pregabalin is a schedule V controlled substance; in clinical studies, abrupt or rapid discontinuation of the drug led to insomnia, nausea, headache, or diarrhea in some patients, suggesting symptoms of dependence. In clinical studies involving a total of more than 5,500 patients, 4% of patients on pregabalin and 1% of patients on placebo reported euphoria as an adverse effect,19 suggesting possible potential for abuse.
Dosing
As a result of the above studies, the recommended starting dose of pregabalin for fibromyalgia is 150 mg/day in two or three divided doses, gradually increased to 300 mg/day within 1 week based on tolerability and efficacy. The dose may be increased to a maximum of 450 mg/day. The 600-mg dose was found to have no significant additional benefit, but it did have more adverse effects and therefore is not recommended. It is important to note that in these studies multiple medications for pain and insomnia were prohibited, so data on drug interactions with pregabalin are limited.
Few achieve complete remission, but most patients feel better
Several studies of the natural history of fibromyalgia have shown that very few patients experience complete remission of the disease, even after many years. Therefore, one should try to set up realistic expectations for patients, with the goal of achieving functional improvement in activities of daily living and a return to one’s predisease state.
In the longest follow-up study, 39 patients in Boston, MA, were prospectively followed for over 10 years. No patient achieved complete remission: all of them reported some fibromyalgia-related symptoms at the end of the study.23 However, 66% of them felt a little to a lot better than when first diagnosed, 55% felt well or very well, and only 7% felt poorly.
Other studies have also shown complete remission to be rare.24,25 A 5-year follow-up study investigating fibromyalgia patients’ perceptions of their symptoms and its impact on everyday life activities demonstrated that the social consequences of fibromyalgia’s symptoms are severe and constant over time.26
Evidence of favorable outcomes was reported in one study in which 47% of patients reported moderate to marked improvement in overall fibromyalgia status upon 3-year follow-up,27 and in another study, in which remission was objectively identified in 24.2% of patients 2 years after diagnosis.28
OTHER THERAPIES
Although there have been many studies of pharmacologic therapies for fibromyalgia to date, the trials had significant limitations, such as short duration, inadequate sample size, nonstandardized measures of efficacy, question of regression to the mean, and inadequate blinding, resulting in insufficient evidence to recommend one drug over another.
Tricyclic antidepressants. Two meta-analyses and a clinical review have supported the efficacy of tricyclic antidepressants in improving symptoms in fibromyalgia patients.29–31
Selective serotonin reuptake inhibitors (SSRIs) have not been well studied, and the small size and methodologic shortcomings of these studies make it difficult to draw conclusions about the efficacy of SSRIs in reducing pain in fibromyalgia patients.30,31
Duloxetine (Cymbalta) and milnacipran (Savella) are serotonin and norepinephrine reuptake inhibitors.32–34 A randomized, double-blind placebo-controlled trial evaluated duloxetine in 520 fibromyalgia patients with and without major depressive disorder. Pain scores improved significantly over 6 months in duloxetine-treated patients at doses of 60 and 120 mg/day.33 Duloxetine became the second drug approved for the treatment of fibromyalgia in 2007, and milnacipran became the third in 2009.
WHAT ROLE FOR PREGABALIN?
Pregabalin may reduce pain in some patients with fibromyalgia. However, the presenting symptoms can vary significantly, and symptoms can vary even in individual patients over time. Therefore, in most patients with fibromyalgia, a multidisciplinary approach is used to treat pain, sleep disturbance, and fatigue, along with comorbidities such as neurally mediated hypotension and psychiatric disorders. Because treatment of fibromyalgia often involves multiple drugs in addition to exercise and behavioral therapies, future studies should examine combinations of drugs and the use of drugs in conjunction with nondrug treatments.
Pregabalin advances our knowledge of fibromyalgia through improving the understanding of central sensitization and how brain neurotransmitters control central pain perceptions. Drug treatment must still be part of the comprehensive management of this disease. Physician and patient education about the current understanding of the disease is paramount in setting realistic goals for treatment.14 Future strategies to manage fibromyalgia will be based on the pathophysiology of this complex condition.
- Berenson A. Drug approved. Is disease real? New York Times, January 14, 2008. http://www.nytimes.com/2008/01/14/health/14pain.html. Accessed February 2, 2009.
- White KP, Harth M. Classification, epidemiology, and natural history of fibromyalgia. Curr Pain Headache Rep 2001; 5:320–329.
- Bennett RM. Fibromyalgia: present to future. Curr Pain Headache Rep 2004; 8:379–384.
- Wolfe F, Smythe HA, Yunus MF, et al. The American College of Rheumatolgy 1990 Criteria for the Classification of Fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis Rheum 1990; 33:160–172.
- Bennett RM. The rational management of fibromyalgia patients. Rheum Dis Clin North Am 2002; 28:181–199.
- Staud R. Evidence of involvement of central neural mechanisms in generating fibromyalgia pain. Curr Rheumatol Rep 2002; 4:299–305.
- Li J, Simone DA, Larson AA. Windup leads to characteristics of central sensitization. Pain 1999; 79:75–82.
- Desmeules JA, Cedraschi C, Rapiti E, et al. Neurophysiologic evidence for a central sensitization in patients with fibromyalgia. Arthritis Rheum 2003; 48:1420–1429.
- Staud R, Vierck CJ, Cannon RL, Mauderli AP, Price DD. Abnormal sensitization and temporal summation of pain (wind-up) in patients with fibromyalgia syndrome. Pain 2001; 91:165–175.
- Russell IJ, Orr MD, Littman B, et al. Elevated cerebrospinal fluid levels of substance P in patients with fibromyalgia syndrome. Arthritis Rheum 1994; 37:1593–1601.
- Harris RE, Sundgren PC, Pang Y, et al. Dynamic levels of glutamate within the insula are associated with improvements in multiple pain domains in fibromyalgia. Arthritis Rheum 2008; 58:903–907.
- Gracely RH, Petzke F, Wolf JM, Clauw DJ. Functional magnetic resonance imaging evidence of augmented pain processing in fibromyalgia. Arthritis Rheum 2002; 46:1333–1343.
- Staud R, Craggs JG, Perlstein WM, Robinson ME, Price DD. Brain activity associated with slow temporal summation of C-fiber evoked pain in fibromyalgia patients and healthy controls. Eur J Pain 2008; 12:1078–1089.
- Baker K, Barkhuizen A. Pharmacologic treatment of fibromyalgia. Curr Pain Headache Rep 2005; 9:301–306.
- Tassone DM, Boyce E, Guyer J, Nuzum D. Pregabalin: a novel gamma-aminobutyric acid analogue in the treatment of neuropathic pain, partial-onset seizures, and anxiety disorders. Clin Ther 2007; 29:26–48.
- Crofford LJ, Rowbotham MC, Mease PJ, et al, and the Pregabalin 1008-105 Study Group. Pregabalin for the treatment of fibromyalgia syndrome. results of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 2005; 52:1264–1273.
- Stahl SM. Anticonvulsants and the relief of chronic pain: pregabalin and gabapentin as alpha(2)delta ligands at voltage-gated calcium channels. J Clin Psychiatry 2004; 65:596–597.
- Hindmarch I, Dawson J, Stanley N. A double-blind study in healthy volunteers to assess the effects of sleep on pregabalin compared with alprazolam and placebo. Sleep 2005; 28:187–193.
- Pfizer Executive Summary. Lyrica (pregabalin) capsules c-v. July 2007. www.fda.gov/OHRMS/DOCKETS/ac/08/briefing/2008-4372b1-02-Pfizer.pdf. Accessed February 2, 2009.
- Arnold LM, Russell IJ, Diri EW, et al. A 14-week, randomized, double-blinded, placebo-controlled mono-therapy trial of pregabalin in patients with fibomyalgia. J Pain 2008; 9:792–805.
- Duan WR, Florian H, Young JP, Martin S, Haig G, Barrett JA. Pregabalin monotherapy for management of fibromyalgia: analysis of two double-blind, randomized, placebo-controlled trials (poster presentation). American College of Rheumatology Annual Scientific Meeting, Boston, MA, November 6–7, 2007.
- Crofford LJ, Mease PJ, Simpson SL, et al. Fibromyalgia relapse evaluation and efficacy for durability of meaningful relief (FREEDOM): a 6-month, double-blind, placebo-controlled trial with pregabalin. Pain 2008; 136:419–431.
- Kennedy M, Felson DT. A prospective long-term study of fibromyalgia syndrome. Arthritis Rheum 1996; 39:682–685.
- Bengtsson A, Backman E. Long-term follow-up of fibro-myalgia patients [abstract]. Scand J Rheumatolology 1992; 21(suppl 94):9.
- Ledingham J, Doherty S, Doherty M. Primary fibromyalgia syndrome—an outcome study. Br J Rheumatol 1993; 32:139–142.
- Henrikkson CM. Longterm effects of fibromyalgia on everyday life: a study of 56 patients. Scand J Rheumatol 1994; 23:36–41.
- Fitzcharles MA, Costa DD, Pöyhiä R. A study of standard care in fibromyalgia syndrome: a favorable outcome. J Rheumatol 2003; 30:154–159.
- Granges G, Zilko P, Littlejohn GO. Fibromyalgia syndrome: assessment of the severity of the condition 2 years after the diagnosis. J Rheumatol 1994; 21:523–529.
- Goldenberg DL, Burckhardt C, Crofford L. Management of fibromyalgia syndrome. JAMA 2004; 292:2388–2395.
- Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia: a meta-analysis and review. Psychosomatics 2000; 41:104–113.
- O’Malley PG, Balden E, Tomkins G, Santoro J, Kroenke K, Jackson JL. Treatment of fibromyalgia with anti-depressants: a meta-analysis. J Gen Intern Med 2000; 15:659–666.
- Arnold LM, Lu Y, Crofford LJ, et al. A double-blind, multicenter trial comparing duloxetine with placebo in the treatment of fibromyalgia patients with or without major depressive disorder. Arthritis Rheum 2004; 50:2974–2984.
- Russell IJ, Mease PJ, Smith TR, et al. Efficacy and safety of duloxetine for treatment of fibromyalgia in patients with or without major depressive disorder: Results from a 6-month, randomized, double-blind, placebo-controlled fixed-dose trial. Pain 2008; 136:432–444.
- Clauw DJ, Mease P, Palmer RH, Gendreau RM, Wang Y. Milnacipran for the treatment of fibromyalgia in adults: a 15-week, multicenter, randomized, double-blind, placebo-controlled, multiple-dose clinical trial. Clin Ther 2008; 30:1988–2004.
Pregabalin (Lyrica) is a novel analogue of the neurotransmitter gamma aminobutyric acid (GABA) with analgesic, anticonvulsant, and anxiolytic activity. Its approval by the US Food and Drug Administration (FDA) in 2007 for the treatment of fibromyalgia made it the first drug approved for this indication. Until then, management of fibromyalgia entailed drugs to treat pain, sleep, fatigue, and psychological disorders, and a strong emphasis on exercise and physical therapy.
Those who still question the validity of fibromyalgia as a diagnosis object to drug companies “benefiting” from the sale of such drugs.1 But many hail pregabalin as an important advance in our understanding of the pathogenesis of fibromyalgia and how to treat it. A key question remains: How will pregabalin fit into the treatment of this often-challenging disease?
FROM FIBROSITIS TO FIBROMYALGIA
Fibromyalgia is a syndrome characterized by widespread pain. Chronic muscular pain is a common problem, but fibromyalgia is distinguished from other pain disorders by additional findings, such as consistent areas of tenderness (tender points), nonrestorative sleep, severe fatigue, and frequent psychological comorbidities such as depression and anxiety.
Fibromyalgia was originally termed “fibrositis” in 1904 by Sir William Gowers, who described it as a painful condition of the fibrous tissue, which he believed was due to inflammation in the muscles.2,3 For several decades, research was dedicated to looking for pathology in the muscle tissue, which was thought to be the major source of pain for most patients with fibromyalgia.
In the mid-1970s, Dr. H. Moldofsky, a noted sleep researcher, reported on abnormalities of the alpha-delta component of nonrapid-eye-movement sleep in these patients. He subsequently collaborated with Dr. Hugh A. Smythe, who helped define the fibromyalgia tender points. Fibrositis was subsequently renamed fibromyalgia syndrome, since it was agreed that there was no true inflammation in muscles or fibrous tissue.
In 1990, the American College of Rheumatology published “classification criteria” for the disease.4 The criteria include two main features:
- A history of widespread pain (“widespread” being defined as in the axial distribution, in both the left and right sides of the body, and above and below the waist), which must be present for 3 months or more, and
- Tenderness in at least 11 of 18 specified points that is elicited when a pressure of 4 kg (the amount of pressure required to blanch a thumbnail) is applied in steady increments starting at 1 kg.
Although pain is subjective and therefore difficult to assess, the classification criteria did make it easier to study the disease in a uniform way and led to an explosion of research in this field.
FUNCTIONAL ABNORMALITIES NI THE CENTRAL NERVOUS SYSTEM
Research to date points to the pain in fibromyalgia as being mediated by changes in the central nervous system rather than in the musculoskeletal system, as was initially thought.
In the dorsal horn of the spinal cord, nociceptive (pain-sensing) neurons from the periphery synapse with the second-order neurons that carry the pain signal to the brain. In fibromyalgia, several processes seem to amplify the signal.
Central sensitization is defined as enhanced excitability of neurons in the dorsal horn. Its features include augmented spontaneous neuronal activity, enlarged receptive field areas, and enhanced responses generated by large- and small-caliber primary afferent fibers. It can result from prolonged or strong activity in the dorsal horn neurons, and it leads to the spread of hyperactivity across multiple spinal segments.5–7
While much of the evidence for central sensitization in fibromyalgia is from animal studies, the phenomenon has also been studied in humans. Desmeules et al8 found that, compared with people without fibromyalgia, those with fibromyalgia had significantly lower thresholds of pain as assessed subjectively and measured objectively using the nociceptive flexion R-III reflex, which the authors described as “a specific physiologic correlate for the objective evaluation of central nociceptive pathways.”
Wind-up. Prolonged stimulation of C fibers in the dorsal horn can result in the phenomenon of wind-up, which refers to the temporal summation of second pain.
A painful stimulus evokes two pain signals. The first signal is brief and travels rapidly to the spinal cord via myelinated fibers (A fibers). The second signal, which is related to chronic pain and is described as dull, aching, or burning, travels more slowly to the dorsal horn via unmyelinated fibers (C fibers), the synapses of which use the neurotransmitter glutamate. Temporal summation is a phenomenon observed in experiments in which a series of painful stimuli are applied at regular intervals of about 2 seconds; although each stimulus is identical in intensity, subjects perceive them as increasing in intensity. The reason: during this repetitive stimulation, N-methyl-d-aspartate (NMDA) receptors become activated, leading to the removal of a magnesium block within the receptor. This results in an influx of calcium into the neuron and activation of protein kinase C, nitric oxide synthase, and cyclooxygenase. Ultimately, the firing rates of the nociceptive neurons are increased and the peripheral pain signal is strongly amplified.6
Wind-up has been shown to lead to characteristics of central sensitization related to C-fiber activity in animals.7 Staud et al9 studied wind-up in patients with and without fibromyalgia using series of repetitive thermal stimulation to produce temporal summation. Though wind-up was evoked in both groups, differences were observed both in the magnitude of sensory response to the first stimulus within a series and in the amount of temporal summation within a series.
Elevated excitatory neurotransmitters. In 1994, Russell et al10 showed that the concentration of substance P, an excitatory neurotransmitter, was three times higher in the cerebrospinal fluid of people with fibromyalgia than in normal controls.
Harris and colleagues11 reported that glutamate, another excitatory neurotransmitter, is elevated within the brain in people with fibromyalgia. They further showed that the levels of glutamate within the insula of the brain are directly associated with the levels of both experimental pressure-evoked pain thresholds and clinical pain ratings in fibromyalgia patients.
Evidence from imaging studies. Other objective evidence of central sensitization in fibromyalgia patients comes from studies using novel imaging.
Gracely et al12 performed functional magnetic resonance imaging (MRI) in people with and without fibromyalgia while applying pressure to their thumbs with a thumbscrew-type device. At equal levels of pressure, the people with fibromyalgia said the pressure hurt more, and specific areas of their brains lit up more on functional MRI. When the experimenters increased the pressure in the people without fibromyalgia until this group subjectively rated the pain as high as the fibromyalgia patients rated the lower level of pressure, their brains lit up to a similar degree in the same areas. These findings provide objective evidence of significantly lower pain thresholds in patients with fibromyalgia than in healthy controls, and they support the theory of central augmentation of pain sensitivity in fibromyalgia.
Staud et al13 also used functional MRI and found greater brain activity associated with temporal summation in fibromyalgia patients compared with controls. (In this experiment, the painful stimulus consisted of heat pulses to the foot.)
Drugs other than pregabalin that modulate the dorsal horn activity of the pain pathway include opioids, tramadol (Ultram), gabapentin (Neurontin), GABA agonists such as baclofen (Lioresal), antidepressants, alpha-2 adrenergic agonists (phenylephrine), and 5-HT3 antagonists such as ondansetron (Zofran), but none has been consistently effective for fibromyalgia.5,14
PREGABALIN
Pregabalin is an alpha-2-delta ligand similar to GABA, but it does not act on GABA receptors. Rather, it binds with high affinity to the alpha-2-delta subunit of voltage-gated presynaptic calcium channels, resulting in reduction of calcium flow through the channels, which subsequently inhibits the release of neurotransmitters including glutamate, norepinephrine, and substance P.15–17 Animal studies suggest that the decrease in the levels of these excitatory neurotransmitters is the mechanism of action of pregabalin, resulting in its analgesic, anticonvulsant, and anxiolytic benefit.15 Another potential mechanism of pregabalin is enhancement of slow-wave sleep, demonstrated in one study in healthy human subjects.18
Besides fibromyalgia, pregabalin is also approved for the treatment of diabetic peripheral neuropathy, postherpetic neuralgia, generalized anxiety disorder, and social anxiety disorder, and as adjunctive therapy for partial-onset seizure in adults.
Pharmacokinetics
Pregabalin is quickly absorbed, primarily in the proximal colon (bioavailability > 90%), and has highly predictable and linear pharmacokinetics.15 Food consumption does not affect its absorption or elimination but can delay its peak plasma concentration, which occurs at 1.5 hours. Its elimination half-life is approximately 6 hours.15 Because it does not bind to plasma proteins, it freely crosses the blood-brain barrier. The drug reaches its steady-state concentration within 2 days of starting therapy.
Its clearance is not affected by the sex or race of the patient, but its total clearance may be lower in the elderly because of age-related loss of renal function. Patients on hemodialysis may require a supplemental dose after dialysis because hemodialysis removes the pregabalin.
The drug is not metabolized by the P450 system in the liver, so it interacts only minimally with drugs that do use the P450 system. However, its clearance may be decreased when it is used concomitantly with drugs that can reduce the glomerular filtration rate, such as nonsteroidal anti-inflammatory drugs, aminoglycosides, and cyclosporine.15
Efficacy
The efficacy of pregabalin in fibromyalgia was evaluated in several recent trials.19
Crofford et al16 assessed pregabalin’s effects on pain, sleep, fatigue, and health-related quality of life. Some 529 patients with fibromyalgia were randomized in a double-blind fashion to four treatment groups: placebo, and pregabalin 150 mg/day, 300 mg/day, and 450 mg/day. The baseline mean pain scores (a 0-to-10 scale derived from daily diary ratings) were 6.9 in the placebo group, 6.9 for the pregabalin 150 mg/day group, 7.3 for the pregabalin 300 mg/day group, and 7.0 for the pregabalin 450 mg/day group.
The pain scores declined in all groups, but at 8 weeks, the mean score had declined 0.93 points more in the group receiving pregabalin 450 mg/day than in the placebo group (P ≤ .001). The scores in the groups taking pregabalin 150 mg/day and 300 mg/day were not significantly different from those in the placebo group. Significantly more patients in the 450-mg/day group (29%, vs 13% in the placebo group) had at least 50% improvement in pain at the end of the study. Patients in both the 300-mg/day group and the 450-mg/day had statistically significant improvement in their quality of sleep, in fatigue, and on the Patient Global Impression of Change (PGIC) scale.
Arnold et al20 conducted a trial with 750 patients in which three doses of pregabalin were compared with placebo: 300 mg/day, 450 mg/day, and 600 mg/day. The primary end point was also the change in pain score from baseline (using the 0-to-10 scale derived from a daily pain diary). The mean baseline pain score was 6.7.
At 14 weeks, the mean pain score was lower than at baseline in all the groups, but it had declined 0.71 more in the pregabalin 300-mg/day group than in the placebo group, 0.98 points more in the 450-mg/day group, and 1.0 points more in the 600-mg/day group. All three pregabalin groups also showed significant improvement on the PGIC scale, and patients in the 450-mg/day and 600-mg/day groups showed statistically significant improvement in the Fibromyalgia Impact Questionnaire (FIQ) score. All three pregabalin treatment groups also had significantly better patient-reported sleep outcomes than in the placebo group, both in measures of overall sleep and quality of sleep. With the exception of a significant improvement of anxiety on 600 mg/day, there was no significant difference between the treatment and placebo groups in the secondary outcomes of depression and anxiety symptoms and fatigue.
Duan et al21 presented a pooled analysis of this and a similarly designed double-blind, placebo-controlled trial (the results of which were not available individually) at the 71st annual meeting of the American College of Rheumatology in November 2007. The analysis included 1,493 patients with a mean baseline pain score of 6.9. Compared with the mean pain score in the placebo group, those in the pregabalin groups had declined more by the end of the study: 0.55 points more with 300 mg/day, 0.71 points more with 450 mg/day, and 0.82 points more with 600 mg/day. This pooled analysis also showed significant improvement in PGIC score with all pregabalin doses and in the FIQ score with 450 mg/day and 600 mg/day.
The FREEDOM trial22 (Fibromyalgia Relapse Evaluation and Efficacy for Durability of Meaningful Relief) evaluated the durability of effect of pregabalin in reducing pain and symptoms associated with fibromyalgia in 1,051 patients who initially responded to the drug.
The patients received 6 weeks of open-label treatment with pregabalin and then 26 weeks of double-blind treatment (dose adjustment was allowed based on efficacy and tolerability for the first 3 weeks). The time to loss of therapeutic response was significantly longer with pregabalin than with placebo. Loss of therapeutic response was defined as worsening of pain for two consecutive visits or worsening of fibromyalgia symptoms requiring alternative therapy.
By the end of the double-blind phase, 61% of those in the placebo group had loss of therapeutic response compared with only 32% in the pregabalin group. The time to worsening of the FIQ score was also significantly longer in the pregabalin group than in the placebo group.
Adverse effects: Dizziness, sleepiness, weight gain
Dizziness and sleepiness were the most common adverse events in these studies.
In the 8-week study by Crofford et al,16 dizziness was dose-related, occurring in 10.7% of those receiving placebo (one patient withdrew because of dizziness), 22.7% of those receiving 150 mg/day (two patients withdrew), 31.3% of those receiving 300 mg/day (four patients withdrew), and 49.2% of those receiving 450 mg/day (five patients withdrew). Somnolence was also dose-related, occurring in 4.6% in the placebo group, 15.9% in the 150-mg/day group (two patients withdrew due to somnolence), 27.6% in the 300-mg/day group (three withdrew), and 28.0% in the 450-mg/day group (five withdrew).
The 14-week study by Arnold et al20 also showed higher frequencies of adverse events with higher doses. The rates of dizziness were 7.6% with placebo, 27.9% with pregabalin 300 mg/day, 37.4% with 450 mg/day, and 42.0% with 600 mg/day. The rates of somnolence were 3.8% with placebo, 12.6% with 300 mg/day of pregabalin, 19.5% with 450 mg/day, and 21.8% with 600 mg/day. Dizziness and somnolence were also the most common adverse effects that led to discontinuation of pregabalin, with rates of 4% and 3%, respectively.
The open-label phase of the FREEDOM trial showed rates of 36% for dizziness and 22% for somnolence among pregabalin-treated patients.
Weight gain and peripheral edema were also common adverse effects in these studies.22 Definitions of weight gain varied, and edema was not accompanied by evidence of cardiac or renal dysfunction.
Less common side effects seen more frequently in the treated groups included dry mouth, blurred vision, and difficulty with concentration and attention. The package insert also warns of angioedema, hypersensitivity reaction, mild asymptomatic creatine kinase elevation, decreased platelet count (without bleeding), and prolongation of the PR interval on electrocardiography.
Pregabalin is a schedule V controlled substance; in clinical studies, abrupt or rapid discontinuation of the drug led to insomnia, nausea, headache, or diarrhea in some patients, suggesting symptoms of dependence. In clinical studies involving a total of more than 5,500 patients, 4% of patients on pregabalin and 1% of patients on placebo reported euphoria as an adverse effect,19 suggesting possible potential for abuse.
Dosing
As a result of the above studies, the recommended starting dose of pregabalin for fibromyalgia is 150 mg/day in two or three divided doses, gradually increased to 300 mg/day within 1 week based on tolerability and efficacy. The dose may be increased to a maximum of 450 mg/day. The 600-mg dose was found to have no significant additional benefit, but it did have more adverse effects and therefore is not recommended. It is important to note that in these studies multiple medications for pain and insomnia were prohibited, so data on drug interactions with pregabalin are limited.
Few achieve complete remission, but most patients feel better
Several studies of the natural history of fibromyalgia have shown that very few patients experience complete remission of the disease, even after many years. Therefore, one should try to set up realistic expectations for patients, with the goal of achieving functional improvement in activities of daily living and a return to one’s predisease state.
In the longest follow-up study, 39 patients in Boston, MA, were prospectively followed for over 10 years. No patient achieved complete remission: all of them reported some fibromyalgia-related symptoms at the end of the study.23 However, 66% of them felt a little to a lot better than when first diagnosed, 55% felt well or very well, and only 7% felt poorly.
Other studies have also shown complete remission to be rare.24,25 A 5-year follow-up study investigating fibromyalgia patients’ perceptions of their symptoms and its impact on everyday life activities demonstrated that the social consequences of fibromyalgia’s symptoms are severe and constant over time.26
Evidence of favorable outcomes was reported in one study in which 47% of patients reported moderate to marked improvement in overall fibromyalgia status upon 3-year follow-up,27 and in another study, in which remission was objectively identified in 24.2% of patients 2 years after diagnosis.28
OTHER THERAPIES
Although there have been many studies of pharmacologic therapies for fibromyalgia to date, the trials had significant limitations, such as short duration, inadequate sample size, nonstandardized measures of efficacy, question of regression to the mean, and inadequate blinding, resulting in insufficient evidence to recommend one drug over another.
Tricyclic antidepressants. Two meta-analyses and a clinical review have supported the efficacy of tricyclic antidepressants in improving symptoms in fibromyalgia patients.29–31
Selective serotonin reuptake inhibitors (SSRIs) have not been well studied, and the small size and methodologic shortcomings of these studies make it difficult to draw conclusions about the efficacy of SSRIs in reducing pain in fibromyalgia patients.30,31
Duloxetine (Cymbalta) and milnacipran (Savella) are serotonin and norepinephrine reuptake inhibitors.32–34 A randomized, double-blind placebo-controlled trial evaluated duloxetine in 520 fibromyalgia patients with and without major depressive disorder. Pain scores improved significantly over 6 months in duloxetine-treated patients at doses of 60 and 120 mg/day.33 Duloxetine became the second drug approved for the treatment of fibromyalgia in 2007, and milnacipran became the third in 2009.
WHAT ROLE FOR PREGABALIN?
Pregabalin may reduce pain in some patients with fibromyalgia. However, the presenting symptoms can vary significantly, and symptoms can vary even in individual patients over time. Therefore, in most patients with fibromyalgia, a multidisciplinary approach is used to treat pain, sleep disturbance, and fatigue, along with comorbidities such as neurally mediated hypotension and psychiatric disorders. Because treatment of fibromyalgia often involves multiple drugs in addition to exercise and behavioral therapies, future studies should examine combinations of drugs and the use of drugs in conjunction with nondrug treatments.
Pregabalin advances our knowledge of fibromyalgia through improving the understanding of central sensitization and how brain neurotransmitters control central pain perceptions. Drug treatment must still be part of the comprehensive management of this disease. Physician and patient education about the current understanding of the disease is paramount in setting realistic goals for treatment.14 Future strategies to manage fibromyalgia will be based on the pathophysiology of this complex condition.
Pregabalin (Lyrica) is a novel analogue of the neurotransmitter gamma aminobutyric acid (GABA) with analgesic, anticonvulsant, and anxiolytic activity. Its approval by the US Food and Drug Administration (FDA) in 2007 for the treatment of fibromyalgia made it the first drug approved for this indication. Until then, management of fibromyalgia entailed drugs to treat pain, sleep, fatigue, and psychological disorders, and a strong emphasis on exercise and physical therapy.
Those who still question the validity of fibromyalgia as a diagnosis object to drug companies “benefiting” from the sale of such drugs.1 But many hail pregabalin as an important advance in our understanding of the pathogenesis of fibromyalgia and how to treat it. A key question remains: How will pregabalin fit into the treatment of this often-challenging disease?
FROM FIBROSITIS TO FIBROMYALGIA
Fibromyalgia is a syndrome characterized by widespread pain. Chronic muscular pain is a common problem, but fibromyalgia is distinguished from other pain disorders by additional findings, such as consistent areas of tenderness (tender points), nonrestorative sleep, severe fatigue, and frequent psychological comorbidities such as depression and anxiety.
Fibromyalgia was originally termed “fibrositis” in 1904 by Sir William Gowers, who described it as a painful condition of the fibrous tissue, which he believed was due to inflammation in the muscles.2,3 For several decades, research was dedicated to looking for pathology in the muscle tissue, which was thought to be the major source of pain for most patients with fibromyalgia.
In the mid-1970s, Dr. H. Moldofsky, a noted sleep researcher, reported on abnormalities of the alpha-delta component of nonrapid-eye-movement sleep in these patients. He subsequently collaborated with Dr. Hugh A. Smythe, who helped define the fibromyalgia tender points. Fibrositis was subsequently renamed fibromyalgia syndrome, since it was agreed that there was no true inflammation in muscles or fibrous tissue.
In 1990, the American College of Rheumatology published “classification criteria” for the disease.4 The criteria include two main features:
- A history of widespread pain (“widespread” being defined as in the axial distribution, in both the left and right sides of the body, and above and below the waist), which must be present for 3 months or more, and
- Tenderness in at least 11 of 18 specified points that is elicited when a pressure of 4 kg (the amount of pressure required to blanch a thumbnail) is applied in steady increments starting at 1 kg.
Although pain is subjective and therefore difficult to assess, the classification criteria did make it easier to study the disease in a uniform way and led to an explosion of research in this field.
FUNCTIONAL ABNORMALITIES NI THE CENTRAL NERVOUS SYSTEM
Research to date points to the pain in fibromyalgia as being mediated by changes in the central nervous system rather than in the musculoskeletal system, as was initially thought.
In the dorsal horn of the spinal cord, nociceptive (pain-sensing) neurons from the periphery synapse with the second-order neurons that carry the pain signal to the brain. In fibromyalgia, several processes seem to amplify the signal.
Central sensitization is defined as enhanced excitability of neurons in the dorsal horn. Its features include augmented spontaneous neuronal activity, enlarged receptive field areas, and enhanced responses generated by large- and small-caliber primary afferent fibers. It can result from prolonged or strong activity in the dorsal horn neurons, and it leads to the spread of hyperactivity across multiple spinal segments.5–7
While much of the evidence for central sensitization in fibromyalgia is from animal studies, the phenomenon has also been studied in humans. Desmeules et al8 found that, compared with people without fibromyalgia, those with fibromyalgia had significantly lower thresholds of pain as assessed subjectively and measured objectively using the nociceptive flexion R-III reflex, which the authors described as “a specific physiologic correlate for the objective evaluation of central nociceptive pathways.”
Wind-up. Prolonged stimulation of C fibers in the dorsal horn can result in the phenomenon of wind-up, which refers to the temporal summation of second pain.
A painful stimulus evokes two pain signals. The first signal is brief and travels rapidly to the spinal cord via myelinated fibers (A fibers). The second signal, which is related to chronic pain and is described as dull, aching, or burning, travels more slowly to the dorsal horn via unmyelinated fibers (C fibers), the synapses of which use the neurotransmitter glutamate. Temporal summation is a phenomenon observed in experiments in which a series of painful stimuli are applied at regular intervals of about 2 seconds; although each stimulus is identical in intensity, subjects perceive them as increasing in intensity. The reason: during this repetitive stimulation, N-methyl-d-aspartate (NMDA) receptors become activated, leading to the removal of a magnesium block within the receptor. This results in an influx of calcium into the neuron and activation of protein kinase C, nitric oxide synthase, and cyclooxygenase. Ultimately, the firing rates of the nociceptive neurons are increased and the peripheral pain signal is strongly amplified.6
Wind-up has been shown to lead to characteristics of central sensitization related to C-fiber activity in animals.7 Staud et al9 studied wind-up in patients with and without fibromyalgia using series of repetitive thermal stimulation to produce temporal summation. Though wind-up was evoked in both groups, differences were observed both in the magnitude of sensory response to the first stimulus within a series and in the amount of temporal summation within a series.
Elevated excitatory neurotransmitters. In 1994, Russell et al10 showed that the concentration of substance P, an excitatory neurotransmitter, was three times higher in the cerebrospinal fluid of people with fibromyalgia than in normal controls.
Harris and colleagues11 reported that glutamate, another excitatory neurotransmitter, is elevated within the brain in people with fibromyalgia. They further showed that the levels of glutamate within the insula of the brain are directly associated with the levels of both experimental pressure-evoked pain thresholds and clinical pain ratings in fibromyalgia patients.
Evidence from imaging studies. Other objective evidence of central sensitization in fibromyalgia patients comes from studies using novel imaging.
Gracely et al12 performed functional magnetic resonance imaging (MRI) in people with and without fibromyalgia while applying pressure to their thumbs with a thumbscrew-type device. At equal levels of pressure, the people with fibromyalgia said the pressure hurt more, and specific areas of their brains lit up more on functional MRI. When the experimenters increased the pressure in the people without fibromyalgia until this group subjectively rated the pain as high as the fibromyalgia patients rated the lower level of pressure, their brains lit up to a similar degree in the same areas. These findings provide objective evidence of significantly lower pain thresholds in patients with fibromyalgia than in healthy controls, and they support the theory of central augmentation of pain sensitivity in fibromyalgia.
Staud et al13 also used functional MRI and found greater brain activity associated with temporal summation in fibromyalgia patients compared with controls. (In this experiment, the painful stimulus consisted of heat pulses to the foot.)
Drugs other than pregabalin that modulate the dorsal horn activity of the pain pathway include opioids, tramadol (Ultram), gabapentin (Neurontin), GABA agonists such as baclofen (Lioresal), antidepressants, alpha-2 adrenergic agonists (phenylephrine), and 5-HT3 antagonists such as ondansetron (Zofran), but none has been consistently effective for fibromyalgia.5,14
PREGABALIN
Pregabalin is an alpha-2-delta ligand similar to GABA, but it does not act on GABA receptors. Rather, it binds with high affinity to the alpha-2-delta subunit of voltage-gated presynaptic calcium channels, resulting in reduction of calcium flow through the channels, which subsequently inhibits the release of neurotransmitters including glutamate, norepinephrine, and substance P.15–17 Animal studies suggest that the decrease in the levels of these excitatory neurotransmitters is the mechanism of action of pregabalin, resulting in its analgesic, anticonvulsant, and anxiolytic benefit.15 Another potential mechanism of pregabalin is enhancement of slow-wave sleep, demonstrated in one study in healthy human subjects.18
Besides fibromyalgia, pregabalin is also approved for the treatment of diabetic peripheral neuropathy, postherpetic neuralgia, generalized anxiety disorder, and social anxiety disorder, and as adjunctive therapy for partial-onset seizure in adults.
Pharmacokinetics
Pregabalin is quickly absorbed, primarily in the proximal colon (bioavailability > 90%), and has highly predictable and linear pharmacokinetics.15 Food consumption does not affect its absorption or elimination but can delay its peak plasma concentration, which occurs at 1.5 hours. Its elimination half-life is approximately 6 hours.15 Because it does not bind to plasma proteins, it freely crosses the blood-brain barrier. The drug reaches its steady-state concentration within 2 days of starting therapy.
Its clearance is not affected by the sex or race of the patient, but its total clearance may be lower in the elderly because of age-related loss of renal function. Patients on hemodialysis may require a supplemental dose after dialysis because hemodialysis removes the pregabalin.
The drug is not metabolized by the P450 system in the liver, so it interacts only minimally with drugs that do use the P450 system. However, its clearance may be decreased when it is used concomitantly with drugs that can reduce the glomerular filtration rate, such as nonsteroidal anti-inflammatory drugs, aminoglycosides, and cyclosporine.15
Efficacy
The efficacy of pregabalin in fibromyalgia was evaluated in several recent trials.19
Crofford et al16 assessed pregabalin’s effects on pain, sleep, fatigue, and health-related quality of life. Some 529 patients with fibromyalgia were randomized in a double-blind fashion to four treatment groups: placebo, and pregabalin 150 mg/day, 300 mg/day, and 450 mg/day. The baseline mean pain scores (a 0-to-10 scale derived from daily diary ratings) were 6.9 in the placebo group, 6.9 for the pregabalin 150 mg/day group, 7.3 for the pregabalin 300 mg/day group, and 7.0 for the pregabalin 450 mg/day group.
The pain scores declined in all groups, but at 8 weeks, the mean score had declined 0.93 points more in the group receiving pregabalin 450 mg/day than in the placebo group (P ≤ .001). The scores in the groups taking pregabalin 150 mg/day and 300 mg/day were not significantly different from those in the placebo group. Significantly more patients in the 450-mg/day group (29%, vs 13% in the placebo group) had at least 50% improvement in pain at the end of the study. Patients in both the 300-mg/day group and the 450-mg/day had statistically significant improvement in their quality of sleep, in fatigue, and on the Patient Global Impression of Change (PGIC) scale.
Arnold et al20 conducted a trial with 750 patients in which three doses of pregabalin were compared with placebo: 300 mg/day, 450 mg/day, and 600 mg/day. The primary end point was also the change in pain score from baseline (using the 0-to-10 scale derived from a daily pain diary). The mean baseline pain score was 6.7.
At 14 weeks, the mean pain score was lower than at baseline in all the groups, but it had declined 0.71 more in the pregabalin 300-mg/day group than in the placebo group, 0.98 points more in the 450-mg/day group, and 1.0 points more in the 600-mg/day group. All three pregabalin groups also showed significant improvement on the PGIC scale, and patients in the 450-mg/day and 600-mg/day groups showed statistically significant improvement in the Fibromyalgia Impact Questionnaire (FIQ) score. All three pregabalin treatment groups also had significantly better patient-reported sleep outcomes than in the placebo group, both in measures of overall sleep and quality of sleep. With the exception of a significant improvement of anxiety on 600 mg/day, there was no significant difference between the treatment and placebo groups in the secondary outcomes of depression and anxiety symptoms and fatigue.
Duan et al21 presented a pooled analysis of this and a similarly designed double-blind, placebo-controlled trial (the results of which were not available individually) at the 71st annual meeting of the American College of Rheumatology in November 2007. The analysis included 1,493 patients with a mean baseline pain score of 6.9. Compared with the mean pain score in the placebo group, those in the pregabalin groups had declined more by the end of the study: 0.55 points more with 300 mg/day, 0.71 points more with 450 mg/day, and 0.82 points more with 600 mg/day. This pooled analysis also showed significant improvement in PGIC score with all pregabalin doses and in the FIQ score with 450 mg/day and 600 mg/day.
The FREEDOM trial22 (Fibromyalgia Relapse Evaluation and Efficacy for Durability of Meaningful Relief) evaluated the durability of effect of pregabalin in reducing pain and symptoms associated with fibromyalgia in 1,051 patients who initially responded to the drug.
The patients received 6 weeks of open-label treatment with pregabalin and then 26 weeks of double-blind treatment (dose adjustment was allowed based on efficacy and tolerability for the first 3 weeks). The time to loss of therapeutic response was significantly longer with pregabalin than with placebo. Loss of therapeutic response was defined as worsening of pain for two consecutive visits or worsening of fibromyalgia symptoms requiring alternative therapy.
By the end of the double-blind phase, 61% of those in the placebo group had loss of therapeutic response compared with only 32% in the pregabalin group. The time to worsening of the FIQ score was also significantly longer in the pregabalin group than in the placebo group.
Adverse effects: Dizziness, sleepiness, weight gain
Dizziness and sleepiness were the most common adverse events in these studies.
In the 8-week study by Crofford et al,16 dizziness was dose-related, occurring in 10.7% of those receiving placebo (one patient withdrew because of dizziness), 22.7% of those receiving 150 mg/day (two patients withdrew), 31.3% of those receiving 300 mg/day (four patients withdrew), and 49.2% of those receiving 450 mg/day (five patients withdrew). Somnolence was also dose-related, occurring in 4.6% in the placebo group, 15.9% in the 150-mg/day group (two patients withdrew due to somnolence), 27.6% in the 300-mg/day group (three withdrew), and 28.0% in the 450-mg/day group (five withdrew).
The 14-week study by Arnold et al20 also showed higher frequencies of adverse events with higher doses. The rates of dizziness were 7.6% with placebo, 27.9% with pregabalin 300 mg/day, 37.4% with 450 mg/day, and 42.0% with 600 mg/day. The rates of somnolence were 3.8% with placebo, 12.6% with 300 mg/day of pregabalin, 19.5% with 450 mg/day, and 21.8% with 600 mg/day. Dizziness and somnolence were also the most common adverse effects that led to discontinuation of pregabalin, with rates of 4% and 3%, respectively.
The open-label phase of the FREEDOM trial showed rates of 36% for dizziness and 22% for somnolence among pregabalin-treated patients.
Weight gain and peripheral edema were also common adverse effects in these studies.22 Definitions of weight gain varied, and edema was not accompanied by evidence of cardiac or renal dysfunction.
Less common side effects seen more frequently in the treated groups included dry mouth, blurred vision, and difficulty with concentration and attention. The package insert also warns of angioedema, hypersensitivity reaction, mild asymptomatic creatine kinase elevation, decreased platelet count (without bleeding), and prolongation of the PR interval on electrocardiography.
Pregabalin is a schedule V controlled substance; in clinical studies, abrupt or rapid discontinuation of the drug led to insomnia, nausea, headache, or diarrhea in some patients, suggesting symptoms of dependence. In clinical studies involving a total of more than 5,500 patients, 4% of patients on pregabalin and 1% of patients on placebo reported euphoria as an adverse effect,19 suggesting possible potential for abuse.
Dosing
As a result of the above studies, the recommended starting dose of pregabalin for fibromyalgia is 150 mg/day in two or three divided doses, gradually increased to 300 mg/day within 1 week based on tolerability and efficacy. The dose may be increased to a maximum of 450 mg/day. The 600-mg dose was found to have no significant additional benefit, but it did have more adverse effects and therefore is not recommended. It is important to note that in these studies multiple medications for pain and insomnia were prohibited, so data on drug interactions with pregabalin are limited.
Few achieve complete remission, but most patients feel better
Several studies of the natural history of fibromyalgia have shown that very few patients experience complete remission of the disease, even after many years. Therefore, one should try to set up realistic expectations for patients, with the goal of achieving functional improvement in activities of daily living and a return to one’s predisease state.
In the longest follow-up study, 39 patients in Boston, MA, were prospectively followed for over 10 years. No patient achieved complete remission: all of them reported some fibromyalgia-related symptoms at the end of the study.23 However, 66% of them felt a little to a lot better than when first diagnosed, 55% felt well or very well, and only 7% felt poorly.
