User login
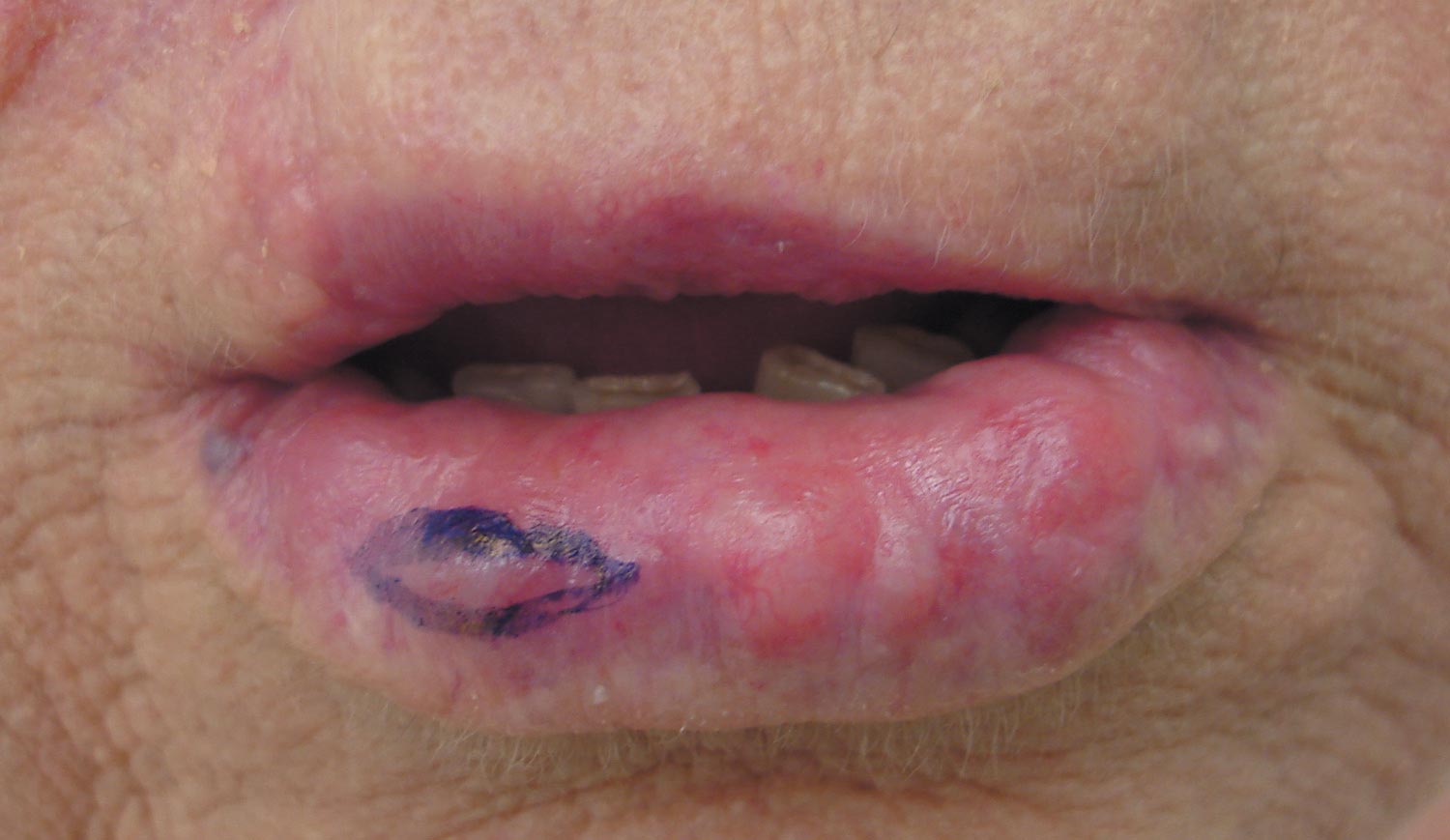
Nontender Nodules on the Lower Lip
The Diagnosis: Primary Systemic Amyloidosis
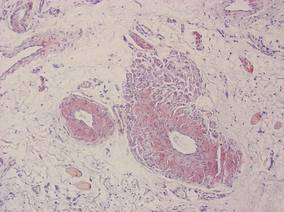
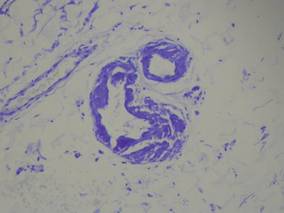
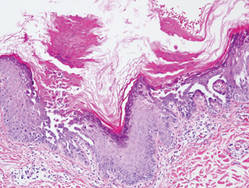
Our patient presented with multiple firm waxy nodules on the mucosal surface of the lower lip. Excision biopsy showed thickening of blood vessel walls with abundant amorphous material that was consistent with amyloid. Further staining with Congo red demonstrated brick red amorphous material within the vessel walls on routine light microscopy (Figure 1), and crystal violet stain showed metachromasia (Figure 2). Fine needle aspiration of the abdominal fat-pad showed amyloid. The final diagnosis was primary systemic amyloidosis (PSA).
|
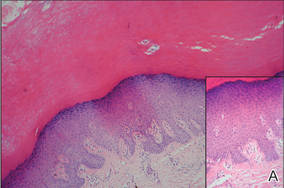
| Figure 1. Congo red staining showed a brick red appearance of amyloid within the vessel walls on routine light microscopy (original magnification ×200). |
|
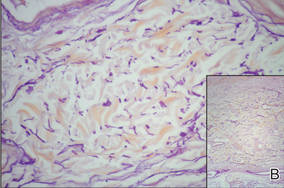
| Figure 2. Crystal violet staining around the blood vessels showed metachromasia (original magnification ×100). |
Amyloid is an ubiquitous fibrillar protein arranged in a cross-beta-pleated sheet that is confirmed with x-ray crystallography.1,2 More than 25 variants of amyloid have been identified.3 Pathologic deposition of amyloid-derived material results in a variable spectrum of clinical findings, collectively known as amyloidosis, with presentations ranging from nonspecific fever or fatigue to frank organ failure, depending on the organ involved. In PSA, immunoglobulin light chains are deposited throughout the body. Associated conditions include malignant or benign monoclonal gammopathy, multiple myeloma, Waldenström macroglobulinemia, malignant lymphoma, heavy chain disease, and chronic lymphocytic leukemia.2,4 The most commonly involved organ systems are the heart, lungs, liver, and kidneys. When patients present with unexplained heart failure, orthostatic hypotension, hepatomegaly, peripheral neuropathy, carpal tunnel syndrome, or renal insufficiency, amyloidosis should always be considered in the differential diagnosis.3
Cutaneous lesions of PSA tend to be vascular due to amyloid infiltration of blood vessel walls, manifesting as petechiae, purpura, ecchymoses, or nonhealing ulcers. Pinch purpura frequently are seen in the periorbital region after minor trauma and are recognized as a clinical indicator of PSA.2,4 Xerostomia from amyloid infiltrates in salivary glands is extremely common, and cases of amyloid in the eyes, bones, and thyroid gland have been reported.2 Macroglossia is seen in 12% to 40% of cases; coupled with xerostomia, it can lead to oropharyngeal dysphagia.2,5
Systemic amyloidosis can be further divided into primary (idiopathic or multiple myeloma associated) or secondary to chronic inflammatory conditions or infections; the key difference is the protein from which the abnormal amyloid is derived.1,4 The presence of cutaneous amyloidosis renders the need to rule out systemic disease because amyloidosis may be a purely localized or systemic process.4 Nodular amyloidosis is a localized form of amyloid that also has immunoglobulin light chain deposits and clinically appears exactly the same as PSA; however, the deposits are restricted to the skin.1,2,4,5
Characteristic biopsy findings in cutaneous amyloidosis include amorphous orange-red amyloid deposits on hematoxylin and eosin–stained sections. The gold standard for amyloid detection is apple green birefringence under polarized light with Congo red stain or electron microscopy.6,7 Other stains used to identify amyloid include crystal violet, methyl violet, periodic acid–Schiff, Sirius red, pagoda red, Dylon stain, and thioflavine T.1,2 Confirmation of systemic disease can be accomplished by fine needle aspiration of abdominal fat-pads or rectal mucosal biopsies.1,3,4 Biopsy of accessory salivary glands also has been reported to be very sensitive and specific.2
Treatment options remain limited; localized cutaneous disease may respond to topical corticosteroids, calcineurin inhibitors, or phototherapy. Primary systemic amyloidosis can be treated with a combination of steroids, melphalan, or colchicine often followed by autologous stem cell transplantation8; however, these regimens are not always curative and patients often have a poor prognosis.1
1. Black MM, Upjohn E, Albert S. Amyloidosis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 1. 2nd ed. Spain: Mosby Elsevier; 2008:623-631.
2. Steciuk A, Dompmartin A, Troussard X, et al. Cutaneous amyloidosis and possible association with systemic amyloidosis. Int J Dermatol. 2002;41:127-132.
3. Picken MM. Amyloidosis-where are we now and where are we heading? Arch Pathol Lab Med. 2010;134:545-551.
4. Schreml S, Szeimies RM, Vogt T, et al. Cutaneous amyloidoses and systemic amyloidoses with cutaneous involvement. Eur J Dermatol. 2010;20:152-160.
5. Breathnach SM. Amyloid and amyloidosis. J Am Acad Dermatol. 1988;18(1, pt 1):1-16.
6. Li WM. Histopathology of primary cutaneous amyloidoses and systemic amyloidosis. Clin Dermatol. 1990;8:30-35.
7. Lin CS, Wong CK. Electron microscopy of primary and secondary cutaneous amyloidoses and systemic amyloidosis. Clin Dermatol. 1990;8:36-45.
8. Dember LM. Modern treatment of amyloidosis: unresolved questions. J Am Soc Nephrol. 2009;20:469-472.
The Diagnosis: Primary Systemic Amyloidosis
Our patient presented with multiple firm waxy nodules on the mucosal surface of the lower lip. Excision biopsy showed thickening of blood vessel walls with abundant amorphous material that was consistent with amyloid. Further staining with Congo red demonstrated brick red amorphous material within the vessel walls on routine light microscopy (Figure 1), and crystal violet stain showed metachromasia (Figure 2). Fine needle aspiration of the abdominal fat-pad showed amyloid. The final diagnosis was primary systemic amyloidosis (PSA).
|
|
| Figure 1. Congo red staining showed a brick red appearance of amyloid within the vessel walls on routine light microscopy (original magnification ×200). |
|
|
| Figure 2. Crystal violet staining around the blood vessels showed metachromasia (original magnification ×100). |
Amyloid is an ubiquitous fibrillar protein arranged in a cross-beta-pleated sheet that is confirmed with x-ray crystallography.1,2 More than 25 variants of amyloid have been identified.3 Pathologic deposition of amyloid-derived material results in a variable spectrum of clinical findings, collectively known as amyloidosis, with presentations ranging from nonspecific fever or fatigue to frank organ failure, depending on the organ involved. In PSA, immunoglobulin light chains are deposited throughout the body. Associated conditions include malignant or benign monoclonal gammopathy, multiple myeloma, Waldenström macroglobulinemia, malignant lymphoma, heavy chain disease, and chronic lymphocytic leukemia.2,4 The most commonly involved organ systems are the heart, lungs, liver, and kidneys. When patients present with unexplained heart failure, orthostatic hypotension, hepatomegaly, peripheral neuropathy, carpal tunnel syndrome, or renal insufficiency, amyloidosis should always be considered in the differential diagnosis.3
Cutaneous lesions of PSA tend to be vascular due to amyloid infiltration of blood vessel walls, manifesting as petechiae, purpura, ecchymoses, or nonhealing ulcers. Pinch purpura frequently are seen in the periorbital region after minor trauma and are recognized as a clinical indicator of PSA.2,4 Xerostomia from amyloid infiltrates in salivary glands is extremely common, and cases of amyloid in the eyes, bones, and thyroid gland have been reported.2 Macroglossia is seen in 12% to 40% of cases; coupled with xerostomia, it can lead to oropharyngeal dysphagia.2,5
Systemic amyloidosis can be further divided into primary (idiopathic or multiple myeloma associated) or secondary to chronic inflammatory conditions or infections; the key difference is the protein from which the abnormal amyloid is derived.1,4 The presence of cutaneous amyloidosis renders the need to rule out systemic disease because amyloidosis may be a purely localized or systemic process.4 Nodular amyloidosis is a localized form of amyloid that also has immunoglobulin light chain deposits and clinically appears exactly the same as PSA; however, the deposits are restricted to the skin.1,2,4,5
Characteristic biopsy findings in cutaneous amyloidosis include amorphous orange-red amyloid deposits on hematoxylin and eosin–stained sections. The gold standard for amyloid detection is apple green birefringence under polarized light with Congo red stain or electron microscopy.6,7 Other stains used to identify amyloid include crystal violet, methyl violet, periodic acid–Schiff, Sirius red, pagoda red, Dylon stain, and thioflavine T.1,2 Confirmation of systemic disease can be accomplished by fine needle aspiration of abdominal fat-pads or rectal mucosal biopsies.1,3,4 Biopsy of accessory salivary glands also has been reported to be very sensitive and specific.2
Treatment options remain limited; localized cutaneous disease may respond to topical corticosteroids, calcineurin inhibitors, or phototherapy. Primary systemic amyloidosis can be treated with a combination of steroids, melphalan, or colchicine often followed by autologous stem cell transplantation8; however, these regimens are not always curative and patients often have a poor prognosis.1
The Diagnosis: Primary Systemic Amyloidosis
Our patient presented with multiple firm waxy nodules on the mucosal surface of the lower lip. Excision biopsy showed thickening of blood vessel walls with abundant amorphous material that was consistent with amyloid. Further staining with Congo red demonstrated brick red amorphous material within the vessel walls on routine light microscopy (Figure 1), and crystal violet stain showed metachromasia (Figure 2). Fine needle aspiration of the abdominal fat-pad showed amyloid. The final diagnosis was primary systemic amyloidosis (PSA).
