User login
Fine-tune staging for better SCC risk stratification
ORLANDO – When caring for individuals with sun-damaged skin, dermatologists need comfort with the full spectrum of photo-related skin disease. From assessment and treatment of actinic keratoses (AKs) and field cancerization, to long-term follow-up of cutaneous squamous cell carcinomas (SCCs), appropriate treatment and staging can improve patient quality of life and reduce health care costs, Vishal Patel, MD, said at the Orlando Dermatology Aesthetic and Clinical Conference.

said Dr. Patel, director of cutaneous oncology at George Washington University Cancer Center, Washington. On the other hand, he added, “field disease can be a marker for invasive squamous cell carcinoma risk, and it requires field treatment.” Treatment that reduces field disease is primary prevention because it decreases the formation of invasive SCC, he noted.
“But this level of disease – AKs and SCC in situ – doesn’t kill people,” he emphasized. “I want to leave you with an ability to stage this disease,” said Dr. Patel, noting that SCC mortality may eventually surpass melanoma mortality as deaths from the latter decline and numbers of older Americans with high ultraviolet light exposure and other risk factors climb.
While the majority of AKs regress within 5 years, he looks at the total burden of AKs as a marker for field cancerization “because having less than five in situ or actinic lesions puts you at less than a 1% risk of squamous cell carcinoma formation. Having more than 20 increases that risk 20-fold to 20%,” he said. “That’s the way we need to start thinking about this: Is this a disease – or a symptom?”
Rather than thinking of each AK or SCC in situ as a separate disease event, “the disease we need to be focusing on and treating is field cancerization,” he continued. Within this context, “we should not be thinking that … we need to be aggressive in our management,” which is what results in high costs.
“The reality is that this is a big quality of life issue for our patients. So what do we do?” Field treatment is appropriate for field disease, he said. Dr. Patel said that at GW only field treatment is used; destructive treatment for AKs and SCC in situ is not used. In the absence of patient and lesion characteristics that elevate risk,“surgery is really not the standard of care for in situ lesions for us,” he commented.
“We start by discerning the field disease from the invasive disease” with an initial round of field treatment and, if needed, adjunctive oral chemoprophylaxis. “We lather, rinse, and repeat” the field therapy, continuously if needed, Dr. Patel said.
“We like to do that because we can then identify those specific lesions we want to go after. No cryosurgery, no destructive therapy, because we run the risk of burying those tumors under the scar. They may recur and make it more difficult to accurately stage them in the future,” he noted.
“I like to be more sophisticated in thinking about our approach to the outcomes of these individual lesions,” he said. When it comes to excising lesions that have been biopsied and show invasive SCC, “disc excision may be a more cost-effective way to treat many low-risk SCCs,” he noted. In any case, “removal with clear surgical margins is key.”
Primary tumors with such low-risk attributes as diameter under a centimeter and thickness under 2 mm; well-defined borders; location on the trunk, neck, or extremities; well-differentiated histology; and lack of perineural invasion can all be considered for a disc technique, especially if the patient is immunocompetent without background chronic inflammation or a history of prior radiation therapy.
Staging SCCs, said Dr. Patel, is where things really get tricky. Older staging systems for SCC “led us to overtreat nonaggressive disease and undertreat aggressive disease. I think we have the responsibility to lead the charge to having a more sophisticated approach.” For example, patients whose tumors were staged T2 in the American Joint Commission on Cancer (AJCC) 7 classification system were most likely to have poor outcomes – in part because so few tumors were staged higher – which meant AJCC 7 didn’t provide adequate differentiation for useful risk prognostication.
A group of researchers at the Brigham and Women’s Hospital (BWH), Boston, “came up with a better system to better differentiate those T2 tumors into a high-risk and a low-risk subtype,” according to Dr. Patel.
With use of validated risk factors, the investigators applied a long list of risk factors to 2,000 tumors to see which risk factors, taken individually, were really contributing to poor outcomes. Eventually, four risk factors that made the most difference were identified: size greater than 2 cm, poor tumor differentiation, perineural invasion greater than 0.1 mm in diameter, and tumor invasion beyond subcutaneous fat. “I really want to highlight the size portion of those risk factors,” said Dr. Patel. “Something I’d like you to do in your clinical practice is to measure and document the size of the lesion. … That really, clearly helps” with risk prognostication.
These four factors were then used to break out a T2a stage for tumors with one risk factor and a T2b stage for tumors with two or three risk factors. Tumors with no risk factors are stage T1, and those with all four risk factors are stage T3. In situ SCC is T0.
Applying this new staging system to a 2,000-patient cohort with SCC yielded clear separation in outcomes including recurrence, nodal metastasis, disease-specific death, and overall survival between patients with the T2a and T2b tumors (P less than .001 for all; J Clin Oncol. 2014 Feb 1;32[4]:327-34).
While AJCC 8 is “significantly better” than AJCC 7 in its incorporation of meaningful risk factors into the SCC staging system, “it still underperforms in comparison” with the BWH staging system using the 2000 patient cohort, he said. Recent work has shown the BWH classification system to have superior specificity and positive predictive value in detecting nodal metastasis and disease-specific death in higher-grade tumors. But both BWH and AJCC 8 need further refinement.
“So what are the staging pearls to take home?” Dr. Patel asked. “First, utilize a staging system.” “Staging of SCC utilizing should be done routinely. Most data seems to suggest that the BWH system appears to outperform AJCC 8, and it is what we currently use routinely at GW,” he said.
Patients who are T1 by BWH criteria, with no risk factors, are at low or even no risk, he noted. He pointed out that of the nearly 1,400 patients who met T1 criteria, there were just eight local recurrences, one nodal metastasis, and no distant metastases or deaths. Knowing this should guide physicians on a treatment path that will reduce costs and provide patients with peace of mind, he said.
In the BWH schema, T2a patients fared almost as well, with a 2% risk of nodal metastasis and an overall 1% risk of disease-specific death. “T2a disease is low risk, in my mind. Most of these patients will go on to do well,” he said.
By contrast, “there may be a number of tumors that you are missing” that are candidates for close follow-up if the BWH criteria are not being used, said Dr. Patel. These are the T2b tumors. “For those patients, we want to aggressively follow them and think about a more aggressive management plan.”
The bottom line is that BWH T2b and T3 tumors are both high risk, and management needs to acknowledge this, he said. The current protocol in our cutaneous oncology program includes using routine radiologic nodal staging in patients with BWH stage 2b and above SCCs and considering sentinel lymph node biopsy for certain individuals.
For patients with BWH T2b and T3 tumors, dermatologists should give consideration to tertiary care or cancer center referrals so they have access to the full spectrum of diagnostic and therapeutic modalities and the opportunity to participate in clinical trials, Dr. Patel said.
Dr. Patel reported that he is a speaker for Regeneron/Sanofi and a cofounder of the Skin Cancer Outcomes (SCOUT) consortium.
This article was updated 2/9/2019
ORLANDO – When caring for individuals with sun-damaged skin, dermatologists need comfort with the full spectrum of photo-related skin disease. From assessment and treatment of actinic keratoses (AKs) and field cancerization, to long-term follow-up of cutaneous squamous cell carcinomas (SCCs), appropriate treatment and staging can improve patient quality of life and reduce health care costs, Vishal Patel, MD, said at the Orlando Dermatology Aesthetic and Clinical Conference.
said Dr. Patel, director of cutaneous oncology at George Washington University Cancer Center, Washington. On the other hand, he added, “field disease can be a marker for invasive squamous cell carcinoma risk, and it requires field treatment.” Treatment that reduces field disease is primary prevention because it decreases the formation of invasive SCC, he noted.
“But this level of disease – AKs and SCC in situ – doesn’t kill people,” he emphasized. “I want to leave you with an ability to stage this disease,” said Dr. Patel, noting that SCC mortality may eventually surpass melanoma mortality as deaths from the latter decline and numbers of older Americans with high ultraviolet light exposure and other risk factors climb.
While the majority of AKs regress within 5 years, he looks at the total burden of AKs as a marker for field cancerization “because having less than five in situ or actinic lesions puts you at less than a 1% risk of squamous cell carcinoma formation. Having more than 20 increases that risk 20-fold to 20%,” he said. “That’s the way we need to start thinking about this: Is this a disease – or a symptom?”
Rather than thinking of each AK or SCC in situ as a separate disease event, “the disease we need to be focusing on and treating is field cancerization,” he continued. Within this context, “we should not be thinking that … we need to be aggressive in our management,” which is what results in high costs.
“The reality is that this is a big quality of life issue for our patients. So what do we do?” Field treatment is appropriate for field disease, he said. Dr. Patel said that at GW only field treatment is used; destructive treatment for AKs and SCC in situ is not used. In the absence of patient and lesion characteristics that elevate risk,“surgery is really not the standard of care for in situ lesions for us,” he commented.
“We start by discerning the field disease from the invasive disease” with an initial round of field treatment and, if needed, adjunctive oral chemoprophylaxis. “We lather, rinse, and repeat” the field therapy, continuously if needed, Dr. Patel said.
“We like to do that because we can then identify those specific lesions we want to go after. No cryosurgery, no destructive therapy, because we run the risk of burying those tumors under the scar. They may recur and make it more difficult to accurately stage them in the future,” he noted.
“I like to be more sophisticated in thinking about our approach to the outcomes of these individual lesions,” he said. When it comes to excising lesions that have been biopsied and show invasive SCC, “disc excision may be a more cost-effective way to treat many low-risk SCCs,” he noted. In any case, “removal with clear surgical margins is key.”
Primary tumors with such low-risk attributes as diameter under a centimeter and thickness under 2 mm; well-defined borders; location on the trunk, neck, or extremities; well-differentiated histology; and lack of perineural invasion can all be considered for a disc technique, especially if the patient is immunocompetent without background chronic inflammation or a history of prior radiation therapy.
Staging SCCs, said Dr. Patel, is where things really get tricky. Older staging systems for SCC “led us to overtreat nonaggressive disease and undertreat aggressive disease. I think we have the responsibility to lead the charge to having a more sophisticated approach.” For example, patients whose tumors were staged T2 in the American Joint Commission on Cancer (AJCC) 7 classification system were most likely to have poor outcomes – in part because so few tumors were staged higher – which meant AJCC 7 didn’t provide adequate differentiation for useful risk prognostication.
A group of researchers at the Brigham and Women’s Hospital (BWH), Boston, “came up with a better system to better differentiate those T2 tumors into a high-risk and a low-risk subtype,” according to Dr. Patel.
With use of validated risk factors, the investigators applied a long list of risk factors to 2,000 tumors to see which risk factors, taken individually, were really contributing to poor outcomes. Eventually, four risk factors that made the most difference were identified: size greater than 2 cm, poor tumor differentiation, perineural invasion greater than 0.1 mm in diameter, and tumor invasion beyond subcutaneous fat. “I really want to highlight the size portion of those risk factors,” said Dr. Patel. “Something I’d like you to do in your clinical practice is to measure and document the size of the lesion. … That really, clearly helps” with risk prognostication.
These four factors were then used to break out a T2a stage for tumors with one risk factor and a T2b stage for tumors with two or three risk factors. Tumors with no risk factors are stage T1, and those with all four risk factors are stage T3. In situ SCC is T0.
Applying this new staging system to a 2,000-patient cohort with SCC yielded clear separation in outcomes including recurrence, nodal metastasis, disease-specific death, and overall survival between patients with the T2a and T2b tumors (P less than .001 for all; J Clin Oncol. 2014 Feb 1;32[4]:327-34).
While AJCC 8 is “significantly better” than AJCC 7 in its incorporation of meaningful risk factors into the SCC staging system, “it still underperforms in comparison” with the BWH staging system using the 2000 patient cohort, he said. Recent work has shown the BWH classification system to have superior specificity and positive predictive value in detecting nodal metastasis and disease-specific death in higher-grade tumors. But both BWH and AJCC 8 need further refinement.
“So what are the staging pearls to take home?” Dr. Patel asked. “First, utilize a staging system.” “Staging of SCC utilizing should be done routinely. Most data seems to suggest that the BWH system appears to outperform AJCC 8, and it is what we currently use routinely at GW,” he said.
Patients who are T1 by BWH criteria, with no risk factors, are at low or even no risk, he noted. He pointed out that of the nearly 1,400 patients who met T1 criteria, there were just eight local recurrences, one nodal metastasis, and no distant metastases or deaths. Knowing this should guide physicians on a treatment path that will reduce costs and provide patients with peace of mind, he said.
In the BWH schema, T2a patients fared almost as well, with a 2% risk of nodal metastasis and an overall 1% risk of disease-specific death. “T2a disease is low risk, in my mind. Most of these patients will go on to do well,” he said.
By contrast, “there may be a number of tumors that you are missing” that are candidates for close follow-up if the BWH criteria are not being used, said Dr. Patel. These are the T2b tumors. “For those patients, we want to aggressively follow them and think about a more aggressive management plan.”
The bottom line is that BWH T2b and T3 tumors are both high risk, and management needs to acknowledge this, he said. The current protocol in our cutaneous oncology program includes using routine radiologic nodal staging in patients with BWH stage 2b and above SCCs and considering sentinel lymph node biopsy for certain individuals.
For patients with BWH T2b and T3 tumors, dermatologists should give consideration to tertiary care or cancer center referrals so they have access to the full spectrum of diagnostic and therapeutic modalities and the opportunity to participate in clinical trials, Dr. Patel said.
Dr. Patel reported that he is a speaker for Regeneron/Sanofi and a cofounder of the Skin Cancer Outcomes (SCOUT) consortium.
This article was updated 2/9/2019
ORLANDO – When caring for individuals with sun-damaged skin, dermatologists need comfort with the full spectrum of photo-related skin disease. From assessment and treatment of actinic keratoses (AKs) and field cancerization, to long-term follow-up of cutaneous squamous cell carcinomas (SCCs), appropriate treatment and staging can improve patient quality of life and reduce health care costs, Vishal Patel, MD, said at the Orlando Dermatology Aesthetic and Clinical Conference.
said Dr. Patel, director of cutaneous oncology at George Washington University Cancer Center, Washington. On the other hand, he added, “field disease can be a marker for invasive squamous cell carcinoma risk, and it requires field treatment.” Treatment that reduces field disease is primary prevention because it decreases the formation of invasive SCC, he noted.
“But this level of disease – AKs and SCC in situ – doesn’t kill people,” he emphasized. “I want to leave you with an ability to stage this disease,” said Dr. Patel, noting that SCC mortality may eventually surpass melanoma mortality as deaths from the latter decline and numbers of older Americans with high ultraviolet light exposure and other risk factors climb.
While the majority of AKs regress within 5 years, he looks at the total burden of AKs as a marker for field cancerization “because having less than five in situ or actinic lesions puts you at less than a 1% risk of squamous cell carcinoma formation. Having more than 20 increases that risk 20-fold to 20%,” he said. “That’s the way we need to start thinking about this: Is this a disease – or a symptom?”
Rather than thinking of each AK or SCC in situ as a separate disease event, “the disease we need to be focusing on and treating is field cancerization,” he continued. Within this context, “we should not be thinking that … we need to be aggressive in our management,” which is what results in high costs.
“The reality is that this is a big quality of life issue for our patients. So what do we do?” Field treatment is appropriate for field disease, he said. Dr. Patel said that at GW only field treatment is used; destructive treatment for AKs and SCC in situ is not used. In the absence of patient and lesion characteristics that elevate risk,“surgery is really not the standard of care for in situ lesions for us,” he commented.
“We start by discerning the field disease from the invasive disease” with an initial round of field treatment and, if needed, adjunctive oral chemoprophylaxis. “We lather, rinse, and repeat” the field therapy, continuously if needed, Dr. Patel said.
“We like to do that because we can then identify those specific lesions we want to go after. No cryosurgery, no destructive therapy, because we run the risk of burying those tumors under the scar. They may recur and make it more difficult to accurately stage them in the future,” he noted.
“I like to be more sophisticated in thinking about our approach to the outcomes of these individual lesions,” he said. When it comes to excising lesions that have been biopsied and show invasive SCC, “disc excision may be a more cost-effective way to treat many low-risk SCCs,” he noted. In any case, “removal with clear surgical margins is key.”
Primary tumors with such low-risk attributes as diameter under a centimeter and thickness under 2 mm; well-defined borders; location on the trunk, neck, or extremities; well-differentiated histology; and lack of perineural invasion can all be considered for a disc technique, especially if the patient is immunocompetent without background chronic inflammation or a history of prior radiation therapy.
Staging SCCs, said Dr. Patel, is where things really get tricky. Older staging systems for SCC “led us to overtreat nonaggressive disease and undertreat aggressive disease. I think we have the responsibility to lead the charge to having a more sophisticated approach.” For example, patients whose tumors were staged T2 in the American Joint Commission on Cancer (AJCC) 7 classification system were most likely to have poor outcomes – in part because so few tumors were staged higher – which meant AJCC 7 didn’t provide adequate differentiation for useful risk prognostication.
A group of researchers at the Brigham and Women’s Hospital (BWH), Boston, “came up with a better system to better differentiate those T2 tumors into a high-risk and a low-risk subtype,” according to Dr. Patel.
With use of validated risk factors, the investigators applied a long list of risk factors to 2,000 tumors to see which risk factors, taken individually, were really contributing to poor outcomes. Eventually, four risk factors that made the most difference were identified: size greater than 2 cm, poor tumor differentiation, perineural invasion greater than 0.1 mm in diameter, and tumor invasion beyond subcutaneous fat. “I really want to highlight the size portion of those risk factors,” said Dr. Patel. “Something I’d like you to do in your clinical practice is to measure and document the size of the lesion. … That really, clearly helps” with risk prognostication.
These four factors were then used to break out a T2a stage for tumors with one risk factor and a T2b stage for tumors with two or three risk factors. Tumors with no risk factors are stage T1, and those with all four risk factors are stage T3. In situ SCC is T0.
Applying this new staging system to a 2,000-patient cohort with SCC yielded clear separation in outcomes including recurrence, nodal metastasis, disease-specific death, and overall survival between patients with the T2a and T2b tumors (P less than .001 for all; J Clin Oncol. 2014 Feb 1;32[4]:327-34).
While AJCC 8 is “significantly better” than AJCC 7 in its incorporation of meaningful risk factors into the SCC staging system, “it still underperforms in comparison” with the BWH staging system using the 2000 patient cohort, he said. Recent work has shown the BWH classification system to have superior specificity and positive predictive value in detecting nodal metastasis and disease-specific death in higher-grade tumors. But both BWH and AJCC 8 need further refinement.
“So what are the staging pearls to take home?” Dr. Patel asked. “First, utilize a staging system.” “Staging of SCC utilizing should be done routinely. Most data seems to suggest that the BWH system appears to outperform AJCC 8, and it is what we currently use routinely at GW,” he said.
Patients who are T1 by BWH criteria, with no risk factors, are at low or even no risk, he noted. He pointed out that of the nearly 1,400 patients who met T1 criteria, there were just eight local recurrences, one nodal metastasis, and no distant metastases or deaths. Knowing this should guide physicians on a treatment path that will reduce costs and provide patients with peace of mind, he said.
In the BWH schema, T2a patients fared almost as well, with a 2% risk of nodal metastasis and an overall 1% risk of disease-specific death. “T2a disease is low risk, in my mind. Most of these patients will go on to do well,” he said.
By contrast, “there may be a number of tumors that you are missing” that are candidates for close follow-up if the BWH criteria are not being used, said Dr. Patel. These are the T2b tumors. “For those patients, we want to aggressively follow them and think about a more aggressive management plan.”
The bottom line is that BWH T2b and T3 tumors are both high risk, and management needs to acknowledge this, he said. The current protocol in our cutaneous oncology program includes using routine radiologic nodal staging in patients with BWH stage 2b and above SCCs and considering sentinel lymph node biopsy for certain individuals.
For patients with BWH T2b and T3 tumors, dermatologists should give consideration to tertiary care or cancer center referrals so they have access to the full spectrum of diagnostic and therapeutic modalities and the opportunity to participate in clinical trials, Dr. Patel said.
Dr. Patel reported that he is a speaker for Regeneron/Sanofi and a cofounder of the Skin Cancer Outcomes (SCOUT) consortium.
This article was updated 2/9/2019
EXPERT ANALYSIS FROM ODAC 2019
Paraneoplastic Dermatomyositis Presenting With Interesting Cutaneous Findings
To the Editor:
We report an interesting clinical case of dermatomyositis (DM) that presented with an associated malignancy (small cell lung cancer). This patient also had an unusual clinical finding of predominantly unilateral, confluent, erythematous papules on the knee, a cutaneous sign that is seldom described in the DM literature. This case serves to reinforce the classic findings and associations of DM, in addition to the uncommon manifestation of predominantly unilateral papules on the knee.
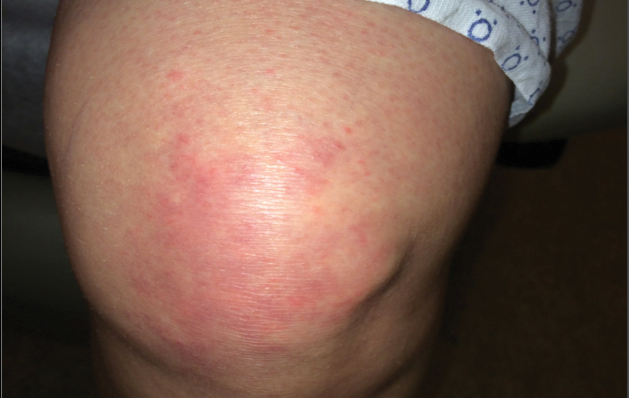
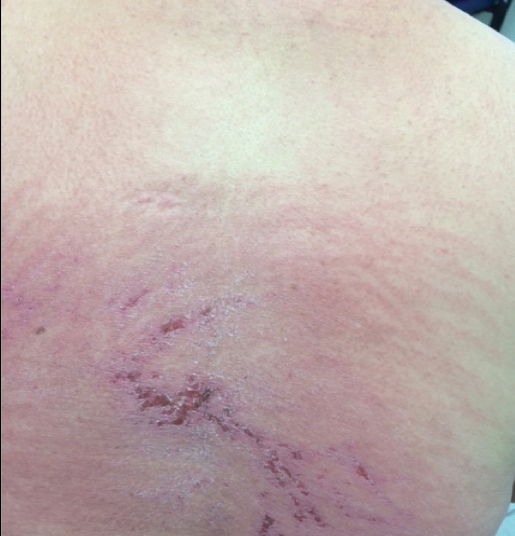
A 68-year-old woman presented with several cutaneous manifestations including the classic findings of photo distributed erythema on the arms and face, a heliotrope rash, Gottron papules, and confluent pink papules on the left knee (Figure 1). The patient also had one of the more rare manifestations of DM, flagellate erythema on the back (Figure 2). She had a history of breast cancer and was found to have metastatic small cell lung cancer at the time of the DM diagnosis.
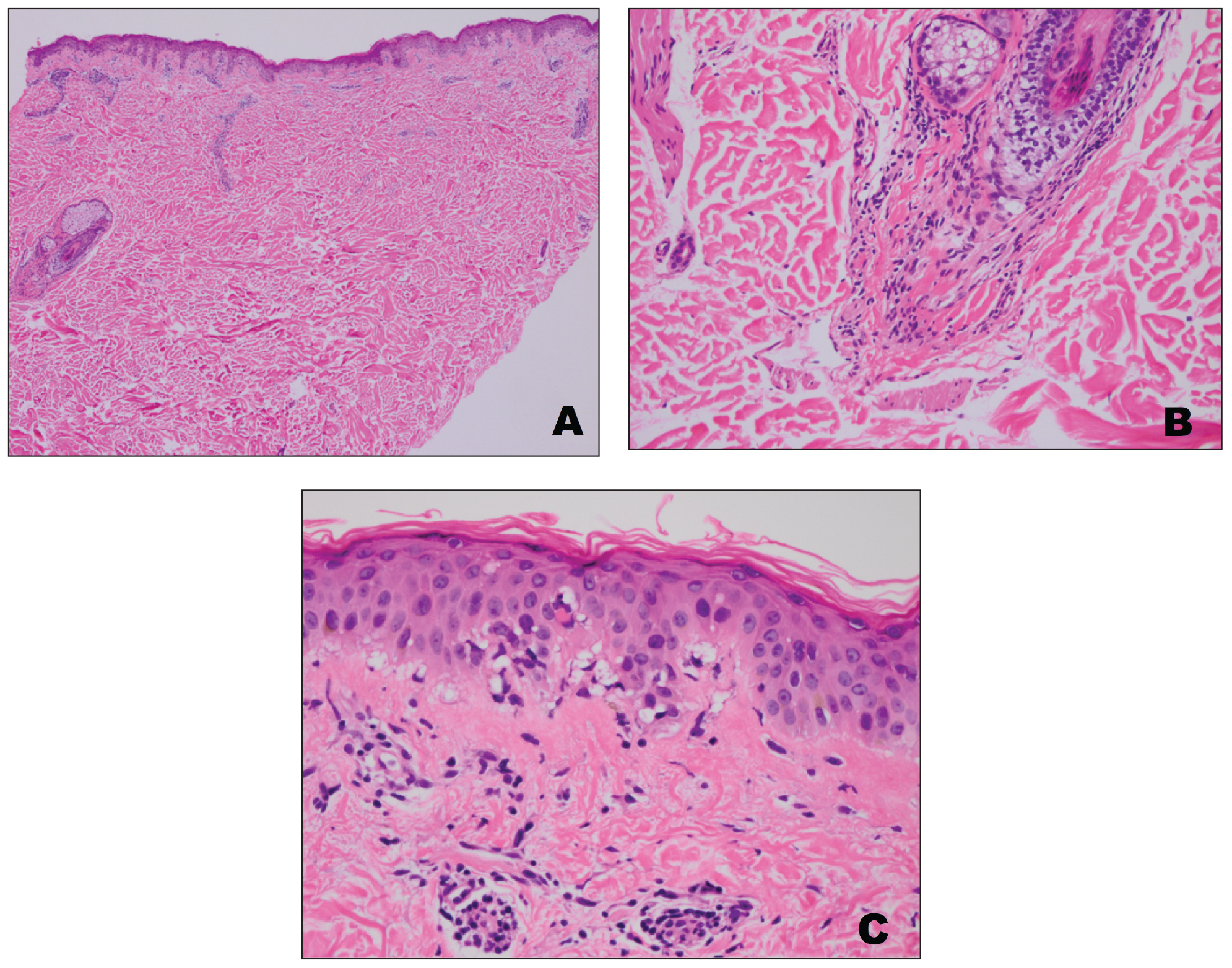
A punch biopsy from an area of flagellate erythema on the back revealed an interface dermatitis with a superficial, perivascular, lymphocyte-predominant inflammatory infiltrate (Figure 3). Alcian blue and colloidal iron stains revealed a marked increase in papillary dermal mucin. With the characteristic changes on skin biopsy and the classic skin findings present in our patient, we felt confident diagnosing her with DM. At the time of diagnosis, the patient also was found to have metastatic small cell lung cancer, suggesting a true paraneoplastic relationship.

The association of DM and amyopathic DM with internal malignancy is well known. Bohan and Peter1 noted an overall figure ranging from 15% to 34% with an increased frequency in patients with skin and muscle involvement.1 Hill et al5 examined this link in a population-based study that identified corresponding malignancies. Specifically, they noted cancers to arise most frequently in the airway (eg, lung, trachea, bronchus), ovaries, breasts, colorectal region, and stomach.5 There also has been work performed to identify if certain dermatologic findings may be associated with a higher risk of malignancy.6,7 A meta-analysis by Wang et al6 showed that Gottron sign did not have an association with cancer, but findings of cutaneous necrosis did have an association. It is unknown if the specific cutaneous findings in our patient, including the predominantly unilateral papules on the knee, may have been a clue to the underlying malignancy.
In summary, we believe that our patient presented with the classic manifestations of DM in addition to the curious cutaneous sign of predominantly unilateral, confluent, erythematous papules on the knee, a clinical finding that may aid in the diagnosis of DM and also may alert the clinician to a possible underlying malignancy.
- Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344-347.
- Santmyire-Rosenberger B, Dugan EM. Skin involvement in dermatomyositis. Curr Opin Rheumatol. 2003;15:714-722.
- Callen JP. Dermatomyositis. Lancet. 2000;355:53-57.
- Lister RK, Cooper ES, Paige DG. Papules and pustules of the elbows and knees: an uncommon clinical sign of dermatomyositis in oriental children. Pediatr Dermatol. 2000;17:37-40.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Wang J, Guo G, Chen G, et al. Meta‐analysis of the association of dermatomyositis and polymyositis with cancer. Br J Dermatol. 2013;169:838-847.
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case–control study. Br J Dermatol. 2001;144:825-831.
To the Editor:
We report an interesting clinical case of dermatomyositis (DM) that presented with an associated malignancy (small cell lung cancer). This patient also had an unusual clinical finding of predominantly unilateral, confluent, erythematous papules on the knee, a cutaneous sign that is seldom described in the DM literature. This case serves to reinforce the classic findings and associations of DM, in addition to the uncommon manifestation of predominantly unilateral papules on the knee.
A 68-year-old woman presented with several cutaneous manifestations including the classic findings of photo distributed erythema on the arms and face, a heliotrope rash, Gottron papules, and confluent pink papules on the left knee (Figure 1). The patient also had one of the more rare manifestations of DM, flagellate erythema on the back (Figure 2). She had a history of breast cancer and was found to have metastatic small cell lung cancer at the time of the DM diagnosis.
A punch biopsy from an area of flagellate erythema on the back revealed an interface dermatitis with a superficial, perivascular, lymphocyte-predominant inflammatory infiltrate (Figure 3). Alcian blue and colloidal iron stains revealed a marked increase in papillary dermal mucin. With the characteristic changes on skin biopsy and the classic skin findings present in our patient, we felt confident diagnosing her with DM. At the time of diagnosis, the patient also was found to have metastatic small cell lung cancer, suggesting a true paraneoplastic relationship.
The association of DM and amyopathic DM with internal malignancy is well known. Bohan and Peter1 noted an overall figure ranging from 15% to 34% with an increased frequency in patients with skin and muscle involvement.1 Hill et al5 examined this link in a population-based study that identified corresponding malignancies. Specifically, they noted cancers to arise most frequently in the airway (eg, lung, trachea, bronchus), ovaries, breasts, colorectal region, and stomach.5 There also has been work performed to identify if certain dermatologic findings may be associated with a higher risk of malignancy.6,7 A meta-analysis by Wang et al6 showed that Gottron sign did not have an association with cancer, but findings of cutaneous necrosis did have an association. It is unknown if the specific cutaneous findings in our patient, including the predominantly unilateral papules on the knee, may have been a clue to the underlying malignancy.
In summary, we believe that our patient presented with the classic manifestations of DM in addition to the curious cutaneous sign of predominantly unilateral, confluent, erythematous papules on the knee, a clinical finding that may aid in the diagnosis of DM and also may alert the clinician to a possible underlying malignancy.
To the Editor:
We report an interesting clinical case of dermatomyositis (DM) that presented with an associated malignancy (small cell lung cancer). This patient also had an unusual clinical finding of predominantly unilateral, confluent, erythematous papules on the knee, a cutaneous sign that is seldom described in the DM literature. This case serves to reinforce the classic findings and associations of DM, in addition to the uncommon manifestation of predominantly unilateral papules on the knee.
A 68-year-old woman presented with several cutaneous manifestations including the classic findings of photo distributed erythema on the arms and face, a heliotrope rash, Gottron papules, and confluent pink papules on the left knee (Figure 1). The patient also had one of the more rare manifestations of DM, flagellate erythema on the back (Figure 2). She had a history of breast cancer and was found to have metastatic small cell lung cancer at the time of the DM diagnosis.
A punch biopsy from an area of flagellate erythema on the back revealed an interface dermatitis with a superficial, perivascular, lymphocyte-predominant inflammatory infiltrate (Figure 3). Alcian blue and colloidal iron stains revealed a marked increase in papillary dermal mucin. With the characteristic changes on skin biopsy and the classic skin findings present in our patient, we felt confident diagnosing her with DM. At the time of diagnosis, the patient also was found to have metastatic small cell lung cancer, suggesting a true paraneoplastic relationship.
The association of DM and amyopathic DM with internal malignancy is well known. Bohan and Peter1 noted an overall figure ranging from 15% to 34% with an increased frequency in patients with skin and muscle involvement.1 Hill et al5 examined this link in a population-based study that identified corresponding malignancies. Specifically, they noted cancers to arise most frequently in the airway (eg, lung, trachea, bronchus), ovaries, breasts, colorectal region, and stomach.5 There also has been work performed to identify if certain dermatologic findings may be associated with a higher risk of malignancy.6,7 A meta-analysis by Wang et al6 showed that Gottron sign did not have an association with cancer, but findings of cutaneous necrosis did have an association. It is unknown if the specific cutaneous findings in our patient, including the predominantly unilateral papules on the knee, may have been a clue to the underlying malignancy.
In summary, we believe that our patient presented with the classic manifestations of DM in addition to the curious cutaneous sign of predominantly unilateral, confluent, erythematous papules on the knee, a clinical finding that may aid in the diagnosis of DM and also may alert the clinician to a possible underlying malignancy.
- Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344-347.
- Santmyire-Rosenberger B, Dugan EM. Skin involvement in dermatomyositis. Curr Opin Rheumatol. 2003;15:714-722.
- Callen JP. Dermatomyositis. Lancet. 2000;355:53-57.
- Lister RK, Cooper ES, Paige DG. Papules and pustules of the elbows and knees: an uncommon clinical sign of dermatomyositis in oriental children. Pediatr Dermatol. 2000;17:37-40.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Wang J, Guo G, Chen G, et al. Meta‐analysis of the association of dermatomyositis and polymyositis with cancer. Br J Dermatol. 2013;169:838-847.
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case–control study. Br J Dermatol. 2001;144:825-831.
- Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344-347.
- Santmyire-Rosenberger B, Dugan EM. Skin involvement in dermatomyositis. Curr Opin Rheumatol. 2003;15:714-722.
- Callen JP. Dermatomyositis. Lancet. 2000;355:53-57.
- Lister RK, Cooper ES, Paige DG. Papules and pustules of the elbows and knees: an uncommon clinical sign of dermatomyositis in oriental children. Pediatr Dermatol. 2000;17:37-40.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Wang J, Guo G, Chen G, et al. Meta‐analysis of the association of dermatomyositis and polymyositis with cancer. Br J Dermatol. 2013;169:838-847.
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case–control study. Br J Dermatol. 2001;144:825-831.
Practice Points
- Dermatomyositis has myriad cutaneous features including the shawl sign, the heliotrope sign, and Gottron papules.
- Less commonly, patients can present with the Holster sign (poikiloderma of the lateral thighs).
- Even less commonly, as in this report, patients can present with a psoriasiform papular eruption on the knees or with flagellate erythema on the back.
Annular Elastolytic Giant Cell Granuloma: Mysterious Enlarging Scarring Lesions
To the Editor:
A 52-year-old woman with a medical history of migraines and cervicalgia presented with lesions on the right arm, back, and right calf. The patient stated that the lesions began as small papules that had grown over 13 months, with the largest papule on the right forearm. She reported no itching, bleeding, pain, discharge, or other symptoms associated with the lesions. She had a multiple-year history of similar lesions that did not respond to treatment with antifungals, moderate-potency steroids, and other over-the-counter creams. The lesions would resolve spontaneously with scarring and subsequently recur. Prior skin biopsies were inconclusive. The patient did not report any systemic symptoms or a personal or family history of connective tissue diseases.
