User login
The ENHANCE trial
To the Editor: Dr. Davidson concludes his article saying “we should remember [that ezetimibe] is safe and well-tolerated.” Yet, he admits there is a lack of outcomes data for the drug. So, how does he know it is safe if we don’t have the mortality outcomes? The just-published Simvastatin and Ezetimibe in Aortic Stenosis (SEAS) trial indicated that there may be an increase in cancer mortality.1 The point is that we need more data. Until we have that outcomes data we should not be saying a drug is safe as a matter of fact. Physicians need to learn the lessons we should have learned from drugs such as torcetrapib2 or erythropoietin3 and so many others. We often think we are doing a good thing by correcting lab values, but we often learn too late that we harmed the patient at a staggering ethical and financial cost.
Dr. Davidson also references the impressive LDL-C lowering of Senator McCain while taking ezetimibe. Senator McCain has a publicized history of melanoma. Hopefully, ezetimibe doesn’t increase his cancer mortality risk because his physicians are proud of his LDL-C lowering. My advice to the senator is to use one of the many other proven methods of LDL-C lowering until there is good mortality outcome data with ezetimibe (but I’m not a Republican, so he may want to get a second opinion).
- Rossebø AB, Pedersen TR, Boman K, et al, for the SEAS Investigators. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med 10.1056/NEJMoa0804602.
- Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 2007; 357:2109–2122.
- Wright JR, Ung YC, Julian JA, et al. Randomized, doubleblind, placebo-controlled trial of erythropoietin in nonsmall-cell lung cancer with disease-related anemia. J Clin Oncol 2007; 25:1027–1032.
To the Editor: Dr. Davidson concludes his article saying “we should remember [that ezetimibe] is safe and well-tolerated.” Yet, he admits there is a lack of outcomes data for the drug. So, how does he know it is safe if we don’t have the mortality outcomes? The just-published Simvastatin and Ezetimibe in Aortic Stenosis (SEAS) trial indicated that there may be an increase in cancer mortality.1 The point is that we need more data. Until we have that outcomes data we should not be saying a drug is safe as a matter of fact. Physicians need to learn the lessons we should have learned from drugs such as torcetrapib2 or erythropoietin3 and so many others. We often think we are doing a good thing by correcting lab values, but we often learn too late that we harmed the patient at a staggering ethical and financial cost.
Dr. Davidson also references the impressive LDL-C lowering of Senator McCain while taking ezetimibe. Senator McCain has a publicized history of melanoma. Hopefully, ezetimibe doesn’t increase his cancer mortality risk because his physicians are proud of his LDL-C lowering. My advice to the senator is to use one of the many other proven methods of LDL-C lowering until there is good mortality outcome data with ezetimibe (but I’m not a Republican, so he may want to get a second opinion).
To the Editor: Dr. Davidson concludes his article saying “we should remember [that ezetimibe] is safe and well-tolerated.” Yet, he admits there is a lack of outcomes data for the drug. So, how does he know it is safe if we don’t have the mortality outcomes? The just-published Simvastatin and Ezetimibe in Aortic Stenosis (SEAS) trial indicated that there may be an increase in cancer mortality.1 The point is that we need more data. Until we have that outcomes data we should not be saying a drug is safe as a matter of fact. Physicians need to learn the lessons we should have learned from drugs such as torcetrapib2 or erythropoietin3 and so many others. We often think we are doing a good thing by correcting lab values, but we often learn too late that we harmed the patient at a staggering ethical and financial cost.
Dr. Davidson also references the impressive LDL-C lowering of Senator McCain while taking ezetimibe. Senator McCain has a publicized history of melanoma. Hopefully, ezetimibe doesn’t increase his cancer mortality risk because his physicians are proud of his LDL-C lowering. My advice to the senator is to use one of the many other proven methods of LDL-C lowering until there is good mortality outcome data with ezetimibe (but I’m not a Republican, so he may want to get a second opinion).
- Rossebø AB, Pedersen TR, Boman K, et al, for the SEAS Investigators. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med 10.1056/NEJMoa0804602.
- Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 2007; 357:2109–2122.
- Wright JR, Ung YC, Julian JA, et al. Randomized, doubleblind, placebo-controlled trial of erythropoietin in nonsmall-cell lung cancer with disease-related anemia. J Clin Oncol 2007; 25:1027–1032.
- Rossebø AB, Pedersen TR, Boman K, et al, for the SEAS Investigators. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med 10.1056/NEJMoa0804602.
- Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 2007; 357:2109–2122.
- Wright JR, Ung YC, Julian JA, et al. Randomized, doubleblind, placebo-controlled trial of erythropoietin in nonsmall-cell lung cancer with disease-related anemia. J Clin Oncol 2007; 25:1027–1032.
In reply: The ENHANCE trial
In Reply: Both Dr. Fee and Dr. Porat recommend cautious utilization of ezetimibe until outcome studies are completed. As I stated in my article, it is unfortunate that for ezetimibe, hard outcome trials are not yet available (the SEAS trial showed a cardiovascular benefit for the combination of simvastatin/ezetimibe, but it was not the primary end point). The main point of my article is that the weight of evidence for the benefits of LDL-C lowering is one of the most proven surrogate measures in clinical medicine. The biology, epidemiology, and clinical trials with multiple LDL-C-lowering therapies (bile-acid resin, niacin, fibrates, diet, ileal bypass surgery, and statins) convincingly demonstrate the validity of this surrogate measure for regulatory approval. In fact, every drug that has been approved for the treatment of hypercholesterolemia has been based on LDL-C reduction and not on outcome trials.
If this requirement was in place, it is doubtful that statins would have been approved. Lovastatin was approved by the US Food and Drug Administration in 1987; the Scandinavian Simvastatin Survival Study (4S) trial1 was completed in 1994. The 4S trial showed, for the first time, a reduction in total mortality with an LDL-C-lowering therapy. Millions of patients were placed on statins prior to 1994, and it is unlikely the 4S trial would have been funded unless there had been prior regulatory approval.
As a researcher, I truly believe hard outcome trials are essential, but as a clinician, I realize that most of our medical care is based on drugs approved utilizing surrogate measures. Hard outcome trials are not required for antihypertensives, oral hypoglycemics, or smoking cessation treatments prior to approval. Ezetimibe lowers LDL-C by a known mechanism and is well tolerated. The ENHANCE trial, with its well-recognized flaws, should not refute the benefits of LDL-C reduction. For patients not at goal on statin therapy, ezetimibe should remain a widely used option.
- The Scandinavian Simvastatin Survival Study (4S). Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease. Lancet 1994; 344:1383–1389.
In Reply: Both Dr. Fee and Dr. Porat recommend cautious utilization of ezetimibe until outcome studies are completed. As I stated in my article, it is unfortunate that for ezetimibe, hard outcome trials are not yet available (the SEAS trial showed a cardiovascular benefit for the combination of simvastatin/ezetimibe, but it was not the primary end point). The main point of my article is that the weight of evidence for the benefits of LDL-C lowering is one of the most proven surrogate measures in clinical medicine. The biology, epidemiology, and clinical trials with multiple LDL-C-lowering therapies (bile-acid resin, niacin, fibrates, diet, ileal bypass surgery, and statins) convincingly demonstrate the validity of this surrogate measure for regulatory approval. In fact, every drug that has been approved for the treatment of hypercholesterolemia has been based on LDL-C reduction and not on outcome trials.
If this requirement was in place, it is doubtful that statins would have been approved. Lovastatin was approved by the US Food and Drug Administration in 1987; the Scandinavian Simvastatin Survival Study (4S) trial1 was completed in 1994. The 4S trial showed, for the first time, a reduction in total mortality with an LDL-C-lowering therapy. Millions of patients were placed on statins prior to 1994, and it is unlikely the 4S trial would have been funded unless there had been prior regulatory approval.
As a researcher, I truly believe hard outcome trials are essential, but as a clinician, I realize that most of our medical care is based on drugs approved utilizing surrogate measures. Hard outcome trials are not required for antihypertensives, oral hypoglycemics, or smoking cessation treatments prior to approval. Ezetimibe lowers LDL-C by a known mechanism and is well tolerated. The ENHANCE trial, with its well-recognized flaws, should not refute the benefits of LDL-C reduction. For patients not at goal on statin therapy, ezetimibe should remain a widely used option.
In Reply: Both Dr. Fee and Dr. Porat recommend cautious utilization of ezetimibe until outcome studies are completed. As I stated in my article, it is unfortunate that for ezetimibe, hard outcome trials are not yet available (the SEAS trial showed a cardiovascular benefit for the combination of simvastatin/ezetimibe, but it was not the primary end point). The main point of my article is that the weight of evidence for the benefits of LDL-C lowering is one of the most proven surrogate measures in clinical medicine. The biology, epidemiology, and clinical trials with multiple LDL-C-lowering therapies (bile-acid resin, niacin, fibrates, diet, ileal bypass surgery, and statins) convincingly demonstrate the validity of this surrogate measure for regulatory approval. In fact, every drug that has been approved for the treatment of hypercholesterolemia has been based on LDL-C reduction and not on outcome trials.
If this requirement was in place, it is doubtful that statins would have been approved. Lovastatin was approved by the US Food and Drug Administration in 1987; the Scandinavian Simvastatin Survival Study (4S) trial1 was completed in 1994. The 4S trial showed, for the first time, a reduction in total mortality with an LDL-C-lowering therapy. Millions of patients were placed on statins prior to 1994, and it is unlikely the 4S trial would have been funded unless there had been prior regulatory approval.
As a researcher, I truly believe hard outcome trials are essential, but as a clinician, I realize that most of our medical care is based on drugs approved utilizing surrogate measures. Hard outcome trials are not required for antihypertensives, oral hypoglycemics, or smoking cessation treatments prior to approval. Ezetimibe lowers LDL-C by a known mechanism and is well tolerated. The ENHANCE trial, with its well-recognized flaws, should not refute the benefits of LDL-C reduction. For patients not at goal on statin therapy, ezetimibe should remain a widely used option.
- The Scandinavian Simvastatin Survival Study (4S). Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease. Lancet 1994; 344:1383–1389.
- The Scandinavian Simvastatin Survival Study (4S). Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease. Lancet 1994; 344:1383–1389.
Update on constipation: One treatment does not fit all
Constipation is both a symptom and, when chronic, a multisymptom disorder, and it can overlap with other gastrointestinal tract disorders such as dyspepsia and gastroesophageal reflux disease. Furthermore, one should keep in mind the possibility of cancer and be alert for its warning signs.
Since constipation has a variety of causes and forms, one treatment does not fit all patients. Conservative measures such as recommending that the patient increase his or her intake of dietary fiber and water and engage in more physical activity are still the cornerstone of treatment, but they do not help all patients. On the other hand, polyethylene glycol and stimulant laxatives, which are traditionally given only for a short time, can be safe and effective when given long-term if other agents fail. New agents have become available or are in development.
In this article we outline our approach to constipation, as a guide for internists.
CONSTIPATION IS COMMON, BUT HOW SHOULD WE DEFINE IT?
Constipation affects 2% to 27% (average 14.8%) of the North American adult population—approximately 63 million people.1 It is more common than many other chronic diseases, including hypertension (48 million people), migraine (33 million), obesity (50 million), and diabetes mellitus (15 million).1–3
Constipation affects more women than men (2.1:1 ratio) and more nonwhites than whites (1.68:1).1 It occurs in all age groups but is more common in those older than 65 years and younger than 4 years.4,5
Constipation accounts for more than 2.5 million office visits and more than $500 million spent on laxatives per year.6,7 Also, people with constipation may report decreased productivity and increased absenteeism.8
The broad range in the prevalence of constipation cited above reflects differences in how it is defined and, in particular, a lack of agreement between how patients and physicians perceive it.1,9 Physicians mainly define constipation on the basis of stool frequency, considering fewer than three bowel movements per week to be abnormal.1 In contrast, patients typically define it on the basis of bothersome symptoms such as straining, passage of hard stool, unproductive urges, inability to defecate at will, and sensations of incomplete evacuation or abdominal bloating.1,9,10
The Rome III diagnostic criteria were developed to provide a consistent diagnostic approach for use in clinical practice and clinical trials.11 The Rome III criteria define functional chronic constipation as a chronic bowel disorder characterized by two or more of the following:
- Straining
- Lumpy or hard stools
- Sensations of incomplete evacuation
- Sensations of anorectal obstruction or blockage
- Use of manual maneuvers to facilitate defecation (eg, digital evacuation, support of the pelvic floor) during at least 25% of defecations
- Fewer than three bowel movements per week.
In addition, loose stools should rarely occur without the use of laxatives, and there should be insufficient criteria for irritable bowel syndrome.11 Chronicity is established by symptom onset within the previous 6 months and symptom duration of at least 3 months.
In contrast, patients with irritable bowel syndrome, also a functional bowel disorder, experience recurrent abdominal pain and discomfort associated with two or more of the following: symptom improvement with defecation, symptom onset associated with a change in the frequency of bowel movements, and a change in the form or appearance of the stool.
THREE TYPES OF IDIOPATHIC CONSTIPATION
There are three types of primary or idiopathic constipation5,9,12,13:
- Functional
- Slow-transit
- Outlet dysfunction.
Functional constipation includes functional chronic idiopathic constipation and constipation-predominant irritable bowel syndrome. It presents with a sense of difficult or delayed evacuation, hard stools, or abdominal bloating or discomfort.6,9,13 The predominant symptom of constipation-predominant irritable bowel syndrome is severe discomfort or pain; in chronic idiopathic constipation, pain and discomfort may be present but are not the primary symptom.
Slow-transit constipation (or delayed-transit constipation) is associated with a prolonged time between bowel movements. Its symptoms include low stool frequency, lack of urge to defecate, abdominal distention, bloating, and abdominal discomfort.14
Outlet dysfunction. Disorders of defecation can be due to mechanical causes such as Hirschsprung disease, anal stricture, cancer, prolapse, and large rectoceles, or from pelvic floor dysfunction. Pelvic floor dysfunction may be due to inadequate or excessive perineal descent or to inadequate propulsive forces, as may occur in neurologic or neuromuscular conditions and dyssynergia.
Pelvic floor dyssynergia, also called anorectal dyssynergia, dyssynergic defecation, and anismus, results from a functional defect in coordinated evacuation. The characteristic symptom is a feeling of being unable to adequately empty the rectum.14 Other symptoms such as excessive straining and manual disimpaction indicate but are not unique to pelvic floor dyssynergia.14,15
Combined forms. Patients may have more than one type of primary constipation and presentation, and pelvic floor dyssynergia has been shown to prolong intestinal transit, which may improve with treatment.
Secondary constipation can be due to causes such as diet, lifestyle, certain medications (calcium channel blockers, beta-blockers, opioids, diuretics, antidepressants, anticonvulsants, antacids, anticholinergics, and antispasmodics),5,16 underlying medical conditions (diabetes, hypothyroidism, multiple sclerosis, parkinsonism),16,17 pregnancy, and advanced age.18
NEUROTRANSMITTERS MAY PLAY A ROLE
Among the mechanisms thought to cause chronic constipation are impaired gastrointestinal motility,19–22 reduced intestinal secretions,21–23 and inadequate reflex relaxation of the pelvic floor muscles.22,24
Neurotransmitters such as serotonin, somatostatin, peptide YY, and vasoactive intestinal peptide affect intestinal secretion and motility.25,26 Hyperactivity of these neurotransmitters associated with increased secretion and motility results in diarrhea, whereas hypoactivity leads to decreased secretion, delayed transit, and constipation.23
Serotonin has a role in regulating visceral pain perception and intestinal motility, as well as secretion.26–28 Clinical trials have shown that activation of serotonin receptors in the gut enhances gastrointestinal motility, inhibits visceral sensitivity, and stimulates intestinal secretion.26,27,29
A hypothesis has recently been proposed that degeneration of enteric neurons may also play a role in the development of severe idiopathic constipation.30
DIAGNOSIS IS MOSTLY CLINICAL
The history and physical examination remain the cornerstones in the diagnosis and subsequent treatment of chronic constipation.
History

Risk factors for primary and secondary constipation to note during the interview include age (< 4 years, > 65 years); low-fiber diet; female sex; lack of physical activity; history of childhood constipation, endocrine and neuromuscular disorders, abuse, depression, or anxiety; family history of cancer; and personal history of pelvic surgery.
Since drugs can also cause chronic constipation, especially in elderly or immobile patients, medication lists should be reviewed and adjustments should be made if necessary (or possible) before recommending laxatives or invasive testing, if no alarm signs are present.
Alarm signs such as weight loss, hematochezia, melena, change in bowel habits, and symptoms refractory to therapy may represent colon cancer and indicate the need for early diagnostic testing.
Physical examination
Physical examination should always include inspection of the perianal area for evidence of hemorrhoids or fissures. Digital rectal examination may reveal a contracted sphincter or a puborectalis muscle that contracts with the Valsalva maneuver, suggesting dysfunction.
Laboratory testing
If the history and physical examination suggest that the constipation may be secondary, or if the patient is 50 years of age or older, then laboratory studies such as a complete blood cell count, serum electrolyte levels, blood sugar level, and thyroid function studies may help rule out a metabolic, endocrine, or organic cause.
Colonoscopy, other tests
At present, little evidence suggests that routine testing is warranted in patients without evidence of secondary constipation and without alarm signs. However, diagnostic studies are indicated in patients 50 years of age and older, as well as in those with alarm symptoms such as hematochezia, anemia, a positive fecal occult blood test, unintentional loss of more than 10 pounds, family history of colon cancer or inflammatory bowel disease, fever, nausea, vomiting, acute onset (especially in the elderly), and lack of improvement with conventional therapies regardless of age.2
The full length of the colon should be inspected by colonoscopy or by flexible sigmoidoscopy paired with a barium enema study to rule out structural disease. Of note, all patients 50 years of age or older should be screened for colon cancer.
If the patient does not respond to therapy, further tests such as colonic transit studies, anorectal manometry with balloon expulsion, and, possibly, defecating proctography or dynamic pelvic magnetic resonance imaging may be considered. These patients would likely also benefit from referral to a gastroenterologist for further management
DIET AND LIFESTYLE AS TREATMENT
For many years, health care providers have provided reassurance and recommended diet and lifestyle modifications as treatment for constipation. Increased water intake, increased activity, and a scheduled attempt at defecation when motor activity in the colon is highest, ie, in the morning or after eating, have all been recommended.
Data on the efficacy of these recommendations are scarce and often contradictory. Studies have shown that increasing water intake or daily exercise is not always helpful.32–34 Nevertheless, many patients who comply with dietary and exercise recommendations have improvement in symptoms. Eating fewer meals per day (and hence taking in fewer calories) has been shown to be associated with constipation in the elderly. However, no relationships between fiber or fluid intake and constipation were noted.35
In a study in which chronically constipated patients were fed a standardized diet that contained 25 g of fiber a day, stool frequency increased significantly and laxative use decreased.36 While on a high-fiber diet, the patients were divided into two groups, one that drank 1.1 L of fluid per day and one that drank 2.1 L of mineral water per day. Both groups experienced further improvements in stool frequency and decreases in laxative use, with the mineral-water group benefiting the most.36
Recently, Murakami and others37 found, in a cross-sectional study in young Japanese women with low daily fiber intake (6.4 g/day), that low water intake from foods and low magnesium intake were associated with an increasing prevalence of functional constipation as defined by the Rome III criteria. Constipation was also found to be significantly associated with low intake of fruits and vegetables in a study from Singapore.38
Moderate physical activity and high fiber intake may be associated with a lower prevalence of constipation in women. In the Nurses’ Health Study, more than 62,000 women between the ages of 36 and 61 were surveyed, and those who said they engaged in daily physical activity had a lower prevalence of constipation (prevalence ratio [PR] = 0.56, 95% confidence interval [CI] 0.44–0.70), as did those with a median fiber intake of 20 g/day (PR = 0.64, 95% CI 0.57–0.73).39
BULK LAXATIVES (FIBER SUPPLEMENTS): THE FIRST-LINE TREATMENT
Fiber remains the first-line treatment for constipation. It may relieve or improve symptoms in functional constipation. However, fewer than 30% of patients with either slow-transit constipation or pelvic floor dysfunction have improvement in symptoms with fiber, and in these types of constipation it can even worsen symptoms.40
There is much confusion about what types of fiber should be recommended and how the various types of fiber perform in resolving constipation.
Insoluble fiber
Insoluble fiber resists bacterial degradation in the colon and can retain more water than soluble fiber can.
Bran 20 g/day increased the frequency of bowel movements by 55%, increased fecal weight by 157%, and decreased intestinal transit time by 50% in women who had three or fewer bowel movements per week.41
Muller-Lissner42 and others performed a meta-analysis and found that bran (25 g/day) increased stool weight and decreased transit time in both healthy controls and patients with chronic constipation. Yet constipated patients taking bran still had lower stool weights and slower transit times than did healthy subjects.
When bran 20 g/day was compared with placebo in chronically constipated patients, bowel frequency and stool weight increased with both treatments,43 suggesting that factors other than intake may affect bowel function and transit time. However, bran was more effective than placebo in decreasing oroanal transit time.
Elderly constipated patients who received bran 10 g twice a day had significantly shorter transit times (89 hours vs 126 hours) than did those who received psyllium (a soluble fiber) 6 g twice daily. They also needed less additional laxative.44
Soluble fiber
Soluble fiber also affects the bowel habits of both healthy and constipated patients.
Methylcellulose, given to healthy volunteers at a dose of 4 g/day, resulted in statistically significant increases in stool weight, fecal water weight, and fecal solids.45 In constipated patients, methylcellulose 1 g/day was as effective as psyllium 3.4 g/day at increasing stool frequency, fecal water weight, and fecal solids.45
Konjac glucomannan was also shown to significantly increase stool frequency, water weight, and fecal solids.46
Psyllium. In a study that randomly assigned 22 patients with chronic constipation to receive either psyllium 5 g twice daily or placebo for 8 weeks, followed by a 4-week washout phase in which placebo was given,47 those who received psyllium reported significant improvements in stool consistency and pain with defecation, as well as significant increases in both stool frequency (3.8 vs 2.9 per week, P < .05) and stool weight (665 g vs 405 g, P < .05). However, colonic transit times and anorectal manometric measurements did not differ significantly between those who received psyllium vs placebo.47
Fiber may not help everyone
Others have also shown that while fiber may improve stool characteristics, it may not significantly alter the sensorimotor functions of the colon and pelvic floor.
Cheskin et al48 performed a crossover study in 10 constipated men and women in the community. Patients received either 24 g of psyllium fiber daily or a placebo fiber for 1 month and then crossed over to the other treatment for the next month. The most common cause of constipation in this study was pelvic floor dysfunction. Total gut transit time was significantly increased by psyllium fiber, and there was a trend toward increased stool frequency, demonstrating that psyllium clinically improved constipation. However, pelvic floor dysfunction, as measured by rectal manometry, was not improved.
It may be that only people with normal-transit constipation, not those with underlying slow-transit constipation or pelvic floor dysfunction, are helped by additional dietary fiber. Voderholzer and others40 studied 149 consecutive patients with chronic constipation and evaluated their response to at least 6 weeks of psyllium (Plantago ovata seeds 15 to 30 g/day) by serial symptom measurements, oroanal transit times, and functional rectoanal evaluation with defecography, manometry, and sigmoidoscopy. Of the patients with no evidence of pelvic floor dysfunction or slow-transit constipation, 85% improved. However, 80% of those with slow-transit constipation and 63% of those with pelvic floor dysfunction did not improve with the use of fiber. The authors concluded that it is reasonable to try dietary fiber in patients with constipation and, if no improvement is noted, to then consider further investigation for other subtypes of constipation (ie, slow-transit or pelvic-floor dysfunction).
Adverse effects may limit the use of fiber and may differ depending on the type of fiber used. Soluble fiber may be better tolerated, especially in patients with constipation-predominant irritable bowel syndrome.49 Side effects include the sensation of bloating and distention, excessive gas production, and abdominal cramping.
Our recommendations on fiber
We recommend the following regarding fiber in constipated patients:
- Increase fiber intake from natural foods up to 20 g/day. This increase should be completed over 2 to 3 weeks to minimize adverse effects.
- Consider adding a fiber supplement, such as psyllium, if increasing the intake of natural fiber does not relieve constipation-related symptoms.
- If symptoms persist despite the use of fiber supplements and diet and lifestyle modification, then further structural and functional investigation of the colon (anorectal manometry, colonoscopy, defecography, colon manometry) should be considered.
OSMOTIC LAXATIVES
Osmotic laxatives are molecules that are either not absorbed or poorly absorbed and that draw water into the intestinal lumen to maintain isotonicity between the intestinal contents and the serum. Examples are polyethylene glycol, sodium phosphate (Fleet phosphosoda), magnesium hydroxide, magnesium citrate, the sugars lactulose and sorbitol, and glycerin.
Certain formulations of this class of laxative can cause bloating, diarrhea, electrolyte disturbances, volume overload, or dehydration. These effects limit their use, and these medications should be used with caution in patients prone to renal insufficiency or cardiac abnormalities.
Polyethylene glycol
Polyethylene glycol is an exception. It is not absorbed and lacks electrolytes, making it an attractive option in patients with underlying renal or cardiac dysfunction. In several placebo-controlled trials,50–52 various formulations significantly increased stool frequency while significantly decreasing straining, use of other laxatives, and colonic transit. No increase in adverse effects was noted compared with placebo.
Compared with lactulose, polyethylene glycol at about 21 g/day significantly increased bowel movement frequency while significantly decreasing the sense of straining with bowel movements and flatus due to laxative use.51 Both polyethylene glycol and lactulose accelerate colonic transit, although polyethylene glycol does so to a greater extent.53
Polyethylene glycol has been safe and effective when used for up to 6 months.54
Lactulose and sorbitol
Carbohydrate or sugar-based laxatives, if taken in sufficient doses, have a cathartic effect through two mechanisms: a primary osmotic effect of the sugar itself and a secondary osmotic effect as a substrate for colonic bacteria to cleave to acid metabolites, which exert an osmotic effect in the colon. This secondary effect will be discussed in a later section.
Lactulose and sorbitol are sugars that are poorly absorbed by the intestine. Lactulose has been shown to be more effective than placebo in increasing stool frequency, volume, weight, and consistency in chronically constipated patients.55 In a head-to-head comparison between sugar laxatives, 70% sorbitol was as effective as lactulose in increasing the frequency of bowel movements, and it was similar in its adverse effects56; 70% sorbitol is a cost-effective alternative to lactulose in the elderly nursing home population.57
Compared with fiber alone, lactulose use leads to a significantly higher number of bowel movements and better stool consistency.58 However, when lactulose was compared with a combination of fiber and a stimulant laxative, it was less effective than the combination therapy.59,60
Sugar laxatives, while effective, may have dose-limiting or use-limiting adverse effects such as abdominal bloating and flatulence.
Phosphate, magnesium
Sodium phosphate, like polyethylene glycol, is often used as a bowel preparation before colonoscopy, for which it is about as good or slightly better than polyethylene glycol.61,62
Although magnesium and sodium phosphate preparations are effective, there are multiple reports of clinically significant electrolyte abnormalities, renal failure, and congestive heart failure occurring with these preparations. Therefore, they must be used with discretion and caution in appropriate patients with frequent monitoring.
STIMULANT (IRRITANT) LAXATIVES
Stimulant laxatives are usually reserved for use when bulking agents and osmotic laxatives fail. Their mechanism of action involves the alteration of intestinal motility and intestinal fluid secretion.
Anthraquinones (cascara, aloe, and senna), castor oil, and diphenylmethanes (bisacodyl) are the most commonly used stimulant laxatives. They work relatively quickly, often eliciting a bowel movement 2 to 8 hours after they are taken.
This class of laxatives has historically been underused or given for only short periods of time, owing to concern about impairing colonic function, damaging the enteric nervous system, causing laxative dependency, causing cathartic colon, and even causing colon cancer. However, there is very little evidence to support these concerns. Stimulant laxatives can be used on a more regular basis when bulking or osmotic agents fail.63
Possibly of greatest concern is the potential for the overuse and abuse of stimulant laxatives. Excessive use can cause electrolyte disturbances brought about by high-volume watery diarrhea. Risk factors for overuse and abuse include underlying psychiatric disturbances and eating disorders. Prescribing other types of laxatives or cathartic agents may reduce risk, but the potential for abuse exists with all categories of laxatives.
TEGASEROD: GONE BUT STILL AVAILABLE, ON A CONTROLLED BASIS
Tegaserod (Zelnorm), a serotonin (5-HT4) agonist, was used predominantly in women with constipation-predominant irritable bowel syndrome and in men and women with chronic constipation. However, it was suspended from the market in the United States in March 2007 owing to concern about a high risk of adverse cardiovascular effects compared with placebo.
In a double-blind, randomized controlled trial, men with chronic constipation who received tegaserod 6 mg twice a day for 12 weeks had more spontaneous bowel movements than those receiving placebo (P = .04).64
Lin et al65 evaluated the use of tegaserod 6 mg twice daily for 4 weeks in both men and women with chronic constipation. Those receiving tegaserod had significantly more spontaneous bowel movements per week, less straining, and better stool consistency than those receiving placebo.
Tegaserod can still be obtained for appropriate patients via a treatment investigational new drug application. Safety data are under further review by the US Food and Drug Administration. Studies of other serotonin agonists are under way.
LUBIPROSTONE
Lubiprostone (Amitiza) is an agonist of the chloride channel subtype 2, found on the apical membrane of intestinal epithelial cells. It causes increased chloride secretion into the intestinal lumen, enhancing intestinal fluid secretion. It has been shown to be effective in chronic constipation by improving stool consistency and increasing the motility of the small intestine and colon.66 It is approved for treating chronic constipation in adults.
In randomized, double-blind trials, patients receiving lubiprostone 24 fig twice daily for 4 weeks had significantly more bowel movements per week, reported significantly better stool consistency and less abdominal bloating and straining, and rated their constipation as less severe than did patients receiving placebo.67–69
More recently, in an open-label study, lubiprostone improved constipation symptoms when taken for up to 48 weeks.70
The drug is well tolerated, but its adverse effects include nausea (which appears to be dose-dependent and may diminish over time or if the drug is taken with food), diarrhea, and headache.68 Of note, the drug appears to be well tolerated by older people (65 years of age and older), in whom adverse effects occur less often than in younger users.71 However, adverse events may cause up to 20% of patients to stop taking the drug.69 When lubiprostone is discontinued, patients may once again revert to their baseline bowel habit.72
Lubiprostone has not been compared with conventional laxatives, and cost may prohibit it from becoming a first-line drug for chronic constipation.73
OTHER PROMOTILITY AGENTS
Several promotility agents have been studied for treating chronic idiopathic constipation.
Cisapride (Propulsid), a 5-HT3 receptor antagonist and 5-HT4 receptor agonist, and prucalopride, a 5-HT4 agonist, were effective in relieving symptoms associated with chronic constipation.74–76 However, safety issues (cardiac arrhythmias) necessitated withdrawal of cisapride from the US market in 2000. Prucalopride is undergoing clinical trials.77
Renzapride, a mixed 5-HT4 receptor agonist and 5-HT3 receptor antagonist, has been shown to improve stool consistency and to increase colonic transit in patients with constipation-predominant irritable bowel syndrome.78 Renzapride has been studied in patients with this condition,78–81 but not in patients with chronic constipation. Renzapride is in phase III clinical development in the United States for treating constipation-predominant irritable bowel syndrome.
EMERGING TREATMENTS
New drugs with novel mechanisms of action are being investigated for the treatment of chronic idiopathic constipation.
Neurotrophin-3, a neurotrophic factor, modulates the development of the nervous system by regulating the survival and differentiation of nerves.82 In patients with functional constipation, subcutaneous doses of neurotrophin-3 improved stool frequency, the number of complete spontaneous bowel movements, and stool consistency.83
Alvimopan is a selective antagonist of the muopioid receptor that is being studied for opiate-related constipation and postoperative ileus.84,85 Little of this drug is systemically absorbed and it does not cross the blood-brain barrier; thus, it relieves the opiate-related side effects, ie, bloating, abdominal discomfort, and reduced stool frequency, without interfering with the central analgesic effects.
Linaclotide (MD 1100), a poorly absorbed guanylate cyclase agonist, is also being investigated as a treatment for chronic constipation.86 Linaclotide increases intestinal fluid secretion and transit via stimulation of cyclic guanosine monophosphate production and activation of the cystic fibrosis transmembrane conductance regulator.86,87 In preliminary studies, linaclotide increased stool frequency and the Bristol Stool Form Scale consistency score (Table 1) by increasing intestinal fluid secretion and transit.86
Chenodeoxycholic acid is a bile acid that is synthesized from cholesterol.88 Treatment of constipation with chenodeoxycholic acid has been proposed, given its laxative effect. A study by Bazzoli et al89 showed increased stool frequency and a decrease in stool consistency in chronic constipation patients given chenodeoxycholic acid 10 mg/kg/day. The main side effect was diarrhea. Chenodeoxycholic acid may be worthwhile in the management of constipation, but more studies are needed.
PROBIOTICS AND PREBIOTICS
The bacteria of the colon influence peristalsis of the colon.90 Probiotics (live bacterial preparations) and prebiotics (nondigestible preparations that stimulate the growth or activity of beneficial colonic bacteria) have been gaining interest as potential therapies for constipation.91,92
Probiotic bacterial preparations are generally composed of strains of Bifodobacterium,93,94Lactobacillus,95 and combinations thereof, and are available as mixed preparations of multiple bacterial strains of Lactobacillus, Bifodobacterium, and Streptococcus species, such as VSL#3.96
Probiotics may help relieve constipation, but their effect may depend on the strain of bacteria used and the population being studied.97 In a double-blind parallel study in 70 healthy adults, ingestion of 375 g/day of milk fermented with B animalis strain DN-173 010 for 11 days reduced colon transit time by 20% from baseline. The effect was more pronounced in women, particularly in those with longer baseline transit.98
Lactic acid-producing bacteria are considered commensal organisms with essentially no pathogenic potential.99 A review of the safety of bifodobacteria and lactobacilli concluded there was no health risk to consumers.100
Prebiotics are short-chain carbohydrates such as lactulose that stimulate the activity of beneficial colonic bacteria.91 They are thought to have a small laxative effect that is likely both osmotic and due to beneficial actions of bacteria for which they are a substrate. Both konjac glucomannan and lactulose, sugar-based laxatives and prebiotics, have been shown to significantly increase the fecal concentrations of lactobacilli and total bacteria, possibly through increases in stool bulk.46 Prebiotics that have been the focus of research include inulin, fructo-oligosaccharides, and galacto-oligosaccharides.91 Evidence on the efficacy of probiotics and prebiotics at relieving symptoms of constipation, however, is inconclusive because few well-controlled clinical studies have been done.91,92
STRATEGIES FOR MANAGING CHRONIC CONSTIPATION
In the absence of secondary causes, treatment of chronic constipation is focused on relieving symptoms.
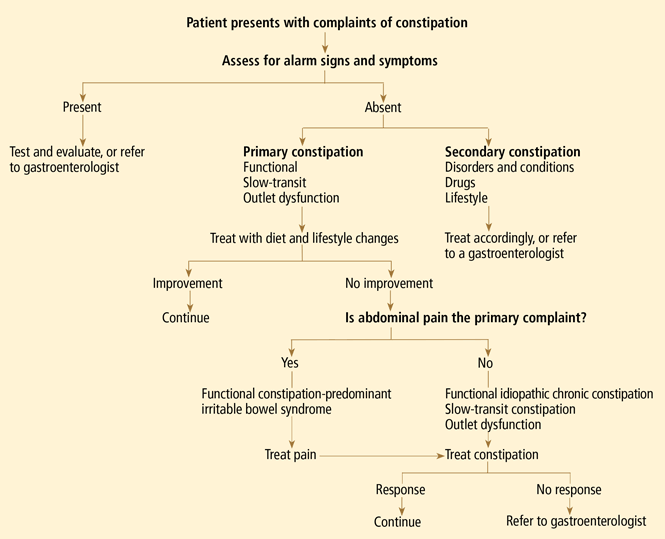
If symptoms are refractory to these traditional treatments, agents such as lactulose and polyethylene glycol may provide relief.11,21 Although they do not address the underlying cause of constipation, these agents increase the fluid content of the intestine, contributing to improved stool consistency, and consequently increase the frequency of bowel movements.
Lubiprostone similarly increases the fluid content of the colon, contributing to improved stool consistency, reduced fecal transit time, and increased frequency of bowel movements.66,70,102 Unlike lactulose and polyethylene glycol, which are indicated only for short-term use, lubiprostone has been found to be safe and effective when used for up to 48 weeks.70,71
Biofeedback is the preferred treatment for pelvic floor dyssynergia, in which it has a success rate of 70% to 81% and in which it is superior to standard treatment (laxatives, fiber, and education).103–105 In an instrument-based training program, patients receive auditory or visual feedback or both to help train the pelvic floor and relax the anal sphincter while simulating defecation. It also improves rectal sensation to assist in proper evacuation. The best outcomes are achieved when committed patients receive instruction from empathetic, properly trained physical therapists or other technicians. Studies show that the benefits of biofeedback are long-lasting.104 It does not improve slow-transit constipation, though pelvic floor dyssynergia and slow-transit constipation can overlap.
- Higgins PD, Johanson JF. Epidemiology of constipation in North America: a systematic review. Am J Gastroenterol 2004; 99:750–759.
- Brandt LJ, Prather CM, Quigley EM, Schiller LR, Schoenfeld P, Talley NJ. Systematic review on the management of chronic constipation in North America. Am J Gastroenterol 2005; 100 suppl 1:S5–S21.
- Pleis JR, Lethbridge-Cejku M. Summary health statistics for U.S. adults: National Health Interview Survey, 2005. Vital Health Stat 10 2006; 232:1–153.
- Dennison C, Prasad M, Lloyd A, Bhattacharyya SK, Dhawan R, Coyne K. The health-related quality of life and economic burden of constipation. Pharmacoeconomics 2005; 23:461–476.
- Arce DA, Ermocilla CA, Costa H. Evaluation of constipation. Am Fam Physician 2002; 65:2283–2290.
- Johanson JF, Sonnenberg A, Koch TR. Clinical epidemiology of chronic constipation. J Clin Gastroenterol 1989; 11:525–536.
- Harari D, Gurwitz JH, Minaker KL. Constipation in the elderly. J Am Geriatr Soc 1993; 41:1130–1140.
- Johanson JF, Kralstein J. Chronic constipation: a survey of the patient perspective. Aliment Pharmacol Ther 2007; 25:599–608.
- Lembo A, Camilleri M. Chronic constipation. N Engl J Med 2003; 349:1360–1368.
- Sandler RS, Drossman DA. Bowel habits in young adults not seeking health care. Dig Dis Sci 1987; 32:841–845.
- Longstreth GF, Thompson WG, Chey WD, Houghton LA, Mearin F, Spiller RC. Functional bowel disorders. Gastroenterology 2006; 130:1480–1491.
- Locke GR, Pemberton JH, Phillips SF. AGA technical review on constipation. Gastroenterology 2000; 119:1766–1778.
- Rao SS. Constipation: evaluation and treatment. Gastroenterol Clin North Am 2003; 32:659–683.
- Locke GR, Pemberton JH, Phillips SF. American Gastroenterological Association Medical Position Statement: guidelines on constipation. Gastroenterology 2000; 119:1761–1766.
- Prather CM. Subtypes of constipation: sorting out the confusion. Rev Gastroenterol Disord 2004; 4 suppl 2:S11–S16.
- Talley NJ, Jones M, Nuyts G, Dubois D. Risk factors for chronic constipation based on a general practice sample. Am J Gastroenterol 2003; 98:1107–1111.
- Bharucha AE. Treatment of severe and intractable constipation. Curr Treat Options Gastroenterol 2004; 7:291–298.
- Borum ML. Constipation: evaluation and management. Prim Care 2001; 28:577–590.
- Camilleri M, Ford MJ. Review article: colonic sensorimotor physiology in health, and its alteration in constipation and diarrhoeal disorders. Aliment Pharmacol Ther 1998; 12:287–302.
- Knowles CH, Martin JE. Slow transit constipation: a model of human gut dysmotility. Review of possible aetiologies. Neurogastroenterology Motility 2000; 12:181–196.
- Schiller LR. Review article: the therapy of constipation. Aliment Pharmacol Ther 2001; 15:749–763.
- Crowell MD. Pathogenesis of slow transit and pelvic floor dysfunction: from bench to bedside. Rev Gastroenterol Disord 2004; 4 suppl 2:S17–S27.
- Wood JD. Neuropathophysiology of irritable bowel syndrome. J Clin Gastroenterol 2002; 35 suppl 1:S11–S22.
- Chitkara DK, Bredenoord AJ, Cremonini F, et al. The role of pelvic floor dysfunction and slow colonic transit in adolescents with refractory constipation. Am J Gastroenterol 2004; 99:1579–1584.
- El-Salhy M. Gastrointestinal transit in an animal model of human diabetes type 2: relationship to gut neuroendocrine peptide contents. Ups J Med Sci 2002; 107:101–110.
- Crowell MD. Role of serotonin in the pathophysiology of the irritable bowel syndrome. Br J Pharmacol 2004; 141:1285–1293.
- Gershon MD. Review article: roles played by 5-hydroxytryptamine in the physiology of the bowel. Aliment Pharmacol Ther 1999; 13 suppl 2:15–30.
- Gershon MD. Serotonin and its implication for the management of irritable bowel syndrome. Rev Gastroenterol Disord 2003; 3 suppl 2:S25–S34.
- Grider JR, Foxx-Orenstein AE, Jin JG. 5-Hydroxytryptamine4 receptor agonists initiate the peristaltic reflex in human, rat, and guinea pig intestine. Gastroenterology 1998; 115:370–380.
- Krishnamurthy S, Schuffler MD, Rohrmann CA, Pope CE. Severe idiopathic constipation is associated with a distinctive abnormality of the colonic myenteric plexus. Gastroenterology 1985; 88:26–34.
- O'Donnell LJ, Virjee J, Heaton KW. Detection of pseudodiarrhoea by simple clinical assessment of intestinal transit rate. BMJ 1990; 300:439–440.
- Meshkinpour H, Selod S, Movahedi H, Nami N, James N, Wilson A. Effects of regular exercise in management of chronic idopathic constipation. Dig Dis Sci 1998; 43:2379–2383.
- Young RJ, Beerman LE, Vanderhoff JA. Increasing oral fluids in chronic constipation in children. Gastroenterol Nurs 1998; 21:156–161.
- Tuteja AK, Talley NJ, Joos SK, Woehl JV, Hickam DH. Is constipation associated with decreased physical activity in normally active subjects? Am J Gastroenterol 2005; 100:124–129.
- Towers AL, Burgio KL, Locher JL, Merkel IS, Safaeian M, Wald A. Constipation in the elderly: influence of dietary, psychological, and physiological factors. J Am Geriatr Soc 1994; 42:701–706.
- Anti J, Pignataro G, Armuzzi A, et al. Water supplementation enhances the effect of high-fiber diet on stool frequency and laxative consumption in adult patients with functional constipation. Hepatogastroenterology 1998; 45:727–732.
- Murakami K, Sasaki S, Okubo H, et al. Association between dietary fiber, water and magnesium intake and functional constipation among young Japanese women. Eur J Clin Nutr 2007; 61:616–622.
- Wong ML, Wee S, Pin CH, Gan GL, Ye HC. Sociodemographic and lifestyle factors associated with constipation in an elderly Asian community. Am J Gastroenterol 1999; 94:1283–1291.
- Dukas L, Willett WC, Giovannucci EL. Association between physical activity, fiber intake, and other lifestyle variables and constipation in a study of women. Am J Gastroenterol 2003; 98:1790–1796.
- Voderholzer WA, Schatke W, Muhldorfer BE, et al. Clinical response to dietary fiber treatment of chronic constipation. Am J Gastroenterol 1997; 92:95–98.
- Graham DY, Moser SE, Estes MK. The effect of bran on bowel function in constipation. Am J Gastroenterol 1982; 77:599–603.
- Muller-Lissner SA. Effect of wheat bran on weight of stool and gastrointestinal transit time: a meta analysis. Br Med J (Clin Res Ed) 1988; 296:615–617.
- Badiali D, Corazziari E, Habib FI, et al. Effect of wheat bran in treatment of chronic nonorganic constipation. A double-blind controlled trial. Dig Dis Sci 1995; 40:349–356.
- Andersson H, Basaeus I, Falkheden T, Melkersson M. Transit time in constipated geriatric patients during treatment with bulk laxative and bran: a comparison. Scand J Gastroenterol 1979; 14:821–826.
- Hamilton JW, Wagner J, Burdick BB, Bass P. Clinical evaluation of methyl-cellulose as a bulk laxative. Dig Dis Sci 1988; 33:993–998.
- Chen HL, Cheng HC, Liu YJ, Liu SY, Wu WT. Konjac acts as a natural laxative by increasing stool bulk and improving colonic ecology in healthy adults. Nutrition 2006; 22:1112–1119.
- Ashraf W, Park F, Lof J, Quigley EM. Effects of psyllium therapy on stool characteristics, colon transit and anorectal function in chronic idiopathic constipation. Aliment Pharmacol Ther 1995; 9:639–647.
- Cheskin LJ, Kamal N, Crowell MD, Schuster MM, Whitehead WE. Mechanisms of constipation in older persons and effects of fiber compared with placebo. J Am Geriatr Soc 1995; 43:666–669.
- Bijkerk CJ, Muris JW, Knottnerus JA, Hoes AW, de Wit NJ. Systematic review: the role of different types of fiber in the treatment of irritable bowel syndrome. Aliment Pharmacol Ther 2004; 19:245–251.
- Corazziari E, Badiali D, Habib FI, et al. Small volume isosmotic polyethylene glycol electrolyte balanced solution (PMF–100) in treatment of chronic nonorganic constipation. Dig Dis Sci 1996; 41:1636–1642.
- Attar A, Lemann M, Ferguson A, et al. Comparison of a low dose polyethylene glycol electrolyte solution with lactulose for treatment of chronic constipation. Gut 1999; 44:226–230.
- Corazziari E, Badiali D, Bazzocchi G, et al. Long term efficacy, safety, and tolerabilitity of low daily doses of isosmotic polyethylene glycol electrolyte balanced solution (PMF–100) in the treatment of functional chronic constipation. Gut 2000; 46:522–526.
- Fritz E, Hammer HF, Lipp RW, Högenauer C, Stauber R, Hammer J. Effects of lactulose and polyethylene glycol on colonic transit. Aliment Pharmacol Ther 2005; 21:259–268.
- DiPalma JA, Cleveland MV, McGowan J, Herrera JL. A randomized, multicenter, placebo-controlled trial of polyethylene glycol laxative for chronic treatment of chronic constipation. Am J Gastroenterol 2007; 102:1436–1441.
- Bass P, Dennis S. The laxative effects of lactulose in normal and constipated subjects. J Clin Gastroenterol 1981; 3 suppl 1:23–28.
- Lederle FA, Busch DL, Mattox KM, West MJ, Aske DM. Cost-effective treatment of constipation in the elderly: a randomized double-blind comparison of sorbitol and lactulose. Am J Med 1990; 89:597–601.
- Volicer L, Lane P, Panke J, Lyman P. Management of constipation in residents with dementia: sorbitol effectiveness and cost. J Am Med Dir Assoc 2004; 5:239–241.
- Quah HM, Ooi BS, Seow-Choen F, Sng KK, Ho KS. Prospective randomized crossover trial comparing fibre with lactulose in the treatment of idiopathic chronic constipation. Tech Coloproctol 2006; 10:111–114.
- Passmore AP, Davies KW, Flanagan PG, Stoker C, Scott MG. A comparison of Agiolax and lactulose in elderly patients with chronic constipation. Pharmacology 1993; 47 suppl 1:249–252.
- Passmore AP, Wilson-Davies K, Stoker C, Scott ME. Chronic constipation in long stay elderly patients: a comparison of lactulose and a senna-fibre combination. BMJ 1993; 307:769–771.
- Poon CM, Lee DW, Mak SK, et al. Two liters of polyethylene glycol-electrolyte lavage solution versus sodium phosphate as bowel cleansing regimen for colonoscopy: a prospective randomized controlled trial. Endoscopy 2002; 34:560–563.
- Rostom A, Jolicoeur E, Dube C, et al. A randomized prospective trial comparing different regimens of oral sodium phosphate and polyethylene glycol-based lavage solution in the preparation of patients for colonoscopy. Gastrointest Endosc 2006; 64:544–552.
- Wald A. Is chronic use of stimulant laxatives harmful to the colon? J Clin Gastroenterol 2003; 36:386–389.
- Fried M, Johanson JF, Gwee KA, Wagner A, Pecher E, Rueegg P. Efficacy of tegaserod in chronic constipation in men. Am J Gastroenterol 2007; 102:362–370.
- Lin SR, Ke MY, Luo JY, et al. A randomized, double-blind, placebo-controlled trial assessing the efficacy and safety of tegaserod in patients from China with chronic constipation. World J Gastroenterol 2007; 13:732–739.
- Camilleri M, Bharucha AE, Ueno R, et al. Effect of a selective chloride channel activator, lubiprostone, on gastrointestinal transit, gastric sensory, and motor functions in healthy volunteers. Am J Physiol Gastrointest Liver Physiol 2006; 290:G942–G947.
- McKeage K, Plosker GL, Siddiqui MA. Lubiprostone. Drugs 2006; 66:873–879.
- Johanson JF, Gargano MA, Patchen ML, Ueno R. Efficacy and safety of a novel compound, RU-0211, for the treatment of constipation [abstract]. Gastroenterology 2002; 122 suppl 1:A315.
- Johanson JF, Gargano MA, Holland PC, Patchen ML, Ueno R. Multicenter open-label study of oral lubiprostone for the treatment of chronic constipation [abstract]. Am J Gastroenterol 2005; 100 suppl:S331.
- Johanson JF, Panas R, Holland P, Ueno R. Long-term efficacy of lubiprostone for the treatment of chronic constipation [abstract]. Gastroenterology 2006; 130 suppl 2:A317.
- Ueno R, Panas R, Wahle A, Zhu Y, Holland P. Long-term safety and efficacy of lubiprostone for the treatment of chronic constipation in the elderly [abstract]. Gastroenterology 2006; 130 suppl 2:A188.
- Johanson JF, Gargano MA, Holland P, Patchen ML, Ueno R. Phase III, randomized withdrawal study of RU-0211, a novel chloride channel activator for the treatment of constipation [abstract]. Gastroenterology 2004; 126 suppl 2:A100.
- Rivkin A, Chagan L. Lubiprostone: chloride channel activator for chronic constipation. Clin Ther 2006; 28:2008–2021.
- Johanson JF, Miner PB, Parkman HP, et al. Prucalopride improves bowel movement frequency and symptoms in patients with chronic constipation: results of two double-blind, placebo-controlled trials [abstract]. Gastroenterology 2000; 118 suppl 2:A175.
- Cash BD, Chey WD. Review article: the role of serotonergic agents in the treatment of patients with primary chronic constipation. Aliment Pharmacol Ther 2005; 22:1047–1060.
- Altabas K, Biliæ A, Jurciæ D, et al. The efficacy of cisapride vs. placebo and diet in patients with chronic constipation. Coll Antropol 2003; 27:197–204.
- Camilleri M, Kerstens R, Rykx A, Vandeplassche L. A placebo-controlled trial of prucalopride for severe chronic constipation. N Engl J Med 2008; 358:2344–2354.
- Camilleri M, McKinzie S, Fox J, et al. Effect of renzapride on transit in constipation-predominant irritable bowel syndrome. Clin Gastroenterol Hepatol 2004; 2:895–904.
- Tack J, Middleton SJ, Horne MC, et al. Pilot study of the efficacy of renzapride on gastrointestinal motility and symptoms in patients with constipation-predominant irritable bowel syndrome. Aliment Pharmacol Ther 2006; 23:1655–1665.
- Henderson JC, Palmer RM, Meyers NL, Spiller RC. A phase IIb clinical study of renzapride in mixed symptom (alternating) irritable bowel syndrome [abstract]. Gastroenterology 2004; 126 suppl 2:A644.
- Meyers NL, Palmer RMJ, George A. Efficacy and safety of renzapride in patients with constipation-predominant IBS: a phase IIb study in the UK primary healthcare setting [abstract]. Gastroenterology 2004; 126 suppl 2:A640.
- Coulie B, Szarka LA, Camilleri M, et al. Recombinant human neurotrophic factors accelerate colonic transit and relieve constipation in humans. Gastroenterology 2000; 119:41–50.
- Parkman HP, Rao SS, Reynolds JC, et al. Neurotrophin-3 improves functional constipation. Am J Gastroenterol 2003; 98:1338–1347.
- Camilleri M. Alvimopan, a selective peripherally acting muopioid antagonist. Neurogastroenterol Motil 2005; 17:157–165.
- Holzer P. Opioids and opioid receptors in the enteric nervous system: from a problem in opioid analgesia to a possible new prokinetic therapy in humans. Neurosci Lett 2004; 361:192–195.
- Kurtz C, Fitch D, Busby R, et al. Effects of multidose administration of MD-1100 on safety, tolerability, exposure and pharmacodynamics in healthy subjects [abstract]. Gastroenterology 2006; 130 suppl 2:A26.
- Eutamene H, Theodorou V, Tondereau V, et al. Influence of guanylate cyclase C binding ligand MD-1100 on TNBS-induced visceral hypersensitivity in WT vs. KO guanylate cyclase C deficient mice [abstract]. Gastroenterology 2006; 130 suppl 2:A597.
- Broughton G. Chenodeoxycholate: the bile acid. The drug. a review. Am J Med Sci 1994; 307:54–63.
- Bazzoli F, Malavolti M, Petronelli A, Barbara L, Roda E. Treatment of constipation with chenodeoxycholic acid. J Int Med Res 1983; 11:120–123.
- Picard C, Fioramonti J, Francois A, Robinson T, Neant F, Matuchansky C. Review article: bifidobacteria as probiotic agents – physiological effects and clinical benefits. Aliment Pharmacol Ther 2005; 22:495–512.
- Macfarlane S, Macfarlane GT, Cummings JH. Review article: prebiotics in the gastrointestinal tract. Aliment Pharmacol Ther 2006; 24:701–714.
- Ouwehand A, Lagstrom H, Suomalainen T, Salminen S. Effect of probiotics on constipation, fecal azoreductase activity and fecal mucin content in the elderly. Ann Nutr Metab 2002; 46:159–162.
- Whorwell P, Altringer L, Morel J, et al. Efficacy of an encapsulated probiotic Bifidobacterium infantis 35624 in women with irritable bowel syndrome. Am J Gastroenterol 2006; 101:1581–1590.
- O'Mahony L, McCarthy J, Kelly P, et al. Lactobacillus and Bifidobacterium in irritable bowel syndrome: symptom responses and relationship to cytokine profiles. Gastroenterology 2005; 128:541–551.
- Koebnick C, Wagner I, Leitzmann P, Stern U, Zunft H. Probiotic beverage containing Lactobacillus casei Shirota improves gastrointestinal symptoms in patients with chronic constipation. Can J Gastroenterol 2003; 17:655–659.
- Kim HJ, Camilleri M, McKinzie S, et al. A randomized controlled trial of a probiotic, VSL#3, on gut transit and symptoms in diarrhoea-predominant irritable bowel syndrome. Aliment Pharmacol Ther 2003; 17:895–904.
- Fernández-Bañares F. Nutritional care of the patient with constipation. Best Pract Res Clin Gastroenterol 2006; 20:575–587.
- Bouvier M, Meance S, Bouley C, Berta J, Grimaud J. Effects of consumption of milk fermented by the probiotic strain Bifidobacterium animalis DN-173 010 on colonit transit times in healthy humans. Biosci Microflor 2001; 20 2:43–48.
- Makelainen H, Tahvonen R, Salminen S, Ouwehand AC. In vivo safety assessment of two Bifidobacterium longum strains. Microbiol Immunol 2003; 47:911–914.
- Borriello SP, Hammes WP, Holzapfel W, et al. Safety of probiotics that contain lactobacilli or bifidobacteria. Clin Infect Dis 2003; 36:775–780.
- Ramkumar D, Rao S. Efficacy and safety of traditional medical therapies for chronic constipation: systematic review. Am J Gastroenterol 2005; 100:936–971.
- Ueno R, Osama H, Habe T, Engelke K, Patchen M. Oral SPI-0211 increases intestinal fluid secretion and chloride concentration without altering serum electrolyte levels [abstract]. Gastroenterology 2004; 126 suppl 2:A298.
- Rao SS, Seaton K, Miller M, et al. Randomized controlled trial of biofeedback, sham biofeedback, and standard therapy for dyssynergic defecation. Clin Gastroenterol Hepatol 2007; 5:331–338.
- Chiarioni G, Whitehead WE, Pezza V, Morelli A, Bassotti G. Biofeedback is superior to laxatives for normal transit constipation due to pelvic floor dyssynergia. Gastroenterology 2006; 130:657–664.
- Heymen S, Scarlett Y, Jones K, Ringel Y, Drossman D, Whitehead WE. Randomized, controlled trial shows biofeedback to be superior to alternative treatments for patients with pelvic floor dyssynergia-type constipation. Dis Colon Rectum 2007; 50:428–441.
Constipation is both a symptom and, when chronic, a multisymptom disorder, and it can overlap with other gastrointestinal tract disorders such as dyspepsia and gastroesophageal reflux disease. Furthermore, one should keep in mind the possibility of cancer and be alert for its warning signs.
Since constipation has a variety of causes and forms, one treatment does not fit all patients. Conservative measures such as recommending that the patient increase his or her intake of dietary fiber and water and engage in more physical activity are still the cornerstone of treatment, but they do not help all patients. On the other hand, polyethylene glycol and stimulant laxatives, which are traditionally given only for a short time, can be safe and effective when given long-term if other agents fail. New agents have become available or are in development.
In this article we outline our approach to constipation, as a guide for internists.
CONSTIPATION IS COMMON, BUT HOW SHOULD WE DEFINE IT?
Constipation affects 2% to 27% (average 14.8%) of the North American adult population—approximately 63 million people.1 It is more common than many other chronic diseases, including hypertension (48 million people), migraine (33 million), obesity (50 million), and diabetes mellitus (15 million).1–3
Constipation affects more women than men (2.1:1 ratio) and more nonwhites than whites (1.68:1).1 It occurs in all age groups but is more common in those older than 65 years and younger than 4 years.4,5
Constipation accounts for more than 2.5 million office visits and more than $500 million spent on laxatives per year.6,7 Also, people with constipation may report decreased productivity and increased absenteeism.8
The broad range in the prevalence of constipation cited above reflects differences in how it is defined and, in particular, a lack of agreement between how patients and physicians perceive it.1,9 Physicians mainly define constipation on the basis of stool frequency, considering fewer than three bowel movements per week to be abnormal.1 In contrast, patients typically define it on the basis of bothersome symptoms such as straining, passage of hard stool, unproductive urges, inability to defecate at will, and sensations of incomplete evacuation or abdominal bloating.1,9,10
The Rome III diagnostic criteria were developed to provide a consistent diagnostic approach for use in clinical practice and clinical trials.11 The Rome III criteria define functional chronic constipation as a chronic bowel disorder characterized by two or more of the following:
- Straining
- Lumpy or hard stools
- Sensations of incomplete evacuation
- Sensations of anorectal obstruction or blockage
- Use of manual maneuvers to facilitate defecation (eg, digital evacuation, support of the pelvic floor) during at least 25% of defecations
- Fewer than three bowel movements per week.
In addition, loose stools should rarely occur without the use of laxatives, and there should be insufficient criteria for irritable bowel syndrome.11 Chronicity is established by symptom onset within the previous 6 months and symptom duration of at least 3 months.
In contrast, patients with irritable bowel syndrome, also a functional bowel disorder, experience recurrent abdominal pain and discomfort associated with two or more of the following: symptom improvement with defecation, symptom onset associated with a change in the frequency of bowel movements, and a change in the form or appearance of the stool.
THREE TYPES OF IDIOPATHIC CONSTIPATION
There are three types of primary or idiopathic constipation5,9,12,13:
- Functional
- Slow-transit
- Outlet dysfunction.
Functional constipation includes functional chronic idiopathic constipation and constipation-predominant irritable bowel syndrome. It presents with a sense of difficult or delayed evacuation, hard stools, or abdominal bloating or discomfort.6,9,13 The predominant symptom of constipation-predominant irritable bowel syndrome is severe discomfort or pain; in chronic idiopathic constipation, pain and discomfort may be present but are not the primary symptom.
Slow-transit constipation (or delayed-transit constipation) is associated with a prolonged time between bowel movements. Its symptoms include low stool frequency, lack of urge to defecate, abdominal distention, bloating, and abdominal discomfort.14
Outlet dysfunction. Disorders of defecation can be due to mechanical causes such as Hirschsprung disease, anal stricture, cancer, prolapse, and large rectoceles, or from pelvic floor dysfunction. Pelvic floor dysfunction may be due to inadequate or excessive perineal descent or to inadequate propulsive forces, as may occur in neurologic or neuromuscular conditions and dyssynergia.
Pelvic floor dyssynergia, also called anorectal dyssynergia, dyssynergic defecation, and anismus, results from a functional defect in coordinated evacuation. The characteristic symptom is a feeling of being unable to adequately empty the rectum.14 Other symptoms such as excessive straining and manual disimpaction indicate but are not unique to pelvic floor dyssynergia.14,15
Combined forms. Patients may have more than one type of primary constipation and presentation, and pelvic floor dyssynergia has been shown to prolong intestinal transit, which may improve with treatment.
Secondary constipation can be due to causes such as diet, lifestyle, certain medications (calcium channel blockers, beta-blockers, opioids, diuretics, antidepressants, anticonvulsants, antacids, anticholinergics, and antispasmodics),5,16 underlying medical conditions (diabetes, hypothyroidism, multiple sclerosis, parkinsonism),16,17 pregnancy, and advanced age.18
NEUROTRANSMITTERS MAY PLAY A ROLE
Among the mechanisms thought to cause chronic constipation are impaired gastrointestinal motility,19–22 reduced intestinal secretions,21–23 and inadequate reflex relaxation of the pelvic floor muscles.22,24
Neurotransmitters such as serotonin, somatostatin, peptide YY, and vasoactive intestinal peptide affect intestinal secretion and motility.25,26 Hyperactivity of these neurotransmitters associated with increased secretion and motility results in diarrhea, whereas hypoactivity leads to decreased secretion, delayed transit, and constipation.23
Serotonin has a role in regulating visceral pain perception and intestinal motility, as well as secretion.26–28 Clinical trials have shown that activation of serotonin receptors in the gut enhances gastrointestinal motility, inhibits visceral sensitivity, and stimulates intestinal secretion.26,27,29
A hypothesis has recently been proposed that degeneration of enteric neurons may also play a role in the development of severe idiopathic constipation.30
DIAGNOSIS IS MOSTLY CLINICAL
The history and physical examination remain the cornerstones in the diagnosis and subsequent treatment of chronic constipation.
History
Risk factors for primary and secondary constipation to note during the interview include age (< 4 years, > 65 years); low-fiber diet; female sex; lack of physical activity; history of childhood constipation, endocrine and neuromuscular disorders, abuse, depression, or anxiety; family history of cancer; and personal history of pelvic surgery.
Since drugs can also cause chronic constipation, especially in elderly or immobile patients, medication lists should be reviewed and adjustments should be made if necessary (or possible) before recommending laxatives or invasive testing, if no alarm signs are present.
Alarm signs such as weight loss, hematochezia, melena, change in bowel habits, and symptoms refractory to therapy may represent colon cancer and indicate the need for early diagnostic testing.
Physical examination
Physical examination should always include inspection of the perianal area for evidence of hemorrhoids or fissures. Digital rectal examination may reveal a contracted sphincter or a puborectalis muscle that contracts with the Valsalva maneuver, suggesting dysfunction.
Laboratory testing
If the history and physical examination suggest that the constipation may be secondary, or if the patient is 50 years of age or older, then laboratory studies such as a complete blood cell count, serum electrolyte levels, blood sugar level, and thyroid function studies may help rule out a metabolic, endocrine, or organic cause.
Colonoscopy, other tests
At present, little evidence suggests that routine testing is warranted in patients without evidence of secondary constipation and without alarm signs. However, diagnostic studies are indicated in patients 50 years of age and older, as well as in those with alarm symptoms such as hematochezia, anemia, a positive fecal occult blood test, unintentional loss of more than 10 pounds, family history of colon cancer or inflammatory bowel disease, fever, nausea, vomiting, acute onset (especially in the elderly), and lack of improvement with conventional therapies regardless of age.2
The full length of the colon should be inspected by colonoscopy or by flexible sigmoidoscopy paired with a barium enema study to rule out structural disease. Of note, all patients 50 years of age or older should be screened for colon cancer.
If the patient does not respond to therapy, further tests such as colonic transit studies, anorectal manometry with balloon expulsion, and, possibly, defecating proctography or dynamic pelvic magnetic resonance imaging may be considered. These patients would likely also benefit from referral to a gastroenterologist for further management
DIET AND LIFESTYLE AS TREATMENT
For many years, health care providers have provided reassurance and recommended diet and lifestyle modifications as treatment for constipation. Increased water intake, increased activity, and a scheduled attempt at defecation when motor activity in the colon is highest, ie, in the morning or after eating, have all been recommended.
Data on the efficacy of these recommendations are scarce and often contradictory. Studies have shown that increasing water intake or daily exercise is not always helpful.32–34 Nevertheless, many patients who comply with dietary and exercise recommendations have improvement in symptoms. Eating fewer meals per day (and hence taking in fewer calories) has been shown to be associated with constipation in the elderly. However, no relationships between fiber or fluid intake and constipation were noted.35
In a study in which chronically constipated patients were fed a standardized diet that contained 25 g of fiber a day, stool frequency increased significantly and laxative use decreased.36 While on a high-fiber diet, the patients were divided into two groups, one that drank 1.1 L of fluid per day and one that drank 2.1 L of mineral water per day. Both groups experienced further improvements in stool frequency and decreases in laxative use, with the mineral-water group benefiting the most.36
Recently, Murakami and others37 found, in a cross-sectional study in young Japanese women with low daily fiber intake (6.4 g/day), that low water intake from foods and low magnesium intake were associated with an increasing prevalence of functional constipation as defined by the Rome III criteria. Constipation was also found to be significantly associated with low intake of fruits and vegetables in a study from Singapore.38
Moderate physical activity and high fiber intake may be associated with a lower prevalence of constipation in women. In the Nurses’ Health Study, more than 62,000 women between the ages of 36 and 61 were surveyed, and those who said they engaged in daily physical activity had a lower prevalence of constipation (prevalence ratio [PR] = 0.56, 95% confidence interval [CI] 0.44–0.70), as did those with a median fiber intake of 20 g/day (PR = 0.64, 95% CI 0.57–0.73).39
BULK LAXATIVES (FIBER SUPPLEMENTS): THE FIRST-LINE TREATMENT
Fiber remains the first-line treatment for constipation. It may relieve or improve symptoms in functional constipation. However, fewer than 30% of patients with either slow-transit constipation or pelvic floor dysfunction have improvement in symptoms with fiber, and in these types of constipation it can even worsen symptoms.40
There is much confusion about what types of fiber should be recommended and how the various types of fiber perform in resolving constipation.
Insoluble fiber
Insoluble fiber resists bacterial degradation in the colon and can retain more water than soluble fiber can.
Bran 20 g/day increased the frequency of bowel movements by 55%, increased fecal weight by 157%, and decreased intestinal transit time by 50% in women who had three or fewer bowel movements per week.41
Muller-Lissner42 and others performed a meta-analysis and found that bran (25 g/day) increased stool weight and decreased transit time in both healthy controls and patients with chronic constipation. Yet constipated patients taking bran still had lower stool weights and slower transit times than did healthy subjects.
When bran 20 g/day was compared with placebo in chronically constipated patients, bowel frequency and stool weight increased with both treatments,43 suggesting that factors other than intake may affect bowel function and transit time. However, bran was more effective than placebo in decreasing oroanal transit time.
Elderly constipated patients who received bran 10 g twice a day had significantly shorter transit times (89 hours vs 126 hours) than did those who received psyllium (a soluble fiber) 6 g twice daily. They also needed less additional laxative.44
Soluble fiber
Soluble fiber also affects the bowel habits of both healthy and constipated patients.
Methylcellulose, given to healthy volunteers at a dose of 4 g/day, resulted in statistically significant increases in stool weight, fecal water weight, and fecal solids.45 In constipated patients, methylcellulose 1 g/day was as effective as psyllium 3.4 g/day at increasing stool frequency, fecal water weight, and fecal solids.45
Konjac glucomannan was also shown to significantly increase stool frequency, water weight, and fecal solids.46
Psyllium. In a study that randomly assigned 22 patients with chronic constipation to receive either psyllium 5 g twice daily or placebo for 8 weeks, followed by a 4-week washout phase in which placebo was given,47 those who received psyllium reported significant improvements in stool consistency and pain with defecation, as well as significant increases in both stool frequency (3.8 vs 2.9 per week, P < .05) and stool weight (665 g vs 405 g, P < .05). However, colonic transit times and anorectal manometric measurements did not differ significantly between those who received psyllium vs placebo.47
Fiber may not help everyone
Others have also shown that while fiber may improve stool characteristics, it may not significantly alter the sensorimotor functions of the colon and pelvic floor.
Cheskin et al48 performed a crossover study in 10 constipated men and women in the community. Patients received either 24 g of psyllium fiber daily or a placebo fiber for 1 month and then crossed over to the other treatment for the next month. The most common cause of constipation in this study was pelvic floor dysfunction. Total gut transit time was significantly increased by psyllium fiber, and there was a trend toward increased stool frequency, demonstrating that psyllium clinically improved constipation. However, pelvic floor dysfunction, as measured by rectal manometry, was not improved.
It may be that only people with normal-transit constipation, not those with underlying slow-transit constipation or pelvic floor dysfunction, are helped by additional dietary fiber. Voderholzer and others40 studied 149 consecutive patients with chronic constipation and evaluated their response to at least 6 weeks of psyllium (Plantago ovata seeds 15 to 30 g/day) by serial symptom measurements, oroanal transit times, and functional rectoanal evaluation with defecography, manometry, and sigmoidoscopy. Of the patients with no evidence of pelvic floor dysfunction or slow-transit constipation, 85% improved. However, 80% of those with slow-transit constipation and 63% of those with pelvic floor dysfunction did not improve with the use of fiber. The authors concluded that it is reasonable to try dietary fiber in patients with constipation and, if no improvement is noted, to then consider further investigation for other subtypes of constipation (ie, slow-transit or pelvic-floor dysfunction).
Adverse effects may limit the use of fiber and may differ depending on the type of fiber used. Soluble fiber may be better tolerated, especially in patients with constipation-predominant irritable bowel syndrome.49 Side effects include the sensation of bloating and distention, excessive gas production, and abdominal cramping.
Our recommendations on fiber
We recommend the following regarding fiber in constipated patients:
- Increase fiber intake from natural foods up to 20 g/day. This increase should be completed over 2 to 3 weeks to minimize adverse effects.
- Consider adding a fiber supplement, such as psyllium, if increasing the intake of natural fiber does not relieve constipation-related symptoms.
- If symptoms persist despite the use of fiber supplements and diet and lifestyle modification, then further structural and functional investigation of the colon (anorectal manometry, colonoscopy, defecography, colon manometry) should be considered.
OSMOTIC LAXATIVES
Osmotic laxatives are molecules that are either not absorbed or poorly absorbed and that draw water into the intestinal lumen to maintain isotonicity between the intestinal contents and the serum. Examples are polyethylene glycol, sodium phosphate (Fleet phosphosoda), magnesium hydroxide, magnesium citrate, the sugars lactulose and sorbitol, and glycerin.
