User login
Cystic Nodule on the Palm
The Diagnosis: Nodular Hidradenoma
Nodular hidradenomas (NHs) are rare benign cutaneous adnexal neoplasms first described in 1949 as clear cell papillary carcinomas.1 Since then, various terms have been used to describe this entity, such as eccrine acrospiroma, solid-cystic hidradenoma, and clear cell hidradenoma.2 Review of the literature revealed a female predominance (2:1 ratio) and a mean age at presentation of 37.2 years.3,4 Nodular hidradenoma presents as an asymptomatic, solitary, mobile, firm nodule with intact overlying skin. Rarely, multiple nodules may occur.3 Some tumors display ulceration and serous fluid leakage.5 They occur most commonly on the scalp, face, and upper extremities with an average size of 2 cm.3 Rapid growth of the tumor may signal a malignant change.6
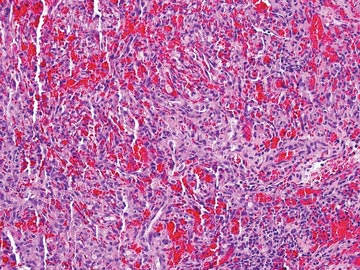
Histopathology reveals a lobulated, circumscribed, symmetrical tumor with dermal nests of epithelial cells that are polygonal with eosinophilic cytoplasm forming ductlike spaces (Figure). However, clear cell changes and squamous differentiation may be prominent features. Cystic spaces may result from tumor cell degeneration. Most tumors are encased by collagenous fibrous tissue and rarely have epidermal attachments.3
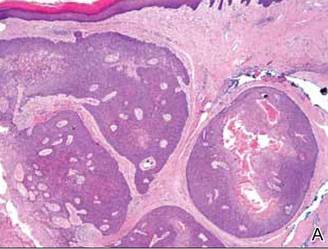
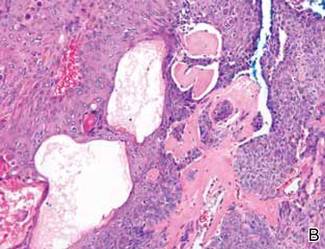
Anastomosing aggregates of squamous cells forming ductlike spaces were viewed on low-power magnification (A)(H&E, original magnification ×10). On higher power there were ductlike spaces and eosinophilic hyalinized stroma entrapped by the bland-appearing squamous proliferation (B)(H&E, original magnification ×20). |
Nodular hidradenoma traditionally has been considered to be of eccrine origin, but more recent literature indicates that the majority of NHs are of apocrine origin. Histologically, apocrine tumors display eosinophilic secretion, mucinous epithelium, squamous or sebaceous differentiation, and decapitation secretion, whereas eccrine tumors are identified by their lack of specific features.3
Nodular hidradenoma may recur after excision. Malignant transformation is rare. In one review, 6.7% (6/89) of NHs were malignant, characterized by abnormal mitoses, nuclear atypia, and necrosis.4 Malignant NH or nodular hidradenocarcinoma behaves aggressively with up to an 86% local recurrence and 60% rate of metastasis within 2 years.6 Survival time is inversely proportional to the size of the tumor and is generally poor, with a 5-year disease-free survival of less than 30%.6,7
Treatment of NH is achieved through primary excision or Mohs micrographic surgery; however, treatment of nodular hidradenocarcinoma is controversial and typically begins with wide local excision but may involve lymph node dissection if necessary. Use of adjuvant chemotherapy and radiation therapy for metastases warrants more clinical studies, as it is a rare occurrence.6 Our patient planned to undergo a total excision of the benign nodule once she healed from the biopsy; however, she was lost to follow-up, as she moved out of state.
1. Lui Y. The histogenesis of clear cell papillary carcinoma of the skin. Am J Pathol. 1949;25:93-103.
2. Obaidat NA, Khaled OA, Ghazarian D. Skin adnexal neoplasms–part 2: an approach to tumours of cutaneous sweat glands. J Clin Pathol. 2007;60:145-159.
3. Nandeesh BN, Rajalakshmi T. A study of histopathologic spectrum of nodular hidradenoma. Am J Dermatopathol. 2012;34:461-470.
4. Hernández-Pérez E, Cestoni-Parducci R. Nodular hidradenoma and hidradenocarcinoma: a 10-year review. J Am Acad Dermatol. 1985;12:15-20.
5. Sirinoglu H, Celebiler O. Benign nodular hidradenoma of the face. J Craniofac Surg. 2011;22:750-751.
6. Souvatzidis P, Sbano P, Mandato F, et al. Malignant nodular hidradenoma of the skin: report of seven cases. J Eur Acad Dermatol Venereol. 2008;22:549-554.
7. Ko CJ, Cochran AJ, Eng W, et al. Hidradenocarcinoma: a histological and immunohistochemical study. J Cutan Pathol. 2006;33:726-730.
The Diagnosis: Nodular Hidradenoma
Nodular hidradenomas (NHs) are rare benign cutaneous adnexal neoplasms first described in 1949 as clear cell papillary carcinomas.1 Since then, various terms have been used to describe this entity, such as eccrine acrospiroma, solid-cystic hidradenoma, and clear cell hidradenoma.2 Review of the literature revealed a female predominance (2:1 ratio) and a mean age at presentation of 37.2 years.3,4 Nodular hidradenoma presents as an asymptomatic, solitary, mobile, firm nodule with intact overlying skin. Rarely, multiple nodules may occur.3 Some tumors display ulceration and serous fluid leakage.5 They occur most commonly on the scalp, face, and upper extremities with an average size of 2 cm.3 Rapid growth of the tumor may signal a malignant change.6
Histopathology reveals a lobulated, circumscribed, symmetrical tumor with dermal nests of epithelial cells that are polygonal with eosinophilic cytoplasm forming ductlike spaces (Figure). However, clear cell changes and squamous differentiation may be prominent features. Cystic spaces may result from tumor cell degeneration. Most tumors are encased by collagenous fibrous tissue and rarely have epidermal attachments.3
|
Anastomosing aggregates of squamous cells forming ductlike spaces were viewed on low-power magnification (A)(H&E, original magnification ×10). On higher power there were ductlike spaces and eosinophilic hyalinized stroma entrapped by the bland-appearing squamous proliferation (B)(H&E, original magnification ×20). |
Nodular hidradenoma traditionally has been considered to be of eccrine origin, but more recent literature indicates that the majority of NHs are of apocrine origin. Histologically, apocrine tumors display eosinophilic secretion, mucinous epithelium, squamous or sebaceous differentiation, and decapitation secretion, whereas eccrine tumors are identified by their lack of specific features.3
Nodular hidradenoma may recur after excision. Malignant transformation is rare. In one review, 6.7% (6/89) of NHs were malignant, characterized by abnormal mitoses, nuclear atypia, and necrosis.4 Malignant NH or nodular hidradenocarcinoma behaves aggressively with up to an 86% local recurrence and 60% rate of metastasis within 2 years.6 Survival time is inversely proportional to the size of the tumor and is generally poor, with a 5-year disease-free survival of less than 30%.6,7
Treatment of NH is achieved through primary excision or Mohs micrographic surgery; however, treatment of nodular hidradenocarcinoma is controversial and typically begins with wide local excision but may involve lymph node dissection if necessary. Use of adjuvant chemotherapy and radiation therapy for metastases warrants more clinical studies, as it is a rare occurrence.6 Our patient planned to undergo a total excision of the benign nodule once she healed from the biopsy; however, she was lost to follow-up, as she moved out of state.
The Diagnosis: Nodular Hidradenoma
Nodular hidradenomas (NHs) are rare benign cutaneous adnexal neoplasms first described in 1949 as clear cell papillary carcinomas.1 Since then, various terms have been used to describe this entity, such as eccrine acrospiroma, solid-cystic hidradenoma, and clear cell hidradenoma.2 Review of the literature revealed a female predominance (2:1 ratio) and a mean age at presentation of 37.2 years.3,4 Nodular hidradenoma presents as an asymptomatic, solitary, mobile, firm nodule with intact overlying skin. Rarely, multiple nodules may occur.3 Some tumors display ulceration and serous fluid leakage.5 They occur most commonly on the scalp, face, and upper extremities with an average size of 2 cm.3 Rapid growth of the tumor may signal a malignant change.6
Histopathology reveals a lobulated, circumscribed, symmetrical tumor with dermal nests of epithelial cells that are polygonal with eosinophilic cytoplasm forming ductlike spaces (Figure). However, clear cell changes and squamous differentiation may be prominent features. Cystic spaces may result from tumor cell degeneration. Most tumors are encased by collagenous fibrous tissue and rarely have epidermal attachments.3
|
Anastomosing aggregates of squamous cells forming ductlike spaces were viewed on low-power magnification (A)(H&E, original magnification ×10). On higher power there were ductlike spaces and eosinophilic hyalinized stroma entrapped by the bland-appearing squamous proliferation (B)(H&E, original magnification ×20). |
Nodular hidradenoma traditionally has been considered to be of eccrine origin, but more recent literature indicates that the majority of NHs are of apocrine origin. Histologically, apocrine tumors display eosinophilic secretion, mucinous epithelium, squamous or sebaceous differentiation, and decapitation secretion, whereas eccrine tumors are identified by their lack of specific features.3
Nodular hidradenoma may recur after excision. Malignant transformation is rare. In one review, 6.7% (6/89) of NHs were malignant, characterized by abnormal mitoses, nuclear atypia, and necrosis.4 Malignant NH or nodular hidradenocarcinoma behaves aggressively with up to an 86% local recurrence and 60% rate of metastasis within 2 years.6 Survival time is inversely proportional to the size of the tumor and is generally poor, with a 5-year disease-free survival of less than 30%.6,7
Treatment of NH is achieved through primary excision or Mohs micrographic surgery; however, treatment of nodular hidradenocarcinoma is controversial and typically begins with wide local excision but may involve lymph node dissection if necessary. Use of adjuvant chemotherapy and radiation therapy for metastases warrants more clinical studies, as it is a rare occurrence.6 Our patient planned to undergo a total excision of the benign nodule once she healed from the biopsy; however, she was lost to follow-up, as she moved out of state.
1. Lui Y. The histogenesis of clear cell papillary carcinoma of the skin. Am J Pathol. 1949;25:93-103.
2. Obaidat NA, Khaled OA, Ghazarian D. Skin adnexal neoplasms–part 2: an approach to tumours of cutaneous sweat glands. J Clin Pathol. 2007;60:145-159.
3. Nandeesh BN, Rajalakshmi T. A study of histopathologic spectrum of nodular hidradenoma. Am J Dermatopathol. 2012;34:461-470.
4. Hernández-Pérez E, Cestoni-Parducci R. Nodular hidradenoma and hidradenocarcinoma: a 10-year review. J Am Acad Dermatol. 1985;12:15-20.
5. Sirinoglu H, Celebiler O. Benign nodular hidradenoma of the face. J Craniofac Surg. 2011;22:750-751.
6. Souvatzidis P, Sbano P, Mandato F, et al. Malignant nodular hidradenoma of the skin: report of seven cases. J Eur Acad Dermatol Venereol. 2008;22:549-554.
7. Ko CJ, Cochran AJ, Eng W, et al. Hidradenocarcinoma: a histological and immunohistochemical study. J Cutan Pathol. 2006;33:726-730.
1. Lui Y. The histogenesis of clear cell papillary carcinoma of the skin. Am J Pathol. 1949;25:93-103.
2. Obaidat NA, Khaled OA, Ghazarian D. Skin adnexal neoplasms–part 2: an approach to tumours of cutaneous sweat glands. J Clin Pathol. 2007;60:145-159.
3. Nandeesh BN, Rajalakshmi T. A study of histopathologic spectrum of nodular hidradenoma. Am J Dermatopathol. 2012;34:461-470.
4. Hernández-Pérez E, Cestoni-Parducci R. Nodular hidradenoma and hidradenocarcinoma: a 10-year review. J Am Acad Dermatol. 1985;12:15-20.
5. Sirinoglu H, Celebiler O. Benign nodular hidradenoma of the face. J Craniofac Surg. 2011;22:750-751.
6. Souvatzidis P, Sbano P, Mandato F, et al. Malignant nodular hidradenoma of the skin: report of seven cases. J Eur Acad Dermatol Venereol. 2008;22:549-554.
7. Ko CJ, Cochran AJ, Eng W, et al. Hidradenocarcinoma: a histological and immunohistochemical study. J Cutan Pathol. 2006;33:726-730.
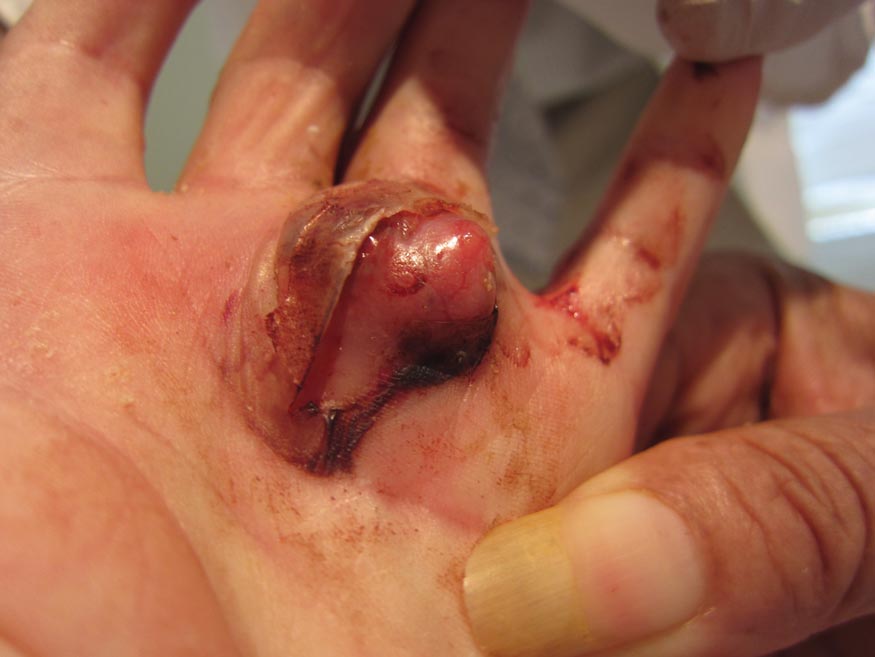
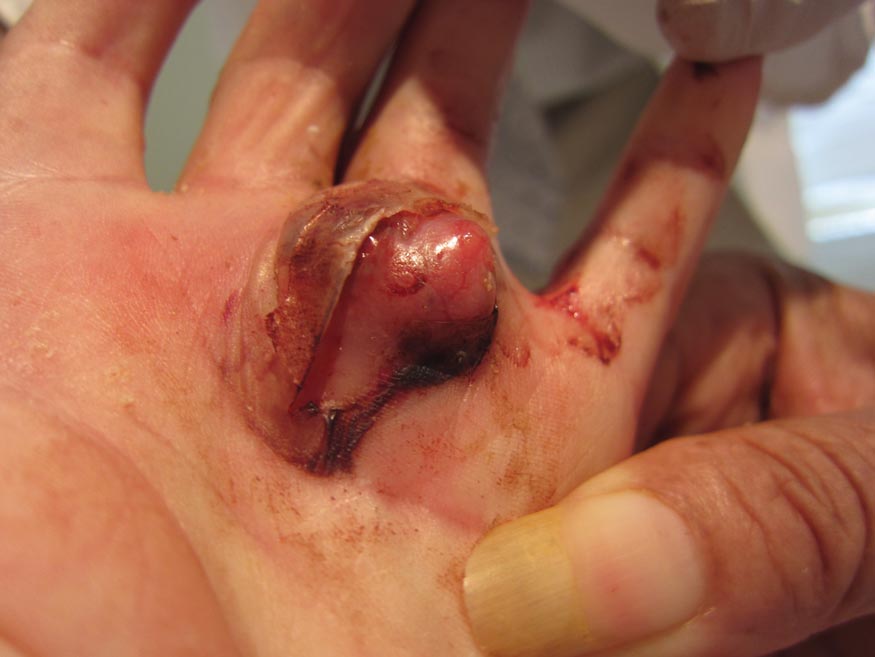
A 73-year-old woman with a history of multiple strokes with residual left-sided motor deficits and resultant left-hand contracture, type 2 diabetes mellitus, hypertension, and a remote history of treated colon cancer and breast cancer presented with hypertensive urgency and neck pain. Upon admission, the nursing staff found an “unusual growth” on the patient’s left hand. Dermatology was consulted and a 2×1.5×1.5-cm multilobulated, malodorous, slightly tender, nonfluctuant, gelatinous, mobile, cystic nodule overlying the fourth metacarpal palmar head was examined. The patient reported the lesion was present for more than a year. Imaging was pursued, but radiography, ultrasonography, and magnetic resonance imaging could not be performed adequately due to the patient’s severe contracture. Given the extensive differential diagnoses, an orthopedic hand surgeon performed a large incisional biopsy to obtain tissue diagnosis.
Non–HIV-Related Kaposi Sarcoma in 2 Hispanic Patients Arising in the Setting of Chronic Venous Insufficiency
Kaposi sarcoma (KS) is a vascular neoplasm associated with human herpesvirus 8 (HHV-8) infection that is conventionally divided into 4 clinical variants: classic, African, transplant associated, and AIDS related. In the United States, classic KS predominantly is observed in elderly white men of Ashkenazi Jewish or Mediterranean descent.1
The clinical and histological changes seen in KS can be confused with those from chronic venous insufficiency. Both conditions tend to present with purple-colored patches, plaques, or nodules on the lower extremities. Histologically, both KS and chronic venous insufficiency are characterized by blood vessel and endothelial cell proliferation in the papillary dermis, red blood cell extravasation, and hemosiderin deposition.2 Nevertheless, KS can be diagnosed based on the presence of neoplastic spindle-shaped cells and positive immunostaining for HHV-8 antigen.3
We describe the cases of 2 elderly Hispanic patients in the United States with no history of human immunodeficiency virus (HIV), immunosuppression, or travel to the Mediterranean region who presented with erythematous to violaceous papules, plaques, and nodules on the distal lower extremities in the setting of chronic venous insufficiency. We review the relationship between KS and chronic venous insufficiency and suggest that these presentations may represent a distinct clinical variant of KS.
Case Reports
Patient 1
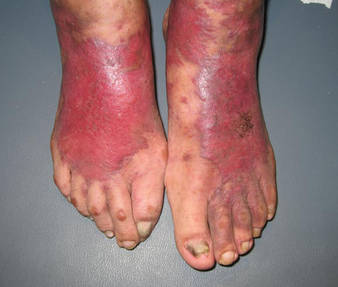
An 83-year-old Hispanic woman with a history of hypertension, atrial fibrillation, and chronic venous insufficiency presented with a chronic painful violaceous eruption on the lower legs of 3 years’ duration. The patient reported that the erythematous patches, which she described as bruiselike, originally developed 3 years prior after starting warfarin therapy for atrial fibrillation. At that time, a biopsy indicated findings of pigmented purpuric dermatosis and negative immunostaining for HHV-8. Her condition had worsened over the last 6 months with the development of tender eroded plaques on the dorsal aspects of the feet (Figure 1) and purple-brown patches and plaques on the legs. Prior treatment with topical corticosteroids and a short course of prednisone was unsuccessful. Of note, the patient had no history of immunosuppression or HIV and was not of Mediterranean or African descent.
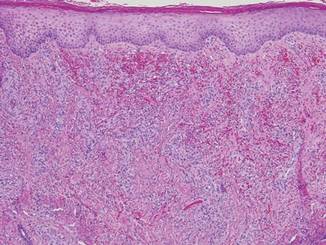
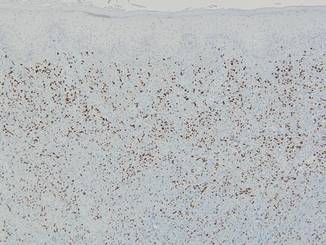
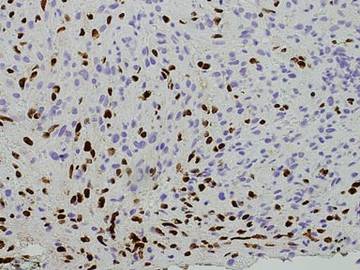
Punch biopsies from the left dorsal foot and left lower leg revealed dermal fibrosis, a proliferation of small blood vessels with plump endothelial cells, and foci of spindled endothelial cells with narrow slitlike spaces containing erythrocytes (Figure 2). There also were extravasated erythrocytes and a sparse inflammatory cell infiltrate comprised of lymphocytes, eosinophils, neutrophils, and plasma cells. Perls Prussian blue stain highlighted numerous siderophages scattered in the dermis. Based on these findings, immunostaining for HHV-8 was performed and highlighted reactivity of the spindled cells (Figure 3). The patient was diagnosed with KS in the setting of chronic venous insufficiency.
|
Patient 2
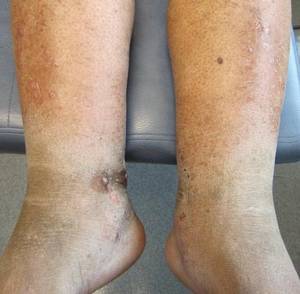
A 67-year-old Hispanic man with a history of diabetes mellitus presented with 10 asymptomatic purple papules and hyperkeratotic and hyperpigmented plaques on the distal aspect of the legs of 1 year’s duration in the setting of chronic venous insufficiency (Figure 4). The patient had no history of immunosuppression or HIV and was not of Mediterranean or African descent. An excisional biopsy from the left fourth toe revealed a cellular dermal vascular proliferation associated with numerous small slitlike vessels and scattered dilated blood vessels with prominent hemorrhage and surrounding dermal fibrosis (Figure 5). The lesion was markedly cellular with nuclear atypia. Many cells showed enlarged oval- to spindle-shaped hyperchromatic nuclei, some with prominent nucleoli and scant amounts of eosinophilic cytoplasm. Although the differential diagnosis included an unusual cellular pyogenic granuloma with atypia in the setting of chronic venous insufficiency, the degree of cellularity and presence of spindled cells associated with slitlike vessels were more consistent with a pyogenic granulomalike variant of KS. This variant is rare but has previously been reported.4 Many of the tumor nucleoli stained strongly for HHV-8, which is a diagnostic finding of KS (Figure 6). The patient subsequently was diagnosed with KS.
Comment
These 2 cases represent unusual clinical presentations of KS in Hispanic patients with no known risk factors for KS. In patient 1, an initial skin biopsy prompted HHV-8 testing due to suspicion for KS. At that time, HHV-8 was negative, perhaps because of technical deficiencies in the staining protocol; alternatively, the patient may have subsequently developed KS. Both patients had known chronic venous insufficiency. However, biopsies revealed spindle-shaped cells forming clefts and positivity for HHV-8. We propose that these cases may represent an additional clinical variant of KS, chronic venous insufficiency–associated KS.
If similar presentations of KS are identified, studies will need to be done to uncover the specific risk factors involved. Human herpesvirus 8 is not sufficient for the development of KS on its own, as oncogenesis of KS requires immunodeficiency or an additional environmental factor such as diabetes mellitus.5 Through impaired microvascular circulation and the release of hypoxia-inducible factor 1a, diabetes can promote KS-herpesvirus replication. Therefore, the risk for KS is increased in individuals with diabetes regardless of a negative history of immunodeficiency, which may have been the case in patient 2.
Our cases suggest that chronic venous insufficiency may be another factor that predisposes immunocompetent individuals to KS. Chronic venous insufficiency can cause hypoxia, promoting the release of cytokines and angiogenic factors responsible for the formation of vascular tumors such as KS.6 Once present, KS can worsen preexisting stasis dermatitis by compressing the external lymphatics and exacerbating lymphedema.7 Stasis dermatitis and KS may be part of a self-perpetuating cycle that involves obstruction due to secondary lymphadenopathy, the development of lymphedema, and the release of cytokines and growth factors that lead to further vascular proliferation.8
| 
|
In summary, we present 2 cases of non–HIV-related KS that may represent an additional clinical variant of KS that mimics and/or arises in chronic venous insufficiency and appears as papules and plaques in elderly patients who are Hispanic, immunocompetent, and HIV negative. We suggest including KS in the differential diagnosis for chronic venous insufficiency, especially in cases with an unusual clinical appearance or course. In these cases, skin biopsy with HHV-8 testing may be warranted.
Acknowledgment
The authors would like to acknowledge William Putnam, BFA, New York, New York, for his assistance with the figures.
1. Hiatt KM, Nelson AM, Lichy JH, et al. Classic Kaposi sarcoma in the United States over the last two decades: a clinicopathologic and molecular study of 438 non-HIV-related Kaposi Sarcoma patients with comparison to HIV-related Kaposi Sarcoma [published online ahead of print March 28, 2008]. Mod Pathol. 2008;21:572-582.
2. Weaver J, Billings SD. Initial presentation of stasis dermatitis mimicking solitary lesions: a previously unrecognized clinical scenario. J Am Acad Dermatol. 2009;61:1028-1032.
3. Bulat V, Lugovic´ L, Šitum M, et al. Acroangiodermatitis (pseudo-Kaposi sarcoma) as part of chronic venous insufficiency. Acta Clin Croat. 2007;46:273-277.
4. Urquhart JL, Uzieblo A, Kohler S. Detection of HHV-8 in pyogenic granuloma-like Kaposi sarcoma. Am J Dermatopathol. 2006;28:317-321.
5. Anderson LA, Lauria C, Romano N, et al. Risk factors for classical Kaposi sarcoma in a population-based case-control study in Sicily. Cancer Epidemiol Biomarkers Prev. 2008;17:3435-3443.
6. Lunardi-Iskandar Y, Bryant JL, Zeman RA, et al. Tumorigenesis and metastasis of neoplastic Kaposi’s sarcoma cell line in immunodeficient mice blocked by a human pregnancy hormone. Nature. 1995;375:64-68.
7. Allen PJ, Gillespie DL, Redfield RR, et al. Lower extremity lymphedema caused by acquired immune deficiency syndrome-related Kaposi’s sarcoma: case report and review of the literature. J Vasc Surg. 1995;22:178-181.
8. Ramdial PK, Chetty R, Singh B, et al. Lymphedematous HIV-associated Kaposi’s sarcoma. J Cutan Pathol. 2006;33:474-481.
Kaposi sarcoma (KS) is a vascular neoplasm associated with human herpesvirus 8 (HHV-8) infection that is conventionally divided into 4 clinical variants: classic, African, transplant associated, and AIDS related. In the United States, classic KS predominantly is observed in elderly white men of Ashkenazi Jewish or Mediterranean descent.1
The clinical and histological changes seen in KS can be confused with those from chronic venous insufficiency. Both conditions tend to present with purple-colored patches, plaques, or nodules on the lower extremities. Histologically, both KS and chronic venous insufficiency are characterized by blood vessel and endothelial cell proliferation in the papillary dermis, red blood cell extravasation, and hemosiderin deposition.2 Nevertheless, KS can be diagnosed based on the presence of neoplastic spindle-shaped cells and positive immunostaining for HHV-8 antigen.3
We describe the cases of 2 elderly Hispanic patients in the United States with no history of human immunodeficiency virus (HIV), immunosuppression, or travel to the Mediterranean region who presented with erythematous to violaceous papules, plaques, and nodules on the distal lower extremities in the setting of chronic venous insufficiency. We review the relationship between KS and chronic venous insufficiency and suggest that these presentations may represent a distinct clinical variant of KS.
Case Reports
Patient 1
An 83-year-old Hispanic woman with a history of hypertension, atrial fibrillation, and chronic venous insufficiency presented with a chronic painful violaceous eruption on the lower legs of 3 years’ duration. The patient reported that the erythematous patches, which she described as bruiselike, originally developed 3 years prior after starting warfarin therapy for atrial fibrillation. At that time, a biopsy indicated findings of pigmented purpuric dermatosis and negative immunostaining for HHV-8. Her condition had worsened over the last 6 months with the development of tender eroded plaques on the dorsal aspects of the feet (Figure 1) and purple-brown patches and plaques on the legs. Prior treatment with topical corticosteroids and a short course of prednisone was unsuccessful. Of note, the patient had no history of immunosuppression or HIV and was not of Mediterranean or African descent.
Punch biopsies from the left dorsal foot and left lower leg revealed dermal fibrosis, a proliferation of small blood vessels with plump endothelial cells, and foci of spindled endothelial cells with narrow slitlike spaces containing erythrocytes (Figure 2). There also were extravasated erythrocytes and a sparse inflammatory cell infiltrate comprised of lymphocytes, eosinophils, neutrophils, and plasma cells. Perls Prussian blue stain highlighted numerous siderophages scattered in the dermis. Based on these findings, immunostaining for HHV-8 was performed and highlighted reactivity of the spindled cells (Figure 3). The patient was diagnosed with KS in the setting of chronic venous insufficiency.
|
|
Patient 2
A 67-year-old Hispanic man with a history of diabetes mellitus presented with 10 asymptomatic purple papules and hyperkeratotic and hyperpigmented plaques on the distal aspect of the legs of 1 year’s duration in the setting of chronic venous insufficiency (Figure 4). The patient had no history of immunosuppression or HIV and was not of Mediterranean or African descent. An excisional biopsy from the left fourth toe revealed a cellular dermal vascular proliferation associated with numerous small slitlike vessels and scattered dilated blood vessels with prominent hemorrhage and surrounding dermal fibrosis (Figure 5). The lesion was markedly cellular with nuclear atypia. Many cells showed enlarged oval- to spindle-shaped hyperchromatic nuclei, some with prominent nucleoli and scant amounts of eosinophilic cytoplasm. Although the differential diagnosis included an unusual cellular pyogenic granuloma with atypia in the setting of chronic venous insufficiency, the degree of cellularity and presence of spindled cells associated with slitlike vessels were more consistent with a pyogenic granulomalike variant of KS. This variant is rare but has previously been reported.4 Many of the tumor nucleoli stained strongly for HHV-8, which is a diagnostic finding of KS (Figure 6). The patient subsequently was diagnosed with KS.
Comment
These 2 cases represent unusual clinical presentations of KS in Hispanic patients with no known risk factors for KS. In patient 1, an initial skin biopsy prompted HHV-8 testing due to suspicion for KS. At that time, HHV-8 was negative, perhaps because of technical deficiencies in the staining protocol; alternatively, the patient may have subsequently developed KS. Both patients had known chronic venous insufficiency. However, biopsies revealed spindle-shaped cells forming clefts and positivity for HHV-8. We propose that these cases may represent an additional clinical variant of KS, chronic venous insufficiency–associated KS.
If similar presentations of KS are identified, studies will need to be done to uncover the specific risk factors involved. Human herpesvirus 8 is not sufficient for the development of KS on its own, as oncogenesis of KS requires immunodeficiency or an additional environmental factor such as diabetes mellitus.5 Through impaired microvascular circulation and the release of hypoxia-inducible factor 1a, diabetes can promote KS-herpesvirus replication. Therefore, the risk for KS is increased in individuals with diabetes regardless of a negative history of immunodeficiency, which may have been the case in patient 2.
Our cases suggest that chronic venous insufficiency may be another factor that predisposes immunocompetent individuals to KS. Chronic venous insufficiency can cause hypoxia, promoting the release of cytokines and angiogenic factors responsible for the formation of vascular tumors such as KS.6 Once present, KS can worsen preexisting stasis dermatitis by compressing the external lymphatics and exacerbating lymphedema.7 Stasis dermatitis and KS may be part of a self-perpetuating cycle that involves obstruction due to secondary lymphadenopathy, the development of lymphedema, and the release of cytokines and growth factors that lead to further vascular proliferation.8
|
|
|
In summary, we present 2 cases of non–HIV-related KS that may represent an additional clinical variant of KS that mimics and/or arises in chronic venous insufficiency and appears as papules and plaques in elderly patients who are Hispanic, immunocompetent, and HIV negative. We suggest including KS in the differential diagnosis for chronic venous insufficiency, especially in cases with an unusual clinical appearance or course. In these cases, skin biopsy with HHV-8 testing may be warranted.
Acknowledgment
The authors would like to acknowledge William Putnam, BFA, New York, New York, for his assistance with the figures.
Kaposi sarcoma (KS) is a vascular neoplasm associated with human herpesvirus 8 (HHV-8) infection that is conventionally divided into 4 clinical variants: classic, African, transplant associated, and AIDS related. In the United States, classic KS predominantly is observed in elderly white men of Ashkenazi Jewish or Mediterranean descent.1
The clinical and histological changes seen in KS can be confused with those from chronic venous insufficiency. Both conditions tend to present with purple-colored patches, plaques, or nodules on the lower extremities. Histologically, both KS and chronic venous insufficiency are characterized by blood vessel and endothelial cell proliferation in the papillary dermis, red blood cell extravasation, and hemosiderin deposition.2 Nevertheless, KS can be diagnosed based on the presence of neoplastic spindle-shaped cells and positive immunostaining for HHV-8 antigen.3
We describe the cases of 2 elderly Hispanic patients in the United States with no history of human immunodeficiency virus (HIV), immunosuppression, or travel to the Mediterranean region who presented with erythematous to violaceous papules, plaques, and nodules on the distal lower extremities in the setting of chronic venous insufficiency. We review the relationship between KS and chronic venous insufficiency and suggest that these presentations may represent a distinct clinical variant of KS.
Case Reports
Patient 1
An 83-year-old Hispanic woman with a history of hypertension, atrial fibrillation, and chronic venous insufficiency presented with a chronic painful violaceous eruption on the lower legs of 3 years’ duration. The patient reported that the erythematous patches, which she described as bruiselike, originally developed 3 years prior after starting warfarin therapy for atrial fibrillation. At that time, a biopsy indicated findings of pigmented purpuric dermatosis and negative immunostaining for HHV-8. Her condition had worsened over the last 6 months with the development of tender eroded plaques on the dorsal aspects of the feet (Figure 1) and purple-brown patches and plaques on the legs. Prior treatment with topical corticosteroids and a short course of prednisone was unsuccessful. Of note, the patient had no history of immunosuppression or HIV and was not of Mediterranean or African descent.
Punch biopsies from the left dorsal foot and left lower leg revealed dermal fibrosis, a proliferation of small blood vessels with plump endothelial cells, and foci of spindled endothelial cells with narrow slitlike spaces containing erythrocytes (Figure 2). There also were extravasated erythrocytes and a sparse inflammatory cell infiltrate comprised of lymphocytes, eosinophils, neutrophils, and plasma cells. Perls Prussian blue stain highlighted numerous siderophages scattered in the dermis. Based on these findings, immunostaining for HHV-8 was performed and highlighted reactivity of the spindled cells (Figure 3). The patient was diagnosed with KS in the setting of chronic venous insufficiency.
|
|
Patient 2
A 67-year-old Hispanic man with a history of diabetes mellitus presented with 10 asymptomatic purple papules and hyperkeratotic and hyperpigmented plaques on the distal aspect of the legs of 1 year’s duration in the setting of chronic venous insufficiency (Figure 4). The patient had no history of immunosuppression or HIV and was not of Mediterranean or African descent. An excisional biopsy from the left fourth toe revealed a cellular dermal vascular proliferation associated with numerous small slitlike vessels and scattered dilated blood vessels with prominent hemorrhage and surrounding dermal fibrosis (Figure 5). The lesion was markedly cellular with nuclear atypia. Many cells showed enlarged oval- to spindle-shaped hyperchromatic nuclei, some with prominent nucleoli and scant amounts of eosinophilic cytoplasm. Although the differential diagnosis included an unusual cellular pyogenic granuloma with atypia in the setting of chronic venous insufficiency, the degree of cellularity and presence of spindled cells associated with slitlike vessels were more consistent with a pyogenic granulomalike variant of KS. This variant is rare but has previously been reported.4 Many of the tumor nucleoli stained strongly for HHV-8, which is a diagnostic finding of KS (Figure 6). The patient subsequently was diagnosed with KS.
Comment
These 2 cases represent unusual clinical presentations of KS in Hispanic patients with no known risk factors for KS. In patient 1, an initial skin biopsy prompted HHV-8 testing due to suspicion for KS. At that time, HHV-8 was negative, perhaps because of technical deficiencies in the staining protocol; alternatively, the patient may have subsequently developed KS. Both patients had known chronic venous insufficiency. However, biopsies revealed spindle-shaped cells forming clefts and positivity for HHV-8. We propose that these cases may represent an additional clinical variant of KS, chronic venous insufficiency–associated KS.
If similar presentations of KS are identified, studies will need to be done to uncover the specific risk factors involved. Human herpesvirus 8 is not sufficient for the development of KS on its own, as oncogenesis of KS requires immunodeficiency or an additional environmental factor such as diabetes mellitus.5 Through impaired microvascular circulation and the release of hypoxia-inducible factor 1a, diabetes can promote KS-herpesvirus replication. Therefore, the risk for KS is increased in individuals with diabetes regardless of a negative history of immunodeficiency, which may have been the case in patient 2.
Our cases suggest that chronic venous insufficiency may be another factor that predisposes immunocompetent individuals to KS. Chronic venous insufficiency can cause hypoxia, promoting the release of cytokines and angiogenic factors responsible for the formation of vascular tumors such as KS.6 Once present, KS can worsen preexisting stasis dermatitis by compressing the external lymphatics and exacerbating lymphedema.7 Stasis dermatitis and KS may be part of a self-perpetuating cycle that involves obstruction due to secondary lymphadenopathy, the development of lymphedema, and the release of cytokines and growth factors that lead to further vascular proliferation.8
|
|
|
In summary, we present 2 cases of non–HIV-related KS that may represent an additional clinical variant of KS that mimics and/or arises in chronic venous insufficiency and appears as papules and plaques in elderly patients who are Hispanic, immunocompetent, and HIV negative. We suggest including KS in the differential diagnosis for chronic venous insufficiency, especially in cases with an unusual clinical appearance or course. In these cases, skin biopsy with HHV-8 testing may be warranted.
Acknowledgment
The authors would like to acknowledge William Putnam, BFA, New York, New York, for his assistance with the figures.
1. Hiatt KM, Nelson AM, Lichy JH, et al. Classic Kaposi sarcoma in the United States over the last two decades: a clinicopathologic and molecular study of 438 non-HIV-related Kaposi Sarcoma patients with comparison to HIV-related Kaposi Sarcoma [published online ahead of print March 28, 2008]. Mod Pathol. 2008;21:572-582.
2. Weaver J, Billings SD. Initial presentation of stasis dermatitis mimicking solitary lesions: a previously unrecognized clinical scenario. J Am Acad Dermatol. 2009;61:1028-1032.
3. Bulat V, Lugovic´ L, Šitum M, et al. Acroangiodermatitis (pseudo-Kaposi sarcoma) as part of chronic venous insufficiency. Acta Clin Croat. 2007;46:273-277.
4. Urquhart JL, Uzieblo A, Kohler S. Detection of HHV-8 in pyogenic granuloma-like Kaposi sarcoma. Am J Dermatopathol. 2006;28:317-321.
5. Anderson LA, Lauria C, Romano N, et al. Risk factors for classical Kaposi sarcoma in a population-based case-control study in Sicily. Cancer Epidemiol Biomarkers Prev. 2008;17:3435-3443.
6. Lunardi-Iskandar Y, Bryant JL, Zeman RA, et al. Tumorigenesis and metastasis of neoplastic Kaposi’s sarcoma cell line in immunodeficient mice blocked by a human pregnancy hormone. Nature. 1995;375:64-68.
7. Allen PJ, Gillespie DL, Redfield RR, et al. Lower extremity lymphedema caused by acquired immune deficiency syndrome-related Kaposi’s sarcoma: case report and review of the literature. J Vasc Surg. 1995;22:178-181.
8. Ramdial PK, Chetty R, Singh B, et al. Lymphedematous HIV-associated Kaposi’s sarcoma. J Cutan Pathol. 2006;33:474-481.
1. Hiatt KM, Nelson AM, Lichy JH, et al. Classic Kaposi sarcoma in the United States over the last two decades: a clinicopathologic and molecular study of 438 non-HIV-related Kaposi Sarcoma patients with comparison to HIV-related Kaposi Sarcoma [published online ahead of print March 28, 2008]. Mod Pathol. 2008;21:572-582.
