User login
Nail Biopsy: 6 Techniques to Biopsy the Nail Matrix
Nail matrix biopsies are performed to confirm a diagnosis or surgically remove a skin lesion that is affecting the growth of the nail plate. The procedure may be used to identify:
- Inflammatory conditions such as nail psoriasis and lichen planus
- Benign tumors
- Solitary melanonychia
- Squamous cell carcinoma (SCC)
- Other nail disorders
Nail biopsy can lead to complications such as bleeding, infection, or scarring. Postoperative scarring can cause permanent nail splitting, dystrophy, or both.
In a Cosmetic Dermatology article, “Matrix Biopsy of Longitudinal Melanonychia and Longitudinal Erythronychia: A Step-by-Step Approach,” Drs. Siobhan C. Collins and Nathaniel J. Jellinek review 6 techniques used to biopsy the nail matrix.
- Punch excision
- Matrix shave
- Lateral longitudinal excision
- Midline/paramedian longitudinal excision
- Transverse excision
- Longitudinal excision of erythronychia
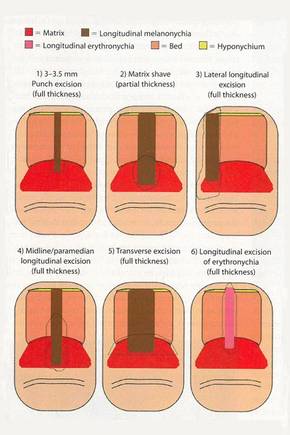
In the setting of longitudinal melanonychia (to diagnose nail melanoma or SCC) and longitudinal erythronychia (to diagnose SCC and rarely amelanotic melanoma or basal cell carcinoma), the techniques they describe accomplish 3 fundamental goals of nail surgery:
- Obtain adequate tissue via an excisional biopsy to make an accurate diagnosis and avoid sampling error
- Avoid unnecessary trauma to surrounding nail tissues by the judicious use of partial plate avulsions whenever feasible
- Avoid unnecessary postoperative nail scarring whenever possible
Dermatologists must be confident when performing nail biopsies and the techniques discussed by the authors will help approach nail surgery with more certainty.
At the 73rd Annual Meeting of the American Academy of Dermatology, Dr. Jellinek provides a hands-on approach to nail surgery. On Saturday, March 21, he will provide tips for nail surgeries at the “Medical and Surgical Management of Nail Disorders” lecture.
For more information, read the Collins and Jellinek article from Cosmetic Dermatology.
Nail matrix biopsies are performed to confirm a diagnosis or surgically remove a skin lesion that is affecting the growth of the nail plate. The procedure may be used to identify:
- Inflammatory conditions such as nail psoriasis and lichen planus
- Benign tumors
- Solitary melanonychia
- Squamous cell carcinoma (SCC)
- Other nail disorders
Nail biopsy can lead to complications such as bleeding, infection, or scarring. Postoperative scarring can cause permanent nail splitting, dystrophy, or both.
In a Cosmetic Dermatology article, “Matrix Biopsy of Longitudinal Melanonychia and Longitudinal Erythronychia: A Step-by-Step Approach,” Drs. Siobhan C. Collins and Nathaniel J. Jellinek review 6 techniques used to biopsy the nail matrix.
- Punch excision
- Matrix shave
- Lateral longitudinal excision
- Midline/paramedian longitudinal excision
- Transverse excision
- Longitudinal excision of erythronychia
In the setting of longitudinal melanonychia (to diagnose nail melanoma or SCC) and longitudinal erythronychia (to diagnose SCC and rarely amelanotic melanoma or basal cell carcinoma), the techniques they describe accomplish 3 fundamental goals of nail surgery:
- Obtain adequate tissue via an excisional biopsy to make an accurate diagnosis and avoid sampling error
- Avoid unnecessary trauma to surrounding nail tissues by the judicious use of partial plate avulsions whenever feasible
- Avoid unnecessary postoperative nail scarring whenever possible
Dermatologists must be confident when performing nail biopsies and the techniques discussed by the authors will help approach nail surgery with more certainty.
At the 73rd Annual Meeting of the American Academy of Dermatology, Dr. Jellinek provides a hands-on approach to nail surgery. On Saturday, March 21, he will provide tips for nail surgeries at the “Medical and Surgical Management of Nail Disorders” lecture.
For more information, read the Collins and Jellinek article from Cosmetic Dermatology.
Nail matrix biopsies are performed to confirm a diagnosis or surgically remove a skin lesion that is affecting the growth of the nail plate. The procedure may be used to identify:
- Inflammatory conditions such as nail psoriasis and lichen planus
- Benign tumors
- Solitary melanonychia
- Squamous cell carcinoma (SCC)
- Other nail disorders
Nail biopsy can lead to complications such as bleeding, infection, or scarring. Postoperative scarring can cause permanent nail splitting, dystrophy, or both.
In a Cosmetic Dermatology article, “Matrix Biopsy of Longitudinal Melanonychia and Longitudinal Erythronychia: A Step-by-Step Approach,” Drs. Siobhan C. Collins and Nathaniel J. Jellinek review 6 techniques used to biopsy the nail matrix.
- Punch excision
- Matrix shave
- Lateral longitudinal excision
- Midline/paramedian longitudinal excision
- Transverse excision
- Longitudinal excision of erythronychia
In the setting of longitudinal melanonychia (to diagnose nail melanoma or SCC) and longitudinal erythronychia (to diagnose SCC and rarely amelanotic melanoma or basal cell carcinoma), the techniques they describe accomplish 3 fundamental goals of nail surgery:
- Obtain adequate tissue via an excisional biopsy to make an accurate diagnosis and avoid sampling error
- Avoid unnecessary trauma to surrounding nail tissues by the judicious use of partial plate avulsions whenever feasible
- Avoid unnecessary postoperative nail scarring whenever possible
Dermatologists must be confident when performing nail biopsies and the techniques discussed by the authors will help approach nail surgery with more certainty.
At the 73rd Annual Meeting of the American Academy of Dermatology, Dr. Jellinek provides a hands-on approach to nail surgery. On Saturday, March 21, he will provide tips for nail surgeries at the “Medical and Surgical Management of Nail Disorders” lecture.
For more information, read the Collins and Jellinek article from Cosmetic Dermatology.
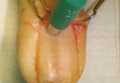
What Is Your Diagnosis? Extramammary Paget Disease

A 70-year-old man presented with a nonpruritic erythematous scaly plaque in the left suprapubic region of 6 months’ duration that had failed to respond to terbinafine cream 1% after 1 month of treatment of suspected tinea cruris. His medical history was remarkable for hypertension, hyperlipidemia, chronic obstructive pulmonary disease, benign prostatic hyperplasia, an abdominal aortic aneurysm, alcohol dependence, tobacco use disorder, and unintentional weight loss of 15 lb over the last year.
The Diagnosis: Extramammary Paget Disease
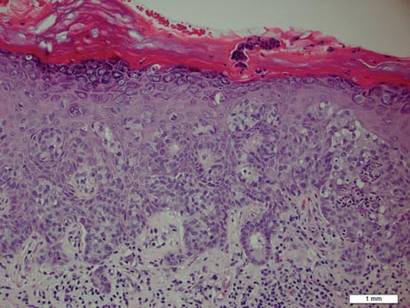
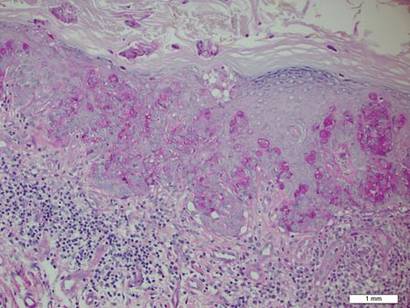
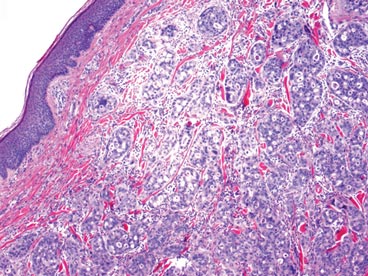
A biopsy of the plaque revealed an intraepidermal proliferation of large cells with abundant clear cytoplasm and large vesicular nuclei distributed throughout the epidermis (Figure 1). The neoplastic cells stained positive for both periodic acid–Schiff stain (Figure 2) and CK7 (Figure 3). Chemistry and liver function panel, urine analysis, carcinoembryonic antigen levels, and prostate-specific antigen levels were within reference range. A complete blood cell count revealed mild megaloblastic anemia. Subsequent computed tomography of the chest, abdomen, and pelvis revealed an abdominal aortic aneurysm and prostatic enlargement without any evidence of potential malignancies. Colonoscopy revealed multiple hyperplastic polyps and a tubular adenoma. Cystoscopy was normal, except for evidence of prostate enlargement. Urine cytology was unremarkable. The patient was referred for excision of the lesion with Mohs micrographic surgery. Follow-up was recommended every 3 months for the first 2 years following surgery and every 6 months thereafter to monitor for recurrence or secondary neoplasms.
|
Sir James Paget first described mammary Paget disease of the nipple in 1874 in his report of 15 women with skin eruptions of the nipple and areola and subsequent carcinoma of the underlying breast.1 Paget also described a patient with a similar eruption on the glans penis and Crocker2 described extramammary Paget disease (EMPD) of the scrotum and penis in 1889. The principle difference between mammary Paget disease and EMPD is the anatomic location.
Extramammary Paget disease is a rare condition that typically affects patients aged 50 to 80 years and is more common in women and white-skinned races.3 Extramammary Paget disease frequently targets cutaneous sites that are rich in apocrine glands. The most commonly affected site is the vulva followed by perineal, perianal, scrotal, and penile skin. Less commonly, the axillae, buttocks, thighs, eyelids, and external auditory canals may be affected.4
Patients with EMPD typically present with well-demarcated, nonresolving, erythematous and eczematous plaques that may have associated crusting, scaling, papillomatous excrescences, lichenification, ulceration, or bleeding. The most common symptom is pruritus, followed by burning, irritation, pain, and tenderness.5 Ten percent of patients are asymptomatic. The average interval between symptom onset and diagnosis is 2 years.5
Histopathology reveals diffusely infiltrating, irregular, neoplastic Paget cells within the epidermis that are large and vacuolated with abundant pale bluish cytoplasm and large vesicular nuclei, which may be centrally or laterally compressed. The cells may be distributed singly or in groups as strands, nests, or glandular patterns within the lower epidermis, rete ridges, and adnexal structures. Hyperkeratosis, acanthosis, and parakeratosis may also be present. Paget cells stain for immunohistochemical markers of apocrine and eccrine derivation including low-molecular-weight cytokeratins, gross cystic disease fluid protein 15, periodic acid–Schiff stain, and carcinoembryonic antigen.5 Perrotto et al6 studied 98 specimens from 61 patients and found that CK7 was positive in all EMPD specimens, while CK20 and gross cystic disease fluid protein 15 were positive in large subsets of both primary and secondary EMPD. Cases of EMPD secondary to anorectal adenocarcinoma were largely ERBB2 (formerly HER2/neu) negative and CDX2 positive.6
Diagnosis of EMPD should be followed by a thorough investigation for underlying carcinomas. In a review of 197 cases of EMPD, 24% of patients with EMPD had an associated underlying in situ or invasive adnexal apocrine carcinoma, which was associated with a higher mortality rate than in patients without this underlying malignancy. Additionally, 12% of EMPD patients had an associated underlying internal malignancy.7 These malignancies may include carcinomas of the urethra, bladder, vagina, cervix, endometrium, prostate, colon, and rectum. Perianal EMPD has a higher frequency of associated malignancies than vulvar EMPD.5 The location of EMPD is related to the location of the underlying malignancy; for example, perianal EMPD is associated with colorectal adenocarcinomas, and EMPD of the penis, scrotum, and groin is associated with genitourinary malignancies. Investigations to search for associated malignancies in patients with EMPD may include pelvic ultrasonography and/or magnetic resonance imaging, hysteroscopy, colonoscopy, sigmoidoscopy, cystoscopy, intravenous pyelogram, mammogram, and/or chest radiograph.
The most effective treatment of EMPD is margin-controlled surgical excision. High local recurrence rates may be due to irregular margins, multicentricity, and the tendency of EMPD to involve clinically normal-appearing skin. Hendi et al8 noted that EMPD may actually be unifocal with subclinical fingerlike projections extending beyond the main body of the tumor, requiring CK7 immunostaining for visualization to ensure complete margin control. The recurrence rate after standard surgical excision is 33% to 60%. The recurrence rate after excision via Mohs micrographic surgery is 16% for primary EMPD and 50% for recurrent EMPD.9 Other treatment modalities include radiotherapy, topical chemotherapy with 5-fluorouracil or imiquimod, and photodynamic therapy.10-13 Combined systemic chemotherapy with trastuzumab and paclitaxel can be considered for the treatment of ERBB2-positive EMPD.14
For patients with chronic genital or perianal lesions that are unresponsive to treatment, dermatologists should maintain a high index of suspicion for EMPD. If a patient is diagnosed with EMPD, a full-body skin examination should be performed with palpation of all lymph nodes. Imaging studies directed at the anatomic location of the involved skin should be utilized to search for an underlying internal malignancy.
1. Paget J. On disease of the mammary areola preceding cancer of the mammary gland. St Bartholomew Hosp Rep. 1874;10:87-89.
2. Crocker H. Paget’s disease affecting the scrotum and penis. Trans Pathol Soc Lond. 1889;40:187-191.
3. Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
4. Heymann WR. Extramammary Paget’s disease. Clin Dermatol. 1993;11:83-87.5. Shepherd V, Davidson EJ, Davies-Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
6. Perrotto J, Abbott JJ, Ceilley RI, et al. The role of immunohistochemistry in discriminating primary from secondary extramammary Paget disease. Am J Dermatopathol. 2010;32:137-143.
7. Chanda JJ. Extramammary Paget’s disease: prognosis and relationship to internal malignancy. J Am Acad Dermatol. 1985;13:1009-1014.
8. Hendi A, Perdikis G, Snow JL. Unifocality of extramammary Paget disease. J Am Acad Dermatol. 2008;59:811-813.
9. Hendi A, Brodland DG, Zitelli JA. Extramammary Paget’s disease: surgical treatment with mohs micrographic surgery. J Am Acad Dermatol. 2004;51:767-773.10. Zampogna JC, Flowers FP, Roth WI, et al. Treatment of primary limited cutaneous extramammary Paget’s disease with topical imiquimod monotherapy: two case reports. J Am Acad Dermatol. 2002;47:S229-S235.
11. Beleznay KM, Levesque MA, Gill S. Response to 5-fluorouracil in metastatic extramammary Paget disease of the scrotum presenting as pancytopenia and back pain. Curr Oncol. 2009;16:81-83.
12. Kitagawa KH, Bogner P, Zeitouni NC. Photodynamic therapy with methyl-aminolevulinate for the treatment of double extramammary Paget’s disease. Dermatol Surg. 2011;37:1043-1046.
13. Hata M, Omura M, Koike I, et al. Role of radiotherapy as curative treatment of extramammary Paget’s disease. Int J Radiat Oncol Biol Phys. 2011;80:47-54.
14. Takahagi S, Noda H, Kamegashira A, et al. Metastatic extramammary Paget’s disease treated with paclitaxel and trastuzumab combination chemotherapy. J Dermatol. 2009;36:457-461.
A 70-year-old man presented with a nonpruritic erythematous scaly plaque in the left suprapubic region of 6 months’ duration that had failed to respond to terbinafine cream 1% after 1 month of treatment of suspected tinea cruris. His medical history was remarkable for hypertension, hyperlipidemia, chronic obstructive pulmonary disease, benign prostatic hyperplasia, an abdominal aortic aneurysm, alcohol dependence, tobacco use disorder, and unintentional weight loss of 15 lb over the last year.
The Diagnosis: Extramammary Paget Disease
A biopsy of the plaque revealed an intraepidermal proliferation of large cells with abundant clear cytoplasm and large vesicular nuclei distributed throughout the epidermis (Figure 1). The neoplastic cells stained positive for both periodic acid–Schiff stain (Figure 2) and CK7 (Figure 3). Chemistry and liver function panel, urine analysis, carcinoembryonic antigen levels, and prostate-specific antigen levels were within reference range. A complete blood cell count revealed mild megaloblastic anemia. Subsequent computed tomography of the chest, abdomen, and pelvis revealed an abdominal aortic aneurysm and prostatic enlargement without any evidence of potential malignancies. Colonoscopy revealed multiple hyperplastic polyps and a tubular adenoma. Cystoscopy was normal, except for evidence of prostate enlargement. Urine cytology was unremarkable. The patient was referred for excision of the lesion with Mohs micrographic surgery. Follow-up was recommended every 3 months for the first 2 years following surgery and every 6 months thereafter to monitor for recurrence or secondary neoplasms.
|
Sir James Paget first described mammary Paget disease of the nipple in 1874 in his report of 15 women with skin eruptions of the nipple and areola and subsequent carcinoma of the underlying breast.1 Paget also described a patient with a similar eruption on the glans penis and Crocker2 described extramammary Paget disease (EMPD) of the scrotum and penis in 1889. The principle difference between mammary Paget disease and EMPD is the anatomic location.
Extramammary Paget disease is a rare condition that typically affects patients aged 50 to 80 years and is more common in women and white-skinned races.3 Extramammary Paget disease frequently targets cutaneous sites that are rich in apocrine glands. The most commonly affected site is the vulva followed by perineal, perianal, scrotal, and penile skin. Less commonly, the axillae, buttocks, thighs, eyelids, and external auditory canals may be affected.4
Patients with EMPD typically present with well-demarcated, nonresolving, erythematous and eczematous plaques that may have associated crusting, scaling, papillomatous excrescences, lichenification, ulceration, or bleeding. The most common symptom is pruritus, followed by burning, irritation, pain, and tenderness.5 Ten percent of patients are asymptomatic. The average interval between symptom onset and diagnosis is 2 years.5
Histopathology reveals diffusely infiltrating, irregular, neoplastic Paget cells within the epidermis that are large and vacuolated with abundant pale bluish cytoplasm and large vesicular nuclei, which may be centrally or laterally compressed. The cells may be distributed singly or in groups as strands, nests, or glandular patterns within the lower epidermis, rete ridges, and adnexal structures. Hyperkeratosis, acanthosis, and parakeratosis may also be present. Paget cells stain for immunohistochemical markers of apocrine and eccrine derivation including low-molecular-weight cytokeratins, gross cystic disease fluid protein 15, periodic acid–Schiff stain, and carcinoembryonic antigen.5 Perrotto et al6 studied 98 specimens from 61 patients and found that CK7 was positive in all EMPD specimens, while CK20 and gross cystic disease fluid protein 15 were positive in large subsets of both primary and secondary EMPD. Cases of EMPD secondary to anorectal adenocarcinoma were largely ERBB2 (formerly HER2/neu) negative and CDX2 positive.6
Diagnosis of EMPD should be followed by a thorough investigation for underlying carcinomas. In a review of 197 cases of EMPD, 24% of patients with EMPD had an associated underlying in situ or invasive adnexal apocrine carcinoma, which was associated with a higher mortality rate than in patients without this underlying malignancy. Additionally, 12% of EMPD patients had an associated underlying internal malignancy.7 These malignancies may include carcinomas of the urethra, bladder, vagina, cervix, endometrium, prostate, colon, and rectum. Perianal EMPD has a higher frequency of associated malignancies than vulvar EMPD.5 The location of EMPD is related to the location of the underlying malignancy; for example, perianal EMPD is associated with colorectal adenocarcinomas, and EMPD of the penis, scrotum, and groin is associated with genitourinary malignancies. Investigations to search for associated malignancies in patients with EMPD may include pelvic ultrasonography and/or magnetic resonance imaging, hysteroscopy, colonoscopy, sigmoidoscopy, cystoscopy, intravenous pyelogram, mammogram, and/or chest radiograph.
The most effective treatment of EMPD is margin-controlled surgical excision. High local recurrence rates may be due to irregular margins, multicentricity, and the tendency of EMPD to involve clinically normal-appearing skin. Hendi et al8 noted that EMPD may actually be unifocal with subclinical fingerlike projections extending beyond the main body of the tumor, requiring CK7 immunostaining for visualization to ensure complete margin control. The recurrence rate after standard surgical excision is 33% to 60%. The recurrence rate after excision via Mohs micrographic surgery is 16% for primary EMPD and 50% for recurrent EMPD.9 Other treatment modalities include radiotherapy, topical chemotherapy with 5-fluorouracil or imiquimod, and photodynamic therapy.10-13 Combined systemic chemotherapy with trastuzumab and paclitaxel can be considered for the treatment of ERBB2-positive EMPD.14
For patients with chronic genital or perianal lesions that are unresponsive to treatment, dermatologists should maintain a high index of suspicion for EMPD. If a patient is diagnosed with EMPD, a full-body skin examination should be performed with palpation of all lymph nodes. Imaging studies directed at the anatomic location of the involved skin should be utilized to search for an underlying internal malignancy.
A 70-year-old man presented with a nonpruritic erythematous scaly plaque in the left suprapubic region of 6 months’ duration that had failed to respond to terbinafine cream 1% after 1 month of treatment of suspected tinea cruris. His medical history was remarkable for hypertension, hyperlipidemia, chronic obstructive pulmonary disease, benign prostatic hyperplasia, an abdominal aortic aneurysm, alcohol dependence, tobacco use disorder, and unintentional weight loss of 15 lb over the last year.
The Diagnosis: Extramammary Paget Disease
A biopsy of the plaque revealed an intraepidermal proliferation of large cells with abundant clear cytoplasm and large vesicular nuclei distributed throughout the epidermis (Figure 1). The neoplastic cells stained positive for both periodic acid–Schiff stain (Figure 2) and CK7 (Figure 3). Chemistry and liver function panel, urine analysis, carcinoembryonic antigen levels, and prostate-specific antigen levels were within reference range. A complete blood cell count revealed mild megaloblastic anemia. Subsequent computed tomography of the chest, abdomen, and pelvis revealed an abdominal aortic aneurysm and prostatic enlargement without any evidence of potential malignancies. Colonoscopy revealed multiple hyperplastic polyps and a tubular adenoma. Cystoscopy was normal, except for evidence of prostate enlargement. Urine cytology was unremarkable. The patient was referred for excision of the lesion with Mohs micrographic surgery. Follow-up was recommended every 3 months for the first 2 years following surgery and every 6 months thereafter to monitor for recurrence or secondary neoplasms.
|
Sir James Paget first described mammary Paget disease of the nipple in 1874 in his report of 15 women with skin eruptions of the nipple and areola and subsequent carcinoma of the underlying breast.1 Paget also described a patient with a similar eruption on the glans penis and Crocker2 described extramammary Paget disease (EMPD) of the scrotum and penis in 1889. The principle difference between mammary Paget disease and EMPD is the anatomic location.
Extramammary Paget disease is a rare condition that typically affects patients aged 50 to 80 years and is more common in women and white-skinned races.3 Extramammary Paget disease frequently targets cutaneous sites that are rich in apocrine glands. The most commonly affected site is the vulva followed by perineal, perianal, scrotal, and penile skin. Less commonly, the axillae, buttocks, thighs, eyelids, and external auditory canals may be affected.4
Patients with EMPD typically present with well-demarcated, nonresolving, erythematous and eczematous plaques that may have associated crusting, scaling, papillomatous excrescences, lichenification, ulceration, or bleeding. The most common symptom is pruritus, followed by burning, irritation, pain, and tenderness.5 Ten percent of patients are asymptomatic. The average interval between symptom onset and diagnosis is 2 years.5
Histopathology reveals diffusely infiltrating, irregular, neoplastic Paget cells within the epidermis that are large and vacuolated with abundant pale bluish cytoplasm and large vesicular nuclei, which may be centrally or laterally compressed. The cells may be distributed singly or in groups as strands, nests, or glandular patterns within the lower epidermis, rete ridges, and adnexal structures. Hyperkeratosis, acanthosis, and parakeratosis may also be present. Paget cells stain for immunohistochemical markers of apocrine and eccrine derivation including low-molecular-weight cytokeratins, gross cystic disease fluid protein 15, periodic acid–Schiff stain, and carcinoembryonic antigen.5 Perrotto et al6 studied 98 specimens from 61 patients and found that CK7 was positive in all EMPD specimens, while CK20 and gross cystic disease fluid protein 15 were positive in large subsets of both primary and secondary EMPD. Cases of EMPD secondary to anorectal adenocarcinoma were largely ERBB2 (formerly HER2/neu) negative and CDX2 positive.6
Diagnosis of EMPD should be followed by a thorough investigation for underlying carcinomas. In a review of 197 cases of EMPD, 24% of patients with EMPD had an associated underlying in situ or invasive adnexal apocrine carcinoma, which was associated with a higher mortality rate than in patients without this underlying malignancy. Additionally, 12% of EMPD patients had an associated underlying internal malignancy.7 These malignancies may include carcinomas of the urethra, bladder, vagina, cervix, endometrium, prostate, colon, and rectum. Perianal EMPD has a higher frequency of associated malignancies than vulvar EMPD.5 The location of EMPD is related to the location of the underlying malignancy; for example, perianal EMPD is associated with colorectal adenocarcinomas, and EMPD of the penis, scrotum, and groin is associated with genitourinary malignancies. Investigations to search for associated malignancies in patients with EMPD may include pelvic ultrasonography and/or magnetic resonance imaging, hysteroscopy, colonoscopy, sigmoidoscopy, cystoscopy, intravenous pyelogram, mammogram, and/or chest radiograph.
