User login
Lipidized Dermatofibroma
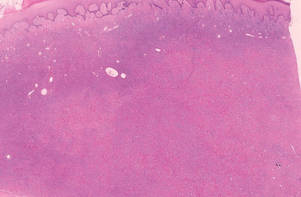
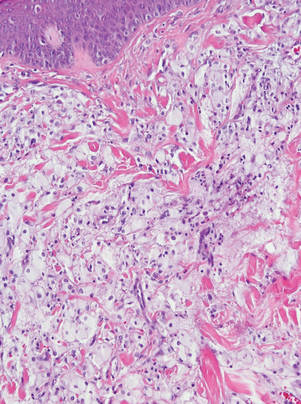
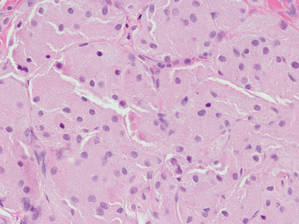
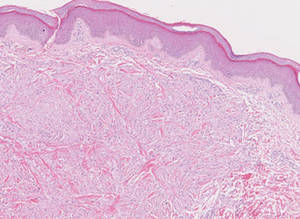
Lipidized dermatofibromas most commonly are found on the ankles, which has led some authors to refer to these lesions as ankle-type fibrous histiocytomas.1 Compared to ordinary dermatofibromas, patients with lipidized dermatofibromas tend to be older, most commonly presenting in the fifth or sixth decades of life, and are predominantly male. Lipidized dermatofibromas typically present as well-circumscribed solitary nodules in the dermis. Characteristic features include numerous xanthomatous cells dissected by distinctive hyalinized wiry collagen fibers (Figures 1 and 2).1 Xanthomatous cells can be round, polygonal, or stellate in shape. These characteristic features in combination with others of dermatofibromas (eg, epidermal acanthosis [Figure 1]) fulfill the criteria for diagnosis of a lipidized dermatofibroma. Additionally, lipidized dermatofibromas tend to be larger than ordinary dermatofibromas, which typically are less than 2 cm in diameter.1
|
Figure 1. Lipidized dermatofibromas are characterized by classic epidermal features of dermatofibromas, such as acanthosis, along with numerous foam cells and extensive stromal hyalinization (H&E, original magnification ×1.5). |
|
Figure 2. Higher-power view of a lipidized dermatofibroma shows the characteristic irregular dissection of hyalinized wiry collagen fibers between the xanthomatous cells (H&E, original magnification ×20). |
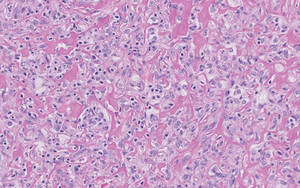
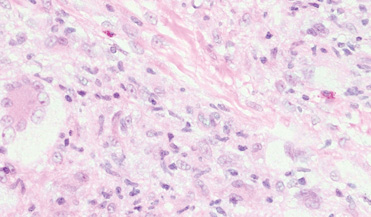
Eruptive xanthomas are characterized by a lacelike infiltrate of extravascular lipid deposits between collagen bundles (Figure 3).2 Granular cell tumors are composed of sheets and/or nests of large cells with abundant eosinophilic cytoplasm and may be confused with lipidized dermatofibromas, as they also may induce overlying pseudoepitheliomatous hyperplasia3; however, on closer examination of the cells, the cytoplasm is found to be granular (Figure 4), which contrasts the finely vacuolated cytoplasm of xanthomatous cells found in lipidized dermatofibromas. Giant lysosomal granules (eg, pustulo-ovoid bodies of Milian) are present in some cases.2 Of note, an unusual variant of dermatofibroma exists that features prominent granular cells.4
|
Figure 3. Lacelike deposition of extravascular lipid deposits is seen infiltrating between collagen bundles in an eruptive xanthoma (H&E, original magnification ×20). |
|
Figure 4. An abundant eosinophilic, finely granular cytoplasm is characteristic of granular cell tumor (H&E, original magnification ×40). |
Tuberous xanthomas most commonly occur around the pressure areas, such as the knees, elbows, and buttocks. Foam cells are a main feature of tuberous xanthomas and are arranged in large aggregates throughout the dermis.2 Tuberous xanthomas lack Touton giant cells or inflammatory cells. Older lesions tend to develop substantial fibrosis (Figure 5). Although foam cells can be present in older lesions, they are never as conspicuous as those found in other xanthomas.
Xanthogranulomas commonly occur on the head and neck. Findings noted on low magnification include a well-circumscribed exophytic nodule and an epidermal collarette, which help to easily distinguish xanthogranulomas from lipidized dermatofibromas. Additionally, the presence of a more prominent inflammatory infiltrate, which often includes eosinophils, as well as multinucleated Touton giant cells (Figure 6) and histiocytes with more eosinophilic and less xanthomatous cytoplasm can help distinguish between the lesions.1,5 Notably, Touton giant cells also can be seen in lipidized dermatofibromas,1 but the presence of unique features such as distinctive stromal hyalinization are clues to the correct diagnosis of a lipidized dermatofibroma.
- Iwata J, Fletcher CD. Lipidized fibrous histiocytoma: clinicopathologic analysis of 22 cases. Am J Dermatopathol. 2000;22:126-134.
- Weedon D. Weedon’s Skin Pathology. 3rd ed. Edinburgh, Scotland: Elsevier Health Sciences; 2009.
- Elston DM, Ferringer T. Dermatopathology. Philadelphia, PA: Saunders Elsevier; 2009.
- Yogesh TL, Sowmya SV. Granules in granular cell lesions of the head and neck: a review. ISRN Pathol. 2011;2011:10.
- Fujita Y, Tsunemi Y, Kadono T, et al. Lipidized fibrous histiocytoma on the left condyle of the tibia. Int J Dermatol. 2011;50:634-636.
Lipidized dermatofibromas most commonly are found on the ankles, which has led some authors to refer to these lesions as ankle-type fibrous histiocytomas.1 Compared to ordinary dermatofibromas, patients with lipidized dermatofibromas tend to be older, most commonly presenting in the fifth or sixth decades of life, and are predominantly male. Lipidized dermatofibromas typically present as well-circumscribed solitary nodules in the dermis. Characteristic features include numerous xanthomatous cells dissected by distinctive hyalinized wiry collagen fibers (Figures 1 and 2).1 Xanthomatous cells can be round, polygonal, or stellate in shape. These characteristic features in combination with others of dermatofibromas (eg, epidermal acanthosis [Figure 1]) fulfill the criteria for diagnosis of a lipidized dermatofibroma. Additionally, lipidized dermatofibromas tend to be larger than ordinary dermatofibromas, which typically are less than 2 cm in diameter.1
|
|
Figure 1. Lipidized dermatofibromas are characterized by classic epidermal features of dermatofibromas, such as acanthosis, along with numerous foam cells and extensive stromal hyalinization (H&E, original magnification ×1.5). |
|
|
Figure 2. Higher-power view of a lipidized dermatofibroma shows the characteristic irregular dissection of hyalinized wiry collagen fibers between the xanthomatous cells (H&E, original magnification ×20). |
Eruptive xanthomas are characterized by a lacelike infiltrate of extravascular lipid deposits between collagen bundles (Figure 3).2 Granular cell tumors are composed of sheets and/or nests of large cells with abundant eosinophilic cytoplasm and may be confused with lipidized dermatofibromas, as they also may induce overlying pseudoepitheliomatous hyperplasia3; however, on closer examination of the cells, the cytoplasm is found to be granular (Figure 4), which contrasts the finely vacuolated cytoplasm of xanthomatous cells found in lipidized dermatofibromas. Giant lysosomal granules (eg, pustulo-ovoid bodies of Milian) are present in some cases.2 Of note, an unusual variant of dermatofibroma exists that features prominent granular cells.4
|
|
Figure 3. Lacelike deposition of extravascular lipid deposits is seen infiltrating between collagen bundles in an eruptive xanthoma (H&E, original magnification ×20). |
|
|
Figure 4. An abundant eosinophilic, finely granular cytoplasm is characteristic of granular cell tumor (H&E, original magnification ×40). |
Tuberous xanthomas most commonly occur around the pressure areas, such as the knees, elbows, and buttocks. Foam cells are a main feature of tuberous xanthomas and are arranged in large aggregates throughout the dermis.2 Tuberous xanthomas lack Touton giant cells or inflammatory cells. Older lesions tend to develop substantial fibrosis (Figure 5). Although foam cells can be present in older lesions, they are never as conspicuous as those found in other xanthomas.
Xanthogranulomas commonly occur on the head and neck. Findings noted on low magnification include a well-circumscribed exophytic nodule and an epidermal collarette, which help to easily distinguish xanthogranulomas from lipidized dermatofibromas. Additionally, the presence of a more prominent inflammatory infiltrate, which often includes eosinophils, as well as multinucleated Touton giant cells (Figure 6) and histiocytes with more eosinophilic and less xanthomatous cytoplasm can help distinguish between the lesions.1,5 Notably, Touton giant cells also can be seen in lipidized dermatofibromas,1 but the presence of unique features such as distinctive stromal hyalinization are clues to the correct diagnosis of a lipidized dermatofibroma.
Lipidized dermatofibromas most commonly are found on the ankles, which has led some authors to refer to these lesions as ankle-type fibrous histiocytomas.1 Compared to ordinary dermatofibromas, patients with lipidized dermatofibromas tend to be older, most commonly presenting in the fifth or sixth decades of life, and are predominantly male. Lipidized dermatofibromas typically present as well-circumscribed solitary nodules in the dermis. Characteristic features include numerous xanthomatous cells dissected by distinctive hyalinized wiry collagen fibers (Figures 1 and 2).1 Xanthomatous cells can be round, polygonal, or stellate in shape. These characteristic features in combination with others of dermatofibromas (eg, epidermal acanthosis [Figure 1]) fulfill the criteria for diagnosis of a lipidized dermatofibroma. Additionally, lipidized dermatofibromas tend to be larger than ordinary dermatofibromas, which typically are less than 2 cm in diameter.1
|
|
Figure 1. Lipidized dermatofibromas are characterized by classic epidermal features of dermatofibromas, such as acanthosis, along with numerous foam cells and extensive stromal hyalinization (H&E, original magnification ×1.5). |
|
|
Figure 2. Higher-power view of a lipidized dermatofibroma shows the characteristic irregular dissection of hyalinized wiry collagen fibers between the xanthomatous cells (H&E, original magnification ×20). |
Eruptive xanthomas are characterized by a lacelike infiltrate of extravascular lipid deposits between collagen bundles (Figure 3).2 Granular cell tumors are composed of sheets and/or nests of large cells with abundant eosinophilic cytoplasm and may be confused with lipidized dermatofibromas, as they also may induce overlying pseudoepitheliomatous hyperplasia3; however, on closer examination of the cells, the cytoplasm is found to be granular (Figure 4), which contrasts the finely vacuolated cytoplasm of xanthomatous cells found in lipidized dermatofibromas. Giant lysosomal granules (eg, pustulo-ovoid bodies of Milian) are present in some cases.2 Of note, an unusual variant of dermatofibroma exists that features prominent granular cells.4
|
|
Figure 3. Lacelike deposition of extravascular lipid deposits is seen infiltrating between collagen bundles in an eruptive xanthoma (H&E, original magnification ×20). |
|
|
Figure 4. An abundant eosinophilic, finely granular cytoplasm is characteristic of granular cell tumor (H&E, original magnification ×40). |
Tuberous xanthomas most commonly occur around the pressure areas, such as the knees, elbows, and buttocks. Foam cells are a main feature of tuberous xanthomas and are arranged in large aggregates throughout the dermis.2 Tuberous xanthomas lack Touton giant cells or inflammatory cells. Older lesions tend to develop substantial fibrosis (Figure 5). Although foam cells can be present in older lesions, they are never as conspicuous as those found in other xanthomas.
Xanthogranulomas commonly occur on the head and neck. Findings noted on low magnification include a well-circumscribed exophytic nodule and an epidermal collarette, which help to easily distinguish xanthogranulomas from lipidized dermatofibromas. Additionally, the presence of a more prominent inflammatory infiltrate, which often includes eosinophils, as well as multinucleated Touton giant cells (Figure 6) and histiocytes with more eosinophilic and less xanthomatous cytoplasm can help distinguish between the lesions.1,5 Notably, Touton giant cells also can be seen in lipidized dermatofibromas,1 but the presence of unique features such as distinctive stromal hyalinization are clues to the correct diagnosis of a lipidized dermatofibroma.
- Iwata J, Fletcher CD. Lipidized fibrous histiocytoma: clinicopathologic analysis of 22 cases. Am J Dermatopathol. 2000;22:126-134.
- Weedon D. Weedon’s Skin Pathology. 3rd ed. Edinburgh, Scotland: Elsevier Health Sciences; 2009.
- Elston DM, Ferringer T. Dermatopathology. Philadelphia, PA: Saunders Elsevier; 2009.
- Yogesh TL, Sowmya SV. Granules in granular cell lesions of the head and neck: a review. ISRN Pathol. 2011;2011:10.
- Fujita Y, Tsunemi Y, Kadono T, et al. Lipidized fibrous histiocytoma on the left condyle of the tibia. Int J Dermatol. 2011;50:634-636.
- Iwata J, Fletcher CD. Lipidized fibrous histiocytoma: clinicopathologic analysis of 22 cases. Am J Dermatopathol. 2000;22:126-134.
- Weedon D. Weedon’s Skin Pathology. 3rd ed. Edinburgh, Scotland: Elsevier Health Sciences; 2009.
- Elston DM, Ferringer T. Dermatopathology. Philadelphia, PA: Saunders Elsevier; 2009.
- Yogesh TL, Sowmya SV. Granules in granular cell lesions of the head and neck: a review. ISRN Pathol. 2011;2011:10.
- Fujita Y, Tsunemi Y, Kadono T, et al. Lipidized fibrous histiocytoma on the left condyle of the tibia. Int J Dermatol. 2011;50:634-636.
Pemphigus Vulgaris in Pregnancy
Pemphigus vulgaris (PV) is a rare autoimmune bullous dermatosis that has not shown a predilection toward a particular race or sex.1 Autoantibodies for desmoglein 1 and desmoglein 3, members of the cadherin family that are involved in cellular adhesion, have been linked to the pathogenesis of PV.2 These autoantibodies play a role in the loss of cell-to-cell adhesion in the basal and suprabasal layers of the deep epidermis while cellular adhesion in the superficial epidermis remains intact, leading to the clinical presentation of epidermal blistering and ulcerations most commonly found on the scalp, face, groin, and axillae. Diagnosis typically is made based on skin biopsy and confirmed by direct immunofluorescence. Histologically, PV displays acantholysis and suprabasal cleft formation. Immunofluorescence may show IgG antibodies against the PV antigen in the epidermis.3 Once a diagnosis has been made, treatment typically consists of systemic steroids, as the use of steroids has had great effect in preventing infections, sepsis, and fatality that were once associated with PV.4 Mortality rates associated with PV have decreased to 10% to 15% with systemic steroids from a mortality rate as high as 70% in the presteroid era.1,5 Treatment of PV during pregnancy, as in our patient, requires obstetric and pediatric consultations before therapy is initiated. Use of corticosteroids during pregnancy can be potentially dangerous to the fetus, particularly if high doses are necessary to control maternal disease.6,7
Case Report
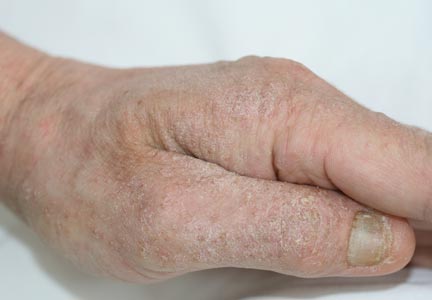
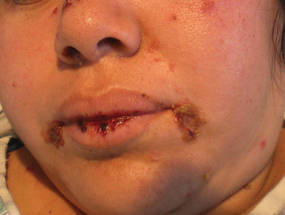
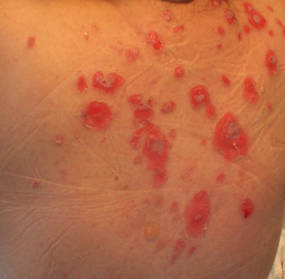
A 34-year-old pregnant woman at 6 weeks’ gestation presented with widespread blistering dermatitis and associated burning and pruritus. Her obstetrical history was gravida 3, para 2. The patient reported a “rash” on the scalp that had developed 9 months prior. She had been treated as an outpatient at an outside institution with topical antibiotics and antifungal medications, yet the dermatitis progressed. Three weeks prior to hospitalization, the rash was present on the skin and mucosal surfaces, including the groin, chest, face, hard palate, buccal mucosa, lips (Figure 1), and back (Figure 2). Nontender bullae ruptured after 3 days, releasing clear, yellow, serous fluid with associated burning and pruritus. The bullae were hemorrhagic and erythematous at the base.
|
| Figure 1. Facial involvement with bullae, crusted hemorrhagic lesions, and eschar in a 34-year-old pregnant woman. |
|
| Figure 2. Involvement of the back with bullae in various stages. Some bullae were intact while others newly erupted. |
|
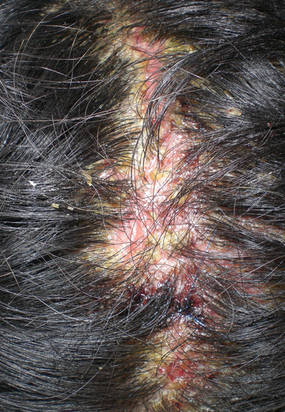
| Figure 3. Superinfected and flaking scalp. |
|
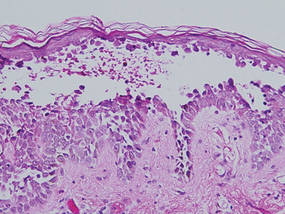
| Figure 4. Biopsy revealed suprabasal acantholysis with a tombstone effect of residual basal cells (H&E, original magnification ×200). |
At the current presentation, the patient had several excoriated 1- to 2-cm oval denudations; some were crusted with eschar. Nikolsky sign was negative. Multiple confluent bullous lesions had erupted on the entire scalp with a thick, impetiginous, yellow crust. She had a wet, boggy, foul-smelling, superinfected scalp that was mildly tender to touch with flaking tissue debris (Figure 3). A white blood cell count was 13.2×109/L (reference range, 4.5–11.0×109/L) with 5% eosinophils (reference range, 2.7%). The differential diagnosis included bullous impetigo, pemphigoid, Stevens-Johnson syndrome, dermatitis herpetiformis, and pemphigus vulgaris.
Biopsies of the scalp and back were taken and showed suprabasal acantholysis with a tombstone effect of residual basal cells standing up on the basement membrane without the characteristic acantholysis into skin appendages (Figure 4). The acantholytic cells in the bullous chamber did not round up as in Hailey-Hailey disease nor was there the dyskeratosis of Grover disease. Direct immunofluorescence on an elbow punch biopsy found diffuse 1+ intercellular IgG in the epidermis and diffuse 1+ basal intercellular C3, and was negative for IgA, IgM, and C1q, thus confirming a diagnosis of PV.
The patient was started on prednisone 20 mg once daily. An increase to prednisone 60 mg led to initial improvement of symptoms, but there was a relapse after several days, which is typical of PV in pregnancy,7 prompting the dose to be increased to 120 mg. Following alleviation of symptoms, the dose was later tapered back to 60 mg. No lesions were present at discharge or for 2.5 months thereafter, as the prednisone was tapered from 60 to 45 mg daily after discharge.
On follow-up, the patient’s PV was well controlled, but the prednisone dose was back up to 60 mg daily because of 2 new skin lesions that had developed since her last visit 2.5 months prior. Ultrasonography showed no fetal abnormalities as the pregnancy progressed to 28 weeks’ gestation. The patient developed hypertension and went into premature labor due to placenta previa. The neonate showed no skin lesions or anomalies while in the neonatal intensive care unit. The mother’s prednisone dose was tapered from 60 to 20 mg daily while the white blood cell count was 7.1×109/L with 2% eosinophils and a new scalp lesion appeared. Seven months after her initial discharge from the hospital for the dermatologic condition, she was no longer nursing and azathioprine was added to prednisone 60 mg daily.
Comment
Pemphigus vulgaris is associated with infertility in its active phase; therefore, PV during pregnancy is rare.8 Pregnancy may exacerbate PV, which has been a similar finding in other well-documented autoimmune diseases.7 One review of PV in pregnancy reported that 11 of 49 patients (22%) experienced an exacerbation of the disease.8 This finding pre-sents 2 problems: (1) severe active disease during pregnancy with high antibody titers has been shown to heighten risk for morbidity and mortality for the fetus, and (2) a patient with active PV during pregnancy may require systemic therapy with doses high enough to subdue the disease. The presence of PV was a challenge throughout our patient’s pregnancy. Transient skin lesions may occasionally appear in the neonate and seem to have an increased association with severe active PV in the mother; however, neonatal PV also has been present in mild cases in the mother.7 These lesions are secondary to passive transplacental transfer of PV antibodies but do not have long-lasting clinical implications because of an antibody’s brief half-life.9 The lesions either spontaneously resolve or can be treated with a topical corticosteroid.
Treatment with high-dose systemic corticosteroids or immunosuppressants can be problematic because of the risks posed to the fetus, especially if the mother must be treated when the embryo is particularly susceptible (eg, during organogenesis).10 If a woman with known PV is planning to become pregnant, it is recommended to first control and suppress the disease so that therapy can be minimal during the pregnancy. It also is recommended to use aggressive topical therapy if possible to control PV in a pregnant woman.8 This option would not have been efficacious in our patient because of her severe widespread disease.
Prednisone is considered one of the first-line treatments of PV and has been historically successful as a treatment for pregnant patients with PV if maintained at a low dosage. Prednisone, similar to other corticosteroids, can cross the placental barrier and can increase the chance of premature birth, infection, and mortality in high doses.7 Similar to prednisone, azathioprine is not recommended during pregnancy, but if use is necessary, it is suggested to keep the dose low to prevent fetal harm.11 Inadequate treatment and control of PV can be life threatening to the patient because of the severe infection that may ensue; thus it is necessary for the health of the patient and fetus to suppress the PV. One alternative to treatment with steroids and immunosuppressants is plasma exchange, which has been successful in the clinical context of pregnancy.12 The cons of plasma exchange are repeat procedures, the need to give the patient more immunosuppressants to prevent a rejection, and the return of the autoantibody.7
Several studies have evaluated the safety and efficacy of rituximab in the treatment of refractory PV. Multiple case reports state that both 1 and 2 courses of intravenous rituximab therapy at a dosage of 375 mg per square meter of body surface area affected once weekly for 4 weeks proved to be useful in clinical improvement for patients with refractory disease.13,14 Studies are currently underway to look at the effects of rituximab on pregnancy and the fetus. Preliminary findings show neonates may have B-cell abnormalities initially yet recover fully without infectious complications or sequelae.15 Rituximab currently is a pregnancy category C drug, and women are counseled to avoid pregnancy for at least 12 months after rituximab exposure and use contraception while actively taking the drug.16
Conclusion
Contrary to traditional thinking, PV itself may be associated with poor neonatal outcome, including prematurity and fetal death. These complications seem to be restricted to pregnancies with clinically severe PV.7 Our patient decided to progress with her pregnancy despite the potential risk to the fetus from the disease and treatment. Ultimately, the infant was delivered prematurely but was free of disease.
1. Fainaru O, Mashiach R, Kupferminc M, et al. Pemphigus vulgaris in pregnancy: a case report and review of literature. Hum Reprod. 2000;15:1195-1197.
2. Joly P, Gilbert D, Thomine E, et al. Identification of a new antibody population directed against a desmosomal plaque antigen in pemphigus vulgaris and pemphigus foliaceus. J Invest Dermatol. 1997;108:469-475.
3. Daniel Y, Shenhav M, Botchan A, et al. Pregnancy associated with pemphigus. Br J Obstet Gynecol. 1995;102:667-669.
4. Ruach M, Ohel G, Rahav D, et al. Pemphigus vulgaris and pregnancy. Obstet Gynecol Surv. 1995;50:755-760.
5. Carson PJ, Hameed A, Ahmed AR. Influence of treatment on clinical course of pemphigus vulgaris. J Am Acad Dermatol. 1996;34:645-652.
6. Goldberg NS, DeFeo C, Kirshenbaum N. Pemphigus and pregnancy: risk factors and recommendations. J Am Acad Dermatol. 1993;28(5, pt 2):877-879.
7. Lehman JS, Mueller KK, Schraith DF. Do safe and effective treatment options exist for patients with active pemphigus vulgaris who plan conception and pregnancy? Arch Dermatol. 2008;144:783-785.
8. Kardos M, Levine D, Gurcan H, et al. Pemphigus vulgaris in pregnancy: analysis of current data on the management and outcomes. Obstet Gynecol Surv. 2009;64:739-749.
9. Fenniche S, Benmously R, Marrak H, et al. Neonatal pemphigus vulgaris in an infant born to a mother with pemphigus vulgaris in remission. Pediatr Dermatol. 2006;23:124-127.
10. Kalayciyan A, Engin B, Serdaroglu S, et al. A retrospective analysis of patients with pemphigus vulgaris associated with pregnancy. Br J Dermatol. 2002;147:396-397.
11. Hup JM, Bruinsma RA, Boersma ER, et al. Neonatal pemphigus vulgaris: transplacental transmission of antibodies. Pediatr Dermatol. 1986;3:468-472.
12. Piontek JO, Borberg H, Sollberg S, et al. Severe exacerbation of pemphigus vulgaris in pregnancy: successful treatment with plasma exchange. Br J Dermatol. 2000;143:455-456.
13. Faurschou A, Gniadecki R. Two courses of rituximab (anti-CD20 monoclonal antibody) for recalcitrant pemphigus vulgaris. Int J Dermatol. 2008;47:292-294.
14. Marzano AV, Fanoni D, Venegoni L, et al. Treatment of refractory pemphigus with the anti-CD20 monoclonal antibody (rituximab). Dermatology. 2007;214:310-318.
15. Braunstein I, Werth V. Treatment of dermatologic connective tissue disease and autoimmune blistering disorders in pregnancy. Dermatol Ther. 2013;26:354-363.
16. Chakravarty EF, Murray ER, Kelman A, et al. Pregnancy outcomes after maternal exposure to rituximab. Blood. 2011;117:1499-1506.
Pemphigus vulgaris (PV) is a rare autoimmune bullous dermatosis that has not shown a predilection toward a particular race or sex.1 Autoantibodies for desmoglein 1 and desmoglein 3, members of the cadherin family that are involved in cellular adhesion, have been linked to the pathogenesis of PV.2 These autoantibodies play a role in the loss of cell-to-cell adhesion in the basal and suprabasal layers of the deep epidermis while cellular adhesion in the superficial epidermis remains intact, leading to the clinical presentation of epidermal blistering and ulcerations most commonly found on the scalp, face, groin, and axillae. Diagnosis typically is made based on skin biopsy and confirmed by direct immunofluorescence. Histologically, PV displays acantholysis and suprabasal cleft formation. Immunofluorescence may show IgG antibodies against the PV antigen in the epidermis.3 Once a diagnosis has been made, treatment typically consists of systemic steroids, as the use of steroids has had great effect in preventing infections, sepsis, and fatality that were once associated with PV.4 Mortality rates associated with PV have decreased to 10% to 15% with systemic steroids from a mortality rate as high as 70% in the presteroid era.1,5 Treatment of PV during pregnancy, as in our patient, requires obstetric and pediatric consultations before therapy is initiated. Use of corticosteroids during pregnancy can be potentially dangerous to the fetus, particularly if high doses are necessary to control maternal disease.6,7
Case Report
A 34-year-old pregnant woman at 6 weeks’ gestation presented with widespread blistering dermatitis and associated burning and pruritus. Her obstetrical history was gravida 3, para 2. The patient reported a “rash” on the scalp that had developed 9 months prior. She had been treated as an outpatient at an outside institution with topical antibiotics and antifungal medications, yet the dermatitis progressed. Three weeks prior to hospitalization, the rash was present on the skin and mucosal surfaces, including the groin, chest, face, hard palate, buccal mucosa, lips (Figure 1), and back (Figure 2). Nontender bullae ruptured after 3 days, releasing clear, yellow, serous fluid with associated burning and pruritus. The bullae were hemorrhagic and erythematous at the base.
|
|
| Figure 1. Facial involvement with bullae, crusted hemorrhagic lesions, and eschar in a 34-year-old pregnant woman. |
|
|
| Figure 2. Involvement of the back with bullae in various stages. Some bullae were intact while others newly erupted. |
|
|
| Figure 3. Superinfected and flaking scalp. |
|
|
| Figure 4. Biopsy revealed suprabasal acantholysis with a tombstone effect of residual basal cells (H&E, original magnification ×200). |
At the current presentation, the patient had several excoriated 1- to 2-cm oval denudations; some were crusted with eschar. Nikolsky sign was negative. Multiple confluent bullous lesions had erupted on the entire scalp with a thick, impetiginous, yellow crust. She had a wet, boggy, foul-smelling, superinfected scalp that was mildly tender to touch with flaking tissue debris (Figure 3). A white blood cell count was 13.2×109/L (reference range, 4.5–11.0×109/L) with 5% eosinophils (reference range, 2.7%). The differential diagnosis included bullous impetigo, pemphigoid, Stevens-Johnson syndrome, dermatitis herpetiformis, and pemphigus vulgaris.
Biopsies of the scalp and back were taken and showed suprabasal acantholysis with a tombstone effect of residual basal cells standing up on the basement membrane without the characteristic acantholysis into skin appendages (Figure 4). The acantholytic cells in the bullous chamber did not round up as in Hailey-Hailey disease nor was there the dyskeratosis of Grover disease. Direct immunofluorescence on an elbow punch biopsy found diffuse 1+ intercellular IgG in the epidermis and diffuse 1+ basal intercellular C3, and was negative for IgA, IgM, and C1q, thus confirming a diagnosis of PV.
The patient was started on prednisone 20 mg once daily. An increase to prednisone 60 mg led to initial improvement of symptoms, but there was a relapse after several days, which is typical of PV in pregnancy,7 prompting the dose to be increased to 120 mg. Following alleviation of symptoms, the dose was later tapered back to 60 mg. No lesions were present at discharge or for 2.5 months thereafter, as the prednisone was tapered from 60 to 45 mg daily after discharge.
On follow-up, the patient’s PV was well controlled, but the prednisone dose was back up to 60 mg daily because of 2 new skin lesions that had developed since her last visit 2.5 months prior. Ultrasonography showed no fetal abnormalities as the pregnancy progressed to 28 weeks’ gestation. The patient developed hypertension and went into premature labor due to placenta previa. The neonate showed no skin lesions or anomalies while in the neonatal intensive care unit. The mother’s prednisone dose was tapered from 60 to 20 mg daily while the white blood cell count was 7.1×109/L with 2% eosinophils and a new scalp lesion appeared. Seven months after her initial discharge from the hospital for the dermatologic condition, she was no longer nursing and azathioprine was added to prednisone 60 mg daily.
