User login
Lupus-like Rash of Chronic Granulomatous Disease Effectively Treated With Hydroxychloroquine
To the Editor:
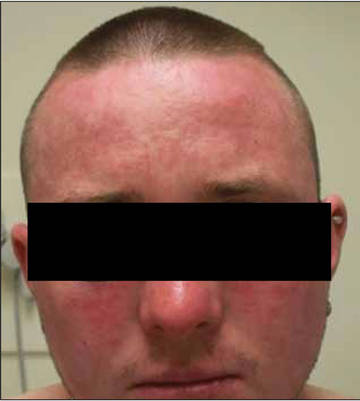
A 26-year-old man was referred to our clinic for evaluation of a persistent red rash to rule out cutaneous lupus erythematosus (LE). The patient was diagnosed at 12 years of age with autosomal-recessive chronic granulomatous disease (CGD)(nitroblue tetrazolium test, 5.0; low normal, 20.6), type p47phox mutation. At that time the patient had recurrent fevers, sinusitis, anemia, and noncaseating granulomatous liver lesions, but he lacked any cutaneous manifestations. The patient was then treated for approximately 2 years with interferon therapy but discontinued therapy given the absence of any signs or symptoms. He remained asymptomatic until approximately 16 years of age when he experienced the onset of an intermittently painful and pruritic rash on the face that slowly spread over the ensuing years to involve the trunk, arms, forearms, and hands. Although he reported that sunlight exacerbated the rash, the rash also persisted through the winter months when the majority of the sun-exposed areas of the trunk and arms were covered. He denied exposure to topical products and denied the use of any oral medications (prescription or over-the-counter).
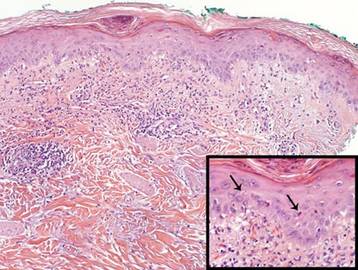
Review of systems was negative for fever, fatigue, malaise, headaches, joint pain, arthritis, oral ulcers, dyspnea, or dysuria. Physical examination revealed a well-defined exanthem comprised of erythematous, mildly indurated papules coalescing into larger plaques with white scale that were exclusively limited to the photodistributed areas of the face (Figure 1), neck, arms, forearms, hands, chest, and back. Laboratory test results included the following: minimally elevated erythrocyte sedimentation rate of 31 mm/h (reference range, 0–15 mm/h) and rheumatoid factor of 45 IU/mL (reference range, <20 IU/mL; negative antinuclear antibody screen, Sjögren syndrome antigens A and B, double-stranded DNA, anti–extractable nuclear antigen antibody test, and anti-Jo-1 antibody; complete blood cell count revealed no abnormalities; basic metabolic panel, C3 and C4, CH50, glucose-6-phosphate dehydrogenase activity, total plasma porphyrins, and testing for hepatitis B and C virus and human immunodeficiency virus serologies were negative. Skin biopsy from a lesion on the lateral arm showed features consistent with interface dermatitis (Figure 2). Additional skin biopsies for direct immunofluorescence showed linear deposition of IgG at the dermoepidermal junction, both from involved and uninvolved neck skin (more focally from the involved site). Extensive photopatch testing did not show any clinically relevant positive reactions.
|
Given the patient’s history of CGD and the extensive negative workup for rheumatologic, photoallergic, and phototoxic causes, the patient was diagnosed with a lupus-like rash of CGD. The rash failed to respond to rigorous sun avoidance and a 3-week on/1-week off regimen of high-potency class 1 topical steroids to the trunk, arms, forearms, and legs, and lower-potency class 4 topical steroid to the face, with disease flaring almost immediately on cessation of treatment during the rest weeks. Given the marked photodistribution resembling subacute cutaneous LE, oral hydroxychloroquine 200 mg (5.7 mg/kg) twice daily was initiated in addition to continued topical steroid therapy.
Four months after the addition of hydroxychloroquine, the patient showed considerable improvement of the rash. Seven months after initiation of hydroxychloroquine, the photodistributed rash was completely resolved and topical steroids were stopped. The rash remained in remission for an additional 24 months with hydroxychloroquine alone, at which time hydroxychloroquine was stopped; however, the rash flared 2 months later and hydroxychloroquine was restarted at 200 mg twice daily, resulting in clearance within 3 months. The patient was maintained on this dose of hydroxychloroquine.
During treatment, the patient had an episode of extensive furunculosis caused by Staphylococcus aureus that was successfully treated with a 14-day course of oral doxycycline 100 mg twice daily. He has since been maintained on prophylactic intranasal mupirocin ointment 2% for the first several days of each month and daily benzoyl peroxide wash 10% without further episodes. He also developed a single lesion of alopecia areata that was successfully treated with intralesional steroid injections.
Chronic granulomatous disease can either be X-linked or, less commonly, autosomal recessive, resulting from a defect in components of the nicotinamide adenine dinucleotide phosphate oxidase complex that is necessary to generate reactive oxygen intermediates for killing phagocytosed microbes. Cutaneous manifestations are relatively common in CGD (60%–70% of cases)1 and include infectious lesions (eg, recurrent mucous membrane infections, impetigo, carbuncles, otitis externa, suppurative lymphadenopathy) as well as the less common chronic inflammatory conditions such as lupus-like eruption, aphthous stomatitis, Raynaud phenomenon, arcuate dermal erythema, and Jessner lymphocytic infiltrate.2 The pathognomonic clinical feature of CGD is the presence of characteristic multinucleated giant cell granulomas distributed in multiple organ systems such as the gastrointestinal system, causing pyloric and/or small bowel obstruction, and the genitourinary system, causing ureter and/or bladder outlet obstruction.3
Importantly, CGD patients also demonstrate immune-related inflammatory disorders, most commonly inflammatory bowel disease, IgA nephropathy, sarcoidosis, and juvenile idiopathic arthritis.3 In addition, both CGD patients and female carriers of X-linked CGD have been reported to demonstrate lupus-like rashes that share overlapping clinical and histologic features with the rashes seen in true discoid LE and tumid LE patients without CGD.4-6 This lupus-like rash is more commonly observed in adulthood and in carriers, possibly secondary to the high childhood mortality rate of CGD patients.4,6
De Ravin et al3 proposed that autoimmune conditions arising in CGD patients who have met established criteria for a particular autoimmune disease should be treated for that condition rather than consider it as a part of the CGD spectrum. This theory has important therapeutic implications, including initiating paradoxical corticosteroid and/or steroid-sparing immunosuppressive agents in this otherwise immunocompromised patient population. They reported a 21-year-old man with cutaneous LE lesions and negative lupus serologies whose lesions were refractory to topical steroids but responded to systemic prednisone, requiring a low-dose alternate-day maintenance regimen.3 Beyond the development of a true autoimmune disease associated with CGD, systemic medications, specifically voriconazole, have been implicated as an alternative etiology for this rash in CGD.7 While important to consider, our patient’s rash presented in the absence of any systemic medications, supporting the former etiology over the latter.
Our case demonstrates the utility of hydroxychloroquine to treat the lupus-like rash of CGD. Similarly, the lupus-like symptoms of female carriers of X-linked CGD, predominantly with negative lupus serologies, also have been reported to respond to hydroxychloroquine and mepacrine.4,5,8-10 Interestingly, the utility of monotherapy with hydroxychloroquine may extend beyond treating cutaneous lupus-like lesions, as this regimen also was reported to successfully treat gastric granulomatous involvement in a CGD patient.11
Chronic granulomatous disease often is fatal in early childhood or adolescence due to sequelae from infections or chronic granulomatous infiltration of internal organs. Residual reactive oxygen intermediate production was shown to be a predictor of overall survival, and CGD patients with 1% of normal reactive oxygen intermediate production by neutrophils had a greater likelihood of survival.12 In this regard, the otherwise good health of our patient at the time of presentation was consistent with his initial nitroblue tetrazolium test showing some residual oxidative activity, emphasizing the phenotypic variability of this rare genetic disorder and the importance of considering CGD in the diagnosis of seronegative cutaneous lupus-like reactions.
1. Dohil M, Prendiville JS, Crawford RI, et al. Cutaneous manifestations of chronic granulomatous disease. a report of four cases and review of the literature. J Am Acad Dermatol. 1997;36(6, pt 1):899-907.
2. Chowdhury MM, Anstey A, Matthews CN. The dermatosis of chronic granulomatous disease. Clin Exp Dermatol. 2000;25:190-194.
3. De Ravin SS, Naumann N, Cowen EW, et al. Chronic granulomatous disease as a risk factor for autoimmune disease [published online ahead of print September 26, 2008]. J Allergy Clin Immunol. 2008;122:1097-1103.
4. Kragballe K, Borregaard N, Brandrup F, et al. Relation of monocyte and neutrophil oxidative metabolism to skin and oral lesions in carriers of chronic granulomatous disease. Clin Exp Immunol. 1981;43:390-398.
5. Barton LL, Johnson CR. Discoid lupus erythematosus and X-linked chronic granulomatous disease. Pediatr Dermatol. 1986;3:376-379.
6. Córdoba-Guijarro S, Feal C, Daudén E, et al. Lupus erythematosus-like lesions in a carrier of X-linked chronic granulomatous disease. J Eur Acad Dermatol Venereol. 2000;14:409-411.
7. Gomez-Moyano E, Vera-Casaño A, Moreno-Perez D, et al. Lupus erythematosus-like lesions by voriconazole in an infant with chronic granulomatous disease. Pediatr Dermatol. 2010;27:105-106.
8. Brandrup F, Koch C, Petri M, et al. Discoid lupus erythematosus-like lesions and stomatitis in female carriers of X-linked chronic granulomatous disease. Br J Dermatol. 1981;104:495-505.
9. Cale CM, Morton L, Goldblatt D. Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin Exp Immunol. 2007;148:79-84.
10. Levinsky RJ, Harvey BA, Roberton DM, et al. A polymorph bactericidal defect and a lupus-like syndrome. Arch Dis Child. 1981;56:382-385.
11. Arlet JB, Aouba A, Suarez F, et al. Efficiency of hydroxychloroquine in the treatment of granulomatous complications in chronic granulomatous disease. Eur J Gastroenterol Hepatol. 2008;20:142-144.
12. Kuhns DB, Alvord WG, Heller T, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363:2600-2610.
To the Editor:
A 26-year-old man was referred to our clinic for evaluation of a persistent red rash to rule out cutaneous lupus erythematosus (LE). The patient was diagnosed at 12 years of age with autosomal-recessive chronic granulomatous disease (CGD)(nitroblue tetrazolium test, 5.0; low normal, 20.6), type p47phox mutation. At that time the patient had recurrent fevers, sinusitis, anemia, and noncaseating granulomatous liver lesions, but he lacked any cutaneous manifestations. The patient was then treated for approximately 2 years with interferon therapy but discontinued therapy given the absence of any signs or symptoms. He remained asymptomatic until approximately 16 years of age when he experienced the onset of an intermittently painful and pruritic rash on the face that slowly spread over the ensuing years to involve the trunk, arms, forearms, and hands. Although he reported that sunlight exacerbated the rash, the rash also persisted through the winter months when the majority of the sun-exposed areas of the trunk and arms were covered. He denied exposure to topical products and denied the use of any oral medications (prescription or over-the-counter).
Review of systems was negative for fever, fatigue, malaise, headaches, joint pain, arthritis, oral ulcers, dyspnea, or dysuria. Physical examination revealed a well-defined exanthem comprised of erythematous, mildly indurated papules coalescing into larger plaques with white scale that were exclusively limited to the photodistributed areas of the face (Figure 1), neck, arms, forearms, hands, chest, and back. Laboratory test results included the following: minimally elevated erythrocyte sedimentation rate of 31 mm/h (reference range, 0–15 mm/h) and rheumatoid factor of 45 IU/mL (reference range, <20 IU/mL; negative antinuclear antibody screen, Sjögren syndrome antigens A and B, double-stranded DNA, anti–extractable nuclear antigen antibody test, and anti-Jo-1 antibody; complete blood cell count revealed no abnormalities; basic metabolic panel, C3 and C4, CH50, glucose-6-phosphate dehydrogenase activity, total plasma porphyrins, and testing for hepatitis B and C virus and human immunodeficiency virus serologies were negative. Skin biopsy from a lesion on the lateral arm showed features consistent with interface dermatitis (Figure 2). Additional skin biopsies for direct immunofluorescence showed linear deposition of IgG at the dermoepidermal junction, both from involved and uninvolved neck skin (more focally from the involved site). Extensive photopatch testing did not show any clinically relevant positive reactions.
|
Given the patient’s history of CGD and the extensive negative workup for rheumatologic, photoallergic, and phototoxic causes, the patient was diagnosed with a lupus-like rash of CGD. The rash failed to respond to rigorous sun avoidance and a 3-week on/1-week off regimen of high-potency class 1 topical steroids to the trunk, arms, forearms, and legs, and lower-potency class 4 topical steroid to the face, with disease flaring almost immediately on cessation of treatment during the rest weeks. Given the marked photodistribution resembling subacute cutaneous LE, oral hydroxychloroquine 200 mg (5.7 mg/kg) twice daily was initiated in addition to continued topical steroid therapy.
Four months after the addition of hydroxychloroquine, the patient showed considerable improvement of the rash. Seven months after initiation of hydroxychloroquine, the photodistributed rash was completely resolved and topical steroids were stopped. The rash remained in remission for an additional 24 months with hydroxychloroquine alone, at which time hydroxychloroquine was stopped; however, the rash flared 2 months later and hydroxychloroquine was restarted at 200 mg twice daily, resulting in clearance within 3 months. The patient was maintained on this dose of hydroxychloroquine.
During treatment, the patient had an episode of extensive furunculosis caused by Staphylococcus aureus that was successfully treated with a 14-day course of oral doxycycline 100 mg twice daily. He has since been maintained on prophylactic intranasal mupirocin ointment 2% for the first several days of each month and daily benzoyl peroxide wash 10% without further episodes. He also developed a single lesion of alopecia areata that was successfully treated with intralesional steroid injections.
Chronic granulomatous disease can either be X-linked or, less commonly, autosomal recessive, resulting from a defect in components of the nicotinamide adenine dinucleotide phosphate oxidase complex that is necessary to generate reactive oxygen intermediates for killing phagocytosed microbes. Cutaneous manifestations are relatively common in CGD (60%–70% of cases)1 and include infectious lesions (eg, recurrent mucous membrane infections, impetigo, carbuncles, otitis externa, suppurative lymphadenopathy) as well as the less common chronic inflammatory conditions such as lupus-like eruption, aphthous stomatitis, Raynaud phenomenon, arcuate dermal erythema, and Jessner lymphocytic infiltrate.2 The pathognomonic clinical feature of CGD is the presence of characteristic multinucleated giant cell granulomas distributed in multiple organ systems such as the gastrointestinal system, causing pyloric and/or small bowel obstruction, and the genitourinary system, causing ureter and/or bladder outlet obstruction.3
Importantly, CGD patients also demonstrate immune-related inflammatory disorders, most commonly inflammatory bowel disease, IgA nephropathy, sarcoidosis, and juvenile idiopathic arthritis.3 In addition, both CGD patients and female carriers of X-linked CGD have been reported to demonstrate lupus-like rashes that share overlapping clinical and histologic features with the rashes seen in true discoid LE and tumid LE patients without CGD.4-6 This lupus-like rash is more commonly observed in adulthood and in carriers, possibly secondary to the high childhood mortality rate of CGD patients.4,6
De Ravin et al3 proposed that autoimmune conditions arising in CGD patients who have met established criteria for a particular autoimmune disease should be treated for that condition rather than consider it as a part of the CGD spectrum. This theory has important therapeutic implications, including initiating paradoxical corticosteroid and/or steroid-sparing immunosuppressive agents in this otherwise immunocompromised patient population. They reported a 21-year-old man with cutaneous LE lesions and negative lupus serologies whose lesions were refractory to topical steroids but responded to systemic prednisone, requiring a low-dose alternate-day maintenance regimen.3 Beyond the development of a true autoimmune disease associated with CGD, systemic medications, specifically voriconazole, have been implicated as an alternative etiology for this rash in CGD.7 While important to consider, our patient’s rash presented in the absence of any systemic medications, supporting the former etiology over the latter.
Our case demonstrates the utility of hydroxychloroquine to treat the lupus-like rash of CGD. Similarly, the lupus-like symptoms of female carriers of X-linked CGD, predominantly with negative lupus serologies, also have been reported to respond to hydroxychloroquine and mepacrine.4,5,8-10 Interestingly, the utility of monotherapy with hydroxychloroquine may extend beyond treating cutaneous lupus-like lesions, as this regimen also was reported to successfully treat gastric granulomatous involvement in a CGD patient.11
Chronic granulomatous disease often is fatal in early childhood or adolescence due to sequelae from infections or chronic granulomatous infiltration of internal organs. Residual reactive oxygen intermediate production was shown to be a predictor of overall survival, and CGD patients with 1% of normal reactive oxygen intermediate production by neutrophils had a greater likelihood of survival.12 In this regard, the otherwise good health of our patient at the time of presentation was consistent with his initial nitroblue tetrazolium test showing some residual oxidative activity, emphasizing the phenotypic variability of this rare genetic disorder and the importance of considering CGD in the diagnosis of seronegative cutaneous lupus-like reactions.
To the Editor:
A 26-year-old man was referred to our clinic for evaluation of a persistent red rash to rule out cutaneous lupus erythematosus (LE). The patient was diagnosed at 12 years of age with autosomal-recessive chronic granulomatous disease (CGD)(nitroblue tetrazolium test, 5.0; low normal, 20.6), type p47phox mutation. At that time the patient had recurrent fevers, sinusitis, anemia, and noncaseating granulomatous liver lesions, but he lacked any cutaneous manifestations. The patient was then treated for approximately 2 years with interferon therapy but discontinued therapy given the absence of any signs or symptoms. He remained asymptomatic until approximately 16 years of age when he experienced the onset of an intermittently painful and pruritic rash on the face that slowly spread over the ensuing years to involve the trunk, arms, forearms, and hands. Although he reported that sunlight exacerbated the rash, the rash also persisted through the winter months when the majority of the sun-exposed areas of the trunk and arms were covered. He denied exposure to topical products and denied the use of any oral medications (prescription or over-the-counter).
Review of systems was negative for fever, fatigue, malaise, headaches, joint pain, arthritis, oral ulcers, dyspnea, or dysuria. Physical examination revealed a well-defined exanthem comprised of erythematous, mildly indurated papules coalescing into larger plaques with white scale that were exclusively limited to the photodistributed areas of the face (Figure 1), neck, arms, forearms, hands, chest, and back. Laboratory test results included the following: minimally elevated erythrocyte sedimentation rate of 31 mm/h (reference range, 0–15 mm/h) and rheumatoid factor of 45 IU/mL (reference range, <20 IU/mL; negative antinuclear antibody screen, Sjögren syndrome antigens A and B, double-stranded DNA, anti–extractable nuclear antigen antibody test, and anti-Jo-1 antibody; complete blood cell count revealed no abnormalities; basic metabolic panel, C3 and C4, CH50, glucose-6-phosphate dehydrogenase activity, total plasma porphyrins, and testing for hepatitis B and C virus and human immunodeficiency virus serologies were negative. Skin biopsy from a lesion on the lateral arm showed features consistent with interface dermatitis (Figure 2). Additional skin biopsies for direct immunofluorescence showed linear deposition of IgG at the dermoepidermal junction, both from involved and uninvolved neck skin (more focally from the involved site). Extensive photopatch testing did not show any clinically relevant positive reactions.
|
Given the patient’s history of CGD and the extensive negative workup for rheumatologic, photoallergic, and phototoxic causes, the patient was diagnosed with a lupus-like rash of CGD. The rash failed to respond to rigorous sun avoidance and a 3-week on/1-week off regimen of high-potency class 1 topical steroids to the trunk, arms, forearms, and legs, and lower-potency class 4 topical steroid to the face, with disease flaring almost immediately on cessation of treatment during the rest weeks. Given the marked photodistribution resembling subacute cutaneous LE, oral hydroxychloroquine 200 mg (5.7 mg/kg) twice daily was initiated in addition to continued topical steroid therapy.
Four months after the addition of hydroxychloroquine, the patient showed considerable improvement of the rash. Seven months after initiation of hydroxychloroquine, the photodistributed rash was completely resolved and topical steroids were stopped. The rash remained in remission for an additional 24 months with hydroxychloroquine alone, at which time hydroxychloroquine was stopped; however, the rash flared 2 months later and hydroxychloroquine was restarted at 200 mg twice daily, resulting in clearance within 3 months. The patient was maintained on this dose of hydroxychloroquine.
During treatment, the patient had an episode of extensive furunculosis caused by Staphylococcus aureus that was successfully treated with a 14-day course of oral doxycycline 100 mg twice daily. He has since been maintained on prophylactic intranasal mupirocin ointment 2% for the first several days of each month and daily benzoyl peroxide wash 10% without further episodes. He also developed a single lesion of alopecia areata that was successfully treated with intralesional steroid injections.
Chronic granulomatous disease can either be X-linked or, less commonly, autosomal recessive, resulting from a defect in components of the nicotinamide adenine dinucleotide phosphate oxidase complex that is necessary to generate reactive oxygen intermediates for killing phagocytosed microbes. Cutaneous manifestations are relatively common in CGD (60%–70% of cases)1 and include infectious lesions (eg, recurrent mucous membrane infections, impetigo, carbuncles, otitis externa, suppurative lymphadenopathy) as well as the less common chronic inflammatory conditions such as lupus-like eruption, aphthous stomatitis, Raynaud phenomenon, arcuate dermal erythema, and Jessner lymphocytic infiltrate.2 The pathognomonic clinical feature of CGD is the presence of characteristic multinucleated giant cell granulomas distributed in multiple organ systems such as the gastrointestinal system, causing pyloric and/or small bowel obstruction, and the genitourinary system, causing ureter and/or bladder outlet obstruction.3
Importantly, CGD patients also demonstrate immune-related inflammatory disorders, most commonly inflammatory bowel disease, IgA nephropathy, sarcoidosis, and juvenile idiopathic arthritis.3 In addition, both CGD patients and female carriers of X-linked CGD have been reported to demonstrate lupus-like rashes that share overlapping clinical and histologic features with the rashes seen in true discoid LE and tumid LE patients without CGD.4-6 This lupus-like rash is more commonly observed in adulthood and in carriers, possibly secondary to the high childhood mortality rate of CGD patients.4,6
De Ravin et al3 proposed that autoimmune conditions arising in CGD patients who have met established criteria for a particular autoimmune disease should be treated for that condition rather than consider it as a part of the CGD spectrum. This theory has important therapeutic implications, including initiating paradoxical corticosteroid and/or steroid-sparing immunosuppressive agents in this otherwise immunocompromised patient population. They reported a 21-year-old man with cutaneous LE lesions and negative lupus serologies whose lesions were refractory to topical steroids but responded to systemic prednisone, requiring a low-dose alternate-day maintenance regimen.3 Beyond the development of a true autoimmune disease associated with CGD, systemic medications, specifically voriconazole, have been implicated as an alternative etiology for this rash in CGD.7 While important to consider, our patient’s rash presented in the absence of any systemic medications, supporting the former etiology over the latter.
Our case demonstrates the utility of hydroxychloroquine to treat the lupus-like rash of CGD. Similarly, the lupus-like symptoms of female carriers of X-linked CGD, predominantly with negative lupus serologies, also have been reported to respond to hydroxychloroquine and mepacrine.4,5,8-10 Interestingly, the utility of monotherapy with hydroxychloroquine may extend beyond treating cutaneous lupus-like lesions, as this regimen also was reported to successfully treat gastric granulomatous involvement in a CGD patient.11
Chronic granulomatous disease often is fatal in early childhood or adolescence due to sequelae from infections or chronic granulomatous infiltration of internal organs. Residual reactive oxygen intermediate production was shown to be a predictor of overall survival, and CGD patients with 1% of normal reactive oxygen intermediate production by neutrophils had a greater likelihood of survival.12 In this regard, the otherwise good health of our patient at the time of presentation was consistent with his initial nitroblue tetrazolium test showing some residual oxidative activity, emphasizing the phenotypic variability of this rare genetic disorder and the importance of considering CGD in the diagnosis of seronegative cutaneous lupus-like reactions.
1. Dohil M, Prendiville JS, Crawford RI, et al. Cutaneous manifestations of chronic granulomatous disease. a report of four cases and review of the literature. J Am Acad Dermatol. 1997;36(6, pt 1):899-907.
2. Chowdhury MM, Anstey A, Matthews CN. The dermatosis of chronic granulomatous disease. Clin Exp Dermatol. 2000;25:190-194.
3. De Ravin SS, Naumann N, Cowen EW, et al. Chronic granulomatous disease as a risk factor for autoimmune disease [published online ahead of print September 26, 2008]. J Allergy Clin Immunol. 2008;122:1097-1103.
4. Kragballe K, Borregaard N, Brandrup F, et al. Relation of monocyte and neutrophil oxidative metabolism to skin and oral lesions in carriers of chronic granulomatous disease. Clin Exp Immunol. 1981;43:390-398.
5. Barton LL, Johnson CR. Discoid lupus erythematosus and X-linked chronic granulomatous disease. Pediatr Dermatol. 1986;3:376-379.
6. Córdoba-Guijarro S, Feal C, Daudén E, et al. Lupus erythematosus-like lesions in a carrier of X-linked chronic granulomatous disease. J Eur Acad Dermatol Venereol. 2000;14:409-411.
7. Gomez-Moyano E, Vera-Casaño A, Moreno-Perez D, et al. Lupus erythematosus-like lesions by voriconazole in an infant with chronic granulomatous disease. Pediatr Dermatol. 2010;27:105-106.
8. Brandrup F, Koch C, Petri M, et al. Discoid lupus erythematosus-like lesions and stomatitis in female carriers of X-linked chronic granulomatous disease. Br J Dermatol. 1981;104:495-505.
9. Cale CM, Morton L, Goldblatt D. Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin Exp Immunol. 2007;148:79-84.
10. Levinsky RJ, Harvey BA, Roberton DM, et al. A polymorph bactericidal defect and a lupus-like syndrome. Arch Dis Child. 1981;56:382-385.
11. Arlet JB, Aouba A, Suarez F, et al. Efficiency of hydroxychloroquine in the treatment of granulomatous complications in chronic granulomatous disease. Eur J Gastroenterol Hepatol. 2008;20:142-144.
12. Kuhns DB, Alvord WG, Heller T, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363:2600-2610.
1. Dohil M, Prendiville JS, Crawford RI, et al. Cutaneous manifestations of chronic granulomatous disease. a report of four cases and review of the literature. J Am Acad Dermatol. 1997;36(6, pt 1):899-907.
2. Chowdhury MM, Anstey A, Matthews CN. The dermatosis of chronic granulomatous disease. Clin Exp Dermatol. 2000;25:190-194.
3. De Ravin SS, Naumann N, Cowen EW, et al. Chronic granulomatous disease as a risk factor for autoimmune disease [published online ahead of print September 26, 2008]. J Allergy Clin Immunol. 2008;122:1097-1103.
4. Kragballe K, Borregaard N, Brandrup F, et al. Relation of monocyte and neutrophil oxidative metabolism to skin and oral lesions in carriers of chronic granulomatous disease. Clin Exp Immunol. 1981;43:390-398.
5. Barton LL, Johnson CR. Discoid lupus erythematosus and X-linked chronic granulomatous disease. Pediatr Dermatol. 1986;3:376-379.
6. Córdoba-Guijarro S, Feal C, Daudén E, et al. Lupus erythematosus-like lesions in a carrier of X-linked chronic granulomatous disease. J Eur Acad Dermatol Venereol. 2000;14:409-411.
7. Gomez-Moyano E, Vera-Casaño A, Moreno-Perez D, et al. Lupus erythematosus-like lesions by voriconazole in an infant with chronic granulomatous disease. Pediatr Dermatol. 2010;27:105-106.
8. Brandrup F, Koch C, Petri M, et al. Discoid lupus erythematosus-like lesions and stomatitis in female carriers of X-linked chronic granulomatous disease. Br J Dermatol. 1981;104:495-505.
9. Cale CM, Morton L, Goldblatt D. Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin Exp Immunol. 2007;148:79-84.
10. Levinsky RJ, Harvey BA, Roberton DM, et al. A polymorph bactericidal defect and a lupus-like syndrome. Arch Dis Child. 1981;56:382-385.
11. Arlet JB, Aouba A, Suarez F, et al. Efficiency of hydroxychloroquine in the treatment of granulomatous complications in chronic granulomatous disease. Eur J Gastroenterol Hepatol. 2008;20:142-144.
12. Kuhns DB, Alvord WG, Heller T, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363:2600-2610.
High-Yield Biopsy Technique for Subepidermal Blisters
The traditional approach for confirming the diagnosis of subepidermal blistering diseases such as bullous pemphigoid (BP), epidermolysis bullosa acquisita (EBA), dermatitis herpetifor-mis (DH), and linear IgA bullous dermatosis (LABD) requires 2 punch biopsies: one from perilesional skin for direct immunofluorescence (DIF) and one from lesional skin for light microscopy using hematoxylin and eosin (H&E) stain.1-4 These conditions are distinguished by a combination of features appreciated on H&E-stained sections, DIF, and indirect immunofluorescence for a subset of disorders. Additional information may be provided by DIF or indirect immunofluorescence utilizing the salt-split skin technique to differentiate BP (in which linear IgG deposition is identified by immunofluorescence on the roof of salt-split skin) from EBA and antiepiligrin cicatricial pemphigoid (in which linear IgG deposition is identified by immunofluorescence along the floor of the salt-split skin), which is more rare.4 One bisected punch biopsy of a subepidermal blister yields salt-split skin–like information through standard DIF and supersedes the need for the more cumbersome salt-split skin technique.
Serologic tests for the presence of circulating antibodies to BP180 and BP230 represent an emerging technology that can confirm the diagnosis of BP, but it has been difficult to identify clinically useful autoantibodies to confirm diagnoses of EBA and LABD.5-7 Serologic tests for tissue transglutaminase IgA antibodies may be useful in the diagnosis of DH.8 We present a cost-effective approach to biopsy in the diagnosis of subepidermal blistering diseases that provides the necessary diagnostic information to distinguish relevant disease processes.
