User login
Atypical case of cutaneous MCL mimics SPTCL
An atypical case of cutaneous mantle cell lymphoma (MCL) with histomorphological features mimicking subcutaneous panniculitis-like T-cell lymphoma (SPTCL) highlights a “potential pitfall,” according to investigators.
This unusual case stresses the importance of molecular cytogenetics and/or immunohistochemistry for panniculitis-type lymphomas, reported lead author Caroline Laggis, MD of the University of Utah, Salt Lake City, and colleagues.
“While morphologic features of SPTCL, specifically rimming of adipocytes by neoplastic lymphoid cells, have been documented in other types of lymphomas, this case is exceptional in that the morphologic features of SPTCL are showed in secondary cutaneous involvement by MCL,” the investigators wrote. Their report is in Journal of Cutaneous Pathology.
The patient was a 69-year-old man who presented with 2-year history of night sweats and fever of unknown origin, and, closer to presentation, weight loss and tender bumps under the skin of his pelvic region.
Subsequent computed tomography and excisional lymph node biopsy led to a diagnosis of MCL, with a Mantle Cell Lymphoma International Prognostic Index of 5, suggesting aggressive, intermediate-risk disease. Further imaging showed involvement of the nasopharynx, and cervical and mediastinal lymph nodes.
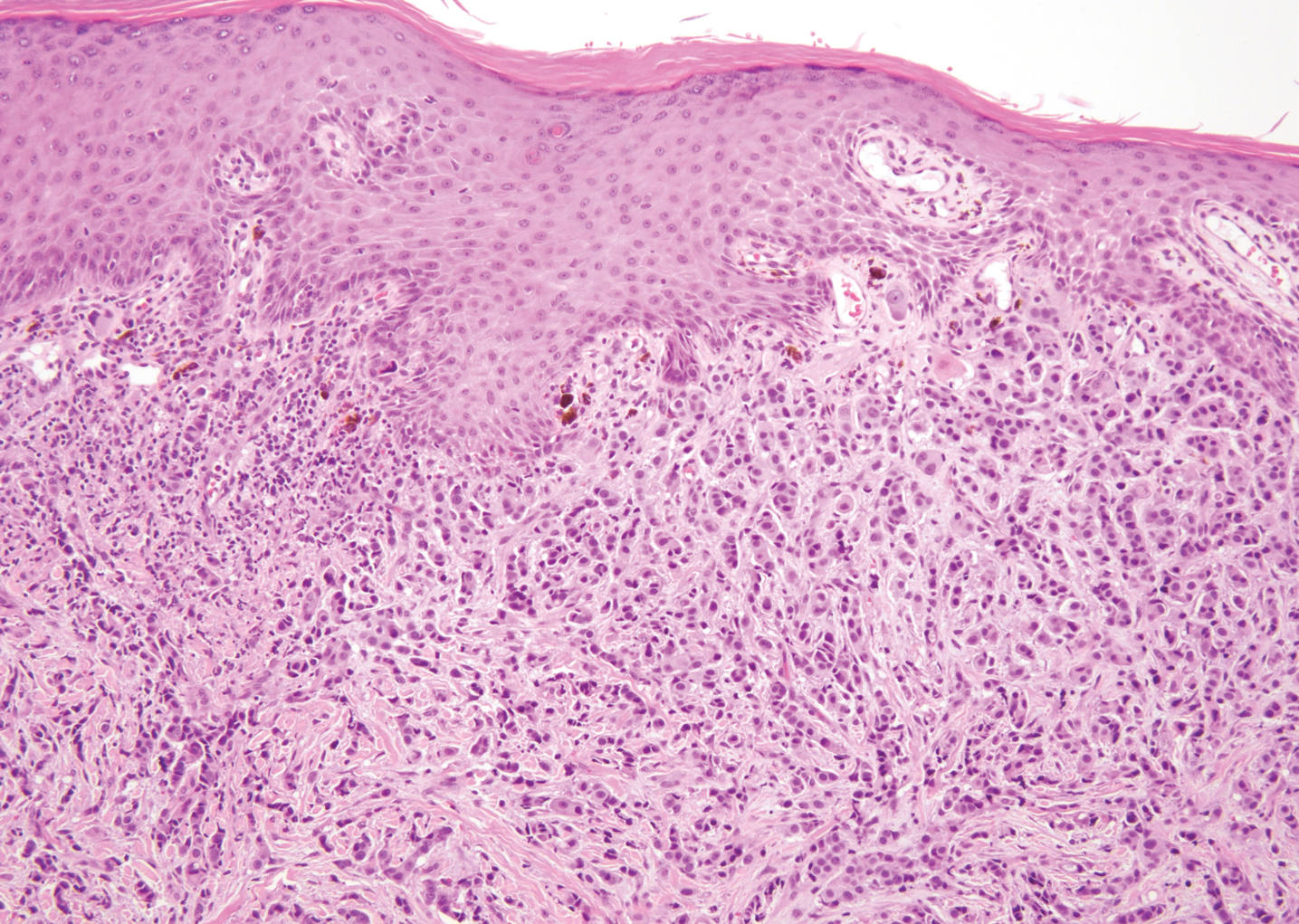
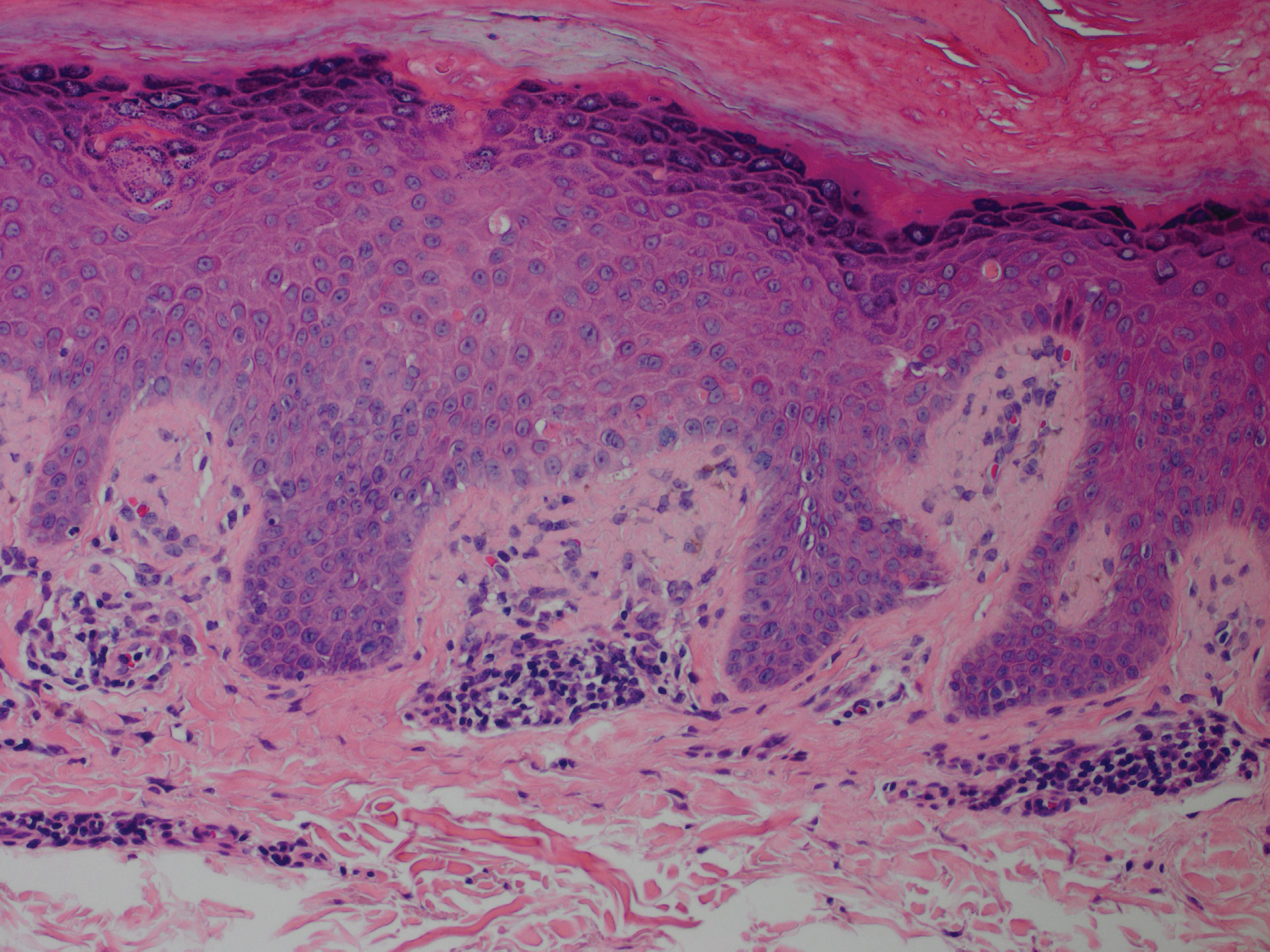
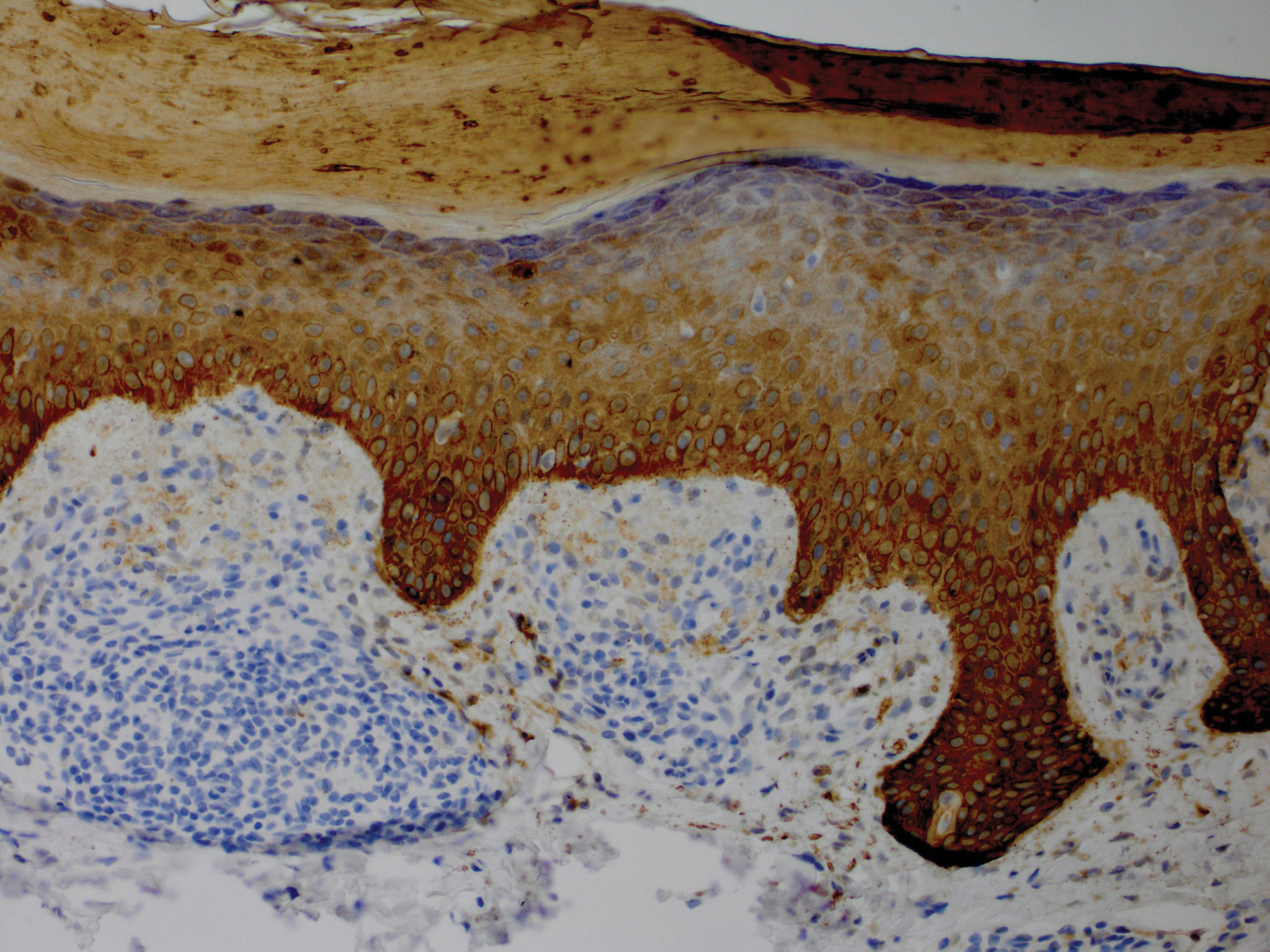
Bendamustine and rituximab chemotherapy was given unremarkably until the final cycle, at which point the patient presented with tender subcutaneous nodules on his lower legs. Histopathology from punch biopsies revealed “a dense infiltrate of monomorphic, mitotically active lymphoid cells with infiltration between the deep dermal collagen and the adipocytes in subcutaneous fat,” the investigators wrote, noting that the infiltrative cells were blastoid and 70% expressed cyclin D1, supporting cutaneous involvement of his systemic MCL.