The most effective treatment of EMPD is margin-controlled surgical excision. High local recurrence rates may be due to irregular margins, multicentricity, and the tendency of EMPD to involve clinically normal-appearing skin. Hendi et al8 noted that EMPD may actually be unifocal with subclinical fingerlike projections extending beyond the main body of the tumor, requiring CK7 immunostaining for visualization to ensure complete margin control. The recurrence rate after standard surgical excision is 33% to 60%. The recurrence rate after excision via Mohs micrographic surgery is 16% for primary EMPD and 50% for recurrent EMPD.9 Other treatment modalities include radiotherapy, topical chemotherapy with 5-fluorouracil or imiquimod, and photodynamic therapy.10-13 Combined systemic chemotherapy with trastuzumab and paclitaxel can be considered for the treatment of ERBB2-positive EMPD.14
For patients with chronic genital or perianal lesions that are unresponsive to treatment, dermatologists should maintain a high index of suspicion for EMPD. If a patient is diagnosed with EMPD, a full-body skin examination should be performed with palpation of all lymph nodes. Imaging studies directed at the anatomic location of the involved skin should be utilized to search for an underlying internal malignancy.
1. Paget J. On disease of the mammary areola preceding cancer of the mammary gland. St Bartholomew Hosp Rep. 1874;10:87-89.
2. Crocker H. Paget’s disease affecting the scrotum and penis. Trans Pathol Soc Lond. 1889;40:187-191.
3. Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
4. Heymann WR. Extramammary Paget’s disease. Clin Dermatol. 1993;11:83-87.5. Shepherd V, Davidson EJ, Davies-Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
6. Perrotto J, Abbott JJ, Ceilley RI, et al. The role of immunohistochemistry in discriminating primary from secondary extramammary Paget disease. Am J Dermatopathol. 2010;32:137-143.
7. Chanda JJ. Extramammary Paget’s disease: prognosis and relationship to internal malignancy. J Am Acad Dermatol. 1985;13:1009-1014.
8. Hendi A, Perdikis G, Snow JL. Unifocality of extramammary Paget disease. J Am Acad Dermatol. 2008;59:811-813.
9. Hendi A, Brodland DG, Zitelli JA. Extramammary Paget’s disease: surgical treatment with mohs micrographic surgery. J Am Acad Dermatol. 2004;51:767-773.10. Zampogna JC, Flowers FP, Roth WI, et al. Treatment of primary limited cutaneous extramammary Paget’s disease with topical imiquimod monotherapy: two case reports. J Am Acad Dermatol. 2002;47:S229-S235.
11. Beleznay KM, Levesque MA, Gill S. Response to 5-fluorouracil in metastatic extramammary Paget disease of the scrotum presenting as pancytopenia and back pain. Curr Oncol. 2009;16:81-83.
12. Kitagawa KH, Bogner P, Zeitouni NC. Photodynamic therapy with methyl-aminolevulinate for the treatment of double extramammary Paget’s disease. Dermatol Surg. 2011;37:1043-1046.
13. Hata M, Omura M, Koike I, et al. Role of radiotherapy as curative treatment of extramammary Paget’s disease. Int J Radiat Oncol Biol Phys. 2011;80:47-54.
14. Takahagi S, Noda H, Kamegashira A, et al. Metastatic extramammary Paget’s disease treated with paclitaxel and trastuzumab combination chemotherapy. J Dermatol. 2009;36:457-461.
1. Paget J. On disease of the mammary areola preceding cancer of the mammary gland. St Bartholomew Hosp Rep. 1874;10:87-89.
2. Crocker H. Paget’s disease affecting the scrotum and penis. Trans Pathol Soc Lond. 1889;40:187-191.
3. Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
4. Heymann WR. Extramammary Paget’s disease. Clin Dermatol. 1993;11:83-87.5. Shepherd V, Davidson EJ, Davies-Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
6. Perrotto J, Abbott JJ, Ceilley RI, et al. The role of immunohistochemistry in discriminating primary from secondary extramammary Paget disease. Am J Dermatopathol. 2010;32:137-143.
7. Chanda JJ. Extramammary Paget’s disease: prognosis and relationship to internal malignancy. J Am Acad Dermatol. 1985;13:1009-1014.
8. Hendi A, Perdikis G, Snow JL. Unifocality of extramammary Paget disease. J Am Acad Dermatol. 2008;59:811-813.
9. Hendi A, Brodland DG, Zitelli JA. Extramammary Paget’s disease: surgical treatment with mohs micrographic surgery. J Am Acad Dermatol. 2004;51:767-773.10. Zampogna JC, Flowers FP, Roth WI, et al. Treatment of primary limited cutaneous extramammary Paget’s disease with topical imiquimod monotherapy: two case reports. J Am Acad Dermatol. 2002;47:S229-S235.
11. Beleznay KM, Levesque MA, Gill S. Response to 5-fluorouracil in metastatic extramammary Paget disease of the scrotum presenting as pancytopenia and back pain. Curr Oncol. 2009;16:81-83.
12. Kitagawa KH, Bogner P, Zeitouni NC. Photodynamic therapy with methyl-aminolevulinate for the treatment of double extramammary Paget’s disease. Dermatol Surg. 2011;37:1043-1046.
13. Hata M, Omura M, Koike I, et al. Role of radiotherapy as curative treatment of extramammary Paget’s disease. Int J Radiat Oncol Biol Phys. 2011;80:47-54.
14. Takahagi S, Noda H, Kamegashira A, et al. Metastatic extramammary Paget’s disease treated with paclitaxel and trastuzumab combination chemotherapy. J Dermatol. 2009;36:457-461.
Onchocerciasis
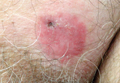
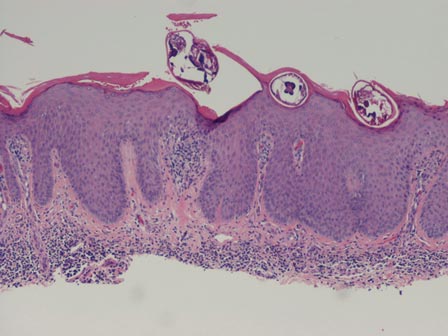
The larvae of Onchocerca volvulus, a nematode that is most commonly found in tropical Africa, Yemen, Central America, and South America, are transmitted by flies of the genus Simulium that breed near fast-flowing rivers.1 The flies bite the host and transmit the larvae, and the larvae then mature into adults within the skin and subcutis, forming nodules that typically are not painful. The worms may reside within the skin for years and produce microfilariae, which can migrate and cause visual impairment, blindness, or a pruritic papular rash.1
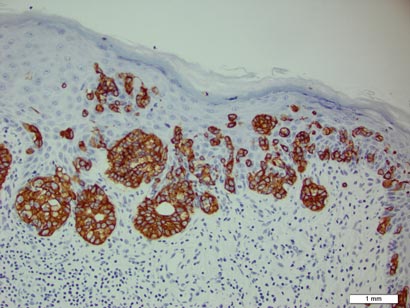
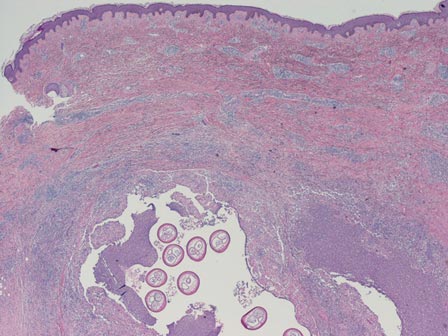
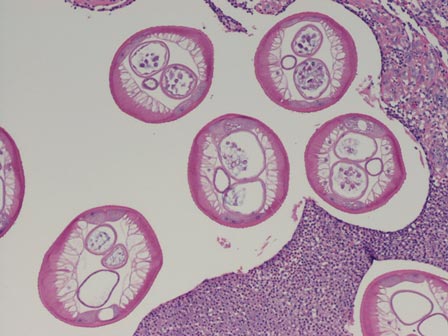
The nematode produces a nodule within the dermis or subcutis with surrounding fibrous tissue and a mixed inflammatory infiltrate with eosinophils (Figure 1). In some cases, microfilariae can be seen within the lymphatics or within the uteri of the worms.1 Male and female worms typically are present and have a corrugated cuticle with a thin underlying layer of striated muscle. The females have paired uteri, which usually contain microfilariae2 (Figure 2).
| 
|
Dirofilaria repens also is a nematode that produces a subcutaneous nodule with an inflammatory reaction. This worm typically has a thick cuticle with longitudinal ridges, long thick muscle, and lateral cords.3 Additionally, because humans are not the usual host, Dirofilaria species do not complete their lifecycle and typically are not gravid, unlike Onchocerca species.
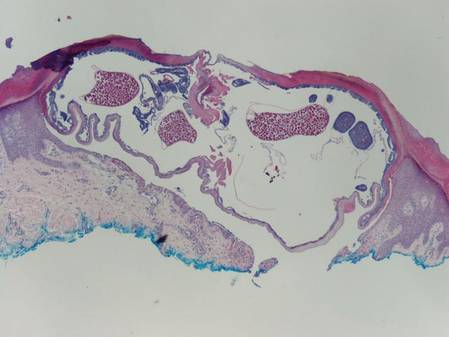
Myiasis is the presence of fly larvae within the skin. The larvae demonstrate a thick hyaline cuticle with pigmented brown-yellow spikes (Figure 3). There is a thick muscular layer under the cuticle and a tubular tracheal system containing vertical striations. The digestive system has an epithelial lining with prominent vessels. Adipose tissue with granulated cytoplasm, prominent nuclei, and coarse chromatin also are present.4

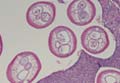
Scabies mites (Figure 4), ova, and scybala are present within the stratum corneum. A mixed inflammatory infiltrate also can be present.1 Tungiasis is caused by burrowing fleas and typically occurs on acral skin; therefore, it is more frequently found in the superficial portion of the skin. Erythrocytes usually are present in the gastrointestinal tract, and the females usually are gravid.2 A surrounding mixed inflammatory infiltrate is present, and necrosis also can occur (Figure 5).1
| 
|
1. Weedon D. Weedon’s Skin Pathology. 3rd ed. Edinburgh, Scotland: Churchill Livingstone Elsevier; 2010.
2. Elston DM, Ferringer T. Dermatopathology: Requisites in Dermatology. Edinburgh, Scotland: Saunders Elsevier; 2008.
3. Tzanetou K, Gasteratos S, Pantazopoulou A, et al. Subcutaneous dirofilariasis caused by Dirofilaria repens in Greece: a case report. J Cutan Pathol. 2009;36:892-895.
4. Fernandez-Flores A, Saeb-Lima M. Pulse granuloma of the lip: morphologic clues in its differential diagnosis. J Cutan Pathol. 2014;41:394-399.
The larvae of Onchocerca volvulus, a nematode that is most commonly found in tropical Africa, Yemen, Central America, and South America, are transmitted by flies of the genus Simulium that breed near fast-flowing rivers.1 The flies bite the host and transmit the larvae, and the larvae then mature into adults within the skin and subcutis, forming nodules that typically are not painful. The worms may reside within the skin for years and produce microfilariae, which can migrate and cause visual impairment, blindness, or a pruritic papular rash.1
The nematode produces a nodule within the dermis or subcutis with surrounding fibrous tissue and a mixed inflammatory infiltrate with eosinophils (Figure 1). In some cases, microfilariae can be seen within the lymphatics or within the uteri of the worms.1 Male and female worms typically are present and have a corrugated cuticle with a thin underlying layer of striated muscle. The females have paired uteri, which usually contain microfilariae2 (Figure 2).
|
|
|
Dirofilaria repens also is a nematode that produces a subcutaneous nodule with an inflammatory reaction. This worm typically has a thick cuticle with longitudinal ridges, long thick muscle, and lateral cords.3 Additionally, because humans are not the usual host, Dirofilaria species do not complete their lifecycle and typically are not gravid, unlike Onchocerca species.
Myiasis is the presence of fly larvae within the skin. The larvae demonstrate a thick hyaline cuticle with pigmented brown-yellow spikes (Figure 3). There is a thick muscular layer under the cuticle and a tubular tracheal system containing vertical striations. The digestive system has an epithelial lining with prominent vessels. Adipose tissue with granulated cytoplasm, prominent nuclei, and coarse chromatin also are present.4
Scabies mites (Figure 4), ova, and scybala are present within the stratum corneum. A mixed inflammatory infiltrate also can be present.1 Tungiasis is caused by burrowing fleas and typically occurs on acral skin; therefore, it is more frequently found in the superficial portion of the skin. Erythrocytes usually are present in the gastrointestinal tract, and the females usually are gravid.2 A surrounding mixed inflammatory infiltrate is present, and necrosis also can occur (Figure 5).1
|
|
|
The larvae of Onchocerca volvulus, a nematode that is most commonly found in tropical Africa, Yemen, Central America, and South America, are transmitted by flies of the genus Simulium that breed near fast-flowing rivers.1 The flies bite the host and transmit the larvae, and the larvae then mature into adults within the skin and subcutis, forming nodules that typically are not painful. The worms may reside within the skin for years and produce microfilariae, which can migrate and cause visual impairment, blindness, or a pruritic papular rash.1
The nematode produces a nodule within the dermis or subcutis with surrounding fibrous tissue and a mixed inflammatory infiltrate with eosinophils (Figure 1). In some cases, microfilariae can be seen within the lymphatics or within the uteri of the worms.1 Male and female worms typically are present and have a corrugated cuticle with a thin underlying layer of striated muscle. The females have paired uteri, which usually contain microfilariae2 (Figure 2).
|
|
|
Dirofilaria repens also is a nematode that produces a subcutaneous nodule with an inflammatory reaction. This worm typically has a thick cuticle with longitudinal ridges, long thick muscle, and lateral cords.3 Additionally, because humans are not the usual host, Dirofilaria species do not complete their lifecycle and typically are not gravid, unlike Onchocerca species.
Myiasis is the presence of fly larvae within the skin. The larvae demonstrate a thick hyaline cuticle with pigmented brown-yellow spikes (Figure 3). There is a thick muscular layer under the cuticle and a tubular tracheal system containing vertical striations. The digestive system has an epithelial lining with prominent vessels. Adipose tissue with granulated cytoplasm, prominent nuclei, and coarse chromatin also are present.4
Scabies mites (Figure 4), ova, and scybala are present within the stratum corneum. A mixed inflammatory infiltrate also can be present.1 Tungiasis is caused by burrowing fleas and typically occurs on acral skin; therefore, it is more frequently found in the superficial portion of the skin. Erythrocytes usually are present in the gastrointestinal tract, and the females usually are gravid.2 A surrounding mixed inflammatory infiltrate is present, and necrosis also can occur (Figure 5).1
|
|
|
1. Weedon D. Weedon’s Skin Pathology. 3rd ed. Edinburgh, Scotland: Churchill Livingstone Elsevier; 2010.
2. Elston DM, Ferringer T. Dermatopathology: Requisites in Dermatology. Edinburgh, Scotland: Saunders Elsevier; 2008.
3. Tzanetou K, Gasteratos S, Pantazopoulou A, et al. Subcutaneous dirofilariasis caused by Dirofilaria repens in Greece: a case report. J Cutan Pathol. 2009;36:892-895.
4. Fernandez-Flores A, Saeb-Lima M. Pulse granuloma of the lip: morphologic clues in its differential diagnosis. J Cutan Pathol. 2014;41:394-399.
1. Weedon D. Weedon’s Skin Pathology. 3rd ed. Edinburgh, Scotland: Churchill Livingstone Elsevier; 2010.
2. Elston DM, Ferringer T. Dermatopathology: Requisites in Dermatology. Edinburgh, Scotland: Saunders Elsevier; 2008.
3. Tzanetou K, Gasteratos S, Pantazopoulou A, et al. Subcutaneous dirofilariasis caused by Dirofilaria repens in Greece: a case report. J Cutan Pathol. 2009;36:892-895.
4. Fernandez-Flores A, Saeb-Lima M. Pulse granuloma of the lip: morphologic clues in its differential diagnosis. J Cutan Pathol. 2014;41:394-399.
Pseudoglandular Squamous Cell Carcinoma
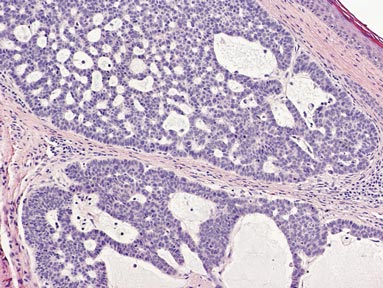
Squamous cell carcinoma (SCC) is the second most common form of skin cancer. Pseudoglandular SCC, also known as adenoid SCC or acantholytic SCC, is an uncommon variant that was first described by Lever1 in 1947 as an adenoacanthoma of the sweat glands. Of the many variants of SCC, pseudoglandular SCC generally is considered to behave aggressively with intermediate (3%–10%) risk for metastasis.2 The metastatic potential of pseudoglandular SCC may be conferred in part by diminished expression of intercellular adhesion molecules, including desmoglein 3, epithelial cadherin, and syn-decan 1.3,4 Pseudoglandular SCC presents most often on sun-damaged skin of elderly patients, especially the face and ears, as a pink or red nodule with central ulceration and a raised indurated border. It may be mistaken clinically for basal cell carcinoma (BCC) or keratoacanthoma.
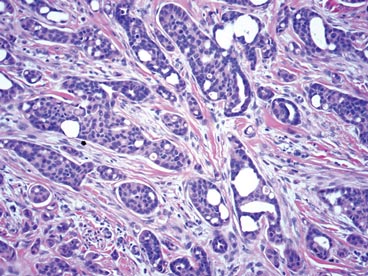
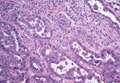
On microscopic examination, the lesion is predominantly located in the dermis and may extend to the subcutis. There usually is connection to the overlying epidermis, which often shows hyperkeratosis and parakeratosis. Epidermal squamous dysplasia may be present. The dermis typically contains nests of squamous cells with a variable degree of central acantholysis. The morphology on low-power magnification consists of tubules of irregular size and shape, which are present either focally or throughout the lesion (Figure 1). The tubules are typically admixed with foci of keratinization. One or more layers of cohesive cells line the tubules. Partial keratinization may be found in the lining of tubules with more than 1 cell layer. The tumor cells are polygonal with eosinophilic cytoplasm, ovoid hyperchromatic or vesicular nuclei, and prominent nucleoli. Mitoses are common. The tubular lumina are filled with acantholytic cells, either singly or in small clusters, which may demonstrate residual bridging to tubular lining cells (Figure 2). The acantholytic cells show some variability in size and may be large, multinucleated, or keratinized. The tubules may contain material that is amorphous, basophilic, periodic acid–Schiff positive, diastase sensitive, and mucicarmine negative.5 Eccrine ducts at the periphery of the tumor may show reactive dilatation and proliferation. Tumor cells show positive immunostaining for epithelial membrane antigen, 34βE12, CK5/6, and tumor protein p63.6-8 There is negative immunostaining for carcinoembryonic antigen, amylase, S-100 protein, and factor VIII.5
|

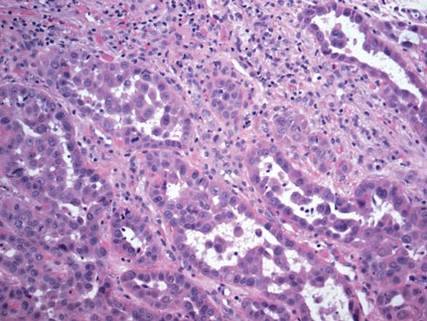
The differential diagnosis includes adenoid BCC, angiosarcoma, eccrine carcinoma, and metastatic adenocarcinoma of the skin. In adenoid BCC, excess stromal mucin imparts pseudoglandular architecture (Figure 3). However, features of conventional BCC, including peripheral nuclear palisading and retraction artifact often are present as well.
Angiosarcoma shows slitlike vascular spaces lined by hyperchromatic endothelial cells (Figure 4). Further, there is positive immunostaining for vascular markers CD31 and CD34.
In eccrine carcinoma, there are invasive ductal structures lined by either a single or double layer of cells that may contain luminal material that is periodic acid–Schiff positive and diastase resistant (Figure 5).9 The tumor cells show positive immunostaining for cytokeratins, epithelial membrane antigen, carcinoembryonic antigen, and S-100 protein.10
Pseudoglandular SCC is susceptible to misdiagnosis as adenocarcinoma by sampling error if biopsies do not capture areas with typical features of SCC, including dysplastic squamous epithelium and keratinization. Metastatic adenocarcinoma of the skin is more likely to present with multiple nodules in older individuals. Lack of epidermal connection of the tumor and minimal to no acantholytic dyskeratosis further support cutaneous metastasis (Figure 6). Review of the patient’s clinical history might be helpful if adenocarcinoma was previously diagnosed. Immunohistochemical evaluation may aid in the prediction of the primary site in patients with metastatic adenocarcinoma of unknown origin.11
1. Lever WF. Adenocanthoma of sweat glands; carcinoma of sweat glands with glandular and epidermal elements: report of four cases. Arch Derm Syphilol. 1947;56:157-171.
2. Bonerandi JJ, Beauvillain C, Caquant L, et al. Guidelines for the diagnosis and treatment of cutaneous squamous cell carcinoma and precursor lesions. J Eur Acad Dermatol Venereol. 2011;25(suppl 5):1-51.
3. Griffin JR, Wriston CC, Peters MS, et al. Decreased expression of intercellular adhesion molecules in acantholytic squamous cell carcinoma compared with invasive well-differentiated squamous cell carcinoma of the skin. Am J Clin Pathol. 2013;139:442-447.
4. Bayer-Garner IB, Smoller BR. The expression of syndecan-1 is preferentially reduced compared with that of E-cadherin in acantholytic squamous cell carcinoma. J Cutan Pathol. 2001;28:83-89.
5. Nappi O, Pettinato G, Wick MR. Adenoid (acantholytic) squamous cell carcinoma of the skin. J Cutan Pathol. 1989;16:114-121.
6. Sajin M, Hodorogea Prisăcaru A, Luchian MC, et al. Acantholytic squamous cell carcinoma: pathological study of nine cases with review of literature. Rom J Morphol Embryol. 2014;55:279-283.
7. Gray Y, Robidoux HJ, Farrell DS, et al. Squamous cell carcinoma detected by high-molecular-weight cytokeratin immunostaining mimicking atypical fibroxanthoma. Arch Pathol Lab Med. 2001;125:799-802.
8. Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
9. Plaza JA, Prieto VG. Neoplastic Lesions of the Skin. New York, NY: Demos Medical Publishing; 2014.
10. Swanson PE, Cherwitz DL, Neumann MP, et al. Eccrine sweat gland carcinoma: an histologic and immunohistochemical study of 32 cases. J Cutan Pathol. 1987;14:65-86.
11. Dennis JL, Hvidsten TR, Wit EC, et al. Markers of adenocarcinoma characteristic of the site of origin: development of a diagnostic algorithm. Clin Cancer Res. 2005;11:3766-3772.
Squamous cell carcinoma (SCC) is the second most common form of skin cancer. Pseudoglandular SCC, also known as adenoid SCC or acantholytic SCC, is an uncommon variant that was first described by Lever1 in 1947 as an adenoacanthoma of the sweat glands. Of the many variants of SCC, pseudoglandular SCC generally is considered to behave aggressively with intermediate (3%–10%) risk for metastasis.2 The metastatic potential of pseudoglandular SCC may be conferred in part by diminished expression of intercellular adhesion molecules, including desmoglein 3, epithelial cadherin, and syn-decan 1.3,4 Pseudoglandular SCC presents most often on sun-damaged skin of elderly patients, especially the face and ears, as a pink or red nodule with central ulceration and a raised indurated border. It may be mistaken clinically for basal cell carcinoma (BCC) or keratoacanthoma.