Subepidermal Blistering Diseases
Bullous pemphigoid commonly presents with widespread tense bullae of varying sizes on an erythematous base or on otherwise normal skin.9 Some cases of BP present not with bullae but with pruritic, urticarial, plaquelike, or papular lesions. Bullous pemphigoid commonly involves flexural surfaces and the trunk but can appear anywhere on the skin. The induction of blisters by shearing with mechanical pressure on perilesional skin (Nikolsky sign) is not characteristically present in BP as it is in pemphigus vulgaris.10 Epidermolysis bullosa acquisita can mimic BP in the development of widespread tense bullae, but blisters typically appear on areas of the skin that are prone to trauma (eg, toes, knees, elbows, hands). Crusted erosions, scarring, and milia also are clinical manifestations of EBA.11 Dermatitis herpetiformis presents with grouped vesicles, papulovesicles, plaques, and excoriations that are symmetrically distributed on extensor surfaces of the skin but also can occur on the buttocks, scalp, and other areas of the skin.12,13 Although it may mimic both BP and DH, LABD frequently is less pruritic than these other conditions.14,15 Linear IgA bullous dermatosis also demonstrates the characteristic finding of multiple bullae that form concentrically around a crusted area of skin. This physical finding is known as a string of pearls. Linear IgA bullous dermatosis typically occurs in childhood and may resolve without treatment in months to years.16
Traditional Biopsy Approach
A review of several articles from the literature and multiple dermatology and dermatopathology textbooks revealed uniform recommendations for biopsy of subepidermal blistering conditions that manifest as tense blisters.1-4,9-23 A biopsy of early lesional skin or of a blister for light microscopy with H&E stain and biopsy of perilesional skin for DIF is recommended.1-4,9-23 Three review articles specifically suggested biopsy of “perilesional skin” for DIF.1-3 The majority of textbooks we reviewed also suggested that perilesional skin, or skin adjacent to a zone of erythema in the case of DH, should be sampled for DIF to assist in the diagnosis of BP, EBA, DH, and LABD.4,9-21 Biopsy of adjacent or nonlesional skin or skin around the lesion for DIF also was recommended by other textbooks for diagnosis of subepidermal blistering diseases.22,23 Perilesional skin is chosen because it is critical that the epidermis be included for adequate immunofluorescence studies.5,20 Biopsy of healed and crusted lesions should be avoided.24
Recommended Alternative Approach
A single punch biopsy produces the best possible specimen for light microscopy with H&E stain and DIF if it is obtained via one of 2 methods.
The first method involves choosing a small, 1- to 2-mm tense blister.25 Use an 8-mm punch centered on the blister that includes at least 3 mm of circumferential perilesional skin (Figures 1 and 2).20 Holding the specimen with forceps, use a no. 15 scalpel blade to bisect the blister with a sawing motion. Place half of the specimen in formalin for H&E staining and the other half in Zeus (or Michel) medium for DIF (Figure 3).
|
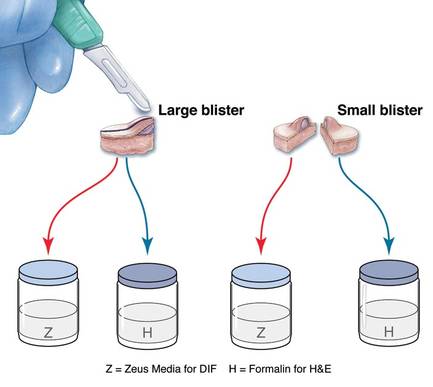 |
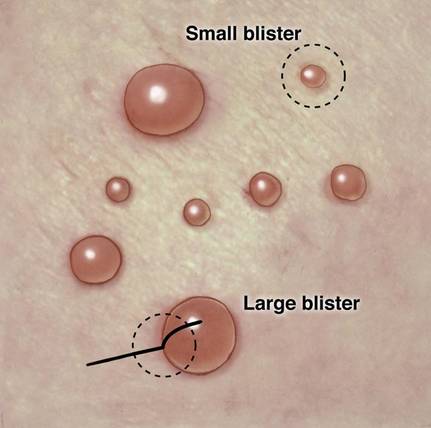
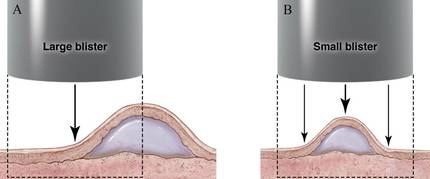
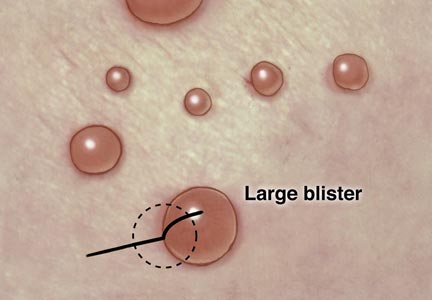
The second method is to choose any large blister and, utilizing a surgical marker, draw a line from the roof of the blister onto the adjacent perilesional skin (Figure 1).20,24 After blotting with an alcohol pad so as not to remove the mark, anesthetize the site with lidocaine 1% with epinephrine,24 then take an 8-mm punch biopsy encompassing 75% perilesional skin and 25% of the blister centered on the line (Figure 2). After separating the punch specimen from the subcutaneous tissue with surgical scissors, hold the tissue with forceps and bisect the specimen with a no. 15 scalpel blade. Use a sawing motion along the line drawn in the prior steps. Submit half of the biopsy for H&E staining in formalin and the other half for DIF in Zeus (or Michel) medium (Figure 3).
Advantages
This approach offers several advantages. First, when biopsying either a small or large tense blister with this technique, only 1 invasive procedure, not 2 separate biopsies, is required. Therefore, our proposed procedures can be done quickly and efficiently with the least morbidity and scarring. Secondly, because the patient is billed for 1 biopsy instead of 2, the single punch biopsy technique is more cost effective.
The bisected specimen resulting from complete excision of a small blister or from biopsy of a larger blister that includes 75% perilesional skin and 25% from the blister cavity also provides the best tissue specimen for interpretation of the subepidermal blistering processes via H&E staining.4,20,24 When traditional unmarked punch specimens of a blister margin are sent to the laboratory in formalin for H&E staining, the technician that grosses the specimen may or may not bisect the specimen demonstrating the edge of the blister at the point where the epidermis is separated from the papillary dermis.
Finally, when the DIF specimen is prepared using either of these 2 approaches, the immunoprecipitants can be seen at the dermoepidermal junction or in the papillary dermis in the perilesional portion of the specimen.2,4 Additionally, the immunoprecipitant may be identified on the roof or floor of the blister. Although this approach has not been studied in a systematic fashion, we believe this technique provides “bonus” information (eg, the same information gained from salt-split skin indirect immunofluorescence to demonstrate if immunoprecipitants are deposited in the roof or floor of the blister).
Limitations
It is critical for the pathologist or technician grossing these specimens to understand this technique and ensure that the cut edge of each half punch specimen is properly embedded for both H&E and DIF specimens. Additionally, with either recommended technique, if the portion of perilesional skin is not sufficient and the epidermis completely separates from the dermis, interpretation of both the H&E staining and DIF sections is substantially compromised.20 Therefore, an 8-mm disposable punch is recommended to avoid mangling the specimens when they are bisected and to ensure that the epithelium is not lost. This technique is less suitable for blistering processes with a positive Nikolsky sign, such as pemphigus and toxic epidermal necrolysis, because the small area of perilesional skin adjacent to the blister may detach completely, requiring the epidermis and dermis to be evaluated separately or, in the worst-case scenario, the epidermis may be lost in processing.
Conclusion
Bisecting a single punch biopsy on subepidermal blisters provides the best specimen for H&E staining and DIF. The single punch biopsy technique also differentiates BP and EBA without utilizing salt-split skin immunofluorescence studies. This technique is more efficient and cost effective than the traditional approach of multiple biopsies on subepidermal blisters.
1. Kasperkiewicz M, Zillikens D, Schmidt E. Pemphigoid diseases: pathogenesis, diagnosis, and treatment. Autoimmunity. 2012;45:55-70.
2. Mihai S, Sitaru C. Immunopathology and molecular diagnosis of autoimmune bullous diseases. J Cell Mol Med. 2007;11:462-481.
3. Yeh SW, Ahmed B, Sami N, et al. Blistering disorders: diagnosis and treatment. Dermatol Ther. 2003;16:214-223.
4. Vesicular and bullous diseases. In: Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Edinburgh, Scotland: Mosby Elsevier; 2010:635-670.
5. Schmidt E, della Torre R, Borradori L. Clinical features and practical diagnosis of bullous pemphigoid. Dermatol Clin. 2011;29:427-438.
6. Caux F. Diagnosis and clinical features of epidermolysis bullosa acquisita. Dermatol Clin. 2011;29:485-491.
7. Terra JB, Jonkman MF, Diercks GF, et al. Low sensitivity of type VII collagen enzyme-linked immunosorbent assay in epidermolysis bullosa acquisita: serration pattern analysis on skin biopsy is required for diagnosis. Br J Dermatol. 2013;169:164-167.
8. Plotnikova N, Miller JL. Dermatitis herpetiformis. Skin Therapy Lett. 2013;18:1-3.
9. Culton DA, Liu Z, Diaz LA. Bullous pemphigoid. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012. http://accessmedicine.mhmedi cal.com.proxy.kcumb.edu/content.aspx?bookid=392& Sectionid=41138755. Accessed June 24, 2013.
10. Bernard P, Borradori L. Pemphigoid group. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Edinburgh, Scotland: Saunders Elsevier; 2012:475-490.
11. Woodley DT, Chen M. Epidermolysis bullosa acquisita. In: Goldsmith LA, Katz SI, Gilchrest, BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012. http://access medicine.mhmedical.com.proxy.kcumb.edu/content.aspx?bookid=392&Sectionid=41138759. Accessed June 24, 2013.
12. Chronic blistering dermatoses. In: James WD, Berger TG, Elston DM. Andrew’s Diseases of the Skin. 11th ed. London, England: Saunders Elsevier; 2011:448-467.
13. Ronaghy A, Katz SI, Hall RP. Dermatitis herpetiformis. In: Goldsmith LA, Katz SI, Gilchrest, BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012. http://accessmedicine.mhmedical.com.proxy.kcumb.edu/content.aspx?bookid=392&Sectionid=41138760. Accessed June 24, 2013.
14. Hull CM, Zone JJ. Dermatitis herpetiformis and linear IgA bullous dermatosis. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Edinburgh, Scotland: Saunders Elsevier; 2012:491-500.
15. Rao CL, Hall RP III. Linear immunoglobulin a dermatosis and chronic bullous disease of childhood. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012. http://accessmedicine.mhmedical.com.proxy.kcumb.edu/content.aspx?bookid=392&Sectionid=41138757. Accessed June 24, 2013.
16. Bullous disorders of childhood. In: Paller AS, Mancini AJ. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. 4th ed. Philadelphia, PA: Saunders Elsevier; 2011:303-320.
17. Inherited and autoimmune subepidermal blistering diseases. In: Caljone E, Brenn T, Lazar A, et al. McKee’s Pathology of the Skin: With Clinical Correlations. 4th ed. Edinburgh, Scotland: Elsevier Saunders; 2012:99-150.
18. Elenitsas R, Ming ME. Biopsy techniques. In: Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever’s Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008:5-6.
19. Wu H, Brandling-Bennett HA, Harrist TJ. Noninfectious vesiculobullous and vesiculopustular diseases. In: Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever’s Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008:235-278.
20. Smoller BR, Kohler S. Subepidermal vesicular dermatitis. In: Barnhill RL, Crowson AN, eds. Textbook of Dermatopathology. New York, NY: McGraw-Hill; 2004:167-194.
21. Junkins-Hopkins JM, Busam KJ. Blistering skin diseases. In: Busam KJ, ed. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2010:207-248.
22. High, WA. Blistering diseases. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. Edinburgh, Scotland: Saunders Elsevier; 2009:161-172.
23. The vesicobullous reaction pattern. In: Weedon D. Weedon’s Skin Pathology. 3rd ed. Edinburgh, Scotland; Churchill Livingstone/Elsevier; 2009:93-148.
24. Alguire PC, Mathes BM. Skin biopsy techniques for the internist. J Gen Intern Med. 1998;13:46-54.
25. Sina B, Kao GF, Deng AC, et al. Skin biopsy for inflammatory and common neoplastic skin diseases: optimum time, best location and preferred techniques. a critical review. J Cutan Pathol. 2009;36:505-510.
The traditional approach for confirming the diagnosis of subepidermal blistering diseases such as bullous pemphigoid (BP), epidermolysis bullosa acquisita (EBA), dermatitis herpetifor-mis (DH), and linear IgA bullous dermatosis (LABD) requires 2 punch biopsies: one from perilesional skin for direct immunofluorescence (DIF) and one from lesional skin for light microscopy using hematoxylin and eosin (H&E) stain.1-4 These conditions are distinguished by a combination of features appreciated on H&E-stained sections, DIF, and indirect immunofluorescence for a subset of disorders. Additional information may be provided by DIF or indirect immunofluorescence utilizing the salt-split skin technique to differentiate BP (in which linear IgG deposition is identified by immunofluorescence on the roof of salt-split skin) from EBA and antiepiligrin cicatricial pemphigoid (in which linear IgG deposition is identified by immunofluorescence along the floor of the salt-split skin), which is more rare.4 One bisected punch biopsy of a subepidermal blister yields salt-split skin–like information through standard DIF and supersedes the need for the more cumbersome salt-split skin technique.
Serologic tests for the presence of circulating antibodies to BP180 and BP230 represent an emerging technology that can confirm the diagnosis of BP, but it has been difficult to identify clinically useful autoantibodies to confirm diagnoses of EBA and LABD.5-7 Serologic tests for tissue transglutaminase IgA antibodies may be useful in the diagnosis of DH.8 We present a cost-effective approach to biopsy in the diagnosis of subepidermal blistering diseases that provides the necessary diagnostic information to distinguish relevant disease processes.
Subepidermal Blistering Diseases
Bullous pemphigoid commonly presents with widespread tense bullae of varying sizes on an erythematous base or on otherwise normal skin.9 Some cases of BP present not with bullae but with pruritic, urticarial, plaquelike, or papular lesions. Bullous pemphigoid commonly involves flexural surfaces and the trunk but can appear anywhere on the skin. The induction of blisters by shearing with mechanical pressure on perilesional skin (Nikolsky sign) is not characteristically present in BP as it is in pemphigus vulgaris.10 Epidermolysis bullosa acquisita can mimic BP in the development of widespread tense bullae, but blisters typically appear on areas of the skin that are prone to trauma (eg, toes, knees, elbows, hands). Crusted erosions, scarring, and milia also are clinical manifestations of EBA.11 Dermatitis herpetiformis presents with grouped vesicles, papulovesicles, plaques, and excoriations that are symmetrically distributed on extensor surfaces of the skin but also can occur on the buttocks, scalp, and other areas of the skin.12,13 Although it may mimic both BP and DH, LABD frequently is less pruritic than these other conditions.14,15 Linear IgA bullous dermatosis also demonstrates the characteristic finding of multiple bullae that form concentrically around a crusted area of skin. This physical finding is known as a string of pearls. Linear IgA bullous dermatosis typically occurs in childhood and may resolve without treatment in months to years.16
Traditional Biopsy Approach
A review of several articles from the literature and multiple dermatology and dermatopathology textbooks revealed uniform recommendations for biopsy of subepidermal blistering conditions that manifest as tense blisters.1-4,9-23 A biopsy of early lesional skin or of a blister for light microscopy with H&E stain and biopsy of perilesional skin for DIF is recommended.1-4,9-23 Three review articles specifically suggested biopsy of “perilesional skin” for DIF.1-3 The majority of textbooks we reviewed also suggested that perilesional skin, or skin adjacent to a zone of erythema in the case of DH, should be sampled for DIF to assist in the diagnosis of BP, EBA, DH, and LABD.4,9-21 Biopsy of adjacent or nonlesional skin or skin around the lesion for DIF also was recommended by other textbooks for diagnosis of subepidermal blistering diseases.22,23 Perilesional skin is chosen because it is critical that the epidermis be included for adequate immunofluorescence studies.5,20 Biopsy of healed and crusted lesions should be avoided.24
Recommended Alternative Approach
A single punch biopsy produces the best possible specimen for light microscopy with H&E stain and DIF if it is obtained via one of 2 methods.
The first method involves choosing a small, 1- to 2-mm tense blister.25 Use an 8-mm punch centered on the blister that includes at least 3 mm of circumferential perilesional skin (Figures 1 and 2).20 Holding the specimen with forceps, use a no. 15 scalpel blade to bisect the blister with a sawing motion. Place half of the specimen in formalin for H&E staining and the other half in Zeus (or Michel) medium for DIF (Figure 3).
|
|
The second method is to choose any large blister and, utilizing a surgical marker, draw a line from the roof of the blister onto the adjacent perilesional skin (Figure 1).20,24 After blotting with an alcohol pad so as not to remove the mark, anesthetize the site with lidocaine 1% with epinephrine,24 then take an 8-mm punch biopsy encompassing 75% perilesional skin and 25% of the blister centered on the line (Figure 2). After separating the punch specimen from the subcutaneous tissue with surgical scissors, hold the tissue with forceps and bisect the specimen with a no. 15 scalpel blade. Use a sawing motion along the line drawn in the prior steps. Submit half of the biopsy for H&E staining in formalin and the other half for DIF in Zeus (or Michel) medium (Figure 3).
Advantages
This approach offers several advantages. First, when biopsying either a small or large tense blister with this technique, only 1 invasive procedure, not 2 separate biopsies, is required. Therefore, our proposed procedures can be done quickly and efficiently with the least morbidity and scarring. Secondly, because the patient is billed for 1 biopsy instead of 2, the single punch biopsy technique is more cost effective.
The bisected specimen resulting from complete excision of a small blister or from biopsy of a larger blister that includes 75% perilesional skin and 25% from the blister cavity also provides the best tissue specimen for interpretation of the subepidermal blistering processes via H&E staining.4,20,24 When traditional unmarked punch specimens of a blister margin are sent to the laboratory in formalin for H&E staining, the technician that grosses the specimen may or may not bisect the specimen demonstrating the edge of the blister at the point where the epidermis is separated from the papillary dermis.
Finally, when the DIF specimen is prepared using either of these 2 approaches, the immunoprecipitants can be seen at the dermoepidermal junction or in the papillary dermis in the perilesional portion of the specimen.2,4 Additionally, the immunoprecipitant may be identified on the roof or floor of the blister. Although this approach has not been studied in a systematic fashion, we believe this technique provides “bonus” information (eg, the same information gained from salt-split skin indirect immunofluorescence to demonstrate if immunoprecipitants are deposited in the roof or floor of the blister).
Limitations
It is critical for the pathologist or technician grossing these specimens to understand this technique and ensure that the cut edge of each half punch specimen is properly embedded for both H&E and DIF specimens. Additionally, with either recommended technique, if the portion of perilesional skin is not sufficient and the epidermis completely separates from the dermis, interpretation of both the H&E staining and DIF sections is substantially compromised.20 Therefore, an 8-mm disposable punch is recommended to avoid mangling the specimens when they are bisected and to ensure that the epithelium is not lost. This technique is less suitable for blistering processes with a positive Nikolsky sign, such as pemphigus and toxic epidermal necrolysis, because the small area of perilesional skin adjacent to the blister may detach completely, requiring the epidermis and dermis to be evaluated separately or, in the worst-case scenario, the epidermis may be lost in processing.
Conclusion
Bisecting a single punch biopsy on subepidermal blisters provides the best specimen for H&E staining and DIF. The single punch biopsy technique also differentiates BP and EBA without utilizing salt-split skin immunofluorescence studies. This technique is more efficient and cost effective than the traditional approach of multiple biopsies on subepidermal blisters.
The traditional approach for confirming the diagnosis of subepidermal blistering diseases such as bullous pemphigoid (BP), epidermolysis bullosa acquisita (EBA), dermatitis herpetifor-mis (DH), and linear IgA bullous dermatosis (LABD) requires 2 punch biopsies: one from perilesional skin for direct immunofluorescence (DIF) and one from lesional skin for light microscopy using hematoxylin and eosin (H&E) stain.1-4 These conditions are distinguished by a combination of features appreciated on H&E-stained sections, DIF, and indirect immunofluorescence for a subset of disorders. Additional information may be provided by DIF or indirect immunofluorescence utilizing the salt-split skin technique to differentiate BP (in which linear IgG deposition is identified by immunofluorescence on the roof of salt-split skin) from EBA and antiepiligrin cicatricial pemphigoid (in which linear IgG deposition is identified by immunofluorescence along the floor of the salt-split skin), which is more rare.4 One bisected punch biopsy of a subepidermal blister yields salt-split skin–like information through standard DIF and supersedes the need for the more cumbersome salt-split skin technique.
Serologic tests for the presence of circulating antibodies to BP180 and BP230 represent an emerging technology that can confirm the diagnosis of BP, but it has been difficult to identify clinically useful autoantibodies to confirm diagnoses of EBA and LABD.5-7 Serologic tests for tissue transglutaminase IgA antibodies may be useful in the diagnosis of DH.8 We present a cost-effective approach to biopsy in the diagnosis of subepidermal blistering diseases that provides the necessary diagnostic information to distinguish relevant disease processes.
Subepidermal Blistering Diseases
Bullous pemphigoid commonly presents with widespread tense bullae of varying sizes on an erythematous base or on otherwise normal skin.9 Some cases of BP present not with bullae but with pruritic, urticarial, plaquelike, or papular lesions. Bullous pemphigoid commonly involves flexural surfaces and the trunk but can appear anywhere on the skin. The induction of blisters by shearing with mechanical pressure on perilesional skin (Nikolsky sign) is not characteristically present in BP as it is in pemphigus vulgaris.10 Epidermolysis bullosa acquisita can mimic BP in the development of widespread tense bullae, but blisters typically appear on areas of the skin that are prone to trauma (eg, toes, knees, elbows, hands). Crusted erosions, scarring, and milia also are clinical manifestations of EBA.11 Dermatitis herpetiformis presents with grouped vesicles, papulovesicles, plaques, and excoriations that are symmetrically distributed on extensor surfaces of the skin but also can occur on the buttocks, scalp, and other areas of the skin.12,13 Although it may mimic both BP and DH, LABD frequently is less pruritic than these other conditions.14,15 Linear IgA bullous dermatosis also demonstrates the characteristic finding of multiple bullae that form concentrically around a crusted area of skin. This physical finding is known as a string of pearls. Linear IgA bullous dermatosis typically occurs in childhood and may resolve without treatment in months to years.16
Traditional Biopsy Approach
A review of several articles from the literature and multiple dermatology and dermatopathology textbooks revealed uniform recommendations for biopsy of subepidermal blistering conditions that manifest as tense blisters.1-4,9-23 A biopsy of early lesional skin or of a blister for light microscopy with H&E stain and biopsy of perilesional skin for DIF is recommended.1-4,9-23 Three review articles specifically suggested biopsy of “perilesional skin” for DIF.1-3 The majority of textbooks we reviewed also suggested that perilesional skin, or skin adjacent to a zone of erythema in the case of DH, should be sampled for DIF to assist in the diagnosis of BP, EBA, DH, and LABD.4,9-21 Biopsy of adjacent or nonlesional skin or skin around the lesion for DIF also was recommended by other textbooks for diagnosis of subepidermal blistering diseases.22,23 Perilesional skin is chosen because it is critical that the epidermis be included for adequate immunofluorescence studies.5,20 Biopsy of healed and crusted lesions should be avoided.24
Recommended Alternative Approach
A single punch biopsy produces the best possible specimen for light microscopy with H&E stain and DIF if it is obtained via one of 2 methods.
The first method involves choosing a small, 1- to 2-mm tense blister.25 Use an 8-mm punch centered on the blister that includes at least 3 mm of circumferential perilesional skin (Figures 1 and 2).20 Holding the specimen with forceps, use a no. 15 scalpel blade to bisect the blister with a sawing motion. Place half of the specimen in formalin for H&E staining and the other half in Zeus (or Michel) medium for DIF (Figure 3).
|
|
The second method is to choose any large blister and, utilizing a surgical marker, draw a line from the roof of the blister onto the adjacent perilesional skin (Figure 1).20,24 After blotting with an alcohol pad so as not to remove the mark, anesthetize the site with lidocaine 1% with epinephrine,24 then take an 8-mm punch biopsy encompassing 75% perilesional skin and 25% of the blister centered on the line (Figure 2). After separating the punch specimen from the subcutaneous tissue with surgical scissors, hold the tissue with forceps and bisect the specimen with a no. 15 scalpel blade. Use a sawing motion along the line drawn in the prior steps. Submit half of the biopsy for H&E staining in formalin and the other half for DIF in Zeus (or Michel) medium (Figure 3).
Advantages
This approach offers several advantages. First, when biopsying either a small or large tense blister with this technique, only 1 invasive procedure, not 2 separate biopsies, is required. Therefore, our proposed procedures can be done quickly and efficiently with the least morbidity and scarring. Secondly, because the patient is billed for 1 biopsy instead of 2, the single punch biopsy technique is more cost effective.
The bisected specimen resulting from complete excision of a small blister or from biopsy of a larger blister that includes 75% perilesional skin and 25% from the blister cavity also provides the best tissue specimen for interpretation of the subepidermal blistering processes via H&E staining.4,20,24 When traditional unmarked punch specimens of a blister margin are sent to the laboratory in formalin for H&E staining, the technician that grosses the specimen may or may not bisect the specimen demonstrating the edge of the blister at the point where the epidermis is separated from the papillary dermis.
Finally, when the DIF specimen is prepared using either of these 2 approaches, the immunoprecipitants can be seen at the dermoepidermal junction or in the papillary dermis in the perilesional portion of the specimen.2,4 Additionally, the immunoprecipitant may be identified on the roof or floor of the blister. Although this approach has not been studied in a systematic fashion, we believe this technique provides “bonus” information (eg, the same information gained from salt-split skin indirect immunofluorescence to demonstrate if immunoprecipitants are deposited in the roof or floor of the blister).
Limitations
It is critical for the pathologist or technician grossing these specimens to understand this technique and ensure that the cut edge of each half punch specimen is properly embedded for both H&E and DIF specimens. Additionally, with either recommended technique, if the portion of perilesional skin is not sufficient and the epidermis completely separates from the dermis, interpretation of both the H&E staining and DIF sections is substantially compromised.20 Therefore, an 8-mm disposable punch is recommended to avoid mangling the specimens when they are bisected and to ensure that the epithelium is not lost. This technique is less suitable for blistering processes with a positive Nikolsky sign, such as pemphigus and toxic epidermal necrolysis, because the small area of perilesional skin adjacent to the blister may detach completely, requiring the epidermis and dermis to be evaluated separately or, in the worst-case scenario, the epidermis may be lost in processing.
Conclusion
Bisecting a single punch biopsy on subepidermal blisters provides the best specimen for H&E staining and DIF. The single punch biopsy technique also differentiates BP and EBA without utilizing salt-split skin immunofluorescence studies. This technique is more efficient and cost effective than the traditional approach of multiple biopsies on subepidermal blisters.
1. Kasperkiewicz M, Zillikens D, Schmidt E. Pemphigoid diseases: pathogenesis, diagnosis, and treatment. Autoimmunity. 2012;45:55-70.
2. Mihai S, Sitaru C. Immunopathology and molecular diagnosis of autoimmune bullous diseases. J Cell Mol Med. 2007;11:462-481.
3. Yeh SW, Ahmed B, Sami N, et al. Blistering disorders: diagnosis and treatment. Dermatol Ther. 2003;16:214-223.
4. Vesicular and bullous diseases. In: Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Edinburgh, Scotland: Mosby Elsevier; 2010:635-670.
5. Schmidt E, della Torre R, Borradori L. Clinical features and practical diagnosis of bullous pemphigoid. Dermatol Clin. 2011;29:427-438.
6. Caux F. Diagnosis and clinical features of epidermolysis bullosa acquisita. Dermatol Clin. 2011;29:485-491.
7. Terra JB, Jonkman MF, Diercks GF, et al. Low sensitivity of type VII collagen enzyme-linked immunosorbent assay in epidermolysis bullosa acquisita: serration pattern analysis on skin biopsy is required for diagnosis. Br J Dermatol. 2013;169:164-167.
8. Plotnikova N, Miller JL. Dermatitis herpetiformis. Skin Therapy Lett. 2013;18:1-3.
9. Culton DA, Liu Z, Diaz LA. Bullous pemphigoid. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012. http://accessmedicine.mhmedi cal.com.proxy.kcumb.edu/content.aspx?bookid=392& Sectionid=41138755. Accessed June 24, 2013.
10. Bernard P, Borradori L. Pemphigoid group. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Edinburgh, Scotland: Saunders Elsevier; 2012:475-490.
11. Woodley DT, Chen M. Epidermolysis bullosa acquisita. In: Goldsmith LA, Katz SI, Gilchrest, BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012. http://access medicine.mhmedical.com.proxy.kcumb.edu/content.aspx?bookid=392&Sectionid=41138759. Accessed June 24, 2013.
12. Chronic blistering dermatoses. In: James WD, Berger TG, Elston DM. Andrew’s Diseases of the Skin. 11th ed. London, England: Saunders Elsevier; 2011:448-467.
13. Ronaghy A, Katz SI, Hall RP. Dermatitis herpetiformis. In: Goldsmith LA, Katz SI, Gilchrest, BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012. http://accessmedicine.mhmedical.com.proxy.kcumb.edu/content.aspx?bookid=392&Sectionid=41138760. Accessed June 24, 2013.
14. Hull CM, Zone JJ. Dermatitis herpetiformis and linear IgA bullous dermatosis. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Edinburgh, Scotland: Saunders Elsevier; 2012:491-500.
15. Rao CL, Hall RP III. Linear immunoglobulin a dermatosis and chronic bullous disease of childhood. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012. http://accessmedicine.mhmedical.com.proxy.kcumb.edu/content.aspx?bookid=392&Sectionid=41138757. Accessed June 24, 2013.
16. Bullous disorders of childhood. In: Paller AS, Mancini AJ. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. 4th ed. Philadelphia, PA: Saunders Elsevier; 2011:303-320.
17. Inherited and autoimmune subepidermal blistering diseases. In: Caljone E, Brenn T, Lazar A, et al. McKee’s Pathology of the Skin: With Clinical Correlations. 4th ed. Edinburgh, Scotland: Elsevier Saunders; 2012:99-150.
18. Elenitsas R, Ming ME. Biopsy techniques. In: Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever’s Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008:5-6.
19. Wu H, Brandling-Bennett HA, Harrist TJ. Noninfectious vesiculobullous and vesiculopustular diseases. In: Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever’s Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008:235-278.
20. Smoller BR, Kohler S. Subepidermal vesicular dermatitis. In: Barnhill RL, Crowson AN, eds. Textbook of Dermatopathology. New York, NY: McGraw-Hill; 2004:167-194.
21. Junkins-Hopkins JM, Busam KJ. Blistering skin diseases. In: Busam KJ, ed. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2010:207-248.
22. High, WA. Blistering diseases. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. Edinburgh, Scotland: Saunders Elsevier; 2009:161-172.
23. The vesicobullous reaction pattern. In: Weedon D. Weedon’s Skin Pathology. 3rd ed. Edinburgh, Scotland; Churchill Livingstone/Elsevier; 2009:93-148.
24. Alguire PC, Mathes BM. Skin biopsy techniques for the internist. J Gen Intern Med. 1998;13:46-54.
25. Sina B, Kao GF, Deng AC, et al. Skin biopsy for inflammatory and common neoplastic skin diseases: optimum time, best location and preferred techniques. a critical review. J Cutan Pathol. 2009;36:505-510.