Comment
Pemphigus vulgaris is associated with infertility in its active phase; therefore, PV during pregnancy is rare.8 Pregnancy may exacerbate PV, which has been a similar finding in other well-documented autoimmune diseases.7 One review of PV in pregnancy reported that 11 of 49 patients (22%) experienced an exacerbation of the disease.8 This finding pre-sents 2 problems: (1) severe active disease during pregnancy with high antibody titers has been shown to heighten risk for morbidity and mortality for the fetus, and (2) a patient with active PV during pregnancy may require systemic therapy with doses high enough to subdue the disease. The presence of PV was a challenge throughout our patient’s pregnancy. Transient skin lesions may occasionally appear in the neonate and seem to have an increased association with severe active PV in the mother; however, neonatal PV also has been present in mild cases in the mother.7 These lesions are secondary to passive transplacental transfer of PV antibodies but do not have long-lasting clinical implications because of an antibody’s brief half-life.9 The lesions either spontaneously resolve or can be treated with a topical corticosteroid.
Treatment with high-dose systemic corticosteroids or immunosuppressants can be problematic because of the risks posed to the fetus, especially if the mother must be treated when the embryo is particularly susceptible (eg, during organogenesis).10 If a woman with known PV is planning to become pregnant, it is recommended to first control and suppress the disease so that therapy can be minimal during the pregnancy. It also is recommended to use aggressive topical therapy if possible to control PV in a pregnant woman.8 This option would not have been efficacious in our patient because of her severe widespread disease.
Prednisone is considered one of the first-line treatments of PV and has been historically successful as a treatment for pregnant patients with PV if maintained at a low dosage. Prednisone, similar to other corticosteroids, can cross the placental barrier and can increase the chance of premature birth, infection, and mortality in high doses.7 Similar to prednisone, azathioprine is not recommended during pregnancy, but if use is necessary, it is suggested to keep the dose low to prevent fetal harm.11 Inadequate treatment and control of PV can be life threatening to the patient because of the severe infection that may ensue; thus it is necessary for the health of the patient and fetus to suppress the PV. One alternative to treatment with steroids and immunosuppressants is plasma exchange, which has been successful in the clinical context of pregnancy.12 The cons of plasma exchange are repeat procedures, the need to give the patient more immunosuppressants to prevent a rejection, and the return of the autoantibody.7
Several studies have evaluated the safety and efficacy of rituximab in the treatment of refractory PV. Multiple case reports state that both 1 and 2 courses of intravenous rituximab therapy at a dosage of 375 mg per square meter of body surface area affected once weekly for 4 weeks proved to be useful in clinical improvement for patients with refractory disease.13,14 Studies are currently underway to look at the effects of rituximab on pregnancy and the fetus. Preliminary findings show neonates may have B-cell abnormalities initially yet recover fully without infectious complications or sequelae.15 Rituximab currently is a pregnancy category C drug, and women are counseled to avoid pregnancy for at least 12 months after rituximab exposure and use contraception while actively taking the drug.16
Conclusion
Contrary to traditional thinking, PV itself may be associated with poor neonatal outcome, including prematurity and fetal death. These complications seem to be restricted to pregnancies with clinically severe PV.7 Our patient decided to progress with her pregnancy despite the potential risk to the fetus from the disease and treatment. Ultimately, the infant was delivered prematurely but was free of disease.
Pemphigus vulgaris (PV) is a rare autoimmune bullous dermatosis that has not shown a predilection toward a particular race or sex.1 Autoantibodies for desmoglein 1 and desmoglein 3, members of the cadherin family that are involved in cellular adhesion, have been linked to the pathogenesis of PV.2 These autoantibodies play a role in the loss of cell-to-cell adhesion in the basal and suprabasal layers of the deep epidermis while cellular adhesion in the superficial epidermis remains intact, leading to the clinical presentation of epidermal blistering and ulcerations most commonly found on the scalp, face, groin, and axillae. Diagnosis typically is made based on skin biopsy and confirmed by direct immunofluorescence. Histologically, PV displays acantholysis and suprabasal cleft formation. Immunofluorescence may show IgG antibodies against the PV antigen in the epidermis.3 Once a diagnosis has been made, treatment typically consists of systemic steroids, as the use of steroids has had great effect in preventing infections, sepsis, and fatality that were once associated with PV.4 Mortality rates associated with PV have decreased to 10% to 15% with systemic steroids from a mortality rate as high as 70% in the presteroid era.1,5 Treatment of PV during pregnancy, as in our patient, requires obstetric and pediatric consultations before therapy is initiated. Use of corticosteroids during pregnancy can be potentially dangerous to the fetus, particularly if high doses are necessary to control maternal disease.6,7
Case Report
A 34-year-old pregnant woman at 6 weeks’ gestation presented with widespread blistering dermatitis and associated burning and pruritus. Her obstetrical history was gravida 3, para 2. The patient reported a “rash” on the scalp that had developed 9 months prior. She had been treated as an outpatient at an outside institution with topical antibiotics and antifungal medications, yet the dermatitis progressed. Three weeks prior to hospitalization, the rash was present on the skin and mucosal surfaces, including the groin, chest, face, hard palate, buccal mucosa, lips (Figure 1), and back (Figure 2). Nontender bullae ruptured after 3 days, releasing clear, yellow, serous fluid with associated burning and pruritus. The bullae were hemorrhagic and erythematous at the base.
|
|
| Figure 1. Facial involvement with bullae, crusted hemorrhagic lesions, and eschar in a 34-year-old pregnant woman. |
|
|
| Figure 2. Involvement of the back with bullae in various stages. Some bullae were intact while others newly erupted. |
|
|
| Figure 3. Superinfected and flaking scalp. |
|
|
| Figure 4. Biopsy revealed suprabasal acantholysis with a tombstone effect of residual basal cells (H&E, original magnification ×200). |
At the current presentation, the patient had several excoriated 1- to 2-cm oval denudations; some were crusted with eschar. Nikolsky sign was negative. Multiple confluent bullous lesions had erupted on the entire scalp with a thick, impetiginous, yellow crust. She had a wet, boggy, foul-smelling, superinfected scalp that was mildly tender to touch with flaking tissue debris (Figure 3). A white blood cell count was 13.2×109/L (reference range, 4.5–11.0×109/L) with 5% eosinophils (reference range, 2.7%). The differential diagnosis included bullous impetigo, pemphigoid, Stevens-Johnson syndrome, dermatitis herpetiformis, and pemphigus vulgaris.
Biopsies of the scalp and back were taken and showed suprabasal acantholysis with a tombstone effect of residual basal cells standing up on the basement membrane without the characteristic acantholysis into skin appendages (Figure 4). The acantholytic cells in the bullous chamber did not round up as in Hailey-Hailey disease nor was there the dyskeratosis of Grover disease. Direct immunofluorescence on an elbow punch biopsy found diffuse 1+ intercellular IgG in the epidermis and diffuse 1+ basal intercellular C3, and was negative for IgA, IgM, and C1q, thus confirming a diagnosis of PV.
The patient was started on prednisone 20 mg once daily. An increase to prednisone 60 mg led to initial improvement of symptoms, but there was a relapse after several days, which is typical of PV in pregnancy,7 prompting the dose to be increased to 120 mg. Following alleviation of symptoms, the dose was later tapered back to 60 mg. No lesions were present at discharge or for 2.5 months thereafter, as the prednisone was tapered from 60 to 45 mg daily after discharge.
On follow-up, the patient’s PV was well controlled, but the prednisone dose was back up to 60 mg daily because of 2 new skin lesions that had developed since her last visit 2.5 months prior. Ultrasonography showed no fetal abnormalities as the pregnancy progressed to 28 weeks’ gestation. The patient developed hypertension and went into premature labor due to placenta previa. The neonate showed no skin lesions or anomalies while in the neonatal intensive care unit. The mother’s prednisone dose was tapered from 60 to 20 mg daily while the white blood cell count was 7.1×109/L with 2% eosinophils and a new scalp lesion appeared. Seven months after her initial discharge from the hospital for the dermatologic condition, she was no longer nursing and azathioprine was added to prednisone 60 mg daily.
Comment
Pemphigus vulgaris is associated with infertility in its active phase; therefore, PV during pregnancy is rare.8 Pregnancy may exacerbate PV, which has been a similar finding in other well-documented autoimmune diseases.7 One review of PV in pregnancy reported that 11 of 49 patients (22%) experienced an exacerbation of the disease.8 This finding pre-sents 2 problems: (1) severe active disease during pregnancy with high antibody titers has been shown to heighten risk for morbidity and mortality for the fetus, and (2) a patient with active PV during pregnancy may require systemic therapy with doses high enough to subdue the disease. The presence of PV was a challenge throughout our patient’s pregnancy. Transient skin lesions may occasionally appear in the neonate and seem to have an increased association with severe active PV in the mother; however, neonatal PV also has been present in mild cases in the mother.7 These lesions are secondary to passive transplacental transfer of PV antibodies but do not have long-lasting clinical implications because of an antibody’s brief half-life.9 The lesions either spontaneously resolve or can be treated with a topical corticosteroid.
Treatment with high-dose systemic corticosteroids or immunosuppressants can be problematic because of the risks posed to the fetus, especially if the mother must be treated when the embryo is particularly susceptible (eg, during organogenesis).10 If a woman with known PV is planning to become pregnant, it is recommended to first control and suppress the disease so that therapy can be minimal during the pregnancy. It also is recommended to use aggressive topical therapy if possible to control PV in a pregnant woman.8 This option would not have been efficacious in our patient because of her severe widespread disease.
Prednisone is considered one of the first-line treatments of PV and has been historically successful as a treatment for pregnant patients with PV if maintained at a low dosage. Prednisone, similar to other corticosteroids, can cross the placental barrier and can increase the chance of premature birth, infection, and mortality in high doses.7 Similar to prednisone, azathioprine is not recommended during pregnancy, but if use is necessary, it is suggested to keep the dose low to prevent fetal harm.11 Inadequate treatment and control of PV can be life threatening to the patient because of the severe infection that may ensue; thus it is necessary for the health of the patient and fetus to suppress the PV. One alternative to treatment with steroids and immunosuppressants is plasma exchange, which has been successful in the clinical context of pregnancy.12 The cons of plasma exchange are repeat procedures, the need to give the patient more immunosuppressants to prevent a rejection, and the return of the autoantibody.7
Several studies have evaluated the safety and efficacy of rituximab in the treatment of refractory PV. Multiple case reports state that both 1 and 2 courses of intravenous rituximab therapy at a dosage of 375 mg per square meter of body surface area affected once weekly for 4 weeks proved to be useful in clinical improvement for patients with refractory disease.13,14 Studies are currently underway to look at the effects of rituximab on pregnancy and the fetus. Preliminary findings show neonates may have B-cell abnormalities initially yet recover fully without infectious complications or sequelae.15 Rituximab currently is a pregnancy category C drug, and women are counseled to avoid pregnancy for at least 12 months after rituximab exposure and use contraception while actively taking the drug.16
Conclusion
Contrary to traditional thinking, PV itself may be associated with poor neonatal outcome, including prematurity and fetal death. These complications seem to be restricted to pregnancies with clinically severe PV.7 Our patient decided to progress with her pregnancy despite the potential risk to the fetus from the disease and treatment. Ultimately, the infant was delivered prematurely but was free of disease.
1. Fainaru O, Mashiach R, Kupferminc M, et al. Pemphigus vulgaris in pregnancy: a case report and review of literature. Hum Reprod. 2000;15:1195-1197.
2. Joly P, Gilbert D, Thomine E, et al. Identification of a new antibody population directed against a desmosomal plaque antigen in pemphigus vulgaris and pemphigus foliaceus. J Invest Dermatol. 1997;108:469-475.
3. Daniel Y, Shenhav M, Botchan A, et al. Pregnancy associated with pemphigus. Br J Obstet Gynecol. 1995;102:667-669.
4. Ruach M, Ohel G, Rahav D, et al. Pemphigus vulgaris and pregnancy. Obstet Gynecol Surv. 1995;50:755-760.
5. Carson PJ, Hameed A, Ahmed AR. Influence of treatment on clinical course of pemphigus vulgaris. J Am Acad Dermatol. 1996;34:645-652.
6. Goldberg NS, DeFeo C, Kirshenbaum N. Pemphigus and pregnancy: risk factors and recommendations. J Am Acad Dermatol. 1993;28(5, pt 2):877-879.
7. Lehman JS, Mueller KK, Schraith DF. Do safe and effective treatment options exist for patients with active pemphigus vulgaris who plan conception and pregnancy? Arch Dermatol. 2008;144:783-785.
8. Kardos M, Levine D, Gurcan H, et al. Pemphigus vulgaris in pregnancy: analysis of current data on the management and outcomes. Obstet Gynecol Surv. 2009;64:739-749.
9. Fenniche S, Benmously R, Marrak H, et al. Neonatal pemphigus vulgaris in an infant born to a mother with pemphigus vulgaris in remission. Pediatr Dermatol. 2006;23:124-127.
10. Kalayciyan A, Engin B, Serdaroglu S, et al. A retrospective analysis of patients with pemphigus vulgaris associated with pregnancy. Br J Dermatol. 2002;147:396-397.
11. Hup JM, Bruinsma RA, Boersma ER, et al. Neonatal pemphigus vulgaris: transplacental transmission of antibodies. Pediatr Dermatol. 1986;3:468-472.
12. Piontek JO, Borberg H, Sollberg S, et al. Severe exacerbation of pemphigus vulgaris in pregnancy: successful treatment with plasma exchange. Br J Dermatol. 2000;143:455-456.
13. Faurschou A, Gniadecki R. Two courses of rituximab (anti-CD20 monoclonal antibody) for recalcitrant pemphigus vulgaris. Int J Dermatol. 2008;47:292-294.
14. Marzano AV, Fanoni D, Venegoni L, et al. Treatment of refractory pemphigus with the anti-CD20 monoclonal antibody (rituximab). Dermatology. 2007;214:310-318.
15. Braunstein I, Werth V. Treatment of dermatologic connective tissue disease and autoimmune blistering disorders in pregnancy. Dermatol Ther. 2013;26:354-363.
16. Chakravarty EF, Murray ER, Kelman A, et al. Pregnancy outcomes after maternal exposure to rituximab. Blood. 2011;117:1499-1506.
1. Fainaru O, Mashiach R, Kupferminc M, et al. Pemphigus vulgaris in pregnancy: a case report and review of literature. Hum Reprod. 2000;15:1195-1197.
2. Joly P, Gilbert D, Thomine E, et al. Identification of a new antibody population directed against a desmosomal plaque antigen in pemphigus vulgaris and pemphigus foliaceus. J Invest Dermatol. 1997;108:469-475.
3. Daniel Y, Shenhav M, Botchan A, et al. Pregnancy associated with pemphigus. Br J Obstet Gynecol. 1995;102:667-669.
4. Ruach M, Ohel G, Rahav D, et al. Pemphigus vulgaris and pregnancy. Obstet Gynecol Surv. 1995;50:755-760.
5. Carson PJ, Hameed A, Ahmed AR. Influence of treatment on clinical course of pemphigus vulgaris. J Am Acad Dermatol. 1996;34:645-652.
6. Goldberg NS, DeFeo C, Kirshenbaum N. Pemphigus and pregnancy: risk factors and recommendations. J Am Acad Dermatol. 1993;28(5, pt 2):877-879.
7. Lehman JS, Mueller KK, Schraith DF. Do safe and effective treatment options exist for patients with active pemphigus vulgaris who plan conception and pregnancy? Arch Dermatol. 2008;144:783-785.
8. Kardos M, Levine D, Gurcan H, et al. Pemphigus vulgaris in pregnancy: analysis of current data on the management and outcomes. Obstet Gynecol Surv. 2009;64:739-749.
9. Fenniche S, Benmously R, Marrak H, et al. Neonatal pemphigus vulgaris in an infant born to a mother with pemphigus vulgaris in remission. Pediatr Dermatol. 2006;23:124-127.
10. Kalayciyan A, Engin B, Serdaroglu S, et al. A retrospective analysis of patients with pemphigus vulgaris associated with pregnancy. Br J Dermatol. 2002;147:396-397.
11. Hup JM, Bruinsma RA, Boersma ER, et al. Neonatal pemphigus vulgaris: transplacental transmission of antibodies. Pediatr Dermatol. 1986;3:468-472.
12. Piontek JO, Borberg H, Sollberg S, et al. Severe exacerbation of pemphigus vulgaris in pregnancy: successful treatment with plasma exchange. Br J Dermatol. 2000;143:455-456.
13. Faurschou A, Gniadecki R. Two courses of rituximab (anti-CD20 monoclonal antibody) for recalcitrant pemphigus vulgaris. Int J Dermatol. 2008;47:292-294.
14. Marzano AV, Fanoni D, Venegoni L, et al. Treatment of refractory pemphigus with the anti-CD20 monoclonal antibody (rituximab). Dermatology. 2007;214:310-318.
15. Braunstein I, Werth V. Treatment of dermatologic connective tissue disease and autoimmune blistering disorders in pregnancy. Dermatol Ther. 2013;26:354-363.
16. Chakravarty EF, Murray ER, Kelman A, et al. Pregnancy outcomes after maternal exposure to rituximab. Blood. 2011;117:1499-1506.
Practice Points
- Early diagnosis and appropriate treatment of pemphigus vulgaris in pregnancy is paramount in protecting the health of the mother and fetus.
- Management of autoimmune diseases during pregnancy continues to present numerous challenges for physicians due to the pathology of the diseases as well as the sensitive nature of pregnancy and lack of robust data in this patient population.
Granulomatous Changes Associated With Pigmented Purpuric Dermatosis
Pigmented purpuric dermatoses (PPDs) are a group of common chronic disorders characterized by speckled, cayenne pepper–like petechiae and orange-brown discoloration of the skin resulting from capillaritis.1 Pigmented purpuric dermatoses typically occur in the absence of underlying vascular insufficiency or other hematologic dysfunction. The 5 well-known clinicopathologic variants of PPD include Schamberg disease; purpura annularis telangiectodes of Majocchi; pigmented purpuric lichenoid dermatitis of Gougerot and Blum; eczematoidlike purpura of Doucas and Kapetanakis; and lichen aureus.2 All PPDs share common characteristic clinical and histologic features. Clinically, patients generally present with symmetric petechiae and/or pigmented macules. All 5 PPD variants share similar histologic findings, including a vasculocentric lymphocytic infiltrate in the papillary dermis, swelling of the endothelial cells, erythrocyte extravasation, and often hemosiderin-laden macrophages.1 Despite these clinical and histopathologic similarities, each variant contains additional distinctive features, such as telangiectasia (purpura annularis telangiectodes of Majocchi), a lichenoid infiltrate (pigmented purpuric lichenoid dermatitis of Gougerot and Blum), eczematous changes (eczematoidlike purpura of Doucas and Kapetanakis), and marked hemosiderin deposition (lichen aureus).
Granulomatous pigmented purpuric dermatosis (GPPD) is a rare variant of PPD.3-7 Clinically, these lesions appear similar to other PPDs; however, in addition to the characteristic changes associated with conventional PPD, histologic examination of GPPD reveals a granulomatous inflammatory reaction pattern. Although the pathogenesis of GPPD is not well understood, its association with hyperlipidemia may suggest a granulomatous response to capillaritis mediated by lipid deposition in the microvasculature.6,7
We present 3 cases of GPPD and provide a review of the literature. In all of our patients, biopsy specimens were fixed in 10% buffered formalin and embedded in paraffin by standard methodologies, and all stains were performed on sections by standard methodologies. Based on a PubMed search of articles indexed for MEDLINE using the terms granulomatous pigmented purpuric dermatosis, sarcoidosis, pigmented purpuric dermatosis, granulomas, and pigmented purpuric dermatosis, we review 5 additional reports describing 10 total patients.3-7
Case Reports
Patient 1
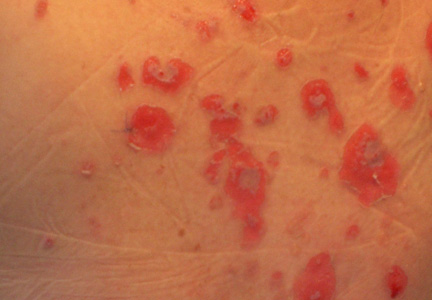
A 9-year-old white boy presented with a 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (Figure, A). The lesion appeared 3 to 4 years prior to presentation but had become progressively darker and centrally indurated over the last 2 years. The patient and his mother denied any history of trauma to the area. His medical history was unremarkable, and his current medications included fish oil and multivitamin tablets.
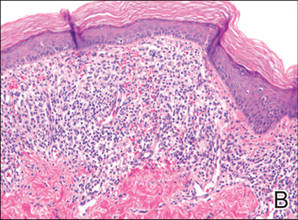
Histologic examination of a punch biopsy specimen taken from the center of the lesion revealed a lichenoid lymphohistiocytic infiltrate with marked red blood cell (RBC) extravasation and associated hemosiderin-laden macrophages. The lymphocytes comprising this infiltrate lacked cytologic atypia and exhibited minimal epidermotropism (Figure, B). Additionally, there was a superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised ofepithelioid histiocytes in the mid and deep dermis (Figure, C). Periodic acid–Schiff, acid-fast bacilli (AFB), and Fite stains were negative for organisms. Polarization was negative for refractile foreign material. Due to the patient’s age, no treatment was performed, and the lesion remains unchanged 1 year after biopsy.
Patient 2
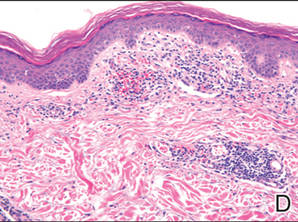
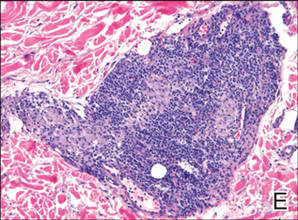
A 49-year-old white woman presented with a 2-cm yellow-brown patch with a faint, nonblanchable, violaceous center on the right lateral thigh of 4 months’ duration. The patch initially appeared as a small asymptomatic purple papule. The patient denied any history of trauma to the area. A purified protein derivative (tuberculin) skin test was negative at the time of examination. The patient’s medical history was remarkable for renal calculi. Her current medications included progesterone; estradiol; lansoprazole; prenatal vitamins; vitamins C and E; zinc; and calcium. The patient had no family history of sarcoidosis. Complete blood cell count, urinalysis, liver function tests, and angiotensin-converting enzyme levels were unremarkable. Pulmonary function tests were normal, and there was no evidence of sarcoidosis on chest radiography. Initial biopsy of the lesion revealed a perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated RBCs in the papillary dermis (Figure, D). Similar to patient 1, the infiltrate lacked cytologic atypia and did not involve the overlying epidermis. There was perivascular granulomatous inflammation in the mid dermis (Figure, E). Periodic acid–Schiff, Warthin-Starry, and AFB stains were negative for organisms. Polarization was negative for refractile foreign material.
The lesion was treated with clobetasol propionate ointment 0.05% twice daily for 6 weeks with transient improvement, but the lesion recurred upon treatment cessation. Subsequent treatment with intralesional triamcinolone resulted in slight improvement of the lesion. A therapeutic trial of targeted pulsed dye laser treatment was ineffective. The lesion gradually increased in size over the next year with no therapy, and a repeat biopsy revealed a lichenoid lymphohistiocytic infiltrate with abundant extravasated RBCs consistent with persistent PPD. A granulomatous infiltrate was not evident in the superficial shave biopsy specimen.
Patient 3
A 75-year-old white woman presented with scattered, speckled, cayenne pepper–like, red-brown macules on the legs. Two years prior to presentation, a few scattered symmetrical macules appeared on the dorsal aspects of the feet, which gradually increased in number to form larger confluent patches that spread to the lower legs. The patient denied itching or burning but reported that the patches became painful when scratched and were aggravated by sun exposure. Her medical history was remarkable for asthma, chronic renal insufficiency, coronary artery disease, Barrett esophagus, obstructive sleep apnea, hypothyroidism, renal calculi, type 2 diabetes mellitus, and hyperlipidemia. Her current medications included carvedilol, valsartan, levothyroxine, aspirin, clopidogrel, furosemide, nitrofurantoin, temazepam, insulin, ezetimibe-simvastatin, and lansoprazole. Computed tomography of the chest revealed no signs of sarcoidosis. Pulmonary function tests revealed moderate obstructive lung disease. An ophthalmology examination showed no evidence of sarcoidosis. Laboratory results revealed an elevated glucose, blood urea nitrogen, creatinine, and triglyceride levels, as well as low hematocrit and vitamin D levels. Urinalysis, thyroid-stimulating hormone (thyrotropin) test, liver function tests, angiotensin-converting enzyme test, hepatitis B surface antigen, and IFN-g release assay were normal.
Histologic examination of a punch biopsy specimen revealed an inflammatory infiltrate confined to the papillary dermis. This infiltrate was comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated RBC extravasation and intimately associated granulomas (Figure, F). Additional inflammation in the deeper aspects of the dermis was not identified. Periodic acid–Schiff, AFB, and Fite stains were negative for organisms. Polarization was negative for refractile foreign material.

| 
| 
| ||
| 
| 
| ||
| A 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (A). Lichenoid lymphohistiocytic infiltrate in the papillary dermis with marked red blood cell extravasation (B)(H&E, original magnification ×20). Superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised of epithelioid histiocytes in the mid and deep dermis (C)(H&E, original magnification ×20). Perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated red blood cells in the papillary dermis (D)(H&E, original magnification ×10). Perivascular lymphohistiocytic inflammation with epithelioid granulomas in the mid dermis (E)(H&E, original magnification ×20). Lymphohistiocytic inflammation in the papillary dermis comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated red blood cell extravasation and intimately associated granulomas (F)(H&E, original magnification ×20). | ||||
The patient was treated with topical steroids and minocycline 50 mg twice daily without improvement. The lesions improved after the patient underwent treatment with oral corticosteroids for pulmonary disease.
Comment
Pigmented purpuric dermatoses comprise a spectrum of clinical and pathologic conditions.1,2 Granulomatous PPD is a much less common variant, characterized by a granulomatous infiltrate admixed with PPD. We report 3 additional cases and review the literature on this rare and interesting variant of PPD.
We noted several unifying clinicopathologic features among our patients and those previously reported in the literature (Table).3-7 Including our cases, our review yielded 13 GPPD patients ranging in age from 9 to 75 years, with a mean age of 49.1 years. Two of our patients—patients 1 and 3—were the youngest and oldest patients, respectively, among the cases we reviewed. The majority of the cases we reviewed included patients of East Asian descent (6 Taiwanese; 2 Japanese; 1 Korean) as well as 4 white patients. No distinctive gender predilection was apparent, as our review included 8 females and 5 males.
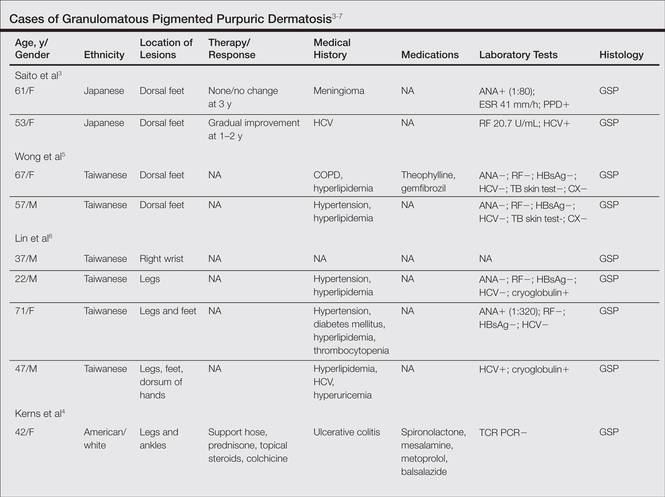
Our review revealed that GPPD lesions typically involve the lower extremities and usually are asymptomatic, with the exception of occasional pruritus. Additional lesions have been reported on the dorsal aspect of the hands, and 1 case noted exclusive involvement of the wrist.6 Lesions of GPPD can range in their clinical appearance. Three of 13 patients presented with purpuric papules and 2 had brown pigmentation with hemorrhagic papules3,4,6; the remaining 8 patients had erythematous or brown macules, papules, or plaques.5-7 The most commonly associated disease condition was hyperlipidemia, which was reported in 7 of 13 cases.5-7 Additional reported comorbidities included meningioma, renal calculi, obesity, hypertension, diabetes mellitus, chronic obstructive pulmonary disease, hepatitis C virus, ulcerative colitis, thrombocytopenia, and hyperuricemia. Reported serologic abnormalities included a rare positive antinuclear antibody, rheumatoid factor, and cryoglobulins.3,6 Therapeutic efficacy in the management of GPPD has not been well described; however, for the rare cases in which therapies were described, they were largely unsuccessful, with 1 patient exhibiting spontaneous improvement.3,4
Granulomatous PPD also appears to exhibit a range of histologic findings. All cases of GPPD shared fundamental components, such as a brisk perivascular infiltrate accompanied by RBC extravasation with variable hemosiderin-laden macrophages and a granulomatous infiltrate. All of the reports we reviewed described an intimate association between these components, with the granulomas being essentially superimposed on typical PPD. As for other types of PPD, obvious vasculitis characterized by a vasculocentric inflammatory infiltrate and evidence of vascular injury, such as fibrinoid necrosis of the vessel wall, has not been described in GPPD.3-7 Finally, histologic changes suggestive of a relationship with cutaneous T-cell lymphoma, cytologic atypia, and epidermotropism have been described for some forms of PPD but have not been described for GPPD.3-8
Our case reports expand the histologic spectrum of GPPD. Although patient 3 exhibited a relatively intimate association of granulomas and PPD, 2 of our cases (patients 1 and 2) demonstrated a granulomatous infiltrate in the mid to deep dermis, which is separate from the more superficially situated lichenoid lymphohistiocytic infiltrate, RBC extravasation, and hemosiderin-laden macrophages noted in the papillary dermis. Considered along with the absence of an obvious clinicopathologic explanation for the granulomatous infiltrates (eg, polarizable material, infectious organisms, systemic disease), these 2 cases (patients 1 and 2) suggest a composite form of PPD that combines the lichenoid pattern of PPD of Gougerot and Blum with a deep granulomatous component of GPPD. The importance of this distinction lies in the potential for physicians to overlook this potentially informative histologic pattern if only a superficial biopsy is performed. The clinical relevance is unclear; however, in our experience, it has been challenging to treat this relatively small subset of patients who have exhibited a limited response to treatment with topical steroids, intralesional steroids, pulsed dye laser, and vitamin supplementation.
The cause of the granulomatous infiltrate in GPPD is poorly understood. Seven of 13 cases included in our review occurred in patients with a history of hyperlipidemia.5-7 Some have postulated that the constellation of findings of GPPD in hyperlipidemic patients reflects an underlying vascular injury process induced by lipid deposition in the endothelial cells with subsequent RBC extravasation and a secondary granulomatous response to the lipid deposits.6,7 However, given the occurrence of GPPD in patients without hyperlipidemia, other mechanisms also must be considered in the pathogenesis of GPPD, including a reaction to medications, systemic diseases, and infectious etiologies (eg, hepatitis B virus).4,6 As additional cases are described in the literature, other unifying clinical etiologies for this histopathologic reaction pattern may emerge.
Conclusion
Granulomatous PPD may comprise an underrecognized variant of PPD in cases when only a superficial biopsy is evaluated. Clinicians and pathologists should be aware of this variant, and in refractory cases of PPD, deeper sampling may be warranted to identify granulomas.
1. Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
2. Piette WW. Purpura: mechanisms and differential diagnosis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby; 2008:321-330.
3. Saito R, Matsuoka Y. Granulomatous pigmented purpuric dermatosis. J Dermatol. 1996;23:551-555.
4. Kerns MJ, Mallatt BD, Shamma HN. Granulomatous pigmented purpura: an unusual histological variant. Am J Dermatopathol. 2009;31:77-80.
5. Wong WR, Kuo TT, Chen MJ, et al. Granulomatous variant of chronic pigmented purpuric dermatosis: report of two cases. Br J Dermatol. 2001;145:162-164.
6. Lin WL, Kou TT, Shih PY, et al. Granulomatous variant of chronic pigmented purpuric dermatoses: report of four new cases and an association with hyperlipidaemia [published online ahead of print May 29, 2007]. Clin Exp Dermatol. 2007;32:513-515.
7. Lee SH, Kwon JE, Lee KG, et al. Granulomatous variant of chronic pigmented purpuric dermatosis associated with hyperlipidemia. J Eur Acad Dermatol Venereol. 2010;24:1243-1245.
8. Toro JR, Sander CA, LeBoit PE. Persistent pigmented dermatoses and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
Pigmented purpuric dermatoses (PPDs) are a group of common chronic disorders characterized by speckled, cayenne pepper–like petechiae and orange-brown discoloration of the skin resulting from capillaritis.1 Pigmented purpuric dermatoses typically occur in the absence of underlying vascular insufficiency or other hematologic dysfunction. The 5 well-known clinicopathologic variants of PPD include Schamberg disease; purpura annularis telangiectodes of Majocchi; pigmented purpuric lichenoid dermatitis of Gougerot and Blum; eczematoidlike purpura of Doucas and Kapetanakis; and lichen aureus.2 All PPDs share common characteristic clinical and histologic features. Clinically, patients generally present with symmetric petechiae and/or pigmented macules. All 5 PPD variants share similar histologic findings, including a vasculocentric lymphocytic infiltrate in the papillary dermis, swelling of the endothelial cells, erythrocyte extravasation, and often hemosiderin-laden macrophages.1 Despite these clinical and histopathologic similarities, each variant contains additional distinctive features, such as telangiectasia (purpura annularis telangiectodes of Majocchi), a lichenoid infiltrate (pigmented purpuric lichenoid dermatitis of Gougerot and Blum), eczematous changes (eczematoidlike purpura of Doucas and Kapetanakis), and marked hemosiderin deposition (lichen aureus).
Granulomatous pigmented purpuric dermatosis (GPPD) is a rare variant of PPD.3-7 Clinically, these lesions appear similar to other PPDs; however, in addition to the characteristic changes associated with conventional PPD, histologic examination of GPPD reveals a granulomatous inflammatory reaction pattern. Although the pathogenesis of GPPD is not well understood, its association with hyperlipidemia may suggest a granulomatous response to capillaritis mediated by lipid deposition in the microvasculature.6,7
We present 3 cases of GPPD and provide a review of the literature. In all of our patients, biopsy specimens were fixed in 10% buffered formalin and embedded in paraffin by standard methodologies, and all stains were performed on sections by standard methodologies. Based on a PubMed search of articles indexed for MEDLINE using the terms granulomatous pigmented purpuric dermatosis, sarcoidosis, pigmented purpuric dermatosis, granulomas, and pigmented purpuric dermatosis, we review 5 additional reports describing 10 total patients.3-7
Case Reports
Patient 1
A 9-year-old white boy presented with a 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (Figure, A). The lesion appeared 3 to 4 years prior to presentation but had become progressively darker and centrally indurated over the last 2 years. The patient and his mother denied any history of trauma to the area. His medical history was unremarkable, and his current medications included fish oil and multivitamin tablets.
Histologic examination of a punch biopsy specimen taken from the center of the lesion revealed a lichenoid lymphohistiocytic infiltrate with marked red blood cell (RBC) extravasation and associated hemosiderin-laden macrophages. The lymphocytes comprising this infiltrate lacked cytologic atypia and exhibited minimal epidermotropism (Figure, B). Additionally, there was a superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised ofepithelioid histiocytes in the mid and deep dermis (Figure, C). Periodic acid–Schiff, acid-fast bacilli (AFB), and Fite stains were negative for organisms. Polarization was negative for refractile foreign material. Due to the patient’s age, no treatment was performed, and the lesion remains unchanged 1 year after biopsy.
Patient 2
A 49-year-old white woman presented with a 2-cm yellow-brown patch with a faint, nonblanchable, violaceous center on the right lateral thigh of 4 months’ duration. The patch initially appeared as a small asymptomatic purple papule. The patient denied any history of trauma to the area. A purified protein derivative (tuberculin) skin test was negative at the time of examination. The patient’s medical history was remarkable for renal calculi. Her current medications included progesterone; estradiol; lansoprazole; prenatal vitamins; vitamins C and E; zinc; and calcium. The patient had no family history of sarcoidosis. Complete blood cell count, urinalysis, liver function tests, and angiotensin-converting enzyme levels were unremarkable. Pulmonary function tests were normal, and there was no evidence of sarcoidosis on chest radiography. Initial biopsy of the lesion revealed a perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated RBCs in the papillary dermis (Figure, D). Similar to patient 1, the infiltrate lacked cytologic atypia and did not involve the overlying epidermis. There was perivascular granulomatous inflammation in the mid dermis (Figure, E). Periodic acid–Schiff, Warthin-Starry, and AFB stains were negative for organisms. Polarization was negative for refractile foreign material.
The lesion was treated with clobetasol propionate ointment 0.05% twice daily for 6 weeks with transient improvement, but the lesion recurred upon treatment cessation. Subsequent treatment with intralesional triamcinolone resulted in slight improvement of the lesion. A therapeutic trial of targeted pulsed dye laser treatment was ineffective. The lesion gradually increased in size over the next year with no therapy, and a repeat biopsy revealed a lichenoid lymphohistiocytic infiltrate with abundant extravasated RBCs consistent with persistent PPD. A granulomatous infiltrate was not evident in the superficial shave biopsy specimen.
Patient 3
A 75-year-old white woman presented with scattered, speckled, cayenne pepper–like, red-brown macules on the legs. Two years prior to presentation, a few scattered symmetrical macules appeared on the dorsal aspects of the feet, which gradually increased in number to form larger confluent patches that spread to the lower legs. The patient denied itching or burning but reported that the patches became painful when scratched and were aggravated by sun exposure. Her medical history was remarkable for asthma, chronic renal insufficiency, coronary artery disease, Barrett esophagus, obstructive sleep apnea, hypothyroidism, renal calculi, type 2 diabetes mellitus, and hyperlipidemia. Her current medications included carvedilol, valsartan, levothyroxine, aspirin, clopidogrel, furosemide, nitrofurantoin, temazepam, insulin, ezetimibe-simvastatin, and lansoprazole. Computed tomography of the chest revealed no signs of sarcoidosis. Pulmonary function tests revealed moderate obstructive lung disease. An ophthalmology examination showed no evidence of sarcoidosis. Laboratory results revealed an elevated glucose, blood urea nitrogen, creatinine, and triglyceride levels, as well as low hematocrit and vitamin D levels. Urinalysis, thyroid-stimulating hormone (thyrotropin) test, liver function tests, angiotensin-converting enzyme test, hepatitis B surface antigen, and IFN-g release assay were normal.
Histologic examination of a punch biopsy specimen revealed an inflammatory infiltrate confined to the papillary dermis. This infiltrate was comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated RBC extravasation and intimately associated granulomas (Figure, F). Additional inflammation in the deeper aspects of the dermis was not identified. Periodic acid–Schiff, AFB, and Fite stains were negative for organisms. Polarization was negative for refractile foreign material.
|
|
|
| ||
|
|
|
| ||
| A 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (A). Lichenoid lymphohistiocytic infiltrate in the papillary dermis with marked red blood cell extravasation (B)(H&E, original magnification ×20). Superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised of epithelioid histiocytes in the mid and deep dermis (C)(H&E, original magnification ×20). Perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated red blood cells in the papillary dermis (D)(H&E, original magnification ×10). Perivascular lymphohistiocytic inflammation with epithelioid granulomas in the mid dermis (E)(H&E, original magnification ×20). Lymphohistiocytic inflammation in the papillary dermis comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated red blood cell extravasation and intimately associated granulomas (F)(H&E, original magnification ×20). | ||||
The patient was treated with topical steroids and minocycline 50 mg twice daily without improvement. The lesions improved after the patient underwent treatment with oral corticosteroids for pulmonary disease.
Comment
Pigmented purpuric dermatoses comprise a spectrum of clinical and pathologic conditions.1,2 Granulomatous PPD is a much less common variant, characterized by a granulomatous infiltrate admixed with PPD. We report 3 additional cases and review the literature on this rare and interesting variant of PPD.
We noted several unifying clinicopathologic features among our patients and those previously reported in the literature (Table).3-7 Including our cases, our review yielded 13 GPPD patients ranging in age from 9 to 75 years, with a mean age of 49.1 years. Two of our patients—patients 1 and 3—were the youngest and oldest patients, respectively, among the cases we reviewed. The majority of the cases we reviewed included patients of East Asian descent (6 Taiwanese; 2 Japanese; 1 Korean) as well as 4 white patients. No distinctive gender predilection was apparent, as our review included 8 females and 5 males.
Our review revealed that GPPD lesions typically involve the lower extremities and usually are asymptomatic, with the exception of occasional pruritus. Additional lesions have been reported on the dorsal aspect of the hands, and 1 case noted exclusive involvement of the wrist.6 Lesions of GPPD can range in their clinical appearance. Three of 13 patients presented with purpuric papules and 2 had brown pigmentation with hemorrhagic papules3,4,6; the remaining 8 patients had erythematous or brown macules, papules, or plaques.5-7 The most commonly associated disease condition was hyperlipidemia, which was reported in 7 of 13 cases.5-7 Additional reported comorbidities included meningioma, renal calculi, obesity, hypertension, diabetes mellitus, chronic obstructive pulmonary disease, hepatitis C virus, ulcerative colitis, thrombocytopenia, and hyperuricemia. Reported serologic abnormalities included a rare positive antinuclear antibody, rheumatoid factor, and cryoglobulins.3,6 Therapeutic efficacy in the management of GPPD has not been well described; however, for the rare cases in which therapies were described, they were largely unsuccessful, with 1 patient exhibiting spontaneous improvement.3,4
Granulomatous PPD also appears to exhibit a range of histologic findings. All cases of GPPD shared fundamental components, such as a brisk perivascular infiltrate accompanied by RBC extravasation with variable hemosiderin-laden macrophages and a granulomatous infiltrate. All of the reports we reviewed described an intimate association between these components, with the granulomas being essentially superimposed on typical PPD. As for other types of PPD, obvious vasculitis characterized by a vasculocentric inflammatory infiltrate and evidence of vascular injury, such as fibrinoid necrosis of the vessel wall, has not been described in GPPD.3-7 Finally, histologic changes suggestive of a relationship with cutaneous T-cell lymphoma, cytologic atypia, and epidermotropism have been described for some forms of PPD but have not been described for GPPD.3-8
Our case reports expand the histologic spectrum of GPPD. Although patient 3 exhibited a relatively intimate association of granulomas and PPD, 2 of our cases (patients 1 and 2) demonstrated a granulomatous infiltrate in the mid to deep dermis, which is separate from the more superficially situated lichenoid lymphohistiocytic infiltrate, RBC extravasation, and hemosiderin-laden macrophages noted in the papillary dermis. Considered along with the absence of an obvious clinicopathologic explanation for the granulomatous infiltrates (eg, polarizable material, infectious organisms, systemic disease), these 2 cases (patients 1 and 2) suggest a composite form of PPD that combines the lichenoid pattern of PPD of Gougerot and Blum with a deep granulomatous component of GPPD. The importance of this distinction lies in the potential for physicians to overlook this potentially informative histologic pattern if only a superficial biopsy is performed. The clinical relevance is unclear; however, in our experience, it has been challenging to treat this relatively small subset of patients who have exhibited a limited response to treatment with topical steroids, intralesional steroids, pulsed dye laser, and vitamin supplementation.
The cause of the granulomatous infiltrate in GPPD is poorly understood. Seven of 13 cases included in our review occurred in patients with a history of hyperlipidemia.5-7 Some have postulated that the constellation of findings of GPPD in hyperlipidemic patients reflects an underlying vascular injury process induced by lipid deposition in the endothelial cells with subsequent RBC extravasation and a secondary granulomatous response to the lipid deposits.6,7 However, given the occurrence of GPPD in patients without hyperlipidemia, other mechanisms also must be considered in the pathogenesis of GPPD, including a reaction to medications, systemic diseases, and infectious etiologies (eg, hepatitis B virus).4,6 As additional cases are described in the literature, other unifying clinical etiologies for this histopathologic reaction pattern may emerge.
Conclusion
Granulomatous PPD may comprise an underrecognized variant of PPD in cases when only a superficial biopsy is evaluated. Clinicians and pathologists should be aware of this variant, and in refractory cases of PPD, deeper sampling may be warranted to identify granulomas.
Pigmented purpuric dermatoses (PPDs) are a group of common chronic disorders characterized by speckled, cayenne pepper–like petechiae and orange-brown discoloration of the skin resulting from capillaritis.1 Pigmented purpuric dermatoses typically occur in the absence of underlying vascular insufficiency or other hematologic dysfunction. The 5 well-known clinicopathologic variants of PPD include Schamberg disease; purpura annularis telangiectodes of Majocchi; pigmented purpuric lichenoid dermatitis of Gougerot and Blum; eczematoidlike purpura of Doucas and Kapetanakis; and lichen aureus.2 All PPDs share common characteristic clinical and histologic features. Clinically, patients generally present with symmetric petechiae and/or pigmented macules. All 5 PPD variants share similar histologic findings, including a vasculocentric lymphocytic infiltrate in the papillary dermis, swelling of the endothelial cells, erythrocyte extravasation, and often hemosiderin-laden macrophages.1 Despite these clinical and histopathologic similarities, each variant contains additional distinctive features, such as telangiectasia (purpura annularis telangiectodes of Majocchi), a lichenoid infiltrate (pigmented purpuric lichenoid dermatitis of Gougerot and Blum), eczematous changes (eczematoidlike purpura of Doucas and Kapetanakis), and marked hemosiderin deposition (lichen aureus).
Granulomatous pigmented purpuric dermatosis (GPPD) is a rare variant of PPD.3-7 Clinically, these lesions appear similar to other PPDs; however, in addition to the characteristic changes associated with conventional PPD, histologic examination of GPPD reveals a granulomatous inflammatory reaction pattern. Although the pathogenesis of GPPD is not well understood, its association with hyperlipidemia may suggest a granulomatous response to capillaritis mediated by lipid deposition in the microvasculature.6,7
We present 3 cases of GPPD and provide a review of the literature. In all of our patients, biopsy specimens were fixed in 10% buffered formalin and embedded in paraffin by standard methodologies, and all stains were performed on sections by standard methodologies. Based on a PubMed search of articles indexed for MEDLINE using the terms granulomatous pigmented purpuric dermatosis, sarcoidosis, pigmented purpuric dermatosis, granulomas, and pigmented purpuric dermatosis, we review 5 additional reports describing 10 total patients.3-7
Case Reports
Patient 1
A 9-year-old white boy presented with a 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (Figure, A). The lesion appeared 3 to 4 years prior to presentation but had become progressively darker and centrally indurated over the last 2 years. The patient and his mother denied any history of trauma to the area. His medical history was unremarkable, and his current medications included fish oil and multivitamin tablets.
Histologic examination of a punch biopsy specimen taken from the center of the lesion revealed a lichenoid lymphohistiocytic infiltrate with marked red blood cell (RBC) extravasation and associated hemosiderin-laden macrophages. The lymphocytes comprising this infiltrate lacked cytologic atypia and exhibited minimal epidermotropism (Figure, B). Additionally, there was a superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised ofepithelioid histiocytes in the mid and deep dermis (Figure, C). Periodic acid–Schiff, acid-fast bacilli (AFB), and Fite stains were negative for organisms. Polarization was negative for refractile foreign material. Due to the patient’s age, no treatment was performed, and the lesion remains unchanged 1 year after biopsy.
Patient 2
A 49-year-old white woman presented with a 2-cm yellow-brown patch with a faint, nonblanchable, violaceous center on the right lateral thigh of 4 months’ duration. The patch initially appeared as a small asymptomatic purple papule. The patient denied any history of trauma to the area. A purified protein derivative (tuberculin) skin test was negative at the time of examination. The patient’s medical history was remarkable for renal calculi. Her current medications included progesterone; estradiol; lansoprazole; prenatal vitamins; vitamins C and E; zinc; and calcium. The patient had no family history of sarcoidosis. Complete blood cell count, urinalysis, liver function tests, and angiotensin-converting enzyme levels were unremarkable. Pulmonary function tests were normal, and there was no evidence of sarcoidosis on chest radiography. Initial biopsy of the lesion revealed a perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated RBCs in the papillary dermis (Figure, D). Similar to patient 1, the infiltrate lacked cytologic atypia and did not involve the overlying epidermis. There was perivascular granulomatous inflammation in the mid dermis (Figure, E). Periodic acid–Schiff, Warthin-Starry, and AFB stains were negative for organisms. Polarization was negative for refractile foreign material.
The lesion was treated with clobetasol propionate ointment 0.05% twice daily for 6 weeks with transient improvement, but the lesion recurred upon treatment cessation. Subsequent treatment with intralesional triamcinolone resulted in slight improvement of the lesion. A therapeutic trial of targeted pulsed dye laser treatment was ineffective. The lesion gradually increased in size over the next year with no therapy, and a repeat biopsy revealed a lichenoid lymphohistiocytic infiltrate with abundant extravasated RBCs consistent with persistent PPD. A granulomatous infiltrate was not evident in the superficial shave biopsy specimen.
Patient 3
A 75-year-old white woman presented with scattered, speckled, cayenne pepper–like, red-brown macules on the legs. Two years prior to presentation, a few scattered symmetrical macules appeared on the dorsal aspects of the feet, which gradually increased in number to form larger confluent patches that spread to the lower legs. The patient denied itching or burning but reported that the patches became painful when scratched and were aggravated by sun exposure. Her medical history was remarkable for asthma, chronic renal insufficiency, coronary artery disease, Barrett esophagus, obstructive sleep apnea, hypothyroidism, renal calculi, type 2 diabetes mellitus, and hyperlipidemia. Her current medications included carvedilol, valsartan, levothyroxine, aspirin, clopidogrel, furosemide, nitrofurantoin, temazepam, insulin, ezetimibe-simvastatin, and lansoprazole. Computed tomography of the chest revealed no signs of sarcoidosis. Pulmonary function tests revealed moderate obstructive lung disease. An ophthalmology examination showed no evidence of sarcoidosis. Laboratory results revealed an elevated glucose, blood urea nitrogen, creatinine, and triglyceride levels, as well as low hematocrit and vitamin D levels. Urinalysis, thyroid-stimulating hormone (thyrotropin) test, liver function tests, angiotensin-converting enzyme test, hepatitis B surface antigen, and IFN-g release assay were normal.
Histologic examination of a punch biopsy specimen revealed an inflammatory infiltrate confined to the papillary dermis. This infiltrate was comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated RBC extravasation and intimately associated granulomas (Figure, F). Additional inflammation in the deeper aspects of the dermis was not identified. Periodic acid–Schiff, AFB, and Fite stains were negative for organisms. Polarization was negative for refractile foreign material.
|
|
|
| ||
|
|
|
| ||
| A 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (A). Lichenoid lymphohistiocytic infiltrate in the papillary dermis with marked red blood cell extravasation (B)(H&E, original magnification ×20). Superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised of epithelioid histiocytes in the mid and deep dermis (C)(H&E, original magnification ×20). Perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated red blood cells in the papillary dermis (D)(H&E, original magnification ×10). Perivascular lymphohistiocytic inflammation with epithelioid granulomas in the mid dermis (E)(H&E, original magnification ×20). Lymphohistiocytic inflammation in the papillary dermis comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated red blood cell extravasation and intimately associated granulomas (F)(H&E, original magnification ×20). | ||||
The patient was treated with topical steroids and minocycline 50 mg twice daily without improvement. The lesions improved after the patient underwent treatment with oral corticosteroids for pulmonary disease.
Comment
Pigmented purpuric dermatoses comprise a spectrum of clinical and pathologic conditions.1,2 Granulomatous PPD is a much less common variant, characterized by a granulomatous infiltrate admixed with PPD. We report 3 additional cases and review the literature on this rare and interesting variant of PPD.
We noted several unifying clinicopathologic features among our patients and those previously reported in the literature (Table).3-7 Including our cases, our review yielded 13 GPPD patients ranging in age from 9 to 75 years, with a mean age of 49.1 years. Two of our patients—patients 1 and 3—were the youngest and oldest patients, respectively, among the cases we reviewed. The majority of the cases we reviewed included patients of East Asian descent (6 Taiwanese; 2 Japanese; 1 Korean) as well as 4 white patients. No distinctive gender predilection was apparent, as our review included 8 females and 5 males.
Our review revealed that GPPD lesions typically involve the lower extremities and usually are asymptomatic, with the exception of occasional pruritus. Additional lesions have been reported on the dorsal aspect of the hands, and 1 case noted exclusive involvement of the wrist.6 Lesions of GPPD can range in their clinical appearance. Three of 13 patients presented with purpuric papules and 2 had brown pigmentation with hemorrhagic papules3,4,6; the remaining 8 patients had erythematous or brown macules, papules, or plaques.5-7 The most commonly associated disease condition was hyperlipidemia, which was reported in 7 of 13 cases.5-7 Additional reported comorbidities included meningioma, renal calculi, obesity, hypertension, diabetes mellitus, chronic obstructive pulmonary disease, hepatitis C virus, ulcerative colitis, thrombocytopenia, and hyperuricemia. Reported serologic abnormalities included a rare positive antinuclear antibody, rheumatoid factor, and cryoglobulins.3,6 Therapeutic efficacy in the management of GPPD has not been well described; however, for the rare cases in which therapies were described, they were largely unsuccessful, with 1 patient exhibiting spontaneous improvement.3,4
Granulomatous PPD also appears to exhibit a range of histologic findings. All cases of GPPD shared fundamental components, such as a brisk perivascular infiltrate accompanied by RBC extravasation with variable hemosiderin-laden macrophages and a granulomatous infiltrate. All of the reports we reviewed described an intimate association between these components, with the granulomas being essentially superimposed on typical PPD. As for other types of PPD, obvious vasculitis characterized by a vasculocentric inflammatory infiltrate and evidence of vascular injury, such as fibrinoid necrosis of the vessel wall, has not been described in GPPD.3-7 Finally, histologic changes suggestive of a relationship with cutaneous T-cell lymphoma, cytologic atypia, and epidermotropism have been described for some forms of PPD but have not been described for GPPD.3-8
Our case reports expand the histologic spectrum of GPPD. Although patient 3 exhibited a relatively intimate association of granulomas and PPD, 2 of our cases (patients 1 and 2) demonstrated a granulomatous infiltrate in the mid to deep dermis, which is separate from the more superficially situated lichenoid lymphohistiocytic infiltrate, RBC extravasation, and hemosiderin-laden macrophages noted in the papillary dermis. Considered along with the absence of an obvious clinicopathologic explanation for the granulomatous infiltrates (eg, polarizable material, infectious organisms, systemic disease), these 2 cases (patients 1 and 2) suggest a composite form of PPD that combines the lichenoid pattern of PPD of Gougerot and Blum with a deep granulomatous component of GPPD. The importance of this distinction lies in the potential for physicians to overlook this potentially informative histologic pattern if only a superficial biopsy is performed. The clinical relevance is unclear; however, in our experience, it has been challenging to treat this relatively small subset of patients who have exhibited a limited response to treatment with topical steroids, intralesional steroids, pulsed dye laser, and vitamin supplementation.
The cause of the granulomatous infiltrate in GPPD is poorly understood. Seven of 13 cases included in our review occurred in patients with a history of hyperlipidemia.5-7 Some have postulated that the constellation of findings of GPPD in hyperlipidemic patients reflects an underlying vascular injury process induced by lipid deposition in the endothelial cells with subsequent RBC extravasation and a secondary granulomatous response to the lipid deposits.6,7 However, given the occurrence of GPPD in patients without hyperlipidemia, other mechanisms also must be considered in the pathogenesis of GPPD, including a reaction to medications, systemic diseases, and infectious etiologies (eg, hepatitis B virus).4,6 As additional cases are described in the literature, other unifying clinical etiologies for this histopathologic reaction pattern may emerge.
Conclusion
Granulomatous PPD may comprise an underrecognized variant of PPD in cases when only a superficial biopsy is evaluated. Clinicians and pathologists should be aware of this variant, and in refractory cases of PPD, deeper sampling may be warranted to identify granulomas.
1. Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
2. Piette WW. Purpura: mechanisms and differential diagnosis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby; 2008:321-330.
3. Saito R, Matsuoka Y. Granulomatous pigmented purpuric dermatosis. J Dermatol. 1996;23:551-555.
4. Kerns MJ, Mallatt BD, Shamma HN. Granulomatous pigmented purpura: an unusual histological variant. Am J Dermatopathol. 2009;31:77-80.
5. Wong WR, Kuo TT, Chen MJ, et al. Granulomatous variant of chronic pigmented purpuric dermatosis: report of two cases. Br J Dermatol. 2001;145:162-164.
6. Lin WL, Kou TT, Shih PY, et al. Granulomatous variant of chronic pigmented purpuric dermatoses: report of four new cases and an association with hyperlipidaemia [published online ahead of print May 29, 2007]. Clin Exp Dermatol. 2007;32:513-515.
7. Lee SH, Kwon JE, Lee KG, et al. Granulomatous variant of chronic pigmented purpuric dermatosis associated with hyperlipidemia. J Eur Acad Dermatol Venereol. 2010;24:1243-1245.
8. Toro JR, Sander CA, LeBoit PE. Persistent pigmented dermatoses and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
1. Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
2. Piette WW. Purpura: mechanisms and differential diagnosis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby; 2008:321-330.
3. Saito R, Matsuoka Y. Granulomatous pigmented purpuric dermatosis. J Dermatol. 1996;23:551-555.
4. Kerns MJ, Mallatt BD, Shamma HN. Granulomatous pigmented purpura: an unusual histological variant. Am J Dermatopathol. 2009;31:77-80.
5. Wong WR, Kuo TT, Chen MJ, et al. Granulomatous variant of chronic pigmented purpuric dermatosis: report of two cases. Br J Dermatol. 2001;145:162-164.
6. Lin WL, Kou TT, Shih PY, et al. Granulomatous variant of chronic pigmented purpuric dermatoses: report of four new cases and an association with hyperlipidaemia [published online ahead of print May 29, 2007]. Clin Exp Dermatol. 2007;32:513-515.
7. Lee SH, Kwon JE, Lee KG, et al. Granulomatous variant of chronic pigmented purpuric dermatosis associated with hyperlipidemia. J Eur Acad Dermatol Venereol. 2010;24:1243-1245.
8. Toro JR, Sander CA, LeBoit PE. Persistent pigmented dermatoses and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
Practice Points
- Consider a punch biopsy when sampling suspected inflammatory dermatoses, such as pigmented purpuric dermatosis, to allow deeper sampling.
- Provide all clinical details to the dermatopathologist to assist with clinicopathologic correlation and diagnostic accuracy.
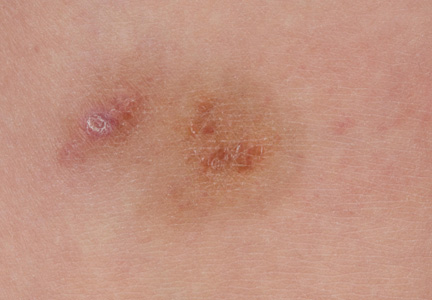
Subcutaneous Panniculitislike T-Cell Lymphoma
Subcutaneous panniculitislike T-cell lymphoma (SPTL) is a cutaneous lymphoma of α and β phenotype cytotoxic T cells in which the neoplastic cells are found almost exclusively in the subcutaneous layer and resemble a panniculitis.1 It affects males and females with equal incidence and is seen in both adults and children. Clinically, this disease presents as a nonspecific panniculitis with indurated but typically nonulcerated erythematous plaques and nodules most commonly located on the extremities. Plaques and nodules may appear on other body sites and may be generalized.1 In some cases, patients present with associated systemic symptoms including fever, malaise, weight loss, and fatigue.2
Histologically, SPTL presents as a predominantly lobular panniculitis (Figure 1) with rimming of adipocytes by neoplastic cells that appear as small and medium-sized atypical lymphocytes with hyperchromatic nuclei (Figure 2A). A less dominant septal component may be present, and neoplastic cells may encroach into the lower reticular dermis, rarely involving the papillary dermis or epidermis.2 Although rimming of adipocytes is classic, it is not specific to this entity, as rimming also can be found in other lymphomas and infectious panniculitis. Reactive lymphocytes and macrophages with ingested lipid material also are seen intermixed with neoplastic cells.2 Necrosis is a common finding, including destructive fragmentation of the nucleus, known as karyorrhexis. If necrosis is extensive, appreciation of other histologic features may be hindered.3 Histiocytes engulfing the nuclear debris known as beanbag cells also can be seen (Figure 2B). The diagnosis can be made on immunohistologic analysis demonstrating neoplastic cells with a cytotoxic α and β T-suppressor phenotype centered around and rimming the adipocytes in the subcutaneous fat.3 Immunohistochemistry reveals positive CD3, CD8 (Figure 2C), and βF1 markers, as well as T-cell intracellular antigen 1 (TIA-1), granzyme B, and perforin.1,2 The neoplastic cells often have a high proliferation index as evidenced by MIB-1 (Ki-67) labeling (Figure 2D). The neoplastic cells are negative for CD4, CD56, and CD30.1,2 Subcutaneous panniculitislike T-cell lymphoma cells are negative for Epstein-Barr virus by in situ hybridization.2
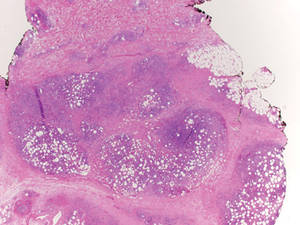
|
| Figure 1. Subcutaneous panniculitislike T-cell lymphoma showing a predominantly lobular panniculitis (H&E, original magnification ×20). |
|
| Figure 2. Rimming of adipocytes by hyperchromatic lymphocytes (A)(H&E, original magnification ×400). Arrowhead indicates a histiocyte (ie, beanbag cell) that has undergone cytophagocytosis of nuclear debris (B)(H&E, original magnification ×600). Immunohistochemistry with CD8 highlights the cells rimming the adipocytes (C)(original magnification ×600). Immunohistochemistry with MIB-1 shows an increased proliferative rate in the lymphocytes rimming the adipocytes (D)(original magnification ×600). |
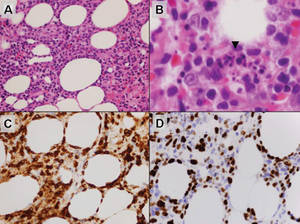
Subcutaneous panniculitislike T-cell lymphoma must be distinguished from lupus erythematosus panniculitis (LEP) and other cutaneous lymphomas. Importantly, LEP and SPTL clinically may appear similar and are not mutually exclusive diagnoses.2 On histology, they may look similar, showing T cell aggregates and necrosis; however, thickening of the basement membrane, vacuolar change at the dermoepidermal junction, plasma cells, hyaline sclerosis, mucin deposition, a lymphocytic perivascular infiltrate, and nodular aggregates of B cells are more common in LEP (Figure 3).2,4 Additionally, in LEP the T cell aggregates typically will not have a high proliferative rate as evidenced by MIB-1.3
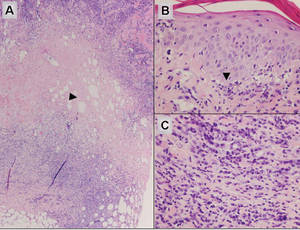
Additionally, other lobular panniculitides can be considered in the differential diagnosis, including erythema induratum (EI), α1-antitrypsin deficiency panniculitis (A1ATDP), and infectious panniculitis. Histologically, EI (Figure 4), also known as nodular vasculitis when not associated with Mycobacterium tuberculosis, has a lobular pattern of inflammation. Early in the disease process there are discrete collections of neutrophils; later, granulomas with histiocytes, giant cells, and foamy macrophages are seen.4 The reactive infiltrate of EI is more mixed than in SPTL, with small lymphocytes, plasma cells, and eosinophils. Leukocytoclastic vasculitis and extravascular caseous or fibrinoid necrosis also may be present.4,5 Substantial caseous necrosis may extend to the dermis and epidermis with EI. Importantly, EI lacks true tuberculoid granulomas and stains negative for acid-fast bacilli, as it is a reactive rather than a local infectious process, but a history of M tuberculosis exposure is common.4 α1-Antitrypsin deficiency panniculitis results from a deficiency of proteinase activity and can be distinguished from SPTL by a neutrophil-rich panniculitis (Figure 5) as well as the classic appearance of splaying of neutrophils between collagen bundles in the deep reticular dermis. Additionally, the panniculitis is characterized by focal areas of necrotic lobules and septa with an infiltrate of neutrophils and macrophages that abut areas of normal-appearing subcutaneous fat without infiltrate.6 Clinically, the A1ATDP lesions may have ulceration and express an oily substance from fat necrosis. Panniculitis with A1ATDP may precede liver and lung disease.4 Panniculitis from bacterial or fungal infection is more common in immunocompromised patients but should be considered when subcutaneous inflammation and/or necrosis is present. Depending on the responsible organism and the status of a patient’s immune system, infectious panniculitis can have variable presentations, including suppurative granulomas with mycobacterial organisms, a dermal focus of infection if the primary source is cutaneous, or a deeper reticular and subcuticular focus in the subcutaneous fat if the infectious panniculitis occurs from hematogenous spread.4 An example of an infectious panniculitis having more of a granulomatous pattern secondary to Cryptococcus can be seen in Figure 6. Ultimately, special stains to identify infectious organisms (eg, Gram, periodic acid–Schiff, Ziehl-Neelsen) can be ordered to aid in the diagnosis if a responsible organism is not visible on hematoxylin and eosin staining.
