User login
Dyspnea, arthralgias, and muscle weakness
Q: Which condition is most likely?
- Rheumatoid arthritis with pulmonary involvement
- Hypertrophic pulmonary osteoarthropathy
- Polymyositis-dermatomyositis with pulmonary involvement
- Systemic lupus erythematosus with pulmonary involvement
A: The patient’s symptoms and physical findings suggest polymyositis-dermatomyositis with associated interstitial lung disease.
Rheumatoid arthritis can also cause lung disease and proximal myopathy, but early physical findings in the hands would include symmetrical joint effusions and soft tissue swelling of the metacarpophalangeal joints.
Patients with hypertrophic pulmonary osteoarthropathy present with arthralgias without weakness. Radiographic findings such as osteophytosis and tufting of terminal processes in the hands would support its diagnosis.
A small number of patients with systemic lupus erythematosus develop deforming arthritis with hand involvement that is either erosive (rhupus hand) or nonerosive (Jaccoud arthropathy, or lupus hand), but interstitial lung disease is rare in lupus, making this combination unlikely.
MULTIPLE PATHS TO DIAGNOSIS
Physical examination, review of systems, laboratory screening, radiographic findings, lung biopsy, electromyography, and muscle biopsy may be used in conjunction.
The criteria of Bohan and Peter are often used to diagnose polymyositis-dermatomyositis: symmetric proximal muscle weakness, elevated muscle enzymes, electromyographic changes consistent with myopathy, and compatible histologic findings on muscle biopsy, with or without the characteristic dermatologic manifestations.1,2 However, the diagnosis can be made in the typical clinical setting on the basis of characteristic levels of anti-Jo-1 antinuclear antibody and elevated serum muscle enzyme.
Depending on the criteria used, the incidence of interstitial lung disease in various studies of polymyositis-dermatomyositis ranged from 5% to 46%.3 Pulmonary involvement can present in one of three forms:
- Sudden onset of dyspnea and fever with alveolar infiltrates on chest radiography and ground-glass opacities on high-resolution chest CT
- Progressive dyspnea with radiographic findings of chronic interstitial lung disease
- No clinical symptoms, but with abnormal findings on chest radiography.4
In a minority of patients, lung disease precedes the onset of muscle or skin disease. Much more commonly, patients present with skin and muscle involvement first. In these patients, pulmonary involvement is typically seen 2 to 5 years after the diagnosis.2 Patients with interstitial lung disease are more likely to have arthralgias and arthritis than are those without lung involvement. Interestingly, the finding of microangiopathy on nail fold capillaroscopy strongly suggests pulmonary disease.3
Laboratory findings
Creatine kinase elevation is a marker of disease activity in the muscles. Aldolase, aspartate aminotransferase, and alanine aminotransferase levels may also be elevated but are not muscle-specific. Anti-Jo-1 antinuclear antibody is characteristic, although it can be negative in some patients.
Pulmonary function testing
Restrictive lung physiology with impaired diffusing capacity is the predominant pattern noted.
Lung biopsy findings
Polymyositis-dermatomyositis-associated interstitial lung disease is not limited to one histologic pattern. Nonspecific interstitial pneumonitis is the most common, but usual interstitial pneumonia, organizing pneumonia, and diffuse alveolar damage are also described.2 Patients with nonspecific interstitial pneumonitis and organizing pneumonia are suspected to have a better response to immunosuppression and better survival, although controlled studies are absent.
CT appearance
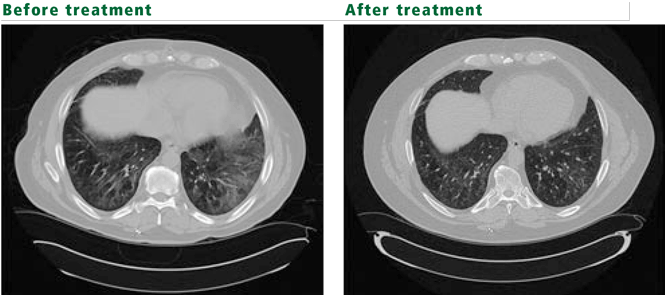
Polymyositis-dermatomyositis complicated by interstitial lung disease does not have a distinct appearance on high-resolution CT. However, the radiographic changes in most cases will suggest the underlying pathology, and this can be used to guide therapy. Most common are bibasilar subpleural ground-glass and reticular opacities that curiously spare the immediate 1 to 2 mm of subpleural parenchyma.1,3 This pattern is very suggestive of fibrotic nonspecific interstitial pneumonitis. Patchy consolidations with air bronchograms suggest organizing pneumonia. Bibasilar, subpleural honeycomb cystic changes and traction bronchiectasis are noted in usual interstitial pneumonia and suggest fibrosis, which will not improve with therapy. In patients whose disease is progressive, the areas of consolidation often evolve into honeycomb cystic changes.2,4
TREATMENT IS WITH STEROIDS AND OTHER IMMUNOSUPPRESSIVES
Oral corticosteroids in dosages of 0.5 to 1 mg/kg are first-line therapy. Clinically, muscle disease often improves before lung disease, and treatment may be extended to several months. The histologic pattern suggested by CT or by pathologic study of surgical lung biopsy specimens is a better predictor of treatment response than clinical presentation. Nonspecific interstitial pneumonitis and organizing pneumonia have the highest steroid response rates.3 However, many patients do not respond to steroids alone—only 44% in one study.5 Furthermore, treatment response does not indicate recovery, as the disease may relapse.
The addition of immunosuppressive therapy with cyclophosphamide (Cytoxan) may halt deterioration in patients with polymyositis-dermatomyositis-associated interstitial lung disease who are steroid-resistant or may be useful as a steroid-sparing agent in recurrent disease after initial steroid withdrawal. In some cases, therapy with cyclophosphamide improved oxygenation and led to resolution of abnormalities on chest radiography.6,7
Azathioprine (Imuran), methotrexate, and hydroxychloroquine (Plaquenil) have all been used as part of a steroid-sparing regimen.1 Tacrolimus (FK 506; Prograf) and rituximab (Rituxan) are emerging therapies, especially for patients who cannot tolerate cytotoxic immunosuppressive agents or who progress despite them.8,9
PROGNOSIS IS WORSE IF LUNG DISEASE IS PRESENT
The presence of interstitial lung disease increases the risk of death in polymyositis-dermatomyositis. Additionally, clinicians must assess interstitial lung disease separately from muscle or skin disease, as there does not have to be correlation between the activity in the separate organs. Fortunately, the treatment for lung, muscle, and skin involvement is often the same.
Several elements suggest poor prognosis. An acute and aggressive presentation often heralds a poor outcome.1 Neutrophil alveolitis on bronchoalveolar lavage and a very low diffusing capacity (< 45%) have both been associated with a poorer prognosis.3 The histologic pattern not only predicts treatment response but also prognosis. Patients whose lung biopsies reveal nonspecific interstitial pneumonitis or organizing pneumonia have a better outcome than do patients with usual interstitial pneumonia or diffuse alveolar damage.
In one study,1 36 patients with polymyositis-dermatomyositis and interstitial lung disease were followed for 5 years. Resolution was noted in 19.4%, improvement in 55.6%, and deterioration in 25%. Overall, the survival rate was 86.5% at 5 years, and the death rate attributable to pulmonary complications was 13.9% in patients with interstitial lung disease.1
- Douglas WW, Tazelaar HD, Hartman TE, et al. Polymyositis-dermatomyositis-associated interstitial lung disease. Am J Respir Crit Care Med 2001; 164:1182–1185.
- Marie I, Hatron PY, Hachulla E, Wallaert B, Michon-Pasturel U, Devulder B. Pulmonary involvement in polymyositis and in dermatomyositis. J Rheumatol 1998; 25:1336–1343.
- Marie I, Hachulla E, Cherin P, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum 2002; 47:614–622.
- Akira M, Hara H, Sakatani M. Interstitial lung disease in association with polymyositis-dermatomyositis: long-term follow-up CT evaluation in seven patients. Radiology 1999; 210:333–338.
- Nawata Y, Kurasawa K, Takabayashi K, et al. Corticosteroid resistant interstitial pneumonitis in dermatomyositis/polymyositis: prediction and treatment with cyclosporine. J Rheumatol 1999; 26:1527–1533.
- Schnabel A, Reuter M, Gross WL. Intravenous pulse cyclophosphamide in the treatment of interstitial lung disease due to collagen vascular disease. Arthritis Rheum 1998; 41:1215–1220.
- Shinohara T, Hidaka T, Matsuki Y, et al. Rapidly progressive interstitial lung disease associated with dermatomyositis responding to intravenous cyclophosphamide pulse therapy. Intern Med 1997; 36:519–523.
- Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum 2005; 52:2439–2446.
- Ytterberg SR. Treatment of refractory polymyositis and dermatomyositis. Curr Rheumatol Rep 2006; 8:167–173.
Q: Which condition is most likely?
- Rheumatoid arthritis with pulmonary involvement
- Hypertrophic pulmonary osteoarthropathy
- Polymyositis-dermatomyositis with pulmonary involvement
- Systemic lupus erythematosus with pulmonary involvement
A: The patient’s symptoms and physical findings suggest polymyositis-dermatomyositis with associated interstitial lung disease.
Rheumatoid arthritis can also cause lung disease and proximal myopathy, but early physical findings in the hands would include symmetrical joint effusions and soft tissue swelling of the metacarpophalangeal joints.
Patients with hypertrophic pulmonary osteoarthropathy present with arthralgias without weakness. Radiographic findings such as osteophytosis and tufting of terminal processes in the hands would support its diagnosis.
A small number of patients with systemic lupus erythematosus develop deforming arthritis with hand involvement that is either erosive (rhupus hand) or nonerosive (Jaccoud arthropathy, or lupus hand), but interstitial lung disease is rare in lupus, making this combination unlikely.
MULTIPLE PATHS TO DIAGNOSIS
Physical examination, review of systems, laboratory screening, radiographic findings, lung biopsy, electromyography, and muscle biopsy may be used in conjunction.
The criteria of Bohan and Peter are often used to diagnose polymyositis-dermatomyositis: symmetric proximal muscle weakness, elevated muscle enzymes, electromyographic changes consistent with myopathy, and compatible histologic findings on muscle biopsy, with or without the characteristic dermatologic manifestations.1,2 However, the diagnosis can be made in the typical clinical setting on the basis of characteristic levels of anti-Jo-1 antinuclear antibody and elevated serum muscle enzyme.
Depending on the criteria used, the incidence of interstitial lung disease in various studies of polymyositis-dermatomyositis ranged from 5% to 46%.3 Pulmonary involvement can present in one of three forms:
- Sudden onset of dyspnea and fever with alveolar infiltrates on chest radiography and ground-glass opacities on high-resolution chest CT
- Progressive dyspnea with radiographic findings of chronic interstitial lung disease
- No clinical symptoms, but with abnormal findings on chest radiography.4
In a minority of patients, lung disease precedes the onset of muscle or skin disease. Much more commonly, patients present with skin and muscle involvement first. In these patients, pulmonary involvement is typically seen 2 to 5 years after the diagnosis.2 Patients with interstitial lung disease are more likely to have arthralgias and arthritis than are those without lung involvement. Interestingly, the finding of microangiopathy on nail fold capillaroscopy strongly suggests pulmonary disease.3
Laboratory findings
Creatine kinase elevation is a marker of disease activity in the muscles. Aldolase, aspartate aminotransferase, and alanine aminotransferase levels may also be elevated but are not muscle-specific. Anti-Jo-1 antinuclear antibody is characteristic, although it can be negative in some patients.
Pulmonary function testing
Restrictive lung physiology with impaired diffusing capacity is the predominant pattern noted.
Lung biopsy findings
Polymyositis-dermatomyositis-associated interstitial lung disease is not limited to one histologic pattern. Nonspecific interstitial pneumonitis is the most common, but usual interstitial pneumonia, organizing pneumonia, and diffuse alveolar damage are also described.2 Patients with nonspecific interstitial pneumonitis and organizing pneumonia are suspected to have a better response to immunosuppression and better survival, although controlled studies are absent.
CT appearance
Polymyositis-dermatomyositis complicated by interstitial lung disease does not have a distinct appearance on high-resolution CT. However, the radiographic changes in most cases will suggest the underlying pathology, and this can be used to guide therapy. Most common are bibasilar subpleural ground-glass and reticular opacities that curiously spare the immediate 1 to 2 mm of subpleural parenchyma.1,3 This pattern is very suggestive of fibrotic nonspecific interstitial pneumonitis. Patchy consolidations with air bronchograms suggest organizing pneumonia. Bibasilar, subpleural honeycomb cystic changes and traction bronchiectasis are noted in usual interstitial pneumonia and suggest fibrosis, which will not improve with therapy. In patients whose disease is progressive, the areas of consolidation often evolve into honeycomb cystic changes.2,4
TREATMENT IS WITH STEROIDS AND OTHER IMMUNOSUPPRESSIVES
Oral corticosteroids in dosages of 0.5 to 1 mg/kg are first-line therapy. Clinically, muscle disease often improves before lung disease, and treatment may be extended to several months. The histologic pattern suggested by CT or by pathologic study of surgical lung biopsy specimens is a better predictor of treatment response than clinical presentation. Nonspecific interstitial pneumonitis and organizing pneumonia have the highest steroid response rates.3 However, many patients do not respond to steroids alone—only 44% in one study.5 Furthermore, treatment response does not indicate recovery, as the disease may relapse.
The addition of immunosuppressive therapy with cyclophosphamide (Cytoxan) may halt deterioration in patients with polymyositis-dermatomyositis-associated interstitial lung disease who are steroid-resistant or may be useful as a steroid-sparing agent in recurrent disease after initial steroid withdrawal. In some cases, therapy with cyclophosphamide improved oxygenation and led to resolution of abnormalities on chest radiography.6,7
Azathioprine (Imuran), methotrexate, and hydroxychloroquine (Plaquenil) have all been used as part of a steroid-sparing regimen.1 Tacrolimus (FK 506; Prograf) and rituximab (Rituxan) are emerging therapies, especially for patients who cannot tolerate cytotoxic immunosuppressive agents or who progress despite them.8,9
PROGNOSIS IS WORSE IF LUNG DISEASE IS PRESENT
The presence of interstitial lung disease increases the risk of death in polymyositis-dermatomyositis. Additionally, clinicians must assess interstitial lung disease separately from muscle or skin disease, as there does not have to be correlation between the activity in the separate organs. Fortunately, the treatment for lung, muscle, and skin involvement is often the same.
Several elements suggest poor prognosis. An acute and aggressive presentation often heralds a poor outcome.1 Neutrophil alveolitis on bronchoalveolar lavage and a very low diffusing capacity (< 45%) have both been associated with a poorer prognosis.3 The histologic pattern not only predicts treatment response but also prognosis. Patients whose lung biopsies reveal nonspecific interstitial pneumonitis or organizing pneumonia have a better outcome than do patients with usual interstitial pneumonia or diffuse alveolar damage.
In one study,1 36 patients with polymyositis-dermatomyositis and interstitial lung disease were followed for 5 years. Resolution was noted in 19.4%, improvement in 55.6%, and deterioration in 25%. Overall, the survival rate was 86.5% at 5 years, and the death rate attributable to pulmonary complications was 13.9% in patients with interstitial lung disease.1
Q: Which condition is most likely?
- Rheumatoid arthritis with pulmonary involvement
- Hypertrophic pulmonary osteoarthropathy
- Polymyositis-dermatomyositis with pulmonary involvement
- Systemic lupus erythematosus with pulmonary involvement
A: The patient’s symptoms and physical findings suggest polymyositis-dermatomyositis with associated interstitial lung disease.
Rheumatoid arthritis can also cause lung disease and proximal myopathy, but early physical findings in the hands would include symmetrical joint effusions and soft tissue swelling of the metacarpophalangeal joints.
Patients with hypertrophic pulmonary osteoarthropathy present with arthralgias without weakness. Radiographic findings such as osteophytosis and tufting of terminal processes in the hands would support its diagnosis.
A small number of patients with systemic lupus erythematosus develop deforming arthritis with hand involvement that is either erosive (rhupus hand) or nonerosive (Jaccoud arthropathy, or lupus hand), but interstitial lung disease is rare in lupus, making this combination unlikely.
MULTIPLE PATHS TO DIAGNOSIS
Physical examination, review of systems, laboratory screening, radiographic findings, lung biopsy, electromyography, and muscle biopsy may be used in conjunction.
The criteria of Bohan and Peter are often used to diagnose polymyositis-dermatomyositis: symmetric proximal muscle weakness, elevated muscle enzymes, electromyographic changes consistent with myopathy, and compatible histologic findings on muscle biopsy, with or without the characteristic dermatologic manifestations.1,2 However, the diagnosis can be made in the typical clinical setting on the basis of characteristic levels of anti-Jo-1 antinuclear antibody and elevated serum muscle enzyme.
Depending on the criteria used, the incidence of interstitial lung disease in various studies of polymyositis-dermatomyositis ranged from 5% to 46%.3 Pulmonary involvement can present in one of three forms:
- Sudden onset of dyspnea and fever with alveolar infiltrates on chest radiography and ground-glass opacities on high-resolution chest CT
- Progressive dyspnea with radiographic findings of chronic interstitial lung disease
- No clinical symptoms, but with abnormal findings on chest radiography.4
In a minority of patients, lung disease precedes the onset of muscle or skin disease. Much more commonly, patients present with skin and muscle involvement first. In these patients, pulmonary involvement is typically seen 2 to 5 years after the diagnosis.2 Patients with interstitial lung disease are more likely to have arthralgias and arthritis than are those without lung involvement. Interestingly, the finding of microangiopathy on nail fold capillaroscopy strongly suggests pulmonary disease.3
Laboratory findings
Creatine kinase elevation is a marker of disease activity in the muscles. Aldolase, aspartate aminotransferase, and alanine aminotransferase levels may also be elevated but are not muscle-specific. Anti-Jo-1 antinuclear antibody is characteristic, although it can be negative in some patients.
Pulmonary function testing
Restrictive lung physiology with impaired diffusing capacity is the predominant pattern noted.
Lung biopsy findings
Polymyositis-dermatomyositis-associated interstitial lung disease is not limited to one histologic pattern. Nonspecific interstitial pneumonitis is the most common, but usual interstitial pneumonia, organizing pneumonia, and diffuse alveolar damage are also described.2 Patients with nonspecific interstitial pneumonitis and organizing pneumonia are suspected to have a better response to immunosuppression and better survival, although controlled studies are absent.
CT appearance
Polymyositis-dermatomyositis complicated by interstitial lung disease does not have a distinct appearance on high-resolution CT. However, the radiographic changes in most cases will suggest the underlying pathology, and this can be used to guide therapy. Most common are bibasilar subpleural ground-glass and reticular opacities that curiously spare the immediate 1 to 2 mm of subpleural parenchyma.1,3 This pattern is very suggestive of fibrotic nonspecific interstitial pneumonitis. Patchy consolidations with air bronchograms suggest organizing pneumonia. Bibasilar, subpleural honeycomb cystic changes and traction bronchiectasis are noted in usual interstitial pneumonia and suggest fibrosis, which will not improve with therapy. In patients whose disease is progressive, the areas of consolidation often evolve into honeycomb cystic changes.2,4
TREATMENT IS WITH STEROIDS AND OTHER IMMUNOSUPPRESSIVES
Oral corticosteroids in dosages of 0.5 to 1 mg/kg are first-line therapy. Clinically, muscle disease often improves before lung disease, and treatment may be extended to several months. The histologic pattern suggested by CT or by pathologic study of surgical lung biopsy specimens is a better predictor of treatment response than clinical presentation. Nonspecific interstitial pneumonitis and organizing pneumonia have the highest steroid response rates.3 However, many patients do not respond to steroids alone—only 44% in one study.5 Furthermore, treatment response does not indicate recovery, as the disease may relapse.
The addition of immunosuppressive therapy with cyclophosphamide (Cytoxan) may halt deterioration in patients with polymyositis-dermatomyositis-associated interstitial lung disease who are steroid-resistant or may be useful as a steroid-sparing agent in recurrent disease after initial steroid withdrawal. In some cases, therapy with cyclophosphamide improved oxygenation and led to resolution of abnormalities on chest radiography.6,7
Azathioprine (Imuran), methotrexate, and hydroxychloroquine (Plaquenil) have all been used as part of a steroid-sparing regimen.1 Tacrolimus (FK 506; Prograf) and rituximab (Rituxan) are emerging therapies, especially for patients who cannot tolerate cytotoxic immunosuppressive agents or who progress despite them.8,9
PROGNOSIS IS WORSE IF LUNG DISEASE IS PRESENT
The presence of interstitial lung disease increases the risk of death in polymyositis-dermatomyositis. Additionally, clinicians must assess interstitial lung disease separately from muscle or skin disease, as there does not have to be correlation between the activity in the separate organs. Fortunately, the treatment for lung, muscle, and skin involvement is often the same.
Several elements suggest poor prognosis. An acute and aggressive presentation often heralds a poor outcome.1 Neutrophil alveolitis on bronchoalveolar lavage and a very low diffusing capacity (< 45%) have both been associated with a poorer prognosis.3 The histologic pattern not only predicts treatment response but also prognosis. Patients whose lung biopsies reveal nonspecific interstitial pneumonitis or organizing pneumonia have a better outcome than do patients with usual interstitial pneumonia or diffuse alveolar damage.
In one study,1 36 patients with polymyositis-dermatomyositis and interstitial lung disease were followed for 5 years. Resolution was noted in 19.4%, improvement in 55.6%, and deterioration in 25%. Overall, the survival rate was 86.5% at 5 years, and the death rate attributable to pulmonary complications was 13.9% in patients with interstitial lung disease.1
- Douglas WW, Tazelaar HD, Hartman TE, et al. Polymyositis-dermatomyositis-associated interstitial lung disease. Am J Respir Crit Care Med 2001; 164:1182–1185.
- Marie I, Hatron PY, Hachulla E, Wallaert B, Michon-Pasturel U, Devulder B. Pulmonary involvement in polymyositis and in dermatomyositis. J Rheumatol 1998; 25:1336–1343.
- Marie I, Hachulla E, Cherin P, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum 2002; 47:614–622.
- Akira M, Hara H, Sakatani M. Interstitial lung disease in association with polymyositis-dermatomyositis: long-term follow-up CT evaluation in seven patients. Radiology 1999; 210:333–338.
- Nawata Y, Kurasawa K, Takabayashi K, et al. Corticosteroid resistant interstitial pneumonitis in dermatomyositis/polymyositis: prediction and treatment with cyclosporine. J Rheumatol 1999; 26:1527–1533.
- Schnabel A, Reuter M, Gross WL. Intravenous pulse cyclophosphamide in the treatment of interstitial lung disease due to collagen vascular disease. Arthritis Rheum 1998; 41:1215–1220.
- Shinohara T, Hidaka T, Matsuki Y, et al. Rapidly progressive interstitial lung disease associated with dermatomyositis responding to intravenous cyclophosphamide pulse therapy. Intern Med 1997; 36:519–523.
- Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum 2005; 52:2439–2446.
- Ytterberg SR. Treatment of refractory polymyositis and dermatomyositis. Curr Rheumatol Rep 2006; 8:167–173.
- Douglas WW, Tazelaar HD, Hartman TE, et al. Polymyositis-dermatomyositis-associated interstitial lung disease. Am J Respir Crit Care Med 2001; 164:1182–1185.
- Marie I, Hatron PY, Hachulla E, Wallaert B, Michon-Pasturel U, Devulder B. Pulmonary involvement in polymyositis and in dermatomyositis. J Rheumatol 1998; 25:1336–1343.
- Marie I, Hachulla E, Cherin P, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum 2002; 47:614–622.
- Akira M, Hara H, Sakatani M. Interstitial lung disease in association with polymyositis-dermatomyositis: long-term follow-up CT evaluation in seven patients. Radiology 1999; 210:333–338.
- Nawata Y, Kurasawa K, Takabayashi K, et al. Corticosteroid resistant interstitial pneumonitis in dermatomyositis/polymyositis: prediction and treatment with cyclosporine. J Rheumatol 1999; 26:1527–1533.
- Schnabel A, Reuter M, Gross WL. Intravenous pulse cyclophosphamide in the treatment of interstitial lung disease due to collagen vascular disease. Arthritis Rheum 1998; 41:1215–1220.
- Shinohara T, Hidaka T, Matsuki Y, et al. Rapidly progressive interstitial lung disease associated with dermatomyositis responding to intravenous cyclophosphamide pulse therapy. Intern Med 1997; 36:519–523.
- Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum 2005; 52:2439–2446.
- Ytterberg SR. Treatment of refractory polymyositis and dermatomyositis. Curr Rheumatol Rep 2006; 8:167–173.
Perioperative beta-blockers in noncardiac surgery: Evolution of the evidence
The pendulum of expert opinion is swinging away from routinely recommending beta-blockers to prevent cardiac events in non-cardiac surgery patients. We won’t be abandoning the perioperative use of beta-blockers altogether, but we will probably be using them more selectively than in the past.
The latest factor driving the trend is the online publication in May 2008 of the results of the Perioperative Ischemic Evaluation (POISE) trial,1 the largest placebo-controlled trial of perioperative beta-blocker use to date. In brief, in a cohort of patients with atherosclerotic disease or at risk for it who were undergoing noncardiac surgery, fewer patients who received extended-release metoprolol succinate had a myocardial infarction, but more of them died or had a stroke compared with those receiving placebo. (Extended-release metoprolol succinate is available in the United States as Toprol-XL and generically.)
Not so long ago, the pendulum was going the other way. After two small trials in the 1990s concluded that beta-blockers reduced the risk of perioperative cardiac events in selected patients with known or suspected coronary disease,2,3 their perioperative use was subsequently endorsed by the Leapfrog Group and the Agency for Healthcare Research and Quality. The National Quality Forum included perioperative beta-blockade in its “Safe Practices for Better Healthcare 2006 update,”4,5 and the Physician Consortium for Performance Improvement and the Surgical Care Improvement Project both listed it as a quality measure.
Since then, this practice has been closely studied, especially as concomitant research has failed to demonstrate that pre-operative coronary revascularization improves outcomes, even in the presence of ischemic disease. But evidence has been accumulating that routine use of beta-blockers may not benefit as many patients as was hoped, and may actually cause harm. The 2007 joint American College of Cardiology (ACC) and American Heart Association (AHA) guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery gives its strongest recommendation (class I: benefit clearly outweighs risk) for perioperative beta-blocker use only for patients at high risk: those with known ischemic heart disease undergoing vascular surgery and those who are already on these drugs before surgery.6
However, there are still gaps in our knowledge. Perhaps, with proper implementation, we may be able to use beta-blockers to improve outcomes in patients at intermediate risk as well. In this paper, we review the rationale and the evidence for and against perioperative use of beta-blockers and provide practical guidance for internists and hospitalists.
WHY CARDIAC EVENTS OCCUR AFTER SURGERY
Adverse cardiovascular events such as myocardial infarction and unstable angina are the leading causes of death after surgery.7 Such events occur in approximately 1% of patients older than 50 years undergoing elective inpatient surgery, but this number may be higher (approximately 5%) in those with known or suspected coronary disease.8,9 Perioperative cardiac events can also be harbingers of further complications, dramatically increasing hospital length of stay.10
Some ischemic events are caused by physiologic derangements involving the balance between inflammatory mediators, sympathetic tone, and oxygen supply and demand that occur under the stress of surgery. Others are more “traditional” in etiology, involving acute plaque rupture, thrombosis, and occlusion. Studies have consistently found a correlation between perioperative ischemia and cardiac events (both in-hospital and long-term) and death.11–17 Other studies suggest that most perioperative cardiac infarcts are non-Q-wave events,18 and most events occur within the first few days after surgery, particularly the first 48 hours, when the effects of anesthetics, pain, fluid shifts, and physiologic derangements are greatest.
Factors that may trigger acute occlusion in the perioperative period include abrupt changes in sympathetic tone, increased levels of cortisol and catecholamines, and tissue hypoxia. Other potential triggers activated or increased by the stress of surgery include coagulation factors such as alterations in platelet function; inflammatory factors such as tumor necrosis factor alpha, interleukin 1, interleukin 6, and C-reactive protein; and metabolism of free fatty acids (which contribute to increased oxygen demand as well as endothelial dysfunction).9,19,20
A 1996 autopsy study found that 38 (90%) of 42 patients who died of a perioperative infarct had evidence of acute plaque rupture or plaque hemorrhage on coronary sectioning, findings corroborated in another, similar study.21,22 These studies suggest that multiple causes contribute to perioperative myocardial infarction, and a single strategy may not suffice for prevention.
IF BETA-BLOCKERS PROTECT, HOW DO THEY DO IT?
Beta-blockers have several effects that should, in theory, protect against cardiac events during and after surgery.23 They reduce cardiac oxygen demand by reducing the force of contraction and the heart rate, and they increase the duration of diastole, when the heart muscle is perfused. They are also antiarrhythmic, and they may limit free radical production, metalloproteinase activity, and myocardial plaque inflammation.24
Some researchers have speculated that using beta-blockers long-term may alter intra-cellular signaling processes, for example decreasing the expression of receptors that receive signals for cell death, which in turn may affect the response to reperfusion cell injury and death. If this is true, there may be an advantage to starting beta-blockers well in advance of surgery.25
EARLY CLINICAL EVIDENCE IN FAVOR OF PERIOPERATIVE BETA-BLOCKER USE
Evidence in patients at high risk
Mangano et al,2 in a study published in 1996, randomized 200 patients with known coronary disease or established risk factors for it who were undergoing noncardiac surgery to receive the beta-blocker atenolol orally and intravenously or placebo in the immediate perioperative period. Fewer patients in the atenolol group died in the first 6 months after hospital discharge (0 vs 8%, P < .001), the first year (3% vs 14%, P = .005), and the first 2 years (10% vs 21%, P = .019). However, there was no difference in short-term outcomes, and the study excluded patients who died in the immediate postoperative period. If these patients had been included in the analysis, the difference in the death rate at 2 years would not have been statistically significant.26 Other critical findings: more patients in the atenolol group were using angiotensin-converting enzyme inhibitors and beta-blockers when they were discharged, and the placebo group had slightly more patients with prior myocardial infarction or diabetes.27 (Atenolol is available in the United Sates as Tenormin and generically.)
Poldermans et al,3 in a study published in 1999, randomized 112 vascular surgery patients to receive either oral bisoprolol or placebo. These patients were selected from a larger cohort of 1,351 patients on the basis of high-risk clinical features and abnormal results on dobutamine echocardiography. Bisoprolol was started at least 1 week before surgery (range 7–89 days, mean 37 days), and patients were reevaluated before surgery so that the dose could be titrated to a goal heart rate of less than 60 beats per minute. After surgery, the drug was continued for another 30 days. The study was stopped early because the bisoprolol group had a 90% lower rate of non-fatal myocardial infarction and cardiac death at 30 days. Despite the study’s limitation (eg, enrolling selected patients and using an unblinded protocol), these compelling findings made a strong case for the use of beta-blockers perioperatively in patients at high risk, ie, those with ischemic heart disease who are undergoing major vascular surgery. (Bisoprolol is available in the United States as Zebeta and generically)
Evidence in patients at intermediate risk
Boersma et al28 performed a follow-up to the study by Poldermans et al, published in 2001, in which they analyzed characteristics of all 1,351 patients who had been originally considered for enrollment. Using regression analysis, they identified seven clinical risk factors that predicted adverse cardiac events: angina, prior myocardial infarction, congestive heart failure, prior stroke, diabetes, renal failure, and age 70 years or older. Furthermore, for the entire cohort, patients receiving beta-blockers had a lower risk of cardiac complications (0.8%) than those not receiving beta-blockers (2.3%). In particular, the patients at intermediate risk (defined as having one or two risk factors) had a very low event rate regardless of stress test results, provided they were on beta-blockers: their risk of death or myocardial infarction was 0.9%, compared with 3.0% for those not on beta-blockers.
The authors concluded that dobutamine stress testing may not be necessary in patients at intermediate risk if beta-blockers are appropriately prescribed. However, others took issue with their data and conclusions, arguing that there have been so few trials that the data are still inconclusive and inadequate to ascertain the benefit of perioperative beta-blockade, particularly in patients not at high risk.26,29
The Revised Cardiac Risk Index. Although the Boersma risk-factor index is not used in general practice, numerous experts27,20–32 recommend a similar one, the Revised Cardiac Risk Index, devised by Lee et al.8 This index consists of six risk factors, each of which is worth one point:
- Congestive heart failure, based on history or examination
- Myocardial infarction, symptomatic ischemic heart disease, or a positive stress test
- Renal insufficiency (ie, serum creatinine level > 2 mg/dL)
- History of stroke or transient ischemic attack
- Diabetes requiring insulin
- High-risk surgery (defined as intrathoracic, intra-abdominal, or suprainguinal vascular surgery).
Patients with three or more points are considered to be at high risk, and those with one or two points are considered to be at intermediate risk. The ACC/AHA 2007 guidelines6 use a modified version of this index that considers the issue of surgical risk separately from the other five clinical conditions.
Devereaux et al33 performed a meta-analysis, published in 2005, of 22 studies of perioperative beta-blockade. They concluded that beta-blockers had no discernable benefit in any outcome measured, including deaths from any cause, deaths from cardiovascular causes, other cardiac events, hypotension, bradycardia, and bronchospasm. However, they based this conclusion on the use of a 99% confidence interval for each relative risk, which they believed was justified because the trials were small and the numbers of events were only moderate. When the outcomes are assessed using the more common 95% confidence interval, benefit was detected in the combined end point of cardiovascular death, nonfatal myocardial infarction, and nonfatal cardiac arrest.
Yang et al,34 Brady et al,35 and Juul et al36 performed three subsequent randomized trials that added to the controversy. Most of the patients in these trials were at intermediate or low risk, and none of the trials found a significant benefit with perioperative beta-blocker use. However, the protocols in these studies were different from the one in the study by Poldermans et al,3 which had found perioperative beta-blockade to be beneficial. Whereas patients in that earlier study started taking a beta-blocker at least 1 week before surgery (and on average much earlier), had their dose aggressively titrated to a target heart rate, and continued taking it for 30 days afterward, the protocols in the later trials called for the drug to be started within 24 hours before surgery and continued for only a short time afterward.
Lindenauer et al,37 in a retrospective study published in 2005, found that fewer surgical patients who received beta-blockers in the hospital died in the hospital. The researchers used an administrative database of more than 780,000 patients who underwent noncardiac surgery, and they used propensity-score matching to compare the postoperative mortality rates of patients who received beta-blockers and a matched group in the same large cohort who did not. Beta-blockers were associated with a lower morality rate in patients in whom the Revised Cardiac Risk Index score was 3 or greater. However, although there was a trend toward a lower rate with beta-blocker use in patients whose score was 2 (ie, at intermediate risk), the difference was not statistically significant, and patients with a score of 0 or 1 saw no benefit and were possibly harmed.
The authors admitted that their study had a number of limitations, including a retrospective design and the use of an administrative database for information regarding risk index conditions and comorbidities. In addition, because they assumed that any patient who received a beta-blocker on the first 2 hospital days was receiving appropriate perioperative treatment, they may have incorrectly estimated the number of patients who actually received these drugs as a risk-reduction strategy. For instance, some patients at low risk could have received beta-blockers for treatment of a specific event, which would be reflected as an increase in event rates for this group. They also had no data on what medications the patients received before they were hospitalized or whether the dose was titrated effectively. The study excluded all patients with congestive heart failure and chronic obstructive pulmonary disease, who may be candidates for beta-blockers in actual practice. In fact, a recent observational study in patients with severe left ventricular dysfunction suggested that these drugs substantially reduced the incidence of death in the short term and the long term.38 Finally, half the surgeries were nonelective, which makes extrapolation of their risk profile by the Revised Cardiac Risk Index difficult, since Lee et al excluded patients undergoing emergency surgery from the cohorts from which they derived and validated their index criteria.
Nevertheless, the authors concluded that patients at intermediate risk derive no benefit from perioperative beta-blocker use, and that the odds ratio for death was actually higher in patients with no risk factors who received a beta-blocker.
DOES PERIOPERATIVE BETA-BLOCKER USE CAUSE HARM?
The published data on whether perioperative beta-blocker use harms patients are conflicting and up to now have been limited.
Stone et al39 reported a substantial incidence of bradycardia requiring atropine in patients treated with a single dose of a beta-blocker preoperatively, but the complications were not clearly characterized.
The Perioperative Beta-Blockade trial.35 Significantly more patients given short-acting metoprolol had intraoperative falls in blood pressure and heart rate, and more required inotropic support during surgery, although the treating anesthesiologists refused to be blinded in that study. (Short-acting metoprolol is available in the United States as Lopressor and generically.)
Devereaux et al,33 in their meta-analysis, found a higher risk of bradycardia requiring treatment (but not a higher risk of hypotension) in beta-blocker users in nine studies, including the study by Stone et al and the Perioperative Beta-Blockade trial (relative risk 2.27, 95% confidence interval 1.36–3.80).
Conversely, at least three other studies found no difference in rates of intraoperative events.36,40,41 There are few data on the incidence of other complications such as perioperative pulmonary edema and bronchospasm.
POISE: THE FIRST LARGE RANDOMIZED TRIAL
In May 2008, results were published from POISE, the first large randomized controlled trial of perioperative beta-blockade.1 An impressive 8,351 patients—most of them at intermediate risk—were randomized to receive extended-release metoprolol succinate or placebo starting just before surgery and continuing for 30 days afterward.
Although the incidence of the primary composite end point (cardiovascular death, nonfatal myocardial infarction, and nonfatal cardiac arrest) was lower at 30 days in the metoprolol group than in the placebo group (5.8% vs 6.9%, hazard ratio 0.83, P = .04), other findings were worrisome: more metoprolol recipients died of any cause (3.1% vs 2.3%, P = .03) or had a stroke (1.0% vs 0.5%, P = .005). The major contributor to the higher mortality rate in this group appears to have been sepsis.
How beta-blockers might promote death by sepsis is unclear. The authors offered two possible explanations: perhaps beta-blocker-induced hypotension predisposes patients to infection and sepsis, or perhaps the slower heart rate and lower force of contraction induced by beta-blockers could mask normal responses to systemic infection, which in turn could delay recognition and treatment or impede the normal immune response. These mechanisms, like others, are speculative.
The risks of other adverse outcomes such as bradycardia and hypotension were substantially higher in the metoprolol group. The authors also pointed out that most of the patients who suffered nonfatal strokes were subsequently disabled or incapacitated, while most of those who suffered nonfatal cardiac events did not progress to further cardiac problems.
This new study has not yet been rigorously debated, but it will likely come under scrutiny for its dosing regimen (extended-release metoprolol succinate 100 mg or placebo 2–4 hours before surgery; another 100 mg or placebo 6 hours after surgery or sooner if the heart rate was 80 beats per minute or more and the systolic blood pressure 100 mm Hg or higher; and then 200 mg or placebo 12 hours after the second dose and every 24 hours thereafter for 30 days). This was fairly aggressive, especially for patients who have never received a beta-blocker before. In contrast, the protocol for the Perioperative Beta-Blockade trial called for only 25 to 50 mg of short-acting metoprolol twice a day. Another criticism is that the medication was started only a few hours before surgery, although there is no current standard practice for either the dose or when the treatment should be started. The population had a fairly high rate of cerebrovascular disease (perhaps predisposing to stroke whenever blood pressure dropped), and 10% of patients were undergoing urgent or emergency surgery, which carries a higher risk of morbidity.
ANY ROLE FOR BETA-BLOCKERS IN THOSE AT INTERMEDIATE RISK?
Thus, in the past decade, the appropriate perioperative use of beta-blockers, which, after the findings by Mangano et al and Poldermans et al, were seen as potentially beneficial for any patient at risk of coronary disease, with little suggestion of harm, has become more clearly defined, and the risks are more evident. The most compelling evidence in favor of using them comes from patients with ischemic heart disease undergoing vascular surgery; the 2007 ACC/AHA guidelines recommend that this group be offered beta-blockers in the absence of a contraindication (class I recommendation: benefit clearly outweighs risk).6 The guidelines also point out that these drugs should be continued in patients already taking them for cardiac indications before surgery, because ischemia may be precipitated if a beta-blocker is abruptly discontinued.42,43
Additionally, the guidelines recommend considering beta-blockers for vascular surgery patients at high cardiac risk (with a Revised Cardiac Risk Index score of 3 or more), even if they are not known to have ischemic heart disease. This is a class IIa recommendation (the benefit outweighs the risk, but more studies are required).
The guidelines also recommend that beta-blockers be considered for patients who have a score of 0 if they are undergoing vascular surgery (class IIb recommendation) or a score of 1 if they are undergoing vascular surgery (class IIa recommendation) or intermediate-risk surgery (class IIb recommendation). However, in view of the POISE results, these recommendations need to be carefully scrutinized.
These data notwithstanding, beta-blockers still might be beneficial in perioperative patients at intermediate risk.
Start beta-blockers sooner?
To help patients at intermediate risk (such as those with diabetes without known heart disease), we may need to do what Poldermans et al did3: instead of seeing patients only once a day or two before surgery, we may need to do the preoperative assessment as much as a month before and, if necessary, start a beta-blocker at a low dose, titrate it to a goal heart rate, and follow the patient closely up until surgery and afterward.
The importance of heart-rate control was illustrated in a recent cohort study of perioperative beta-blockers in vascular surgery patients,44 in which higher beta-blocker doses, carefully monitored, were associated with less ischemia and cardiac enzyme release. In addition, long-term mortality rates were lower in patients with lower heart rates. And Poldermans et al45 recently performed a study in more than 700 intermediate-risk patients who were divided into two groups, one that underwent preoperative stress testing and one that did not. Beta-blockers were given to both groups, and doses were titrated to a goal heart rate of less than 65. The patients with optimally controlled heart rates had the lowest event rates.
However, the logistics of such a program would be challenging. For the most part, internists and hospitalists involved in perioperative assessment do not control the timing of referral or surgery, and adjustments cannot be made for patients whose preoperative clinic visit falls only a few days before surgery. Instituting a second or third visit to assess the efficacy of beta-blockade burdens the patient and may not be practical.
Are all beta-blockers equivalent?
An additional factor is the choice of agent. While the most significant studies of perioperative beta-blockade have used beta-1 receptor-selective agents (ie, metoprolol, atenolol, and bisoprolol), there is no prospective evidence that any particular agent is superior. However, a recent retrospective analysis of elderly surgical patients did suggest that longer-acting beta-blockers may be preferable: patients who had been on atenolol in the year before surgery had a 20% lower risk of postoperative myocardial infarction or death than those who had been on short-acting metoprolol, with no difference in noncardiac outcomes.46
- POISE Study Group. Effect of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet 2008; published online May 13. DOI: 10.1016/S0140-6736(08)60601-7.
- Mangano DT, Layug EL, Wallace A, Tateo I. Effect of atenolol on mortality and cardiovascular morbidity after noncardiac surgery. Multicenter Study of Perioperative Ischemia Research Group. N Engl J Med 1996; 335:1713–1720.
- Poldermans D, Boersma E, Bax JJ, et al. The effect of bisoprolol on perioperative mortality and myocardial infarction in high-risk patients undergoing vascular surgery. Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group. N Engl J Med 1999; 341:1789–1794.
- Shojania KG, Duncan BW, McDonald KM, Wachter RM, Markowitz AJ. Making health care safer: a critical analysis of patient safety practices. Evid Rep Technol Assess (Summ) 2001; ( 43):i–x,1–668.
- National Quality Forum. Safe Practices for Better Healthcare—2006 update. Washington, DC: National Quality Forum, 2006.
- Fleisher LA, Beckman JA, Brown KA, et al. ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery). Circulation 2007; 116:1971–1996.
- Mangano DT. Perioperative cardiac morbidity. Anesthesiology 1990; 72:153–184.
- Lee TH, Marcantonio ER, Mangione CM, et al. Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery. Circulation 1999; 100:1043–1049.
- Devereaux PJ, Goldman L, Cook DJ, Gilbert K, Leslie K, Guyatt GH. Perioperative cardiac events in patients undergoing noncardiac surgery: a review of the magnitude of the problem, the pathophysiology of the events and methods to estimate and communicate risk. Can Med Assoc J 2005; 173:627–634.
- Fleischmann KE, Goldman L, Young B, Lee TH. Association between cardiac and noncardiac complications in patients undergoing noncardiac surgery: outcomes and effects on length of stay. Am J Med 2003; 115:515–520.
- Landesberg G, Luria MH, Cotev S, et al. Importance of long-duration postoperative ST-segment depression in cardiac morbidity after vascular surgery. Lancet 1993; 341:715–719.
- Mangano DT, Browner WS, Hollenberg M, London MJ, Tubau JF, Tateo IM. Association of perioperative myocardial ischemia with cardiac morbidity and mortality in men undergoing noncardiac surgery. The Study of Perioperative Ischemia Research Group. N Engl J Med 1990; 323:1781–1788.
- Raby KE, Goldman L, Creager MA, et al. Correlation between preoperative ischemia and major cardiac events after peripheral vascular surgery. N Engl J Med 1989; 321:1296–1300.
- Landesberg G, Mosseri M, Zahger D, et al. Myocardial infarction after vascular surgery: the role of prolonged stress-induced, ST depression-type ischemia. J Am Coll Cardiol 2001; 37:1839–1845.
- Mangano DT, Browner WS, Hollenberg M, Li J, Tateo IM. Long-term cardiac prognosis following noncardiac surgery. The Study of Perioperative Ischemia Research Group. JAMA 1992; 268:233–239.
- Kim LJ, Martinez EA, Faraday N, et al. Cardiac troponin I predicts short-term mortality in vascular surgery patients. Circulation 2002; 106:2366–2371.
- Landesberg G, Shatz V, Akopnik I, et al. Association of cardiac troponin, CK-MB, and postoperative myocardial ischemia with long-term survival after major vascular surgery. J Am Coll Cardiol 2003; 42:1547–1554.
- Badner NH, Knill RL, Brown JE, Novick TV, Gelb AW. Myocardial infarction after noncardiac surgery. Anesthesiology 1998; 88:572–578.
- Priebe HJ. Triggers of perioperative myocardial ischaemia and infarction. Br J Anaesth 2004; 93:9–20.
- Zaugg M, Schaub MC, Foex P. Myocardial injury and its prevention in the perioperative setting. Br J Anaesth 2004; 93:21–33.
- Dawood MM, Gutpa DK, Southern J, Walia A, Atkinson JB, Eagle KA. Pathology of fatal perioperative myocardial infarction: implications regarding pathophysiology and prevention. Int J Cardiol 1996; 57:37–44.
- Cohen MC, Aretz TH. Histological analysis of coronary artery lesions in fatal postoperative myocardial infarction. Cardiovasc Pathol 1999; 8:133–139.
- London MJ, Zaugg M, Schaub MC, Spahn DR. Perioperative beta-adrenergic receptor blockade: physiologic foundations and clinical controversies. Anesthesiology 2004; 100:170–175.
- Yeager MP, Fillinger MP, Hettleman BD, Hartman GS. Perioperative beta-blockade and late cardiac outcomes: a complementary hypothesis. J Cardiothorac Vasc Anesth 2005; 19:237–241.
- Zaugg M, Schaub MC, Pasch T, Spahn DR. Modulation of beta-adrenergic receptor subtype activities in perioperative medicine: mechanisms and sites of action. Br J Anaesth 2002; 88:101–123.
- Devereaux PJ, Yusuf S, Yang H, Choi PT, Guyatt GH. Are the recommendations to use perioperative beta-blocker therapy in patients undergoing non-cardiac surgery based on reliable evidence? Can Med Assoc J 2004; 171:245–247.
- Eagle KA, Froehlich JB. Reducing cardiovascular risk in patients undergoing noncardiac surgery. N Engl J Med 1996; 335:1761–1763.
- Boersma E, Poldermans D, Bax JJ, et al. Predictors of cardiac events after major vascular surgery: role of clinical characteristics, dobutamine echocardiography, and beta-blocker therapy. JAMA 2001; 285:1865–1873.
- Stevens RD, Burri H, Tramer MR. Pharmacologic myocardial protection in patients undergoing noncardiac surgery: a quantitative systematic review. Anesth Analg 2003; 97:623–633.
- Goldman L. Assessing and reducing cardiac risks of noncardiac surgery. Am J Med 2001; 110:320–323.
- Wesorick DH, Eagle KA. The preoperative cardiovascular evaluation of the intermediate-risk patient: new data, changing strategies. Am J Med 2005; 118:1413.
- Auerbach AD, Goldman L. Beta-blockers and reduction of cardiac events in noncardiac surgery: scientific review. JAMA 2002; 287:1435–1444.
- Devereaux PJ, Beattie WS, Choi PT, et al. How strong is the evidence for the use of perioperative beta blockers in non-cardiac surgery? Systematic review and meta–analysis of randomized controlled trials. BMJ 2005; 331:313–321.
- Yang H, Raymer K, Butler R, Parlow JL, Roberts RS. Metoprolol after vascular surgery (MaVS). Can J Anaesth 2004; 51( suppl 1):A7.
- Brady AR, Gibbs JS, Greenhalgh RM, Powell JT, Sydes MR. Perioperative beta-blockade (POBBLE) for patients undergoing infrarenal vascular surgery: results of a randomized double-blind controlled trial. J Vasc Surg 2005; 41:602–609.
- Juul AB, Wetterslev J, Gluud C, et al. Effect of perioperative βblockade in patients with diabetes undergoing major non-cardiac surgery: randomised placebo controlled, blinded multicentre trial. BMJ 2006; 332:1482.
- Lindenauer PK, Pekow P, Wang K, Mamidi DK, Gutierrez B, Benjamin EM. Perioperative beta-blocker therapy and mortality after major noncardiac surgery. N Engl J Med 2005; 353:349–361.
- Feringa HH, Bax JJ, Schouten O, et al. Beta-blockers improve in-hospital and long-term survival in patients with severe left ventricular dysfunction undergoing major vascular surgery. Eur J Vasc Endovasc Surg 2005; 31:351–358.
- Stone JG, Foex P, Sear JW, Johnson LL, Khambatta HJ, Triner L. Myocardial ischemia in untreated hypertensive patients: effect of a single small oral dose of a beta-adrenergic blocking agent. Anesthesiology 1988; 68:495–500.
- Zaugg M, Tagliente T, Lucchinetti E, et al. Beneficial effects from beta-adrenergic blockade in elderly patients undergoing noncardiac surgery. Anesthesiology 1999; 91:1674–1686.
- Wallace A, Layug B, Tateo I, et al. Prophylactic atenolol reduces postoperative myocardial ischemia. McSPI Research Group. Anesthesiology 1998; 88:7–17.
- Psaty BM, Koepsell TD, Wagner EH, LoGerfo JP, Inui TS. The relative risk of incident coronary heart disease associated with recently stopping the use of beta-blockers. JAMA 1990; 263:1653–1657.
- Shammash JB, Trost JC, Gold JM, Berlin JA, Golden MA, Kimmel SE. Perioperative beta-blocker withdrawal and mortality in vascular surgical patients. Am Heart J 2001; 141:148–153.
- Feringa HHH, Bax JJ, Boersma E, et al. High-dose beta-blockers and tight heart rate control reduce myocardial ischemia and troponin T release in vascular surgery patients. Circulation 2006; 114( suppl 1):I-344–I-349.
- Poldermans D, Bax JJ, Schouten O, et al. Should major vascular surgery be delayed because of preoperative cardiac testing in intermediate-risk patients receiving beta-blocker therapy with tight heart rate control? J Am Coll Cardiol 2006; 48:964–969.
- Redelmeier D, Scales D, Kopp A. Beta blockers for elective surgery in elderly patients: population based, retrospective cohort study. BMJ 2005; 331:932.
The pendulum of expert opinion is swinging away from routinely recommending beta-blockers to prevent cardiac events in non-cardiac surgery patients. We won’t be abandoning the perioperative use of beta-blockers altogether, but we will probably be using them more selectively than in the past.
The latest factor driving the trend is the online publication in May 2008 of the results of the Perioperative Ischemic Evaluation (POISE) trial,1 the largest placebo-controlled trial of perioperative beta-blocker use to date. In brief, in a cohort of patients with atherosclerotic disease or at risk for it who were undergoing noncardiac surgery, fewer patients who received extended-release metoprolol succinate had a myocardial infarction, but more of them died or had a stroke compared with those receiving placebo. (Extended-release metoprolol succinate is available in the United States as Toprol-XL and generically.)
Not so long ago, the pendulum was going the other way. After two small trials in the 1990s concluded that beta-blockers reduced the risk of perioperative cardiac events in selected patients with known or suspected coronary disease,2,3 their perioperative use was subsequently endorsed by the Leapfrog Group and the Agency for Healthcare Research and Quality. The National Quality Forum included perioperative beta-blockade in its “Safe Practices for Better Healthcare 2006 update,”4,5 and the Physician Consortium for Performance Improvement and the Surgical Care Improvement Project both listed it as a quality measure.
Since then, this practice has been closely studied, especially as concomitant research has failed to demonstrate that pre-operative coronary revascularization improves outcomes, even in the presence of ischemic disease. But evidence has been accumulating that routine use of beta-blockers may not benefit as many patients as was hoped, and may actually cause harm. The 2007 joint American College of Cardiology (ACC) and American Heart Association (AHA) guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery gives its strongest recommendation (class I: benefit clearly outweighs risk) for perioperative beta-blocker use only for patients at high risk: those with known ischemic heart disease undergoing vascular surgery and those who are already on these drugs before surgery.6
However, there are still gaps in our knowledge. Perhaps, with proper implementation, we may be able to use beta-blockers to improve outcomes in patients at intermediate risk as well. In this paper, we review the rationale and the evidence for and against perioperative use of beta-blockers and provide practical guidance for internists and hospitalists.
WHY CARDIAC EVENTS OCCUR AFTER SURGERY
Adverse cardiovascular events such as myocardial infarction and unstable angina are the leading causes of death after surgery.7 Such events occur in approximately 1% of patients older than 50 years undergoing elective inpatient surgery, but this number may be higher (approximately 5%) in those with known or suspected coronary disease.8,9 Perioperative cardiac events can also be harbingers of further complications, dramatically increasing hospital length of stay.10
Some ischemic events are caused by physiologic derangements involving the balance between inflammatory mediators, sympathetic tone, and oxygen supply and demand that occur under the stress of surgery. Others are more “traditional” in etiology, involving acute plaque rupture, thrombosis, and occlusion. Studies have consistently found a correlation between perioperative ischemia and cardiac events (both in-hospital and long-term) and death.11–17 Other studies suggest that most perioperative cardiac infarcts are non-Q-wave events,18 and most events occur within the first few days after surgery, particularly the first 48 hours, when the effects of anesthetics, pain, fluid shifts, and physiologic derangements are greatest.
Factors that may trigger acute occlusion in the perioperative period include abrupt changes in sympathetic tone, increased levels of cortisol and catecholamines, and tissue hypoxia. Other potential triggers activated or increased by the stress of surgery include coagulation factors such as alterations in platelet function; inflammatory factors such as tumor necrosis factor alpha, interleukin 1, interleukin 6, and C-reactive protein; and metabolism of free fatty acids (which contribute to increased oxygen demand as well as endothelial dysfunction).9,19,20
A 1996 autopsy study found that 38 (90%) of 42 patients who died of a perioperative infarct had evidence of acute plaque rupture or plaque hemorrhage on coronary sectioning, findings corroborated in another, similar study.21,22 These studies suggest that multiple causes contribute to perioperative myocardial infarction, and a single strategy may not suffice for prevention.
IF BETA-BLOCKERS PROTECT, HOW DO THEY DO IT?
Beta-blockers have several effects that should, in theory, protect against cardiac events during and after surgery.23 They reduce cardiac oxygen demand by reducing the force of contraction and the heart rate, and they increase the duration of diastole, when the heart muscle is perfused. They are also antiarrhythmic, and they may limit free radical production, metalloproteinase activity, and myocardial plaque inflammation.24
Some researchers have speculated that using beta-blockers long-term may alter intra-cellular signaling processes, for example decreasing the expression of receptors that receive signals for cell death, which in turn may affect the response to reperfusion cell injury and death. If this is true, there may be an advantage to starting beta-blockers well in advance of surgery.25
EARLY CLINICAL EVIDENCE IN FAVOR OF PERIOPERATIVE BETA-BLOCKER USE
Evidence in patients at high risk
Mangano et al,2 in a study published in 1996, randomized 200 patients with known coronary disease or established risk factors for it who were undergoing noncardiac surgery to receive the beta-blocker atenolol orally and intravenously or placebo in the immediate perioperative period. Fewer patients in the atenolol group died in the first 6 months after hospital discharge (0 vs 8%, P < .001), the first year (3% vs 14%, P = .005), and the first 2 years (10% vs 21%, P = .019). However, there was no difference in short-term outcomes, and the study excluded patients who died in the immediate postoperative period. If these patients had been included in the analysis, the difference in the death rate at 2 years would not have been statistically significant.26 Other critical findings: more patients in the atenolol group were using angiotensin-converting enzyme inhibitors and beta-blockers when they were discharged, and the placebo group had slightly more patients with prior myocardial infarction or diabetes.27 (Atenolol is available in the United Sates as Tenormin and generically.)
Poldermans et al,3 in a study published in 1999, randomized 112 vascular surgery patients to receive either oral bisoprolol or placebo. These patients were selected from a larger cohort of 1,351 patients on the basis of high-risk clinical features and abnormal results on dobutamine echocardiography. Bisoprolol was started at least 1 week before surgery (range 7–89 days, mean 37 days), and patients were reevaluated before surgery so that the dose could be titrated to a goal heart rate of less than 60 beats per minute. After surgery, the drug was continued for another 30 days. The study was stopped early because the bisoprolol group had a 90% lower rate of non-fatal myocardial infarction and cardiac death at 30 days. Despite the study’s limitation (eg, enrolling selected patients and using an unblinded protocol), these compelling findings made a strong case for the use of beta-blockers perioperatively in patients at high risk, ie, those with ischemic heart disease who are undergoing major vascular surgery. (Bisoprolol is available in the United States as Zebeta and generically)
Evidence in patients at intermediate risk
Boersma et al28 performed a follow-up to the study by Poldermans et al, published in 2001, in which they analyzed characteristics of all 1,351 patients who had been originally considered for enrollment. Using regression analysis, they identified seven clinical risk factors that predicted adverse cardiac events: angina, prior myocardial infarction, congestive heart failure, prior stroke, diabetes, renal failure, and age 70 years or older. Furthermore, for the entire cohort, patients receiving beta-blockers had a lower risk of cardiac complications (0.8%) than those not receiving beta-blockers (2.3%). In particular, the patients at intermediate risk (defined as having one or two risk factors) had a very low event rate regardless of stress test results, provided they were on beta-blockers: their risk of death or myocardial infarction was 0.9%, compared with 3.0% for those not on beta-blockers.
The authors concluded that dobutamine stress testing may not be necessary in patients at intermediate risk if beta-blockers are appropriately prescribed. However, others took issue with their data and conclusions, arguing that there have been so few trials that the data are still inconclusive and inadequate to ascertain the benefit of perioperative beta-blockade, particularly in patients not at high risk.26,29
The Revised Cardiac Risk Index. Although the Boersma risk-factor index is not used in general practice, numerous experts27,20–32 recommend a similar one, the Revised Cardiac Risk Index, devised by Lee et al.8 This index consists of six risk factors, each of which is worth one point:
- Congestive heart failure, based on history or examination
- Myocardial infarction, symptomatic ischemic heart disease, or a positive stress test
- Renal insufficiency (ie, serum creatinine level > 2 mg/dL)
- History of stroke or transient ischemic attack
- Diabetes requiring insulin
- High-risk surgery (defined as intrathoracic, intra-abdominal, or suprainguinal vascular surgery).
Patients with three or more points are considered to be at high risk, and those with one or two points are considered to be at intermediate risk. The ACC/AHA 2007 guidelines6 use a modified version of this index that considers the issue of surgical risk separately from the other five clinical conditions.
Devereaux et al33 performed a meta-analysis, published in 2005, of 22 studies of perioperative beta-blockade. They concluded that beta-blockers had no discernable benefit in any outcome measured, including deaths from any cause, deaths from cardiovascular causes, other cardiac events, hypotension, bradycardia, and bronchospasm. However, they based this conclusion on the use of a 99% confidence interval for each relative risk, which they believed was justified because the trials were small and the numbers of events were only moderate. When the outcomes are assessed using the more common 95% confidence interval, benefit was detected in the combined end point of cardiovascular death, nonfatal myocardial infarction, and nonfatal cardiac arrest.
Yang et al,34 Brady et al,35 and Juul et al36 performed three subsequent randomized trials that added to the controversy. Most of the patients in these trials were at intermediate or low risk, and none of the trials found a significant benefit with perioperative beta-blocker use. However, the protocols in these studies were different from the one in the study by Poldermans et al,3 which had found perioperative beta-blockade to be beneficial. Whereas patients in that earlier study started taking a beta-blocker at least 1 week before surgery (and on average much earlier), had their dose aggressively titrated to a target heart rate, and continued taking it for 30 days afterward, the protocols in the later trials called for the drug to be started within 24 hours before surgery and continued for only a short time afterward.
Lindenauer et al,37 in a retrospective study published in 2005, found that fewer surgical patients who received beta-blockers in the hospital died in the hospital. The researchers used an administrative database of more than 780,000 patients who underwent noncardiac surgery, and they used propensity-score matching to compare the postoperative mortality rates of patients who received beta-blockers and a matched group in the same large cohort who did not. Beta-blockers were associated with a lower morality rate in patients in whom the Revised Cardiac Risk Index score was 3 or greater. However, although there was a trend toward a lower rate with beta-blocker use in patients whose score was 2 (ie, at intermediate risk), the difference was not statistically significant, and patients with a score of 0 or 1 saw no benefit and were possibly harmed.
The authors admitted that their study had a number of limitations, including a retrospective design and the use of an administrative database for information regarding risk index conditions and comorbidities. In addition, because they assumed that any patient who received a beta-blocker on the first 2 hospital days was receiving appropriate perioperative treatment, they may have incorrectly estimated the number of patients who actually received these drugs as a risk-reduction strategy. For instance, some patients at low risk could have received beta-blockers for treatment of a specific event, which would be reflected as an increase in event rates for this group. They also had no data on what medications the patients received before they were hospitalized or whether the dose was titrated effectively. The study excluded all patients with congestive heart failure and chronic obstructive pulmonary disease, who may be candidates for beta-blockers in actual practice. In fact, a recent observational study in patients with severe left ventricular dysfunction suggested that these drugs substantially reduced the incidence of death in the short term and the long term.38 Finally, half the surgeries were nonelective, which makes extrapolation of their risk profile by the Revised Cardiac Risk Index difficult, since Lee et al excluded patients undergoing emergency surgery from the cohorts from which they derived and validated their index criteria.
Nevertheless, the authors concluded that patients at intermediate risk derive no benefit from perioperative beta-blocker use, and that the odds ratio for death was actually higher in patients with no risk factors who received a beta-blocker.
DOES PERIOPERATIVE BETA-BLOCKER USE CAUSE HARM?
The published data on whether perioperative beta-blocker use harms patients are conflicting and up to now have been limited.
Stone et al39 reported a substantial incidence of bradycardia requiring atropine in patients treated with a single dose of a beta-blocker preoperatively, but the complications were not clearly characterized.