Other studies have also shown complete remission to be rare.24,25 A 5-year follow-up study investigating fibromyalgia patients’ perceptions of their symptoms and its impact on everyday life activities demonstrated that the social consequences of fibromyalgia’s symptoms are severe and constant over time.26
Evidence of favorable outcomes was reported in one study in which 47% of patients reported moderate to marked improvement in overall fibromyalgia status upon 3-year follow-up,27 and in another study, in which remission was objectively identified in 24.2% of patients 2 years after diagnosis.28
OTHER THERAPIES
Although there have been many studies of pharmacologic therapies for fibromyalgia to date, the trials had significant limitations, such as short duration, inadequate sample size, nonstandardized measures of efficacy, question of regression to the mean, and inadequate blinding, resulting in insufficient evidence to recommend one drug over another.
Tricyclic antidepressants. Two meta-analyses and a clinical review have supported the efficacy of tricyclic antidepressants in improving symptoms in fibromyalgia patients.29–31
Selective serotonin reuptake inhibitors (SSRIs) have not been well studied, and the small size and methodologic shortcomings of these studies make it difficult to draw conclusions about the efficacy of SSRIs in reducing pain in fibromyalgia patients.30,31
Duloxetine (Cymbalta) and milnacipran (Savella) are serotonin and norepinephrine reuptake inhibitors.32–34 A randomized, double-blind placebo-controlled trial evaluated duloxetine in 520 fibromyalgia patients with and without major depressive disorder. Pain scores improved significantly over 6 months in duloxetine-treated patients at doses of 60 and 120 mg/day.33 Duloxetine became the second drug approved for the treatment of fibromyalgia in 2007, and milnacipran became the third in 2009.
WHAT ROLE FOR PREGABALIN?
Pregabalin may reduce pain in some patients with fibromyalgia. However, the presenting symptoms can vary significantly, and symptoms can vary even in individual patients over time. Therefore, in most patients with fibromyalgia, a multidisciplinary approach is used to treat pain, sleep disturbance, and fatigue, along with comorbidities such as neurally mediated hypotension and psychiatric disorders. Because treatment of fibromyalgia often involves multiple drugs in addition to exercise and behavioral therapies, future studies should examine combinations of drugs and the use of drugs in conjunction with nondrug treatments.
Pregabalin advances our knowledge of fibromyalgia through improving the understanding of central sensitization and how brain neurotransmitters control central pain perceptions. Drug treatment must still be part of the comprehensive management of this disease. Physician and patient education about the current understanding of the disease is paramount in setting realistic goals for treatment.14 Future strategies to manage fibromyalgia will be based on the pathophysiology of this complex condition.
- Berenson A. Drug approved. Is disease real? New York Times, January 14, 2008. http://www.nytimes.com/2008/01/14/health/14pain.html. Accessed February 2, 2009.
- White KP, Harth M. Classification, epidemiology, and natural history of fibromyalgia. Curr Pain Headache Rep 2001; 5:320–329.
- Bennett RM. Fibromyalgia: present to future. Curr Pain Headache Rep 2004; 8:379–384.
- Wolfe F, Smythe HA, Yunus MF, et al. The American College of Rheumatolgy 1990 Criteria for the Classification of Fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis Rheum 1990; 33:160–172.
- Bennett RM. The rational management of fibromyalgia patients. Rheum Dis Clin North Am 2002; 28:181–199.
- Staud R. Evidence of involvement of central neural mechanisms in generating fibromyalgia pain. Curr Rheumatol Rep 2002; 4:299–305.
- Li J, Simone DA, Larson AA. Windup leads to characteristics of central sensitization. Pain 1999; 79:75–82.
- Desmeules JA, Cedraschi C, Rapiti E, et al. Neurophysiologic evidence for a central sensitization in patients with fibromyalgia. Arthritis Rheum 2003; 48:1420–1429.
- Staud R, Vierck CJ, Cannon RL, Mauderli AP, Price DD. Abnormal sensitization and temporal summation of pain (wind-up) in patients with fibromyalgia syndrome. Pain 2001; 91:165–175.
- Russell IJ, Orr MD, Littman B, et al. Elevated cerebrospinal fluid levels of substance P in patients with fibromyalgia syndrome. Arthritis Rheum 1994; 37:1593–1601.
- Harris RE, Sundgren PC, Pang Y, et al. Dynamic levels of glutamate within the insula are associated with improvements in multiple pain domains in fibromyalgia. Arthritis Rheum 2008; 58:903–907.
- Gracely RH, Petzke F, Wolf JM, Clauw DJ. Functional magnetic resonance imaging evidence of augmented pain processing in fibromyalgia. Arthritis Rheum 2002; 46:1333–1343.
- Staud R, Craggs JG, Perlstein WM, Robinson ME, Price DD. Brain activity associated with slow temporal summation of C-fiber evoked pain in fibromyalgia patients and healthy controls. Eur J Pain 2008; 12:1078–1089.
- Baker K, Barkhuizen A. Pharmacologic treatment of fibromyalgia. Curr Pain Headache Rep 2005; 9:301–306.
- Tassone DM, Boyce E, Guyer J, Nuzum D. Pregabalin: a novel gamma-aminobutyric acid analogue in the treatment of neuropathic pain, partial-onset seizures, and anxiety disorders. Clin Ther 2007; 29:26–48.
- Crofford LJ, Rowbotham MC, Mease PJ, et al, and the Pregabalin 1008-105 Study Group. Pregabalin for the treatment of fibromyalgia syndrome. results of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 2005; 52:1264–1273.
- Stahl SM. Anticonvulsants and the relief of chronic pain: pregabalin and gabapentin as alpha(2)delta ligands at voltage-gated calcium channels. J Clin Psychiatry 2004; 65:596–597.
- Hindmarch I, Dawson J, Stanley N. A double-blind study in healthy volunteers to assess the effects of sleep on pregabalin compared with alprazolam and placebo. Sleep 2005; 28:187–193.
- Pfizer Executive Summary. Lyrica (pregabalin) capsules c-v. July 2007. www.fda.gov/OHRMS/DOCKETS/ac/08/briefing/2008-4372b1-02-Pfizer.pdf. Accessed February 2, 2009.
- Arnold LM, Russell IJ, Diri EW, et al. A 14-week, randomized, double-blinded, placebo-controlled mono-therapy trial of pregabalin in patients with fibomyalgia. J Pain 2008; 9:792–805.
- Duan WR, Florian H, Young JP, Martin S, Haig G, Barrett JA. Pregabalin monotherapy for management of fibromyalgia: analysis of two double-blind, randomized, placebo-controlled trials (poster presentation). American College of Rheumatology Annual Scientific Meeting, Boston, MA, November 6–7, 2007.
- Crofford LJ, Mease PJ, Simpson SL, et al. Fibromyalgia relapse evaluation and efficacy for durability of meaningful relief (FREEDOM): a 6-month, double-blind, placebo-controlled trial with pregabalin. Pain 2008; 136:419–431.
- Kennedy M, Felson DT. A prospective long-term study of fibromyalgia syndrome. Arthritis Rheum 1996; 39:682–685.
- Bengtsson A, Backman E. Long-term follow-up of fibro-myalgia patients [abstract]. Scand J Rheumatolology 1992; 21(suppl 94):9.
- Ledingham J, Doherty S, Doherty M. Primary fibromyalgia syndrome—an outcome study. Br J Rheumatol 1993; 32:139–142.
- Henrikkson CM. Longterm effects of fibromyalgia on everyday life: a study of 56 patients. Scand J Rheumatol 1994; 23:36–41.
- Fitzcharles MA, Costa DD, Pöyhiä R. A study of standard care in fibromyalgia syndrome: a favorable outcome. J Rheumatol 2003; 30:154–159.
- Granges G, Zilko P, Littlejohn GO. Fibromyalgia syndrome: assessment of the severity of the condition 2 years after the diagnosis. J Rheumatol 1994; 21:523–529.
- Goldenberg DL, Burckhardt C, Crofford L. Management of fibromyalgia syndrome. JAMA 2004; 292:2388–2395.
- Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia: a meta-analysis and review. Psychosomatics 2000; 41:104–113.
- O’Malley PG, Balden E, Tomkins G, Santoro J, Kroenke K, Jackson JL. Treatment of fibromyalgia with anti-depressants: a meta-analysis. J Gen Intern Med 2000; 15:659–666.
- Arnold LM, Lu Y, Crofford LJ, et al. A double-blind, multicenter trial comparing duloxetine with placebo in the treatment of fibromyalgia patients with or without major depressive disorder. Arthritis Rheum 2004; 50:2974–2984.
- Russell IJ, Mease PJ, Smith TR, et al. Efficacy and safety of duloxetine for treatment of fibromyalgia in patients with or without major depressive disorder: Results from a 6-month, randomized, double-blind, placebo-controlled fixed-dose trial. Pain 2008; 136:432–444.
- Clauw DJ, Mease P, Palmer RH, Gendreau RM, Wang Y. Milnacipran for the treatment of fibromyalgia in adults: a 15-week, multicenter, randomized, double-blind, placebo-controlled, multiple-dose clinical trial. Clin Ther 2008; 30:1988–2004.
- Berenson A. Drug approved. Is disease real? New York Times, January 14, 2008. http://www.nytimes.com/2008/01/14/health/14pain.html. Accessed February 2, 2009.
- White KP, Harth M. Classification, epidemiology, and natural history of fibromyalgia. Curr Pain Headache Rep 2001; 5:320–329.
- Bennett RM. Fibromyalgia: present to future. Curr Pain Headache Rep 2004; 8:379–384.
- Wolfe F, Smythe HA, Yunus MF, et al. The American College of Rheumatolgy 1990 Criteria for the Classification of Fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis Rheum 1990; 33:160–172.
- Bennett RM. The rational management of fibromyalgia patients. Rheum Dis Clin North Am 2002; 28:181–199.
- Staud R. Evidence of involvement of central neural mechanisms in generating fibromyalgia pain. Curr Rheumatol Rep 2002; 4:299–305.
- Li J, Simone DA, Larson AA. Windup leads to characteristics of central sensitization. Pain 1999; 79:75–82.
- Desmeules JA, Cedraschi C, Rapiti E, et al. Neurophysiologic evidence for a central sensitization in patients with fibromyalgia. Arthritis Rheum 2003; 48:1420–1429.
- Staud R, Vierck CJ, Cannon RL, Mauderli AP, Price DD. Abnormal sensitization and temporal summation of pain (wind-up) in patients with fibromyalgia syndrome. Pain 2001; 91:165–175.
- Russell IJ, Orr MD, Littman B, et al. Elevated cerebrospinal fluid levels of substance P in patients with fibromyalgia syndrome. Arthritis Rheum 1994; 37:1593–1601.
- Harris RE, Sundgren PC, Pang Y, et al. Dynamic levels of glutamate within the insula are associated with improvements in multiple pain domains in fibromyalgia. Arthritis Rheum 2008; 58:903–907.
- Gracely RH, Petzke F, Wolf JM, Clauw DJ. Functional magnetic resonance imaging evidence of augmented pain processing in fibromyalgia. Arthritis Rheum 2002; 46:1333–1343.
- Staud R, Craggs JG, Perlstein WM, Robinson ME, Price DD. Brain activity associated with slow temporal summation of C-fiber evoked pain in fibromyalgia patients and healthy controls. Eur J Pain 2008; 12:1078–1089.
- Baker K, Barkhuizen A. Pharmacologic treatment of fibromyalgia. Curr Pain Headache Rep 2005; 9:301–306.
- Tassone DM, Boyce E, Guyer J, Nuzum D. Pregabalin: a novel gamma-aminobutyric acid analogue in the treatment of neuropathic pain, partial-onset seizures, and anxiety disorders. Clin Ther 2007; 29:26–48.
- Crofford LJ, Rowbotham MC, Mease PJ, et al, and the Pregabalin 1008-105 Study Group. Pregabalin for the treatment of fibromyalgia syndrome. results of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 2005; 52:1264–1273.
- Stahl SM. Anticonvulsants and the relief of chronic pain: pregabalin and gabapentin as alpha(2)delta ligands at voltage-gated calcium channels. J Clin Psychiatry 2004; 65:596–597.
- Hindmarch I, Dawson J, Stanley N. A double-blind study in healthy volunteers to assess the effects of sleep on pregabalin compared with alprazolam and placebo. Sleep 2005; 28:187–193.
- Pfizer Executive Summary. Lyrica (pregabalin) capsules c-v. July 2007. www.fda.gov/OHRMS/DOCKETS/ac/08/briefing/2008-4372b1-02-Pfizer.pdf. Accessed February 2, 2009.
- Arnold LM, Russell IJ, Diri EW, et al. A 14-week, randomized, double-blinded, placebo-controlled mono-therapy trial of pregabalin in patients with fibomyalgia. J Pain 2008; 9:792–805.
- Duan WR, Florian H, Young JP, Martin S, Haig G, Barrett JA. Pregabalin monotherapy for management of fibromyalgia: analysis of two double-blind, randomized, placebo-controlled trials (poster presentation). American College of Rheumatology Annual Scientific Meeting, Boston, MA, November 6–7, 2007.
- Crofford LJ, Mease PJ, Simpson SL, et al. Fibromyalgia relapse evaluation and efficacy for durability of meaningful relief (FREEDOM): a 6-month, double-blind, placebo-controlled trial with pregabalin. Pain 2008; 136:419–431.
- Kennedy M, Felson DT. A prospective long-term study of fibromyalgia syndrome. Arthritis Rheum 1996; 39:682–685.
- Bengtsson A, Backman E. Long-term follow-up of fibro-myalgia patients [abstract]. Scand J Rheumatolology 1992; 21(suppl 94):9.
- Ledingham J, Doherty S, Doherty M. Primary fibromyalgia syndrome—an outcome study. Br J Rheumatol 1993; 32:139–142.
- Henrikkson CM. Longterm effects of fibromyalgia on everyday life: a study of 56 patients. Scand J Rheumatol 1994; 23:36–41.
- Fitzcharles MA, Costa DD, Pöyhiä R. A study of standard care in fibromyalgia syndrome: a favorable outcome. J Rheumatol 2003; 30:154–159.
- Granges G, Zilko P, Littlejohn GO. Fibromyalgia syndrome: assessment of the severity of the condition 2 years after the diagnosis. J Rheumatol 1994; 21:523–529.
- Goldenberg DL, Burckhardt C, Crofford L. Management of fibromyalgia syndrome. JAMA 2004; 292:2388–2395.
- Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia: a meta-analysis and review. Psychosomatics 2000; 41:104–113.
- O’Malley PG, Balden E, Tomkins G, Santoro J, Kroenke K, Jackson JL. Treatment of fibromyalgia with anti-depressants: a meta-analysis. J Gen Intern Med 2000; 15:659–666.
- Arnold LM, Lu Y, Crofford LJ, et al. A double-blind, multicenter trial comparing duloxetine with placebo in the treatment of fibromyalgia patients with or without major depressive disorder. Arthritis Rheum 2004; 50:2974–2984.
- Russell IJ, Mease PJ, Smith TR, et al. Efficacy and safety of duloxetine for treatment of fibromyalgia in patients with or without major depressive disorder: Results from a 6-month, randomized, double-blind, placebo-controlled fixed-dose trial. Pain 2008; 136:432–444.
- Clauw DJ, Mease P, Palmer RH, Gendreau RM, Wang Y. Milnacipran for the treatment of fibromyalgia in adults: a 15-week, multicenter, randomized, double-blind, placebo-controlled, multiple-dose clinical trial. Clin Ther 2008; 30:1988–2004.
KEY POINTS
- Several lines of evidence point to functional abnormalities in the central nervous system as being responsible for fibromyalgia.
- Clinical trials found pregabalin superior to placebo. Nevertheless, patients need to have reasonable expectations of its possible benefit.
- In most patients with fibromyalgia, a multidisciplinary approach is used to treat pain, sleep disturbance, and fatigue, along with comorbidities such as neurally mediated hypotension and psychiatric disorders.
- Research with pregabalin enhances our understanding of fibromyalgia and may point the way to future treatments.
What can we expect from omega-3 fatty acids?
Many patients are taking fish oil supplements, which contain omega-3 fatty acids, either on their own initiative or on their physician’s advice. Driving this trend are accumulating data from observational and epidemiologic studies and clinical trials that these lipids actually reduce cardiovascular risk.
In the following article, we review available studies of omega-3 fatty acids in cardiovascular disease.
WHAT ARE OMEGA-3 FATTY ACIDS?
Omega-3 fatty acids are a class of polyunsaturated fatty acids. Their name means that they all have a double carbon-to-carbon bond in the third position from the omega (or methyl, or n) end of the fatty acid chain.
Most of the cardiovascular research on the omega-3 family has been on eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and alpha-linolenic acid (ALA). EPA and DHA are found primarily in fatty fish; ALA is abundant in flaxseed, walnuts, and soybeans.1 The human body can convert small amounts of ALA into EPA and DHA: only about 5% of ALA is converted to EPA and less than 0.5% is converted to DHA. Currently, it is not known whether ALA is active itself or only via these metabolites. In this review, the term omega-3 fatty acid refers to EPA and DHA only.
GETTING ENOUGH FISH OIL
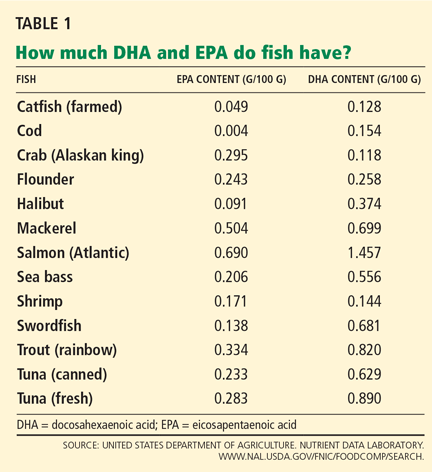
Healthy people should consume fish (preferably oily fish) at least twice a week, according to the American Heart Association.1 However, not all fish contain the same amount of oil. Some, such as cod and catfish, contain only 0.2 g of EPA/DHA per 100-g serving; others, such as Atlantic salmon, contain about 10 times as much (Table 1).2
People with known coronary artery disease should take in 1 g of EPA/DHA per day, according to the American Heart Association.1 This recommendation is based on clinical trials that found omega-3 fatty acids to have beneficial effects.
For most people with coronary artery disease, this means taking supplements. However, buyers need to carefully examine the label of over-the-counter fish oil supplements to see if they contain the recommended amounts of both DHA and EPA. Generic 1-g fish oil supplements may contain variable amounts of DHA and EPA, and some may have less than 300 mg.
People with hypertriglyceridemia. The US Food and Drug Administration (FDA) has approved Lovaza (formerly Omacor), which contains EPA/DHA in higher concentrations than over-the-counter preparations, for the treatment of hypertriglyceridemia in people with triglyceride levels higher than 500 mg/ dL, along with a regimen of diet and regular exercise.3 It is currently the only FDA-approved prescription form of omega-3 fatty acid ethyl esters. Each 1-g capsule contains 375 mg of DHA and 465 mg of EPA; the recommended dose is 2 to 4 g/day. To take in an equivalent amount of these substances with over-the counter-preparations, patients might have to take many capsules a day.
Safety of omega-3 fatty acids
Generally, omega-3 fatty acids are very well tolerated, and their adverse effects are limited to gastrointestinal complaints (discomfort, upset stomach) and a fishy odor. Common ways to prevent these effects are to freeze the capsules or take them at bedtime or with meals.
Mercury advisory on fish. Nursing or pregnant women should limit their consumption of certain fish, as some fish (but not fish oil) contain high levels of mercury. The highest levels of mercury are usually found in the larger, older predatory fish such as swordfish, tilefish, and mackerel, and the FDA advises women who are nursing or pregnant to avoid these fish completely. Tuna, red snapper, and orange roughy are lower in mercury, but nursing or pregnant women should still limit consumption of these fish to 12 oz per week.4
Theoretical risk of bleeding. In theory, high doses of omega-3 fatty acids may increase the bleeding time by inhibiting the arachidonic acid pathway. Clinically, this effect is minimal. In a trial in 511 patients undergoing coronary artery bypass grafting who were receiving aspirin or warfarin (Coumadin), the bleeding time and the number of bleeding episodes were no higher in those who were randomized to receive 4 g/day of omega-3 fatty acids daily than in a control group.5
Harris6 reviewed 19 studies of omega-3 fatty acids in patients undergoing coronary artery bypass grafting, carotid endarterectomy, or femoral artery catheterization, and none of the studies found a significantly increased risk of bleeding.
HOW DO OMEGA-3 FATTY ACIDS REDUCE RISK?
After epidemiologic studies found that Greenland Eskimos (who consume diets rich in omega-3 fatty acids) have low rates of cardiovascular disease,7 omega-3 fatty acids were hypothesized to reduce cardiovascular risk. Over the past 3 decades, their potential benefit in lowering lipid levels, blood pressure, and the risk of death in patients with known heart disease has been widely researched.
Lower triglyceride levels
The growing problem of obesity in the United States has led to more patients presenting with hypertriglyceridemia, a risk factor for coronary heart disease.
In 2001, the National Cholesterol Education Program’s third Adult Treatment Panel (ATP III)8 redefined normal triglyceride levels as less than 150 mg/dL; previously, normal was defined as less than 200 mg/dL. For people with borderline-high triglyceride levels (150–200 mg/dL), the ATP III recommends focusing on lowering the level of low-density lipoprotein cholesterol (LDL-C). For those with high to very high triglyceride levels (> 500 mg/dL), the current treatment options are niacin, fibrates, and omega-3 fatty acids.
Hypertriglyceridemia is thought to increase the risk of coronary heart disease by two mechanisms. First, and more important, triglyceride-rich lipoproteins such as very-low-density lipoprotein (VLDL) and intermediate-density lipoprotein (IDL) are thought to be atherogenic. Secondly, triglyceride-lipoprotein metabolism involves competition with high-density lipoprotein (HDL), leading to a decrease in HDL production and to denser LDL particles.9
How omega-3 fatty acids lower triglyceride levels has been inferred from preclinical studies. One mechanism, seen in animal studies, is by decreasing hepatic synthesis and secretion of VLDL particles by inhibiting various enzyme transcription factors. Another proposed mechanism is that EPA and DHA increase the activity of lipoprotein lipase, leading to an increase in chylomicron clearance.10 This was validated by Khan et al,11 who showed that lipoprotein lipase activity increased in patients who received omega-3 fatty acids 3 g/day for 6 weeks.
How much do they lower triglycerides? Data from the makers of Lovaza3 indicate that in a patient population with a mean baseline triglyceride level of 816 mg/dL, 4 g/day of omega-3 fatty acids lowered triglyceride levels to 488 mg/dL, a 45% reduction (P < .0001). In addition, HDL cholesterol (HDL-C) levels increased by 9%.
The higher the dose and the higher the baseline triglyceride level, the greater the effect. Balk et al12 performed a meta-analysis of 25 randomized trials and calculated that each 1-g increase in fish oil dose per day lowered the triglyceride level by about 8 mg/dL. However, patients with high baseline triglyceride levels had more dramatic reduction of triglycerides with fish oil. The average reduction in triglyceride levels was 27 mg/dL, accompanied by an increase in HDL-C of 1.6 mg/dL, an increase in LDL-C of 6 mg/dL, and no change in total cholesterol levels.
Pownall et al13 report that, in 19 patients with hypertriglyceridemia (median baseline level 801 mg/dL), omega-3 fatty acids 4 g/day reduced triglyceride levels to 512 mg/dL, a 38.9% change (P = .001). In 21 patients receiving placebo, triglyceride levels decreased by 7.8% (P = .001 compared with active therapy). The effect on HDL-C was minimal, but the median LDL-C level increased by 16.7% (from 43 to 53 mg/dL, P = .007) with fish oil therapy.
Fish oil plus a statin may have advantages
Most patients seen in clinical practice present with mixed dyslipidemias. The current ATP III guidelines aim for stricter triglyceride and LDL-C targets than in the past, which monotherapy alone may not be able to achieve.
Statin therapy by itself effectively lowers LDL-C but has modest effects on triglycerides. Omega-3 fatty acids effectively reduce triglycerides but have been known to increase LDL-C levels. This net LDL-C increase averaged around 10 mg/dL as reported in a review by Harris et al,14 and 6 mg/dL as reported by Balk et al.12 However, despite the net effect of an increase in LDL-C, it is hypothesized that the larger LDL particles produced by omega-3 fatty acid treatment may be less atherogenic.15
The effectiveness of combined therapy in reducing triglycerides has been widely studied.
Chan et al,16 in a randomized, placebo-controlled trial, looked at the effectiveness of atorvastatin (Lipitor) and EPA/DHA. Fifty-two obese men were randomized to receive atorvastatin 40 mg/day, EPA/DHA 4 g/day, both in combination, or placebo. After 6 weeks, triglyceride levels had decreased by 26% from baseline in the atorvastatin group, 25% in the EPA/DHA group, and 40% in the combination therapy group (P = .002). LDL-C levels decreased to a similar degree with either atorvastatin monotherapy or combination therapy. Similar studies show similar results.
Combination therapy may also lower the rate of major coronary events (see below).
The Japan EPA Lipid Intervention Study (JELIS)17 randomized more than 18,000 patients to receive either a statin alone or a statin plus EPA 1,800 mg daily, in an open-label fashion. The statins used were pravastatin (Pravachol) 10 mg daily or simvastatin (Zocor) 5 mg daily; if hypercholesterolemia remained uncontrolled, these doses were doubled. The patients were 5,859 men and 12,786 postmenopausal women (mean age 61) with or without coronary artery disease who had total cholesterol levels of 251 mg/dL or greater. The mean baseline LDL-C level was 180 mg/ dL. People who had had an acute myocardial infarction in the past 6 months or unstable angina were excluded. The primary end point examined was any major coronary event, defined as sudden death, fatal or nonfatal myocardial infarction, unstable angina, angioplasty, or coronary artery bypass grafting.
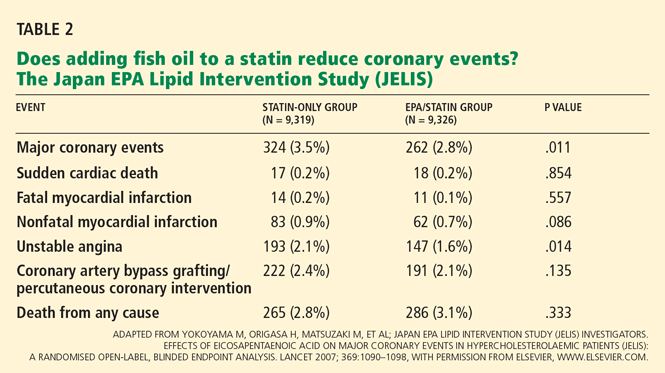
The JELIS trial showed that combination therapy may reduce the risk of coronary events, the aim of treating dyslipidemia. It was the largest randomized trial to date comparing statin use alone and in combination with omega-3 fatty acids. However, it was performed in Japan, where people already have a high intake of fatty fish, and the results may not be applicable to other countries.
May prevent arrhythmias
The Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico-Prevenzione (GISSI-Prevention) trial18 was the largest randomized trial to date of fish oil therapy as secondary prevention. In this trial, 11,323 patients who had had a myocardial infarction less than 3 months before enrollment were randomized to receive either EPA/DHA 850 mg daily, vitamin E, both, or no treatment. The primary end points were death from any cause, nonfatal myocardial infarction, and nonfatal stroke.
At 3 months, 63 (1.1%) of the patients in the EPA/DHA group had died, compared with 88 (1.6%) of those in the no-treatment group, for a relative risk of 0.59 (P = .037), and the benefit persisted for the duration of the study. However, the difference between the groups in the rates of nonfatal myocardial infarction did not reach statistical significance. Vitamin E seemed to have no effect.
EPA/DHA is thought to have prevented deaths in this study, not by reversing atherosclerosis, but rather by suppressing arrhythmias and inflammation. In support of this theory, Getz and Reardon19 noted that in GISSI the treatment showed its maximal benefit on the incidence of sudden death by 9 months, whereas statin treatment takes 1 to 2 years to reach its maximal effect. This point suggests that the role of omega-3 fatty acids in secondary prevention will be different from that of statins.
Extensive clinical studies have looked at the possibility of using omega-3 fatty acids as part of the treatment for reducing arrhythmic events. Several animal and human studies have shown that these drugs reduce the incidence of sudden death and ventricular fibrillation.20
Omega-3 fatty acids are thought to prevent arrhythmias by stabilizing the myocardial membrane through interaction with voltage-gated sodium and L-type calcium channels. During an ischemic event, the affected heart cells allow potassium ions to escape. Since potassium ions carry a positive charge, the resting membrane potential (ie, the difference in electrical charge between the inside and outside of the cell) is increased, lowering the threshold for initiating an action potential through sodium channels and increasing the risk of fatal arrhythmias. It is hypothesized that omega-3 fatty acids inhibit sodium channels by being incorporated into the membrane phospholipid bilayer, increasing its fluidity and thereby affecting the sodium channel. This reduces membrane excitability and arrhythmic potential.20
This premise was examined in three large randomized clinical trials specifically looking at ventricular arrhythmias in patients with an implanted cardioverter-defibrillator (ICD).21–23 The results were mixed.

In another study, Calo and colleagues25 randomized 160 patients to receive omega-3 fatty acids 2 g per day or placebo starting at least 5 days before elective coronary artery bypass surgery and continuing until discharge. The primary end point measured was the development of atrial fibrillation after surgery. The incidence of atrial fibrillation in the omega-3 fatty acid group was 15.2%, compared with 33% in the control group (P = .013).
Despite the differences in the results of these studies, experts generally believe that these agents reduce arrhythmic events. Nevertheless, we lack clear evidence of their clinical effectiveness, and their use for such purposes is off-label.
May reduce inflammation and platelet aggregation
Arachidonic acid is an omega-6 fatty acid that is metabolized into prostaglandins, leukotrienes, and thromboxanes, which are important for cell function. Many of these by-products (eg, leukotriene B4) have inflammatory effects, and others (eg, prostaglandin I2 E2) promote arrhythmias. EPA and DHA competitively inhibit the arachidonic acid cascade, leading to different by-products that promote vasodilation and inhibit platelet aggregation, among other effects.26 The impact of this effect in clinical practice is still unclear.
The evidence still conflicts as to whether omega-3 fatty acids reduce markers of inflammation such as C-reactive protein (CRP). Balk et al,12 in their meta-analysis, looked for studies that examined the effect of these agents on CRP and cardiovascular disease (either known risk factors or coronary artery disease). They excluded studies that were less than 4 weeks in duration, did not specify the dose of fish oil, or used doses higher than 6 g/day. Four trials were found that met their criteria, with dosages of omega-3 fatty acids ranging from 1.6 g/day to 5.9 g/day and from 12 to 20 patients in each study. Although baseline CRP levels in these studies varied, the net change in CRP was minimal, ranging from −0.15 to +1.7 mg/L.
May stabilize plaque
Thies et al27 randomized 188 patients to receive fish oil supplements before carotid endarterectomy. They found that the carotid plaque of patients who received the supplements had higher levels of EPA and DHA and had thicker fibrous caps and fewer signs of inflammation (eg, macrophages) compared with a control group and a group that received sunflower oil.
These findings show that omega-3 fatty acids are readily incorporated into atheromatous plaque and can help stabilize it. An inference from this study is that fish oil could also play a role in stabilizing coronary artery plaque.
No effect on restenosis
These agents, however, have no effect on restenosis rates after coronary angioplasty, as restenosis is mediated less by plaque formation than by intimal hyperplasia and negative remodeling within the endothelium. Even at high doses of 5 mg/day before angioplasty, omega-3 fatty acids failed to reduce the incidence of restenosis at 6 months.28
Modest effect on blood pressure
Omega-3 fatty acids are incorporated into the phospholipid bilayer of the endothelial membrane, increasing its fluidity and promoting vasodilation via an increase in nitric oxide production. These effects suggest they could be used to help control blood pressure, but studies have shown this effect to be minimal.
In a meta-analysis of 36 trials, Geleijnse et al29 estimated the reduction in blood pressure to be 2.1 mm Hg systolic and 1.6 mm Hg diastolic. The median intake of fish oil was 3.7 g/day. The largest reductions were in patients with known hypertension and those over age 45.
These findings seem consistent with the hypothesis that omega-3 fatty acids affect the endothelium, given that the arterial wall tends to become stiffer with age. Overall, however, the results of a number of studies show that fish oil supplementation is of limited clinical use in lowering blood pressure.
Confounding factors among studies
The variability in the results of different studies may be due to confounding factors such as the patients’ baseline diet, the doses of EPA and DHA given, the duration of treatment, and patient compliance. These factors must be considered when examining evidence supporting the use of omega-3 fatty acids.
- American Heart Association. Fish and Omega-3 Fatty Acids. www.americanheart.org/presenter.jhtml?identifier=4632. Accessed March 3, 2009.
- United States Department of Agriculture. Nutrient Data Laboratory. www.nal.usda.gov/fnic/foodcomp/search. Accessed March 3, 2009.
- GlaxoSmithKline. Patient Information: Lovaza. www.lovaza.com. Accessed March 3, 2009.
- US Food and Drug Administration. Mercury in fish: cause for concern? www.fda.gov/fdac/reprints/mercury.html. Accessed March 3, 2009.
- Eritsland J, Arnesen H, Seljeflot I, Kierulf P. Long-term effects of n-3 polyunsaturated fatty acids on haemostatic variables and bleeding episodes in patients with coronary artery disease. Blood Coagul Fibrinolysis 1995; 6:17–22.
- Harris WS. Expert opinion: omega-3 fatty acids and bleeding—cause for concern? Am J Cardiol 2007; 99:44C–46C.
- Bjerregaard P, Johansen LG. Mortality pattern in Greenland. Arctic Med Res 1987; 46:71–77.
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2001; 285:2486–2497.
- Jacobson TA. Secondary prevention of coronary artery disease with omega-3 fatty acids. Am J Cardiol 2006; 98:61i–70i.
- Harris WS, Miller M, Tighe AP, Davidson MH, Schaefer EJ. Omega-3 fatty acids and coronary heart disease risk: clinical and mechanistic perspectives. Atherosclerosis 2008; 197:12–24.
- Khan S, Minihane AM, Talmud PJ, et al. Dietary long-chain n-3 PUFAs increase LPL gene expression in adipose tissue of subjects with an atherogenic lipoprotein phenotype. J Lipid Res 2002; 43:979–985.
- Balk EM, Lichtenstein AH, Chung M, Kupelnick B, Chew P, Lau J. Effects of omega-3 fatty acids on serum markers of cardiovascular disease risk: a systematic review. Atherosclerosis 2006; 189:19–30.
- Pownall HJ, Brauchi D, Kilinc C, et al. Correlation of serum triglyceride and its reduction by omega-3 fatty acids with lipid transfer activity and the neutral lipid compositions of high-density and low-density lipoproteins. Atherosclerosis 1999; 143:285–297.
- Harris WS. N-3 fatty acids and serum lipoproteins: human studies. Am J Clin Nutr 1997; 65 suppl 5:1645S–1654S.
- Robinson JG, Stone NJ. Antiatherosclerotic and antithrombotic effects of omega-3 fatty acids. Am J Cardiol 2006; 98:39i–49i.
- Chan DC, Watts GF, Mori TA, Barrett PH, Beilin LJ, Redgrave TG. Factorial study of the effects of atorvastatin and fish oil on dyslipidaemia in visceral obesity. Eur J Clin Invest 2002; 32:429–436.
- Yokoyama M, Origasa H, Matsuzaki M, et al; Japan EPA lipid intervention study (JELIS) Investigators. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet 2007; 369:1090–1098.
- Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza Nell’Infarto Miocardico. Lancet 1999; 354:447–455.
- Getz GS, Reardon CA. Nutrition and cardiovascular disease. Arterioscler Thromb Vasc Biol 2007; 27:2499–2506.
- Reiffel JA, McDonald A. Antiarrhythmic effects of omega-3 fatty acids. Am J Cardiol 2006; 98:50i–60i.
- Raitt MH, Connor WE, Morris C, et al. Fish oil supplementation and risk of ventricular tachycardia and ventricular fibrillation in patients with implantable defibrillators: a randomized controlled trial. JAMA 2005; 293:2884–2891.
- Leaf A, Albert CM, Josephson M, et al; Fatty Acid Antiarrhythmia Trial Investigators. Prevention of fatal arrhythmias in high-risk subjects by fish oil n-3 fatty acid intake. Circulation 2005; 112:2762–2768.
- Brouwer IA, Zock PL, Wever EF, et al. Rationale and design of a randomised controlled clinical trial on supplemental intake of n-3 fatty acids and incidence of cardiac arrhythmia: SOFA. Eur J Clin Nutr 2003; 57:1323–1330.
- London B, Albert C, Anderson ME, et al. Omega-3 fatty acids and cardiac arrhythmias: prior studies and recommendations for future research: a report from the National Heart, Lung, and Blood Institute and Office of Dietary Supplements Omega-3 Fatty Acids and their Role in Cardiac Arrhythmogenesis Workshop. Circulation 2007; 116:e320–e335.
- Calo L, Bianconi L, Colivicchi F, et al. N-3 fatty acids for the prevention of atrial fibrillation after coronary artery bypass surgery: a randomized, controlled trial. J Am Coll Cardiol 2005; 45:1723–1728.
- Harris WS, Assaad B, Poston WC. Tissue omega-6/omega-3 fatty acid ratio and risk for coronary artery disease. Am J Cardiol 2006; 98:19i–26i.
- Thies F, Garry JM, Yaqoob P, et al. Association of n-3 polyunsaturated fatty acids with stability of atherosclerotic plaques: a randomised controlled trial. Lancet 2003; 361:477–485.
- Johansen O, Brekke M, Seljeflot I, Abdelnoor M, Arnesen H. N-3 fatty acids do not prevent restenosis after coronary angioplasty: results from the CART study. Coronary Angioplasty Restenosis Trial. J Am Coll Cardiol 1999; 33:1619–1626.
- Geleijnse JM, Giltay EJ, Grobbee DE, Donders AR, Kok FJ. Blood pressure response to fish oil supplementation: metaregression analysis of randomized trials. J Hypertens 2002; 20:1493–1499.
Many patients are taking fish oil supplements, which contain omega-3 fatty acids, either on their own initiative or on their physician’s advice. Driving this trend are accumulating data from observational and epidemiologic studies and clinical trials that these lipids actually reduce cardiovascular risk.
In the following article, we review available studies of omega-3 fatty acids in cardiovascular disease.
WHAT ARE OMEGA-3 FATTY ACIDS?
Omega-3 fatty acids are a class of polyunsaturated fatty acids. Their name means that they all have a double carbon-to-carbon bond in the third position from the omega (or methyl, or n) end of the fatty acid chain.
Most of the cardiovascular research on the omega-3 family has been on eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and alpha-linolenic acid (ALA). EPA and DHA are found primarily in fatty fish; ALA is abundant in flaxseed, walnuts, and soybeans.1 The human body can convert small amounts of ALA into EPA and DHA: only about 5% of ALA is converted to EPA and less than 0.5% is converted to DHA. Currently, it is not known whether ALA is active itself or only via these metabolites. In this review, the term omega-3 fatty acid refers to EPA and DHA only.
GETTING ENOUGH FISH OIL
Healthy people should consume fish (preferably oily fish) at least twice a week, according to the American Heart Association.1 However, not all fish contain the same amount of oil. Some, such as cod and catfish, contain only 0.2 g of EPA/DHA per 100-g serving; others, such as Atlantic salmon, contain about 10 times as much (Table 1).2
People with known coronary artery disease should take in 1 g of EPA/DHA per day, according to the American Heart Association.1 This recommendation is based on clinical trials that found omega-3 fatty acids to have beneficial effects.
For most people with coronary artery disease, this means taking supplements. However, buyers need to carefully examine the label of over-the-counter fish oil supplements to see if they contain the recommended amounts of both DHA and EPA. Generic 1-g fish oil supplements may contain variable amounts of DHA and EPA, and some may have less than 300 mg.
People with hypertriglyceridemia. The US Food and Drug Administration (FDA) has approved Lovaza (formerly Omacor), which contains EPA/DHA in higher concentrations than over-the-counter preparations, for the treatment of hypertriglyceridemia in people with triglyceride levels higher than 500 mg/ dL, along with a regimen of diet and regular exercise.3 It is currently the only FDA-approved prescription form of omega-3 fatty acid ethyl esters. Each 1-g capsule contains 375 mg of DHA and 465 mg of EPA; the recommended dose is 2 to 4 g/day. To take in an equivalent amount of these substances with over-the counter-preparations, patients might have to take many capsules a day.
Safety of omega-3 fatty acids
Generally, omega-3 fatty acids are very well tolerated, and their adverse effects are limited to gastrointestinal complaints (discomfort, upset stomach) and a fishy odor. Common ways to prevent these effects are to freeze the capsules or take them at bedtime or with meals.
Mercury advisory on fish. Nursing or pregnant women should limit their consumption of certain fish, as some fish (but not fish oil) contain high levels of mercury. The highest levels of mercury are usually found in the larger, older predatory fish such as swordfish, tilefish, and mackerel, and the FDA advises women who are nursing or pregnant to avoid these fish completely. Tuna, red snapper, and orange roughy are lower in mercury, but nursing or pregnant women should still limit consumption of these fish to 12 oz per week.4
Theoretical risk of bleeding. In theory, high doses of omega-3 fatty acids may increase the bleeding time by inhibiting the arachidonic acid pathway. Clinically, this effect is minimal. In a trial in 511 patients undergoing coronary artery bypass grafting who were receiving aspirin or warfarin (Coumadin), the bleeding time and the number of bleeding episodes were no higher in those who were randomized to receive 4 g/day of omega-3 fatty acids daily than in a control group.5
Harris6 reviewed 19 studies of omega-3 fatty acids in patients undergoing coronary artery bypass grafting, carotid endarterectomy, or femoral artery catheterization, and none of the studies found a significantly increased risk of bleeding.
HOW DO OMEGA-3 FATTY ACIDS REDUCE RISK?
After epidemiologic studies found that Greenland Eskimos (who consume diets rich in omega-3 fatty acids) have low rates of cardiovascular disease,7 omega-3 fatty acids were hypothesized to reduce cardiovascular risk. Over the past 3 decades, their potential benefit in lowering lipid levels, blood pressure, and the risk of death in patients with known heart disease has been widely researched.
Lower triglyceride levels
The growing problem of obesity in the United States has led to more patients presenting with hypertriglyceridemia, a risk factor for coronary heart disease.
In 2001, the National Cholesterol Education Program’s third Adult Treatment Panel (ATP III)8 redefined normal triglyceride levels as less than 150 mg/dL; previously, normal was defined as less than 200 mg/dL. For people with borderline-high triglyceride levels (150–200 mg/dL), the ATP III recommends focusing on lowering the level of low-density lipoprotein cholesterol (LDL-C). For those with high to very high triglyceride levels (> 500 mg/dL), the current treatment options are niacin, fibrates, and omega-3 fatty acids.
Hypertriglyceridemia is thought to increase the risk of coronary heart disease by two mechanisms. First, and more important, triglyceride-rich lipoproteins such as very-low-density lipoprotein (VLDL) and intermediate-density lipoprotein (IDL) are thought to be atherogenic. Secondly, triglyceride-lipoprotein metabolism involves competition with high-density lipoprotein (HDL), leading to a decrease in HDL production and to denser LDL particles.9
How omega-3 fatty acids lower triglyceride levels has been inferred from preclinical studies. One mechanism, seen in animal studies, is by decreasing hepatic synthesis and secretion of VLDL particles by inhibiting various enzyme transcription factors. Another proposed mechanism is that EPA and DHA increase the activity of lipoprotein lipase, leading to an increase in chylomicron clearance.10 This was validated by Khan et al,11 who showed that lipoprotein lipase activity increased in patients who received omega-3 fatty acids 3 g/day for 6 weeks.