1. Kasperkiewicz M, Zillikens D, Schmidt E. Pemphigoid diseases: pathogenesis, diagnosis, and treatment. Autoimmunity. 2012;45:55-70.
2. Mihai S, Sitaru C. Immunopathology and molecular diagnosis of autoimmune bullous diseases. J Cell Mol Med. 2007;11:462-481.
3. Yeh SW, Ahmed B, Sami N, et al. Blistering disorders: diagnosis and treatment. Dermatol Ther. 2003;16:214-223.
4. Vesicular and bullous diseases. In: Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Edinburgh, Scotland: Mosby Elsevier; 2010:635-670.
5. Schmidt E, della Torre R, Borradori L. Clinical features and practical diagnosis of bullous pemphigoid. Dermatol Clin. 2011;29:427-438.
6. Caux F. Diagnosis and clinical features of epidermolysis bullosa acquisita. Dermatol Clin. 2011;29:485-491.
7. Terra JB, Jonkman MF, Diercks GF, et al. Low sensitivity of type VII collagen enzyme-linked immunosorbent assay in epidermolysis bullosa acquisita: serration pattern analysis on skin biopsy is required for diagnosis. Br J Dermatol. 2013;169:164-167.
8. Plotnikova N, Miller JL. Dermatitis herpetiformis. Skin Therapy Lett. 2013;18:1-3.
9. Culton DA, Liu Z, Diaz LA. Bullous pemphigoid. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012. http://accessmedicine.mhmedi cal.com.proxy.kcumb.edu/content.aspx?bookid=392& Sectionid=41138755. Accessed June 24, 2013.
10. Bernard P, Borradori L. Pemphigoid group. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Edinburgh, Scotland: Saunders Elsevier; 2012:475-490.
11. Woodley DT, Chen M. Epidermolysis bullosa acquisita. In: Goldsmith LA, Katz SI, Gilchrest, BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012. http://access medicine.mhmedical.com.proxy.kcumb.edu/content.aspx?bookid=392&Sectionid=41138759. Accessed June 24, 2013.
12. Chronic blistering dermatoses. In: James WD, Berger TG, Elston DM. Andrew’s Diseases of the Skin. 11th ed. London, England: Saunders Elsevier; 2011:448-467.
13. Ronaghy A, Katz SI, Hall RP. Dermatitis herpetiformis. In: Goldsmith LA, Katz SI, Gilchrest, BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012. http://accessmedicine.mhmedical.com.proxy.kcumb.edu/content.aspx?bookid=392&Sectionid=41138760. Accessed June 24, 2013.
14. Hull CM, Zone JJ. Dermatitis herpetiformis and linear IgA bullous dermatosis. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Edinburgh, Scotland: Saunders Elsevier; 2012:491-500.
15. Rao CL, Hall RP III. Linear immunoglobulin a dermatosis and chronic bullous disease of childhood. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012. http://accessmedicine.mhmedical.com.proxy.kcumb.edu/content.aspx?bookid=392&Sectionid=41138757. Accessed June 24, 2013.
16. Bullous disorders of childhood. In: Paller AS, Mancini AJ. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. 4th ed. Philadelphia, PA: Saunders Elsevier; 2011:303-320.
17. Inherited and autoimmune subepidermal blistering diseases. In: Caljone E, Brenn T, Lazar A, et al. McKee’s Pathology of the Skin: With Clinical Correlations. 4th ed. Edinburgh, Scotland: Elsevier Saunders; 2012:99-150.
18. Elenitsas R, Ming ME. Biopsy techniques. In: Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever’s Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008:5-6.
19. Wu H, Brandling-Bennett HA, Harrist TJ. Noninfectious vesiculobullous and vesiculopustular diseases. In: Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever’s Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008:235-278.
20. Smoller BR, Kohler S. Subepidermal vesicular dermatitis. In: Barnhill RL, Crowson AN, eds. Textbook of Dermatopathology. New York, NY: McGraw-Hill; 2004:167-194.
21. Junkins-Hopkins JM, Busam KJ. Blistering skin diseases. In: Busam KJ, ed. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2010:207-248.
22. High, WA. Blistering diseases. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. Edinburgh, Scotland: Saunders Elsevier; 2009:161-172.
23. The vesicobullous reaction pattern. In: Weedon D. Weedon’s Skin Pathology. 3rd ed. Edinburgh, Scotland; Churchill Livingstone/Elsevier; 2009:93-148.
24. Alguire PC, Mathes BM. Skin biopsy techniques for the internist. J Gen Intern Med. 1998;13:46-54.
25. Sina B, Kao GF, Deng AC, et al. Skin biopsy for inflammatory and common neoplastic skin diseases: optimum time, best location and preferred techniques. a critical review. J Cutan Pathol. 2009;36:505-510.
Practice Points
- Allergic contact dermatitis, pemphigoid, dermatitis herpetiformis, acquired epidermolysis bullosa, and porphyria cutanea tarda produce tense blisters.
- Biopsy of the edge point of a tense blister localizes immunoglobulin to the floor versus the roof on
direct immunofluorescence. - A punch biopsy should include 75% perilesional skin at the edge of a blister.
- Marking the blister with a skin marker will assure that the tissue is properly oriented when bisected at the bedside.
Trichoepithelioma and Spiradenoma Collision Tumor
The coexistence of more than one cutaneous adnexal neoplasm in a single biopsy specimen is unusual and is most frequently recognized in the context of a nevus sebaceous or Brooke-Spiegler syndrome, an autosomal-dominant inherited disease characterized by cutaneous adnexal neoplasms, most commonly cylindromas and trichoepitheliomas.1-3 Brooke-Spiegler syndrome is caused by germline mutations in the cylindromatosis gene, CYLD, located on band 16q12; it functions as a tumor suppressor gene and has regulatory roles in development, immunity, and inflammation.1 Weyers et al3 first recognized the tendency for adnexal collision tumors to present in patients with Brooke-Spiegler syndrome; they reported a patient with Brooke-Spiegler syndrome with spiradenomas found in the immediate vicinity of trichoepitheliomas and in continuity with hair follicles.
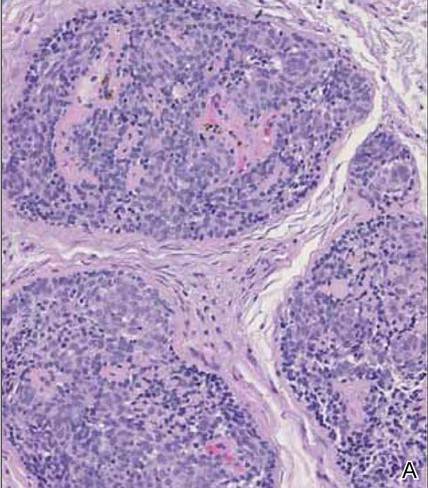
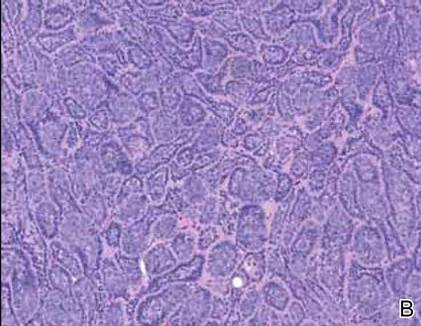
Spiradenomas are composed of large, sharply demarcated, rounded nodules of basaloid cells with little cytoplasm (Figure 1).4 The basaloid nodules may demonstrate a trabecular architecture, and on close inspection 2 cell types—paler cells with more cytoplasm and darker cells with less cytoplasm—are distinguishable (Figure 2A). Lymphocytes often are scattered within the tumor nodules and/or stroma. In Brooke-Spiegler syndrome, collision tumors containing a spiradenomatous component in collision with trichoepithelioma are not uncommon.1 Spiradenomas in Brooke-Spiegler syndrome have been reported to contain sebaceous differentiation or foci with an adenoid cystic carcinoma (ACC)–like pattern and are known to occur as hybrid lesions of spiradenoma and cylindroma or trichoepithelioma (as in this case).
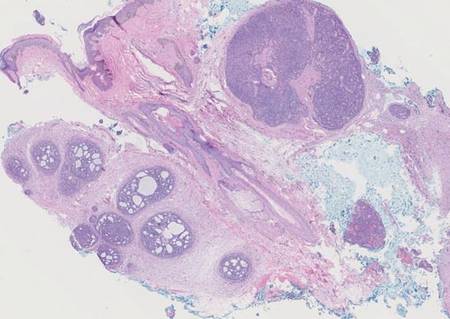
In this case, 2 distinct neoplasms (spiradenoma and trichoepithelioma) are apparent, side by side, with an intervening hair follicle (Figure 1). Trichoepitheliomas, also known as cribriform trichoblastomas,5 are characterized by lobules of basaloid cells resembling basal cell carcinoma surrounded by a fibroblast-rich stroma. They often contain fingerlike projections and adopt a cribriform morphology within the tumor lobules (Figure 2B).4 Numerous horn cysts may be present, but their absence does not preclude the diagnosis. Mucin may be present within the cribriform tumor islands (Figure 2B) but not in the stroma. Characteristically, trichoepitheliomas are distinctly negative for CK7 (Figure 3), and unlike spiradenomas, they lack a myoepithelial component.6 This staining pattern in combination with the tumor’s proximity to an adjacent hair follicle makes a diagnosis of trichoepithelioma and spiradenoma collision tumor most likely and supports a clinical suspicion for Brooke-Spiegler syndrome.
 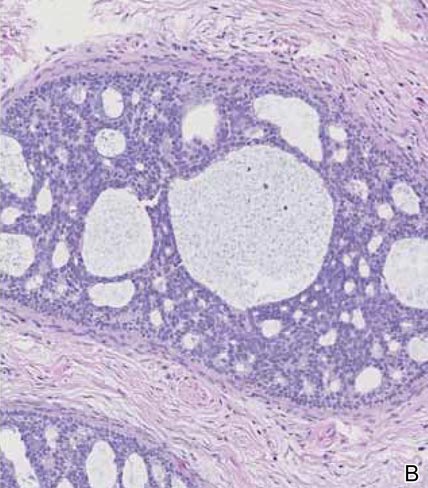 |
Although spiradenomas sometimes contain cystic cavities (microcystic change), they typically are filled with finely granular eosinophilic material, not mucin, that is diastase resistant and periodic acid–Schiff positive (Figure 4).7 Spiradenomas classically stain positive with CK7 (Figure 3), epithelial membrane antigen, and carcinoembryonic antigen, and have a substantial myoepithelial component, as evidenced by the myoepithelial component staining with p63, S-100, and smooth muscle actin (SMA).7-9 The distinct lack of staining with CK7 and SMA in the tumor on the left in Figure 3 confirms that these tumors are of different lineage, rather than representing cystic change within a spiradenoma.
 |  |
Adenoid cystic carcinoma is a rare neoplasm that may occur in a primary cutaneous form, as a direct extension from an underlying salivary gland neoplasm, or rarely as a focal pattern within spiradenomas occurring both sporadically or in the context of Brooke-Spiegler syndrome.2,7 The tumor is composed of variably sized cribriform islands of basaloid to pink cells concentrically arranged around glandlike spaces filled with mucin (Figure 5A). In contrast to trichoepithelioma, ACC occurs in the mid to deep dermis, often extending into subcutaneous fat with an infiltrative border, and is not often found in close proximity to hair follicles.7 Characteristically, hyaline basement membrane–like material that is periodic acid–Schiff positive is found between the tumor cells and also surrounding the individual lobules. Immunohistochemically, ACC has a myoepithelial component that stains positive with SMA, S-100, and p63; additionally, the tumor cells express low- and high-molecular-weight keratin and demonstrate variable epithelial membrane antigen positivity.10 In the current case, the superficial location, close association with a hair follicle, and lack of staining with both CK7 (Figure 3) and SMA (not shown) make ACC arising within a spiradenoma a less likely diagnosis.
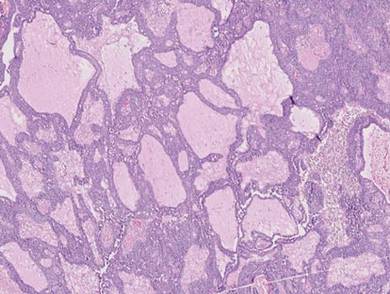
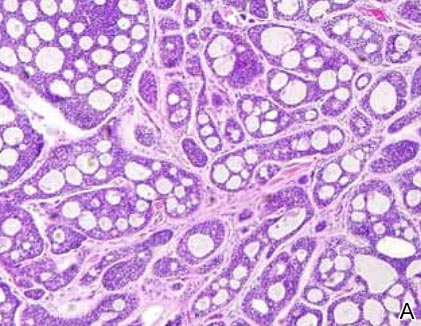
Cylindromas are composed of basaloid islands interconnected in a jigsaw puzzle configuration (Figure 5B).4 Similar to spiradenomas, they also are composed of 2 cell populations. Characteristically, the tumor islands are outlined by a hyalinized eosinophilic basement membrane. Hyalinized droplets of basement membrane zone material also may be noted in the islands. Unlike spiradenomas, they lack both intratumoral lymphocytes and a trabecular growth pattern. Although spiradenocylindromas (cylindroma and spiradenoma collision tumors) are perhaps the most common collision tumor associated with Brooke-Spiegler syndrome, there is no evidence suggesting the presence of a cylindroma in the current case.
  |
Primary cutaneous mucinous carcinoma is a rare neoplasm with a predilection for the eyelids; lesions occurring outside of this facial distribution, particularly of the breast, warrant a workup for metastatic disease.7 It typically occurs in the deeper dermis with involvement of the subcutaneous fat and is characterized by delicate fibrous septa enveloping large lakes of mucin, which contain islands of tumor cells (Figure 6). It has not been reported in association with spiradenomas. In addition, the tumor cells typically are CK7 positive.
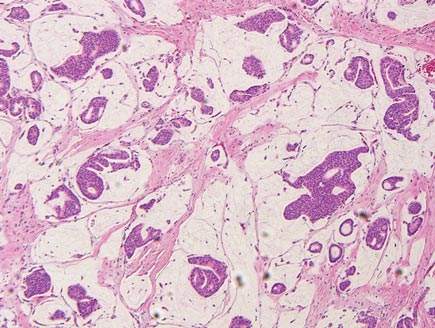
1. Kazakov DV, Soukup R, Mukensnabl P, et al. Brooke-Spiegler syndrome: report of a case with combined lesions containing cylindromatous, spiradenomatous, trichoblastomatous, and sebaceous differentiation. Am J Dermatopathol. 2005;27:27-33.
2. Petersson F, Kutzner H, Spagnolo DV, et al. Adenoid cystic carcinoma-like pattern in spiradenoma and spiradenocylindroma: a rare feature in sporadic neoplasms and those associated with Brooke-Spiegler syndrome. Am J Dermatopathol. 2009;31:642-648.
3. Weyers W, Nilles M, Eckert F, et al. Spiradenomas in Brooke-Spiegler syndrome. Am J Dermatopathol. 1993;15:156-161.
4. Elston DM, Ferringer T. Dermatopathology. Edinburgh, Scotland: Elsevier Saunders; 2009.
5. Ackerman AB, de Viragh PA, Chongchitnant N. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
6. Yamamoto O, Asahi M. Cytokeratin expression in trichoblastic fibroma (small nodular type trichoblastoma), trichoepithelioma and basal cell carcinoma. Br J Dermatol. 1999;140:8-16.
7. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin with Clinical Correlations. 4th ed. St Louis, MO: Elsevier Saunders; 2012.
8. Meybehm M, Fischer HP. Spiradenoma and dermal cylindroma: comparative immunohistochemical analysis and histogenetic considerations. Am J Dermatopathol. 1997;19:154-161.
9. Kurokawa I, Nishimura K, Tarumi C, et al. Eccrinespiradenoma: co-expression of cytokeratin and smooth muscle actin suggesting differentiation toward myoepithelial cells. J Eur Acad Dermatol Venereol. 2007;21:121-123.
10. Thompson LD, Penner C, Ho NJ, et al. Sinonasal tract and nasopharyngeal adenoid cystic carcinoma: a clinicopathologic and immunophenotypic study of 86 cases. Head Neck Pathol. 2014;8:88-109.
The coexistence of more than one cutaneous adnexal neoplasm in a single biopsy specimen is unusual and is most frequently recognized in the context of a nevus sebaceous or Brooke-Spiegler syndrome, an autosomal-dominant inherited disease characterized by cutaneous adnexal neoplasms, most commonly cylindromas and trichoepitheliomas.1-3 Brooke-Spiegler syndrome is caused by germline mutations in the cylindromatosis gene, CYLD, located on band 16q12; it functions as a tumor suppressor gene and has regulatory roles in development, immunity, and inflammation.1 Weyers et al3 first recognized the tendency for adnexal collision tumors to present in patients with Brooke-Spiegler syndrome; they reported a patient with Brooke-Spiegler syndrome with spiradenomas found in the immediate vicinity of trichoepitheliomas and in continuity with hair follicles.
Spiradenomas are composed of large, sharply demarcated, rounded nodules of basaloid cells with little cytoplasm (Figure 1).4 The basaloid nodules may demonstrate a trabecular architecture, and on close inspection 2 cell types—paler cells with more cytoplasm and darker cells with less cytoplasm—are distinguishable (Figure 2A). Lymphocytes often are scattered within the tumor nodules and/or stroma. In Brooke-Spiegler syndrome, collision tumors containing a spiradenomatous component in collision with trichoepithelioma are not uncommon.1 Spiradenomas in Brooke-Spiegler syndrome have been reported to contain sebaceous differentiation or foci with an adenoid cystic carcinoma (ACC)–like pattern and are known to occur as hybrid lesions of spiradenoma and cylindroma or trichoepithelioma (as in this case).
In this case, 2 distinct neoplasms (spiradenoma and trichoepithelioma) are apparent, side by side, with an intervening hair follicle (Figure 1). Trichoepitheliomas, also known as cribriform trichoblastomas,5 are characterized by lobules of basaloid cells resembling basal cell carcinoma surrounded by a fibroblast-rich stroma. They often contain fingerlike projections and adopt a cribriform morphology within the tumor lobules (Figure 2B).4 Numerous horn cysts may be present, but their absence does not preclude the diagnosis. Mucin may be present within the cribriform tumor islands (Figure 2B) but not in the stroma. Characteristically, trichoepitheliomas are distinctly negative for CK7 (Figure 3), and unlike spiradenomas, they lack a myoepithelial component.6 This staining pattern in combination with the tumor’s proximity to an adjacent hair follicle makes a diagnosis of trichoepithelioma and spiradenoma collision tumor most likely and supports a clinical suspicion for Brooke-Spiegler syndrome.
|
Although spiradenomas sometimes contain cystic cavities (microcystic change), they typically are filled with finely granular eosinophilic material, not mucin, that is diastase resistant and periodic acid–Schiff positive (Figure 4).7 Spiradenomas classically stain positive with CK7 (Figure 3), epithelial membrane antigen, and carcinoembryonic antigen, and have a substantial myoepithelial component, as evidenced by the myoepithelial component staining with p63, S-100, and smooth muscle actin (SMA).7-9 The distinct lack of staining with CK7 and SMA in the tumor on the left in Figure 3 confirms that these tumors are of different lineage, rather than representing cystic change within a spiradenoma.
| |
Adenoid cystic carcinoma is a rare neoplasm that may occur in a primary cutaneous form, as a direct extension from an underlying salivary gland neoplasm, or rarely as a focal pattern within spiradenomas occurring both sporadically or in the context of Brooke-Spiegler syndrome.2,7 The tumor is composed of variably sized cribriform islands of basaloid to pink cells concentrically arranged around glandlike spaces filled with mucin (Figure 5A). In contrast to trichoepithelioma, ACC occurs in the mid to deep dermis, often extending into subcutaneous fat with an infiltrative border, and is not often found in close proximity to hair follicles.7 Characteristically, hyaline basement membrane–like material that is periodic acid–Schiff positive is found between the tumor cells and also surrounding the individual lobules. Immunohistochemically, ACC has a myoepithelial component that stains positive with SMA, S-100, and p63; additionally, the tumor cells express low- and high-molecular-weight keratin and demonstrate variable epithelial membrane antigen positivity.10 In the current case, the superficial location, close association with a hair follicle, and lack of staining with both CK7 (Figure 3) and SMA (not shown) make ACC arising within a spiradenoma a less likely diagnosis.
Cylindromas are composed of basaloid islands interconnected in a jigsaw puzzle configuration (Figure 5B).4 Similar to spiradenomas, they also are composed of 2 cell populations. Characteristically, the tumor islands are outlined by a hyalinized eosinophilic basement membrane. Hyalinized droplets of basement membrane zone material also may be noted in the islands. Unlike spiradenomas, they lack both intratumoral lymphocytes and a trabecular growth pattern. Although spiradenocylindromas (cylindroma and spiradenoma collision tumors) are perhaps the most common collision tumor associated with Brooke-Spiegler syndrome, there is no evidence suggesting the presence of a cylindroma in the current case.
|
Primary cutaneous mucinous carcinoma is a rare neoplasm with a predilection for the eyelids; lesions occurring outside of this facial distribution, particularly of the breast, warrant a workup for metastatic disease.7 It typically occurs in the deeper dermis with involvement of the subcutaneous fat and is characterized by delicate fibrous septa enveloping large lakes of mucin, which contain islands of tumor cells (Figure 6). It has not been reported in association with spiradenomas. In addition, the tumor cells typically are CK7 positive.
The coexistence of more than one cutaneous adnexal neoplasm in a single biopsy specimen is unusual and is most frequently recognized in the context of a nevus sebaceous or Brooke-Spiegler syndrome, an autosomal-dominant inherited disease characterized by cutaneous adnexal neoplasms, most commonly cylindromas and trichoepitheliomas.1-3 Brooke-Spiegler syndrome is caused by germline mutations in the cylindromatosis gene, CYLD, located on band 16q12; it functions as a tumor suppressor gene and has regulatory roles in development, immunity, and inflammation.1 Weyers et al3 first recognized the tendency for adnexal collision tumors to present in patients with Brooke-Spiegler syndrome; they reported a patient with Brooke-Spiegler syndrome with spiradenomas found in the immediate vicinity of trichoepitheliomas and in continuity with hair follicles.
Spiradenomas are composed of large, sharply demarcated, rounded nodules of basaloid cells with little cytoplasm (Figure 1).4 The basaloid nodules may demonstrate a trabecular architecture, and on close inspection 2 cell types—paler cells with more cytoplasm and darker cells with less cytoplasm—are distinguishable (Figure 2A). Lymphocytes often are scattered within the tumor nodules and/or stroma. In Brooke-Spiegler syndrome, collision tumors containing a spiradenomatous component in collision with trichoepithelioma are not uncommon.1 Spiradenomas in Brooke-Spiegler syndrome have been reported to contain sebaceous differentiation or foci with an adenoid cystic carcinoma (ACC)–like pattern and are known to occur as hybrid lesions of spiradenoma and cylindroma or trichoepithelioma (as in this case).
In this case, 2 distinct neoplasms (spiradenoma and trichoepithelioma) are apparent, side by side, with an intervening hair follicle (Figure 1). Trichoepitheliomas, also known as cribriform trichoblastomas,5 are characterized by lobules of basaloid cells resembling basal cell carcinoma surrounded by a fibroblast-rich stroma. They often contain fingerlike projections and adopt a cribriform morphology within the tumor lobules (Figure 2B).4 Numerous horn cysts may be present, but their absence does not preclude the diagnosis. Mucin may be present within the cribriform tumor islands (Figure 2B) but not in the stroma. Characteristically, trichoepitheliomas are distinctly negative for CK7 (Figure 3), and unlike spiradenomas, they lack a myoepithelial component.6 This staining pattern in combination with the tumor’s proximity to an adjacent hair follicle makes a diagnosis of trichoepithelioma and spiradenoma collision tumor most likely and supports a clinical suspicion for Brooke-Spiegler syndrome.
|
Although spiradenomas sometimes contain cystic cavities (microcystic change), they typically are filled with finely granular eosinophilic material, not mucin, that is diastase resistant and periodic acid–Schiff positive (Figure 4).7 Spiradenomas classically stain positive with CK7 (Figure 3), epithelial membrane antigen, and carcinoembryonic antigen, and have a substantial myoepithelial component, as evidenced by the myoepithelial component staining with p63, S-100, and smooth muscle actin (SMA).7-9 The distinct lack of staining with CK7 and SMA in the tumor on the left in Figure 3 confirms that these tumors are of different lineage, rather than representing cystic change within a spiradenoma.
| |
Adenoid cystic carcinoma is a rare neoplasm that may occur in a primary cutaneous form, as a direct extension from an underlying salivary gland neoplasm, or rarely as a focal pattern within spiradenomas occurring both sporadically or in the context of Brooke-Spiegler syndrome.2,7 The tumor is composed of variably sized cribriform islands of basaloid to pink cells concentrically arranged around glandlike spaces filled with mucin (Figure 5A). In contrast to trichoepithelioma, ACC occurs in the mid to deep dermis, often extending into subcutaneous fat with an infiltrative border, and is not often found in close proximity to hair follicles.7 Characteristically, hyaline basement membrane–like material that is periodic acid–Schiff positive is found between the tumor cells and also surrounding the individual lobules. Immunohistochemically, ACC has a myoepithelial component that stains positive with SMA, S-100, and p63; additionally, the tumor cells express low- and high-molecular-weight keratin and demonstrate variable epithelial membrane antigen positivity.10 In the current case, the superficial location, close association with a hair follicle, and lack of staining with both CK7 (Figure 3) and SMA (not shown) make ACC arising within a spiradenoma a less likely diagnosis.
Cylindromas are composed of basaloid islands interconnected in a jigsaw puzzle configuration (Figure 5B).4 Similar to spiradenomas, they also are composed of 2 cell populations. Characteristically, the tumor islands are outlined by a hyalinized eosinophilic basement membrane. Hyalinized droplets of basement membrane zone material also may be noted in the islands. Unlike spiradenomas, they lack both intratumoral lymphocytes and a trabecular growth pattern. Although spiradenocylindromas (cylindroma and spiradenoma collision tumors) are perhaps the most common collision tumor associated with Brooke-Spiegler syndrome, there is no evidence suggesting the presence of a cylindroma in the current case.
|
Primary cutaneous mucinous carcinoma is a rare neoplasm with a predilection for the eyelids; lesions occurring outside of this facial distribution, particularly of the breast, warrant a workup for metastatic disease.7 It typically occurs in the deeper dermis with involvement of the subcutaneous fat and is characterized by delicate fibrous septa enveloping large lakes of mucin, which contain islands of tumor cells (Figure 6). It has not been reported in association with spiradenomas. In addition, the tumor cells typically are CK7 positive.
1. Kazakov DV, Soukup R, Mukensnabl P, et al. Brooke-Spiegler syndrome: report of a case with combined lesions containing cylindromatous, spiradenomatous, trichoblastomatous, and sebaceous differentiation. Am J Dermatopathol. 2005;27:27-33.
2. Petersson F, Kutzner H, Spagnolo DV, et al. Adenoid cystic carcinoma-like pattern in spiradenoma and spiradenocylindroma: a rare feature in sporadic neoplasms and those associated with Brooke-Spiegler syndrome. Am J Dermatopathol. 2009;31:642-648.
3. Weyers W, Nilles M, Eckert F, et al. Spiradenomas in Brooke-Spiegler syndrome. Am J Dermatopathol. 1993;15:156-161.
4. Elston DM, Ferringer T. Dermatopathology. Edinburgh, Scotland: Elsevier Saunders; 2009.
5. Ackerman AB, de Viragh PA, Chongchitnant N. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
6. Yamamoto O, Asahi M. Cytokeratin expression in trichoblastic fibroma (small nodular type trichoblastoma), trichoepithelioma and basal cell carcinoma. Br J Dermatol. 1999;140:8-16.
7. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin with Clinical Correlations. 4th ed. St Louis, MO: Elsevier Saunders; 2012.
8. Meybehm M, Fischer HP. Spiradenoma and dermal cylindroma: comparative immunohistochemical analysis and histogenetic considerations. Am J Dermatopathol. 1997;19:154-161.
9. Kurokawa I, Nishimura K, Tarumi C, et al. Eccrinespiradenoma: co-expression of cytokeratin and smooth muscle actin suggesting differentiation toward myoepithelial cells. J Eur Acad Dermatol Venereol. 2007;21:121-123.
10. Thompson LD, Penner C, Ho NJ, et al. Sinonasal tract and nasopharyngeal adenoid cystic carcinoma: a clinicopathologic and immunophenotypic study of 86 cases. Head Neck Pathol. 2014;8:88-109.
1. Kazakov DV, Soukup R, Mukensnabl P, et al. Brooke-Spiegler syndrome: report of a case with combined lesions containing cylindromatous, spiradenomatous, trichoblastomatous, and sebaceous differentiation. Am J Dermatopathol. 2005;27:27-33.
2. Petersson F, Kutzner H, Spagnolo DV, et al. Adenoid cystic carcinoma-like pattern in spiradenoma and spiradenocylindroma: a rare feature in sporadic neoplasms and those associated with Brooke-Spiegler syndrome. Am J Dermatopathol. 2009;31:642-648.
3. Weyers W, Nilles M, Eckert F, et al. Spiradenomas in Brooke-Spiegler syndrome. Am J Dermatopathol. 1993;15:156-161.
4. Elston DM, Ferringer T. Dermatopathology. Edinburgh, Scotland: Elsevier Saunders; 2009.
5. Ackerman AB, de Viragh PA, Chongchitnant N. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
6. Yamamoto O, Asahi M. Cytokeratin expression in trichoblastic fibroma (small nodular type trichoblastoma), trichoepithelioma and basal cell carcinoma. Br J Dermatol. 1999;140:8-16.
7. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin with Clinical Correlations. 4th ed. St Louis, MO: Elsevier Saunders; 2012.
8. Meybehm M, Fischer HP. Spiradenoma and dermal cylindroma: comparative immunohistochemical analysis and histogenetic considerations. Am J Dermatopathol. 1997;19:154-161.
9. Kurokawa I, Nishimura K, Tarumi C, et al. Eccrinespiradenoma: co-expression of cytokeratin and smooth muscle actin suggesting differentiation toward myoepithelial cells. J Eur Acad Dermatol Venereol. 2007;21:121-123.
10. Thompson LD, Penner C, Ho NJ, et al. Sinonasal tract and nasopharyngeal adenoid cystic carcinoma: a clinicopathologic and immunophenotypic study of 86 cases. Head Neck Pathol. 2014;8:88-109.
How Can Dermatologists Help Dermatopathologists Work “Smarter” for Them?
In dermatology and dermatopathology, some histologic diagnoses are incontrovertible and reveal features that are readily diagnostic; however, in many cases clinical correlation is essential, as many diseases have similar histologic reaction patterns and the correct diagnosis is based on additional clinical information. Accuracy of diagnosis has been shown to improve when cases were evaluated at a clinical conference where histology and clinical features were correlated or when digital clinical photographs were evaluated with histologic findings.1
Because clinical features usually are not available to the dermatopathologist when evaluating histologic specimens, he/she must rely on the information provided by the referring clinician on the pathology requisition form. It is important for clinicians to include as much information as is reasonably possible on the form in a legible fashion. If the specimen is a pigmented lesion, it should be described by its diameter and any additional information that is available, such as dermoscopic findings. If the process is an eruption, the extent, distribution, color, duration, symptoms, and any other relevant information should be provided.