The Perioperative Beta-Blockade trial.35 Significantly more patients given short-acting metoprolol had intraoperative falls in blood pressure and heart rate, and more required inotropic support during surgery, although the treating anesthesiologists refused to be blinded in that study. (Short-acting metoprolol is available in the United States as Lopressor and generically.)
Devereaux et al,33 in their meta-analysis, found a higher risk of bradycardia requiring treatment (but not a higher risk of hypotension) in beta-blocker users in nine studies, including the study by Stone et al and the Perioperative Beta-Blockade trial (relative risk 2.27, 95% confidence interval 1.36–3.80).
Conversely, at least three other studies found no difference in rates of intraoperative events.36,40,41 There are few data on the incidence of other complications such as perioperative pulmonary edema and bronchospasm.
POISE: THE FIRST LARGE RANDOMIZED TRIAL
In May 2008, results were published from POISE, the first large randomized controlled trial of perioperative beta-blockade.1 An impressive 8,351 patients—most of them at intermediate risk—were randomized to receive extended-release metoprolol succinate or placebo starting just before surgery and continuing for 30 days afterward.
Although the incidence of the primary composite end point (cardiovascular death, nonfatal myocardial infarction, and nonfatal cardiac arrest) was lower at 30 days in the metoprolol group than in the placebo group (5.8% vs 6.9%, hazard ratio 0.83, P = .04), other findings were worrisome: more metoprolol recipients died of any cause (3.1% vs 2.3%, P = .03) or had a stroke (1.0% vs 0.5%, P = .005). The major contributor to the higher mortality rate in this group appears to have been sepsis.
How beta-blockers might promote death by sepsis is unclear. The authors offered two possible explanations: perhaps beta-blocker-induced hypotension predisposes patients to infection and sepsis, or perhaps the slower heart rate and lower force of contraction induced by beta-blockers could mask normal responses to systemic infection, which in turn could delay recognition and treatment or impede the normal immune response. These mechanisms, like others, are speculative.
The risks of other adverse outcomes such as bradycardia and hypotension were substantially higher in the metoprolol group. The authors also pointed out that most of the patients who suffered nonfatal strokes were subsequently disabled or incapacitated, while most of those who suffered nonfatal cardiac events did not progress to further cardiac problems.
This new study has not yet been rigorously debated, but it will likely come under scrutiny for its dosing regimen (extended-release metoprolol succinate 100 mg or placebo 2–4 hours before surgery; another 100 mg or placebo 6 hours after surgery or sooner if the heart rate was 80 beats per minute or more and the systolic blood pressure 100 mm Hg or higher; and then 200 mg or placebo 12 hours after the second dose and every 24 hours thereafter for 30 days). This was fairly aggressive, especially for patients who have never received a beta-blocker before. In contrast, the protocol for the Perioperative Beta-Blockade trial called for only 25 to 50 mg of short-acting metoprolol twice a day. Another criticism is that the medication was started only a few hours before surgery, although there is no current standard practice for either the dose or when the treatment should be started. The population had a fairly high rate of cerebrovascular disease (perhaps predisposing to stroke whenever blood pressure dropped), and 10% of patients were undergoing urgent or emergency surgery, which carries a higher risk of morbidity.
ANY ROLE FOR BETA-BLOCKERS IN THOSE AT INTERMEDIATE RISK?
Thus, in the past decade, the appropriate perioperative use of beta-blockers, which, after the findings by Mangano et al and Poldermans et al, were seen as potentially beneficial for any patient at risk of coronary disease, with little suggestion of harm, has become more clearly defined, and the risks are more evident. The most compelling evidence in favor of using them comes from patients with ischemic heart disease undergoing vascular surgery; the 2007 ACC/AHA guidelines recommend that this group be offered beta-blockers in the absence of a contraindication (class I recommendation: benefit clearly outweighs risk).6 The guidelines also point out that these drugs should be continued in patients already taking them for cardiac indications before surgery, because ischemia may be precipitated if a beta-blocker is abruptly discontinued.42,43
Additionally, the guidelines recommend considering beta-blockers for vascular surgery patients at high cardiac risk (with a Revised Cardiac Risk Index score of 3 or more), even if they are not known to have ischemic heart disease. This is a class IIa recommendation (the benefit outweighs the risk, but more studies are required).
The guidelines also recommend that beta-blockers be considered for patients who have a score of 0 if they are undergoing vascular surgery (class IIb recommendation) or a score of 1 if they are undergoing vascular surgery (class IIa recommendation) or intermediate-risk surgery (class IIb recommendation). However, in view of the POISE results, these recommendations need to be carefully scrutinized.
These data notwithstanding, beta-blockers still might be beneficial in perioperative patients at intermediate risk.
Start beta-blockers sooner?
To help patients at intermediate risk (such as those with diabetes without known heart disease), we may need to do what Poldermans et al did3: instead of seeing patients only once a day or two before surgery, we may need to do the preoperative assessment as much as a month before and, if necessary, start a beta-blocker at a low dose, titrate it to a goal heart rate, and follow the patient closely up until surgery and afterward.
The importance of heart-rate control was illustrated in a recent cohort study of perioperative beta-blockers in vascular surgery patients,44 in which higher beta-blocker doses, carefully monitored, were associated with less ischemia and cardiac enzyme release. In addition, long-term mortality rates were lower in patients with lower heart rates. And Poldermans et al45 recently performed a study in more than 700 intermediate-risk patients who were divided into two groups, one that underwent preoperative stress testing and one that did not. Beta-blockers were given to both groups, and doses were titrated to a goal heart rate of less than 65. The patients with optimally controlled heart rates had the lowest event rates.
However, the logistics of such a program would be challenging. For the most part, internists and hospitalists involved in perioperative assessment do not control the timing of referral or surgery, and adjustments cannot be made for patients whose preoperative clinic visit falls only a few days before surgery. Instituting a second or third visit to assess the efficacy of beta-blockade burdens the patient and may not be practical.
Are all beta-blockers equivalent?
An additional factor is the choice of agent. While the most significant studies of perioperative beta-blockade have used beta-1 receptor-selective agents (ie, metoprolol, atenolol, and bisoprolol), there is no prospective evidence that any particular agent is superior. However, a recent retrospective analysis of elderly surgical patients did suggest that longer-acting beta-blockers may be preferable: patients who had been on atenolol in the year before surgery had a 20% lower risk of postoperative myocardial infarction or death than those who had been on short-acting metoprolol, with no difference in noncardiac outcomes.46
The pendulum of expert opinion is swinging away from routinely recommending beta-blockers to prevent cardiac events in non-cardiac surgery patients. We won’t be abandoning the perioperative use of beta-blockers altogether, but we will probably be using them more selectively than in the past.
The latest factor driving the trend is the online publication in May 2008 of the results of the Perioperative Ischemic Evaluation (POISE) trial,1 the largest placebo-controlled trial of perioperative beta-blocker use to date. In brief, in a cohort of patients with atherosclerotic disease or at risk for it who were undergoing noncardiac surgery, fewer patients who received extended-release metoprolol succinate had a myocardial infarction, but more of them died or had a stroke compared with those receiving placebo. (Extended-release metoprolol succinate is available in the United States as Toprol-XL and generically.)
Not so long ago, the pendulum was going the other way. After two small trials in the 1990s concluded that beta-blockers reduced the risk of perioperative cardiac events in selected patients with known or suspected coronary disease,2,3 their perioperative use was subsequently endorsed by the Leapfrog Group and the Agency for Healthcare Research and Quality. The National Quality Forum included perioperative beta-blockade in its “Safe Practices for Better Healthcare 2006 update,”4,5 and the Physician Consortium for Performance Improvement and the Surgical Care Improvement Project both listed it as a quality measure.
Since then, this practice has been closely studied, especially as concomitant research has failed to demonstrate that pre-operative coronary revascularization improves outcomes, even in the presence of ischemic disease. But evidence has been accumulating that routine use of beta-blockers may not benefit as many patients as was hoped, and may actually cause harm. The 2007 joint American College of Cardiology (ACC) and American Heart Association (AHA) guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery gives its strongest recommendation (class I: benefit clearly outweighs risk) for perioperative beta-blocker use only for patients at high risk: those with known ischemic heart disease undergoing vascular surgery and those who are already on these drugs before surgery.6
However, there are still gaps in our knowledge. Perhaps, with proper implementation, we may be able to use beta-blockers to improve outcomes in patients at intermediate risk as well. In this paper, we review the rationale and the evidence for and against perioperative use of beta-blockers and provide practical guidance for internists and hospitalists.
WHY CARDIAC EVENTS OCCUR AFTER SURGERY
Adverse cardiovascular events such as myocardial infarction and unstable angina are the leading causes of death after surgery.7 Such events occur in approximately 1% of patients older than 50 years undergoing elective inpatient surgery, but this number may be higher (approximately 5%) in those with known or suspected coronary disease.8,9 Perioperative cardiac events can also be harbingers of further complications, dramatically increasing hospital length of stay.10
Some ischemic events are caused by physiologic derangements involving the balance between inflammatory mediators, sympathetic tone, and oxygen supply and demand that occur under the stress of surgery. Others are more “traditional” in etiology, involving acute plaque rupture, thrombosis, and occlusion. Studies have consistently found a correlation between perioperative ischemia and cardiac events (both in-hospital and long-term) and death.11–17 Other studies suggest that most perioperative cardiac infarcts are non-Q-wave events,18 and most events occur within the first few days after surgery, particularly the first 48 hours, when the effects of anesthetics, pain, fluid shifts, and physiologic derangements are greatest.
Factors that may trigger acute occlusion in the perioperative period include abrupt changes in sympathetic tone, increased levels of cortisol and catecholamines, and tissue hypoxia. Other potential triggers activated or increased by the stress of surgery include coagulation factors such as alterations in platelet function; inflammatory factors such as tumor necrosis factor alpha, interleukin 1, interleukin 6, and C-reactive protein; and metabolism of free fatty acids (which contribute to increased oxygen demand as well as endothelial dysfunction).9,19,20
A 1996 autopsy study found that 38 (90%) of 42 patients who died of a perioperative infarct had evidence of acute plaque rupture or plaque hemorrhage on coronary sectioning, findings corroborated in another, similar study.21,22 These studies suggest that multiple causes contribute to perioperative myocardial infarction, and a single strategy may not suffice for prevention.
IF BETA-BLOCKERS PROTECT, HOW DO THEY DO IT?
Beta-blockers have several effects that should, in theory, protect against cardiac events during and after surgery.23 They reduce cardiac oxygen demand by reducing the force of contraction and the heart rate, and they increase the duration of diastole, when the heart muscle is perfused. They are also antiarrhythmic, and they may limit free radical production, metalloproteinase activity, and myocardial plaque inflammation.24
Some researchers have speculated that using beta-blockers long-term may alter intra-cellular signaling processes, for example decreasing the expression of receptors that receive signals for cell death, which in turn may affect the response to reperfusion cell injury and death. If this is true, there may be an advantage to starting beta-blockers well in advance of surgery.25
EARLY CLINICAL EVIDENCE IN FAVOR OF PERIOPERATIVE BETA-BLOCKER USE
Evidence in patients at high risk
Mangano et al,2 in a study published in 1996, randomized 200 patients with known coronary disease or established risk factors for it who were undergoing noncardiac surgery to receive the beta-blocker atenolol orally and intravenously or placebo in the immediate perioperative period. Fewer patients in the atenolol group died in the first 6 months after hospital discharge (0 vs 8%, P < .001), the first year (3% vs 14%, P = .005), and the first 2 years (10% vs 21%, P = .019). However, there was no difference in short-term outcomes, and the study excluded patients who died in the immediate postoperative period. If these patients had been included in the analysis, the difference in the death rate at 2 years would not have been statistically significant.26 Other critical findings: more patients in the atenolol group were using angiotensin-converting enzyme inhibitors and beta-blockers when they were discharged, and the placebo group had slightly more patients with prior myocardial infarction or diabetes.27 (Atenolol is available in the United Sates as Tenormin and generically.)
Poldermans et al,3 in a study published in 1999, randomized 112 vascular surgery patients to receive either oral bisoprolol or placebo. These patients were selected from a larger cohort of 1,351 patients on the basis of high-risk clinical features and abnormal results on dobutamine echocardiography. Bisoprolol was started at least 1 week before surgery (range 7–89 days, mean 37 days), and patients were reevaluated before surgery so that the dose could be titrated to a goal heart rate of less than 60 beats per minute. After surgery, the drug was continued for another 30 days. The study was stopped early because the bisoprolol group had a 90% lower rate of non-fatal myocardial infarction and cardiac death at 30 days. Despite the study’s limitation (eg, enrolling selected patients and using an unblinded protocol), these compelling findings made a strong case for the use of beta-blockers perioperatively in patients at high risk, ie, those with ischemic heart disease who are undergoing major vascular surgery. (Bisoprolol is available in the United States as Zebeta and generically)
Evidence in patients at intermediate risk
Boersma et al28 performed a follow-up to the study by Poldermans et al, published in 2001, in which they analyzed characteristics of all 1,351 patients who had been originally considered for enrollment. Using regression analysis, they identified seven clinical risk factors that predicted adverse cardiac events: angina, prior myocardial infarction, congestive heart failure, prior stroke, diabetes, renal failure, and age 70 years or older. Furthermore, for the entire cohort, patients receiving beta-blockers had a lower risk of cardiac complications (0.8%) than those not receiving beta-blockers (2.3%). In particular, the patients at intermediate risk (defined as having one or two risk factors) had a very low event rate regardless of stress test results, provided they were on beta-blockers: their risk of death or myocardial infarction was 0.9%, compared with 3.0% for those not on beta-blockers.
The authors concluded that dobutamine stress testing may not be necessary in patients at intermediate risk if beta-blockers are appropriately prescribed. However, others took issue with their data and conclusions, arguing that there have been so few trials that the data are still inconclusive and inadequate to ascertain the benefit of perioperative beta-blockade, particularly in patients not at high risk.26,29
The Revised Cardiac Risk Index. Although the Boersma risk-factor index is not used in general practice, numerous experts27,20–32 recommend a similar one, the Revised Cardiac Risk Index, devised by Lee et al.8 This index consists of six risk factors, each of which is worth one point:
- Congestive heart failure, based on history or examination
- Myocardial infarction, symptomatic ischemic heart disease, or a positive stress test
- Renal insufficiency (ie, serum creatinine level > 2 mg/dL)
- History of stroke or transient ischemic attack
- Diabetes requiring insulin
- High-risk surgery (defined as intrathoracic, intra-abdominal, or suprainguinal vascular surgery).
Patients with three or more points are considered to be at high risk, and those with one or two points are considered to be at intermediate risk. The ACC/AHA 2007 guidelines6 use a modified version of this index that considers the issue of surgical risk separately from the other five clinical conditions.
Devereaux et al33 performed a meta-analysis, published in 2005, of 22 studies of perioperative beta-blockade. They concluded that beta-blockers had no discernable benefit in any outcome measured, including deaths from any cause, deaths from cardiovascular causes, other cardiac events, hypotension, bradycardia, and bronchospasm. However, they based this conclusion on the use of a 99% confidence interval for each relative risk, which they believed was justified because the trials were small and the numbers of events were only moderate. When the outcomes are assessed using the more common 95% confidence interval, benefit was detected in the combined end point of cardiovascular death, nonfatal myocardial infarction, and nonfatal cardiac arrest.
Yang et al,34 Brady et al,35 and Juul et al36 performed three subsequent randomized trials that added to the controversy. Most of the patients in these trials were at intermediate or low risk, and none of the trials found a significant benefit with perioperative beta-blocker use. However, the protocols in these studies were different from the one in the study by Poldermans et al,3 which had found perioperative beta-blockade to be beneficial. Whereas patients in that earlier study started taking a beta-blocker at least 1 week before surgery (and on average much earlier), had their dose aggressively titrated to a target heart rate, and continued taking it for 30 days afterward, the protocols in the later trials called for the drug to be started within 24 hours before surgery and continued for only a short time afterward.
Lindenauer et al,37 in a retrospective study published in 2005, found that fewer surgical patients who received beta-blockers in the hospital died in the hospital. The researchers used an administrative database of more than 780,000 patients who underwent noncardiac surgery, and they used propensity-score matching to compare the postoperative mortality rates of patients who received beta-blockers and a matched group in the same large cohort who did not. Beta-blockers were associated with a lower morality rate in patients in whom the Revised Cardiac Risk Index score was 3 or greater. However, although there was a trend toward a lower rate with beta-blocker use in patients whose score was 2 (ie, at intermediate risk), the difference was not statistically significant, and patients with a score of 0 or 1 saw no benefit and were possibly harmed.
The authors admitted that their study had a number of limitations, including a retrospective design and the use of an administrative database for information regarding risk index conditions and comorbidities. In addition, because they assumed that any patient who received a beta-blocker on the first 2 hospital days was receiving appropriate perioperative treatment, they may have incorrectly estimated the number of patients who actually received these drugs as a risk-reduction strategy. For instance, some patients at low risk could have received beta-blockers for treatment of a specific event, which would be reflected as an increase in event rates for this group. They also had no data on what medications the patients received before they were hospitalized or whether the dose was titrated effectively. The study excluded all patients with congestive heart failure and chronic obstructive pulmonary disease, who may be candidates for beta-blockers in actual practice. In fact, a recent observational study in patients with severe left ventricular dysfunction suggested that these drugs substantially reduced the incidence of death in the short term and the long term.38 Finally, half the surgeries were nonelective, which makes extrapolation of their risk profile by the Revised Cardiac Risk Index difficult, since Lee et al excluded patients undergoing emergency surgery from the cohorts from which they derived and validated their index criteria.
Nevertheless, the authors concluded that patients at intermediate risk derive no benefit from perioperative beta-blocker use, and that the odds ratio for death was actually higher in patients with no risk factors who received a beta-blocker.
DOES PERIOPERATIVE BETA-BLOCKER USE CAUSE HARM?
The published data on whether perioperative beta-blocker use harms patients are conflicting and up to now have been limited.
Stone et al39 reported a substantial incidence of bradycardia requiring atropine in patients treated with a single dose of a beta-blocker preoperatively, but the complications were not clearly characterized.
The Perioperative Beta-Blockade trial.35 Significantly more patients given short-acting metoprolol had intraoperative falls in blood pressure and heart rate, and more required inotropic support during surgery, although the treating anesthesiologists refused to be blinded in that study. (Short-acting metoprolol is available in the United States as Lopressor and generically.)
Devereaux et al,33 in their meta-analysis, found a higher risk of bradycardia requiring treatment (but not a higher risk of hypotension) in beta-blocker users in nine studies, including the study by Stone et al and the Perioperative Beta-Blockade trial (relative risk 2.27, 95% confidence interval 1.36–3.80).
Conversely, at least three other studies found no difference in rates of intraoperative events.36,40,41 There are few data on the incidence of other complications such as perioperative pulmonary edema and bronchospasm.
POISE: THE FIRST LARGE RANDOMIZED TRIAL
In May 2008, results were published from POISE, the first large randomized controlled trial of perioperative beta-blockade.1 An impressive 8,351 patients—most of them at intermediate risk—were randomized to receive extended-release metoprolol succinate or placebo starting just before surgery and continuing for 30 days afterward.
Although the incidence of the primary composite end point (cardiovascular death, nonfatal myocardial infarction, and nonfatal cardiac arrest) was lower at 30 days in the metoprolol group than in the placebo group (5.8% vs 6.9%, hazard ratio 0.83, P = .04), other findings were worrisome: more metoprolol recipients died of any cause (3.1% vs 2.3%, P = .03) or had a stroke (1.0% vs 0.5%, P = .005). The major contributor to the higher mortality rate in this group appears to have been sepsis.
How beta-blockers might promote death by sepsis is unclear. The authors offered two possible explanations: perhaps beta-blocker-induced hypotension predisposes patients to infection and sepsis, or perhaps the slower heart rate and lower force of contraction induced by beta-blockers could mask normal responses to systemic infection, which in turn could delay recognition and treatment or impede the normal immune response. These mechanisms, like others, are speculative.
The risks of other adverse outcomes such as bradycardia and hypotension were substantially higher in the metoprolol group. The authors also pointed out that most of the patients who suffered nonfatal strokes were subsequently disabled or incapacitated, while most of those who suffered nonfatal cardiac events did not progress to further cardiac problems.
This new study has not yet been rigorously debated, but it will likely come under scrutiny for its dosing regimen (extended-release metoprolol succinate 100 mg or placebo 2–4 hours before surgery; another 100 mg or placebo 6 hours after surgery or sooner if the heart rate was 80 beats per minute or more and the systolic blood pressure 100 mm Hg or higher; and then 200 mg or placebo 12 hours after the second dose and every 24 hours thereafter for 30 days). This was fairly aggressive, especially for patients who have never received a beta-blocker before. In contrast, the protocol for the Perioperative Beta-Blockade trial called for only 25 to 50 mg of short-acting metoprolol twice a day. Another criticism is that the medication was started only a few hours before surgery, although there is no current standard practice for either the dose or when the treatment should be started. The population had a fairly high rate of cerebrovascular disease (perhaps predisposing to stroke whenever blood pressure dropped), and 10% of patients were undergoing urgent or emergency surgery, which carries a higher risk of morbidity.
ANY ROLE FOR BETA-BLOCKERS IN THOSE AT INTERMEDIATE RISK?
Thus, in the past decade, the appropriate perioperative use of beta-blockers, which, after the findings by Mangano et al and Poldermans et al, were seen as potentially beneficial for any patient at risk of coronary disease, with little suggestion of harm, has become more clearly defined, and the risks are more evident. The most compelling evidence in favor of using them comes from patients with ischemic heart disease undergoing vascular surgery; the 2007 ACC/AHA guidelines recommend that this group be offered beta-blockers in the absence of a contraindication (class I recommendation: benefit clearly outweighs risk).6 The guidelines also point out that these drugs should be continued in patients already taking them for cardiac indications before surgery, because ischemia may be precipitated if a beta-blocker is abruptly discontinued.42,43
Additionally, the guidelines recommend considering beta-blockers for vascular surgery patients at high cardiac risk (with a Revised Cardiac Risk Index score of 3 or more), even if they are not known to have ischemic heart disease. This is a class IIa recommendation (the benefit outweighs the risk, but more studies are required).
The guidelines also recommend that beta-blockers be considered for patients who have a score of 0 if they are undergoing vascular surgery (class IIb recommendation) or a score of 1 if they are undergoing vascular surgery (class IIa recommendation) or intermediate-risk surgery (class IIb recommendation). However, in view of the POISE results, these recommendations need to be carefully scrutinized.
These data notwithstanding, beta-blockers still might be beneficial in perioperative patients at intermediate risk.
Start beta-blockers sooner?
To help patients at intermediate risk (such as those with diabetes without known heart disease), we may need to do what Poldermans et al did3: instead of seeing patients only once a day or two before surgery, we may need to do the preoperative assessment as much as a month before and, if necessary, start a beta-blocker at a low dose, titrate it to a goal heart rate, and follow the patient closely up until surgery and afterward.
The importance of heart-rate control was illustrated in a recent cohort study of perioperative beta-blockers in vascular surgery patients,44 in which higher beta-blocker doses, carefully monitored, were associated with less ischemia and cardiac enzyme release. In addition, long-term mortality rates were lower in patients with lower heart rates. And Poldermans et al45 recently performed a study in more than 700 intermediate-risk patients who were divided into two groups, one that underwent preoperative stress testing and one that did not. Beta-blockers were given to both groups, and doses were titrated to a goal heart rate of less than 65. The patients with optimally controlled heart rates had the lowest event rates.
However, the logistics of such a program would be challenging. For the most part, internists and hospitalists involved in perioperative assessment do not control the timing of referral or surgery, and adjustments cannot be made for patients whose preoperative clinic visit falls only a few days before surgery. Instituting a second or third visit to assess the efficacy of beta-blockade burdens the patient and may not be practical.
Are all beta-blockers equivalent?
An additional factor is the choice of agent. While the most significant studies of perioperative beta-blockade have used beta-1 receptor-selective agents (ie, metoprolol, atenolol, and bisoprolol), there is no prospective evidence that any particular agent is superior. However, a recent retrospective analysis of elderly surgical patients did suggest that longer-acting beta-blockers may be preferable: patients who had been on atenolol in the year before surgery had a 20% lower risk of postoperative myocardial infarction or death than those who had been on short-acting metoprolol, with no difference in noncardiac outcomes.46
- POISE Study Group. Effect of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet 2008; published online May 13. DOI: 10.1016/S0140-6736(08)60601-7.
- Mangano DT, Layug EL, Wallace A, Tateo I. Effect of atenolol on mortality and cardiovascular morbidity after noncardiac surgery. Multicenter Study of Perioperative Ischemia Research Group. N Engl J Med 1996; 335:1713–1720.
- Poldermans D, Boersma E, Bax JJ, et al. The effect of bisoprolol on perioperative mortality and myocardial infarction in high-risk patients undergoing vascular surgery. Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group. N Engl J Med 1999; 341:1789–1794.
- Shojania KG, Duncan BW, McDonald KM, Wachter RM, Markowitz AJ. Making health care safer: a critical analysis of patient safety practices. Evid Rep Technol Assess (Summ) 2001; ( 43):i–x,1–668.
- National Quality Forum. Safe Practices for Better Healthcare—2006 update. Washington, DC: National Quality Forum, 2006.
- Fleisher LA, Beckman JA, Brown KA, et al. ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery). Circulation 2007; 116:1971–1996.
- Mangano DT. Perioperative cardiac morbidity. Anesthesiology 1990; 72:153–184.
- Lee TH, Marcantonio ER, Mangione CM, et al. Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery. Circulation 1999; 100:1043–1049.
- Devereaux PJ, Goldman L, Cook DJ, Gilbert K, Leslie K, Guyatt GH. Perioperative cardiac events in patients undergoing noncardiac surgery: a review of the magnitude of the problem, the pathophysiology of the events and methods to estimate and communicate risk. Can Med Assoc J 2005; 173:627–634.
- Fleischmann KE, Goldman L, Young B, Lee TH. Association between cardiac and noncardiac complications in patients undergoing noncardiac surgery: outcomes and effects on length of stay. Am J Med 2003; 115:515–520.
- Landesberg G, Luria MH, Cotev S, et al. Importance of long-duration postoperative ST-segment depression in cardiac morbidity after vascular surgery. Lancet 1993; 341:715–719.
- Mangano DT, Browner WS, Hollenberg M, London MJ, Tubau JF, Tateo IM. Association of perioperative myocardial ischemia with cardiac morbidity and mortality in men undergoing noncardiac surgery. The Study of Perioperative Ischemia Research Group. N Engl J Med 1990; 323:1781–1788.
- Raby KE, Goldman L, Creager MA, et al. Correlation between preoperative ischemia and major cardiac events after peripheral vascular surgery. N Engl J Med 1989; 321:1296–1300.
- Landesberg G, Mosseri M, Zahger D, et al. Myocardial infarction after vascular surgery: the role of prolonged stress-induced, ST depression-type ischemia. J Am Coll Cardiol 2001; 37:1839–1845.
- Mangano DT, Browner WS, Hollenberg M, Li J, Tateo IM. Long-term cardiac prognosis following noncardiac surgery. The Study of Perioperative Ischemia Research Group. JAMA 1992; 268:233–239.
- Kim LJ, Martinez EA, Faraday N, et al. Cardiac troponin I predicts short-term mortality in vascular surgery patients. Circulation 2002; 106:2366–2371.
- Landesberg G, Shatz V, Akopnik I, et al. Association of cardiac troponin, CK-MB, and postoperative myocardial ischemia with long-term survival after major vascular surgery. J Am Coll Cardiol 2003; 42:1547–1554.
- Badner NH, Knill RL, Brown JE, Novick TV, Gelb AW. Myocardial infarction after noncardiac surgery. Anesthesiology 1998; 88:572–578.
- Priebe HJ. Triggers of perioperative myocardial ischaemia and infarction. Br J Anaesth 2004; 93:9–20.
- Zaugg M, Schaub MC, Foex P. Myocardial injury and its prevention in the perioperative setting. Br J Anaesth 2004; 93:21–33.
- Dawood MM, Gutpa DK, Southern J, Walia A, Atkinson JB, Eagle KA. Pathology of fatal perioperative myocardial infarction: implications regarding pathophysiology and prevention. Int J Cardiol 1996; 57:37–44.
- Cohen MC, Aretz TH. Histological analysis of coronary artery lesions in fatal postoperative myocardial infarction. Cardiovasc Pathol 1999; 8:133–139.
- London MJ, Zaugg M, Schaub MC, Spahn DR. Perioperative beta-adrenergic receptor blockade: physiologic foundations and clinical controversies. Anesthesiology 2004; 100:170–175.
- Yeager MP, Fillinger MP, Hettleman BD, Hartman GS. Perioperative beta-blockade and late cardiac outcomes: a complementary hypothesis. J Cardiothorac Vasc Anesth 2005; 19:237–241.
- Zaugg M, Schaub MC, Pasch T, Spahn DR. Modulation of beta-adrenergic receptor subtype activities in perioperative medicine: mechanisms and sites of action. Br J Anaesth 2002; 88:101–123.
- Devereaux PJ, Yusuf S, Yang H, Choi PT, Guyatt GH. Are the recommendations to use perioperative beta-blocker therapy in patients undergoing non-cardiac surgery based on reliable evidence? Can Med Assoc J 2004; 171:245–247.
- Eagle KA, Froehlich JB. Reducing cardiovascular risk in patients undergoing noncardiac surgery. N Engl J Med 1996; 335:1761–1763.
- Boersma E, Poldermans D, Bax JJ, et al. Predictors of cardiac events after major vascular surgery: role of clinical characteristics, dobutamine echocardiography, and beta-blocker therapy. JAMA 2001; 285:1865–1873.
- Stevens RD, Burri H, Tramer MR. Pharmacologic myocardial protection in patients undergoing noncardiac surgery: a quantitative systematic review. Anesth Analg 2003; 97:623–633.
- Goldman L. Assessing and reducing cardiac risks of noncardiac surgery. Am J Med 2001; 110:320–323.
- Wesorick DH, Eagle KA. The preoperative cardiovascular evaluation of the intermediate-risk patient: new data, changing strategies. Am J Med 2005; 118:1413.
- Auerbach AD, Goldman L. Beta-blockers and reduction of cardiac events in noncardiac surgery: scientific review. JAMA 2002; 287:1435–1444.
- Devereaux PJ, Beattie WS, Choi PT, et al. How strong is the evidence for the use of perioperative beta blockers in non-cardiac surgery? Systematic review and meta–analysis of randomized controlled trials. BMJ 2005; 331:313–321.
- Yang H, Raymer K, Butler R, Parlow JL, Roberts RS. Metoprolol after vascular surgery (MaVS). Can J Anaesth 2004; 51( suppl 1):A7.
- Brady AR, Gibbs JS, Greenhalgh RM, Powell JT, Sydes MR. Perioperative beta-blockade (POBBLE) for patients undergoing infrarenal vascular surgery: results of a randomized double-blind controlled trial. J Vasc Surg 2005; 41:602–609.
- Juul AB, Wetterslev J, Gluud C, et al. Effect of perioperative βblockade in patients with diabetes undergoing major non-cardiac surgery: randomised placebo controlled, blinded multicentre trial. BMJ 2006; 332:1482.
- Lindenauer PK, Pekow P, Wang K, Mamidi DK, Gutierrez B, Benjamin EM. Perioperative beta-blocker therapy and mortality after major noncardiac surgery. N Engl J Med 2005; 353:349–361.
- Feringa HH, Bax JJ, Schouten O, et al. Beta-blockers improve in-hospital and long-term survival in patients with severe left ventricular dysfunction undergoing major vascular surgery. Eur J Vasc Endovasc Surg 2005; 31:351–358.
- Stone JG, Foex P, Sear JW, Johnson LL, Khambatta HJ, Triner L. Myocardial ischemia in untreated hypertensive patients: effect of a single small oral dose of a beta-adrenergic blocking agent. Anesthesiology 1988; 68:495–500.
- Zaugg M, Tagliente T, Lucchinetti E, et al. Beneficial effects from beta-adrenergic blockade in elderly patients undergoing noncardiac surgery. Anesthesiology 1999; 91:1674–1686.
- Wallace A, Layug B, Tateo I, et al. Prophylactic atenolol reduces postoperative myocardial ischemia. McSPI Research Group. Anesthesiology 1998; 88:7–17.
- Psaty BM, Koepsell TD, Wagner EH, LoGerfo JP, Inui TS. The relative risk of incident coronary heart disease associated with recently stopping the use of beta-blockers. JAMA 1990; 263:1653–1657.
- Shammash JB, Trost JC, Gold JM, Berlin JA, Golden MA, Kimmel SE. Perioperative beta-blocker withdrawal and mortality in vascular surgical patients. Am Heart J 2001; 141:148–153.
- Feringa HHH, Bax JJ, Boersma E, et al. High-dose beta-blockers and tight heart rate control reduce myocardial ischemia and troponin T release in vascular surgery patients. Circulation 2006; 114( suppl 1):I-344–I-349.
- Poldermans D, Bax JJ, Schouten O, et al. Should major vascular surgery be delayed because of preoperative cardiac testing in intermediate-risk patients receiving beta-blocker therapy with tight heart rate control? J Am Coll Cardiol 2006; 48:964–969.
- Redelmeier D, Scales D, Kopp A. Beta blockers for elective surgery in elderly patients: population based, retrospective cohort study. BMJ 2005; 331:932.
- POISE Study Group. Effect of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet 2008; published online May 13. DOI: 10.1016/S0140-6736(08)60601-7.
- Mangano DT, Layug EL, Wallace A, Tateo I. Effect of atenolol on mortality and cardiovascular morbidity after noncardiac surgery. Multicenter Study of Perioperative Ischemia Research Group. N Engl J Med 1996; 335:1713–1720.
- Poldermans D, Boersma E, Bax JJ, et al. The effect of bisoprolol on perioperative mortality and myocardial infarction in high-risk patients undergoing vascular surgery. Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group. N Engl J Med 1999; 341:1789–1794.
- Shojania KG, Duncan BW, McDonald KM, Wachter RM, Markowitz AJ. Making health care safer: a critical analysis of patient safety practices. Evid Rep Technol Assess (Summ) 2001; ( 43):i–x,1–668.
- National Quality Forum. Safe Practices for Better Healthcare—2006 update. Washington, DC: National Quality Forum, 2006.
- Fleisher LA, Beckman JA, Brown KA, et al. ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery). Circulation 2007; 116:1971–1996.
- Mangano DT. Perioperative cardiac morbidity. Anesthesiology 1990; 72:153–184.
- Lee TH, Marcantonio ER, Mangione CM, et al. Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery. Circulation 1999; 100:1043–1049.
- Devereaux PJ, Goldman L, Cook DJ, Gilbert K, Leslie K, Guyatt GH. Perioperative cardiac events in patients undergoing noncardiac surgery: a review of the magnitude of the problem, the pathophysiology of the events and methods to estimate and communicate risk. Can Med Assoc J 2005; 173:627–634.
- Fleischmann KE, Goldman L, Young B, Lee TH. Association between cardiac and noncardiac complications in patients undergoing noncardiac surgery: outcomes and effects on length of stay. Am J Med 2003; 115:515–520.
- Landesberg G, Luria MH, Cotev S, et al. Importance of long-duration postoperative ST-segment depression in cardiac morbidity after vascular surgery. Lancet 1993; 341:715–719.
- Mangano DT, Browner WS, Hollenberg M, London MJ, Tubau JF, Tateo IM. Association of perioperative myocardial ischemia with cardiac morbidity and mortality in men undergoing noncardiac surgery. The Study of Perioperative Ischemia Research Group. N Engl J Med 1990; 323:1781–1788.
- Raby KE, Goldman L, Creager MA, et al. Correlation between preoperative ischemia and major cardiac events after peripheral vascular surgery. N Engl J Med 1989; 321:1296–1300.
- Landesberg G, Mosseri M, Zahger D, et al. Myocardial infarction after vascular surgery: the role of prolonged stress-induced, ST depression-type ischemia. J Am Coll Cardiol 2001; 37:1839–1845.
- Mangano DT, Browner WS, Hollenberg M, Li J, Tateo IM. Long-term cardiac prognosis following noncardiac surgery. The Study of Perioperative Ischemia Research Group. JAMA 1992; 268:233–239.
- Kim LJ, Martinez EA, Faraday N, et al. Cardiac troponin I predicts short-term mortality in vascular surgery patients. Circulation 2002; 106:2366–2371.
- Landesberg G, Shatz V, Akopnik I, et al. Association of cardiac troponin, CK-MB, and postoperative myocardial ischemia with long-term survival after major vascular surgery. J Am Coll Cardiol 2003; 42:1547–1554.
- Badner NH, Knill RL, Brown JE, Novick TV, Gelb AW. Myocardial infarction after noncardiac surgery. Anesthesiology 1998; 88:572–578.
- Priebe HJ. Triggers of perioperative myocardial ischaemia and infarction. Br J Anaesth 2004; 93:9–20.
- Zaugg M, Schaub MC, Foex P. Myocardial injury and its prevention in the perioperative setting. Br J Anaesth 2004; 93:21–33.
- Dawood MM, Gutpa DK, Southern J, Walia A, Atkinson JB, Eagle KA. Pathology of fatal perioperative myocardial infarction: implications regarding pathophysiology and prevention. Int J Cardiol 1996; 57:37–44.
- Cohen MC, Aretz TH. Histological analysis of coronary artery lesions in fatal postoperative myocardial infarction. Cardiovasc Pathol 1999; 8:133–139.
- London MJ, Zaugg M, Schaub MC, Spahn DR. Perioperative beta-adrenergic receptor blockade: physiologic foundations and clinical controversies. Anesthesiology 2004; 100:170–175.
- Yeager MP, Fillinger MP, Hettleman BD, Hartman GS. Perioperative beta-blockade and late cardiac outcomes: a complementary hypothesis. J Cardiothorac Vasc Anesth 2005; 19:237–241.
- Zaugg M, Schaub MC, Pasch T, Spahn DR. Modulation of beta-adrenergic receptor subtype activities in perioperative medicine: mechanisms and sites of action. Br J Anaesth 2002; 88:101–123.
- Devereaux PJ, Yusuf S, Yang H, Choi PT, Guyatt GH. Are the recommendations to use perioperative beta-blocker therapy in patients undergoing non-cardiac surgery based on reliable evidence? Can Med Assoc J 2004; 171:245–247.
- Eagle KA, Froehlich JB. Reducing cardiovascular risk in patients undergoing noncardiac surgery. N Engl J Med 1996; 335:1761–1763.
- Boersma E, Poldermans D, Bax JJ, et al. Predictors of cardiac events after major vascular surgery: role of clinical characteristics, dobutamine echocardiography, and beta-blocker therapy. JAMA 2001; 285:1865–1873.
- Stevens RD, Burri H, Tramer MR. Pharmacologic myocardial protection in patients undergoing noncardiac surgery: a quantitative systematic review. Anesth Analg 2003; 97:623–633.
- Goldman L. Assessing and reducing cardiac risks of noncardiac surgery. Am J Med 2001; 110:320–323.
- Wesorick DH, Eagle KA. The preoperative cardiovascular evaluation of the intermediate-risk patient: new data, changing strategies. Am J Med 2005; 118:1413.
- Auerbach AD, Goldman L. Beta-blockers and reduction of cardiac events in noncardiac surgery: scientific review. JAMA 2002; 287:1435–1444.
- Devereaux PJ, Beattie WS, Choi PT, et al. How strong is the evidence for the use of perioperative beta blockers in non-cardiac surgery? Systematic review and meta–analysis of randomized controlled trials. BMJ 2005; 331:313–321.
- Yang H, Raymer K, Butler R, Parlow JL, Roberts RS. Metoprolol after vascular surgery (MaVS). Can J Anaesth 2004; 51( suppl 1):A7.
- Brady AR, Gibbs JS, Greenhalgh RM, Powell JT, Sydes MR. Perioperative beta-blockade (POBBLE) for patients undergoing infrarenal vascular surgery: results of a randomized double-blind controlled trial. J Vasc Surg 2005; 41:602–609.
- Juul AB, Wetterslev J, Gluud C, et al. Effect of perioperative βblockade in patients with diabetes undergoing major non-cardiac surgery: randomised placebo controlled, blinded multicentre trial. BMJ 2006; 332:1482.
- Lindenauer PK, Pekow P, Wang K, Mamidi DK, Gutierrez B, Benjamin EM. Perioperative beta-blocker therapy and mortality after major noncardiac surgery. N Engl J Med 2005; 353:349–361.
- Feringa HH, Bax JJ, Schouten O, et al. Beta-blockers improve in-hospital and long-term survival in patients with severe left ventricular dysfunction undergoing major vascular surgery. Eur J Vasc Endovasc Surg 2005; 31:351–358.
- Stone JG, Foex P, Sear JW, Johnson LL, Khambatta HJ, Triner L. Myocardial ischemia in untreated hypertensive patients: effect of a single small oral dose of a beta-adrenergic blocking agent. Anesthesiology 1988; 68:495–500.
- Zaugg M, Tagliente T, Lucchinetti E, et al. Beneficial effects from beta-adrenergic blockade in elderly patients undergoing noncardiac surgery. Anesthesiology 1999; 91:1674–1686.
- Wallace A, Layug B, Tateo I, et al. Prophylactic atenolol reduces postoperative myocardial ischemia. McSPI Research Group. Anesthesiology 1998; 88:7–17.
- Psaty BM, Koepsell TD, Wagner EH, LoGerfo JP, Inui TS. The relative risk of incident coronary heart disease associated with recently stopping the use of beta-blockers. JAMA 1990; 263:1653–1657.
- Shammash JB, Trost JC, Gold JM, Berlin JA, Golden MA, Kimmel SE. Perioperative beta-blocker withdrawal and mortality in vascular surgical patients. Am Heart J 2001; 141:148–153.
- Feringa HHH, Bax JJ, Boersma E, et al. High-dose beta-blockers and tight heart rate control reduce myocardial ischemia and troponin T release in vascular surgery patients. Circulation 2006; 114( suppl 1):I-344–I-349.
- Poldermans D, Bax JJ, Schouten O, et al. Should major vascular surgery be delayed because of preoperative cardiac testing in intermediate-risk patients receiving beta-blocker therapy with tight heart rate control? J Am Coll Cardiol 2006; 48:964–969.
- Redelmeier D, Scales D, Kopp A. Beta blockers for elective surgery in elderly patients: population based, retrospective cohort study. BMJ 2005; 331:932.
KEY POINTS
- Beta-blockers reduce perioperative ischemia, but the benefit may be only in high-risk patients undergoing high-risk surgery. Currently, the best evidence supports their use in two groups: patients undergoing vascular surgery who have known ischemic heart disease or multiple risk factors for it, and patients who are already on beta-blockers.
- The Perioperative Ischemic Evaluation (POISE) findings suggest that beta-blockers should be used in the immediate preoperative period only with great caution, after ensuring that the patient is clinically stable and without evidence of infection, hypovolemia, anemia, or other conditions that could make heart-rate titration misleading or use of the drug dangerous.
- When feasible, beta-blockers should be started a month before surgery, titrated to a heart rate of 60 beats per minute, and continued for approximately a month. If the drug is then to be discontinued, it should be tapered slowly.
A drug, a concept, and a clinical trial on trial

Many trials are funded by industry and carried out by clinical investigators in academic and private practice. Drug companies must perform these trials to win approval from the US Food and Drug Administration (FDA) for their new drugs and package inserts, which dictates what they can and can’t say in their advertising. This latter requirement often leads to trials after a drug is approved in an effort to aid drug promotion and improve its position in the marketplace.
The FDA is increasingly demanding that new drug studies use “hard” measures of efficacy and less reliance on surrogate end points. This requires larger, longer, more expensive trials.
A recent trial that relied on surrogate end points was the ENHANCE trial, which evaluated the addition of a second approved cholesterol-lowering drug (ezetimibe) to a statin in a relatively small number of mostly pretreated patients. The surrogates were lipid-lowering and carotid intima-media thickness. At the time the study was designed, I’m sure it seemed obvious that lowering low-density lipoprotein cholesterol (LDL-C) or reducing the measured burden of atherosclerosis would reduce the consequences of hypercholesterolemia, including myocardial infarction and stroke. Therefore, the use of surrogate markers seemed an acceptable expediency.
However, the ENHANCE results hit the national news when the two surrogates didn’t coincide as anticipated. Although ezetimibe/simvastatin (Vytorin) lowered the LDL-C level more than simvastatin alone (Zocor), it did not reduce carotid intima-media thickness.
The response was intense. Trialists, drug safety pundits, industry representatives, politicians, and clinicians all weighed in. Some patients apparently stopped taking their lipid-lowering medications. Without any striking evidence of worse outcome, doubt has been cast on the safety and efficacy of the drug and—perhaps inappropriately—on the entire LDL-C hypothesis of atherosclerosis.
In this issue, Dr. Michael Davidson and Dr. Allen Taylor, two clinical experts in atherosclerosis, present widely divergent views on the conduct, results, and implications of the ENHANCE trial. Dr. Taylor was afforded the opportunity to read Dr. Davidson’s manuscript before writing his own editorial. I’m not sure their discussion will settle this intense debate, but they outline the issues clearly.
Many trials are funded by industry and carried out by clinical investigators in academic and private practice. Drug companies must perform these trials to win approval from the US Food and Drug Administration (FDA) for their new drugs and package inserts, which dictates what they can and can’t say in their advertising. This latter requirement often leads to trials after a drug is approved in an effort to aid drug promotion and improve its position in the marketplace.
The FDA is increasingly demanding that new drug studies use “hard” measures of efficacy and less reliance on surrogate end points. This requires larger, longer, more expensive trials.
A recent trial that relied on surrogate end points was the ENHANCE trial, which evaluated the addition of a second approved cholesterol-lowering drug (ezetimibe) to a statin in a relatively small number of mostly pretreated patients. The surrogates were lipid-lowering and carotid intima-media thickness. At the time the study was designed, I’m sure it seemed obvious that lowering low-density lipoprotein cholesterol (LDL-C) or reducing the measured burden of atherosclerosis would reduce the consequences of hypercholesterolemia, including myocardial infarction and stroke. Therefore, the use of surrogate markers seemed an acceptable expediency.
However, the ENHANCE results hit the national news when the two surrogates didn’t coincide as anticipated. Although ezetimibe/simvastatin (Vytorin) lowered the LDL-C level more than simvastatin alone (Zocor), it did not reduce carotid intima-media thickness.
The response was intense. Trialists, drug safety pundits, industry representatives, politicians, and clinicians all weighed in. Some patients apparently stopped taking their lipid-lowering medications. Without any striking evidence of worse outcome, doubt has been cast on the safety and efficacy of the drug and—perhaps inappropriately—on the entire LDL-C hypothesis of atherosclerosis.
In this issue, Dr. Michael Davidson and Dr. Allen Taylor, two clinical experts in atherosclerosis, present widely divergent views on the conduct, results, and implications of the ENHANCE trial. Dr. Taylor was afforded the opportunity to read Dr. Davidson’s manuscript before writing his own editorial. I’m not sure their discussion will settle this intense debate, but they outline the issues clearly.
Many trials are funded by industry and carried out by clinical investigators in academic and private practice. Drug companies must perform these trials to win approval from the US Food and Drug Administration (FDA) for their new drugs and package inserts, which dictates what they can and can’t say in their advertising. This latter requirement often leads to trials after a drug is approved in an effort to aid drug promotion and improve its position in the marketplace.
The FDA is increasingly demanding that new drug studies use “hard” measures of efficacy and less reliance on surrogate end points. This requires larger, longer, more expensive trials.
A recent trial that relied on surrogate end points was the ENHANCE trial, which evaluated the addition of a second approved cholesterol-lowering drug (ezetimibe) to a statin in a relatively small number of mostly pretreated patients. The surrogates were lipid-lowering and carotid intima-media thickness. At the time the study was designed, I’m sure it seemed obvious that lowering low-density lipoprotein cholesterol (LDL-C) or reducing the measured burden of atherosclerosis would reduce the consequences of hypercholesterolemia, including myocardial infarction and stroke. Therefore, the use of surrogate markers seemed an acceptable expediency.
However, the ENHANCE results hit the national news when the two surrogates didn’t coincide as anticipated. Although ezetimibe/simvastatin (Vytorin) lowered the LDL-C level more than simvastatin alone (Zocor), it did not reduce carotid intima-media thickness.
The response was intense. Trialists, drug safety pundits, industry representatives, politicians, and clinicians all weighed in. Some patients apparently stopped taking their lipid-lowering medications. Without any striking evidence of worse outcome, doubt has been cast on the safety and efficacy of the drug and—perhaps inappropriately—on the entire LDL-C hypothesis of atherosclerosis.
In this issue, Dr. Michael Davidson and Dr. Allen Taylor, two clinical experts in atherosclerosis, present widely divergent views on the conduct, results, and implications of the ENHANCE trial. Dr. Taylor was afforded the opportunity to read Dr. Davidson’s manuscript before writing his own editorial. I’m not sure their discussion will settle this intense debate, but they outline the issues clearly.
Given the ENHANCE trial results, ezetimibe is still unproven
Ezetimibe (Zetia) was licensed by the US Food and Drug Administration in 2002 on the basis of its ability to reduce low-density lipoprotein cholesterol (LDL-C) levels. The reductions are mild, approximately 15%,1 which is comparable to the effects of a stringent diet and exercise or of a statin in titrated doses.
However, there was no evidence that ezetimbe, which has a unique mechanism of action, delivers a benefit in terms of clinical outcomes. Despite this, the use of ezetimibe (alone or in fixed-dose combination with simvastatin, a preparation sold as Vytorin) grew rapidly, generating annual sales of $5.2 billion. Clinicians and the manufacturer (Merck/Schering-Plough) broadly assumed that LDL-C reduction would carry ezetimibe’s day as clinical trials emerged.
The assumption seemed reasonable, since evidence from the past 3 decades has established a clear link between lowering LDL-C levels via diverse mechanisms and positive clinical outcomes, particularly lower rates of cardiovascular disease and death. Indeed, LDL-C measurement is now a focus of cardiovascular risk assessment and management, as reflected in national treatment guidelines.
THE ENHANCE TRIAL: EZETIMIBE FAILS A KEY TEST
Unexpectedly, ezetimibe failed its first step in clinical trial validation, the Ezetimibe and Simvastatin in Hypercholesterolemia Enhances Atherosclerosis Regression (ENHANCE) trial.2 Apart from the scientifically irrelevant political regulatory intrigue generated by the sponsor’s conduct in this trial, ENHANCE’s findings challenge us to confront issues of what we assume vs what we really know, and how to interpret the complex results of clinical trials.
To be fair to the trial’s investigators, ENHANCE achieved its objective of enrolling a population with a very high LDL-C level, which is ezetimibe’s target and has been widely used in the study of atherosclerosis progression as a marker of potential drug benefit. Nevertheless, and even though the LDL-C level 2 years later was 52 mg/dL lower in the group receiving ezetimibe/simvastatin than in the group receiving simvastatin alone (Zocor), at LDL-C levels that are typically associated with atherosclerosis progression (140–190 mg/dL), ezetimibe failed to reduce the progression of atherosclerosis.
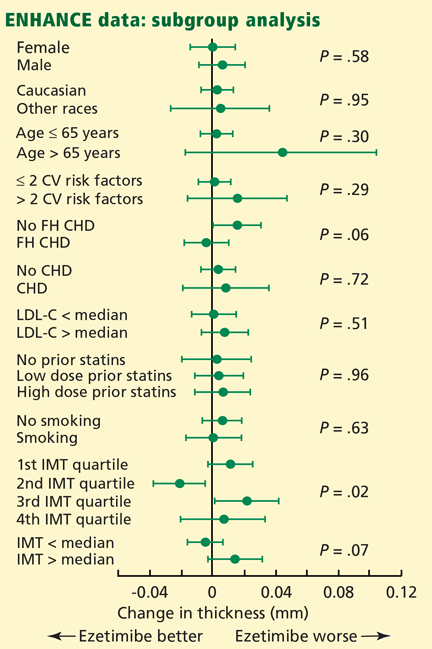
These trends are particularly worrisome, given that the ezetimibe/simvastatin group achieved a greater reduction in C-reactive protein levels, which typically has resulted in superior outcomes in atherosclerosis3 and clinical effects4 in combination with LDL-C reduction.
In view of these findings, should clinicians stand firm and continue to use ezetimibe? Or should we reevaluate our position and await more data about this unique, first-in-class compound?
WISHFUL POST HOC HYPOTHESES
In this issue of the Cleveland Clinic Journal of Medicine, Dr. Michael Davidson,5 a respected lipid expert but one invested in ezetimibe’s development, assures us that all is in order and that the results of ENHANCE can be explained away by several arguments, most notably that most of the trial’s participants had previously received lipid-lowering treatment, which obscured the effects of ezetimibe. Moreover, he argues that ezetimibe’s mechanism of action is well understood and that the drug is safe and well tolerated and thus should remain a first-line treatment for hyperlipidemia.
These arguments may eventually prove to be correct, but as of now they are merely wishful post hoc hypotheses awaiting more data apart from ENHANCE. Negative clinical trials do occur as a matter of chance, but we should be cautious in any attempts to explain away a trial that was designed, executed, and reported as conceived simply because the results do not match our expectations.
Confronted with ENHANCE, the astute clinician should ask three questions: Do we really understand ezetimibe’s mechanism of action? Do other lines of evidence indicate the drug is beneficial? And how reliable is the arterial thickness as a surrogate end point?
DO WE UNDERSTAND EZETIMIBE’S MECHANISM OF ACTION?
Do we understand ezetimibe’s full mechanism of action? Not really.
True, ezetimibe inhibits cholesterol transport, a process that is integral both to cholesterol’s enteric absorption and to its systemic clearance. But although Dr. Davidson asserts that ezetimibe has cellular effects similar to those of statins, in fact it has the opposite effect on HMG-coA reductase, and no effects on LDL receptors.6
Furthermore, although initial studies suggested that ezetimibe inhibits enteric cholesterol absorption by inhibiting the Niemann-Pick C1L1 (NPC1L1) receptor, more recent investigations call this into serious question and point more definitively at a receptor known as scavenger receptor-B1 (SR-B1). As stated in a recent editorial, “SR-B1 in the apical site of enterocytes is the primary high-affinity site of cholesterol uptake and ezetimibe can inhibit this process. Moreover, the [possibility is ruled out] of NPC1L1 being a major player in this cholesterol uptake. This is at variance with the view of the colleagues from Schering-Plough who claim the same for NPC1L1.”7
SR-B1 is also a high-affinity receptor for high-density lipoprotein8 and thus is active in the antiatherosclerotic process of reverse cholesterol transport, inhibition of which significantly accelerates the development of atherosclerosis.9
Additionally, in vitro and thus unrelated to the effects of changing cholesterol concentration, ezetimibe down-regulates SR-B1 and another key cholesterol transporter protein called ABCA1.10 Further, ezetimibe induces down-regulation of raft protein domains, including CD36,11 another effect opposite to that of statins.
These little-recognized effects of ezetimibe are among many that are completely unrelated to enteric cholesterol absorption. Yet, they are likely to be active within the liver and systemically where these proteins reside, and they are putatively proatherosclerotic. Contrary to often-cited opinion, ezetimibe is systemically absorbed, with 11% of the compound excreted in the urine.12 Thus, the compound is systemically available to exert these same actions in the liver and elsewhere. Moreover, the absorbed drug is glucuronidated and is extensively recirculated in the liver in a form (its glucuronide) that is more potent than the parent compound.
In sum, present opinion is that ezetimibe inhibits lipid transport and interacts with a variety of receptors, not only in the gut but also systemically at the cell membrane and also inside the cell, focally disrupting several tightly regulated biologic processes.7 Thus, although ezetimibe reduces serum LDL-C levels via its effect in the gut, this effect may well be offset or even overridden systemically by other, unmeasurable effects, leading to counterintuitive results in terms of atherosclerosis or clinical events.
This would not be the first time a lipid-lowering drug has disappointed us: torcetrapib, another transport inhibitor, dramatically raises serum high-density lipoprotein cholesterol levels and reduces LDL-C but was found not only to have no effect on atherosclerosis, but also to potentiate adverse clinical outcomes.
The net impact of these other actions of ezetimibe is not known. We will discover its true clinical effects only through studies of endothelial function, atherosclerosis, and clinical cardiovascular outcomes. ENHANCE, which looked at atherosclerosis, is thus our strongest signal to date on the net effect of ezetimibe.
DO OTHER LINES OF EVIDENCE INDICATE EZETIMIBE IS BENEFICIAL?
Can we be reassured that ENHANCE’s results are spurious on the basis of other lines of evidence? Again, not really.
Experiments in animals, particularly in mice,13 have shown that ezetimibe may be antiatherosclerotic, although mice are considered the “worst model”7 for the study of ezetimibe, and notably, LDL-C levels were lowered far more in these experiments than they are clinically. Enthusiasm for these animal models should be tempered by interspecies variability in ezetimibe’s “off-target” effects and in the recent failure of other lipid transport drugs in human trials (torcetrapib and ACAT inhibitors) that had shown initial success in animals. No animal model is established for evaluating drugs of ezetimibe’s class, given its complex mechanism of action.
In human studies, the only other surrogate of the net effect of ezetimibe is endothelial function. Among several randomized clinical trials of ezetimibe,14–18 only one was designed to compare the effects of ezetimibe alone, ezetimibe plus a statin, and a statin by itself in titrated or in maximum doses.15 After 4 weeks of therapy, all groups had lower LDL-C levels. However, ezetimibe monotherapy and ezetimibe/simvastatin combination therapy had no detectable effect on the arterial response to acetylcholine, but atorvastatin (Lipitor) monotherapy did. To be fair, the other (very small) trials showed mixed results, thus keeping the hypothesis of ezetimibe’s benefit alive, but with nothing close to a clear signal of benefit.
IS ARTERIAL THICKNESS RELIABLE AS A SURROGATE END POINT?
Was the principal problem in ENHANCE the use of carotid intima-media thickness as the primary end point? No.
This issue has received a lot of attention, much of which I believe is misinformed. No trial end point is infallible, including carotid intima-media thickness, and one must remain open to the possibility of chance findings. However, it has been a relatively reasonable end point in trials of diverse cardiovascular preventive strategies, including lipid-lowering, blood-pressure-lowering, and lifestyle interventions and as a directional biomarker of clinical atherosclerotic events.
We should be cautious about comparing data on carotid intima-media thickness from different trials, as Dr. Davidson attempts to do, in view of methodologic and population differences: each trial must be considered independently. Of greatest concern in ENHANCE is the consistency among intima-media thickness end points, including strong trends toward adverse effects in the most diseased carotid and femoral segments.
Moreover, ENHANCE’s detractors contend that the carotid intima-media thickness of the studied population was normal, citing this as evidence of delipidation from prior treatment. Although not impossible (as shown by the work of Zhao and colleagues in the setting of prolonged, intense lipid-lowering therapy19), at the moment this hypothesis is a matter of conjecture in the ENHANCE participants, particularly because their LDL-C levels were still quite elevated during the trial and conceivably even before randomization.
But these patients were not normal: they were typical patients with familial hypercholesterolemia with extremely elevated LDL-C levels and abnormally thick arteries for their age. Population screening estimates show that, for age and sex, the carotid intima-media thickness values in ENHANCE would lie in the upper quartile of those in the general population.20 Moreover, their mean value is consistent with that in similar-aged groups of patients with familial hypercholesterolemia, even with lower rates of prior statin pretreatment.21
The most convincing evidence for the validity of the ENHANCE findings comes from the published subgroup data (Figure 1). In participants whose baseline carotid intima-media thickness was above the median at baseline, the thickness increased more with ezetimibe/simvastatin than with simvastatin alone. The same was true in the subgroup with above-average LDL-C levels at baseline. The subgroups with no prior statin treatment, low-dose prior statin treatment, and high-dose prior statin showed no heterogeneity of response: their carotid intima-media thickness increased more with ezetimibe/simvastatin than with simvastatin alone. None of these differences was statistically significant; however, these prespecified subgroup data seemingly invalidate arguments against the ENHANCE results based on carotid intima-media thickness findings.
In this context, ENHANCE can only be interpreted as a strong initial negative signal, a “red flag” about ezetimibe’s net health benefits.
WHAT NEXT?
The proper present focus of this debate is not on LDL-C but rather on ezetimibe, its unique mechanism of action, and on the need for more evidence about this complex compound.
At present, ezetimibe’s mechanism of action is not fully understood, and its benefit—for now, only mild LDL-C reduction—is too uncertain for us to be spending $5.2 billion a year for it. Its manufacturer is fortunate that the drug is even licensed, given the current and seemingly appropriate regulatory changes under which drugs introducing new therapeutic classes are scrutinized more closely for benefits and risks. “Safe and well tolerated,” as contended by Dr. Davidson, is not nearly enough: drugs must show clinically important benefits. We still know too little about this drug, the manufacturer of which has invested far more in marketing than in science, a point on which Dr. Davidson and I agree.
In 2008, ezetimibe is an appropriate candidate for testing in clinical trials, and in years to come it may be worthy of clinical attention—if rigorous and objectively conducted clinical trials prove its worth. At present, clinical equipoise dictates that ezetimibe is not an appropriate alternative to a statin in titrated doses, to the addition of other lipid-lowering drugs to a statin, to greater attention to drug adherence, or to lifestyle modification.
For the moment, given the ENHANCE results, the clinical usefulness of ezetimibe still remains to be proven. Much more evidence is needed before we can confidently reembrace the clinical use of ezetimibe.
- Ballantyne CM, Houri J, Notarbartolo A, et al. Effect of ezetimibe coadministered with atorvastatin in 628 patients with primary hypercholesterolemia: a prospective, randomized, double-blind trial. Circulation 2003; 107:2409–2415.
- Kastelein JJ, Akdim F, Stroes ES, et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med 2008; 358:1431–1443.
- Kent SM, Taylor AJ. Usefulness of lowering low-density lipoprotein cholesterol to < 70 mg/dL and usefulness of C-reactive protein in patient selection. Am J Cardiol 2003; 92:1224–1227.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N Engl J Med 2005; 352:29–38.
- Davidson MH. Interpreting the ENHANCE trial. Is ezetimibe/simvastatin no better than simvastatin alone? Leessons learned and clinical implications. Cleve Clin J Med 2008; 75:479–491.
- Gouni-Berthold I, Berthold HK, Gylling H, et al. Effects of ezetimibe and/or simvastatin on LDL receptor protein expression and on LDL receptor and HMG-CoA reductase gene expression: a randomized trial in healthy men. Atherosclerosis 2008; 198:198–207.
- Spener F. Ezetimibe in search of receptor(s)—still a never-ending challenge in cholesterol absorption and transport. Biochim Biophys Acta 2007; 1771:1113–1116.
- Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996; 271:518–520.
- Kitayama K, Nishizawa T, Abe K, et al. Blockade of scavenger receptor class B type I raises high density lipoprotein cholesterol levels but exacerbates atherosclerotic lesion formation in apolipoprotein E deficient mice. J Pharm Pharmacol 2006; 58:1629–1638.
- During A, Dawson HD, Harrison EH. Carotenoid transport is decreased and expression of the lipid transporters SR-BI, NPC1L1, and ABCA1 is downregulated in Caco-2 cells treated with ezetimibe. J Nutr 2005; 135:2305–2312.