How much do they lower triglycerides? Data from the makers of Lovaza3 indicate that in a patient population with a mean baseline triglyceride level of 816 mg/dL, 4 g/day of omega-3 fatty acids lowered triglyceride levels to 488 mg/dL, a 45% reduction (P < .0001). In addition, HDL cholesterol (HDL-C) levels increased by 9%.
The higher the dose and the higher the baseline triglyceride level, the greater the effect. Balk et al12 performed a meta-analysis of 25 randomized trials and calculated that each 1-g increase in fish oil dose per day lowered the triglyceride level by about 8 mg/dL. However, patients with high baseline triglyceride levels had more dramatic reduction of triglycerides with fish oil. The average reduction in triglyceride levels was 27 mg/dL, accompanied by an increase in HDL-C of 1.6 mg/dL, an increase in LDL-C of 6 mg/dL, and no change in total cholesterol levels.
Pownall et al13 report that, in 19 patients with hypertriglyceridemia (median baseline level 801 mg/dL), omega-3 fatty acids 4 g/day reduced triglyceride levels to 512 mg/dL, a 38.9% change (P = .001). In 21 patients receiving placebo, triglyceride levels decreased by 7.8% (P = .001 compared with active therapy). The effect on HDL-C was minimal, but the median LDL-C level increased by 16.7% (from 43 to 53 mg/dL, P = .007) with fish oil therapy.
Fish oil plus a statin may have advantages
Most patients seen in clinical practice present with mixed dyslipidemias. The current ATP III guidelines aim for stricter triglyceride and LDL-C targets than in the past, which monotherapy alone may not be able to achieve.
Statin therapy by itself effectively lowers LDL-C but has modest effects on triglycerides. Omega-3 fatty acids effectively reduce triglycerides but have been known to increase LDL-C levels. This net LDL-C increase averaged around 10 mg/dL as reported in a review by Harris et al,14 and 6 mg/dL as reported by Balk et al.12 However, despite the net effect of an increase in LDL-C, it is hypothesized that the larger LDL particles produced by omega-3 fatty acid treatment may be less atherogenic.15
The effectiveness of combined therapy in reducing triglycerides has been widely studied.
Chan et al,16 in a randomized, placebo-controlled trial, looked at the effectiveness of atorvastatin (Lipitor) and EPA/DHA. Fifty-two obese men were randomized to receive atorvastatin 40 mg/day, EPA/DHA 4 g/day, both in combination, or placebo. After 6 weeks, triglyceride levels had decreased by 26% from baseline in the atorvastatin group, 25% in the EPA/DHA group, and 40% in the combination therapy group (P = .002). LDL-C levels decreased to a similar degree with either atorvastatin monotherapy or combination therapy. Similar studies show similar results.
Combination therapy may also lower the rate of major coronary events (see below).
The Japan EPA Lipid Intervention Study (JELIS)17 randomized more than 18,000 patients to receive either a statin alone or a statin plus EPA 1,800 mg daily, in an open-label fashion. The statins used were pravastatin (Pravachol) 10 mg daily or simvastatin (Zocor) 5 mg daily; if hypercholesterolemia remained uncontrolled, these doses were doubled. The patients were 5,859 men and 12,786 postmenopausal women (mean age 61) with or without coronary artery disease who had total cholesterol levels of 251 mg/dL or greater. The mean baseline LDL-C level was 180 mg/ dL. People who had had an acute myocardial infarction in the past 6 months or unstable angina were excluded. The primary end point examined was any major coronary event, defined as sudden death, fatal or nonfatal myocardial infarction, unstable angina, angioplasty, or coronary artery bypass grafting.
The JELIS trial showed that combination therapy may reduce the risk of coronary events, the aim of treating dyslipidemia. It was the largest randomized trial to date comparing statin use alone and in combination with omega-3 fatty acids. However, it was performed in Japan, where people already have a high intake of fatty fish, and the results may not be applicable to other countries.
May prevent arrhythmias
The Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico-Prevenzione (GISSI-Prevention) trial18 was the largest randomized trial to date of fish oil therapy as secondary prevention. In this trial, 11,323 patients who had had a myocardial infarction less than 3 months before enrollment were randomized to receive either EPA/DHA 850 mg daily, vitamin E, both, or no treatment. The primary end points were death from any cause, nonfatal myocardial infarction, and nonfatal stroke.
At 3 months, 63 (1.1%) of the patients in the EPA/DHA group had died, compared with 88 (1.6%) of those in the no-treatment group, for a relative risk of 0.59 (P = .037), and the benefit persisted for the duration of the study. However, the difference between the groups in the rates of nonfatal myocardial infarction did not reach statistical significance. Vitamin E seemed to have no effect.
EPA/DHA is thought to have prevented deaths in this study, not by reversing atherosclerosis, but rather by suppressing arrhythmias and inflammation. In support of this theory, Getz and Reardon19 noted that in GISSI the treatment showed its maximal benefit on the incidence of sudden death by 9 months, whereas statin treatment takes 1 to 2 years to reach its maximal effect. This point suggests that the role of omega-3 fatty acids in secondary prevention will be different from that of statins.
Extensive clinical studies have looked at the possibility of using omega-3 fatty acids as part of the treatment for reducing arrhythmic events. Several animal and human studies have shown that these drugs reduce the incidence of sudden death and ventricular fibrillation.20
Omega-3 fatty acids are thought to prevent arrhythmias by stabilizing the myocardial membrane through interaction with voltage-gated sodium and L-type calcium channels. During an ischemic event, the affected heart cells allow potassium ions to escape. Since potassium ions carry a positive charge, the resting membrane potential (ie, the difference in electrical charge between the inside and outside of the cell) is increased, lowering the threshold for initiating an action potential through sodium channels and increasing the risk of fatal arrhythmias. It is hypothesized that omega-3 fatty acids inhibit sodium channels by being incorporated into the membrane phospholipid bilayer, increasing its fluidity and thereby affecting the sodium channel. This reduces membrane excitability and arrhythmic potential.20
This premise was examined in three large randomized clinical trials specifically looking at ventricular arrhythmias in patients with an implanted cardioverter-defibrillator (ICD).21–23 The results were mixed.
In another study, Calo and colleagues25 randomized 160 patients to receive omega-3 fatty acids 2 g per day or placebo starting at least 5 days before elective coronary artery bypass surgery and continuing until discharge. The primary end point measured was the development of atrial fibrillation after surgery. The incidence of atrial fibrillation in the omega-3 fatty acid group was 15.2%, compared with 33% in the control group (P = .013).
Despite the differences in the results of these studies, experts generally believe that these agents reduce arrhythmic events. Nevertheless, we lack clear evidence of their clinical effectiveness, and their use for such purposes is off-label.
May reduce inflammation and platelet aggregation
Arachidonic acid is an omega-6 fatty acid that is metabolized into prostaglandins, leukotrienes, and thromboxanes, which are important for cell function. Many of these by-products (eg, leukotriene B4) have inflammatory effects, and others (eg, prostaglandin I2 E2) promote arrhythmias. EPA and DHA competitively inhibit the arachidonic acid cascade, leading to different by-products that promote vasodilation and inhibit platelet aggregation, among other effects.26 The impact of this effect in clinical practice is still unclear.
The evidence still conflicts as to whether omega-3 fatty acids reduce markers of inflammation such as C-reactive protein (CRP). Balk et al,12 in their meta-analysis, looked for studies that examined the effect of these agents on CRP and cardiovascular disease (either known risk factors or coronary artery disease). They excluded studies that were less than 4 weeks in duration, did not specify the dose of fish oil, or used doses higher than 6 g/day. Four trials were found that met their criteria, with dosages of omega-3 fatty acids ranging from 1.6 g/day to 5.9 g/day and from 12 to 20 patients in each study. Although baseline CRP levels in these studies varied, the net change in CRP was minimal, ranging from −0.15 to +1.7 mg/L.
May stabilize plaque
Thies et al27 randomized 188 patients to receive fish oil supplements before carotid endarterectomy. They found that the carotid plaque of patients who received the supplements had higher levels of EPA and DHA and had thicker fibrous caps and fewer signs of inflammation (eg, macrophages) compared with a control group and a group that received sunflower oil.
These findings show that omega-3 fatty acids are readily incorporated into atheromatous plaque and can help stabilize it. An inference from this study is that fish oil could also play a role in stabilizing coronary artery plaque.
No effect on restenosis
These agents, however, have no effect on restenosis rates after coronary angioplasty, as restenosis is mediated less by plaque formation than by intimal hyperplasia and negative remodeling within the endothelium. Even at high doses of 5 mg/day before angioplasty, omega-3 fatty acids failed to reduce the incidence of restenosis at 6 months.28
Modest effect on blood pressure
Omega-3 fatty acids are incorporated into the phospholipid bilayer of the endothelial membrane, increasing its fluidity and promoting vasodilation via an increase in nitric oxide production. These effects suggest they could be used to help control blood pressure, but studies have shown this effect to be minimal.
In a meta-analysis of 36 trials, Geleijnse et al29 estimated the reduction in blood pressure to be 2.1 mm Hg systolic and 1.6 mm Hg diastolic. The median intake of fish oil was 3.7 g/day. The largest reductions were in patients with known hypertension and those over age 45.
These findings seem consistent with the hypothesis that omega-3 fatty acids affect the endothelium, given that the arterial wall tends to become stiffer with age. Overall, however, the results of a number of studies show that fish oil supplementation is of limited clinical use in lowering blood pressure.
Confounding factors among studies
The variability in the results of different studies may be due to confounding factors such as the patients’ baseline diet, the doses of EPA and DHA given, the duration of treatment, and patient compliance. These factors must be considered when examining evidence supporting the use of omega-3 fatty acids.
Many patients are taking fish oil supplements, which contain omega-3 fatty acids, either on their own initiative or on their physician’s advice. Driving this trend are accumulating data from observational and epidemiologic studies and clinical trials that these lipids actually reduce cardiovascular risk.
In the following article, we review available studies of omega-3 fatty acids in cardiovascular disease.
WHAT ARE OMEGA-3 FATTY ACIDS?
Omega-3 fatty acids are a class of polyunsaturated fatty acids. Their name means that they all have a double carbon-to-carbon bond in the third position from the omega (or methyl, or n) end of the fatty acid chain.
Most of the cardiovascular research on the omega-3 family has been on eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and alpha-linolenic acid (ALA). EPA and DHA are found primarily in fatty fish; ALA is abundant in flaxseed, walnuts, and soybeans.1 The human body can convert small amounts of ALA into EPA and DHA: only about 5% of ALA is converted to EPA and less than 0.5% is converted to DHA. Currently, it is not known whether ALA is active itself or only via these metabolites. In this review, the term omega-3 fatty acid refers to EPA and DHA only.
GETTING ENOUGH FISH OIL
Healthy people should consume fish (preferably oily fish) at least twice a week, according to the American Heart Association.1 However, not all fish contain the same amount of oil. Some, such as cod and catfish, contain only 0.2 g of EPA/DHA per 100-g serving; others, such as Atlantic salmon, contain about 10 times as much (Table 1).2
People with known coronary artery disease should take in 1 g of EPA/DHA per day, according to the American Heart Association.1 This recommendation is based on clinical trials that found omega-3 fatty acids to have beneficial effects.
For most people with coronary artery disease, this means taking supplements. However, buyers need to carefully examine the label of over-the-counter fish oil supplements to see if they contain the recommended amounts of both DHA and EPA. Generic 1-g fish oil supplements may contain variable amounts of DHA and EPA, and some may have less than 300 mg.
People with hypertriglyceridemia. The US Food and Drug Administration (FDA) has approved Lovaza (formerly Omacor), which contains EPA/DHA in higher concentrations than over-the-counter preparations, for the treatment of hypertriglyceridemia in people with triglyceride levels higher than 500 mg/ dL, along with a regimen of diet and regular exercise.3 It is currently the only FDA-approved prescription form of omega-3 fatty acid ethyl esters. Each 1-g capsule contains 375 mg of DHA and 465 mg of EPA; the recommended dose is 2 to 4 g/day. To take in an equivalent amount of these substances with over-the counter-preparations, patients might have to take many capsules a day.
Safety of omega-3 fatty acids
Generally, omega-3 fatty acids are very well tolerated, and their adverse effects are limited to gastrointestinal complaints (discomfort, upset stomach) and a fishy odor. Common ways to prevent these effects are to freeze the capsules or take them at bedtime or with meals.
Mercury advisory on fish. Nursing or pregnant women should limit their consumption of certain fish, as some fish (but not fish oil) contain high levels of mercury. The highest levels of mercury are usually found in the larger, older predatory fish such as swordfish, tilefish, and mackerel, and the FDA advises women who are nursing or pregnant to avoid these fish completely. Tuna, red snapper, and orange roughy are lower in mercury, but nursing or pregnant women should still limit consumption of these fish to 12 oz per week.4
Theoretical risk of bleeding. In theory, high doses of omega-3 fatty acids may increase the bleeding time by inhibiting the arachidonic acid pathway. Clinically, this effect is minimal. In a trial in 511 patients undergoing coronary artery bypass grafting who were receiving aspirin or warfarin (Coumadin), the bleeding time and the number of bleeding episodes were no higher in those who were randomized to receive 4 g/day of omega-3 fatty acids daily than in a control group.5
Harris6 reviewed 19 studies of omega-3 fatty acids in patients undergoing coronary artery bypass grafting, carotid endarterectomy, or femoral artery catheterization, and none of the studies found a significantly increased risk of bleeding.
HOW DO OMEGA-3 FATTY ACIDS REDUCE RISK?
After epidemiologic studies found that Greenland Eskimos (who consume diets rich in omega-3 fatty acids) have low rates of cardiovascular disease,7 omega-3 fatty acids were hypothesized to reduce cardiovascular risk. Over the past 3 decades, their potential benefit in lowering lipid levels, blood pressure, and the risk of death in patients with known heart disease has been widely researched.
Lower triglyceride levels
The growing problem of obesity in the United States has led to more patients presenting with hypertriglyceridemia, a risk factor for coronary heart disease.
In 2001, the National Cholesterol Education Program’s third Adult Treatment Panel (ATP III)8 redefined normal triglyceride levels as less than 150 mg/dL; previously, normal was defined as less than 200 mg/dL. For people with borderline-high triglyceride levels (150–200 mg/dL), the ATP III recommends focusing on lowering the level of low-density lipoprotein cholesterol (LDL-C). For those with high to very high triglyceride levels (> 500 mg/dL), the current treatment options are niacin, fibrates, and omega-3 fatty acids.
Hypertriglyceridemia is thought to increase the risk of coronary heart disease by two mechanisms. First, and more important, triglyceride-rich lipoproteins such as very-low-density lipoprotein (VLDL) and intermediate-density lipoprotein (IDL) are thought to be atherogenic. Secondly, triglyceride-lipoprotein metabolism involves competition with high-density lipoprotein (HDL), leading to a decrease in HDL production and to denser LDL particles.9
How omega-3 fatty acids lower triglyceride levels has been inferred from preclinical studies. One mechanism, seen in animal studies, is by decreasing hepatic synthesis and secretion of VLDL particles by inhibiting various enzyme transcription factors. Another proposed mechanism is that EPA and DHA increase the activity of lipoprotein lipase, leading to an increase in chylomicron clearance.10 This was validated by Khan et al,11 who showed that lipoprotein lipase activity increased in patients who received omega-3 fatty acids 3 g/day for 6 weeks.
How much do they lower triglycerides? Data from the makers of Lovaza3 indicate that in a patient population with a mean baseline triglyceride level of 816 mg/dL, 4 g/day of omega-3 fatty acids lowered triglyceride levels to 488 mg/dL, a 45% reduction (P < .0001). In addition, HDL cholesterol (HDL-C) levels increased by 9%.
The higher the dose and the higher the baseline triglyceride level, the greater the effect. Balk et al12 performed a meta-analysis of 25 randomized trials and calculated that each 1-g increase in fish oil dose per day lowered the triglyceride level by about 8 mg/dL. However, patients with high baseline triglyceride levels had more dramatic reduction of triglycerides with fish oil. The average reduction in triglyceride levels was 27 mg/dL, accompanied by an increase in HDL-C of 1.6 mg/dL, an increase in LDL-C of 6 mg/dL, and no change in total cholesterol levels.
Pownall et al13 report that, in 19 patients with hypertriglyceridemia (median baseline level 801 mg/dL), omega-3 fatty acids 4 g/day reduced triglyceride levels to 512 mg/dL, a 38.9% change (P = .001). In 21 patients receiving placebo, triglyceride levels decreased by 7.8% (P = .001 compared with active therapy). The effect on HDL-C was minimal, but the median LDL-C level increased by 16.7% (from 43 to 53 mg/dL, P = .007) with fish oil therapy.
Fish oil plus a statin may have advantages
Most patients seen in clinical practice present with mixed dyslipidemias. The current ATP III guidelines aim for stricter triglyceride and LDL-C targets than in the past, which monotherapy alone may not be able to achieve.
Statin therapy by itself effectively lowers LDL-C but has modest effects on triglycerides. Omega-3 fatty acids effectively reduce triglycerides but have been known to increase LDL-C levels. This net LDL-C increase averaged around 10 mg/dL as reported in a review by Harris et al,14 and 6 mg/dL as reported by Balk et al.12 However, despite the net effect of an increase in LDL-C, it is hypothesized that the larger LDL particles produced by omega-3 fatty acid treatment may be less atherogenic.15
The effectiveness of combined therapy in reducing triglycerides has been widely studied.
Chan et al,16 in a randomized, placebo-controlled trial, looked at the effectiveness of atorvastatin (Lipitor) and EPA/DHA. Fifty-two obese men were randomized to receive atorvastatin 40 mg/day, EPA/DHA 4 g/day, both in combination, or placebo. After 6 weeks, triglyceride levels had decreased by 26% from baseline in the atorvastatin group, 25% in the EPA/DHA group, and 40% in the combination therapy group (P = .002). LDL-C levels decreased to a similar degree with either atorvastatin monotherapy or combination therapy. Similar studies show similar results.
Combination therapy may also lower the rate of major coronary events (see below).
The Japan EPA Lipid Intervention Study (JELIS)17 randomized more than 18,000 patients to receive either a statin alone or a statin plus EPA 1,800 mg daily, in an open-label fashion. The statins used were pravastatin (Pravachol) 10 mg daily or simvastatin (Zocor) 5 mg daily; if hypercholesterolemia remained uncontrolled, these doses were doubled. The patients were 5,859 men and 12,786 postmenopausal women (mean age 61) with or without coronary artery disease who had total cholesterol levels of 251 mg/dL or greater. The mean baseline LDL-C level was 180 mg/ dL. People who had had an acute myocardial infarction in the past 6 months or unstable angina were excluded. The primary end point examined was any major coronary event, defined as sudden death, fatal or nonfatal myocardial infarction, unstable angina, angioplasty, or coronary artery bypass grafting.
The JELIS trial showed that combination therapy may reduce the risk of coronary events, the aim of treating dyslipidemia. It was the largest randomized trial to date comparing statin use alone and in combination with omega-3 fatty acids. However, it was performed in Japan, where people already have a high intake of fatty fish, and the results may not be applicable to other countries.
May prevent arrhythmias
The Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico-Prevenzione (GISSI-Prevention) trial18 was the largest randomized trial to date of fish oil therapy as secondary prevention. In this trial, 11,323 patients who had had a myocardial infarction less than 3 months before enrollment were randomized to receive either EPA/DHA 850 mg daily, vitamin E, both, or no treatment. The primary end points were death from any cause, nonfatal myocardial infarction, and nonfatal stroke.
At 3 months, 63 (1.1%) of the patients in the EPA/DHA group had died, compared with 88 (1.6%) of those in the no-treatment group, for a relative risk of 0.59 (P = .037), and the benefit persisted for the duration of the study. However, the difference between the groups in the rates of nonfatal myocardial infarction did not reach statistical significance. Vitamin E seemed to have no effect.
EPA/DHA is thought to have prevented deaths in this study, not by reversing atherosclerosis, but rather by suppressing arrhythmias and inflammation. In support of this theory, Getz and Reardon19 noted that in GISSI the treatment showed its maximal benefit on the incidence of sudden death by 9 months, whereas statin treatment takes 1 to 2 years to reach its maximal effect. This point suggests that the role of omega-3 fatty acids in secondary prevention will be different from that of statins.
Extensive clinical studies have looked at the possibility of using omega-3 fatty acids as part of the treatment for reducing arrhythmic events. Several animal and human studies have shown that these drugs reduce the incidence of sudden death and ventricular fibrillation.20
Omega-3 fatty acids are thought to prevent arrhythmias by stabilizing the myocardial membrane through interaction with voltage-gated sodium and L-type calcium channels. During an ischemic event, the affected heart cells allow potassium ions to escape. Since potassium ions carry a positive charge, the resting membrane potential (ie, the difference in electrical charge between the inside and outside of the cell) is increased, lowering the threshold for initiating an action potential through sodium channels and increasing the risk of fatal arrhythmias. It is hypothesized that omega-3 fatty acids inhibit sodium channels by being incorporated into the membrane phospholipid bilayer, increasing its fluidity and thereby affecting the sodium channel. This reduces membrane excitability and arrhythmic potential.20
This premise was examined in three large randomized clinical trials specifically looking at ventricular arrhythmias in patients with an implanted cardioverter-defibrillator (ICD).21–23 The results were mixed.
In another study, Calo and colleagues25 randomized 160 patients to receive omega-3 fatty acids 2 g per day or placebo starting at least 5 days before elective coronary artery bypass surgery and continuing until discharge. The primary end point measured was the development of atrial fibrillation after surgery. The incidence of atrial fibrillation in the omega-3 fatty acid group was 15.2%, compared with 33% in the control group (P = .013).
Despite the differences in the results of these studies, experts generally believe that these agents reduce arrhythmic events. Nevertheless, we lack clear evidence of their clinical effectiveness, and their use for such purposes is off-label.
May reduce inflammation and platelet aggregation
Arachidonic acid is an omega-6 fatty acid that is metabolized into prostaglandins, leukotrienes, and thromboxanes, which are important for cell function. Many of these by-products (eg, leukotriene B4) have inflammatory effects, and others (eg, prostaglandin I2 E2) promote arrhythmias. EPA and DHA competitively inhibit the arachidonic acid cascade, leading to different by-products that promote vasodilation and inhibit platelet aggregation, among other effects.26 The impact of this effect in clinical practice is still unclear.
The evidence still conflicts as to whether omega-3 fatty acids reduce markers of inflammation such as C-reactive protein (CRP). Balk et al,12 in their meta-analysis, looked for studies that examined the effect of these agents on CRP and cardiovascular disease (either known risk factors or coronary artery disease). They excluded studies that were less than 4 weeks in duration, did not specify the dose of fish oil, or used doses higher than 6 g/day. Four trials were found that met their criteria, with dosages of omega-3 fatty acids ranging from 1.6 g/day to 5.9 g/day and from 12 to 20 patients in each study. Although baseline CRP levels in these studies varied, the net change in CRP was minimal, ranging from −0.15 to +1.7 mg/L.
May stabilize plaque
Thies et al27 randomized 188 patients to receive fish oil supplements before carotid endarterectomy. They found that the carotid plaque of patients who received the supplements had higher levels of EPA and DHA and had thicker fibrous caps and fewer signs of inflammation (eg, macrophages) compared with a control group and a group that received sunflower oil.
These findings show that omega-3 fatty acids are readily incorporated into atheromatous plaque and can help stabilize it. An inference from this study is that fish oil could also play a role in stabilizing coronary artery plaque.
No effect on restenosis
These agents, however, have no effect on restenosis rates after coronary angioplasty, as restenosis is mediated less by plaque formation than by intimal hyperplasia and negative remodeling within the endothelium. Even at high doses of 5 mg/day before angioplasty, omega-3 fatty acids failed to reduce the incidence of restenosis at 6 months.28
Modest effect on blood pressure
Omega-3 fatty acids are incorporated into the phospholipid bilayer of the endothelial membrane, increasing its fluidity and promoting vasodilation via an increase in nitric oxide production. These effects suggest they could be used to help control blood pressure, but studies have shown this effect to be minimal.
In a meta-analysis of 36 trials, Geleijnse et al29 estimated the reduction in blood pressure to be 2.1 mm Hg systolic and 1.6 mm Hg diastolic. The median intake of fish oil was 3.7 g/day. The largest reductions were in patients with known hypertension and those over age 45.
These findings seem consistent with the hypothesis that omega-3 fatty acids affect the endothelium, given that the arterial wall tends to become stiffer with age. Overall, however, the results of a number of studies show that fish oil supplementation is of limited clinical use in lowering blood pressure.
Confounding factors among studies
The variability in the results of different studies may be due to confounding factors such as the patients’ baseline diet, the doses of EPA and DHA given, the duration of treatment, and patient compliance. These factors must be considered when examining evidence supporting the use of omega-3 fatty acids.
- American Heart Association. Fish and Omega-3 Fatty Acids. www.americanheart.org/presenter.jhtml?identifier=4632. Accessed March 3, 2009.
- United States Department of Agriculture. Nutrient Data Laboratory. www.nal.usda.gov/fnic/foodcomp/search. Accessed March 3, 2009.
- GlaxoSmithKline. Patient Information: Lovaza. www.lovaza.com. Accessed March 3, 2009.
- US Food and Drug Administration. Mercury in fish: cause for concern? www.fda.gov/fdac/reprints/mercury.html. Accessed March 3, 2009.
- Eritsland J, Arnesen H, Seljeflot I, Kierulf P. Long-term effects of n-3 polyunsaturated fatty acids on haemostatic variables and bleeding episodes in patients with coronary artery disease. Blood Coagul Fibrinolysis 1995; 6:17–22.
- Harris WS. Expert opinion: omega-3 fatty acids and bleeding—cause for concern? Am J Cardiol 2007; 99:44C–46C.
- Bjerregaard P, Johansen LG. Mortality pattern in Greenland. Arctic Med Res 1987; 46:71–77.
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2001; 285:2486–2497.
- Jacobson TA. Secondary prevention of coronary artery disease with omega-3 fatty acids. Am J Cardiol 2006; 98:61i–70i.
- Harris WS, Miller M, Tighe AP, Davidson MH, Schaefer EJ. Omega-3 fatty acids and coronary heart disease risk: clinical and mechanistic perspectives. Atherosclerosis 2008; 197:12–24.
- Khan S, Minihane AM, Talmud PJ, et al. Dietary long-chain n-3 PUFAs increase LPL gene expression in adipose tissue of subjects with an atherogenic lipoprotein phenotype. J Lipid Res 2002; 43:979–985.
- Balk EM, Lichtenstein AH, Chung M, Kupelnick B, Chew P, Lau J. Effects of omega-3 fatty acids on serum markers of cardiovascular disease risk: a systematic review. Atherosclerosis 2006; 189:19–30.
- Pownall HJ, Brauchi D, Kilinc C, et al. Correlation of serum triglyceride and its reduction by omega-3 fatty acids with lipid transfer activity and the neutral lipid compositions of high-density and low-density lipoproteins. Atherosclerosis 1999; 143:285–297.
- Harris WS. N-3 fatty acids and serum lipoproteins: human studies. Am J Clin Nutr 1997; 65 suppl 5:1645S–1654S.
- Robinson JG, Stone NJ. Antiatherosclerotic and antithrombotic effects of omega-3 fatty acids. Am J Cardiol 2006; 98:39i–49i.
- Chan DC, Watts GF, Mori TA, Barrett PH, Beilin LJ, Redgrave TG. Factorial study of the effects of atorvastatin and fish oil on dyslipidaemia in visceral obesity. Eur J Clin Invest 2002; 32:429–436.
- Yokoyama M, Origasa H, Matsuzaki M, et al; Japan EPA lipid intervention study (JELIS) Investigators. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet 2007; 369:1090–1098.
- Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza Nell’Infarto Miocardico. Lancet 1999; 354:447–455.
- Getz GS, Reardon CA. Nutrition and cardiovascular disease. Arterioscler Thromb Vasc Biol 2007; 27:2499–2506.
- Reiffel JA, McDonald A. Antiarrhythmic effects of omega-3 fatty acids. Am J Cardiol 2006; 98:50i–60i.
- Raitt MH, Connor WE, Morris C, et al. Fish oil supplementation and risk of ventricular tachycardia and ventricular fibrillation in patients with implantable defibrillators: a randomized controlled trial. JAMA 2005; 293:2884–2891.
- Leaf A, Albert CM, Josephson M, et al; Fatty Acid Antiarrhythmia Trial Investigators. Prevention of fatal arrhythmias in high-risk subjects by fish oil n-3 fatty acid intake. Circulation 2005; 112:2762–2768.
- Brouwer IA, Zock PL, Wever EF, et al. Rationale and design of a randomised controlled clinical trial on supplemental intake of n-3 fatty acids and incidence of cardiac arrhythmia: SOFA. Eur J Clin Nutr 2003; 57:1323–1330.
- London B, Albert C, Anderson ME, et al. Omega-3 fatty acids and cardiac arrhythmias: prior studies and recommendations for future research: a report from the National Heart, Lung, and Blood Institute and Office of Dietary Supplements Omega-3 Fatty Acids and their Role in Cardiac Arrhythmogenesis Workshop. Circulation 2007; 116:e320–e335.
- Calo L, Bianconi L, Colivicchi F, et al. N-3 fatty acids for the prevention of atrial fibrillation after coronary artery bypass surgery: a randomized, controlled trial. J Am Coll Cardiol 2005; 45:1723–1728.
- Harris WS, Assaad B, Poston WC. Tissue omega-6/omega-3 fatty acid ratio and risk for coronary artery disease. Am J Cardiol 2006; 98:19i–26i.
- Thies F, Garry JM, Yaqoob P, et al. Association of n-3 polyunsaturated fatty acids with stability of atherosclerotic plaques: a randomised controlled trial. Lancet 2003; 361:477–485.
- Johansen O, Brekke M, Seljeflot I, Abdelnoor M, Arnesen H. N-3 fatty acids do not prevent restenosis after coronary angioplasty: results from the CART study. Coronary Angioplasty Restenosis Trial. J Am Coll Cardiol 1999; 33:1619–1626.
- Geleijnse JM, Giltay EJ, Grobbee DE, Donders AR, Kok FJ. Blood pressure response to fish oil supplementation: metaregression analysis of randomized trials. J Hypertens 2002; 20:1493–1499.
- American Heart Association. Fish and Omega-3 Fatty Acids. www.americanheart.org/presenter.jhtml?identifier=4632. Accessed March 3, 2009.
- United States Department of Agriculture. Nutrient Data Laboratory. www.nal.usda.gov/fnic/foodcomp/search. Accessed March 3, 2009.
- GlaxoSmithKline. Patient Information: Lovaza. www.lovaza.com. Accessed March 3, 2009.
- US Food and Drug Administration. Mercury in fish: cause for concern? www.fda.gov/fdac/reprints/mercury.html. Accessed March 3, 2009.
- Eritsland J, Arnesen H, Seljeflot I, Kierulf P. Long-term effects of n-3 polyunsaturated fatty acids on haemostatic variables and bleeding episodes in patients with coronary artery disease. Blood Coagul Fibrinolysis 1995; 6:17–22.
- Harris WS. Expert opinion: omega-3 fatty acids and bleeding—cause for concern? Am J Cardiol 2007; 99:44C–46C.
- Bjerregaard P, Johansen LG. Mortality pattern in Greenland. Arctic Med Res 1987; 46:71–77.
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2001; 285:2486–2497.
- Jacobson TA. Secondary prevention of coronary artery disease with omega-3 fatty acids. Am J Cardiol 2006; 98:61i–70i.
- Harris WS, Miller M, Tighe AP, Davidson MH, Schaefer EJ. Omega-3 fatty acids and coronary heart disease risk: clinical and mechanistic perspectives. Atherosclerosis 2008; 197:12–24.
- Khan S, Minihane AM, Talmud PJ, et al. Dietary long-chain n-3 PUFAs increase LPL gene expression in adipose tissue of subjects with an atherogenic lipoprotein phenotype. J Lipid Res 2002; 43:979–985.
- Balk EM, Lichtenstein AH, Chung M, Kupelnick B, Chew P, Lau J. Effects of omega-3 fatty acids on serum markers of cardiovascular disease risk: a systematic review. Atherosclerosis 2006; 189:19–30.
- Pownall HJ, Brauchi D, Kilinc C, et al. Correlation of serum triglyceride and its reduction by omega-3 fatty acids with lipid transfer activity and the neutral lipid compositions of high-density and low-density lipoproteins. Atherosclerosis 1999; 143:285–297.
- Harris WS. N-3 fatty acids and serum lipoproteins: human studies. Am J Clin Nutr 1997; 65 suppl 5:1645S–1654S.
- Robinson JG, Stone NJ. Antiatherosclerotic and antithrombotic effects of omega-3 fatty acids. Am J Cardiol 2006; 98:39i–49i.
- Chan DC, Watts GF, Mori TA, Barrett PH, Beilin LJ, Redgrave TG. Factorial study of the effects of atorvastatin and fish oil on dyslipidaemia in visceral obesity. Eur J Clin Invest 2002; 32:429–436.
- Yokoyama M, Origasa H, Matsuzaki M, et al; Japan EPA lipid intervention study (JELIS) Investigators. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet 2007; 369:1090–1098.
- Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza Nell’Infarto Miocardico. Lancet 1999; 354:447–455.
- Getz GS, Reardon CA. Nutrition and cardiovascular disease. Arterioscler Thromb Vasc Biol 2007; 27:2499–2506.
- Reiffel JA, McDonald A. Antiarrhythmic effects of omega-3 fatty acids. Am J Cardiol 2006; 98:50i–60i.
- Raitt MH, Connor WE, Morris C, et al. Fish oil supplementation and risk of ventricular tachycardia and ventricular fibrillation in patients with implantable defibrillators: a randomized controlled trial. JAMA 2005; 293:2884–2891.
- Leaf A, Albert CM, Josephson M, et al; Fatty Acid Antiarrhythmia Trial Investigators. Prevention of fatal arrhythmias in high-risk subjects by fish oil n-3 fatty acid intake. Circulation 2005; 112:2762–2768.
- Brouwer IA, Zock PL, Wever EF, et al. Rationale and design of a randomised controlled clinical trial on supplemental intake of n-3 fatty acids and incidence of cardiac arrhythmia: SOFA. Eur J Clin Nutr 2003; 57:1323–1330.
- London B, Albert C, Anderson ME, et al. Omega-3 fatty acids and cardiac arrhythmias: prior studies and recommendations for future research: a report from the National Heart, Lung, and Blood Institute and Office of Dietary Supplements Omega-3 Fatty Acids and their Role in Cardiac Arrhythmogenesis Workshop. Circulation 2007; 116:e320–e335.
- Calo L, Bianconi L, Colivicchi F, et al. N-3 fatty acids for the prevention of atrial fibrillation after coronary artery bypass surgery: a randomized, controlled trial. J Am Coll Cardiol 2005; 45:1723–1728.
- Harris WS, Assaad B, Poston WC. Tissue omega-6/omega-3 fatty acid ratio and risk for coronary artery disease. Am J Cardiol 2006; 98:19i–26i.
- Thies F, Garry JM, Yaqoob P, et al. Association of n-3 polyunsaturated fatty acids with stability of atherosclerotic plaques: a randomised controlled trial. Lancet 2003; 361:477–485.
- Johansen O, Brekke M, Seljeflot I, Abdelnoor M, Arnesen H. N-3 fatty acids do not prevent restenosis after coronary angioplasty: results from the CART study. Coronary Angioplasty Restenosis Trial. J Am Coll Cardiol 1999; 33:1619–1626.
- Geleijnse JM, Giltay EJ, Grobbee DE, Donders AR, Kok FJ. Blood pressure response to fish oil supplementation: metaregression analysis of randomized trials. J Hypertens 2002; 20:1493–1499.
KEY POINTS
- The American Heart Association recommends that healthy people consume fatty fish at least twice a week. The recommendation for people with coronary artery disease is 1 g of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) per day.
- A formulation of EPA 465 mg plus DHA 375 mg is available by prescription and is approved for treating triglyceridemia in excess of 500 mg/dL. The dose is 2 to 4 capsules per day.
- Experts generally believe that omega-3 fatty acids reduce arrhythmic events. Nevertheless, we lack clear evidence of their clinical effectiveness, and their use for such purposes is off-label.
- Overall, omega-3 fatty acids have minimal side effects.
Multiple huge bullae after renal transplant
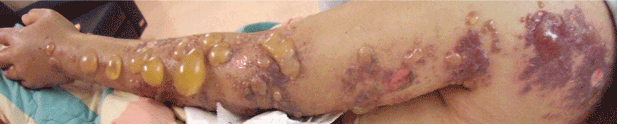
Q: What is the most likely diagnosis?
- Contact dermatitis
- Herpes zoster
- Herpes simplex
- Pemphigus
- Bullous pemphigoid
- Graft-vs-host disease
A: The correct answer is herpes zoster (shingles), which represents reactivation of varicella-zoster virus.
The diagnosis of herpes zoster is usually based solely on the clinical presentation. It is typically characterized in immunocompetent patients by a unilateral vesicular eruption with a well-defined dermatomal distribution. But occasionally, as in this patient on immunosuppressant drugs, it presents with atypical skin lesions such as multiple huge bullae involving multiple dermatomes.1,2
Patients treated with immunosuppressive agents after organ transplantation are at high risk of herpes zoster. A recent published retrospective study of adult kidney transplant recipients showed an average incidence of approximately 28 per 1,000 person-years.3
Treatment involves analgesics and sometimes antiviral drugs, and the decisions should take into account the patient’s age and immune status.1
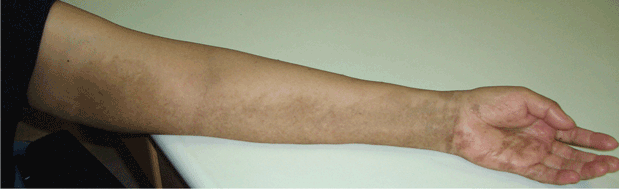
- Nagel MA, Gilden DH. The protean neurologic manifestations of varicella-zoster virus infection. Cleve Clin J Med 2007; 74:489–504.
- Albrecht MA. Clinical manifestations of varicella-zoster virus infection: Herpes zoster. InRose BD, editor: UpToDate. Waltham, MA: UpToDate, 2008.
- Arness T, Pedersen R, Dierkhising R, Kremers W, Patel R. Varicella zoster virus-associated disease in adult kidney transplant recipients: incidence and risk-factor analysis. Transpl Infect Dis 2008; 10:260–268.
Q: What is the most likely diagnosis?
- Contact dermatitis
- Herpes zoster
- Herpes simplex
- Pemphigus
- Bullous pemphigoid
- Graft-vs-host disease
A: The correct answer is herpes zoster (shingles), which represents reactivation of varicella-zoster virus.
The diagnosis of herpes zoster is usually based solely on the clinical presentation. It is typically characterized in immunocompetent patients by a unilateral vesicular eruption with a well-defined dermatomal distribution. But occasionally, as in this patient on immunosuppressant drugs, it presents with atypical skin lesions such as multiple huge bullae involving multiple dermatomes.1,2
Patients treated with immunosuppressive agents after organ transplantation are at high risk of herpes zoster. A recent published retrospective study of adult kidney transplant recipients showed an average incidence of approximately 28 per 1,000 person-years.3
Treatment involves analgesics and sometimes antiviral drugs, and the decisions should take into account the patient’s age and immune status.1
Q: What is the most likely diagnosis?
- Contact dermatitis
- Herpes zoster
- Herpes simplex
- Pemphigus
- Bullous pemphigoid
- Graft-vs-host disease
A: The correct answer is herpes zoster (shingles), which represents reactivation of varicella-zoster virus.
The diagnosis of herpes zoster is usually based solely on the clinical presentation. It is typically characterized in immunocompetent patients by a unilateral vesicular eruption with a well-defined dermatomal distribution. But occasionally, as in this patient on immunosuppressant drugs, it presents with atypical skin lesions such as multiple huge bullae involving multiple dermatomes.1,2
Patients treated with immunosuppressive agents after organ transplantation are at high risk of herpes zoster. A recent published retrospective study of adult kidney transplant recipients showed an average incidence of approximately 28 per 1,000 person-years.3
Treatment involves analgesics and sometimes antiviral drugs, and the decisions should take into account the patient’s age and immune status.1
- Nagel MA, Gilden DH. The protean neurologic manifestations of varicella-zoster virus infection. Cleve Clin J Med 2007; 74:489–504.
- Albrecht MA. Clinical manifestations of varicella-zoster virus infection: Herpes zoster. InRose BD, editor: UpToDate. Waltham, MA: UpToDate, 2008.
- Arness T, Pedersen R, Dierkhising R, Kremers W, Patel R. Varicella zoster virus-associated disease in adult kidney transplant recipients: incidence and risk-factor analysis. Transpl Infect Dis 2008; 10:260–268.
- Nagel MA, Gilden DH. The protean neurologic manifestations of varicella-zoster virus infection. Cleve Clin J Med 2007; 74:489–504.
- Albrecht MA. Clinical manifestations of varicella-zoster virus infection: Herpes zoster. InRose BD, editor: UpToDate. Waltham, MA: UpToDate, 2008.
- Arness T, Pedersen R, Dierkhising R, Kremers W, Patel R. Varicella zoster virus-associated disease in adult kidney transplant recipients: incidence and risk-factor analysis. Transpl Infect Dis 2008; 10:260–268.
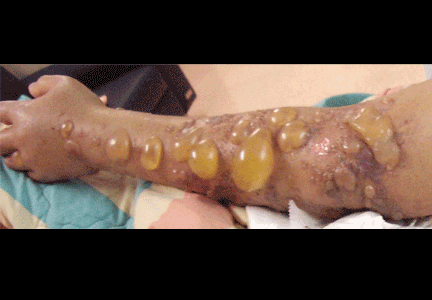
Psychiatric symptoms of dementia: Treatable, but no silver bullet
Your 84-year-old patient's son is distraught. “I know Mom has dementia, but I don’t understand why she cannot relax. She is busy all night long, taking out the silverware, packing her clothes, and trying to leave the house. Sometimes she tells me that there are little children in the room. These hallucinations scare me, although they do not seem to bother her very much. She keeps me awake; I’m often late to work because I’m up much of the night. I’m afraid I’m going to lose my job; and I don’t want to put Mom into a nursing home. Please give her a medication for this behavior.”
Another of your patients, an 82-year-old man, is admitted to a nursing home after an emergency hospitalization in the geriatric psychiatry unit. His daughter left him alone with her boyfriend one morning while she went to work. Not recognizing him, your patient attacked the young man with a kitchen knife. The police initially arrested your patient and then had him admitted to the psychiatric unit. He is discharged 2 weeks later to the nursing home.
Can anything be done for these patients?
A GROWING PROBLEM
Dementia is a growing problem with the aging of the population. At the time of the 2000 census there were 4.5 million people in the United States with Alzheimer disease, the most common type of dementia,1 and the prevalence is expected to increase to 13.2 million by the year 2050.1

CONSERVATIVE MEASURES ARE THE MAINSTAY OF TREATMENT

As for offending drugs, removing an antimuscarinic or anticholinergic drug may resolve hallucinations; stopping propoxyphene (Darvon) may improve sleep.
No drugs are approved for treating hallucinations, agitation, or other distressing behavior in neurodegenerative diseases such as Alzheimer dementia. Rather, the mainstay of treatment is behavioral and environmental modification.7 In an environment optimized to maximize comfort, reduce stress, and permit safe wandering, behavioral medications may be unnecessary.