Clinicians should always avoid “cryptic” allusions. Occasionally, dermatopathologists receive biopsy specimens with no information other than to rule out leukemia cutis. There is obviously more to that story. Do not expect the dermatopathologist to be a mind reader. We appreciate a request when a clinician wants a special stain or a margin rather than assuming we know when those are desired. We also would prefer for margin requests not to be made in an automatic manner when it does not matter whether the process involves the margins, such as in inflammatory conditions.
One should not dilute the value of the clinical impression. For example, do not write “rule out melanoma” for all pigmented lesions or “neoplasm of uncertain behavior” for all cutaneous neoplasms. If there is a prior biopsy, submit the prior number and the diagnosis if possible. Fill out all demographic information (eg, sex, race, age) and other important information such as pregnancy status, medication history, underlying condition, or history of neoplasia, as they all have bearing on the diagnosis.
Inflammatory skin diseases can be challenging to diagnose, as classic examples described in textbooks usually are not sampled. Consider performing more than one biopsy from lesions at different stages of evolution or from different body sites. In difficult cases, bring the patient to a conference, send the patient for consultation, or submit a clinical photograph or digital image. It also may be beneficial to call and discuss the case with the dermatopathologist. If the diagnosis does not make sense, ask for recuts, special stains, or a second expert opinion.
Regarding the biopsy process itself, always harvest a good piece of tissue and place it into the proper medium for the appropriate test. Formalin solution 10% is used for routine specimens, while Michel’s transport medium and saline are used for immunofluorescence studies. Make sure the specimen is floating in the liquid, as specimens may adhere to the side of the bottle or the lid. Beware of specimens remaining on a scalpel blade or within the barrel of a punch before inadvertently submitting a bottle containing no material. Also be sure to fill out the information on the bottle label, as the bottle and requisition form may get separated. Do not write on the lid in case it happens to come off.
It also is important not to put more than 1 specimen in the same bottle, especially when dealing with multiple different neoplasms. A possible reasonable exception is multiple skin tags, but note in the chart that you are submitting representative specimens. Beware of throwing away tissue instead of submitting it for biopsy whenever something is removed from the skin.
Keep a biopsy logbook or other records and review all pathology reports systematically. Communicate with the laboratory to check on the status of a biopsy if results are not back as soon as expected to ensure that there is not an inadvertent problem. Legal action can result if the patient is not notified in a timely fashion or if treatment is delayed. If the patient does not follow-up in a timely fashion, a certified letter should be sent to the patient.
Extremely small specimens (ie, <1 mm in diameter) or friable specimens may not survive processing. Occasionally, a specimen gets lost, either on the way to the laboratory or otherwise. If there is still a neoplasm at the site or the patient has a widespread process, another biopsy can be performed. My laboratory has a policy of not charging the patient given the circumstances. If nothing is left at the site and it is a neoplasm that could possibly have been malignant, a reasonable approach would be to conservatively re-excise the area and document everything in the medical record.
Regarding biopsy technique, punches of broad neoplasms, especially possible melanoma, may give false-negative results, which includes sampling the darkest area, a practice thought to be more sensitive at detecting malignancy. In actuality, it may be an area of hemorrhage, an associated solar lentigo, or a seborrheic keratosis. Furthermore, this may produce the phenomenon of “biopsy sculpture,” turning a large asymmetrical neoplasm into a smaller sample that looks symmetrical and more benign than it actually is. If a punch biopsy is performed, it should be a broad punch (ie, >5 mm in diameter) or the entire lesion should be punched out, which represents an excision. Multiple small punches are not optimal, as a broad specimen that provides a panoramic view of the entire process is preferable. Although incision or excision specimens are excellent, they often are impractical; rather, a broad saucerization biopsy is an excellent method that provides a representative specimen in the majority of cases.
Shave specimens of inflammatory processes do not sample the lower dermis or subcutis and often are inadequate, leading to reports of “tissue insufficient for diagnosis” or “descriptive” rather than specific diagnoses. Inadequate biopsies increase expenses as well as patient inconvenience and anger. In my laboratory, we teach residents to treat the biopsy as a treasure. Some clinicians think that performing a biopsy is analogous to doing a blood test and that any tissue at all will be sufficient for a diagnosis to be rendered; however, small curettings or tiny fragments of tissue often are inadequate and place both the clinician and the pathologist at medicolegal risk.
Clinicians requesting margins on shave biopsies must understand that they are not equivalent to margins reported on elliptical excision specimens or those performed using Mohs micrographic surgery. The pathologist can only report that a neoplasm removed by shave technique “seems to be removed in these sections,” as it cannot be determined with certainty that the lesion has been completely removed using this technique.
All biopsies are prone to sampling error. Dermatopathologists often put a note on a report saying that if this specimen represents part of a larger lesion, clinical correlation is recommended to exclude sampling error, which should be known by clinicians. It is extremely risky to ask for margins on a melanoma biopsy, and one should never assume a shave biopsy of a melanoma is adequate treatment.
If a clinician is clinically concerned about a diagnosis, especially melanoma, and the histologic diagnosis is benign, it is recommended that the lesion be excised nonetheless. A final diagnosis depends on a number of clinical, histological, historical, and genetic elements and possibly others. In some cases, the clinical diagnosis is more important than the histologic diagnosis. Clinicians should always feel free to call their dermatopathologist, ask questions, and refute a diagnosis. The dermatopathologist seeks to arrive at the best diagnosis for the patient, not to be “right.”
Diagnoses should be simple and differential diagnoses few. The longer the report and the more stains that are performed, generally the less is known about the diagnosis. Diagnoses should be rendered in terms easily understood by clinical dermatologists. Get a consultant dermatopathologist that you know and trust.
Reference
1. Cerroni L, Argenyi Z, Cerio R, et al. Influence of evaluation of clinical pictures on the histopathologic diagnosis of inflammatory skin diseases. J Am Acad Dermatol. 2010;63:647-652.
In dermatology and dermatopathology, some histologic diagnoses are incontrovertible and reveal features that are readily diagnostic; however, in many cases clinical correlation is essential, as many diseases have similar histologic reaction patterns and the correct diagnosis is based on additional clinical information. Accuracy of diagnosis has been shown to improve when cases were evaluated at a clinical conference where histology and clinical features were correlated or when digital clinical photographs were evaluated with histologic findings.1
Because clinical features usually are not available to the dermatopathologist when evaluating histologic specimens, he/she must rely on the information provided by the referring clinician on the pathology requisition form. It is important for clinicians to include as much information as is reasonably possible on the form in a legible fashion. If the specimen is a pigmented lesion, it should be described by its diameter and any additional information that is available, such as dermoscopic findings. If the process is an eruption, the extent, distribution, color, duration, symptoms, and any other relevant information should be provided.
Clinicians should always avoid “cryptic” allusions. Occasionally, dermatopathologists receive biopsy specimens with no information other than to rule out leukemia cutis. There is obviously more to that story. Do not expect the dermatopathologist to be a mind reader. We appreciate a request when a clinician wants a special stain or a margin rather than assuming we know when those are desired. We also would prefer for margin requests not to be made in an automatic manner when it does not matter whether the process involves the margins, such as in inflammatory conditions.
One should not dilute the value of the clinical impression. For example, do not write “rule out melanoma” for all pigmented lesions or “neoplasm of uncertain behavior” for all cutaneous neoplasms. If there is a prior biopsy, submit the prior number and the diagnosis if possible. Fill out all demographic information (eg, sex, race, age) and other important information such as pregnancy status, medication history, underlying condition, or history of neoplasia, as they all have bearing on the diagnosis.
Inflammatory skin diseases can be challenging to diagnose, as classic examples described in textbooks usually are not sampled. Consider performing more than one biopsy from lesions at different stages of evolution or from different body sites. In difficult cases, bring the patient to a conference, send the patient for consultation, or submit a clinical photograph or digital image. It also may be beneficial to call and discuss the case with the dermatopathologist. If the diagnosis does not make sense, ask for recuts, special stains, or a second expert opinion.
Regarding the biopsy process itself, always harvest a good piece of tissue and place it into the proper medium for the appropriate test. Formalin solution 10% is used for routine specimens, while Michel’s transport medium and saline are used for immunofluorescence studies. Make sure the specimen is floating in the liquid, as specimens may adhere to the side of the bottle or the lid. Beware of specimens remaining on a scalpel blade or within the barrel of a punch before inadvertently submitting a bottle containing no material. Also be sure to fill out the information on the bottle label, as the bottle and requisition form may get separated. Do not write on the lid in case it happens to come off.
It also is important not to put more than 1 specimen in the same bottle, especially when dealing with multiple different neoplasms. A possible reasonable exception is multiple skin tags, but note in the chart that you are submitting representative specimens. Beware of throwing away tissue instead of submitting it for biopsy whenever something is removed from the skin.
Keep a biopsy logbook or other records and review all pathology reports systematically. Communicate with the laboratory to check on the status of a biopsy if results are not back as soon as expected to ensure that there is not an inadvertent problem. Legal action can result if the patient is not notified in a timely fashion or if treatment is delayed. If the patient does not follow-up in a timely fashion, a certified letter should be sent to the patient.
Extremely small specimens (ie, <1 mm in diameter) or friable specimens may not survive processing. Occasionally, a specimen gets lost, either on the way to the laboratory or otherwise. If there is still a neoplasm at the site or the patient has a widespread process, another biopsy can be performed. My laboratory has a policy of not charging the patient given the circumstances. If nothing is left at the site and it is a neoplasm that could possibly have been malignant, a reasonable approach would be to conservatively re-excise the area and document everything in the medical record.
Regarding biopsy technique, punches of broad neoplasms, especially possible melanoma, may give false-negative results, which includes sampling the darkest area, a practice thought to be more sensitive at detecting malignancy. In actuality, it may be an area of hemorrhage, an associated solar lentigo, or a seborrheic keratosis. Furthermore, this may produce the phenomenon of “biopsy sculpture,” turning a large asymmetrical neoplasm into a smaller sample that looks symmetrical and more benign than it actually is. If a punch biopsy is performed, it should be a broad punch (ie, >5 mm in diameter) or the entire lesion should be punched out, which represents an excision. Multiple small punches are not optimal, as a broad specimen that provides a panoramic view of the entire process is preferable. Although incision or excision specimens are excellent, they often are impractical; rather, a broad saucerization biopsy is an excellent method that provides a representative specimen in the majority of cases.
Shave specimens of inflammatory processes do not sample the lower dermis or subcutis and often are inadequate, leading to reports of “tissue insufficient for diagnosis” or “descriptive” rather than specific diagnoses. Inadequate biopsies increase expenses as well as patient inconvenience and anger. In my laboratory, we teach residents to treat the biopsy as a treasure. Some clinicians think that performing a biopsy is analogous to doing a blood test and that any tissue at all will be sufficient for a diagnosis to be rendered; however, small curettings or tiny fragments of tissue often are inadequate and place both the clinician and the pathologist at medicolegal risk.
Clinicians requesting margins on shave biopsies must understand that they are not equivalent to margins reported on elliptical excision specimens or those performed using Mohs micrographic surgery. The pathologist can only report that a neoplasm removed by shave technique “seems to be removed in these sections,” as it cannot be determined with certainty that the lesion has been completely removed using this technique.
All biopsies are prone to sampling error. Dermatopathologists often put a note on a report saying that if this specimen represents part of a larger lesion, clinical correlation is recommended to exclude sampling error, which should be known by clinicians. It is extremely risky to ask for margins on a melanoma biopsy, and one should never assume a shave biopsy of a melanoma is adequate treatment.
If a clinician is clinically concerned about a diagnosis, especially melanoma, and the histologic diagnosis is benign, it is recommended that the lesion be excised nonetheless. A final diagnosis depends on a number of clinical, histological, historical, and genetic elements and possibly others. In some cases, the clinical diagnosis is more important than the histologic diagnosis. Clinicians should always feel free to call their dermatopathologist, ask questions, and refute a diagnosis. The dermatopathologist seeks to arrive at the best diagnosis for the patient, not to be “right.”
Diagnoses should be simple and differential diagnoses few. The longer the report and the more stains that are performed, generally the less is known about the diagnosis. Diagnoses should be rendered in terms easily understood by clinical dermatologists. Get a consultant dermatopathologist that you know and trust.
In dermatology and dermatopathology, some histologic diagnoses are incontrovertible and reveal features that are readily diagnostic; however, in many cases clinical correlation is essential, as many diseases have similar histologic reaction patterns and the correct diagnosis is based on additional clinical information. Accuracy of diagnosis has been shown to improve when cases were evaluated at a clinical conference where histology and clinical features were correlated or when digital clinical photographs were evaluated with histologic findings.1
Because clinical features usually are not available to the dermatopathologist when evaluating histologic specimens, he/she must rely on the information provided by the referring clinician on the pathology requisition form. It is important for clinicians to include as much information as is reasonably possible on the form in a legible fashion. If the specimen is a pigmented lesion, it should be described by its diameter and any additional information that is available, such as dermoscopic findings. If the process is an eruption, the extent, distribution, color, duration, symptoms, and any other relevant information should be provided.
Clinicians should always avoid “cryptic” allusions. Occasionally, dermatopathologists receive biopsy specimens with no information other than to rule out leukemia cutis. There is obviously more to that story. Do not expect the dermatopathologist to be a mind reader. We appreciate a request when a clinician wants a special stain or a margin rather than assuming we know when those are desired. We also would prefer for margin requests not to be made in an automatic manner when it does not matter whether the process involves the margins, such as in inflammatory conditions.
One should not dilute the value of the clinical impression. For example, do not write “rule out melanoma” for all pigmented lesions or “neoplasm of uncertain behavior” for all cutaneous neoplasms. If there is a prior biopsy, submit the prior number and the diagnosis if possible. Fill out all demographic information (eg, sex, race, age) and other important information such as pregnancy status, medication history, underlying condition, or history of neoplasia, as they all have bearing on the diagnosis.
Inflammatory skin diseases can be challenging to diagnose, as classic examples described in textbooks usually are not sampled. Consider performing more than one biopsy from lesions at different stages of evolution or from different body sites. In difficult cases, bring the patient to a conference, send the patient for consultation, or submit a clinical photograph or digital image. It also may be beneficial to call and discuss the case with the dermatopathologist. If the diagnosis does not make sense, ask for recuts, special stains, or a second expert opinion.
Regarding the biopsy process itself, always harvest a good piece of tissue and place it into the proper medium for the appropriate test. Formalin solution 10% is used for routine specimens, while Michel’s transport medium and saline are used for immunofluorescence studies. Make sure the specimen is floating in the liquid, as specimens may adhere to the side of the bottle or the lid. Beware of specimens remaining on a scalpel blade or within the barrel of a punch before inadvertently submitting a bottle containing no material. Also be sure to fill out the information on the bottle label, as the bottle and requisition form may get separated. Do not write on the lid in case it happens to come off.
It also is important not to put more than 1 specimen in the same bottle, especially when dealing with multiple different neoplasms. A possible reasonable exception is multiple skin tags, but note in the chart that you are submitting representative specimens. Beware of throwing away tissue instead of submitting it for biopsy whenever something is removed from the skin.
Keep a biopsy logbook or other records and review all pathology reports systematically. Communicate with the laboratory to check on the status of a biopsy if results are not back as soon as expected to ensure that there is not an inadvertent problem. Legal action can result if the patient is not notified in a timely fashion or if treatment is delayed. If the patient does not follow-up in a timely fashion, a certified letter should be sent to the patient.
Extremely small specimens (ie, <1 mm in diameter) or friable specimens may not survive processing. Occasionally, a specimen gets lost, either on the way to the laboratory or otherwise. If there is still a neoplasm at the site or the patient has a widespread process, another biopsy can be performed. My laboratory has a policy of not charging the patient given the circumstances. If nothing is left at the site and it is a neoplasm that could possibly have been malignant, a reasonable approach would be to conservatively re-excise the area and document everything in the medical record.
Regarding biopsy technique, punches of broad neoplasms, especially possible melanoma, may give false-negative results, which includes sampling the darkest area, a practice thought to be more sensitive at detecting malignancy. In actuality, it may be an area of hemorrhage, an associated solar lentigo, or a seborrheic keratosis. Furthermore, this may produce the phenomenon of “biopsy sculpture,” turning a large asymmetrical neoplasm into a smaller sample that looks symmetrical and more benign than it actually is. If a punch biopsy is performed, it should be a broad punch (ie, >5 mm in diameter) or the entire lesion should be punched out, which represents an excision. Multiple small punches are not optimal, as a broad specimen that provides a panoramic view of the entire process is preferable. Although incision or excision specimens are excellent, they often are impractical; rather, a broad saucerization biopsy is an excellent method that provides a representative specimen in the majority of cases.
Shave specimens of inflammatory processes do not sample the lower dermis or subcutis and often are inadequate, leading to reports of “tissue insufficient for diagnosis” or “descriptive” rather than specific diagnoses. Inadequate biopsies increase expenses as well as patient inconvenience and anger. In my laboratory, we teach residents to treat the biopsy as a treasure. Some clinicians think that performing a biopsy is analogous to doing a blood test and that any tissue at all will be sufficient for a diagnosis to be rendered; however, small curettings or tiny fragments of tissue often are inadequate and place both the clinician and the pathologist at medicolegal risk.
Clinicians requesting margins on shave biopsies must understand that they are not equivalent to margins reported on elliptical excision specimens or those performed using Mohs micrographic surgery. The pathologist can only report that a neoplasm removed by shave technique “seems to be removed in these sections,” as it cannot be determined with certainty that the lesion has been completely removed using this technique.
All biopsies are prone to sampling error. Dermatopathologists often put a note on a report saying that if this specimen represents part of a larger lesion, clinical correlation is recommended to exclude sampling error, which should be known by clinicians. It is extremely risky to ask for margins on a melanoma biopsy, and one should never assume a shave biopsy of a melanoma is adequate treatment.
If a clinician is clinically concerned about a diagnosis, especially melanoma, and the histologic diagnosis is benign, it is recommended that the lesion be excised nonetheless. A final diagnosis depends on a number of clinical, histological, historical, and genetic elements and possibly others. In some cases, the clinical diagnosis is more important than the histologic diagnosis. Clinicians should always feel free to call their dermatopathologist, ask questions, and refute a diagnosis. The dermatopathologist seeks to arrive at the best diagnosis for the patient, not to be “right.”
Diagnoses should be simple and differential diagnoses few. The longer the report and the more stains that are performed, generally the less is known about the diagnosis. Diagnoses should be rendered in terms easily understood by clinical dermatologists. Get a consultant dermatopathologist that you know and trust.
Reference
1. Cerroni L, Argenyi Z, Cerio R, et al. Influence of evaluation of clinical pictures on the histopathologic diagnosis of inflammatory skin diseases. J Am Acad Dermatol. 2010;63:647-652.
Reference
1. Cerroni L, Argenyi Z, Cerio R, et al. Influence of evaluation of clinical pictures on the histopathologic diagnosis of inflammatory skin diseases. J Am Acad Dermatol. 2010;63:647-652.

Fellowships After Dermatology Residency: The Traditional and Beyond
Dermatology residents, such as myself, often wonder what we will do after graduation. There are many resources for finding job opportunities, and many of us have received solicitation e-mails from various headhunters and medical groups that are looking to hire. The American Academy of Dermatology (AAD) has a resource called the AAD Career Compass (http://www.healthecareers.com/aad), which is an exhaustive database of job listings for dermatologists. However, I could not locate a definitive resource containing information that might be useful for dermatology residents who are interested in subspecializing or pursuing fellowships.
Subspecialty training is typically pursued after successful completion of a dermatology residency training program. Fellowships are traditionally offered in dermatopathology, pediatric dermatology, micrographic surgery and dermatologic oncology (procedural dermatology), and cosmetic dermatologic surgery. Fellowships also are available in other subspecialties or for those pursuing an academic career. The goal of this article is to help dermatology residents learn more about traditional and nontraditional opportunities for graduate education and certification in various dermatologic subspecialties, with links to sources for more detailed information.
Traditional Fellowship Programs by Subspecialty
Dermatopathology
One- to 2-year dermatopathology fellowship programs are certified by both the American Board of Dermatology (ABD) and the American Board of Pathology and are available to graduates of either dermatology or pathology residency programs. These programs offer combined training in either anatomic pathology (for dermatologists) or clinical dermatology (for pathologists), along with dermatopathology; the majority of time is devoted to the latter. The Accreditation Council for Graduate Medical Education and the ABD have issued specific requirements for graduate medical education and subspecialty certification in dermatopathology.1,2 Fellowship matches are institution dependent, and the application process and match generally takes place during the second year of dermatology residency for those residents who want to start a fellowship program immediately following graduation. The American Society of Dermatopathology offers a dermatopathology fellowship program finder on its Web site.
Pediatric Dermatology
Fellowships in pediatric dermatology are typically 1- to 2-year programs that focus on dermatologic diseases in the pediatric population. Applicants are matched to these programs through the San Francisco Matching Program (SF Match) and the programs are ABD accredited.3,4 (There also are a number of non–ABD-approved training opportunities available.5) On completion of the training program, fellows may qualify for subspecialty board certification in pediatric dermatology. Applications are open starting in January, and the rank order list and match occur in August of the same year. As of 2012, there were 20 participating programs with 28 available positions, while the match included 22 applicants; of these applicants, 15 matched formally into pediatric dermatology fellowships.6
Micrographic Surgery and Dermatologic Oncology (Procedural Dermatology)
There are specific requirements issued by the Accreditation Council for Graduate Medical Education for dermatologic surgery fellowships,7 which are typically 1- to 2-year programs. Many fellowship programs also are accredited by the American College of Mohs Surgery. This subspecialty is not ABD accredited; therefore, there is no certification process upon completion of a fellowship program. The American College of Mohs Surgery sponsors the match process through SF Match. Applicant registration begins in July and the match occurs in December of the same year. As of 2013, there were 47 participating programs offering 55 positions. Of 77 applicants, 49 obtained fellowship positions formally through the match.8 The American Society for Dermatologic Surgery (ASDS) Web site provides the DermSurg Fellowship Finder, which includes information about independent fellowship programs.
Cosmetic Dermatologic Surgery
The ASDS has an accreditation program for fellowships in cosmetic dermatologic surgery,9 which are generally 1-year programs. Certification in this subspecialty is not ABD accredited. Fellowship opportunities can be found using the ASDS DermSurg Fellowship Finder.
Nontraditional Fellowship Programs
The following are fellowship programs that are in nontraditional subspecialties, are only available at certain institutions, and are not accredited. This list is not exhaustive of all available programs but are those that may be of interest to dermatology residents who are drawn to a particular dermatologic subspecialty or have an interest in academic dermatology. There is no formal match process and applications vary by institution.
Clinician Educator Fellowship
The clinician educator fellowship is available at the Department of Dermatology at the University of Pennsylvania (Philadelphia, Pennsylvania) and is intended to foster dermatologic clinician educators. More information can be found on the program’s Web site.
Cutaneous Oncology Fellowship
This 1- to 2-year fellowship program focuses on diagnosis and management of melanoma and nonmelanoma skin cancers as well as cutaneous lymphomas. Fellowships in cutaneous oncology are offered at the University of California, San Francisco (San Francisco, California), Brigham and Women’s Hospital (Boston, Massachusetts), the University of Pennsylvania (Philadelphia, Pennsylvania), Case Western Reserve University (Cleveland, Ohio), the University of Pittsburgh (Pittsburgh, Pennsylvania), and Stanford University Medical Center (Stanford, California).
Dermatology/Rheumatology
The dermatology/rheumatology fellowship offered by Brigham and Women’s Hospital is a 1-year program that focuses on the management of connective-tissue diseases in a multidisciplinary fashion with rheumatology.
Advanced Medical Dermatology/Complex Medical Dermatology
Several programs offer fellowships in medical dermatology under different titles but with a similar curriculum and goal: to foster dermatologists interested in careers as academic medical dermatologists or as future clinician scientists by means of specialized training and mentorship in complex medical and dermatological issues in the outpatient and inpatient settings. The 2-year program at New York University School of Medicine (New York, New York) also gives fellows the opportunity to earn a master of science in clinical investigation degree. The University of California, San Francisco, program offers protected time for career development.
Epidemiology Training Program
Fellows and residents in the University of Pennsylvania’s dermatology training program may elect to work with the Center for Clinical Epidemiology and Biostatistics and have the opportunity to earn a graduate degree (MSCE or PhD).
Contact Dermatitis and Patch Testing Fellowship
The Dermatology Department at the Cleveland Clinic (Cleveland, Ohio) offers a 1-year contact dermatitis and patch testing fellowship that includes clinical research.
Conclusion
Both traditional and nontraditional fellowship opportunities exist after dermatology residency. This guide serves as an overview of the training programs in dermatopathology, pediatric dermatology, micrographic surgery and dermatologic oncology (procedural dermatology), and cosmetic dermatologic surgery, as well as the fellowships offered at certain institutions for those interested in more specific subspecialties or academia.

1. Accreditation Council for Graduate Medical Education. ACGME program requirements for graduate medical education in dermatopathology. http://www.acgme.org/acgmeweb/Portals/0/PFAssets/ProgramRequirements/100_dermatopathology_2016_1-YR.pdf. Revised April 2014. Effective July 1, 2015. Accessed on February 26, 2015.
2. Subspecialty certification in dermatopathology. The American Board of Dermatology, Inc Web site. http://www.abderm.org/subspecialties/derm.html. Accessed February 26, 2015.
3. American Board of Dermatology (ABD) approved pediatric dermatology fellowship programs. The Society for Pediatric Dermatology Web site. http://pedsderm.net/training/fellowships/abd-approved-pediatric-dermatology-fellowship-programs/. Updated June 9, 2014. Accessed February 26, 2015.
4. Subspecialty certification in pediatric dermatology. The American Board of Dermatology, Inc Web site. http://www.abderm.org/subspecialties/pediatric.html. Accessed February 26, 2015.
5. Non-ABD pediatric dermatology fellowship programs. The Society for Pediatric Dermatology Web site. https://pedsderm.net/training/fellowships/non-abd-pediatric-dermatology-fellowship-programs/. Accessed February 26, 2015.
6. Pediatric dermatology match report. SF Match Web site. https://www.sfmatch.org/SpecialtyInsideAll.aspx?id=16&typ=1&name=Pediatric%20Dermatology#. Accessed March 4, 2015.
7. Accreditation Council for Graduate Medical Education. ACGME program requirements for graduate medical education in procedural dermatology. https://www.acgme.org/acgmeweb/Portals/0/PFAssets/ProgramRequirements/081_procedural_derm_1-YR_07012014.pdf. Effective July 2014. Accessed February 26, 2015.
8. Statistics: micrographic surgery & dermatologic oncology fellowship. SF Match Web site. https://www.sfmatch.org/SpecialtyInsideAll.aspx?id=10&typ=1&name=Micrographic%20Surgery%20and%20Dermatologic%20Oncology#. Accessed March 4, 2015.
9. ASDS cosmetic dermatologic surgery fellowship accreditation program. American Society for Dermatologic Surgery Web site. http://www.asds.net/cosmetic-accreditation/. Accessed February 26, 2015.
Dermatology residents, such as myself, often wonder what we will do after graduation. There are many resources for finding job opportunities, and many of us have received solicitation e-mails from various headhunters and medical groups that are looking to hire. The American Academy of Dermatology (AAD) has a resource called the AAD Career Compass (http://www.healthecareers.com/aad), which is an exhaustive database of job listings for dermatologists. However, I could not locate a definitive resource containing information that might be useful for dermatology residents who are interested in subspecializing or pursuing fellowships.
Subspecialty training is typically pursued after successful completion of a dermatology residency training program. Fellowships are traditionally offered in dermatopathology, pediatric dermatology, micrographic surgery and dermatologic oncology (procedural dermatology), and cosmetic dermatologic surgery. Fellowships also are available in other subspecialties or for those pursuing an academic career. The goal of this article is to help dermatology residents learn more about traditional and nontraditional opportunities for graduate education and certification in various dermatologic subspecialties, with links to sources for more detailed information.
Traditional Fellowship Programs by Subspecialty
Dermatopathology
One- to 2-year dermatopathology fellowship programs are certified by both the American Board of Dermatology (ABD) and the American Board of Pathology and are available to graduates of either dermatology or pathology residency programs. These programs offer combined training in either anatomic pathology (for dermatologists) or clinical dermatology (for pathologists), along with dermatopathology; the majority of time is devoted to the latter. The Accreditation Council for Graduate Medical Education and the ABD have issued specific requirements for graduate medical education and subspecialty certification in dermatopathology.1,2 Fellowship matches are institution dependent, and the application process and match generally takes place during the second year of dermatology residency for those residents who want to start a fellowship program immediately following graduation. The American Society of Dermatopathology offers a dermatopathology fellowship program finder on its Web site.
Pediatric Dermatology
Fellowships in pediatric dermatology are typically 1- to 2-year programs that focus on dermatologic diseases in the pediatric population. Applicants are matched to these programs through the San Francisco Matching Program (SF Match) and the programs are ABD accredited.3,4 (There also are a number of non–ABD-approved training opportunities available.5) On completion of the training program, fellows may qualify for subspecialty board certification in pediatric dermatology. Applications are open starting in January, and the rank order list and match occur in August of the same year. As of 2012, there were 20 participating programs with 28 available positions, while the match included 22 applicants; of these applicants, 15 matched formally into pediatric dermatology fellowships.6
Micrographic Surgery and Dermatologic Oncology (Procedural Dermatology)
There are specific requirements issued by the Accreditation Council for Graduate Medical Education for dermatologic surgery fellowships,7 which are typically 1- to 2-year programs. Many fellowship programs also are accredited by the American College of Mohs Surgery. This subspecialty is not ABD accredited; therefore, there is no certification process upon completion of a fellowship program. The American College of Mohs Surgery sponsors the match process through SF Match. Applicant registration begins in July and the match occurs in December of the same year. As of 2013, there were 47 participating programs offering 55 positions. Of 77 applicants, 49 obtained fellowship positions formally through the match.8 The American Society for Dermatologic Surgery (ASDS) Web site provides the DermSurg Fellowship Finder, which includes information about independent fellowship programs.
Cosmetic Dermatologic Surgery
The ASDS has an accreditation program for fellowships in cosmetic dermatologic surgery,9 which are generally 1-year programs. Certification in this subspecialty is not ABD accredited. Fellowship opportunities can be found using the ASDS DermSurg Fellowship Finder.
Nontraditional Fellowship Programs
The following are fellowship programs that are in nontraditional subspecialties, are only available at certain institutions, and are not accredited. This list is not exhaustive of all available programs but are those that may be of interest to dermatology residents who are drawn to a particular dermatologic subspecialty or have an interest in academic dermatology. There is no formal match process and applications vary by institution.