|
|
| Figure 1. Congo red staining showed a brick red appearance of amyloid within the vessel walls on routine light microscopy (original magnification ×200). |
|
|
| Figure 2. Crystal violet staining around the blood vessels showed metachromasia (original magnification ×100). |
Amyloid is an ubiquitous fibrillar protein arranged in a cross-beta-pleated sheet that is confirmed with x-ray crystallography.1,2 More than 25 variants of amyloid have been identified.3 Pathologic deposition of amyloid-derived material results in a variable spectrum of clinical findings, collectively known as amyloidosis, with presentations ranging from nonspecific fever or fatigue to frank organ failure, depending on the organ involved. In PSA, immunoglobulin light chains are deposited throughout the body. Associated conditions include malignant or benign monoclonal gammopathy, multiple myeloma, Waldenström macroglobulinemia, malignant lymphoma, heavy chain disease, and chronic lymphocytic leukemia.2,4 The most commonly involved organ systems are the heart, lungs, liver, and kidneys. When patients present with unexplained heart failure, orthostatic hypotension, hepatomegaly, peripheral neuropathy, carpal tunnel syndrome, or renal insufficiency, amyloidosis should always be considered in the differential diagnosis.3
Cutaneous lesions of PSA tend to be vascular due to amyloid infiltration of blood vessel walls, manifesting as petechiae, purpura, ecchymoses, or nonhealing ulcers. Pinch purpura frequently are seen in the periorbital region after minor trauma and are recognized as a clinical indicator of PSA.2,4 Xerostomia from amyloid infiltrates in salivary glands is extremely common, and cases of amyloid in the eyes, bones, and thyroid gland have been reported.2 Macroglossia is seen in 12% to 40% of cases; coupled with xerostomia, it can lead to oropharyngeal dysphagia.2,5
Systemic amyloidosis can be further divided into primary (idiopathic or multiple myeloma associated) or secondary to chronic inflammatory conditions or infections; the key difference is the protein from which the abnormal amyloid is derived.1,4 The presence of cutaneous amyloidosis renders the need to rule out systemic disease because amyloidosis may be a purely localized or systemic process.4 Nodular amyloidosis is a localized form of amyloid that also has immunoglobulin light chain deposits and clinically appears exactly the same as PSA; however, the deposits are restricted to the skin.1,2,4,5
Characteristic biopsy findings in cutaneous amyloidosis include amorphous orange-red amyloid deposits on hematoxylin and eosin–stained sections. The gold standard for amyloid detection is apple green birefringence under polarized light with Congo red stain or electron microscopy.6,7 Other stains used to identify amyloid include crystal violet, methyl violet, periodic acid–Schiff, Sirius red, pagoda red, Dylon stain, and thioflavine T.1,2 Confirmation of systemic disease can be accomplished by fine needle aspiration of abdominal fat-pads or rectal mucosal biopsies.1,3,4 Biopsy of accessory salivary glands also has been reported to be very sensitive and specific.2
Treatment options remain limited; localized cutaneous disease may respond to topical corticosteroids, calcineurin inhibitors, or phototherapy. Primary systemic amyloidosis can be treated with a combination of steroids, melphalan, or colchicine often followed by autologous stem cell transplantation8; however, these regimens are not always curative and patients often have a poor prognosis.1
1. Black MM, Upjohn E, Albert S. Amyloidosis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 1. 2nd ed. Spain: Mosby Elsevier; 2008:623-631.
2. Steciuk A, Dompmartin A, Troussard X, et al. Cutaneous amyloidosis and possible association with systemic amyloidosis. Int J Dermatol. 2002;41:127-132.
3. Picken MM. Amyloidosis-where are we now and where are we heading? Arch Pathol Lab Med. 2010;134:545-551.
4. Schreml S, Szeimies RM, Vogt T, et al. Cutaneous amyloidoses and systemic amyloidoses with cutaneous involvement. Eur J Dermatol. 2010;20:152-160.
5. Breathnach SM. Amyloid and amyloidosis. J Am Acad Dermatol. 1988;18(1, pt 1):1-16.
6. Li WM. Histopathology of primary cutaneous amyloidoses and systemic amyloidosis. Clin Dermatol. 1990;8:30-35.
7. Lin CS, Wong CK. Electron microscopy of primary and secondary cutaneous amyloidoses and systemic amyloidosis. Clin Dermatol. 1990;8:36-45.
8. Dember LM. Modern treatment of amyloidosis: unresolved questions. J Am Soc Nephrol. 2009;20:469-472.
1. Black MM, Upjohn E, Albert S. Amyloidosis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 1. 2nd ed. Spain: Mosby Elsevier; 2008:623-631.
2. Steciuk A, Dompmartin A, Troussard X, et al. Cutaneous amyloidosis and possible association with systemic amyloidosis. Int J Dermatol. 2002;41:127-132.
3. Picken MM. Amyloidosis-where are we now and where are we heading? Arch Pathol Lab Med. 2010;134:545-551.
4. Schreml S, Szeimies RM, Vogt T, et al. Cutaneous amyloidoses and systemic amyloidoses with cutaneous involvement. Eur J Dermatol. 2010;20:152-160.
5. Breathnach SM. Amyloid and amyloidosis. J Am Acad Dermatol. 1988;18(1, pt 1):1-16.
6. Li WM. Histopathology of primary cutaneous amyloidoses and systemic amyloidosis. Clin Dermatol. 1990;8:30-35.
7. Lin CS, Wong CK. Electron microscopy of primary and secondary cutaneous amyloidoses and systemic amyloidosis. Clin Dermatol. 1990;8:36-45.
8. Dember LM. Modern treatment of amyloidosis: unresolved questions. J Am Soc Nephrol. 2009;20:469-472.
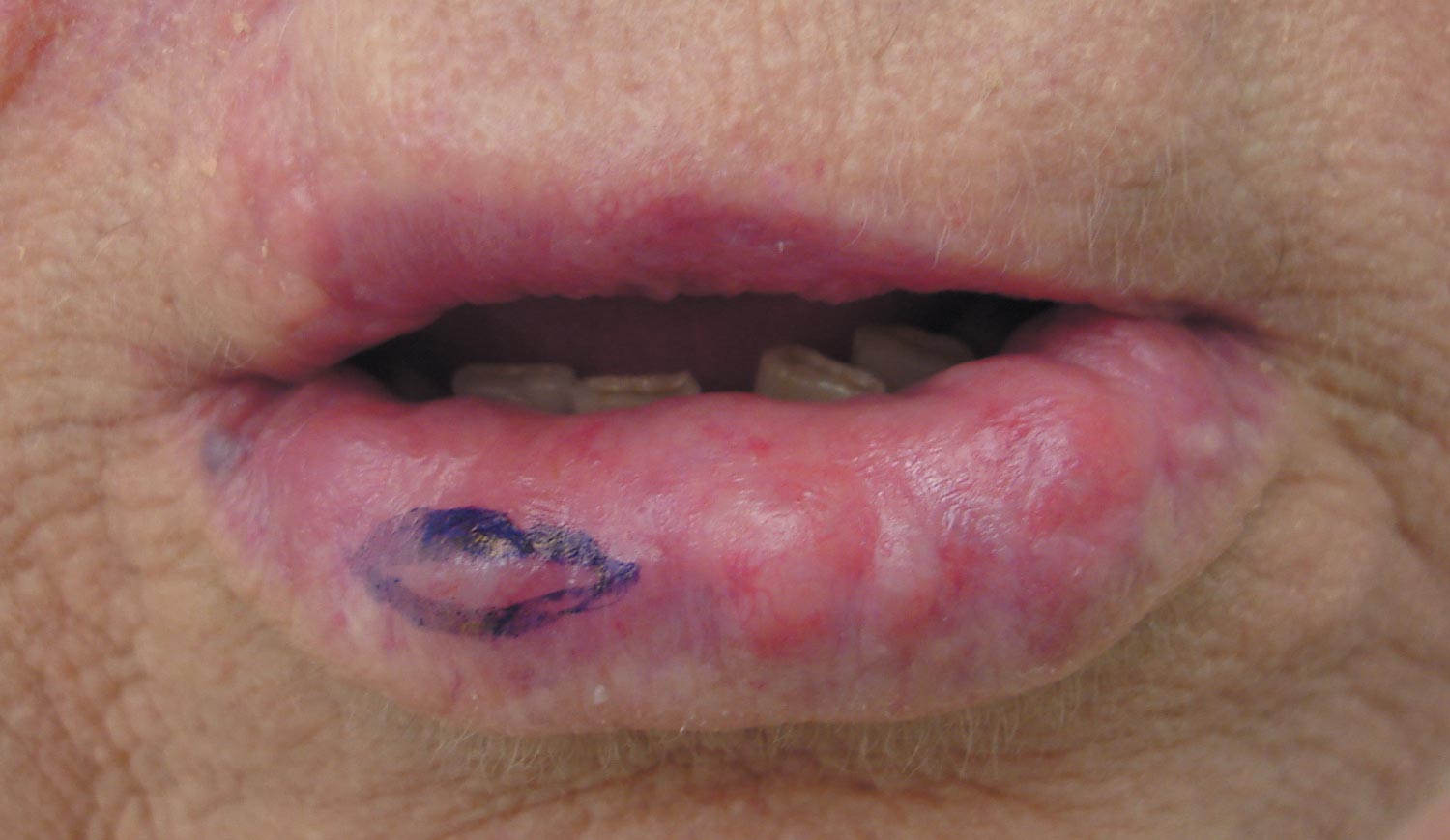
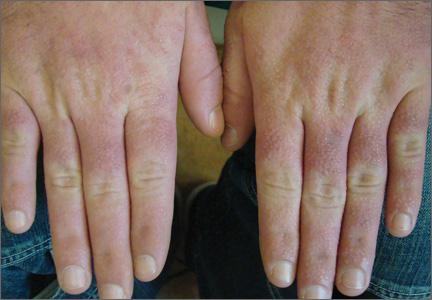
A 71-year-old woman presented with multiple 3×3-mm, firm, nontender nodules of 3 years’ duration on the mucosal surface of the lower lip that gradually enlarged. There was no macroglossia on presentation. The lip nodules were asymptomatic; however, they did interfere with eating and drinking, necessitating the use of a straw. Her medical history was remarkable for emphysema that required supplemental oxygen, rheumatoid arthritis, orthostatic hypotension, renal insufficiency, autonomic nervous system dysfunction, and Sjögren syndrome. She also had a recent thyroidectomy due to multiple thyroid nodules. An excision biopsy was performed of the lip nodules.
Plaques: A Rare Presentation of Acrokeratoelastoidosis
To the Editor:
Acrokeratoelastoidosis (AKE) is a rare disease first described by Costa1 in 1953. Typically it is only a cosmetic nuisance in the majority of patients and presents as asymptomatic, small, firm, flesh-colored to yellowish, round to polygonal papules with occasional keratosis or umbilication on the radial and ulnar margins of the hands and/or feet.1-3 In some cases, the lesions occur on the anterior aspects of the wrists, fingers, or lower legs.1 The lesions are always bilaterally distributed. Acrokeratoelastoidosis is a chronic skin disorder that commonly presents during childhood or adolescence, but presentation in adulthood also has been described.3 Histologically, AKE always shows hyperkeratosis, acanthosis, decrease of elastic tissue, and elastorrhexis of remaining elastic fibers. Plaque-type lesions are rare. We describe a patient who presented with plaques on the radial and ulnar margins of the hands.
A 36-year-old Chinese woman presented with asymptomatic, small, firm papules of 6 months’ duration that initially developed on the hands and gradually increased in number, coalescing into plaques. The feet were spared. She had no medical history of hyperhidrosis, chronic trauma, friction, or excessive sun exposure, and no family history of similar symptoms. No prior therapy had been attempted.