Nevertheless, environments are not always optimal, and physicians may offer medications to treat behavioral symptoms to improve quality of life and to let patients keep living in the community.
Below, we discuss the drugs used to treat behavioral problems in dementia, evidence for the efficacy of these drugs, and their potential for adverse effects.
ANTIPSYCHOTIC DRUGS: SMALL BENEFIT, BIG RISK
Although antipsychotic drugs, both typical and atypical, are often used to treat dementia- related behaviors, their beneficial effects are minimal and adverse effects are common.8,9
Aggression has been considered a symptom that might respond to an atypical antipsychotic drug.10 However, the Clinical Antipsychotic Trials of Intervention Effectiveness—Alzheimer’s Disease (CATIE-AD) trial11 found no differences in efficacy between placebo and the atypical antipsychotics olanzapine (Zyprexa), quetiapine (Seroquel), and risperidone (Risperdal) in treating psychosis, aggression, and agitation in dementia. In that study, rates of drug discontinuation due to adverse effects ranged from 5% for placebo to 24% for olanzapine. Overall, 82% of the patients stopped taking their initially assigned medications during the 36-week period of the trial.11
Antipsychotic drugs may cause more adverse effects in patients with Parkinson disease or dementia with Lewy bodies, and medications with the least dopamine D2 receptor blockade are chosen to reduce the impact on the parkinsonism. Patients with movement disorders were excluded from the CATIE-AD study, and data on this topic are very limited. Quetiapine and olanzapine are often used as alternatives to clozapine (Clozaril) for treating psychosis in Parkinson disease and may have a role in dementia with Lewy bodies.12,13
Atypical antipsychotics carry significant risk of illness and even death. The US Food and Drug Administration (FDA) has published advisories about hyperglycemia, cerebrovascular events, and death.14 Returning to the older, “typical” antipsychotics is not a solution either, given their high incidence of extrapyramidal symptoms15 and potentially higher risk of death.16,17
Even if effective, try stopping the drug
Even in the few situations in dementia in which antipsychotics prove efficacious, a trial of dose-reduction and possible discontinuation is a part of the appropriate plan of care. Symptoms such as aggression and delusions may decrease as the underlying dementia progresses.2 A consensus statement on antipsychotic drug use in the elderly18 recommended stopping antipsychotic drugs as follows:
- If given for delirium—discontinue the drug after 1 week
- For agitated dementia—taper within 3 to 6 months to determine the lowest effective maintenance dose
- For psychotic major depression—discontinue after 6 months
- For mania with psychosis—discontinue after 3 months.18
Disorders for which antipsychotics are not recommended are irritability, hostility, generalized anxiety, and insomnia. In contrast with recommendations for dementia-related behaviors, the psychosis of schizophrenia is treated lifelong at the lowest effective dose of medication.
ANTIDEPRESSANTS: MANY CHOICES, LITTLE EVIDENCE
Depression is hard to assess in a patient with dementia, particularly since apathy is a common symptom in both dementia and depression and may confuse the presentation. Additionally, screening tests for depression have not been validated in the demented elderly.
Depression in dementia is associated with poorer quality of life, greater disability in activities of daily living, a faster cognitive decline, a high rate of nursing home placement, a higher death rate, and a higher frequency of depression and burden in caregivers.19 Quality of life may improve with antidepressant treatment even if the patient does not meet all the criteria for a major depressive disorder. Provisional recommendations for diagnosing depression in dementia suggest using three (instead of five) or more criteria, and include irritability or social isolation as additional criteria.20
Choosing an antidepressant
Only a few randomized controlled trials of antidepressants for depression with dementia have been completed, each with a small number of patients.
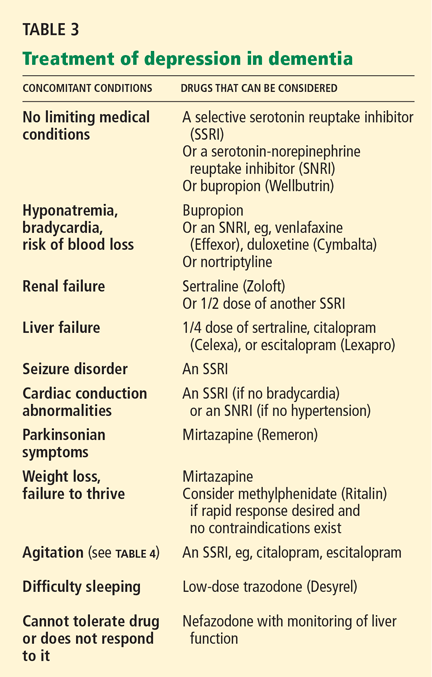
Mirtazapine (Remeron) is what we recommend to improve sleep and appetite and restore lost weight.21 It can be used in patients with Parkinson disease or parkinsonian symptoms who experience increased tremors or bradykinesia with selective serotonin reuptake inhibitors (SSRIs). On the other hand, it may not be the best option for those with diabetes mellitus, metabolic syndrome, hyperlipidemia, or obesity. It may rarely also cause a reversible agranulocytosis.
Venlafaxine (Effexor) and duloxetine (Cymbalta) are serotonin-norepinephrine reuptake inhibitors (SNRIs) and may help in concomitant pain syndromes.22 Either drug can cause anorexia at any dose and can elevate blood pressure at higher doses. Venlafaxine may also cause insomnia in some patients.
Bupropion (Wellbutrin) can be difficult to titrate to an effective dose in an older person with unsuspected renal insufficiency, and it may interact at the P450 complex.23 The risk of seizures is greater at higher bupropion serum levels. There is also a high incidence of weight loss. Frail elderly patients, those with hypertension, and those vulnerable to hallucinations will likely do better with another drug.
Nefazodone is a third- or fourth-line antidepressive choice because of the risk of hepatic failure. However, it can help reduce disabling anxiety associated with depression. The FDA requires periodic liver function testing if this drug is used.
Trazodone in low doses (≤ 100 mg) each evening may help with sleep, but it cannot be titrated to antidepressive doses in older adults because of orthostatic effects.
Nortriptyline is recommended by some geriatricians for depression or pathologic crying in patients with mixed vascular dementia. However, it often causes cardiac conduction delays with reflex sympathetic tachycardia and anticholinergic side effects.
Monoamine oxidase inhibitors interact with many foods and drugs, limiting their use in older adults.
Methylphenidate (Ritalin) at low doses is used off-label for depression in palliative care, with noted rapid improvements in mood and appetite.24 Monitoring for increases in blood pressure, heart rate, and respiratory rate is essential if this stimulant is chosen. Patients who respond may make a transition to other traditional drugs after 2 to 4 weeks.
Caveats with SSRIs
- Despite the safety profile of SSRIs in older adults, care must be taken when prescribing them to frail elderly patients, given recent data associating SSRIs with falls and fragility fractures25,26 and urinary incontinence.27
- SSRIs may decrease appetite during initial treatment.
- Sertraline (Zoloft) may have to be started at a very low dose to decrease possible adverse gastrointestinal symptoms, such as diarrhea.
- Paroxetine (Paxil) has multiple interactions at the cytochrome P450 complex and has the most anticholinergic properties of the SSRIs, rendering it more likely to cause adverse drug reactions, constipation, and delirium.
- Daily fluoxetine (Prozac) may not be appropriate in older adults because of its long half-life and the risk of insomnia and agitation.28
- Tremors can emerge with all the SSRIs; akathisia, dystonia, and parkinsonism are also possible.29
- Hyponatremia, bruising, and increased bleeding time can occur with any SSRI.
- Abrupt cessation of any SSRI except fluoxetine (due to its long half-life) or of SNRIs may cause a very unpleasant flu-like withdrawal syndrome.
- Apathy can be a reversible, dose-dependent adverse effect of SSRIs in young persons30; there are no data on the dose at which this adverse effect might emerge in demented elderly patients.
In a systematic review, Sink et al31 found citalopram (Celexa) to help reduce nondepressive agitation.
How long should depression be treated?
Antidepressant treatment is typically for 6 to 12 months. However, the optimal duration in an older adult with dementia is not known and is not addressed in either the American Psychiatric Association practice guideline on dementia32 or the position statement of the American Association for Geriatric Psychiatry.33
Patients with executive dysfunction, particularly those with perseveration and diminished inhibition, may be less likely to respond to antidepressants, and the symptoms are more likely to recur if they do respond.34 It may be appropriate to treat them for a year and then withdraw the drug and monitor for recurrence. Some patients may need indefinite treatment.
No data on treating apathy
Apathy in elderly patients with dementia is common. It is found in nearly half of elderly patients with mild dementia and in nearly all of those with severe dementia. If accompanied by depressive symptoms such as sadness, guilt, feelings of worthlessness, passive or active death wish, changes in sleep or appetite, or tearfulness, apathy and other depressive symptoms may respond to antidepressive treatment—both behavioral and pharmacologic. When present in dementia without depressive symptomatology, apathy is unlikely to respond to antidepressants. In particular, SSRIs may actually induce or worsen apathy through their effect on the angular gyrus. Apathy can be very frustrating to family members but not troublesome at all to the patient.
No medication carries an indication for apathy in dementia. Although stimulants such as methylphenidate and modafinil (Provigil) have been used, there is no evidence to date from any controlled study of efficacy and safety in this population.
Try nondrug measures concomitantly
Given the limited evidence of efficacy of antidepressive therapy in demented elderly patients, nonpharmacologic therapy should be offered concomitantly.
Evidence-based nonpharmacologic treatment for depression in dementia includes increasing enjoyable activities and socialization with people and pets, reducing the need to perform frustrating activities, redirecting perseverative behaviors and speech, and addressing caregiver needs.34 Exercise may improve physical functioning in depression with dementia.35 A comprehensive sleep program may improve associated sleep disorders.36
An intensive collaborative-care intervention37 resulted in more demented elderly patients in the intervention group receiving a cholinesterase inhibitor and an antidepressive than in the usual-care group. Outcomes included fewer behavioral symptoms, less caregiver distress, and less caregiver depression.
So far, no randomized trial has shown electroconvulsive therapy to be effective in elderly patients with depression and dementia.38
ANTICONVULSANT DRUGS MAY STABILIZE MOOD
On the basis of small studies with some contradictory outcomes,39 both older and newer anticonvulsants have been used in nonpsychotic agitation, aggression, and impulsivity in a variety of psychiatric disorders, brain injury, and dementia.40 Most of the data are on the older drugs such as valproic acid and carbamazepine (Tegretol).
Valproic acid is associated with an adverse metabolic profile (hyperglycemia, weight gain, and hyperlipidemia),41,42 dose-related orthostasis, sedation, and worsening cognitive performance. In addition, the possibility of thrombocytopenia and blood level fluctuations requires monitoring. Older adults may tolerate 250 to 500 mg/day with minimal adverse effects.
Carbamazepine reduced aggression in a blinded, placebo-controlled study in nursing home patients.43 Use of carbamazepine requires monitoring of hematologic and liver profiles, alters the metabolism of itself and other drugs, and is associated with dose-related sedation.
Lamotrigine (Lamictal) takes a long time to titrate but may help with nonpsychotic agitation and impulsivity; it is a relatively new drug, and there are limited data to support its use at this time in the elderly.
Gabapentin (Gabarone), in case reports at doses primarily from 600 to 1,200 mg/day, reduced behavioral and psychological problems of patients with dementia and with good renal clearance.44 Some patients may experience tremors or oversedation.
Phenytoin (Dilantin) is not a good choice for behavioral problems because of unwanted effects on teeth, bones, and balance.
Levetiracetam (Keppra) may cause behavioral disturbances to emerge or worsen.45
Emerging evidence suggests that all anticonvulsants may also be associated with an increased risk of depressive symptoms.
COGNITIVE ENHANCERS MAY IMPROVE BEHAVIOR
Acetylcholinesterase inhibitors may improve some behavioral symptoms of dementia. In an open-label retrospective trial, delusionality, irritability, anxiety, disinhibition, and agitation improved in some patients on these drugs.46 Patients most likely to respond were those with the most impairment from these behaviors and those with depressive or apathetic symptoms.46 A Cochrane review found a modest beneficial effect on behavior.47
Acetylcholinesterase inhibitors may reduce symptoms of apathy. Additionally, they actually improve depressive symptoms in mild to moderate dementia independent of any effect on cognition.48
Memantine (Namenda), approved for the treatment of moderate to severe dementia, may reduce the prevalence and incidence of agitation, particularly in more advanced dementia.49
The cognitive enhancers all require several weeks for titration and are not helpful for the acute management of behavioral or depressive symptoms.
OTHER DRUGS
Beta-blockers50 and estrogen51 have been studied as off-label, nonneuroleptic treatments for male aggression. Use of progesterone in men with inappropriate sexual behavior52 may have benefit; further interventions are reviewed by Srinivasan and Weinberg.53 These recommendations are based on small case series. In addition, the hormonal treatments may carry significant morbidity.
Sedative hypnotics were evaluated for sleep difficulties in demented patients in a meta-analysis by Glass et al,54 who found adverse cognitive events, psychomotor events, and daytime fatigue more common (5, 2.6, and 3.8 times, respectively) in the sedative group than in the placebo group.
For agitation in delirium, haloperidol (Haldol) is preferable to benzodiazepines, based on studies from the 1970s.55 Although benzodiazepines carry an indication for anxiety, newly prescribed benzodiazepines and those with a longer half-life are associated with hip fractures in older adults,56 possibly from sedation.
WHAT TO DO FOR YOUR PATIENTS
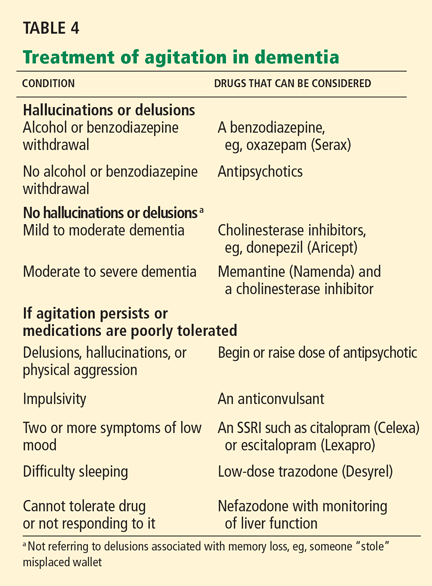
The 84-year-old woman
For the 84-year-old woman who is keeping her son awake all night, recommend making the environment safe for her to wander, including placing a bolt on the doors leading to the basement and outdoors and moving the knives to an area that she cannot reach, to avoid accidents. Recommend that she be given things to do that are repetitive, such as folding towels and arranging drawers. Referring her to day care may improve socialization and increase physical activity during the day, possibly improving her sleep time at night.
The 82-year-old man
Let’s assume the 82-year-old man arrested and then hospitalized is placed on risperidone 1 mg twice daily prior to discharge to the nursing home. In the nursing home, he becomes irritable with any change in his routine: the door has to be open by exactly 6 inches; his meals have to be identical and served on time; the newspaper needs to arrive by 8 AM. Since routine is paramount in the nursing home, the staff accommodates his need for a very regular schedule. Donepezil (Aricept) and memantine can be added as cognitive enhancers, and citalopram can be added for possible depression and obsessive features. The daughter should then be approached about reducing the risperidone dose and, hopefully, discontinuing it in the future.
Comment. A stable, routine environment is the most important intervention for managing this aggressive resident’s behavior, although he may have been helped to some degree by the adjunct medications. Once he is stable, the daughter may be able to bring him home for weekends and holidays, as long as she is advised never to surprise him with an unexpected visit or to bring home unexpected guests.
- Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 2003; 60:1119–1122.
- Holtzer R, Tang MX, Devanand DP, et al. Psychopathological features in Alzheimer's disease: course and relationship with cognitive status. J Am Geriatr Soc 2003; 51:953–960.
- Hart DJ, Craig D, Compton SA, et al. A retrospective study of the behavioural and psychological symptoms of mid and late phase Alzheimer's disease. Int J Geriatr Psychiatry 2003; 18:1037–1042.
- McKeith I, Cummings J. Behavioural changes and psychological symptoms in dementia disorders. Lancet Neurol 2005; 4:735–742.
- Stern Y, Albert M, Brandt J, et al. Utility of extrapyramidal signs and psychosis as predictors of cognitive and functional decline, nursing home admission, and death in Alzheimer's disease: prospective analyses from the Predictors Study. Neurology 1994; 44:2300–2307.
- Chibnall JT, Tait RC, Harman B, Luebbert RA. Effect of acetaminophen on behavior, well-being, and psychotropic medication use in nursing home residents with moderate-to-severe dementia. J Am Geriatr Soc 2005; 53:1921–1929.
- Doody RS, Stevens JC, Beck C, et al. Practice parameter: management of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2001; 56:1154–1166.
- Schneider LS, Dagerman K, Insel PS. Efficacy and adverse effects of atypical antipsychotics for dementia: meta-analysis of randomized, placebo-controlled trials. Am J Geriatr Psychiatry 2006; 14:191–210.
- Jeste DV, Dolder CR, Nayak GV, Salzman C. Atypical antipsychotics in elderly patients with dementia or schizophrenia: review of recent literature. Harv Rev Psychiatry 2005; 13:340–351.
- Rabinowitz J, Katz IR, De Deyn PP, Brodaty H, Greenspan A, Davidson M. Behavioral and psychological symptoms in patients with dementia as a target for pharmacotherapy with risperidone. J Clin Psychiatry 2004; 65:1329–1334.
- Schneider LS, Tariot PN, Dagerman KS, et al. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer's disease. N Engl J Med 2006; 355:1525–1538.
- Fernandez HH, Trieschmann ME, Burke MA, Friedman JH. Quetiapine for psychosis in Parkinson's disease versus dementia with Lewy bodies. J Clin Psychiatry 2002; 63:513–515.
- Cummings JL, Street J, Masterman D, Clark WS. Efficacy of olanzapine in the treatment of psychosis in dementia with Lewy bodies. Dement Geriatr Cogn Disord 2002; 13:67–73.
- US Food and Drug Administration. FDA Public Health Advisory—Deaths with Antipsychotics in Elderly Patients with Behavioral Disturbances: FDA/Center for Drug Evaluation and Research; April 11 2005.
- Lonergan E, Luxenberg J, Colford J. Haloperidol for agitation in dementia. Cochrane Database Syst Rev 2001; (4):CD002852.
- Wang PS, Schneeweiss S, Avorn J, et al. Risk of death in elderly users of conventional vs. atypical antipsychotic medications. N Engl J Med 2005; 353:2335–2341.
- Gill SS, Bronskill SE, Normand SL, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med 2007; 146:775–786.
- Alexopoulos GS, Streim J, Carpenter D, Docherty JP; Expert Consensus Panel for Using Antipsychotic Drugs in Older Patients. Using antipsychotic agents in older patients. J Clin Psychiatry 2004; 65(suppl 2):5–99.
- Starkstein SE, Mizrahi R. Depression in Alzheimer's disease. Expert Rev Neurother 2006; 6:887–895.
- Olin JT, Katz IR, Meyers BS, Schneider LS, Lebowitz BD. Provisional diagnostic criteria for depression of Alzheimer disease: rationale and background. Am J Geriatr Psychiatry 2002; 10:129–141.
- Aronne LJ, Segal KR. Weight gain in the treatment of mood disorders. J Clin Psychiatry 2003; 64(suppl 8):22–29.
- Barkin RL, Barkin S. The role of venlafaxine and duloxetine in the treatment of depression with decremental changes in somatic symptoms of pain, chronic pain, and the pharmacokinetics and clinical considerations of duloxetine pharmacotherapy. Am J Ther 2005; 12:431–438.
- Wilkes S. Bupropion. Drugs Today (Barc) 2006; 42:671–681.
- Homsi J, Walsh D, Nelson KA, LeGrand S, Davis M. Methylphenidate for depression in hospice practice: a case series. Am J Hosp Palliat Care 2000; 17:393–398.
- Kallin K, Lundin-Olsson L, Jensen J, Nyberg L, Gustafson Y. Predisposing and precipitating factors for falls among older people in residential care. Public Health 2002; 116:263–271.
- Richards JB, Papaioannou A, Adachi JD, et al; Canadian Multicentre Osteoporosis Study Research Group. Effect of selective serotonin reuptake inhibitors on the risk of fracture. Arch Intern Med 2007; 167:188–194.
- Movig KL, Leufkens HG, Belitser SV, Lenderink AW, Egberts ACG. Selective serotonin reuptake inhibitor-induced urinary incontinence. Pharmacoepidemiol Drug Saf 2002; 11:271–279.
- Fick DM, Cooper JW, Wade WE, Waller JL, Maclean JR, Beers MH. Updating the Beers criteria for potentially inappropriate medication use in older adults: results of a US consensus panel of experts. Arch Intern Med 2003; 163:2716–2724.
- Leo RJ. Movement disorders associated with the serotonin selective reuptake inhibitors. J Clin Psychiatry 1996; 57:449–454.
- Barnhart WJ, Makela EH, Latocha MJ. SSRI-induced apathy syndrome: a clinical review. J Psychiatr Pract 2004; 10:196–199.
- Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA 2005; 293:596–608.
- American Psychiatric Association. Practice Guideline and Resources for Treatment of Patients with Alzheimer's Disease and Other Dementias, 2nd Edition. October 2007. www.psychiatryonline.com/pracGuide/pracGuideTopic_3.aspx. Accessed 2/2/2009.
- Lyketsos CG, Colenda CC, Beck C, et al; Task Force of American Association for Geriatric Psychiatry. Position Statement of the American Association for Geriatric Psychiatry regarding principles of care for patients with dementia resulting from Alzheimer disease. Am J Geriatr Psychiatry 2006; 14:561–572.
- Potter GG, Steffens DC. Contribution of depression to cognitive impairment and dementia in older adults. Neurologist 2007; 13:105– 117.
- Teri L, Gibbons LE, McCurry SM, et al. Exercise plus behavioral management in patients with Alzheimer disease: a randomized controlled trial. JAMA 2003; 290:2015–2022.
- McCurry SM, Gibbons LE, Logsdon RG, Vitiello MV, Teri L. Nighttime insomnia treatment and education for Alzheimer's disease: a randomized, controlled trial. J Am Geriatr Soc 2005; 53:793–802.
- Callahan CM, Boustani MA, Unverzagt FW, et al. Effectiveness of collaborative care for older adults with Alzheimer disease in primary care: a randomized controlled trial. JAMA 2006; 295:2148–2157.
- Van der Wurff FB, Stek ML, Hoogendijk WL, Beekman AT. Electroconvulsive therapy for the depressed elderly. Cochrane Database Syst Rev 2003; ( 2):CD003593.
- Tariot PN, Raman R, Jakimovich L, et al. Divalproex sodium in nursing home residents with possible or probable Alzheimer disease complicated by agitation: a randomized, controlled trial. Am J Geriatr Psychiatry 2005; 13:942–949.
- Kim E. The use of newer anticonvulsants in neuropsychiatric disorders. Curr Psychiatry Rep 2002; 4:331–337.
- Ness-Abramof R, Apovian CM. Drug-induced weight gain. Drugs Today (Barc) 2005; 41:547–555.
- Biton V. Weight change and antiepileptic drugs: health issues and criteria for appropriate selection of an antiepileptic agent. Neurologist 2006; 12:163–167.
- Tariot PN, Erb R, Podgorski CA, et al. Efficacy and tolerability of carbamazepine for agitation and aggression in dementia. Am J Psychiatry 1998; 155:54–61.
- Miller LJ. Gabapentin for treatment of behavioral and psychological symptoms of dementia. Ann Pharmacother 2001; 35:427–431.
- White JR, Walczak TS, Leppik IE, et al. Discontinuation of levetiracetam because of behavioral side effects: a case-control study. Neurology 2003; 61:1218–1221.
- Mega S, Masterman DM, O'Connor SM, Barclay TR, Cummings JL. The spectrum of behavioral responses to cholinesterase inhibitor therapy in Alzheimer disease. Arch Neurol 1999; 56:1388–1393.
- Birks J. Cholinesterase inhibitors for Alzheimer's disease. Cochrane Database of Syst Rev 2006; (1):CD005593.
- Rozzini L, Vicini Chilovi B, Bertoletti E, Trabucchi M, Padovani A. Acetyl-cholinesterase inhibitors and depressive symptoms in patients with mild to moderate Alzheimer's disease. Aging Clin Exp Res 2007; 19:220–223.
- McShane R, Areosa Sastre A, Minakaran N. Memantine for dementia. Cochrane Database Syst Rev 2006; (2):CD003154.
- Peskind ER, Tsuang DW, Bonner LT, et al. Propranolol for disruptive behaviors in nursing home residents with probable or possible Alzheimer disease: a placebo-controlled study. Alzheimer Dis Assoc Disord 2005; 19:23–28.
- Hall KA, Keks NA, O'Connor DW. Transdermal estrogen patches for aggressive behavior in male patients with dementia: a randomized, controlled trial. Int Psychogeriatr 2005; 17:165–178.
- Light SA, Holroyd S. The use of medroxyprogesterone acetate for the treatment of sexually inappropriate behaviour in patients with dementia. J Psychiatry Neurosci 2006; 31:132–134.
- Srinivasan S, Weinberg A. Pharmacologic treatment of sexual inappropriateness in long-term care residents with dementia. Ann Long-Term Care: Clin Care Aging 2006; 14:20–28.
- Glass J, Lanctot KL, Herrmann N, Sproule BA, Busto UE. Sedative hypnotics in older people with insomnia: meta-analysis of risks and benefits. BMJ 2005; 331:1169.
- Kirven LE, Montero EF. Comparison of thioridazine and diazepam in the control of nonpsychotic symptoms associated with senility: double-blind study. J Am Geriatr Soc 1973; 21:546–551.
- Cumming RG, Le Couteur DG. Benzodiazepines and risk of hip fractures in older people: a review of the evidence. CNS Drugs 2003; 17:825–837.
Your 84-year-old patient's son is distraught. “I know Mom has dementia, but I don’t understand why she cannot relax. She is busy all night long, taking out the silverware, packing her clothes, and trying to leave the house. Sometimes she tells me that there are little children in the room. These hallucinations scare me, although they do not seem to bother her very much. She keeps me awake; I’m often late to work because I’m up much of the night. I’m afraid I’m going to lose my job; and I don’t want to put Mom into a nursing home. Please give her a medication for this behavior.”
Another of your patients, an 82-year-old man, is admitted to a nursing home after an emergency hospitalization in the geriatric psychiatry unit. His daughter left him alone with her boyfriend one morning while she went to work. Not recognizing him, your patient attacked the young man with a kitchen knife. The police initially arrested your patient and then had him admitted to the psychiatric unit. He is discharged 2 weeks later to the nursing home.
Can anything be done for these patients?
A GROWING PROBLEM
Dementia is a growing problem with the aging of the population. At the time of the 2000 census there were 4.5 million people in the United States with Alzheimer disease, the most common type of dementia,1 and the prevalence is expected to increase to 13.2 million by the year 2050.1
CONSERVATIVE MEASURES ARE THE MAINSTAY OF TREATMENT
As for offending drugs, removing an antimuscarinic or anticholinergic drug may resolve hallucinations; stopping propoxyphene (Darvon) may improve sleep.
No drugs are approved for treating hallucinations, agitation, or other distressing behavior in neurodegenerative diseases such as Alzheimer dementia. Rather, the mainstay of treatment is behavioral and environmental modification.7 In an environment optimized to maximize comfort, reduce stress, and permit safe wandering, behavioral medications may be unnecessary.
Nevertheless, environments are not always optimal, and physicians may offer medications to treat behavioral symptoms to improve quality of life and to let patients keep living in the community.
Below, we discuss the drugs used to treat behavioral problems in dementia, evidence for the efficacy of these drugs, and their potential for adverse effects.
ANTIPSYCHOTIC DRUGS: SMALL BENEFIT, BIG RISK
Although antipsychotic drugs, both typical and atypical, are often used to treat dementia- related behaviors, their beneficial effects are minimal and adverse effects are common.8,9
Aggression has been considered a symptom that might respond to an atypical antipsychotic drug.10 However, the Clinical Antipsychotic Trials of Intervention Effectiveness—Alzheimer’s Disease (CATIE-AD) trial11 found no differences in efficacy between placebo and the atypical antipsychotics olanzapine (Zyprexa), quetiapine (Seroquel), and risperidone (Risperdal) in treating psychosis, aggression, and agitation in dementia. In that study, rates of drug discontinuation due to adverse effects ranged from 5% for placebo to 24% for olanzapine. Overall, 82% of the patients stopped taking their initially assigned medications during the 36-week period of the trial.11
Antipsychotic drugs may cause more adverse effects in patients with Parkinson disease or dementia with Lewy bodies, and medications with the least dopamine D2 receptor blockade are chosen to reduce the impact on the parkinsonism. Patients with movement disorders were excluded from the CATIE-AD study, and data on this topic are very limited. Quetiapine and olanzapine are often used as alternatives to clozapine (Clozaril) for treating psychosis in Parkinson disease and may have a role in dementia with Lewy bodies.12,13
Atypical antipsychotics carry significant risk of illness and even death. The US Food and Drug Administration (FDA) has published advisories about hyperglycemia, cerebrovascular events, and death.14 Returning to the older, “typical” antipsychotics is not a solution either, given their high incidence of extrapyramidal symptoms15 and potentially higher risk of death.16,17
Even if effective, try stopping the drug
Even in the few situations in dementia in which antipsychotics prove efficacious, a trial of dose-reduction and possible discontinuation is a part of the appropriate plan of care. Symptoms such as aggression and delusions may decrease as the underlying dementia progresses.2 A consensus statement on antipsychotic drug use in the elderly18 recommended stopping antipsychotic drugs as follows:
- If given for delirium—discontinue the drug after 1 week
- For agitated dementia—taper within 3 to 6 months to determine the lowest effective maintenance dose
- For psychotic major depression—discontinue after 6 months
- For mania with psychosis—discontinue after 3 months.18
Disorders for which antipsychotics are not recommended are irritability, hostility, generalized anxiety, and insomnia. In contrast with recommendations for dementia-related behaviors, the psychosis of schizophrenia is treated lifelong at the lowest effective dose of medication.
ANTIDEPRESSANTS: MANY CHOICES, LITTLE EVIDENCE
Depression is hard to assess in a patient with dementia, particularly since apathy is a common symptom in both dementia and depression and may confuse the presentation. Additionally, screening tests for depression have not been validated in the demented elderly.
Depression in dementia is associated with poorer quality of life, greater disability in activities of daily living, a faster cognitive decline, a high rate of nursing home placement, a higher death rate, and a higher frequency of depression and burden in caregivers.19 Quality of life may improve with antidepressant treatment even if the patient does not meet all the criteria for a major depressive disorder. Provisional recommendations for diagnosing depression in dementia suggest using three (instead of five) or more criteria, and include irritability or social isolation as additional criteria.20
Choosing an antidepressant
Only a few randomized controlled trials of antidepressants for depression with dementia have been completed, each with a small number of patients.
Mirtazapine (Remeron) is what we recommend to improve sleep and appetite and restore lost weight.21 It can be used in patients with Parkinson disease or parkinsonian symptoms who experience increased tremors or bradykinesia with selective serotonin reuptake inhibitors (SSRIs). On the other hand, it may not be the best option for those with diabetes mellitus, metabolic syndrome, hyperlipidemia, or obesity. It may rarely also cause a reversible agranulocytosis.
Venlafaxine (Effexor) and duloxetine (Cymbalta) are serotonin-norepinephrine reuptake inhibitors (SNRIs) and may help in concomitant pain syndromes.22 Either drug can cause anorexia at any dose and can elevate blood pressure at higher doses. Venlafaxine may also cause insomnia in some patients.
Bupropion (Wellbutrin) can be difficult to titrate to an effective dose in an older person with unsuspected renal insufficiency, and it may interact at the P450 complex.23 The risk of seizures is greater at higher bupropion serum levels. There is also a high incidence of weight loss. Frail elderly patients, those with hypertension, and those vulnerable to hallucinations will likely do better with another drug.
Nefazodone is a third- or fourth-line antidepressive choice because of the risk of hepatic failure. However, it can help reduce disabling anxiety associated with depression. The FDA requires periodic liver function testing if this drug is used.
Trazodone in low doses (≤ 100 mg) each evening may help with sleep, but it cannot be titrated to antidepressive doses in older adults because of orthostatic effects.
Nortriptyline is recommended by some geriatricians for depression or pathologic crying in patients with mixed vascular dementia. However, it often causes cardiac conduction delays with reflex sympathetic tachycardia and anticholinergic side effects.
Monoamine oxidase inhibitors interact with many foods and drugs, limiting their use in older adults.
Methylphenidate (Ritalin) at low doses is used off-label for depression in palliative care, with noted rapid improvements in mood and appetite.24 Monitoring for increases in blood pressure, heart rate, and respiratory rate is essential if this stimulant is chosen. Patients who respond may make a transition to other traditional drugs after 2 to 4 weeks.
Caveats with SSRIs
- Despite the safety profile of SSRIs in older adults, care must be taken when prescribing them to frail elderly patients, given recent data associating SSRIs with falls and fragility fractures25,26 and urinary incontinence.27
- SSRIs may decrease appetite during initial treatment.
- Sertraline (Zoloft) may have to be started at a very low dose to decrease possible adverse gastrointestinal symptoms, such as diarrhea.
- Paroxetine (Paxil) has multiple interactions at the cytochrome P450 complex and has the most anticholinergic properties of the SSRIs, rendering it more likely to cause adverse drug reactions, constipation, and delirium.
- Daily fluoxetine (Prozac) may not be appropriate in older adults because of its long half-life and the risk of insomnia and agitation.28
- Tremors can emerge with all the SSRIs; akathisia, dystonia, and parkinsonism are also possible.29
- Hyponatremia, bruising, and increased bleeding time can occur with any SSRI.
- Abrupt cessation of any SSRI except fluoxetine (due to its long half-life) or of SNRIs may cause a very unpleasant flu-like withdrawal syndrome.
- Apathy can be a reversible, dose-dependent adverse effect of SSRIs in young persons30; there are no data on the dose at which this adverse effect might emerge in demented elderly patients.
In a systematic review, Sink et al31 found citalopram (Celexa) to help reduce nondepressive agitation.
How long should depression be treated?
Antidepressant treatment is typically for 6 to 12 months. However, the optimal duration in an older adult with dementia is not known and is not addressed in either the American Psychiatric Association practice guideline on dementia32 or the position statement of the American Association for Geriatric Psychiatry.33
Patients with executive dysfunction, particularly those with perseveration and diminished inhibition, may be less likely to respond to antidepressants, and the symptoms are more likely to recur if they do respond.34 It may be appropriate to treat them for a year and then withdraw the drug and monitor for recurrence. Some patients may need indefinite treatment.
No data on treating apathy
Apathy in elderly patients with dementia is common. It is found in nearly half of elderly patients with mild dementia and in nearly all of those with severe dementia. If accompanied by depressive symptoms such as sadness, guilt, feelings of worthlessness, passive or active death wish, changes in sleep or appetite, or tearfulness, apathy and other depressive symptoms may respond to antidepressive treatment—both behavioral and pharmacologic. When present in dementia without depressive symptomatology, apathy is unlikely to respond to antidepressants. In particular, SSRIs may actually induce or worsen apathy through their effect on the angular gyrus. Apathy can be very frustrating to family members but not troublesome at all to the patient.
No medication carries an indication for apathy in dementia. Although stimulants such as methylphenidate and modafinil (Provigil) have been used, there is no evidence to date from any controlled study of efficacy and safety in this population.
Try nondrug measures concomitantly
Given the limited evidence of efficacy of antidepressive therapy in demented elderly patients, nonpharmacologic therapy should be offered concomitantly.
Evidence-based nonpharmacologic treatment for depression in dementia includes increasing enjoyable activities and socialization with people and pets, reducing the need to perform frustrating activities, redirecting perseverative behaviors and speech, and addressing caregiver needs.34 Exercise may improve physical functioning in depression with dementia.35 A comprehensive sleep program may improve associated sleep disorders.36
An intensive collaborative-care intervention37 resulted in more demented elderly patients in the intervention group receiving a cholinesterase inhibitor and an antidepressive than in the usual-care group. Outcomes included fewer behavioral symptoms, less caregiver distress, and less caregiver depression.
So far, no randomized trial has shown electroconvulsive therapy to be effective in elderly patients with depression and dementia.38
ANTICONVULSANT DRUGS MAY STABILIZE MOOD
On the basis of small studies with some contradictory outcomes,39 both older and newer anticonvulsants have been used in nonpsychotic agitation, aggression, and impulsivity in a variety of psychiatric disorders, brain injury, and dementia.40 Most of the data are on the older drugs such as valproic acid and carbamazepine (Tegretol).
Valproic acid is associated with an adverse metabolic profile (hyperglycemia, weight gain, and hyperlipidemia),41,42 dose-related orthostasis, sedation, and worsening cognitive performance. In addition, the possibility of thrombocytopenia and blood level fluctuations requires monitoring. Older adults may tolerate 250 to 500 mg/day with minimal adverse effects.
Carbamazepine reduced aggression in a blinded, placebo-controlled study in nursing home patients.43 Use of carbamazepine requires monitoring of hematologic and liver profiles, alters the metabolism of itself and other drugs, and is associated with dose-related sedation.
Lamotrigine (Lamictal) takes a long time to titrate but may help with nonpsychotic agitation and impulsivity; it is a relatively new drug, and there are limited data to support its use at this time in the elderly.
Gabapentin (Gabarone), in case reports at doses primarily from 600 to 1,200 mg/day, reduced behavioral and psychological problems of patients with dementia and with good renal clearance.44 Some patients may experience tremors or oversedation.
Phenytoin (Dilantin) is not a good choice for behavioral problems because of unwanted effects on teeth, bones, and balance.
Levetiracetam (Keppra) may cause behavioral disturbances to emerge or worsen.45
Emerging evidence suggests that all anticonvulsants may also be associated with an increased risk of depressive symptoms.
COGNITIVE ENHANCERS MAY IMPROVE BEHAVIOR
Acetylcholinesterase inhibitors may improve some behavioral symptoms of dementia. In an open-label retrospective trial, delusionality, irritability, anxiety, disinhibition, and agitation improved in some patients on these drugs.46 Patients most likely to respond were those with the most impairment from these behaviors and those with depressive or apathetic symptoms.46 A Cochrane review found a modest beneficial effect on behavior.47
Acetylcholinesterase inhibitors may reduce symptoms of apathy. Additionally, they actually improve depressive symptoms in mild to moderate dementia independent of any effect on cognition.48
Memantine (Namenda), approved for the treatment of moderate to severe dementia, may reduce the prevalence and incidence of agitation, particularly in more advanced dementia.49
The cognitive enhancers all require several weeks for titration and are not helpful for the acute management of behavioral or depressive symptoms.
OTHER DRUGS
Beta-blockers50 and estrogen51 have been studied as off-label, nonneuroleptic treatments for male aggression. Use of progesterone in men with inappropriate sexual behavior52 may have benefit; further interventions are reviewed by Srinivasan and Weinberg.53 These recommendations are based on small case series. In addition, the hormonal treatments may carry significant morbidity.
Sedative hypnotics were evaluated for sleep difficulties in demented patients in a meta-analysis by Glass et al,54 who found adverse cognitive events, psychomotor events, and daytime fatigue more common (5, 2.6, and 3.8 times, respectively) in the sedative group than in the placebo group.
For agitation in delirium, haloperidol (Haldol) is preferable to benzodiazepines, based on studies from the 1970s.55 Although benzodiazepines carry an indication for anxiety, newly prescribed benzodiazepines and those with a longer half-life are associated with hip fractures in older adults,56 possibly from sedation.
WHAT TO DO FOR YOUR PATIENTS
The 84-year-old woman
For the 84-year-old woman who is keeping her son awake all night, recommend making the environment safe for her to wander, including placing a bolt on the doors leading to the basement and outdoors and moving the knives to an area that she cannot reach, to avoid accidents. Recommend that she be given things to do that are repetitive, such as folding towels and arranging drawers. Referring her to day care may improve socialization and increase physical activity during the day, possibly improving her sleep time at night.
The 82-year-old man
Let’s assume the 82-year-old man arrested and then hospitalized is placed on risperidone 1 mg twice daily prior to discharge to the nursing home. In the nursing home, he becomes irritable with any change in his routine: the door has to be open by exactly 6 inches; his meals have to be identical and served on time; the newspaper needs to arrive by 8 AM. Since routine is paramount in the nursing home, the staff accommodates his need for a very regular schedule. Donepezil (Aricept) and memantine can be added as cognitive enhancers, and citalopram can be added for possible depression and obsessive features. The daughter should then be approached about reducing the risperidone dose and, hopefully, discontinuing it in the future.
Comment. A stable, routine environment is the most important intervention for managing this aggressive resident’s behavior, although he may have been helped to some degree by the adjunct medications. Once he is stable, the daughter may be able to bring him home for weekends and holidays, as long as she is advised never to surprise him with an unexpected visit or to bring home unexpected guests.
Your 84-year-old patient's son is distraught. “I know Mom has dementia, but I don’t understand why she cannot relax. She is busy all night long, taking out the silverware, packing her clothes, and trying to leave the house. Sometimes she tells me that there are little children in the room. These hallucinations scare me, although they do not seem to bother her very much. She keeps me awake; I’m often late to work because I’m up much of the night. I’m afraid I’m going to lose my job; and I don’t want to put Mom into a nursing home. Please give her a medication for this behavior.”
Another of your patients, an 82-year-old man, is admitted to a nursing home after an emergency hospitalization in the geriatric psychiatry unit. His daughter left him alone with her boyfriend one morning while she went to work. Not recognizing him, your patient attacked the young man with a kitchen knife. The police initially arrested your patient and then had him admitted to the psychiatric unit. He is discharged 2 weeks later to the nursing home.
Can anything be done for these patients?
A GROWING PROBLEM
Dementia is a growing problem with the aging of the population. At the time of the 2000 census there were 4.5 million people in the United States with Alzheimer disease, the most common type of dementia,1 and the prevalence is expected to increase to 13.2 million by the year 2050.1
CONSERVATIVE MEASURES ARE THE MAINSTAY OF TREATMENT
As for offending drugs, removing an antimuscarinic or anticholinergic drug may resolve hallucinations; stopping propoxyphene (Darvon) may improve sleep.
No drugs are approved for treating hallucinations, agitation, or other distressing behavior in neurodegenerative diseases such as Alzheimer dementia. Rather, the mainstay of treatment is behavioral and environmental modification.7 In an environment optimized to maximize comfort, reduce stress, and permit safe wandering, behavioral medications may be unnecessary.
Nevertheless, environments are not always optimal, and physicians may offer medications to treat behavioral symptoms to improve quality of life and to let patients keep living in the community.
Below, we discuss the drugs used to treat behavioral problems in dementia, evidence for the efficacy of these drugs, and their potential for adverse effects.