Treatment was switched to ibrutinib and selinexor via a clinical trial, which led to temporary improvement of leg lesions; when the lesions returned, biopsy was performed with the same histopathological result. Lenalidomide and rituximab were started, but without success, and disease spread to the central nervous system.
Another biopsy of the skin lesions again supported cutaneous MCL, with tumor cells rimming individual adipocytes.
Because of this atypical morphology, fluorescence in situ hybridization (FISH) was conducted, revealing t(11;14)(q13:32) positivity, thereby “confirming the diagnosis of cutaneous involvement by systemic MCL,” the investigators wrote.
Genomic sequencing revealed abnormalities of “ataxia-telangiectasia mutated, mechanistic target of rapamycin kinase (mTOR), BCL6 corepressor, and FAS-associated factor 1, as well as the expected mutation in IGH-CCND1, leading to cyclin D1 upregulation.”
Subsequent treatment was unsuccessful, and the patient died from his disease.
“The complex and central role that mTOR plays in adipose homeostasis may link our tumor to its preference to the adipose tissue, although further investigation is warranted regarding specific genomic alterations in lymphomas and the implications these mutations have in the involvement of tumor cells with cutaneous and adipose environments,” the investigators wrote.
The investigators did not report conflicts of interest.
SOURCE: Laggis C et al. 2019 Apr 8. doi:10.1111/cup.13471.
An atypical case of cutaneous mantle cell lymphoma (MCL) with histomorphological features mimicking subcutaneous panniculitis-like T-cell lymphoma (SPTCL) highlights a “potential pitfall,” according to investigators.
This unusual case stresses the importance of molecular cytogenetics and/or immunohistochemistry for panniculitis-type lymphomas, reported lead author Caroline Laggis, MD of the University of Utah, Salt Lake City, and colleagues.
“While morphologic features of SPTCL, specifically rimming of adipocytes by neoplastic lymphoid cells, have been documented in other types of lymphomas, this case is exceptional in that the morphologic features of SPTCL are showed in secondary cutaneous involvement by MCL,” the investigators wrote. Their report is in Journal of Cutaneous Pathology.
The patient was a 69-year-old man who presented with 2-year history of night sweats and fever of unknown origin, and, closer to presentation, weight loss and tender bumps under the skin of his pelvic region.
Subsequent computed tomography and excisional lymph node biopsy led to a diagnosis of MCL, with a Mantle Cell Lymphoma International Prognostic Index of 5, suggesting aggressive, intermediate-risk disease. Further imaging showed involvement of the nasopharynx, and cervical and mediastinal lymph nodes.
Bendamustine and rituximab chemotherapy was given unremarkably until the final cycle, at which point the patient presented with tender subcutaneous nodules on his lower legs. Histopathology from punch biopsies revealed “a dense infiltrate of monomorphic, mitotically active lymphoid cells with infiltration between the deep dermal collagen and the adipocytes in subcutaneous fat,” the investigators wrote, noting that the infiltrative cells were blastoid and 70% expressed cyclin D1, supporting cutaneous involvement of his systemic MCL.
Treatment was switched to ibrutinib and selinexor via a clinical trial, which led to temporary improvement of leg lesions; when the lesions returned, biopsy was performed with the same histopathological result. Lenalidomide and rituximab were started, but without success, and disease spread to the central nervous system.
Another biopsy of the skin lesions again supported cutaneous MCL, with tumor cells rimming individual adipocytes.
Because of this atypical morphology, fluorescence in situ hybridization (FISH) was conducted, revealing t(11;14)(q13:32) positivity, thereby “confirming the diagnosis of cutaneous involvement by systemic MCL,” the investigators wrote.
Genomic sequencing revealed abnormalities of “ataxia-telangiectasia mutated, mechanistic target of rapamycin kinase (mTOR), BCL6 corepressor, and FAS-associated factor 1, as well as the expected mutation in IGH-CCND1, leading to cyclin D1 upregulation.”
Subsequent treatment was unsuccessful, and the patient died from his disease.
“The complex and central role that mTOR plays in adipose homeostasis may link our tumor to its preference to the adipose tissue, although further investigation is warranted regarding specific genomic alterations in lymphomas and the implications these mutations have in the involvement of tumor cells with cutaneous and adipose environments,” the investigators wrote.
The investigators did not report conflicts of interest.
SOURCE: Laggis C et al. 2019 Apr 8. doi:10.1111/cup.13471.
An atypical case of cutaneous mantle cell lymphoma (MCL) with histomorphological features mimicking subcutaneous panniculitis-like T-cell lymphoma (SPTCL) highlights a “potential pitfall,” according to investigators.
This unusual case stresses the importance of molecular cytogenetics and/or immunohistochemistry for panniculitis-type lymphomas, reported lead author Caroline Laggis, MD of the University of Utah, Salt Lake City, and colleagues.
“While morphologic features of SPTCL, specifically rimming of adipocytes by neoplastic lymphoid cells, have been documented in other types of lymphomas, this case is exceptional in that the morphologic features of SPTCL are showed in secondary cutaneous involvement by MCL,” the investigators wrote. Their report is in Journal of Cutaneous Pathology.
The patient was a 69-year-old man who presented with 2-year history of night sweats and fever of unknown origin, and, closer to presentation, weight loss and tender bumps under the skin of his pelvic region.
Subsequent computed tomography and excisional lymph node biopsy led to a diagnosis of MCL, with a Mantle Cell Lymphoma International Prognostic Index of 5, suggesting aggressive, intermediate-risk disease. Further imaging showed involvement of the nasopharynx, and cervical and mediastinal lymph nodes.