Physical examination showed nonconfluent, firm, flesh-colored to yellowish, translucent, smooth papules with wavy edges that were symmetrically distributed on the radial and ulnar margins of the hands; some papules had coalesced into plaques (Figure 1). A biopsy specimen taken from a plaque on the hypothenar eminence of the right hand revealed focal hyperkeratosis, hypergranulosis, acanthosis, and mild chronic inflammation with hematoxylin and eosin stain (Figure 2A). Aldehyde fuchsin staining showed fragmented and rarefied elastic fibers in the reticular dermis (Figure 2B). The patient was diagnosed with AKE. Oral tretinoin 10 mg twice daily was initiated and resulted in an evident response after 2 weeks of treatment. However, the patient stopped taking the medication because of pruritus and dry skin and the lesions then reappeared.

|
| Figure 1. Nonconfluent, firm, flesh-colored to yellowish, translucent, smooth papules distributed on the radial margin of the hand; some of papules coalesced into plaques. |
|
|
| Figure 2. Histopathology revealed hyperkeratosis, hypergranulosis, acanthosis, and mild chronic inflammation (A) (H&E, original magnification ×250 [inset, original magnification ×400]), as well as fragmented elastic fibers in the reticular dermis (B) (Aldehyde fuchsin, original magnification ×400 [inset, original magnification ×100]). |
Acrokeratoelastoidosis is a rare keratotic disorder. It seems to have no racial or ethnic predilection and occurs more frequently in women.4,5 It also is rare in China, with few cases reported, all women.5 The reason for the gender predilection in China remains unknown. The course is chronic, but it may rapidly progress during pregnancy.6
The pathogenesis of AKE is still unresolved.2,3 Although many cases are sporadic,5 it appears to be inherited in an autosomal-dominant fashion, most likely related to chromosome 2.7 Typically, AKE presents as papules that are discrete and bilaterally distributed in the palmoplantar margins,2,3 but some of the papules in our patient coalesced into plaques, which is unique. The histologic hallmarks indicated that the lesions were AKE.
The differential diagnosis of AKE includes hereditary papulotranslucent acrokeratoderma, focal acral hyperkeratosis, and keratoelastoidosis marginalis.8 Hereditary papulotranslucent acrokeratoderma also is inherited in an autosomal-dominant fashion and shares similar acral, translucent, keratotic papules with AKE, but there is no chronic inflammatory cell infiltrate, degeneration of collagenous fibers, or fragmentation of elastic fibers. The clinical appearance of focal acral hyperkeratosis is similar to AKE, but no changes are revealed in the elastic tissue.9 Because AKE, focal acral hyperkeratosis, and hereditary papulotranslucent acrokeratoderma have similar lesions and overlapping histologic changes, they may be considered variants of the same entity.4 Keratoelastoidosis marginalis, also called degenerative collagenous plaques of the hand, mainly affects white individuals aged 40 to 60 years with a history of prolonged sun exposure. Papules often are distributed over the junction of the dorsal and palmar skin and less often on the ulnar sides of the hands. The clinical lesions are similar to those in our patient, but histopathology of keratoelastoidosis marginalis shows amorphous, basophilic, elastotic masses and thickened, fragmented, calcified elastic fibers in the upper and mid dermis.
Therapies including liquid nitrogen, topical salicylic acid, methotrexate, dapsone, tar, cryotherapy, systemic prednisone, retinoic acid, clobetasone cream,5 and erbium:YAG laser10 have been applied. Thus far, no optimal treatment has been recommended and no tendency of spontaneous resolution has been previously reported in the literature. Our patient responded to tretinoin, but the lesions recurred after withdrawal of the medication; therefore, tretinoin may not be an optimal treatment option. Because the lesions are limited to the skin and AKE is only considered a cosmetic problem with a good prognosis, we recommend a wait-and-watch approach.
Acknowledgment—We thank Rashmi Sarkar, MD, New Delhi, India, for her assistance.
1. Costa OG. Akrokerato-elastoidosis; a hitherto undescribed skin disease. Dermatologica. 1953;107:164-168.
2. Bogle MA, Huang LY, Tschen JA. Acrokeratoelastoidosis. J Am Acad Dermatol. 2002;47:448-451.
3. Highet AS, Rook A, Anderson JR. Acrokeratoelastoidosis. Br J Dermatol. 1982;106:337-344.
4. Abulafia J, Vignale RA. Degenerative collagenous
plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
5. Luo DQ, Zhang B, Huang YB, et al. Papules on a young woman’s hands and feet. Clin Exp Dermatol. 2010;35:451-452.
6. Nelson-Adesokan P, Mallory SB, Lombardi C, et al. Acrokeratoelastoidosis of Costa [published correction appears in Int J Dermatol. 1996;35:380]. Int J Dermatol. 1995;34:431-433.
7. Greiner J, Krüger J, Palden L, et al. A linkage study of acrokeratoelastoidosis. possible mapping to chromosome 2. Hum Genet. 1983;63:222-227.
8. Hu W, Cook TF, Vicki GJ, et al. Acrokeratoelastoidosis. Pediatr Dermatol. 2002;19:320-322.
9. Dowd PM, Harman RR, Black MM. Focal acral hyperkeratosis. Br J Dermatol. 1983;109:97-103.
10. Erbil AH, Sezer E, Koç E, et al. Acrokeratoelastoidosis treated with the erbium:YAG laser [published online ahead of print November 3, 2007]. Clin Exp Dermatol. 2008;33:30-31.
To the Editor:
Acrokeratoelastoidosis (AKE) is a rare disease first described by Costa1 in 1953. Typically it is only a cosmetic nuisance in the majority of patients and presents as asymptomatic, small, firm, flesh-colored to yellowish, round to polygonal papules with occasional keratosis or umbilication on the radial and ulnar margins of the hands and/or feet.1-3 In some cases, the lesions occur on the anterior aspects of the wrists, fingers, or lower legs.1 The lesions are always bilaterally distributed. Acrokeratoelastoidosis is a chronic skin disorder that commonly presents during childhood or adolescence, but presentation in adulthood also has been described.3 Histologically, AKE always shows hyperkeratosis, acanthosis, decrease of elastic tissue, and elastorrhexis of remaining elastic fibers. Plaque-type lesions are rare. We describe a patient who presented with plaques on the radial and ulnar margins of the hands.
A 36-year-old Chinese woman presented with asymptomatic, small, firm papules of 6 months’ duration that initially developed on the hands and gradually increased in number, coalescing into plaques. The feet were spared. She had no medical history of hyperhidrosis, chronic trauma, friction, or excessive sun exposure, and no family history of similar symptoms. No prior therapy had been attempted.
Physical examination showed nonconfluent, firm, flesh-colored to yellowish, translucent, smooth papules with wavy edges that were symmetrically distributed on the radial and ulnar margins of the hands; some papules had coalesced into plaques (Figure 1). A biopsy specimen taken from a plaque on the hypothenar eminence of the right hand revealed focal hyperkeratosis, hypergranulosis, acanthosis, and mild chronic inflammation with hematoxylin and eosin stain (Figure 2A). Aldehyde fuchsin staining showed fragmented and rarefied elastic fibers in the reticular dermis (Figure 2B). The patient was diagnosed with AKE. Oral tretinoin 10 mg twice daily was initiated and resulted in an evident response after 2 weeks of treatment. However, the patient stopped taking the medication because of pruritus and dry skin and the lesions then reappeared.
|
|
| Figure 1. Nonconfluent, firm, flesh-colored to yellowish, translucent, smooth papules distributed on the radial margin of the hand; some of papules coalesced into plaques. |
|
|
|
|
| Figure 2. Histopathology revealed hyperkeratosis, hypergranulosis, acanthosis, and mild chronic inflammation (A) (H&E, original magnification ×250 [inset, original magnification ×400]), as well as fragmented elastic fibers in the reticular dermis (B) (Aldehyde fuchsin, original magnification ×400 [inset, original magnification ×100]). |
Acrokeratoelastoidosis is a rare keratotic disorder. It seems to have no racial or ethnic predilection and occurs more frequently in women.4,5 It also is rare in China, with few cases reported, all women.5 The reason for the gender predilection in China remains unknown. The course is chronic, but it may rapidly progress during pregnancy.6
The pathogenesis of AKE is still unresolved.2,3 Although many cases are sporadic,5 it appears to be inherited in an autosomal-dominant fashion, most likely related to chromosome 2.7 Typically, AKE presents as papules that are discrete and bilaterally distributed in the palmoplantar margins,2,3 but some of the papules in our patient coalesced into plaques, which is unique. The histologic hallmarks indicated that the lesions were AKE.
The differential diagnosis of AKE includes hereditary papulotranslucent acrokeratoderma, focal acral hyperkeratosis, and keratoelastoidosis marginalis.8 Hereditary papulotranslucent acrokeratoderma also is inherited in an autosomal-dominant fashion and shares similar acral, translucent, keratotic papules with AKE, but there is no chronic inflammatory cell infiltrate, degeneration of collagenous fibers, or fragmentation of elastic fibers. The clinical appearance of focal acral hyperkeratosis is similar to AKE, but no changes are revealed in the elastic tissue.9 Because AKE, focal acral hyperkeratosis, and hereditary papulotranslucent acrokeratoderma have similar lesions and overlapping histologic changes, they may be considered variants of the same entity.4 Keratoelastoidosis marginalis, also called degenerative collagenous plaques of the hand, mainly affects white individuals aged 40 to 60 years with a history of prolonged sun exposure. Papules often are distributed over the junction of the dorsal and palmar skin and less often on the ulnar sides of the hands. The clinical lesions are similar to those in our patient, but histopathology of keratoelastoidosis marginalis shows amorphous, basophilic, elastotic masses and thickened, fragmented, calcified elastic fibers in the upper and mid dermis.
Therapies including liquid nitrogen, topical salicylic acid, methotrexate, dapsone, tar, cryotherapy, systemic prednisone, retinoic acid, clobetasone cream,5 and erbium:YAG laser10 have been applied. Thus far, no optimal treatment has been recommended and no tendency of spontaneous resolution has been previously reported in the literature. Our patient responded to tretinoin, but the lesions recurred after withdrawal of the medication; therefore, tretinoin may not be an optimal treatment option. Because the lesions are limited to the skin and AKE is only considered a cosmetic problem with a good prognosis, we recommend a wait-and-watch approach.
Acknowledgment—We thank Rashmi Sarkar, MD, New Delhi, India, for her assistance.
To the Editor:
Acrokeratoelastoidosis (AKE) is a rare disease first described by Costa1 in 1953. Typically it is only a cosmetic nuisance in the majority of patients and presents as asymptomatic, small, firm, flesh-colored to yellowish, round to polygonal papules with occasional keratosis or umbilication on the radial and ulnar margins of the hands and/or feet.1-3 In some cases, the lesions occur on the anterior aspects of the wrists, fingers, or lower legs.1 The lesions are always bilaterally distributed. Acrokeratoelastoidosis is a chronic skin disorder that commonly presents during childhood or adolescence, but presentation in adulthood also has been described.3 Histologically, AKE always shows hyperkeratosis, acanthosis, decrease of elastic tissue, and elastorrhexis of remaining elastic fibers. Plaque-type lesions are rare. We describe a patient who presented with plaques on the radial and ulnar margins of the hands.
A 36-year-old Chinese woman presented with asymptomatic, small, firm papules of 6 months’ duration that initially developed on the hands and gradually increased in number, coalescing into plaques. The feet were spared. She had no medical history of hyperhidrosis, chronic trauma, friction, or excessive sun exposure, and no family history of similar symptoms. No prior therapy had been attempted.
Physical examination showed nonconfluent, firm, flesh-colored to yellowish, translucent, smooth papules with wavy edges that were symmetrically distributed on the radial and ulnar margins of the hands; some papules had coalesced into plaques (Figure 1). A biopsy specimen taken from a plaque on the hypothenar eminence of the right hand revealed focal hyperkeratosis, hypergranulosis, acanthosis, and mild chronic inflammation with hematoxylin and eosin stain (Figure 2A). Aldehyde fuchsin staining showed fragmented and rarefied elastic fibers in the reticular dermis (Figure 2B). The patient was diagnosed with AKE. Oral tretinoin 10 mg twice daily was initiated and resulted in an evident response after 2 weeks of treatment. However, the patient stopped taking the medication because of pruritus and dry skin and the lesions then reappeared.
|
|
| Figure 1. Nonconfluent, firm, flesh-colored to yellowish, translucent, smooth papules distributed on the radial margin of the hand; some of papules coalesced into plaques. |
|
|
|
|
| Figure 2. Histopathology revealed hyperkeratosis, hypergranulosis, acanthosis, and mild chronic inflammation (A) (H&E, original magnification ×250 [inset, original magnification ×400]), as well as fragmented elastic fibers in the reticular dermis (B) (Aldehyde fuchsin, original magnification ×400 [inset, original magnification ×100]). |
Acrokeratoelastoidosis is a rare keratotic disorder. It seems to have no racial or ethnic predilection and occurs more frequently in women.4,5 It also is rare in China, with few cases reported, all women.5 The reason for the gender predilection in China remains unknown. The course is chronic, but it may rapidly progress during pregnancy.6
The pathogenesis of AKE is still unresolved.2,3 Although many cases are sporadic,5 it appears to be inherited in an autosomal-dominant fashion, most likely related to chromosome 2.7 Typically, AKE presents as papules that are discrete and bilaterally distributed in the palmoplantar margins,2,3 but some of the papules in our patient coalesced into plaques, which is unique. The histologic hallmarks indicated that the lesions were AKE.