Certain formulations of this class of laxative can cause bloating, diarrhea, electrolyte disturbances, volume overload, or dehydration. These effects limit their use, and these medications should be used with caution in patients prone to renal insufficiency or cardiac abnormalities.
Polyethylene glycol
Polyethylene glycol is an exception. It is not absorbed and lacks electrolytes, making it an attractive option in patients with underlying renal or cardiac dysfunction. In several placebo-controlled trials,50–52 various formulations significantly increased stool frequency while significantly decreasing straining, use of other laxatives, and colonic transit. No increase in adverse effects was noted compared with placebo.
Compared with lactulose, polyethylene glycol at about 21 g/day significantly increased bowel movement frequency while significantly decreasing the sense of straining with bowel movements and flatus due to laxative use.51 Both polyethylene glycol and lactulose accelerate colonic transit, although polyethylene glycol does so to a greater extent.53
Polyethylene glycol has been safe and effective when used for up to 6 months.54
Lactulose and sorbitol
Carbohydrate or sugar-based laxatives, if taken in sufficient doses, have a cathartic effect through two mechanisms: a primary osmotic effect of the sugar itself and a secondary osmotic effect as a substrate for colonic bacteria to cleave to acid metabolites, which exert an osmotic effect in the colon. This secondary effect will be discussed in a later section.
Lactulose and sorbitol are sugars that are poorly absorbed by the intestine. Lactulose has been shown to be more effective than placebo in increasing stool frequency, volume, weight, and consistency in chronically constipated patients.55 In a head-to-head comparison between sugar laxatives, 70% sorbitol was as effective as lactulose in increasing the frequency of bowel movements, and it was similar in its adverse effects56; 70% sorbitol is a cost-effective alternative to lactulose in the elderly nursing home population.57
Compared with fiber alone, lactulose use leads to a significantly higher number of bowel movements and better stool consistency.58 However, when lactulose was compared with a combination of fiber and a stimulant laxative, it was less effective than the combination therapy.59,60
Sugar laxatives, while effective, may have dose-limiting or use-limiting adverse effects such as abdominal bloating and flatulence.
Phosphate, magnesium
Sodium phosphate, like polyethylene glycol, is often used as a bowel preparation before colonoscopy, for which it is about as good or slightly better than polyethylene glycol.61,62
Although magnesium and sodium phosphate preparations are effective, there are multiple reports of clinically significant electrolyte abnormalities, renal failure, and congestive heart failure occurring with these preparations. Therefore, they must be used with discretion and caution in appropriate patients with frequent monitoring.
STIMULANT (IRRITANT) LAXATIVES
Stimulant laxatives are usually reserved for use when bulking agents and osmotic laxatives fail. Their mechanism of action involves the alteration of intestinal motility and intestinal fluid secretion.
Anthraquinones (cascara, aloe, and senna), castor oil, and diphenylmethanes (bisacodyl) are the most commonly used stimulant laxatives. They work relatively quickly, often eliciting a bowel movement 2 to 8 hours after they are taken.
This class of laxatives has historically been underused or given for only short periods of time, owing to concern about impairing colonic function, damaging the enteric nervous system, causing laxative dependency, causing cathartic colon, and even causing colon cancer. However, there is very little evidence to support these concerns. Stimulant laxatives can be used on a more regular basis when bulking or osmotic agents fail.63
Possibly of greatest concern is the potential for the overuse and abuse of stimulant laxatives. Excessive use can cause electrolyte disturbances brought about by high-volume watery diarrhea. Risk factors for overuse and abuse include underlying psychiatric disturbances and eating disorders. Prescribing other types of laxatives or cathartic agents may reduce risk, but the potential for abuse exists with all categories of laxatives.
TEGASEROD: GONE BUT STILL AVAILABLE, ON A CONTROLLED BASIS
Tegaserod (Zelnorm), a serotonin (5-HT4) agonist, was used predominantly in women with constipation-predominant irritable bowel syndrome and in men and women with chronic constipation. However, it was suspended from the market in the United States in March 2007 owing to concern about a high risk of adverse cardiovascular effects compared with placebo.
In a double-blind, randomized controlled trial, men with chronic constipation who received tegaserod 6 mg twice a day for 12 weeks had more spontaneous bowel movements than those receiving placebo (P = .04).64
Lin et al65 evaluated the use of tegaserod 6 mg twice daily for 4 weeks in both men and women with chronic constipation. Those receiving tegaserod had significantly more spontaneous bowel movements per week, less straining, and better stool consistency than those receiving placebo.
Tegaserod can still be obtained for appropriate patients via a treatment investigational new drug application. Safety data are under further review by the US Food and Drug Administration. Studies of other serotonin agonists are under way.
LUBIPROSTONE
Lubiprostone (Amitiza) is an agonist of the chloride channel subtype 2, found on the apical membrane of intestinal epithelial cells. It causes increased chloride secretion into the intestinal lumen, enhancing intestinal fluid secretion. It has been shown to be effective in chronic constipation by improving stool consistency and increasing the motility of the small intestine and colon.66 It is approved for treating chronic constipation in adults.
In randomized, double-blind trials, patients receiving lubiprostone 24 fig twice daily for 4 weeks had significantly more bowel movements per week, reported significantly better stool consistency and less abdominal bloating and straining, and rated their constipation as less severe than did patients receiving placebo.67–69
More recently, in an open-label study, lubiprostone improved constipation symptoms when taken for up to 48 weeks.70
The drug is well tolerated, but its adverse effects include nausea (which appears to be dose-dependent and may diminish over time or if the drug is taken with food), diarrhea, and headache.68 Of note, the drug appears to be well tolerated by older people (65 years of age and older), in whom adverse effects occur less often than in younger users.71 However, adverse events may cause up to 20% of patients to stop taking the drug.69 When lubiprostone is discontinued, patients may once again revert to their baseline bowel habit.72
Lubiprostone has not been compared with conventional laxatives, and cost may prohibit it from becoming a first-line drug for chronic constipation.73
OTHER PROMOTILITY AGENTS
Several promotility agents have been studied for treating chronic idiopathic constipation.
Cisapride (Propulsid), a 5-HT3 receptor antagonist and 5-HT4 receptor agonist, and prucalopride, a 5-HT4 agonist, were effective in relieving symptoms associated with chronic constipation.74–76 However, safety issues (cardiac arrhythmias) necessitated withdrawal of cisapride from the US market in 2000. Prucalopride is undergoing clinical trials.77
Renzapride, a mixed 5-HT4 receptor agonist and 5-HT3 receptor antagonist, has been shown to improve stool consistency and to increase colonic transit in patients with constipation-predominant irritable bowel syndrome.78 Renzapride has been studied in patients with this condition,78–81 but not in patients with chronic constipation. Renzapride is in phase III clinical development in the United States for treating constipation-predominant irritable bowel syndrome.
EMERGING TREATMENTS
New drugs with novel mechanisms of action are being investigated for the treatment of chronic idiopathic constipation.
Neurotrophin-3, a neurotrophic factor, modulates the development of the nervous system by regulating the survival and differentiation of nerves.82 In patients with functional constipation, subcutaneous doses of neurotrophin-3 improved stool frequency, the number of complete spontaneous bowel movements, and stool consistency.83
Alvimopan is a selective antagonist of the muopioid receptor that is being studied for opiate-related constipation and postoperative ileus.84,85 Little of this drug is systemically absorbed and it does not cross the blood-brain barrier; thus, it relieves the opiate-related side effects, ie, bloating, abdominal discomfort, and reduced stool frequency, without interfering with the central analgesic effects.
Linaclotide (MD 1100), a poorly absorbed guanylate cyclase agonist, is also being investigated as a treatment for chronic constipation.86 Linaclotide increases intestinal fluid secretion and transit via stimulation of cyclic guanosine monophosphate production and activation of the cystic fibrosis transmembrane conductance regulator.86,87 In preliminary studies, linaclotide increased stool frequency and the Bristol Stool Form Scale consistency score (Table 1) by increasing intestinal fluid secretion and transit.86
Chenodeoxycholic acid is a bile acid that is synthesized from cholesterol.88 Treatment of constipation with chenodeoxycholic acid has been proposed, given its laxative effect. A study by Bazzoli et al89 showed increased stool frequency and a decrease in stool consistency in chronic constipation patients given chenodeoxycholic acid 10 mg/kg/day. The main side effect was diarrhea. Chenodeoxycholic acid may be worthwhile in the management of constipation, but more studies are needed.
PROBIOTICS AND PREBIOTICS
The bacteria of the colon influence peristalsis of the colon.90 Probiotics (live bacterial preparations) and prebiotics (nondigestible preparations that stimulate the growth or activity of beneficial colonic bacteria) have been gaining interest as potential therapies for constipation.91,92
Probiotic bacterial preparations are generally composed of strains of Bifodobacterium,93,94Lactobacillus,95 and combinations thereof, and are available as mixed preparations of multiple bacterial strains of Lactobacillus, Bifodobacterium, and Streptococcus species, such as VSL#3.96
Probiotics may help relieve constipation, but their effect may depend on the strain of bacteria used and the population being studied.97 In a double-blind parallel study in 70 healthy adults, ingestion of 375 g/day of milk fermented with B animalis strain DN-173 010 for 11 days reduced colon transit time by 20% from baseline. The effect was more pronounced in women, particularly in those with longer baseline transit.98
Lactic acid-producing bacteria are considered commensal organisms with essentially no pathogenic potential.99 A review of the safety of bifodobacteria and lactobacilli concluded there was no health risk to consumers.100
Prebiotics are short-chain carbohydrates such as lactulose that stimulate the activity of beneficial colonic bacteria.91 They are thought to have a small laxative effect that is likely both osmotic and due to beneficial actions of bacteria for which they are a substrate. Both konjac glucomannan and lactulose, sugar-based laxatives and prebiotics, have been shown to significantly increase the fecal concentrations of lactobacilli and total bacteria, possibly through increases in stool bulk.46 Prebiotics that have been the focus of research include inulin, fructo-oligosaccharides, and galacto-oligosaccharides.91 Evidence on the efficacy of probiotics and prebiotics at relieving symptoms of constipation, however, is inconclusive because few well-controlled clinical studies have been done.91,92
STRATEGIES FOR MANAGING CHRONIC CONSTIPATION
In the absence of secondary causes, treatment of chronic constipation is focused on relieving symptoms.
If symptoms are refractory to these traditional treatments, agents such as lactulose and polyethylene glycol may provide relief.11,21 Although they do not address the underlying cause of constipation, these agents increase the fluid content of the intestine, contributing to improved stool consistency, and consequently increase the frequency of bowel movements.
Lubiprostone similarly increases the fluid content of the colon, contributing to improved stool consistency, reduced fecal transit time, and increased frequency of bowel movements.66,70,102 Unlike lactulose and polyethylene glycol, which are indicated only for short-term use, lubiprostone has been found to be safe and effective when used for up to 48 weeks.70,71
Biofeedback is the preferred treatment for pelvic floor dyssynergia, in which it has a success rate of 70% to 81% and in which it is superior to standard treatment (laxatives, fiber, and education).103–105 In an instrument-based training program, patients receive auditory or visual feedback or both to help train the pelvic floor and relax the anal sphincter while simulating defecation. It also improves rectal sensation to assist in proper evacuation. The best outcomes are achieved when committed patients receive instruction from empathetic, properly trained physical therapists or other technicians. Studies show that the benefits of biofeedback are long-lasting.104 It does not improve slow-transit constipation, though pelvic floor dyssynergia and slow-transit constipation can overlap.
Constipation is both a symptom and, when chronic, a multisymptom disorder, and it can overlap with other gastrointestinal tract disorders such as dyspepsia and gastroesophageal reflux disease. Furthermore, one should keep in mind the possibility of cancer and be alert for its warning signs.
Since constipation has a variety of causes and forms, one treatment does not fit all patients. Conservative measures such as recommending that the patient increase his or her intake of dietary fiber and water and engage in more physical activity are still the cornerstone of treatment, but they do not help all patients. On the other hand, polyethylene glycol and stimulant laxatives, which are traditionally given only for a short time, can be safe and effective when given long-term if other agents fail. New agents have become available or are in development.
In this article we outline our approach to constipation, as a guide for internists.
CONSTIPATION IS COMMON, BUT HOW SHOULD WE DEFINE IT?
Constipation affects 2% to 27% (average 14.8%) of the North American adult population—approximately 63 million people.1 It is more common than many other chronic diseases, including hypertension (48 million people), migraine (33 million), obesity (50 million), and diabetes mellitus (15 million).1–3
Constipation affects more women than men (2.1:1 ratio) and more nonwhites than whites (1.68:1).1 It occurs in all age groups but is more common in those older than 65 years and younger than 4 years.4,5
Constipation accounts for more than 2.5 million office visits and more than $500 million spent on laxatives per year.6,7 Also, people with constipation may report decreased productivity and increased absenteeism.8
The broad range in the prevalence of constipation cited above reflects differences in how it is defined and, in particular, a lack of agreement between how patients and physicians perceive it.1,9 Physicians mainly define constipation on the basis of stool frequency, considering fewer than three bowel movements per week to be abnormal.1 In contrast, patients typically define it on the basis of bothersome symptoms such as straining, passage of hard stool, unproductive urges, inability to defecate at will, and sensations of incomplete evacuation or abdominal bloating.1,9,10
The Rome III diagnostic criteria were developed to provide a consistent diagnostic approach for use in clinical practice and clinical trials.11 The Rome III criteria define functional chronic constipation as a chronic bowel disorder characterized by two or more of the following:
- Straining
- Lumpy or hard stools
- Sensations of incomplete evacuation
- Sensations of anorectal obstruction or blockage
- Use of manual maneuvers to facilitate defecation (eg, digital evacuation, support of the pelvic floor) during at least 25% of defecations
- Fewer than three bowel movements per week.
In addition, loose stools should rarely occur without the use of laxatives, and there should be insufficient criteria for irritable bowel syndrome.11 Chronicity is established by symptom onset within the previous 6 months and symptom duration of at least 3 months.
In contrast, patients with irritable bowel syndrome, also a functional bowel disorder, experience recurrent abdominal pain and discomfort associated with two or more of the following: symptom improvement with defecation, symptom onset associated with a change in the frequency of bowel movements, and a change in the form or appearance of the stool.
THREE TYPES OF IDIOPATHIC CONSTIPATION
There are three types of primary or idiopathic constipation5,9,12,13:
- Functional
- Slow-transit
- Outlet dysfunction.
Functional constipation includes functional chronic idiopathic constipation and constipation-predominant irritable bowel syndrome. It presents with a sense of difficult or delayed evacuation, hard stools, or abdominal bloating or discomfort.6,9,13 The predominant symptom of constipation-predominant irritable bowel syndrome is severe discomfort or pain; in chronic idiopathic constipation, pain and discomfort may be present but are not the primary symptom.
Slow-transit constipation (or delayed-transit constipation) is associated with a prolonged time between bowel movements. Its symptoms include low stool frequency, lack of urge to defecate, abdominal distention, bloating, and abdominal discomfort.14
Outlet dysfunction. Disorders of defecation can be due to mechanical causes such as Hirschsprung disease, anal stricture, cancer, prolapse, and large rectoceles, or from pelvic floor dysfunction. Pelvic floor dysfunction may be due to inadequate or excessive perineal descent or to inadequate propulsive forces, as may occur in neurologic or neuromuscular conditions and dyssynergia.
Pelvic floor dyssynergia, also called anorectal dyssynergia, dyssynergic defecation, and anismus, results from a functional defect in coordinated evacuation. The characteristic symptom is a feeling of being unable to adequately empty the rectum.14 Other symptoms such as excessive straining and manual disimpaction indicate but are not unique to pelvic floor dyssynergia.14,15
Combined forms. Patients may have more than one type of primary constipation and presentation, and pelvic floor dyssynergia has been shown to prolong intestinal transit, which may improve with treatment.
Secondary constipation can be due to causes such as diet, lifestyle, certain medications (calcium channel blockers, beta-blockers, opioids, diuretics, antidepressants, anticonvulsants, antacids, anticholinergics, and antispasmodics),5,16 underlying medical conditions (diabetes, hypothyroidism, multiple sclerosis, parkinsonism),16,17 pregnancy, and advanced age.18
NEUROTRANSMITTERS MAY PLAY A ROLE
Among the mechanisms thought to cause chronic constipation are impaired gastrointestinal motility,19–22 reduced intestinal secretions,21–23 and inadequate reflex relaxation of the pelvic floor muscles.22,24
Neurotransmitters such as serotonin, somatostatin, peptide YY, and vasoactive intestinal peptide affect intestinal secretion and motility.25,26 Hyperactivity of these neurotransmitters associated with increased secretion and motility results in diarrhea, whereas hypoactivity leads to decreased secretion, delayed transit, and constipation.23
Serotonin has a role in regulating visceral pain perception and intestinal motility, as well as secretion.26–28 Clinical trials have shown that activation of serotonin receptors in the gut enhances gastrointestinal motility, inhibits visceral sensitivity, and stimulates intestinal secretion.26,27,29
A hypothesis has recently been proposed that degeneration of enteric neurons may also play a role in the development of severe idiopathic constipation.30
DIAGNOSIS IS MOSTLY CLINICAL
The history and physical examination remain the cornerstones in the diagnosis and subsequent treatment of chronic constipation.
History
Risk factors for primary and secondary constipation to note during the interview include age (< 4 years, > 65 years); low-fiber diet; female sex; lack of physical activity; history of childhood constipation, endocrine and neuromuscular disorders, abuse, depression, or anxiety; family history of cancer; and personal history of pelvic surgery.
Since drugs can also cause chronic constipation, especially in elderly or immobile patients, medication lists should be reviewed and adjustments should be made if necessary (or possible) before recommending laxatives or invasive testing, if no alarm signs are present.
Alarm signs such as weight loss, hematochezia, melena, change in bowel habits, and symptoms refractory to therapy may represent colon cancer and indicate the need for early diagnostic testing.
Physical examination
Physical examination should always include inspection of the perianal area for evidence of hemorrhoids or fissures. Digital rectal examination may reveal a contracted sphincter or a puborectalis muscle that contracts with the Valsalva maneuver, suggesting dysfunction.
Laboratory testing
If the history and physical examination suggest that the constipation may be secondary, or if the patient is 50 years of age or older, then laboratory studies such as a complete blood cell count, serum electrolyte levels, blood sugar level, and thyroid function studies may help rule out a metabolic, endocrine, or organic cause.
Colonoscopy, other tests
At present, little evidence suggests that routine testing is warranted in patients without evidence of secondary constipation and without alarm signs. However, diagnostic studies are indicated in patients 50 years of age and older, as well as in those with alarm symptoms such as hematochezia, anemia, a positive fecal occult blood test, unintentional loss of more than 10 pounds, family history of colon cancer or inflammatory bowel disease, fever, nausea, vomiting, acute onset (especially in the elderly), and lack of improvement with conventional therapies regardless of age.2
The full length of the colon should be inspected by colonoscopy or by flexible sigmoidoscopy paired with a barium enema study to rule out structural disease. Of note, all patients 50 years of age or older should be screened for colon cancer.
If the patient does not respond to therapy, further tests such as colonic transit studies, anorectal manometry with balloon expulsion, and, possibly, defecating proctography or dynamic pelvic magnetic resonance imaging may be considered. These patients would likely also benefit from referral to a gastroenterologist for further management
DIET AND LIFESTYLE AS TREATMENT
For many years, health care providers have provided reassurance and recommended diet and lifestyle modifications as treatment for constipation. Increased water intake, increased activity, and a scheduled attempt at defecation when motor activity in the colon is highest, ie, in the morning or after eating, have all been recommended.
Data on the efficacy of these recommendations are scarce and often contradictory. Studies have shown that increasing water intake or daily exercise is not always helpful.32–34 Nevertheless, many patients who comply with dietary and exercise recommendations have improvement in symptoms. Eating fewer meals per day (and hence taking in fewer calories) has been shown to be associated with constipation in the elderly. However, no relationships between fiber or fluid intake and constipation were noted.35
In a study in which chronically constipated patients were fed a standardized diet that contained 25 g of fiber a day, stool frequency increased significantly and laxative use decreased.36 While on a high-fiber diet, the patients were divided into two groups, one that drank 1.1 L of fluid per day and one that drank 2.1 L of mineral water per day. Both groups experienced further improvements in stool frequency and decreases in laxative use, with the mineral-water group benefiting the most.36
Recently, Murakami and others37 found, in a cross-sectional study in young Japanese women with low daily fiber intake (6.4 g/day), that low water intake from foods and low magnesium intake were associated with an increasing prevalence of functional constipation as defined by the Rome III criteria. Constipation was also found to be significantly associated with low intake of fruits and vegetables in a study from Singapore.38
Moderate physical activity and high fiber intake may be associated with a lower prevalence of constipation in women. In the Nurses’ Health Study, more than 62,000 women between the ages of 36 and 61 were surveyed, and those who said they engaged in daily physical activity had a lower prevalence of constipation (prevalence ratio [PR] = 0.56, 95% confidence interval [CI] 0.44–0.70), as did those with a median fiber intake of 20 g/day (PR = 0.64, 95% CI 0.57–0.73).39
BULK LAXATIVES (FIBER SUPPLEMENTS): THE FIRST-LINE TREATMENT
Fiber remains the first-line treatment for constipation. It may relieve or improve symptoms in functional constipation. However, fewer than 30% of patients with either slow-transit constipation or pelvic floor dysfunction have improvement in symptoms with fiber, and in these types of constipation it can even worsen symptoms.40
There is much confusion about what types of fiber should be recommended and how the various types of fiber perform in resolving constipation.
Insoluble fiber
Insoluble fiber resists bacterial degradation in the colon and can retain more water than soluble fiber can.
Bran 20 g/day increased the frequency of bowel movements by 55%, increased fecal weight by 157%, and decreased intestinal transit time by 50% in women who had three or fewer bowel movements per week.41
Muller-Lissner42 and others performed a meta-analysis and found that bran (25 g/day) increased stool weight and decreased transit time in both healthy controls and patients with chronic constipation. Yet constipated patients taking bran still had lower stool weights and slower transit times than did healthy subjects.
When bran 20 g/day was compared with placebo in chronically constipated patients, bowel frequency and stool weight increased with both treatments,43 suggesting that factors other than intake may affect bowel function and transit time. However, bran was more effective than placebo in decreasing oroanal transit time.
Elderly constipated patients who received bran 10 g twice a day had significantly shorter transit times (89 hours vs 126 hours) than did those who received psyllium (a soluble fiber) 6 g twice daily. They also needed less additional laxative.44
Soluble fiber
Soluble fiber also affects the bowel habits of both healthy and constipated patients.
Methylcellulose, given to healthy volunteers at a dose of 4 g/day, resulted in statistically significant increases in stool weight, fecal water weight, and fecal solids.45 In constipated patients, methylcellulose 1 g/day was as effective as psyllium 3.4 g/day at increasing stool frequency, fecal water weight, and fecal solids.45
Konjac glucomannan was also shown to significantly increase stool frequency, water weight, and fecal solids.46
Psyllium. In a study that randomly assigned 22 patients with chronic constipation to receive either psyllium 5 g twice daily or placebo for 8 weeks, followed by a 4-week washout phase in which placebo was given,47 those who received psyllium reported significant improvements in stool consistency and pain with defecation, as well as significant increases in both stool frequency (3.8 vs 2.9 per week, P < .05) and stool weight (665 g vs 405 g, P < .05). However, colonic transit times and anorectal manometric measurements did not differ significantly between those who received psyllium vs placebo.47
Fiber may not help everyone
Others have also shown that while fiber may improve stool characteristics, it may not significantly alter the sensorimotor functions of the colon and pelvic floor.
Cheskin et al48 performed a crossover study in 10 constipated men and women in the community. Patients received either 24 g of psyllium fiber daily or a placebo fiber for 1 month and then crossed over to the other treatment for the next month. The most common cause of constipation in this study was pelvic floor dysfunction. Total gut transit time was significantly increased by psyllium fiber, and there was a trend toward increased stool frequency, demonstrating that psyllium clinically improved constipation. However, pelvic floor dysfunction, as measured by rectal manometry, was not improved.
It may be that only people with normal-transit constipation, not those with underlying slow-transit constipation or pelvic floor dysfunction, are helped by additional dietary fiber. Voderholzer and others40 studied 149 consecutive patients with chronic constipation and evaluated their response to at least 6 weeks of psyllium (Plantago ovata seeds 15 to 30 g/day) by serial symptom measurements, oroanal transit times, and functional rectoanal evaluation with defecography, manometry, and sigmoidoscopy. Of the patients with no evidence of pelvic floor dysfunction or slow-transit constipation, 85% improved. However, 80% of those with slow-transit constipation and 63% of those with pelvic floor dysfunction did not improve with the use of fiber. The authors concluded that it is reasonable to try dietary fiber in patients with constipation and, if no improvement is noted, to then consider further investigation for other subtypes of constipation (ie, slow-transit or pelvic-floor dysfunction).
Adverse effects may limit the use of fiber and may differ depending on the type of fiber used. Soluble fiber may be better tolerated, especially in patients with constipation-predominant irritable bowel syndrome.49 Side effects include the sensation of bloating and distention, excessive gas production, and abdominal cramping.
Our recommendations on fiber
We recommend the following regarding fiber in constipated patients:
- Increase fiber intake from natural foods up to 20 g/day. This increase should be completed over 2 to 3 weeks to minimize adverse effects.
- Consider adding a fiber supplement, such as psyllium, if increasing the intake of natural fiber does not relieve constipation-related symptoms.
- If symptoms persist despite the use of fiber supplements and diet and lifestyle modification, then further structural and functional investigation of the colon (anorectal manometry, colonoscopy, defecography, colon manometry) should be considered.
OSMOTIC LAXATIVES
Osmotic laxatives are molecules that are either not absorbed or poorly absorbed and that draw water into the intestinal lumen to maintain isotonicity between the intestinal contents and the serum. Examples are polyethylene glycol, sodium phosphate (Fleet phosphosoda), magnesium hydroxide, magnesium citrate, the sugars lactulose and sorbitol, and glycerin.
Certain formulations of this class of laxative can cause bloating, diarrhea, electrolyte disturbances, volume overload, or dehydration. These effects limit their use, and these medications should be used with caution in patients prone to renal insufficiency or cardiac abnormalities.
Polyethylene glycol
Polyethylene glycol is an exception. It is not absorbed and lacks electrolytes, making it an attractive option in patients with underlying renal or cardiac dysfunction. In several placebo-controlled trials,50–52 various formulations significantly increased stool frequency while significantly decreasing straining, use of other laxatives, and colonic transit. No increase in adverse effects was noted compared with placebo.
Compared with lactulose, polyethylene glycol at about 21 g/day significantly increased bowel movement frequency while significantly decreasing the sense of straining with bowel movements and flatus due to laxative use.51 Both polyethylene glycol and lactulose accelerate colonic transit, although polyethylene glycol does so to a greater extent.53
Polyethylene glycol has been safe and effective when used for up to 6 months.54
Lactulose and sorbitol
Carbohydrate or sugar-based laxatives, if taken in sufficient doses, have a cathartic effect through two mechanisms: a primary osmotic effect of the sugar itself and a secondary osmotic effect as a substrate for colonic bacteria to cleave to acid metabolites, which exert an osmotic effect in the colon. This secondary effect will be discussed in a later section.
Lactulose and sorbitol are sugars that are poorly absorbed by the intestine. Lactulose has been shown to be more effective than placebo in increasing stool frequency, volume, weight, and consistency in chronically constipated patients.55 In a head-to-head comparison between sugar laxatives, 70% sorbitol was as effective as lactulose in increasing the frequency of bowel movements, and it was similar in its adverse effects56; 70% sorbitol is a cost-effective alternative to lactulose in the elderly nursing home population.57
Compared with fiber alone, lactulose use leads to a significantly higher number of bowel movements and better stool consistency.58 However, when lactulose was compared with a combination of fiber and a stimulant laxative, it was less effective than the combination therapy.59,60
Sugar laxatives, while effective, may have dose-limiting or use-limiting adverse effects such as abdominal bloating and flatulence.
Phosphate, magnesium
Sodium phosphate, like polyethylene glycol, is often used as a bowel preparation before colonoscopy, for which it is about as good or slightly better than polyethylene glycol.61,62
Although magnesium and sodium phosphate preparations are effective, there are multiple reports of clinically significant electrolyte abnormalities, renal failure, and congestive heart failure occurring with these preparations. Therefore, they must be used with discretion and caution in appropriate patients with frequent monitoring.
STIMULANT (IRRITANT) LAXATIVES
Stimulant laxatives are usually reserved for use when bulking agents and osmotic laxatives fail. Their mechanism of action involves the alteration of intestinal motility and intestinal fluid secretion.
Anthraquinones (cascara, aloe, and senna), castor oil, and diphenylmethanes (bisacodyl) are the most commonly used stimulant laxatives. They work relatively quickly, often eliciting a bowel movement 2 to 8 hours after they are taken.
This class of laxatives has historically been underused or given for only short periods of time, owing to concern about impairing colonic function, damaging the enteric nervous system, causing laxative dependency, causing cathartic colon, and even causing colon cancer. However, there is very little evidence to support these concerns. Stimulant laxatives can be used on a more regular basis when bulking or osmotic agents fail.63
Possibly of greatest concern is the potential for the overuse and abuse of stimulant laxatives. Excessive use can cause electrolyte disturbances brought about by high-volume watery diarrhea. Risk factors for overuse and abuse include underlying psychiatric disturbances and eating disorders. Prescribing other types of laxatives or cathartic agents may reduce risk, but the potential for abuse exists with all categories of laxatives.
TEGASEROD: GONE BUT STILL AVAILABLE, ON A CONTROLLED BASIS
Tegaserod (Zelnorm), a serotonin (5-HT4) agonist, was used predominantly in women with constipation-predominant irritable bowel syndrome and in men and women with chronic constipation. However, it was suspended from the market in the United States in March 2007 owing to concern about a high risk of adverse cardiovascular effects compared with placebo.
In a double-blind, randomized controlled trial, men with chronic constipation who received tegaserod 6 mg twice a day for 12 weeks had more spontaneous bowel movements than those receiving placebo (P = .04).64
Lin et al65 evaluated the use of tegaserod 6 mg twice daily for 4 weeks in both men and women with chronic constipation. Those receiving tegaserod had significantly more spontaneous bowel movements per week, less straining, and better stool consistency than those receiving placebo.
Tegaserod can still be obtained for appropriate patients via a treatment investigational new drug application. Safety data are under further review by the US Food and Drug Administration. Studies of other serotonin agonists are under way.
LUBIPROSTONE
Lubiprostone (Amitiza) is an agonist of the chloride channel subtype 2, found on the apical membrane of intestinal epithelial cells. It causes increased chloride secretion into the intestinal lumen, enhancing intestinal fluid secretion. It has been shown to be effective in chronic constipation by improving stool consistency and increasing the motility of the small intestine and colon.66 It is approved for treating chronic constipation in adults.
In randomized, double-blind trials, patients receiving lubiprostone 24 fig twice daily for 4 weeks had significantly more bowel movements per week, reported significantly better stool consistency and less abdominal bloating and straining, and rated their constipation as less severe than did patients receiving placebo.67–69
More recently, in an open-label study, lubiprostone improved constipation symptoms when taken for up to 48 weeks.70
The drug is well tolerated, but its adverse effects include nausea (which appears to be dose-dependent and may diminish over time or if the drug is taken with food), diarrhea, and headache.68 Of note, the drug appears to be well tolerated by older people (65 years of age and older), in whom adverse effects occur less often than in younger users.71 However, adverse events may cause up to 20% of patients to stop taking the drug.69 When lubiprostone is discontinued, patients may once again revert to their baseline bowel habit.72
Lubiprostone has not been compared with conventional laxatives, and cost may prohibit it from becoming a first-line drug for chronic constipation.73
OTHER PROMOTILITY AGENTS
Several promotility agents have been studied for treating chronic idiopathic constipation.
Cisapride (Propulsid), a 5-HT3 receptor antagonist and 5-HT4 receptor agonist, and prucalopride, a 5-HT4 agonist, were effective in relieving symptoms associated with chronic constipation.74–76 However, safety issues (cardiac arrhythmias) necessitated withdrawal of cisapride from the US market in 2000. Prucalopride is undergoing clinical trials.77
Renzapride, a mixed 5-HT4 receptor agonist and 5-HT3 receptor antagonist, has been shown to improve stool consistency and to increase colonic transit in patients with constipation-predominant irritable bowel syndrome.78 Renzapride has been studied in patients with this condition,78–81 but not in patients with chronic constipation. Renzapride is in phase III clinical development in the United States for treating constipation-predominant irritable bowel syndrome.
EMERGING TREATMENTS
New drugs with novel mechanisms of action are being investigated for the treatment of chronic idiopathic constipation.
Neurotrophin-3, a neurotrophic factor, modulates the development of the nervous system by regulating the survival and differentiation of nerves.82 In patients with functional constipation, subcutaneous doses of neurotrophin-3 improved stool frequency, the number of complete spontaneous bowel movements, and stool consistency.83
Alvimopan is a selective antagonist of the muopioid receptor that is being studied for opiate-related constipation and postoperative ileus.84,85 Little of this drug is systemically absorbed and it does not cross the blood-brain barrier; thus, it relieves the opiate-related side effects, ie, bloating, abdominal discomfort, and reduced stool frequency, without interfering with the central analgesic effects.
Linaclotide (MD 1100), a poorly absorbed guanylate cyclase agonist, is also being investigated as a treatment for chronic constipation.86 Linaclotide increases intestinal fluid secretion and transit via stimulation of cyclic guanosine monophosphate production and activation of the cystic fibrosis transmembrane conductance regulator.86,87 In preliminary studies, linaclotide increased stool frequency and the Bristol Stool Form Scale consistency score (Table 1) by increasing intestinal fluid secretion and transit.86
Chenodeoxycholic acid is a bile acid that is synthesized from cholesterol.88 Treatment of constipation with chenodeoxycholic acid has been proposed, given its laxative effect. A study by Bazzoli et al89 showed increased stool frequency and a decrease in stool consistency in chronic constipation patients given chenodeoxycholic acid 10 mg/kg/day. The main side effect was diarrhea. Chenodeoxycholic acid may be worthwhile in the management of constipation, but more studies are needed.
PROBIOTICS AND PREBIOTICS
The bacteria of the colon influence peristalsis of the colon.90 Probiotics (live bacterial preparations) and prebiotics (nondigestible preparations that stimulate the growth or activity of beneficial colonic bacteria) have been gaining interest as potential therapies for constipation.91,92
Probiotic bacterial preparations are generally composed of strains of Bifodobacterium,93,94Lactobacillus,95 and combinations thereof, and are available as mixed preparations of multiple bacterial strains of Lactobacillus, Bifodobacterium, and Streptococcus species, such as VSL#3.96
Probiotics may help relieve constipation, but their effect may depend on the strain of bacteria used and the population being studied.97 In a double-blind parallel study in 70 healthy adults, ingestion of 375 g/day of milk fermented with B animalis strain DN-173 010 for 11 days reduced colon transit time by 20% from baseline. The effect was more pronounced in women, particularly in those with longer baseline transit.98
Lactic acid-producing bacteria are considered commensal organisms with essentially no pathogenic potential.99 A review of the safety of bifodobacteria and lactobacilli concluded there was no health risk to consumers.100
Prebiotics are short-chain carbohydrates such as lactulose that stimulate the activity of beneficial colonic bacteria.91 They are thought to have a small laxative effect that is likely both osmotic and due to beneficial actions of bacteria for which they are a substrate. Both konjac glucomannan and lactulose, sugar-based laxatives and prebiotics, have been shown to significantly increase the fecal concentrations of lactobacilli and total bacteria, possibly through increases in stool bulk.46 Prebiotics that have been the focus of research include inulin, fructo-oligosaccharides, and galacto-oligosaccharides.91 Evidence on the efficacy of probiotics and prebiotics at relieving symptoms of constipation, however, is inconclusive because few well-controlled clinical studies have been done.91,92
STRATEGIES FOR MANAGING CHRONIC CONSTIPATION
In the absence of secondary causes, treatment of chronic constipation is focused on relieving symptoms.
If symptoms are refractory to these traditional treatments, agents such as lactulose and polyethylene glycol may provide relief.11,21 Although they do not address the underlying cause of constipation, these agents increase the fluid content of the intestine, contributing to improved stool consistency, and consequently increase the frequency of bowel movements.
Lubiprostone similarly increases the fluid content of the colon, contributing to improved stool consistency, reduced fecal transit time, and increased frequency of bowel movements.66,70,102 Unlike lactulose and polyethylene glycol, which are indicated only for short-term use, lubiprostone has been found to be safe and effective when used for up to 48 weeks.70,71
Biofeedback is the preferred treatment for pelvic floor dyssynergia, in which it has a success rate of 70% to 81% and in which it is superior to standard treatment (laxatives, fiber, and education).103–105 In an instrument-based training program, patients receive auditory or visual feedback or both to help train the pelvic floor and relax the anal sphincter while simulating defecation. It also improves rectal sensation to assist in proper evacuation. The best outcomes are achieved when committed patients receive instruction from empathetic, properly trained physical therapists or other technicians. Studies show that the benefits of biofeedback are long-lasting.104 It does not improve slow-transit constipation, though pelvic floor dyssynergia and slow-transit constipation can overlap.
- Higgins PD, Johanson JF. Epidemiology of constipation in North America: a systematic review. Am J Gastroenterol 2004; 99:750–759.
- Brandt LJ, Prather CM, Quigley EM, Schiller LR, Schoenfeld P, Talley NJ. Systematic review on the management of chronic constipation in North America. Am J Gastroenterol 2005; 100 suppl 1:S5–S21.
- Pleis JR, Lethbridge-Cejku M. Summary health statistics for U.S. adults: National Health Interview Survey, 2005. Vital Health Stat 10 2006; 232:1–153.
- Dennison C, Prasad M, Lloyd A, Bhattacharyya SK, Dhawan R, Coyne K. The health-related quality of life and economic burden of constipation. Pharmacoeconomics 2005; 23:461–476.
- Arce DA, Ermocilla CA, Costa H. Evaluation of constipation. Am Fam Physician 2002; 65:2283–2290.
- Johanson JF, Sonnenberg A, Koch TR. Clinical epidemiology of chronic constipation. J Clin Gastroenterol 1989; 11:525–536.
- Harari D, Gurwitz JH, Minaker KL. Constipation in the elderly. J Am Geriatr Soc 1993; 41:1130–1140.
- Johanson JF, Kralstein J. Chronic constipation: a survey of the patient perspective. Aliment Pharmacol Ther 2007; 25:599–608.
- Lembo A, Camilleri M. Chronic constipation. N Engl J Med 2003; 349:1360–1368.
- Sandler RS, Drossman DA. Bowel habits in young adults not seeking health care. Dig Dis Sci 1987; 32:841–845.
- Longstreth GF, Thompson WG, Chey WD, Houghton LA, Mearin F, Spiller RC. Functional bowel disorders. Gastroenterology 2006; 130:1480–1491.
- Locke GR, Pemberton JH, Phillips SF. AGA technical review on constipation. Gastroenterology 2000; 119:1766–1778.
- Rao SS. Constipation: evaluation and treatment. Gastroenterol Clin North Am 2003; 32:659–683.
- Locke GR, Pemberton JH, Phillips SF. American Gastroenterological Association Medical Position Statement: guidelines on constipation. Gastroenterology 2000; 119:1761–1766.
- Prather CM. Subtypes of constipation: sorting out the confusion. Rev Gastroenterol Disord 2004; 4 suppl 2:S11–S16.
- Talley NJ, Jones M, Nuyts G, Dubois D. Risk factors for chronic constipation based on a general practice sample. Am J Gastroenterol 2003; 98:1107–1111.
- Bharucha AE. Treatment of severe and intractable constipation. Curr Treat Options Gastroenterol 2004; 7:291–298.
- Borum ML. Constipation: evaluation and management. Prim Care 2001; 28:577–590.
- Camilleri M, Ford MJ. Review article: colonic sensorimotor physiology in health, and its alteration in constipation and diarrhoeal disorders. Aliment Pharmacol Ther 1998; 12:287–302.
- Knowles CH, Martin JE. Slow transit constipation: a model of human gut dysmotility. Review of possible aetiologies. Neurogastroenterology Motility 2000; 12:181–196.
- Schiller LR. Review article: the therapy of constipation. Aliment Pharmacol Ther 2001; 15:749–763.
- Crowell MD. Pathogenesis of slow transit and pelvic floor dysfunction: from bench to bedside. Rev Gastroenterol Disord 2004; 4 suppl 2:S17–S27.
- Wood JD. Neuropathophysiology of irritable bowel syndrome. J Clin Gastroenterol 2002; 35 suppl 1:S11–S22.
- Chitkara DK, Bredenoord AJ, Cremonini F, et al. The role of pelvic floor dysfunction and slow colonic transit in adolescents with refractory constipation. Am J Gastroenterol 2004; 99:1579–1584.
- El-Salhy M. Gastrointestinal transit in an animal model of human diabetes type 2: relationship to gut neuroendocrine peptide contents. Ups J Med Sci 2002; 107:101–110.
- Crowell MD. Role of serotonin in the pathophysiology of the irritable bowel syndrome. Br J Pharmacol 2004; 141:1285–1293.
- Gershon MD. Review article: roles played by 5-hydroxytryptamine in the physiology of the bowel. Aliment Pharmacol Ther 1999; 13 suppl 2:15–30.
- Gershon MD. Serotonin and its implication for the management of irritable bowel syndrome. Rev Gastroenterol Disord 2003; 3 suppl 2:S25–S34.
- Grider JR, Foxx-Orenstein AE, Jin JG. 5-Hydroxytryptamine4 receptor agonists initiate the peristaltic reflex in human, rat, and guinea pig intestine. Gastroenterology 1998; 115:370–380.
- Krishnamurthy S, Schuffler MD, Rohrmann CA, Pope CE. Severe idiopathic constipation is associated with a distinctive abnormality of the colonic myenteric plexus. Gastroenterology 1985; 88:26–34.
- O'Donnell LJ, Virjee J, Heaton KW. Detection of pseudodiarrhoea by simple clinical assessment of intestinal transit rate. BMJ 1990; 300:439–440.
- Meshkinpour H, Selod S, Movahedi H, Nami N, James N, Wilson A. Effects of regular exercise in management of chronic idopathic constipation. Dig Dis Sci 1998; 43:2379–2383.
- Young RJ, Beerman LE, Vanderhoff JA. Increasing oral fluids in chronic constipation in children. Gastroenterol Nurs 1998; 21:156–161.
- Tuteja AK, Talley NJ, Joos SK, Woehl JV, Hickam DH. Is constipation associated with decreased physical activity in normally active subjects? Am J Gastroenterol 2005; 100:124–129.
- Towers AL, Burgio KL, Locher JL, Merkel IS, Safaeian M, Wald A. Constipation in the elderly: influence of dietary, psychological, and physiological factors. J Am Geriatr Soc 1994; 42:701–706.
- Anti J, Pignataro G, Armuzzi A, et al. Water supplementation enhances the effect of high-fiber diet on stool frequency and laxative consumption in adult patients with functional constipation. Hepatogastroenterology 1998; 45:727–732.
- Murakami K, Sasaki S, Okubo H, et al. Association between dietary fiber, water and magnesium intake and functional constipation among young Japanese women. Eur J Clin Nutr 2007; 61:616–622.
- Wong ML, Wee S, Pin CH, Gan GL, Ye HC. Sociodemographic and lifestyle factors associated with constipation in an elderly Asian community. Am J Gastroenterol 1999; 94:1283–1291.
- Dukas L, Willett WC, Giovannucci EL. Association between physical activity, fiber intake, and other lifestyle variables and constipation in a study of women. Am J Gastroenterol 2003; 98:1790–1796.
- Voderholzer WA, Schatke W, Muhldorfer BE, et al. Clinical response to dietary fiber treatment of chronic constipation. Am J Gastroenterol 1997; 92:95–98.
- Graham DY, Moser SE, Estes MK. The effect of bran on bowel function in constipation. Am J Gastroenterol 1982; 77:599–603.
- Muller-Lissner SA. Effect of wheat bran on weight of stool and gastrointestinal transit time: a meta analysis. Br Med J (Clin Res Ed) 1988; 296:615–617.
- Badiali D, Corazziari E, Habib FI, et al. Effect of wheat bran in treatment of chronic nonorganic constipation. A double-blind controlled trial. Dig Dis Sci 1995; 40:349–356.
- Andersson H, Basaeus I, Falkheden T, Melkersson M. Transit time in constipated geriatric patients during treatment with bulk laxative and bran: a comparison. Scand J Gastroenterol 1979; 14:821–826.
- Hamilton JW, Wagner J, Burdick BB, Bass P. Clinical evaluation of methyl-cellulose as a bulk laxative. Dig Dis Sci 1988; 33:993–998.
- Chen HL, Cheng HC, Liu YJ, Liu SY, Wu WT. Konjac acts as a natural laxative by increasing stool bulk and improving colonic ecology in healthy adults. Nutrition 2006; 22:1112–1119.
- Ashraf W, Park F, Lof J, Quigley EM. Effects of psyllium therapy on stool characteristics, colon transit and anorectal function in chronic idiopathic constipation. Aliment Pharmacol Ther 1995; 9:639–647.
- Cheskin LJ, Kamal N, Crowell MD, Schuster MM, Whitehead WE. Mechanisms of constipation in older persons and effects of fiber compared with placebo. J Am Geriatr Soc 1995; 43:666–669.
- Bijkerk CJ, Muris JW, Knottnerus JA, Hoes AW, de Wit NJ. Systematic review: the role of different types of fiber in the treatment of irritable bowel syndrome. Aliment Pharmacol Ther 2004; 19:245–251.
- Corazziari E, Badiali D, Habib FI, et al. Small volume isosmotic polyethylene glycol electrolyte balanced solution (PMF–100) in treatment of chronic nonorganic constipation. Dig Dis Sci 1996; 41:1636–1642.
- Attar A, Lemann M, Ferguson A, et al. Comparison of a low dose polyethylene glycol electrolyte solution with lactulose for treatment of chronic constipation. Gut 1999; 44:226–230.
- Corazziari E, Badiali D, Bazzocchi G, et al. Long term efficacy, safety, and tolerabilitity of low daily doses of isosmotic polyethylene glycol electrolyte balanced solution (PMF–100) in the treatment of functional chronic constipation. Gut 2000; 46:522–526.
- Fritz E, Hammer HF, Lipp RW, Högenauer C, Stauber R, Hammer J. Effects of lactulose and polyethylene glycol on colonic transit. Aliment Pharmacol Ther 2005; 21:259–268.
- DiPalma JA, Cleveland MV, McGowan J, Herrera JL. A randomized, multicenter, placebo-controlled trial of polyethylene glycol laxative for chronic treatment of chronic constipation. Am J Gastroenterol 2007; 102:1436–1441.
- Bass P, Dennis S. The laxative effects of lactulose in normal and constipated subjects. J Clin Gastroenterol 1981; 3 suppl 1:23–28.
- Lederle FA, Busch DL, Mattox KM, West MJ, Aske DM. Cost-effective treatment of constipation in the elderly: a randomized double-blind comparison of sorbitol and lactulose. Am J Med 1990; 89:597–601.
- Volicer L, Lane P, Panke J, Lyman P. Management of constipation in residents with dementia: sorbitol effectiveness and cost. J Am Med Dir Assoc 2004; 5:239–241.
- Quah HM, Ooi BS, Seow-Choen F, Sng KK, Ho KS. Prospective randomized crossover trial comparing fibre with lactulose in the treatment of idiopathic chronic constipation. Tech Coloproctol 2006; 10:111–114.
- Passmore AP, Davies KW, Flanagan PG, Stoker C, Scott MG. A comparison of Agiolax and lactulose in elderly patients with chronic constipation. Pharmacology 1993; 47 suppl 1:249–252.
- Passmore AP, Wilson-Davies K, Stoker C, Scott ME. Chronic constipation in long stay elderly patients: a comparison of lactulose and a senna-fibre combination. BMJ 1993; 307:769–771.
- Poon CM, Lee DW, Mak SK, et al. Two liters of polyethylene glycol-electrolyte lavage solution versus sodium phosphate as bowel cleansing regimen for colonoscopy: a prospective randomized controlled trial. Endoscopy 2002; 34:560–563.
- Rostom A, Jolicoeur E, Dube C, et al. A randomized prospective trial comparing different regimens of oral sodium phosphate and polyethylene glycol-based lavage solution in the preparation of patients for colonoscopy. Gastrointest Endosc 2006; 64:544–552.
- Wald A. Is chronic use of stimulant laxatives harmful to the colon? J Clin Gastroenterol 2003; 36:386–389.
- Fried M, Johanson JF, Gwee KA, Wagner A, Pecher E, Rueegg P. Efficacy of tegaserod in chronic constipation in men. Am J Gastroenterol 2007; 102:362–370.
- Lin SR, Ke MY, Luo JY, et al. A randomized, double-blind, placebo-controlled trial assessing the efficacy and safety of tegaserod in patients from China with chronic constipation. World J Gastroenterol 2007; 13:732–739.
- Camilleri M, Bharucha AE, Ueno R, et al. Effect of a selective chloride channel activator, lubiprostone, on gastrointestinal transit, gastric sensory, and motor functions in healthy volunteers. Am J Physiol Gastrointest Liver Physiol 2006; 290:G942–G947.
- McKeage K, Plosker GL, Siddiqui MA. Lubiprostone. Drugs 2006; 66:873–879.
- Johanson JF, Gargano MA, Patchen ML, Ueno R. Efficacy and safety of a novel compound, RU-0211, for the treatment of constipation [abstract]. Gastroenterology 2002; 122 suppl 1:A315.
- Johanson JF, Gargano MA, Holland PC, Patchen ML, Ueno R. Multicenter open-label study of oral lubiprostone for the treatment of chronic constipation [abstract]. Am J Gastroenterol 2005; 100 suppl:S331.
- Johanson JF, Panas R, Holland P, Ueno R. Long-term efficacy of lubiprostone for the treatment of chronic constipation [abstract]. Gastroenterology 2006; 130 suppl 2:A317.
- Ueno R, Panas R, Wahle A, Zhu Y, Holland P. Long-term safety and efficacy of lubiprostone for the treatment of chronic constipation in the elderly [abstract]. Gastroenterology 2006; 130 suppl 2:A188.
- Johanson JF, Gargano MA, Holland P, Patchen ML, Ueno R. Phase III, randomized withdrawal study of RU-0211, a novel chloride channel activator for the treatment of constipation [abstract]. Gastroenterology 2004; 126 suppl 2:A100.
- Rivkin A, Chagan L. Lubiprostone: chloride channel activator for chronic constipation. Clin Ther 2006; 28:2008–2021.
- Johanson JF, Miner PB, Parkman HP, et al. Prucalopride improves bowel movement frequency and symptoms in patients with chronic constipation: results of two double-blind, placebo-controlled trials [abstract]. Gastroenterology 2000; 118 suppl 2:A175.
- Cash BD, Chey WD. Review article: the role of serotonergic agents in the treatment of patients with primary chronic constipation. Aliment Pharmacol Ther 2005; 22:1047–1060.
- Altabas K, Biliæ A, Jurciæ D, et al. The efficacy of cisapride vs. placebo and diet in patients with chronic constipation. Coll Antropol 2003; 27:197–204.
- Camilleri M, Kerstens R, Rykx A, Vandeplassche L. A placebo-controlled trial of prucalopride for severe chronic constipation. N Engl J Med 2008; 358:2344–2354.
- Camilleri M, McKinzie S, Fox J, et al. Effect of renzapride on transit in constipation-predominant irritable bowel syndrome. Clin Gastroenterol Hepatol 2004; 2:895–904.
- Tack J, Middleton SJ, Horne MC, et al. Pilot study of the efficacy of renzapride on gastrointestinal motility and symptoms in patients with constipation-predominant irritable bowel syndrome. Aliment Pharmacol Ther 2006; 23:1655–1665.
- Henderson JC, Palmer RM, Meyers NL, Spiller RC. A phase IIb clinical study of renzapride in mixed symptom (alternating) irritable bowel syndrome [abstract]. Gastroenterology 2004; 126 suppl 2:A644.
- Meyers NL, Palmer RMJ, George A. Efficacy and safety of renzapride in patients with constipation-predominant IBS: a phase IIb study in the UK primary healthcare setting [abstract]. Gastroenterology 2004; 126 suppl 2:A640.
- Coulie B, Szarka LA, Camilleri M, et al. Recombinant human neurotrophic factors accelerate colonic transit and relieve constipation in humans. Gastroenterology 2000; 119:41–50.
- Parkman HP, Rao SS, Reynolds JC, et al. Neurotrophin-3 improves functional constipation. Am J Gastroenterol 2003; 98:1338–1347.
- Camilleri M. Alvimopan, a selective peripherally acting muopioid antagonist. Neurogastroenterol Motil 2005; 17:157–165.
- Holzer P. Opioids and opioid receptors in the enteric nervous system: from a problem in opioid analgesia to a possible new prokinetic therapy in humans. Neurosci Lett 2004; 361:192–195.
- Kurtz C, Fitch D, Busby R, et al. Effects of multidose administration of MD-1100 on safety, tolerability, exposure and pharmacodynamics in healthy subjects [abstract]. Gastroenterology 2006; 130 suppl 2:A26.
- Eutamene H, Theodorou V, Tondereau V, et al. Influence of guanylate cyclase C binding ligand MD-1100 on TNBS-induced visceral hypersensitivity in WT vs. KO guanylate cyclase C deficient mice [abstract]. Gastroenterology 2006; 130 suppl 2:A597.
- Broughton G. Chenodeoxycholate: the bile acid. The drug. a review. Am J Med Sci 1994; 307:54–63.
- Bazzoli F, Malavolti M, Petronelli A, Barbara L, Roda E. Treatment of constipation with chenodeoxycholic acid. J Int Med Res 1983; 11:120–123.
- Picard C, Fioramonti J, Francois A, Robinson T, Neant F, Matuchansky C. Review article: bifidobacteria as probiotic agents – physiological effects and clinical benefits. Aliment Pharmacol Ther 2005; 22:495–512.
- Macfarlane S, Macfarlane GT, Cummings JH. Review article: prebiotics in the gastrointestinal tract. Aliment Pharmacol Ther 2006; 24:701–714.
- Ouwehand A, Lagstrom H, Suomalainen T, Salminen S. Effect of probiotics on constipation, fecal azoreductase activity and fecal mucin content in the elderly. Ann Nutr Metab 2002; 46:159–162.
- Whorwell P, Altringer L, Morel J, et al. Efficacy of an encapsulated probiotic Bifidobacterium infantis 35624 in women with irritable bowel syndrome. Am J Gastroenterol 2006; 101:1581–1590.
- O'Mahony L, McCarthy J, Kelly P, et al. Lactobacillus and Bifidobacterium in irritable bowel syndrome: symptom responses and relationship to cytokine profiles. Gastroenterology 2005; 128:541–551.
- Koebnick C, Wagner I, Leitzmann P, Stern U, Zunft H. Probiotic beverage containing Lactobacillus casei Shirota improves gastrointestinal symptoms in patients with chronic constipation. Can J Gastroenterol 2003; 17:655–659.
- Kim HJ, Camilleri M, McKinzie S, et al. A randomized controlled trial of a probiotic, VSL#3, on gut transit and symptoms in diarrhoea-predominant irritable bowel syndrome. Aliment Pharmacol Ther 2003; 17:895–904.
- Fernández-Bañares F. Nutritional care of the patient with constipation. Best Pract Res Clin Gastroenterol 2006; 20:575–587.
- Bouvier M, Meance S, Bouley C, Berta J, Grimaud J. Effects of consumption of milk fermented by the probiotic strain Bifidobacterium animalis DN-173 010 on colonit transit times in healthy humans. Biosci Microflor 2001; 20 2:43–48.
- Makelainen H, Tahvonen R, Salminen S, Ouwehand AC. In vivo safety assessment of two Bifidobacterium longum strains. Microbiol Immunol 2003; 47:911–914.
- Borriello SP, Hammes WP, Holzapfel W, et al. Safety of probiotics that contain lactobacilli or bifidobacteria. Clin Infect Dis 2003; 36:775–780.
- Ramkumar D, Rao S. Efficacy and safety of traditional medical therapies for chronic constipation: systematic review. Am J Gastroenterol 2005; 100:936–971.
- Ueno R, Osama H, Habe T, Engelke K, Patchen M. Oral SPI-0211 increases intestinal fluid secretion and chloride concentration without altering serum electrolyte levels [abstract]. Gastroenterology 2004; 126 suppl 2:A298.
- Rao SS, Seaton K, Miller M, et al. Randomized controlled trial of biofeedback, sham biofeedback, and standard therapy for dyssynergic defecation. Clin Gastroenterol Hepatol 2007; 5:331–338.
- Chiarioni G, Whitehead WE, Pezza V, Morelli A, Bassotti G. Biofeedback is superior to laxatives for normal transit constipation due to pelvic floor dyssynergia. Gastroenterology 2006; 130:657–664.
- Heymen S, Scarlett Y, Jones K, Ringel Y, Drossman D, Whitehead WE. Randomized, controlled trial shows biofeedback to be superior to alternative treatments for patients with pelvic floor dyssynergia-type constipation. Dis Colon Rectum 2007; 50:428–441.
- Higgins PD, Johanson JF. Epidemiology of constipation in North America: a systematic review. Am J Gastroenterol 2004; 99:750–759.
- Brandt LJ, Prather CM, Quigley EM, Schiller LR, Schoenfeld P, Talley NJ. Systematic review on the management of chronic constipation in North America. Am J Gastroenterol 2005; 100 suppl 1:S5–S21.
- Pleis JR, Lethbridge-Cejku M. Summary health statistics for U.S. adults: National Health Interview Survey, 2005. Vital Health Stat 10 2006; 232:1–153.
- Dennison C, Prasad M, Lloyd A, Bhattacharyya SK, Dhawan R, Coyne K. The health-related quality of life and economic burden of constipation. Pharmacoeconomics 2005; 23:461–476.
- Arce DA, Ermocilla CA, Costa H. Evaluation of constipation. Am Fam Physician 2002; 65:2283–2290.
- Johanson JF, Sonnenberg A, Koch TR. Clinical epidemiology of chronic constipation. J Clin Gastroenterol 1989; 11:525–536.
- Harari D, Gurwitz JH, Minaker KL. Constipation in the elderly. J Am Geriatr Soc 1993; 41:1130–1140.
- Johanson JF, Kralstein J. Chronic constipation: a survey of the patient perspective. Aliment Pharmacol Ther 2007; 25:599–608.
- Lembo A, Camilleri M. Chronic constipation. N Engl J Med 2003; 349:1360–1368.
- Sandler RS, Drossman DA. Bowel habits in young adults not seeking health care. Dig Dis Sci 1987; 32:841–845.
- Longstreth GF, Thompson WG, Chey WD, Houghton LA, Mearin F, Spiller RC. Functional bowel disorders. Gastroenterology 2006; 130:1480–1491.
- Locke GR, Pemberton JH, Phillips SF. AGA technical review on constipation. Gastroenterology 2000; 119:1766–1778.
- Rao SS. Constipation: evaluation and treatment. Gastroenterol Clin North Am 2003; 32:659–683.
- Locke GR, Pemberton JH, Phillips SF. American Gastroenterological Association Medical Position Statement: guidelines on constipation. Gastroenterology 2000; 119:1761–1766.
- Prather CM. Subtypes of constipation: sorting out the confusion. Rev Gastroenterol Disord 2004; 4 suppl 2:S11–S16.
- Talley NJ, Jones M, Nuyts G, Dubois D. Risk factors for chronic constipation based on a general practice sample. Am J Gastroenterol 2003; 98:1107–1111.
- Bharucha AE. Treatment of severe and intractable constipation. Curr Treat Options Gastroenterol 2004; 7:291–298.
- Borum ML. Constipation: evaluation and management. Prim Care 2001; 28:577–590.
- Camilleri M, Ford MJ. Review article: colonic sensorimotor physiology in health, and its alteration in constipation and diarrhoeal disorders. Aliment Pharmacol Ther 1998; 12:287–302.
- Knowles CH, Martin JE. Slow transit constipation: a model of human gut dysmotility. Review of possible aetiologies. Neurogastroenterology Motility 2000; 12:181–196.
- Schiller LR. Review article: the therapy of constipation. Aliment Pharmacol Ther 2001; 15:749–763.
- Crowell MD. Pathogenesis of slow transit and pelvic floor dysfunction: from bench to bedside. Rev Gastroenterol Disord 2004; 4 suppl 2:S17–S27.
- Wood JD. Neuropathophysiology of irritable bowel syndrome. J Clin Gastroenterol 2002; 35 suppl 1:S11–S22.
- Chitkara DK, Bredenoord AJ, Cremonini F, et al. The role of pelvic floor dysfunction and slow colonic transit in adolescents with refractory constipation. Am J Gastroenterol 2004; 99:1579–1584.
- El-Salhy M. Gastrointestinal transit in an animal model of human diabetes type 2: relationship to gut neuroendocrine peptide contents. Ups J Med Sci 2002; 107:101–110.
- Crowell MD. Role of serotonin in the pathophysiology of the irritable bowel syndrome. Br J Pharmacol 2004; 141:1285–1293.
- Gershon MD. Review article: roles played by 5-hydroxytryptamine in the physiology of the bowel. Aliment Pharmacol Ther 1999; 13 suppl 2:15–30.
- Gershon MD. Serotonin and its implication for the management of irritable bowel syndrome. Rev Gastroenterol Disord 2003; 3 suppl 2:S25–S34.
- Grider JR, Foxx-Orenstein AE, Jin JG. 5-Hydroxytryptamine4 receptor agonists initiate the peristaltic reflex in human, rat, and guinea pig intestine. Gastroenterology 1998; 115:370–380.
- Krishnamurthy S, Schuffler MD, Rohrmann CA, Pope CE. Severe idiopathic constipation is associated with a distinctive abnormality of the colonic myenteric plexus. Gastroenterology 1985; 88:26–34.
- O'Donnell LJ, Virjee J, Heaton KW. Detection of pseudodiarrhoea by simple clinical assessment of intestinal transit rate. BMJ 1990; 300:439–440.
- Meshkinpour H, Selod S, Movahedi H, Nami N, James N, Wilson A. Effects of regular exercise in management of chronic idopathic constipation. Dig Dis Sci 1998; 43:2379–2383.
- Young RJ, Beerman LE, Vanderhoff JA. Increasing oral fluids in chronic constipation in children. Gastroenterol Nurs 1998; 21:156–161.
- Tuteja AK, Talley NJ, Joos SK, Woehl JV, Hickam DH. Is constipation associated with decreased physical activity in normally active subjects? Am J Gastroenterol 2005; 100:124–129.
- Towers AL, Burgio KL, Locher JL, Merkel IS, Safaeian M, Wald A. Constipation in the elderly: influence of dietary, psychological, and physiological factors. J Am Geriatr Soc 1994; 42:701–706.
- Anti J, Pignataro G, Armuzzi A, et al. Water supplementation enhances the effect of high-fiber diet on stool frequency and laxative consumption in adult patients with functional constipation. Hepatogastroenterology 1998; 45:727–732.
- Murakami K, Sasaki S, Okubo H, et al. Association between dietary fiber, water and magnesium intake and functional constipation among young Japanese women. Eur J Clin Nutr 2007; 61:616–622.
- Wong ML, Wee S, Pin CH, Gan GL, Ye HC. Sociodemographic and lifestyle factors associated with constipation in an elderly Asian community. Am J Gastroenterol 1999; 94:1283–1291.
- Dukas L, Willett WC, Giovannucci EL. Association between physical activity, fiber intake, and other lifestyle variables and constipation in a study of women. Am J Gastroenterol 2003; 98:1790–1796.
- Voderholzer WA, Schatke W, Muhldorfer BE, et al. Clinical response to dietary fiber treatment of chronic constipation. Am J Gastroenterol 1997; 92:95–98.
- Graham DY, Moser SE, Estes MK. The effect of bran on bowel function in constipation. Am J Gastroenterol 1982; 77:599–603.
- Muller-Lissner SA. Effect of wheat bran on weight of stool and gastrointestinal transit time: a meta analysis. Br Med J (Clin Res Ed) 1988; 296:615–617.
- Badiali D, Corazziari E, Habib FI, et al. Effect of wheat bran in treatment of chronic nonorganic constipation. A double-blind controlled trial. Dig Dis Sci 1995; 40:349–356.
- Andersson H, Basaeus I, Falkheden T, Melkersson M. Transit time in constipated geriatric patients during treatment with bulk laxative and bran: a comparison. Scand J Gastroenterol 1979; 14:821–826.
- Hamilton JW, Wagner J, Burdick BB, Bass P. Clinical evaluation of methyl-cellulose as a bulk laxative. Dig Dis Sci 1988; 33:993–998.
- Chen HL, Cheng HC, Liu YJ, Liu SY, Wu WT. Konjac acts as a natural laxative by increasing stool bulk and improving colonic ecology in healthy adults. Nutrition 2006; 22:1112–1119.
- Ashraf W, Park F, Lof J, Quigley EM. Effects of psyllium therapy on stool characteristics, colon transit and anorectal function in chronic idiopathic constipation. Aliment Pharmacol Ther 1995; 9:639–647.
- Cheskin LJ, Kamal N, Crowell MD, Schuster MM, Whitehead WE. Mechanisms of constipation in older persons and effects of fiber compared with placebo. J Am Geriatr Soc 1995; 43:666–669.
- Bijkerk CJ, Muris JW, Knottnerus JA, Hoes AW, de Wit NJ. Systematic review: the role of different types of fiber in the treatment of irritable bowel syndrome. Aliment Pharmacol Ther 2004; 19:245–251.
- Corazziari E, Badiali D, Habib FI, et al. Small volume isosmotic polyethylene glycol electrolyte balanced solution (PMF–100) in treatment of chronic nonorganic constipation. Dig Dis Sci 1996; 41:1636–1642.
- Attar A, Lemann M, Ferguson A, et al. Comparison of a low dose polyethylene glycol electrolyte solution with lactulose for treatment of chronic constipation. Gut 1999; 44:226–230.
- Corazziari E, Badiali D, Bazzocchi G, et al. Long term efficacy, safety, and tolerabilitity of low daily doses of isosmotic polyethylene glycol electrolyte balanced solution (PMF–100) in the treatment of functional chronic constipation. Gut 2000; 46:522–526.
- Fritz E, Hammer HF, Lipp RW, Högenauer C, Stauber R, Hammer J. Effects of lactulose and polyethylene glycol on colonic transit. Aliment Pharmacol Ther 2005; 21:259–268.
- DiPalma JA, Cleveland MV, McGowan J, Herrera JL. A randomized, multicenter, placebo-controlled trial of polyethylene glycol laxative for chronic treatment of chronic constipation. Am J Gastroenterol 2007; 102:1436–1441.
- Bass P, Dennis S. The laxative effects of lactulose in normal and constipated subjects. J Clin Gastroenterol 1981; 3 suppl 1:23–28.
- Lederle FA, Busch DL, Mattox KM, West MJ, Aske DM. Cost-effective treatment of constipation in the elderly: a randomized double-blind comparison of sorbitol and lactulose. Am J Med 1990; 89:597–601.
- Volicer L, Lane P, Panke J, Lyman P. Management of constipation in residents with dementia: sorbitol effectiveness and cost. J Am Med Dir Assoc 2004; 5:239–241.
- Quah HM, Ooi BS, Seow-Choen F, Sng KK, Ho KS. Prospective randomized crossover trial comparing fibre with lactulose in the treatment of idiopathic chronic constipation. Tech Coloproctol 2006; 10:111–114.
- Passmore AP, Davies KW, Flanagan PG, Stoker C, Scott MG. A comparison of Agiolax and lactulose in elderly patients with chronic constipation. Pharmacology 1993; 47 suppl 1:249–252.
- Passmore AP, Wilson-Davies K, Stoker C, Scott ME. Chronic constipation in long stay elderly patients: a comparison of lactulose and a senna-fibre combination. BMJ 1993; 307:769–771.
- Poon CM, Lee DW, Mak SK, et al. Two liters of polyethylene glycol-electrolyte lavage solution versus sodium phosphate as bowel cleansing regimen for colonoscopy: a prospective randomized controlled trial. Endoscopy 2002; 34:560–563.
- Rostom A, Jolicoeur E, Dube C, et al. A randomized prospective trial comparing different regimens of oral sodium phosphate and polyethylene glycol-based lavage solution in the preparation of patients for colonoscopy. Gastrointest Endosc 2006; 64:544–552.
- Wald A. Is chronic use of stimulant laxatives harmful to the colon? J Clin Gastroenterol 2003; 36:386–389.
- Fried M, Johanson JF, Gwee KA, Wagner A, Pecher E, Rueegg P. Efficacy of tegaserod in chronic constipation in men. Am J Gastroenterol 2007; 102:362–370.
- Lin SR, Ke MY, Luo JY, et al. A randomized, double-blind, placebo-controlled trial assessing the efficacy and safety of tegaserod in patients from China with chronic constipation. World J Gastroenterol 2007; 13:732–739.
- Camilleri M, Bharucha AE, Ueno R, et al. Effect of a selective chloride channel activator, lubiprostone, on gastrointestinal transit, gastric sensory, and motor functions in healthy volunteers. Am J Physiol Gastrointest Liver Physiol 2006; 290:G942–G947.
- McKeage K, Plosker GL, Siddiqui MA. Lubiprostone. Drugs 2006; 66:873–879.
- Johanson JF, Gargano MA, Patchen ML, Ueno R. Efficacy and safety of a novel compound, RU-0211, for the treatment of constipation [abstract]. Gastroenterology 2002; 122 suppl 1:A315.
- Johanson JF, Gargano MA, Holland PC, Patchen ML, Ueno R. Multicenter open-label study of oral lubiprostone for the treatment of chronic constipation [abstract]. Am J Gastroenterol 2005; 100 suppl:S331.
- Johanson JF, Panas R, Holland P, Ueno R. Long-term efficacy of lubiprostone for the treatment of chronic constipation [abstract]. Gastroenterology 2006; 130 suppl 2:A317.
- Ueno R, Panas R, Wahle A, Zhu Y, Holland P. Long-term safety and efficacy of lubiprostone for the treatment of chronic constipation in the elderly [abstract]. Gastroenterology 2006; 130 suppl 2:A188.
- Johanson JF, Gargano MA, Holland P, Patchen ML, Ueno R. Phase III, randomized withdrawal study of RU-0211, a novel chloride channel activator for the treatment of constipation [abstract]. Gastroenterology 2004; 126 suppl 2:A100.
- Rivkin A, Chagan L. Lubiprostone: chloride channel activator for chronic constipation. Clin Ther 2006; 28:2008–2021.
- Johanson JF, Miner PB, Parkman HP, et al. Prucalopride improves bowel movement frequency and symptoms in patients with chronic constipation: results of two double-blind, placebo-controlled trials [abstract]. Gastroenterology 2000; 118 suppl 2:A175.
- Cash BD, Chey WD. Review article: the role of serotonergic agents in the treatment of patients with primary chronic constipation. Aliment Pharmacol Ther 2005; 22:1047–1060.
- Altabas K, Biliæ A, Jurciæ D, et al. The efficacy of cisapride vs. placebo and diet in patients with chronic constipation. Coll Antropol 2003; 27:197–204.
- Camilleri M, Kerstens R, Rykx A, Vandeplassche L. A placebo-controlled trial of prucalopride for severe chronic constipation. N Engl J Med 2008; 358:2344–2354.
- Camilleri M, McKinzie S, Fox J, et al. Effect of renzapride on transit in constipation-predominant irritable bowel syndrome. Clin Gastroenterol Hepatol 2004; 2:895–904.