- Orso E, Werner T, Wolf Z, Bandulik S, Kramer W, Schmitz G. Ezetimib influences the expression of raft-associated antigens in human monocytes. Cytometry A 2006; 69:206–208.
- Patrick JE, Kosoglou T, Stauber KL, et al. Disposition of the selective cholesterol absorption inhibitor ezetimibe in healthy male subjects. Drug Metab Dispos 2002; 30:430–437.
- Kuhlencordt PJ, Padmapriya P, Rutzel S, et al. Ezetimibe potently reduces vascular inflammation and arteriosclerosis in eNOS-deficient ApoE ko mice. Atherosclerosis 2008; April 6.
- Bulut D, Hanefeld C, Bulut-Streich N, Graf C, Mugge A, Spiecker M. Endothelial function in the forearm circulation of patients with the metabolic syndrome—effect of different lipid-lowering regimens. Cardiology 2005; 104:176–180.
- Fichtlscherer S, Schmidt-Lucke C, Bojunga S, et al. Differential effects of short-term lipid lowering with ezetimibe and statins on endothelial function in patients with CAD: clinical evidence for ‘pleiotropic’ functions of statin therapy. Eur Heart J 2006; 27:1182–1190.
- Landmesser U, Bahlmann F, Mueller M, et al. Simvastatin versus ezetimibe: pleiotropic and lipid-lowering effects on endothelial function in humans. Circulation 2005; 111:2356–2363.
- Maki-Petaja KM, Booth AD, Hall FC, et al. Ezetimibe and simvastatin reduce inflammation, disease activity, and aortic stiffness and improve endothelial function in rheumatoid arthritis. J Am Coll Cardiol 2007; 50:852–858.
- Settergren M, Bohm F, Ryden L, Pernow J. Cholesterol lowering is more important than pleiotropic effects of statins for endothelial function in patients with dysglycaemia and coronary artery disease. Eur Heart J 2008 April 25.
- Zhao XQ, Yuan C, Hatsukami TS, et al. Effects of prolonged intensive lipid-lowering therapy on the characteristics of carotid atherosclerotic plaques in vivo by MRI: a case-control study. Arterioscler Thromb Vasc Biol 2001; 21:1623–1629.
- Stein JH, Korcarz CE, Hurst RT, et al. Use of carotid ultrasound to identify subclinical vascular disease and evaluate cardiovascular disease risk: a consensus statement from the American Society of Echocardiography Carotid Intima-Media Thickness Task Force. Endorsed by the Society for Vascular Medicine. J Am Soc Echocardiogr 2008; 21:93–111.
- Junyent M, Cofan M, Nunez I, Gilabert R, Zambon D, Ros E. Influence of HDL cholesterol on preclinical carotid atherosclerosis in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol 2006; 26:1107–1113.
Ezetimibe (Zetia) was licensed by the US Food and Drug Administration in 2002 on the basis of its ability to reduce low-density lipoprotein cholesterol (LDL-C) levels. The reductions are mild, approximately 15%,1 which is comparable to the effects of a stringent diet and exercise or of a statin in titrated doses.
However, there was no evidence that ezetimbe, which has a unique mechanism of action, delivers a benefit in terms of clinical outcomes. Despite this, the use of ezetimibe (alone or in fixed-dose combination with simvastatin, a preparation sold as Vytorin) grew rapidly, generating annual sales of $5.2 billion. Clinicians and the manufacturer (Merck/Schering-Plough) broadly assumed that LDL-C reduction would carry ezetimibe’s day as clinical trials emerged.
The assumption seemed reasonable, since evidence from the past 3 decades has established a clear link between lowering LDL-C levels via diverse mechanisms and positive clinical outcomes, particularly lower rates of cardiovascular disease and death. Indeed, LDL-C measurement is now a focus of cardiovascular risk assessment and management, as reflected in national treatment guidelines.
THE ENHANCE TRIAL: EZETIMIBE FAILS A KEY TEST
Unexpectedly, ezetimibe failed its first step in clinical trial validation, the Ezetimibe and Simvastatin in Hypercholesterolemia Enhances Atherosclerosis Regression (ENHANCE) trial.2 Apart from the scientifically irrelevant political regulatory intrigue generated by the sponsor’s conduct in this trial, ENHANCE’s findings challenge us to confront issues of what we assume vs what we really know, and how to interpret the complex results of clinical trials.
To be fair to the trial’s investigators, ENHANCE achieved its objective of enrolling a population with a very high LDL-C level, which is ezetimibe’s target and has been widely used in the study of atherosclerosis progression as a marker of potential drug benefit. Nevertheless, and even though the LDL-C level 2 years later was 52 mg/dL lower in the group receiving ezetimibe/simvastatin than in the group receiving simvastatin alone (Zocor), at LDL-C levels that are typically associated with atherosclerosis progression (140–190 mg/dL), ezetimibe failed to reduce the progression of atherosclerosis.
These trends are particularly worrisome, given that the ezetimibe/simvastatin group achieved a greater reduction in C-reactive protein levels, which typically has resulted in superior outcomes in atherosclerosis3 and clinical effects4 in combination with LDL-C reduction.
In view of these findings, should clinicians stand firm and continue to use ezetimibe? Or should we reevaluate our position and await more data about this unique, first-in-class compound?
WISHFUL POST HOC HYPOTHESES
In this issue of the Cleveland Clinic Journal of Medicine, Dr. Michael Davidson,5 a respected lipid expert but one invested in ezetimibe’s development, assures us that all is in order and that the results of ENHANCE can be explained away by several arguments, most notably that most of the trial’s participants had previously received lipid-lowering treatment, which obscured the effects of ezetimibe. Moreover, he argues that ezetimibe’s mechanism of action is well understood and that the drug is safe and well tolerated and thus should remain a first-line treatment for hyperlipidemia.
These arguments may eventually prove to be correct, but as of now they are merely wishful post hoc hypotheses awaiting more data apart from ENHANCE. Negative clinical trials do occur as a matter of chance, but we should be cautious in any attempts to explain away a trial that was designed, executed, and reported as conceived simply because the results do not match our expectations.
Confronted with ENHANCE, the astute clinician should ask three questions: Do we really understand ezetimibe’s mechanism of action? Do other lines of evidence indicate the drug is beneficial? And how reliable is the arterial thickness as a surrogate end point?
DO WE UNDERSTAND EZETIMIBE’S MECHANISM OF ACTION?
Do we understand ezetimibe’s full mechanism of action? Not really.
True, ezetimibe inhibits cholesterol transport, a process that is integral both to cholesterol’s enteric absorption and to its systemic clearance. But although Dr. Davidson asserts that ezetimibe has cellular effects similar to those of statins, in fact it has the opposite effect on HMG-coA reductase, and no effects on LDL receptors.6
Furthermore, although initial studies suggested that ezetimibe inhibits enteric cholesterol absorption by inhibiting the Niemann-Pick C1L1 (NPC1L1) receptor, more recent investigations call this into serious question and point more definitively at a receptor known as scavenger receptor-B1 (SR-B1). As stated in a recent editorial, “SR-B1 in the apical site of enterocytes is the primary high-affinity site of cholesterol uptake and ezetimibe can inhibit this process. Moreover, the [possibility is ruled out] of NPC1L1 being a major player in this cholesterol uptake. This is at variance with the view of the colleagues from Schering-Plough who claim the same for NPC1L1.”7
SR-B1 is also a high-affinity receptor for high-density lipoprotein8 and thus is active in the antiatherosclerotic process of reverse cholesterol transport, inhibition of which significantly accelerates the development of atherosclerosis.9
Additionally, in vitro and thus unrelated to the effects of changing cholesterol concentration, ezetimibe down-regulates SR-B1 and another key cholesterol transporter protein called ABCA1.10 Further, ezetimibe induces down-regulation of raft protein domains, including CD36,11 another effect opposite to that of statins.
These little-recognized effects of ezetimibe are among many that are completely unrelated to enteric cholesterol absorption. Yet, they are likely to be active within the liver and systemically where these proteins reside, and they are putatively proatherosclerotic. Contrary to often-cited opinion, ezetimibe is systemically absorbed, with 11% of the compound excreted in the urine.12 Thus, the compound is systemically available to exert these same actions in the liver and elsewhere. Moreover, the absorbed drug is glucuronidated and is extensively recirculated in the liver in a form (its glucuronide) that is more potent than the parent compound.
In sum, present opinion is that ezetimibe inhibits lipid transport and interacts with a variety of receptors, not only in the gut but also systemically at the cell membrane and also inside the cell, focally disrupting several tightly regulated biologic processes.7 Thus, although ezetimibe reduces serum LDL-C levels via its effect in the gut, this effect may well be offset or even overridden systemically by other, unmeasurable effects, leading to counterintuitive results in terms of atherosclerosis or clinical events.
This would not be the first time a lipid-lowering drug has disappointed us: torcetrapib, another transport inhibitor, dramatically raises serum high-density lipoprotein cholesterol levels and reduces LDL-C but was found not only to have no effect on atherosclerosis, but also to potentiate adverse clinical outcomes.
The net impact of these other actions of ezetimibe is not known. We will discover its true clinical effects only through studies of endothelial function, atherosclerosis, and clinical cardiovascular outcomes. ENHANCE, which looked at atherosclerosis, is thus our strongest signal to date on the net effect of ezetimibe.
DO OTHER LINES OF EVIDENCE INDICATE EZETIMIBE IS BENEFICIAL?
Can we be reassured that ENHANCE’s results are spurious on the basis of other lines of evidence? Again, not really.
Experiments in animals, particularly in mice,13 have shown that ezetimibe may be antiatherosclerotic, although mice are considered the “worst model”7 for the study of ezetimibe, and notably, LDL-C levels were lowered far more in these experiments than they are clinically. Enthusiasm for these animal models should be tempered by interspecies variability in ezetimibe’s “off-target” effects and in the recent failure of other lipid transport drugs in human trials (torcetrapib and ACAT inhibitors) that had shown initial success in animals. No animal model is established for evaluating drugs of ezetimibe’s class, given its complex mechanism of action.
In human studies, the only other surrogate of the net effect of ezetimibe is endothelial function. Among several randomized clinical trials of ezetimibe,14–18 only one was designed to compare the effects of ezetimibe alone, ezetimibe plus a statin, and a statin by itself in titrated or in maximum doses.15 After 4 weeks of therapy, all groups had lower LDL-C levels. However, ezetimibe monotherapy and ezetimibe/simvastatin combination therapy had no detectable effect on the arterial response to acetylcholine, but atorvastatin (Lipitor) monotherapy did. To be fair, the other (very small) trials showed mixed results, thus keeping the hypothesis of ezetimibe’s benefit alive, but with nothing close to a clear signal of benefit.
IS ARTERIAL THICKNESS RELIABLE AS A SURROGATE END POINT?
Was the principal problem in ENHANCE the use of carotid intima-media thickness as the primary end point? No.
This issue has received a lot of attention, much of which I believe is misinformed. No trial end point is infallible, including carotid intima-media thickness, and one must remain open to the possibility of chance findings. However, it has been a relatively reasonable end point in trials of diverse cardiovascular preventive strategies, including lipid-lowering, blood-pressure-lowering, and lifestyle interventions and as a directional biomarker of clinical atherosclerotic events.
We should be cautious about comparing data on carotid intima-media thickness from different trials, as Dr. Davidson attempts to do, in view of methodologic and population differences: each trial must be considered independently. Of greatest concern in ENHANCE is the consistency among intima-media thickness end points, including strong trends toward adverse effects in the most diseased carotid and femoral segments.
Moreover, ENHANCE’s detractors contend that the carotid intima-media thickness of the studied population was normal, citing this as evidence of delipidation from prior treatment. Although not impossible (as shown by the work of Zhao and colleagues in the setting of prolonged, intense lipid-lowering therapy19), at the moment this hypothesis is a matter of conjecture in the ENHANCE participants, particularly because their LDL-C levels were still quite elevated during the trial and conceivably even before randomization.
But these patients were not normal: they were typical patients with familial hypercholesterolemia with extremely elevated LDL-C levels and abnormally thick arteries for their age. Population screening estimates show that, for age and sex, the carotid intima-media thickness values in ENHANCE would lie in the upper quartile of those in the general population.20 Moreover, their mean value is consistent with that in similar-aged groups of patients with familial hypercholesterolemia, even with lower rates of prior statin pretreatment.21
The most convincing evidence for the validity of the ENHANCE findings comes from the published subgroup data (Figure 1). In participants whose baseline carotid intima-media thickness was above the median at baseline, the thickness increased more with ezetimibe/simvastatin than with simvastatin alone. The same was true in the subgroup with above-average LDL-C levels at baseline. The subgroups with no prior statin treatment, low-dose prior statin treatment, and high-dose prior statin showed no heterogeneity of response: their carotid intima-media thickness increased more with ezetimibe/simvastatin than with simvastatin alone. None of these differences was statistically significant; however, these prespecified subgroup data seemingly invalidate arguments against the ENHANCE results based on carotid intima-media thickness findings.
In this context, ENHANCE can only be interpreted as a strong initial negative signal, a “red flag” about ezetimibe’s net health benefits.
WHAT NEXT?
The proper present focus of this debate is not on LDL-C but rather on ezetimibe, its unique mechanism of action, and on the need for more evidence about this complex compound.
At present, ezetimibe’s mechanism of action is not fully understood, and its benefit—for now, only mild LDL-C reduction—is too uncertain for us to be spending $5.2 billion a year for it. Its manufacturer is fortunate that the drug is even licensed, given the current and seemingly appropriate regulatory changes under which drugs introducing new therapeutic classes are scrutinized more closely for benefits and risks. “Safe and well tolerated,” as contended by Dr. Davidson, is not nearly enough: drugs must show clinically important benefits. We still know too little about this drug, the manufacturer of which has invested far more in marketing than in science, a point on which Dr. Davidson and I agree.
In 2008, ezetimibe is an appropriate candidate for testing in clinical trials, and in years to come it may be worthy of clinical attention—if rigorous and objectively conducted clinical trials prove its worth. At present, clinical equipoise dictates that ezetimibe is not an appropriate alternative to a statin in titrated doses, to the addition of other lipid-lowering drugs to a statin, to greater attention to drug adherence, or to lifestyle modification.
For the moment, given the ENHANCE results, the clinical usefulness of ezetimibe still remains to be proven. Much more evidence is needed before we can confidently reembrace the clinical use of ezetimibe.
Ezetimibe (Zetia) was licensed by the US Food and Drug Administration in 2002 on the basis of its ability to reduce low-density lipoprotein cholesterol (LDL-C) levels. The reductions are mild, approximately 15%,1 which is comparable to the effects of a stringent diet and exercise or of a statin in titrated doses.
However, there was no evidence that ezetimbe, which has a unique mechanism of action, delivers a benefit in terms of clinical outcomes. Despite this, the use of ezetimibe (alone or in fixed-dose combination with simvastatin, a preparation sold as Vytorin) grew rapidly, generating annual sales of $5.2 billion. Clinicians and the manufacturer (Merck/Schering-Plough) broadly assumed that LDL-C reduction would carry ezetimibe’s day as clinical trials emerged.
The assumption seemed reasonable, since evidence from the past 3 decades has established a clear link between lowering LDL-C levels via diverse mechanisms and positive clinical outcomes, particularly lower rates of cardiovascular disease and death. Indeed, LDL-C measurement is now a focus of cardiovascular risk assessment and management, as reflected in national treatment guidelines.
THE ENHANCE TRIAL: EZETIMIBE FAILS A KEY TEST
Unexpectedly, ezetimibe failed its first step in clinical trial validation, the Ezetimibe and Simvastatin in Hypercholesterolemia Enhances Atherosclerosis Regression (ENHANCE) trial.2 Apart from the scientifically irrelevant political regulatory intrigue generated by the sponsor’s conduct in this trial, ENHANCE’s findings challenge us to confront issues of what we assume vs what we really know, and how to interpret the complex results of clinical trials.
To be fair to the trial’s investigators, ENHANCE achieved its objective of enrolling a population with a very high LDL-C level, which is ezetimibe’s target and has been widely used in the study of atherosclerosis progression as a marker of potential drug benefit. Nevertheless, and even though the LDL-C level 2 years later was 52 mg/dL lower in the group receiving ezetimibe/simvastatin than in the group receiving simvastatin alone (Zocor), at LDL-C levels that are typically associated with atherosclerosis progression (140–190 mg/dL), ezetimibe failed to reduce the progression of atherosclerosis.
These trends are particularly worrisome, given that the ezetimibe/simvastatin group achieved a greater reduction in C-reactive protein levels, which typically has resulted in superior outcomes in atherosclerosis3 and clinical effects4 in combination with LDL-C reduction.
In view of these findings, should clinicians stand firm and continue to use ezetimibe? Or should we reevaluate our position and await more data about this unique, first-in-class compound?
WISHFUL POST HOC HYPOTHESES
In this issue of the Cleveland Clinic Journal of Medicine, Dr. Michael Davidson,5 a respected lipid expert but one invested in ezetimibe’s development, assures us that all is in order and that the results of ENHANCE can be explained away by several arguments, most notably that most of the trial’s participants had previously received lipid-lowering treatment, which obscured the effects of ezetimibe. Moreover, he argues that ezetimibe’s mechanism of action is well understood and that the drug is safe and well tolerated and thus should remain a first-line treatment for hyperlipidemia.
These arguments may eventually prove to be correct, but as of now they are merely wishful post hoc hypotheses awaiting more data apart from ENHANCE. Negative clinical trials do occur as a matter of chance, but we should be cautious in any attempts to explain away a trial that was designed, executed, and reported as conceived simply because the results do not match our expectations.
Confronted with ENHANCE, the astute clinician should ask three questions: Do we really understand ezetimibe’s mechanism of action? Do other lines of evidence indicate the drug is beneficial? And how reliable is the arterial thickness as a surrogate end point?
DO WE UNDERSTAND EZETIMIBE’S MECHANISM OF ACTION?
Do we understand ezetimibe’s full mechanism of action? Not really.
True, ezetimibe inhibits cholesterol transport, a process that is integral both to cholesterol’s enteric absorption and to its systemic clearance. But although Dr. Davidson asserts that ezetimibe has cellular effects similar to those of statins, in fact it has the opposite effect on HMG-coA reductase, and no effects on LDL receptors.6
Furthermore, although initial studies suggested that ezetimibe inhibits enteric cholesterol absorption by inhibiting the Niemann-Pick C1L1 (NPC1L1) receptor, more recent investigations call this into serious question and point more definitively at a receptor known as scavenger receptor-B1 (SR-B1). As stated in a recent editorial, “SR-B1 in the apical site of enterocytes is the primary high-affinity site of cholesterol uptake and ezetimibe can inhibit this process. Moreover, the [possibility is ruled out] of NPC1L1 being a major player in this cholesterol uptake. This is at variance with the view of the colleagues from Schering-Plough who claim the same for NPC1L1.”7
SR-B1 is also a high-affinity receptor for high-density lipoprotein8 and thus is active in the antiatherosclerotic process of reverse cholesterol transport, inhibition of which significantly accelerates the development of atherosclerosis.9
Additionally, in vitro and thus unrelated to the effects of changing cholesterol concentration, ezetimibe down-regulates SR-B1 and another key cholesterol transporter protein called ABCA1.10 Further, ezetimibe induces down-regulation of raft protein domains, including CD36,11 another effect opposite to that of statins.
These little-recognized effects of ezetimibe are among many that are completely unrelated to enteric cholesterol absorption. Yet, they are likely to be active within the liver and systemically where these proteins reside, and they are putatively proatherosclerotic. Contrary to often-cited opinion, ezetimibe is systemically absorbed, with 11% of the compound excreted in the urine.12 Thus, the compound is systemically available to exert these same actions in the liver and elsewhere. Moreover, the absorbed drug is glucuronidated and is extensively recirculated in the liver in a form (its glucuronide) that is more potent than the parent compound.
In sum, present opinion is that ezetimibe inhibits lipid transport and interacts with a variety of receptors, not only in the gut but also systemically at the cell membrane and also inside the cell, focally disrupting several tightly regulated biologic processes.7 Thus, although ezetimibe reduces serum LDL-C levels via its effect in the gut, this effect may well be offset or even overridden systemically by other, unmeasurable effects, leading to counterintuitive results in terms of atherosclerosis or clinical events.
This would not be the first time a lipid-lowering drug has disappointed us: torcetrapib, another transport inhibitor, dramatically raises serum high-density lipoprotein cholesterol levels and reduces LDL-C but was found not only to have no effect on atherosclerosis, but also to potentiate adverse clinical outcomes.
The net impact of these other actions of ezetimibe is not known. We will discover its true clinical effects only through studies of endothelial function, atherosclerosis, and clinical cardiovascular outcomes. ENHANCE, which looked at atherosclerosis, is thus our strongest signal to date on the net effect of ezetimibe.
DO OTHER LINES OF EVIDENCE INDICATE EZETIMIBE IS BENEFICIAL?
Can we be reassured that ENHANCE’s results are spurious on the basis of other lines of evidence? Again, not really.
Experiments in animals, particularly in mice,13 have shown that ezetimibe may be antiatherosclerotic, although mice are considered the “worst model”7 for the study of ezetimibe, and notably, LDL-C levels were lowered far more in these experiments than they are clinically. Enthusiasm for these animal models should be tempered by interspecies variability in ezetimibe’s “off-target” effects and in the recent failure of other lipid transport drugs in human trials (torcetrapib and ACAT inhibitors) that had shown initial success in animals. No animal model is established for evaluating drugs of ezetimibe’s class, given its complex mechanism of action.
In human studies, the only other surrogate of the net effect of ezetimibe is endothelial function. Among several randomized clinical trials of ezetimibe,14–18 only one was designed to compare the effects of ezetimibe alone, ezetimibe plus a statin, and a statin by itself in titrated or in maximum doses.15 After 4 weeks of therapy, all groups had lower LDL-C levels. However, ezetimibe monotherapy and ezetimibe/simvastatin combination therapy had no detectable effect on the arterial response to acetylcholine, but atorvastatin (Lipitor) monotherapy did. To be fair, the other (very small) trials showed mixed results, thus keeping the hypothesis of ezetimibe’s benefit alive, but with nothing close to a clear signal of benefit.
IS ARTERIAL THICKNESS RELIABLE AS A SURROGATE END POINT?
Was the principal problem in ENHANCE the use of carotid intima-media thickness as the primary end point? No.
This issue has received a lot of attention, much of which I believe is misinformed. No trial end point is infallible, including carotid intima-media thickness, and one must remain open to the possibility of chance findings. However, it has been a relatively reasonable end point in trials of diverse cardiovascular preventive strategies, including lipid-lowering, blood-pressure-lowering, and lifestyle interventions and as a directional biomarker of clinical atherosclerotic events.
We should be cautious about comparing data on carotid intima-media thickness from different trials, as Dr. Davidson attempts to do, in view of methodologic and population differences: each trial must be considered independently. Of greatest concern in ENHANCE is the consistency among intima-media thickness end points, including strong trends toward adverse effects in the most diseased carotid and femoral segments.
Moreover, ENHANCE’s detractors contend that the carotid intima-media thickness of the studied population was normal, citing this as evidence of delipidation from prior treatment. Although not impossible (as shown by the work of Zhao and colleagues in the setting of prolonged, intense lipid-lowering therapy19), at the moment this hypothesis is a matter of conjecture in the ENHANCE participants, particularly because their LDL-C levels were still quite elevated during the trial and conceivably even before randomization.
But these patients were not normal: they were typical patients with familial hypercholesterolemia with extremely elevated LDL-C levels and abnormally thick arteries for their age. Population screening estimates show that, for age and sex, the carotid intima-media thickness values in ENHANCE would lie in the upper quartile of those in the general population.20 Moreover, their mean value is consistent with that in similar-aged groups of patients with familial hypercholesterolemia, even with lower rates of prior statin pretreatment.21
The most convincing evidence for the validity of the ENHANCE findings comes from the published subgroup data (Figure 1). In participants whose baseline carotid intima-media thickness was above the median at baseline, the thickness increased more with ezetimibe/simvastatin than with simvastatin alone. The same was true in the subgroup with above-average LDL-C levels at baseline. The subgroups with no prior statin treatment, low-dose prior statin treatment, and high-dose prior statin showed no heterogeneity of response: their carotid intima-media thickness increased more with ezetimibe/simvastatin than with simvastatin alone. None of these differences was statistically significant; however, these prespecified subgroup data seemingly invalidate arguments against the ENHANCE results based on carotid intima-media thickness findings.
In this context, ENHANCE can only be interpreted as a strong initial negative signal, a “red flag” about ezetimibe’s net health benefits.
WHAT NEXT?
The proper present focus of this debate is not on LDL-C but rather on ezetimibe, its unique mechanism of action, and on the need for more evidence about this complex compound.
At present, ezetimibe’s mechanism of action is not fully understood, and its benefit—for now, only mild LDL-C reduction—is too uncertain for us to be spending $5.2 billion a year for it. Its manufacturer is fortunate that the drug is even licensed, given the current and seemingly appropriate regulatory changes under which drugs introducing new therapeutic classes are scrutinized more closely for benefits and risks. “Safe and well tolerated,” as contended by Dr. Davidson, is not nearly enough: drugs must show clinically important benefits. We still know too little about this drug, the manufacturer of which has invested far more in marketing than in science, a point on which Dr. Davidson and I agree.
In 2008, ezetimibe is an appropriate candidate for testing in clinical trials, and in years to come it may be worthy of clinical attention—if rigorous and objectively conducted clinical trials prove its worth. At present, clinical equipoise dictates that ezetimibe is not an appropriate alternative to a statin in titrated doses, to the addition of other lipid-lowering drugs to a statin, to greater attention to drug adherence, or to lifestyle modification.
For the moment, given the ENHANCE results, the clinical usefulness of ezetimibe still remains to be proven. Much more evidence is needed before we can confidently reembrace the clinical use of ezetimibe.
- Ballantyne CM, Houri J, Notarbartolo A, et al. Effect of ezetimibe coadministered with atorvastatin in 628 patients with primary hypercholesterolemia: a prospective, randomized, double-blind trial. Circulation 2003; 107:2409–2415.
- Kastelein JJ, Akdim F, Stroes ES, et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med 2008; 358:1431–1443.
- Kent SM, Taylor AJ. Usefulness of lowering low-density lipoprotein cholesterol to < 70 mg/dL and usefulness of C-reactive protein in patient selection. Am J Cardiol 2003; 92:1224–1227.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N Engl J Med 2005; 352:29–38.
- Davidson MH. Interpreting the ENHANCE trial. Is ezetimibe/simvastatin no better than simvastatin alone? Leessons learned and clinical implications. Cleve Clin J Med 2008; 75:479–491.
- Gouni-Berthold I, Berthold HK, Gylling H, et al. Effects of ezetimibe and/or simvastatin on LDL receptor protein expression and on LDL receptor and HMG-CoA reductase gene expression: a randomized trial in healthy men. Atherosclerosis 2008; 198:198–207.
- Spener F. Ezetimibe in search of receptor(s)—still a never-ending challenge in cholesterol absorption and transport. Biochim Biophys Acta 2007; 1771:1113–1116.
- Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996; 271:518–520.
- Kitayama K, Nishizawa T, Abe K, et al. Blockade of scavenger receptor class B type I raises high density lipoprotein cholesterol levels but exacerbates atherosclerotic lesion formation in apolipoprotein E deficient mice. J Pharm Pharmacol 2006; 58:1629–1638.
- During A, Dawson HD, Harrison EH. Carotenoid transport is decreased and expression of the lipid transporters SR-BI, NPC1L1, and ABCA1 is downregulated in Caco-2 cells treated with ezetimibe. J Nutr 2005; 135:2305–2312.
- Orso E, Werner T, Wolf Z, Bandulik S, Kramer W, Schmitz G. Ezetimib influences the expression of raft-associated antigens in human monocytes. Cytometry A 2006; 69:206–208.
- Patrick JE, Kosoglou T, Stauber KL, et al. Disposition of the selective cholesterol absorption inhibitor ezetimibe in healthy male subjects. Drug Metab Dispos 2002; 30:430–437.
- Kuhlencordt PJ, Padmapriya P, Rutzel S, et al. Ezetimibe potently reduces vascular inflammation and arteriosclerosis in eNOS-deficient ApoE ko mice. Atherosclerosis 2008; April 6.
- Bulut D, Hanefeld C, Bulut-Streich N, Graf C, Mugge A, Spiecker M. Endothelial function in the forearm circulation of patients with the metabolic syndrome—effect of different lipid-lowering regimens. Cardiology 2005; 104:176–180.
- Fichtlscherer S, Schmidt-Lucke C, Bojunga S, et al. Differential effects of short-term lipid lowering with ezetimibe and statins on endothelial function in patients with CAD: clinical evidence for ‘pleiotropic’ functions of statin therapy. Eur Heart J 2006; 27:1182–1190.
- Landmesser U, Bahlmann F, Mueller M, et al. Simvastatin versus ezetimibe: pleiotropic and lipid-lowering effects on endothelial function in humans. Circulation 2005; 111:2356–2363.
- Maki-Petaja KM, Booth AD, Hall FC, et al. Ezetimibe and simvastatin reduce inflammation, disease activity, and aortic stiffness and improve endothelial function in rheumatoid arthritis. J Am Coll Cardiol 2007; 50:852–858.
- Settergren M, Bohm F, Ryden L, Pernow J. Cholesterol lowering is more important than pleiotropic effects of statins for endothelial function in patients with dysglycaemia and coronary artery disease. Eur Heart J 2008 April 25.
- Zhao XQ, Yuan C, Hatsukami TS, et al. Effects of prolonged intensive lipid-lowering therapy on the characteristics of carotid atherosclerotic plaques in vivo by MRI: a case-control study. Arterioscler Thromb Vasc Biol 2001; 21:1623–1629.
- Stein JH, Korcarz CE, Hurst RT, et al. Use of carotid ultrasound to identify subclinical vascular disease and evaluate cardiovascular disease risk: a consensus statement from the American Society of Echocardiography Carotid Intima-Media Thickness Task Force. Endorsed by the Society for Vascular Medicine. J Am Soc Echocardiogr 2008; 21:93–111.
- Junyent M, Cofan M, Nunez I, Gilabert R, Zambon D, Ros E. Influence of HDL cholesterol on preclinical carotid atherosclerosis in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol 2006; 26:1107–1113.
- Ballantyne CM, Houri J, Notarbartolo A, et al. Effect of ezetimibe coadministered with atorvastatin in 628 patients with primary hypercholesterolemia: a prospective, randomized, double-blind trial. Circulation 2003; 107:2409–2415.
- Kastelein JJ, Akdim F, Stroes ES, et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med 2008; 358:1431–1443.
- Kent SM, Taylor AJ. Usefulness of lowering low-density lipoprotein cholesterol to < 70 mg/dL and usefulness of C-reactive protein in patient selection. Am J Cardiol 2003; 92:1224–1227.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N Engl J Med 2005; 352:29–38.
- Davidson MH. Interpreting the ENHANCE trial. Is ezetimibe/simvastatin no better than simvastatin alone? Leessons learned and clinical implications. Cleve Clin J Med 2008; 75:479–491.
- Gouni-Berthold I, Berthold HK, Gylling H, et al. Effects of ezetimibe and/or simvastatin on LDL receptor protein expression and on LDL receptor and HMG-CoA reductase gene expression: a randomized trial in healthy men. Atherosclerosis 2008; 198:198–207.
- Spener F. Ezetimibe in search of receptor(s)—still a never-ending challenge in cholesterol absorption and transport. Biochim Biophys Acta 2007; 1771:1113–1116.
- Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996; 271:518–520.
- Kitayama K, Nishizawa T, Abe K, et al. Blockade of scavenger receptor class B type I raises high density lipoprotein cholesterol levels but exacerbates atherosclerotic lesion formation in apolipoprotein E deficient mice. J Pharm Pharmacol 2006; 58:1629–1638.
- During A, Dawson HD, Harrison EH. Carotenoid transport is decreased and expression of the lipid transporters SR-BI, NPC1L1, and ABCA1 is downregulated in Caco-2 cells treated with ezetimibe. J Nutr 2005; 135:2305–2312.
- Orso E, Werner T, Wolf Z, Bandulik S, Kramer W, Schmitz G. Ezetimib influences the expression of raft-associated antigens in human monocytes. Cytometry A 2006; 69:206–208.
- Patrick JE, Kosoglou T, Stauber KL, et al. Disposition of the selective cholesterol absorption inhibitor ezetimibe in healthy male subjects. Drug Metab Dispos 2002; 30:430–437.
- Kuhlencordt PJ, Padmapriya P, Rutzel S, et al. Ezetimibe potently reduces vascular inflammation and arteriosclerosis in eNOS-deficient ApoE ko mice. Atherosclerosis 2008; April 6.
- Bulut D, Hanefeld C, Bulut-Streich N, Graf C, Mugge A, Spiecker M. Endothelial function in the forearm circulation of patients with the metabolic syndrome—effect of different lipid-lowering regimens. Cardiology 2005; 104:176–180.
- Fichtlscherer S, Schmidt-Lucke C, Bojunga S, et al. Differential effects of short-term lipid lowering with ezetimibe and statins on endothelial function in patients with CAD: clinical evidence for ‘pleiotropic’ functions of statin therapy. Eur Heart J 2006; 27:1182–1190.
- Landmesser U, Bahlmann F, Mueller M, et al. Simvastatin versus ezetimibe: pleiotropic and lipid-lowering effects on endothelial function in humans. Circulation 2005; 111:2356–2363.
- Maki-Petaja KM, Booth AD, Hall FC, et al. Ezetimibe and simvastatin reduce inflammation, disease activity, and aortic stiffness and improve endothelial function in rheumatoid arthritis. J Am Coll Cardiol 2007; 50:852–858.
- Settergren M, Bohm F, Ryden L, Pernow J. Cholesterol lowering is more important than pleiotropic effects of statins for endothelial function in patients with dysglycaemia and coronary artery disease. Eur Heart J 2008 April 25.
- Zhao XQ, Yuan C, Hatsukami TS, et al. Effects of prolonged intensive lipid-lowering therapy on the characteristics of carotid atherosclerotic plaques in vivo by MRI: a case-control study. Arterioscler Thromb Vasc Biol 2001; 21:1623–1629.
- Stein JH, Korcarz CE, Hurst RT, et al. Use of carotid ultrasound to identify subclinical vascular disease and evaluate cardiovascular disease risk: a consensus statement from the American Society of Echocardiography Carotid Intima-Media Thickness Task Force. Endorsed by the Society for Vascular Medicine. J Am Soc Echocardiogr 2008; 21:93–111.
- Junyent M, Cofan M, Nunez I, Gilabert R, Zambon D, Ros E. Influence of HDL cholesterol on preclinical carotid atherosclerosis in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol 2006; 26:1107–1113.
Is ezetimibe/simvastatin no better than simvastatin alone? Lessons learned and clinical implications
The Ezetimibe and Simvastatin in Hypercholesterolemia Enhances Atherosclerosis Regression (ENHANCE) trial1 was probably the most widely publicized clinical study of the past decade. How did a 720-patient imaging trial with a neutral result in patients with severe hypercholesterolemia rise to a level warranting massive media attention, a congressional investigation, and a recommendation to curtail the use of a drug widely used to reduce levels of low-density-lipoprotein cholesterol (LDL-C)?
The reaction to the ENHANCE trial reveals more about the political climate and the relationship between the pharmaceutical industry and the American public than it does about the effects of ezetimibe (available combined with simvastatin as Vytorin and by itself as Zetia) on the progression of atherosclerosis.
SOME SELF-DISCLOSURE
Before I discuss the clinical implications of the ENHANCE trial, I must describe both my financial conflicts and intellectual biases. I am a paid consultant, speaker, and researcher on behalf of Merck/Schering-Plough, the sponsor of the ENHANCE trial. I was a principal investigator in the first phase II trial of ezetimibe and have conducted more than 10 clinical trials of either ezetimibe or ezetimibe/simvastatin. I also have been a strong advocate for imaging trials to assist in the clinical development of novel therapeutic agents and to support regulatory approval.
Therefore, I believe that the thickness of the intima and media layers of the carotid arteries is a useful surrogate to evaluate the potential antiatherosclerotic effects of drugs (more on this topic below). Also, I believe that the LDL-C-lowering hypothesis has been proven: ie, that all drugs that lower LDL-C safely, without off-target adverse effects, should reduce cardiovascular events. I support the goal levels of LDL-C and non-high-density-lipoprotein cholesterol set by the National Cholesterol Education Program’s third Adult Treatment Panel (ATP III) guidelines,2,3 which specify LDL-C targets rather than the use of specific drugs. In spite of these conflicts and potential biases, I believe I have always served the best interests of patient care.
HISTORY OF THE ENHANCE TRIAL

The end point defined as the mean of six measurements
The primary end point was the change in the thickness of the intima and media layers of the carotid arteries over a 2-year period, measured by ultrasonography. A composite measure was used: the mean of the thicknesses in the far walls of the right and left common carotid arteries, the right and left carotid bulbs, and the right and left internal carotid arteries. Secondary end points included the change in the mean maximal carotid artery intima-media thickness (ie, the thickest of the six baseline measurements), the proportion of participants who developed new carotid artery plaque (defined arbitrarily as an intima-media thickness > 1.3 mm), and changes in the mean of the intima-media thickness of the six carotid sites plus the common femoral arteries.
The last participant completed the trial in April 2006. Reading of the almost 30,000 scans was not started until the last participant was finished, so that all scans for each participant could be read in a blinded, randomized order by five separate readers. A significant proportion of the images that the protocol called for could not be obtained or analyzed, particularly in the internal carotid artery and the carotid bulb, which are often difficult to visualize. As a result, 17% of the internal carotid or carotid bulb measurements were discarded.
To change the end point post hoc, or not to change the end point?
The sponsor of the trial was concerned about the missing data points and convened a special advisory board to review the blinded data. This group suggested a solution: changing the primary end point from the six-site composite value to the mean value in just the common carotid arteries. They based this suggestion on the greater success rate in measuring the common carotids (97%) than in measuring all six sites (88%), as well as on recent trials that indicated that the common carotid artery measurement correlates better with clinical outcomes (because the internal carotid and the bulb measurements vary more). On November 26, 2007, Merck/Schering-Plough announced the primary end point would be changed to the mean change in the common carotid arteries.
However, during a separate meeting on November 30, 2007, some members of the Merck/Schering-Plough advisory board objected to the change. On December 11, 2007, the company announced that the original primary end point would not be changed.
Neutral results, negative publicity
On December 31, 2007, the ENHANCE study was unblinded, and on January 14, 2008, Merck/Schering-Plough issued a press release announcing the results. The press release stated that there were no statistically significant differences between the treatment groups in the primary end point or in any of the secondary end points, despite a 16.5% greater reduction in LDL-C (about 50 mg/dL) in the group receiving the ezetimibe/simvastatin combination. The composite intima-media thickness had increased by an average of 0.0111 mm in the combined-therapy group vs 0.0058 mm in the simvastatin-only group (P = .29) over the 24-month treatment period.5
The press release received unprecedented international media attention. One leading cardiologist commented to the media that ENHANCE showed “millions of patients may be taking a drug [ezetimibe] that does not benefit them, raising their risk of heart attacks and exposing them to potential side effects.”6 The perceived message that ezetimibe/simvastatin is harmful resulted in thousands of phone calls from concerned patients to their physicians throughout the United States. The American Heart Association (AHA) and the American College of Cardiology (ACC) issued a joint statement the next day saying that ezetimibe/simvastatin does not appear to be unsafe and that patients should not stop taking the drug on their own. In the following days, Merck/Schering-Plough placed advertisements in newspapers reaffirming the safety of ezetimibe and quoting the AHA/ACC statement.
But the full results of the study were not available at that point. In fact, Senator Charles Grassley (R-Iowa) had launched a congressional investigation into the delays in releasing the results of the ENHANCE trial in December 2007. A focus of the investigation was whether the sponsor was delaying the release either because the data reflected negatively on its product or because it was legitimately concerned about the quality of the measurements of the carotid intima-media thickness. After Merck/Schering-Plough placed the advertisements quoting the AHA/ACC statement, these organizations were criticized for touting the safety of ezetimibe while receiving educational grants and other funds from Merck/Schering-Plough. Senator Grassley sent a letter to the ACC in late March requesting information about the amount of funds the ACC had received.
Full results are published, and the ACC is misquoted
The ENHANCE study was selected for a special presentation at the ACC annual scientific session on March 30, 2008. The full ENHANCE results were presented by Dr. Kastelein, after which an expert panel led by Harlan M. Krumholz, MD, discussed the trial’s implications. The ENHANCE results were simultaneously published in the New England Journal of Medicine,1 accompanied by an editorial by B. Greg Brown, MD, and Allen J. Taylor, MD,7 and another editorial by the editors of that journal, Jeffrey M. Drazen, MD, and colleagues.8 The expert panel and the editorialists concluded that the ENHANCE trial data raised concerns about the cardiovascular benefits of ezetimibe; that statins should be used as initial therapy for hyperlipidemia and titrated to the goal LDL-C level or to the maximally tolerated dose; and that other drugs such as bile acid sequestrants, fibrates, and niacin should be used in combination with statins before considering ezetimibe.9
The next day, stories appeared in the media mistakenly stating that the ACC had recommended that ezetimibe/simvastatin be discontinued. This view was fueled by an article in the ACC’s Scientific Session News, penned by a contract writer and editor, with the headline, “ACC on Vytorin: Go Back to Statins” that said, “After waiting for 18 months for the results of the ENHANCE study, an ACC panel on Sunday encouraged physicians to use statins as a first line and prescribe Vytorin only as a last resort for patients unable to tolerate other cholesterol-lowering agents.”10
The ACC later clarified that this was the opinion of the panelists and not that of the ACC, and they reiterated statements from the AHA/ACC Secondary Prevention Guidelines11 recommending statins in maximally tolerated doses or titrated to a goal LDL-C level for first-line drug treatment of coronary artery disease, and recommending that patients speak with their physicians before discontinuing any therapy.
WHY WERE THE ENHANCE STUDY RESULTS NEUTRAL?
The ACC expert panel concluded that the most likely reason for the neutral ENHANCE results was that ezetimibe lowers LDL-C but does not confer a cardiovascular benefit. In the words of Dr. Krumholz (as quoted by Shannon Pettypiece and Michelle Fay Cortez on bloomberg.com), ezetimibe is “just an expensive placebo.”12
There are at least three potential explanations for the lack of benefit with ezetimibe in the ENHANCE trial. I list them below in order of lowest to highest probability, in my opinion:
Theory 1: Ezetimibe lowers LDL-C but is not antiatherogenic
Since almost all experts agree that lowering LDL-C confers cardiovascular benefits, if ezetimibe does not inhibit atherosclerosis it must have some “off-target” effect that negates its LDL-C-lowering benefit. Critics of ezetimibe point out that oral estrogen and torcetrapib also lower LDL-C but do not improve cardiovascular outcomes.13,14
The lack of benefit with these two other agents can be explained. Oral estrogen does not lower apolipoprotein B (an indication of the number of atherogenic particles), but rather it increases the levels of both triglycerides and C-reactive protein, and it is prothrombotic in some people.15 Torcetrapib increases aldosterone production and substantially raises blood pressure.16 Therefore, both drugs have true off-target effects that could explain their failure to reduce cardiovascular risk despite reductions in LDL-C. (Interestingly, though, oral estrogen has been shown to slow the progression of carotid intima-media thickness in newly postmenopausal women.17
Ezetimibe, however, lowers LDL-C by an ultimate mechanism similar to that of statins and bile acid sequestrants, ie, by up-regulating LDL receptors, although these drugs reach this mechanism via different pathways. Statins inhibit cholesterol synthesis, thereby lowering hepatic intracellular cholesterol and thus up-regulating LDL-receptors and enhancing LDL-C clearance from the plasma. Bile acid sequestrants interrupt bile acid reabsorption in the ileum, thereby decreasing intracellular hepatic cholesterol and up-regulating LDL receptors. Ezetimibe, like bile acid sequestrants, also decreases cholesterol return to the liver, lowering hepatic intracellular levels and thus up-regulating LDL receptors.18
Ezetimibe is unlikely to have an off-target effect because it is only fractionally absorbed systemically, and a recent animal study showed that it enhances macrophage efflux of cholesterol, thereby potentially increasing reverse cholesterol transport.19 Ezetimibe has also been shown to reduce atherosclerosis in animal models.20
In their editorial, Drs. Brown and Taylor7 noted that ezetimibe reduces the expression of adenosine triphosphate binding cassette A1 (ABCA1) in Caco-2 (an intestinal cell line), and this may be an example of an off-target effect. However, statins also reduce ABCA1 expression in macrophages.21 ABCA1 is sensitive to intracellular cholesterol, and when cholesterol levels are decreased, whether by statins or by ezetimibe, ABCA1 expression is down-regulated.22
Theory 2: Intima-media thickness does not reflect the true benefits of lowering LDL-C
The carotid intima-media thickness is a surrogate end point that predicts coronary events and the rate of progression of coronary atherosclerosis.23 In trials of lovastatin (Mevacor),24 pravastatin (Pravachol),25 and rosuvastatin (Crestor),26 the carotid intima-media was thinner at 24 months with the active drug than with placebo. In two relatively small trials—ARBITER 1 (n = 161),27 which was open-label, and ASAP (n = 325)28,29—aggressive lipid-lowering reduced the progression of intima-media thickness better than less-aggressive therapy. However, this measure has been used to evaluate the effects of differing degrees of LDL-C reduction between active treatments in fewer than 500 research participants.
Furthermore, what part or parts of the carotid system are we talking about? In recent trials led by Dr. Kastelein, the intima-media thickness of the common carotid arteries increased with pactimibe (an acyl-coenzyme A:cholesterol O-acyltransferase, or ACAT, inhibitor)30 and torcetrapib,31 but the six-site composite measure (which was the primary end point in these trials, as in ENHANCE) did not increase more than in the control groups. Pactimibe was also shown to increase atheroma volume as measured by intravascular ultrasonography in the ACTIVATE trial.32 Therefore, the thickness of the common carotid arteries has been shown to be a better predictor of harm from a therapy than the composite measurement.
The advantage of measuring the common carotid artery is that it is easier to visualize and measure, and therefore the measurements vary less. In the METEOR trial,26 the six-site measurement increased significantly less with rosuvastatin than with placebo, but the common carotid measurement alone was more strongly associated with a difference in progression. In the ENHANCE trial, the thickness of the common carotid arteries increased by 0.0024 mm with simvastatin alone vs 0.0019 mm with simvastatin/ezetimibe, a difference of 0.005 mm that was not statistically significant (P = .93).1
Although the six-site measurement appears to be good for predicting coronary events and evaluating therapies, the measurement in the common carotid arteries appears to be a more reliable surrogate end point for predicting both benefit and harm from antiatherogenic agents. However, trials of statins and other lipid-lowering therapies that assessed clinical events have shown that the reduction in risk associated with a given reduction in cholesterol is similar regardless of the mechanism by which cholesterol is lowered.33 Therefore, the LDL-C level is far superior as a marker of clinical benefit.
Theory 3: Previous statin treatment affected the ENHANCE results
By far the most likely explanation for the neutral findings in ENHANCE is that the patients were so well treated before entry that it was impossible to detect a difference between the two treatment groups in carotid intima-media thickness at the end of the study. Eighty percent of the patients had received statins previously, and at baseline the mean intima-media thickness of the common carotid arteries was only 0.68 mm.1 In contrast, most other trials required a thickness greater than 0.7 mm for entry.
The two main reasons for selecting a population with familial hypercholesterolemia were the assumptions that these participants would have a greater-than-average carotid intima-media thickness at baseline and that they would show an above-average progression rate, even on high-dose statin therapy.4 Both of these assumptions were incorrect: the baseline thickness was normal and the progression rate was negligible in both groups.
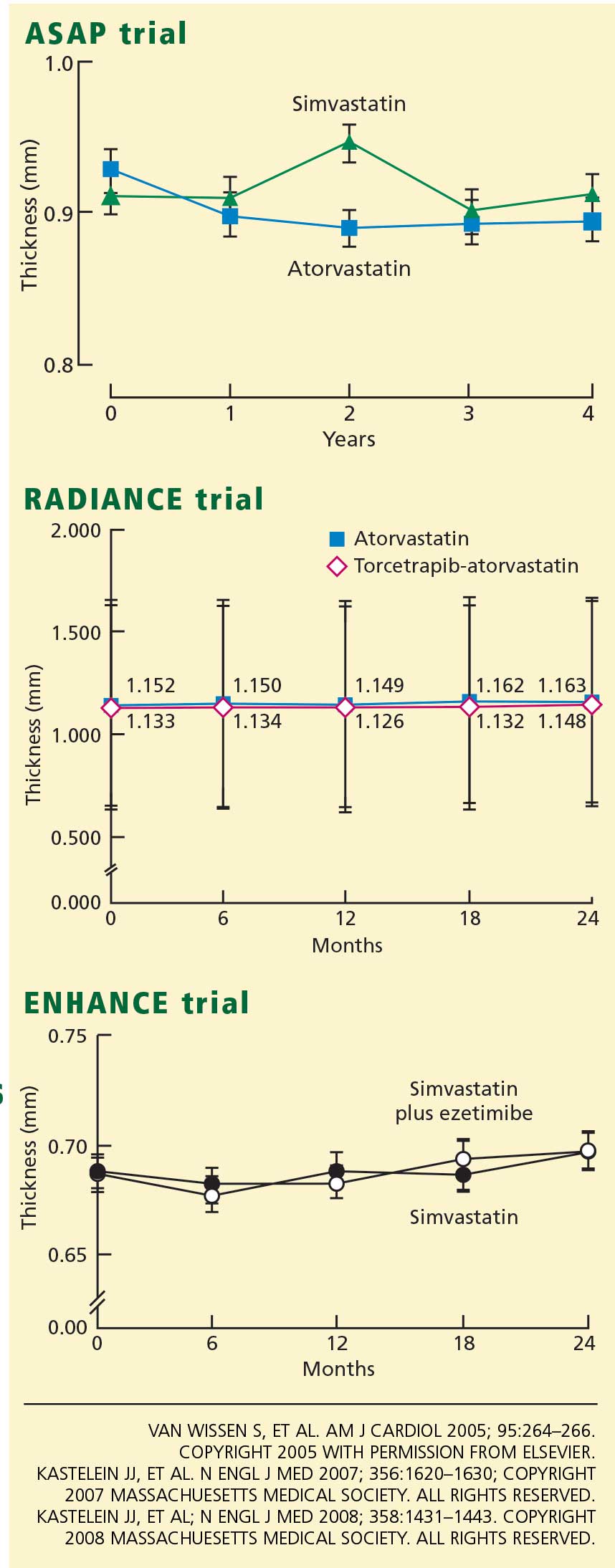
Accordingly, the high prevalence of statin pretreatment and the near-normal carotid intima-media thickness at baseline may have prevented the 16.5% greater reduction in LDL-C due to ezetimibe from producing a difference in progression over 24 months of treatment. This conclusion is supported by the long-term follow-up results from ASAP, RADIANCE 1, and CAPTIVATE, all of which showed that in patients with familial hypercholesterolemia well treated with statins, progression of carotid intima-media thickness is negligible.30,31
Further supporting this view, in a previous trial by Dr. Kastelein’s group in patients with familial hypercholesterolemia,34 giving simvastatin 80 mg for 2 years decreased the intima-medial thickness by .081 mm (P < .001), compared with 0.0058 mm in ENHANCE (a 14-fold difference). In the previous trial, the baseline measurement was 1.07 mm (vs 0.68 mm in ENHANCE), and the extent of the change was significantly associated with the baseline measurement (r = .53, P < .001) but not with the change in LDL-C levels.
This is powerful evidence that, in two similar studies that used the same methodology and the same drug, the thinner arteries in the ENHANCE trial are by far the most likely explanation for the lack of change with the addition of ezetimibe to high-dose simvastatin. The METEOR trial enrolled only patients who had never received statins and whose carotid intima-media was thicker than 1.2 mm. In retrospect, a similar design would have been preferable for ENHANCE.35
LESSONS LEARNED AND CLINICAL IMPLICATIONS
For Merck/Schering-Plough, missed opportunities
Although Dr. Krumholz (the spokesman for the ACC panel discussion) and I disagree on the clinical implications of the ENHANCE trial, we do agree on an important point. Dr. Krumholz posed the question that if the LDL-C-lowering hypothesis was already proven for ezetimibe, why was the ENHANCE trial conducted? After 6 years on the market, the efficacy of ezetimibe on cardiovascular outcomes should already have been established. It should not take this long to determine the clinical outcome benefit for a drug.
Merck/Schering-Plough’s outcome program for ezetimibe was inadequately designed to demonstrate the clinical value of this novel compound. Rather than assuming the LDL-C-lowering hypothesis was already established, they conducted another “lower-is-better” trial with the carotid intima-media thickness as the end point, and they succeeded only in raising doubt about the benefits of ezetimibe rather than showing that dual therapy is at least equivalent to high-dose statin therapy.
A preferable approach would have been to compare the effects of a statin in low doses plus ezetimibe vs high-dose statin monotherapy on either surrogate or hard outcomes. If the low-dose statin/ezetimibe combination, which should lower the LDL-C level as much as high-dose statin monotherapy, could provide similar or better outcomes with fewer side effects, this trial would change our practice.
One had hoped that dual therapy, by reducing both intestinal cholesterol absorption and hepatic synthesis of cholesterol, would improve outcomes by modifying postprandial chylomicron composition or by reducing plant sterol absorption.36 Unfortunately, other outcome trials of ezetimibe/simvastatin will not provide an answer regarding the potential advantages of dual therapy. The SEAS study is comparing the number of clinical events in patients with aortic stenosis who receive ezetimibe/simvastatin or placebo; SHARP is being conducted in patients with chronic kidney disease. Although both groups of patients have high rates of coronary events, these trials will not address whether adding ezetimibe provides additional benefits. In fact, if the results of these trials turn out neutral, as in ENHANCE, then ezetimibe will be blamed for potentially offsetting the benefits of simvastatin, and if the trials show a benefit, the simvastatin component of ezetimibe/simvastatin will be given the credit.
The answer may come in 3 to 4 years with the results of IMPROVE-IT, a study of 18,000 patients with acute coronary syndrome treated with ezetimibe/simvastatin or simvastatin. The simvastatin monotherapy group will have a target LDL-C level of less than 80 mg/dL and the ezetimibe/simvastatin group will have an LDL-C target about 15% less. Although this trial is testing the lower-is-better hypothesis with ezetimibe, if the study does not show a benefit, it may not be because ezetimibe lacks clinical efficacy but rather because the LDL-C effect is curvilinear, and there is minimal further benefit of lowering the LDL-C level past 70 mg/dL. If the results of the IMPROVE-IT trial are negative, it may mean the end of ezetimibe as an LDL-C-lowering drug.
Merck/Schering-Plough has lost valuable time in not demonstrating the benefits of ezetimibe on clinical events. In contrast, consider rosuvastatin, an AstraZeneca product. Rosuvastatin was approved about the same time as ezetimibe/simvastatin, and 6 years later it has already received a label change for the reduction of progression of atherosclerosis, based on positive outcomes in the METEOR trial,35 the ASTEROID intravascular ultrasonography trial,37 and the CORONA trial (an important trial that examined hard clinical end points).38 More importantly, the JUPITER trial was recently stopped early owing to a reduction in cardiovascular deaths. Initially, rosuvastatin received an unfair media portrayal as an unsafe drug. Now, because of its proven benefits in outcome trials, it will receive more widespread consideration for clinical use.
For preventive cardiologists, a painful reminder to focus on LDL-C
For the preventive cardiologist or lipidologist, the ENHANCE trial has been a painful reminder that despite overwhelming evidence, the mantra of “the lower the LDL-C the better” is still not universally accepted. We acknowledge the great benefits of statins, but the lure of “pleiotropic effects” distracts many of us from the necessity of more aggressive LDL-C reduction.
The pleiotropic benefits of statins were first raised as a means of supporting increased clinical use of pravastatin vis-a-vis other, more efficacious statins. It was not until the PROVE-IT study that pravastatin’s pleiotropic effects were found not to translate into a benefit equivalent to that of the more efficacious statin, atorvastatin.39
The success of ezetimibe was its ability to safely and easily lower LDL-C in combination with statins to achieve treatment goals. For many patients, a lower-dose statin and ezetimibe together provide a well-tolerated and efficacious approach to treating hyperlipidemia. The fallout from the ENHANCE trial is that many patients who were well treated or who could be better treated with ezetimibe in combination with a statin will not receive the best tolerated regimen. In fact, preliminary prescription data after the release of the ENHANCE study support our worse fear, ie, that patients at high risk will receive less aggressive LDL-C reduction. Since the ENHANCE data were released, more than 300,000 patients have stopped taking either ezetimibe/simvastatin or ezetimibe, and nearly all have continued on generic simvastatin or on a dose of statin with less overall efficacy.
An example is Senator John McCain, who, according to his recently released medical records, has a Framingham 10-year risk of more than 20% and was on ezetimibe/simvastatin to treat an elevated cholesterol level. After release of the ENHANCE trial, he was switched to generic simvastatin, and his LDL-C increased from 82 mg/dL to 122 mg/dL. He most likely has an LDL-C goal of less than 100 mg/dL according to the ATP III guidelines, and he is therefore no longer at his target.
For physicians in the community, questions from concerned patients
For the physicians who have received hundreds of phone calls and e-mails from concerned patients, the ENHANCE trial results must have been both discouraging and confusing. At present, I think we should remember the following:
- Ezetimibe’s mechanism of action is well understood
- It is safe and well-tolerated
- It still has a role as an add-on to statin therapy (or as monotherapy or combined with other agents in those who cannot tolerate statins) for patients who have not yet achieved their LDL-C target.
For the pharmaceutical industry, enormous challenges
The neutral ENHANCE trial results created an uncomfortable situation for the trial sponsor. A heavily marketed drug failed to achieve its expected result after the study results were delayed for a few months. The pharmaceutical industry ranks 14th out of 17 industries in public trust among the American public, and this study provided an opportunity for its critics to attack what is, in their opinion, an overly marketed drug.
Enormous challenges are on the horizon for the pharmaceutical industry, with a shrinking pipeline of potential new drugs, increasing regulatory hurdles, greater liability risk, political pressure for price controls, enhanced scrutiny of sales practices, and a growing media bias. As a cardiologist and clinical researcher whose father died at age 47 of a myocardial infarction, I am concerned that, unless change occurs, a vibrant pharmaceutical industry with the financial and intellectual capital to find and develop new, more effective treatments will cease to exist.
- Kastelein JJ, Akdim F, Stroes ES, et al ENHANCE Investigators. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med 2008; 358:1431–1443.
- Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Grundy SM, Cleeman JI, Bairey Merz N, et al for the Coordinating Committee of the National Cholesterol Education Program. Circulation 2004; 110:227–239.
- Kastelein JJ, Sager PT, de Groot E, Veltri E. Comparison of ezetimibe plus simvastatin versus simvastatin monotherapy on atherosclerosis progression in familial hypercholesterolemia. Design and rationale of the Ezetimibe and Simvastatin in Hypercholesterolemia Enhances Atherosclerosis Regression (ENHANCE) trial. Am Heart J 2005; 49:234–239.
- Merck/Schering-Plough Pharmaceutical Press Release, January 14, 2008.
- Berenson A. Study reveals doubt on drug for cholesterol. New York Times January 15, 2008.
- Brown BG, Taylor AJ. Does ENHANCE diminish confidence in lowering LDL or in ezetimibe? N Engl J Med 2008; 358:1504–1507.
- Drazen JM, Jarcho JA, Morrissey S, Curfman GD. Cholesterol lowering and ezetimibe. N Engl J Med 2008; 358:1507–1508.
- American College of Cardiology. ENHANCED analysis of ezetimibe. ACC News, April 2, 2008. www.acc.org/emails/myacc/accnews%5Fapril%5F02%5F08.htm. Accessed 6/2/2008.
- American College of Cardiology. ACC panel on Vytorin: Go back to statins. Scientific Session News 3/31/2008. http://www.acc08.acc.org/SSN/Documents/ACC%20Monday%20v2.pdf. Accessed 6/2/2008.
- Smith SC, Allen J, Blair SN, et al. AHA/ACC guidelines for secondary prevention for patients with coronary and other atherosclerotic vascular disease: 2006 update. Endorsed by the National Heart, Lung, and Blood Institute. J Am Coll Cardiol 2006; 47:2130–2139.
- Pettypiece S, Cortez MF. Merck, Schering plunge as doctors discourage Vytorin. www.bloomberg.com/apps/news?pid=20601103&refer=news&sid=aV_T9WirgAkI. Accessed 6/2/2008.
- Barter PJ, Caulfield M, Eriksson M, et al ILLUMINATE Investigators. . Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 2007; 357:2109–2122.
- Hulley S, Grady D, Bush T, et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in post-menopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA 1998; 280:605–613.
- Rader DJ. Illuminating HDL—is it still a viable therapeutic target? N Engl J Med 2007; 357:2180–2183.
- Davidson MH, Maki KC, Marx P, et al. Effects of continuous estrogen and estrogen-progestin replacement regimens on cardiovascular risk markers in postmenopausal women. Arch Intern Med 2000; 160:3315–3325.
- Hodis HN, Mack WJ, Lobo RA, et al Estrogen in the Prevention of Atherosclerosis Trial Research Group. . Estrogen in the prevention of atherosclerosis. A randomized, double-blind, placebo-controlled trial. Ann Intern Med 2001; 135:939–953.
- Turley SD. Cholesterol metabolism and therapeutic targets: rationale for targeting multiple metabolic pathways. Clin Cardiol 2004; 27( suppl 3):III16–III21.
- Sehayek E, Hazen SL. Cholesterol absorption from the intestine is a major determinant of reverse cholesterol transport from peripheral tissue macrophages. Arterioscler Thromb Vasc Biol 2008;27 (Epub ahead of print].
- Davis HR, Compton DS, Hoos L, Tetzloff G. Ezetimibe, a potent cholesterol absorption inhibitor, inhibits the development of atherosclerosis in ApoE knockout mice. Arterioscler Thromb Vasc Biol 2001; 21:2032–2038.
- Wong J, Quinn CM, Gelissen IC, Jessup W, Brown AJ. The effect of statins on ABCA1 and ABCG1 expression in human macrophages is influenced by cellular cholesterol levels and extent of differentiation. Atherosclerosis 2008; 196:180–189.
- Wang N, Tall AR. Regulation and mechanisms of ATP-binding cassette transporter A1-mediated cellular cholesterol efflux. Arterioscler Thromb Vasc Biol 2003; 23:1178–1184.
- Bots ML. Carotid intima-media thickness as a surrogate marker for cardiovascular disease in intervention studies. Curr Med Res Opin 2006; 22:2181–2190.
- Byington RP, Evans GW, Espeland MA, et al. Effects of lovastatin and warfarin on early carotid atherosclerosis: sex-specific analyses. Asymptomatic Carotid Artery Progression Study (ACAPS) Research Group. Circulation 1999; 100:e14–e17.
- Byington RP, Furberg CD, Crouse JR, Espeland MA, Bond MG. Pravastatin, Lipids, and Atherosclerosis in the Carotid Arteries (PLAC-II). Am J Cardiol 1995; 76:54C–59C.
- Crouse JR, Raichlen JS, Riley WA, et al METEOR Study Group. . Effect of rosuvastatin on progression of carotid intima-media thickness in low-risk individuals with subclinical atherosclerosis: the METEOR trial. JAMA 2007; 297:1344–1353.