On microscopic examination, the lesion is predominantly located in the dermis and may extend to the subcutis. There usually is connection to the overlying epidermis, which often shows hyperkeratosis and parakeratosis. Epidermal squamous dysplasia may be present. The dermis typically contains nests of squamous cells with a variable degree of central acantholysis. The morphology on low-power magnification consists of tubules of irregular size and shape, which are present either focally or throughout the lesion (Figure 1). The tubules are typically admixed with foci of keratinization. One or more layers of cohesive cells line the tubules. Partial keratinization may be found in the lining of tubules with more than 1 cell layer. The tumor cells are polygonal with eosinophilic cytoplasm, ovoid hyperchromatic or vesicular nuclei, and prominent nucleoli. Mitoses are common. The tubular lumina are filled with acantholytic cells, either singly or in small clusters, which may demonstrate residual bridging to tubular lining cells (Figure 2). The acantholytic cells show some variability in size and may be large, multinucleated, or keratinized. The tubules may contain material that is amorphous, basophilic, periodic acid–Schiff positive, diastase sensitive, and mucicarmine negative.5 Eccrine ducts at the periphery of the tumor may show reactive dilatation and proliferation. Tumor cells show positive immunostaining for epithelial membrane antigen, 34βE12, CK5/6, and tumor protein p63.6-8 There is negative immunostaining for carcinoembryonic antigen, amylase, S-100 protein, and factor VIII.5
|
The differential diagnosis includes adenoid BCC, angiosarcoma, eccrine carcinoma, and metastatic adenocarcinoma of the skin. In adenoid BCC, excess stromal mucin imparts pseudoglandular architecture (Figure 3). However, features of conventional BCC, including peripheral nuclear palisading and retraction artifact often are present as well.
Angiosarcoma shows slitlike vascular spaces lined by hyperchromatic endothelial cells (Figure 4). Further, there is positive immunostaining for vascular markers CD31 and CD34.
In eccrine carcinoma, there are invasive ductal structures lined by either a single or double layer of cells that may contain luminal material that is periodic acid–Schiff positive and diastase resistant (Figure 5).9 The tumor cells show positive immunostaining for cytokeratins, epithelial membrane antigen, carcinoembryonic antigen, and S-100 protein.10
Pseudoglandular SCC is susceptible to misdiagnosis as adenocarcinoma by sampling error if biopsies do not capture areas with typical features of SCC, including dysplastic squamous epithelium and keratinization. Metastatic adenocarcinoma of the skin is more likely to present with multiple nodules in older individuals. Lack of epidermal connection of the tumor and minimal to no acantholytic dyskeratosis further support cutaneous metastasis (Figure 6). Review of the patient’s clinical history might be helpful if adenocarcinoma was previously diagnosed. Immunohistochemical evaluation may aid in the prediction of the primary site in patients with metastatic adenocarcinoma of unknown origin.11
Squamous cell carcinoma (SCC) is the second most common form of skin cancer. Pseudoglandular SCC, also known as adenoid SCC or acantholytic SCC, is an uncommon variant that was first described by Lever1 in 1947 as an adenoacanthoma of the sweat glands. Of the many variants of SCC, pseudoglandular SCC generally is considered to behave aggressively with intermediate (3%–10%) risk for metastasis.2 The metastatic potential of pseudoglandular SCC may be conferred in part by diminished expression of intercellular adhesion molecules, including desmoglein 3, epithelial cadherin, and syn-decan 1.3,4 Pseudoglandular SCC presents most often on sun-damaged skin of elderly patients, especially the face and ears, as a pink or red nodule with central ulceration and a raised indurated border. It may be mistaken clinically for basal cell carcinoma (BCC) or keratoacanthoma.
On microscopic examination, the lesion is predominantly located in the dermis and may extend to the subcutis. There usually is connection to the overlying epidermis, which often shows hyperkeratosis and parakeratosis. Epidermal squamous dysplasia may be present. The dermis typically contains nests of squamous cells with a variable degree of central acantholysis. The morphology on low-power magnification consists of tubules of irregular size and shape, which are present either focally or throughout the lesion (Figure 1). The tubules are typically admixed with foci of keratinization. One or more layers of cohesive cells line the tubules. Partial keratinization may be found in the lining of tubules with more than 1 cell layer. The tumor cells are polygonal with eosinophilic cytoplasm, ovoid hyperchromatic or vesicular nuclei, and prominent nucleoli. Mitoses are common. The tubular lumina are filled with acantholytic cells, either singly or in small clusters, which may demonstrate residual bridging to tubular lining cells (Figure 2). The acantholytic cells show some variability in size and may be large, multinucleated, or keratinized. The tubules may contain material that is amorphous, basophilic, periodic acid–Schiff positive, diastase sensitive, and mucicarmine negative.5 Eccrine ducts at the periphery of the tumor may show reactive dilatation and proliferation. Tumor cells show positive immunostaining for epithelial membrane antigen, 34βE12, CK5/6, and tumor protein p63.6-8 There is negative immunostaining for carcinoembryonic antigen, amylase, S-100 protein, and factor VIII.5
|
The differential diagnosis includes adenoid BCC, angiosarcoma, eccrine carcinoma, and metastatic adenocarcinoma of the skin. In adenoid BCC, excess stromal mucin imparts pseudoglandular architecture (Figure 3). However, features of conventional BCC, including peripheral nuclear palisading and retraction artifact often are present as well.
Angiosarcoma shows slitlike vascular spaces lined by hyperchromatic endothelial cells (Figure 4). Further, there is positive immunostaining for vascular markers CD31 and CD34.
In eccrine carcinoma, there are invasive ductal structures lined by either a single or double layer of cells that may contain luminal material that is periodic acid–Schiff positive and diastase resistant (Figure 5).9 The tumor cells show positive immunostaining for cytokeratins, epithelial membrane antigen, carcinoembryonic antigen, and S-100 protein.10
Pseudoglandular SCC is susceptible to misdiagnosis as adenocarcinoma by sampling error if biopsies do not capture areas with typical features of SCC, including dysplastic squamous epithelium and keratinization. Metastatic adenocarcinoma of the skin is more likely to present with multiple nodules in older individuals. Lack of epidermal connection of the tumor and minimal to no acantholytic dyskeratosis further support cutaneous metastasis (Figure 6). Review of the patient’s clinical history might be helpful if adenocarcinoma was previously diagnosed. Immunohistochemical evaluation may aid in the prediction of the primary site in patients with metastatic adenocarcinoma of unknown origin.11
1. Lever WF. Adenocanthoma of sweat glands; carcinoma of sweat glands with glandular and epidermal elements: report of four cases. Arch Derm Syphilol. 1947;56:157-171.
2. Bonerandi JJ, Beauvillain C, Caquant L, et al. Guidelines for the diagnosis and treatment of cutaneous squamous cell carcinoma and precursor lesions. J Eur Acad Dermatol Venereol. 2011;25(suppl 5):1-51.
3. Griffin JR, Wriston CC, Peters MS, et al. Decreased expression of intercellular adhesion molecules in acantholytic squamous cell carcinoma compared with invasive well-differentiated squamous cell carcinoma of the skin. Am J Clin Pathol. 2013;139:442-447.
4. Bayer-Garner IB, Smoller BR. The expression of syndecan-1 is preferentially reduced compared with that of E-cadherin in acantholytic squamous cell carcinoma. J Cutan Pathol. 2001;28:83-89.
5. Nappi O, Pettinato G, Wick MR. Adenoid (acantholytic) squamous cell carcinoma of the skin. J Cutan Pathol. 1989;16:114-121.
6. Sajin M, Hodorogea Prisăcaru A, Luchian MC, et al. Acantholytic squamous cell carcinoma: pathological study of nine cases with review of literature. Rom J Morphol Embryol. 2014;55:279-283.
7. Gray Y, Robidoux HJ, Farrell DS, et al. Squamous cell carcinoma detected by high-molecular-weight cytokeratin immunostaining mimicking atypical fibroxanthoma. Arch Pathol Lab Med. 2001;125:799-802.
8. Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
9. Plaza JA, Prieto VG. Neoplastic Lesions of the Skin. New York, NY: Demos Medical Publishing; 2014.
10. Swanson PE, Cherwitz DL, Neumann MP, et al. Eccrine sweat gland carcinoma: an histologic and immunohistochemical study of 32 cases. J Cutan Pathol. 1987;14:65-86.
11. Dennis JL, Hvidsten TR, Wit EC, et al. Markers of adenocarcinoma characteristic of the site of origin: development of a diagnostic algorithm. Clin Cancer Res. 2005;11:3766-3772.
1. Lever WF. Adenocanthoma of sweat glands; carcinoma of sweat glands with glandular and epidermal elements: report of four cases. Arch Derm Syphilol. 1947;56:157-171.
2. Bonerandi JJ, Beauvillain C, Caquant L, et al. Guidelines for the diagnosis and treatment of cutaneous squamous cell carcinoma and precursor lesions. J Eur Acad Dermatol Venereol. 2011;25(suppl 5):1-51.
3. Griffin JR, Wriston CC, Peters MS, et al. Decreased expression of intercellular adhesion molecules in acantholytic squamous cell carcinoma compared with invasive well-differentiated squamous cell carcinoma of the skin. Am J Clin Pathol. 2013;139:442-447.
4. Bayer-Garner IB, Smoller BR. The expression of syndecan-1 is preferentially reduced compared with that of E-cadherin in acantholytic squamous cell carcinoma. J Cutan Pathol. 2001;28:83-89.
5. Nappi O, Pettinato G, Wick MR. Adenoid (acantholytic) squamous cell carcinoma of the skin. J Cutan Pathol. 1989;16:114-121.
6. Sajin M, Hodorogea Prisăcaru A, Luchian MC, et al. Acantholytic squamous cell carcinoma: pathological study of nine cases with review of literature. Rom J Morphol Embryol. 2014;55:279-283.
7. Gray Y, Robidoux HJ, Farrell DS, et al. Squamous cell carcinoma detected by high-molecular-weight cytokeratin immunostaining mimicking atypical fibroxanthoma. Arch Pathol Lab Med. 2001;125:799-802.
8. Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
9. Plaza JA, Prieto VG. Neoplastic Lesions of the Skin. New York, NY: Demos Medical Publishing; 2014.
10. Swanson PE, Cherwitz DL, Neumann MP, et al. Eccrine sweat gland carcinoma: an histologic and immunohistochemical study of 32 cases. J Cutan Pathol. 1987;14:65-86.
11. Dennis JL, Hvidsten TR, Wit EC, et al. Markers of adenocarcinoma characteristic of the site of origin: development of a diagnostic algorithm. Clin Cancer Res. 2005;11:3766-3772.
What Is Your Diagnosis? Acquired Lymphangiectasia
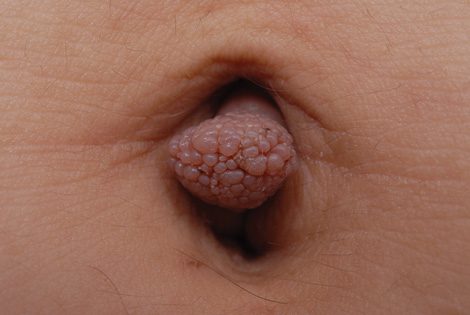
A 19-year-old woman presented with an umbilical mass of 5 months’ duration that had grown in size. Physical examination revealed a 1×1-cm brownish, pedunculated, cauliflower-shaped lesion on the umbilicus. There were no other signs or symptoms of disease. The patient’s personal and family disease history were unremarkable. An excisional biopsy was performed.
The Diagnosis: Acquired Lymphangiectasia
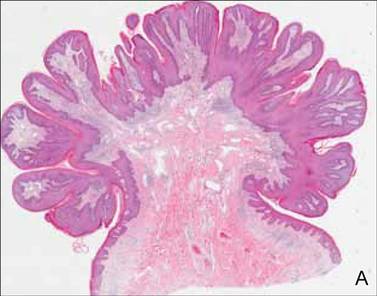
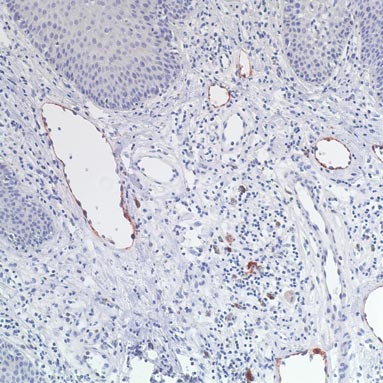
On histopathology numerous dilated channels lined by a single flat layer of endothelial cells were noted within the dermis. The overlying epidermis was papillomatous and acanthotic (Figure 1). The endothelial cells lining the dilated channels were D2-40 positive (Figure 2). Furthermore, the channels contained a pinkish amorphous material and a few red blood cells. The surrounding stroma showed scattered lymphocyte infiltration. These findings were consistent with lymphangiectasia. The lesion has not recurred 4 years following total excision.
|

Acquired lymphangiectasia is known by various names, including lymphangioma, acquired lymphangioma, and acquired lymphangioma circumscriptum, which has led to confusion.1 Acquired lymphangiectasia, which is characterized by dilated superficial lymphatics, develops following damage to previously normal lymphatic channels, leading to a buildup of lymph pressure and backflow.2 Acquired lymphangiectasia has been reported as clinically and histologically indistinguishable from lymphangioma circumscriptum2; however, unlike in lymphangiectasia, the suffix -oma denotes a tumor. Our case matched more closely with the typical concept of lymphangiectasia rather than lymphangioma.
Clinical findings of acquired lymphangiectasia usually include translucent, flat or slightly raised, 2- to 5-mm, flesh-colored papules and vesicles.3,4 Acquired lymphangiectasia has been described with lesions that have verrucous surfaces mimicking warts, condyloma acuminata, or molluscum contagiosum.5,6 Our case suggests that acquired lymphangiectasia also can present with a pedunculated cauliflowerlike appearance. In general, it develops secondary to certain conditions such as recovery from trauma or surgery, postsurgical fibrosis, and irradiation. Lymphangiectasia often is seen on the arms, axillae, chest wall, and genital area in women and the scrotum, penis, thighs, and pubic region in men, both who have undergone radical surgery and irradiation for treatment of breast and prostate cancer, respectively.3 Our patient did not report any history of trauma to the umbilicus.
On histopathology acquired lymphangiectasia typically shows edematous polypoid nodules with dilated lymphatics. The overlying epidermis usually shows a spectrum of proliferation ranging from mild acanthosis to florid pseudoepitheliomatous hyperplasia with marked hyperkeratosis and parakeratosis. The distinctive finding of lymphangiectasia is the presence of dilated lymphatic spaces within the dermis. The dilated channels are filled with lymphatic fluid and often red and white blood cells. The single layer of flattened endothelial cells generally exhibits immunoreactivity to D2-40 and CD31.1
Treatment of lymphangiectasia is focused on reducing the pressure within the lymph vessels and managing consequent lymphedema with compression dressings. Simple surgical excision of lesions on sites such as the vulva or legs often is effective.3 If surgical intervention is not an option, cryotherapy, sclerotherapy, cauterization, and treatment with CO2 lasers also have been utilized with good outcomes.7 In the current case, total surgical excision was performed, which provided good results.
1. Stewart CJ, Chan T, Platten M. Acquired lymphangiectasia (‘lymphangioma circumscriptum’) of the vulva: a report of eight cases. Pathology. 2009;41:448-453.
2. Celis AV, Gaughf CN, Sangueza OP, et al. Acquired lymphangiectasis. South Med J. 1999;92:69-72.
3. Verma SB. Lymphangiectasias of the skin: victims of confusing nomenclature. Clin Exp Dermatol. 2009;34:566-569.
4. Mortimer PS. Disorder of lymphatic vessels. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. Vol 3. 8th ed. Hoboken, NJ: Wiley-Blackwell; 2010:48.28-48.29.
5. Sharma R, Tomar S, Chandra M. Acquired vulval lymphangiectases mimicking genital warts. Indian J Dermatol Venereol Leprol. 2002;68:166-167.
6. Horn LC, Kühndel K, Pawlowitsch T, et al. Acquired lymphangioma circumscriptum of the vulva mimicking genital warts. Eur J Obstet Gynecol Reprod Biol. 2005;123:118-120.
7. Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295.
A 19-year-old woman presented with an umbilical mass of 5 months’ duration that had grown in size. Physical examination revealed a 1×1-cm brownish, pedunculated, cauliflower-shaped lesion on the umbilicus. There were no other signs or symptoms of disease. The patient’s personal and family disease history were unremarkable. An excisional biopsy was performed.
The Diagnosis: Acquired Lymphangiectasia
On histopathology numerous dilated channels lined by a single flat layer of endothelial cells were noted within the dermis. The overlying epidermis was papillomatous and acanthotic (Figure 1). The endothelial cells lining the dilated channels were D2-40 positive (Figure 2). Furthermore, the channels contained a pinkish amorphous material and a few red blood cells. The surrounding stroma showed scattered lymphocyte infiltration. These findings were consistent with lymphangiectasia. The lesion has not recurred 4 years following total excision.
|
Acquired lymphangiectasia is known by various names, including lymphangioma, acquired lymphangioma, and acquired lymphangioma circumscriptum, which has led to confusion.1 Acquired lymphangiectasia, which is characterized by dilated superficial lymphatics, develops following damage to previously normal lymphatic channels, leading to a buildup of lymph pressure and backflow.2 Acquired lymphangiectasia has been reported as clinically and histologically indistinguishable from lymphangioma circumscriptum2; however, unlike in lymphangiectasia, the suffix -oma denotes a tumor. Our case matched more closely with the typical concept of lymphangiectasia rather than lymphangioma.
Clinical findings of acquired lymphangiectasia usually include translucent, flat or slightly raised, 2- to 5-mm, flesh-colored papules and vesicles.3,4 Acquired lymphangiectasia has been described with lesions that have verrucous surfaces mimicking warts, condyloma acuminata, or molluscum contagiosum.5,6 Our case suggests that acquired lymphangiectasia also can present with a pedunculated cauliflowerlike appearance. In general, it develops secondary to certain conditions such as recovery from trauma or surgery, postsurgical fibrosis, and irradiation. Lymphangiectasia often is seen on the arms, axillae, chest wall, and genital area in women and the scrotum, penis, thighs, and pubic region in men, both who have undergone radical surgery and irradiation for treatment of breast and prostate cancer, respectively.3 Our patient did not report any history of trauma to the umbilicus.
On histopathology acquired lymphangiectasia typically shows edematous polypoid nodules with dilated lymphatics. The overlying epidermis usually shows a spectrum of proliferation ranging from mild acanthosis to florid pseudoepitheliomatous hyperplasia with marked hyperkeratosis and parakeratosis. The distinctive finding of lymphangiectasia is the presence of dilated lymphatic spaces within the dermis. The dilated channels are filled with lymphatic fluid and often red and white blood cells. The single layer of flattened endothelial cells generally exhibits immunoreactivity to D2-40 and CD31.1
Treatment of lymphangiectasia is focused on reducing the pressure within the lymph vessels and managing consequent lymphedema with compression dressings. Simple surgical excision of lesions on sites such as the vulva or legs often is effective.3 If surgical intervention is not an option, cryotherapy, sclerotherapy, cauterization, and treatment with CO2 lasers also have been utilized with good outcomes.7 In the current case, total surgical excision was performed, which provided good results.
A 19-year-old woman presented with an umbilical mass of 5 months’ duration that had grown in size. Physical examination revealed a 1×1-cm brownish, pedunculated, cauliflower-shaped lesion on the umbilicus. There were no other signs or symptoms of disease. The patient’s personal and family disease history were unremarkable. An excisional biopsy was performed.
The Diagnosis: Acquired Lymphangiectasia
On histopathology numerous dilated channels lined by a single flat layer of endothelial cells were noted within the dermis. The overlying epidermis was papillomatous and acanthotic (Figure 1). The endothelial cells lining the dilated channels were D2-40 positive (Figure 2). Furthermore, the channels contained a pinkish amorphous material and a few red blood cells. The surrounding stroma showed scattered lymphocyte infiltration. These findings were consistent with lymphangiectasia. The lesion has not recurred 4 years following total excision.
|
Acquired lymphangiectasia is known by various names, including lymphangioma, acquired lymphangioma, and acquired lymphangioma circumscriptum, which has led to confusion.1 Acquired lymphangiectasia, which is characterized by dilated superficial lymphatics, develops following damage to previously normal lymphatic channels, leading to a buildup of lymph pressure and backflow.2 Acquired lymphangiectasia has been reported as clinically and histologically indistinguishable from lymphangioma circumscriptum2; however, unlike in lymphangiectasia, the suffix -oma denotes a tumor. Our case matched more closely with the typical concept of lymphangiectasia rather than lymphangioma.
Clinical findings of acquired lymphangiectasia usually include translucent, flat or slightly raised, 2- to 5-mm, flesh-colored papules and vesicles.3,4 Acquired lymphangiectasia has been described with lesions that have verrucous surfaces mimicking warts, condyloma acuminata, or molluscum contagiosum.5,6 Our case suggests that acquired lymphangiectasia also can present with a pedunculated cauliflowerlike appearance. In general, it develops secondary to certain conditions such as recovery from trauma or surgery, postsurgical fibrosis, and irradiation. Lymphangiectasia often is seen on the arms, axillae, chest wall, and genital area in women and the scrotum, penis, thighs, and pubic region in men, both who have undergone radical surgery and irradiation for treatment of breast and prostate cancer, respectively.3 Our patient did not report any history of trauma to the umbilicus.
On histopathology acquired lymphangiectasia typically shows edematous polypoid nodules with dilated lymphatics. The overlying epidermis usually shows a spectrum of proliferation ranging from mild acanthosis to florid pseudoepitheliomatous hyperplasia with marked hyperkeratosis and parakeratosis. The distinctive finding of lymphangiectasia is the presence of dilated lymphatic spaces within the dermis. The dilated channels are filled with lymphatic fluid and often red and white blood cells. The single layer of flattened endothelial cells generally exhibits immunoreactivity to D2-40 and CD31.1
Treatment of lymphangiectasia is focused on reducing the pressure within the lymph vessels and managing consequent lymphedema with compression dressings. Simple surgical excision of lesions on sites such as the vulva or legs often is effective.3 If surgical intervention is not an option, cryotherapy, sclerotherapy, cauterization, and treatment with CO2 lasers also have been utilized with good outcomes.7 In the current case, total surgical excision was performed, which provided good results.
1. Stewart CJ, Chan T, Platten M. Acquired lymphangiectasia (‘lymphangioma circumscriptum’) of the vulva: a report of eight cases. Pathology. 2009;41:448-453.
2. Celis AV, Gaughf CN, Sangueza OP, et al. Acquired lymphangiectasis. South Med J. 1999;92:69-72.
3. Verma SB. Lymphangiectasias of the skin: victims of confusing nomenclature. Clin Exp Dermatol. 2009;34:566-569.
4. Mortimer PS. Disorder of lymphatic vessels. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. Vol 3. 8th ed. Hoboken, NJ: Wiley-Blackwell; 2010:48.28-48.29.
5. Sharma R, Tomar S, Chandra M. Acquired vulval lymphangiectases mimicking genital warts. Indian J Dermatol Venereol Leprol. 2002;68:166-167.
6. Horn LC, Kühndel K, Pawlowitsch T, et al. Acquired lymphangioma circumscriptum of the vulva mimicking genital warts. Eur J Obstet Gynecol Reprod Biol. 2005;123:118-120.
7. Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295.
1. Stewart CJ, Chan T, Platten M. Acquired lymphangiectasia (‘lymphangioma circumscriptum’) of the vulva: a report of eight cases. Pathology. 2009;41:448-453.
2. Celis AV, Gaughf CN, Sangueza OP, et al. Acquired lymphangiectasis. South Med J. 1999;92:69-72.
3. Verma SB. Lymphangiectasias of the skin: victims of confusing nomenclature. Clin Exp Dermatol. 2009;34:566-569.
4. Mortimer PS. Disorder of lymphatic vessels. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. Vol 3. 8th ed. Hoboken, NJ: Wiley-Blackwell; 2010:48.28-48.29.
5. Sharma R, Tomar S, Chandra M. Acquired vulval lymphangiectases mimicking genital warts. Indian J Dermatol Venereol Leprol. 2002;68:166-167.
6. Horn LC, Kühndel K, Pawlowitsch T, et al. Acquired lymphangioma circumscriptum of the vulva mimicking genital warts. Eur J Obstet Gynecol Reprod Biol. 2005;123:118-120.
7. Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295.

Angioimmunoblastic T-cell Lymphoma Presenting as Purpura Fulminans
Purpura fulminans is a hematologic emergency, with clinical skin necrosis and laboratory testing showing disseminated intravascular coagulation. The thrombotic occlusion usually affects small and medium-sized blood vessels and may involve any organ. Purpura fulminans has been implicated with sepsis, most commonly meningococcal infections; other infections such as Staphylococcus aureus, groups A and B β-hemolytic streptococci, Streptococcus pneumoniae, and Haemophilus influenzae; and as a sequela to benign childhood infections, such as varicella. Other associations with purpura fulminans include autoimmune disease and heritable or acquired deficiency of anticoagulant proteins, most commonly protein C. We present a rare case of purpura fulminans as the presenting sign of angioimmunoblastic T-cell lymphoma (AITL), an aggressive primary nodal peripheral T-cell lymphoma with a high mortality rate and nonspecific skin manifestations in roughly half of all patients involved.