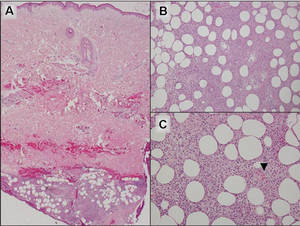
|
Figure 4. Erythema induratum is characterized by a lobular panniculitis (A and B)(both H&E, original magnifications ×40 and ×200). Vascular changes (arrowhead) are present in a majority of cases with endothelial swelling and extravasation of erythrocytes (C)(H&E, original magnification ×400). |
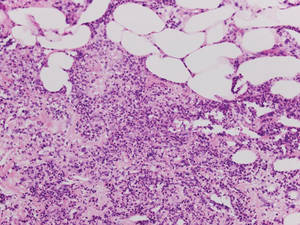
|
| Figure 5. Neutrophilic panniculitis that can be seen in α1-antitrypsin deficiency panniculitis (H&E, original magnification ×400). |
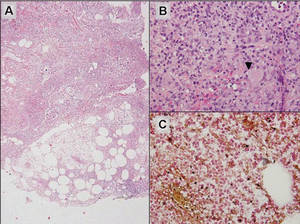
|
Figure 6. Infectious panniculitis secondary to Cryptococcus showing a granulomatous reaction in the subcutis (A)(H&E, original magnification ×40). Closer inspection shows a dense infiltrate of chronic inflammatory cells including numerous histiocytes and multinucleated giant cells. Some of the giant cells contain refractile organisms (arrowhead)(B)(H&E, original magnification ×400). Mucicarmine histochemical stain highlights the capsule of the organism (C)(original magnification ×400). |
Acknowledgment
The authors would like to thank Drake Poeschl, MD, St. Louis, Missouri, for proofreading the manuscript.
1. Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3765-3785.
2. Willemze R, Jansen PM, Cerroni L, et al. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group study of 83 cases. Blood. 2008;111:838-845.
3. Cerroni L, Gatter K, Kerl H. Subcutaneous “panniculitis-like” T-cell lymphoma. In: Cerroni L, Gatter K, Kerl H. Skin Lymphoma: The Illustrated Guide. 3rd ed. Hoboken, NJ: Wiley-Blackwell Publishing; 2011:87-96.
4. Requena L, Sánchez Yus E. Panniculitis. part II. mostly lobular panniculitis. J Am Acad Dermatol. 2001;45:325-361.
5. Sharon V, Goodarzi H, Chambers CJ, et al. Erythema induratum of Bazin. Dermatol Online J. 2010;16:1.
6. Rajagopal R, Malik AK, Murthy PS, et al. Alpha-1 antitrypsin deficiency panniculitis. Indian J Dermatol Venereol Leprol. 2002;68:362-364.
Subcutaneous panniculitislike T-cell lymphoma (SPTL) is a cutaneous lymphoma of α and β phenotype cytotoxic T cells in which the neoplastic cells are found almost exclusively in the subcutaneous layer and resemble a panniculitis.1 It affects males and females with equal incidence and is seen in both adults and children. Clinically, this disease presents as a nonspecific panniculitis with indurated but typically nonulcerated erythematous plaques and nodules most commonly located on the extremities. Plaques and nodules may appear on other body sites and may be generalized.1 In some cases, patients present with associated systemic symptoms including fever, malaise, weight loss, and fatigue.2
Histologically, SPTL presents as a predominantly lobular panniculitis (Figure 1) with rimming of adipocytes by neoplastic cells that appear as small and medium-sized atypical lymphocytes with hyperchromatic nuclei (Figure 2A). A less dominant septal component may be present, and neoplastic cells may encroach into the lower reticular dermis, rarely involving the papillary dermis or epidermis.2 Although rimming of adipocytes is classic, it is not specific to this entity, as rimming also can be found in other lymphomas and infectious panniculitis. Reactive lymphocytes and macrophages with ingested lipid material also are seen intermixed with neoplastic cells.2 Necrosis is a common finding, including destructive fragmentation of the nucleus, known as karyorrhexis. If necrosis is extensive, appreciation of other histologic features may be hindered.3 Histiocytes engulfing the nuclear debris known as beanbag cells also can be seen (Figure 2B). The diagnosis can be made on immunohistologic analysis demonstrating neoplastic cells with a cytotoxic α and β T-suppressor phenotype centered around and rimming the adipocytes in the subcutaneous fat.3 Immunohistochemistry reveals positive CD3, CD8 (Figure 2C), and βF1 markers, as well as T-cell intracellular antigen 1 (TIA-1), granzyme B, and perforin.1,2 The neoplastic cells often have a high proliferation index as evidenced by MIB-1 (Ki-67) labeling (Figure 2D). The neoplastic cells are negative for CD4, CD56, and CD30.1,2 Subcutaneous panniculitislike T-cell lymphoma cells are negative for Epstein-Barr virus by in situ hybridization.2
|
|
| Figure 1. Subcutaneous panniculitislike T-cell lymphoma showing a predominantly lobular panniculitis (H&E, original magnification ×20). |
|
| Figure 2. Rimming of adipocytes by hyperchromatic lymphocytes (A)(H&E, original magnification ×400). Arrowhead indicates a histiocyte (ie, beanbag cell) that has undergone cytophagocytosis of nuclear debris (B)(H&E, original magnification ×600). Immunohistochemistry with CD8 highlights the cells rimming the adipocytes (C)(original magnification ×600). Immunohistochemistry with MIB-1 shows an increased proliferative rate in the lymphocytes rimming the adipocytes (D)(original magnification ×600). |
Subcutaneous panniculitislike T-cell lymphoma must be distinguished from lupus erythematosus panniculitis (LEP) and other cutaneous lymphomas. Importantly, LEP and SPTL clinically may appear similar and are not mutually exclusive diagnoses.2 On histology, they may look similar, showing T cell aggregates and necrosis; however, thickening of the basement membrane, vacuolar change at the dermoepidermal junction, plasma cells, hyaline sclerosis, mucin deposition, a lymphocytic perivascular infiltrate, and nodular aggregates of B cells are more common in LEP (Figure 3).2,4 Additionally, in LEP the T cell aggregates typically will not have a high proliferative rate as evidenced by MIB-1.3
Additionally, other lobular panniculitides can be considered in the differential diagnosis, including erythema induratum (EI), α1-antitrypsin deficiency panniculitis (A1ATDP), and infectious panniculitis. Histologically, EI (Figure 4), also known as nodular vasculitis when not associated with Mycobacterium tuberculosis, has a lobular pattern of inflammation. Early in the disease process there are discrete collections of neutrophils; later, granulomas with histiocytes, giant cells, and foamy macrophages are seen.4 The reactive infiltrate of EI is more mixed than in SPTL, with small lymphocytes, plasma cells, and eosinophils. Leukocytoclastic vasculitis and extravascular caseous or fibrinoid necrosis also may be present.4,5 Substantial caseous necrosis may extend to the dermis and epidermis with EI. Importantly, EI lacks true tuberculoid granulomas and stains negative for acid-fast bacilli, as it is a reactive rather than a local infectious process, but a history of M tuberculosis exposure is common.4 α1-Antitrypsin deficiency panniculitis results from a deficiency of proteinase activity and can be distinguished from SPTL by a neutrophil-rich panniculitis (Figure 5) as well as the classic appearance of splaying of neutrophils between collagen bundles in the deep reticular dermis. Additionally, the panniculitis is characterized by focal areas of necrotic lobules and septa with an infiltrate of neutrophils and macrophages that abut areas of normal-appearing subcutaneous fat without infiltrate.6 Clinically, the A1ATDP lesions may have ulceration and express an oily substance from fat necrosis. Panniculitis with A1ATDP may precede liver and lung disease.4 Panniculitis from bacterial or fungal infection is more common in immunocompromised patients but should be considered when subcutaneous inflammation and/or necrosis is present. Depending on the responsible organism and the status of a patient’s immune system, infectious panniculitis can have variable presentations, including suppurative granulomas with mycobacterial organisms, a dermal focus of infection if the primary source is cutaneous, or a deeper reticular and subcuticular focus in the subcutaneous fat if the infectious panniculitis occurs from hematogenous spread.4 An example of an infectious panniculitis having more of a granulomatous pattern secondary to Cryptococcus can be seen in Figure 6. Ultimately, special stains to identify infectious organisms (eg, Gram, periodic acid–Schiff, Ziehl-Neelsen) can be ordered to aid in the diagnosis if a responsible organism is not visible on hematoxylin and eosin staining.
|
|
Figure 4. Erythema induratum is characterized by a lobular panniculitis (A and B)(both H&E, original magnifications ×40 and ×200). Vascular changes (arrowhead) are present in a majority of cases with endothelial swelling and extravasation of erythrocytes (C)(H&E, original magnification ×400). |
|
|
| Figure 5. Neutrophilic panniculitis that can be seen in α1-antitrypsin deficiency panniculitis (H&E, original magnification ×400). |
|
|
Figure 6. Infectious panniculitis secondary to Cryptococcus showing a granulomatous reaction in the subcutis (A)(H&E, original magnification ×40). Closer inspection shows a dense infiltrate of chronic inflammatory cells including numerous histiocytes and multinucleated giant cells. Some of the giant cells contain refractile organisms (arrowhead)(B)(H&E, original magnification ×400). Mucicarmine histochemical stain highlights the capsule of the organism (C)(original magnification ×400). |
Acknowledgment
The authors would like to thank Drake Poeschl, MD, St. Louis, Missouri, for proofreading the manuscript.
Subcutaneous panniculitislike T-cell lymphoma (SPTL) is a cutaneous lymphoma of α and β phenotype cytotoxic T cells in which the neoplastic cells are found almost exclusively in the subcutaneous layer and resemble a panniculitis.1 It affects males and females with equal incidence and is seen in both adults and children. Clinically, this disease presents as a nonspecific panniculitis with indurated but typically nonulcerated erythematous plaques and nodules most commonly located on the extremities. Plaques and nodules may appear on other body sites and may be generalized.1 In some cases, patients present with associated systemic symptoms including fever, malaise, weight loss, and fatigue.2
Histologically, SPTL presents as a predominantly lobular panniculitis (Figure 1) with rimming of adipocytes by neoplastic cells that appear as small and medium-sized atypical lymphocytes with hyperchromatic nuclei (Figure 2A). A less dominant septal component may be present, and neoplastic cells may encroach into the lower reticular dermis, rarely involving the papillary dermis or epidermis.2 Although rimming of adipocytes is classic, it is not specific to this entity, as rimming also can be found in other lymphomas and infectious panniculitis. Reactive lymphocytes and macrophages with ingested lipid material also are seen intermixed with neoplastic cells.2 Necrosis is a common finding, including destructive fragmentation of the nucleus, known as karyorrhexis. If necrosis is extensive, appreciation of other histologic features may be hindered.3 Histiocytes engulfing the nuclear debris known as beanbag cells also can be seen (Figure 2B). The diagnosis can be made on immunohistologic analysis demonstrating neoplastic cells with a cytotoxic α and β T-suppressor phenotype centered around and rimming the adipocytes in the subcutaneous fat.3 Immunohistochemistry reveals positive CD3, CD8 (Figure 2C), and βF1 markers, as well as T-cell intracellular antigen 1 (TIA-1), granzyme B, and perforin.1,2 The neoplastic cells often have a high proliferation index as evidenced by MIB-1 (Ki-67) labeling (Figure 2D). The neoplastic cells are negative for CD4, CD56, and CD30.1,2 Subcutaneous panniculitislike T-cell lymphoma cells are negative for Epstein-Barr virus by in situ hybridization.2
|
|
| Figure 1. Subcutaneous panniculitislike T-cell lymphoma showing a predominantly lobular panniculitis (H&E, original magnification ×20). |
|
| Figure 2. Rimming of adipocytes by hyperchromatic lymphocytes (A)(H&E, original magnification ×400). Arrowhead indicates a histiocyte (ie, beanbag cell) that has undergone cytophagocytosis of nuclear debris (B)(H&E, original magnification ×600). Immunohistochemistry with CD8 highlights the cells rimming the adipocytes (C)(original magnification ×600). Immunohistochemistry with MIB-1 shows an increased proliferative rate in the lymphocytes rimming the adipocytes (D)(original magnification ×600). |
Subcutaneous panniculitislike T-cell lymphoma must be distinguished from lupus erythematosus panniculitis (LEP) and other cutaneous lymphomas. Importantly, LEP and SPTL clinically may appear similar and are not mutually exclusive diagnoses.2 On histology, they may look similar, showing T cell aggregates and necrosis; however, thickening of the basement membrane, vacuolar change at the dermoepidermal junction, plasma cells, hyaline sclerosis, mucin deposition, a lymphocytic perivascular infiltrate, and nodular aggregates of B cells are more common in LEP (Figure 3).2,4 Additionally, in LEP the T cell aggregates typically will not have a high proliferative rate as evidenced by MIB-1.3
Additionally, other lobular panniculitides can be considered in the differential diagnosis, including erythema induratum (EI), α1-antitrypsin deficiency panniculitis (A1ATDP), and infectious panniculitis. Histologically, EI (Figure 4), also known as nodular vasculitis when not associated with Mycobacterium tuberculosis, has a lobular pattern of inflammation. Early in the disease process there are discrete collections of neutrophils; later, granulomas with histiocytes, giant cells, and foamy macrophages are seen.4 The reactive infiltrate of EI is more mixed than in SPTL, with small lymphocytes, plasma cells, and eosinophils. Leukocytoclastic vasculitis and extravascular caseous or fibrinoid necrosis also may be present.4,5 Substantial caseous necrosis may extend to the dermis and epidermis with EI. Importantly, EI lacks true tuberculoid granulomas and stains negative for acid-fast bacilli, as it is a reactive rather than a local infectious process, but a history of M tuberculosis exposure is common.4 α1-Antitrypsin deficiency panniculitis results from a deficiency of proteinase activity and can be distinguished from SPTL by a neutrophil-rich panniculitis (Figure 5) as well as the classic appearance of splaying of neutrophils between collagen bundles in the deep reticular dermis. Additionally, the panniculitis is characterized by focal areas of necrotic lobules and septa with an infiltrate of neutrophils and macrophages that abut areas of normal-appearing subcutaneous fat without infiltrate.6 Clinically, the A1ATDP lesions may have ulceration and express an oily substance from fat necrosis. Panniculitis with A1ATDP may precede liver and lung disease.4 Panniculitis from bacterial or fungal infection is more common in immunocompromised patients but should be considered when subcutaneous inflammation and/or necrosis is present. Depending on the responsible organism and the status of a patient’s immune system, infectious panniculitis can have variable presentations, including suppurative granulomas with mycobacterial organisms, a dermal focus of infection if the primary source is cutaneous, or a deeper reticular and subcuticular focus in the subcutaneous fat if the infectious panniculitis occurs from hematogenous spread.4 An example of an infectious panniculitis having more of a granulomatous pattern secondary to Cryptococcus can be seen in Figure 6. Ultimately, special stains to identify infectious organisms (eg, Gram, periodic acid–Schiff, Ziehl-Neelsen) can be ordered to aid in the diagnosis if a responsible organism is not visible on hematoxylin and eosin staining.
|
|
Figure 4. Erythema induratum is characterized by a lobular panniculitis (A and B)(both H&E, original magnifications ×40 and ×200). Vascular changes (arrowhead) are present in a majority of cases with endothelial swelling and extravasation of erythrocytes (C)(H&E, original magnification ×400). |
|
|
| Figure 5. Neutrophilic panniculitis that can be seen in α1-antitrypsin deficiency panniculitis (H&E, original magnification ×400). |
|
|
Figure 6. Infectious panniculitis secondary to Cryptococcus showing a granulomatous reaction in the subcutis (A)(H&E, original magnification ×40). Closer inspection shows a dense infiltrate of chronic inflammatory cells including numerous histiocytes and multinucleated giant cells. Some of the giant cells contain refractile organisms (arrowhead)(B)(H&E, original magnification ×400). Mucicarmine histochemical stain highlights the capsule of the organism (C)(original magnification ×400). |
Acknowledgment
The authors would like to thank Drake Poeschl, MD, St. Louis, Missouri, for proofreading the manuscript.
1. Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3765-3785.
2. Willemze R, Jansen PM, Cerroni L, et al. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group study of 83 cases. Blood. 2008;111:838-845.
3. Cerroni L, Gatter K, Kerl H. Subcutaneous “panniculitis-like” T-cell lymphoma. In: Cerroni L, Gatter K, Kerl H. Skin Lymphoma: The Illustrated Guide. 3rd ed. Hoboken, NJ: Wiley-Blackwell Publishing; 2011:87-96.
4. Requena L, Sánchez Yus E. Panniculitis. part II. mostly lobular panniculitis. J Am Acad Dermatol. 2001;45:325-361.
5. Sharon V, Goodarzi H, Chambers CJ, et al. Erythema induratum of Bazin. Dermatol Online J. 2010;16:1.
6. Rajagopal R, Malik AK, Murthy PS, et al. Alpha-1 antitrypsin deficiency panniculitis. Indian J Dermatol Venereol Leprol. 2002;68:362-364.
1. Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3765-3785.
2. Willemze R, Jansen PM, Cerroni L, et al. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group study of 83 cases. Blood. 2008;111:838-845.
3. Cerroni L, Gatter K, Kerl H. Subcutaneous “panniculitis-like” T-cell lymphoma. In: Cerroni L, Gatter K, Kerl H. Skin Lymphoma: The Illustrated Guide. 3rd ed. Hoboken, NJ: Wiley-Blackwell Publishing; 2011:87-96.
4. Requena L, Sánchez Yus E. Panniculitis. part II. mostly lobular panniculitis. J Am Acad Dermatol. 2001;45:325-361.
5. Sharon V, Goodarzi H, Chambers CJ, et al. Erythema induratum of Bazin. Dermatol Online J. 2010;16:1.
6. Rajagopal R, Malik AK, Murthy PS, et al. Alpha-1 antitrypsin deficiency panniculitis. Indian J Dermatol Venereol Leprol. 2002;68:362-364.
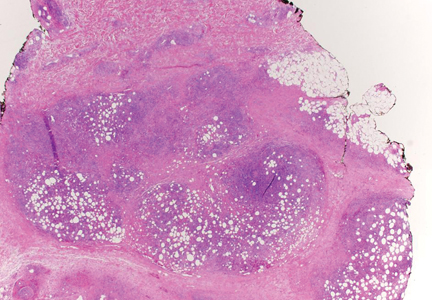
Yellowish Papulonodular Periorbital Eruption
The Diagnosis: Adult-Onset Xanthogranuloma
Biopsies of the lesions on the neck (Figure 1) and back were performed. Histologic analyses revealed a diffuse dermatitis consisting of foamy histiocytes admixed with a few Touton-type giant cells in the dermis (Figure 2), which was associated with an inflammatory infiltrate of eosinophils and lymphocytes. Laboratory investigations revealed mild thrombocytopenia with a platelet count of 134×109/L (reference range, 140–440×109/L). Other investigations including biochemistry, lipid, serum electrophoresis, and chest radiogram were normal. A bone marrow trephine biopsy and flow cytometry were performed and were normal. Magnetic resonance imaging revealed periorbital soft-tissue masses that did not extend into the orbits.
|
| Figure 1. Firm plaques with a yellowish tinge over the side of the neck |
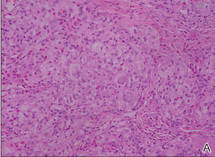
|
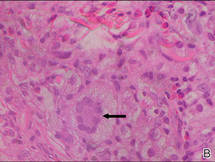
|
| Figure 2. A dense infiltrate of foamy histiocytes in the dermis (A) associated with a Touton-type giant cell (arrow) and an inflammatory infiltrate consisting of eosinophils and lymphocytes (B)(both H&E, original magnifications ×200 and ×400). |
Adult-onset xanthogranuloma (AXG) is a rare disease entity, usually presenting in the third to fourth decades of life. The condition typically presents as a red to yellow-brown nodular cutaneous lesion located on the scalp, face, neck, trunk, or limbs. The presentation typically consists of a solitary lesion, occurring in 70% to 89% of cases,1 but more rarely, as in this case, lesions can be multiple or even disseminated.
Histologically, AXG presents as a dense, well-circumscribed, histiocytic infiltrate consisting of lipophages possessing foamy cytoplasm and giant cells. The presence of histiocytic giant cells differentiates AXG from xanthelasma, a clinical differential diagnosis in this case, and xanthoma. In AXG, there are 4 main types of histiocytes: xanthomatized, spindle shaped, vacuolated, and oncocytic.2 They can be seen in variable proportions, together with different types of giant cells (eg, Touton, foreign body, ground glass, nonspecific). A mixed infiltrate of eosinophils, lymphocytes, plasma cells, and neutrophils also may be seen scattered throughout the lesion.2
Correlating with the clinical and histological features of xanthogranuloma, the firm plaques and nodules represent the dense dermal infiltration of histiocytes that may extend into the subcutis. The lesions demonstrate a time-dependent progression both clinically and histologically. Early lesions are comprised of a dense monomorphous nonlipid histiocytic inflammatory infiltrate, and they clinically appear more erythematous. In mature lesions, as in our patient, the infiltrate is predominantly composed of lipid-laden histiocytes with associated Touton giant cells. They appear more yellowish on clinical presentation.
Adult-onset xanthogranuloma is part of a rare heterogenous group of disorders termed adult orbital xanthogranulomatous disease, which includes 3 other syndromes: necrobiotic xanthogranuloma, adult-onset asthma and periocular xanthogranuloma, and Erdheim-Chester disease.
Necrobiotic xanthogranuloma is clinically characterized by the presence of subcutaneous lesions that ulcerate in approximately 40% to 50% of cases and is histologically characterized by necrobiosis with palisading epithelioid histiocytes. It also is systemically associated with paraproteinemia and multiple myeloma.3
Adult-onset asthma and periocular xanthogranuloma is characterized by yellowish papules and nodules predominantly over the lower eyelids that are histologically comprised of lymphoid follicles with reactive germinal centers. It is associated with asthma, which normally is severe and often occurs almost simultaneously with the periorbital lesions.4
There are no definite diagnostic criteria for Erdheim-Chester disease and the diagnosis is usually based on radiologic findings of osteosclerosis and histopathologic evidence of foamy histiocytic infiltration. Systemic manifestations are common with lymphohistiocytic infiltration of the heart, lungs, pericardium, bones, and intestines. Prognosis is uniformly dismal.
Based on the clinical presentation of a nonulcerative papulonodular eruption and the absence of systemic involvement including asthma, we made the diagnosis of AXG. In view of the heterogeneity among these clinical entities as well as the time-based evolution of the lesions involved, we continued to monitor the patient for 2 years and there was no development of other systemic manifestations and hematologic abnormalities.
In contrast to the more common form of juvenile-onset xanthogranuloma, the adult type is not associated with widespread visceral lesions. Hence extensive screening investigations for systemic disease generally are not necessary. Another difference is that AXG has been associated with hematologic abnormalities, including essential thrombocytosis, chronic lymphocytic leukemia, large B-cell lymphoma, and monoclonal gammopathy.5,6 In our patient, the presence of thrombocytopenia and older age caused us to be concerned about an associated hematologic malignancy; therefore, a bone marrow biopsy and flow cytometry were performed.
Adult-onset xanthogranuloma typically is an asymptomatic and self-healing disease and therefore treatment usually is not required. Spontaneous regression of xanthogranuloma was observed to occur in 54% of cases with a median duration of 22 months,7 though lesions were noted to last as long as 15 years.8 When treatment is necessary, a combination of local and systemic corticosteroids, cytotoxic agents, and radiotherapy have been routinely used. In particular, intralesional corticosteroid therapy has been found to be efficacious in controlling symptoms and signs of AXG affecting the eyelids and orbits while avoiding the systemic side effects of other agents.9
Because our patient’s lesions were longstanding and disfiguring, we opted for active intervention with intralesional triamcinolone, which resulted in only a slight reduction in size of the lesions. The lesions remain largely unchanged in 2 years of follow-up.
1. Chang SE, Cho S, Choi JC, et al. Clinicohistopathologic comparison of adult type and juvenile type xanthogranulomas in Korea. J Dermatol. 2001;28:413-418.
2. Gelmetti C. Non-Langerhans cell histiocytosis. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
3. Mehregan DA, Winkelmann RK. Necrobiotic xanthogranuloma. Arch Dermatol. 1992;128:94-100.
4. Jakobiec FA, Mills MD, Hidayat AA, et al. Periocular xanthogranulomas associated with severe adult-onset asthma. Trans Am Ophthalmol Soc. 1993;91:99-125.
5. Shoo BA, Shinkai K, McCalmont TH, et al. Xanthogranulomas associated with hematologic malignancy in adulthood. J Am Acad Dermatol. 2008;59:488-493.
6. Chiou CC, Wang PN, Yang LC, et al. Disseminated xanthogranulomas associated with adult T-cell leukaemia/lymphoma: a case report and review the association of haematologic malignancies. J Eur Acad Dermatol Venereol. 2007;21:532-535.
7. Robinson HM, Harmon CE, Firminger HI. Multiple lipoidal histiocytomas with regression. Arch Dermatol. 1963;88:660-667.
8. Winkelmann RK. Cutaneous syndromes of non-X histiocytosis: a review of the macrophage-histiocyte diseases of the skin. Arch Dermatol. 1981;117:667-672.
9. Elner VM, Mintz R, Demirci H, et al. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Ophthal Plast Reconstr Surg. 2006;22:36-40.
The Diagnosis: Adult-Onset Xanthogranuloma
Biopsies of the lesions on the neck (Figure 1) and back were performed. Histologic analyses revealed a diffuse dermatitis consisting of foamy histiocytes admixed with a few Touton-type giant cells in the dermis (Figure 2), which was associated with an inflammatory infiltrate of eosinophils and lymphocytes. Laboratory investigations revealed mild thrombocytopenia with a platelet count of 134×109/L (reference range, 140–440×109/L). Other investigations including biochemistry, lipid, serum electrophoresis, and chest radiogram were normal. A bone marrow trephine biopsy and flow cytometry were performed and were normal. Magnetic resonance imaging revealed periorbital soft-tissue masses that did not extend into the orbits.
|
|
| Figure 1. Firm plaques with a yellowish tinge over the side of the neck |
|
|
|
|
| Figure 2. A dense infiltrate of foamy histiocytes in the dermis (A) associated with a Touton-type giant cell (arrow) and an inflammatory infiltrate consisting of eosinophils and lymphocytes (B)(both H&E, original magnifications ×200 and ×400). |
Adult-onset xanthogranuloma (AXG) is a rare disease entity, usually presenting in the third to fourth decades of life. The condition typically presents as a red to yellow-brown nodular cutaneous lesion located on the scalp, face, neck, trunk, or limbs. The presentation typically consists of a solitary lesion, occurring in 70% to 89% of cases,1 but more rarely, as in this case, lesions can be multiple or even disseminated.
Histologically, AXG presents as a dense, well-circumscribed, histiocytic infiltrate consisting of lipophages possessing foamy cytoplasm and giant cells. The presence of histiocytic giant cells differentiates AXG from xanthelasma, a clinical differential diagnosis in this case, and xanthoma. In AXG, there are 4 main types of histiocytes: xanthomatized, spindle shaped, vacuolated, and oncocytic.2 They can be seen in variable proportions, together with different types of giant cells (eg, Touton, foreign body, ground glass, nonspecific). A mixed infiltrate of eosinophils, lymphocytes, plasma cells, and neutrophils also may be seen scattered throughout the lesion.2
Correlating with the clinical and histological features of xanthogranuloma, the firm plaques and nodules represent the dense dermal infiltration of histiocytes that may extend into the subcutis. The lesions demonstrate a time-dependent progression both clinically and histologically. Early lesions are comprised of a dense monomorphous nonlipid histiocytic inflammatory infiltrate, and they clinically appear more erythematous. In mature lesions, as in our patient, the infiltrate is predominantly composed of lipid-laden histiocytes with associated Touton giant cells. They appear more yellowish on clinical presentation.
Adult-onset xanthogranuloma is part of a rare heterogenous group of disorders termed adult orbital xanthogranulomatous disease, which includes 3 other syndromes: necrobiotic xanthogranuloma, adult-onset asthma and periocular xanthogranuloma, and Erdheim-Chester disease.