Bendamustine and rituximab chemotherapy was given unremarkably until the final cycle, at which point the patient presented with tender subcutaneous nodules on his lower legs. Histopathology from punch biopsies revealed “a dense infiltrate of monomorphic, mitotically active lymphoid cells with infiltration between the deep dermal collagen and the adipocytes in subcutaneous fat,” the investigators wrote, noting that the infiltrative cells were blastoid and 70% expressed cyclin D1, supporting cutaneous involvement of his systemic MCL.
Treatment was switched to ibrutinib and selinexor via a clinical trial, which led to temporary improvement of leg lesions; when the lesions returned, biopsy was performed with the same histopathological result. Lenalidomide and rituximab were started, but without success, and disease spread to the central nervous system.
Another biopsy of the skin lesions again supported cutaneous MCL, with tumor cells rimming individual adipocytes.
Because of this atypical morphology, fluorescence in situ hybridization (FISH) was conducted, revealing t(11;14)(q13:32) positivity, thereby “confirming the diagnosis of cutaneous involvement by systemic MCL,” the investigators wrote.
Genomic sequencing revealed abnormalities of “ataxia-telangiectasia mutated, mechanistic target of rapamycin kinase (mTOR), BCL6 corepressor, and FAS-associated factor 1, as well as the expected mutation in IGH-CCND1, leading to cyclin D1 upregulation.”
Subsequent treatment was unsuccessful, and the patient died from his disease.
“The complex and central role that mTOR plays in adipose homeostasis may link our tumor to its preference to the adipose tissue, although further investigation is warranted regarding specific genomic alterations in lymphomas and the implications these mutations have in the involvement of tumor cells with cutaneous and adipose environments,” the investigators wrote.
The investigators did not report conflicts of interest.
SOURCE: Laggis C et al. 2019 Apr 8. doi:10.1111/cup.13471.
FROM JOURNAL OF CUTANEOUS PATHOLOGY
Enlarging Nodule on the Thigh
The Diagnosis: Metastatic Adenocarcinoma of the Colon
Cutaneous adenocarcinomas are uncommon, whether they present as a primary lesion or metastatic disease. In our patient, the histologic findings and immunohistochemical staining pattern were consistent with metastatic adenocarcinoma of the colon, an uncommon clinical presentation.
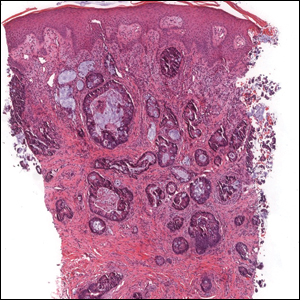
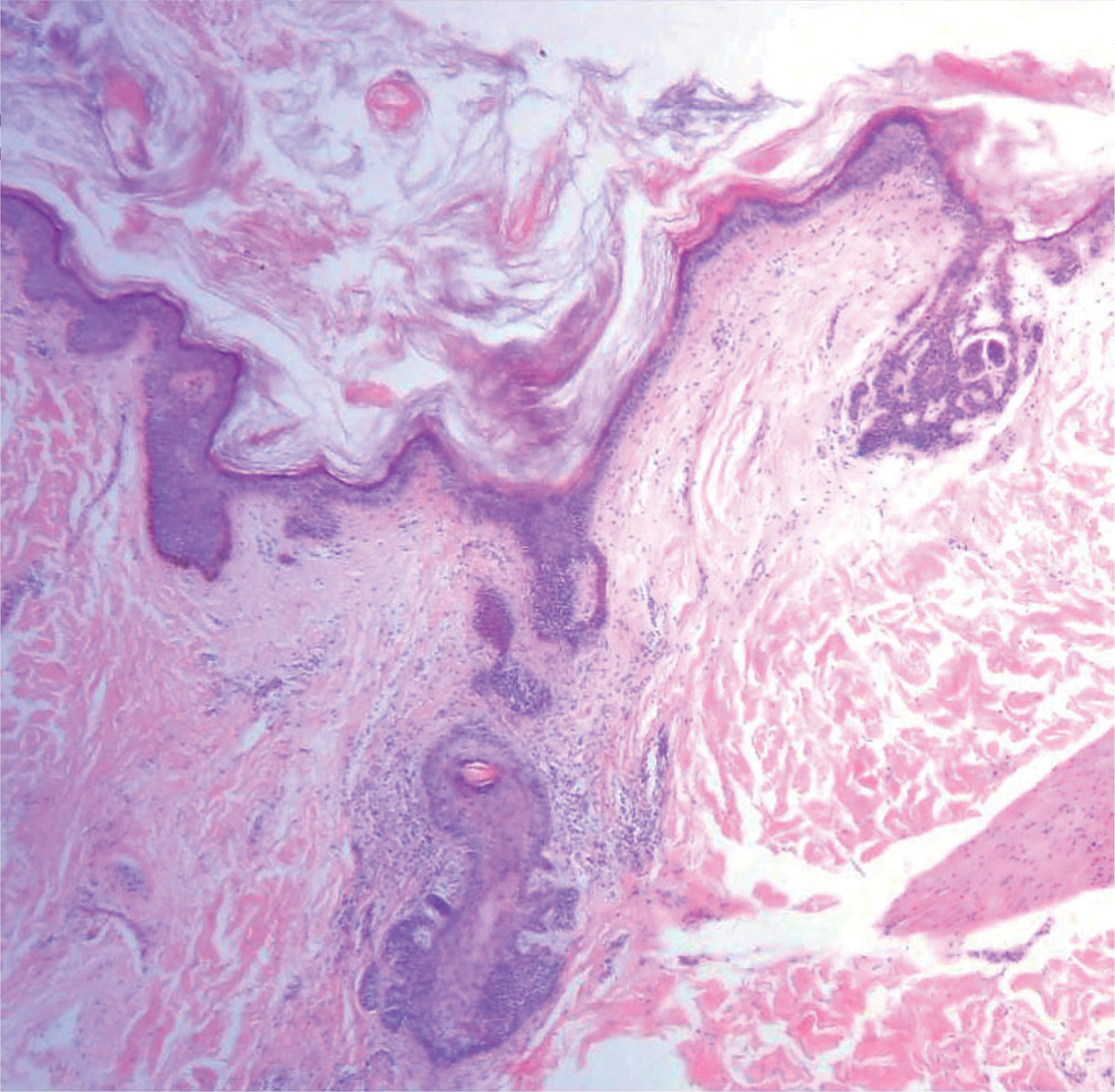