The differential diagnosis of AKE includes hereditary papulotranslucent acrokeratoderma, focal acral hyperkeratosis, and keratoelastoidosis marginalis.8 Hereditary papulotranslucent acrokeratoderma also is inherited in an autosomal-dominant fashion and shares similar acral, translucent, keratotic papules with AKE, but there is no chronic inflammatory cell infiltrate, degeneration of collagenous fibers, or fragmentation of elastic fibers. The clinical appearance of focal acral hyperkeratosis is similar to AKE, but no changes are revealed in the elastic tissue.9 Because AKE, focal acral hyperkeratosis, and hereditary papulotranslucent acrokeratoderma have similar lesions and overlapping histologic changes, they may be considered variants of the same entity.4 Keratoelastoidosis marginalis, also called degenerative collagenous plaques of the hand, mainly affects white individuals aged 40 to 60 years with a history of prolonged sun exposure. Papules often are distributed over the junction of the dorsal and palmar skin and less often on the ulnar sides of the hands. The clinical lesions are similar to those in our patient, but histopathology of keratoelastoidosis marginalis shows amorphous, basophilic, elastotic masses and thickened, fragmented, calcified elastic fibers in the upper and mid dermis.
Therapies including liquid nitrogen, topical salicylic acid, methotrexate, dapsone, tar, cryotherapy, systemic prednisone, retinoic acid, clobetasone cream,5 and erbium:YAG laser10 have been applied. Thus far, no optimal treatment has been recommended and no tendency of spontaneous resolution has been previously reported in the literature. Our patient responded to tretinoin, but the lesions recurred after withdrawal of the medication; therefore, tretinoin may not be an optimal treatment option. Because the lesions are limited to the skin and AKE is only considered a cosmetic problem with a good prognosis, we recommend a wait-and-watch approach.
Acknowledgment—We thank Rashmi Sarkar, MD, New Delhi, India, for her assistance.
1. Costa OG. Akrokerato-elastoidosis; a hitherto undescribed skin disease. Dermatologica. 1953;107:164-168.
2. Bogle MA, Huang LY, Tschen JA. Acrokeratoelastoidosis. J Am Acad Dermatol. 2002;47:448-451.
3. Highet AS, Rook A, Anderson JR. Acrokeratoelastoidosis. Br J Dermatol. 1982;106:337-344.
4. Abulafia J, Vignale RA. Degenerative collagenous
plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
5. Luo DQ, Zhang B, Huang YB, et al. Papules on a young woman’s hands and feet. Clin Exp Dermatol. 2010;35:451-452.
6. Nelson-Adesokan P, Mallory SB, Lombardi C, et al. Acrokeratoelastoidosis of Costa [published correction appears in Int J Dermatol. 1996;35:380]. Int J Dermatol. 1995;34:431-433.
7. Greiner J, Krüger J, Palden L, et al. A linkage study of acrokeratoelastoidosis. possible mapping to chromosome 2. Hum Genet. 1983;63:222-227.
8. Hu W, Cook TF, Vicki GJ, et al. Acrokeratoelastoidosis. Pediatr Dermatol. 2002;19:320-322.
9. Dowd PM, Harman RR, Black MM. Focal acral hyperkeratosis. Br J Dermatol. 1983;109:97-103.
10. Erbil AH, Sezer E, Koç E, et al. Acrokeratoelastoidosis treated with the erbium:YAG laser [published online ahead of print November 3, 2007]. Clin Exp Dermatol. 2008;33:30-31.
1. Costa OG. Akrokerato-elastoidosis; a hitherto undescribed skin disease. Dermatologica. 1953;107:164-168.
2. Bogle MA, Huang LY, Tschen JA. Acrokeratoelastoidosis. J Am Acad Dermatol. 2002;47:448-451.
3. Highet AS, Rook A, Anderson JR. Acrokeratoelastoidosis. Br J Dermatol. 1982;106:337-344.
4. Abulafia J, Vignale RA. Degenerative collagenous
plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
5. Luo DQ, Zhang B, Huang YB, et al. Papules on a young woman’s hands and feet. Clin Exp Dermatol. 2010;35:451-452.
6. Nelson-Adesokan P, Mallory SB, Lombardi C, et al. Acrokeratoelastoidosis of Costa [published correction appears in Int J Dermatol. 1996;35:380]. Int J Dermatol. 1995;34:431-433.
7. Greiner J, Krüger J, Palden L, et al. A linkage study of acrokeratoelastoidosis. possible mapping to chromosome 2. Hum Genet. 1983;63:222-227.
8. Hu W, Cook TF, Vicki GJ, et al. Acrokeratoelastoidosis. Pediatr Dermatol. 2002;19:320-322.
9. Dowd PM, Harman RR, Black MM. Focal acral hyperkeratosis. Br J Dermatol. 1983;109:97-103.
10. Erbil AH, Sezer E, Koç E, et al. Acrokeratoelastoidosis treated with the erbium:YAG laser [published online ahead of print November 3, 2007]. Clin Exp Dermatol. 2008;33:30-31.

Hailey-Hailey Disease
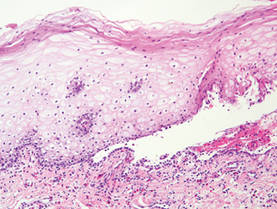
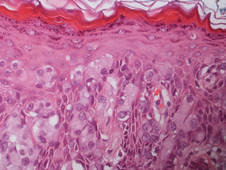
Hailey-Hailey disease (HHD), or benign familial chronic pemphigus, typically presents as suprabasal blisters with a perivascular and interstitial lymphocytic infiltrate (Figure 1).1 Villi, or elongated dermal papillae lined with a single layer of basal cells, protrude into the bullae (Figure 2). In HHD lesions, the epidermis is thickened with scale-crust, and at least the lower half of the epidermis shows acantholysis. Despite the acantholytic changes, a few intact intercellular bridges remain, giving the appearance of a dilapidated brick wall (Figure 2). There may be dyskeratotic cells among the acantholytic cells, though they are scant in many cases. These acantholytic dyskeratotic cells have eosinophilic polygonal-shaped cytoplasm. Hailey-Hailey disease typically does not show adnexal extension of the acantholysis. Direct immunofluorescence is negative in HHD.
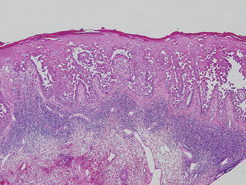
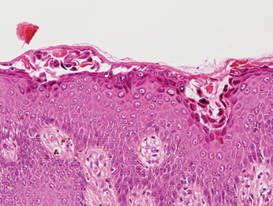
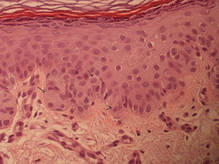
Pemphigus vulgaris is an autoimmune intraepidermal bullous disease that presents with suprabasal acantholysis (Figure 3).2 The epidermis is not thickened and acantholysis is limited to the suprabasal layer. Acantholytic cells with eosinophils and/or neutrophils are found within the bullae. Perivascular and interstitial infiltrates of lymphocytes, eosinophils, and occasionally neutrophils are seen; however, the inflammatory cell infiltrate can vary from extensive to scant. Direct immunofluorescence usually reveals IgG and/or C3 deposition on the surface of the keratinocytes throughout the epidermis.
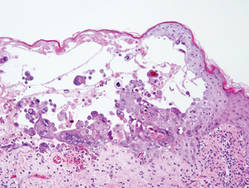
Pemphigus foliaceus is another autoimmune intraepidermal bullous disease that is characterized by acantholysis in the granular or upper spinous layers (Figure 4).3 The epidermis is not thickened. Sometimes acantholytic cells show dyskeratotic change (Figure 4). Some biopsy specimens do not contain the roof of the bullae; therefore, only erosion is seen and the diagnosis may be missed. Moreover, when only the adnexal epithelium shows acantholysis without epidermal involvement, diagnosis can be difficult.4 Acantholysis is accompanied with a superficial perivascular and interstitial inflammatory cell infiltrate consisting of lymphocytes, eosinophils, and occasionally neutrophils. The amount of inflammatory cell infiltrate may vary. Bullous impetigo and staphylococcal scalded skin syndrome reveal a similar histopathologic pattern. Direct immunofluorescence usually discloses IgG and/or C3 deposition on cell surfaces of keratinocytes in the entire or upper epidermis.
Herpesvirus infection shows ballooning (intracellular edema) of keratinocytes. Eventually acantholysis occurs and intraepidermal bullae are formed. In the bullae, virus-associated acantholytic keratinocytes, some that are multinucleated, can be easily found (Figure 5).5 These cells are larger than normal keratinocytes and have steel gray nuclei with peripheral accentuation. Some of these cells are necrotic, and the remains of necrotic multinucleated acantholytic cells are easily recognized. Adnexal epithelial cells occasionally are affected by herpesvirus infection; nuclear change is similar to the epidermis. A perivascular and interstitial infiltrate of lymphocytes and neutrophils is seen. Neutrophils accumulate within the old bullae, clinically manifesting as a pustule.
Darier disease is characterized by suprabasal clefts and acantholysis above the basal layer (Figure 6).6 Similar to HHD, villi protrude within the clefts (Figure 6). Conspicuous columns of parakeratosis above the acantholytic epidermis often are observed. Dyskeratotic cells exist among acantholytic ke-ratinocytes in the granular layer and parakeratotic column, which are known as corps ronds and crops grains, respectively. A scant to moderate lymphocytic infiltrate is found in the upper dermis.
- Hernandez-Perez E. Familial benign chronic pemphigus. Cutis. 1987;39:75-77.
- Venugopal SS, Murrell DF. Diagnosis and clinical features of pemphigus vulgaris. Dermatol Clin. 2011;29:373-380, vii.
- Dasher D, Rubenstein D, Diaz LA. Pemphigus foliaceus. Curr Dir Autoimmun. 2008;10:182-194.
- Ohata C, Akamatsu K, Imai N, et al. Localized pemphigus foliaceus exclusively involving the follicular infundibulum: a novel peau d’orange appearance. Eur J Dermatol. 2011;21:392-395.
- King DF, King LA. Giant cells in lesions of varicella and herpes zoster. Am J Dermatopathol. 1986;8:456-458.
- Burge S. Management of Darier’s disease. Clin Exp Dermatol. 1999;24:53-56.
Hailey-Hailey disease (HHD), or benign familial chronic pemphigus, typically presents as suprabasal blisters with a perivascular and interstitial lymphocytic infiltrate (Figure 1).1 Villi, or elongated dermal papillae lined with a single layer of basal cells, protrude into the bullae (Figure 2). In HHD lesions, the epidermis is thickened with scale-crust, and at least the lower half of the epidermis shows acantholysis. Despite the acantholytic changes, a few intact intercellular bridges remain, giving the appearance of a dilapidated brick wall (Figure 2). There may be dyskeratotic cells among the acantholytic cells, though they are scant in many cases. These acantholytic dyskeratotic cells have eosinophilic polygonal-shaped cytoplasm. Hailey-Hailey disease typically does not show adnexal extension of the acantholysis. Direct immunofluorescence is negative in HHD.
Pemphigus vulgaris is an autoimmune intraepidermal bullous disease that presents with suprabasal acantholysis (Figure 3).2 The epidermis is not thickened and acantholysis is limited to the suprabasal layer. Acantholytic cells with eosinophils and/or neutrophils are found within the bullae. Perivascular and interstitial infiltrates of lymphocytes, eosinophils, and occasionally neutrophils are seen; however, the inflammatory cell infiltrate can vary from extensive to scant. Direct immunofluorescence usually reveals IgG and/or C3 deposition on the surface of the keratinocytes throughout the epidermis.
Pemphigus foliaceus is another autoimmune intraepidermal bullous disease that is characterized by acantholysis in the granular or upper spinous layers (Figure 4).3 The epidermis is not thickened. Sometimes acantholytic cells show dyskeratotic change (Figure 4). Some biopsy specimens do not contain the roof of the bullae; therefore, only erosion is seen and the diagnosis may be missed. Moreover, when only the adnexal epithelium shows acantholysis without epidermal involvement, diagnosis can be difficult.4 Acantholysis is accompanied with a superficial perivascular and interstitial inflammatory cell infiltrate consisting of lymphocytes, eosinophils, and occasionally neutrophils. The amount of inflammatory cell infiltrate may vary. Bullous impetigo and staphylococcal scalded skin syndrome reveal a similar histopathologic pattern. Direct immunofluorescence usually discloses IgG and/or C3 deposition on cell surfaces of keratinocytes in the entire or upper epidermis.
Herpesvirus infection shows ballooning (intracellular edema) of keratinocytes. Eventually acantholysis occurs and intraepidermal bullae are formed. In the bullae, virus-associated acantholytic keratinocytes, some that are multinucleated, can be easily found (Figure 5).5 These cells are larger than normal keratinocytes and have steel gray nuclei with peripheral accentuation. Some of these cells are necrotic, and the remains of necrotic multinucleated acantholytic cells are easily recognized. Adnexal epithelial cells occasionally are affected by herpesvirus infection; nuclear change is similar to the epidermis. A perivascular and interstitial infiltrate of lymphocytes and neutrophils is seen. Neutrophils accumulate within the old bullae, clinically manifesting as a pustule.