Case Report
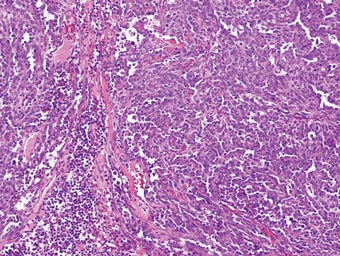
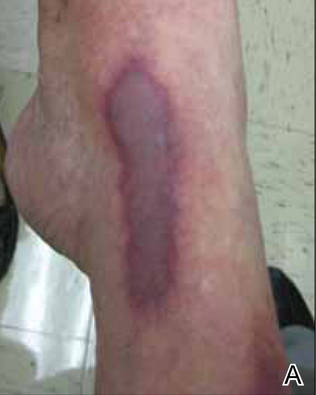
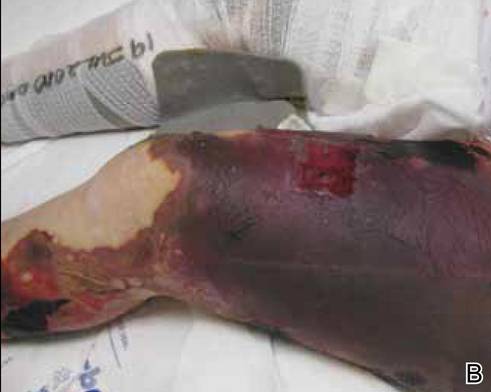
A 56-year-old woman presented with purpuric patches on the left foot (Figure 1A). Seven days after presentation the lesion progressed into ecchymotic geographic plaques and hemorrhagic bullae that spread upward and contralaterally, sparing the digits, trunk, head, neck, and mucous membranes. Ultimately, the involved skin became necrotic and involved 20% of the body surface area (Figure 1B). The lesions were painful with a burning sensation but were not pruritic. The patient also reported intermittent fevers, chills, myalgia, nausea, and shortness of breath. Enlarged lymph nodes were present in the right cervical chain. She denied new medications; stated she had been in good health prior to this episode; and had no history of spontaneous abortion, neurologic symptoms, or other serious illness.
|
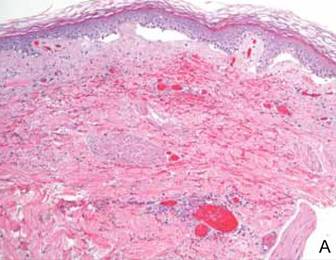
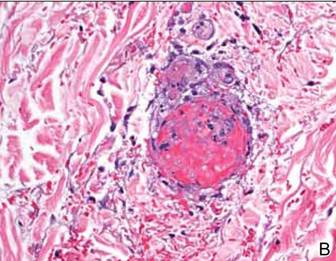
Computed tomography showed prominent diffuse mediastinal, mesenteric, retroperitoneal, and pelvic lymphadenopathy with involvement of the cervical and inguinal areas. Laboratory values showed thrombocytopenia and increased fibrin degradation products. Blood and tissue cultures were negative; the patient also had a negative viral serology, except for Epstein-Barr virus IgG titers (>1:2560). A skin biopsy of the left thigh demonstrated venules and capillaries in the mid and superficial dermis filled with fibrin thrombi without vasculitis (Figure 2). A lymph node biopsy was consistent with a diagnosis of AITL. The lymph node architecture was largely effaced by a polymorphous lymphoid infiltrate that predominantly expanded into paracortical areas and was associated with a prominent arborizing vascular proliferation. The infiltrate was composed of lymphocytes ranging in size from small to medium, with ample cytoplasm, coarsely clumped chromatin, and mildly irregular nuclear membranes. Large atypical lymphocytes with features of immunoblasts were easily identified. An associated inflammatory background composed of eosinophils, plasma cells, and histiocytes was present (Figure 3). The atypical lymphocytes stained positive for CD3and CD10 on immunohistochemistry. Additionally, a subset of large immunoblastlike lymphocytes was positive for Epstein-Barr–encoded small RNAs by in situ hybridization.
|
The patient was started on rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone. She received 2 cycles with positive response based on subsequent computed tomography and positron emission tomography scans that showed regression of her disease as well as the lack of formation of new skin lesions. She was transferred to a burn unit where she had continuing treatment and skin grafts. Despite 2 cycles of chemotherapy, broad-spectrum antibiotics, and daily wound care management, the patient died secondary to sepsis 6 months after presentation.
Comment
Angioimmunoblastic T-cell lymphoma is a primary nodal lymphoma with occasional cutaneous involvement. Cutaneous manifestations occur in roughly half of all patients with AITL1 and have mainly been described as erythematous macules and papules that can resemble a viral exanthem or a drug reaction.2 However, other skin manifestations include urticaria, papulovesicular lesions, nodules, erythroderma,3 and to a lesser degree purpura.4 The lesions have been noted to occur prior to, concurrent with, or anytime during the disease.3,5,6 This aggressive lymphoma has mortality rates ranging from 50% to 72%, and median survival ranges from 11 to 30 months.6
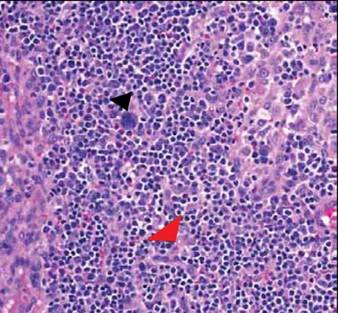
To arrive at the correct diagnosis of AITL, a nodal biopsy with immunochemistry is necessary. Classic findings on histopathology include effacement of normal architecture, marked vascular proliferation, and aggregates of atypical lymphoid cells. CD10 has been shown to be a good objective criterion for the diagnosis of AITL,4 with characteristic tumor cells expressing CD10. Nodal Epstein-Barr virus–positive lymphocytes often are present.2 Other T-cell lymphomas with primarily nodal presentation along with peripheral T-cell lymphoma include peripheral T-cell lymphoma unspecified type and anaplastic large cell lymphoma, according to the World Health Organization classification.7 Anaplastic large cell lymphoma is easily distinguished from AITL based on histopathology, immunostaining, and clinical presentation. Until recently, peripheral T-cell lymphoma unspecified type and reactive lymphoid hyperplasia presented a challenge to differentiate from AITL, especially in the early phases of the disease; however, the introduction of CD10 as a phenotypic marker has been instrumental in distinguishing AITL from other T-cell lymphomas with primary nodal involvement.1,4
The development of purpura fulminans and disseminated intravascular coagulation in a patient with AITL is rare. Although the exact mechanism for the thrombus formation in the skin has not been elucidated, purpura fulminans typically develops secondary to a severe infection. The exact incidence of purpura fulminans in the setting of AITL is unknown, but purpura as a cutaneous eruption has been associated as a clinical finding in AITL.6 Although our case may be a rare presentation of AITL, a prompt and accurate diagnosis can drastically change the prognosis of this aggressive disease.
1. Ferry JA. Angioimmunoblastic T-cell lymphoma. Adv Anat Pathol. 2002;9:273-279.
2. Brown HA, Macon WR, Kurtin PJ, et al. Cutaneous involvement by angioimmunoblastic T-cell lymphoma with remarkable heterogeneous Epstein-Barr virus expression. J Cutan Pathol. 2001;28:432-438.
3. Bernstein JE, Soltani K, Lorincz AL. Cutaneous manifestations of angioimmunoblastic lymphadenopathy. J Am Acad Dermatol. 1979;1:227-232.
4. Attygalle A, Al-Jehani R, Diss TC, et al. Neoplastic T cells in angioimmunoblastic T-cell lymphoma express CD10. Blood. 2002;99:627-633.
5. Jayaramna AG, Cassarino D, Advani R, et al. Cutaneous involvement by angioimmunoblastic T-cell lymphoma: a unique histologic presentation, mimicking an infectious etiology. J Cutan Pathol. 2006;33(suppl 2):6-11.
6. Martel P, Laroche L, Courville P, et al. Cutaneous involvement in patients with angioimmunoblastic lymphadenopathy with dysproteinemia: a clinical, immunohistological, and molecular analysis. Archives of Dermatology. 2000;136:881-886.
7. Jaffe ES, Harris NL, Stein H, et al, eds. Tumours of Haematopoietic and Lymphoid Tissues. 1st ed. Bethesda, MD: International Agency for Research on Cancer; 2001.
Purpura fulminans is a hematologic emergency, with clinical skin necrosis and laboratory testing showing disseminated intravascular coagulation. The thrombotic occlusion usually affects small and medium-sized blood vessels and may involve any organ. Purpura fulminans has been implicated with sepsis, most commonly meningococcal infections; other infections such as Staphylococcus aureus, groups A and B β-hemolytic streptococci, Streptococcus pneumoniae, and Haemophilus influenzae; and as a sequela to benign childhood infections, such as varicella. Other associations with purpura fulminans include autoimmune disease and heritable or acquired deficiency of anticoagulant proteins, most commonly protein C. We present a rare case of purpura fulminans as the presenting sign of angioimmunoblastic T-cell lymphoma (AITL), an aggressive primary nodal peripheral T-cell lymphoma with a high mortality rate and nonspecific skin manifestations in roughly half of all patients involved.
Case Report
A 56-year-old woman presented with purpuric patches on the left foot (Figure 1A). Seven days after presentation the lesion progressed into ecchymotic geographic plaques and hemorrhagic bullae that spread upward and contralaterally, sparing the digits, trunk, head, neck, and mucous membranes. Ultimately, the involved skin became necrotic and involved 20% of the body surface area (Figure 1B). The lesions were painful with a burning sensation but were not pruritic. The patient also reported intermittent fevers, chills, myalgia, nausea, and shortness of breath. Enlarged lymph nodes were present in the right cervical chain. She denied new medications; stated she had been in good health prior to this episode; and had no history of spontaneous abortion, neurologic symptoms, or other serious illness.
|
|
Computed tomography showed prominent diffuse mediastinal, mesenteric, retroperitoneal, and pelvic lymphadenopathy with involvement of the cervical and inguinal areas. Laboratory values showed thrombocytopenia and increased fibrin degradation products. Blood and tissue cultures were negative; the patient also had a negative viral serology, except for Epstein-Barr virus IgG titers (>1:2560). A skin biopsy of the left thigh demonstrated venules and capillaries in the mid and superficial dermis filled with fibrin thrombi without vasculitis (Figure 2). A lymph node biopsy was consistent with a diagnosis of AITL. The lymph node architecture was largely effaced by a polymorphous lymphoid infiltrate that predominantly expanded into paracortical areas and was associated with a prominent arborizing vascular proliferation. The infiltrate was composed of lymphocytes ranging in size from small to medium, with ample cytoplasm, coarsely clumped chromatin, and mildly irregular nuclear membranes. Large atypical lymphocytes with features of immunoblasts were easily identified. An associated inflammatory background composed of eosinophils, plasma cells, and histiocytes was present (Figure 3). The atypical lymphocytes stained positive for CD3and CD10 on immunohistochemistry. Additionally, a subset of large immunoblastlike lymphocytes was positive for Epstein-Barr–encoded small RNAs by in situ hybridization.
|
The patient was started on rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone. She received 2 cycles with positive response based on subsequent computed tomography and positron emission tomography scans that showed regression of her disease as well as the lack of formation of new skin lesions. She was transferred to a burn unit where she had continuing treatment and skin grafts. Despite 2 cycles of chemotherapy, broad-spectrum antibiotics, and daily wound care management, the patient died secondary to sepsis 6 months after presentation.
Comment
Angioimmunoblastic T-cell lymphoma is a primary nodal lymphoma with occasional cutaneous involvement. Cutaneous manifestations occur in roughly half of all patients with AITL1 and have mainly been described as erythematous macules and papules that can resemble a viral exanthem or a drug reaction.2 However, other skin manifestations include urticaria, papulovesicular lesions, nodules, erythroderma,3 and to a lesser degree purpura.4 The lesions have been noted to occur prior to, concurrent with, or anytime during the disease.3,5,6 This aggressive lymphoma has mortality rates ranging from 50% to 72%, and median survival ranges from 11 to 30 months.6
To arrive at the correct diagnosis of AITL, a nodal biopsy with immunochemistry is necessary. Classic findings on histopathology include effacement of normal architecture, marked vascular proliferation, and aggregates of atypical lymphoid cells. CD10 has been shown to be a good objective criterion for the diagnosis of AITL,4 with characteristic tumor cells expressing CD10. Nodal Epstein-Barr virus–positive lymphocytes often are present.2 Other T-cell lymphomas with primarily nodal presentation along with peripheral T-cell lymphoma include peripheral T-cell lymphoma unspecified type and anaplastic large cell lymphoma, according to the World Health Organization classification.7 Anaplastic large cell lymphoma is easily distinguished from AITL based on histopathology, immunostaining, and clinical presentation. Until recently, peripheral T-cell lymphoma unspecified type and reactive lymphoid hyperplasia presented a challenge to differentiate from AITL, especially in the early phases of the disease; however, the introduction of CD10 as a phenotypic marker has been instrumental in distinguishing AITL from other T-cell lymphomas with primary nodal involvement.1,4
The development of purpura fulminans and disseminated intravascular coagulation in a patient with AITL is rare. Although the exact mechanism for the thrombus formation in the skin has not been elucidated, purpura fulminans typically develops secondary to a severe infection. The exact incidence of purpura fulminans in the setting of AITL is unknown, but purpura as a cutaneous eruption has been associated as a clinical finding in AITL.6 Although our case may be a rare presentation of AITL, a prompt and accurate diagnosis can drastically change the prognosis of this aggressive disease.
Purpura fulminans is a hematologic emergency, with clinical skin necrosis and laboratory testing showing disseminated intravascular coagulation. The thrombotic occlusion usually affects small and medium-sized blood vessels and may involve any organ. Purpura fulminans has been implicated with sepsis, most commonly meningococcal infections; other infections such as Staphylococcus aureus, groups A and B β-hemolytic streptococci, Streptococcus pneumoniae, and Haemophilus influenzae; and as a sequela to benign childhood infections, such as varicella. Other associations with purpura fulminans include autoimmune disease and heritable or acquired deficiency of anticoagulant proteins, most commonly protein C. We present a rare case of purpura fulminans as the presenting sign of angioimmunoblastic T-cell lymphoma (AITL), an aggressive primary nodal peripheral T-cell lymphoma with a high mortality rate and nonspecific skin manifestations in roughly half of all patients involved.
Case Report
A 56-year-old woman presented with purpuric patches on the left foot (Figure 1A). Seven days after presentation the lesion progressed into ecchymotic geographic plaques and hemorrhagic bullae that spread upward and contralaterally, sparing the digits, trunk, head, neck, and mucous membranes. Ultimately, the involved skin became necrotic and involved 20% of the body surface area (Figure 1B). The lesions were painful with a burning sensation but were not pruritic. The patient also reported intermittent fevers, chills, myalgia, nausea, and shortness of breath. Enlarged lymph nodes were present in the right cervical chain. She denied new medications; stated she had been in good health prior to this episode; and had no history of spontaneous abortion, neurologic symptoms, or other serious illness.
|
|
Computed tomography showed prominent diffuse mediastinal, mesenteric, retroperitoneal, and pelvic lymphadenopathy with involvement of the cervical and inguinal areas. Laboratory values showed thrombocytopenia and increased fibrin degradation products. Blood and tissue cultures were negative; the patient also had a negative viral serology, except for Epstein-Barr virus IgG titers (>1:2560). A skin biopsy of the left thigh demonstrated venules and capillaries in the mid and superficial dermis filled with fibrin thrombi without vasculitis (Figure 2). A lymph node biopsy was consistent with a diagnosis of AITL. The lymph node architecture was largely effaced by a polymorphous lymphoid infiltrate that predominantly expanded into paracortical areas and was associated with a prominent arborizing vascular proliferation. The infiltrate was composed of lymphocytes ranging in size from small to medium, with ample cytoplasm, coarsely clumped chromatin, and mildly irregular nuclear membranes. Large atypical lymphocytes with features of immunoblasts were easily identified. An associated inflammatory background composed of eosinophils, plasma cells, and histiocytes was present (Figure 3). The atypical lymphocytes stained positive for CD3and CD10 on immunohistochemistry. Additionally, a subset of large immunoblastlike lymphocytes was positive for Epstein-Barr–encoded small RNAs by in situ hybridization.
|
The patient was started on rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone. She received 2 cycles with positive response based on subsequent computed tomography and positron emission tomography scans that showed regression of her disease as well as the lack of formation of new skin lesions. She was transferred to a burn unit where she had continuing treatment and skin grafts. Despite 2 cycles of chemotherapy, broad-spectrum antibiotics, and daily wound care management, the patient died secondary to sepsis 6 months after presentation.
Comment
Angioimmunoblastic T-cell lymphoma is a primary nodal lymphoma with occasional cutaneous involvement. Cutaneous manifestations occur in roughly half of all patients with AITL1 and have mainly been described as erythematous macules and papules that can resemble a viral exanthem or a drug reaction.2 However, other skin manifestations include urticaria, papulovesicular lesions, nodules, erythroderma,3 and to a lesser degree purpura.4 The lesions have been noted to occur prior to, concurrent with, or anytime during the disease.3,5,6 This aggressive lymphoma has mortality rates ranging from 50% to 72%, and median survival ranges from 11 to 30 months.6
To arrive at the correct diagnosis of AITL, a nodal biopsy with immunochemistry is necessary. Classic findings on histopathology include effacement of normal architecture, marked vascular proliferation, and aggregates of atypical lymphoid cells. CD10 has been shown to be a good objective criterion for the diagnosis of AITL,4 with characteristic tumor cells expressing CD10. Nodal Epstein-Barr virus–positive lymphocytes often are present.2 Other T-cell lymphomas with primarily nodal presentation along with peripheral T-cell lymphoma include peripheral T-cell lymphoma unspecified type and anaplastic large cell lymphoma, according to the World Health Organization classification.7 Anaplastic large cell lymphoma is easily distinguished from AITL based on histopathology, immunostaining, and clinical presentation. Until recently, peripheral T-cell lymphoma unspecified type and reactive lymphoid hyperplasia presented a challenge to differentiate from AITL, especially in the early phases of the disease; however, the introduction of CD10 as a phenotypic marker has been instrumental in distinguishing AITL from other T-cell lymphomas with primary nodal involvement.1,4
The development of purpura fulminans and disseminated intravascular coagulation in a patient with AITL is rare. Although the exact mechanism for the thrombus formation in the skin has not been elucidated, purpura fulminans typically develops secondary to a severe infection. The exact incidence of purpura fulminans in the setting of AITL is unknown, but purpura as a cutaneous eruption has been associated as a clinical finding in AITL.6 Although our case may be a rare presentation of AITL, a prompt and accurate diagnosis can drastically change the prognosis of this aggressive disease.
1. Ferry JA. Angioimmunoblastic T-cell lymphoma. Adv Anat Pathol. 2002;9:273-279.
2. Brown HA, Macon WR, Kurtin PJ, et al. Cutaneous involvement by angioimmunoblastic T-cell lymphoma with remarkable heterogeneous Epstein-Barr virus expression. J Cutan Pathol. 2001;28:432-438.
3. Bernstein JE, Soltani K, Lorincz AL. Cutaneous manifestations of angioimmunoblastic lymphadenopathy. J Am Acad Dermatol. 1979;1:227-232.
4. Attygalle A, Al-Jehani R, Diss TC, et al. Neoplastic T cells in angioimmunoblastic T-cell lymphoma express CD10. Blood. 2002;99:627-633.
5. Jayaramna AG, Cassarino D, Advani R, et al. Cutaneous involvement by angioimmunoblastic T-cell lymphoma: a unique histologic presentation, mimicking an infectious etiology. J Cutan Pathol. 2006;33(suppl 2):6-11.
6. Martel P, Laroche L, Courville P, et al. Cutaneous involvement in patients with angioimmunoblastic lymphadenopathy with dysproteinemia: a clinical, immunohistological, and molecular analysis. Archives of Dermatology. 2000;136:881-886.
7. Jaffe ES, Harris NL, Stein H, et al, eds. Tumours of Haematopoietic and Lymphoid Tissues. 1st ed. Bethesda, MD: International Agency for Research on Cancer; 2001.
1. Ferry JA. Angioimmunoblastic T-cell lymphoma. Adv Anat Pathol. 2002;9:273-279.
2. Brown HA, Macon WR, Kurtin PJ, et al. Cutaneous involvement by angioimmunoblastic T-cell lymphoma with remarkable heterogeneous Epstein-Barr virus expression. J Cutan Pathol. 2001;28:432-438.
3. Bernstein JE, Soltani K, Lorincz AL. Cutaneous manifestations of angioimmunoblastic lymphadenopathy. J Am Acad Dermatol. 1979;1:227-232.
4. Attygalle A, Al-Jehani R, Diss TC, et al. Neoplastic T cells in angioimmunoblastic T-cell lymphoma express CD10. Blood. 2002;99:627-633.
5. Jayaramna AG, Cassarino D, Advani R, et al. Cutaneous involvement by angioimmunoblastic T-cell lymphoma: a unique histologic presentation, mimicking an infectious etiology. J Cutan Pathol. 2006;33(suppl 2):6-11.
6. Martel P, Laroche L, Courville P, et al. Cutaneous involvement in patients with angioimmunoblastic lymphadenopathy with dysproteinemia: a clinical, immunohistological, and molecular analysis. Archives of Dermatology. 2000;136:881-886.
7. Jaffe ES, Harris NL, Stein H, et al, eds. Tumours of Haematopoietic and Lymphoid Tissues. 1st ed. Bethesda, MD: International Agency for Research on Cancer; 2001.
Practice Points
- Angioimmunoblastic T-cell lymphoma (AITL) is a primary nodal lymphoma with occasional nonspecific cutaneous involvement that may be morbilliform, maculopapular, erythrodermic, or rarely purpuric.
- To arrive at the correct diagnosis of AITL, a nodal biopsy with immunochemistry is necessary.
- CD10 positivity is a good objective criterion for the diagnosis of AITL, and Epstein-Barr virus–positive lymphocytes are nearly always present.
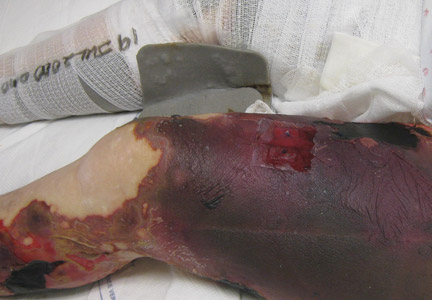
Epithelioid Sarcoma Resembling Benign Fibrous Histiocytoma
Epithelioid sarcoma (ES) is a rare malignant soft tissue neoplasm that is most often encountered on the distal extremities of young adults.1 Epithelioid sarcoma is notorious for its tendency to mimic palisading granulomatous processes such as granuloma annulare. We report a case of ES on the right hand of a 23-year-old man that resembled a benign fibrous histiocytoma (dermatofibroma) on incisional biopsy. The typical histopathologic features of ES were identified after amputation of the hand and evaluation of the deeper regions of the tumor. The tendency for ES to mimic granulomatous processes is a common diagnostic pitfall, but the potential for its close resemblance to benign fibrous histiocytoma is less recognized.
|
Case Report
A 23-year-old man presented with a nonhealing lesion on the right palm. His medical history was remarkable for a giant cell tumor of the tendon sheath involving the right fifth finger that had been treated via excision at an outside institution 2 years prior. Clinical examination revealed a 0.8×0.6-cm painful, firm, ulcerated dermal nodule with a hemorrhagic crust on the palmar surface of the right hand (Figure 1A). The clinical differential diagnosis included melanoma, traumatized verruca vulgaris, thrombosed pyogenic granuloma, and foreign body. A shave biopsy demonstrated verrucous epidermal hyperplasia, but the specimen did not include the dermis. Cultures of the lesion were positive for Staphylococcus aureus, and antibiotic therapy was initiated. In light of the clinical findings and the patient’s history of a giant cell tumor, imaging studies were performed. Magnetic resonance angiography showed abnormal masslike infiltrative enhancement throughout the soft tissues surrounding the right fifth metacarpal bone. The differential included a recurrent giant cell tumor, fibromatosis, and other soft tissue neoplasms.