Clinician Educator Fellowship
The clinician educator fellowship is available at the Department of Dermatology at the University of Pennsylvania (Philadelphia, Pennsylvania) and is intended to foster dermatologic clinician educators. More information can be found on the program’s Web site.
Cutaneous Oncology Fellowship
This 1- to 2-year fellowship program focuses on diagnosis and management of melanoma and nonmelanoma skin cancers as well as cutaneous lymphomas. Fellowships in cutaneous oncology are offered at the University of California, San Francisco (San Francisco, California), Brigham and Women’s Hospital (Boston, Massachusetts), the University of Pennsylvania (Philadelphia, Pennsylvania), Case Western Reserve University (Cleveland, Ohio), the University of Pittsburgh (Pittsburgh, Pennsylvania), and Stanford University Medical Center (Stanford, California).
Dermatology/Rheumatology
The dermatology/rheumatology fellowship offered by Brigham and Women’s Hospital is a 1-year program that focuses on the management of connective-tissue diseases in a multidisciplinary fashion with rheumatology.
Advanced Medical Dermatology/Complex Medical Dermatology
Several programs offer fellowships in medical dermatology under different titles but with a similar curriculum and goal: to foster dermatologists interested in careers as academic medical dermatologists or as future clinician scientists by means of specialized training and mentorship in complex medical and dermatological issues in the outpatient and inpatient settings. The 2-year program at New York University School of Medicine (New York, New York) also gives fellows the opportunity to earn a master of science in clinical investigation degree. The University of California, San Francisco, program offers protected time for career development.
Epidemiology Training Program
Fellows and residents in the University of Pennsylvania’s dermatology training program may elect to work with the Center for Clinical Epidemiology and Biostatistics and have the opportunity to earn a graduate degree (MSCE or PhD).
Contact Dermatitis and Patch Testing Fellowship
The Dermatology Department at the Cleveland Clinic (Cleveland, Ohio) offers a 1-year contact dermatitis and patch testing fellowship that includes clinical research.
Conclusion
Both traditional and nontraditional fellowship opportunities exist after dermatology residency. This guide serves as an overview of the training programs in dermatopathology, pediatric dermatology, micrographic surgery and dermatologic oncology (procedural dermatology), and cosmetic dermatologic surgery, as well as the fellowships offered at certain institutions for those interested in more specific subspecialties or academia.
Dermatology residents, such as myself, often wonder what we will do after graduation. There are many resources for finding job opportunities, and many of us have received solicitation e-mails from various headhunters and medical groups that are looking to hire. The American Academy of Dermatology (AAD) has a resource called the AAD Career Compass (http://www.healthecareers.com/aad), which is an exhaustive database of job listings for dermatologists. However, I could not locate a definitive resource containing information that might be useful for dermatology residents who are interested in subspecializing or pursuing fellowships.
Subspecialty training is typically pursued after successful completion of a dermatology residency training program. Fellowships are traditionally offered in dermatopathology, pediatric dermatology, micrographic surgery and dermatologic oncology (procedural dermatology), and cosmetic dermatologic surgery. Fellowships also are available in other subspecialties or for those pursuing an academic career. The goal of this article is to help dermatology residents learn more about traditional and nontraditional opportunities for graduate education and certification in various dermatologic subspecialties, with links to sources for more detailed information.
Traditional Fellowship Programs by Subspecialty
Dermatopathology
One- to 2-year dermatopathology fellowship programs are certified by both the American Board of Dermatology (ABD) and the American Board of Pathology and are available to graduates of either dermatology or pathology residency programs. These programs offer combined training in either anatomic pathology (for dermatologists) or clinical dermatology (for pathologists), along with dermatopathology; the majority of time is devoted to the latter. The Accreditation Council for Graduate Medical Education and the ABD have issued specific requirements for graduate medical education and subspecialty certification in dermatopathology.1,2 Fellowship matches are institution dependent, and the application process and match generally takes place during the second year of dermatology residency for those residents who want to start a fellowship program immediately following graduation. The American Society of Dermatopathology offers a dermatopathology fellowship program finder on its Web site.
Pediatric Dermatology
Fellowships in pediatric dermatology are typically 1- to 2-year programs that focus on dermatologic diseases in the pediatric population. Applicants are matched to these programs through the San Francisco Matching Program (SF Match) and the programs are ABD accredited.3,4 (There also are a number of non–ABD-approved training opportunities available.5) On completion of the training program, fellows may qualify for subspecialty board certification in pediatric dermatology. Applications are open starting in January, and the rank order list and match occur in August of the same year. As of 2012, there were 20 participating programs with 28 available positions, while the match included 22 applicants; of these applicants, 15 matched formally into pediatric dermatology fellowships.6
Micrographic Surgery and Dermatologic Oncology (Procedural Dermatology)
There are specific requirements issued by the Accreditation Council for Graduate Medical Education for dermatologic surgery fellowships,7 which are typically 1- to 2-year programs. Many fellowship programs also are accredited by the American College of Mohs Surgery. This subspecialty is not ABD accredited; therefore, there is no certification process upon completion of a fellowship program. The American College of Mohs Surgery sponsors the match process through SF Match. Applicant registration begins in July and the match occurs in December of the same year. As of 2013, there were 47 participating programs offering 55 positions. Of 77 applicants, 49 obtained fellowship positions formally through the match.8 The American Society for Dermatologic Surgery (ASDS) Web site provides the DermSurg Fellowship Finder, which includes information about independent fellowship programs.
Cosmetic Dermatologic Surgery
The ASDS has an accreditation program for fellowships in cosmetic dermatologic surgery,9 which are generally 1-year programs. Certification in this subspecialty is not ABD accredited. Fellowship opportunities can be found using the ASDS DermSurg Fellowship Finder.
Nontraditional Fellowship Programs
The following are fellowship programs that are in nontraditional subspecialties, are only available at certain institutions, and are not accredited. This list is not exhaustive of all available programs but are those that may be of interest to dermatology residents who are drawn to a particular dermatologic subspecialty or have an interest in academic dermatology. There is no formal match process and applications vary by institution.
Clinician Educator Fellowship
The clinician educator fellowship is available at the Department of Dermatology at the University of Pennsylvania (Philadelphia, Pennsylvania) and is intended to foster dermatologic clinician educators. More information can be found on the program’s Web site.
Cutaneous Oncology Fellowship
This 1- to 2-year fellowship program focuses on diagnosis and management of melanoma and nonmelanoma skin cancers as well as cutaneous lymphomas. Fellowships in cutaneous oncology are offered at the University of California, San Francisco (San Francisco, California), Brigham and Women’s Hospital (Boston, Massachusetts), the University of Pennsylvania (Philadelphia, Pennsylvania), Case Western Reserve University (Cleveland, Ohio), the University of Pittsburgh (Pittsburgh, Pennsylvania), and Stanford University Medical Center (Stanford, California).
Dermatology/Rheumatology
The dermatology/rheumatology fellowship offered by Brigham and Women’s Hospital is a 1-year program that focuses on the management of connective-tissue diseases in a multidisciplinary fashion with rheumatology.
Advanced Medical Dermatology/Complex Medical Dermatology
Several programs offer fellowships in medical dermatology under different titles but with a similar curriculum and goal: to foster dermatologists interested in careers as academic medical dermatologists or as future clinician scientists by means of specialized training and mentorship in complex medical and dermatological issues in the outpatient and inpatient settings. The 2-year program at New York University School of Medicine (New York, New York) also gives fellows the opportunity to earn a master of science in clinical investigation degree. The University of California, San Francisco, program offers protected time for career development.
Epidemiology Training Program
Fellows and residents in the University of Pennsylvania’s dermatology training program may elect to work with the Center for Clinical Epidemiology and Biostatistics and have the opportunity to earn a graduate degree (MSCE or PhD).
Contact Dermatitis and Patch Testing Fellowship
The Dermatology Department at the Cleveland Clinic (Cleveland, Ohio) offers a 1-year contact dermatitis and patch testing fellowship that includes clinical research.
Conclusion
Both traditional and nontraditional fellowship opportunities exist after dermatology residency. This guide serves as an overview of the training programs in dermatopathology, pediatric dermatology, micrographic surgery and dermatologic oncology (procedural dermatology), and cosmetic dermatologic surgery, as well as the fellowships offered at certain institutions for those interested in more specific subspecialties or academia.
1. Accreditation Council for Graduate Medical Education. ACGME program requirements for graduate medical education in dermatopathology. http://www.acgme.org/acgmeweb/Portals/0/PFAssets/ProgramRequirements/100_dermatopathology_2016_1-YR.pdf. Revised April 2014. Effective July 1, 2015. Accessed on February 26, 2015.
2. Subspecialty certification in dermatopathology. The American Board of Dermatology, Inc Web site. http://www.abderm.org/subspecialties/derm.html. Accessed February 26, 2015.
3. American Board of Dermatology (ABD) approved pediatric dermatology fellowship programs. The Society for Pediatric Dermatology Web site. http://pedsderm.net/training/fellowships/abd-approved-pediatric-dermatology-fellowship-programs/. Updated June 9, 2014. Accessed February 26, 2015.
4. Subspecialty certification in pediatric dermatology. The American Board of Dermatology, Inc Web site. http://www.abderm.org/subspecialties/pediatric.html. Accessed February 26, 2015.
5. Non-ABD pediatric dermatology fellowship programs. The Society for Pediatric Dermatology Web site. https://pedsderm.net/training/fellowships/non-abd-pediatric-dermatology-fellowship-programs/. Accessed February 26, 2015.
6. Pediatric dermatology match report. SF Match Web site. https://www.sfmatch.org/SpecialtyInsideAll.aspx?id=16&typ=1&name=Pediatric%20Dermatology#. Accessed March 4, 2015.
7. Accreditation Council for Graduate Medical Education. ACGME program requirements for graduate medical education in procedural dermatology. https://www.acgme.org/acgmeweb/Portals/0/PFAssets/ProgramRequirements/081_procedural_derm_1-YR_07012014.pdf. Effective July 2014. Accessed February 26, 2015.
8. Statistics: micrographic surgery & dermatologic oncology fellowship. SF Match Web site. https://www.sfmatch.org/SpecialtyInsideAll.aspx?id=10&typ=1&name=Micrographic%20Surgery%20and%20Dermatologic%20Oncology#. Accessed March 4, 2015.
9. ASDS cosmetic dermatologic surgery fellowship accreditation program. American Society for Dermatologic Surgery Web site. http://www.asds.net/cosmetic-accreditation/. Accessed February 26, 2015.
1. Accreditation Council for Graduate Medical Education. ACGME program requirements for graduate medical education in dermatopathology. http://www.acgme.org/acgmeweb/Portals/0/PFAssets/ProgramRequirements/100_dermatopathology_2016_1-YR.pdf. Revised April 2014. Effective July 1, 2015. Accessed on February 26, 2015.
2. Subspecialty certification in dermatopathology. The American Board of Dermatology, Inc Web site. http://www.abderm.org/subspecialties/derm.html. Accessed February 26, 2015.
3. American Board of Dermatology (ABD) approved pediatric dermatology fellowship programs. The Society for Pediatric Dermatology Web site. http://pedsderm.net/training/fellowships/abd-approved-pediatric-dermatology-fellowship-programs/. Updated June 9, 2014. Accessed February 26, 2015.
4. Subspecialty certification in pediatric dermatology. The American Board of Dermatology, Inc Web site. http://www.abderm.org/subspecialties/pediatric.html. Accessed February 26, 2015.
5. Non-ABD pediatric dermatology fellowship programs. The Society for Pediatric Dermatology Web site. https://pedsderm.net/training/fellowships/non-abd-pediatric-dermatology-fellowship-programs/. Accessed February 26, 2015.
6. Pediatric dermatology match report. SF Match Web site. https://www.sfmatch.org/SpecialtyInsideAll.aspx?id=16&typ=1&name=Pediatric%20Dermatology#. Accessed March 4, 2015.
7. Accreditation Council for Graduate Medical Education. ACGME program requirements for graduate medical education in procedural dermatology. https://www.acgme.org/acgmeweb/Portals/0/PFAssets/ProgramRequirements/081_procedural_derm_1-YR_07012014.pdf. Effective July 2014. Accessed February 26, 2015.
8. Statistics: micrographic surgery & dermatologic oncology fellowship. SF Match Web site. https://www.sfmatch.org/SpecialtyInsideAll.aspx?id=10&typ=1&name=Micrographic%20Surgery%20and%20Dermatologic%20Oncology#. Accessed March 4, 2015.
9. ASDS cosmetic dermatologic surgery fellowship accreditation program. American Society for Dermatologic Surgery Web site. http://www.asds.net/cosmetic-accreditation/. Accessed February 26, 2015.

Trigeminal Trophic Syndrome With Histopathologic Correlation
Case Report
A 49-year-old woman presented to the dermatology department with a concern of itching distributed along the V1 branch of the trigeminal nerve on the left frontoparietal scalp following a herpes zoster (HZ) outbreak in the same dermatome 2 months prior. She initially presented to the emergency department 2 months earlier with vesicular lesions distributed along the V1 branch of the trigeminal nerve, along with facial swelling, periorbital edema, inability to open the left eye, and “excruciating” pain. Her left eye was “itchy” but no ophthalmologic pathology was noted on examination. She was diagnosed with HZ and was treated with valacyclovir and prednisone. Oxycodone-acetaminophen followed by hydromorphone was prescribed for the severe pain with limited benefit. After completing treatment with valacyclovir, oral gabapentin was added for additional pain management, with an initial dose of 100 mg 3 times daily.
At the current presentation, the patient reported profound pruritus in the left frontoparietal scalp region that was intractable and debilitating. Some improvement of the itching was achieved with scratching that resulted in deep ulcerations of the scalp with moderate associated pain. In addition to the prior HZ outbreak, her medical history was remarkable for recurrent lymphoma, uterine cancer, chronic bronchitis, depression, hypothyroidism, osteoarthritis, and primary varicella-zoster virus infection in childhood. Her current medications included oral gabapentin (600 mg 3 times daily), diphenhydramine, levothyroxine, simvastatin, and topical ointments for itching.
On dermatologic evaluation, the patient rated her pain as a 5 on a 10-point scale of intensity. Alopecia involving the left frontoparietal scalp with a 2×3-cm ulceration in a geometric pattern with surrounding erythema was noted (Figure 1A). There also was hyperpigmentation on the forehead distributed along the V1 branch of the trigeminal nerve (Figure 1B). The patient also had been seen in the pain clinic where examination revealed sensory loss to both light touch and sharp stimulus along the left V1 branch of the trigeminal nerve. Visual fields were full, ocular movements were intact, and the face was symmetric with lower cranial nerves intact.
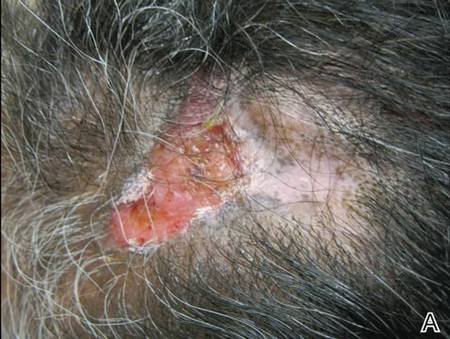 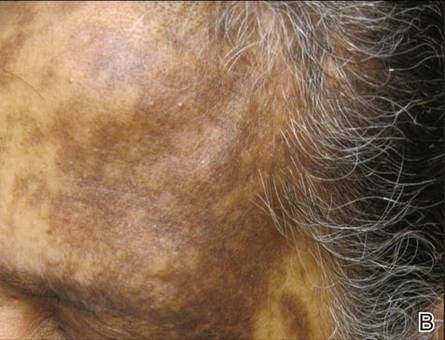 |
A diagnosis of trigeminal trophic syndrome (TTS) with chronic pain and pruritus due to a complex sensory neural disorder associated with HZ reactivation was made. Treatment included an increase in the dosage of oral gabapentin (1200 mg 3 times daily), oral oxycodone (5 mg every 4 to 6 hours as needed), and sphenopalatine ganglion block on the left side in an attempt to decrease pain and pruritus. At 6-week follow-up, the patient had no improvement in symptoms.
Three scalp punch biopsies were performed on presentation to the dermatology clinic including 2 from the affected area on the left frontoparietal scalp, and one from normal skin on the right side to assess the small nerve fibers affected. Protein gene product 9.5 (PGP 9.5) immunostaining was performed to assess epidermal nerve fiber density. The left scalp biopsies were consistent with a complete focal sensory neuropathy affecting sensory and autonomic axons (Figure 2A). The right scalp biopsy revealed well-innervated skin (Figure 2B).
 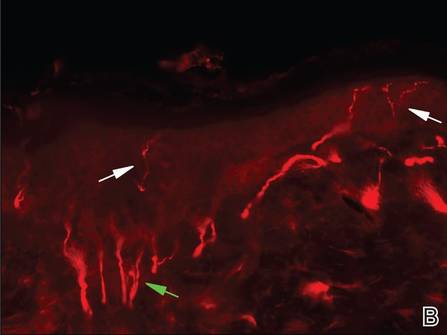 |
One year after the original HZ outbreak, the patient continued to have debilitating pruritus and pain in the affected dermatome. On physical examination at 1-year follow-up, the hyperpigmentation on the left side of the forehead showed minimal improvement. The ulcerations were healed, but excoriations were noted in the area. Having experienced some relief from titration of the dose of gabapentin 800 mg 3 times daily and doxepin 25 mg nightly at 1-year follow-up, the patient returned to work but remained highly distressed by her symptoms. Neurosurgery was consulted for possible balloon rhizotomy of the left trigeminal nerve, which she ultimately refused due to concerns about side effects.
Comment
Trophic trigeminal syndrome is characterized by unilateral ulceration of the face with anesthesia, paresthesia, and a crescent-shaped erosion or ulcer.1,2 It is one of 2 causes of self-induced facial ulcerations, the other being factitial dermatitis.1,3,4 A 2008 retrospective medical chart review and report of 14 cases helped elucidate the epidemiology of TTS.2 In this case series, the female to male ratio was 6 to 1, and the mean age of TTS onset was 45 years (age range, 6–82 years). The cause of disease in most patients was iatrogenic and the latent period to onset ranged from days to almost one decade. Most patients self-manipulated the face (n=9), and most ulcers affected the second trigeminal division. Pain intensity was severe in most (n=6), and gabapentin offered relief in only 2 cases.2
The etiologies of TTS are wide ranging, and the differential diagnosis should be contemplated when patients present with facial ulcers. Most cases are iatrogenic secondary to trigeminal rhizotomy,5 alcohol injections into the gasserian ganglion, or electrocoagulation. Also common are cases caused by ischemic damage to the trigeminal ganglion6 or Wallenberg syndrome.7 More rare etiologies include trauma,7 craniotomy,7 astrocytoma, acoustic neuroma, meningioma,8 idiopathic causes, basal cell carcinoma, infectious diseases (eg, tertiary syphilis, recurrent herpes simplex virus, leishmaniasis, cutaneous tuberculosis, leprosy, HZ),9-11 or systemic disease (eg, Wegener granulomatosis, Horton arteritis).
Trigeminal trophic syndrome is rare and there is little agreement on a treatment algorithm. As in our case, a methodical trial-and-error approach is suggested while encouraging the patient not to abandon treatment when efforts are not fruitful. The most important treatment strategy is behavioral modification; patients must become aware of the role of self-manipulation and assiduously avoid it. Using occlusive dressings at the affected site also may be helpful3,12 Transcutaneous electrical nerve stimulation may lead to improvement, but relapse is common with treatment discontinuation. Therapies directed at reducing paresthesia (eg, carbamazepine, diazepam, amitriptyline, chlorpromazine, pimozide) are sometimes successful, but relapse is common.1,3 Transplantation of in vitro–cultured epidermal cells is a new experimental treatment that offers hope for future success.13 Facial reconstruction of the affected area may help patients who can restrain themselves from self-manipulation.4
Skin biopsy findings in our case revealed an interesting aspect of the disease process of TTS. Skin biopsies are helpful in ruling out malignancy and specific stains can be used to further elucidate disease or pathologic processes occurring in the skin. In TTS, no specific changes are seen on hematoxylin and eosin staining, revealing only nonspecific inflammatory changes.1,5 Strikingly, the pathology of affected skin in patients with postherpetic neuralgia often reveals distal nociceptive axon loss,9 as was seen in the skin biopsies from our patient’s left scalp. It has been proven by many researchers in many neuropathic pain conditions that the pathological signature of chronic neuropathic pain is reduction in the density of cutaneous nociceptive innervation.9 The most common method for visualizing cutaneous neuritis is using an immunohistochemical labeling method in which antibodies are directed against PGP 9.5. A pan-axonal neurofilament marker, PGP 9.5 allows for visualization of small sensory nerve endings in the skin. As nociceptive axons degenerate in neuropathic pain conditions, it is believed that initiation of proalgesic changes within remaining peripheral nerves and the central nervous system (CNS) occur. Another interesting aspect of our case was the patient’s persistent intractable itching and chronic pain 2 months following the initial HZ outbreak. Although pain and itching can be evoked by similar stimuli and injuries, it has been shown that both have separate neuronal pathways because they produce different conscious and reflex motor actions.14 For instance, pain causes a withdrawal reflex, while itching causes mechanical stimulation of the affected area. The act of itching is thought to have evolved to protect against threats by the act of dislodging the stimulus rather than withdrawing as seen in pain.14 It has been hypothesized that postherpetic itching (chronic pruritus following an HZ outbreak) is due to spontaneous firing of denervated CNS itch neurons.9
Postherpetic neuralgia–related pain seems to be most closely correlated with degeneration of varicella-zoster virus–infected primary afferent neurons. With deceased afferent neurons sending signals to the CNS and death or dysfunction of inhibitory interneurons in the dorsal horn of the spinal cord due to peripheral nerve injury, there is increased paradoxical electrical activity in specific CNS neurons. This CNS plasticity results in neuropathic pain and other altered sensory abnormalities in patients with TTS.9
Conclusion
We present a case of TTS distributed along the V1 branch of the trigeminal nerve on the left frontoparietal scalp following an HZ outbreak in a 49-year-old woman. Skin biopsies were consistent with this diagnosis, which revealed no neuronal innervation of the affected scalp despite intractable itching and chronic pain. Further research of TTS and postherpetic neuralgia is necessary to find appropriate treatment for patients with these conditions.
1. Kautz O, Bruckner-Tuderman L, Müller ML, et al. Trigeminal trophic syndrome with extensive ulceration following herpes zoster. Eur J Dermatol. 2009;19:61-63.
2. Garza I. The trigeminal trophic syndrome: an unusual cause of face pain, dysaesthesias, anaesthesia and skin/soft tissue lesions. Cephalalgia. 2008;28:980-985.
3. Farahani RM, Marsee DK, Baden LR, et al. Trigeminal trophic syndrome with features of oral CMV disease. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2008;106:15-18.
4. Tollefson TT, Kriet JD, Wang TD, et al. Self-induced nasal ulceration. Arch Facial Plast Surg. 2004;6:162-166.
5. Monrad SU, Terrell JE, Aronoff DM. The trigeminal trophic syndrome: an unusual cause of nasal ulceration. J Am Acad Dermatol. 2004;50:949-952.
6. Elloumi-Jellouli A, Ben Ammar S, Fenniche S, et al. Trigeminal trophic syndrome: a report of two cases with review of literature. Dermatol Online J. 2003;9:26.
7. Sadeghi P, Papay FA, Vidimos AT. Trigeminal trophic syndrome—report of four cases and review of the literature. Dermatol Surg. 2004;30:807-812.
8. Luksi´c I, Luksi´c I, Sestan-Crnek S, et al. Trigeminal trophic syndrome of all three nerve branches: an underrecognized complication after brain surgery. J Neurosurg. 2008;108:170-173.
9. Oaklander AL. Mechanisms of pain and itch caused by herpes zoster (shingles). J Pain. 2008;9(1 suppl 1):S10-S18.
10. Gawande A. The itch. The New Yorker. June 2008:58-67.
11. Oaklander AL, Cohen SP, Raju SV. Intractable postherpetic itch and cutaneous deafferentation after facial shingles. Pain. 2002;96:9-12.
12. Preston PW, Orpin SD, Tucker WF, et al. Successful use of a thermoplastic dressing in two cases of the trigeminal trophic syndrome. Clin Exp Dermatol. 2006;31:525-527.
13. Schwerdtner O, Damaskos T, Kage A, et al. Autologous epidermal cells can induce wound closure of neurotrophic ulceration caused by trigeminal trophic syndrome. Int J Oral Maxillofac Surg. 2005;34:443-445.
14. Oaklander AL, Siegel SM. Cutaneous innervation: form and function. J Am Acad Dermatol. 2005;53:1027-1037.
Case Report
A 49-year-old woman presented to the dermatology department with a concern of itching distributed along the V1 branch of the trigeminal nerve on the left frontoparietal scalp following a herpes zoster (HZ) outbreak in the same dermatome 2 months prior. She initially presented to the emergency department 2 months earlier with vesicular lesions distributed along the V1 branch of the trigeminal nerve, along with facial swelling, periorbital edema, inability to open the left eye, and “excruciating” pain. Her left eye was “itchy” but no ophthalmologic pathology was noted on examination. She was diagnosed with HZ and was treated with valacyclovir and prednisone. Oxycodone-acetaminophen followed by hydromorphone was prescribed for the severe pain with limited benefit. After completing treatment with valacyclovir, oral gabapentin was added for additional pain management, with an initial dose of 100 mg 3 times daily.
At the current presentation, the patient reported profound pruritus in the left frontoparietal scalp region that was intractable and debilitating. Some improvement of the itching was achieved with scratching that resulted in deep ulcerations of the scalp with moderate associated pain. In addition to the prior HZ outbreak, her medical history was remarkable for recurrent lymphoma, uterine cancer, chronic bronchitis, depression, hypothyroidism, osteoarthritis, and primary varicella-zoster virus infection in childhood. Her current medications included oral gabapentin (600 mg 3 times daily), diphenhydramine, levothyroxine, simvastatin, and topical ointments for itching.
On dermatologic evaluation, the patient rated her pain as a 5 on a 10-point scale of intensity. Alopecia involving the left frontoparietal scalp with a 2×3-cm ulceration in a geometric pattern with surrounding erythema was noted (Figure 1A). There also was hyperpigmentation on the forehead distributed along the V1 branch of the trigeminal nerve (Figure 1B). The patient also had been seen in the pain clinic where examination revealed sensory loss to both light touch and sharp stimulus along the left V1 branch of the trigeminal nerve. Visual fields were full, ocular movements were intact, and the face was symmetric with lower cranial nerves intact.
|
A diagnosis of trigeminal trophic syndrome (TTS) with chronic pain and pruritus due to a complex sensory neural disorder associated with HZ reactivation was made. Treatment included an increase in the dosage of oral gabapentin (1200 mg 3 times daily), oral oxycodone (5 mg every 4 to 6 hours as needed), and sphenopalatine ganglion block on the left side in an attempt to decrease pain and pruritus. At 6-week follow-up, the patient had no improvement in symptoms.
Three scalp punch biopsies were performed on presentation to the dermatology clinic including 2 from the affected area on the left frontoparietal scalp, and one from normal skin on the right side to assess the small nerve fibers affected. Protein gene product 9.5 (PGP 9.5) immunostaining was performed to assess epidermal nerve fiber density. The left scalp biopsies were consistent with a complete focal sensory neuropathy affecting sensory and autonomic axons (Figure 2A). The right scalp biopsy revealed well-innervated skin (Figure 2B).
|
One year after the original HZ outbreak, the patient continued to have debilitating pruritus and pain in the affected dermatome. On physical examination at 1-year follow-up, the hyperpigmentation on the left side of the forehead showed minimal improvement. The ulcerations were healed, but excoriations were noted in the area. Having experienced some relief from titration of the dose of gabapentin 800 mg 3 times daily and doxepin 25 mg nightly at 1-year follow-up, the patient returned to work but remained highly distressed by her symptoms. Neurosurgery was consulted for possible balloon rhizotomy of the left trigeminal nerve, which she ultimately refused due to concerns about side effects.
Comment
Trophic trigeminal syndrome is characterized by unilateral ulceration of the face with anesthesia, paresthesia, and a crescent-shaped erosion or ulcer.1,2 It is one of 2 causes of self-induced facial ulcerations, the other being factitial dermatitis.1,3,4 A 2008 retrospective medical chart review and report of 14 cases helped elucidate the epidemiology of TTS.2 In this case series, the female to male ratio was 6 to 1, and the mean age of TTS onset was 45 years (age range, 6–82 years). The cause of disease in most patients was iatrogenic and the latent period to onset ranged from days to almost one decade. Most patients self-manipulated the face (n=9), and most ulcers affected the second trigeminal division. Pain intensity was severe in most (n=6), and gabapentin offered relief in only 2 cases.2
The etiologies of TTS are wide ranging, and the differential diagnosis should be contemplated when patients present with facial ulcers. Most cases are iatrogenic secondary to trigeminal rhizotomy,5 alcohol injections into the gasserian ganglion, or electrocoagulation. Also common are cases caused by ischemic damage to the trigeminal ganglion6 or Wallenberg syndrome.7 More rare etiologies include trauma,7 craniotomy,7 astrocytoma, acoustic neuroma, meningioma,8 idiopathic causes, basal cell carcinoma, infectious diseases (eg, tertiary syphilis, recurrent herpes simplex virus, leishmaniasis, cutaneous tuberculosis, leprosy, HZ),9-11 or systemic disease (eg, Wegener granulomatosis, Horton arteritis).