Darier disease is characterized by suprabasal clefts and acantholysis above the basal layer (Figure 6).6 Similar to HHD, villi protrude within the clefts (Figure 6). Conspicuous columns of parakeratosis above the acantholytic epidermis often are observed. Dyskeratotic cells exist among acantholytic ke-ratinocytes in the granular layer and parakeratotic column, which are known as corps ronds and crops grains, respectively. A scant to moderate lymphocytic infiltrate is found in the upper dermis.
Hailey-Hailey disease (HHD), or benign familial chronic pemphigus, typically presents as suprabasal blisters with a perivascular and interstitial lymphocytic infiltrate (Figure 1).1 Villi, or elongated dermal papillae lined with a single layer of basal cells, protrude into the bullae (Figure 2). In HHD lesions, the epidermis is thickened with scale-crust, and at least the lower half of the epidermis shows acantholysis. Despite the acantholytic changes, a few intact intercellular bridges remain, giving the appearance of a dilapidated brick wall (Figure 2). There may be dyskeratotic cells among the acantholytic cells, though they are scant in many cases. These acantholytic dyskeratotic cells have eosinophilic polygonal-shaped cytoplasm. Hailey-Hailey disease typically does not show adnexal extension of the acantholysis. Direct immunofluorescence is negative in HHD.
Pemphigus vulgaris is an autoimmune intraepidermal bullous disease that presents with suprabasal acantholysis (Figure 3).2 The epidermis is not thickened and acantholysis is limited to the suprabasal layer. Acantholytic cells with eosinophils and/or neutrophils are found within the bullae. Perivascular and interstitial infiltrates of lymphocytes, eosinophils, and occasionally neutrophils are seen; however, the inflammatory cell infiltrate can vary from extensive to scant. Direct immunofluorescence usually reveals IgG and/or C3 deposition on the surface of the keratinocytes throughout the epidermis.
Pemphigus foliaceus is another autoimmune intraepidermal bullous disease that is characterized by acantholysis in the granular or upper spinous layers (Figure 4).3 The epidermis is not thickened. Sometimes acantholytic cells show dyskeratotic change (Figure 4). Some biopsy specimens do not contain the roof of the bullae; therefore, only erosion is seen and the diagnosis may be missed. Moreover, when only the adnexal epithelium shows acantholysis without epidermal involvement, diagnosis can be difficult.4 Acantholysis is accompanied with a superficial perivascular and interstitial inflammatory cell infiltrate consisting of lymphocytes, eosinophils, and occasionally neutrophils. The amount of inflammatory cell infiltrate may vary. Bullous impetigo and staphylococcal scalded skin syndrome reveal a similar histopathologic pattern. Direct immunofluorescence usually discloses IgG and/or C3 deposition on cell surfaces of keratinocytes in the entire or upper epidermis.
Herpesvirus infection shows ballooning (intracellular edema) of keratinocytes. Eventually acantholysis occurs and intraepidermal bullae are formed. In the bullae, virus-associated acantholytic keratinocytes, some that are multinucleated, can be easily found (Figure 5).5 These cells are larger than normal keratinocytes and have steel gray nuclei with peripheral accentuation. Some of these cells are necrotic, and the remains of necrotic multinucleated acantholytic cells are easily recognized. Adnexal epithelial cells occasionally are affected by herpesvirus infection; nuclear change is similar to the epidermis. A perivascular and interstitial infiltrate of lymphocytes and neutrophils is seen. Neutrophils accumulate within the old bullae, clinically manifesting as a pustule.
Darier disease is characterized by suprabasal clefts and acantholysis above the basal layer (Figure 6).6 Similar to HHD, villi protrude within the clefts (Figure 6). Conspicuous columns of parakeratosis above the acantholytic epidermis often are observed. Dyskeratotic cells exist among acantholytic ke-ratinocytes in the granular layer and parakeratotic column, which are known as corps ronds and crops grains, respectively. A scant to moderate lymphocytic infiltrate is found in the upper dermis.
- Hernandez-Perez E. Familial benign chronic pemphigus. Cutis. 1987;39:75-77.
- Venugopal SS, Murrell DF. Diagnosis and clinical features of pemphigus vulgaris. Dermatol Clin. 2011;29:373-380, vii.
- Dasher D, Rubenstein D, Diaz LA. Pemphigus foliaceus. Curr Dir Autoimmun. 2008;10:182-194.
- Ohata C, Akamatsu K, Imai N, et al. Localized pemphigus foliaceus exclusively involving the follicular infundibulum: a novel peau d’orange appearance. Eur J Dermatol. 2011;21:392-395.
- King DF, King LA. Giant cells in lesions of varicella and herpes zoster. Am J Dermatopathol. 1986;8:456-458.
- Burge S. Management of Darier’s disease. Clin Exp Dermatol. 1999;24:53-56.
- Hernandez-Perez E. Familial benign chronic pemphigus. Cutis. 1987;39:75-77.
- Venugopal SS, Murrell DF. Diagnosis and clinical features of pemphigus vulgaris. Dermatol Clin. 2011;29:373-380, vii.
- Dasher D, Rubenstein D, Diaz LA. Pemphigus foliaceus. Curr Dir Autoimmun. 2008;10:182-194.
- Ohata C, Akamatsu K, Imai N, et al. Localized pemphigus foliaceus exclusively involving the follicular infundibulum: a novel peau d’orange appearance. Eur J Dermatol. 2011;21:392-395.
- King DF, King LA. Giant cells in lesions of varicella and herpes zoster. Am J Dermatopathol. 1986;8:456-458.
- Burge S. Management of Darier’s disease. Clin Exp Dermatol. 1999;24:53-56.

Multicentric Primary Extramammary Paget Disease: A Toker Cell Disorder?
Extramammary Paget disease (EMPD), which was first described by Crocker1 in a patient with erythematous patches on the penis and scrotum, is morphologically identical to mammary Paget disease (MPD) of the nipple. The principal difference between EMPD and MPD is anatomic location. Extramammary Paget disease predominantly affects apocrine gland–bearing areas including the vulva, scrotum, and perianal areas. Although EMPD is not a common condition, it must be considered in the differential diagnosis for patients with chronic genital or perianal dermatitis. Primary EMPD must be distinguished from secondary epithelial involvement by an underlying invasive carcinoma that originates from sites such as the gastrointestinal or genitourinary systems (secondary EMPD).
Although multicentric primary EMPD is not uncommon among Eastern Asians, as there have been several reports in the literature from Japan,2-5 multicentric EMPD in white individuals is rare.6 We report a case of primary EMPD that was established when no underlying malignancies were detected.
Case Report
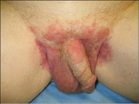
A 63-year-old white man presented to the dermatology clinic with a pruritic rash involving the groin of 8 years’ duration. Over-the-counter antifungal agents provided no improvement. Confluent erythematous and macerated plaques on the scrotum, shaft of the penis, bilateral inguinal areas, and perineum were noted on clinical examination (Figure 1). There also were well-demarcated, 4×5-cm, erythematous, velvety plaques on the right axilla and a small erythematous plaque on the left axilla. Systemic workup including colonoscopy, cystoscopy, and magnetic resonance imaging of the abdomen and pelvis were unremarkable. A positron emission tomography–computed tomography scan revealed foci of hypermetabolic activity in the bilateral inguinal nodes that were interpreted as evidence of an inflammatory process.
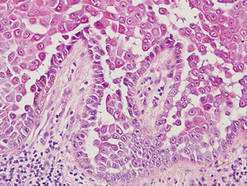
Fourteen biopsies were taken from the clinically involved regions in the genital area to delineate the extent of involvement. They all showed intraepidermal carcinoma characterized by large cells with clear cytoplasm and large hyperchromatic nuclei throughout the epidermis, which were more abundant in the basilar and lower portions of the epidermis (Figure 2). There was no evidence of underlying carcinoma in the dermis.
On immunohistochemical staining, the neoplastic cells were diffusely and strongly positive for cytokeratin 7 (Figure 3) and moderately positive for carcinoembryonic antigen, estrogen and progesterone receptors, and ERBB2 (formerly HER-2/neu). Additional immunostaining for cytokeratin 20 and gross cystic disease fluid protein 15 were negative. High-molecular-weight keratin staining highlighted background epithelium and spared neoplastic cells. Mucicarmine staining highlighted mucin within the cytoplasm of neoplastic cells. Ultrastructural studies showed evidence of apocrine differentiation in the neoplastic cells with deeply indented, bean-shaped nuclei and clear cytoplasm with inconspicuous organelles. Clinically unaffected skin from the periumbilical region also was biopsied. The histology demonstrated Toker cells characterized by clear cytoplasm, small vesicular nuclei, and the absence of hyperchromasia (Figure 4).

The patient was treated with 6 cycles of 5-aminolevulinic acid 20% photodynamic therapy but showed no improvement. Imiquimod cream 5% resulted in mild clinical improvement in the axillae but caused severe irritation in the groin and was discontinued after 3 months. A radiology consultation obtained 6 years prior to presentation determined that the risk associated with radiation therapy outweighed any possible benefits. Although localized disease persisted, repeated biopsies and positron emission tomography–computed tomography scans did not show any evidence of invasion or metastasis, respectively. Three years after initial presentation, groin lesions progressed to become more erythematous and vegetative. The corresponding histology demonstrated EMPD. An ancillary test (polymerase chain reaction analysis) for low- and high-risk human papillomavirus was negative.
Comment
Primary EMPD has been well documented in the literature to have a favorable prognosis if adequately treated. The few cases reported from Japan on EMPD without underlying adenocarcinoma had good outcomes.2,3 Further reports of multicentric EMPD involving 2 or 3 sites in the axillae and/or genital region did not reveal any progression to invasive carcinomas after adequate follow-up.3-9 Indeed, many of these cases, particularly those in the genital region, were aggressively treated with surgery, topical chemotherapy, immunomodulatory agents, and radiation; therefore, their natural course is not known.2 A Japanese studyconducted at 75 medical institutions (1987-1991) included 46 EMPD patients with multiple (ie, 2 or 3) sites of involvement.10 Some of the patients with a combination of genital and axillary lesions were followed without any treatment. None of the EMPD patients with axillary involvement developed invasive carcinoma after 4- to 12-year follow-up.10
Toker cells, which were first described in 1970,11 have been recognized as precursors to EMPD.12-14 Toker cells are intraepithelial cells with clear to pale-staining cytoplasm that are smaller in size than Paget cells but larger than neighboring keratinocytes. They are found in approximately 10% of normal nipples.11,14 Toker cells show vesicular chromatin, whereas Paget cells are hyperchromatic with prominent nucleoli.12 Willman et al15 examined 11 vulvectomies for the presence of Toker cells in association with mammarylike glands of the vulva. They demonstrated the presence of Toker cells in 4 (36%) of the samples.15 Additionally, Van der Putte et al16 observed Toker cells in an areolar lesion in a 47-year-old woman with MPD without underlying adenocarcinoma, suggesting that cases of MPD and EMPD confined to epithelial cells may be derived from Toker cells. Toker cells have been associated in the pathogenesis of 2 other benign entities, including clear cell papulosis17,18 and cutaneous hamartoma with pagetoid cells.19
Primary EMPD has the potential to develop in several regions of the skin that contain apocrine glands. Our patient presented with persistent genital lesions for many years without any concerns of axillary disease. Interestingly, biopsies from normal-appearing skin in the periumbilical region revealed clear cells (Figure 4). Likewise, Toker cells have been described in biopsies from clinically unaffected skin of the axillae in some cases where EMPD was identified in the genital regions.3,5 Genital lesions were the main clinical presentation and preceded axillary involvement in most instances.3,5,9 Therefore, biopsies from uninvolved apocrine sites (ie, axillary and periumbilical skin) should be considered in these patients. Based on our observation, we speculate that multicentric primary EMPD starts with Toker cell hyperplasia with a propensity to evolve into neoplasia in sites with apocrine or mammarylike glands.
Primary and secondary EMPD cannot be distinguished by histopathology and immunohistochemistry has a limited role.20,21 However, immunostaining with CDX2 (a caudal-type homeobox protein) might be helpful in differentiating primary EMPD from secondary EMPD extending from underlying anorectal adenocarcinomas.22 Primary EMPD has been treated with surgical excision.2,3,8 Despite the high recurrence rate after surgery, the prognosis for localized primary EMPD disease has been favorable.
Conclusion
Our case suggests that Toker cell hyperplasia is a precursor to primary EMPD. In patients with established EMPD, other apocrine gland–bearing areas should be examined for multicentric disease. Lastly, the clinical course in our patient supports the hypothesis that primary multicentric EMPD has a favorable outcome. The Table lists the characteristics of MPD and EMPD and describes their development with respect to Toker cells. More studies are required to further outline the cytologic characteristics of Toker cells and to distinguish primary EMPD from secondary EMPD.