Necrobiotic xanthogranuloma is clinically characterized by the presence of subcutaneous lesions that ulcerate in approximately 40% to 50% of cases and is histologically characterized by necrobiosis with palisading epithelioid histiocytes. It also is systemically associated with paraproteinemia and multiple myeloma.3
Adult-onset asthma and periocular xanthogranuloma is characterized by yellowish papules and nodules predominantly over the lower eyelids that are histologically comprised of lymphoid follicles with reactive germinal centers. It is associated with asthma, which normally is severe and often occurs almost simultaneously with the periorbital lesions.4
There are no definite diagnostic criteria for Erdheim-Chester disease and the diagnosis is usually based on radiologic findings of osteosclerosis and histopathologic evidence of foamy histiocytic infiltration. Systemic manifestations are common with lymphohistiocytic infiltration of the heart, lungs, pericardium, bones, and intestines. Prognosis is uniformly dismal.
Based on the clinical presentation of a nonulcerative papulonodular eruption and the absence of systemic involvement including asthma, we made the diagnosis of AXG. In view of the heterogeneity among these clinical entities as well as the time-based evolution of the lesions involved, we continued to monitor the patient for 2 years and there was no development of other systemic manifestations and hematologic abnormalities.
In contrast to the more common form of juvenile-onset xanthogranuloma, the adult type is not associated with widespread visceral lesions. Hence extensive screening investigations for systemic disease generally are not necessary. Another difference is that AXG has been associated with hematologic abnormalities, including essential thrombocytosis, chronic lymphocytic leukemia, large B-cell lymphoma, and monoclonal gammopathy.5,6 In our patient, the presence of thrombocytopenia and older age caused us to be concerned about an associated hematologic malignancy; therefore, a bone marrow biopsy and flow cytometry were performed.
Adult-onset xanthogranuloma typically is an asymptomatic and self-healing disease and therefore treatment usually is not required. Spontaneous regression of xanthogranuloma was observed to occur in 54% of cases with a median duration of 22 months,7 though lesions were noted to last as long as 15 years.8 When treatment is necessary, a combination of local and systemic corticosteroids, cytotoxic agents, and radiotherapy have been routinely used. In particular, intralesional corticosteroid therapy has been found to be efficacious in controlling symptoms and signs of AXG affecting the eyelids and orbits while avoiding the systemic side effects of other agents.9
Because our patient’s lesions were longstanding and disfiguring, we opted for active intervention with intralesional triamcinolone, which resulted in only a slight reduction in size of the lesions. The lesions remain largely unchanged in 2 years of follow-up.
The Diagnosis: Adult-Onset Xanthogranuloma
Biopsies of the lesions on the neck (Figure 1) and back were performed. Histologic analyses revealed a diffuse dermatitis consisting of foamy histiocytes admixed with a few Touton-type giant cells in the dermis (Figure 2), which was associated with an inflammatory infiltrate of eosinophils and lymphocytes. Laboratory investigations revealed mild thrombocytopenia with a platelet count of 134×109/L (reference range, 140–440×109/L). Other investigations including biochemistry, lipid, serum electrophoresis, and chest radiogram were normal. A bone marrow trephine biopsy and flow cytometry were performed and were normal. Magnetic resonance imaging revealed periorbital soft-tissue masses that did not extend into the orbits.
|
|
| Figure 1. Firm plaques with a yellowish tinge over the side of the neck |
|
|
|
|
| Figure 2. A dense infiltrate of foamy histiocytes in the dermis (A) associated with a Touton-type giant cell (arrow) and an inflammatory infiltrate consisting of eosinophils and lymphocytes (B)(both H&E, original magnifications ×200 and ×400). |
Adult-onset xanthogranuloma (AXG) is a rare disease entity, usually presenting in the third to fourth decades of life. The condition typically presents as a red to yellow-brown nodular cutaneous lesion located on the scalp, face, neck, trunk, or limbs. The presentation typically consists of a solitary lesion, occurring in 70% to 89% of cases,1 but more rarely, as in this case, lesions can be multiple or even disseminated.
Histologically, AXG presents as a dense, well-circumscribed, histiocytic infiltrate consisting of lipophages possessing foamy cytoplasm and giant cells. The presence of histiocytic giant cells differentiates AXG from xanthelasma, a clinical differential diagnosis in this case, and xanthoma. In AXG, there are 4 main types of histiocytes: xanthomatized, spindle shaped, vacuolated, and oncocytic.2 They can be seen in variable proportions, together with different types of giant cells (eg, Touton, foreign body, ground glass, nonspecific). A mixed infiltrate of eosinophils, lymphocytes, plasma cells, and neutrophils also may be seen scattered throughout the lesion.2
Correlating with the clinical and histological features of xanthogranuloma, the firm plaques and nodules represent the dense dermal infiltration of histiocytes that may extend into the subcutis. The lesions demonstrate a time-dependent progression both clinically and histologically. Early lesions are comprised of a dense monomorphous nonlipid histiocytic inflammatory infiltrate, and they clinically appear more erythematous. In mature lesions, as in our patient, the infiltrate is predominantly composed of lipid-laden histiocytes with associated Touton giant cells. They appear more yellowish on clinical presentation.
Adult-onset xanthogranuloma is part of a rare heterogenous group of disorders termed adult orbital xanthogranulomatous disease, which includes 3 other syndromes: necrobiotic xanthogranuloma, adult-onset asthma and periocular xanthogranuloma, and Erdheim-Chester disease.
Necrobiotic xanthogranuloma is clinically characterized by the presence of subcutaneous lesions that ulcerate in approximately 40% to 50% of cases and is histologically characterized by necrobiosis with palisading epithelioid histiocytes. It also is systemically associated with paraproteinemia and multiple myeloma.3
Adult-onset asthma and periocular xanthogranuloma is characterized by yellowish papules and nodules predominantly over the lower eyelids that are histologically comprised of lymphoid follicles with reactive germinal centers. It is associated with asthma, which normally is severe and often occurs almost simultaneously with the periorbital lesions.4
There are no definite diagnostic criteria for Erdheim-Chester disease and the diagnosis is usually based on radiologic findings of osteosclerosis and histopathologic evidence of foamy histiocytic infiltration. Systemic manifestations are common with lymphohistiocytic infiltration of the heart, lungs, pericardium, bones, and intestines. Prognosis is uniformly dismal.
Based on the clinical presentation of a nonulcerative papulonodular eruption and the absence of systemic involvement including asthma, we made the diagnosis of AXG. In view of the heterogeneity among these clinical entities as well as the time-based evolution of the lesions involved, we continued to monitor the patient for 2 years and there was no development of other systemic manifestations and hematologic abnormalities.
In contrast to the more common form of juvenile-onset xanthogranuloma, the adult type is not associated with widespread visceral lesions. Hence extensive screening investigations for systemic disease generally are not necessary. Another difference is that AXG has been associated with hematologic abnormalities, including essential thrombocytosis, chronic lymphocytic leukemia, large B-cell lymphoma, and monoclonal gammopathy.5,6 In our patient, the presence of thrombocytopenia and older age caused us to be concerned about an associated hematologic malignancy; therefore, a bone marrow biopsy and flow cytometry were performed.
Adult-onset xanthogranuloma typically is an asymptomatic and self-healing disease and therefore treatment usually is not required. Spontaneous regression of xanthogranuloma was observed to occur in 54% of cases with a median duration of 22 months,7 though lesions were noted to last as long as 15 years.8 When treatment is necessary, a combination of local and systemic corticosteroids, cytotoxic agents, and radiotherapy have been routinely used. In particular, intralesional corticosteroid therapy has been found to be efficacious in controlling symptoms and signs of AXG affecting the eyelids and orbits while avoiding the systemic side effects of other agents.9
Because our patient’s lesions were longstanding and disfiguring, we opted for active intervention with intralesional triamcinolone, which resulted in only a slight reduction in size of the lesions. The lesions remain largely unchanged in 2 years of follow-up.
1. Chang SE, Cho S, Choi JC, et al. Clinicohistopathologic comparison of adult type and juvenile type xanthogranulomas in Korea. J Dermatol. 2001;28:413-418.
2. Gelmetti C. Non-Langerhans cell histiocytosis. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
3. Mehregan DA, Winkelmann RK. Necrobiotic xanthogranuloma. Arch Dermatol. 1992;128:94-100.
4. Jakobiec FA, Mills MD, Hidayat AA, et al. Periocular xanthogranulomas associated with severe adult-onset asthma. Trans Am Ophthalmol Soc. 1993;91:99-125.
5. Shoo BA, Shinkai K, McCalmont TH, et al. Xanthogranulomas associated with hematologic malignancy in adulthood. J Am Acad Dermatol. 2008;59:488-493.
6. Chiou CC, Wang PN, Yang LC, et al. Disseminated xanthogranulomas associated with adult T-cell leukaemia/lymphoma: a case report and review the association of haematologic malignancies. J Eur Acad Dermatol Venereol. 2007;21:532-535.
7. Robinson HM, Harmon CE, Firminger HI. Multiple lipoidal histiocytomas with regression. Arch Dermatol. 1963;88:660-667.
8. Winkelmann RK. Cutaneous syndromes of non-X histiocytosis: a review of the macrophage-histiocyte diseases of the skin. Arch Dermatol. 1981;117:667-672.
9. Elner VM, Mintz R, Demirci H, et al. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Ophthal Plast Reconstr Surg. 2006;22:36-40.
1. Chang SE, Cho S, Choi JC, et al. Clinicohistopathologic comparison of adult type and juvenile type xanthogranulomas in Korea. J Dermatol. 2001;28:413-418.
2. Gelmetti C. Non-Langerhans cell histiocytosis. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
3. Mehregan DA, Winkelmann RK. Necrobiotic xanthogranuloma. Arch Dermatol. 1992;128:94-100.
4. Jakobiec FA, Mills MD, Hidayat AA, et al. Periocular xanthogranulomas associated with severe adult-onset asthma. Trans Am Ophthalmol Soc. 1993;91:99-125.
5. Shoo BA, Shinkai K, McCalmont TH, et al. Xanthogranulomas associated with hematologic malignancy in adulthood. J Am Acad Dermatol. 2008;59:488-493.
6. Chiou CC, Wang PN, Yang LC, et al. Disseminated xanthogranulomas associated with adult T-cell leukaemia/lymphoma: a case report and review the association of haematologic malignancies. J Eur Acad Dermatol Venereol. 2007;21:532-535.
7. Robinson HM, Harmon CE, Firminger HI. Multiple lipoidal histiocytomas with regression. Arch Dermatol. 1963;88:660-667.
8. Winkelmann RK. Cutaneous syndromes of non-X histiocytosis: a review of the macrophage-histiocyte diseases of the skin. Arch Dermatol. 1981;117:667-672.
9. Elner VM, Mintz R, Demirci H, et al. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Ophthal Plast Reconstr Surg. 2006;22:36-40.
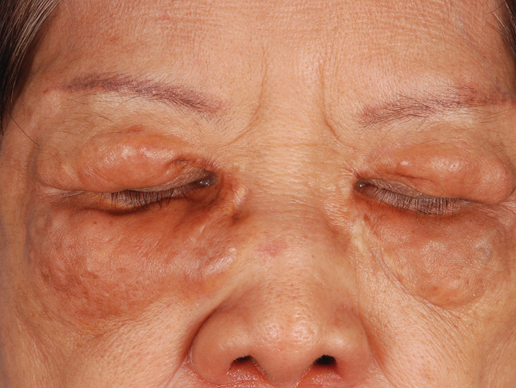
A 66-year-old woman with a history of type 2 diabetes mellitus and mild dyslipidemia presented with persistent lesions over the eyelids and cheeks of 10 years’ duration. Systemic review was unremarkable. There was no family or personal history of atopy, asthma, or other dermatologic disorders. Physical examination revealed confluent yellowish plaques and nodules over the periorbital regions as well as yellowish plaques over the neck and back. The lesions were firm to palpation and the epidermis appeared unaffected. The ophthalmic examination was normal and other mucosal surfaces were unaffected.
Glomus Tumor of Uncertain Malignant Potential on the Forehead
Glomus tumors (GTs) are uncommon benign tumors originating in the neuromyoarterial elements of the glomus body, an arteriovenous shunt specialized in thermoregulation.1 Glomus tumors usually occur in the distal extremities of young adults2 and rarely are seen in the deep soft tissue or viscera. Malignant GTs are rare and highly aggressive tumors that have been associated with both local recurrence and distant metastasis.1-11 Glomus tumors have been subdivided into 3 groups with different prognoses2: (1) malignant GT with metastatic potential (subfascial or visceral location, >2 cm in size, atypical mitotic figures, >5 mitoses per 50 high-power fields [HPFs], marked nuclear atypia); (2) symplastic GT (benign tumor with nuclear pleomorphism without mitotic activity); and (3) GT of uncertain malignant potential (GTUMP)(absence of metastatic disease, favorable prognosis, at least 1 feature of malignant GTs other than marked nuclear atypia [eg, high mitotic activity, >2 cm in size, deep location]).1,2
We report a case of GTUMP with unusual clinicopathologic features in a 74-year-old man that was treated via wide surgical excision. No local recurrence or distant metastasis was noted at 3-year follow-up.
Case Report
A 74-year-old man presented to the Unit of Surgery with a slowly progressing, painful, ulcerated, 2.5-cm, red-blue nodule on the forehead (Figure 1). An excisional biopsy of the nodule was performed. Histologically, the dermis and superficial subcutis were filled with a proliferation of atypical epithelioid cells to slightly spindled cells. Both cells displayed a weakly eosinophilic cytoplasm with indistinct membranes and larger ovoid nuclei, some with prominent nucleoli. Neoplastic cells showed a disordered arrangement or were organized in short fascicles separated by slitlike spaces, vascular lumens of various sizes, or hemorrhagic stroma resembling angiosarcoma or Kaposi sarcoma (Figure 2). No areas of necrosis were noted. Pleomorphic nuclei and some mitotic figures also were identified, but they were not atypical and showed fewer than 5 mitoses per 50 HPFs. Immunohistochemically, the neoplastic cells stained positive for vimentin, caldesmon (Figure 3), and a–smooth muscle actin, and they stained negative for cytokeratins, desmin, CD34, factor VIII–related antigen, S-100 protein, and the latent nuclear antigen of Kaposi sarcoma–associated herpesvirus. The Ki-67 labeling index revealed less than 20% positive cells.
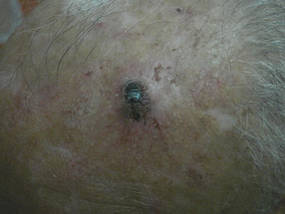
|
| Figure 1. A painful, red-blue, ulcerated nodule on the forehead of a 74-year-old man. |
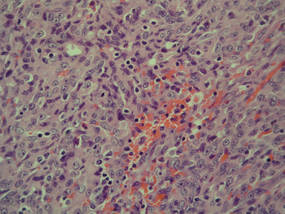
|
| Figure 2. The tumor was composed of atypical epithelioid cells to slightly spindled cells with indistinct membranes and larger ovoid nuclei, some with prominent nucleoli in a disordered arrangement or rather in short fascicles separated in a hemorrhagic background (H&E, original magnification ×40). |
|
| Figure 3. The neoplastic cells stained positive for caldesmon (original magnification ×20). |
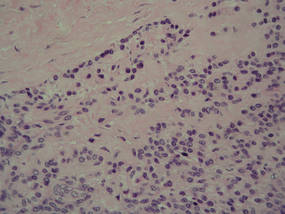
|
| Figure 4. A focus of round to polygonal tumor cells reminiscent of a preexisting benign-appearing glomus tumor was found on biopsy following reexcision (H&E, original magnification ×20). |
Because the tumor involved margins of excision, the patient successfully underwent wide reexcision with adequate margins. Histological examination of the reexcised specimen showed a focus of bland, round to polygonal tumor cells with the features of glomus cells (Figure 4). On the basis of the histologic features of both specimens, the lesion was classified as a GT. The biggest problem in our case was the classification of the lesion according to established pathologic criteria. We considered this case to be borderline because the lesion was greater than 2 cm but the location was superficial; marked atypia with sarcomatoid features also were present, but there was an absence of necrosis and fewer than 5 mitoses per 50 HPFs. For these reasons, we diagnosed this problematic lesion as a GTUMP. Following reexcision, the patient underwent strict follow-up. Wound healing was uncomplicated and the patient showed no local recurrence or distant metastasis at 3-year follow-up.
Comment
Clinically metastatic and histologically malignant GTs are exceptional.3-11 The classification system for GTs based on histologic criteria subdivided these tumors into 3 groups with different prognoses.2 Malignant GTs are highly aggressive tumors with metastatic potential, symplastic GTs are considered to be a degenerative phenomenon, and GTUMPs have a favorable clinical outcome and absence of metastatic disease.12
Diagnosis of malignant GTs and symplastic GTs is relatively easy in the presence of typical uniform, small, round epithelioid cells (glomus cells) located around blood vessels. Immunohistochemistry may be useful, as GTs express smooth muscle actin and caldesmon. Over the years the existence and diagnosis of malignant GT has been questioned because a residual component of benign GT in the surgical biopsy is useful in diagnosis but is not always present1-3 and because an unusual pattern may be present in malignant tumors with prevalent spindle cells resembling fibrosarcoma, leiomyosarcoma, spindle cell angiosarcoma, and spindle cell melanoma.1 In the absence of a preexisting GT, the differential diagnosis may be difficult; in such cases, a panel of immunohistochemical markers including smooth muscle actin, caldesmon, desmin, S-100, human melanoma black-45 (HMB-45), CD34, and CD31 is always necessary. The GTUMP category was introduced for GTs that demonstrate marked nuclear atypia but do not fulfill histologic criteria for malignancy. Along with other tumors of uncertain malignant potential, borderline cases should be considered GTUMPs to guarantee wide excision of the tumor with negative margins and an adequate follow-up due to the possibility of local recurrence or distant metastasis. In our patient, a diagnosis of GTUMP was made. Additionally, our case demonstrates some previously unreported features of GTUMPs, such as spindled cells in short fascicles separated by slitlike spaces, small vessels, and hemorrhagic stroma resembling Kaposi sarcoma. Along with these unusual sarcomatous features, the superficial location of the lesion, absence of necrosis, and a mitotic count of less than 5 per 50 HPFs were suggestive of an uncertain malignant potential for this tumor.
Distinction between malignant GTs and GTUMPs in the presence of unusual histologic features may be difficult.12 Glomus tumors that do not fulfill criteria for malignancy but have at least 1 atypical feature other than nuclear pleomorphism should be named GTUMPs. According to classification criteria, a true malignant GT is a highly aggressive tumor with metastatic potential. In a case series reported by Folpe et al,2 38% (20/52) of malignant GTs showed metastases, while metastatic disease was not observed in the tumors classified as GTUMPs. Wide surgical excision or Mohs micrographic surgery13 are the treatments of choice for malignant GTs and GTUMPs. Complete excision of the lesion with negative margins is always necessary in cases of GTUMPs. After the diagnosis of GTUMP, adequate follow-up should berecommended due to the possibility of local recurrence or distant metastasis.
Conclusion
Malignant GTs and GTUMPs are rare, and the nomenclature and classification of these tumors is controversial. These findings and the difficulty of differential diagnosis in a continuum between benignity and malignancy prompted our report.
1. Weiss SW, Goldblum JR. Perivascular tumors. In: Enzinger FM, Weiss SW, eds. Enzinger and Weiss’s Soft Tissue Tumors. 5th ed. St Louis, MO: Mosby; 2008:751-768.
2. Folpe AL, Fanburg-Smith JC, Miettinen M, et al. Atypical and malignant glomus tumors: analysis of 52 cases, with a proposal for the reclassification of glomus tumors. Am J Surg Pathol. 2001;25:1-12.
3. Aiba M, Hirayama A, Kuramochi S. Glomangiosarcoma in a glomus tumor. an immunohistochemical and ultrastructural study. Cancer. 1988;61:1467-1471.
4. Gould EW, Manivel JC, Albores-Saavedra J, et al. Locally infiltrative glomus tumors and glomangiosarcomas. a clinical, ultrastructural, and immunohistochemical study. Cancer. 1990;65:310-318.
5. Noer H, Krogdahl A. Glomangiosarcoma of the lower extremity. Histopathology. 1991;18:365-366.
6. Hiruta N, Kameda N, Tokudome T, et al. Malignant glomus tumor: a case report and review of the literature. Am J Surg Pathol. 1997;21:1096-1103.
7. Watanabe K, Sugino T, Saito A, et al. Glomangiosarcoma of the hip: report of a highly aggressive tumour with widespread distant metastases. Br J Dermatol. 1998;139:1097-1101.
8. Park JH, Oh SH, Yang MH, et al. Glomangiosarcoma of the hand: a case report and review of the literature. J Dermatol. 2003;30:827-833.
9. Kayal JD, Hampton RW, Sheehan DJ, et al. Malignant glomus tumor: a case report and review of the literature. Dermatol Surg. 2001;27:837-840.
10. Pérez de la Fuente T, Vega C, Gutierrez Palacios A, et al. Glomangiosarcoma of the hypothenar eminence: a case report. Chir Main. 2005;24:199-202.
11. Terada T, Fujimoto J, Shirakashi Y, et al. Malignant glomus tumor of the palm: a case report. J Cutan Pathol. 2011;38:381-384.
12. Gill J, Van Vliet C. Infiltrating glomus tumor of uncertain malignant potential arising in the kidney. Hum Pathol. 2010;41:145-149.
13. Cecchi R, Pavesi M, Apicella P. Malignant glomus tumor of the trunk treated with Mohs micrographic surgery [in English, German]. J Dtsch Dermatol Ges. 2011;9:391-392.
Glomus tumors (GTs) are uncommon benign tumors originating in the neuromyoarterial elements of the glomus body, an arteriovenous shunt specialized in thermoregulation.1 Glomus tumors usually occur in the distal extremities of young adults2 and rarely are seen in the deep soft tissue or viscera. Malignant GTs are rare and highly aggressive tumors that have been associated with both local recurrence and distant metastasis.1-11 Glomus tumors have been subdivided into 3 groups with different prognoses2: (1) malignant GT with metastatic potential (subfascial or visceral location, >2 cm in size, atypical mitotic figures, >5 mitoses per 50 high-power fields [HPFs], marked nuclear atypia); (2) symplastic GT (benign tumor with nuclear pleomorphism without mitotic activity); and (3) GT of uncertain malignant potential (GTUMP)(absence of metastatic disease, favorable prognosis, at least 1 feature of malignant GTs other than marked nuclear atypia [eg, high mitotic activity, >2 cm in size, deep location]).1,2
We report a case of GTUMP with unusual clinicopathologic features in a 74-year-old man that was treated via wide surgical excision. No local recurrence or distant metastasis was noted at 3-year follow-up.
Case Report
A 74-year-old man presented to the Unit of Surgery with a slowly progressing, painful, ulcerated, 2.5-cm, red-blue nodule on the forehead (Figure 1). An excisional biopsy of the nodule was performed. Histologically, the dermis and superficial subcutis were filled with a proliferation of atypical epithelioid cells to slightly spindled cells. Both cells displayed a weakly eosinophilic cytoplasm with indistinct membranes and larger ovoid nuclei, some with prominent nucleoli. Neoplastic cells showed a disordered arrangement or were organized in short fascicles separated by slitlike spaces, vascular lumens of various sizes, or hemorrhagic stroma resembling angiosarcoma or Kaposi sarcoma (Figure 2). No areas of necrosis were noted. Pleomorphic nuclei and some mitotic figures also were identified, but they were not atypical and showed fewer than 5 mitoses per 50 HPFs. Immunohistochemically, the neoplastic cells stained positive for vimentin, caldesmon (Figure 3), and a–smooth muscle actin, and they stained negative for cytokeratins, desmin, CD34, factor VIII–related antigen, S-100 protein, and the latent nuclear antigen of Kaposi sarcoma–associated herpesvirus. The Ki-67 labeling index revealed less than 20% positive cells.
|
|
| Figure 1. A painful, red-blue, ulcerated nodule on the forehead of a 74-year-old man. |
|
|
| Figure 2. The tumor was composed of atypical epithelioid cells to slightly spindled cells with indistinct membranes and larger ovoid nuclei, some with prominent nucleoli in a disordered arrangement or rather in short fascicles separated in a hemorrhagic background (H&E, original magnification ×40). |
|
|
| Figure 3. The neoplastic cells stained positive for caldesmon (original magnification ×20). |
|
|
| Figure 4. A focus of round to polygonal tumor cells reminiscent of a preexisting benign-appearing glomus tumor was found on biopsy following reexcision (H&E, original magnification ×20). |
Because the tumor involved margins of excision, the patient successfully underwent wide reexcision with adequate margins. Histological examination of the reexcised specimen showed a focus of bland, round to polygonal tumor cells with the features of glomus cells (Figure 4). On the basis of the histologic features of both specimens, the lesion was classified as a GT. The biggest problem in our case was the classification of the lesion according to established pathologic criteria. We considered this case to be borderline because the lesion was greater than 2 cm but the location was superficial; marked atypia with sarcomatoid features also were present, but there was an absence of necrosis and fewer than 5 mitoses per 50 HPFs. For these reasons, we diagnosed this problematic lesion as a GTUMP. Following reexcision, the patient underwent strict follow-up. Wound healing was uncomplicated and the patient showed no local recurrence or distant metastasis at 3-year follow-up.
Comment
Clinically metastatic and histologically malignant GTs are exceptional.3-11 The classification system for GTs based on histologic criteria subdivided these tumors into 3 groups with different prognoses.2 Malignant GTs are highly aggressive tumors with metastatic potential, symplastic GTs are considered to be a degenerative phenomenon, and GTUMPs have a favorable clinical outcome and absence of metastatic disease.12
Diagnosis of malignant GTs and symplastic GTs is relatively easy in the presence of typical uniform, small, round epithelioid cells (glomus cells) located around blood vessels. Immunohistochemistry may be useful, as GTs express smooth muscle actin and caldesmon. Over the years the existence and diagnosis of malignant GT has been questioned because a residual component of benign GT in the surgical biopsy is useful in diagnosis but is not always present1-3 and because an unusual pattern may be present in malignant tumors with prevalent spindle cells resembling fibrosarcoma, leiomyosarcoma, spindle cell angiosarcoma, and spindle cell melanoma.1 In the absence of a preexisting GT, the differential diagnosis may be difficult; in such cases, a panel of immunohistochemical markers including smooth muscle actin, caldesmon, desmin, S-100, human melanoma black-45 (HMB-45), CD34, and CD31 is always necessary. The GTUMP category was introduced for GTs that demonstrate marked nuclear atypia but do not fulfill histologic criteria for malignancy. Along with other tumors of uncertain malignant potential, borderline cases should be considered GTUMPs to guarantee wide excision of the tumor with negative margins and an adequate follow-up due to the possibility of local recurrence or distant metastasis. In our patient, a diagnosis of GTUMP was made. Additionally, our case demonstrates some previously unreported features of GTUMPs, such as spindled cells in short fascicles separated by slitlike spaces, small vessels, and hemorrhagic stroma resembling Kaposi sarcoma. Along with these unusual sarcomatous features, the superficial location of the lesion, absence of necrosis, and a mitotic count of less than 5 per 50 HPFs were suggestive of an uncertain malignant potential for this tumor.
Distinction between malignant GTs and GTUMPs in the presence of unusual histologic features may be difficult.12 Glomus tumors that do not fulfill criteria for malignancy but have at least 1 atypical feature other than nuclear pleomorphism should be named GTUMPs. According to classification criteria, a true malignant GT is a highly aggressive tumor with metastatic potential. In a case series reported by Folpe et al,2 38% (20/52) of malignant GTs showed metastases, while metastatic disease was not observed in the tumors classified as GTUMPs. Wide surgical excision or Mohs micrographic surgery13 are the treatments of choice for malignant GTs and GTUMPs. Complete excision of the lesion with negative margins is always necessary in cases of GTUMPs. After the diagnosis of GTUMP, adequate follow-up should berecommended due to the possibility of local recurrence or distant metastasis.
Conclusion
Malignant GTs and GTUMPs are rare, and the nomenclature and classification of these tumors is controversial. These findings and the difficulty of differential diagnosis in a continuum between benignity and malignancy prompted our report.
Glomus tumors (GTs) are uncommon benign tumors originating in the neuromyoarterial elements of the glomus body, an arteriovenous shunt specialized in thermoregulation.1 Glomus tumors usually occur in the distal extremities of young adults2 and rarely are seen in the deep soft tissue or viscera. Malignant GTs are rare and highly aggressive tumors that have been associated with both local recurrence and distant metastasis.1-11 Glomus tumors have been subdivided into 3 groups with different prognoses2: (1) malignant GT with metastatic potential (subfascial or visceral location, >2 cm in size, atypical mitotic figures, >5 mitoses per 50 high-power fields [HPFs], marked nuclear atypia); (2) symplastic GT (benign tumor with nuclear pleomorphism without mitotic activity); and (3) GT of uncertain malignant potential (GTUMP)(absence of metastatic disease, favorable prognosis, at least 1 feature of malignant GTs other than marked nuclear atypia [eg, high mitotic activity, >2 cm in size, deep location]).1,2
We report a case of GTUMP with unusual clinicopathologic features in a 74-year-old man that was treated via wide surgical excision. No local recurrence or distant metastasis was noted at 3-year follow-up.
Case Report
A 74-year-old man presented to the Unit of Surgery with a slowly progressing, painful, ulcerated, 2.5-cm, red-blue nodule on the forehead (Figure 1). An excisional biopsy of the nodule was performed. Histologically, the dermis and superficial subcutis were filled with a proliferation of atypical epithelioid cells to slightly spindled cells. Both cells displayed a weakly eosinophilic cytoplasm with indistinct membranes and larger ovoid nuclei, some with prominent nucleoli. Neoplastic cells showed a disordered arrangement or were organized in short fascicles separated by slitlike spaces, vascular lumens of various sizes, or hemorrhagic stroma resembling angiosarcoma or Kaposi sarcoma (Figure 2). No areas of necrosis were noted. Pleomorphic nuclei and some mitotic figures also were identified, but they were not atypical and showed fewer than 5 mitoses per 50 HPFs. Immunohistochemically, the neoplastic cells stained positive for vimentin, caldesmon (Figure 3), and a–smooth muscle actin, and they stained negative for cytokeratins, desmin, CD34, factor VIII–related antigen, S-100 protein, and the latent nuclear antigen of Kaposi sarcoma–associated herpesvirus. The Ki-67 labeling index revealed less than 20% positive cells.
|
|
| Figure 1. A painful, red-blue, ulcerated nodule on the forehead of a 74-year-old man. |
|
|
| Figure 2. The tumor was composed of atypical epithelioid cells to slightly spindled cells with indistinct membranes and larger ovoid nuclei, some with prominent nucleoli in a disordered arrangement or rather in short fascicles separated in a hemorrhagic background (H&E, original magnification ×40). |
|
|
| Figure 3. The neoplastic cells stained positive for caldesmon (original magnification ×20). |
|
|
| Figure 4. A focus of round to polygonal tumor cells reminiscent of a preexisting benign-appearing glomus tumor was found on biopsy following reexcision (H&E, original magnification ×20). |
Because the tumor involved margins of excision, the patient successfully underwent wide reexcision with adequate margins. Histological examination of the reexcised specimen showed a focus of bland, round to polygonal tumor cells with the features of glomus cells (Figure 4). On the basis of the histologic features of both specimens, the lesion was classified as a GT. The biggest problem in our case was the classification of the lesion according to established pathologic criteria. We considered this case to be borderline because the lesion was greater than 2 cm but the location was superficial; marked atypia with sarcomatoid features also were present, but there was an absence of necrosis and fewer than 5 mitoses per 50 HPFs. For these reasons, we diagnosed this problematic lesion as a GTUMP. Following reexcision, the patient underwent strict follow-up. Wound healing was uncomplicated and the patient showed no local recurrence or distant metastasis at 3-year follow-up.