- Taylor AJ, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation 2004; 110:3512–3517.
- Smilde TJ, van Wissen S, Wollersheim H, Trip MD, Kastelein JJ, Stalenhoef AF. Effect of aggressive versus conventional lipid lowering on atherosclerosis progression in familial hypercholesterolaemia (ASAP): a prospective, randomised, double-blind trial. Lancet 2001; 357:577–581.
- van Wissen S, Smilde TJ, Trip MD, Stalenhoef AFH, Kastelein JJP. Long-term safety and efficacy of high-dose atorvastatin treatment in patients with familial hypercholesterolemia. Am J Cardiol 2005; 95:264–266.
- Meuwese MC, Franssen R, Stroes ES, Kastelein JJ. And then there were acyl coenzyme A:cholesterol acyl transferase inhibitors. Curr Opin Lipidol 2006; 17:426–430.
- Kastelein JJ, van Leuven SI, Burgess L, et al RADIANCE 1 Investigators. . Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia. N Engl J Med 2007; 356:1620–1630.
- Nissen SE, Tuzcu EM, Brewer HB, et al ACAT Intravascular Atherosclerosis Treatment Evaluation (ACTIVATE) Investigators. Effect of ACAT inhibition on the progression of coronary atherosclerosis. N Engl J Med 2006; 354:1253–1263.
- Davidson MH. Clinical significance of statin pleiotropic effects: hypotheses versus evidence. Circulation 2005; 111:2280–2281.
- Nolting PR, de Groot E, Zwinderman AH, Buirma RJ, Trip MD, Kastelein JJ. Regression of carotid and femoral artery intima-media thickness in familial hypercholesterolemia. Arch Intern Med 2003; 163:1837–1841.
- Crouse JR, Grobbee DE, O’Leary DH, et al Measuring Effects on intima media Thickness: an Evaluation Of Rosuvastatin Study Group. . Measuring effects on intima media thickness: an evaluation of rosuvastatin in subclinical atherosclerosis—the rationale and methodology of the METEOR study. Cardiovasc Drugs Ther 2004; 18:231–238.
- Toth PP, Davidson MH. Cholesterol absorption blockade with ezetimibe. Curr Drug Targets Cardiovasc Haematol Disord 2005; 5:455–462.
- Nissen SE, Nicholls SJ, Sipahi I, et al ASTEROID Investigators. . Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006; 295:1556–1565.
- Kjekshus J, Apetrei E, Barrios V, et al CORONA Group. . Rosuvastatin in older patients with systolic heart failure. N Engl J Med 2007; 357:2248–2261.
- Cannon CP, Braunwald E, McCabe CH, et al Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. . Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
The Ezetimibe and Simvastatin in Hypercholesterolemia Enhances Atherosclerosis Regression (ENHANCE) trial1 was probably the most widely publicized clinical study of the past decade. How did a 720-patient imaging trial with a neutral result in patients with severe hypercholesterolemia rise to a level warranting massive media attention, a congressional investigation, and a recommendation to curtail the use of a drug widely used to reduce levels of low-density-lipoprotein cholesterol (LDL-C)?
The reaction to the ENHANCE trial reveals more about the political climate and the relationship between the pharmaceutical industry and the American public than it does about the effects of ezetimibe (available combined with simvastatin as Vytorin and by itself as Zetia) on the progression of atherosclerosis.
SOME SELF-DISCLOSURE
Before I discuss the clinical implications of the ENHANCE trial, I must describe both my financial conflicts and intellectual biases. I am a paid consultant, speaker, and researcher on behalf of Merck/Schering-Plough, the sponsor of the ENHANCE trial. I was a principal investigator in the first phase II trial of ezetimibe and have conducted more than 10 clinical trials of either ezetimibe or ezetimibe/simvastatin. I also have been a strong advocate for imaging trials to assist in the clinical development of novel therapeutic agents and to support regulatory approval.
Therefore, I believe that the thickness of the intima and media layers of the carotid arteries is a useful surrogate to evaluate the potential antiatherosclerotic effects of drugs (more on this topic below). Also, I believe that the LDL-C-lowering hypothesis has been proven: ie, that all drugs that lower LDL-C safely, without off-target adverse effects, should reduce cardiovascular events. I support the goal levels of LDL-C and non-high-density-lipoprotein cholesterol set by the National Cholesterol Education Program’s third Adult Treatment Panel (ATP III) guidelines,2,3 which specify LDL-C targets rather than the use of specific drugs. In spite of these conflicts and potential biases, I believe I have always served the best interests of patient care.
HISTORY OF THE ENHANCE TRIAL
The end point defined as the mean of six measurements
The primary end point was the change in the thickness of the intima and media layers of the carotid arteries over a 2-year period, measured by ultrasonography. A composite measure was used: the mean of the thicknesses in the far walls of the right and left common carotid arteries, the right and left carotid bulbs, and the right and left internal carotid arteries. Secondary end points included the change in the mean maximal carotid artery intima-media thickness (ie, the thickest of the six baseline measurements), the proportion of participants who developed new carotid artery plaque (defined arbitrarily as an intima-media thickness > 1.3 mm), and changes in the mean of the intima-media thickness of the six carotid sites plus the common femoral arteries.
The last participant completed the trial in April 2006. Reading of the almost 30,000 scans was not started until the last participant was finished, so that all scans for each participant could be read in a blinded, randomized order by five separate readers. A significant proportion of the images that the protocol called for could not be obtained or analyzed, particularly in the internal carotid artery and the carotid bulb, which are often difficult to visualize. As a result, 17% of the internal carotid or carotid bulb measurements were discarded.
To change the end point post hoc, or not to change the end point?
The sponsor of the trial was concerned about the missing data points and convened a special advisory board to review the blinded data. This group suggested a solution: changing the primary end point from the six-site composite value to the mean value in just the common carotid arteries. They based this suggestion on the greater success rate in measuring the common carotids (97%) than in measuring all six sites (88%), as well as on recent trials that indicated that the common carotid artery measurement correlates better with clinical outcomes (because the internal carotid and the bulb measurements vary more). On November 26, 2007, Merck/Schering-Plough announced the primary end point would be changed to the mean change in the common carotid arteries.
However, during a separate meeting on November 30, 2007, some members of the Merck/Schering-Plough advisory board objected to the change. On December 11, 2007, the company announced that the original primary end point would not be changed.
Neutral results, negative publicity
On December 31, 2007, the ENHANCE study was unblinded, and on January 14, 2008, Merck/Schering-Plough issued a press release announcing the results. The press release stated that there were no statistically significant differences between the treatment groups in the primary end point or in any of the secondary end points, despite a 16.5% greater reduction in LDL-C (about 50 mg/dL) in the group receiving the ezetimibe/simvastatin combination. The composite intima-media thickness had increased by an average of 0.0111 mm in the combined-therapy group vs 0.0058 mm in the simvastatin-only group (P = .29) over the 24-month treatment period.5
The press release received unprecedented international media attention. One leading cardiologist commented to the media that ENHANCE showed “millions of patients may be taking a drug [ezetimibe] that does not benefit them, raising their risk of heart attacks and exposing them to potential side effects.”6 The perceived message that ezetimibe/simvastatin is harmful resulted in thousands of phone calls from concerned patients to their physicians throughout the United States. The American Heart Association (AHA) and the American College of Cardiology (ACC) issued a joint statement the next day saying that ezetimibe/simvastatin does not appear to be unsafe and that patients should not stop taking the drug on their own. In the following days, Merck/Schering-Plough placed advertisements in newspapers reaffirming the safety of ezetimibe and quoting the AHA/ACC statement.
But the full results of the study were not available at that point. In fact, Senator Charles Grassley (R-Iowa) had launched a congressional investigation into the delays in releasing the results of the ENHANCE trial in December 2007. A focus of the investigation was whether the sponsor was delaying the release either because the data reflected negatively on its product or because it was legitimately concerned about the quality of the measurements of the carotid intima-media thickness. After Merck/Schering-Plough placed the advertisements quoting the AHA/ACC statement, these organizations were criticized for touting the safety of ezetimibe while receiving educational grants and other funds from Merck/Schering-Plough. Senator Grassley sent a letter to the ACC in late March requesting information about the amount of funds the ACC had received.
Full results are published, and the ACC is misquoted
The ENHANCE study was selected for a special presentation at the ACC annual scientific session on March 30, 2008. The full ENHANCE results were presented by Dr. Kastelein, after which an expert panel led by Harlan M. Krumholz, MD, discussed the trial’s implications. The ENHANCE results were simultaneously published in the New England Journal of Medicine,1 accompanied by an editorial by B. Greg Brown, MD, and Allen J. Taylor, MD,7 and another editorial by the editors of that journal, Jeffrey M. Drazen, MD, and colleagues.8 The expert panel and the editorialists concluded that the ENHANCE trial data raised concerns about the cardiovascular benefits of ezetimibe; that statins should be used as initial therapy for hyperlipidemia and titrated to the goal LDL-C level or to the maximally tolerated dose; and that other drugs such as bile acid sequestrants, fibrates, and niacin should be used in combination with statins before considering ezetimibe.9
The next day, stories appeared in the media mistakenly stating that the ACC had recommended that ezetimibe/simvastatin be discontinued. This view was fueled by an article in the ACC’s Scientific Session News, penned by a contract writer and editor, with the headline, “ACC on Vytorin: Go Back to Statins” that said, “After waiting for 18 months for the results of the ENHANCE study, an ACC panel on Sunday encouraged physicians to use statins as a first line and prescribe Vytorin only as a last resort for patients unable to tolerate other cholesterol-lowering agents.”10
The ACC later clarified that this was the opinion of the panelists and not that of the ACC, and they reiterated statements from the AHA/ACC Secondary Prevention Guidelines11 recommending statins in maximally tolerated doses or titrated to a goal LDL-C level for first-line drug treatment of coronary artery disease, and recommending that patients speak with their physicians before discontinuing any therapy.
WHY WERE THE ENHANCE STUDY RESULTS NEUTRAL?
The ACC expert panel concluded that the most likely reason for the neutral ENHANCE results was that ezetimibe lowers LDL-C but does not confer a cardiovascular benefit. In the words of Dr. Krumholz (as quoted by Shannon Pettypiece and Michelle Fay Cortez on bloomberg.com), ezetimibe is “just an expensive placebo.”12
There are at least three potential explanations for the lack of benefit with ezetimibe in the ENHANCE trial. I list them below in order of lowest to highest probability, in my opinion:
Theory 1: Ezetimibe lowers LDL-C but is not antiatherogenic
Since almost all experts agree that lowering LDL-C confers cardiovascular benefits, if ezetimibe does not inhibit atherosclerosis it must have some “off-target” effect that negates its LDL-C-lowering benefit. Critics of ezetimibe point out that oral estrogen and torcetrapib also lower LDL-C but do not improve cardiovascular outcomes.13,14
The lack of benefit with these two other agents can be explained. Oral estrogen does not lower apolipoprotein B (an indication of the number of atherogenic particles), but rather it increases the levels of both triglycerides and C-reactive protein, and it is prothrombotic in some people.15 Torcetrapib increases aldosterone production and substantially raises blood pressure.16 Therefore, both drugs have true off-target effects that could explain their failure to reduce cardiovascular risk despite reductions in LDL-C. (Interestingly, though, oral estrogen has been shown to slow the progression of carotid intima-media thickness in newly postmenopausal women.17
Ezetimibe, however, lowers LDL-C by an ultimate mechanism similar to that of statins and bile acid sequestrants, ie, by up-regulating LDL receptors, although these drugs reach this mechanism via different pathways. Statins inhibit cholesterol synthesis, thereby lowering hepatic intracellular cholesterol and thus up-regulating LDL-receptors and enhancing LDL-C clearance from the plasma. Bile acid sequestrants interrupt bile acid reabsorption in the ileum, thereby decreasing intracellular hepatic cholesterol and up-regulating LDL receptors. Ezetimibe, like bile acid sequestrants, also decreases cholesterol return to the liver, lowering hepatic intracellular levels and thus up-regulating LDL receptors.18
Ezetimibe is unlikely to have an off-target effect because it is only fractionally absorbed systemically, and a recent animal study showed that it enhances macrophage efflux of cholesterol, thereby potentially increasing reverse cholesterol transport.19 Ezetimibe has also been shown to reduce atherosclerosis in animal models.20
In their editorial, Drs. Brown and Taylor7 noted that ezetimibe reduces the expression of adenosine triphosphate binding cassette A1 (ABCA1) in Caco-2 (an intestinal cell line), and this may be an example of an off-target effect. However, statins also reduce ABCA1 expression in macrophages.21 ABCA1 is sensitive to intracellular cholesterol, and when cholesterol levels are decreased, whether by statins or by ezetimibe, ABCA1 expression is down-regulated.22
Theory 2: Intima-media thickness does not reflect the true benefits of lowering LDL-C
The carotid intima-media thickness is a surrogate end point that predicts coronary events and the rate of progression of coronary atherosclerosis.23 In trials of lovastatin (Mevacor),24 pravastatin (Pravachol),25 and rosuvastatin (Crestor),26 the carotid intima-media was thinner at 24 months with the active drug than with placebo. In two relatively small trials—ARBITER 1 (n = 161),27 which was open-label, and ASAP (n = 325)28,29—aggressive lipid-lowering reduced the progression of intima-media thickness better than less-aggressive therapy. However, this measure has been used to evaluate the effects of differing degrees of LDL-C reduction between active treatments in fewer than 500 research participants.
Furthermore, what part or parts of the carotid system are we talking about? In recent trials led by Dr. Kastelein, the intima-media thickness of the common carotid arteries increased with pactimibe (an acyl-coenzyme A:cholesterol O-acyltransferase, or ACAT, inhibitor)30 and torcetrapib,31 but the six-site composite measure (which was the primary end point in these trials, as in ENHANCE) did not increase more than in the control groups. Pactimibe was also shown to increase atheroma volume as measured by intravascular ultrasonography in the ACTIVATE trial.32 Therefore, the thickness of the common carotid arteries has been shown to be a better predictor of harm from a therapy than the composite measurement.
The advantage of measuring the common carotid artery is that it is easier to visualize and measure, and therefore the measurements vary less. In the METEOR trial,26 the six-site measurement increased significantly less with rosuvastatin than with placebo, but the common carotid measurement alone was more strongly associated with a difference in progression. In the ENHANCE trial, the thickness of the common carotid arteries increased by 0.0024 mm with simvastatin alone vs 0.0019 mm with simvastatin/ezetimibe, a difference of 0.005 mm that was not statistically significant (P = .93).1
Although the six-site measurement appears to be good for predicting coronary events and evaluating therapies, the measurement in the common carotid arteries appears to be a more reliable surrogate end point for predicting both benefit and harm from antiatherogenic agents. However, trials of statins and other lipid-lowering therapies that assessed clinical events have shown that the reduction in risk associated with a given reduction in cholesterol is similar regardless of the mechanism by which cholesterol is lowered.33 Therefore, the LDL-C level is far superior as a marker of clinical benefit.
Theory 3: Previous statin treatment affected the ENHANCE results
By far the most likely explanation for the neutral findings in ENHANCE is that the patients were so well treated before entry that it was impossible to detect a difference between the two treatment groups in carotid intima-media thickness at the end of the study. Eighty percent of the patients had received statins previously, and at baseline the mean intima-media thickness of the common carotid arteries was only 0.68 mm.1 In contrast, most other trials required a thickness greater than 0.7 mm for entry.
The two main reasons for selecting a population with familial hypercholesterolemia were the assumptions that these participants would have a greater-than-average carotid intima-media thickness at baseline and that they would show an above-average progression rate, even on high-dose statin therapy.4 Both of these assumptions were incorrect: the baseline thickness was normal and the progression rate was negligible in both groups.
Accordingly, the high prevalence of statin pretreatment and the near-normal carotid intima-media thickness at baseline may have prevented the 16.5% greater reduction in LDL-C due to ezetimibe from producing a difference in progression over 24 months of treatment. This conclusion is supported by the long-term follow-up results from ASAP, RADIANCE 1, and CAPTIVATE, all of which showed that in patients with familial hypercholesterolemia well treated with statins, progression of carotid intima-media thickness is negligible.30,31
Further supporting this view, in a previous trial by Dr. Kastelein’s group in patients with familial hypercholesterolemia,34 giving simvastatin 80 mg for 2 years decreased the intima-medial thickness by .081 mm (P < .001), compared with 0.0058 mm in ENHANCE (a 14-fold difference). In the previous trial, the baseline measurement was 1.07 mm (vs 0.68 mm in ENHANCE), and the extent of the change was significantly associated with the baseline measurement (r = .53, P < .001) but not with the change in LDL-C levels.
This is powerful evidence that, in two similar studies that used the same methodology and the same drug, the thinner arteries in the ENHANCE trial are by far the most likely explanation for the lack of change with the addition of ezetimibe to high-dose simvastatin. The METEOR trial enrolled only patients who had never received statins and whose carotid intima-media was thicker than 1.2 mm. In retrospect, a similar design would have been preferable for ENHANCE.35
LESSONS LEARNED AND CLINICAL IMPLICATIONS
For Merck/Schering-Plough, missed opportunities
Although Dr. Krumholz (the spokesman for the ACC panel discussion) and I disagree on the clinical implications of the ENHANCE trial, we do agree on an important point. Dr. Krumholz posed the question that if the LDL-C-lowering hypothesis was already proven for ezetimibe, why was the ENHANCE trial conducted? After 6 years on the market, the efficacy of ezetimibe on cardiovascular outcomes should already have been established. It should not take this long to determine the clinical outcome benefit for a drug.
Merck/Schering-Plough’s outcome program for ezetimibe was inadequately designed to demonstrate the clinical value of this novel compound. Rather than assuming the LDL-C-lowering hypothesis was already established, they conducted another “lower-is-better” trial with the carotid intima-media thickness as the end point, and they succeeded only in raising doubt about the benefits of ezetimibe rather than showing that dual therapy is at least equivalent to high-dose statin therapy.
A preferable approach would have been to compare the effects of a statin in low doses plus ezetimibe vs high-dose statin monotherapy on either surrogate or hard outcomes. If the low-dose statin/ezetimibe combination, which should lower the LDL-C level as much as high-dose statin monotherapy, could provide similar or better outcomes with fewer side effects, this trial would change our practice.
One had hoped that dual therapy, by reducing both intestinal cholesterol absorption and hepatic synthesis of cholesterol, would improve outcomes by modifying postprandial chylomicron composition or by reducing plant sterol absorption.36 Unfortunately, other outcome trials of ezetimibe/simvastatin will not provide an answer regarding the potential advantages of dual therapy. The SEAS study is comparing the number of clinical events in patients with aortic stenosis who receive ezetimibe/simvastatin or placebo; SHARP is being conducted in patients with chronic kidney disease. Although both groups of patients have high rates of coronary events, these trials will not address whether adding ezetimibe provides additional benefits. In fact, if the results of these trials turn out neutral, as in ENHANCE, then ezetimibe will be blamed for potentially offsetting the benefits of simvastatin, and if the trials show a benefit, the simvastatin component of ezetimibe/simvastatin will be given the credit.
The answer may come in 3 to 4 years with the results of IMPROVE-IT, a study of 18,000 patients with acute coronary syndrome treated with ezetimibe/simvastatin or simvastatin. The simvastatin monotherapy group will have a target LDL-C level of less than 80 mg/dL and the ezetimibe/simvastatin group will have an LDL-C target about 15% less. Although this trial is testing the lower-is-better hypothesis with ezetimibe, if the study does not show a benefit, it may not be because ezetimibe lacks clinical efficacy but rather because the LDL-C effect is curvilinear, and there is minimal further benefit of lowering the LDL-C level past 70 mg/dL. If the results of the IMPROVE-IT trial are negative, it may mean the end of ezetimibe as an LDL-C-lowering drug.
Merck/Schering-Plough has lost valuable time in not demonstrating the benefits of ezetimibe on clinical events. In contrast, consider rosuvastatin, an AstraZeneca product. Rosuvastatin was approved about the same time as ezetimibe/simvastatin, and 6 years later it has already received a label change for the reduction of progression of atherosclerosis, based on positive outcomes in the METEOR trial,35 the ASTEROID intravascular ultrasonography trial,37 and the CORONA trial (an important trial that examined hard clinical end points).38 More importantly, the JUPITER trial was recently stopped early owing to a reduction in cardiovascular deaths. Initially, rosuvastatin received an unfair media portrayal as an unsafe drug. Now, because of its proven benefits in outcome trials, it will receive more widespread consideration for clinical use.
For preventive cardiologists, a painful reminder to focus on LDL-C
For the preventive cardiologist or lipidologist, the ENHANCE trial has been a painful reminder that despite overwhelming evidence, the mantra of “the lower the LDL-C the better” is still not universally accepted. We acknowledge the great benefits of statins, but the lure of “pleiotropic effects” distracts many of us from the necessity of more aggressive LDL-C reduction.
The pleiotropic benefits of statins were first raised as a means of supporting increased clinical use of pravastatin vis-a-vis other, more efficacious statins. It was not until the PROVE-IT study that pravastatin’s pleiotropic effects were found not to translate into a benefit equivalent to that of the more efficacious statin, atorvastatin.39
The success of ezetimibe was its ability to safely and easily lower LDL-C in combination with statins to achieve treatment goals. For many patients, a lower-dose statin and ezetimibe together provide a well-tolerated and efficacious approach to treating hyperlipidemia. The fallout from the ENHANCE trial is that many patients who were well treated or who could be better treated with ezetimibe in combination with a statin will not receive the best tolerated regimen. In fact, preliminary prescription data after the release of the ENHANCE study support our worse fear, ie, that patients at high risk will receive less aggressive LDL-C reduction. Since the ENHANCE data were released, more than 300,000 patients have stopped taking either ezetimibe/simvastatin or ezetimibe, and nearly all have continued on generic simvastatin or on a dose of statin with less overall efficacy.
An example is Senator John McCain, who, according to his recently released medical records, has a Framingham 10-year risk of more than 20% and was on ezetimibe/simvastatin to treat an elevated cholesterol level. After release of the ENHANCE trial, he was switched to generic simvastatin, and his LDL-C increased from 82 mg/dL to 122 mg/dL. He most likely has an LDL-C goal of less than 100 mg/dL according to the ATP III guidelines, and he is therefore no longer at his target.
For physicians in the community, questions from concerned patients
For the physicians who have received hundreds of phone calls and e-mails from concerned patients, the ENHANCE trial results must have been both discouraging and confusing. At present, I think we should remember the following:
- Ezetimibe’s mechanism of action is well understood
- It is safe and well-tolerated
- It still has a role as an add-on to statin therapy (or as monotherapy or combined with other agents in those who cannot tolerate statins) for patients who have not yet achieved their LDL-C target.
For the pharmaceutical industry, enormous challenges
The neutral ENHANCE trial results created an uncomfortable situation for the trial sponsor. A heavily marketed drug failed to achieve its expected result after the study results were delayed for a few months. The pharmaceutical industry ranks 14th out of 17 industries in public trust among the American public, and this study provided an opportunity for its critics to attack what is, in their opinion, an overly marketed drug.
Enormous challenges are on the horizon for the pharmaceutical industry, with a shrinking pipeline of potential new drugs, increasing regulatory hurdles, greater liability risk, political pressure for price controls, enhanced scrutiny of sales practices, and a growing media bias. As a cardiologist and clinical researcher whose father died at age 47 of a myocardial infarction, I am concerned that, unless change occurs, a vibrant pharmaceutical industry with the financial and intellectual capital to find and develop new, more effective treatments will cease to exist.
The Ezetimibe and Simvastatin in Hypercholesterolemia Enhances Atherosclerosis Regression (ENHANCE) trial1 was probably the most widely publicized clinical study of the past decade. How did a 720-patient imaging trial with a neutral result in patients with severe hypercholesterolemia rise to a level warranting massive media attention, a congressional investigation, and a recommendation to curtail the use of a drug widely used to reduce levels of low-density-lipoprotein cholesterol (LDL-C)?
The reaction to the ENHANCE trial reveals more about the political climate and the relationship between the pharmaceutical industry and the American public than it does about the effects of ezetimibe (available combined with simvastatin as Vytorin and by itself as Zetia) on the progression of atherosclerosis.
SOME SELF-DISCLOSURE
Before I discuss the clinical implications of the ENHANCE trial, I must describe both my financial conflicts and intellectual biases. I am a paid consultant, speaker, and researcher on behalf of Merck/Schering-Plough, the sponsor of the ENHANCE trial. I was a principal investigator in the first phase II trial of ezetimibe and have conducted more than 10 clinical trials of either ezetimibe or ezetimibe/simvastatin. I also have been a strong advocate for imaging trials to assist in the clinical development of novel therapeutic agents and to support regulatory approval.
Therefore, I believe that the thickness of the intima and media layers of the carotid arteries is a useful surrogate to evaluate the potential antiatherosclerotic effects of drugs (more on this topic below). Also, I believe that the LDL-C-lowering hypothesis has been proven: ie, that all drugs that lower LDL-C safely, without off-target adverse effects, should reduce cardiovascular events. I support the goal levels of LDL-C and non-high-density-lipoprotein cholesterol set by the National Cholesterol Education Program’s third Adult Treatment Panel (ATP III) guidelines,2,3 which specify LDL-C targets rather than the use of specific drugs. In spite of these conflicts and potential biases, I believe I have always served the best interests of patient care.
HISTORY OF THE ENHANCE TRIAL
The end point defined as the mean of six measurements
The primary end point was the change in the thickness of the intima and media layers of the carotid arteries over a 2-year period, measured by ultrasonography. A composite measure was used: the mean of the thicknesses in the far walls of the right and left common carotid arteries, the right and left carotid bulbs, and the right and left internal carotid arteries. Secondary end points included the change in the mean maximal carotid artery intima-media thickness (ie, the thickest of the six baseline measurements), the proportion of participants who developed new carotid artery plaque (defined arbitrarily as an intima-media thickness > 1.3 mm), and changes in the mean of the intima-media thickness of the six carotid sites plus the common femoral arteries.
The last participant completed the trial in April 2006. Reading of the almost 30,000 scans was not started until the last participant was finished, so that all scans for each participant could be read in a blinded, randomized order by five separate readers. A significant proportion of the images that the protocol called for could not be obtained or analyzed, particularly in the internal carotid artery and the carotid bulb, which are often difficult to visualize. As a result, 17% of the internal carotid or carotid bulb measurements were discarded.
To change the end point post hoc, or not to change the end point?
The sponsor of the trial was concerned about the missing data points and convened a special advisory board to review the blinded data. This group suggested a solution: changing the primary end point from the six-site composite value to the mean value in just the common carotid arteries. They based this suggestion on the greater success rate in measuring the common carotids (97%) than in measuring all six sites (88%), as well as on recent trials that indicated that the common carotid artery measurement correlates better with clinical outcomes (because the internal carotid and the bulb measurements vary more). On November 26, 2007, Merck/Schering-Plough announced the primary end point would be changed to the mean change in the common carotid arteries.
However, during a separate meeting on November 30, 2007, some members of the Merck/Schering-Plough advisory board objected to the change. On December 11, 2007, the company announced that the original primary end point would not be changed.
Neutral results, negative publicity
On December 31, 2007, the ENHANCE study was unblinded, and on January 14, 2008, Merck/Schering-Plough issued a press release announcing the results. The press release stated that there were no statistically significant differences between the treatment groups in the primary end point or in any of the secondary end points, despite a 16.5% greater reduction in LDL-C (about 50 mg/dL) in the group receiving the ezetimibe/simvastatin combination. The composite intima-media thickness had increased by an average of 0.0111 mm in the combined-therapy group vs 0.0058 mm in the simvastatin-only group (P = .29) over the 24-month treatment period.5
The press release received unprecedented international media attention. One leading cardiologist commented to the media that ENHANCE showed “millions of patients may be taking a drug [ezetimibe] that does not benefit them, raising their risk of heart attacks and exposing them to potential side effects.”6 The perceived message that ezetimibe/simvastatin is harmful resulted in thousands of phone calls from concerned patients to their physicians throughout the United States. The American Heart Association (AHA) and the American College of Cardiology (ACC) issued a joint statement the next day saying that ezetimibe/simvastatin does not appear to be unsafe and that patients should not stop taking the drug on their own. In the following days, Merck/Schering-Plough placed advertisements in newspapers reaffirming the safety of ezetimibe and quoting the AHA/ACC statement.
But the full results of the study were not available at that point. In fact, Senator Charles Grassley (R-Iowa) had launched a congressional investigation into the delays in releasing the results of the ENHANCE trial in December 2007. A focus of the investigation was whether the sponsor was delaying the release either because the data reflected negatively on its product or because it was legitimately concerned about the quality of the measurements of the carotid intima-media thickness. After Merck/Schering-Plough placed the advertisements quoting the AHA/ACC statement, these organizations were criticized for touting the safety of ezetimibe while receiving educational grants and other funds from Merck/Schering-Plough. Senator Grassley sent a letter to the ACC in late March requesting information about the amount of funds the ACC had received.
Full results are published, and the ACC is misquoted
The ENHANCE study was selected for a special presentation at the ACC annual scientific session on March 30, 2008. The full ENHANCE results were presented by Dr. Kastelein, after which an expert panel led by Harlan M. Krumholz, MD, discussed the trial’s implications. The ENHANCE results were simultaneously published in the New England Journal of Medicine,1 accompanied by an editorial by B. Greg Brown, MD, and Allen J. Taylor, MD,7 and another editorial by the editors of that journal, Jeffrey M. Drazen, MD, and colleagues.8 The expert panel and the editorialists concluded that the ENHANCE trial data raised concerns about the cardiovascular benefits of ezetimibe; that statins should be used as initial therapy for hyperlipidemia and titrated to the goal LDL-C level or to the maximally tolerated dose; and that other drugs such as bile acid sequestrants, fibrates, and niacin should be used in combination with statins before considering ezetimibe.9
The next day, stories appeared in the media mistakenly stating that the ACC had recommended that ezetimibe/simvastatin be discontinued. This view was fueled by an article in the ACC’s Scientific Session News, penned by a contract writer and editor, with the headline, “ACC on Vytorin: Go Back to Statins” that said, “After waiting for 18 months for the results of the ENHANCE study, an ACC panel on Sunday encouraged physicians to use statins as a first line and prescribe Vytorin only as a last resort for patients unable to tolerate other cholesterol-lowering agents.”10
The ACC later clarified that this was the opinion of the panelists and not that of the ACC, and they reiterated statements from the AHA/ACC Secondary Prevention Guidelines11 recommending statins in maximally tolerated doses or titrated to a goal LDL-C level for first-line drug treatment of coronary artery disease, and recommending that patients speak with their physicians before discontinuing any therapy.
WHY WERE THE ENHANCE STUDY RESULTS NEUTRAL?
The ACC expert panel concluded that the most likely reason for the neutral ENHANCE results was that ezetimibe lowers LDL-C but does not confer a cardiovascular benefit. In the words of Dr. Krumholz (as quoted by Shannon Pettypiece and Michelle Fay Cortez on bloomberg.com), ezetimibe is “just an expensive placebo.”12
There are at least three potential explanations for the lack of benefit with ezetimibe in the ENHANCE trial. I list them below in order of lowest to highest probability, in my opinion:
Theory 1: Ezetimibe lowers LDL-C but is not antiatherogenic
Since almost all experts agree that lowering LDL-C confers cardiovascular benefits, if ezetimibe does not inhibit atherosclerosis it must have some “off-target” effect that negates its LDL-C-lowering benefit. Critics of ezetimibe point out that oral estrogen and torcetrapib also lower LDL-C but do not improve cardiovascular outcomes.13,14
The lack of benefit with these two other agents can be explained. Oral estrogen does not lower apolipoprotein B (an indication of the number of atherogenic particles), but rather it increases the levels of both triglycerides and C-reactive protein, and it is prothrombotic in some people.15 Torcetrapib increases aldosterone production and substantially raises blood pressure.16 Therefore, both drugs have true off-target effects that could explain their failure to reduce cardiovascular risk despite reductions in LDL-C. (Interestingly, though, oral estrogen has been shown to slow the progression of carotid intima-media thickness in newly postmenopausal women.17
Ezetimibe, however, lowers LDL-C by an ultimate mechanism similar to that of statins and bile acid sequestrants, ie, by up-regulating LDL receptors, although these drugs reach this mechanism via different pathways. Statins inhibit cholesterol synthesis, thereby lowering hepatic intracellular cholesterol and thus up-regulating LDL-receptors and enhancing LDL-C clearance from the plasma. Bile acid sequestrants interrupt bile acid reabsorption in the ileum, thereby decreasing intracellular hepatic cholesterol and up-regulating LDL receptors. Ezetimibe, like bile acid sequestrants, also decreases cholesterol return to the liver, lowering hepatic intracellular levels and thus up-regulating LDL receptors.18
Ezetimibe is unlikely to have an off-target effect because it is only fractionally absorbed systemically, and a recent animal study showed that it enhances macrophage efflux of cholesterol, thereby potentially increasing reverse cholesterol transport.19 Ezetimibe has also been shown to reduce atherosclerosis in animal models.20
In their editorial, Drs. Brown and Taylor7 noted that ezetimibe reduces the expression of adenosine triphosphate binding cassette A1 (ABCA1) in Caco-2 (an intestinal cell line), and this may be an example of an off-target effect. However, statins also reduce ABCA1 expression in macrophages.21 ABCA1 is sensitive to intracellular cholesterol, and when cholesterol levels are decreased, whether by statins or by ezetimibe, ABCA1 expression is down-regulated.22
Theory 2: Intima-media thickness does not reflect the true benefits of lowering LDL-C
The carotid intima-media thickness is a surrogate end point that predicts coronary events and the rate of progression of coronary atherosclerosis.23 In trials of lovastatin (Mevacor),24 pravastatin (Pravachol),25 and rosuvastatin (Crestor),26 the carotid intima-media was thinner at 24 months with the active drug than with placebo. In two relatively small trials—ARBITER 1 (n = 161),27 which was open-label, and ASAP (n = 325)28,29—aggressive lipid-lowering reduced the progression of intima-media thickness better than less-aggressive therapy. However, this measure has been used to evaluate the effects of differing degrees of LDL-C reduction between active treatments in fewer than 500 research participants.
Furthermore, what part or parts of the carotid system are we talking about? In recent trials led by Dr. Kastelein, the intima-media thickness of the common carotid arteries increased with pactimibe (an acyl-coenzyme A:cholesterol O-acyltransferase, or ACAT, inhibitor)30 and torcetrapib,31 but the six-site composite measure (which was the primary end point in these trials, as in ENHANCE) did not increase more than in the control groups. Pactimibe was also shown to increase atheroma volume as measured by intravascular ultrasonography in the ACTIVATE trial.32 Therefore, the thickness of the common carotid arteries has been shown to be a better predictor of harm from a therapy than the composite measurement.
The advantage of measuring the common carotid artery is that it is easier to visualize and measure, and therefore the measurements vary less. In the METEOR trial,26 the six-site measurement increased significantly less with rosuvastatin than with placebo, but the common carotid measurement alone was more strongly associated with a difference in progression. In the ENHANCE trial, the thickness of the common carotid arteries increased by 0.0024 mm with simvastatin alone vs 0.0019 mm with simvastatin/ezetimibe, a difference of 0.005 mm that was not statistically significant (P = .93).1
Although the six-site measurement appears to be good for predicting coronary events and evaluating therapies, the measurement in the common carotid arteries appears to be a more reliable surrogate end point for predicting both benefit and harm from antiatherogenic agents. However, trials of statins and other lipid-lowering therapies that assessed clinical events have shown that the reduction in risk associated with a given reduction in cholesterol is similar regardless of the mechanism by which cholesterol is lowered.33 Therefore, the LDL-C level is far superior as a marker of clinical benefit.
Theory 3: Previous statin treatment affected the ENHANCE results
By far the most likely explanation for the neutral findings in ENHANCE is that the patients were so well treated before entry that it was impossible to detect a difference between the two treatment groups in carotid intima-media thickness at the end of the study. Eighty percent of the patients had received statins previously, and at baseline the mean intima-media thickness of the common carotid arteries was only 0.68 mm.1 In contrast, most other trials required a thickness greater than 0.7 mm for entry.
The two main reasons for selecting a population with familial hypercholesterolemia were the assumptions that these participants would have a greater-than-average carotid intima-media thickness at baseline and that they would show an above-average progression rate, even on high-dose statin therapy.4 Both of these assumptions were incorrect: the baseline thickness was normal and the progression rate was negligible in both groups.
Accordingly, the high prevalence of statin pretreatment and the near-normal carotid intima-media thickness at baseline may have prevented the 16.5% greater reduction in LDL-C due to ezetimibe from producing a difference in progression over 24 months of treatment. This conclusion is supported by the long-term follow-up results from ASAP, RADIANCE 1, and CAPTIVATE, all of which showed that in patients with familial hypercholesterolemia well treated with statins, progression of carotid intima-media thickness is negligible.30,31
Further supporting this view, in a previous trial by Dr. Kastelein’s group in patients with familial hypercholesterolemia,34 giving simvastatin 80 mg for 2 years decreased the intima-medial thickness by .081 mm (P < .001), compared with 0.0058 mm in ENHANCE (a 14-fold difference). In the previous trial, the baseline measurement was 1.07 mm (vs 0.68 mm in ENHANCE), and the extent of the change was significantly associated with the baseline measurement (r = .53, P < .001) but not with the change in LDL-C levels.
This is powerful evidence that, in two similar studies that used the same methodology and the same drug, the thinner arteries in the ENHANCE trial are by far the most likely explanation for the lack of change with the addition of ezetimibe to high-dose simvastatin. The METEOR trial enrolled only patients who had never received statins and whose carotid intima-media was thicker than 1.2 mm. In retrospect, a similar design would have been preferable for ENHANCE.35
LESSONS LEARNED AND CLINICAL IMPLICATIONS
For Merck/Schering-Plough, missed opportunities
Although Dr. Krumholz (the spokesman for the ACC panel discussion) and I disagree on the clinical implications of the ENHANCE trial, we do agree on an important point. Dr. Krumholz posed the question that if the LDL-C-lowering hypothesis was already proven for ezetimibe, why was the ENHANCE trial conducted? After 6 years on the market, the efficacy of ezetimibe on cardiovascular outcomes should already have been established. It should not take this long to determine the clinical outcome benefit for a drug.
Merck/Schering-Plough’s outcome program for ezetimibe was inadequately designed to demonstrate the clinical value of this novel compound. Rather than assuming the LDL-C-lowering hypothesis was already established, they conducted another “lower-is-better” trial with the carotid intima-media thickness as the end point, and they succeeded only in raising doubt about the benefits of ezetimibe rather than showing that dual therapy is at least equivalent to high-dose statin therapy.
A preferable approach would have been to compare the effects of a statin in low doses plus ezetimibe vs high-dose statin monotherapy on either surrogate or hard outcomes. If the low-dose statin/ezetimibe combination, which should lower the LDL-C level as much as high-dose statin monotherapy, could provide similar or better outcomes with fewer side effects, this trial would change our practice.
One had hoped that dual therapy, by reducing both intestinal cholesterol absorption and hepatic synthesis of cholesterol, would improve outcomes by modifying postprandial chylomicron composition or by reducing plant sterol absorption.36 Unfortunately, other outcome trials of ezetimibe/simvastatin will not provide an answer regarding the potential advantages of dual therapy. The SEAS study is comparing the number of clinical events in patients with aortic stenosis who receive ezetimibe/simvastatin or placebo; SHARP is being conducted in patients with chronic kidney disease. Although both groups of patients have high rates of coronary events, these trials will not address whether adding ezetimibe provides additional benefits. In fact, if the results of these trials turn out neutral, as in ENHANCE, then ezetimibe will be blamed for potentially offsetting the benefits of simvastatin, and if the trials show a benefit, the simvastatin component of ezetimibe/simvastatin will be given the credit.
The answer may come in 3 to 4 years with the results of IMPROVE-IT, a study of 18,000 patients with acute coronary syndrome treated with ezetimibe/simvastatin or simvastatin. The simvastatin monotherapy group will have a target LDL-C level of less than 80 mg/dL and the ezetimibe/simvastatin group will have an LDL-C target about 15% less. Although this trial is testing the lower-is-better hypothesis with ezetimibe, if the study does not show a benefit, it may not be because ezetimibe lacks clinical efficacy but rather because the LDL-C effect is curvilinear, and there is minimal further benefit of lowering the LDL-C level past 70 mg/dL. If the results of the IMPROVE-IT trial are negative, it may mean the end of ezetimibe as an LDL-C-lowering drug.
Merck/Schering-Plough has lost valuable time in not demonstrating the benefits of ezetimibe on clinical events. In contrast, consider rosuvastatin, an AstraZeneca product. Rosuvastatin was approved about the same time as ezetimibe/simvastatin, and 6 years later it has already received a label change for the reduction of progression of atherosclerosis, based on positive outcomes in the METEOR trial,35 the ASTEROID intravascular ultrasonography trial,37 and the CORONA trial (an important trial that examined hard clinical end points).38 More importantly, the JUPITER trial was recently stopped early owing to a reduction in cardiovascular deaths. Initially, rosuvastatin received an unfair media portrayal as an unsafe drug. Now, because of its proven benefits in outcome trials, it will receive more widespread consideration for clinical use.
For preventive cardiologists, a painful reminder to focus on LDL-C
For the preventive cardiologist or lipidologist, the ENHANCE trial has been a painful reminder that despite overwhelming evidence, the mantra of “the lower the LDL-C the better” is still not universally accepted. We acknowledge the great benefits of statins, but the lure of “pleiotropic effects” distracts many of us from the necessity of more aggressive LDL-C reduction.
The pleiotropic benefits of statins were first raised as a means of supporting increased clinical use of pravastatin vis-a-vis other, more efficacious statins. It was not until the PROVE-IT study that pravastatin’s pleiotropic effects were found not to translate into a benefit equivalent to that of the more efficacious statin, atorvastatin.39
The success of ezetimibe was its ability to safely and easily lower LDL-C in combination with statins to achieve treatment goals. For many patients, a lower-dose statin and ezetimibe together provide a well-tolerated and efficacious approach to treating hyperlipidemia. The fallout from the ENHANCE trial is that many patients who were well treated or who could be better treated with ezetimibe in combination with a statin will not receive the best tolerated regimen. In fact, preliminary prescription data after the release of the ENHANCE study support our worse fear, ie, that patients at high risk will receive less aggressive LDL-C reduction. Since the ENHANCE data were released, more than 300,000 patients have stopped taking either ezetimibe/simvastatin or ezetimibe, and nearly all have continued on generic simvastatin or on a dose of statin with less overall efficacy.
An example is Senator John McCain, who, according to his recently released medical records, has a Framingham 10-year risk of more than 20% and was on ezetimibe/simvastatin to treat an elevated cholesterol level. After release of the ENHANCE trial, he was switched to generic simvastatin, and his LDL-C increased from 82 mg/dL to 122 mg/dL. He most likely has an LDL-C goal of less than 100 mg/dL according to the ATP III guidelines, and he is therefore no longer at his target.
For physicians in the community, questions from concerned patients
For the physicians who have received hundreds of phone calls and e-mails from concerned patients, the ENHANCE trial results must have been both discouraging and confusing. At present, I think we should remember the following:
- Ezetimibe’s mechanism of action is well understood
- It is safe and well-tolerated
- It still has a role as an add-on to statin therapy (or as monotherapy or combined with other agents in those who cannot tolerate statins) for patients who have not yet achieved their LDL-C target.
For the pharmaceutical industry, enormous challenges
The neutral ENHANCE trial results created an uncomfortable situation for the trial sponsor. A heavily marketed drug failed to achieve its expected result after the study results were delayed for a few months. The pharmaceutical industry ranks 14th out of 17 industries in public trust among the American public, and this study provided an opportunity for its critics to attack what is, in their opinion, an overly marketed drug.
Enormous challenges are on the horizon for the pharmaceutical industry, with a shrinking pipeline of potential new drugs, increasing regulatory hurdles, greater liability risk, political pressure for price controls, enhanced scrutiny of sales practices, and a growing media bias. As a cardiologist and clinical researcher whose father died at age 47 of a myocardial infarction, I am concerned that, unless change occurs, a vibrant pharmaceutical industry with the financial and intellectual capital to find and develop new, more effective treatments will cease to exist.
- Kastelein JJ, Akdim F, Stroes ES, et al ENHANCE Investigators. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med 2008; 358:1431–1443.
- Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Grundy SM, Cleeman JI, Bairey Merz N, et al for the Coordinating Committee of the National Cholesterol Education Program. Circulation 2004; 110:227–239.
- Kastelein JJ, Sager PT, de Groot E, Veltri E. Comparison of ezetimibe plus simvastatin versus simvastatin monotherapy on atherosclerosis progression in familial hypercholesterolemia. Design and rationale of the Ezetimibe and Simvastatin in Hypercholesterolemia Enhances Atherosclerosis Regression (ENHANCE) trial. Am Heart J 2005; 49:234–239.
- Merck/Schering-Plough Pharmaceutical Press Release, January 14, 2008.
- Berenson A. Study reveals doubt on drug for cholesterol. New York Times January 15, 2008.
- Brown BG, Taylor AJ. Does ENHANCE diminish confidence in lowering LDL or in ezetimibe? N Engl J Med 2008; 358:1504–1507.
- Drazen JM, Jarcho JA, Morrissey S, Curfman GD. Cholesterol lowering and ezetimibe. N Engl J Med 2008; 358:1507–1508.
- American College of Cardiology. ENHANCED analysis of ezetimibe. ACC News, April 2, 2008. www.acc.org/emails/myacc/accnews%5Fapril%5F02%5F08.htm. Accessed 6/2/2008.
- American College of Cardiology. ACC panel on Vytorin: Go back to statins. Scientific Session News 3/31/2008. http://www.acc08.acc.org/SSN/Documents/ACC%20Monday%20v2.pdf. Accessed 6/2/2008.
- Smith SC, Allen J, Blair SN, et al. AHA/ACC guidelines for secondary prevention for patients with coronary and other atherosclerotic vascular disease: 2006 update. Endorsed by the National Heart, Lung, and Blood Institute. J Am Coll Cardiol 2006; 47:2130–2139.
- Pettypiece S, Cortez MF. Merck, Schering plunge as doctors discourage Vytorin. www.bloomberg.com/apps/news?pid=20601103&refer=news&sid=aV_T9WirgAkI. Accessed 6/2/2008.
- Barter PJ, Caulfield M, Eriksson M, et al ILLUMINATE Investigators. . Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 2007; 357:2109–2122.
- Hulley S, Grady D, Bush T, et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in post-menopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA 1998; 280:605–613.
- Rader DJ. Illuminating HDL—is it still a viable therapeutic target? N Engl J Med 2007; 357:2180–2183.
- Davidson MH, Maki KC, Marx P, et al. Effects of continuous estrogen and estrogen-progestin replacement regimens on cardiovascular risk markers in postmenopausal women. Arch Intern Med 2000; 160:3315–3325.
- Hodis HN, Mack WJ, Lobo RA, et al Estrogen in the Prevention of Atherosclerosis Trial Research Group. . Estrogen in the prevention of atherosclerosis. A randomized, double-blind, placebo-controlled trial. Ann Intern Med 2001; 135:939–953.
- Turley SD. Cholesterol metabolism and therapeutic targets: rationale for targeting multiple metabolic pathways. Clin Cardiol 2004; 27( suppl 3):III16–III21.
- Sehayek E, Hazen SL. Cholesterol absorption from the intestine is a major determinant of reverse cholesterol transport from peripheral tissue macrophages. Arterioscler Thromb Vasc Biol 2008;27 (Epub ahead of print].
- Davis HR, Compton DS, Hoos L, Tetzloff G. Ezetimibe, a potent cholesterol absorption inhibitor, inhibits the development of atherosclerosis in ApoE knockout mice. Arterioscler Thromb Vasc Biol 2001; 21:2032–2038.
- Wong J, Quinn CM, Gelissen IC, Jessup W, Brown AJ. The effect of statins on ABCA1 and ABCG1 expression in human macrophages is influenced by cellular cholesterol levels and extent of differentiation. Atherosclerosis 2008; 196:180–189.
- Wang N, Tall AR. Regulation and mechanisms of ATP-binding cassette transporter A1-mediated cellular cholesterol efflux. Arterioscler Thromb Vasc Biol 2003; 23:1178–1184.
- Bots ML. Carotid intima-media thickness as a surrogate marker for cardiovascular disease in intervention studies. Curr Med Res Opin 2006; 22:2181–2190.
- Byington RP, Evans GW, Espeland MA, et al. Effects of lovastatin and warfarin on early carotid atherosclerosis: sex-specific analyses. Asymptomatic Carotid Artery Progression Study (ACAPS) Research Group. Circulation 1999; 100:e14–e17.
- Byington RP, Furberg CD, Crouse JR, Espeland MA, Bond MG. Pravastatin, Lipids, and Atherosclerosis in the Carotid Arteries (PLAC-II). Am J Cardiol 1995; 76:54C–59C.
- Crouse JR, Raichlen JS, Riley WA, et al METEOR Study Group. . Effect of rosuvastatin on progression of carotid intima-media thickness in low-risk individuals with subclinical atherosclerosis: the METEOR trial. JAMA 2007; 297:1344–1353.
- Taylor AJ, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation 2004; 110:3512–3517.
- Smilde TJ, van Wissen S, Wollersheim H, Trip MD, Kastelein JJ, Stalenhoef AF. Effect of aggressive versus conventional lipid lowering on atherosclerosis progression in familial hypercholesterolaemia (ASAP): a prospective, randomised, double-blind trial. Lancet 2001; 357:577–581.
- van Wissen S, Smilde TJ, Trip MD, Stalenhoef AFH, Kastelein JJP. Long-term safety and efficacy of high-dose atorvastatin treatment in patients with familial hypercholesterolemia. Am J Cardiol 2005; 95:264–266.
- Meuwese MC, Franssen R, Stroes ES, Kastelein JJ. And then there were acyl coenzyme A:cholesterol acyl transferase inhibitors. Curr Opin Lipidol 2006; 17:426–430.
- Kastelein JJ, van Leuven SI, Burgess L, et al RADIANCE 1 Investigators. . Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia. N Engl J Med 2007; 356:1620–1630.
- Nissen SE, Tuzcu EM, Brewer HB, et al ACAT Intravascular Atherosclerosis Treatment Evaluation (ACTIVATE) Investigators. Effect of ACAT inhibition on the progression of coronary atherosclerosis. N Engl J Med 2006; 354:1253–1263.
- Davidson MH. Clinical significance of statin pleiotropic effects: hypotheses versus evidence. Circulation 2005; 111:2280–2281.
- Nolting PR, de Groot E, Zwinderman AH, Buirma RJ, Trip MD, Kastelein JJ. Regression of carotid and femoral artery intima-media thickness in familial hypercholesterolemia. Arch Intern Med 2003; 163:1837–1841.
- Crouse JR, Grobbee DE, O’Leary DH, et al Measuring Effects on intima media Thickness: an Evaluation Of Rosuvastatin Study Group. . Measuring effects on intima media thickness: an evaluation of rosuvastatin in subclinical atherosclerosis—the rationale and methodology of the METEOR study. Cardiovasc Drugs Ther 2004; 18:231–238.
- Toth PP, Davidson MH. Cholesterol absorption blockade with ezetimibe. Curr Drug Targets Cardiovasc Haematol Disord 2005; 5:455–462.
- Nissen SE, Nicholls SJ, Sipahi I, et al ASTEROID Investigators. . Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006; 295:1556–1565.
- Kjekshus J, Apetrei E, Barrios V, et al CORONA Group. . Rosuvastatin in older patients with systolic heart failure. N Engl J Med 2007; 357:2248–2261.
- Cannon CP, Braunwald E, McCabe CH, et al Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. . Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Kastelein JJ, Akdim F, Stroes ES, et al ENHANCE Investigators. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med 2008; 358:1431–1443.
- Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Grundy SM, Cleeman JI, Bairey Merz N, et al for the Coordinating Committee of the National Cholesterol Education Program. Circulation 2004; 110:227–239.
- Kastelein JJ, Sager PT, de Groot E, Veltri E. Comparison of ezetimibe plus simvastatin versus simvastatin monotherapy on atherosclerosis progression in familial hypercholesterolemia. Design and rationale of the Ezetimibe and Simvastatin in Hypercholesterolemia Enhances Atherosclerosis Regression (ENHANCE) trial. Am Heart J 2005; 49:234–239.
- Merck/Schering-Plough Pharmaceutical Press Release, January 14, 2008.
- Berenson A. Study reveals doubt on drug for cholesterol. New York Times January 15, 2008.
- Brown BG, Taylor AJ. Does ENHANCE diminish confidence in lowering LDL or in ezetimibe? N Engl J Med 2008; 358:1504–1507.
- Drazen JM, Jarcho JA, Morrissey S, Curfman GD. Cholesterol lowering and ezetimibe. N Engl J Med 2008; 358:1507–1508.
- American College of Cardiology. ENHANCED analysis of ezetimibe. ACC News, April 2, 2008. www.acc.org/emails/myacc/accnews%5Fapril%5F02%5F08.htm. Accessed 6/2/2008.
- American College of Cardiology. ACC panel on Vytorin: Go back to statins. Scientific Session News 3/31/2008. http://www.acc08.acc.org/SSN/Documents/ACC%20Monday%20v2.pdf. Accessed 6/2/2008.
- Smith SC, Allen J, Blair SN, et al. AHA/ACC guidelines for secondary prevention for patients with coronary and other atherosclerotic vascular disease: 2006 update. Endorsed by the National Heart, Lung, and Blood Institute. J Am Coll Cardiol 2006; 47:2130–2139.
- Pettypiece S, Cortez MF. Merck, Schering plunge as doctors discourage Vytorin. www.bloomberg.com/apps/news?pid=20601103&refer=news&sid=aV_T9WirgAkI. Accessed 6/2/2008.
- Barter PJ, Caulfield M, Eriksson M, et al ILLUMINATE Investigators. . Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 2007; 357:2109–2122.
- Hulley S, Grady D, Bush T, et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in post-menopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA 1998; 280:605–613.
- Rader DJ. Illuminating HDL—is it still a viable therapeutic target? N Engl J Med 2007; 357:2180–2183.
- Davidson MH, Maki KC, Marx P, et al. Effects of continuous estrogen and estrogen-progestin replacement regimens on cardiovascular risk markers in postmenopausal women. Arch Intern Med 2000; 160:3315–3325.
- Hodis HN, Mack WJ, Lobo RA, et al Estrogen in the Prevention of Atherosclerosis Trial Research Group. . Estrogen in the prevention of atherosclerosis. A randomized, double-blind, placebo-controlled trial. Ann Intern Med 2001; 135:939–953.
- Turley SD. Cholesterol metabolism and therapeutic targets: rationale for targeting multiple metabolic pathways. Clin Cardiol 2004; 27( suppl 3):III16–III21.
- Sehayek E, Hazen SL. Cholesterol absorption from the intestine is a major determinant of reverse cholesterol transport from peripheral tissue macrophages. Arterioscler Thromb Vasc Biol 2008;27 (Epub ahead of print].
- Davis HR, Compton DS, Hoos L, Tetzloff G. Ezetimibe, a potent cholesterol absorption inhibitor, inhibits the development of atherosclerosis in ApoE knockout mice. Arterioscler Thromb Vasc Biol 2001; 21:2032–2038.
- Wong J, Quinn CM, Gelissen IC, Jessup W, Brown AJ. The effect of statins on ABCA1 and ABCG1 expression in human macrophages is influenced by cellular cholesterol levels and extent of differentiation. Atherosclerosis 2008; 196:180–189.
- Wang N, Tall AR. Regulation and mechanisms of ATP-binding cassette transporter A1-mediated cellular cholesterol efflux. Arterioscler Thromb Vasc Biol 2003; 23:1178–1184.
- Bots ML. Carotid intima-media thickness as a surrogate marker for cardiovascular disease in intervention studies. Curr Med Res Opin 2006; 22:2181–2190.
- Byington RP, Evans GW, Espeland MA, et al. Effects of lovastatin and warfarin on early carotid atherosclerosis: sex-specific analyses. Asymptomatic Carotid Artery Progression Study (ACAPS) Research Group. Circulation 1999; 100:e14–e17.
- Byington RP, Furberg CD, Crouse JR, Espeland MA, Bond MG. Pravastatin, Lipids, and Atherosclerosis in the Carotid Arteries (PLAC-II). Am J Cardiol 1995; 76:54C–59C.
- Crouse JR, Raichlen JS, Riley WA, et al METEOR Study Group. . Effect of rosuvastatin on progression of carotid intima-media thickness in low-risk individuals with subclinical atherosclerosis: the METEOR trial. JAMA 2007; 297:1344–1353.
- Taylor AJ, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation 2004; 110:3512–3517.
- Smilde TJ, van Wissen S, Wollersheim H, Trip MD, Kastelein JJ, Stalenhoef AF. Effect of aggressive versus conventional lipid lowering on atherosclerosis progression in familial hypercholesterolaemia (ASAP): a prospective, randomised, double-blind trial. Lancet 2001; 357:577–581.
- van Wissen S, Smilde TJ, Trip MD, Stalenhoef AFH, Kastelein JJP. Long-term safety and efficacy of high-dose atorvastatin treatment in patients with familial hypercholesterolemia. Am J Cardiol 2005; 95:264–266.
- Meuwese MC, Franssen R, Stroes ES, Kastelein JJ. And then there were acyl coenzyme A:cholesterol acyl transferase inhibitors. Curr Opin Lipidol 2006; 17:426–430.
- Kastelein JJ, van Leuven SI, Burgess L, et al RADIANCE 1 Investigators. . Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia. N Engl J Med 2007; 356:1620–1630.
- Nissen SE, Tuzcu EM, Brewer HB, et al ACAT Intravascular Atherosclerosis Treatment Evaluation (ACTIVATE) Investigators. Effect of ACAT inhibition on the progression of coronary atherosclerosis. N Engl J Med 2006; 354:1253–1263.
- Davidson MH. Clinical significance of statin pleiotropic effects: hypotheses versus evidence. Circulation 2005; 111:2280–2281.
- Nolting PR, de Groot E, Zwinderman AH, Buirma RJ, Trip MD, Kastelein JJ. Regression of carotid and femoral artery intima-media thickness in familial hypercholesterolemia. Arch Intern Med 2003; 163:1837–1841.
- Crouse JR, Grobbee DE, O’Leary DH, et al Measuring Effects on intima media Thickness: an Evaluation Of Rosuvastatin Study Group. . Measuring effects on intima media thickness: an evaluation of rosuvastatin in subclinical atherosclerosis—the rationale and methodology of the METEOR study. Cardiovasc Drugs Ther 2004; 18:231–238.
- Toth PP, Davidson MH. Cholesterol absorption blockade with ezetimibe. Curr Drug Targets Cardiovasc Haematol Disord 2005; 5:455–462.
- Nissen SE, Nicholls SJ, Sipahi I, et al ASTEROID Investigators. . Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006; 295:1556–1565.
- Kjekshus J, Apetrei E, Barrios V, et al CORONA Group. . Rosuvastatin in older patients with systolic heart failure. N Engl J Med 2007; 357:2248–2261.
- Cannon CP, Braunwald E, McCabe CH, et al Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. . Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
Movement disorder emergencies in the elderly: Recognizing and treating an often-iatrogenic problem
Although we tend to think of movement disorders as chronic conditions, some of them can present as true emergencies in which failure to diagnose the condition and treat it promptly can result in significant sickness or even death.
Many cases are iatrogenic, occurring in patients with Parkinson disease or those taking antipsychotic or antidepressant medications when their regimen is started or altered. Elderly patients are particularly at risk, as they take more drugs and have less physiologic reserve.
Movement disorder emergencies in elderly patients can be difficult to diagnose and treat, since many patients are taking more than one medication: polypharmacy raises the possibility of interactions, and different drugs can cause different movement disorder syndromes. Moreover, because so many patients are at risk—for example, more than 1 million people in the United States now have Parkinson disease, and the number is growing—it is important for physicians who take care of the elderly to be more informed about these disorders, especially the presenting symptoms.
SCOPE OF THIS ARTICLE

- Disorders presenting with rigidity or stiffness
- Disorders presenting with dystonia
- Disorders presenting with hyperkinetic movements
- Disorders presenting with psychiatric disturbances.
Of these, the scenarios most likely to require emergency evaluation in the elderly are acute hypokinetic and hyperkinetic syndromes and psychiatric presentations. This article discusses movement disorder emergencies in the elderly, focusing on the more common disorders with common presentations.
DISORDERS PRESENTING WITH RIGIDITY OR STIFFNESS
Serotonin syndrome

Chief among the offenders are the selective serotonin-reuptake inhibitors (SSRIs), either alone or in combination. This syndrome occurs in 14% to 16% of patients who overdose on SSRIs.1 Examples of combinations that can lead to serotonin syndrome are an SSRI plus any of the following:
- An anxiolytic such as buspirone (BuSpar; this combination is popular for the treatment of depression and anxiety)
- A tricyclic agent such as imipramine (Tofranil)
- A serotonin and norepinephrine reuptake inhibitor such as venlafaxine (Effexor).
In addition, antiparkinson drugs such as levodopa and selegiline (Eldepryl) enhance serotonin release.
Signs and symptoms. Serotonin syndrome is characterized by:
- Severe rigidity
- Dysautonomia
- Change in mental status.
Other clinical findings include fever, gastrointestinal disturbances, and motor restlessness. Clonus is the most important finding in establishing the diagnosis.2
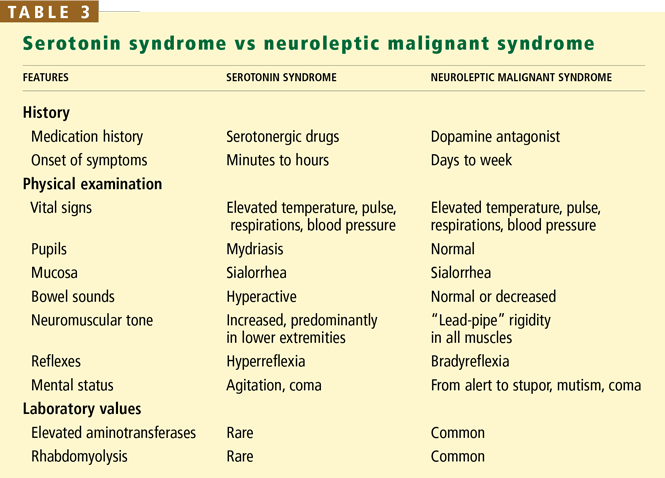
The syndrome may initially go unrecognized and can be mistaken for viral illness or anxiety.4 Manifestations range from mild to life-threatening; initially, it may present with akathisia and tremor. The symptoms progress rapidly over hours and can range from myoclonus, hyperreflexia, and seizures to severe forms of rhabdomyolysis, renal failure, and respiratory failure. The hyperreflexia and clonus seen in moderate cases may be considerably greater in the lower extremities than in the upper extremities.5
No laboratory test confirms the diagnosis, but tremor, clonus, or akathisia without additional extrapyramidal signs should lead to the diagnosis if the patient was taking a serotonergic medication.5 The onset of symptoms is usually rapid. The majority of patients present within 6 hours after initial use of the medication, an overdose, or a change in dosing.5
Treatment. The first steps are to stop the serotonergic medication and to hydrate and cool the patient to counteract the hyperpyrexic state. Benzodiazepine drugs are important in controlling agitation, regardless of its severity.5 Propranolol (Inderal) is not recommended, as it may cause hypotension and shock in patients with autonomic instability.5
Patients with moderate cases may additionally benefit from cyproheptadine (Periactin), an antihistamine that antagonizes serotonin. The initial dose is 4 to 8 mg orally, with a repeat dose after 2 hours.6 Whether to continue this treatment depends on the response after two doses.