1. Crocker HR. Paget’s disease affecting the scrotum and penis. Trans Path Soc London. 1889;40:187-191.
2. Kawatsu T, Miki Y. Triple extramammary Paget’s disease. Arch Dermatol. 1971;104:316-319.
3. Makino T, Nakamura S, Nakayama H, et al. Genital Paget’s disease with clear cells in the epidermis of the axilla. J Cutan Pathol. 1998;25:568-571.
4. Inui S, Fukuhara S, Asada H, et al. Double involvement of extramammary Paget’s disease in the genitalia and axilla. J Dermatol. 2000;27:409-412.
5. Kitajima S, Yamamoto K, Tsuji T, et al. Triple extramammary Paget’s disease. Dermatol Surg. 1997;23:1035-1038.
6. Van Hamme C, Marot L, Dachelet C, et al. Paget’s extramammary disease of the axillae and perineum [in French]. Ann Dermatol Venereol. 2002;129(5, pt 1):717-719.
7. Kao GF, Graham JH, Helwig EB. Paget’s disease of the ectopic breast with an underlying intraductal carcinoma: report of a case. J Cutan Pathol. 1986;13:59-66.
8. Murrell TW Jr, McMullan FH. Extramammary Paget’s disease. a report of two cases. Arch Dermatol. 1962;85:600-613.
9. Koseki S, Mitsuhashi Y, Yoshikawa K, et al. A case of triple extramammary Paget’s disease. J Dermatol. 1997;24:535-538.
10. Ishihara K. Statistical study of extramammary Paget’s disease in Japan [in Japanese]. Skin Cancer. 1994;9:38.
11. Toker C. Clear cells of the nipple epidermis. Cancer. 1970;25:601-610.
12. Marucci G, Betts CM, Golouh R, et al. Toker cells are probably precursors of Paget cell carcinoma: a morphological and ultrastructural description. Virchows Archiv. 2002;441:117-123.
13. Belousova IE, Kazakov DV, Michal M, et al. Vulvar toker cells: the long-awaited missing link: a proposal for an origin-based histogenetic classification of extramammary Paget disease. Am J Dermatopathol. 2006;28:84-86.
14. Val-Bernal JF. Diego C, Rodriguez-Villar D, et al. The nipple-areola complex epidermis: a prospective systemic study in adult autopsies. Am J Dermatopathology. 2010;32:787-93.
15. Willman JH, Golitz LE, Fitzpatrick JE. Vulvar clear cells of Toker: precursors of extramammary Paget’s disease. Am J Dermatopathol. 2005;27:185-188.
16. Van der Putte SC, Toonstra J, Hennipman A. Mammary Paget’s disease confined to the areola and associated with multifocal Toker cell hyperplasia. Am J Dermatopathol. 1995;17:487-493.
17. Kuo TT, Chan HL, Hsueh S. Clear cell papulosis of the skin. a new entity with histogenetic implications for cutaneous Paget’s disease. Am J Surg Pathol. 1987;11:827-834.
18. Kuo TT, Huang CL, Chan HL, et al. Clear cell papulosis: report of three cases of a newly recognized disease. J Am Acad Dermatol. 1995;33(2, pt 1):230-233.
19. Piérard-Franchimont C, Dosal FL, Estrada JA, et al. Cutaneous hamartoma with pagetoid cells. Am J Dermatopathol. 1991;13:158-161.
20. Belcher RW. Extramammary Paget’s disease. enzyme histochemical and electron microscopic study. Arch Pathol. 1972;94:59-64.
21. Ordóñez NG, Awalt H, Mackay B. Mammary and extramammary Paget’s disease. an immunocytochemical and ultrastructural study. Cancer. 1987;59:1173-1183.
22. Perrotto J, Abbott JJ, Ceilley RI, et al. The role of immunohistochemistry in discriminating primary from secondary extramammary Paget disease. Am J Dermatopathol. 2010;32:137-143.
23. Jones RE Jr, Austin C, Ackerman AB. Extramammary Paget’s disease. a critical reexamination. Am J Dermatopathol. 1979;1:101-132.
Extramammary Paget disease (EMPD), which was first described by Crocker1 in a patient with erythematous patches on the penis and scrotum, is morphologically identical to mammary Paget disease (MPD) of the nipple. The principal difference between EMPD and MPD is anatomic location. Extramammary Paget disease predominantly affects apocrine gland–bearing areas including the vulva, scrotum, and perianal areas. Although EMPD is not a common condition, it must be considered in the differential diagnosis for patients with chronic genital or perianal dermatitis. Primary EMPD must be distinguished from secondary epithelial involvement by an underlying invasive carcinoma that originates from sites such as the gastrointestinal or genitourinary systems (secondary EMPD).
Although multicentric primary EMPD is not uncommon among Eastern Asians, as there have been several reports in the literature from Japan,2-5 multicentric EMPD in white individuals is rare.6 We report a case of primary EMPD that was established when no underlying malignancies were detected.
Case Report
A 63-year-old white man presented to the dermatology clinic with a pruritic rash involving the groin of 8 years’ duration. Over-the-counter antifungal agents provided no improvement. Confluent erythematous and macerated plaques on the scrotum, shaft of the penis, bilateral inguinal areas, and perineum were noted on clinical examination (Figure 1). There also were well-demarcated, 4×5-cm, erythematous, velvety plaques on the right axilla and a small erythematous plaque on the left axilla. Systemic workup including colonoscopy, cystoscopy, and magnetic resonance imaging of the abdomen and pelvis were unremarkable. A positron emission tomography–computed tomography scan revealed foci of hypermetabolic activity in the bilateral inguinal nodes that were interpreted as evidence of an inflammatory process.
Fourteen biopsies were taken from the clinically involved regions in the genital area to delineate the extent of involvement. They all showed intraepidermal carcinoma characterized by large cells with clear cytoplasm and large hyperchromatic nuclei throughout the epidermis, which were more abundant in the basilar and lower portions of the epidermis (Figure 2). There was no evidence of underlying carcinoma in the dermis.
On immunohistochemical staining, the neoplastic cells were diffusely and strongly positive for cytokeratin 7 (Figure 3) and moderately positive for carcinoembryonic antigen, estrogen and progesterone receptors, and ERBB2 (formerly HER-2/neu). Additional immunostaining for cytokeratin 20 and gross cystic disease fluid protein 15 were negative. High-molecular-weight keratin staining highlighted background epithelium and spared neoplastic cells. Mucicarmine staining highlighted mucin within the cytoplasm of neoplastic cells. Ultrastructural studies showed evidence of apocrine differentiation in the neoplastic cells with deeply indented, bean-shaped nuclei and clear cytoplasm with inconspicuous organelles. Clinically unaffected skin from the periumbilical region also was biopsied. The histology demonstrated Toker cells characterized by clear cytoplasm, small vesicular nuclei, and the absence of hyperchromasia (Figure 4).
The patient was treated with 6 cycles of 5-aminolevulinic acid 20% photodynamic therapy but showed no improvement. Imiquimod cream 5% resulted in mild clinical improvement in the axillae but caused severe irritation in the groin and was discontinued after 3 months. A radiology consultation obtained 6 years prior to presentation determined that the risk associated with radiation therapy outweighed any possible benefits. Although localized disease persisted, repeated biopsies and positron emission tomography–computed tomography scans did not show any evidence of invasion or metastasis, respectively. Three years after initial presentation, groin lesions progressed to become more erythematous and vegetative. The corresponding histology demonstrated EMPD. An ancillary test (polymerase chain reaction analysis) for low- and high-risk human papillomavirus was negative.
Comment
Primary EMPD has been well documented in the literature to have a favorable prognosis if adequately treated. The few cases reported from Japan on EMPD without underlying adenocarcinoma had good outcomes.2,3 Further reports of multicentric EMPD involving 2 or 3 sites in the axillae and/or genital region did not reveal any progression to invasive carcinomas after adequate follow-up.3-9 Indeed, many of these cases, particularly those in the genital region, were aggressively treated with surgery, topical chemotherapy, immunomodulatory agents, and radiation; therefore, their natural course is not known.2 A Japanese studyconducted at 75 medical institutions (1987-1991) included 46 EMPD patients with multiple (ie, 2 or 3) sites of involvement.10 Some of the patients with a combination of genital and axillary lesions were followed without any treatment. None of the EMPD patients with axillary involvement developed invasive carcinoma after 4- to 12-year follow-up.10
Toker cells, which were first described in 1970,11 have been recognized as precursors to EMPD.12-14 Toker cells are intraepithelial cells with clear to pale-staining cytoplasm that are smaller in size than Paget cells but larger than neighboring keratinocytes. They are found in approximately 10% of normal nipples.11,14 Toker cells show vesicular chromatin, whereas Paget cells are hyperchromatic with prominent nucleoli.12 Willman et al15 examined 11 vulvectomies for the presence of Toker cells in association with mammarylike glands of the vulva. They demonstrated the presence of Toker cells in 4 (36%) of the samples.15 Additionally, Van der Putte et al16 observed Toker cells in an areolar lesion in a 47-year-old woman with MPD without underlying adenocarcinoma, suggesting that cases of MPD and EMPD confined to epithelial cells may be derived from Toker cells. Toker cells have been associated in the pathogenesis of 2 other benign entities, including clear cell papulosis17,18 and cutaneous hamartoma with pagetoid cells.19
Primary EMPD has the potential to develop in several regions of the skin that contain apocrine glands. Our patient presented with persistent genital lesions for many years without any concerns of axillary disease. Interestingly, biopsies from normal-appearing skin in the periumbilical region revealed clear cells (Figure 4). Likewise, Toker cells have been described in biopsies from clinically unaffected skin of the axillae in some cases where EMPD was identified in the genital regions.3,5 Genital lesions were the main clinical presentation and preceded axillary involvement in most instances.3,5,9 Therefore, biopsies from uninvolved apocrine sites (ie, axillary and periumbilical skin) should be considered in these patients. Based on our observation, we speculate that multicentric primary EMPD starts with Toker cell hyperplasia with a propensity to evolve into neoplasia in sites with apocrine or mammarylike glands.
Primary and secondary EMPD cannot be distinguished by histopathology and immunohistochemistry has a limited role.20,21 However, immunostaining with CDX2 (a caudal-type homeobox protein) might be helpful in differentiating primary EMPD from secondary EMPD extending from underlying anorectal adenocarcinomas.22 Primary EMPD has been treated with surgical excision.2,3,8 Despite the high recurrence rate after surgery, the prognosis for localized primary EMPD disease has been favorable.
Conclusion
Our case suggests that Toker cell hyperplasia is a precursor to primary EMPD. In patients with established EMPD, other apocrine gland–bearing areas should be examined for multicentric disease. Lastly, the clinical course in our patient supports the hypothesis that primary multicentric EMPD has a favorable outcome. The Table lists the characteristics of MPD and EMPD and describes their development with respect to Toker cells. More studies are required to further outline the cytologic characteristics of Toker cells and to distinguish primary EMPD from secondary EMPD.
Extramammary Paget disease (EMPD), which was first described by Crocker1 in a patient with erythematous patches on the penis and scrotum, is morphologically identical to mammary Paget disease (MPD) of the nipple. The principal difference between EMPD and MPD is anatomic location. Extramammary Paget disease predominantly affects apocrine gland–bearing areas including the vulva, scrotum, and perianal areas. Although EMPD is not a common condition, it must be considered in the differential diagnosis for patients with chronic genital or perianal dermatitis. Primary EMPD must be distinguished from secondary epithelial involvement by an underlying invasive carcinoma that originates from sites such as the gastrointestinal or genitourinary systems (secondary EMPD).
Although multicentric primary EMPD is not uncommon among Eastern Asians, as there have been several reports in the literature from Japan,2-5 multicentric EMPD in white individuals is rare.6 We report a case of primary EMPD that was established when no underlying malignancies were detected.
Case Report
A 63-year-old white man presented to the dermatology clinic with a pruritic rash involving the groin of 8 years’ duration. Over-the-counter antifungal agents provided no improvement. Confluent erythematous and macerated plaques on the scrotum, shaft of the penis, bilateral inguinal areas, and perineum were noted on clinical examination (Figure 1). There also were well-demarcated, 4×5-cm, erythematous, velvety plaques on the right axilla and a small erythematous plaque on the left axilla. Systemic workup including colonoscopy, cystoscopy, and magnetic resonance imaging of the abdomen and pelvis were unremarkable. A positron emission tomography–computed tomography scan revealed foci of hypermetabolic activity in the bilateral inguinal nodes that were interpreted as evidence of an inflammatory process.
Fourteen biopsies were taken from the clinically involved regions in the genital area to delineate the extent of involvement. They all showed intraepidermal carcinoma characterized by large cells with clear cytoplasm and large hyperchromatic nuclei throughout the epidermis, which were more abundant in the basilar and lower portions of the epidermis (Figure 2). There was no evidence of underlying carcinoma in the dermis.