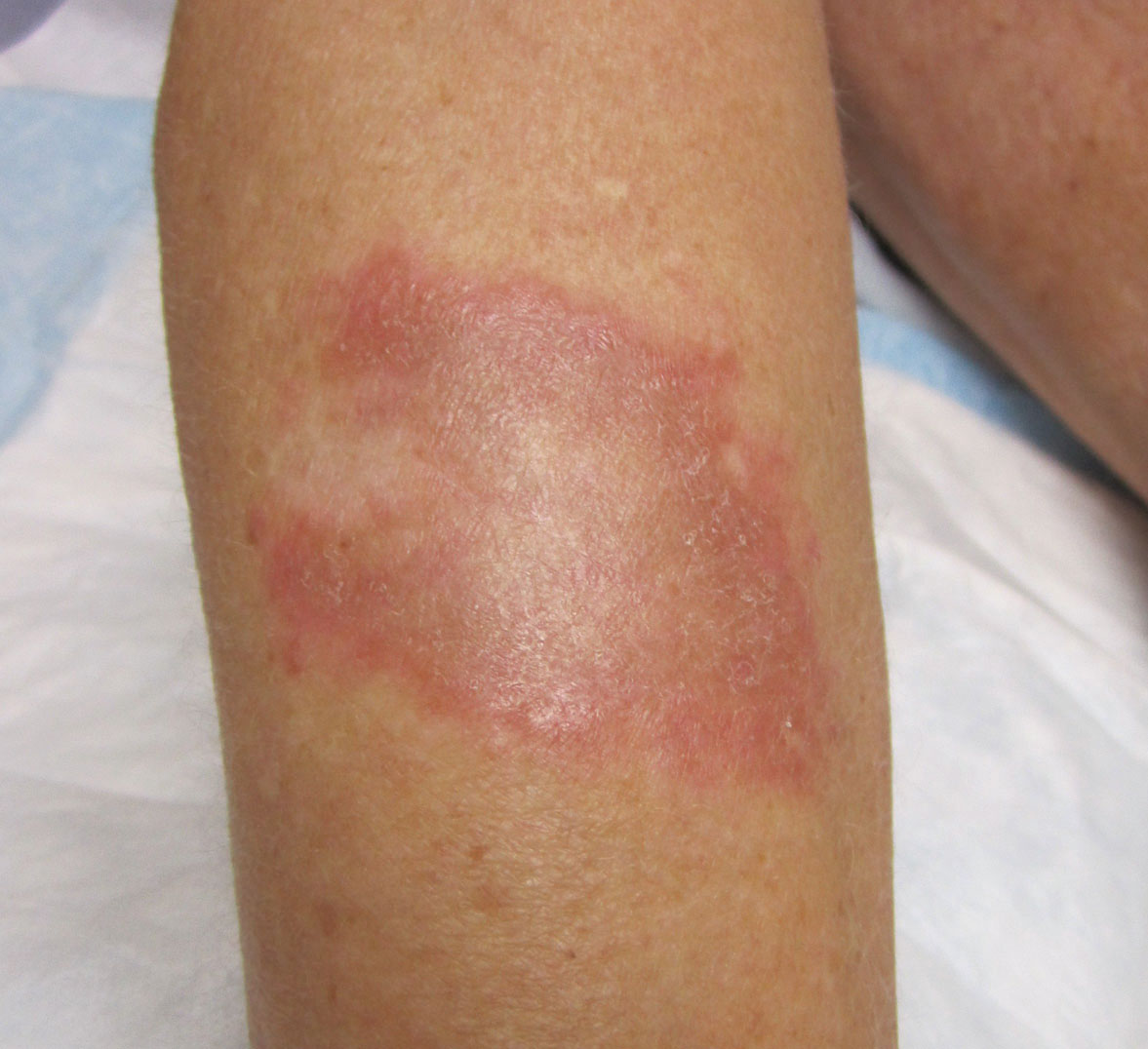
Physical examination revealed a 4-cm asymmetric, annular, erythematous plaque with central clearing on the right dorsal forearm with defined margins except over the distal aspect (Figure 1). She also had several 1- to 2-cm erythematous, nummular, asymmetric plaques on the right upper arm with well-defined margins. She had several lesions over the central and left sides of the upper back that were similar to the lesions on the upper arm.
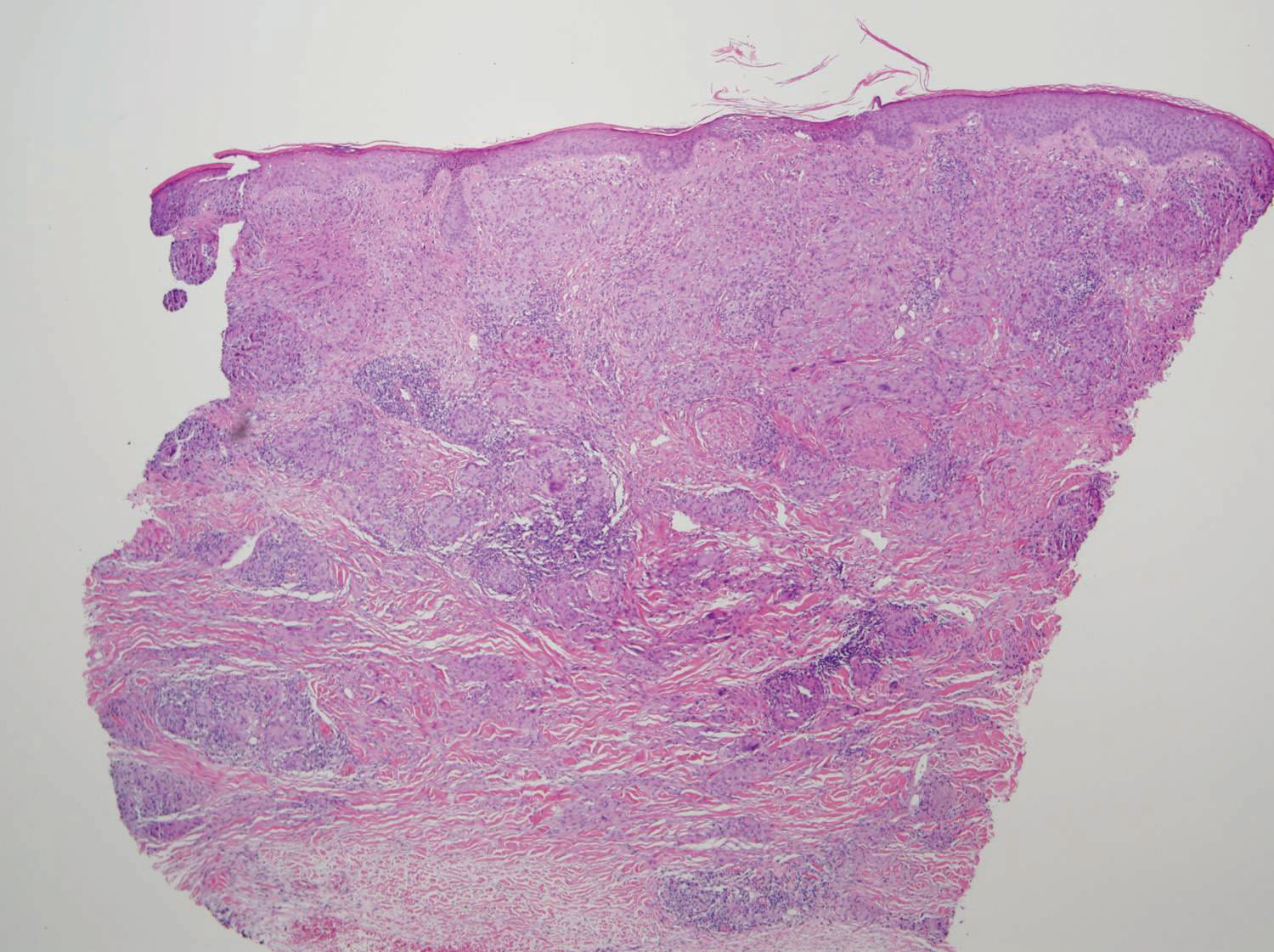
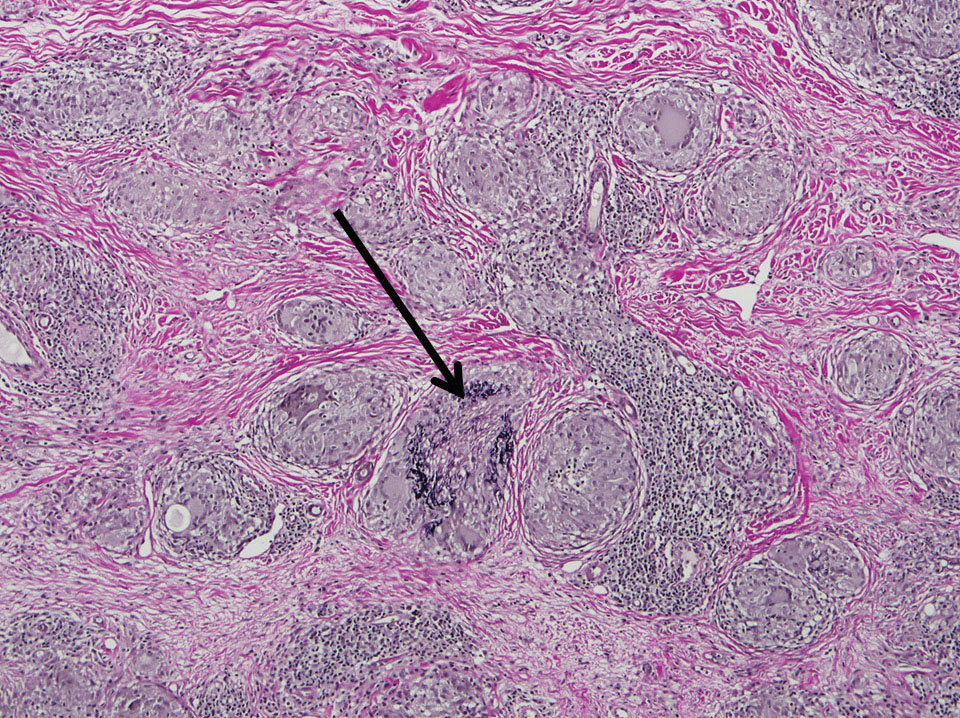
Two 4-mm punch biopsies of the right dorsal forearm and left side of the upper back revealed similar histologic features with a predominantly unremarkable epidermis. The dermis revealed a lymphohistiocytic infiltrate with prominent multinucleated giant cells organized into foreign body–type granulomas that extended into the deep dermis and subcutaneous tissue (Figure 2). In the granulomatous areas, there was a near-complete loss of elastic fibers with focal elastophagocytosis highlighted with Verhoeff-van Gieson (elastin) stain (Figure 3). Grocott-Gomori methenamine-silver and Fite stains for microorganisms were negative, and there was an absence of necrobiosis, lipids, and mucin.
The histologic findings of a granulomatous dermatitis with loss of elastic fibers and elastophagocytosis in addition to the patient’s clinical presentation and history were consistent with the diagnosis of annular elastolytic giant cell granuloma (AEGCG). Infectious and other granulomatous diseases including sarcoidosis were ruled out via clinical history, unremarkable laboratory analysis (ie, complete blood cell count, chemistry panel, antinuclear antibody, urinalysis), and a normal chest radiograph. The histologic findings via the various stains were instrumental to the diagnosis. The patient was treated with fluocinonide and subsequently lost to follow-up.
Annular elastolytic giant cell granuloma is an uncommon cutaneous disease that presents with recurring annular plaques with raised erythematous borders and subsequent residual scarring.1 O’Brien2 originally described this condition in 1975 as an actinic granuloma due to similar histologic findings in areas of the patient’s sun-exposed skin. Ragaz and Ackerman3 disputed O’Brien’s2 description, claiming granulomatous inflammation was a primary pathologic process and not a consequence to damaged elastotic material. In 1979, Hanke et al4 termed the lesions as AEGCG because he did not find a correlation to the sun-exposed areas of the patients and did not see solar elastosis.
Although AEGCG has an unclear pathogenesis, cellular immunologic reactions induced by modified function of elastic fibers’ antigenicity contribute to AEGCG formation.5 Therefore, environmental and host factors may play a role in its etiopathogenesis. In one study, 37% of 38 Japanese patients with AEGCG were found to have definitive or latent diabetes mellitus, raising the possible role of diabetes in the structural damage of the elastic fibers.6
Patients typically are middle-aged women who present clinically with red or atrophic plaques that have slightly elevated borders. They have centripetal spread with a resulting atrophic center.7 Clinically, the differential diagnosis of this condition includes actinic granuloma, granuloma annulare, and granuloma multiforme.8
Histologically, AEGCG has a granulomatous component with multinucleated giant cells in the upper and mid dermis. This component typically is distributed peripherally to a central zone that lacks elastic tissue. Elastophagocytosis, a classic finding in AEGCG, is the phagocytosis of elastic fibers that can microscopically be seen in the cytoplasm of histiocytes and multinucleated giant cells. There also is an absence of necrobiosis, lipids, mucin, and a palisading arrangement of the granulomas. These findings distinguish AEGCG from granuloma annulare and necrobiosis lipoidica, the primary histologic differential diagnoses.9 In addition, consideration of entities consistently exhibiting elastophagocytosis such as mid-dermal elastolysis, papillary dermal elastolysis, actinic granuloma, and granulomatous slack skin should be considered.5,10,11
Therapy for AEGCG is broad and includes topical, intralesional, and systemic corticosteroids. Hydroxychloroquine, isotretinoin, clofazimine, dapsone, photochemotherapy, and cyclosporine also have been utilized with varying results. Other reports show improvement with surgical excision, cryotherapy, or cauterization of small lesions.12-15
1. Tock CL, Cohen PR. Annular elastolytic giant cell granuloma. Cutis. 1998;62:181-187.
2. O’Brien JP. Actinic granuloma: an annular connective tissue disorder affecting sun- and heat-damaged (elastotic) skin. Arch Dermatol. 1975;111:460-466.
3. Ragaz A, Ackerman AB. Is actinic granuloma a specific condition? Am J Dermatopathol. 1979;1:43-50.
4. Hanke CW, Bailin PL, Roenigk HH Jr. Annular elastolytic giant cell granuloma. a clinicopathologic study of five cases and a review of similar entities. J Am Acad Dermatol. 1979;1:413-421.
5. El-Khoury J, Kurban M, Abbas O. Elastophagocytosis: underlying mechanisms and associated cutaneous entities. J Am Acad Dermatol. 2014;70:934-44.
6. Aso Y, Izaki Y, Teraki Y. Annular elastolytic giant cell granuloma associated with diabetes mellitus: a case report and review of the Japanese literature. Clin Exp Dermatol. 2011;36:917-919.
7. Pestoni C, Pereiro M Jr, Toribio J. Annular elastolytic giant cell granuloma produced on an old burn scar and spreading after a mechanical trauma. Acta Derm Venereol. 2003;83:312-313.
8. Oka M, Kunisada M, Nishigori C. Generalized annular elastolytic giant cell granuloma with sparing of striae distensae. J Dermatol. 2013;40:220-222.
9. Limas C. The spectrum of primary cutaneous elastolytic granulomas and their distinction from granuloma annulare: a clinicopathological analysis. Histopathology. 2004;44:277-282.
10. McGrae JD Jr. Actinic granuloma: a clinical, histopathologic, and immunocytochemical study. Arch Dermatol. 1986;122:43-47.
11. Shah A, Safaya A. Granulomatous slack skin disease: a review, in comparison with mycosis fungoides. J Eur Acad Dermatol Venereol. 2012;26:1472-1478.
12. Chou WT, Tsai TF, Hung CM, et al. Multiple annular erythematous plaques on the back. Annular elastolytic giant cell granuloma (AEGCG). Indian J Dermatol Venereol Leprol. 2011;77:727-728.
13. Pérez-Pérez L, Garcia-Gavin J, Alleque F, et al. Successful treatment of generalized elastolytic giant cell granuloma with psoralen-ultraviolet A. Photodermatol Photoimmunol Photomed. 2012;28:264-266.
14. Babuna G, Buyukbabani N, Yazganoglu KD, et al. Effective treatment with hydroxychloroquine in a case of annular elastolytic giant cell granuloma. Indian J Dermatol Venereol Leprol. 2011;77:110-111.
15. Can B, Kavala M, Türkoglu Z, et al. Successful treatment of annular elastolytic giant cell granuloma with hydroxylchloroquine. Int J Dermatol. 2013;52:509-511.
To the Editor:
A 52-year-old woman with a medical history of migraines and cervicalgia presented with lesions on the right arm, back, and right calf. The patient stated that the lesions began as small papules that had grown over 13 months, with the largest papule on the right forearm. She reported no itching, bleeding, pain, discharge, or other symptoms associated with the lesions. She had a multiple-year history of similar lesions that did not respond to treatment with antifungals, moderate-potency steroids, and other over-the-counter creams. The lesions would resolve spontaneously with scarring and subsequently recur. Prior skin biopsies were inconclusive. The patient did not report any systemic symptoms or a personal or family history of connective tissue diseases.
Physical examination revealed a 4-cm asymmetric, annular, erythematous plaque with central clearing on the right dorsal forearm with defined margins except over the distal aspect (Figure 1). She also had several 1- to 2-cm erythematous, nummular, asymmetric plaques on the right upper arm with well-defined margins. She had several lesions over the central and left sides of the upper back that were similar to the lesions on the upper arm.
Two 4-mm punch biopsies of the right dorsal forearm and left side of the upper back revealed similar histologic features with a predominantly unremarkable epidermis. The dermis revealed a lymphohistiocytic infiltrate with prominent multinucleated giant cells organized into foreign body–type granulomas that extended into the deep dermis and subcutaneous tissue (Figure 2). In the granulomatous areas, there was a near-complete loss of elastic fibers with focal elastophagocytosis highlighted with Verhoeff-van Gieson (elastin) stain (Figure 3). Grocott-Gomori methenamine-silver and Fite stains for microorganisms were negative, and there was an absence of necrobiosis, lipids, and mucin.
The histologic findings of a granulomatous dermatitis with loss of elastic fibers and elastophagocytosis in addition to the patient’s clinical presentation and history were consistent with the diagnosis of annular elastolytic giant cell granuloma (AEGCG). Infectious and other granulomatous diseases including sarcoidosis were ruled out via clinical history, unremarkable laboratory analysis (ie, complete blood cell count, chemistry panel, antinuclear antibody, urinalysis), and a normal chest radiograph. The histologic findings via the various stains were instrumental to the diagnosis. The patient was treated with fluocinonide and subsequently lost to follow-up.
Annular elastolytic giant cell granuloma is an uncommon cutaneous disease that presents with recurring annular plaques with raised erythematous borders and subsequent residual scarring.1 O’Brien2 originally described this condition in 1975 as an actinic granuloma due to similar histologic findings in areas of the patient’s sun-exposed skin. Ragaz and Ackerman3 disputed O’Brien’s2 description, claiming granulomatous inflammation was a primary pathologic process and not a consequence to damaged elastotic material. In 1979, Hanke et al4 termed the lesions as AEGCG because he did not find a correlation to the sun-exposed areas of the patients and did not see solar elastosis.
Although AEGCG has an unclear pathogenesis, cellular immunologic reactions induced by modified function of elastic fibers’ antigenicity contribute to AEGCG formation.5 Therefore, environmental and host factors may play a role in its etiopathogenesis. In one study, 37% of 38 Japanese patients with AEGCG were found to have definitive or latent diabetes mellitus, raising the possible role of diabetes in the structural damage of the elastic fibers.6
Patients typically are middle-aged women who present clinically with red or atrophic plaques that have slightly elevated borders. They have centripetal spread with a resulting atrophic center.7 Clinically, the differential diagnosis of this condition includes actinic granuloma, granuloma annulare, and granuloma multiforme.8
Histologically, AEGCG has a granulomatous component with multinucleated giant cells in the upper and mid dermis. This component typically is distributed peripherally to a central zone that lacks elastic tissue. Elastophagocytosis, a classic finding in AEGCG, is the phagocytosis of elastic fibers that can microscopically be seen in the cytoplasm of histiocytes and multinucleated giant cells. There also is an absence of necrobiosis, lipids, mucin, and a palisading arrangement of the granulomas. These findings distinguish AEGCG from granuloma annulare and necrobiosis lipoidica, the primary histologic differential diagnoses.9 In addition, consideration of entities consistently exhibiting elastophagocytosis such as mid-dermal elastolysis, papillary dermal elastolysis, actinic granuloma, and granulomatous slack skin should be considered.5,10,11
Therapy for AEGCG is broad and includes topical, intralesional, and systemic corticosteroids. Hydroxychloroquine, isotretinoin, clofazimine, dapsone, photochemotherapy, and cyclosporine also have been utilized with varying results. Other reports show improvement with surgical excision, cryotherapy, or cauterization of small lesions.12-15
To the Editor:
A 52-year-old woman with a medical history of migraines and cervicalgia presented with lesions on the right arm, back, and right calf. The patient stated that the lesions began as small papules that had grown over 13 months, with the largest papule on the right forearm. She reported no itching, bleeding, pain, discharge, or other symptoms associated with the lesions. She had a multiple-year history of similar lesions that did not respond to treatment with antifungals, moderate-potency steroids, and other over-the-counter creams. The lesions would resolve spontaneously with scarring and subsequently recur. Prior skin biopsies were inconclusive. The patient did not report any systemic symptoms or a personal or family history of connective tissue diseases.
Physical examination revealed a 4-cm asymmetric, annular, erythematous plaque with central clearing on the right dorsal forearm with defined margins except over the distal aspect (Figure 1). She also had several 1- to 2-cm erythematous, nummular, asymmetric plaques on the right upper arm with well-defined margins. She had several lesions over the central and left sides of the upper back that were similar to the lesions on the upper arm.
Two 4-mm punch biopsies of the right dorsal forearm and left side of the upper back revealed similar histologic features with a predominantly unremarkable epidermis. The dermis revealed a lymphohistiocytic infiltrate with prominent multinucleated giant cells organized into foreign body–type granulomas that extended into the deep dermis and subcutaneous tissue (Figure 2). In the granulomatous areas, there was a near-complete loss of elastic fibers with focal elastophagocytosis highlighted with Verhoeff-van Gieson (elastin) stain (Figure 3). Grocott-Gomori methenamine-silver and Fite stains for microorganisms were negative, and there was an absence of necrobiosis, lipids, and mucin.
The histologic findings of a granulomatous dermatitis with loss of elastic fibers and elastophagocytosis in addition to the patient’s clinical presentation and history were consistent with the diagnosis of annular elastolytic giant cell granuloma (AEGCG). Infectious and other granulomatous diseases including sarcoidosis were ruled out via clinical history, unremarkable laboratory analysis (ie, complete blood cell count, chemistry panel, antinuclear antibody, urinalysis), and a normal chest radiograph. The histologic findings via the various stains were instrumental to the diagnosis. The patient was treated with fluocinonide and subsequently lost to follow-up.
Annular elastolytic giant cell granuloma is an uncommon cutaneous disease that presents with recurring annular plaques with raised erythematous borders and subsequent residual scarring.1 O’Brien2 originally described this condition in 1975 as an actinic granuloma due to similar histologic findings in areas of the patient’s sun-exposed skin. Ragaz and Ackerman3 disputed O’Brien’s2 description, claiming granulomatous inflammation was a primary pathologic process and not a consequence to damaged elastotic material. In 1979, Hanke et al4 termed the lesions as AEGCG because he did not find a correlation to the sun-exposed areas of the patients and did not see solar elastosis.
Although AEGCG has an unclear pathogenesis, cellular immunologic reactions induced by modified function of elastic fibers’ antigenicity contribute to AEGCG formation.5 Therefore, environmental and host factors may play a role in its etiopathogenesis. In one study, 37% of 38 Japanese patients with AEGCG were found to have definitive or latent diabetes mellitus, raising the possible role of diabetes in the structural damage of the elastic fibers.6
Patients typically are middle-aged women who present clinically with red or atrophic plaques that have slightly elevated borders. They have centripetal spread with a resulting atrophic center.7 Clinically, the differential diagnosis of this condition includes actinic granuloma, granuloma annulare, and granuloma multiforme.8
Histologically, AEGCG has a granulomatous component with multinucleated giant cells in the upper and mid dermis. This component typically is distributed peripherally to a central zone that lacks elastic tissue. Elastophagocytosis, a classic finding in AEGCG, is the phagocytosis of elastic fibers that can microscopically be seen in the cytoplasm of histiocytes and multinucleated giant cells. There also is an absence of necrobiosis, lipids, mucin, and a palisading arrangement of the granulomas. These findings distinguish AEGCG from granuloma annulare and necrobiosis lipoidica, the primary histologic differential diagnoses.9 In addition, consideration of entities consistently exhibiting elastophagocytosis such as mid-dermal elastolysis, papillary dermal elastolysis, actinic granuloma, and granulomatous slack skin should be considered.5,10,11
Therapy for AEGCG is broad and includes topical, intralesional, and systemic corticosteroids. Hydroxychloroquine, isotretinoin, clofazimine, dapsone, photochemotherapy, and cyclosporine also have been utilized with varying results. Other reports show improvement with surgical excision, cryotherapy, or cauterization of small lesions.12-15
1. Tock CL, Cohen PR. Annular elastolytic giant cell granuloma. Cutis. 1998;62:181-187.
2. O’Brien JP. Actinic granuloma: an annular connective tissue disorder affecting sun- and heat-damaged (elastotic) skin. Arch Dermatol. 1975;111:460-466.
3. Ragaz A, Ackerman AB. Is actinic granuloma a specific condition? Am J Dermatopathol. 1979;1:43-50.
4. Hanke CW, Bailin PL, Roenigk HH Jr. Annular elastolytic giant cell granuloma. a clinicopathologic study of five cases and a review of similar entities. J Am Acad Dermatol. 1979;1:413-421.
5. El-Khoury J, Kurban M, Abbas O. Elastophagocytosis: underlying mechanisms and associated cutaneous entities. J Am Acad Dermatol. 2014;70:934-44.
6. Aso Y, Izaki Y, Teraki Y. Annular elastolytic giant cell granuloma associated with diabetes mellitus: a case report and review of the Japanese literature. Clin Exp Dermatol. 2011;36:917-919.
7. Pestoni C, Pereiro M Jr, Toribio J. Annular elastolytic giant cell granuloma produced on an old burn scar and spreading after a mechanical trauma. Acta Derm Venereol. 2003;83:312-313.
8. Oka M, Kunisada M, Nishigori C. Generalized annular elastolytic giant cell granuloma with sparing of striae distensae. J Dermatol. 2013;40:220-222.
9. Limas C. The spectrum of primary cutaneous elastolytic granulomas and their distinction from granuloma annulare: a clinicopathological analysis. Histopathology. 2004;44:277-282.
10. McGrae JD Jr. Actinic granuloma: a clinical, histopathologic, and immunocytochemical study. Arch Dermatol. 1986;122:43-47.
11. Shah A, Safaya A. Granulomatous slack skin disease: a review, in comparison with mycosis fungoides. J Eur Acad Dermatol Venereol. 2012;26:1472-1478.
12. Chou WT, Tsai TF, Hung CM, et al. Multiple annular erythematous plaques on the back. Annular elastolytic giant cell granuloma (AEGCG). Indian J Dermatol Venereol Leprol. 2011;77:727-728.
13. Pérez-Pérez L, Garcia-Gavin J, Alleque F, et al. Successful treatment of generalized elastolytic giant cell granuloma with psoralen-ultraviolet A. Photodermatol Photoimmunol Photomed. 2012;28:264-266.
14. Babuna G, Buyukbabani N, Yazganoglu KD, et al. Effective treatment with hydroxychloroquine in a case of annular elastolytic giant cell granuloma. Indian J Dermatol Venereol Leprol. 2011;77:110-111.
15. Can B, Kavala M, Türkoglu Z, et al. Successful treatment of annular elastolytic giant cell granuloma with hydroxylchloroquine. Int J Dermatol. 2013;52:509-511.
1. Tock CL, Cohen PR. Annular elastolytic giant cell granuloma. Cutis. 1998;62:181-187.
2. O’Brien JP. Actinic granuloma: an annular connective tissue disorder affecting sun- and heat-damaged (elastotic) skin. Arch Dermatol. 1975;111:460-466.
3. Ragaz A, Ackerman AB. Is actinic granuloma a specific condition? Am J Dermatopathol. 1979;1:43-50.
4. Hanke CW, Bailin PL, Roenigk HH Jr. Annular elastolytic giant cell granuloma. a clinicopathologic study of five cases and a review of similar entities. J Am Acad Dermatol. 1979;1:413-421.
5. El-Khoury J, Kurban M, Abbas O. Elastophagocytosis: underlying mechanisms and associated cutaneous entities. J Am Acad Dermatol. 2014;70:934-44.
6. Aso Y, Izaki Y, Teraki Y. Annular elastolytic giant cell granuloma associated with diabetes mellitus: a case report and review of the Japanese literature. Clin Exp Dermatol. 2011;36:917-919.
7. Pestoni C, Pereiro M Jr, Toribio J. Annular elastolytic giant cell granuloma produced on an old burn scar and spreading after a mechanical trauma. Acta Derm Venereol. 2003;83:312-313.
8. Oka M, Kunisada M, Nishigori C. Generalized annular elastolytic giant cell granuloma with sparing of striae distensae. J Dermatol. 2013;40:220-222.
9. Limas C. The spectrum of primary cutaneous elastolytic granulomas and their distinction from granuloma annulare: a clinicopathological analysis. Histopathology. 2004;44:277-282.
10. McGrae JD Jr. Actinic granuloma: a clinical, histopathologic, and immunocytochemical study. Arch Dermatol. 1986;122:43-47.
11. Shah A, Safaya A. Granulomatous slack skin disease: a review, in comparison with mycosis fungoides. J Eur Acad Dermatol Venereol. 2012;26:1472-1478.
12. Chou WT, Tsai TF, Hung CM, et al. Multiple annular erythematous plaques on the back. Annular elastolytic giant cell granuloma (AEGCG). Indian J Dermatol Venereol Leprol. 2011;77:727-728.
13. Pérez-Pérez L, Garcia-Gavin J, Alleque F, et al. Successful treatment of generalized elastolytic giant cell granuloma with psoralen-ultraviolet A. Photodermatol Photoimmunol Photomed. 2012;28:264-266.
14. Babuna G, Buyukbabani N, Yazganoglu KD, et al. Effective treatment with hydroxychloroquine in a case of annular elastolytic giant cell granuloma. Indian J Dermatol Venereol Leprol. 2011;77:110-111.
15. Can B, Kavala M, Türkoglu Z, et al. Successful treatment of annular elastolytic giant cell granuloma with hydroxylchloroquine. Int J Dermatol. 2013;52:509-511.
Practice Points
- Annular elastolytic giant cell granuloma (AEGCG) should be kept in the differential diagnosis when assessing a middle-aged woman with recurring annular plaques with a raised border and an atrophic center on both sun-exposed and sun-protected areas of the body.
- Histologically, AEGCG classically has a granulomatous component in the dermis that lacks elastic tissue and has no necrobiosis, lipids, or mucin. Staining with elastin may be necessary to highlight these areas as well as demonstrate elastophagocytosis.
Solitary Nodule on the Thigh
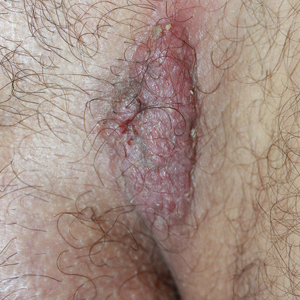
The Diagnosis: Ruptured Molluscum
Molluscum contagiosum (MC) is caused by a DNA virus (MC virus) belonging to the poxvirus family. Molluscum contagiosum is common and predominantly seen in children and young adults. In sexually active adults, the lesions commonly occur in the genital region, abdomen, and inner thighs. In immunocompromised individuals, including those with AIDS, the lesions are more extensive and may cause disfigurement.1 Molluscum contagiosum involving epidermoid cysts has been reported.2
Histopathologically, MC can be classified as noninflammatory or inflammatory. In noninflamed lesions, multiple large, intracytoplasmic, eosinophilic inclusions (Henderson-Paterson bodies) appear within the lobulated endophytic and hyperplastic epidermis. Ultrastructurally, these bodies show membrane-bound collections of MC virus.1 Replicating Henderson-Paterson bodies can result in rupture and inflammation. This case demonstrates a palisading granuloma containing keratin with few Henderson-Paterson bodies (quiz image) due to prior rupture of a molluscum or molluscoid cyst.
Rheumatoid nodules, the most characteristic histopathologic lesions of rheumatoid arthritis, are most commonly found in the subcutis at points of pressure and may occur in connective tissue of numerous organs. Rheumatoid nodules are firm, nontender, and mobile within the subcutaneous tissue but may be fixed to underlying structures including the periosteum, tendons, or bursae.3,4 Occasionally, superficial nodules may perforate the epidermis.5 The inner central necrobiotic zone appears as intensely eosinophilic, amorphous fibrin and other cellular debris. This central area is surrounded by histiocytes in a palisaded configuration (Figure 1). Multinucleated foreign body giant cells also may be present. Occasionally, mast cells, eosinophils, and neutrophils are present.6,7
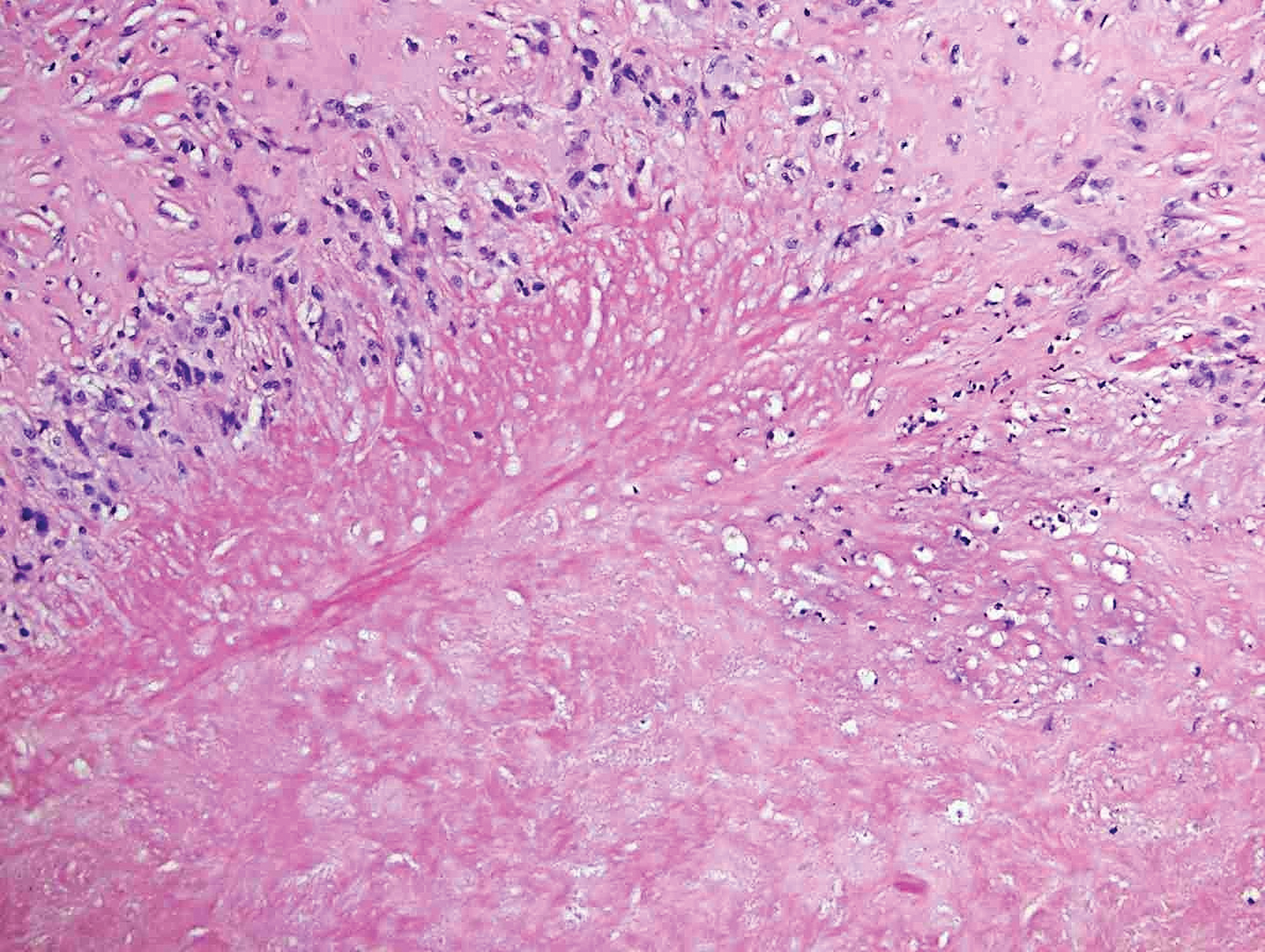
Lupus miliaris disseminatus faciei presents with multiple discrete, smooth, yellow-brown to red, dome-shaped papules. The lesions typically are located on the central and lateral sides of the face and infrequently involve the neck. Other sites including the axillae, arms, hands, legs, and groin occasionally can be involved. Diascopy may reveal an apple jelly color.8,9 The histopathologic hallmark of lupus miliaris disseminatus faciei is an epithelioid cell granuloma with central necrosis (Figure 2).