- Tack J, Middleton SJ, Horne MC, et al. Pilot study of the efficacy of renzapride on gastrointestinal motility and symptoms in patients with constipation-predominant irritable bowel syndrome. Aliment Pharmacol Ther 2006; 23:1655–1665.
- Henderson JC, Palmer RM, Meyers NL, Spiller RC. A phase IIb clinical study of renzapride in mixed symptom (alternating) irritable bowel syndrome [abstract]. Gastroenterology 2004; 126 suppl 2:A644.
- Meyers NL, Palmer RMJ, George A. Efficacy and safety of renzapride in patients with constipation-predominant IBS: a phase IIb study in the UK primary healthcare setting [abstract]. Gastroenterology 2004; 126 suppl 2:A640.
- Coulie B, Szarka LA, Camilleri M, et al. Recombinant human neurotrophic factors accelerate colonic transit and relieve constipation in humans. Gastroenterology 2000; 119:41–50.
- Parkman HP, Rao SS, Reynolds JC, et al. Neurotrophin-3 improves functional constipation. Am J Gastroenterol 2003; 98:1338–1347.
- Camilleri M. Alvimopan, a selective peripherally acting muopioid antagonist. Neurogastroenterol Motil 2005; 17:157–165.
- Holzer P. Opioids and opioid receptors in the enteric nervous system: from a problem in opioid analgesia to a possible new prokinetic therapy in humans. Neurosci Lett 2004; 361:192–195.
- Kurtz C, Fitch D, Busby R, et al. Effects of multidose administration of MD-1100 on safety, tolerability, exposure and pharmacodynamics in healthy subjects [abstract]. Gastroenterology 2006; 130 suppl 2:A26.
- Eutamene H, Theodorou V, Tondereau V, et al. Influence of guanylate cyclase C binding ligand MD-1100 on TNBS-induced visceral hypersensitivity in WT vs. KO guanylate cyclase C deficient mice [abstract]. Gastroenterology 2006; 130 suppl 2:A597.
- Broughton G. Chenodeoxycholate: the bile acid. The drug. a review. Am J Med Sci 1994; 307:54–63.
- Bazzoli F, Malavolti M, Petronelli A, Barbara L, Roda E. Treatment of constipation with chenodeoxycholic acid. J Int Med Res 1983; 11:120–123.
- Picard C, Fioramonti J, Francois A, Robinson T, Neant F, Matuchansky C. Review article: bifidobacteria as probiotic agents – physiological effects and clinical benefits. Aliment Pharmacol Ther 2005; 22:495–512.
- Macfarlane S, Macfarlane GT, Cummings JH. Review article: prebiotics in the gastrointestinal tract. Aliment Pharmacol Ther 2006; 24:701–714.
- Ouwehand A, Lagstrom H, Suomalainen T, Salminen S. Effect of probiotics on constipation, fecal azoreductase activity and fecal mucin content in the elderly. Ann Nutr Metab 2002; 46:159–162.
- Whorwell P, Altringer L, Morel J, et al. Efficacy of an encapsulated probiotic Bifidobacterium infantis 35624 in women with irritable bowel syndrome. Am J Gastroenterol 2006; 101:1581–1590.
- O'Mahony L, McCarthy J, Kelly P, et al. Lactobacillus and Bifidobacterium in irritable bowel syndrome: symptom responses and relationship to cytokine profiles. Gastroenterology 2005; 128:541–551.
- Koebnick C, Wagner I, Leitzmann P, Stern U, Zunft H. Probiotic beverage containing Lactobacillus casei Shirota improves gastrointestinal symptoms in patients with chronic constipation. Can J Gastroenterol 2003; 17:655–659.
- Kim HJ, Camilleri M, McKinzie S, et al. A randomized controlled trial of a probiotic, VSL#3, on gut transit and symptoms in diarrhoea-predominant irritable bowel syndrome. Aliment Pharmacol Ther 2003; 17:895–904.
- Fernández-Bañares F. Nutritional care of the patient with constipation. Best Pract Res Clin Gastroenterol 2006; 20:575–587.
- Bouvier M, Meance S, Bouley C, Berta J, Grimaud J. Effects of consumption of milk fermented by the probiotic strain Bifidobacterium animalis DN-173 010 on colonit transit times in healthy humans. Biosci Microflor 2001; 20 2:43–48.
- Makelainen H, Tahvonen R, Salminen S, Ouwehand AC. In vivo safety assessment of two Bifidobacterium longum strains. Microbiol Immunol 2003; 47:911–914.
- Borriello SP, Hammes WP, Holzapfel W, et al. Safety of probiotics that contain lactobacilli or bifidobacteria. Clin Infect Dis 2003; 36:775–780.
- Ramkumar D, Rao S. Efficacy and safety of traditional medical therapies for chronic constipation: systematic review. Am J Gastroenterol 2005; 100:936–971.
- Ueno R, Osama H, Habe T, Engelke K, Patchen M. Oral SPI-0211 increases intestinal fluid secretion and chloride concentration without altering serum electrolyte levels [abstract]. Gastroenterology 2004; 126 suppl 2:A298.
- Rao SS, Seaton K, Miller M, et al. Randomized controlled trial of biofeedback, sham biofeedback, and standard therapy for dyssynergic defecation. Clin Gastroenterol Hepatol 2007; 5:331–338.
- Chiarioni G, Whitehead WE, Pezza V, Morelli A, Bassotti G. Biofeedback is superior to laxatives for normal transit constipation due to pelvic floor dyssynergia. Gastroenterology 2006; 130:657–664.
- Heymen S, Scarlett Y, Jones K, Ringel Y, Drossman D, Whitehead WE. Randomized, controlled trial shows biofeedback to be superior to alternative treatments for patients with pelvic floor dyssynergia-type constipation. Dis Colon Rectum 2007; 50:428–441.
KEY POINTS
- A high-fiber diet often improves functional constipation, but it may worsen slow-transit constipation or dyssynergia (a failure of the pelvic floor muscles to relax). Nevertheless, fiber remains a mainstay of treatment for its ability to provide homogeneous stool consistency.
- Drugs approved for treating constipation increase fluid in the lumen, speed intestinal transit, and improve stool consistency, while tegaserod (Zelnorm) additionally acts as a serotonin agonist.
- Colonoscopy and other tests are reserved for patients with refractory constipation and those with symptoms suggesting colon cancer.
- Prebiotics (short-chain carbohydrates that stimulate activity of beneficial colonic bacterial flora) and probiotics (live bacterial preparations) are under evaluation as treatments for chronic constipation.
Preventing renal disease progression: Can complete renin-angiotensin-aldosterone blockade work?
Perhaps the most daunting challenge for any primary care physician, nephrologist, or other internal medicine specialist is how to prevent the progression of chronic kidney disease.
A MAJOR HEALTH CARE CRISIS
Ten to 20 million people in the United States have chronic kidney disease, with diabetic nephropathy and arterial hypertension accounting for two-thirds of cases. In 2007, the US Renal Data System1 reported that, at the end of 2005, 341,319 patients were receiving dialysis and another 143,693 had received renal transplants.
The National Kidney Foundation’s Kidney Disease Outcomes Quality Initiatives2 has raised the level of awareness of chronic kidney disease among physicians and the general public. We have become more adept at diagnosing chronic kidney disease, in particular by calculating the estimated glomerular filtration rate, and we are starting to learn how to sort out the patients designated as having chronic kidney disease by this calculation but without “true” kidney disease. Nevertheless, the medical profession is still struggling to determine the best way to prevent progression in chronic kidney disease, and no single innovative approach currently exists. Should the emphasis be on the blood pressure target, the level of proteinuria reduction, the classes of medications to be used, or on other factors such as lipid control, vitamin D repletion,3 or glycemic control?
WHY INHIBIT THE RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM?
Over the last 20 years, investigators have devoted much effort to controlling the adverse effects of the renin-angiotensin-aldosterone system on the renal vasculature and parenchyma. We now understand that this system is a complex cascade and that angiotensin II plays a key role.
Angiotensin II enhances the vascular tone of both the afferent and the efferent glomerular arterioles, helps regulate intraglomerular pressure and glomerular filtration, and stimulates the adrenal cortex to release aldosterone. In addition, it has several nonhemodynamic effects. In particular, it may alter the selective permeability of the glomerular capillary barrier by influencing podocyte morphology and by directing a reorganization of its actin cytostructure.
Podocytes are highly differentiated pericyte-like cells that are essential for normal kidney function, but they have limited regenerative ability. Angiotensin II stimulation can lead to podocyte injury via mechanical stress due to increased intraglomerular pressure or an increase in cytosolic calcium,4 formation of bridging between the parietal basement membrane and the glomerular basement membrane,5 and extension of the extracapillary disease process to the glomerular-proximal tubular junction.6 These alterations can result in progressive atrophy, cell death, subsequent fibrosis, and irreversible loss in functioning renal parenchyma.
EVIDENCE FOR AND AGAINST COMBINATION THERAPY
In theory, by completely inhibiting the renin-angiotensin-aldosterone system in some patients with proteinuric chronic kidney disease (as Dr. Sheldon Hirsch suggests in this issue of the Cleveland Clinic Journal of Medicine7), we might be better able to prevent progressive renal injury than with an incomplete blockade of this system.
The rationale for complete blockade stems from evidence that long-term treatment with an angiotensin-converting enzyme (ACE) inhibitor results in the accumulation of angiotensin I, the escape of angiotensin II generation by ACE-independent enzymes (chymases), and the inhibition of angiotensin-(1–7) formation that partially antagonizes the effects of angiotensin II. In addition, aldosterone may injure the kidney by its rapid nongenomic effect on the renal vasculature, resulting in increased renal vascular resistance, with afferent and efferent vasoconstriction. Therefore, treatment with either an ACE inhibitor or an angiotensin receptor blocker (ARB) by itself may delay but not prevent end-stage renal disease for most patients with proteinuric chronic kidney disease.8
Combining an ACE inhibitor and an ARB
Regimens in which an ACE inhibitor is combined with an ARB may achieve their therapeutic benefit of lowering proteinuria by modulating the compensatory events in kidney injury that stress “normal” nephrons, inhibiting the podocyte injury responsible for contiguous damage in the tubulointerstitial area, and limiting fibrosis and inflammation. However, few trials actually showed that combining an ACE inhibitor with an ARB leads to greater renal protection in the long term than with either agent alone, despite a greater chance of lowering the protein excretion rate.9,10
The COOPERATE study. The Combination Treatment of Angiotensin II Receptor Blocker and Angiotensin-Converting-Enzyme Inhibitor in Non-diabetic Renal Disease (COOPERATE) study11 evaluated the renoprotective effects of the combination of trandolapril (Mavik, an ACE inhibitor) and losartan (Cozaar, an ARB). Significantly fewer patients reached one of the end points (doubling of the serum creatinine concentration or end-stage renal disease) with the combined therapy than with either agent alone.
Kunz et al12 recently performed a meta-analysis, which indicated that the combination of an ACE inhibitor and an ARB reduces proteinuria to a greater extent than either drug alone. However, the total number of patients in each trial was less than 30 on average, the duration of therapy rarely exceeded 1 year, and the effect on changes in the glomerular filtration rate or the need for dialysis was not reported.
ONTARGET. In the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET),13 combination therapy had no clear benefit in the group at the highest renal risk (ie, with overt diabetic nephropathy), and it was associated with a trend toward worse results in the low-risk group. Most participants in ONTARGET did not have microalbuminuria or macroalbuminuria, and of interest, these patients without protein excretion were at increased risk for renal events, such as acute renal failure requiring dialysis.
Phillips et al14 recently reported on the safety profile of patients with symptomatic left ventricular dysfunction treated with the combination of an ACE inhibitor and an ARB. Even in these nonrenal patients there was a significantly higher risk of worsening renal dysfunction (relative risk 4.87, 95% confidence interval 2.39–9.94) and hyperkalemia (relative risk 4.87, 95% confidence interval 2.39–9.94) with combination therapy.
Adding an aldosterone blocker to an ACE inhibitor, ARB, or both
There is little evidence that aldosterone plays a role in the progression of chronic kidney disease. However, several studies found that combining an aldosterone blocker with an ACE inhibitor, ARB, or both had an additional impact on reducing proteinuria and modulating the rate of change in the glomerular filtration rate.15–17
When aldosterone antagonists were added to an ACE inhibitor, an ARB, or both combined, proteinuria was reduced, but there was little effect on preserving the glomerular filtration rate.17 However, most of the studies were small, with short observation periods. Hyperkalemia is a risk when using aldosterone antagonists in combination with ACE inhibitors and ARBs, especially in patients with glomerular filtration rates less than 30 mL/minute.18
Adding a renin inhibitor to an ACE inhibitor or an ARB
Few studies have examined combination therapy with either an ACE inhibitor or ARB plus a renin inhibitor, the newest class of agents that block this system.
Parving et al19 recently reported the results of combining aliskiren (Tekturna, a renin inhibitor) with losartan in 599 patients with type 2 diabetes and nephropathy. At 6 months, the renin inhibitor showed a renoprotective effect that was independent of its blood-pressure-lowering effect in those who were receiving maximal recommended doses of the ARB.
OTHER FACTORS ALSO INFLUENCE PROGRESSION
Even though there is broad agreement that an approach that neutralizes the effects of the renin-angiotensin-aldosterone system on the kidney would lower blood pressure and protein excretion rates, whether it would change the natural history of chronic kidney disease and prevent progression is less clear. In reality, a number of factors other than the renin-angiotensin-aldosterone system are responsible for the progression of chronic kidney disease. These other factors may help explain why control of this system does not totally prevent deterioration of chronic kidney disease, although the rate may be slowed.
MORE QUESTIONS THAN ANSWERS
A number of provocative questions arise from Dr. Hirsch’s discussion of complete renin-angiotensin-aldosterone system blockade to prevent disease progression:
- Will decreasing proteinuria to a specific target (< 500 mg/day) prevent progression?
- How low should the blood pressure target be set to modulate progression, and should it be the same in all age groups?
- Should complete blockade be applied all at once or in a stepwise fashion depending on the glomerular filtration rate, the level of proteinuria, or both?
- Which patients would benefit most from complete blockade?
- Is direct renin inhibition a critical component of complete blockade?
- What model of chronic disease management is required to avoid unexpected complications if this treatment approach is embraced?
Currently, therefore, there are more questions than answers. This strategy is an intriguing, opinion-based option, but for now it should only be applied to patients with proteinuria and evidence of early progression despite standard therapy who can be closely monitored, and it is not for the faint of heart. In view of the risks of hyperkalemia, hypotension, and perhaps even worsening renal function, more data from carefully designed trials are needed before the general medical community widely applies a complete blockade of the renin-angiotensin-aldosterone pathway to prevent progressive chronic kidney disease.
- United States Renal Data System. Annual data report. www.usrds.org/adr.htm. Accessed 9/5/2008.
- National Kidney Foundation. NKF K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. www.kidney.org/Professionals/Kdoqi/guidelines_ckd/toc.htm. Accessed 9/5/2008.
- Remuzzi A. Vitamin D, insulin resistance, and renal disease. Kidney Int. 2007; 71:96–98.
- Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003; 83:253–307.
- Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int 1998; 54:687–697.
- Endlich N, Endlich K. Stretch, tension and adhesion—adaptive mechanisms of the actin cytoskeleton in podocytes. Eur J Cell Biol. 2006; 85:229–234.
- Hirsch S. An update on proteinuric chronic kidney disease: the dual-goal approach. Cleve Clin J Med. 2008; 75:705–713.
- Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993; 329:1456–1462.
- Wolf G, Ritz E. Combination therapy with ACE inhibitors and angiotensin II receptor blockers to halt progression of chronic renal disease: pathophysiology and indications. Kidney Int. 2005; 67:799–812.
- Campbell R, Sangalli F, Perticucci E, et al. Effects of combined ACE inhibitor and angiotensin II antagonist treatment in human chronic nephropathies. Kidney Int. 2003; 63:1094–1103.
- Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomised controlled trial. Lancet. 2003; 361:117–124.
- Kunz R, Friedrich C, Wolbers M, Mann JF. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med. 2008; 148:30–48.
- Mann JF, Schmieder RE, McQueen M, et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet. 2008; 372:547–553.
- Phillips CO, Kashani A, Ko DK, Francis G, Krumholz HM. Adverse effects of combination angiotensin II receptor blockers plus angiotensin-converting enzyme inhibitors for left ventricular dysfunction: a quantitative review of data from randomized clinical trials. Arch Intern Med. 2007; 167:1930–1936.
- Epstein M. Adding spironolactone to conventional antihypertensives reduces albuminuria in patients with diabetic nephropathy. Nat Clin Pract Nephrol. 2006; 2:310–311.
- Rossing K, Schjoedt KJ, Smidt UM, Boomsma F, Parving HH. Beneficial effects of adding spironolactone to recommended antihypertensive treatment in diabetic nephropathy: a randomized, double-masked, cross-over study. Diabetes Care. 2005; 28:2106–2112.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006; 70:2116–2123.
- Bomback AS, Kshirsagar AV, Amamoo MA, Klemmer PJ. Change in proteinuria after adding aldosterone blockers to ACE inhibitors or angiotensin receptor blockers in CKD: a systematic review. Am J Kidney Dis. 2008; 51:199–211.
- Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med. 2008; 358:2433–2446.
Perhaps the most daunting challenge for any primary care physician, nephrologist, or other internal medicine specialist is how to prevent the progression of chronic kidney disease.
A MAJOR HEALTH CARE CRISIS
Ten to 20 million people in the United States have chronic kidney disease, with diabetic nephropathy and arterial hypertension accounting for two-thirds of cases. In 2007, the US Renal Data System1 reported that, at the end of 2005, 341,319 patients were receiving dialysis and another 143,693 had received renal transplants.
The National Kidney Foundation’s Kidney Disease Outcomes Quality Initiatives2 has raised the level of awareness of chronic kidney disease among physicians and the general public. We have become more adept at diagnosing chronic kidney disease, in particular by calculating the estimated glomerular filtration rate, and we are starting to learn how to sort out the patients designated as having chronic kidney disease by this calculation but without “true” kidney disease. Nevertheless, the medical profession is still struggling to determine the best way to prevent progression in chronic kidney disease, and no single innovative approach currently exists. Should the emphasis be on the blood pressure target, the level of proteinuria reduction, the classes of medications to be used, or on other factors such as lipid control, vitamin D repletion,3 or glycemic control?
WHY INHIBIT THE RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM?
Over the last 20 years, investigators have devoted much effort to controlling the adverse effects of the renin-angiotensin-aldosterone system on the renal vasculature and parenchyma. We now understand that this system is a complex cascade and that angiotensin II plays a key role.
Angiotensin II enhances the vascular tone of both the afferent and the efferent glomerular arterioles, helps regulate intraglomerular pressure and glomerular filtration, and stimulates the adrenal cortex to release aldosterone. In addition, it has several nonhemodynamic effects. In particular, it may alter the selective permeability of the glomerular capillary barrier by influencing podocyte morphology and by directing a reorganization of its actin cytostructure.
Podocytes are highly differentiated pericyte-like cells that are essential for normal kidney function, but they have limited regenerative ability. Angiotensin II stimulation can lead to podocyte injury via mechanical stress due to increased intraglomerular pressure or an increase in cytosolic calcium,4 formation of bridging between the parietal basement membrane and the glomerular basement membrane,5 and extension of the extracapillary disease process to the glomerular-proximal tubular junction.6 These alterations can result in progressive atrophy, cell death, subsequent fibrosis, and irreversible loss in functioning renal parenchyma.
EVIDENCE FOR AND AGAINST COMBINATION THERAPY
In theory, by completely inhibiting the renin-angiotensin-aldosterone system in some patients with proteinuric chronic kidney disease (as Dr. Sheldon Hirsch suggests in this issue of the Cleveland Clinic Journal of Medicine7), we might be better able to prevent progressive renal injury than with an incomplete blockade of this system.
The rationale for complete blockade stems from evidence that long-term treatment with an angiotensin-converting enzyme (ACE) inhibitor results in the accumulation of angiotensin I, the escape of angiotensin II generation by ACE-independent enzymes (chymases), and the inhibition of angiotensin-(1–7) formation that partially antagonizes the effects of angiotensin II. In addition, aldosterone may injure the kidney by its rapid nongenomic effect on the renal vasculature, resulting in increased renal vascular resistance, with afferent and efferent vasoconstriction. Therefore, treatment with either an ACE inhibitor or an angiotensin receptor blocker (ARB) by itself may delay but not prevent end-stage renal disease for most patients with proteinuric chronic kidney disease.8
Combining an ACE inhibitor and an ARB
Regimens in which an ACE inhibitor is combined with an ARB may achieve their therapeutic benefit of lowering proteinuria by modulating the compensatory events in kidney injury that stress “normal” nephrons, inhibiting the podocyte injury responsible for contiguous damage in the tubulointerstitial area, and limiting fibrosis and inflammation. However, few trials actually showed that combining an ACE inhibitor with an ARB leads to greater renal protection in the long term than with either agent alone, despite a greater chance of lowering the protein excretion rate.9,10
The COOPERATE study. The Combination Treatment of Angiotensin II Receptor Blocker and Angiotensin-Converting-Enzyme Inhibitor in Non-diabetic Renal Disease (COOPERATE) study11 evaluated the renoprotective effects of the combination of trandolapril (Mavik, an ACE inhibitor) and losartan (Cozaar, an ARB). Significantly fewer patients reached one of the end points (doubling of the serum creatinine concentration or end-stage renal disease) with the combined therapy than with either agent alone.
Kunz et al12 recently performed a meta-analysis, which indicated that the combination of an ACE inhibitor and an ARB reduces proteinuria to a greater extent than either drug alone. However, the total number of patients in each trial was less than 30 on average, the duration of therapy rarely exceeded 1 year, and the effect on changes in the glomerular filtration rate or the need for dialysis was not reported.
ONTARGET. In the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET),13 combination therapy had no clear benefit in the group at the highest renal risk (ie, with overt diabetic nephropathy), and it was associated with a trend toward worse results in the low-risk group. Most participants in ONTARGET did not have microalbuminuria or macroalbuminuria, and of interest, these patients without protein excretion were at increased risk for renal events, such as acute renal failure requiring dialysis.
Phillips et al14 recently reported on the safety profile of patients with symptomatic left ventricular dysfunction treated with the combination of an ACE inhibitor and an ARB. Even in these nonrenal patients there was a significantly higher risk of worsening renal dysfunction (relative risk 4.87, 95% confidence interval 2.39–9.94) and hyperkalemia (relative risk 4.87, 95% confidence interval 2.39–9.94) with combination therapy.
Adding an aldosterone blocker to an ACE inhibitor, ARB, or both
There is little evidence that aldosterone plays a role in the progression of chronic kidney disease. However, several studies found that combining an aldosterone blocker with an ACE inhibitor, ARB, or both had an additional impact on reducing proteinuria and modulating the rate of change in the glomerular filtration rate.15–17
When aldosterone antagonists were added to an ACE inhibitor, an ARB, or both combined, proteinuria was reduced, but there was little effect on preserving the glomerular filtration rate.17 However, most of the studies were small, with short observation periods. Hyperkalemia is a risk when using aldosterone antagonists in combination with ACE inhibitors and ARBs, especially in patients with glomerular filtration rates less than 30 mL/minute.18
Adding a renin inhibitor to an ACE inhibitor or an ARB
Few studies have examined combination therapy with either an ACE inhibitor or ARB plus a renin inhibitor, the newest class of agents that block this system.
Parving et al19 recently reported the results of combining aliskiren (Tekturna, a renin inhibitor) with losartan in 599 patients with type 2 diabetes and nephropathy. At 6 months, the renin inhibitor showed a renoprotective effect that was independent of its blood-pressure-lowering effect in those who were receiving maximal recommended doses of the ARB.
OTHER FACTORS ALSO INFLUENCE PROGRESSION
Even though there is broad agreement that an approach that neutralizes the effects of the renin-angiotensin-aldosterone system on the kidney would lower blood pressure and protein excretion rates, whether it would change the natural history of chronic kidney disease and prevent progression is less clear. In reality, a number of factors other than the renin-angiotensin-aldosterone system are responsible for the progression of chronic kidney disease. These other factors may help explain why control of this system does not totally prevent deterioration of chronic kidney disease, although the rate may be slowed.
MORE QUESTIONS THAN ANSWERS
A number of provocative questions arise from Dr. Hirsch’s discussion of complete renin-angiotensin-aldosterone system blockade to prevent disease progression:
- Will decreasing proteinuria to a specific target (< 500 mg/day) prevent progression?
- How low should the blood pressure target be set to modulate progression, and should it be the same in all age groups?
- Should complete blockade be applied all at once or in a stepwise fashion depending on the glomerular filtration rate, the level of proteinuria, or both?
- Which patients would benefit most from complete blockade?
- Is direct renin inhibition a critical component of complete blockade?
- What model of chronic disease management is required to avoid unexpected complications if this treatment approach is embraced?
Currently, therefore, there are more questions than answers. This strategy is an intriguing, opinion-based option, but for now it should only be applied to patients with proteinuria and evidence of early progression despite standard therapy who can be closely monitored, and it is not for the faint of heart. In view of the risks of hyperkalemia, hypotension, and perhaps even worsening renal function, more data from carefully designed trials are needed before the general medical community widely applies a complete blockade of the renin-angiotensin-aldosterone pathway to prevent progressive chronic kidney disease.
Perhaps the most daunting challenge for any primary care physician, nephrologist, or other internal medicine specialist is how to prevent the progression of chronic kidney disease.
A MAJOR HEALTH CARE CRISIS
Ten to 20 million people in the United States have chronic kidney disease, with diabetic nephropathy and arterial hypertension accounting for two-thirds of cases. In 2007, the US Renal Data System1 reported that, at the end of 2005, 341,319 patients were receiving dialysis and another 143,693 had received renal transplants.
The National Kidney Foundation’s Kidney Disease Outcomes Quality Initiatives2 has raised the level of awareness of chronic kidney disease among physicians and the general public. We have become more adept at diagnosing chronic kidney disease, in particular by calculating the estimated glomerular filtration rate, and we are starting to learn how to sort out the patients designated as having chronic kidney disease by this calculation but without “true” kidney disease. Nevertheless, the medical profession is still struggling to determine the best way to prevent progression in chronic kidney disease, and no single innovative approach currently exists. Should the emphasis be on the blood pressure target, the level of proteinuria reduction, the classes of medications to be used, or on other factors such as lipid control, vitamin D repletion,3 or glycemic control?
WHY INHIBIT THE RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM?
Over the last 20 years, investigators have devoted much effort to controlling the adverse effects of the renin-angiotensin-aldosterone system on the renal vasculature and parenchyma. We now understand that this system is a complex cascade and that angiotensin II plays a key role.
Angiotensin II enhances the vascular tone of both the afferent and the efferent glomerular arterioles, helps regulate intraglomerular pressure and glomerular filtration, and stimulates the adrenal cortex to release aldosterone. In addition, it has several nonhemodynamic effects. In particular, it may alter the selective permeability of the glomerular capillary barrier by influencing podocyte morphology and by directing a reorganization of its actin cytostructure.
Podocytes are highly differentiated pericyte-like cells that are essential for normal kidney function, but they have limited regenerative ability. Angiotensin II stimulation can lead to podocyte injury via mechanical stress due to increased intraglomerular pressure or an increase in cytosolic calcium,4 formation of bridging between the parietal basement membrane and the glomerular basement membrane,5 and extension of the extracapillary disease process to the glomerular-proximal tubular junction.6 These alterations can result in progressive atrophy, cell death, subsequent fibrosis, and irreversible loss in functioning renal parenchyma.
EVIDENCE FOR AND AGAINST COMBINATION THERAPY
In theory, by completely inhibiting the renin-angiotensin-aldosterone system in some patients with proteinuric chronic kidney disease (as Dr. Sheldon Hirsch suggests in this issue of the Cleveland Clinic Journal of Medicine7), we might be better able to prevent progressive renal injury than with an incomplete blockade of this system.
The rationale for complete blockade stems from evidence that long-term treatment with an angiotensin-converting enzyme (ACE) inhibitor results in the accumulation of angiotensin I, the escape of angiotensin II generation by ACE-independent enzymes (chymases), and the inhibition of angiotensin-(1–7) formation that partially antagonizes the effects of angiotensin II. In addition, aldosterone may injure the kidney by its rapid nongenomic effect on the renal vasculature, resulting in increased renal vascular resistance, with afferent and efferent vasoconstriction. Therefore, treatment with either an ACE inhibitor or an angiotensin receptor blocker (ARB) by itself may delay but not prevent end-stage renal disease for most patients with proteinuric chronic kidney disease.8
Combining an ACE inhibitor and an ARB
Regimens in which an ACE inhibitor is combined with an ARB may achieve their therapeutic benefit of lowering proteinuria by modulating the compensatory events in kidney injury that stress “normal” nephrons, inhibiting the podocyte injury responsible for contiguous damage in the tubulointerstitial area, and limiting fibrosis and inflammation. However, few trials actually showed that combining an ACE inhibitor with an ARB leads to greater renal protection in the long term than with either agent alone, despite a greater chance of lowering the protein excretion rate.9,10
The COOPERATE study. The Combination Treatment of Angiotensin II Receptor Blocker and Angiotensin-Converting-Enzyme Inhibitor in Non-diabetic Renal Disease (COOPERATE) study11 evaluated the renoprotective effects of the combination of trandolapril (Mavik, an ACE inhibitor) and losartan (Cozaar, an ARB). Significantly fewer patients reached one of the end points (doubling of the serum creatinine concentration or end-stage renal disease) with the combined therapy than with either agent alone.
Kunz et al12 recently performed a meta-analysis, which indicated that the combination of an ACE inhibitor and an ARB reduces proteinuria to a greater extent than either drug alone. However, the total number of patients in each trial was less than 30 on average, the duration of therapy rarely exceeded 1 year, and the effect on changes in the glomerular filtration rate or the need for dialysis was not reported.
ONTARGET. In the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET),13 combination therapy had no clear benefit in the group at the highest renal risk (ie, with overt diabetic nephropathy), and it was associated with a trend toward worse results in the low-risk group. Most participants in ONTARGET did not have microalbuminuria or macroalbuminuria, and of interest, these patients without protein excretion were at increased risk for renal events, such as acute renal failure requiring dialysis.
Phillips et al14 recently reported on the safety profile of patients with symptomatic left ventricular dysfunction treated with the combination of an ACE inhibitor and an ARB. Even in these nonrenal patients there was a significantly higher risk of worsening renal dysfunction (relative risk 4.87, 95% confidence interval 2.39–9.94) and hyperkalemia (relative risk 4.87, 95% confidence interval 2.39–9.94) with combination therapy.
Adding an aldosterone blocker to an ACE inhibitor, ARB, or both
There is little evidence that aldosterone plays a role in the progression of chronic kidney disease. However, several studies found that combining an aldosterone blocker with an ACE inhibitor, ARB, or both had an additional impact on reducing proteinuria and modulating the rate of change in the glomerular filtration rate.15–17
When aldosterone antagonists were added to an ACE inhibitor, an ARB, or both combined, proteinuria was reduced, but there was little effect on preserving the glomerular filtration rate.17 However, most of the studies were small, with short observation periods. Hyperkalemia is a risk when using aldosterone antagonists in combination with ACE inhibitors and ARBs, especially in patients with glomerular filtration rates less than 30 mL/minute.18
Adding a renin inhibitor to an ACE inhibitor or an ARB
Few studies have examined combination therapy with either an ACE inhibitor or ARB plus a renin inhibitor, the newest class of agents that block this system.
Parving et al19 recently reported the results of combining aliskiren (Tekturna, a renin inhibitor) with losartan in 599 patients with type 2 diabetes and nephropathy. At 6 months, the renin inhibitor showed a renoprotective effect that was independent of its blood-pressure-lowering effect in those who were receiving maximal recommended doses of the ARB.
OTHER FACTORS ALSO INFLUENCE PROGRESSION
Even though there is broad agreement that an approach that neutralizes the effects of the renin-angiotensin-aldosterone system on the kidney would lower blood pressure and protein excretion rates, whether it would change the natural history of chronic kidney disease and prevent progression is less clear. In reality, a number of factors other than the renin-angiotensin-aldosterone system are responsible for the progression of chronic kidney disease. These other factors may help explain why control of this system does not totally prevent deterioration of chronic kidney disease, although the rate may be slowed.
MORE QUESTIONS THAN ANSWERS
A number of provocative questions arise from Dr. Hirsch’s discussion of complete renin-angiotensin-aldosterone system blockade to prevent disease progression:
- Will decreasing proteinuria to a specific target (< 500 mg/day) prevent progression?
- How low should the blood pressure target be set to modulate progression, and should it be the same in all age groups?
- Should complete blockade be applied all at once or in a stepwise fashion depending on the glomerular filtration rate, the level of proteinuria, or both?
- Which patients would benefit most from complete blockade?
- Is direct renin inhibition a critical component of complete blockade?
- What model of chronic disease management is required to avoid unexpected complications if this treatment approach is embraced?
Currently, therefore, there are more questions than answers. This strategy is an intriguing, opinion-based option, but for now it should only be applied to patients with proteinuria and evidence of early progression despite standard therapy who can be closely monitored, and it is not for the faint of heart. In view of the risks of hyperkalemia, hypotension, and perhaps even worsening renal function, more data from carefully designed trials are needed before the general medical community widely applies a complete blockade of the renin-angiotensin-aldosterone pathway to prevent progressive chronic kidney disease.
- United States Renal Data System. Annual data report. www.usrds.org/adr.htm. Accessed 9/5/2008.
- National Kidney Foundation. NKF K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. www.kidney.org/Professionals/Kdoqi/guidelines_ckd/toc.htm. Accessed 9/5/2008.
- Remuzzi A. Vitamin D, insulin resistance, and renal disease. Kidney Int. 2007; 71:96–98.
- Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003; 83:253–307.
- Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int 1998; 54:687–697.
- Endlich N, Endlich K. Stretch, tension and adhesion—adaptive mechanisms of the actin cytoskeleton in podocytes. Eur J Cell Biol. 2006; 85:229–234.
- Hirsch S. An update on proteinuric chronic kidney disease: the dual-goal approach. Cleve Clin J Med. 2008; 75:705–713.
- Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993; 329:1456–1462.
- Wolf G, Ritz E. Combination therapy with ACE inhibitors and angiotensin II receptor blockers to halt progression of chronic renal disease: pathophysiology and indications. Kidney Int. 2005; 67:799–812.
- Campbell R, Sangalli F, Perticucci E, et al. Effects of combined ACE inhibitor and angiotensin II antagonist treatment in human chronic nephropathies. Kidney Int. 2003; 63:1094–1103.
- Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomised controlled trial. Lancet. 2003; 361:117–124.
- Kunz R, Friedrich C, Wolbers M, Mann JF. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med. 2008; 148:30–48.
- Mann JF, Schmieder RE, McQueen M, et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet. 2008; 372:547–553.
- Phillips CO, Kashani A, Ko DK, Francis G, Krumholz HM. Adverse effects of combination angiotensin II receptor blockers plus angiotensin-converting enzyme inhibitors for left ventricular dysfunction: a quantitative review of data from randomized clinical trials. Arch Intern Med. 2007; 167:1930–1936.
- Epstein M. Adding spironolactone to conventional antihypertensives reduces albuminuria in patients with diabetic nephropathy. Nat Clin Pract Nephrol. 2006; 2:310–311.
- Rossing K, Schjoedt KJ, Smidt UM, Boomsma F, Parving HH. Beneficial effects of adding spironolactone to recommended antihypertensive treatment in diabetic nephropathy: a randomized, double-masked, cross-over study. Diabetes Care. 2005; 28:2106–2112.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006; 70:2116–2123.
- Bomback AS, Kshirsagar AV, Amamoo MA, Klemmer PJ. Change in proteinuria after adding aldosterone blockers to ACE inhibitors or angiotensin receptor blockers in CKD: a systematic review. Am J Kidney Dis. 2008; 51:199–211.
- Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med. 2008; 358:2433–2446.
- United States Renal Data System. Annual data report. www.usrds.org/adr.htm. Accessed 9/5/2008.
- National Kidney Foundation. NKF K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. www.kidney.org/Professionals/Kdoqi/guidelines_ckd/toc.htm. Accessed 9/5/2008.
- Remuzzi A. Vitamin D, insulin resistance, and renal disease. Kidney Int. 2007; 71:96–98.
- Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003; 83:253–307.
- Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int 1998; 54:687–697.
- Endlich N, Endlich K. Stretch, tension and adhesion—adaptive mechanisms of the actin cytoskeleton in podocytes. Eur J Cell Biol. 2006; 85:229–234.
- Hirsch S. An update on proteinuric chronic kidney disease: the dual-goal approach. Cleve Clin J Med. 2008; 75:705–713.
- Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993; 329:1456–1462.
- Wolf G, Ritz E. Combination therapy with ACE inhibitors and angiotensin II receptor blockers to halt progression of chronic renal disease: pathophysiology and indications. Kidney Int. 2005; 67:799–812.
- Campbell R, Sangalli F, Perticucci E, et al. Effects of combined ACE inhibitor and angiotensin II antagonist treatment in human chronic nephropathies. Kidney Int. 2003; 63:1094–1103.
- Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomised controlled trial. Lancet. 2003; 361:117–124.
- Kunz R, Friedrich C, Wolbers M, Mann JF. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med. 2008; 148:30–48.
- Mann JF, Schmieder RE, McQueen M, et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet. 2008; 372:547–553.
- Phillips CO, Kashani A, Ko DK, Francis G, Krumholz HM. Adverse effects of combination angiotensin II receptor blockers plus angiotensin-converting enzyme inhibitors for left ventricular dysfunction: a quantitative review of data from randomized clinical trials. Arch Intern Med. 2007; 167:1930–1936.
- Epstein M. Adding spironolactone to conventional antihypertensives reduces albuminuria in patients with diabetic nephropathy. Nat Clin Pract Nephrol. 2006; 2:310–311.
- Rossing K, Schjoedt KJ, Smidt UM, Boomsma F, Parving HH. Beneficial effects of adding spironolactone to recommended antihypertensive treatment in diabetic nephropathy: a randomized, double-masked, cross-over study. Diabetes Care. 2005; 28:2106–2112.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006; 70:2116–2123.
- Bomback AS, Kshirsagar AV, Amamoo MA, Klemmer PJ. Change in proteinuria after adding aldosterone blockers to ACE inhibitors or angiotensin receptor blockers in CKD: a systematic review. Am J Kidney Dis. 2008; 51:199–211.
- Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med. 2008; 358:2433–2446.
An update on proteinuric chronic kidney disease: The dual-goal approach
When angiotensin-converting enzyme (ACE) inhibitors and angiotensin II type 1 receptor blockers (ARBs) were introduced, we hoped that these drugs would slow or stop the inexorable progression of chronic kidney disease. This hasn’t come to pass: the incidence of end-stage renal disease continued to increase throughout the 1990s, and although it may have finally reached a plateau, it remains unacceptably high.1 One reason may be that, used singly, drugs that block the renin-angiotensin-aldosterone system are only moderately successful, as approximately 20% to 40% of patients still reach unfavorable renal end points such as doubling of the serum creatinine level or dialysis.2–7
In view of these disappointing results, some experts are advocating a new strategy in which they advise that both blood pressure and urinary albumin excretion be lowered to specific goals. To achieve these goals, we will generally have to give higher doses of ACE inhibitors and ARBs alone or use a combination of these and other drugs that block the renin-angiotensin-aldosterone system at various sites.
This article describes how the dual-goal approach, with a focus on renin-angiotensin-aldosterone system inhibition, may be applied in the therapy of proteinuric chronic kidney disease. This appears to be a reasonable approach, based on current evidence, to address the epidemic of renal failure. However, further studies are needed to establish the effectiveness of this approach, and the risk of hyperkalemia following aggressive inhibition of the renin-angiotensin-aldosterone system poses a significant management problem.
ALBUMIN MAY BE TOXIC
While hypertension has long been associated with poor renal outcomes, urinary albumin has more recently been implicated by observational and experimental evidence as a tubular-interstitial toxin that may also accelerate the progression of renal disease.
For example, in both the Reduction of Endpoints in NIDDM With the Angiotensin II Antagonist Losartan (RENAAL) study8 and the Ramipril Efficacy in Nephropathy study,4 baseline proteinuria was almost linearly related to worse renal outcomes. In RENAAL, patients who excreted more than about 3 g of albumin per day had an 8.1-fold higher risk of progressing to end-stage renal disease.8 Moreover, the more that protein excretion could be reduced, the better the renal outcomes, down to a level of about 500 mg/day.8
Of importance, lowering blood pressure did not always decrease protein excretion—nearly 40% of patients had a dissociation between the two.9 In fact, prescribing a single ACE inhibitor or ARB while targeting only blood pressure has not predictably reduced protein excretion to 500 mg/day (the proposed goal).2–7

Although albumin has not been conclusively proven to be a renal toxin, targeting the reduction of proteinuria may also succeed if urinary albumin simply serves as a marker of the success of chronic kidney disease treatment and reflects prognosis.
A DUAL-GOAL APPROACH
In view of the observational and experimental evidence, many experts10,15–18 are advocating a dual-goal approach that stresses lowering both blood pressure and urinary protein (albumin) excretion. The recommended goal for systolic blood pressure is less than 120 to 125 mm Hg; the goal for proteinuria is less than 300 to 500 mg/24 hours,16,17,19 aiming to slow the decline in glomerular filtration rate to less than 2 mL/ min/year.11,20
The strategy of targeting both proteinuria and blood pressure has recently received further support. In a prospective randomized controlled study,21 nondiabetic patients with proteinuria received either an ACE inhibitor or an ARB. In one group, the dose was adjusted to lower the blood pressure to less than 130/80 mm/Hg; in the other group, the dose was adjusted to lower the blood pressure to 130/80 and to reduce protein excretion maximally. Only about half as many patients in the group with the dual-goal strategy reached the composite primary end point (doubling of serum creatinine, end-stage renal disease, or death) over a median of 3.7 years of follow-up, as compared with those treated by targeting the blood pressure alone.
In retrospect, the suboptimal success in the earlier landmark studies2–7 may have derived from the failure of ACE inhibitors and ARBs, used by themselves at moderate doses, to either lower the blood pressure to the recently advised goal (the actual results obtained varied from about 128 to about 145 mm Hg systolic) or, perhaps, to reduce proteinuria to the goal level.
Not all nephrologists currently pursue the stringent proteinuria goal of 500 mg per day—the targeted reduction of proteinuria requires further prospective evidence to support it. However, nephrologists do commonly follow the broad theme that antihypertensive therapy in proteinuric chronic kidney disease should accentuate medicines that protect the kidney beyond their antihypertensive effect (Table 1), and that proteinuria is an important metric that, at the very least, reflects the response to therapy and prognosis.
BLOCKING RENIN-ANGIOTENSIN- ALDOSTERONE MORE COMPLETELY
These issues may be addressed by more complete inhibition of the renin-angiotensin-aldosterone system, now achievable with the addition of aldosterone receptor antagonists and direct renin inhibitors to the ACE inhibitors and ARBs. Although we lack long-term studies of the relative efficacy of these medicines alone or in various combinations, the multistep sequence of the renin-angiotensin-aldosterone system allows for the possibility that more complete suppression via coordinated pharmacologic attention to multiple sites will yield beneficial results.
Combining an ACE inhibitor and an ARB
Even in the absence of ACE, angiotensin II is also produced by other kinases and therefore is not completely suppressed by an ACE inhibitor. For this and other reasons, there are theoretical advantages to adding an ARB to an ACE inhibitor.
In the Combination Treatment of Angiotensin 2 Receptor Blocker and Angiotensin-Converting-Enzyme Inhibitor in Non-Diabetic Renal Disease (COOPERATE) study,20 the combination of an ACE inhibitor and an ARB protected the kidneys better than either medicine alone, not only in terms of less protein in the urine but also in terms of significantly fewer patients progressing to the primary end points of doubling of serum creatinine or end-stage renal disease after 3 years of follow-up (11% of patients on combination therapy vs 23% on single therapy).
Aldosterone receptor antagonists or renin inhibitors plus ACE inhibitors and ARBs
Aldosterone escape is common during long-term therapy with ACE inhibitors and ARBs, and an aldosterone-receptor antagonist reduces proteinuria11–13 and stabilizes kidney function13 in a manner additive to that of ACE inhibitors and ARBs.
Direct renin inhibitors overcome the reactive rises in renin activity and in angiotensin II that complicate therapy with ACE inhibitors and ARBs, and they also reduce urinary aldosterone excretion.14
When to consider combination therapy
Inhibition of the renin-angiotensin-aldosterone system at multiple sites may be considered in cases of persistent hypertension or proteinuria, or of progression of chronic kidney disease despite single-drug therapy, or more broadly, with increasing evidence that combination therapy may preserve the glomerular filtration rate.13,20 This article suggests one way to apply the several available renin-angiotensin-aldosterone inhibitors, keeping in mind extensive interindividual variations, uncertain responses, and the absence of a linear evidence-based strategy known to be broadly successful.
INITIAL CONSIDERATION: WHAT IS THE BLOOD PRESSURE GOAL?
Determining the blood pressure goal for a patient may not be as straightforward as usually assumed. Typically, advisories suggest a discrete goal; for example, the Seventh Joint National Committee22 recommended a systolic blood pressure of 130 mm Hg or lower for patients with chronic kidney disease or diabetes. However, if we weigh the risks and benefits, we find that the situation is more nuanced. The blood pressure goal should vary among patients, depending on age, amount of proteinuria, whether the patient can tolerate the lowered blood pressure, and whether lowering the blood pressure to this goal stabilizes kidney function.
Long-term follow-up of the Modification of Diet in Renal Disease (MDRD) study demonstrated a benefit of setting the goal mean arterial pressure to less than 92 mm Hg (about 125 mm Hg systolic) regardless of proteinuria.23 In addition, a meta-analysis suggested that nondiabetic proteinuric patients benefit from even lower systolic blood pressures (110–119 mm Hg).19
In older patients
However, in the MDRD study, the goal of approximately 125 mm Hg systolic pertained only to patients no older than 60 years.23 The goal was increased to about 130 mm Hg for patients 61 to 70 years old. In addition, major clinical studies of chronic kidney disease have excluded patients older than 70 years.2–7,23
Therapy for chronic kidney disease in this older age group is essentially unstudied, and we should be cautious about extrapolating results of aggressive blood pressure-lowering (and renin-angiotensin-aldosterone inhibition) from younger patients to older patients, who may have extensive vascular disease.24,25
For patients older than 70 years, guidance is perhaps best provided by the Systolic Hypertension in the Elderly Program (SHEP), which found that lowering systolic blood pressure to an average of 143 mm Hg reduced the incidence of stroke and cardiovascular disease.26 The SHEP study does not establish the optimal blood pressure goal for preventing progressive chronic kidney disease (or even cardiovascular disease) in the older age group. However, this is the lowest systolic pressure yet shown to be generally safe and associated with any improved outcome for these patients.
Additional studies are needed to evaluate whether this blood pressure level provides the best outcomes in patients with chronic kidney disease, or whether even lower blood pressures in the elderly are safe and will further improve either renal or cardiovascular outcomes.
In younger patients
In contrast, younger patients without diabetes or vascular disease may, in theory, be candidates for even lower blood pressure. No major study of chronic kidney disease isolated patients from about 20 to 40 years old for analysis, precluding direct evidence-based guidelines for this cohort at this time.
However, some of these patients may have had premorbid systolic blood pressures of 90 to 110 mm Hg, so systolic pressures of 110 to 120 mm Hg would be “hypertensive” by 10 to 30 mm Hg for them. It is possible that some patients in this cohort will tolerate a systolic pressure lower than 110 mm Hg, and that the lower blood pressure may provide additional long-term renal protection for them. This notion is theoretical, however, and has not been verified by clinical studies.
No one pressure fits all
In summary, an initial target systolic pressure for proteinuric patients, based on available evidence, might be less than 130 mm Hg for patients 61 to 70 years old,23 less than 125 mm Hg for patients younger than 61 years,23 and perhaps as low as 110 to 119 mm Hg for non-diabetic patients.19 Caution is advised against targeting systolic blood pressure less than 140 mm Hg for patients older than 70 years.
These are only initial goals and should be reevaluated as treatment progresses. The achieved blood pressure must be clinically tolerated—symptoms of tissue hypoperfusion indicate that the blood pressure is too low for the patient. In addition, the blood pressure goal (like the proteinuria goal) is only a surrogate end point, and if kidney function declines even though the surrogate end points are attained, then those end points should be reevaluated.
Tailoring blood pressure goals to the individual patient dovetails with the recent suggestion that blood pressure should not be perceived as a rigid dichotomy of “hypertension” vs “normal.”27 There is, in general, a continuous correlation between blood pressure, beginning at low levels, and the risk of cardiorenal disease, and choosing an optimal blood pressure goal for an individual patient requires an ongoing assessment of benefits, risks, and side effects.
STARTING ANTIHYPERTENSIVE THERAPY
The question of which antihypertensive drug to try first is moot in chronic kidney disease because almost all patients need multiple medicines to reach their blood pressure goals.
The Seventh Joint National Committee recommended an ACE inhibitor for initial therapy in hypertensive patients with chronic kidney disease,22 although an ARB is a reasonable first choice for those with type 2 diabetes.5,6
Diuretics potentiate the effects of ACE inhibitors and ARBs and are generally prescribed concomitantly or as the second choice.
A beta-blocker may be recommended as a third medicine (when needed), to provide a complementary class of antihypertensive, to address the high incidence of concomitant coronary artery disease and systolic dysfunction, and because of evidence that sympathetic excess contributes to the hypertension and progression of chronic kidney disease.28,29 The National Kidney Foundation30 suggests that the dose of beta-blocker be increased if the heart rate is greater than 84.
INTENSIFYING RENIN-ANGIOTENSIN-ALDOSTERONE INHIBITION: WHICH DRUGS, AND WHEN?
When hypertension and proteinuria persist despite the use of an ACE inhibitor or an ARB, additional inhibition of the renin-angiotensin-aldosterone system is generally recommended to lower both the blood pressure and the protein excretion. Increasing the dose of ACE inhibitor or ARB,31–34 combining an ACE inhibitor and an ARB,20 or adding an aldosterone receptor antagonist to either an ACE inhibitor or an ARB11–13 have all been shown to reduce proteinuria (as a surrogate end point), and several studies have, importantly, found that these combinations preserve kidney function over time.13,20
However, lacking long-term studies that compare these options, we cannot insist upon specific treatment choices or sequences in these situations.
An approach based on serum potassium and volume status

For example, if a patient has obvious signs of volume excess (eg, edema, jugular venous distention, rales) and the serum potassium concentration is less than about 5.0 or 5.5 mEq/L, then an aldosterone receptor antagonist may logically be added or increased in dose.
Aldosterone is more than a kidney hormone

Increasing the diuretic or renin-angiotensin-aldosterone inhibition
For patients who have obvious signs of volume excess and a serum potassium level greater than 5.0 mEq/L, the dosage of kaliuretic (potassium-excreting) diuretic (usually a loop diuretic in chronic kidney disease) can be increased. Although kaliuretic diuretics do not specifically lower proteinuria, they will help control volume and blood pressure and, by lowering the serum potassium level, facilitate the subsequent augmention of renin-angiotensin-aldosterone inhibition.
When a hypertensive patient does not seem to have excess volume or tachycardia and the serum potassium level is less than about 5.5 mEq/L, then additional renin-angiotensin-aldosterone inhibition is indicated.16 This may be accomplished either by increasing the ACE inhibitor or the ARB to its maximal antihypertensive dose or by starting combination therapy.
Starting a calcium channel blocker
When the serum potassium level is higher than about 5.5 mEq/L, further inhibition of the renin-angiotensin-aldosterone system is contraindicated, and a nondihydropyridine calcium channel blocker can be added for its anti-hypertensive and antiproteinuric effects.16,36
When nondihydropyridine calcium channel blockers are contraindicated due to their antiinotropic effect, an attractive alternative may be to cautiously increase the dose of kaliuretic diuretics. Given the high prevalence of (often covert) volume excess in chronic kidney disease, empiric diuresis may lower blood pressure, particularly in patients already receiving several vasodilators.37 Moreover, as mentioned, by reducing serum potassium, kaliuretic diuretics help allow for a subsequent increase in renin-angiotensin-aldosterone inhibition.
IF BLOOD PRESSURE IS NORMAL, BUT PROTEINURIA PERSISTS
Because lowering blood pressure does not necessarily reduce protein excretion, some patients achieve their blood pressure goal but still have excessive proteinuria. Proponents of the dual-goal approach suggest that these patients require further treatment modifications to reach the proteinuria goal and their optimal renal prognosis.

A cautious increase in renin-angiotensin-aldosterone inhibition is possible but is likely to be limited by low blood pressure. When applicable, any nonessential antihypertensive drug that does not specifically reduce proteinuria (ie, dihydropyridine calcium channel blockers and central and direct vasodilators) should first be discontinued. This allows additional renin-angiotensin-aldosterone inhibition to reduce proteinuria without causing hypotension.
In addition, “ultra-high” doses of these drugs—two or more times the maximal antihypertensive dose—appear to reduce proteinuria without further reducing blood pressure.31–34
Various combinations of an ACE inhibitor, an ARB, and an aldosterone receptor antagonist (and possibly a renin inhibitor) may also be prescribed, striving for more complete suppression of the renin-angiotensin-aldosterone system, with dose adjustments to prevent hypotension.
KEEPING SERUM POTASSIUM AT SAFE LEVELS
Intensive inhibition of the renin-angiotensin-aldosterone system, via higher doses or combination therapy, increases the risk of hyperkalemia. This risk must be addressed energetically to prevent a potentially life-threatening complication.
When prescribed by nephrologists in clinical studies, renin-angiotensin-aldosterone inhibition has proven safe, with minimal adverse events (including hyperkalemia), even with high doses,32–34 in stage 4 chronic kidney disease (ie, with a glomerular filtration rate of 15 to 29 mL/min/1.73m2, inclusively)7 and with combination therapy.11–13,20
However, the increased incidence of hyperkalemia reported with spironolactone in patients with congestive heart failure following publication of the Randomized Aldactone Evaluation Study38 suggests that safety in clinical studies should not be extrapolated to mean safety in routine, community use. Patients with chronic kidney disease should not be given high doses or combinations of these drugs unless the treating physician is experienced in the prevention and treatment of hyperkalemia; typically such therapy should be guided by a nephrologist.
When serum potassium levels exceed 5.6 mEq/L, renin-angiotensin-aldosterone inhibitors should be decreased in dose or discontinued.39 Ideally, the drug or drugs should be restarted (to provide the potential benefits of these classes of drugs) when hyperkalemia has resolved, but this requires not only resolution of hyperkalemia but also steps to prevent this serious problem from recurring. The serum potassium level should be checked frequently, particularly after any increase in renin-angiotensin-aldosterone inhibition.
Treating hyperkalemia
Potential treatments for hyperkalemia include dietary restriction, sodium bicarbonate,39 fludrocortisone (Florinef),40 kaliuretic diuretics, and sodium polystyrene sulfonate (Kayexalate). Nonsteroidal anti-inflammatory drugs should be avoided.
Dietary restriction should be particularly emphasized: if potassium intake is decreased to the same extent as renin-angiotensin-aldosterone inhibitors reduce its excretion, then the serum potassium level will remain acceptable. All dietary supplements whose contents are not precisely known should be proscribed. A list of high-potassium foods to avoid should be given with the initial prescription for the drug. If briefly reviewed at each visit, with feedback given based on measured serum potassium levels, dietary treatment is typically effective (personal observation).
Fludrocortisone is an option when dietary potassium restriction fails.
An increase in the dose of diuretic is typically required with fludrocortisone to prevent sodium retention. The combination of dietary potassium restriction, fludrocortisone (0.1 mg/day, 3–5 days a week), and furosemide (Lasix) allowed high doses of an ACE inhibitor or a combination of an ACE inhibitor and an ARB to be given in 132 patients with chronic kidney disease.40 Over several years, their mean peak potassium level was 4.87 mEq/L, and no instance of acute hyperkalemia requiried stopping the ACE inhibitor or ARB.
However, fludrocortisone is an aldosterone analogue with potentially long-term aldosterone-mediated injurious effects on heart and renal function, even though only low doses were required in the three-pronged approach to hyperkalemia.40 The long-term effect of a regimen of an ACE inhibitor plus an ARB plus fludrocortisone on cardiac and renal outcomes is unknown and of concern.
Therefore, fludrocortisone should probably be avoided in patients with systolic heart dysfunction and should be used cautiously in general. Its use might be limited to patients with proteinuric chronic kidney disease that progresses despite therapy, particularly when that progression is in the context of inability to give significant renin-angiotensin-aldosterone inhibition because of hyperkalemia.
MORE STUDY NEEDED
Chronic kidney disease treatment is becoming increasingly complex, with a lengthening list of potentially effective drugs, difficult-to-reach goals, and a less structured approach. This complexity is magnified by issues of potassium homeostasis and interindividual variations in response to renin-angiotensin-aldosterone inhibition.
More prospective studies are needed to confirm the benefits of targeting proteinuria along with blood pressure and the metrics of the goals in tandem, but, based on available information, the dual-goal approach has been recommended for proteinuric patients,10,15–18 and evidence is accumulating for greater renal protection from larger doses of renin-angiotensin-aldosterone inhibitors and from using these drugs in combination.
- US Renal Data System. Excerpts from the USRDS 2005 Annual Data Report. Am J Kidney Dis 2006; 47(suppl 1):S1–S286.
- Lewis E, Hunsicker L, Bain R, Rohde Rfor the Collaborative Study Group. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. N Engl J Med. 1993; 329:1456–1462.
- Maschio G, Alberti D, Janin G, et al. Effect of the angiotensin-converting enzyme inhibitor benazepril on the progression of chronic renal insufficiency. The Angiotensin-Converting Enzyme Inhibition in Progressive Renal Insufficiency Study Group. N Engl J Med. 1996; 334:939–945.
- The GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia). Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. Lancet. 1997; 349:1857–1863.
- Brenner B, Cooper M, De Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001; 345:861–869.
- Lewis E, Hunsicker L, Clarke W, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001; 345:851–860.
- Hou F, Zhang X, Zhang G, et al. Efficacy and safety of benazepril for advanced chronic renal insufficiency. N Engl J Med. 2006; 354:131–140.
- De Zeeuw D, Remuzzi G, Parving H-H, et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int. 2004; 65:2309–2320.
- Eijkelkamp W, Zhang Z, Remuzzi G, et al. Albuminuria is a target for renoprotective therapy independent from blood pressure in patients with type 2 diabetic nephropathy: post hoc analysis from the Reduction of Endpoints in NIDDM with the Angiotension 2 Antagonist Losartan (RENAAL) trial. J Am Soc Nephrol. 2007; 18:1540–1546.
- Khosla N, Bakris G. Lessons learned from recent hypertension trials about kidney disease. Clin J Am Soc Nephrol. 2006; 1:229–235.
- Chrysostomou A, Pedagogoa E, MacGregor L, Becker G. Double-blind, placebo-controlled study on the effect of the aldosterone receptor antagonist spironolactone in patients who have persistent proteinuria and are on long-term angiotensin-converting enzyme inhibitor therapy, with or without an angiotensin 2 receptor blocker. Clin J Am Soc Nephrol. 2006; 1:256–262.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int. 2006; 70:536–542.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006; 70:2116–2123.
- Azizi M, Menard J, Bissery A, Guyene T-T, Bura-Riviere A. Hormonal and hemodynamic effects of aliskiren and valsartan and their combinations in sodium-replete normotensive individuals Clin J Am Soc Nephrol 2007; 2:947–955.
- Hebert L, Wilmer W, Falkenhain M, Ladson-Wofford S, Nahman S, Rovin B. Renoprotection: one or many therapies? Kidney Int 2001; 59:1211–1226.
- Shieppate A, Remuzzi G. The future of renoprotection: frustration and promises. Kidney Int. 2003; 64:1947–1955.
- Zandi-Nejad K, Brenner B. Strategies to retard the progression of chronic kidney disease. Med Clin North Am. 2005; 89:489–509.
- Ritz E, Dikow R. Hypertension and antihypertensive treatment of diabetic nephropathy. Nat Clinl Pract Nephrol. 2006; 2:562–567.
- Jafar T, Stark P, Schmid C, et al., for the AIPRD Study Group. Progression of chronic kidney disease: the role of blood pressure control, proteinuria, and angiotensin-converting enzyme inhibition. A patient-level meta-analysis. Ann Intern Med. 2003; 139:244–252.
- Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin 2 receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomized controlled trial. Lancet. 2003; 361:117–124.
- Hou F, Xie D, Zhang X, et al. Renoprotection of optimal antiproteinuric doses (ROAD) study: a randomized controlled study of benazepril and losartan in chronic renal insufficiency. J Am Soc Nephrol. 2007; 18:1889–1898.
- Chobanian AV, Bakris GL, Black HR. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003; 289:2560–2572.
- Sarnak M, Greene T, Wang X, et al. The effect of a lower target blood pressure on the progression of kidney disease: long-term follow-up of the Modification of Diet in Renal Disease Study. Ann Intern Med. 2005; 142:342–351.
- Hemmelgarn BR, Zhang J, Manns BJ, et al. Progression of kidney dysfunction in the community-dwelling elderly. Kidney Int. 2006; 69:2155–2161.
- Locatelli F, Pozzoni P. Chronic kidney disease in the elderly: is it really a premise for overwhelming renal failure? Kidney Int 2006; 69:2118–2120.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA. 1991; 265:3255–3264.
- Forman JP, Brenner BM. ‘Hypertension’ and ‘microalbuminuria’: the bell tolls for thee. Kidney Int. 2006; 69:22–28.
- Bakris G, Hart P, Ritz E. Beta blockers in the management of chronic kidney disease. Kidney Int. 2006; 70:1905–1913.
- UKPD Study Group. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. UK Prospective diabetes study group. BMJ. 1998; 317:713–720.
- Bakris G, Williams M, Dworkin L, et al. Preserving renal function in adults with hypertension and diabetes: a consensus approach. National Kidney Foundation Hypertensive and Diabetes Executive Committees Working Group. Am J Kidney Dis. 2000; 36:646–661.
- Navis G, Kramer A, de Jong P. High-dose ACE inhibition: can it improve renoprotection? Am J Kidney Dis 2002; 40:664–666.
- Rossing K, Schjoedt K, Jensin B, Boomsma F, Parving H-H. Enhanced renoprotective effects of ultrahigh doses of irbesartan in patients with type 2 diabetes and microalbuminuria. Kidney Int. 2005; 68:1190–1198.
- Schmieder R, Klingbeil A, Fleischman E, Veelken R, Delles C. Additional antiproteinuric effect of ultrahigh dose candesartan: a double-blind, randomized, prospective study. J Am Soc Nephrol. 2005; 16:3038–3045.
- Aranda P, Segura J, Ruilope L, et al. Long-term renoprotective effects of standard versus high doses of telmisartan in hypertensive nondiabetic nephropathies. Am J Kidney Dis. 2005; 46:1074–1079.
- Calhoun D. Aldosteronism and hypertension. Clin J Am Soc Nephrol. 2006; 1:1039–1045.
- Bakris G, Weir M, Secic M, Campbell B, Weis-McNulty A. Differential effects of calcium antagonist subclasses on markers of nephropathy progression. Kidney Int. 2004; 65:1991–2002.
- Hirsch S. A different approach to resistant hypertension. Cleve Clin J Med 2007: 74;449–456.
- Juurling D, Mamdani M, Lee D, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med. 2004; 351:543–551.
- Palmer B. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med. 2004; 351:585–592.
- Moskowitz D. From pharmacogenomics to improved patient outcomes: angiotensin 1-converting enzyme as an example. Diabetes Tech Ther. 2002; 4:519–532.
When angiotensin-converting enzyme (ACE) inhibitors and angiotensin II type 1 receptor blockers (ARBs) were introduced, we hoped that these drugs would slow or stop the inexorable progression of chronic kidney disease. This hasn’t come to pass: the incidence of end-stage renal disease continued to increase throughout the 1990s, and although it may have finally reached a plateau, it remains unacceptably high.1 One reason may be that, used singly, drugs that block the renin-angiotensin-aldosterone system are only moderately successful, as approximately 20% to 40% of patients still reach unfavorable renal end points such as doubling of the serum creatinine level or dialysis.2–7
In view of these disappointing results, some experts are advocating a new strategy in which they advise that both blood pressure and urinary albumin excretion be lowered to specific goals. To achieve these goals, we will generally have to give higher doses of ACE inhibitors and ARBs alone or use a combination of these and other drugs that block the renin-angiotensin-aldosterone system at various sites.
This article describes how the dual-goal approach, with a focus on renin-angiotensin-aldosterone system inhibition, may be applied in the therapy of proteinuric chronic kidney disease. This appears to be a reasonable approach, based on current evidence, to address the epidemic of renal failure. However, further studies are needed to establish the effectiveness of this approach, and the risk of hyperkalemia following aggressive inhibition of the renin-angiotensin-aldosterone system poses a significant management problem.
ALBUMIN MAY BE TOXIC
While hypertension has long been associated with poor renal outcomes, urinary albumin has more recently been implicated by observational and experimental evidence as a tubular-interstitial toxin that may also accelerate the progression of renal disease.
For example, in both the Reduction of Endpoints in NIDDM With the Angiotensin II Antagonist Losartan (RENAAL) study8 and the Ramipril Efficacy in Nephropathy study,4 baseline proteinuria was almost linearly related to worse renal outcomes. In RENAAL, patients who excreted more than about 3 g of albumin per day had an 8.1-fold higher risk of progressing to end-stage renal disease.8 Moreover, the more that protein excretion could be reduced, the better the renal outcomes, down to a level of about 500 mg/day.8
Of importance, lowering blood pressure did not always decrease protein excretion—nearly 40% of patients had a dissociation between the two.9 In fact, prescribing a single ACE inhibitor or ARB while targeting only blood pressure has not predictably reduced protein excretion to 500 mg/day (the proposed goal).2–7
Although albumin has not been conclusively proven to be a renal toxin, targeting the reduction of proteinuria may also succeed if urinary albumin simply serves as a marker of the success of chronic kidney disease treatment and reflects prognosis.
A DUAL-GOAL APPROACH
In view of the observational and experimental evidence, many experts10,15–18 are advocating a dual-goal approach that stresses lowering both blood pressure and urinary protein (albumin) excretion. The recommended goal for systolic blood pressure is less than 120 to 125 mm Hg; the goal for proteinuria is less than 300 to 500 mg/24 hours,16,17,19 aiming to slow the decline in glomerular filtration rate to less than 2 mL/ min/year.11,20
The strategy of targeting both proteinuria and blood pressure has recently received further support. In a prospective randomized controlled study,21 nondiabetic patients with proteinuria received either an ACE inhibitor or an ARB. In one group, the dose was adjusted to lower the blood pressure to less than 130/80 mm/Hg; in the other group, the dose was adjusted to lower the blood pressure to 130/80 and to reduce protein excretion maximally. Only about half as many patients in the group with the dual-goal strategy reached the composite primary end point (doubling of serum creatinine, end-stage renal disease, or death) over a median of 3.7 years of follow-up, as compared with those treated by targeting the blood pressure alone.
In retrospect, the suboptimal success in the earlier landmark studies2–7 may have derived from the failure of ACE inhibitors and ARBs, used by themselves at moderate doses, to either lower the blood pressure to the recently advised goal (the actual results obtained varied from about 128 to about 145 mm Hg systolic) or, perhaps, to reduce proteinuria to the goal level.
Not all nephrologists currently pursue the stringent proteinuria goal of 500 mg per day—the targeted reduction of proteinuria requires further prospective evidence to support it. However, nephrologists do commonly follow the broad theme that antihypertensive therapy in proteinuric chronic kidney disease should accentuate medicines that protect the kidney beyond their antihypertensive effect (Table 1), and that proteinuria is an important metric that, at the very least, reflects the response to therapy and prognosis.
BLOCKING RENIN-ANGIOTENSIN- ALDOSTERONE MORE COMPLETELY
These issues may be addressed by more complete inhibition of the renin-angiotensin-aldosterone system, now achievable with the addition of aldosterone receptor antagonists and direct renin inhibitors to the ACE inhibitors and ARBs. Although we lack long-term studies of the relative efficacy of these medicines alone or in various combinations, the multistep sequence of the renin-angiotensin-aldosterone system allows for the possibility that more complete suppression via coordinated pharmacologic attention to multiple sites will yield beneficial results.
Combining an ACE inhibitor and an ARB
Even in the absence of ACE, angiotensin II is also produced by other kinases and therefore is not completely suppressed by an ACE inhibitor. For this and other reasons, there are theoretical advantages to adding an ARB to an ACE inhibitor.
In the Combination Treatment of Angiotensin 2 Receptor Blocker and Angiotensin-Converting-Enzyme Inhibitor in Non-Diabetic Renal Disease (COOPERATE) study,20 the combination of an ACE inhibitor and an ARB protected the kidneys better than either medicine alone, not only in terms of less protein in the urine but also in terms of significantly fewer patients progressing to the primary end points of doubling of serum creatinine or end-stage renal disease after 3 years of follow-up (11% of patients on combination therapy vs 23% on single therapy).
Aldosterone receptor antagonists or renin inhibitors plus ACE inhibitors and ARBs
Aldosterone escape is common during long-term therapy with ACE inhibitors and ARBs, and an aldosterone-receptor antagonist reduces proteinuria11–13 and stabilizes kidney function13 in a manner additive to that of ACE inhibitors and ARBs.
Direct renin inhibitors overcome the reactive rises in renin activity and in angiotensin II that complicate therapy with ACE inhibitors and ARBs, and they also reduce urinary aldosterone excretion.14
When to consider combination therapy
Inhibition of the renin-angiotensin-aldosterone system at multiple sites may be considered in cases of persistent hypertension or proteinuria, or of progression of chronic kidney disease despite single-drug therapy, or more broadly, with increasing evidence that combination therapy may preserve the glomerular filtration rate.13,20 This article suggests one way to apply the several available renin-angiotensin-aldosterone inhibitors, keeping in mind extensive interindividual variations, uncertain responses, and the absence of a linear evidence-based strategy known to be broadly successful.
INITIAL CONSIDERATION: WHAT IS THE BLOOD PRESSURE GOAL?
Determining the blood pressure goal for a patient may not be as straightforward as usually assumed. Typically, advisories suggest a discrete goal; for example, the Seventh Joint National Committee22 recommended a systolic blood pressure of 130 mm Hg or lower for patients with chronic kidney disease or diabetes. However, if we weigh the risks and benefits, we find that the situation is more nuanced. The blood pressure goal should vary among patients, depending on age, amount of proteinuria, whether the patient can tolerate the lowered blood pressure, and whether lowering the blood pressure to this goal stabilizes kidney function.
Long-term follow-up of the Modification of Diet in Renal Disease (MDRD) study demonstrated a benefit of setting the goal mean arterial pressure to less than 92 mm Hg (about 125 mm Hg systolic) regardless of proteinuria.23 In addition, a meta-analysis suggested that nondiabetic proteinuric patients benefit from even lower systolic blood pressures (110–119 mm Hg).19
In older patients
However, in the MDRD study, the goal of approximately 125 mm Hg systolic pertained only to patients no older than 60 years.23 The goal was increased to about 130 mm Hg for patients 61 to 70 years old. In addition, major clinical studies of chronic kidney disease have excluded patients older than 70 years.2–7,23
Therapy for chronic kidney disease in this older age group is essentially unstudied, and we should be cautious about extrapolating results of aggressive blood pressure-lowering (and renin-angiotensin-aldosterone inhibition) from younger patients to older patients, who may have extensive vascular disease.24,25
For patients older than 70 years, guidance is perhaps best provided by the Systolic Hypertension in the Elderly Program (SHEP), which found that lowering systolic blood pressure to an average of 143 mm Hg reduced the incidence of stroke and cardiovascular disease.26 The SHEP study does not establish the optimal blood pressure goal for preventing progressive chronic kidney disease (or even cardiovascular disease) in the older age group. However, this is the lowest systolic pressure yet shown to be generally safe and associated with any improved outcome for these patients.