On immunohistochemical staining, the neoplastic cells were diffusely and strongly positive for cytokeratin 7 (Figure 3) and moderately positive for carcinoembryonic antigen, estrogen and progesterone receptors, and ERBB2 (formerly HER-2/neu). Additional immunostaining for cytokeratin 20 and gross cystic disease fluid protein 15 were negative. High-molecular-weight keratin staining highlighted background epithelium and spared neoplastic cells. Mucicarmine staining highlighted mucin within the cytoplasm of neoplastic cells. Ultrastructural studies showed evidence of apocrine differentiation in the neoplastic cells with deeply indented, bean-shaped nuclei and clear cytoplasm with inconspicuous organelles. Clinically unaffected skin from the periumbilical region also was biopsied. The histology demonstrated Toker cells characterized by clear cytoplasm, small vesicular nuclei, and the absence of hyperchromasia (Figure 4).
The patient was treated with 6 cycles of 5-aminolevulinic acid 20% photodynamic therapy but showed no improvement. Imiquimod cream 5% resulted in mild clinical improvement in the axillae but caused severe irritation in the groin and was discontinued after 3 months. A radiology consultation obtained 6 years prior to presentation determined that the risk associated with radiation therapy outweighed any possible benefits. Although localized disease persisted, repeated biopsies and positron emission tomography–computed tomography scans did not show any evidence of invasion or metastasis, respectively. Three years after initial presentation, groin lesions progressed to become more erythematous and vegetative. The corresponding histology demonstrated EMPD. An ancillary test (polymerase chain reaction analysis) for low- and high-risk human papillomavirus was negative.
Comment
Primary EMPD has been well documented in the literature to have a favorable prognosis if adequately treated. The few cases reported from Japan on EMPD without underlying adenocarcinoma had good outcomes.2,3 Further reports of multicentric EMPD involving 2 or 3 sites in the axillae and/or genital region did not reveal any progression to invasive carcinomas after adequate follow-up.3-9 Indeed, many of these cases, particularly those in the genital region, were aggressively treated with surgery, topical chemotherapy, immunomodulatory agents, and radiation; therefore, their natural course is not known.2 A Japanese studyconducted at 75 medical institutions (1987-1991) included 46 EMPD patients with multiple (ie, 2 or 3) sites of involvement.10 Some of the patients with a combination of genital and axillary lesions were followed without any treatment. None of the EMPD patients with axillary involvement developed invasive carcinoma after 4- to 12-year follow-up.10
Toker cells, which were first described in 1970,11 have been recognized as precursors to EMPD.12-14 Toker cells are intraepithelial cells with clear to pale-staining cytoplasm that are smaller in size than Paget cells but larger than neighboring keratinocytes. They are found in approximately 10% of normal nipples.11,14 Toker cells show vesicular chromatin, whereas Paget cells are hyperchromatic with prominent nucleoli.12 Willman et al15 examined 11 vulvectomies for the presence of Toker cells in association with mammarylike glands of the vulva. They demonstrated the presence of Toker cells in 4 (36%) of the samples.15 Additionally, Van der Putte et al16 observed Toker cells in an areolar lesion in a 47-year-old woman with MPD without underlying adenocarcinoma, suggesting that cases of MPD and EMPD confined to epithelial cells may be derived from Toker cells. Toker cells have been associated in the pathogenesis of 2 other benign entities, including clear cell papulosis17,18 and cutaneous hamartoma with pagetoid cells.19
Primary EMPD has the potential to develop in several regions of the skin that contain apocrine glands. Our patient presented with persistent genital lesions for many years without any concerns of axillary disease. Interestingly, biopsies from normal-appearing skin in the periumbilical region revealed clear cells (Figure 4). Likewise, Toker cells have been described in biopsies from clinically unaffected skin of the axillae in some cases where EMPD was identified in the genital regions.3,5 Genital lesions were the main clinical presentation and preceded axillary involvement in most instances.3,5,9 Therefore, biopsies from uninvolved apocrine sites (ie, axillary and periumbilical skin) should be considered in these patients. Based on our observation, we speculate that multicentric primary EMPD starts with Toker cell hyperplasia with a propensity to evolve into neoplasia in sites with apocrine or mammarylike glands.
Primary and secondary EMPD cannot be distinguished by histopathology and immunohistochemistry has a limited role.20,21 However, immunostaining with CDX2 (a caudal-type homeobox protein) might be helpful in differentiating primary EMPD from secondary EMPD extending from underlying anorectal adenocarcinomas.22 Primary EMPD has been treated with surgical excision.2,3,8 Despite the high recurrence rate after surgery, the prognosis for localized primary EMPD disease has been favorable.
Conclusion
Our case suggests that Toker cell hyperplasia is a precursor to primary EMPD. In patients with established EMPD, other apocrine gland–bearing areas should be examined for multicentric disease. Lastly, the clinical course in our patient supports the hypothesis that primary multicentric EMPD has a favorable outcome. The Table lists the characteristics of MPD and EMPD and describes their development with respect to Toker cells. More studies are required to further outline the cytologic characteristics of Toker cells and to distinguish primary EMPD from secondary EMPD.
1. Crocker HR. Paget’s disease affecting the scrotum and penis. Trans Path Soc London. 1889;40:187-191.
2. Kawatsu T, Miki Y. Triple extramammary Paget’s disease. Arch Dermatol. 1971;104:316-319.
3. Makino T, Nakamura S, Nakayama H, et al. Genital Paget’s disease with clear cells in the epidermis of the axilla. J Cutan Pathol. 1998;25:568-571.
4. Inui S, Fukuhara S, Asada H, et al. Double involvement of extramammary Paget’s disease in the genitalia and axilla. J Dermatol. 2000;27:409-412.
5. Kitajima S, Yamamoto K, Tsuji T, et al. Triple extramammary Paget’s disease. Dermatol Surg. 1997;23:1035-1038.
6. Van Hamme C, Marot L, Dachelet C, et al. Paget’s extramammary disease of the axillae and perineum [in French]. Ann Dermatol Venereol. 2002;129(5, pt 1):717-719.
7. Kao GF, Graham JH, Helwig EB. Paget’s disease of the ectopic breast with an underlying intraductal carcinoma: report of a case. J Cutan Pathol. 1986;13:59-66.
8. Murrell TW Jr, McMullan FH. Extramammary Paget’s disease. a report of two cases. Arch Dermatol. 1962;85:600-613.
9. Koseki S, Mitsuhashi Y, Yoshikawa K, et al. A case of triple extramammary Paget’s disease. J Dermatol. 1997;24:535-538.
10. Ishihara K. Statistical study of extramammary Paget’s disease in Japan [in Japanese]. Skin Cancer. 1994;9:38.
11. Toker C. Clear cells of the nipple epidermis. Cancer. 1970;25:601-610.
12. Marucci G, Betts CM, Golouh R, et al. Toker cells are probably precursors of Paget cell carcinoma: a morphological and ultrastructural description. Virchows Archiv. 2002;441:117-123.
13. Belousova IE, Kazakov DV, Michal M, et al. Vulvar toker cells: the long-awaited missing link: a proposal for an origin-based histogenetic classification of extramammary Paget disease. Am J Dermatopathol. 2006;28:84-86.
14. Val-Bernal JF. Diego C, Rodriguez-Villar D, et al. The nipple-areola complex epidermis: a prospective systemic study in adult autopsies. Am J Dermatopathology. 2010;32:787-93.
15. Willman JH, Golitz LE, Fitzpatrick JE. Vulvar clear cells of Toker: precursors of extramammary Paget’s disease. Am J Dermatopathol. 2005;27:185-188.
16. Van der Putte SC, Toonstra J, Hennipman A. Mammary Paget’s disease confined to the areola and associated with multifocal Toker cell hyperplasia. Am J Dermatopathol. 1995;17:487-493.
17. Kuo TT, Chan HL, Hsueh S. Clear cell papulosis of the skin. a new entity with histogenetic implications for cutaneous Paget’s disease. Am J Surg Pathol. 1987;11:827-834.
18. Kuo TT, Huang CL, Chan HL, et al. Clear cell papulosis: report of three cases of a newly recognized disease. J Am Acad Dermatol. 1995;33(2, pt 1):230-233.
19. Piérard-Franchimont C, Dosal FL, Estrada JA, et al. Cutaneous hamartoma with pagetoid cells. Am J Dermatopathol. 1991;13:158-161.
20. Belcher RW. Extramammary Paget’s disease. enzyme histochemical and electron microscopic study. Arch Pathol. 1972;94:59-64.
21. Ordóñez NG, Awalt H, Mackay B. Mammary and extramammary Paget’s disease. an immunocytochemical and ultrastructural study. Cancer. 1987;59:1173-1183.
22. Perrotto J, Abbott JJ, Ceilley RI, et al. The role of immunohistochemistry in discriminating primary from secondary extramammary Paget disease. Am J Dermatopathol. 2010;32:137-143.
23. Jones RE Jr, Austin C, Ackerman AB. Extramammary Paget’s disease. a critical reexamination. Am J Dermatopathol. 1979;1:101-132.
1. Crocker HR. Paget’s disease affecting the scrotum and penis. Trans Path Soc London. 1889;40:187-191.
2. Kawatsu T, Miki Y. Triple extramammary Paget’s disease. Arch Dermatol. 1971;104:316-319.
3. Makino T, Nakamura S, Nakayama H, et al. Genital Paget’s disease with clear cells in the epidermis of the axilla. J Cutan Pathol. 1998;25:568-571.
4. Inui S, Fukuhara S, Asada H, et al. Double involvement of extramammary Paget’s disease in the genitalia and axilla. J Dermatol. 2000;27:409-412.
5. Kitajima S, Yamamoto K, Tsuji T, et al. Triple extramammary Paget’s disease. Dermatol Surg. 1997;23:1035-1038.
6. Van Hamme C, Marot L, Dachelet C, et al. Paget’s extramammary disease of the axillae and perineum [in French]. Ann Dermatol Venereol. 2002;129(5, pt 1):717-719.
7. Kao GF, Graham JH, Helwig EB. Paget’s disease of the ectopic breast with an underlying intraductal carcinoma: report of a case. J Cutan Pathol. 1986;13:59-66.
8. Murrell TW Jr, McMullan FH. Extramammary Paget’s disease. a report of two cases. Arch Dermatol. 1962;85:600-613.
9. Koseki S, Mitsuhashi Y, Yoshikawa K, et al. A case of triple extramammary Paget’s disease. J Dermatol. 1997;24:535-538.
10. Ishihara K. Statistical study of extramammary Paget’s disease in Japan [in Japanese]. Skin Cancer. 1994;9:38.
11. Toker C. Clear cells of the nipple epidermis. Cancer. 1970;25:601-610.
12. Marucci G, Betts CM, Golouh R, et al. Toker cells are probably precursors of Paget cell carcinoma: a morphological and ultrastructural description. Virchows Archiv. 2002;441:117-123.
13. Belousova IE, Kazakov DV, Michal M, et al. Vulvar toker cells: the long-awaited missing link: a proposal for an origin-based histogenetic classification of extramammary Paget disease. Am J Dermatopathol. 2006;28:84-86.
14. Val-Bernal JF. Diego C, Rodriguez-Villar D, et al. The nipple-areola complex epidermis: a prospective systemic study in adult autopsies. Am J Dermatopathology. 2010;32:787-93.
15. Willman JH, Golitz LE, Fitzpatrick JE. Vulvar clear cells of Toker: precursors of extramammary Paget’s disease. Am J Dermatopathol. 2005;27:185-188.
16. Van der Putte SC, Toonstra J, Hennipman A. Mammary Paget’s disease confined to the areola and associated with multifocal Toker cell hyperplasia. Am J Dermatopathol. 1995;17:487-493.
17. Kuo TT, Chan HL, Hsueh S. Clear cell papulosis of the skin. a new entity with histogenetic implications for cutaneous Paget’s disease. Am J Surg Pathol. 1987;11:827-834.
18. Kuo TT, Huang CL, Chan HL, et al. Clear cell papulosis: report of three cases of a newly recognized disease. J Am Acad Dermatol. 1995;33(2, pt 1):230-233.
19. Piérard-Franchimont C, Dosal FL, Estrada JA, et al. Cutaneous hamartoma with pagetoid cells. Am J Dermatopathol. 1991;13:158-161.
20. Belcher RW. Extramammary Paget’s disease. enzyme histochemical and electron microscopic study. Arch Pathol. 1972;94:59-64.
21. Ordóñez NG, Awalt H, Mackay B. Mammary and extramammary Paget’s disease. an immunocytochemical and ultrastructural study. Cancer. 1987;59:1173-1183.
22. Perrotto J, Abbott JJ, Ceilley RI, et al. The role of immunohistochemistry in discriminating primary from secondary extramammary Paget disease. Am J Dermatopathol. 2010;32:137-143.
23. Jones RE Jr, Austin C, Ackerman AB. Extramammary Paget’s disease. a critical reexamination. Am J Dermatopathol. 1979;1:101-132.
Practice Points
- Toker cells are epithelial clear cells found in apocrine gland–bearing areas of the skin.
- Toker cell hyperplasia may be a precursor to primary extramammary Paget disease (EMPD).
- In patients with established EMPD, it is important to examine other parts of the skin with apocrine glands for multicentric disease.
- Primary multicentric EMPD has a favorable outcome.