2. Weaver J, Billings SD. Initial presentation of stasis dermatitis mimicking solitary lesions: a previously unrecognized clinical scenario. J Am Acad Dermatol. 2009;61:1028-1032.
3. Bulat V, Lugovic´ L, Šitum M, et al. Acroangiodermatitis (pseudo-Kaposi sarcoma) as part of chronic venous insufficiency. Acta Clin Croat. 2007;46:273-277.
4. Urquhart JL, Uzieblo A, Kohler S. Detection of HHV-8 in pyogenic granuloma-like Kaposi sarcoma. Am J Dermatopathol. 2006;28:317-321.
5. Anderson LA, Lauria C, Romano N, et al. Risk factors for classical Kaposi sarcoma in a population-based case-control study in Sicily. Cancer Epidemiol Biomarkers Prev. 2008;17:3435-3443.
6. Lunardi-Iskandar Y, Bryant JL, Zeman RA, et al. Tumorigenesis and metastasis of neoplastic Kaposi’s sarcoma cell line in immunodeficient mice blocked by a human pregnancy hormone. Nature. 1995;375:64-68.
7. Allen PJ, Gillespie DL, Redfield RR, et al. Lower extremity lymphedema caused by acquired immune deficiency syndrome-related Kaposi’s sarcoma: case report and review of the literature. J Vasc Surg. 1995;22:178-181.
8. Ramdial PK, Chetty R, Singh B, et al. Lymphedematous HIV-associated Kaposi’s sarcoma. J Cutan Pathol. 2006;33:474-481.
Practice Points
- The 4 clinical variants of Kaposi sarcoma (KS) are classic, African, transplant associated, and AIDS related.
- Human herpesvirus 8 (HHV-8) staining can be used to confirm the presence of KS.
- A subset of patients with chronic venous insufficiency with no other risk factors for KS have been found to have violaceous plaques that test positive for HHV-8.
- Chronic venous insufficiency may be a predisposing factor to KS in immunocompetent individuals and may constitute an additional clinical variant of KS.
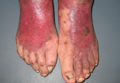
Firm Plaques and Nodules Over the Body
The Diagnosis: Pancreatic Panniculitis
The biopsy specimen revealed necrosis of the panniculus with “ghost” cells (Figure). Calcification was encountered. Changes of vasculitis were not identified and fungal organisms were not noted. The histopathologic findings supported a diagnosis of pancreatic panniculitis.

Pancreatic panniculitis has been associated with pancreatitis, pancreatic carcinoma, pancreatic pseudocysts, congenital abnormalities of the pancreas, and drug-induced pancreatitis.1 Skin lesions may herald a diagnosis of pancreatic disease in an outpatient and should prompt thorough clinical evaluation when encountered in an outpatient setting. Our patient first developed tender nodules on the left shin 2 to 3 weeks prior to presentation. She reported that her initial nodules were flesh colored but then became erythematous and tender over 1 week’s time. The patient’s history was remarkable for ovarian cancer. She had been hospitalized 2 weeks prior to presentation for abdominal pain and ascites. Imaging studies revealed a cystic lesion in the head of the pancreas. The pancreas was traumatized during a peritoneal tap. Her nodules developed shortly thereafter and were distributed on the arms, legs, back, and abdomen.
Pancreatic tumors or inflammation are thought to trigger pancreatic panniculitis by releasing enzymes. Pancreatic enzymes such as lipase are thought to play a role in the development of pancreatic panniculitis by entering the vascular system and leading to fat necrosis.2,3 Biopsy reveals necrosis of adipocytes in the center of fat lobules.4 Ghost cells result from hydrolytic activity of enzymes on the fat cells followed by calcium deposition. A report indicates that fungal infection or gout also can cause changes that mimic pancreatic panniculitis.5
Other entities in the differential diagnosis can be excluded by biopsy. Polyarteritis nodosa is a vasculitis. Although panniculitis may be seen in polyarteritis as a secondary phenomenon, lesions are centered around blood vessels and often eventuate in ulceration.6 Subcutaneous fungal infection typically reveals organisms on periodic acid–Schiff stain.7 Pyoderma gangrenosum is associated with ulceration and a neutrophilic infiltrate that is often centered around a central pilosebaceous unit in developing lesions.8 Erythema nodosum is a panniculitis in which septal inflammation predominates.9 These differential diagnoses of pancreatic panniculitis are summarized in the Table.
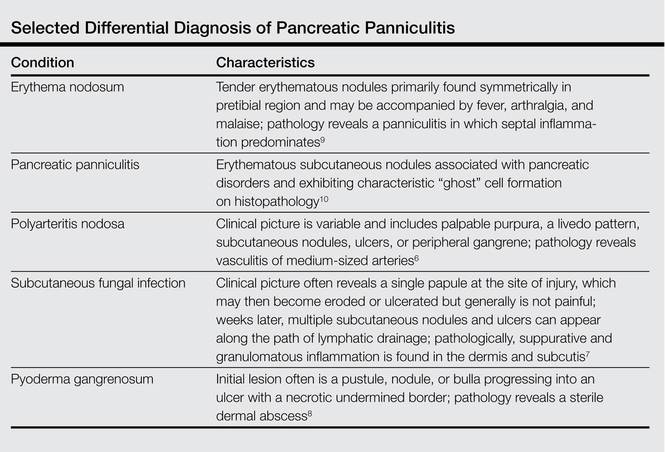
Pancreatic panniculitis can be associated with acute arthritis and inflammation of periarticular fat.10 Treatment of pancreatic panniculitis is usually focused on the underlying pancreatic disease.11,12 Our patient benefited from analgesic therapy and her lesions improved on follow-up. Clinicians encountering a patient with new tender nodules should be prompted to perform a biopsy. When histopathologic evaluation reveals ghosted adipocytes, pancreatic panniculitis should be suspected and clinical evaluation undertaken.
1. Garcia-Romero D, Vanaclocha F. Pancreatic panniculitis. Dermatol Clin. 2008;26:465-470.
2. Berman B, Conteas C, Smith B, et al. Fatal pancreatitis presenting with subcutaneous fat necrosis. J Am Acad Dermatol. 1987;17:359-364.
3. Dhawan SS, Jimenez-Acosta F, Poppiti RJ Jr, et al. Subcutaneous fat necrosis associated with pancreatitis: histochemical and electron microscopic findings. Am J Gastroenterol. 1990;85:1025-1028.
4. Cannon JR, Pitha JV, Everett MA. Subcutaneous fat necrosis in pancreatitis. J Cutan Pathol. 1979;6:501-506.
5. Requena L, Sitthinamsuwan P, Santonja C, et al. Cutaneous and mucosal mucormycosis mimicking pancreatic panniculitis and gouty panniculitis. J Am Acad Dermatol. 2012;66:975-984.
6. Grattan CEH. Polyarteritis nodosa. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 1. 3rd ed. China: Elsevier Saunders; 2012:405-407.
7. Millett CR, Halpern AV, Heymann WR. Subcutaneous mycoses. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 3rd ed. China: Elsevier Saunders; 2012:1266-1273.
8. Moschella SL, Davis MDP. Pyoderma gangrenosum. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 1. 3rd ed. China: Elsevier Saunders; 2012:427-431.
9. Patterson JW. Erythema nodosum. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 3rd ed. China: Elsevier Saunders; 2012:1641-1645.
10. Patterson JW. Pancreatic panniculitis. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 3rd ed. China: Elsevier Saunders; 2012:1649-1650.
11. Requena L, Sanchez Yus E. Panniculitis. part II. mostly lobular panniculitis. J Am Acad Dermatol. 2001;45:325-361.
12. Dahl PR, Su WP, Cullimore KC, et al. Pancreatic panniculitis. J Am Acad Dermatol. 1995;33:413-417.
The Diagnosis: Pancreatic Panniculitis
The biopsy specimen revealed necrosis of the panniculus with “ghost” cells (Figure). Calcification was encountered. Changes of vasculitis were not identified and fungal organisms were not noted. The histopathologic findings supported a diagnosis of pancreatic panniculitis.
Pancreatic panniculitis has been associated with pancreatitis, pancreatic carcinoma, pancreatic pseudocysts, congenital abnormalities of the pancreas, and drug-induced pancreatitis.1 Skin lesions may herald a diagnosis of pancreatic disease in an outpatient and should prompt thorough clinical evaluation when encountered in an outpatient setting. Our patient first developed tender nodules on the left shin 2 to 3 weeks prior to presentation. She reported that her initial nodules were flesh colored but then became erythematous and tender over 1 week’s time. The patient’s history was remarkable for ovarian cancer. She had been hospitalized 2 weeks prior to presentation for abdominal pain and ascites. Imaging studies revealed a cystic lesion in the head of the pancreas. The pancreas was traumatized during a peritoneal tap. Her nodules developed shortly thereafter and were distributed on the arms, legs, back, and abdomen.
Pancreatic tumors or inflammation are thought to trigger pancreatic panniculitis by releasing enzymes. Pancreatic enzymes such as lipase are thought to play a role in the development of pancreatic panniculitis by entering the vascular system and leading to fat necrosis.2,3 Biopsy reveals necrosis of adipocytes in the center of fat lobules.4 Ghost cells result from hydrolytic activity of enzymes on the fat cells followed by calcium deposition. A report indicates that fungal infection or gout also can cause changes that mimic pancreatic panniculitis.5
Other entities in the differential diagnosis can be excluded by biopsy. Polyarteritis nodosa is a vasculitis. Although panniculitis may be seen in polyarteritis as a secondary phenomenon, lesions are centered around blood vessels and often eventuate in ulceration.6 Subcutaneous fungal infection typically reveals organisms on periodic acid–Schiff stain.7 Pyoderma gangrenosum is associated with ulceration and a neutrophilic infiltrate that is often centered around a central pilosebaceous unit in developing lesions.8 Erythema nodosum is a panniculitis in which septal inflammation predominates.9 These differential diagnoses of pancreatic panniculitis are summarized in the Table.
Pancreatic panniculitis can be associated with acute arthritis and inflammation of periarticular fat.10 Treatment of pancreatic panniculitis is usually focused on the underlying pancreatic disease.11,12 Our patient benefited from analgesic therapy and her lesions improved on follow-up. Clinicians encountering a patient with new tender nodules should be prompted to perform a biopsy. When histopathologic evaluation reveals ghosted adipocytes, pancreatic panniculitis should be suspected and clinical evaluation undertaken.
The Diagnosis: Pancreatic Panniculitis
The biopsy specimen revealed necrosis of the panniculus with “ghost” cells (Figure). Calcification was encountered. Changes of vasculitis were not identified and fungal organisms were not noted. The histopathologic findings supported a diagnosis of pancreatic panniculitis.
Pancreatic panniculitis has been associated with pancreatitis, pancreatic carcinoma, pancreatic pseudocysts, congenital abnormalities of the pancreas, and drug-induced pancreatitis.1 Skin lesions may herald a diagnosis of pancreatic disease in an outpatient and should prompt thorough clinical evaluation when encountered in an outpatient setting. Our patient first developed tender nodules on the left shin 2 to 3 weeks prior to presentation. She reported that her initial nodules were flesh colored but then became erythematous and tender over 1 week’s time. The patient’s history was remarkable for ovarian cancer. She had been hospitalized 2 weeks prior to presentation for abdominal pain and ascites. Imaging studies revealed a cystic lesion in the head of the pancreas. The pancreas was traumatized during a peritoneal tap. Her nodules developed shortly thereafter and were distributed on the arms, legs, back, and abdomen.
Pancreatic tumors or inflammation are thought to trigger pancreatic panniculitis by releasing enzymes. Pancreatic enzymes such as lipase are thought to play a role in the development of pancreatic panniculitis by entering the vascular system and leading to fat necrosis.2,3 Biopsy reveals necrosis of adipocytes in the center of fat lobules.4 Ghost cells result from hydrolytic activity of enzymes on the fat cells followed by calcium deposition. A report indicates that fungal infection or gout also can cause changes that mimic pancreatic panniculitis.5
Other entities in the differential diagnosis can be excluded by biopsy. Polyarteritis nodosa is a vasculitis. Although panniculitis may be seen in polyarteritis as a secondary phenomenon, lesions are centered around blood vessels and often eventuate in ulceration.6 Subcutaneous fungal infection typically reveals organisms on periodic acid–Schiff stain.7 Pyoderma gangrenosum is associated with ulceration and a neutrophilic infiltrate that is often centered around a central pilosebaceous unit in developing lesions.8 Erythema nodosum is a panniculitis in which septal inflammation predominates.9 These differential diagnoses of pancreatic panniculitis are summarized in the Table.
Pancreatic panniculitis can be associated with acute arthritis and inflammation of periarticular fat.10 Treatment of pancreatic panniculitis is usually focused on the underlying pancreatic disease.11,12 Our patient benefited from analgesic therapy and her lesions improved on follow-up. Clinicians encountering a patient with new tender nodules should be prompted to perform a biopsy. When histopathologic evaluation reveals ghosted adipocytes, pancreatic panniculitis should be suspected and clinical evaluation undertaken.
1. Garcia-Romero D, Vanaclocha F. Pancreatic panniculitis. Dermatol Clin. 2008;26:465-470.
2. Berman B, Conteas C, Smith B, et al. Fatal pancreatitis presenting with subcutaneous fat necrosis. J Am Acad Dermatol. 1987;17:359-364.
3. Dhawan SS, Jimenez-Acosta F, Poppiti RJ Jr, et al. Subcutaneous fat necrosis associated with pancreatitis: histochemical and electron microscopic findings. Am J Gastroenterol. 1990;85:1025-1028.
4. Cannon JR, Pitha JV, Everett MA. Subcutaneous fat necrosis in pancreatitis. J Cutan Pathol. 1979;6:501-506.
5. Requena L, Sitthinamsuwan P, Santonja C, et al. Cutaneous and mucosal mucormycosis mimicking pancreatic panniculitis and gouty panniculitis. J Am Acad Dermatol. 2012;66:975-984.
6. Grattan CEH. Polyarteritis nodosa. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 1. 3rd ed. China: Elsevier Saunders; 2012:405-407.
7. Millett CR, Halpern AV, Heymann WR. Subcutaneous mycoses. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 3rd ed. China: Elsevier Saunders; 2012:1266-1273.
8. Moschella SL, Davis MDP. Pyoderma gangrenosum. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 1. 3rd ed. China: Elsevier Saunders; 2012:427-431.
9. Patterson JW. Erythema nodosum. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 3rd ed. China: Elsevier Saunders; 2012:1641-1645.
10. Patterson JW. Pancreatic panniculitis. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 3rd ed. China: Elsevier Saunders; 2012:1649-1650.
11. Requena L, Sanchez Yus E. Panniculitis. part II. mostly lobular panniculitis. J Am Acad Dermatol. 2001;45:325-361.
12. Dahl PR, Su WP, Cullimore KC, et al. Pancreatic panniculitis. J Am Acad Dermatol. 1995;33:413-417.
1. Garcia-Romero D, Vanaclocha F. Pancreatic panniculitis. Dermatol Clin. 2008;26:465-470.
2. Berman B, Conteas C, Smith B, et al. Fatal pancreatitis presenting with subcutaneous fat necrosis. J Am Acad Dermatol. 1987;17:359-364.
3. Dhawan SS, Jimenez-Acosta F, Poppiti RJ Jr, et al. Subcutaneous fat necrosis associated with pancreatitis: histochemical and electron microscopic findings. Am J Gastroenterol. 1990;85:1025-1028.
4. Cannon JR, Pitha JV, Everett MA. Subcutaneous fat necrosis in pancreatitis. J Cutan Pathol. 1979;6:501-506.
5. Requena L, Sitthinamsuwan P, Santonja C, et al. Cutaneous and mucosal mucormycosis mimicking pancreatic panniculitis and gouty panniculitis. J Am Acad Dermatol. 2012;66:975-984.
6. Grattan CEH. Polyarteritis nodosa. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 1. 3rd ed. China: Elsevier Saunders; 2012:405-407.
7. Millett CR, Halpern AV, Heymann WR. Subcutaneous mycoses. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 3rd ed. China: Elsevier Saunders; 2012:1266-1273.
8. Moschella SL, Davis MDP. Pyoderma gangrenosum. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 1. 3rd ed. China: Elsevier Saunders; 2012:427-431.
9. Patterson JW. Erythema nodosum. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 3rd ed. China: Elsevier Saunders; 2012:1641-1645.
10. Patterson JW. Pancreatic panniculitis. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 3rd ed. China: Elsevier Saunders; 2012:1649-1650.
11. Requena L, Sanchez Yus E. Panniculitis. part II. mostly lobular panniculitis. J Am Acad Dermatol. 2001;45:325-361.
12. Dahl PR, Su WP, Cullimore KC, et al. Pancreatic panniculitis. J Am Acad Dermatol. 1995;33:413-417.
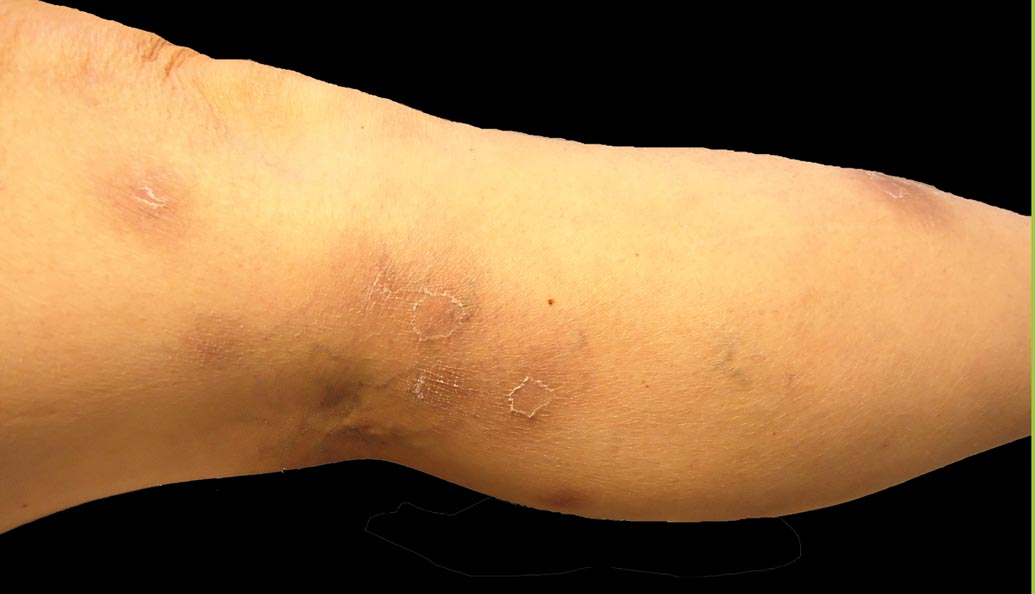
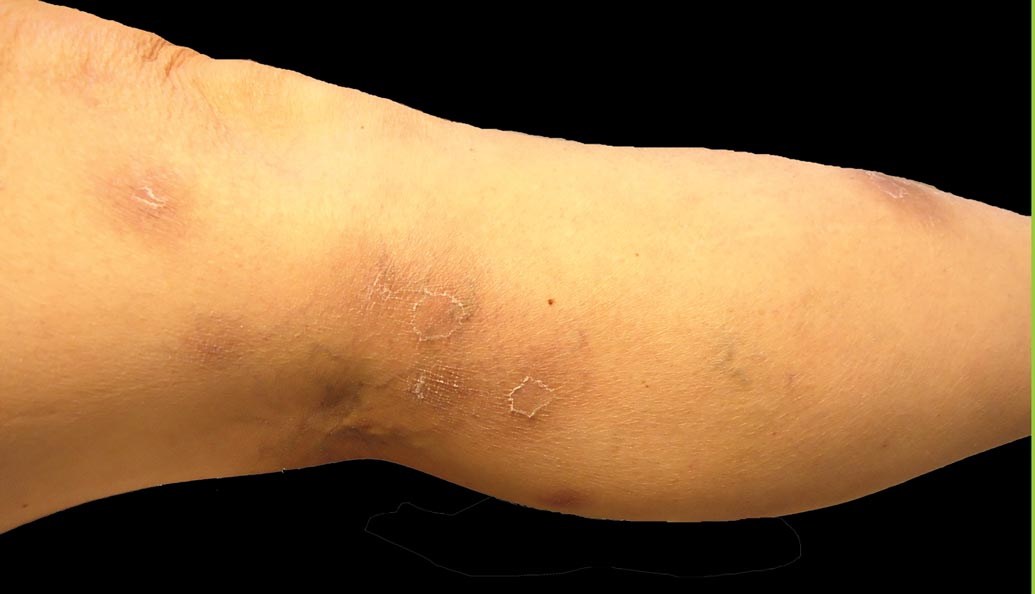
A 52-year-old woman presented with painful erythematous nodules of 2 weeks’ duration that began as a single lesion on the left shin and spread rapidly to involve the trunk, arms, and legs. A punch biopsy was performed. Pertinent history included a recent hospitalization for drainage of malignant ascites secondary to metastatic ovarian cancer. The lesions did not drain and were not pruritic. The patient did not have a history of fever, night sweats, nausea, headache, neurologic change, muscle aching, or recent weight loss.
Imiquimod-Induced Subacute Cutaneous Lupus Erythematosus–like Changes
Drug-induced lupus accounts for up to 10% of lupus erythematosus cases. Subacute cutaneous lupus erythematosus (SCLE) is a distinct clinical variant of lupus erythematosus that typically presents as annular, often scaly, erythematous plaques in photodistributed areas. Subacute cutaneous lupus erythematosus has been reported in association with multiple systemic medications including docetaxel, terbinafine, leuprolide acetate, etanercept, and efalizumab1-5; however, the induction of SCLE by topical agents has not been widely reported. We report a case of local induction of lesions that clinically and histologically resembled SCLE in a 50-year-old woman following treatment with topical imiquimod.
Case Report
A 50-year-old woman presented with an adverse inflammatory reaction on the right side of the upper chest secondary to application of topical imiquimod. Prior to the current presentation the patient was diagnosed with a biopsy-proven superficial basal cell carcinoma (BCC) on the sun-exposed area of the upper right breast, and topical imiquimod therapy was initiated. Several days after starting treatment, the patient began applying imiquimod on areas of clinically normal skin on the upper chest as advised by her dermatologist. After 7 to 10 days of application the patient reported intense erythema, pain, and crusting in the treated area on the right side of the upper chest. She also experienced systemic symptoms including fatigue, arthralgia, malaise, and fever.
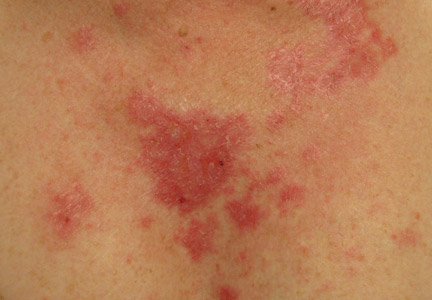
Clinical examination revealed erythematous to violaceous annular and polycyclic plaques on the upper chest (Figure 1). Erythema and scaling were noted at the site of the superficial BCC. A biopsy showed a superficial and mid-dermal perivascular and periadnexal lymphocytic infiltrate with overlying vacuolar interface dermatitis and scattered necrotic keratinocytes (Figures 2A and 2B). There was slightly increased dermal edema and mucin. Anti-CD123 immunohistochemical staining revealed nodular aggregates of plasmacytoid dendritic cells (pDCs) accompanying lymphocytes around dermal vessels and adnexa (Figure 2C). The patient’s family history was remarkable for SCLE in her daughter. Antinuclear antibody, anti-Ro (Sjögren syndrome antigen A), and anti-La (Sjögren syndrome antigen B) were negative. On follow-up examination 1 month after discontinuation of imiquimod, the patient’s skin lesions had completely cleared. Two years later, the patient continued to be free of skin lesions.
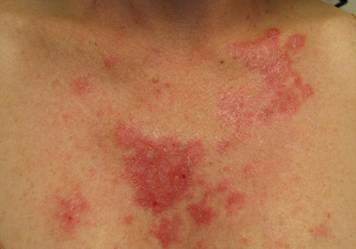
Figure 1. Annular plaques on the upper chest with overlying scale and focal central clearing. 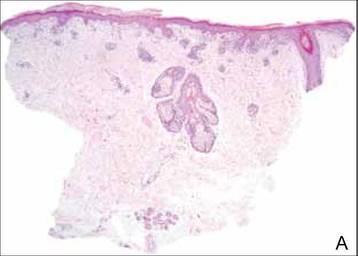
| ||
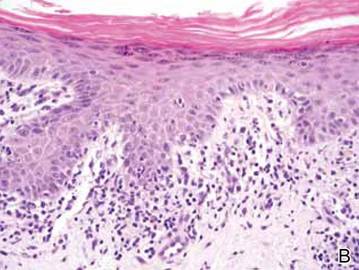
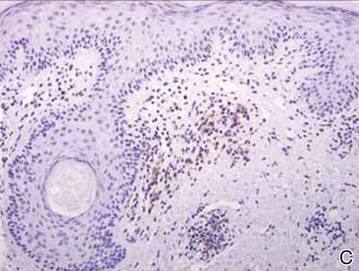
Figure 2. Vacuolar interface dermatitis with a perivascular and periadnexal lymphocytic infiltrate (A)(H&E, original magnification ×40). Scattered necrotic keratinocytes were evident (B)(H&E, original magnification ×400). Anti-CD123 immunohistochemical staining revealed nodular aggregates of plasmacytoid dendritic cells with accompanying lymphocytes centered around dermal vessels and adnexa (C)(original magnification ×200). | ||
Comment
Imiquimod is a topical immunomodulator used for the treatment of genital warts and cutaneous malignancies. It exerts its effect via induction of proinflammatory cytokines (eg, IFN-a, tumor necrosis factor a [TNF-α]) through activation of toll-like receptor (TLR) 7, an intracytoplasmic receptor that is found on several cell types including pDCs and B cells. When the cell surface receptor is bound by activating ligands (eg, single-stranded RNA, imiquimod), downstream signaling is initiated, resulting in the production of large amounts of IFN-a and TNF-α.6,7 Both IFN-a and TNF-α are upregulated in patient serum and lesional skin in SCLE.8,9 Additionally, pDCs have been shown to accumulate in lesions of cutaneous lupus erythematosus (CLE) in a distinct dermal pattern, as demonstrated by CD123 staining.9,10 This pattern is identical to the one seen in our case. Although pDCs also are present in cutaneous dermatomyositis lesions, the pattern is distinct from CLE.10 These findings indicate that IFN-a produced by pDCs may play an integral role in the pathogenesis of CLE. Several observations implicating TLR signaling in the pathogenesis of lupus have been described. In a lupus-prone mouse model of lupus erythematosus, transgenic overexpression of TLR-7 resulted in increased severity of clinical disease and accelerated mortality. Antimalarials improve lupus erythematosus via blockade of TLR-7 and TLR-9 signaling.11
Imiquimod, which acts as an inducer of IFN-α expression through TLR-7 signaling, may have been an inciting factor in the development of SCLE-like lesions in this genetically predisposed patient. Histopathology of cutaneous malignancies treated with topical imiquimod typically does not show lupuslike features.12 It is possible that a subset of predisposed patients have increased numbers of pDCs primed in their skin or that they exhibit a more robust TLR-7 reaction to imiquimod, resulting in abundant IFN-a production. Other autoimmune diseases, such as pemphigus foliaceus and vitiligo, also have been reported to occur locally after the application of imiquimod,13,14 which suggests that a localized autoimmune reaction can be induced by activation of TLR-7; however, a case of chronic discoid lupus erythematosus of the scalp improving after treatment with imiquimod has been reported.15
The use of imiquimod in patients with a personal or family history of lupus erythematosus or those with a personal history of an autoimmune blistering disorder should be undertaken with caution until more is known.
1. Chen M, Crowson AN, Woofter M, et al. Docetaxel (taxotere) induced subacute cutaneous lupus erythematosus: report of 4 cases. J Rheumatol. 2004;31:818-820.
2. Farhi D, Viguier M, Cosnes A, et al. Terbinafine-induced subacute cutaneous lupus erythematosus. Dermatology. 2006;212:59-65.
3. Wiechert A, Tüting T, Bieber T, et al. Subacute cutaneous lupus erythematosus in a leuprorelin-treated patient with prostate carcinoma. Br J Dermatol. 2008;159:231-233.
4. Bleumink GS, ter Borg EJ, Ramselaar CG, et al. Etanercept-induced subcutaneous lupus erythematosus. Rheumatology (Oxford). 2001;40:1317-1319.
5. Bentley DD, Graves JE, Smith DI, et al. Efalizumab-induced subacute cutaneous lupus erythematosus. J Am Acad Dermatol. 2006;54(suppl 5):S242-S243.
6. Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823-835.
7. Hurwitz DJ, Pincus L, Kupper TS. Imiquimod: a topically applied link between innate and acquired immunity. Arch Dermatol. 2003;139:1347-1350.
8. Zampieri S, Alaibac M, Iaccarino L, et al. Tumour necrosis factor alpha is expressed in refractory skin lesions from patients with subacute cutaneous lupus erythematosus. Ann Rheum Dis. 2006;65:545-548.
9. Farkas L, Beiske K, Lund-Johansen F, et al. Plasmacytoid dendritic cells (natural interferon-alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol. 2001;159:237-243.
10. McNiff JM, Kaplan DH. Plasmacytoid dendritic cells are present in cutaneous dermatomyositis lesions in a pattern distinct from lupus erythematosus. J Cutan Pathol. 2008;35:452-456.
11. Pisitkun P, Deane JA, Difilippantonio MJ, et al. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669-1672.
12. Wolf IH, Kodama K, Cerroni L, et al. Nature of inflammatory infiltrate in superficial cutaneous malignancies during topical imiquimod treatment. Am J Dermatopathol. 2007;29:237-241.
13. Lin R, Ladd DJ Jr, Powell DJ, et al. Localized pemphigus foliaceus induced by topical imiquimod treatment. Arch Dermatol. 2004;140:889-890.
14. Brown T, Zirvi M, Cotsarelis G, et al. Vitiligo-like hypopigmentation associated with imiquimod treatment of genital warts. J Am Acad Dermatol. 2005;52:715-716.
15. Gersden R, Wenzel J, Uerlich M, et al. Successful treatment of chronic discoid lupus erythematosus of the scalp with imiquimod. Dermatology. 2002;205:416-418.
Drug-induced lupus accounts for up to 10% of lupus erythematosus cases. Subacute cutaneous lupus erythematosus (SCLE) is a distinct clinical variant of lupus erythematosus that typically presents as annular, often scaly, erythematous plaques in photodistributed areas. Subacute cutaneous lupus erythematosus has been reported in association with multiple systemic medications including docetaxel, terbinafine, leuprolide acetate, etanercept, and efalizumab1-5; however, the induction of SCLE by topical agents has not been widely reported. We report a case of local induction of lesions that clinically and histologically resembled SCLE in a 50-year-old woman following treatment with topical imiquimod.
Case Report
A 50-year-old woman presented with an adverse inflammatory reaction on the right side of the upper chest secondary to application of topical imiquimod. Prior to the current presentation the patient was diagnosed with a biopsy-proven superficial basal cell carcinoma (BCC) on the sun-exposed area of the upper right breast, and topical imiquimod therapy was initiated. Several days after starting treatment, the patient began applying imiquimod on areas of clinically normal skin on the upper chest as advised by her dermatologist. After 7 to 10 days of application the patient reported intense erythema, pain, and crusting in the treated area on the right side of the upper chest. She also experienced systemic symptoms including fatigue, arthralgia, malaise, and fever.
Clinical examination revealed erythematous to violaceous annular and polycyclic plaques on the upper chest (Figure 1). Erythema and scaling were noted at the site of the superficial BCC. A biopsy showed a superficial and mid-dermal perivascular and periadnexal lymphocytic infiltrate with overlying vacuolar interface dermatitis and scattered necrotic keratinocytes (Figures 2A and 2B). There was slightly increased dermal edema and mucin. Anti-CD123 immunohistochemical staining revealed nodular aggregates of plasmacytoid dendritic cells (pDCs) accompanying lymphocytes around dermal vessels and adnexa (Figure 2C). The patient’s family history was remarkable for SCLE in her daughter. Antinuclear antibody, anti-Ro (Sjögren syndrome antigen A), and anti-La (Sjögren syndrome antigen B) were negative. On follow-up examination 1 month after discontinuation of imiquimod, the patient’s skin lesions had completely cleared. Two years later, the patient continued to be free of skin lesions.
|
Figure 1. Annular plaques on the upper chest with overlying scale and focal central clearing.
| ||
|
Figure 2. Vacuolar interface dermatitis with a perivascular and periadnexal lymphocytic infiltrate (A)(H&E, original magnification ×40). Scattered necrotic keratinocytes were evident (B)(H&E, original magnification ×400). Anti-CD123 immunohistochemical staining revealed nodular aggregates of plasmacytoid dendritic cells with accompanying lymphocytes centered around dermal vessels and adnexa (C)(original magnification ×200). | ||
Comment
Imiquimod is a topical immunomodulator used for the treatment of genital warts and cutaneous malignancies. It exerts its effect via induction of proinflammatory cytokines (eg, IFN-a, tumor necrosis factor a [TNF-α]) through activation of toll-like receptor (TLR) 7, an intracytoplasmic receptor that is found on several cell types including pDCs and B cells. When the cell surface receptor is bound by activating ligands (eg, single-stranded RNA, imiquimod), downstream signaling is initiated, resulting in the production of large amounts of IFN-a and TNF-α.6,7 Both IFN-a and TNF-α are upregulated in patient serum and lesional skin in SCLE.8,9 Additionally, pDCs have been shown to accumulate in lesions of cutaneous lupus erythematosus (CLE) in a distinct dermal pattern, as demonstrated by CD123 staining.9,10 This pattern is identical to the one seen in our case. Although pDCs also are present in cutaneous dermatomyositis lesions, the pattern is distinct from CLE.10 These findings indicate that IFN-a produced by pDCs may play an integral role in the pathogenesis of CLE. Several observations implicating TLR signaling in the pathogenesis of lupus have been described. In a lupus-prone mouse model of lupus erythematosus, transgenic overexpression of TLR-7 resulted in increased severity of clinical disease and accelerated mortality. Antimalarials improve lupus erythematosus via blockade of TLR-7 and TLR-9 signaling.11
Imiquimod, which acts as an inducer of IFN-α expression through TLR-7 signaling, may have been an inciting factor in the development of SCLE-like lesions in this genetically predisposed patient. Histopathology of cutaneous malignancies treated with topical imiquimod typically does not show lupuslike features.12 It is possible that a subset of predisposed patients have increased numbers of pDCs primed in their skin or that they exhibit a more robust TLR-7 reaction to imiquimod, resulting in abundant IFN-a production. Other autoimmune diseases, such as pemphigus foliaceus and vitiligo, also have been reported to occur locally after the application of imiquimod,13,14 which suggests that a localized autoimmune reaction can be induced by activation of TLR-7; however, a case of chronic discoid lupus erythematosus of the scalp improving after treatment with imiquimod has been reported.15
The use of imiquimod in patients with a personal or family history of lupus erythematosus or those with a personal history of an autoimmune blistering disorder should be undertaken with caution until more is known.
Drug-induced lupus accounts for up to 10% of lupus erythematosus cases. Subacute cutaneous lupus erythematosus (SCLE) is a distinct clinical variant of lupus erythematosus that typically presents as annular, often scaly, erythematous plaques in photodistributed areas. Subacute cutaneous lupus erythematosus has been reported in association with multiple systemic medications including docetaxel, terbinafine, leuprolide acetate, etanercept, and efalizumab1-5; however, the induction of SCLE by topical agents has not been widely reported. We report a case of local induction of lesions that clinically and histologically resembled SCLE in a 50-year-old woman following treatment with topical imiquimod.
Case Report
A 50-year-old woman presented with an adverse inflammatory reaction on the right side of the upper chest secondary to application of topical imiquimod. Prior to the current presentation the patient was diagnosed with a biopsy-proven superficial basal cell carcinoma (BCC) on the sun-exposed area of the upper right breast, and topical imiquimod therapy was initiated. Several days after starting treatment, the patient began applying imiquimod on areas of clinically normal skin on the upper chest as advised by her dermatologist. After 7 to 10 days of application the patient reported intense erythema, pain, and crusting in the treated area on the right side of the upper chest. She also experienced systemic symptoms including fatigue, arthralgia, malaise, and fever.
Clinical examination revealed erythematous to violaceous annular and polycyclic plaques on the upper chest (Figure 1). Erythema and scaling were noted at the site of the superficial BCC. A biopsy showed a superficial and mid-dermal perivascular and periadnexal lymphocytic infiltrate with overlying vacuolar interface dermatitis and scattered necrotic keratinocytes (Figures 2A and 2B). There was slightly increased dermal edema and mucin. Anti-CD123 immunohistochemical staining revealed nodular aggregates of plasmacytoid dendritic cells (pDCs) accompanying lymphocytes around dermal vessels and adnexa (Figure 2C). The patient’s family history was remarkable for SCLE in her daughter. Antinuclear antibody, anti-Ro (Sjögren syndrome antigen A), and anti-La (Sjögren syndrome antigen B) were negative. On follow-up examination 1 month after discontinuation of imiquimod, the patient’s skin lesions had completely cleared. Two years later, the patient continued to be free of skin lesions.
|
Figure 1. Annular plaques on the upper chest with overlying scale and focal central clearing.
| ||
|
Figure 2. Vacuolar interface dermatitis with a perivascular and periadnexal lymphocytic infiltrate (A)(H&E, original magnification ×40). Scattered necrotic keratinocytes were evident (B)(H&E, original magnification ×400). Anti-CD123 immunohistochemical staining revealed nodular aggregates of plasmacytoid dendritic cells with accompanying lymphocytes centered around dermal vessels and adnexa (C)(original magnification ×200). | ||
Comment
Imiquimod is a topical immunomodulator used for the treatment of genital warts and cutaneous malignancies. It exerts its effect via induction of proinflammatory cytokines (eg, IFN-a, tumor necrosis factor a [TNF-α]) through activation of toll-like receptor (TLR) 7, an intracytoplasmic receptor that is found on several cell types including pDCs and B cells. When the cell surface receptor is bound by activating ligands (eg, single-stranded RNA, imiquimod), downstream signaling is initiated, resulting in the production of large amounts of IFN-a and TNF-α.6,7 Both IFN-a and TNF-α are upregulated in patient serum and lesional skin in SCLE.8,9 Additionally, pDCs have been shown to accumulate in lesions of cutaneous lupus erythematosus (CLE) in a distinct dermal pattern, as demonstrated by CD123 staining.9,10 This pattern is identical to the one seen in our case. Although pDCs also are present in cutaneous dermatomyositis lesions, the pattern is distinct from CLE.10 These findings indicate that IFN-a produced by pDCs may play an integral role in the pathogenesis of CLE. Several observations implicating TLR signaling in the pathogenesis of lupus have been described. In a lupus-prone mouse model of lupus erythematosus, transgenic overexpression of TLR-7 resulted in increased severity of clinical disease and accelerated mortality. Antimalarials improve lupus erythematosus via blockade of TLR-7 and TLR-9 signaling.11
Imiquimod, which acts as an inducer of IFN-α expression through TLR-7 signaling, may have been an inciting factor in the development of SCLE-like lesions in this genetically predisposed patient. Histopathology of cutaneous malignancies treated with topical imiquimod typically does not show lupuslike features.12 It is possible that a subset of predisposed patients have increased numbers of pDCs primed in their skin or that they exhibit a more robust TLR-7 reaction to imiquimod, resulting in abundant IFN-a production. Other autoimmune diseases, such as pemphigus foliaceus and vitiligo, also have been reported to occur locally after the application of imiquimod,13,14 which suggests that a localized autoimmune reaction can be induced by activation of TLR-7; however, a case of chronic discoid lupus erythematosus of the scalp improving after treatment with imiquimod has been reported.15
The use of imiquimod in patients with a personal or family history of lupus erythematosus or those with a personal history of an autoimmune blistering disorder should be undertaken with caution until more is known.