Trigeminal trophic syndrome is rare and there is little agreement on a treatment algorithm. As in our case, a methodical trial-and-error approach is suggested while encouraging the patient not to abandon treatment when efforts are not fruitful. The most important treatment strategy is behavioral modification; patients must become aware of the role of self-manipulation and assiduously avoid it. Using occlusive dressings at the affected site also may be helpful3,12 Transcutaneous electrical nerve stimulation may lead to improvement, but relapse is common with treatment discontinuation. Therapies directed at reducing paresthesia (eg, carbamazepine, diazepam, amitriptyline, chlorpromazine, pimozide) are sometimes successful, but relapse is common.1,3 Transplantation of in vitro–cultured epidermal cells is a new experimental treatment that offers hope for future success.13 Facial reconstruction of the affected area may help patients who can restrain themselves from self-manipulation.4
Skin biopsy findings in our case revealed an interesting aspect of the disease process of TTS. Skin biopsies are helpful in ruling out malignancy and specific stains can be used to further elucidate disease or pathologic processes occurring in the skin. In TTS, no specific changes are seen on hematoxylin and eosin staining, revealing only nonspecific inflammatory changes.1,5 Strikingly, the pathology of affected skin in patients with postherpetic neuralgia often reveals distal nociceptive axon loss,9 as was seen in the skin biopsies from our patient’s left scalp. It has been proven by many researchers in many neuropathic pain conditions that the pathological signature of chronic neuropathic pain is reduction in the density of cutaneous nociceptive innervation.9 The most common method for visualizing cutaneous neuritis is using an immunohistochemical labeling method in which antibodies are directed against PGP 9.5. A pan-axonal neurofilament marker, PGP 9.5 allows for visualization of small sensory nerve endings in the skin. As nociceptive axons degenerate in neuropathic pain conditions, it is believed that initiation of proalgesic changes within remaining peripheral nerves and the central nervous system (CNS) occur. Another interesting aspect of our case was the patient’s persistent intractable itching and chronic pain 2 months following the initial HZ outbreak. Although pain and itching can be evoked by similar stimuli and injuries, it has been shown that both have separate neuronal pathways because they produce different conscious and reflex motor actions.14 For instance, pain causes a withdrawal reflex, while itching causes mechanical stimulation of the affected area. The act of itching is thought to have evolved to protect against threats by the act of dislodging the stimulus rather than withdrawing as seen in pain.14 It has been hypothesized that postherpetic itching (chronic pruritus following an HZ outbreak) is due to spontaneous firing of denervated CNS itch neurons.9
Postherpetic neuralgia–related pain seems to be most closely correlated with degeneration of varicella-zoster virus–infected primary afferent neurons. With deceased afferent neurons sending signals to the CNS and death or dysfunction of inhibitory interneurons in the dorsal horn of the spinal cord due to peripheral nerve injury, there is increased paradoxical electrical activity in specific CNS neurons. This CNS plasticity results in neuropathic pain and other altered sensory abnormalities in patients with TTS.9
Conclusion
We present a case of TTS distributed along the V1 branch of the trigeminal nerve on the left frontoparietal scalp following an HZ outbreak in a 49-year-old woman. Skin biopsies were consistent with this diagnosis, which revealed no neuronal innervation of the affected scalp despite intractable itching and chronic pain. Further research of TTS and postherpetic neuralgia is necessary to find appropriate treatment for patients with these conditions.
Case Report
A 49-year-old woman presented to the dermatology department with a concern of itching distributed along the V1 branch of the trigeminal nerve on the left frontoparietal scalp following a herpes zoster (HZ) outbreak in the same dermatome 2 months prior. She initially presented to the emergency department 2 months earlier with vesicular lesions distributed along the V1 branch of the trigeminal nerve, along with facial swelling, periorbital edema, inability to open the left eye, and “excruciating” pain. Her left eye was “itchy” but no ophthalmologic pathology was noted on examination. She was diagnosed with HZ and was treated with valacyclovir and prednisone. Oxycodone-acetaminophen followed by hydromorphone was prescribed for the severe pain with limited benefit. After completing treatment with valacyclovir, oral gabapentin was added for additional pain management, with an initial dose of 100 mg 3 times daily.
At the current presentation, the patient reported profound pruritus in the left frontoparietal scalp region that was intractable and debilitating. Some improvement of the itching was achieved with scratching that resulted in deep ulcerations of the scalp with moderate associated pain. In addition to the prior HZ outbreak, her medical history was remarkable for recurrent lymphoma, uterine cancer, chronic bronchitis, depression, hypothyroidism, osteoarthritis, and primary varicella-zoster virus infection in childhood. Her current medications included oral gabapentin (600 mg 3 times daily), diphenhydramine, levothyroxine, simvastatin, and topical ointments for itching.
On dermatologic evaluation, the patient rated her pain as a 5 on a 10-point scale of intensity. Alopecia involving the left frontoparietal scalp with a 2×3-cm ulceration in a geometric pattern with surrounding erythema was noted (Figure 1A). There also was hyperpigmentation on the forehead distributed along the V1 branch of the trigeminal nerve (Figure 1B). The patient also had been seen in the pain clinic where examination revealed sensory loss to both light touch and sharp stimulus along the left V1 branch of the trigeminal nerve. Visual fields were full, ocular movements were intact, and the face was symmetric with lower cranial nerves intact.
|
A diagnosis of trigeminal trophic syndrome (TTS) with chronic pain and pruritus due to a complex sensory neural disorder associated with HZ reactivation was made. Treatment included an increase in the dosage of oral gabapentin (1200 mg 3 times daily), oral oxycodone (5 mg every 4 to 6 hours as needed), and sphenopalatine ganglion block on the left side in an attempt to decrease pain and pruritus. At 6-week follow-up, the patient had no improvement in symptoms.
Three scalp punch biopsies were performed on presentation to the dermatology clinic including 2 from the affected area on the left frontoparietal scalp, and one from normal skin on the right side to assess the small nerve fibers affected. Protein gene product 9.5 (PGP 9.5) immunostaining was performed to assess epidermal nerve fiber density. The left scalp biopsies were consistent with a complete focal sensory neuropathy affecting sensory and autonomic axons (Figure 2A). The right scalp biopsy revealed well-innervated skin (Figure 2B).
|
One year after the original HZ outbreak, the patient continued to have debilitating pruritus and pain in the affected dermatome. On physical examination at 1-year follow-up, the hyperpigmentation on the left side of the forehead showed minimal improvement. The ulcerations were healed, but excoriations were noted in the area. Having experienced some relief from titration of the dose of gabapentin 800 mg 3 times daily and doxepin 25 mg nightly at 1-year follow-up, the patient returned to work but remained highly distressed by her symptoms. Neurosurgery was consulted for possible balloon rhizotomy of the left trigeminal nerve, which she ultimately refused due to concerns about side effects.
Comment
Trophic trigeminal syndrome is characterized by unilateral ulceration of the face with anesthesia, paresthesia, and a crescent-shaped erosion or ulcer.1,2 It is one of 2 causes of self-induced facial ulcerations, the other being factitial dermatitis.1,3,4 A 2008 retrospective medical chart review and report of 14 cases helped elucidate the epidemiology of TTS.2 In this case series, the female to male ratio was 6 to 1, and the mean age of TTS onset was 45 years (age range, 6–82 years). The cause of disease in most patients was iatrogenic and the latent period to onset ranged from days to almost one decade. Most patients self-manipulated the face (n=9), and most ulcers affected the second trigeminal division. Pain intensity was severe in most (n=6), and gabapentin offered relief in only 2 cases.2
The etiologies of TTS are wide ranging, and the differential diagnosis should be contemplated when patients present with facial ulcers. Most cases are iatrogenic secondary to trigeminal rhizotomy,5 alcohol injections into the gasserian ganglion, or electrocoagulation. Also common are cases caused by ischemic damage to the trigeminal ganglion6 or Wallenberg syndrome.7 More rare etiologies include trauma,7 craniotomy,7 astrocytoma, acoustic neuroma, meningioma,8 idiopathic causes, basal cell carcinoma, infectious diseases (eg, tertiary syphilis, recurrent herpes simplex virus, leishmaniasis, cutaneous tuberculosis, leprosy, HZ),9-11 or systemic disease (eg, Wegener granulomatosis, Horton arteritis).
Trigeminal trophic syndrome is rare and there is little agreement on a treatment algorithm. As in our case, a methodical trial-and-error approach is suggested while encouraging the patient not to abandon treatment when efforts are not fruitful. The most important treatment strategy is behavioral modification; patients must become aware of the role of self-manipulation and assiduously avoid it. Using occlusive dressings at the affected site also may be helpful3,12 Transcutaneous electrical nerve stimulation may lead to improvement, but relapse is common with treatment discontinuation. Therapies directed at reducing paresthesia (eg, carbamazepine, diazepam, amitriptyline, chlorpromazine, pimozide) are sometimes successful, but relapse is common.1,3 Transplantation of in vitro–cultured epidermal cells is a new experimental treatment that offers hope for future success.13 Facial reconstruction of the affected area may help patients who can restrain themselves from self-manipulation.4
Skin biopsy findings in our case revealed an interesting aspect of the disease process of TTS. Skin biopsies are helpful in ruling out malignancy and specific stains can be used to further elucidate disease or pathologic processes occurring in the skin. In TTS, no specific changes are seen on hematoxylin and eosin staining, revealing only nonspecific inflammatory changes.1,5 Strikingly, the pathology of affected skin in patients with postherpetic neuralgia often reveals distal nociceptive axon loss,9 as was seen in the skin biopsies from our patient’s left scalp. It has been proven by many researchers in many neuropathic pain conditions that the pathological signature of chronic neuropathic pain is reduction in the density of cutaneous nociceptive innervation.9 The most common method for visualizing cutaneous neuritis is using an immunohistochemical labeling method in which antibodies are directed against PGP 9.5. A pan-axonal neurofilament marker, PGP 9.5 allows for visualization of small sensory nerve endings in the skin. As nociceptive axons degenerate in neuropathic pain conditions, it is believed that initiation of proalgesic changes within remaining peripheral nerves and the central nervous system (CNS) occur. Another interesting aspect of our case was the patient’s persistent intractable itching and chronic pain 2 months following the initial HZ outbreak. Although pain and itching can be evoked by similar stimuli and injuries, it has been shown that both have separate neuronal pathways because they produce different conscious and reflex motor actions.14 For instance, pain causes a withdrawal reflex, while itching causes mechanical stimulation of the affected area. The act of itching is thought to have evolved to protect against threats by the act of dislodging the stimulus rather than withdrawing as seen in pain.14 It has been hypothesized that postherpetic itching (chronic pruritus following an HZ outbreak) is due to spontaneous firing of denervated CNS itch neurons.9
Postherpetic neuralgia–related pain seems to be most closely correlated with degeneration of varicella-zoster virus–infected primary afferent neurons. With deceased afferent neurons sending signals to the CNS and death or dysfunction of inhibitory interneurons in the dorsal horn of the spinal cord due to peripheral nerve injury, there is increased paradoxical electrical activity in specific CNS neurons. This CNS plasticity results in neuropathic pain and other altered sensory abnormalities in patients with TTS.9
Conclusion
We present a case of TTS distributed along the V1 branch of the trigeminal nerve on the left frontoparietal scalp following an HZ outbreak in a 49-year-old woman. Skin biopsies were consistent with this diagnosis, which revealed no neuronal innervation of the affected scalp despite intractable itching and chronic pain. Further research of TTS and postherpetic neuralgia is necessary to find appropriate treatment for patients with these conditions.
1. Kautz O, Bruckner-Tuderman L, Müller ML, et al. Trigeminal trophic syndrome with extensive ulceration following herpes zoster. Eur J Dermatol. 2009;19:61-63.
2. Garza I. The trigeminal trophic syndrome: an unusual cause of face pain, dysaesthesias, anaesthesia and skin/soft tissue lesions. Cephalalgia. 2008;28:980-985.
3. Farahani RM, Marsee DK, Baden LR, et al. Trigeminal trophic syndrome with features of oral CMV disease. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2008;106:15-18.
4. Tollefson TT, Kriet JD, Wang TD, et al. Self-induced nasal ulceration. Arch Facial Plast Surg. 2004;6:162-166.
5. Monrad SU, Terrell JE, Aronoff DM. The trigeminal trophic syndrome: an unusual cause of nasal ulceration. J Am Acad Dermatol. 2004;50:949-952.
6. Elloumi-Jellouli A, Ben Ammar S, Fenniche S, et al. Trigeminal trophic syndrome: a report of two cases with review of literature. Dermatol Online J. 2003;9:26.
7. Sadeghi P, Papay FA, Vidimos AT. Trigeminal trophic syndrome—report of four cases and review of the literature. Dermatol Surg. 2004;30:807-812.
8. Luksi´c I, Luksi´c I, Sestan-Crnek S, et al. Trigeminal trophic syndrome of all three nerve branches: an underrecognized complication after brain surgery. J Neurosurg. 2008;108:170-173.
9. Oaklander AL. Mechanisms of pain and itch caused by herpes zoster (shingles). J Pain. 2008;9(1 suppl 1):S10-S18.
10. Gawande A. The itch. The New Yorker. June 2008:58-67.
11. Oaklander AL, Cohen SP, Raju SV. Intractable postherpetic itch and cutaneous deafferentation after facial shingles. Pain. 2002;96:9-12.
12. Preston PW, Orpin SD, Tucker WF, et al. Successful use of a thermoplastic dressing in two cases of the trigeminal trophic syndrome. Clin Exp Dermatol. 2006;31:525-527.
13. Schwerdtner O, Damaskos T, Kage A, et al. Autologous epidermal cells can induce wound closure of neurotrophic ulceration caused by trigeminal trophic syndrome. Int J Oral Maxillofac Surg. 2005;34:443-445.
14. Oaklander AL, Siegel SM. Cutaneous innervation: form and function. J Am Acad Dermatol. 2005;53:1027-1037.
1. Kautz O, Bruckner-Tuderman L, Müller ML, et al. Trigeminal trophic syndrome with extensive ulceration following herpes zoster. Eur J Dermatol. 2009;19:61-63.
2. Garza I. The trigeminal trophic syndrome: an unusual cause of face pain, dysaesthesias, anaesthesia and skin/soft tissue lesions. Cephalalgia. 2008;28:980-985.
3. Farahani RM, Marsee DK, Baden LR, et al. Trigeminal trophic syndrome with features of oral CMV disease. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2008;106:15-18.
4. Tollefson TT, Kriet JD, Wang TD, et al. Self-induced nasal ulceration. Arch Facial Plast Surg. 2004;6:162-166.
5. Monrad SU, Terrell JE, Aronoff DM. The trigeminal trophic syndrome: an unusual cause of nasal ulceration. J Am Acad Dermatol. 2004;50:949-952.
6. Elloumi-Jellouli A, Ben Ammar S, Fenniche S, et al. Trigeminal trophic syndrome: a report of two cases with review of literature. Dermatol Online J. 2003;9:26.
7. Sadeghi P, Papay FA, Vidimos AT. Trigeminal trophic syndrome—report of four cases and review of the literature. Dermatol Surg. 2004;30:807-812.
8. Luksi´c I, Luksi´c I, Sestan-Crnek S, et al. Trigeminal trophic syndrome of all three nerve branches: an underrecognized complication after brain surgery. J Neurosurg. 2008;108:170-173.
9. Oaklander AL. Mechanisms of pain and itch caused by herpes zoster (shingles). J Pain. 2008;9(1 suppl 1):S10-S18.
10. Gawande A. The itch. The New Yorker. June 2008:58-67.
11. Oaklander AL, Cohen SP, Raju SV. Intractable postherpetic itch and cutaneous deafferentation after facial shingles. Pain. 2002;96:9-12.
12. Preston PW, Orpin SD, Tucker WF, et al. Successful use of a thermoplastic dressing in two cases of the trigeminal trophic syndrome. Clin Exp Dermatol. 2006;31:525-527.
13. Schwerdtner O, Damaskos T, Kage A, et al. Autologous epidermal cells can induce wound closure of neurotrophic ulceration caused by trigeminal trophic syndrome. Int J Oral Maxillofac Surg. 2005;34:443-445.
14. Oaklander AL, Siegel SM. Cutaneous innervation: form and function. J Am Acad Dermatol. 2005;53:1027-1037.
Practice Points
- Clinicians should remember to include trigeminal trophic syndrome in the differential diagnosis of patients with facial ulcers.
- Trigeminal trophic syndrome is a rare syndrome with a variety of treatment options, though no gold standard for treatment exists.
Glatiramer Acetate–Induced Lobular Panniculitis and Skin Necrosis
Glatiramer acetate (GA), a synthetic polypeptide that is injected subcutaneously, has proven effective in the treatment of relapsing-remitting multiple sclerosis (RRMS) and is now considered a first-line agent in the treatment of this condition. Adverse effects associated with GA primarily include local injection-site reactions (LISRs)(eg, erythema, pruritus, burning, pain, inflammation). Transient acute systemic reactions such as flushing and dyspnea also are commonly reported. Lipoatrophy at the injection site frequently has been reported in the literature as a cutaneous adverse effect of GA, but lobular panniculitis and necrosis at the site of injection rarely have been noted.
We report the case of a 36-year-old woman who experienced a severe adverse reaction to a single injection of GA after nearly 1 year of daily use to control symptoms of RRMS. Review of the current literature revealed few reports of the severe reaction of panniculitis and necrosis occurring at the injection site of GA.
Case Report
A 36-year-old woman was referred by her neurologist to the emergency department of our institution’s allergy and immunology clinic for treatment of an allergic reaction to a 20-mg GA injection, which she had been receiving daily for nearly 1 year as therapy for RRMS. A nodule immediately formed at the injection site and eventually became ulcerated. The patient also reported intense chest tightness, shortness of breath, and flushing following the injection. Physical examination revealed a large 8- to 9-cm erythematous area at the injection site on the left buttock. Necrosis and eschar formation also were evident (Figure 1).
Figure 1. Panniculitis with central ulceration and necrosis at the site of a glatiramer acetate injection on the left buttock (A). Closer view of an irregularly shaped necrotic lesion with surrounding erythema (B). |
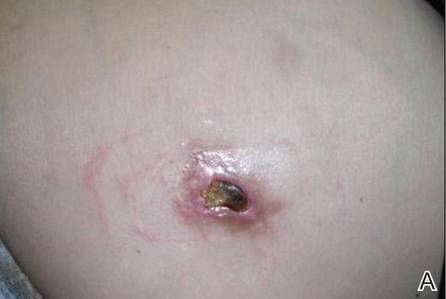
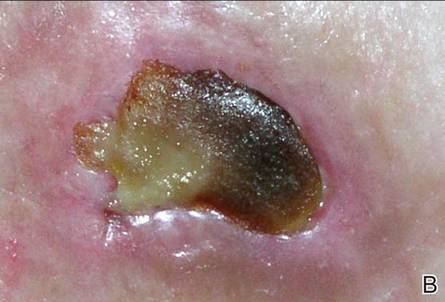
A punch biopsy from the edge of the lesion revealed predominantly lobular panniculitis (Figure 2A) with fat necrosis and numerous foamy macrophages (Figures 2B and 2C). Scattered lymphocytes also were present but no neutrophils or eosinophils were noted (Figure 2B). Interlobular septa were widened secondary to fibrosis (Figure 2A). No lymphoid follicles were identified. A subcutaneous artery was sampled but was negative for vasculitis (Figure 2D).
|
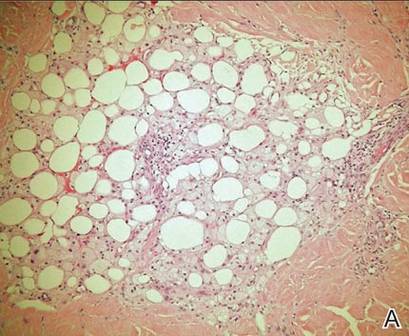
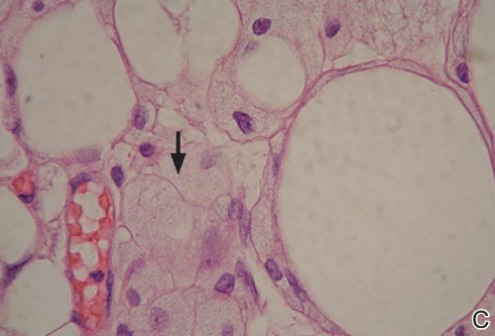

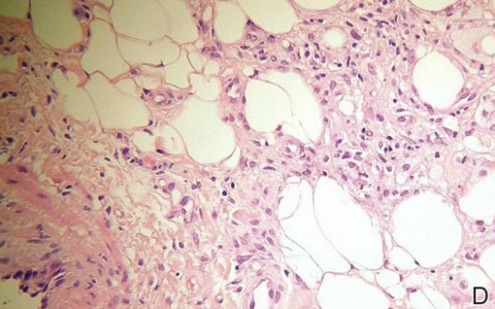
The necrotic lesion on the left buttock was present for more than 2 months before complete healing occurred. The patient had a history of intolerance or unresponsiveness to all prior medications for RRMS. Several years prior she responded well to treatment with GA for a few months and had been responding well to the injections over the last year. Incremental challenge testing with GA for desensitization was offered to the patient, but she declined treatment out of fear of a recurrent episode, particularly the severe systemic symptoms she had experienced. Unfortunately, she was lost to follow-up.
Comment
Glatiramer acetate, formerly known as copolymer-1, is a first-line treatment of patients with RRMS.1 Daily administration of subcutaneous injections of GA (20 mg/mL) has proven effective in relapse rate reduction and reduced morbidity in patients with RRMS.2 Long-term studies support a relapse rate reduction of more than 50% in patients using GA.3 The most common adverse effects are LISRs.2 Systemic reactions following GA injection also are common. A much less common reaction is panniculitis followed by lipoatrophy and/or skin necrosis. Only a few instances of panniculitis-associated necrosis have been reported.
The occurrence of LISRs was reported in 20% to 90% of patients using GA to control RRMS.2,4 Local injection-site reactions typically resolve within hours to days and have been reported to decrease in frequency over time.5 Acute systemic reactions (eg, anxiety, flushing, palpitations, dyspnea) to GA injection are described in approximately 15% of patients.6 Systemic reactions usually resolve in 5 to 15 minutes but can last for more than 1 hour.5 These reactions are mostly benign and generally are not considered to be allergic or anaphylactic in nature. True systemic anaphylaxis associated with administration of GA is extremely rare.7
Lipoatrophy, or localized loss of subcutaneous adipose tissue without evidence of inflammation, has been reported fairly frequently in association with GA (up to 45% of patients receiving GA injections).2,6,8,9 Lipoatrophy also has been seen following subcutaneous injection of many other drugs, including steroids and insulin. Unlike LISRs, the likelihood of developing lipoatrophy at the injection site increases with longer durations of GA injections.5 Lipoatrophy also develops following panniculitis at the site of GA injection.
Based on a search of the MeSH (Medical Subject Headings) database using the terms panniculitis and glatiramer acetate, there only are 10 reported cases of panniculitis as an adverse effect of GA injections.2,6,10 Lesions were described as either subcutaneous erythematous nodules or atrophic areas that demonstrated panniculitis on histologic examination. Injections preceding the development of panniculitis often were described as remarkably painful.4 Residual lipoatrophy and/or hyperpigmentation at the site of panniculitis development is common.2 It has been suggested that GA-induced panniculitis may be an early underlying mechanism for the development of lipoatrophy, and thus may be more common than originally suspected.10
Histopathologic examination of GA-induced panniculitis typically reveals a localized, mostly lobular, panniculitis with lipophagic granulomas, lymphocytes, and thickened septa. The lipophagic granulomas (a characteristic finding in panniculitis) form from local macrophages that engulf the lipids released from necrotic adipocytes.11 A large, pale, granular or vacuolated cytoplasm typically can be observed on microscopic examination of the macrophages (Figure 2C). Connective tissue septa typically are widened with cell infiltrates, usually lymphocytes. Other cell types, including macrophages, eosinophils, and neutrophils, also have been identified in both the septa and fat lobules. These histologic elements may change and evolve over time.
Necrosis in association with panniculitis, as seen in our patient, rarely has been reported.4,12 All of the necrotic reactions described occurred after at least 1 year of GA therapy and took several weeks to resolve.4,12 When presented with the development of skin necrosis at the site of GA injection, it is essential to distinguish between an adverse effect associated with the drug itself and Nicolau syndrome (embolia cutis medicamentosa).13 Necrosis at multiple injection sites or recurrence with later injections supports a GA-specific effect.12
Nicolau syndrome is a well-known traumatic reaction that leads to microembolization and resultant vasospasm as well as necrosis throughout the skin and possibly the underlying muscular layer.14 Although more commonly associated with intramuscular injections, Nicolau syndrome has been described with subcutaneous injections of GA in a few rare instances.13,15 Because of the associated severe systemic reaction as well as the histologic examination (Figure 2D), we believe the skin necrosis seen in our patient was from a reaction to GA rather than Nicolau syndrome. Our patient was not interested in restarting GA therapy; therefore, it is unknown if this reaction would have recurred, but we suspect high probability of recurrence without desensitization attempts.
Preventative measures can be taken to decrease the risk for LISRs, and patients should be educated on these techniques. Applying ice to the injection site for at least 30 seconds before cleaning the skin for injection may reduce local adverse effects.4 Proper instruction on injection techniques should be provided by a knowledgeable health care professional and topical anesthetics and/or steroids may be offered to reduce pain associated with injection. There have been no proven measures for prevention of lipoatrophy, panniculitis, or necrosis, and these adverse effects are not thought to be attributed to improper injection techniques.14 Rotation of injection sites is the only suggested means of decreasing the potential risk for more severely and permanently disfiguring local reactions.
If panniculitis following GA injection is suspected, a large biopsy that encompasses the entire subcutaneous fat layer is necessary for proper dermatopathologic classification.11 Glatiramer acetate injections should be stopped immediately. These reactions disappear when the injections are stopped but recur when restarting treatment.2 The efficacy of GA in the treatment of RRMS has led to the possible use of this drug in the treatment of other autoimmune diseases.16 Thus, it is important for clinicians to be aware of all adverse effects of subcutaneous injections of GA, including the rare occurrence of panniculitis and necrosis, and when discontinuation of therapy is indicated.
Conclusion
Daily subcutaneous injection of GA for the treatment of RRMS can result in the rare but characteristic development of localized panniculitis and necrosis. Glatiramer acetate is a common and highly effective therapy used for the treatment of RRMS. Common adverse effects include LISRs and transient acute systemic reactions. Less commonly observed but characteristic of GA injections is localized lipoatrophy and mostly lobular panniculitis. Necrosis rarely can develop in association with these cutaneous reactions. It is essential to differentiate between necrosis secondary to Nicolau syndrome and skin necrosis as a unique reaction to GA; the latter is an indication for discontinuation of GA injections. Dermatologists should be made aware of adverse cutaneous reactions seen with GA therapy, especially with the potential for expansion of the use of GA to treat other autoimmune processes. Further research is needed regarding the histopathologic evolution and mechanisms behind the development of lipoatrophy, panniculitis, and necrosis at the site of GA injection.
1. Anderson G, Meyer D, Herrman CE, et al. Tolerability and safety of novel half milliliter formulation of glatiramer acetate for subcutaneous injection: an open-label, multicenter, randomized comparative study. J Neurol. 2010;257:1917-1923.
2. Soares Almeida LM, Requena L, Kutzner H, et al. Localized panniculitis secondary to subcutaneous glatiramer acetate injections for the treatment of multiple sclerosis: a clinicopathologic and immunohistochemical study. J Am Acad Dermatol. 2006;55:968-974.
3. Ford CC, Johnson KP, Lisak RP, et al. A prospective open-label study of glatiramer acetate: over a decade of continuous use in multiple sclerosis patients. Mult Scler. 2006;12:309-320.
4. Frohman EM, Brannon K, Alexander S, et al. Disease modifying agent related skin reactions in multiple sclerosis: prevention, assessment, and management. Mult Scler. 2004;10:302-307.
5. Ziemssen T, Neuhaus O, Hohlfeld R. Risk-benefit assessment of glatiramer acetate in multiple sclerosis. Drug Saf. 2001;24:979-990.
6. Ball NJ, Cowan BJ, Moore GR, et al. Lobular panniculitis at the site of glatiramer acetate injections for the treatment of relapsing-remitting multiple sclerosis. a report of two cases. J Cutan Pathol. 2008;35:407-410.
7. Rauschka H, Farina C, Sator P, et al. Severe anaphylactic reaction to glatiramer acetate with specific IgE. Neurology. 2005;64:1481-1482.
8. Hwang L, Orengo I. Lipoatrophy associated with glatiramer acetate injections for the treatment of multiple sclerosis. Cutis. 2001;68:287-288.
9. Edgar CM, Brunet DG, Fenton P, et al. Lipoatrophy in patients with multiple sclerosis on glatiramer acetate. Can J Neurol Sci. 2004;31:58-63.
10. Soós N, Shakery K, Mrowietz U. Localized panniculitis and subsequent lipoatrophy with subcutaneous glatiramer acetate (Copaxone) injection for the treatment of multiple sclerosis. Am J Clin Dermatol. 2004;5:357-359.
11. Segura S, Requena L. Anatomy and histology of normal subcutaneous fat, necrosis of adipocytes, and classification of the panniculitides. Dermatol Clin. 2008;26:419-424, v.
12. Bosca I, Bosca M, Belenguer A, et al. Necrotising cutaneous lesions as a side effect of glatiramer acetate. J Neurol. 2006;253:1370-1371.
13. Feldmann R, Schierl M, Rauschka H, et al. Necrotizing skin lesions with involvement of muscle tissue after subcutaneous injection of glatiramer acetate. Eur J Dermatol. 2009;19:385.
14. Kluger N, Thouvenot E, Camu W, et al. Cutaneous adverse events related to glatiramer acetate injection (copolymer-1, Copaxone). J Eur Acad Dermatol Venereol. 2009;23:1332-1333.
15. Harde V, Schwarz T. Embolia cutis medicamentosa following subcutaneous injection of glatiramer acetate [in English, German]. J Dtsch Dermatol Ges. 2007;5:1122-1123.
16. Racke MK, Lovett-Racke AE. Glatiramer acetate treatment of multiple sclerosis: an immunological perspective. J Immunol. 2011;186:1887-1890.
Glatiramer acetate (GA), a synthetic polypeptide that is injected subcutaneously, has proven effective in the treatment of relapsing-remitting multiple sclerosis (RRMS) and is now considered a first-line agent in the treatment of this condition. Adverse effects associated with GA primarily include local injection-site reactions (LISRs)(eg, erythema, pruritus, burning, pain, inflammation). Transient acute systemic reactions such as flushing and dyspnea also are commonly reported. Lipoatrophy at the injection site frequently has been reported in the literature as a cutaneous adverse effect of GA, but lobular panniculitis and necrosis at the site of injection rarely have been noted.
We report the case of a 36-year-old woman who experienced a severe adverse reaction to a single injection of GA after nearly 1 year of daily use to control symptoms of RRMS. Review of the current literature revealed few reports of the severe reaction of panniculitis and necrosis occurring at the injection site of GA.
Case Report
A 36-year-old woman was referred by her neurologist to the emergency department of our institution’s allergy and immunology clinic for treatment of an allergic reaction to a 20-mg GA injection, which she had been receiving daily for nearly 1 year as therapy for RRMS. A nodule immediately formed at the injection site and eventually became ulcerated. The patient also reported intense chest tightness, shortness of breath, and flushing following the injection. Physical examination revealed a large 8- to 9-cm erythematous area at the injection site on the left buttock. Necrosis and eschar formation also were evident (Figure 1).
Figure 1. Panniculitis with central ulceration and necrosis at the site of a glatiramer acetate injection on the left buttock (A). Closer view of an irregularly shaped necrotic lesion with surrounding erythema (B). |
A punch biopsy from the edge of the lesion revealed predominantly lobular panniculitis (Figure 2A) with fat necrosis and numerous foamy macrophages (Figures 2B and 2C). Scattered lymphocytes also were present but no neutrophils or eosinophils were noted (Figure 2B). Interlobular septa were widened secondary to fibrosis (Figure 2A). No lymphoid follicles were identified. A subcutaneous artery was sampled but was negative for vasculitis (Figure 2D).
|
The necrotic lesion on the left buttock was present for more than 2 months before complete healing occurred. The patient had a history of intolerance or unresponsiveness to all prior medications for RRMS. Several years prior she responded well to treatment with GA for a few months and had been responding well to the injections over the last year. Incremental challenge testing with GA for desensitization was offered to the patient, but she declined treatment out of fear of a recurrent episode, particularly the severe systemic symptoms she had experienced. Unfortunately, she was lost to follow-up.