Additional studies are needed to evaluate whether this blood pressure level provides the best outcomes in patients with chronic kidney disease, or whether even lower blood pressures in the elderly are safe and will further improve either renal or cardiovascular outcomes.
In younger patients
In contrast, younger patients without diabetes or vascular disease may, in theory, be candidates for even lower blood pressure. No major study of chronic kidney disease isolated patients from about 20 to 40 years old for analysis, precluding direct evidence-based guidelines for this cohort at this time.
However, some of these patients may have had premorbid systolic blood pressures of 90 to 110 mm Hg, so systolic pressures of 110 to 120 mm Hg would be “hypertensive” by 10 to 30 mm Hg for them. It is possible that some patients in this cohort will tolerate a systolic pressure lower than 110 mm Hg, and that the lower blood pressure may provide additional long-term renal protection for them. This notion is theoretical, however, and has not been verified by clinical studies.
No one pressure fits all
In summary, an initial target systolic pressure for proteinuric patients, based on available evidence, might be less than 130 mm Hg for patients 61 to 70 years old,23 less than 125 mm Hg for patients younger than 61 years,23 and perhaps as low as 110 to 119 mm Hg for non-diabetic patients.19 Caution is advised against targeting systolic blood pressure less than 140 mm Hg for patients older than 70 years.
These are only initial goals and should be reevaluated as treatment progresses. The achieved blood pressure must be clinically tolerated—symptoms of tissue hypoperfusion indicate that the blood pressure is too low for the patient. In addition, the blood pressure goal (like the proteinuria goal) is only a surrogate end point, and if kidney function declines even though the surrogate end points are attained, then those end points should be reevaluated.
Tailoring blood pressure goals to the individual patient dovetails with the recent suggestion that blood pressure should not be perceived as a rigid dichotomy of “hypertension” vs “normal.”27 There is, in general, a continuous correlation between blood pressure, beginning at low levels, and the risk of cardiorenal disease, and choosing an optimal blood pressure goal for an individual patient requires an ongoing assessment of benefits, risks, and side effects.
STARTING ANTIHYPERTENSIVE THERAPY
The question of which antihypertensive drug to try first is moot in chronic kidney disease because almost all patients need multiple medicines to reach their blood pressure goals.
The Seventh Joint National Committee recommended an ACE inhibitor for initial therapy in hypertensive patients with chronic kidney disease,22 although an ARB is a reasonable first choice for those with type 2 diabetes.5,6
Diuretics potentiate the effects of ACE inhibitors and ARBs and are generally prescribed concomitantly or as the second choice.
A beta-blocker may be recommended as a third medicine (when needed), to provide a complementary class of antihypertensive, to address the high incidence of concomitant coronary artery disease and systolic dysfunction, and because of evidence that sympathetic excess contributes to the hypertension and progression of chronic kidney disease.28,29 The National Kidney Foundation30 suggests that the dose of beta-blocker be increased if the heart rate is greater than 84.
INTENSIFYING RENIN-ANGIOTENSIN-ALDOSTERONE INHIBITION: WHICH DRUGS, AND WHEN?
When hypertension and proteinuria persist despite the use of an ACE inhibitor or an ARB, additional inhibition of the renin-angiotensin-aldosterone system is generally recommended to lower both the blood pressure and the protein excretion. Increasing the dose of ACE inhibitor or ARB,31–34 combining an ACE inhibitor and an ARB,20 or adding an aldosterone receptor antagonist to either an ACE inhibitor or an ARB11–13 have all been shown to reduce proteinuria (as a surrogate end point), and several studies have, importantly, found that these combinations preserve kidney function over time.13,20
However, lacking long-term studies that compare these options, we cannot insist upon specific treatment choices or sequences in these situations.
An approach based on serum potassium and volume status
For example, if a patient has obvious signs of volume excess (eg, edema, jugular venous distention, rales) and the serum potassium concentration is less than about 5.0 or 5.5 mEq/L, then an aldosterone receptor antagonist may logically be added or increased in dose.
Aldosterone is more than a kidney hormone
Increasing the diuretic or renin-angiotensin-aldosterone inhibition
For patients who have obvious signs of volume excess and a serum potassium level greater than 5.0 mEq/L, the dosage of kaliuretic (potassium-excreting) diuretic (usually a loop diuretic in chronic kidney disease) can be increased. Although kaliuretic diuretics do not specifically lower proteinuria, they will help control volume and blood pressure and, by lowering the serum potassium level, facilitate the subsequent augmention of renin-angiotensin-aldosterone inhibition.
When a hypertensive patient does not seem to have excess volume or tachycardia and the serum potassium level is less than about 5.5 mEq/L, then additional renin-angiotensin-aldosterone inhibition is indicated.16 This may be accomplished either by increasing the ACE inhibitor or the ARB to its maximal antihypertensive dose or by starting combination therapy.
Starting a calcium channel blocker
When the serum potassium level is higher than about 5.5 mEq/L, further inhibition of the renin-angiotensin-aldosterone system is contraindicated, and a nondihydropyridine calcium channel blocker can be added for its anti-hypertensive and antiproteinuric effects.16,36
When nondihydropyridine calcium channel blockers are contraindicated due to their antiinotropic effect, an attractive alternative may be to cautiously increase the dose of kaliuretic diuretics. Given the high prevalence of (often covert) volume excess in chronic kidney disease, empiric diuresis may lower blood pressure, particularly in patients already receiving several vasodilators.37 Moreover, as mentioned, by reducing serum potassium, kaliuretic diuretics help allow for a subsequent increase in renin-angiotensin-aldosterone inhibition.
IF BLOOD PRESSURE IS NORMAL, BUT PROTEINURIA PERSISTS
Because lowering blood pressure does not necessarily reduce protein excretion, some patients achieve their blood pressure goal but still have excessive proteinuria. Proponents of the dual-goal approach suggest that these patients require further treatment modifications to reach the proteinuria goal and their optimal renal prognosis.
A cautious increase in renin-angiotensin-aldosterone inhibition is possible but is likely to be limited by low blood pressure. When applicable, any nonessential antihypertensive drug that does not specifically reduce proteinuria (ie, dihydropyridine calcium channel blockers and central and direct vasodilators) should first be discontinued. This allows additional renin-angiotensin-aldosterone inhibition to reduce proteinuria without causing hypotension.
In addition, “ultra-high” doses of these drugs—two or more times the maximal antihypertensive dose—appear to reduce proteinuria without further reducing blood pressure.31–34
Various combinations of an ACE inhibitor, an ARB, and an aldosterone receptor antagonist (and possibly a renin inhibitor) may also be prescribed, striving for more complete suppression of the renin-angiotensin-aldosterone system, with dose adjustments to prevent hypotension.
KEEPING SERUM POTASSIUM AT SAFE LEVELS
Intensive inhibition of the renin-angiotensin-aldosterone system, via higher doses or combination therapy, increases the risk of hyperkalemia. This risk must be addressed energetically to prevent a potentially life-threatening complication.
When prescribed by nephrologists in clinical studies, renin-angiotensin-aldosterone inhibition has proven safe, with minimal adverse events (including hyperkalemia), even with high doses,32–34 in stage 4 chronic kidney disease (ie, with a glomerular filtration rate of 15 to 29 mL/min/1.73m2, inclusively)7 and with combination therapy.11–13,20
However, the increased incidence of hyperkalemia reported with spironolactone in patients with congestive heart failure following publication of the Randomized Aldactone Evaluation Study38 suggests that safety in clinical studies should not be extrapolated to mean safety in routine, community use. Patients with chronic kidney disease should not be given high doses or combinations of these drugs unless the treating physician is experienced in the prevention and treatment of hyperkalemia; typically such therapy should be guided by a nephrologist.
When serum potassium levels exceed 5.6 mEq/L, renin-angiotensin-aldosterone inhibitors should be decreased in dose or discontinued.39 Ideally, the drug or drugs should be restarted (to provide the potential benefits of these classes of drugs) when hyperkalemia has resolved, but this requires not only resolution of hyperkalemia but also steps to prevent this serious problem from recurring. The serum potassium level should be checked frequently, particularly after any increase in renin-angiotensin-aldosterone inhibition.
Treating hyperkalemia
Potential treatments for hyperkalemia include dietary restriction, sodium bicarbonate,39 fludrocortisone (Florinef),40 kaliuretic diuretics, and sodium polystyrene sulfonate (Kayexalate). Nonsteroidal anti-inflammatory drugs should be avoided.
Dietary restriction should be particularly emphasized: if potassium intake is decreased to the same extent as renin-angiotensin-aldosterone inhibitors reduce its excretion, then the serum potassium level will remain acceptable. All dietary supplements whose contents are not precisely known should be proscribed. A list of high-potassium foods to avoid should be given with the initial prescription for the drug. If briefly reviewed at each visit, with feedback given based on measured serum potassium levels, dietary treatment is typically effective (personal observation).
Fludrocortisone is an option when dietary potassium restriction fails.
An increase in the dose of diuretic is typically required with fludrocortisone to prevent sodium retention. The combination of dietary potassium restriction, fludrocortisone (0.1 mg/day, 3–5 days a week), and furosemide (Lasix) allowed high doses of an ACE inhibitor or a combination of an ACE inhibitor and an ARB to be given in 132 patients with chronic kidney disease.40 Over several years, their mean peak potassium level was 4.87 mEq/L, and no instance of acute hyperkalemia requiried stopping the ACE inhibitor or ARB.
However, fludrocortisone is an aldosterone analogue with potentially long-term aldosterone-mediated injurious effects on heart and renal function, even though only low doses were required in the three-pronged approach to hyperkalemia.40 The long-term effect of a regimen of an ACE inhibitor plus an ARB plus fludrocortisone on cardiac and renal outcomes is unknown and of concern.
Therefore, fludrocortisone should probably be avoided in patients with systolic heart dysfunction and should be used cautiously in general. Its use might be limited to patients with proteinuric chronic kidney disease that progresses despite therapy, particularly when that progression is in the context of inability to give significant renin-angiotensin-aldosterone inhibition because of hyperkalemia.
MORE STUDY NEEDED
Chronic kidney disease treatment is becoming increasingly complex, with a lengthening list of potentially effective drugs, difficult-to-reach goals, and a less structured approach. This complexity is magnified by issues of potassium homeostasis and interindividual variations in response to renin-angiotensin-aldosterone inhibition.
More prospective studies are needed to confirm the benefits of targeting proteinuria along with blood pressure and the metrics of the goals in tandem, but, based on available information, the dual-goal approach has been recommended for proteinuric patients,10,15–18 and evidence is accumulating for greater renal protection from larger doses of renin-angiotensin-aldosterone inhibitors and from using these drugs in combination.
When angiotensin-converting enzyme (ACE) inhibitors and angiotensin II type 1 receptor blockers (ARBs) were introduced, we hoped that these drugs would slow or stop the inexorable progression of chronic kidney disease. This hasn’t come to pass: the incidence of end-stage renal disease continued to increase throughout the 1990s, and although it may have finally reached a plateau, it remains unacceptably high.1 One reason may be that, used singly, drugs that block the renin-angiotensin-aldosterone system are only moderately successful, as approximately 20% to 40% of patients still reach unfavorable renal end points such as doubling of the serum creatinine level or dialysis.2–7
In view of these disappointing results, some experts are advocating a new strategy in which they advise that both blood pressure and urinary albumin excretion be lowered to specific goals. To achieve these goals, we will generally have to give higher doses of ACE inhibitors and ARBs alone or use a combination of these and other drugs that block the renin-angiotensin-aldosterone system at various sites.
This article describes how the dual-goal approach, with a focus on renin-angiotensin-aldosterone system inhibition, may be applied in the therapy of proteinuric chronic kidney disease. This appears to be a reasonable approach, based on current evidence, to address the epidemic of renal failure. However, further studies are needed to establish the effectiveness of this approach, and the risk of hyperkalemia following aggressive inhibition of the renin-angiotensin-aldosterone system poses a significant management problem.
ALBUMIN MAY BE TOXIC
While hypertension has long been associated with poor renal outcomes, urinary albumin has more recently been implicated by observational and experimental evidence as a tubular-interstitial toxin that may also accelerate the progression of renal disease.
For example, in both the Reduction of Endpoints in NIDDM With the Angiotensin II Antagonist Losartan (RENAAL) study8 and the Ramipril Efficacy in Nephropathy study,4 baseline proteinuria was almost linearly related to worse renal outcomes. In RENAAL, patients who excreted more than about 3 g of albumin per day had an 8.1-fold higher risk of progressing to end-stage renal disease.8 Moreover, the more that protein excretion could be reduced, the better the renal outcomes, down to a level of about 500 mg/day.8
Of importance, lowering blood pressure did not always decrease protein excretion—nearly 40% of patients had a dissociation between the two.9 In fact, prescribing a single ACE inhibitor or ARB while targeting only blood pressure has not predictably reduced protein excretion to 500 mg/day (the proposed goal).2–7
Although albumin has not been conclusively proven to be a renal toxin, targeting the reduction of proteinuria may also succeed if urinary albumin simply serves as a marker of the success of chronic kidney disease treatment and reflects prognosis.
A DUAL-GOAL APPROACH
In view of the observational and experimental evidence, many experts10,15–18 are advocating a dual-goal approach that stresses lowering both blood pressure and urinary protein (albumin) excretion. The recommended goal for systolic blood pressure is less than 120 to 125 mm Hg; the goal for proteinuria is less than 300 to 500 mg/24 hours,16,17,19 aiming to slow the decline in glomerular filtration rate to less than 2 mL/ min/year.11,20
The strategy of targeting both proteinuria and blood pressure has recently received further support. In a prospective randomized controlled study,21 nondiabetic patients with proteinuria received either an ACE inhibitor or an ARB. In one group, the dose was adjusted to lower the blood pressure to less than 130/80 mm/Hg; in the other group, the dose was adjusted to lower the blood pressure to 130/80 and to reduce protein excretion maximally. Only about half as many patients in the group with the dual-goal strategy reached the composite primary end point (doubling of serum creatinine, end-stage renal disease, or death) over a median of 3.7 years of follow-up, as compared with those treated by targeting the blood pressure alone.
In retrospect, the suboptimal success in the earlier landmark studies2–7 may have derived from the failure of ACE inhibitors and ARBs, used by themselves at moderate doses, to either lower the blood pressure to the recently advised goal (the actual results obtained varied from about 128 to about 145 mm Hg systolic) or, perhaps, to reduce proteinuria to the goal level.
Not all nephrologists currently pursue the stringent proteinuria goal of 500 mg per day—the targeted reduction of proteinuria requires further prospective evidence to support it. However, nephrologists do commonly follow the broad theme that antihypertensive therapy in proteinuric chronic kidney disease should accentuate medicines that protect the kidney beyond their antihypertensive effect (Table 1), and that proteinuria is an important metric that, at the very least, reflects the response to therapy and prognosis.
BLOCKING RENIN-ANGIOTENSIN- ALDOSTERONE MORE COMPLETELY
These issues may be addressed by more complete inhibition of the renin-angiotensin-aldosterone system, now achievable with the addition of aldosterone receptor antagonists and direct renin inhibitors to the ACE inhibitors and ARBs. Although we lack long-term studies of the relative efficacy of these medicines alone or in various combinations, the multistep sequence of the renin-angiotensin-aldosterone system allows for the possibility that more complete suppression via coordinated pharmacologic attention to multiple sites will yield beneficial results.
Combining an ACE inhibitor and an ARB
Even in the absence of ACE, angiotensin II is also produced by other kinases and therefore is not completely suppressed by an ACE inhibitor. For this and other reasons, there are theoretical advantages to adding an ARB to an ACE inhibitor.
In the Combination Treatment of Angiotensin 2 Receptor Blocker and Angiotensin-Converting-Enzyme Inhibitor in Non-Diabetic Renal Disease (COOPERATE) study,20 the combination of an ACE inhibitor and an ARB protected the kidneys better than either medicine alone, not only in terms of less protein in the urine but also in terms of significantly fewer patients progressing to the primary end points of doubling of serum creatinine or end-stage renal disease after 3 years of follow-up (11% of patients on combination therapy vs 23% on single therapy).
Aldosterone receptor antagonists or renin inhibitors plus ACE inhibitors and ARBs
Aldosterone escape is common during long-term therapy with ACE inhibitors and ARBs, and an aldosterone-receptor antagonist reduces proteinuria11–13 and stabilizes kidney function13 in a manner additive to that of ACE inhibitors and ARBs.
Direct renin inhibitors overcome the reactive rises in renin activity and in angiotensin II that complicate therapy with ACE inhibitors and ARBs, and they also reduce urinary aldosterone excretion.14
When to consider combination therapy
Inhibition of the renin-angiotensin-aldosterone system at multiple sites may be considered in cases of persistent hypertension or proteinuria, or of progression of chronic kidney disease despite single-drug therapy, or more broadly, with increasing evidence that combination therapy may preserve the glomerular filtration rate.13,20 This article suggests one way to apply the several available renin-angiotensin-aldosterone inhibitors, keeping in mind extensive interindividual variations, uncertain responses, and the absence of a linear evidence-based strategy known to be broadly successful.
INITIAL CONSIDERATION: WHAT IS THE BLOOD PRESSURE GOAL?
Determining the blood pressure goal for a patient may not be as straightforward as usually assumed. Typically, advisories suggest a discrete goal; for example, the Seventh Joint National Committee22 recommended a systolic blood pressure of 130 mm Hg or lower for patients with chronic kidney disease or diabetes. However, if we weigh the risks and benefits, we find that the situation is more nuanced. The blood pressure goal should vary among patients, depending on age, amount of proteinuria, whether the patient can tolerate the lowered blood pressure, and whether lowering the blood pressure to this goal stabilizes kidney function.
Long-term follow-up of the Modification of Diet in Renal Disease (MDRD) study demonstrated a benefit of setting the goal mean arterial pressure to less than 92 mm Hg (about 125 mm Hg systolic) regardless of proteinuria.23 In addition, a meta-analysis suggested that nondiabetic proteinuric patients benefit from even lower systolic blood pressures (110–119 mm Hg).19
In older patients
However, in the MDRD study, the goal of approximately 125 mm Hg systolic pertained only to patients no older than 60 years.23 The goal was increased to about 130 mm Hg for patients 61 to 70 years old. In addition, major clinical studies of chronic kidney disease have excluded patients older than 70 years.2–7,23
Therapy for chronic kidney disease in this older age group is essentially unstudied, and we should be cautious about extrapolating results of aggressive blood pressure-lowering (and renin-angiotensin-aldosterone inhibition) from younger patients to older patients, who may have extensive vascular disease.24,25
For patients older than 70 years, guidance is perhaps best provided by the Systolic Hypertension in the Elderly Program (SHEP), which found that lowering systolic blood pressure to an average of 143 mm Hg reduced the incidence of stroke and cardiovascular disease.26 The SHEP study does not establish the optimal blood pressure goal for preventing progressive chronic kidney disease (or even cardiovascular disease) in the older age group. However, this is the lowest systolic pressure yet shown to be generally safe and associated with any improved outcome for these patients.
Additional studies are needed to evaluate whether this blood pressure level provides the best outcomes in patients with chronic kidney disease, or whether even lower blood pressures in the elderly are safe and will further improve either renal or cardiovascular outcomes.
In younger patients
In contrast, younger patients without diabetes or vascular disease may, in theory, be candidates for even lower blood pressure. No major study of chronic kidney disease isolated patients from about 20 to 40 years old for analysis, precluding direct evidence-based guidelines for this cohort at this time.
However, some of these patients may have had premorbid systolic blood pressures of 90 to 110 mm Hg, so systolic pressures of 110 to 120 mm Hg would be “hypertensive” by 10 to 30 mm Hg for them. It is possible that some patients in this cohort will tolerate a systolic pressure lower than 110 mm Hg, and that the lower blood pressure may provide additional long-term renal protection for them. This notion is theoretical, however, and has not been verified by clinical studies.
No one pressure fits all
In summary, an initial target systolic pressure for proteinuric patients, based on available evidence, might be less than 130 mm Hg for patients 61 to 70 years old,23 less than 125 mm Hg for patients younger than 61 years,23 and perhaps as low as 110 to 119 mm Hg for non-diabetic patients.19 Caution is advised against targeting systolic blood pressure less than 140 mm Hg for patients older than 70 years.
These are only initial goals and should be reevaluated as treatment progresses. The achieved blood pressure must be clinically tolerated—symptoms of tissue hypoperfusion indicate that the blood pressure is too low for the patient. In addition, the blood pressure goal (like the proteinuria goal) is only a surrogate end point, and if kidney function declines even though the surrogate end points are attained, then those end points should be reevaluated.
Tailoring blood pressure goals to the individual patient dovetails with the recent suggestion that blood pressure should not be perceived as a rigid dichotomy of “hypertension” vs “normal.”27 There is, in general, a continuous correlation between blood pressure, beginning at low levels, and the risk of cardiorenal disease, and choosing an optimal blood pressure goal for an individual patient requires an ongoing assessment of benefits, risks, and side effects.
STARTING ANTIHYPERTENSIVE THERAPY
The question of which antihypertensive drug to try first is moot in chronic kidney disease because almost all patients need multiple medicines to reach their blood pressure goals.
The Seventh Joint National Committee recommended an ACE inhibitor for initial therapy in hypertensive patients with chronic kidney disease,22 although an ARB is a reasonable first choice for those with type 2 diabetes.5,6
Diuretics potentiate the effects of ACE inhibitors and ARBs and are generally prescribed concomitantly or as the second choice.
A beta-blocker may be recommended as a third medicine (when needed), to provide a complementary class of antihypertensive, to address the high incidence of concomitant coronary artery disease and systolic dysfunction, and because of evidence that sympathetic excess contributes to the hypertension and progression of chronic kidney disease.28,29 The National Kidney Foundation30 suggests that the dose of beta-blocker be increased if the heart rate is greater than 84.
INTENSIFYING RENIN-ANGIOTENSIN-ALDOSTERONE INHIBITION: WHICH DRUGS, AND WHEN?
When hypertension and proteinuria persist despite the use of an ACE inhibitor or an ARB, additional inhibition of the renin-angiotensin-aldosterone system is generally recommended to lower both the blood pressure and the protein excretion. Increasing the dose of ACE inhibitor or ARB,31–34 combining an ACE inhibitor and an ARB,20 or adding an aldosterone receptor antagonist to either an ACE inhibitor or an ARB11–13 have all been shown to reduce proteinuria (as a surrogate end point), and several studies have, importantly, found that these combinations preserve kidney function over time.13,20
However, lacking long-term studies that compare these options, we cannot insist upon specific treatment choices or sequences in these situations.
An approach based on serum potassium and volume status
For example, if a patient has obvious signs of volume excess (eg, edema, jugular venous distention, rales) and the serum potassium concentration is less than about 5.0 or 5.5 mEq/L, then an aldosterone receptor antagonist may logically be added or increased in dose.
Aldosterone is more than a kidney hormone
Increasing the diuretic or renin-angiotensin-aldosterone inhibition
For patients who have obvious signs of volume excess and a serum potassium level greater than 5.0 mEq/L, the dosage of kaliuretic (potassium-excreting) diuretic (usually a loop diuretic in chronic kidney disease) can be increased. Although kaliuretic diuretics do not specifically lower proteinuria, they will help control volume and blood pressure and, by lowering the serum potassium level, facilitate the subsequent augmention of renin-angiotensin-aldosterone inhibition.
When a hypertensive patient does not seem to have excess volume or tachycardia and the serum potassium level is less than about 5.5 mEq/L, then additional renin-angiotensin-aldosterone inhibition is indicated.16 This may be accomplished either by increasing the ACE inhibitor or the ARB to its maximal antihypertensive dose or by starting combination therapy.
Starting a calcium channel blocker
When the serum potassium level is higher than about 5.5 mEq/L, further inhibition of the renin-angiotensin-aldosterone system is contraindicated, and a nondihydropyridine calcium channel blocker can be added for its anti-hypertensive and antiproteinuric effects.16,36
When nondihydropyridine calcium channel blockers are contraindicated due to their antiinotropic effect, an attractive alternative may be to cautiously increase the dose of kaliuretic diuretics. Given the high prevalence of (often covert) volume excess in chronic kidney disease, empiric diuresis may lower blood pressure, particularly in patients already receiving several vasodilators.37 Moreover, as mentioned, by reducing serum potassium, kaliuretic diuretics help allow for a subsequent increase in renin-angiotensin-aldosterone inhibition.
IF BLOOD PRESSURE IS NORMAL, BUT PROTEINURIA PERSISTS
Because lowering blood pressure does not necessarily reduce protein excretion, some patients achieve their blood pressure goal but still have excessive proteinuria. Proponents of the dual-goal approach suggest that these patients require further treatment modifications to reach the proteinuria goal and their optimal renal prognosis.
A cautious increase in renin-angiotensin-aldosterone inhibition is possible but is likely to be limited by low blood pressure. When applicable, any nonessential antihypertensive drug that does not specifically reduce proteinuria (ie, dihydropyridine calcium channel blockers and central and direct vasodilators) should first be discontinued. This allows additional renin-angiotensin-aldosterone inhibition to reduce proteinuria without causing hypotension.
In addition, “ultra-high” doses of these drugs—two or more times the maximal antihypertensive dose—appear to reduce proteinuria without further reducing blood pressure.31–34
Various combinations of an ACE inhibitor, an ARB, and an aldosterone receptor antagonist (and possibly a renin inhibitor) may also be prescribed, striving for more complete suppression of the renin-angiotensin-aldosterone system, with dose adjustments to prevent hypotension.
KEEPING SERUM POTASSIUM AT SAFE LEVELS
Intensive inhibition of the renin-angiotensin-aldosterone system, via higher doses or combination therapy, increases the risk of hyperkalemia. This risk must be addressed energetically to prevent a potentially life-threatening complication.
When prescribed by nephrologists in clinical studies, renin-angiotensin-aldosterone inhibition has proven safe, with minimal adverse events (including hyperkalemia), even with high doses,32–34 in stage 4 chronic kidney disease (ie, with a glomerular filtration rate of 15 to 29 mL/min/1.73m2, inclusively)7 and with combination therapy.11–13,20
However, the increased incidence of hyperkalemia reported with spironolactone in patients with congestive heart failure following publication of the Randomized Aldactone Evaluation Study38 suggests that safety in clinical studies should not be extrapolated to mean safety in routine, community use. Patients with chronic kidney disease should not be given high doses or combinations of these drugs unless the treating physician is experienced in the prevention and treatment of hyperkalemia; typically such therapy should be guided by a nephrologist.
When serum potassium levels exceed 5.6 mEq/L, renin-angiotensin-aldosterone inhibitors should be decreased in dose or discontinued.39 Ideally, the drug or drugs should be restarted (to provide the potential benefits of these classes of drugs) when hyperkalemia has resolved, but this requires not only resolution of hyperkalemia but also steps to prevent this serious problem from recurring. The serum potassium level should be checked frequently, particularly after any increase in renin-angiotensin-aldosterone inhibition.
Treating hyperkalemia
Potential treatments for hyperkalemia include dietary restriction, sodium bicarbonate,39 fludrocortisone (Florinef),40 kaliuretic diuretics, and sodium polystyrene sulfonate (Kayexalate). Nonsteroidal anti-inflammatory drugs should be avoided.
Dietary restriction should be particularly emphasized: if potassium intake is decreased to the same extent as renin-angiotensin-aldosterone inhibitors reduce its excretion, then the serum potassium level will remain acceptable. All dietary supplements whose contents are not precisely known should be proscribed. A list of high-potassium foods to avoid should be given with the initial prescription for the drug. If briefly reviewed at each visit, with feedback given based on measured serum potassium levels, dietary treatment is typically effective (personal observation).
Fludrocortisone is an option when dietary potassium restriction fails.
An increase in the dose of diuretic is typically required with fludrocortisone to prevent sodium retention. The combination of dietary potassium restriction, fludrocortisone (0.1 mg/day, 3–5 days a week), and furosemide (Lasix) allowed high doses of an ACE inhibitor or a combination of an ACE inhibitor and an ARB to be given in 132 patients with chronic kidney disease.40 Over several years, their mean peak potassium level was 4.87 mEq/L, and no instance of acute hyperkalemia requiried stopping the ACE inhibitor or ARB.
However, fludrocortisone is an aldosterone analogue with potentially long-term aldosterone-mediated injurious effects on heart and renal function, even though only low doses were required in the three-pronged approach to hyperkalemia.40 The long-term effect of a regimen of an ACE inhibitor plus an ARB plus fludrocortisone on cardiac and renal outcomes is unknown and of concern.
Therefore, fludrocortisone should probably be avoided in patients with systolic heart dysfunction and should be used cautiously in general. Its use might be limited to patients with proteinuric chronic kidney disease that progresses despite therapy, particularly when that progression is in the context of inability to give significant renin-angiotensin-aldosterone inhibition because of hyperkalemia.
MORE STUDY NEEDED
Chronic kidney disease treatment is becoming increasingly complex, with a lengthening list of potentially effective drugs, difficult-to-reach goals, and a less structured approach. This complexity is magnified by issues of potassium homeostasis and interindividual variations in response to renin-angiotensin-aldosterone inhibition.
More prospective studies are needed to confirm the benefits of targeting proteinuria along with blood pressure and the metrics of the goals in tandem, but, based on available information, the dual-goal approach has been recommended for proteinuric patients,10,15–18 and evidence is accumulating for greater renal protection from larger doses of renin-angiotensin-aldosterone inhibitors and from using these drugs in combination.
- US Renal Data System. Excerpts from the USRDS 2005 Annual Data Report. Am J Kidney Dis 2006; 47(suppl 1):S1–S286.
- Lewis E, Hunsicker L, Bain R, Rohde Rfor the Collaborative Study Group. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. N Engl J Med. 1993; 329:1456–1462.
- Maschio G, Alberti D, Janin G, et al. Effect of the angiotensin-converting enzyme inhibitor benazepril on the progression of chronic renal insufficiency. The Angiotensin-Converting Enzyme Inhibition in Progressive Renal Insufficiency Study Group. N Engl J Med. 1996; 334:939–945.
- The GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia). Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. Lancet. 1997; 349:1857–1863.
- Brenner B, Cooper M, De Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001; 345:861–869.
- Lewis E, Hunsicker L, Clarke W, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001; 345:851–860.
- Hou F, Zhang X, Zhang G, et al. Efficacy and safety of benazepril for advanced chronic renal insufficiency. N Engl J Med. 2006; 354:131–140.
- De Zeeuw D, Remuzzi G, Parving H-H, et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int. 2004; 65:2309–2320.
- Eijkelkamp W, Zhang Z, Remuzzi G, et al. Albuminuria is a target for renoprotective therapy independent from blood pressure in patients with type 2 diabetic nephropathy: post hoc analysis from the Reduction of Endpoints in NIDDM with the Angiotension 2 Antagonist Losartan (RENAAL) trial. J Am Soc Nephrol. 2007; 18:1540–1546.
- Khosla N, Bakris G. Lessons learned from recent hypertension trials about kidney disease. Clin J Am Soc Nephrol. 2006; 1:229–235.
- Chrysostomou A, Pedagogoa E, MacGregor L, Becker G. Double-blind, placebo-controlled study on the effect of the aldosterone receptor antagonist spironolactone in patients who have persistent proteinuria and are on long-term angiotensin-converting enzyme inhibitor therapy, with or without an angiotensin 2 receptor blocker. Clin J Am Soc Nephrol. 2006; 1:256–262.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int. 2006; 70:536–542.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006; 70:2116–2123.
- Azizi M, Menard J, Bissery A, Guyene T-T, Bura-Riviere A. Hormonal and hemodynamic effects of aliskiren and valsartan and their combinations in sodium-replete normotensive individuals Clin J Am Soc Nephrol 2007; 2:947–955.
- Hebert L, Wilmer W, Falkenhain M, Ladson-Wofford S, Nahman S, Rovin B. Renoprotection: one or many therapies? Kidney Int 2001; 59:1211–1226.
- Shieppate A, Remuzzi G. The future of renoprotection: frustration and promises. Kidney Int. 2003; 64:1947–1955.
- Zandi-Nejad K, Brenner B. Strategies to retard the progression of chronic kidney disease. Med Clin North Am. 2005; 89:489–509.
- Ritz E, Dikow R. Hypertension and antihypertensive treatment of diabetic nephropathy. Nat Clinl Pract Nephrol. 2006; 2:562–567.
- Jafar T, Stark P, Schmid C, et al., for the AIPRD Study Group. Progression of chronic kidney disease: the role of blood pressure control, proteinuria, and angiotensin-converting enzyme inhibition. A patient-level meta-analysis. Ann Intern Med. 2003; 139:244–252.
- Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin 2 receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomized controlled trial. Lancet. 2003; 361:117–124.
- Hou F, Xie D, Zhang X, et al. Renoprotection of optimal antiproteinuric doses (ROAD) study: a randomized controlled study of benazepril and losartan in chronic renal insufficiency. J Am Soc Nephrol. 2007; 18:1889–1898.
- Chobanian AV, Bakris GL, Black HR. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003; 289:2560–2572.
- Sarnak M, Greene T, Wang X, et al. The effect of a lower target blood pressure on the progression of kidney disease: long-term follow-up of the Modification of Diet in Renal Disease Study. Ann Intern Med. 2005; 142:342–351.
- Hemmelgarn BR, Zhang J, Manns BJ, et al. Progression of kidney dysfunction in the community-dwelling elderly. Kidney Int. 2006; 69:2155–2161.
- Locatelli F, Pozzoni P. Chronic kidney disease in the elderly: is it really a premise for overwhelming renal failure? Kidney Int 2006; 69:2118–2120.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA. 1991; 265:3255–3264.
- Forman JP, Brenner BM. ‘Hypertension’ and ‘microalbuminuria’: the bell tolls for thee. Kidney Int. 2006; 69:22–28.
- Bakris G, Hart P, Ritz E. Beta blockers in the management of chronic kidney disease. Kidney Int. 2006; 70:1905–1913.
- UKPD Study Group. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. UK Prospective diabetes study group. BMJ. 1998; 317:713–720.
- Bakris G, Williams M, Dworkin L, et al. Preserving renal function in adults with hypertension and diabetes: a consensus approach. National Kidney Foundation Hypertensive and Diabetes Executive Committees Working Group. Am J Kidney Dis. 2000; 36:646–661.
- Navis G, Kramer A, de Jong P. High-dose ACE inhibition: can it improve renoprotection? Am J Kidney Dis 2002; 40:664–666.
- Rossing K, Schjoedt K, Jensin B, Boomsma F, Parving H-H. Enhanced renoprotective effects of ultrahigh doses of irbesartan in patients with type 2 diabetes and microalbuminuria. Kidney Int. 2005; 68:1190–1198.
- Schmieder R, Klingbeil A, Fleischman E, Veelken R, Delles C. Additional antiproteinuric effect of ultrahigh dose candesartan: a double-blind, randomized, prospective study. J Am Soc Nephrol. 2005; 16:3038–3045.
- Aranda P, Segura J, Ruilope L, et al. Long-term renoprotective effects of standard versus high doses of telmisartan in hypertensive nondiabetic nephropathies. Am J Kidney Dis. 2005; 46:1074–1079.
- Calhoun D. Aldosteronism and hypertension. Clin J Am Soc Nephrol. 2006; 1:1039–1045.
- Bakris G, Weir M, Secic M, Campbell B, Weis-McNulty A. Differential effects of calcium antagonist subclasses on markers of nephropathy progression. Kidney Int. 2004; 65:1991–2002.
- Hirsch S. A different approach to resistant hypertension. Cleve Clin J Med 2007: 74;449–456.
- Juurling D, Mamdani M, Lee D, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med. 2004; 351:543–551.
- Palmer B. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med. 2004; 351:585–592.
- Moskowitz D. From pharmacogenomics to improved patient outcomes: angiotensin 1-converting enzyme as an example. Diabetes Tech Ther. 2002; 4:519–532.
- US Renal Data System. Excerpts from the USRDS 2005 Annual Data Report. Am J Kidney Dis 2006; 47(suppl 1):S1–S286.
- Lewis E, Hunsicker L, Bain R, Rohde Rfor the Collaborative Study Group. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. N Engl J Med. 1993; 329:1456–1462.
- Maschio G, Alberti D, Janin G, et al. Effect of the angiotensin-converting enzyme inhibitor benazepril on the progression of chronic renal insufficiency. The Angiotensin-Converting Enzyme Inhibition in Progressive Renal Insufficiency Study Group. N Engl J Med. 1996; 334:939–945.
- The GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia). Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. Lancet. 1997; 349:1857–1863.
- Brenner B, Cooper M, De Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001; 345:861–869.
- Lewis E, Hunsicker L, Clarke W, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001; 345:851–860.
- Hou F, Zhang X, Zhang G, et al. Efficacy and safety of benazepril for advanced chronic renal insufficiency. N Engl J Med. 2006; 354:131–140.
- De Zeeuw D, Remuzzi G, Parving H-H, et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int. 2004; 65:2309–2320.
- Eijkelkamp W, Zhang Z, Remuzzi G, et al. Albuminuria is a target for renoprotective therapy independent from blood pressure in patients with type 2 diabetic nephropathy: post hoc analysis from the Reduction of Endpoints in NIDDM with the Angiotension 2 Antagonist Losartan (RENAAL) trial. J Am Soc Nephrol. 2007; 18:1540–1546.
- Khosla N, Bakris G. Lessons learned from recent hypertension trials about kidney disease. Clin J Am Soc Nephrol. 2006; 1:229–235.
- Chrysostomou A, Pedagogoa E, MacGregor L, Becker G. Double-blind, placebo-controlled study on the effect of the aldosterone receptor antagonist spironolactone in patients who have persistent proteinuria and are on long-term angiotensin-converting enzyme inhibitor therapy, with or without an angiotensin 2 receptor blocker. Clin J Am Soc Nephrol. 2006; 1:256–262.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int. 2006; 70:536–542.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006; 70:2116–2123.
- Azizi M, Menard J, Bissery A, Guyene T-T, Bura-Riviere A. Hormonal and hemodynamic effects of aliskiren and valsartan and their combinations in sodium-replete normotensive individuals Clin J Am Soc Nephrol 2007; 2:947–955.
- Hebert L, Wilmer W, Falkenhain M, Ladson-Wofford S, Nahman S, Rovin B. Renoprotection: one or many therapies? Kidney Int 2001; 59:1211–1226.
- Shieppate A, Remuzzi G. The future of renoprotection: frustration and promises. Kidney Int. 2003; 64:1947–1955.
- Zandi-Nejad K, Brenner B. Strategies to retard the progression of chronic kidney disease. Med Clin North Am. 2005; 89:489–509.
- Ritz E, Dikow R. Hypertension and antihypertensive treatment of diabetic nephropathy. Nat Clinl Pract Nephrol. 2006; 2:562–567.
- Jafar T, Stark P, Schmid C, et al., for the AIPRD Study Group. Progression of chronic kidney disease: the role of blood pressure control, proteinuria, and angiotensin-converting enzyme inhibition. A patient-level meta-analysis. Ann Intern Med. 2003; 139:244–252.
- Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin 2 receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomized controlled trial. Lancet. 2003; 361:117–124.
- Hou F, Xie D, Zhang X, et al. Renoprotection of optimal antiproteinuric doses (ROAD) study: a randomized controlled study of benazepril and losartan in chronic renal insufficiency. J Am Soc Nephrol. 2007; 18:1889–1898.
- Chobanian AV, Bakris GL, Black HR. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003; 289:2560–2572.
- Sarnak M, Greene T, Wang X, et al. The effect of a lower target blood pressure on the progression of kidney disease: long-term follow-up of the Modification of Diet in Renal Disease Study. Ann Intern Med. 2005; 142:342–351.
- Hemmelgarn BR, Zhang J, Manns BJ, et al. Progression of kidney dysfunction in the community-dwelling elderly. Kidney Int. 2006; 69:2155–2161.
- Locatelli F, Pozzoni P. Chronic kidney disease in the elderly: is it really a premise for overwhelming renal failure? Kidney Int 2006; 69:2118–2120.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA. 1991; 265:3255–3264.
- Forman JP, Brenner BM. ‘Hypertension’ and ‘microalbuminuria’: the bell tolls for thee. Kidney Int. 2006; 69:22–28.
- Bakris G, Hart P, Ritz E. Beta blockers in the management of chronic kidney disease. Kidney Int. 2006; 70:1905–1913.
- UKPD Study Group. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. UK Prospective diabetes study group. BMJ. 1998; 317:713–720.
- Bakris G, Williams M, Dworkin L, et al. Preserving renal function in adults with hypertension and diabetes: a consensus approach. National Kidney Foundation Hypertensive and Diabetes Executive Committees Working Group. Am J Kidney Dis. 2000; 36:646–661.
- Navis G, Kramer A, de Jong P. High-dose ACE inhibition: can it improve renoprotection? Am J Kidney Dis 2002; 40:664–666.
- Rossing K, Schjoedt K, Jensin B, Boomsma F, Parving H-H. Enhanced renoprotective effects of ultrahigh doses of irbesartan in patients with type 2 diabetes and microalbuminuria. Kidney Int. 2005; 68:1190–1198.
- Schmieder R, Klingbeil A, Fleischman E, Veelken R, Delles C. Additional antiproteinuric effect of ultrahigh dose candesartan: a double-blind, randomized, prospective study. J Am Soc Nephrol. 2005; 16:3038–3045.
- Aranda P, Segura J, Ruilope L, et al. Long-term renoprotective effects of standard versus high doses of telmisartan in hypertensive nondiabetic nephropathies. Am J Kidney Dis. 2005; 46:1074–1079.
- Calhoun D. Aldosteronism and hypertension. Clin J Am Soc Nephrol. 2006; 1:1039–1045.
- Bakris G, Weir M, Secic M, Campbell B, Weis-McNulty A. Differential effects of calcium antagonist subclasses on markers of nephropathy progression. Kidney Int. 2004; 65:1991–2002.
- Hirsch S. A different approach to resistant hypertension. Cleve Clin J Med 2007: 74;449–456.
- Juurling D, Mamdani M, Lee D, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med. 2004; 351:543–551.
- Palmer B. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med. 2004; 351:585–592.
- Moskowitz D. From pharmacogenomics to improved patient outcomes: angiotensin 1-converting enzyme as an example. Diabetes Tech Ther. 2002; 4:519–532.
KEY POINTS
- Evidence is emerging that urinary albumin is toxic to the kidney.
- Lowering both blood pressure and urinary albumin excretion, as a means to prevent progressive renal disease, appears to require aggressive inhibition of the renin-angiotensin-aldosterone system, often with several complementary drugs, ie, angiotensin-converting enzyme inhibitors, angiotensin II type 1 receptor blockers, aldosterone receptor antagonists, and possibly, direct renin inhibitors.
- Volume status and potassium levels may help suggest which of several available drugs could be added at different times.
- Serum potassium levels must be managed aggressively when using renin-angiotensin-aldosterone inhibitors in combination.
When a quick sound bite won’t do

The sound bites about these trials in the news have confused physicians and patients alike. But, as we have all experienced during this election year, to understand complex problems requires an in-depth analysis instead of a sound bite.
I was troubled by the results of the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial,2 in which more patients who were treated with an intense hemoglobin A1c-lowering strategy died (mostly of macrovascular events) than those treated with a standard strategy. Older data showing a beneficial effect of glucose-lowering on the microvascular complications of diabetes are solid. I did not understand the mechanistic basis of the ACCORD results, unless the very aggressive therapy caused many hypoglycemic events with catecholamine surges, resulting in stroke or myocardial infarction, or whether a problem with a specific drug arose more often in the intensive-treatment group. There has been similar dialogue surrounding intensity of glucose control in critically ill inpatients3; here, the data suggest that hypoglycemic episodes may limit other benefits of aggressive treatment in the intensive care unit, such as reduced infection rates.
Not to be ignored is that the patients in all arms of the ACCORD trial fared far better than historical diabetic controls. The meticulous attention to management of blood pressure and LDL-C that all patients in the ACCORD trial received paid off. (If only we could do as well in our practices!) But what do we do about the sugar?
This large, well-done, ongoing trial deserves a detailed analysis for those of us who need to translate the conclusions regarding glucose control to our patients. This month in the Journal, I have invited Byron Hoogwerf, a clinical diabetologist, former internal medicine program director, well-published clinical trialist, and ACCORD investigator, to provide this analysis.4 His discussion is more detailed than what we often print purposefully, and it is well worth reading. Some issues simply can’t be understood as a sound bite.
- Kastelein JJ, Akdim F, Stroes ES, et alENHANCE Investigators. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008; 358:1431–1443.
- Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008; 358:2545–2559.
- Soylemez Wiener R, Wiener DC, Larson RJ. Benefits and risks of tight glucose control in critically ill adults: a meta-analysis. JAMA. 2008; 300:933–944.
- Hoogwerf BF. A clinician and clinical trialist’s perspective: does intensive therapy of type 2 diabetes help or harm? Seeking accord on ACCORD. Cleve Clin J Med. 2008; 75:729–737.
The sound bites about these trials in the news have confused physicians and patients alike. But, as we have all experienced during this election year, to understand complex problems requires an in-depth analysis instead of a sound bite.
I was troubled by the results of the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial,2 in which more patients who were treated with an intense hemoglobin A1c-lowering strategy died (mostly of macrovascular events) than those treated with a standard strategy. Older data showing a beneficial effect of glucose-lowering on the microvascular complications of diabetes are solid. I did not understand the mechanistic basis of the ACCORD results, unless the very aggressive therapy caused many hypoglycemic events with catecholamine surges, resulting in stroke or myocardial infarction, or whether a problem with a specific drug arose more often in the intensive-treatment group. There has been similar dialogue surrounding intensity of glucose control in critically ill inpatients3; here, the data suggest that hypoglycemic episodes may limit other benefits of aggressive treatment in the intensive care unit, such as reduced infection rates.
Not to be ignored is that the patients in all arms of the ACCORD trial fared far better than historical diabetic controls. The meticulous attention to management of blood pressure and LDL-C that all patients in the ACCORD trial received paid off. (If only we could do as well in our practices!) But what do we do about the sugar?
This large, well-done, ongoing trial deserves a detailed analysis for those of us who need to translate the conclusions regarding glucose control to our patients. This month in the Journal, I have invited Byron Hoogwerf, a clinical diabetologist, former internal medicine program director, well-published clinical trialist, and ACCORD investigator, to provide this analysis.4 His discussion is more detailed than what we often print purposefully, and it is well worth reading. Some issues simply can’t be understood as a sound bite.
The sound bites about these trials in the news have confused physicians and patients alike. But, as we have all experienced during this election year, to understand complex problems requires an in-depth analysis instead of a sound bite.
I was troubled by the results of the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial,2 in which more patients who were treated with an intense hemoglobin A1c-lowering strategy died (mostly of macrovascular events) than those treated with a standard strategy. Older data showing a beneficial effect of glucose-lowering on the microvascular complications of diabetes are solid. I did not understand the mechanistic basis of the ACCORD results, unless the very aggressive therapy caused many hypoglycemic events with catecholamine surges, resulting in stroke or myocardial infarction, or whether a problem with a specific drug arose more often in the intensive-treatment group. There has been similar dialogue surrounding intensity of glucose control in critically ill inpatients3; here, the data suggest that hypoglycemic episodes may limit other benefits of aggressive treatment in the intensive care unit, such as reduced infection rates.
Not to be ignored is that the patients in all arms of the ACCORD trial fared far better than historical diabetic controls. The meticulous attention to management of blood pressure and LDL-C that all patients in the ACCORD trial received paid off. (If only we could do as well in our practices!) But what do we do about the sugar?
This large, well-done, ongoing trial deserves a detailed analysis for those of us who need to translate the conclusions regarding glucose control to our patients. This month in the Journal, I have invited Byron Hoogwerf, a clinical diabetologist, former internal medicine program director, well-published clinical trialist, and ACCORD investigator, to provide this analysis.4 His discussion is more detailed than what we often print purposefully, and it is well worth reading. Some issues simply can’t be understood as a sound bite.
- Kastelein JJ, Akdim F, Stroes ES, et alENHANCE Investigators. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008; 358:1431–1443.
- Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008; 358:2545–2559.
- Soylemez Wiener R, Wiener DC, Larson RJ. Benefits and risks of tight glucose control in critically ill adults: a meta-analysis. JAMA. 2008; 300:933–944.
- Hoogwerf BF. A clinician and clinical trialist’s perspective: does intensive therapy of type 2 diabetes help or harm? Seeking accord on ACCORD. Cleve Clin J Med. 2008; 75:729–737.
- Kastelein JJ, Akdim F, Stroes ES, et alENHANCE Investigators. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008; 358:1431–1443.
- Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008; 358:2545–2559.
- Soylemez Wiener R, Wiener DC, Larson RJ. Benefits and risks of tight glucose control in critically ill adults: a meta-analysis. JAMA. 2008; 300:933–944.
- Hoogwerf BF. A clinician and clinical trialist’s perspective: does intensive therapy of type 2 diabetes help or harm? Seeking accord on ACCORD. Cleve Clin J Med. 2008; 75:729–737.
Does intensive therapy of type 2 diabetes help or harm? Seeking accord on ACCORD
The Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial1–5 was designed primarily to address, in patients with type 2 diabetes at high risk of cardiovascular events, whether intensive glucose control would result in a lower risk of atherosclerotic disease events or death than would standard treatment.
It was widely expected that intensive treatment would confer either modest benefit or, at worst, no benefit. However, the glucose-lowering arm of the trial was terminated early because of a higher mortality rate in the intensively treated group. (The ACCORD trial has two other arms, which concern blood pressure and lipid-lowering, and these are continuing.)
In earlier trials in type 2 diabetes, concerns had been raised about an increased risk of cardiovascular events and possibly death associated with glucose-lowering drugs, hypoglycemia itself, or both, and these were well known when ACCORD was convened. ACCORD was very carefully designed and included careful adjudication of each cardiovascular event and death, including whether hypoglycemia might have been a proximate cause of some sudden deaths.5
Therefore, the surprising result of the higher mortality rate with intensive glycemic control in ACCORD will be fodder for discussion in many arenas over the next several years, and it poses some challenges for physicians and patients in determining treatment goals, as well as for organizations that write clinical practice guidelines (and perhaps organizations involved in pay-for-performance based on these guidelines).
Still, I believe that the ACCORD results should not substantially change our approach to treatment goals in type 2 diabetes, although hemoglobin A1c targets below 6% may not have much added value for cardiovascular risk reduction. The low overall mortality rate in all the arms of the ACCORD trial emphasizes the importance of lifestyle modification, lipid and blood pressure therapy, and encouragement of aspirin use in all patients with type 2 diabetes.
This article reflects my views as a practicing diabetologist and clinical trialist (I was an investigator in the ACCORD trial) with a long-standing interest in clinical trials and in how the results influence clinical practice. The views I express herein may not reflect the views of other ACCORD investigators, the National Heart, Lung, and Blood Institute (NHLBI), the ACCORD trial coordinating center at Wake Forest University, or its data safety and monitoring board.
RISK OF CORONARY DISEASE INCREASES WITH GLUCOSE
Many observational studies6–10 have shown that the risk of cardiovascular disease, especially coronary heart disease, is two to five times higher in people with diabetes mellitus than in people without diabetes. The risk appears to be continuous, so the higher one’s glucose or hemoglobin A1c, the higher the risk.6 This risk even extends to glucose values well below the threshold values currently used to diagnose diabetes mellitus.6 Since there is no glucose threshold for coronary heart disease, the term dysglycemia (rather than hyperglycemia) has been proposed to note the relationship between glucose and coronary heart disease. (The glucose threshold for microvascular complications of diabetes, such as retinopathy and nephropathy, appears to be between 110 and 126 mg/dL).
The clustering of multiple coronary risk factors such as obesity, dyslipidemia, and hypertension has always raised the question of whether glucose is a culprit in coronary risk or whether it simply “runs in bad company.”
EARLIER CLINICAL TRIALS SUGGEST INTENSIVE TREATMENT RAISES RISK
Even though it has been widely believed that intensive glucose-lowering would reduce cardiovascular risk in type 2 diabetes, there have been hints in previous studies that some intensive-treatment regimens might increase risk.
Two large randomized clinical trials and one small one (discussed below) addressed whether glucose control would reduce the risk of atherosclerotic vascular disease events. In each of them, an increased risk of cardiovascular events and possibly of death was seen in at least one intensively treated group.
In the following discussion, I have calculated all of the death rates as the number of deaths per 1,000 patients per year, based on published study results. In this way, we can compare the rates in the various studies (including ACCORD), regardless of the trial duration.
The university group Diabetes Program: Controvery about tolbutamide therapy
The University Group Diabetes Program (UGDP)11–16 included about 1,000 participants randomized to five treatments: tolbutamide (Orinase, a sulfonylurea), insulin in a fixed dose based on body weight, insulin in adjusted doses based on fasting glucose levels, placebo, and (later) phenformin.
In the 1970s, when the UGDP was carried out, randomized clinical trials were uncommon. Like other trials from that era, the UGDP was underpowered by today’s standards and did not have a data safety and monitoring board.
Rates of cardiovascular events and deaths (per 1,000 patient-years):
- 25 (tolbutamide group)
- 12 (placebo group).
The two insulin groups did not differ from the placebo group in their rates of cardiovascular events or death.15 The tolbutamide arm was stopped, and the ensuing controversy about how to interpret the trial results lasted for more than a decade. It also resulted in a black-box warning for tolbutamide and all subsequent sulfonylureas.
United Kingdom Prospective Diabetes Study: Method of glucose-lowering an issue
The United Kingdom Prospective Diabetes Study (UKPDS)17–27 was launched in 1977. A cohort of 5,102 patients (mean age 54 years) with newly diagnosed type 2 diabetes mellitus followed a “prudent diet” for the first 3 to 4 months. Then, if their fasting glucose levels were in the range of 6.1 to 15 mmol/L (110–270 mg/dL), they were randomized to receive various treatments.
Patients who were not obese were randomized to receive either intensive treatment or conventional treatment. The intensive-treatment group received either insulin or a sulfonylurea (chlorpropamide [Diabinese], glibenclamide, or glipizide [Glucotrol]); the conventional-treatment group received diet therapy. The sulfonylurea arm was included partly to address the UGDP results.
Patients who were obese were randomized to receive one of three treatments: intensive treatment (with the agents listed above), conventional treatment, or metformin (Fortamet, Glucophage).
The mean in-trial hemoglobin A1c level in the intensive-treatment group was 7.0%, compared with 7.9% in the conventional-treatment group.
After a mean follow-up of more than 10 years, the incidence of myocardial infarction was 16% lower in the intensive-treatment group, but the difference was not statistically significant (P = .052).
Rates of death from all causes among nonobese subjects (per 1,000 patient-years):
- 18.2–20.5 (intensive-treatment group)
- 19.9 (conventional-treatment group).
In the obese patients who received metformin, the incidence of myocardial infarction was lower than in the conventional-treatment group but not the intensive-treatment group.
Rates of death among obese patients (per 1,000 patient-years):
- 13.5 (metformin group)
- 18.9 (intensive-treatment group)
- 20.6 (conventional-treatment group).
However, a small subset (n = 587) of the original group assigned to sulfonylurea therapy whose glycemic control deteriorated during the trial were rerandomized to continue to receive a sulfonylurea alone or to have metformin added. There was a statistically significantly higher rate of cardiovascular events and a nonsignificantly higher rate of total mortality in the metformin-plus-sulfonylurea group (30.3 per 1,000 patient-years) than in the sulfonylurea-only group (19.1 per 1,000 patient-years).
These data suggested that the way glucose-lowering was achieved might be as important as the glucose levels actually achieved. However, no definite conclusions could be drawn.
In an editorial on the UKPDS, Nathan26 made a comment that may have been prescient in terms of the ACCORD trial: “Professional organizations will now scramble to decide how to translate the UKPDS results … Whether the UKPDS firmly establishes the choice of any one therapy…or any combination of therapies for the long-term treatment of type 2 diabetes is more questionable.”26
Veterans Administration feasibility study
A Veterans Administration feasibility study28,29 included 153 men (mean age 60) with type 2 diabetes (mean duration 7.8 years) who received either conventional therapy (a single daily dose of insulin) or intensive therapy (multiple doses of insulin plus a sulfonylurea). Over a mean of 27 months, the intensive-therapy group achieved a hemoglobin A1c level that was 2 percentage points lower than in the conventional-therapy group.
At 2.25 years of follow-up, cardiovascular events had occurred in 24 (24%) of the intensive-therapy group and in 16 (20%) of the standard-therapy group (P = .10).
Rates of death from all causes (per 1,000 patient-years):
- 28.9 (intensive-treatment group)
- 17.5 (conventional-treatment group).
ACCORD TRIAL DESIGN
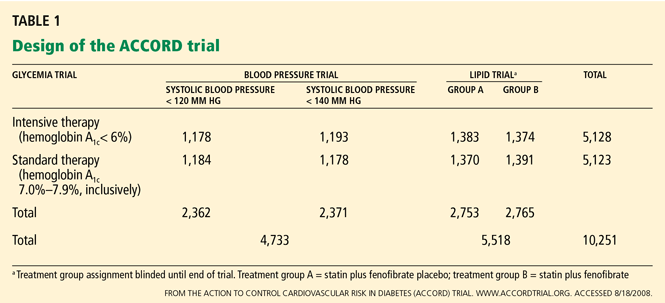
The primary outcome measured was the combined incidence of nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes. Secondary outcomes included death from any cause. The study is also evaluating the effect of intensive treatment on microvascular disease, hypoglycemia, cognition, quality of life, and cost-effectiveness.
The ACCORD study was designed to have 89% power to detect a 15% treatment effect of intensive glycemic control compared with standard glycemic control for the primary end point.
ACCORD RESULTS
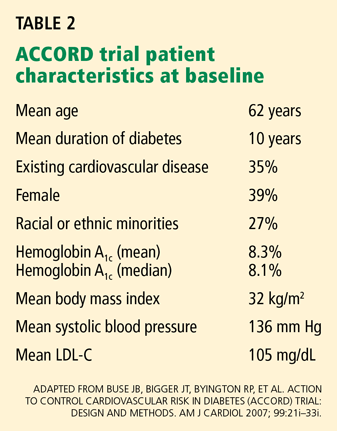
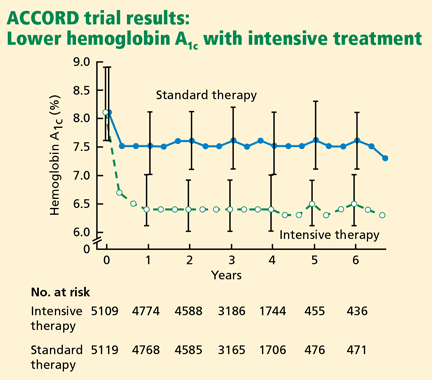
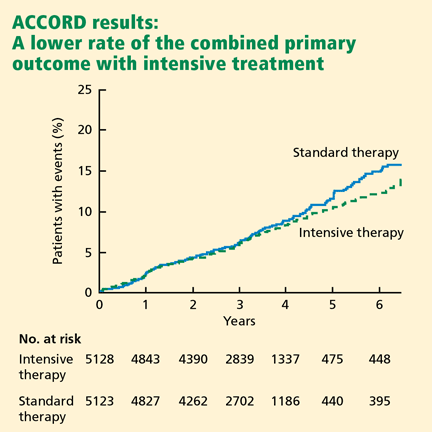
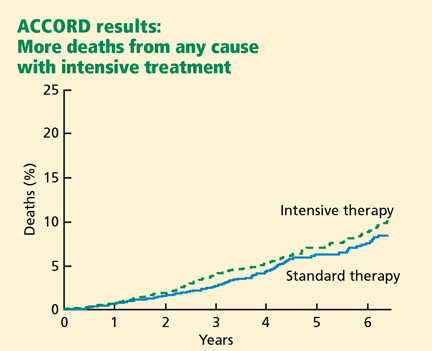
Rates of death from any cause (per 1,000 patient-years):
- 14 (intensive-treatment group)
- 11 (standard-treatment group).
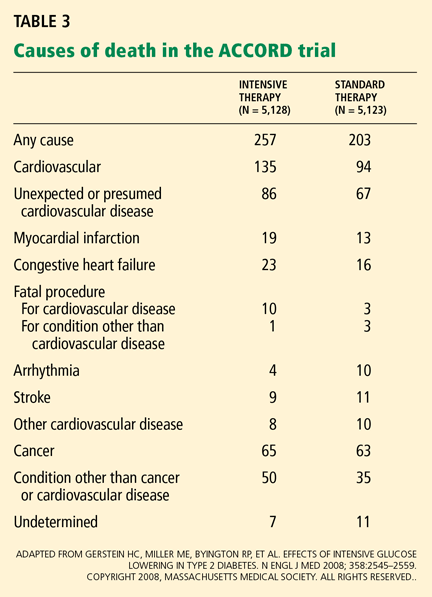
In the analyses available at the time that this study arm closed, the excess mortality was not attributable to any particular treatment regimen. In particular, rosiglitazone (Avandia) use did not contribute to the excess mortality. (Of note, 91.2% of the intensive-treatment group and 57.5% of the conventional-treatment group had been treated with rosiglitazone, with more than 19,000 patient-years of rosiglitazone exposure). The excess mortality was also not attributable to hypoglycemia immediately proximate to the death.
The ACCORD trial’s data safety and monitoring board recommended that this arm of the study be discontinued for safety reasons, and this recommendation was accepted by the NHLBI project office. All participants were notified by letter before the trial results were announced publicly, and all intensive-therapy group participants are now being treated by the protocol used in the standard-therapy group.1
FEWER DEATHS IN ACCORD THAN IN OTHER STUDIES IN DIABETES
The mortality rates in both arms of ACCORD were much lower than in other observational studies and clinical trials in type 2 diabetes.
The National Health and Nutrition Education Survey (NHANES),30 conducted from 1971 to 1975, included 14,374 people with diabetes between the ages of 25 and 74. Many of them were younger than the ACCORD patients, but two NHANES age-groups overlapped the ACCORD cohort. Rates of death from any cause at 22 years (per 1,000 patient-years):
- 39.7 (ages 45–64)
- 89.7 (ages 65–74).
The NHANES cohort would not have been treated as vigorously for coronary risk and other common causes of death.
UGDP, UKPDS. Death rates in the glucose-lowering trials of type 2 diabetes mellitus cited above were typically in the range of 20 deaths per 1,000 patient-years but were as high as 30 deaths per 1,000 patient-years in the UGDP tolbutamide group16 and the UK-PDS sulfonylurea-plus-metformin group.20,22,26
Steno-2.31 Half of 160 patients with type 2 diabetes were randomized to intensive strategies for controlling glucose, lipids, and blood pressure and for taking aspirin and angiotensin-converting enzyme inhibitors and following a healthy lifestyle. The other half received conventional therapy. Even in the intensive-treatment group, the mortality rate at 13 years was higher than in ACCORD. Rates of death from any cause (per 1,000 patient years):
- 22.5 (intensive-treatment group)
- 37.6 (conventional-treatment group).
After the ACCORD results were presented, two other trials addressing the question of whether lower hemoglobin A1c would reduce cardiovascular risk in type 2 diabetes have reported their outcomes:
The ADVANCE trial (Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation),32,33 with 11,140 patients, had a target hemoglobin A1c of 6.5% in an intensive-treatment group and 7.3% in a usual-treatment group. The intensive-treatment group showed no difference in the rates of major macrovascular events (HR 0.94, 95% CI 0.84–1.06, P = .32) or all-cause mortality (HR 0.93, 95% CI 0.83–1.06, P = .32). The overall death rate in ADVANCE (about 18 deaths per 1,000 patient-years) was higher than in ACCORD.
The Veterans Administration Diabetes Trial included 1,791 patients.34 Like the ADVANCE trial, it also found no difference in major cardiovascular outcomes (HR 0.868, P = .11) or cardiovascular mortality rates (HR 1.258, P = .36) with intensive therapy vs conventional therapy, ie, achieved hemoglobin A1c levels of 6.9% vs 8.4% (presented at the American Diabetes Association 2008 Scientific Sessions). Hypoglycemia was associated with an increased risk of death in the standard-treatment group.
An analysis suggested that patients with a shorter duration of diabetes may have had cardiovascular benefit from intensive glucose-lowering, while those who had had it longer may have had increased risk associated with the more intensive therapy. The rate of death from all causes appears to have been higher than in ACCORD, but this could not be determined accurately from the presentations.
Comment. Thus, the ACCORD cohort as a whole has had strikingly lower death rates than in these other studies. The fact that all participants had lower glucose levels on therapy than at baseline may possibly contribute to these lower death rates. In addition, all ACCORD participants in the lipid arm received a statin; all participants in the blood pressure arm had their blood pressure lowered to levels below those commonly seen in clinical practice; participants were encouraged to exercise regularly; most participants were given diet instruction; and other healthy behaviors such as aspirin use, regular follow-up with primary care physicians, and recommendations about smoking were encouraged throughout the study. These comprehensive strategies may represent better care and thus result in lower death rates than in other studies.
POSSIBLE EXPLANATIONS FOR THE ACCORD OUTCOMES
The ACCORD trial has already stimulated fierce debate about the reasons for the higher mortality rate in the intensive-treatment group. With longer follow-up, some new risk factors for death may be identified that are not evident in the analyses of the current 460 deaths. What follows are some of my thoughts, with the caveat that they are not confirmed (supported statistically) by any currently available analyses from ACCORD.
It seems unlikely that lower glucose values as reflected by lower hemoglobin A1c values in the intensive-treatment group are an a priori explanation for the observed differences in mortality rates—especially since the mortality rates were lower than in the NHANES and clinical trial data sets cited above. If we assume that a type 1 statistical error (finding a difference where no difference actually exists) does not explain the findings, then at least four reasonable postulates exist:
Hypoglycemia may have some adverse effect, either acutely or from recurrent events that trigger a catecholamine response with associated risk for arrhythmia or increased coronary heart disease risk. However, the investigators analyzed each death to determine whether hypoglycemia was a contributing cause, and they found no statistically significant relationship between hypoglycemia and death in the intensive-treatment group.
Weight gain is common with intensive therapy. Obesity may be associated with greater cytokine production, higher concentrations of clotting factors, higher levels of free fatty acids, and other potential contributors to the risk of coronary heart disease and death. Currently, the ACCORD analyses do not suggest that weight gain explains the higher death rate.
Medications such as rosiglitazone, sulfonylureas, and the combination of a sulfonylurea plus metformin have been previously associated with increased death rates in some observational and intervention trials. These studies had some serious methodologic limitations (eg, absence of risk adjustment, events not adjudicated, small study cohorts, wide variation in study cohort characteristics) and small numbers of events.11–13,16,26,35 ACCORD analyses have not shown that any single glucose-lowering agent—including rosiglitazone—or combination of agents explains the death rates.
The stress of maintaining glycemic control has been speculated to have in some way contributed to an increased risk. To achieve intensive control, patients had to have frequent contact with their health care providers, they were often told that their hemoglobin A1c values were “too high” even when they were well below those in the American Diabetes Association guidelines, and they had to follow complex glucose-lowering regimens.
Semiquantitative measures of overall attitudes about health exist (eg, the “Feeling Thermometer” scale), but stress was not measured quantitatively in the ACCORD trial.
IMPLICATIONS OF ACCORD
In practice, most clinicians believe that the target glucose level in patients with type 2 diabetes should be as low as safely possible. This approach does not need to be modified on the basis of current information from ACCORD.
To be safe, regimens should be associated with a low risk of hypoglycemia and a low risk of weight gain. Use of combinations of medications that work by different mechanisms is still prudent. Agents should be used that may have favorable effects on other cardiovascular risk factors (eg, lipids, blood pressure, visceral fat).
Hemoglobin A1c targets below 7% are not precluded in all patients on the basis of the ACCORD results, though values lower than 6% may not have much added benefit for cardiovascular risk reduction. We should note that hemoglobin A1c was reduced in all ACCORD participants and that death rates were lower than in many other type 2 diabetic cohorts. Pending data on other outcomes in ACCORD (nephropathy, retinopathy, dementia, fracture risk), I believe it is premature for organizations to change their proposed hemoglobin A1c targets,36,37 as none have proposed values as low as the target in the ACCORD intensive-treatment group. At present, no class of glucose-lowering agents needs to be excluded from consideration on the basis of the ACCORD data.
The overall low rates of death in this population at high risk of coronary heart disease deserve comment. Not only are they lower than in other glucose-lowering trials, but they are also lower than in a number of studies of mortality in diabetes cohorts. As noted above, multiple risk factors for coronary heart disease and death were (and are) addressed in the ACCORD study participants, including repeated recommendation for lifestyle modification, intervention arms with lipid and blood pressure therapy, encouragement of aspirin use, and regular follow-up with health care providers for risk factors not managed by the ACCORD trial protocol. It is likely that multiple approaches to reducing the risk of cardiovascular disease contributed to this low mortality rate and that similar approaches will reduce the risk of coronary disease and death in regular clinical practice.
The ACCORD lipid and blood pressure arms are continuing, with results expected in 2010. The future results from ACCORD as well as from several glucose-lowering trials currently in progress (ADVANCE,32,33 Veteran’s Administration,34 Bypass Angioplasty Revascularization Investigation 2 Diabetes [BARI-2D]38) will likely help refine our understanding of the effects of glucose-lowering, glucose-lowering strategies and targets, and multiple interventions on coronary events and all-cause mortality.
For now, any strategy that lowers glucose and is associated with a low risk of hypoglycemia and does not cause excessive weight gain should be considered appropriate in patients with type 2 diabetes.
- Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008; 358:2545–2559.
- Goff DC, Gerstein HC, Ginsberg HN, et al. Prevention of cardiovascular disease in persons with type 2 diabetes mellitus: current knowledge and rationale for the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007; 99:4i–20i.
- Buse JB, Bigger JT, Byington RP, et al. Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial: design and methods. Am J Cardiol 2007; 99:21i–33i.
- Gerstein HC, Riddle MC, Kendall DM, et al. Glycemia treatment strategies in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007; 99:34i–43i.
- Bonds DE, Kurashige EM, Bergenstal R, et al. Severe hypoglycemia monitoring and risk management procedures in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007; 99:80i–89i.
- Gerstein HC. Dysglycemia, not just diabetes, is a continuous risk factor for cardiovascular disease. Evid Based Cardiovasc Med. 1997; 1:87–88.
- Gerstein HC, Pais P, Pogue J, Yusuf S. Relationship of glucose and insulin levels to the risk of myocardial infarction: a case-control study. J Am Coll Cardiol. 1999; 33:612–619.
- Gerstein HC, Capes SE. Dysglycemia: a key cardiovascular risk factor. Semin Vasc Med. 2002; 2:165–174.
- Gerstein HC, Santaguida P, Raina P, et al. Annual incidence and relative risk of diabetes in people with various categories of dysglycemia: a systematic overview and meta-analysis of prospective studies. Diabetes Res Clin Pract. 2007; 78:305–312.
- American Diabetes Association. Role of cardiovascular risk factors in prevention and treatment of macrovascular disease in diabetes. Diabetes Care. 1989; 12:573–579.
- Schor S. The University Group Diabetes Program. A statistician looks at the mortality results. JAMA. 1971; 217:1671–1675.
- Cornfield JThe University Group Diabetes Program. A further statistical analysis of the mortality findings. JAMA. 1971; 217:1676–1687.
- Feinstein AR. Clinical biostatistics. 8. An analytic appraisal of the University Group Diabetes Program (UGDP) study. Clin Pharmacol Ther. 1971; 12:167–191.
- The University Group Diabetes Program. A study of the effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. V. Evaluation of pheniformin therapy. Diabetes 1975; 24( suppl 1):65–184.