1. Chen M, Crowson AN, Woofter M, et al. Docetaxel (taxotere) induced subacute cutaneous lupus erythematosus: report of 4 cases. J Rheumatol. 2004;31:818-820.
2. Farhi D, Viguier M, Cosnes A, et al. Terbinafine-induced subacute cutaneous lupus erythematosus. Dermatology. 2006;212:59-65.
3. Wiechert A, Tüting T, Bieber T, et al. Subacute cutaneous lupus erythematosus in a leuprorelin-treated patient with prostate carcinoma. Br J Dermatol. 2008;159:231-233.
4. Bleumink GS, ter Borg EJ, Ramselaar CG, et al. Etanercept-induced subcutaneous lupus erythematosus. Rheumatology (Oxford). 2001;40:1317-1319.
5. Bentley DD, Graves JE, Smith DI, et al. Efalizumab-induced subacute cutaneous lupus erythematosus. J Am Acad Dermatol. 2006;54(suppl 5):S242-S243.
6. Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823-835.
7. Hurwitz DJ, Pincus L, Kupper TS. Imiquimod: a topically applied link between innate and acquired immunity. Arch Dermatol. 2003;139:1347-1350.
8. Zampieri S, Alaibac M, Iaccarino L, et al. Tumour necrosis factor alpha is expressed in refractory skin lesions from patients with subacute cutaneous lupus erythematosus. Ann Rheum Dis. 2006;65:545-548.
9. Farkas L, Beiske K, Lund-Johansen F, et al. Plasmacytoid dendritic cells (natural interferon-alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol. 2001;159:237-243.
10. McNiff JM, Kaplan DH. Plasmacytoid dendritic cells are present in cutaneous dermatomyositis lesions in a pattern distinct from lupus erythematosus. J Cutan Pathol. 2008;35:452-456.
11. Pisitkun P, Deane JA, Difilippantonio MJ, et al. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669-1672.
12. Wolf IH, Kodama K, Cerroni L, et al. Nature of inflammatory infiltrate in superficial cutaneous malignancies during topical imiquimod treatment. Am J Dermatopathol. 2007;29:237-241.
13. Lin R, Ladd DJ Jr, Powell DJ, et al. Localized pemphigus foliaceus induced by topical imiquimod treatment. Arch Dermatol. 2004;140:889-890.
14. Brown T, Zirvi M, Cotsarelis G, et al. Vitiligo-like hypopigmentation associated with imiquimod treatment of genital warts. J Am Acad Dermatol. 2005;52:715-716.
15. Gersden R, Wenzel J, Uerlich M, et al. Successful treatment of chronic discoid lupus erythematosus of the scalp with imiquimod. Dermatology. 2002;205:416-418.
1. Chen M, Crowson AN, Woofter M, et al. Docetaxel (taxotere) induced subacute cutaneous lupus erythematosus: report of 4 cases. J Rheumatol. 2004;31:818-820.
2. Farhi D, Viguier M, Cosnes A, et al. Terbinafine-induced subacute cutaneous lupus erythematosus. Dermatology. 2006;212:59-65.
3. Wiechert A, Tüting T, Bieber T, et al. Subacute cutaneous lupus erythematosus in a leuprorelin-treated patient with prostate carcinoma. Br J Dermatol. 2008;159:231-233.
4. Bleumink GS, ter Borg EJ, Ramselaar CG, et al. Etanercept-induced subcutaneous lupus erythematosus. Rheumatology (Oxford). 2001;40:1317-1319.
5. Bentley DD, Graves JE, Smith DI, et al. Efalizumab-induced subacute cutaneous lupus erythematosus. J Am Acad Dermatol. 2006;54(suppl 5):S242-S243.
6. Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823-835.
7. Hurwitz DJ, Pincus L, Kupper TS. Imiquimod: a topically applied link between innate and acquired immunity. Arch Dermatol. 2003;139:1347-1350.
8. Zampieri S, Alaibac M, Iaccarino L, et al. Tumour necrosis factor alpha is expressed in refractory skin lesions from patients with subacute cutaneous lupus erythematosus. Ann Rheum Dis. 2006;65:545-548.
9. Farkas L, Beiske K, Lund-Johansen F, et al. Plasmacytoid dendritic cells (natural interferon-alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol. 2001;159:237-243.
10. McNiff JM, Kaplan DH. Plasmacytoid dendritic cells are present in cutaneous dermatomyositis lesions in a pattern distinct from lupus erythematosus. J Cutan Pathol. 2008;35:452-456.
11. Pisitkun P, Deane JA, Difilippantonio MJ, et al. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669-1672.
12. Wolf IH, Kodama K, Cerroni L, et al. Nature of inflammatory infiltrate in superficial cutaneous malignancies during topical imiquimod treatment. Am J Dermatopathol. 2007;29:237-241.
13. Lin R, Ladd DJ Jr, Powell DJ, et al. Localized pemphigus foliaceus induced by topical imiquimod treatment. Arch Dermatol. 2004;140:889-890.
14. Brown T, Zirvi M, Cotsarelis G, et al. Vitiligo-like hypopigmentation associated with imiquimod treatment of genital warts. J Am Acad Dermatol. 2005;52:715-716.
15. Gersden R, Wenzel J, Uerlich M, et al. Successful treatment of chronic discoid lupus erythematosus of the scalp with imiquimod. Dermatology. 2002;205:416-418.
Practice Points
- Subacute cutaneous lupus erythematosus has been reported in association with multiple systemic medications; however, association of this disorder with topical agents has not been widely reported.
- The use of topical imiquimod in patients with a personal or family history of lupus erythematosus should be undertaken with caution until more is known.
Rare Angioinvasive Fungal Infection in Association With Leukemia Cutis
Leukemia cutis (LC) is characterized by the infiltration of malignant neoplastic leukocytes or their precursors into the skin, most often in conjunction with systemic leukemia.1 Acute myelogenous leukemia (AML) is the second most common cause of LC and the most common form of leukemia among adults.1 Patients with leukemia often are in a relative or absolute immunocompromised state, which may be secondary to neutropenia, chemotherapy regimens, or immunosuppressive regimens following stem cell transplant (SCT). Thus, when evaluating cutaneous lesions consistent with LC in immunocompromised patients, there must be a high index of suspicion for concomitant opportunistic infections.
We report the case of a 52-year-old man with primary refractory AML following allogeneic SCT with relapse who presented with an LC lesion below the knee with concomitant invasive fungal infection despite being on prophylactic oral antifungal therapy.
Case Report
A 52-year-old man with primary refractory AML (M1) of 1 year’s duration presented for evaluation of a slowly progressing reddish purple nodule on the right knee of 2 to 4 months’ duration. The patient had undergone a matched unrelated donor allogeneic SCT 6 months following diagnosis of AML with subsequent disease progression despite reduction of posttransplant graft-versus-host disease prophylactic immune suppression and a cycle of clofarabine. The patient was hospitalized 2 months after the SCT for neutropenic fever and was found to have vancomycin-resistant enterococcal bacteremia, Clostridium difficile colitis, and possible fungal pneumonia. He was treated with voriconazole 200 mg twice daily, which he continued following discharge for antifungal prophylaxis. At the time of discharge, the patient reported that he noticed an asymptomatic “purple papule” on the right knee but did not seek further workup.
Two months later, the patient presented with a fever (temperature, 38.6°C) and leukocytosis (white blood cell count, 130 cells/mL [increased from 53 cells/mL 1 week prior to admission]). Due to his history of immunosuppression and neutropenia, the patient was placed on a broad-spectrum antibiotic regimen of cefepime, daptomycin, and linezolid on admission. Later, vancomycin and gentamicin were added and voriconazole was switched to caspofungin. The patient also received granulocyte-macrophage colony-stimulating factor for neutropenia. During the current hospitalization, blood cultures demonstrated vancomycin-resistant enterococcemia, and computed tomography of the chest revealed findings consistent with multilobar pneumonia.
Dermatology was consulted to evaluate the purple nodule on the right knee, which had slowly progressed since his last admission. Physical examination revealed a violaceous, 1.5×1.5-cm nodule with central necrosis covered by black eschar with surrounding erythema (Figure 1). Biopsy specimens for routine histology and a tissue culture were obtained. Histopathologic examination revealed a dense diffuse infiltrate of large hyperchromatic mononuclear cells extending through the dermis, which was consistent with the patient’s known AML (Figure 2). Acid-fast bacillus staining was negative for mycobacterial organisms. Grocott-Gomori methenamine-silver stain demonstrated an overwhelming number of septate fungal hyphae with acute-angle branching, concerning for Aspergillus species (Figure 3). Of note, an Aspergillus serum antigen test was performed at this time and was negative. On repeat review of the routine histologic sections, angioinvasion by hyphae was detected amidst the dense lymphocytic infiltrate (Figure 4).
Given the patient’s immunocompromised state, the presence of angioinvasive fungi on the skin biopsy, and unresolved pneumonia, the patient was restarted on voriconazole for treatment of likely Aspergillus infection. He was continued on chemotherapy for the primary refractory AML and received a donor lymphocyte infusion prior to discharge. After the patient was discharged, the tissue culture grew Paecilomyces, a rare fungal species. The patient died 1 week after discharge.
|
|
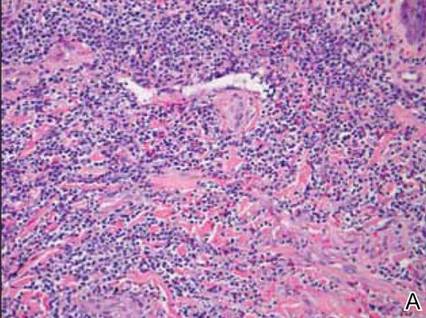
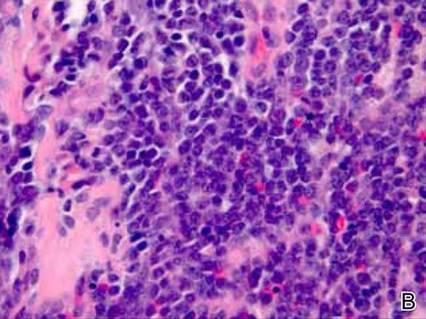
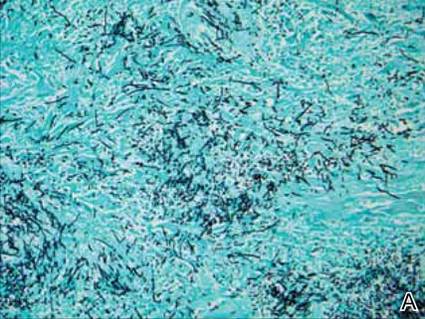
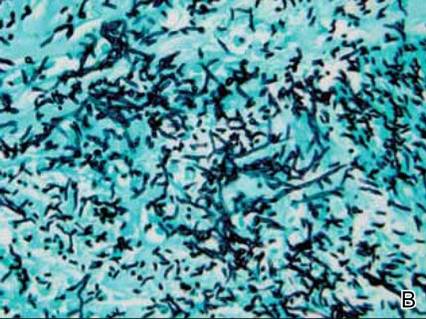
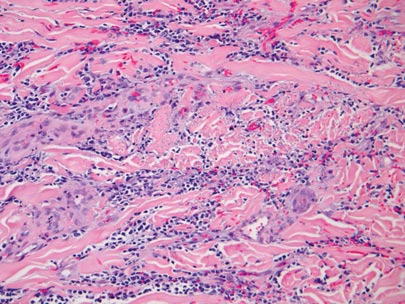
Comment
Leukemia cutis is an extramedullary manifestation of leukemia that appears in 10% to 15% of patients with AML.2 The frequency of LC differs widely for the various types of AML, with the majority of cases occurring in the acute myelomonocytic leukemia (M4) or acute monocytic leukemia (M5) subtypes.3,4 One large study of AML patients (N=381) demonstrated an incidence of LC in 28.6% of patients with the M4 subtype and 42.9% of those with the M5 subtype, with an incidence of only 7.1% of patients with the M1 subtype.3 It occurs less frequently in chronic myeloproliferative diseases.2,4
Leukemia cutis has a wide range of cutaneous manifestations and may present with solitary or multiple papular, nodular, or plaquelike lesions that are red-brown, blue, violaceous, or hemorrhagic.4 Leukemia cutis occurs most commonly on the legs, followed by the arms, back, chest, scalp, and face.2,4 Leukemia cutis may be hard to distinguish clinically from other conditions such as cutaneous metastases of visceral malignancies, lymphoma, drug eruptions, and opportunistic infections. Leukemia cutis ulcers often measure only a few centimeters in diameter with a firmly adherent purulent or hemorrhagic crust and may occur in unusual locations. These lesions usually are treatment resistant and their persistence may help to lead to diagnosis.4
Microscopically, most LC lesions show a perivascular or periadnexal pattern of involvement or a dense diffuse, interstitial, or nodular atypical lymphocytic infiltrate involving the dermis and subcutis with sparing of the upper papillary dermis.2 The cytologic appearance of M1 and M2 subtypes of AML are characterized by medium-sized to large mononuclear cells with a light cytoplasm and large basophilic cell nuclei. The M4 and M5 subtypes of AML generally are dominated by medium-sized, round or oval-shaped mononuclear cells that may have eosinophilic cytoplasm and segmented or kidney-shaped basophilic nuclei.4 Immunophenotyping is crucial for diagnosis. In myeloid disorders, there is positive staining with markers of myeloid lineage such as myeloperoxidase, lysozyme, CD34, CD15, CD68, CD43, and CD117.5
In our patient, there was an ulcerated dense diffuse dermal infiltrate of large atypical lymphocytes consistent with LC and positive immunostaining consistent with LC associated with AML. Additionally, septate hyphae with acute-angle branching also were noted in the dermal blood vessels on hematoxylin and eosin and fungal staining, demonstrating concomitant fungal infection. Angioinvasion of organisms demonstrated on skin biopsy and persistent pneumonia noted on chest imaging suggested a disseminated infectious process.
Invasive fungal infections are an increasing cause of morbidity and mortality in immunocompromised individuals, including those with hematologic malignancies and hematologic SCTs. Despite an increasing number of antifungal therapies, outcomes are frequently suboptimal with mortality rates often greater than 50% depending on the pathogen and disease.6 Thus, there must be a high index of suspicion of infection even when a separate histopathologic diagnosis is available, such as the finding of leukemic infiltrates in this patient’s biopsy specimen. A similar case of LC has been reported with concomitant fungal infection involving Fusarium and Enterococcus.7 Patients with leukemic cells may develop leukemic infiltrates in response to cutaneous infection, and a high index of suspicion for 2 related but distinct processes is necessary.
Paecilomyces species are an emerging cause of opportunistic and usually severe human infections.8-11 The Paecilomyces species are saprophytic filamentous fungi that are found worldwide in soil as well as contaminants in the air and water.12Paecilomyces infection is generally associated with the use of immunosuppressive therapies, implants, or ocular surgery. Among species in this genus, Paecilomyces lilacinus and Paecilomyces variotii are of clinical importance. Most species have high susceptibility to the newer azoles such as voriconazole.6
Conclusion
Despite continued treatment with voriconazole, our patient still developed a rare fungal infection arising in a lesion of LC. He had signs of infection, including an elevated white blood cell count, fever, and malaise, which are nonspecific clinical findings that could have been attributed to known relapse of systemic leukemia or to known enterococcemia. Even in patients on antifungal prophylaxis and with other possible causes of leukocytosis, this case illustrates that there must be a high index of suspicion for angioinvasive fungal infection.
1. Aquilera SB, Zarraga M, Rosen L. Leukemia cutis in a patient with acute myelogenous leukemia: a case report and review of the literature. Cutis. 2010;85:31-36.
2. Cho-Vega JH, Medeiros J, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130.
3. Agis H, Weltermann A, Fonatsch C, et al. A comparative study on demographic, hematological, and cytogenetic findings and prognosis in acute myeloid leukemia with and without leukemia cutis. Ann Hematol. 2002;81:90-95.
4. Wagner G, Fenchel K, Back W, et al. Leukemia cutis—epidemiology, clinical presentation, and differential diagnoses. J Dtsch Dermatol Ges. 2012;10:27-36.
5. Hejmadi RK, Thompson D, Shah F, et al. Cutaneous presentation of aleukemic monoblastic leukemia cutis: a case report and review of literature with focus on immunohistochemistry. J Cutan Pathol. 2008;35(suppl 1):46.
6. Kontoyiannis DP. Invasive mycoses: strategies for effective management. Am J Med. 2012;125(suppl 1):25-38.
7. Feramisco JD, Hsiao JL, Fox LP, et al. Angioinvasive Fusarium and concomitant Enterococcus infection arising in association with leukemia cutis. J Cutan Pathol. 2011;38:926-929.
8. Antachopoulos C, Walsh TJ, Roilides E. Fungal infections in primary immunodeficiencies. Eur J Pediatr. 2007;166:1099-1117.
9. Carey J, D’Amico R, Sutton DA, et al. Paecilomyces lilacinus vaginitis in an immuno-competent patient. Emerg Infect Dis. 2003;9:1155-1158.
10. Castro LG, Salebian A, Sotto MN. Hyalohyphomycosis by Paecilomyces lilacinus in a renal transplant patient and a review of human Paecilomyces species infections. J Med Vet Mycol. 1990;28:15-26.
11. Pastor FJ, Guarro J. Clinical manifestations, treatment and outcome of Paecilomyces lilacinus infections. Clin Microbiol Infect. 2006;12:948-960.
12. Castelli MV, Alastruey-Izquierdo A, Cuesta I, et al. Susceptibility testing and molecular classification of Paecilomyces spp. Antimicrob Agents Chemother. 2008;52:2926-2928.
Leukemia cutis (LC) is characterized by the infiltration of malignant neoplastic leukocytes or their precursors into the skin, most often in conjunction with systemic leukemia.1 Acute myelogenous leukemia (AML) is the second most common cause of LC and the most common form of leukemia among adults.1 Patients with leukemia often are in a relative or absolute immunocompromised state, which may be secondary to neutropenia, chemotherapy regimens, or immunosuppressive regimens following stem cell transplant (SCT). Thus, when evaluating cutaneous lesions consistent with LC in immunocompromised patients, there must be a high index of suspicion for concomitant opportunistic infections.
We report the case of a 52-year-old man with primary refractory AML following allogeneic SCT with relapse who presented with an LC lesion below the knee with concomitant invasive fungal infection despite being on prophylactic oral antifungal therapy.
Case Report
A 52-year-old man with primary refractory AML (M1) of 1 year’s duration presented for evaluation of a slowly progressing reddish purple nodule on the right knee of 2 to 4 months’ duration. The patient had undergone a matched unrelated donor allogeneic SCT 6 months following diagnosis of AML with subsequent disease progression despite reduction of posttransplant graft-versus-host disease prophylactic immune suppression and a cycle of clofarabine. The patient was hospitalized 2 months after the SCT for neutropenic fever and was found to have vancomycin-resistant enterococcal bacteremia, Clostridium difficile colitis, and possible fungal pneumonia. He was treated with voriconazole 200 mg twice daily, which he continued following discharge for antifungal prophylaxis. At the time of discharge, the patient reported that he noticed an asymptomatic “purple papule” on the right knee but did not seek further workup.
Two months later, the patient presented with a fever (temperature, 38.6°C) and leukocytosis (white blood cell count, 130 cells/mL [increased from 53 cells/mL 1 week prior to admission]). Due to his history of immunosuppression and neutropenia, the patient was placed on a broad-spectrum antibiotic regimen of cefepime, daptomycin, and linezolid on admission. Later, vancomycin and gentamicin were added and voriconazole was switched to caspofungin. The patient also received granulocyte-macrophage colony-stimulating factor for neutropenia. During the current hospitalization, blood cultures demonstrated vancomycin-resistant enterococcemia, and computed tomography of the chest revealed findings consistent with multilobar pneumonia.
Dermatology was consulted to evaluate the purple nodule on the right knee, which had slowly progressed since his last admission. Physical examination revealed a violaceous, 1.5×1.5-cm nodule with central necrosis covered by black eschar with surrounding erythema (Figure 1). Biopsy specimens for routine histology and a tissue culture were obtained. Histopathologic examination revealed a dense diffuse infiltrate of large hyperchromatic mononuclear cells extending through the dermis, which was consistent with the patient’s known AML (Figure 2). Acid-fast bacillus staining was negative for mycobacterial organisms. Grocott-Gomori methenamine-silver stain demonstrated an overwhelming number of septate fungal hyphae with acute-angle branching, concerning for Aspergillus species (Figure 3). Of note, an Aspergillus serum antigen test was performed at this time and was negative. On repeat review of the routine histologic sections, angioinvasion by hyphae was detected amidst the dense lymphocytic infiltrate (Figure 4).
Given the patient’s immunocompromised state, the presence of angioinvasive fungi on the skin biopsy, and unresolved pneumonia, the patient was restarted on voriconazole for treatment of likely Aspergillus infection. He was continued on chemotherapy for the primary refractory AML and received a donor lymphocyte infusion prior to discharge. After the patient was discharged, the tissue culture grew Paecilomyces, a rare fungal species. The patient died 1 week after discharge.
|
|
Comment
Leukemia cutis is an extramedullary manifestation of leukemia that appears in 10% to 15% of patients with AML.2 The frequency of LC differs widely for the various types of AML, with the majority of cases occurring in the acute myelomonocytic leukemia (M4) or acute monocytic leukemia (M5) subtypes.3,4 One large study of AML patients (N=381) demonstrated an incidence of LC in 28.6% of patients with the M4 subtype and 42.9% of those with the M5 subtype, with an incidence of only 7.1% of patients with the M1 subtype.3 It occurs less frequently in chronic myeloproliferative diseases.2,4
Leukemia cutis has a wide range of cutaneous manifestations and may present with solitary or multiple papular, nodular, or plaquelike lesions that are red-brown, blue, violaceous, or hemorrhagic.4 Leukemia cutis occurs most commonly on the legs, followed by the arms, back, chest, scalp, and face.2,4 Leukemia cutis may be hard to distinguish clinically from other conditions such as cutaneous metastases of visceral malignancies, lymphoma, drug eruptions, and opportunistic infections. Leukemia cutis ulcers often measure only a few centimeters in diameter with a firmly adherent purulent or hemorrhagic crust and may occur in unusual locations. These lesions usually are treatment resistant and their persistence may help to lead to diagnosis.4
Microscopically, most LC lesions show a perivascular or periadnexal pattern of involvement or a dense diffuse, interstitial, or nodular atypical lymphocytic infiltrate involving the dermis and subcutis with sparing of the upper papillary dermis.2 The cytologic appearance of M1 and M2 subtypes of AML are characterized by medium-sized to large mononuclear cells with a light cytoplasm and large basophilic cell nuclei. The M4 and M5 subtypes of AML generally are dominated by medium-sized, round or oval-shaped mononuclear cells that may have eosinophilic cytoplasm and segmented or kidney-shaped basophilic nuclei.4 Immunophenotyping is crucial for diagnosis. In myeloid disorders, there is positive staining with markers of myeloid lineage such as myeloperoxidase, lysozyme, CD34, CD15, CD68, CD43, and CD117.5
In our patient, there was an ulcerated dense diffuse dermal infiltrate of large atypical lymphocytes consistent with LC and positive immunostaining consistent with LC associated with AML. Additionally, septate hyphae with acute-angle branching also were noted in the dermal blood vessels on hematoxylin and eosin and fungal staining, demonstrating concomitant fungal infection. Angioinvasion of organisms demonstrated on skin biopsy and persistent pneumonia noted on chest imaging suggested a disseminated infectious process.
Invasive fungal infections are an increasing cause of morbidity and mortality in immunocompromised individuals, including those with hematologic malignancies and hematologic SCTs. Despite an increasing number of antifungal therapies, outcomes are frequently suboptimal with mortality rates often greater than 50% depending on the pathogen and disease.6 Thus, there must be a high index of suspicion of infection even when a separate histopathologic diagnosis is available, such as the finding of leukemic infiltrates in this patient’s biopsy specimen. A similar case of LC has been reported with concomitant fungal infection involving Fusarium and Enterococcus.7 Patients with leukemic cells may develop leukemic infiltrates in response to cutaneous infection, and a high index of suspicion for 2 related but distinct processes is necessary.
Paecilomyces species are an emerging cause of opportunistic and usually severe human infections.8-11 The Paecilomyces species are saprophytic filamentous fungi that are found worldwide in soil as well as contaminants in the air and water.12Paecilomyces infection is generally associated with the use of immunosuppressive therapies, implants, or ocular surgery. Among species in this genus, Paecilomyces lilacinus and Paecilomyces variotii are of clinical importance. Most species have high susceptibility to the newer azoles such as voriconazole.6
Conclusion
Despite continued treatment with voriconazole, our patient still developed a rare fungal infection arising in a lesion of LC. He had signs of infection, including an elevated white blood cell count, fever, and malaise, which are nonspecific clinical findings that could have been attributed to known relapse of systemic leukemia or to known enterococcemia. Even in patients on antifungal prophylaxis and with other possible causes of leukocytosis, this case illustrates that there must be a high index of suspicion for angioinvasive fungal infection.
Leukemia cutis (LC) is characterized by the infiltration of malignant neoplastic leukocytes or their precursors into the skin, most often in conjunction with systemic leukemia.1 Acute myelogenous leukemia (AML) is the second most common cause of LC and the most common form of leukemia among adults.1 Patients with leukemia often are in a relative or absolute immunocompromised state, which may be secondary to neutropenia, chemotherapy regimens, or immunosuppressive regimens following stem cell transplant (SCT). Thus, when evaluating cutaneous lesions consistent with LC in immunocompromised patients, there must be a high index of suspicion for concomitant opportunistic infections.
We report the case of a 52-year-old man with primary refractory AML following allogeneic SCT with relapse who presented with an LC lesion below the knee with concomitant invasive fungal infection despite being on prophylactic oral antifungal therapy.
Case Report
A 52-year-old man with primary refractory AML (M1) of 1 year’s duration presented for evaluation of a slowly progressing reddish purple nodule on the right knee of 2 to 4 months’ duration. The patient had undergone a matched unrelated donor allogeneic SCT 6 months following diagnosis of AML with subsequent disease progression despite reduction of posttransplant graft-versus-host disease prophylactic immune suppression and a cycle of clofarabine. The patient was hospitalized 2 months after the SCT for neutropenic fever and was found to have vancomycin-resistant enterococcal bacteremia, Clostridium difficile colitis, and possible fungal pneumonia. He was treated with voriconazole 200 mg twice daily, which he continued following discharge for antifungal prophylaxis. At the time of discharge, the patient reported that he noticed an asymptomatic “purple papule” on the right knee but did not seek further workup.
Two months later, the patient presented with a fever (temperature, 38.6°C) and leukocytosis (white blood cell count, 130 cells/mL [increased from 53 cells/mL 1 week prior to admission]). Due to his history of immunosuppression and neutropenia, the patient was placed on a broad-spectrum antibiotic regimen of cefepime, daptomycin, and linezolid on admission. Later, vancomycin and gentamicin were added and voriconazole was switched to caspofungin. The patient also received granulocyte-macrophage colony-stimulating factor for neutropenia. During the current hospitalization, blood cultures demonstrated vancomycin-resistant enterococcemia, and computed tomography of the chest revealed findings consistent with multilobar pneumonia.
Dermatology was consulted to evaluate the purple nodule on the right knee, which had slowly progressed since his last admission. Physical examination revealed a violaceous, 1.5×1.5-cm nodule with central necrosis covered by black eschar with surrounding erythema (Figure 1). Biopsy specimens for routine histology and a tissue culture were obtained. Histopathologic examination revealed a dense diffuse infiltrate of large hyperchromatic mononuclear cells extending through the dermis, which was consistent with the patient’s known AML (Figure 2). Acid-fast bacillus staining was negative for mycobacterial organisms. Grocott-Gomori methenamine-silver stain demonstrated an overwhelming number of septate fungal hyphae with acute-angle branching, concerning for Aspergillus species (Figure 3). Of note, an Aspergillus serum antigen test was performed at this time and was negative. On repeat review of the routine histologic sections, angioinvasion by hyphae was detected amidst the dense lymphocytic infiltrate (Figure 4).
Given the patient’s immunocompromised state, the presence of angioinvasive fungi on the skin biopsy, and unresolved pneumonia, the patient was restarted on voriconazole for treatment of likely Aspergillus infection. He was continued on chemotherapy for the primary refractory AML and received a donor lymphocyte infusion prior to discharge. After the patient was discharged, the tissue culture grew Paecilomyces, a rare fungal species. The patient died 1 week after discharge.
|
|
Comment
Leukemia cutis is an extramedullary manifestation of leukemia that appears in 10% to 15% of patients with AML.2 The frequency of LC differs widely for the various types of AML, with the majority of cases occurring in the acute myelomonocytic leukemia (M4) or acute monocytic leukemia (M5) subtypes.3,4 One large study of AML patients (N=381) demonstrated an incidence of LC in 28.6% of patients with the M4 subtype and 42.9% of those with the M5 subtype, with an incidence of only 7.1% of patients with the M1 subtype.3 It occurs less frequently in chronic myeloproliferative diseases.2,4
Leukemia cutis has a wide range of cutaneous manifestations and may present with solitary or multiple papular, nodular, or plaquelike lesions that are red-brown, blue, violaceous, or hemorrhagic.4 Leukemia cutis occurs most commonly on the legs, followed by the arms, back, chest, scalp, and face.2,4 Leukemia cutis may be hard to distinguish clinically from other conditions such as cutaneous metastases of visceral malignancies, lymphoma, drug eruptions, and opportunistic infections. Leukemia cutis ulcers often measure only a few centimeters in diameter with a firmly adherent purulent or hemorrhagic crust and may occur in unusual locations. These lesions usually are treatment resistant and their persistence may help to lead to diagnosis.4
Microscopically, most LC lesions show a perivascular or periadnexal pattern of involvement or a dense diffuse, interstitial, or nodular atypical lymphocytic infiltrate involving the dermis and subcutis with sparing of the upper papillary dermis.2 The cytologic appearance of M1 and M2 subtypes of AML are characterized by medium-sized to large mononuclear cells with a light cytoplasm and large basophilic cell nuclei. The M4 and M5 subtypes of AML generally are dominated by medium-sized, round or oval-shaped mononuclear cells that may have eosinophilic cytoplasm and segmented or kidney-shaped basophilic nuclei.4 Immunophenotyping is crucial for diagnosis. In myeloid disorders, there is positive staining with markers of myeloid lineage such as myeloperoxidase, lysozyme, CD34, CD15, CD68, CD43, and CD117.5
In our patient, there was an ulcerated dense diffuse dermal infiltrate of large atypical lymphocytes consistent with LC and positive immunostaining consistent with LC associated with AML. Additionally, septate hyphae with acute-angle branching also were noted in the dermal blood vessels on hematoxylin and eosin and fungal staining, demonstrating concomitant fungal infection. Angioinvasion of organisms demonstrated on skin biopsy and persistent pneumonia noted on chest imaging suggested a disseminated infectious process.
Invasive fungal infections are an increasing cause of morbidity and mortality in immunocompromised individuals, including those with hematologic malignancies and hematologic SCTs. Despite an increasing number of antifungal therapies, outcomes are frequently suboptimal with mortality rates often greater than 50% depending on the pathogen and disease.6 Thus, there must be a high index of suspicion of infection even when a separate histopathologic diagnosis is available, such as the finding of leukemic infiltrates in this patient’s biopsy specimen. A similar case of LC has been reported with concomitant fungal infection involving Fusarium and Enterococcus.7 Patients with leukemic cells may develop leukemic infiltrates in response to cutaneous infection, and a high index of suspicion for 2 related but distinct processes is necessary.
Paecilomyces species are an emerging cause of opportunistic and usually severe human infections.8-11 The Paecilomyces species are saprophytic filamentous fungi that are found worldwide in soil as well as contaminants in the air and water.12Paecilomyces infection is generally associated with the use of immunosuppressive therapies, implants, or ocular surgery. Among species in this genus, Paecilomyces lilacinus and Paecilomyces variotii are of clinical importance. Most species have high susceptibility to the newer azoles such as voriconazole.6
Conclusion
Despite continued treatment with voriconazole, our patient still developed a rare fungal infection arising in a lesion of LC. He had signs of infection, including an elevated white blood cell count, fever, and malaise, which are nonspecific clinical findings that could have been attributed to known relapse of systemic leukemia or to known enterococcemia. Even in patients on antifungal prophylaxis and with other possible causes of leukocytosis, this case illustrates that there must be a high index of suspicion for angioinvasive fungal infection.
1. Aquilera SB, Zarraga M, Rosen L. Leukemia cutis in a patient with acute myelogenous leukemia: a case report and review of the literature. Cutis. 2010;85:31-36.
2. Cho-Vega JH, Medeiros J, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130.
3. Agis H, Weltermann A, Fonatsch C, et al. A comparative study on demographic, hematological, and cytogenetic findings and prognosis in acute myeloid leukemia with and without leukemia cutis. Ann Hematol. 2002;81:90-95.
4. Wagner G, Fenchel K, Back W, et al. Leukemia cutis—epidemiology, clinical presentation, and differential diagnoses. J Dtsch Dermatol Ges. 2012;10:27-36.
5. Hejmadi RK, Thompson D, Shah F, et al. Cutaneous presentation of aleukemic monoblastic leukemia cutis: a case report and review of literature with focus on immunohistochemistry. J Cutan Pathol. 2008;35(suppl 1):46.
6. Kontoyiannis DP. Invasive mycoses: strategies for effective management. Am J Med. 2012;125(suppl 1):25-38.
7. Feramisco JD, Hsiao JL, Fox LP, et al. Angioinvasive Fusarium and concomitant Enterococcus infection arising in association with leukemia cutis. J Cutan Pathol. 2011;38:926-929.
8. Antachopoulos C, Walsh TJ, Roilides E. Fungal infections in primary immunodeficiencies. Eur J Pediatr. 2007;166:1099-1117.
9. Carey J, D’Amico R, Sutton DA, et al. Paecilomyces lilacinus vaginitis in an immuno-competent patient. Emerg Infect Dis. 2003;9:1155-1158.
10. Castro LG, Salebian A, Sotto MN. Hyalohyphomycosis by Paecilomyces lilacinus in a renal transplant patient and a review of human Paecilomyces species infections. J Med Vet Mycol. 1990;28:15-26.
11. Pastor FJ, Guarro J. Clinical manifestations, treatment and outcome of Paecilomyces lilacinus infections. Clin Microbiol Infect. 2006;12:948-960.
12. Castelli MV, Alastruey-Izquierdo A, Cuesta I, et al. Susceptibility testing and molecular classification of Paecilomyces spp. Antimicrob Agents Chemother. 2008;52:2926-2928.
1. Aquilera SB, Zarraga M, Rosen L. Leukemia cutis in a patient with acute myelogenous leukemia: a case report and review of the literature. Cutis. 2010;85:31-36.
2. Cho-Vega JH, Medeiros J, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130.
3. Agis H, Weltermann A, Fonatsch C, et al. A comparative study on demographic, hematological, and cytogenetic findings and prognosis in acute myeloid leukemia with and without leukemia cutis. Ann Hematol. 2002;81:90-95.
4. Wagner G, Fenchel K, Back W, et al. Leukemia cutis—epidemiology, clinical presentation, and differential diagnoses. J Dtsch Dermatol Ges. 2012;10:27-36.
5. Hejmadi RK, Thompson D, Shah F, et al. Cutaneous presentation of aleukemic monoblastic leukemia cutis: a case report and review of literature with focus on immunohistochemistry. J Cutan Pathol. 2008;35(suppl 1):46.
6. Kontoyiannis DP. Invasive mycoses: strategies for effective management. Am J Med. 2012;125(suppl 1):25-38.
7. Feramisco JD, Hsiao JL, Fox LP, et al. Angioinvasive Fusarium and concomitant Enterococcus infection arising in association with leukemia cutis. J Cutan Pathol. 2011;38:926-929.
8. Antachopoulos C, Walsh TJ, Roilides E. Fungal infections in primary immunodeficiencies. Eur J Pediatr. 2007;166:1099-1117.
9. Carey J, D’Amico R, Sutton DA, et al. Paecilomyces lilacinus vaginitis in an immuno-competent patient. Emerg Infect Dis. 2003;9:1155-1158.
10. Castro LG, Salebian A, Sotto MN. Hyalohyphomycosis by Paecilomyces lilacinus in a renal transplant patient and a review of human Paecilomyces species infections. J Med Vet Mycol. 1990;28:15-26.
11. Pastor FJ, Guarro J. Clinical manifestations, treatment and outcome of Paecilomyces lilacinus infections. Clin Microbiol Infect. 2006;12:948-960.
12. Castelli MV, Alastruey-Izquierdo A, Cuesta I, et al. Susceptibility testing and molecular classification of Paecilomyces spp. Antimicrob Agents Chemother. 2008;52:2926-2928.
Practice Points
- Immunosuppressed patients are at risk for atypical presentations of common infections as well as infection with rare pathogens.
- Skin biopsy and tissue culture play an important role in identifying infectious agents in immunosuppressed patients.
- Leukemic infiltrates may mask pathogens, and pathologists should strongly consider additional stains when indicated.