Comment
Clinically metastatic and histologically malignant GTs are exceptional.3-11 The classification system for GTs based on histologic criteria subdivided these tumors into 3 groups with different prognoses.2 Malignant GTs are highly aggressive tumors with metastatic potential, symplastic GTs are considered to be a degenerative phenomenon, and GTUMPs have a favorable clinical outcome and absence of metastatic disease.12
Diagnosis of malignant GTs and symplastic GTs is relatively easy in the presence of typical uniform, small, round epithelioid cells (glomus cells) located around blood vessels. Immunohistochemistry may be useful, as GTs express smooth muscle actin and caldesmon. Over the years the existence and diagnosis of malignant GT has been questioned because a residual component of benign GT in the surgical biopsy is useful in diagnosis but is not always present1-3 and because an unusual pattern may be present in malignant tumors with prevalent spindle cells resembling fibrosarcoma, leiomyosarcoma, spindle cell angiosarcoma, and spindle cell melanoma.1 In the absence of a preexisting GT, the differential diagnosis may be difficult; in such cases, a panel of immunohistochemical markers including smooth muscle actin, caldesmon, desmin, S-100, human melanoma black-45 (HMB-45), CD34, and CD31 is always necessary. The GTUMP category was introduced for GTs that demonstrate marked nuclear atypia but do not fulfill histologic criteria for malignancy. Along with other tumors of uncertain malignant potential, borderline cases should be considered GTUMPs to guarantee wide excision of the tumor with negative margins and an adequate follow-up due to the possibility of local recurrence or distant metastasis. In our patient, a diagnosis of GTUMP was made. Additionally, our case demonstrates some previously unreported features of GTUMPs, such as spindled cells in short fascicles separated by slitlike spaces, small vessels, and hemorrhagic stroma resembling Kaposi sarcoma. Along with these unusual sarcomatous features, the superficial location of the lesion, absence of necrosis, and a mitotic count of less than 5 per 50 HPFs were suggestive of an uncertain malignant potential for this tumor.
Distinction between malignant GTs and GTUMPs in the presence of unusual histologic features may be difficult.12 Glomus tumors that do not fulfill criteria for malignancy but have at least 1 atypical feature other than nuclear pleomorphism should be named GTUMPs. According to classification criteria, a true malignant GT is a highly aggressive tumor with metastatic potential. In a case series reported by Folpe et al,2 38% (20/52) of malignant GTs showed metastases, while metastatic disease was not observed in the tumors classified as GTUMPs. Wide surgical excision or Mohs micrographic surgery13 are the treatments of choice for malignant GTs and GTUMPs. Complete excision of the lesion with negative margins is always necessary in cases of GTUMPs. After the diagnosis of GTUMP, adequate follow-up should berecommended due to the possibility of local recurrence or distant metastasis.
Conclusion
Malignant GTs and GTUMPs are rare, and the nomenclature and classification of these tumors is controversial. These findings and the difficulty of differential diagnosis in a continuum between benignity and malignancy prompted our report.
1. Weiss SW, Goldblum JR. Perivascular tumors. In: Enzinger FM, Weiss SW, eds. Enzinger and Weiss’s Soft Tissue Tumors. 5th ed. St Louis, MO: Mosby; 2008:751-768.
2. Folpe AL, Fanburg-Smith JC, Miettinen M, et al. Atypical and malignant glomus tumors: analysis of 52 cases, with a proposal for the reclassification of glomus tumors. Am J Surg Pathol. 2001;25:1-12.
3. Aiba M, Hirayama A, Kuramochi S. Glomangiosarcoma in a glomus tumor. an immunohistochemical and ultrastructural study. Cancer. 1988;61:1467-1471.
4. Gould EW, Manivel JC, Albores-Saavedra J, et al. Locally infiltrative glomus tumors and glomangiosarcomas. a clinical, ultrastructural, and immunohistochemical study. Cancer. 1990;65:310-318.
5. Noer H, Krogdahl A. Glomangiosarcoma of the lower extremity. Histopathology. 1991;18:365-366.
6. Hiruta N, Kameda N, Tokudome T, et al. Malignant glomus tumor: a case report and review of the literature. Am J Surg Pathol. 1997;21:1096-1103.
7. Watanabe K, Sugino T, Saito A, et al. Glomangiosarcoma of the hip: report of a highly aggressive tumour with widespread distant metastases. Br J Dermatol. 1998;139:1097-1101.
8. Park JH, Oh SH, Yang MH, et al. Glomangiosarcoma of the hand: a case report and review of the literature. J Dermatol. 2003;30:827-833.
9. Kayal JD, Hampton RW, Sheehan DJ, et al. Malignant glomus tumor: a case report and review of the literature. Dermatol Surg. 2001;27:837-840.
10. Pérez de la Fuente T, Vega C, Gutierrez Palacios A, et al. Glomangiosarcoma of the hypothenar eminence: a case report. Chir Main. 2005;24:199-202.
11. Terada T, Fujimoto J, Shirakashi Y, et al. Malignant glomus tumor of the palm: a case report. J Cutan Pathol. 2011;38:381-384.
12. Gill J, Van Vliet C. Infiltrating glomus tumor of uncertain malignant potential arising in the kidney. Hum Pathol. 2010;41:145-149.
13. Cecchi R, Pavesi M, Apicella P. Malignant glomus tumor of the trunk treated with Mohs micrographic surgery [in English, German]. J Dtsch Dermatol Ges. 2011;9:391-392.
1. Weiss SW, Goldblum JR. Perivascular tumors. In: Enzinger FM, Weiss SW, eds. Enzinger and Weiss’s Soft Tissue Tumors. 5th ed. St Louis, MO: Mosby; 2008:751-768.
2. Folpe AL, Fanburg-Smith JC, Miettinen M, et al. Atypical and malignant glomus tumors: analysis of 52 cases, with a proposal for the reclassification of glomus tumors. Am J Surg Pathol. 2001;25:1-12.
3. Aiba M, Hirayama A, Kuramochi S. Glomangiosarcoma in a glomus tumor. an immunohistochemical and ultrastructural study. Cancer. 1988;61:1467-1471.
4. Gould EW, Manivel JC, Albores-Saavedra J, et al. Locally infiltrative glomus tumors and glomangiosarcomas. a clinical, ultrastructural, and immunohistochemical study. Cancer. 1990;65:310-318.
5. Noer H, Krogdahl A. Glomangiosarcoma of the lower extremity. Histopathology. 1991;18:365-366.
6. Hiruta N, Kameda N, Tokudome T, et al. Malignant glomus tumor: a case report and review of the literature. Am J Surg Pathol. 1997;21:1096-1103.
7. Watanabe K, Sugino T, Saito A, et al. Glomangiosarcoma of the hip: report of a highly aggressive tumour with widespread distant metastases. Br J Dermatol. 1998;139:1097-1101.
8. Park JH, Oh SH, Yang MH, et al. Glomangiosarcoma of the hand: a case report and review of the literature. J Dermatol. 2003;30:827-833.
9. Kayal JD, Hampton RW, Sheehan DJ, et al. Malignant glomus tumor: a case report and review of the literature. Dermatol Surg. 2001;27:837-840.
10. Pérez de la Fuente T, Vega C, Gutierrez Palacios A, et al. Glomangiosarcoma of the hypothenar eminence: a case report. Chir Main. 2005;24:199-202.
11. Terada T, Fujimoto J, Shirakashi Y, et al. Malignant glomus tumor of the palm: a case report. J Cutan Pathol. 2011;38:381-384.
12. Gill J, Van Vliet C. Infiltrating glomus tumor of uncertain malignant potential arising in the kidney. Hum Pathol. 2010;41:145-149.
13. Cecchi R, Pavesi M, Apicella P. Malignant glomus tumor of the trunk treated with Mohs micrographic surgery [in English, German]. J Dtsch Dermatol Ges. 2011;9:391-392.
Practice Points
- Glomus tumors have been subdivided into 3 groups with different prognoses.
- The term glomus tumor of uncertain malignant potential (GTUMP) was introduced to describe glomus tumors that demonstrate marked nuclear atypia but do not fulfill histologic criteria for malignancy.
- Complete excision with negative margins is always necessary in cases of GTUMPs.
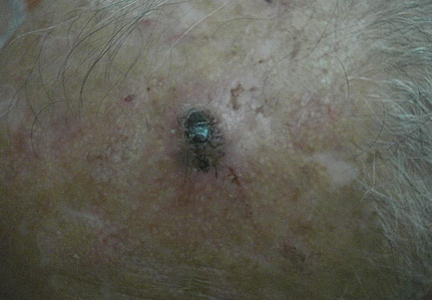
Adult-Type Langerhans Cell Histiocytosis: Minimal Treatment for Maximal Results
To the Editor:
A 78-year-old man presented with erythematous circular skin papules that were widely scattered over the trunk. He denied recent contact with ill individuals and denied any systemic symptoms indicating internal involvement or malignancy leading to possible paraneoplastic presentation. Physical examination showed erythematous, circular, slightly elevated plaques of varying sizes scattered over the trunk (Figure 1) and right axilla.

|
| Figure 1. Nontender, erythematous, brown nodules scattered over the trunk. |
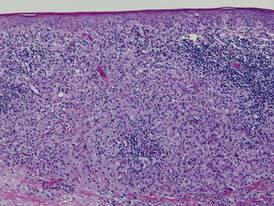
|
| Figure 2. Light microscopy revealed Langerhans cells filling the superficial dermis, abutting the epidermis, and extending into the deep dermis with surrounding inflammatory infiltrates (CD1a, original magnification ×100). |
|
| Figure 3. Langerhans cells appeared strongly positive for CD1a (original magnification ×400). |
Biopsies of lesions were taken and stained with immunoperoxidase. On light microscopy there was a reticular and papillary dermal dense infiltrate of cells with indented nuclei (Figure 2). At higher magnification, cells appeared strongly positive for CD1a (Figure 3) and S-100 protein, which was histologically consistent with adult-type Langerhans cell histiocytosis (ALCH).
Computed tomography of the head, chest, abdomen, and pelvis were ordered to rule out spread of ALCH to other organ sites. Results were clear of evidence of systemic spread. Additionally, a complete blood cell count and comprehensive metabolic panel were within reference range.
He was started on topical tacrolimus; however, most of the lesions resolved on their own. As a result, tacrolimus was discontinued due to its propensity to cause skin irritation and lack of change in disease progression. At 3-month follow-up, he was prescribed triamcinolone acetonide cream 0.1% for minor outbreaks. After 2 years, he was completely clear of all skin signs of ALCH.
Adult-type Langerhans cell histiocytosis is characterized as a group of disorders associated with abnormal spread and proliferation of dendritic cells of the epidermis. The disease primarily affects children aged 1 to 4 years. It is estimated that only 1 to 2 cases of ALCH per million occur.1 The pathophysiology of ALCH is unknown; it is speculated that it may be associated with a reactive inflammatory process triggered by proliferation of Langerhans-type dendritic cells. It is possible that the release of multiple cytokines by dendritic cells and T cells in ALCH lesions leads to erythematous eruptions and can contribute to spontaneous remission of the disorder.2 Various cases of ALCH have reported high serum levels of IL-17 and IL-10 proinflammatory cytokines, supporting the theory of an inflammatory etiology of ALCH.3
Comparative genomic hybridization with loss of heterozygosity of pulmonary lesions has provided further evidence to suggest that chromosomal aberrations also may contribute to the pathophysiology of ALCH.4 One study evaluated 14 cases of pulmonary ALCH for loss of heterozygosity and found allelic loss of 1p, 1q, 3p, 5p, 17p, and 22q.5 In addition, allelic loss of 1 or more tumor suppressor genes was identified in 19 of 24 specimens, suggesting a neoplastic type of pathology through uncontrolled cellular proliferation.6
Lesions of ALCH can be broad but typically present as red-brown maculopapular lesions with petechiae that erupt over the trunk, axilla, and perivulvar or retroauricular regions.7 The papules may unify to form an erythematous, weeping, or crusted eruption that appears similar to seborrheic dermatitis. Typically the lesions remit on their own; however, lesions can recur with the same or decreased severity as the primary eruption. Complications have been noted with lesions, particularly secondary infection and ulceration.7
Systemic involvement has been noted in adults, particularly in the lungs. Patients typically present with chronic cough, dyspnea, and chest pain with evidence of a solitary nodular lesion on radiologic testing. In addition, bone involvement has been noted as eosinophilic granulomas that can produce osteolytic lesions that lead to spontaneous fractures. Use of corticosteroids and immunosuppressive agents, as opposed to just observation, is warranted in cases of systemic involvement, according to the National Cancer Institute.7
Exact treatment modalities have not yet been elucidated due to the ambiguity of pathogenesis. In addition, ALCH is known to remit and relapse in patients, which increases the difficulty in evaluating the efficacy of particular treatments. Trials conducted by the Histiocyte Society have shown that treatment regimens should be tailored to disease severity. Epidermal involvement of ALCH typically responds to corticosteroid creams, whereas patients with systemic involvement respond well to strong chemotherapeutic agents such as vincristine and prednisone with mercaptopurine.8 However, as demonstrated in our case, lesions may remit on their own and use of corticosteroids and immunosuppressive agents may lead to further detriment without treating disease progression.
Because of a low prevalence among adults, ALCH is difficult to recognize and diagnose, and the uncertainty of the pathogenesis of ALCH limits treatment alternatives. Further study into proper treatment modalities is warranted given that the remitting and relapsing course of the disease and cosmetic quandaries are detrimental to patient well-being. Our case illustrates that it is appropriate to simply monitor lesions for cases limited to cutaneous involvement. Systemic agents may be used when there are signs of organ involvement outside the skin, but providers must proceed to do so with caution.
1. Baumgartner I, von Hochstetter A, Baumert B, et al. Langerhans’-cell histiocytosis in adults. Med Pediatr Oncol. 1997;28:9-14.
2. Egeler RM, Favara BE, van Meurs M, et al. Differential in situ cytokine profiles of Langerhans-like cells and T cells in Langerhans cell histiocytosis: abundant expression of cytokines relevant to disease and treatment. Blood. 1999;94:4195-4201.
3. da Costa CE, Szuhai K, van Eijk R, et al. No genomic aberrations in Langerhans cell histiocytosis as assessed by diverse molecular technologies. Genes Chromosomes Cancer. 2009;48:239-249.
4. Murakami I, Gogusev J, Fournet JC, et al. Detection of molecular cytogenetic aberrations in Langerhans cell histiocytosis of bone. Hum Pathol. 2002;33:555-560.
5. Dacic S, Trusky C, Bakker A, et al. Genotypic analysis of pulmonary Langerhans cell histiocytosis. Hum Pathol. 2003;34:1345-1349.
6. Chikwava KR, Hunt JL, Mantha GS, et al. Analysis of loss of heterozygosity in single-system and multisystem Langerhans’ cell histiocytosis. Pediatr Dev Pathol. 2007;10:18-24.
7. Langerhans cell histiocytosis treatment. National Cancer Institute Web site. http://www.cancer.gov/cancertopics/pdq/treatment/lchistio/HealthProfessional/page5. Updated June 4, 2014. Accessed August 27, 2014.
8. Weitzman S, Wayne AS, Arceci R, et al. Nucleoside analogues in the therapy of Langerhans cell histiocytosis: a survey of members of the histiocyte society and review of the literature. Med Pediatr Oncol. 1999;33:476-481.
To the Editor:
A 78-year-old man presented with erythematous circular skin papules that were widely scattered over the trunk. He denied recent contact with ill individuals and denied any systemic symptoms indicating internal involvement or malignancy leading to possible paraneoplastic presentation. Physical examination showed erythematous, circular, slightly elevated plaques of varying sizes scattered over the trunk (Figure 1) and right axilla.
|
|
| Figure 1. Nontender, erythematous, brown nodules scattered over the trunk. |
|
|
| Figure 2. Light microscopy revealed Langerhans cells filling the superficial dermis, abutting the epidermis, and extending into the deep dermis with surrounding inflammatory infiltrates (CD1a, original magnification ×100). |
|
|
| Figure 3. Langerhans cells appeared strongly positive for CD1a (original magnification ×400). |
Biopsies of lesions were taken and stained with immunoperoxidase. On light microscopy there was a reticular and papillary dermal dense infiltrate of cells with indented nuclei (Figure 2). At higher magnification, cells appeared strongly positive for CD1a (Figure 3) and S-100 protein, which was histologically consistent with adult-type Langerhans cell histiocytosis (ALCH).
Computed tomography of the head, chest, abdomen, and pelvis were ordered to rule out spread of ALCH to other organ sites. Results were clear of evidence of systemic spread. Additionally, a complete blood cell count and comprehensive metabolic panel were within reference range.
He was started on topical tacrolimus; however, most of the lesions resolved on their own. As a result, tacrolimus was discontinued due to its propensity to cause skin irritation and lack of change in disease progression. At 3-month follow-up, he was prescribed triamcinolone acetonide cream 0.1% for minor outbreaks. After 2 years, he was completely clear of all skin signs of ALCH.
Adult-type Langerhans cell histiocytosis is characterized as a group of disorders associated with abnormal spread and proliferation of dendritic cells of the epidermis. The disease primarily affects children aged 1 to 4 years. It is estimated that only 1 to 2 cases of ALCH per million occur.1 The pathophysiology of ALCH is unknown; it is speculated that it may be associated with a reactive inflammatory process triggered by proliferation of Langerhans-type dendritic cells. It is possible that the release of multiple cytokines by dendritic cells and T cells in ALCH lesions leads to erythematous eruptions and can contribute to spontaneous remission of the disorder.2 Various cases of ALCH have reported high serum levels of IL-17 and IL-10 proinflammatory cytokines, supporting the theory of an inflammatory etiology of ALCH.3
Comparative genomic hybridization with loss of heterozygosity of pulmonary lesions has provided further evidence to suggest that chromosomal aberrations also may contribute to the pathophysiology of ALCH.4 One study evaluated 14 cases of pulmonary ALCH for loss of heterozygosity and found allelic loss of 1p, 1q, 3p, 5p, 17p, and 22q.5 In addition, allelic loss of 1 or more tumor suppressor genes was identified in 19 of 24 specimens, suggesting a neoplastic type of pathology through uncontrolled cellular proliferation.6
Lesions of ALCH can be broad but typically present as red-brown maculopapular lesions with petechiae that erupt over the trunk, axilla, and perivulvar or retroauricular regions.7 The papules may unify to form an erythematous, weeping, or crusted eruption that appears similar to seborrheic dermatitis. Typically the lesions remit on their own; however, lesions can recur with the same or decreased severity as the primary eruption. Complications have been noted with lesions, particularly secondary infection and ulceration.7
Systemic involvement has been noted in adults, particularly in the lungs. Patients typically present with chronic cough, dyspnea, and chest pain with evidence of a solitary nodular lesion on radiologic testing. In addition, bone involvement has been noted as eosinophilic granulomas that can produce osteolytic lesions that lead to spontaneous fractures. Use of corticosteroids and immunosuppressive agents, as opposed to just observation, is warranted in cases of systemic involvement, according to the National Cancer Institute.7
Exact treatment modalities have not yet been elucidated due to the ambiguity of pathogenesis. In addition, ALCH is known to remit and relapse in patients, which increases the difficulty in evaluating the efficacy of particular treatments. Trials conducted by the Histiocyte Society have shown that treatment regimens should be tailored to disease severity. Epidermal involvement of ALCH typically responds to corticosteroid creams, whereas patients with systemic involvement respond well to strong chemotherapeutic agents such as vincristine and prednisone with mercaptopurine.8 However, as demonstrated in our case, lesions may remit on their own and use of corticosteroids and immunosuppressive agents may lead to further detriment without treating disease progression.
Because of a low prevalence among adults, ALCH is difficult to recognize and diagnose, and the uncertainty of the pathogenesis of ALCH limits treatment alternatives. Further study into proper treatment modalities is warranted given that the remitting and relapsing course of the disease and cosmetic quandaries are detrimental to patient well-being. Our case illustrates that it is appropriate to simply monitor lesions for cases limited to cutaneous involvement. Systemic agents may be used when there are signs of organ involvement outside the skin, but providers must proceed to do so with caution.
To the Editor:
A 78-year-old man presented with erythematous circular skin papules that were widely scattered over the trunk. He denied recent contact with ill individuals and denied any systemic symptoms indicating internal involvement or malignancy leading to possible paraneoplastic presentation. Physical examination showed erythematous, circular, slightly elevated plaques of varying sizes scattered over the trunk (Figure 1) and right axilla.
|
|
| Figure 1. Nontender, erythematous, brown nodules scattered over the trunk. |
|
|
| Figure 2. Light microscopy revealed Langerhans cells filling the superficial dermis, abutting the epidermis, and extending into the deep dermis with surrounding inflammatory infiltrates (CD1a, original magnification ×100). |
|
|
| Figure 3. Langerhans cells appeared strongly positive for CD1a (original magnification ×400). |
Biopsies of lesions were taken and stained with immunoperoxidase. On light microscopy there was a reticular and papillary dermal dense infiltrate of cells with indented nuclei (Figure 2). At higher magnification, cells appeared strongly positive for CD1a (Figure 3) and S-100 protein, which was histologically consistent with adult-type Langerhans cell histiocytosis (ALCH).
Computed tomography of the head, chest, abdomen, and pelvis were ordered to rule out spread of ALCH to other organ sites. Results were clear of evidence of systemic spread. Additionally, a complete blood cell count and comprehensive metabolic panel were within reference range.
He was started on topical tacrolimus; however, most of the lesions resolved on their own. As a result, tacrolimus was discontinued due to its propensity to cause skin irritation and lack of change in disease progression. At 3-month follow-up, he was prescribed triamcinolone acetonide cream 0.1% for minor outbreaks. After 2 years, he was completely clear of all skin signs of ALCH.
Adult-type Langerhans cell histiocytosis is characterized as a group of disorders associated with abnormal spread and proliferation of dendritic cells of the epidermis. The disease primarily affects children aged 1 to 4 years. It is estimated that only 1 to 2 cases of ALCH per million occur.1 The pathophysiology of ALCH is unknown; it is speculated that it may be associated with a reactive inflammatory process triggered by proliferation of Langerhans-type dendritic cells. It is possible that the release of multiple cytokines by dendritic cells and T cells in ALCH lesions leads to erythematous eruptions and can contribute to spontaneous remission of the disorder.2 Various cases of ALCH have reported high serum levels of IL-17 and IL-10 proinflammatory cytokines, supporting the theory of an inflammatory etiology of ALCH.3
Comparative genomic hybridization with loss of heterozygosity of pulmonary lesions has provided further evidence to suggest that chromosomal aberrations also may contribute to the pathophysiology of ALCH.4 One study evaluated 14 cases of pulmonary ALCH for loss of heterozygosity and found allelic loss of 1p, 1q, 3p, 5p, 17p, and 22q.5 In addition, allelic loss of 1 or more tumor suppressor genes was identified in 19 of 24 specimens, suggesting a neoplastic type of pathology through uncontrolled cellular proliferation.6
Lesions of ALCH can be broad but typically present as red-brown maculopapular lesions with petechiae that erupt over the trunk, axilla, and perivulvar or retroauricular regions.7 The papules may unify to form an erythematous, weeping, or crusted eruption that appears similar to seborrheic dermatitis. Typically the lesions remit on their own; however, lesions can recur with the same or decreased severity as the primary eruption. Complications have been noted with lesions, particularly secondary infection and ulceration.7
Systemic involvement has been noted in adults, particularly in the lungs. Patients typically present with chronic cough, dyspnea, and chest pain with evidence of a solitary nodular lesion on radiologic testing. In addition, bone involvement has been noted as eosinophilic granulomas that can produce osteolytic lesions that lead to spontaneous fractures. Use of corticosteroids and immunosuppressive agents, as opposed to just observation, is warranted in cases of systemic involvement, according to the National Cancer Institute.7
Exact treatment modalities have not yet been elucidated due to the ambiguity of pathogenesis. In addition, ALCH is known to remit and relapse in patients, which increases the difficulty in evaluating the efficacy of particular treatments. Trials conducted by the Histiocyte Society have shown that treatment regimens should be tailored to disease severity. Epidermal involvement of ALCH typically responds to corticosteroid creams, whereas patients with systemic involvement respond well to strong chemotherapeutic agents such as vincristine and prednisone with mercaptopurine.8 However, as demonstrated in our case, lesions may remit on their own and use of corticosteroids and immunosuppressive agents may lead to further detriment without treating disease progression.
Because of a low prevalence among adults, ALCH is difficult to recognize and diagnose, and the uncertainty of the pathogenesis of ALCH limits treatment alternatives. Further study into proper treatment modalities is warranted given that the remitting and relapsing course of the disease and cosmetic quandaries are detrimental to patient well-being. Our case illustrates that it is appropriate to simply monitor lesions for cases limited to cutaneous involvement. Systemic agents may be used when there are signs of organ involvement outside the skin, but providers must proceed to do so with caution.
1. Baumgartner I, von Hochstetter A, Baumert B, et al. Langerhans’-cell histiocytosis in adults. Med Pediatr Oncol. 1997;28:9-14.
2. Egeler RM, Favara BE, van Meurs M, et al. Differential in situ cytokine profiles of Langerhans-like cells and T cells in Langerhans cell histiocytosis: abundant expression of cytokines relevant to disease and treatment. Blood. 1999;94:4195-4201.
3. da Costa CE, Szuhai K, van Eijk R, et al. No genomic aberrations in Langerhans cell histiocytosis as assessed by diverse molecular technologies. Genes Chromosomes Cancer. 2009;48:239-249.
4. Murakami I, Gogusev J, Fournet JC, et al. Detection of molecular cytogenetic aberrations in Langerhans cell histiocytosis of bone. Hum Pathol. 2002;33:555-560.
5. Dacic S, Trusky C, Bakker A, et al. Genotypic analysis of pulmonary Langerhans cell histiocytosis. Hum Pathol. 2003;34:1345-1349.
6. Chikwava KR, Hunt JL, Mantha GS, et al. Analysis of loss of heterozygosity in single-system and multisystem Langerhans’ cell histiocytosis. Pediatr Dev Pathol. 2007;10:18-24.
7. Langerhans cell histiocytosis treatment. National Cancer Institute Web site. http://www.cancer.gov/cancertopics/pdq/treatment/lchistio/HealthProfessional/page5. Updated June 4, 2014. Accessed August 27, 2014.
8. Weitzman S, Wayne AS, Arceci R, et al. Nucleoside analogues in the therapy of Langerhans cell histiocytosis: a survey of members of the histiocyte society and review of the literature. Med Pediatr Oncol. 1999;33:476-481.
1. Baumgartner I, von Hochstetter A, Baumert B, et al. Langerhans’-cell histiocytosis in adults. Med Pediatr Oncol. 1997;28:9-14.
2. Egeler RM, Favara BE, van Meurs M, et al. Differential in situ cytokine profiles of Langerhans-like cells and T cells in Langerhans cell histiocytosis: abundant expression of cytokines relevant to disease and treatment. Blood. 1999;94:4195-4201.
3. da Costa CE, Szuhai K, van Eijk R, et al. No genomic aberrations in Langerhans cell histiocytosis as assessed by diverse molecular technologies. Genes Chromosomes Cancer. 2009;48:239-249.
4. Murakami I, Gogusev J, Fournet JC, et al. Detection of molecular cytogenetic aberrations in Langerhans cell histiocytosis of bone. Hum Pathol. 2002;33:555-560.
5. Dacic S, Trusky C, Bakker A, et al. Genotypic analysis of pulmonary Langerhans cell histiocytosis. Hum Pathol. 2003;34:1345-1349.
6. Chikwava KR, Hunt JL, Mantha GS, et al. Analysis of loss of heterozygosity in single-system and multisystem Langerhans’ cell histiocytosis. Pediatr Dev Pathol. 2007;10:18-24.
7. Langerhans cell histiocytosis treatment. National Cancer Institute Web site. http://www.cancer.gov/cancertopics/pdq/treatment/lchistio/HealthProfessional/page5. Updated June 4, 2014. Accessed August 27, 2014.
8. Weitzman S, Wayne AS, Arceci R, et al. Nucleoside analogues in the therapy of Langerhans cell histiocytosis: a survey of members of the histiocyte society and review of the literature. Med Pediatr Oncol. 1999;33:476-481.
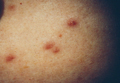
Pretibial Myxedema
Pretibial myxedema (PM) is a cutaneous mucinosis associated with thyroid dysfunction. It most commonly presents in the setting of Graves disease and is seen less often in patients with hypothyroidism and euthyroidism.1 The anterior tibia is most frequently affected, but lesions also may present on the feet, thighs, and upper extremities. Physical examination generally demonstrates skin thickening, hyperkeratosis, hyperpigmentation, yellow-red discoloration, and hyperhidrosis. Classically, the term peau d’orange has been used to characterize these clinical features.1 Histologic examination of PM typically reveals marked deposition of mucin throughout the reticular dermis with sparing of the papillary dermis (Figure 1) and may be accompanied by overlying hyperkeratosis. Collagen fibers are splayed and appear decreased in density (Figure 2). Alcian blue, periodic acid–Schiff, colloidal iron, and toluidine blue staining can be used to highlight dermal mucin.

| 
|
Figure 1. Prominent mucin deposition throughout the reticular dermis in pretibial myxedema (H&E, original magnification ×40). | Figure 2. Increased mucin deposition and collagen fiber splaying in pretibial myxedema (H&E, original magnification ×200). |
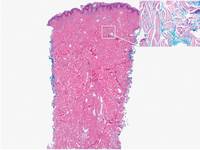
| 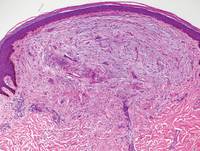
|
Figure 3. Scleredema with increased dermal thickness (H&E, original magnification ×20) and interstitial mucin on colloidal iron–stained sections (inset in upper right corner, original magnification ×400). | Figure 4. Scleromyxedema with mucin deposition primarily in the superficial dermis as well as increased cellularity and fibrosis (H&E, original magnification ×200). |
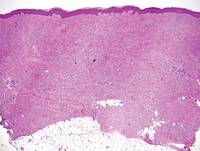
| 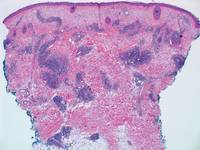
|
Figure 5. Nephrogenic systemic fibrosis with prominent mucinous fibrosis (H&E, original magnification ×40). | Figure 6. Tumid lupus erythematosus with a superficial and perivascular lymphoid infiltrate as well as increased dermal mucin (H&E, original magnification ×40). |
Similar to PM, histologic examination of scleredema and scleromyxedema usually demonstrate prominent mucin deposition. In scleredema, mucin primarily is visualized in the deep dermis between thick collagen bundles and typically is localized to the back (Figure 3). Scleromyxedema is distinguished by mucin deposition in the superficial dermis with associated fibroblast proliferation and fibrosis (Figure 4).