If medications must be given parenterally, physicians can consider chlorpromazine (Thorazine) 50 to 100 mg intramuscularly.5
Vital signs should be monitored. In severe cases, intensive care may be required with immediate sedation, neuromuscular paralysis, and intubation.
In most cases, patients improve rapidly.
Comment. Serotonin syndrome can be avoided by educating physicians and by modifying prescribing practices.5 Avoiding multidrug regimens is critical to preventing serotonin syndrome. Computer-based ordering systems and personal digital assistants can help one avoid drug interactions.5
Neuroleptic malignant syndrome
This syndrome is an infrequent but potentially lethal complication associated with therapy with antipsychotic drugs such as haloperidol (Haldol) and lithium (Eskalith) and with other medications with dopamine type-2 receptor antagonist activity such as metoclopramide (Reglan) and prochlorperazine (Compazine). It has become rare since the introduction of atypical antipsychotics and now occurs in 0.2% of patients receiving atypical antipsychotics.7 Its pathogenesis is not fully understood.
This syndrome occurs mainly in young or middle-aged patients receiving doses of neuroleptics within the usual therapeutic range, but it also appears to occur in elderly patients who receive higher doses.8 Although most cases develop in the first 2 weeks of treatment, it can develop at any time during therapy.
Signs and symptoms. Four features characterize neuroleptic malignant syndrome9:
- Muscle rigidity—generalized (“lead-pipe”) muscular rigidity is accompanied by bradykinesia or akinesia.
- Autonomic dysregulation, with tachycardia, tachypnea, alterations in blood pressure, excessive sweating, and incontinence.
- Hyperthermia—fever can begin hours to days after initiating or increasing the dose of a dopamine antagonist.
- Altered sensorium, ranging from confusion to disorientation and coma.
Symptoms of neuroleptic malignant syndrome typically evolve over several days, in contrast to the rapid onset of the serotonin syndrome. Knowing the precipitating drug also helps distinguish the syndromes: dopamine antagonists produce bradykinesia, whereas serotonin agonists produce hyperkinesia.5
Laboratory abnormalities include elevated serum creatine kinase concentrations and white blood cell counts. Renal function should be assessed when renal failure and rhabdomyolysis are suspected.
Treatment involves stopping the causative medication, cooling the patient, and supporting vital functions.
In mild cases (eg, low-grade fever) benzodiazepines such as lorazepam (Ativan) can stabilize the condition. In moderate cases (eg, more significant rigidity), dopaminergic agonists such as bromocriptine (Parlodel) can be given, although there is no strong clinical evidence for their use. Bromocriptine is usually started at 2.5 mg three times a day and gradually increased in dose if tolerated.
In severe cases, muscle rigidity can be reduced with dantrolene (Dantrium), a muscle relaxant. Dantrolene is started at 1 mg/kg intravenously every 6 hours and gradually increased up to 10 mg/kg total per day.
Some patients remain rigid and febrile up to 4 weeks after the causative agent has been withdrawn. Therefore, these treatments can be continued for a few weeks. After the patient has recovered fully, if it is necessary to resume antipsychotic therapy, an atypical antipsychotic such as quetiapine (Seroquel) can be started after 2 weeks.8
Comment. Although uncommon, neuroleptic malignant syndrome is the most serious adverse effect of neuroleptic drugs, and it is potentially fatal. When neuroleptic malignant syndrome is suspected, treatment should be prompt, and the neuroleptic medication should be immediately stopped.
Parkinsonism-hyperpyrexia syndrome
Withdrawing or decreasing the dose of dopaminergic medications in patients with Parkinson disease can cause parkinsonism-hyperpyrexia syndrome, a condition that is similar to neuroleptic malignant syndrome. It can also arise after sudden withdrawal of amantadine (Symmetrel) or anticholinergics. In view of this concern, adjustments to antiparkinson drugs may need to be more gradual in some elderly patients.
Patients present with fever, rigidity, and autonomic instability and are at risk of aspiration pneumonia.
Treatment includes resuming dopaminergic therapy and giving supportive care.
Apomorphine (Apokyn), a dopaminergic agonist, was used in a 71-year-old female parkinsonian patient who developed parkinsonism-hyperpyrexia syndrome after abrupt reduction of chronic levodopa treatment.10 The symptoms resolved within 24 hours of the addition of apomorphine to her previous levodopa therapy. If the patient is taking apomorphine for the first time, the injections should be given in low doses, 0.2 mL subcutaneously. Apomorphine can induce vomiting, and if this occurs an antiemetic such as trimethobenzamide (Tigan) should be given before subsequent injections. In the elderly, caution is advised as apomorphine may cause severe orthostasis.
Methylprednisolone (Solu-Medrol) pulse therapy has been shown to shorten the duration of this syndrome in a randomized, controlled study.11
Akinetic syndrome after failure of deep brain stimulator
Deep brain stimulation involves surgical placement of a pacemaker with electrodes in specific areas of the brain. It is used to control Parkinson disease, tremor, and, less commonly, dystonia, and a number of other uses are under investigation. Continuous electrical stimulation of different nuclei in the brain has been shown to alleviate some symptoms of Parkinson disease (eg, rigidity) and to enable some patients to decrease the dose of their antiparkinson medications.
Several cases have been reported of sudden, unexpected reappearance of freezing, gait disturbance, or severe akinesia in Parkinson disease patients whose stimulators had been turned off inadvertently (eg, by a magnet in a dicating machine that was placed too close to the stimulator) and who presented to an emergency room.12
Treatment is easy if this diagnosis is considered. Checking the neurostimulator and switching it to “on” are all that is needed. Since patients and their caregivers are trained how to check and turn on the stimulator, the role of the geriatrician is simply to remind the caregiver of this possibility.
FDA warning. The US Food and Drug Administration has issued a warning against use of shortwave or microwave diathermy for patients with deep brain stimulation or other implanted leads (www.fda.gov/cdrh/safety/121902.html), stating: “There are three types of diathermy equipment used by physicians, dentists, physical therapists, chiropractors, sports therapists, and others: radio frequency (shortwave) diathermy, microwave diathermy, and ultrasound diathermy. Shortwave and microwave diathermy, in both heating and nonheating modes, can result in serious injury or death to patients with implanted devices with leads. This kind of interaction is not expected with ultrasound diathermy. Electrocautery devices are not included in this notification.” If a patient has an implanted deep brain stimulator, magnetic resonance imaging should be done only if absolutely needed and then only if the guidelines are followed.
DISORDERS PRESENTING WITH DYSTONIA
Acute dystonic reaction
Medications are a common cause of acute focal dystonia. The symptoms, which can be life-threatening, usually occur within 24 hours after taking the medication.12 The most common offenders are neuroleptic drugs and antiemetic drugs with dopamine-blocking activity (eg, metoclopramide), although in older patients, they are more likely to cause tardive dyskinesia and parkinsonism.13,14
Metoclopramide accounts for nearly one-third of all drug-induced movement disorders, and this adverse effect is a common reason for malpractice suits. The entire spectrum of drug-induced movement disorders, ranging from subtle to life-threatening, can ensue from its use; akathisia and dystonia are generally seen early in the course of metoclopramide-induced movement disorders, whereas tardive dyskinesia and parkinsonism seem to be more prevalent in long-term users.15
Treatment includes stopping the precipitating medication and reversing dystonia with anticholinergic medications such as benztropine (Cogentin). Anticholinergic therapy is given intravenously or intramuscularly followed by oral therapy for few days, as the acute dystonic reaction may recur after the effect of parenteral medication wears off.
Intravenous diphenhydramine (Benadryl), an antihistamine with additional anticholiner-gic effects, can abort dystonia in a few minutes.16
Laryngeal dystonia accompanied by multiple system atrophy
Multiple system atrophy, a Parkinson-plus syndrome, is characterized by parkinsonism (mostly with poor response to levodopa) and early onset of dysfunction of the autonomic nervous system, urinary tract, cerebellum, and corticospinal tract (hyperreflexia).17
In the course of the disease, about one-third of patients develop respiratory stridor due to abnormal movements of the vocal cords.18 Nocturnal stridor portends a poor prognosis,19 with an increased risk of sudden death. Geriatricians should be aware of these symptoms, as these patients may seek care because of hoarseness or difficulty swallowing.
Treatment. Laryngeal dystonia can be improved with continuous positive airway pressure. In some cases, tracheostomy may be needed.19
Sudden withdrawal of baclofen
Baclofen (Lioresal), a treatment for spasticity and dystonia, is delivered via a pump through a catheter into an intrathecal space. The pump needs to be refilled every 3 to 6 months. Sudden discontinuation of medication caused by a dislodged catheter tip or forgetting to refill the pump provokes withdrawal symptoms. Patients with this life-threatening syndrome can present with rigidity, fever, change in mental status, and worsening dystonic symptoms.
Treatment involves high doses of baclofen (up to 120 mg/day in divided doses).6
DISORDERS PRESENTING WITH HYPERKINETIC MOVEMENTS
Chorea, ballism (ballismus), and athetosis constitute a range of involuntary, hyperkinetic movement disorders. Chorea consists of involuntary, continuous, sudden, brief, unsustained, irregular movements that flow from one part of the body to another. Hemiballism presents as forceful flinging movements of the limbs or high-amplitude chorea that affects one side of the body.
Acute hemichorea and hemiballism
Acute hemichorea and hemiballism commonly result from infarction or hemorrhage of the basal ganglia.20 Computed tomography and especially magnetic resonance imaging can show the lesions in patients with ballism. Stroke-induced ballism is usually self-limited and resolves after a few weeks. Acute hemiballism generally evolves to hemichorea or hemiathetosis in a few days, which requires only protective measures.
Treatment. Mild cases do not need treatment but severe cases call for medical therapy. Antidopaminergics are the drugs of choice. A dopamine depletor such as reserpine (Serpasil) 0.1 mg once or twice daily or dopamine receptor blockers such as neuroleptics are considered.16 The combination of a benzodiazepine plus an antipsychotic such as olanzapine (Zyprexa) has been suggested.6
Severe parkinsonian dyskinesia
Dyskinesia is common in Parkinson disease, and patients may present to an emergency room with severe levodopa-induced dyskinesia. Dyskinesia can be exhausting if prolonged and severe. Elevated levels of creatine kinase raise the concern of rhabdomyolysis. In rare cases, the patient develops respiratory dyskinesia when respiratory muscles such as those in the diaphragm become involved.21
The risk of levodopa-induced dyskinesia increases with disease severity and higher levodopa doses. Using a dopamine agonist as initial therapy delays the onset of levodopa-induced dyskinesia in early Parkinson disease. However, Factor and Molho,21 in a case series, reported that adding dopamine agonists to the regimen was a precipitating factor; another was infection.
Treatment. A reasonable approach to treating peak-dose dyskinesia is to lower the doses of dopaminergic medications.
A mild sedative such as lorazepam, alprazolam (Xanax, Niravam), or clonazepam (Klonopin) may reduce the severity of dyski-nesia.21 These drugs are particularly useful if the dyskinesia is worse at night, and they can be used in the emergency department while waiting for the effect of the dopaminergic medications to wear off.
Amantadine ameliorates levodopa-induced peak-dose dyskinesia without worsening parkinsonian symptoms in some patients.22
Drug-induced myoclonus
Myoclonus is sudden, jerky, brief involuntary movement of the face, limbs, or trunk. Unlike tics, myoclonus cannot be controlled by the patient.
Myoclonus has various pathophysiologic mechanisms. Most myoclonic emergencies are epileptic myoclonic seizures, which are beyond the scope of this article. Often, myoclonus is caused by opiate overdose or withdrawal. It can also be a side effect of SSRIs, tricyclic anti-depressants, lithium, amantadine, and rarely, antibiotics such as imipenem (Primaxin).23
Treatment. Opiate-induced myoclonus may respond to naloxone (Narcan), whereas opiate withdrawal responds to benzodi-azepines.6
Acute akathisia
Acute akathisia occurs in susceptible patients after exposure to dopamine receptor blockers or dopamine depletors. It is characterized by subjective restless feelings accompanied by objective restless movements. The course is usually self-limited after the causative medication is discontinued.
Treatment. Symptomatic treatment may be needed in most cases for several days. Anticholinergics are effective. Additionally, vitamin B6, mianserine, propranolol, and mirtazapine (Remeron) in a low dose (15 mg/day) have been shown to be effective16,24,25
DISORDERS WITH PSYCHIATRIC PRESENTATIONS
Hallucinations and psychosis in Parkinson disease
Neuropsychiatric or behavioral complications of Parkinson disease include hallucinations, dementia, depression, psychosis, and sleep disorders.21,26 Psychosis is the leading reason for nursing home placement in advanced cases.27 Psychosis can present as hallucinations or a paranoid delusional state in association with clear sensorium.28 However, hallucinations accounted for only 3% of emergency admissions to the hospital for Parkinson disease patients in one series.29
Risk factors for hallucinations in parkinsonian patients include dementia, long-term therapy with dopaminergic drugs, long duration of disease, advanced age, anticholinergic drugs, and sleep disorders. Severe cognitive impairment or dementia is a major and independent predictive factor for visual hallucinations.30
Most hallucinations are visual; auditory, tactile, and olfactory hallucinations are rare.30
Treatment initially should be the same as in any patient with delirium. The systemic disorders that can aggravate or cause hallucinations such as electrolyte abnormalities, urinary or respiratory infection, and systemic illness should be ruled out.
The next step is to reduce or discontinue the adjunctive drugs that have the least antiparkinsonian effect and the greatest potential of inducing hallucination or psychosis. Examples of such medications include histamine-2 antagonists (eg, cimetidine [Tagamet], amantadine, selegeline, and anti-cholinergics). Selegeline can be discontinued abruptly because it has a long duration of action in the brain, but amantadine and anti-cholinergics should be tapered. Dopamine agonists can be discontinued. Levodopa can be reduced until the side effects begin to subside without significant worsening of motor symptoms.
If all the above adjustments fail, an antipsychotic medication can be considered.26 Clozapine (Clozaril) has the best result and is nearly free of extrapyramidal side effects but can cause agranulocytosis, which requires frequent blood counts. The Parkinson Study Group suggested that clozapine, at daily doses of 50 mg or less, is safe and significantly improves drug-induced psychosis without worsening parkinsonism.31 Clozapine may be impractical for elderly patients due to its side effect profile.
Quetiapine is a good alternative to cloza-pine and is less likely to worsen parkinsonian symptoms than other atypical antipsy-chotics.32 Olanzapine and risperidone (Risperdal) are reported to worsen parkinsonian symptoms.33 Not enough data have been published about the efficacy of the newer medications such as ziprasidone (Geodon) and aripiprazole (Abilify) to advocate their routine clinical use.
Rivastigmine (Exelon) was reported to improve hallucinations, sleep disturbance, and caregiver distress in addition to enhancing cognitive performance in advanced Parkinson disease in a small study.34 Burn and colleagues35 reported that rivastigmine was beneficial in patients with dementia associated with Parkinson disease, with or without hallucinations. Efficacy measures were cognitive scales, activities of daily living, behavioral symptoms, and executive and attentional functions. The differences in these measures between rivastigmine and placebo recipients tended to be larger in patients with visual hallucinations than in those without hallucinations. The study was not designed to assess the effect of treatment on psychosis or hallucination.
WHEN PATIENTS WITH MOVEMENT DISORDERS NEED SURGERY
Some of these syndromes can be prevented, especially in patients who are known to have movement disorders and are undergoing surgery.
One problem is stopping oral dopaminergic drugs before the operation. Parkinson disease patients on dopaminergic drugs can develop parkinsonism-hyperpyrexia syndrome or akinetic crisis if the drug is stopped suddenly. Restarting dopaminergic therapy and supportive measures are the main treatments. Patients who have Parkinson disease should receive their usual dose of levodopa, dopamine agonist, or amantadine up until the time of surgery and then again as soon as they awaken in the recovery room.36 That goal can be achieved more easily now that these drugs come in transdermal patches and long-acting formulas.37 Droperidol (Inapsine) and metoclopramide worsen parkinsonism and should be avoided.
Myoclonus is the most common movement disorder seen in the postoperative period. In fact, myoclonic shivering is common as patients awaken from general anesthesia.36 The anesthetic agents etomidate (Amidate) and enflurane (Ethrane) and the opioids fentanyl (Actiq, Duralgesic, Sublimaze) and meperidine (Demerol) can cause myoclonus.38
Occasionally, a patient in the recovery room suddenly develops a neurologic deficit that is inconsistent with the history and physical findings. Psychogenic movement disorders should be considered in the differential diagnosis. Reassurance and occasionally psychiatric intervention are required in these cases.36
IN THE ELDERLY, GO EASY
Polypharmacy is a huge issue in the elderly. Some of the principles in prescribing medications in the elderly can be helpful in preventing movement disorder emergencies:
- Assess the current regimen, including over-the-counter drugs, before prescribing a new drug.
- Begin with a low dose and increase as necessary. “Start low, go slow.”
- Consider the possibility that any new symptoms can be a drug side effect or due to withdrawal of a drug.
- Discuss with the patient or caregiver what kind of side effect to expect and advise him or her to report serious ones.
- Isbister GK, Bowe SJ, Dawson A, Whyte IM. Relative toxicity of selective serotonin reuptake inhibitors (SSRIs) in overdose. J Toxicol Clin Toxicol 2004; 42:277–285.
- Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003; 96:635–642.
- Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2000; 79:201–209.
- LoCurto MJ. The serotonin syndrome. Emerg Med Clin North Am 1997; 15( 3):665–675.
- Boyer E, Shannon S. The serotonin syndrome. N Engl J Med 2005; 352:1112–1120.
- Kipps CM, Fung VS, Grattan-Smith P, de Moore GM, Morris JG. Movement disorder emergencies. Mov Disord 2005; 20:322–334.
- Shalev A, Munitz H. The neuroleptic malignant syndrome: agent and host interaction. Acta Psychiatr Scand 1986; 73:337–347.
- Rosebush PI, Stewart TD, Gelenberg AJ. Twenty neuroleptic rechallenges after neuroleptic malignant syndrome in 15 patients. J Clin Psychiatry 1989; 50:295–298.
- Adityanjee , Singh S, Singh G, Ong S. Spectrum concept of neuroleptic malignant syndrome. Br J Psychiatry 1988; 153:107–111.
- Bonuccelli U, Piccini P, Corsini GU, Muratorio A. Apomorphine in malignant syndrome due to levodopa withdrawal. Ital J Neurol Sci 1992; 13:169–170.
- Sato Y, Asoh T, Metoki N, et al. Efficacy of methylprednisolone pulse therapy on neuroleptic malignant syndrome in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2004; 74:574–576.
- Hariz MI, Johansson F. Hardware failure in parkinsonian patients with chronic subthalamic nucleus stimulation is a medical emergency. Mov Disord 2001; 16:166–168.
- Pollera CF, Cognetli F, Nardi M, Mozza D. Sudden death after acute dystonic reaction to high-dose metoclopramide. Lancet 1984; 2:460–461.
- Bateman DN, Rawlins MD, Simpson JM. Extrapyramidal reactions with metoclopramide. Br Med J 1985; 291:930–932.
- Pasricha PJ, Pehlivanov N, Sugumar A, Jankovic J. Drug insight: from disturbed motility to disordered movement—a review of the clinical benefits and medicolegal risks of metoclopramide. Nat Clin Pract Gastroenterol Hepatol 2006; 3:138–148.
- Hu S, Frucht S. Emergency treatment of movement disorders. Curr Treat Options Neurol 2007; 9:103–114.
- Tousi B, Schuele SU, Subramanian T. A 46-year-old woman with rigidity and frequent falls. Cleve Clin J Med 2005; 72:57–63.
- Merlo IM, Occhini A, Pacchetti C, Alfonsi E. Not paralysis, but dystonia causes stridor in multiple system atrophy. Neurology 2002; 58:649–652.
- Silber MH, Levine S. Stridor and death in multiple system atrophy. Mov Disord 2000; 15:699–704.
- Bhatia KP, Marsden CD. The behavioural and motor consequences of focal lesions of the basal ganglia in man. Brain 1994; 117:859–876.
- Factor SA, Molho ES. Emergency department presentations of patients with Parkinson’s disease. Am J Emerg Med 2000; 18:209–215.
- Verhagen Metman L, Del Dotto P, van den Munckhof P, Fang J, Mouradian MM, Chase TN. Amantadine as a treatment for dyskinesia and motor fluctuations in Parkinson’s disease. Neurology 1998; 50:1323–1326.
- Frucht S, Eidelberg D. Imipenem-induced myoclonus. Mov Disord 1997; 12:621–622.
- Miodownik C, Lerner V, Statsenko N, et al. Vitamin B6 versus mianserin and placebo in acute neuroleptic-induced akathisia: a randomized, double-blind, controlled study. Clin Neuropharmacol 2006; 29:68–72.
- Poyurovsky M, Pashinian A, Weizman R, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry 2006; 59:1071–1077.
- Tousi B, Subramanian T. Hallucinations in Parkinson’s disease: approach and management. Clin Geriatr 2004: 12:19–24.
- Goetz CG, Stebbins GT. Risk factors for nursing home placement in advanced Parkinson’s disease. Neurology 1993; 43:2227–2229.
- Factor SA, Molho ES, Podskalny GD, Brown D. Parkinson’s disease: drug-induced psychiatric states. Adv Neurol 1995; 65:115–138.
- Woodford H, Walker R. Emergency hospital admissions in idiopathic Parkinson’s disease. Mov Disord 2005; 20:1104–1108.
- Tousi B, Frankel M. Olfactory and visual hallucinations in Parkinson’s disease. Parkinsonism Relat Disord 2004; 10:253–254.
- The Parkinson Study Group. Low-dose clozapine for the treatment of drug-induced psychosis in Parkinson’s disease. N Engl J Med 1999; 340:757–763.
- Merims D, Balas M, Pertez C, Shabtai H, Giladi N. Rater-blinded, prospective comparison: quetiapine versus clozapine for Parkinson’s disease psychosis. Clin Neuropharmacol 2006; 29:331–337.
- Goetz CG, Blasucci LM, Leurgans S, Pappert EJ. Olanzapine and clozapine: comparative effects on motor function in hallucinating PD patients. Neurology 2000; 55:789–794.
- Reading PJ, Luce AK, McKeith IG. Rivastigmine in the treatment of parkinsonian psychosis and cognitive impairment: preliminary findings from an open trial. Mov Disord 2001; 16:1171–1174.
- Burn D, Emre M, McKeith I, et al. Effects of rivastigmine in patients with and without visual hallucinations in dementia associated with Parkinson’s disease. Mov Disord 2006; 21:1899–1907.
- Frucht SJ. Movement disorder emergencies in the perioperative period. Neurol Clin 2004; 22:379–387.
- Korczyn AD, Reichmann H, Boroojerdi B, et al. Rotigotin trans-dermal system for perioperative administration. J Neural Transm 2007; 114:219–221.
- Gordon MF. Toxin and drug-induced myoclonus. Adv Neurol 2002; 89:49–76.
Although we tend to think of movement disorders as chronic conditions, some of them can present as true emergencies in which failure to diagnose the condition and treat it promptly can result in significant sickness or even death.
Many cases are iatrogenic, occurring in patients with Parkinson disease or those taking antipsychotic or antidepressant medications when their regimen is started or altered. Elderly patients are particularly at risk, as they take more drugs and have less physiologic reserve.
Movement disorder emergencies in elderly patients can be difficult to diagnose and treat, since many patients are taking more than one medication: polypharmacy raises the possibility of interactions, and different drugs can cause different movement disorder syndromes. Moreover, because so many patients are at risk—for example, more than 1 million people in the United States now have Parkinson disease, and the number is growing—it is important for physicians who take care of the elderly to be more informed about these disorders, especially the presenting symptoms.
SCOPE OF THIS ARTICLE
- Disorders presenting with rigidity or stiffness
- Disorders presenting with dystonia
- Disorders presenting with hyperkinetic movements
- Disorders presenting with psychiatric disturbances.
Of these, the scenarios most likely to require emergency evaluation in the elderly are acute hypokinetic and hyperkinetic syndromes and psychiatric presentations. This article discusses movement disorder emergencies in the elderly, focusing on the more common disorders with common presentations.
DISORDERS PRESENTING WITH RIGIDITY OR STIFFNESS
Serotonin syndrome
Chief among the offenders are the selective serotonin-reuptake inhibitors (SSRIs), either alone or in combination. This syndrome occurs in 14% to 16% of patients who overdose on SSRIs.1 Examples of combinations that can lead to serotonin syndrome are an SSRI plus any of the following:
- An anxiolytic such as buspirone (BuSpar; this combination is popular for the treatment of depression and anxiety)
- A tricyclic agent such as imipramine (Tofranil)
- A serotonin and norepinephrine reuptake inhibitor such as venlafaxine (Effexor).
In addition, antiparkinson drugs such as levodopa and selegiline (Eldepryl) enhance serotonin release.
Signs and symptoms. Serotonin syndrome is characterized by:
- Severe rigidity
- Dysautonomia
- Change in mental status.
Other clinical findings include fever, gastrointestinal disturbances, and motor restlessness. Clonus is the most important finding in establishing the diagnosis.2
The syndrome may initially go unrecognized and can be mistaken for viral illness or anxiety.4 Manifestations range from mild to life-threatening; initially, it may present with akathisia and tremor. The symptoms progress rapidly over hours and can range from myoclonus, hyperreflexia, and seizures to severe forms of rhabdomyolysis, renal failure, and respiratory failure. The hyperreflexia and clonus seen in moderate cases may be considerably greater in the lower extremities than in the upper extremities.5
No laboratory test confirms the diagnosis, but tremor, clonus, or akathisia without additional extrapyramidal signs should lead to the diagnosis if the patient was taking a serotonergic medication.5 The onset of symptoms is usually rapid. The majority of patients present within 6 hours after initial use of the medication, an overdose, or a change in dosing.5
Treatment. The first steps are to stop the serotonergic medication and to hydrate and cool the patient to counteract the hyperpyrexic state. Benzodiazepine drugs are important in controlling agitation, regardless of its severity.5 Propranolol (Inderal) is not recommended, as it may cause hypotension and shock in patients with autonomic instability.5
Patients with moderate cases may additionally benefit from cyproheptadine (Periactin), an antihistamine that antagonizes serotonin. The initial dose is 4 to 8 mg orally, with a repeat dose after 2 hours.6 Whether to continue this treatment depends on the response after two doses.
If medications must be given parenterally, physicians can consider chlorpromazine (Thorazine) 50 to 100 mg intramuscularly.5
Vital signs should be monitored. In severe cases, intensive care may be required with immediate sedation, neuromuscular paralysis, and intubation.
In most cases, patients improve rapidly.
Comment. Serotonin syndrome can be avoided by educating physicians and by modifying prescribing practices.5 Avoiding multidrug regimens is critical to preventing serotonin syndrome. Computer-based ordering systems and personal digital assistants can help one avoid drug interactions.5
Neuroleptic malignant syndrome
This syndrome is an infrequent but potentially lethal complication associated with therapy with antipsychotic drugs such as haloperidol (Haldol) and lithium (Eskalith) and with other medications with dopamine type-2 receptor antagonist activity such as metoclopramide (Reglan) and prochlorperazine (Compazine). It has become rare since the introduction of atypical antipsychotics and now occurs in 0.2% of patients receiving atypical antipsychotics.7 Its pathogenesis is not fully understood.
This syndrome occurs mainly in young or middle-aged patients receiving doses of neuroleptics within the usual therapeutic range, but it also appears to occur in elderly patients who receive higher doses.8 Although most cases develop in the first 2 weeks of treatment, it can develop at any time during therapy.
Signs and symptoms. Four features characterize neuroleptic malignant syndrome9:
- Muscle rigidity—generalized (“lead-pipe”) muscular rigidity is accompanied by bradykinesia or akinesia.
- Autonomic dysregulation, with tachycardia, tachypnea, alterations in blood pressure, excessive sweating, and incontinence.
- Hyperthermia—fever can begin hours to days after initiating or increasing the dose of a dopamine antagonist.
- Altered sensorium, ranging from confusion to disorientation and coma.
Symptoms of neuroleptic malignant syndrome typically evolve over several days, in contrast to the rapid onset of the serotonin syndrome. Knowing the precipitating drug also helps distinguish the syndromes: dopamine antagonists produce bradykinesia, whereas serotonin agonists produce hyperkinesia.5
Laboratory abnormalities include elevated serum creatine kinase concentrations and white blood cell counts. Renal function should be assessed when renal failure and rhabdomyolysis are suspected.
Treatment involves stopping the causative medication, cooling the patient, and supporting vital functions.
In mild cases (eg, low-grade fever) benzodiazepines such as lorazepam (Ativan) can stabilize the condition. In moderate cases (eg, more significant rigidity), dopaminergic agonists such as bromocriptine (Parlodel) can be given, although there is no strong clinical evidence for their use. Bromocriptine is usually started at 2.5 mg three times a day and gradually increased in dose if tolerated.
In severe cases, muscle rigidity can be reduced with dantrolene (Dantrium), a muscle relaxant. Dantrolene is started at 1 mg/kg intravenously every 6 hours and gradually increased up to 10 mg/kg total per day.
Some patients remain rigid and febrile up to 4 weeks after the causative agent has been withdrawn. Therefore, these treatments can be continued for a few weeks. After the patient has recovered fully, if it is necessary to resume antipsychotic therapy, an atypical antipsychotic such as quetiapine (Seroquel) can be started after 2 weeks.8
Comment. Although uncommon, neuroleptic malignant syndrome is the most serious adverse effect of neuroleptic drugs, and it is potentially fatal. When neuroleptic malignant syndrome is suspected, treatment should be prompt, and the neuroleptic medication should be immediately stopped.
Parkinsonism-hyperpyrexia syndrome
Withdrawing or decreasing the dose of dopaminergic medications in patients with Parkinson disease can cause parkinsonism-hyperpyrexia syndrome, a condition that is similar to neuroleptic malignant syndrome. It can also arise after sudden withdrawal of amantadine (Symmetrel) or anticholinergics. In view of this concern, adjustments to antiparkinson drugs may need to be more gradual in some elderly patients.
Patients present with fever, rigidity, and autonomic instability and are at risk of aspiration pneumonia.
Treatment includes resuming dopaminergic therapy and giving supportive care.
Apomorphine (Apokyn), a dopaminergic agonist, was used in a 71-year-old female parkinsonian patient who developed parkinsonism-hyperpyrexia syndrome after abrupt reduction of chronic levodopa treatment.10 The symptoms resolved within 24 hours of the addition of apomorphine to her previous levodopa therapy. If the patient is taking apomorphine for the first time, the injections should be given in low doses, 0.2 mL subcutaneously. Apomorphine can induce vomiting, and if this occurs an antiemetic such as trimethobenzamide (Tigan) should be given before subsequent injections. In the elderly, caution is advised as apomorphine may cause severe orthostasis.
Methylprednisolone (Solu-Medrol) pulse therapy has been shown to shorten the duration of this syndrome in a randomized, controlled study.11
Akinetic syndrome after failure of deep brain stimulator
Deep brain stimulation involves surgical placement of a pacemaker with electrodes in specific areas of the brain. It is used to control Parkinson disease, tremor, and, less commonly, dystonia, and a number of other uses are under investigation. Continuous electrical stimulation of different nuclei in the brain has been shown to alleviate some symptoms of Parkinson disease (eg, rigidity) and to enable some patients to decrease the dose of their antiparkinson medications.
Several cases have been reported of sudden, unexpected reappearance of freezing, gait disturbance, or severe akinesia in Parkinson disease patients whose stimulators had been turned off inadvertently (eg, by a magnet in a dicating machine that was placed too close to the stimulator) and who presented to an emergency room.12
Treatment is easy if this diagnosis is considered. Checking the neurostimulator and switching it to “on” are all that is needed. Since patients and their caregivers are trained how to check and turn on the stimulator, the role of the geriatrician is simply to remind the caregiver of this possibility.
FDA warning. The US Food and Drug Administration has issued a warning against use of shortwave or microwave diathermy for patients with deep brain stimulation or other implanted leads (www.fda.gov/cdrh/safety/121902.html), stating: “There are three types of diathermy equipment used by physicians, dentists, physical therapists, chiropractors, sports therapists, and others: radio frequency (shortwave) diathermy, microwave diathermy, and ultrasound diathermy. Shortwave and microwave diathermy, in both heating and nonheating modes, can result in serious injury or death to patients with implanted devices with leads. This kind of interaction is not expected with ultrasound diathermy. Electrocautery devices are not included in this notification.” If a patient has an implanted deep brain stimulator, magnetic resonance imaging should be done only if absolutely needed and then only if the guidelines are followed.
DISORDERS PRESENTING WITH DYSTONIA
Acute dystonic reaction
Medications are a common cause of acute focal dystonia. The symptoms, which can be life-threatening, usually occur within 24 hours after taking the medication.12 The most common offenders are neuroleptic drugs and antiemetic drugs with dopamine-blocking activity (eg, metoclopramide), although in older patients, they are more likely to cause tardive dyskinesia and parkinsonism.13,14
Metoclopramide accounts for nearly one-third of all drug-induced movement disorders, and this adverse effect is a common reason for malpractice suits. The entire spectrum of drug-induced movement disorders, ranging from subtle to life-threatening, can ensue from its use; akathisia and dystonia are generally seen early in the course of metoclopramide-induced movement disorders, whereas tardive dyskinesia and parkinsonism seem to be more prevalent in long-term users.15
Treatment includes stopping the precipitating medication and reversing dystonia with anticholinergic medications such as benztropine (Cogentin). Anticholinergic therapy is given intravenously or intramuscularly followed by oral therapy for few days, as the acute dystonic reaction may recur after the effect of parenteral medication wears off.
Intravenous diphenhydramine (Benadryl), an antihistamine with additional anticholiner-gic effects, can abort dystonia in a few minutes.16
Laryngeal dystonia accompanied by multiple system atrophy
Multiple system atrophy, a Parkinson-plus syndrome, is characterized by parkinsonism (mostly with poor response to levodopa) and early onset of dysfunction of the autonomic nervous system, urinary tract, cerebellum, and corticospinal tract (hyperreflexia).17
In the course of the disease, about one-third of patients develop respiratory stridor due to abnormal movements of the vocal cords.18 Nocturnal stridor portends a poor prognosis,19 with an increased risk of sudden death. Geriatricians should be aware of these symptoms, as these patients may seek care because of hoarseness or difficulty swallowing.
Treatment. Laryngeal dystonia can be improved with continuous positive airway pressure. In some cases, tracheostomy may be needed.19
Sudden withdrawal of baclofen
Baclofen (Lioresal), a treatment for spasticity and dystonia, is delivered via a pump through a catheter into an intrathecal space. The pump needs to be refilled every 3 to 6 months. Sudden discontinuation of medication caused by a dislodged catheter tip or forgetting to refill the pump provokes withdrawal symptoms. Patients with this life-threatening syndrome can present with rigidity, fever, change in mental status, and worsening dystonic symptoms.
Treatment involves high doses of baclofen (up to 120 mg/day in divided doses).6
DISORDERS PRESENTING WITH HYPERKINETIC MOVEMENTS
Chorea, ballism (ballismus), and athetosis constitute a range of involuntary, hyperkinetic movement disorders. Chorea consists of involuntary, continuous, sudden, brief, unsustained, irregular movements that flow from one part of the body to another. Hemiballism presents as forceful flinging movements of the limbs or high-amplitude chorea that affects one side of the body.
Acute hemichorea and hemiballism
Acute hemichorea and hemiballism commonly result from infarction or hemorrhage of the basal ganglia.20 Computed tomography and especially magnetic resonance imaging can show the lesions in patients with ballism. Stroke-induced ballism is usually self-limited and resolves after a few weeks. Acute hemiballism generally evolves to hemichorea or hemiathetosis in a few days, which requires only protective measures.
Treatment. Mild cases do not need treatment but severe cases call for medical therapy. Antidopaminergics are the drugs of choice. A dopamine depletor such as reserpine (Serpasil) 0.1 mg once or twice daily or dopamine receptor blockers such as neuroleptics are considered.16 The combination of a benzodiazepine plus an antipsychotic such as olanzapine (Zyprexa) has been suggested.6
Severe parkinsonian dyskinesia
Dyskinesia is common in Parkinson disease, and patients may present to an emergency room with severe levodopa-induced dyskinesia. Dyskinesia can be exhausting if prolonged and severe. Elevated levels of creatine kinase raise the concern of rhabdomyolysis. In rare cases, the patient develops respiratory dyskinesia when respiratory muscles such as those in the diaphragm become involved.21
The risk of levodopa-induced dyskinesia increases with disease severity and higher levodopa doses. Using a dopamine agonist as initial therapy delays the onset of levodopa-induced dyskinesia in early Parkinson disease. However, Factor and Molho,21 in a case series, reported that adding dopamine agonists to the regimen was a precipitating factor; another was infection.
Treatment. A reasonable approach to treating peak-dose dyskinesia is to lower the doses of dopaminergic medications.
A mild sedative such as lorazepam, alprazolam (Xanax, Niravam), or clonazepam (Klonopin) may reduce the severity of dyski-nesia.21 These drugs are particularly useful if the dyskinesia is worse at night, and they can be used in the emergency department while waiting for the effect of the dopaminergic medications to wear off.
Amantadine ameliorates levodopa-induced peak-dose dyskinesia without worsening parkinsonian symptoms in some patients.22
Drug-induced myoclonus
Myoclonus is sudden, jerky, brief involuntary movement of the face, limbs, or trunk. Unlike tics, myoclonus cannot be controlled by the patient.
Myoclonus has various pathophysiologic mechanisms. Most myoclonic emergencies are epileptic myoclonic seizures, which are beyond the scope of this article. Often, myoclonus is caused by opiate overdose or withdrawal. It can also be a side effect of SSRIs, tricyclic anti-depressants, lithium, amantadine, and rarely, antibiotics such as imipenem (Primaxin).23
Treatment. Opiate-induced myoclonus may respond to naloxone (Narcan), whereas opiate withdrawal responds to benzodi-azepines.6
Acute akathisia
Acute akathisia occurs in susceptible patients after exposure to dopamine receptor blockers or dopamine depletors. It is characterized by subjective restless feelings accompanied by objective restless movements. The course is usually self-limited after the causative medication is discontinued.
Treatment. Symptomatic treatment may be needed in most cases for several days. Anticholinergics are effective. Additionally, vitamin B6, mianserine, propranolol, and mirtazapine (Remeron) in a low dose (15 mg/day) have been shown to be effective16,24,25
DISORDERS WITH PSYCHIATRIC PRESENTATIONS
Hallucinations and psychosis in Parkinson disease
Neuropsychiatric or behavioral complications of Parkinson disease include hallucinations, dementia, depression, psychosis, and sleep disorders.21,26 Psychosis is the leading reason for nursing home placement in advanced cases.27 Psychosis can present as hallucinations or a paranoid delusional state in association with clear sensorium.28 However, hallucinations accounted for only 3% of emergency admissions to the hospital for Parkinson disease patients in one series.29
Risk factors for hallucinations in parkinsonian patients include dementia, long-term therapy with dopaminergic drugs, long duration of disease, advanced age, anticholinergic drugs, and sleep disorders. Severe cognitive impairment or dementia is a major and independent predictive factor for visual hallucinations.30
Most hallucinations are visual; auditory, tactile, and olfactory hallucinations are rare.30
Treatment initially should be the same as in any patient with delirium. The systemic disorders that can aggravate or cause hallucinations such as electrolyte abnormalities, urinary or respiratory infection, and systemic illness should be ruled out.
The next step is to reduce or discontinue the adjunctive drugs that have the least antiparkinsonian effect and the greatest potential of inducing hallucination or psychosis. Examples of such medications include histamine-2 antagonists (eg, cimetidine [Tagamet], amantadine, selegeline, and anti-cholinergics). Selegeline can be discontinued abruptly because it has a long duration of action in the brain, but amantadine and anti-cholinergics should be tapered. Dopamine agonists can be discontinued. Levodopa can be reduced until the side effects begin to subside without significant worsening of motor symptoms.
If all the above adjustments fail, an antipsychotic medication can be considered.26 Clozapine (Clozaril) has the best result and is nearly free of extrapyramidal side effects but can cause agranulocytosis, which requires frequent blood counts. The Parkinson Study Group suggested that clozapine, at daily doses of 50 mg or less, is safe and significantly improves drug-induced psychosis without worsening parkinsonism.31 Clozapine may be impractical for elderly patients due to its side effect profile.
Quetiapine is a good alternative to cloza-pine and is less likely to worsen parkinsonian symptoms than other atypical antipsy-chotics.32 Olanzapine and risperidone (Risperdal) are reported to worsen parkinsonian symptoms.33 Not enough data have been published about the efficacy of the newer medications such as ziprasidone (Geodon) and aripiprazole (Abilify) to advocate their routine clinical use.
Rivastigmine (Exelon) was reported to improve hallucinations, sleep disturbance, and caregiver distress in addition to enhancing cognitive performance in advanced Parkinson disease in a small study.34 Burn and colleagues35 reported that rivastigmine was beneficial in patients with dementia associated with Parkinson disease, with or without hallucinations. Efficacy measures were cognitive scales, activities of daily living, behavioral symptoms, and executive and attentional functions. The differences in these measures between rivastigmine and placebo recipients tended to be larger in patients with visual hallucinations than in those without hallucinations. The study was not designed to assess the effect of treatment on psychosis or hallucination.
WHEN PATIENTS WITH MOVEMENT DISORDERS NEED SURGERY
Some of these syndromes can be prevented, especially in patients who are known to have movement disorders and are undergoing surgery.
One problem is stopping oral dopaminergic drugs before the operation. Parkinson disease patients on dopaminergic drugs can develop parkinsonism-hyperpyrexia syndrome or akinetic crisis if the drug is stopped suddenly. Restarting dopaminergic therapy and supportive measures are the main treatments. Patients who have Parkinson disease should receive their usual dose of levodopa, dopamine agonist, or amantadine up until the time of surgery and then again as soon as they awaken in the recovery room.36 That goal can be achieved more easily now that these drugs come in transdermal patches and long-acting formulas.37 Droperidol (Inapsine) and metoclopramide worsen parkinsonism and should be avoided.
Myoclonus is the most common movement disorder seen in the postoperative period. In fact, myoclonic shivering is common as patients awaken from general anesthesia.36 The anesthetic agents etomidate (Amidate) and enflurane (Ethrane) and the opioids fentanyl (Actiq, Duralgesic, Sublimaze) and meperidine (Demerol) can cause myoclonus.38
Occasionally, a patient in the recovery room suddenly develops a neurologic deficit that is inconsistent with the history and physical findings. Psychogenic movement disorders should be considered in the differential diagnosis. Reassurance and occasionally psychiatric intervention are required in these cases.36
IN THE ELDERLY, GO EASY
Polypharmacy is a huge issue in the elderly. Some of the principles in prescribing medications in the elderly can be helpful in preventing movement disorder emergencies:
- Assess the current regimen, including over-the-counter drugs, before prescribing a new drug.
- Begin with a low dose and increase as necessary. “Start low, go slow.”
- Consider the possibility that any new symptoms can be a drug side effect or due to withdrawal of a drug.
- Discuss with the patient or caregiver what kind of side effect to expect and advise him or her to report serious ones.
Although we tend to think of movement disorders as chronic conditions, some of them can present as true emergencies in which failure to diagnose the condition and treat it promptly can result in significant sickness or even death.
Many cases are iatrogenic, occurring in patients with Parkinson disease or those taking antipsychotic or antidepressant medications when their regimen is started or altered. Elderly patients are particularly at risk, as they take more drugs and have less physiologic reserve.
Movement disorder emergencies in elderly patients can be difficult to diagnose and treat, since many patients are taking more than one medication: polypharmacy raises the possibility of interactions, and different drugs can cause different movement disorder syndromes. Moreover, because so many patients are at risk—for example, more than 1 million people in the United States now have Parkinson disease, and the number is growing—it is important for physicians who take care of the elderly to be more informed about these disorders, especially the presenting symptoms.
SCOPE OF THIS ARTICLE
- Disorders presenting with rigidity or stiffness
- Disorders presenting with dystonia
- Disorders presenting with hyperkinetic movements
- Disorders presenting with psychiatric disturbances.
Of these, the scenarios most likely to require emergency evaluation in the elderly are acute hypokinetic and hyperkinetic syndromes and psychiatric presentations. This article discusses movement disorder emergencies in the elderly, focusing on the more common disorders with common presentations.
DISORDERS PRESENTING WITH RIGIDITY OR STIFFNESS
Serotonin syndrome
Chief among the offenders are the selective serotonin-reuptake inhibitors (SSRIs), either alone or in combination. This syndrome occurs in 14% to 16% of patients who overdose on SSRIs.1 Examples of combinations that can lead to serotonin syndrome are an SSRI plus any of the following:
- An anxiolytic such as buspirone (BuSpar; this combination is popular for the treatment of depression and anxiety)
- A tricyclic agent such as imipramine (Tofranil)
- A serotonin and norepinephrine reuptake inhibitor such as venlafaxine (Effexor).
In addition, antiparkinson drugs such as levodopa and selegiline (Eldepryl) enhance serotonin release.
Signs and symptoms. Serotonin syndrome is characterized by:
- Severe rigidity
- Dysautonomia
- Change in mental status.
Other clinical findings include fever, gastrointestinal disturbances, and motor restlessness. Clonus is the most important finding in establishing the diagnosis.2
The syndrome may initially go unrecognized and can be mistaken for viral illness or anxiety.4 Manifestations range from mild to life-threatening; initially, it may present with akathisia and tremor. The symptoms progress rapidly over hours and can range from myoclonus, hyperreflexia, and seizures to severe forms of rhabdomyolysis, renal failure, and respiratory failure. The hyperreflexia and clonus seen in moderate cases may be considerably greater in the lower extremities than in the upper extremities.5
No laboratory test confirms the diagnosis, but tremor, clonus, or akathisia without additional extrapyramidal signs should lead to the diagnosis if the patient was taking a serotonergic medication.5 The onset of symptoms is usually rapid. The majority of patients present within 6 hours after initial use of the medication, an overdose, or a change in dosing.5
Treatment. The first steps are to stop the serotonergic medication and to hydrate and cool the patient to counteract the hyperpyrexic state. Benzodiazepine drugs are important in controlling agitation, regardless of its severity.5 Propranolol (Inderal) is not recommended, as it may cause hypotension and shock in patients with autonomic instability.5
Patients with moderate cases may additionally benefit from cyproheptadine (Periactin), an antihistamine that antagonizes serotonin. The initial dose is 4 to 8 mg orally, with a repeat dose after 2 hours.6 Whether to continue this treatment depends on the response after two doses.
If medications must be given parenterally, physicians can consider chlorpromazine (Thorazine) 50 to 100 mg intramuscularly.5
Vital signs should be monitored. In severe cases, intensive care may be required with immediate sedation, neuromuscular paralysis, and intubation.
In most cases, patients improve rapidly.
Comment. Serotonin syndrome can be avoided by educating physicians and by modifying prescribing practices.5 Avoiding multidrug regimens is critical to preventing serotonin syndrome. Computer-based ordering systems and personal digital assistants can help one avoid drug interactions.5
Neuroleptic malignant syndrome
This syndrome is an infrequent but potentially lethal complication associated with therapy with antipsychotic drugs such as haloperidol (Haldol) and lithium (Eskalith) and with other medications with dopamine type-2 receptor antagonist activity such as metoclopramide (Reglan) and prochlorperazine (Compazine). It has become rare since the introduction of atypical antipsychotics and now occurs in 0.2% of patients receiving atypical antipsychotics.7 Its pathogenesis is not fully understood.
This syndrome occurs mainly in young or middle-aged patients receiving doses of neuroleptics within the usual therapeutic range, but it also appears to occur in elderly patients who receive higher doses.8 Although most cases develop in the first 2 weeks of treatment, it can develop at any time during therapy.
Signs and symptoms. Four features characterize neuroleptic malignant syndrome9:
- Muscle rigidity—generalized (“lead-pipe”) muscular rigidity is accompanied by bradykinesia or akinesia.
- Autonomic dysregulation, with tachycardia, tachypnea, alterations in blood pressure, excessive sweating, and incontinence.
- Hyperthermia—fever can begin hours to days after initiating or increasing the dose of a dopamine antagonist.
- Altered sensorium, ranging from confusion to disorientation and coma.
Symptoms of neuroleptic malignant syndrome typically evolve over several days, in contrast to the rapid onset of the serotonin syndrome. Knowing the precipitating drug also helps distinguish the syndromes: dopamine antagonists produce bradykinesia, whereas serotonin agonists produce hyperkinesia.5
Laboratory abnormalities include elevated serum creatine kinase concentrations and white blood cell counts. Renal function should be assessed when renal failure and rhabdomyolysis are suspected.
Treatment involves stopping the causative medication, cooling the patient, and supporting vital functions.
In mild cases (eg, low-grade fever) benzodiazepines such as lorazepam (Ativan) can stabilize the condition. In moderate cases (eg, more significant rigidity), dopaminergic agonists such as bromocriptine (Parlodel) can be given, although there is no strong clinical evidence for their use. Bromocriptine is usually started at 2.5 mg three times a day and gradually increased in dose if tolerated.
In severe cases, muscle rigidity can be reduced with dantrolene (Dantrium), a muscle relaxant. Dantrolene is started at 1 mg/kg intravenously every 6 hours and gradually increased up to 10 mg/kg total per day.
Some patients remain rigid and febrile up to 4 weeks after the causative agent has been withdrawn. Therefore, these treatments can be continued for a few weeks. After the patient has recovered fully, if it is necessary to resume antipsychotic therapy, an atypical antipsychotic such as quetiapine (Seroquel) can be started after 2 weeks.8
Comment. Although uncommon, neuroleptic malignant syndrome is the most serious adverse effect of neuroleptic drugs, and it is potentially fatal. When neuroleptic malignant syndrome is suspected, treatment should be prompt, and the neuroleptic medication should be immediately stopped.
Parkinsonism-hyperpyrexia syndrome
Withdrawing or decreasing the dose of dopaminergic medications in patients with Parkinson disease can cause parkinsonism-hyperpyrexia syndrome, a condition that is similar to neuroleptic malignant syndrome. It can also arise after sudden withdrawal of amantadine (Symmetrel) or anticholinergics. In view of this concern, adjustments to antiparkinson drugs may need to be more gradual in some elderly patients.
Patients present with fever, rigidity, and autonomic instability and are at risk of aspiration pneumonia.
Treatment includes resuming dopaminergic therapy and giving supportive care.
Apomorphine (Apokyn), a dopaminergic agonist, was used in a 71-year-old female parkinsonian patient who developed parkinsonism-hyperpyrexia syndrome after abrupt reduction of chronic levodopa treatment.10 The symptoms resolved within 24 hours of the addition of apomorphine to her previous levodopa therapy. If the patient is taking apomorphine for the first time, the injections should be given in low doses, 0.2 mL subcutaneously. Apomorphine can induce vomiting, and if this occurs an antiemetic such as trimethobenzamide (Tigan) should be given before subsequent injections. In the elderly, caution is advised as apomorphine may cause severe orthostasis.
Methylprednisolone (Solu-Medrol) pulse therapy has been shown to shorten the duration of this syndrome in a randomized, controlled study.11
Akinetic syndrome after failure of deep brain stimulator
Deep brain stimulation involves surgical placement of a pacemaker with electrodes in specific areas of the brain. It is used to control Parkinson disease, tremor, and, less commonly, dystonia, and a number of other uses are under investigation. Continuous electrical stimulation of different nuclei in the brain has been shown to alleviate some symptoms of Parkinson disease (eg, rigidity) and to enable some patients to decrease the dose of their antiparkinson medications.
Several cases have been reported of sudden, unexpected reappearance of freezing, gait disturbance, or severe akinesia in Parkinson disease patients whose stimulators had been turned off inadvertently (eg, by a magnet in a dicating machine that was placed too close to the stimulator) and who presented to an emergency room.12
Treatment is easy if this diagnosis is considered. Checking the neurostimulator and switching it to “on” are all that is needed. Since patients and their caregivers are trained how to check and turn on the stimulator, the role of the geriatrician is simply to remind the caregiver of this possibility.
FDA warning. The US Food and Drug Administration has issued a warning against use of shortwave or microwave diathermy for patients with deep brain stimulation or other implanted leads (www.fda.gov/cdrh/safety/121902.html), stating: “There are three types of diathermy equipment used by physicians, dentists, physical therapists, chiropractors, sports therapists, and others: radio frequency (shortwave) diathermy, microwave diathermy, and ultrasound diathermy. Shortwave and microwave diathermy, in both heating and nonheating modes, can result in serious injury or death to patients with implanted devices with leads. This kind of interaction is not expected with ultrasound diathermy. Electrocautery devices are not included in this notification.” If a patient has an implanted deep brain stimulator, magnetic resonance imaging should be done only if absolutely needed and then only if the guidelines are followed.
DISORDERS PRESENTING WITH DYSTONIA
Acute dystonic reaction
Medications are a common cause of acute focal dystonia. The symptoms, which can be life-threatening, usually occur within 24 hours after taking the medication.12 The most common offenders are neuroleptic drugs and antiemetic drugs with dopamine-blocking activity (eg, metoclopramide), although in older patients, they are more likely to cause tardive dyskinesia and parkinsonism.13,14
Metoclopramide accounts for nearly one-third of all drug-induced movement disorders, and this adverse effect is a common reason for malpractice suits. The entire spectrum of drug-induced movement disorders, ranging from subtle to life-threatening, can ensue from its use; akathisia and dystonia are generally seen early in the course of metoclopramide-induced movement disorders, whereas tardive dyskinesia and parkinsonism seem to be more prevalent in long-term users.15
Treatment includes stopping the precipitating medication and reversing dystonia with anticholinergic medications such as benztropine (Cogentin). Anticholinergic therapy is given intravenously or intramuscularly followed by oral therapy for few days, as the acute dystonic reaction may recur after the effect of parenteral medication wears off.
Intravenous diphenhydramine (Benadryl), an antihistamine with additional anticholiner-gic effects, can abort dystonia in a few minutes.16
Laryngeal dystonia accompanied by multiple system atrophy
Multiple system atrophy, a Parkinson-plus syndrome, is characterized by parkinsonism (mostly with poor response to levodopa) and early onset of dysfunction of the autonomic nervous system, urinary tract, cerebellum, and corticospinal tract (hyperreflexia).17
In the course of the disease, about one-third of patients develop respiratory stridor due to abnormal movements of the vocal cords.18 Nocturnal stridor portends a poor prognosis,19 with an increased risk of sudden death. Geriatricians should be aware of these symptoms, as these patients may seek care because of hoarseness or difficulty swallowing.
Treatment. Laryngeal dystonia can be improved with continuous positive airway pressure. In some cases, tracheostomy may be needed.19
Sudden withdrawal of baclofen
Baclofen (Lioresal), a treatment for spasticity and dystonia, is delivered via a pump through a catheter into an intrathecal space. The pump needs to be refilled every 3 to 6 months. Sudden discontinuation of medication caused by a dislodged catheter tip or forgetting to refill the pump provokes withdrawal symptoms. Patients with this life-threatening syndrome can present with rigidity, fever, change in mental status, and worsening dystonic symptoms.
Treatment involves high doses of baclofen (up to 120 mg/day in divided doses).6
DISORDERS PRESENTING WITH HYPERKINETIC MOVEMENTS
Chorea, ballism (ballismus), and athetosis constitute a range of involuntary, hyperkinetic movement disorders. Chorea consists of involuntary, continuous, sudden, brief, unsustained, irregular movements that flow from one part of the body to another. Hemiballism presents as forceful flinging movements of the limbs or high-amplitude chorea that affects one side of the body.
Acute hemichorea and hemiballism
Acute hemichorea and hemiballism commonly result from infarction or hemorrhage of the basal ganglia.20 Computed tomography and especially magnetic resonance imaging can show the lesions in patients with ballism. Stroke-induced ballism is usually self-limited and resolves after a few weeks. Acute hemiballism generally evolves to hemichorea or hemiathetosis in a few days, which requires only protective measures.
Treatment. Mild cases do not need treatment but severe cases call for medical therapy. Antidopaminergics are the drugs of choice. A dopamine depletor such as reserpine (Serpasil) 0.1 mg once or twice daily or dopamine receptor blockers such as neuroleptics are considered.16 The combination of a benzodiazepine plus an antipsychotic such as olanzapine (Zyprexa) has been suggested.6
Severe parkinsonian dyskinesia
Dyskinesia is common in Parkinson disease, and patients may present to an emergency room with severe levodopa-induced dyskinesia. Dyskinesia can be exhausting if prolonged and severe. Elevated levels of creatine kinase raise the concern of rhabdomyolysis. In rare cases, the patient develops respiratory dyskinesia when respiratory muscles such as those in the diaphragm become involved.21
The risk of levodopa-induced dyskinesia increases with disease severity and higher levodopa doses. Using a dopamine agonist as initial therapy delays the onset of levodopa-induced dyskinesia in early Parkinson disease. However, Factor and Molho,21 in a case series, reported that adding dopamine agonists to the regimen was a precipitating factor; another was infection.
Treatment. A reasonable approach to treating peak-dose dyskinesia is to lower the doses of dopaminergic medications.
A mild sedative such as lorazepam, alprazolam (Xanax, Niravam), or clonazepam (Klonopin) may reduce the severity of dyski-nesia.21 These drugs are particularly useful if the dyskinesia is worse at night, and they can be used in the emergency department while waiting for the effect of the dopaminergic medications to wear off.
Amantadine ameliorates levodopa-induced peak-dose dyskinesia without worsening parkinsonian symptoms in some patients.22
Drug-induced myoclonus
Myoclonus is sudden, jerky, brief involuntary movement of the face, limbs, or trunk. Unlike tics, myoclonus cannot be controlled by the patient.
Myoclonus has various pathophysiologic mechanisms. Most myoclonic emergencies are epileptic myoclonic seizures, which are beyond the scope of this article. Often, myoclonus is caused by opiate overdose or withdrawal. It can also be a side effect of SSRIs, tricyclic anti-depressants, lithium, amantadine, and rarely, antibiotics such as imipenem (Primaxin).23
Treatment. Opiate-induced myoclonus may respond to naloxone (Narcan), whereas opiate withdrawal responds to benzodi-azepines.6
Acute akathisia
Acute akathisia occurs in susceptible patients after exposure to dopamine receptor blockers or dopamine depletors. It is characterized by subjective restless feelings accompanied by objective restless movements. The course is usually self-limited after the causative medication is discontinued.
Treatment. Symptomatic treatment may be needed in most cases for several days. Anticholinergics are effective. Additionally, vitamin B6, mianserine, propranolol, and mirtazapine (Remeron) in a low dose (15 mg/day) have been shown to be effective16,24,25
DISORDERS WITH PSYCHIATRIC PRESENTATIONS
Hallucinations and psychosis in Parkinson disease
Neuropsychiatric or behavioral complications of Parkinson disease include hallucinations, dementia, depression, psychosis, and sleep disorders.21,26 Psychosis is the leading reason for nursing home placement in advanced cases.27 Psychosis can present as hallucinations or a paranoid delusional state in association with clear sensorium.28 However, hallucinations accounted for only 3% of emergency admissions to the hospital for Parkinson disease patients in one series.29
Risk factors for hallucinations in parkinsonian patients include dementia, long-term therapy with dopaminergic drugs, long duration of disease, advanced age, anticholinergic drugs, and sleep disorders. Severe cognitive impairment or dementia is a major and independent predictive factor for visual hallucinations.30
Most hallucinations are visual; auditory, tactile, and olfactory hallucinations are rare.30
Treatment initially should be the same as in any patient with delirium. The systemic disorders that can aggravate or cause hallucinations such as electrolyte abnormalities, urinary or respiratory infection, and systemic illness should be ruled out.
The next step is to reduce or discontinue the adjunctive drugs that have the least antiparkinsonian effect and the greatest potential of inducing hallucination or psychosis. Examples of such medications include histamine-2 antagonists (eg, cimetidine [Tagamet], amantadine, selegeline, and anti-cholinergics). Selegeline can be discontinued abruptly because it has a long duration of action in the brain, but amantadine and anti-cholinergics should be tapered. Dopamine agonists can be discontinued. Levodopa can be reduced until the side effects begin to subside without significant worsening of motor symptoms.
If all the above adjustments fail, an antipsychotic medication can be considered.26 Clozapine (Clozaril) has the best result and is nearly free of extrapyramidal side effects but can cause agranulocytosis, which requires frequent blood counts. The Parkinson Study Group suggested that clozapine, at daily doses of 50 mg or less, is safe and significantly improves drug-induced psychosis without worsening parkinsonism.31 Clozapine may be impractical for elderly patients due to its side effect profile.
Quetiapine is a good alternative to cloza-pine and is less likely to worsen parkinsonian symptoms than other atypical antipsy-chotics.32 Olanzapine and risperidone (Risperdal) are reported to worsen parkinsonian symptoms.33 Not enough data have been published about the efficacy of the newer medications such as ziprasidone (Geodon) and aripiprazole (Abilify) to advocate their routine clinical use.
Rivastigmine (Exelon) was reported to improve hallucinations, sleep disturbance, and caregiver distress in addition to enhancing cognitive performance in advanced Parkinson disease in a small study.34 Burn and colleagues35 reported that rivastigmine was beneficial in patients with dementia associated with Parkinson disease, with or without hallucinations. Efficacy measures were cognitive scales, activities of daily living, behavioral symptoms, and executive and attentional functions. The differences in these measures between rivastigmine and placebo recipients tended to be larger in patients with visual hallucinations than in those without hallucinations. The study was not designed to assess the effect of treatment on psychosis or hallucination.
WHEN PATIENTS WITH MOVEMENT DISORDERS NEED SURGERY
Some of these syndromes can be prevented, especially in patients who are known to have movement disorders and are undergoing surgery.
One problem is stopping oral dopaminergic drugs before the operation. Parkinson disease patients on dopaminergic drugs can develop parkinsonism-hyperpyrexia syndrome or akinetic crisis if the drug is stopped suddenly. Restarting dopaminergic therapy and supportive measures are the main treatments. Patients who have Parkinson disease should receive their usual dose of levodopa, dopamine agonist, or amantadine up until the time of surgery and then again as soon as they awaken in the recovery room.36 That goal can be achieved more easily now that these drugs come in transdermal patches and long-acting formulas.37 Droperidol (Inapsine) and metoclopramide worsen parkinsonism and should be avoided.
Myoclonus is the most common movement disorder seen in the postoperative period. In fact, myoclonic shivering is common as patients awaken from general anesthesia.36 The anesthetic agents etomidate (Amidate) and enflurane (Ethrane) and the opioids fentanyl (Actiq, Duralgesic, Sublimaze) and meperidine (Demerol) can cause myoclonus.38
Occasionally, a patient in the recovery room suddenly develops a neurologic deficit that is inconsistent with the history and physical findings. Psychogenic movement disorders should be considered in the differential diagnosis. Reassurance and occasionally psychiatric intervention are required in these cases.36
IN THE ELDERLY, GO EASY
Polypharmacy is a huge issue in the elderly. Some of the principles in prescribing medications in the elderly can be helpful in preventing movement disorder emergencies:
- Assess the current regimen, including over-the-counter drugs, before prescribing a new drug.