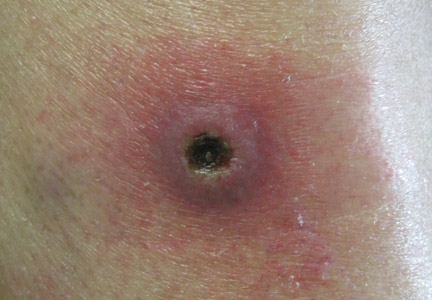
Extramammary Paget Disease
Extramammary Paget disease (EMPD) is an uncommon condition that usually presents in apocrine sweat gland–rich areas, most commonly the vulva followed by the perianal region. Lesions clinically present as erythematous, well-demarcated plaques that may become ulcerated, erosive, scaly, or eczematous. Extramammary Paget disease has a female predominance and usually occurs in the sixth to eighth decades of life.1 Histologically, EMPD displays intraepidermal spread of large cells with plentiful amphophilic cytoplasm and large nuclei (Figure 1). These atypical cells may be seen “spit out” whole into the stratum corneum rather than keratinizing into parakeratotic cells (Figure 2). Frequently, the cytoplasm of these tumor cells is positive on mucicarmine staining, which indicates the presence of mucin, giving the cytoplasm a bluish gray color on hematoxylin and eosin–stained sections. Typically, EMPD cells can be found alone or in nests throughout the epithelium. The basal layer of the epithelium will appear crushed but not infiltrated by these atypical cells in some areas.2 Extramammary Paget disease is epithelial membrane antigen and cytokeratin 7 positive, unlike other conditions in the differential diagnosis such as benign acral nevus, Bowen disease, mycosis fungoides, and superficial spreading melanoma in situ, with the rare exception of cytokeratin 7 positivity in Bowen disease.3
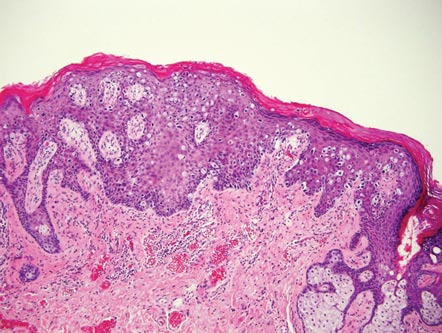
| 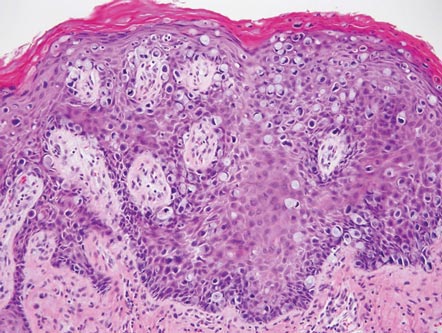
|
Benign acral nevi, similar to melanoma in situ, can have melanocytes scattered above the basal layer, but they usually appear in the lower half of the epidermis without cytologic atypia.4 When present, these pagetoid cells are most often limited to the center of a well-delineated lesion. The compact thick stratum corneum characteristic of acral skin also is helpful in distinguishing a benign acral nevus from EMPD, which does not involve acral sites (Figure 3).2
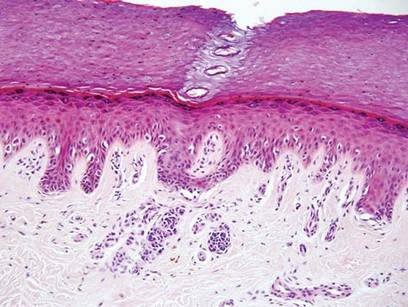
Bowen disease (squamous cell carcinoma in situ) may have pagetoid spread (or buckshot scatter) through the epidermis similar to EMPD and melanoma in situ. However, in Bowen disease the malignant cells are keratinocytes that keratinize and become incorporated into the stratum corneum as parakeratotic nuclei rather than intact “spit out” cells, as seen in melanoma in situ and EMPD. Usually the pagetoid spread is only focal in Bowen disease with other areas of more characteristic full-thickness keratinocyte atypia (Figure 4).2
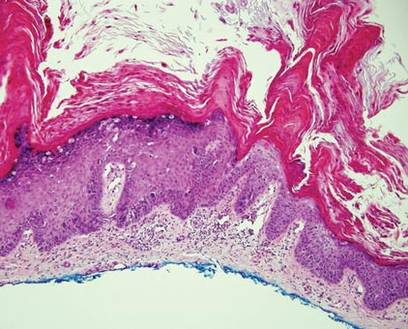
Mycosis fungoides displays atypical lymphocytes with large dark nuclei and minimal to no cytoplasm scattered throughout the epidermis. The atypical cells have irregular nuclear contours and often a clear perinuclear space (Figure 5). These cells tend to line up along the dermoepidermal junction and form intraepidermal clusters known as Pautrier microabscesses. Papillary dermal fibroplasia also is usually present in mycosis fungoides.2
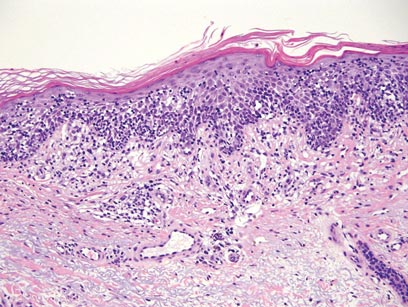
Similar to EMPD, superficial spreading melanoma in situ shows single or nested atypical cells scattered throughout all levels of the epithelium and may be “spit out” whole into the stratum corneum rather than keratinizing into parakeratotic cells. However, in melanoma, nests of atypical melanocytes predominate and involve the basal layer (Figure 6), whereas clusters of cells in EMPD typically are located superficial to the basal layer. The cells of melanoma also lack the amphophilic mucinous cytoplasm of EMPD.1
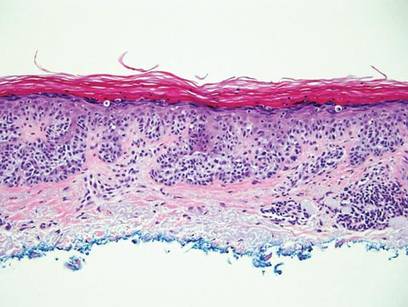
1. Calonje E, Brenn T, Lazar A, et al. McKee’s Pathology of the Skin. 4th ed. London, England: Elsevier Saunders; 2011.
2. Ferringer T, Elston D, eds. Dermatopathology. 2nd ed. London, England: Elsevier; 2014.
3. Sah SP, Kelly PJ, McManus DT, et al. Diffuse CK7, CAM5.2 and BerEP4 positivity in pagetoid squamous cell carcinoma in situ (pagetoid Bowen’s disease) of the perianal region: a mimic of extramammary Paget’s disease. Histopathology. 2013;62:511-514.
4. LeBoit PE. A diagnosis for maniacs. Am J Dermatopathol. 2000;22:556-558.
Extramammary Paget disease (EMPD) is an uncommon condition that usually presents in apocrine sweat gland–rich areas, most commonly the vulva followed by the perianal region. Lesions clinically present as erythematous, well-demarcated plaques that may become ulcerated, erosive, scaly, or eczematous. Extramammary Paget disease has a female predominance and usually occurs in the sixth to eighth decades of life.1 Histologically, EMPD displays intraepidermal spread of large cells with plentiful amphophilic cytoplasm and large nuclei (Figure 1). These atypical cells may be seen “spit out” whole into the stratum corneum rather than keratinizing into parakeratotic cells (Figure 2). Frequently, the cytoplasm of these tumor cells is positive on mucicarmine staining, which indicates the presence of mucin, giving the cytoplasm a bluish gray color on hematoxylin and eosin–stained sections. Typically, EMPD cells can be found alone or in nests throughout the epithelium. The basal layer of the epithelium will appear crushed but not infiltrated by these atypical cells in some areas.2 Extramammary Paget disease is epithelial membrane antigen and cytokeratin 7 positive, unlike other conditions in the differential diagnosis such as benign acral nevus, Bowen disease, mycosis fungoides, and superficial spreading melanoma in situ, with the rare exception of cytokeratin 7 positivity in Bowen disease.3
|
|
|
Benign acral nevi, similar to melanoma in situ, can have melanocytes scattered above the basal layer, but they usually appear in the lower half of the epidermis without cytologic atypia.4 When present, these pagetoid cells are most often limited to the center of a well-delineated lesion. The compact thick stratum corneum characteristic of acral skin also is helpful in distinguishing a benign acral nevus from EMPD, which does not involve acral sites (Figure 3).2
Bowen disease (squamous cell carcinoma in situ) may have pagetoid spread (or buckshot scatter) through the epidermis similar to EMPD and melanoma in situ. However, in Bowen disease the malignant cells are keratinocytes that keratinize and become incorporated into the stratum corneum as parakeratotic nuclei rather than intact “spit out” cells, as seen in melanoma in situ and EMPD. Usually the pagetoid spread is only focal in Bowen disease with other areas of more characteristic full-thickness keratinocyte atypia (Figure 4).2
Mycosis fungoides displays atypical lymphocytes with large dark nuclei and minimal to no cytoplasm scattered throughout the epidermis. The atypical cells have irregular nuclear contours and often a clear perinuclear space (Figure 5). These cells tend to line up along the dermoepidermal junction and form intraepidermal clusters known as Pautrier microabscesses. Papillary dermal fibroplasia also is usually present in mycosis fungoides.2
Similar to EMPD, superficial spreading melanoma in situ shows single or nested atypical cells scattered throughout all levels of the epithelium and may be “spit out” whole into the stratum corneum rather than keratinizing into parakeratotic cells. However, in melanoma, nests of atypical melanocytes predominate and involve the basal layer (Figure 6), whereas clusters of cells in EMPD typically are located superficial to the basal layer. The cells of melanoma also lack the amphophilic mucinous cytoplasm of EMPD.1
Extramammary Paget disease (EMPD) is an uncommon condition that usually presents in apocrine sweat gland–rich areas, most commonly the vulva followed by the perianal region. Lesions clinically present as erythematous, well-demarcated plaques that may become ulcerated, erosive, scaly, or eczematous. Extramammary Paget disease has a female predominance and usually occurs in the sixth to eighth decades of life.1 Histologically, EMPD displays intraepidermal spread of large cells with plentiful amphophilic cytoplasm and large nuclei (Figure 1). These atypical cells may be seen “spit out” whole into the stratum corneum rather than keratinizing into parakeratotic cells (Figure 2). Frequently, the cytoplasm of these tumor cells is positive on mucicarmine staining, which indicates the presence of mucin, giving the cytoplasm a bluish gray color on hematoxylin and eosin–stained sections. Typically, EMPD cells can be found alone or in nests throughout the epithelium. The basal layer of the epithelium will appear crushed but not infiltrated by these atypical cells in some areas.2 Extramammary Paget disease is epithelial membrane antigen and cytokeratin 7 positive, unlike other conditions in the differential diagnosis such as benign acral nevus, Bowen disease, mycosis fungoides, and superficial spreading melanoma in situ, with the rare exception of cytokeratin 7 positivity in Bowen disease.3
|
|
|
Benign acral nevi, similar to melanoma in situ, can have melanocytes scattered above the basal layer, but they usually appear in the lower half of the epidermis without cytologic atypia.4 When present, these pagetoid cells are most often limited to the center of a well-delineated lesion. The compact thick stratum corneum characteristic of acral skin also is helpful in distinguishing a benign acral nevus from EMPD, which does not involve acral sites (Figure 3).2
Bowen disease (squamous cell carcinoma in situ) may have pagetoid spread (or buckshot scatter) through the epidermis similar to EMPD and melanoma in situ. However, in Bowen disease the malignant cells are keratinocytes that keratinize and become incorporated into the stratum corneum as parakeratotic nuclei rather than intact “spit out” cells, as seen in melanoma in situ and EMPD. Usually the pagetoid spread is only focal in Bowen disease with other areas of more characteristic full-thickness keratinocyte atypia (Figure 4).2
Mycosis fungoides displays atypical lymphocytes with large dark nuclei and minimal to no cytoplasm scattered throughout the epidermis. The atypical cells have irregular nuclear contours and often a clear perinuclear space (Figure 5). These cells tend to line up along the dermoepidermal junction and form intraepidermal clusters known as Pautrier microabscesses. Papillary dermal fibroplasia also is usually present in mycosis fungoides.2
Similar to EMPD, superficial spreading melanoma in situ shows single or nested atypical cells scattered throughout all levels of the epithelium and may be “spit out” whole into the stratum corneum rather than keratinizing into parakeratotic cells. However, in melanoma, nests of atypical melanocytes predominate and involve the basal layer (Figure 6), whereas clusters of cells in EMPD typically are located superficial to the basal layer. The cells of melanoma also lack the amphophilic mucinous cytoplasm of EMPD.1
1. Calonje E, Brenn T, Lazar A, et al. McKee’s Pathology of the Skin. 4th ed. London, England: Elsevier Saunders; 2011.
2. Ferringer T, Elston D, eds. Dermatopathology. 2nd ed. London, England: Elsevier; 2014.
3. Sah SP, Kelly PJ, McManus DT, et al. Diffuse CK7, CAM5.2 and BerEP4 positivity in pagetoid squamous cell carcinoma in situ (pagetoid Bowen’s disease) of the perianal region: a mimic of extramammary Paget’s disease. Histopathology. 2013;62:511-514.
4. LeBoit PE. A diagnosis for maniacs. Am J Dermatopathol. 2000;22:556-558.
1. Calonje E, Brenn T, Lazar A, et al. McKee’s Pathology of the Skin. 4th ed. London, England: Elsevier Saunders; 2011.
2. Ferringer T, Elston D, eds. Dermatopathology. 2nd ed. London, England: Elsevier; 2014.
3. Sah SP, Kelly PJ, McManus DT, et al. Diffuse CK7, CAM5.2 and BerEP4 positivity in pagetoid squamous cell carcinoma in situ (pagetoid Bowen’s disease) of the perianal region: a mimic of extramammary Paget’s disease. Histopathology. 2013;62:511-514.
4. LeBoit PE. A diagnosis for maniacs. Am J Dermatopathol. 2000;22:556-558.
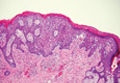
Primary Localized Cutaneous Nodular Amyloidosis of the Thighs
Case Report
Clinical Findings
A 65-year-old woman presented with multiple asymptomatic discrete nodules and atrophic plaques on the thighs of 4 years’ duration. The lesions had started as 2 small, asymptomatic, madder red plaques symmetrically located on the anterior aspect of each thigh that had gradually increased in number and size, particularly on the right thigh. Two years later, 2 new atrophic plaques appeared on the anterior aspect of each. The lesions developed slowly but never remitted and had been misdiagnosed as primary macular atrophy of skin by several outpatient clinics. The patient’s general health was good and her personal and family history was unremarkable.
Physical examination revealed multiple madder red plaques and nodules of various shapes and sizes (ie, 1–3 cm in diameter) on the anterior aspect of the right thigh. The lesions were slightly elevated with a waxy surface, firm, and painless to palpation. One similar lesion was noted on the anterior aspect of the left thigh. Two 2-cm, brown-red, atrophic plaques also were noted in a symmetrical distribution on the anterior aspect of each thigh. The plaque surfaces were slightly crinkly and shiny, and anetodermalike lesions produced a buttonhole sign identical to a neurofibroma on palpation (Figure 1).
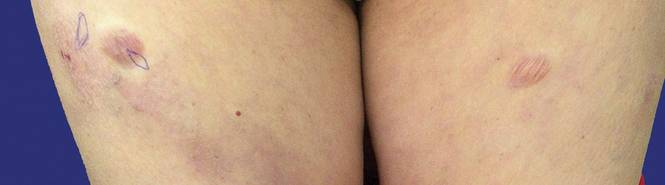
Histopathologic Findings
Two biopsy specimens were taken from a nodule and an atrophic plaque on the right thigh. Microscopic examination revealed deposition of homogeneous eosinophilic material in the reticular dermis and subcutis as well as around the fine vessels (Figure 2A). There was mild cellular infiltration of lymphocytes, plasma cells, and giant cells in the dermis, especially adjacent to deposits and around the vessels (Figure 2B). The homogeneous material appeared salmon pink on Congo red staining and bright green by thioflavin T staining using a fluorescent microscope (Figures 3 and 4). These results suggested the characteristic features of cutaneous nodular amyloidosis.
Figure 2. Histopathologically, homogeneous eosinophilic material deposited in the reticular dermis and subcutis was noted (A)(H&E, original magnification ×25). Microscopic examination showed lymphocytes and plasma cells infiltrated in the dermis, especially adjacent to deposits and around the vessels (B)(H&E, original magnification ×200). |
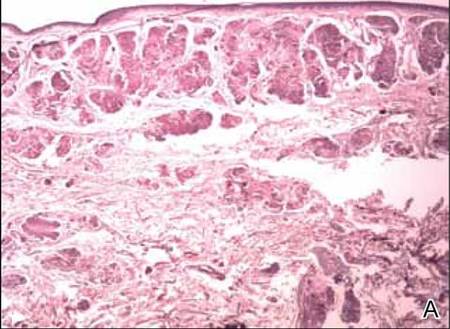
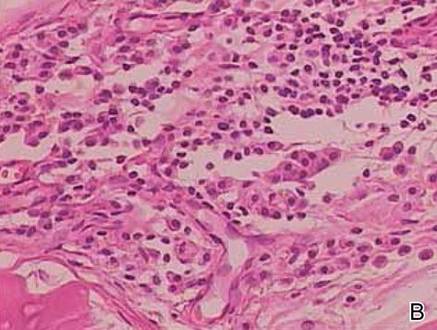
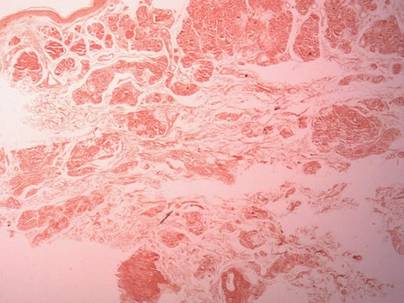
|
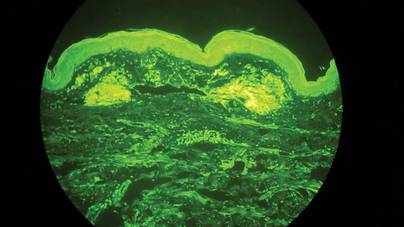
|
Laboratory Findings
Laboratory studies showed normal results for complete blood cell count, urinalysis, liver and renal function tests, blood glucose levels, lipid panel, and erythrocyte sedimentation rate. Serum protein electrophoresis was normal and no Bence Jones proteins were detected. Serum IgA, IgG, and IgM levels showed no abnormalities. Electrocardiogram, chest radiography, and abdominal ultrasound were normal.
A diagnosis of primary localized cutaneous nodular amyloidosis (PLCNA) was made based on clinical, histopathologic, and laboratory findings. Although surgical excision in stages was proposed, the patient refused treatment because the lesions were asymptomatic. There was no obvious progression of the skin lesions and no abnormal systemic findings during 2.5 years’ follow-up.
Comment
Amyloidosis is a spectrum of diseases consisting of deposition of amyloid proteins in various tissues. Clinically, amyloidosis is divided into both primary and secondary forms of systemic amyloidosis, hemodialysis-associated amyloidosis, heredofamilial amyloidosis, and cutaneous amyloidosis. Primary cutaneous amyloidosis is localized to the skin without other organ involvement and does not occur in systemic amyloidosis. Secondary cutaneous involvement in systemic amyloidosis is rare. Most cases of primary localized cutaneous amyloidosis (PLCA) are sporadic but approximately 10% of cases may be familial.1 There are 3 main forms of localized cutaneous amyloidosis: macular, lichen, and nodular amyloidosis. Nodular cutaneous amyloidosis is the rarest form of PLCA.
Nodular amyloidosis was first described by Gottron in 1950.2 Its cutaneous lesions may present as single or multiple nodules, occasionally with overlying atrophic plaques. The lesions consist of firm, smooth-surfaced, waxy or rubbery, pink to tan papules, plaques, or nodules measuring up to several centimeters. On some lesions, surface telangiectasia may be seen. Bullous-appearing and anetodermalike lesions have been reported.3 The acral region is the most common location, followed by the legs, head, trunk, arms, and genitalia, respectively.4 In some cases the lesions can spontaneously improve over time. In our patient, the lesions were composed of both multiple nodules and atrophic plaques, which is uncommon.
The pathogenesis of amyloid deposition is still unknown. Cutaneous macular and lichen amyloidosis may originate from degenerated keratinocyte intermediate filaments. Nodular amyloidosis may represent a localized plasma cell dyscrasia that can be associated with a monoclonal gammopathy or multiple myeloma.5 Some components of amyloid in some cases of PLCNA may consist of κ and λ immunoglobulin light chains, with most reported cases being of the l subtype.6 The results of one study indicated that β2-microglobulin was another major component of amyloid fibrils and that β2-microglobulin was partly subjected to the modification of advanced glycation end product in PLCNA.7
The histopathologic examination of PLCNA is characterized by large deposits of amorphous, sometimes fissured, pale, eosinophilic material in the papillary dermis, reticular dermis, and subcutaneous fat. The overlying epidermis may exhibit flattened rete ridges. Amyloid may occur within vessel walls and adnexal structures, sometimes in a ring surrounding individual fat cells. Clinically, the lesions of PLCNA may be indistinguishable from nodular deposits of amyloid occurring in primary systemic amyloidosis or myeloma-associated amyloidosis. Histopathologically, PLCNA usually has a variable infiltrate of plasma cells and lymphocytes at the periphery or within the amyloid deposits,6 but no single stain is highly sensitive and specific. Congo red–stained deposits showed salmon pink amorphous material or apple green birefringence with polarizing microscopy.8 Amyloid derived from immunoglobulin light chains, including cutaneous nodular amyloid, also stain positive for anti-human λ light chain antibody on immunohistochemistry. Additionally, amyloid can stain positively with methyl violet and crystal violet Gram stains, Picrosirius red, thioflavin T, Dylon, and periodic acid–Schiff stains.
Clinically, some PLCNA lesions can be removed via surgical excision or laser if they are cosmetically disfiguring or symptomatic. Other methods have been attempted to improve the appearance of the lesions, such as intralesional corticosteroids, cryotherapy, and dermabrasion,9 but they usually are not helpful and have a high rate of recurrence. Although PLCNA often is a benign cutaneous disorder and some cases of PLCNA could be reactive diseases rather than neoplastic ones, some patients may develop underlying systemic amyloidosis or even paraproteinemia.10 Northcutt and Vanover11 indicated that systemic amyloidosis may be expected in less than 15% of 47 patients with localized cutaneous amyloidosis during follow-up by reviewing most of the related literature. Some cases may be associated with Sjögren syndrome (SJS), CREST (calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, and telangiectasia) syndrome, dermatomyositis, and diabetes mellitus.12-14 Polyclonal immunoglobulin amyloid has been reported only in PLCNA with SJS, which may be due to the fact that a certain population of SJS develops polyclonal B-cell proliferation and hyperglobulinemia.12 Woollons and Black15 estimated the rate of progression of PLCNA to systemic amyloidosis to be only 7%, which is much lower than the rate in the literature by a large clinical follow-up study on PLCNA.16 However, all patients with PLCNA should have a systemic evaluation and should be advised to undergo long-term clinical follow-up to help prevent progression to systemic amyloidosis or plasma cell dyscrasia.
1. Sakuma TH, Hans-Filho G, Arita K, et al. Familial primary localized cutaneous amyloidosis in Brazil. Arch Dermatol. 2009;145:695-699.
2. Rodermund OE. On amyloidosis cutis nodularis atrophicans (Gottron 1950). at the same time a contribution to the classification of amyloidosis [in German]. Arch Klin Exp Dermatol. 1967;230:153-171.
3. Chapel TA, Birmingham DJ, Malinowski YE. Nodular primary localized cutaneous amyloidosis. Arch Dermatol. 1977;113:1248-1249.
4. Criado PR, Silva CS, Vasconcellos C, et al. Extensive nodular cutaneous amyloidosis: an unusual presentation. J Eur Acad Dermatol Venereol. 2005;19:481-483.
5. Touart DM, Sau P. Cutaneous deposition diseases. part I [published correction appears in J Am Acad Dermatol. 1998;39:1042]. J Am Acad Dermatol. 1998;39(2, pt 1):149-171; quiz 172-174.
6. Borrowman TA, Lutz ME, Walsh JS. Cutaneous nodular amyloidosis masquerading as a foot callus. J Am Acad Dermatol. 2003;49:307-310.
7. Fujimoto N, Yajima M, Ohnishi Y, et al. Advanced glycation end product-modified beta2-microglobulin is a component of amyloid fibrils of primary localized cutaneous nodular amyloidosis. J Invest Dermatol. 2002;118:479-484.
8. Clement CG, Truong LD. An evaluation of Congo red fluorescence for the diagnosis of amyloidosis. Hum Pathol. 2014;45:1766-1772.
9. Lien MH, Railan D, Nelson BR. The efficacy of dermabrasion in the treatment of nodular amyloidosis. J Am Acad Dermatol. 1997;36(2, pt 2):315-316.
10. Taylor SC, Baker E, Grossman ME. Nodular vulvar amyloid as a presentation of systemic amyloidosis. J Am Acad Dermatol. 1991;24:139.
11. Northcutt AD, Vanover MJ. Nodular cutaneous amyloidosis involving the vulva. case report and literature review. Arch Dermatol. 1985;121:518-521.
12. Yoneyama K, Tochigi N, Oikawa A, et al. Primary localized cutaneous nodular amyloidosis in a patient with Sjögren’s syndrome: a review of the literature. J Dermatol. 2005;32:120-123.
13. Summers EM, Kendrick CG. Primary localized cutaneous nodular amyloidosis and CREST syndrome: a case report and review of the literature. Cutis. 2008;82:55-59.
14. Taniguchi Y, Horino T, Terada Y. Cutaneous amyloidosis associated with amyopathic dermatomyositis. J Rheumatol. 2009;36:1088-1089.
15. Woollons A, Black MM. Nodular localized primary cutaneous amyloidosis: a long-term follow-up study. Br J Dermatol. 2001;145:105-109.
16. Brownstein MH, Helwig EB. The cutaneous amyloidoses. I. localized forms. Arch Dermatol. 1970;102:8-19.
Case Report
Clinical Findings
A 65-year-old woman presented with multiple asymptomatic discrete nodules and atrophic plaques on the thighs of 4 years’ duration. The lesions had started as 2 small, asymptomatic, madder red plaques symmetrically located on the anterior aspect of each thigh that had gradually increased in number and size, particularly on the right thigh. Two years later, 2 new atrophic plaques appeared on the anterior aspect of each. The lesions developed slowly but never remitted and had been misdiagnosed as primary macular atrophy of skin by several outpatient clinics. The patient’s general health was good and her personal and family history was unremarkable.
Physical examination revealed multiple madder red plaques and nodules of various shapes and sizes (ie, 1–3 cm in diameter) on the anterior aspect of the right thigh. The lesions were slightly elevated with a waxy surface, firm, and painless to palpation. One similar lesion was noted on the anterior aspect of the left thigh. Two 2-cm, brown-red, atrophic plaques also were noted in a symmetrical distribution on the anterior aspect of each thigh. The plaque surfaces were slightly crinkly and shiny, and anetodermalike lesions produced a buttonhole sign identical to a neurofibroma on palpation (Figure 1).
Histopathologic Findings
Two biopsy specimens were taken from a nodule and an atrophic plaque on the right thigh. Microscopic examination revealed deposition of homogeneous eosinophilic material in the reticular dermis and subcutis as well as around the fine vessels (Figure 2A). There was mild cellular infiltration of lymphocytes, plasma cells, and giant cells in the dermis, especially adjacent to deposits and around the vessels (Figure 2B). The homogeneous material appeared salmon pink on Congo red staining and bright green by thioflavin T staining using a fluorescent microscope (Figures 3 and 4). These results suggested the characteristic features of cutaneous nodular amyloidosis.
Figure 2. Histopathologically, homogeneous eosinophilic material deposited in the reticular dermis and subcutis was noted (A)(H&E, original magnification ×25). Microscopic examination showed lymphocytes and plasma cells infiltrated in the dermis, especially adjacent to deposits and around the vessels (B)(H&E, original magnification ×200). |
|
|
|
|
Laboratory Findings
Laboratory studies showed normal results for complete blood cell count, urinalysis, liver and renal function tests, blood glucose levels, lipid panel, and erythrocyte sedimentation rate. Serum protein electrophoresis was normal and no Bence Jones proteins were detected. Serum IgA, IgG, and IgM levels showed no abnormalities. Electrocardiogram, chest radiography, and abdominal ultrasound were normal.
A diagnosis of primary localized cutaneous nodular amyloidosis (PLCNA) was made based on clinical, histopathologic, and laboratory findings. Although surgical excision in stages was proposed, the patient refused treatment because the lesions were asymptomatic. There was no obvious progression of the skin lesions and no abnormal systemic findings during 2.5 years’ follow-up.
Comment
Amyloidosis is a spectrum of diseases consisting of deposition of amyloid proteins in various tissues. Clinically, amyloidosis is divided into both primary and secondary forms of systemic amyloidosis, hemodialysis-associated amyloidosis, heredofamilial amyloidosis, and cutaneous amyloidosis. Primary cutaneous amyloidosis is localized to the skin without other organ involvement and does not occur in systemic amyloidosis. Secondary cutaneous involvement in systemic amyloidosis is rare. Most cases of primary localized cutaneous amyloidosis (PLCA) are sporadic but approximately 10% of cases may be familial.1 There are 3 main forms of localized cutaneous amyloidosis: macular, lichen, and nodular amyloidosis. Nodular cutaneous amyloidosis is the rarest form of PLCA.
Nodular amyloidosis was first described by Gottron in 1950.2 Its cutaneous lesions may present as single or multiple nodules, occasionally with overlying atrophic plaques. The lesions consist of firm, smooth-surfaced, waxy or rubbery, pink to tan papules, plaques, or nodules measuring up to several centimeters. On some lesions, surface telangiectasia may be seen. Bullous-appearing and anetodermalike lesions have been reported.3 The acral region is the most common location, followed by the legs, head, trunk, arms, and genitalia, respectively.4 In some cases the lesions can spontaneously improve over time. In our patient, the lesions were composed of both multiple nodules and atrophic plaques, which is uncommon.
The pathogenesis of amyloid deposition is still unknown. Cutaneous macular and lichen amyloidosis may originate from degenerated keratinocyte intermediate filaments. Nodular amyloidosis may represent a localized plasma cell dyscrasia that can be associated with a monoclonal gammopathy or multiple myeloma.5 Some components of amyloid in some cases of PLCNA may consist of κ and λ immunoglobulin light chains, with most reported cases being of the l subtype.6 The results of one study indicated that β2-microglobulin was another major component of amyloid fibrils and that β2-microglobulin was partly subjected to the modification of advanced glycation end product in PLCNA.7
The histopathologic examination of PLCNA is characterized by large deposits of amorphous, sometimes fissured, pale, eosinophilic material in the papillary dermis, reticular dermis, and subcutaneous fat. The overlying epidermis may exhibit flattened rete ridges. Amyloid may occur within vessel walls and adnexal structures, sometimes in a ring surrounding individual fat cells. Clinically, the lesions of PLCNA may be indistinguishable from nodular deposits of amyloid occurring in primary systemic amyloidosis or myeloma-associated amyloidosis. Histopathologically, PLCNA usually has a variable infiltrate of plasma cells and lymphocytes at the periphery or within the amyloid deposits,6 but no single stain is highly sensitive and specific. Congo red–stained deposits showed salmon pink amorphous material or apple green birefringence with polarizing microscopy.8 Amyloid derived from immunoglobulin light chains, including cutaneous nodular amyloid, also stain positive for anti-human λ light chain antibody on immunohistochemistry. Additionally, amyloid can stain positively with methyl violet and crystal violet Gram stains, Picrosirius red, thioflavin T, Dylon, and periodic acid–Schiff stains.
Clinically, some PLCNA lesions can be removed via surgical excision or laser if they are cosmetically disfiguring or symptomatic. Other methods have been attempted to improve the appearance of the lesions, such as intralesional corticosteroids, cryotherapy, and dermabrasion,9 but they usually are not helpful and have a high rate of recurrence. Although PLCNA often is a benign cutaneous disorder and some cases of PLCNA could be reactive diseases rather than neoplastic ones, some patients may develop underlying systemic amyloidosis or even paraproteinemia.10 Northcutt and Vanover11 indicated that systemic amyloidosis may be expected in less than 15% of 47 patients with localized cutaneous amyloidosis during follow-up by reviewing most of the related literature. Some cases may be associated with Sjögren syndrome (SJS), CREST (calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, and telangiectasia) syndrome, dermatomyositis, and diabetes mellitus.12-14 Polyclonal immunoglobulin amyloid has been reported only in PLCNA with SJS, which may be due to the fact that a certain population of SJS develops polyclonal B-cell proliferation and hyperglobulinemia.12 Woollons and Black15 estimated the rate of progression of PLCNA to systemic amyloidosis to be only 7%, which is much lower than the rate in the literature by a large clinical follow-up study on PLCNA.16 However, all patients with PLCNA should have a systemic evaluation and should be advised to undergo long-term clinical follow-up to help prevent progression to systemic amyloidosis or plasma cell dyscrasia.
Case Report
Clinical Findings
A 65-year-old woman presented with multiple asymptomatic discrete nodules and atrophic plaques on the thighs of 4 years’ duration. The lesions had started as 2 small, asymptomatic, madder red plaques symmetrically located on the anterior aspect of each thigh that had gradually increased in number and size, particularly on the right thigh. Two years later, 2 new atrophic plaques appeared on the anterior aspect of each. The lesions developed slowly but never remitted and had been misdiagnosed as primary macular atrophy of skin by several outpatient clinics. The patient’s general health was good and her personal and family history was unremarkable.
Physical examination revealed multiple madder red plaques and nodules of various shapes and sizes (ie, 1–3 cm in diameter) on the anterior aspect of the right thigh. The lesions were slightly elevated with a waxy surface, firm, and painless to palpation. One similar lesion was noted on the anterior aspect of the left thigh. Two 2-cm, brown-red, atrophic plaques also were noted in a symmetrical distribution on the anterior aspect of each thigh. The plaque surfaces were slightly crinkly and shiny, and anetodermalike lesions produced a buttonhole sign identical to a neurofibroma on palpation (Figure 1).
Histopathologic Findings
Two biopsy specimens were taken from a nodule and an atrophic plaque on the right thigh. Microscopic examination revealed deposition of homogeneous eosinophilic material in the reticular dermis and subcutis as well as around the fine vessels (Figure 2A). There was mild cellular infiltration of lymphocytes, plasma cells, and giant cells in the dermis, especially adjacent to deposits and around the vessels (Figure 2B). The homogeneous material appeared salmon pink on Congo red staining and bright green by thioflavin T staining using a fluorescent microscope (Figures 3 and 4). These results suggested the characteristic features of cutaneous nodular amyloidosis.
Figure 2. Histopathologically, homogeneous eosinophilic material deposited in the reticular dermis and subcutis was noted (A)(H&E, original magnification ×25). Microscopic examination showed lymphocytes and plasma cells infiltrated in the dermis, especially adjacent to deposits and around the vessels (B)(H&E, original magnification ×200). |
|
|
|
|
Laboratory Findings
Laboratory studies showed normal results for complete blood cell count, urinalysis, liver and renal function tests, blood glucose levels, lipid panel, and erythrocyte sedimentation rate. Serum protein electrophoresis was normal and no Bence Jones proteins were detected. Serum IgA, IgG, and IgM levels showed no abnormalities. Electrocardiogram, chest radiography, and abdominal ultrasound were normal.
A diagnosis of primary localized cutaneous nodular amyloidosis (PLCNA) was made based on clinical, histopathologic, and laboratory findings. Although surgical excision in stages was proposed, the patient refused treatment because the lesions were asymptomatic. There was no obvious progression of the skin lesions and no abnormal systemic findings during 2.5 years’ follow-up.
Comment
Amyloidosis is a spectrum of diseases consisting of deposition of amyloid proteins in various tissues. Clinically, amyloidosis is divided into both primary and secondary forms of systemic amyloidosis, hemodialysis-associated amyloidosis, heredofamilial amyloidosis, and cutaneous amyloidosis. Primary cutaneous amyloidosis is localized to the skin without other organ involvement and does not occur in systemic amyloidosis. Secondary cutaneous involvement in systemic amyloidosis is rare. Most cases of primary localized cutaneous amyloidosis (PLCA) are sporadic but approximately 10% of cases may be familial.1 There are 3 main forms of localized cutaneous amyloidosis: macular, lichen, and nodular amyloidosis. Nodular cutaneous amyloidosis is the rarest form of PLCA.
Nodular amyloidosis was first described by Gottron in 1950.2 Its cutaneous lesions may present as single or multiple nodules, occasionally with overlying atrophic plaques. The lesions consist of firm, smooth-surfaced, waxy or rubbery, pink to tan papules, plaques, or nodules measuring up to several centimeters. On some lesions, surface telangiectasia may be seen. Bullous-appearing and anetodermalike lesions have been reported.3 The acral region is the most common location, followed by the legs, head, trunk, arms, and genitalia, respectively.4 In some cases the lesions can spontaneously improve over time. In our patient, the lesions were composed of both multiple nodules and atrophic plaques, which is uncommon.
The pathogenesis of amyloid deposition is still unknown. Cutaneous macular and lichen amyloidosis may originate from degenerated keratinocyte intermediate filaments. Nodular amyloidosis may represent a localized plasma cell dyscrasia that can be associated with a monoclonal gammopathy or multiple myeloma.5 Some components of amyloid in some cases of PLCNA may consist of κ and λ immunoglobulin light chains, with most reported cases being of the l subtype.6 The results of one study indicated that β2-microglobulin was another major component of amyloid fibrils and that β2-microglobulin was partly subjected to the modification of advanced glycation end product in PLCNA.7
The histopathologic examination of PLCNA is characterized by large deposits of amorphous, sometimes fissured, pale, eosinophilic material in the papillary dermis, reticular dermis, and subcutaneous fat. The overlying epidermis may exhibit flattened rete ridges. Amyloid may occur within vessel walls and adnexal structures, sometimes in a ring surrounding individual fat cells. Clinically, the lesions of PLCNA may be indistinguishable from nodular deposits of amyloid occurring in primary systemic amyloidosis or myeloma-associated amyloidosis. Histopathologically, PLCNA usually has a variable infiltrate of plasma cells and lymphocytes at the periphery or within the amyloid deposits,6 but no single stain is highly sensitive and specific. Congo red–stained deposits showed salmon pink amorphous material or apple green birefringence with polarizing microscopy.8 Amyloid derived from immunoglobulin light chains, including cutaneous nodular amyloid, also stain positive for anti-human λ light chain antibody on immunohistochemistry. Additionally, amyloid can stain positively with methyl violet and crystal violet Gram stains, Picrosirius red, thioflavin T, Dylon, and periodic acid–Schiff stains.
Clinically, some PLCNA lesions can be removed via surgical excision or laser if they are cosmetically disfiguring or symptomatic. Other methods have been attempted to improve the appearance of the lesions, such as intralesional corticosteroids, cryotherapy, and dermabrasion,9 but they usually are not helpful and have a high rate of recurrence. Although PLCNA often is a benign cutaneous disorder and some cases of PLCNA could be reactive diseases rather than neoplastic ones, some patients may develop underlying systemic amyloidosis or even paraproteinemia.10 Northcutt and Vanover11 indicated that systemic amyloidosis may be expected in less than 15% of 47 patients with localized cutaneous amyloidosis during follow-up by reviewing most of the related literature. Some cases may be associated with Sjögren syndrome (SJS), CREST (calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, and telangiectasia) syndrome, dermatomyositis, and diabetes mellitus.12-14 Polyclonal immunoglobulin amyloid has been reported only in PLCNA with SJS, which may be due to the fact that a certain population of SJS develops polyclonal B-cell proliferation and hyperglobulinemia.12 Woollons and Black15 estimated the rate of progression of PLCNA to systemic amyloidosis to be only 7%, which is much lower than the rate in the literature by a large clinical follow-up study on PLCNA.16 However, all patients with PLCNA should have a systemic evaluation and should be advised to undergo long-term clinical follow-up to help prevent progression to systemic amyloidosis or plasma cell dyscrasia.
1. Sakuma TH, Hans-Filho G, Arita K, et al. Familial primary localized cutaneous amyloidosis in Brazil. Arch Dermatol. 2009;145:695-699.
2. Rodermund OE. On amyloidosis cutis nodularis atrophicans (Gottron 1950). at the same time a contribution to the classification of amyloidosis [in German]. Arch Klin Exp Dermatol. 1967;230:153-171.
3. Chapel TA, Birmingham DJ, Malinowski YE. Nodular primary localized cutaneous amyloidosis. Arch Dermatol. 1977;113:1248-1249.
4. Criado PR, Silva CS, Vasconcellos C, et al. Extensive nodular cutaneous amyloidosis: an unusual presentation. J Eur Acad Dermatol Venereol. 2005;19:481-483.
5. Touart DM, Sau P. Cutaneous deposition diseases. part I [published correction appears in J Am Acad Dermatol. 1998;39:1042]. J Am Acad Dermatol. 1998;39(2, pt 1):149-171; quiz 172-174.
6. Borrowman TA, Lutz ME, Walsh JS. Cutaneous nodular amyloidosis masquerading as a foot callus. J Am Acad Dermatol. 2003;49:307-310.
7. Fujimoto N, Yajima M, Ohnishi Y, et al. Advanced glycation end product-modified beta2-microglobulin is a component of amyloid fibrils of primary localized cutaneous nodular amyloidosis. J Invest Dermatol. 2002;118:479-484.
8. Clement CG, Truong LD. An evaluation of Congo red fluorescence for the diagnosis of amyloidosis. Hum Pathol. 2014;45:1766-1772.
9. Lien MH, Railan D, Nelson BR. The efficacy of dermabrasion in the treatment of nodular amyloidosis. J Am Acad Dermatol. 1997;36(2, pt 2):315-316.
10. Taylor SC, Baker E, Grossman ME. Nodular vulvar amyloid as a presentation of systemic amyloidosis. J Am Acad Dermatol. 1991;24:139.
11. Northcutt AD, Vanover MJ. Nodular cutaneous amyloidosis involving the vulva. case report and literature review. Arch Dermatol. 1985;121:518-521.
12. Yoneyama K, Tochigi N, Oikawa A, et al. Primary localized cutaneous nodular amyloidosis in a patient with Sjögren’s syndrome: a review of the literature. J Dermatol. 2005;32:120-123.
13. Summers EM, Kendrick CG. Primary localized cutaneous nodular amyloidosis and CREST syndrome: a case report and review of the literature. Cutis. 2008;82:55-59.
14. Taniguchi Y, Horino T, Terada Y. Cutaneous amyloidosis associated with amyopathic dermatomyositis. J Rheumatol. 2009;36:1088-1089.
15. Woollons A, Black MM. Nodular localized primary cutaneous amyloidosis: a long-term follow-up study. Br J Dermatol. 2001;145:105-109.