ANTIPSYCHOTIC DRUGS: SMALL BENEFIT, BIG RISK
Although antipsychotic drugs, both typical and atypical, are often used to treat dementia- related behaviors, their beneficial effects are minimal and adverse effects are common.8,9
Aggression has been considered a symptom that might respond to an atypical antipsychotic drug.10 However, the Clinical Antipsychotic Trials of Intervention Effectiveness—Alzheimer’s Disease (CATIE-AD) trial11 found no differences in efficacy between placebo and the atypical antipsychotics olanzapine (Zyprexa), quetiapine (Seroquel), and risperidone (Risperdal) in treating psychosis, aggression, and agitation in dementia. In that study, rates of drug discontinuation due to adverse effects ranged from 5% for placebo to 24% for olanzapine. Overall, 82% of the patients stopped taking their initially assigned medications during the 36-week period of the trial.11
Antipsychotic drugs may cause more adverse effects in patients with Parkinson disease or dementia with Lewy bodies, and medications with the least dopamine D2 receptor blockade are chosen to reduce the impact on the parkinsonism. Patients with movement disorders were excluded from the CATIE-AD study, and data on this topic are very limited. Quetiapine and olanzapine are often used as alternatives to clozapine (Clozaril) for treating psychosis in Parkinson disease and may have a role in dementia with Lewy bodies.12,13
Atypical antipsychotics carry significant risk of illness and even death. The US Food and Drug Administration (FDA) has published advisories about hyperglycemia, cerebrovascular events, and death.14 Returning to the older, “typical” antipsychotics is not a solution either, given their high incidence of extrapyramidal symptoms15 and potentially higher risk of death.16,17
Even if effective, try stopping the drug
Even in the few situations in dementia in which antipsychotics prove efficacious, a trial of dose-reduction and possible discontinuation is a part of the appropriate plan of care. Symptoms such as aggression and delusions may decrease as the underlying dementia progresses.2 A consensus statement on antipsychotic drug use in the elderly18 recommended stopping antipsychotic drugs as follows:
- If given for delirium—discontinue the drug after 1 week
- For agitated dementia—taper within 3 to 6 months to determine the lowest effective maintenance dose
- For psychotic major depression—discontinue after 6 months
- For mania with psychosis—discontinue after 3 months.18
Disorders for which antipsychotics are not recommended are irritability, hostility, generalized anxiety, and insomnia. In contrast with recommendations for dementia-related behaviors, the psychosis of schizophrenia is treated lifelong at the lowest effective dose of medication.
ANTIDEPRESSANTS: MANY CHOICES, LITTLE EVIDENCE
Depression is hard to assess in a patient with dementia, particularly since apathy is a common symptom in both dementia and depression and may confuse the presentation. Additionally, screening tests for depression have not been validated in the demented elderly.
Depression in dementia is associated with poorer quality of life, greater disability in activities of daily living, a faster cognitive decline, a high rate of nursing home placement, a higher death rate, and a higher frequency of depression and burden in caregivers.19 Quality of life may improve with antidepressant treatment even if the patient does not meet all the criteria for a major depressive disorder. Provisional recommendations for diagnosing depression in dementia suggest using three (instead of five) or more criteria, and include irritability or social isolation as additional criteria.20
Choosing an antidepressant
Only a few randomized controlled trials of antidepressants for depression with dementia have been completed, each with a small number of patients.
Mirtazapine (Remeron) is what we recommend to improve sleep and appetite and restore lost weight.21 It can be used in patients with Parkinson disease or parkinsonian symptoms who experience increased tremors or bradykinesia with selective serotonin reuptake inhibitors (SSRIs). On the other hand, it may not be the best option for those with diabetes mellitus, metabolic syndrome, hyperlipidemia, or obesity. It may rarely also cause a reversible agranulocytosis.
Venlafaxine (Effexor) and duloxetine (Cymbalta) are serotonin-norepinephrine reuptake inhibitors (SNRIs) and may help in concomitant pain syndromes.22 Either drug can cause anorexia at any dose and can elevate blood pressure at higher doses. Venlafaxine may also cause insomnia in some patients.
Bupropion (Wellbutrin) can be difficult to titrate to an effective dose in an older person with unsuspected renal insufficiency, and it may interact at the P450 complex.23 The risk of seizures is greater at higher bupropion serum levels. There is also a high incidence of weight loss. Frail elderly patients, those with hypertension, and those vulnerable to hallucinations will likely do better with another drug.
Nefazodone is a third- or fourth-line antidepressive choice because of the risk of hepatic failure. However, it can help reduce disabling anxiety associated with depression. The FDA requires periodic liver function testing if this drug is used.
Trazodone in low doses (≤ 100 mg) each evening may help with sleep, but it cannot be titrated to antidepressive doses in older adults because of orthostatic effects.
Nortriptyline is recommended by some geriatricians for depression or pathologic crying in patients with mixed vascular dementia. However, it often causes cardiac conduction delays with reflex sympathetic tachycardia and anticholinergic side effects.
Monoamine oxidase inhibitors interact with many foods and drugs, limiting their use in older adults.
Methylphenidate (Ritalin) at low doses is used off-label for depression in palliative care, with noted rapid improvements in mood and appetite.24 Monitoring for increases in blood pressure, heart rate, and respiratory rate is essential if this stimulant is chosen. Patients who respond may make a transition to other traditional drugs after 2 to 4 weeks.
Caveats with SSRIs
- Despite the safety profile of SSRIs in older adults, care must be taken when prescribing them to frail elderly patients, given recent data associating SSRIs with falls and fragility fractures25,26 and urinary incontinence.27
- SSRIs may decrease appetite during initial treatment.
- Sertraline (Zoloft) may have to be started at a very low dose to decrease possible adverse gastrointestinal symptoms, such as diarrhea.
- Paroxetine (Paxil) has multiple interactions at the cytochrome P450 complex and has the most anticholinergic properties of the SSRIs, rendering it more likely to cause adverse drug reactions, constipation, and delirium.
- Daily fluoxetine (Prozac) may not be appropriate in older adults because of its long half-life and the risk of insomnia and agitation.28
- Tremors can emerge with all the SSRIs; akathisia, dystonia, and parkinsonism are also possible.29
- Hyponatremia, bruising, and increased bleeding time can occur with any SSRI.
- Abrupt cessation of any SSRI except fluoxetine (due to its long half-life) or of SNRIs may cause a very unpleasant flu-like withdrawal syndrome.
- Apathy can be a reversible, dose-dependent adverse effect of SSRIs in young persons30; there are no data on the dose at which this adverse effect might emerge in demented elderly patients.
In a systematic review, Sink et al31 found citalopram (Celexa) to help reduce nondepressive agitation.
How long should depression be treated?
Antidepressant treatment is typically for 6 to 12 months. However, the optimal duration in an older adult with dementia is not known and is not addressed in either the American Psychiatric Association practice guideline on dementia32 or the position statement of the American Association for Geriatric Psychiatry.33
Patients with executive dysfunction, particularly those with perseveration and diminished inhibition, may be less likely to respond to antidepressants, and the symptoms are more likely to recur if they do respond.34 It may be appropriate to treat them for a year and then withdraw the drug and monitor for recurrence. Some patients may need indefinite treatment.
No data on treating apathy
Apathy in elderly patients with dementia is common. It is found in nearly half of elderly patients with mild dementia and in nearly all of those with severe dementia. If accompanied by depressive symptoms such as sadness, guilt, feelings of worthlessness, passive or active death wish, changes in sleep or appetite, or tearfulness, apathy and other depressive symptoms may respond to antidepressive treatment—both behavioral and pharmacologic. When present in dementia without depressive symptomatology, apathy is unlikely to respond to antidepressants. In particular, SSRIs may actually induce or worsen apathy through their effect on the angular gyrus. Apathy can be very frustrating to family members but not troublesome at all to the patient.
No medication carries an indication for apathy in dementia. Although stimulants such as methylphenidate and modafinil (Provigil) have been used, there is no evidence to date from any controlled study of efficacy and safety in this population.
Try nondrug measures concomitantly
Given the limited evidence of efficacy of antidepressive therapy in demented elderly patients, nonpharmacologic therapy should be offered concomitantly.
Evidence-based nonpharmacologic treatment for depression in dementia includes increasing enjoyable activities and socialization with people and pets, reducing the need to perform frustrating activities, redirecting perseverative behaviors and speech, and addressing caregiver needs.34 Exercise may improve physical functioning in depression with dementia.35 A comprehensive sleep program may improve associated sleep disorders.36
An intensive collaborative-care intervention37 resulted in more demented elderly patients in the intervention group receiving a cholinesterase inhibitor and an antidepressive than in the usual-care group. Outcomes included fewer behavioral symptoms, less caregiver distress, and less caregiver depression.
So far, no randomized trial has shown electroconvulsive therapy to be effective in elderly patients with depression and dementia.38
ANTICONVULSANT DRUGS MAY STABILIZE MOOD
On the basis of small studies with some contradictory outcomes,39 both older and newer anticonvulsants have been used in nonpsychotic agitation, aggression, and impulsivity in a variety of psychiatric disorders, brain injury, and dementia.40 Most of the data are on the older drugs such as valproic acid and carbamazepine (Tegretol).
Valproic acid is associated with an adverse metabolic profile (hyperglycemia, weight gain, and hyperlipidemia),41,42 dose-related orthostasis, sedation, and worsening cognitive performance. In addition, the possibility of thrombocytopenia and blood level fluctuations requires monitoring. Older adults may tolerate 250 to 500 mg/day with minimal adverse effects.
Carbamazepine reduced aggression in a blinded, placebo-controlled study in nursing home patients.43 Use of carbamazepine requires monitoring of hematologic and liver profiles, alters the metabolism of itself and other drugs, and is associated with dose-related sedation.
Lamotrigine (Lamictal) takes a long time to titrate but may help with nonpsychotic agitation and impulsivity; it is a relatively new drug, and there are limited data to support its use at this time in the elderly.
Gabapentin (Gabarone), in case reports at doses primarily from 600 to 1,200 mg/day, reduced behavioral and psychological problems of patients with dementia and with good renal clearance.44 Some patients may experience tremors or oversedation.
Phenytoin (Dilantin) is not a good choice for behavioral problems because of unwanted effects on teeth, bones, and balance.
Levetiracetam (Keppra) may cause behavioral disturbances to emerge or worsen.45
Emerging evidence suggests that all anticonvulsants may also be associated with an increased risk of depressive symptoms.
COGNITIVE ENHANCERS MAY IMPROVE BEHAVIOR
Acetylcholinesterase inhibitors may improve some behavioral symptoms of dementia. In an open-label retrospective trial, delusionality, irritability, anxiety, disinhibition, and agitation improved in some patients on these drugs.46 Patients most likely to respond were those with the most impairment from these behaviors and those with depressive or apathetic symptoms.46 A Cochrane review found a modest beneficial effect on behavior.47
Acetylcholinesterase inhibitors may reduce symptoms of apathy. Additionally, they actually improve depressive symptoms in mild to moderate dementia independent of any effect on cognition.48
Memantine (Namenda), approved for the treatment of moderate to severe dementia, may reduce the prevalence and incidence of agitation, particularly in more advanced dementia.49
The cognitive enhancers all require several weeks for titration and are not helpful for the acute management of behavioral or depressive symptoms.
OTHER DRUGS
Beta-blockers50 and estrogen51 have been studied as off-label, nonneuroleptic treatments for male aggression. Use of progesterone in men with inappropriate sexual behavior52 may have benefit; further interventions are reviewed by Srinivasan and Weinberg.53 These recommendations are based on small case series. In addition, the hormonal treatments may carry significant morbidity.
Sedative hypnotics were evaluated for sleep difficulties in demented patients in a meta-analysis by Glass et al,54 who found adverse cognitive events, psychomotor events, and daytime fatigue more common (5, 2.6, and 3.8 times, respectively) in the sedative group than in the placebo group.
For agitation in delirium, haloperidol (Haldol) is preferable to benzodiazepines, based on studies from the 1970s.55 Although benzodiazepines carry an indication for anxiety, newly prescribed benzodiazepines and those with a longer half-life are associated with hip fractures in older adults,56 possibly from sedation.
WHAT TO DO FOR YOUR PATIENTS
The 84-year-old woman
For the 84-year-old woman who is keeping her son awake all night, recommend making the environment safe for her to wander, including placing a bolt on the doors leading to the basement and outdoors and moving the knives to an area that she cannot reach, to avoid accidents. Recommend that she be given things to do that are repetitive, such as folding towels and arranging drawers. Referring her to day care may improve socialization and increase physical activity during the day, possibly improving her sleep time at night.
The 82-year-old man
Let’s assume the 82-year-old man arrested and then hospitalized is placed on risperidone 1 mg twice daily prior to discharge to the nursing home. In the nursing home, he becomes irritable with any change in his routine: the door has to be open by exactly 6 inches; his meals have to be identical and served on time; the newspaper needs to arrive by 8 AM. Since routine is paramount in the nursing home, the staff accommodates his need for a very regular schedule. Donepezil (Aricept) and memantine can be added as cognitive enhancers, and citalopram can be added for possible depression and obsessive features. The daughter should then be approached about reducing the risperidone dose and, hopefully, discontinuing it in the future.
Comment. A stable, routine environment is the most important intervention for managing this aggressive resident’s behavior, although he may have been helped to some degree by the adjunct medications. Once he is stable, the daughter may be able to bring him home for weekends and holidays, as long as she is advised never to surprise him with an unexpected visit or to bring home unexpected guests.
- Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 2003; 60:1119–1122.
- Holtzer R, Tang MX, Devanand DP, et al. Psychopathological features in Alzheimer's disease: course and relationship with cognitive status. J Am Geriatr Soc 2003; 51:953–960.
- Hart DJ, Craig D, Compton SA, et al. A retrospective study of the behavioural and psychological symptoms of mid and late phase Alzheimer's disease. Int J Geriatr Psychiatry 2003; 18:1037–1042.
- McKeith I, Cummings J. Behavioural changes and psychological symptoms in dementia disorders. Lancet Neurol 2005; 4:735–742.
- Stern Y, Albert M, Brandt J, et al. Utility of extrapyramidal signs and psychosis as predictors of cognitive and functional decline, nursing home admission, and death in Alzheimer's disease: prospective analyses from the Predictors Study. Neurology 1994; 44:2300–2307.
- Chibnall JT, Tait RC, Harman B, Luebbert RA. Effect of acetaminophen on behavior, well-being, and psychotropic medication use in nursing home residents with moderate-to-severe dementia. J Am Geriatr Soc 2005; 53:1921–1929.
- Doody RS, Stevens JC, Beck C, et al. Practice parameter: management of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2001; 56:1154–1166.
- Schneider LS, Dagerman K, Insel PS. Efficacy and adverse effects of atypical antipsychotics for dementia: meta-analysis of randomized, placebo-controlled trials. Am J Geriatr Psychiatry 2006; 14:191–210.
- Jeste DV, Dolder CR, Nayak GV, Salzman C. Atypical antipsychotics in elderly patients with dementia or schizophrenia: review of recent literature. Harv Rev Psychiatry 2005; 13:340–351.
- Rabinowitz J, Katz IR, De Deyn PP, Brodaty H, Greenspan A, Davidson M. Behavioral and psychological symptoms in patients with dementia as a target for pharmacotherapy with risperidone. J Clin Psychiatry 2004; 65:1329–1334.
- Schneider LS, Tariot PN, Dagerman KS, et al. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer's disease. N Engl J Med 2006; 355:1525–1538.
- Fernandez HH, Trieschmann ME, Burke MA, Friedman JH. Quetiapine for psychosis in Parkinson's disease versus dementia with Lewy bodies. J Clin Psychiatry 2002; 63:513–515.
- Cummings JL, Street J, Masterman D, Clark WS. Efficacy of olanzapine in the treatment of psychosis in dementia with Lewy bodies. Dement Geriatr Cogn Disord 2002; 13:67–73.
- US Food and Drug Administration. FDA Public Health Advisory—Deaths with Antipsychotics in Elderly Patients with Behavioral Disturbances: FDA/Center for Drug Evaluation and Research; April 11 2005.
- Lonergan E, Luxenberg J, Colford J. Haloperidol for agitation in dementia. Cochrane Database Syst Rev 2001; (4):CD002852.
- Wang PS, Schneeweiss S, Avorn J, et al. Risk of death in elderly users of conventional vs. atypical antipsychotic medications. N Engl J Med 2005; 353:2335–2341.
- Gill SS, Bronskill SE, Normand SL, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med 2007; 146:775–786.
- Alexopoulos GS, Streim J, Carpenter D, Docherty JP; Expert Consensus Panel for Using Antipsychotic Drugs in Older Patients. Using antipsychotic agents in older patients. J Clin Psychiatry 2004; 65(suppl 2):5–99.
- Starkstein SE, Mizrahi R. Depression in Alzheimer's disease. Expert Rev Neurother 2006; 6:887–895.
- Olin JT, Katz IR, Meyers BS, Schneider LS, Lebowitz BD. Provisional diagnostic criteria for depression of Alzheimer disease: rationale and background. Am J Geriatr Psychiatry 2002; 10:129–141.
- Aronne LJ, Segal KR. Weight gain in the treatment of mood disorders. J Clin Psychiatry 2003; 64(suppl 8):22–29.
- Barkin RL, Barkin S. The role of venlafaxine and duloxetine in the treatment of depression with decremental changes in somatic symptoms of pain, chronic pain, and the pharmacokinetics and clinical considerations of duloxetine pharmacotherapy. Am J Ther 2005; 12:431–438.
- Wilkes S. Bupropion. Drugs Today (Barc) 2006; 42:671–681.
- Homsi J, Walsh D, Nelson KA, LeGrand S, Davis M. Methylphenidate for depression in hospice practice: a case series. Am J Hosp Palliat Care 2000; 17:393–398.
- Kallin K, Lundin-Olsson L, Jensen J, Nyberg L, Gustafson Y. Predisposing and precipitating factors for falls among older people in residential care. Public Health 2002; 116:263–271.
- Richards JB, Papaioannou A, Adachi JD, et al; Canadian Multicentre Osteoporosis Study Research Group. Effect of selective serotonin reuptake inhibitors on the risk of fracture. Arch Intern Med 2007; 167:188–194.
- Movig KL, Leufkens HG, Belitser SV, Lenderink AW, Egberts ACG. Selective serotonin reuptake inhibitor-induced urinary incontinence. Pharmacoepidemiol Drug Saf 2002; 11:271–279.
- Fick DM, Cooper JW, Wade WE, Waller JL, Maclean JR, Beers MH. Updating the Beers criteria for potentially inappropriate medication use in older adults: results of a US consensus panel of experts. Arch Intern Med 2003; 163:2716–2724.
- Leo RJ. Movement disorders associated with the serotonin selective reuptake inhibitors. J Clin Psychiatry 1996; 57:449–454.
- Barnhart WJ, Makela EH, Latocha MJ. SSRI-induced apathy syndrome: a clinical review. J Psychiatr Pract 2004; 10:196–199.
- Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA 2005; 293:596–608.
- American Psychiatric Association. Practice Guideline and Resources for Treatment of Patients with Alzheimer's Disease and Other Dementias, 2nd Edition. October 2007. www.psychiatryonline.com/pracGuide/pracGuideTopic_3.aspx. Accessed 2/2/2009.
- Lyketsos CG, Colenda CC, Beck C, et al; Task Force of American Association for Geriatric Psychiatry. Position Statement of the American Association for Geriatric Psychiatry regarding principles of care for patients with dementia resulting from Alzheimer disease. Am J Geriatr Psychiatry 2006; 14:561–572.
- Potter GG, Steffens DC. Contribution of depression to cognitive impairment and dementia in older adults. Neurologist 2007; 13:105– 117.
- Teri L, Gibbons LE, McCurry SM, et al. Exercise plus behavioral management in patients with Alzheimer disease: a randomized controlled trial. JAMA 2003; 290:2015–2022.
- McCurry SM, Gibbons LE, Logsdon RG, Vitiello MV, Teri L. Nighttime insomnia treatment and education for Alzheimer's disease: a randomized, controlled trial. J Am Geriatr Soc 2005; 53:793–802.
- Callahan CM, Boustani MA, Unverzagt FW, et al. Effectiveness of collaborative care for older adults with Alzheimer disease in primary care: a randomized controlled trial. JAMA 2006; 295:2148–2157.
- Van der Wurff FB, Stek ML, Hoogendijk WL, Beekman AT. Electroconvulsive therapy for the depressed elderly. Cochrane Database Syst Rev 2003; ( 2):CD003593.
- Tariot PN, Raman R, Jakimovich L, et al. Divalproex sodium in nursing home residents with possible or probable Alzheimer disease complicated by agitation: a randomized, controlled trial. Am J Geriatr Psychiatry 2005; 13:942–949.
- Kim E. The use of newer anticonvulsants in neuropsychiatric disorders. Curr Psychiatry Rep 2002; 4:331–337.
- Ness-Abramof R, Apovian CM. Drug-induced weight gain. Drugs Today (Barc) 2005; 41:547–555.
- Biton V. Weight change and antiepileptic drugs: health issues and criteria for appropriate selection of an antiepileptic agent. Neurologist 2006; 12:163–167.
- Tariot PN, Erb R, Podgorski CA, et al. Efficacy and tolerability of carbamazepine for agitation and aggression in dementia. Am J Psychiatry 1998; 155:54–61.
- Miller LJ. Gabapentin for treatment of behavioral and psychological symptoms of dementia. Ann Pharmacother 2001; 35:427–431.
- White JR, Walczak TS, Leppik IE, et al. Discontinuation of levetiracetam because of behavioral side effects: a case-control study. Neurology 2003; 61:1218–1221.
- Mega S, Masterman DM, O'Connor SM, Barclay TR, Cummings JL. The spectrum of behavioral responses to cholinesterase inhibitor therapy in Alzheimer disease. Arch Neurol 1999; 56:1388–1393.
- Birks J. Cholinesterase inhibitors for Alzheimer's disease. Cochrane Database of Syst Rev 2006; (1):CD005593.
- Rozzini L, Vicini Chilovi B, Bertoletti E, Trabucchi M, Padovani A. Acetyl-cholinesterase inhibitors and depressive symptoms in patients with mild to moderate Alzheimer's disease. Aging Clin Exp Res 2007; 19:220–223.
- McShane R, Areosa Sastre A, Minakaran N. Memantine for dementia. Cochrane Database Syst Rev 2006; (2):CD003154.
- Peskind ER, Tsuang DW, Bonner LT, et al. Propranolol for disruptive behaviors in nursing home residents with probable or possible Alzheimer disease: a placebo-controlled study. Alzheimer Dis Assoc Disord 2005; 19:23–28.
- Hall KA, Keks NA, O'Connor DW. Transdermal estrogen patches for aggressive behavior in male patients with dementia: a randomized, controlled trial. Int Psychogeriatr 2005; 17:165–178.
- Light SA, Holroyd S. The use of medroxyprogesterone acetate for the treatment of sexually inappropriate behaviour in patients with dementia. J Psychiatry Neurosci 2006; 31:132–134.
- Srinivasan S, Weinberg A. Pharmacologic treatment of sexual inappropriateness in long-term care residents with dementia. Ann Long-Term Care: Clin Care Aging 2006; 14:20–28.
- Glass J, Lanctot KL, Herrmann N, Sproule BA, Busto UE. Sedative hypnotics in older people with insomnia: meta-analysis of risks and benefits. BMJ 2005; 331:1169.
- Kirven LE, Montero EF. Comparison of thioridazine and diazepam in the control of nonpsychotic symptoms associated with senility: double-blind study. J Am Geriatr Soc 1973; 21:546–551.
- Cumming RG, Le Couteur DG. Benzodiazepines and risk of hip fractures in older people: a review of the evidence. CNS Drugs 2003; 17:825–837.
- Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 2003; 60:1119–1122.
- Holtzer R, Tang MX, Devanand DP, et al. Psychopathological features in Alzheimer's disease: course and relationship with cognitive status. J Am Geriatr Soc 2003; 51:953–960.
- Hart DJ, Craig D, Compton SA, et al. A retrospective study of the behavioural and psychological symptoms of mid and late phase Alzheimer's disease. Int J Geriatr Psychiatry 2003; 18:1037–1042.
- McKeith I, Cummings J. Behavioural changes and psychological symptoms in dementia disorders. Lancet Neurol 2005; 4:735–742.
- Stern Y, Albert M, Brandt J, et al. Utility of extrapyramidal signs and psychosis as predictors of cognitive and functional decline, nursing home admission, and death in Alzheimer's disease: prospective analyses from the Predictors Study. Neurology 1994; 44:2300–2307.
- Chibnall JT, Tait RC, Harman B, Luebbert RA. Effect of acetaminophen on behavior, well-being, and psychotropic medication use in nursing home residents with moderate-to-severe dementia. J Am Geriatr Soc 2005; 53:1921–1929.
- Doody RS, Stevens JC, Beck C, et al. Practice parameter: management of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2001; 56:1154–1166.
- Schneider LS, Dagerman K, Insel PS. Efficacy and adverse effects of atypical antipsychotics for dementia: meta-analysis of randomized, placebo-controlled trials. Am J Geriatr Psychiatry 2006; 14:191–210.
- Jeste DV, Dolder CR, Nayak GV, Salzman C. Atypical antipsychotics in elderly patients with dementia or schizophrenia: review of recent literature. Harv Rev Psychiatry 2005; 13:340–351.
- Rabinowitz J, Katz IR, De Deyn PP, Brodaty H, Greenspan A, Davidson M. Behavioral and psychological symptoms in patients with dementia as a target for pharmacotherapy with risperidone. J Clin Psychiatry 2004; 65:1329–1334.
- Schneider LS, Tariot PN, Dagerman KS, et al. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer's disease. N Engl J Med 2006; 355:1525–1538.
- Fernandez HH, Trieschmann ME, Burke MA, Friedman JH. Quetiapine for psychosis in Parkinson's disease versus dementia with Lewy bodies. J Clin Psychiatry 2002; 63:513–515.
- Cummings JL, Street J, Masterman D, Clark WS. Efficacy of olanzapine in the treatment of psychosis in dementia with Lewy bodies. Dement Geriatr Cogn Disord 2002; 13:67–73.
- US Food and Drug Administration. FDA Public Health Advisory—Deaths with Antipsychotics in Elderly Patients with Behavioral Disturbances: FDA/Center for Drug Evaluation and Research; April 11 2005.
- Lonergan E, Luxenberg J, Colford J. Haloperidol for agitation in dementia. Cochrane Database Syst Rev 2001; (4):CD002852.
- Wang PS, Schneeweiss S, Avorn J, et al. Risk of death in elderly users of conventional vs. atypical antipsychotic medications. N Engl J Med 2005; 353:2335–2341.
- Gill SS, Bronskill SE, Normand SL, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med 2007; 146:775–786.
- Alexopoulos GS, Streim J, Carpenter D, Docherty JP; Expert Consensus Panel for Using Antipsychotic Drugs in Older Patients. Using antipsychotic agents in older patients. J Clin Psychiatry 2004; 65(suppl 2):5–99.
- Starkstein SE, Mizrahi R. Depression in Alzheimer's disease. Expert Rev Neurother 2006; 6:887–895.
- Olin JT, Katz IR, Meyers BS, Schneider LS, Lebowitz BD. Provisional diagnostic criteria for depression of Alzheimer disease: rationale and background. Am J Geriatr Psychiatry 2002; 10:129–141.
- Aronne LJ, Segal KR. Weight gain in the treatment of mood disorders. J Clin Psychiatry 2003; 64(suppl 8):22–29.
- Barkin RL, Barkin S. The role of venlafaxine and duloxetine in the treatment of depression with decremental changes in somatic symptoms of pain, chronic pain, and the pharmacokinetics and clinical considerations of duloxetine pharmacotherapy. Am J Ther 2005; 12:431–438.
- Wilkes S. Bupropion. Drugs Today (Barc) 2006; 42:671–681.
- Homsi J, Walsh D, Nelson KA, LeGrand S, Davis M. Methylphenidate for depression in hospice practice: a case series. Am J Hosp Palliat Care 2000; 17:393–398.
- Kallin K, Lundin-Olsson L, Jensen J, Nyberg L, Gustafson Y. Predisposing and precipitating factors for falls among older people in residential care. Public Health 2002; 116:263–271.
- Richards JB, Papaioannou A, Adachi JD, et al; Canadian Multicentre Osteoporosis Study Research Group. Effect of selective serotonin reuptake inhibitors on the risk of fracture. Arch Intern Med 2007; 167:188–194.
- Movig KL, Leufkens HG, Belitser SV, Lenderink AW, Egberts ACG. Selective serotonin reuptake inhibitor-induced urinary incontinence. Pharmacoepidemiol Drug Saf 2002; 11:271–279.
- Fick DM, Cooper JW, Wade WE, Waller JL, Maclean JR, Beers MH. Updating the Beers criteria for potentially inappropriate medication use in older adults: results of a US consensus panel of experts. Arch Intern Med 2003; 163:2716–2724.
- Leo RJ. Movement disorders associated with the serotonin selective reuptake inhibitors. J Clin Psychiatry 1996; 57:449–454.
- Barnhart WJ, Makela EH, Latocha MJ. SSRI-induced apathy syndrome: a clinical review. J Psychiatr Pract 2004; 10:196–199.
- Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA 2005; 293:596–608.
- American Psychiatric Association. Practice Guideline and Resources for Treatment of Patients with Alzheimer's Disease and Other Dementias, 2nd Edition. October 2007. www.psychiatryonline.com/pracGuide/pracGuideTopic_3.aspx. Accessed 2/2/2009.
- Lyketsos CG, Colenda CC, Beck C, et al; Task Force of American Association for Geriatric Psychiatry. Position Statement of the American Association for Geriatric Psychiatry regarding principles of care for patients with dementia resulting from Alzheimer disease. Am J Geriatr Psychiatry 2006; 14:561–572.
- Potter GG, Steffens DC. Contribution of depression to cognitive impairment and dementia in older adults. Neurologist 2007; 13:105– 117.
- Teri L, Gibbons LE, McCurry SM, et al. Exercise plus behavioral management in patients with Alzheimer disease: a randomized controlled trial. JAMA 2003; 290:2015–2022.
- McCurry SM, Gibbons LE, Logsdon RG, Vitiello MV, Teri L. Nighttime insomnia treatment and education for Alzheimer's disease: a randomized, controlled trial. J Am Geriatr Soc 2005; 53:793–802.
- Callahan CM, Boustani MA, Unverzagt FW, et al. Effectiveness of collaborative care for older adults with Alzheimer disease in primary care: a randomized controlled trial. JAMA 2006; 295:2148–2157.
- Van der Wurff FB, Stek ML, Hoogendijk WL, Beekman AT. Electroconvulsive therapy for the depressed elderly. Cochrane Database Syst Rev 2003; ( 2):CD003593.
- Tariot PN, Raman R, Jakimovich L, et al. Divalproex sodium in nursing home residents with possible or probable Alzheimer disease complicated by agitation: a randomized, controlled trial. Am J Geriatr Psychiatry 2005; 13:942–949.
- Kim E. The use of newer anticonvulsants in neuropsychiatric disorders. Curr Psychiatry Rep 2002; 4:331–337.
- Ness-Abramof R, Apovian CM. Drug-induced weight gain. Drugs Today (Barc) 2005; 41:547–555.
- Biton V. Weight change and antiepileptic drugs: health issues and criteria for appropriate selection of an antiepileptic agent. Neurologist 2006; 12:163–167.
- Tariot PN, Erb R, Podgorski CA, et al. Efficacy and tolerability of carbamazepine for agitation and aggression in dementia. Am J Psychiatry 1998; 155:54–61.
- Miller LJ. Gabapentin for treatment of behavioral and psychological symptoms of dementia. Ann Pharmacother 2001; 35:427–431.
- White JR, Walczak TS, Leppik IE, et al. Discontinuation of levetiracetam because of behavioral side effects: a case-control study. Neurology 2003; 61:1218–1221.
- Mega S, Masterman DM, O'Connor SM, Barclay TR, Cummings JL. The spectrum of behavioral responses to cholinesterase inhibitor therapy in Alzheimer disease. Arch Neurol 1999; 56:1388–1393.
- Birks J. Cholinesterase inhibitors for Alzheimer's disease. Cochrane Database of Syst Rev 2006; (1):CD005593.
- Rozzini L, Vicini Chilovi B, Bertoletti E, Trabucchi M, Padovani A. Acetyl-cholinesterase inhibitors and depressive symptoms in patients with mild to moderate Alzheimer's disease. Aging Clin Exp Res 2007; 19:220–223.
- McShane R, Areosa Sastre A, Minakaran N. Memantine for dementia. Cochrane Database Syst Rev 2006; (2):CD003154.
- Peskind ER, Tsuang DW, Bonner LT, et al. Propranolol for disruptive behaviors in nursing home residents with probable or possible Alzheimer disease: a placebo-controlled study. Alzheimer Dis Assoc Disord 2005; 19:23–28.
- Hall KA, Keks NA, O'Connor DW. Transdermal estrogen patches for aggressive behavior in male patients with dementia: a randomized, controlled trial. Int Psychogeriatr 2005; 17:165–178.
- Light SA, Holroyd S. The use of medroxyprogesterone acetate for the treatment of sexually inappropriate behaviour in patients with dementia. J Psychiatry Neurosci 2006; 31:132–134.
- Srinivasan S, Weinberg A. Pharmacologic treatment of sexual inappropriateness in long-term care residents with dementia. Ann Long-Term Care: Clin Care Aging 2006; 14:20–28.
- Glass J, Lanctot KL, Herrmann N, Sproule BA, Busto UE. Sedative hypnotics in older people with insomnia: meta-analysis of risks and benefits. BMJ 2005; 331:1169.
- Kirven LE, Montero EF. Comparison of thioridazine and diazepam in the control of nonpsychotic symptoms associated with senility: double-blind study. J Am Geriatr Soc 1973; 21:546–551.
- Cumming RG, Le Couteur DG. Benzodiazepines and risk of hip fractures in older people: a review of the evidence. CNS Drugs 2003; 17:825–837.
KEY POINTS
- No drug specifically addresses wandering, hoarding, or resistance to care, behaviors that are particularly frustrating to caregivers.
- Many drugs are sedating and increase the risk of falling and injury; antipsychotic use is off-label for dementia and carries significant and possibly lethal adverse effects.
- Managing the behavioral symptoms of dementia requires attention to the environmental and psychosocial context in which they occur, as well as to comorbidities and potential adverse drug effects.
- Evidence for the efficacy of antidepressants for depression in dementia is limited.
Depression and heart disease: What do we know, and where are we headed?
Depression is a risk factor for heart disease, and in patients with heart disease, it is a risk factor for complications and death. Unfortunately, in the trials performed to date, treating depression in cardiac patients did not lead to lower rates of recurrent cardiovascular events or death. Nevertheless, we recommend that clinicians systematically screen for it in their heart patients, in view of the benefits of antidepressant therapy.
In this article we review key epidemiologic and psychosocial studies, the mechanistic links between depression and heart disease, and recent intervention trials. We also offer practical management advice and address the continued need for guidelines and risk stratification in the treatment of depressed cardiac patients.
After we submitted our review article, the American Heart Association (AHA)1 released a consensus document recommending that health care providers screen for and treat depression in patients with coronary heart disease. We will discuss the same screening tests that have been recommended by the AHA.
DEPRESSION AND HEART DISEASE: COMMON AND LINKED
Depression and heart disease are very common and often coexist: the prevalence of depression in various heart conditions ranges from 15% to 20%.1–3 According to data from the World Health Organization, by the year 2020 depression will be the second-leading cause of disability in developed countries (after heart disease).4
The World Health Survey5 showed that depression worsens health more than angina, arthritis, asthma, or diabetes. Furthermore, patients with severe mental illness have a higher risk of dying from heart disease and stroke.6
SOME HEART DISEASE RISK FACTORS ARE PSYCHOSOCIAL
In the 1980s, the “type A” personality (ambitious, aggressive, hostile, and competitive, with a chronic sense of urgency) was linked to heart disease.7 Later studies differed as to whether the entire set of features is valid as a collective risk factor for progressive heart disease,8 but hostility remains a validated risk factor and a focus of behavior modification.9,10
Other psychosocial risk factors have been implicated,11,12 one of which is social isolation.9,13 Another is the “type D” personality, which includes a tendency to experience negative emotions across time and situations coupled with social inhibition and which is believed to be more valid than the type A personality as a risk factor for cardiac disease.14,15
The INTERHEART study16 gathered data about attributable risk in the development of myocardial infarction (MI) in 52 countries in a case-control fashion. Psychosocial factors including stress, low generalized locus of control (ie, the perceived inability to control one’s life), and depression accounted for 32.5% of the attributable risk for an MI.17 This would mean that they account for slightly less attributable risk than that of lifetime smoking but more than that of hypertension and obesity.
Job stress increases the risk of initial coronary heart disease18 and also the risk of recurrent cardiac events after a first MI.19 Even though numerous psychosocial risk factors have been associated with coronary heart disease, including anxiety,20,21 depression is perhaps the best studied.
PROSPECTIVE STUDIES OF DEPRESSION AND HEART DISEASE
To examine the impact of depression in coronary heart disease, prospective studies have been done in healthy people and in patients with established cardiovascular disease who develop depression.22
In healthy people, depression increases the risk of coronary disease
The 1996 Epidemiologic Catchment Area study23 found that people with major depression had a risk of MI four times higher than the norm, and people with 2 weeks of sadness or dysphoria had a risk two times higher.
A subsequent meta-analysis of 11 studies,24 which included 36,000 patients, found that the overall relative risk of developing heart disease in depressed but healthy people was 1.64.
A meta-analysis by Van der Kooy et al25 of 28 epidemiologic studies with nearly 80,000 patients showed depression to be an independent risk factor for cardiovascular disease.
Wulsin and Singal26 performed a systematic review to see if depression increases the risk of coronary disease. In 10 studies with a follow-up of more than 4 years, the relative risk in people with depression was 1.64, which was less than that in active smokers (2.5) but more than that in passive smokers (1.25).
Depression can also exacerbate the classic risk factors for coronary disease, such as smoking, diabetes, obesity, and physical inactivity. 27
A 2007 study from Sweden28 prospectively followed patients who were hospitalized for depression. The odds ratio of developing an acute MI was 2.9, and the risk persisted for decades after the initial hospitalization.
A prospective United Kingdom cohort study of people initially free of heart disease revealed major depression to be associated with a higher rate of death from ischemic heart disease.29 Specifically, patients who had depression currently or in the past 12 months had a 2.7 times higher risk of dying than those who had never had depression or who had had it more than 12 months previously.
In existing heart disease, depression predicts recurrent events, death
Carney et el30 found that patients with major depressive disorder had a higher incidence of new cardiac events in the 12 months after undergoing cardiac catheterization than those without major depressive disorder.
Frasure-Smith et al,31 in a landmark study, showed that patients who were depressed at 1 week after an MI were three to four times more likely to die in the next 6 months than nondepressed post-MI patients. Even after 18 months, depression remained an independent risk factor for cardiac-related death.32
In longer studies (with up to 19.4 years of follow-up), depression was associated with higher rates of death from cardiac and all causes in patients with coronary artery disease.33 Lespérance et al34 found that in MI patients, the higher the Beck Depression Inventory score at the time of hospital admission, the higher the 5-year death rate.
Using meta-analysis, Barth et al35 found the risk of dying in the first 2 years after initial assessment to be twice as high in depressed cardiac patients as in nondepressed cardiac patients (odds ratio 2.24).
Van Melle et al36 reviewed 22 studies and found that in the 2 years after an MI, depressed patients had a 2 to 2.5 times higher risk of dying of a cardiac or any other cause than did nondepressed patients.
Depression also predicts higher morbidity and mortality rates in patients undergoing coronary artery bypass grafting,37,38 patients with congestive heart failure,39 and heart transplant recipients.40
MEDICAL ILLNESS CAN PREDISPOSE TO DEPRESSION, AND VICE VERSA
Medical illnesses can predispose a patient to develop depression. Specifically, compared with healthy people, cardiac patients appear to be at greater risk of developing depression for many years after the initial medical diagnosis is made.41
Katon et al42 reviewed 31 studies involving 16,922 patients, that assessed the impact of depression and anxiety in chronic medical illnesses such as heart disease, diabetes, pulmonary disease, and arthritis. After the severity of the medical disorder was controlled for, patients with depression and anxiety reported a higher number of medical symptoms.
DEPRESSION WORSENS QUALITY OF LIFE AND ADHERENCE TO TREATMENT
Depressed patients perceive their health status and quality of life negatively. In the Heart and Soul study,43 depressive symptoms and low exercise capacity—but not low ejection fraction or ischemia—were significantly associated with perceived deterioration of health in patients with coronary artery disease.
After an MI, patients who take their cardiac drugs properly have a better chance of survival.44,45 Clinical depression can worsen compliance with cardiac medication regimens,46 and reducing depression increases medication adherence overall.47 Not surprisingly, depressed patients also adhere less well to other recommendations,48 including modifying the diet, exercising, stopping smoking, and attending cardiac rehabilitation programs. 49
PLAUSIBLE MECHANISMS LINK DEPRESSION AND HEART DISEASE
Traditional cardiac risk factors such as smoking, high cholesterol, hypertension, diabetes, and obesity tend to cluster in depressed patients. 50 Other mechanisms linking depression and heart disease are reviewed below.51,52
Autonomic imbalance
Excessive sympathetic stimulation or diminished vagal stimulation or both are associated with higher rates of morbidity and death.53
Lack of variability in the heart rate reflects a sympathetic-vagal imbalance and is a risk factor for ventricular arrhythmias and sudden cardiac death in patients with cardiovascular disease.54 Carney et al55 reported that patients with coronary artery disease and depression had significantly less heart rate variability than nondepressed cardiac patients. Similarly, after an MI, depressed patients had significantly less heart rate variability than nondepressed patients,56 implying that low heart rate variability may mediate the adverse effect of depression on survival after an MI.57
In the Heart and Soul study, Gehi et al58 found no distinct relationship between heart rate variability and depression. However, in the same study, de Jong et al59 did find specific somatic symptoms of depression to be associated with lower heart rate variability, although cognitive symptoms were not.
Platelet activation, endothelial dysfunction
Depressed patients have been found to have exaggerated platelet reactivity.60 Plasma levels of platelet factor IV and beta-thromboglobulin, markers of platelet activation, are higher in depressed patients with ischemic heart disease than in nondepressed patients with ischemic heart disease and in control patients.61 This activation of platelets can lead to vascular damage and thrombosis.
In a subset study of the Sertraline Anti-Depressant Heart Attack Randomized Trial (SADHART), depressed MI patients were treated with sertraline (Zoloft), a selective serotonin reuptake inhibitor (SSRI), and had substantially less platelet and endothelial biomarker release.62
Depressed cardiac patients also have impaired flow-mediated dilation of the brachial artery, a sign of endothelial dysfunction.63 Although a recent study did not find coronary endothelial dysfunction in depressed patients who did not have cardiac disease, these patients had more clustering of other cardiac risk factors.64
Hypothalamic-pituitary-adrenocortical and sympathetic adrenal medullary activation
High cortisol levels can accelerate the development of hypertension and atherosclerosis and result in endothelial vascular injury. Sympathoadrenal activation in turn can lead to higher levels of catecholamines, predisposing to vasoconstriction, a rapid heart rate, and platelet activation. Depressed patients have more activation of the hypothalamic-pituitary-adrenocortical and sympathetic adrenal medullary systems,51,65 yet another plausible mechanism for worse clinical outcomes in depressed cardiac patients.
Sudden emotional stress can cause transient left ventricular dysfunction, even in people without coronary disease, an effect that may be mediated by elevated plasma catecholamine levels.66
Inflammatory cytokines
Inflammatory cytokines play a key role in the development of atherosclerosis.67 C-reactive protein, an acute-phase reactant produced in hepatocytes, can be induced by cytokines such as interleukin 6. Damage to endothelial tissues leads to the release of inflammatory cytokines, including interleukin 1, interleukin 6, and tumor tumor necrosis factor alpha.