- Begin with a low dose and increase as necessary. “Start low, go slow.”
- Consider the possibility that any new symptoms can be a drug side effect or due to withdrawal of a drug.
- Discuss with the patient or caregiver what kind of side effect to expect and advise him or her to report serious ones.
- Isbister GK, Bowe SJ, Dawson A, Whyte IM. Relative toxicity of selective serotonin reuptake inhibitors (SSRIs) in overdose. J Toxicol Clin Toxicol 2004; 42:277–285.
- Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003; 96:635–642.
- Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2000; 79:201–209.
- LoCurto MJ. The serotonin syndrome. Emerg Med Clin North Am 1997; 15( 3):665–675.
- Boyer E, Shannon S. The serotonin syndrome. N Engl J Med 2005; 352:1112–1120.
- Kipps CM, Fung VS, Grattan-Smith P, de Moore GM, Morris JG. Movement disorder emergencies. Mov Disord 2005; 20:322–334.
- Shalev A, Munitz H. The neuroleptic malignant syndrome: agent and host interaction. Acta Psychiatr Scand 1986; 73:337–347.
- Rosebush PI, Stewart TD, Gelenberg AJ. Twenty neuroleptic rechallenges after neuroleptic malignant syndrome in 15 patients. J Clin Psychiatry 1989; 50:295–298.
- Adityanjee , Singh S, Singh G, Ong S. Spectrum concept of neuroleptic malignant syndrome. Br J Psychiatry 1988; 153:107–111.
- Bonuccelli U, Piccini P, Corsini GU, Muratorio A. Apomorphine in malignant syndrome due to levodopa withdrawal. Ital J Neurol Sci 1992; 13:169–170.
- Sato Y, Asoh T, Metoki N, et al. Efficacy of methylprednisolone pulse therapy on neuroleptic malignant syndrome in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2004; 74:574–576.
- Hariz MI, Johansson F. Hardware failure in parkinsonian patients with chronic subthalamic nucleus stimulation is a medical emergency. Mov Disord 2001; 16:166–168.
- Pollera CF, Cognetli F, Nardi M, Mozza D. Sudden death after acute dystonic reaction to high-dose metoclopramide. Lancet 1984; 2:460–461.
- Bateman DN, Rawlins MD, Simpson JM. Extrapyramidal reactions with metoclopramide. Br Med J 1985; 291:930–932.
- Pasricha PJ, Pehlivanov N, Sugumar A, Jankovic J. Drug insight: from disturbed motility to disordered movement—a review of the clinical benefits and medicolegal risks of metoclopramide. Nat Clin Pract Gastroenterol Hepatol 2006; 3:138–148.
- Hu S, Frucht S. Emergency treatment of movement disorders. Curr Treat Options Neurol 2007; 9:103–114.
- Tousi B, Schuele SU, Subramanian T. A 46-year-old woman with rigidity and frequent falls. Cleve Clin J Med 2005; 72:57–63.
- Merlo IM, Occhini A, Pacchetti C, Alfonsi E. Not paralysis, but dystonia causes stridor in multiple system atrophy. Neurology 2002; 58:649–652.
- Silber MH, Levine S. Stridor and death in multiple system atrophy. Mov Disord 2000; 15:699–704.
- Bhatia KP, Marsden CD. The behavioural and motor consequences of focal lesions of the basal ganglia in man. Brain 1994; 117:859–876.
- Factor SA, Molho ES. Emergency department presentations of patients with Parkinson’s disease. Am J Emerg Med 2000; 18:209–215.
- Verhagen Metman L, Del Dotto P, van den Munckhof P, Fang J, Mouradian MM, Chase TN. Amantadine as a treatment for dyskinesia and motor fluctuations in Parkinson’s disease. Neurology 1998; 50:1323–1326.
- Frucht S, Eidelberg D. Imipenem-induced myoclonus. Mov Disord 1997; 12:621–622.
- Miodownik C, Lerner V, Statsenko N, et al. Vitamin B6 versus mianserin and placebo in acute neuroleptic-induced akathisia: a randomized, double-blind, controlled study. Clin Neuropharmacol 2006; 29:68–72.
- Poyurovsky M, Pashinian A, Weizman R, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry 2006; 59:1071–1077.
- Tousi B, Subramanian T. Hallucinations in Parkinson’s disease: approach and management. Clin Geriatr 2004: 12:19–24.
- Goetz CG, Stebbins GT. Risk factors for nursing home placement in advanced Parkinson’s disease. Neurology 1993; 43:2227–2229.
- Factor SA, Molho ES, Podskalny GD, Brown D. Parkinson’s disease: drug-induced psychiatric states. Adv Neurol 1995; 65:115–138.
- Woodford H, Walker R. Emergency hospital admissions in idiopathic Parkinson’s disease. Mov Disord 2005; 20:1104–1108.
- Tousi B, Frankel M. Olfactory and visual hallucinations in Parkinson’s disease. Parkinsonism Relat Disord 2004; 10:253–254.
- The Parkinson Study Group. Low-dose clozapine for the treatment of drug-induced psychosis in Parkinson’s disease. N Engl J Med 1999; 340:757–763.
- Merims D, Balas M, Pertez C, Shabtai H, Giladi N. Rater-blinded, prospective comparison: quetiapine versus clozapine for Parkinson’s disease psychosis. Clin Neuropharmacol 2006; 29:331–337.
- Goetz CG, Blasucci LM, Leurgans S, Pappert EJ. Olanzapine and clozapine: comparative effects on motor function in hallucinating PD patients. Neurology 2000; 55:789–794.
- Reading PJ, Luce AK, McKeith IG. Rivastigmine in the treatment of parkinsonian psychosis and cognitive impairment: preliminary findings from an open trial. Mov Disord 2001; 16:1171–1174.
- Burn D, Emre M, McKeith I, et al. Effects of rivastigmine in patients with and without visual hallucinations in dementia associated with Parkinson’s disease. Mov Disord 2006; 21:1899–1907.
- Frucht SJ. Movement disorder emergencies in the perioperative period. Neurol Clin 2004; 22:379–387.
- Korczyn AD, Reichmann H, Boroojerdi B, et al. Rotigotin trans-dermal system for perioperative administration. J Neural Transm 2007; 114:219–221.
- Gordon MF. Toxin and drug-induced myoclonus. Adv Neurol 2002; 89:49–76.
- Isbister GK, Bowe SJ, Dawson A, Whyte IM. Relative toxicity of selective serotonin reuptake inhibitors (SSRIs) in overdose. J Toxicol Clin Toxicol 2004; 42:277–285.
- Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003; 96:635–642.
- Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2000; 79:201–209.
- LoCurto MJ. The serotonin syndrome. Emerg Med Clin North Am 1997; 15( 3):665–675.
- Boyer E, Shannon S. The serotonin syndrome. N Engl J Med 2005; 352:1112–1120.
- Kipps CM, Fung VS, Grattan-Smith P, de Moore GM, Morris JG. Movement disorder emergencies. Mov Disord 2005; 20:322–334.
- Shalev A, Munitz H. The neuroleptic malignant syndrome: agent and host interaction. Acta Psychiatr Scand 1986; 73:337–347.
- Rosebush PI, Stewart TD, Gelenberg AJ. Twenty neuroleptic rechallenges after neuroleptic malignant syndrome in 15 patients. J Clin Psychiatry 1989; 50:295–298.
- Adityanjee , Singh S, Singh G, Ong S. Spectrum concept of neuroleptic malignant syndrome. Br J Psychiatry 1988; 153:107–111.
- Bonuccelli U, Piccini P, Corsini GU, Muratorio A. Apomorphine in malignant syndrome due to levodopa withdrawal. Ital J Neurol Sci 1992; 13:169–170.
- Sato Y, Asoh T, Metoki N, et al. Efficacy of methylprednisolone pulse therapy on neuroleptic malignant syndrome in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2004; 74:574–576.
- Hariz MI, Johansson F. Hardware failure in parkinsonian patients with chronic subthalamic nucleus stimulation is a medical emergency. Mov Disord 2001; 16:166–168.
- Pollera CF, Cognetli F, Nardi M, Mozza D. Sudden death after acute dystonic reaction to high-dose metoclopramide. Lancet 1984; 2:460–461.
- Bateman DN, Rawlins MD, Simpson JM. Extrapyramidal reactions with metoclopramide. Br Med J 1985; 291:930–932.
- Pasricha PJ, Pehlivanov N, Sugumar A, Jankovic J. Drug insight: from disturbed motility to disordered movement—a review of the clinical benefits and medicolegal risks of metoclopramide. Nat Clin Pract Gastroenterol Hepatol 2006; 3:138–148.
- Hu S, Frucht S. Emergency treatment of movement disorders. Curr Treat Options Neurol 2007; 9:103–114.
- Tousi B, Schuele SU, Subramanian T. A 46-year-old woman with rigidity and frequent falls. Cleve Clin J Med 2005; 72:57–63.
- Merlo IM, Occhini A, Pacchetti C, Alfonsi E. Not paralysis, but dystonia causes stridor in multiple system atrophy. Neurology 2002; 58:649–652.
- Silber MH, Levine S. Stridor and death in multiple system atrophy. Mov Disord 2000; 15:699–704.
- Bhatia KP, Marsden CD. The behavioural and motor consequences of focal lesions of the basal ganglia in man. Brain 1994; 117:859–876.
- Factor SA, Molho ES. Emergency department presentations of patients with Parkinson’s disease. Am J Emerg Med 2000; 18:209–215.
- Verhagen Metman L, Del Dotto P, van den Munckhof P, Fang J, Mouradian MM, Chase TN. Amantadine as a treatment for dyskinesia and motor fluctuations in Parkinson’s disease. Neurology 1998; 50:1323–1326.
- Frucht S, Eidelberg D. Imipenem-induced myoclonus. Mov Disord 1997; 12:621–622.
- Miodownik C, Lerner V, Statsenko N, et al. Vitamin B6 versus mianserin and placebo in acute neuroleptic-induced akathisia: a randomized, double-blind, controlled study. Clin Neuropharmacol 2006; 29:68–72.
- Poyurovsky M, Pashinian A, Weizman R, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry 2006; 59:1071–1077.
- Tousi B, Subramanian T. Hallucinations in Parkinson’s disease: approach and management. Clin Geriatr 2004: 12:19–24.
- Goetz CG, Stebbins GT. Risk factors for nursing home placement in advanced Parkinson’s disease. Neurology 1993; 43:2227–2229.
- Factor SA, Molho ES, Podskalny GD, Brown D. Parkinson’s disease: drug-induced psychiatric states. Adv Neurol 1995; 65:115–138.
- Woodford H, Walker R. Emergency hospital admissions in idiopathic Parkinson’s disease. Mov Disord 2005; 20:1104–1108.
- Tousi B, Frankel M. Olfactory and visual hallucinations in Parkinson’s disease. Parkinsonism Relat Disord 2004; 10:253–254.
- The Parkinson Study Group. Low-dose clozapine for the treatment of drug-induced psychosis in Parkinson’s disease. N Engl J Med 1999; 340:757–763.
- Merims D, Balas M, Pertez C, Shabtai H, Giladi N. Rater-blinded, prospective comparison: quetiapine versus clozapine for Parkinson’s disease psychosis. Clin Neuropharmacol 2006; 29:331–337.
- Goetz CG, Blasucci LM, Leurgans S, Pappert EJ. Olanzapine and clozapine: comparative effects on motor function in hallucinating PD patients. Neurology 2000; 55:789–794.
- Reading PJ, Luce AK, McKeith IG. Rivastigmine in the treatment of parkinsonian psychosis and cognitive impairment: preliminary findings from an open trial. Mov Disord 2001; 16:1171–1174.
- Burn D, Emre M, McKeith I, et al. Effects of rivastigmine in patients with and without visual hallucinations in dementia associated with Parkinson’s disease. Mov Disord 2006; 21:1899–1907.
- Frucht SJ. Movement disorder emergencies in the perioperative period. Neurol Clin 2004; 22:379–387.
- Korczyn AD, Reichmann H, Boroojerdi B, et al. Rotigotin trans-dermal system for perioperative administration. J Neural Transm 2007; 114:219–221.
- Gordon MF. Toxin and drug-induced myoclonus. Adv Neurol 2002; 89:49–76.
KEY POINTS
- Supportive measures must be taken immediately to maintain the functions of vital organs.
- Serotonin syndrome, which can cause rigidity or stiffness, can be prevented by avoiding multidrug regimens.
- Withdrawing or decreasing the dose of dopaminergic drugs in patients with Parkinson disease can cause parkinsonism-hyperpyrexia syndrome, a condition similar to neuroleptic malignant syndrome.
- Metoclopramide (Reglan) accounts for nearly one-third of all drug-induced movement disorders. The entire spectrum of drug-induced movement disorders, ranging from subtle to life-threatening, can ensue from its use.
- Complications of Parkinson disease include hallucinations, dementia, depression, psychosis, and sleep disorders.
The Women’s Health Initiative: Implications for clinicians
More than 2 years have passed since we published the results of the Women’s Health Initiative (WHI), which caused a storm of information—and misinformation—about the effect of long-term dietary intervention on disease outcomes in postmenopausal women. Now that the dust has long settled, what have we learned from this landmark study?
The WHI results led to numerous additional analyses of all aspects of the study.1–7 What are the implications of all the analyses to clinical practice?
In this article, we summarize key aspects of the clinical trial, including study design, interventions, main results, and future plans. We also discuss potential clinical applications and practical considerations for public health efforts.
WHO WAS ELIGIBLE, WHO WAS NOT
A total of 48,835 postmenopausal women were randomly assigned to either no dietary intervention (n = 29,294) or a dietary intervention (n = 19,541) (see below).7 Participants were followed at 40 clinical centers between 1993 and 2005.4 Their mean age was 62.3 years; 18.6% were members of minorities.
Women were eligible if they were post-menopausal and had a daily dietary fat intake of at least 32% of total calories, based on assessment via a food-frequency questionnaire. They were excluded from the study if they had any of the following: a history of breast cancer, colorectal cancer, or other cancer except skin cancer during the past 10 years; type 1 diabetes; a medical condition in which the predicted survival was less than 3 years; and a potential barrier to adherence to the study regimen, including alcoholism or a lifestyle that involved often eating meals away from home.
THE WHI DIET: LESS FAT, BUT MORE FRUITS, VEGETABLES, GRAINS
The WHI dietary intervention was designed to prevent breast cancer, based on the evidence available when the study was planned. The targets included a total fat intake of less than 20% of energy (in kilocalories), increasing the intake of fruits and vegetables to at least five servings per day, and increasing the intake of grains to at least six servings per day.
Although reduction in saturated fat intake per se was not part of the WHI protocol, we assumed from previous pilot studies8 that the reduction of total fat intake would simultaneously produce a reduction in saturated fat intake to 7% of total calories.
A simpler dietary intervention
Unlike the 2006 American Heart Association guidelines and the US Department of Agriculture’s Dietary Guidelines for Americans 2005, the WHI dietary intervention had no specifications for dietary fiber, specific fatty acids (trans-fatty acids, omega-3 fatty acids, conjugated linoleic acid), complex carbohydrates, whole grains, vegetable protein, or other factors that have emerged as potential risk factors for cardiovascular and other chronic diseases since the study began. The WHI intervention also included no specific recommendation for total calorie intake, nor were patients in the intervention group encouraged to lose weight, as this could have confounded the results of the dietary intervention.
Education and encouragement
Those in the intervention group were each assigned a fat-gram goal, calculated on the basis of height. They were taught how to monitor their intake of total fat, fruits, vegetables, and grains. They attended intensive behavioral modification sessions to encourage them to keep to the dietary program: 18 group sessions in the first year and quarterly maintenance sessions thereafter, touching on a wide variety of nutrition- and behavior-related topics.7,9 Specially trained and certified nutritionists supervised the dietary intervention and the behavioral modification sessions according to the WHI study protocol.
Control-group participants received a copy of the US Department of Agriculture’s Dietary Guidelines for Americans10 and other health-related materials. They had no contact with the study nutritionists.
Other arms of the study
The WHI trial design included several arms,4,11–13 and many participants joined more than one arm: 20,592 postmenopausal women (42.2% of the total enrollment) chose dietary modification only, 8,050 (16.5%) chose diet plus hormone replacement therapy, 25,210 (51.6%) chose diet plus calcium and vitamin D supplementation, and 5,017 (10.3%) enrolled in all three.
Length of follow-up
Participants were followed from enrollment until they died, were lost to follow-up, or requested no further contact, or until the trial’s planned completion date, regardless of adherence to the dietary intervention, according to intention-to-treat analysis. All participants were contacted by clinic staff at 6-month intervals to provide updates on their health outcomes.
Factors assessed
Height, weight, waist circumference, and blood pressure were measured at annual visits using standardized procedures. Fasting blood samples were collected at baseline and at year 1 from all participants and from a subsample of 2,816 women (5.8% of the study population) at years 3 and 6. This subsample was randomly chosen with oversampling of minority women, for whom the odds for selection were six times higher than for white women.
Physical activity was assessed at baseline and at years 1, 3, 6, and 9. Walking and participation in sports and hours of activity per week were calculated for each participant. Physical activity was expressed as metabolic equivalent tasks per week for the analyses.
A food-frequency questionnaire6 to assess average dietary intake in the past 3 months was given at baseline and at year 1 for all participants. A third of all participants completed the questionnaire each year in a rotating sample. Completion rates were 100% at baseline and 81% thereafter. Follow-up data were collected from years 5 through 7. Also, 4-day food records were provided by all women before randomization.
HOW OUTCOMES WERE ASSESSED
The primary assessments of clinical outcome1–3 were mammographic screening, a self-reported medical history documented by a review of medical records, and electrocardiograms digitally obtained every 3 years. Mammograms and electrocardiograms were centrally adjudicated. The diagnosis of acute myocardial infarction was based on an algorithm that included cardiac pain, enzyme levels, and electrocardiographic readings.
OVERALL RESULTS
At 8.1 years, the incidence of breast cancer was 9% lower in the intervention group than in the comparison group (95% confidence interval [CI] = 0.83–1.01; P = .07, P = .09 weighted for length of follow-up).3 Subgroup analysis further showed that women who reported higher intakes of total dietary fat at baseline reduced their risk of breast cancer by 22% (95% CI = 0.64–0.96). Whether extended follow-up will show a significant association has yet to be determined.
Colon cancer rates did not differ between groups, but the number of polyps and adenomas reported was significantly lower in the dietary intervention group.1 The rate of colon cancer will also be included in the extended follow-up study of the WHI.
Risk factors for coronary heart disease in both groups—including levels of serum total cholesterol and serum low-density lipoprotein cholesterol, body weight, body mass index, diastolic blood pressure, and factor VIIc—improved slightly, but at year 3 of the trial, differences in overall rates of coronary heart disease and stroke in the two groups were not statistically significant.2 In addition, the low-fat diet intervention was associated with a reduction in blood estradiol concentrations between baseline and year 1.3 At the end of the study, however, differences in rates of breast cancer, colorectal cancer, and heart disease between the two groups were not statistically significant.
RESULTS OF DIETARY MODIFICATIONS
Fat as a percentage of total calories
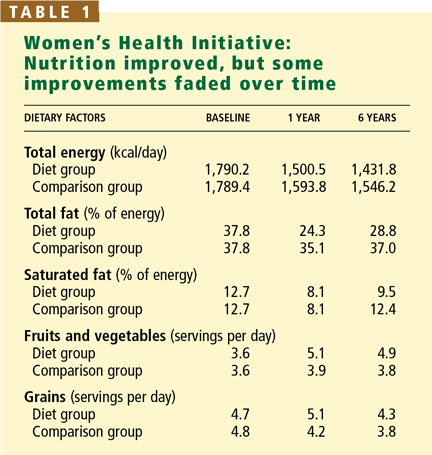
At the beginning of the WHI, all participants reported consuming an average of 35% of their caloric intake from fat (Table 1). At 1 year from baseline, the fat intake decreased to 24.3% in the intervention group (short of the study goal of 20%); this level had risen again to 26.7% by year 3 and to 28.8% at the end of the study. Stratified by quartile, women who achieved the greatest reductions in saturated and trans-fatty acids or the largest increases in their intake of fruits and vegetables appeared to have a moderate reduction in the risk of coronary heart disease.2 Women in the comparison group also decreased their fat intake initially, but to a lesser degree, and gradually increased it again thereafter. The mean net difference in self-reported total fat intake between the intervention group and the comparison group at 6 years was 8.2% (P < .001) (study goal, 13%).1–3
Intake of fruits, vegetables, and grains
At baseline, fruit and vegetable intake averaged 3.6 servings per day (Table 1). In the intervention group, this increased to 5.1 servings per day at year 1, and to 5.2 servings at year 3, but at the end of the study it had decreased to 4.9 servings.
Women in the intervention group were eating 4.7 servings of grains per day at baseline. This increased to 5.1 servings at year 1 and then decreased to 4.6 servings at year 3 and to 4.3 servings at the end of the study. It seems that as the women grew older their determination to increase servings of these foods diminished.
Proponents of some currently popular diets blame weight gain on a higher intake of carbohydrates, but the women following the WHI low-fat diet did not gain weight.2
Total fat vs saturated fat
Intake of total fat and saturated fat decreased in the intervention group during the study, but the difference between fat intake in the intervention group and that in the comparison group did not reach the degree expected.
At year 1, total fat as a percentage of total caloric intake was 10.8 percentage points below that of the comparison group, whereas the study expected difference was 13.0. At the end of the trial, the difference was only 8.2 percentage points, whereas the expected difference was 11.0.
Intake of all fatty acids (saturated and unsaturated) decreased at year 1, but then went back up slightly by the end of the trial but did not exceed baseline levels, and saturated fatty acids remained well below baseline levels: 9.5% vs 12.5% of caloric intake at baseline.4
INTERPRETING THE RESULTS
It might be tempting to dismiss the results of the WHI dietary intervention trial as not significant and therefore not meaningful. This would be unfortunate. The trial had some remarkable accomplishments and offers important lessons for future investigations.
The initial reductions in total fat intake were impressive, and women who had the highest total fat intake at baseline achieved the greatest reduction of total fat (to less than 22% of total calories).3 Nonetheless, the dietary intervention goal of less than 20% of calories from fat was not achieved despite intensive dietary counseling and a highly motivated study population. Thus, this dietary fat target may not be reasonable in the general population.
Also, despite the absence of targeted intervention on specific fatty acids, the observed blood cholesterol levels were as expected based on the well-known formula of Mensink and Katan,14 which incorporates information on changes in saturated fat, polyunsaturated fat, and dietary cholesterol intake. The predicted reduction in low-density lipoprotein cholesterol was 2.7 mg/dL; the observed reduction was 2.3 mg/dL.2 This illustrates that with greater modifications in specific known dietary risk factors for cardiovascular disease, such as saturated fatty acids, cholesterol, and unsaturated fatty acids, blood cholesterol levels respond in a predictable fashion. This was presumably not observed in WHI precisely because no goals and objectives were provided to participants for intake of saturated or polyunsaturated fatty acids.
Recent findings from the Optimal Macronutrient Intake Trial to Prevent Heart Disease (OmniHeart)15 further highlight differences in the total cholesterol response to diets of varying macronutrient (carbohydrate, protein, fat) content compared with the WHI dietary intervention.15 Participants in OmniHeart had reductions in levels of low-density lipoprotein cholesterol that were predictable from the changes reported in intake of saturated fatty acids. Presumably, the results of the WHI intervention would have been similar if the study had included this level of detail.
QUESTIONS REMAIN
Questions from the WHI that need consideration for future clinical applications include whether the study population may have already been “too old” to achieve a benefit from dietary modification, and whether the best timing for dietary intervention might be earlier adulthood with sustained changes in saturated fat, cholesterol, and unsaturated fat intake throughout life. Future subgroup analyses based on age at baseline will need to address these questions. Likewise, a longer follow-up period may be needed for a definitive evaluation of the impact of a regular low-fat diet on different health outcomes.
As reported by Patterson et al,16 the major contributors to total dietary fat intake at baseline were “added fats” such as sauces, gravies, butter, and margarines (25.1% of fat intake), followed by meats (20.9% of fat intake), and desserts (12.8% of fat intake). These findings highlight target areas for future interventions in women of this age group.
Another issue is how to standardize the dietary intervention from one clinical center to another—ie, to minimize differences in how each clinical center manages the study patients. Such differences were noted in WHI and other studies.17 Despite standardized training in delivering the dietary intervention, nutritionists encountered regional and cultural differences that required tailoring the dietary intervention to their patients’ needs. Staff turnover, an unavoidable phenomenon in long-term studies, has previously been reported to negatively influence dietary adherence.18
LIMITATIONS
A major limitation of diet modification research in general is the self-reporting of dietary intake, primarily by a food-frequency questionnaire. Although the use of a questionnaire is the most practical way to obtain dietary data for large studies, systematic biases may exist that obscure true nutrient-outcome relationships.19 Biomarker studies of energy balance suggest that people who are overweight or obese may under-report energy intake to a greater degree than people who are not overweight.20 Also, we still do not know how to get people to follow a healthy diet, although theories and models abound, such as social learning and cognitive-behavioral theory, and a lack of data limits our understanding of factors related to dietary adherence.21,22
FUTURE DIRECTIONS IN WHI
The WHI Extension Study is under way and has been funded through the year 2010. Outcomes ascertainment is the primary focus with no ongoing intervention, although the intervention group participants continue to receive a WHI newsletter that simply reiterates the importance of the study and encourages ongoing participation. As of 2006, an estimated 84% of the cohort, including both observational study and clinical trial participants, are involved. Efforts continue to recruit the remaining 16%, but many of these participants now consider themselves too old or too feeble to respond reliably.
In regard to breast cancer, the results published in 2006 are promising, albeit not statistically significant, and definitive statements cannot yet be made. However, postmenopausal women who are eating the diets highest in fat may have the greatest benefit from reductions in total fat.
Other considerations regarding the lack of statistically significant differences between groups may include the possibility that women in the intervention group may have been at lower risk for breast cancer at baseline. Likewise, although the results of the WHI dietary intervention do not include a statistically significant impact on colorectal cancer outcomes, the significant reduction in polyps and adenomas may later translate into a reduction in invasive cancer risk.
Finally, although no significant reduction was seen in the rate of death due to cardiovascular causes, greater reductions in saturated and trans-fatty acid intake were associated with greater reductions in blood cholesterol and cardiovascular risk.
Numerous subgroup analyses and ongoing assessments of the long-term impact of the diet modification are planned. Further associations are expected to emerge. The current and future results will continue to provide new insights that may lead to new clinical and public health recommendations in the future.
The WHI has raised additional issues that warrant further investigation:
- Will earlier dietary intervention, eg, during premenopausal years or even childhood, alter these results?
- Does the low-fat, high-carbohydrate diet used in WHI facilitate weight maintenance or even weight loss, as proposed by Howard et al23?
- Do quantitative changes in physical activity and weight control attenuate morbidity and mortality rates beyond changes in diet alone?
- Do vitamin and mineral supplements or hormone therapy alter disease outcomes or quality of life?
- Which behavioral approaches are best suited to the recruitment of patients for dietary intervention trials?
- Beresford SA, Johnson KC, Ritenbaugh C, et al. Low-fat dietary pattern and risk of colorectal cancer: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 2006; 295:643–654.
- Howard BV, Van Horn L, Hsia J, et al. Low-fat dietary pattern and risk of cardiovascular disease: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 2006; 295:655–666.
- Prentice RL, Caan B, Chlebowski RT, et al. Low-fat dietary pattern and risk of invasive breast cancer: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 2006; 295:629–642.
- The Women’s Health Initiative Study Group. Design of the Women’s Health Initiative clinical trial and observational study. Control Clin Trials 1998; 19:61–109.
- Ritenbaugh C, Patterson RE, Chlebowski RT, et al. The Women’s Health Initiative Dietary Modification trial: overview and baseline characteristics of participants. Ann Epidemiol 2003; 13 9 suppl:S87–97.
- Patterson RE, Kristal AR, Tinker LF, Carter RA, Bolton MP, Agurs-Collins T. Measurement characteristics of the Women’s Health Initiative food frequency questionnaire. Ann Epidemiol 1999; 9:178–187.
- Tinker LF, Burrows ER, Henry H, Patterson RE, Rupp JW, Van Horn LV. The Women’s Health Initiative: overview of the nutrition components. In:Krummel DA, Kris-Etherton PM, editors. Nutrition in Women’s Health. Gaithersburg, MD: Aspen, 1996:510–542.
- Henderson MM, Kushi LH, Thompson DJ, et al. Feasibility of a randomized trial of a low-fat diet for the prevention of breast cancer: dietary compliance in the Women’s Health Trial Vanguard Study. Prev Med 1990; 19:115–133.
- Bowen D, Ehret C, Pedersen M, et al. Results of an adjunct dietary intervention program in the Women’s Health Initiative. J Am Diet Assoc 2002; 102:1631–1637.
- US Department of Agriculture. Dietary Guidelines for Americans. 6. Washington, DC: US Dept of Health and Human Services, 2005.
- Jackson RD, LaCroix AZ, Cauley JA, McGowan J. The Women’s Health Initiative calcium-vitamin D trial: overview and baseline characteristics of participants. Ann Epidemiol 2003; 13 9 Suppl:S98–106.
- Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288:321–333.
- Stefanick ML, Cochrane BB, Hsia J, Barad DH, Liu JH, Johnson SR. The Women’s Health Initiative postmenopausal hormone trials: overview and baseline characteristics of participants. Ann Epidemiol 2003; 13 9 suppl:S78–86.
- Mensink RP, Katan MB. Effect of dietary fatty acids on serum lipids and lipoproteins. A meta-analysis of 27 trials. Arterioscler Thromb 1992; 12:911–919.
- Appel LJ, Sacks FM, Carey VJ, et al. Effects of protein, monounsaturated fat, and carbohydrate intake on blood pressure and serum lipids: results of the OmniHeart randomized trial. JAMA 2005; 294:2455–2464.
- Patterson RE, Kristal A, Rodabough R, et al. Changes in food sources of dietary fat in response to an intensive low-fat dietary intervention: early results from the Women’s Health Initiative. J Am Diet Assoc 2003; 103:454–460.
- Lichtman JH, Roumanis SA, Radford MJ, Riedinger MS, Weingarten S, Krumholz HM. Can practice guidelines be transported effectively to different settings? Results from a multicenter interventional study. Jt Comm J Qual Improv 2001; 27:42–53.
- Jackson M, Berman N, Huber M, et al. Research staff turnover and participant adherence in the Women’s Health Initiative. Control Clin Trials 2003; 24:422–435.
- Willett W, Lenart E. Reproducibility and validity of food-frequency questionnaires. In:Willett W, ed. Nutritional Epidemiology. 2. New York: Oxford University Press, 1998:101–147.
- Subar AF, Kipnis V, Troiano RP, et al. Using intake biomarkers to evaluate the extent of dietary misreporting in a large sample of adults: the OPEN study. Am J Epidemiol 2003; 158:1–13.
- Bowen D, Raczynski J, George V, Feng Z, Fouad M. The role of participation in the women’s health trial: feasibility study in minority populations. Prev Med 2000; 31:474–480.
- Patterson RE, Kristal AR, White E. Do beliefs, knowledge, and perceived norms about diet and cancer predict dietary change? Am J Public Health 1996; 86:1394–1400.
- Howard BV, Manson JE, Stefanick ML, et al. Low-fat dietary pattern and weight change over 7 years: the Women’s Health Initiative Dietary Modification Trial. JAMA 2006; 295:35–49.
More than 2 years have passed since we published the results of the Women’s Health Initiative (WHI), which caused a storm of information—and misinformation—about the effect of long-term dietary intervention on disease outcomes in postmenopausal women. Now that the dust has long settled, what have we learned from this landmark study?
The WHI results led to numerous additional analyses of all aspects of the study.1–7 What are the implications of all the analyses to clinical practice?
In this article, we summarize key aspects of the clinical trial, including study design, interventions, main results, and future plans. We also discuss potential clinical applications and practical considerations for public health efforts.
WHO WAS ELIGIBLE, WHO WAS NOT
A total of 48,835 postmenopausal women were randomly assigned to either no dietary intervention (n = 29,294) or a dietary intervention (n = 19,541) (see below).7 Participants were followed at 40 clinical centers between 1993 and 2005.4 Their mean age was 62.3 years; 18.6% were members of minorities.
Women were eligible if they were post-menopausal and had a daily dietary fat intake of at least 32% of total calories, based on assessment via a food-frequency questionnaire. They were excluded from the study if they had any of the following: a history of breast cancer, colorectal cancer, or other cancer except skin cancer during the past 10 years; type 1 diabetes; a medical condition in which the predicted survival was less than 3 years; and a potential barrier to adherence to the study regimen, including alcoholism or a lifestyle that involved often eating meals away from home.
THE WHI DIET: LESS FAT, BUT MORE FRUITS, VEGETABLES, GRAINS
The WHI dietary intervention was designed to prevent breast cancer, based on the evidence available when the study was planned. The targets included a total fat intake of less than 20% of energy (in kilocalories), increasing the intake of fruits and vegetables to at least five servings per day, and increasing the intake of grains to at least six servings per day.
Although reduction in saturated fat intake per se was not part of the WHI protocol, we assumed from previous pilot studies8 that the reduction of total fat intake would simultaneously produce a reduction in saturated fat intake to 7% of total calories.
A simpler dietary intervention
Unlike the 2006 American Heart Association guidelines and the US Department of Agriculture’s Dietary Guidelines for Americans 2005, the WHI dietary intervention had no specifications for dietary fiber, specific fatty acids (trans-fatty acids, omega-3 fatty acids, conjugated linoleic acid), complex carbohydrates, whole grains, vegetable protein, or other factors that have emerged as potential risk factors for cardiovascular and other chronic diseases since the study began. The WHI intervention also included no specific recommendation for total calorie intake, nor were patients in the intervention group encouraged to lose weight, as this could have confounded the results of the dietary intervention.
Education and encouragement
Those in the intervention group were each assigned a fat-gram goal, calculated on the basis of height. They were taught how to monitor their intake of total fat, fruits, vegetables, and grains. They attended intensive behavioral modification sessions to encourage them to keep to the dietary program: 18 group sessions in the first year and quarterly maintenance sessions thereafter, touching on a wide variety of nutrition- and behavior-related topics.7,9 Specially trained and certified nutritionists supervised the dietary intervention and the behavioral modification sessions according to the WHI study protocol.
Control-group participants received a copy of the US Department of Agriculture’s Dietary Guidelines for Americans10 and other health-related materials. They had no contact with the study nutritionists.
Other arms of the study
The WHI trial design included several arms,4,11–13 and many participants joined more than one arm: 20,592 postmenopausal women (42.2% of the total enrollment) chose dietary modification only, 8,050 (16.5%) chose diet plus hormone replacement therapy, 25,210 (51.6%) chose diet plus calcium and vitamin D supplementation, and 5,017 (10.3%) enrolled in all three.
Length of follow-up
Participants were followed from enrollment until they died, were lost to follow-up, or requested no further contact, or until the trial’s planned completion date, regardless of adherence to the dietary intervention, according to intention-to-treat analysis. All participants were contacted by clinic staff at 6-month intervals to provide updates on their health outcomes.
Factors assessed
Height, weight, waist circumference, and blood pressure were measured at annual visits using standardized procedures. Fasting blood samples were collected at baseline and at year 1 from all participants and from a subsample of 2,816 women (5.8% of the study population) at years 3 and 6. This subsample was randomly chosen with oversampling of minority women, for whom the odds for selection were six times higher than for white women.
Physical activity was assessed at baseline and at years 1, 3, 6, and 9. Walking and participation in sports and hours of activity per week were calculated for each participant. Physical activity was expressed as metabolic equivalent tasks per week for the analyses.
A food-frequency questionnaire6 to assess average dietary intake in the past 3 months was given at baseline and at year 1 for all participants. A third of all participants completed the questionnaire each year in a rotating sample. Completion rates were 100% at baseline and 81% thereafter. Follow-up data were collected from years 5 through 7. Also, 4-day food records were provided by all women before randomization.
HOW OUTCOMES WERE ASSESSED
The primary assessments of clinical outcome1–3 were mammographic screening, a self-reported medical history documented by a review of medical records, and electrocardiograms digitally obtained every 3 years. Mammograms and electrocardiograms were centrally adjudicated. The diagnosis of acute myocardial infarction was based on an algorithm that included cardiac pain, enzyme levels, and electrocardiographic readings.
OVERALL RESULTS
At 8.1 years, the incidence of breast cancer was 9% lower in the intervention group than in the comparison group (95% confidence interval [CI] = 0.83–1.01; P = .07, P = .09 weighted for length of follow-up).3 Subgroup analysis further showed that women who reported higher intakes of total dietary fat at baseline reduced their risk of breast cancer by 22% (95% CI = 0.64–0.96). Whether extended follow-up will show a significant association has yet to be determined.
Colon cancer rates did not differ between groups, but the number of polyps and adenomas reported was significantly lower in the dietary intervention group.1 The rate of colon cancer will also be included in the extended follow-up study of the WHI.
Risk factors for coronary heart disease in both groups—including levels of serum total cholesterol and serum low-density lipoprotein cholesterol, body weight, body mass index, diastolic blood pressure, and factor VIIc—improved slightly, but at year 3 of the trial, differences in overall rates of coronary heart disease and stroke in the two groups were not statistically significant.2 In addition, the low-fat diet intervention was associated with a reduction in blood estradiol concentrations between baseline and year 1.3 At the end of the study, however, differences in rates of breast cancer, colorectal cancer, and heart disease between the two groups were not statistically significant.
RESULTS OF DIETARY MODIFICATIONS
Fat as a percentage of total calories
At the beginning of the WHI, all participants reported consuming an average of 35% of their caloric intake from fat (Table 1). At 1 year from baseline, the fat intake decreased to 24.3% in the intervention group (short of the study goal of 20%); this level had risen again to 26.7% by year 3 and to 28.8% at the end of the study. Stratified by quartile, women who achieved the greatest reductions in saturated and trans-fatty acids or the largest increases in their intake of fruits and vegetables appeared to have a moderate reduction in the risk of coronary heart disease.2 Women in the comparison group also decreased their fat intake initially, but to a lesser degree, and gradually increased it again thereafter. The mean net difference in self-reported total fat intake between the intervention group and the comparison group at 6 years was 8.2% (P < .001) (study goal, 13%).1–3
Intake of fruits, vegetables, and grains
At baseline, fruit and vegetable intake averaged 3.6 servings per day (Table 1). In the intervention group, this increased to 5.1 servings per day at year 1, and to 5.2 servings at year 3, but at the end of the study it had decreased to 4.9 servings.
Women in the intervention group were eating 4.7 servings of grains per day at baseline. This increased to 5.1 servings at year 1 and then decreased to 4.6 servings at year 3 and to 4.3 servings at the end of the study. It seems that as the women grew older their determination to increase servings of these foods diminished.
Proponents of some currently popular diets blame weight gain on a higher intake of carbohydrates, but the women following the WHI low-fat diet did not gain weight.2
Total fat vs saturated fat
Intake of total fat and saturated fat decreased in the intervention group during the study, but the difference between fat intake in the intervention group and that in the comparison group did not reach the degree expected.
At year 1, total fat as a percentage of total caloric intake was 10.8 percentage points below that of the comparison group, whereas the study expected difference was 13.0. At the end of the trial, the difference was only 8.2 percentage points, whereas the expected difference was 11.0.
Intake of all fatty acids (saturated and unsaturated) decreased at year 1, but then went back up slightly by the end of the trial but did not exceed baseline levels, and saturated fatty acids remained well below baseline levels: 9.5% vs 12.5% of caloric intake at baseline.4
INTERPRETING THE RESULTS
It might be tempting to dismiss the results of the WHI dietary intervention trial as not significant and therefore not meaningful. This would be unfortunate. The trial had some remarkable accomplishments and offers important lessons for future investigations.
The initial reductions in total fat intake were impressive, and women who had the highest total fat intake at baseline achieved the greatest reduction of total fat (to less than 22% of total calories).3 Nonetheless, the dietary intervention goal of less than 20% of calories from fat was not achieved despite intensive dietary counseling and a highly motivated study population. Thus, this dietary fat target may not be reasonable in the general population.
Also, despite the absence of targeted intervention on specific fatty acids, the observed blood cholesterol levels were as expected based on the well-known formula of Mensink and Katan,14 which incorporates information on changes in saturated fat, polyunsaturated fat, and dietary cholesterol intake. The predicted reduction in low-density lipoprotein cholesterol was 2.7 mg/dL; the observed reduction was 2.3 mg/dL.2 This illustrates that with greater modifications in specific known dietary risk factors for cardiovascular disease, such as saturated fatty acids, cholesterol, and unsaturated fatty acids, blood cholesterol levels respond in a predictable fashion. This was presumably not observed in WHI precisely because no goals and objectives were provided to participants for intake of saturated or polyunsaturated fatty acids.
Recent findings from the Optimal Macronutrient Intake Trial to Prevent Heart Disease (OmniHeart)15 further highlight differences in the total cholesterol response to diets of varying macronutrient (carbohydrate, protein, fat) content compared with the WHI dietary intervention.15 Participants in OmniHeart had reductions in levels of low-density lipoprotein cholesterol that were predictable from the changes reported in intake of saturated fatty acids. Presumably, the results of the WHI intervention would have been similar if the study had included this level of detail.
QUESTIONS REMAIN
Questions from the WHI that need consideration for future clinical applications include whether the study population may have already been “too old” to achieve a benefit from dietary modification, and whether the best timing for dietary intervention might be earlier adulthood with sustained changes in saturated fat, cholesterol, and unsaturated fat intake throughout life. Future subgroup analyses based on age at baseline will need to address these questions. Likewise, a longer follow-up period may be needed for a definitive evaluation of the impact of a regular low-fat diet on different health outcomes.
As reported by Patterson et al,16 the major contributors to total dietary fat intake at baseline were “added fats” such as sauces, gravies, butter, and margarines (25.1% of fat intake), followed by meats (20.9% of fat intake), and desserts (12.8% of fat intake). These findings highlight target areas for future interventions in women of this age group.
Another issue is how to standardize the dietary intervention from one clinical center to another—ie, to minimize differences in how each clinical center manages the study patients. Such differences were noted in WHI and other studies.17 Despite standardized training in delivering the dietary intervention, nutritionists encountered regional and cultural differences that required tailoring the dietary intervention to their patients’ needs. Staff turnover, an unavoidable phenomenon in long-term studies, has previously been reported to negatively influence dietary adherence.18
LIMITATIONS
A major limitation of diet modification research in general is the self-reporting of dietary intake, primarily by a food-frequency questionnaire. Although the use of a questionnaire is the most practical way to obtain dietary data for large studies, systematic biases may exist that obscure true nutrient-outcome relationships.19 Biomarker studies of energy balance suggest that people who are overweight or obese may under-report energy intake to a greater degree than people who are not overweight.20 Also, we still do not know how to get people to follow a healthy diet, although theories and models abound, such as social learning and cognitive-behavioral theory, and a lack of data limits our understanding of factors related to dietary adherence.21,22
FUTURE DIRECTIONS IN WHI
The WHI Extension Study is under way and has been funded through the year 2010. Outcomes ascertainment is the primary focus with no ongoing intervention, although the intervention group participants continue to receive a WHI newsletter that simply reiterates the importance of the study and encourages ongoing participation. As of 2006, an estimated 84% of the cohort, including both observational study and clinical trial participants, are involved. Efforts continue to recruit the remaining 16%, but many of these participants now consider themselves too old or too feeble to respond reliably.
In regard to breast cancer, the results published in 2006 are promising, albeit not statistically significant, and definitive statements cannot yet be made. However, postmenopausal women who are eating the diets highest in fat may have the greatest benefit from reductions in total fat.
Other considerations regarding the lack of statistically significant differences between groups may include the possibility that women in the intervention group may have been at lower risk for breast cancer at baseline. Likewise, although the results of the WHI dietary intervention do not include a statistically significant impact on colorectal cancer outcomes, the significant reduction in polyps and adenomas may later translate into a reduction in invasive cancer risk.
Finally, although no significant reduction was seen in the rate of death due to cardiovascular causes, greater reductions in saturated and trans-fatty acid intake were associated with greater reductions in blood cholesterol and cardiovascular risk.
Numerous subgroup analyses and ongoing assessments of the long-term impact of the diet modification are planned. Further associations are expected to emerge. The current and future results will continue to provide new insights that may lead to new clinical and public health recommendations in the future.
The WHI has raised additional issues that warrant further investigation:
- Will earlier dietary intervention, eg, during premenopausal years or even childhood, alter these results?
- Does the low-fat, high-carbohydrate diet used in WHI facilitate weight maintenance or even weight loss, as proposed by Howard et al23?
- Do quantitative changes in physical activity and weight control attenuate morbidity and mortality rates beyond changes in diet alone?
- Do vitamin and mineral supplements or hormone therapy alter disease outcomes or quality of life?
- Which behavioral approaches are best suited to the recruitment of patients for dietary intervention trials?
More than 2 years have passed since we published the results of the Women’s Health Initiative (WHI), which caused a storm of information—and misinformation—about the effect of long-term dietary intervention on disease outcomes in postmenopausal women. Now that the dust has long settled, what have we learned from this landmark study?
The WHI results led to numerous additional analyses of all aspects of the study.1–7 What are the implications of all the analyses to clinical practice?
In this article, we summarize key aspects of the clinical trial, including study design, interventions, main results, and future plans. We also discuss potential clinical applications and practical considerations for public health efforts.
WHO WAS ELIGIBLE, WHO WAS NOT
A total of 48,835 postmenopausal women were randomly assigned to either no dietary intervention (n = 29,294) or a dietary intervention (n = 19,541) (see below).7 Participants were followed at 40 clinical centers between 1993 and 2005.4 Their mean age was 62.3 years; 18.6% were members of minorities.
Women were eligible if they were post-menopausal and had a daily dietary fat intake of at least 32% of total calories, based on assessment via a food-frequency questionnaire. They were excluded from the study if they had any of the following: a history of breast cancer, colorectal cancer, or other cancer except skin cancer during the past 10 years; type 1 diabetes; a medical condition in which the predicted survival was less than 3 years; and a potential barrier to adherence to the study regimen, including alcoholism or a lifestyle that involved often eating meals away from home.
THE WHI DIET: LESS FAT, BUT MORE FRUITS, VEGETABLES, GRAINS
The WHI dietary intervention was designed to prevent breast cancer, based on the evidence available when the study was planned. The targets included a total fat intake of less than 20% of energy (in kilocalories), increasing the intake of fruits and vegetables to at least five servings per day, and increasing the intake of grains to at least six servings per day.
Although reduction in saturated fat intake per se was not part of the WHI protocol, we assumed from previous pilot studies8 that the reduction of total fat intake would simultaneously produce a reduction in saturated fat intake to 7% of total calories.
A simpler dietary intervention
Unlike the 2006 American Heart Association guidelines and the US Department of Agriculture’s Dietary Guidelines for Americans 2005, the WHI dietary intervention had no specifications for dietary fiber, specific fatty acids (trans-fatty acids, omega-3 fatty acids, conjugated linoleic acid), complex carbohydrates, whole grains, vegetable protein, or other factors that have emerged as potential risk factors for cardiovascular and other chronic diseases since the study began. The WHI intervention also included no specific recommendation for total calorie intake, nor were patients in the intervention group encouraged to lose weight, as this could have confounded the results of the dietary intervention.
Education and encouragement
Those in the intervention group were each assigned a fat-gram goal, calculated on the basis of height. They were taught how to monitor their intake of total fat, fruits, vegetables, and grains. They attended intensive behavioral modification sessions to encourage them to keep to the dietary program: 18 group sessions in the first year and quarterly maintenance sessions thereafter, touching on a wide variety of nutrition- and behavior-related topics.7,9 Specially trained and certified nutritionists supervised the dietary intervention and the behavioral modification sessions according to the WHI study protocol.
Control-group participants received a copy of the US Department of Agriculture’s Dietary Guidelines for Americans10 and other health-related materials. They had no contact with the study nutritionists.
Other arms of the study
The WHI trial design included several arms,4,11–13 and many participants joined more than one arm: 20,592 postmenopausal women (42.2% of the total enrollment) chose dietary modification only, 8,050 (16.5%) chose diet plus hormone replacement therapy, 25,210 (51.6%) chose diet plus calcium and vitamin D supplementation, and 5,017 (10.3%) enrolled in all three.
Length of follow-up
Participants were followed from enrollment until they died, were lost to follow-up, or requested no further contact, or until the trial’s planned completion date, regardless of adherence to the dietary intervention, according to intention-to-treat analysis. All participants were contacted by clinic staff at 6-month intervals to provide updates on their health outcomes.
Factors assessed
Height, weight, waist circumference, and blood pressure were measured at annual visits using standardized procedures. Fasting blood samples were collected at baseline and at year 1 from all participants and from a subsample of 2,816 women (5.8% of the study population) at years 3 and 6. This subsample was randomly chosen with oversampling of minority women, for whom the odds for selection were six times higher than for white women.
Physical activity was assessed at baseline and at years 1, 3, 6, and 9. Walking and participation in sports and hours of activity per week were calculated for each participant. Physical activity was expressed as metabolic equivalent tasks per week for the analyses.
A food-frequency questionnaire6 to assess average dietary intake in the past 3 months was given at baseline and at year 1 for all participants. A third of all participants completed the questionnaire each year in a rotating sample. Completion rates were 100% at baseline and 81% thereafter. Follow-up data were collected from years 5 through 7. Also, 4-day food records were provided by all women before randomization.
HOW OUTCOMES WERE ASSESSED
The primary assessments of clinical outcome1–3 were mammographic screening, a self-reported medical history documented by a review of medical records, and electrocardiograms digitally obtained every 3 years. Mammograms and electrocardiograms were centrally adjudicated. The diagnosis of acute myocardial infarction was based on an algorithm that included cardiac pain, enzyme levels, and electrocardiographic readings.
OVERALL RESULTS
At 8.1 years, the incidence of breast cancer was 9% lower in the intervention group than in the comparison group (95% confidence interval [CI] = 0.83–1.01; P = .07, P = .09 weighted for length of follow-up).3 Subgroup analysis further showed that women who reported higher intakes of total dietary fat at baseline reduced their risk of breast cancer by 22% (95% CI = 0.64–0.96). Whether extended follow-up will show a significant association has yet to be determined.
Colon cancer rates did not differ between groups, but the number of polyps and adenomas reported was significantly lower in the dietary intervention group.1 The rate of colon cancer will also be included in the extended follow-up study of the WHI.
Risk factors for coronary heart disease in both groups—including levels of serum total cholesterol and serum low-density lipoprotein cholesterol, body weight, body mass index, diastolic blood pressure, and factor VIIc—improved slightly, but at year 3 of the trial, differences in overall rates of coronary heart disease and stroke in the two groups were not statistically significant.2 In addition, the low-fat diet intervention was associated with a reduction in blood estradiol concentrations between baseline and year 1.3 At the end of the study, however, differences in rates of breast cancer, colorectal cancer, and heart disease between the two groups were not statistically significant.
RESULTS OF DIETARY MODIFICATIONS
Fat as a percentage of total calories
At the beginning of the WHI, all participants reported consuming an average of 35% of their caloric intake from fat (Table 1). At 1 year from baseline, the fat intake decreased to 24.3% in the intervention group (short of the study goal of 20%); this level had risen again to 26.7% by year 3 and to 28.8% at the end of the study. Stratified by quartile, women who achieved the greatest reductions in saturated and trans-fatty acids or the largest increases in their intake of fruits and vegetables appeared to have a moderate reduction in the risk of coronary heart disease.2 Women in the comparison group also decreased their fat intake initially, but to a lesser degree, and gradually increased it again thereafter. The mean net difference in self-reported total fat intake between the intervention group and the comparison group at 6 years was 8.2% (P < .001) (study goal, 13%).1–3
Intake of fruits, vegetables, and grains
At baseline, fruit and vegetable intake averaged 3.6 servings per day (Table 1). In the intervention group, this increased to 5.1 servings per day at year 1, and to 5.2 servings at year 3, but at the end of the study it had decreased to 4.9 servings.
Women in the intervention group were eating 4.7 servings of grains per day at baseline. This increased to 5.1 servings at year 1 and then decreased to 4.6 servings at year 3 and to 4.3 servings at the end of the study. It seems that as the women grew older their determination to increase servings of these foods diminished.
Proponents of some currently popular diets blame weight gain on a higher intake of carbohydrates, but the women following the WHI low-fat diet did not gain weight.2
Total fat vs saturated fat
Intake of total fat and saturated fat decreased in the intervention group during the study, but the difference between fat intake in the intervention group and that in the comparison group did not reach the degree expected.
At year 1, total fat as a percentage of total caloric intake was 10.8 percentage points below that of the comparison group, whereas the study expected difference was 13.0. At the end of the trial, the difference was only 8.2 percentage points, whereas the expected difference was 11.0.
Intake of all fatty acids (saturated and unsaturated) decreased at year 1, but then went back up slightly by the end of the trial but did not exceed baseline levels, and saturated fatty acids remained well below baseline levels: 9.5% vs 12.5% of caloric intake at baseline.4
INTERPRETING THE RESULTS
It might be tempting to dismiss the results of the WHI dietary intervention trial as not significant and therefore not meaningful. This would be unfortunate. The trial had some remarkable accomplishments and offers important lessons for future investigations.
The initial reductions in total fat intake were impressive, and women who had the highest total fat intake at baseline achieved the greatest reduction of total fat (to less than 22% of total calories).3 Nonetheless, the dietary intervention goal of less than 20% of calories from fat was not achieved despite intensive dietary counseling and a highly motivated study population. Thus, this dietary fat target may not be reasonable in the general population.
Also, despite the absence of targeted intervention on specific fatty acids, the observed blood cholesterol levels were as expected based on the well-known formula of Mensink and Katan,14 which incorporates information on changes in saturated fat, polyunsaturated fat, and dietary cholesterol intake. The predicted reduction in low-density lipoprotein cholesterol was 2.7 mg/dL; the observed reduction was 2.3 mg/dL.2 This illustrates that with greater modifications in specific known dietary risk factors for cardiovascular disease, such as saturated fatty acids, cholesterol, and unsaturated fatty acids, blood cholesterol levels respond in a predictable fashion. This was presumably not observed in WHI precisely because no goals and objectives were provided to participants for intake of saturated or polyunsaturated fatty acids.
Recent findings from the Optimal Macronutrient Intake Trial to Prevent Heart Disease (OmniHeart)15 further highlight differences in the total cholesterol response to diets of varying macronutrient (carbohydrate, protein, fat) content compared with the WHI dietary intervention.15 Participants in OmniHeart had reductions in levels of low-density lipoprotein cholesterol that were predictable from the changes reported in intake of saturated fatty acids. Presumably, the results of the WHI intervention would have been similar if the study had included this level of detail.
QUESTIONS REMAIN
Questions from the WHI that need consideration for future clinical applications include whether the study population may have already been “too old” to achieve a benefit from dietary modification, and whether the best timing for dietary intervention might be earlier adulthood with sustained changes in saturated fat, cholesterol, and unsaturated fat intake throughout life. Future subgroup analyses based on age at baseline will need to address these questions. Likewise, a longer follow-up period may be needed for a definitive evaluation of the impact of a regular low-fat diet on different health outcomes.
As reported by Patterson et al,16 the major contributors to total dietary fat intake at baseline were “added fats” such as sauces, gravies, butter, and margarines (25.1% of fat intake), followed by meats (20.9% of fat intake), and desserts (12.8% of fat intake). These findings highlight target areas for future interventions in women of this age group.
Another issue is how to standardize the dietary intervention from one clinical center to another—ie, to minimize differences in how each clinical center manages the study patients. Such differences were noted in WHI and other studies.17 Despite standardized training in delivering the dietary intervention, nutritionists encountered regional and cultural differences that required tailoring the dietary intervention to their patients’ needs. Staff turnover, an unavoidable phenomenon in long-term studies, has previously been reported to negatively influence dietary adherence.18
LIMITATIONS
A major limitation of diet modification research in general is the self-reporting of dietary intake, primarily by a food-frequency questionnaire. Although the use of a questionnaire is the most practical way to obtain dietary data for large studies, systematic biases may exist that obscure true nutrient-outcome relationships.19 Biomarker studies of energy balance suggest that people who are overweight or obese may under-report energy intake to a greater degree than people who are not overweight.20 Also, we still do not know how to get people to follow a healthy diet, although theories and models abound, such as social learning and cognitive-behavioral theory, and a lack of data limits our understanding of factors related to dietary adherence.21,22
FUTURE DIRECTIONS IN WHI
The WHI Extension Study is under way and has been funded through the year 2010. Outcomes ascertainment is the primary focus with no ongoing intervention, although the intervention group participants continue to receive a WHI newsletter that simply reiterates the importance of the study and encourages ongoing participation. As of 2006, an estimated 84% of the cohort, including both observational study and clinical trial participants, are involved. Efforts continue to recruit the remaining 16%, but many of these participants now consider themselves too old or too feeble to respond reliably.
In regard to breast cancer, the results published in 2006 are promising, albeit not statistically significant, and definitive statements cannot yet be made. However, postmenopausal women who are eating the diets highest in fat may have the greatest benefit from reductions in total fat.
Other considerations regarding the lack of statistically significant differences between groups may include the possibility that women in the intervention group may have been at lower risk for breast cancer at baseline. Likewise, although the results of the WHI dietary intervention do not include a statistically significant impact on colorectal cancer outcomes, the significant reduction in polyps and adenomas may later translate into a reduction in invasive cancer risk.
Finally, although no significant reduction was seen in the rate of death due to cardiovascular causes, greater reductions in saturated and trans-fatty acid intake were associated with greater reductions in blood cholesterol and cardiovascular risk.
Numerous subgroup analyses and ongoing assessments of the long-term impact of the diet modification are planned. Further associations are expected to emerge. The current and future results will continue to provide new insights that may lead to new clinical and public health recommendations in the future.
The WHI has raised additional issues that warrant further investigation:
- Will earlier dietary intervention, eg, during premenopausal years or even childhood, alter these results?
- Does the low-fat, high-carbohydrate diet used in WHI facilitate weight maintenance or even weight loss, as proposed by Howard et al23?
- Do quantitative changes in physical activity and weight control attenuate morbidity and mortality rates beyond changes in diet alone?
- Do vitamin and mineral supplements or hormone therapy alter disease outcomes or quality of life?
- Which behavioral approaches are best suited to the recruitment of patients for dietary intervention trials?
- Beresford SA, Johnson KC, Ritenbaugh C, et al. Low-fat dietary pattern and risk of colorectal cancer: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 2006; 295:643–654.
- Howard BV, Van Horn L, Hsia J, et al. Low-fat dietary pattern and risk of cardiovascular disease: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 2006; 295:655–666.
- Prentice RL, Caan B, Chlebowski RT, et al. Low-fat dietary pattern and risk of invasive breast cancer: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 2006; 295:629–642.
- The Women’s Health Initiative Study Group. Design of the Women’s Health Initiative clinical trial and observational study. Control Clin Trials 1998; 19:61–109.
- Ritenbaugh C, Patterson RE, Chlebowski RT, et al. The Women’s Health Initiative Dietary Modification trial: overview and baseline characteristics of participants. Ann Epidemiol 2003; 13 9 suppl:S87–97.
- Patterson RE, Kristal AR, Tinker LF, Carter RA, Bolton MP, Agurs-Collins T. Measurement characteristics of the Women’s Health Initiative food frequency questionnaire. Ann Epidemiol 1999; 9:178–187.
- Tinker LF, Burrows ER, Henry H, Patterson RE, Rupp JW, Van Horn LV. The Women’s Health Initiative: overview of the nutrition components. In:Krummel DA, Kris-Etherton PM, editors. Nutrition in Women’s Health. Gaithersburg, MD: Aspen, 1996:510–542.
- Henderson MM, Kushi LH, Thompson DJ, et al. Feasibility of a randomized trial of a low-fat diet for the prevention of breast cancer: dietary compliance in the Women’s Health Trial Vanguard Study. Prev Med 1990; 19:115–133.
- Bowen D, Ehret C, Pedersen M, et al. Results of an adjunct dietary intervention program in the Women’s Health Initiative. J Am Diet Assoc 2002; 102:1631–1637.
- US Department of Agriculture. Dietary Guidelines for Americans. 6. Washington, DC: US Dept of Health and Human Services, 2005.
- Jackson RD, LaCroix AZ, Cauley JA, McGowan J. The Women’s Health Initiative calcium-vitamin D trial: overview and baseline characteristics of participants. Ann Epidemiol 2003; 13 9 Suppl:S98–106.
- Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288:321–333.
- Stefanick ML, Cochrane BB, Hsia J, Barad DH, Liu JH, Johnson SR. The Women’s Health Initiative postmenopausal hormone trials: overview and baseline characteristics of participants. Ann Epidemiol 2003; 13 9 suppl:S78–86.
- Mensink RP, Katan MB. Effect of dietary fatty acids on serum lipids and lipoproteins. A meta-analysis of 27 trials. Arterioscler Thromb 1992; 12:911–919.
- Appel LJ, Sacks FM, Carey VJ, et al. Effects of protein, monounsaturated fat, and carbohydrate intake on blood pressure and serum lipids: results of the OmniHeart randomized trial. JAMA 2005; 294:2455–2464.
- Patterson RE, Kristal A, Rodabough R, et al. Changes in food sources of dietary fat in response to an intensive low-fat dietary intervention: early results from the Women’s Health Initiative. J Am Diet Assoc 2003; 103:454–460.
- Lichtman JH, Roumanis SA, Radford MJ, Riedinger MS, Weingarten S, Krumholz HM. Can practice guidelines be transported effectively to different settings? Results from a multicenter interventional study. Jt Comm J Qual Improv 2001; 27:42–53.
- Jackson M, Berman N, Huber M, et al. Research staff turnover and participant adherence in the Women’s Health Initiative. Control Clin Trials 2003; 24:422–435.
- Willett W, Lenart E. Reproducibility and validity of food-frequency questionnaires. In:Willett W, ed. Nutritional Epidemiology. 2. New York: Oxford University Press, 1998:101–147.
- Subar AF, Kipnis V, Troiano RP, et al. Using intake biomarkers to evaluate the extent of dietary misreporting in a large sample of adults: the OPEN study. Am J Epidemiol 2003; 158:1–13.
- Bowen D, Raczynski J, George V, Feng Z, Fouad M. The role of participation in the women’s health trial: feasibility study in minority populations. Prev Med 2000; 31:474–480.
- Patterson RE, Kristal AR, White E. Do beliefs, knowledge, and perceived norms about diet and cancer predict dietary change? Am J Public Health 1996; 86:1394–1400.
- Howard BV, Manson JE, Stefanick ML, et al. Low-fat dietary pattern and weight change over 7 years: the Women’s Health Initiative Dietary Modification Trial. JAMA 2006; 295:35–49.
- Beresford SA, Johnson KC, Ritenbaugh C, et al. Low-fat dietary pattern and risk of colorectal cancer: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 2006; 295:643–654.
- Howard BV, Van Horn L, Hsia J, et al. Low-fat dietary pattern and risk of cardiovascular disease: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 2006; 295:655–666.