- Knatterud GL, Klimt CR, Levin ME, Jacobson ME, Goldner MG. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. VII. Mortality and selected nonfatal events with insulin treatment. JAMA. 1978; 240:37–42.
- Schwartz TB, Meinert CL. The UGDP controversy: thirty-four years of contentious ambiguity laid to rest. Perspect Biol Med. 2004; 47:564–574.
- Turner RC, Holman RR. Lessons from UK Prospective Diabetes Study. Diabetes Res Clin Pract 1995; 28( suppl):S151–S157.
- UKPDS Research Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998; 352:854–865.
- UKPDS Study Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998; 352:837–853.
- UK Prospective Diabetes Study Group. UKPDS 28: a randomized trial of efficacy of early addition of metformin in sulfonylurea-treated type 2 diabetes. Diabetes Care. 1998; 21:87–92.
- Bretzel RG, Voigt K, Schatz H. The United Kingdom Prospective Diabetes Study (UKPDS) implications for the pharmacotherapy of type 2 diabetes mellitus. Exp Clin Endocrinol Diabetes. 1998; 106:369–372.
- Turner RC, Cull CA, Frighi V, Holman RR. Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. JAMA. 1999; 281:2005–2012.
- Leslie RD. United Kingdom prospective diabetes study (UKPDS): what now or so what? Diabetes Metab Res Rev 1999; 15:65–71.
- Stratton IM, Adler AI, Neil HA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000; 321:405–412.
- Mooradian AD, Chehade J. Implications of the UK Prospective Diabetes Study: questions answered and issues remaining. Drugs Aging. 2000; 16:159–164.
- Nathan DM. Some answers, more controversy, from UKPDS. United Kingdom Prospective Diabetes Study. Lancet. 1998; 352:832–833.
- Srimanunthiphol J, Beddow R, Arakaki R. A review of the United Kingdom Prospective Diabetes Study (UKPDS) and a discussion of the implications for patient care. Hawaii Med J. 2000; 59:295–298.
- Duckworth WC, McCarren M, Abraira C. Glucose control and cardiovascular complications: the VA Diabetes Trial. Diabetes Care. 2001; 24:942–945.
- Abraira C, Colwell JA, Nuttall FQ, et al. Veterans Affairs Cooperative Study on glycemic control and complications in type II diabetes (VA CSDM). Results of the feasibility trial. Veterans Affairs Cooperative Study in Type II Diabetes. Diabetes Care. 1995; 18:1113–1123.
- Gu K, Cowie CC, Harris MI. Mortality in adults with and without diabetes in a national cohort of the U.S. population, 1971–1993. Diabetes Care. 1998; 21:1138–1145. NHANES
- Gaede P, Lund-Andersen H, Parving HH, Pedersen O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med. 2008; 358:580–591.
- Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008; 358:2560–2572.
- Action in Diabetes and Vascular Disease: PreterAx and DiamicroN Modified-Release Controlled Evaluation. Rationale and design of the ADVANCE study: a randomised trial of blood pressure lowering and intensive glucose control in high-risk individuals with type 2 diabetes mellitus. J Hypertens 2001; 19(suppl):S21–S28.
- Abraira C, Duckworth W, McCarren M, et al. Design of the cooperative study on glycemic control and complications in diabetes mellitus type 2: Veterans Affairs Diabetes Trial. J Diabetes Complications. 2003; 17:314–322.
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007; 356:2457–2471.
- American Association of Clinical Endocrinologists. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the management of diabetes mellitus. Endocr Pract 2007; 13(suppl 1):1–68.
- American Diabetes Association. Standards of medical care in diabetes—2008. Diabetes Care 2008; 31(suppl 1):S12–S54.
- Magee MF, Isley WL. Rationale, design, and methods for glycemic control in the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) Trial. Am J Cardiol 2006; 97:20G–30G.
The Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial1–5 was designed primarily to address, in patients with type 2 diabetes at high risk of cardiovascular events, whether intensive glucose control would result in a lower risk of atherosclerotic disease events or death than would standard treatment.
It was widely expected that intensive treatment would confer either modest benefit or, at worst, no benefit. However, the glucose-lowering arm of the trial was terminated early because of a higher mortality rate in the intensively treated group. (The ACCORD trial has two other arms, which concern blood pressure and lipid-lowering, and these are continuing.)
In earlier trials in type 2 diabetes, concerns had been raised about an increased risk of cardiovascular events and possibly death associated with glucose-lowering drugs, hypoglycemia itself, or both, and these were well known when ACCORD was convened. ACCORD was very carefully designed and included careful adjudication of each cardiovascular event and death, including whether hypoglycemia might have been a proximate cause of some sudden deaths.5
Therefore, the surprising result of the higher mortality rate with intensive glycemic control in ACCORD will be fodder for discussion in many arenas over the next several years, and it poses some challenges for physicians and patients in determining treatment goals, as well as for organizations that write clinical practice guidelines (and perhaps organizations involved in pay-for-performance based on these guidelines).
Still, I believe that the ACCORD results should not substantially change our approach to treatment goals in type 2 diabetes, although hemoglobin A1c targets below 6% may not have much added value for cardiovascular risk reduction. The low overall mortality rate in all the arms of the ACCORD trial emphasizes the importance of lifestyle modification, lipid and blood pressure therapy, and encouragement of aspirin use in all patients with type 2 diabetes.
This article reflects my views as a practicing diabetologist and clinical trialist (I was an investigator in the ACCORD trial) with a long-standing interest in clinical trials and in how the results influence clinical practice. The views I express herein may not reflect the views of other ACCORD investigators, the National Heart, Lung, and Blood Institute (NHLBI), the ACCORD trial coordinating center at Wake Forest University, or its data safety and monitoring board.
RISK OF CORONARY DISEASE INCREASES WITH GLUCOSE
Many observational studies6–10 have shown that the risk of cardiovascular disease, especially coronary heart disease, is two to five times higher in people with diabetes mellitus than in people without diabetes. The risk appears to be continuous, so the higher one’s glucose or hemoglobin A1c, the higher the risk.6 This risk even extends to glucose values well below the threshold values currently used to diagnose diabetes mellitus.6 Since there is no glucose threshold for coronary heart disease, the term dysglycemia (rather than hyperglycemia) has been proposed to note the relationship between glucose and coronary heart disease. (The glucose threshold for microvascular complications of diabetes, such as retinopathy and nephropathy, appears to be between 110 and 126 mg/dL).
The clustering of multiple coronary risk factors such as obesity, dyslipidemia, and hypertension has always raised the question of whether glucose is a culprit in coronary risk or whether it simply “runs in bad company.”
EARLIER CLINICAL TRIALS SUGGEST INTENSIVE TREATMENT RAISES RISK
Even though it has been widely believed that intensive glucose-lowering would reduce cardiovascular risk in type 2 diabetes, there have been hints in previous studies that some intensive-treatment regimens might increase risk.
Two large randomized clinical trials and one small one (discussed below) addressed whether glucose control would reduce the risk of atherosclerotic vascular disease events. In each of them, an increased risk of cardiovascular events and possibly of death was seen in at least one intensively treated group.
In the following discussion, I have calculated all of the death rates as the number of deaths per 1,000 patients per year, based on published study results. In this way, we can compare the rates in the various studies (including ACCORD), regardless of the trial duration.
The university group Diabetes Program: Controvery about tolbutamide therapy
The University Group Diabetes Program (UGDP)11–16 included about 1,000 participants randomized to five treatments: tolbutamide (Orinase, a sulfonylurea), insulin in a fixed dose based on body weight, insulin in adjusted doses based on fasting glucose levels, placebo, and (later) phenformin.
In the 1970s, when the UGDP was carried out, randomized clinical trials were uncommon. Like other trials from that era, the UGDP was underpowered by today’s standards and did not have a data safety and monitoring board.
Rates of cardiovascular events and deaths (per 1,000 patient-years):
- 25 (tolbutamide group)
- 12 (placebo group).
The two insulin groups did not differ from the placebo group in their rates of cardiovascular events or death.15 The tolbutamide arm was stopped, and the ensuing controversy about how to interpret the trial results lasted for more than a decade. It also resulted in a black-box warning for tolbutamide and all subsequent sulfonylureas.
United Kingdom Prospective Diabetes Study: Method of glucose-lowering an issue
The United Kingdom Prospective Diabetes Study (UKPDS)17–27 was launched in 1977. A cohort of 5,102 patients (mean age 54 years) with newly diagnosed type 2 diabetes mellitus followed a “prudent diet” for the first 3 to 4 months. Then, if their fasting glucose levels were in the range of 6.1 to 15 mmol/L (110–270 mg/dL), they were randomized to receive various treatments.
Patients who were not obese were randomized to receive either intensive treatment or conventional treatment. The intensive-treatment group received either insulin or a sulfonylurea (chlorpropamide [Diabinese], glibenclamide, or glipizide [Glucotrol]); the conventional-treatment group received diet therapy. The sulfonylurea arm was included partly to address the UGDP results.
Patients who were obese were randomized to receive one of three treatments: intensive treatment (with the agents listed above), conventional treatment, or metformin (Fortamet, Glucophage).
The mean in-trial hemoglobin A1c level in the intensive-treatment group was 7.0%, compared with 7.9% in the conventional-treatment group.
After a mean follow-up of more than 10 years, the incidence of myocardial infarction was 16% lower in the intensive-treatment group, but the difference was not statistically significant (P = .052).
Rates of death from all causes among nonobese subjects (per 1,000 patient-years):
- 18.2–20.5 (intensive-treatment group)
- 19.9 (conventional-treatment group).
In the obese patients who received metformin, the incidence of myocardial infarction was lower than in the conventional-treatment group but not the intensive-treatment group.
Rates of death among obese patients (per 1,000 patient-years):
- 13.5 (metformin group)
- 18.9 (intensive-treatment group)
- 20.6 (conventional-treatment group).
However, a small subset (n = 587) of the original group assigned to sulfonylurea therapy whose glycemic control deteriorated during the trial were rerandomized to continue to receive a sulfonylurea alone or to have metformin added. There was a statistically significantly higher rate of cardiovascular events and a nonsignificantly higher rate of total mortality in the metformin-plus-sulfonylurea group (30.3 per 1,000 patient-years) than in the sulfonylurea-only group (19.1 per 1,000 patient-years).
These data suggested that the way glucose-lowering was achieved might be as important as the glucose levels actually achieved. However, no definite conclusions could be drawn.
In an editorial on the UKPDS, Nathan26 made a comment that may have been prescient in terms of the ACCORD trial: “Professional organizations will now scramble to decide how to translate the UKPDS results … Whether the UKPDS firmly establishes the choice of any one therapy…or any combination of therapies for the long-term treatment of type 2 diabetes is more questionable.”26
Veterans Administration feasibility study
A Veterans Administration feasibility study28,29 included 153 men (mean age 60) with type 2 diabetes (mean duration 7.8 years) who received either conventional therapy (a single daily dose of insulin) or intensive therapy (multiple doses of insulin plus a sulfonylurea). Over a mean of 27 months, the intensive-therapy group achieved a hemoglobin A1c level that was 2 percentage points lower than in the conventional-therapy group.
At 2.25 years of follow-up, cardiovascular events had occurred in 24 (24%) of the intensive-therapy group and in 16 (20%) of the standard-therapy group (P = .10).
Rates of death from all causes (per 1,000 patient-years):
- 28.9 (intensive-treatment group)
- 17.5 (conventional-treatment group).
ACCORD TRIAL DESIGN
The primary outcome measured was the combined incidence of nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes. Secondary outcomes included death from any cause. The study is also evaluating the effect of intensive treatment on microvascular disease, hypoglycemia, cognition, quality of life, and cost-effectiveness.
The ACCORD study was designed to have 89% power to detect a 15% treatment effect of intensive glycemic control compared with standard glycemic control for the primary end point.
ACCORD RESULTS
Rates of death from any cause (per 1,000 patient-years):
- 14 (intensive-treatment group)
- 11 (standard-treatment group).
In the analyses available at the time that this study arm closed, the excess mortality was not attributable to any particular treatment regimen. In particular, rosiglitazone (Avandia) use did not contribute to the excess mortality. (Of note, 91.2% of the intensive-treatment group and 57.5% of the conventional-treatment group had been treated with rosiglitazone, with more than 19,000 patient-years of rosiglitazone exposure). The excess mortality was also not attributable to hypoglycemia immediately proximate to the death.
The ACCORD trial’s data safety and monitoring board recommended that this arm of the study be discontinued for safety reasons, and this recommendation was accepted by the NHLBI project office. All participants were notified by letter before the trial results were announced publicly, and all intensive-therapy group participants are now being treated by the protocol used in the standard-therapy group.1
FEWER DEATHS IN ACCORD THAN IN OTHER STUDIES IN DIABETES
The mortality rates in both arms of ACCORD were much lower than in other observational studies and clinical trials in type 2 diabetes.
The National Health and Nutrition Education Survey (NHANES),30 conducted from 1971 to 1975, included 14,374 people with diabetes between the ages of 25 and 74. Many of them were younger than the ACCORD patients, but two NHANES age-groups overlapped the ACCORD cohort. Rates of death from any cause at 22 years (per 1,000 patient-years):
- 39.7 (ages 45–64)
- 89.7 (ages 65–74).
The NHANES cohort would not have been treated as vigorously for coronary risk and other common causes of death.
UGDP, UKPDS. Death rates in the glucose-lowering trials of type 2 diabetes mellitus cited above were typically in the range of 20 deaths per 1,000 patient-years but were as high as 30 deaths per 1,000 patient-years in the UGDP tolbutamide group16 and the UK-PDS sulfonylurea-plus-metformin group.20,22,26
Steno-2.31 Half of 160 patients with type 2 diabetes were randomized to intensive strategies for controlling glucose, lipids, and blood pressure and for taking aspirin and angiotensin-converting enzyme inhibitors and following a healthy lifestyle. The other half received conventional therapy. Even in the intensive-treatment group, the mortality rate at 13 years was higher than in ACCORD. Rates of death from any cause (per 1,000 patient years):
- 22.5 (intensive-treatment group)
- 37.6 (conventional-treatment group).
After the ACCORD results were presented, two other trials addressing the question of whether lower hemoglobin A1c would reduce cardiovascular risk in type 2 diabetes have reported their outcomes:
The ADVANCE trial (Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation),32,33 with 11,140 patients, had a target hemoglobin A1c of 6.5% in an intensive-treatment group and 7.3% in a usual-treatment group. The intensive-treatment group showed no difference in the rates of major macrovascular events (HR 0.94, 95% CI 0.84–1.06, P = .32) or all-cause mortality (HR 0.93, 95% CI 0.83–1.06, P = .32). The overall death rate in ADVANCE (about 18 deaths per 1,000 patient-years) was higher than in ACCORD.
The Veterans Administration Diabetes Trial included 1,791 patients.34 Like the ADVANCE trial, it also found no difference in major cardiovascular outcomes (HR 0.868, P = .11) or cardiovascular mortality rates (HR 1.258, P = .36) with intensive therapy vs conventional therapy, ie, achieved hemoglobin A1c levels of 6.9% vs 8.4% (presented at the American Diabetes Association 2008 Scientific Sessions). Hypoglycemia was associated with an increased risk of death in the standard-treatment group.
An analysis suggested that patients with a shorter duration of diabetes may have had cardiovascular benefit from intensive glucose-lowering, while those who had had it longer may have had increased risk associated with the more intensive therapy. The rate of death from all causes appears to have been higher than in ACCORD, but this could not be determined accurately from the presentations.
Comment. Thus, the ACCORD cohort as a whole has had strikingly lower death rates than in these other studies. The fact that all participants had lower glucose levels on therapy than at baseline may possibly contribute to these lower death rates. In addition, all ACCORD participants in the lipid arm received a statin; all participants in the blood pressure arm had their blood pressure lowered to levels below those commonly seen in clinical practice; participants were encouraged to exercise regularly; most participants were given diet instruction; and other healthy behaviors such as aspirin use, regular follow-up with primary care physicians, and recommendations about smoking were encouraged throughout the study. These comprehensive strategies may represent better care and thus result in lower death rates than in other studies.
POSSIBLE EXPLANATIONS FOR THE ACCORD OUTCOMES
The ACCORD trial has already stimulated fierce debate about the reasons for the higher mortality rate in the intensive-treatment group. With longer follow-up, some new risk factors for death may be identified that are not evident in the analyses of the current 460 deaths. What follows are some of my thoughts, with the caveat that they are not confirmed (supported statistically) by any currently available analyses from ACCORD.
It seems unlikely that lower glucose values as reflected by lower hemoglobin A1c values in the intensive-treatment group are an a priori explanation for the observed differences in mortality rates—especially since the mortality rates were lower than in the NHANES and clinical trial data sets cited above. If we assume that a type 1 statistical error (finding a difference where no difference actually exists) does not explain the findings, then at least four reasonable postulates exist:
Hypoglycemia may have some adverse effect, either acutely or from recurrent events that trigger a catecholamine response with associated risk for arrhythmia or increased coronary heart disease risk. However, the investigators analyzed each death to determine whether hypoglycemia was a contributing cause, and they found no statistically significant relationship between hypoglycemia and death in the intensive-treatment group.
Weight gain is common with intensive therapy. Obesity may be associated with greater cytokine production, higher concentrations of clotting factors, higher levels of free fatty acids, and other potential contributors to the risk of coronary heart disease and death. Currently, the ACCORD analyses do not suggest that weight gain explains the higher death rate.
Medications such as rosiglitazone, sulfonylureas, and the combination of a sulfonylurea plus metformin have been previously associated with increased death rates in some observational and intervention trials. These studies had some serious methodologic limitations (eg, absence of risk adjustment, events not adjudicated, small study cohorts, wide variation in study cohort characteristics) and small numbers of events.11–13,16,26,35 ACCORD analyses have not shown that any single glucose-lowering agent—including rosiglitazone—or combination of agents explains the death rates.
The stress of maintaining glycemic control has been speculated to have in some way contributed to an increased risk. To achieve intensive control, patients had to have frequent contact with their health care providers, they were often told that their hemoglobin A1c values were “too high” even when they were well below those in the American Diabetes Association guidelines, and they had to follow complex glucose-lowering regimens.
Semiquantitative measures of overall attitudes about health exist (eg, the “Feeling Thermometer” scale), but stress was not measured quantitatively in the ACCORD trial.
IMPLICATIONS OF ACCORD
In practice, most clinicians believe that the target glucose level in patients with type 2 diabetes should be as low as safely possible. This approach does not need to be modified on the basis of current information from ACCORD.
To be safe, regimens should be associated with a low risk of hypoglycemia and a low risk of weight gain. Use of combinations of medications that work by different mechanisms is still prudent. Agents should be used that may have favorable effects on other cardiovascular risk factors (eg, lipids, blood pressure, visceral fat).
Hemoglobin A1c targets below 7% are not precluded in all patients on the basis of the ACCORD results, though values lower than 6% may not have much added benefit for cardiovascular risk reduction. We should note that hemoglobin A1c was reduced in all ACCORD participants and that death rates were lower than in many other type 2 diabetic cohorts. Pending data on other outcomes in ACCORD (nephropathy, retinopathy, dementia, fracture risk), I believe it is premature for organizations to change their proposed hemoglobin A1c targets,36,37 as none have proposed values as low as the target in the ACCORD intensive-treatment group. At present, no class of glucose-lowering agents needs to be excluded from consideration on the basis of the ACCORD data.
The overall low rates of death in this population at high risk of coronary heart disease deserve comment. Not only are they lower than in other glucose-lowering trials, but they are also lower than in a number of studies of mortality in diabetes cohorts. As noted above, multiple risk factors for coronary heart disease and death were (and are) addressed in the ACCORD study participants, including repeated recommendation for lifestyle modification, intervention arms with lipid and blood pressure therapy, encouragement of aspirin use, and regular follow-up with health care providers for risk factors not managed by the ACCORD trial protocol. It is likely that multiple approaches to reducing the risk of cardiovascular disease contributed to this low mortality rate and that similar approaches will reduce the risk of coronary disease and death in regular clinical practice.
The ACCORD lipid and blood pressure arms are continuing, with results expected in 2010. The future results from ACCORD as well as from several glucose-lowering trials currently in progress (ADVANCE,32,33 Veteran’s Administration,34 Bypass Angioplasty Revascularization Investigation 2 Diabetes [BARI-2D]38) will likely help refine our understanding of the effects of glucose-lowering, glucose-lowering strategies and targets, and multiple interventions on coronary events and all-cause mortality.
For now, any strategy that lowers glucose and is associated with a low risk of hypoglycemia and does not cause excessive weight gain should be considered appropriate in patients with type 2 diabetes.
The Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial1–5 was designed primarily to address, in patients with type 2 diabetes at high risk of cardiovascular events, whether intensive glucose control would result in a lower risk of atherosclerotic disease events or death than would standard treatment.
It was widely expected that intensive treatment would confer either modest benefit or, at worst, no benefit. However, the glucose-lowering arm of the trial was terminated early because of a higher mortality rate in the intensively treated group. (The ACCORD trial has two other arms, which concern blood pressure and lipid-lowering, and these are continuing.)
In earlier trials in type 2 diabetes, concerns had been raised about an increased risk of cardiovascular events and possibly death associated with glucose-lowering drugs, hypoglycemia itself, or both, and these were well known when ACCORD was convened. ACCORD was very carefully designed and included careful adjudication of each cardiovascular event and death, including whether hypoglycemia might have been a proximate cause of some sudden deaths.5
Therefore, the surprising result of the higher mortality rate with intensive glycemic control in ACCORD will be fodder for discussion in many arenas over the next several years, and it poses some challenges for physicians and patients in determining treatment goals, as well as for organizations that write clinical practice guidelines (and perhaps organizations involved in pay-for-performance based on these guidelines).
Still, I believe that the ACCORD results should not substantially change our approach to treatment goals in type 2 diabetes, although hemoglobin A1c targets below 6% may not have much added value for cardiovascular risk reduction. The low overall mortality rate in all the arms of the ACCORD trial emphasizes the importance of lifestyle modification, lipid and blood pressure therapy, and encouragement of aspirin use in all patients with type 2 diabetes.
This article reflects my views as a practicing diabetologist and clinical trialist (I was an investigator in the ACCORD trial) with a long-standing interest in clinical trials and in how the results influence clinical practice. The views I express herein may not reflect the views of other ACCORD investigators, the National Heart, Lung, and Blood Institute (NHLBI), the ACCORD trial coordinating center at Wake Forest University, or its data safety and monitoring board.
RISK OF CORONARY DISEASE INCREASES WITH GLUCOSE
Many observational studies6–10 have shown that the risk of cardiovascular disease, especially coronary heart disease, is two to five times higher in people with diabetes mellitus than in people without diabetes. The risk appears to be continuous, so the higher one’s glucose or hemoglobin A1c, the higher the risk.6 This risk even extends to glucose values well below the threshold values currently used to diagnose diabetes mellitus.6 Since there is no glucose threshold for coronary heart disease, the term dysglycemia (rather than hyperglycemia) has been proposed to note the relationship between glucose and coronary heart disease. (The glucose threshold for microvascular complications of diabetes, such as retinopathy and nephropathy, appears to be between 110 and 126 mg/dL).
The clustering of multiple coronary risk factors such as obesity, dyslipidemia, and hypertension has always raised the question of whether glucose is a culprit in coronary risk or whether it simply “runs in bad company.”
EARLIER CLINICAL TRIALS SUGGEST INTENSIVE TREATMENT RAISES RISK
Even though it has been widely believed that intensive glucose-lowering would reduce cardiovascular risk in type 2 diabetes, there have been hints in previous studies that some intensive-treatment regimens might increase risk.
Two large randomized clinical trials and one small one (discussed below) addressed whether glucose control would reduce the risk of atherosclerotic vascular disease events. In each of them, an increased risk of cardiovascular events and possibly of death was seen in at least one intensively treated group.
In the following discussion, I have calculated all of the death rates as the number of deaths per 1,000 patients per year, based on published study results. In this way, we can compare the rates in the various studies (including ACCORD), regardless of the trial duration.
The university group Diabetes Program: Controvery about tolbutamide therapy
The University Group Diabetes Program (UGDP)11–16 included about 1,000 participants randomized to five treatments: tolbutamide (Orinase, a sulfonylurea), insulin in a fixed dose based on body weight, insulin in adjusted doses based on fasting glucose levels, placebo, and (later) phenformin.
In the 1970s, when the UGDP was carried out, randomized clinical trials were uncommon. Like other trials from that era, the UGDP was underpowered by today’s standards and did not have a data safety and monitoring board.
Rates of cardiovascular events and deaths (per 1,000 patient-years):
- 25 (tolbutamide group)
- 12 (placebo group).
The two insulin groups did not differ from the placebo group in their rates of cardiovascular events or death.15 The tolbutamide arm was stopped, and the ensuing controversy about how to interpret the trial results lasted for more than a decade. It also resulted in a black-box warning for tolbutamide and all subsequent sulfonylureas.
United Kingdom Prospective Diabetes Study: Method of glucose-lowering an issue
The United Kingdom Prospective Diabetes Study (UKPDS)17–27 was launched in 1977. A cohort of 5,102 patients (mean age 54 years) with newly diagnosed type 2 diabetes mellitus followed a “prudent diet” for the first 3 to 4 months. Then, if their fasting glucose levels were in the range of 6.1 to 15 mmol/L (110–270 mg/dL), they were randomized to receive various treatments.
Patients who were not obese were randomized to receive either intensive treatment or conventional treatment. The intensive-treatment group received either insulin or a sulfonylurea (chlorpropamide [Diabinese], glibenclamide, or glipizide [Glucotrol]); the conventional-treatment group received diet therapy. The sulfonylurea arm was included partly to address the UGDP results.
Patients who were obese were randomized to receive one of three treatments: intensive treatment (with the agents listed above), conventional treatment, or metformin (Fortamet, Glucophage).
The mean in-trial hemoglobin A1c level in the intensive-treatment group was 7.0%, compared with 7.9% in the conventional-treatment group.
After a mean follow-up of more than 10 years, the incidence of myocardial infarction was 16% lower in the intensive-treatment group, but the difference was not statistically significant (P = .052).
Rates of death from all causes among nonobese subjects (per 1,000 patient-years):
- 18.2–20.5 (intensive-treatment group)
- 19.9 (conventional-treatment group).
In the obese patients who received metformin, the incidence of myocardial infarction was lower than in the conventional-treatment group but not the intensive-treatment group.
Rates of death among obese patients (per 1,000 patient-years):
- 13.5 (metformin group)
- 18.9 (intensive-treatment group)
- 20.6 (conventional-treatment group).
However, a small subset (n = 587) of the original group assigned to sulfonylurea therapy whose glycemic control deteriorated during the trial were rerandomized to continue to receive a sulfonylurea alone or to have metformin added. There was a statistically significantly higher rate of cardiovascular events and a nonsignificantly higher rate of total mortality in the metformin-plus-sulfonylurea group (30.3 per 1,000 patient-years) than in the sulfonylurea-only group (19.1 per 1,000 patient-years).
These data suggested that the way glucose-lowering was achieved might be as important as the glucose levels actually achieved. However, no definite conclusions could be drawn.
In an editorial on the UKPDS, Nathan26 made a comment that may have been prescient in terms of the ACCORD trial: “Professional organizations will now scramble to decide how to translate the UKPDS results … Whether the UKPDS firmly establishes the choice of any one therapy…or any combination of therapies for the long-term treatment of type 2 diabetes is more questionable.”26
Veterans Administration feasibility study
A Veterans Administration feasibility study28,29 included 153 men (mean age 60) with type 2 diabetes (mean duration 7.8 years) who received either conventional therapy (a single daily dose of insulin) or intensive therapy (multiple doses of insulin plus a sulfonylurea). Over a mean of 27 months, the intensive-therapy group achieved a hemoglobin A1c level that was 2 percentage points lower than in the conventional-therapy group.
At 2.25 years of follow-up, cardiovascular events had occurred in 24 (24%) of the intensive-therapy group and in 16 (20%) of the standard-therapy group (P = .10).
Rates of death from all causes (per 1,000 patient-years):
- 28.9 (intensive-treatment group)
- 17.5 (conventional-treatment group).
ACCORD TRIAL DESIGN
The primary outcome measured was the combined incidence of nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes. Secondary outcomes included death from any cause. The study is also evaluating the effect of intensive treatment on microvascular disease, hypoglycemia, cognition, quality of life, and cost-effectiveness.
The ACCORD study was designed to have 89% power to detect a 15% treatment effect of intensive glycemic control compared with standard glycemic control for the primary end point.
ACCORD RESULTS
Rates of death from any cause (per 1,000 patient-years):
- 14 (intensive-treatment group)
- 11 (standard-treatment group).
In the analyses available at the time that this study arm closed, the excess mortality was not attributable to any particular treatment regimen. In particular, rosiglitazone (Avandia) use did not contribute to the excess mortality. (Of note, 91.2% of the intensive-treatment group and 57.5% of the conventional-treatment group had been treated with rosiglitazone, with more than 19,000 patient-years of rosiglitazone exposure). The excess mortality was also not attributable to hypoglycemia immediately proximate to the death.
The ACCORD trial’s data safety and monitoring board recommended that this arm of the study be discontinued for safety reasons, and this recommendation was accepted by the NHLBI project office. All participants were notified by letter before the trial results were announced publicly, and all intensive-therapy group participants are now being treated by the protocol used in the standard-therapy group.1
FEWER DEATHS IN ACCORD THAN IN OTHER STUDIES IN DIABETES
The mortality rates in both arms of ACCORD were much lower than in other observational studies and clinical trials in type 2 diabetes.
The National Health and Nutrition Education Survey (NHANES),30 conducted from 1971 to 1975, included 14,374 people with diabetes between the ages of 25 and 74. Many of them were younger than the ACCORD patients, but two NHANES age-groups overlapped the ACCORD cohort. Rates of death from any cause at 22 years (per 1,000 patient-years):
- 39.7 (ages 45–64)
- 89.7 (ages 65–74).
The NHANES cohort would not have been treated as vigorously for coronary risk and other common causes of death.
UGDP, UKPDS. Death rates in the glucose-lowering trials of type 2 diabetes mellitus cited above were typically in the range of 20 deaths per 1,000 patient-years but were as high as 30 deaths per 1,000 patient-years in the UGDP tolbutamide group16 and the UK-PDS sulfonylurea-plus-metformin group.20,22,26
Steno-2.31 Half of 160 patients with type 2 diabetes were randomized to intensive strategies for controlling glucose, lipids, and blood pressure and for taking aspirin and angiotensin-converting enzyme inhibitors and following a healthy lifestyle. The other half received conventional therapy. Even in the intensive-treatment group, the mortality rate at 13 years was higher than in ACCORD. Rates of death from any cause (per 1,000 patient years):
- 22.5 (intensive-treatment group)
- 37.6 (conventional-treatment group).
After the ACCORD results were presented, two other trials addressing the question of whether lower hemoglobin A1c would reduce cardiovascular risk in type 2 diabetes have reported their outcomes:
The ADVANCE trial (Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation),32,33 with 11,140 patients, had a target hemoglobin A1c of 6.5% in an intensive-treatment group and 7.3% in a usual-treatment group. The intensive-treatment group showed no difference in the rates of major macrovascular events (HR 0.94, 95% CI 0.84–1.06, P = .32) or all-cause mortality (HR 0.93, 95% CI 0.83–1.06, P = .32). The overall death rate in ADVANCE (about 18 deaths per 1,000 patient-years) was higher than in ACCORD.
The Veterans Administration Diabetes Trial included 1,791 patients.34 Like the ADVANCE trial, it also found no difference in major cardiovascular outcomes (HR 0.868, P = .11) or cardiovascular mortality rates (HR 1.258, P = .36) with intensive therapy vs conventional therapy, ie, achieved hemoglobin A1c levels of 6.9% vs 8.4% (presented at the American Diabetes Association 2008 Scientific Sessions). Hypoglycemia was associated with an increased risk of death in the standard-treatment group.
An analysis suggested that patients with a shorter duration of diabetes may have had cardiovascular benefit from intensive glucose-lowering, while those who had had it longer may have had increased risk associated with the more intensive therapy. The rate of death from all causes appears to have been higher than in ACCORD, but this could not be determined accurately from the presentations.
Comment. Thus, the ACCORD cohort as a whole has had strikingly lower death rates than in these other studies. The fact that all participants had lower glucose levels on therapy than at baseline may possibly contribute to these lower death rates. In addition, all ACCORD participants in the lipid arm received a statin; all participants in the blood pressure arm had their blood pressure lowered to levels below those commonly seen in clinical practice; participants were encouraged to exercise regularly; most participants were given diet instruction; and other healthy behaviors such as aspirin use, regular follow-up with primary care physicians, and recommendations about smoking were encouraged throughout the study. These comprehensive strategies may represent better care and thus result in lower death rates than in other studies.
POSSIBLE EXPLANATIONS FOR THE ACCORD OUTCOMES
The ACCORD trial has already stimulated fierce debate about the reasons for the higher mortality rate in the intensive-treatment group. With longer follow-up, some new risk factors for death may be identified that are not evident in the analyses of the current 460 deaths. What follows are some of my thoughts, with the caveat that they are not confirmed (supported statistically) by any currently available analyses from ACCORD.
It seems unlikely that lower glucose values as reflected by lower hemoglobin A1c values in the intensive-treatment group are an a priori explanation for the observed differences in mortality rates—especially since the mortality rates were lower than in the NHANES and clinical trial data sets cited above. If we assume that a type 1 statistical error (finding a difference where no difference actually exists) does not explain the findings, then at least four reasonable postulates exist:
Hypoglycemia may have some adverse effect, either acutely or from recurrent events that trigger a catecholamine response with associated risk for arrhythmia or increased coronary heart disease risk. However, the investigators analyzed each death to determine whether hypoglycemia was a contributing cause, and they found no statistically significant relationship between hypoglycemia and death in the intensive-treatment group.
Weight gain is common with intensive therapy. Obesity may be associated with greater cytokine production, higher concentrations of clotting factors, higher levels of free fatty acids, and other potential contributors to the risk of coronary heart disease and death. Currently, the ACCORD analyses do not suggest that weight gain explains the higher death rate.
Medications such as rosiglitazone, sulfonylureas, and the combination of a sulfonylurea plus metformin have been previously associated with increased death rates in some observational and intervention trials. These studies had some serious methodologic limitations (eg, absence of risk adjustment, events not adjudicated, small study cohorts, wide variation in study cohort characteristics) and small numbers of events.11–13,16,26,35 ACCORD analyses have not shown that any single glucose-lowering agent—including rosiglitazone—or combination of agents explains the death rates.
The stress of maintaining glycemic control has been speculated to have in some way contributed to an increased risk. To achieve intensive control, patients had to have frequent contact with their health care providers, they were often told that their hemoglobin A1c values were “too high” even when they were well below those in the American Diabetes Association guidelines, and they had to follow complex glucose-lowering regimens.
Semiquantitative measures of overall attitudes about health exist (eg, the “Feeling Thermometer” scale), but stress was not measured quantitatively in the ACCORD trial.
IMPLICATIONS OF ACCORD
In practice, most clinicians believe that the target glucose level in patients with type 2 diabetes should be as low as safely possible. This approach does not need to be modified on the basis of current information from ACCORD.
To be safe, regimens should be associated with a low risk of hypoglycemia and a low risk of weight gain. Use of combinations of medications that work by different mechanisms is still prudent. Agents should be used that may have favorable effects on other cardiovascular risk factors (eg, lipids, blood pressure, visceral fat).
Hemoglobin A1c targets below 7% are not precluded in all patients on the basis of the ACCORD results, though values lower than 6% may not have much added benefit for cardiovascular risk reduction. We should note that hemoglobin A1c was reduced in all ACCORD participants and that death rates were lower than in many other type 2 diabetic cohorts. Pending data on other outcomes in ACCORD (nephropathy, retinopathy, dementia, fracture risk), I believe it is premature for organizations to change their proposed hemoglobin A1c targets,36,37 as none have proposed values as low as the target in the ACCORD intensive-treatment group. At present, no class of glucose-lowering agents needs to be excluded from consideration on the basis of the ACCORD data.
The overall low rates of death in this population at high risk of coronary heart disease deserve comment. Not only are they lower than in other glucose-lowering trials, but they are also lower than in a number of studies of mortality in diabetes cohorts. As noted above, multiple risk factors for coronary heart disease and death were (and are) addressed in the ACCORD study participants, including repeated recommendation for lifestyle modification, intervention arms with lipid and blood pressure therapy, encouragement of aspirin use, and regular follow-up with health care providers for risk factors not managed by the ACCORD trial protocol. It is likely that multiple approaches to reducing the risk of cardiovascular disease contributed to this low mortality rate and that similar approaches will reduce the risk of coronary disease and death in regular clinical practice.
The ACCORD lipid and blood pressure arms are continuing, with results expected in 2010. The future results from ACCORD as well as from several glucose-lowering trials currently in progress (ADVANCE,32,33 Veteran’s Administration,34 Bypass Angioplasty Revascularization Investigation 2 Diabetes [BARI-2D]38) will likely help refine our understanding of the effects of glucose-lowering, glucose-lowering strategies and targets, and multiple interventions on coronary events and all-cause mortality.
For now, any strategy that lowers glucose and is associated with a low risk of hypoglycemia and does not cause excessive weight gain should be considered appropriate in patients with type 2 diabetes.
- Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008; 358:2545–2559.
- Goff DC, Gerstein HC, Ginsberg HN, et al. Prevention of cardiovascular disease in persons with type 2 diabetes mellitus: current knowledge and rationale for the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007; 99:4i–20i.
- Buse JB, Bigger JT, Byington RP, et al. Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial: design and methods. Am J Cardiol 2007; 99:21i–33i.
- Gerstein HC, Riddle MC, Kendall DM, et al. Glycemia treatment strategies in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007; 99:34i–43i.
- Bonds DE, Kurashige EM, Bergenstal R, et al. Severe hypoglycemia monitoring and risk management procedures in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007; 99:80i–89i.
- Gerstein HC. Dysglycemia, not just diabetes, is a continuous risk factor for cardiovascular disease. Evid Based Cardiovasc Med. 1997; 1:87–88.
- Gerstein HC, Pais P, Pogue J, Yusuf S. Relationship of glucose and insulin levels to the risk of myocardial infarction: a case-control study. J Am Coll Cardiol. 1999; 33:612–619.
- Gerstein HC, Capes SE. Dysglycemia: a key cardiovascular risk factor. Semin Vasc Med. 2002; 2:165–174.
- Gerstein HC, Santaguida P, Raina P, et al. Annual incidence and relative risk of diabetes in people with various categories of dysglycemia: a systematic overview and meta-analysis of prospective studies. Diabetes Res Clin Pract. 2007; 78:305–312.
- American Diabetes Association. Role of cardiovascular risk factors in prevention and treatment of macrovascular disease in diabetes. Diabetes Care. 1989; 12:573–579.
- Schor S. The University Group Diabetes Program. A statistician looks at the mortality results. JAMA. 1971; 217:1671–1675.
- Cornfield JThe University Group Diabetes Program. A further statistical analysis of the mortality findings. JAMA. 1971; 217:1676–1687.
- Feinstein AR. Clinical biostatistics. 8. An analytic appraisal of the University Group Diabetes Program (UGDP) study. Clin Pharmacol Ther. 1971; 12:167–191.
- The University Group Diabetes Program. A study of the effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. V. Evaluation of pheniformin therapy. Diabetes 1975; 24( suppl 1):65–184.
- Knatterud GL, Klimt CR, Levin ME, Jacobson ME, Goldner MG. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. VII. Mortality and selected nonfatal events with insulin treatment. JAMA. 1978; 240:37–42.
- Schwartz TB, Meinert CL. The UGDP controversy: thirty-four years of contentious ambiguity laid to rest. Perspect Biol Med. 2004; 47:564–574.
- Turner RC, Holman RR. Lessons from UK Prospective Diabetes Study. Diabetes Res Clin Pract 1995; 28( suppl):S151–S157.
- UKPDS Research Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998; 352:854–865.
- UKPDS Study Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998; 352:837–853.
- UK Prospective Diabetes Study Group. UKPDS 28: a randomized trial of efficacy of early addition of metformin in sulfonylurea-treated type 2 diabetes. Diabetes Care. 1998; 21:87–92.
- Bretzel RG, Voigt K, Schatz H. The United Kingdom Prospective Diabetes Study (UKPDS) implications for the pharmacotherapy of type 2 diabetes mellitus. Exp Clin Endocrinol Diabetes. 1998; 106:369–372.
- Turner RC, Cull CA, Frighi V, Holman RR. Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. JAMA. 1999; 281:2005–2012.
- Leslie RD. United Kingdom prospective diabetes study (UKPDS): what now or so what? Diabetes Metab Res Rev 1999; 15:65–71.
- Stratton IM, Adler AI, Neil HA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000; 321:405–412.
- Mooradian AD, Chehade J. Implications of the UK Prospective Diabetes Study: questions answered and issues remaining. Drugs Aging. 2000; 16:159–164.
- Nathan DM. Some answers, more controversy, from UKPDS. United Kingdom Prospective Diabetes Study. Lancet. 1998; 352:832–833.
- Srimanunthiphol J, Beddow R, Arakaki R. A review of the United Kingdom Prospective Diabetes Study (UKPDS) and a discussion of the implications for patient care. Hawaii Med J. 2000; 59:295–298.
- Duckworth WC, McCarren M, Abraira C. Glucose control and cardiovascular complications: the VA Diabetes Trial. Diabetes Care. 2001; 24:942–945.
- Abraira C, Colwell JA, Nuttall FQ, et al. Veterans Affairs Cooperative Study on glycemic control and complications in type II diabetes (VA CSDM). Results of the feasibility trial. Veterans Affairs Cooperative Study in Type II Diabetes. Diabetes Care. 1995; 18:1113–1123.
- Gu K, Cowie CC, Harris MI. Mortality in adults with and without diabetes in a national cohort of the U.S. population, 1971–1993. Diabetes Care. 1998; 21:1138–1145. NHANES
- Gaede P, Lund-Andersen H, Parving HH, Pedersen O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med. 2008; 358:580–591.
- Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008; 358:2560–2572.
- Action in Diabetes and Vascular Disease: PreterAx and DiamicroN Modified-Release Controlled Evaluation. Rationale and design of the ADVANCE study: a randomised trial of blood pressure lowering and intensive glucose control in high-risk individuals with type 2 diabetes mellitus. J Hypertens 2001; 19(suppl):S21–S28.
- Abraira C, Duckworth W, McCarren M, et al. Design of the cooperative study on glycemic control and complications in diabetes mellitus type 2: Veterans Affairs Diabetes Trial. J Diabetes Complications. 2003; 17:314–322.
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007; 356:2457–2471.
- American Association of Clinical Endocrinologists. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the management of diabetes mellitus. Endocr Pract 2007; 13(suppl 1):1–68.
- American Diabetes Association. Standards of medical care in diabetes—2008. Diabetes Care 2008; 31(suppl 1):S12–S54.
- Magee MF, Isley WL. Rationale, design, and methods for glycemic control in the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) Trial. Am J Cardiol 2006; 97:20G–30G.
- Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008; 358:2545–2559.
- Goff DC, Gerstein HC, Ginsberg HN, et al. Prevention of cardiovascular disease in persons with type 2 diabetes mellitus: current knowledge and rationale for the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007; 99:4i–20i.
- Buse JB, Bigger JT, Byington RP, et al. Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial: design and methods. Am J Cardiol 2007; 99:21i–33i.
- Gerstein HC, Riddle MC, Kendall DM, et al. Glycemia treatment strategies in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007; 99:34i–43i.
- Bonds DE, Kurashige EM, Bergenstal R, et al. Severe hypoglycemia monitoring and risk management procedures in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007; 99:80i–89i.
- Gerstein HC. Dysglycemia, not just diabetes, is a continuous risk factor for cardiovascular disease. Evid Based Cardiovasc Med. 1997; 1:87–88.
- Gerstein HC, Pais P, Pogue J, Yusuf S. Relationship of glucose and insulin levels to the risk of myocardial infarction: a case-control study. J Am Coll Cardiol. 1999; 33:612–619.
- Gerstein HC, Capes SE. Dysglycemia: a key cardiovascular risk factor. Semin Vasc Med. 2002; 2:165–174.
- Gerstein HC, Santaguida P, Raina P, et al. Annual incidence and relative risk of diabetes in people with various categories of dysglycemia: a systematic overview and meta-analysis of prospective studies. Diabetes Res Clin Pract. 2007; 78:305–312.
- American Diabetes Association. Role of cardiovascular risk factors in prevention and treatment of macrovascular disease in diabetes. Diabetes Care. 1989; 12:573–579.
- Schor S. The University Group Diabetes Program. A statistician looks at the mortality results. JAMA. 1971; 217:1671–1675.
- Cornfield JThe University Group Diabetes Program. A further statistical analysis of the mortality findings. JAMA. 1971; 217:1676–1687.
- Feinstein AR. Clinical biostatistics. 8. An analytic appraisal of the University Group Diabetes Program (UGDP) study. Clin Pharmacol Ther. 1971; 12:167–191.
- The University Group Diabetes Program. A study of the effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. V. Evaluation of pheniformin therapy. Diabetes 1975; 24( suppl 1):65–184.
- Knatterud GL, Klimt CR, Levin ME, Jacobson ME, Goldner MG. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. VII. Mortality and selected nonfatal events with insulin treatment. JAMA. 1978; 240:37–42.
- Schwartz TB, Meinert CL. The UGDP controversy: thirty-four years of contentious ambiguity laid to rest. Perspect Biol Med. 2004; 47:564–574.
- Turner RC, Holman RR. Lessons from UK Prospective Diabetes Study. Diabetes Res Clin Pract 1995; 28( suppl):S151–S157.
- UKPDS Research Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998; 352:854–865.
- UKPDS Study Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998; 352:837–853.
- UK Prospective Diabetes Study Group. UKPDS 28: a randomized trial of efficacy of early addition of metformin in sulfonylurea-treated type 2 diabetes. Diabetes Care. 1998; 21:87–92.
- Bretzel RG, Voigt K, Schatz H. The United Kingdom Prospective Diabetes Study (UKPDS) implications for the pharmacotherapy of type 2 diabetes mellitus. Exp Clin Endocrinol Diabetes. 1998; 106:369–372.
- Turner RC, Cull CA, Frighi V, Holman RR. Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. JAMA. 1999; 281:2005–2012.
- Leslie RD. United Kingdom prospective diabetes study (UKPDS): what now or so what? Diabetes Metab Res Rev 1999; 15:65–71.
- Stratton IM, Adler AI, Neil HA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000; 321:405–412.
- Mooradian AD, Chehade J. Implications of the UK Prospective Diabetes Study: questions answered and issues remaining. Drugs Aging. 2000; 16:159–164.
- Nathan DM. Some answers, more controversy, from UKPDS. United Kingdom Prospective Diabetes Study. Lancet. 1998; 352:832–833.
- Srimanunthiphol J, Beddow R, Arakaki R. A review of the United Kingdom Prospective Diabetes Study (UKPDS) and a discussion of the implications for patient care. Hawaii Med J. 2000; 59:295–298.
- Duckworth WC, McCarren M, Abraira C. Glucose control and cardiovascular complications: the VA Diabetes Trial. Diabetes Care. 2001; 24:942–945.
- Abraira C, Colwell JA, Nuttall FQ, et al. Veterans Affairs Cooperative Study on glycemic control and complications in type II diabetes (VA CSDM). Results of the feasibility trial. Veterans Affairs Cooperative Study in Type II Diabetes. Diabetes Care. 1995; 18:1113–1123.
- Gu K, Cowie CC, Harris MI. Mortality in adults with and without diabetes in a national cohort of the U.S. population, 1971–1993. Diabetes Care. 1998; 21:1138–1145. NHANES
- Gaede P, Lund-Andersen H, Parving HH, Pedersen O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med. 2008; 358:580–591.
- Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008; 358:2560–2572.
- Action in Diabetes and Vascular Disease: PreterAx and DiamicroN Modified-Release Controlled Evaluation. Rationale and design of the ADVANCE study: a randomised trial of blood pressure lowering and intensive glucose control in high-risk individuals with type 2 diabetes mellitus. J Hypertens 2001; 19(suppl):S21–S28.
- Abraira C, Duckworth W, McCarren M, et al. Design of the cooperative study on glycemic control and complications in diabetes mellitus type 2: Veterans Affairs Diabetes Trial. J Diabetes Complications. 2003; 17:314–322.
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007; 356:2457–2471.
- American Association of Clinical Endocrinologists. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the management of diabetes mellitus. Endocr Pract 2007; 13(suppl 1):1–68.
- American Diabetes Association. Standards of medical care in diabetes—2008. Diabetes Care 2008; 31(suppl 1):S12–S54.
- Magee MF, Isley WL. Rationale, design, and methods for glycemic control in the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) Trial. Am J Cardiol 2006; 97:20G–30G.
KEY POINTS
- No obvious cause, including hypoglycemia proximate to death or the use of any particular medication, clearly explained the excess deaths, although hypoglycemia occurred more often in intensively treated participants.
- The death rates in ACCORD were lower than in population studies and in other intervention trials. It is likely that multiple approaches to reducing the risk of cardiovascular disease contributed to this low mortality rate.
Perioperative statins: More than lipid-lowering?
Soon, the checklist for internists seeing patients about to undergo surgery may include prescribing one of the lipid-lowering hydroxymethylglutaryl-CoA reductase inhibitors, also called statins.
Statins? Not long ago, we were debating whether patients who take statins should stop taking them before surgery, based on the manufacturers’ recommendations.1 The discussion, however, has changed to whether patients who have never received a statin should be started on one before surgery to provide immediate prophylaxis against cardiac morbidity, and how much harm long-term statin users face if these drugs are withheld perioperatively.
The evidence is still very preliminary and based mostly on studies in animals and retrospective studies in people. However, an expanding body of indirect evidence suggests that these drugs are beneficial in this situation.
In this review, we discuss the mechanisms by which statins may protect the heart in the short term, drawing on data from animal and human studies of acute myocardial infarction, and we review the current (albeit limited) data from the perioperative setting.
FEW INTERVENTIONS DECREASE RISK
Each year, approximately 50,000 patients suffer a perioperative cardiovascular event; the incidence of myocardial infarction during or after noncardiac surgery is 2% to 3%.2 The primary goal of preoperative cardiovascular risk assessment is to predict and avert these events.
But short of canceling surgery, few interventions have been found to reduce a patient’s risk. For example, a landmark study in 2004 cast doubt on the efficacy of preoperative coronary revascularization.3 Similarly, although early studies of beta-blockers were promising4,5 and although most internists prescribe these drugs before surgery, more recent studies have cast doubt on their efficacy, particularly in patients at low risk undergoing intermediate-risk (rather than vascular) surgery.6–8
This changing clinical landscape has prompted a search for new strategies for perioperative risk-reduction. Several recent studies have placed statins in the spotlight.
POTENTIAL MECHANISMS OF SHORT-TERM BENEFIT
Statins have been proven to save lives when used long-term, but how could this class of drugs, designed to prevent the accumulation of arterial plaques by lowering low-density lipoprotein cholesterol (LDL-C) levels, have any short-term impact on operative outcomes? Although LDL-C reduction is the principal mechanism of action of statins, not all of the benefit can be ascribed to this mechanism.9 The answer may lie in their “pleiotropic” effects—ie, actions other than LDL-C reduction.
The more immediate pleiotropic effects of statins in the proinflammatory and prothrombotic environment of the perioperative period are thought to include improved endothelial function (both antithrombotic function and vasomotor function in response to ischemic stress), enhanced stability of atherosclerotic plaques, decreased oxidative stress, and decreased vascular inflammation.10–12
EVIDENCE FROM ANIMAL STUDIES
Experiments in animals suggest that statins, given shortly before or after a cardiovascular event, confer benefit before any changes in LDL-C are measurable.
Lefer et al13 found that simvastatin (Zocor), given 18 hours before an ischemic episode in rats, blunted the inflammatory response in cardiac reperfusion injury. Not only was reperfusion injury significantly less in the hearts of the rats that received simvastatin than in the saline control group, but the simvastatin-treated hearts also expressed fewer neutrophil adhesion molecules such as P-selectin, and they had more basal release of nitric oxide, the potent endothelial-derived vasodilator with antithrombotic, anti-inflammatory, and antiproliferative effects.14 These results suggest that statins may improve endothelial function acutely, particularly during ischemic stress.
Osborne et al15 fed rabbits a cholesterol-rich diet plus either lovastatin (Mevacor) or placebo. After 2 weeks, the rabbits underwent either surgery to induce a myocardial infarction or a sham procedure. Regardless of the pretreatment, biopsies of the aorta did not reveal any atherosclerosis; yet the lovastatin-treated rabbits sustained less myocardial ischemic damage and they had more endothelium-mediated vasodilatation.
Statin therapy also may improve cerebral ischemia outcomes in animal models.14,16
Sironi et al16 induced strokes in rats by occluding the middle cerebral artery. The rats received either simvastatin or vehicle for 3 days before the stroke or immediately afterwards. Even though simvastatin did not have enough time to affect the total cholesterol level, rats treated with simvastatin had smaller infarcts (as measured by magnetic resonance imaging) and produced more nitric oxide.
Comment. Taken together, these studies offer tantalizing evidence that statins have short-term, beneficial nonlipid effects and may reduce not only the likelihood of an ischemic event, but—should one occur—the degree of tissue damage that ensues.
EFFECTS OF STATINS IN ACUTE CORONARY SYNDROME
The National Registry of Myocardial Infarction17 is a prospective, observational database of all patients with acute myocardial infarction admitted to 1,230 participating hospitals throughout the United States. In an analysis from this cohort, patients were divided into four groups: those receiving statins before and after admission, those receiving statins only before admission, those receiving statins only after admission, and those who never received statins.
Compared with those who never received statins, fewer patients who received them both before and after admission died while in the hospital (unadjusted odds ratio 0.23, 95% confidence interval [CI] 0.22–0.25), and the odds ratio for those who received statins for the first time was 0.31 (95% CI 0.29–0.33). Patients who stopped receiving a statin on admission were more likely to die than were patients who never received statins (odds ratio 1.09, 95% CI 1.03–1.15). These trends held true even when adjustments were made for potential confounding factors.
Comment. Unmeasured confounding factors (such as the inability to take pills due to altered mental status or the different practice styles of the providers who chose to discontinue statins) might have affected the results. Nevertheless, these results suggest that the protective effects of statins stop almost immediately when these drugs are discontinued, and that there may even be an adverse “rebound” effect when patients who have been taking these drugs for a long time stop taking them temporarily.
The Platelet Receptor Inhibition in Ischemic Syndrome Management trial,18 in a subgroup analysis, had nearly identical findings. In the main part of this trial, patients with coronary artery disease and chest pain at rest or accelerating pain in the last 24 hours were randomized to receive tirofiban (Aggrastat) or heparin. Complete data on statin use were available for 1,616 (50%) of the 3,232 patients in this trial, and the rate of the primary end point (death, myocardial infarction, or recurrent ischemia) was analyzed on the basis of statin therapy in this subgroup.
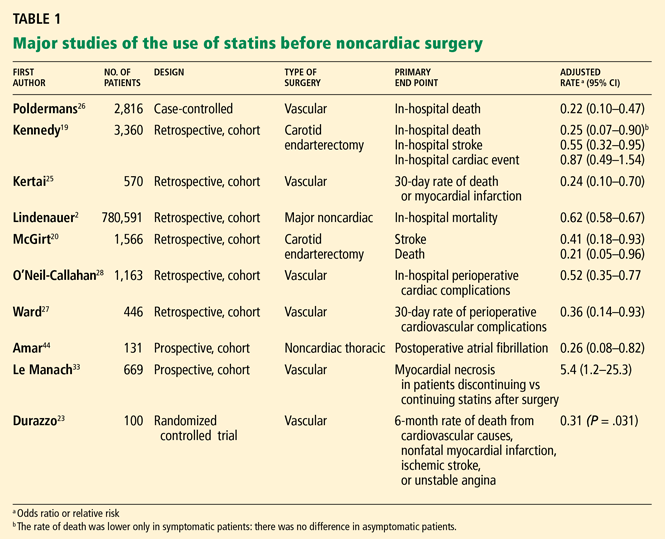
Comment. Together, these data lead to the conclusion that, when admitted for either acute myocardial infarction or acute coronary syndrome, patients already receiving statins should not have them stopped, and those who had not been receiving statins should receive them immediately. The safety of these medications in the acute setting appears excellent: in the Myocardial Ischemia Reduction With Acute Cholesterol Lowering (MIRACL)12 and the Pravastatin or Atorvastatin Evaluation and Infection Therapy (PROVE-IT)11 trials, fewer than 5% of statin-treated patients had transient elevations in transaminase levels, and no cases of rhabdomyolysis were reported.
PERIOPERATIVE STATIN STUDIES
The data on perioperative statin use are mostly observational and retrospective and fall into essentially four surgical categories: coronary artery bypass grafting (CABG), carotid endarterectomy,19,20 noncardiac vascular surgery, and major noncardiac surgery. Two meta-analyses have also evaluated the data.21,22 The only randomized controlled trial (performed by Durazzo et al23) was small and was carried out at a single center in vascular surgery patients, and the event rate was low.
Current recommendations from the National Cholesterol Education Program (NCEP)24 say that patients who need CABG, have peripheral arterial disease, have an abdominal aortic aneurysm, or have cerebrovascular disease should already be on a statin to achieve an LDL-C goal level of less than 100 mg/dL, with an optional goal of less than 70 mg/dL, independent of surgery.
Since not all patients who should be on statins are actually on them, questions arise:
- Is it important (and safe) to start statin treatment preoperatively?
- Will patients with cardiovascular risk factors but without known cardiovascular disease benefit from statins perioperatively?
Noncardiac vascular surgery
Multiple retrospective studies have evaluated the effect of statins in patients undergoing major noncardiac vascular surgery.25–32
Kertai et al25 evaluated 570 patients in Holland who underwent elective open surgery for infrarenal abdominal aortic aneurysms between 1991 and 2001, looking for an association between statin use and the incidence of perioperative death from myocardial infarction. Only 162 of the 570 patients had been on long-term statin therapy before the surgery. The use of statins was only one of many known baseline characteristics that were significantly different between the two groups, including age, body mass index, known coronary artery disease, and use of angiotensin-converting enzyme inhibitors and beta-blockers. In univariate analysis, statins appeared to be protective: 6 (3.7%) of the patients in the statin group died of a myocardial infarction, compared with 45 (11%) of those in the nostatin group. A multivariate analysis yielded similar findings, with an odds ratio of 0.24 (95% CI 0.11–0.54).
Ward et al27 performed a very similar retrospective study, with similar findings. In 446 patients who underwent surgery for infrarenal abdominal aortic aneurysm, statin therapy was associated with a significantly lower incidence of the combined end point of death, myocardial infarction, stroke, and major peripheral vascular complications, with an adjusted odds ratio of 0.36 (95% CI 0.14–0.93).
Poldermans et al26 noted similar findings in a case-control study of noncardiac vascular surgery patients. Statin users had a much lower perioperative risk of death than did nonusers, with an adjusted odds ratio of 0.22 (95% CI 0.10–0.47).
O’Neil-Callahan et al,28 in a cohort study, found that statin users had fewer perioperative cardiac complications, with an adjusted odds ratio of 0.49 (95% CI 0.28–0.84, P = .009).
Dogma of withdrawing statins before major surgery is challenged
Le Manach et al33 reviewed the outcomes for all patients of a single hospital in Paris who underwent nonemergency infrarenal aortic procedures between January 2001 and December 2004. In January 2004, the hospital instituted guidelines to ensure that patients on statins continue taking them up to the evening before surgery and that statins be restarted on the first postoperative day (via nasogastric tube if necessary). Before 2004, there had been no specific guidelines, and patients on statins did not receive them for a median of 4 days postoperatively. Types of procedures were similar during the two time periods, as were the rates of beta-blocker use, preoperative revascularization, venous thromboembolism prophylaxis, and perioperative blood pressure control. After surgery, topononin I levels were measured in all patients as surveillance for cardiac events, and were defined as elevated when greater than 0.2 ng/mL.
Compared with patients not on statins at all, those treated with statins continuously throughout the perioperative period (after January 2004) had a lower rate of elevated troponin (relative risk 0.38). In contrast, those who had their statins transiently discontinued perioperatively (prior to 2004) had troponin elevations more often than those who had never been treated (relative risk 2.1). This suggested an over fivefold risk reduction (P < .001) conferred by not discontinuing statins in the immediate postoperative period. This finding was maintained after multivariate adjustment: statin withdrawal was associated with a 2.9-fold (95% CI 1.6–5.5) increase in the risk of cardiac enzyme elevations postoperatively. No fewer deaths were noted, but the study was not powered to detect a mortality difference.
Comment. Although secular trends cannot be entirely discounted as contributing to these findings, the prompt increase in cardiac events after just 4 days of statin withdrawal adds to the growing body of evidence suggesting that statin discontinuation can have harmful acute effects. It also brings up the question: Can starting statins benefit patients in the same time period?
Should statins be started before vascular surgery?
Schouten et al32 evaluated the effects of newly started or continued statin treatment in patients undergoing major elective vascular surgery. Patients were screened before surgery and started on statins if they were not already receiving them and their total cholesterol levels were elevated; new users received the medication for about 40 days before surgery. Of the 981 screened patients, 44 (5%) were newly started on statins and 182 (19%) were continued on their therapy. Perioperative death or myocardial infarction occurred in 22 (8.8%) of the statin users and 111 (14.7%) of the nonusers, a statistically significant difference. Temporary discontinuation (median 1 day) of statins in this study due to the inability to take an oral medication did not appear to affect the likelihood of a myocardial infarction.
Durazzo et al23 performed a single-center, randomized, prospective, placebo-controlled, double-blind clinical trial of atorvastatin (Lipitor) 20 mg daily vs placebo in 100 patients undergoing noncardiac arterial vascular surgery. Patients were excluded if they had previously used medications to treat dyslipidemia, recently had a cardiovascular event, or had contraindications to statin treatment such as a baseline creatinine level greater than 2.0 mg/dL or severe hepatic disease. The intervention group received atorvastatin starting at least 2 weeks before surgery for a total of 45 days. Patients were then continued or started on a statin after surgery if their LDL-C level was greater than 100 mg/dL. Beta-blocker use was recommended “on the basis of current guidelines.”
One month after surgery, the LDL-C level was statistically significantly lower in the atorvastatin group. Since most patients did not continue or start statin therapy after the 45-day treatment period, the LDL-C levels were not statistically different at 3 and 6 months after surgery.
At 6 months, the rate of the primary end point (death from cardiovascular causes, nonfatal acute myocardial infarction, ischemic stroke, or unstable angina) was 26.0% in the placebo group and 8.0% in the atorvastatin group, a statistically significant difference. Three patients in the atorvastatin group had cardiac events in the first 10 days after surgery, compared with 11 patients in the placebo group. Thirteen of the 17 total cardiac events took place within 10 days after surgery.
One of the atorvastatin patients developed rhabdomyolysis and elevated aminotransferase levels.
Major noncardiac surgery
Lindenauer et al2 performed a retrospective cohort study of surgical patients who were at least 18 years old and survived beyond the second hospital day. Patients were divided into a group receiving any form of lipid-lowering treatment (of whom more than 90% were taking statins) and a group that had never never received a lipid-lowering drug or only started one on the third day of the hospitalization or later. The period of study was from January 1, 2000, to December 31, 2001.
In all, 780,591 patients from 329 hospitals throughout the United States were included, of whom only 77,082 (9.9%) received lipid-lowering therapy. Eight percent of the patients underwent vascular surgery. Not surprisingly, the treated patients were more likely to have a history of hypertension, diabetes, ischemic heart disease, or hyperlipidemia. They also were more likely to have a vascular procedure performed, to have two or more cardiac risk factors (high-risk surgery, ischemic heart disease, congestive heart failure, cerebrovascular disease, renal insufficiency, or diabetes mellitus), and to be treated with beta-blockers and angiotensin-converting enzyme inhibitors, but they were less likely to have high-risk and emergency surgery performed.
The primary end point, perioperative death, occurred in 2.13% of the treated patients and 3.05% of the nontreated group. Compared with the rate in a propensity-matched cohort, the odds ratio adjusted for unbalanced covariates was 0.62 (95% CI 0.58–0.67) in favor of lipid treatment. Stratification by cardiac risk index revealed a number needed to treat of 186 for those with no risk factors, 60 for those with two risk factors, and 30 for those with four or more risk factors.
Unfortunately, this analysis was not able to take into account whether and for how long patients were receiving lipid-lowering therapy before hospitalization. It therefore does not answer the questions of whether starting lipid-lowering therapy before surgery is beneficial or whether stopping it is harmful. It also does not shed light on whether perioperative lipid-lowering increases the risk of rhabdomyolysis or liver disease.
Carotid endarterectomy
Two recent retrospective cohort studies evaluated the outcomes in patients undergoing carotid endarterectomy.19,20
Kennedy et al19 found that patients on a statin at the time of admission who had symptomatic carotid disease had lower rates of inhospital death (adjusted odds ratio 0.24, 95% CI 0.06–0.91) and ischemic stroke or death (adjusted odds ratio 0.55, 95% CI 0.31–0.97). However, cardiac outcomes among these symptomatic patients were not significantly improved (odds ratio 0.82, 95% CI 0.45–1.50), nor was there benefit for asymptomatic patients, raising the possibility that the positive findings were due to chance or that patients at lower baseline risk for vascular events may have less benefit.
McGirt et al20 performed a similar study; they did not, however, distinguish whether patients had symptomatic vs asymptomatic carotid disease. The 30-day risk of perioperative stroke was lower in patients treated with a statin, with an odds ratio of 0.41 (95% CI 0.18–0.93); the odds ratio for death was 0.21 (95% CI 0.05–0.96). Cardiac outcomes were not significantly affected.
Coronary artery bypass graft surgery
According to the NCEP recommendations, nearly all patients undergoing CABG should already be on a statin before surgery since they all have known coronary artery disease. Multiple observational studies have offered confirmatory evidence that statins are beneficial in this setting.34–38
Liakopoulos et al39 evaluated whether the anti-inflammatory effects of statins may, in part, account for their beneficial effect in the perioperative period. The authors prospectively matched 18 patients who were taking statins and were referred for elective CABG with 18 patients who were not prescribed statins previously. The only major measured baseline characteristic that differed between the two groups was a statistically significantly lower LDL-C level in the statin group. The operative characteristics did not differ, and cytokine levels at baseline were similar.
Tumor necrosis factor alpha levels increased significantly in the control group but did not change significantly in the statin group. Interleukin 8 increased in both groups by a similar amount. Interleukin 6 (the major inducer of C-reactive protein) increased from baseline in both groups but did not increase nearly as much in the statin group as in the control group; the intergroup difference was statistically significant. The anti-inflammatory cytokine interleukin 10 increased minimally from baseline in the control group, while the statin group’s levels increased significantly above baseline and those of the control group.
Christenson40 also found that inflammatory markers were improved with pre-CABG statin treatment in a small randomized trial in which patients received simvastatin 20 mg 4 weeks prior to CABG surgery vs no statin. Interestingly, far fewer statin-treated patients developed thrombocytosis (platelet count > 400 × 109/L) than did control patients (3% vs 81%, P < .0001).
RISKS OF PERIOPERATIVE STATINS
The risks associated with statin therapy in general appear low, but specific perioperative risks have not been well studied.
Baigent et al,41 in a meta-analysis of randomized trials of nonperioperative statin therapy, found that rhabdomyolysis occurred in 9 (0.023%) of 39,884 patients receiving statins vs 6 (0.015%) of the 39,817 controls, with a number needed to harm of 12,500. Moreover, the rates of nonvascular death and cancer did not increase. It is plausible that the risk is somewhat greater in the perioperative setting but is likely not enough to outweigh the potential benefits, especially since the risk of ischemic vascular events is particularly high then.
Some of the perioperative studies cited above specifically addressed potential risks. For example, in the study by Schouten et al,32 mild creatine kinase elevations were more common in the statin-treated group, but the incidence of moderate and severe creatine kinase elevations did not differ significantly. No case of rhabdomyolysis occurred, and length of surgery was the only predictor of myopathy. MIRACL and PROVE-IT revealed similar safety profiles; aminotransferase levels normalized when statins were stopped, and no cases of rhabdomyolysis occurred.11,12 In the vascular surgery study by Durazzo et al,23 1 (2%) of the 50 atorvastatin-treated patients developed both rhabdomyolysis and elevated aminotransferase levels that prompted discontinuation of the statin.
Overall, the observational studies do not indicate that statin continuation or treatment is harmful in perioperative patients. However, these studies did not specifically evaluate patients with acute insults from surgery such as sepsis, renal failure, or hepatitis. It is unknown what effect statin therapy would have in those patients and whether statins should be selectively discontinued in patients who develop major hepatic, musculoskeletal, or renal complications after surgery.
OUR RECOMMENDATIONS
Before CABG or vascular surgery
Given the NCEP recommendations, existing primary and secondary prevention studies, observational studies of CABG and noncardiac vascular surgery patients, and the one randomized trial of vascular surgery patients, data support the use of statins in nearly all patients undergoing cardiac or vascular surgery. We advocate starting statins in the perioperative period to take advantage of their rapid-acting pleiotropic effects, and continuing them long-term to take advantage of their lipid-lowering effects. This recommendation is in line with the recently released American College of Cardiology/American Heart Association (ACC/AHA) 2007 perioperative guidelines that state “for patients undergoing vascular surgery with or without clinical risk factors, statin use is reasonable.”42
Although the ideal time to start statins is not certain, the study by Durazzo et al23 suggests that they should be started at least 2 weeks before surgery if possible. Moreover, patients already taking statins should definitely not have their statins discontinued if at all possible.
Before major nonvascular surgery
For patients undergoing major nonvascular (intermediate-risk) surgery, physicians should first ascertain if the patient has an indication for statin therapy based on current nonsurgical lipid level recommendations. However, even if there is no clear indication for statin therapy based on NCEP guidelines, we endorse the recently released ACC/AHA perioperative guidelines that state that statin therapy can be considered in patients with a risk factor who are undergoing intermediate-risk procedures. Moreover, we wholeheartedly support the ACC/AHA’s strongest recommendation that patients who are already receiving statins and are undergoing noncardiac surgery should not have their statins discontinued.
When to discontinue statins?
The risk of harm overall appears to be minimal and certainly less than the likelihood of benefit. It is reasonable to observe patients postoperatively for adverse clinical events that may increase the risk of perioperative statin treatment, such as acute renal failure, hepatic failure, or sepsis, but whether statins should be stopped in patients with these complications remains unknown; we advocate individualizing the decision.
More studies needed
We need more data on whether moderate-risk patients undergoing moderate-risk surgery benefit from perioperative statin therapy, when therapy should be started, whether therapy should be started on the day of surgery if it was not started earlier, which statin and what doses are optimal, how long therapy should be continued, and what degree of risk is associated with perioperative statin therapy.
Fortunately, important data should be forthcoming in the next few years: the Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography (DECREASE-IV) study43 is a 4-year two-by-two factorial placebo-controlled study evaluating the use of fluvastatin (Lescol) and bisoprolol (Zebeta, a beta-blocker) separately and together in patients who are older than 40 years, are undergoing elective noncardiac surgery, have an estimated risk of cardiovascular death of more than 1%, have not used statins previously, and do not have elevated cholesterol.
- Grant PJ, Kedia N. Should statins be discontinued preoperatively? IMPACT consults. Proceedings of the 2nd Annual Cleveland Clinic Perioperative Medicine Summit. Cleve Clin J Med 2006; 73 Electronic suppl 1:S9–S10.
- Lindenauer PK, Pekow P, Wang K, Gutierrez B, Benjamin EM. Lipid-lowering therapy and in-hospital mortality following major noncardiac surgery. JAMA 2004; 291:2092–2099.