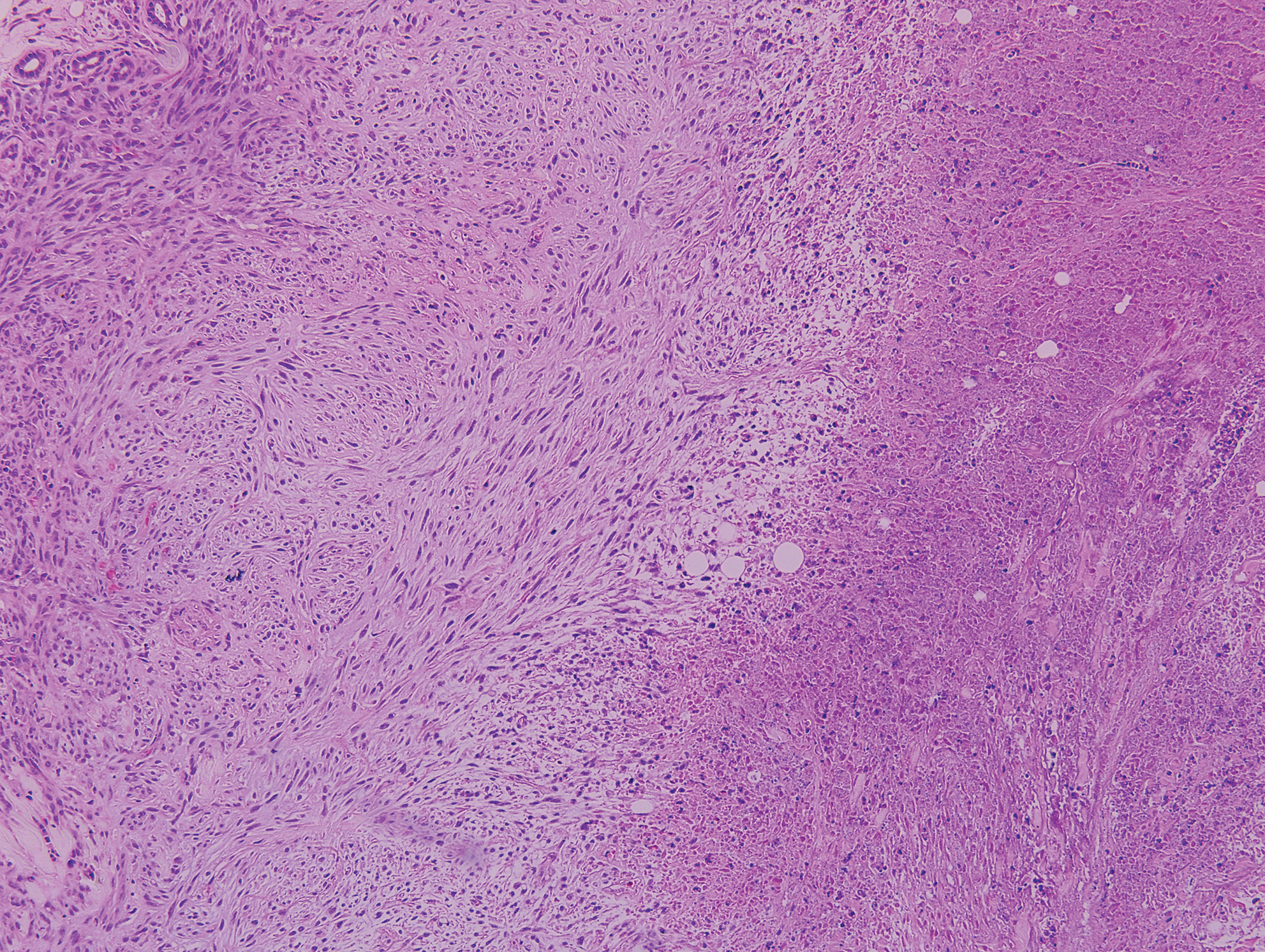
Epithelioid sarcoma (ES) is a soft tissue tumor with a known propensity for local recurrence, regional lymph node involvement, sporotrichoid spread, and distant metastases.10 The name was coined by Enzinger11 in 1970 during a review of 62 cases of a “peculiar form of sarcoma that has repeatedly been confused with a chronic inflammatory process, a necrotizing granuloma, and a squamous cell carcinoma.” Epithelioid sarcoma tends to grow slowly in a nodular or multinodular manner along fascial structures and tendons, often with central necrosis and ulceration of the overlying skin. Histopathologically, classic ES shows nodular masses of uniform plump epithelioid cells with abundant eosinophilic cytoplasm and prominent central necrosis. A biphasic pattern is typical with spindle cells merging with epithelioid cells. Cellular atypia is relatively mild and mitoses are rare (Figure 3). Recurrent or metastatic lesions can show a greater degree of pleomorphism.12 Given the low-grade atypia in early lesions, this sarcoma is easily misdiagnosed as granulomatous dermatitis. Immunohistochemically, the majority of ES cases are positive for cytokeratins and epithelial membrane antigen; SMARCB1/INI-1 expression is characteristically lost.13
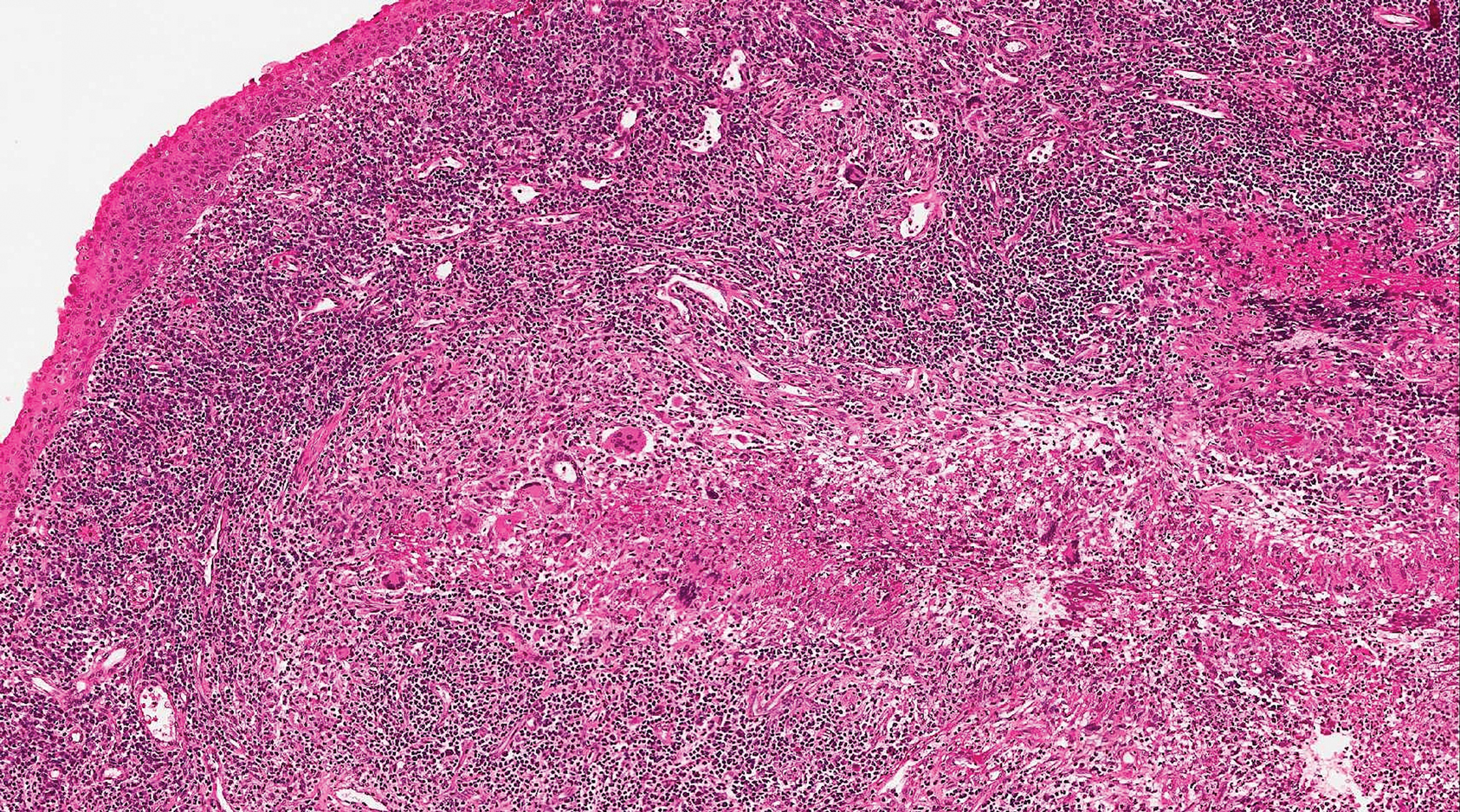
Granulomatosis with polyangiitis (formerly Wegener granulomatosis) is an autoimmune vasculitis highly associated with antineutrophil cytoplasmic antibodies. Clinical manifestations include systemic necrotizing vasculitis; necrotizing glomerulonephritis; and granulomatous inflammation, which predominantly involves the upper respiratory tract, skin, and mucosa.14,15 Skin involvement may be the initial manifestation of the disease and consists of palpable purpura, papules, ulcerations, vesicles, subcutaneous nodules, necrotizing ulcerations, papulonecrotic lesions, and petechiae. None of the findings are pathognomonic. The cutaneous histopathologic spectrum includes leukocytoclastic vasculitis, extravascular palisading granulomas, and granulomatous vasculitis.16 In the acute lesions of granulomatosis with polyangiitis, the predominant pattern of inflammation is not granulomatous but purulent with the appearance of an abscess. As it evolves, it develops a central zone of necrosis with extensive karyorrhectic debris and palisades of macrophages with scattered multinucleated giant cells (Figure 4).17
1. Nandhini G, Rajkumar K, Kanth KS, et al. Molluscum contagiosum in a 12-year-old child—report of a case and review of literature. J Int Oral Health. 2015;7:63-66.
2. Phelps A, Murphy M, Elaba Z, et al. Molluscum contagiosum virus infection in benign cutaneous epithelial cystic lesions-report of 2 cases with different pathogenesis? Am J Dermatopathol. 2010;32:740-742.
3. Sayah A, English JC 3rd. Rheumatoid arthritis: a review of the cutaneous manifestations. J Am Acad Dermatol. 2005;53:191-209; quiz 210-192.
4. Sibbitt WL Jr, Williams RC Jr. Cutaneous manifestations of rheumatoid arthritis. Int J Dermatol. 1982;21:563-572.
5. Barzilai A, Huszar M, Shpiro D, et al. Pseudorheumatoid nodules in adults: a juxta-articular form of nodular granuloma annulare. Am J Dermatopathol. 2005;27:1-5.
6. Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg. 2007;26:100-107.
7. Patterson JW. Rheumatoid nodule and subcutaneous granuloma annulare. a comparative histologic study. Am J Dermatopathol. 1988;10:1-8.
8. Sehgal VN, Srivastava G, Aggarwal AK, et al. Lupus miliaris disseminatus faciei part II: an overview. Skinmed. 2005;4:234-238.
9. Cymerman R, Rosenstein R, Shvartsbeyn M, et al. Lupus miliaris disseminatus faciei. Dermatol Online J. 2015;21. pii:13030/qt6b83q5gp.
10. Sobanko JF, Meijer L, Nigra TP. Epithelioid sarcoma: a review and update. J Clin Aesthet Dermatol. 2009;2:49-54.
11. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
12. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
13. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
14. Lutalo PM, D’Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis)[published online January 29, 2014]. J Autoimmun. 2014;48-49:94-98.
15. Frances C, Du LT, Piette JC, et al. Wegener’s granulomatosis. dermatological manifestations in 75 cases with clinicopathologic correlation. Arch Dermatol. 1994;130:861-867.
16. Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener’s granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
17. Jennette JC. Nomenclature and classification of vasculitis: lessons learned from granulomatosis with polyangiitis (Wegener’s granulomatosis). Clin Exp Immunol. 2011;164 (suppl 1):7-10.
The Diagnosis: Ruptured Molluscum
Molluscum contagiosum (MC) is caused by a DNA virus (MC virus) belonging to the poxvirus family. Molluscum contagiosum is common and predominantly seen in children and young adults. In sexually active adults, the lesions commonly occur in the genital region, abdomen, and inner thighs. In immunocompromised individuals, including those with AIDS, the lesions are more extensive and may cause disfigurement.1 Molluscum contagiosum involving epidermoid cysts has been reported.2
Histopathologically, MC can be classified as noninflammatory or inflammatory. In noninflamed lesions, multiple large, intracytoplasmic, eosinophilic inclusions (Henderson-Paterson bodies) appear within the lobulated endophytic and hyperplastic epidermis. Ultrastructurally, these bodies show membrane-bound collections of MC virus.1 Replicating Henderson-Paterson bodies can result in rupture and inflammation. This case demonstrates a palisading granuloma containing keratin with few Henderson-Paterson bodies (quiz image) due to prior rupture of a molluscum or molluscoid cyst.
Rheumatoid nodules, the most characteristic histopathologic lesions of rheumatoid arthritis, are most commonly found in the subcutis at points of pressure and may occur in connective tissue of numerous organs. Rheumatoid nodules are firm, nontender, and mobile within the subcutaneous tissue but may be fixed to underlying structures including the periosteum, tendons, or bursae.3,4 Occasionally, superficial nodules may perforate the epidermis.5 The inner central necrobiotic zone appears as intensely eosinophilic, amorphous fibrin and other cellular debris. This central area is surrounded by histiocytes in a palisaded configuration (Figure 1). Multinucleated foreign body giant cells also may be present. Occasionally, mast cells, eosinophils, and neutrophils are present.6,7
Lupus miliaris disseminatus faciei presents with multiple discrete, smooth, yellow-brown to red, dome-shaped papules. The lesions typically are located on the central and lateral sides of the face and infrequently involve the neck. Other sites including the axillae, arms, hands, legs, and groin occasionally can be involved. Diascopy may reveal an apple jelly color.8,9 The histopathologic hallmark of lupus miliaris disseminatus faciei is an epithelioid cell granuloma with central necrosis (Figure 2).
Epithelioid sarcoma (ES) is a soft tissue tumor with a known propensity for local recurrence, regional lymph node involvement, sporotrichoid spread, and distant metastases.10 The name was coined by Enzinger11 in 1970 during a review of 62 cases of a “peculiar form of sarcoma that has repeatedly been confused with a chronic inflammatory process, a necrotizing granuloma, and a squamous cell carcinoma.” Epithelioid sarcoma tends to grow slowly in a nodular or multinodular manner along fascial structures and tendons, often with central necrosis and ulceration of the overlying skin. Histopathologically, classic ES shows nodular masses of uniform plump epithelioid cells with abundant eosinophilic cytoplasm and prominent central necrosis. A biphasic pattern is typical with spindle cells merging with epithelioid cells. Cellular atypia is relatively mild and mitoses are rare (Figure 3). Recurrent or metastatic lesions can show a greater degree of pleomorphism.12 Given the low-grade atypia in early lesions, this sarcoma is easily misdiagnosed as granulomatous dermatitis. Immunohistochemically, the majority of ES cases are positive for cytokeratins and epithelial membrane antigen; SMARCB1/INI-1 expression is characteristically lost.13
Granulomatosis with polyangiitis (formerly Wegener granulomatosis) is an autoimmune vasculitis highly associated with antineutrophil cytoplasmic antibodies. Clinical manifestations include systemic necrotizing vasculitis; necrotizing glomerulonephritis; and granulomatous inflammation, which predominantly involves the upper respiratory tract, skin, and mucosa.14,15 Skin involvement may be the initial manifestation of the disease and consists of palpable purpura, papules, ulcerations, vesicles, subcutaneous nodules, necrotizing ulcerations, papulonecrotic lesions, and petechiae. None of the findings are pathognomonic. The cutaneous histopathologic spectrum includes leukocytoclastic vasculitis, extravascular palisading granulomas, and granulomatous vasculitis.16 In the acute lesions of granulomatosis with polyangiitis, the predominant pattern of inflammation is not granulomatous but purulent with the appearance of an abscess. As it evolves, it develops a central zone of necrosis with extensive karyorrhectic debris and palisades of macrophages with scattered multinucleated giant cells (Figure 4).17
The Diagnosis: Ruptured Molluscum
Molluscum contagiosum (MC) is caused by a DNA virus (MC virus) belonging to the poxvirus family. Molluscum contagiosum is common and predominantly seen in children and young adults. In sexually active adults, the lesions commonly occur in the genital region, abdomen, and inner thighs. In immunocompromised individuals, including those with AIDS, the lesions are more extensive and may cause disfigurement.1 Molluscum contagiosum involving epidermoid cysts has been reported.2
Histopathologically, MC can be classified as noninflammatory or inflammatory. In noninflamed lesions, multiple large, intracytoplasmic, eosinophilic inclusions (Henderson-Paterson bodies) appear within the lobulated endophytic and hyperplastic epidermis. Ultrastructurally, these bodies show membrane-bound collections of MC virus.1 Replicating Henderson-Paterson bodies can result in rupture and inflammation. This case demonstrates a palisading granuloma containing keratin with few Henderson-Paterson bodies (quiz image) due to prior rupture of a molluscum or molluscoid cyst.
Rheumatoid nodules, the most characteristic histopathologic lesions of rheumatoid arthritis, are most commonly found in the subcutis at points of pressure and may occur in connective tissue of numerous organs. Rheumatoid nodules are firm, nontender, and mobile within the subcutaneous tissue but may be fixed to underlying structures including the periosteum, tendons, or bursae.3,4 Occasionally, superficial nodules may perforate the epidermis.5 The inner central necrobiotic zone appears as intensely eosinophilic, amorphous fibrin and other cellular debris. This central area is surrounded by histiocytes in a palisaded configuration (Figure 1). Multinucleated foreign body giant cells also may be present. Occasionally, mast cells, eosinophils, and neutrophils are present.6,7
Lupus miliaris disseminatus faciei presents with multiple discrete, smooth, yellow-brown to red, dome-shaped papules. The lesions typically are located on the central and lateral sides of the face and infrequently involve the neck. Other sites including the axillae, arms, hands, legs, and groin occasionally can be involved. Diascopy may reveal an apple jelly color.8,9 The histopathologic hallmark of lupus miliaris disseminatus faciei is an epithelioid cell granuloma with central necrosis (Figure 2).
Epithelioid sarcoma (ES) is a soft tissue tumor with a known propensity for local recurrence, regional lymph node involvement, sporotrichoid spread, and distant metastases.10 The name was coined by Enzinger11 in 1970 during a review of 62 cases of a “peculiar form of sarcoma that has repeatedly been confused with a chronic inflammatory process, a necrotizing granuloma, and a squamous cell carcinoma.” Epithelioid sarcoma tends to grow slowly in a nodular or multinodular manner along fascial structures and tendons, often with central necrosis and ulceration of the overlying skin. Histopathologically, classic ES shows nodular masses of uniform plump epithelioid cells with abundant eosinophilic cytoplasm and prominent central necrosis. A biphasic pattern is typical with spindle cells merging with epithelioid cells. Cellular atypia is relatively mild and mitoses are rare (Figure 3). Recurrent or metastatic lesions can show a greater degree of pleomorphism.12 Given the low-grade atypia in early lesions, this sarcoma is easily misdiagnosed as granulomatous dermatitis. Immunohistochemically, the majority of ES cases are positive for cytokeratins and epithelial membrane antigen; SMARCB1/INI-1 expression is characteristically lost.13
Granulomatosis with polyangiitis (formerly Wegener granulomatosis) is an autoimmune vasculitis highly associated with antineutrophil cytoplasmic antibodies. Clinical manifestations include systemic necrotizing vasculitis; necrotizing glomerulonephritis; and granulomatous inflammation, which predominantly involves the upper respiratory tract, skin, and mucosa.14,15 Skin involvement may be the initial manifestation of the disease and consists of palpable purpura, papules, ulcerations, vesicles, subcutaneous nodules, necrotizing ulcerations, papulonecrotic lesions, and petechiae. None of the findings are pathognomonic. The cutaneous histopathologic spectrum includes leukocytoclastic vasculitis, extravascular palisading granulomas, and granulomatous vasculitis.16 In the acute lesions of granulomatosis with polyangiitis, the predominant pattern of inflammation is not granulomatous but purulent with the appearance of an abscess. As it evolves, it develops a central zone of necrosis with extensive karyorrhectic debris and palisades of macrophages with scattered multinucleated giant cells (Figure 4).17
1. Nandhini G, Rajkumar K, Kanth KS, et al. Molluscum contagiosum in a 12-year-old child—report of a case and review of literature. J Int Oral Health. 2015;7:63-66.
2. Phelps A, Murphy M, Elaba Z, et al. Molluscum contagiosum virus infection in benign cutaneous epithelial cystic lesions-report of 2 cases with different pathogenesis? Am J Dermatopathol. 2010;32:740-742.
3. Sayah A, English JC 3rd. Rheumatoid arthritis: a review of the cutaneous manifestations. J Am Acad Dermatol. 2005;53:191-209; quiz 210-192.
4. Sibbitt WL Jr, Williams RC Jr. Cutaneous manifestations of rheumatoid arthritis. Int J Dermatol. 1982;21:563-572.
5. Barzilai A, Huszar M, Shpiro D, et al. Pseudorheumatoid nodules in adults: a juxta-articular form of nodular granuloma annulare. Am J Dermatopathol. 2005;27:1-5.
6. Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg. 2007;26:100-107.
7. Patterson JW. Rheumatoid nodule and subcutaneous granuloma annulare. a comparative histologic study. Am J Dermatopathol. 1988;10:1-8.
8. Sehgal VN, Srivastava G, Aggarwal AK, et al. Lupus miliaris disseminatus faciei part II: an overview. Skinmed. 2005;4:234-238.
9. Cymerman R, Rosenstein R, Shvartsbeyn M, et al. Lupus miliaris disseminatus faciei. Dermatol Online J. 2015;21. pii:13030/qt6b83q5gp.
10. Sobanko JF, Meijer L, Nigra TP. Epithelioid sarcoma: a review and update. J Clin Aesthet Dermatol. 2009;2:49-54.
11. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
12. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
13. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
14. Lutalo PM, D’Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis)[published online January 29, 2014]. J Autoimmun. 2014;48-49:94-98.
15. Frances C, Du LT, Piette JC, et al. Wegener’s granulomatosis. dermatological manifestations in 75 cases with clinicopathologic correlation. Arch Dermatol. 1994;130:861-867.
16. Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener’s granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
17. Jennette JC. Nomenclature and classification of vasculitis: lessons learned from granulomatosis with polyangiitis (Wegener’s granulomatosis). Clin Exp Immunol. 2011;164 (suppl 1):7-10.
1. Nandhini G, Rajkumar K, Kanth KS, et al. Molluscum contagiosum in a 12-year-old child—report of a case and review of literature. J Int Oral Health. 2015;7:63-66.
2. Phelps A, Murphy M, Elaba Z, et al. Molluscum contagiosum virus infection in benign cutaneous epithelial cystic lesions-report of 2 cases with different pathogenesis? Am J Dermatopathol. 2010;32:740-742.
3. Sayah A, English JC 3rd. Rheumatoid arthritis: a review of the cutaneous manifestations. J Am Acad Dermatol. 2005;53:191-209; quiz 210-192.
4. Sibbitt WL Jr, Williams RC Jr. Cutaneous manifestations of rheumatoid arthritis. Int J Dermatol. 1982;21:563-572.
5. Barzilai A, Huszar M, Shpiro D, et al. Pseudorheumatoid nodules in adults: a juxta-articular form of nodular granuloma annulare. Am J Dermatopathol. 2005;27:1-5.
6. Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg. 2007;26:100-107.
7. Patterson JW. Rheumatoid nodule and subcutaneous granuloma annulare. a comparative histologic study. Am J Dermatopathol. 1988;10:1-8.
8. Sehgal VN, Srivastava G, Aggarwal AK, et al. Lupus miliaris disseminatus faciei part II: an overview. Skinmed. 2005;4:234-238.
9. Cymerman R, Rosenstein R, Shvartsbeyn M, et al. Lupus miliaris disseminatus faciei. Dermatol Online J. 2015;21. pii:13030/qt6b83q5gp.
10. Sobanko JF, Meijer L, Nigra TP. Epithelioid sarcoma: a review and update. J Clin Aesthet Dermatol. 2009;2:49-54.
11. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
12. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
13. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
14. Lutalo PM, D’Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis)[published online January 29, 2014]. J Autoimmun. 2014;48-49:94-98.
15. Frances C, Du LT, Piette JC, et al. Wegener’s granulomatosis. dermatological manifestations in 75 cases with clinicopathologic correlation. Arch Dermatol. 1994;130:861-867.
16. Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener’s granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
17. Jennette JC. Nomenclature and classification of vasculitis: lessons learned from granulomatosis with polyangiitis (Wegener’s granulomatosis). Clin Exp Immunol. 2011;164 (suppl 1):7-10.
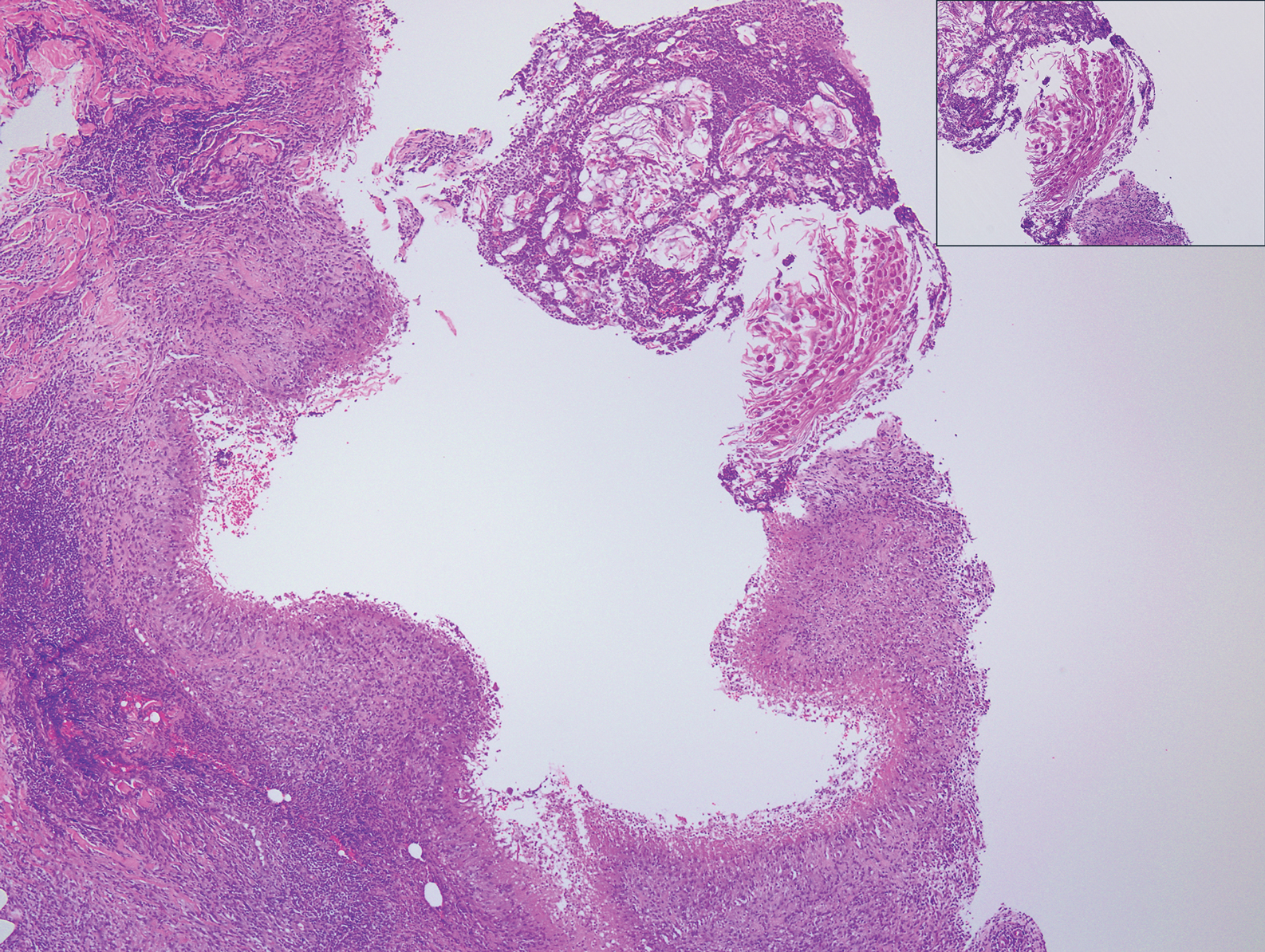
A 17-year-old adolescent girl presented with a discrete nodule on the thigh.
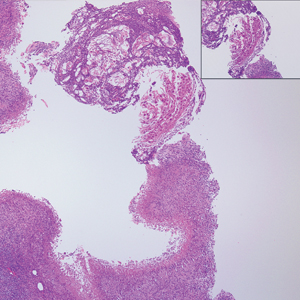
Primary Cutaneous Epstein-Barr Virus–Positive Diffuse Large B-Cell Lymphoma: A Rare and Aggressive Cutaneous Lymphoma
Cutaneous B-cell lymphomas represent a group of lymphomas derived from B lymphocytes in various stages of differentiation. The skin can be the site of primary or secondary involvement of any of the B-cell lymphomas. Primary cutaneous B-cell lymphomas present in the skin without evidence of extracutaneous disease at the time of diagnosis.1 The World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues recognizes 5 distinct primary cutaneous B-cell lymphoma subtypes: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone lymphoma; primary cutaneous diffuse large B-cell lymphoma (DLBCL), leg type; DLBCL, not otherwise specified; and intravascular DLBCL.1-3 The DLBCL, not otherwise specified, category includes less common provisional entities with insufficient evidence to be recognized as distinct diseases. Epstein-Barr virus (EBV)–positive DLBCL is a rare subtype in this group.4
This article reviews the different clinicopathologic subtypes of primary cutaneous B-cell lymphoma. It also serves to help dermatologists recognize primary cutaneous EBV-positive DLBCL as a rare and aggressive form of this disease.
Case Report
An 84-year-old white man presented with a pruritic eruption on the arms, legs, back, neck, and face of 5 months’ duration. His medical history was notable for prostate cancer that was successfully treated with radiation therapy 6 years prior. The patient denied any constitutional symptoms such as fever, chills, night sweats, or weight loss, and review of systems was negative. The patient was taking prednisone, which alleviated the pruritus, but the lesions persisted.
Physical examination revealed multiple pink to erythematous papules and subcutaneous nodules involving the face, neck, back, arms, and legs (Figure 1). No scale, crust, or ulceration was present. Palpation of the cervical, supraclavicular, axillary, and inguinal lymph nodes was negative for lymphadenopathy.