After several missed appointments and surgery cancellations, the patient returned 4 months later for an incisional biopsy. Physical examination revealed a persistent palmar ulcer that had grown to 1.4×1 cm in size, along with an indurated purple plaque wrapping around the ulnar aspect of the right hand (Figure 1B). The biopsy demonstrated a proliferation of spindled and ovoid cells with scant cytoplasm that surrounded sclerotic collagen bundles resembling a dermatofibroma (Figure 2A). Cytologic atypia and mitotic activity were absent (Figure 2B). Glass slides of the original biopsy, which ultimately led to the diagnosis of the giant cell tumor of the tendon sheath more than 2 years earlier, were obtained and showed similar features. The proliferating cells were strongly and diffusely immunoreactive for vimentin, CD34, and cancer antigen 125 (CA 125). Scattered tumor cells strongly expressed cytokeratins (CKs) AE1/AE3 and cell adhesion molecule 5.2 (Figure 3). Staining for CD99 and epithelial membrane antigen was diffuse but weak. Factor XIIIa, S-100, CK7, smooth muscle actin, muscle-specific actin (HHF35), CD31, CD68, and B-cell lymphoma 2 were negative within the proliferating cells. Based on the clinical examination and results of the immunohistochemical staining, a diagnosis of ES was favored.
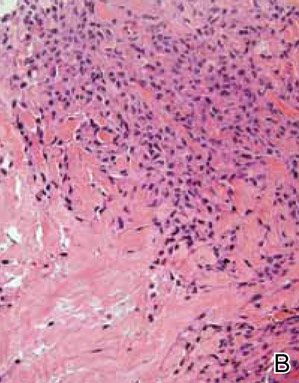
|
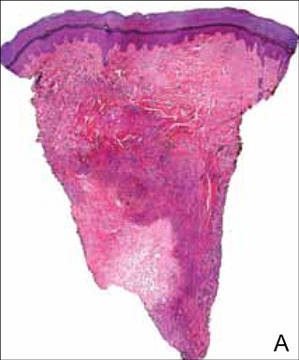
After a negative metastatic workup, amputation of the right hand was performed. The amputation specimen showed a tumor that extended through the entire hand with encasement of large vessels and tendons. Although the more superficial regions were cytologically bland, deep-seated regions of the tumor exhibited greater cellularity, nuclear pleomorphism, and mitotic activity (Figure 4). There was no bone involvement. Right axillary sentinel lymph nodes were negative for metastasis. Eighteen months later the patient developed chest and back pain with dyspnea. Thorascopic surgery was performed for a left pleural effusion and metastases to the left parietal pleura and adjacent soft tissue were identified. The patient was subsequently lost to follow-up.
Comment
First described by Enzinger1 in 1970, ES is a rare malignant soft tissue neoplasm that most frequently arises on the hands, forearms, and pretibial soft tissues of young adults.1-3 It is an aggressive tumor characterized by frequent recurrences and a high metastatic rate, with lung and regional lymph nodes being favored metastatic sites.1-5 Periods of several months or even years often pass between the initial presentation and establishment of a correct diagnosis, as ES frequently is mistaken for other benign conditions. The tendency for ES to mimic granulomatous processes is a common diagnostic pitfall, but the potential for its close resemblance to benign fibrous histiocytoma is less recognized.6,7 In his original series of 62 cases, Enzinger1 noted that 17 patients were referred for treatment with a diagnosis of a benign fibrohistiocytic neoplasm, and other reports have described a resemblance to fibrous and fibrohistiocytic neoplasms.8-11 Mirra et al10 designated these tumors as fibromalike variants of ES. Additional subtypes of ES have subsequently been recognized, including those described as angiomatoid or angiosarcomalike, reflecting the potential of ES to resemble vascular tumors.12 A proximal type of ES also has been described. This lesion presents as a deep-seated tumor on the proximal limbs and is associated with more aggressive behavior. It lacks the granulomalike pattern and has more prominent epithelioid and rhabdoid histological presentation.13-15
Epithelioid sarcoma is a mesenchymal tumor that can display multidirectional differentiation that is primarily epithelial.16 The precise histogenesis of ES remains unclear, but studies have demonstrated a spectrum of differentiation that ranges from primitive myofibroblast or fibrohistiocytelike cells to those with well-developed epithelial properties.16,17 Epithelioid sarcoma characteristically coexpresses vimentin and low-molecular-weight CKs such as cell adhesion molecule 5.2. The tumor cells often are immunoreactive for epithelial membrane antigen and more than 50% of cases exhibit remarkable CD34 positivity.16 More recent studies have further refined the immunophenotype, demonstrating frequent expression of CK8 and CK19 but less commonly CK7, CK20, CK34bE12, and CK5/6.18-20 Additional studies reported that in 10 of 11 cases, ES was positive for CA 125 on immunohistochemical staining, and 3 of 5 patients also had elevated serum CA 125 levels.21,22 More recently, Hoshino et al23 showed elevated serum CA 125 levels in 5 of 7 patients with ES. Cancer antigen 125 is a high-molecular-weight glycoprotein commonly used in the identification of epithelial ovarian carcinomas; however, it also has been described in a number of other neoplasms including carcinomas of the breast, lungs, and colon and lymphoma.24-27 Although it appears that the addition of CA 125 to a panel of other immunohistochemical stains may be helpful in differentiating ES from other soft tissue sarcomas and serum CA 125 levels may help determine tumor burden, currently the number of cases studied is too small to definitively make that conclusion.21,23 In our case, the tumor cells were strongly and diffusely positive for CA 125. Serum CA 125 levels were not available.
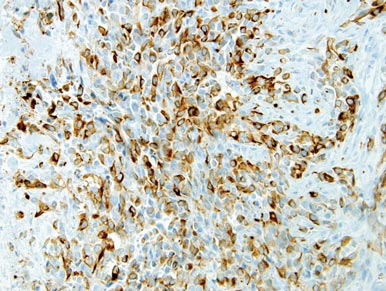
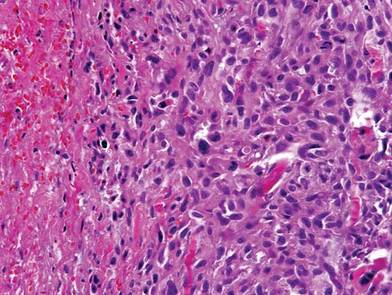
Cytogenetic studies have failed to identify a consistent chromosomal abnormality in ES.5 Some analyses performed by comparative genomic hybridization on isolated cases and small case series indicate that the most frequent alterations involve 8q, 18q11, and 22q11.13,28,29 The tumor suppressor gene SMARCB1/INI1 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily B, member 1/integrase interactor 1) has been mapped to 22q11, and ES commonly shows absence of nuclear staining for this protein, indicating inactivation.13-15
Conclusion
Benign fibrohistiocytic proliferations should be included in the differential of histological mimickers of ES. Deep biopsies are essential to differentiate these benign tumors from fibrous histiocytomalike or fibromalike lesions of ES because superficial portions of ES may be well differentiated.
1. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
2. Spillane AJ, Thomas JM, Fisher C. Epithelioid sarcoma: the clinicopathological complexities of this rare soft tissue sarcoma. Ann Surg Oncol. 2000;7:218-225.
3. Chase DR, Enzinger FM. Epithelioid sarcoma. diagnosis, prognostic indicators, and treatment. Am J Surg Pathol. 1985;9:241-263.
4. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
5. Evans HL, Baer SC. Epithelioid sarcoma: a clinicopathologic and prognostic study of 26 cases. Semin Diagn Pathol. 1993;10:286-291.
6. Heenan PJ, Quirk CJ, Papadimitriou JM. Epithelioid sarcoma. a diagnostic problem. Am J Dermatopathol. 1986;8:95-104.
7. DiCaudo DJ, McCalmont TH, Wick MR. Selected diagnostic problems in neoplastic dermatopathology. Arch Pathol Lab Med. 2007;131:434-439.
8. Ormsby AH, Liou LS, Oriba HA, et al. Epithelioid sarcoma of the penis: report of an unusual case and review of the literature. Ann Diagn Pathol. 2000;4:88-94.
9. Lowentritt B, Parsons JK, Argani P, et al. Pediatric epithelioid sarcoma of the penis. J Urol. 2004;172:296-297.
10. Mirra JM, Kessler S, Bhuta S, et al. The fibroma-like variant of epithelioid sarcoma. a fibrohistiocytic/myoid cell lesion often confused with benign and malignant spindle cell tumors. Cancer. 1992;69:1382-1395.
11. Tan SH, Ong BH. Spindle cell variant of epithelioid sarcoma: an easily misdiagnosed tumour. Australas J Dermatol. 2001;42:139-141.
12. von Hochstetter AR, Grant JW, Meyer VE, et al. Angiomatoid variant of epithelioid sarcoma. the value of immunohistochemistry in the differential diagnosis. Chir Organi Mov. 1990;75(suppl 1):158-162.
13. Modena P, Lualdi E, Facchinetti F, et al. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res. 2005;65:4012-4019.
14. Lualdi E, Modena P, Debiec-Rychter M, et al. Molecular cytogenetic characterization of proximal-type epithelioid sarcoma. Genes Chromosomes Cancer. 2004;41:283-290.
15. Kosemehmetoglu K, Kaygusuz G, Bahrami A, et al. Intra-articular epithelioid sarcoma showing mixed classic and proximal-type features: report of 2 cases, with immunohistochemical and molecular cytogenetic INI-1 study. Am J Surg Pathol. 2011;35:891-897.
16. Armah HB, Parwani AV. Epithelioid sarcoma. Arch Pathol Lab Med. 2009;133:814-819.
17. Fisher C. Epithelioid sarcoma: the spectrum of ultrastructural differentiation in seven immunohistochemically defined cases. Hum Pathol. 1988;19:265-275.
18. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
19. Humble SD, Prieto VG, Horenstein MG. Cytokeratin 7 and 20 expression in epithelioid sarcoma. J Cutan Pathol. 2003;30:242-246.
20. Lin L, Skacel M, Sigel JE, et al. Epithelioid sarcoma: an immunohistochemical analysis evaluating the utility of cytokeratin 5/6 in distinguishing superficial epithelioid sarcoma from spindled squamous cell carcinoma. J Cutan Pathol. 2003;30:114-117.
21. Kato H, Hatori M, Kokubun S, et al. CA125 expression in epithelioid sarcoma. Jpn J Clin Oncol. 2004;34:149-154.
22. Kato H, Hatori M, Watanabe M, et al. Epithelioid sarcomas with elevated serum CA125: report of two cases. Jpn J Clin Oncol. 2003;33:141-144.
23. Hoshino M, Kawashima H, Ogose A, et al. Serum CA 125 expression as a tumor marker for the diagnosis and monitoring the clinical course of epithelioid sarcoma [published online ahead of print September 16, 2009]. J Cancer Res Clin Oncol. 2010;136:457-464.
24. Lee AH, Paish EC, Marchio C, et al. The expression of Wilm’s tumour-1 and CA125 in invasive micropapillary carcinoma of the breast. Histopathology. 2007;51:824-828.
25. Homma S, Satoh H, Kagohashi K, et al. Production of CA125 by human lung cancer cell lines. Clin Exp Med. 2004;4:139-141.
26. Streppel MM, Vincent A, Mukherjee R, et al. Mucin 16 (cancer antigen 125) expression in human tissues and cell lines and correlation with clinical outcome in adenocarcinomas of the pancreas, esophagus, stomach, and colon. Hum Pathol. 2012;42:1755-1763.
27. Wei G, Yuping Z, Jun W, et al. CA125 expression in patients with non-Hodgkin’s lymphoma. Leuk Lymphoma. 2006; 47:1322-1326.
28. Feely MG, Fidler ME, Nelson M, et al. Cytogenetic findings in a case of epithelioid sarcoma and a review of the literature. Cancer Genet Cytogenet. 2000;119:155-157.
29. Lushnikova T, Knuutila S, Miettinen M. DNA copy number changes in epithelioid sarcoma and its variants: a comparative genomic hybridization study. Mod Pathol. 2000;13:1092-1096.
Epithelioid sarcoma (ES) is a rare malignant soft tissue neoplasm that is most often encountered on the distal extremities of young adults.1 Epithelioid sarcoma is notorious for its tendency to mimic palisading granulomatous processes such as granuloma annulare. We report a case of ES on the right hand of a 23-year-old man that resembled a benign fibrous histiocytoma (dermatofibroma) on incisional biopsy. The typical histopathologic features of ES were identified after amputation of the hand and evaluation of the deeper regions of the tumor. The tendency for ES to mimic granulomatous processes is a common diagnostic pitfall, but the potential for its close resemblance to benign fibrous histiocytoma is less recognized.
|
Case Report
A 23-year-old man presented with a nonhealing lesion on the right palm. His medical history was remarkable for a giant cell tumor of the tendon sheath involving the right fifth finger that had been treated via excision at an outside institution 2 years prior. Clinical examination revealed a 0.8×0.6-cm painful, firm, ulcerated dermal nodule with a hemorrhagic crust on the palmar surface of the right hand (Figure 1A). The clinical differential diagnosis included melanoma, traumatized verruca vulgaris, thrombosed pyogenic granuloma, and foreign body. A shave biopsy demonstrated verrucous epidermal hyperplasia, but the specimen did not include the dermis. Cultures of the lesion were positive for Staphylococcus aureus, and antibiotic therapy was initiated. In light of the clinical findings and the patient’s history of a giant cell tumor, imaging studies were performed. Magnetic resonance angiography showed abnormal masslike infiltrative enhancement throughout the soft tissues surrounding the right fifth metacarpal bone. The differential included a recurrent giant cell tumor, fibromatosis, and other soft tissue neoplasms.
After several missed appointments and surgery cancellations, the patient returned 4 months later for an incisional biopsy. Physical examination revealed a persistent palmar ulcer that had grown to 1.4×1 cm in size, along with an indurated purple plaque wrapping around the ulnar aspect of the right hand (Figure 1B). The biopsy demonstrated a proliferation of spindled and ovoid cells with scant cytoplasm that surrounded sclerotic collagen bundles resembling a dermatofibroma (Figure 2A). Cytologic atypia and mitotic activity were absent (Figure 2B). Glass slides of the original biopsy, which ultimately led to the diagnosis of the giant cell tumor of the tendon sheath more than 2 years earlier, were obtained and showed similar features. The proliferating cells were strongly and diffusely immunoreactive for vimentin, CD34, and cancer antigen 125 (CA 125). Scattered tumor cells strongly expressed cytokeratins (CKs) AE1/AE3 and cell adhesion molecule 5.2 (Figure 3). Staining for CD99 and epithelial membrane antigen was diffuse but weak. Factor XIIIa, S-100, CK7, smooth muscle actin, muscle-specific actin (HHF35), CD31, CD68, and B-cell lymphoma 2 were negative within the proliferating cells. Based on the clinical examination and results of the immunohistochemical staining, a diagnosis of ES was favored.
|
After a negative metastatic workup, amputation of the right hand was performed. The amputation specimen showed a tumor that extended through the entire hand with encasement of large vessels and tendons. Although the more superficial regions were cytologically bland, deep-seated regions of the tumor exhibited greater cellularity, nuclear pleomorphism, and mitotic activity (Figure 4). There was no bone involvement. Right axillary sentinel lymph nodes were negative for metastasis. Eighteen months later the patient developed chest and back pain with dyspnea. Thorascopic surgery was performed for a left pleural effusion and metastases to the left parietal pleura and adjacent soft tissue were identified. The patient was subsequently lost to follow-up.
Comment
First described by Enzinger1 in 1970, ES is a rare malignant soft tissue neoplasm that most frequently arises on the hands, forearms, and pretibial soft tissues of young adults.1-3 It is an aggressive tumor characterized by frequent recurrences and a high metastatic rate, with lung and regional lymph nodes being favored metastatic sites.1-5 Periods of several months or even years often pass between the initial presentation and establishment of a correct diagnosis, as ES frequently is mistaken for other benign conditions. The tendency for ES to mimic granulomatous processes is a common diagnostic pitfall, but the potential for its close resemblance to benign fibrous histiocytoma is less recognized.6,7 In his original series of 62 cases, Enzinger1 noted that 17 patients were referred for treatment with a diagnosis of a benign fibrohistiocytic neoplasm, and other reports have described a resemblance to fibrous and fibrohistiocytic neoplasms.8-11 Mirra et al10 designated these tumors as fibromalike variants of ES. Additional subtypes of ES have subsequently been recognized, including those described as angiomatoid or angiosarcomalike, reflecting the potential of ES to resemble vascular tumors.12 A proximal type of ES also has been described. This lesion presents as a deep-seated tumor on the proximal limbs and is associated with more aggressive behavior. It lacks the granulomalike pattern and has more prominent epithelioid and rhabdoid histological presentation.13-15
Epithelioid sarcoma is a mesenchymal tumor that can display multidirectional differentiation that is primarily epithelial.16 The precise histogenesis of ES remains unclear, but studies have demonstrated a spectrum of differentiation that ranges from primitive myofibroblast or fibrohistiocytelike cells to those with well-developed epithelial properties.16,17 Epithelioid sarcoma characteristically coexpresses vimentin and low-molecular-weight CKs such as cell adhesion molecule 5.2. The tumor cells often are immunoreactive for epithelial membrane antigen and more than 50% of cases exhibit remarkable CD34 positivity.16 More recent studies have further refined the immunophenotype, demonstrating frequent expression of CK8 and CK19 but less commonly CK7, CK20, CK34bE12, and CK5/6.18-20 Additional studies reported that in 10 of 11 cases, ES was positive for CA 125 on immunohistochemical staining, and 3 of 5 patients also had elevated serum CA 125 levels.21,22 More recently, Hoshino et al23 showed elevated serum CA 125 levels in 5 of 7 patients with ES. Cancer antigen 125 is a high-molecular-weight glycoprotein commonly used in the identification of epithelial ovarian carcinomas; however, it also has been described in a number of other neoplasms including carcinomas of the breast, lungs, and colon and lymphoma.24-27 Although it appears that the addition of CA 125 to a panel of other immunohistochemical stains may be helpful in differentiating ES from other soft tissue sarcomas and serum CA 125 levels may help determine tumor burden, currently the number of cases studied is too small to definitively make that conclusion.21,23 In our case, the tumor cells were strongly and diffusely positive for CA 125. Serum CA 125 levels were not available.
Cytogenetic studies have failed to identify a consistent chromosomal abnormality in ES.5 Some analyses performed by comparative genomic hybridization on isolated cases and small case series indicate that the most frequent alterations involve 8q, 18q11, and 22q11.13,28,29 The tumor suppressor gene SMARCB1/INI1 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily B, member 1/integrase interactor 1) has been mapped to 22q11, and ES commonly shows absence of nuclear staining for this protein, indicating inactivation.13-15
Conclusion
Benign fibrohistiocytic proliferations should be included in the differential of histological mimickers of ES. Deep biopsies are essential to differentiate these benign tumors from fibrous histiocytomalike or fibromalike lesions of ES because superficial portions of ES may be well differentiated.
Epithelioid sarcoma (ES) is a rare malignant soft tissue neoplasm that is most often encountered on the distal extremities of young adults.1 Epithelioid sarcoma is notorious for its tendency to mimic palisading granulomatous processes such as granuloma annulare. We report a case of ES on the right hand of a 23-year-old man that resembled a benign fibrous histiocytoma (dermatofibroma) on incisional biopsy. The typical histopathologic features of ES were identified after amputation of the hand and evaluation of the deeper regions of the tumor. The tendency for ES to mimic granulomatous processes is a common diagnostic pitfall, but the potential for its close resemblance to benign fibrous histiocytoma is less recognized.
|
Case Report
A 23-year-old man presented with a nonhealing lesion on the right palm. His medical history was remarkable for a giant cell tumor of the tendon sheath involving the right fifth finger that had been treated via excision at an outside institution 2 years prior. Clinical examination revealed a 0.8×0.6-cm painful, firm, ulcerated dermal nodule with a hemorrhagic crust on the palmar surface of the right hand (Figure 1A). The clinical differential diagnosis included melanoma, traumatized verruca vulgaris, thrombosed pyogenic granuloma, and foreign body. A shave biopsy demonstrated verrucous epidermal hyperplasia, but the specimen did not include the dermis. Cultures of the lesion were positive for Staphylococcus aureus, and antibiotic therapy was initiated. In light of the clinical findings and the patient’s history of a giant cell tumor, imaging studies were performed. Magnetic resonance angiography showed abnormal masslike infiltrative enhancement throughout the soft tissues surrounding the right fifth metacarpal bone. The differential included a recurrent giant cell tumor, fibromatosis, and other soft tissue neoplasms.
After several missed appointments and surgery cancellations, the patient returned 4 months later for an incisional biopsy. Physical examination revealed a persistent palmar ulcer that had grown to 1.4×1 cm in size, along with an indurated purple plaque wrapping around the ulnar aspect of the right hand (Figure 1B). The biopsy demonstrated a proliferation of spindled and ovoid cells with scant cytoplasm that surrounded sclerotic collagen bundles resembling a dermatofibroma (Figure 2A). Cytologic atypia and mitotic activity were absent (Figure 2B). Glass slides of the original biopsy, which ultimately led to the diagnosis of the giant cell tumor of the tendon sheath more than 2 years earlier, were obtained and showed similar features. The proliferating cells were strongly and diffusely immunoreactive for vimentin, CD34, and cancer antigen 125 (CA 125). Scattered tumor cells strongly expressed cytokeratins (CKs) AE1/AE3 and cell adhesion molecule 5.2 (Figure 3). Staining for CD99 and epithelial membrane antigen was diffuse but weak. Factor XIIIa, S-100, CK7, smooth muscle actin, muscle-specific actin (HHF35), CD31, CD68, and B-cell lymphoma 2 were negative within the proliferating cells. Based on the clinical examination and results of the immunohistochemical staining, a diagnosis of ES was favored.
|
After a negative metastatic workup, amputation of the right hand was performed. The amputation specimen showed a tumor that extended through the entire hand with encasement of large vessels and tendons. Although the more superficial regions were cytologically bland, deep-seated regions of the tumor exhibited greater cellularity, nuclear pleomorphism, and mitotic activity (Figure 4). There was no bone involvement. Right axillary sentinel lymph nodes were negative for metastasis. Eighteen months later the patient developed chest and back pain with dyspnea. Thorascopic surgery was performed for a left pleural effusion and metastases to the left parietal pleura and adjacent soft tissue were identified. The patient was subsequently lost to follow-up.
Comment
First described by Enzinger1 in 1970, ES is a rare malignant soft tissue neoplasm that most frequently arises on the hands, forearms, and pretibial soft tissues of young adults.1-3 It is an aggressive tumor characterized by frequent recurrences and a high metastatic rate, with lung and regional lymph nodes being favored metastatic sites.1-5 Periods of several months or even years often pass between the initial presentation and establishment of a correct diagnosis, as ES frequently is mistaken for other benign conditions. The tendency for ES to mimic granulomatous processes is a common diagnostic pitfall, but the potential for its close resemblance to benign fibrous histiocytoma is less recognized.6,7 In his original series of 62 cases, Enzinger1 noted that 17 patients were referred for treatment with a diagnosis of a benign fibrohistiocytic neoplasm, and other reports have described a resemblance to fibrous and fibrohistiocytic neoplasms.8-11 Mirra et al10 designated these tumors as fibromalike variants of ES. Additional subtypes of ES have subsequently been recognized, including those described as angiomatoid or angiosarcomalike, reflecting the potential of ES to resemble vascular tumors.12 A proximal type of ES also has been described. This lesion presents as a deep-seated tumor on the proximal limbs and is associated with more aggressive behavior. It lacks the granulomalike pattern and has more prominent epithelioid and rhabdoid histological presentation.13-15
Epithelioid sarcoma is a mesenchymal tumor that can display multidirectional differentiation that is primarily epithelial.16 The precise histogenesis of ES remains unclear, but studies have demonstrated a spectrum of differentiation that ranges from primitive myofibroblast or fibrohistiocytelike cells to those with well-developed epithelial properties.16,17 Epithelioid sarcoma characteristically coexpresses vimentin and low-molecular-weight CKs such as cell adhesion molecule 5.2. The tumor cells often are immunoreactive for epithelial membrane antigen and more than 50% of cases exhibit remarkable CD34 positivity.16 More recent studies have further refined the immunophenotype, demonstrating frequent expression of CK8 and CK19 but less commonly CK7, CK20, CK34bE12, and CK5/6.18-20 Additional studies reported that in 10 of 11 cases, ES was positive for CA 125 on immunohistochemical staining, and 3 of 5 patients also had elevated serum CA 125 levels.21,22 More recently, Hoshino et al23 showed elevated serum CA 125 levels in 5 of 7 patients with ES. Cancer antigen 125 is a high-molecular-weight glycoprotein commonly used in the identification of epithelial ovarian carcinomas; however, it also has been described in a number of other neoplasms including carcinomas of the breast, lungs, and colon and lymphoma.24-27 Although it appears that the addition of CA 125 to a panel of other immunohistochemical stains may be helpful in differentiating ES from other soft tissue sarcomas and serum CA 125 levels may help determine tumor burden, currently the number of cases studied is too small to definitively make that conclusion.21,23 In our case, the tumor cells were strongly and diffusely positive for CA 125. Serum CA 125 levels were not available.