- McFalls EO, Ward HB, Moritz TE, et al. Coronary-artery revascularization before elective major vascular surgery. N Engl J Med 2004; 351:2795–2804.
- Mangano DT, Layug EL, Wallace A, Tateo I. Effect of atenolol on mortality and cardiovascular morbidity after noncardiac surgery. Multicenter Study of Perioperative Ischemia Research Group. N Engl J Med 1996; 335:1713–1720.
- Poldermans D, Boersma E, Bax JJ, et al. The effect of bisoprolol on perioperative mortality and myocardial infarction in high-risk patients undergoing vascular surgery. Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group. N Engl J Med 1999; 341:1789–1794.
- Brady AR, Gibbs JS, Greenhalgh RM, Powell JT, Sydes MR. Perioperative beta-blockade (POBBLE) for patients undergoing infrarenal vascular surgery: results of a randomized double-blind controlled trial. J Vasc Surg 2005; 41:602–609.
- Juul AB, Wetterslev J, Gluud C, et al. Effect of perioperative beta blockade in patients with diabetes undergoing major non-cardiac surgery: randomised placebo controlled, blinded multicentre trial. BMJ 2006; 332:1482.
- Yang H, Raymer K, Butler R, Parlow J, Roberts R. The effects of perioperative beta-blockade: results of the Metoprolol after Vascular Surgery (MaVS) study, a randomized controlled trial. Am Heart J 2006; 152:983–990.
- Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med 2005; 352:20–28.
- Ito MK, Talbert RL, Tsimikas S. Statin-associated pleiotropy: possible beneficial effects beyond cholesterol reduction. Pharmacotherapy 2006; 26:85S–97S.
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
- Lefer AM, Campbell B, Shin YK, Scalia R, Hayward R, Lefer DJ. Simvastatin preserves the ischemic-reperfused myocardium in normocholesterolemic rat hearts. Circulation 1999; 100:178–184.
- Endres M, Laufs U, Liao JK, Moskowitz MA. Targeting eNOS for stroke protection. Trends Neurosci 2004; 27:283–289.
- Osborne JA, Lento PH, Siegfried MR, Stahl GL, Fusman B, Lefer AM. Cardiovascular effects of acute hypercholesterolemia in rabbits. Reversal with lovastatin treatment. J Clin Invest 1989; 83:465–473.
- Sironi L, Cimino M, Guerrini U, et al. Treatment with statins after induction of focal ischemia in rats reduces the extent of brain damage. Arterioscler Thromb Vasc Biol 2003; 23:322–327.
- Fonarow GC, Wright RS, Spencer FA, et al. Effect of statin use within the first 24 hours of admission for acute myocardial infarction on early morbidity and mortality. Am J Cardiol 2005; 96:611–616.
- Heeschen C, Hamm CW, Laufs U, Snapinn S, Bohm M, White HD. Withdrawal of statins increases event rates in patients with acute coronary syndromes. Circulation 2002; 105:1446–1452.
- Kennedy J, Quan H, Buchan AM, Ghali WA, Feasby TE. Statins are associated with better outcomes after carotid endarterectomy in symptomatic patients. Stroke 2005; 36:2072–2076.
- McGirt MJ, Perler BA, Brooke BS, et al. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors reduce the risk of perioperative stroke and mortality after carotid endarterectomy. J Vasc Surg 2005; 42:829–836.
- Hindler K, Shaw AD, Samuels J, Fulton S, Collard CD, Riedel B. Improved postoperative outcomes associated with preoperative statin therapy. Anesthesiology 2006; 105:1260–1272.
- Kapoor AS, Kanji H, Buckingham J, Devereaux PJ, McAlister FA. Strength of evidence for perioperative use of statins to reduce cardiovascular risk: systematic review of controlled studies. BMJ 2006; 333:1149.
- Durazzo AE, Machado FS, Ikeoka DT, et al. Reduction in cardiovascular events after vascular surgery with atorvastatin: a randomized trial. J Vasc Surg 2004; 39:967–975.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- Kertai MD, Boersma E, Westerhout CM, et al. A combination of statins and beta-blockers is independently associated with a reduction in the incidence of perioperative mortality and nonfatal myocardial infarction in patients undergoing abdominal aortic aneurysm surgery. Eur J Vasc Endovasc Surg 2004; 28:343–352.
- Poldermans D, Bax JJ, Kertai MD, et al. Statins are associated with a reduced incidence of perioperative mortality in patients undergoing major noncardiac vascular surgery. Circulation 2003; 107:1848–1851.
- Ward RP, Leeper NJ, Kirkpatrick JN, Lang RM, Sorrentino MJ, Williams KA. The effect of preoperative statin therapy on cardiovascular outcomes in patients undergoing infrainguinal vascular surgery. Int J Cardiol 2005; 104:264–268.
- O’Neil-Callahan K, Katsimaglis G, Tepper MR, et al. Statins decrease perioperative cardiac complications in patients undergoing non-cardiac vascular surgery: the Statins for Risk Reduction in Surgery (StaRRS) study. J Am Coll Cardiol 2005; 45:336–342.
- Abbruzzese TA, Havens J, Belkin M, et al. Statin therapy is associated with improved patency of autogenous infrainguinal bypass grafts. J Vasc Surg 2004; 39:1178–1185.
- Boersma E, Poldermans D, Bax JJ, et al. Predictors of cardiac events after major vascular surgery: role of clinical characteristics, dobutamine echocardiography, and beta-blocker therapy. JAMA 2001; 285:1865–1873.
- Landesberg G, Mosseri M, Wolf YG, et al. Preoperative thallium scanning, selective coronary revascularization, and long-term survival after major vascular surgery. Circulation 2003; 108:177–183.
- Schouten O, Kertai MD, Bax JJ, et al. Safety of perioperative statin use in high-risk patients undergoing major vascular surgery. Am J Cardiol 2005; 95:658–660.
- Le Manach Y, Godet G, Coriat P, et al. The impact of postoperative discontinuation or continuation of chronic statin therapy on cardiac outcome after major vascular surgery. Anesth Analg 2007; 104:1326–1333.
- Ali IS, Buth KJ. Preoperative statin use and outcomes following cardiac surgery. Int J Cardiol 2005; 103:12–18.
- Clark LL, Ikonomidis JS, Crawford FA, et al. Preoperative statin treatment is associated with reduced postoperative mortality and morbidity in patients undergoing cardiac surgery: an 8-year retrospective cohort study. J Thorac Cardiovasc Surg 2006; 131:679–685.
- Pan W, Pintar T, Anton J, Lee VV, Vaughn WK, Collard CD. Statins are associated with a reduced incidence of perioperative mortality after coronary artery bypass graft surgery. Circulation 2004; 110(suppl 2):II45–II49.
- Pascual DA, Arribas JM, Tornel PL, et al. Preoperative statin therapy and troponin T predict early complications of coronary artery surgery. Ann Thorac Surg 2006; 81:78–83.
- Dotani MI, Elnicki DM, Jain AC, Gibson CM. Effect of preoperative statin therapy and cardiac outcomes after coronary artery bypass grafting. Am J Cardiol 2000; 86:1128–1130.
- Liakopoulos OJ, Dorge H, Schmitto JD, Nagorsnik U, Grabedunkel J, Schoendube FA. Effects of preoperative statin therapy on cytokines after cardiac surgery. Thorac Cardiovasc Surg 2006; 54:250–254.
- Christenson JT. Preoperative lipid-control with simvastatin reduces the risk of postoperative thrombocytosis and thrombotic complications following CABG. Eur J Cardiothorac Surg 1999; 15:394–399.
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Fleisher LA, Beckman JA, Brown KA, et al. ACC/AHA 2007 Guidelines on Perioperative Cardiovascular Evaluation and Care for Noncardiac Surgery. A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery). Circulation 2007; 116:e418–e499.
- Schouten O, Poldermans D, Visser L, et al. Fluvastatin and bisoprolol for the reduction of perioperative cardiac mortality and morbidity in high-risk patients undergoing non-cardiac surgery: rationale and design of the DECREASE-IV study. Am Heart J 2004; 148:1047–1052.
- Amar D, Zhang H, Heerdt PM, Park B, Fleisher M, Thaler HT. Statin use is associated with a reduction in atrial fibrillation after noncardiac thoracic surgery independent of C-reactive protein. Chest 2005; 128:3421–3427.
Soon, the checklist for internists seeing patients about to undergo surgery may include prescribing one of the lipid-lowering hydroxymethylglutaryl-CoA reductase inhibitors, also called statins.
Statins? Not long ago, we were debating whether patients who take statins should stop taking them before surgery, based on the manufacturers’ recommendations.1 The discussion, however, has changed to whether patients who have never received a statin should be started on one before surgery to provide immediate prophylaxis against cardiac morbidity, and how much harm long-term statin users face if these drugs are withheld perioperatively.
The evidence is still very preliminary and based mostly on studies in animals and retrospective studies in people. However, an expanding body of indirect evidence suggests that these drugs are beneficial in this situation.
In this review, we discuss the mechanisms by which statins may protect the heart in the short term, drawing on data from animal and human studies of acute myocardial infarction, and we review the current (albeit limited) data from the perioperative setting.
FEW INTERVENTIONS DECREASE RISK
Each year, approximately 50,000 patients suffer a perioperative cardiovascular event; the incidence of myocardial infarction during or after noncardiac surgery is 2% to 3%.2 The primary goal of preoperative cardiovascular risk assessment is to predict and avert these events.
But short of canceling surgery, few interventions have been found to reduce a patient’s risk. For example, a landmark study in 2004 cast doubt on the efficacy of preoperative coronary revascularization.3 Similarly, although early studies of beta-blockers were promising4,5 and although most internists prescribe these drugs before surgery, more recent studies have cast doubt on their efficacy, particularly in patients at low risk undergoing intermediate-risk (rather than vascular) surgery.6–8
This changing clinical landscape has prompted a search for new strategies for perioperative risk-reduction. Several recent studies have placed statins in the spotlight.
POTENTIAL MECHANISMS OF SHORT-TERM BENEFIT
Statins have been proven to save lives when used long-term, but how could this class of drugs, designed to prevent the accumulation of arterial plaques by lowering low-density lipoprotein cholesterol (LDL-C) levels, have any short-term impact on operative outcomes? Although LDL-C reduction is the principal mechanism of action of statins, not all of the benefit can be ascribed to this mechanism.9 The answer may lie in their “pleiotropic” effects—ie, actions other than LDL-C reduction.
The more immediate pleiotropic effects of statins in the proinflammatory and prothrombotic environment of the perioperative period are thought to include improved endothelial function (both antithrombotic function and vasomotor function in response to ischemic stress), enhanced stability of atherosclerotic plaques, decreased oxidative stress, and decreased vascular inflammation.10–12
EVIDENCE FROM ANIMAL STUDIES
Experiments in animals suggest that statins, given shortly before or after a cardiovascular event, confer benefit before any changes in LDL-C are measurable.
Lefer et al13 found that simvastatin (Zocor), given 18 hours before an ischemic episode in rats, blunted the inflammatory response in cardiac reperfusion injury. Not only was reperfusion injury significantly less in the hearts of the rats that received simvastatin than in the saline control group, but the simvastatin-treated hearts also expressed fewer neutrophil adhesion molecules such as P-selectin, and they had more basal release of nitric oxide, the potent endothelial-derived vasodilator with antithrombotic, anti-inflammatory, and antiproliferative effects.14 These results suggest that statins may improve endothelial function acutely, particularly during ischemic stress.
Osborne et al15 fed rabbits a cholesterol-rich diet plus either lovastatin (Mevacor) or placebo. After 2 weeks, the rabbits underwent either surgery to induce a myocardial infarction or a sham procedure. Regardless of the pretreatment, biopsies of the aorta did not reveal any atherosclerosis; yet the lovastatin-treated rabbits sustained less myocardial ischemic damage and they had more endothelium-mediated vasodilatation.
Statin therapy also may improve cerebral ischemia outcomes in animal models.14,16
Sironi et al16 induced strokes in rats by occluding the middle cerebral artery. The rats received either simvastatin or vehicle for 3 days before the stroke or immediately afterwards. Even though simvastatin did not have enough time to affect the total cholesterol level, rats treated with simvastatin had smaller infarcts (as measured by magnetic resonance imaging) and produced more nitric oxide.
Comment. Taken together, these studies offer tantalizing evidence that statins have short-term, beneficial nonlipid effects and may reduce not only the likelihood of an ischemic event, but—should one occur—the degree of tissue damage that ensues.
EFFECTS OF STATINS IN ACUTE CORONARY SYNDROME
The National Registry of Myocardial Infarction17 is a prospective, observational database of all patients with acute myocardial infarction admitted to 1,230 participating hospitals throughout the United States. In an analysis from this cohort, patients were divided into four groups: those receiving statins before and after admission, those receiving statins only before admission, those receiving statins only after admission, and those who never received statins.
Compared with those who never received statins, fewer patients who received them both before and after admission died while in the hospital (unadjusted odds ratio 0.23, 95% confidence interval [CI] 0.22–0.25), and the odds ratio for those who received statins for the first time was 0.31 (95% CI 0.29–0.33). Patients who stopped receiving a statin on admission were more likely to die than were patients who never received statins (odds ratio 1.09, 95% CI 1.03–1.15). These trends held true even when adjustments were made for potential confounding factors.
Comment. Unmeasured confounding factors (such as the inability to take pills due to altered mental status or the different practice styles of the providers who chose to discontinue statins) might have affected the results. Nevertheless, these results suggest that the protective effects of statins stop almost immediately when these drugs are discontinued, and that there may even be an adverse “rebound” effect when patients who have been taking these drugs for a long time stop taking them temporarily.
The Platelet Receptor Inhibition in Ischemic Syndrome Management trial,18 in a subgroup analysis, had nearly identical findings. In the main part of this trial, patients with coronary artery disease and chest pain at rest or accelerating pain in the last 24 hours were randomized to receive tirofiban (Aggrastat) or heparin. Complete data on statin use were available for 1,616 (50%) of the 3,232 patients in this trial, and the rate of the primary end point (death, myocardial infarction, or recurrent ischemia) was analyzed on the basis of statin therapy in this subgroup.
Comment. Together, these data lead to the conclusion that, when admitted for either acute myocardial infarction or acute coronary syndrome, patients already receiving statins should not have them stopped, and those who had not been receiving statins should receive them immediately. The safety of these medications in the acute setting appears excellent: in the Myocardial Ischemia Reduction With Acute Cholesterol Lowering (MIRACL)12 and the Pravastatin or Atorvastatin Evaluation and Infection Therapy (PROVE-IT)11 trials, fewer than 5% of statin-treated patients had transient elevations in transaminase levels, and no cases of rhabdomyolysis were reported.
PERIOPERATIVE STATIN STUDIES
The data on perioperative statin use are mostly observational and retrospective and fall into essentially four surgical categories: coronary artery bypass grafting (CABG), carotid endarterectomy,19,20 noncardiac vascular surgery, and major noncardiac surgery. Two meta-analyses have also evaluated the data.21,22 The only randomized controlled trial (performed by Durazzo et al23) was small and was carried out at a single center in vascular surgery patients, and the event rate was low.
Current recommendations from the National Cholesterol Education Program (NCEP)24 say that patients who need CABG, have peripheral arterial disease, have an abdominal aortic aneurysm, or have cerebrovascular disease should already be on a statin to achieve an LDL-C goal level of less than 100 mg/dL, with an optional goal of less than 70 mg/dL, independent of surgery.
Since not all patients who should be on statins are actually on them, questions arise:
- Is it important (and safe) to start statin treatment preoperatively?
- Will patients with cardiovascular risk factors but without known cardiovascular disease benefit from statins perioperatively?
Noncardiac vascular surgery
Multiple retrospective studies have evaluated the effect of statins in patients undergoing major noncardiac vascular surgery.25–32
Kertai et al25 evaluated 570 patients in Holland who underwent elective open surgery for infrarenal abdominal aortic aneurysms between 1991 and 2001, looking for an association between statin use and the incidence of perioperative death from myocardial infarction. Only 162 of the 570 patients had been on long-term statin therapy before the surgery. The use of statins was only one of many known baseline characteristics that were significantly different between the two groups, including age, body mass index, known coronary artery disease, and use of angiotensin-converting enzyme inhibitors and beta-blockers. In univariate analysis, statins appeared to be protective: 6 (3.7%) of the patients in the statin group died of a myocardial infarction, compared with 45 (11%) of those in the nostatin group. A multivariate analysis yielded similar findings, with an odds ratio of 0.24 (95% CI 0.11–0.54).
Ward et al27 performed a very similar retrospective study, with similar findings. In 446 patients who underwent surgery for infrarenal abdominal aortic aneurysm, statin therapy was associated with a significantly lower incidence of the combined end point of death, myocardial infarction, stroke, and major peripheral vascular complications, with an adjusted odds ratio of 0.36 (95% CI 0.14–0.93).
Poldermans et al26 noted similar findings in a case-control study of noncardiac vascular surgery patients. Statin users had a much lower perioperative risk of death than did nonusers, with an adjusted odds ratio of 0.22 (95% CI 0.10–0.47).
O’Neil-Callahan et al,28 in a cohort study, found that statin users had fewer perioperative cardiac complications, with an adjusted odds ratio of 0.49 (95% CI 0.28–0.84, P = .009).
Dogma of withdrawing statins before major surgery is challenged
Le Manach et al33 reviewed the outcomes for all patients of a single hospital in Paris who underwent nonemergency infrarenal aortic procedures between January 2001 and December 2004. In January 2004, the hospital instituted guidelines to ensure that patients on statins continue taking them up to the evening before surgery and that statins be restarted on the first postoperative day (via nasogastric tube if necessary). Before 2004, there had been no specific guidelines, and patients on statins did not receive them for a median of 4 days postoperatively. Types of procedures were similar during the two time periods, as were the rates of beta-blocker use, preoperative revascularization, venous thromboembolism prophylaxis, and perioperative blood pressure control. After surgery, topononin I levels were measured in all patients as surveillance for cardiac events, and were defined as elevated when greater than 0.2 ng/mL.
Compared with patients not on statins at all, those treated with statins continuously throughout the perioperative period (after January 2004) had a lower rate of elevated troponin (relative risk 0.38). In contrast, those who had their statins transiently discontinued perioperatively (prior to 2004) had troponin elevations more often than those who had never been treated (relative risk 2.1). This suggested an over fivefold risk reduction (P < .001) conferred by not discontinuing statins in the immediate postoperative period. This finding was maintained after multivariate adjustment: statin withdrawal was associated with a 2.9-fold (95% CI 1.6–5.5) increase in the risk of cardiac enzyme elevations postoperatively. No fewer deaths were noted, but the study was not powered to detect a mortality difference.
Comment. Although secular trends cannot be entirely discounted as contributing to these findings, the prompt increase in cardiac events after just 4 days of statin withdrawal adds to the growing body of evidence suggesting that statin discontinuation can have harmful acute effects. It also brings up the question: Can starting statins benefit patients in the same time period?
Should statins be started before vascular surgery?
Schouten et al32 evaluated the effects of newly started or continued statin treatment in patients undergoing major elective vascular surgery. Patients were screened before surgery and started on statins if they were not already receiving them and their total cholesterol levels were elevated; new users received the medication for about 40 days before surgery. Of the 981 screened patients, 44 (5%) were newly started on statins and 182 (19%) were continued on their therapy. Perioperative death or myocardial infarction occurred in 22 (8.8%) of the statin users and 111 (14.7%) of the nonusers, a statistically significant difference. Temporary discontinuation (median 1 day) of statins in this study due to the inability to take an oral medication did not appear to affect the likelihood of a myocardial infarction.
Durazzo et al23 performed a single-center, randomized, prospective, placebo-controlled, double-blind clinical trial of atorvastatin (Lipitor) 20 mg daily vs placebo in 100 patients undergoing noncardiac arterial vascular surgery. Patients were excluded if they had previously used medications to treat dyslipidemia, recently had a cardiovascular event, or had contraindications to statin treatment such as a baseline creatinine level greater than 2.0 mg/dL or severe hepatic disease. The intervention group received atorvastatin starting at least 2 weeks before surgery for a total of 45 days. Patients were then continued or started on a statin after surgery if their LDL-C level was greater than 100 mg/dL. Beta-blocker use was recommended “on the basis of current guidelines.”
One month after surgery, the LDL-C level was statistically significantly lower in the atorvastatin group. Since most patients did not continue or start statin therapy after the 45-day treatment period, the LDL-C levels were not statistically different at 3 and 6 months after surgery.
At 6 months, the rate of the primary end point (death from cardiovascular causes, nonfatal acute myocardial infarction, ischemic stroke, or unstable angina) was 26.0% in the placebo group and 8.0% in the atorvastatin group, a statistically significant difference. Three patients in the atorvastatin group had cardiac events in the first 10 days after surgery, compared with 11 patients in the placebo group. Thirteen of the 17 total cardiac events took place within 10 days after surgery.
One of the atorvastatin patients developed rhabdomyolysis and elevated aminotransferase levels.
Major noncardiac surgery
Lindenauer et al2 performed a retrospective cohort study of surgical patients who were at least 18 years old and survived beyond the second hospital day. Patients were divided into a group receiving any form of lipid-lowering treatment (of whom more than 90% were taking statins) and a group that had never never received a lipid-lowering drug or only started one on the third day of the hospitalization or later. The period of study was from January 1, 2000, to December 31, 2001.
In all, 780,591 patients from 329 hospitals throughout the United States were included, of whom only 77,082 (9.9%) received lipid-lowering therapy. Eight percent of the patients underwent vascular surgery. Not surprisingly, the treated patients were more likely to have a history of hypertension, diabetes, ischemic heart disease, or hyperlipidemia. They also were more likely to have a vascular procedure performed, to have two or more cardiac risk factors (high-risk surgery, ischemic heart disease, congestive heart failure, cerebrovascular disease, renal insufficiency, or diabetes mellitus), and to be treated with beta-blockers and angiotensin-converting enzyme inhibitors, but they were less likely to have high-risk and emergency surgery performed.
The primary end point, perioperative death, occurred in 2.13% of the treated patients and 3.05% of the nontreated group. Compared with the rate in a propensity-matched cohort, the odds ratio adjusted for unbalanced covariates was 0.62 (95% CI 0.58–0.67) in favor of lipid treatment. Stratification by cardiac risk index revealed a number needed to treat of 186 for those with no risk factors, 60 for those with two risk factors, and 30 for those with four or more risk factors.
Unfortunately, this analysis was not able to take into account whether and for how long patients were receiving lipid-lowering therapy before hospitalization. It therefore does not answer the questions of whether starting lipid-lowering therapy before surgery is beneficial or whether stopping it is harmful. It also does not shed light on whether perioperative lipid-lowering increases the risk of rhabdomyolysis or liver disease.
Carotid endarterectomy
Two recent retrospective cohort studies evaluated the outcomes in patients undergoing carotid endarterectomy.19,20
Kennedy et al19 found that patients on a statin at the time of admission who had symptomatic carotid disease had lower rates of inhospital death (adjusted odds ratio 0.24, 95% CI 0.06–0.91) and ischemic stroke or death (adjusted odds ratio 0.55, 95% CI 0.31–0.97). However, cardiac outcomes among these symptomatic patients were not significantly improved (odds ratio 0.82, 95% CI 0.45–1.50), nor was there benefit for asymptomatic patients, raising the possibility that the positive findings were due to chance or that patients at lower baseline risk for vascular events may have less benefit.
McGirt et al20 performed a similar study; they did not, however, distinguish whether patients had symptomatic vs asymptomatic carotid disease. The 30-day risk of perioperative stroke was lower in patients treated with a statin, with an odds ratio of 0.41 (95% CI 0.18–0.93); the odds ratio for death was 0.21 (95% CI 0.05–0.96). Cardiac outcomes were not significantly affected.
Coronary artery bypass graft surgery
According to the NCEP recommendations, nearly all patients undergoing CABG should already be on a statin before surgery since they all have known coronary artery disease. Multiple observational studies have offered confirmatory evidence that statins are beneficial in this setting.34–38
Liakopoulos et al39 evaluated whether the anti-inflammatory effects of statins may, in part, account for their beneficial effect in the perioperative period. The authors prospectively matched 18 patients who were taking statins and were referred for elective CABG with 18 patients who were not prescribed statins previously. The only major measured baseline characteristic that differed between the two groups was a statistically significantly lower LDL-C level in the statin group. The operative characteristics did not differ, and cytokine levels at baseline were similar.
Tumor necrosis factor alpha levels increased significantly in the control group but did not change significantly in the statin group. Interleukin 8 increased in both groups by a similar amount. Interleukin 6 (the major inducer of C-reactive protein) increased from baseline in both groups but did not increase nearly as much in the statin group as in the control group; the intergroup difference was statistically significant. The anti-inflammatory cytokine interleukin 10 increased minimally from baseline in the control group, while the statin group’s levels increased significantly above baseline and those of the control group.
Christenson40 also found that inflammatory markers were improved with pre-CABG statin treatment in a small randomized trial in which patients received simvastatin 20 mg 4 weeks prior to CABG surgery vs no statin. Interestingly, far fewer statin-treated patients developed thrombocytosis (platelet count > 400 × 109/L) than did control patients (3% vs 81%, P < .0001).
RISKS OF PERIOPERATIVE STATINS
The risks associated with statin therapy in general appear low, but specific perioperative risks have not been well studied.
Baigent et al,41 in a meta-analysis of randomized trials of nonperioperative statin therapy, found that rhabdomyolysis occurred in 9 (0.023%) of 39,884 patients receiving statins vs 6 (0.015%) of the 39,817 controls, with a number needed to harm of 12,500. Moreover, the rates of nonvascular death and cancer did not increase. It is plausible that the risk is somewhat greater in the perioperative setting but is likely not enough to outweigh the potential benefits, especially since the risk of ischemic vascular events is particularly high then.
Some of the perioperative studies cited above specifically addressed potential risks. For example, in the study by Schouten et al,32 mild creatine kinase elevations were more common in the statin-treated group, but the incidence of moderate and severe creatine kinase elevations did not differ significantly. No case of rhabdomyolysis occurred, and length of surgery was the only predictor of myopathy. MIRACL and PROVE-IT revealed similar safety profiles; aminotransferase levels normalized when statins were stopped, and no cases of rhabdomyolysis occurred.11,12 In the vascular surgery study by Durazzo et al,23 1 (2%) of the 50 atorvastatin-treated patients developed both rhabdomyolysis and elevated aminotransferase levels that prompted discontinuation of the statin.
Overall, the observational studies do not indicate that statin continuation or treatment is harmful in perioperative patients. However, these studies did not specifically evaluate patients with acute insults from surgery such as sepsis, renal failure, or hepatitis. It is unknown what effect statin therapy would have in those patients and whether statins should be selectively discontinued in patients who develop major hepatic, musculoskeletal, or renal complications after surgery.
OUR RECOMMENDATIONS
Before CABG or vascular surgery
Given the NCEP recommendations, existing primary and secondary prevention studies, observational studies of CABG and noncardiac vascular surgery patients, and the one randomized trial of vascular surgery patients, data support the use of statins in nearly all patients undergoing cardiac or vascular surgery. We advocate starting statins in the perioperative period to take advantage of their rapid-acting pleiotropic effects, and continuing them long-term to take advantage of their lipid-lowering effects. This recommendation is in line with the recently released American College of Cardiology/American Heart Association (ACC/AHA) 2007 perioperative guidelines that state “for patients undergoing vascular surgery with or without clinical risk factors, statin use is reasonable.”42
Although the ideal time to start statins is not certain, the study by Durazzo et al23 suggests that they should be started at least 2 weeks before surgery if possible. Moreover, patients already taking statins should definitely not have their statins discontinued if at all possible.
Before major nonvascular surgery
For patients undergoing major nonvascular (intermediate-risk) surgery, physicians should first ascertain if the patient has an indication for statin therapy based on current nonsurgical lipid level recommendations. However, even if there is no clear indication for statin therapy based on NCEP guidelines, we endorse the recently released ACC/AHA perioperative guidelines that state that statin therapy can be considered in patients with a risk factor who are undergoing intermediate-risk procedures. Moreover, we wholeheartedly support the ACC/AHA’s strongest recommendation that patients who are already receiving statins and are undergoing noncardiac surgery should not have their statins discontinued.
When to discontinue statins?
The risk of harm overall appears to be minimal and certainly less than the likelihood of benefit. It is reasonable to observe patients postoperatively for adverse clinical events that may increase the risk of perioperative statin treatment, such as acute renal failure, hepatic failure, or sepsis, but whether statins should be stopped in patients with these complications remains unknown; we advocate individualizing the decision.
More studies needed
We need more data on whether moderate-risk patients undergoing moderate-risk surgery benefit from perioperative statin therapy, when therapy should be started, whether therapy should be started on the day of surgery if it was not started earlier, which statin and what doses are optimal, how long therapy should be continued, and what degree of risk is associated with perioperative statin therapy.
Fortunately, important data should be forthcoming in the next few years: the Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography (DECREASE-IV) study43 is a 4-year two-by-two factorial placebo-controlled study evaluating the use of fluvastatin (Lescol) and bisoprolol (Zebeta, a beta-blocker) separately and together in patients who are older than 40 years, are undergoing elective noncardiac surgery, have an estimated risk of cardiovascular death of more than 1%, have not used statins previously, and do not have elevated cholesterol.
Soon, the checklist for internists seeing patients about to undergo surgery may include prescribing one of the lipid-lowering hydroxymethylglutaryl-CoA reductase inhibitors, also called statins.
Statins? Not long ago, we were debating whether patients who take statins should stop taking them before surgery, based on the manufacturers’ recommendations.1 The discussion, however, has changed to whether patients who have never received a statin should be started on one before surgery to provide immediate prophylaxis against cardiac morbidity, and how much harm long-term statin users face if these drugs are withheld perioperatively.
The evidence is still very preliminary and based mostly on studies in animals and retrospective studies in people. However, an expanding body of indirect evidence suggests that these drugs are beneficial in this situation.
In this review, we discuss the mechanisms by which statins may protect the heart in the short term, drawing on data from animal and human studies of acute myocardial infarction, and we review the current (albeit limited) data from the perioperative setting.
FEW INTERVENTIONS DECREASE RISK
Each year, approximately 50,000 patients suffer a perioperative cardiovascular event; the incidence of myocardial infarction during or after noncardiac surgery is 2% to 3%.2 The primary goal of preoperative cardiovascular risk assessment is to predict and avert these events.
But short of canceling surgery, few interventions have been found to reduce a patient’s risk. For example, a landmark study in 2004 cast doubt on the efficacy of preoperative coronary revascularization.3 Similarly, although early studies of beta-blockers were promising4,5 and although most internists prescribe these drugs before surgery, more recent studies have cast doubt on their efficacy, particularly in patients at low risk undergoing intermediate-risk (rather than vascular) surgery.6–8
This changing clinical landscape has prompted a search for new strategies for perioperative risk-reduction. Several recent studies have placed statins in the spotlight.
POTENTIAL MECHANISMS OF SHORT-TERM BENEFIT
Statins have been proven to save lives when used long-term, but how could this class of drugs, designed to prevent the accumulation of arterial plaques by lowering low-density lipoprotein cholesterol (LDL-C) levels, have any short-term impact on operative outcomes? Although LDL-C reduction is the principal mechanism of action of statins, not all of the benefit can be ascribed to this mechanism.9 The answer may lie in their “pleiotropic” effects—ie, actions other than LDL-C reduction.
The more immediate pleiotropic effects of statins in the proinflammatory and prothrombotic environment of the perioperative period are thought to include improved endothelial function (both antithrombotic function and vasomotor function in response to ischemic stress), enhanced stability of atherosclerotic plaques, decreased oxidative stress, and decreased vascular inflammation.10–12
EVIDENCE FROM ANIMAL STUDIES
Experiments in animals suggest that statins, given shortly before or after a cardiovascular event, confer benefit before any changes in LDL-C are measurable.
Lefer et al13 found that simvastatin (Zocor), given 18 hours before an ischemic episode in rats, blunted the inflammatory response in cardiac reperfusion injury. Not only was reperfusion injury significantly less in the hearts of the rats that received simvastatin than in the saline control group, but the simvastatin-treated hearts also expressed fewer neutrophil adhesion molecules such as P-selectin, and they had more basal release of nitric oxide, the potent endothelial-derived vasodilator with antithrombotic, anti-inflammatory, and antiproliferative effects.14 These results suggest that statins may improve endothelial function acutely, particularly during ischemic stress.
Osborne et al15 fed rabbits a cholesterol-rich diet plus either lovastatin (Mevacor) or placebo. After 2 weeks, the rabbits underwent either surgery to induce a myocardial infarction or a sham procedure. Regardless of the pretreatment, biopsies of the aorta did not reveal any atherosclerosis; yet the lovastatin-treated rabbits sustained less myocardial ischemic damage and they had more endothelium-mediated vasodilatation.
Statin therapy also may improve cerebral ischemia outcomes in animal models.14,16
Sironi et al16 induced strokes in rats by occluding the middle cerebral artery. The rats received either simvastatin or vehicle for 3 days before the stroke or immediately afterwards. Even though simvastatin did not have enough time to affect the total cholesterol level, rats treated with simvastatin had smaller infarcts (as measured by magnetic resonance imaging) and produced more nitric oxide.
Comment. Taken together, these studies offer tantalizing evidence that statins have short-term, beneficial nonlipid effects and may reduce not only the likelihood of an ischemic event, but—should one occur—the degree of tissue damage that ensues.
EFFECTS OF STATINS IN ACUTE CORONARY SYNDROME
The National Registry of Myocardial Infarction17 is a prospective, observational database of all patients with acute myocardial infarction admitted to 1,230 participating hospitals throughout the United States. In an analysis from this cohort, patients were divided into four groups: those receiving statins before and after admission, those receiving statins only before admission, those receiving statins only after admission, and those who never received statins.
Compared with those who never received statins, fewer patients who received them both before and after admission died while in the hospital (unadjusted odds ratio 0.23, 95% confidence interval [CI] 0.22–0.25), and the odds ratio for those who received statins for the first time was 0.31 (95% CI 0.29–0.33). Patients who stopped receiving a statin on admission were more likely to die than were patients who never received statins (odds ratio 1.09, 95% CI 1.03–1.15). These trends held true even when adjustments were made for potential confounding factors.
Comment. Unmeasured confounding factors (such as the inability to take pills due to altered mental status or the different practice styles of the providers who chose to discontinue statins) might have affected the results. Nevertheless, these results suggest that the protective effects of statins stop almost immediately when these drugs are discontinued, and that there may even be an adverse “rebound” effect when patients who have been taking these drugs for a long time stop taking them temporarily.
The Platelet Receptor Inhibition in Ischemic Syndrome Management trial,18 in a subgroup analysis, had nearly identical findings. In the main part of this trial, patients with coronary artery disease and chest pain at rest or accelerating pain in the last 24 hours were randomized to receive tirofiban (Aggrastat) or heparin. Complete data on statin use were available for 1,616 (50%) of the 3,232 patients in this trial, and the rate of the primary end point (death, myocardial infarction, or recurrent ischemia) was analyzed on the basis of statin therapy in this subgroup.
Comment. Together, these data lead to the conclusion that, when admitted for either acute myocardial infarction or acute coronary syndrome, patients already receiving statins should not have them stopped, and those who had not been receiving statins should receive them immediately. The safety of these medications in the acute setting appears excellent: in the Myocardial Ischemia Reduction With Acute Cholesterol Lowering (MIRACL)12 and the Pravastatin or Atorvastatin Evaluation and Infection Therapy (PROVE-IT)11 trials, fewer than 5% of statin-treated patients had transient elevations in transaminase levels, and no cases of rhabdomyolysis were reported.
PERIOPERATIVE STATIN STUDIES
The data on perioperative statin use are mostly observational and retrospective and fall into essentially four surgical categories: coronary artery bypass grafting (CABG), carotid endarterectomy,19,20 noncardiac vascular surgery, and major noncardiac surgery. Two meta-analyses have also evaluated the data.21,22 The only randomized controlled trial (performed by Durazzo et al23) was small and was carried out at a single center in vascular surgery patients, and the event rate was low.
Current recommendations from the National Cholesterol Education Program (NCEP)24 say that patients who need CABG, have peripheral arterial disease, have an abdominal aortic aneurysm, or have cerebrovascular disease should already be on a statin to achieve an LDL-C goal level of less than 100 mg/dL, with an optional goal of less than 70 mg/dL, independent of surgery.
Since not all patients who should be on statins are actually on them, questions arise:
- Is it important (and safe) to start statin treatment preoperatively?
- Will patients with cardiovascular risk factors but without known cardiovascular disease benefit from statins perioperatively?
Noncardiac vascular surgery
Multiple retrospective studies have evaluated the effect of statins in patients undergoing major noncardiac vascular surgery.25–32
Kertai et al25 evaluated 570 patients in Holland who underwent elective open surgery for infrarenal abdominal aortic aneurysms between 1991 and 2001, looking for an association between statin use and the incidence of perioperative death from myocardial infarction. Only 162 of the 570 patients had been on long-term statin therapy before the surgery. The use of statins was only one of many known baseline characteristics that were significantly different between the two groups, including age, body mass index, known coronary artery disease, and use of angiotensin-converting enzyme inhibitors and beta-blockers. In univariate analysis, statins appeared to be protective: 6 (3.7%) of the patients in the statin group died of a myocardial infarction, compared with 45 (11%) of those in the nostatin group. A multivariate analysis yielded similar findings, with an odds ratio of 0.24 (95% CI 0.11–0.54).
Ward et al27 performed a very similar retrospective study, with similar findings. In 446 patients who underwent surgery for infrarenal abdominal aortic aneurysm, statin therapy was associated with a significantly lower incidence of the combined end point of death, myocardial infarction, stroke, and major peripheral vascular complications, with an adjusted odds ratio of 0.36 (95% CI 0.14–0.93).
Poldermans et al26 noted similar findings in a case-control study of noncardiac vascular surgery patients. Statin users had a much lower perioperative risk of death than did nonusers, with an adjusted odds ratio of 0.22 (95% CI 0.10–0.47).
O’Neil-Callahan et al,28 in a cohort study, found that statin users had fewer perioperative cardiac complications, with an adjusted odds ratio of 0.49 (95% CI 0.28–0.84, P = .009).
Dogma of withdrawing statins before major surgery is challenged
Le Manach et al33 reviewed the outcomes for all patients of a single hospital in Paris who underwent nonemergency infrarenal aortic procedures between January 2001 and December 2004. In January 2004, the hospital instituted guidelines to ensure that patients on statins continue taking them up to the evening before surgery and that statins be restarted on the first postoperative day (via nasogastric tube if necessary). Before 2004, there had been no specific guidelines, and patients on statins did not receive them for a median of 4 days postoperatively. Types of procedures were similar during the two time periods, as were the rates of beta-blocker use, preoperative revascularization, venous thromboembolism prophylaxis, and perioperative blood pressure control. After surgery, topononin I levels were measured in all patients as surveillance for cardiac events, and were defined as elevated when greater than 0.2 ng/mL.
Compared with patients not on statins at all, those treated with statins continuously throughout the perioperative period (after January 2004) had a lower rate of elevated troponin (relative risk 0.38). In contrast, those who had their statins transiently discontinued perioperatively (prior to 2004) had troponin elevations more often than those who had never been treated (relative risk 2.1). This suggested an over fivefold risk reduction (P < .001) conferred by not discontinuing statins in the immediate postoperative period. This finding was maintained after multivariate adjustment: statin withdrawal was associated with a 2.9-fold (95% CI 1.6–5.5) increase in the risk of cardiac enzyme elevations postoperatively. No fewer deaths were noted, but the study was not powered to detect a mortality difference.
Comment. Although secular trends cannot be entirely discounted as contributing to these findings, the prompt increase in cardiac events after just 4 days of statin withdrawal adds to the growing body of evidence suggesting that statin discontinuation can have harmful acute effects. It also brings up the question: Can starting statins benefit patients in the same time period?
Should statins be started before vascular surgery?
Schouten et al32 evaluated the effects of newly started or continued statin treatment in patients undergoing major elective vascular surgery. Patients were screened before surgery and started on statins if they were not already receiving them and their total cholesterol levels were elevated; new users received the medication for about 40 days before surgery. Of the 981 screened patients, 44 (5%) were newly started on statins and 182 (19%) were continued on their therapy. Perioperative death or myocardial infarction occurred in 22 (8.8%) of the statin users and 111 (14.7%) of the nonusers, a statistically significant difference. Temporary discontinuation (median 1 day) of statins in this study due to the inability to take an oral medication did not appear to affect the likelihood of a myocardial infarction.
Durazzo et al23 performed a single-center, randomized, prospective, placebo-controlled, double-blind clinical trial of atorvastatin (Lipitor) 20 mg daily vs placebo in 100 patients undergoing noncardiac arterial vascular surgery. Patients were excluded if they had previously used medications to treat dyslipidemia, recently had a cardiovascular event, or had contraindications to statin treatment such as a baseline creatinine level greater than 2.0 mg/dL or severe hepatic disease. The intervention group received atorvastatin starting at least 2 weeks before surgery for a total of 45 days. Patients were then continued or started on a statin after surgery if their LDL-C level was greater than 100 mg/dL. Beta-blocker use was recommended “on the basis of current guidelines.”
One month after surgery, the LDL-C level was statistically significantly lower in the atorvastatin group. Since most patients did not continue or start statin therapy after the 45-day treatment period, the LDL-C levels were not statistically different at 3 and 6 months after surgery.
At 6 months, the rate of the primary end point (death from cardiovascular causes, nonfatal acute myocardial infarction, ischemic stroke, or unstable angina) was 26.0% in the placebo group and 8.0% in the atorvastatin group, a statistically significant difference. Three patients in the atorvastatin group had cardiac events in the first 10 days after surgery, compared with 11 patients in the placebo group. Thirteen of the 17 total cardiac events took place within 10 days after surgery.
One of the atorvastatin patients developed rhabdomyolysis and elevated aminotransferase levels.
Major noncardiac surgery
Lindenauer et al2 performed a retrospective cohort study of surgical patients who were at least 18 years old and survived beyond the second hospital day. Patients were divided into a group receiving any form of lipid-lowering treatment (of whom more than 90% were taking statins) and a group that had never never received a lipid-lowering drug or only started one on the third day of the hospitalization or later. The period of study was from January 1, 2000, to December 31, 2001.
In all, 780,591 patients from 329 hospitals throughout the United States were included, of whom only 77,082 (9.9%) received lipid-lowering therapy. Eight percent of the patients underwent vascular surgery. Not surprisingly, the treated patients were more likely to have a history of hypertension, diabetes, ischemic heart disease, or hyperlipidemia. They also were more likely to have a vascular procedure performed, to have two or more cardiac risk factors (high-risk surgery, ischemic heart disease, congestive heart failure, cerebrovascular disease, renal insufficiency, or diabetes mellitus), and to be treated with beta-blockers and angiotensin-converting enzyme inhibitors, but they were less likely to have high-risk and emergency surgery performed.
The primary end point, perioperative death, occurred in 2.13% of the treated patients and 3.05% of the nontreated group. Compared with the rate in a propensity-matched cohort, the odds ratio adjusted for unbalanced covariates was 0.62 (95% CI 0.58–0.67) in favor of lipid treatment. Stratification by cardiac risk index revealed a number needed to treat of 186 for those with no risk factors, 60 for those with two risk factors, and 30 for those with four or more risk factors.
Unfortunately, this analysis was not able to take into account whether and for how long patients were receiving lipid-lowering therapy before hospitalization. It therefore does not answer the questions of whether starting lipid-lowering therapy before surgery is beneficial or whether stopping it is harmful. It also does not shed light on whether perioperative lipid-lowering increases the risk of rhabdomyolysis or liver disease.
Carotid endarterectomy
Two recent retrospective cohort studies evaluated the outcomes in patients undergoing carotid endarterectomy.19,20
Kennedy et al19 found that patients on a statin at the time of admission who had symptomatic carotid disease had lower rates of inhospital death (adjusted odds ratio 0.24, 95% CI 0.06–0.91) and ischemic stroke or death (adjusted odds ratio 0.55, 95% CI 0.31–0.97). However, cardiac outcomes among these symptomatic patients were not significantly improved (odds ratio 0.82, 95% CI 0.45–1.50), nor was there benefit for asymptomatic patients, raising the possibility that the positive findings were due to chance or that patients at lower baseline risk for vascular events may have less benefit.
McGirt et al20 performed a similar study; they did not, however, distinguish whether patients had symptomatic vs asymptomatic carotid disease. The 30-day risk of perioperative stroke was lower in patients treated with a statin, with an odds ratio of 0.41 (95% CI 0.18–0.93); the odds ratio for death was 0.21 (95% CI 0.05–0.96). Cardiac outcomes were not significantly affected.
Coronary artery bypass graft surgery
According to the NCEP recommendations, nearly all patients undergoing CABG should already be on a statin before surgery since they all have known coronary artery disease. Multiple observational studies have offered confirmatory evidence that statins are beneficial in this setting.34–38
Liakopoulos et al39 evaluated whether the anti-inflammatory effects of statins may, in part, account for their beneficial effect in the perioperative period. The authors prospectively matched 18 patients who were taking statins and were referred for elective CABG with 18 patients who were not prescribed statins previously. The only major measured baseline characteristic that differed between the two groups was a statistically significantly lower LDL-C level in the statin group. The operative characteristics did not differ, and cytokine levels at baseline were similar.
Tumor necrosis factor alpha levels increased significantly in the control group but did not change significantly in the statin group. Interleukin 8 increased in both groups by a similar amount. Interleukin 6 (the major inducer of C-reactive protein) increased from baseline in both groups but did not increase nearly as much in the statin group as in the control group; the intergroup difference was statistically significant. The anti-inflammatory cytokine interleukin 10 increased minimally from baseline in the control group, while the statin group’s levels increased significantly above baseline and those of the control group.
Christenson40 also found that inflammatory markers were improved with pre-CABG statin treatment in a small randomized trial in which patients received simvastatin 20 mg 4 weeks prior to CABG surgery vs no statin. Interestingly, far fewer statin-treated patients developed thrombocytosis (platelet count > 400 × 109/L) than did control patients (3% vs 81%, P < .0001).
RISKS OF PERIOPERATIVE STATINS
The risks associated with statin therapy in general appear low, but specific perioperative risks have not been well studied.
Baigent et al,41 in a meta-analysis of randomized trials of nonperioperative statin therapy, found that rhabdomyolysis occurred in 9 (0.023%) of 39,884 patients receiving statins vs 6 (0.015%) of the 39,817 controls, with a number needed to harm of 12,500. Moreover, the rates of nonvascular death and cancer did not increase. It is plausible that the risk is somewhat greater in the perioperative setting but is likely not enough to outweigh the potential benefits, especially since the risk of ischemic vascular events is particularly high then.
Some of the perioperative studies cited above specifically addressed potential risks. For example, in the study by Schouten et al,32 mild creatine kinase elevations were more common in the statin-treated group, but the incidence of moderate and severe creatine kinase elevations did not differ significantly. No case of rhabdomyolysis occurred, and length of surgery was the only predictor of myopathy. MIRACL and PROVE-IT revealed similar safety profiles; aminotransferase levels normalized when statins were stopped, and no cases of rhabdomyolysis occurred.11,12 In the vascular surgery study by Durazzo et al,23 1 (2%) of the 50 atorvastatin-treated patients developed both rhabdomyolysis and elevated aminotransferase levels that prompted discontinuation of the statin.
Overall, the observational studies do not indicate that statin continuation or treatment is harmful in perioperative patients. However, these studies did not specifically evaluate patients with acute insults from surgery such as sepsis, renal failure, or hepatitis. It is unknown what effect statin therapy would have in those patients and whether statins should be selectively discontinued in patients who develop major hepatic, musculoskeletal, or renal complications after surgery.
OUR RECOMMENDATIONS
Before CABG or vascular surgery
Given the NCEP recommendations, existing primary and secondary prevention studies, observational studies of CABG and noncardiac vascular surgery patients, and the one randomized trial of vascular surgery patients, data support the use of statins in nearly all patients undergoing cardiac or vascular surgery. We advocate starting statins in the perioperative period to take advantage of their rapid-acting pleiotropic effects, and continuing them long-term to take advantage of their lipid-lowering effects. This recommendation is in line with the recently released American College of Cardiology/American Heart Association (ACC/AHA) 2007 perioperative guidelines that state “for patients undergoing vascular surgery with or without clinical risk factors, statin use is reasonable.”42
Although the ideal time to start statins is not certain, the study by Durazzo et al23 suggests that they should be started at least 2 weeks before surgery if possible. Moreover, patients already taking statins should definitely not have their statins discontinued if at all possible.
Before major nonvascular surgery
For patients undergoing major nonvascular (intermediate-risk) surgery, physicians should first ascertain if the patient has an indication for statin therapy based on current nonsurgical lipid level recommendations. However, even if there is no clear indication for statin therapy based on NCEP guidelines, we endorse the recently released ACC/AHA perioperative guidelines that state that statin therapy can be considered in patients with a risk factor who are undergoing intermediate-risk procedures. Moreover, we wholeheartedly support the ACC/AHA’s strongest recommendation that patients who are already receiving statins and are undergoing noncardiac surgery should not have their statins discontinued.
When to discontinue statins?
The risk of harm overall appears to be minimal and certainly less than the likelihood of benefit. It is reasonable to observe patients postoperatively for adverse clinical events that may increase the risk of perioperative statin treatment, such as acute renal failure, hepatic failure, or sepsis, but whether statins should be stopped in patients with these complications remains unknown; we advocate individualizing the decision.
More studies needed
We need more data on whether moderate-risk patients undergoing moderate-risk surgery benefit from perioperative statin therapy, when therapy should be started, whether therapy should be started on the day of surgery if it was not started earlier, which statin and what doses are optimal, how long therapy should be continued, and what degree of risk is associated with perioperative statin therapy.
Fortunately, important data should be forthcoming in the next few years: the Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography (DECREASE-IV) study43 is a 4-year two-by-two factorial placebo-controlled study evaluating the use of fluvastatin (Lescol) and bisoprolol (Zebeta, a beta-blocker) separately and together in patients who are older than 40 years, are undergoing elective noncardiac surgery, have an estimated risk of cardiovascular death of more than 1%, have not used statins previously, and do not have elevated cholesterol.
- Grant PJ, Kedia N. Should statins be discontinued preoperatively? IMPACT consults. Proceedings of the 2nd Annual Cleveland Clinic Perioperative Medicine Summit. Cleve Clin J Med 2006; 73 Electronic suppl 1:S9–S10.
- Lindenauer PK, Pekow P, Wang K, Gutierrez B, Benjamin EM. Lipid-lowering therapy and in-hospital mortality following major noncardiac surgery. JAMA 2004; 291:2092–2099.
- McFalls EO, Ward HB, Moritz TE, et al. Coronary-artery revascularization before elective major vascular surgery. N Engl J Med 2004; 351:2795–2804.
- Mangano DT, Layug EL, Wallace A, Tateo I. Effect of atenolol on mortality and cardiovascular morbidity after noncardiac surgery. Multicenter Study of Perioperative Ischemia Research Group. N Engl J Med 1996; 335:1713–1720.
- Poldermans D, Boersma E, Bax JJ, et al. The effect of bisoprolol on perioperative mortality and myocardial infarction in high-risk patients undergoing vascular surgery. Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group. N Engl J Med 1999; 341:1789–1794.
- Brady AR, Gibbs JS, Greenhalgh RM, Powell JT, Sydes MR. Perioperative beta-blockade (POBBLE) for patients undergoing infrarenal vascular surgery: results of a randomized double-blind controlled trial. J Vasc Surg 2005; 41:602–609.
- Juul AB, Wetterslev J, Gluud C, et al. Effect of perioperative beta blockade in patients with diabetes undergoing major non-cardiac surgery: randomised placebo controlled, blinded multicentre trial. BMJ 2006; 332:1482.
- Yang H, Raymer K, Butler R, Parlow J, Roberts R. The effects of perioperative beta-blockade: results of the Metoprolol after Vascular Surgery (MaVS) study, a randomized controlled trial. Am Heart J 2006; 152:983–990.
- Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med 2005; 352:20–28.
- Ito MK, Talbert RL, Tsimikas S. Statin-associated pleiotropy: possible beneficial effects beyond cholesterol reduction. Pharmacotherapy 2006; 26:85S–97S.
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
- Lefer AM, Campbell B, Shin YK, Scalia R, Hayward R, Lefer DJ. Simvastatin preserves the ischemic-reperfused myocardium in normocholesterolemic rat hearts. Circulation 1999; 100:178–184.
- Endres M, Laufs U, Liao JK, Moskowitz MA. Targeting eNOS for stroke protection. Trends Neurosci 2004; 27:283–289.
- Osborne JA, Lento PH, Siegfried MR, Stahl GL, Fusman B, Lefer AM. Cardiovascular effects of acute hypercholesterolemia in rabbits. Reversal with lovastatin treatment. J Clin Invest 1989; 83:465–473.
- Sironi L, Cimino M, Guerrini U, et al. Treatment with statins after induction of focal ischemia in rats reduces the extent of brain damage. Arterioscler Thromb Vasc Biol 2003; 23:322–327.
- Fonarow GC, Wright RS, Spencer FA, et al. Effect of statin use within the first 24 hours of admission for acute myocardial infarction on early morbidity and mortality. Am J Cardiol 2005; 96:611–616.
- Heeschen C, Hamm CW, Laufs U, Snapinn S, Bohm M, White HD. Withdrawal of statins increases event rates in patients with acute coronary syndromes. Circulation 2002; 105:1446–1452.
- Kennedy J, Quan H, Buchan AM, Ghali WA, Feasby TE. Statins are associated with better outcomes after carotid endarterectomy in symptomatic patients. Stroke 2005; 36:2072–2076.
- McGirt MJ, Perler BA, Brooke BS, et al. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors reduce the risk of perioperative stroke and mortality after carotid endarterectomy. J Vasc Surg 2005; 42:829–836.
- Hindler K, Shaw AD, Samuels J, Fulton S, Collard CD, Riedel B. Improved postoperative outcomes associated with preoperative statin therapy. Anesthesiology 2006; 105:1260–1272.
- Kapoor AS, Kanji H, Buckingham J, Devereaux PJ, McAlister FA. Strength of evidence for perioperative use of statins to reduce cardiovascular risk: systematic review of controlled studies. BMJ 2006; 333:1149.
- Durazzo AE, Machado FS, Ikeoka DT, et al. Reduction in cardiovascular events after vascular surgery with atorvastatin: a randomized trial. J Vasc Surg 2004; 39:967–975.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- Kertai MD, Boersma E, Westerhout CM, et al. A combination of statins and beta-blockers is independently associated with a reduction in the incidence of perioperative mortality and nonfatal myocardial infarction in patients undergoing abdominal aortic aneurysm surgery. Eur J Vasc Endovasc Surg 2004; 28:343–352.
- Poldermans D, Bax JJ, Kertai MD, et al. Statins are associated with a reduced incidence of perioperative mortality in patients undergoing major noncardiac vascular surgery. Circulation 2003; 107:1848–1851.
- Ward RP, Leeper NJ, Kirkpatrick JN, Lang RM, Sorrentino MJ, Williams KA. The effect of preoperative statin therapy on cardiovascular outcomes in patients undergoing infrainguinal vascular surgery. Int J Cardiol 2005; 104:264–268.
- O’Neil-Callahan K, Katsimaglis G, Tepper MR, et al. Statins decrease perioperative cardiac complications in patients undergoing non-cardiac vascular surgery: the Statins for Risk Reduction in Surgery (StaRRS) study. J Am Coll Cardiol 2005; 45:336–342.
- Abbruzzese TA, Havens J, Belkin M, et al. Statin therapy is associated with improved patency of autogenous infrainguinal bypass grafts. J Vasc Surg 2004; 39:1178–1185.
- Boersma E, Poldermans D, Bax JJ, et al. Predictors of cardiac events after major vascular surgery: role of clinical characteristics, dobutamine echocardiography, and beta-blocker therapy. JAMA 2001; 285:1865–1873.
- Landesberg G, Mosseri M, Wolf YG, et al. Preoperative thallium scanning, selective coronary revascularization, and long-term survival after major vascular surgery. Circulation 2003; 108:177–183.
- Schouten O, Kertai MD, Bax JJ, et al. Safety of perioperative statin use in high-risk patients undergoing major vascular surgery. Am J Cardiol 2005; 95:658–660.
- Le Manach Y, Godet G, Coriat P, et al. The impact of postoperative discontinuation or continuation of chronic statin therapy on cardiac outcome after major vascular surgery. Anesth Analg 2007; 104:1326–1333.
- Ali IS, Buth KJ. Preoperative statin use and outcomes following cardiac surgery. Int J Cardiol 2005; 103:12–18.
- Clark LL, Ikonomidis JS, Crawford FA, et al. Preoperative statin treatment is associated with reduced postoperative mortality and morbidity in patients undergoing cardiac surgery: an 8-year retrospective cohort study. J Thorac Cardiovasc Surg 2006; 131:679–685.
- Pan W, Pintar T, Anton J, Lee VV, Vaughn WK, Collard CD. Statins are associated with a reduced incidence of perioperative mortality after coronary artery bypass graft surgery. Circulation 2004; 110(suppl 2):II45–II49.
- Pascual DA, Arribas JM, Tornel PL, et al. Preoperative statin therapy and troponin T predict early complications of coronary artery surgery. Ann Thorac Surg 2006; 81:78–83.
- Dotani MI, Elnicki DM, Jain AC, Gibson CM. Effect of preoperative statin therapy and cardiac outcomes after coronary artery bypass grafting. Am J Cardiol 2000; 86:1128–1130.
- Liakopoulos OJ, Dorge H, Schmitto JD, Nagorsnik U, Grabedunkel J, Schoendube FA. Effects of preoperative statin therapy on cytokines after cardiac surgery. Thorac Cardiovasc Surg 2006; 54:250–254.
- Christenson JT. Preoperative lipid-control with simvastatin reduces the risk of postoperative thrombocytosis and thrombotic complications following CABG. Eur J Cardiothorac Surg 1999; 15:394–399.
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Fleisher LA, Beckman JA, Brown KA, et al. ACC/AHA 2007 Guidelines on Perioperative Cardiovascular Evaluation and Care for Noncardiac Surgery. A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery). Circulation 2007; 116:e418–e499.
- Schouten O, Poldermans D, Visser L, et al. Fluvastatin and bisoprolol for the reduction of perioperative cardiac mortality and morbidity in high-risk patients undergoing non-cardiac surgery: rationale and design of the DECREASE-IV study. Am Heart J 2004; 148:1047–1052.
- Amar D, Zhang H, Heerdt PM, Park B, Fleisher M, Thaler HT. Statin use is associated with a reduction in atrial fibrillation after noncardiac thoracic surgery independent of C-reactive protein. Chest 2005; 128:3421–3427.
- Grant PJ, Kedia N. Should statins be discontinued preoperatively? IMPACT consults. Proceedings of the 2nd Annual Cleveland Clinic Perioperative Medicine Summit. Cleve Clin J Med 2006; 73 Electronic suppl 1:S9–S10.
- Lindenauer PK, Pekow P, Wang K, Gutierrez B, Benjamin EM. Lipid-lowering therapy and in-hospital mortality following major noncardiac surgery. JAMA 2004; 291:2092–2099.
- McFalls EO, Ward HB, Moritz TE, et al. Coronary-artery revascularization before elective major vascular surgery. N Engl J Med 2004; 351:2795–2804.
- Mangano DT, Layug EL, Wallace A, Tateo I. Effect of atenolol on mortality and cardiovascular morbidity after noncardiac surgery. Multicenter Study of Perioperative Ischemia Research Group. N Engl J Med 1996; 335:1713–1720.
- Poldermans D, Boersma E, Bax JJ, et al. The effect of bisoprolol on perioperative mortality and myocardial infarction in high-risk patients undergoing vascular surgery. Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group. N Engl J Med 1999; 341:1789–1794.
- Brady AR, Gibbs JS, Greenhalgh RM, Powell JT, Sydes MR. Perioperative beta-blockade (POBBLE) for patients undergoing infrarenal vascular surgery: results of a randomized double-blind controlled trial. J Vasc Surg 2005; 41:602–609.
- Juul AB, Wetterslev J, Gluud C, et al. Effect of perioperative beta blockade in patients with diabetes undergoing major non-cardiac surgery: randomised placebo controlled, blinded multicentre trial. BMJ 2006; 332:1482.
- Yang H, Raymer K, Butler R, Parlow J, Roberts R. The effects of perioperative beta-blockade: results of the Metoprolol after Vascular Surgery (MaVS) study, a randomized controlled trial. Am Heart J 2006; 152:983–990.
- Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med 2005; 352:20–28.
- Ito MK, Talbert RL, Tsimikas S. Statin-associated pleiotropy: possible beneficial effects beyond cholesterol reduction. Pharmacotherapy 2006; 26:85S–97S.
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
- Lefer AM, Campbell B, Shin YK, Scalia R, Hayward R, Lefer DJ. Simvastatin preserves the ischemic-reperfused myocardium in normocholesterolemic rat hearts. Circulation 1999; 100:178–184.
- Endres M, Laufs U, Liao JK, Moskowitz MA. Targeting eNOS for stroke protection. Trends Neurosci 2004; 27:283–289.
- Osborne JA, Lento PH, Siegfried MR, Stahl GL, Fusman B, Lefer AM. Cardiovascular effects of acute hypercholesterolemia in rabbits. Reversal with lovastatin treatment. J Clin Invest 1989; 83:465–473.
- Sironi L, Cimino M, Guerrini U, et al. Treatment with statins after induction of focal ischemia in rats reduces the extent of brain damage. Arterioscler Thromb Vasc Biol 2003; 23:322–327.
- Fonarow GC, Wright RS, Spencer FA, et al. Effect of statin use within the first 24 hours of admission for acute myocardial infarction on early morbidity and mortality. Am J Cardiol 2005; 96:611–616.
- Heeschen C, Hamm CW, Laufs U, Snapinn S, Bohm M, White HD. Withdrawal of statins increases event rates in patients with acute coronary syndromes. Circulation 2002; 105:1446–1452.
- Kennedy J, Quan H, Buchan AM, Ghali WA, Feasby TE. Statins are associated with better outcomes after carotid endarterectomy in symptomatic patients. Stroke 2005; 36:2072–2076.
- McGirt MJ, Perler BA, Brooke BS, et al. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors reduce the risk of perioperative stroke and mortality after carotid endarterectomy. J Vasc Surg 2005; 42:829–836.
- Hindler K, Shaw AD, Samuels J, Fulton S, Collard CD, Riedel B. Improved postoperative outcomes associated with preoperative statin therapy. Anesthesiology 2006; 105:1260–1272.
- Kapoor AS, Kanji H, Buckingham J, Devereaux PJ, McAlister FA. Strength of evidence for perioperative use of statins to reduce cardiovascular risk: systematic review of controlled studies. BMJ 2006; 333:1149.
- Durazzo AE, Machado FS, Ikeoka DT, et al. Reduction in cardiovascular events after vascular surgery with atorvastatin: a randomized trial. J Vasc Surg 2004; 39:967–975.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- Kertai MD, Boersma E, Westerhout CM, et al. A combination of statins and beta-blockers is independently associated with a reduction in the incidence of perioperative mortality and nonfatal myocardial infarction in patients undergoing abdominal aortic aneurysm surgery. Eur J Vasc Endovasc Surg 2004; 28:343–352.
- Poldermans D, Bax JJ, Kertai MD, et al. Statins are associated with a reduced incidence of perioperative mortality in patients undergoing major noncardiac vascular surgery. Circulation 2003; 107:1848–1851.
- Ward RP, Leeper NJ, Kirkpatrick JN, Lang RM, Sorrentino MJ, Williams KA. The effect of preoperative statin therapy on cardiovascular outcomes in patients undergoing infrainguinal vascular surgery. Int J Cardiol 2005; 104:264–268.
- O’Neil-Callahan K, Katsimaglis G, Tepper MR, et al. Statins decrease perioperative cardiac complications in patients undergoing non-cardiac vascular surgery: the Statins for Risk Reduction in Surgery (StaRRS) study. J Am Coll Cardiol 2005; 45:336–342.
- Abbruzzese TA, Havens J, Belkin M, et al. Statin therapy is associated with improved patency of autogenous infrainguinal bypass grafts. J Vasc Surg 2004; 39:1178–1185.
- Boersma E, Poldermans D, Bax JJ, et al. Predictors of cardiac events after major vascular surgery: role of clinical characteristics, dobutamine echocardiography, and beta-blocker therapy. JAMA 2001; 285:1865–1873.
- Landesberg G, Mosseri M, Wolf YG, et al. Preoperative thallium scanning, selective coronary revascularization, and long-term survival after major vascular surgery. Circulation 2003; 108:177–183.
- Schouten O, Kertai MD, Bax JJ, et al. Safety of perioperative statin use in high-risk patients undergoing major vascular surgery. Am J Cardiol 2005; 95:658–660.
- Le Manach Y, Godet G, Coriat P, et al. The impact of postoperative discontinuation or continuation of chronic statin therapy on cardiac outcome after major vascular surgery. Anesth Analg 2007; 104:1326–1333.
- Ali IS, Buth KJ. Preoperative statin use and outcomes following cardiac surgery. Int J Cardiol 2005; 103:12–18.
- Clark LL, Ikonomidis JS, Crawford FA, et al. Preoperative statin treatment is associated with reduced postoperative mortality and morbidity in patients undergoing cardiac surgery: an 8-year retrospective cohort study. J Thorac Cardiovasc Surg 2006; 131:679–685.
- Pan W, Pintar T, Anton J, Lee VV, Vaughn WK, Collard CD. Statins are associated with a reduced incidence of perioperative mortality after coronary artery bypass graft surgery. Circulation 2004; 110(suppl 2):II45–II49.
- Pascual DA, Arribas JM, Tornel PL, et al. Preoperative statin therapy and troponin T predict early complications of coronary artery surgery. Ann Thorac Surg 2006; 81:78–83.
- Dotani MI, Elnicki DM, Jain AC, Gibson CM. Effect of preoperative statin therapy and cardiac outcomes after coronary artery bypass grafting. Am J Cardiol 2000; 86:1128–1130.
- Liakopoulos OJ, Dorge H, Schmitto JD, Nagorsnik U, Grabedunkel J, Schoendube FA. Effects of preoperative statin therapy on cytokines after cardiac surgery. Thorac Cardiovasc Surg 2006; 54:250–254.
- Christenson JT. Preoperative lipid-control with simvastatin reduces the risk of postoperative thrombocytosis and thrombotic complications following CABG. Eur J Cardiothorac Surg 1999; 15:394–399.
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Fleisher LA, Beckman JA, Brown KA, et al. ACC/AHA 2007 Guidelines on Perioperative Cardiovascular Evaluation and Care for Noncardiac Surgery. A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery). Circulation 2007; 116:e418–e499.
- Schouten O, Poldermans D, Visser L, et al. Fluvastatin and bisoprolol for the reduction of perioperative cardiac mortality and morbidity in high-risk patients undergoing non-cardiac surgery: rationale and design of the DECREASE-IV study. Am Heart J 2004; 148:1047–1052.
- Amar D, Zhang H, Heerdt PM, Park B, Fleisher M, Thaler HT. Statin use is associated with a reduction in atrial fibrillation after noncardiac thoracic surgery independent of C-reactive protein. Chest 2005; 128:3421–3427.
KEY POINTS
- Experiments in animals suggest that statins, given shortly before or after a cardiovascular event, confer benefit before any changes in lipids are measurable.
- Retrospective and prospective studies indicate that patients with either acute myocardial infarction or acute coronary syndrome who are already receiving statins should not have them stopped, and those who had not been receiving statins should receive them immediately.
- Most patients undergoing coronary artery bypass grafting or noncardiac vascular surgery should already be receiving a statin. These drugs can also be considered in patients undergoing intermediate-risk nonvascular surgery. Patients who have been receiving statins prior to surgery should not have them stopped for surgery.
New asthma guidelines emphasize control, regular monitoring
This review focuses on several elements in the National Asthma Education and Prevention Program’s new guidelines, the third Expert Panel Report (EPR3),1 that differ substantially from those in EPR2,2 issued in 1997 and updated in 2002.3 These differences in approach to the management of asthma described in EPR3 offer a clear potential for reducing the gap between optimal asthma care outcomes as described in guidelines and normative asthma care outcomes in the “real world.”
GREATER EMPHASIS ON CONTROL
The EPR2 guidelines2 recommended that asthma management be carried out in an algorithmic manner. Patients were classified into four severity categories: mild intermittent, mild persistent, moderate persistent, and severe persistent asthma, based on assessment of the level of symptoms (day/night), reliance on “reliever” medication, and lung function at the time of presentation. Pharmacologic management was then assigned according to each respective categorization in an evidence-based fashion.
In an ideal world, this would result in patients with asthma receiving appropriate pharmacotherapeutic agents associated with favorable asthma care outcomes, which were also advantageous from both cost- and risk-benefit standpoints. In the real world, however, this paradigm was flawed, as it relied on accurate categorization of patients in order for pharmacotherapy to be prescribed appropriately. Both providers and patients are prone to underestimate asthma severity,4,5 and for this reason many patients managed on the basis of this paradigm were undertreated.
A new paradigm, based on the assessment of asthma control, has been encouraged in the EPR3 guidelines.1
Severity and control are not synonymous
More than a decade ago, Cockroft and Swystun6 pointed out that asthma control (or lack thereof) is often used inappropriately to define asthma severity: ie, well-controlled asthma is seen as synonymous with mild asthma, and poorly controlled asthma with severe asthma.
Asthma severity can be defined as the intrinsic intensity of the disease process, while asthma control is the degree to which the manifestations of asthma are minimized. Asthma severity is clearly a determinant of asthma control, but its impact is affected by a variety of factors, including but not limited to:
- Whether appropriate medication is prescribed
- Patterns of therapeutic adherence
- The degree to which recommended measures for avoiding for clinically relevant aeroallergens are pursued.
Health care utilization, including hospitalizations and emergency department visits, correlates more closely with asthma control than with asthma severity.7–9 Indeed, a patient with severe persistent asthma who is treated appropriately with multiple “controller” medications and who takes his or her medications and avoids allergens as directed can achieve well-controlled or totally controlled asthma, and is not likely to require hospitalization or emergency department management, to miss school or work, or to experience nocturnal awakening or limitation in routine activities due to asthma. This patient has severe persistent asthma that is well controlled.
In contrast, a patient with mild or moderate persistent asthma who does not receive appropriate instructions for avoiding allergens or taking controller medication regularly or who is poorly adherent will likely have poor asthma control. This patient is more likely to require hospitalization or emergency department management, to miss school or work, and to experience nocturnal awakening or limitation in routine activities due to asthma. This patient has mild persistent asthma that is poorly controlled.