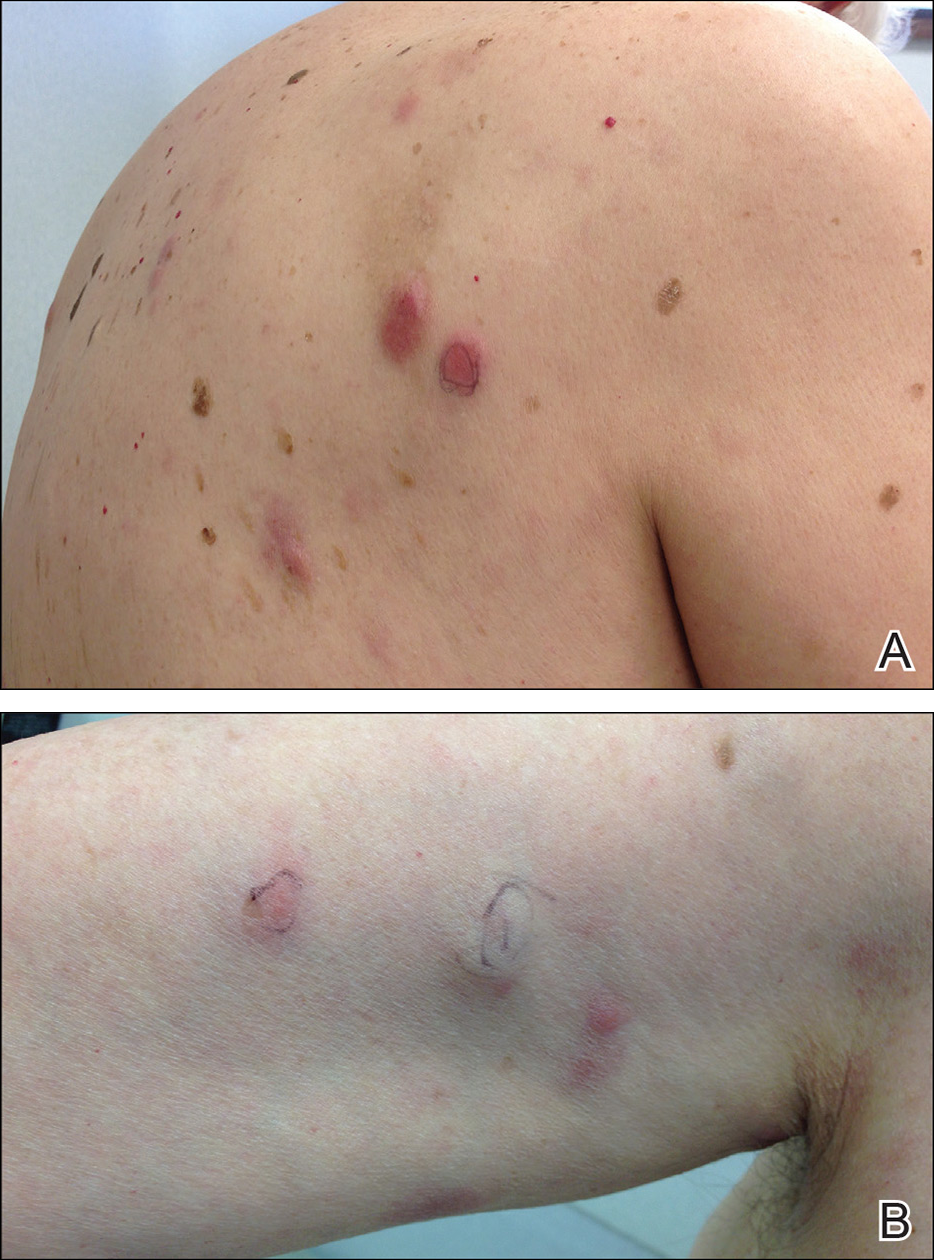
Punch biopsies of representative lesions on the upper back and right arm revealed diffuse and nodular infiltrates of large atypical lymphoid cells with scattered centroblasts and immunoblasts (Figures 2 and 3). Immunohistochemical staining demonstrated CD79, MUM-1, and EBV-encoded RNA positivity among the neoplastic cells. The Ki-67 proliferative index was greater than 90%. The neoplastic cells were negative for CD5, CD10, CD20, CD21, CD30, CD56, CD123, CD138, PAX5, C-MYC, BCL-2, BCL-6, cyclin D1, TCL-1A, and terminal deoxynucleotidyl transferase
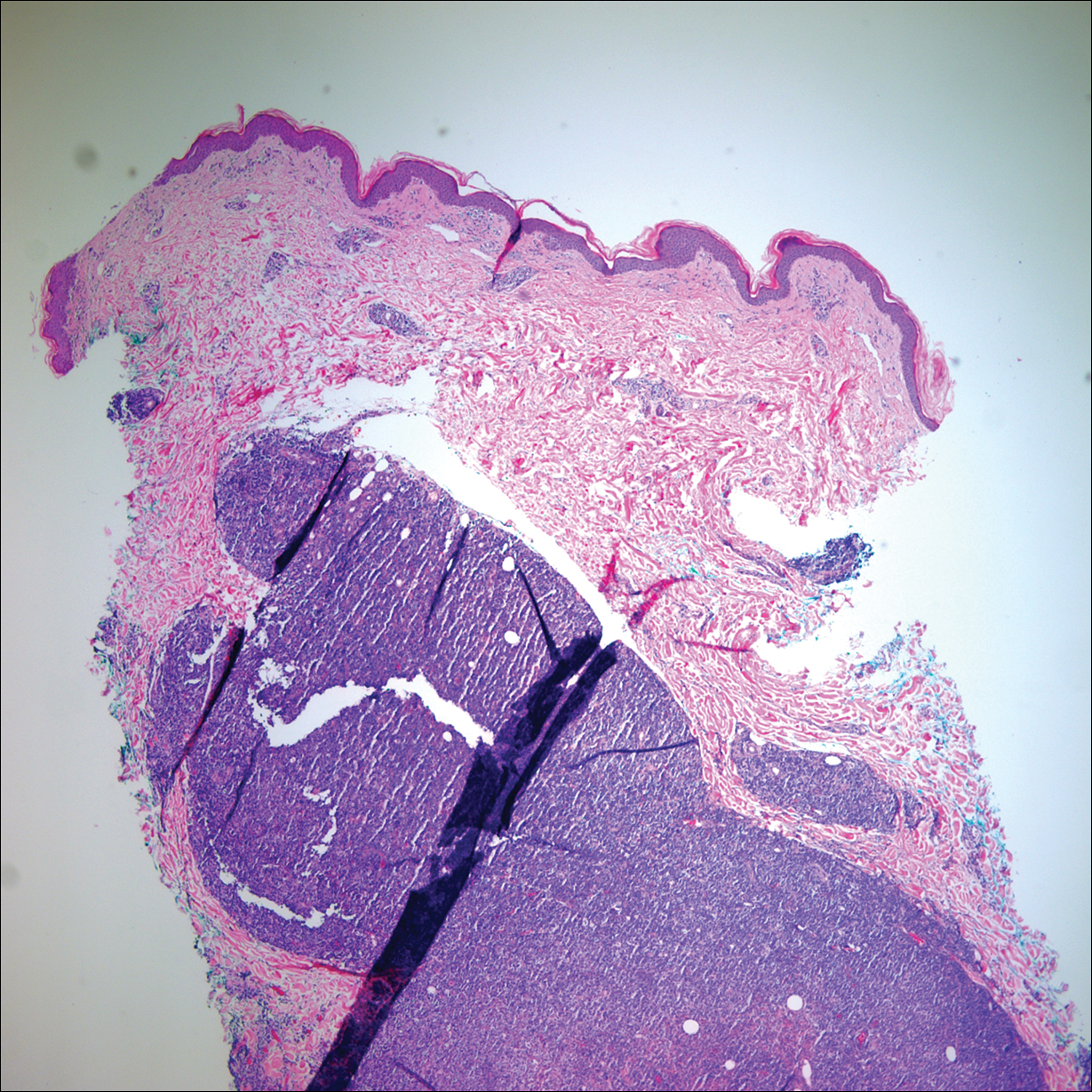
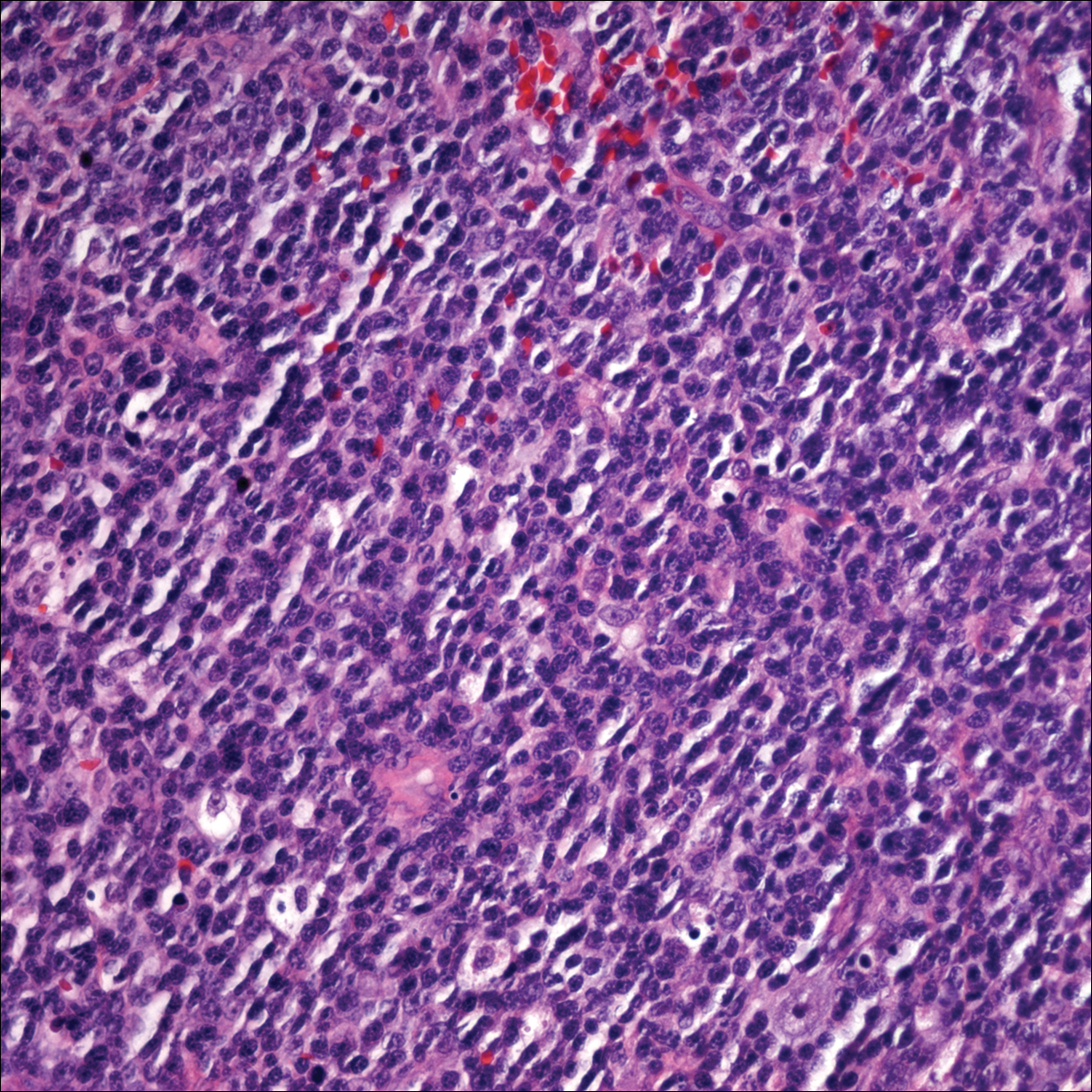
A peripheral blood smear did not show evidence of a B-cell lymphoproliferative process. A bone marrow biopsy was performed and did not show evidence of B-cell lymphoid neoplasia but did show reactive lymphoid aggregates composed of CD4+ and CD10+ T cells. Peripheral blood T-cell rearrangement and JAK2 were negative.
Based on clinical and histologic findings, the patient was diagnosed with primary cutaneous EBV-positive DLBCL. The patient was started on CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy for treatment of this aggressive cutaneous lymphoma, which initially resulted in clinical improvement of the lesions and complete involution of the subcutaneous nodules. After the sixth cycle of CHOP, he developed faintly erythematous indurated papules on the upper arms, chest, and back. Biopsy confirmed recurrence of the EBV-positive cutaneous lymphoma, and he started salvage chemotherapy with gemcitabine, oxaliplatin, and rituximab every 2 weeks; however, 4 months later (9 months after the initial presentation) he died from complications of the disease.
Comment
Etiology
Epstein-Barr virus–positive DLBCL, also called EBV-positive DLBCL of the elderly, was initially described in 2003 by Oyama et al5 and was included as a provisional entity in the 2008 World Health Organization classification system as a rare subtype of the DLBCL, not otherwise specified, category.2 It is defined as an EBV-positive monoclonal large B-cell proliferation that occurs in immunocompetent patients older than 50 years.6 Epstein-Barr virus is a human herpesvirus that demonstrates tropism for lymphocytes and survives in human hosts by establishing latency in B cells. Under normal immune conditions, the proliferation of EBV-infected B cells is prevented by cytotoxic T cells.7 It is important to recognize that patients with EBV-positive DLBCL do not have a known immunodeficiency state; therefore, it has been postulated that EBV-positive DLBCL might be caused by age-related senescence of the immune system.4,8
Epidemiology and Clinical Features
Epstein-Barr virus–positive DLBCL is more common in Asian countries than in Western countries, and there is a slight male predominance.6 A majority of patients present with extranodal disease at the time of diagnosis, and the skin is the most common extranodal site of involvement.6,9 Rare cases of primary cutaneous involvement also have been described.7,9,10 Cutaneous manifestations include erythematous papules and subcutaneous nodules. Other sites of extranodal involvement include the lungs, oral cavity, pharynx, gastrointestinal tract, and bone marrow.8,9 However, EBV-positive DLBCL is an aggressive lymphoma and prognosis is poor irrespective of the primary site of involvement.
Histopathology
Two morphologic subtypes can be seen on histology. The polymorphic pattern is characterized by a broad range of B-cell maturation with admixed reactive cells (eg, lymphocytes, histiocytes, plasma cells). The monomorphic or large-cell pattern is characterized by monotonous sheets of large transformed B cells.4,11 Many cases show both histologic patterns, and these morphologic variants do not impart any clinical or prognostic significance. Regardless of the histologic subtype, the neoplastic cells express pan B-cell antigens (eg, CD19, CD20, CD79a, PAX5), as well as MUM-1, BCL-2, and EBV-encoded RNA.4 Cases with plasmablastic features, as in our patient, may show weak or absent CD20 staining.12 Detection of EBV by in situ hybridization is required for the diagnosis.
Diagnosis
Workup for a suspected cutaneous lymphoma should include a complete history and physical examination; laboratory studies; and relevant imaging evaluation such as computed tomography of the chest, abdomen, and pelvis with or without whole-body positron emission tomography. A bone marrow biopsy and aspirate also should be performed in all cutaneous lymphomas with intermediate to aggressive clinical behavior. Accurate staging evaluation is integral to confirm the absence of extracutaneous involvement and to provide prognostic and anatomic information for the appropriate selection of treatment.13
Prognosis and Management
Primary cutaneous lymphomas tend to have different clinical behaviors and prognoses compared to histologically similar systemic lymphomas; therefore, different therapeutic strategies are warranted.14 Epstein-Barr virus–positive DLBCL has an aggressive clinical course with a median survival of 2 years.8 Patients with EBV-positive DLBCL have a poorer overall survival and treatment response when compared to patients with EBV-negative DLBCLs.4 Primary cutaneous B-cell lymphomas with indolent behavior, such as primary cutaneous marginal zone lymphoma and primary cutaneous follicle center lymphoma, can be treated with surgical excision, radiation therapy, or observation.15 No standard treatment exists for EBV-positive DLBCL, but R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone), which is the standard treatment of primary cutaneous DLBCL, leg type, may provide a survival benefit.13,15 Further studies are required to determine optimal treatment strategies.
Conclusion
Although rare, EBV-positive DLBCL is an important entity to consider when evaluating a patient with a suspected primary cutaneous lymphoma. Workup to rule out an underlying systemic lymphoma with relevant laboratory evaluation, imaging studies, and bone marrow biopsy is critical. Prognosis is poor and treatment is difficult, as standard treatment protocols have yet to be determined.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nakmura S, Jaffe ES, Swerdlow SH. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2008:243-244.
- Kempf W, Sander CA. Classification of cutaneous lymphomas—an update. Histopathology. 2010;56:57-70.
- Castillo JJ, Beltran BE, Miranda RN, et al. Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: what we know so far. Oncologist. 2011;16:87-96.
- Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16-26.
- Ok CY, Papathomas TG, Medeiros LJ, et al. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328-340.
- Tokuda Y, Fukushima M, Nakazawa K, et al. A case of primary Epstein-Barr virus-associated cutaneous diffuse large B-cell lymphoma unassociated with iatrogenic or endogenous immune dysregulation. J Cutan Pathol. 2008;35:666-671.
- Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124-5132.
- Eminger LA, Hall LD, Hesterman KS, et al. Epstein-Barr virus: dermatologic associations and implications. J Am Acad Dermatol. 2015;72:21-34.
- Martin B, Whittaker S, Morris S, et al. A case of primary cutaneous senile EBV-related diffuse large B-cell lymphoma. Am J Dermatopathol. 2010;32:190-193.
- Gibson SE, Hsi ED. Epstein-Barr virus-positive B-cell lymphoma of the elderly at a United States tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol. 2009;40:653-661.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-329.e13; quiz 341-342.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:343.e1-343.e11; quiz 355-356.
Cutaneous B-cell lymphomas represent a group of lymphomas derived from B lymphocytes in various stages of differentiation. The skin can be the site of primary or secondary involvement of any of the B-cell lymphomas. Primary cutaneous B-cell lymphomas present in the skin without evidence of extracutaneous disease at the time of diagnosis.1 The World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues recognizes 5 distinct primary cutaneous B-cell lymphoma subtypes: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone lymphoma; primary cutaneous diffuse large B-cell lymphoma (DLBCL), leg type; DLBCL, not otherwise specified; and intravascular DLBCL.1-3 The DLBCL, not otherwise specified, category includes less common provisional entities with insufficient evidence to be recognized as distinct diseases. Epstein-Barr virus (EBV)–positive DLBCL is a rare subtype in this group.4
This article reviews the different clinicopathologic subtypes of primary cutaneous B-cell lymphoma. It also serves to help dermatologists recognize primary cutaneous EBV-positive DLBCL as a rare and aggressive form of this disease.
Case Report
An 84-year-old white man presented with a pruritic eruption on the arms, legs, back, neck, and face of 5 months’ duration. His medical history was notable for prostate cancer that was successfully treated with radiation therapy 6 years prior. The patient denied any constitutional symptoms such as fever, chills, night sweats, or weight loss, and review of systems was negative. The patient was taking prednisone, which alleviated the pruritus, but the lesions persisted.
Physical examination revealed multiple pink to erythematous papules and subcutaneous nodules involving the face, neck, back, arms, and legs (Figure 1). No scale, crust, or ulceration was present. Palpation of the cervical, supraclavicular, axillary, and inguinal lymph nodes was negative for lymphadenopathy.
Punch biopsies of representative lesions on the upper back and right arm revealed diffuse and nodular infiltrates of large atypical lymphoid cells with scattered centroblasts and immunoblasts (Figures 2 and 3). Immunohistochemical staining demonstrated CD79, MUM-1, and EBV-encoded RNA positivity among the neoplastic cells. The Ki-67 proliferative index was greater than 90%. The neoplastic cells were negative for CD5, CD10, CD20, CD21, CD30, CD56, CD123, CD138, PAX5, C-MYC, BCL-2, BCL-6, cyclin D1, TCL-1A, and terminal deoxynucleotidyl transferase
A peripheral blood smear did not show evidence of a B-cell lymphoproliferative process. A bone marrow biopsy was performed and did not show evidence of B-cell lymphoid neoplasia but did show reactive lymphoid aggregates composed of CD4+ and CD10+ T cells. Peripheral blood T-cell rearrangement and JAK2 were negative.
Based on clinical and histologic findings, the patient was diagnosed with primary cutaneous EBV-positive DLBCL. The patient was started on CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy for treatment of this aggressive cutaneous lymphoma, which initially resulted in clinical improvement of the lesions and complete involution of the subcutaneous nodules. After the sixth cycle of CHOP, he developed faintly erythematous indurated papules on the upper arms, chest, and back. Biopsy confirmed recurrence of the EBV-positive cutaneous lymphoma, and he started salvage chemotherapy with gemcitabine, oxaliplatin, and rituximab every 2 weeks; however, 4 months later (9 months after the initial presentation) he died from complications of the disease.
Comment
Etiology
Epstein-Barr virus–positive DLBCL, also called EBV-positive DLBCL of the elderly, was initially described in 2003 by Oyama et al5 and was included as a provisional entity in the 2008 World Health Organization classification system as a rare subtype of the DLBCL, not otherwise specified, category.2 It is defined as an EBV-positive monoclonal large B-cell proliferation that occurs in immunocompetent patients older than 50 years.6 Epstein-Barr virus is a human herpesvirus that demonstrates tropism for lymphocytes and survives in human hosts by establishing latency in B cells. Under normal immune conditions, the proliferation of EBV-infected B cells is prevented by cytotoxic T cells.7 It is important to recognize that patients with EBV-positive DLBCL do not have a known immunodeficiency state; therefore, it has been postulated that EBV-positive DLBCL might be caused by age-related senescence of the immune system.4,8
Epidemiology and Clinical Features
Epstein-Barr virus–positive DLBCL is more common in Asian countries than in Western countries, and there is a slight male predominance.6 A majority of patients present with extranodal disease at the time of diagnosis, and the skin is the most common extranodal site of involvement.6,9 Rare cases of primary cutaneous involvement also have been described.7,9,10 Cutaneous manifestations include erythematous papules and subcutaneous nodules. Other sites of extranodal involvement include the lungs, oral cavity, pharynx, gastrointestinal tract, and bone marrow.8,9 However, EBV-positive DLBCL is an aggressive lymphoma and prognosis is poor irrespective of the primary site of involvement.
Histopathology
Two morphologic subtypes can be seen on histology. The polymorphic pattern is characterized by a broad range of B-cell maturation with admixed reactive cells (eg, lymphocytes, histiocytes, plasma cells). The monomorphic or large-cell pattern is characterized by monotonous sheets of large transformed B cells.4,11 Many cases show both histologic patterns, and these morphologic variants do not impart any clinical or prognostic significance. Regardless of the histologic subtype, the neoplastic cells express pan B-cell antigens (eg, CD19, CD20, CD79a, PAX5), as well as MUM-1, BCL-2, and EBV-encoded RNA.4 Cases with plasmablastic features, as in our patient, may show weak or absent CD20 staining.12 Detection of EBV by in situ hybridization is required for the diagnosis.
Diagnosis
Workup for a suspected cutaneous lymphoma should include a complete history and physical examination; laboratory studies; and relevant imaging evaluation such as computed tomography of the chest, abdomen, and pelvis with or without whole-body positron emission tomography. A bone marrow biopsy and aspirate also should be performed in all cutaneous lymphomas with intermediate to aggressive clinical behavior. Accurate staging evaluation is integral to confirm the absence of extracutaneous involvement and to provide prognostic and anatomic information for the appropriate selection of treatment.13
Prognosis and Management
Primary cutaneous lymphomas tend to have different clinical behaviors and prognoses compared to histologically similar systemic lymphomas; therefore, different therapeutic strategies are warranted.14 Epstein-Barr virus–positive DLBCL has an aggressive clinical course with a median survival of 2 years.8 Patients with EBV-positive DLBCL have a poorer overall survival and treatment response when compared to patients with EBV-negative DLBCLs.4 Primary cutaneous B-cell lymphomas with indolent behavior, such as primary cutaneous marginal zone lymphoma and primary cutaneous follicle center lymphoma, can be treated with surgical excision, radiation therapy, or observation.15 No standard treatment exists for EBV-positive DLBCL, but R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone), which is the standard treatment of primary cutaneous DLBCL, leg type, may provide a survival benefit.13,15 Further studies are required to determine optimal treatment strategies.
Conclusion
Although rare, EBV-positive DLBCL is an important entity to consider when evaluating a patient with a suspected primary cutaneous lymphoma. Workup to rule out an underlying systemic lymphoma with relevant laboratory evaluation, imaging studies, and bone marrow biopsy is critical. Prognosis is poor and treatment is difficult, as standard treatment protocols have yet to be determined.
Cutaneous B-cell lymphomas represent a group of lymphomas derived from B lymphocytes in various stages of differentiation. The skin can be the site of primary or secondary involvement of any of the B-cell lymphomas. Primary cutaneous B-cell lymphomas present in the skin without evidence of extracutaneous disease at the time of diagnosis.1 The World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues recognizes 5 distinct primary cutaneous B-cell lymphoma subtypes: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone lymphoma; primary cutaneous diffuse large B-cell lymphoma (DLBCL), leg type; DLBCL, not otherwise specified; and intravascular DLBCL.1-3 The DLBCL, not otherwise specified, category includes less common provisional entities with insufficient evidence to be recognized as distinct diseases. Epstein-Barr virus (EBV)–positive DLBCL is a rare subtype in this group.4
This article reviews the different clinicopathologic subtypes of primary cutaneous B-cell lymphoma. It also serves to help dermatologists recognize primary cutaneous EBV-positive DLBCL as a rare and aggressive form of this disease.
Case Report
An 84-year-old white man presented with a pruritic eruption on the arms, legs, back, neck, and face of 5 months’ duration. His medical history was notable for prostate cancer that was successfully treated with radiation therapy 6 years prior. The patient denied any constitutional symptoms such as fever, chills, night sweats, or weight loss, and review of systems was negative. The patient was taking prednisone, which alleviated the pruritus, but the lesions persisted.
Physical examination revealed multiple pink to erythematous papules and subcutaneous nodules involving the face, neck, back, arms, and legs (Figure 1). No scale, crust, or ulceration was present. Palpation of the cervical, supraclavicular, axillary, and inguinal lymph nodes was negative for lymphadenopathy.
Punch biopsies of representative lesions on the upper back and right arm revealed diffuse and nodular infiltrates of large atypical lymphoid cells with scattered centroblasts and immunoblasts (Figures 2 and 3). Immunohistochemical staining demonstrated CD79, MUM-1, and EBV-encoded RNA positivity among the neoplastic cells. The Ki-67 proliferative index was greater than 90%. The neoplastic cells were negative for CD5, CD10, CD20, CD21, CD30, CD56, CD123, CD138, PAX5, C-MYC, BCL-2, BCL-6, cyclin D1, TCL-1A, and terminal deoxynucleotidyl transferase
A peripheral blood smear did not show evidence of a B-cell lymphoproliferative process. A bone marrow biopsy was performed and did not show evidence of B-cell lymphoid neoplasia but did show reactive lymphoid aggregates composed of CD4+ and CD10+ T cells. Peripheral blood T-cell rearrangement and JAK2 were negative.
Based on clinical and histologic findings, the patient was diagnosed with primary cutaneous EBV-positive DLBCL. The patient was started on CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy for treatment of this aggressive cutaneous lymphoma, which initially resulted in clinical improvement of the lesions and complete involution of the subcutaneous nodules. After the sixth cycle of CHOP, he developed faintly erythematous indurated papules on the upper arms, chest, and back. Biopsy confirmed recurrence of the EBV-positive cutaneous lymphoma, and he started salvage chemotherapy with gemcitabine, oxaliplatin, and rituximab every 2 weeks; however, 4 months later (9 months after the initial presentation) he died from complications of the disease.
Comment
Etiology
Epstein-Barr virus–positive DLBCL, also called EBV-positive DLBCL of the elderly, was initially described in 2003 by Oyama et al5 and was included as a provisional entity in the 2008 World Health Organization classification system as a rare subtype of the DLBCL, not otherwise specified, category.2 It is defined as an EBV-positive monoclonal large B-cell proliferation that occurs in immunocompetent patients older than 50 years.6 Epstein-Barr virus is a human herpesvirus that demonstrates tropism for lymphocytes and survives in human hosts by establishing latency in B cells. Under normal immune conditions, the proliferation of EBV-infected B cells is prevented by cytotoxic T cells.7 It is important to recognize that patients with EBV-positive DLBCL do not have a known immunodeficiency state; therefore, it has been postulated that EBV-positive DLBCL might be caused by age-related senescence of the immune system.4,8
Epidemiology and Clinical Features
Epstein-Barr virus–positive DLBCL is more common in Asian countries than in Western countries, and there is a slight male predominance.6 A majority of patients present with extranodal disease at the time of diagnosis, and the skin is the most common extranodal site of involvement.6,9 Rare cases of primary cutaneous involvement also have been described.7,9,10 Cutaneous manifestations include erythematous papules and subcutaneous nodules. Other sites of extranodal involvement include the lungs, oral cavity, pharynx, gastrointestinal tract, and bone marrow.8,9 However, EBV-positive DLBCL is an aggressive lymphoma and prognosis is poor irrespective of the primary site of involvement.
Histopathology
Two morphologic subtypes can be seen on histology. The polymorphic pattern is characterized by a broad range of B-cell maturation with admixed reactive cells (eg, lymphocytes, histiocytes, plasma cells). The monomorphic or large-cell pattern is characterized by monotonous sheets of large transformed B cells.4,11 Many cases show both histologic patterns, and these morphologic variants do not impart any clinical or prognostic significance. Regardless of the histologic subtype, the neoplastic cells express pan B-cell antigens (eg, CD19, CD20, CD79a, PAX5), as well as MUM-1, BCL-2, and EBV-encoded RNA.4 Cases with plasmablastic features, as in our patient, may show weak or absent CD20 staining.12 Detection of EBV by in situ hybridization is required for the diagnosis.
Diagnosis
Workup for a suspected cutaneous lymphoma should include a complete history and physical examination; laboratory studies; and relevant imaging evaluation such as computed tomography of the chest, abdomen, and pelvis with or without whole-body positron emission tomography. A bone marrow biopsy and aspirate also should be performed in all cutaneous lymphomas with intermediate to aggressive clinical behavior. Accurate staging evaluation is integral to confirm the absence of extracutaneous involvement and to provide prognostic and anatomic information for the appropriate selection of treatment.13
Prognosis and Management
Primary cutaneous lymphomas tend to have different clinical behaviors and prognoses compared to histologically similar systemic lymphomas; therefore, different therapeutic strategies are warranted.14 Epstein-Barr virus–positive DLBCL has an aggressive clinical course with a median survival of 2 years.8 Patients with EBV-positive DLBCL have a poorer overall survival and treatment response when compared to patients with EBV-negative DLBCLs.4 Primary cutaneous B-cell lymphomas with indolent behavior, such as primary cutaneous marginal zone lymphoma and primary cutaneous follicle center lymphoma, can be treated with surgical excision, radiation therapy, or observation.15 No standard treatment exists for EBV-positive DLBCL, but R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone), which is the standard treatment of primary cutaneous DLBCL, leg type, may provide a survival benefit.13,15 Further studies are required to determine optimal treatment strategies.
Conclusion
Although rare, EBV-positive DLBCL is an important entity to consider when evaluating a patient with a suspected primary cutaneous lymphoma. Workup to rule out an underlying systemic lymphoma with relevant laboratory evaluation, imaging studies, and bone marrow biopsy is critical. Prognosis is poor and treatment is difficult, as standard treatment protocols have yet to be determined.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nakmura S, Jaffe ES, Swerdlow SH. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2008:243-244.
- Kempf W, Sander CA. Classification of cutaneous lymphomas—an update. Histopathology. 2010;56:57-70.
- Castillo JJ, Beltran BE, Miranda RN, et al. Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: what we know so far. Oncologist. 2011;16:87-96.
- Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16-26.
- Ok CY, Papathomas TG, Medeiros LJ, et al. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328-340.
- Tokuda Y, Fukushima M, Nakazawa K, et al. A case of primary Epstein-Barr virus-associated cutaneous diffuse large B-cell lymphoma unassociated with iatrogenic or endogenous immune dysregulation. J Cutan Pathol. 2008;35:666-671.
- Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124-5132.
- Eminger LA, Hall LD, Hesterman KS, et al. Epstein-Barr virus: dermatologic associations and implications. J Am Acad Dermatol. 2015;72:21-34.
- Martin B, Whittaker S, Morris S, et al. A case of primary cutaneous senile EBV-related diffuse large B-cell lymphoma. Am J Dermatopathol. 2010;32:190-193.
- Gibson SE, Hsi ED. Epstein-Barr virus-positive B-cell lymphoma of the elderly at a United States tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol. 2009;40:653-661.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-329.e13; quiz 341-342.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:343.e1-343.e11; quiz 355-356.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nakmura S, Jaffe ES, Swerdlow SH. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2008:243-244.
- Kempf W, Sander CA. Classification of cutaneous lymphomas—an update. Histopathology. 2010;56:57-70.
- Castillo JJ, Beltran BE, Miranda RN, et al. Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: what we know so far. Oncologist. 2011;16:87-96.
- Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16-26.
- Ok CY, Papathomas TG, Medeiros LJ, et al. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328-340.
- Tokuda Y, Fukushima M, Nakazawa K, et al. A case of primary Epstein-Barr virus-associated cutaneous diffuse large B-cell lymphoma unassociated with iatrogenic or endogenous immune dysregulation. J Cutan Pathol. 2008;35:666-671.
- Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124-5132.
- Eminger LA, Hall LD, Hesterman KS, et al. Epstein-Barr virus: dermatologic associations and implications. J Am Acad Dermatol. 2015;72:21-34.
- Martin B, Whittaker S, Morris S, et al. A case of primary cutaneous senile EBV-related diffuse large B-cell lymphoma. Am J Dermatopathol. 2010;32:190-193.
- Gibson SE, Hsi ED. Epstein-Barr virus-positive B-cell lymphoma of the elderly at a United States tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol. 2009;40:653-661.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-329.e13; quiz 341-342.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:343.e1-343.e11; quiz 355-356.
Practice Points
- Primary cutaneous lymphomas are malignant lymphomas confined to the skin.
- Complete staging workup is necessary to rule out secondary involvement of the skin from a nodal lymphoma.
- Epstein-Barr virus-positive diffuse large B-cell lymphoma is a rare and aggressive primary cutaneous lymphoma.
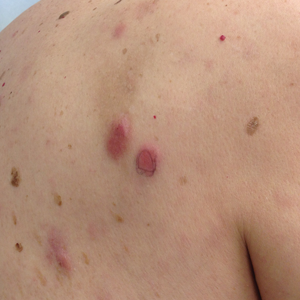
Diffuse Pustular Eruption Following Computed Tomography
The Diagnosis: Acute Generalized Exanthematous Pustulosis
Histopathology demonstrated spongiosis with subcorneal pustules and an overlying basket-weave pattern stratum corneum. There was mild papillary dermal edema with scattered dermal neutrophils and rare eosinophils (Figure). The patient's clinical presentation and histopathology were consistent with acute generalized exanthematous pustulosis (AGEP). The inciting agent in this case was the contrast medium iopamidol. The patient was treated with a short course of prednisone, triamcinolone cream, diphenhydramine, and acetaminophen. Within 1 week the pustules and erythema had resolved.