- Prentice RL, Caan B, Chlebowski RT, et al. Low-fat dietary pattern and risk of invasive breast cancer: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 2006; 295:629–642.
- The Women’s Health Initiative Study Group. Design of the Women’s Health Initiative clinical trial and observational study. Control Clin Trials 1998; 19:61–109.
- Ritenbaugh C, Patterson RE, Chlebowski RT, et al. The Women’s Health Initiative Dietary Modification trial: overview and baseline characteristics of participants. Ann Epidemiol 2003; 13 9 suppl:S87–97.
- Patterson RE, Kristal AR, Tinker LF, Carter RA, Bolton MP, Agurs-Collins T. Measurement characteristics of the Women’s Health Initiative food frequency questionnaire. Ann Epidemiol 1999; 9:178–187.
- Tinker LF, Burrows ER, Henry H, Patterson RE, Rupp JW, Van Horn LV. The Women’s Health Initiative: overview of the nutrition components. In:Krummel DA, Kris-Etherton PM, editors. Nutrition in Women’s Health. Gaithersburg, MD: Aspen, 1996:510–542.
- Henderson MM, Kushi LH, Thompson DJ, et al. Feasibility of a randomized trial of a low-fat diet for the prevention of breast cancer: dietary compliance in the Women’s Health Trial Vanguard Study. Prev Med 1990; 19:115–133.
- Bowen D, Ehret C, Pedersen M, et al. Results of an adjunct dietary intervention program in the Women’s Health Initiative. J Am Diet Assoc 2002; 102:1631–1637.
- US Department of Agriculture. Dietary Guidelines for Americans. 6. Washington, DC: US Dept of Health and Human Services, 2005.
- Jackson RD, LaCroix AZ, Cauley JA, McGowan J. The Women’s Health Initiative calcium-vitamin D trial: overview and baseline characteristics of participants. Ann Epidemiol 2003; 13 9 Suppl:S98–106.
- Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288:321–333.
- Stefanick ML, Cochrane BB, Hsia J, Barad DH, Liu JH, Johnson SR. The Women’s Health Initiative postmenopausal hormone trials: overview and baseline characteristics of participants. Ann Epidemiol 2003; 13 9 suppl:S78–86.
- Mensink RP, Katan MB. Effect of dietary fatty acids on serum lipids and lipoproteins. A meta-analysis of 27 trials. Arterioscler Thromb 1992; 12:911–919.
- Appel LJ, Sacks FM, Carey VJ, et al. Effects of protein, monounsaturated fat, and carbohydrate intake on blood pressure and serum lipids: results of the OmniHeart randomized trial. JAMA 2005; 294:2455–2464.
- Patterson RE, Kristal A, Rodabough R, et al. Changes in food sources of dietary fat in response to an intensive low-fat dietary intervention: early results from the Women’s Health Initiative. J Am Diet Assoc 2003; 103:454–460.
- Lichtman JH, Roumanis SA, Radford MJ, Riedinger MS, Weingarten S, Krumholz HM. Can practice guidelines be transported effectively to different settings? Results from a multicenter interventional study. Jt Comm J Qual Improv 2001; 27:42–53.
- Jackson M, Berman N, Huber M, et al. Research staff turnover and participant adherence in the Women’s Health Initiative. Control Clin Trials 2003; 24:422–435.
- Willett W, Lenart E. Reproducibility and validity of food-frequency questionnaires. In:Willett W, ed. Nutritional Epidemiology. 2. New York: Oxford University Press, 1998:101–147.
- Subar AF, Kipnis V, Troiano RP, et al. Using intake biomarkers to evaluate the extent of dietary misreporting in a large sample of adults: the OPEN study. Am J Epidemiol 2003; 158:1–13.
- Bowen D, Raczynski J, George V, Feng Z, Fouad M. The role of participation in the women’s health trial: feasibility study in minority populations. Prev Med 2000; 31:474–480.
- Patterson RE, Kristal AR, White E. Do beliefs, knowledge, and perceived norms about diet and cancer predict dietary change? Am J Public Health 1996; 86:1394–1400.
- Howard BV, Manson JE, Stefanick ML, et al. Low-fat dietary pattern and weight change over 7 years: the Women’s Health Initiative Dietary Modification Trial. JAMA 2006; 295:35–49.
KEY POINTS
- Colon cancer rates did not differ between the dietary intervention group and the comparison group, but the number of polyps and adenomas reported was significantly lower in the dietary intervention group.
- Risk factors for coronary heart disease improved slightly with the diet, but by trial year 3, differences in overall rates of coronary heart disease and stroke in the two groups were not statistically significant.
- When stratified by quartiles, those who reduced their intake of saturated and trans-fatty acids the most, or who increased their intake of fruits and vegetables the most, appeared to have a moderate reduction in the risk of coronary heart disease.
How safe are erythropoiesis-stimulating agents?
The year 2007 was a rough one for erythropoiesis-stimulating agents (ESAs). Increasing concerns about their safety were raised in important meta-analyses of previously published data, specifically, the possibility that these agents increase the risk of venous thromboembolic phenomenona and shorten survival. These trends were seen primarily in studies of cancer patients.1 Meanwhile, front-page headlines in The New York Times were unkind: “Doctors reaping millions for use of anemia drugs.”2 However, the signals came earlier than 2007.
THE RISE AND POSSIBLE FALL OF ESAs
1989—These costly drugs are introduced and start their ascent to becoming one of the most widely used drug classes, helped along by direct-to-consumer advertising. (In one ad, Grandpa can run after the grandchildren despite being on chemotherapy because he uses erythropoietin!)
2001—A study declares that the higher the hemoglobin level rises in response to ESAs, the better the quality of life. It also hints that these agents improve survival.3
2002—The American Society of Hematology/American Society of Clinical Oncology Practice Guideline Writing Committee reviews the medical literature, performs a systematic review, and recommends that patients with low-risk myelodysplasia and those on chemotherapy who become anemic (with a hemoglobin level approaching 10 g/dL) have the option of receiving ESAs to raise their hemoglobin and decrease the need for transfusion.4
2003—Henke et al5 report that anemic patients with head and neck cancer who underwent radiotherapy and received erythropoietin in a randomized study had poorer survival and progression-free survival.
2005—Leyland-Jones et al,6 in another randomized study, report that patients with metastatic breast cancer receiving first-line chemotherapy (most of whom were not anemic) had a higher mortality rate if they received epoetin alfa.
2006—The guideline authors meet again to start the process of writing an update. A meta-analysis shows the thromboembolic risk and survival problems in a more systematic way, covering multiple studies.7
2007—The Centers for Medicare and Medicaid Services cuts back the reimbursement for the use of erythropoietin. The US Food and Drug Administration (FDA) publishes a black box warning suggesting that any hemoglobin level greater than 12 g/dL would be detrimental to a patient. The Guideline Writing Committee works on its document with this backdrop of turmoil.
2008—The updated guidelines are published. They recommend continuing to use ESAs for patients with low-risk myelodysplasia, and as an option to raise hemoglobin levels and prevent the need for transfusion in cancer patients undergoing chemotherapy whose hemoglobin level falls to 10 g/dL or less.8 The document includes stronger language against the use of ESAs in patients with anemia from cancer who are not undergoing chemotherapy. Meanwhile, some editorialists have suggested that it may be time to abandon ESAs because these drugs may promote more rapid tumor growth or pose a prohibitive risk of thromboembolic disease.9,10
In mid-March 2008, after reviewing the most recent data, an FDA panel calls for new limits on the use of ESAs: cancer patients who are undergoing treatment that could cure their cancer should not receive them, and neither should patients with advanced breast cancer or head and neck cancer. Furthermore, the FDA panel stipulates that when doctors do prescribe these drugs, they should warn patients of the possible dangers and seek their informed consent.
WHAT HAPPENS NOW?
Will the FDA take ESAs off the market? That is unlikely. Nephrologists need these drugs to avoid the need for transfusion in dialysis patients (see the accompanying article by Drs. Demirjian and Nurko on the use of ESAs in patients with chronic kidney disease on page 353 of this issue of the Journal). Some of the very first signals of harm with raising the hemoglobin too high came from the nephrology field.
Hematologists should still have the option of using ESAs in some settings, particularly in patients with low-risk myelodysplasia who are becoming more and more anemic and in those who have comorbid conditions in which lower hemoglobin levels are unsafe, particularly if they have coronary artery disease. They also should still be able to use ESAs for selected patients who develop severe chemotherapy-induced anemia and who become so weakened from their anemic state that their life and quality of life are threatened. If ESAs are taken away from the formulary of hematologists and oncologists, these specialists will rely on transfusions to treat their anemic patients, whether the anemia be due to myelodysplasia or to chemotherapy. Some say that the blood supply is so safe that we should not have the same worries about using blood transfusions as we did in the late 1980s. However, we all have seen patients who adamantly do not want transfusions.
Certainly, more events will transpire in the next year with the ESAs. There will probably be more data on erythropoietin receptors on the surface of tumor cells and what happens when pharmacologic doses of erythropoietin interact with these receptors. Patient-specific meta-analyses will probably shed more light on individual patients’ risk of thromboembolic disease and early death from the use of these agents.
The ESA story is far from over.
- Bohlius J, Wilson J, Seidenfeld J, et al. Erythropoietin or darbopoietin for patients with cancer. Cochrane Database Syst Rev 2007; 2:CD 003407.
- Berenson A, Pollack A. Doctors reaping millions for use of anemia drugs. New York Times, May 9, 2007, page1A.
- Littlewood TJ, Bajette E, Nortier JW, et al. Effects of epoetin alfa on hematologic parameters & quality of life in cancer patients receiving nonplatinum chemotherapy: results of a randomized, double-blind, placebo-controlled trial. J Clin Oncol 2001; 19:2865–2874.
- Rizzo JD, Lichtin AE, Woolf SH, et al. Use of epoetin in patients with cancer: evidence based clinical practice guidelines of the American Society of Clinical Oncology and the American Society of Hematology. J Clin Oncol 2002; 20:4083–4107.
- Henke M, Laszig R, Rube C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double–blind, placebo-controlled trial. Lancet 2003; 362:1255–1260.
- Leyland-Jones B, Semiglazov V, Pawlicki M, et al. Maintaining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: a survival study. J Clin Oncol 2005; 23:5960–5972.
- Bohlius J, Wilson J, Seidenfeld J, et al. Recombinant human erythropoietins and cancer patients: updated meta-analysis of 57 studies including 9353 patients. J Natl Cancer Inst 2006; 98:708–714.
- Rizzo JD, Somerfield MR, Hagerty K, et al. Use of epoetin and darbepoetin in patients with cancer: 2007 American Society of Hematology/American Society of Clinical Oncology clinical practice guideline update. Blood 2008; 111:25–41.
- Tefferi A. Pharmaceutical erythropoietin use in patients with cancer: is it time to abandon ship or just drop anchor? [editorial] Mayo Clin Proc 2007; 82:1316–1318.
- Anonymous. Erythropoietin analogues: an unnecessary class of drugs [editorial]. Lancet Oncol 2008; 9:81.
The year 2007 was a rough one for erythropoiesis-stimulating agents (ESAs). Increasing concerns about their safety were raised in important meta-analyses of previously published data, specifically, the possibility that these agents increase the risk of venous thromboembolic phenomenona and shorten survival. These trends were seen primarily in studies of cancer patients.1 Meanwhile, front-page headlines in The New York Times were unkind: “Doctors reaping millions for use of anemia drugs.”2 However, the signals came earlier than 2007.
THE RISE AND POSSIBLE FALL OF ESAs
1989—These costly drugs are introduced and start their ascent to becoming one of the most widely used drug classes, helped along by direct-to-consumer advertising. (In one ad, Grandpa can run after the grandchildren despite being on chemotherapy because he uses erythropoietin!)
2001—A study declares that the higher the hemoglobin level rises in response to ESAs, the better the quality of life. It also hints that these agents improve survival.3
2002—The American Society of Hematology/American Society of Clinical Oncology Practice Guideline Writing Committee reviews the medical literature, performs a systematic review, and recommends that patients with low-risk myelodysplasia and those on chemotherapy who become anemic (with a hemoglobin level approaching 10 g/dL) have the option of receiving ESAs to raise their hemoglobin and decrease the need for transfusion.4
2003—Henke et al5 report that anemic patients with head and neck cancer who underwent radiotherapy and received erythropoietin in a randomized study had poorer survival and progression-free survival.
2005—Leyland-Jones et al,6 in another randomized study, report that patients with metastatic breast cancer receiving first-line chemotherapy (most of whom were not anemic) had a higher mortality rate if they received epoetin alfa.
2006—The guideline authors meet again to start the process of writing an update. A meta-analysis shows the thromboembolic risk and survival problems in a more systematic way, covering multiple studies.7
2007—The Centers for Medicare and Medicaid Services cuts back the reimbursement for the use of erythropoietin. The US Food and Drug Administration (FDA) publishes a black box warning suggesting that any hemoglobin level greater than 12 g/dL would be detrimental to a patient. The Guideline Writing Committee works on its document with this backdrop of turmoil.
2008—The updated guidelines are published. They recommend continuing to use ESAs for patients with low-risk myelodysplasia, and as an option to raise hemoglobin levels and prevent the need for transfusion in cancer patients undergoing chemotherapy whose hemoglobin level falls to 10 g/dL or less.8 The document includes stronger language against the use of ESAs in patients with anemia from cancer who are not undergoing chemotherapy. Meanwhile, some editorialists have suggested that it may be time to abandon ESAs because these drugs may promote more rapid tumor growth or pose a prohibitive risk of thromboembolic disease.9,10
In mid-March 2008, after reviewing the most recent data, an FDA panel calls for new limits on the use of ESAs: cancer patients who are undergoing treatment that could cure their cancer should not receive them, and neither should patients with advanced breast cancer or head and neck cancer. Furthermore, the FDA panel stipulates that when doctors do prescribe these drugs, they should warn patients of the possible dangers and seek their informed consent.
WHAT HAPPENS NOW?
Will the FDA take ESAs off the market? That is unlikely. Nephrologists need these drugs to avoid the need for transfusion in dialysis patients (see the accompanying article by Drs. Demirjian and Nurko on the use of ESAs in patients with chronic kidney disease on page 353 of this issue of the Journal). Some of the very first signals of harm with raising the hemoglobin too high came from the nephrology field.
Hematologists should still have the option of using ESAs in some settings, particularly in patients with low-risk myelodysplasia who are becoming more and more anemic and in those who have comorbid conditions in which lower hemoglobin levels are unsafe, particularly if they have coronary artery disease. They also should still be able to use ESAs for selected patients who develop severe chemotherapy-induced anemia and who become so weakened from their anemic state that their life and quality of life are threatened. If ESAs are taken away from the formulary of hematologists and oncologists, these specialists will rely on transfusions to treat their anemic patients, whether the anemia be due to myelodysplasia or to chemotherapy. Some say that the blood supply is so safe that we should not have the same worries about using blood transfusions as we did in the late 1980s. However, we all have seen patients who adamantly do not want transfusions.
Certainly, more events will transpire in the next year with the ESAs. There will probably be more data on erythropoietin receptors on the surface of tumor cells and what happens when pharmacologic doses of erythropoietin interact with these receptors. Patient-specific meta-analyses will probably shed more light on individual patients’ risk of thromboembolic disease and early death from the use of these agents.
The ESA story is far from over.
The year 2007 was a rough one for erythropoiesis-stimulating agents (ESAs). Increasing concerns about their safety were raised in important meta-analyses of previously published data, specifically, the possibility that these agents increase the risk of venous thromboembolic phenomenona and shorten survival. These trends were seen primarily in studies of cancer patients.1 Meanwhile, front-page headlines in The New York Times were unkind: “Doctors reaping millions for use of anemia drugs.”2 However, the signals came earlier than 2007.
THE RISE AND POSSIBLE FALL OF ESAs
1989—These costly drugs are introduced and start their ascent to becoming one of the most widely used drug classes, helped along by direct-to-consumer advertising. (In one ad, Grandpa can run after the grandchildren despite being on chemotherapy because he uses erythropoietin!)
2001—A study declares that the higher the hemoglobin level rises in response to ESAs, the better the quality of life. It also hints that these agents improve survival.3
2002—The American Society of Hematology/American Society of Clinical Oncology Practice Guideline Writing Committee reviews the medical literature, performs a systematic review, and recommends that patients with low-risk myelodysplasia and those on chemotherapy who become anemic (with a hemoglobin level approaching 10 g/dL) have the option of receiving ESAs to raise their hemoglobin and decrease the need for transfusion.4
2003—Henke et al5 report that anemic patients with head and neck cancer who underwent radiotherapy and received erythropoietin in a randomized study had poorer survival and progression-free survival.
2005—Leyland-Jones et al,6 in another randomized study, report that patients with metastatic breast cancer receiving first-line chemotherapy (most of whom were not anemic) had a higher mortality rate if they received epoetin alfa.
2006—The guideline authors meet again to start the process of writing an update. A meta-analysis shows the thromboembolic risk and survival problems in a more systematic way, covering multiple studies.7
2007—The Centers for Medicare and Medicaid Services cuts back the reimbursement for the use of erythropoietin. The US Food and Drug Administration (FDA) publishes a black box warning suggesting that any hemoglobin level greater than 12 g/dL would be detrimental to a patient. The Guideline Writing Committee works on its document with this backdrop of turmoil.
2008—The updated guidelines are published. They recommend continuing to use ESAs for patients with low-risk myelodysplasia, and as an option to raise hemoglobin levels and prevent the need for transfusion in cancer patients undergoing chemotherapy whose hemoglobin level falls to 10 g/dL or less.8 The document includes stronger language against the use of ESAs in patients with anemia from cancer who are not undergoing chemotherapy. Meanwhile, some editorialists have suggested that it may be time to abandon ESAs because these drugs may promote more rapid tumor growth or pose a prohibitive risk of thromboembolic disease.9,10
In mid-March 2008, after reviewing the most recent data, an FDA panel calls for new limits on the use of ESAs: cancer patients who are undergoing treatment that could cure their cancer should not receive them, and neither should patients with advanced breast cancer or head and neck cancer. Furthermore, the FDA panel stipulates that when doctors do prescribe these drugs, they should warn patients of the possible dangers and seek their informed consent.
WHAT HAPPENS NOW?
Will the FDA take ESAs off the market? That is unlikely. Nephrologists need these drugs to avoid the need for transfusion in dialysis patients (see the accompanying article by Drs. Demirjian and Nurko on the use of ESAs in patients with chronic kidney disease on page 353 of this issue of the Journal). Some of the very first signals of harm with raising the hemoglobin too high came from the nephrology field.
Hematologists should still have the option of using ESAs in some settings, particularly in patients with low-risk myelodysplasia who are becoming more and more anemic and in those who have comorbid conditions in which lower hemoglobin levels are unsafe, particularly if they have coronary artery disease. They also should still be able to use ESAs for selected patients who develop severe chemotherapy-induced anemia and who become so weakened from their anemic state that their life and quality of life are threatened. If ESAs are taken away from the formulary of hematologists and oncologists, these specialists will rely on transfusions to treat their anemic patients, whether the anemia be due to myelodysplasia or to chemotherapy. Some say that the blood supply is so safe that we should not have the same worries about using blood transfusions as we did in the late 1980s. However, we all have seen patients who adamantly do not want transfusions.
Certainly, more events will transpire in the next year with the ESAs. There will probably be more data on erythropoietin receptors on the surface of tumor cells and what happens when pharmacologic doses of erythropoietin interact with these receptors. Patient-specific meta-analyses will probably shed more light on individual patients’ risk of thromboembolic disease and early death from the use of these agents.
The ESA story is far from over.
- Bohlius J, Wilson J, Seidenfeld J, et al. Erythropoietin or darbopoietin for patients with cancer. Cochrane Database Syst Rev 2007; 2:CD 003407.
- Berenson A, Pollack A. Doctors reaping millions for use of anemia drugs. New York Times, May 9, 2007, page1A.
- Littlewood TJ, Bajette E, Nortier JW, et al. Effects of epoetin alfa on hematologic parameters & quality of life in cancer patients receiving nonplatinum chemotherapy: results of a randomized, double-blind, placebo-controlled trial. J Clin Oncol 2001; 19:2865–2874.
- Rizzo JD, Lichtin AE, Woolf SH, et al. Use of epoetin in patients with cancer: evidence based clinical practice guidelines of the American Society of Clinical Oncology and the American Society of Hematology. J Clin Oncol 2002; 20:4083–4107.
- Henke M, Laszig R, Rube C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double–blind, placebo-controlled trial. Lancet 2003; 362:1255–1260.
- Leyland-Jones B, Semiglazov V, Pawlicki M, et al. Maintaining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: a survival study. J Clin Oncol 2005; 23:5960–5972.
- Bohlius J, Wilson J, Seidenfeld J, et al. Recombinant human erythropoietins and cancer patients: updated meta-analysis of 57 studies including 9353 patients. J Natl Cancer Inst 2006; 98:708–714.
- Rizzo JD, Somerfield MR, Hagerty K, et al. Use of epoetin and darbepoetin in patients with cancer: 2007 American Society of Hematology/American Society of Clinical Oncology clinical practice guideline update. Blood 2008; 111:25–41.
- Tefferi A. Pharmaceutical erythropoietin use in patients with cancer: is it time to abandon ship or just drop anchor? [editorial] Mayo Clin Proc 2007; 82:1316–1318.
- Anonymous. Erythropoietin analogues: an unnecessary class of drugs [editorial]. Lancet Oncol 2008; 9:81.
- Bohlius J, Wilson J, Seidenfeld J, et al. Erythropoietin or darbopoietin for patients with cancer. Cochrane Database Syst Rev 2007; 2:CD 003407.
- Berenson A, Pollack A. Doctors reaping millions for use of anemia drugs. New York Times, May 9, 2007, page1A.
- Littlewood TJ, Bajette E, Nortier JW, et al. Effects of epoetin alfa on hematologic parameters & quality of life in cancer patients receiving nonplatinum chemotherapy: results of a randomized, double-blind, placebo-controlled trial. J Clin Oncol 2001; 19:2865–2874.
- Rizzo JD, Lichtin AE, Woolf SH, et al. Use of epoetin in patients with cancer: evidence based clinical practice guidelines of the American Society of Clinical Oncology and the American Society of Hematology. J Clin Oncol 2002; 20:4083–4107.
- Henke M, Laszig R, Rube C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double–blind, placebo-controlled trial. Lancet 2003; 362:1255–1260.
- Leyland-Jones B, Semiglazov V, Pawlicki M, et al. Maintaining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: a survival study. J Clin Oncol 2005; 23:5960–5972.
- Bohlius J, Wilson J, Seidenfeld J, et al. Recombinant human erythropoietins and cancer patients: updated meta-analysis of 57 studies including 9353 patients. J Natl Cancer Inst 2006; 98:708–714.
- Rizzo JD, Somerfield MR, Hagerty K, et al. Use of epoetin and darbepoetin in patients with cancer: 2007 American Society of Hematology/American Society of Clinical Oncology clinical practice guideline update. Blood 2008; 111:25–41.
- Tefferi A. Pharmaceutical erythropoietin use in patients with cancer: is it time to abandon ship or just drop anchor? [editorial] Mayo Clin Proc 2007; 82:1316–1318.
- Anonymous. Erythropoietin analogues: an unnecessary class of drugs [editorial]. Lancet Oncol 2008; 9:81.
What role will ‘gliptins’ play in glycemic control?
The “gliptins”—the nickname for dipeptidyl peptidase 4 (DPP-4) inhibitors—are one of the newest classes of drugs for the treatment of type 2 diabetes mellitus.
These drugs work by prolonging the action of gut hormones called incretins, which boost insulin levels. The greatest advantage of the gliptins appears to be their ability to stimulate insulin production with little risk of corresponding hypoglycemia.
Sitagliptin (Januvia), the first commercially available DPP-4 inhibitor, has been approved by the US Food and Drug Administration (FDA) and is currently in clinical use, and vildagliptin (Galvus) awaits FDA approval at the time of this writing. Other drugs of this class are in development.
However, because these drugs are so new, a number of questions remain about their use. In this article, we discuss the rationale behind gliptin drugs, the evidence to date on their use alone or in combination with current oral hypoglycemic drugs (and even with insulin), and when and how to use them in daily practice.
THE NEED FOR MORE EFFECTIVE DIABETES TREATMENT
As the number of patients with type 2 diabetes continues its steep and steady rise,1,2 much work has gone into studying treatment goals and how to achieve them. Although experts generally agree on glycemic goals,3 we currently fail to achieve those goals in close to two-thirds of patients: only 37% have a hemo-globin A1c (HbA1c) value at or below the goal of 7%, and the same number have levels exceeding 8%.4
Part of the problem is that treatment regimens are not adjusted in a timely fashion. In a prescribing database of almost 4,000 patients with type 2 diabetes,5 the mean time from the first HbA1c reading above 8% to an actual change in therapy was about 15 months for those taking metformin (Glucophage) alone, and 21 months for those taking a sulfonylurea alone. Another part of the problem is that, on average, patients with an HbA1c of 8.0% to 8.9% can expect only a 0.6% lowering with the addition of one agent.6 Clearly, we need new pharmacologic approaches and new management paradigms. One new approach is the use of gliptins.
HOW GLIPTINS WORK
Incretins promote insulin secretion
We have known for more than 20 years that insulin levels rise considerably higher in response to an oral glucose load than to an intravenous glucose infusion, even though the plasma glucose concentrations may be similar.7 This phenomenon involves a myriad of neural and nutritional factors, but the gut hormones glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP) appear to be key.
These peptides—called incretins—have a high degree of homology, and both promote insulin secretion. However, GLP-1, produced by the L cells of the ileum and colon, inhibits glucagon secretion and slows gastric emptying, whereas GIP, secreted from the K cells of the duodenum, has no effect on glucagon and little effect on gastric emptying. Both peptides appear to promote pancreatic beta cell growth and survival,8,9 an effect that in theory might allow us to slow the progressive loss of insulin secretory capacity in type 2 diabetes.
Furthermore, the effect of GLP-1 on insulin secretion depends on the plasma glucose concentration, with a greater insulin secretory effect at higher glucose levels and minimal effect at euglycemic levels.10 This phenomenon suggests that drugs that boost GLP-1 activity should not cause the troublesome hypoglycemia typically seen in patients taking insulin, insulin secretagogues, sulfonyl-ureas, or the meglitinides repaglinide (Prandin) or nateglinide (Starlix). Studies of combination treatment with metformin and the GLP-1 receptor agonist exenatide (Byetta) have shown little risk of hypoglycemia,11 offering evidence favoring this conjecture.
Inhibition of DPP-4 boosts incretin action
The challenge for creating treatments that take advantage of the beneficial effects of GLP-1 and GIP is that they have very short physiologic half-lives, ie, less than 10 minutes. GLP-1 and GIP both have two N-terminal amino acids that are quickly cleaved by DPP-4,12 an enzyme present in the circulation13 and on endothelial cells.14
Currently, there are two classes of drugs based on incretins. One class, the incretin mimetics or GLP-1 receptor agonists, includes drugs that mimic the effect of GLP-1 but are not so quickly degraded by DPP-4. Examples of these drugs are exenatide, which is currently FDA-approved, and liraglutide, which is not yet approved.
On the other hand, by inhibiting the cleaving action of DPP-4, the gliptins can prolong the half-life of endogenous GLP-1, increasing its physiologic effects.
Studies comparing gliptins with GLP-1 receptor agonists are only at the preclinical phase. Liraglutide showed an antiglycemic effect similar to that of vildagliptin in an animal model of glucose intolerance.15 This and other16,17 preclinical studies have shown evidence of improved beta cell growth and survival with DPP-4 inhibitor treatment, to an extent similar to that reported with thiazo-lidinediones, whereas sulfonylureas show no evidence either of increase in beta cells or of improved intrinsic beta cell secretory function in these models. Of course, animal studies can only be cautiously extrapolated to potential effects in humans, and it is uncertain whether such benefits will occur with the therapeutic use of DPP-4 inhibitors.
RANDOMIZED CLINICAL TRIALS OF SITAGLIPTIN
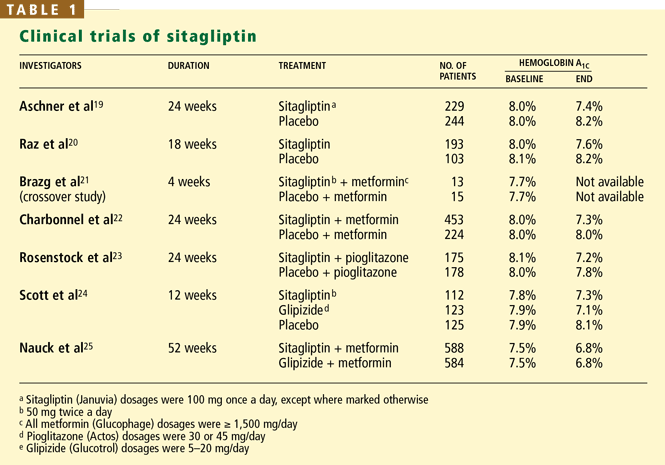
Sitagliptin is effective when used by itself,reducing a baseline HbA1c level of about 8% by 0.6% to 0.8%,19,20,24 and is similarly effective when combined with metformin21,22,25 or pioglitazone (Actos, a thiazolidinedione).23 It also decreases fasting blood glucose levels and improves other measures of glucose control.
A study comparing sitagliptin and the sul-fonylurea glipizide (Glucotrol) showed identical glucose-lowering over a 1-year period, with less hypoglycemia and weight gain with sitagliptin.25 Hypoglycemic episodes occurred in 32% of patients taking glipizide but in only 5% of those taking sitagliptin.
Studies noted several trends in laboratory values, though none was associated with clinical evidence of adverse outcome:
- White blood cell counts were noted to increase in three of the studies by 4.7% to 10%, owing to increases in neutro-phils19,20,22
- Alkaline phosphatase concentrations decreased in four studies19,20,22,23
- Uric acid levels increased in four studies.19,20,22,23
RENAL INSUFFICIENCY SLOWS SITAGLIPTIN CLEARANCE
Lower doses and periodic monitoring of renal function are recommended in patients taking sitagliptin who have some degree of renal insufficiency. Clearance of sitagliptin is delayed in patients with renal insufficiency (creatinine clearance < 50 mL/minute).
In a placebo-controlled study of sitagliptin safety, Scott et al26 found that the area under the sitagliptin concentration-time curve was 2.3 times greater in patients with moderate renal insufficiency (creatinine clearance rate 30–49.9 mL/minute), 3.8 times greater in those with severe renal insufficiency (15–29.9 mL/minute), and 4.5 times greater in those with end-stage renal disease (< 15 mL/minute).
The Januvia package insert27 recommends that the daily dose be decreased to 50 mg in patients with creatinine clearance rates of 30 to 49.9 mL/minute (serum creatinine > 1.7 mg/dL in men, > 1.5 mg/dL in women), and that the dose be decreased to 25 mg per day in those with creatinine clearance rates below 30 mL/minute (creatinine > 3.0/2.5 mg/dL).
CLINICAL TRIALS OF VILDAGLIPTIN BEGIN
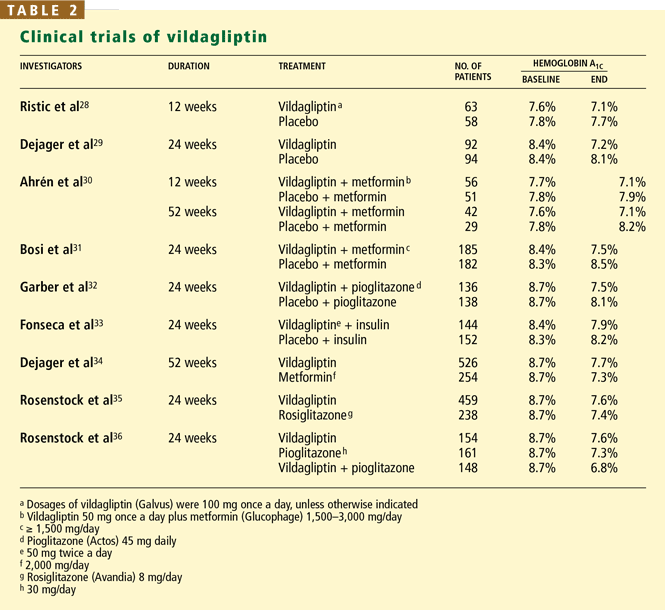
A study comparing vildagliptin against metformin34 showed less glucose-lowering over a 1-year period with vildagliptin, albeit with fewer gastrointestinal side effects, while comparisons with rosiglitazone (Avandia)35 and with pioglitazone36 showed similar glucose-lowering ability.
In a 24-week study,33 256 patients with type 2 diabetes who had a mean body mass index of 33 kg/m2 and who were taking more than 30 units of insulin daily (an average of 82 units) were randomized to additionally receive either vildagliptin 50 mg twice daily or placebo. The HbA1c decreased by 0.5% with vildagliptin and by 0.2% with placebo, from a baseline level of 8.5%. Of interest, 33 patients receiving vildagliptin had a hypo-glycemic episode (a total of 113 events), compared with 45 patients in the placebo group (185 events). None of the episodes in the vildagliptin group was classified as severe, whereas six episodes in the placebo group were classified as severe. This suggests that adding vildagliptin in patients taking insulin can improve glycemia without causing excessive hypoglycemia.
A weakness of the design of this study is that it did not include patients who were receiving an insulin sensitizer, an approach that is typically taken. Given this, it is understandable that overall glycemic control was relatively poor. More effort is needed to explore the use of gliptins with insulin.
WHAT ROLE FOR GLIPTINS?
The evidence from the studies reviewed in this article suggests that gliptins can play an important role in the treatment of type 2 diabetes. In certain patient groups such as the elderly, who cannot take either metformin or a thiazolidinedione and in whom concerns about hypoglycemia are greatest, thus precluding sulfonylurea therapy, gliptins may be the agents of choice. The trials reviewed here suggest that gliptins have glucose-lowering efficacy similar to that of these classes of agents. Gliptins are also effective when combined with metformin or a thiazolidinedione and, as discussed above, may prove to be useful in combination with insulin.
The eventual role of gliptins in the treatment of type 2 diabetes will depend on the answers to several questions. For example, do they preserve beta cell function and reverse the progression of diabetes? Do they affect insulin resistance? Do they have cardiovascular benefits beyond glucose-lowering? Also, since DPP-4 is widely distributed in the body, and since we do not yet know the effects of all the proteins cleaved by this enzyme, will this affect the long-term safety of these drugs?
For now, we can state with reasonable certainty that gliptins lower blood sugar levels to a degree similar to that of other oral hypo-glycemic therapies, with minimal risk of hypo-glycemia, with few immediate adverse effects, and without requiring dose titration. These characteristics suggest that gliptins should be considered useful agents in monotherapy and combination therapy for the treatment of type 2 diabetes.
- National Diabetes Surveillance System. www.cdc.gov/diabetes/statistics/prev/national/figpersons.htm. Last accessed February 28, 2008.
- Narayan KM, Boyle JP, Geiss LS, Saaddine JB, Thompson TJ. Impact of recent increase in incidence on future diabetes burden: US, 2005–2050. Diabetes Care 2006; 29:2114–2116.
- American Diabetes Association. Standards of medical care in diabetes—2007. Diabetes Care 2007; 30 suppl 1:S4–S41.
- Saydah SH, Fradkin J, Cowie CC. Poor control of risk factors for vascular disease among adults with previously diagnosed diabetes. JAMA 2004; 291:335–342.
- Brown JB, Nichols GA, Perry A. The burden of treatment failure in type 2 diabetes. Diabetes Care 2004; 27:1535–1540.
- Bloomgarden ZT, Dodis R, Viscoli CM, Holmboe ES, Inzucchi SE. Lower baseline glycemia reduces apparent oral agent glucose-lowering efficacy: a meta-regression analysis. Diabetes Care 2006; 29:2137–2139.
- Nauck M, Stockmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 1986; 29:46–52.
- Drucker DJ. Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care 2003; 26:2929–2940.
- Bloomgarden ZT. Gut hormones and related concepts. Diabetes Care 2006; 29:2319–2324.
- Nauck MA, Kleine N, Orskov C, et al. Normalization of fasting hyper-glycaemia by exogenous glucagon-like peptide 1 (7–36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 1993; 36:741–744.
- DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care 2005; 28:1092–1100.
- Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes 1995; 44:1126–1131.
- Holst JJ, Deacon CF. Glucagon-like peptide-1 mediates the therapeutic actions of DPP-4 inhibitors. Diabetologia 2005; 48:612–615.
- Hansen L, Deacon CF, Orskov C, Holst JJ. Glucagon-like peptide-1-(7–36)amide is transformed to glucagon-like peptide-1-(9–36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology 1999; 140:5356–5363.
- Raun K, von Voss P, Gotfredsen CF, Golozoubova V, Rolin B, Knudsen LB. Liraglutide, a long-acting glucagon-like peptide-1 analog, reduces body weight and food intake in obese candy-fed rats, whereas a dipeptidyl peptidase-IV inhibitor, vildagliptin, does not. Diabetes 2007; 56:8–15.
- Mu J, Woods J, Zhou YP, et al. Chronic inhibition of dipeptidyl peptidase IV with a sitagliptin analog preserves pancreatic beta-cell mass and function in a rodent model of type 2 diabetes. Diabetes 2006; 55:1695–1704.
- Pospisilik JA, Martin J, Doty T, et al. Dipeptidyl peptidase IV inhibitor treatment stimulates beta-cell survival and islet neogenesis in streptozotocin-induced diabetic rats. Diabetes 2003; 52:741–750.
- Herman GA, Bergman A, Stevens C, et al. Effect of single oral doses of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on incretin and plasma glucose levels after an oral glucose tolerance test in patients with type 2 diabetes. J Clin Endocrinol Metab 2006; 91:4612–4619.
- Aschner P, Kipnes MS, Lunceford JK, Sanchez M, Mickel C, Williams-Herman DE. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care 2006; 29:2632–2637.
- Raz I, Hanefeld M, Xu L, Caria C, Williams-Herman D, Khatami H. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia 2006; 49:2564–2571.
- Brazg R, Xu L, Dalla Man C, Cobelli C, Thomas K, Stein PP. Effect of adding sitagliptin, a dipeptidyl peptidase-4 inhibitor, to metformin on 24-h glycaemic control and beta-cell function in patients with type 2 diabetes. Diabetes Obes Metab 2007; 9:186–193.
- Charbonnel B, Karasik A, Liu J, Wu M, Meininger G. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 2006; 29:2638–2643.
- Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P Sitagliptin Study 019 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2006; 28:1556–1568.
- Scott R, Wu M, Sanchez M, Stein P. Efficacy and tolerability of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy over 12 weeks in patients with type 2 diabetes. Int J Clin Pract 2007; 61:171–180.
- Nauck MA, Meininger JG, Sheng D, Terranella L, Stein PP. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 2007; 9:194–205.
- Scott RS, Hartley P, Luo E, et al. Use of sitagliptin in patients with type 2 diabetes and renal insufficiency [abtract]. Diabetes 2006; 55 suppl 1:A462.
- Januvia prescribing information. www.merck.com/product/usa/pi_circulars/j/products_j.html. Last accessed February 28, 2008.
- Ristic S, Byiers S, Foley J, Holmes D. Improved glycaemic control with dipeptidyl peptidase-4 inhibition in patients with type 2 diabetes: vildagliptin (LAF237) dose response. Diabetes Obes Metab 2005; 7:692–698.
- Dejager S, Baron M, Razac S, Foley JE, Dickinson S, Schweizer S. Effect of vildagliptin on drug-naïve patients with type 2 diabetes. Diabetologia 2006; 49 suppl 1:479–480.
- Ahrén B, Gomis R, Standl E, Mills D, Schweizer A. Twelve- and 52-week efficacy of the dipeptidyl peptidase iv inhibitor laf237 in metformin-treated patients with type 2 diabetes. Diabetes Care 2004; 27:2874–2880.
- Bosi E, Camisasca RP, Collober C, Rochotte E, Garber AJ. Effects of vildagliptin on glucose control over 24 weeks in patients with type 2 diabetes inadequately controlled with metformin. Diabetes Care 2007; 30:890–895.
- Garber A, Schweizer A, Baron MA, Rochotte E, Dejager S. Vildagliptin in combination with pioglitazone improves glycaemic control in patients with type 2 diabetes failing thiazolidinedione monotherapy: a randomized, placebo-controlled study. Diabetes Obes Metab 2007; 9:166–174.
- Fonseca V, Schweizer A, Albrecht D, Baron MA, Chang I, Dejager S. Addition of vildagliptin to insulin improves glycaemic control in type 2 diabetes. Diabetologia 2007; 50:1148–1155.
- Dejager S, LeBeaut A, Couturier A, Schweizer A. Sustained reduction in HbA1c during one-year treatment with vildagliptin in patients with type 2 diabetes (T2DM) [abstract]. Diabetes 2006; 55 suppl 1:A29.
- Rosenstock J, Baron MA, Dejager S, Mills D, Schweizer A. Comparison of vildagliptin and rosiglitazone monotherapy in patients with type 2 diabetes. Diabetes Care 2007; 30:217–223.
- Rosenstock J, Baron MA, Camisasca R-P, Cressier F, Couturier A, Dejager S. Efficacy and tolerability of initial combination therapy with vildagliptin and pioglitazone compared with component monotherapy in patients with type 2 diabetes. Diabetes Obes Metab 2007; 9:175–185.
The “gliptins”—the nickname for dipeptidyl peptidase 4 (DPP-4) inhibitors—are one of the newest classes of drugs for the treatment of type 2 diabetes mellitus.
These drugs work by prolonging the action of gut hormones called incretins, which boost insulin levels. The greatest advantage of the gliptins appears to be their ability to stimulate insulin production with little risk of corresponding hypoglycemia.
Sitagliptin (Januvia), the first commercially available DPP-4 inhibitor, has been approved by the US Food and Drug Administration (FDA) and is currently in clinical use, and vildagliptin (Galvus) awaits FDA approval at the time of this writing. Other drugs of this class are in development.
However, because these drugs are so new, a number of questions remain about their use. In this article, we discuss the rationale behind gliptin drugs, the evidence to date on their use alone or in combination with current oral hypoglycemic drugs (and even with insulin), and when and how to use them in daily practice.
THE NEED FOR MORE EFFECTIVE DIABETES TREATMENT
As the number of patients with type 2 diabetes continues its steep and steady rise,1,2 much work has gone into studying treatment goals and how to achieve them. Although experts generally agree on glycemic goals,3 we currently fail to achieve those goals in close to two-thirds of patients: only 37% have a hemo-globin A1c (HbA1c) value at or below the goal of 7%, and the same number have levels exceeding 8%.4
Part of the problem is that treatment regimens are not adjusted in a timely fashion. In a prescribing database of almost 4,000 patients with type 2 diabetes,5 the mean time from the first HbA1c reading above 8% to an actual change in therapy was about 15 months for those taking metformin (Glucophage) alone, and 21 months for those taking a sulfonylurea alone. Another part of the problem is that, on average, patients with an HbA1c of 8.0% to 8.9% can expect only a 0.6% lowering with the addition of one agent.6 Clearly, we need new pharmacologic approaches and new management paradigms. One new approach is the use of gliptins.
HOW GLIPTINS WORK
Incretins promote insulin secretion
We have known for more than 20 years that insulin levels rise considerably higher in response to an oral glucose load than to an intravenous glucose infusion, even though the plasma glucose concentrations may be similar.7 This phenomenon involves a myriad of neural and nutritional factors, but the gut hormones glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP) appear to be key.
These peptides—called incretins—have a high degree of homology, and both promote insulin secretion. However, GLP-1, produced by the L cells of the ileum and colon, inhibits glucagon secretion and slows gastric emptying, whereas GIP, secreted from the K cells of the duodenum, has no effect on glucagon and little effect on gastric emptying. Both peptides appear to promote pancreatic beta cell growth and survival,8,9 an effect that in theory might allow us to slow the progressive loss of insulin secretory capacity in type 2 diabetes.
Furthermore, the effect of GLP-1 on insulin secretion depends on the plasma glucose concentration, with a greater insulin secretory effect at higher glucose levels and minimal effect at euglycemic levels.10 This phenomenon suggests that drugs that boost GLP-1 activity should not cause the troublesome hypoglycemia typically seen in patients taking insulin, insulin secretagogues, sulfonyl-ureas, or the meglitinides repaglinide (Prandin) or nateglinide (Starlix). Studies of combination treatment with metformin and the GLP-1 receptor agonist exenatide (Byetta) have shown little risk of hypoglycemia,11 offering evidence favoring this conjecture.
Inhibition of DPP-4 boosts incretin action
The challenge for creating treatments that take advantage of the beneficial effects of GLP-1 and GIP is that they have very short physiologic half-lives, ie, less than 10 minutes. GLP-1 and GIP both have two N-terminal amino acids that are quickly cleaved by DPP-4,12 an enzyme present in the circulation13 and on endothelial cells.14
Currently, there are two classes of drugs based on incretins. One class, the incretin mimetics or GLP-1 receptor agonists, includes drugs that mimic the effect of GLP-1 but are not so quickly degraded by DPP-4. Examples of these drugs are exenatide, which is currently FDA-approved, and liraglutide, which is not yet approved.
On the other hand, by inhibiting the cleaving action of DPP-4, the gliptins can prolong the half-life of endogenous GLP-1, increasing its physiologic effects.
Studies comparing gliptins with GLP-1 receptor agonists are only at the preclinical phase. Liraglutide showed an antiglycemic effect similar to that of vildagliptin in an animal model of glucose intolerance.15 This and other16,17 preclinical studies have shown evidence of improved beta cell growth and survival with DPP-4 inhibitor treatment, to an extent similar to that reported with thiazo-lidinediones, whereas sulfonylureas show no evidence either of increase in beta cells or of improved intrinsic beta cell secretory function in these models. Of course, animal studies can only be cautiously extrapolated to potential effects in humans, and it is uncertain whether such benefits will occur with the therapeutic use of DPP-4 inhibitors.
RANDOMIZED CLINICAL TRIALS OF SITAGLIPTIN
Sitagliptin is effective when used by itself,reducing a baseline HbA1c level of about 8% by 0.6% to 0.8%,19,20,24 and is similarly effective when combined with metformin21,22,25 or pioglitazone (Actos, a thiazolidinedione).23 It also decreases fasting blood glucose levels and improves other measures of glucose control.
A study comparing sitagliptin and the sul-fonylurea glipizide (Glucotrol) showed identical glucose-lowering over a 1-year period, with less hypoglycemia and weight gain with sitagliptin.25 Hypoglycemic episodes occurred in 32% of patients taking glipizide but in only 5% of those taking sitagliptin.
Studies noted several trends in laboratory values, though none was associated with clinical evidence of adverse outcome:
- White blood cell counts were noted to increase in three of the studies by 4.7% to 10%, owing to increases in neutro-phils19,20,22
- Alkaline phosphatase concentrations decreased in four studies19,20,22,23
- Uric acid levels increased in four studies.19,20,22,23
RENAL INSUFFICIENCY SLOWS SITAGLIPTIN CLEARANCE
Lower doses and periodic monitoring of renal function are recommended in patients taking sitagliptin who have some degree of renal insufficiency. Clearance of sitagliptin is delayed in patients with renal insufficiency (creatinine clearance < 50 mL/minute).
In a placebo-controlled study of sitagliptin safety, Scott et al26 found that the area under the sitagliptin concentration-time curve was 2.3 times greater in patients with moderate renal insufficiency (creatinine clearance rate 30–49.9 mL/minute), 3.8 times greater in those with severe renal insufficiency (15–29.9 mL/minute), and 4.5 times greater in those with end-stage renal disease (< 15 mL/minute).
The Januvia package insert27 recommends that the daily dose be decreased to 50 mg in patients with creatinine clearance rates of 30 to 49.9 mL/minute (serum creatinine > 1.7 mg/dL in men, > 1.5 mg/dL in women), and that the dose be decreased to 25 mg per day in those with creatinine clearance rates below 30 mL/minute (creatinine > 3.0/2.5 mg/dL).
CLINICAL TRIALS OF VILDAGLIPTIN BEGIN
A study comparing vildagliptin against metformin34 showed less glucose-lowering over a 1-year period with vildagliptin, albeit with fewer gastrointestinal side effects, while comparisons with rosiglitazone (Avandia)35 and with pioglitazone36 showed similar glucose-lowering ability.
In a 24-week study,33 256 patients with type 2 diabetes who had a mean body mass index of 33 kg/m2 and who were taking more than 30 units of insulin daily (an average of 82 units) were randomized to additionally receive either vildagliptin 50 mg twice daily or placebo. The HbA1c decreased by 0.5% with vildagliptin and by 0.2% with placebo, from a baseline level of 8.5%. Of interest, 33 patients receiving vildagliptin had a hypo-glycemic episode (a total of 113 events), compared with 45 patients in the placebo group (185 events). None of the episodes in the vildagliptin group was classified as severe, whereas six episodes in the placebo group were classified as severe. This suggests that adding vildagliptin in patients taking insulin can improve glycemia without causing excessive hypoglycemia.
A weakness of the design of this study is that it did not include patients who were receiving an insulin sensitizer, an approach that is typically taken. Given this, it is understandable that overall glycemic control was relatively poor. More effort is needed to explore the use of gliptins with insulin.
WHAT ROLE FOR GLIPTINS?
The evidence from the studies reviewed in this article suggests that gliptins can play an important role in the treatment of type 2 diabetes. In certain patient groups such as the elderly, who cannot take either metformin or a thiazolidinedione and in whom concerns about hypoglycemia are greatest, thus precluding sulfonylurea therapy, gliptins may be the agents of choice. The trials reviewed here suggest that gliptins have glucose-lowering efficacy similar to that of these classes of agents. Gliptins are also effective when combined with metformin or a thiazolidinedione and, as discussed above, may prove to be useful in combination with insulin.
The eventual role of gliptins in the treatment of type 2 diabetes will depend on the answers to several questions. For example, do they preserve beta cell function and reverse the progression of diabetes? Do they affect insulin resistance? Do they have cardiovascular benefits beyond glucose-lowering? Also, since DPP-4 is widely distributed in the body, and since we do not yet know the effects of all the proteins cleaved by this enzyme, will this affect the long-term safety of these drugs?
For now, we can state with reasonable certainty that gliptins lower blood sugar levels to a degree similar to that of other oral hypo-glycemic therapies, with minimal risk of hypo-glycemia, with few immediate adverse effects, and without requiring dose titration. These characteristics suggest that gliptins should be considered useful agents in monotherapy and combination therapy for the treatment of type 2 diabetes.
The “gliptins”—the nickname for dipeptidyl peptidase 4 (DPP-4) inhibitors—are one of the newest classes of drugs for the treatment of type 2 diabetes mellitus.
These drugs work by prolonging the action of gut hormones called incretins, which boost insulin levels. The greatest advantage of the gliptins appears to be their ability to stimulate insulin production with little risk of corresponding hypoglycemia.
Sitagliptin (Januvia), the first commercially available DPP-4 inhibitor, has been approved by the US Food and Drug Administration (FDA) and is currently in clinical use, and vildagliptin (Galvus) awaits FDA approval at the time of this writing. Other drugs of this class are in development.
However, because these drugs are so new, a number of questions remain about their use. In this article, we discuss the rationale behind gliptin drugs, the evidence to date on their use alone or in combination with current oral hypoglycemic drugs (and even with insulin), and when and how to use them in daily practice.
THE NEED FOR MORE EFFECTIVE DIABETES TREATMENT
As the number of patients with type 2 diabetes continues its steep and steady rise,1,2 much work has gone into studying treatment goals and how to achieve them. Although experts generally agree on glycemic goals,3 we currently fail to achieve those goals in close to two-thirds of patients: only 37% have a hemo-globin A1c (HbA1c) value at or below the goal of 7%, and the same number have levels exceeding 8%.4
Part of the problem is that treatment regimens are not adjusted in a timely fashion. In a prescribing database of almost 4,000 patients with type 2 diabetes,5 the mean time from the first HbA1c reading above 8% to an actual change in therapy was about 15 months for those taking metformin (Glucophage) alone, and 21 months for those taking a sulfonylurea alone. Another part of the problem is that, on average, patients with an HbA1c of 8.0% to 8.9% can expect only a 0.6% lowering with the addition of one agent.6 Clearly, we need new pharmacologic approaches and new management paradigms. One new approach is the use of gliptins.
HOW GLIPTINS WORK
Incretins promote insulin secretion
We have known for more than 20 years that insulin levels rise considerably higher in response to an oral glucose load than to an intravenous glucose infusion, even though the plasma glucose concentrations may be similar.7 This phenomenon involves a myriad of neural and nutritional factors, but the gut hormones glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP) appear to be key.
These peptides—called incretins—have a high degree of homology, and both promote insulin secretion. However, GLP-1, produced by the L cells of the ileum and colon, inhibits glucagon secretion and slows gastric emptying, whereas GIP, secreted from the K cells of the duodenum, has no effect on glucagon and little effect on gastric emptying. Both peptides appear to promote pancreatic beta cell growth and survival,8,9 an effect that in theory might allow us to slow the progressive loss of insulin secretory capacity in type 2 diabetes.
Furthermore, the effect of GLP-1 on insulin secretion depends on the plasma glucose concentration, with a greater insulin secretory effect at higher glucose levels and minimal effect at euglycemic levels.10 This phenomenon suggests that drugs that boost GLP-1 activity should not cause the troublesome hypoglycemia typically seen in patients taking insulin, insulin secretagogues, sulfonyl-ureas, or the meglitinides repaglinide (Prandin) or nateglinide (Starlix). Studies of combination treatment with metformin and the GLP-1 receptor agonist exenatide (Byetta) have shown little risk of hypoglycemia,11 offering evidence favoring this conjecture.
Inhibition of DPP-4 boosts incretin action
The challenge for creating treatments that take advantage of the beneficial effects of GLP-1 and GIP is that they have very short physiologic half-lives, ie, less than 10 minutes. GLP-1 and GIP both have two N-terminal amino acids that are quickly cleaved by DPP-4,12 an enzyme present in the circulation13 and on endothelial cells.14
Currently, there are two classes of drugs based on incretins. One class, the incretin mimetics or GLP-1 receptor agonists, includes drugs that mimic the effect of GLP-1 but are not so quickly degraded by DPP-4. Examples of these drugs are exenatide, which is currently FDA-approved, and liraglutide, which is not yet approved.
On the other hand, by inhibiting the cleaving action of DPP-4, the gliptins can prolong the half-life of endogenous GLP-1, increasing its physiologic effects.
Studies comparing gliptins with GLP-1 receptor agonists are only at the preclinical phase. Liraglutide showed an antiglycemic effect similar to that of vildagliptin in an animal model of glucose intolerance.15 This and other16,17 preclinical studies have shown evidence of improved beta cell growth and survival with DPP-4 inhibitor treatment, to an extent similar to that reported with thiazo-lidinediones, whereas sulfonylureas show no evidence either of increase in beta cells or of improved intrinsic beta cell secretory function in these models. Of course, animal studies can only be cautiously extrapolated to potential effects in humans, and it is uncertain whether such benefits will occur with the therapeutic use of DPP-4 inhibitors.
RANDOMIZED CLINICAL TRIALS OF SITAGLIPTIN
Sitagliptin is effective when used by itself,reducing a baseline HbA1c level of about 8% by 0.6% to 0.8%,19,20,24 and is similarly effective when combined with metformin21,22,25 or pioglitazone (Actos, a thiazolidinedione).23 It also decreases fasting blood glucose levels and improves other measures of glucose control.
A study comparing sitagliptin and the sul-fonylurea glipizide (Glucotrol) showed identical glucose-lowering over a 1-year period, with less hypoglycemia and weight gain with sitagliptin.25 Hypoglycemic episodes occurred in 32% of patients taking glipizide but in only 5% of those taking sitagliptin.
Studies noted several trends in laboratory values, though none was associated with clinical evidence of adverse outcome:
- White blood cell counts were noted to increase in three of the studies by 4.7% to 10%, owing to increases in neutro-phils19,20,22
- Alkaline phosphatase concentrations decreased in four studies19,20,22,23
- Uric acid levels increased in four studies.19,20,22,23
RENAL INSUFFICIENCY SLOWS SITAGLIPTIN CLEARANCE
Lower doses and periodic monitoring of renal function are recommended in patients taking sitagliptin who have some degree of renal insufficiency. Clearance of sitagliptin is delayed in patients with renal insufficiency (creatinine clearance < 50 mL/minute).
In a placebo-controlled study of sitagliptin safety, Scott et al26 found that the area under the sitagliptin concentration-time curve was 2.3 times greater in patients with moderate renal insufficiency (creatinine clearance rate 30–49.9 mL/minute), 3.8 times greater in those with severe renal insufficiency (15–29.9 mL/minute), and 4.5 times greater in those with end-stage renal disease (< 15 mL/minute).
The Januvia package insert27 recommends that the daily dose be decreased to 50 mg in patients with creatinine clearance rates of 30 to 49.9 mL/minute (serum creatinine > 1.7 mg/dL in men, > 1.5 mg/dL in women), and that the dose be decreased to 25 mg per day in those with creatinine clearance rates below 30 mL/minute (creatinine > 3.0/2.5 mg/dL).
CLINICAL TRIALS OF VILDAGLIPTIN BEGIN
A study comparing vildagliptin against metformin34 showed less glucose-lowering over a 1-year period with vildagliptin, albeit with fewer gastrointestinal side effects, while comparisons with rosiglitazone (Avandia)35 and with pioglitazone36 showed similar glucose-lowering ability.
In a 24-week study,33 256 patients with type 2 diabetes who had a mean body mass index of 33 kg/m2 and who were taking more than 30 units of insulin daily (an average of 82 units) were randomized to additionally receive either vildagliptin 50 mg twice daily or placebo. The HbA1c decreased by 0.5% with vildagliptin and by 0.2% with placebo, from a baseline level of 8.5%. Of interest, 33 patients receiving vildagliptin had a hypo-glycemic episode (a total of 113 events), compared with 45 patients in the placebo group (185 events). None of the episodes in the vildagliptin group was classified as severe, whereas six episodes in the placebo group were classified as severe. This suggests that adding vildagliptin in patients taking insulin can improve glycemia without causing excessive hypoglycemia.
A weakness of the design of this study is that it did not include patients who were receiving an insulin sensitizer, an approach that is typically taken. Given this, it is understandable that overall glycemic control was relatively poor. More effort is needed to explore the use of gliptins with insulin.
WHAT ROLE FOR GLIPTINS?
The evidence from the studies reviewed in this article suggests that gliptins can play an important role in the treatment of type 2 diabetes. In certain patient groups such as the elderly, who cannot take either metformin or a thiazolidinedione and in whom concerns about hypoglycemia are greatest, thus precluding sulfonylurea therapy, gliptins may be the agents of choice. The trials reviewed here suggest that gliptins have glucose-lowering efficacy similar to that of these classes of agents. Gliptins are also effective when combined with metformin or a thiazolidinedione and, as discussed above, may prove to be useful in combination with insulin.
The eventual role of gliptins in the treatment of type 2 diabetes will depend on the answers to several questions. For example, do they preserve beta cell function and reverse the progression of diabetes? Do they affect insulin resistance? Do they have cardiovascular benefits beyond glucose-lowering? Also, since DPP-4 is widely distributed in the body, and since we do not yet know the effects of all the proteins cleaved by this enzyme, will this affect the long-term safety of these drugs?
For now, we can state with reasonable certainty that gliptins lower blood sugar levels to a degree similar to that of other oral hypo-glycemic therapies, with minimal risk of hypo-glycemia, with few immediate adverse effects, and without requiring dose titration. These characteristics suggest that gliptins should be considered useful agents in monotherapy and combination therapy for the treatment of type 2 diabetes.
- National Diabetes Surveillance System. www.cdc.gov/diabetes/statistics/prev/national/figpersons.htm. Last accessed February 28, 2008.
- Narayan KM, Boyle JP, Geiss LS, Saaddine JB, Thompson TJ. Impact of recent increase in incidence on future diabetes burden: US, 2005–2050. Diabetes Care 2006; 29:2114–2116.
- American Diabetes Association. Standards of medical care in diabetes—2007. Diabetes Care 2007; 30 suppl 1:S4–S41.
- Saydah SH, Fradkin J, Cowie CC. Poor control of risk factors for vascular disease among adults with previously diagnosed diabetes. JAMA 2004; 291:335–342.
- Brown JB, Nichols GA, Perry A. The burden of treatment failure in type 2 diabetes. Diabetes Care 2004; 27:1535–1540.
- Bloomgarden ZT, Dodis R, Viscoli CM, Holmboe ES, Inzucchi SE. Lower baseline glycemia reduces apparent oral agent glucose-lowering efficacy: a meta-regression analysis. Diabetes Care 2006; 29:2137–2139.
- Nauck M, Stockmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 1986; 29:46–52.
- Drucker DJ. Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care 2003; 26:2929–2940.
- Bloomgarden ZT. Gut hormones and related concepts. Diabetes Care 2006; 29:2319–2324.
- Nauck MA, Kleine N, Orskov C, et al. Normalization of fasting hyper-glycaemia by exogenous glucagon-like peptide 1 (7–36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 1993; 36:741–744.
- DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care 2005; 28:1092–1100.
- Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes 1995; 44:1126–1131.
- Holst JJ, Deacon CF. Glucagon-like peptide-1 mediates the therapeutic actions of DPP-4 inhibitors. Diabetologia 2005; 48:612–615.
- Hansen L, Deacon CF, Orskov C, Holst JJ. Glucagon-like peptide-1-(7–36)amide is transformed to glucagon-like peptide-1-(9–36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology 1999; 140:5356–5363.
- Raun K, von Voss P, Gotfredsen CF, Golozoubova V, Rolin B, Knudsen LB. Liraglutide, a long-acting glucagon-like peptide-1 analog, reduces body weight and food intake in obese candy-fed rats, whereas a dipeptidyl peptidase-IV inhibitor, vildagliptin, does not. Diabetes 2007; 56:8–15.
- Mu J, Woods J, Zhou YP, et al. Chronic inhibition of dipeptidyl peptidase IV with a sitagliptin analog preserves pancreatic beta-cell mass and function in a rodent model of type 2 diabetes. Diabetes 2006; 55:1695–1704.
- Pospisilik JA, Martin J, Doty T, et al. Dipeptidyl peptidase IV inhibitor treatment stimulates beta-cell survival and islet neogenesis in streptozotocin-induced diabetic rats. Diabetes 2003; 52:741–750.
- Herman GA, Bergman A, Stevens C, et al. Effect of single oral doses of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on incretin and plasma glucose levels after an oral glucose tolerance test in patients with type 2 diabetes. J Clin Endocrinol Metab 2006; 91:4612–4619.
- Aschner P, Kipnes MS, Lunceford JK, Sanchez M, Mickel C, Williams-Herman DE. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care 2006; 29:2632–2637.
- Raz I, Hanefeld M, Xu L, Caria C, Williams-Herman D, Khatami H. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia 2006; 49:2564–2571.
- Brazg R, Xu L, Dalla Man C, Cobelli C, Thomas K, Stein PP. Effect of adding sitagliptin, a dipeptidyl peptidase-4 inhibitor, to metformin on 24-h glycaemic control and beta-cell function in patients with type 2 diabetes. Diabetes Obes Metab 2007; 9:186–193.
- Charbonnel B, Karasik A, Liu J, Wu M, Meininger G. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 2006; 29:2638–2643.
- Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P Sitagliptin Study 019 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2006; 28:1556–1568.
- Scott R, Wu M, Sanchez M, Stein P. Efficacy and tolerability of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy over 12 weeks in patients with type 2 diabetes. Int J Clin Pract 2007; 61:171–180.
- Nauck MA, Meininger JG, Sheng D, Terranella L, Stein PP. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 2007; 9:194–205.
- Scott RS, Hartley P, Luo E, et al. Use of sitagliptin in patients with type 2 diabetes and renal insufficiency [abtract]. Diabetes 2006; 55 suppl 1:A462.
- Januvia prescribing information. www.merck.com/product/usa/pi_circulars/j/products_j.html. Last accessed February 28, 2008.
- Ristic S, Byiers S, Foley J, Holmes D. Improved glycaemic control with dipeptidyl peptidase-4 inhibition in patients with type 2 diabetes: vildagliptin (LAF237) dose response. Diabetes Obes Metab 2005; 7:692–698.
- Dejager S, Baron M, Razac S, Foley JE, Dickinson S, Schweizer S. Effect of vildagliptin on drug-naïve patients with type 2 diabetes. Diabetologia 2006; 49 suppl 1:479–480.
- Ahrén B, Gomis R, Standl E, Mills D, Schweizer A. Twelve- and 52-week efficacy of the dipeptidyl peptidase iv inhibitor laf237 in metformin-treated patients with type 2 diabetes. Diabetes Care 2004; 27:2874–2880.
- Bosi E, Camisasca RP, Collober C, Rochotte E, Garber AJ. Effects of vildagliptin on glucose control over 24 weeks in patients with type 2 diabetes inadequately controlled with metformin. Diabetes Care 2007; 30:890–895.
- Garber A, Schweizer A, Baron MA, Rochotte E, Dejager S. Vildagliptin in combination with pioglitazone improves glycaemic control in patients with type 2 diabetes failing thiazolidinedione monotherapy: a randomized, placebo-controlled study. Diabetes Obes Metab 2007; 9:166–174.
- Fonseca V, Schweizer A, Albrecht D, Baron MA, Chang I, Dejager S. Addition of vildagliptin to insulin improves glycaemic control in type 2 diabetes. Diabetologia 2007; 50:1148–1155.
- Dejager S, LeBeaut A, Couturier A, Schweizer A. Sustained reduction in HbA1c during one-year treatment with vildagliptin in patients with type 2 diabetes (T2DM) [abstract]. Diabetes 2006; 55 suppl 1:A29.
- Rosenstock J, Baron MA, Dejager S, Mills D, Schweizer A. Comparison of vildagliptin and rosiglitazone monotherapy in patients with type 2 diabetes. Diabetes Care 2007; 30:217–223.
- Rosenstock J, Baron MA, Camisasca R-P, Cressier F, Couturier A, Dejager S. Efficacy and tolerability of initial combination therapy with vildagliptin and pioglitazone compared with component monotherapy in patients with type 2 diabetes. Diabetes Obes Metab 2007; 9:175–185.
- National Diabetes Surveillance System. www.cdc.gov/diabetes/statistics/prev/national/figpersons.htm. Last accessed February 28, 2008.
- Narayan KM, Boyle JP, Geiss LS, Saaddine JB, Thompson TJ. Impact of recent increase in incidence on future diabetes burden: US, 2005–2050. Diabetes Care 2006; 29:2114–2116.
- American Diabetes Association. Standards of medical care in diabetes—2007. Diabetes Care 2007; 30 suppl 1:S4–S41.
- Saydah SH, Fradkin J, Cowie CC. Poor control of risk factors for vascular disease among adults with previously diagnosed diabetes. JAMA 2004; 291:335–342.
- Brown JB, Nichols GA, Perry A. The burden of treatment failure in type 2 diabetes. Diabetes Care 2004; 27:1535–1540.
- Bloomgarden ZT, Dodis R, Viscoli CM, Holmboe ES, Inzucchi SE. Lower baseline glycemia reduces apparent oral agent glucose-lowering efficacy: a meta-regression analysis. Diabetes Care 2006; 29:2137–2139.
- Nauck M, Stockmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 1986; 29:46–52.
- Drucker DJ. Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care 2003; 26:2929–2940.
- Bloomgarden ZT. Gut hormones and related concepts. Diabetes Care 2006; 29:2319–2324.
- Nauck MA, Kleine N, Orskov C, et al. Normalization of fasting hyper-glycaemia by exogenous glucagon-like peptide 1 (7–36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 1993; 36:741–744.
- DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care 2005; 28:1092–1100.
- Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes 1995; 44:1126–1131.
- Holst JJ, Deacon CF. Glucagon-like peptide-1 mediates the therapeutic actions of DPP-4 inhibitors. Diabetologia 2005; 48:612–615.
- Hansen L, Deacon CF, Orskov C, Holst JJ. Glucagon-like peptide-1-(7–36)amide is transformed to glucagon-like peptide-1-(9–36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology 1999; 140:5356–5363.
- Raun K, von Voss P, Gotfredsen CF, Golozoubova V, Rolin B, Knudsen LB. Liraglutide, a long-acting glucagon-like peptide-1 analog, reduces body weight and food intake in obese candy-fed rats, whereas a dipeptidyl peptidase-IV inhibitor, vildagliptin, does not. Diabetes 2007; 56:8–15.
- Mu J, Woods J, Zhou YP, et al. Chronic inhibition of dipeptidyl peptidase IV with a sitagliptin analog preserves pancreatic beta-cell mass and function in a rodent model of type 2 diabetes. Diabetes 2006; 55:1695–1704.
- Pospisilik JA, Martin J, Doty T, et al. Dipeptidyl peptidase IV inhibitor treatment stimulates beta-cell survival and islet neogenesis in streptozotocin-induced diabetic rats. Diabetes 2003; 52:741–750.
- Herman GA, Bergman A, Stevens C, et al. Effect of single oral doses of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on incretin and plasma glucose levels after an oral glucose tolerance test in patients with type 2 diabetes. J Clin Endocrinol Metab 2006; 91:4612–4619.
- Aschner P, Kipnes MS, Lunceford JK, Sanchez M, Mickel C, Williams-Herman DE. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care 2006; 29:2632–2637.
- Raz I, Hanefeld M, Xu L, Caria C, Williams-Herman D, Khatami H. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia 2006; 49:2564–2571.
- Brazg R, Xu L, Dalla Man C, Cobelli C, Thomas K, Stein PP. Effect of adding sitagliptin, a dipeptidyl peptidase-4 inhibitor, to metformin on 24-h glycaemic control and beta-cell function in patients with type 2 diabetes. Diabetes Obes Metab 2007; 9:186–193.
- Charbonnel B, Karasik A, Liu J, Wu M, Meininger G. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 2006; 29:2638–2643.
- Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P Sitagliptin Study 019 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2006; 28:1556–1568.
- Scott R, Wu M, Sanchez M, Stein P. Efficacy and tolerability of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy over 12 weeks in patients with type 2 diabetes. Int J Clin Pract 2007; 61:171–180.
- Nauck MA, Meininger JG, Sheng D, Terranella L, Stein PP. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 2007; 9:194–205.
- Scott RS, Hartley P, Luo E, et al. Use of sitagliptin in patients with type 2 diabetes and renal insufficiency [abtract]. Diabetes 2006; 55 suppl 1:A462.
- Januvia prescribing information. www.merck.com/product/usa/pi_circulars/j/products_j.html. Last accessed February 28, 2008.
- Ristic S, Byiers S, Foley J, Holmes D. Improved glycaemic control with dipeptidyl peptidase-4 inhibition in patients with type 2 diabetes: vildagliptin (LAF237) dose response. Diabetes Obes Metab 2005; 7:692–698.
- Dejager S, Baron M, Razac S, Foley JE, Dickinson S, Schweizer S. Effect of vildagliptin on drug-naïve patients with type 2 diabetes. Diabetologia 2006; 49 suppl 1:479–480.
- Ahrén B, Gomis R, Standl E, Mills D, Schweizer A. Twelve- and 52-week efficacy of the dipeptidyl peptidase iv inhibitor laf237 in metformin-treated patients with type 2 diabetes. Diabetes Care 2004; 27:2874–2880.
- Bosi E, Camisasca RP, Collober C, Rochotte E, Garber AJ. Effects of vildagliptin on glucose control over 24 weeks in patients with type 2 diabetes inadequately controlled with metformin. Diabetes Care 2007; 30:890–895.
- Garber A, Schweizer A, Baron MA, Rochotte E, Dejager S. Vildagliptin in combination with pioglitazone improves glycaemic control in patients with type 2 diabetes failing thiazolidinedione monotherapy: a randomized, placebo-controlled study. Diabetes Obes Metab 2007; 9:166–174.
- Fonseca V, Schweizer A, Albrecht D, Baron MA, Chang I, Dejager S. Addition of vildagliptin to insulin improves glycaemic control in type 2 diabetes. Diabetologia 2007; 50:1148–1155.
- Dejager S, LeBeaut A, Couturier A, Schweizer A. Sustained reduction in HbA1c during one-year treatment with vildagliptin in patients with type 2 diabetes (T2DM) [abstract]. Diabetes 2006; 55 suppl 1:A29.
- Rosenstock J, Baron MA, Dejager S, Mills D, Schweizer A. Comparison of vildagliptin and rosiglitazone monotherapy in patients with type 2 diabetes. Diabetes Care 2007; 30:217–223.
- Rosenstock J, Baron MA, Camisasca R-P, Cressier F, Couturier A, Dejager S. Efficacy and tolerability of initial combination therapy with vildagliptin and pioglitazone compared with component monotherapy in patients with type 2 diabetes. Diabetes Obes Metab 2007; 9:175–185.
KEY POINTS
- Sitagliptin (Januvia) is now available, and vildagliptin (Galvus) is awaiting approval. Other gliptins are under development.
- The gliptins effectively lower blood glucose levels, do not require titration, are unlikely to cause hypoglycemia, do not cause weight gain or loss, and are well tolerated.
- Gliptins can be used alone or in combination with metformin (Glucophage) or a thiazolidinedione. Preliminary studies also show evidence of benefit when they are used in combination with insulin.
- Comparative studies suggest that gliptins lower blood glucose levels by about the same amount as other oral hypoglycemic agents.
What is the role of dual antiplatelet therapy with clopidogrel and aspirin?
In patients at risk of myocardial infarction or stroke, two antiplatelet drugs are not always better than one. In a large recent trial,1,2 adding clopidogrel (Plavix) to aspirin therapy did not offer much benefit to a cohort of patients at risk of cardiovascular events, although a subgroup did appear to benefit: those at even higher risk because they already had a history of myocardial infarction, ischemic stroke, or peripheral arterial disease.
These were the principal findings in the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) study,1,2 in which one of us (D.L.B.) was principal investigator.
These findings further our understanding of who should receive dual antiplatelet therapy, and who would be better served with aspirin therapy alone. In this article, we discuss important studies that led up to the CHARISMA trial, review CHARISMA’s purpose and study design, and interpret its results.
PREVENTING ATHEROTHROMBOSIS BY BLOCKING PLATELETS
Platelets are key players in the atherothrom-botic process.3–5 The Antiplatelet Trialists’ Collaboration,6 in a meta-analysis of trials performed up to 1997, calculated that antiplatelet therapy (mostly with aspirin) reduced the vascular mortality rate by 15% in patients with acute or previous vascular disease or some other predisposing condition. Thus, aspirin has already been shown to be effective as primary prevention (ie, in patients at risk but without established vascular disease) and as secondary prevention (ie, in those with established disease).7,8
Yet many patients have significant vascular events in spite of taking aspirin.6 Aspirin failure is thought to be multifactorial, with causes that include weak platelet inhibition, noncompliance, discontinuation due to adverse effects (including severe bleeding), and drug interactions. In addition, aspirin resistance has been linked to worse prognosis and may prove to be another cause of aspirin failure.9–11
Clopidogrel, an adenosine diphosphate (ADP) receptor antagonist, has also been studied extensively as an antiplatelet agent.5,12 Several studies have indicated that clopidogrel and ticlopidine (Ticlid, a related drug) may be more potent than aspirin, both in the test tube and in real patients.13–15
KEY TRIALS LEADING TO CHARISMA

- Clopidogrel is more effective and slightly safer than aspirin as secondary prevention, as shown in the Clopidogrel Versus Aspirin in Patients at Risk of Ischemic Events (CAPRIE) trial.16–21
- The combination of clopidogrel plus aspirin is more beneficial than placebo plus aspirin in patients with acute coronary syndromes, as shown in the Clopidogrel in Unstable Angina to Prevent Recurrent Ischemic Events (CURE) trial,22–24 the Clopidogrel as Adjunctive Reperfusion Therapy-Thrombolysis in Myo-car-dial Infarction (CLARITY-TIMI 28) trial,25 and the Clopidogrel and Metoprolol in Myocardial Infarction Trial (COMMIT).26
- The combination of clopidogrel plus aspirin is beneficial in patients undergoing percutaneous coronary interventions, with or without drug-eluting stent placement,27–30 as shown in the Clopidogrel for the Reduction of Events During Observation (CREDO) trial,28 the Effect of Clopidogrel Pretreatment Before Percutaneous Coronary Intervention in Patients With ST-Elevation Myocardial Infarction With Fibrinolytics (PCI-CLARITY) study,29 and the Effects of Pre-treatment With Clopidogrel and Aspirin Followed by Long-term Therapy in Patients Undergoing Percutaneous Coronary Intervention (PCI-CURE) study.30 In fact, most patients undergoing percutaneous interventions now receive a loading dose of clopidogrel before the procedure and continue to take it for up to 1 year afterward. However, the ideal long-term duration of clopidogrel treatment is still under debate.
In view of these previous studies, we wanted to test dual antiplatelet therapy in a broader population at high risk of atherothrombosis, ie, in patients with either established vascular disease or with multiple risk factors for it.
CHARISMA STUDY DESIGN
CHARISMA was a prospective, randomized, double-blind, placebo-controlled study of the efficacy and safety of clopidogrel plus aspirin vs placebo plus aspirin in patients at high risk of cardiovascular events.
A total of 15,603 patients, all older than 45 years, were randomly assigned to receive clopidogrel 75 mg/day plus aspirin 75 to 162 mg/day or placebo plus aspirin, in addition to standard therapy as directed by individual clinicians (eg, statins, beta-blockers). Patients were followed up at 1, 3, and 6 months and every 6 months thereafter until study completion, which occurred after 1,040 primary efficacy end points. The median duration of follow-up was 28 months.1

Patients had to have one of the following to be included: multiple atherothrombotic risk factors, documented coronary disease, documented cerebrovascular disease, or documented peripheral arterial disease (Table 2). Specific exclusion criteria included the use of oral antithrombotic or chronic nonsteroidal anti-inflammatory medications.1
End points
The primary end point was the combined incidence of the first episode of myocardial infarction or stroke, or death from cardiovascular causes.
The secondary end point was the combined incidence of myocardial infarction, stroke, death from cardiovascular causes, or hospitalization for unstable angina, a transient ischemic attack, or revascularization procedure.
The primary safety end point was severe bleeding, as defined in the Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries (GUSTO) study31 as intracranial hemorrhage, fatal bleeding, or bleeding leading to hemody-namic compromise. Moderate bleeding was defined as bleeding that required transfusion but did not meet the GUSTO definition of severe bleeding.
OVERALL, NO BENEFIT
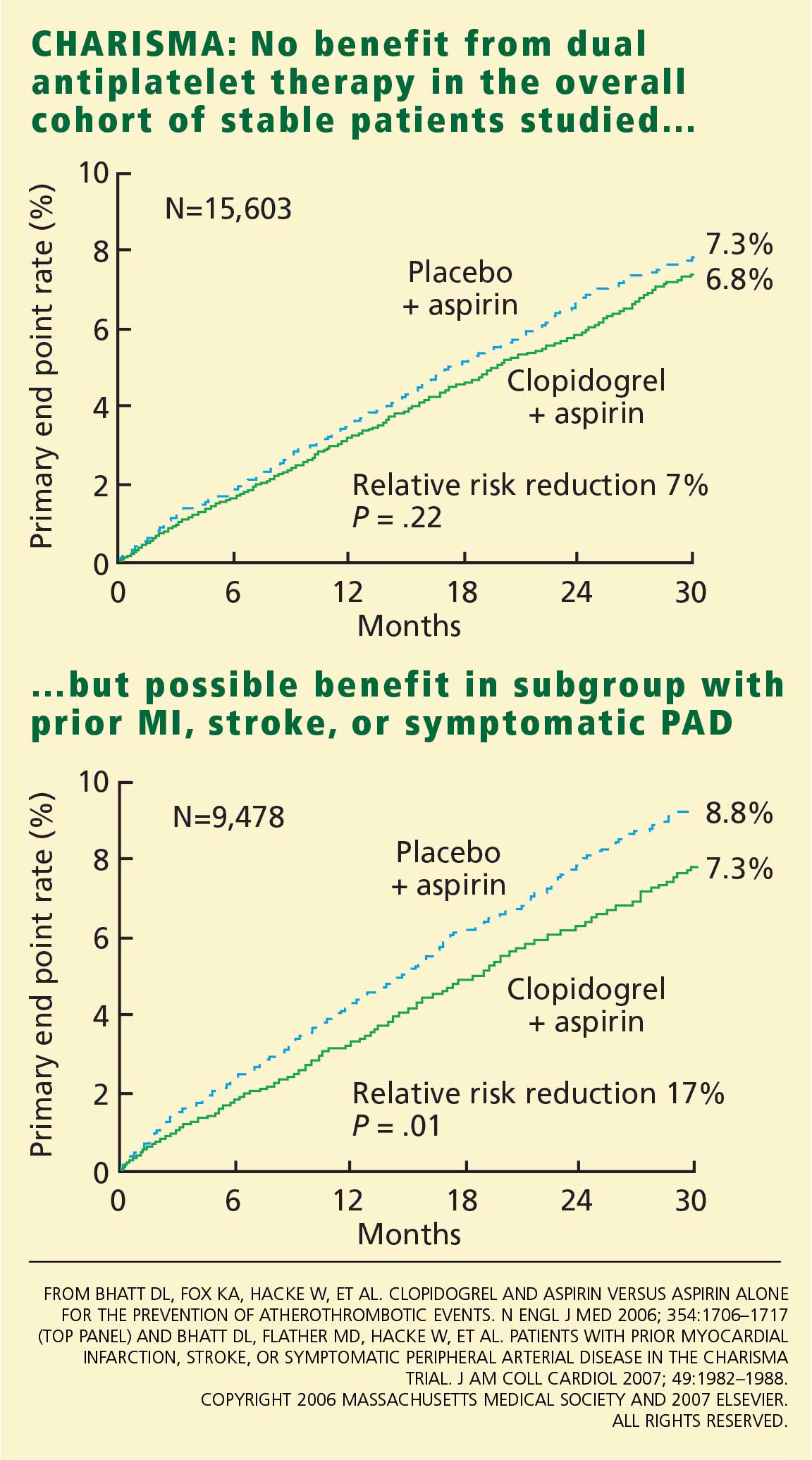
The rates of the secondary end point were 16.7% vs 17.9% (absolute risk reduction 1.2%; relative risk reduction 8%; P = .04).
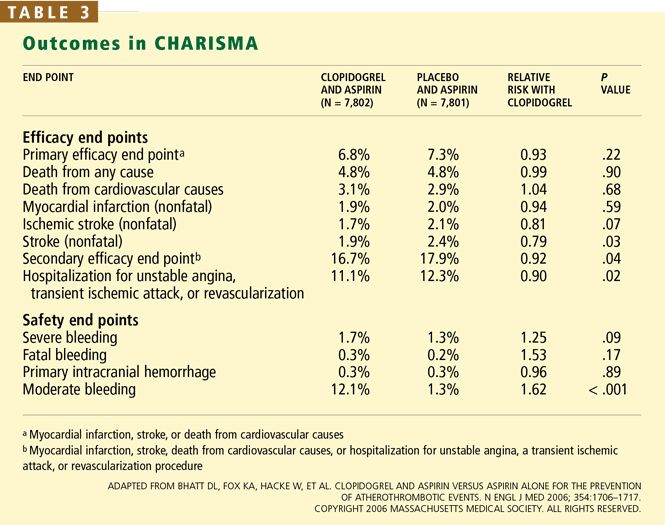
Possible benefit in symptomatic patients
In a prespecified analysis, patients were classified as being “symptomatic” (having documented cardiovascular disease, ie, coronary, cerebrovascular, or symptomatic peripheral arterial disease) or “asymptomatic” (having multiple risk factors without established cardiovascular disease).1
In the symptomatic group (n = 12,153), the primary end point was reached in 6.9% of patients treated with clopidogrel vs 7.9% with placebo (absolute risk reduction 1.0%; relative risk reduction 13%; P = .046). The 3,284 asymptomatic patients showed no benefit; the rate of the primary end point for the clopido-grel group was 6.6% vs 5.5% in the placebo group (P = .20).
In a post hoc analysis, we examined the data from 9,478 patients who were similar to those in the CAPRIE study (ie, with documented prior myocardial infarction, prior ischemic stroke, or symptomatic peripheral arterial disease). The rate of cardiovascular death, myocardial infarction, or stroke was 8.8% in the placebo-plus-aspirin group and 7.3% in the clopidogrel-plus-aspirin group (absolute risk reduction 1.5%; relative risk reduction 17%; P = .01; Figure 1).2
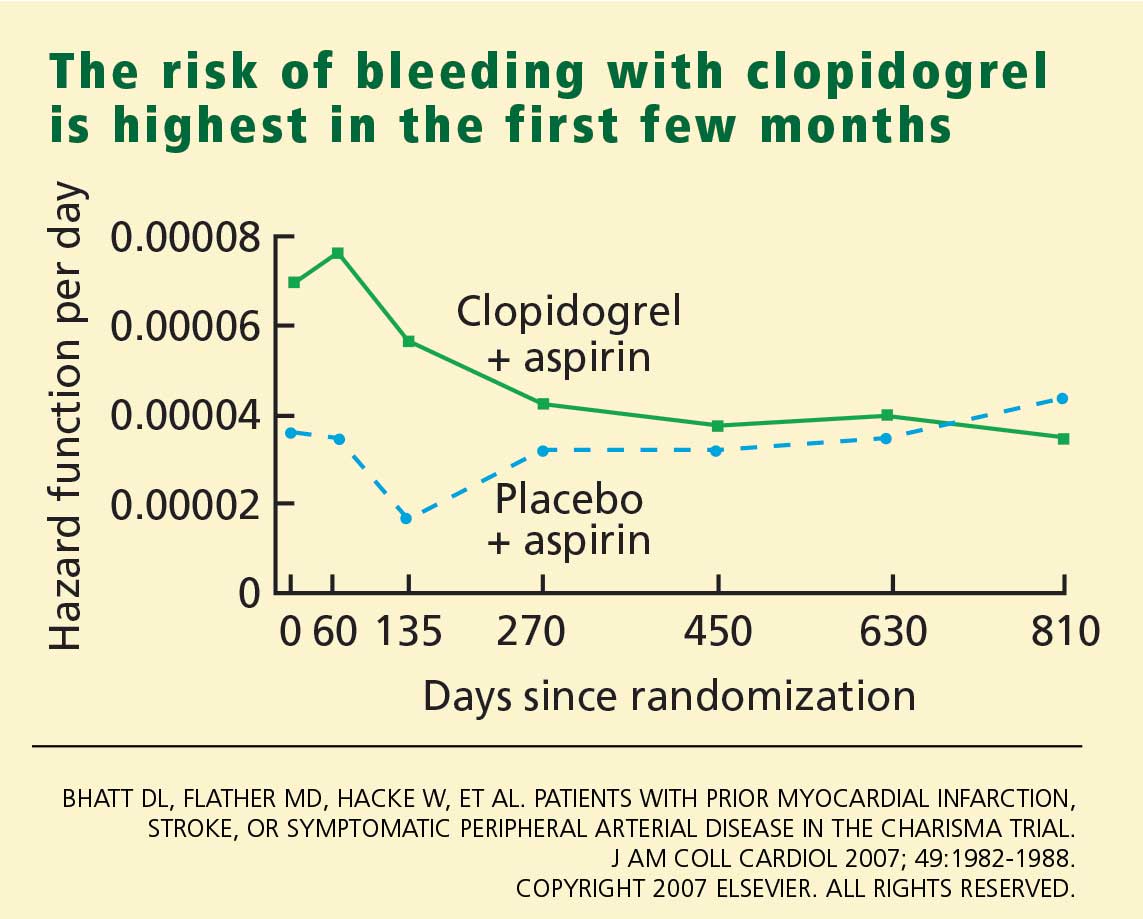
HOW SHOULD WE INTERPRET THESE FINDINGS?
CHARISMA was the first trial to evaluate whether adding clopidogrel to aspirin therapy would reduce the rates of vascular events and death from cardiovascular causes in stable patients at risk of ischemic events. As in other trials, the benefit of clopidogrel-plus-aspirin therapy was weighed against the risk of bleeding with this regimen. How are we to interpret the findings?
- In the group with multiple risk factors but without clearly documented cardiovascular disease, there was no benefit—and there was an increase in moderate bleeding. Given these findings, physicians should not prescribe dual antiplatelet therapy for primary prevention in patients without known vascular disease.
- A potential benefit was seen in a prespecified subgroup who had documented cardiovascular disease. Given the limitations of subgroup analysis, however, and given the increased risk of moderate bleeding, this positive result should be interpreted with some degree of caution.
- CHARISMA suggests that there may be benefit of protracted dual antiplatelet therapy in stable patients with documented prior ischemic events.
A possible reason for the observed lack of benefit in the overall cohort but the positive results in the subgroups with established vascular disease is that plaque rupture and thrombosis may be a precondition for dual antiplatelet therapy to work.
Another possibility is that, although we have been saying that diabetes mellitus (one of the possible entry criteria in CHARISMA) is a “coronary risk equivalent,” this may not be absolutely true. Although it had been demonstrated that patients with certain risk factors, such as diabetes, have an incidence of ischemic events similar to that in patients with prior MI and should be considered for antiplatelet therapy to prevent vascular events,32 more recent data have shown that patients with prior ischemic events are at much higher risk than patients without ischemic events, even if the latter have diabetes.33,34
- The observation in CHARISMA that the incremental bleeding risk of dual antiplatelet therapy vs aspirin does not persist beyond a year in patients who have tolerated therapy for a year without a bleeding event may affect the decision to continue clopidogrel beyond 1 year, such as in patients with acute coronary syndromes or patients who have received drug-eluting stents.35,36
- Another important consideration is cost-effectiveness. Several studies have analyzed the impact of cost and found clopidogrel to be cost-effective by preventing ischemic events and adding years of life.37,38 A recent analysis from CHARISMA also shows cost-effectiveness in the subgroup of patients enrolled with established cardiovascular disease.39 Once clopidogrel becomes generic, the cost-effectiveness will become even better.
Further studies should better define which stable patients with cardiovascular disease should be on more than aspirin alone.
- Bhatt DL, Fox KA, Hacke W, et al. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Ruggeri ZM. Platelets in atherothrombosis. Nat Med 2002; 8:1227–1234.
- Fuster V, Moreno PR, Fayad ZA, Corti R, Badimon JJ. Atherothrombosis and high-risk plaque: part I: evolving concepts. J Am Coll Cardiol 2005; 46:937–954.
- Meadows TA, Bhatt DL. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 2007; 100:1261–1275.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Sanmuganathan PS, Ghahramani P, Jackson PR, Wallis EJ, Ramsay LE. Aspirin for primary prevention of coronary heart disease: safety and absolute benefit related to coronary risk derived from meta-analysis of randomised trials. Heart 2001; 85:265–271.
- Hayden M, Pignone M, Phillips C, Mulrow C. Aspirin for the primary prevention of cardiovascular events: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med 2002; 136:161–172.
- Helgason CM, Bolin KM, Hoff JA, et al. Development of aspirin resistance in persons with previous ischemic stroke. Stroke 1994; 25:2331–2336.
- Helgason CM, Tortorice KL, Winkler SR, et al. Aspirin response and failure in cerebral infarction. Stroke 1993; 24:345–350.
- Gum PA, Kottke-Marchant K, Poggio ED, et al. Profile and prevalence of aspirin resistance in patients with cardiovascular disease. Am J Cardiol 2001; 88:230–235.
- Coukell AJ, Markham A. Clopidogrel. Drugs 1997; 54:745–750.
- Humbert M, Nurden P, Bihour C, et al. Ultrastructural studies of platelet aggregates from human subjects receiving clopidogrel and from a patient with an inherited defect of an ADP-dependent pathway of platelet activation. Arterioscler Thromb Vasc Biol 1996; 16:1532–1543.
- Hass WK, Easton JD, Adams HP, et al. A randomized trial comparing ticlopidine hydrochloride with aspirin for the prevention of stroke in high-risk patients. Ticlopidine Aspirin Stroke Study Group. N Engl J Med 1989; 321:501–507.
- Savi P, Bernat A, Dumas A, Ait-Chek L, Herbert JM. Effect of aspirin and clopidogrel on platelet-dependent tissue factor expression in endothelial cells. Thromb Res 1994; 73:117–124.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopido-grel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet 1996; 348:1329–1339.
- Bhatt DL, Marso SP, Hirsch AT, Ringleb PA, Hacke W, Topol EJ. Amplified benefit of clopidogrel versus aspirin in patients with diabetes mellitus. Am J Cardiol 2002; 90:625–628.
- Bhatt DL, Hirsch AT, Ringleb PA, Hacke W, Topol EJ. Reduction in the need for hospitalization for recurrent ischemic events and bleeding with clopidogrel instead of aspirin. CAPRIE investigators. Am Heart J 2000; 140:67–73.
- Bhatt DL, Topol EJ. Antiplatelet and anticoagulant therapy in the secondary prevention of ischemic heart disease. Med Clin North Am 2000; 84 1:163–179.
- Ringleb PA, Bhatt DL, Hirsch AT, Topol EJ, Hacke W Clopidogrel Versus Aspirin in Patients at Risk of Ischemic Events Investigators. Benefit of clopidogrel over aspirin is amplified in patients with a history of ischemic events. Stroke 2004; 35:528–532.
- Bhatt DL, Chew DP, Hirsch AT, Ringleb PA, Hacke W, Topol EJ. Superiority of clopidogrel versus aspirin in patients with prior cardiac surgery. Circulation 2001; 103:363–368.
- Yusuf S, Zhao F, Mehta SR, et al. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Budaj A, Yusuf S, Mehta SR, et al. Benefit of clopidogrel in patients with acute coronary syndromes without ST-segment elevation in various risk groups. Circulation 2002; 106:1622–1626.
- Fox KA, Mehta SR, Peters R, et al. Benefits and risks of the combination of clopidogrel and aspirin in patients undergoing surgical revascularization for non–ST-elevation acute coronary syndrome: the Clopidogrel in Unstable angina to prevent Recurrent ischemic Events (CURE) Trial. Circulation 2004; 110:1202–1208.
- Sabatine MS, Cannon CP, Gibson CM, et al. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation. N Engl J Med 2005; 352:1179–1189.
- Chen ZM, Jiang LX, Chen YP, et al. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Bhatt DL, Kapadia SR, Bajzer CT, et al. Dual antiplatelet therapy with clopidogrel and aspirin after carotid artery stenting. J Invasive Cardiol 2001; 13:767–771.
- Steinhubl SR, Berger PB, Mann JT, et al. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 288:2411–2420.
- Sabatine MS, Cannon CP, Gibson CM, et al. Effect of clopidogrel pre-treatment before percutaneous coronary intervention in patients with ST-elevation myocardial infarction treated with fibrinolytics: the PCI-CLARITY study. JAMA 2005; 294:1224–1232.
- Mehta SR, Yusuf S, Peters RJ, et al. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- The GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med 1993; 329:673–682.
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339:229–234.
- Bhatt DL, Steg PG, Ohman EM, et al. International prevalence, recognition, and treatment of cardiovascular risk factors in outpatients with atherothrombosis. JAMA 2006; 295:180–189.
- Steg PG, Bhatt DL, Wilson PW, et al. One-year cardiovascular event rates in outpatients with atherothrombosis. JAMA 2007; 297:1197–1206.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Rabbat MG, Bavry AA, Bhatt DL, Ellis SG. Understanding and minimizing late thrombosis of drug-eluting stents. Cleve Clin J Med 2007; 74:129–136.
- Gaspoz JM, Coxson PG, Goldman PA, et al. Cost effectiveness of aspirin, clopidogrel, or both for secondary prevention of coronary heart disease. N Engl J Med 2002; 346:1800–1806.
- Beinart SC, Kolm P, Veledar E, et al. Longterm cost effectiveness of early and sustained dual oral antiplatelet therapy with clopidogrel given for up to one year after percutaneous coronary intervention results: from the Clopidogrel for the Reduction of Events During Observation (CREDO) trial. J Am Coll Cardiol 2005; 46:761–769.
- Chen J, Bhatt DL, Schneider E, et al. Cost-effectiveness of clopidogrel + aspirin vs. aspirin alone for secondary prevention of cardiovascular events: results from the CHARISMA Trial Session; APS.96.1; Presentation 3855; American Heart Association Scientific Sessions; Nov 12–15, 2006; Chicago IL.
In patients at risk of myocardial infarction or stroke, two antiplatelet drugs are not always better than one. In a large recent trial,1,2 adding clopidogrel (Plavix) to aspirin therapy did not offer much benefit to a cohort of patients at risk of cardiovascular events, although a subgroup did appear to benefit: those at even higher risk because they already had a history of myocardial infarction, ischemic stroke, or peripheral arterial disease.
These were the principal findings in the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) study,1,2 in which one of us (D.L.B.) was principal investigator.
These findings further our understanding of who should receive dual antiplatelet therapy, and who would be better served with aspirin therapy alone. In this article, we discuss important studies that led up to the CHARISMA trial, review CHARISMA’s purpose and study design, and interpret its results.
PREVENTING ATHEROTHROMBOSIS BY BLOCKING PLATELETS
Platelets are key players in the atherothrom-botic process.3–5 The Antiplatelet Trialists’ Collaboration,6 in a meta-analysis of trials performed up to 1997, calculated that antiplatelet therapy (mostly with aspirin) reduced the vascular mortality rate by 15% in patients with acute or previous vascular disease or some other predisposing condition. Thus, aspirin has already been shown to be effective as primary prevention (ie, in patients at risk but without established vascular disease) and as secondary prevention (ie, in those with established disease).7,8
Yet many patients have significant vascular events in spite of taking aspirin.6 Aspirin failure is thought to be multifactorial, with causes that include weak platelet inhibition, noncompliance, discontinuation due to adverse effects (including severe bleeding), and drug interactions. In addition, aspirin resistance has been linked to worse prognosis and may prove to be another cause of aspirin failure.9–11
Clopidogrel, an adenosine diphosphate (ADP) receptor antagonist, has also been studied extensively as an antiplatelet agent.5,12 Several studies have indicated that clopidogrel and ticlopidine (Ticlid, a related drug) may be more potent than aspirin, both in the test tube and in real patients.13–15
KEY TRIALS LEADING TO CHARISMA
- Clopidogrel is more effective and slightly safer than aspirin as secondary prevention, as shown in the Clopidogrel Versus Aspirin in Patients at Risk of Ischemic Events (CAPRIE) trial.16–21
- The combination of clopidogrel plus aspirin is more beneficial than placebo plus aspirin in patients with acute coronary syndromes, as shown in the Clopidogrel in Unstable Angina to Prevent Recurrent Ischemic Events (CURE) trial,22–24 the Clopidogrel as Adjunctive Reperfusion Therapy-Thrombolysis in Myo-car-dial Infarction (CLARITY-TIMI 28) trial,25 and the Clopidogrel and Metoprolol in Myocardial Infarction Trial (COMMIT).26
- The combination of clopidogrel plus aspirin is beneficial in patients undergoing percutaneous coronary interventions, with or without drug-eluting stent placement,27–30 as shown in the Clopidogrel for the Reduction of Events During Observation (CREDO) trial,28 the Effect of Clopidogrel Pretreatment Before Percutaneous Coronary Intervention in Patients With ST-Elevation Myocardial Infarction With Fibrinolytics (PCI-CLARITY) study,29 and the Effects of Pre-treatment With Clopidogrel and Aspirin Followed by Long-term Therapy in Patients Undergoing Percutaneous Coronary Intervention (PCI-CURE) study.30 In fact, most patients undergoing percutaneous interventions now receive a loading dose of clopidogrel before the procedure and continue to take it for up to 1 year afterward. However, the ideal long-term duration of clopidogrel treatment is still under debate.
In view of these previous studies, we wanted to test dual antiplatelet therapy in a broader population at high risk of atherothrombosis, ie, in patients with either established vascular disease or with multiple risk factors for it.
CHARISMA STUDY DESIGN
CHARISMA was a prospective, randomized, double-blind, placebo-controlled study of the efficacy and safety of clopidogrel plus aspirin vs placebo plus aspirin in patients at high risk of cardiovascular events.
A total of 15,603 patients, all older than 45 years, were randomly assigned to receive clopidogrel 75 mg/day plus aspirin 75 to 162 mg/day or placebo plus aspirin, in addition to standard therapy as directed by individual clinicians (eg, statins, beta-blockers). Patients were followed up at 1, 3, and 6 months and every 6 months thereafter until study completion, which occurred after 1,040 primary efficacy end points. The median duration of follow-up was 28 months.1
Patients had to have one of the following to be included: multiple atherothrombotic risk factors, documented coronary disease, documented cerebrovascular disease, or documented peripheral arterial disease (Table 2). Specific exclusion criteria included the use of oral antithrombotic or chronic nonsteroidal anti-inflammatory medications.1
End points
The primary end point was the combined incidence of the first episode of myocardial infarction or stroke, or death from cardiovascular causes.
The secondary end point was the combined incidence of myocardial infarction, stroke, death from cardiovascular causes, or hospitalization for unstable angina, a transient ischemic attack, or revascularization procedure.
The primary safety end point was severe bleeding, as defined in the Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries (GUSTO) study31 as intracranial hemorrhage, fatal bleeding, or bleeding leading to hemody-namic compromise. Moderate bleeding was defined as bleeding that required transfusion but did not meet the GUSTO definition of severe bleeding.
OVERALL, NO BENEFIT
The rates of the secondary end point were 16.7% vs 17.9% (absolute risk reduction 1.2%; relative risk reduction 8%; P = .04).
Possible benefit in symptomatic patients
In a prespecified analysis, patients were classified as being “symptomatic” (having documented cardiovascular disease, ie, coronary, cerebrovascular, or symptomatic peripheral arterial disease) or “asymptomatic” (having multiple risk factors without established cardiovascular disease).1
In the symptomatic group (n = 12,153), the primary end point was reached in 6.9% of patients treated with clopidogrel vs 7.9% with placebo (absolute risk reduction 1.0%; relative risk reduction 13%; P = .046). The 3,284 asymptomatic patients showed no benefit; the rate of the primary end point for the clopido-grel group was 6.6% vs 5.5% in the placebo group (P = .20).
In a post hoc analysis, we examined the data from 9,478 patients who were similar to those in the CAPRIE study (ie, with documented prior myocardial infarction, prior ischemic stroke, or symptomatic peripheral arterial disease). The rate of cardiovascular death, myocardial infarction, or stroke was 8.8% in the placebo-plus-aspirin group and 7.3% in the clopidogrel-plus-aspirin group (absolute risk reduction 1.5%; relative risk reduction 17%; P = .01; Figure 1).2
HOW SHOULD WE INTERPRET THESE FINDINGS?
CHARISMA was the first trial to evaluate whether adding clopidogrel to aspirin therapy would reduce the rates of vascular events and death from cardiovascular causes in stable patients at risk of ischemic events. As in other trials, the benefit of clopidogrel-plus-aspirin therapy was weighed against the risk of bleeding with this regimen. How are we to interpret the findings?
- In the group with multiple risk factors but without clearly documented cardiovascular disease, there was no benefit—and there was an increase in moderate bleeding. Given these findings, physicians should not prescribe dual antiplatelet therapy for primary prevention in patients without known vascular disease.
- A potential benefit was seen in a prespecified subgroup who had documented cardiovascular disease. Given the limitations of subgroup analysis, however, and given the increased risk of moderate bleeding, this positive result should be interpreted with some degree of caution.
- CHARISMA suggests that there may be benefit of protracted dual antiplatelet therapy in stable patients with documented prior ischemic events.
A possible reason for the observed lack of benefit in the overall cohort but the positive results in the subgroups with established vascular disease is that plaque rupture and thrombosis may be a precondition for dual antiplatelet therapy to work.
Another possibility is that, although we have been saying that diabetes mellitus (one of the possible entry criteria in CHARISMA) is a “coronary risk equivalent,” this may not be absolutely true. Although it had been demonstrated that patients with certain risk factors, such as diabetes, have an incidence of ischemic events similar to that in patients with prior MI and should be considered for antiplatelet therapy to prevent vascular events,32 more recent data have shown that patients with prior ischemic events are at much higher risk than patients without ischemic events, even if the latter have diabetes.33,34
- The observation in CHARISMA that the incremental bleeding risk of dual antiplatelet therapy vs aspirin does not persist beyond a year in patients who have tolerated therapy for a year without a bleeding event may affect the decision to continue clopidogrel beyond 1 year, such as in patients with acute coronary syndromes or patients who have received drug-eluting stents.35,36
- Another important consideration is cost-effectiveness. Several studies have analyzed the impact of cost and found clopidogrel to be cost-effective by preventing ischemic events and adding years of life.37,38 A recent analysis from CHARISMA also shows cost-effectiveness in the subgroup of patients enrolled with established cardiovascular disease.39 Once clopidogrel becomes generic, the cost-effectiveness will become even better.
Further studies should better define which stable patients with cardiovascular disease should be on more than aspirin alone.
In patients at risk of myocardial infarction or stroke, two antiplatelet drugs are not always better than one. In a large recent trial,1,2 adding clopidogrel (Plavix) to aspirin therapy did not offer much benefit to a cohort of patients at risk of cardiovascular events, although a subgroup did appear to benefit: those at even higher risk because they already had a history of myocardial infarction, ischemic stroke, or peripheral arterial disease.
These were the principal findings in the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) study,1,2 in which one of us (D.L.B.) was principal investigator.
These findings further our understanding of who should receive dual antiplatelet therapy, and who would be better served with aspirin therapy alone. In this article, we discuss important studies that led up to the CHARISMA trial, review CHARISMA’s purpose and study design, and interpret its results.
PREVENTING ATHEROTHROMBOSIS BY BLOCKING PLATELETS
Platelets are key players in the atherothrom-botic process.3–5 The Antiplatelet Trialists’ Collaboration,6 in a meta-analysis of trials performed up to 1997, calculated that antiplatelet therapy (mostly with aspirin) reduced the vascular mortality rate by 15% in patients with acute or previous vascular disease or some other predisposing condition. Thus, aspirin has already been shown to be effective as primary prevention (ie, in patients at risk but without established vascular disease) and as secondary prevention (ie, in those with established disease).7,8
Yet many patients have significant vascular events in spite of taking aspirin.6 Aspirin failure is thought to be multifactorial, with causes that include weak platelet inhibition, noncompliance, discontinuation due to adverse effects (including severe bleeding), and drug interactions. In addition, aspirin resistance has been linked to worse prognosis and may prove to be another cause of aspirin failure.9–11
Clopidogrel, an adenosine diphosphate (ADP) receptor antagonist, has also been studied extensively as an antiplatelet agent.5,12 Several studies have indicated that clopidogrel and ticlopidine (Ticlid, a related drug) may be more potent than aspirin, both in the test tube and in real patients.13–15
KEY TRIALS LEADING TO CHARISMA
- Clopidogrel is more effective and slightly safer than aspirin as secondary prevention, as shown in the Clopidogrel Versus Aspirin in Patients at Risk of Ischemic Events (CAPRIE) trial.16–21
- The combination of clopidogrel plus aspirin is more beneficial than placebo plus aspirin in patients with acute coronary syndromes, as shown in the Clopidogrel in Unstable Angina to Prevent Recurrent Ischemic Events (CURE) trial,22–24 the Clopidogrel as Adjunctive Reperfusion Therapy-Thrombolysis in Myo-car-dial Infarction (CLARITY-TIMI 28) trial,25 and the Clopidogrel and Metoprolol in Myocardial Infarction Trial (COMMIT).26
- The combination of clopidogrel plus aspirin is beneficial in patients undergoing percutaneous coronary interventions, with or without drug-eluting stent placement,27–30 as shown in the Clopidogrel for the Reduction of Events During Observation (CREDO) trial,28 the Effect of Clopidogrel Pretreatment Before Percutaneous Coronary Intervention in Patients With ST-Elevation Myocardial Infarction With Fibrinolytics (PCI-CLARITY) study,29 and the Effects of Pre-treatment With Clopidogrel and Aspirin Followed by Long-term Therapy in Patients Undergoing Percutaneous Coronary Intervention (PCI-CURE) study.30 In fact, most patients undergoing percutaneous interventions now receive a loading dose of clopidogrel before the procedure and continue to take it for up to 1 year afterward. However, the ideal long-term duration of clopidogrel treatment is still under debate.
In view of these previous studies, we wanted to test dual antiplatelet therapy in a broader population at high risk of atherothrombosis, ie, in patients with either established vascular disease or with multiple risk factors for it.
CHARISMA STUDY DESIGN
CHARISMA was a prospective, randomized, double-blind, placebo-controlled study of the efficacy and safety of clopidogrel plus aspirin vs placebo plus aspirin in patients at high risk of cardiovascular events.
A total of 15,603 patients, all older than 45 years, were randomly assigned to receive clopidogrel 75 mg/day plus aspirin 75 to 162 mg/day or placebo plus aspirin, in addition to standard therapy as directed by individual clinicians (eg, statins, beta-blockers). Patients were followed up at 1, 3, and 6 months and every 6 months thereafter until study completion, which occurred after 1,040 primary efficacy end points. The median duration of follow-up was 28 months.1
Patients had to have one of the following to be included: multiple atherothrombotic risk factors, documented coronary disease, documented cerebrovascular disease, or documented peripheral arterial disease (Table 2). Specific exclusion criteria included the use of oral antithrombotic or chronic nonsteroidal anti-inflammatory medications.1
End points
The primary end point was the combined incidence of the first episode of myocardial infarction or stroke, or death from cardiovascular causes.
The secondary end point was the combined incidence of myocardial infarction, stroke, death from cardiovascular causes, or hospitalization for unstable angina, a transient ischemic attack, or revascularization procedure.
The primary safety end point was severe bleeding, as defined in the Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries (GUSTO) study31 as intracranial hemorrhage, fatal bleeding, or bleeding leading to hemody-namic compromise. Moderate bleeding was defined as bleeding that required transfusion but did not meet the GUSTO definition of severe bleeding.
OVERALL, NO BENEFIT
The rates of the secondary end point were 16.7% vs 17.9% (absolute risk reduction 1.2%; relative risk reduction 8%; P = .04).
Possible benefit in symptomatic patients
In a prespecified analysis, patients were classified as being “symptomatic” (having documented cardiovascular disease, ie, coronary, cerebrovascular, or symptomatic peripheral arterial disease) or “asymptomatic” (having multiple risk factors without established cardiovascular disease).1
In the symptomatic group (n = 12,153), the primary end point was reached in 6.9% of patients treated with clopidogrel vs 7.9% with placebo (absolute risk reduction 1.0%; relative risk reduction 13%; P = .046). The 3,284 asymptomatic patients showed no benefit; the rate of the primary end point for the clopido-grel group was 6.6% vs 5.5% in the placebo group (P = .20).
In a post hoc analysis, we examined the data from 9,478 patients who were similar to those in the CAPRIE study (ie, with documented prior myocardial infarction, prior ischemic stroke, or symptomatic peripheral arterial disease). The rate of cardiovascular death, myocardial infarction, or stroke was 8.8% in the placebo-plus-aspirin group and 7.3% in the clopidogrel-plus-aspirin group (absolute risk reduction 1.5%; relative risk reduction 17%; P = .01; Figure 1).2
HOW SHOULD WE INTERPRET THESE FINDINGS?
CHARISMA was the first trial to evaluate whether adding clopidogrel to aspirin therapy would reduce the rates of vascular events and death from cardiovascular causes in stable patients at risk of ischemic events. As in other trials, the benefit of clopidogrel-plus-aspirin therapy was weighed against the risk of bleeding with this regimen. How are we to interpret the findings?
- In the group with multiple risk factors but without clearly documented cardiovascular disease, there was no benefit—and there was an increase in moderate bleeding. Given these findings, physicians should not prescribe dual antiplatelet therapy for primary prevention in patients without known vascular disease.
- A potential benefit was seen in a prespecified subgroup who had documented cardiovascular disease. Given the limitations of subgroup analysis, however, and given the increased risk of moderate bleeding, this positive result should be interpreted with some degree of caution.
- CHARISMA suggests that there may be benefit of protracted dual antiplatelet therapy in stable patients with documented prior ischemic events.
A possible reason for the observed lack of benefit in the overall cohort but the positive results in the subgroups with established vascular disease is that plaque rupture and thrombosis may be a precondition for dual antiplatelet therapy to work.
Another possibility is that, although we have been saying that diabetes mellitus (one of the possible entry criteria in CHARISMA) is a “coronary risk equivalent,” this may not be absolutely true. Although it had been demonstrated that patients with certain risk factors, such as diabetes, have an incidence of ischemic events similar to that in patients with prior MI and should be considered for antiplatelet therapy to prevent vascular events,32 more recent data have shown that patients with prior ischemic events are at much higher risk than patients without ischemic events, even if the latter have diabetes.33,34
- The observation in CHARISMA that the incremental bleeding risk of dual antiplatelet therapy vs aspirin does not persist beyond a year in patients who have tolerated therapy for a year without a bleeding event may affect the decision to continue clopidogrel beyond 1 year, such as in patients with acute coronary syndromes or patients who have received drug-eluting stents.35,36
- Another important consideration is cost-effectiveness. Several studies have analyzed the impact of cost and found clopidogrel to be cost-effective by preventing ischemic events and adding years of life.37,38 A recent analysis from CHARISMA also shows cost-effectiveness in the subgroup of patients enrolled with established cardiovascular disease.39 Once clopidogrel becomes generic, the cost-effectiveness will become even better.
Further studies should better define which stable patients with cardiovascular disease should be on more than aspirin alone.
- Bhatt DL, Fox KA, Hacke W, et al. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Ruggeri ZM. Platelets in atherothrombosis. Nat Med 2002; 8:1227–1234.
- Fuster V, Moreno PR, Fayad ZA, Corti R, Badimon JJ. Atherothrombosis and high-risk plaque: part I: evolving concepts. J Am Coll Cardiol 2005; 46:937–954.
- Meadows TA, Bhatt DL. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 2007; 100:1261–1275.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Sanmuganathan PS, Ghahramani P, Jackson PR, Wallis EJ, Ramsay LE. Aspirin for primary prevention of coronary heart disease: safety and absolute benefit related to coronary risk derived from meta-analysis of randomised trials. Heart 2001; 85:265–271.
- Hayden M, Pignone M, Phillips C, Mulrow C. Aspirin for the primary prevention of cardiovascular events: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med 2002; 136:161–172.
- Helgason CM, Bolin KM, Hoff JA, et al. Development of aspirin resistance in persons with previous ischemic stroke. Stroke 1994; 25:2331–2336.
- Helgason CM, Tortorice KL, Winkler SR, et al. Aspirin response and failure in cerebral infarction. Stroke 1993; 24:345–350.
- Gum PA, Kottke-Marchant K, Poggio ED, et al. Profile and prevalence of aspirin resistance in patients with cardiovascular disease. Am J Cardiol 2001; 88:230–235.
- Coukell AJ, Markham A. Clopidogrel. Drugs 1997; 54:745–750.
- Humbert M, Nurden P, Bihour C, et al. Ultrastructural studies of platelet aggregates from human subjects receiving clopidogrel and from a patient with an inherited defect of an ADP-dependent pathway of platelet activation. Arterioscler Thromb Vasc Biol 1996; 16:1532–1543.
- Hass WK, Easton JD, Adams HP, et al. A randomized trial comparing ticlopidine hydrochloride with aspirin for the prevention of stroke in high-risk patients. Ticlopidine Aspirin Stroke Study Group. N Engl J Med 1989; 321:501–507.
- Savi P, Bernat A, Dumas A, Ait-Chek L, Herbert JM. Effect of aspirin and clopidogrel on platelet-dependent tissue factor expression in endothelial cells. Thromb Res 1994; 73:117–124.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopido-grel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet 1996; 348:1329–1339.
- Bhatt DL, Marso SP, Hirsch AT, Ringleb PA, Hacke W, Topol EJ. Amplified benefit of clopidogrel versus aspirin in patients with diabetes mellitus. Am J Cardiol 2002; 90:625–628.
- Bhatt DL, Hirsch AT, Ringleb PA, Hacke W, Topol EJ. Reduction in the need for hospitalization for recurrent ischemic events and bleeding with clopidogrel instead of aspirin. CAPRIE investigators. Am Heart J 2000; 140:67–73.
- Bhatt DL, Topol EJ. Antiplatelet and anticoagulant therapy in the secondary prevention of ischemic heart disease. Med Clin North Am 2000; 84 1:163–179.
- Ringleb PA, Bhatt DL, Hirsch AT, Topol EJ, Hacke W Clopidogrel Versus Aspirin in Patients at Risk of Ischemic Events Investigators. Benefit of clopidogrel over aspirin is amplified in patients with a history of ischemic events. Stroke 2004; 35:528–532.
- Bhatt DL, Chew DP, Hirsch AT, Ringleb PA, Hacke W, Topol EJ. Superiority of clopidogrel versus aspirin in patients with prior cardiac surgery. Circulation 2001; 103:363–368.
- Yusuf S, Zhao F, Mehta SR, et al. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Budaj A, Yusuf S, Mehta SR, et al. Benefit of clopidogrel in patients with acute coronary syndromes without ST-segment elevation in various risk groups. Circulation 2002; 106:1622–1626.
- Fox KA, Mehta SR, Peters R, et al. Benefits and risks of the combination of clopidogrel and aspirin in patients undergoing surgical revascularization for non–ST-elevation acute coronary syndrome: the Clopidogrel in Unstable angina to prevent Recurrent ischemic Events (CURE) Trial. Circulation 2004; 110:1202–1208.
- Sabatine MS, Cannon CP, Gibson CM, et al. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation. N Engl J Med 2005; 352:1179–1189.
- Chen ZM, Jiang LX, Chen YP, et al. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Bhatt DL, Kapadia SR, Bajzer CT, et al. Dual antiplatelet therapy with clopidogrel and aspirin after carotid artery stenting. J Invasive Cardiol 2001; 13:767–771.
- Steinhubl SR, Berger PB, Mann JT, et al. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 288:2411–2420.
- Sabatine MS, Cannon CP, Gibson CM, et al. Effect of clopidogrel pre-treatment before percutaneous coronary intervention in patients with ST-elevation myocardial infarction treated with fibrinolytics: the PCI-CLARITY study. JAMA 2005; 294:1224–1232.
- Mehta SR, Yusuf S, Peters RJ, et al. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- The GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med 1993; 329:673–682.
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339:229–234.
- Bhatt DL, Steg PG, Ohman EM, et al. International prevalence, recognition, and treatment of cardiovascular risk factors in outpatients with atherothrombosis. JAMA 2006; 295:180–189.
- Steg PG, Bhatt DL, Wilson PW, et al. One-year cardiovascular event rates in outpatients with atherothrombosis. JAMA 2007; 297:1197–1206.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Rabbat MG, Bavry AA, Bhatt DL, Ellis SG. Understanding and minimizing late thrombosis of drug-eluting stents. Cleve Clin J Med 2007; 74:129–136.
- Gaspoz JM, Coxson PG, Goldman PA, et al. Cost effectiveness of aspirin, clopidogrel, or both for secondary prevention of coronary heart disease. N Engl J Med 2002; 346:1800–1806.
- Beinart SC, Kolm P, Veledar E, et al. Longterm cost effectiveness of early and sustained dual oral antiplatelet therapy with clopidogrel given for up to one year after percutaneous coronary intervention results: from the Clopidogrel for the Reduction of Events During Observation (CREDO) trial. J Am Coll Cardiol 2005; 46:761–769.
- Chen J, Bhatt DL, Schneider E, et al. Cost-effectiveness of clopidogrel + aspirin vs. aspirin alone for secondary prevention of cardiovascular events: results from the CHARISMA Trial Session; APS.96.1; Presentation 3855; American Heart Association Scientific Sessions; Nov 12–15, 2006; Chicago IL.
- Bhatt DL, Fox KA, Hacke W, et al. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Ruggeri ZM. Platelets in atherothrombosis. Nat Med 2002; 8:1227–1234.
- Fuster V, Moreno PR, Fayad ZA, Corti R, Badimon JJ. Atherothrombosis and high-risk plaque: part I: evolving concepts. J Am Coll Cardiol 2005; 46:937–954.
- Meadows TA, Bhatt DL. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 2007; 100:1261–1275.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Sanmuganathan PS, Ghahramani P, Jackson PR, Wallis EJ, Ramsay LE. Aspirin for primary prevention of coronary heart disease: safety and absolute benefit related to coronary risk derived from meta-analysis of randomised trials. Heart 2001; 85:265–271.
- Hayden M, Pignone M, Phillips C, Mulrow C. Aspirin for the primary prevention of cardiovascular events: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med 2002; 136:161–172.
- Helgason CM, Bolin KM, Hoff JA, et al. Development of aspirin resistance in persons with previous ischemic stroke. Stroke 1994; 25:2331–2336.
- Helgason CM, Tortorice KL, Winkler SR, et al. Aspirin response and failure in cerebral infarction. Stroke 1993; 24:345–350.
- Gum PA, Kottke-Marchant K, Poggio ED, et al. Profile and prevalence of aspirin resistance in patients with cardiovascular disease. Am J Cardiol 2001; 88:230–235.
- Coukell AJ, Markham A. Clopidogrel. Drugs 1997; 54:745–750.
- Humbert M, Nurden P, Bihour C, et al. Ultrastructural studies of platelet aggregates from human subjects receiving clopidogrel and from a patient with an inherited defect of an ADP-dependent pathway of platelet activation. Arterioscler Thromb Vasc Biol 1996; 16:1532–1543.
- Hass WK, Easton JD, Adams HP, et al. A randomized trial comparing ticlopidine hydrochloride with aspirin for the prevention of stroke in high-risk patients. Ticlopidine Aspirin Stroke Study Group. N Engl J Med 1989; 321:501–507.
- Savi P, Bernat A, Dumas A, Ait-Chek L, Herbert JM. Effect of aspirin and clopidogrel on platelet-dependent tissue factor expression in endothelial cells. Thromb Res 1994; 73:117–124.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopido-grel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet 1996; 348:1329–1339.
- Bhatt DL, Marso SP, Hirsch AT, Ringleb PA, Hacke W, Topol EJ. Amplified benefit of clopidogrel versus aspirin in patients with diabetes mellitus. Am J Cardiol 2002; 90:625–628.
- Bhatt DL, Hirsch AT, Ringleb PA, Hacke W, Topol EJ. Reduction in the need for hospitalization for recurrent ischemic events and bleeding with clopidogrel instead of aspirin. CAPRIE investigators. Am Heart J 2000; 140:67–73.
- Bhatt DL, Topol EJ. Antiplatelet and anticoagulant therapy in the secondary prevention of ischemic heart disease. Med Clin North Am 2000; 84 1:163–179.
- Ringleb PA, Bhatt DL, Hirsch AT, Topol EJ, Hacke W Clopidogrel Versus Aspirin in Patients at Risk of Ischemic Events Investigators. Benefit of clopidogrel over aspirin is amplified in patients with a history of ischemic events. Stroke 2004; 35:528–532.
- Bhatt DL, Chew DP, Hirsch AT, Ringleb PA, Hacke W, Topol EJ. Superiority of clopidogrel versus aspirin in patients with prior cardiac surgery. Circulation 2001; 103:363–368.
- Yusuf S, Zhao F, Mehta SR, et al. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Budaj A, Yusuf S, Mehta SR, et al. Benefit of clopidogrel in patients with acute coronary syndromes without ST-segment elevation in various risk groups. Circulation 2002; 106:1622–1626.
- Fox KA, Mehta SR, Peters R, et al. Benefits and risks of the combination of clopidogrel and aspirin in patients undergoing surgical revascularization for non–ST-elevation acute coronary syndrome: the Clopidogrel in Unstable angina to prevent Recurrent ischemic Events (CURE) Trial. Circulation 2004; 110:1202–1208.
- Sabatine MS, Cannon CP, Gibson CM, et al. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation. N Engl J Med 2005; 352:1179–1189.
- Chen ZM, Jiang LX, Chen YP, et al. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Bhatt DL, Kapadia SR, Bajzer CT, et al. Dual antiplatelet therapy with clopidogrel and aspirin after carotid artery stenting. J Invasive Cardiol 2001; 13:767–771.
- Steinhubl SR, Berger PB, Mann JT, et al. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 288:2411–2420.
- Sabatine MS, Cannon CP, Gibson CM, et al. Effect of clopidogrel pre-treatment before percutaneous coronary intervention in patients with ST-elevation myocardial infarction treated with fibrinolytics: the PCI-CLARITY study. JAMA 2005; 294:1224–1232.
- Mehta SR, Yusuf S, Peters RJ, et al. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- The GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med 1993; 329:673–682.
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339:229–234.
- Bhatt DL, Steg PG, Ohman EM, et al. International prevalence, recognition, and treatment of cardiovascular risk factors in outpatients with atherothrombosis. JAMA 2006; 295:180–189.
- Steg PG, Bhatt DL, Wilson PW, et al. One-year cardiovascular event rates in outpatients with atherothrombosis. JAMA 2007; 297:1197–1206.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Rabbat MG, Bavry AA, Bhatt DL, Ellis SG. Understanding and minimizing late thrombosis of drug-eluting stents. Cleve Clin J Med 2007; 74:129–136.
- Gaspoz JM, Coxson PG, Goldman PA, et al. Cost effectiveness of aspirin, clopidogrel, or both for secondary prevention of coronary heart disease. N Engl J Med 2002; 346:1800–1806.
- Beinart SC, Kolm P, Veledar E, et al. Longterm cost effectiveness of early and sustained dual oral antiplatelet therapy with clopidogrel given for up to one year after percutaneous coronary intervention results: from the Clopidogrel for the Reduction of Events During Observation (CREDO) trial. J Am Coll Cardiol 2005; 46:761–769.
- Chen J, Bhatt DL, Schneider E, et al. Cost-effectiveness of clopidogrel + aspirin vs. aspirin alone for secondary prevention of cardiovascular events: results from the CHARISMA Trial Session; APS.96.1; Presentation 3855; American Heart Association Scientific Sessions; Nov 12–15, 2006; Chicago IL.
KEY POINTS
- Platelets are key players in atherothrombosis, and antiplatelet drugs such as aspirin and clopidogrel prevent events in patients at risk.
- In studies leading up to CHARISMA, the combination of clopidogrel and aspirin was found to be beneficial in patients with acute coronary syndromes and in those undergoing percutaneous coronary interventions.
- Clopidogrel should not be combined with aspirin as a primary preventive therapy (ie, for people without established vascular disease). How dual antiplatelet therapy should be used as secondary prevention in stable patients needs further study.