16. Brownstein MH, Helwig EB. The cutaneous amyloidoses. I. localized forms. Arch Dermatol. 1970;102:8-19.
1. Sakuma TH, Hans-Filho G, Arita K, et al. Familial primary localized cutaneous amyloidosis in Brazil. Arch Dermatol. 2009;145:695-699.
2. Rodermund OE. On amyloidosis cutis nodularis atrophicans (Gottron 1950). at the same time a contribution to the classification of amyloidosis [in German]. Arch Klin Exp Dermatol. 1967;230:153-171.
3. Chapel TA, Birmingham DJ, Malinowski YE. Nodular primary localized cutaneous amyloidosis. Arch Dermatol. 1977;113:1248-1249.
4. Criado PR, Silva CS, Vasconcellos C, et al. Extensive nodular cutaneous amyloidosis: an unusual presentation. J Eur Acad Dermatol Venereol. 2005;19:481-483.
5. Touart DM, Sau P. Cutaneous deposition diseases. part I [published correction appears in J Am Acad Dermatol. 1998;39:1042]. J Am Acad Dermatol. 1998;39(2, pt 1):149-171; quiz 172-174.
6. Borrowman TA, Lutz ME, Walsh JS. Cutaneous nodular amyloidosis masquerading as a foot callus. J Am Acad Dermatol. 2003;49:307-310.
7. Fujimoto N, Yajima M, Ohnishi Y, et al. Advanced glycation end product-modified beta2-microglobulin is a component of amyloid fibrils of primary localized cutaneous nodular amyloidosis. J Invest Dermatol. 2002;118:479-484.
8. Clement CG, Truong LD. An evaluation of Congo red fluorescence for the diagnosis of amyloidosis. Hum Pathol. 2014;45:1766-1772.
9. Lien MH, Railan D, Nelson BR. The efficacy of dermabrasion in the treatment of nodular amyloidosis. J Am Acad Dermatol. 1997;36(2, pt 2):315-316.
10. Taylor SC, Baker E, Grossman ME. Nodular vulvar amyloid as a presentation of systemic amyloidosis. J Am Acad Dermatol. 1991;24:139.
11. Northcutt AD, Vanover MJ. Nodular cutaneous amyloidosis involving the vulva. case report and literature review. Arch Dermatol. 1985;121:518-521.
12. Yoneyama K, Tochigi N, Oikawa A, et al. Primary localized cutaneous nodular amyloidosis in a patient with Sjögren’s syndrome: a review of the literature. J Dermatol. 2005;32:120-123.
13. Summers EM, Kendrick CG. Primary localized cutaneous nodular amyloidosis and CREST syndrome: a case report and review of the literature. Cutis. 2008;82:55-59.
14. Taniguchi Y, Horino T, Terada Y. Cutaneous amyloidosis associated with amyopathic dermatomyositis. J Rheumatol. 2009;36:1088-1089.
15. Woollons A, Black MM. Nodular localized primary cutaneous amyloidosis: a long-term follow-up study. Br J Dermatol. 2001;145:105-109.
16. Brownstein MH, Helwig EB. The cutaneous amyloidoses. I. localized forms. Arch Dermatol. 1970;102:8-19.
Practice Points
- The cutaneous lesions of primary localized cutaneous nodular amyloidosis (PLCNA) may present as single or multiple nodules, occasionally with overlying atrophic plaques and some with surface telangiectasia.
- Primary localized cutaneous nodular amyloidosis may represent a localized plasma cell dyscrasia that can be associated with a monoclonal gammopathy or multiple myeloma.
- Although PLCNA often is a benign cutaneous disorder, some patients can develop underlying systemic amyloidosis or even paraproteinemia.
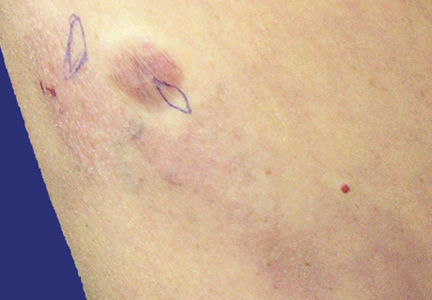
Benign Cephalic Histiocytosis
To the Editor:
Benign cephalic histiocytosis (BCH) falls into the group of non–Langerhans cell histiocytosis (non-LCH), which is characterized by a benign course and tendency toward spontaneous remission. Apart from BCH, the main types of non-LCH include juvenile xanthogranuloma, generalized eruptive histiocytoma, and xanthoma disseminatum.1
Benign cephalic histiocytosis is a rare form of cutaneous histiocytosis in young children. It presents as a papular eruption on the head and has not been associated with internal organ involvement.2-4 It was described in 1971 by Gianotti et al5 and was named infantile histiocytosis with intracytoplasmic worm-like bodies because electron microscopy revealed histiocytes with large cytoplasmatic inclusions composed of wormlike membranous profiles and absence of Birbeck granules. In BCH, skin lesions are located on the head including the face and sometimes on the neck. Lesions occasionally may appear on the trunk, buttocks, and thighs. Mucous membranes are not involved. The onset of disease is typical in the first year of life; however, the disease may begin within the first 3 years of life. An eruption is characterized by small, 2- to 8-mm, discrete, asymptomatic, tan to red-brown macules and papules. The lesions may persist for several months or years and subsequently flatten, becoming hyperpigmented briefly. They often completely resolve with time. Most children are otherwise healthy and developmentally normal6-9; however, diabetes insipidus has been reported in some children with BCH.10
Histologic examination of skin samples reveals the infiltrate of histiocytes, which closely approaches the epidermis, accompanied by scattered lymphocytes and a few eosinophils.1,2,11 The histiocytes express a typical macrophage marker CD68, whereas immunostaining for Langerhans cell markers such as CD1a and S-100 is negative.3,9,12,13
A 2.5-year-old boy was admitted to our dermatology department with suspected cutaneous mastocytosis (CM). Since the age of 13 months, he had developed small, 4- to 8-mm, dark pinkish macules and papules localized on the cheeks (Figure 1). Physical examination performed in our center revealed yellowish macules and flat papules limited to the cheeks. Darier sign was negative. The boy was otherwise healthy and developmentally normal. All laboratory tests were within reference range and his family history was uneventful.
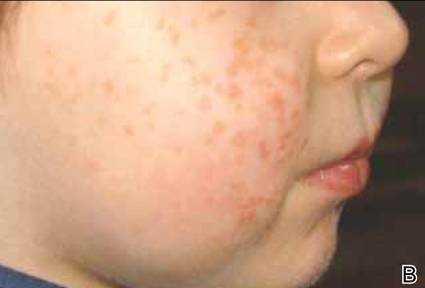
Figure 1. Maculopapular eruption of benign cephalic histiocytosis on the cheeks (A and B). |

Histopathologic examination of the skin sample revealed a normotypic epidermis and scattered subepidermal infiltrates in the upper dermis. The infiltrates were composed of predominating histiocytes and a few mast cells and eosinophils (Figure 2). The histiocytes were slightly pleomorphic and had abundant clear cytoplasm, vesicular nuclei, and prominent nucleoli. Mitoses were absent in these cells. The majority of cells within the infiltrate expressed CD68 and were CD1a- and S-100-. However, occasional CD1a+ cells were seen. Immunostaining for mast cell marker CD117 was negative. Cutaneous mastocytosis was excluded and non-LCH was recognized. Based on the typical location, morphology, and immunophenotype of skin lesions, BCH was diagnosed. At 24-month follow-up, spontaneous regression of skin lesions was observed.
Gianotti et al7 described BCH as a separate entity of the non-LCH group of disorders and established its diagnostic criteria: (1) onset of disease within the first 3 years of life; (2) location of skin lesions on the scalp and lack of lesions on the hands, feet, mucous membranes, and internal organ involvement; (3) spontaneous complete remission of symptoms; and (4) monomorphic infiltration of histiocytes that do not express S-100 and CD1a.
The macular and flat, papular, pink-yellow or orange lesions visible on the face of our patient are characteristic of BCH. Moreover, the cheeks are the most typical location of a BCH eruption, as noted in our patient.6,7,12 The presence of histiocytic infiltrates composed of CD68+ cells strongly support the diagnosis.3,4,9-13 In contrast to other reports, occasional CD1a+ cells of Langerhans phenotype were found in our case.3,9,11,12 The proliferation of Langerhans cells in the skin and internal organs and presence of langerin are characteristic of Langerhans cell histiocytosis (LCH).1,4,14 The presence of a few CD1a cells in cases with clinical features compatible with non-LCH may suggest that some of these cases may represent a papular self-healing variant of LCH or may indicate that there is an overlap among the histiocytic syndromes (eg, non-LCH and LCH). Furthermore, many of benign histiocytic lesions may evolve over the course of time into the others.12,13 Differential diagnosis of BCH should include other benign forms of cutaneous histiocytosis, particularly the small nodular variant of juvenile xanthogranuloma and generalized eruptive histiocytoma (GEH). Juvenile xanthogranuloma pre-sents as disseminated, yellowish, nodular lesions and may be associated with ocular involvement, whereas GEH is characterized by rapid onset of the disease and disseminated nodular eruption.1,4
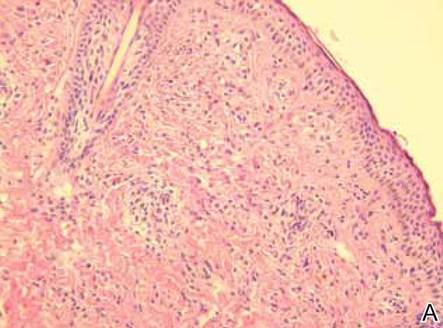
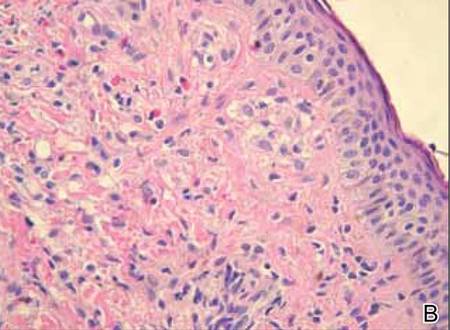
Figure 2. Skin section showing the upper and mid dermis infiltrated with slightly pleomorphic epithelioid histiocytic cells with clear cytoplasm and vesicular nuclei. Few accompanying lymphocytes and eosinophils were visible (H&E, original magnifications ×200 and ×400). |
A close histologic relationship and presence of overlapping symptoms observed among BCH, GEH, and juvenile xanthogranuloma indicate that these entities fall into a spectrum of the same disorder. However, the presence of a uniform infiltrate of large foamy histiocytes readily distinguishes xanthomas from BCH.4 In some unusual clinical presentations of CM or in cases of the nodular form of the condition, there is a need to distinguish between non-LCH and CM, as in our patient. Darier sign, consisting of urtication and erythema appearing after mechanical irritation of the skin lesion, is pathognomonic for CM. Nevertheless, Darier sign is not sufficient to confirm CM when it is not pronounced. Therefore, histologic examination with the use of immunostaining plays a key role in the differential diagnosis of these disorders in children.15 Treatment of BCH is not recommended because of spontaneous remission of the disease.1-5
Benign cephalic histiocytosis is a rare clinical form of non-LCH. No systemic or mucosal involvement has been described. Lesions often are confused with plane warts, but a biopsy is definitive. Therapy is not effective but fortunately none is necessary.
1. Gianotti F, Caputo R. Histiocytic syndromes: a review. J Am Acad Dermatol. 1985;13:383-404.
2. Jih DM, Salcedo SL, Jaworsky C. Benign cephalic histiocytosis: a case report and review. J Am Acad Dermatol. 2002;47:908-913.
3. Dadzie O, Hopster D, Cerio R, et al. Benign cephalic histiocytosis in a British-African child. Pediatr Dermatol. 2005;22:444-446.
4. Goodman WT, Barret TL. Histiocytoses. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Hong Kong, China: Elsevier Saunders; 2012:1527-1546.
5. Gianotti F, Caputo R, Ermacora E. Singular infantile histiocytosis with cells with intracytoplasmic vermiform particles [in French]. Bull Soc Fr Dermatol Syphiligr. 1971;78:232-233.
6. Barsky B, Lao I, Barsky S, et al. Benign cephalic histiocytosis. Arch Dermatol. 1984;120:650-655.
7. Gianotti F, Caputo R, Ermacora E, et al. Benign cephalic histiocytosis. Arch Dermatol. 1986;122:1038-1043.
8. Zelger BW, Sidoroff A, Orchard G, et al. Non-Langerhans cell histiocytosis. a new unifying concept. Am J Dermatopathol. 1996;18:490-504.
9. Hasegawa S, Deguchi M, Chiba-Okada S, et al. Japanese case of benign cephalic histiocytosis. J Dermatol. 2009;36:69-71.
10. Weston WL, Travers SH, Mierau GW, et al. Benign cephalic histiocytosis with diabetes insipidus. Pediatr Dermatol. 2000;17:296-298.
11. Gianotti R, Alessi E, Caputo R. Benign cephalic histiocytosis: a distinct entity or a part of a wide spectrum of histiocytic proliferative disorders of children? a histopathological study. Am J Dermatopathol. 1993;15:315-319.
12. Rodriguez-Jurado R, Duran-McKinster C, Ruiz-Maldonado R. Benign cephalic histiocytosis progressing into juvenile xanthogranuloma: a non-Langerhans cell histiocytosis transforming under the influence of a virus? Am J Dermatopathol. 2000;22:70-74.
13. Sidwell RU, Francis N, Slater DN, et al. Is disseminated juvenile xanthogranulomatosis benign cephalic histiocytosis? Pediatr Dermatol. 2005;22:40-43.
14. Favara BE, Jaffe R. The histopathology of Langerhans cell histiocytosis. Br J Cancer Suppl. 1994;23:S17-S23.
15. Heide R, Beishuizen A, De Groot H, et al. Mastocytosis in children: a protocol for management. Pediatr Dermatol. 2008;25:493-500.
To the Editor:
Benign cephalic histiocytosis (BCH) falls into the group of non–Langerhans cell histiocytosis (non-LCH), which is characterized by a benign course and tendency toward spontaneous remission. Apart from BCH, the main types of non-LCH include juvenile xanthogranuloma, generalized eruptive histiocytoma, and xanthoma disseminatum.1
Benign cephalic histiocytosis is a rare form of cutaneous histiocytosis in young children. It presents as a papular eruption on the head and has not been associated with internal organ involvement.2-4 It was described in 1971 by Gianotti et al5 and was named infantile histiocytosis with intracytoplasmic worm-like bodies because electron microscopy revealed histiocytes with large cytoplasmatic inclusions composed of wormlike membranous profiles and absence of Birbeck granules. In BCH, skin lesions are located on the head including the face and sometimes on the neck. Lesions occasionally may appear on the trunk, buttocks, and thighs. Mucous membranes are not involved. The onset of disease is typical in the first year of life; however, the disease may begin within the first 3 years of life. An eruption is characterized by small, 2- to 8-mm, discrete, asymptomatic, tan to red-brown macules and papules. The lesions may persist for several months or years and subsequently flatten, becoming hyperpigmented briefly. They often completely resolve with time. Most children are otherwise healthy and developmentally normal6-9; however, diabetes insipidus has been reported in some children with BCH.10
Histologic examination of skin samples reveals the infiltrate of histiocytes, which closely approaches the epidermis, accompanied by scattered lymphocytes and a few eosinophils.1,2,11 The histiocytes express a typical macrophage marker CD68, whereas immunostaining for Langerhans cell markers such as CD1a and S-100 is negative.3,9,12,13
A 2.5-year-old boy was admitted to our dermatology department with suspected cutaneous mastocytosis (CM). Since the age of 13 months, he had developed small, 4- to 8-mm, dark pinkish macules and papules localized on the cheeks (Figure 1). Physical examination performed in our center revealed yellowish macules and flat papules limited to the cheeks. Darier sign was negative. The boy was otherwise healthy and developmentally normal. All laboratory tests were within reference range and his family history was uneventful.
Figure 1. Maculopapular eruption of benign cephalic histiocytosis on the cheeks (A and B). |
Histopathologic examination of the skin sample revealed a normotypic epidermis and scattered subepidermal infiltrates in the upper dermis. The infiltrates were composed of predominating histiocytes and a few mast cells and eosinophils (Figure 2). The histiocytes were slightly pleomorphic and had abundant clear cytoplasm, vesicular nuclei, and prominent nucleoli. Mitoses were absent in these cells. The majority of cells within the infiltrate expressed CD68 and were CD1a- and S-100-. However, occasional CD1a+ cells were seen. Immunostaining for mast cell marker CD117 was negative. Cutaneous mastocytosis was excluded and non-LCH was recognized. Based on the typical location, morphology, and immunophenotype of skin lesions, BCH was diagnosed. At 24-month follow-up, spontaneous regression of skin lesions was observed.
Gianotti et al7 described BCH as a separate entity of the non-LCH group of disorders and established its diagnostic criteria: (1) onset of disease within the first 3 years of life; (2) location of skin lesions on the scalp and lack of lesions on the hands, feet, mucous membranes, and internal organ involvement; (3) spontaneous complete remission of symptoms; and (4) monomorphic infiltration of histiocytes that do not express S-100 and CD1a.
The macular and flat, papular, pink-yellow or orange lesions visible on the face of our patient are characteristic of BCH. Moreover, the cheeks are the most typical location of a BCH eruption, as noted in our patient.6,7,12 The presence of histiocytic infiltrates composed of CD68+ cells strongly support the diagnosis.3,4,9-13 In contrast to other reports, occasional CD1a+ cells of Langerhans phenotype were found in our case.3,9,11,12 The proliferation of Langerhans cells in the skin and internal organs and presence of langerin are characteristic of Langerhans cell histiocytosis (LCH).1,4,14 The presence of a few CD1a cells in cases with clinical features compatible with non-LCH may suggest that some of these cases may represent a papular self-healing variant of LCH or may indicate that there is an overlap among the histiocytic syndromes (eg, non-LCH and LCH). Furthermore, many of benign histiocytic lesions may evolve over the course of time into the others.12,13 Differential diagnosis of BCH should include other benign forms of cutaneous histiocytosis, particularly the small nodular variant of juvenile xanthogranuloma and generalized eruptive histiocytoma (GEH). Juvenile xanthogranuloma pre-sents as disseminated, yellowish, nodular lesions and may be associated with ocular involvement, whereas GEH is characterized by rapid onset of the disease and disseminated nodular eruption.1,4
|
Figure 2. Skin section showing the upper and mid dermis infiltrated with slightly pleomorphic epithelioid histiocytic cells with clear cytoplasm and vesicular nuclei. Few accompanying lymphocytes and eosinophils were visible (H&E, original magnifications ×200 and ×400). |
A close histologic relationship and presence of overlapping symptoms observed among BCH, GEH, and juvenile xanthogranuloma indicate that these entities fall into a spectrum of the same disorder. However, the presence of a uniform infiltrate of large foamy histiocytes readily distinguishes xanthomas from BCH.4 In some unusual clinical presentations of CM or in cases of the nodular form of the condition, there is a need to distinguish between non-LCH and CM, as in our patient. Darier sign, consisting of urtication and erythema appearing after mechanical irritation of the skin lesion, is pathognomonic for CM. Nevertheless, Darier sign is not sufficient to confirm CM when it is not pronounced. Therefore, histologic examination with the use of immunostaining plays a key role in the differential diagnosis of these disorders in children.15 Treatment of BCH is not recommended because of spontaneous remission of the disease.1-5
Benign cephalic histiocytosis is a rare clinical form of non-LCH. No systemic or mucosal involvement has been described. Lesions often are confused with plane warts, but a biopsy is definitive. Therapy is not effective but fortunately none is necessary.
To the Editor:
Benign cephalic histiocytosis (BCH) falls into the group of non–Langerhans cell histiocytosis (non-LCH), which is characterized by a benign course and tendency toward spontaneous remission. Apart from BCH, the main types of non-LCH include juvenile xanthogranuloma, generalized eruptive histiocytoma, and xanthoma disseminatum.1
Benign cephalic histiocytosis is a rare form of cutaneous histiocytosis in young children. It presents as a papular eruption on the head and has not been associated with internal organ involvement.2-4 It was described in 1971 by Gianotti et al5 and was named infantile histiocytosis with intracytoplasmic worm-like bodies because electron microscopy revealed histiocytes with large cytoplasmatic inclusions composed of wormlike membranous profiles and absence of Birbeck granules. In BCH, skin lesions are located on the head including the face and sometimes on the neck. Lesions occasionally may appear on the trunk, buttocks, and thighs. Mucous membranes are not involved. The onset of disease is typical in the first year of life; however, the disease may begin within the first 3 years of life. An eruption is characterized by small, 2- to 8-mm, discrete, asymptomatic, tan to red-brown macules and papules. The lesions may persist for several months or years and subsequently flatten, becoming hyperpigmented briefly. They often completely resolve with time. Most children are otherwise healthy and developmentally normal6-9; however, diabetes insipidus has been reported in some children with BCH.10
Histologic examination of skin samples reveals the infiltrate of histiocytes, which closely approaches the epidermis, accompanied by scattered lymphocytes and a few eosinophils.1,2,11 The histiocytes express a typical macrophage marker CD68, whereas immunostaining for Langerhans cell markers such as CD1a and S-100 is negative.3,9,12,13
A 2.5-year-old boy was admitted to our dermatology department with suspected cutaneous mastocytosis (CM). Since the age of 13 months, he had developed small, 4- to 8-mm, dark pinkish macules and papules localized on the cheeks (Figure 1). Physical examination performed in our center revealed yellowish macules and flat papules limited to the cheeks. Darier sign was negative. The boy was otherwise healthy and developmentally normal. All laboratory tests were within reference range and his family history was uneventful.
Figure 1. Maculopapular eruption of benign cephalic histiocytosis on the cheeks (A and B). |
Histopathologic examination of the skin sample revealed a normotypic epidermis and scattered subepidermal infiltrates in the upper dermis. The infiltrates were composed of predominating histiocytes and a few mast cells and eosinophils (Figure 2). The histiocytes were slightly pleomorphic and had abundant clear cytoplasm, vesicular nuclei, and prominent nucleoli. Mitoses were absent in these cells. The majority of cells within the infiltrate expressed CD68 and were CD1a- and S-100-. However, occasional CD1a+ cells were seen. Immunostaining for mast cell marker CD117 was negative. Cutaneous mastocytosis was excluded and non-LCH was recognized. Based on the typical location, morphology, and immunophenotype of skin lesions, BCH was diagnosed. At 24-month follow-up, spontaneous regression of skin lesions was observed.
Gianotti et al7 described BCH as a separate entity of the non-LCH group of disorders and established its diagnostic criteria: (1) onset of disease within the first 3 years of life; (2) location of skin lesions on the scalp and lack of lesions on the hands, feet, mucous membranes, and internal organ involvement; (3) spontaneous complete remission of symptoms; and (4) monomorphic infiltration of histiocytes that do not express S-100 and CD1a.
The macular and flat, papular, pink-yellow or orange lesions visible on the face of our patient are characteristic of BCH. Moreover, the cheeks are the most typical location of a BCH eruption, as noted in our patient.6,7,12 The presence of histiocytic infiltrates composed of CD68+ cells strongly support the diagnosis.3,4,9-13 In contrast to other reports, occasional CD1a+ cells of Langerhans phenotype were found in our case.3,9,11,12 The proliferation of Langerhans cells in the skin and internal organs and presence of langerin are characteristic of Langerhans cell histiocytosis (LCH).1,4,14 The presence of a few CD1a cells in cases with clinical features compatible with non-LCH may suggest that some of these cases may represent a papular self-healing variant of LCH or may indicate that there is an overlap among the histiocytic syndromes (eg, non-LCH and LCH). Furthermore, many of benign histiocytic lesions may evolve over the course of time into the others.12,13 Differential diagnosis of BCH should include other benign forms of cutaneous histiocytosis, particularly the small nodular variant of juvenile xanthogranuloma and generalized eruptive histiocytoma (GEH). Juvenile xanthogranuloma pre-sents as disseminated, yellowish, nodular lesions and may be associated with ocular involvement, whereas GEH is characterized by rapid onset of the disease and disseminated nodular eruption.1,4
|
Figure 2. Skin section showing the upper and mid dermis infiltrated with slightly pleomorphic epithelioid histiocytic cells with clear cytoplasm and vesicular nuclei. Few accompanying lymphocytes and eosinophils were visible (H&E, original magnifications ×200 and ×400). |
A close histologic relationship and presence of overlapping symptoms observed among BCH, GEH, and juvenile xanthogranuloma indicate that these entities fall into a spectrum of the same disorder. However, the presence of a uniform infiltrate of large foamy histiocytes readily distinguishes xanthomas from BCH.4 In some unusual clinical presentations of CM or in cases of the nodular form of the condition, there is a need to distinguish between non-LCH and CM, as in our patient. Darier sign, consisting of urtication and erythema appearing after mechanical irritation of the skin lesion, is pathognomonic for CM. Nevertheless, Darier sign is not sufficient to confirm CM when it is not pronounced. Therefore, histologic examination with the use of immunostaining plays a key role in the differential diagnosis of these disorders in children.15 Treatment of BCH is not recommended because of spontaneous remission of the disease.1-5
Benign cephalic histiocytosis is a rare clinical form of non-LCH. No systemic or mucosal involvement has been described. Lesions often are confused with plane warts, but a biopsy is definitive. Therapy is not effective but fortunately none is necessary.
1. Gianotti F, Caputo R. Histiocytic syndromes: a review. J Am Acad Dermatol. 1985;13:383-404.
2. Jih DM, Salcedo SL, Jaworsky C. Benign cephalic histiocytosis: a case report and review. J Am Acad Dermatol. 2002;47:908-913.
3. Dadzie O, Hopster D, Cerio R, et al. Benign cephalic histiocytosis in a British-African child. Pediatr Dermatol. 2005;22:444-446.
4. Goodman WT, Barret TL. Histiocytoses. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Hong Kong, China: Elsevier Saunders; 2012:1527-1546.
5. Gianotti F, Caputo R, Ermacora E. Singular infantile histiocytosis with cells with intracytoplasmic vermiform particles [in French]. Bull Soc Fr Dermatol Syphiligr. 1971;78:232-233.
6. Barsky B, Lao I, Barsky S, et al. Benign cephalic histiocytosis. Arch Dermatol. 1984;120:650-655.
7. Gianotti F, Caputo R, Ermacora E, et al. Benign cephalic histiocytosis. Arch Dermatol. 1986;122:1038-1043.
8. Zelger BW, Sidoroff A, Orchard G, et al. Non-Langerhans cell histiocytosis. a new unifying concept. Am J Dermatopathol. 1996;18:490-504.
9. Hasegawa S, Deguchi M, Chiba-Okada S, et al. Japanese case of benign cephalic histiocytosis. J Dermatol. 2009;36:69-71.
10. Weston WL, Travers SH, Mierau GW, et al. Benign cephalic histiocytosis with diabetes insipidus. Pediatr Dermatol. 2000;17:296-298.
11. Gianotti R, Alessi E, Caputo R. Benign cephalic histiocytosis: a distinct entity or a part of a wide spectrum of histiocytic proliferative disorders of children? a histopathological study. Am J Dermatopathol. 1993;15:315-319.
12. Rodriguez-Jurado R, Duran-McKinster C, Ruiz-Maldonado R. Benign cephalic histiocytosis progressing into juvenile xanthogranuloma: a non-Langerhans cell histiocytosis transforming under the influence of a virus? Am J Dermatopathol. 2000;22:70-74.
13. Sidwell RU, Francis N, Slater DN, et al. Is disseminated juvenile xanthogranulomatosis benign cephalic histiocytosis? Pediatr Dermatol. 2005;22:40-43.
14. Favara BE, Jaffe R. The histopathology of Langerhans cell histiocytosis. Br J Cancer Suppl. 1994;23:S17-S23.
15. Heide R, Beishuizen A, De Groot H, et al. Mastocytosis in children: a protocol for management. Pediatr Dermatol. 2008;25:493-500.
1. Gianotti F, Caputo R. Histiocytic syndromes: a review. J Am Acad Dermatol. 1985;13:383-404.
2. Jih DM, Salcedo SL, Jaworsky C. Benign cephalic histiocytosis: a case report and review. J Am Acad Dermatol. 2002;47:908-913.
3. Dadzie O, Hopster D, Cerio R, et al. Benign cephalic histiocytosis in a British-African child. Pediatr Dermatol. 2005;22:444-446.
4. Goodman WT, Barret TL. Histiocytoses. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Hong Kong, China: Elsevier Saunders; 2012:1527-1546.
5. Gianotti F, Caputo R, Ermacora E. Singular infantile histiocytosis with cells with intracytoplasmic vermiform particles [in French]. Bull Soc Fr Dermatol Syphiligr. 1971;78:232-233.
6. Barsky B, Lao I, Barsky S, et al. Benign cephalic histiocytosis. Arch Dermatol. 1984;120:650-655.
7. Gianotti F, Caputo R, Ermacora E, et al. Benign cephalic histiocytosis. Arch Dermatol. 1986;122:1038-1043.
8. Zelger BW, Sidoroff A, Orchard G, et al. Non-Langerhans cell histiocytosis. a new unifying concept. Am J Dermatopathol. 1996;18:490-504.
9. Hasegawa S, Deguchi M, Chiba-Okada S, et al. Japanese case of benign cephalic histiocytosis. J Dermatol. 2009;36:69-71.
10. Weston WL, Travers SH, Mierau GW, et al. Benign cephalic histiocytosis with diabetes insipidus. Pediatr Dermatol. 2000;17:296-298.
11. Gianotti R, Alessi E, Caputo R. Benign cephalic histiocytosis: a distinct entity or a part of a wide spectrum of histiocytic proliferative disorders of children? a histopathological study. Am J Dermatopathol. 1993;15:315-319.
12. Rodriguez-Jurado R, Duran-McKinster C, Ruiz-Maldonado R. Benign cephalic histiocytosis progressing into juvenile xanthogranuloma: a non-Langerhans cell histiocytosis transforming under the influence of a virus? Am J Dermatopathol. 2000;22:70-74.
13. Sidwell RU, Francis N, Slater DN, et al. Is disseminated juvenile xanthogranulomatosis benign cephalic histiocytosis? Pediatr Dermatol. 2005;22:40-43.
14. Favara BE, Jaffe R. The histopathology of Langerhans cell histiocytosis. Br J Cancer Suppl. 1994;23:S17-S23.
15. Heide R, Beishuizen A, De Groot H, et al. Mastocytosis in children: a protocol for management. Pediatr Dermatol. 2008;25:493-500.
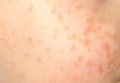
Idiopathic Follicular Mucinosis or Mycosis Fungoides? Classification and Diagnostic Challenges
When follicular mucinosis (FM) is defined as an epithelial reaction pattern characterized by intrafollicular and perifollicular mucin accumulation, it cannot be considered a distinct disease entity, as this pattern is ubiquitously present in various inflammatory and neoplastic skin conditions.1,2 The distinction between idiopathic FM and lymphoma-associated follicular mucinosis (LAFM) was made several years ago by authors who evaluated the differences in the clinical presentation of these entities, including patient age at onset, number of lesions, pattern of distribution, and most importantly clinical progression.1 In this article, we discuss the importance of close clinical follow-up in patients with FM or patch-stage mycosis fungoides (MF) in whom histopathologic evaluation is ambiguous or nondiagnostic. We also highlight the value of ancillary testing, including T-cell receptor gene rearrangement, flow cytometry, and immunohistochemistry, as a component in the diagnostic process rather than the sole diagnostic moiety. A review of the pertinent literature also is performed.
History of FM and MF
Pinkus3 first described an entity he termed alopecia mucinosa in 1957. Pinkus noted 3 distinct patterns: an idiopathic form of alopecia mucinosa, lymphoblastoma with associated FM, and alopecia mucinosa that later transformed into lymphoblastoma.4 In 1983, however, Pinkus4 described uncertainty if alopecia mucinosa represented the first stage of MF or if patients with alopecia mucinosa were simply at an increased risk for developing lymphoma. He believed there were too many cases of lymphoma following a diagnosis of alopecia mucinosa for the relation to be coincidental, yet he noted that many of the cases resolved either spontaneously or following treatment with x-rays or topical steroids. He concluded his report with a sentiment that is echoed in many current studies regarding this entity: “Many questions surrounding this entity are as unanswerable today as they were 25 years ago.”4
Jablonska et al5 were the first to coin the term mucinosis follicularis, now known as FM, to replace alopecia mucinosa because they felt the description was more accurate, as lesions also arise on non–hair-bearing skin. Although there is general agreement that there is a form of MF that has associated FM, this is where the agreement ends with regard to the diagnosis of MF versus FM. Böer et al6 discussed the historic evolution of these terms, mostly to highlight the origins of the confusion. The investigators proposed that FM should only be used as a descriptive term and that all cases of alopecia mucinosa represent MF. They also concluded that many benign dermatoses associated with a risk for evolution to MF (eg, small and large plaque psoriasis [LPP]) should simply be diagnosed as MF.6 Subsequently, the proposal that idiopathic FM and LAFM are not 2 distinct entities but rather a clinicopathologic continuum and that idiopathic FM is simply a variant of MF along this spectrum has gained some approval.6,7 However, this belief is not shared among all authorities in the field, and attempts to define diagnostic criteria that distinguish between a benign clinical course and a course that is more progressive and fatal continue. Currently, it is agreed upon that when distinguishing between these 2 clinical courses, primary (idiopathic) follicular mucinosis refers to a benign course with no overt sign of malignancy, and lymphoma-associated follicular mucinosis refers to a diagnostic malignant condition. Lymphoma-associated follicular mucinosis refers to FM associated with cutaneous T-cell lymphoma, the most common form of which is FM. Many authors8-15 have sought ancillary methodologies in addition to clinical parameters to assist in the evaluation between both disease courses. Methodologies have included assessment of T-cell receptor gene rearrangements, flow cytometry, and immunohistochemical staining, mostly as an effort to establish monoclonality as a defining characteristic of LAFM; however, monoclonality in cutaneous T-cell infiltrates should be interpreted with caution and should not be considered as a confirmation of malignancy due to recent findings of monoclonality in benign inflammatory dermatoses such as lichen planus. The Table outlines several of the most common benign inflammatory dermatoses that demonstrate monoclonality, but this list should not be considered exhaustive, as there are many others in which monoclonality is sometimes seen.8-15 The lack of definitive criteria to distinguish between the 2 groups has led to confusion and consternation regarding the diagnosis of idiopathic FM versus LAFM and has led many in the field to consider the 2 conditions to be one and the same.

Diagnosis of FM and MF: Clinicopathologic Features
The World Health Organization (WHO) defined MF as an epidermotropic primary cutaneous T-cell lymphoma (CTCL) characterized by infiltrates of small- to medium-sized T lymphocytes with cerebriform nuclei. Further, the WHO stated that the term mycosis fungoides should be exclusively reserved for classical cases typified by the evolution of cutaneous patches, plaques, and tumors, or for variants that show a similar clinical course.16 Mycosis fungoides is divided into 3 stages—patch, plaque, and tumor—which are solely clinical descriptors.17 The WHO also described a clinical staging system with pathologic emphasis placed only on lymph node involvement and identification of Sézary cells.16 It lists folliculotropic MF as a variant, with only some cases presenting with mucinous degeneration of hair follicles. A lack of consensus among pathologists regarding criteria for diagnosis in patch-stage MF remains, but diagnosis of plaque-stage disease is not regularly debated due to its more reliably present, well-developed histologic features (eg, haloed lymphocytes, epidermotropism of lymphocytes, lymphocytes with convoluted nuceli, Pautrier microabscesses).18 Although there have been specific histologic findings reported to be associated with patch-stage MF, they have only been present in a few cases and are therefore of limited usefulness in practice.1,19 The categorization of patients with subtle histologic features common to both MF and inflammatory conditions such as parapsoriasis en plaques (the term plaque in this case is a misnomer because the word plaque means patch in French) continues to be elusive. A lack of agreement regarding LPP persists in the current literature in the same manner as FM. Some researchers have contended for many years that LPP is a type of MF, while others remain unconvinced, mainly due to the lack of evidence that lumping a benign condition (LPP) with an increased risk for malignant transformation and a known malignancy (MF) together is of any benefit to the patient. Assessment of clinicopathologic correlation, immunohistochemistry, clonality, and T-cell gene rearrangement have failed to positively identify patients who are at risk for disease progression, whether the diagnosis is called LPP or early patch-stage MF.20
Mycosis fungoides is more common in males and its incidence increases with age; however, diagnosis should not be ruled out based on age or gender. Typical presentation of early-stage disease includes erythematous patches or plaques, often with light scaling.19 Lesions routinely are of long-standing duration (months to years), are located in areas that are infrequently exposed to sunlight, and often are 5 cm in diameter or larger with irregular borders.21 Associated poikiloderma is relatively specific to MF but rarely is seen in other CTCLs, connective-tissue diseases, and some genodermatoses. Poikiloderma commonly is identified in LPP, which shows the same telangiectasia, mottled pigmentation, and epidermal atrophy as MF-associated poikiloderma, leading some to believe that there is no separation between the 2 conditions. In all stages of MF, lesions frequently are numerous and occur on multiple sites. Plaques and tumors can show spontaneous ulceration. When lesions are folliculotropic, they can cause localized alopecia, follicular-based papules, and fungating pseudotumors in more advanced stages.1 The clinical presentation of FM substantially overlaps with folliculotropic MF, and although FM lesions often are solitary and are located on the face or scalp, they also can present as multiple lesions located elsewhere on the body. It also has been proposed that folliculotropic MF should not be separated from FM-associated MF (or LAFM).22
The characteristic histologic picture of LAFM in patch or plaque stage shows mucin deposition within hair follicles, similar to idiopathic FM. On histology, both conditions demonstrate dense lymphoid infiltrates around and within hair follicles as well as in the dermis (Figure). Most cases of LAFM show epidermotropism of lymphocytes between follicles, but this finding is not present in every case and often disappears when the disease advances to the tumor stage.1,19 Although Pautrier microabscesses (collections of lymphocytes within the superficial epidermis) are considered to be somewhat specific to MF, they are only present in a minority of cases.20 In a study by the International Society for Cutaneous Lymphomas,21 the only histopathologic criteria that showed any appreciable sensitivity or specificity in the diagnosis of MF were the presence of lymphoid cells with variable nuclear and cytoplasmic features and/or strikingly irregular nuclear contours with the presence of lymphocytes larger than those usually seen in inflammatory dermatoses. Despite these criteria, the study reported a high misclassification rate. A complicated scoring system for diagnosis of MF in patch- or early plaque-stage disease was proposed by the International Society for Cutaneous Lymphomas,21 which integrates clinical, histopathologic, molecular, and immunophenotypic criteria. However, these criteria have been continually debated in the literature and are only discussed in this article in relation to the association between MF and FM. Diagnosis of tumor-stage MF is not addressed in this article, as it is readily identified as lymphoma and is not easily confused with idiopathic FM.
|

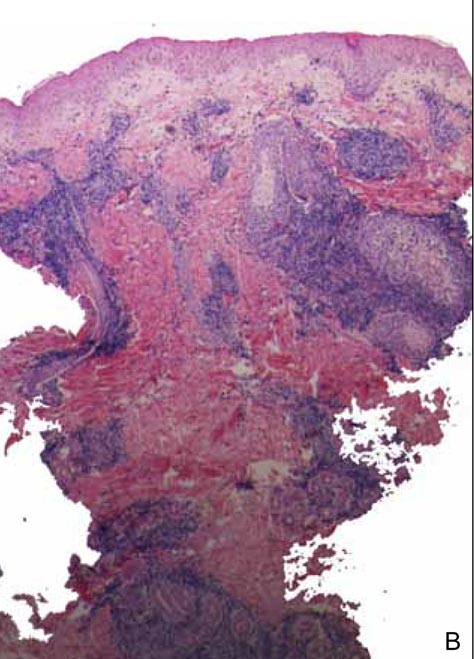
Clinical assessment of a patient’s medical history to identify persistent and progressive disease is paramount to the diagnosis of MF. Although MF lesions tend to increase in size and number over time, this presentation is not without exception.21 In early patch-stage disease, eliminating some of the patient’s current medications may be sufficient in clearing cutaneous patches that cannot be conclusively identified as either MF or a benign inflammatory lymphoid infiltrate, which further emphasizes the importance of clinical assessment of the patient’s medical history in the diagnosis of MF. The shape of the lesions also is helpful in distinguishing between MF and other skin disorders, such as digitate dermatosis or LPP; unlike the latter, the waxing and waning nature of MF lesions often produces irregularly shaped patches with little coalescence. Again, there are some investigators who believe that these lesions represent varying presentations of MF.6
In a study by Cerroni et al,1 44 patients with FM were divided into 2 groups: (1) a cohort of 16 patients with no history or clinical evidence of MF or Sézary syndrome (ie, LAFM), and (2) a cohort of 28 patients with clinicopathologic evidence of CTCL. Patients in both groups were followed for a maximum of 20 years. Results indicated that that the presence of perifollicular or intrafollicular mucin, epidermotropism of lymphocytes, monoclonality, and epidemiologic characteristics (eg, age, sex, race) cannot reliably distinguish the 2 disease forms. Furthermore, it was suggested that these conditions are not mutually exclusive entities and are actually variants of CTCL. The observation that the 2 diseases share prognostic overlap adds further credence to the already puzzling conundrum. Nineteen of 28 patients with MF were alive and well at follow-up, and all patients in the idiopathic FM group were alive, with only 9 of 16 patients showing residual disease and none with CTCL.1
Other clinical factors that may be helpful in the diagnosis of MF are the presentation of lesions in non–sun-exposed areas of the skin and multiple lesions, as unilesional MF is exceedingly uncommon.21 No histologic features have been proven to predict which early patch- or plaque-stage MFs will progress to full-blown CTCL versus benign idiopathic FM; thus, great caution should be taken in patients with early-stage disease to ensure they are not prematurely and/or incorrectly classified as CTCL. Such a diagnosis has medical, social, and economical ramifications that should not be overlooked.