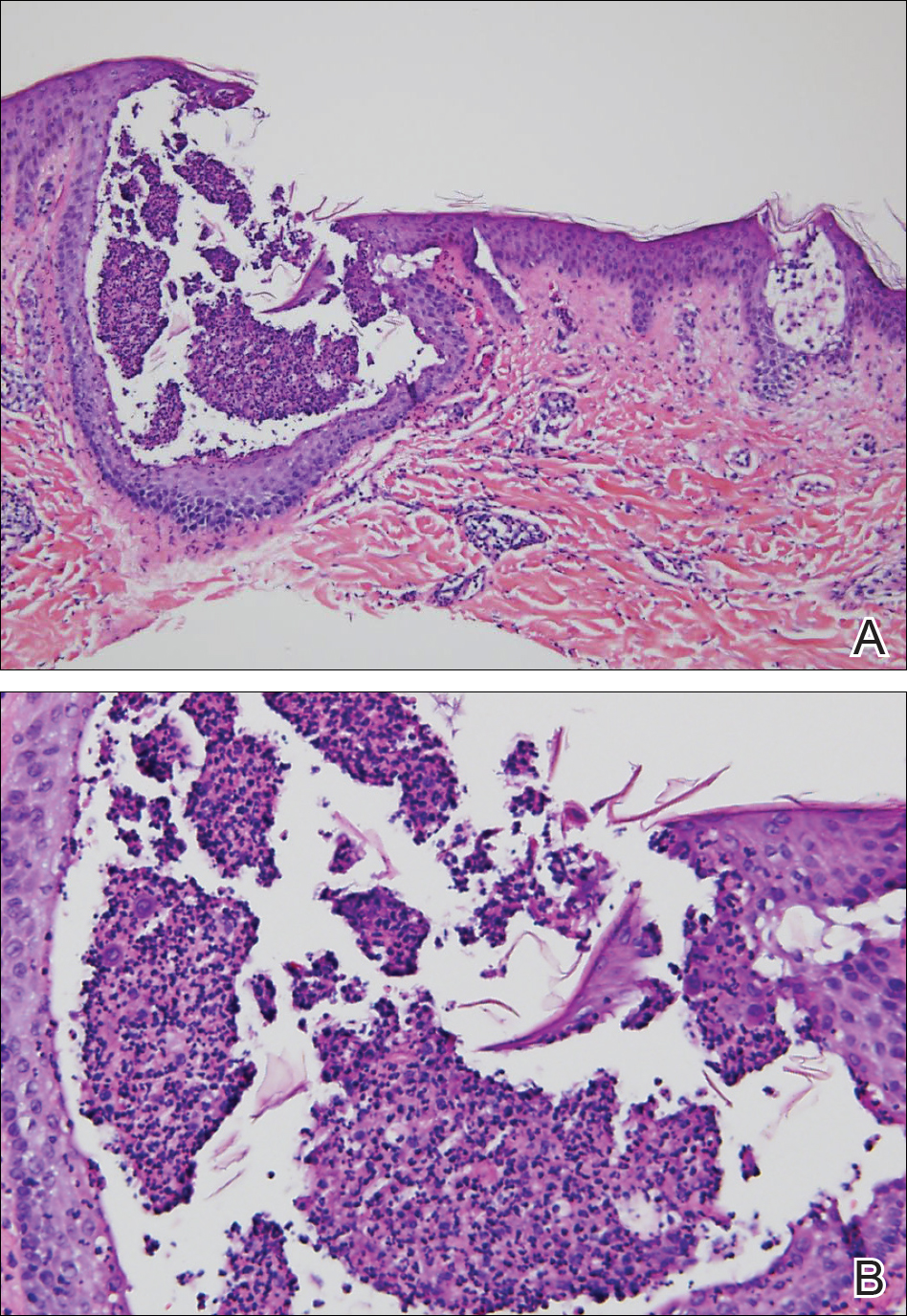
Acute generalized exanthematous pustulosis is an uncommon T cell-mediated cutaneous reaction characterized by widespread progressive erythema with numerous nonfollicular pinpoint pustules. The patient usually is well appearing; however, he/she often will have concurrent fever and facial edema. Mucous membranes rarely are involved. Laboratory results typically are notable only for leukocytosis with neutrophilia.
The pustular eruption typically occurs within 1 to 2 days after exposure to an inciting agent1; however, this latent period can range from 1 hour to nearly 4 weeks in some studies.2 Systemic medications are the cause in approximately 90% of cases, with antibiotics being the most common category. Frequently implicated medications include β-lactams, macrolides, quinolones, sulfonamides, proton pump inhibitors, hydroxychloroquine, terbinafine, nonsteroidal anti-inflammatory drugs, diltiazem, ketoconazole, and fluconazole. Acute generalized exanthematous pustulosis also has been rarely reported following contact with mercury, viral and bacterial infections, and spider bites.3
Iodinated contrast agents have long been known to cause immediate and delayed adverse cutaneous reactions. However, one consensus study indicated that these reactions occur in only 0.05% to 0.10% of patients.4 Although rare, iodinated contrast media (eg, iopamidol, iohexol, ioversol, iodixanol, iomeprol, iobitridol, iopromide) have been reported as a cause of AGEP. A PubMed search of articles indexed for MEDLINE using the terms acute generalized exanthematous pustulosis, contrast, iodine, and iodinated revealed 10 adult cases reported in 6 articles in the English-language literature.1,5-9 The most recent articles focus on methods to identify the causative agent. If the etiology of the reaction is unclear, patch or intradermal testing can help to confirm the causative agent. These tests also can help determine similar agents to which the patient may cross-react.4,5
It can be difficult to differentiate AGEP from other cutaneous drug reactions and other nonfollicular pustular conditions. Drug-induced hypersensitivity syndrome typically presents with facial edema and a morbilliform rash. Although it can present with pustules, the latent period is longer (2-6 weeks), and there frequently are signs of multiorgan involvement including hepatic dysfunction, eosinophilia, atypical lymphocytosis, and lymphadenopathy. Patients with generalized pustular psoriasis often have a history of plaque psoriasis; the pustules are more concentrated in flexural sites; the eruption is gradual in onset; and histologically there tends to be features of psoriasis including parakeratosis, Munro microabscesses, and dilated blood vessels.10 Subcorneal pustular dermatosis also is more concentrated in flexural sites and frequently has an annular or serpiginous configuration. The onset also is gradual, and it follows a more chronic course than AGEP. Exfoliative erythroderma presents with widespread erythema and superficial desquamating scale. It often occurs in association with systemic symptoms and can be the result of a drug reaction or underlying inflammatory dermatosis such as psoriasis, mycosis fungoides, or pityriasis rubra pilaris.
Acute generalized exanthematous pustulosis usually resolves spontaneously within 2 weeks and is associated with a superficial desquamation as it clears. Appropriate treatment includes discontinuing the offending agent; monitoring for systemic involvement; and treating the patient's symptoms with antihistamines, analgesics, topical steroids, and emollients. In more severe or persistent cases, treatment with systemic steroids and tumor necrosis factor α inhibitors has been attempted, though their efficacy remains unclear. We report a case of iopamidol-induced AGEP that highlights the importance of eliciting a history of contrast exposure from a patient with suspected AGEP.
- Hammerbeck AA, Daniels NH, Callen JP. Ioversol-induced acute generalized exanthematous pustulosis. Arch Dermatol. 2009;145:683-687.
- Thienvibul C, Vachiramon V, Chanprapaph K. Five-year retrospective review of acute generalized exanthematous pustulosis. Dermatol Res Pract. 2015;2015:1-8.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2016;73:843-848.
- Rosado Ingelmo A, Doña Diaz I, Cabañas Moreno R, et al. Clinical practice guidelines for diagnosis and management of hypersensitivity reactions to contrast media. J Investig Allergol Clin Immunol. 2016;26:144-155.
- Grandvuillemin A, Ripert C, Sgro C, et al. Iodinated contrast media-induced acute generalized exanthematous pustulosis confirmed by delayed skin tests. J Allergy Clin Immunol Pract. 2014;2:805-806.
- Bavbek S, Sözener ZÇ, Aydin Ö, et al. First case report of acute generalized exanthematous pustulosis due to intravenous iopromide. J Investig Allergol Clin Immunol. 2014;24:66-67.
- Kim SJ, Lee T, Lee YS, et al. Acute generalized exanthematous pustulosis caused by radiocontrast media. Ann Allergy Asthma Immunol. 2010;105:492-493.
- Peterson A, Katzberg RW, Fung MA, et al. Acute generalized exanthematous pustulosis as a delayed dermatotoxic reaction to IV-administered nonionic contrast media. Am J Roentgenol. 2006;187:198-201.
- Atasoy M, Erdem T, Sari RA. A case of acute generalized exanthematous pustulosis (AGEP) possibly induced by iohexol. J Dermatol. 2003;30:723-726.
- Halevy S, Kardaun S, Davidovici B, et al; EuroSCAR and RegiSCAR Study Group. The spectrum of histopathological features in acute generalized exanthematous pustulosis: a study of 102 cases. Br J Dermatol. 2010:163:1245-1252.
The Diagnosis: Acute Generalized Exanthematous Pustulosis
Histopathology demonstrated spongiosis with subcorneal pustules and an overlying basket-weave pattern stratum corneum. There was mild papillary dermal edema with scattered dermal neutrophils and rare eosinophils (Figure). The patient's clinical presentation and histopathology were consistent with acute generalized exanthematous pustulosis (AGEP). The inciting agent in this case was the contrast medium iopamidol. The patient was treated with a short course of prednisone, triamcinolone cream, diphenhydramine, and acetaminophen. Within 1 week the pustules and erythema had resolved.
Acute generalized exanthematous pustulosis is an uncommon T cell-mediated cutaneous reaction characterized by widespread progressive erythema with numerous nonfollicular pinpoint pustules. The patient usually is well appearing; however, he/she often will have concurrent fever and facial edema. Mucous membranes rarely are involved. Laboratory results typically are notable only for leukocytosis with neutrophilia.
The pustular eruption typically occurs within 1 to 2 days after exposure to an inciting agent1; however, this latent period can range from 1 hour to nearly 4 weeks in some studies.2 Systemic medications are the cause in approximately 90% of cases, with antibiotics being the most common category. Frequently implicated medications include β-lactams, macrolides, quinolones, sulfonamides, proton pump inhibitors, hydroxychloroquine, terbinafine, nonsteroidal anti-inflammatory drugs, diltiazem, ketoconazole, and fluconazole. Acute generalized exanthematous pustulosis also has been rarely reported following contact with mercury, viral and bacterial infections, and spider bites.3
Iodinated contrast agents have long been known to cause immediate and delayed adverse cutaneous reactions. However, one consensus study indicated that these reactions occur in only 0.05% to 0.10% of patients.4 Although rare, iodinated contrast media (eg, iopamidol, iohexol, ioversol, iodixanol, iomeprol, iobitridol, iopromide) have been reported as a cause of AGEP. A PubMed search of articles indexed for MEDLINE using the terms acute generalized exanthematous pustulosis, contrast, iodine, and iodinated revealed 10 adult cases reported in 6 articles in the English-language literature.1,5-9 The most recent articles focus on methods to identify the causative agent. If the etiology of the reaction is unclear, patch or intradermal testing can help to confirm the causative agent. These tests also can help determine similar agents to which the patient may cross-react.4,5
It can be difficult to differentiate AGEP from other cutaneous drug reactions and other nonfollicular pustular conditions. Drug-induced hypersensitivity syndrome typically presents with facial edema and a morbilliform rash. Although it can present with pustules, the latent period is longer (2-6 weeks), and there frequently are signs of multiorgan involvement including hepatic dysfunction, eosinophilia, atypical lymphocytosis, and lymphadenopathy. Patients with generalized pustular psoriasis often have a history of plaque psoriasis; the pustules are more concentrated in flexural sites; the eruption is gradual in onset; and histologically there tends to be features of psoriasis including parakeratosis, Munro microabscesses, and dilated blood vessels.10 Subcorneal pustular dermatosis also is more concentrated in flexural sites and frequently has an annular or serpiginous configuration. The onset also is gradual, and it follows a more chronic course than AGEP. Exfoliative erythroderma presents with widespread erythema and superficial desquamating scale. It often occurs in association with systemic symptoms and can be the result of a drug reaction or underlying inflammatory dermatosis such as psoriasis, mycosis fungoides, or pityriasis rubra pilaris.
Acute generalized exanthematous pustulosis usually resolves spontaneously within 2 weeks and is associated with a superficial desquamation as it clears. Appropriate treatment includes discontinuing the offending agent; monitoring for systemic involvement; and treating the patient's symptoms with antihistamines, analgesics, topical steroids, and emollients. In more severe or persistent cases, treatment with systemic steroids and tumor necrosis factor α inhibitors has been attempted, though their efficacy remains unclear. We report a case of iopamidol-induced AGEP that highlights the importance of eliciting a history of contrast exposure from a patient with suspected AGEP.
The Diagnosis: Acute Generalized Exanthematous Pustulosis
Histopathology demonstrated spongiosis with subcorneal pustules and an overlying basket-weave pattern stratum corneum. There was mild papillary dermal edema with scattered dermal neutrophils and rare eosinophils (Figure). The patient's clinical presentation and histopathology were consistent with acute generalized exanthematous pustulosis (AGEP). The inciting agent in this case was the contrast medium iopamidol. The patient was treated with a short course of prednisone, triamcinolone cream, diphenhydramine, and acetaminophen. Within 1 week the pustules and erythema had resolved.
Acute generalized exanthematous pustulosis is an uncommon T cell-mediated cutaneous reaction characterized by widespread progressive erythema with numerous nonfollicular pinpoint pustules. The patient usually is well appearing; however, he/she often will have concurrent fever and facial edema. Mucous membranes rarely are involved. Laboratory results typically are notable only for leukocytosis with neutrophilia.
The pustular eruption typically occurs within 1 to 2 days after exposure to an inciting agent1; however, this latent period can range from 1 hour to nearly 4 weeks in some studies.2 Systemic medications are the cause in approximately 90% of cases, with antibiotics being the most common category. Frequently implicated medications include β-lactams, macrolides, quinolones, sulfonamides, proton pump inhibitors, hydroxychloroquine, terbinafine, nonsteroidal anti-inflammatory drugs, diltiazem, ketoconazole, and fluconazole. Acute generalized exanthematous pustulosis also has been rarely reported following contact with mercury, viral and bacterial infections, and spider bites.3
Iodinated contrast agents have long been known to cause immediate and delayed adverse cutaneous reactions. However, one consensus study indicated that these reactions occur in only 0.05% to 0.10% of patients.4 Although rare, iodinated contrast media (eg, iopamidol, iohexol, ioversol, iodixanol, iomeprol, iobitridol, iopromide) have been reported as a cause of AGEP. A PubMed search of articles indexed for MEDLINE using the terms acute generalized exanthematous pustulosis, contrast, iodine, and iodinated revealed 10 adult cases reported in 6 articles in the English-language literature.1,5-9 The most recent articles focus on methods to identify the causative agent. If the etiology of the reaction is unclear, patch or intradermal testing can help to confirm the causative agent. These tests also can help determine similar agents to which the patient may cross-react.4,5
It can be difficult to differentiate AGEP from other cutaneous drug reactions and other nonfollicular pustular conditions. Drug-induced hypersensitivity syndrome typically presents with facial edema and a morbilliform rash. Although it can present with pustules, the latent period is longer (2-6 weeks), and there frequently are signs of multiorgan involvement including hepatic dysfunction, eosinophilia, atypical lymphocytosis, and lymphadenopathy. Patients with generalized pustular psoriasis often have a history of plaque psoriasis; the pustules are more concentrated in flexural sites; the eruption is gradual in onset; and histologically there tends to be features of psoriasis including parakeratosis, Munro microabscesses, and dilated blood vessels.10 Subcorneal pustular dermatosis also is more concentrated in flexural sites and frequently has an annular or serpiginous configuration. The onset also is gradual, and it follows a more chronic course than AGEP. Exfoliative erythroderma presents with widespread erythema and superficial desquamating scale. It often occurs in association with systemic symptoms and can be the result of a drug reaction or underlying inflammatory dermatosis such as psoriasis, mycosis fungoides, or pityriasis rubra pilaris.
Acute generalized exanthematous pustulosis usually resolves spontaneously within 2 weeks and is associated with a superficial desquamation as it clears. Appropriate treatment includes discontinuing the offending agent; monitoring for systemic involvement; and treating the patient's symptoms with antihistamines, analgesics, topical steroids, and emollients. In more severe or persistent cases, treatment with systemic steroids and tumor necrosis factor α inhibitors has been attempted, though their efficacy remains unclear. We report a case of iopamidol-induced AGEP that highlights the importance of eliciting a history of contrast exposure from a patient with suspected AGEP.
- Hammerbeck AA, Daniels NH, Callen JP. Ioversol-induced acute generalized exanthematous pustulosis. Arch Dermatol. 2009;145:683-687.
- Thienvibul C, Vachiramon V, Chanprapaph K. Five-year retrospective review of acute generalized exanthematous pustulosis. Dermatol Res Pract. 2015;2015:1-8.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2016;73:843-848.
- Rosado Ingelmo A, Doña Diaz I, Cabañas Moreno R, et al. Clinical practice guidelines for diagnosis and management of hypersensitivity reactions to contrast media. J Investig Allergol Clin Immunol. 2016;26:144-155.
- Grandvuillemin A, Ripert C, Sgro C, et al. Iodinated contrast media-induced acute generalized exanthematous pustulosis confirmed by delayed skin tests. J Allergy Clin Immunol Pract. 2014;2:805-806.
- Bavbek S, Sözener ZÇ, Aydin Ö, et al. First case report of acute generalized exanthematous pustulosis due to intravenous iopromide. J Investig Allergol Clin Immunol. 2014;24:66-67.
- Kim SJ, Lee T, Lee YS, et al. Acute generalized exanthematous pustulosis caused by radiocontrast media. Ann Allergy Asthma Immunol. 2010;105:492-493.
- Peterson A, Katzberg RW, Fung MA, et al. Acute generalized exanthematous pustulosis as a delayed dermatotoxic reaction to IV-administered nonionic contrast media. Am J Roentgenol. 2006;187:198-201.
- Atasoy M, Erdem T, Sari RA. A case of acute generalized exanthematous pustulosis (AGEP) possibly induced by iohexol. J Dermatol. 2003;30:723-726.
- Halevy S, Kardaun S, Davidovici B, et al; EuroSCAR and RegiSCAR Study Group. The spectrum of histopathological features in acute generalized exanthematous pustulosis: a study of 102 cases. Br J Dermatol. 2010:163:1245-1252.
- Hammerbeck AA, Daniels NH, Callen JP. Ioversol-induced acute generalized exanthematous pustulosis. Arch Dermatol. 2009;145:683-687.
- Thienvibul C, Vachiramon V, Chanprapaph K. Five-year retrospective review of acute generalized exanthematous pustulosis. Dermatol Res Pract. 2015;2015:1-8.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2016;73:843-848.
- Rosado Ingelmo A, Doña Diaz I, Cabañas Moreno R, et al. Clinical practice guidelines for diagnosis and management of hypersensitivity reactions to contrast media. J Investig Allergol Clin Immunol. 2016;26:144-155.
- Grandvuillemin A, Ripert C, Sgro C, et al. Iodinated contrast media-induced acute generalized exanthematous pustulosis confirmed by delayed skin tests. J Allergy Clin Immunol Pract. 2014;2:805-806.
- Bavbek S, Sözener ZÇ, Aydin Ö, et al. First case report of acute generalized exanthematous pustulosis due to intravenous iopromide. J Investig Allergol Clin Immunol. 2014;24:66-67.
- Kim SJ, Lee T, Lee YS, et al. Acute generalized exanthematous pustulosis caused by radiocontrast media. Ann Allergy Asthma Immunol. 2010;105:492-493.
- Peterson A, Katzberg RW, Fung MA, et al. Acute generalized exanthematous pustulosis as a delayed dermatotoxic reaction to IV-administered nonionic contrast media. Am J Roentgenol. 2006;187:198-201.
- Atasoy M, Erdem T, Sari RA. A case of acute generalized exanthematous pustulosis (AGEP) possibly induced by iohexol. J Dermatol. 2003;30:723-726.
- Halevy S, Kardaun S, Davidovici B, et al; EuroSCAR and RegiSCAR Study Group. The spectrum of histopathological features in acute generalized exanthematous pustulosis: a study of 102 cases. Br J Dermatol. 2010:163:1245-1252.
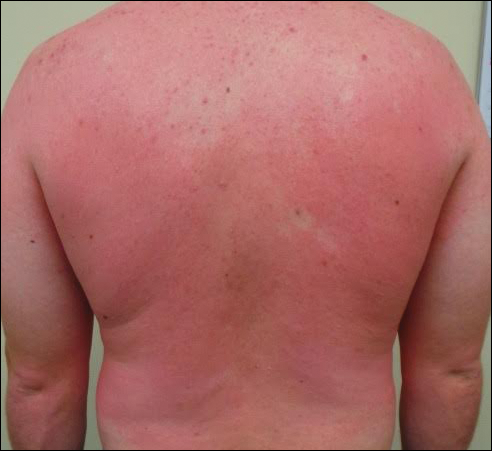
A 31-year-old man presented with a rapidly progressive, burning rash of 1 day's duration, along with malaise, nausea, and dizziness. At the time of presentation, he was hemodynamically stable and afebrile. Laboratory analysis revealed mild leukocytosis with neutrophilia. A complete metabolic panel was within normal limits. He had no chronic medical conditions and was taking no medications or supplements. One day prior to onset of the rash, he underwent contrast-enhanced (iopamidol) computed tomography of the abdomen. Physical examination revealed large edematous plaques on the face, neck, and trunk (top) that were studded with numerous pinpoint pustules (bottom). He also had subtle facial edema. There was relative sparing of the flexural sites and no involvement of the palms, soles, or mucous membranes. A shave biopsy was obtained from a pustular area on the neck.
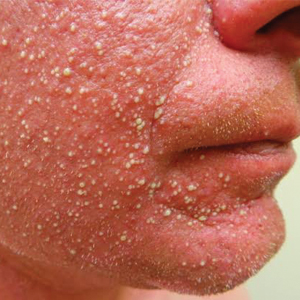
Concomitant Fibrofolliculoma and Trichodiscoma on the Abdomen
Fibrofolliculomas and trichodiscomas typically present on the head or neck as smooth, flesh-colored, dome-shaped papules. These two entities are considered to constitute two separate time points on a spectrum of histopathologic changes in mantleoma differentiation.1 Histologically, both are benign hamartomas of the pilosebaceous subunit and collectively are known as mantleomas. We present an unusual case of a concomitant fibrofolliculoma and trichodiscoma on the abdomen.
Case Report
An asymptomatic 54-year-old man presented for a routine full-body skin examination. A solitary, 2×1-cm, subcutaneous, doughy, mobile nodule was found on the left side of the abdomen with an overlying 2-mm yellow fleshy papule. The patient declined excision of the lesion, and it was recommended that he return for follow-up 3 months later.
The patient did not present for follow-up until 4.5 years later, at which point the lesion had grown to 3.0×2.5 cm in size. An excision was performed, at which time the lesion was noted to be cystic, extruding an oily, yellow-white liquid. Bacterial culture was negative. Histopathologic sections showed a dome-shaped papule with connection to the overlying epidermis. Epithelial extensions from the infundibular epithelium formed a fenestrated pattern surrounding a fibrous and mucinous stroma (Figure, A and B). The differential diagnosis at this time included an epidermal inclusion cyst, fibroma, intradermal nevus, verruca, hemangioma, angiofibroma, and lipoma.2-4
The same lesion cut in a different plane of sectioning showed an expansile dermal nodule comprising clusters of sebaceous lobules surrounding a fibrous and mucinous stroma. Within the second lesion, fibrous and stromal components predominated over epithelial components (Figure, C). A diagnosis of fibrofolliculoma showing features of a trichodiscoma arising in the unusual location of the abdomen was made.
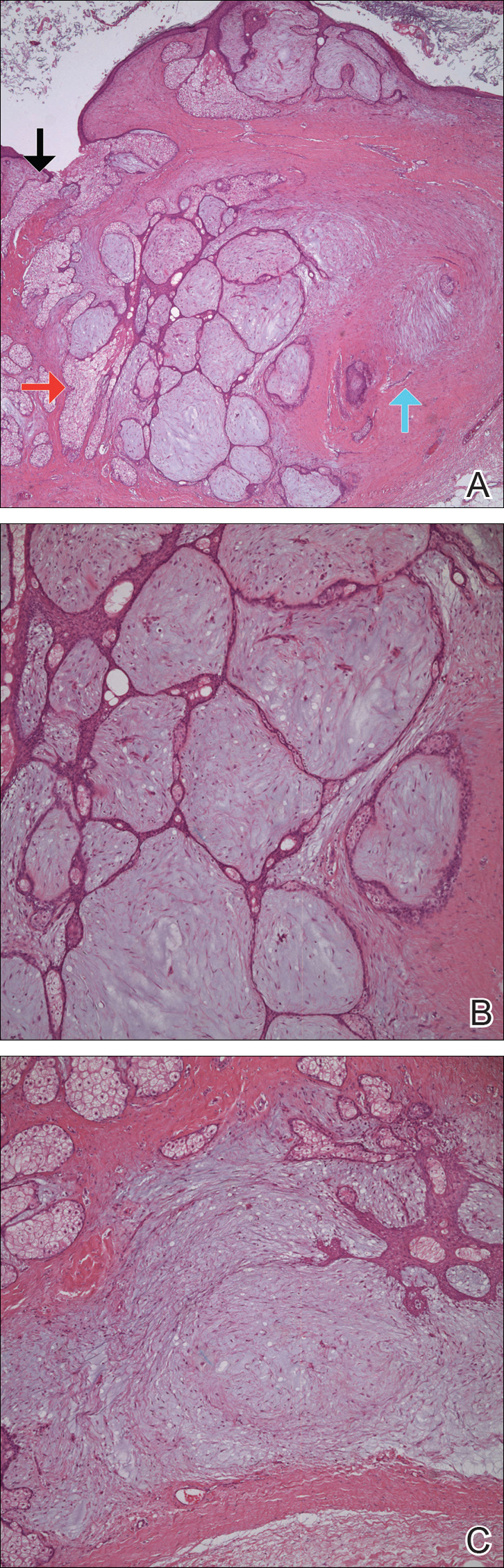
Comment
Solitary fibrofolliculomas and trichodiscomas are flesh-colored, dome-shaped papules that generally present on the face, specifically on the chin, nose, cheeks, ears, and eyebrows without considerable symptoms.2,4,5 Clinically, fibrofolliculomas are indistinguishable from trichodiscomas but demonstrate different features on biopsy.1,5
Fibrofolliculomas and trichodiscomas are well known for their association with Birt-Hogg-Dubé (BHD) syndrome when they present concomitantly and typically arise earlier in the third decade of life than solitary fibrofolliculomas; however, there have been reports of solitary fibrofolliculomas in patients aged 1 to 36 years.4,6 The triad of BHD syndrome consists of multiple fibrofolliculomas, trichodiscomas, and acrochordons, and it is acquired in an autosomal-dominant manner, unlike solitary fibrofolliculomas, which typically are not inherited. Birt-Hogg-Dubé syndrome is caused by a mutation in the FLCN gene that codes for the tumor-suppressor protein folliculin, which when mutated can cause unregulated proliferation of cells.7 Solitary fibrofolliculomas and the multiple fibrofolliculomas seen in BHD syndrome are histologically similar.
Fibrofolliculoma can be clinically indistinguishable from fibroepithelioma of Pinkus, perifollicular fibroma, trichilemmoma, trichodiscoma, trichoepithelioma, and trichofolliculoma. All typically present clinically as flesh-colored papules,1 although histologic distinction can be made (Table).5,8-13
Fibrofolliculoma is a benign hamartoma that arises from the pilosebaceous follicle and consists of an expansion of the fibrous root sheath, which typically surrounds the hair follicle along with proliferating bands or ribbons of perifollicular connective tissue. As such, the hair follicle may be dilated and filled with keratin in the expanded infundibulum.8 Follicles also may be surrounded by a myxoid stroma.2 In contrast, trichodiscoma is characterized by connective tissue with mature sebaceous lobules in the periphery. It has a myxoid stroma, as opposed to the more fibrous stroma seen in fibrofolliculomas.

Reports have examined the staining patterns of fibrofolliculomas, which show characteristics similar to those of other hair follicle hamartomas, including trichodiscomas.10 The connective tissue and epithelial components that constitute a fibrofolliculoma show different staining patterns. The connective tissue component stains positive for CD34 spindle cells, factor XIIIa, and nestin (a marker of angiogenesis). CD117 (c-kit) expression in the stroma, a marker of fibrocytes, is a feature of both fibrofolliculoma and perifollicular fibromas. The epithelial component, consisting of the hair follicle itself, stains positive for CK15. CK15 expression has been reported in undifferentiated sebocytes of the mantle and in the hair follicle.10 Immunohistochemical staining supports the notion that fibrofolliculomas contain connective tissue and epithelial components and helps to compare and contrast them to those of other hair follicle hamartomas.
Ackerman et al1 considered both fibrofolliculomas and trichodiscomas to be hamartomas of the epithelial hair follicle. The exact etiology of each of these hamartomas is unknown, but the undifferentiated epithelial strands protruding from the hair follicle in a fibrofolliculoma lie in close proximity to sebaceous glands. Furthermore, the authors postulated that fibrofolliculomas and trichodiscomas constitute a spectrum that encompasses the differentiation process of a mantleoma, with fibrofolliculoma representing the beginning of mantleoma differentiation and trichodiscoma representing the end. This end stage of follicular differentiation is one in which there is a predominant stroma and the previously undifferentiated epithelium has formed into sebaceous ducts and lobules in the stroma.1
Most cases of fibrofolliculoma and/or trichodiscoma arise in areas of dense sebaceous follicle concentration (eg, face), further supporting the hypothesis that sebaceous gland proliferation contributes to fibrofolliculoma.14 The case described here, with the fibrofolliculoma arising on the abdomen in conjunction with a trichodiscoma, is therefore worth noting because its location differs from what has been observed in previously reported cases.4
There are both surgical and medical options for treatment of fibrofolliculoma. Although surgical excision is an option for a single lesion, patients with multiple fibrofolliculomas or BHD may prefer removal with the combined CO2 laser and erbium-doped YAG laser.15
Conclusion
We present a rare case of concomitant fibrofolliculoma and trichodiscoma arising on the unusual location of the abdomen. This report highlights the histopathologic features of multiple adnexal tumors and emphasizes the importance of biopsy for differentiating fibrofolliculoma and trichodiscoma.
- Ackerman AB, Chongchitnant N, DeViragh P. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
- Scully K, Bargman H, Assaad D. Solitary fibrofolliculoma. J Am Acad Dermatol. 1984;11:361-363.
- Chang JK, Lee DC, Chang MH. A solitary fibrofolliculoma in the eyelid. Korean J Ophthalmol. 2007;21:169-171.
- Starink TM, Brownstein MH. Fibrofolliculoma: solitary and multiple types. J Am Acad Dermatol. 1987;17:493-496.
- Cho EU, Lee JD, Cho SH. A solitary fibrofolliculoma on the concha of the ear. Int J Dermatol. 2012;51:616-628.
- Mo HJ, Park CK, Yi JY. A case of solitary fibrofolliculoma. Korean J Dermatol. 2001;39:602-604.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157-164.
- Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674-1677.
- Foucar K, Rosen TH, Foucar E, et al. Fibrofolliculoma: a clinicopathologic study. Cutis. 1981;28:429-432.
- Misago NO, Kimura TE, Narisawa YU. Fibrofolliculoma/trichodiscoma and fibrous papule (perifollicular fibroma/angiofibroma): a revaluation of the histopathological and immunohistochemical features. J Cutan Pathol. 2009;36:943-951.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dubé syndrome. J Am Acad Dermatol. 2005;53(2 suppl 1):S108-S111.
- Lee Y, Su H, Chen H. Fibroepithelioma of Pinkus. a case report. Dermatologica Sinica. 2002;20:142-146.
- Nam JH, Min JH, Lee GY, et al. A case of perifollicular fibroma. Ann Dermatol. 2011:23:236-238.
- Vernooij M, Claessens T, Luijten M, et al. Birt-Hogg-Dubé syndrome and the skin. Fam Cancer. 2013;12:381-385.
- Jacob CI, Dover JS. Birt-Hogg-Dubé syndrome: treatment of cutaneous manifestations with laser skin resurfacing. Arch Dermatol. 2001;137:98-99.
Fibrofolliculomas and trichodiscomas typically present on the head or neck as smooth, flesh-colored, dome-shaped papules. These two entities are considered to constitute two separate time points on a spectrum of histopathologic changes in mantleoma differentiation.1 Histologically, both are benign hamartomas of the pilosebaceous subunit and collectively are known as mantleomas. We present an unusual case of a concomitant fibrofolliculoma and trichodiscoma on the abdomen.
Case Report
An asymptomatic 54-year-old man presented for a routine full-body skin examination. A solitary, 2×1-cm, subcutaneous, doughy, mobile nodule was found on the left side of the abdomen with an overlying 2-mm yellow fleshy papule. The patient declined excision of the lesion, and it was recommended that he return for follow-up 3 months later.
The patient did not present for follow-up until 4.5 years later, at which point the lesion had grown to 3.0×2.5 cm in size. An excision was performed, at which time the lesion was noted to be cystic, extruding an oily, yellow-white liquid. Bacterial culture was negative. Histopathologic sections showed a dome-shaped papule with connection to the overlying epidermis. Epithelial extensions from the infundibular epithelium formed a fenestrated pattern surrounding a fibrous and mucinous stroma (Figure, A and B). The differential diagnosis at this time included an epidermal inclusion cyst, fibroma, intradermal nevus, verruca, hemangioma, angiofibroma, and lipoma.2-4
The same lesion cut in a different plane of sectioning showed an expansile dermal nodule comprising clusters of sebaceous lobules surrounding a fibrous and mucinous stroma. Within the second lesion, fibrous and stromal components predominated over epithelial components (Figure, C). A diagnosis of fibrofolliculoma showing features of a trichodiscoma arising in the unusual location of the abdomen was made.
Comment
Solitary fibrofolliculomas and trichodiscomas are flesh-colored, dome-shaped papules that generally present on the face, specifically on the chin, nose, cheeks, ears, and eyebrows without considerable symptoms.2,4,5 Clinically, fibrofolliculomas are indistinguishable from trichodiscomas but demonstrate different features on biopsy.1,5
Fibrofolliculomas and trichodiscomas are well known for their association with Birt-Hogg-Dubé (BHD) syndrome when they present concomitantly and typically arise earlier in the third decade of life than solitary fibrofolliculomas; however, there have been reports of solitary fibrofolliculomas in patients aged 1 to 36 years.4,6 The triad of BHD syndrome consists of multiple fibrofolliculomas, trichodiscomas, and acrochordons, and it is acquired in an autosomal-dominant manner, unlike solitary fibrofolliculomas, which typically are not inherited. Birt-Hogg-Dubé syndrome is caused by a mutation in the FLCN gene that codes for the tumor-suppressor protein folliculin, which when mutated can cause unregulated proliferation of cells.7 Solitary fibrofolliculomas and the multiple fibrofolliculomas seen in BHD syndrome are histologically similar.
Fibrofolliculoma can be clinically indistinguishable from fibroepithelioma of Pinkus, perifollicular fibroma, trichilemmoma, trichodiscoma, trichoepithelioma, and trichofolliculoma. All typically present clinically as flesh-colored papules,1 although histologic distinction can be made (Table).5,8-13
Fibrofolliculoma is a benign hamartoma that arises from the pilosebaceous follicle and consists of an expansion of the fibrous root sheath, which typically surrounds the hair follicle along with proliferating bands or ribbons of perifollicular connective tissue. As such, the hair follicle may be dilated and filled with keratin in the expanded infundibulum.8 Follicles also may be surrounded by a myxoid stroma.2 In contrast, trichodiscoma is characterized by connective tissue with mature sebaceous lobules in the periphery. It has a myxoid stroma, as opposed to the more fibrous stroma seen in fibrofolliculomas.
Reports have examined the staining patterns of fibrofolliculomas, which show characteristics similar to those of other hair follicle hamartomas, including trichodiscomas.10 The connective tissue and epithelial components that constitute a fibrofolliculoma show different staining patterns. The connective tissue component stains positive for CD34 spindle cells, factor XIIIa, and nestin (a marker of angiogenesis). CD117 (c-kit) expression in the stroma, a marker of fibrocytes, is a feature of both fibrofolliculoma and perifollicular fibromas. The epithelial component, consisting of the hair follicle itself, stains positive for CK15. CK15 expression has been reported in undifferentiated sebocytes of the mantle and in the hair follicle.10 Immunohistochemical staining supports the notion that fibrofolliculomas contain connective tissue and epithelial components and helps to compare and contrast them to those of other hair follicle hamartomas.