Nephrogenic systemic fibrosis is characterized by proliferation of CD34+ dermal spindle cells, fibroblasts, interstitial mucin, altered elastic fibers, and thickened collagen bundles that involve the dermis and subcutaneous septa (Figure 5).2 Tumid lupus erythematosus classically demonstrates perivascular and periadnexal superficial and deep lymphocytic inflammation (Figure 6). Similar to scleredema and PM, mucin deposition in tumid lupus erythematosus is interspersed between collagen bundles in the reticular dermis.3
1. Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309.
2. Cowper SE, Lyndon DS, Bhawan J, et al. Nephro-genic fibrosing dermopathy. Am J Dermatopathol.2001;23:383-393.
3. Kuhn A, Dagmar RH, Oslislo C, et al. Lupus erythematosus tumidus: a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
Pretibial myxedema (PM) is a cutaneous mucinosis associated with thyroid dysfunction. It most commonly presents in the setting of Graves disease and is seen less often in patients with hypothyroidism and euthyroidism.1 The anterior tibia is most frequently affected, but lesions also may present on the feet, thighs, and upper extremities. Physical examination generally demonstrates skin thickening, hyperkeratosis, hyperpigmentation, yellow-red discoloration, and hyperhidrosis. Classically, the term peau d’orange has been used to characterize these clinical features.1 Histologic examination of PM typically reveals marked deposition of mucin throughout the reticular dermis with sparing of the papillary dermis (Figure 1) and may be accompanied by overlying hyperkeratosis. Collagen fibers are splayed and appear decreased in density (Figure 2). Alcian blue, periodic acid–Schiff, colloidal iron, and toluidine blue staining can be used to highlight dermal mucin.
|
|
|
Figure 1. Prominent mucin deposition throughout the reticular dermis in pretibial myxedema (H&E, original magnification ×40). | Figure 2. Increased mucin deposition and collagen fiber splaying in pretibial myxedema (H&E, original magnification ×200). |
|
|
|
Figure 3. Scleredema with increased dermal thickness (H&E, original magnification ×20) and interstitial mucin on colloidal iron–stained sections (inset in upper right corner, original magnification ×400). | Figure 4. Scleromyxedema with mucin deposition primarily in the superficial dermis as well as increased cellularity and fibrosis (H&E, original magnification ×200). |
|
|
|
Figure 5. Nephrogenic systemic fibrosis with prominent mucinous fibrosis (H&E, original magnification ×40). | Figure 6. Tumid lupus erythematosus with a superficial and perivascular lymphoid infiltrate as well as increased dermal mucin (H&E, original magnification ×40). |
Similar to PM, histologic examination of scleredema and scleromyxedema usually demonstrate prominent mucin deposition. In scleredema, mucin primarily is visualized in the deep dermis between thick collagen bundles and typically is localized to the back (Figure 3). Scleromyxedema is distinguished by mucin deposition in the superficial dermis with associated fibroblast proliferation and fibrosis (Figure 4).
Nephrogenic systemic fibrosis is characterized by proliferation of CD34+ dermal spindle cells, fibroblasts, interstitial mucin, altered elastic fibers, and thickened collagen bundles that involve the dermis and subcutaneous septa (Figure 5).2 Tumid lupus erythematosus classically demonstrates perivascular and periadnexal superficial and deep lymphocytic inflammation (Figure 6). Similar to scleredema and PM, mucin deposition in tumid lupus erythematosus is interspersed between collagen bundles in the reticular dermis.3
Pretibial myxedema (PM) is a cutaneous mucinosis associated with thyroid dysfunction. It most commonly presents in the setting of Graves disease and is seen less often in patients with hypothyroidism and euthyroidism.1 The anterior tibia is most frequently affected, but lesions also may present on the feet, thighs, and upper extremities. Physical examination generally demonstrates skin thickening, hyperkeratosis, hyperpigmentation, yellow-red discoloration, and hyperhidrosis. Classically, the term peau d’orange has been used to characterize these clinical features.1 Histologic examination of PM typically reveals marked deposition of mucin throughout the reticular dermis with sparing of the papillary dermis (Figure 1) and may be accompanied by overlying hyperkeratosis. Collagen fibers are splayed and appear decreased in density (Figure 2). Alcian blue, periodic acid–Schiff, colloidal iron, and toluidine blue staining can be used to highlight dermal mucin.
|
|
|
Figure 1. Prominent mucin deposition throughout the reticular dermis in pretibial myxedema (H&E, original magnification ×40). | Figure 2. Increased mucin deposition and collagen fiber splaying in pretibial myxedema (H&E, original magnification ×200). |
|
|
|
Figure 3. Scleredema with increased dermal thickness (H&E, original magnification ×20) and interstitial mucin on colloidal iron–stained sections (inset in upper right corner, original magnification ×400). | Figure 4. Scleromyxedema with mucin deposition primarily in the superficial dermis as well as increased cellularity and fibrosis (H&E, original magnification ×200). |
|
|
|
Figure 5. Nephrogenic systemic fibrosis with prominent mucinous fibrosis (H&E, original magnification ×40). | Figure 6. Tumid lupus erythematosus with a superficial and perivascular lymphoid infiltrate as well as increased dermal mucin (H&E, original magnification ×40). |
Similar to PM, histologic examination of scleredema and scleromyxedema usually demonstrate prominent mucin deposition. In scleredema, mucin primarily is visualized in the deep dermis between thick collagen bundles and typically is localized to the back (Figure 3). Scleromyxedema is distinguished by mucin deposition in the superficial dermis with associated fibroblast proliferation and fibrosis (Figure 4).
Nephrogenic systemic fibrosis is characterized by proliferation of CD34+ dermal spindle cells, fibroblasts, interstitial mucin, altered elastic fibers, and thickened collagen bundles that involve the dermis and subcutaneous septa (Figure 5).2 Tumid lupus erythematosus classically demonstrates perivascular and periadnexal superficial and deep lymphocytic inflammation (Figure 6). Similar to scleredema and PM, mucin deposition in tumid lupus erythematosus is interspersed between collagen bundles in the reticular dermis.3
1. Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309.
2. Cowper SE, Lyndon DS, Bhawan J, et al. Nephro-genic fibrosing dermopathy. Am J Dermatopathol.2001;23:383-393.
3. Kuhn A, Dagmar RH, Oslislo C, et al. Lupus erythematosus tumidus: a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
1. Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309.
2. Cowper SE, Lyndon DS, Bhawan J, et al. Nephro-genic fibrosing dermopathy. Am J Dermatopathol.2001;23:383-393.
3. Kuhn A, Dagmar RH, Oslislo C, et al. Lupus erythematosus tumidus: a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
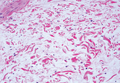
Buschke-Ollendorff Syndrome: Sparing Unnecessary Investigations
Buschke-Ollendorff syndrome (BOS) is a rare disease that is inherited in an autosomal-dominant fashion with high penetrance. It is characterized by osteopoikilosis associated with skin manifestations. The approximate incidence of the disease is 1:20,000, with few cases reported in the literature since 1928.1 Skeletal lesions known as osteopoikilosis are areas of increased bone density that can be seen on radiographic imaging and typically are located in the substantia spongiosa of the epiphyses and metaphyses of long bones and the pelvis. In BOS, cutaneous lesions consist of elastic or collagen nevi. Phenotypic expression of the disease is variable, and skeletal and cutaneous lesions may occur separately. Gene mutations of proteins involved in bone and connective tissue morphogenesis have been described in patients with BOS.2-5
Case Reports
Patient 1
A 17-year-old adolescent girl was referred to our hospital for evaluation of an incidental finding of osteopoikilosis that had been noted in the setting of a traumatic event. Clinical examination revealed cutaneous lesions characterized by asymptomatic, linear, stringlike and atrophic fibrotic plaques localized symmetrically on the trunk, right buttock, and right thigh (Figure 1). The lesions on the thigh were present at birth and had spread progressively to the other areas. There was no known history of inflammatory skin disease to explain the presence of the lesions, and no family history of similar signs or symptoms was reported. Histopathologic examination of a punch biopsy of a plaque from the trunk revealed increased collagen bundles associated with thick interlacing elastic fibers. The epidermis did not show any specific histologic alterations. These histopathologic features were diagnostic of a connective tissue nevus (Figure 2).
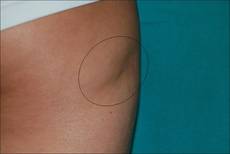
|
Figure 1. An asymptomatic atrophic fibrotic plaque localized on the right thigh. |
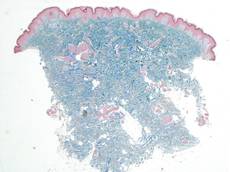
|
Figure 2. Low-power view of a punch biopsy showing a normal epidermis. Collagen bundles of the dermis were thickened and somewhat homogenized, as highlighted by the Masson stain (original magnification ×2.5). |
|
Figure 3. Radiograph of the legs revealed numerous small, ovoid or round foci of sclerosis on the substantia spongiosa of the metaphyses and the epiphyses of the femur, tibia, and fibula on both sides of the body. |
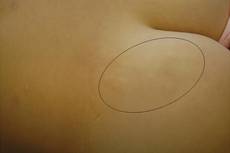
|
Figure 4. Multiple asymptomatic flesh-colored papules with elastic consistency on the back that were characteristic of dermatofibrosis lenticularis disseminata. |
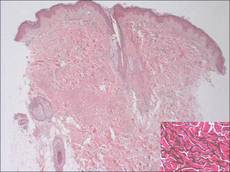
|
Figure 5. The epidermis and dermis appeared normal on histopathology; however, the elastic fibers were markedly increased in both size and number up to the deep dermis in the absence of degenerating changes (H&E, original magnification ×2.5; vascular endothelial growth factor, original magnification ×20 [inset in bottom right corner]). |
|
| Figure 6. A radiograph of the left hand revealed small sporadic areas of sclerosis in the substantia spongiosa on the heads of some phalanges (white arrows). |
Radiographic imaging of the hands and feet revealed numerous small, ovoid or round foci of sclerosis that were a few millimeters in diameter and were occasionally confluent. This finding was prevalent on the carpal and tarsal bones but less evident on the phalanges and the epiphysis of the metatarsal and metacarpal bones. Full radiographic imaging of both arms and legs subsequently was obtained and showed similar lesions predominantly in the substantia spongiosa of the metaphyses and the epiphyses of the humerus, femur, tibia, and fibula bilaterally (Figure 3). Evaluation of the patient’s parents revealed that her mother had sporadic lenticular areas of increased bone density seen on radiography of the carpal and tarsal bones, particularly at the level of calcaneus, and the proximal and distal humeral epiphyses.
These lesions in our patient were consistent with an incomplete form of osteopoikilosis, which reflects the known variable expression of BOS. The radiologic findings in addition to the cutaneous lesions and the positive family history supported a diagnosis of BOS.
Patient 2
A 5-year-old boy presented with multiple congenital, asymptomatic, flesh-colored papules with elastic consistency on the left thigh, back, and pubic area that were characteristic of dermatofibrosis lenticularis disseminata (Figure 4). The patient’s 7-year-old sister had similar cutaneous lesions. The patient underwent a skin biopsy from the back that revealed thickening of collagen bundles in the dermis with an increase in elastic fibers (Figure 5). On radiographic imaging, small sporadic areas of sclerosis were noted in the substantia spongiosa of the lateral aspect of the humeral capitulum, the radial neck, the capitate, the head of the proximal phalanx of the fourth finger on the left hand, and the heads of the third and fifth middle phalanges on the left hand (Figure 6). Radiography revealed osteopoikilosis on both humeral heads in the patient’s mother. These findings in addition to the dermatologic and histopathologic features suggested a diagnosis of BOS.
Comment
In 1928, Buschke and Ollendorff1 first reported the association between osteopoikilosis and dermatofibrosis lenticularis disseminata, which showed autosomal-dominant inheritance. Skin and bone lesions can occur independently in different family members. Two different clinical cutaneous patterns are described in BOS.2-4 The first and most frequent form is characterized by yellowish nodules often grouped in plaques that are asymmetrically distributed, such as those seen in patient 1. The second form is known as dermatofibrosis lenticularis disseminata, which was seen in patient 2. Histologically, most lesions show normal collagen fibrils and an increased number of elastic fibers.
Osteopoikilosis, also known as spotted bone disease or osteopathia condensans, is a rare asymptomatic bone dysplasia of unknown etiology. It is characterized by an abnormality in the bone maturation process and often is found incidentally on radiologic examination, as seen in patient 1. Radiologic signs of osteopoikilosis consist of small, disseminated, well-circumscribed areas of increased radiodensity located in the epiphyses and metaphyses of long bones, as well as the pelvis, hands, and feet. These lesions, which typically are asymptomatic, could sometimes be associated with bone and/or joint pain but do not cause a predisposition to fractures. Documentation of these bone lesions by early adult life is important to avoid confusion with osteoblastic metastases on the skeleton.6
Buschke-Ollendorff syndrome is characterized by a variable expression. Loss-of-function mutations of the LEMD3 gene have been described in association with this disorder.7 This gene encodes an inner nuclear protein membrane with a C-terminal domain that binds SMAD2 and SMAD3 and antagonizes the bone morphogenetic proteins and transforming growth factor b. These proteins are involved in connective tissue morphogenesis, inducing elastin production from fibroblasts.7 Seven novel loss-of-function mutations of LEMD3 have been identified.8 The segmental manifestation of BOS could be a consequence of the mosaicism resulting from a somatic mutation.9,10 A case describing the absence of LEMD3 mutation in an affected family suggested the genetic heterogeneity of BOS.11
Conclusion
Buschke-Ollendorff syndrome is a benign condition with no repercussions on the patient’s health or quality of life. It does not require any specific treatment because the lesions generally remain asymptomatic and do not generate any substantial cosmetic burden. Mutation analysis was not performed in our patients because BOS has a good prognosis and the parents refused further investigation in both patients; therefore, a correct diagnosis was essential in our cases to rule out malignant bone disease in patient 1 whose osteopoikilosis prompted the workup and other disorders (eg, tuberous sclerosis, pseudoxanthoma elasticum) in patient 2 whose cutaneous lesions were the primary cause for presentation. A correct diagnosis of BOS is necessary to spare patients from expensive investigations and to provide reassurance about the benign nature of the disease.
1. Buschke A, Ollendorff H. Ein fall von dermatofibrosis lenticularis disseminata und osteopathia condensas disseminata. Derm Worchenschr. 1928;86:257-262.
2. Ramme K, Kolde G, Stadler R. Dermatofibrosis lenticularis disseminata with osteopoikilosis. Buschke-Ollendorff syndrome [in German]. Hautarzt. 1993;44:312-314.
3. Schena D, Germi L, Zamperetti MR, et al. Buschke-Ollendorff syndrome. Int J Dermatol. 2008;47:1159-1161.
4. Kawamura A, Ochiai T, Tan-Kinoshita M, et al. Buschke-Ollendorff syndrome: three generations in a Japanese family. Pediatr Dermatol. 2005;22:133-137.
5. Woodrow SL, Pope FM, Handfield-Jones SE. The Buschke-Ollendorff syndrome presenting as familial elastic tissue naevi. Br J Dermatol. 2001;144:890-893.
6. Whyte MP, Murphy WA, Siegel BA. 99mTc-pyrophosphate bone imaging in osteopoikilosis, osteopathia striata, and melorheostosis. Radiology. 1978;127:439-443.
7. Giro MG, Duvic M, Smith LT, et al. Buschke-Ollendorff syndrome associated with elevated elastin production by affected skin fibroblasts in culture. J Invest Dermatol. 1992;99:129-137.
8. Hellemans J, Preobrazhenska O, Willaert A, et al. Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis [published online ahead of print October 17, 2004]. Nat Genet. 2004;36:1213-1218.
9. Ehrig T, Cockerell CJ. Buschke-Ollendorff syndrome: report of a case and interpretation of the clinical phenotype as a type 2 segmental manifestation of an autosomal dominant skin disease. J Am Acad Dermatol. 2003;49:1163-1166.
10. Hellemans J, Debeer P, Wright M, et al. Germline LEMD3 mutations are rare in sporadic patients with isolated melorheostosis. Hum Mutat. 2006;27:290.
11. Yadegari M, Whyte MP, Mumm S, et al. Buschke-Ollendorff syndrome: absence of LEMD3 mutation in an affected family. Arch Dermatol. 2010;146:63-68.
Buschke-Ollendorff syndrome (BOS) is a rare disease that is inherited in an autosomal-dominant fashion with high penetrance. It is characterized by osteopoikilosis associated with skin manifestations. The approximate incidence of the disease is 1:20,000, with few cases reported in the literature since 1928.1 Skeletal lesions known as osteopoikilosis are areas of increased bone density that can be seen on radiographic imaging and typically are located in the substantia spongiosa of the epiphyses and metaphyses of long bones and the pelvis. In BOS, cutaneous lesions consist of elastic or collagen nevi. Phenotypic expression of the disease is variable, and skeletal and cutaneous lesions may occur separately. Gene mutations of proteins involved in bone and connective tissue morphogenesis have been described in patients with BOS.2-5
Case Reports
Patient 1
A 17-year-old adolescent girl was referred to our hospital for evaluation of an incidental finding of osteopoikilosis that had been noted in the setting of a traumatic event. Clinical examination revealed cutaneous lesions characterized by asymptomatic, linear, stringlike and atrophic fibrotic plaques localized symmetrically on the trunk, right buttock, and right thigh (Figure 1). The lesions on the thigh were present at birth and had spread progressively to the other areas. There was no known history of inflammatory skin disease to explain the presence of the lesions, and no family history of similar signs or symptoms was reported. Histopathologic examination of a punch biopsy of a plaque from the trunk revealed increased collagen bundles associated with thick interlacing elastic fibers. The epidermis did not show any specific histologic alterations. These histopathologic features were diagnostic of a connective tissue nevus (Figure 2).
|
|
Figure 1. An asymptomatic atrophic fibrotic plaque localized on the right thigh. |
|
|
Figure 2. Low-power view of a punch biopsy showing a normal epidermis. Collagen bundles of the dermis were thickened and somewhat homogenized, as highlighted by the Masson stain (original magnification ×2.5). |
|
|
Figure 3. Radiograph of the legs revealed numerous small, ovoid or round foci of sclerosis on the substantia spongiosa of the metaphyses and the epiphyses of the femur, tibia, and fibula on both sides of the body. |
|
|
Figure 4. Multiple asymptomatic flesh-colored papules with elastic consistency on the back that were characteristic of dermatofibrosis lenticularis disseminata. |
|
|
Figure 5. The epidermis and dermis appeared normal on histopathology; however, the elastic fibers were markedly increased in both size and number up to the deep dermis in the absence of degenerating changes (H&E, original magnification ×2.5; vascular endothelial growth factor, original magnification ×20 [inset in bottom right corner]). |
|
|
| Figure 6. A radiograph of the left hand revealed small sporadic areas of sclerosis in the substantia spongiosa on the heads of some phalanges (white arrows). |
Radiographic imaging of the hands and feet revealed numerous small, ovoid or round foci of sclerosis that were a few millimeters in diameter and were occasionally confluent. This finding was prevalent on the carpal and tarsal bones but less evident on the phalanges and the epiphysis of the metatarsal and metacarpal bones. Full radiographic imaging of both arms and legs subsequently was obtained and showed similar lesions predominantly in the substantia spongiosa of the metaphyses and the epiphyses of the humerus, femur, tibia, and fibula bilaterally (Figure 3). Evaluation of the patient’s parents revealed that her mother had sporadic lenticular areas of increased bone density seen on radiography of the carpal and tarsal bones, particularly at the level of calcaneus, and the proximal and distal humeral epiphyses.
These lesions in our patient were consistent with an incomplete form of osteopoikilosis, which reflects the known variable expression of BOS. The radiologic findings in addition to the cutaneous lesions and the positive family history supported a diagnosis of BOS.
Patient 2
A 5-year-old boy presented with multiple congenital, asymptomatic, flesh-colored papules with elastic consistency on the left thigh, back, and pubic area that were characteristic of dermatofibrosis lenticularis disseminata (Figure 4). The patient’s 7-year-old sister had similar cutaneous lesions. The patient underwent a skin biopsy from the back that revealed thickening of collagen bundles in the dermis with an increase in elastic fibers (Figure 5). On radiographic imaging, small sporadic areas of sclerosis were noted in the substantia spongiosa of the lateral aspect of the humeral capitulum, the radial neck, the capitate, the head of the proximal phalanx of the fourth finger on the left hand, and the heads of the third and fifth middle phalanges on the left hand (Figure 6). Radiography revealed osteopoikilosis on both humeral heads in the patient’s mother. These findings in addition to the dermatologic and histopathologic features suggested a diagnosis of BOS.
Comment
In 1928, Buschke and Ollendorff1 first reported the association between osteopoikilosis and dermatofibrosis lenticularis disseminata, which showed autosomal-dominant inheritance. Skin and bone lesions can occur independently in different family members. Two different clinical cutaneous patterns are described in BOS.2-4 The first and most frequent form is characterized by yellowish nodules often grouped in plaques that are asymmetrically distributed, such as those seen in patient 1. The second form is known as dermatofibrosis lenticularis disseminata, which was seen in patient 2. Histologically, most lesions show normal collagen fibrils and an increased number of elastic fibers.
Osteopoikilosis, also known as spotted bone disease or osteopathia condensans, is a rare asymptomatic bone dysplasia of unknown etiology. It is characterized by an abnormality in the bone maturation process and often is found incidentally on radiologic examination, as seen in patient 1. Radiologic signs of osteopoikilosis consist of small, disseminated, well-circumscribed areas of increased radiodensity located in the epiphyses and metaphyses of long bones, as well as the pelvis, hands, and feet. These lesions, which typically are asymptomatic, could sometimes be associated with bone and/or joint pain but do not cause a predisposition to fractures. Documentation of these bone lesions by early adult life is important to avoid confusion with osteoblastic metastases on the skeleton.6
Buschke-Ollendorff syndrome is characterized by a variable expression. Loss-of-function mutations of the LEMD3 gene have been described in association with this disorder.7 This gene encodes an inner nuclear protein membrane with a C-terminal domain that binds SMAD2 and SMAD3 and antagonizes the bone morphogenetic proteins and transforming growth factor b. These proteins are involved in connective tissue morphogenesis, inducing elastin production from fibroblasts.7 Seven novel loss-of-function mutations of LEMD3 have been identified.8 The segmental manifestation of BOS could be a consequence of the mosaicism resulting from a somatic mutation.9,10 A case describing the absence of LEMD3 mutation in an affected family suggested the genetic heterogeneity of BOS.11
Conclusion
Buschke-Ollendorff syndrome is a benign condition with no repercussions on the patient’s health or quality of life. It does not require any specific treatment because the lesions generally remain asymptomatic and do not generate any substantial cosmetic burden. Mutation analysis was not performed in our patients because BOS has a good prognosis and the parents refused further investigation in both patients; therefore, a correct diagnosis was essential in our cases to rule out malignant bone disease in patient 1 whose osteopoikilosis prompted the workup and other disorders (eg, tuberous sclerosis, pseudoxanthoma elasticum) in patient 2 whose cutaneous lesions were the primary cause for presentation. A correct diagnosis of BOS is necessary to spare patients from expensive investigations and to provide reassurance about the benign nature of the disease.
Buschke-Ollendorff syndrome (BOS) is a rare disease that is inherited in an autosomal-dominant fashion with high penetrance. It is characterized by osteopoikilosis associated with skin manifestations. The approximate incidence of the disease is 1:20,000, with few cases reported in the literature since 1928.1 Skeletal lesions known as osteopoikilosis are areas of increased bone density that can be seen on radiographic imaging and typically are located in the substantia spongiosa of the epiphyses and metaphyses of long bones and the pelvis. In BOS, cutaneous lesions consist of elastic or collagen nevi. Phenotypic expression of the disease is variable, and skeletal and cutaneous lesions may occur separately. Gene mutations of proteins involved in bone and connective tissue morphogenesis have been described in patients with BOS.2-5
Case Reports
Patient 1
A 17-year-old adolescent girl was referred to our hospital for evaluation of an incidental finding of osteopoikilosis that had been noted in the setting of a traumatic event. Clinical examination revealed cutaneous lesions characterized by asymptomatic, linear, stringlike and atrophic fibrotic plaques localized symmetrically on the trunk, right buttock, and right thigh (Figure 1). The lesions on the thigh were present at birth and had spread progressively to the other areas. There was no known history of inflammatory skin disease to explain the presence of the lesions, and no family history of similar signs or symptoms was reported. Histopathologic examination of a punch biopsy of a plaque from the trunk revealed increased collagen bundles associated with thick interlacing elastic fibers. The epidermis did not show any specific histologic alterations. These histopathologic features were diagnostic of a connective tissue nevus (Figure 2).
|
|
Figure 1. An asymptomatic atrophic fibrotic plaque localized on the right thigh. |
|
|
Figure 2. Low-power view of a punch biopsy showing a normal epidermis. Collagen bundles of the dermis were thickened and somewhat homogenized, as highlighted by the Masson stain (original magnification ×2.5). |
|
|
Figure 3. Radiograph of the legs revealed numerous small, ovoid or round foci of sclerosis on the substantia spongiosa of the metaphyses and the epiphyses of the femur, tibia, and fibula on both sides of the body. |
|
|
Figure 4. Multiple asymptomatic flesh-colored papules with elastic consistency on the back that were characteristic of dermatofibrosis lenticularis disseminata. |
|
|
Figure 5. The epidermis and dermis appeared normal on histopathology; however, the elastic fibers were markedly increased in both size and number up to the deep dermis in the absence of degenerating changes (H&E, original magnification ×2.5; vascular endothelial growth factor, original magnification ×20 [inset in bottom right corner]). |
|
|
| Figure 6. A radiograph of the left hand revealed small sporadic areas of sclerosis in the substantia spongiosa on the heads of some phalanges (white arrows). |
Radiographic imaging of the hands and feet revealed numerous small, ovoid or round foci of sclerosis that were a few millimeters in diameter and were occasionally confluent. This finding was prevalent on the carpal and tarsal bones but less evident on the phalanges and the epiphysis of the metatarsal and metacarpal bones. Full radiographic imaging of both arms and legs subsequently was obtained and showed similar lesions predominantly in the substantia spongiosa of the metaphyses and the epiphyses of the humerus, femur, tibia, and fibula bilaterally (Figure 3). Evaluation of the patient’s parents revealed that her mother had sporadic lenticular areas of increased bone density seen on radiography of the carpal and tarsal bones, particularly at the level of calcaneus, and the proximal and distal humeral epiphyses.
These lesions in our patient were consistent with an incomplete form of osteopoikilosis, which reflects the known variable expression of BOS. The radiologic findings in addition to the cutaneous lesions and the positive family history supported a diagnosis of BOS.
Patient 2
A 5-year-old boy presented with multiple congenital, asymptomatic, flesh-colored papules with elastic consistency on the left thigh, back, and pubic area that were characteristic of dermatofibrosis lenticularis disseminata (Figure 4). The patient’s 7-year-old sister had similar cutaneous lesions. The patient underwent a skin biopsy from the back that revealed thickening of collagen bundles in the dermis with an increase in elastic fibers (Figure 5). On radiographic imaging, small sporadic areas of sclerosis were noted in the substantia spongiosa of the lateral aspect of the humeral capitulum, the radial neck, the capitate, the head of the proximal phalanx of the fourth finger on the left hand, and the heads of the third and fifth middle phalanges on the left hand (Figure 6). Radiography revealed osteopoikilosis on both humeral heads in the patient’s mother. These findings in addition to the dermatologic and histopathologic features suggested a diagnosis of BOS.
Comment
In 1928, Buschke and Ollendorff1 first reported the association between osteopoikilosis and dermatofibrosis lenticularis disseminata, which showed autosomal-dominant inheritance. Skin and bone lesions can occur independently in different family members. Two different clinical cutaneous patterns are described in BOS.2-4 The first and most frequent form is characterized by yellowish nodules often grouped in plaques that are asymmetrically distributed, such as those seen in patient 1. The second form is known as dermatofibrosis lenticularis disseminata, which was seen in patient 2. Histologically, most lesions show normal collagen fibrils and an increased number of elastic fibers.
Osteopoikilosis, also known as spotted bone disease or osteopathia condensans, is a rare asymptomatic bone dysplasia of unknown etiology. It is characterized by an abnormality in the bone maturation process and often is found incidentally on radiologic examination, as seen in patient 1. Radiologic signs of osteopoikilosis consist of small, disseminated, well-circumscribed areas of increased radiodensity located in the epiphyses and metaphyses of long bones, as well as the pelvis, hands, and feet. These lesions, which typically are asymptomatic, could sometimes be associated with bone and/or joint pain but do not cause a predisposition to fractures. Documentation of these bone lesions by early adult life is important to avoid confusion with osteoblastic metastases on the skeleton.6
Buschke-Ollendorff syndrome is characterized by a variable expression. Loss-of-function mutations of the LEMD3 gene have been described in association with this disorder.7 This gene encodes an inner nuclear protein membrane with a C-terminal domain that binds SMAD2 and SMAD3 and antagonizes the bone morphogenetic proteins and transforming growth factor b. These proteins are involved in connective tissue morphogenesis, inducing elastin production from fibroblasts.7 Seven novel loss-of-function mutations of LEMD3 have been identified.8 The segmental manifestation of BOS could be a consequence of the mosaicism resulting from a somatic mutation.9,10 A case describing the absence of LEMD3 mutation in an affected family suggested the genetic heterogeneity of BOS.11
Conclusion
Buschke-Ollendorff syndrome is a benign condition with no repercussions on the patient’s health or quality of life. It does not require any specific treatment because the lesions generally remain asymptomatic and do not generate any substantial cosmetic burden. Mutation analysis was not performed in our patients because BOS has a good prognosis and the parents refused further investigation in both patients; therefore, a correct diagnosis was essential in our cases to rule out malignant bone disease in patient 1 whose osteopoikilosis prompted the workup and other disorders (eg, tuberous sclerosis, pseudoxanthoma elasticum) in patient 2 whose cutaneous lesions were the primary cause for presentation. A correct diagnosis of BOS is necessary to spare patients from expensive investigations and to provide reassurance about the benign nature of the disease.
1. Buschke A, Ollendorff H. Ein fall von dermatofibrosis lenticularis disseminata und osteopathia condensas disseminata. Derm Worchenschr. 1928;86:257-262.
2. Ramme K, Kolde G, Stadler R. Dermatofibrosis lenticularis disseminata with osteopoikilosis. Buschke-Ollendorff syndrome [in German]. Hautarzt. 1993;44:312-314.
3. Schena D, Germi L, Zamperetti MR, et al. Buschke-Ollendorff syndrome. Int J Dermatol. 2008;47:1159-1161.
4. Kawamura A, Ochiai T, Tan-Kinoshita M, et al. Buschke-Ollendorff syndrome: three generations in a Japanese family. Pediatr Dermatol. 2005;22:133-137.