If idiopathic FM and LAFM were considered distinct disease processes, the ambiguity in making a definitive diagnosis should give the physician pause, and a diagnosis of LAFM may only be appropriate when there is unequivocal clinicopathologic evidence. Otherwise, a lymphoma diagnosis is somewhat superfluous and potentially harmful. Definitive diagnosis of LAFM also is complicated by reports of other hematologic malignancies presenting with FM-like histopathologic findings, such as chronic myelogenous leukemia, leukemia-associated eosinophilic folliculitis, and acute myeloblastic leukemia.23,24 Although MF is the most common malignancy associated with FM, it is important to consider other less common malignancies that also may be present.
Diagnosis: Patient Consequences
Accurate diagnosis of idiopathic FM versus LAFM is critical, as the ramifications of a cancer diagnosis can have broad implications. For example, patients who receive cancer diagnoses often experience emotional trauma and social stigma, even when adequate patient education has been provided. The incidence of depression and anxiety also can increase following a cancer diagnosis and can be complicated by medical treatments (eg, systemic steroids, interferon),25 which are known to increase the frequency of these psychological disturbances. Health insurance premiums likely will be higher if a patient is diagnosed with cancer versus a benign inflammatory condition. Hesitation of the pathologist to assign a cancer diagnosis when unequivocal evidence is not present should not be regarded as trickery, malpractice, or deceit of the health care bylaws, as benign language with suggestion of close clinical follow-up in the setting of diagnostic uncertainty will “first, do no harm” and secondly, serve as a vehicle for patient advocacy.
If there is a definitive distinction between idiopathic FM and LAFM, it requires further research before it can be fully understood. Currently, the WHO does not recognize a diagnosis of FM-associated MF (or LAFM) and acknowledges that folliculotropic MF is not always associated with FM.16,26 Given uncertainty and repercussions associated with a cancer diagnosis, however indolent, it may be morally responsible and medically favorable for physicians to consider FM in the differential diagnosis when applicable rather than making a diagnosis of MF outright. Given the importance of both clinical and histologic factors, it may be beneficial for definitive diagnosis of FM versus MF to lie with the clinician, while the pathologist serves as an adjunct in the diagnostic process. Because the prognosis of idiopathic FM often is marred by possible transformation into MF or other CTCLs, therapeutic decisions should be dictated by close clinical follow-up. Additionally, stage of disease, patient age, treatment compliance, comorbidities, and possible side effects of medications should all be considered when evaluating potential therapeutic regimens.27
Conclusion
Research is underway to more accurately identify patients with FM who are at risk for progression to LAFM versus those with benign remitting FM. Once the required diagnostic criteria are established to accurately classify these patients, with an emphasis on prognosis and suggested treatments, it might be necessary to establish new, less debated terminology so pathologists and clinicians alike can improve patient care. Continued histopathologic scrutiny, use of sophisticated molecular techniques, and knowledge of other currently undiscovered modalities will shed light on this important disease process and aid in proper disease management, which may be advantageous to both patients and physicians.
1. Cerroni L, Fink-Puches R, Bäck B, et al. Follicular mucinosis: a critical reappraisal of clinicopathologic features and association with mycosis fungoides and Sézary syndrome. Arch Dermatol. 2002;138:182-189.
2. Parker SR, Murad E. Follicular mucinosis: clinical, histologic, and molecular remission with minocycline [published online ahead of print July 25, 2009]. J Am Acad Dermatol. 2010;62:139-141.
3. Pinkus H. Alopecia mucinosa; inflammatory plaques with alopecia characterized by root-sheath mucinosis. AMA Arch Dermatol. 1957;76:419-424, 424-426.
4. Pinkus H. Alopecia mucinosa. additional data in 1983. Arch Dermatol. 1983;119:698-699.
5. Jablonska S, Chorzelski T, Lancucki J. Mucinosis follicularis [in German]. Hautarzt. 1959;10:27-33.
6. Böer A, Guo Y, Ackerman AB. Alopecia mucinosa is mycosis fungoides. Am J Dermatopathol. 2004;26:33-52.
7. Brown HA, Gibson LE, Pujol RM, et al. Primary follicular mucinosis: long-term follow-up of patients younger than 40 years with and withoutclonal T-cell receptor gene rearrangement. J Am Acad Dermatol. 2002;47:856-862.
8. Schiller PI, Flaig MJ, Puchta U, et al. Detection of clonal T cells in lichen planus. Arch Dermatol Res. 2000;292:568-569.
9. Cerroni L, Kerl H. Primary follicular mucinosis and association with mycosis fungoides and other cutaneous T-cell lymphomas. J Am Acad Dermatol. 2004;51:146-147.
10. Dereure O, Levi E, Kadin ME. T-Cell clonality in pityriasis lichenoides et varioliformis acuta: a heteroduplex analysis of 20 cases. Arch Dermatol. 2000;136:1483-1486.
11. Haeffner AC, Smoller BR, Zepter K, et al. Differentiation and clonality of lesional lymphocytes in small plaque parapsoriasis. Arch Dermatol. 1995;131:321-324.
12. Schultz JC, Granados S, Vonderheid EC, et al. T-cell clonality of peripheral blood lymphocytes in patients with lymphomatoid papulosis. J Am Acad Dermatol. 2005;53:152-155.
13. Pfaltz K, Kerl K, Palmedo G, et al. Clonality in sarcoidosis, granuloma annulare, and granulomatous mycosis fungoides. Am J Dermatopathol. 2011;33:659-662.
14. Weinberg JM, Kristal L, Chooback L, et al. The clonal nature of pityriasis lichenoides. Arch Dermatol. 2002;138:1063-1067.
15. Guitart J, Magro C. Cutaneous T-cell lymphoid dyscrasia: a unifying term for idiopathic chronic dermatoses with persistent T-cell clones. Arch Dermatol. 2007;143:921-932.
16. Swerdlow SH, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008.
17. Zinzani PL, Ferreri AJ, Cerroni L. Mycosis fungoides [published online ahead of print October 22, 2007]. Crit Rev Oncol Hematol. 2008;65:172-182.
18. Smoller BR, Bishop K, Glusac E, et al. Reassessment of histologic parameters in the diagnosis of mycosis fungoides. Am J Surg Pathol. 1995;19:1423-1430.
19. Hwang ST, Janik JE, Jaffe ES, et al. Mycosis fungoides and Sézary syndrome. Lancet. 2008;371:945-957.
20. Sarveswari KN, Yesudian P. The conundrum of parapsoriasis versus patch stage of mycosis fungoides. Indian J Dermatol Venereol Leprol. 2009;75:229-235.
21. Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063.
22. Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides. a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525-530.
23. Rashid R, Hymes S. Folliculitis, follicular mucinosis, and papular mucinosis as a presentation of chronic myelomonocytic leukemia. Dermatol Online J. 2009;15:16.
24. Wada T, Yoshinaga E, Oiso N, et al. Adult T-cell leukemia-lymphoma associated with follicular mucinosis. J Dermatol. 2009;36:638-642.
25. Sampogna F, Frontani M, Baliva G, et al. Quality of life and psychological distress in patients with cutaneous lymphoma [published online ahead of print December 16, 2008]. Br J Dermatol. 2009;160:815-822.
26. Boone SL, Guitart J, Gerami P. Follicular mycosis fungoides: a histopathologic, immunohistochemical, and genotypic review. G Ital Dermatol Venereol. 2008;143:409-414.
27. Prince HM, Whittaker S, Hoppe RT. How I treat mycosis fungoides and Sézary syndrome [published online ahead of print August 20, 2009]. Blood. 2009;114:4337-4353.
When follicular mucinosis (FM) is defined as an epithelial reaction pattern characterized by intrafollicular and perifollicular mucin accumulation, it cannot be considered a distinct disease entity, as this pattern is ubiquitously present in various inflammatory and neoplastic skin conditions.1,2 The distinction between idiopathic FM and lymphoma-associated follicular mucinosis (LAFM) was made several years ago by authors who evaluated the differences in the clinical presentation of these entities, including patient age at onset, number of lesions, pattern of distribution, and most importantly clinical progression.1 In this article, we discuss the importance of close clinical follow-up in patients with FM or patch-stage mycosis fungoides (MF) in whom histopathologic evaluation is ambiguous or nondiagnostic. We also highlight the value of ancillary testing, including T-cell receptor gene rearrangement, flow cytometry, and immunohistochemistry, as a component in the diagnostic process rather than the sole diagnostic moiety. A review of the pertinent literature also is performed.
History of FM and MF
Pinkus3 first described an entity he termed alopecia mucinosa in 1957. Pinkus noted 3 distinct patterns: an idiopathic form of alopecia mucinosa, lymphoblastoma with associated FM, and alopecia mucinosa that later transformed into lymphoblastoma.4 In 1983, however, Pinkus4 described uncertainty if alopecia mucinosa represented the first stage of MF or if patients with alopecia mucinosa were simply at an increased risk for developing lymphoma. He believed there were too many cases of lymphoma following a diagnosis of alopecia mucinosa for the relation to be coincidental, yet he noted that many of the cases resolved either spontaneously or following treatment with x-rays or topical steroids. He concluded his report with a sentiment that is echoed in many current studies regarding this entity: “Many questions surrounding this entity are as unanswerable today as they were 25 years ago.”4
Jablonska et al5 were the first to coin the term mucinosis follicularis, now known as FM, to replace alopecia mucinosa because they felt the description was more accurate, as lesions also arise on non–hair-bearing skin. Although there is general agreement that there is a form of MF that has associated FM, this is where the agreement ends with regard to the diagnosis of MF versus FM. Böer et al6 discussed the historic evolution of these terms, mostly to highlight the origins of the confusion. The investigators proposed that FM should only be used as a descriptive term and that all cases of alopecia mucinosa represent MF. They also concluded that many benign dermatoses associated with a risk for evolution to MF (eg, small and large plaque psoriasis [LPP]) should simply be diagnosed as MF.6 Subsequently, the proposal that idiopathic FM and LAFM are not 2 distinct entities but rather a clinicopathologic continuum and that idiopathic FM is simply a variant of MF along this spectrum has gained some approval.6,7 However, this belief is not shared among all authorities in the field, and attempts to define diagnostic criteria that distinguish between a benign clinical course and a course that is more progressive and fatal continue. Currently, it is agreed upon that when distinguishing between these 2 clinical courses, primary (idiopathic) follicular mucinosis refers to a benign course with no overt sign of malignancy, and lymphoma-associated follicular mucinosis refers to a diagnostic malignant condition. Lymphoma-associated follicular mucinosis refers to FM associated with cutaneous T-cell lymphoma, the most common form of which is FM. Many authors8-15 have sought ancillary methodologies in addition to clinical parameters to assist in the evaluation between both disease courses. Methodologies have included assessment of T-cell receptor gene rearrangements, flow cytometry, and immunohistochemical staining, mostly as an effort to establish monoclonality as a defining characteristic of LAFM; however, monoclonality in cutaneous T-cell infiltrates should be interpreted with caution and should not be considered as a confirmation of malignancy due to recent findings of monoclonality in benign inflammatory dermatoses such as lichen planus. The Table outlines several of the most common benign inflammatory dermatoses that demonstrate monoclonality, but this list should not be considered exhaustive, as there are many others in which monoclonality is sometimes seen.8-15 The lack of definitive criteria to distinguish between the 2 groups has led to confusion and consternation regarding the diagnosis of idiopathic FM versus LAFM and has led many in the field to consider the 2 conditions to be one and the same.
Diagnosis of FM and MF: Clinicopathologic Features
The World Health Organization (WHO) defined MF as an epidermotropic primary cutaneous T-cell lymphoma (CTCL) characterized by infiltrates of small- to medium-sized T lymphocytes with cerebriform nuclei. Further, the WHO stated that the term mycosis fungoides should be exclusively reserved for classical cases typified by the evolution of cutaneous patches, plaques, and tumors, or for variants that show a similar clinical course.16 Mycosis fungoides is divided into 3 stages—patch, plaque, and tumor—which are solely clinical descriptors.17 The WHO also described a clinical staging system with pathologic emphasis placed only on lymph node involvement and identification of Sézary cells.16 It lists folliculotropic MF as a variant, with only some cases presenting with mucinous degeneration of hair follicles. A lack of consensus among pathologists regarding criteria for diagnosis in patch-stage MF remains, but diagnosis of plaque-stage disease is not regularly debated due to its more reliably present, well-developed histologic features (eg, haloed lymphocytes, epidermotropism of lymphocytes, lymphocytes with convoluted nuceli, Pautrier microabscesses).18 Although there have been specific histologic findings reported to be associated with patch-stage MF, they have only been present in a few cases and are therefore of limited usefulness in practice.1,19 The categorization of patients with subtle histologic features common to both MF and inflammatory conditions such as parapsoriasis en plaques (the term plaque in this case is a misnomer because the word plaque means patch in French) continues to be elusive. A lack of agreement regarding LPP persists in the current literature in the same manner as FM. Some researchers have contended for many years that LPP is a type of MF, while others remain unconvinced, mainly due to the lack of evidence that lumping a benign condition (LPP) with an increased risk for malignant transformation and a known malignancy (MF) together is of any benefit to the patient. Assessment of clinicopathologic correlation, immunohistochemistry, clonality, and T-cell gene rearrangement have failed to positively identify patients who are at risk for disease progression, whether the diagnosis is called LPP or early patch-stage MF.20
Mycosis fungoides is more common in males and its incidence increases with age; however, diagnosis should not be ruled out based on age or gender. Typical presentation of early-stage disease includes erythematous patches or plaques, often with light scaling.19 Lesions routinely are of long-standing duration (months to years), are located in areas that are infrequently exposed to sunlight, and often are 5 cm in diameter or larger with irregular borders.21 Associated poikiloderma is relatively specific to MF but rarely is seen in other CTCLs, connective-tissue diseases, and some genodermatoses. Poikiloderma commonly is identified in LPP, which shows the same telangiectasia, mottled pigmentation, and epidermal atrophy as MF-associated poikiloderma, leading some to believe that there is no separation between the 2 conditions. In all stages of MF, lesions frequently are numerous and occur on multiple sites. Plaques and tumors can show spontaneous ulceration. When lesions are folliculotropic, they can cause localized alopecia, follicular-based papules, and fungating pseudotumors in more advanced stages.1 The clinical presentation of FM substantially overlaps with folliculotropic MF, and although FM lesions often are solitary and are located on the face or scalp, they also can present as multiple lesions located elsewhere on the body. It also has been proposed that folliculotropic MF should not be separated from FM-associated MF (or LAFM).22
The characteristic histologic picture of LAFM in patch or plaque stage shows mucin deposition within hair follicles, similar to idiopathic FM. On histology, both conditions demonstrate dense lymphoid infiltrates around and within hair follicles as well as in the dermis (Figure). Most cases of LAFM show epidermotropism of lymphocytes between follicles, but this finding is not present in every case and often disappears when the disease advances to the tumor stage.1,19 Although Pautrier microabscesses (collections of lymphocytes within the superficial epidermis) are considered to be somewhat specific to MF, they are only present in a minority of cases.20 In a study by the International Society for Cutaneous Lymphomas,21 the only histopathologic criteria that showed any appreciable sensitivity or specificity in the diagnosis of MF were the presence of lymphoid cells with variable nuclear and cytoplasmic features and/or strikingly irregular nuclear contours with the presence of lymphocytes larger than those usually seen in inflammatory dermatoses. Despite these criteria, the study reported a high misclassification rate. A complicated scoring system for diagnosis of MF in patch- or early plaque-stage disease was proposed by the International Society for Cutaneous Lymphomas,21 which integrates clinical, histopathologic, molecular, and immunophenotypic criteria. However, these criteria have been continually debated in the literature and are only discussed in this article in relation to the association between MF and FM. Diagnosis of tumor-stage MF is not addressed in this article, as it is readily identified as lymphoma and is not easily confused with idiopathic FM.
|
Clinical assessment of a patient’s medical history to identify persistent and progressive disease is paramount to the diagnosis of MF. Although MF lesions tend to increase in size and number over time, this presentation is not without exception.21 In early patch-stage disease, eliminating some of the patient’s current medications may be sufficient in clearing cutaneous patches that cannot be conclusively identified as either MF or a benign inflammatory lymphoid infiltrate, which further emphasizes the importance of clinical assessment of the patient’s medical history in the diagnosis of MF. The shape of the lesions also is helpful in distinguishing between MF and other skin disorders, such as digitate dermatosis or LPP; unlike the latter, the waxing and waning nature of MF lesions often produces irregularly shaped patches with little coalescence. Again, there are some investigators who believe that these lesions represent varying presentations of MF.6
In a study by Cerroni et al,1 44 patients with FM were divided into 2 groups: (1) a cohort of 16 patients with no history or clinical evidence of MF or Sézary syndrome (ie, LAFM), and (2) a cohort of 28 patients with clinicopathologic evidence of CTCL. Patients in both groups were followed for a maximum of 20 years. Results indicated that that the presence of perifollicular or intrafollicular mucin, epidermotropism of lymphocytes, monoclonality, and epidemiologic characteristics (eg, age, sex, race) cannot reliably distinguish the 2 disease forms. Furthermore, it was suggested that these conditions are not mutually exclusive entities and are actually variants of CTCL. The observation that the 2 diseases share prognostic overlap adds further credence to the already puzzling conundrum. Nineteen of 28 patients with MF were alive and well at follow-up, and all patients in the idiopathic FM group were alive, with only 9 of 16 patients showing residual disease and none with CTCL.1
Other clinical factors that may be helpful in the diagnosis of MF are the presentation of lesions in non–sun-exposed areas of the skin and multiple lesions, as unilesional MF is exceedingly uncommon.21 No histologic features have been proven to predict which early patch- or plaque-stage MFs will progress to full-blown CTCL versus benign idiopathic FM; thus, great caution should be taken in patients with early-stage disease to ensure they are not prematurely and/or incorrectly classified as CTCL. Such a diagnosis has medical, social, and economical ramifications that should not be overlooked.
If idiopathic FM and LAFM were considered distinct disease processes, the ambiguity in making a definitive diagnosis should give the physician pause, and a diagnosis of LAFM may only be appropriate when there is unequivocal clinicopathologic evidence. Otherwise, a lymphoma diagnosis is somewhat superfluous and potentially harmful. Definitive diagnosis of LAFM also is complicated by reports of other hematologic malignancies presenting with FM-like histopathologic findings, such as chronic myelogenous leukemia, leukemia-associated eosinophilic folliculitis, and acute myeloblastic leukemia.23,24 Although MF is the most common malignancy associated with FM, it is important to consider other less common malignancies that also may be present.
Diagnosis: Patient Consequences
Accurate diagnosis of idiopathic FM versus LAFM is critical, as the ramifications of a cancer diagnosis can have broad implications. For example, patients who receive cancer diagnoses often experience emotional trauma and social stigma, even when adequate patient education has been provided. The incidence of depression and anxiety also can increase following a cancer diagnosis and can be complicated by medical treatments (eg, systemic steroids, interferon),25 which are known to increase the frequency of these psychological disturbances. Health insurance premiums likely will be higher if a patient is diagnosed with cancer versus a benign inflammatory condition. Hesitation of the pathologist to assign a cancer diagnosis when unequivocal evidence is not present should not be regarded as trickery, malpractice, or deceit of the health care bylaws, as benign language with suggestion of close clinical follow-up in the setting of diagnostic uncertainty will “first, do no harm” and secondly, serve as a vehicle for patient advocacy.
If there is a definitive distinction between idiopathic FM and LAFM, it requires further research before it can be fully understood. Currently, the WHO does not recognize a diagnosis of FM-associated MF (or LAFM) and acknowledges that folliculotropic MF is not always associated with FM.16,26 Given uncertainty and repercussions associated with a cancer diagnosis, however indolent, it may be morally responsible and medically favorable for physicians to consider FM in the differential diagnosis when applicable rather than making a diagnosis of MF outright. Given the importance of both clinical and histologic factors, it may be beneficial for definitive diagnosis of FM versus MF to lie with the clinician, while the pathologist serves as an adjunct in the diagnostic process. Because the prognosis of idiopathic FM often is marred by possible transformation into MF or other CTCLs, therapeutic decisions should be dictated by close clinical follow-up. Additionally, stage of disease, patient age, treatment compliance, comorbidities, and possible side effects of medications should all be considered when evaluating potential therapeutic regimens.27
Conclusion
Research is underway to more accurately identify patients with FM who are at risk for progression to LAFM versus those with benign remitting FM. Once the required diagnostic criteria are established to accurately classify these patients, with an emphasis on prognosis and suggested treatments, it might be necessary to establish new, less debated terminology so pathologists and clinicians alike can improve patient care. Continued histopathologic scrutiny, use of sophisticated molecular techniques, and knowledge of other currently undiscovered modalities will shed light on this important disease process and aid in proper disease management, which may be advantageous to both patients and physicians.
When follicular mucinosis (FM) is defined as an epithelial reaction pattern characterized by intrafollicular and perifollicular mucin accumulation, it cannot be considered a distinct disease entity, as this pattern is ubiquitously present in various inflammatory and neoplastic skin conditions.1,2 The distinction between idiopathic FM and lymphoma-associated follicular mucinosis (LAFM) was made several years ago by authors who evaluated the differences in the clinical presentation of these entities, including patient age at onset, number of lesions, pattern of distribution, and most importantly clinical progression.1 In this article, we discuss the importance of close clinical follow-up in patients with FM or patch-stage mycosis fungoides (MF) in whom histopathologic evaluation is ambiguous or nondiagnostic. We also highlight the value of ancillary testing, including T-cell receptor gene rearrangement, flow cytometry, and immunohistochemistry, as a component in the diagnostic process rather than the sole diagnostic moiety. A review of the pertinent literature also is performed.
History of FM and MF
Pinkus3 first described an entity he termed alopecia mucinosa in 1957. Pinkus noted 3 distinct patterns: an idiopathic form of alopecia mucinosa, lymphoblastoma with associated FM, and alopecia mucinosa that later transformed into lymphoblastoma.4 In 1983, however, Pinkus4 described uncertainty if alopecia mucinosa represented the first stage of MF or if patients with alopecia mucinosa were simply at an increased risk for developing lymphoma. He believed there were too many cases of lymphoma following a diagnosis of alopecia mucinosa for the relation to be coincidental, yet he noted that many of the cases resolved either spontaneously or following treatment with x-rays or topical steroids. He concluded his report with a sentiment that is echoed in many current studies regarding this entity: “Many questions surrounding this entity are as unanswerable today as they were 25 years ago.”4
Jablonska et al5 were the first to coin the term mucinosis follicularis, now known as FM, to replace alopecia mucinosa because they felt the description was more accurate, as lesions also arise on non–hair-bearing skin. Although there is general agreement that there is a form of MF that has associated FM, this is where the agreement ends with regard to the diagnosis of MF versus FM. Böer et al6 discussed the historic evolution of these terms, mostly to highlight the origins of the confusion. The investigators proposed that FM should only be used as a descriptive term and that all cases of alopecia mucinosa represent MF. They also concluded that many benign dermatoses associated with a risk for evolution to MF (eg, small and large plaque psoriasis [LPP]) should simply be diagnosed as MF.6 Subsequently, the proposal that idiopathic FM and LAFM are not 2 distinct entities but rather a clinicopathologic continuum and that idiopathic FM is simply a variant of MF along this spectrum has gained some approval.6,7 However, this belief is not shared among all authorities in the field, and attempts to define diagnostic criteria that distinguish between a benign clinical course and a course that is more progressive and fatal continue. Currently, it is agreed upon that when distinguishing between these 2 clinical courses, primary (idiopathic) follicular mucinosis refers to a benign course with no overt sign of malignancy, and lymphoma-associated follicular mucinosis refers to a diagnostic malignant condition. Lymphoma-associated follicular mucinosis refers to FM associated with cutaneous T-cell lymphoma, the most common form of which is FM. Many authors8-15 have sought ancillary methodologies in addition to clinical parameters to assist in the evaluation between both disease courses. Methodologies have included assessment of T-cell receptor gene rearrangements, flow cytometry, and immunohistochemical staining, mostly as an effort to establish monoclonality as a defining characteristic of LAFM; however, monoclonality in cutaneous T-cell infiltrates should be interpreted with caution and should not be considered as a confirmation of malignancy due to recent findings of monoclonality in benign inflammatory dermatoses such as lichen planus. The Table outlines several of the most common benign inflammatory dermatoses that demonstrate monoclonality, but this list should not be considered exhaustive, as there are many others in which monoclonality is sometimes seen.8-15 The lack of definitive criteria to distinguish between the 2 groups has led to confusion and consternation regarding the diagnosis of idiopathic FM versus LAFM and has led many in the field to consider the 2 conditions to be one and the same.
Diagnosis of FM and MF: Clinicopathologic Features
The World Health Organization (WHO) defined MF as an epidermotropic primary cutaneous T-cell lymphoma (CTCL) characterized by infiltrates of small- to medium-sized T lymphocytes with cerebriform nuclei. Further, the WHO stated that the term mycosis fungoides should be exclusively reserved for classical cases typified by the evolution of cutaneous patches, plaques, and tumors, or for variants that show a similar clinical course.16 Mycosis fungoides is divided into 3 stages—patch, plaque, and tumor—which are solely clinical descriptors.17 The WHO also described a clinical staging system with pathologic emphasis placed only on lymph node involvement and identification of Sézary cells.16 It lists folliculotropic MF as a variant, with only some cases presenting with mucinous degeneration of hair follicles. A lack of consensus among pathologists regarding criteria for diagnosis in patch-stage MF remains, but diagnosis of plaque-stage disease is not regularly debated due to its more reliably present, well-developed histologic features (eg, haloed lymphocytes, epidermotropism of lymphocytes, lymphocytes with convoluted nuceli, Pautrier microabscesses).18 Although there have been specific histologic findings reported to be associated with patch-stage MF, they have only been present in a few cases and are therefore of limited usefulness in practice.1,19 The categorization of patients with subtle histologic features common to both MF and inflammatory conditions such as parapsoriasis en plaques (the term plaque in this case is a misnomer because the word plaque means patch in French) continues to be elusive. A lack of agreement regarding LPP persists in the current literature in the same manner as FM. Some researchers have contended for many years that LPP is a type of MF, while others remain unconvinced, mainly due to the lack of evidence that lumping a benign condition (LPP) with an increased risk for malignant transformation and a known malignancy (MF) together is of any benefit to the patient. Assessment of clinicopathologic correlation, immunohistochemistry, clonality, and T-cell gene rearrangement have failed to positively identify patients who are at risk for disease progression, whether the diagnosis is called LPP or early patch-stage MF.20
Mycosis fungoides is more common in males and its incidence increases with age; however, diagnosis should not be ruled out based on age or gender. Typical presentation of early-stage disease includes erythematous patches or plaques, often with light scaling.19 Lesions routinely are of long-standing duration (months to years), are located in areas that are infrequently exposed to sunlight, and often are 5 cm in diameter or larger with irregular borders.21 Associated poikiloderma is relatively specific to MF but rarely is seen in other CTCLs, connective-tissue diseases, and some genodermatoses. Poikiloderma commonly is identified in LPP, which shows the same telangiectasia, mottled pigmentation, and epidermal atrophy as MF-associated poikiloderma, leading some to believe that there is no separation between the 2 conditions. In all stages of MF, lesions frequently are numerous and occur on multiple sites. Plaques and tumors can show spontaneous ulceration. When lesions are folliculotropic, they can cause localized alopecia, follicular-based papules, and fungating pseudotumors in more advanced stages.1 The clinical presentation of FM substantially overlaps with folliculotropic MF, and although FM lesions often are solitary and are located on the face or scalp, they also can present as multiple lesions located elsewhere on the body. It also has been proposed that folliculotropic MF should not be separated from FM-associated MF (or LAFM).22
The characteristic histologic picture of LAFM in patch or plaque stage shows mucin deposition within hair follicles, similar to idiopathic FM. On histology, both conditions demonstrate dense lymphoid infiltrates around and within hair follicles as well as in the dermis (Figure). Most cases of LAFM show epidermotropism of lymphocytes between follicles, but this finding is not present in every case and often disappears when the disease advances to the tumor stage.1,19 Although Pautrier microabscesses (collections of lymphocytes within the superficial epidermis) are considered to be somewhat specific to MF, they are only present in a minority of cases.20 In a study by the International Society for Cutaneous Lymphomas,21 the only histopathologic criteria that showed any appreciable sensitivity or specificity in the diagnosis of MF were the presence of lymphoid cells with variable nuclear and cytoplasmic features and/or strikingly irregular nuclear contours with the presence of lymphocytes larger than those usually seen in inflammatory dermatoses. Despite these criteria, the study reported a high misclassification rate. A complicated scoring system for diagnosis of MF in patch- or early plaque-stage disease was proposed by the International Society for Cutaneous Lymphomas,21 which integrates clinical, histopathologic, molecular, and immunophenotypic criteria. However, these criteria have been continually debated in the literature and are only discussed in this article in relation to the association between MF and FM. Diagnosis of tumor-stage MF is not addressed in this article, as it is readily identified as lymphoma and is not easily confused with idiopathic FM.
|
Clinical assessment of a patient’s medical history to identify persistent and progressive disease is paramount to the diagnosis of MF. Although MF lesions tend to increase in size and number over time, this presentation is not without exception.21 In early patch-stage disease, eliminating some of the patient’s current medications may be sufficient in clearing cutaneous patches that cannot be conclusively identified as either MF or a benign inflammatory lymphoid infiltrate, which further emphasizes the importance of clinical assessment of the patient’s medical history in the diagnosis of MF. The shape of the lesions also is helpful in distinguishing between MF and other skin disorders, such as digitate dermatosis or LPP; unlike the latter, the waxing and waning nature of MF lesions often produces irregularly shaped patches with little coalescence. Again, there are some investigators who believe that these lesions represent varying presentations of MF.6
In a study by Cerroni et al,1 44 patients with FM were divided into 2 groups: (1) a cohort of 16 patients with no history or clinical evidence of MF or Sézary syndrome (ie, LAFM), and (2) a cohort of 28 patients with clinicopathologic evidence of CTCL. Patients in both groups were followed for a maximum of 20 years. Results indicated that that the presence of perifollicular or intrafollicular mucin, epidermotropism of lymphocytes, monoclonality, and epidemiologic characteristics (eg, age, sex, race) cannot reliably distinguish the 2 disease forms. Furthermore, it was suggested that these conditions are not mutually exclusive entities and are actually variants of CTCL. The observation that the 2 diseases share prognostic overlap adds further credence to the already puzzling conundrum. Nineteen of 28 patients with MF were alive and well at follow-up, and all patients in the idiopathic FM group were alive, with only 9 of 16 patients showing residual disease and none with CTCL.1
Other clinical factors that may be helpful in the diagnosis of MF are the presentation of lesions in non–sun-exposed areas of the skin and multiple lesions, as unilesional MF is exceedingly uncommon.21 No histologic features have been proven to predict which early patch- or plaque-stage MFs will progress to full-blown CTCL versus benign idiopathic FM; thus, great caution should be taken in patients with early-stage disease to ensure they are not prematurely and/or incorrectly classified as CTCL. Such a diagnosis has medical, social, and economical ramifications that should not be overlooked.
If idiopathic FM and LAFM were considered distinct disease processes, the ambiguity in making a definitive diagnosis should give the physician pause, and a diagnosis of LAFM may only be appropriate when there is unequivocal clinicopathologic evidence. Otherwise, a lymphoma diagnosis is somewhat superfluous and potentially harmful. Definitive diagnosis of LAFM also is complicated by reports of other hematologic malignancies presenting with FM-like histopathologic findings, such as chronic myelogenous leukemia, leukemia-associated eosinophilic folliculitis, and acute myeloblastic leukemia.23,24 Although MF is the most common malignancy associated with FM, it is important to consider other less common malignancies that also may be present.
Diagnosis: Patient Consequences
Accurate diagnosis of idiopathic FM versus LAFM is critical, as the ramifications of a cancer diagnosis can have broad implications. For example, patients who receive cancer diagnoses often experience emotional trauma and social stigma, even when adequate patient education has been provided. The incidence of depression and anxiety also can increase following a cancer diagnosis and can be complicated by medical treatments (eg, systemic steroids, interferon),25 which are known to increase the frequency of these psychological disturbances. Health insurance premiums likely will be higher if a patient is diagnosed with cancer versus a benign inflammatory condition. Hesitation of the pathologist to assign a cancer diagnosis when unequivocal evidence is not present should not be regarded as trickery, malpractice, or deceit of the health care bylaws, as benign language with suggestion of close clinical follow-up in the setting of diagnostic uncertainty will “first, do no harm” and secondly, serve as a vehicle for patient advocacy.
If there is a definitive distinction between idiopathic FM and LAFM, it requires further research before it can be fully understood. Currently, the WHO does not recognize a diagnosis of FM-associated MF (or LAFM) and acknowledges that folliculotropic MF is not always associated with FM.16,26 Given uncertainty and repercussions associated with a cancer diagnosis, however indolent, it may be morally responsible and medically favorable for physicians to consider FM in the differential diagnosis when applicable rather than making a diagnosis of MF outright. Given the importance of both clinical and histologic factors, it may be beneficial for definitive diagnosis of FM versus MF to lie with the clinician, while the pathologist serves as an adjunct in the diagnostic process. Because the prognosis of idiopathic FM often is marred by possible transformation into MF or other CTCLs, therapeutic decisions should be dictated by close clinical follow-up. Additionally, stage of disease, patient age, treatment compliance, comorbidities, and possible side effects of medications should all be considered when evaluating potential therapeutic regimens.27
Conclusion
Research is underway to more accurately identify patients with FM who are at risk for progression to LAFM versus those with benign remitting FM. Once the required diagnostic criteria are established to accurately classify these patients, with an emphasis on prognosis and suggested treatments, it might be necessary to establish new, less debated terminology so pathologists and clinicians alike can improve patient care. Continued histopathologic scrutiny, use of sophisticated molecular techniques, and knowledge of other currently undiscovered modalities will shed light on this important disease process and aid in proper disease management, which may be advantageous to both patients and physicians.
1. Cerroni L, Fink-Puches R, Bäck B, et al. Follicular mucinosis: a critical reappraisal of clinicopathologic features and association with mycosis fungoides and Sézary syndrome. Arch Dermatol. 2002;138:182-189.
2. Parker SR, Murad E. Follicular mucinosis: clinical, histologic, and molecular remission with minocycline [published online ahead of print July 25, 2009]. J Am Acad Dermatol. 2010;62:139-141.
3. Pinkus H. Alopecia mucinosa; inflammatory plaques with alopecia characterized by root-sheath mucinosis. AMA Arch Dermatol. 1957;76:419-424, 424-426.
4. Pinkus H. Alopecia mucinosa. additional data in 1983. Arch Dermatol. 1983;119:698-699.
5. Jablonska S, Chorzelski T, Lancucki J. Mucinosis follicularis [in German]. Hautarzt. 1959;10:27-33.
6. Böer A, Guo Y, Ackerman AB. Alopecia mucinosa is mycosis fungoides. Am J Dermatopathol. 2004;26:33-52.
7. Brown HA, Gibson LE, Pujol RM, et al. Primary follicular mucinosis: long-term follow-up of patients younger than 40 years with and withoutclonal T-cell receptor gene rearrangement. J Am Acad Dermatol. 2002;47:856-862.
8. Schiller PI, Flaig MJ, Puchta U, et al. Detection of clonal T cells in lichen planus. Arch Dermatol Res. 2000;292:568-569.
9. Cerroni L, Kerl H. Primary follicular mucinosis and association with mycosis fungoides and other cutaneous T-cell lymphomas. J Am Acad Dermatol. 2004;51:146-147.
10. Dereure O, Levi E, Kadin ME. T-Cell clonality in pityriasis lichenoides et varioliformis acuta: a heteroduplex analysis of 20 cases. Arch Dermatol. 2000;136:1483-1486.
11. Haeffner AC, Smoller BR, Zepter K, et al. Differentiation and clonality of lesional lymphocytes in small plaque parapsoriasis. Arch Dermatol. 1995;131:321-324.
12. Schultz JC, Granados S, Vonderheid EC, et al. T-cell clonality of peripheral blood lymphocytes in patients with lymphomatoid papulosis. J Am Acad Dermatol. 2005;53:152-155.
13. Pfaltz K, Kerl K, Palmedo G, et al. Clonality in sarcoidosis, granuloma annulare, and granulomatous mycosis fungoides. Am J Dermatopathol. 2011;33:659-662.
14. Weinberg JM, Kristal L, Chooback L, et al. The clonal nature of pityriasis lichenoides. Arch Dermatol. 2002;138:1063-1067.
15. Guitart J, Magro C. Cutaneous T-cell lymphoid dyscrasia: a unifying term for idiopathic chronic dermatoses with persistent T-cell clones. Arch Dermatol. 2007;143:921-932.
16. Swerdlow SH, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008.
17. Zinzani PL, Ferreri AJ, Cerroni L. Mycosis fungoides [published online ahead of print October 22, 2007]. Crit Rev Oncol Hematol. 2008;65:172-182.
18. Smoller BR, Bishop K, Glusac E, et al. Reassessment of histologic parameters in the diagnosis of mycosis fungoides. Am J Surg Pathol. 1995;19:1423-1430.
19. Hwang ST, Janik JE, Jaffe ES, et al. Mycosis fungoides and Sézary syndrome. Lancet. 2008;371:945-957.
20. Sarveswari KN, Yesudian P. The conundrum of parapsoriasis versus patch stage of mycosis fungoides. Indian J Dermatol Venereol Leprol. 2009;75:229-235.
21. Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063.
22. Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides. a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525-530.
23. Rashid R, Hymes S. Folliculitis, follicular mucinosis, and papular mucinosis as a presentation of chronic myelomonocytic leukemia. Dermatol Online J. 2009;15:16.
24. Wada T, Yoshinaga E, Oiso N, et al. Adult T-cell leukemia-lymphoma associated with follicular mucinosis. J Dermatol. 2009;36:638-642.
25. Sampogna F, Frontani M, Baliva G, et al. Quality of life and psychological distress in patients with cutaneous lymphoma [published online ahead of print December 16, 2008]. Br J Dermatol. 2009;160:815-822.
26. Boone SL, Guitart J, Gerami P. Follicular mycosis fungoides: a histopathologic, immunohistochemical, and genotypic review. G Ital Dermatol Venereol. 2008;143:409-414.