Ackerman et al1 considered both fibrofolliculomas and trichodiscomas to be hamartomas of the epithelial hair follicle. The exact etiology of each of these hamartomas is unknown, but the undifferentiated epithelial strands protruding from the hair follicle in a fibrofolliculoma lie in close proximity to sebaceous glands. Furthermore, the authors postulated that fibrofolliculomas and trichodiscomas constitute a spectrum that encompasses the differentiation process of a mantleoma, with fibrofolliculoma representing the beginning of mantleoma differentiation and trichodiscoma representing the end. This end stage of follicular differentiation is one in which there is a predominant stroma and the previously undifferentiated epithelium has formed into sebaceous ducts and lobules in the stroma.1
Most cases of fibrofolliculoma and/or trichodiscoma arise in areas of dense sebaceous follicle concentration (eg, face), further supporting the hypothesis that sebaceous gland proliferation contributes to fibrofolliculoma.14 The case described here, with the fibrofolliculoma arising on the abdomen in conjunction with a trichodiscoma, is therefore worth noting because its location differs from what has been observed in previously reported cases.4
There are both surgical and medical options for treatment of fibrofolliculoma. Although surgical excision is an option for a single lesion, patients with multiple fibrofolliculomas or BHD may prefer removal with the combined CO2 laser and erbium-doped YAG laser.15
Conclusion
We present a rare case of concomitant fibrofolliculoma and trichodiscoma arising on the unusual location of the abdomen. This report highlights the histopathologic features of multiple adnexal tumors and emphasizes the importance of biopsy for differentiating fibrofolliculoma and trichodiscoma.
Fibrofolliculomas and trichodiscomas typically present on the head or neck as smooth, flesh-colored, dome-shaped papules. These two entities are considered to constitute two separate time points on a spectrum of histopathologic changes in mantleoma differentiation.1 Histologically, both are benign hamartomas of the pilosebaceous subunit and collectively are known as mantleomas. We present an unusual case of a concomitant fibrofolliculoma and trichodiscoma on the abdomen.
Case Report
An asymptomatic 54-year-old man presented for a routine full-body skin examination. A solitary, 2×1-cm, subcutaneous, doughy, mobile nodule was found on the left side of the abdomen with an overlying 2-mm yellow fleshy papule. The patient declined excision of the lesion, and it was recommended that he return for follow-up 3 months later.
The patient did not present for follow-up until 4.5 years later, at which point the lesion had grown to 3.0×2.5 cm in size. An excision was performed, at which time the lesion was noted to be cystic, extruding an oily, yellow-white liquid. Bacterial culture was negative. Histopathologic sections showed a dome-shaped papule with connection to the overlying epidermis. Epithelial extensions from the infundibular epithelium formed a fenestrated pattern surrounding a fibrous and mucinous stroma (Figure, A and B). The differential diagnosis at this time included an epidermal inclusion cyst, fibroma, intradermal nevus, verruca, hemangioma, angiofibroma, and lipoma.2-4
The same lesion cut in a different plane of sectioning showed an expansile dermal nodule comprising clusters of sebaceous lobules surrounding a fibrous and mucinous stroma. Within the second lesion, fibrous and stromal components predominated over epithelial components (Figure, C). A diagnosis of fibrofolliculoma showing features of a trichodiscoma arising in the unusual location of the abdomen was made.
Comment
Solitary fibrofolliculomas and trichodiscomas are flesh-colored, dome-shaped papules that generally present on the face, specifically on the chin, nose, cheeks, ears, and eyebrows without considerable symptoms.2,4,5 Clinically, fibrofolliculomas are indistinguishable from trichodiscomas but demonstrate different features on biopsy.1,5
Fibrofolliculomas and trichodiscomas are well known for their association with Birt-Hogg-Dubé (BHD) syndrome when they present concomitantly and typically arise earlier in the third decade of life than solitary fibrofolliculomas; however, there have been reports of solitary fibrofolliculomas in patients aged 1 to 36 years.4,6 The triad of BHD syndrome consists of multiple fibrofolliculomas, trichodiscomas, and acrochordons, and it is acquired in an autosomal-dominant manner, unlike solitary fibrofolliculomas, which typically are not inherited. Birt-Hogg-Dubé syndrome is caused by a mutation in the FLCN gene that codes for the tumor-suppressor protein folliculin, which when mutated can cause unregulated proliferation of cells.7 Solitary fibrofolliculomas and the multiple fibrofolliculomas seen in BHD syndrome are histologically similar.
Fibrofolliculoma can be clinically indistinguishable from fibroepithelioma of Pinkus, perifollicular fibroma, trichilemmoma, trichodiscoma, trichoepithelioma, and trichofolliculoma. All typically present clinically as flesh-colored papules,1 although histologic distinction can be made (Table).5,8-13
Fibrofolliculoma is a benign hamartoma that arises from the pilosebaceous follicle and consists of an expansion of the fibrous root sheath, which typically surrounds the hair follicle along with proliferating bands or ribbons of perifollicular connective tissue. As such, the hair follicle may be dilated and filled with keratin in the expanded infundibulum.8 Follicles also may be surrounded by a myxoid stroma.2 In contrast, trichodiscoma is characterized by connective tissue with mature sebaceous lobules in the periphery. It has a myxoid stroma, as opposed to the more fibrous stroma seen in fibrofolliculomas.
Reports have examined the staining patterns of fibrofolliculomas, which show characteristics similar to those of other hair follicle hamartomas, including trichodiscomas.10 The connective tissue and epithelial components that constitute a fibrofolliculoma show different staining patterns. The connective tissue component stains positive for CD34 spindle cells, factor XIIIa, and nestin (a marker of angiogenesis). CD117 (c-kit) expression in the stroma, a marker of fibrocytes, is a feature of both fibrofolliculoma and perifollicular fibromas. The epithelial component, consisting of the hair follicle itself, stains positive for CK15. CK15 expression has been reported in undifferentiated sebocytes of the mantle and in the hair follicle.10 Immunohistochemical staining supports the notion that fibrofolliculomas contain connective tissue and epithelial components and helps to compare and contrast them to those of other hair follicle hamartomas.
Ackerman et al1 considered both fibrofolliculomas and trichodiscomas to be hamartomas of the epithelial hair follicle. The exact etiology of each of these hamartomas is unknown, but the undifferentiated epithelial strands protruding from the hair follicle in a fibrofolliculoma lie in close proximity to sebaceous glands. Furthermore, the authors postulated that fibrofolliculomas and trichodiscomas constitute a spectrum that encompasses the differentiation process of a mantleoma, with fibrofolliculoma representing the beginning of mantleoma differentiation and trichodiscoma representing the end. This end stage of follicular differentiation is one in which there is a predominant stroma and the previously undifferentiated epithelium has formed into sebaceous ducts and lobules in the stroma.1
Most cases of fibrofolliculoma and/or trichodiscoma arise in areas of dense sebaceous follicle concentration (eg, face), further supporting the hypothesis that sebaceous gland proliferation contributes to fibrofolliculoma.14 The case described here, with the fibrofolliculoma arising on the abdomen in conjunction with a trichodiscoma, is therefore worth noting because its location differs from what has been observed in previously reported cases.4
There are both surgical and medical options for treatment of fibrofolliculoma. Although surgical excision is an option for a single lesion, patients with multiple fibrofolliculomas or BHD may prefer removal with the combined CO2 laser and erbium-doped YAG laser.15
Conclusion
We present a rare case of concomitant fibrofolliculoma and trichodiscoma arising on the unusual location of the abdomen. This report highlights the histopathologic features of multiple adnexal tumors and emphasizes the importance of biopsy for differentiating fibrofolliculoma and trichodiscoma.
- Ackerman AB, Chongchitnant N, DeViragh P. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
- Scully K, Bargman H, Assaad D. Solitary fibrofolliculoma. J Am Acad Dermatol. 1984;11:361-363.
- Chang JK, Lee DC, Chang MH. A solitary fibrofolliculoma in the eyelid. Korean J Ophthalmol. 2007;21:169-171.
- Starink TM, Brownstein MH. Fibrofolliculoma: solitary and multiple types. J Am Acad Dermatol. 1987;17:493-496.
- Cho EU, Lee JD, Cho SH. A solitary fibrofolliculoma on the concha of the ear. Int J Dermatol. 2012;51:616-628.
- Mo HJ, Park CK, Yi JY. A case of solitary fibrofolliculoma. Korean J Dermatol. 2001;39:602-604.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157-164.
- Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674-1677.
- Foucar K, Rosen TH, Foucar E, et al. Fibrofolliculoma: a clinicopathologic study. Cutis. 1981;28:429-432.
- Misago NO, Kimura TE, Narisawa YU. Fibrofolliculoma/trichodiscoma and fibrous papule (perifollicular fibroma/angiofibroma): a revaluation of the histopathological and immunohistochemical features. J Cutan Pathol. 2009;36:943-951.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dubé syndrome. J Am Acad Dermatol. 2005;53(2 suppl 1):S108-S111.
- Lee Y, Su H, Chen H. Fibroepithelioma of Pinkus. a case report. Dermatologica Sinica. 2002;20:142-146.
- Nam JH, Min JH, Lee GY, et al. A case of perifollicular fibroma. Ann Dermatol. 2011:23:236-238.
- Vernooij M, Claessens T, Luijten M, et al. Birt-Hogg-Dubé syndrome and the skin. Fam Cancer. 2013;12:381-385.
- Jacob CI, Dover JS. Birt-Hogg-Dubé syndrome: treatment of cutaneous manifestations with laser skin resurfacing. Arch Dermatol. 2001;137:98-99.
- Ackerman AB, Chongchitnant N, DeViragh P. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
- Scully K, Bargman H, Assaad D. Solitary fibrofolliculoma. J Am Acad Dermatol. 1984;11:361-363.
- Chang JK, Lee DC, Chang MH. A solitary fibrofolliculoma in the eyelid. Korean J Ophthalmol. 2007;21:169-171.
- Starink TM, Brownstein MH. Fibrofolliculoma: solitary and multiple types. J Am Acad Dermatol. 1987;17:493-496.
- Cho EU, Lee JD, Cho SH. A solitary fibrofolliculoma on the concha of the ear. Int J Dermatol. 2012;51:616-628.
- Mo HJ, Park CK, Yi JY. A case of solitary fibrofolliculoma. Korean J Dermatol. 2001;39:602-604.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157-164.
- Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674-1677.
- Foucar K, Rosen TH, Foucar E, et al. Fibrofolliculoma: a clinicopathologic study. Cutis. 1981;28:429-432.
- Misago NO, Kimura TE, Narisawa YU. Fibrofolliculoma/trichodiscoma and fibrous papule (perifollicular fibroma/angiofibroma): a revaluation of the histopathological and immunohistochemical features. J Cutan Pathol. 2009;36:943-951.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dubé syndrome. J Am Acad Dermatol. 2005;53(2 suppl 1):S108-S111.
- Lee Y, Su H, Chen H. Fibroepithelioma of Pinkus. a case report. Dermatologica Sinica. 2002;20:142-146.
- Nam JH, Min JH, Lee GY, et al. A case of perifollicular fibroma. Ann Dermatol. 2011:23:236-238.
- Vernooij M, Claessens T, Luijten M, et al. Birt-Hogg-Dubé syndrome and the skin. Fam Cancer. 2013;12:381-385.
- Jacob CI, Dover JS. Birt-Hogg-Dubé syndrome: treatment of cutaneous manifestations with laser skin resurfacing. Arch Dermatol. 2001;137:98-99.
Practice Points
- Fibrofolliculoma and trichodiscoma are flesh-colored adnexal tumors that arise from or around hair follicles.
- It is important to recognize these entities, as they can be related to Birt-Hogg-Dubé syndrome.
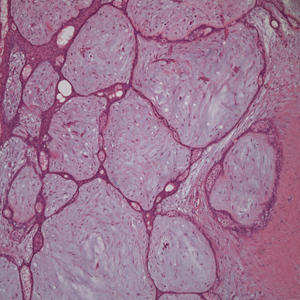
Progressive and Translucent Plaques on the Soles
The Diagnosis: Cutaneous Macroglobulinosis
Waldenström macroglobulinemia is a lymphoplasmacytic lymphoma that produces a circulating monoclonal IgM. Incidence in the United States is 1500 patients annually, most commonly men in their 70s.1 The disease process is largely indolent, with early symptoms consisting of generalized weakness, weight loss, and fatigue. Signs of lymphadenopathy, hepatosplenomegaly, and cytopenia may emerge as the disease progresses. Diagnostic criteria include bone marrow biopsy with plasmacytoid/plasmacellular infiltrate; IgM monoclonal gammopathy; and end-organ damage, which may include cutaneous manifestations.2
Cutaneous findings in Waldenström macroglobulinemia are nonspecific and secondary to the disease's hematologic manifestations, presenting as livedo reticularis, purpura, and mucosal bleeding.3 True cutaneous involvement of the disease is rare and was first described in 1978 by Tichenor.4 Specific cutaneous lesions have 2 separate clinical presentations: (1) a primary cutaneous infiltrate of lymphoplasmacytic cells, and (2) deposition of IgM in the dermis.5 Although the primary infiltrate of neoplastic cells appears as erythematous firm papules or plaques on the face and trunk, similar to other manifestations of leukemia cutis, deposition of IgM presents as translucent papules and plaques and is located more distally, particularly on the extensor extremities.6 These depositional plaques are not pruritic but may be tender if located over sites of pressure, as seen with the plantar presentation in our patient.
Histologically, cutaneous macroglobulinosis demonstrates IgM deposition in perieccrine, perivascular, or intravascular tissue that is periodic acid-Schiff (PAS) positive.7 Staining with Congo red and Alcian blue is negative. In our case, biopsy showed a nodular deposition of hypocellular globular material that stained brightly with PAS and PAS diastase. With Masson trichome stain, intensity of staining diminished, suggesting that the deposition was not composed of collagen; rather, this deposition appeared to consist of IgM storage papules on immunohistochemistry (Figure 1). Further workup revealed borderline pancytopenia and elevated globulins with a monoclonal peak on serum protein electrophoresis, confirming the diagnosis of cutaneous macroglobulinosis secondary to Waldenström macroglobulinemia.
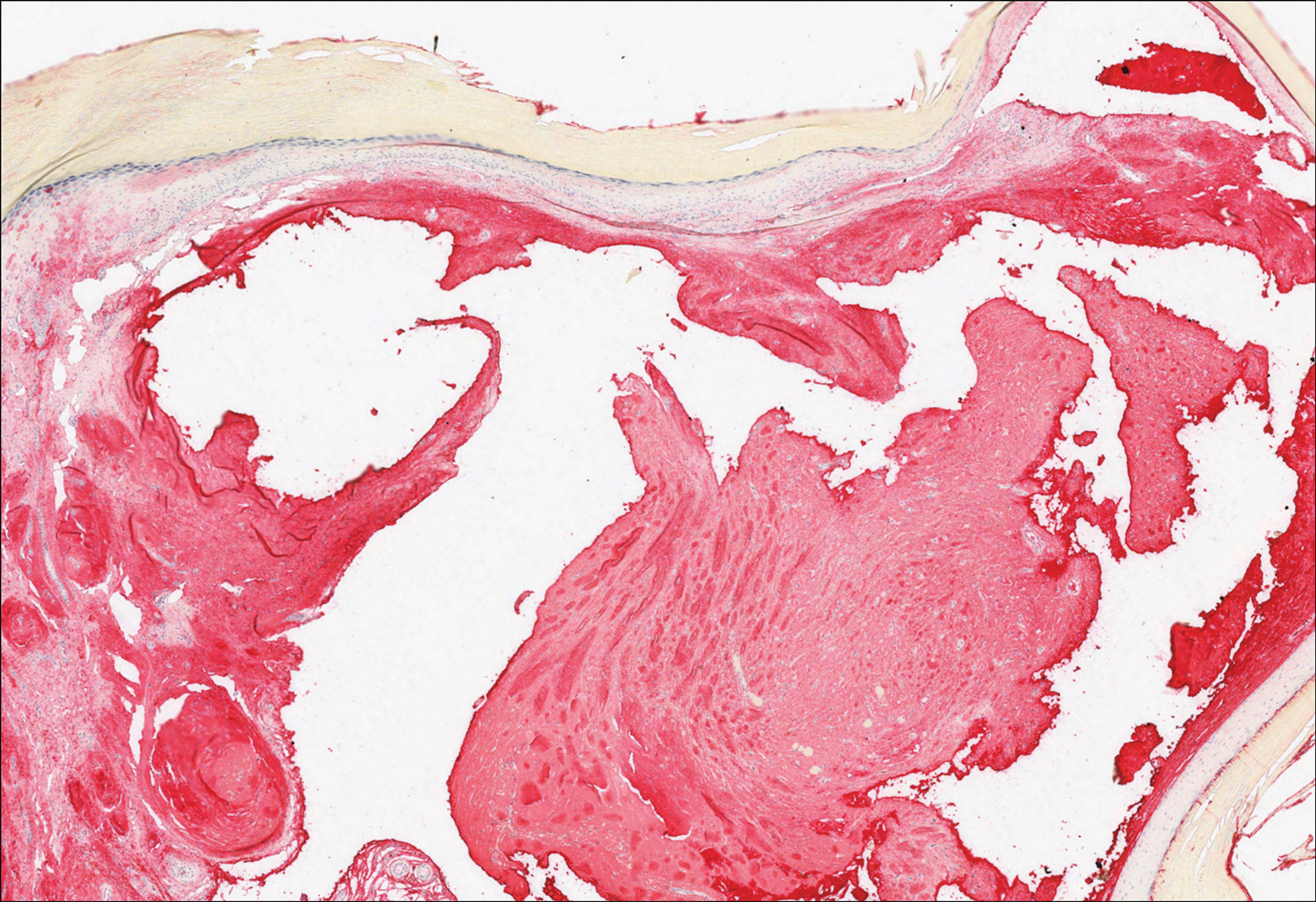
A PubMed search of articles indexed for MEDLINE using the terms cutaneous, macroglobulinosis, macroglobulinemia, Waldenström's macroglobulinemia, Waldenström's macroglobulinaemia, and macroglobulinemia cutis revealed a total of 19 cases of cutaneous macroglobulinosis (including this case). The average age of presentation in these cases is 60 years (range, 29-83 years) with a predisposition for men (68% [13/19]). The development of cutaneous macroglobulinosis primarily has been noted following diagnosis of Waldenström macroglobulinemia (53% [10/19]), with some cases prior to diagnosis (37% [7/19]) or at the time of diagnosis (11% [2/19]). The presence of cutaneous lesions does not correlate with prognosis of the underlying malignancy.5,8,9
Systemic treatment of the underlying macroglobulinemia has been suggested for symptomatic cases of cutaneous macroglobulinosis.3 Prior therapy has consisted primarily of chlorambucil; however, treatment with rituximab, occasionally in conjunction with the proteasome inhibitor bortezomib, recently has been reported.10 Because of the symptomatic nature of our patient's lesions, she was referred to the oncology department and started on rituximab therapy. The lesions improved with therapy and have remained stable following treatment.
The differential diagnosis for tender pink papules and plaques on the arms and legs includes tophaceous gout, plantar fibromatosis, erythropoietic protoporphyria, and acral fibrokeratoma.
Gouty tophi commonly accumulate as painful, edematous, yellow to whitish nodules and tumors with erythema, often overlying joints or extensor surfaces. Histopathologic examination after formalin fixation shows needle-shaped clefts within feathery amorphous pink areas surrounded by granuloma (Figure 2).11 Yellow, needle-shaped, negatively birefringent crystals can be viewed under polarized microscopy in alcohol-fixed samples.
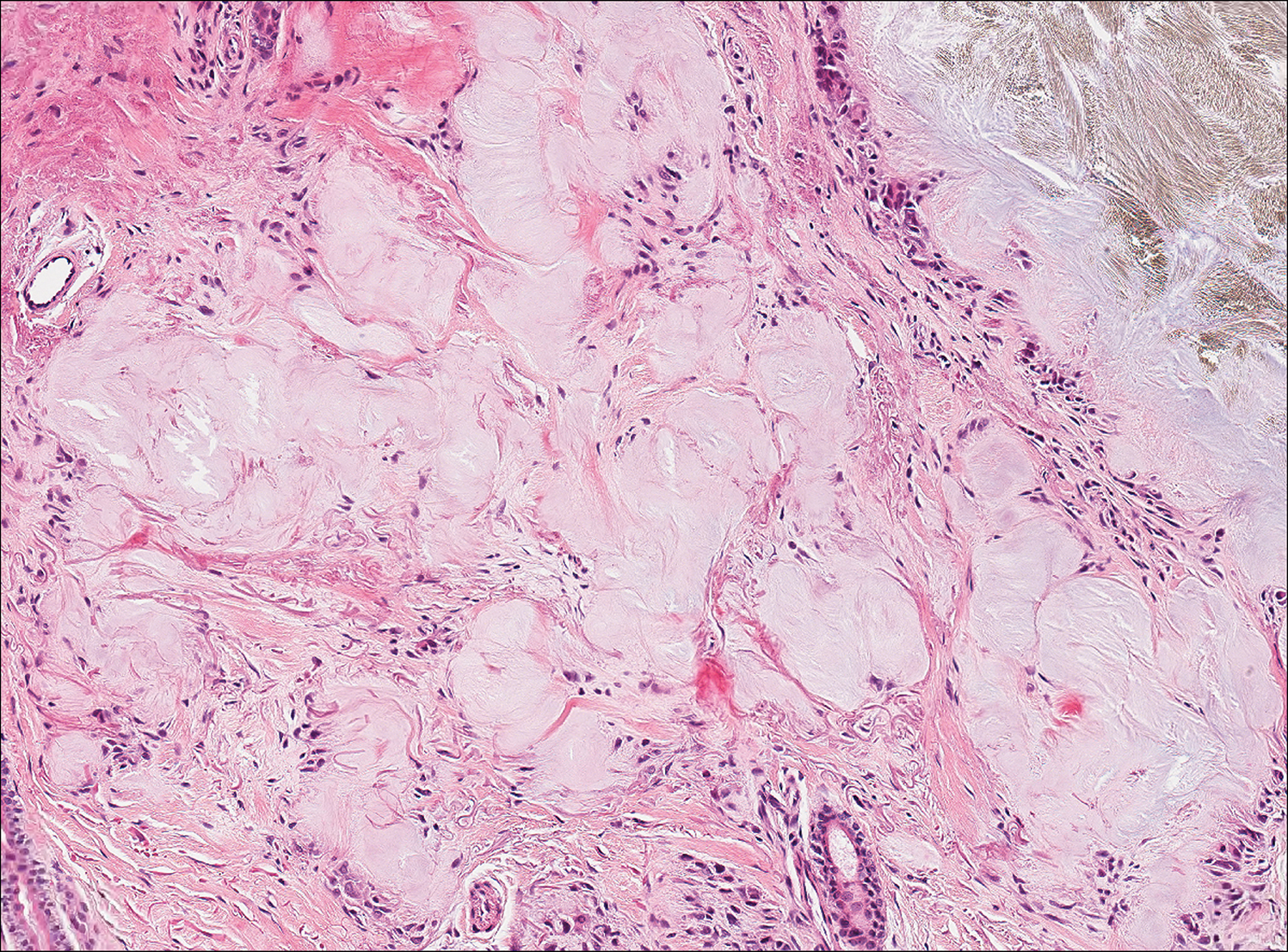
Plantar fibromatosis (Ledderhose disease) is a benign proliferation of the plantar aponeurosis linked to alcohol use; liver disease; and notably epilepsy,12 a component of our patient's medical history. Large nodules appear grossly on the plantar feet and may progress to contractures in more advanced lesions. Biopsy reveals bland hyperproliferation of fibroblasts in a background of fascial fibrous tissue (Figure 3).12 Clinically, this diagnosis is part of the differential diagnosis of plantar nodules but appears histologically different than cutaneous macroglobulinosis because there are no hyaline deposits in plantar fibromatosis.
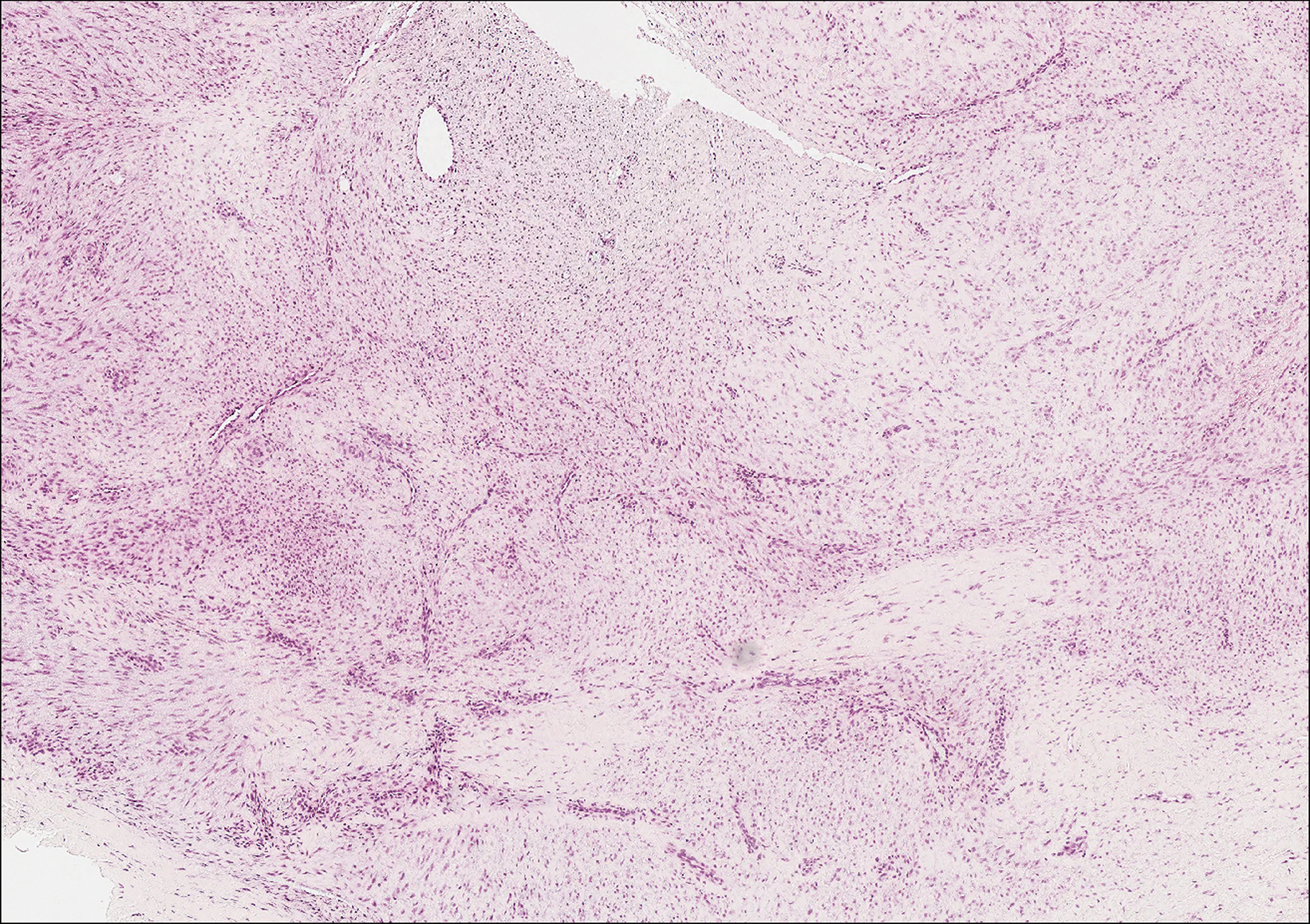
Erythropoietic protoporphyria is a rare disorder that primarily arises due to a congenital deficiency in the ferrochelatase enzyme involved in heme biosynthesis. Erythropoietic protoporphyria is the most common porphyria among children and typically presents in infancy or early childhood as a painful photosensitivity with ensuing cutaneous manifestations and possible hepatobiliary disease. Edema and severe burning pain can be noted within minutes of sun exposure in a dose-response relationship.13 Histologic findings of erythropoietic protoporphyria differ based on acute or chronic skin changes. Acute lesions exhibit a predominantly neutrophilic interstitial dermal infiltrate with vacuoles and intercellular edema. Chronic changes include the accumulation of a PAS-positive, amorphous, hyalinelike substance, similar to the microscopic findings of cutaneous macroglobulinosis (Figure 4).13
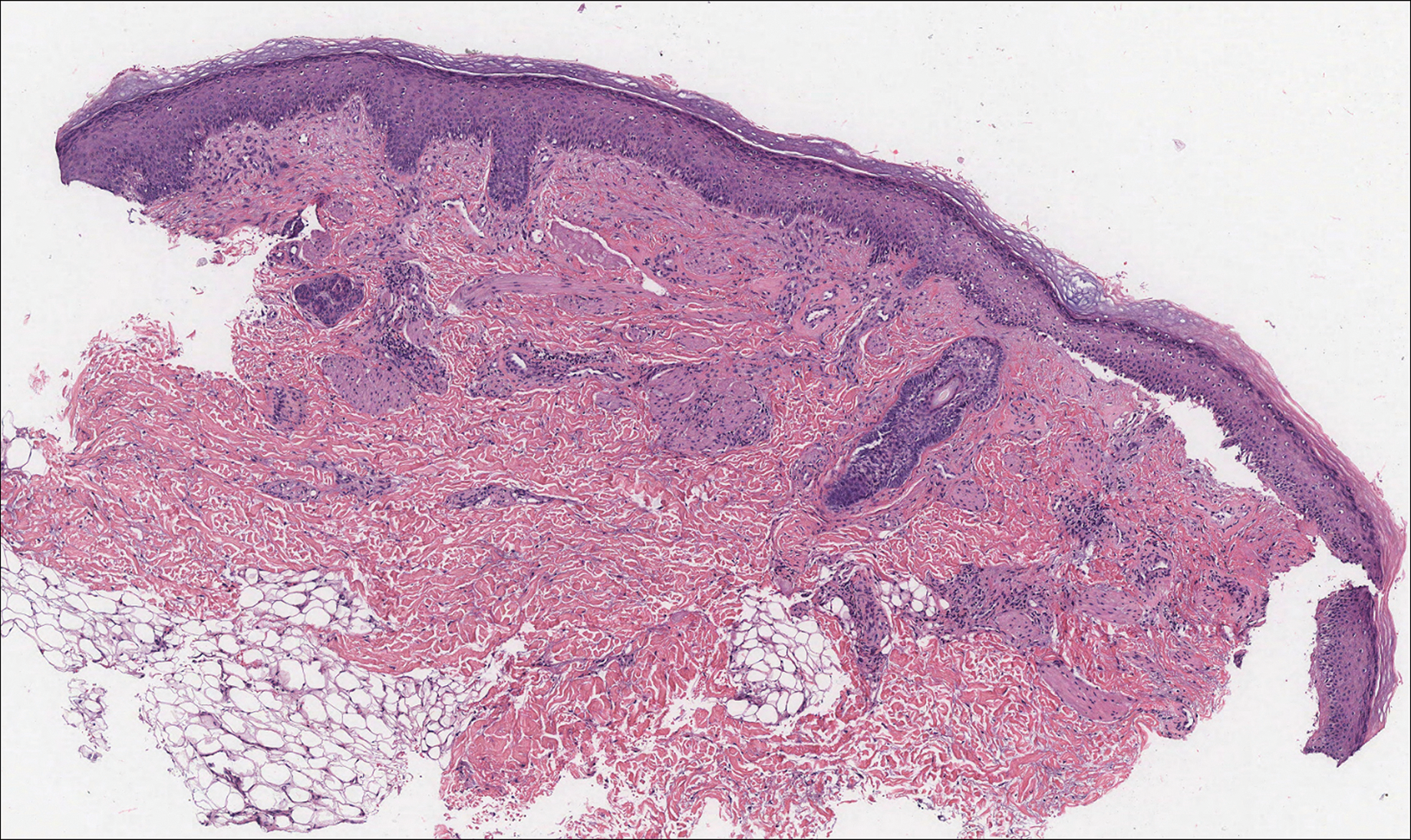
An acral fibrokeratoma is a benign fibroepithelial tumor that clinically appears as a flesh-colored or slightly erythematous exophytic nodule that most commonly is found on the fingers or toes. Thought to arise from trauma to the affected area, it is histologically characterized by interwoven collagenous bundles with overlying epidermal hyperkeratosis, acanthosis, and deep thickened rete ridges14 (Figure 5). Although multiple acral fibrokeratomas have been reported (similar to presentations of prurigo nodularis),15 they more commonly appear as solitary lesions as opposed to the numerous translucent papules seen in our patient.
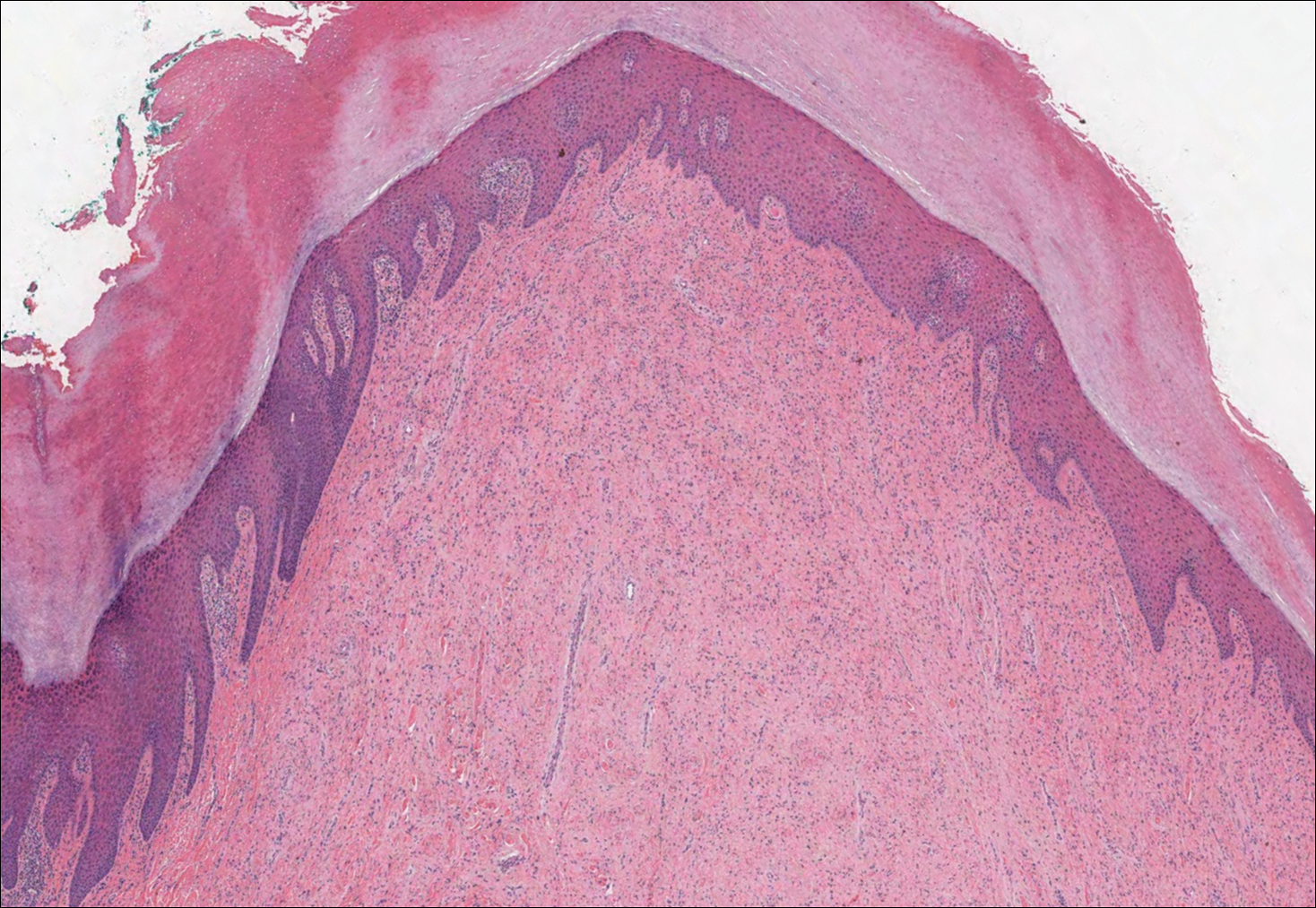
- Camp BJ, Magro CM. Cutaneous macroglobulinosis: a case series. J Cutan Pathol. 2012;39:962-970.