Cytogenetic studies have failed to identify a consistent chromosomal abnormality in ES.5 Some analyses performed by comparative genomic hybridization on isolated cases and small case series indicate that the most frequent alterations involve 8q, 18q11, and 22q11.13,28,29 The tumor suppressor gene SMARCB1/INI1 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily B, member 1/integrase interactor 1) has been mapped to 22q11, and ES commonly shows absence of nuclear staining for this protein, indicating inactivation.13-15
Conclusion
Benign fibrohistiocytic proliferations should be included in the differential of histological mimickers of ES. Deep biopsies are essential to differentiate these benign tumors from fibrous histiocytomalike or fibromalike lesions of ES because superficial portions of ES may be well differentiated.
1. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
2. Spillane AJ, Thomas JM, Fisher C. Epithelioid sarcoma: the clinicopathological complexities of this rare soft tissue sarcoma. Ann Surg Oncol. 2000;7:218-225.
3. Chase DR, Enzinger FM. Epithelioid sarcoma. diagnosis, prognostic indicators, and treatment. Am J Surg Pathol. 1985;9:241-263.
4. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
5. Evans HL, Baer SC. Epithelioid sarcoma: a clinicopathologic and prognostic study of 26 cases. Semin Diagn Pathol. 1993;10:286-291.
6. Heenan PJ, Quirk CJ, Papadimitriou JM. Epithelioid sarcoma. a diagnostic problem. Am J Dermatopathol. 1986;8:95-104.
7. DiCaudo DJ, McCalmont TH, Wick MR. Selected diagnostic problems in neoplastic dermatopathology. Arch Pathol Lab Med. 2007;131:434-439.
8. Ormsby AH, Liou LS, Oriba HA, et al. Epithelioid sarcoma of the penis: report of an unusual case and review of the literature. Ann Diagn Pathol. 2000;4:88-94.
9. Lowentritt B, Parsons JK, Argani P, et al. Pediatric epithelioid sarcoma of the penis. J Urol. 2004;172:296-297.
10. Mirra JM, Kessler S, Bhuta S, et al. The fibroma-like variant of epithelioid sarcoma. a fibrohistiocytic/myoid cell lesion often confused with benign and malignant spindle cell tumors. Cancer. 1992;69:1382-1395.
11. Tan SH, Ong BH. Spindle cell variant of epithelioid sarcoma: an easily misdiagnosed tumour. Australas J Dermatol. 2001;42:139-141.
12. von Hochstetter AR, Grant JW, Meyer VE, et al. Angiomatoid variant of epithelioid sarcoma. the value of immunohistochemistry in the differential diagnosis. Chir Organi Mov. 1990;75(suppl 1):158-162.
13. Modena P, Lualdi E, Facchinetti F, et al. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res. 2005;65:4012-4019.
14. Lualdi E, Modena P, Debiec-Rychter M, et al. Molecular cytogenetic characterization of proximal-type epithelioid sarcoma. Genes Chromosomes Cancer. 2004;41:283-290.
15. Kosemehmetoglu K, Kaygusuz G, Bahrami A, et al. Intra-articular epithelioid sarcoma showing mixed classic and proximal-type features: report of 2 cases, with immunohistochemical and molecular cytogenetic INI-1 study. Am J Surg Pathol. 2011;35:891-897.
16. Armah HB, Parwani AV. Epithelioid sarcoma. Arch Pathol Lab Med. 2009;133:814-819.
17. Fisher C. Epithelioid sarcoma: the spectrum of ultrastructural differentiation in seven immunohistochemically defined cases. Hum Pathol. 1988;19:265-275.
18. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
19. Humble SD, Prieto VG, Horenstein MG. Cytokeratin 7 and 20 expression in epithelioid sarcoma. J Cutan Pathol. 2003;30:242-246.
20. Lin L, Skacel M, Sigel JE, et al. Epithelioid sarcoma: an immunohistochemical analysis evaluating the utility of cytokeratin 5/6 in distinguishing superficial epithelioid sarcoma from spindled squamous cell carcinoma. J Cutan Pathol. 2003;30:114-117.
21. Kato H, Hatori M, Kokubun S, et al. CA125 expression in epithelioid sarcoma. Jpn J Clin Oncol. 2004;34:149-154.
22. Kato H, Hatori M, Watanabe M, et al. Epithelioid sarcomas with elevated serum CA125: report of two cases. Jpn J Clin Oncol. 2003;33:141-144.
23. Hoshino M, Kawashima H, Ogose A, et al. Serum CA 125 expression as a tumor marker for the diagnosis and monitoring the clinical course of epithelioid sarcoma [published online ahead of print September 16, 2009]. J Cancer Res Clin Oncol. 2010;136:457-464.
24. Lee AH, Paish EC, Marchio C, et al. The expression of Wilm’s tumour-1 and CA125 in invasive micropapillary carcinoma of the breast. Histopathology. 2007;51:824-828.
25. Homma S, Satoh H, Kagohashi K, et al. Production of CA125 by human lung cancer cell lines. Clin Exp Med. 2004;4:139-141.
26. Streppel MM, Vincent A, Mukherjee R, et al. Mucin 16 (cancer antigen 125) expression in human tissues and cell lines and correlation with clinical outcome in adenocarcinomas of the pancreas, esophagus, stomach, and colon. Hum Pathol. 2012;42:1755-1763.
27. Wei G, Yuping Z, Jun W, et al. CA125 expression in patients with non-Hodgkin’s lymphoma. Leuk Lymphoma. 2006; 47:1322-1326.
28. Feely MG, Fidler ME, Nelson M, et al. Cytogenetic findings in a case of epithelioid sarcoma and a review of the literature. Cancer Genet Cytogenet. 2000;119:155-157.
29. Lushnikova T, Knuutila S, Miettinen M. DNA copy number changes in epithelioid sarcoma and its variants: a comparative genomic hybridization study. Mod Pathol. 2000;13:1092-1096.
1. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
2. Spillane AJ, Thomas JM, Fisher C. Epithelioid sarcoma: the clinicopathological complexities of this rare soft tissue sarcoma. Ann Surg Oncol. 2000;7:218-225.
3. Chase DR, Enzinger FM. Epithelioid sarcoma. diagnosis, prognostic indicators, and treatment. Am J Surg Pathol. 1985;9:241-263.
4. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
5. Evans HL, Baer SC. Epithelioid sarcoma: a clinicopathologic and prognostic study of 26 cases. Semin Diagn Pathol. 1993;10:286-291.
6. Heenan PJ, Quirk CJ, Papadimitriou JM. Epithelioid sarcoma. a diagnostic problem. Am J Dermatopathol. 1986;8:95-104.
7. DiCaudo DJ, McCalmont TH, Wick MR. Selected diagnostic problems in neoplastic dermatopathology. Arch Pathol Lab Med. 2007;131:434-439.
8. Ormsby AH, Liou LS, Oriba HA, et al. Epithelioid sarcoma of the penis: report of an unusual case and review of the literature. Ann Diagn Pathol. 2000;4:88-94.
9. Lowentritt B, Parsons JK, Argani P, et al. Pediatric epithelioid sarcoma of the penis. J Urol. 2004;172:296-297.
10. Mirra JM, Kessler S, Bhuta S, et al. The fibroma-like variant of epithelioid sarcoma. a fibrohistiocytic/myoid cell lesion often confused with benign and malignant spindle cell tumors. Cancer. 1992;69:1382-1395.
11. Tan SH, Ong BH. Spindle cell variant of epithelioid sarcoma: an easily misdiagnosed tumour. Australas J Dermatol. 2001;42:139-141.
12. von Hochstetter AR, Grant JW, Meyer VE, et al. Angiomatoid variant of epithelioid sarcoma. the value of immunohistochemistry in the differential diagnosis. Chir Organi Mov. 1990;75(suppl 1):158-162.
13. Modena P, Lualdi E, Facchinetti F, et al. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res. 2005;65:4012-4019.
14. Lualdi E, Modena P, Debiec-Rychter M, et al. Molecular cytogenetic characterization of proximal-type epithelioid sarcoma. Genes Chromosomes Cancer. 2004;41:283-290.
15. Kosemehmetoglu K, Kaygusuz G, Bahrami A, et al. Intra-articular epithelioid sarcoma showing mixed classic and proximal-type features: report of 2 cases, with immunohistochemical and molecular cytogenetic INI-1 study. Am J Surg Pathol. 2011;35:891-897.
16. Armah HB, Parwani AV. Epithelioid sarcoma. Arch Pathol Lab Med. 2009;133:814-819.
17. Fisher C. Epithelioid sarcoma: the spectrum of ultrastructural differentiation in seven immunohistochemically defined cases. Hum Pathol. 1988;19:265-275.
18. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
19. Humble SD, Prieto VG, Horenstein MG. Cytokeratin 7 and 20 expression in epithelioid sarcoma. J Cutan Pathol. 2003;30:242-246.
20. Lin L, Skacel M, Sigel JE, et al. Epithelioid sarcoma: an immunohistochemical analysis evaluating the utility of cytokeratin 5/6 in distinguishing superficial epithelioid sarcoma from spindled squamous cell carcinoma. J Cutan Pathol. 2003;30:114-117.
21. Kato H, Hatori M, Kokubun S, et al. CA125 expression in epithelioid sarcoma. Jpn J Clin Oncol. 2004;34:149-154.
22. Kato H, Hatori M, Watanabe M, et al. Epithelioid sarcomas with elevated serum CA125: report of two cases. Jpn J Clin Oncol. 2003;33:141-144.
23. Hoshino M, Kawashima H, Ogose A, et al. Serum CA 125 expression as a tumor marker for the diagnosis and monitoring the clinical course of epithelioid sarcoma [published online ahead of print September 16, 2009]. J Cancer Res Clin Oncol. 2010;136:457-464.
24. Lee AH, Paish EC, Marchio C, et al. The expression of Wilm’s tumour-1 and CA125 in invasive micropapillary carcinoma of the breast. Histopathology. 2007;51:824-828.
25. Homma S, Satoh H, Kagohashi K, et al. Production of CA125 by human lung cancer cell lines. Clin Exp Med. 2004;4:139-141.
26. Streppel MM, Vincent A, Mukherjee R, et al. Mucin 16 (cancer antigen 125) expression in human tissues and cell lines and correlation with clinical outcome in adenocarcinomas of the pancreas, esophagus, stomach, and colon. Hum Pathol. 2012;42:1755-1763.
27. Wei G, Yuping Z, Jun W, et al. CA125 expression in patients with non-Hodgkin’s lymphoma. Leuk Lymphoma. 2006; 47:1322-1326.
28. Feely MG, Fidler ME, Nelson M, et al. Cytogenetic findings in a case of epithelioid sarcoma and a review of the literature. Cancer Genet Cytogenet. 2000;119:155-157.
29. Lushnikova T, Knuutila S, Miettinen M. DNA copy number changes in epithelioid sarcoma and its variants: a comparative genomic hybridization study. Mod Pathol. 2000;13:1092-1096.
Practice Points
- Epithelioid sarcoma should be considered in the clinical differential diagnosis of nonhealing recurrent lesions of the distal extremities in a young adult.
- Histological presentation of epithelioid sarcoma can mimic a number of benign granulomatous and fibrohistiocytic processes, including benign fibrous histiocytoma.
- Deeper biopsies may be needed to demonstrate the overtly malignant morphology characteristic of epithelioid sarcoma.
- Inactivation of SMARCB1/INI1 is a common molecular aberration identified in epithelioid sarcoma and can be demonstrated immunohistochemically by absence of nuclear staining in tumor cells.
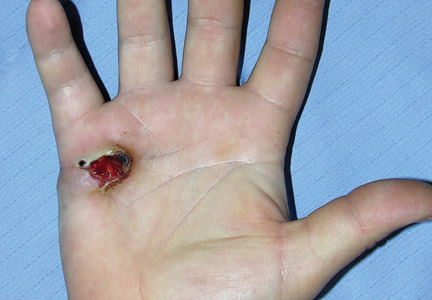
Superficial Acral Fibromyxoma and Other Slow-Growing Tumors in Acral Areas
First described by Fetsch et al1 in 2001, superficial acral fibromyxoma (SAFM) is a rare fibromyxoid mesenchymal tumor that typically affects the fingers and toes with frequent involvement of the nail unit. It is not widely recognized and remains poorly understood. We describe a series of 3 cases of SAFM encountered at our institution and provide a review of the literature on this unique tumor.
Case Reports
Patient 1
A 35-year-old man presented for treatment of a “wart” on the right fifth toe that had increased in size over the last year. He reported that the lesion was mildly painful and occasionally bled or drained clear fluid. He also noted cracking of the nail plate on the same toe. Physical examination revealed a firm, flesh-colored, 3-mm dermal papule on the proximal nail fold of the right fifth toe with subtle flattening of the underlying nail plate (Figure 1). The patient underwent biopsy of the involved proximal nail fold. Histopathology revealed a proliferation of small oval and spindle cells arranged in fascicles and bundles in the dermis (Figure 2). There was extensive mucin deposition associated with the spindle cell proliferation. Additionally, spindle cells and mucin surrounded and entrapped collagen bundles on the periphery of the lesion. Lesional cells were diffusely positive for CD34 and extended to the deep surgical margin (Figure 3). S-100 and factor XIIIa stains were negative. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
|
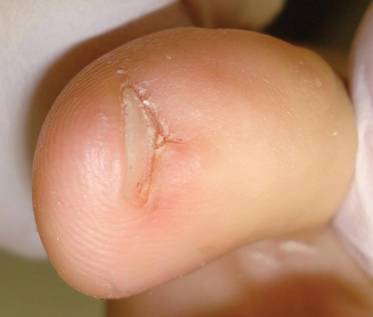

Patient 2
A 47-year-old man presented with an asymptomatic growth on the left fourth toe that had increased in size over the last year. Physical examination revealed an 8-mm, firm, fleshy, flesh-colored, smooth and slightly pedunculated papule on the distal aspect of the left fourth toe. The nail plate and periungual region were not involved. A shave biopsy of the papule was obtained. Histopathology demonstrated dermal stellate spindle cells arranged in a loose fascicular pattern with marked mucin deposition throughout the dermis (Figure 4). Lesional cells were positive for CD34. An S-100 stain highlighted dermal dendritic cells, but lesional cells were negative. No further excision was undertaken, and there was no evidence of recurrence at 1-year follow-up. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
Patient 3
A 45-year-old woman presented with asymptomatic distal onycholysis of the right thumbnail of 1 year’s duration. She denied any history of trauma, and no bleeding or pigmentary changes were noted. Physical examination revealed a 5-mm flesh-colored papule on the hyponychium of the right thumb with focal onycholysis (Figure 5). A wedge biopsy of the lesion was performed. Histopathology showed an intradermal nodular proliferation of bland spindle cells arranged in loose fascicles and bundles and embedded in a myxoid stroma (Figure 6). CD34 staining strongly highlighted lesional cells. S-100 and neurofilament stains were negative. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
Comment
Clinically, SAFM typically presents as a slow-growing solitary nodule on the distal fingers or toes. The great toe is the most commonly affected digit, and the tumor may be subungual in up to two-thirds of cases.1 Unusual locations, such as the heel, also have been reported.2 Onset typically occurs in the fifth or sixth decade, and there is an approximately 2-fold higher incidence in men than women.1-3
Histopathologically, SAFM is a characteristically well-circumscribed but unencapsulated dermal tumor composed of spindle and stellate cells in a loose storiform or fascicular arrangement embedded in a myxoid, myxocollagenous, or collagenous stroma.4 The tumor often occupies the entire dermis and may extend into the subcutis or occasionally the underlying fascia and bone.4,5 Mast cells often are prominent, and microvascular accentuation also may be seen. Inflammatory infiltrates and multinucleated giant cells typically are not seen.6 Although 2 cases of atypical SAFM have been described,2 cellular atypia is not a characteristic feature of SAFM.
The immunohistochemical profile of SAFM is characterized by diffuse or focal expression of CD34, focal expression of epithelial membrane antigen (EMA), CD99 expression, and varying numbers of factor XIIIa–positive histiocytes.2,3 Positive staining for vimentin also is common. Staining typically is negative for S-100, human melanoma black 45, keratin, smooth muscle actin, and desmin.
The standard treatment of SAFM is complete local resection of the tumor, though some patients have been treated with partial excision or biopsy and partial or complete digital amputation.1 Local recurrence may occur in up to 20% of cases; however, approximately two-thirds of the reported recurrences in the literature occurred after incomplete tumor excision.1,2 It may be more appropriate to consider these cases as persistent rather than recurrent tumors. Superficial acral fibromyxoma is considered a benign tumor, with no known cases of metastases.4
|
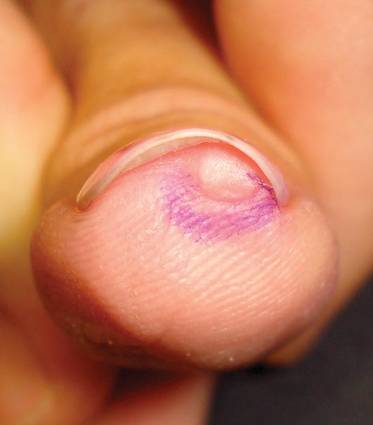
A broad differential diagnosis exists for SAFM and it can be difficult to differentiate it from a wide variety of benign and malignant tumors that may be seen on the nail unit and distal extremities (Table). Myxoid neurofibromas typically present as solitary lesions on the hands and feet. Similar to SAFM, myxoid neurofibromas are unencapsulated dermal tumors composed of spindle-shaped cells in which mast cells often are conspicuous.2,7 However, tumor cells in myxoid neurofibromas are S-100 positive, and the lesions typically do not show vasculature accentuation.4,7
Sclerosing perineuriomas are benign fibrous tumors of the fingers and palms. Histopathologically, bland spindle cells arranged in fascicles and whorls are observed in a hyalinized collagen matrix.8 Immunohistochemically, sclerosing perineuriomas are positive for EMA and negative for S-100, but unlike SAFM, these tumors usually are CD34 negative.8
Superficial angiomyxomas typically are located on the head and neck but also may be found in other locations such as the trunk. They present as cutaneous papules or polypoid lesions. Histopathologically, superficial angiomyxomas are poorly circumscribed with a lobular pattern. Spindle-shaped fibroblasts exist in a myxoid matrix with neutrophils and thin-walled capillaries. The fibroblasts are variably positive for CD34 but also are S-100 positive.1,9
Myxoid dermatofibrosarcoma protuberans is a rare, locally aggressive, mesenchymal tumor of the skin and subcutis2 that typically presents on the trunk, proximal extremities, or head and neck; occurrence on the fingers or toes is exceedingly rare.2,10 Histopathologically, a myxoid stroma contains sheets of bland spindle-shaped cells with minimal to no atypia, sometimes arranged in a storiform pattern. The tumor characteristically invades deeply into the subcutaneous tissues. CD34 is characteristically positive and S-100 is negative.2,10
Low-grade myxofibrosarcoma is a soft tissue sarcoma easily confused with other spindle cell tumors. It is one of the most common sarcomas in adults but rarely arises in acral areas.2 It is characterized by a nodular growth pattern with marked nuclear atypia and perivascular clustering of tumor cells. CD34 staining may be positive in some cases.11
Similar to SAFM, myxoinflammatory fibroblastic sarcoma has a predilection for the extremities.4 However, it typically presents as a subcutaneous mass and has no documented tendency for nail bed involvement. Also unlike SAFM, it has a remarkable inflammatory infiltrate and characteristic virocyte or Reed-Sternberg cells.12
Acquired digital fibrokeratomas are benign neoplasms that occur on fingers and toes; the classic clinical presentation is a solitary smooth nodule or dome, often with a characteristic projecting configuration and horn shape.1 Histopathologically, these tumors are paucicellular with thick, vertically oriented, interwoven collagen bundles; cells may be positive for CD34 but are negative for EMA.1,13 Related to acquired digital fibrokeratomas are Koenen tumors, which share a similar histology but are distinguished by their clinical characteristics. For example, Koenen tumors tend to be multifocal and are strongly associated with tuberous sclerosis. These tumors also have a tendency to recur.1
Conclusion
Our report of 3 typical cases of SAFM highlights the need to keep this increasingly recognized and well-defined clinicopathological entity in the differential for slow-growing tumors in acral locations, particularly those in the periungual and subungual regions.
1. Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma: a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes. Hum Pathol. 2001;32:704-714.
2. Al-Daraji WI, Miettinen M. Superficial acral fibromyxoma: a clinicopathological analysis of 32 tumors including 4 in the heel. J Cutan Pathol. 2008;35:1020-1026.
3. Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
4. André J, Theunis A, Richert B, et al. Superficial acral fibromyxoma: clinical and pathological features. Am J Dermatopathol. 2004;26:472-474.
5. Kazakov DV, Mentzel T, Burg G, et al. Superficial acral fibromyxoma: report of two cases. Dermatology. 2002;205:285-288.
6. Meyerle JH, Keller RA, Krivda SJ. Superficial acral fibromyxoma of the index finger. J Am Acad Dermatol. 2004;50:134-136.
7. Graadt van Roggen JF, Hogendoorn PC, Fletcher CD. Myxoid tumours of soft tissue. Histopathology. 1999;35:291-312.
8. Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
9. Calonje E, Guerin D, McCormick D, et al. Superficial angiomyxoma: clinicopathologic analysis of a series of distinctive but poorly recognized cutaneous tumors with tendency for recurrence. Am J Surg Pathol. 1999;23:910-917.
10. Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans. a study of 115 cases. Cancer. 1962;15:717-725.
11. Wada T, Hasegawa T, Nagoya S, et al. Myxofibrosarcoma with an infiltrative growth pattern: a case report. Jpn J Clin Oncol. 2000;30:458-462.
12. Meis-Kindblom JM, Kindblom LG. Acral myxoinflammatory fibroblastic sarcoma: a low-grade tumor of the hands and feet. Am J Surg Pathol. 1998;22:911-924.
13. Bart RS, Andrade R, Kopf AW, et al. Acquired digital fibrokeratomas. Arch Dermatol. 1968;97:120-129.
First described by Fetsch et al1 in 2001, superficial acral fibromyxoma (SAFM) is a rare fibromyxoid mesenchymal tumor that typically affects the fingers and toes with frequent involvement of the nail unit. It is not widely recognized and remains poorly understood. We describe a series of 3 cases of SAFM encountered at our institution and provide a review of the literature on this unique tumor.
Case Reports
Patient 1
A 35-year-old man presented for treatment of a “wart” on the right fifth toe that had increased in size over the last year. He reported that the lesion was mildly painful and occasionally bled or drained clear fluid. He also noted cracking of the nail plate on the same toe. Physical examination revealed a firm, flesh-colored, 3-mm dermal papule on the proximal nail fold of the right fifth toe with subtle flattening of the underlying nail plate (Figure 1). The patient underwent biopsy of the involved proximal nail fold. Histopathology revealed a proliferation of small oval and spindle cells arranged in fascicles and bundles in the dermis (Figure 2). There was extensive mucin deposition associated with the spindle cell proliferation. Additionally, spindle cells and mucin surrounded and entrapped collagen bundles on the periphery of the lesion. Lesional cells were diffusely positive for CD34 and extended to the deep surgical margin (Figure 3). S-100 and factor XIIIa stains were negative. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
|
Patient 2
A 47-year-old man presented with an asymptomatic growth on the left fourth toe that had increased in size over the last year. Physical examination revealed an 8-mm, firm, fleshy, flesh-colored, smooth and slightly pedunculated papule on the distal aspect of the left fourth toe. The nail plate and periungual region were not involved. A shave biopsy of the papule was obtained. Histopathology demonstrated dermal stellate spindle cells arranged in a loose fascicular pattern with marked mucin deposition throughout the dermis (Figure 4). Lesional cells were positive for CD34. An S-100 stain highlighted dermal dendritic cells, but lesional cells were negative. No further excision was undertaken, and there was no evidence of recurrence at 1-year follow-up. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
Patient 3
A 45-year-old woman presented with asymptomatic distal onycholysis of the right thumbnail of 1 year’s duration. She denied any history of trauma, and no bleeding or pigmentary changes were noted. Physical examination revealed a 5-mm flesh-colored papule on the hyponychium of the right thumb with focal onycholysis (Figure 5). A wedge biopsy of the lesion was performed. Histopathology showed an intradermal nodular proliferation of bland spindle cells arranged in loose fascicles and bundles and embedded in a myxoid stroma (Figure 6). CD34 staining strongly highlighted lesional cells. S-100 and neurofilament stains were negative. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
Comment
Clinically, SAFM typically presents as a slow-growing solitary nodule on the distal fingers or toes. The great toe is the most commonly affected digit, and the tumor may be subungual in up to two-thirds of cases.1 Unusual locations, such as the heel, also have been reported.2 Onset typically occurs in the fifth or sixth decade, and there is an approximately 2-fold higher incidence in men than women.1-3
Histopathologically, SAFM is a characteristically well-circumscribed but unencapsulated dermal tumor composed of spindle and stellate cells in a loose storiform or fascicular arrangement embedded in a myxoid, myxocollagenous, or collagenous stroma.4 The tumor often occupies the entire dermis and may extend into the subcutis or occasionally the underlying fascia and bone.4,5 Mast cells often are prominent, and microvascular accentuation also may be seen. Inflammatory infiltrates and multinucleated giant cells typically are not seen.6 Although 2 cases of atypical SAFM have been described,2 cellular atypia is not a characteristic feature of SAFM.