27. Prince HM, Whittaker S, Hoppe RT. How I treat mycosis fungoides and Sézary syndrome [published online ahead of print August 20, 2009]. Blood. 2009;114:4337-4353.
1. Cerroni L, Fink-Puches R, Bäck B, et al. Follicular mucinosis: a critical reappraisal of clinicopathologic features and association with mycosis fungoides and Sézary syndrome. Arch Dermatol. 2002;138:182-189.
2. Parker SR, Murad E. Follicular mucinosis: clinical, histologic, and molecular remission with minocycline [published online ahead of print July 25, 2009]. J Am Acad Dermatol. 2010;62:139-141.
3. Pinkus H. Alopecia mucinosa; inflammatory plaques with alopecia characterized by root-sheath mucinosis. AMA Arch Dermatol. 1957;76:419-424, 424-426.
4. Pinkus H. Alopecia mucinosa. additional data in 1983. Arch Dermatol. 1983;119:698-699.
5. Jablonska S, Chorzelski T, Lancucki J. Mucinosis follicularis [in German]. Hautarzt. 1959;10:27-33.
6. Böer A, Guo Y, Ackerman AB. Alopecia mucinosa is mycosis fungoides. Am J Dermatopathol. 2004;26:33-52.
7. Brown HA, Gibson LE, Pujol RM, et al. Primary follicular mucinosis: long-term follow-up of patients younger than 40 years with and withoutclonal T-cell receptor gene rearrangement. J Am Acad Dermatol. 2002;47:856-862.
8. Schiller PI, Flaig MJ, Puchta U, et al. Detection of clonal T cells in lichen planus. Arch Dermatol Res. 2000;292:568-569.
9. Cerroni L, Kerl H. Primary follicular mucinosis and association with mycosis fungoides and other cutaneous T-cell lymphomas. J Am Acad Dermatol. 2004;51:146-147.
10. Dereure O, Levi E, Kadin ME. T-Cell clonality in pityriasis lichenoides et varioliformis acuta: a heteroduplex analysis of 20 cases. Arch Dermatol. 2000;136:1483-1486.
11. Haeffner AC, Smoller BR, Zepter K, et al. Differentiation and clonality of lesional lymphocytes in small plaque parapsoriasis. Arch Dermatol. 1995;131:321-324.
12. Schultz JC, Granados S, Vonderheid EC, et al. T-cell clonality of peripheral blood lymphocytes in patients with lymphomatoid papulosis. J Am Acad Dermatol. 2005;53:152-155.
13. Pfaltz K, Kerl K, Palmedo G, et al. Clonality in sarcoidosis, granuloma annulare, and granulomatous mycosis fungoides. Am J Dermatopathol. 2011;33:659-662.
14. Weinberg JM, Kristal L, Chooback L, et al. The clonal nature of pityriasis lichenoides. Arch Dermatol. 2002;138:1063-1067.
15. Guitart J, Magro C. Cutaneous T-cell lymphoid dyscrasia: a unifying term for idiopathic chronic dermatoses with persistent T-cell clones. Arch Dermatol. 2007;143:921-932.
16. Swerdlow SH, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008.
17. Zinzani PL, Ferreri AJ, Cerroni L. Mycosis fungoides [published online ahead of print October 22, 2007]. Crit Rev Oncol Hematol. 2008;65:172-182.
18. Smoller BR, Bishop K, Glusac E, et al. Reassessment of histologic parameters in the diagnosis of mycosis fungoides. Am J Surg Pathol. 1995;19:1423-1430.
19. Hwang ST, Janik JE, Jaffe ES, et al. Mycosis fungoides and Sézary syndrome. Lancet. 2008;371:945-957.
20. Sarveswari KN, Yesudian P. The conundrum of parapsoriasis versus patch stage of mycosis fungoides. Indian J Dermatol Venereol Leprol. 2009;75:229-235.
21. Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063.
22. Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides. a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525-530.
23. Rashid R, Hymes S. Folliculitis, follicular mucinosis, and papular mucinosis as a presentation of chronic myelomonocytic leukemia. Dermatol Online J. 2009;15:16.
24. Wada T, Yoshinaga E, Oiso N, et al. Adult T-cell leukemia-lymphoma associated with follicular mucinosis. J Dermatol. 2009;36:638-642.
25. Sampogna F, Frontani M, Baliva G, et al. Quality of life and psychological distress in patients with cutaneous lymphoma [published online ahead of print December 16, 2008]. Br J Dermatol. 2009;160:815-822.
26. Boone SL, Guitart J, Gerami P. Follicular mycosis fungoides: a histopathologic, immunohistochemical, and genotypic review. G Ital Dermatol Venereol. 2008;143:409-414.
27. Prince HM, Whittaker S, Hoppe RT. How I treat mycosis fungoides and Sézary syndrome [published online ahead of print August 20, 2009]. Blood. 2009;114:4337-4353.
Practice Points
- An isolated patch in the head or neck area is much more likely to be follicular mucinosis (FM) than mycosis fungoides (MF).
- Monoclonality does not reliably distinguish FM from MF.
- Younger patients are more likely to have FM with spontaneous remission, and older patients are more likely to develop MF.
- None of the clinicopathologic features of FM or MF are without overlap.
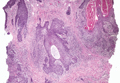
Reflectance Confocal Microscopy: An Overview of Technology and Advances in Telepathology
Reflectance confocal microscopy (RCM) creates an image by detecting backscattered light from illuminated tissue and displaying it on a monitor at high resolution and contrast. The grayscale images, oriented in a horizontal (en face) plane, reveal cellular and morphologic architecture in progressive depths from the epidermis to the papillary dermis.1,2 Analyses of confocal features have shown good correlation with histologic and dermoscopic findings, allowing key features of normal skin topography as well as cutaneous lesions to be delineated.1-15 Most research has focused on differentiating benign and malignant lesions, but RCM also has proven utility in presurgical mapping and in monitoring therapeutic efficacy of topical treatments of malignancies.16-19 Most recently, this US Food and Drug Administration 510(k)-cleared tool for imaging in vivo unstained epithelium (including blood, collagen, and pigment) has an added adjunct: a telepathology network dedicated to the transfer of confocal images from a private practice to a remote confocal diagnostic reader for image interpretation. As a noninvasive technique, RCM is a promising tool, not only in the field of dermatology but also in primary care.
Comparison of Diagnostic Modalities
Historically, diagnostic modalities have included visual and histopathologic examination; however, basing a diagnosis solely on clinical grounds may not be reliable, and obtaining a tissue specimen may not be feasible or practical. Thus, noninvasive modalities as adjuncts for evaluation were developed, including high-frequency ultrasound, high-definition optical coherence tomography, dermoscopy, and in vivo RCM. Sonography was not reliable in clinical practice due to poor echogenicity and insufficient resolution, and although high-definition optical coherence tomography is emerging as an important tool for the evaluation of lesions with high clinical suspicion for nonmelanoma skin cancers (eg, basal cell carcinoma [BCC]), resolution is still insufficient for definitive diagnosis; therefore, these devices cannot be reliably used on pigmented lesions suspicious for melanoma.20-23
Reflectance confocal microscopy has many properties similar to both dermoscopy and histology (Table).1-3,24-28 Dermoscopy and RCM are used by physicians to noninvasively analyze lesions en face in real time; both modalities operate through optical magnification and liquid immersion without exogenous contrast agents and can be used to monitor lesion progression over time.25,29 However, when comparing these modalities for melanoma identification among equivocal melanocytic lesions, they revealed statistically similar sensitivities (dermoscopy, 88%; RCM, 91%) but notably different specificities with RCM achieving more than double the specificity (dermoscopy, 32%; RCM, 68%).30

Similar to histology images, RCM images provide high axial and lateral resolution, delineating cellular and morphologic architecture in both vertical and horizontal planes.29,31 Unlike histology, RCM does not require tissue removal and processing, thus the images are immediately available for analysis. Although RCM is noninvasive similar to dermoscopy and has resolutions comparable to histology, it uniquely demonstrates the dynamic processes of living skin in real time.1-3
Technical Properties of RCM
There are 7 components to the microscope: a laser light source, scanning elements, a relay telescope, a beam splitter, a pinhole aperture, an objective lens, and a detector (Figure 1).1,2,32 A low-power laser beam illuminates a point on or within the skin. The scattered light reflected back into the optical system is imaged on a detector. A pinhole aperture in front of this detector filters out the scattered light and allows only the light from the image plane (a thin, in-focus plane in the tissue) to pass through, creating a high-resolution image (3–5 μm horizontal optical sections) of the target lesion. The optical parameters include an 830-nm laser with an operating power of 22 mW and an immersion objective lens with a 0.9 numerical aperture. Each image has a 500-μm field of view with approximately ×30 magnification. A larger 2-dimensional image is created when the laser rapidly scans across the plane of the skin lesion, sequentially capturing multiple images. These individual images are stitched together to create a mosaic with a field of up to 8×8 mm.1,2,32
The maximum imaging depth extends into the papillary dermis (up to 350 mm, depending on tissue thickness).1,2,27,32 Depth of light penetration is limited by wavelength and intensity to maximize resolution of discernible structures and to avoid tissue damage. Contrast is dependent on light scattering, which is generated in 2 ways: (1) by differences between the refractive index of water and tissue constituents, and (2) by diffraction of light by structures similar in size to the illuminating light wavelength. Thus, highly reflective or diffractive structures such as melanin, keratin, hydrated collagen, and melanosomes produce backscattered light that appears bright (white) compared to their surroundings.1,2,27,32
RCM Image Acquisition and Interpretation
After patient and lesion history are obtained, visual and dermoscopic evaluation of the lesion is performed. Index fluid (mineral oil) is applied to the lesion and a metal ring with an optically clear, nonbirefringent polymer window is attached to the skin.32 A 5-megapixel dermoscopic-quality image is captured through this ring and window. A water-based immersion medium (ultrasound gel) for the objective lens is then placed on the window and the confocal microscope is magnetically attached to the ring. The index fluid has a refractive index similar to the stratum corneum and the window, allowing for optimal imaging down to the papillary dermis. The adhesive and magnetic attachments stabilize the skin lesion as the confocal microscope captures sequential 2-dimensional images. A mosaic is then created at the specified anatomic level. Levels of imaging are determined by the depth (in micrometers) from the stratum corneum and correspond to each anatomic layer. En face images also can be vertically stacked, generally in 3- to 5-μm increments.32
The number of mosaics and stacks obtained are based on a preset standardized protocol. Once captured, images are saved and then sent over the telemedicine network to a remote confocal diagnostic reader for image interpretation, which can be done immediately or the images can be stored and sent (known as store and forward) at a later time.
Lesion evaluation begins with a review of patient and lesion history and dermoscopic images, followed by review of confocal images for additional information through visualization of cellular structures and architecture. Confocal interpretation commonly begins with examination of the mosaic at the level of the dermoepidermal junction, as it often provides the most diagnostic information. The papillary epidermal layers can then be used to confirm the working diagnosis, to further enhance the description, or to refine the diagnosis. Areas of interest may then be further examined in a vertical plane, which is achieved by observing a specific spot at different depths. Image interpretation can be approached in a variety of ways; however, the most critical aspect to any method is the recognition of skin morphology.
Confocal Images
Epidermal and dermal structures identified with in vivo confocal images are comparable to histology. The first consensus terminology glossary with illustrative images was published in 2007 with descriptions and definitions of image quality, normal skin morphology, lesional architecture, and cellular details of melanocytic lesions.33 Figure 2 shows the normal structures that comprise the different layers of skin as seen on RCM.
Stratum Corneum
At a thickness of 0 to 15 μm, the stratum corneum is the first bright image seen on RCM.1,2 The individual anucleate corneocytes often cannot be delineated; thus, sheets of cells appear as islands separated by dark furrows (wrinkles).1,2
Stratum Granulosum
The first layer of viable cells is located 15 to 20 μm below the skin’s surface.1,2 The large 25- to 35-μm polygonal structures (granular keratinocytes) contain bright border zones (cytoplasm), a central dark oval (nucleus), and a grainy appearance (keratohyalin granules, organelles, and melanosomes).1-3 A honeycomb pattern is seen within the normal epidermis, whereas in darkly pigmented lesions where keratinocytes may contain some pigment, it has been described as cobblestone pattern.3
Stratum Spinosum
At a depth of 20 to 50 μm, the honeycomb pattern consists of smaller 15- to 25-μm polygonal structures (spinous keratinocytes) with thinner bright borders (cytoplasm) and a darker oval nucleus.1-3
Stratum Basale
Below the spinous cells is a single layer of brighter round structures (basal keratinocytes), each 7 to 12 mm in diameter.1,2 Due to the supranuclear melanin caps, the basal keratinocytes have increased reflectivity, appearing brighter than granular and spinous cells. The more abundant the melanin within the basal keratinocytes, the brighter the appearance.1,2
Dermoepidermal Junction
Below the stratum corneum (50–150 μm) is the dermoepidermal junction.1 The “peaks” of dermal papillae emerge as clusters of bright cells (basal keratinocytes). With deeper sectioning, the dark round-oval spaces rimmed by bright basal cells (dermal papillae) progressively enlarge. They continue to enlarge until neighboring papillae touch each other tangentially, corresponding to the valleys of rete ridges.1
Papillary Dermis
At a depth of 60 to 80 μm, blood vessels and collagen fibers are seen.1-3 Collagen and elastin fibers present as thin, delicately intertwined, highly reflective fibrillar structures (1–5 μm). Blood vessels appear as weakly reflective, round or canalicular structures within dermal papillae. Within the lumina, serum appears dark, but blood cells can be seen in real time as continually moving, weakly reflective or bright round structures corresponding to leukocytes, erythrocytes, and platelets. With real-time imaging, cells also can be identified based on their movement; leukocytes can fill or distend the lumen and roll slowly along vessel walls, whereas erythrocytes move rapidly within vessel lumina.1-3
Reticular Dermis
Further below the stratum corneum (100–350 μm), similar highly reflective collagen fibers and bundles are present, with diameters of 1 to 5 μm and 5 to 25 μm, respectively.2,3
Adnexal Structures
The limitation of imaging depth by wavelength and intensity restricts visualization to upper portions of sebaceous glands, sweat ducts, and hair shafts within hair follicles.2,32
Clinical Applications of RCM
Diagnosis of Lesions
Since the inception of RCM, confocal-based diagnostic criteria have been established for allergic and irritant contact dermatitis,4,5 malignant melanoma,6 BCC,7 actinic keratosis,8 and squamous cell carcinoma.8 Much of the research has focused on skin cancers, including the differentiation of benign and malignant skin lesions,34-38 to help improve clinical diagnostic accuracy, reducing the number of biopsies of benign lesions.10,11,28,35,38 In 2008 Guitera et al39 used RCM and dermoscopy to detect melanoma with a sensitivity of 98% and in 2012 determined that biopsies of benign nevi and lesions clinically suspicious for BCC could be reduced by as much as 68% in a series of 710 equivocal lesions.35 In 2014, in a prospective study including more than 1000 patients, Pellacani et al38 demonstrated that biopsies of equivocal benign lesions were reduced by more that 50%, and all of the melanomas and BCCs excised in the study were correctly detected by RCM interpretation. Additionally, in both studies, the sensitivity of the RCM interpretation for detecting BCC was 100%. Amelanotic melanoma can be diagnostically challenging because clinical and dermoscopic features often are nondescript. In 2001, Busam et al17 successfully used RCM for amelanotic melanoma detection and margin assessment. A subsequent study by Braga et al24 positively demonstrated that RCM may aid in the detection and diagnosis of various solitary pink lesions.
Adjunct to Mohs Micrographic Surgery
When excisional biopsies are impractical, incisional biopsies may be performed, which may lead to sampling errors. Atypical lesions with poorly defined clinical borders dictates standard of care with surgical excision and microscopic evaluation of margins. For malignancies requiring treatment with Mohs micrographic surgery, further staging often is required. These limitations may be overcome with RCM. Early detection of amelanotic malignant melanoma with margin assessment has been successfully demonstrated.17 Curiel-Lewandroski et al16 reported 3 successful cases wherein RCM was used for diagnosis and monitoring of topical treatment, delineation of surgical margins, and guidance in tissue-sparing surgical excision with amelanotic melanoma, locally recurrent melanoma, and lentigo maligna melanoma, respectively. In 2013, Guitera et al40 demonstrated that mapping lentigo maligna margins prior to Mohs surgery changed the surgical management of 73% of patients in a study that included 37 patients with clinically or dermoscopically visible lesions.
Monitoring Topical Treatment
Unlike conventional histology, RCM does not involve tissue destruction, allowing for longitudinal surveillance when treating a malignancy with topical therapy. In a 2003 case study, RCM was used to confirm a previously diagnosed BCC, map tumor periphery, visualize the inflammatory response to imiquimod cream 5%, and confirm posttreatment clearance. Reflectance confocal microscopy features were confirmed with biopsy before and after treatment, and clinical findings during treatment precisely correlated with RCM findings.18 A similar study the following year demonstrated the efficacy of imiquimod cream 5% as an adjunct to BCC treatment by reducing or eliminating the lesion size prior to Mohs micrographic surgery, thereby decreasing the resulting surgical defect.19 To date, several studies have been performed by physicians throughout the world that have used RCM to monitor therapeutic outcomes of topically applied treatments such as imiquimod and hyaluronic acid as well as photodynamic therapy.41-43
A Clinical Tool
In vivo reflectance confocal microscopy, previously used only in the research setting, is now being used as a clinical tool for the evaluation of lesions suspicious for skin cancer by several academic centers and private practices throughout the United States. With clearance from the US Food and Drug Administration, physicians can use the device clinically for in vivo microscopic examination of skin lesions. The telepathology network allows for images to be acquired by a trained technician in a clinician’s office and then to be evaluated remotely by a diagnostic reader. The clinician can receive a diagnosis in as little as 30 minutes. The potential to noninvasively monitor tumor response to topical therapies, to delineate tumor margins prior to surgery, and to monitor lesions over time is an attractive option to patients.
The technology and telepathology network of RCM continues to be developed as diagnostic criteria are established and diagnostic readers are trained; however, diagnostic confocal features of various lesions have yet to be described, refined, or validated. Consequently, an extensive library of reference images has not yet been constructed.
Practical Application
A dermatology practice collaborated with a dermatopathology office to examine the feasibility of incorporating RCM and the telepathology network into the workflow of a private practice while creating a comprehensive library of cutaneous pathologies. A physician who did not have prior knowledge of RCM was selected for training with the goal to become proficient at operating the confocal microscope and interpreting the images. A dermatopathologist (also a confocal diagnostic reader) performed the histopathologic diagnoses of the lesions and correlated findings to confocal images.
Once images were captured using a standardized protocol, the lesion was biopsied according to standard of care. The images were sent over the telepathology network for interpretation and correlation to the histologic specimen by the dermatopathologist. These images were then stored on a secure server for use as a reference and educational tool for other diagnostic readers. We successfully achieved our goal of assisting with the development and integration of RCM and the telepathology network into the workflow of a busy private practice while building an extensive image library, thus showing potential use for other private practitioners.
Limitations of RCM
Although RCM may provide diagnostic information for many epidermal and papillary dermal lesions, it is not practical for predominantly dermal lesions or for providing prognostic information of invasive malignancies. Maximal imaging depth is 350 mm, but structures can truly be delineated at only approximately 250 μm (papillary dermis).2 Evaluation is further challenged with hypertrophic or hyperkeratotic lesions as well as those located on glabrous skin. Compared to histology, RCM resolution is slightly lower and nuclear features are not easily seen due to their weak backscattering effect.2 There are no adverse effects related to operator use; however, use may be limited if the patient has an allergy to the mediums used or to adhesive tape.
Challenges faced in integrating the technology into our practice include the machine size, time constraints, and reimbursement issues. Although not available in our office, smaller clinical devices exist (including a handheld RCM device that launched in 2007) and continue to be developed for future implementation. In our practice, capturing an image of 1 lesion took up to 20 minutes, but other protocols may necessitate only 10 minutes. Reimbursement for the imaging and image-reading procedures currently is being pursued.
Conclusion
In vivo RCM was developed as a noninvasive modality for the assessment of physiologic and pathologic conditions of the skin. Cellular and subcellular structures as well as dynamic processes are observed without destruction of tissue. The morphologic features seen in RCM are comparable to those demonstrated with histology and dermoscopy. Despite current challenges, RCM has been shown to be an advantageous diagnostic tool, a guide to evaluating benign and malignant lesions, an adjunct to Mohs micrographic surgery via presurgical mapping of tumor margins, and a monitoring tool to establish treatment responses and efficacy. Reflectance confocal microscopy has steadily gained acceptance in clinical dermatology over the last decade, and the number of users continues to grow. With the continued efforts in advancing research, including usage of the telepathology network, we believe these tools will prove to be valuable in the private practice setting, both in the fields of dermatology and primary care.
Acknowledgment
The authors thank Caliber Imaging & Diagnostics, Inc (Rochester, New York), for providing the RCM imaging system with associated disposable supplies and the reader workstation for this review.
1. Rajadhyaksha M, Grossman M, Esterowitz D, et al. In vivo confocal scanning laser microscopy of human skin: melanin provides strong contrast. J Invest Dermatol. 1995;104:946-952.
2. Rajadhyaksha M, González S, Zavislan JM, et al. In vivo confocal scanning laser microscopy of human skin II: advances in instrumentation and comparison to histology. J Invest Dermatol. 1999;113:293-303.
3. Huzaira M, Rius F, Rajadhyaksha M, et al. Topographic variations in normal skin, as viewed by in vivo reflectance confocal microscopy. J Invest Dermatol. 2001;116:846-852.
4. Swindells K, Burnett N, Rius-Diaz F, et al. Reflectance confocal microscopy may differentiate acute allergic and irritant contact dermatitis in vivo. J Am Acad Dermatol. 2004;50:220-228.
5. Astner S, González E, Cheung AC, et al. Non-invasive evaluation of the kinetics of allergic and irritant contact dermatitis. J Invest Dermatol. 2005;124:351-359.
6. Pellacani G, Cesinaro AM, Seidenari S. Reflectance-mode confocal microscopy of pigmented skin lesions—improvement in melanoma diagnostic specificity. J Am Acad Dermatol. 2005;53:979-985.
7. González S, Tannous Z. Real-time, in vivo confocal reflectance microscopy of basal cell carcinoma. J Am Acad Dermatol. 2002;47:869-874.
8. Rishpon A, Kim N, Scope A, et al. Reflectance confocal microscopy criteria for squamous cell carcinomas and actinic keratoses. Arch Dermatol. 2009;145:766-772.
9. Busam KJ, Charles C, Lee G, et al. Morphologic features of melanocytes, pigmented keratinocytes, and melanophages by in vivo confocal scanning laser microscopy. Mod Pathol. 2001;14:862-868.
10. Langley RG, Rajadhyaksha M, Dwyer PJ, et al. Confocal scanning laser microscopy of benign and malignant melanocytic skin lesions in vivo. J Am Acad Dermatol. 2001;45:365-376.
11. Pellacani G, Cesinaro AM, Seidenari S. In vivo assessment of melanocytic nests in nevi and melanomas by reflectance confocal microscopy. Mod Pathol. 2005;18:469-474.
12. Pellacani G, Cesinaro AM, Seidenari S. Reflectance-mode confocal microscopy for the in vivo characterization of pagetoid melanocytosis in melanomas and nevi. J Invest Dermatol. 2005;125:532-537.
13. Scope A, Benvenuto-Andrade C, Agero AL, et al. Correlation of dermoscopic structures of melanocytic lesions to reflectance confocal microscopy. Arch Dermatol. 2007;143:176-185.
14. Pellacani G, Longo C, Malvehy J, et al. In vivo confocal microscopic and histopathologic correlations of dermoscopic features in 202 melanocytic lesions. Arch Dermatol. 2008:144:1597-1608.
15. Scope A, Gill M, Benveuto-Andrade C, et al. Correlation of dermoscopy with in vivo reflectance confocal microscopy of streaks in melanocytic lesions. Arch Dermatol. 2007;143:727-734.
16. Curiel-Lewandrowski C, Williams CM, Swindells KJ, et al. Use of in vivo confocal microscopy in malignant melanoma: an aid in diagnosis and assessment of surgical and nonsurgical therapeutic approaches. Arch Dermatol. 2004;140:1127-1132.
17. Busam KJ, Hester K, Charles C, et al. Detection of clinically amelanotic malignant melanoma and assessment of its margins by in vivo confocal scanning laser microscopy. Arch Dermatol. 2001;137:923-929.
18. Goldgeier M, Fox CA, Zavislan JM, et al. Noninvasive imaging, treatment, and microscopic confirmation of clearance of basal cell carcinoma. Dermatol Surg. 2003;29:205-210.
19. Torres A, Niemeyer A, Berkes B, et al. 5% Imiquimod cream and reflectance-mode confocal microscopy as adjunct modalities to Mohs micrographic surgery for treatment of basal cell carcinoma. Dermatol Surg. 2004;30(12, pt 1):1462-1469.
20. Lassau N, Mercier S, Koscielny S, et al. Prognostic value of high-frequency sonography and color Doppler sonography for the preoperative assessment of melanomas. AJR Am J Roentgenol. 1999;172:457-461.
21. Ruocco E, Argenziano G, Pellacani G, et al. Noninvasive imaging of skin tumors. Dermatol Surg. 2004;30(2, pt 2):301-310.
22. Welzel J. Optical coherence tomography in dermatology: a review. Skin Res Technol. 2001;7:1-9.
23. Gambichler T, Plura I, Schmid-Wendtner M, et al. High-definition optical coherence tomography of melanocytic skin lesions [published online ahead of print September 18, 2014]. J Biophotonics. doi:10.1002/jbio.201400085.
24. Braga JC, Scope A, Klaz I, et al. The significance of reflectance confocal microscopy in the assessment of solitary pink skin lesions. J Am Acad Dermatol. 2009;61:230-241.
25. Argenyl ZB. Dermoscopy (epiluminescence microscopy) of pigmented skin lesions. current status and evolving trends. Dermatologic Clin. 1997;15:79-95.
26. Grin CM, Kopf AW, Welkovich B, et al. Accuracy in the clinical diagnosis of malignant melanoma. Arch Dermatol. 1990;126:763-766.
27. Rajadhyaksha M, Gonzalez S, Zavislan JM. Detectability of contrast agents for confocal reflectance imaging of skin and microcirculation. J Biomed Opt. 2004;9:323-331.
28. Langley RG, Burton E, Walsh N, et al. In vivo confocal scanning laser microscopy of benign lentigines: comparison to conventional histology and in vivo characteristics of lentigo maligna. J Am Acad Dermatol. 2006;55:88-97.
29. Scope A, Halpern AC. Diagnostic procedures and devices. In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2008:40-42.
30. Guitera P, Pellacani G, Longo C, et al. In vivo reflectance microscopy enhances secondary evaluation of melanocytic lesions. J Invest Dermatol. 2008;129:131-138.
31. Junqueira LC, Carneiro J. Histology and its methods of study. In: Junqueira LC, Carneiro J, eds. Basic Histology: Text and Atlas. 11 ed. New York, NY: McGraw-Hill; 2005:1-21.
32. González S, Gill M, Halpern A, eds. Reflectance Confocal Microscopy of Cutaneous Tumors: An Atlas With Clinical, Dermoscopic and Histological Correlations. London, UK: Informa Healthcare; 2008.
33. Scope A, Benvenuto-Andrade C, Agero AL, et al. In vivo reflectance confocal microscopy imaging of melanocytic lesions: consensus terminology glossary and illustrative images. J Am Acad Dermatol. 2007;57:644-658.
34. Langley RG, Walsh N, Sutherland AE, et al. The diagnostic accuracy of in vivo confocal scanning laser microscopy compared to dermoscopy of benign and malignant melanocytic lesions: a prospective study. Dermatology. 2007;215:365-372.
35. Guitera P, Menzies SW, Longo C, et al. In vivo confocal microscopy for diagnosis of melanoma and basal cell carcinoma using a two-step method: analysis of 710 consecutive clinically equivocal cases [published online ahead of print June 21, 2012]. J Invest Dermatol. 2012;132:2386-2394.
36. Guitera P, Pellacani G, Crotty KA, et al. The impact of in vivo reflectance confocal microscopy on the diagnostic accuracy of lentigo maligna and equivocal pigmented and nonpigmented macules of the face [published online ahead of print April 15, 2010]. J Invest Dermatol. 2010;130:2080-2091.
37. Pellacani G, Guitera P, Longo C, et al. The impact of in vivo reflectance confocal microscopy for the diagnostic accuracy of melanoma and equivocal melanocytic lesions [published online ahead of print July 26, 2007]. J Invest Dermatol. 2007;127:2759-2765.
38. Pellacani G, Pepe P, Casari A, et al. Reflectance confocal microscopy as a second-level examination in skin oncology improves diagnostic accuracy and saves unnecessary excisions: a longitudinal prospective study [published online ahead of print October 19, 2014]. Br J Dermatol. 2014;171:1044-1051.
39. Guitera P, Pellacani G, Longo C, et al. In vivo reflectance confocal microscopy enhances secondary evaluation of melanocytic lesions [published online ahead of print July 17, 2008]. J Invest Dermatol. 2009;129:131-138
40. Guitera P, Moloney FJ, Menzies SW, et al. Improving management and patient care in lentigo maligna by mapping with in vivo confocal microscopy. JAMA Dermatol. 2013;149:692-698.
41. Malvehy J, Roldán-Marín R, Iglesias-García P, et al. Monitoring treatment of field cancerisation with 3% diclofenac sodium 2.5% hyaluronic acid by reflectance confocal microscopy: a histologic correlation. Acta Derm Venereol. 2015;95:45-50.
42. Zalaudek I, Piana S, Moscarella E, et al. Morphologic grading and treatment of facial actinic keratosis. Clin Dermatol. 2014;32:80-87.
43. Longo C, Casari A, Pepe P, et al. Confocal microscopy insights into the treatment and cellular immune response of basal cell carcinoma tophotodynamic therapy [published online ahead of print December 13, 2012]. Dermatology. 2012;225:264-270.
Reflectance confocal microscopy (RCM) creates an image by detecting backscattered light from illuminated tissue and displaying it on a monitor at high resolution and contrast. The grayscale images, oriented in a horizontal (en face) plane, reveal cellular and morphologic architecture in progressive depths from the epidermis to the papillary dermis.1,2 Analyses of confocal features have shown good correlation with histologic and dermoscopic findings, allowing key features of normal skin topography as well as cutaneous lesions to be delineated.1-15 Most research has focused on differentiating benign and malignant lesions, but RCM also has proven utility in presurgical mapping and in monitoring therapeutic efficacy of topical treatments of malignancies.16-19 Most recently, this US Food and Drug Administration 510(k)-cleared tool for imaging in vivo unstained epithelium (including blood, collagen, and pigment) has an added adjunct: a telepathology network dedicated to the transfer of confocal images from a private practice to a remote confocal diagnostic reader for image interpretation. As a noninvasive technique, RCM is a promising tool, not only in the field of dermatology but also in primary care.
Comparison of Diagnostic Modalities
Historically, diagnostic modalities have included visual and histopathologic examination; however, basing a diagnosis solely on clinical grounds may not be reliable, and obtaining a tissue specimen may not be feasible or practical. Thus, noninvasive modalities as adjuncts for evaluation were developed, including high-frequency ultrasound, high-definition optical coherence tomography, dermoscopy, and in vivo RCM. Sonography was not reliable in clinical practice due to poor echogenicity and insufficient resolution, and although high-definition optical coherence tomography is emerging as an important tool for the evaluation of lesions with high clinical suspicion for nonmelanoma skin cancers (eg, basal cell carcinoma [BCC]), resolution is still insufficient for definitive diagnosis; therefore, these devices cannot be reliably used on pigmented lesions suspicious for melanoma.20-23
Reflectance confocal microscopy has many properties similar to both dermoscopy and histology (Table).1-3,24-28 Dermoscopy and RCM are used by physicians to noninvasively analyze lesions en face in real time; both modalities operate through optical magnification and liquid immersion without exogenous contrast agents and can be used to monitor lesion progression over time.25,29 However, when comparing these modalities for melanoma identification among equivocal melanocytic lesions, they revealed statistically similar sensitivities (dermoscopy, 88%; RCM, 91%) but notably different specificities with RCM achieving more than double the specificity (dermoscopy, 32%; RCM, 68%).30
Similar to histology images, RCM images provide high axial and lateral resolution, delineating cellular and morphologic architecture in both vertical and horizontal planes.29,31 Unlike histology, RCM does not require tissue removal and processing, thus the images are immediately available for analysis. Although RCM is noninvasive similar to dermoscopy and has resolutions comparable to histology, it uniquely demonstrates the dynamic processes of living skin in real time.1-3
Technical Properties of RCM
There are 7 components to the microscope: a laser light source, scanning elements, a relay telescope, a beam splitter, a pinhole aperture, an objective lens, and a detector (Figure 1).1,2,32 A low-power laser beam illuminates a point on or within the skin. The scattered light reflected back into the optical system is imaged on a detector. A pinhole aperture in front of this detector filters out the scattered light and allows only the light from the image plane (a thin, in-focus plane in the tissue) to pass through, creating a high-resolution image (3–5 μm horizontal optical sections) of the target lesion. The optical parameters include an 830-nm laser with an operating power of 22 mW and an immersion objective lens with a 0.9 numerical aperture. Each image has a 500-μm field of view with approximately ×30 magnification. A larger 2-dimensional image is created when the laser rapidly scans across the plane of the skin lesion, sequentially capturing multiple images. These individual images are stitched together to create a mosaic with a field of up to 8×8 mm.1,2,32
The maximum imaging depth extends into the papillary dermis (up to 350 mm, depending on tissue thickness).1,2,27,32 Depth of light penetration is limited by wavelength and intensity to maximize resolution of discernible structures and to avoid tissue damage. Contrast is dependent on light scattering, which is generated in 2 ways: (1) by differences between the refractive index of water and tissue constituents, and (2) by diffraction of light by structures similar in size to the illuminating light wavelength. Thus, highly reflective or diffractive structures such as melanin, keratin, hydrated collagen, and melanosomes produce backscattered light that appears bright (white) compared to their surroundings.1,2,27,32
RCM Image Acquisition and Interpretation
After patient and lesion history are obtained, visual and dermoscopic evaluation of the lesion is performed. Index fluid (mineral oil) is applied to the lesion and a metal ring with an optically clear, nonbirefringent polymer window is attached to the skin.32 A 5-megapixel dermoscopic-quality image is captured through this ring and window. A water-based immersion medium (ultrasound gel) for the objective lens is then placed on the window and the confocal microscope is magnetically attached to the ring. The index fluid has a refractive index similar to the stratum corneum and the window, allowing for optimal imaging down to the papillary dermis. The adhesive and magnetic attachments stabilize the skin lesion as the confocal microscope captures sequential 2-dimensional images. A mosaic is then created at the specified anatomic level. Levels of imaging are determined by the depth (in micrometers) from the stratum corneum and correspond to each anatomic layer. En face images also can be vertically stacked, generally in 3- to 5-μm increments.32
The number of mosaics and stacks obtained are based on a preset standardized protocol. Once captured, images are saved and then sent over the telemedicine network to a remote confocal diagnostic reader for image interpretation, which can be done immediately or the images can be stored and sent (known as store and forward) at a later time.
Lesion evaluation begins with a review of patient and lesion history and dermoscopic images, followed by review of confocal images for additional information through visualization of cellular structures and architecture. Confocal interpretation commonly begins with examination of the mosaic at the level of the dermoepidermal junction, as it often provides the most diagnostic information. The papillary epidermal layers can then be used to confirm the working diagnosis, to further enhance the description, or to refine the diagnosis. Areas of interest may then be further examined in a vertical plane, which is achieved by observing a specific spot at different depths. Image interpretation can be approached in a variety of ways; however, the most critical aspect to any method is the recognition of skin morphology.
Confocal Images
Epidermal and dermal structures identified with in vivo confocal images are comparable to histology. The first consensus terminology glossary with illustrative images was published in 2007 with descriptions and definitions of image quality, normal skin morphology, lesional architecture, and cellular details of melanocytic lesions.33 Figure 2 shows the normal structures that comprise the different layers of skin as seen on RCM.
Stratum Corneum
At a thickness of 0 to 15 μm, the stratum corneum is the first bright image seen on RCM.1,2 The individual anucleate corneocytes often cannot be delineated; thus, sheets of cells appear as islands separated by dark furrows (wrinkles).1,2
Stratum Granulosum
The first layer of viable cells is located 15 to 20 μm below the skin’s surface.1,2 The large 25- to 35-μm polygonal structures (granular keratinocytes) contain bright border zones (cytoplasm), a central dark oval (nucleus), and a grainy appearance (keratohyalin granules, organelles, and melanosomes).1-3 A honeycomb pattern is seen within the normal epidermis, whereas in darkly pigmented lesions where keratinocytes may contain some pigment, it has been described as cobblestone pattern.3
Stratum Spinosum
At a depth of 20 to 50 μm, the honeycomb pattern consists of smaller 15- to 25-μm polygonal structures (spinous keratinocytes) with thinner bright borders (cytoplasm) and a darker oval nucleus.1-3
Stratum Basale
Below the spinous cells is a single layer of brighter round structures (basal keratinocytes), each 7 to 12 mm in diameter.1,2 Due to the supranuclear melanin caps, the basal keratinocytes have increased reflectivity, appearing brighter than granular and spinous cells. The more abundant the melanin within the basal keratinocytes, the brighter the appearance.1,2
Dermoepidermal Junction
Below the stratum corneum (50–150 μm) is the dermoepidermal junction.1 The “peaks” of dermal papillae emerge as clusters of bright cells (basal keratinocytes). With deeper sectioning, the dark round-oval spaces rimmed by bright basal cells (dermal papillae) progressively enlarge. They continue to enlarge until neighboring papillae touch each other tangentially, corresponding to the valleys of rete ridges.1
Papillary Dermis
At a depth of 60 to 80 μm, blood vessels and collagen fibers are seen.1-3 Collagen and elastin fibers present as thin, delicately intertwined, highly reflective fibrillar structures (1–5 μm). Blood vessels appear as weakly reflective, round or canalicular structures within dermal papillae. Within the lumina, serum appears dark, but blood cells can be seen in real time as continually moving, weakly reflective or bright round structures corresponding to leukocytes, erythrocytes, and platelets. With real-time imaging, cells also can be identified based on their movement; leukocytes can fill or distend the lumen and roll slowly along vessel walls, whereas erythrocytes move rapidly within vessel lumina.1-3
Reticular Dermis
Further below the stratum corneum (100–350 μm), similar highly reflective collagen fibers and bundles are present, with diameters of 1 to 5 μm and 5 to 25 μm, respectively.2,3
Adnexal Structures
The limitation of imaging depth by wavelength and intensity restricts visualization to upper portions of sebaceous glands, sweat ducts, and hair shafts within hair follicles.2,32
Clinical Applications of RCM
Diagnosis of Lesions
Since the inception of RCM, confocal-based diagnostic criteria have been established for allergic and irritant contact dermatitis,4,5 malignant melanoma,6 BCC,7 actinic keratosis,8 and squamous cell carcinoma.8 Much of the research has focused on skin cancers, including the differentiation of benign and malignant skin lesions,34-38 to help improve clinical diagnostic accuracy, reducing the number of biopsies of benign lesions.10,11,28,35,38 In 2008 Guitera et al39 used RCM and dermoscopy to detect melanoma with a sensitivity of 98% and in 2012 determined that biopsies of benign nevi and lesions clinically suspicious for BCC could be reduced by as much as 68% in a series of 710 equivocal lesions.35 In 2014, in a prospective study including more than 1000 patients, Pellacani et al38 demonstrated that biopsies of equivocal benign lesions were reduced by more that 50%, and all of the melanomas and BCCs excised in the study were correctly detected by RCM interpretation. Additionally, in both studies, the sensitivity of the RCM interpretation for detecting BCC was 100%. Amelanotic melanoma can be diagnostically challenging because clinical and dermoscopic features often are nondescript. In 2001, Busam et al17 successfully used RCM for amelanotic melanoma detection and margin assessment. A subsequent study by Braga et al24 positively demonstrated that RCM may aid in the detection and diagnosis of various solitary pink lesions.