Comment
Glatiramer acetate, formerly known as copolymer-1, is a first-line treatment of patients with RRMS.1 Daily administration of subcutaneous injections of GA (20 mg/mL) has proven effective in relapse rate reduction and reduced morbidity in patients with RRMS.2 Long-term studies support a relapse rate reduction of more than 50% in patients using GA.3 The most common adverse effects are LISRs.2 Systemic reactions following GA injection also are common. A much less common reaction is panniculitis followed by lipoatrophy and/or skin necrosis. Only a few instances of panniculitis-associated necrosis have been reported.
The occurrence of LISRs was reported in 20% to 90% of patients using GA to control RRMS.2,4 Local injection-site reactions typically resolve within hours to days and have been reported to decrease in frequency over time.5 Acute systemic reactions (eg, anxiety, flushing, palpitations, dyspnea) to GA injection are described in approximately 15% of patients.6 Systemic reactions usually resolve in 5 to 15 minutes but can last for more than 1 hour.5 These reactions are mostly benign and generally are not considered to be allergic or anaphylactic in nature. True systemic anaphylaxis associated with administration of GA is extremely rare.7
Lipoatrophy, or localized loss of subcutaneous adipose tissue without evidence of inflammation, has been reported fairly frequently in association with GA (up to 45% of patients receiving GA injections).2,6,8,9 Lipoatrophy also has been seen following subcutaneous injection of many other drugs, including steroids and insulin. Unlike LISRs, the likelihood of developing lipoatrophy at the injection site increases with longer durations of GA injections.5 Lipoatrophy also develops following panniculitis at the site of GA injection.
Based on a search of the MeSH (Medical Subject Headings) database using the terms panniculitis and glatiramer acetate, there only are 10 reported cases of panniculitis as an adverse effect of GA injections.2,6,10 Lesions were described as either subcutaneous erythematous nodules or atrophic areas that demonstrated panniculitis on histologic examination. Injections preceding the development of panniculitis often were described as remarkably painful.4 Residual lipoatrophy and/or hyperpigmentation at the site of panniculitis development is common.2 It has been suggested that GA-induced panniculitis may be an early underlying mechanism for the development of lipoatrophy, and thus may be more common than originally suspected.10
Histopathologic examination of GA-induced panniculitis typically reveals a localized, mostly lobular, panniculitis with lipophagic granulomas, lymphocytes, and thickened septa. The lipophagic granulomas (a characteristic finding in panniculitis) form from local macrophages that engulf the lipids released from necrotic adipocytes.11 A large, pale, granular or vacuolated cytoplasm typically can be observed on microscopic examination of the macrophages (Figure 2C). Connective tissue septa typically are widened with cell infiltrates, usually lymphocytes. Other cell types, including macrophages, eosinophils, and neutrophils, also have been identified in both the septa and fat lobules. These histologic elements may change and evolve over time.
Necrosis in association with panniculitis, as seen in our patient, rarely has been reported.4,12 All of the necrotic reactions described occurred after at least 1 year of GA therapy and took several weeks to resolve.4,12 When presented with the development of skin necrosis at the site of GA injection, it is essential to distinguish between an adverse effect associated with the drug itself and Nicolau syndrome (embolia cutis medicamentosa).13 Necrosis at multiple injection sites or recurrence with later injections supports a GA-specific effect.12
Nicolau syndrome is a well-known traumatic reaction that leads to microembolization and resultant vasospasm as well as necrosis throughout the skin and possibly the underlying muscular layer.14 Although more commonly associated with intramuscular injections, Nicolau syndrome has been described with subcutaneous injections of GA in a few rare instances.13,15 Because of the associated severe systemic reaction as well as the histologic examination (Figure 2D), we believe the skin necrosis seen in our patient was from a reaction to GA rather than Nicolau syndrome. Our patient was not interested in restarting GA therapy; therefore, it is unknown if this reaction would have recurred, but we suspect high probability of recurrence without desensitization attempts.
Preventative measures can be taken to decrease the risk for LISRs, and patients should be educated on these techniques. Applying ice to the injection site for at least 30 seconds before cleaning the skin for injection may reduce local adverse effects.4 Proper instruction on injection techniques should be provided by a knowledgeable health care professional and topical anesthetics and/or steroids may be offered to reduce pain associated with injection. There have been no proven measures for prevention of lipoatrophy, panniculitis, or necrosis, and these adverse effects are not thought to be attributed to improper injection techniques.14 Rotation of injection sites is the only suggested means of decreasing the potential risk for more severely and permanently disfiguring local reactions.
If panniculitis following GA injection is suspected, a large biopsy that encompasses the entire subcutaneous fat layer is necessary for proper dermatopathologic classification.11 Glatiramer acetate injections should be stopped immediately. These reactions disappear when the injections are stopped but recur when restarting treatment.2 The efficacy of GA in the treatment of RRMS has led to the possible use of this drug in the treatment of other autoimmune diseases.16 Thus, it is important for clinicians to be aware of all adverse effects of subcutaneous injections of GA, including the rare occurrence of panniculitis and necrosis, and when discontinuation of therapy is indicated.
Conclusion
Daily subcutaneous injection of GA for the treatment of RRMS can result in the rare but characteristic development of localized panniculitis and necrosis. Glatiramer acetate is a common and highly effective therapy used for the treatment of RRMS. Common adverse effects include LISRs and transient acute systemic reactions. Less commonly observed but characteristic of GA injections is localized lipoatrophy and mostly lobular panniculitis. Necrosis rarely can develop in association with these cutaneous reactions. It is essential to differentiate between necrosis secondary to Nicolau syndrome and skin necrosis as a unique reaction to GA; the latter is an indication for discontinuation of GA injections. Dermatologists should be made aware of adverse cutaneous reactions seen with GA therapy, especially with the potential for expansion of the use of GA to treat other autoimmune processes. Further research is needed regarding the histopathologic evolution and mechanisms behind the development of lipoatrophy, panniculitis, and necrosis at the site of GA injection.
Glatiramer acetate (GA), a synthetic polypeptide that is injected subcutaneously, has proven effective in the treatment of relapsing-remitting multiple sclerosis (RRMS) and is now considered a first-line agent in the treatment of this condition. Adverse effects associated with GA primarily include local injection-site reactions (LISRs)(eg, erythema, pruritus, burning, pain, inflammation). Transient acute systemic reactions such as flushing and dyspnea also are commonly reported. Lipoatrophy at the injection site frequently has been reported in the literature as a cutaneous adverse effect of GA, but lobular panniculitis and necrosis at the site of injection rarely have been noted.
We report the case of a 36-year-old woman who experienced a severe adverse reaction to a single injection of GA after nearly 1 year of daily use to control symptoms of RRMS. Review of the current literature revealed few reports of the severe reaction of panniculitis and necrosis occurring at the injection site of GA.
Case Report
A 36-year-old woman was referred by her neurologist to the emergency department of our institution’s allergy and immunology clinic for treatment of an allergic reaction to a 20-mg GA injection, which she had been receiving daily for nearly 1 year as therapy for RRMS. A nodule immediately formed at the injection site and eventually became ulcerated. The patient also reported intense chest tightness, shortness of breath, and flushing following the injection. Physical examination revealed a large 8- to 9-cm erythematous area at the injection site on the left buttock. Necrosis and eschar formation also were evident (Figure 1).
Figure 1. Panniculitis with central ulceration and necrosis at the site of a glatiramer acetate injection on the left buttock (A). Closer view of an irregularly shaped necrotic lesion with surrounding erythema (B). |
A punch biopsy from the edge of the lesion revealed predominantly lobular panniculitis (Figure 2A) with fat necrosis and numerous foamy macrophages (Figures 2B and 2C). Scattered lymphocytes also were present but no neutrophils or eosinophils were noted (Figure 2B). Interlobular septa were widened secondary to fibrosis (Figure 2A). No lymphoid follicles were identified. A subcutaneous artery was sampled but was negative for vasculitis (Figure 2D).
|
The necrotic lesion on the left buttock was present for more than 2 months before complete healing occurred. The patient had a history of intolerance or unresponsiveness to all prior medications for RRMS. Several years prior she responded well to treatment with GA for a few months and had been responding well to the injections over the last year. Incremental challenge testing with GA for desensitization was offered to the patient, but she declined treatment out of fear of a recurrent episode, particularly the severe systemic symptoms she had experienced. Unfortunately, she was lost to follow-up.
Comment
Glatiramer acetate, formerly known as copolymer-1, is a first-line treatment of patients with RRMS.1 Daily administration of subcutaneous injections of GA (20 mg/mL) has proven effective in relapse rate reduction and reduced morbidity in patients with RRMS.2 Long-term studies support a relapse rate reduction of more than 50% in patients using GA.3 The most common adverse effects are LISRs.2 Systemic reactions following GA injection also are common. A much less common reaction is panniculitis followed by lipoatrophy and/or skin necrosis. Only a few instances of panniculitis-associated necrosis have been reported.
The occurrence of LISRs was reported in 20% to 90% of patients using GA to control RRMS.2,4 Local injection-site reactions typically resolve within hours to days and have been reported to decrease in frequency over time.5 Acute systemic reactions (eg, anxiety, flushing, palpitations, dyspnea) to GA injection are described in approximately 15% of patients.6 Systemic reactions usually resolve in 5 to 15 minutes but can last for more than 1 hour.5 These reactions are mostly benign and generally are not considered to be allergic or anaphylactic in nature. True systemic anaphylaxis associated with administration of GA is extremely rare.7
Lipoatrophy, or localized loss of subcutaneous adipose tissue without evidence of inflammation, has been reported fairly frequently in association with GA (up to 45% of patients receiving GA injections).2,6,8,9 Lipoatrophy also has been seen following subcutaneous injection of many other drugs, including steroids and insulin. Unlike LISRs, the likelihood of developing lipoatrophy at the injection site increases with longer durations of GA injections.5 Lipoatrophy also develops following panniculitis at the site of GA injection.
Based on a search of the MeSH (Medical Subject Headings) database using the terms panniculitis and glatiramer acetate, there only are 10 reported cases of panniculitis as an adverse effect of GA injections.2,6,10 Lesions were described as either subcutaneous erythematous nodules or atrophic areas that demonstrated panniculitis on histologic examination. Injections preceding the development of panniculitis often were described as remarkably painful.4 Residual lipoatrophy and/or hyperpigmentation at the site of panniculitis development is common.2 It has been suggested that GA-induced panniculitis may be an early underlying mechanism for the development of lipoatrophy, and thus may be more common than originally suspected.10
Histopathologic examination of GA-induced panniculitis typically reveals a localized, mostly lobular, panniculitis with lipophagic granulomas, lymphocytes, and thickened septa. The lipophagic granulomas (a characteristic finding in panniculitis) form from local macrophages that engulf the lipids released from necrotic adipocytes.11 A large, pale, granular or vacuolated cytoplasm typically can be observed on microscopic examination of the macrophages (Figure 2C). Connective tissue septa typically are widened with cell infiltrates, usually lymphocytes. Other cell types, including macrophages, eosinophils, and neutrophils, also have been identified in both the septa and fat lobules. These histologic elements may change and evolve over time.
Necrosis in association with panniculitis, as seen in our patient, rarely has been reported.4,12 All of the necrotic reactions described occurred after at least 1 year of GA therapy and took several weeks to resolve.4,12 When presented with the development of skin necrosis at the site of GA injection, it is essential to distinguish between an adverse effect associated with the drug itself and Nicolau syndrome (embolia cutis medicamentosa).13 Necrosis at multiple injection sites or recurrence with later injections supports a GA-specific effect.12
Nicolau syndrome is a well-known traumatic reaction that leads to microembolization and resultant vasospasm as well as necrosis throughout the skin and possibly the underlying muscular layer.14 Although more commonly associated with intramuscular injections, Nicolau syndrome has been described with subcutaneous injections of GA in a few rare instances.13,15 Because of the associated severe systemic reaction as well as the histologic examination (Figure 2D), we believe the skin necrosis seen in our patient was from a reaction to GA rather than Nicolau syndrome. Our patient was not interested in restarting GA therapy; therefore, it is unknown if this reaction would have recurred, but we suspect high probability of recurrence without desensitization attempts.
Preventative measures can be taken to decrease the risk for LISRs, and patients should be educated on these techniques. Applying ice to the injection site for at least 30 seconds before cleaning the skin for injection may reduce local adverse effects.4 Proper instruction on injection techniques should be provided by a knowledgeable health care professional and topical anesthetics and/or steroids may be offered to reduce pain associated with injection. There have been no proven measures for prevention of lipoatrophy, panniculitis, or necrosis, and these adverse effects are not thought to be attributed to improper injection techniques.14 Rotation of injection sites is the only suggested means of decreasing the potential risk for more severely and permanently disfiguring local reactions.
If panniculitis following GA injection is suspected, a large biopsy that encompasses the entire subcutaneous fat layer is necessary for proper dermatopathologic classification.11 Glatiramer acetate injections should be stopped immediately. These reactions disappear when the injections are stopped but recur when restarting treatment.2 The efficacy of GA in the treatment of RRMS has led to the possible use of this drug in the treatment of other autoimmune diseases.16 Thus, it is important for clinicians to be aware of all adverse effects of subcutaneous injections of GA, including the rare occurrence of panniculitis and necrosis, and when discontinuation of therapy is indicated.
Conclusion
Daily subcutaneous injection of GA for the treatment of RRMS can result in the rare but characteristic development of localized panniculitis and necrosis. Glatiramer acetate is a common and highly effective therapy used for the treatment of RRMS. Common adverse effects include LISRs and transient acute systemic reactions. Less commonly observed but characteristic of GA injections is localized lipoatrophy and mostly lobular panniculitis. Necrosis rarely can develop in association with these cutaneous reactions. It is essential to differentiate between necrosis secondary to Nicolau syndrome and skin necrosis as a unique reaction to GA; the latter is an indication for discontinuation of GA injections. Dermatologists should be made aware of adverse cutaneous reactions seen with GA therapy, especially with the potential for expansion of the use of GA to treat other autoimmune processes. Further research is needed regarding the histopathologic evolution and mechanisms behind the development of lipoatrophy, panniculitis, and necrosis at the site of GA injection.
1. Anderson G, Meyer D, Herrman CE, et al. Tolerability and safety of novel half milliliter formulation of glatiramer acetate for subcutaneous injection: an open-label, multicenter, randomized comparative study. J Neurol. 2010;257:1917-1923.
2. Soares Almeida LM, Requena L, Kutzner H, et al. Localized panniculitis secondary to subcutaneous glatiramer acetate injections for the treatment of multiple sclerosis: a clinicopathologic and immunohistochemical study. J Am Acad Dermatol. 2006;55:968-974.
3. Ford CC, Johnson KP, Lisak RP, et al. A prospective open-label study of glatiramer acetate: over a decade of continuous use in multiple sclerosis patients. Mult Scler. 2006;12:309-320.
4. Frohman EM, Brannon K, Alexander S, et al. Disease modifying agent related skin reactions in multiple sclerosis: prevention, assessment, and management. Mult Scler. 2004;10:302-307.
5. Ziemssen T, Neuhaus O, Hohlfeld R. Risk-benefit assessment of glatiramer acetate in multiple sclerosis. Drug Saf. 2001;24:979-990.
6. Ball NJ, Cowan BJ, Moore GR, et al. Lobular panniculitis at the site of glatiramer acetate injections for the treatment of relapsing-remitting multiple sclerosis. a report of two cases. J Cutan Pathol. 2008;35:407-410.
7. Rauschka H, Farina C, Sator P, et al. Severe anaphylactic reaction to glatiramer acetate with specific IgE. Neurology. 2005;64:1481-1482.
8. Hwang L, Orengo I. Lipoatrophy associated with glatiramer acetate injections for the treatment of multiple sclerosis. Cutis. 2001;68:287-288.
9. Edgar CM, Brunet DG, Fenton P, et al. Lipoatrophy in patients with multiple sclerosis on glatiramer acetate. Can J Neurol Sci. 2004;31:58-63.
10. Soós N, Shakery K, Mrowietz U. Localized panniculitis and subsequent lipoatrophy with subcutaneous glatiramer acetate (Copaxone) injection for the treatment of multiple sclerosis. Am J Clin Dermatol. 2004;5:357-359.
11. Segura S, Requena L. Anatomy and histology of normal subcutaneous fat, necrosis of adipocytes, and classification of the panniculitides. Dermatol Clin. 2008;26:419-424, v.
12. Bosca I, Bosca M, Belenguer A, et al. Necrotising cutaneous lesions as a side effect of glatiramer acetate. J Neurol. 2006;253:1370-1371.
13. Feldmann R, Schierl M, Rauschka H, et al. Necrotizing skin lesions with involvement of muscle tissue after subcutaneous injection of glatiramer acetate. Eur J Dermatol. 2009;19:385.
14. Kluger N, Thouvenot E, Camu W, et al. Cutaneous adverse events related to glatiramer acetate injection (copolymer-1, Copaxone). J Eur Acad Dermatol Venereol. 2009;23:1332-1333.
15. Harde V, Schwarz T. Embolia cutis medicamentosa following subcutaneous injection of glatiramer acetate [in English, German]. J Dtsch Dermatol Ges. 2007;5:1122-1123.
16. Racke MK, Lovett-Racke AE. Glatiramer acetate treatment of multiple sclerosis: an immunological perspective. J Immunol. 2011;186:1887-1890.
1. Anderson G, Meyer D, Herrman CE, et al. Tolerability and safety of novel half milliliter formulation of glatiramer acetate for subcutaneous injection: an open-label, multicenter, randomized comparative study. J Neurol. 2010;257:1917-1923.
2. Soares Almeida LM, Requena L, Kutzner H, et al. Localized panniculitis secondary to subcutaneous glatiramer acetate injections for the treatment of multiple sclerosis: a clinicopathologic and immunohistochemical study. J Am Acad Dermatol. 2006;55:968-974.
3. Ford CC, Johnson KP, Lisak RP, et al. A prospective open-label study of glatiramer acetate: over a decade of continuous use in multiple sclerosis patients. Mult Scler. 2006;12:309-320.
4. Frohman EM, Brannon K, Alexander S, et al. Disease modifying agent related skin reactions in multiple sclerosis: prevention, assessment, and management. Mult Scler. 2004;10:302-307.
5. Ziemssen T, Neuhaus O, Hohlfeld R. Risk-benefit assessment of glatiramer acetate in multiple sclerosis. Drug Saf. 2001;24:979-990.
6. Ball NJ, Cowan BJ, Moore GR, et al. Lobular panniculitis at the site of glatiramer acetate injections for the treatment of relapsing-remitting multiple sclerosis. a report of two cases. J Cutan Pathol. 2008;35:407-410.
7. Rauschka H, Farina C, Sator P, et al. Severe anaphylactic reaction to glatiramer acetate with specific IgE. Neurology. 2005;64:1481-1482.
8. Hwang L, Orengo I. Lipoatrophy associated with glatiramer acetate injections for the treatment of multiple sclerosis. Cutis. 2001;68:287-288.
9. Edgar CM, Brunet DG, Fenton P, et al. Lipoatrophy in patients with multiple sclerosis on glatiramer acetate. Can J Neurol Sci. 2004;31:58-63.
10. Soós N, Shakery K, Mrowietz U. Localized panniculitis and subsequent lipoatrophy with subcutaneous glatiramer acetate (Copaxone) injection for the treatment of multiple sclerosis. Am J Clin Dermatol. 2004;5:357-359.
11. Segura S, Requena L. Anatomy and histology of normal subcutaneous fat, necrosis of adipocytes, and classification of the panniculitides. Dermatol Clin. 2008;26:419-424, v.
12. Bosca I, Bosca M, Belenguer A, et al. Necrotising cutaneous lesions as a side effect of glatiramer acetate. J Neurol. 2006;253:1370-1371.
13. Feldmann R, Schierl M, Rauschka H, et al. Necrotizing skin lesions with involvement of muscle tissue after subcutaneous injection of glatiramer acetate. Eur J Dermatol. 2009;19:385.
14. Kluger N, Thouvenot E, Camu W, et al. Cutaneous adverse events related to glatiramer acetate injection (copolymer-1, Copaxone). J Eur Acad Dermatol Venereol. 2009;23:1332-1333.
15. Harde V, Schwarz T. Embolia cutis medicamentosa following subcutaneous injection of glatiramer acetate [in English, German]. J Dtsch Dermatol Ges. 2007;5:1122-1123.
16. Racke MK, Lovett-Racke AE. Glatiramer acetate treatment of multiple sclerosis: an immunological perspective. J Immunol. 2011;186:1887-1890.
Practice Points
- Glatiramer acetate is a common and highly effective therapy administered subcutaneously for the treatment of relapsing-remitting multiple sclerosis.
- Common adverse effects include local injection-site reactions and transient acute systemic reactions.
- Rarely, localized lipoatrophy and mostly lobular panniculitis with occasional necrosis can be observed at the site of glatiramer acetate injections. This reaction is specific to the medication and can recur with subsequent injections.
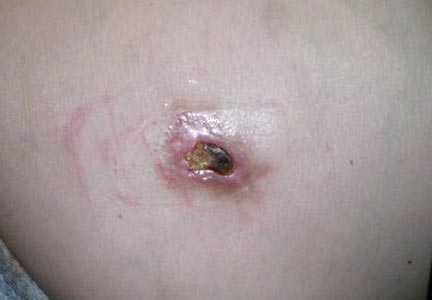
Nail Biopsy: 6 Techniques to Biopsy the Nail Matrix
Nail matrix biopsies are performed to confirm a diagnosis or surgically remove a skin lesion that is affecting the growth of the nail plate. The procedure may be used to identify:
- Inflammatory conditions such as nail psoriasis and lichen planus
- Benign tumors
- Solitary melanonychia
- Squamous cell carcinoma (SCC)
- Other nail disorders
Nail biopsy can lead to complications such as bleeding, infection, or scarring. Postoperative scarring can cause permanent nail splitting, dystrophy, or both.
In a Cosmetic Dermatology article, “Matrix Biopsy of Longitudinal Melanonychia and Longitudinal Erythronychia: A Step-by-Step Approach,” Drs. Siobhan C. Collins and Nathaniel J. Jellinek review 6 techniques used to biopsy the nail matrix.
- Punch excision
- Matrix shave
- Lateral longitudinal excision
- Midline/paramedian longitudinal excision
- Transverse excision
- Longitudinal excision of erythronychia
In the setting of longitudinal melanonychia (to diagnose nail melanoma or SCC) and longitudinal erythronychia (to diagnose SCC and rarely amelanotic melanoma or basal cell carcinoma), the techniques they describe accomplish 3 fundamental goals of nail surgery:
- Obtain adequate tissue via an excisional biopsy to make an accurate diagnosis and avoid sampling error
- Avoid unnecessary trauma to surrounding nail tissues by the judicious use of partial plate avulsions whenever feasible
- Avoid unnecessary postoperative nail scarring whenever possible
Dermatologists must be confident when performing nail biopsies and the techniques discussed by the authors will help approach nail surgery with more certainty.
At the 73rd Annual Meeting of the American Academy of Dermatology, Dr. Jellinek provides a hands-on approach to nail surgery. On Saturday, March 21, he will provide tips for nail surgeries at the “Medical and Surgical Management of Nail Disorders” lecture.
For more information, read the Collins and Jellinek article from Cosmetic Dermatology.
Nail matrix biopsies are performed to confirm a diagnosis or surgically remove a skin lesion that is affecting the growth of the nail plate. The procedure may be used to identify:
- Inflammatory conditions such as nail psoriasis and lichen planus
- Benign tumors
- Solitary melanonychia
- Squamous cell carcinoma (SCC)
- Other nail disorders
Nail biopsy can lead to complications such as bleeding, infection, or scarring. Postoperative scarring can cause permanent nail splitting, dystrophy, or both.
In a Cosmetic Dermatology article, “Matrix Biopsy of Longitudinal Melanonychia and Longitudinal Erythronychia: A Step-by-Step Approach,” Drs. Siobhan C. Collins and Nathaniel J. Jellinek review 6 techniques used to biopsy the nail matrix.
- Punch excision
- Matrix shave
- Lateral longitudinal excision
- Midline/paramedian longitudinal excision
- Transverse excision
- Longitudinal excision of erythronychia
In the setting of longitudinal melanonychia (to diagnose nail melanoma or SCC) and longitudinal erythronychia (to diagnose SCC and rarely amelanotic melanoma or basal cell carcinoma), the techniques they describe accomplish 3 fundamental goals of nail surgery:
- Obtain adequate tissue via an excisional biopsy to make an accurate diagnosis and avoid sampling error
- Avoid unnecessary trauma to surrounding nail tissues by the judicious use of partial plate avulsions whenever feasible
- Avoid unnecessary postoperative nail scarring whenever possible
Dermatologists must be confident when performing nail biopsies and the techniques discussed by the authors will help approach nail surgery with more certainty.
At the 73rd Annual Meeting of the American Academy of Dermatology, Dr. Jellinek provides a hands-on approach to nail surgery. On Saturday, March 21, he will provide tips for nail surgeries at the “Medical and Surgical Management of Nail Disorders” lecture.
For more information, read the Collins and Jellinek article from Cosmetic Dermatology.
Nail matrix biopsies are performed to confirm a diagnosis or surgically remove a skin lesion that is affecting the growth of the nail plate. The procedure may be used to identify:
- Inflammatory conditions such as nail psoriasis and lichen planus
- Benign tumors
- Solitary melanonychia
- Squamous cell carcinoma (SCC)
- Other nail disorders
Nail biopsy can lead to complications such as bleeding, infection, or scarring. Postoperative scarring can cause permanent nail splitting, dystrophy, or both.
In a Cosmetic Dermatology article, “Matrix Biopsy of Longitudinal Melanonychia and Longitudinal Erythronychia: A Step-by-Step Approach,” Drs. Siobhan C. Collins and Nathaniel J. Jellinek review 6 techniques used to biopsy the nail matrix.
- Punch excision
- Matrix shave
- Lateral longitudinal excision
- Midline/paramedian longitudinal excision
- Transverse excision
- Longitudinal excision of erythronychia
In the setting of longitudinal melanonychia (to diagnose nail melanoma or SCC) and longitudinal erythronychia (to diagnose SCC and rarely amelanotic melanoma or basal cell carcinoma), the techniques they describe accomplish 3 fundamental goals of nail surgery:
- Obtain adequate tissue via an excisional biopsy to make an accurate diagnosis and avoid sampling error
- Avoid unnecessary trauma to surrounding nail tissues by the judicious use of partial plate avulsions whenever feasible
- Avoid unnecessary postoperative nail scarring whenever possible
Dermatologists must be confident when performing nail biopsies and the techniques discussed by the authors will help approach nail surgery with more certainty.
At the 73rd Annual Meeting of the American Academy of Dermatology, Dr. Jellinek provides a hands-on approach to nail surgery. On Saturday, March 21, he will provide tips for nail surgeries at the “Medical and Surgical Management of Nail Disorders” lecture.
For more information, read the Collins and Jellinek article from Cosmetic Dermatology.
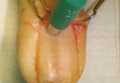
What Is Your Diagnosis? Extramammary Paget Disease

A 70-year-old man presented with a nonpruritic erythematous scaly plaque in the left suprapubic region of 6 months’ duration that had failed to respond to terbinafine cream 1% after 1 month of treatment of suspected tinea cruris. His medical history was remarkable for hypertension, hyperlipidemia, chronic obstructive pulmonary disease, benign prostatic hyperplasia, an abdominal aortic aneurysm, alcohol dependence, tobacco use disorder, and unintentional weight loss of 15 lb over the last year.
The Diagnosis: Extramammary Paget Disease
A biopsy of the plaque revealed an intraepidermal proliferation of large cells with abundant clear cytoplasm and large vesicular nuclei distributed throughout the epidermis (Figure 1). The neoplastic cells stained positive for both periodic acid–Schiff stain (Figure 2) and CK7 (Figure 3). Chemistry and liver function panel, urine analysis, carcinoembryonic antigen levels, and prostate-specific antigen levels were within reference range. A complete blood cell count revealed mild megaloblastic anemia. Subsequent computed tomography of the chest, abdomen, and pelvis revealed an abdominal aortic aneurysm and prostatic enlargement without any evidence of potential malignancies. Colonoscopy revealed multiple hyperplastic polyps and a tubular adenoma. Cystoscopy was normal, except for evidence of prostate enlargement. Urine cytology was unremarkable. The patient was referred for excision of the lesion with Mohs micrographic surgery. Follow-up was recommended every 3 months for the first 2 years following surgery and every 6 months thereafter to monitor for recurrence or secondary neoplasms.
|
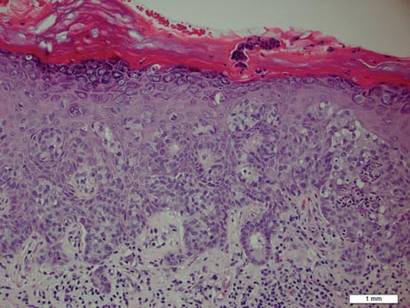
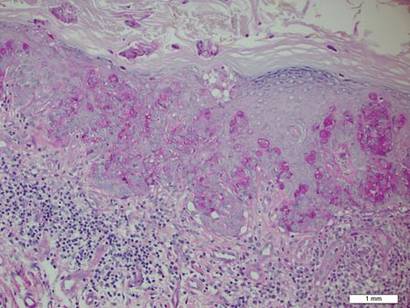
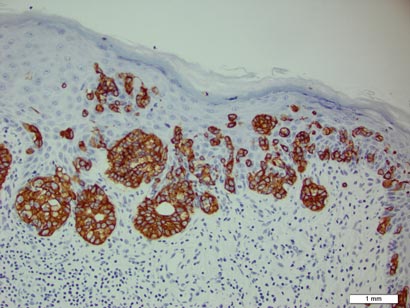
Sir James Paget first described mammary Paget disease of the nipple in 1874 in his report of 15 women with skin eruptions of the nipple and areola and subsequent carcinoma of the underlying breast.1 Paget also described a patient with a similar eruption on the glans penis and Crocker2 described extramammary Paget disease (EMPD) of the scrotum and penis in 1889. The principle difference between mammary Paget disease and EMPD is the anatomic location.