Colonic adenocarcinoma can cause cutaneous metastasis in 3% of cases. The most common sites of metastases include the abdomen, chest, and back.1 On histologic examination, hematoxylin and eosin (H&E)-stained sections of cutaneous metastatic adenocarcinoma illustrate a malignant gland-forming neoplasm in the dermis with luminal mucin and necrotic debris (quiz image). The glands are lined by tall columnar epithelial cells with hyperchromatic nuclei. Alternatively, poorly differentiated morphology can be seen with fewer glands and more infiltrating nests of tumor cells.2 Immunohistochemically, colonic adenocarcinoma typically is negative for cytokeratin (CK) 7 and positive for CK20 and caudal type homeobox transcription factor 2 (CDX-2).3
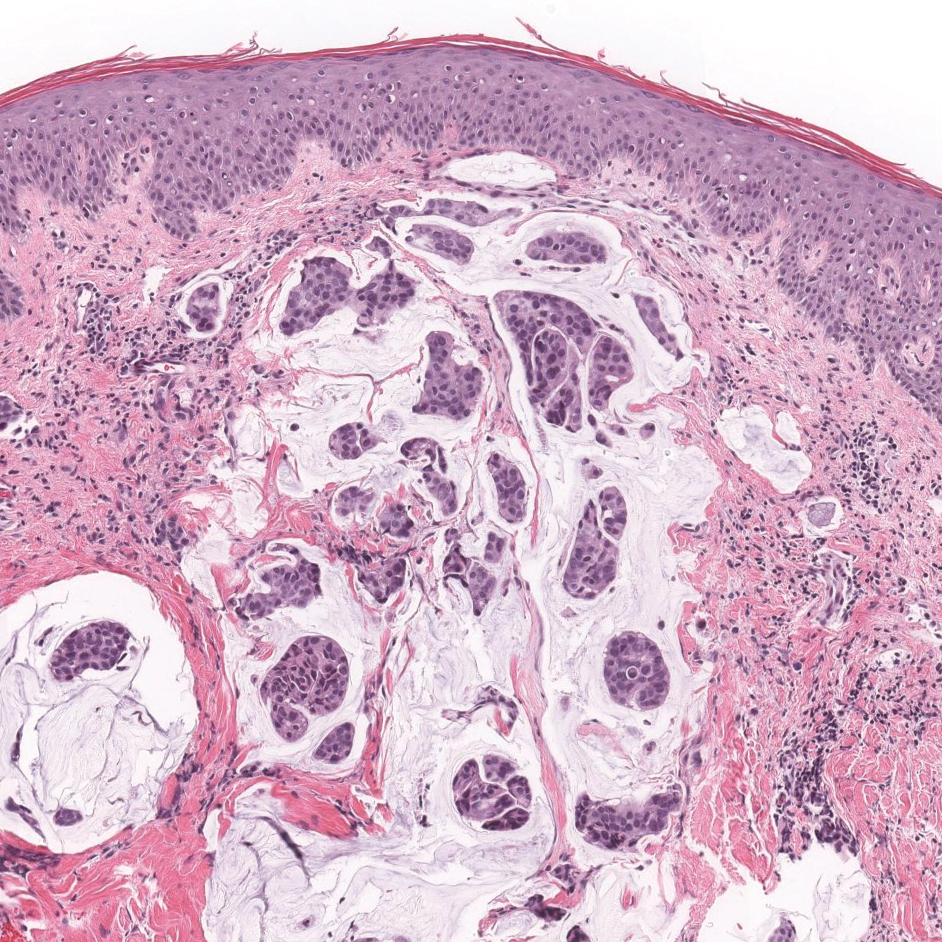
Primary cutaneous mucinous carcinoma is characterized by islands of neoplastic cells floating in pools of mucin (Figure 1). It may be indistinguishable from metastatic mucinous carcinomas of the colon or breast. Immunohistochemistry can be helpful in differentiating metastatic breast vs colon carcinoma. Cytokeratin 7, GATA binding protein 3, gross cystic disease fluid protein 15, and estrogen receptor will be positive in carcinomas of the breast and will be negative in colonic adenocarcinomas.4-6 Furthermore, lesional cells in metastatic adenocarcinoma of the colon are positive for CDX-2 and CK20, while those in metastatic carcinoma of the breast are negative.2 Immunohistochemistry also can differentiate primary cutaneous carcinoma from metastatic adenocarcinoma. When used in combination, p63 and podoplanin (D2-40) offer a highly sensitive and specific indicator of a primary cutaneous neoplasm, as both demonstrate either focal or diffuse positivity in this setting. In contrast, these stains typically are negative in metastatic adenocarcinomas of the skin.7
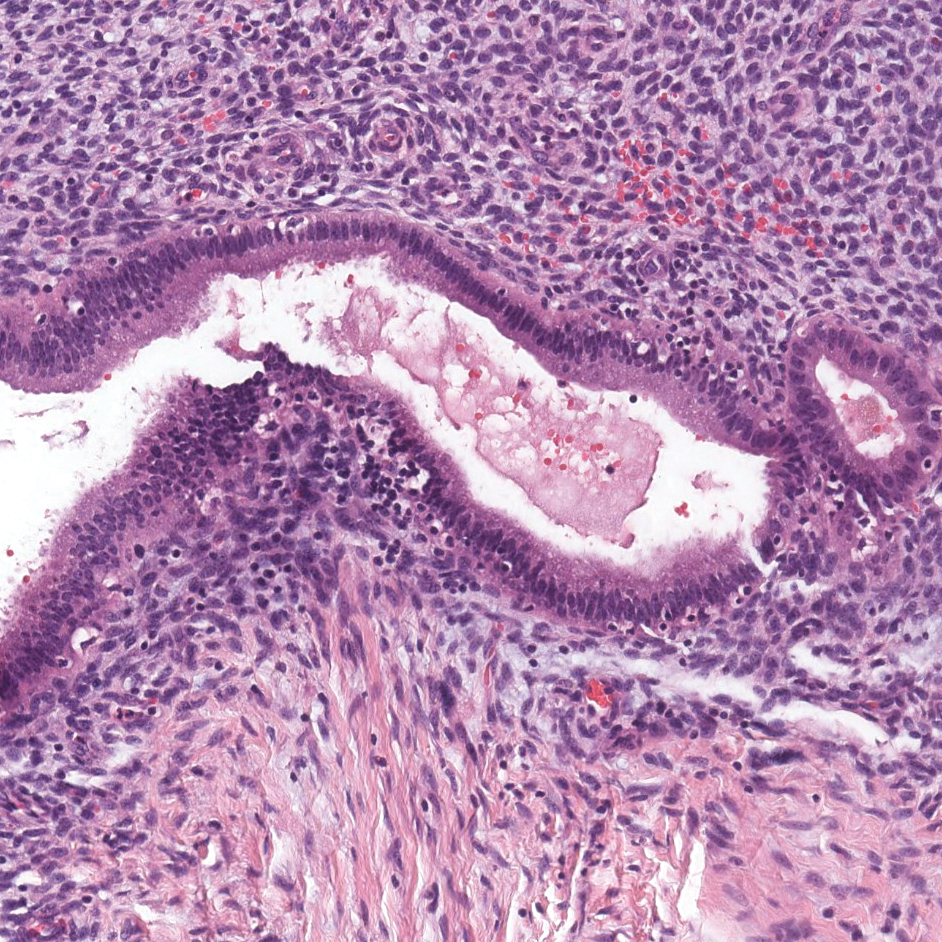
Endometriosis affects 1% to 2% of all reproductive-age females, of which extrapelvic manifestations account for only 0.5% to 1.0% of cases.8 Histologically, extrapelvic endometriosis is characterized by the triad of endometrial-type glands, endometrial stroma, and hemorrhage or hemosiderin deposition (Figure 2). The glands can enlarge and demonstrate architectural distortion with partial lack of polarity. These features initially can be concerning for adenocarcinoma, but on closer examination, nuclear morphology is regular and mitoses are absent.8,9 The diagnosis usually can be rendered with H&E alone; however, immunohistochemical stains for CD10 and estrogen receptor can highlight the endometrial stroma.10 Furthermore, endometrial glands will stain positive for paired box gene 8 (PAX8), a marker that is not expressed within the gastrointestinal tract and associated malignancies.11
cytoplasm (H&E, original magnification ×100).