Depressed patients have higher levels of these inflammatory markers.68,69 A prospective study reported direct correlations between depression scores and C-reactive protein levels in post-MI patients.70 The Heart and Soul study, however, did not confirm that coronary patients have more inflammation if they have depression,71 indicating that the relationship is complex and is perhaps more evident in specific types of depression.72
Anticholinergic inflammatory pathway
Tracey73 proposed a theory that vagal tone inhibits the release of inflammatory cytokines. This has important implications for treatment, as exercise, biofeedback, and meditation can stimulate the vagus nerve and therefore have beneficial anti-inflammatory effects.74
Polymorphism in the serotonin transport promoter region gene
Research is focusing on the serotonin transport promoter region gene (5-HTTLPR).75 The gene exists in two forms, a long one and a less-effective short one that appears to predispose to depression.76
Nakatani et al77 showed that MI patients were more likely to become depressed and to have subsequent cardiac events if one or both of their alleles of this gene were short. Otte et al,78 using Heart and Soul study data, found that patients with a short allele had a higher likelihood of depression, higher perceived levels of stress, and higher urinary norepinephrine secretion. However, the long allele genotype may be associated with a higher risk of developing an MI.79
Our knowledge of the genetic interplay of depression and cardiovascular disease is still in its infancy, and further studies are needed to clarify these findings.
IN TRIALS, LESS DEPRESSION BUT NO EFFECT ON DEATHS, RECURRENT MI
Major behavioral and drug trials conducted in the last 15 years have focused on how to best treat depression in cardiac patients.80–85
The Montreal Heart Attack Readjustment Trial (MHART)81 used telephone calls and home nursing visits to explore and monitor psychological distress for up to 1 year after an MI. The overall trial did not show these interventions to have any impact on survival compared with usual care. In fact, in women receiving the telephone intervention, there was a trend toward higher rates of cardiac and all-cause death, which was quite unexpected. Uncovering stresses and problems without resolving them, rather than encouraging patients to place these on the “back burner,” may partially explain these results.
SADHART82 studied the safety of sertraline in depressed post-MI patients. No major differences in cardiac function were noted between the sertraline and placebo groups, showing that sertraline was safe for these patients. The sertraline group had fewer cardiovascular events, but the difference was not statistically significant.
The Enhancing Recovery in Coronary Heart Disease (ENRICHD) study83 was primarily designed to see whether a psychosocial intervention would decrease deaths in depressed cardiac patients. Much to the chagrin of behavioral medicine, the group undergoing cognitive behavioral therapy did not have a higher rate of event-free survival, although the intervention had a favorable impact on depression and social support.
The Myocardial Infarction Depression Intervention Trial (MIND-IT)84 looked at whether the antidepressant mirtazapine (Remeron) would improve long-term depression and cardiovascular outcomes in depressed post-MI patients. In 18 months of follow-up, neither objective was obtained.
The Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial85 tested the efficacy of the SSRI citalopram (Celexa) and interpersonal therapy in a short-term intervention. Here, the antidepressant was superior to placebo in the primary outcome of treating depression, but interpersonal therapy had no advantage over “clinical management,” ie, a shorter, 20-minute supportive intervention.
Common threads in these studies.
- In ENRICHD and MIND-IT, patients whose depression did not respond to treatment were at higher risk of cardiac events during follow-up.86–88
- In SADHART and CREATE, which used drug treatment, the antidepressant response was more robust in patients with a history of depression before their heart attacks, suggesting that a patient with recurrent depression at the time of a cardiac event should receive medication for it.85,89
CLINICAL RECOMMENDATIONS
Use a depression screening tool
Ziegelstein et al90 recently studied the ability of clinical personnel to detect depression in hospitalized MI patients. If a screening tool was not used, the results were abysmal, indicating the need to use formal screening for symptoms of depression in acute MI patients.
Many self-rating scales are available, among which are the Beck Depression Inventory (BDI) and the Hospital Anxiety and Depression Scale (HADS). Others are:
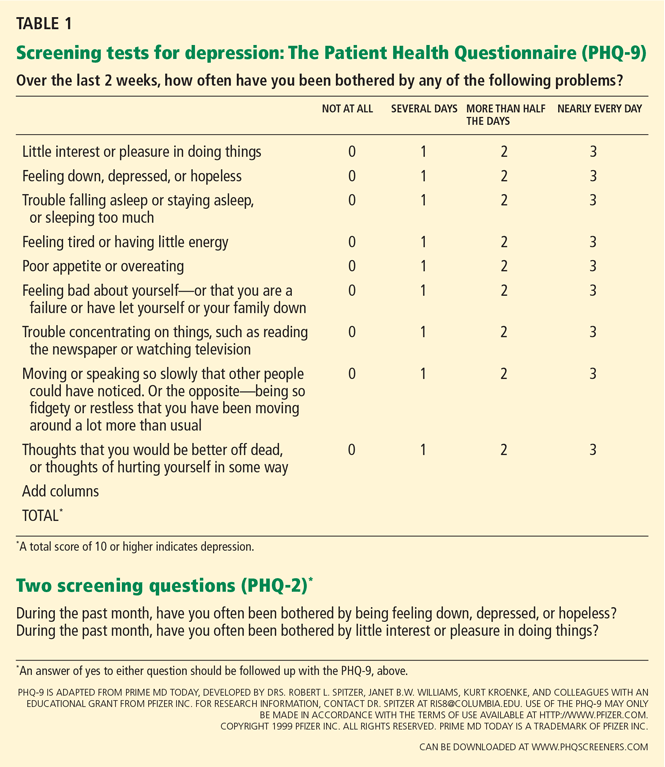
The PHQ-2 consists of the two first questions of the PHQ-9, which deal with mood and lack of pleasure. A cut-off score of 3 or higher has a sensitivity of 83% and a specificity of 92%,96 fulfilling the need for a quick and reliable depression screening tool. The clinician can also ask for a yes-or-no answer to the two questions of the PHQ-2 (Table 1). A yes to either of the two questions is up to 90% sensitive and 75% specific.92,97
When to suspect depression in cardiac patients

Which type of psychotherapy is best?
The negative results of psychosocial interventions (phone calls and home visits from a nurse) in MHART and of cognitive behavioral therapy in ENRICHD raise questions about which type of psychotherapy is best for depression in heart disease. CREATE found that 50-minute weekly sessions of interpersonal psychotherapy were no more beneficial than clinical management, ie, 20-minute weekly sessions that focused on compliance with treatment and education about depression and overall management. Perhaps a type of therapy akin to “clinical management” in this study or the brief behavior-based and targeted therapy used in the Improving Mood Promoting Access to Collaborative Care Treatment (IMPACT) trials of depression in primary care99 could be designed specifically to treat depression in cardiac disease. However, it is also quite possible that treatments that focus on uncovering stresses or problems may not be timely for these patients.
Which therapy is best for women is another area of consideration. In MHART, even after 5 years of follow-up,100 women who received the psychosocial support intervention did marginally worse. In the ENRICHD study, women did not experience a benefit from cognitive behavioral therapy. Further studies must address sex differences in response to different therapies.
SSRIs seem to be better than other antidepressants for cardiac patients
Before SSRIs were available, tricyclic antidepressants were the mainstays. Subsequent analysis showed the tricyclics to have an unfavorable risk-benefit profile in cardiac patients,101 and since other types of antidepressants are available, tricyclics should be avoided altogether in cardiac patients.102
Whether the SSRIs actually decrease one’s risk of death in heart disease is still an issue of debate, but there are encouraging signs. In SADHART, the rate of death and recurrent nonfatal MI was 20% lower in the patients randomized to receive sertraline, although the difference was not statistically significant.82 In ENRICHD, patients who did not respond to cognitive behavioral treatment or had severe depression could receive sertraline or other antidepressant drugs on a nonrandomized basis, and those who did had a 42% lower incidence of death or recurrent MI.103
The SADHART and CREATE trials provide convincing evidence of the cardiac safety and antidepressant efficacy of two SSRIs (sertraline and citalopram) in depressed cardiac patients. Mirtazapine, studied in MIND-IT, was not effective in treating depression in cardiac patients, although it had a better adverse effect and safety profile than tricyclic antidepressants. 104
Clinical observations indicate that SSRIs are associated with less risk of MI than non-SSRI drugs.105,106 During hospitalization for acute coronary syndromes, patients on SSRIs had lower rates of recurrent ischemia and heart failure but higher bleeding rates than patients not taking SSRIs.107 In a retrospective study of patients undergoing coronary artery bypass grafting, those on an SSRI before surgery had higher rates of death and rehospitalization.108 Being on antidepressant medication could be interpreted as a surrogate marker of having more severe depression before surgery; this issue clearly requires further study.
Given current observations and recent data from interventional trials coupled with the safe drug-interaction profile of sertraline and citalopram, these two SSRIs are recommended for treating depression in cardiac patients. If the patient is also receiving an anticoagulant, one should monitor for bleeding, as all SSRIs are associated with a prolonged bleeding time. Monitoring for rare cases of hyponatremia and bradycardia should also be part of early follow-up.
Do cardiac drugs have psychiatric effects?
Some concerns have arisen about cardiovascular drugs causing or aggravating psychiatric conditions.
Statins were once suspected of causing clinical depression or even suicide. However, subsequent studies have not substantiated this.109,110 In fact, long-term statin use has been associated with improved psychological wellbeing. 111 Whether the favorable psychological profile is due to an improved lifestyle, a direct noncholesterol effect, or an immunomodulatory effect has yet to be determined.
Beta-blockers have been suspected of increasing depression and fatigue. Robust metaanalyses have shown no increased risk of depressive symptoms but a small increased risk of fatigue and sexual dysfunction.112 Observational trials in the first year post-MI have shown no differences between beta-blocker users and nonusers in depressive symptoms or depressive disorders.113
Statins and beta-blockers offer both immense cardiac benefit and low risk, and both may be prescribed with confidence in depressed cardiac patients.
Refer patients for cardiac rehabilitation
The American Association of Cardiovascular and Pulmonary Rehabilitation strongly recommends screening cardiac patients for depression and referring them to cardiac rehabilitation programs.114 Typical programs run 12 weeks, affording an opportunity to further listen to and assess the patient and to promote general wellness via nutrition, stress management, and exercise.
These interventions by themselves can favorably affect depression. Blumenthal and colleagues,115 in the Standard Medical Intervention and Long-Term Exercise (SMILE) study, found that exercise was as effective as drug treatment in reducing depression. In addition, stress management as a psychosocial treatment in cardiac rehabilitation can reduce death rates in cardiac patients.116
Unfortunately, many patients who are eligible for cardiac rehabilitation programs do not avail themselves of them.117
Our algorithm

FUTURE DIRECTIONS FOR RESEARCH
Can we predict the course of depression?
We need to identify better which patients will have a spontaneous remission of their depressive symptoms after a cardiac event, which patients will linger with depression, and which patients will best respond to treatment. Risk stratification, using the psychiatric history, symptoms and severity of depression, and genetic predisposition118 might allow improved targeted therapies.
Does depression cause cardiac disease?
The link between depression and heart disease can be seen as merely an association. In the interventional trials performed to date, we have not yet seen a reduction in cardiac deaths when depression was treated, challenging any assumption of a causal relationship between depression and heart disease. The debate about association vs cause is germane to behavioral medicine,119 and the better we understand the mechanistic pathways, the better we can advise patients and treat depression comorbid with heart disease.
Behavioral medicine is currently measuring the aspects of depression associated with cardiac disease, including the spectrum of somatic (body) and affective (mood) symptoms120 and specific areas such as sympathetic arousal and early morning insomnia.121 If we can determine the depression subtype that carries a worse cardiac prognosis, we may untangle the biobehavioral links that bidirectionally bridge clinical depression and cardiac disease.
Another area of interest, emotional vitality (a positive state associated with interest, enthusiasm, excitement, and energy for living) has been shown to protect against coronary heart disease122 and holds much promise.
In the plenary lecture of the Academy of Psychosomatic Medicine in 2006, Frasure-Smith spoke of the “pleiotropism” of our antidepressant interventions on the various risk factors in depressed cardiac patients.123 We need behavioral medicine studies that elucidate these mechanisms, guiding more precise treatments as well as novel therapies. Omega-3 fatty acids, which benefit heart disease and clinical depression,124 will be used in a randomized controlled trial by Lespérance and colleagues.125 We await the results of this exciting research.
Will treating depression help in other types of heart disease?
The SADHART-CHF trial is examining whether 12 weeks of sertraline therapy is better than placebo in preventing death and improving cardiac outcomes in patients with chronic heart failure and comorbid major depressive disorder. It was to be completed in the fall of 2008. The results and experience of this study will help in designing future interventional trials to reduce the risk of depression in cardiovascular diseases.
We also await the results of a National Heart, Lung, and Blood Institute (NHLBI) trial, “Bypassing the Blues,” which is studying the treatment of depression after cardiac bypass surgery. This study should provide further insights into management of the depressed cardiac patient. Further prognostic studies in cardiac patients are also needed using the PHQ-9 and its shorter version, PHQ-2.
Current and future guidelines
For years our European colleagues have been ahead of us in recognizing depression screening and stress management as key to cardiac disease-prevention strategies.126 The NHLBI nicely outlined recommendations on the assessment and treatment of depression in cardiovascular patients.127 The just-published AHA Science Advisory should further encourage clinicians to screen and treat depression in the patient population.1 As our knowledge grows, we look forward to future evidence-based guidelines for depressed cardiac patients.
- Lichtman JH, Bigger JT, Blumenthal JA, et al. Depression and coronary heart disease. Recommendations for screening, referral, and treatment. Circulation 2008; 118:1768–1775.
- Jiang W, Glassman A, Krishnan R, O’Connor CM, Califf RM. Depression and ischemic heart disease: what have we learned so far and what must we do in the future? Am Heart J 2005; 150:54–78.
- Freedland KE, Rich MW, Skala JA, Carney RM, Davila-Roman VG, Jaffe AS. Prevalence of depression in hospitalized patients with congestive heart failure. Psychosom Med 2003; 65:119–128.
- Murray CJ, Lopez AD, Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet 1997; 349:1436–1442.
- Moussavi S, Chatterji S, Verdes E, Tandon A, Patel V, Ustun B. Depression, chronic diseases, and decrements in health: results from the World Health Surveys. Lancet 2007; 370:851–858.
- Osborn DP, Levy G, Nazareth I, Petersen I, Islam A, King MB. Relative risk of cardiovascular and cancer mortality in people with severe mental illness from the United Kingdom’s general practice research database. Arch Gen Psychiatry 2007; 64:242–249.
- Friedman M, Thoresen CE, Gill JJ, et al. Alteration of type A behavior and its effect on cardiac recurrences in post myocardial infarction patients: summary results of the recurrent coronary prevention project. Am Heart J 1986; 112:653–665.
- Ragland DR, Brand RJ. Type A behavior and mortality from coronary heart disease. N Engl J Med 1988; 318:65–69.
- Rozanski A, Blumenthal JA, Kaplan J. Impact of psychological factors on the pathogenesis of cardiovascular disease and implications for therapy. Circulation 1999; 99:2192–2217.
- Iribarren C, Sidney S, Bild DE, et al. Association of hostility with coronary artery calcification in young adults: the CARDIA study. Coronary Artery Risk Development in Young Adults. JAMA 2000; 283:2546–2551.
- Williams RB, Barefoot JC, Schneiderman N. Psychosocial risk factors for cardiovascular disease: more than one culprit at work. JAMA 2003; 290:2190–2192.
- Rozanski A, Blumenthal JA, Davidson KW, Saab PG, Kubzansky L. The epidemiology, pathophysiology, and management of psychosocial risk factors in cardiac practice: the emerging field of behavioral cardiology. J Am Coll Cardiol 2005; 45:637–651.
- Hemingway H, Marmot M. Evidence based cardiology: psychosocial factors in the aetiology and prognosis of coronary heart disease: systematic review of prospective cohort studies. BMJ 1999; 318:1460–1467.
- Denollet J. DS14: Standard assessment of negative affectivity, social inhibition, and type D personality. Psychosom Med 2005; 67:89–97.
- Steptoe A, Molloy GJ. Personality and heart disease. Heart 2007; 93:783–784.
- Yusuf S, Hawken S, Ôunpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet 2004; 364:937–952.
- Rosengren A, Hawken S, Ounpuu S, et al. Association of psychosocial risk factors with risk of acute myocardial infarction in 11119 cases and 13648 controls from 52 countries (the INTERHEART study): case-control study. Lancet 2004; 364:953–962.
- Kuper H, Marmot M. Job strain, job demands, decision latitude, and risk of coronary heart disease within the Whitehall II study. J Epidemiol Community Health 2003; 57:147–153.
- Aboa-Eboule C, Brisson C, Maunsell E, et al. Job strain and risk of acute recurrent coronary heart disease events. JAMA 2007; 298:1652–1660.
- Frasure-Smith N, Lespérance F. Depression and anxiety as predictors of 2-year cardiac events in patients with stable coronary artery disease. Arch Gen Psychiatry 2008; 65:62–71.
- Shen B-J, Avivi YE, Todaro JF, et al. Anxiety characteristics independently and prospectively predict myocardial infarction in men. J Am Coll Cardiol 2008; 51:113–119.
- Carney RM, Freedland KE. Depression, mortality, and medical morbidity in patients with coronary heart disease. Biol Psychiatry 2003; 54:241–247.
- Pratt LA, Ford DE, Crum RM, Armenian HK, Gallo JJ, Eaton WW. Depression, psychotropic medication, and risk of myocardial infarction: prospective data from the Baltimore ECA follow-up. Circulation 1996; 94:3123–3129.
- Rugulies R. Depression as a predictor for coronary heart disease. A review and meta-analysis. Am J Prev Med 2002; 23:51–61.
- Van der Kooy K, van Hout H, Marwijk H, Marten H, Stehouwer C, Beekman A. Depression and the risk for cardiovascular diseases: systematic review and meta analysis. Int J Geriatr Psychiatry 2007; 22:613–626.
- Wulsin LR, Singal BM. Do depressive symptoms increase the risk for the onset of coronary disease? A systematic quantitative review. Psychosom Med 2003; 65:201–210.
- Wulsin LR. Is depression a major risk factor for coronary disease? A systematic review of the epidemiologic evidence. Harv Rev Psychiatry 2004; 12:79–93.
- Janszky I, Ahlbom A, Hallqvist J, Ahnve S. Hospitalization for depression is associated with an increased risk for myocardial infarction not explained by lifestyle, lipids, coagulation, and inflammation: The SHEEP Study. Biol Psychiatry 2007; 62:25–32.
- Surtees PG, Wainwright NWJ, Luben RN, Wareham NJ, Bingham SA, Khaw K-T. Depression and ischemic heart disease mortality: evidence from the EPIC-Norfolk United Kingdom Prospective Cohort Study. Am J Psychiatry 2008; 165:515–523.
- Carney RM, Rich MW, Freedland KE, et al. Major depressive disorder predicts cardiac events in patients with coronary artery disease. Psychosom Med 1988; 50:627–633.
- Frasure-Smith N, Lespérance F, Talajic M. Depression following myocardial infarction. Impact on 6-month survival. JAMA 1993; 270:1819–1825.
- Frasure-Smith N, Lespérance F, Talajic M. Depression and 18-month prognosis after myocardial infarction. Circulation 1995; 91:999–1005.
- Barefoot JC, Helms MJ, Mark DB, et al. Depression and long-term mortality risk in patients with coronary artery disease. Am J Cardiol 1996; 78:613–617.
- Lespérance F, Frasure-Smith N, Talajic M, Bourassa MG. Five-year risk of cardiac mortality in relation to initial severity and one-year changes in depression symptoms after myocardial infarction. Circulation 2002; 105:1049–1053.
- Barth J, Schumacher M, Herrmann-Lingen C. Depression as a risk factor for mortality in patients with coronary heart disease: a meta-analysis. Psychosom Med 2004; 66:802–813.
- van Melle JP, de Jonge P, Spijkerman TA, et al. Prognostic association of depression following myocardial infarction with mortality and cardiovascular events: a meta-analysis. Psychosom Med 2004; 66:814–822.
- Blumenthal JA, Lett HS, Babyak MA, et al. Depression as a risk factor for mortality after coronary artery bypass surgery. Lancet 2003; 362:604–609.
- Sullivan MD, LaCroix AZ, Spertus JA, Hecht J, Russo J. Depression predicts revascularization procedures for 5 years after coronary angiography. Psychosom Med 2003; 65:229–236.
- Jiang W, Alexander J, Christopher E, et al. Relationship of depression to increased risk of mortality and rehospitalization in patients with congestive heart failure. Arch Intern Med 2001; 161:1849–1856.
- Zipfel S, Schneider A, Wild B, et al. Effect of depressive symptoms on survival after heart transplantation. Psychosom Med 2002; 64:740–747.
- Polsky D, Doshi JA, Marcus S, et al. Long-term risk for depressive symptoms after a medical diagnosis. Arch Intern Med 2005; 165:1260–1266.
- Katon W, Lin EHB, Kroenke K. The association of depression and anxiety with medical symptom burden in patients with chronic medical illness. Gen Hosp Psychiatry 2007; 29:147–155.
- Ruo B, Rumsfeld JS, Hlatky MA, Liu H, Browner WS, Whooley MA. Depressive symptoms and health-related quality of life: the Heart and Soul Study. JAMA 2003; 290:215–221.
- Rasmussen JN, Chong A, Alter DA. Relationship between adherence to evidence-based pharmacotherapy and long-term mortality after acute myocardial infarction. JAMA 2007; 297:177–186.
- Gehi AK, Ali S, Na B, Whooley MA. Self-reported medication adherence and cardiovascular events in patients with stable coronary heart disease: the Heart and Soul Study. Arch Intern Med 2007; 167:1798–1803.
- Gehi A, Haas D, Pipkin S, Whooley MA. Depression and medication adherence in outpatients with coronary heart disease: findings from the Heart and Soul Study. Arch Intern Med 2005; 165:2508–2513.
- Rieckmann N, Gerin W, Kronish IM, et al. Course of depressive symptoms and medication adherence after acute coronary syndromes: an electronic medication monitoring study. J Am Coll Cardiol 2006; 48:2218–2222.
- Ziegelstein RC, Fauerbach JA, Stevens SS, Romanelli J, Richter DP, Bush DE. Patients with depression are less likely to follow recommendations to reduce cardiac risk during recovery from a myocardial infarction. Arch Intern Med 2000; 160:1818–1823.
- Kronish IM, Rieckmann N, Halm EA, et al. Persistent depression affects adherence to secondary prevention behaviors after acute coronary syndromes. J Gen Intern Med 2006; 21:1178–1183.
- Joynt KE, Whellan DJ, O’Connor CM. Depression and cardiovascular disease: mechanisms of interaction. Biol Psychiatry 2003; 54:248–261.
- Musselman DL, Evans DL, Nemeroff CB. The relationship of depression to cardiovascular disease: epidemiology, biology, and treatment. Arch Gen Psychiatry 1998; 55:580–592.
- Lespérance F, Frasure-Smith N. Depression and heart disease. Cleve Clin J Med 2007; 74(suppl 1):S63–S66.
- Curtis BM. Autonomic tone as a cardiovascular risk factor: the dangers of chronic fight or flight. Mayo Clin Proc 2002; 77:45–54.
- Topol EJ. Textbook of Cardiovascular Medicine, 2nd ed. Philadelphia: Lippincott Williams & Williams 2002.
- Carney RM, Saunders RD, Freedland KE, Stein P, Rich MW, Jaffe AS. Association of depression with reduced heart rate variability in coronary artery disease. Am J Cardiol 1995; 76:562–564.
- Carney RM, Blumenthal JA, Stein PK, et al. Depression, heart rate variability, and acute myocardial infarction. Circulation 2001; 104:2024–2028.
- Carney RM, Blumenthal JA, Freedland KE, et al. Low heart rate variability and the effect of depression on post-myocardial infarction mortality. Arch Intern Med 2005; 165:1486–1491.
- Gehi A, Mangano D, Pipkin S, Browner WS, Whooley MA. Depression and heart rate variability in patients with stable coronary heart disease: findings from the Heart and Soul Study. Arch Gen Psychiatry 2005; 62:661–666.
- de Jonge P, Mangano D, Whooley MA. Differential association of cognitive and somatic depressive symptoms with heart rate variability in patients with stable coronary heart disease: findings from the Heart and Soul Study. Psychosom Med 2007; 69:735–739.
- Musselman DL, Tomer A, Manatunga AK, et al. Exaggerated platelet reactivity in major depression. Am J Psychiatry 1996; 153:1313–1317.
- Laghrissi-Thode F, Wagner WR, Pollock BG, Johnson PC, Finkel MS. Elevated platelet factor 4 and beta-thromboglobulin plasma levels in depressed patients with ischemic heart disease. Biol Psychiatry 1997; 42:290–295.
- Serebruany VL, Glassman AH, Malinin AI, et al. Platelet/endothelial biomarkers in depressed patients treated with the selective serotonin reuptake inhibitor sertraline after acute coronary events: the Sertraline AntiDepressant Heart Attack Randomized Trial (SADHART) Platelet Substudy. Circulation 2003; 108:939–944.
- Sherwood A, Hinderliter AL, Watkins LL, Waugh RA, Blumenthal JA. Impaired endothelial function in coronary heart disease patients with depressive symptomatology. J Am Coll Cardiol 2005; 46:656–659.
- Yang EH, Lerman S, Lennon RJ, Simari RD, Lerman LO, Lerman A. Relation of depression to coronary endothelial function. Am J Cardiol 2007; 99:1134–1136.
- Otte C, Neylan TC, Pipkin SS, Browner WS, Whooley MA. Depressive symptoms and 24-hour urinary norepinephrine excretion levels in patients with coronary disease: findings from the Heart and Soul Study. Am J Psychiatry 2005; 162:2139–2145.
- Wittstein IS, Thiemann DR, Lima JAC, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med 2005; 352:539–548.
- Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 2005; 352:1685–1695.
- Miller GE, Stetler CA, Carney RM, Freedland KE, Banks WA. Clinical depression and inflammatory risk markers for coronary heart disease. Am J Cardiol 2002; 90:1279–1283.
- Empana JP, Sykes DH, Luc G, et al. Contributions of depressive mood and circulating inflammatory markers to coronary heart disease in healthy European men: the Prospective Epidemiological Study of Myocardial Infarction (PRIME). Circulation 2005; 111:2299–2305.
- Frasure-Smith N, Lespérance F, Irwin MR, Sauvé C, Lespérance J, Théroux P. Depression, C-reactive protein and two-year major adverse cardiac events in men after acute coronary syndromes. Biol Psychiatry 2007; 62:302–308.
- Whooley MA, Caska CM, Hendrickson BE, Rourke MA, Ho J, Ali S. Depression and inflammation in patients with coronary heart disease: findings from the Heart and Soul Study. Biol Psychiatry 2007; 62:314–320.
- Glassman AH, Miller GE. Where there is depression, there is inflammation…sometimes! Biol Psychiatry 2007; 62:280–281.
- Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest 2007; 117:289–296.
- Tracey KJ. The inflammatory reflex. Nature 2002; 420:853–859.
- McCaffery JM, Frasure-Smith N, Dubé M-P, et al. Common genetic vulnerability to depressive symptoms and coronary artery disease: a review and development of candidate genes related to inflammation and serotonin. Psychosom Med 2006; 68:187–200.
- Caspi A, Sugden K, Moffitt TE, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 2003; 301:386–389.
- Nakatani D, Sato H, Sakata Y, et al. Influence of serotonin transporter gene polymorphism on depressive symptoms and new cardiac events after acute myocardial infarction. Am Heart J 2005; 150:652–658.
- Otte C, McCaffery J, Ali S, Whooley MA. Association of a serotonin transporter polymorphism (5-HTTLPR) with depression, perceived stress, and norepinephrine in patients with coronary disease: the Heart and Soul Study. Am J Psychiatry 2007; 164:1379–1384.
- Fumeron F, Betoulle D, Nicaud V, et al. Serotonin transporter gene polymorphism and myocardial infarction: Etude Cas-Témoins de l’Infarctus du Myocarde (ECTIM). Circulation 2002; 105:2943–2945.
- Rivelli S, Jiang W. Depression and ischemic heart disease: what have we learned from clinical trials? Curr Opin Cardiol 2007; 22:286–291.
- Frasure-Smith N, Lespérance F, Prince RH, et al. Randomised trial of home-based psychosocial nursing intervention for patients recovering from myocardial infarction. Lancet 1997; 350:473–479.
- Glassman AH, O’Connor CM, Califf RM, et al. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002; 288:701–709.
- Writing Committee for the EI. Effects of treating depression and low perceived social support on clinical events after myocardial infarction: the Enhancing Recovery in Coronary Heart Disease Patients (ENRICHD) randomized trial. JAMA 2003; 289:3106–3116.
- van Melle JP, de Jonge P, Honig A, et al. Effects of antidepressant treatment following myocardial infarction. Br J Psychiatry 2007; 190:460–466.
- Lespérance F, Frasure-Smith N, Koszycki D, et al. Effects of citalopram and interpersonal psychotherapy on depression in patients with coronary artery disease: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial. JAMA 2007; 297:367–379.
- Carney RM, Blumenthal JA, Freedland KE, et al. Depression and late mortality after myocardial infarction in the Enhancing Recovery in Coronary Heart Disease (ENRICHD) Study. Psychosom Med 2004; 66:466–474.
- de Jonge P, Honig A, van Melle JP, et al. Nonresponse to treatment for depression following myocardial infarction: association with subsequent cardiac events. Am J Psychiatry 2007; 164:1371–1378.
- Carney RM, Freedland KE. Depression and coronary heart disease: more pieces of the puzzle. Am J Psychiatry 2007; 164:1307–1309.
- Glassman AH, Bigger JT, Gaffney M, Shapiro PA, Swenson JR. Onset of major depression associated with acute coronary syndromes: relationship of onset, major depressive disorder history, and episode severity to sertraline benefit. Arch Gen Psychiatry 2006; 63:283–288.
- Ziegelstein RC, Kim SY, Kao D, et al. Can doctors and nurses recognize depression in patients hospitalized with an acute myocardial infarction in the absence of formal screening? Psychosom Med 2005; 67:393–397.
- Kroenke K, Spitzer RL, Williams JBW. The PHQ-9. Validity of a brief depression severity measure. J Gen Intern Med 2001; 16:606–613.
- Löwe B, Unutzer J, Callahan CM, Perkins AJ, Kroenke K. Monitoring depression treatment outcomes with the Patient Health Questionnaire-9. Med Care 2004; 42:1194–1201.
- Parashar S, Rumsfeld JS, Spertus JA, et al. Time course of depression and outcome of myocardial infarction. Arch Intern Med 2006; 166:2035–2043.
- Amin AA, Jones AMH, Nugent K, Rumsfeld JS, Spertus JA. The prevalence of unrecognized depression in patients with acute coronary syndrome. Am Heart J 2006; 152:928–934.
- McManus D, Pipkin SS, Whooley MA. Screening for depression in patients with coronary heart disease (data from the Heart and Soul Study). Am J Cardiol 2005; 96:1076–1081.
- Kroenke K, Spitzer RL, Williams JB. The Patient Health Questionnaire-2: validity of a two-item depression screener. Med Care 2003; 41:1284–1292.
- Spitzer RL, Williams JB, Kroenke K, et al. Utility of a new procedure for diagnosing mental disorders in primary care. The PRIME-MD 1000 study. JAMA 1994; 272:1749–1756.
- Lespérance F, Frasure-Smith N. Depression in patients with cardiac disease. J Psychosom Res 2000; 48:379–391.
- Hunkeler EM, Katon W, Tang L, et al. Long term outcomes from the IMPACT randomised trial for depressed elderly patients in primary care. BMJ 2006; 332:259–263.
- Frasure-Smith N, Lespérance F, Gravel G, Masson A, Juneau M, Bourassa MG. Long-term survival differences among low-anxious, high-anxious and repressive copers enrolled in the Montreal Heart Attack Readjustment Trial. Psychosom Med 2002; 64:571–579.
- Glassman AH, Roose SP, Bigger JT. The safety of tricyclic antidepressants in cardiac patients: risk-benefit reconsidered. JAMA 1993; 269:2673–2675.
- Cohen HW, Gibson G, Alderman MH. Excess risk of myocardial infarction in patients treated with antidepressant medications: association with use of tricyclic agents. Am J Med 2000; 108:2–8.
- Taylor CB, Youngblood ME, Catellier D, et al. Effects of antidepressant medication on morbidity and mortality in depressed patients after myocardial infarction. Arch Gen Psychiatry 2005; 62:792–798.
- Montgomery SA. Safety of mirtazapine: a review. Int Clin Psychopharmacol 1995; 10(suppl 4):37–45.
- Sauer WH, Berlin JA, Kimmel SE. Selective serotonin reuptake inhibitors and myocardial infarction. Circulation 2001; 104:1894–1898.
- Sauer WH, Berlin JA, Kimmel SE. Effect of antidepressants and their relative affinity for the serotonin transporter on the risk of myocardial infarction. Circulation 2003; 108:32–36.
- Ziegelstein RC, Meuchel J, Kim TJ, et al. Selective serotonin reuptake inhibitor use by patients with acute coronary syndromes. Am J Med 2007; 120:525–530.
- Xiong GL, Jiang W, Clare R, et al. Prognosis of patients taking selective serotonin reuptake inhibitors before coronary artery bypass grafting. Am J Cardiol 2006; 98:42–47.
- Yang C-C, Jick SS, Jick H. Lipid-lowering drugs and the risk of depression and suicidal behavior. Arch Intern Med 2003; 163:1926–1932.
- Callreus T, Agerskov Andersen U, Hallas J, Andersen M. Cardiovascular drugs and the risk of suicide: a nested case-control study. Eur J Clin Pharmacol 2007; 63:591–596.
- Young-Xu Y, Chan KA, Liao JK, Ravid S, Blatt CM. Long-term statin use and psychological well-being. J Am Coll Cardiol 2003; 42:690–697.
- Ko DT, Hebert PR, Coffey CS, Sedrakyan A, Curtis JP, Krumholz HM. Beta-blocker therapy and symptoms of depression, fatigue, and sexual dysfunction. JAMA 2002; 288:351–357.
- van Melle JP, Verbeek D, van den Berg MP, Ormel J, van der Line MR, de Jonge P. Beta-blockers and depression after myocardial infarction. J Am Coll Cardiol 2006; 48:2209–2214.
- Thomas RJ, King M, Lui K, et al. AACVPR/ACC/AHA 2007 performance measures on cardiac rehabilitation for referral to and delivery of cardiac rehabilitation/secondary prevention services. J Am Coll Cardiol 2007; 50:1400–1433.
- Blumenthal JA, Babyak MA, Doraiswamy PM, et al. Exercise and pharmacotherapy in the treatment of major depressive disorder. Psychosom Med 2007; 69:587–596.
- Linden W, Phillips MJ, Leclerc J. Psychological treatment of cardiac patients: a meta-analysis. Eur Heart J 2007; 28:2972–2984.
- Centers for Disease Control and Prevention. Receipt of outpatient cardiac rehabilitation among heart attack survivors—United States, 2005. JAMA 2008; 299:1534–1536.
- Williams RB. Treating depression after myocardial infarction: can selecting patients on the basis of genetic susceptibility improve psychiatric and medical outcomes? Am Heart J 2005; 150:617–619.
- Schneiderman N, Williams RB. The great debate editorial, revisited. Psychosom Med 2006; 68:636–638.
- de Jonge P, Ormel J, van den Brink RHS, et al. Symptom dimensions of depression following myocardial infarction and their relationship with somatic health status and cardiovascular prognosis. Am J Psychiatry 2006; 163:138–144.
- Fraguas R, Iosifescu DV, Alpert J, et al. Major depressive disorder and comorbid cardiac disease: is there a depressive subtype with greater cardiovascular morbidity? Results from the STAR*D Study. Psychosomatics 2007; 48:418–425.
- Kubzansky LD, Thurston RC. Emotional vitality and incident coronary heart disease: benefits of healthy psychological functioning. Arch Gen Psychiatry 2007; 64:1393–1401.
- Frasure-Smith N. Reflections on depression as a cardiac risk factor Academy of Psychosomatic Medicine, 53rd Annual Meeting, Tucson, Arizona, 2006.
- Frasure-Smith N, Lespérance F. Major depression is associated with lower omega-3 fatty acid levels in patients with recent acute coronary syndromes. Biol Psychiatry 2004; 55:891–896.
- Lespérance F. Annual Research Award Lecture Academy of Psychosomatic Medicine, Amelia Island, Florida, 2007.
- Graham I, Atar D, Borch-Johnsen K, et al. European guidelines on cardiovascular disease prevention in clinical practice: executive summary. Eur Heart J 2007; 28:2375–2414.
- Davidson KW, Kupfer DJ, Bigger JT, et al. Assessment and treatment of depression in patients with cardiovascular disease: National Heart, Lung, and Blood Institute Working Group Report. Psychosom Med 2006; 68:645–650.
Depression is a risk factor for heart disease, and in patients with heart disease, it is a risk factor for complications and death. Unfortunately, in the trials performed to date, treating depression in cardiac patients did not lead to lower rates of recurrent cardiovascular events or death. Nevertheless, we recommend that clinicians systematically screen for it in their heart patients, in view of the benefits of antidepressant therapy.
In this article we review key epidemiologic and psychosocial studies, the mechanistic links between depression and heart disease, and recent intervention trials. We also offer practical management advice and address the continued need for guidelines and risk stratification in the treatment of depressed cardiac patients.
After we submitted our review article, the American Heart Association (AHA)1 released a consensus document recommending that health care providers screen for and treat depression in patients with coronary heart disease. We will discuss the same screening tests that have been recommended by the AHA.
DEPRESSION AND HEART DISEASE: COMMON AND LINKED
Depression and heart disease are very common and often coexist: the prevalence of depression in various heart conditions ranges from 15% to 20%.1–3 According to data from the World Health Organization, by the year 2020 depression will be the second-leading cause of disability in developed countries (after heart disease).4
The World Health Survey5 showed that depression worsens health more than angina, arthritis, asthma, or diabetes. Furthermore, patients with severe mental illness have a higher risk of dying from heart disease and stroke.6
SOME HEART DISEASE RISK FACTORS ARE PSYCHOSOCIAL
In the 1980s, the “type A” personality (ambitious, aggressive, hostile, and competitive, with a chronic sense of urgency) was linked to heart disease.7 Later studies differed as to whether the entire set of features is valid as a collective risk factor for progressive heart disease,8 but hostility remains a validated risk factor and a focus of behavior modification.9,10
Other psychosocial risk factors have been implicated,11,12 one of which is social isolation.9,13 Another is the “type D” personality, which includes a tendency to experience negative emotions across time and situations coupled with social inhibition and which is believed to be more valid than the type A personality as a risk factor for cardiac disease.14,15
The INTERHEART study16 gathered data about attributable risk in the development of myocardial infarction (MI) in 52 countries in a case-control fashion. Psychosocial factors including stress, low generalized locus of control (ie, the perceived inability to control one’s life), and depression accounted for 32.5% of the attributable risk for an MI.17 This would mean that they account for slightly less attributable risk than that of lifetime smoking but more than that of hypertension and obesity.
Job stress increases the risk of initial coronary heart disease18 and also the risk of recurrent cardiac events after a first MI.19 Even though numerous psychosocial risk factors have been associated with coronary heart disease, including anxiety,20,21 depression is perhaps the best studied.
PROSPECTIVE STUDIES OF DEPRESSION AND HEART DISEASE
To examine the impact of depression in coronary heart disease, prospective studies have been done in healthy people and in patients with established cardiovascular disease who develop depression.22
In healthy people, depression increases the risk of coronary disease
The 1996 Epidemiologic Catchment Area study23 found that people with major depression had a risk of MI four times higher than the norm, and people with 2 weeks of sadness or dysphoria had a risk two times higher.
A subsequent meta-analysis of 11 studies,24 which included 36,000 patients, found that the overall relative risk of developing heart disease in depressed but healthy people was 1.64.
A meta-analysis by Van der Kooy et al25 of 28 epidemiologic studies with nearly 80,000 patients showed depression to be an independent risk factor for cardiovascular disease.
Wulsin and Singal26 performed a systematic review to see if depression increases the risk of coronary disease. In 10 studies with a follow-up of more than 4 years, the relative risk in people with depression was 1.64, which was less than that in active smokers (2.5) but more than that in passive smokers (1.25).
Depression can also exacerbate the classic risk factors for coronary disease, such as smoking, diabetes, obesity, and physical inactivity. 27
A 2007 study from Sweden28 prospectively followed patients who were hospitalized for depression. The odds ratio of developing an acute MI was 2.9, and the risk persisted for decades after the initial hospitalization.
A prospective United Kingdom cohort study of people initially free of heart disease revealed major depression to be associated with a higher rate of death from ischemic heart disease.29 Specifically, patients who had depression currently or in the past 12 months had a 2.7 times higher risk of dying than those who had never had depression or who had had it more than 12 months previously.
In existing heart disease, depression predicts recurrent events, death
Carney et el30 found that patients with major depressive disorder had a higher incidence of new cardiac events in the 12 months after undergoing cardiac catheterization than those without major depressive disorder.
Frasure-Smith et al,31 in a landmark study, showed that patients who were depressed at 1 week after an MI were three to four times more likely to die in the next 6 months than nondepressed post-MI patients. Even after 18 months, depression remained an independent risk factor for cardiac-related death.32
In longer studies (with up to 19.4 years of follow-up), depression was associated with higher rates of death from cardiac and all causes in patients with coronary artery disease.33 Lespérance et al34 found that in MI patients, the higher the Beck Depression Inventory score at the time of hospital admission, the higher the 5-year death rate.
Using meta-analysis, Barth et al35 found the risk of dying in the first 2 years after initial assessment to be twice as high in depressed cardiac patients as in nondepressed cardiac patients (odds ratio 2.24).
Van Melle et al36 reviewed 22 studies and found that in the 2 years after an MI, depressed patients had a 2 to 2.5 times higher risk of dying of a cardiac or any other cause than did nondepressed patients.
Depression also predicts higher morbidity and mortality rates in patients undergoing coronary artery bypass grafting,37,38 patients with congestive heart failure,39 and heart transplant recipients.40
MEDICAL ILLNESS CAN PREDISPOSE TO DEPRESSION, AND VICE VERSA
Medical illnesses can predispose a patient to develop depression. Specifically, compared with healthy people, cardiac patients appear to be at greater risk of developing depression for many years after the initial medical diagnosis is made.41
Katon et al42 reviewed 31 studies involving 16,922 patients, that assessed the impact of depression and anxiety in chronic medical illnesses such as heart disease, diabetes, pulmonary disease, and arthritis. After the severity of the medical disorder was controlled for, patients with depression and anxiety reported a higher number of medical symptoms.
DEPRESSION WORSENS QUALITY OF LIFE AND ADHERENCE TO TREATMENT
Depressed patients perceive their health status and quality of life negatively. In the Heart and Soul study,43 depressive symptoms and low exercise capacity—but not low ejection fraction or ischemia—were significantly associated with perceived deterioration of health in patients with coronary artery disease.
After an MI, patients who take their cardiac drugs properly have a better chance of survival.44,45 Clinical depression can worsen compliance with cardiac medication regimens,46 and reducing depression increases medication adherence overall.47 Not surprisingly, depressed patients also adhere less well to other recommendations,48 including modifying the diet, exercising, stopping smoking, and attending cardiac rehabilitation programs. 49
PLAUSIBLE MECHANISMS LINK DEPRESSION AND HEART DISEASE
Traditional cardiac risk factors such as smoking, high cholesterol, hypertension, diabetes, and obesity tend to cluster in depressed patients. 50 Other mechanisms linking depression and heart disease are reviewed below.51,52
Autonomic imbalance
Excessive sympathetic stimulation or diminished vagal stimulation or both are associated with higher rates of morbidity and death.53
Lack of variability in the heart rate reflects a sympathetic-vagal imbalance and is a risk factor for ventricular arrhythmias and sudden cardiac death in patients with cardiovascular disease.54 Carney et al55 reported that patients with coronary artery disease and depression had significantly less heart rate variability than nondepressed cardiac patients. Similarly, after an MI, depressed patients had significantly less heart rate variability than nondepressed patients,56 implying that low heart rate variability may mediate the adverse effect of depression on survival after an MI.57
In the Heart and Soul study, Gehi et al58 found no distinct relationship between heart rate variability and depression. However, in the same study, de Jong et al59 did find specific somatic symptoms of depression to be associated with lower heart rate variability, although cognitive symptoms were not.