Assess asthma severity in the first visit, and control in subsequent visits
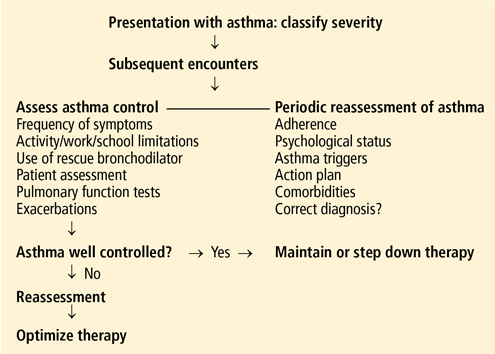
How to assess severity
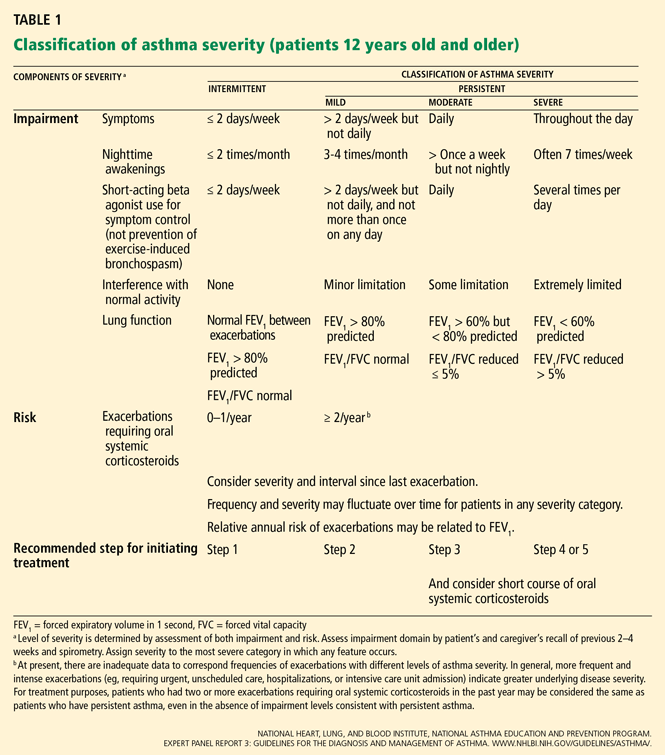
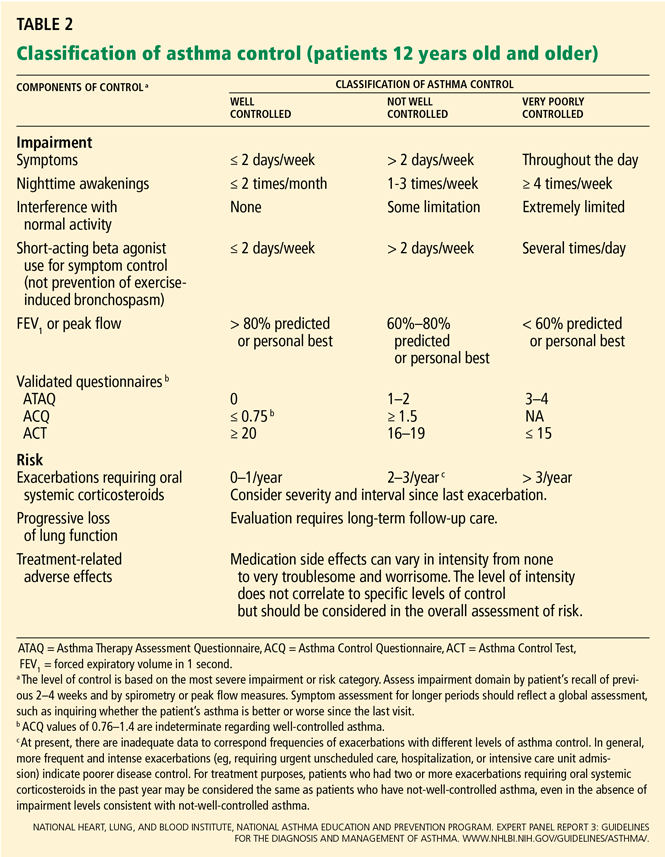
How to measure control
For all patients with asthma, regardless of severity, the goal is the same: to achieve control by reducing both impairment and risk. Asthma is classified as well controlled, not well controlled, or poorly controlled (Table 2).1
Validated tests are available to assess control
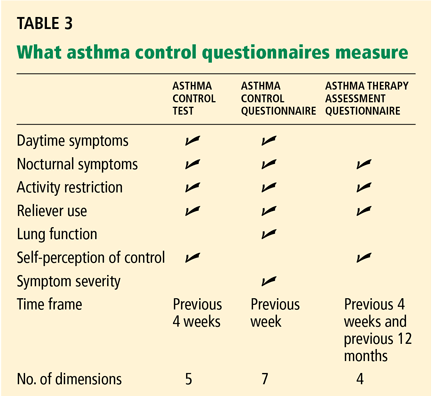
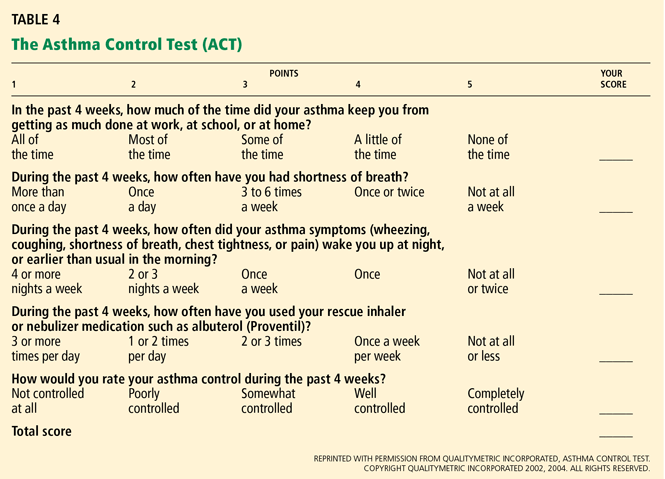
Serial testing as a quality indicator
Serial ACT scores give an objective measure of the degree to which the goals of management1 are being achieved, and in so doing can encourage optimal outcomes.14
Another use of these tests is to document whether asthma control improves over time when patients receive care from a particular physician or group. This use may become increasingly important in view of efforts underway to implement a pay-for-performance model for asthma care, in which providers will be financially rewarded for improved patient care outcomes and adherence to standards of practice based on Health Plan Employer Data and Information Set measures.15
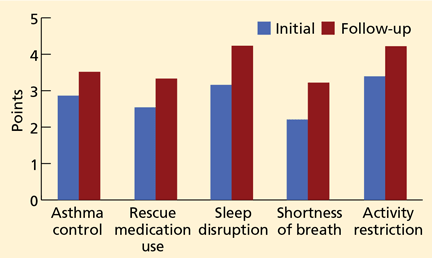
We have used the ACT in the Section of Allergy/Immunology at Cleveland Clinic for 3 years on a routine basis. All patients with asthma being seen either for the first time or as follow-up complete the ACT, which has been entered in a flow sheet in our electronic medical record, at the same time they undergo spirometry. We have shown that care in the Section of Allergy/Immunology is associated with improvement in asthma control over time, in patients who have completed serial ACT measurements at initial visits and at follow-up visits (Figure 2).
Objective measurement of lung function is also important
Serial monitoring of lung function at every patient visit with spirometry is also important, as some patients may be “poor perceivers,”16 ie, they may have little or no subjective awareness of moderate or even severe ventilatory impairment. A number of studies17,18 support the contention that symptoms and lung function are separate and independent dimensions of asthma control, and that both of them need to be assessed.
Responding to changes in control

THE STEP 3 CONTROVERSY
Salmeterol Multicenter Asthma Research Trial
In the Salmeterol Multicenter Asthma Research Trial (SMART), patients randomized to the long-acting beta agonist (LABA) salmeterol (Serevent)—particularly African Americans—had a statistically significant increase in the risk of untoward asthma care outcomes.20
SMART was launched in 1996. Patients were randomized in a double-blind fashion to receive either salmeterol 42 μg twice a day or placebo in addition to their usual asthma therapy for 28 weeks. The rate of the primary outcome (respiratory-related deaths or life-threatening experiences) was not significantly different with salmeterol than with placebo (relative risk [RR] = 1.40, 95% confidence interval [CI] 0.91–2.14). However, in 2003, the study was halted prematurely because of difficulty enrolling the targeted number of 60,000 patients, and an interim analysis that revealed significantly higher rates of secondary outcomes in subjects randomized to salmeterol. Compared with the placebo group, the salmeterol group had significantly higher rates of respiratory-related deaths (RR 2.16, 95% CI 1.06–4.41), asthma-related deaths (RR = 4.37, 95% CI = 1.25–15.34), and combined asthma-related deaths or life-threatening experiences (RR = 1.71, 95% CI 1.01–2.89). There were 13 asthma-related deaths and 37 combined asthma-related deaths or life-threatening experiences in the salmeterol group, compared with 3 and 22, respectively, in the placebo group. Of the 16 asthma deaths in the study, 13 (81%) occurred in the initial phase of SMART, when patients were recruited via print, radio, and television advertising; afterward, patients were recruited directly by investigators.
Statistically significant differences in outcomes occurred primarily in African Americans. African Americans who received salmeterol had higher rates of respiratory death or life-threatening experiences (RR = 4.10, 95% CI 1.54–10.90), the primary end point for the study, as well as higher rates of combined asthma-related deaths or life-threatening experiences (RR = 10.46, 95% CI 1.34–81.58), a secondary end point. No statistically significant differences were observed in white patients randomized to salmeterol with respect to the primary end point (RR = 1.05, 95% = 0.62–1.76); the secondary end point of combined asthma-related deaths or life-threatening experiences (RR = 1.08, 95% CI 0.55–2.14); or other end points.
Medication exposures were not tracked during the study, and allocation to inhaled corticosteroids combined with salmeterol was not randomized, so the effect of concomitant inhaled corticosteroid use cannot be determined from these data.
As a result of SMART, medications that contain either of the two LABAs, salmeterol or formoterol (Foradil), carry a black-box warning.
LABAs: Risks and benefits
Two studies21,22 have suggested that asthmatic patients who are homozygous for Arg/Arg at codon 16 of the beta-2 adrenergic receptor are predisposed to untoward asthma outcomes with regular exposure to LABAs. However, other data23–25 do not support the contention that B16 Arg/Arg patients experience adverse asthma outcomes with LABA exposure. In two recently published studies, no difference in rates of exacerbations, severe exacerbations, lung function, frequency of reliance on SABA, or nocturnal awakenings was observed in patients receiving formoterol combined with budesonide24 or salmeterol combined with fluticasone25 according to genotype. A prospective study26 also found no statistically significant difference in exacerbation rates according to beta adrenergic receptor genotype in individuals randomized to LABA monotherapy, or LABA combined with inhaled corticosteroids.
The updated EPR2 asthma guidelines,3 published in November 2002, stipulated that LABAs were the preferred controller agent to “add on” to low-dose inhaled corticosteroids for patients with moderate persistent asthma, and that the combination of low-dose inhaled corticosteroids and LABA was associated with superior outcomes: reduction of symptoms, including nocturnal awakening, increase in lung function, improvement in health-related quality of life, decreased use of “rescue” medication, and reduced rate of exacerbations and severe exacerbations, compared with higher-dose inhaled corticosteroid monotherapy. This management recommendation was categorized as level A, on the basis of data from multiple randomized, controlled, double-blinded trials.27–29 Additional evidence14,30 and data from two meta-analyses31,32 have provided further support for this recommendation, while no evidence linking LABA exposure to risk for fatal or near-fatal asthma has been found in cohort or case-control studies.33–38
Based on safety concerns, the EPR3 guidelines1 recommend that medium-dose inhaled corticosteroids be regarded as equivalent to adding LABAs to low-dose inhaled corticosteroids, and state: “the established, beneficial effects of LABA for the great majority of patients whose asthma is not well controlled with [inhaled corticosteroids] alone should be weighed against the increased risk for severe exacerbations, although uncommon, associated with daily use of LABA.”1
There is currently an honest difference of opinion39,40 among asthma specialists as to how this management recommendation for moderate persistent asthma—now depicted at “step 3” in the EPR3 guidelines (Table 4)—should be implemented. The LABA controversy was reviewed previously in the Cleveland Clinic Journal of Medicine.41
THE ROLE OF OMALIZUMAB: WEIGHING COST VS BENEFIT
The 2002 update to the EPR2 guidelines3 was issued before omalizumab (Xolair) was approved in June 2003.
Patients with severe persistent asthma are categorized in steps 5 or 6 in the EPR3 guidelines (Table 5).1 Preferred management for these patients includes inhaled corticosteroids in high doses combined with long-acting beta agonists and, for step 6 patients, oral corticosteroids.
Omalizumab was approved for management of patients with moderate or severe persistent asthma who are not achieving the goals of asthma management on inhaled corticosteroids, who exhibit a wheal-flare reaction to a perennial allergen, and whose immunoglobulin E (IgE) level is in the range of 30 to 700 IU/mL.42 Omalizumab dosing is based on the serum IgE level and on body weight.
Omalizumab, an anti-IgE monoclonal antibody
Omalizumab is a recombinant, humanized, monoclonal anti-IgE antibody that binds to IgE at the same Fc site as the high-affinity IgE receptor. Its primary mechanism of action is the binding of free IgE in the circulation, forming biologically inert, small complexes that do not activate complement and are cleared by the reticuloendothelial system.42 Its secondary mechanism of action entails a reduction in the number of high-affinity receptors on basophils, from approximately 220,000 to 8,300 receptors per cell. The latter effect was associated with a 90% reduction in histamine release from basophils in response to ex vivo challenge with dust mite allergen.43
Benefit in randomized trials
Omalizumab has been associated with statistically and clinically significant benefit in randomized, double-blind, placebo-controlled trials.44,45
Humbert et al46 randomized 419 patients whose asthma was not adequately controlled on high-dose inhaled corticosteroids and long-acting beta agonists, who were 12 to 75 years old, with reduced lung function and a history of recent asthma exacerbation, to treatment with omalizumab or placebo. Omalizumab was associated with a statistically significant reduction in the rate of asthma exacerbations and severe asthma exacerbations, as well as statistically significant improvements in asthma-related quality of life, morning peak expiratory flow rate, and asthma symptom scores.
These data support the recommendation in EPR3 to consider a trial of omalizumab in properly selected patients with severe, persistent allergic asthma.
Omalizumab is cost-beneficial in properly selected patients
The current wholesale acquisition cost of omalizumab is $532 for one 150-mg vial (David Zito, personal communication). The cost of treatment varies based on body weight and IgE level but may range from a wholesale cost of $6,388 to $38,326 per year.
However, as asthma severity increases, both direct and indirect medical expenditures increase substantially.47,48 Annual costs are approximately four times higher for severe asthma compared with mild asthma49; not only are treatment and exacerbation costs higher, but indirect costs are also disproportionately greater. Annual costs for severe asthma are significantly greater if the disease is inadequately controlled.50 For these reasons, an intervention that leads to improved outcomes for severe, poorly controlled asthma carries the potential for the greatest cost-utility for society, as it can lower direct costs by reducing the frequency and severity of exacerbations, in addition to reducing indirect medical expenditures on the basis of increased productivity and fewer days of missed work or school. The cost of omalizumab in quality-adjusted life years compares favorably with that of biologicals used in managing rheumatoid arthritis, Crohn disease, and multiple sclerosis.50
Adverse effects of omalizumab
In pivotal trials,43,44 omalizumab was associated with a substantial rate of local reactions. The rate of anaphylaxis was slightly less than 1 in 1,000, and this has been confirmed by surveillance data recorded since approval of the drug in 2003. Based on the observed risk of anaphylaxis, in July 2007, the US Food and Drug Administration added a black-box warning to the omalizumab label and stipulated that a medication guide should be provided for patients.51 The warning indicates that health care providers administering omalizumab should be prepared to manage anaphylaxis and that patients should be closely observed for an appropriate period after omalizumab administration.
The package insert also describes a numerical, but not statistically significant, increase in the rate of malignancy in patients receiving omalizumab.42 Malignancy developed in 0.5% of patients receiving omalizumab, compared with 0.2% of patients who received placebo. Because these malignancies were diagnosed over a shorter period than the time required for oncogenesis (ie, 6 months in 60% of cases), and because a heterogeneous variety of tumors was observed, there is reason to doubt these tumors were causally associated with omalizumab.
Postmarketing surveillance studies are in progress that will provide more definitive data on the potential relationship between malignancy and omalizumab exposure.
Omalizumab: Guideline recommendations
The EPR3 guidelines1 state that omalizumab is the only adjunctive therapy to demonstrate efficacy when added to high-dose inhaled corticosteroids plus long-acting beta agonists in patients with severe, persistent, allergic asthma and that evidence does not support use of the following agents, which in some cases are approved for managing other conditions and have been advocated for management of severe, refractory asthma: methotrexate, soluble interleukin (IL)-4 receptor, anti-IL-5, anti-IL-12, cyclosporine A, intravenous immune globulin, gold, troleandomycin, and colchicine. The data supporting use of macrolides were characterized as “encouraging but insufficient to support a recommendation.”
The strength of evidence for the use of omalizumab for patients in steps 5 and 6 who fulfill the criteria for its use (see above) was classified in the EPR3 guidelines1 as category B. The guidelines also say that omalizumab may be considered for adjunctive therapy in properly selected patients in step 4, as a means to avoid higher doses of inhaled corticosteroids, but that additional studies are needed to establish its utility for such patients. This recommendation was classified as category D because of the lack of published comparator trials.
ALLERGEN IMMUNOTHERAPY FOR PATIENTS WITH ASTHMA
Many patients with asthma have clinically relevant, IgE-mediated (allergic) potential to inhaled allergens.1 For patients with persistent asthma (steps 2–6 in Table 5), allergic reactions can contribute to airway inflammation, provoke symptoms, and lead to more use of medications. For this reason, identification and management of clinically relevant allergy merits consideration.52
The EPR3 guidelines1 recommend considering allergen immunotherapy for patients with mild or moderate persistent asthma (steps 2–4) who have a clinically relevant component of allergy to inhaled substances.
Changing the immune response
Allergen immunotherapy entails the incremental administration of inhalant allergens by subcutaneous injection for the purpose of inducing immune system changes in the host response. The goal of immunotherapy is to protect against allergic reactions that can be expected to occur with ongoing exposure to clinically relevant allergens.53
The immunologic changes that develop with allergen immunotherapy are complex.53,54 Successful immunotherapy results in generation of a population of CD4+/CD25+ T lymphocytes producing IL-10, transforming growth factor beta, or both. Allergen immunotherapy has been shown to block the immediate- and late-phase allergic response; to decrease recruitment of mast cells, basophils, and eosinophils on provocation or natural exposure to allergens in the skin, nose, eye, and bronchial mucosa; to blunt the seasonal rise in specific IgE; and to suppress late-phase inflammatory responses in the skin and respiratory tract. However, the efficacy of immunotherapy in relation to these immunologic changes is not completely understood.54
Many patients need skin testing
Allergen immunotherapy may be considered for patients with asthma for whom a clear relationship exists between symptoms and exposure to an allergen to which the patient is sensitive.53 Because it is often not possible to determine whether a patient is sensitive to a perennial indoor allergen (eg, dust mite) on the basis of the medical history alone,55 many patients with asthma benefit from immediate hypersensitivity skin testing to objectively assess or rule out allergy to common inhalants. In certain situations, in vitro testing may be performed, but skin testing has a higher negative predictive value and is recommended as a better screening test.56
Benefits of allergen immunotherapy
Numerous randomized, double-blind, placebo-controlled trials have shown that allergen immunotherapy is associated with benefit for reducing symptoms and medication reliance.57–63
A meta-analysis of 75 randomized, placebo-controlled studies confirmed the effectiveness of immunotherapy in asthma, with a significant reduction in asthma symptoms and medication use and with improvement in bronchial hyperreactivity.64 This meta-analysis included 36 trials of dust mite allergen, 20 of pollen, and 10 of animal dander. Immunotherapy is efficacious for pollen, mold, dust mite, cockroach, and animal allergens; however, its effectiveness is more established for dust mite, animal dander, and pollen allergens, as fewer studies have been published demonstrating efficacy using mold and cockroach allergens.53
In addition, several studies have found that children with allergic rhinitis who receive allergen immunotherapy are significantly less likely to develop asthma.65–67 Immunotherapy has also been associated with a statistically significant reduction in future sensitization to other aeroallergens.68,69
Risk of systemic reaction from allergen immunotherapy
The decision to begin allergen immunotherapy should be individualized on the basis of symptom severity, relative benefit compared with drug therapy, and whether comorbid conditions such as cardiovascular disease or beta-blocker exposure are present. These comorbid conditions are associated with heightened risk of (more serious) anaphylaxis—the major hazard of allergen immunotherapy.70 Systemic reactions during allergen immunotherapy occur at a rate of approximately 3 to 5 per 1,000 injections; for this reason, allergen immunotherapy should only be administered in a medical facility where personnel, supplies, and equipment are available to treat anaphylaxis.5
- National Heart, Lung, and Blood institute, National Asthma education and Prevention Program. Expert Panel Report 3: guidelines for the diagnosis and management of asthma. www.nhlbi.nih.gov/guidelines/asthma. Accessed 8/7/08.
- Expert Panel Report 2: Guidelines for the diagnosis and management of asthma. U.S. Department of Health and Human Services. Publication No. 97-4051; 1997.
- Expert Panel Report: Guidelines for the diagnosis and management of asthma. Update on Selected Topics—2002. J Allergy Clin Immunol 2002; 110:S141–S207.
- FitzGerald JM, Boulet LP, McIvor RA, Zimmerman S, Chapman KR. Asthma control in Canada remains suboptimal: the Reality of Asthma Control (TRAC) study. Can Respir J 2006; 13:253–259.
- Braganza S, Sharif I, Ozuah P. Documenting asthma severity: do we get it right? J Asthma 2003; 40:661–665.
- Cockcroft DW, Swystun VA. Asthma control versus asthma severity. J Allergy Clin Immunol 1996; 98:1016–1018.
- Peters SP, Jones CA, Haselkorn T, Mink DR, Valacer DJ, Weiss ST. Real-world Evaluation of Asthma Control and Treatment (REACT): findings from a national Web-based survey. J Allergy Clin Immunol. 2007; 119:1454–1461.
- Osborne ML, Vollmer WM, Pedula KL, Wilkins J, Buist AS, O’Hollaren M. Lack of correlation of symptoms with specialist-assessed long-term asthma severity. Chest 1999; 115:85–91.
- Li JT, Oppenheimer J, Bernstein IL, et al. Attaining optimal asthma control: a practice parameter. J Allergy Clin Immunol 2005; 116:S3–S11.
- Nathan RA, Sorkness C, Kosinski M, et al. Development of the Asthma Control Test: a survey for assessing asthma control. J Allergy Clin Immunol 2004; 113:59–65.
- Schatz M, Zeiger RS, Drane A, et al. Reliability and predictive validity of the Asthma Control Test administered by telephone calls using speech recognition technology. J Allergy Clin Immunol 2007; 119:336–343.
- Peters D, Chen C, Markson LE, Allen-Ramey FC, Vollmer WM. Using an asthma control questionnaire and administrative data to predict healthcare utilization. Chest 2006; 129:918–924.
- Schatz M, Sorkness C, Li JT, et al. Asthma Control Test: reliability, validity, and responsiveness in patients not previously followed by asthma specialists. J Allergy Clin Immunol 2006; 117:549–556.
- Bateman E, Boushey H, Bousquet J, et al. Can guideline-defined asthma control be achieved? Am J Respir Crit Care Med 2004; 170:836–844.
- Davies TJ, Bunn WB, Fromer L, Gelfand EW, Colice GL. A focus on the asthma HEDIS measure and its implications for clinical practice. Manag Care Interface 2006; 19:29–36.
- Rubinfeld AR, Pain MC. Perception of asthma. Lancet 1976; 1:882–884.
- Teeter J, Bleecker E. Relationship between airway obstruction and respiratory symptoms in adult asthmatics. Chest 1998; 113:272–277.
- Shingo S, Zhang J, Reiss T. Correlation of airway obstruction and patient reported endpoints in clinical studies. Eur Resp J 2001; 17:220–224.
- Juniper EF, Bousquet J, Abetz L, Bateman ED; GOAL Committee. Identifying ‘well-controlled’ and ‘not well-controlled’ asthma using the Asthma Control Questionnaire. Respir Med 2006; 100:616–621.
- Nelson H, Weiss S, Bleecker E, Yancey S, Dorinsky P. The Salmeterol Multicenter Asthma Research Trial. Chest 2006; 129:15–26.
- Wechsler M, Lehman E, Lazarus S, et al. ß-Adrenergic receptor polymorphisms and response to salmeterol. Am J Respir Crit Care Med 2006; 173:519–526.
- Palmer CNA, Lipworth BJ, Lee S, Ismail T, MacGregor DF, Mukhopadhyay S. Arginine-16 beta-2 adrenoceptor genotype predisposes to exacerbations in young asthmatics taking regular salmeterol. Thorax 2006; 61:940–944.
- Taylor DR, Drazen JM, Herbison GP, Yandava CN, Hancox RJ, Town GI. Asthma exacerbations during long term beta agonist use: influence of beta 2 adrenoceptor polymorphism. Thorax 2000; 55:762–727.
- Bleecker E, Postma D, Lawrance R, Meyers D, Ambrose H, Goldman M. Effect of ADRB2 polymorphisms on response to long-acting beta2-agonist therapy: a pharmacogenetic analysis of two randomized studies. Lancet 2007; 370:2118–2125.
- Bleecker E, Yancey S, Baitinger L, et al. Salmeterol response is not affected by beta-2 adrenergic receptor genotype in subjects with persistent asthma. J Allergy Clin Immunol 2006; 118:809–816.
- Nelson H, Bleecker E, Corren J, et al. Characterization of asthma exacerbations by Arg16Gly genotype in subjects with asthma receiving salmeterol alone or with fluticasone propionate. J Allergy Clin Immunol 2008; 121:S131.
- O’Byrne P, Barnes P, Rodriguez-Roisin R, et al. Low dose Inhaled budesonide and formoterol in mild persistent asthma. The OPTIMA Randomized Trial. Am J Respir Crit Care Med 2001; 164:1392–1397.
- Greening AP, Ind PW, Northfield M, Shaw G. Added salmeterol versus higher dose corticosteroid in asthma patients with symptoms on existing inhaled corticosteroid. Lancet 1994; 344:219–224.
- Woolcock A, Lundback B, Ringdal N, Jacques LA. Comparison of addition of salmeterol to inhaled steroids with doubling of the dose of inhaled steroids. Am J Respir Crit Care Med 1996; 153:1481–1488.
- Walters EH, Walters JAE, Gibson MDP. Long-acting beta2-agonists for stable chronic asthma. Cochrane Database Syst Rev 2003; (3):CD001385. doi:10.1002/14651858.CD001385.
- Masoli M, Weatherall M, Holt S, Beasley R. Moderate dose inhaled corticosteroids plus salmeterol versus higher doses of inhaled corticosteroid in symptomatic asthma. Thorax 2005; 60:730–734.
- Sin DD, Man J, Sharpe H, Gan WQ, Man SFP. Pharmacological management to reduce exacerbations in adults with asthma. A systematic review and meta-analysis. JAMA 2004; 292:367–376.
- Mann RD, Kubota K, Pearce G, Wilton L. Salmeterol: a study by prescription event monitoring in a UK cohort of 15,407 patients. J Clin Epidemiol 1996; 49:247–250.
- Lanes S, Lanza L, Wentworth C. Risk of emergency care, hospitalization, and ICU stays for acute asthma among recipients of salmeterol. Am J Respir Crit Care Med 1998; 158:857–861.
- Meier CR, Jick H. Drug use and pulmonary death rates in increasingly symptomatic asthma patients in the UK. Thorax 1997; 52:612–617.
- Williams C, Crossland L, Finnerty J, et al. A case control study of salmeterol and near-fatal attacks of asthma. Thorax 1998; 53:7–13.
- Lanes S, Garcia Rodriguez LA, Herta C. Respiratory medications and risk of asthma death. Thorax 2002; 57:683–686.
- Anderson HR, Ayres JG, Sturdy PM, et al. Bronchodilator treatment and deaths from asthma: case control study. Br Med J 2005; 330:117–124.
- Martinez FD. Safety of long-acting beta agonists—an urgent need to clear the air. N Engl J Med 2005; 353:2637–2639.
- Nelson HS. Long-acting beta-agonists in adult asthma: evidence that these drugs are safe. Prim Care Respir J 2006; 15:271–277.
- Lang DM. The long-acting beta agonist controversy: a critical examination of the evidence. Cleve Clin J Med 2006; 73:973–992.
- Rambasek T, Lang DM, Kavuru M. Omalizumab: where does it fit in current asthma management? Cleve Clin J Med 2004; 71:251–261.
- McGlashan D, Bochner B, Adelman D, et al. Down regulation of Fc(epsilon)RI expression on human basophils during in vivo treatment of atopic patients with anti-IgE antibody. J Immunol 1997; 158:1438–1445.
- Busse W, Corren J, Lanier B, et al. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol 2001; 108:184–190.
- Soler M, Matz J, Townley R, et al. The anti-IgE antibody omalizumab reduces asthma exacerbations and steroid requirement in allergic asthmatics. Eur Respir J 2001; 18:254–261.
- Humbert M, Beasley R, Ayres J, et al. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy 2005; 60:309–316.
- Van Ganse E, Antonicelli L, Zhang Q, et al. Asthma-related resource use and cost by GINA classification of severity in three European countries. Respir Med 2006; 100:140–147.
- Godard P, Chanez P, Siraudin L, Nicoloyannis N, Duru G. Costs of asthma are correlated with severity: a 1-yr prospective study. Eur Respir J 2002; 19:61–67.
- Cisternas MG, Blanc PH, Yen IH, et al. A comprehensive study of the direct and indirect costs of adult asthma. J Allergy Clin Immunol 2003; 111:1212–1218.
- Sullivan S, Turk F. An evaluation of the cost effectiveness of omalizumab for the treatment of severe persistent asthma. Allergy 2008; 63:670–684.
- US Food and Drug Administration. Omalizumab (marketed as Xolair) information. www.fda.gov/cder/drug/infopage/omalizumab/default.htm. Accessed August 31, 2007.
- Williams SG, Schmidt DK, Redd SC, Storms W. Key clinical activities for quality asthma care. Recommendations of the National Asthma Education and Prevention Program. MMWR Recomm Rep 2003; 52 RR-6:1–8.
- Cox L, Li J, Nelson H, Lockey R, et al. Allergy Immunotherapy: a practice parameter second update. J Allergy Clin Immunol 2007; 120:S25–S85.
- Akdis M, Akdis CA. Mechanisms of allergen-specific immunotherapy. J Allergy Clin Immunol 2007; 119:780–789.
- Murray AB, Milner RA. The accuracy of features in the clinical history for predicting atopic sensitization to airborne allergens in children. J Allergy Clin Immunol 1995; 96:588–596.
- Bernstein IL, Li JT, Bernstein DI, et al. Allergy diagnostic testing: an updated practice parameter. Ann Allergy Asthma Immunol 2008; 100 suppl 3:1S–148S.
- Walker S, Pajno GB, Lima MT, Wilson DR, Durham SR. Grass pollen immunotherapy for seasonal rhinitis and asthma: a randomized, controlled trial. J Allergy Clin Immunol 2001; 107:87–93.
- Varney VA, Edwards J, Tabbah K, Brewster H, Mavroleon G, Frew AJ. Clinical efficacy of specific immunotherapy to cat dander: a double-blind placebo-controlled trial. Clin Exp Allergy 1997; 27:860–867.
- Cantani A, Arcese G, Lucenti P, Gagliesi D, Bartolucci M. A three-year prospective study of specific immunotherapy to inhalant allergens: evidence of safety and efficacy in 300 children with allergic asthma. J Investig Allergol Clin Immunol 1997; 7:90–97.
- Hedlin G, Wille S, Browaldh L, et al. Immunotherapy in children with allergic asthma: effect on bronchial hyperreactivity and pharmacotherapy. J Allergy Clin Immunol 1999; 103:609–614.
- Arvidsson MB, Löwhagen O, Rak S. Allergen specific immunotherapy attenuates early and late phase reactions in lower airways of birch pollen asthmatic patients: a double blind placebo-controlled study. Allergy 2004; 59:74–80.
- Pichler CE, Helbling A, Pichler WJ. Three years of specific immunotherapy with house-dust-mite extracts in patients with rhinitis and asthma: significant improvement of allergen-specific parameters and of nonspecific bronchial hyperreactivity. Allergy 2001; 56:301–306.
- Mirone C, Albert F, Tosi A, et al. Efficacy and safety of subcutaneous immunotherapy with a biologically standardized extract of Ambrosia artemisiifolia pollen: a double-blind, placebo-controlled study. Clin Exp Allergy 2004; 34:1408–1414.
- Abramson MJ, Puy RM, Weiner JM. Allergen immunotherapy for asthma. Cochrane Database Syst Rev 2003; (4):CD001186.
- Jacobsen L. Preventive aspects of immunotherapy: prevention for children at risk of developing asthma. Ann Allergy Asthma Immunol 2001; 87:43–46.
- Moller C, Dreborg S, Ferdousi HA, et al. Pollen immunotherapy reduces the development of asthma in children with seasonal rhinoconjunctivitis (the PAT study). J Allergy Clin Immunol 2002; 109:251–256.
- Niggemann B, Jacobsen L, Dreborg S, et al; PAT Investigator Group. Five year follow-up on the PAT study: specific immunotherapy and long-term prevention of asthma in children. Allergy 2006: 61:855–859.
- Des Roches A, Paradis L, Menardo JL, et al. Immunotherapy with a standardized Dermatophagoides pteronyssinus extract VI: specific immunotherapy prevents the onset of new sensitizations in children. J Allergy Clin Immunol 1997; 99:450–453.
- Pajno GB, Barberio G, DeLuca F, et al. Prevention of new sensitizations in asthmatic children monosensitized to the house dust mite by specific immunotherapy: a six year follow up study. Clin Exp Allergy 2001; 31:1392–1397.
- Lang DM. Do beta blockers really enhance the risk of anaphylaxis during immunotherapy? Curr Allergy Asthma Rep 2008; 8:37–44.
This review focuses on several elements in the National Asthma Education and Prevention Program’s new guidelines, the third Expert Panel Report (EPR3),1 that differ substantially from those in EPR2,2 issued in 1997 and updated in 2002.3 These differences in approach to the management of asthma described in EPR3 offer a clear potential for reducing the gap between optimal asthma care outcomes as described in guidelines and normative asthma care outcomes in the “real world.”
GREATER EMPHASIS ON CONTROL
The EPR2 guidelines2 recommended that asthma management be carried out in an algorithmic manner. Patients were classified into four severity categories: mild intermittent, mild persistent, moderate persistent, and severe persistent asthma, based on assessment of the level of symptoms (day/night), reliance on “reliever” medication, and lung function at the time of presentation. Pharmacologic management was then assigned according to each respective categorization in an evidence-based fashion.
In an ideal world, this would result in patients with asthma receiving appropriate pharmacotherapeutic agents associated with favorable asthma care outcomes, which were also advantageous from both cost- and risk-benefit standpoints. In the real world, however, this paradigm was flawed, as it relied on accurate categorization of patients in order for pharmacotherapy to be prescribed appropriately. Both providers and patients are prone to underestimate asthma severity,4,5 and for this reason many patients managed on the basis of this paradigm were undertreated.
A new paradigm, based on the assessment of asthma control, has been encouraged in the EPR3 guidelines.1
Severity and control are not synonymous
More than a decade ago, Cockroft and Swystun6 pointed out that asthma control (or lack thereof) is often used inappropriately to define asthma severity: ie, well-controlled asthma is seen as synonymous with mild asthma, and poorly controlled asthma with severe asthma.
Asthma severity can be defined as the intrinsic intensity of the disease process, while asthma control is the degree to which the manifestations of asthma are minimized. Asthma severity is clearly a determinant of asthma control, but its impact is affected by a variety of factors, including but not limited to:
- Whether appropriate medication is prescribed
- Patterns of therapeutic adherence
- The degree to which recommended measures for avoiding for clinically relevant aeroallergens are pursued.
Health care utilization, including hospitalizations and emergency department visits, correlates more closely with asthma control than with asthma severity.7–9 Indeed, a patient with severe persistent asthma who is treated appropriately with multiple “controller” medications and who takes his or her medications and avoids allergens as directed can achieve well-controlled or totally controlled asthma, and is not likely to require hospitalization or emergency department management, to miss school or work, or to experience nocturnal awakening or limitation in routine activities due to asthma. This patient has severe persistent asthma that is well controlled.
In contrast, a patient with mild or moderate persistent asthma who does not receive appropriate instructions for avoiding allergens or taking controller medication regularly or who is poorly adherent will likely have poor asthma control. This patient is more likely to require hospitalization or emergency department management, to miss school or work, and to experience nocturnal awakening or limitation in routine activities due to asthma. This patient has mild persistent asthma that is poorly controlled.
Assess asthma severity in the first visit, and control in subsequent visits
How to assess severity
How to measure control
For all patients with asthma, regardless of severity, the goal is the same: to achieve control by reducing both impairment and risk. Asthma is classified as well controlled, not well controlled, or poorly controlled (Table 2).1
Validated tests are available to assess control
Serial testing as a quality indicator
Serial ACT scores give an objective measure of the degree to which the goals of management1 are being achieved, and in so doing can encourage optimal outcomes.14
Another use of these tests is to document whether asthma control improves over time when patients receive care from a particular physician or group. This use may become increasingly important in view of efforts underway to implement a pay-for-performance model for asthma care, in which providers will be financially rewarded for improved patient care outcomes and adherence to standards of practice based on Health Plan Employer Data and Information Set measures.15
We have used the ACT in the Section of Allergy/Immunology at Cleveland Clinic for 3 years on a routine basis. All patients with asthma being seen either for the first time or as follow-up complete the ACT, which has been entered in a flow sheet in our electronic medical record, at the same time they undergo spirometry. We have shown that care in the Section of Allergy/Immunology is associated with improvement in asthma control over time, in patients who have completed serial ACT measurements at initial visits and at follow-up visits (Figure 2).
Objective measurement of lung function is also important
Serial monitoring of lung function at every patient visit with spirometry is also important, as some patients may be “poor perceivers,”16 ie, they may have little or no subjective awareness of moderate or even severe ventilatory impairment. A number of studies17,18 support the contention that symptoms and lung function are separate and independent dimensions of asthma control, and that both of them need to be assessed.
Responding to changes in control
THE STEP 3 CONTROVERSY
Salmeterol Multicenter Asthma Research Trial
In the Salmeterol Multicenter Asthma Research Trial (SMART), patients randomized to the long-acting beta agonist (LABA) salmeterol (Serevent)—particularly African Americans—had a statistically significant increase in the risk of untoward asthma care outcomes.20
SMART was launched in 1996. Patients were randomized in a double-blind fashion to receive either salmeterol 42 μg twice a day or placebo in addition to their usual asthma therapy for 28 weeks. The rate of the primary outcome (respiratory-related deaths or life-threatening experiences) was not significantly different with salmeterol than with placebo (relative risk [RR] = 1.40, 95% confidence interval [CI] 0.91–2.14). However, in 2003, the study was halted prematurely because of difficulty enrolling the targeted number of 60,000 patients, and an interim analysis that revealed significantly higher rates of secondary outcomes in subjects randomized to salmeterol. Compared with the placebo group, the salmeterol group had significantly higher rates of respiratory-related deaths (RR 2.16, 95% CI 1.06–4.41), asthma-related deaths (RR = 4.37, 95% CI = 1.25–15.34), and combined asthma-related deaths or life-threatening experiences (RR = 1.71, 95% CI 1.01–2.89). There were 13 asthma-related deaths and 37 combined asthma-related deaths or life-threatening experiences in the salmeterol group, compared with 3 and 22, respectively, in the placebo group. Of the 16 asthma deaths in the study, 13 (81%) occurred in the initial phase of SMART, when patients were recruited via print, radio, and television advertising; afterward, patients were recruited directly by investigators.
Statistically significant differences in outcomes occurred primarily in African Americans. African Americans who received salmeterol had higher rates of respiratory death or life-threatening experiences (RR = 4.10, 95% CI 1.54–10.90), the primary end point for the study, as well as higher rates of combined asthma-related deaths or life-threatening experiences (RR = 10.46, 95% CI 1.34–81.58), a secondary end point. No statistically significant differences were observed in white patients randomized to salmeterol with respect to the primary end point (RR = 1.05, 95% = 0.62–1.76); the secondary end point of combined asthma-related deaths or life-threatening experiences (RR = 1.08, 95% CI 0.55–2.14); or other end points.
Medication exposures were not tracked during the study, and allocation to inhaled corticosteroids combined with salmeterol was not randomized, so the effect of concomitant inhaled corticosteroid use cannot be determined from these data.
As a result of SMART, medications that contain either of the two LABAs, salmeterol or formoterol (Foradil), carry a black-box warning.
LABAs: Risks and benefits
Two studies21,22 have suggested that asthmatic patients who are homozygous for Arg/Arg at codon 16 of the beta-2 adrenergic receptor are predisposed to untoward asthma outcomes with regular exposure to LABAs. However, other data23–25 do not support the contention that B16 Arg/Arg patients experience adverse asthma outcomes with LABA exposure. In two recently published studies, no difference in rates of exacerbations, severe exacerbations, lung function, frequency of reliance on SABA, or nocturnal awakenings was observed in patients receiving formoterol combined with budesonide24 or salmeterol combined with fluticasone25 according to genotype. A prospective study26 also found no statistically significant difference in exacerbation rates according to beta adrenergic receptor genotype in individuals randomized to LABA monotherapy, or LABA combined with inhaled corticosteroids.
The updated EPR2 asthma guidelines,3 published in November 2002, stipulated that LABAs were the preferred controller agent to “add on” to low-dose inhaled corticosteroids for patients with moderate persistent asthma, and that the combination of low-dose inhaled corticosteroids and LABA was associated with superior outcomes: reduction of symptoms, including nocturnal awakening, increase in lung function, improvement in health-related quality of life, decreased use of “rescue” medication, and reduced rate of exacerbations and severe exacerbations, compared with higher-dose inhaled corticosteroid monotherapy. This management recommendation was categorized as level A, on the basis of data from multiple randomized, controlled, double-blinded trials.27–29 Additional evidence14,30 and data from two meta-analyses31,32 have provided further support for this recommendation, while no evidence linking LABA exposure to risk for fatal or near-fatal asthma has been found in cohort or case-control studies.33–38
Based on safety concerns, the EPR3 guidelines1 recommend that medium-dose inhaled corticosteroids be regarded as equivalent to adding LABAs to low-dose inhaled corticosteroids, and state: “the established, beneficial effects of LABA for the great majority of patients whose asthma is not well controlled with [inhaled corticosteroids] alone should be weighed against the increased risk for severe exacerbations, although uncommon, associated with daily use of LABA.”1
There is currently an honest difference of opinion39,40 among asthma specialists as to how this management recommendation for moderate persistent asthma—now depicted at “step 3” in the EPR3 guidelines (Table 4)—should be implemented. The LABA controversy was reviewed previously in the Cleveland Clinic Journal of Medicine.41
THE ROLE OF OMALIZUMAB: WEIGHING COST VS BENEFIT
The 2002 update to the EPR2 guidelines3 was issued before omalizumab (Xolair) was approved in June 2003.
Patients with severe persistent asthma are categorized in steps 5 or 6 in the EPR3 guidelines (Table 5).1 Preferred management for these patients includes inhaled corticosteroids in high doses combined with long-acting beta agonists and, for step 6 patients, oral corticosteroids.
Omalizumab was approved for management of patients with moderate or severe persistent asthma who are not achieving the goals of asthma management on inhaled corticosteroids, who exhibit a wheal-flare reaction to a perennial allergen, and whose immunoglobulin E (IgE) level is in the range of 30 to 700 IU/mL.42 Omalizumab dosing is based on the serum IgE level and on body weight.
Omalizumab, an anti-IgE monoclonal antibody
Omalizumab is a recombinant, humanized, monoclonal anti-IgE antibody that binds to IgE at the same Fc site as the high-affinity IgE receptor. Its primary mechanism of action is the binding of free IgE in the circulation, forming biologically inert, small complexes that do not activate complement and are cleared by the reticuloendothelial system.42 Its secondary mechanism of action entails a reduction in the number of high-affinity receptors on basophils, from approximately 220,000 to 8,300 receptors per cell. The latter effect was associated with a 90% reduction in histamine release from basophils in response to ex vivo challenge with dust mite allergen.43
Benefit in randomized trials
Omalizumab has been associated with statistically and clinically significant benefit in randomized, double-blind, placebo-controlled trials.44,45
Humbert et al46 randomized 419 patients whose asthma was not adequately controlled on high-dose inhaled corticosteroids and long-acting beta agonists, who were 12 to 75 years old, with reduced lung function and a history of recent asthma exacerbation, to treatment with omalizumab or placebo. Omalizumab was associated with a statistically significant reduction in the rate of asthma exacerbations and severe asthma exacerbations, as well as statistically significant improvements in asthma-related quality of life, morning peak expiratory flow rate, and asthma symptom scores.
These data support the recommendation in EPR3 to consider a trial of omalizumab in properly selected patients with severe, persistent allergic asthma.
Omalizumab is cost-beneficial in properly selected patients
The current wholesale acquisition cost of omalizumab is $532 for one 150-mg vial (David Zito, personal communication). The cost of treatment varies based on body weight and IgE level but may range from a wholesale cost of $6,388 to $38,326 per year.
However, as asthma severity increases, both direct and indirect medical expenditures increase substantially.47,48 Annual costs are approximately four times higher for severe asthma compared with mild asthma49; not only are treatment and exacerbation costs higher, but indirect costs are also disproportionately greater. Annual costs for severe asthma are significantly greater if the disease is inadequately controlled.50 For these reasons, an intervention that leads to improved outcomes for severe, poorly controlled asthma carries the potential for the greatest cost-utility for society, as it can lower direct costs by reducing the frequency and severity of exacerbations, in addition to reducing indirect medical expenditures on the basis of increased productivity and fewer days of missed work or school. The cost of omalizumab in quality-adjusted life years compares favorably with that of biologicals used in managing rheumatoid arthritis, Crohn disease, and multiple sclerosis.50
Adverse effects of omalizumab
In pivotal trials,43,44 omalizumab was associated with a substantial rate of local reactions. The rate of anaphylaxis was slightly less than 1 in 1,000, and this has been confirmed by surveillance data recorded since approval of the drug in 2003. Based on the observed risk of anaphylaxis, in July 2007, the US Food and Drug Administration added a black-box warning to the omalizumab label and stipulated that a medication guide should be provided for patients.51 The warning indicates that health care providers administering omalizumab should be prepared to manage anaphylaxis and that patients should be closely observed for an appropriate period after omalizumab administration.
The package insert also describes a numerical, but not statistically significant, increase in the rate of malignancy in patients receiving omalizumab.42 Malignancy developed in 0.5% of patients receiving omalizumab, compared with 0.2% of patients who received placebo. Because these malignancies were diagnosed over a shorter period than the time required for oncogenesis (ie, 6 months in 60% of cases), and because a heterogeneous variety of tumors was observed, there is reason to doubt these tumors were causally associated with omalizumab.
Postmarketing surveillance studies are in progress that will provide more definitive data on the potential relationship between malignancy and omalizumab exposure.
Omalizumab: Guideline recommendations
The EPR3 guidelines1 state that omalizumab is the only adjunctive therapy to demonstrate efficacy when added to high-dose inhaled corticosteroids plus long-acting beta agonists in patients with severe, persistent, allergic asthma and that evidence does not support use of the following agents, which in some cases are approved for managing other conditions and have been advocated for management of severe, refractory asthma: methotrexate, soluble interleukin (IL)-4 receptor, anti-IL-5, anti-IL-12, cyclosporine A, intravenous immune globulin, gold, troleandomycin, and colchicine. The data supporting use of macrolides were characterized as “encouraging but insufficient to support a recommendation.”
The strength of evidence for the use of omalizumab for patients in steps 5 and 6 who fulfill the criteria for its use (see above) was classified in the EPR3 guidelines1 as category B. The guidelines also say that omalizumab may be considered for adjunctive therapy in properly selected patients in step 4, as a means to avoid higher doses of inhaled corticosteroids, but that additional studies are needed to establish its utility for such patients. This recommendation was classified as category D because of the lack of published comparator trials.
ALLERGEN IMMUNOTHERAPY FOR PATIENTS WITH ASTHMA
Many patients with asthma have clinically relevant, IgE-mediated (allergic) potential to inhaled allergens.1 For patients with persistent asthma (steps 2–6 in Table 5), allergic reactions can contribute to airway inflammation, provoke symptoms, and lead to more use of medications. For this reason, identification and management of clinically relevant allergy merits consideration.52
The EPR3 guidelines1 recommend considering allergen immunotherapy for patients with mild or moderate persistent asthma (steps 2–4) who have a clinically relevant component of allergy to inhaled substances.
Changing the immune response
Allergen immunotherapy entails the incremental administration of inhalant allergens by subcutaneous injection for the purpose of inducing immune system changes in the host response. The goal of immunotherapy is to protect against allergic reactions that can be expected to occur with ongoing exposure to clinically relevant allergens.53
The immunologic changes that develop with allergen immunotherapy are complex.53,54 Successful immunotherapy results in generation of a population of CD4+/CD25+ T lymphocytes producing IL-10, transforming growth factor beta, or both. Allergen immunotherapy has been shown to block the immediate- and late-phase allergic response; to decrease recruitment of mast cells, basophils, and eosinophils on provocation or natural exposure to allergens in the skin, nose, eye, and bronchial mucosa; to blunt the seasonal rise in specific IgE; and to suppress late-phase inflammatory responses in the skin and respiratory tract. However, the efficacy of immunotherapy in relation to these immunologic changes is not completely understood.54
Many patients need skin testing
Allergen immunotherapy may be considered for patients with asthma for whom a clear relationship exists between symptoms and exposure to an allergen to which the patient is sensitive.53 Because it is often not possible to determine whether a patient is sensitive to a perennial indoor allergen (eg, dust mite) on the basis of the medical history alone,55 many patients with asthma benefit from immediate hypersensitivity skin testing to objectively assess or rule out allergy to common inhalants. In certain situations, in vitro testing may be performed, but skin testing has a higher negative predictive value and is recommended as a better screening test.56
Benefits of allergen immunotherapy
Numerous randomized, double-blind, placebo-controlled trials have shown that allergen immunotherapy is associated with benefit for reducing symptoms and medication reliance.57–63
A meta-analysis of 75 randomized, placebo-controlled studies confirmed the effectiveness of immunotherapy in asthma, with a significant reduction in asthma symptoms and medication use and with improvement in bronchial hyperreactivity.64 This meta-analysis included 36 trials of dust mite allergen, 20 of pollen, and 10 of animal dander. Immunotherapy is efficacious for pollen, mold, dust mite, cockroach, and animal allergens; however, its effectiveness is more established for dust mite, animal dander, and pollen allergens, as fewer studies have been published demonstrating efficacy using mold and cockroach allergens.53
In addition, several studies have found that children with allergic rhinitis who receive allergen immunotherapy are significantly less likely to develop asthma.65–67 Immunotherapy has also been associated with a statistically significant reduction in future sensitization to other aeroallergens.68,69
Risk of systemic reaction from allergen immunotherapy
The decision to begin allergen immunotherapy should be individualized on the basis of symptom severity, relative benefit compared with drug therapy, and whether comorbid conditions such as cardiovascular disease or beta-blocker exposure are present. These comorbid conditions are associated with heightened risk of (more serious) anaphylaxis—the major hazard of allergen immunotherapy.70 Systemic reactions during allergen immunotherapy occur at a rate of approximately 3 to 5 per 1,000 injections; for this reason, allergen immunotherapy should only be administered in a medical facility where personnel, supplies, and equipment are available to treat anaphylaxis.5
This review focuses on several elements in the National Asthma Education and Prevention Program’s new guidelines, the third Expert Panel Report (EPR3),1 that differ substantially from those in EPR2,2 issued in 1997 and updated in 2002.3 These differences in approach to the management of asthma described in EPR3 offer a clear potential for reducing the gap between optimal asthma care outcomes as described in guidelines and normative asthma care outcomes in the “real world.”
GREATER EMPHASIS ON CONTROL
The EPR2 guidelines2 recommended that asthma management be carried out in an algorithmic manner. Patients were classified into four severity categories: mild intermittent, mild persistent, moderate persistent, and severe persistent asthma, based on assessment of the level of symptoms (day/night), reliance on “reliever” medication, and lung function at the time of presentation. Pharmacologic management was then assigned according to each respective categorization in an evidence-based fashion.
In an ideal world, this would result in patients with asthma receiving appropriate pharmacotherapeutic agents associated with favorable asthma care outcomes, which were also advantageous from both cost- and risk-benefit standpoints. In the real world, however, this paradigm was flawed, as it relied on accurate categorization of patients in order for pharmacotherapy to be prescribed appropriately. Both providers and patients are prone to underestimate asthma severity,4,5 and for this reason many patients managed on the basis of this paradigm were undertreated.
A new paradigm, based on the assessment of asthma control, has been encouraged in the EPR3 guidelines.1
Severity and control are not synonymous
More than a decade ago, Cockroft and Swystun6 pointed out that asthma control (or lack thereof) is often used inappropriately to define asthma severity: ie, well-controlled asthma is seen as synonymous with mild asthma, and poorly controlled asthma with severe asthma.
Asthma severity can be defined as the intrinsic intensity of the disease process, while asthma control is the degree to which the manifestations of asthma are minimized. Asthma severity is clearly a determinant of asthma control, but its impact is affected by a variety of factors, including but not limited to:
- Whether appropriate medication is prescribed
- Patterns of therapeutic adherence
- The degree to which recommended measures for avoiding for clinically relevant aeroallergens are pursued.
Health care utilization, including hospitalizations and emergency department visits, correlates more closely with asthma control than with asthma severity.7–9 Indeed, a patient with severe persistent asthma who is treated appropriately with multiple “controller” medications and who takes his or her medications and avoids allergens as directed can achieve well-controlled or totally controlled asthma, and is not likely to require hospitalization or emergency department management, to miss school or work, or to experience nocturnal awakening or limitation in routine activities due to asthma. This patient has severe persistent asthma that is well controlled.
In contrast, a patient with mild or moderate persistent asthma who does not receive appropriate instructions for avoiding allergens or taking controller medication regularly or who is poorly adherent will likely have poor asthma control. This patient is more likely to require hospitalization or emergency department management, to miss school or work, and to experience nocturnal awakening or limitation in routine activities due to asthma. This patient has mild persistent asthma that is poorly controlled.
Assess asthma severity in the first visit, and control in subsequent visits
How to assess severity
How to measure control
For all patients with asthma, regardless of severity, the goal is the same: to achieve control by reducing both impairment and risk. Asthma is classified as well controlled, not well controlled, or poorly controlled (Table 2).1
Validated tests are available to assess control
Serial testing as a quality indicator
Serial ACT scores give an objective measure of the degree to which the goals of management1 are being achieved, and in so doing can encourage optimal outcomes.14
Another use of these tests is to document whether asthma control improves over time when patients receive care from a particular physician or group. This use may become increasingly important in view of efforts underway to implement a pay-for-performance model for asthma care, in which providers will be financially rewarded for improved patient care outcomes and adherence to standards of practice based on Health Plan Employer Data and Information Set measures.15
We have used the ACT in the Section of Allergy/Immunology at Cleveland Clinic for 3 years on a routine basis. All patients with asthma being seen either for the first time or as follow-up complete the ACT, which has been entered in a flow sheet in our electronic medical record, at the same time they undergo spirometry. We have shown that care in the Section of Allergy/Immunology is associated with improvement in asthma control over time, in patients who have completed serial ACT measurements at initial visits and at follow-up visits (Figure 2).
Objective measurement of lung function is also important
Serial monitoring of lung function at every patient visit with spirometry is also important, as some patients may be “poor perceivers,”16 ie, they may have little or no subjective awareness of moderate or even severe ventilatory impairment. A number of studies17,18 support the contention that symptoms and lung function are separate and independent dimensions of asthma control, and that both of them need to be assessed.
Responding to changes in control
THE STEP 3 CONTROVERSY
Salmeterol Multicenter Asthma Research Trial
In the Salmeterol Multicenter Asthma Research Trial (SMART), patients randomized to the long-acting beta agonist (LABA) salmeterol (Serevent)—particularly African Americans—had a statistically significant increase in the risk of untoward asthma care outcomes.20
SMART was launched in 1996. Patients were randomized in a double-blind fashion to receive either salmeterol 42 μg twice a day or placebo in addition to their usual asthma therapy for 28 weeks. The rate of the primary outcome (respiratory-related deaths or life-threatening experiences) was not significantly different with salmeterol than with placebo (relative risk [RR] = 1.40, 95% confidence interval [CI] 0.91–2.14). However, in 2003, the study was halted prematurely because of difficulty enrolling the targeted number of 60,000 patients, and an interim analysis that revealed significantly higher rates of secondary outcomes in subjects randomized to salmeterol. Compared with the placebo group, the salmeterol group had significantly higher rates of respiratory-related deaths (RR 2.16, 95% CI 1.06–4.41), asthma-related deaths (RR = 4.37, 95% CI = 1.25–15.34), and combined asthma-related deaths or life-threatening experiences (RR = 1.71, 95% CI 1.01–2.89). There were 13 asthma-related deaths and 37 combined asthma-related deaths or life-threatening experiences in the salmeterol group, compared with 3 and 22, respectively, in the placebo group. Of the 16 asthma deaths in the study, 13 (81%) occurred in the initial phase of SMART, when patients were recruited via print, radio, and television advertising; afterward, patients were recruited directly by investigators.
Statistically significant differences in outcomes occurred primarily in African Americans. African Americans who received salmeterol had higher rates of respiratory death or life-threatening experiences (RR = 4.10, 95% CI 1.54–10.90), the primary end point for the study, as well as higher rates of combined asthma-related deaths or life-threatening experiences (RR = 10.46, 95% CI 1.34–81.58), a secondary end point. No statistically significant differences were observed in white patients randomized to salmeterol with respect to the primary end point (RR = 1.05, 95% = 0.62–1.76); the secondary end point of combined asthma-related deaths or life-threatening experiences (RR = 1.08, 95% CI 0.55–2.14); or other end points.
Medication exposures were not tracked during the study, and allocation to inhaled corticosteroids combined with salmeterol was not randomized, so the effect of concomitant inhaled corticosteroid use cannot be determined from these data.
As a result of SMART, medications that contain either of the two LABAs, salmeterol or formoterol (Foradil), carry a black-box warning.
LABAs: Risks and benefits
Two studies21,22 have suggested that asthmatic patients who are homozygous for Arg/Arg at codon 16 of the beta-2 adrenergic receptor are predisposed to untoward asthma outcomes with regular exposure to LABAs. However, other data23–25 do not support the contention that B16 Arg/Arg patients experience adverse asthma outcomes with LABA exposure. In two recently published studies, no difference in rates of exacerbations, severe exacerbations, lung function, frequency of reliance on SABA, or nocturnal awakenings was observed in patients receiving formoterol combined with budesonide24 or salmeterol combined with fluticasone25 according to genotype. A prospective study26 also found no statistically significant difference in exacerbation rates according to beta adrenergic receptor genotype in individuals randomized to LABA monotherapy, or LABA combined with inhaled corticosteroids.
The updated EPR2 asthma guidelines,3 published in November 2002, stipulated that LABAs were the preferred controller agent to “add on” to low-dose inhaled corticosteroids for patients with moderate persistent asthma, and that the combination of low-dose inhaled corticosteroids and LABA was associated with superior outcomes: reduction of symptoms, including nocturnal awakening, increase in lung function, improvement in health-related quality of life, decreased use of “rescue” medication, and reduced rate of exacerbations and severe exacerbations, compared with higher-dose inhaled corticosteroid monotherapy. This management recommendation was categorized as level A, on the basis of data from multiple randomized, controlled, double-blinded trials.27–29 Additional evidence14,30 and data from two meta-analyses31,32 have provided further support for this recommendation, while no evidence linking LABA exposure to risk for fatal or near-fatal asthma has been found in cohort or case-control studies.33–38
Based on safety concerns, the EPR3 guidelines1 recommend that medium-dose inhaled corticosteroids be regarded as equivalent to adding LABAs to low-dose inhaled corticosteroids, and state: “the established, beneficial effects of LABA for the great majority of patients whose asthma is not well controlled with [inhaled corticosteroids] alone should be weighed against the increased risk for severe exacerbations, although uncommon, associated with daily use of LABA.”1
There is currently an honest difference of opinion39,40 among asthma specialists as to how this management recommendation for moderate persistent asthma—now depicted at “step 3” in the EPR3 guidelines (Table 4)—should be implemented. The LABA controversy was reviewed previously in the Cleveland Clinic Journal of Medicine.41
THE ROLE OF OMALIZUMAB: WEIGHING COST VS BENEFIT
The 2002 update to the EPR2 guidelines3 was issued before omalizumab (Xolair) was approved in June 2003.
Patients with severe persistent asthma are categorized in steps 5 or 6 in the EPR3 guidelines (Table 5).1 Preferred management for these patients includes inhaled corticosteroids in high doses combined with long-acting beta agonists and, for step 6 patients, oral corticosteroids.
Omalizumab was approved for management of patients with moderate or severe persistent asthma who are not achieving the goals of asthma management on inhaled corticosteroids, who exhibit a wheal-flare reaction to a perennial allergen, and whose immunoglobulin E (IgE) level is in the range of 30 to 700 IU/mL.42 Omalizumab dosing is based on the serum IgE level and on body weight.
Omalizumab, an anti-IgE monoclonal antibody
Omalizumab is a recombinant, humanized, monoclonal anti-IgE antibody that binds to IgE at the same Fc site as the high-affinity IgE receptor. Its primary mechanism of action is the binding of free IgE in the circulation, forming biologically inert, small complexes that do not activate complement and are cleared by the reticuloendothelial system.42 Its secondary mechanism of action entails a reduction in the number of high-affinity receptors on basophils, from approximately 220,000 to 8,300 receptors per cell. The latter effect was associated with a 90% reduction in histamine release from basophils in response to ex vivo challenge with dust mite allergen.43
Benefit in randomized trials
Omalizumab has been associated with statistically and clinically significant benefit in randomized, double-blind, placebo-controlled trials.44,45
Humbert et al46 randomized 419 patients whose asthma was not adequately controlled on high-dose inhaled corticosteroids and long-acting beta agonists, who were 12 to 75 years old, with reduced lung function and a history of recent asthma exacerbation, to treatment with omalizumab or placebo. Omalizumab was associated with a statistically significant reduction in the rate of asthma exacerbations and severe asthma exacerbations, as well as statistically significant improvements in asthma-related quality of life, morning peak expiratory flow rate, and asthma symptom scores.
These data support the recommendation in EPR3 to consider a trial of omalizumab in properly selected patients with severe, persistent allergic asthma.
Omalizumab is cost-beneficial in properly selected patients
The current wholesale acquisition cost of omalizumab is $532 for one 150-mg vial (David Zito, personal communication). The cost of treatment varies based on body weight and IgE level but may range from a wholesale cost of $6,388 to $38,326 per year.
However, as asthma severity increases, both direct and indirect medical expenditures increase substantially.47,48 Annual costs are approximately four times higher for severe asthma compared with mild asthma49; not only are treatment and exacerbation costs higher, but indirect costs are also disproportionately greater. Annual costs for severe asthma are significantly greater if the disease is inadequately controlled.50 For these reasons, an intervention that leads to improved outcomes for severe, poorly controlled asthma carries the potential for the greatest cost-utility for society, as it can lower direct costs by reducing the frequency and severity of exacerbations, in addition to reducing indirect medical expenditures on the basis of increased productivity and fewer days of missed work or school. The cost of omalizumab in quality-adjusted life years compares favorably with that of biologicals used in managing rheumatoid arthritis, Crohn disease, and multiple sclerosis.50
Adverse effects of omalizumab
In pivotal trials,43,44 omalizumab was associated with a substantial rate of local reactions. The rate of anaphylaxis was slightly less than 1 in 1,000, and this has been confirmed by surveillance data recorded since approval of the drug in 2003. Based on the observed risk of anaphylaxis, in July 2007, the US Food and Drug Administration added a black-box warning to the omalizumab label and stipulated that a medication guide should be provided for patients.51 The warning indicates that health care providers administering omalizumab should be prepared to manage anaphylaxis and that patients should be closely observed for an appropriate period after omalizumab administration.
The package insert also describes a numerical, but not statistically significant, increase in the rate of malignancy in patients receiving omalizumab.42 Malignancy developed in 0.5% of patients receiving omalizumab, compared with 0.2% of patients who received placebo. Because these malignancies were diagnosed over a shorter period than the time required for oncogenesis (ie, 6 months in 60% of cases), and because a heterogeneous variety of tumors was observed, there is reason to doubt these tumors were causally associated with omalizumab.
Postmarketing surveillance studies are in progress that will provide more definitive data on the potential relationship between malignancy and omalizumab exposure.
Omalizumab: Guideline recommendations
The EPR3 guidelines1 state that omalizumab is the only adjunctive therapy to demonstrate efficacy when added to high-dose inhaled corticosteroids plus long-acting beta agonists in patients with severe, persistent, allergic asthma and that evidence does not support use of the following agents, which in some cases are approved for managing other conditions and have been advocated for management of severe, refractory asthma: methotrexate, soluble interleukin (IL)-4 receptor, anti-IL-5, anti-IL-12, cyclosporine A, intravenous immune globulin, gold, troleandomycin, and colchicine. The data supporting use of macrolides were characterized as “encouraging but insufficient to support a recommendation.”
The strength of evidence for the use of omalizumab for patients in steps 5 and 6 who fulfill the criteria for its use (see above) was classified in the EPR3 guidelines1 as category B. The guidelines also say that omalizumab may be considered for adjunctive therapy in properly selected patients in step 4, as a means to avoid higher doses of inhaled corticosteroids, but that additional studies are needed to establish its utility for such patients. This recommendation was classified as category D because of the lack of published comparator trials.
ALLERGEN IMMUNOTHERAPY FOR PATIENTS WITH ASTHMA
Many patients with asthma have clinically relevant, IgE-mediated (allergic) potential to inhaled allergens.1 For patients with persistent asthma (steps 2–6 in Table 5), allergic reactions can contribute to airway inflammation, provoke symptoms, and lead to more use of medications. For this reason, identification and management of clinically relevant allergy merits consideration.52
The EPR3 guidelines1 recommend considering allergen immunotherapy for patients with mild or moderate persistent asthma (steps 2–4) who have a clinically relevant component of allergy to inhaled substances.
Changing the immune response
Allergen immunotherapy entails the incremental administration of inhalant allergens by subcutaneous injection for the purpose of inducing immune system changes in the host response. The goal of immunotherapy is to protect against allergic reactions that can be expected to occur with ongoing exposure to clinically relevant allergens.53
The immunologic changes that develop with allergen immunotherapy are complex.53,54 Successful immunotherapy results in generation of a population of CD4+/CD25+ T lymphocytes producing IL-10, transforming growth factor beta, or both. Allergen immunotherapy has been shown to block the immediate- and late-phase allergic response; to decrease recruitment of mast cells, basophils, and eosinophils on provocation or natural exposure to allergens in the skin, nose, eye, and bronchial mucosa; to blunt the seasonal rise in specific IgE; and to suppress late-phase inflammatory responses in the skin and respiratory tract. However, the efficacy of immunotherapy in relation to these immunologic changes is not completely understood.54
Many patients need skin testing
Allergen immunotherapy may be considered for patients with asthma for whom a clear relationship exists between symptoms and exposure to an allergen to which the patient is sensitive.53 Because it is often not possible to determine whether a patient is sensitive to a perennial indoor allergen (eg, dust mite) on the basis of the medical history alone,55 many patients with asthma benefit from immediate hypersensitivity skin testing to objectively assess or rule out allergy to common inhalants. In certain situations, in vitro testing may be performed, but skin testing has a higher negative predictive value and is recommended as a better screening test.56
Benefits of allergen immunotherapy
Numerous randomized, double-blind, placebo-controlled trials have shown that allergen immunotherapy is associated with benefit for reducing symptoms and medication reliance.57–63
A meta-analysis of 75 randomized, placebo-controlled studies confirmed the effectiveness of immunotherapy in asthma, with a significant reduction in asthma symptoms and medication use and with improvement in bronchial hyperreactivity.64 This meta-analysis included 36 trials of dust mite allergen, 20 of pollen, and 10 of animal dander. Immunotherapy is efficacious for pollen, mold, dust mite, cockroach, and animal allergens; however, its effectiveness is more established for dust mite, animal dander, and pollen allergens, as fewer studies have been published demonstrating efficacy using mold and cockroach allergens.53
In addition, several studies have found that children with allergic rhinitis who receive allergen immunotherapy are significantly less likely to develop asthma.65–67 Immunotherapy has also been associated with a statistically significant reduction in future sensitization to other aeroallergens.68,69
Risk of systemic reaction from allergen immunotherapy
The decision to begin allergen immunotherapy should be individualized on the basis of symptom severity, relative benefit compared with drug therapy, and whether comorbid conditions such as cardiovascular disease or beta-blocker exposure are present. These comorbid conditions are associated with heightened risk of (more serious) anaphylaxis—the major hazard of allergen immunotherapy.70 Systemic reactions during allergen immunotherapy occur at a rate of approximately 3 to 5 per 1,000 injections; for this reason, allergen immunotherapy should only be administered in a medical facility where personnel, supplies, and equipment are available to treat anaphylaxis.5
- National Heart, Lung, and Blood institute, National Asthma education and Prevention Program. Expert Panel Report 3: guidelines for the diagnosis and management of asthma. www.nhlbi.nih.gov/guidelines/asthma. Accessed 8/7/08.
- Expert Panel Report 2: Guidelines for the diagnosis and management of asthma. U.S. Department of Health and Human Services. Publication No. 97-4051; 1997.
- Expert Panel Report: Guidelines for the diagnosis and management of asthma. Update on Selected Topics—2002. J Allergy Clin Immunol 2002; 110:S141–S207.
- FitzGerald JM, Boulet LP, McIvor RA, Zimmerman S, Chapman KR. Asthma control in Canada remains suboptimal: the Reality of Asthma Control (TRAC) study. Can Respir J 2006; 13:253–259.
- Braganza S, Sharif I, Ozuah P. Documenting asthma severity: do we get it right? J Asthma 2003; 40:661–665.
- Cockcroft DW, Swystun VA. Asthma control versus asthma severity. J Allergy Clin Immunol 1996; 98:1016–1018.
- Peters SP, Jones CA, Haselkorn T, Mink DR, Valacer DJ, Weiss ST. Real-world Evaluation of Asthma Control and Treatment (REACT): findings from a national Web-based survey. J Allergy Clin Immunol. 2007; 119:1454–1461.
- Osborne ML, Vollmer WM, Pedula KL, Wilkins J, Buist AS, O’Hollaren M. Lack of correlation of symptoms with specialist-assessed long-term asthma severity. Chest 1999; 115:85–91.
- Li JT, Oppenheimer J, Bernstein IL, et al. Attaining optimal asthma control: a practice parameter. J Allergy Clin Immunol 2005; 116:S3–S11.
- Nathan RA, Sorkness C, Kosinski M, et al. Development of the Asthma Control Test: a survey for assessing asthma control. J Allergy Clin Immunol 2004; 113:59–65.
- Schatz M, Zeiger RS, Drane A, et al. Reliability and predictive validity of the Asthma Control Test administered by telephone calls using speech recognition technology. J Allergy Clin Immunol 2007; 119:336–343.
- Peters D, Chen C, Markson LE, Allen-Ramey FC, Vollmer WM. Using an asthma control questionnaire and administrative data to predict healthcare utilization. Chest 2006; 129:918–924.
- Schatz M, Sorkness C, Li JT, et al. Asthma Control Test: reliability, validity, and responsiveness in patients not previously followed by asthma specialists. J Allergy Clin Immunol 2006; 117:549–556.
- Bateman E, Boushey H, Bousquet J, et al. Can guideline-defined asthma control be achieved? Am J Respir Crit Care Med 2004; 170:836–844.
- Davies TJ, Bunn WB, Fromer L, Gelfand EW, Colice GL. A focus on the asthma HEDIS measure and its implications for clinical practice. Manag Care Interface 2006; 19:29–36.
- Rubinfeld AR, Pain MC. Perception of asthma. Lancet 1976; 1:882–884.
- Teeter J, Bleecker E. Relationship between airway obstruction and respiratory symptoms in adult asthmatics. Chest 1998; 113:272–277.