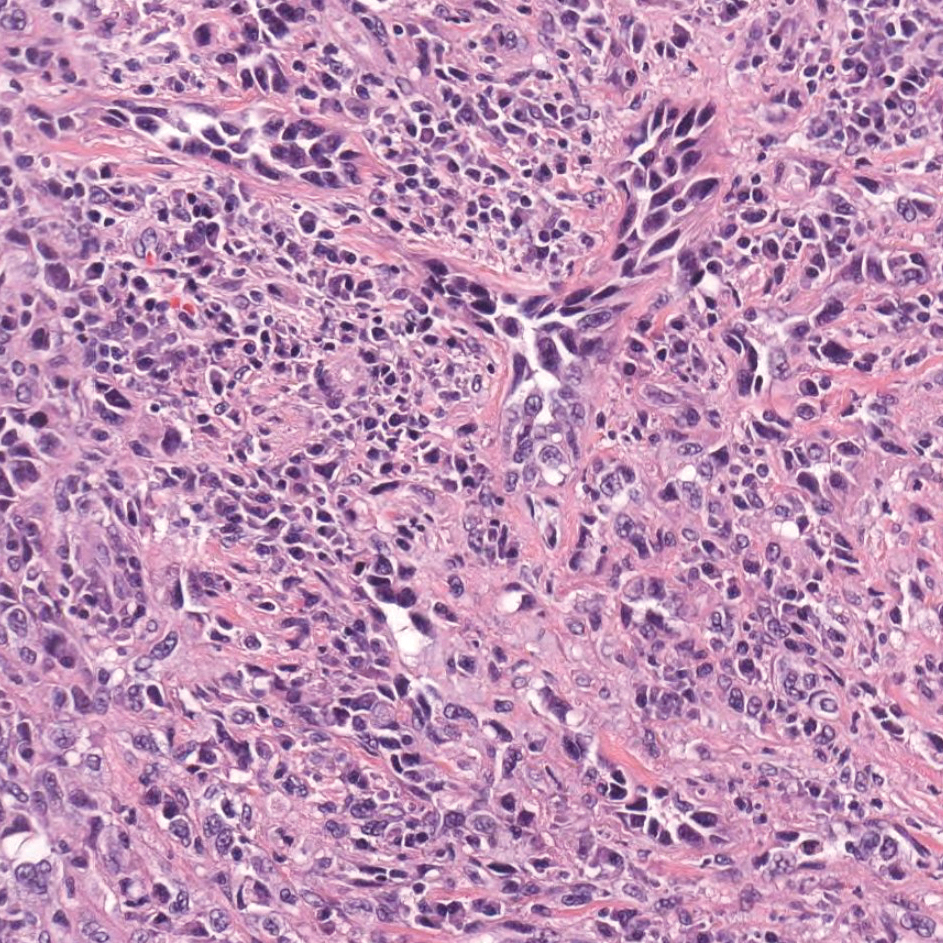
Primary cutaneous angiosarcoma may mimic adenocarcinoma, as the endothelial-lined vessels can be confused as malignant glands (Figure 3). Angiosarcoma often is seen in 1 of 3 clinical presentations: the head and neck of elderly patients, postradiation treatment, and chronic lymphedema.12,13 Regardless of the location, the disease carries a poor prognosis, with a 5-year survival rate of 12% following initial diagnosis.13 Angiosarcoma is characterized by malignant endothelial cells dissecting through the dermis. Although the histology can be deceptively bland in some cases, the neoplasm most commonly demonstrates notable atypia with a multilayered endothelium and occasional intravascular atypical cells ("fish in the creek appearance").13,14 There can be frequent mitoses, and the atypical cells may show intracytoplasmic lumina containing red blood cells. The lesional cells are positive for endothelial markers such as erythroblast transformation specific related gene (ERG), CD31, CD34, and friend leukemia integration factor 1 (FLI-1).15,16
Breast cancer also can cause cutaneous metastases in approximately 20% of cases, with the most common presenting site being the anterior chest wall.17 Macroscopically, these lesions appear most commonly as painless nodules but also as telangiectatic, erysipeloid, fibrotic, and alopecic lesions.17-19 The histologic findings from H&E-stained sections of a cutaneous metastasis of breast cancer are variable and depend on the specific tumor subtype (eg, ductal, lobular, mucinous). However, the classic histologic presentation is that of nests and cords of malignant epithelial cells with variable gland formation. Often, tumor cells infiltrate in a single-file fashion (Figure 4).17 Although inflammatory breast carcinoma is a strictly clinical diagnosis, the presence of tumor cells in the lymphovascular spaces is a histologic clue to this diagnosis. Immunohistochemically, GATA binding protein 3 is helpful in identifying both hormone receptor-positive and -negative breast cancer subtypes that have metastasized.20
Within the histologic differential diagnoses, the most useful tool to diagnose metastatic adenocarcinoma of the colon often is a thorough clinical history. In the absence of a clinical history of adenocarcinoma, immunohistochemistry can be a useful adjunct to aid in the correct characterization and classification of a malignant gland-forming tumor.2,3,6
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29:228-236.