Disseminated Waxy Papules: A Sign of Systemic Disease
To the Editor:
A 35-year-old man presented with asymptomatic multiple waxy firm papules on the dorsal aspect of the hands (Figure 1). On complete clinical examination, multiple similar lesions were found on the face and the lateral aspect of the neck; slightly pruritic, clustered, erythematous papules were seen in the pubic area. Deep longitudinal furrows on the glabella and swelling of the ears (Figure 2) also were present. Histology revealed a dermal infiltrate of fibroblasts (Figure 3). Abundant mucin deposition between collagen bundles stained positive with Alcian blue, confirming the diagnosis of generalized lichen myxedematosus (GLM). A thorough laboratory evaluation revealed λ light chain gammopathy. Bone marrow examination was normal. Thyroid function tests and radiography did not detect any abnormalities.
Being a musician, the patient traveled frequently and therefore did not present for follow-up. The disease ran its course. Approximately 6 months later he presented with a generalized eruption with weakness and intense bone pain. Progression to multiple myeloma was confirmed by serum protein electrophoresis and plain radiography, which revealed lytic lesions of the skull.
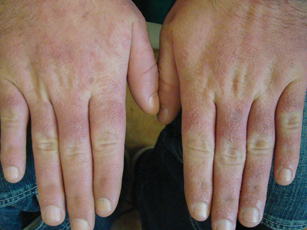
Generalized lichen myxedematosus, or scleromyxedema, is a form of papular mucinosis. It is a cutaneous mucinosis characterized by a generalized papular and sclerodermoid eruption, mucin deposition, increased fibroblast proliferation, fibrosis, and monoclonal gammopathy in the absence of thyroid disease. It is a rare disease that does not have a gender predilection and affects men and women aged 30 to 80 years.1,2
Clinically, GLM is characterized by a widespread symmetric eruption of small, closely spaced, waxy, firm, dome-shaped or flat-topped papules that measure 2 to 3 mm in diameter. Papules commonly form clusters on the face, neck, distal forearms, and hands. The affected skin may have a shiny sclerodermatous appearance.2
Histologically, there is a pronounced mucin deposition within the upper and mid reticular dermis, along with a proliferation of irregularly arranged fibroblasts and fibrosis.3 Collagen bundles appear pushed together into fascicles by the mucinous deposits.4
Generalized lichen myxedematosus should be distinguished from nephrogenic systemic fibrosis, granuloma annulare, amyloidosis, and Hansen disease. In nearly 83% of cases there is an accompanying paraproteinemia, most commonly an IgG λ light chain elevation.2 Monoclonal gammopathy of IgG with λ light chain has been considered as a fibroblast growth factor. Paraprotein levels do not correlate with the extent or progression of the disease.1 Nonparaprotein factors also can be responsible for excess fibroblast proliferation.2 It still is not elucidated as to how the abnormal mucin production results from the proliferation of fibroblasts.
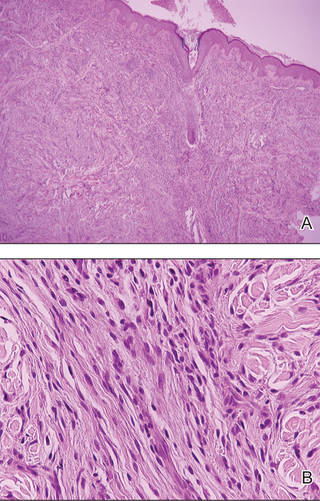
Patients with GLM must be closely followed. The course of the disease is chronic and progressive. Extracutaneous manifestations may involve the gastrointestinal, musculoskeletal, cardiovascular, pulmonary, and central nervous systems, most likely because of mucin deposition in various organs.1,4 In these cases, the life expectancy is limited. Possibility of progression to multiple myeloma is less than 10%.1 Waldenström macroglobulinemia, Hodgkin lymphoma, and non-Hodgkin lymphomas all have been associated with GLM.2 Although extremely uncommon, spontaneous resolution has been reported after as long as a decade.5
Treatment of GLM, including melphalan, plasmapheresis, corticosteroids, cyclophosphamide, methotrexate, thalidomide, electron beam therapy, intravenous immunoglobulin therapy, extracorporeal photochemotherapy, high-dose chemotherapy with autologous stem cell rescue, and melphalan with autologous peripheral blood stem cell transplant are considered to improve the prognosis of this noncurable disease.6 As there is still no consensus on the treatment of scleromyxedema, multicenter studies on therapeutic schemes and responses are anticipated.
Generalized lichen myxedematosus is an instructive example of skin lesions being the presenting sign of a systemic disease. Progression to multiple myeloma in our patient made the prognosis less favorable.
- Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25:100-104.
- Cokonis Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Pomann JJ, Rudner EJ. Scleromyxedema revisited. Int J Dermatol. 2003;42:31-35.
- Dinneen AM, Dicken CH. Scleromyxedema. J Am Acad Dermatol. 1995;33:37-43.
- Heymann WR. Scleromyxedema. J Am Acad Dermatol. 2007;57:890-891.
To the Editor:
A 35-year-old man presented with asymptomatic multiple waxy firm papules on the dorsal aspect of the hands (Figure 1). On complete clinical examination, multiple similar lesions were found on the face and the lateral aspect of the neck; slightly pruritic, clustered, erythematous papules were seen in the pubic area. Deep longitudinal furrows on the glabella and swelling of the ears (Figure 2) also were present. Histology revealed a dermal infiltrate of fibroblasts (Figure 3). Abundant mucin deposition between collagen bundles stained positive with Alcian blue, confirming the diagnosis of generalized lichen myxedematosus (GLM). A thorough laboratory evaluation revealed λ light chain gammopathy. Bone marrow examination was normal. Thyroid function tests and radiography did not detect any abnormalities.
Being a musician, the patient traveled frequently and therefore did not present for follow-up. The disease ran its course. Approximately 6 months later he presented with a generalized eruption with weakness and intense bone pain. Progression to multiple myeloma was confirmed by serum protein electrophoresis and plain radiography, which revealed lytic lesions of the skull.
Generalized lichen myxedematosus, or scleromyxedema, is a form of papular mucinosis. It is a cutaneous mucinosis characterized by a generalized papular and sclerodermoid eruption, mucin deposition, increased fibroblast proliferation, fibrosis, and monoclonal gammopathy in the absence of thyroid disease. It is a rare disease that does not have a gender predilection and affects men and women aged 30 to 80 years.1,2
Clinically, GLM is characterized by a widespread symmetric eruption of small, closely spaced, waxy, firm, dome-shaped or flat-topped papules that measure 2 to 3 mm in diameter. Papules commonly form clusters on the face, neck, distal forearms, and hands. The affected skin may have a shiny sclerodermatous appearance.2
Histologically, there is a pronounced mucin deposition within the upper and mid reticular dermis, along with a proliferation of irregularly arranged fibroblasts and fibrosis.3 Collagen bundles appear pushed together into fascicles by the mucinous deposits.4
Generalized lichen myxedematosus should be distinguished from nephrogenic systemic fibrosis, granuloma annulare, amyloidosis, and Hansen disease. In nearly 83% of cases there is an accompanying paraproteinemia, most commonly an IgG λ light chain elevation.2 Monoclonal gammopathy of IgG with λ light chain has been considered as a fibroblast growth factor. Paraprotein levels do not correlate with the extent or progression of the disease.1 Nonparaprotein factors also can be responsible for excess fibroblast proliferation.2 It still is not elucidated as to how the abnormal mucin production results from the proliferation of fibroblasts.
Patients with GLM must be closely followed. The course of the disease is chronic and progressive. Extracutaneous manifestations may involve the gastrointestinal, musculoskeletal, cardiovascular, pulmonary, and central nervous systems, most likely because of mucin deposition in various organs.1,4 In these cases, the life expectancy is limited. Possibility of progression to multiple myeloma is less than 10%.1 Waldenström macroglobulinemia, Hodgkin lymphoma, and non-Hodgkin lymphomas all have been associated with GLM.2 Although extremely uncommon, spontaneous resolution has been reported after as long as a decade.5
Treatment of GLM, including melphalan, plasmapheresis, corticosteroids, cyclophosphamide, methotrexate, thalidomide, electron beam therapy, intravenous immunoglobulin therapy, extracorporeal photochemotherapy, high-dose chemotherapy with autologous stem cell rescue, and melphalan with autologous peripheral blood stem cell transplant are considered to improve the prognosis of this noncurable disease.6 As there is still no consensus on the treatment of scleromyxedema, multicenter studies on therapeutic schemes and responses are anticipated.
Generalized lichen myxedematosus is an instructive example of skin lesions being the presenting sign of a systemic disease. Progression to multiple myeloma in our patient made the prognosis less favorable.
To the Editor:
A 35-year-old man presented with asymptomatic multiple waxy firm papules on the dorsal aspect of the hands (Figure 1). On complete clinical examination, multiple similar lesions were found on the face and the lateral aspect of the neck; slightly pruritic, clustered, erythematous papules were seen in the pubic area. Deep longitudinal furrows on the glabella and swelling of the ears (Figure 2) also were present. Histology revealed a dermal infiltrate of fibroblasts (Figure 3). Abundant mucin deposition between collagen bundles stained positive with Alcian blue, confirming the diagnosis of generalized lichen myxedematosus (GLM). A thorough laboratory evaluation revealed λ light chain gammopathy. Bone marrow examination was normal. Thyroid function tests and radiography did not detect any abnormalities.
Being a musician, the patient traveled frequently and therefore did not present for follow-up. The disease ran its course. Approximately 6 months later he presented with a generalized eruption with weakness and intense bone pain. Progression to multiple myeloma was confirmed by serum protein electrophoresis and plain radiography, which revealed lytic lesions of the skull.
Generalized lichen myxedematosus, or scleromyxedema, is a form of papular mucinosis. It is a cutaneous mucinosis characterized by a generalized papular and sclerodermoid eruption, mucin deposition, increased fibroblast proliferation, fibrosis, and monoclonal gammopathy in the absence of thyroid disease. It is a rare disease that does not have a gender predilection and affects men and women aged 30 to 80 years.1,2
Clinically, GLM is characterized by a widespread symmetric eruption of small, closely spaced, waxy, firm, dome-shaped or flat-topped papules that measure 2 to 3 mm in diameter. Papules commonly form clusters on the face, neck, distal forearms, and hands. The affected skin may have a shiny sclerodermatous appearance.2
Histologically, there is a pronounced mucin deposition within the upper and mid reticular dermis, along with a proliferation of irregularly arranged fibroblasts and fibrosis.3 Collagen bundles appear pushed together into fascicles by the mucinous deposits.4
Generalized lichen myxedematosus should be distinguished from nephrogenic systemic fibrosis, granuloma annulare, amyloidosis, and Hansen disease. In nearly 83% of cases there is an accompanying paraproteinemia, most commonly an IgG λ light chain elevation.2 Monoclonal gammopathy of IgG with λ light chain has been considered as a fibroblast growth factor. Paraprotein levels do not correlate with the extent or progression of the disease.1 Nonparaprotein factors also can be responsible for excess fibroblast proliferation.2 It still is not elucidated as to how the abnormal mucin production results from the proliferation of fibroblasts.
Patients with GLM must be closely followed. The course of the disease is chronic and progressive. Extracutaneous manifestations may involve the gastrointestinal, musculoskeletal, cardiovascular, pulmonary, and central nervous systems, most likely because of mucin deposition in various organs.1,4 In these cases, the life expectancy is limited. Possibility of progression to multiple myeloma is less than 10%.1 Waldenström macroglobulinemia, Hodgkin lymphoma, and non-Hodgkin lymphomas all have been associated with GLM.2 Although extremely uncommon, spontaneous resolution has been reported after as long as a decade.5
Treatment of GLM, including melphalan, plasmapheresis, corticosteroids, cyclophosphamide, methotrexate, thalidomide, electron beam therapy, intravenous immunoglobulin therapy, extracorporeal photochemotherapy, high-dose chemotherapy with autologous stem cell rescue, and melphalan with autologous peripheral blood stem cell transplant are considered to improve the prognosis of this noncurable disease.6 As there is still no consensus on the treatment of scleromyxedema, multicenter studies on therapeutic schemes and responses are anticipated.
Generalized lichen myxedematosus is an instructive example of skin lesions being the presenting sign of a systemic disease. Progression to multiple myeloma in our patient made the prognosis less favorable.
- Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25:100-104.
- Cokonis Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Pomann JJ, Rudner EJ. Scleromyxedema revisited. Int J Dermatol. 2003;42:31-35.
- Dinneen AM, Dicken CH. Scleromyxedema. J Am Acad Dermatol. 1995;33:37-43.
- Heymann WR. Scleromyxedema. J Am Acad Dermatol. 2007;57:890-891.
- Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25:100-104.
- Cokonis Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Pomann JJ, Rudner EJ. Scleromyxedema revisited. Int J Dermatol. 2003;42:31-35.
- Dinneen AM, Dicken CH. Scleromyxedema. J Am Acad Dermatol. 1995;33:37-43.
- Heymann WR. Scleromyxedema. J Am Acad Dermatol. 2007;57:890-891.