The immunohistochemical profile of SAFM is characterized by diffuse or focal expression of CD34, focal expression of epithelial membrane antigen (EMA), CD99 expression, and varying numbers of factor XIIIa–positive histiocytes.2,3 Positive staining for vimentin also is common. Staining typically is negative for S-100, human melanoma black 45, keratin, smooth muscle actin, and desmin.
The standard treatment of SAFM is complete local resection of the tumor, though some patients have been treated with partial excision or biopsy and partial or complete digital amputation.1 Local recurrence may occur in up to 20% of cases; however, approximately two-thirds of the reported recurrences in the literature occurred after incomplete tumor excision.1,2 It may be more appropriate to consider these cases as persistent rather than recurrent tumors. Superficial acral fibromyxoma is considered a benign tumor, with no known cases of metastases.4
|
A broad differential diagnosis exists for SAFM and it can be difficult to differentiate it from a wide variety of benign and malignant tumors that may be seen on the nail unit and distal extremities (Table). Myxoid neurofibromas typically present as solitary lesions on the hands and feet. Similar to SAFM, myxoid neurofibromas are unencapsulated dermal tumors composed of spindle-shaped cells in which mast cells often are conspicuous.2,7 However, tumor cells in myxoid neurofibromas are S-100 positive, and the lesions typically do not show vasculature accentuation.4,7
Sclerosing perineuriomas are benign fibrous tumors of the fingers and palms. Histopathologically, bland spindle cells arranged in fascicles and whorls are observed in a hyalinized collagen matrix.8 Immunohistochemically, sclerosing perineuriomas are positive for EMA and negative for S-100, but unlike SAFM, these tumors usually are CD34 negative.8
Superficial angiomyxomas typically are located on the head and neck but also may be found in other locations such as the trunk. They present as cutaneous papules or polypoid lesions. Histopathologically, superficial angiomyxomas are poorly circumscribed with a lobular pattern. Spindle-shaped fibroblasts exist in a myxoid matrix with neutrophils and thin-walled capillaries. The fibroblasts are variably positive for CD34 but also are S-100 positive.1,9
Myxoid dermatofibrosarcoma protuberans is a rare, locally aggressive, mesenchymal tumor of the skin and subcutis2 that typically presents on the trunk, proximal extremities, or head and neck; occurrence on the fingers or toes is exceedingly rare.2,10 Histopathologically, a myxoid stroma contains sheets of bland spindle-shaped cells with minimal to no atypia, sometimes arranged in a storiform pattern. The tumor characteristically invades deeply into the subcutaneous tissues. CD34 is characteristically positive and S-100 is negative.2,10
Low-grade myxofibrosarcoma is a soft tissue sarcoma easily confused with other spindle cell tumors. It is one of the most common sarcomas in adults but rarely arises in acral areas.2 It is characterized by a nodular growth pattern with marked nuclear atypia and perivascular clustering of tumor cells. CD34 staining may be positive in some cases.11
Similar to SAFM, myxoinflammatory fibroblastic sarcoma has a predilection for the extremities.4 However, it typically presents as a subcutaneous mass and has no documented tendency for nail bed involvement. Also unlike SAFM, it has a remarkable inflammatory infiltrate and characteristic virocyte or Reed-Sternberg cells.12
Acquired digital fibrokeratomas are benign neoplasms that occur on fingers and toes; the classic clinical presentation is a solitary smooth nodule or dome, often with a characteristic projecting configuration and horn shape.1 Histopathologically, these tumors are paucicellular with thick, vertically oriented, interwoven collagen bundles; cells may be positive for CD34 but are negative for EMA.1,13 Related to acquired digital fibrokeratomas are Koenen tumors, which share a similar histology but are distinguished by their clinical characteristics. For example, Koenen tumors tend to be multifocal and are strongly associated with tuberous sclerosis. These tumors also have a tendency to recur.1
Conclusion
Our report of 3 typical cases of SAFM highlights the need to keep this increasingly recognized and well-defined clinicopathological entity in the differential for slow-growing tumors in acral locations, particularly those in the periungual and subungual regions.
First described by Fetsch et al1 in 2001, superficial acral fibromyxoma (SAFM) is a rare fibromyxoid mesenchymal tumor that typically affects the fingers and toes with frequent involvement of the nail unit. It is not widely recognized and remains poorly understood. We describe a series of 3 cases of SAFM encountered at our institution and provide a review of the literature on this unique tumor.
Case Reports
Patient 1
A 35-year-old man presented for treatment of a “wart” on the right fifth toe that had increased in size over the last year. He reported that the lesion was mildly painful and occasionally bled or drained clear fluid. He also noted cracking of the nail plate on the same toe. Physical examination revealed a firm, flesh-colored, 3-mm dermal papule on the proximal nail fold of the right fifth toe with subtle flattening of the underlying nail plate (Figure 1). The patient underwent biopsy of the involved proximal nail fold. Histopathology revealed a proliferation of small oval and spindle cells arranged in fascicles and bundles in the dermis (Figure 2). There was extensive mucin deposition associated with the spindle cell proliferation. Additionally, spindle cells and mucin surrounded and entrapped collagen bundles on the periphery of the lesion. Lesional cells were diffusely positive for CD34 and extended to the deep surgical margin (Figure 3). S-100 and factor XIIIa stains were negative. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
|
Patient 2
A 47-year-old man presented with an asymptomatic growth on the left fourth toe that had increased in size over the last year. Physical examination revealed an 8-mm, firm, fleshy, flesh-colored, smooth and slightly pedunculated papule on the distal aspect of the left fourth toe. The nail plate and periungual region were not involved. A shave biopsy of the papule was obtained. Histopathology demonstrated dermal stellate spindle cells arranged in a loose fascicular pattern with marked mucin deposition throughout the dermis (Figure 4). Lesional cells were positive for CD34. An S-100 stain highlighted dermal dendritic cells, but lesional cells were negative. No further excision was undertaken, and there was no evidence of recurrence at 1-year follow-up. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
Patient 3
A 45-year-old woman presented with asymptomatic distal onycholysis of the right thumbnail of 1 year’s duration. She denied any history of trauma, and no bleeding or pigmentary changes were noted. Physical examination revealed a 5-mm flesh-colored papule on the hyponychium of the right thumb with focal onycholysis (Figure 5). A wedge biopsy of the lesion was performed. Histopathology showed an intradermal nodular proliferation of bland spindle cells arranged in loose fascicles and bundles and embedded in a myxoid stroma (Figure 6). CD34 staining strongly highlighted lesional cells. S-100 and neurofilament stains were negative. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
Comment
Clinically, SAFM typically presents as a slow-growing solitary nodule on the distal fingers or toes. The great toe is the most commonly affected digit, and the tumor may be subungual in up to two-thirds of cases.1 Unusual locations, such as the heel, also have been reported.2 Onset typically occurs in the fifth or sixth decade, and there is an approximately 2-fold higher incidence in men than women.1-3
Histopathologically, SAFM is a characteristically well-circumscribed but unencapsulated dermal tumor composed of spindle and stellate cells in a loose storiform or fascicular arrangement embedded in a myxoid, myxocollagenous, or collagenous stroma.4 The tumor often occupies the entire dermis and may extend into the subcutis or occasionally the underlying fascia and bone.4,5 Mast cells often are prominent, and microvascular accentuation also may be seen. Inflammatory infiltrates and multinucleated giant cells typically are not seen.6 Although 2 cases of atypical SAFM have been described,2 cellular atypia is not a characteristic feature of SAFM.
The immunohistochemical profile of SAFM is characterized by diffuse or focal expression of CD34, focal expression of epithelial membrane antigen (EMA), CD99 expression, and varying numbers of factor XIIIa–positive histiocytes.2,3 Positive staining for vimentin also is common. Staining typically is negative for S-100, human melanoma black 45, keratin, smooth muscle actin, and desmin.
The standard treatment of SAFM is complete local resection of the tumor, though some patients have been treated with partial excision or biopsy and partial or complete digital amputation.1 Local recurrence may occur in up to 20% of cases; however, approximately two-thirds of the reported recurrences in the literature occurred after incomplete tumor excision.1,2 It may be more appropriate to consider these cases as persistent rather than recurrent tumors. Superficial acral fibromyxoma is considered a benign tumor, with no known cases of metastases.4
|
A broad differential diagnosis exists for SAFM and it can be difficult to differentiate it from a wide variety of benign and malignant tumors that may be seen on the nail unit and distal extremities (Table). Myxoid neurofibromas typically present as solitary lesions on the hands and feet. Similar to SAFM, myxoid neurofibromas are unencapsulated dermal tumors composed of spindle-shaped cells in which mast cells often are conspicuous.2,7 However, tumor cells in myxoid neurofibromas are S-100 positive, and the lesions typically do not show vasculature accentuation.4,7
Sclerosing perineuriomas are benign fibrous tumors of the fingers and palms. Histopathologically, bland spindle cells arranged in fascicles and whorls are observed in a hyalinized collagen matrix.8 Immunohistochemically, sclerosing perineuriomas are positive for EMA and negative for S-100, but unlike SAFM, these tumors usually are CD34 negative.8
Superficial angiomyxomas typically are located on the head and neck but also may be found in other locations such as the trunk. They present as cutaneous papules or polypoid lesions. Histopathologically, superficial angiomyxomas are poorly circumscribed with a lobular pattern. Spindle-shaped fibroblasts exist in a myxoid matrix with neutrophils and thin-walled capillaries. The fibroblasts are variably positive for CD34 but also are S-100 positive.1,9
Myxoid dermatofibrosarcoma protuberans is a rare, locally aggressive, mesenchymal tumor of the skin and subcutis2 that typically presents on the trunk, proximal extremities, or head and neck; occurrence on the fingers or toes is exceedingly rare.2,10 Histopathologically, a myxoid stroma contains sheets of bland spindle-shaped cells with minimal to no atypia, sometimes arranged in a storiform pattern. The tumor characteristically invades deeply into the subcutaneous tissues. CD34 is characteristically positive and S-100 is negative.2,10
Low-grade myxofibrosarcoma is a soft tissue sarcoma easily confused with other spindle cell tumors. It is one of the most common sarcomas in adults but rarely arises in acral areas.2 It is characterized by a nodular growth pattern with marked nuclear atypia and perivascular clustering of tumor cells. CD34 staining may be positive in some cases.11
Similar to SAFM, myxoinflammatory fibroblastic sarcoma has a predilection for the extremities.4 However, it typically presents as a subcutaneous mass and has no documented tendency for nail bed involvement. Also unlike SAFM, it has a remarkable inflammatory infiltrate and characteristic virocyte or Reed-Sternberg cells.12
Acquired digital fibrokeratomas are benign neoplasms that occur on fingers and toes; the classic clinical presentation is a solitary smooth nodule or dome, often with a characteristic projecting configuration and horn shape.1 Histopathologically, these tumors are paucicellular with thick, vertically oriented, interwoven collagen bundles; cells may be positive for CD34 but are negative for EMA.1,13 Related to acquired digital fibrokeratomas are Koenen tumors, which share a similar histology but are distinguished by their clinical characteristics. For example, Koenen tumors tend to be multifocal and are strongly associated with tuberous sclerosis. These tumors also have a tendency to recur.1
Conclusion
Our report of 3 typical cases of SAFM highlights the need to keep this increasingly recognized and well-defined clinicopathological entity in the differential for slow-growing tumors in acral locations, particularly those in the periungual and subungual regions.
1. Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma: a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes. Hum Pathol. 2001;32:704-714.
2. Al-Daraji WI, Miettinen M. Superficial acral fibromyxoma: a clinicopathological analysis of 32 tumors including 4 in the heel. J Cutan Pathol. 2008;35:1020-1026.
3. Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
4. André J, Theunis A, Richert B, et al. Superficial acral fibromyxoma: clinical and pathological features. Am J Dermatopathol. 2004;26:472-474.
5. Kazakov DV, Mentzel T, Burg G, et al. Superficial acral fibromyxoma: report of two cases. Dermatology. 2002;205:285-288.
6. Meyerle JH, Keller RA, Krivda SJ. Superficial acral fibromyxoma of the index finger. J Am Acad Dermatol. 2004;50:134-136.
7. Graadt van Roggen JF, Hogendoorn PC, Fletcher CD. Myxoid tumours of soft tissue. Histopathology. 1999;35:291-312.
8. Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
9. Calonje E, Guerin D, McCormick D, et al. Superficial angiomyxoma: clinicopathologic analysis of a series of distinctive but poorly recognized cutaneous tumors with tendency for recurrence. Am J Surg Pathol. 1999;23:910-917.
10. Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans. a study of 115 cases. Cancer. 1962;15:717-725.
11. Wada T, Hasegawa T, Nagoya S, et al. Myxofibrosarcoma with an infiltrative growth pattern: a case report. Jpn J Clin Oncol. 2000;30:458-462.
12. Meis-Kindblom JM, Kindblom LG. Acral myxoinflammatory fibroblastic sarcoma: a low-grade tumor of the hands and feet. Am J Surg Pathol. 1998;22:911-924.
13. Bart RS, Andrade R, Kopf AW, et al. Acquired digital fibrokeratomas. Arch Dermatol. 1968;97:120-129.
1. Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma: a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes. Hum Pathol. 2001;32:704-714.
2. Al-Daraji WI, Miettinen M. Superficial acral fibromyxoma: a clinicopathological analysis of 32 tumors including 4 in the heel. J Cutan Pathol. 2008;35:1020-1026.
3. Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
4. André J, Theunis A, Richert B, et al. Superficial acral fibromyxoma: clinical and pathological features. Am J Dermatopathol. 2004;26:472-474.
5. Kazakov DV, Mentzel T, Burg G, et al. Superficial acral fibromyxoma: report of two cases. Dermatology. 2002;205:285-288.
6. Meyerle JH, Keller RA, Krivda SJ. Superficial acral fibromyxoma of the index finger. J Am Acad Dermatol. 2004;50:134-136.
7. Graadt van Roggen JF, Hogendoorn PC, Fletcher CD. Myxoid tumours of soft tissue. Histopathology. 1999;35:291-312.
8. Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
9. Calonje E, Guerin D, McCormick D, et al. Superficial angiomyxoma: clinicopathologic analysis of a series of distinctive but poorly recognized cutaneous tumors with tendency for recurrence. Am J Surg Pathol. 1999;23:910-917.
10. Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans. a study of 115 cases. Cancer. 1962;15:717-725.
11. Wada T, Hasegawa T, Nagoya S, et al. Myxofibrosarcoma with an infiltrative growth pattern: a case report. Jpn J Clin Oncol. 2000;30:458-462.
12. Meis-Kindblom JM, Kindblom LG. Acral myxoinflammatory fibroblastic sarcoma: a low-grade tumor of the hands and feet. Am J Surg Pathol. 1998;22:911-924.
13. Bart RS, Andrade R, Kopf AW, et al. Acquired digital fibrokeratomas. Arch Dermatol. 1968;97:120-129.
Practice Points
- Superficial acral fibromyxoma (SAFM) is a rare but distinct tumor that may affect the nail bed and nail plate, and it may clinically or histopathologically mimic other tumors of the distal extremities.
- Although SAFM is considered a benign tumor, it frequently persists or recurs after incomplete excision, and therefore complete local resection may be recommended, particularly for symptomatic lesions.
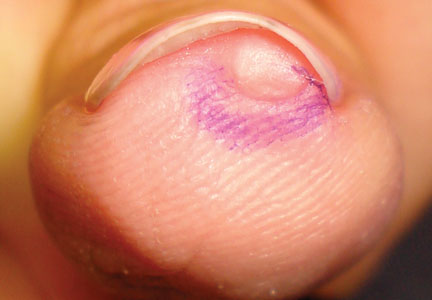
Tense Bullae With Widespread Erosions
The Diagnosis: Linear IgA Bullous Dermatosis
A biopsy specimen from an intact vesicle was obtained. Histologic findings showed a basket weave stratum corneum suggestive of an acute process. There was subepidermal separation with an inflammatory infiltrate of neutrophils (Figure 1). Direct immunofluorescence yielded a pattern of IgA deposition along the dermoepidermal junction (Figure 2). A diagnosis of linear IgA bullous dermatosis (LABD) was made. The patient was started on 100 mg daily of dapsone. The dose was subsequently increased to 175 mg twice daily, resulting in complete clearance. He became dermatologically disease free after 10 months and the dapsone was successfully tapered.
|
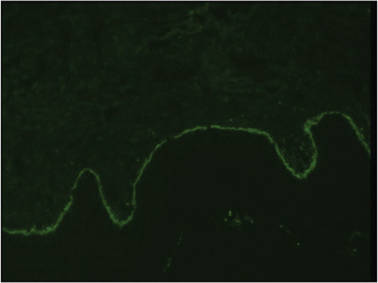
Linear IgA bullous dermatosis is an autoimmune subepidermal blistering disease with linear IgA deposits found along the basement membrane of the skin. There are 3 major categories of LABD: drug induced, systemic disorder related, and idiopathic.1 Patients with LABD present with a pruritic vesicobullous eruption that tends to favor the trunk, proximal extremities, and acral regions of the body. Mucous membrane lesions are present in less than 50% of patients.2 Linear IgA bullous dermatosis may resemble bullous pemphigoid, erythema multiforme, dermatitis herpetiformis, or toxic epidermal necrolysis. The gold standard for diagnosis is immunofluorescence staining that shows linear IgA deposition along the skin’s basement membrane.1 Prognosis for LABD is variable; there is risk for persistence and scarring.2 The drug-induced form of LABD is associated with clearance with the removal of the inciting agent.1
There are several autoimmune disorders that have been described in association with human immunodeficiency virus (HIV).3 Autoimmune bullous dermatoses, while described, are very uncommon in the setting of HIV infection. Previously reported cases include bullous pemphigoid, epidermolysis bullosa acquisita, pemphigus herpetiformis, pemphigus vegetans, pemphigus vulgaris, and cicatricial pemphigoid.4-12 The presentation of LABD in an HIV-positive patient is extremely rare.
There are 3 proposed mechanisms by which HIV and autoimmune bullous dermatoses coexist: unregulated B-cell activation, loss of T-suppressor cell regulation, and molecular mimicry. In patients with HIV, infected macrophages increase production of IL-1 and IL-6, causing nonspecific stimulation of B cells. Further production of tumor necrosis factor and other lymphotoxins may kill CD8+ T-suppressor cells, which further reduces B-cell regulation and production of nonspecific antibodies. Unregulated B-cell activation could lead to proliferation of antiself-specific B cells and autoantibodies. Additionally, various autoantibodies may arise due to mimicry between HIV antigens and human proteins. Some of the antibodies produced may be cytotoxic antilymphocyte antibodies that further disrupt B-cell regulation.13,14
Zandman-Goddard and Shoenfeld14 proposed a staging system of autoimmune disease and HIV with respect to CD4 count and viral load. Stage I is clinical latency of HIV, with a high CD4 count (>500 cells/mm3) and high viral load, which correlates with an acute infection of HIV and an intact immune system. Autoimmune disease can be seen in this stage. Stage II is cellular response, a quiescent period without overt manifestations of AIDS. The CD4 count is declining (200–499 cells/mm3), indicating immunosuppression, and the viral count is high. Autoimmune disease can occur and typically includes immune complex–mediated disease and vasculitis. Stage III is immune deficiency. The CD4 count is low (<200 cells/mm3), viral load is high, and AIDS develops. Autoimmune disease is not seen during this stage. Stage IV is the period of immune restoration following the advent of highly active antiretroviral therapy. There is a high CD4 count (>500 cells/mm3) and low viral load. There is a resurgence of autoimmune disease in this stage. Autoimmune disease can occur with an immune system capable of B- and T-cell interactions and a normal CD4 count. Autoimmunity is possible in stages I, II, and IV.14 Our patient developed bullous disease in stage II.
Although uncommon, autoimmune disease is possible in the setting of immune deficiency. The presence of autoimmune disease in a patient with HIV can only be seen during certain stages of infection. Knowledge of the possible scenarios of autoimmune disease can assist the clinician with monitoring status of the HIV infection or immune reconstitution.
1. Bouldin MB, Clowers-Webb HE, Davis JL, et al. Naproxen-associated linear IgA bullous dermatosis: case report and review. Mayo Clin Proc. 2000;75:967-970.
2. Nousari HC, Kimyai-Asadi A, Caeiro JP, et al. Clinical, demographic, and immunohistologic features of vancomycin-induced linear IgA bullous disease of the skin: report of 2 cases and review of the literature. Medicine. 1999;78:1-8.
3. Gala S, Fulcher DA. How HIV leads to autoimmune disorders. Med J Aust. 1996;164:224-226.
4. Lateef A, Packles MR, White SM, et al. Pemphigus vegetans in association with human immunodeficiency virus. Int J Dermatol. 1999;38:778-781.
5. Levy PM, Balavoine JF, Salomon D, et al. Ritodrine-responsive bullous pemphigoing in a patient with AIDS-related complex. Br J Dermatol. 1986;114:635-636.
6. Bull RH, Fallowfield ME, Marsden RA. Autoimmune blistering diseases associated with HIV infection. Clin Exp Dermatol. 1994;19:47-50.
7. Chou K, Kauh YC, Jacoby RA, et al. Autoimmune bullous disease in a patient with HIV infection. J Am Acad Dermatol. 1991;24:1022-1023.
8. Mahé A, Flageul B, Prost C, et al. Pemphigus vegetans in an HIV-1-infected man. Clin Exp Dermatol. 1994;19:447.
9. Capizzi R, Marasca G, De Luca A, et al. Pemphigus vulgaris in a human-immunodeficiency-virus-infected patient. Dermatology. 1998;197:97-98.
10. Splaver A, Silos S, Lowell B, et al. Case report: pemphigus vulgaris in a patient infected with HIV. AIDS Patient Care STDS. 2000;14:295-296.
11. Hodgson TA, Fidler SJ, Speight PM, et al. Oral pemphigus vulgaris associated with HIV infection. J Am Acad Dermatol. 2003;49:313-315.
12. Demathé A, Arede LT, Miyahara GI. Mucous membrane pemphigoid in HIV patient: a case report. Cases J. 2008;1:345.
13. Etzioni A. Immune deficiency and autoimmunity. Autoimmun Rev. 2003;2:364-369.
14. Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329-337.
The Diagnosis: Linear IgA Bullous Dermatosis
A biopsy specimen from an intact vesicle was obtained. Histologic findings showed a basket weave stratum corneum suggestive of an acute process. There was subepidermal separation with an inflammatory infiltrate of neutrophils (Figure 1). Direct immunofluorescence yielded a pattern of IgA deposition along the dermoepidermal junction (Figure 2). A diagnosis of linear IgA bullous dermatosis (LABD) was made. The patient was started on 100 mg daily of dapsone. The dose was subsequently increased to 175 mg twice daily, resulting in complete clearance. He became dermatologically disease free after 10 months and the dapsone was successfully tapered.
|
Linear IgA bullous dermatosis is an autoimmune subepidermal blistering disease with linear IgA deposits found along the basement membrane of the skin. There are 3 major categories of LABD: drug induced, systemic disorder related, and idiopathic.1 Patients with LABD present with a pruritic vesicobullous eruption that tends to favor the trunk, proximal extremities, and acral regions of the body. Mucous membrane lesions are present in less than 50% of patients.2 Linear IgA bullous dermatosis may resemble bullous pemphigoid, erythema multiforme, dermatitis herpetiformis, or toxic epidermal necrolysis. The gold standard for diagnosis is immunofluorescence staining that shows linear IgA deposition along the skin’s basement membrane.1 Prognosis for LABD is variable; there is risk for persistence and scarring.2 The drug-induced form of LABD is associated with clearance with the removal of the inciting agent.1
There are several autoimmune disorders that have been described in association with human immunodeficiency virus (HIV).3 Autoimmune bullous dermatoses, while described, are very uncommon in the setting of HIV infection. Previously reported cases include bullous pemphigoid, epidermolysis bullosa acquisita, pemphigus herpetiformis, pemphigus vegetans, pemphigus vulgaris, and cicatricial pemphigoid.4-12 The presentation of LABD in an HIV-positive patient is extremely rare.