- Kumar V, Robbins SL. Robbins Basic Pathology. 8th ed. Philadelphia, PA: Saunders/Elsevier; 2007.
- Taliano RJ, LeGolvan M, Resnick MB. Immunohistochemistry of colorectal carcinoma: current practice and evolving applications. Hum Pathol. 2013;44:151-163.
- Kamalpour L, Brindise RT, Nodzenski M, et al. Primary cutaneous mucinous carcinoma: a systematic review and meta-analysis of outcomes after surgery. JAMA Dermatol. 2014;150:380-384.
- Roshan MH, Tambo A, Pace NP. The role of testosterone in colorectal carcinoma: pathomechanisms and open questions. EPMA J. 2016;7:22.
- Mazoujian G, Pinkus GS, Davis S, et al. Immunohistochemistry of a gross cystic disease fluid protein (GCDFP-15) of the breast. a marker of apocrine epithelium and breast carcinomas with apocrine features. Am J Pathol. 1983;110:105-112.
- Plaza JA, Ortega PF, Stockman DL, et al. Value of p63 and podoplanin (D2-40) immunoreactivity in the distinction between primary cutaneous tumors and adenocarcinomas metastatic to the skin: a clinicopathologic and immunohistochemical study of 79 cases. J Cutan Pathol. 2010;37:403-410.
- Machairiotis N, Stylianaki A, Dryllis G, et al. Extrapelvic endometriosis: a rare entity or an under diagnosed condition? Diagn Pathol. 2013;8:194.
- Chen H, Luo Q, Liu S, et al. Rectal mucosal endometriosis primarily misinterpreted as adenocarcinoma: a case report and review of literature. Int J Clin Exp Pathol. 2015;8:5902-5907.
- Terada S, Miyata Y, Nakazawa H, et al. Immunohistochemical analysis of an ectopic endometriosis in the uterine round ligament. Diagn Pathol. 2006;1:27.
- Yemelyanova A, Gown AM, Wu LS, et al. PAX8 expression in uterine adenocarcinomas and mesonephric proliferations. Int J Gynecol Pathol. 2014;33:492-499.
- Farid M, Ong WS, Lee MJ, et al. Cutaneous versus non-cutaneous angiosarcoma: clinicopathologic features and treatment outcomes in 60 patients at a single Asian cancer centre. Oncology. 2013;85:182-190.
- Requena C, Sendra E, Llombart B, et al. Cutaneous angiosarcoma: clinical and pathology study of 16 cases. Actas Dermosifiliogr. 2017;108:457-465.
- Schmidt AP, Tjarks BJ, Lynch DW. Gone fishing: a unique histologic pattern in cutaneous angiosarcoma. Cutis. 2018;101:270-272.
- Sullivan HC, Edgar MA, Cohen C, et al. The utility of ERG, CD31 and CD34 in the cytological diagnosis of angiosarcoma: an analysis of 25 cases. J Clin Pathol. 2015;68:44-50.
- Rossi S, Orvieto E, Furlanetto A, et al. Utility of the immunohistochemical detection of FLI-1 expression in round cell and vascular neoplasm using a monoclonal antibody. Mod Pathol. 2004;17:547-552.
- Tan AR. Cutaneous manifestations of breast cancer. Semin Oncol. 2016;43:331-334.
- Schwartz RA, Wiederkehr M, Lambert WC. Secondary mucinous carcinoma of the skin: metastatic breast cancer. Dermatol Surg. 2004;30(2, pt 1):234-235.
- Mallon E, Dawber RP. Alopecia neoplastica without alopecia: a unique presentation of breast carcinoma scalp metastasis. J Am Acad Dermatol. 1994;31(2, pt 2):319-321.
- Braxton DR, Cohen C, Siddiqui MT. Utility of GATA3 immunohistochemistry for diagnosis of metastatic breast carcinoma in cytology specimens. Diagn Cytopathol. 2015;43:271-277.
The Diagnosis: Metastatic Adenocarcinoma of the Colon
Cutaneous adenocarcinomas are uncommon, whether they present as a primary lesion or metastatic disease. In our patient, the histologic findings and immunohistochemical staining pattern were consistent with metastatic adenocarcinoma of the colon, an uncommon clinical presentation.
Colonic adenocarcinoma can cause cutaneous metastasis in 3% of cases. The most common sites of metastases include the abdomen, chest, and back.1 On histologic examination, hematoxylin and eosin (H&E)-stained sections of cutaneous metastatic adenocarcinoma illustrate a malignant gland-forming neoplasm in the dermis with luminal mucin and necrotic debris (quiz image). The glands are lined by tall columnar epithelial cells with hyperchromatic nuclei. Alternatively, poorly differentiated morphology can be seen with fewer glands and more infiltrating nests of tumor cells.2 Immunohistochemically, colonic adenocarcinoma typically is negative for cytokeratin (CK) 7 and positive for CK20 and caudal type homeobox transcription factor 2 (CDX-2).3
Primary cutaneous mucinous carcinoma is characterized by islands of neoplastic cells floating in pools of mucin (Figure 1). It may be indistinguishable from metastatic mucinous carcinomas of the colon or breast. Immunohistochemistry can be helpful in differentiating metastatic breast vs colon carcinoma. Cytokeratin 7, GATA binding protein 3, gross cystic disease fluid protein 15, and estrogen receptor will be positive in carcinomas of the breast and will be negative in colonic adenocarcinomas.4-6 Furthermore, lesional cells in metastatic adenocarcinoma of the colon are positive for CDX-2 and CK20, while those in metastatic carcinoma of the breast are negative.2 Immunohistochemistry also can differentiate primary cutaneous carcinoma from metastatic adenocarcinoma. When used in combination, p63 and podoplanin (D2-40) offer a highly sensitive and specific indicator of a primary cutaneous neoplasm, as both demonstrate either focal or diffuse positivity in this setting. In contrast, these stains typically are negative in metastatic adenocarcinomas of the skin.7
Endometriosis affects 1% to 2% of all reproductive-age females, of which extrapelvic manifestations account for only 0.5% to 1.0% of cases.8 Histologically, extrapelvic endometriosis is characterized by the triad of endometrial-type glands, endometrial stroma, and hemorrhage or hemosiderin deposition (Figure 2). The glands can enlarge and demonstrate architectural distortion with partial lack of polarity. These features initially can be concerning for adenocarcinoma, but on closer examination, nuclear morphology is regular and mitoses are absent.8,9 The diagnosis usually can be rendered with H&E alone; however, immunohistochemical stains for CD10 and estrogen receptor can highlight the endometrial stroma.10 Furthermore, endometrial glands will stain positive for paired box gene 8 (PAX8), a marker that is not expressed within the gastrointestinal tract and associated malignancies.11
cytoplasm (H&E, original magnification ×100).