Extramammary Paget disease is a rare condition that typically affects patients aged 50 to 80 years and is more common in women and white-skinned races.3 Extramammary Paget disease frequently targets cutaneous sites that are rich in apocrine glands. The most commonly affected site is the vulva followed by perineal, perianal, scrotal, and penile skin. Less commonly, the axillae, buttocks, thighs, eyelids, and external auditory canals may be affected.4
Patients with EMPD typically present with well-demarcated, nonresolving, erythematous and eczematous plaques that may have associated crusting, scaling, papillomatous excrescences, lichenification, ulceration, or bleeding. The most common symptom is pruritus, followed by burning, irritation, pain, and tenderness.5 Ten percent of patients are asymptomatic. The average interval between symptom onset and diagnosis is 2 years.5
Histopathology reveals diffusely infiltrating, irregular, neoplastic Paget cells within the epidermis that are large and vacuolated with abundant pale bluish cytoplasm and large vesicular nuclei, which may be centrally or laterally compressed. The cells may be distributed singly or in groups as strands, nests, or glandular patterns within the lower epidermis, rete ridges, and adnexal structures. Hyperkeratosis, acanthosis, and parakeratosis may also be present. Paget cells stain for immunohistochemical markers of apocrine and eccrine derivation including low-molecular-weight cytokeratins, gross cystic disease fluid protein 15, periodic acid–Schiff stain, and carcinoembryonic antigen.5 Perrotto et al6 studied 98 specimens from 61 patients and found that CK7 was positive in all EMPD specimens, while CK20 and gross cystic disease fluid protein 15 were positive in large subsets of both primary and secondary EMPD. Cases of EMPD secondary to anorectal adenocarcinoma were largely ERBB2 (formerly HER2/neu) negative and CDX2 positive.6
Diagnosis of EMPD should be followed by a thorough investigation for underlying carcinomas. In a review of 197 cases of EMPD, 24% of patients with EMPD had an associated underlying in situ or invasive adnexal apocrine carcinoma, which was associated with a higher mortality rate than in patients without this underlying malignancy. Additionally, 12% of EMPD patients had an associated underlying internal malignancy.7 These malignancies may include carcinomas of the urethra, bladder, vagina, cervix, endometrium, prostate, colon, and rectum. Perianal EMPD has a higher frequency of associated malignancies than vulvar EMPD.5 The location of EMPD is related to the location of the underlying malignancy; for example, perianal EMPD is associated with colorectal adenocarcinomas, and EMPD of the penis, scrotum, and groin is associated with genitourinary malignancies. Investigations to search for associated malignancies in patients with EMPD may include pelvic ultrasonography and/or magnetic resonance imaging, hysteroscopy, colonoscopy, sigmoidoscopy, cystoscopy, intravenous pyelogram, mammogram, and/or chest radiograph.
The most effective treatment of EMPD is margin-controlled surgical excision. High local recurrence rates may be due to irregular margins, multicentricity, and the tendency of EMPD to involve clinically normal-appearing skin. Hendi et al8 noted that EMPD may actually be unifocal with subclinical fingerlike projections extending beyond the main body of the tumor, requiring CK7 immunostaining for visualization to ensure complete margin control. The recurrence rate after standard surgical excision is 33% to 60%. The recurrence rate after excision via Mohs micrographic surgery is 16% for primary EMPD and 50% for recurrent EMPD.9 Other treatment modalities include radiotherapy, topical chemotherapy with 5-fluorouracil or imiquimod, and photodynamic therapy.10-13 Combined systemic chemotherapy with trastuzumab and paclitaxel can be considered for the treatment of ERBB2-positive EMPD.14
For patients with chronic genital or perianal lesions that are unresponsive to treatment, dermatologists should maintain a high index of suspicion for EMPD. If a patient is diagnosed with EMPD, a full-body skin examination should be performed with palpation of all lymph nodes. Imaging studies directed at the anatomic location of the involved skin should be utilized to search for an underlying internal malignancy.
1. Paget J. On disease of the mammary areola preceding cancer of the mammary gland. St Bartholomew Hosp Rep. 1874;10:87-89.
2. Crocker H. Paget’s disease affecting the scrotum and penis. Trans Pathol Soc Lond. 1889;40:187-191.
3. Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
4. Heymann WR. Extramammary Paget’s disease. Clin Dermatol. 1993;11:83-87.5. Shepherd V, Davidson EJ, Davies-Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
6. Perrotto J, Abbott JJ, Ceilley RI, et al. The role of immunohistochemistry in discriminating primary from secondary extramammary Paget disease. Am J Dermatopathol. 2010;32:137-143.
7. Chanda JJ. Extramammary Paget’s disease: prognosis and relationship to internal malignancy. J Am Acad Dermatol. 1985;13:1009-1014.
8. Hendi A, Perdikis G, Snow JL. Unifocality of extramammary Paget disease. J Am Acad Dermatol. 2008;59:811-813.
9. Hendi A, Brodland DG, Zitelli JA. Extramammary Paget’s disease: surgical treatment with mohs micrographic surgery. J Am Acad Dermatol. 2004;51:767-773.10. Zampogna JC, Flowers FP, Roth WI, et al. Treatment of primary limited cutaneous extramammary Paget’s disease with topical imiquimod monotherapy: two case reports. J Am Acad Dermatol. 2002;47:S229-S235.
11. Beleznay KM, Levesque MA, Gill S. Response to 5-fluorouracil in metastatic extramammary Paget disease of the scrotum presenting as pancytopenia and back pain. Curr Oncol. 2009;16:81-83.
12. Kitagawa KH, Bogner P, Zeitouni NC. Photodynamic therapy with methyl-aminolevulinate for the treatment of double extramammary Paget’s disease. Dermatol Surg. 2011;37:1043-1046.
13. Hata M, Omura M, Koike I, et al. Role of radiotherapy as curative treatment of extramammary Paget’s disease. Int J Radiat Oncol Biol Phys. 2011;80:47-54.
14. Takahagi S, Noda H, Kamegashira A, et al. Metastatic extramammary Paget’s disease treated with paclitaxel and trastuzumab combination chemotherapy. J Dermatol. 2009;36:457-461.
A 70-year-old man presented with a nonpruritic erythematous scaly plaque in the left suprapubic region of 6 months’ duration that had failed to respond to terbinafine cream 1% after 1 month of treatment of suspected tinea cruris. His medical history was remarkable for hypertension, hyperlipidemia, chronic obstructive pulmonary disease, benign prostatic hyperplasia, an abdominal aortic aneurysm, alcohol dependence, tobacco use disorder, and unintentional weight loss of 15 lb over the last year.
The Diagnosis: Extramammary Paget Disease
A biopsy of the plaque revealed an intraepidermal proliferation of large cells with abundant clear cytoplasm and large vesicular nuclei distributed throughout the epidermis (Figure 1). The neoplastic cells stained positive for both periodic acid–Schiff stain (Figure 2) and CK7 (Figure 3). Chemistry and liver function panel, urine analysis, carcinoembryonic antigen levels, and prostate-specific antigen levels were within reference range. A complete blood cell count revealed mild megaloblastic anemia. Subsequent computed tomography of the chest, abdomen, and pelvis revealed an abdominal aortic aneurysm and prostatic enlargement without any evidence of potential malignancies. Colonoscopy revealed multiple hyperplastic polyps and a tubular adenoma. Cystoscopy was normal, except for evidence of prostate enlargement. Urine cytology was unremarkable. The patient was referred for excision of the lesion with Mohs micrographic surgery. Follow-up was recommended every 3 months for the first 2 years following surgery and every 6 months thereafter to monitor for recurrence or secondary neoplasms.
|
Sir James Paget first described mammary Paget disease of the nipple in 1874 in his report of 15 women with skin eruptions of the nipple and areola and subsequent carcinoma of the underlying breast.1 Paget also described a patient with a similar eruption on the glans penis and Crocker2 described extramammary Paget disease (EMPD) of the scrotum and penis in 1889. The principle difference between mammary Paget disease and EMPD is the anatomic location.
Extramammary Paget disease is a rare condition that typically affects patients aged 50 to 80 years and is more common in women and white-skinned races.3 Extramammary Paget disease frequently targets cutaneous sites that are rich in apocrine glands. The most commonly affected site is the vulva followed by perineal, perianal, scrotal, and penile skin. Less commonly, the axillae, buttocks, thighs, eyelids, and external auditory canals may be affected.4
Patients with EMPD typically present with well-demarcated, nonresolving, erythematous and eczematous plaques that may have associated crusting, scaling, papillomatous excrescences, lichenification, ulceration, or bleeding. The most common symptom is pruritus, followed by burning, irritation, pain, and tenderness.5 Ten percent of patients are asymptomatic. The average interval between symptom onset and diagnosis is 2 years.5
Histopathology reveals diffusely infiltrating, irregular, neoplastic Paget cells within the epidermis that are large and vacuolated with abundant pale bluish cytoplasm and large vesicular nuclei, which may be centrally or laterally compressed. The cells may be distributed singly or in groups as strands, nests, or glandular patterns within the lower epidermis, rete ridges, and adnexal structures. Hyperkeratosis, acanthosis, and parakeratosis may also be present. Paget cells stain for immunohistochemical markers of apocrine and eccrine derivation including low-molecular-weight cytokeratins, gross cystic disease fluid protein 15, periodic acid–Schiff stain, and carcinoembryonic antigen.5 Perrotto et al6 studied 98 specimens from 61 patients and found that CK7 was positive in all EMPD specimens, while CK20 and gross cystic disease fluid protein 15 were positive in large subsets of both primary and secondary EMPD. Cases of EMPD secondary to anorectal adenocarcinoma were largely ERBB2 (formerly HER2/neu) negative and CDX2 positive.6
Diagnosis of EMPD should be followed by a thorough investigation for underlying carcinomas. In a review of 197 cases of EMPD, 24% of patients with EMPD had an associated underlying in situ or invasive adnexal apocrine carcinoma, which was associated with a higher mortality rate than in patients without this underlying malignancy. Additionally, 12% of EMPD patients had an associated underlying internal malignancy.7 These malignancies may include carcinomas of the urethra, bladder, vagina, cervix, endometrium, prostate, colon, and rectum. Perianal EMPD has a higher frequency of associated malignancies than vulvar EMPD.5 The location of EMPD is related to the location of the underlying malignancy; for example, perianal EMPD is associated with colorectal adenocarcinomas, and EMPD of the penis, scrotum, and groin is associated with genitourinary malignancies. Investigations to search for associated malignancies in patients with EMPD may include pelvic ultrasonography and/or magnetic resonance imaging, hysteroscopy, colonoscopy, sigmoidoscopy, cystoscopy, intravenous pyelogram, mammogram, and/or chest radiograph.
The most effective treatment of EMPD is margin-controlled surgical excision. High local recurrence rates may be due to irregular margins, multicentricity, and the tendency of EMPD to involve clinically normal-appearing skin. Hendi et al8 noted that EMPD may actually be unifocal with subclinical fingerlike projections extending beyond the main body of the tumor, requiring CK7 immunostaining for visualization to ensure complete margin control. The recurrence rate after standard surgical excision is 33% to 60%. The recurrence rate after excision via Mohs micrographic surgery is 16% for primary EMPD and 50% for recurrent EMPD.9 Other treatment modalities include radiotherapy, topical chemotherapy with 5-fluorouracil or imiquimod, and photodynamic therapy.10-13 Combined systemic chemotherapy with trastuzumab and paclitaxel can be considered for the treatment of ERBB2-positive EMPD.14
For patients with chronic genital or perianal lesions that are unresponsive to treatment, dermatologists should maintain a high index of suspicion for EMPD. If a patient is diagnosed with EMPD, a full-body skin examination should be performed with palpation of all lymph nodes. Imaging studies directed at the anatomic location of the involved skin should be utilized to search for an underlying internal malignancy.
A 70-year-old man presented with a nonpruritic erythematous scaly plaque in the left suprapubic region of 6 months’ duration that had failed to respond to terbinafine cream 1% after 1 month of treatment of suspected tinea cruris. His medical history was remarkable for hypertension, hyperlipidemia, chronic obstructive pulmonary disease, benign prostatic hyperplasia, an abdominal aortic aneurysm, alcohol dependence, tobacco use disorder, and unintentional weight loss of 15 lb over the last year.
The Diagnosis: Extramammary Paget Disease
A biopsy of the plaque revealed an intraepidermal proliferation of large cells with abundant clear cytoplasm and large vesicular nuclei distributed throughout the epidermis (Figure 1). The neoplastic cells stained positive for both periodic acid–Schiff stain (Figure 2) and CK7 (Figure 3). Chemistry and liver function panel, urine analysis, carcinoembryonic antigen levels, and prostate-specific antigen levels were within reference range. A complete blood cell count revealed mild megaloblastic anemia. Subsequent computed tomography of the chest, abdomen, and pelvis revealed an abdominal aortic aneurysm and prostatic enlargement without any evidence of potential malignancies. Colonoscopy revealed multiple hyperplastic polyps and a tubular adenoma. Cystoscopy was normal, except for evidence of prostate enlargement. Urine cytology was unremarkable. The patient was referred for excision of the lesion with Mohs micrographic surgery. Follow-up was recommended every 3 months for the first 2 years following surgery and every 6 months thereafter to monitor for recurrence or secondary neoplasms.
|
Sir James Paget first described mammary Paget disease of the nipple in 1874 in his report of 15 women with skin eruptions of the nipple and areola and subsequent carcinoma of the underlying breast.1 Paget also described a patient with a similar eruption on the glans penis and Crocker2 described extramammary Paget disease (EMPD) of the scrotum and penis in 1889. The principle difference between mammary Paget disease and EMPD is the anatomic location.
Extramammary Paget disease is a rare condition that typically affects patients aged 50 to 80 years and is more common in women and white-skinned races.3 Extramammary Paget disease frequently targets cutaneous sites that are rich in apocrine glands. The most commonly affected site is the vulva followed by perineal, perianal, scrotal, and penile skin. Less commonly, the axillae, buttocks, thighs, eyelids, and external auditory canals may be affected.4
Patients with EMPD typically present with well-demarcated, nonresolving, erythematous and eczematous plaques that may have associated crusting, scaling, papillomatous excrescences, lichenification, ulceration, or bleeding. The most common symptom is pruritus, followed by burning, irritation, pain, and tenderness.5 Ten percent of patients are asymptomatic. The average interval between symptom onset and diagnosis is 2 years.5
Histopathology reveals diffusely infiltrating, irregular, neoplastic Paget cells within the epidermis that are large and vacuolated with abundant pale bluish cytoplasm and large vesicular nuclei, which may be centrally or laterally compressed. The cells may be distributed singly or in groups as strands, nests, or glandular patterns within the lower epidermis, rete ridges, and adnexal structures. Hyperkeratosis, acanthosis, and parakeratosis may also be present. Paget cells stain for immunohistochemical markers of apocrine and eccrine derivation including low-molecular-weight cytokeratins, gross cystic disease fluid protein 15, periodic acid–Schiff stain, and carcinoembryonic antigen.5 Perrotto et al6 studied 98 specimens from 61 patients and found that CK7 was positive in all EMPD specimens, while CK20 and gross cystic disease fluid protein 15 were positive in large subsets of both primary and secondary EMPD. Cases of EMPD secondary to anorectal adenocarcinoma were largely ERBB2 (formerly HER2/neu) negative and CDX2 positive.6
Diagnosis of EMPD should be followed by a thorough investigation for underlying carcinomas. In a review of 197 cases of EMPD, 24% of patients with EMPD had an associated underlying in situ or invasive adnexal apocrine carcinoma, which was associated with a higher mortality rate than in patients without this underlying malignancy. Additionally, 12% of EMPD patients had an associated underlying internal malignancy.7 These malignancies may include carcinomas of the urethra, bladder, vagina, cervix, endometrium, prostate, colon, and rectum. Perianal EMPD has a higher frequency of associated malignancies than vulvar EMPD.5 The location of EMPD is related to the location of the underlying malignancy; for example, perianal EMPD is associated with colorectal adenocarcinomas, and EMPD of the penis, scrotum, and groin is associated with genitourinary malignancies. Investigations to search for associated malignancies in patients with EMPD may include pelvic ultrasonography and/or magnetic resonance imaging, hysteroscopy, colonoscopy, sigmoidoscopy, cystoscopy, intravenous pyelogram, mammogram, and/or chest radiograph.
The most effective treatment of EMPD is margin-controlled surgical excision. High local recurrence rates may be due to irregular margins, multicentricity, and the tendency of EMPD to involve clinically normal-appearing skin. Hendi et al8 noted that EMPD may actually be unifocal with subclinical fingerlike projections extending beyond the main body of the tumor, requiring CK7 immunostaining for visualization to ensure complete margin control. The recurrence rate after standard surgical excision is 33% to 60%. The recurrence rate after excision via Mohs micrographic surgery is 16% for primary EMPD and 50% for recurrent EMPD.9 Other treatment modalities include radiotherapy, topical chemotherapy with 5-fluorouracil or imiquimod, and photodynamic therapy.10-13 Combined systemic chemotherapy with trastuzumab and paclitaxel can be considered for the treatment of ERBB2-positive EMPD.14
For patients with chronic genital or perianal lesions that are unresponsive to treatment, dermatologists should maintain a high index of suspicion for EMPD. If a patient is diagnosed with EMPD, a full-body skin examination should be performed with palpation of all lymph nodes. Imaging studies directed at the anatomic location of the involved skin should be utilized to search for an underlying internal malignancy.
1. Paget J. On disease of the mammary areola preceding cancer of the mammary gland. St Bartholomew Hosp Rep. 1874;10:87-89.
2. Crocker H. Paget’s disease affecting the scrotum and penis. Trans Pathol Soc Lond. 1889;40:187-191.
3. Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
4. Heymann WR. Extramammary Paget’s disease. Clin Dermatol. 1993;11:83-87.5. Shepherd V, Davidson EJ, Davies-Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
6. Perrotto J, Abbott JJ, Ceilley RI, et al. The role of immunohistochemistry in discriminating primary from secondary extramammary Paget disease. Am J Dermatopathol. 2010;32:137-143.
7. Chanda JJ. Extramammary Paget’s disease: prognosis and relationship to internal malignancy. J Am Acad Dermatol. 1985;13:1009-1014.
8. Hendi A, Perdikis G, Snow JL. Unifocality of extramammary Paget disease. J Am Acad Dermatol. 2008;59:811-813.
9. Hendi A, Brodland DG, Zitelli JA. Extramammary Paget’s disease: surgical treatment with mohs micrographic surgery. J Am Acad Dermatol. 2004;51:767-773.10. Zampogna JC, Flowers FP, Roth WI, et al. Treatment of primary limited cutaneous extramammary Paget’s disease with topical imiquimod monotherapy: two case reports. J Am Acad Dermatol. 2002;47:S229-S235.
11. Beleznay KM, Levesque MA, Gill S. Response to 5-fluorouracil in metastatic extramammary Paget disease of the scrotum presenting as pancytopenia and back pain. Curr Oncol. 2009;16:81-83.
12. Kitagawa KH, Bogner P, Zeitouni NC. Photodynamic therapy with methyl-aminolevulinate for the treatment of double extramammary Paget’s disease. Dermatol Surg. 2011;37:1043-1046.
13. Hata M, Omura M, Koike I, et al. Role of radiotherapy as curative treatment of extramammary Paget’s disease. Int J Radiat Oncol Biol Phys. 2011;80:47-54.
14. Takahagi S, Noda H, Kamegashira A, et al. Metastatic extramammary Paget’s disease treated with paclitaxel and trastuzumab combination chemotherapy. J Dermatol. 2009;36:457-461.
1. Paget J. On disease of the mammary areola preceding cancer of the mammary gland. St Bartholomew Hosp Rep. 1874;10:87-89.
2. Crocker H. Paget’s disease affecting the scrotum and penis. Trans Pathol Soc Lond. 1889;40:187-191.
3. Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
4. Heymann WR. Extramammary Paget’s disease. Clin Dermatol. 1993;11:83-87.5. Shepherd V, Davidson EJ, Davies-Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
6. Perrotto J, Abbott JJ, Ceilley RI, et al. The role of immunohistochemistry in discriminating primary from secondary extramammary Paget disease. Am J Dermatopathol. 2010;32:137-143.
7. Chanda JJ. Extramammary Paget’s disease: prognosis and relationship to internal malignancy. J Am Acad Dermatol. 1985;13:1009-1014.
8. Hendi A, Perdikis G, Snow JL. Unifocality of extramammary Paget disease. J Am Acad Dermatol. 2008;59:811-813.
9. Hendi A, Brodland DG, Zitelli JA. Extramammary Paget’s disease: surgical treatment with mohs micrographic surgery. J Am Acad Dermatol. 2004;51:767-773.10. Zampogna JC, Flowers FP, Roth WI, et al. Treatment of primary limited cutaneous extramammary Paget’s disease with topical imiquimod monotherapy: two case reports. J Am Acad Dermatol. 2002;47:S229-S235.
11. Beleznay KM, Levesque MA, Gill S. Response to 5-fluorouracil in metastatic extramammary Paget disease of the scrotum presenting as pancytopenia and back pain. Curr Oncol. 2009;16:81-83.
12. Kitagawa KH, Bogner P, Zeitouni NC. Photodynamic therapy with methyl-aminolevulinate for the treatment of double extramammary Paget’s disease. Dermatol Surg. 2011;37:1043-1046.
13. Hata M, Omura M, Koike I, et al. Role of radiotherapy as curative treatment of extramammary Paget’s disease. Int J Radiat Oncol Biol Phys. 2011;80:47-54.
14. Takahagi S, Noda H, Kamegashira A, et al. Metastatic extramammary Paget’s disease treated with paclitaxel and trastuzumab combination chemotherapy. J Dermatol. 2009;36:457-461.
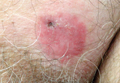
Onchocerciasis
The larvae of Onchocerca volvulus, a nematode that is most commonly found in tropical Africa, Yemen, Central America, and South America, are transmitted by flies of the genus Simulium that breed near fast-flowing rivers.1 The flies bite the host and transmit the larvae, and the larvae then mature into adults within the skin and subcutis, forming nodules that typically are not painful. The worms may reside within the skin for years and produce microfilariae, which can migrate and cause visual impairment, blindness, or a pruritic papular rash.1
The nematode produces a nodule within the dermis or subcutis with surrounding fibrous tissue and a mixed inflammatory infiltrate with eosinophils (Figure 1). In some cases, microfilariae can be seen within the lymphatics or within the uteri of the worms.1 Male and female worms typically are present and have a corrugated cuticle with a thin underlying layer of striated muscle. The females have paired uteri, which usually contain microfilariae2 (Figure 2).
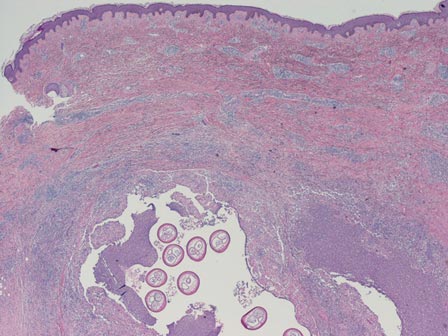 | 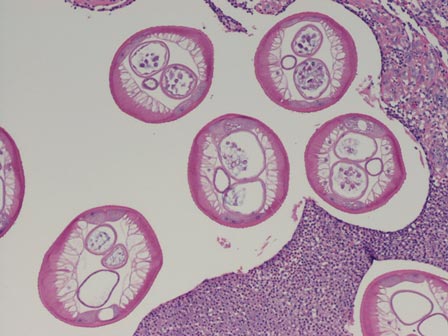 |
Dirofilaria repens also is a nematode that produces a subcutaneous nodule with an inflammatory reaction. This worm typically has a thick cuticle with longitudinal ridges, long thick muscle, and lateral cords.3 Additionally, because humans are not the usual host, Dirofilaria species do not complete their lifecycle and typically are not gravid, unlike Onchocerca species.
Myiasis is the presence of fly larvae within the skin. The larvae demonstrate a thick hyaline cuticle with pigmented brown-yellow spikes (Figure 3). There is a thick muscular layer under the cuticle and a tubular tracheal system containing vertical striations. The digestive system has an epithelial lining with prominent vessels. Adipose tissue with granulated cytoplasm, prominent nuclei, and coarse chromatin also are present.4

Scabies mites (Figure 4), ova, and scybala are present within the stratum corneum. A mixed inflammatory infiltrate also can be present.1 Tungiasis is caused by burrowing fleas and typically occurs on acral skin; therefore, it is more frequently found in the superficial portion of the skin. Erythrocytes usually are present in the gastrointestinal tract, and the females usually are gravid.2 A surrounding mixed inflammatory infiltrate is present, and necrosis also can occur (Figure 5).1
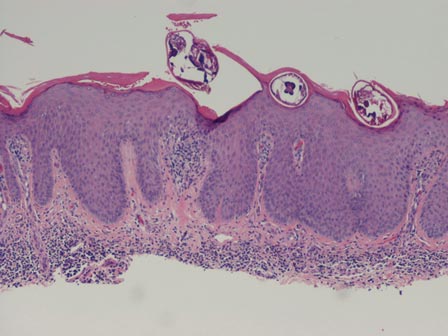 | 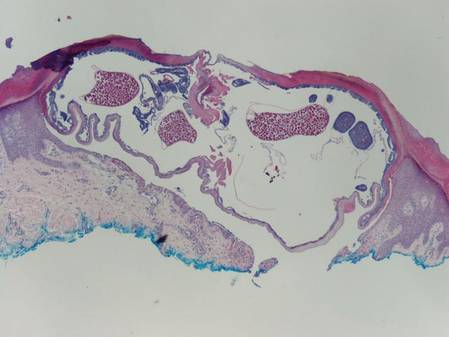 |
1. Weedon D. Weedon’s Skin Pathology. 3rd ed. Edinburgh, Scotland: Churchill Livingstone Elsevier; 2010.
2. Elston DM, Ferringer T. Dermatopathology: Requisites in Dermatology. Edinburgh, Scotland: Saunders Elsevier; 2008.
3. Tzanetou K, Gasteratos S, Pantazopoulou A, et al. Subcutaneous dirofilariasis caused by Dirofilaria repens in Greece: a case report. J Cutan Pathol. 2009;36:892-895.
4. Fernandez-Flores A, Saeb-Lima M. Pulse granuloma of the lip: morphologic clues in its differential diagnosis. J Cutan Pathol. 2014;41:394-399.
The larvae of Onchocerca volvulus, a nematode that is most commonly found in tropical Africa, Yemen, Central America, and South America, are transmitted by flies of the genus Simulium that breed near fast-flowing rivers.1 The flies bite the host and transmit the larvae, and the larvae then mature into adults within the skin and subcutis, forming nodules that typically are not painful. The worms may reside within the skin for years and produce microfilariae, which can migrate and cause visual impairment, blindness, or a pruritic papular rash.1
The nematode produces a nodule within the dermis or subcutis with surrounding fibrous tissue and a mixed inflammatory infiltrate with eosinophils (Figure 1). In some cases, microfilariae can be seen within the lymphatics or within the uteri of the worms.1 Male and female worms typically are present and have a corrugated cuticle with a thin underlying layer of striated muscle. The females have paired uteri, which usually contain microfilariae2 (Figure 2).
| |
Dirofilaria repens also is a nematode that produces a subcutaneous nodule with an inflammatory reaction. This worm typically has a thick cuticle with longitudinal ridges, long thick muscle, and lateral cords.3 Additionally, because humans are not the usual host, Dirofilaria species do not complete their lifecycle and typically are not gravid, unlike Onchocerca species.
Myiasis is the presence of fly larvae within the skin. The larvae demonstrate a thick hyaline cuticle with pigmented brown-yellow spikes (Figure 3). There is a thick muscular layer under the cuticle and a tubular tracheal system containing vertical striations. The digestive system has an epithelial lining with prominent vessels. Adipose tissue with granulated cytoplasm, prominent nuclei, and coarse chromatin also are present.4
Scabies mites (Figure 4), ova, and scybala are present within the stratum corneum. A mixed inflammatory infiltrate also can be present.1 Tungiasis is caused by burrowing fleas and typically occurs on acral skin; therefore, it is more frequently found in the superficial portion of the skin. Erythrocytes usually are present in the gastrointestinal tract, and the females usually are gravid.2 A surrounding mixed inflammatory infiltrate is present, and necrosis also can occur (Figure 5).1
| |
The larvae of Onchocerca volvulus, a nematode that is most commonly found in tropical Africa, Yemen, Central America, and South America, are transmitted by flies of the genus Simulium that breed near fast-flowing rivers.1 The flies bite the host and transmit the larvae, and the larvae then mature into adults within the skin and subcutis, forming nodules that typically are not painful. The worms may reside within the skin for years and produce microfilariae, which can migrate and cause visual impairment, blindness, or a pruritic papular rash.1
The nematode produces a nodule within the dermis or subcutis with surrounding fibrous tissue and a mixed inflammatory infiltrate with eosinophils (Figure 1). In some cases, microfilariae can be seen within the lymphatics or within the uteri of the worms.1 Male and female worms typically are present and have a corrugated cuticle with a thin underlying layer of striated muscle. The females have paired uteri, which usually contain microfilariae2 (Figure 2).
| |
Dirofilaria repens also is a nematode that produces a subcutaneous nodule with an inflammatory reaction. This worm typically has a thick cuticle with longitudinal ridges, long thick muscle, and lateral cords.3 Additionally, because humans are not the usual host, Dirofilaria species do not complete their lifecycle and typically are not gravid, unlike Onchocerca species.
Myiasis is the presence of fly larvae within the skin. The larvae demonstrate a thick hyaline cuticle with pigmented brown-yellow spikes (Figure 3). There is a thick muscular layer under the cuticle and a tubular tracheal system containing vertical striations. The digestive system has an epithelial lining with prominent vessels. Adipose tissue with granulated cytoplasm, prominent nuclei, and coarse chromatin also are present.4
Scabies mites (Figure 4), ova, and scybala are present within the stratum corneum. A mixed inflammatory infiltrate also can be present.1 Tungiasis is caused by burrowing fleas and typically occurs on acral skin; therefore, it is more frequently found in the superficial portion of the skin. Erythrocytes usually are present in the gastrointestinal tract, and the females usually are gravid.2 A surrounding mixed inflammatory infiltrate is present, and necrosis also can occur (Figure 5).1
| |
1. Weedon D. Weedon’s Skin Pathology. 3rd ed. Edinburgh, Scotland: Churchill Livingstone Elsevier; 2010.
2. Elston DM, Ferringer T. Dermatopathology: Requisites in Dermatology. Edinburgh, Scotland: Saunders Elsevier; 2008.
3. Tzanetou K, Gasteratos S, Pantazopoulou A, et al. Subcutaneous dirofilariasis caused by Dirofilaria repens in Greece: a case report. J Cutan Pathol. 2009;36:892-895.
4. Fernandez-Flores A, Saeb-Lima M. Pulse granuloma of the lip: morphologic clues in its differential diagnosis. J Cutan Pathol. 2014;41:394-399.
1. Weedon D. Weedon’s Skin Pathology. 3rd ed. Edinburgh, Scotland: Churchill Livingstone Elsevier; 2010.
2. Elston DM, Ferringer T. Dermatopathology: Requisites in Dermatology. Edinburgh, Scotland: Saunders Elsevier; 2008.
3. Tzanetou K, Gasteratos S, Pantazopoulou A, et al. Subcutaneous dirofilariasis caused by Dirofilaria repens in Greece: a case report. J Cutan Pathol. 2009;36:892-895.
4. Fernandez-Flores A, Saeb-Lima M. Pulse granuloma of the lip: morphologic clues in its differential diagnosis. J Cutan Pathol. 2014;41:394-399.