Adjunct to Mohs Micrographic Surgery
When excisional biopsies are impractical, incisional biopsies may be performed, which may lead to sampling errors. Atypical lesions with poorly defined clinical borders dictates standard of care with surgical excision and microscopic evaluation of margins. For malignancies requiring treatment with Mohs micrographic surgery, further staging often is required. These limitations may be overcome with RCM. Early detection of amelanotic malignant melanoma with margin assessment has been successfully demonstrated.17 Curiel-Lewandroski et al16 reported 3 successful cases wherein RCM was used for diagnosis and monitoring of topical treatment, delineation of surgical margins, and guidance in tissue-sparing surgical excision with amelanotic melanoma, locally recurrent melanoma, and lentigo maligna melanoma, respectively. In 2013, Guitera et al40 demonstrated that mapping lentigo maligna margins prior to Mohs surgery changed the surgical management of 73% of patients in a study that included 37 patients with clinically or dermoscopically visible lesions.
Monitoring Topical Treatment
Unlike conventional histology, RCM does not involve tissue destruction, allowing for longitudinal surveillance when treating a malignancy with topical therapy. In a 2003 case study, RCM was used to confirm a previously diagnosed BCC, map tumor periphery, visualize the inflammatory response to imiquimod cream 5%, and confirm posttreatment clearance. Reflectance confocal microscopy features were confirmed with biopsy before and after treatment, and clinical findings during treatment precisely correlated with RCM findings.18 A similar study the following year demonstrated the efficacy of imiquimod cream 5% as an adjunct to BCC treatment by reducing or eliminating the lesion size prior to Mohs micrographic surgery, thereby decreasing the resulting surgical defect.19 To date, several studies have been performed by physicians throughout the world that have used RCM to monitor therapeutic outcomes of topically applied treatments such as imiquimod and hyaluronic acid as well as photodynamic therapy.41-43
A Clinical Tool
In vivo reflectance confocal microscopy, previously used only in the research setting, is now being used as a clinical tool for the evaluation of lesions suspicious for skin cancer by several academic centers and private practices throughout the United States. With clearance from the US Food and Drug Administration, physicians can use the device clinically for in vivo microscopic examination of skin lesions. The telepathology network allows for images to be acquired by a trained technician in a clinician’s office and then to be evaluated remotely by a diagnostic reader. The clinician can receive a diagnosis in as little as 30 minutes. The potential to noninvasively monitor tumor response to topical therapies, to delineate tumor margins prior to surgery, and to monitor lesions over time is an attractive option to patients.
The technology and telepathology network of RCM continues to be developed as diagnostic criteria are established and diagnostic readers are trained; however, diagnostic confocal features of various lesions have yet to be described, refined, or validated. Consequently, an extensive library of reference images has not yet been constructed.
Practical Application
A dermatology practice collaborated with a dermatopathology office to examine the feasibility of incorporating RCM and the telepathology network into the workflow of a private practice while creating a comprehensive library of cutaneous pathologies. A physician who did not have prior knowledge of RCM was selected for training with the goal to become proficient at operating the confocal microscope and interpreting the images. A dermatopathologist (also a confocal diagnostic reader) performed the histopathologic diagnoses of the lesions and correlated findings to confocal images.
Once images were captured using a standardized protocol, the lesion was biopsied according to standard of care. The images were sent over the telepathology network for interpretation and correlation to the histologic specimen by the dermatopathologist. These images were then stored on a secure server for use as a reference and educational tool for other diagnostic readers. We successfully achieved our goal of assisting with the development and integration of RCM and the telepathology network into the workflow of a busy private practice while building an extensive image library, thus showing potential use for other private practitioners.
Limitations of RCM
Although RCM may provide diagnostic information for many epidermal and papillary dermal lesions, it is not practical for predominantly dermal lesions or for providing prognostic information of invasive malignancies. Maximal imaging depth is 350 mm, but structures can truly be delineated at only approximately 250 μm (papillary dermis).2 Evaluation is further challenged with hypertrophic or hyperkeratotic lesions as well as those located on glabrous skin. Compared to histology, RCM resolution is slightly lower and nuclear features are not easily seen due to their weak backscattering effect.2 There are no adverse effects related to operator use; however, use may be limited if the patient has an allergy to the mediums used or to adhesive tape.
Challenges faced in integrating the technology into our practice include the machine size, time constraints, and reimbursement issues. Although not available in our office, smaller clinical devices exist (including a handheld RCM device that launched in 2007) and continue to be developed for future implementation. In our practice, capturing an image of 1 lesion took up to 20 minutes, but other protocols may necessitate only 10 minutes. Reimbursement for the imaging and image-reading procedures currently is being pursued.
Conclusion
In vivo RCM was developed as a noninvasive modality for the assessment of physiologic and pathologic conditions of the skin. Cellular and subcellular structures as well as dynamic processes are observed without destruction of tissue. The morphologic features seen in RCM are comparable to those demonstrated with histology and dermoscopy. Despite current challenges, RCM has been shown to be an advantageous diagnostic tool, a guide to evaluating benign and malignant lesions, an adjunct to Mohs micrographic surgery via presurgical mapping of tumor margins, and a monitoring tool to establish treatment responses and efficacy. Reflectance confocal microscopy has steadily gained acceptance in clinical dermatology over the last decade, and the number of users continues to grow. With the continued efforts in advancing research, including usage of the telepathology network, we believe these tools will prove to be valuable in the private practice setting, both in the fields of dermatology and primary care.
Acknowledgment
The authors thank Caliber Imaging & Diagnostics, Inc (Rochester, New York), for providing the RCM imaging system with associated disposable supplies and the reader workstation for this review.
Reflectance confocal microscopy (RCM) creates an image by detecting backscattered light from illuminated tissue and displaying it on a monitor at high resolution and contrast. The grayscale images, oriented in a horizontal (en face) plane, reveal cellular and morphologic architecture in progressive depths from the epidermis to the papillary dermis.1,2 Analyses of confocal features have shown good correlation with histologic and dermoscopic findings, allowing key features of normal skin topography as well as cutaneous lesions to be delineated.1-15 Most research has focused on differentiating benign and malignant lesions, but RCM also has proven utility in presurgical mapping and in monitoring therapeutic efficacy of topical treatments of malignancies.16-19 Most recently, this US Food and Drug Administration 510(k)-cleared tool for imaging in vivo unstained epithelium (including blood, collagen, and pigment) has an added adjunct: a telepathology network dedicated to the transfer of confocal images from a private practice to a remote confocal diagnostic reader for image interpretation. As a noninvasive technique, RCM is a promising tool, not only in the field of dermatology but also in primary care.
Comparison of Diagnostic Modalities
Historically, diagnostic modalities have included visual and histopathologic examination; however, basing a diagnosis solely on clinical grounds may not be reliable, and obtaining a tissue specimen may not be feasible or practical. Thus, noninvasive modalities as adjuncts for evaluation were developed, including high-frequency ultrasound, high-definition optical coherence tomography, dermoscopy, and in vivo RCM. Sonography was not reliable in clinical practice due to poor echogenicity and insufficient resolution, and although high-definition optical coherence tomography is emerging as an important tool for the evaluation of lesions with high clinical suspicion for nonmelanoma skin cancers (eg, basal cell carcinoma [BCC]), resolution is still insufficient for definitive diagnosis; therefore, these devices cannot be reliably used on pigmented lesions suspicious for melanoma.20-23
Reflectance confocal microscopy has many properties similar to both dermoscopy and histology (Table).1-3,24-28 Dermoscopy and RCM are used by physicians to noninvasively analyze lesions en face in real time; both modalities operate through optical magnification and liquid immersion without exogenous contrast agents and can be used to monitor lesion progression over time.25,29 However, when comparing these modalities for melanoma identification among equivocal melanocytic lesions, they revealed statistically similar sensitivities (dermoscopy, 88%; RCM, 91%) but notably different specificities with RCM achieving more than double the specificity (dermoscopy, 32%; RCM, 68%).30
Similar to histology images, RCM images provide high axial and lateral resolution, delineating cellular and morphologic architecture in both vertical and horizontal planes.29,31 Unlike histology, RCM does not require tissue removal and processing, thus the images are immediately available for analysis. Although RCM is noninvasive similar to dermoscopy and has resolutions comparable to histology, it uniquely demonstrates the dynamic processes of living skin in real time.1-3
Technical Properties of RCM
There are 7 components to the microscope: a laser light source, scanning elements, a relay telescope, a beam splitter, a pinhole aperture, an objective lens, and a detector (Figure 1).1,2,32 A low-power laser beam illuminates a point on or within the skin. The scattered light reflected back into the optical system is imaged on a detector. A pinhole aperture in front of this detector filters out the scattered light and allows only the light from the image plane (a thin, in-focus plane in the tissue) to pass through, creating a high-resolution image (3–5 μm horizontal optical sections) of the target lesion. The optical parameters include an 830-nm laser with an operating power of 22 mW and an immersion objective lens with a 0.9 numerical aperture. Each image has a 500-μm field of view with approximately ×30 magnification. A larger 2-dimensional image is created when the laser rapidly scans across the plane of the skin lesion, sequentially capturing multiple images. These individual images are stitched together to create a mosaic with a field of up to 8×8 mm.1,2,32
The maximum imaging depth extends into the papillary dermis (up to 350 mm, depending on tissue thickness).1,2,27,32 Depth of light penetration is limited by wavelength and intensity to maximize resolution of discernible structures and to avoid tissue damage. Contrast is dependent on light scattering, which is generated in 2 ways: (1) by differences between the refractive index of water and tissue constituents, and (2) by diffraction of light by structures similar in size to the illuminating light wavelength. Thus, highly reflective or diffractive structures such as melanin, keratin, hydrated collagen, and melanosomes produce backscattered light that appears bright (white) compared to their surroundings.1,2,27,32
RCM Image Acquisition and Interpretation
After patient and lesion history are obtained, visual and dermoscopic evaluation of the lesion is performed. Index fluid (mineral oil) is applied to the lesion and a metal ring with an optically clear, nonbirefringent polymer window is attached to the skin.32 A 5-megapixel dermoscopic-quality image is captured through this ring and window. A water-based immersion medium (ultrasound gel) for the objective lens is then placed on the window and the confocal microscope is magnetically attached to the ring. The index fluid has a refractive index similar to the stratum corneum and the window, allowing for optimal imaging down to the papillary dermis. The adhesive and magnetic attachments stabilize the skin lesion as the confocal microscope captures sequential 2-dimensional images. A mosaic is then created at the specified anatomic level. Levels of imaging are determined by the depth (in micrometers) from the stratum corneum and correspond to each anatomic layer. En face images also can be vertically stacked, generally in 3- to 5-μm increments.32
The number of mosaics and stacks obtained are based on a preset standardized protocol. Once captured, images are saved and then sent over the telemedicine network to a remote confocal diagnostic reader for image interpretation, which can be done immediately or the images can be stored and sent (known as store and forward) at a later time.
Lesion evaluation begins with a review of patient and lesion history and dermoscopic images, followed by review of confocal images for additional information through visualization of cellular structures and architecture. Confocal interpretation commonly begins with examination of the mosaic at the level of the dermoepidermal junction, as it often provides the most diagnostic information. The papillary epidermal layers can then be used to confirm the working diagnosis, to further enhance the description, or to refine the diagnosis. Areas of interest may then be further examined in a vertical plane, which is achieved by observing a specific spot at different depths. Image interpretation can be approached in a variety of ways; however, the most critical aspect to any method is the recognition of skin morphology.
Confocal Images
Epidermal and dermal structures identified with in vivo confocal images are comparable to histology. The first consensus terminology glossary with illustrative images was published in 2007 with descriptions and definitions of image quality, normal skin morphology, lesional architecture, and cellular details of melanocytic lesions.33 Figure 2 shows the normal structures that comprise the different layers of skin as seen on RCM.
Stratum Corneum
At a thickness of 0 to 15 μm, the stratum corneum is the first bright image seen on RCM.1,2 The individual anucleate corneocytes often cannot be delineated; thus, sheets of cells appear as islands separated by dark furrows (wrinkles).1,2
Stratum Granulosum
The first layer of viable cells is located 15 to 20 μm below the skin’s surface.1,2 The large 25- to 35-μm polygonal structures (granular keratinocytes) contain bright border zones (cytoplasm), a central dark oval (nucleus), and a grainy appearance (keratohyalin granules, organelles, and melanosomes).1-3 A honeycomb pattern is seen within the normal epidermis, whereas in darkly pigmented lesions where keratinocytes may contain some pigment, it has been described as cobblestone pattern.3
Stratum Spinosum
At a depth of 20 to 50 μm, the honeycomb pattern consists of smaller 15- to 25-μm polygonal structures (spinous keratinocytes) with thinner bright borders (cytoplasm) and a darker oval nucleus.1-3
Stratum Basale
Below the spinous cells is a single layer of brighter round structures (basal keratinocytes), each 7 to 12 mm in diameter.1,2 Due to the supranuclear melanin caps, the basal keratinocytes have increased reflectivity, appearing brighter than granular and spinous cells. The more abundant the melanin within the basal keratinocytes, the brighter the appearance.1,2
Dermoepidermal Junction
Below the stratum corneum (50–150 μm) is the dermoepidermal junction.1 The “peaks” of dermal papillae emerge as clusters of bright cells (basal keratinocytes). With deeper sectioning, the dark round-oval spaces rimmed by bright basal cells (dermal papillae) progressively enlarge. They continue to enlarge until neighboring papillae touch each other tangentially, corresponding to the valleys of rete ridges.1
Papillary Dermis
At a depth of 60 to 80 μm, blood vessels and collagen fibers are seen.1-3 Collagen and elastin fibers present as thin, delicately intertwined, highly reflective fibrillar structures (1–5 μm). Blood vessels appear as weakly reflective, round or canalicular structures within dermal papillae. Within the lumina, serum appears dark, but blood cells can be seen in real time as continually moving, weakly reflective or bright round structures corresponding to leukocytes, erythrocytes, and platelets. With real-time imaging, cells also can be identified based on their movement; leukocytes can fill or distend the lumen and roll slowly along vessel walls, whereas erythrocytes move rapidly within vessel lumina.1-3
Reticular Dermis
Further below the stratum corneum (100–350 μm), similar highly reflective collagen fibers and bundles are present, with diameters of 1 to 5 μm and 5 to 25 μm, respectively.2,3
Adnexal Structures
The limitation of imaging depth by wavelength and intensity restricts visualization to upper portions of sebaceous glands, sweat ducts, and hair shafts within hair follicles.2,32
Clinical Applications of RCM
Diagnosis of Lesions
Since the inception of RCM, confocal-based diagnostic criteria have been established for allergic and irritant contact dermatitis,4,5 malignant melanoma,6 BCC,7 actinic keratosis,8 and squamous cell carcinoma.8 Much of the research has focused on skin cancers, including the differentiation of benign and malignant skin lesions,34-38 to help improve clinical diagnostic accuracy, reducing the number of biopsies of benign lesions.10,11,28,35,38 In 2008 Guitera et al39 used RCM and dermoscopy to detect melanoma with a sensitivity of 98% and in 2012 determined that biopsies of benign nevi and lesions clinically suspicious for BCC could be reduced by as much as 68% in a series of 710 equivocal lesions.35 In 2014, in a prospective study including more than 1000 patients, Pellacani et al38 demonstrated that biopsies of equivocal benign lesions were reduced by more that 50%, and all of the melanomas and BCCs excised in the study were correctly detected by RCM interpretation. Additionally, in both studies, the sensitivity of the RCM interpretation for detecting BCC was 100%. Amelanotic melanoma can be diagnostically challenging because clinical and dermoscopic features often are nondescript. In 2001, Busam et al17 successfully used RCM for amelanotic melanoma detection and margin assessment. A subsequent study by Braga et al24 positively demonstrated that RCM may aid in the detection and diagnosis of various solitary pink lesions.
Adjunct to Mohs Micrographic Surgery
When excisional biopsies are impractical, incisional biopsies may be performed, which may lead to sampling errors. Atypical lesions with poorly defined clinical borders dictates standard of care with surgical excision and microscopic evaluation of margins. For malignancies requiring treatment with Mohs micrographic surgery, further staging often is required. These limitations may be overcome with RCM. Early detection of amelanotic malignant melanoma with margin assessment has been successfully demonstrated.17 Curiel-Lewandroski et al16 reported 3 successful cases wherein RCM was used for diagnosis and monitoring of topical treatment, delineation of surgical margins, and guidance in tissue-sparing surgical excision with amelanotic melanoma, locally recurrent melanoma, and lentigo maligna melanoma, respectively. In 2013, Guitera et al40 demonstrated that mapping lentigo maligna margins prior to Mohs surgery changed the surgical management of 73% of patients in a study that included 37 patients with clinically or dermoscopically visible lesions.
Monitoring Topical Treatment
Unlike conventional histology, RCM does not involve tissue destruction, allowing for longitudinal surveillance when treating a malignancy with topical therapy. In a 2003 case study, RCM was used to confirm a previously diagnosed BCC, map tumor periphery, visualize the inflammatory response to imiquimod cream 5%, and confirm posttreatment clearance. Reflectance confocal microscopy features were confirmed with biopsy before and after treatment, and clinical findings during treatment precisely correlated with RCM findings.18 A similar study the following year demonstrated the efficacy of imiquimod cream 5% as an adjunct to BCC treatment by reducing or eliminating the lesion size prior to Mohs micrographic surgery, thereby decreasing the resulting surgical defect.19 To date, several studies have been performed by physicians throughout the world that have used RCM to monitor therapeutic outcomes of topically applied treatments such as imiquimod and hyaluronic acid as well as photodynamic therapy.41-43
A Clinical Tool
In vivo reflectance confocal microscopy, previously used only in the research setting, is now being used as a clinical tool for the evaluation of lesions suspicious for skin cancer by several academic centers and private practices throughout the United States. With clearance from the US Food and Drug Administration, physicians can use the device clinically for in vivo microscopic examination of skin lesions. The telepathology network allows for images to be acquired by a trained technician in a clinician’s office and then to be evaluated remotely by a diagnostic reader. The clinician can receive a diagnosis in as little as 30 minutes. The potential to noninvasively monitor tumor response to topical therapies, to delineate tumor margins prior to surgery, and to monitor lesions over time is an attractive option to patients.
The technology and telepathology network of RCM continues to be developed as diagnostic criteria are established and diagnostic readers are trained; however, diagnostic confocal features of various lesions have yet to be described, refined, or validated. Consequently, an extensive library of reference images has not yet been constructed.
Practical Application
A dermatology practice collaborated with a dermatopathology office to examine the feasibility of incorporating RCM and the telepathology network into the workflow of a private practice while creating a comprehensive library of cutaneous pathologies. A physician who did not have prior knowledge of RCM was selected for training with the goal to become proficient at operating the confocal microscope and interpreting the images. A dermatopathologist (also a confocal diagnostic reader) performed the histopathologic diagnoses of the lesions and correlated findings to confocal images.
Once images were captured using a standardized protocol, the lesion was biopsied according to standard of care. The images were sent over the telepathology network for interpretation and correlation to the histologic specimen by the dermatopathologist. These images were then stored on a secure server for use as a reference and educational tool for other diagnostic readers. We successfully achieved our goal of assisting with the development and integration of RCM and the telepathology network into the workflow of a busy private practice while building an extensive image library, thus showing potential use for other private practitioners.
Limitations of RCM
Although RCM may provide diagnostic information for many epidermal and papillary dermal lesions, it is not practical for predominantly dermal lesions or for providing prognostic information of invasive malignancies. Maximal imaging depth is 350 mm, but structures can truly be delineated at only approximately 250 μm (papillary dermis).2 Evaluation is further challenged with hypertrophic or hyperkeratotic lesions as well as those located on glabrous skin. Compared to histology, RCM resolution is slightly lower and nuclear features are not easily seen due to their weak backscattering effect.2 There are no adverse effects related to operator use; however, use may be limited if the patient has an allergy to the mediums used or to adhesive tape.
Challenges faced in integrating the technology into our practice include the machine size, time constraints, and reimbursement issues. Although not available in our office, smaller clinical devices exist (including a handheld RCM device that launched in 2007) and continue to be developed for future implementation. In our practice, capturing an image of 1 lesion took up to 20 minutes, but other protocols may necessitate only 10 minutes. Reimbursement for the imaging and image-reading procedures currently is being pursued.
Conclusion
In vivo RCM was developed as a noninvasive modality for the assessment of physiologic and pathologic conditions of the skin. Cellular and subcellular structures as well as dynamic processes are observed without destruction of tissue. The morphologic features seen in RCM are comparable to those demonstrated with histology and dermoscopy. Despite current challenges, RCM has been shown to be an advantageous diagnostic tool, a guide to evaluating benign and malignant lesions, an adjunct to Mohs micrographic surgery via presurgical mapping of tumor margins, and a monitoring tool to establish treatment responses and efficacy. Reflectance confocal microscopy has steadily gained acceptance in clinical dermatology over the last decade, and the number of users continues to grow. With the continued efforts in advancing research, including usage of the telepathology network, we believe these tools will prove to be valuable in the private practice setting, both in the fields of dermatology and primary care.
Acknowledgment
The authors thank Caliber Imaging & Diagnostics, Inc (Rochester, New York), for providing the RCM imaging system with associated disposable supplies and the reader workstation for this review.
1. Rajadhyaksha M, Grossman M, Esterowitz D, et al. In vivo confocal scanning laser microscopy of human skin: melanin provides strong contrast. J Invest Dermatol. 1995;104:946-952.
2. Rajadhyaksha M, González S, Zavislan JM, et al. In vivo confocal scanning laser microscopy of human skin II: advances in instrumentation and comparison to histology. J Invest Dermatol. 1999;113:293-303.
3. Huzaira M, Rius F, Rajadhyaksha M, et al. Topographic variations in normal skin, as viewed by in vivo reflectance confocal microscopy. J Invest Dermatol. 2001;116:846-852.
4. Swindells K, Burnett N, Rius-Diaz F, et al. Reflectance confocal microscopy may differentiate acute allergic and irritant contact dermatitis in vivo. J Am Acad Dermatol. 2004;50:220-228.
5. Astner S, González E, Cheung AC, et al. Non-invasive evaluation of the kinetics of allergic and irritant contact dermatitis. J Invest Dermatol. 2005;124:351-359.
6. Pellacani G, Cesinaro AM, Seidenari S. Reflectance-mode confocal microscopy of pigmented skin lesions—improvement in melanoma diagnostic specificity. J Am Acad Dermatol. 2005;53:979-985.
7. González S, Tannous Z. Real-time, in vivo confocal reflectance microscopy of basal cell carcinoma. J Am Acad Dermatol. 2002;47:869-874.
8. Rishpon A, Kim N, Scope A, et al. Reflectance confocal microscopy criteria for squamous cell carcinomas and actinic keratoses. Arch Dermatol. 2009;145:766-772.
9. Busam KJ, Charles C, Lee G, et al. Morphologic features of melanocytes, pigmented keratinocytes, and melanophages by in vivo confocal scanning laser microscopy. Mod Pathol. 2001;14:862-868.
10. Langley RG, Rajadhyaksha M, Dwyer PJ, et al. Confocal scanning laser microscopy of benign and malignant melanocytic skin lesions in vivo. J Am Acad Dermatol. 2001;45:365-376.
11. Pellacani G, Cesinaro AM, Seidenari S. In vivo assessment of melanocytic nests in nevi and melanomas by reflectance confocal microscopy. Mod Pathol. 2005;18:469-474.
12. Pellacani G, Cesinaro AM, Seidenari S. Reflectance-mode confocal microscopy for the in vivo characterization of pagetoid melanocytosis in melanomas and nevi. J Invest Dermatol. 2005;125:532-537.
13. Scope A, Benvenuto-Andrade C, Agero AL, et al. Correlation of dermoscopic structures of melanocytic lesions to reflectance confocal microscopy. Arch Dermatol. 2007;143:176-185.
14. Pellacani G, Longo C, Malvehy J, et al. In vivo confocal microscopic and histopathologic correlations of dermoscopic features in 202 melanocytic lesions. Arch Dermatol. 2008:144:1597-1608.
15. Scope A, Gill M, Benveuto-Andrade C, et al. Correlation of dermoscopy with in vivo reflectance confocal microscopy of streaks in melanocytic lesions. Arch Dermatol. 2007;143:727-734.
16. Curiel-Lewandrowski C, Williams CM, Swindells KJ, et al. Use of in vivo confocal microscopy in malignant melanoma: an aid in diagnosis and assessment of surgical and nonsurgical therapeutic approaches. Arch Dermatol. 2004;140:1127-1132.
17. Busam KJ, Hester K, Charles C, et al. Detection of clinically amelanotic malignant melanoma and assessment of its margins by in vivo confocal scanning laser microscopy. Arch Dermatol. 2001;137:923-929.
18. Goldgeier M, Fox CA, Zavislan JM, et al. Noninvasive imaging, treatment, and microscopic confirmation of clearance of basal cell carcinoma. Dermatol Surg. 2003;29:205-210.
19. Torres A, Niemeyer A, Berkes B, et al. 5% Imiquimod cream and reflectance-mode confocal microscopy as adjunct modalities to Mohs micrographic surgery for treatment of basal cell carcinoma. Dermatol Surg. 2004;30(12, pt 1):1462-1469.
20. Lassau N, Mercier S, Koscielny S, et al. Prognostic value of high-frequency sonography and color Doppler sonography for the preoperative assessment of melanomas. AJR Am J Roentgenol. 1999;172:457-461.
21. Ruocco E, Argenziano G, Pellacani G, et al. Noninvasive imaging of skin tumors. Dermatol Surg. 2004;30(2, pt 2):301-310.
22. Welzel J. Optical coherence tomography in dermatology: a review. Skin Res Technol. 2001;7:1-9.
23. Gambichler T, Plura I, Schmid-Wendtner M, et al. High-definition optical coherence tomography of melanocytic skin lesions [published online ahead of print September 18, 2014]. J Biophotonics. doi:10.1002/jbio.201400085.
24. Braga JC, Scope A, Klaz I, et al. The significance of reflectance confocal microscopy in the assessment of solitary pink skin lesions. J Am Acad Dermatol. 2009;61:230-241.
25. Argenyl ZB. Dermoscopy (epiluminescence microscopy) of pigmented skin lesions. current status and evolving trends. Dermatologic Clin. 1997;15:79-95.
26. Grin CM, Kopf AW, Welkovich B, et al. Accuracy in the clinical diagnosis of malignant melanoma. Arch Dermatol. 1990;126:763-766.
27. Rajadhyaksha M, Gonzalez S, Zavislan JM. Detectability of contrast agents for confocal reflectance imaging of skin and microcirculation. J Biomed Opt. 2004;9:323-331.
28. Langley RG, Burton E, Walsh N, et al. In vivo confocal scanning laser microscopy of benign lentigines: comparison to conventional histology and in vivo characteristics of lentigo maligna. J Am Acad Dermatol. 2006;55:88-97.
29. Scope A, Halpern AC. Diagnostic procedures and devices. In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2008:40-42.
30. Guitera P, Pellacani G, Longo C, et al. In vivo reflectance microscopy enhances secondary evaluation of melanocytic lesions. J Invest Dermatol. 2008;129:131-138.
31. Junqueira LC, Carneiro J. Histology and its methods of study. In: Junqueira LC, Carneiro J, eds. Basic Histology: Text and Atlas. 11 ed. New York, NY: McGraw-Hill; 2005:1-21.
32. González S, Gill M, Halpern A, eds. Reflectance Confocal Microscopy of Cutaneous Tumors: An Atlas With Clinical, Dermoscopic and Histological Correlations. London, UK: Informa Healthcare; 2008.
33. Scope A, Benvenuto-Andrade C, Agero AL, et al. In vivo reflectance confocal microscopy imaging of melanocytic lesions: consensus terminology glossary and illustrative images. J Am Acad Dermatol. 2007;57:644-658.
34. Langley RG, Walsh N, Sutherland AE, et al. The diagnostic accuracy of in vivo confocal scanning laser microscopy compared to dermoscopy of benign and malignant melanocytic lesions: a prospective study. Dermatology. 2007;215:365-372.
35. Guitera P, Menzies SW, Longo C, et al. In vivo confocal microscopy for diagnosis of melanoma and basal cell carcinoma using a two-step method: analysis of 710 consecutive clinically equivocal cases [published online ahead of print June 21, 2012]. J Invest Dermatol. 2012;132:2386-2394.
36. Guitera P, Pellacani G, Crotty KA, et al. The impact of in vivo reflectance confocal microscopy on the diagnostic accuracy of lentigo maligna and equivocal pigmented and nonpigmented macules of the face [published online ahead of print April 15, 2010]. J Invest Dermatol. 2010;130:2080-2091.
37. Pellacani G, Guitera P, Longo C, et al. The impact of in vivo reflectance confocal microscopy for the diagnostic accuracy of melanoma and equivocal melanocytic lesions [published online ahead of print July 26, 2007]. J Invest Dermatol. 2007;127:2759-2765.
38. Pellacani G, Pepe P, Casari A, et al. Reflectance confocal microscopy as a second-level examination in skin oncology improves diagnostic accuracy and saves unnecessary excisions: a longitudinal prospective study [published online ahead of print October 19, 2014]. Br J Dermatol. 2014;171:1044-1051.
39. Guitera P, Pellacani G, Longo C, et al. In vivo reflectance confocal microscopy enhances secondary evaluation of melanocytic lesions [published online ahead of print July 17, 2008]. J Invest Dermatol. 2009;129:131-138
40. Guitera P, Moloney FJ, Menzies SW, et al. Improving management and patient care in lentigo maligna by mapping with in vivo confocal microscopy. JAMA Dermatol. 2013;149:692-698.
41. Malvehy J, Roldán-Marín R, Iglesias-García P, et al. Monitoring treatment of field cancerisation with 3% diclofenac sodium 2.5% hyaluronic acid by reflectance confocal microscopy: a histologic correlation. Acta Derm Venereol. 2015;95:45-50.
42. Zalaudek I, Piana S, Moscarella E, et al. Morphologic grading and treatment of facial actinic keratosis. Clin Dermatol. 2014;32:80-87.
43. Longo C, Casari A, Pepe P, et al. Confocal microscopy insights into the treatment and cellular immune response of basal cell carcinoma tophotodynamic therapy [published online ahead of print December 13, 2012]. Dermatology. 2012;225:264-270.
1. Rajadhyaksha M, Grossman M, Esterowitz D, et al. In vivo confocal scanning laser microscopy of human skin: melanin provides strong contrast. J Invest Dermatol. 1995;104:946-952.
2. Rajadhyaksha M, González S, Zavislan JM, et al. In vivo confocal scanning laser microscopy of human skin II: advances in instrumentation and comparison to histology. J Invest Dermatol. 1999;113:293-303.
3. Huzaira M, Rius F, Rajadhyaksha M, et al. Topographic variations in normal skin, as viewed by in vivo reflectance confocal microscopy. J Invest Dermatol. 2001;116:846-852.
4. Swindells K, Burnett N, Rius-Diaz F, et al. Reflectance confocal microscopy may differentiate acute allergic and irritant contact dermatitis in vivo. J Am Acad Dermatol. 2004;50:220-228.
5. Astner S, González E, Cheung AC, et al. Non-invasive evaluation of the kinetics of allergic and irritant contact dermatitis. J Invest Dermatol. 2005;124:351-359.
6. Pellacani G, Cesinaro AM, Seidenari S. Reflectance-mode confocal microscopy of pigmented skin lesions—improvement in melanoma diagnostic specificity. J Am Acad Dermatol. 2005;53:979-985.
7. González S, Tannous Z. Real-time, in vivo confocal reflectance microscopy of basal cell carcinoma. J Am Acad Dermatol. 2002;47:869-874.
8. Rishpon A, Kim N, Scope A, et al. Reflectance confocal microscopy criteria for squamous cell carcinomas and actinic keratoses. Arch Dermatol. 2009;145:766-772.
9. Busam KJ, Charles C, Lee G, et al. Morphologic features of melanocytes, pigmented keratinocytes, and melanophages by in vivo confocal scanning laser microscopy. Mod Pathol. 2001;14:862-868.
10. Langley RG, Rajadhyaksha M, Dwyer PJ, et al. Confocal scanning laser microscopy of benign and malignant melanocytic skin lesions in vivo. J Am Acad Dermatol. 2001;45:365-376.
11. Pellacani G, Cesinaro AM, Seidenari S. In vivo assessment of melanocytic nests in nevi and melanomas by reflectance confocal microscopy. Mod Pathol. 2005;18:469-474.
12. Pellacani G, Cesinaro AM, Seidenari S. Reflectance-mode confocal microscopy for the in vivo characterization of pagetoid melanocytosis in melanomas and nevi. J Invest Dermatol. 2005;125:532-537.
13. Scope A, Benvenuto-Andrade C, Agero AL, et al. Correlation of dermoscopic structures of melanocytic lesions to reflectance confocal microscopy. Arch Dermatol. 2007;143:176-185.
14. Pellacani G, Longo C, Malvehy J, et al. In vivo confocal microscopic and histopathologic correlations of dermoscopic features in 202 melanocytic lesions. Arch Dermatol. 2008:144:1597-1608.
15. Scope A, Gill M, Benveuto-Andrade C, et al. Correlation of dermoscopy with in vivo reflectance confocal microscopy of streaks in melanocytic lesions. Arch Dermatol. 2007;143:727-734.
16. Curiel-Lewandrowski C, Williams CM, Swindells KJ, et al. Use of in vivo confocal microscopy in malignant melanoma: an aid in diagnosis and assessment of surgical and nonsurgical therapeutic approaches. Arch Dermatol. 2004;140:1127-1132.
17. Busam KJ, Hester K, Charles C, et al. Detection of clinically amelanotic malignant melanoma and assessment of its margins by in vivo confocal scanning laser microscopy. Arch Dermatol. 2001;137:923-929.
18. Goldgeier M, Fox CA, Zavislan JM, et al. Noninvasive imaging, treatment, and microscopic confirmation of clearance of basal cell carcinoma. Dermatol Surg. 2003;29:205-210.
19. Torres A, Niemeyer A, Berkes B, et al. 5% Imiquimod cream and reflectance-mode confocal microscopy as adjunct modalities to Mohs micrographic surgery for treatment of basal cell carcinoma. Dermatol Surg. 2004;30(12, pt 1):1462-1469.
20. Lassau N, Mercier S, Koscielny S, et al. Prognostic value of high-frequency sonography and color Doppler sonography for the preoperative assessment of melanomas. AJR Am J Roentgenol. 1999;172:457-461.
21. Ruocco E, Argenziano G, Pellacani G, et al. Noninvasive imaging of skin tumors. Dermatol Surg. 2004;30(2, pt 2):301-310.
22. Welzel J. Optical coherence tomography in dermatology: a review. Skin Res Technol. 2001;7:1-9.
23. Gambichler T, Plura I, Schmid-Wendtner M, et al. High-definition optical coherence tomography of melanocytic skin lesions [published online ahead of print September 18, 2014]. J Biophotonics. doi:10.1002/jbio.201400085.
24. Braga JC, Scope A, Klaz I, et al. The significance of reflectance confocal microscopy in the assessment of solitary pink skin lesions. J Am Acad Dermatol. 2009;61:230-241.
25. Argenyl ZB. Dermoscopy (epiluminescence microscopy) of pigmented skin lesions. current status and evolving trends. Dermatologic Clin. 1997;15:79-95.
26. Grin CM, Kopf AW, Welkovich B, et al. Accuracy in the clinical diagnosis of malignant melanoma. Arch Dermatol. 1990;126:763-766.
27. Rajadhyaksha M, Gonzalez S, Zavislan JM. Detectability of contrast agents for confocal reflectance imaging of skin and microcirculation. J Biomed Opt. 2004;9:323-331.
28. Langley RG, Burton E, Walsh N, et al. In vivo confocal scanning laser microscopy of benign lentigines: comparison to conventional histology and in vivo characteristics of lentigo maligna. J Am Acad Dermatol. 2006;55:88-97.
29. Scope A, Halpern AC. Diagnostic procedures and devices. In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2008:40-42.
30. Guitera P, Pellacani G, Longo C, et al. In vivo reflectance microscopy enhances secondary evaluation of melanocytic lesions. J Invest Dermatol. 2008;129:131-138.
31. Junqueira LC, Carneiro J. Histology and its methods of study. In: Junqueira LC, Carneiro J, eds. Basic Histology: Text and Atlas. 11 ed. New York, NY: McGraw-Hill; 2005:1-21.
32. González S, Gill M, Halpern A, eds. Reflectance Confocal Microscopy of Cutaneous Tumors: An Atlas With Clinical, Dermoscopic and Histological Correlations. London, UK: Informa Healthcare; 2008.
33. Scope A, Benvenuto-Andrade C, Agero AL, et al. In vivo reflectance confocal microscopy imaging of melanocytic lesions: consensus terminology glossary and illustrative images. J Am Acad Dermatol. 2007;57:644-658.
34. Langley RG, Walsh N, Sutherland AE, et al. The diagnostic accuracy of in vivo confocal scanning laser microscopy compared to dermoscopy of benign and malignant melanocytic lesions: a prospective study. Dermatology. 2007;215:365-372.
35. Guitera P, Menzies SW, Longo C, et al. In vivo confocal microscopy for diagnosis of melanoma and basal cell carcinoma using a two-step method: analysis of 710 consecutive clinically equivocal cases [published online ahead of print June 21, 2012]. J Invest Dermatol. 2012;132:2386-2394.
36. Guitera P, Pellacani G, Crotty KA, et al. The impact of in vivo reflectance confocal microscopy on the diagnostic accuracy of lentigo maligna and equivocal pigmented and nonpigmented macules of the face [published online ahead of print April 15, 2010]. J Invest Dermatol. 2010;130:2080-2091.
37. Pellacani G, Guitera P, Longo C, et al. The impact of in vivo reflectance confocal microscopy for the diagnostic accuracy of melanoma and equivocal melanocytic lesions [published online ahead of print July 26, 2007]. J Invest Dermatol. 2007;127:2759-2765.
38. Pellacani G, Pepe P, Casari A, et al. Reflectance confocal microscopy as a second-level examination in skin oncology improves diagnostic accuracy and saves unnecessary excisions: a longitudinal prospective study [published online ahead of print October 19, 2014]. Br J Dermatol. 2014;171:1044-1051.
39. Guitera P, Pellacani G, Longo C, et al. In vivo reflectance confocal microscopy enhances secondary evaluation of melanocytic lesions [published online ahead of print July 17, 2008]. J Invest Dermatol. 2009;129:131-138
40. Guitera P, Moloney FJ, Menzies SW, et al. Improving management and patient care in lentigo maligna by mapping with in vivo confocal microscopy. JAMA Dermatol. 2013;149:692-698.
41. Malvehy J, Roldán-Marín R, Iglesias-García P, et al. Monitoring treatment of field cancerisation with 3% diclofenac sodium 2.5% hyaluronic acid by reflectance confocal microscopy: a histologic correlation. Acta Derm Venereol. 2015;95:45-50.
42. Zalaudek I, Piana S, Moscarella E, et al. Morphologic grading and treatment of facial actinic keratosis. Clin Dermatol. 2014;32:80-87.
43. Longo C, Casari A, Pepe P, et al. Confocal microscopy insights into the treatment and cellular immune response of basal cell carcinoma tophotodynamic therapy [published online ahead of print December 13, 2012]. Dermatology. 2012;225:264-270.
Practice Points
- In vivo reflectance confocal microscopy (RCM) is a noninvasive modality for the assessment of physiologic and pathologic conditions of the skin.
- Similar to dermoscopy, RCM allows lesions to be analyzed noninvasively, and similar to histology, RCM images provide high resolution in both vertical and horizontal planes.
- Utilizing RCM as an adjunctive tool can help improve clinical diagnostic accuracy, reducing the number of biopsies of benign lesions.
- Incorporating RCM and a telepathology network into the workflow of a private practice may be valuable for dermatologists and primary care physicians.