- Shingo S, Zhang J, Reiss T. Correlation of airway obstruction and patient reported endpoints in clinical studies. Eur Resp J 2001; 17:220–224.
- Juniper EF, Bousquet J, Abetz L, Bateman ED; GOAL Committee. Identifying ‘well-controlled’ and ‘not well-controlled’ asthma using the Asthma Control Questionnaire. Respir Med 2006; 100:616–621.
- Nelson H, Weiss S, Bleecker E, Yancey S, Dorinsky P. The Salmeterol Multicenter Asthma Research Trial. Chest 2006; 129:15–26.
- Wechsler M, Lehman E, Lazarus S, et al. ß-Adrenergic receptor polymorphisms and response to salmeterol. Am J Respir Crit Care Med 2006; 173:519–526.
- Palmer CNA, Lipworth BJ, Lee S, Ismail T, MacGregor DF, Mukhopadhyay S. Arginine-16 beta-2 adrenoceptor genotype predisposes to exacerbations in young asthmatics taking regular salmeterol. Thorax 2006; 61:940–944.
- Taylor DR, Drazen JM, Herbison GP, Yandava CN, Hancox RJ, Town GI. Asthma exacerbations during long term beta agonist use: influence of beta 2 adrenoceptor polymorphism. Thorax 2000; 55:762–727.
- Bleecker E, Postma D, Lawrance R, Meyers D, Ambrose H, Goldman M. Effect of ADRB2 polymorphisms on response to long-acting beta2-agonist therapy: a pharmacogenetic analysis of two randomized studies. Lancet 2007; 370:2118–2125.
- Bleecker E, Yancey S, Baitinger L, et al. Salmeterol response is not affected by beta-2 adrenergic receptor genotype in subjects with persistent asthma. J Allergy Clin Immunol 2006; 118:809–816.
- Nelson H, Bleecker E, Corren J, et al. Characterization of asthma exacerbations by Arg16Gly genotype in subjects with asthma receiving salmeterol alone or with fluticasone propionate. J Allergy Clin Immunol 2008; 121:S131.
- O’Byrne P, Barnes P, Rodriguez-Roisin R, et al. Low dose Inhaled budesonide and formoterol in mild persistent asthma. The OPTIMA Randomized Trial. Am J Respir Crit Care Med 2001; 164:1392–1397.
- Greening AP, Ind PW, Northfield M, Shaw G. Added salmeterol versus higher dose corticosteroid in asthma patients with symptoms on existing inhaled corticosteroid. Lancet 1994; 344:219–224.
- Woolcock A, Lundback B, Ringdal N, Jacques LA. Comparison of addition of salmeterol to inhaled steroids with doubling of the dose of inhaled steroids. Am J Respir Crit Care Med 1996; 153:1481–1488.
- Walters EH, Walters JAE, Gibson MDP. Long-acting beta2-agonists for stable chronic asthma. Cochrane Database Syst Rev 2003; (3):CD001385. doi:10.1002/14651858.CD001385.
- Masoli M, Weatherall M, Holt S, Beasley R. Moderate dose inhaled corticosteroids plus salmeterol versus higher doses of inhaled corticosteroid in symptomatic asthma. Thorax 2005; 60:730–734.
- Sin DD, Man J, Sharpe H, Gan WQ, Man SFP. Pharmacological management to reduce exacerbations in adults with asthma. A systematic review and meta-analysis. JAMA 2004; 292:367–376.
- Mann RD, Kubota K, Pearce G, Wilton L. Salmeterol: a study by prescription event monitoring in a UK cohort of 15,407 patients. J Clin Epidemiol 1996; 49:247–250.
- Lanes S, Lanza L, Wentworth C. Risk of emergency care, hospitalization, and ICU stays for acute asthma among recipients of salmeterol. Am J Respir Crit Care Med 1998; 158:857–861.
- Meier CR, Jick H. Drug use and pulmonary death rates in increasingly symptomatic asthma patients in the UK. Thorax 1997; 52:612–617.
- Williams C, Crossland L, Finnerty J, et al. A case control study of salmeterol and near-fatal attacks of asthma. Thorax 1998; 53:7–13.
- Lanes S, Garcia Rodriguez LA, Herta C. Respiratory medications and risk of asthma death. Thorax 2002; 57:683–686.
- Anderson HR, Ayres JG, Sturdy PM, et al. Bronchodilator treatment and deaths from asthma: case control study. Br Med J 2005; 330:117–124.
- Martinez FD. Safety of long-acting beta agonists—an urgent need to clear the air. N Engl J Med 2005; 353:2637–2639.
- Nelson HS. Long-acting beta-agonists in adult asthma: evidence that these drugs are safe. Prim Care Respir J 2006; 15:271–277.
- Lang DM. The long-acting beta agonist controversy: a critical examination of the evidence. Cleve Clin J Med 2006; 73:973–992.
- Rambasek T, Lang DM, Kavuru M. Omalizumab: where does it fit in current asthma management? Cleve Clin J Med 2004; 71:251–261.
- McGlashan D, Bochner B, Adelman D, et al. Down regulation of Fc(epsilon)RI expression on human basophils during in vivo treatment of atopic patients with anti-IgE antibody. J Immunol 1997; 158:1438–1445.
- Busse W, Corren J, Lanier B, et al. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol 2001; 108:184–190.
- Soler M, Matz J, Townley R, et al. The anti-IgE antibody omalizumab reduces asthma exacerbations and steroid requirement in allergic asthmatics. Eur Respir J 2001; 18:254–261.
- Humbert M, Beasley R, Ayres J, et al. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy 2005; 60:309–316.
- Van Ganse E, Antonicelli L, Zhang Q, et al. Asthma-related resource use and cost by GINA classification of severity in three European countries. Respir Med 2006; 100:140–147.
- Godard P, Chanez P, Siraudin L, Nicoloyannis N, Duru G. Costs of asthma are correlated with severity: a 1-yr prospective study. Eur Respir J 2002; 19:61–67.
- Cisternas MG, Blanc PH, Yen IH, et al. A comprehensive study of the direct and indirect costs of adult asthma. J Allergy Clin Immunol 2003; 111:1212–1218.
- Sullivan S, Turk F. An evaluation of the cost effectiveness of omalizumab for the treatment of severe persistent asthma. Allergy 2008; 63:670–684.
- US Food and Drug Administration. Omalizumab (marketed as Xolair) information. www.fda.gov/cder/drug/infopage/omalizumab/default.htm. Accessed August 31, 2007.
- Williams SG, Schmidt DK, Redd SC, Storms W. Key clinical activities for quality asthma care. Recommendations of the National Asthma Education and Prevention Program. MMWR Recomm Rep 2003; 52 RR-6:1–8.
- Cox L, Li J, Nelson H, Lockey R, et al. Allergy Immunotherapy: a practice parameter second update. J Allergy Clin Immunol 2007; 120:S25–S85.
- Akdis M, Akdis CA. Mechanisms of allergen-specific immunotherapy. J Allergy Clin Immunol 2007; 119:780–789.
- Murray AB, Milner RA. The accuracy of features in the clinical history for predicting atopic sensitization to airborne allergens in children. J Allergy Clin Immunol 1995; 96:588–596.
- Bernstein IL, Li JT, Bernstein DI, et al. Allergy diagnostic testing: an updated practice parameter. Ann Allergy Asthma Immunol 2008; 100 suppl 3:1S–148S.
- Walker S, Pajno GB, Lima MT, Wilson DR, Durham SR. Grass pollen immunotherapy for seasonal rhinitis and asthma: a randomized, controlled trial. J Allergy Clin Immunol 2001; 107:87–93.
- Varney VA, Edwards J, Tabbah K, Brewster H, Mavroleon G, Frew AJ. Clinical efficacy of specific immunotherapy to cat dander: a double-blind placebo-controlled trial. Clin Exp Allergy 1997; 27:860–867.
- Cantani A, Arcese G, Lucenti P, Gagliesi D, Bartolucci M. A three-year prospective study of specific immunotherapy to inhalant allergens: evidence of safety and efficacy in 300 children with allergic asthma. J Investig Allergol Clin Immunol 1997; 7:90–97.
- Hedlin G, Wille S, Browaldh L, et al. Immunotherapy in children with allergic asthma: effect on bronchial hyperreactivity and pharmacotherapy. J Allergy Clin Immunol 1999; 103:609–614.
- Arvidsson MB, Löwhagen O, Rak S. Allergen specific immunotherapy attenuates early and late phase reactions in lower airways of birch pollen asthmatic patients: a double blind placebo-controlled study. Allergy 2004; 59:74–80.
- Pichler CE, Helbling A, Pichler WJ. Three years of specific immunotherapy with house-dust-mite extracts in patients with rhinitis and asthma: significant improvement of allergen-specific parameters and of nonspecific bronchial hyperreactivity. Allergy 2001; 56:301–306.
- Mirone C, Albert F, Tosi A, et al. Efficacy and safety of subcutaneous immunotherapy with a biologically standardized extract of Ambrosia artemisiifolia pollen: a double-blind, placebo-controlled study. Clin Exp Allergy 2004; 34:1408–1414.
- Abramson MJ, Puy RM, Weiner JM. Allergen immunotherapy for asthma. Cochrane Database Syst Rev 2003; (4):CD001186.
- Jacobsen L. Preventive aspects of immunotherapy: prevention for children at risk of developing asthma. Ann Allergy Asthma Immunol 2001; 87:43–46.
- Moller C, Dreborg S, Ferdousi HA, et al. Pollen immunotherapy reduces the development of asthma in children with seasonal rhinoconjunctivitis (the PAT study). J Allergy Clin Immunol 2002; 109:251–256.
- Niggemann B, Jacobsen L, Dreborg S, et al; PAT Investigator Group. Five year follow-up on the PAT study: specific immunotherapy and long-term prevention of asthma in children. Allergy 2006: 61:855–859.
- Des Roches A, Paradis L, Menardo JL, et al. Immunotherapy with a standardized Dermatophagoides pteronyssinus extract VI: specific immunotherapy prevents the onset of new sensitizations in children. J Allergy Clin Immunol 1997; 99:450–453.
- Pajno GB, Barberio G, DeLuca F, et al. Prevention of new sensitizations in asthmatic children monosensitized to the house dust mite by specific immunotherapy: a six year follow up study. Clin Exp Allergy 2001; 31:1392–1397.
- Lang DM. Do beta blockers really enhance the risk of anaphylaxis during immunotherapy? Curr Allergy Asthma Rep 2008; 8:37–44.
- National Heart, Lung, and Blood institute, National Asthma education and Prevention Program. Expert Panel Report 3: guidelines for the diagnosis and management of asthma. www.nhlbi.nih.gov/guidelines/asthma. Accessed 8/7/08.
- Expert Panel Report 2: Guidelines for the diagnosis and management of asthma. U.S. Department of Health and Human Services. Publication No. 97-4051; 1997.
- Expert Panel Report: Guidelines for the diagnosis and management of asthma. Update on Selected Topics—2002. J Allergy Clin Immunol 2002; 110:S141–S207.
- FitzGerald JM, Boulet LP, McIvor RA, Zimmerman S, Chapman KR. Asthma control in Canada remains suboptimal: the Reality of Asthma Control (TRAC) study. Can Respir J 2006; 13:253–259.
- Braganza S, Sharif I, Ozuah P. Documenting asthma severity: do we get it right? J Asthma 2003; 40:661–665.
- Cockcroft DW, Swystun VA. Asthma control versus asthma severity. J Allergy Clin Immunol 1996; 98:1016–1018.
- Peters SP, Jones CA, Haselkorn T, Mink DR, Valacer DJ, Weiss ST. Real-world Evaluation of Asthma Control and Treatment (REACT): findings from a national Web-based survey. J Allergy Clin Immunol. 2007; 119:1454–1461.
- Osborne ML, Vollmer WM, Pedula KL, Wilkins J, Buist AS, O’Hollaren M. Lack of correlation of symptoms with specialist-assessed long-term asthma severity. Chest 1999; 115:85–91.
- Li JT, Oppenheimer J, Bernstein IL, et al. Attaining optimal asthma control: a practice parameter. J Allergy Clin Immunol 2005; 116:S3–S11.
- Nathan RA, Sorkness C, Kosinski M, et al. Development of the Asthma Control Test: a survey for assessing asthma control. J Allergy Clin Immunol 2004; 113:59–65.
- Schatz M, Zeiger RS, Drane A, et al. Reliability and predictive validity of the Asthma Control Test administered by telephone calls using speech recognition technology. J Allergy Clin Immunol 2007; 119:336–343.
- Peters D, Chen C, Markson LE, Allen-Ramey FC, Vollmer WM. Using an asthma control questionnaire and administrative data to predict healthcare utilization. Chest 2006; 129:918–924.
- Schatz M, Sorkness C, Li JT, et al. Asthma Control Test: reliability, validity, and responsiveness in patients not previously followed by asthma specialists. J Allergy Clin Immunol 2006; 117:549–556.
- Bateman E, Boushey H, Bousquet J, et al. Can guideline-defined asthma control be achieved? Am J Respir Crit Care Med 2004; 170:836–844.
- Davies TJ, Bunn WB, Fromer L, Gelfand EW, Colice GL. A focus on the asthma HEDIS measure and its implications for clinical practice. Manag Care Interface 2006; 19:29–36.
- Rubinfeld AR, Pain MC. Perception of asthma. Lancet 1976; 1:882–884.
- Teeter J, Bleecker E. Relationship between airway obstruction and respiratory symptoms in adult asthmatics. Chest 1998; 113:272–277.
- Shingo S, Zhang J, Reiss T. Correlation of airway obstruction and patient reported endpoints in clinical studies. Eur Resp J 2001; 17:220–224.
- Juniper EF, Bousquet J, Abetz L, Bateman ED; GOAL Committee. Identifying ‘well-controlled’ and ‘not well-controlled’ asthma using the Asthma Control Questionnaire. Respir Med 2006; 100:616–621.
- Nelson H, Weiss S, Bleecker E, Yancey S, Dorinsky P. The Salmeterol Multicenter Asthma Research Trial. Chest 2006; 129:15–26.
- Wechsler M, Lehman E, Lazarus S, et al. ß-Adrenergic receptor polymorphisms and response to salmeterol. Am J Respir Crit Care Med 2006; 173:519–526.
- Palmer CNA, Lipworth BJ, Lee S, Ismail T, MacGregor DF, Mukhopadhyay S. Arginine-16 beta-2 adrenoceptor genotype predisposes to exacerbations in young asthmatics taking regular salmeterol. Thorax 2006; 61:940–944.
- Taylor DR, Drazen JM, Herbison GP, Yandava CN, Hancox RJ, Town GI. Asthma exacerbations during long term beta agonist use: influence of beta 2 adrenoceptor polymorphism. Thorax 2000; 55:762–727.
- Bleecker E, Postma D, Lawrance R, Meyers D, Ambrose H, Goldman M. Effect of ADRB2 polymorphisms on response to long-acting beta2-agonist therapy: a pharmacogenetic analysis of two randomized studies. Lancet 2007; 370:2118–2125.
- Bleecker E, Yancey S, Baitinger L, et al. Salmeterol response is not affected by beta-2 adrenergic receptor genotype in subjects with persistent asthma. J Allergy Clin Immunol 2006; 118:809–816.
- Nelson H, Bleecker E, Corren J, et al. Characterization of asthma exacerbations by Arg16Gly genotype in subjects with asthma receiving salmeterol alone or with fluticasone propionate. J Allergy Clin Immunol 2008; 121:S131.
- O’Byrne P, Barnes P, Rodriguez-Roisin R, et al. Low dose Inhaled budesonide and formoterol in mild persistent asthma. The OPTIMA Randomized Trial. Am J Respir Crit Care Med 2001; 164:1392–1397.
- Greening AP, Ind PW, Northfield M, Shaw G. Added salmeterol versus higher dose corticosteroid in asthma patients with symptoms on existing inhaled corticosteroid. Lancet 1994; 344:219–224.
- Woolcock A, Lundback B, Ringdal N, Jacques LA. Comparison of addition of salmeterol to inhaled steroids with doubling of the dose of inhaled steroids. Am J Respir Crit Care Med 1996; 153:1481–1488.
- Walters EH, Walters JAE, Gibson MDP. Long-acting beta2-agonists for stable chronic asthma. Cochrane Database Syst Rev 2003; (3):CD001385. doi:10.1002/14651858.CD001385.
- Masoli M, Weatherall M, Holt S, Beasley R. Moderate dose inhaled corticosteroids plus salmeterol versus higher doses of inhaled corticosteroid in symptomatic asthma. Thorax 2005; 60:730–734.
- Sin DD, Man J, Sharpe H, Gan WQ, Man SFP. Pharmacological management to reduce exacerbations in adults with asthma. A systematic review and meta-analysis. JAMA 2004; 292:367–376.
- Mann RD, Kubota K, Pearce G, Wilton L. Salmeterol: a study by prescription event monitoring in a UK cohort of 15,407 patients. J Clin Epidemiol 1996; 49:247–250.
- Lanes S, Lanza L, Wentworth C. Risk of emergency care, hospitalization, and ICU stays for acute asthma among recipients of salmeterol. Am J Respir Crit Care Med 1998; 158:857–861.
- Meier CR, Jick H. Drug use and pulmonary death rates in increasingly symptomatic asthma patients in the UK. Thorax 1997; 52:612–617.
- Williams C, Crossland L, Finnerty J, et al. A case control study of salmeterol and near-fatal attacks of asthma. Thorax 1998; 53:7–13.
- Lanes S, Garcia Rodriguez LA, Herta C. Respiratory medications and risk of asthma death. Thorax 2002; 57:683–686.
- Anderson HR, Ayres JG, Sturdy PM, et al. Bronchodilator treatment and deaths from asthma: case control study. Br Med J 2005; 330:117–124.
- Martinez FD. Safety of long-acting beta agonists—an urgent need to clear the air. N Engl J Med 2005; 353:2637–2639.
- Nelson HS. Long-acting beta-agonists in adult asthma: evidence that these drugs are safe. Prim Care Respir J 2006; 15:271–277.
- Lang DM. The long-acting beta agonist controversy: a critical examination of the evidence. Cleve Clin J Med 2006; 73:973–992.
- Rambasek T, Lang DM, Kavuru M. Omalizumab: where does it fit in current asthma management? Cleve Clin J Med 2004; 71:251–261.
- McGlashan D, Bochner B, Adelman D, et al. Down regulation of Fc(epsilon)RI expression on human basophils during in vivo treatment of atopic patients with anti-IgE antibody. J Immunol 1997; 158:1438–1445.
- Busse W, Corren J, Lanier B, et al. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol 2001; 108:184–190.
- Soler M, Matz J, Townley R, et al. The anti-IgE antibody omalizumab reduces asthma exacerbations and steroid requirement in allergic asthmatics. Eur Respir J 2001; 18:254–261.
- Humbert M, Beasley R, Ayres J, et al. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy 2005; 60:309–316.
- Van Ganse E, Antonicelli L, Zhang Q, et al. Asthma-related resource use and cost by GINA classification of severity in three European countries. Respir Med 2006; 100:140–147.
- Godard P, Chanez P, Siraudin L, Nicoloyannis N, Duru G. Costs of asthma are correlated with severity: a 1-yr prospective study. Eur Respir J 2002; 19:61–67.
- Cisternas MG, Blanc PH, Yen IH, et al. A comprehensive study of the direct and indirect costs of adult asthma. J Allergy Clin Immunol 2003; 111:1212–1218.
- Sullivan S, Turk F. An evaluation of the cost effectiveness of omalizumab for the treatment of severe persistent asthma. Allergy 2008; 63:670–684.
- US Food and Drug Administration. Omalizumab (marketed as Xolair) information. www.fda.gov/cder/drug/infopage/omalizumab/default.htm. Accessed August 31, 2007.
- Williams SG, Schmidt DK, Redd SC, Storms W. Key clinical activities for quality asthma care. Recommendations of the National Asthma Education and Prevention Program. MMWR Recomm Rep 2003; 52 RR-6:1–8.
- Cox L, Li J, Nelson H, Lockey R, et al. Allergy Immunotherapy: a practice parameter second update. J Allergy Clin Immunol 2007; 120:S25–S85.
- Akdis M, Akdis CA. Mechanisms of allergen-specific immunotherapy. J Allergy Clin Immunol 2007; 119:780–789.
- Murray AB, Milner RA. The accuracy of features in the clinical history for predicting atopic sensitization to airborne allergens in children. J Allergy Clin Immunol 1995; 96:588–596.
- Bernstein IL, Li JT, Bernstein DI, et al. Allergy diagnostic testing: an updated practice parameter. Ann Allergy Asthma Immunol 2008; 100 suppl 3:1S–148S.
- Walker S, Pajno GB, Lima MT, Wilson DR, Durham SR. Grass pollen immunotherapy for seasonal rhinitis and asthma: a randomized, controlled trial. J Allergy Clin Immunol 2001; 107:87–93.
- Varney VA, Edwards J, Tabbah K, Brewster H, Mavroleon G, Frew AJ. Clinical efficacy of specific immunotherapy to cat dander: a double-blind placebo-controlled trial. Clin Exp Allergy 1997; 27:860–867.
- Cantani A, Arcese G, Lucenti P, Gagliesi D, Bartolucci M. A three-year prospective study of specific immunotherapy to inhalant allergens: evidence of safety and efficacy in 300 children with allergic asthma. J Investig Allergol Clin Immunol 1997; 7:90–97.
- Hedlin G, Wille S, Browaldh L, et al. Immunotherapy in children with allergic asthma: effect on bronchial hyperreactivity and pharmacotherapy. J Allergy Clin Immunol 1999; 103:609–614.
- Arvidsson MB, Löwhagen O, Rak S. Allergen specific immunotherapy attenuates early and late phase reactions in lower airways of birch pollen asthmatic patients: a double blind placebo-controlled study. Allergy 2004; 59:74–80.
- Pichler CE, Helbling A, Pichler WJ. Three years of specific immunotherapy with house-dust-mite extracts in patients with rhinitis and asthma: significant improvement of allergen-specific parameters and of nonspecific bronchial hyperreactivity. Allergy 2001; 56:301–306.
- Mirone C, Albert F, Tosi A, et al. Efficacy and safety of subcutaneous immunotherapy with a biologically standardized extract of Ambrosia artemisiifolia pollen: a double-blind, placebo-controlled study. Clin Exp Allergy 2004; 34:1408–1414.
- Abramson MJ, Puy RM, Weiner JM. Allergen immunotherapy for asthma. Cochrane Database Syst Rev 2003; (4):CD001186.
- Jacobsen L. Preventive aspects of immunotherapy: prevention for children at risk of developing asthma. Ann Allergy Asthma Immunol 2001; 87:43–46.
- Moller C, Dreborg S, Ferdousi HA, et al. Pollen immunotherapy reduces the development of asthma in children with seasonal rhinoconjunctivitis (the PAT study). J Allergy Clin Immunol 2002; 109:251–256.
- Niggemann B, Jacobsen L, Dreborg S, et al; PAT Investigator Group. Five year follow-up on the PAT study: specific immunotherapy and long-term prevention of asthma in children. Allergy 2006: 61:855–859.
- Des Roches A, Paradis L, Menardo JL, et al. Immunotherapy with a standardized Dermatophagoides pteronyssinus extract VI: specific immunotherapy prevents the onset of new sensitizations in children. J Allergy Clin Immunol 1997; 99:450–453.
- Pajno GB, Barberio G, DeLuca F, et al. Prevention of new sensitizations in asthmatic children monosensitized to the house dust mite by specific immunotherapy: a six year follow up study. Clin Exp Allergy 2001; 31:1392–1397.
- Lang DM. Do beta blockers really enhance the risk of anaphylaxis during immunotherapy? Curr Allergy Asthma Rep 2008; 8:37–44.
KEY POINTS
- The EPR3 recommends that management decisions be based initially on asthma severity, and subsequently on asthma control as assessed serially by validated tests.
- Omalizumab, a monoclonal antibody against immunoglobulin E, is the only adjunctive therapy to demonstrate efficacy when added to high-dose inhaled corticosteroids plus long-acting beta agonists in patients with severe, persistent, allergic asthma.
- The EPR3 guidelines recommend consideration of allergen immunotherapy for patients with mild or moderate persistent allergic asthma.
A case of refractory diarrhea
A 68-year-old white woman with irritable bowel syndrome has had worsening symptoms of right-sided abdominal pain, excessive bloating, and loose stools. Her bowel movements have increased from one a day to two or three a day. She has not noted any mucus or blood in the stool. She cannot identify any alleviating or aggravating factors, and the pain is not related to eating.
She consumes a normal diet, including meat and dairy. Over-the-counter antidiarrheal medications do not relieve the symptoms. She has had no fevers, chills, or night sweats, and she has not lost weight over the past year.
Her medical history includes breast cancer (in remission), alcohol abuse (in remission), and hypothyroidism, osteoporosis, and supraventricular tachycardia, all controlled with treatment as noted below. She has never undergone abdominal surgery.
A general review of systems is normal. Her current medications include oxybutynin (available as Ditropan, others), calcium polycarbophil (FiberCon, others), risedronate (Actonel), levothyroxine (Synthroid, others), simethicone (Maalox Anti-Gas, others), atenolol (Tenormin), trazodone (Desyrel), a calcium supplement, and aspirin. She began taking duloxetine (Cymbalta) 18 months ago, and the dose was increased from 60 mg to 90 mg 1 week before this visit.
She has never smoked, and she has abstained from alcohol for 10 years. She has no family history of colon cancer, celiac disease, or inflammatory bowel disease. She has not traveled outside the country in the past several years, and she notes no change in her source of drinking water.
On physical examination, she does not appear to be in acute distress. Her pulse is 64 and her blood pressure is 112/78 mm Hg. The cardiopulmonary examination is normal. Her abdomen is soft, symmetrical, nondistended, and nontender. Bowel sounds are normal. No abdominal masses, palpable organomegaly, or abdominal bruits are noted.
Results of basic laboratory tests, including thyroid-stimulating hormone (TSH), complete blood count, blood chemistries, renal function, and liver function, are normal. Colonoscopy shows normal mucosa as far as the cecum.
DIFFERENTIAL DIAGNOSIS
1. In addition to irritable bowel syndrome, which of these can explain her symptoms?
- Ulcerative colitis
- Celiac disease
- Microscopic colitis
- Hyperthyroidism
- Lactase deficiency
Ulcerative colitis typically presents with blood and mucus in the stool and gross abnormalities on colonoscopy, none of which is present in this patient.
Hyperthyroidism can be ruled out by the normal TSH level.
Lactase deficiency or lactose intolerance is unlikely because it is present in only 15% of people of northern European descent (compared with 80% of blacks and Hispanics and up to 100% of Native Americans and Asians).1 Furthermore, her pain is apparently not related to consuming dairy products.
The hydrogen breath test can aid in the diagnosis of lactase deficiency. This test relies on the breakdown of malabsorbed lactose by colonic flora. This is the most widely used test for this deficiency, but its high false-negative rate of 25% means that a negative result does not exclude the diagnosis and should not be relied on in working up a patient with chronic diarrhea.2 Simply noting whether symptoms develop after ingesting 50 g of lactose is clinically useful when lactase deficiency is suspected.
Based on the information so far, it is reasonable in this patient to evaluate for celiac disease and for microscopic colitis.
Celiac disease, also called gluten-sensitive enteropathy, has a varied presentation that includes nonspecific symptoms such as those in this patient. Classically, it causes diarrhea, but patients may present with a single nutrient deficiency and no diarrhea.
This patient lacks the elevated alkaline phosphatase or evidence of vitamin deficiencies characteristic of malabsorption in celiac disease (ie, vitamins A, B12, D, K, and folate)3. She also lacks evidence of malnutrition, such as iron deficiency anemia, weight loss, or low serum albumin. Finally, she does not have the dermatitis herpetiformis rash to suggest autoimmune gluten-sensitive enteropathy, nor does she have evidence of follicular hyperplasia or petechiae due to vitamin malabsorption.3
Because no single serologic test is ideal for diagnosing gluten-sensitive enteropathy, several tests are typically used: immunoglobulin A (IgA) antigliadin antibody, IgG antigliadin antibody, IgA antitransglutaminase antibody, and IgA antiendomysial antibody. IgA antitransglutaminase antibody is 92% to 98% sensitive and 91% to 100% specific for celiac disease. IgG antigliadin antibody is 92% to 97% sensitive and 99% specific. The positive predictive value of the IgA and IgG antigliadin antibody tests is less than 2% in the general population, whereas the positive predictive value for antiendomysial antibody and antitransglutaminase antibody are 15.7% and 21.8%, respectively.4 A positive serologic test for antiendomysial antibody is nearly 100% specific.
Our patient’s entire celiac antibody panel is negative, and thus celiac disease is unlikely.
Case continued: Features of microscopic colitis
In our patient, colonic biopsy reveals a mildly expanded lamina propria, intraepithelial lymphocytes, and a patchy but prominent thickening of the subepithelial collagen table. This set of features is consistent with collagenous colitis, a variant of microscopic colitis. Histologic signs on biopsy specimens are fairly specific for the disease.5
Chronic, intermittent, secretory diarrhea without bleeding is the hallmark of microscopic colitis. Associated symptoms may include abdominal pain, weight loss, and fatigue. If biopsies are not taken at the time of the initial evaluation, and the colonic pathology is overlooked, patients with collagenous colitis may be diagnosed with irritable bowel syndrome with diarrhea.6 The sedimentation rate is often elevated, and the antinuclear antibody test can be positive.7 Steatorrhea or protein-losing enteropathy can occur, and fecal leukocytes are present in more than 50% of patients.8
This patient fits well the demographics of the typical collagenous colitis patient: ie, a middle-aged woman in her 6th decade in otherwise good general health. The female-to-male ratio is 15:1 overall, although the relative frequency of collagenous colitis in women is greater than that of lymphocytic colitis.9 In a population-based study, the incidence of collagenous colitis was 5.1 per 100,000 per year, with a prevalence of 36 per 100,000; the incidence of lymphocytic colitis was 9.8 per 100,000 per year, with a prevalence of 64 per 100,000.10
Symptoms are typically vague and range from an annoyance to more than 20 non-bloody stools per day. The course of the disease also varies. Case series have reported a spontaneous remission rate of 15% to 20%,11 though flare-ups are common. Microscopic colitis is largely a benign disease. It does not increase a person’s risk of colon cancer.
CAUSES OF COLLAGENOUS COLITIS
2. What causes of collagenous colitis have been identified?
- Alcohol abuse
- Previous gastrointestinal surgery
- Drug-induced injury to colon
Neither alcohol use nor previous gastrointestinal surgery has been associated with the development of collagenous colitis.
Collagenous colitis has, however, been linked to several causes. Abnormal collagen metabolism has been demonstrated in patients as a result of increased expression of procollagen I and metalloproteinase inhibitor TIMP-1.12 Bacterial toxins and a bile-acid malabsorption defect in the terminal ileum and subsequent exposure of the colon to high concentrations of bile acids have also been linked to the development of collagenous colitis.
Many drugs have been linked to the development of collagenous colitis. Damage to the large intestine related to the use of non-steroidal anti-inflammatory drugs has been attributed to the blockage of prostaglandin synthesis.13 Simvastatin (Zocor), lansoprazole (Prilosec), and ticlopidine (Ticlid) have been linked to collagenous colitis; ticlopidine, flutamide (Eulexin), gold salts, lansoprazole, and sertraline (Zoloft) have been linked to the development of lymphocytic colitis.14 In one small series, patients developed colitis after switching from omeprazole (Prevacid) to lansoprazole. All patients had their symptoms and biopsy findings resolve within 1 week of stopping the drug.15
WHICH DRUG IS BEST?
3. Which drug is best for microscopic colitis, based on the current evidence?
- Bismuth (eg, Kaopectate, Pepto-Bismol)
- Sulfasalazine (Sulfazine)
- Budesonide (Entocort)
- Prednisolone
Studies have evaluated bismuth subsalicylate, Boswellia serrata extract, probiotics, prednisolone, budesonide, and other drugs for treating collagenous colitis.16
Bismuth trials have been small. In an open-label study of bismuth,17 symptoms improved in 11 of 12 patients.
Prednisolone recipients had a trend towards clinical response with treatment vs placebo, but it was not statistically significant, and there was incomplete remission of disease.18
Boswellia serrataextract19 and probiotics20 showed no clinical improvement.
Cholestyramine has been shown to be helpful when used in conjunction with an anti-inflammatory agent,21 and it may be helpful when used alone.
Aminosalicylate compounds have not been tested in prospective randomized trials, even though they are the cornerstone of treatment for ulcerative colitis. Retrospective trials have been equivocal.22
Budesonide currently has the best evidence of efficacy in collagenous colitis,23,24 and some evidence suggests it is also effective for other variants of microscopic colitis.
A total of 94 patients were enrolled in three placebo-controlled trials of budesonide at 9 mg daily or on a tapering schedule for 6 to 8 weeks. The pooled odds ratio for clinical response to treatment with budesonide was 12.32 (95% confidence interval 5.53–27.46), with a number needed to treat of 1.58. Significant histologic improvement with treatment was noted in all three trials.23
Quality of life has also been studied in patients with microscopic colitis who take budesonide. Symptoms, emotional functioning, and physical functioning are improved. Budesonide also improved stool consistency and significantly reduced the mean stool frequency compared with placebo.24
Compared with cortisol, budesonide has a 200 times greater affinity for the glucocorticoid receptor, and a 1,000 times greater topical anti-inflammatory potency. It is also well absorbed in the gastrointestinal tract but is substantially modified into very weak metabolites as a result of first-pass metabolism in the liver.25 This localized effect further supports the use of budesonide in patients with any form of microscopic colitis.
Although studies have shown budesonide to be effective, not every patient with a histologic diagnosis of microscopic colitis needs it. It is reasonable to try antidiarrheal agents, bismuth, or both as a first step because they are inexpensive and have few side effects. If budesonide is used, it should be given for 6 to 8 weeks, then stopped, and the patient should then be monitored for symptom recurrence. If a flare does occur, budesonide can be restarted and continued as maintenance therapy.
KEY CONSIDERATIONS
Microscopic colitis is diagnosed histologically, while irritable bowel syndrome is a clinical diagnosis. In population-based cohorts of histologically confirmed microscopic colitis, 50% to 70% met symptom-based Rome criteria for the diagnosis of irritable bowel syndrome. The clinical symptom-based criteria for irritable bowel syndrome are not specific enough to rule out the diagnosis of microscopic colitis. Therefore, patients with suspected diarrhea-predominant irritable bowel syndrome should undergo colonoscopy with biopsy to investigate microscopic colitis if symptoms are not well controlled by antidiarrheal therapy.26 The patient’s management may be very different depending on whether colonoscopy is done.
Management of microscopic colitis should include stopping any drugs associated with it. Simple antidiarrheal agents should be tried first to manage symptoms. If symptoms persist, patients can be treated with budesonide (Entocort EC) 9 mg by mouth daily for 8 weeks to induce remission, or 6 mg by mouth daily for 3 months as maintenance therapy.
OUR PATIENT’S COURSE
Our patient’s medication list includes duloxetine, a serotonin-norepinephrine reuptake inhibitor related to drugs that have been associated with the development of microscopic colitis. We tapered the duloxetine, and her symptoms improved by 50%. Her symptoms were eventually controlled after an 8-week course of oral budesonide 9 mg and ongoing intermittent use of loperamide (Imodium).
- Swagerty DL, Walling AD, Klein RM. Lactose intolerance. Am Fam Physician 2002; 65:1845–1856.
- Thomas PD, Forbes A, Green J, et al. Guidelines for the investigation of chronic diarrhea, 2nd edition. Gut 2003; 52(suppl 5):1–5.
- Nelsen DA. Gluten-sensitive enteropathy (celiac disease): more common than you think. Am Fam Physician 2002; 66:2259–2266.
- Bardella MT, Trovato C, Cesana BM, Pagliari C, Gebbia C, Peracchi M. Serological markers for coeliac disease: is it time to change? Dig Liver Dis 2001; 33:426–431.
- Barta Z, Mekkel G, Csipo I, et al. Micropscopic colitis: a retrospective study of clinical presentation in 53 patients. World J Gastroenterol 2005; 11:1351–1355.
- Tremaine WJ. Diagnosing collagenous colitis: does it make a difference? Eur J Gastroenterol Hepatol 1999; 11:477–479.
- Bohr J, Tysk C, Yang P, Danielsson D, Järnerot G. Autoantibodies and immunoglobulins in collagenous colitis. Gut 1996; 39:77–81.
- Zins BJ, Tremaine WJ, Carpenter HA. Collagenous colitis: mucosal biopsies and association with fecal leukocytes. Mayo Clin Proc 1995; 70:430–433.
- Olsen M, Eriksson S, Bohr J, Järnerot G, Tysk C. Lymphocytic colitis: a retrospective clinical study of 199 Swedish patients. Gut 2004; 53:536–541.
- Pardi DS. Microscopic colitis: an update. Inflamm Bowel Dis 2004; 10:860–870.
- Fernandez-Banares F, Salas A, Esteve M, Espinos J, Forne M, Viver JM. Collagenous and lymphocytic colitis: evaluation of clinical and histological features, response to treatment, and long-term follow-up. Am J Gastroenterol 2003; 98:340–347.
- Aignet T, Neureiter D, Müller S, Küspert G, Belke J, Kirchner T. Extracellular matrix composition and gene expression in collagenous colitis. Gastroenterology 1997; 113:136–143.
- Parfitt JR, Driman DK. Pathological effects of drugs on the gastrointestinal tract: a review. Hum Pathol 2007; 38:527–536.
- Fernández-Bañares F, Esteve M, Espinós JC, et al. Drug consumption and the risk of microscopic colitis. Am J Gastroenterol 2007; 102:324–330.
- Thomson RD, Lestine LS, Bensen SP, et al. Lansoprazole-associated microscopic colitis: a case series. Am J Gastroenterol 2002; 97:2908–2913.
- Chande N, McDonald JWD, MacDonald JK. Cochrane Inflammatory Bowel Disease and Functional Bowel Disorders Group. Interventions for treating collagenous colitis. Cochrane Database Syst Rev 2007 Jan 24;(1):CD006096.
- Fine K, Lee E. Efficacy of open-label bismuth subsalicylate for the treatment of microscopic colitis. Gastroenterology 1998; 114:29–36.
- Munck LK, Kjeldsen J, Philipsen E, Fscher Hansen B. Incomplete remission with short-term prednisolone treatment in collagenous colitis: a randomized study. Scand J Gastroenterol 2003; 38:606–610.
- Madisch A, Miehlke S, Eichele E, et al. Boswellia serrata extract for the treatment of collagenous colitis: a randomized, double-blind, placebo-controlled, multicenter trial. Int J Colorectal Dis 2007; 22:1445–1451.
- Wildt S, Munck LK, Vinter-Jensen L, et al. Probiotic treatment of collagenous colitis: a randomized, double-blind, placebo-controlled trial with Lactobacillus acidophilus and Bifidobacterium animalis subsp. lactis. Inflamm Bowel Dis 2006; 12:395–401.
- Calabrese C, Fabbri A, Areni A, Zahlane D, Scialpi C, Di Febo G. Mesalazine with or without cholestyramine in the treatment of microscopic colitis: randomized controlled trial. J Gastroenterol Hepatol 2007; 22:809–814.
- Wall GC, Schirmer LL, Page MJ. Pharmacotherapy for microscopic colitis. Pharmacotherapy 2007; 27:425–433.
- Feyen B, Wall GC, Finnerty EP, DeWitt JE, Reyes RS. Meta-analysis: budesonide treatment for collagenous colitis. Aliment Pharmacol Ther 2004; 20:745–749.
- Madisch A, Heymer P, Voss C, et al. Oral budesonide therapy improves quality of life in patients with collagenous colitis. Int J Colorectal Dis 2005; 20:312–316.
- Craig CR, editor. Modern Pharmacology With Clinical Application. 6th edition. Philadelphia: Lippincott Williams and Wilkins, 2003:481.
- Limsui D, Pardi DS, Camilleri M, et al. Symptomatic overlap between irritable bowel syndrome and microscopic colitis. Inflamm Bowel Dis 2007; 13:175–181.
A 68-year-old white woman with irritable bowel syndrome has had worsening symptoms of right-sided abdominal pain, excessive bloating, and loose stools. Her bowel movements have increased from one a day to two or three a day. She has not noted any mucus or blood in the stool. She cannot identify any alleviating or aggravating factors, and the pain is not related to eating.
She consumes a normal diet, including meat and dairy. Over-the-counter antidiarrheal medications do not relieve the symptoms. She has had no fevers, chills, or night sweats, and she has not lost weight over the past year.
Her medical history includes breast cancer (in remission), alcohol abuse (in remission), and hypothyroidism, osteoporosis, and supraventricular tachycardia, all controlled with treatment as noted below. She has never undergone abdominal surgery.
A general review of systems is normal. Her current medications include oxybutynin (available as Ditropan, others), calcium polycarbophil (FiberCon, others), risedronate (Actonel), levothyroxine (Synthroid, others), simethicone (Maalox Anti-Gas, others), atenolol (Tenormin), trazodone (Desyrel), a calcium supplement, and aspirin. She began taking duloxetine (Cymbalta) 18 months ago, and the dose was increased from 60 mg to 90 mg 1 week before this visit.
She has never smoked, and she has abstained from alcohol for 10 years. She has no family history of colon cancer, celiac disease, or inflammatory bowel disease. She has not traveled outside the country in the past several years, and she notes no change in her source of drinking water.
On physical examination, she does not appear to be in acute distress. Her pulse is 64 and her blood pressure is 112/78 mm Hg. The cardiopulmonary examination is normal. Her abdomen is soft, symmetrical, nondistended, and nontender. Bowel sounds are normal. No abdominal masses, palpable organomegaly, or abdominal bruits are noted.
Results of basic laboratory tests, including thyroid-stimulating hormone (TSH), complete blood count, blood chemistries, renal function, and liver function, are normal. Colonoscopy shows normal mucosa as far as the cecum.
DIFFERENTIAL DIAGNOSIS
1. In addition to irritable bowel syndrome, which of these can explain her symptoms?
- Ulcerative colitis
- Celiac disease
- Microscopic colitis
- Hyperthyroidism
- Lactase deficiency
Ulcerative colitis typically presents with blood and mucus in the stool and gross abnormalities on colonoscopy, none of which is present in this patient.
Hyperthyroidism can be ruled out by the normal TSH level.
Lactase deficiency or lactose intolerance is unlikely because it is present in only 15% of people of northern European descent (compared with 80% of blacks and Hispanics and up to 100% of Native Americans and Asians).1 Furthermore, her pain is apparently not related to consuming dairy products.
The hydrogen breath test can aid in the diagnosis of lactase deficiency. This test relies on the breakdown of malabsorbed lactose by colonic flora. This is the most widely used test for this deficiency, but its high false-negative rate of 25% means that a negative result does not exclude the diagnosis and should not be relied on in working up a patient with chronic diarrhea.2 Simply noting whether symptoms develop after ingesting 50 g of lactose is clinically useful when lactase deficiency is suspected.
Based on the information so far, it is reasonable in this patient to evaluate for celiac disease and for microscopic colitis.
Celiac disease, also called gluten-sensitive enteropathy, has a varied presentation that includes nonspecific symptoms such as those in this patient. Classically, it causes diarrhea, but patients may present with a single nutrient deficiency and no diarrhea.
This patient lacks the elevated alkaline phosphatase or evidence of vitamin deficiencies characteristic of malabsorption in celiac disease (ie, vitamins A, B12, D, K, and folate)3. She also lacks evidence of malnutrition, such as iron deficiency anemia, weight loss, or low serum albumin. Finally, she does not have the dermatitis herpetiformis rash to suggest autoimmune gluten-sensitive enteropathy, nor does she have evidence of follicular hyperplasia or petechiae due to vitamin malabsorption.3
Because no single serologic test is ideal for diagnosing gluten-sensitive enteropathy, several tests are typically used: immunoglobulin A (IgA) antigliadin antibody, IgG antigliadin antibody, IgA antitransglutaminase antibody, and IgA antiendomysial antibody. IgA antitransglutaminase antibody is 92% to 98% sensitive and 91% to 100% specific for celiac disease. IgG antigliadin antibody is 92% to 97% sensitive and 99% specific. The positive predictive value of the IgA and IgG antigliadin antibody tests is less than 2% in the general population, whereas the positive predictive value for antiendomysial antibody and antitransglutaminase antibody are 15.7% and 21.8%, respectively.4 A positive serologic test for antiendomysial antibody is nearly 100% specific.
Our patient’s entire celiac antibody panel is negative, and thus celiac disease is unlikely.
Case continued: Features of microscopic colitis
In our patient, colonic biopsy reveals a mildly expanded lamina propria, intraepithelial lymphocytes, and a patchy but prominent thickening of the subepithelial collagen table. This set of features is consistent with collagenous colitis, a variant of microscopic colitis. Histologic signs on biopsy specimens are fairly specific for the disease.5
Chronic, intermittent, secretory diarrhea without bleeding is the hallmark of microscopic colitis. Associated symptoms may include abdominal pain, weight loss, and fatigue. If biopsies are not taken at the time of the initial evaluation, and the colonic pathology is overlooked, patients with collagenous colitis may be diagnosed with irritable bowel syndrome with diarrhea.6 The sedimentation rate is often elevated, and the antinuclear antibody test can be positive.7 Steatorrhea or protein-losing enteropathy can occur, and fecal leukocytes are present in more than 50% of patients.8
This patient fits well the demographics of the typical collagenous colitis patient: ie, a middle-aged woman in her 6th decade in otherwise good general health. The female-to-male ratio is 15:1 overall, although the relative frequency of collagenous colitis in women is greater than that of lymphocytic colitis.9 In a population-based study, the incidence of collagenous colitis was 5.1 per 100,000 per year, with a prevalence of 36 per 100,000; the incidence of lymphocytic colitis was 9.8 per 100,000 per year, with a prevalence of 64 per 100,000.10
Symptoms are typically vague and range from an annoyance to more than 20 non-bloody stools per day. The course of the disease also varies. Case series have reported a spontaneous remission rate of 15% to 20%,11 though flare-ups are common. Microscopic colitis is largely a benign disease. It does not increase a person’s risk of colon cancer.
CAUSES OF COLLAGENOUS COLITIS
2. What causes of collagenous colitis have been identified?
- Alcohol abuse
- Previous gastrointestinal surgery
- Drug-induced injury to colon
Neither alcohol use nor previous gastrointestinal surgery has been associated with the development of collagenous colitis.
Collagenous colitis has, however, been linked to several causes. Abnormal collagen metabolism has been demonstrated in patients as a result of increased expression of procollagen I and metalloproteinase inhibitor TIMP-1.12 Bacterial toxins and a bile-acid malabsorption defect in the terminal ileum and subsequent exposure of the colon to high concentrations of bile acids have also been linked to the development of collagenous colitis.
Many drugs have been linked to the development of collagenous colitis. Damage to the large intestine related to the use of non-steroidal anti-inflammatory drugs has been attributed to the blockage of prostaglandin synthesis.13 Simvastatin (Zocor), lansoprazole (Prilosec), and ticlopidine (Ticlid) have been linked to collagenous colitis; ticlopidine, flutamide (Eulexin), gold salts, lansoprazole, and sertraline (Zoloft) have been linked to the development of lymphocytic colitis.14 In one small series, patients developed colitis after switching from omeprazole (Prevacid) to lansoprazole. All patients had their symptoms and biopsy findings resolve within 1 week of stopping the drug.15
WHICH DRUG IS BEST?
3. Which drug is best for microscopic colitis, based on the current evidence?
- Bismuth (eg, Kaopectate, Pepto-Bismol)
- Sulfasalazine (Sulfazine)
- Budesonide (Entocort)
- Prednisolone
Studies have evaluated bismuth subsalicylate, Boswellia serrata extract, probiotics, prednisolone, budesonide, and other drugs for treating collagenous colitis.16
Bismuth trials have been small. In an open-label study of bismuth,17 symptoms improved in 11 of 12 patients.
Prednisolone recipients had a trend towards clinical response with treatment vs placebo, but it was not statistically significant, and there was incomplete remission of disease.18
Boswellia serrataextract19 and probiotics20 showed no clinical improvement.
Cholestyramine has been shown to be helpful when used in conjunction with an anti-inflammatory agent,21 and it may be helpful when used alone.
Aminosalicylate compounds have not been tested in prospective randomized trials, even though they are the cornerstone of treatment for ulcerative colitis. Retrospective trials have been equivocal.22
Budesonide currently has the best evidence of efficacy in collagenous colitis,23,24 and some evidence suggests it is also effective for other variants of microscopic colitis.
A total of 94 patients were enrolled in three placebo-controlled trials of budesonide at 9 mg daily or on a tapering schedule for 6 to 8 weeks. The pooled odds ratio for clinical response to treatment with budesonide was 12.32 (95% confidence interval 5.53–27.46), with a number needed to treat of 1.58. Significant histologic improvement with treatment was noted in all three trials.23
Quality of life has also been studied in patients with microscopic colitis who take budesonide. Symptoms, emotional functioning, and physical functioning are improved. Budesonide also improved stool consistency and significantly reduced the mean stool frequency compared with placebo.24
Compared with cortisol, budesonide has a 200 times greater affinity for the glucocorticoid receptor, and a 1,000 times greater topical anti-inflammatory potency. It is also well absorbed in the gastrointestinal tract but is substantially modified into very weak metabolites as a result of first-pass metabolism in the liver.25 This localized effect further supports the use of budesonide in patients with any form of microscopic colitis.
Although studies have shown budesonide to be effective, not every patient with a histologic diagnosis of microscopic colitis needs it. It is reasonable to try antidiarrheal agents, bismuth, or both as a first step because they are inexpensive and have few side effects. If budesonide is used, it should be given for 6 to 8 weeks, then stopped, and the patient should then be monitored for symptom recurrence. If a flare does occur, budesonide can be restarted and continued as maintenance therapy.
KEY CONSIDERATIONS
Microscopic colitis is diagnosed histologically, while irritable bowel syndrome is a clinical diagnosis. In population-based cohorts of histologically confirmed microscopic colitis, 50% to 70% met symptom-based Rome criteria for the diagnosis of irritable bowel syndrome. The clinical symptom-based criteria for irritable bowel syndrome are not specific enough to rule out the diagnosis of microscopic colitis. Therefore, patients with suspected diarrhea-predominant irritable bowel syndrome should undergo colonoscopy with biopsy to investigate microscopic colitis if symptoms are not well controlled by antidiarrheal therapy.26 The patient’s management may be very different depending on whether colonoscopy is done.
Management of microscopic colitis should include stopping any drugs associated with it. Simple antidiarrheal agents should be tried first to manage symptoms. If symptoms persist, patients can be treated with budesonide (Entocort EC) 9 mg by mouth daily for 8 weeks to induce remission, or 6 mg by mouth daily for 3 months as maintenance therapy.
OUR PATIENT’S COURSE
Our patient’s medication list includes duloxetine, a serotonin-norepinephrine reuptake inhibitor related to drugs that have been associated with the development of microscopic colitis. We tapered the duloxetine, and her symptoms improved by 50%. Her symptoms were eventually controlled after an 8-week course of oral budesonide 9 mg and ongoing intermittent use of loperamide (Imodium).
A 68-year-old white woman with irritable bowel syndrome has had worsening symptoms of right-sided abdominal pain, excessive bloating, and loose stools. Her bowel movements have increased from one a day to two or three a day. She has not noted any mucus or blood in the stool. She cannot identify any alleviating or aggravating factors, and the pain is not related to eating.
She consumes a normal diet, including meat and dairy. Over-the-counter antidiarrheal medications do not relieve the symptoms. She has had no fevers, chills, or night sweats, and she has not lost weight over the past year.
Her medical history includes breast cancer (in remission), alcohol abuse (in remission), and hypothyroidism, osteoporosis, and supraventricular tachycardia, all controlled with treatment as noted below. She has never undergone abdominal surgery.
A general review of systems is normal. Her current medications include oxybutynin (available as Ditropan, others), calcium polycarbophil (FiberCon, others), risedronate (Actonel), levothyroxine (Synthroid, others), simethicone (Maalox Anti-Gas, others), atenolol (Tenormin), trazodone (Desyrel), a calcium supplement, and aspirin. She began taking duloxetine (Cymbalta) 18 months ago, and the dose was increased from 60 mg to 90 mg 1 week before this visit.
She has never smoked, and she has abstained from alcohol for 10 years. She has no family history of colon cancer, celiac disease, or inflammatory bowel disease. She has not traveled outside the country in the past several years, and she notes no change in her source of drinking water.
On physical examination, she does not appear to be in acute distress. Her pulse is 64 and her blood pressure is 112/78 mm Hg. The cardiopulmonary examination is normal. Her abdomen is soft, symmetrical, nondistended, and nontender. Bowel sounds are normal. No abdominal masses, palpable organomegaly, or abdominal bruits are noted.
Results of basic laboratory tests, including thyroid-stimulating hormone (TSH), complete blood count, blood chemistries, renal function, and liver function, are normal. Colonoscopy shows normal mucosa as far as the cecum.
DIFFERENTIAL DIAGNOSIS
1. In addition to irritable bowel syndrome, which of these can explain her symptoms?
- Ulcerative colitis
- Celiac disease
- Microscopic colitis
- Hyperthyroidism
- Lactase deficiency
Ulcerative colitis typically presents with blood and mucus in the stool and gross abnormalities on colonoscopy, none of which is present in this patient.
Hyperthyroidism can be ruled out by the normal TSH level.
Lactase deficiency or lactose intolerance is unlikely because it is present in only 15% of people of northern European descent (compared with 80% of blacks and Hispanics and up to 100% of Native Americans and Asians).1 Furthermore, her pain is apparently not related to consuming dairy products.
The hydrogen breath test can aid in the diagnosis of lactase deficiency. This test relies on the breakdown of malabsorbed lactose by colonic flora. This is the most widely used test for this deficiency, but its high false-negative rate of 25% means that a negative result does not exclude the diagnosis and should not be relied on in working up a patient with chronic diarrhea.2 Simply noting whether symptoms develop after ingesting 50 g of lactose is clinically useful when lactase deficiency is suspected.
Based on the information so far, it is reasonable in this patient to evaluate for celiac disease and for microscopic colitis.
Celiac disease, also called gluten-sensitive enteropathy, has a varied presentation that includes nonspecific symptoms such as those in this patient. Classically, it causes diarrhea, but patients may present with a single nutrient deficiency and no diarrhea.
This patient lacks the elevated alkaline phosphatase or evidence of vitamin deficiencies characteristic of malabsorption in celiac disease (ie, vitamins A, B12, D, K, and folate)3. She also lacks evidence of malnutrition, such as iron deficiency anemia, weight loss, or low serum albumin. Finally, she does not have the dermatitis herpetiformis rash to suggest autoimmune gluten-sensitive enteropathy, nor does she have evidence of follicular hyperplasia or petechiae due to vitamin malabsorption.3
Because no single serologic test is ideal for diagnosing gluten-sensitive enteropathy, several tests are typically used: immunoglobulin A (IgA) antigliadin antibody, IgG antigliadin antibody, IgA antitransglutaminase antibody, and IgA antiendomysial antibody. IgA antitransglutaminase antibody is 92% to 98% sensitive and 91% to 100% specific for celiac disease. IgG antigliadin antibody is 92% to 97% sensitive and 99% specific. The positive predictive value of the IgA and IgG antigliadin antibody tests is less than 2% in the general population, whereas the positive predictive value for antiendomysial antibody and antitransglutaminase antibody are 15.7% and 21.8%, respectively.4 A positive serologic test for antiendomysial antibody is nearly 100% specific.
Our patient’s entire celiac antibody panel is negative, and thus celiac disease is unlikely.
Case continued: Features of microscopic colitis
In our patient, colonic biopsy reveals a mildly expanded lamina propria, intraepithelial lymphocytes, and a patchy but prominent thickening of the subepithelial collagen table. This set of features is consistent with collagenous colitis, a variant of microscopic colitis. Histologic signs on biopsy specimens are fairly specific for the disease.5
Chronic, intermittent, secretory diarrhea without bleeding is the hallmark of microscopic colitis. Associated symptoms may include abdominal pain, weight loss, and fatigue. If biopsies are not taken at the time of the initial evaluation, and the colonic pathology is overlooked, patients with collagenous colitis may be diagnosed with irritable bowel syndrome with diarrhea.6 The sedimentation rate is often elevated, and the antinuclear antibody test can be positive.7 Steatorrhea or protein-losing enteropathy can occur, and fecal leukocytes are present in more than 50% of patients.8
This patient fits well the demographics of the typical collagenous colitis patient: ie, a middle-aged woman in her 6th decade in otherwise good general health. The female-to-male ratio is 15:1 overall, although the relative frequency of collagenous colitis in women is greater than that of lymphocytic colitis.9 In a population-based study, the incidence of collagenous colitis was 5.1 per 100,000 per year, with a prevalence of 36 per 100,000; the incidence of lymphocytic colitis was 9.8 per 100,000 per year, with a prevalence of 64 per 100,000.10
Symptoms are typically vague and range from an annoyance to more than 20 non-bloody stools per day. The course of the disease also varies. Case series have reported a spontaneous remission rate of 15% to 20%,11 though flare-ups are common. Microscopic colitis is largely a benign disease. It does not increase a person’s risk of colon cancer.
CAUSES OF COLLAGENOUS COLITIS
2. What causes of collagenous colitis have been identified?
- Alcohol abuse
- Previous gastrointestinal surgery
- Drug-induced injury to colon
Neither alcohol use nor previous gastrointestinal surgery has been associated with the development of collagenous colitis.
Collagenous colitis has, however, been linked to several causes. Abnormal collagen metabolism has been demonstrated in patients as a result of increased expression of procollagen I and metalloproteinase inhibitor TIMP-1.12 Bacterial toxins and a bile-acid malabsorption defect in the terminal ileum and subsequent exposure of the colon to high concentrations of bile acids have also been linked to the development of collagenous colitis.
Many drugs have been linked to the development of collagenous colitis. Damage to the large intestine related to the use of non-steroidal anti-inflammatory drugs has been attributed to the blockage of prostaglandin synthesis.13 Simvastatin (Zocor), lansoprazole (Prilosec), and ticlopidine (Ticlid) have been linked to collagenous colitis; ticlopidine, flutamide (Eulexin), gold salts, lansoprazole, and sertraline (Zoloft) have been linked to the development of lymphocytic colitis.14 In one small series, patients developed colitis after switching from omeprazole (Prevacid) to lansoprazole. All patients had their symptoms and biopsy findings resolve within 1 week of stopping the drug.15
WHICH DRUG IS BEST?
3. Which drug is best for microscopic colitis, based on the current evidence?
- Bismuth (eg, Kaopectate, Pepto-Bismol)
- Sulfasalazine (Sulfazine)
- Budesonide (Entocort)
- Prednisolone
Studies have evaluated bismuth subsalicylate, Boswellia serrata extract, probiotics, prednisolone, budesonide, and other drugs for treating collagenous colitis.16
Bismuth trials have been small. In an open-label study of bismuth,17 symptoms improved in 11 of 12 patients.
Prednisolone recipients had a trend towards clinical response with treatment vs placebo, but it was not statistically significant, and there was incomplete remission of disease.18
Boswellia serrataextract19 and probiotics20 showed no clinical improvement.
Cholestyramine has been shown to be helpful when used in conjunction with an anti-inflammatory agent,21 and it may be helpful when used alone.
Aminosalicylate compounds have not been tested in prospective randomized trials, even though they are the cornerstone of treatment for ulcerative colitis. Retrospective trials have been equivocal.22
Budesonide currently has the best evidence of efficacy in collagenous colitis,23,24 and some evidence suggests it is also effective for other variants of microscopic colitis.
A total of 94 patients were enrolled in three placebo-controlled trials of budesonide at 9 mg daily or on a tapering schedule for 6 to 8 weeks. The pooled odds ratio for clinical response to treatment with budesonide was 12.32 (95% confidence interval 5.53–27.46), with a number needed to treat of 1.58. Significant histologic improvement with treatment was noted in all three trials.23
Quality of life has also been studied in patients with microscopic colitis who take budesonide. Symptoms, emotional functioning, and physical functioning are improved. Budesonide also improved stool consistency and significantly reduced the mean stool frequency compared with placebo.24
Compared with cortisol, budesonide has a 200 times greater affinity for the glucocorticoid receptor, and a 1,000 times greater topical anti-inflammatory potency. It is also well absorbed in the gastrointestinal tract but is substantially modified into very weak metabolites as a result of first-pass metabolism in the liver.25 This localized effect further supports the use of budesonide in patients with any form of microscopic colitis.
Although studies have shown budesonide to be effective, not every patient with a histologic diagnosis of microscopic colitis needs it. It is reasonable to try antidiarrheal agents, bismuth, or both as a first step because they are inexpensive and have few side effects. If budesonide is used, it should be given for 6 to 8 weeks, then stopped, and the patient should then be monitored for symptom recurrence. If a flare does occur, budesonide can be restarted and continued as maintenance therapy.
KEY CONSIDERATIONS
Microscopic colitis is diagnosed histologically, while irritable bowel syndrome is a clinical diagnosis. In population-based cohorts of histologically confirmed microscopic colitis, 50% to 70% met symptom-based Rome criteria for the diagnosis of irritable bowel syndrome. The clinical symptom-based criteria for irritable bowel syndrome are not specific enough to rule out the diagnosis of microscopic colitis. Therefore, patients with suspected diarrhea-predominant irritable bowel syndrome should undergo colonoscopy with biopsy to investigate microscopic colitis if symptoms are not well controlled by antidiarrheal therapy.26 The patient’s management may be very different depending on whether colonoscopy is done.
Management of microscopic colitis should include stopping any drugs associated with it. Simple antidiarrheal agents should be tried first to manage symptoms. If symptoms persist, patients can be treated with budesonide (Entocort EC) 9 mg by mouth daily for 8 weeks to induce remission, or 6 mg by mouth daily for 3 months as maintenance therapy.
OUR PATIENT’S COURSE
Our patient’s medication list includes duloxetine, a serotonin-norepinephrine reuptake inhibitor related to drugs that have been associated with the development of microscopic colitis. We tapered the duloxetine, and her symptoms improved by 50%. Her symptoms were eventually controlled after an 8-week course of oral budesonide 9 mg and ongoing intermittent use of loperamide (Imodium).
- Swagerty DL, Walling AD, Klein RM. Lactose intolerance. Am Fam Physician 2002; 65:1845–1856.
- Thomas PD, Forbes A, Green J, et al. Guidelines for the investigation of chronic diarrhea, 2nd edition. Gut 2003; 52(suppl 5):1–5.
- Nelsen DA. Gluten-sensitive enteropathy (celiac disease): more common than you think. Am Fam Physician 2002; 66:2259–2266.
- Bardella MT, Trovato C, Cesana BM, Pagliari C, Gebbia C, Peracchi M. Serological markers for coeliac disease: is it time to change? Dig Liver Dis 2001; 33:426–431.
- Barta Z, Mekkel G, Csipo I, et al. Micropscopic colitis: a retrospective study of clinical presentation in 53 patients. World J Gastroenterol 2005; 11:1351–1355.
- Tremaine WJ. Diagnosing collagenous colitis: does it make a difference? Eur J Gastroenterol Hepatol 1999; 11:477–479.
- Bohr J, Tysk C, Yang P, Danielsson D, Järnerot G. Autoantibodies and immunoglobulins in collagenous colitis. Gut 1996; 39:77–81.
- Zins BJ, Tremaine WJ, Carpenter HA. Collagenous colitis: mucosal biopsies and association with fecal leukocytes. Mayo Clin Proc 1995; 70:430–433.
- Olsen M, Eriksson S, Bohr J, Järnerot G, Tysk C. Lymphocytic colitis: a retrospective clinical study of 199 Swedish patients. Gut 2004; 53:536–541.
- Pardi DS. Microscopic colitis: an update. Inflamm Bowel Dis 2004; 10:860–870.
- Fernandez-Banares F, Salas A, Esteve M, Espinos J, Forne M, Viver JM. Collagenous and lymphocytic colitis: evaluation of clinical and histological features, response to treatment, and long-term follow-up. Am J Gastroenterol 2003; 98:340–347.
- Aignet T, Neureiter D, Müller S, Küspert G, Belke J, Kirchner T. Extracellular matrix composition and gene expression in collagenous colitis. Gastroenterology 1997; 113:136–143.
- Parfitt JR, Driman DK. Pathological effects of drugs on the gastrointestinal tract: a review. Hum Pathol 2007; 38:527–536.
- Fernández-Bañares F, Esteve M, Espinós JC, et al. Drug consumption and the risk of microscopic colitis. Am J Gastroenterol 2007; 102:324–330.
- Thomson RD, Lestine LS, Bensen SP, et al. Lansoprazole-associated microscopic colitis: a case series. Am J Gastroenterol 2002; 97:2908–2913.
- Chande N, McDonald JWD, MacDonald JK. Cochrane Inflammatory Bowel Disease and Functional Bowel Disorders Group. Interventions for treating collagenous colitis. Cochrane Database Syst Rev 2007 Jan 24;(1):CD006096.
- Fine K, Lee E. Efficacy of open-label bismuth subsalicylate for the treatment of microscopic colitis. Gastroenterology 1998; 114:29–36.
- Munck LK, Kjeldsen J, Philipsen E, Fscher Hansen B. Incomplete remission with short-term prednisolone treatment in collagenous colitis: a randomized study. Scand J Gastroenterol 2003; 38:606–610.
- Madisch A, Miehlke S, Eichele E, et al. Boswellia serrata extract for the treatment of collagenous colitis: a randomized, double-blind, placebo-controlled, multicenter trial. Int J Colorectal Dis 2007; 22:1445–1451.
- Wildt S, Munck LK, Vinter-Jensen L, et al. Probiotic treatment of collagenous colitis: a randomized, double-blind, placebo-controlled trial with Lactobacillus acidophilus and Bifidobacterium animalis subsp. lactis. Inflamm Bowel Dis 2006; 12:395–401.
- Calabrese C, Fabbri A, Areni A, Zahlane D, Scialpi C, Di Febo G. Mesalazine with or without cholestyramine in the treatment of microscopic colitis: randomized controlled trial. J Gastroenterol Hepatol 2007; 22:809–814.
- Wall GC, Schirmer LL, Page MJ. Pharmacotherapy for microscopic colitis. Pharmacotherapy 2007; 27:425–433.
- Feyen B, Wall GC, Finnerty EP, DeWitt JE, Reyes RS. Meta-analysis: budesonide treatment for collagenous colitis. Aliment Pharmacol Ther 2004; 20:745–749.
- Madisch A, Heymer P, Voss C, et al. Oral budesonide therapy improves quality of life in patients with collagenous colitis. Int J Colorectal Dis 2005; 20:312–316.
- Craig CR, editor. Modern Pharmacology With Clinical Application. 6th edition. Philadelphia: Lippincott Williams and Wilkins, 2003:481.
- Limsui D, Pardi DS, Camilleri M, et al. Symptomatic overlap between irritable bowel syndrome and microscopic colitis. Inflamm Bowel Dis 2007; 13:175–181.
- Swagerty DL, Walling AD, Klein RM. Lactose intolerance. Am Fam Physician 2002; 65:1845–1856.
- Thomas PD, Forbes A, Green J, et al. Guidelines for the investigation of chronic diarrhea, 2nd edition. Gut 2003; 52(suppl 5):1–5.
- Nelsen DA. Gluten-sensitive enteropathy (celiac disease): more common than you think. Am Fam Physician 2002; 66:2259–2266.
- Bardella MT, Trovato C, Cesana BM, Pagliari C, Gebbia C, Peracchi M. Serological markers for coeliac disease: is it time to change? Dig Liver Dis 2001; 33:426–431.
- Barta Z, Mekkel G, Csipo I, et al. Micropscopic colitis: a retrospective study of clinical presentation in 53 patients. World J Gastroenterol 2005; 11:1351–1355.
- Tremaine WJ. Diagnosing collagenous colitis: does it make a difference? Eur J Gastroenterol Hepatol 1999; 11:477–479.
- Bohr J, Tysk C, Yang P, Danielsson D, Järnerot G. Autoantibodies and immunoglobulins in collagenous colitis. Gut 1996; 39:77–81.
- Zins BJ, Tremaine WJ, Carpenter HA. Collagenous colitis: mucosal biopsies and association with fecal leukocytes. Mayo Clin Proc 1995; 70:430–433.
- Olsen M, Eriksson S, Bohr J, Järnerot G, Tysk C. Lymphocytic colitis: a retrospective clinical study of 199 Swedish patients. Gut 2004; 53:536–541.
- Pardi DS. Microscopic colitis: an update. Inflamm Bowel Dis 2004; 10:860–870.
- Fernandez-Banares F, Salas A, Esteve M, Espinos J, Forne M, Viver JM. Collagenous and lymphocytic colitis: evaluation of clinical and histological features, response to treatment, and long-term follow-up. Am J Gastroenterol 2003; 98:340–347.
- Aignet T, Neureiter D, Müller S, Küspert G, Belke J, Kirchner T. Extracellular matrix composition and gene expression in collagenous colitis. Gastroenterology 1997; 113:136–143.
- Parfitt JR, Driman DK. Pathological effects of drugs on the gastrointestinal tract: a review. Hum Pathol 2007; 38:527–536.
- Fernández-Bañares F, Esteve M, Espinós JC, et al. Drug consumption and the risk of microscopic colitis. Am J Gastroenterol 2007; 102:324–330.
- Thomson RD, Lestine LS, Bensen SP, et al. Lansoprazole-associated microscopic colitis: a case series. Am J Gastroenterol 2002; 97:2908–2913.
- Chande N, McDonald JWD, MacDonald JK. Cochrane Inflammatory Bowel Disease and Functional Bowel Disorders Group. Interventions for treating collagenous colitis. Cochrane Database Syst Rev 2007 Jan 24;(1):CD006096.
- Fine K, Lee E. Efficacy of open-label bismuth subsalicylate for the treatment of microscopic colitis. Gastroenterology 1998; 114:29–36.
- Munck LK, Kjeldsen J, Philipsen E, Fscher Hansen B. Incomplete remission with short-term prednisolone treatment in collagenous colitis: a randomized study. Scand J Gastroenterol 2003; 38:606–610.
- Madisch A, Miehlke S, Eichele E, et al. Boswellia serrata extract for the treatment of collagenous colitis: a randomized, double-blind, placebo-controlled, multicenter trial. Int J Colorectal Dis 2007; 22:1445–1451.
- Wildt S, Munck LK, Vinter-Jensen L, et al. Probiotic treatment of collagenous colitis: a randomized, double-blind, placebo-controlled trial with Lactobacillus acidophilus and Bifidobacterium animalis subsp. lactis. Inflamm Bowel Dis 2006; 12:395–401.
- Calabrese C, Fabbri A, Areni A, Zahlane D, Scialpi C, Di Febo G. Mesalazine with or without cholestyramine in the treatment of microscopic colitis: randomized controlled trial. J Gastroenterol Hepatol 2007; 22:809–814.
- Wall GC, Schirmer LL, Page MJ. Pharmacotherapy for microscopic colitis. Pharmacotherapy 2007; 27:425–433.
- Feyen B, Wall GC, Finnerty EP, DeWitt JE, Reyes RS. Meta-analysis: budesonide treatment for collagenous colitis. Aliment Pharmacol Ther 2004; 20:745–749.
- Madisch A, Heymer P, Voss C, et al. Oral budesonide therapy improves quality of life in patients with collagenous colitis. Int J Colorectal Dis 2005; 20:312–316.
- Craig CR, editor. Modern Pharmacology With Clinical Application. 6th edition. Philadelphia: Lippincott Williams and Wilkins, 2003:481.
- Limsui D, Pardi DS, Camilleri M, et al. Symptomatic overlap between irritable bowel syndrome and microscopic colitis. Inflamm Bowel Dis 2007; 13:175–181.