There are 3 proposed mechanisms by which HIV and autoimmune bullous dermatoses coexist: unregulated B-cell activation, loss of T-suppressor cell regulation, and molecular mimicry. In patients with HIV, infected macrophages increase production of IL-1 and IL-6, causing nonspecific stimulation of B cells. Further production of tumor necrosis factor and other lymphotoxins may kill CD8+ T-suppressor cells, which further reduces B-cell regulation and production of nonspecific antibodies. Unregulated B-cell activation could lead to proliferation of antiself-specific B cells and autoantibodies. Additionally, various autoantibodies may arise due to mimicry between HIV antigens and human proteins. Some of the antibodies produced may be cytotoxic antilymphocyte antibodies that further disrupt B-cell regulation.13,14
Zandman-Goddard and Shoenfeld14 proposed a staging system of autoimmune disease and HIV with respect to CD4 count and viral load. Stage I is clinical latency of HIV, with a high CD4 count (>500 cells/mm3) and high viral load, which correlates with an acute infection of HIV and an intact immune system. Autoimmune disease can be seen in this stage. Stage II is cellular response, a quiescent period without overt manifestations of AIDS. The CD4 count is declining (200–499 cells/mm3), indicating immunosuppression, and the viral count is high. Autoimmune disease can occur and typically includes immune complex–mediated disease and vasculitis. Stage III is immune deficiency. The CD4 count is low (<200 cells/mm3), viral load is high, and AIDS develops. Autoimmune disease is not seen during this stage. Stage IV is the period of immune restoration following the advent of highly active antiretroviral therapy. There is a high CD4 count (>500 cells/mm3) and low viral load. There is a resurgence of autoimmune disease in this stage. Autoimmune disease can occur with an immune system capable of B- and T-cell interactions and a normal CD4 count. Autoimmunity is possible in stages I, II, and IV.14 Our patient developed bullous disease in stage II.
Although uncommon, autoimmune disease is possible in the setting of immune deficiency. The presence of autoimmune disease in a patient with HIV can only be seen during certain stages of infection. Knowledge of the possible scenarios of autoimmune disease can assist the clinician with monitoring status of the HIV infection or immune reconstitution.
The Diagnosis: Linear IgA Bullous Dermatosis
A biopsy specimen from an intact vesicle was obtained. Histologic findings showed a basket weave stratum corneum suggestive of an acute process. There was subepidermal separation with an inflammatory infiltrate of neutrophils (Figure 1). Direct immunofluorescence yielded a pattern of IgA deposition along the dermoepidermal junction (Figure 2). A diagnosis of linear IgA bullous dermatosis (LABD) was made. The patient was started on 100 mg daily of dapsone. The dose was subsequently increased to 175 mg twice daily, resulting in complete clearance. He became dermatologically disease free after 10 months and the dapsone was successfully tapered.
|
Linear IgA bullous dermatosis is an autoimmune subepidermal blistering disease with linear IgA deposits found along the basement membrane of the skin. There are 3 major categories of LABD: drug induced, systemic disorder related, and idiopathic.1 Patients with LABD present with a pruritic vesicobullous eruption that tends to favor the trunk, proximal extremities, and acral regions of the body. Mucous membrane lesions are present in less than 50% of patients.2 Linear IgA bullous dermatosis may resemble bullous pemphigoid, erythema multiforme, dermatitis herpetiformis, or toxic epidermal necrolysis. The gold standard for diagnosis is immunofluorescence staining that shows linear IgA deposition along the skin’s basement membrane.1 Prognosis for LABD is variable; there is risk for persistence and scarring.2 The drug-induced form of LABD is associated with clearance with the removal of the inciting agent.1
There are several autoimmune disorders that have been described in association with human immunodeficiency virus (HIV).3 Autoimmune bullous dermatoses, while described, are very uncommon in the setting of HIV infection. Previously reported cases include bullous pemphigoid, epidermolysis bullosa acquisita, pemphigus herpetiformis, pemphigus vegetans, pemphigus vulgaris, and cicatricial pemphigoid.4-12 The presentation of LABD in an HIV-positive patient is extremely rare.
There are 3 proposed mechanisms by which HIV and autoimmune bullous dermatoses coexist: unregulated B-cell activation, loss of T-suppressor cell regulation, and molecular mimicry. In patients with HIV, infected macrophages increase production of IL-1 and IL-6, causing nonspecific stimulation of B cells. Further production of tumor necrosis factor and other lymphotoxins may kill CD8+ T-suppressor cells, which further reduces B-cell regulation and production of nonspecific antibodies. Unregulated B-cell activation could lead to proliferation of antiself-specific B cells and autoantibodies. Additionally, various autoantibodies may arise due to mimicry between HIV antigens and human proteins. Some of the antibodies produced may be cytotoxic antilymphocyte antibodies that further disrupt B-cell regulation.13,14
Zandman-Goddard and Shoenfeld14 proposed a staging system of autoimmune disease and HIV with respect to CD4 count and viral load. Stage I is clinical latency of HIV, with a high CD4 count (>500 cells/mm3) and high viral load, which correlates with an acute infection of HIV and an intact immune system. Autoimmune disease can be seen in this stage. Stage II is cellular response, a quiescent period without overt manifestations of AIDS. The CD4 count is declining (200–499 cells/mm3), indicating immunosuppression, and the viral count is high. Autoimmune disease can occur and typically includes immune complex–mediated disease and vasculitis. Stage III is immune deficiency. The CD4 count is low (<200 cells/mm3), viral load is high, and AIDS develops. Autoimmune disease is not seen during this stage. Stage IV is the period of immune restoration following the advent of highly active antiretroviral therapy. There is a high CD4 count (>500 cells/mm3) and low viral load. There is a resurgence of autoimmune disease in this stage. Autoimmune disease can occur with an immune system capable of B- and T-cell interactions and a normal CD4 count. Autoimmunity is possible in stages I, II, and IV.14 Our patient developed bullous disease in stage II.
Although uncommon, autoimmune disease is possible in the setting of immune deficiency. The presence of autoimmune disease in a patient with HIV can only be seen during certain stages of infection. Knowledge of the possible scenarios of autoimmune disease can assist the clinician with monitoring status of the HIV infection or immune reconstitution.
1. Bouldin MB, Clowers-Webb HE, Davis JL, et al. Naproxen-associated linear IgA bullous dermatosis: case report and review. Mayo Clin Proc. 2000;75:967-970.
2. Nousari HC, Kimyai-Asadi A, Caeiro JP, et al. Clinical, demographic, and immunohistologic features of vancomycin-induced linear IgA bullous disease of the skin: report of 2 cases and review of the literature. Medicine. 1999;78:1-8.
3. Gala S, Fulcher DA. How HIV leads to autoimmune disorders. Med J Aust. 1996;164:224-226.
4. Lateef A, Packles MR, White SM, et al. Pemphigus vegetans in association with human immunodeficiency virus. Int J Dermatol. 1999;38:778-781.
5. Levy PM, Balavoine JF, Salomon D, et al. Ritodrine-responsive bullous pemphigoing in a patient with AIDS-related complex. Br J Dermatol. 1986;114:635-636.
6. Bull RH, Fallowfield ME, Marsden RA. Autoimmune blistering diseases associated with HIV infection. Clin Exp Dermatol. 1994;19:47-50.
7. Chou K, Kauh YC, Jacoby RA, et al. Autoimmune bullous disease in a patient with HIV infection. J Am Acad Dermatol. 1991;24:1022-1023.
8. Mahé A, Flageul B, Prost C, et al. Pemphigus vegetans in an HIV-1-infected man. Clin Exp Dermatol. 1994;19:447.
9. Capizzi R, Marasca G, De Luca A, et al. Pemphigus vulgaris in a human-immunodeficiency-virus-infected patient. Dermatology. 1998;197:97-98.
10. Splaver A, Silos S, Lowell B, et al. Case report: pemphigus vulgaris in a patient infected with HIV. AIDS Patient Care STDS. 2000;14:295-296.
11. Hodgson TA, Fidler SJ, Speight PM, et al. Oral pemphigus vulgaris associated with HIV infection. J Am Acad Dermatol. 2003;49:313-315.
12. Demathé A, Arede LT, Miyahara GI. Mucous membrane pemphigoid in HIV patient: a case report. Cases J. 2008;1:345.
13. Etzioni A. Immune deficiency and autoimmunity. Autoimmun Rev. 2003;2:364-369.
14. Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329-337.
1. Bouldin MB, Clowers-Webb HE, Davis JL, et al. Naproxen-associated linear IgA bullous dermatosis: case report and review. Mayo Clin Proc. 2000;75:967-970.
2. Nousari HC, Kimyai-Asadi A, Caeiro JP, et al. Clinical, demographic, and immunohistologic features of vancomycin-induced linear IgA bullous disease of the skin: report of 2 cases and review of the literature. Medicine. 1999;78:1-8.
3. Gala S, Fulcher DA. How HIV leads to autoimmune disorders. Med J Aust. 1996;164:224-226.
4. Lateef A, Packles MR, White SM, et al. Pemphigus vegetans in association with human immunodeficiency virus. Int J Dermatol. 1999;38:778-781.
5. Levy PM, Balavoine JF, Salomon D, et al. Ritodrine-responsive bullous pemphigoing in a patient with AIDS-related complex. Br J Dermatol. 1986;114:635-636.
6. Bull RH, Fallowfield ME, Marsden RA. Autoimmune blistering diseases associated with HIV infection. Clin Exp Dermatol. 1994;19:47-50.
7. Chou K, Kauh YC, Jacoby RA, et al. Autoimmune bullous disease in a patient with HIV infection. J Am Acad Dermatol. 1991;24:1022-1023.
8. Mahé A, Flageul B, Prost C, et al. Pemphigus vegetans in an HIV-1-infected man. Clin Exp Dermatol. 1994;19:447.
9. Capizzi R, Marasca G, De Luca A, et al. Pemphigus vulgaris in a human-immunodeficiency-virus-infected patient. Dermatology. 1998;197:97-98.
10. Splaver A, Silos S, Lowell B, et al. Case report: pemphigus vulgaris in a patient infected with HIV. AIDS Patient Care STDS. 2000;14:295-296.
11. Hodgson TA, Fidler SJ, Speight PM, et al. Oral pemphigus vulgaris associated with HIV infection. J Am Acad Dermatol. 2003;49:313-315.
12. Demathé A, Arede LT, Miyahara GI. Mucous membrane pemphigoid in HIV patient: a case report. Cases J. 2008;1:345.
13. Etzioni A. Immune deficiency and autoimmunity. Autoimmun Rev. 2003;2:364-369.
14. Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329-337.
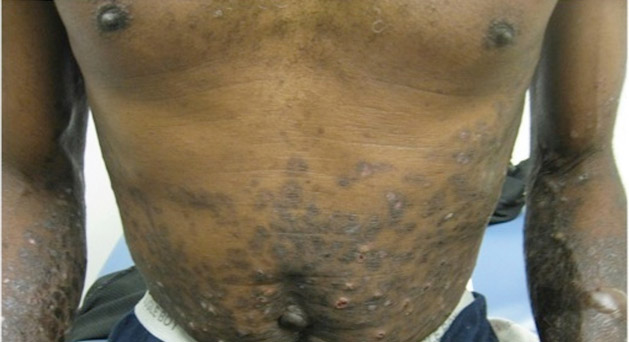
A 50-year-old black man presented with a new-onset widespread pruritic bullous eruption 7 months after being diagnosed with human immunodeficiency virus. The CD4 lymphocyte count was 421 cells/mm3 and viral load was 7818 copies/mL. Results of a viral culture were negative for herpes simplex virus. Dermatologic examination revealed numerous intact tense bullae as well as scattered erosions on the trunk and extremities. Postinflammatory hyperpigmentation was prominent, with some areas of hypopigmentation and depigmentation.
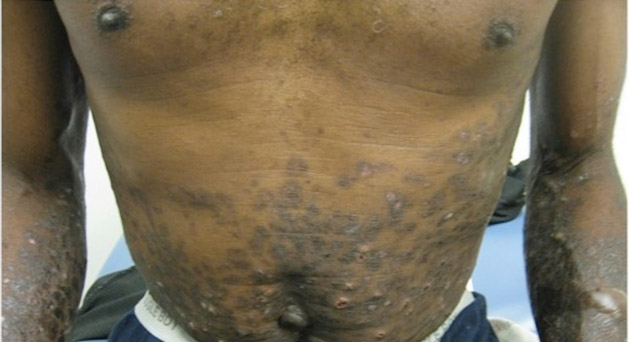
Lobular-Appearing Nodule on the Scalp
The Diagnosis: Dermal Cylindroma
Microsopic evaluation of a tangential biopsy revealed findings of a dermal process consisting of well-circumscribed islands of pale and darker blue cells with little cytoplasm outlined by a hyaline basement membrane (Figure). These cellular islands were arranged in a jigsawlike configuration. These findings were thought to be consistent with a diagnosis of cylindroma.
|
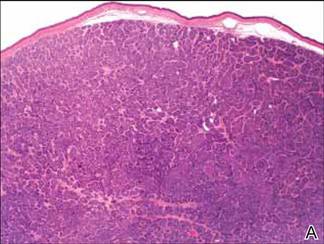
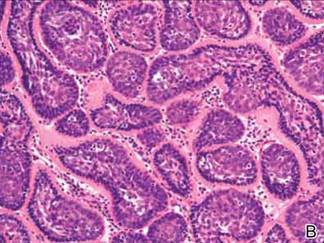
Cylindromas are benign appendageal neoplasms with a somewhat controversial histogenesis. Munger and colleagues1 investigated the pattern of acid mucopolysaccharide secretion by these tumors in association with prosecretory vacuoles in proximity to the Golgi apparatus, which led to their impression that cylindromas most resemble eccrine rather than apocrine sweat glands. Other researchers, however, have concluded that cylindromas are of apocrine derivation.2
Clinically, cylindromas appear most often in 2 settings: isolated or as a manifestation of one of several inherited familial syndromes. One such syndrome is familial cylindromatosis, a rare autosomal-dominant disorder in which affected individuals develop multiple cylindromas, usually on the head and neck. The merging of multiple lesions gives rise to the often-employed term turban tumor.3 This syndrome has been linked to mutations in the cylindromatosis gene, CYLD.4 Brooke-Spiegler syndrome also has been associated with the development of multiple cylindromas. Similar to familial cylindromatosis, it is inherited in an autosomal-dominant fashion. Brooke-Spiegler syndrome is typified by the appearance of multiple cylindromas, trichoepitheliomas, and less commonly spiradenomas. Mutations in the CYLD gene also have been linked to Brooke-Spiegler syndrome in some cases.5
Although considered a benign entity, in rare cases cylindromas have shown evidence of malignant transformation to cylindrocarcinoma. This more aggressive tumor may occur in the setting of isolated cylindromas or more commonly in individuals with numerous lesions, as with both familial cylindromatosis and Brooke-Spiegler syndrome. These lesions may appear to grow rapidly, ulcerate, or bleed, traits that are not associated with their benign counterparts.
Diagnosis of cylindromas rests on histopathologic confirmation, which demonstrates well-defined dermal islands of epithelial cells comprised of dark- and pale-staining nuclei. These tumor islands are surrounded by a hyaline basement membrane and often take on the appearance of a jigsaw puzzle. Cylindrocarcinomas exhibit greater cellular pleomorphism and higher mitotic rates.
Dermal cylindromas require no further treatment but can be electively excised, while treatment of cylindrocarcinoma with excision is curative.6 Definitive excision was offered to our patient, but she declined treatment.
1. Munger BL, Graham JH, Helwig EB. Ultrastructure and histochemical characteristics of dermal eccrine cylindroma (turban tumor). J Invest Dermatol. 1962;39:577-595.
2. Tellechea O, Reis JP, Ilheu O, et al. Dermal cylindroma. an immunohistochemical study of thirteen cases. Am J Dermatopathol. 1995;17:260-265.
3. Biggs PJ, Wooster R, Ford D, et al. Familial cylindromatosis (turban tumour syndrome) gene localised to chromosome 16q12-q13: evidence for its role as a tumour suppressor gene. Nat Genet. 1995;11:441-443.
4. Bignell GR, Warren W, Seal S, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet. 2000;25:160-165.
5. Bowen S, Gill M, Lee DA, et al. Mutations in the CYLD gene in Brooke-Spiegler syndrome, familial cylindromatosis, and multiple familial trichoepithelioma: lack of genotype-phenotype correlation. J Invest Dermatol. 2005;124:919-920.
6. Gerretsen AL, van der Putte SC, Deenstra W, et al. Cutaneous cylindroma with malignant transformation. Cancer. 1993;72:1618-1623.
The Diagnosis: Dermal Cylindroma
Microsopic evaluation of a tangential biopsy revealed findings of a dermal process consisting of well-circumscribed islands of pale and darker blue cells with little cytoplasm outlined by a hyaline basement membrane (Figure). These cellular islands were arranged in a jigsawlike configuration. These findings were thought to be consistent with a diagnosis of cylindroma.
|
Cylindromas are benign appendageal neoplasms with a somewhat controversial histogenesis. Munger and colleagues1 investigated the pattern of acid mucopolysaccharide secretion by these tumors in association with prosecretory vacuoles in proximity to the Golgi apparatus, which led to their impression that cylindromas most resemble eccrine rather than apocrine sweat glands. Other researchers, however, have concluded that cylindromas are of apocrine derivation.2
Clinically, cylindromas appear most often in 2 settings: isolated or as a manifestation of one of several inherited familial syndromes. One such syndrome is familial cylindromatosis, a rare autosomal-dominant disorder in which affected individuals develop multiple cylindromas, usually on the head and neck. The merging of multiple lesions gives rise to the often-employed term turban tumor.3 This syndrome has been linked to mutations in the cylindromatosis gene, CYLD.4 Brooke-Spiegler syndrome also has been associated with the development of multiple cylindromas. Similar to familial cylindromatosis, it is inherited in an autosomal-dominant fashion. Brooke-Spiegler syndrome is typified by the appearance of multiple cylindromas, trichoepitheliomas, and less commonly spiradenomas. Mutations in the CYLD gene also have been linked to Brooke-Spiegler syndrome in some cases.5
Although considered a benign entity, in rare cases cylindromas have shown evidence of malignant transformation to cylindrocarcinoma. This more aggressive tumor may occur in the setting of isolated cylindromas or more commonly in individuals with numerous lesions, as with both familial cylindromatosis and Brooke-Spiegler syndrome. These lesions may appear to grow rapidly, ulcerate, or bleed, traits that are not associated with their benign counterparts.
Diagnosis of cylindromas rests on histopathologic confirmation, which demonstrates well-defined dermal islands of epithelial cells comprised of dark- and pale-staining nuclei. These tumor islands are surrounded by a hyaline basement membrane and often take on the appearance of a jigsaw puzzle. Cylindrocarcinomas exhibit greater cellular pleomorphism and higher mitotic rates.
Dermal cylindromas require no further treatment but can be electively excised, while treatment of cylindrocarcinoma with excision is curative.6 Definitive excision was offered to our patient, but she declined treatment.
The Diagnosis: Dermal Cylindroma
Microsopic evaluation of a tangential biopsy revealed findings of a dermal process consisting of well-circumscribed islands of pale and darker blue cells with little cytoplasm outlined by a hyaline basement membrane (Figure). These cellular islands were arranged in a jigsawlike configuration. These findings were thought to be consistent with a diagnosis of cylindroma.
|
Cylindromas are benign appendageal neoplasms with a somewhat controversial histogenesis. Munger and colleagues1 investigated the pattern of acid mucopolysaccharide secretion by these tumors in association with prosecretory vacuoles in proximity to the Golgi apparatus, which led to their impression that cylindromas most resemble eccrine rather than apocrine sweat glands. Other researchers, however, have concluded that cylindromas are of apocrine derivation.2
Clinically, cylindromas appear most often in 2 settings: isolated or as a manifestation of one of several inherited familial syndromes. One such syndrome is familial cylindromatosis, a rare autosomal-dominant disorder in which affected individuals develop multiple cylindromas, usually on the head and neck. The merging of multiple lesions gives rise to the often-employed term turban tumor.3 This syndrome has been linked to mutations in the cylindromatosis gene, CYLD.4 Brooke-Spiegler syndrome also has been associated with the development of multiple cylindromas. Similar to familial cylindromatosis, it is inherited in an autosomal-dominant fashion. Brooke-Spiegler syndrome is typified by the appearance of multiple cylindromas, trichoepitheliomas, and less commonly spiradenomas. Mutations in the CYLD gene also have been linked to Brooke-Spiegler syndrome in some cases.5
Although considered a benign entity, in rare cases cylindromas have shown evidence of malignant transformation to cylindrocarcinoma. This more aggressive tumor may occur in the setting of isolated cylindromas or more commonly in individuals with numerous lesions, as with both familial cylindromatosis and Brooke-Spiegler syndrome. These lesions may appear to grow rapidly, ulcerate, or bleed, traits that are not associated with their benign counterparts.
Diagnosis of cylindromas rests on histopathologic confirmation, which demonstrates well-defined dermal islands of epithelial cells comprised of dark- and pale-staining nuclei. These tumor islands are surrounded by a hyaline basement membrane and often take on the appearance of a jigsaw puzzle. Cylindrocarcinomas exhibit greater cellular pleomorphism and higher mitotic rates.
Dermal cylindromas require no further treatment but can be electively excised, while treatment of cylindrocarcinoma with excision is curative.6 Definitive excision was offered to our patient, but she declined treatment.
1. Munger BL, Graham JH, Helwig EB. Ultrastructure and histochemical characteristics of dermal eccrine cylindroma (turban tumor). J Invest Dermatol. 1962;39:577-595.
2. Tellechea O, Reis JP, Ilheu O, et al. Dermal cylindroma. an immunohistochemical study of thirteen cases. Am J Dermatopathol. 1995;17:260-265.
3. Biggs PJ, Wooster R, Ford D, et al. Familial cylindromatosis (turban tumour syndrome) gene localised to chromosome 16q12-q13: evidence for its role as a tumour suppressor gene. Nat Genet. 1995;11:441-443.
4. Bignell GR, Warren W, Seal S, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet. 2000;25:160-165.
5. Bowen S, Gill M, Lee DA, et al. Mutations in the CYLD gene in Brooke-Spiegler syndrome, familial cylindromatosis, and multiple familial trichoepithelioma: lack of genotype-phenotype correlation. J Invest Dermatol. 2005;124:919-920.
6. Gerretsen AL, van der Putte SC, Deenstra W, et al. Cutaneous cylindroma with malignant transformation. Cancer. 1993;72:1618-1623.
1. Munger BL, Graham JH, Helwig EB. Ultrastructure and histochemical characteristics of dermal eccrine cylindroma (turban tumor). J Invest Dermatol. 1962;39:577-595.
2. Tellechea O, Reis JP, Ilheu O, et al. Dermal cylindroma. an immunohistochemical study of thirteen cases. Am J Dermatopathol. 1995;17:260-265.
3. Biggs PJ, Wooster R, Ford D, et al. Familial cylindromatosis (turban tumour syndrome) gene localised to chromosome 16q12-q13: evidence for its role as a tumour suppressor gene. Nat Genet. 1995;11:441-443.
4. Bignell GR, Warren W, Seal S, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet. 2000;25:160-165.
5. Bowen S, Gill M, Lee DA, et al. Mutations in the CYLD gene in Brooke-Spiegler syndrome, familial cylindromatosis, and multiple familial trichoepithelioma: lack of genotype-phenotype correlation. J Invest Dermatol. 2005;124:919-920.
6. Gerretsen AL, van der Putte SC, Deenstra W, et al. Cutaneous cylindroma with malignant transformation. Cancer. 1993;72:1618-1623.
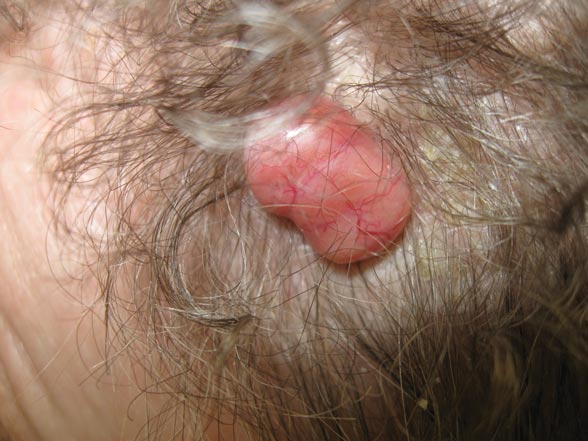
A 79-year-old woman presented with a lesion on the left side of the scalp of several years’ duration that had slowly increased in size. Despite its growth, the lesion remained asymptomatic. Physical examination revealed an exophytic, lobular-appearing nodule on the left side of the temporoparietal scalp, measuring 1.5 cm in size.