Platelet activation, endothelial dysfunction
Depressed patients have been found to have exaggerated platelet reactivity.60 Plasma levels of platelet factor IV and beta-thromboglobulin, markers of platelet activation, are higher in depressed patients with ischemic heart disease than in nondepressed patients with ischemic heart disease and in control patients.61 This activation of platelets can lead to vascular damage and thrombosis.
In a subset study of the Sertraline Anti-Depressant Heart Attack Randomized Trial (SADHART), depressed MI patients were treated with sertraline (Zoloft), a selective serotonin reuptake inhibitor (SSRI), and had substantially less platelet and endothelial biomarker release.62
Depressed cardiac patients also have impaired flow-mediated dilation of the brachial artery, a sign of endothelial dysfunction.63 Although a recent study did not find coronary endothelial dysfunction in depressed patients who did not have cardiac disease, these patients had more clustering of other cardiac risk factors.64
Hypothalamic-pituitary-adrenocortical and sympathetic adrenal medullary activation
High cortisol levels can accelerate the development of hypertension and atherosclerosis and result in endothelial vascular injury. Sympathoadrenal activation in turn can lead to higher levels of catecholamines, predisposing to vasoconstriction, a rapid heart rate, and platelet activation. Depressed patients have more activation of the hypothalamic-pituitary-adrenocortical and sympathetic adrenal medullary systems,51,65 yet another plausible mechanism for worse clinical outcomes in depressed cardiac patients.
Sudden emotional stress can cause transient left ventricular dysfunction, even in people without coronary disease, an effect that may be mediated by elevated plasma catecholamine levels.66
Inflammatory cytokines
Inflammatory cytokines play a key role in the development of atherosclerosis.67 C-reactive protein, an acute-phase reactant produced in hepatocytes, can be induced by cytokines such as interleukin 6. Damage to endothelial tissues leads to the release of inflammatory cytokines, including interleukin 1, interleukin 6, and tumor tumor necrosis factor alpha.
Depressed patients have higher levels of these inflammatory markers.68,69 A prospective study reported direct correlations between depression scores and C-reactive protein levels in post-MI patients.70 The Heart and Soul study, however, did not confirm that coronary patients have more inflammation if they have depression,71 indicating that the relationship is complex and is perhaps more evident in specific types of depression.72
Anticholinergic inflammatory pathway
Tracey73 proposed a theory that vagal tone inhibits the release of inflammatory cytokines. This has important implications for treatment, as exercise, biofeedback, and meditation can stimulate the vagus nerve and therefore have beneficial anti-inflammatory effects.74
Polymorphism in the serotonin transport promoter region gene
Research is focusing on the serotonin transport promoter region gene (5-HTTLPR).75 The gene exists in two forms, a long one and a less-effective short one that appears to predispose to depression.76
Nakatani et al77 showed that MI patients were more likely to become depressed and to have subsequent cardiac events if one or both of their alleles of this gene were short. Otte et al,78 using Heart and Soul study data, found that patients with a short allele had a higher likelihood of depression, higher perceived levels of stress, and higher urinary norepinephrine secretion. However, the long allele genotype may be associated with a higher risk of developing an MI.79
Our knowledge of the genetic interplay of depression and cardiovascular disease is still in its infancy, and further studies are needed to clarify these findings.
IN TRIALS, LESS DEPRESSION BUT NO EFFECT ON DEATHS, RECURRENT MI
Major behavioral and drug trials conducted in the last 15 years have focused on how to best treat depression in cardiac patients.80–85
The Montreal Heart Attack Readjustment Trial (MHART)81 used telephone calls and home nursing visits to explore and monitor psychological distress for up to 1 year after an MI. The overall trial did not show these interventions to have any impact on survival compared with usual care. In fact, in women receiving the telephone intervention, there was a trend toward higher rates of cardiac and all-cause death, which was quite unexpected. Uncovering stresses and problems without resolving them, rather than encouraging patients to place these on the “back burner,” may partially explain these results.
SADHART82 studied the safety of sertraline in depressed post-MI patients. No major differences in cardiac function were noted between the sertraline and placebo groups, showing that sertraline was safe for these patients. The sertraline group had fewer cardiovascular events, but the difference was not statistically significant.
The Enhancing Recovery in Coronary Heart Disease (ENRICHD) study83 was primarily designed to see whether a psychosocial intervention would decrease deaths in depressed cardiac patients. Much to the chagrin of behavioral medicine, the group undergoing cognitive behavioral therapy did not have a higher rate of event-free survival, although the intervention had a favorable impact on depression and social support.
The Myocardial Infarction Depression Intervention Trial (MIND-IT)84 looked at whether the antidepressant mirtazapine (Remeron) would improve long-term depression and cardiovascular outcomes in depressed post-MI patients. In 18 months of follow-up, neither objective was obtained.
The Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial85 tested the efficacy of the SSRI citalopram (Celexa) and interpersonal therapy in a short-term intervention. Here, the antidepressant was superior to placebo in the primary outcome of treating depression, but interpersonal therapy had no advantage over “clinical management,” ie, a shorter, 20-minute supportive intervention.
Common threads in these studies.
- In ENRICHD and MIND-IT, patients whose depression did not respond to treatment were at higher risk of cardiac events during follow-up.86–88
- In SADHART and CREATE, which used drug treatment, the antidepressant response was more robust in patients with a history of depression before their heart attacks, suggesting that a patient with recurrent depression at the time of a cardiac event should receive medication for it.85,89
CLINICAL RECOMMENDATIONS
Use a depression screening tool
Ziegelstein et al90 recently studied the ability of clinical personnel to detect depression in hospitalized MI patients. If a screening tool was not used, the results were abysmal, indicating the need to use formal screening for symptoms of depression in acute MI patients.
Many self-rating scales are available, among which are the Beck Depression Inventory (BDI) and the Hospital Anxiety and Depression Scale (HADS). Others are:
The PHQ-2 consists of the two first questions of the PHQ-9, which deal with mood and lack of pleasure. A cut-off score of 3 or higher has a sensitivity of 83% and a specificity of 92%,96 fulfilling the need for a quick and reliable depression screening tool. The clinician can also ask for a yes-or-no answer to the two questions of the PHQ-2 (Table 1). A yes to either of the two questions is up to 90% sensitive and 75% specific.92,97
When to suspect depression in cardiac patients
Which type of psychotherapy is best?
The negative results of psychosocial interventions (phone calls and home visits from a nurse) in MHART and of cognitive behavioral therapy in ENRICHD raise questions about which type of psychotherapy is best for depression in heart disease. CREATE found that 50-minute weekly sessions of interpersonal psychotherapy were no more beneficial than clinical management, ie, 20-minute weekly sessions that focused on compliance with treatment and education about depression and overall management. Perhaps a type of therapy akin to “clinical management” in this study or the brief behavior-based and targeted therapy used in the Improving Mood Promoting Access to Collaborative Care Treatment (IMPACT) trials of depression in primary care99 could be designed specifically to treat depression in cardiac disease. However, it is also quite possible that treatments that focus on uncovering stresses or problems may not be timely for these patients.
Which therapy is best for women is another area of consideration. In MHART, even after 5 years of follow-up,100 women who received the psychosocial support intervention did marginally worse. In the ENRICHD study, women did not experience a benefit from cognitive behavioral therapy. Further studies must address sex differences in response to different therapies.
SSRIs seem to be better than other antidepressants for cardiac patients
Before SSRIs were available, tricyclic antidepressants were the mainstays. Subsequent analysis showed the tricyclics to have an unfavorable risk-benefit profile in cardiac patients,101 and since other types of antidepressants are available, tricyclics should be avoided altogether in cardiac patients.102
Whether the SSRIs actually decrease one’s risk of death in heart disease is still an issue of debate, but there are encouraging signs. In SADHART, the rate of death and recurrent nonfatal MI was 20% lower in the patients randomized to receive sertraline, although the difference was not statistically significant.82 In ENRICHD, patients who did not respond to cognitive behavioral treatment or had severe depression could receive sertraline or other antidepressant drugs on a nonrandomized basis, and those who did had a 42% lower incidence of death or recurrent MI.103
The SADHART and CREATE trials provide convincing evidence of the cardiac safety and antidepressant efficacy of two SSRIs (sertraline and citalopram) in depressed cardiac patients. Mirtazapine, studied in MIND-IT, was not effective in treating depression in cardiac patients, although it had a better adverse effect and safety profile than tricyclic antidepressants. 104
Clinical observations indicate that SSRIs are associated with less risk of MI than non-SSRI drugs.105,106 During hospitalization for acute coronary syndromes, patients on SSRIs had lower rates of recurrent ischemia and heart failure but higher bleeding rates than patients not taking SSRIs.107 In a retrospective study of patients undergoing coronary artery bypass grafting, those on an SSRI before surgery had higher rates of death and rehospitalization.108 Being on antidepressant medication could be interpreted as a surrogate marker of having more severe depression before surgery; this issue clearly requires further study.
Given current observations and recent data from interventional trials coupled with the safe drug-interaction profile of sertraline and citalopram, these two SSRIs are recommended for treating depression in cardiac patients. If the patient is also receiving an anticoagulant, one should monitor for bleeding, as all SSRIs are associated with a prolonged bleeding time. Monitoring for rare cases of hyponatremia and bradycardia should also be part of early follow-up.
Do cardiac drugs have psychiatric effects?
Some concerns have arisen about cardiovascular drugs causing or aggravating psychiatric conditions.
Statins were once suspected of causing clinical depression or even suicide. However, subsequent studies have not substantiated this.109,110 In fact, long-term statin use has been associated with improved psychological wellbeing. 111 Whether the favorable psychological profile is due to an improved lifestyle, a direct noncholesterol effect, or an immunomodulatory effect has yet to be determined.
Beta-blockers have been suspected of increasing depression and fatigue. Robust metaanalyses have shown no increased risk of depressive symptoms but a small increased risk of fatigue and sexual dysfunction.112 Observational trials in the first year post-MI have shown no differences between beta-blocker users and nonusers in depressive symptoms or depressive disorders.113
Statins and beta-blockers offer both immense cardiac benefit and low risk, and both may be prescribed with confidence in depressed cardiac patients.
Refer patients for cardiac rehabilitation
The American Association of Cardiovascular and Pulmonary Rehabilitation strongly recommends screening cardiac patients for depression and referring them to cardiac rehabilitation programs.114 Typical programs run 12 weeks, affording an opportunity to further listen to and assess the patient and to promote general wellness via nutrition, stress management, and exercise.
These interventions by themselves can favorably affect depression. Blumenthal and colleagues,115 in the Standard Medical Intervention and Long-Term Exercise (SMILE) study, found that exercise was as effective as drug treatment in reducing depression. In addition, stress management as a psychosocial treatment in cardiac rehabilitation can reduce death rates in cardiac patients.116
Unfortunately, many patients who are eligible for cardiac rehabilitation programs do not avail themselves of them.117
Our algorithm
FUTURE DIRECTIONS FOR RESEARCH
Can we predict the course of depression?
We need to identify better which patients will have a spontaneous remission of their depressive symptoms after a cardiac event, which patients will linger with depression, and which patients will best respond to treatment. Risk stratification, using the psychiatric history, symptoms and severity of depression, and genetic predisposition118 might allow improved targeted therapies.
Does depression cause cardiac disease?
The link between depression and heart disease can be seen as merely an association. In the interventional trials performed to date, we have not yet seen a reduction in cardiac deaths when depression was treated, challenging any assumption of a causal relationship between depression and heart disease. The debate about association vs cause is germane to behavioral medicine,119 and the better we understand the mechanistic pathways, the better we can advise patients and treat depression comorbid with heart disease.
Behavioral medicine is currently measuring the aspects of depression associated with cardiac disease, including the spectrum of somatic (body) and affective (mood) symptoms120 and specific areas such as sympathetic arousal and early morning insomnia.121 If we can determine the depression subtype that carries a worse cardiac prognosis, we may untangle the biobehavioral links that bidirectionally bridge clinical depression and cardiac disease.
Another area of interest, emotional vitality (a positive state associated with interest, enthusiasm, excitement, and energy for living) has been shown to protect against coronary heart disease122 and holds much promise.
In the plenary lecture of the Academy of Psychosomatic Medicine in 2006, Frasure-Smith spoke of the “pleiotropism” of our antidepressant interventions on the various risk factors in depressed cardiac patients.123 We need behavioral medicine studies that elucidate these mechanisms, guiding more precise treatments as well as novel therapies. Omega-3 fatty acids, which benefit heart disease and clinical depression,124 will be used in a randomized controlled trial by Lespérance and colleagues.125 We await the results of this exciting research.
Will treating depression help in other types of heart disease?
The SADHART-CHF trial is examining whether 12 weeks of sertraline therapy is better than placebo in preventing death and improving cardiac outcomes in patients with chronic heart failure and comorbid major depressive disorder. It was to be completed in the fall of 2008. The results and experience of this study will help in designing future interventional trials to reduce the risk of depression in cardiovascular diseases.
We also await the results of a National Heart, Lung, and Blood Institute (NHLBI) trial, “Bypassing the Blues,” which is studying the treatment of depression after cardiac bypass surgery. This study should provide further insights into management of the depressed cardiac patient. Further prognostic studies in cardiac patients are also needed using the PHQ-9 and its shorter version, PHQ-2.
Current and future guidelines
For years our European colleagues have been ahead of us in recognizing depression screening and stress management as key to cardiac disease-prevention strategies.126 The NHLBI nicely outlined recommendations on the assessment and treatment of depression in cardiovascular patients.127 The just-published AHA Science Advisory should further encourage clinicians to screen and treat depression in the patient population.1 As our knowledge grows, we look forward to future evidence-based guidelines for depressed cardiac patients.
Depression is a risk factor for heart disease, and in patients with heart disease, it is a risk factor for complications and death. Unfortunately, in the trials performed to date, treating depression in cardiac patients did not lead to lower rates of recurrent cardiovascular events or death. Nevertheless, we recommend that clinicians systematically screen for it in their heart patients, in view of the benefits of antidepressant therapy.
In this article we review key epidemiologic and psychosocial studies, the mechanistic links between depression and heart disease, and recent intervention trials. We also offer practical management advice and address the continued need for guidelines and risk stratification in the treatment of depressed cardiac patients.
After we submitted our review article, the American Heart Association (AHA)1 released a consensus document recommending that health care providers screen for and treat depression in patients with coronary heart disease. We will discuss the same screening tests that have been recommended by the AHA.
DEPRESSION AND HEART DISEASE: COMMON AND LINKED
Depression and heart disease are very common and often coexist: the prevalence of depression in various heart conditions ranges from 15% to 20%.1–3 According to data from the World Health Organization, by the year 2020 depression will be the second-leading cause of disability in developed countries (after heart disease).4
The World Health Survey5 showed that depression worsens health more than angina, arthritis, asthma, or diabetes. Furthermore, patients with severe mental illness have a higher risk of dying from heart disease and stroke.6
SOME HEART DISEASE RISK FACTORS ARE PSYCHOSOCIAL
In the 1980s, the “type A” personality (ambitious, aggressive, hostile, and competitive, with a chronic sense of urgency) was linked to heart disease.7 Later studies differed as to whether the entire set of features is valid as a collective risk factor for progressive heart disease,8 but hostility remains a validated risk factor and a focus of behavior modification.9,10
Other psychosocial risk factors have been implicated,11,12 one of which is social isolation.9,13 Another is the “type D” personality, which includes a tendency to experience negative emotions across time and situations coupled with social inhibition and which is believed to be more valid than the type A personality as a risk factor for cardiac disease.14,15
The INTERHEART study16 gathered data about attributable risk in the development of myocardial infarction (MI) in 52 countries in a case-control fashion. Psychosocial factors including stress, low generalized locus of control (ie, the perceived inability to control one’s life), and depression accounted for 32.5% of the attributable risk for an MI.17 This would mean that they account for slightly less attributable risk than that of lifetime smoking but more than that of hypertension and obesity.
Job stress increases the risk of initial coronary heart disease18 and also the risk of recurrent cardiac events after a first MI.19 Even though numerous psychosocial risk factors have been associated with coronary heart disease, including anxiety,20,21 depression is perhaps the best studied.
PROSPECTIVE STUDIES OF DEPRESSION AND HEART DISEASE
To examine the impact of depression in coronary heart disease, prospective studies have been done in healthy people and in patients with established cardiovascular disease who develop depression.22
In healthy people, depression increases the risk of coronary disease
The 1996 Epidemiologic Catchment Area study23 found that people with major depression had a risk of MI four times higher than the norm, and people with 2 weeks of sadness or dysphoria had a risk two times higher.
A subsequent meta-analysis of 11 studies,24 which included 36,000 patients, found that the overall relative risk of developing heart disease in depressed but healthy people was 1.64.
A meta-analysis by Van der Kooy et al25 of 28 epidemiologic studies with nearly 80,000 patients showed depression to be an independent risk factor for cardiovascular disease.
Wulsin and Singal26 performed a systematic review to see if depression increases the risk of coronary disease. In 10 studies with a follow-up of more than 4 years, the relative risk in people with depression was 1.64, which was less than that in active smokers (2.5) but more than that in passive smokers (1.25).
Depression can also exacerbate the classic risk factors for coronary disease, such as smoking, diabetes, obesity, and physical inactivity. 27
A 2007 study from Sweden28 prospectively followed patients who were hospitalized for depression. The odds ratio of developing an acute MI was 2.9, and the risk persisted for decades after the initial hospitalization.
A prospective United Kingdom cohort study of people initially free of heart disease revealed major depression to be associated with a higher rate of death from ischemic heart disease.29 Specifically, patients who had depression currently or in the past 12 months had a 2.7 times higher risk of dying than those who had never had depression or who had had it more than 12 months previously.
In existing heart disease, depression predicts recurrent events, death
Carney et el30 found that patients with major depressive disorder had a higher incidence of new cardiac events in the 12 months after undergoing cardiac catheterization than those without major depressive disorder.
Frasure-Smith et al,31 in a landmark study, showed that patients who were depressed at 1 week after an MI were three to four times more likely to die in the next 6 months than nondepressed post-MI patients. Even after 18 months, depression remained an independent risk factor for cardiac-related death.32
In longer studies (with up to 19.4 years of follow-up), depression was associated with higher rates of death from cardiac and all causes in patients with coronary artery disease.33 Lespérance et al34 found that in MI patients, the higher the Beck Depression Inventory score at the time of hospital admission, the higher the 5-year death rate.
Using meta-analysis, Barth et al35 found the risk of dying in the first 2 years after initial assessment to be twice as high in depressed cardiac patients as in nondepressed cardiac patients (odds ratio 2.24).
Van Melle et al36 reviewed 22 studies and found that in the 2 years after an MI, depressed patients had a 2 to 2.5 times higher risk of dying of a cardiac or any other cause than did nondepressed patients.
Depression also predicts higher morbidity and mortality rates in patients undergoing coronary artery bypass grafting,37,38 patients with congestive heart failure,39 and heart transplant recipients.40
MEDICAL ILLNESS CAN PREDISPOSE TO DEPRESSION, AND VICE VERSA
Medical illnesses can predispose a patient to develop depression. Specifically, compared with healthy people, cardiac patients appear to be at greater risk of developing depression for many years after the initial medical diagnosis is made.41
Katon et al42 reviewed 31 studies involving 16,922 patients, that assessed the impact of depression and anxiety in chronic medical illnesses such as heart disease, diabetes, pulmonary disease, and arthritis. After the severity of the medical disorder was controlled for, patients with depression and anxiety reported a higher number of medical symptoms.
DEPRESSION WORSENS QUALITY OF LIFE AND ADHERENCE TO TREATMENT
Depressed patients perceive their health status and quality of life negatively. In the Heart and Soul study,43 depressive symptoms and low exercise capacity—but not low ejection fraction or ischemia—were significantly associated with perceived deterioration of health in patients with coronary artery disease.
After an MI, patients who take their cardiac drugs properly have a better chance of survival.44,45 Clinical depression can worsen compliance with cardiac medication regimens,46 and reducing depression increases medication adherence overall.47 Not surprisingly, depressed patients also adhere less well to other recommendations,48 including modifying the diet, exercising, stopping smoking, and attending cardiac rehabilitation programs. 49
PLAUSIBLE MECHANISMS LINK DEPRESSION AND HEART DISEASE
Traditional cardiac risk factors such as smoking, high cholesterol, hypertension, diabetes, and obesity tend to cluster in depressed patients. 50 Other mechanisms linking depression and heart disease are reviewed below.51,52
Autonomic imbalance
Excessive sympathetic stimulation or diminished vagal stimulation or both are associated with higher rates of morbidity and death.53
Lack of variability in the heart rate reflects a sympathetic-vagal imbalance and is a risk factor for ventricular arrhythmias and sudden cardiac death in patients with cardiovascular disease.54 Carney et al55 reported that patients with coronary artery disease and depression had significantly less heart rate variability than nondepressed cardiac patients. Similarly, after an MI, depressed patients had significantly less heart rate variability than nondepressed patients,56 implying that low heart rate variability may mediate the adverse effect of depression on survival after an MI.57
In the Heart and Soul study, Gehi et al58 found no distinct relationship between heart rate variability and depression. However, in the same study, de Jong et al59 did find specific somatic symptoms of depression to be associated with lower heart rate variability, although cognitive symptoms were not.
Platelet activation, endothelial dysfunction
Depressed patients have been found to have exaggerated platelet reactivity.60 Plasma levels of platelet factor IV and beta-thromboglobulin, markers of platelet activation, are higher in depressed patients with ischemic heart disease than in nondepressed patients with ischemic heart disease and in control patients.61 This activation of platelets can lead to vascular damage and thrombosis.
In a subset study of the Sertraline Anti-Depressant Heart Attack Randomized Trial (SADHART), depressed MI patients were treated with sertraline (Zoloft), a selective serotonin reuptake inhibitor (SSRI), and had substantially less platelet and endothelial biomarker release.62
Depressed cardiac patients also have impaired flow-mediated dilation of the brachial artery, a sign of endothelial dysfunction.63 Although a recent study did not find coronary endothelial dysfunction in depressed patients who did not have cardiac disease, these patients had more clustering of other cardiac risk factors.64
Hypothalamic-pituitary-adrenocortical and sympathetic adrenal medullary activation
High cortisol levels can accelerate the development of hypertension and atherosclerosis and result in endothelial vascular injury. Sympathoadrenal activation in turn can lead to higher levels of catecholamines, predisposing to vasoconstriction, a rapid heart rate, and platelet activation. Depressed patients have more activation of the hypothalamic-pituitary-adrenocortical and sympathetic adrenal medullary systems,51,65 yet another plausible mechanism for worse clinical outcomes in depressed cardiac patients.
Sudden emotional stress can cause transient left ventricular dysfunction, even in people without coronary disease, an effect that may be mediated by elevated plasma catecholamine levels.66
Inflammatory cytokines
Inflammatory cytokines play a key role in the development of atherosclerosis.67 C-reactive protein, an acute-phase reactant produced in hepatocytes, can be induced by cytokines such as interleukin 6. Damage to endothelial tissues leads to the release of inflammatory cytokines, including interleukin 1, interleukin 6, and tumor tumor necrosis factor alpha.
Depressed patients have higher levels of these inflammatory markers.68,69 A prospective study reported direct correlations between depression scores and C-reactive protein levels in post-MI patients.70 The Heart and Soul study, however, did not confirm that coronary patients have more inflammation if they have depression,71 indicating that the relationship is complex and is perhaps more evident in specific types of depression.72
Anticholinergic inflammatory pathway
Tracey73 proposed a theory that vagal tone inhibits the release of inflammatory cytokines. This has important implications for treatment, as exercise, biofeedback, and meditation can stimulate the vagus nerve and therefore have beneficial anti-inflammatory effects.74
Polymorphism in the serotonin transport promoter region gene
Research is focusing on the serotonin transport promoter region gene (5-HTTLPR).75 The gene exists in two forms, a long one and a less-effective short one that appears to predispose to depression.76
Nakatani et al77 showed that MI patients were more likely to become depressed and to have subsequent cardiac events if one or both of their alleles of this gene were short. Otte et al,78 using Heart and Soul study data, found that patients with a short allele had a higher likelihood of depression, higher perceived levels of stress, and higher urinary norepinephrine secretion. However, the long allele genotype may be associated with a higher risk of developing an MI.79
Our knowledge of the genetic interplay of depression and cardiovascular disease is still in its infancy, and further studies are needed to clarify these findings.
IN TRIALS, LESS DEPRESSION BUT NO EFFECT ON DEATHS, RECURRENT MI
Major behavioral and drug trials conducted in the last 15 years have focused on how to best treat depression in cardiac patients.80–85
The Montreal Heart Attack Readjustment Trial (MHART)81 used telephone calls and home nursing visits to explore and monitor psychological distress for up to 1 year after an MI. The overall trial did not show these interventions to have any impact on survival compared with usual care. In fact, in women receiving the telephone intervention, there was a trend toward higher rates of cardiac and all-cause death, which was quite unexpected. Uncovering stresses and problems without resolving them, rather than encouraging patients to place these on the “back burner,” may partially explain these results.
SADHART82 studied the safety of sertraline in depressed post-MI patients. No major differences in cardiac function were noted between the sertraline and placebo groups, showing that sertraline was safe for these patients. The sertraline group had fewer cardiovascular events, but the difference was not statistically significant.
The Enhancing Recovery in Coronary Heart Disease (ENRICHD) study83 was primarily designed to see whether a psychosocial intervention would decrease deaths in depressed cardiac patients. Much to the chagrin of behavioral medicine, the group undergoing cognitive behavioral therapy did not have a higher rate of event-free survival, although the intervention had a favorable impact on depression and social support.
The Myocardial Infarction Depression Intervention Trial (MIND-IT)84 looked at whether the antidepressant mirtazapine (Remeron) would improve long-term depression and cardiovascular outcomes in depressed post-MI patients. In 18 months of follow-up, neither objective was obtained.
The Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial85 tested the efficacy of the SSRI citalopram (Celexa) and interpersonal therapy in a short-term intervention. Here, the antidepressant was superior to placebo in the primary outcome of treating depression, but interpersonal therapy had no advantage over “clinical management,” ie, a shorter, 20-minute supportive intervention.
Common threads in these studies.
- In ENRICHD and MIND-IT, patients whose depression did not respond to treatment were at higher risk of cardiac events during follow-up.86–88
- In SADHART and CREATE, which used drug treatment, the antidepressant response was more robust in patients with a history of depression before their heart attacks, suggesting that a patient with recurrent depression at the time of a cardiac event should receive medication for it.85,89
CLINICAL RECOMMENDATIONS
Use a depression screening tool
Ziegelstein et al90 recently studied the ability of clinical personnel to detect depression in hospitalized MI patients. If a screening tool was not used, the results were abysmal, indicating the need to use formal screening for symptoms of depression in acute MI patients.
Many self-rating scales are available, among which are the Beck Depression Inventory (BDI) and the Hospital Anxiety and Depression Scale (HADS). Others are:
The PHQ-2 consists of the two first questions of the PHQ-9, which deal with mood and lack of pleasure. A cut-off score of 3 or higher has a sensitivity of 83% and a specificity of 92%,96 fulfilling the need for a quick and reliable depression screening tool. The clinician can also ask for a yes-or-no answer to the two questions of the PHQ-2 (Table 1). A yes to either of the two questions is up to 90% sensitive and 75% specific.92,97
When to suspect depression in cardiac patients
Which type of psychotherapy is best?
The negative results of psychosocial interventions (phone calls and home visits from a nurse) in MHART and of cognitive behavioral therapy in ENRICHD raise questions about which type of psychotherapy is best for depression in heart disease. CREATE found that 50-minute weekly sessions of interpersonal psychotherapy were no more beneficial than clinical management, ie, 20-minute weekly sessions that focused on compliance with treatment and education about depression and overall management. Perhaps a type of therapy akin to “clinical management” in this study or the brief behavior-based and targeted therapy used in the Improving Mood Promoting Access to Collaborative Care Treatment (IMPACT) trials of depression in primary care99 could be designed specifically to treat depression in cardiac disease. However, it is also quite possible that treatments that focus on uncovering stresses or problems may not be timely for these patients.
Which therapy is best for women is another area of consideration. In MHART, even after 5 years of follow-up,100 women who received the psychosocial support intervention did marginally worse. In the ENRICHD study, women did not experience a benefit from cognitive behavioral therapy. Further studies must address sex differences in response to different therapies.
SSRIs seem to be better than other antidepressants for cardiac patients
Before SSRIs were available, tricyclic antidepressants were the mainstays. Subsequent analysis showed the tricyclics to have an unfavorable risk-benefit profile in cardiac patients,101 and since other types of antidepressants are available, tricyclics should be avoided altogether in cardiac patients.102
Whether the SSRIs actually decrease one’s risk of death in heart disease is still an issue of debate, but there are encouraging signs. In SADHART, the rate of death and recurrent nonfatal MI was 20% lower in the patients randomized to receive sertraline, although the difference was not statistically significant.82 In ENRICHD, patients who did not respond to cognitive behavioral treatment or had severe depression could receive sertraline or other antidepressant drugs on a nonrandomized basis, and those who did had a 42% lower incidence of death or recurrent MI.103
The SADHART and CREATE trials provide convincing evidence of the cardiac safety and antidepressant efficacy of two SSRIs (sertraline and citalopram) in depressed cardiac patients. Mirtazapine, studied in MIND-IT, was not effective in treating depression in cardiac patients, although it had a better adverse effect and safety profile than tricyclic antidepressants. 104
Clinical observations indicate that SSRIs are associated with less risk of MI than non-SSRI drugs.105,106 During hospitalization for acute coronary syndromes, patients on SSRIs had lower rates of recurrent ischemia and heart failure but higher bleeding rates than patients not taking SSRIs.107 In a retrospective study of patients undergoing coronary artery bypass grafting, those on an SSRI before surgery had higher rates of death and rehospitalization.108 Being on antidepressant medication could be interpreted as a surrogate marker of having more severe depression before surgery; this issue clearly requires further study.
Given current observations and recent data from interventional trials coupled with the safe drug-interaction profile of sertraline and citalopram, these two SSRIs are recommended for treating depression in cardiac patients. If the patient is also receiving an anticoagulant, one should monitor for bleeding, as all SSRIs are associated with a prolonged bleeding time. Monitoring for rare cases of hyponatremia and bradycardia should also be part of early follow-up.
Do cardiac drugs have psychiatric effects?
Some concerns have arisen about cardiovascular drugs causing or aggravating psychiatric conditions.
Statins were once suspected of causing clinical depression or even suicide. However, subsequent studies have not substantiated this.109,110 In fact, long-term statin use has been associated with improved psychological wellbeing. 111 Whether the favorable psychological profile is due to an improved lifestyle, a direct noncholesterol effect, or an immunomodulatory effect has yet to be determined.
Beta-blockers have been suspected of increasing depression and fatigue. Robust metaanalyses have shown no increased risk of depressive symptoms but a small increased risk of fatigue and sexual dysfunction.112 Observational trials in the first year post-MI have shown no differences between beta-blocker users and nonusers in depressive symptoms or depressive disorders.113
Statins and beta-blockers offer both immense cardiac benefit and low risk, and both may be prescribed with confidence in depressed cardiac patients.
Refer patients for cardiac rehabilitation
The American Association of Cardiovascular and Pulmonary Rehabilitation strongly recommends screening cardiac patients for depression and referring them to cardiac rehabilitation programs.114 Typical programs run 12 weeks, affording an opportunity to further listen to and assess the patient and to promote general wellness via nutrition, stress management, and exercise.
These interventions by themselves can favorably affect depression. Blumenthal and colleagues,115 in the Standard Medical Intervention and Long-Term Exercise (SMILE) study, found that exercise was as effective as drug treatment in reducing depression. In addition, stress management as a psychosocial treatment in cardiac rehabilitation can reduce death rates in cardiac patients.116
Unfortunately, many patients who are eligible for cardiac rehabilitation programs do not avail themselves of them.117
Our algorithm
FUTURE DIRECTIONS FOR RESEARCH
Can we predict the course of depression?
We need to identify better which patients will have a spontaneous remission of their depressive symptoms after a cardiac event, which patients will linger with depression, and which patients will best respond to treatment. Risk stratification, using the psychiatric history, symptoms and severity of depression, and genetic predisposition118 might allow improved targeted therapies.
Does depression cause cardiac disease?
The link between depression and heart disease can be seen as merely an association. In the interventional trials performed to date, we have not yet seen a reduction in cardiac deaths when depression was treated, challenging any assumption of a causal relationship between depression and heart disease. The debate about association vs cause is germane to behavioral medicine,119 and the better we understand the mechanistic pathways, the better we can advise patients and treat depression comorbid with heart disease.
Behavioral medicine is currently measuring the aspects of depression associated with cardiac disease, including the spectrum of somatic (body) and affective (mood) symptoms120 and specific areas such as sympathetic arousal and early morning insomnia.121 If we can determine the depression subtype that carries a worse cardiac prognosis, we may untangle the biobehavioral links that bidirectionally bridge clinical depression and cardiac disease.
Another area of interest, emotional vitality (a positive state associated with interest, enthusiasm, excitement, and energy for living) has been shown to protect against coronary heart disease122 and holds much promise.
In the plenary lecture of the Academy of Psychosomatic Medicine in 2006, Frasure-Smith spoke of the “pleiotropism” of our antidepressant interventions on the various risk factors in depressed cardiac patients.123 We need behavioral medicine studies that elucidate these mechanisms, guiding more precise treatments as well as novel therapies. Omega-3 fatty acids, which benefit heart disease and clinical depression,124 will be used in a randomized controlled trial by Lespérance and colleagues.125 We await the results of this exciting research.
Will treating depression help in other types of heart disease?
The SADHART-CHF trial is examining whether 12 weeks of sertraline therapy is better than placebo in preventing death and improving cardiac outcomes in patients with chronic heart failure and comorbid major depressive disorder. It was to be completed in the fall of 2008. The results and experience of this study will help in designing future interventional trials to reduce the risk of depression in cardiovascular diseases.
We also await the results of a National Heart, Lung, and Blood Institute (NHLBI) trial, “Bypassing the Blues,” which is studying the treatment of depression after cardiac bypass surgery. This study should provide further insights into management of the depressed cardiac patient. Further prognostic studies in cardiac patients are also needed using the PHQ-9 and its shorter version, PHQ-2.
Current and future guidelines
For years our European colleagues have been ahead of us in recognizing depression screening and stress management as key to cardiac disease-prevention strategies.126 The NHLBI nicely outlined recommendations on the assessment and treatment of depression in cardiovascular patients.127 The just-published AHA Science Advisory should further encourage clinicians to screen and treat depression in the patient population.1 As our knowledge grows, we look forward to future evidence-based guidelines for depressed cardiac patients.
- Lichtman JH, Bigger JT, Blumenthal JA, et al. Depression and coronary heart disease. Recommendations for screening, referral, and treatment. Circulation 2008; 118:1768–1775.
- Jiang W, Glassman A, Krishnan R, O’Connor CM, Califf RM. Depression and ischemic heart disease: what have we learned so far and what must we do in the future? Am Heart J 2005; 150:54–78.
- Freedland KE, Rich MW, Skala JA, Carney RM, Davila-Roman VG, Jaffe AS. Prevalence of depression in hospitalized patients with congestive heart failure. Psychosom Med 2003; 65:119–128.
- Murray CJ, Lopez AD, Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet 1997; 349:1436–1442.
- Moussavi S, Chatterji S, Verdes E, Tandon A, Patel V, Ustun B. Depression, chronic diseases, and decrements in health: results from the World Health Surveys. Lancet 2007; 370:851–858.
- Osborn DP, Levy G, Nazareth I, Petersen I, Islam A, King MB. Relative risk of cardiovascular and cancer mortality in people with severe mental illness from the United Kingdom’s general practice research database. Arch Gen Psychiatry 2007; 64:242–249.
- Friedman M, Thoresen CE, Gill JJ, et al. Alteration of type A behavior and its effect on cardiac recurrences in post myocardial infarction patients: summary results of the recurrent coronary prevention project. Am Heart J 1986; 112:653–665.
- Ragland DR, Brand RJ. Type A behavior and mortality from coronary heart disease. N Engl J Med 1988; 318:65–69.
- Rozanski A, Blumenthal JA, Kaplan J. Impact of psychological factors on the pathogenesis of cardiovascular disease and implications for therapy. Circulation 1999; 99:2192–2217.
- Iribarren C, Sidney S, Bild DE, et al. Association of hostility with coronary artery calcification in young adults: the CARDIA study. Coronary Artery Risk Development in Young Adults. JAMA 2000; 283:2546–2551.
- Williams RB, Barefoot JC, Schneiderman N. Psychosocial risk factors for cardiovascular disease: more than one culprit at work. JAMA 2003; 290:2190–2192.
- Rozanski A, Blumenthal JA, Davidson KW, Saab PG, Kubzansky L. The epidemiology, pathophysiology, and management of psychosocial risk factors in cardiac practice: the emerging field of behavioral cardiology. J Am Coll Cardiol 2005; 45:637–651.
- Hemingway H, Marmot M. Evidence based cardiology: psychosocial factors in the aetiology and prognosis of coronary heart disease: systematic review of prospective cohort studies. BMJ 1999; 318:1460–1467.
- Denollet J. DS14: Standard assessment of negative affectivity, social inhibition, and type D personality. Psychosom Med 2005; 67:89–97.
- Steptoe A, Molloy GJ. Personality and heart disease. Heart 2007; 93:783–784.
- Yusuf S, Hawken S, Ôunpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet 2004; 364:937–952.
- Rosengren A, Hawken S, Ounpuu S, et al. Association of psychosocial risk factors with risk of acute myocardial infarction in 11119 cases and 13648 controls from 52 countries (the INTERHEART study): case-control study. Lancet 2004; 364:953–962.
- Kuper H, Marmot M. Job strain, job demands, decision latitude, and risk of coronary heart disease within the Whitehall II study. J Epidemiol Community Health 2003; 57:147–153.
- Aboa-Eboule C, Brisson C, Maunsell E, et al. Job strain and risk of acute recurrent coronary heart disease events. JAMA 2007; 298:1652–1660.
- Frasure-Smith N, Lespérance F. Depression and anxiety as predictors of 2-year cardiac events in patients with stable coronary artery disease. Arch Gen Psychiatry 2008; 65:62–71.
- Shen B-J, Avivi YE, Todaro JF, et al. Anxiety characteristics independently and prospectively predict myocardial infarction in men. J Am Coll Cardiol 2008; 51:113–119.
- Carney RM, Freedland KE. Depression, mortality, and medical morbidity in patients with coronary heart disease. Biol Psychiatry 2003; 54:241–247.
- Pratt LA, Ford DE, Crum RM, Armenian HK, Gallo JJ, Eaton WW. Depression, psychotropic medication, and risk of myocardial infarction: prospective data from the Baltimore ECA follow-up. Circulation 1996; 94:3123–3129.
- Rugulies R. Depression as a predictor for coronary heart disease. A review and meta-analysis. Am J Prev Med 2002; 23:51–61.
- Van der Kooy K, van Hout H, Marwijk H, Marten H, Stehouwer C, Beekman A. Depression and the risk for cardiovascular diseases: systematic review and meta analysis. Int J Geriatr Psychiatry 2007; 22:613–626.
- Wulsin LR, Singal BM. Do depressive symptoms increase the risk for the onset of coronary disease? A systematic quantitative review. Psychosom Med 2003; 65:201–210.
- Wulsin LR. Is depression a major risk factor for coronary disease? A systematic review of the epidemiologic evidence. Harv Rev Psychiatry 2004; 12:79–93.
- Janszky I, Ahlbom A, Hallqvist J, Ahnve S. Hospitalization for depression is associated with an increased risk for myocardial infarction not explained by lifestyle, lipids, coagulation, and inflammation: The SHEEP Study. Biol Psychiatry 2007; 62:25–32.
- Surtees PG, Wainwright NWJ, Luben RN, Wareham NJ, Bingham SA, Khaw K-T. Depression and ischemic heart disease mortality: evidence from the EPIC-Norfolk United Kingdom Prospective Cohort Study. Am J Psychiatry 2008; 165:515–523.
- Carney RM, Rich MW, Freedland KE, et al. Major depressive disorder predicts cardiac events in patients with coronary artery disease. Psychosom Med 1988; 50:627–633.
- Frasure-Smith N, Lespérance F, Talajic M. Depression following myocardial infarction. Impact on 6-month survival. JAMA 1993; 270:1819–1825.
- Frasure-Smith N, Lespérance F, Talajic M. Depression and 18-month prognosis after myocardial infarction. Circulation 1995; 91:999–1005.
- Barefoot JC, Helms MJ, Mark DB, et al. Depression and long-term mortality risk in patients with coronary artery disease. Am J Cardiol 1996; 78:613–617.
- Lespérance F, Frasure-Smith N, Talajic M, Bourassa MG. Five-year risk of cardiac mortality in relation to initial severity and one-year changes in depression symptoms after myocardial infarction. Circulation 2002; 105:1049–1053.
- Barth J, Schumacher M, Herrmann-Lingen C. Depression as a risk factor for mortality in patients with coronary heart disease: a meta-analysis. Psychosom Med 2004; 66:802–813.
- van Melle JP, de Jonge P, Spijkerman TA, et al. Prognostic association of depression following myocardial infarction with mortality and cardiovascular events: a meta-analysis. Psychosom Med 2004; 66:814–822.
- Blumenthal JA, Lett HS, Babyak MA, et al. Depression as a risk factor for mortality after coronary artery bypass surgery. Lancet 2003; 362:604–609.
- Sullivan MD, LaCroix AZ, Spertus JA, Hecht J, Russo J. Depression predicts revascularization procedures for 5 years after coronary angiography. Psychosom Med 2003; 65:229–236.
- Jiang W, Alexander J, Christopher E, et al. Relationship of depression to increased risk of mortality and rehospitalization in patients with congestive heart failure. Arch Intern Med 2001; 161:1849–1856.
- Zipfel S, Schneider A, Wild B, et al. Effect of depressive symptoms on survival after heart transplantation. Psychosom Med 2002; 64:740–747.
- Polsky D, Doshi JA, Marcus S, et al. Long-term risk for depressive symptoms after a medical diagnosis. Arch Intern Med 2005; 165:1260–1266.
- Katon W, Lin EHB, Kroenke K. The association of depression and anxiety with medical symptom burden in patients with chronic medical illness. Gen Hosp Psychiatry 2007; 29:147–155.
- Ruo B, Rumsfeld JS, Hlatky MA, Liu H, Browner WS, Whooley MA. Depressive symptoms and health-related quality of life: the Heart and Soul Study. JAMA 2003; 290:215–221.
- Rasmussen JN, Chong A, Alter DA. Relationship between adherence to evidence-based pharmacotherapy and long-term mortality after acute myocardial infarction. JAMA 2007; 297:177–186.
- Gehi AK, Ali S, Na B, Whooley MA. Self-reported medication adherence and cardiovascular events in patients with stable coronary heart disease: the Heart and Soul Study. Arch Intern Med 2007; 167:1798–1803.
- Gehi A, Haas D, Pipkin S, Whooley MA. Depression and medication adherence in outpatients with coronary heart disease: findings from the Heart and Soul Study. Arch Intern Med 2005; 165:2508–2513.