5. Woodrow SL, Pope FM, Handfield-Jones SE. The Buschke-Ollendorff syndrome presenting as familial elastic tissue naevi. Br J Dermatol. 2001;144:890-893.
6. Whyte MP, Murphy WA, Siegel BA. 99mTc-pyrophosphate bone imaging in osteopoikilosis, osteopathia striata, and melorheostosis. Radiology. 1978;127:439-443.
7. Giro MG, Duvic M, Smith LT, et al. Buschke-Ollendorff syndrome associated with elevated elastin production by affected skin fibroblasts in culture. J Invest Dermatol. 1992;99:129-137.
8. Hellemans J, Preobrazhenska O, Willaert A, et al. Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis [published online ahead of print October 17, 2004]. Nat Genet. 2004;36:1213-1218.
9. Ehrig T, Cockerell CJ. Buschke-Ollendorff syndrome: report of a case and interpretation of the clinical phenotype as a type 2 segmental manifestation of an autosomal dominant skin disease. J Am Acad Dermatol. 2003;49:1163-1166.
10. Hellemans J, Debeer P, Wright M, et al. Germline LEMD3 mutations are rare in sporadic patients with isolated melorheostosis. Hum Mutat. 2006;27:290.
11. Yadegari M, Whyte MP, Mumm S, et al. Buschke-Ollendorff syndrome: absence of LEMD3 mutation in an affected family. Arch Dermatol. 2010;146:63-68.
1. Buschke A, Ollendorff H. Ein fall von dermatofibrosis lenticularis disseminata und osteopathia condensas disseminata. Derm Worchenschr. 1928;86:257-262.
2. Ramme K, Kolde G, Stadler R. Dermatofibrosis lenticularis disseminata with osteopoikilosis. Buschke-Ollendorff syndrome [in German]. Hautarzt. 1993;44:312-314.
3. Schena D, Germi L, Zamperetti MR, et al. Buschke-Ollendorff syndrome. Int J Dermatol. 2008;47:1159-1161.
4. Kawamura A, Ochiai T, Tan-Kinoshita M, et al. Buschke-Ollendorff syndrome: three generations in a Japanese family. Pediatr Dermatol. 2005;22:133-137.
5. Woodrow SL, Pope FM, Handfield-Jones SE. The Buschke-Ollendorff syndrome presenting as familial elastic tissue naevi. Br J Dermatol. 2001;144:890-893.
6. Whyte MP, Murphy WA, Siegel BA. 99mTc-pyrophosphate bone imaging in osteopoikilosis, osteopathia striata, and melorheostosis. Radiology. 1978;127:439-443.
7. Giro MG, Duvic M, Smith LT, et al. Buschke-Ollendorff syndrome associated with elevated elastin production by affected skin fibroblasts in culture. J Invest Dermatol. 1992;99:129-137.
8. Hellemans J, Preobrazhenska O, Willaert A, et al. Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis [published online ahead of print October 17, 2004]. Nat Genet. 2004;36:1213-1218.
9. Ehrig T, Cockerell CJ. Buschke-Ollendorff syndrome: report of a case and interpretation of the clinical phenotype as a type 2 segmental manifestation of an autosomal dominant skin disease. J Am Acad Dermatol. 2003;49:1163-1166.
10. Hellemans J, Debeer P, Wright M, et al. Germline LEMD3 mutations are rare in sporadic patients with isolated melorheostosis. Hum Mutat. 2006;27:290.
11. Yadegari M, Whyte MP, Mumm S, et al. Buschke-Ollendorff syndrome: absence of LEMD3 mutation in an affected family. Arch Dermatol. 2010;146:63-68.
Practice Points
- Buschke-Ollendorff syndrome (BOS) is an autosomal-dominant disease characterized by the rare association of skin lesions consisting of collagen or elastic nevi and bone lesions known as osteopoikilosis that are reported radiologically.
- Buschke-Ollendorff syndrome is characterized by elevated genetic heterogeneity and is transmitted with a variable expression.
- Although BOS is a benign condition and does not require any treatment, a correct diagnosis is important to spare patients from unnecessary investigations.
Eleven Years of Itching: A Case Report of Crusted Scabies
Case Report
A 48-year-old man presented to our dermatology clinic with pruritus of 11 years’ duration that worsened at night. He had been followed at a different clinic for several years and was unsuccessfully treated with topical permethrin and oral antihistamines on multiple occasions for scabies. He also had been intermittently treated for contact dermatitis with topical and systemic steroids, which also brought no relief. Just prior to his presentation, the patient’s wife and 8-year-old son had sought medical attention at our institution for chronic pruritus and elevated IgE levels. They had been unsuccessfully treated with topical permethrin, topical steroids, and oral antihistamines for atopic dermatitis at a different clinic. When they presented to our clinic, they were both diagnosed with and treated for scabies. At this visit the patient revealed similar concerns and subsequently underwent examination.
Physical examination revealed large erythematous, hyperkeratotic, scaly plaques on the gluteal fold, um-bilicus, glans penis, scrotum, bilateral elbows, knees, nipples, and ear helices (Figure 1). Numerous small erythematous papules and wavy threadlike gray burrows measuring 1 to 10 mm in diameter were distributed primarily around the wrists, ankles, proximal extremities, abdominal and pubic area (Figure 2), and interdigital spaces. Wide oval patches of nonscarring alopecia developed on the scalp, and atrophic glossitis and angular cheilitis were noted on the oral mucosa, along with a white pseudomembranous exudate on the palatum. The patient’s nails also were thickened and discolored (Figure 3). His medical history was remarkable for hypoparathyroidism (42 years), alopecia areata (15 years), oral candidiasis and angular cheilitis (10 years), and primary hypothyroidism (1 year) that was currently being treated with levothyroxine.
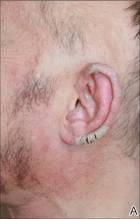
| 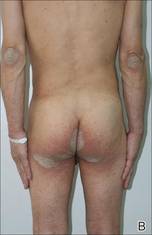
|
| Figure 1. Large erythematous, hyperkeratotic, scaly plaques on the ear helices (A), gluteal fold, and bilateral elbows (B) in a 48-year-old man with crusted scabies. | |
| |
| Figure 2. Numerous small erythematous papules and wavy, threadlike, grayish, 1- to 10-mm burrows distributed in the abdominal and pubic area. | |
| |
| Figure 3. Thickened and discolored nails in a 48-year-old man with crusted scabies. | |
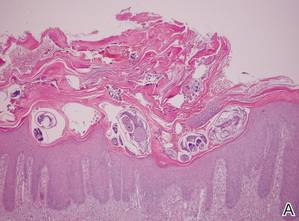
| |
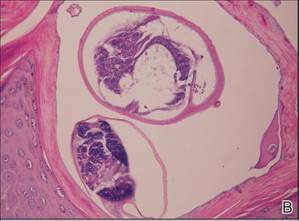
| |
| Figure 4. The psoriasiform epidermis showed massive hyperkeratosis and burrows in the subcorneal layer containing a large number of female mites and feces (A)(H&E, original magnification ×40). A substantial lymphohistiocytic infiltrate with numerous eosinophils was seen diffusely throughout the dermis, though most prominently within the upper half. High-power examination demonstrated mites in the burrow (B)(H&E, original magnification ×400). | |
Laboratory studies revealed normal hemogram with an eosinophil level of 0% (reference range, 0.9%–6%). Biochemistry and hormone profiles were consistent with hypoparathyroidism, with the following levels: serum calcium, 7.6 mg/dL (reference range, 8.4–10.2 mg/dL); phosphorus, 5.1 mg/dL (reference range, 2.4–4.4 mg/dL); and parathyroid hormone, 5.23 pg/mL (reference range, 15–65 pg/mL). The patient tested negative for human immunodeficiency virus.
Dermoscopic examination of the gray threadlike burrows revealed distinctive brown-colored triangular structures that were removed with a fine scalpel. Microscopic examination of the tissue revealed moving mites, eggs, and red-brown scybala.
A biopsy was taken from the thick, scaly, crusted white plaques in the gluteal area. The epidermis showed massive hyperkeratosis and burrows in the subcorneal layer containing a large number of female mites and feces, while the remainder of the epidermis was substantially psoriasiform. A substantial lymphohistiocytic infiltrate with numerous eosinophils was seen diffusely throughout the dermis, though it was most prominent within the upper half (Figure 4). The patient subsequently was diagnosed with crusted scabies.
Crusted scabies typically develops in patients with defective T-cell immunity or a reduced ability to mechanically debride the mites. Because our patient had a history of persistent oral candidiasis, further investigation for immunosuppression was initiated. An immunosuppression panel revealed low IgA levels (46 mg/dL [reference range, 82–453 mg/dL]), low absolute CD8 level (145 cells/µL [reference range, 300–1800 cells/µL), and CD4:CD8 ratio of 4.1. Serum IgG, IgM, IgE, complement levels, and immunoelectrophoresis were within reference range. Abdominal ultrasonography was unremarkable. Taking into account the patient’s history of autoimmune hypoparathyroidism, hypothyroidism, oral candidiasis, and alopecia areata, he was diagnosed with autoimmune polyglandular syndrome.
The patient and his family were successfully treated with modified Wilkinson ointment (goudron végétal 12.5%; sulfur 12.5% in petrolatum) for 3 consecutive days. Within 1 week, the scaly plaques had disappeared and the erythematous papules had faded. The pruritus had resolved and no new papules emerged. Treatment was reapplied once more the following week and complete cure was achieved. We have been following this family for 12 months and no recurrences have occurred.
Comment
Crusted scabies is a rare and highly contagious form of scabies that is characterized by uncontrolled proliferation of mites in the skin, extensive hyperkeratotic scaling, crusted lesions, and variable pruritus.1 The stratum corneum thickens and forms warty crusts as a reaction to the high mite burden.2 The uncontrolled proliferation of mites in the skin typically develops in patients with a defective T-cell response or decreased cutaneous sensation and reduced ability to mechanically debride the mites.3 Patients should be investigated for a predisposition to crusted scabies due to an underlying condition. Crusted scabies also has been shown to develop in Australian natives with normal immunity, though the etiology of the increased susceptibility in this patient population remains unclear. Some studies have shown an association with HLA-A11.4,5 It also has been hypothesized that these patients may have a specific immunodeficiency predisposing them to hyperinfestation.6
Unlike classic scabies, crusted scabies usually does not present acutely, and it usually is insidious at onset. The eruption typically has 2 components: localized horny plaques and a more distinct erythema.3 Crusted scabies can mimic a variety of conditions such as psoriasis, eczema, seborrheic dermatitis, Darier disease, contact dermatitis, and pityriasis rubra pilaris.7 When pruritus is resistant to permethrin therapy, as in our patient, crusted scabies often is misdiagnosed as eczema or contact dermatitis. Topical and systemic corticosteroids often are prescribed, causing progression to scabies incognito.
The diagnosis of crusted scabies is confirmed by examination of scrapings and biopsies, as in classic scabies; however, treatment can be challenging due to compromised immunity, a large mite burden, and limited penetration of topical medications into the hyperkeratotic lesions. Thus treatment should include both keratolytic and scabicidal agents to remove the crusts, reduce the mite load, and enhance the scabicidal therapy.1 Our patient and his affected family members had previously been treated with topical permethrin several times without any benefit. Oral ivermectin has been proven to be effective but is not available in Turkey. Therefore, we treated the patient and his household contacts (other extended family members treated separately) with modified Wilkinson ointment (goudron végétal 12.5%; sulfur 12.5% in petrolatum) for 3 consecutive days, which is known to have both a keratolytic and scabicidal effect.8-11
Conclusion
This case highlights the importance of obtaining a complete family history, skin examination, and thorough investigation for underlying immunodeficiencies that can lead to a predisposition for crusted scabies. It is important to note that the treatment of crusted scabies can be challenging, and effective management of the condition requires a keratolytic agent in conjunction with a scabicidal agent.
1. Douri T, Shawaf AZ. Treatment of crusted scabies with albenzdazole: a case report. Dermatol Online J. 2009;15:17.
2. Burns DA. Diseases caused by arthropods and other noxious animals. In: Champion RH, Burton JL, Burns DA, et al, eds. Textbook of Dermatology. 6th ed. Oxford, England: Wiley-Blackwell; 1998:1423-1482.
3. Karthiyekan K. Crusted scabies. Indian J Dermatol Venereol Leprol. 2009;75:340-347.
4. Falk ES, Thorsby E. HLA antigens in patients with scabies. Br J Dermatol. 1981;104:317-320.
5. Morsy TA, Romia SA, al-Ganayni GA, et al. Histocompatibility (HLA) antigens in Egyptians with two parasitic skin diseases (scabies and leishmaniasis). J Egypt Soc Parasitol. 1990;20:565-572.
6. Roberts LJ, Huffam SE, Walton SF, et al. Crusted scabies: clinical and immunological findings in seventy-eight patients and a review of the literature. J Infect. 2005;50:375-381.
7. Jucowics P, Ramon ME, Don PC, et al. Norwegian scabies in an infant with acquired immunodeficiency syndrome. Arch Dermatol. 1989;125:1670-1671.
8. Goldsmith WN. Wilkinson’s ointment. Br Med J. 1945;1:347-348.
9. Lin AN, Reimer RJ, Carter DM. Sulfur revisited. J Am Acad Dermatol. 1988;18:553-558.
10. Gupta AK, Nikol K. The use of sulfur in dermatology. J Drugs Dermatol. 2004;3:427-431.
11. Lin AN, Moses K. Tar revisited. Int J Dermatol. 1985;24:216-219.
Case Report
A 48-year-old man presented to our dermatology clinic with pruritus of 11 years’ duration that worsened at night. He had been followed at a different clinic for several years and was unsuccessfully treated with topical permethrin and oral antihistamines on multiple occasions for scabies. He also had been intermittently treated for contact dermatitis with topical and systemic steroids, which also brought no relief. Just prior to his presentation, the patient’s wife and 8-year-old son had sought medical attention at our institution for chronic pruritus and elevated IgE levels. They had been unsuccessfully treated with topical permethrin, topical steroids, and oral antihistamines for atopic dermatitis at a different clinic. When they presented to our clinic, they were both diagnosed with and treated for scabies. At this visit the patient revealed similar concerns and subsequently underwent examination.
Physical examination revealed large erythematous, hyperkeratotic, scaly plaques on the gluteal fold, um-bilicus, glans penis, scrotum, bilateral elbows, knees, nipples, and ear helices (Figure 1). Numerous small erythematous papules and wavy threadlike gray burrows measuring 1 to 10 mm in diameter were distributed primarily around the wrists, ankles, proximal extremities, abdominal and pubic area (Figure 2), and interdigital spaces. Wide oval patches of nonscarring alopecia developed on the scalp, and atrophic glossitis and angular cheilitis were noted on the oral mucosa, along with a white pseudomembranous exudate on the palatum. The patient’s nails also were thickened and discolored (Figure 3). His medical history was remarkable for hypoparathyroidism (42 years), alopecia areata (15 years), oral candidiasis and angular cheilitis (10 years), and primary hypothyroidism (1 year) that was currently being treated with levothyroxine.
|
|
|
| Figure 1. Large erythematous, hyperkeratotic, scaly plaques on the ear helices (A), gluteal fold, and bilateral elbows (B) in a 48-year-old man with crusted scabies. | |
|
| |
| Figure 2. Numerous small erythematous papules and wavy, threadlike, grayish, 1- to 10-mm burrows distributed in the abdominal and pubic area. | |
|
| |
| Figure 3. Thickened and discolored nails in a 48-year-old man with crusted scabies. | |
|
| |
|
| |
| Figure 4. The psoriasiform epidermis showed massive hyperkeratosis and burrows in the subcorneal layer containing a large number of female mites and feces (A)(H&E, original magnification ×40). A substantial lymphohistiocytic infiltrate with numerous eosinophils was seen diffusely throughout the dermis, though most prominently within the upper half. High-power examination demonstrated mites in the burrow (B)(H&E, original magnification ×400). | |
Laboratory studies revealed normal hemogram with an eosinophil level of 0% (reference range, 0.9%–6%). Biochemistry and hormone profiles were consistent with hypoparathyroidism, with the following levels: serum calcium, 7.6 mg/dL (reference range, 8.4–10.2 mg/dL); phosphorus, 5.1 mg/dL (reference range, 2.4–4.4 mg/dL); and parathyroid hormone, 5.23 pg/mL (reference range, 15–65 pg/mL). The patient tested negative for human immunodeficiency virus.
Dermoscopic examination of the gray threadlike burrows revealed distinctive brown-colored triangular structures that were removed with a fine scalpel. Microscopic examination of the tissue revealed moving mites, eggs, and red-brown scybala.
A biopsy was taken from the thick, scaly, crusted white plaques in the gluteal area. The epidermis showed massive hyperkeratosis and burrows in the subcorneal layer containing a large number of female mites and feces, while the remainder of the epidermis was substantially psoriasiform. A substantial lymphohistiocytic infiltrate with numerous eosinophils was seen diffusely throughout the dermis, though it was most prominent within the upper half (Figure 4). The patient subsequently was diagnosed with crusted scabies.
Crusted scabies typically develops in patients with defective T-cell immunity or a reduced ability to mechanically debride the mites. Because our patient had a history of persistent oral candidiasis, further investigation for immunosuppression was initiated. An immunosuppression panel revealed low IgA levels (46 mg/dL [reference range, 82–453 mg/dL]), low absolute CD8 level (145 cells/µL [reference range, 300–1800 cells/µL), and CD4:CD8 ratio of 4.1. Serum IgG, IgM, IgE, complement levels, and immunoelectrophoresis were within reference range. Abdominal ultrasonography was unremarkable. Taking into account the patient’s history of autoimmune hypoparathyroidism, hypothyroidism, oral candidiasis, and alopecia areata, he was diagnosed with autoimmune polyglandular syndrome.
The patient and his family were successfully treated with modified Wilkinson ointment (goudron végétal 12.5%; sulfur 12.5% in petrolatum) for 3 consecutive days. Within 1 week, the scaly plaques had disappeared and the erythematous papules had faded. The pruritus had resolved and no new papules emerged. Treatment was reapplied once more the following week and complete cure was achieved. We have been following this family for 12 months and no recurrences have occurred.
Comment
Crusted scabies is a rare and highly contagious form of scabies that is characterized by uncontrolled proliferation of mites in the skin, extensive hyperkeratotic scaling, crusted lesions, and variable pruritus.1 The stratum corneum thickens and forms warty crusts as a reaction to the high mite burden.2 The uncontrolled proliferation of mites in the skin typically develops in patients with a defective T-cell response or decreased cutaneous sensation and reduced ability to mechanically debride the mites.3 Patients should be investigated for a predisposition to crusted scabies due to an underlying condition. Crusted scabies also has been shown to develop in Australian natives with normal immunity, though the etiology of the increased susceptibility in this patient population remains unclear. Some studies have shown an association with HLA-A11.4,5 It also has been hypothesized that these patients may have a specific immunodeficiency predisposing them to hyperinfestation.6
Unlike classic scabies, crusted scabies usually does not present acutely, and it usually is insidious at onset. The eruption typically has 2 components: localized horny plaques and a more distinct erythema.3 Crusted scabies can mimic a variety of conditions such as psoriasis, eczema, seborrheic dermatitis, Darier disease, contact dermatitis, and pityriasis rubra pilaris.7 When pruritus is resistant to permethrin therapy, as in our patient, crusted scabies often is misdiagnosed as eczema or contact dermatitis. Topical and systemic corticosteroids often are prescribed, causing progression to scabies incognito.
The diagnosis of crusted scabies is confirmed by examination of scrapings and biopsies, as in classic scabies; however, treatment can be challenging due to compromised immunity, a large mite burden, and limited penetration of topical medications into the hyperkeratotic lesions. Thus treatment should include both keratolytic and scabicidal agents to remove the crusts, reduce the mite load, and enhance the scabicidal therapy.1 Our patient and his affected family members had previously been treated with topical permethrin several times without any benefit. Oral ivermectin has been proven to be effective but is not available in Turkey. Therefore, we treated the patient and his household contacts (other extended family members treated separately) with modified Wilkinson ointment (goudron végétal 12.5%; sulfur 12.5% in petrolatum) for 3 consecutive days, which is known to have both a keratolytic and scabicidal effect.8-11
Conclusion
This case highlights the importance of obtaining a complete family history, skin examination, and thorough investigation for underlying immunodeficiencies that can lead to a predisposition for crusted scabies. It is important to note that the treatment of crusted scabies can be challenging, and effective management of the condition requires a keratolytic agent in conjunction with a scabicidal agent.
Case Report
A 48-year-old man presented to our dermatology clinic with pruritus of 11 years’ duration that worsened at night. He had been followed at a different clinic for several years and was unsuccessfully treated with topical permethrin and oral antihistamines on multiple occasions for scabies. He also had been intermittently treated for contact dermatitis with topical and systemic steroids, which also brought no relief. Just prior to his presentation, the patient’s wife and 8-year-old son had sought medical attention at our institution for chronic pruritus and elevated IgE levels. They had been unsuccessfully treated with topical permethrin, topical steroids, and oral antihistamines for atopic dermatitis at a different clinic. When they presented to our clinic, they were both diagnosed with and treated for scabies. At this visit the patient revealed similar concerns and subsequently underwent examination.
Physical examination revealed large erythematous, hyperkeratotic, scaly plaques on the gluteal fold, um-bilicus, glans penis, scrotum, bilateral elbows, knees, nipples, and ear helices (Figure 1). Numerous small erythematous papules and wavy threadlike gray burrows measuring 1 to 10 mm in diameter were distributed primarily around the wrists, ankles, proximal extremities, abdominal and pubic area (Figure 2), and interdigital spaces. Wide oval patches of nonscarring alopecia developed on the scalp, and atrophic glossitis and angular cheilitis were noted on the oral mucosa, along with a white pseudomembranous exudate on the palatum. The patient’s nails also were thickened and discolored (Figure 3). His medical history was remarkable for hypoparathyroidism (42 years), alopecia areata (15 years), oral candidiasis and angular cheilitis (10 years), and primary hypothyroidism (1 year) that was currently being treated with levothyroxine.
|
|
|
| Figure 1. Large erythematous, hyperkeratotic, scaly plaques on the ear helices (A), gluteal fold, and bilateral elbows (B) in a 48-year-old man with crusted scabies. | |
|
| |
| Figure 2. Numerous small erythematous papules and wavy, threadlike, grayish, 1- to 10-mm burrows distributed in the abdominal and pubic area. | |
|
| |
| Figure 3. Thickened and discolored nails in a 48-year-old man with crusted scabies. | |
|
| |
|
| |
| Figure 4. The psoriasiform epidermis showed massive hyperkeratosis and burrows in the subcorneal layer containing a large number of female mites and feces (A)(H&E, original magnification ×40). A substantial lymphohistiocytic infiltrate with numerous eosinophils was seen diffusely throughout the dermis, though most prominently within the upper half. High-power examination demonstrated mites in the burrow (B)(H&E, original magnification ×400). | |
Laboratory studies revealed normal hemogram with an eosinophil level of 0% (reference range, 0.9%–6%). Biochemistry and hormone profiles were consistent with hypoparathyroidism, with the following levels: serum calcium, 7.6 mg/dL (reference range, 8.4–10.2 mg/dL); phosphorus, 5.1 mg/dL (reference range, 2.4–4.4 mg/dL); and parathyroid hormone, 5.23 pg/mL (reference range, 15–65 pg/mL). The patient tested negative for human immunodeficiency virus.
Dermoscopic examination of the gray threadlike burrows revealed distinctive brown-colored triangular structures that were removed with a fine scalpel. Microscopic examination of the tissue revealed moving mites, eggs, and red-brown scybala.
A biopsy was taken from the thick, scaly, crusted white plaques in the gluteal area. The epidermis showed massive hyperkeratosis and burrows in the subcorneal layer containing a large number of female mites and feces, while the remainder of the epidermis was substantially psoriasiform. A substantial lymphohistiocytic infiltrate with numerous eosinophils was seen diffusely throughout the dermis, though it was most prominent within the upper half (Figure 4). The patient subsequently was diagnosed with crusted scabies.
Crusted scabies typically develops in patients with defective T-cell immunity or a reduced ability to mechanically debride the mites. Because our patient had a history of persistent oral candidiasis, further investigation for immunosuppression was initiated. An immunosuppression panel revealed low IgA levels (46 mg/dL [reference range, 82–453 mg/dL]), low absolute CD8 level (145 cells/µL [reference range, 300–1800 cells/µL), and CD4:CD8 ratio of 4.1. Serum IgG, IgM, IgE, complement levels, and immunoelectrophoresis were within reference range. Abdominal ultrasonography was unremarkable. Taking into account the patient’s history of autoimmune hypoparathyroidism, hypothyroidism, oral candidiasis, and alopecia areata, he was diagnosed with autoimmune polyglandular syndrome.
The patient and his family were successfully treated with modified Wilkinson ointment (goudron végétal 12.5%; sulfur 12.5% in petrolatum) for 3 consecutive days. Within 1 week, the scaly plaques had disappeared and the erythematous papules had faded. The pruritus had resolved and no new papules emerged. Treatment was reapplied once more the following week and complete cure was achieved. We have been following this family for 12 months and no recurrences have occurred.
Comment
Crusted scabies is a rare and highly contagious form of scabies that is characterized by uncontrolled proliferation of mites in the skin, extensive hyperkeratotic scaling, crusted lesions, and variable pruritus.1 The stratum corneum thickens and forms warty crusts as a reaction to the high mite burden.2 The uncontrolled proliferation of mites in the skin typically develops in patients with a defective T-cell response or decreased cutaneous sensation and reduced ability to mechanically debride the mites.3 Patients should be investigated for a predisposition to crusted scabies due to an underlying condition. Crusted scabies also has been shown to develop in Australian natives with normal immunity, though the etiology of the increased susceptibility in this patient population remains unclear. Some studies have shown an association with HLA-A11.4,5 It also has been hypothesized that these patients may have a specific immunodeficiency predisposing them to hyperinfestation.6
Unlike classic scabies, crusted scabies usually does not present acutely, and it usually is insidious at onset. The eruption typically has 2 components: localized horny plaques and a more distinct erythema.3 Crusted scabies can mimic a variety of conditions such as psoriasis, eczema, seborrheic dermatitis, Darier disease, contact dermatitis, and pityriasis rubra pilaris.7 When pruritus is resistant to permethrin therapy, as in our patient, crusted scabies often is misdiagnosed as eczema or contact dermatitis. Topical and systemic corticosteroids often are prescribed, causing progression to scabies incognito.
The diagnosis of crusted scabies is confirmed by examination of scrapings and biopsies, as in classic scabies; however, treatment can be challenging due to compromised immunity, a large mite burden, and limited penetration of topical medications into the hyperkeratotic lesions. Thus treatment should include both keratolytic and scabicidal agents to remove the crusts, reduce the mite load, and enhance the scabicidal therapy.1 Our patient and his affected family members had previously been treated with topical permethrin several times without any benefit. Oral ivermectin has been proven to be effective but is not available in Turkey. Therefore, we treated the patient and his household contacts (other extended family members treated separately) with modified Wilkinson ointment (goudron végétal 12.5%; sulfur 12.5% in petrolatum) for 3 consecutive days, which is known to have both a keratolytic and scabicidal effect.8-11
Conclusion
This case highlights the importance of obtaining a complete family history, skin examination, and thorough investigation for underlying immunodeficiencies that can lead to a predisposition for crusted scabies. It is important to note that the treatment of crusted scabies can be challenging, and effective management of the condition requires a keratolytic agent in conjunction with a scabicidal agent.
1. Douri T, Shawaf AZ. Treatment of crusted scabies with albenzdazole: a case report. Dermatol Online J. 2009;15:17.
2. Burns DA. Diseases caused by arthropods and other noxious animals. In: Champion RH, Burton JL, Burns DA, et al, eds. Textbook of Dermatology. 6th ed. Oxford, England: Wiley-Blackwell; 1998:1423-1482.
3. Karthiyekan K. Crusted scabies. Indian J Dermatol Venereol Leprol. 2009;75:340-347.
4. Falk ES, Thorsby E. HLA antigens in patients with scabies. Br J Dermatol. 1981;104:317-320.
5. Morsy TA, Romia SA, al-Ganayni GA, et al. Histocompatibility (HLA) antigens in Egyptians with two parasitic skin diseases (scabies and leishmaniasis). J Egypt Soc Parasitol. 1990;20:565-572.
6. Roberts LJ, Huffam SE, Walton SF, et al. Crusted scabies: clinical and immunological findings in seventy-eight patients and a review of the literature. J Infect. 2005;50:375-381.
7. Jucowics P, Ramon ME, Don PC, et al. Norwegian scabies in an infant with acquired immunodeficiency syndrome. Arch Dermatol. 1989;125:1670-1671.
8. Goldsmith WN. Wilkinson’s ointment. Br Med J. 1945;1:347-348.
9. Lin AN, Reimer RJ, Carter DM. Sulfur revisited. J Am Acad Dermatol. 1988;18:553-558.
10. Gupta AK, Nikol K. The use of sulfur in dermatology. J Drugs Dermatol. 2004;3:427-431.
11. Lin AN, Moses K. Tar revisited. Int J Dermatol. 1985;24:216-219.
1. Douri T, Shawaf AZ. Treatment of crusted scabies with albenzdazole: a case report. Dermatol Online J. 2009;15:17.
2. Burns DA. Diseases caused by arthropods and other noxious animals. In: Champion RH, Burton JL, Burns DA, et al, eds. Textbook of Dermatology. 6th ed. Oxford, England: Wiley-Blackwell; 1998:1423-1482.
3. Karthiyekan K. Crusted scabies. Indian J Dermatol Venereol Leprol. 2009;75:340-347.
4. Falk ES, Thorsby E. HLA antigens in patients with scabies. Br J Dermatol. 1981;104:317-320.
5. Morsy TA, Romia SA, al-Ganayni GA, et al. Histocompatibility (HLA) antigens in Egyptians with two parasitic skin diseases (scabies and leishmaniasis). J Egypt Soc Parasitol. 1990;20:565-572.
6. Roberts LJ, Huffam SE, Walton SF, et al. Crusted scabies: clinical and immunological findings in seventy-eight patients and a review of the literature. J Infect. 2005;50:375-381.
7. Jucowics P, Ramon ME, Don PC, et al. Norwegian scabies in an infant with acquired immunodeficiency syndrome. Arch Dermatol. 1989;125:1670-1671.
8. Goldsmith WN. Wilkinson’s ointment. Br Med J. 1945;1:347-348.
9. Lin AN, Reimer RJ, Carter DM. Sulfur revisited. J Am Acad Dermatol. 1988;18:553-558.
10. Gupta AK, Nikol K. The use of sulfur in dermatology. J Drugs Dermatol. 2004;3:427-431.
11. Lin AN, Moses K. Tar revisited. Int J Dermatol. 1985;24:216-219.
Practice Points
- Crusted scabies can mimic a variety of conditions such as psoriasis, eczema, seborrheic dermatitis, and contact dermatitis. Therefore, suspicion is the prerequisite for disease control.
- Scabies usually is found in individuals with a compromised immune system as well as those with decreased sensory functions. Thus patients should be investigated for an underlying immunodeficiency.
- Treatment can be challenging, and effective management of the condition requires a keratolytic agent in conjunction with a scabicidal agent.