- Dimopoulos MA, Alexanian R. Waldenstrom's macroglobulinemia. Blood. 1994;83:1452-1459.
- D'Acunto C, Nigrisoli E, Liardo EV, et al. Painful plantar nodules: a specific manifestation of cutaneous macroglobulinosis. J Am Acad Dermatol. 2014;71:E251-E252.
- Tichenor RE. Macroglobulinemia cutis. Arch Dermatol. 1978;114:280-281.
- Gressier L, Hotz C, Lelièvre JD, et al. Cutaneous macroglobulinosis: a report of 2 cases. Arch Dermatol. 2010;146:165-169.
- Spicknall KE, Dubas LE, Mutasim DF. Cutaneous macroglobulinosis with monotypic plasma cells: a specific manifestation of Waldenström macroglobulinemia. J Cutan Pathol. 2013;40:442-444.
- Lüftl M, Sauter-Jenne B, Gramatzki M, et al. Cutaneous macroglobulinosis deposits in a patient with IgM paraproteinemia/incipient Waldenström macroglobulinemia. J Dtsch Dermatol Ges. 2010;8:1000-1003.
- Mascaro JM, Montserrat E, Estrach T, et al. Specific cutaneous manifestations of Waldenstrom macroglobulinaemia: a report of two cases. Br J Dermatol. 1982;106:217-222.
- Hanke CW, Steck WD, Bergfeld WF, et al. Cutaneous macroglobulinosis. Arch Dermatol. 1980;116:575-577.
- Oshio-Yoshii A, Fujimoto N, Shiba Y, et al. Cutaneous macroglobulinosis: successful treatment with rituximab. J Eur Acad Dermatol Venereol. 2017;31:E30-E31.
- Gupta A, Rai S, Sinha R, et al. Tophi as an initial manifestation of gout. J Cytol. 2009;26:165-166.
- Carroll P, Henshaw RM, Garwood C, et al. Plantar fibromatosis: pathophysiology, surgical and nonsurgical therapies: an evidence-based review. Foot Ankle Spec. 2018;11:168-176.
- Michaels BD, Del Rosso JQ, Mobini N, et al. Erythropoietic protoporphyria: a case report and literature review. J Clin Aesthet Dermatol. 2010;3:44-48.
- Boffeli TJ, Abben KW. Acral fibrokeratoma of the foot treated with excision and trap door flap closure: a case report. J Foot Ankle Surg. 2014;53:449-452.
- Reed RJ. Multiple acral fibrokeratomas (a variant of prurigo nodularis). discussion of classification of acral fibrous nodules and of histogenesis of acral fibrokeratomas. Arch Dermatol. 1971;103:287-297.
The Diagnosis: Cutaneous Macroglobulinosis
Waldenström macroglobulinemia is a lymphoplasmacytic lymphoma that produces a circulating monoclonal IgM. Incidence in the United States is 1500 patients annually, most commonly men in their 70s.1 The disease process is largely indolent, with early symptoms consisting of generalized weakness, weight loss, and fatigue. Signs of lymphadenopathy, hepatosplenomegaly, and cytopenia may emerge as the disease progresses. Diagnostic criteria include bone marrow biopsy with plasmacytoid/plasmacellular infiltrate; IgM monoclonal gammopathy; and end-organ damage, which may include cutaneous manifestations.2
Cutaneous findings in Waldenström macroglobulinemia are nonspecific and secondary to the disease's hematologic manifestations, presenting as livedo reticularis, purpura, and mucosal bleeding.3 True cutaneous involvement of the disease is rare and was first described in 1978 by Tichenor.4 Specific cutaneous lesions have 2 separate clinical presentations: (1) a primary cutaneous infiltrate of lymphoplasmacytic cells, and (2) deposition of IgM in the dermis.5 Although the primary infiltrate of neoplastic cells appears as erythematous firm papules or plaques on the face and trunk, similar to other manifestations of leukemia cutis, deposition of IgM presents as translucent papules and plaques and is located more distally, particularly on the extensor extremities.6 These depositional plaques are not pruritic but may be tender if located over sites of pressure, as seen with the plantar presentation in our patient.
Histologically, cutaneous macroglobulinosis demonstrates IgM deposition in perieccrine, perivascular, or intravascular tissue that is periodic acid-Schiff (PAS) positive.7 Staining with Congo red and Alcian blue is negative. In our case, biopsy showed a nodular deposition of hypocellular globular material that stained brightly with PAS and PAS diastase. With Masson trichome stain, intensity of staining diminished, suggesting that the deposition was not composed of collagen; rather, this deposition appeared to consist of IgM storage papules on immunohistochemistry (Figure 1). Further workup revealed borderline pancytopenia and elevated globulins with a monoclonal peak on serum protein electrophoresis, confirming the diagnosis of cutaneous macroglobulinosis secondary to Waldenström macroglobulinemia.
A PubMed search of articles indexed for MEDLINE using the terms cutaneous, macroglobulinosis, macroglobulinemia, Waldenström's macroglobulinemia, Waldenström's macroglobulinaemia, and macroglobulinemia cutis revealed a total of 19 cases of cutaneous macroglobulinosis (including this case). The average age of presentation in these cases is 60 years (range, 29-83 years) with a predisposition for men (68% [13/19]). The development of cutaneous macroglobulinosis primarily has been noted following diagnosis of Waldenström macroglobulinemia (53% [10/19]), with some cases prior to diagnosis (37% [7/19]) or at the time of diagnosis (11% [2/19]). The presence of cutaneous lesions does not correlate with prognosis of the underlying malignancy.5,8,9
Systemic treatment of the underlying macroglobulinemia has been suggested for symptomatic cases of cutaneous macroglobulinosis.3 Prior therapy has consisted primarily of chlorambucil; however, treatment with rituximab, occasionally in conjunction with the proteasome inhibitor bortezomib, recently has been reported.10 Because of the symptomatic nature of our patient's lesions, she was referred to the oncology department and started on rituximab therapy. The lesions improved with therapy and have remained stable following treatment.
The differential diagnosis for tender pink papules and plaques on the arms and legs includes tophaceous gout, plantar fibromatosis, erythropoietic protoporphyria, and acral fibrokeratoma.
Gouty tophi commonly accumulate as painful, edematous, yellow to whitish nodules and tumors with erythema, often overlying joints or extensor surfaces. Histopathologic examination after formalin fixation shows needle-shaped clefts within feathery amorphous pink areas surrounded by granuloma (Figure 2).11 Yellow, needle-shaped, negatively birefringent crystals can be viewed under polarized microscopy in alcohol-fixed samples.
Plantar fibromatosis (Ledderhose disease) is a benign proliferation of the plantar aponeurosis linked to alcohol use; liver disease; and notably epilepsy,12 a component of our patient's medical history. Large nodules appear grossly on the plantar feet and may progress to contractures in more advanced lesions. Biopsy reveals bland hyperproliferation of fibroblasts in a background of fascial fibrous tissue (Figure 3).12 Clinically, this diagnosis is part of the differential diagnosis of plantar nodules but appears histologically different than cutaneous macroglobulinosis because there are no hyaline deposits in plantar fibromatosis.
Erythropoietic protoporphyria is a rare disorder that primarily arises due to a congenital deficiency in the ferrochelatase enzyme involved in heme biosynthesis. Erythropoietic protoporphyria is the most common porphyria among children and typically presents in infancy or early childhood as a painful photosensitivity with ensuing cutaneous manifestations and possible hepatobiliary disease. Edema and severe burning pain can be noted within minutes of sun exposure in a dose-response relationship.13 Histologic findings of erythropoietic protoporphyria differ based on acute or chronic skin changes. Acute lesions exhibit a predominantly neutrophilic interstitial dermal infiltrate with vacuoles and intercellular edema. Chronic changes include the accumulation of a PAS-positive, amorphous, hyalinelike substance, similar to the microscopic findings of cutaneous macroglobulinosis (Figure 4).13
An acral fibrokeratoma is a benign fibroepithelial tumor that clinically appears as a flesh-colored or slightly erythematous exophytic nodule that most commonly is found on the fingers or toes. Thought to arise from trauma to the affected area, it is histologically characterized by interwoven collagenous bundles with overlying epidermal hyperkeratosis, acanthosis, and deep thickened rete ridges14 (Figure 5). Although multiple acral fibrokeratomas have been reported (similar to presentations of prurigo nodularis),15 they more commonly appear as solitary lesions as opposed to the numerous translucent papules seen in our patient.
The Diagnosis: Cutaneous Macroglobulinosis
Waldenström macroglobulinemia is a lymphoplasmacytic lymphoma that produces a circulating monoclonal IgM. Incidence in the United States is 1500 patients annually, most commonly men in their 70s.1 The disease process is largely indolent, with early symptoms consisting of generalized weakness, weight loss, and fatigue. Signs of lymphadenopathy, hepatosplenomegaly, and cytopenia may emerge as the disease progresses. Diagnostic criteria include bone marrow biopsy with plasmacytoid/plasmacellular infiltrate; IgM monoclonal gammopathy; and end-organ damage, which may include cutaneous manifestations.2
Cutaneous findings in Waldenström macroglobulinemia are nonspecific and secondary to the disease's hematologic manifestations, presenting as livedo reticularis, purpura, and mucosal bleeding.3 True cutaneous involvement of the disease is rare and was first described in 1978 by Tichenor.4 Specific cutaneous lesions have 2 separate clinical presentations: (1) a primary cutaneous infiltrate of lymphoplasmacytic cells, and (2) deposition of IgM in the dermis.5 Although the primary infiltrate of neoplastic cells appears as erythematous firm papules or plaques on the face and trunk, similar to other manifestations of leukemia cutis, deposition of IgM presents as translucent papules and plaques and is located more distally, particularly on the extensor extremities.6 These depositional plaques are not pruritic but may be tender if located over sites of pressure, as seen with the plantar presentation in our patient.
Histologically, cutaneous macroglobulinosis demonstrates IgM deposition in perieccrine, perivascular, or intravascular tissue that is periodic acid-Schiff (PAS) positive.7 Staining with Congo red and Alcian blue is negative. In our case, biopsy showed a nodular deposition of hypocellular globular material that stained brightly with PAS and PAS diastase. With Masson trichome stain, intensity of staining diminished, suggesting that the deposition was not composed of collagen; rather, this deposition appeared to consist of IgM storage papules on immunohistochemistry (Figure 1). Further workup revealed borderline pancytopenia and elevated globulins with a monoclonal peak on serum protein electrophoresis, confirming the diagnosis of cutaneous macroglobulinosis secondary to Waldenström macroglobulinemia.
A PubMed search of articles indexed for MEDLINE using the terms cutaneous, macroglobulinosis, macroglobulinemia, Waldenström's macroglobulinemia, Waldenström's macroglobulinaemia, and macroglobulinemia cutis revealed a total of 19 cases of cutaneous macroglobulinosis (including this case). The average age of presentation in these cases is 60 years (range, 29-83 years) with a predisposition for men (68% [13/19]). The development of cutaneous macroglobulinosis primarily has been noted following diagnosis of Waldenström macroglobulinemia (53% [10/19]), with some cases prior to diagnosis (37% [7/19]) or at the time of diagnosis (11% [2/19]). The presence of cutaneous lesions does not correlate with prognosis of the underlying malignancy.5,8,9
Systemic treatment of the underlying macroglobulinemia has been suggested for symptomatic cases of cutaneous macroglobulinosis.3 Prior therapy has consisted primarily of chlorambucil; however, treatment with rituximab, occasionally in conjunction with the proteasome inhibitor bortezomib, recently has been reported.10 Because of the symptomatic nature of our patient's lesions, she was referred to the oncology department and started on rituximab therapy. The lesions improved with therapy and have remained stable following treatment.
The differential diagnosis for tender pink papules and plaques on the arms and legs includes tophaceous gout, plantar fibromatosis, erythropoietic protoporphyria, and acral fibrokeratoma.
Gouty tophi commonly accumulate as painful, edematous, yellow to whitish nodules and tumors with erythema, often overlying joints or extensor surfaces. Histopathologic examination after formalin fixation shows needle-shaped clefts within feathery amorphous pink areas surrounded by granuloma (Figure 2).11 Yellow, needle-shaped, negatively birefringent crystals can be viewed under polarized microscopy in alcohol-fixed samples.
Plantar fibromatosis (Ledderhose disease) is a benign proliferation of the plantar aponeurosis linked to alcohol use; liver disease; and notably epilepsy,12 a component of our patient's medical history. Large nodules appear grossly on the plantar feet and may progress to contractures in more advanced lesions. Biopsy reveals bland hyperproliferation of fibroblasts in a background of fascial fibrous tissue (Figure 3).12 Clinically, this diagnosis is part of the differential diagnosis of plantar nodules but appears histologically different than cutaneous macroglobulinosis because there are no hyaline deposits in plantar fibromatosis.
Erythropoietic protoporphyria is a rare disorder that primarily arises due to a congenital deficiency in the ferrochelatase enzyme involved in heme biosynthesis. Erythropoietic protoporphyria is the most common porphyria among children and typically presents in infancy or early childhood as a painful photosensitivity with ensuing cutaneous manifestations and possible hepatobiliary disease. Edema and severe burning pain can be noted within minutes of sun exposure in a dose-response relationship.13 Histologic findings of erythropoietic protoporphyria differ based on acute or chronic skin changes. Acute lesions exhibit a predominantly neutrophilic interstitial dermal infiltrate with vacuoles and intercellular edema. Chronic changes include the accumulation of a PAS-positive, amorphous, hyalinelike substance, similar to the microscopic findings of cutaneous macroglobulinosis (Figure 4).13
An acral fibrokeratoma is a benign fibroepithelial tumor that clinically appears as a flesh-colored or slightly erythematous exophytic nodule that most commonly is found on the fingers or toes. Thought to arise from trauma to the affected area, it is histologically characterized by interwoven collagenous bundles with overlying epidermal hyperkeratosis, acanthosis, and deep thickened rete ridges14 (Figure 5). Although multiple acral fibrokeratomas have been reported (similar to presentations of prurigo nodularis),15 they more commonly appear as solitary lesions as opposed to the numerous translucent papules seen in our patient.
- Camp BJ, Magro CM. Cutaneous macroglobulinosis: a case series. J Cutan Pathol. 2012;39:962-970.
- Dimopoulos MA, Alexanian R. Waldenstrom's macroglobulinemia. Blood. 1994;83:1452-1459.
- D'Acunto C, Nigrisoli E, Liardo EV, et al. Painful plantar nodules: a specific manifestation of cutaneous macroglobulinosis. J Am Acad Dermatol. 2014;71:E251-E252.
- Tichenor RE. Macroglobulinemia cutis. Arch Dermatol. 1978;114:280-281.
- Gressier L, Hotz C, Lelièvre JD, et al. Cutaneous macroglobulinosis: a report of 2 cases. Arch Dermatol. 2010;146:165-169.
- Spicknall KE, Dubas LE, Mutasim DF. Cutaneous macroglobulinosis with monotypic plasma cells: a specific manifestation of Waldenström macroglobulinemia. J Cutan Pathol. 2013;40:442-444.
- Lüftl M, Sauter-Jenne B, Gramatzki M, et al. Cutaneous macroglobulinosis deposits in a patient with IgM paraproteinemia/incipient Waldenström macroglobulinemia. J Dtsch Dermatol Ges. 2010;8:1000-1003.
- Mascaro JM, Montserrat E, Estrach T, et al. Specific cutaneous manifestations of Waldenstrom macroglobulinaemia: a report of two cases. Br J Dermatol. 1982;106:217-222.
- Hanke CW, Steck WD, Bergfeld WF, et al. Cutaneous macroglobulinosis. Arch Dermatol. 1980;116:575-577.
- Oshio-Yoshii A, Fujimoto N, Shiba Y, et al. Cutaneous macroglobulinosis: successful treatment with rituximab. J Eur Acad Dermatol Venereol. 2017;31:E30-E31.
- Gupta A, Rai S, Sinha R, et al. Tophi as an initial manifestation of gout. J Cytol. 2009;26:165-166.
- Carroll P, Henshaw RM, Garwood C, et al. Plantar fibromatosis: pathophysiology, surgical and nonsurgical therapies: an evidence-based review. Foot Ankle Spec. 2018;11:168-176.
- Michaels BD, Del Rosso JQ, Mobini N, et al. Erythropoietic protoporphyria: a case report and literature review. J Clin Aesthet Dermatol. 2010;3:44-48.
- Boffeli TJ, Abben KW. Acral fibrokeratoma of the foot treated with excision and trap door flap closure: a case report. J Foot Ankle Surg. 2014;53:449-452.
- Reed RJ. Multiple acral fibrokeratomas (a variant of prurigo nodularis). discussion of classification of acral fibrous nodules and of histogenesis of acral fibrokeratomas. Arch Dermatol. 1971;103:287-297.
- Camp BJ, Magro CM. Cutaneous macroglobulinosis: a case series. J Cutan Pathol. 2012;39:962-970.
- Dimopoulos MA, Alexanian R. Waldenstrom's macroglobulinemia. Blood. 1994;83:1452-1459.
- D'Acunto C, Nigrisoli E, Liardo EV, et al. Painful plantar nodules: a specific manifestation of cutaneous macroglobulinosis. J Am Acad Dermatol. 2014;71:E251-E252.
- Tichenor RE. Macroglobulinemia cutis. Arch Dermatol. 1978;114:280-281.
- Gressier L, Hotz C, Lelièvre JD, et al. Cutaneous macroglobulinosis: a report of 2 cases. Arch Dermatol. 2010;146:165-169.
- Spicknall KE, Dubas LE, Mutasim DF. Cutaneous macroglobulinosis with monotypic plasma cells: a specific manifestation of Waldenström macroglobulinemia. J Cutan Pathol. 2013;40:442-444.
- Lüftl M, Sauter-Jenne B, Gramatzki M, et al. Cutaneous macroglobulinosis deposits in a patient with IgM paraproteinemia/incipient Waldenström macroglobulinemia. J Dtsch Dermatol Ges. 2010;8:1000-1003.
- Mascaro JM, Montserrat E, Estrach T, et al. Specific cutaneous manifestations of Waldenstrom macroglobulinaemia: a report of two cases. Br J Dermatol. 1982;106:217-222.
- Hanke CW, Steck WD, Bergfeld WF, et al. Cutaneous macroglobulinosis. Arch Dermatol. 1980;116:575-577.
- Oshio-Yoshii A, Fujimoto N, Shiba Y, et al. Cutaneous macroglobulinosis: successful treatment with rituximab. J Eur Acad Dermatol Venereol. 2017;31:E30-E31.
- Gupta A, Rai S, Sinha R, et al. Tophi as an initial manifestation of gout. J Cytol. 2009;26:165-166.
- Carroll P, Henshaw RM, Garwood C, et al. Plantar fibromatosis: pathophysiology, surgical and nonsurgical therapies: an evidence-based review. Foot Ankle Spec. 2018;11:168-176.
- Michaels BD, Del Rosso JQ, Mobini N, et al. Erythropoietic protoporphyria: a case report and literature review. J Clin Aesthet Dermatol. 2010;3:44-48.
- Boffeli TJ, Abben KW. Acral fibrokeratoma of the foot treated with excision and trap door flap closure: a case report. J Foot Ankle Surg. 2014;53:449-452.
- Reed RJ. Multiple acral fibrokeratomas (a variant of prurigo nodularis). discussion of classification of acral fibrous nodules and of histogenesis of acral fibrokeratomas. Arch Dermatol. 1971;103:287-297.
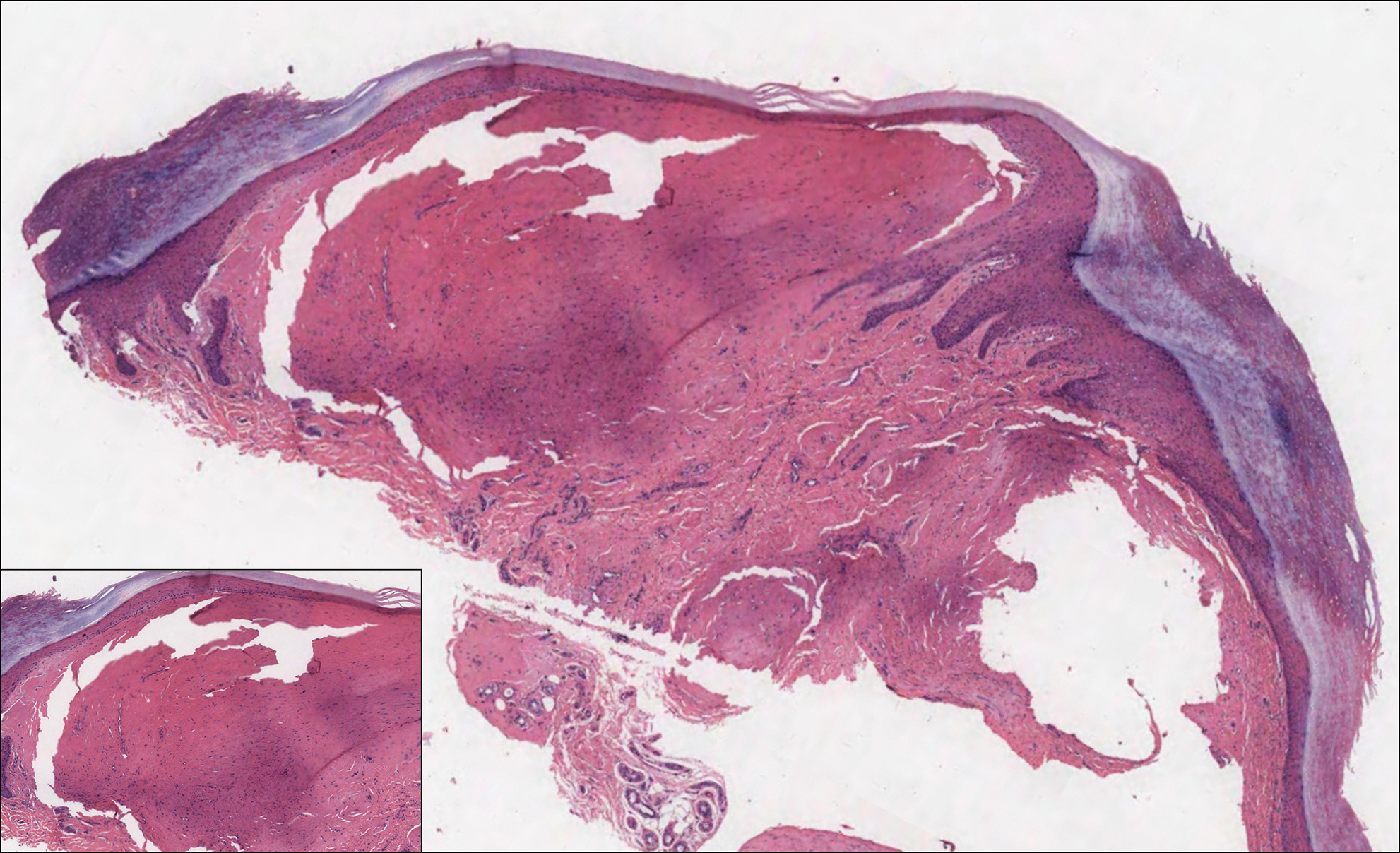
A 64-year-old woman with a medical history of Waldenström macroglobulinemia, multiple sclerosis, and epilepsy presented with slowly growing papules on the plantar feet of 21 months' duration. She was diagnosed with Waldenström macroglobulinemia incidentally on routine blood work 3 years prior and declined treatment because she was asymptomatic. Physical examination revealed a total of 20 firm, variably sized, light pink to purple, partially translucent and telangiectatic papules and plaques bilaterally on the plantar feet. A plaque from the right sole was biopsied.
Cross-contamination of Pathology Specimens: A Cautionary Tale
Cross-contamination of pathology specimens is a rare but nonnegligible source of potential morbidity in clinical practice. Contaminant tissue fragments, colloquially referred to as floaters, typically are readily identifiable based on obvious cytomorphologic differences, especially if the tissues arise from different organs; however, one cannot rely on such distinctions in a pathology laboratory dedicated to a single organ system (eg, dermatopathology). The inability to identify quickly and confidently the presence of a contaminant puts the patient at risk for misdiagnosis, which can lead to unnecessary morbidity or even mortality in the case of cancer misdiagnosis. Studies that have been conducted to estimate the incidence of this type of error have suggested an overall incidence rate between approximately 1% and 3%.1,2 Awareness of this phenomenon and careful scrutiny when the histopathologic evidence diverges considerably from the clinical impression is critical for minimizing the negative outcomes that could result from the presence of contaminant tissue. We present a case in which cross-contamination of a pathology specimen led to an initial erroneous diagnosis of an aggressive cutaneous melanoma in a patient with a benign adnexal neoplasm.
Case Report
A 72-year-old man was referred to the Pigmented Lesion and Melanoma Program at Stanford University Medical Center and Cancer Institute (Palo Alto, California) for evaluation and treatment of a presumed stage IIB melanoma on the right preauricular cheek based on a shave biopsy that had been performed (<1 month prior) by his local dermatology provider and subsequently read by an affiliated out-of-state dermatopathology laboratory. Per the clinical history that was gathered at the current presentation, neither the patient nor his wife had noticed the lesion prior to his dermatology provider pointing it out on the day of the biopsy. Additionally, he denied associated pain, bleeding, or ulceration. According to outside medical records, the referring dermatology provider described the lesion as a 4-mm pink pearly papule with telangiectasia favoring a diagnosis of basal cell carcinoma, and a diagnostic shave biopsy was performed. On presentation to our clinic, physical examination of the right preauricular cheek revealed a 4×3-mm depressed erythematous scar with no evidence of residual pigmentation or nodularity (Figure 1). There was no clinically appreciable regional lymphadenopathy.
The original dermatopathology report indicated an invasive melanoma with the following pathologic characteristics: superficial spreading type, Breslow depth of at least 2.16 mm, ulceration, and a mitotic index of 8 mitotic figures/mm2 with transection of the invasive component at the peripheral and deep margins. There was no evidence of regression, perineural invasion, lymphovascular invasion, or microsatellites. Interestingly, the report indicated that there also was a basaloid proliferation with features of cylindroma in the same pathology slide adjacent to the aggressive invasive melanoma that was described. Given the complexity of cases referred to our academic center, the standard of care includes internal dermatopathology review of all outside pathology specimens. This review proved critical to this patient’s care in light of the considerable divergence of the initial pathologic diagnosis and the reported clinical features of the lesion.
Internal review of the single pathology slide received from the referring provider showed a total of 4 sections, 3 of which are shown here (Figure 2A). Three sections, including the one not shown, were all consistent with a diagnosis of cylindroma and showed no evidence of a melanocytic proliferation (Figure 2B). However, the fourth section demonstrated marked morphologic dissimilarity compared to the other 3 sections. This outlier section showed a thick cutaneous melanoma with a Breslow depth of at least 2.1 mm, ulceration, a mitotic rate of 12 mitotic figures/mm2, and broad transection of the invasive component at the peripheral and deep margins (Figures 2C and 2D). Correlation with the gross description of tissue processing on the original pathology report indicating that the specimen had been trisected raised suspicion that the fourth and very dissimilar section could be a contaminant from another source that was incorporated into our patient’s histologic sections during processing. Taken together, these discrepancies made the diagnosis of cylindroma alone far more likely than cutaneous melanoma, but we needed conclusive evidence given the dramatic difference in prognosis and management between a cylindroma and an aggressive cutaneous melanoma.
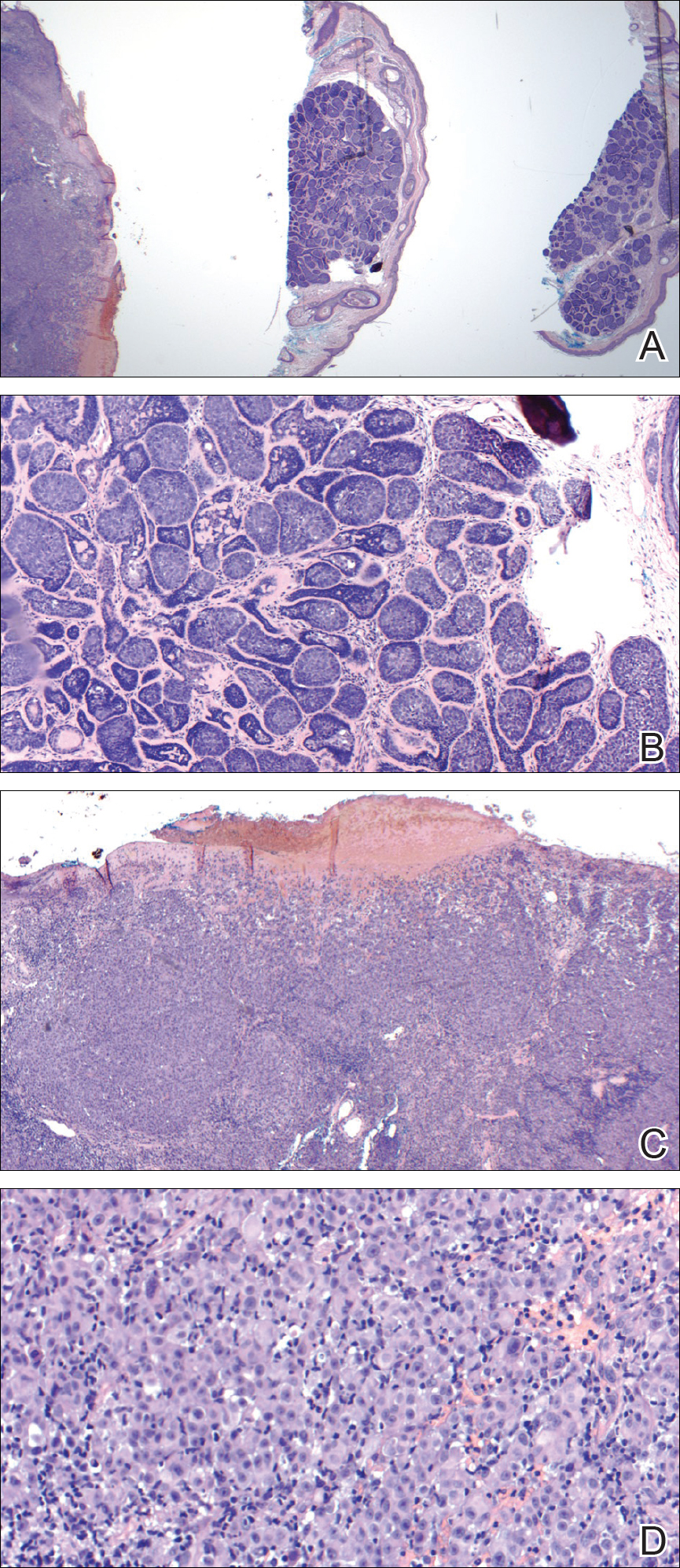
For further diagnostic clarification, we performed polymorphic short tandem repeat (STR) analysis, a well-described forensic pathology technique, to determine if the melanoma and cylindroma specimens derived from different patients, as we hypothesized. This analysis revealed differences in all but one DNA locus tested between the cylindroma specimen and the melanoma specimen, confirming our hypothesis (Figure 3). Subsequent discussion of the case with staff from the dermatopathology laboratory that processed this specimen provided further support for our suspicion that the invasive melanoma specimen was part of a case processed prior to our patient’s benign lesion. Therefore, the wide local excision for treatment of the suspected melanoma fortunately was canceled, and the patient did not require further treatment of the benign cylindroma. The patient expressed relief and gratitude for this critical clarification and change in management.
Comment
Shah et al3 reported a similar case in which a benign granuloma of the lung masqueraded as a squamous cell carcinoma due to histopathologic contamination. Although few similar cases have been described in the literature, the risk posed by such contamination is remarkable, regardless of whether it occurs during specimen grossing, embedding, sectioning, or staining.1,4,5 This risk is amplified in facilities that process specimens originating predominantly from a single organ system or tissue type, as is often the case in dedicated dermatopathology laboratories. In this scenario, it is unlikely that one could use the presence of tissues from 2 different organ systems on a single slide as a way of easily recognizing the presence of a contaminant and rectifying the error. Additionally, the presence of malignant cells in the contaminant further complicates the problem and requires an investigation that can conclusively distinguish the contaminant from the patient’s actual tissue.
In our case, our dermatology and dermatopathology teams partnered with our molecular pathology team to find a solution. Polymorphic STR analysis via polymerase chain reaction amplification is a sensitive method employed commonly in forensic DNA laboratories for determining whether a sample submitted as evidence belongs to a given suspect.6 Although much more commonly used in forensics, STR analysis does have known roles in clinical medicine, such as chimerism testing after bone marrow or allogeneic stem cell transplantation.7 Given the relatively short period of time it takes along with the convenience of commercially available kits, a high discriminative ability, and well-validated interpretation procedures, STR analysis is an excellent method for determining if a given tissue sample came from a given patient, which is what was needed in our case.
The combined clinical, histopathologic, and molecular data in our case allowed for confident clarification of our patient’s diagnosis, sparing him the morbidity of wide local excision on the face, sentinel lymph node biopsy, and emotional distress associated with a diagnosis of aggressive cutaneous melanoma. Our case highlights the critical importance of internal review of pathology specimens in ensuring proper diagnosis and management and reminds us that, though rare, accidental contamination during processing of pathology specimens is a potential adverse event that must be considered, especially when a pathologic finding diverges considerably from what is anticipated based on the patient’s history and physical examination.
Acknowledgment
The authors express gratitude to the patient described herein who graciously provided permission for us to publish his case and clinical photography.
- Gephardt GN, Zarbo RJ. Extraneous tissue in surgical pathology: a College of American Pathologists Q-Probes study of 275 laboratories. Arch Pathol Lab Med. 1996;120:1009-1014.
- Alam M, Shah AD, Ali S, et al. Floaters in Mohs micrographic surgery [published online June 27, 2013]. Dermatol Surg. 2013;39:1317-1322.
- Shah PA, Prat MP, Hostler DC. Benign granuloma masquerading as squamous cell carcinoma due to a “floater.” Hawaii J Med Public Health. 2017;76(11, suppl 2):19-21.
- Platt E, Sommer P, McDonald L, et al. Tissue floaters and contaminants in the histology laboratory. Arch Pathol Lab Med. 2009;133:973-978.
- Layfield LJ, Witt BL, Metzger KG, et al. Extraneous tissue: a potential source for diagnostic error in surgical pathology. Am J Clin Pathol. 2011;136:767-772.