- Rieckmann N, Gerin W, Kronish IM, et al. Course of depressive symptoms and medication adherence after acute coronary syndromes: an electronic medication monitoring study. J Am Coll Cardiol 2006; 48:2218–2222.
- Ziegelstein RC, Fauerbach JA, Stevens SS, Romanelli J, Richter DP, Bush DE. Patients with depression are less likely to follow recommendations to reduce cardiac risk during recovery from a myocardial infarction. Arch Intern Med 2000; 160:1818–1823.
- Kronish IM, Rieckmann N, Halm EA, et al. Persistent depression affects adherence to secondary prevention behaviors after acute coronary syndromes. J Gen Intern Med 2006; 21:1178–1183.
- Joynt KE, Whellan DJ, O’Connor CM. Depression and cardiovascular disease: mechanisms of interaction. Biol Psychiatry 2003; 54:248–261.
- Musselman DL, Evans DL, Nemeroff CB. The relationship of depression to cardiovascular disease: epidemiology, biology, and treatment. Arch Gen Psychiatry 1998; 55:580–592.
- Lespérance F, Frasure-Smith N. Depression and heart disease. Cleve Clin J Med 2007; 74(suppl 1):S63–S66.
- Curtis BM. Autonomic tone as a cardiovascular risk factor: the dangers of chronic fight or flight. Mayo Clin Proc 2002; 77:45–54.
- Topol EJ. Textbook of Cardiovascular Medicine, 2nd ed. Philadelphia: Lippincott Williams & Williams 2002.
- Carney RM, Saunders RD, Freedland KE, Stein P, Rich MW, Jaffe AS. Association of depression with reduced heart rate variability in coronary artery disease. Am J Cardiol 1995; 76:562–564.
- Carney RM, Blumenthal JA, Stein PK, et al. Depression, heart rate variability, and acute myocardial infarction. Circulation 2001; 104:2024–2028.
- Carney RM, Blumenthal JA, Freedland KE, et al. Low heart rate variability and the effect of depression on post-myocardial infarction mortality. Arch Intern Med 2005; 165:1486–1491.
- Gehi A, Mangano D, Pipkin S, Browner WS, Whooley MA. Depression and heart rate variability in patients with stable coronary heart disease: findings from the Heart and Soul Study. Arch Gen Psychiatry 2005; 62:661–666.
- de Jonge P, Mangano D, Whooley MA. Differential association of cognitive and somatic depressive symptoms with heart rate variability in patients with stable coronary heart disease: findings from the Heart and Soul Study. Psychosom Med 2007; 69:735–739.
- Musselman DL, Tomer A, Manatunga AK, et al. Exaggerated platelet reactivity in major depression. Am J Psychiatry 1996; 153:1313–1317.
- Laghrissi-Thode F, Wagner WR, Pollock BG, Johnson PC, Finkel MS. Elevated platelet factor 4 and beta-thromboglobulin plasma levels in depressed patients with ischemic heart disease. Biol Psychiatry 1997; 42:290–295.
- Serebruany VL, Glassman AH, Malinin AI, et al. Platelet/endothelial biomarkers in depressed patients treated with the selective serotonin reuptake inhibitor sertraline after acute coronary events: the Sertraline AntiDepressant Heart Attack Randomized Trial (SADHART) Platelet Substudy. Circulation 2003; 108:939–944.
- Sherwood A, Hinderliter AL, Watkins LL, Waugh RA, Blumenthal JA. Impaired endothelial function in coronary heart disease patients with depressive symptomatology. J Am Coll Cardiol 2005; 46:656–659.
- Yang EH, Lerman S, Lennon RJ, Simari RD, Lerman LO, Lerman A. Relation of depression to coronary endothelial function. Am J Cardiol 2007; 99:1134–1136.
- Otte C, Neylan TC, Pipkin SS, Browner WS, Whooley MA. Depressive symptoms and 24-hour urinary norepinephrine excretion levels in patients with coronary disease: findings from the Heart and Soul Study. Am J Psychiatry 2005; 162:2139–2145.
- Wittstein IS, Thiemann DR, Lima JAC, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med 2005; 352:539–548.
- Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 2005; 352:1685–1695.
- Miller GE, Stetler CA, Carney RM, Freedland KE, Banks WA. Clinical depression and inflammatory risk markers for coronary heart disease. Am J Cardiol 2002; 90:1279–1283.
- Empana JP, Sykes DH, Luc G, et al. Contributions of depressive mood and circulating inflammatory markers to coronary heart disease in healthy European men: the Prospective Epidemiological Study of Myocardial Infarction (PRIME). Circulation 2005; 111:2299–2305.
- Frasure-Smith N, Lespérance F, Irwin MR, Sauvé C, Lespérance J, Théroux P. Depression, C-reactive protein and two-year major adverse cardiac events in men after acute coronary syndromes. Biol Psychiatry 2007; 62:302–308.
- Whooley MA, Caska CM, Hendrickson BE, Rourke MA, Ho J, Ali S. Depression and inflammation in patients with coronary heart disease: findings from the Heart and Soul Study. Biol Psychiatry 2007; 62:314–320.
- Glassman AH, Miller GE. Where there is depression, there is inflammation…sometimes! Biol Psychiatry 2007; 62:280–281.
- Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest 2007; 117:289–296.
- Tracey KJ. The inflammatory reflex. Nature 2002; 420:853–859.
- McCaffery JM, Frasure-Smith N, Dubé M-P, et al. Common genetic vulnerability to depressive symptoms and coronary artery disease: a review and development of candidate genes related to inflammation and serotonin. Psychosom Med 2006; 68:187–200.
- Caspi A, Sugden K, Moffitt TE, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 2003; 301:386–389.
- Nakatani D, Sato H, Sakata Y, et al. Influence of serotonin transporter gene polymorphism on depressive symptoms and new cardiac events after acute myocardial infarction. Am Heart J 2005; 150:652–658.
- Otte C, McCaffery J, Ali S, Whooley MA. Association of a serotonin transporter polymorphism (5-HTTLPR) with depression, perceived stress, and norepinephrine in patients with coronary disease: the Heart and Soul Study. Am J Psychiatry 2007; 164:1379–1384.
- Fumeron F, Betoulle D, Nicaud V, et al. Serotonin transporter gene polymorphism and myocardial infarction: Etude Cas-Témoins de l’Infarctus du Myocarde (ECTIM). Circulation 2002; 105:2943–2945.
- Rivelli S, Jiang W. Depression and ischemic heart disease: what have we learned from clinical trials? Curr Opin Cardiol 2007; 22:286–291.
- Frasure-Smith N, Lespérance F, Prince RH, et al. Randomised trial of home-based psychosocial nursing intervention for patients recovering from myocardial infarction. Lancet 1997; 350:473–479.
- Glassman AH, O’Connor CM, Califf RM, et al. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002; 288:701–709.
- Writing Committee for the EI. Effects of treating depression and low perceived social support on clinical events after myocardial infarction: the Enhancing Recovery in Coronary Heart Disease Patients (ENRICHD) randomized trial. JAMA 2003; 289:3106–3116.
- van Melle JP, de Jonge P, Honig A, et al. Effects of antidepressant treatment following myocardial infarction. Br J Psychiatry 2007; 190:460–466.
- Lespérance F, Frasure-Smith N, Koszycki D, et al. Effects of citalopram and interpersonal psychotherapy on depression in patients with coronary artery disease: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial. JAMA 2007; 297:367–379.
- Carney RM, Blumenthal JA, Freedland KE, et al. Depression and late mortality after myocardial infarction in the Enhancing Recovery in Coronary Heart Disease (ENRICHD) Study. Psychosom Med 2004; 66:466–474.
- de Jonge P, Honig A, van Melle JP, et al. Nonresponse to treatment for depression following myocardial infarction: association with subsequent cardiac events. Am J Psychiatry 2007; 164:1371–1378.
- Carney RM, Freedland KE. Depression and coronary heart disease: more pieces of the puzzle. Am J Psychiatry 2007; 164:1307–1309.
- Glassman AH, Bigger JT, Gaffney M, Shapiro PA, Swenson JR. Onset of major depression associated with acute coronary syndromes: relationship of onset, major depressive disorder history, and episode severity to sertraline benefit. Arch Gen Psychiatry 2006; 63:283–288.
- Ziegelstein RC, Kim SY, Kao D, et al. Can doctors and nurses recognize depression in patients hospitalized with an acute myocardial infarction in the absence of formal screening? Psychosom Med 2005; 67:393–397.
- Kroenke K, Spitzer RL, Williams JBW. The PHQ-9. Validity of a brief depression severity measure. J Gen Intern Med 2001; 16:606–613.
- Löwe B, Unutzer J, Callahan CM, Perkins AJ, Kroenke K. Monitoring depression treatment outcomes with the Patient Health Questionnaire-9. Med Care 2004; 42:1194–1201.
- Parashar S, Rumsfeld JS, Spertus JA, et al. Time course of depression and outcome of myocardial infarction. Arch Intern Med 2006; 166:2035–2043.
- Amin AA, Jones AMH, Nugent K, Rumsfeld JS, Spertus JA. The prevalence of unrecognized depression in patients with acute coronary syndrome. Am Heart J 2006; 152:928–934.
- McManus D, Pipkin SS, Whooley MA. Screening for depression in patients with coronary heart disease (data from the Heart and Soul Study). Am J Cardiol 2005; 96:1076–1081.
- Kroenke K, Spitzer RL, Williams JB. The Patient Health Questionnaire-2: validity of a two-item depression screener. Med Care 2003; 41:1284–1292.
- Spitzer RL, Williams JB, Kroenke K, et al. Utility of a new procedure for diagnosing mental disorders in primary care. The PRIME-MD 1000 study. JAMA 1994; 272:1749–1756.
- Lespérance F, Frasure-Smith N. Depression in patients with cardiac disease. J Psychosom Res 2000; 48:379–391.
- Hunkeler EM, Katon W, Tang L, et al. Long term outcomes from the IMPACT randomised trial for depressed elderly patients in primary care. BMJ 2006; 332:259–263.
- Frasure-Smith N, Lespérance F, Gravel G, Masson A, Juneau M, Bourassa MG. Long-term survival differences among low-anxious, high-anxious and repressive copers enrolled in the Montreal Heart Attack Readjustment Trial. Psychosom Med 2002; 64:571–579.
- Glassman AH, Roose SP, Bigger JT. The safety of tricyclic antidepressants in cardiac patients: risk-benefit reconsidered. JAMA 1993; 269:2673–2675.
- Cohen HW, Gibson G, Alderman MH. Excess risk of myocardial infarction in patients treated with antidepressant medications: association with use of tricyclic agents. Am J Med 2000; 108:2–8.
- Taylor CB, Youngblood ME, Catellier D, et al. Effects of antidepressant medication on morbidity and mortality in depressed patients after myocardial infarction. Arch Gen Psychiatry 2005; 62:792–798.
- Montgomery SA. Safety of mirtazapine: a review. Int Clin Psychopharmacol 1995; 10(suppl 4):37–45.
- Sauer WH, Berlin JA, Kimmel SE. Selective serotonin reuptake inhibitors and myocardial infarction. Circulation 2001; 104:1894–1898.
- Sauer WH, Berlin JA, Kimmel SE. Effect of antidepressants and their relative affinity for the serotonin transporter on the risk of myocardial infarction. Circulation 2003; 108:32–36.
- Ziegelstein RC, Meuchel J, Kim TJ, et al. Selective serotonin reuptake inhibitor use by patients with acute coronary syndromes. Am J Med 2007; 120:525–530.
- Xiong GL, Jiang W, Clare R, et al. Prognosis of patients taking selective serotonin reuptake inhibitors before coronary artery bypass grafting. Am J Cardiol 2006; 98:42–47.
- Yang C-C, Jick SS, Jick H. Lipid-lowering drugs and the risk of depression and suicidal behavior. Arch Intern Med 2003; 163:1926–1932.
- Callreus T, Agerskov Andersen U, Hallas J, Andersen M. Cardiovascular drugs and the risk of suicide: a nested case-control study. Eur J Clin Pharmacol 2007; 63:591–596.
- Young-Xu Y, Chan KA, Liao JK, Ravid S, Blatt CM. Long-term statin use and psychological well-being. J Am Coll Cardiol 2003; 42:690–697.
- Ko DT, Hebert PR, Coffey CS, Sedrakyan A, Curtis JP, Krumholz HM. Beta-blocker therapy and symptoms of depression, fatigue, and sexual dysfunction. JAMA 2002; 288:351–357.
- van Melle JP, Verbeek D, van den Berg MP, Ormel J, van der Line MR, de Jonge P. Beta-blockers and depression after myocardial infarction. J Am Coll Cardiol 2006; 48:2209–2214.
- Thomas RJ, King M, Lui K, et al. AACVPR/ACC/AHA 2007 performance measures on cardiac rehabilitation for referral to and delivery of cardiac rehabilitation/secondary prevention services. J Am Coll Cardiol 2007; 50:1400–1433.
- Blumenthal JA, Babyak MA, Doraiswamy PM, et al. Exercise and pharmacotherapy in the treatment of major depressive disorder. Psychosom Med 2007; 69:587–596.
- Linden W, Phillips MJ, Leclerc J. Psychological treatment of cardiac patients: a meta-analysis. Eur Heart J 2007; 28:2972–2984.
- Centers for Disease Control and Prevention. Receipt of outpatient cardiac rehabilitation among heart attack survivors—United States, 2005. JAMA 2008; 299:1534–1536.
- Williams RB. Treating depression after myocardial infarction: can selecting patients on the basis of genetic susceptibility improve psychiatric and medical outcomes? Am Heart J 2005; 150:617–619.
- Schneiderman N, Williams RB. The great debate editorial, revisited. Psychosom Med 2006; 68:636–638.
- de Jonge P, Ormel J, van den Brink RHS, et al. Symptom dimensions of depression following myocardial infarction and their relationship with somatic health status and cardiovascular prognosis. Am J Psychiatry 2006; 163:138–144.
- Fraguas R, Iosifescu DV, Alpert J, et al. Major depressive disorder and comorbid cardiac disease: is there a depressive subtype with greater cardiovascular morbidity? Results from the STAR*D Study. Psychosomatics 2007; 48:418–425.
- Kubzansky LD, Thurston RC. Emotional vitality and incident coronary heart disease: benefits of healthy psychological functioning. Arch Gen Psychiatry 2007; 64:1393–1401.
- Frasure-Smith N. Reflections on depression as a cardiac risk factor Academy of Psychosomatic Medicine, 53rd Annual Meeting, Tucson, Arizona, 2006.
- Frasure-Smith N, Lespérance F. Major depression is associated with lower omega-3 fatty acid levels in patients with recent acute coronary syndromes. Biol Psychiatry 2004; 55:891–896.
- Lespérance F. Annual Research Award Lecture Academy of Psychosomatic Medicine, Amelia Island, Florida, 2007.
- Graham I, Atar D, Borch-Johnsen K, et al. European guidelines on cardiovascular disease prevention in clinical practice: executive summary. Eur Heart J 2007; 28:2375–2414.
- Davidson KW, Kupfer DJ, Bigger JT, et al. Assessment and treatment of depression in patients with cardiovascular disease: National Heart, Lung, and Blood Institute Working Group Report. Psychosom Med 2006; 68:645–650.
- Lichtman JH, Bigger JT, Blumenthal JA, et al. Depression and coronary heart disease. Recommendations for screening, referral, and treatment. Circulation 2008; 118:1768–1775.
- Jiang W, Glassman A, Krishnan R, O’Connor CM, Califf RM. Depression and ischemic heart disease: what have we learned so far and what must we do in the future? Am Heart J 2005; 150:54–78.
- Freedland KE, Rich MW, Skala JA, Carney RM, Davila-Roman VG, Jaffe AS. Prevalence of depression in hospitalized patients with congestive heart failure. Psychosom Med 2003; 65:119–128.
- Murray CJ, Lopez AD, Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet 1997; 349:1436–1442.
- Moussavi S, Chatterji S, Verdes E, Tandon A, Patel V, Ustun B. Depression, chronic diseases, and decrements in health: results from the World Health Surveys. Lancet 2007; 370:851–858.
- Osborn DP, Levy G, Nazareth I, Petersen I, Islam A, King MB. Relative risk of cardiovascular and cancer mortality in people with severe mental illness from the United Kingdom’s general practice research database. Arch Gen Psychiatry 2007; 64:242–249.
- Friedman M, Thoresen CE, Gill JJ, et al. Alteration of type A behavior and its effect on cardiac recurrences in post myocardial infarction patients: summary results of the recurrent coronary prevention project. Am Heart J 1986; 112:653–665.
- Ragland DR, Brand RJ. Type A behavior and mortality from coronary heart disease. N Engl J Med 1988; 318:65–69.
- Rozanski A, Blumenthal JA, Kaplan J. Impact of psychological factors on the pathogenesis of cardiovascular disease and implications for therapy. Circulation 1999; 99:2192–2217.
- Iribarren C, Sidney S, Bild DE, et al. Association of hostility with coronary artery calcification in young adults: the CARDIA study. Coronary Artery Risk Development in Young Adults. JAMA 2000; 283:2546–2551.
- Williams RB, Barefoot JC, Schneiderman N. Psychosocial risk factors for cardiovascular disease: more than one culprit at work. JAMA 2003; 290:2190–2192.
- Rozanski A, Blumenthal JA, Davidson KW, Saab PG, Kubzansky L. The epidemiology, pathophysiology, and management of psychosocial risk factors in cardiac practice: the emerging field of behavioral cardiology. J Am Coll Cardiol 2005; 45:637–651.
- Hemingway H, Marmot M. Evidence based cardiology: psychosocial factors in the aetiology and prognosis of coronary heart disease: systematic review of prospective cohort studies. BMJ 1999; 318:1460–1467.
- Denollet J. DS14: Standard assessment of negative affectivity, social inhibition, and type D personality. Psychosom Med 2005; 67:89–97.
- Steptoe A, Molloy GJ. Personality and heart disease. Heart 2007; 93:783–784.
- Yusuf S, Hawken S, Ôunpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet 2004; 364:937–952.
- Rosengren A, Hawken S, Ounpuu S, et al. Association of psychosocial risk factors with risk of acute myocardial infarction in 11119 cases and 13648 controls from 52 countries (the INTERHEART study): case-control study. Lancet 2004; 364:953–962.
- Kuper H, Marmot M. Job strain, job demands, decision latitude, and risk of coronary heart disease within the Whitehall II study. J Epidemiol Community Health 2003; 57:147–153.
- Aboa-Eboule C, Brisson C, Maunsell E, et al. Job strain and risk of acute recurrent coronary heart disease events. JAMA 2007; 298:1652–1660.
- Frasure-Smith N, Lespérance F. Depression and anxiety as predictors of 2-year cardiac events in patients with stable coronary artery disease. Arch Gen Psychiatry 2008; 65:62–71.
- Shen B-J, Avivi YE, Todaro JF, et al. Anxiety characteristics independently and prospectively predict myocardial infarction in men. J Am Coll Cardiol 2008; 51:113–119.
- Carney RM, Freedland KE. Depression, mortality, and medical morbidity in patients with coronary heart disease. Biol Psychiatry 2003; 54:241–247.
- Pratt LA, Ford DE, Crum RM, Armenian HK, Gallo JJ, Eaton WW. Depression, psychotropic medication, and risk of myocardial infarction: prospective data from the Baltimore ECA follow-up. Circulation 1996; 94:3123–3129.
- Rugulies R. Depression as a predictor for coronary heart disease. A review and meta-analysis. Am J Prev Med 2002; 23:51–61.
- Van der Kooy K, van Hout H, Marwijk H, Marten H, Stehouwer C, Beekman A. Depression and the risk for cardiovascular diseases: systematic review and meta analysis. Int J Geriatr Psychiatry 2007; 22:613–626.
- Wulsin LR, Singal BM. Do depressive symptoms increase the risk for the onset of coronary disease? A systematic quantitative review. Psychosom Med 2003; 65:201–210.
- Wulsin LR. Is depression a major risk factor for coronary disease? A systematic review of the epidemiologic evidence. Harv Rev Psychiatry 2004; 12:79–93.
- Janszky I, Ahlbom A, Hallqvist J, Ahnve S. Hospitalization for depression is associated with an increased risk for myocardial infarction not explained by lifestyle, lipids, coagulation, and inflammation: The SHEEP Study. Biol Psychiatry 2007; 62:25–32.
- Surtees PG, Wainwright NWJ, Luben RN, Wareham NJ, Bingham SA, Khaw K-T. Depression and ischemic heart disease mortality: evidence from the EPIC-Norfolk United Kingdom Prospective Cohort Study. Am J Psychiatry 2008; 165:515–523.
- Carney RM, Rich MW, Freedland KE, et al. Major depressive disorder predicts cardiac events in patients with coronary artery disease. Psychosom Med 1988; 50:627–633.
- Frasure-Smith N, Lespérance F, Talajic M. Depression following myocardial infarction. Impact on 6-month survival. JAMA 1993; 270:1819–1825.
- Frasure-Smith N, Lespérance F, Talajic M. Depression and 18-month prognosis after myocardial infarction. Circulation 1995; 91:999–1005.
- Barefoot JC, Helms MJ, Mark DB, et al. Depression and long-term mortality risk in patients with coronary artery disease. Am J Cardiol 1996; 78:613–617.
- Lespérance F, Frasure-Smith N, Talajic M, Bourassa MG. Five-year risk of cardiac mortality in relation to initial severity and one-year changes in depression symptoms after myocardial infarction. Circulation 2002; 105:1049–1053.
- Barth J, Schumacher M, Herrmann-Lingen C. Depression as a risk factor for mortality in patients with coronary heart disease: a meta-analysis. Psychosom Med 2004; 66:802–813.
- van Melle JP, de Jonge P, Spijkerman TA, et al. Prognostic association of depression following myocardial infarction with mortality and cardiovascular events: a meta-analysis. Psychosom Med 2004; 66:814–822.
- Blumenthal JA, Lett HS, Babyak MA, et al. Depression as a risk factor for mortality after coronary artery bypass surgery. Lancet 2003; 362:604–609.
- Sullivan MD, LaCroix AZ, Spertus JA, Hecht J, Russo J. Depression predicts revascularization procedures for 5 years after coronary angiography. Psychosom Med 2003; 65:229–236.
- Jiang W, Alexander J, Christopher E, et al. Relationship of depression to increased risk of mortality and rehospitalization in patients with congestive heart failure. Arch Intern Med 2001; 161:1849–1856.
- Zipfel S, Schneider A, Wild B, et al. Effect of depressive symptoms on survival after heart transplantation. Psychosom Med 2002; 64:740–747.
- Polsky D, Doshi JA, Marcus S, et al. Long-term risk for depressive symptoms after a medical diagnosis. Arch Intern Med 2005; 165:1260–1266.
- Katon W, Lin EHB, Kroenke K. The association of depression and anxiety with medical symptom burden in patients with chronic medical illness. Gen Hosp Psychiatry 2007; 29:147–155.
- Ruo B, Rumsfeld JS, Hlatky MA, Liu H, Browner WS, Whooley MA. Depressive symptoms and health-related quality of life: the Heart and Soul Study. JAMA 2003; 290:215–221.
- Rasmussen JN, Chong A, Alter DA. Relationship between adherence to evidence-based pharmacotherapy and long-term mortality after acute myocardial infarction. JAMA 2007; 297:177–186.
- Gehi AK, Ali S, Na B, Whooley MA. Self-reported medication adherence and cardiovascular events in patients with stable coronary heart disease: the Heart and Soul Study. Arch Intern Med 2007; 167:1798–1803.
- Gehi A, Haas D, Pipkin S, Whooley MA. Depression and medication adherence in outpatients with coronary heart disease: findings from the Heart and Soul Study. Arch Intern Med 2005; 165:2508–2513.
- Rieckmann N, Gerin W, Kronish IM, et al. Course of depressive symptoms and medication adherence after acute coronary syndromes: an electronic medication monitoring study. J Am Coll Cardiol 2006; 48:2218–2222.
- Ziegelstein RC, Fauerbach JA, Stevens SS, Romanelli J, Richter DP, Bush DE. Patients with depression are less likely to follow recommendations to reduce cardiac risk during recovery from a myocardial infarction. Arch Intern Med 2000; 160:1818–1823.
- Kronish IM, Rieckmann N, Halm EA, et al. Persistent depression affects adherence to secondary prevention behaviors after acute coronary syndromes. J Gen Intern Med 2006; 21:1178–1183.
- Joynt KE, Whellan DJ, O’Connor CM. Depression and cardiovascular disease: mechanisms of interaction. Biol Psychiatry 2003; 54:248–261.
- Musselman DL, Evans DL, Nemeroff CB. The relationship of depression to cardiovascular disease: epidemiology, biology, and treatment. Arch Gen Psychiatry 1998; 55:580–592.
- Lespérance F, Frasure-Smith N. Depression and heart disease. Cleve Clin J Med 2007; 74(suppl 1):S63–S66.
- Curtis BM. Autonomic tone as a cardiovascular risk factor: the dangers of chronic fight or flight. Mayo Clin Proc 2002; 77:45–54.
- Topol EJ. Textbook of Cardiovascular Medicine, 2nd ed. Philadelphia: Lippincott Williams & Williams 2002.
- Carney RM, Saunders RD, Freedland KE, Stein P, Rich MW, Jaffe AS. Association of depression with reduced heart rate variability in coronary artery disease. Am J Cardiol 1995; 76:562–564.
- Carney RM, Blumenthal JA, Stein PK, et al. Depression, heart rate variability, and acute myocardial infarction. Circulation 2001; 104:2024–2028.
- Carney RM, Blumenthal JA, Freedland KE, et al. Low heart rate variability and the effect of depression on post-myocardial infarction mortality. Arch Intern Med 2005; 165:1486–1491.
- Gehi A, Mangano D, Pipkin S, Browner WS, Whooley MA. Depression and heart rate variability in patients with stable coronary heart disease: findings from the Heart and Soul Study. Arch Gen Psychiatry 2005; 62:661–666.
- de Jonge P, Mangano D, Whooley MA. Differential association of cognitive and somatic depressive symptoms with heart rate variability in patients with stable coronary heart disease: findings from the Heart and Soul Study. Psychosom Med 2007; 69:735–739.
- Musselman DL, Tomer A, Manatunga AK, et al. Exaggerated platelet reactivity in major depression. Am J Psychiatry 1996; 153:1313–1317.
- Laghrissi-Thode F, Wagner WR, Pollock BG, Johnson PC, Finkel MS. Elevated platelet factor 4 and beta-thromboglobulin plasma levels in depressed patients with ischemic heart disease. Biol Psychiatry 1997; 42:290–295.
- Serebruany VL, Glassman AH, Malinin AI, et al. Platelet/endothelial biomarkers in depressed patients treated with the selective serotonin reuptake inhibitor sertraline after acute coronary events: the Sertraline AntiDepressant Heart Attack Randomized Trial (SADHART) Platelet Substudy. Circulation 2003; 108:939–944.
- Sherwood A, Hinderliter AL, Watkins LL, Waugh RA, Blumenthal JA. Impaired endothelial function in coronary heart disease patients with depressive symptomatology. J Am Coll Cardiol 2005; 46:656–659.
- Yang EH, Lerman S, Lennon RJ, Simari RD, Lerman LO, Lerman A. Relation of depression to coronary endothelial function. Am J Cardiol 2007; 99:1134–1136.
- Otte C, Neylan TC, Pipkin SS, Browner WS, Whooley MA. Depressive symptoms and 24-hour urinary norepinephrine excretion levels in patients with coronary disease: findings from the Heart and Soul Study. Am J Psychiatry 2005; 162:2139–2145.
- Wittstein IS, Thiemann DR, Lima JAC, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med 2005; 352:539–548.
- Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 2005; 352:1685–1695.
- Miller GE, Stetler CA, Carney RM, Freedland KE, Banks WA. Clinical depression and inflammatory risk markers for coronary heart disease. Am J Cardiol 2002; 90:1279–1283.
- Empana JP, Sykes DH, Luc G, et al. Contributions of depressive mood and circulating inflammatory markers to coronary heart disease in healthy European men: the Prospective Epidemiological Study of Myocardial Infarction (PRIME). Circulation 2005; 111:2299–2305.
- Frasure-Smith N, Lespérance F, Irwin MR, Sauvé C, Lespérance J, Théroux P. Depression, C-reactive protein and two-year major adverse cardiac events in men after acute coronary syndromes. Biol Psychiatry 2007; 62:302–308.
- Whooley MA, Caska CM, Hendrickson BE, Rourke MA, Ho J, Ali S. Depression and inflammation in patients with coronary heart disease: findings from the Heart and Soul Study. Biol Psychiatry 2007; 62:314–320.
- Glassman AH, Miller GE. Where there is depression, there is inflammation…sometimes! Biol Psychiatry 2007; 62:280–281.
- Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest 2007; 117:289–296.
- Tracey KJ. The inflammatory reflex. Nature 2002; 420:853–859.
- McCaffery JM, Frasure-Smith N, Dubé M-P, et al. Common genetic vulnerability to depressive symptoms and coronary artery disease: a review and development of candidate genes related to inflammation and serotonin. Psychosom Med 2006; 68:187–200.
- Caspi A, Sugden K, Moffitt TE, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 2003; 301:386–389.
- Nakatani D, Sato H, Sakata Y, et al. Influence of serotonin transporter gene polymorphism on depressive symptoms and new cardiac events after acute myocardial infarction. Am Heart J 2005; 150:652–658.
- Otte C, McCaffery J, Ali S, Whooley MA. Association of a serotonin transporter polymorphism (5-HTTLPR) with depression, perceived stress, and norepinephrine in patients with coronary disease: the Heart and Soul Study. Am J Psychiatry 2007; 164:1379–1384.
- Fumeron F, Betoulle D, Nicaud V, et al. Serotonin transporter gene polymorphism and myocardial infarction: Etude Cas-Témoins de l’Infarctus du Myocarde (ECTIM). Circulation 2002; 105:2943–2945.
- Rivelli S, Jiang W. Depression and ischemic heart disease: what have we learned from clinical trials? Curr Opin Cardiol 2007; 22:286–291.
- Frasure-Smith N, Lespérance F, Prince RH, et al. Randomised trial of home-based psychosocial nursing intervention for patients recovering from myocardial infarction. Lancet 1997; 350:473–479.
- Glassman AH, O’Connor CM, Califf RM, et al. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002; 288:701–709.
- Writing Committee for the EI. Effects of treating depression and low perceived social support on clinical events after myocardial infarction: the Enhancing Recovery in Coronary Heart Disease Patients (ENRICHD) randomized trial. JAMA 2003; 289:3106–3116.
- van Melle JP, de Jonge P, Honig A, et al. Effects of antidepressant treatment following myocardial infarction. Br J Psychiatry 2007; 190:460–466.
- Lespérance F, Frasure-Smith N, Koszycki D, et al. Effects of citalopram and interpersonal psychotherapy on depression in patients with coronary artery disease: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial. JAMA 2007; 297:367–379.
- Carney RM, Blumenthal JA, Freedland KE, et al. Depression and late mortality after myocardial infarction in the Enhancing Recovery in Coronary Heart Disease (ENRICHD) Study. Psychosom Med 2004; 66:466–474.
- de Jonge P, Honig A, van Melle JP, et al. Nonresponse to treatment for depression following myocardial infarction: association with subsequent cardiac events. Am J Psychiatry 2007; 164:1371–1378.
- Carney RM, Freedland KE. Depression and coronary heart disease: more pieces of the puzzle. Am J Psychiatry 2007; 164:1307–1309.
- Glassman AH, Bigger JT, Gaffney M, Shapiro PA, Swenson JR. Onset of major depression associated with acute coronary syndromes: relationship of onset, major depressive disorder history, and episode severity to sertraline benefit. Arch Gen Psychiatry 2006; 63:283–288.
- Ziegelstein RC, Kim SY, Kao D, et al. Can doctors and nurses recognize depression in patients hospitalized with an acute myocardial infarction in the absence of formal screening? Psychosom Med 2005; 67:393–397.
- Kroenke K, Spitzer RL, Williams JBW. The PHQ-9. Validity of a brief depression severity measure. J Gen Intern Med 2001; 16:606–613.
- Löwe B, Unutzer J, Callahan CM, Perkins AJ, Kroenke K. Monitoring depression treatment outcomes with the Patient Health Questionnaire-9. Med Care 2004; 42:1194–1201.
- Parashar S, Rumsfeld JS, Spertus JA, et al. Time course of depression and outcome of myocardial infarction. Arch Intern Med 2006; 166:2035–2043.
- Amin AA, Jones AMH, Nugent K, Rumsfeld JS, Spertus JA. The prevalence of unrecognized depression in patients with acute coronary syndrome. Am Heart J 2006; 152:928–934.
- McManus D, Pipkin SS, Whooley MA. Screening for depression in patients with coronary heart disease (data from the Heart and Soul Study). Am J Cardiol 2005; 96:1076–1081.
- Kroenke K, Spitzer RL, Williams JB. The Patient Health Questionnaire-2: validity of a two-item depression screener. Med Care 2003; 41:1284–1292.
- Spitzer RL, Williams JB, Kroenke K, et al. Utility of a new procedure for diagnosing mental disorders in primary care. The PRIME-MD 1000 study. JAMA 1994; 272:1749–1756.
- Lespérance F, Frasure-Smith N. Depression in patients with cardiac disease. J Psychosom Res 2000; 48:379–391.
- Hunkeler EM, Katon W, Tang L, et al. Long term outcomes from the IMPACT randomised trial for depressed elderly patients in primary care. BMJ 2006; 332:259–263.
- Frasure-Smith N, Lespérance F, Gravel G, Masson A, Juneau M, Bourassa MG. Long-term survival differences among low-anxious, high-anxious and repressive copers enrolled in the Montreal Heart Attack Readjustment Trial. Psychosom Med 2002; 64:571–579.
- Glassman AH, Roose SP, Bigger JT. The safety of tricyclic antidepressants in cardiac patients: risk-benefit reconsidered. JAMA 1993; 269:2673–2675.
- Cohen HW, Gibson G, Alderman MH. Excess risk of myocardial infarction in patients treated with antidepressant medications: association with use of tricyclic agents. Am J Med 2000; 108:2–8.
- Taylor CB, Youngblood ME, Catellier D, et al. Effects of antidepressant medication on morbidity and mortality in depressed patients after myocardial infarction. Arch Gen Psychiatry 2005; 62:792–798.
- Montgomery SA. Safety of mirtazapine: a review. Int Clin Psychopharmacol 1995; 10(suppl 4):37–45.
- Sauer WH, Berlin JA, Kimmel SE. Selective serotonin reuptake inhibitors and myocardial infarction. Circulation 2001; 104:1894–1898.
- Sauer WH, Berlin JA, Kimmel SE. Effect of antidepressants and their relative affinity for the serotonin transporter on the risk of myocardial infarction. Circulation 2003; 108:32–36.
- Ziegelstein RC, Meuchel J, Kim TJ, et al. Selective serotonin reuptake inhibitor use by patients with acute coronary syndromes. Am J Med 2007; 120:525–530.
- Xiong GL, Jiang W, Clare R, et al. Prognosis of patients taking selective serotonin reuptake inhibitors before coronary artery bypass grafting. Am J Cardiol 2006; 98:42–47.
- Yang C-C, Jick SS, Jick H. Lipid-lowering drugs and the risk of depression and suicidal behavior. Arch Intern Med 2003; 163:1926–1932.
- Callreus T, Agerskov Andersen U, Hallas J, Andersen M. Cardiovascular drugs and the risk of suicide: a nested case-control study. Eur J Clin Pharmacol 2007; 63:591–596.
- Young-Xu Y, Chan KA, Liao JK, Ravid S, Blatt CM. Long-term statin use and psychological well-being. J Am Coll Cardiol 2003; 42:690–697.
- Ko DT, Hebert PR, Coffey CS, Sedrakyan A, Curtis JP, Krumholz HM. Beta-blocker therapy and symptoms of depression, fatigue, and sexual dysfunction. JAMA 2002; 288:351–357.
- van Melle JP, Verbeek D, van den Berg MP, Ormel J, van der Line MR, de Jonge P. Beta-blockers and depression after myocardial infarction. J Am Coll Cardiol 2006; 48:2209–2214.
- Thomas RJ, King M, Lui K, et al. AACVPR/ACC/AHA 2007 performance measures on cardiac rehabilitation for referral to and delivery of cardiac rehabilitation/secondary prevention services. J Am Coll Cardiol 2007; 50:1400–1433.
- Blumenthal JA, Babyak MA, Doraiswamy PM, et al. Exercise and pharmacotherapy in the treatment of major depressive disorder. Psychosom Med 2007; 69:587–596.
- Linden W, Phillips MJ, Leclerc J. Psychological treatment of cardiac patients: a meta-analysis. Eur Heart J 2007; 28:2972–2984.
- Centers for Disease Control and Prevention. Receipt of outpatient cardiac rehabilitation among heart attack survivors—United States, 2005. JAMA 2008; 299:1534–1536.
- Williams RB. Treating depression after myocardial infarction: can selecting patients on the basis of genetic susceptibility improve psychiatric and medical outcomes? Am Heart J 2005; 150:617–619.
- Schneiderman N, Williams RB. The great debate editorial, revisited. Psychosom Med 2006; 68:636–638.
- de Jonge P, Ormel J, van den Brink RHS, et al. Symptom dimensions of depression following myocardial infarction and their relationship with somatic health status and cardiovascular prognosis. Am J Psychiatry 2006; 163:138–144.
- Fraguas R, Iosifescu DV, Alpert J, et al. Major depressive disorder and comorbid cardiac disease: is there a depressive subtype with greater cardiovascular morbidity? Results from the STAR*D Study. Psychosomatics 2007; 48:418–425.
- Kubzansky LD, Thurston RC. Emotional vitality and incident coronary heart disease: benefits of healthy psychological functioning. Arch Gen Psychiatry 2007; 64:1393–1401.
- Frasure-Smith N. Reflections on depression as a cardiac risk factor Academy of Psychosomatic Medicine, 53rd Annual Meeting, Tucson, Arizona, 2006.
- Frasure-Smith N, Lespérance F. Major depression is associated with lower omega-3 fatty acid levels in patients with recent acute coronary syndromes. Biol Psychiatry 2004; 55:891–896.
- Lespérance F. Annual Research Award Lecture Academy of Psychosomatic Medicine, Amelia Island, Florida, 2007.
- Graham I, Atar D, Borch-Johnsen K, et al. European guidelines on cardiovascular disease prevention in clinical practice: executive summary. Eur Heart J 2007; 28:2375–2414.
- Davidson KW, Kupfer DJ, Bigger JT, et al. Assessment and treatment of depression in patients with cardiovascular disease: National Heart, Lung, and Blood Institute Working Group Report. Psychosom Med 2006; 68:645–650.
KEY POINTS
- Depression is a risk factor for new cardiac disease and has a detrimental impact in established cardiac disease.
- Numerous mechanistic pathways have been implicated.
- In clinical trials, drug therapy and psychotherapy have not clearly decreased the rate of cardiac death in depressed cardiac patients, but they did improve depression, adherence to drug therapy, and quality of life.
- Clinicians should routinely screen for depression in cardiac patients and should not hesitate to treat it.
- Eligible patients should routinely be referred to cardiac rehabilitation programs.
The ENHANCE trial
To the Editor: I read with great interest Dr. Davidson’s commentary article1 about the Ezetimibe and Simvastatin in Hypercholesterolemia Enhances Atherosclerosis Regression (ENHANCE) trial.2 However, his conclusion that ezetimibe (Zetia) still has a role as an add-on to statin therapy for patients who have not achieved their low-density lipoprotein cholesterol (LDL-C) target is of great concern to me and my patients. Based on this trial, I have taken many of my patients off of ezetimibe and have wondered if this is the right decision. I also have several physician patients who have told me that ezetimibe causes muscle cramping and other symptoms often found in patients who cannot tolerate statins, and in fact one of these patients was found to have congenital cirrhosis of the liver.
Ezetimibe is mainly active in the GI tract. What relationship does this medication have in those patients who have liver disease, ie, cirrhosis? Is it safe to give ezetimibe to patients who cannot take statins? I doubt it.
Consequently, I agree with Dr. Taylor’s editorial,3 which in essence states unless you are in a clinical trial, beware of ezetimibe!
- Davidson MH. Interpreting the ENHANCE trial. Is ezetimibe/simvastatin no better than simvastatin alone? Lessons learned and clinical implications. Cleve Clin J Med 2008; 75:479–491.
- Kastelein JJ, Akdim F, Stroes ES, et al; ENHANCE Investigators. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med 2008; 358:1431–1443.
- Taylor AJ. Given the enhance trial results, ezetimibe is still unproven. Cleve Clin J Med 2008; 75:497–506.
To the Editor: I read with great interest Dr. Davidson’s commentary article1 about the Ezetimibe and Simvastatin in Hypercholesterolemia Enhances Atherosclerosis Regression (ENHANCE) trial.2 However, his conclusion that ezetimibe (Zetia) still has a role as an add-on to statin therapy for patients who have not achieved their low-density lipoprotein cholesterol (LDL-C) target is of great concern to me and my patients. Based on this trial, I have taken many of my patients off of ezetimibe and have wondered if this is the right decision. I also have several physician patients who have told me that ezetimibe causes muscle cramping and other symptoms often found in patients who cannot tolerate statins, and in fact one of these patients was found to have congenital cirrhosis of the liver.
Ezetimibe is mainly active in the GI tract. What relationship does this medication have in those patients who have liver disease, ie, cirrhosis? Is it safe to give ezetimibe to patients who cannot take statins? I doubt it.
Consequently, I agree with Dr. Taylor’s editorial,3 which in essence states unless you are in a clinical trial, beware of ezetimibe!
To the Editor: I read with great interest Dr. Davidson’s commentary article1 about the Ezetimibe and Simvastatin in Hypercholesterolemia Enhances Atherosclerosis Regression (ENHANCE) trial.2 However, his conclusion that ezetimibe (Zetia) still has a role as an add-on to statin therapy for patients who have not achieved their low-density lipoprotein cholesterol (LDL-C) target is of great concern to me and my patients. Based on this trial, I have taken many of my patients off of ezetimibe and have wondered if this is the right decision. I also have several physician patients who have told me that ezetimibe causes muscle cramping and other symptoms often found in patients who cannot tolerate statins, and in fact one of these patients was found to have congenital cirrhosis of the liver.
Ezetimibe is mainly active in the GI tract. What relationship does this medication have in those patients who have liver disease, ie, cirrhosis? Is it safe to give ezetimibe to patients who cannot take statins? I doubt it.
Consequently, I agree with Dr. Taylor’s editorial,3 which in essence states unless you are in a clinical trial, beware of ezetimibe!
- Davidson MH. Interpreting the ENHANCE trial. Is ezetimibe/simvastatin no better than simvastatin alone? Lessons learned and clinical implications. Cleve Clin J Med 2008; 75:479–491.
- Kastelein JJ, Akdim F, Stroes ES, et al; ENHANCE Investigators. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med 2008; 358:1431–1443.
- Taylor AJ. Given the enhance trial results, ezetimibe is still unproven. Cleve Clin J Med 2008; 75:497–506.
- Davidson MH. Interpreting the ENHANCE trial. Is ezetimibe/simvastatin no better than simvastatin alone? Lessons learned and clinical implications. Cleve Clin J Med 2008; 75:479–491.
- Kastelein JJ, Akdim F, Stroes ES, et al; ENHANCE Investigators. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med 2008; 358:1431–1443.
- Taylor AJ. Given the enhance trial results, ezetimibe is still unproven. Cleve Clin J Med 2008; 75:497–506.