- Butler JM. Forensic DNA testing. Cold Spring Harb Protoc. 2011;2011:1438-1450.
- Manasatienkij C, Ra-ngabpai C. Clinical application of forensic DNA analysis: a literature review. J Med Assoc Thai. 2012;95:1357-1363.
Cross-contamination of pathology specimens is a rare but nonnegligible source of potential morbidity in clinical practice. Contaminant tissue fragments, colloquially referred to as floaters, typically are readily identifiable based on obvious cytomorphologic differences, especially if the tissues arise from different organs; however, one cannot rely on such distinctions in a pathology laboratory dedicated to a single organ system (eg, dermatopathology). The inability to identify quickly and confidently the presence of a contaminant puts the patient at risk for misdiagnosis, which can lead to unnecessary morbidity or even mortality in the case of cancer misdiagnosis. Studies that have been conducted to estimate the incidence of this type of error have suggested an overall incidence rate between approximately 1% and 3%.1,2 Awareness of this phenomenon and careful scrutiny when the histopathologic evidence diverges considerably from the clinical impression is critical for minimizing the negative outcomes that could result from the presence of contaminant tissue. We present a case in which cross-contamination of a pathology specimen led to an initial erroneous diagnosis of an aggressive cutaneous melanoma in a patient with a benign adnexal neoplasm.
Case Report
A 72-year-old man was referred to the Pigmented Lesion and Melanoma Program at Stanford University Medical Center and Cancer Institute (Palo Alto, California) for evaluation and treatment of a presumed stage IIB melanoma on the right preauricular cheek based on a shave biopsy that had been performed (<1 month prior) by his local dermatology provider and subsequently read by an affiliated out-of-state dermatopathology laboratory. Per the clinical history that was gathered at the current presentation, neither the patient nor his wife had noticed the lesion prior to his dermatology provider pointing it out on the day of the biopsy. Additionally, he denied associated pain, bleeding, or ulceration. According to outside medical records, the referring dermatology provider described the lesion as a 4-mm pink pearly papule with telangiectasia favoring a diagnosis of basal cell carcinoma, and a diagnostic shave biopsy was performed. On presentation to our clinic, physical examination of the right preauricular cheek revealed a 4×3-mm depressed erythematous scar with no evidence of residual pigmentation or nodularity (Figure 1). There was no clinically appreciable regional lymphadenopathy.
The original dermatopathology report indicated an invasive melanoma with the following pathologic characteristics: superficial spreading type, Breslow depth of at least 2.16 mm, ulceration, and a mitotic index of 8 mitotic figures/mm2 with transection of the invasive component at the peripheral and deep margins. There was no evidence of regression, perineural invasion, lymphovascular invasion, or microsatellites. Interestingly, the report indicated that there also was a basaloid proliferation with features of cylindroma in the same pathology slide adjacent to the aggressive invasive melanoma that was described. Given the complexity of cases referred to our academic center, the standard of care includes internal dermatopathology review of all outside pathology specimens. This review proved critical to this patient’s care in light of the considerable divergence of the initial pathologic diagnosis and the reported clinical features of the lesion.
Internal review of the single pathology slide received from the referring provider showed a total of 4 sections, 3 of which are shown here (Figure 2A). Three sections, including the one not shown, were all consistent with a diagnosis of cylindroma and showed no evidence of a melanocytic proliferation (Figure 2B). However, the fourth section demonstrated marked morphologic dissimilarity compared to the other 3 sections. This outlier section showed a thick cutaneous melanoma with a Breslow depth of at least 2.1 mm, ulceration, a mitotic rate of 12 mitotic figures/mm2, and broad transection of the invasive component at the peripheral and deep margins (Figures 2C and 2D). Correlation with the gross description of tissue processing on the original pathology report indicating that the specimen had been trisected raised suspicion that the fourth and very dissimilar section could be a contaminant from another source that was incorporated into our patient’s histologic sections during processing. Taken together, these discrepancies made the diagnosis of cylindroma alone far more likely than cutaneous melanoma, but we needed conclusive evidence given the dramatic difference in prognosis and management between a cylindroma and an aggressive cutaneous melanoma.
For further diagnostic clarification, we performed polymorphic short tandem repeat (STR) analysis, a well-described forensic pathology technique, to determine if the melanoma and cylindroma specimens derived from different patients, as we hypothesized. This analysis revealed differences in all but one DNA locus tested between the cylindroma specimen and the melanoma specimen, confirming our hypothesis (Figure 3). Subsequent discussion of the case with staff from the dermatopathology laboratory that processed this specimen provided further support for our suspicion that the invasive melanoma specimen was part of a case processed prior to our patient’s benign lesion. Therefore, the wide local excision for treatment of the suspected melanoma fortunately was canceled, and the patient did not require further treatment of the benign cylindroma. The patient expressed relief and gratitude for this critical clarification and change in management.
Comment
Shah et al3 reported a similar case in which a benign granuloma of the lung masqueraded as a squamous cell carcinoma due to histopathologic contamination. Although few similar cases have been described in the literature, the risk posed by such contamination is remarkable, regardless of whether it occurs during specimen grossing, embedding, sectioning, or staining.1,4,5 This risk is amplified in facilities that process specimens originating predominantly from a single organ system or tissue type, as is often the case in dedicated dermatopathology laboratories. In this scenario, it is unlikely that one could use the presence of tissues from 2 different organ systems on a single slide as a way of easily recognizing the presence of a contaminant and rectifying the error. Additionally, the presence of malignant cells in the contaminant further complicates the problem and requires an investigation that can conclusively distinguish the contaminant from the patient’s actual tissue.
In our case, our dermatology and dermatopathology teams partnered with our molecular pathology team to find a solution. Polymorphic STR analysis via polymerase chain reaction amplification is a sensitive method employed commonly in forensic DNA laboratories for determining whether a sample submitted as evidence belongs to a given suspect.6 Although much more commonly used in forensics, STR analysis does have known roles in clinical medicine, such as chimerism testing after bone marrow or allogeneic stem cell transplantation.7 Given the relatively short period of time it takes along with the convenience of commercially available kits, a high discriminative ability, and well-validated interpretation procedures, STR analysis is an excellent method for determining if a given tissue sample came from a given patient, which is what was needed in our case.
The combined clinical, histopathologic, and molecular data in our case allowed for confident clarification of our patient’s diagnosis, sparing him the morbidity of wide local excision on the face, sentinel lymph node biopsy, and emotional distress associated with a diagnosis of aggressive cutaneous melanoma. Our case highlights the critical importance of internal review of pathology specimens in ensuring proper diagnosis and management and reminds us that, though rare, accidental contamination during processing of pathology specimens is a potential adverse event that must be considered, especially when a pathologic finding diverges considerably from what is anticipated based on the patient’s history and physical examination.
Acknowledgment
The authors express gratitude to the patient described herein who graciously provided permission for us to publish his case and clinical photography.
Cross-contamination of pathology specimens is a rare but nonnegligible source of potential morbidity in clinical practice. Contaminant tissue fragments, colloquially referred to as floaters, typically are readily identifiable based on obvious cytomorphologic differences, especially if the tissues arise from different organs; however, one cannot rely on such distinctions in a pathology laboratory dedicated to a single organ system (eg, dermatopathology). The inability to identify quickly and confidently the presence of a contaminant puts the patient at risk for misdiagnosis, which can lead to unnecessary morbidity or even mortality in the case of cancer misdiagnosis. Studies that have been conducted to estimate the incidence of this type of error have suggested an overall incidence rate between approximately 1% and 3%.1,2 Awareness of this phenomenon and careful scrutiny when the histopathologic evidence diverges considerably from the clinical impression is critical for minimizing the negative outcomes that could result from the presence of contaminant tissue. We present a case in which cross-contamination of a pathology specimen led to an initial erroneous diagnosis of an aggressive cutaneous melanoma in a patient with a benign adnexal neoplasm.
Case Report
A 72-year-old man was referred to the Pigmented Lesion and Melanoma Program at Stanford University Medical Center and Cancer Institute (Palo Alto, California) for evaluation and treatment of a presumed stage IIB melanoma on the right preauricular cheek based on a shave biopsy that had been performed (<1 month prior) by his local dermatology provider and subsequently read by an affiliated out-of-state dermatopathology laboratory. Per the clinical history that was gathered at the current presentation, neither the patient nor his wife had noticed the lesion prior to his dermatology provider pointing it out on the day of the biopsy. Additionally, he denied associated pain, bleeding, or ulceration. According to outside medical records, the referring dermatology provider described the lesion as a 4-mm pink pearly papule with telangiectasia favoring a diagnosis of basal cell carcinoma, and a diagnostic shave biopsy was performed. On presentation to our clinic, physical examination of the right preauricular cheek revealed a 4×3-mm depressed erythematous scar with no evidence of residual pigmentation or nodularity (Figure 1). There was no clinically appreciable regional lymphadenopathy.
The original dermatopathology report indicated an invasive melanoma with the following pathologic characteristics: superficial spreading type, Breslow depth of at least 2.16 mm, ulceration, and a mitotic index of 8 mitotic figures/mm2 with transection of the invasive component at the peripheral and deep margins. There was no evidence of regression, perineural invasion, lymphovascular invasion, or microsatellites. Interestingly, the report indicated that there also was a basaloid proliferation with features of cylindroma in the same pathology slide adjacent to the aggressive invasive melanoma that was described. Given the complexity of cases referred to our academic center, the standard of care includes internal dermatopathology review of all outside pathology specimens. This review proved critical to this patient’s care in light of the considerable divergence of the initial pathologic diagnosis and the reported clinical features of the lesion.
Internal review of the single pathology slide received from the referring provider showed a total of 4 sections, 3 of which are shown here (Figure 2A). Three sections, including the one not shown, were all consistent with a diagnosis of cylindroma and showed no evidence of a melanocytic proliferation (Figure 2B). However, the fourth section demonstrated marked morphologic dissimilarity compared to the other 3 sections. This outlier section showed a thick cutaneous melanoma with a Breslow depth of at least 2.1 mm, ulceration, a mitotic rate of 12 mitotic figures/mm2, and broad transection of the invasive component at the peripheral and deep margins (Figures 2C and 2D). Correlation with the gross description of tissue processing on the original pathology report indicating that the specimen had been trisected raised suspicion that the fourth and very dissimilar section could be a contaminant from another source that was incorporated into our patient’s histologic sections during processing. Taken together, these discrepancies made the diagnosis of cylindroma alone far more likely than cutaneous melanoma, but we needed conclusive evidence given the dramatic difference in prognosis and management between a cylindroma and an aggressive cutaneous melanoma.
For further diagnostic clarification, we performed polymorphic short tandem repeat (STR) analysis, a well-described forensic pathology technique, to determine if the melanoma and cylindroma specimens derived from different patients, as we hypothesized. This analysis revealed differences in all but one DNA locus tested between the cylindroma specimen and the melanoma specimen, confirming our hypothesis (Figure 3). Subsequent discussion of the case with staff from the dermatopathology laboratory that processed this specimen provided further support for our suspicion that the invasive melanoma specimen was part of a case processed prior to our patient’s benign lesion. Therefore, the wide local excision for treatment of the suspected melanoma fortunately was canceled, and the patient did not require further treatment of the benign cylindroma. The patient expressed relief and gratitude for this critical clarification and change in management.
Comment
Shah et al3 reported a similar case in which a benign granuloma of the lung masqueraded as a squamous cell carcinoma due to histopathologic contamination. Although few similar cases have been described in the literature, the risk posed by such contamination is remarkable, regardless of whether it occurs during specimen grossing, embedding, sectioning, or staining.1,4,5 This risk is amplified in facilities that process specimens originating predominantly from a single organ system or tissue type, as is often the case in dedicated dermatopathology laboratories. In this scenario, it is unlikely that one could use the presence of tissues from 2 different organ systems on a single slide as a way of easily recognizing the presence of a contaminant and rectifying the error. Additionally, the presence of malignant cells in the contaminant further complicates the problem and requires an investigation that can conclusively distinguish the contaminant from the patient’s actual tissue.
In our case, our dermatology and dermatopathology teams partnered with our molecular pathology team to find a solution. Polymorphic STR analysis via polymerase chain reaction amplification is a sensitive method employed commonly in forensic DNA laboratories for determining whether a sample submitted as evidence belongs to a given suspect.6 Although much more commonly used in forensics, STR analysis does have known roles in clinical medicine, such as chimerism testing after bone marrow or allogeneic stem cell transplantation.7 Given the relatively short period of time it takes along with the convenience of commercially available kits, a high discriminative ability, and well-validated interpretation procedures, STR analysis is an excellent method for determining if a given tissue sample came from a given patient, which is what was needed in our case.
The combined clinical, histopathologic, and molecular data in our case allowed for confident clarification of our patient’s diagnosis, sparing him the morbidity of wide local excision on the face, sentinel lymph node biopsy, and emotional distress associated with a diagnosis of aggressive cutaneous melanoma. Our case highlights the critical importance of internal review of pathology specimens in ensuring proper diagnosis and management and reminds us that, though rare, accidental contamination during processing of pathology specimens is a potential adverse event that must be considered, especially when a pathologic finding diverges considerably from what is anticipated based on the patient’s history and physical examination.
Acknowledgment
The authors express gratitude to the patient described herein who graciously provided permission for us to publish his case and clinical photography.
- Gephardt GN, Zarbo RJ. Extraneous tissue in surgical pathology: a College of American Pathologists Q-Probes study of 275 laboratories. Arch Pathol Lab Med. 1996;120:1009-1014.
- Alam M, Shah AD, Ali S, et al. Floaters in Mohs micrographic surgery [published online June 27, 2013]. Dermatol Surg. 2013;39:1317-1322.
- Shah PA, Prat MP, Hostler DC. Benign granuloma masquerading as squamous cell carcinoma due to a “floater.” Hawaii J Med Public Health. 2017;76(11, suppl 2):19-21.
- Platt E, Sommer P, McDonald L, et al. Tissue floaters and contaminants in the histology laboratory. Arch Pathol Lab Med. 2009;133:973-978.
- Layfield LJ, Witt BL, Metzger KG, et al. Extraneous tissue: a potential source for diagnostic error in surgical pathology. Am J Clin Pathol. 2011;136:767-772.
- Butler JM. Forensic DNA testing. Cold Spring Harb Protoc. 2011;2011:1438-1450.
- Manasatienkij C, Ra-ngabpai C. Clinical application of forensic DNA analysis: a literature review. J Med Assoc Thai. 2012;95:1357-1363.
- Gephardt GN, Zarbo RJ. Extraneous tissue in surgical pathology: a College of American Pathologists Q-Probes study of 275 laboratories. Arch Pathol Lab Med. 1996;120:1009-1014.
- Alam M, Shah AD, Ali S, et al. Floaters in Mohs micrographic surgery [published online June 27, 2013]. Dermatol Surg. 2013;39:1317-1322.
- Shah PA, Prat MP, Hostler DC. Benign granuloma masquerading as squamous cell carcinoma due to a “floater.” Hawaii J Med Public Health. 2017;76(11, suppl 2):19-21.
- Platt E, Sommer P, McDonald L, et al. Tissue floaters and contaminants in the histology laboratory. Arch Pathol Lab Med. 2009;133:973-978.
- Layfield LJ, Witt BL, Metzger KG, et al. Extraneous tissue: a potential source for diagnostic error in surgical pathology. Am J Clin Pathol. 2011;136:767-772.
- Butler JM. Forensic DNA testing. Cold Spring Harb Protoc. 2011;2011:1438-1450.
- Manasatienkij C, Ra-ngabpai C. Clinical application of forensic DNA analysis: a literature review. J Med Assoc Thai. 2012;95:1357-1363.
Resident Pearl
- Although cross-contamination of pathology specimens is rare, it does occur and can impact diagnosis and management if detected early.
Solitary Exophytic Plaque on the Left Groin
The Diagnosis: Pemphigus Vegetans
A punch biopsy was taken from the verrucous plaque, and microscopic examination demonstrated prominent epidermal hyperplasia with intraepidermal eosinophilic microabscesses and a superficial dermatitis with abundant eosinophils (Figure 1A). Suprabasal acantholytic cleft formation was noted in a focal area (Figure 1B). Another punch biopsy was performed from the perilesional skin for direct immunofluorescence examination, which revealed intercellular deposits of IgG and C3 throughout the lower half of the epidermis (Figure 1C). Indirect immunofluorescence performed on monkey esophagus substrate showed circulating intercellular IgG antibodies in all the titers of up to 1/160 and an elevated level of IgG antidesmoglein 3 (anti-Dsg3) antibody (enzyme-linked immunosorbent assay index value, >200 RU/mL [reference range, <20 RU/mL]).
Because there was a solitary lesion, the decision was made to perform local treatment. One intralesional triamcinolone acetonide injection (20 mg/mL) resulted in remarkable flattening of the lesion (Figure 2). Subsequently, treatment was continued with clobetasol propionate ointment 3 times weekly for 1 month. During a follow-up period of 2 years, no signs of local relapse or new lesions elsewhere were noted, and the patient continued to be on long-term longitudinal evaluation.
Pemphigus vegetans (PV) is an uncommon variant of pemphigus, typically manifesting with vegetating erosions and plaques localized to the intertriginous areas of the body. Local factors such as semiocclusion, maceration, and/or bacterial or fungal colonization have been hypothesized to account for the distinctive localization and vegetation of the lesions.1,2 Traditionally, 2 clinical subtypes of PV have been described: (1) Hallopeau type presenting with pustules that later evolve into vegetating plaques, and (2) Neumann type that initially manifests as vesicles and bullae with a more disseminated distribution, transforming into hypertrophic masses with erosions.1-5 However, this distinction may not always be clear, and patients with features of both forms have been reported.2,5
At present, our case would best be regarded as a localized form of PV presenting with a solitary lesion. It may progress to more disseminated disease or remain localized during its course; the literature contains reports exemplifying both possibilities. In a large retrospective study from Tunisia encompassing almost 3 decades, the majority of the patients initially presented with unifocal involvement; however, the disease eventually became multifocal in almost all patients during the study period, emphasizing the need for long-term follow-up.2 There also are reports of PV confined to a single anatomic site, such as the scalp, sole, or vulva, that remained localized for years.2,4,6,7 Involvement of the oral mucosa is an important finding of PV and the presenting concern in approximately three-quarters of patients.2 Interestingly, the oral mucosa was not involved in our patient despite the high titer of anti-Dsg3 antibody, which suggests the need for the presence of other factors for clinical expression of the disease.
Although PV is considered a vegetating clinicomorphologic variant of pemphigus vulgaris, PV is histopathologically distinguished from pemphigus vulgaris by the presence of epidermal hyperplasia and intraepidermal eosinophilic microabscesses. Importantly, the epidermis displays signs of exuberant proliferation such as pseudoepitheliomatous hyperplasia and/or papillomatosis of a varying degree.1,2,5 Of note, suprabasal acantholysis is usually overshadowed by the changes in PV and presents only focally, as in our patient. The most common autoantibody profile is IgG targeting Dsg3; however, a spectrum of other autoantibodies has been identified, such as IgG antidesmocollin 3, IgA anti-Dsg3, and IgG anti-Dsg1.8,9
The most important differential diagnosis of PV is pyodermatitis-pyostomatitis vegetans. These 2 entities share many clinical and histopathological features; however, direct immunofluorescence is helpfulfor differentiation because it generally is negative in pyodermatitis-pyostomatitis vegetans.2,10 Furthermore, there is a well-established association between pyodermatitis-pyostomatitis vegetans and inflammatory bowel disorders, whereas PV has anecdotally been linked to malignancy, human immunodeficiency virus infection, and heroin abuse.1,2,10 Our patient was seronegative for human immunodeficiency virus and denied weight loss or loss of appetite. For those cases of PV involving a single anatomic site, the differential diagnosis is broader and encompasses dermatoses such as verrucae, syphilitic chancre, condylomata lata, granuloma inguinale, herpes simplex virus infection, and Kaposi sarcoma.
Treatment of PV is similar to pemphigus vulgaris and consists of a combination of systemic corticosteroids and steroid-sparing agents.1,5 On the other hand, more limited presentations of PV may be suitable for intralesional treatment with triamcinolone acetonide, thus avoiding potential adverse effects of systemic therapy.1,2 In our case with localized involvement, a favorable response was obtained with intralesional triamcinolone acetonide, and we plan to utilize systemic corticosteroids if the disease becomes generalized during follow-up.
- Ruocco V, Ruocco E, Caccavale S, et al. Pemphigus vegetans of the folds (intertriginous areas). Clin Dermatol. 2015;33:471-476.
- Zaraa I, Sellami A, Bouguerra C, et al. Pemphigus vegetans: a clinical, histological, immunopathological and prognostic study. J Eur Acad Dermatol Venereol. 2011;25:1160-1167.
- Madan V, August PJ. Exophytic plaques, blisters, and mouth ulcers. pemphigus vegetans (PV), Neumann type. Arch Dermatol. 2009;145:715-720.
- Mori M, Mariotti G, Grandi V, et al. Pemphigus vegetans of the scalp. J Eur Acad Dermatol Venereol. 2016;30:368-370.
- Monshi B, Marker M, Feichtinger H, et al. Pemphigus vegetans--immunopathological findings in a rare variant of pemphigus vulgaris. J Dtsch Dermatol Ges. 2010;8:179-183.
- Jain VK, Dixit VB, Mohan H. Pemphigus vegetans in an unusual site. Int J Dermatol. 1989;28:352-353.
- Wong KT, Wong KK. A case of acantholytic dermatosis of the vulva with features of pemphigus vegetans. J Cutan Pathol. 1994;21:453-456.
- Morizane S, Yamamoto T, Hisamatsu Y, et al. Pemphigus vegetans with IgG and IgA antidesmoglein 3 antibodies. Br J Dermatol. 2005;153:1236-1237.
- Saruta H, Ishii N, Teye K, et al. Two cases of pemphigus vegetans with IgG anti-desmocollin 3 antibodies. JAMA Dermatol. 2013;149:1209-1213.
- Mehravaran M, Kemény L, Husz S, et al. Pyodermatitis-pyostomatitis vegetans. Br J Dermatol. 1997;137:266-269.
The Diagnosis: Pemphigus Vegetans
A punch biopsy was taken from the verrucous plaque, and microscopic examination demonstrated prominent epidermal hyperplasia with intraepidermal eosinophilic microabscesses and a superficial dermatitis with abundant eosinophils (Figure 1A). Suprabasal acantholytic cleft formation was noted in a focal area (Figure 1B). Another punch biopsy was performed from the perilesional skin for direct immunofluorescence examination, which revealed intercellular deposits of IgG and C3 throughout the lower half of the epidermis (Figure 1C). Indirect immunofluorescence performed on monkey esophagus substrate showed circulating intercellular IgG antibodies in all the titers of up to 1/160 and an elevated level of IgG antidesmoglein 3 (anti-Dsg3) antibody (enzyme-linked immunosorbent assay index value, >200 RU/mL [reference range, <20 RU/mL]).
Because there was a solitary lesion, the decision was made to perform local treatment. One intralesional triamcinolone acetonide injection (20 mg/mL) resulted in remarkable flattening of the lesion (Figure 2). Subsequently, treatment was continued with clobetasol propionate ointment 3 times weekly for 1 month. During a follow-up period of 2 years, no signs of local relapse or new lesions elsewhere were noted, and the patient continued to be on long-term longitudinal evaluation.
Pemphigus vegetans (PV) is an uncommon variant of pemphigus, typically manifesting with vegetating erosions and plaques localized to the intertriginous areas of the body. Local factors such as semiocclusion, maceration, and/or bacterial or fungal colonization have been hypothesized to account for the distinctive localization and vegetation of the lesions.1,2 Traditionally, 2 clinical subtypes of PV have been described: (1) Hallopeau type presenting with pustules that later evolve into vegetating plaques, and (2) Neumann type that initially manifests as vesicles and bullae with a more disseminated distribution, transforming into hypertrophic masses with erosions.1-5 However, this distinction may not always be clear, and patients with features of both forms have been reported.2,5
At present, our case would best be regarded as a localized form of PV presenting with a solitary lesion. It may progress to more disseminated disease or remain localized during its course; the literature contains reports exemplifying both possibilities. In a large retrospective study from Tunisia encompassing almost 3 decades, the majority of the patients initially presented with unifocal involvement; however, the disease eventually became multifocal in almost all patients during the study period, emphasizing the need for long-term follow-up.2 There also are reports of PV confined to a single anatomic site, such as the scalp, sole, or vulva, that remained localized for years.2,4,6,7 Involvement of the oral mucosa is an important finding of PV and the presenting concern in approximately three-quarters of patients.2 Interestingly, the oral mucosa was not involved in our patient despite the high titer of anti-Dsg3 antibody, which suggests the need for the presence of other factors for clinical expression of the disease.
Although PV is considered a vegetating clinicomorphologic variant of pemphigus vulgaris, PV is histopathologically distinguished from pemphigus vulgaris by the presence of epidermal hyperplasia and intraepidermal eosinophilic microabscesses. Importantly, the epidermis displays signs of exuberant proliferation such as pseudoepitheliomatous hyperplasia and/or papillomatosis of a varying degree.1,2,5 Of note, suprabasal acantholysis is usually overshadowed by the changes in PV and presents only focally, as in our patient. The most common autoantibody profile is IgG targeting Dsg3; however, a spectrum of other autoantibodies has been identified, such as IgG antidesmocollin 3, IgA anti-Dsg3, and IgG anti-Dsg1.8,9
The most important differential diagnosis of PV is pyodermatitis-pyostomatitis vegetans. These 2 entities share many clinical and histopathological features; however, direct immunofluorescence is helpfulfor differentiation because it generally is negative in pyodermatitis-pyostomatitis vegetans.2,10 Furthermore, there is a well-established association between pyodermatitis-pyostomatitis vegetans and inflammatory bowel disorders, whereas PV has anecdotally been linked to malignancy, human immunodeficiency virus infection, and heroin abuse.1,2,10 Our patient was seronegative for human immunodeficiency virus and denied weight loss or loss of appetite. For those cases of PV involving a single anatomic site, the differential diagnosis is broader and encompasses dermatoses such as verrucae, syphilitic chancre, condylomata lata, granuloma inguinale, herpes simplex virus infection, and Kaposi sarcoma.
Treatment of PV is similar to pemphigus vulgaris and consists of a combination of systemic corticosteroids and steroid-sparing agents.1,5 On the other hand, more limited presentations of PV may be suitable for intralesional treatment with triamcinolone acetonide, thus avoiding potential adverse effects of systemic therapy.1,2 In our case with localized involvement, a favorable response was obtained with intralesional triamcinolone acetonide, and we plan to utilize systemic corticosteroids if the disease becomes generalized during follow-up.
The Diagnosis: Pemphigus Vegetans
A punch biopsy was taken from the verrucous plaque, and microscopic examination demonstrated prominent epidermal hyperplasia with intraepidermal eosinophilic microabscesses and a superficial dermatitis with abundant eosinophils (Figure 1A). Suprabasal acantholytic cleft formation was noted in a focal area (Figure 1B). Another punch biopsy was performed from the perilesional skin for direct immunofluorescence examination, which revealed intercellular deposits of IgG and C3 throughout the lower half of the epidermis (Figure 1C). Indirect immunofluorescence performed on monkey esophagus substrate showed circulating intercellular IgG antibodies in all the titers of up to 1/160 and an elevated level of IgG antidesmoglein 3 (anti-Dsg3) antibody (enzyme-linked immunosorbent assay index value, >200 RU/mL [reference range, <20 RU/mL]).
Because there was a solitary lesion, the decision was made to perform local treatment. One intralesional triamcinolone acetonide injection (20 mg/mL) resulted in remarkable flattening of the lesion (Figure 2). Subsequently, treatment was continued with clobetasol propionate ointment 3 times weekly for 1 month. During a follow-up period of 2 years, no signs of local relapse or new lesions elsewhere were noted, and the patient continued to be on long-term longitudinal evaluation.
Pemphigus vegetans (PV) is an uncommon variant of pemphigus, typically manifesting with vegetating erosions and plaques localized to the intertriginous areas of the body. Local factors such as semiocclusion, maceration, and/or bacterial or fungal colonization have been hypothesized to account for the distinctive localization and vegetation of the lesions.1,2 Traditionally, 2 clinical subtypes of PV have been described: (1) Hallopeau type presenting with pustules that later evolve into vegetating plaques, and (2) Neumann type that initially manifests as vesicles and bullae with a more disseminated distribution, transforming into hypertrophic masses with erosions.1-5 However, this distinction may not always be clear, and patients with features of both forms have been reported.2,5
At present, our case would best be regarded as a localized form of PV presenting with a solitary lesion. It may progress to more disseminated disease or remain localized during its course; the literature contains reports exemplifying both possibilities. In a large retrospective study from Tunisia encompassing almost 3 decades, the majority of the patients initially presented with unifocal involvement; however, the disease eventually became multifocal in almost all patients during the study period, emphasizing the need for long-term follow-up.2 There also are reports of PV confined to a single anatomic site, such as the scalp, sole, or vulva, that remained localized for years.2,4,6,7 Involvement of the oral mucosa is an important finding of PV and the presenting concern in approximately three-quarters of patients.2 Interestingly, the oral mucosa was not involved in our patient despite the high titer of anti-Dsg3 antibody, which suggests the need for the presence of other factors for clinical expression of the disease.
Although PV is considered a vegetating clinicomorphologic variant of pemphigus vulgaris, PV is histopathologically distinguished from pemphigus vulgaris by the presence of epidermal hyperplasia and intraepidermal eosinophilic microabscesses. Importantly, the epidermis displays signs of exuberant proliferation such as pseudoepitheliomatous hyperplasia and/or papillomatosis of a varying degree.1,2,5 Of note, suprabasal acantholysis is usually overshadowed by the changes in PV and presents only focally, as in our patient. The most common autoantibody profile is IgG targeting Dsg3; however, a spectrum of other autoantibodies has been identified, such as IgG antidesmocollin 3, IgA anti-Dsg3, and IgG anti-Dsg1.8,9
The most important differential diagnosis of PV is pyodermatitis-pyostomatitis vegetans. These 2 entities share many clinical and histopathological features; however, direct immunofluorescence is helpfulfor differentiation because it generally is negative in pyodermatitis-pyostomatitis vegetans.2,10 Furthermore, there is a well-established association between pyodermatitis-pyostomatitis vegetans and inflammatory bowel disorders, whereas PV has anecdotally been linked to malignancy, human immunodeficiency virus infection, and heroin abuse.1,2,10 Our patient was seronegative for human immunodeficiency virus and denied weight loss or loss of appetite. For those cases of PV involving a single anatomic site, the differential diagnosis is broader and encompasses dermatoses such as verrucae, syphilitic chancre, condylomata lata, granuloma inguinale, herpes simplex virus infection, and Kaposi sarcoma.
Treatment of PV is similar to pemphigus vulgaris and consists of a combination of systemic corticosteroids and steroid-sparing agents.1,5 On the other hand, more limited presentations of PV may be suitable for intralesional treatment with triamcinolone acetonide, thus avoiding potential adverse effects of systemic therapy.1,2 In our case with localized involvement, a favorable response was obtained with intralesional triamcinolone acetonide, and we plan to utilize systemic corticosteroids if the disease becomes generalized during follow-up.
- Ruocco V, Ruocco E, Caccavale S, et al. Pemphigus vegetans of the folds (intertriginous areas). Clin Dermatol. 2015;33:471-476.
- Zaraa I, Sellami A, Bouguerra C, et al. Pemphigus vegetans: a clinical, histological, immunopathological and prognostic study. J Eur Acad Dermatol Venereol. 2011;25:1160-1167.
- Madan V, August PJ. Exophytic plaques, blisters, and mouth ulcers. pemphigus vegetans (PV), Neumann type. Arch Dermatol. 2009;145:715-720.
- Mori M, Mariotti G, Grandi V, et al. Pemphigus vegetans of the scalp. J Eur Acad Dermatol Venereol. 2016;30:368-370.
- Monshi B, Marker M, Feichtinger H, et al. Pemphigus vegetans--immunopathological findings in a rare variant of pemphigus vulgaris. J Dtsch Dermatol Ges. 2010;8:179-183.
- Jain VK, Dixit VB, Mohan H. Pemphigus vegetans in an unusual site. Int J Dermatol. 1989;28:352-353.
- Wong KT, Wong KK. A case of acantholytic dermatosis of the vulva with features of pemphigus vegetans. J Cutan Pathol. 1994;21:453-456.
- Morizane S, Yamamoto T, Hisamatsu Y, et al. Pemphigus vegetans with IgG and IgA antidesmoglein 3 antibodies. Br J Dermatol. 2005;153:1236-1237.
- Saruta H, Ishii N, Teye K, et al. Two cases of pemphigus vegetans with IgG anti-desmocollin 3 antibodies. JAMA Dermatol. 2013;149:1209-1213.
- Mehravaran M, Kemény L, Husz S, et al. Pyodermatitis-pyostomatitis vegetans. Br J Dermatol. 1997;137:266-269.
- Ruocco V, Ruocco E, Caccavale S, et al. Pemphigus vegetans of the folds (intertriginous areas). Clin Dermatol. 2015;33:471-476.
- Zaraa I, Sellami A, Bouguerra C, et al. Pemphigus vegetans: a clinical, histological, immunopathological and prognostic study. J Eur Acad Dermatol Venereol. 2011;25:1160-1167.
- Madan V, August PJ. Exophytic plaques, blisters, and mouth ulcers. pemphigus vegetans (PV), Neumann type. Arch Dermatol. 2009;145:715-720.
- Mori M, Mariotti G, Grandi V, et al. Pemphigus vegetans of the scalp. J Eur Acad Dermatol Venereol. 2016;30:368-370.
- Monshi B, Marker M, Feichtinger H, et al. Pemphigus vegetans--immunopathological findings in a rare variant of pemphigus vulgaris. J Dtsch Dermatol Ges. 2010;8:179-183.
- Jain VK, Dixit VB, Mohan H. Pemphigus vegetans in an unusual site. Int J Dermatol. 1989;28:352-353.
- Wong KT, Wong KK. A case of acantholytic dermatosis of the vulva with features of pemphigus vegetans. J Cutan Pathol. 1994;21:453-456.
- Morizane S, Yamamoto T, Hisamatsu Y, et al. Pemphigus vegetans with IgG and IgA antidesmoglein 3 antibodies. Br J Dermatol. 2005;153:1236-1237.
- Saruta H, Ishii N, Teye K, et al. Two cases of pemphigus vegetans with IgG anti-desmocollin 3 antibodies. JAMA Dermatol. 2013;149:1209-1213.
- Mehravaran M, Kemény L, Husz S, et al. Pyodermatitis-pyostomatitis vegetans. Br J Dermatol. 1997;137:266-269.
A 40-year-old otherwise healthy man presented with an exophytic plaque on the left groin of 1 month's duration. The lesion reportedly emerged as pustules that slowly expanded and coalesced. At an outside institution, cryotherapy was planned for a presumed diagnosis of condyloma acuminatum; however, the patient decided to get a second opinion. He denied recent intake of new drugs. Six months prior he had traveled to China and engaged in unprotected sexual intercourse. Physical examination revealed an approximately 4×2-cm exophytic plaque with a partially eroded and exudative surface on the left inguinal fold. Dermatologic examination, including the oral mucosa, was otherwise normal. Complete blood cell count and sexually transmitted disease panel were unremarkable.