Primary cutaneous angiosarcoma may mimic adenocarcinoma, as the endothelial-lined vessels can be confused as malignant glands (Figure 3). Angiosarcoma often is seen in 1 of 3 clinical presentations: the head and neck of elderly patients, postradiation treatment, and chronic lymphedema.12,13 Regardless of the location, the disease carries a poor prognosis, with a 5-year survival rate of 12% following initial diagnosis.13 Angiosarcoma is characterized by malignant endothelial cells dissecting through the dermis. Although the histology can be deceptively bland in some cases, the neoplasm most commonly demonstrates notable atypia with a multilayered endothelium and occasional intravascular atypical cells ("fish in the creek appearance").13,14 There can be frequent mitoses, and the atypical cells may show intracytoplasmic lumina containing red blood cells. The lesional cells are positive for endothelial markers such as erythroblast transformation specific related gene (ERG), CD31, CD34, and friend leukemia integration factor 1 (FLI-1).15,16
Breast cancer also can cause cutaneous metastases in approximately 20% of cases, with the most common presenting site being the anterior chest wall.17 Macroscopically, these lesions appear most commonly as painless nodules but also as telangiectatic, erysipeloid, fibrotic, and alopecic lesions.17-19 The histologic findings from H&E-stained sections of a cutaneous metastasis of breast cancer are variable and depend on the specific tumor subtype (eg, ductal, lobular, mucinous). However, the classic histologic presentation is that of nests and cords of malignant epithelial cells with variable gland formation. Often, tumor cells infiltrate in a single-file fashion (Figure 4).17 Although inflammatory breast carcinoma is a strictly clinical diagnosis, the presence of tumor cells in the lymphovascular spaces is a histologic clue to this diagnosis. Immunohistochemically, GATA binding protein 3 is helpful in identifying both hormone receptor-positive and -negative breast cancer subtypes that have metastasized.20
Within the histologic differential diagnoses, the most useful tool to diagnose metastatic adenocarcinoma of the colon often is a thorough clinical history. In the absence of a clinical history of adenocarcinoma, immunohistochemistry can be a useful adjunct to aid in the correct characterization and classification of a malignant gland-forming tumor.2,3,6
The Diagnosis: Metastatic Adenocarcinoma of the Colon
Cutaneous adenocarcinomas are uncommon, whether they present as a primary lesion or metastatic disease. In our patient, the histologic findings and immunohistochemical staining pattern were consistent with metastatic adenocarcinoma of the colon, an uncommon clinical presentation.
Colonic adenocarcinoma can cause cutaneous metastasis in 3% of cases. The most common sites of metastases include the abdomen, chest, and back.1 On histologic examination, hematoxylin and eosin (H&E)-stained sections of cutaneous metastatic adenocarcinoma illustrate a malignant gland-forming neoplasm in the dermis with luminal mucin and necrotic debris (quiz image). The glands are lined by tall columnar epithelial cells with hyperchromatic nuclei. Alternatively, poorly differentiated morphology can be seen with fewer glands and more infiltrating nests of tumor cells.2 Immunohistochemically, colonic adenocarcinoma typically is negative for cytokeratin (CK) 7 and positive for CK20 and caudal type homeobox transcription factor 2 (CDX-2).3
Primary cutaneous mucinous carcinoma is characterized by islands of neoplastic cells floating in pools of mucin (Figure 1). It may be indistinguishable from metastatic mucinous carcinomas of the colon or breast. Immunohistochemistry can be helpful in differentiating metastatic breast vs colon carcinoma. Cytokeratin 7, GATA binding protein 3, gross cystic disease fluid protein 15, and estrogen receptor will be positive in carcinomas of the breast and will be negative in colonic adenocarcinomas.4-6 Furthermore, lesional cells in metastatic adenocarcinoma of the colon are positive for CDX-2 and CK20, while those in metastatic carcinoma of the breast are negative.2 Immunohistochemistry also can differentiate primary cutaneous carcinoma from metastatic adenocarcinoma. When used in combination, p63 and podoplanin (D2-40) offer a highly sensitive and specific indicator of a primary cutaneous neoplasm, as both demonstrate either focal or diffuse positivity in this setting. In contrast, these stains typically are negative in metastatic adenocarcinomas of the skin.7
Endometriosis affects 1% to 2% of all reproductive-age females, of which extrapelvic manifestations account for only 0.5% to 1.0% of cases.8 Histologically, extrapelvic endometriosis is characterized by the triad of endometrial-type glands, endometrial stroma, and hemorrhage or hemosiderin deposition (Figure 2). The glands can enlarge and demonstrate architectural distortion with partial lack of polarity. These features initially can be concerning for adenocarcinoma, but on closer examination, nuclear morphology is regular and mitoses are absent.8,9 The diagnosis usually can be rendered with H&E alone; however, immunohistochemical stains for CD10 and estrogen receptor can highlight the endometrial stroma.10 Furthermore, endometrial glands will stain positive for paired box gene 8 (PAX8), a marker that is not expressed within the gastrointestinal tract and associated malignancies.11
cytoplasm (H&E, original magnification ×100).
Primary cutaneous angiosarcoma may mimic adenocarcinoma, as the endothelial-lined vessels can be confused as malignant glands (Figure 3). Angiosarcoma often is seen in 1 of 3 clinical presentations: the head and neck of elderly patients, postradiation treatment, and chronic lymphedema.12,13 Regardless of the location, the disease carries a poor prognosis, with a 5-year survival rate of 12% following initial diagnosis.13 Angiosarcoma is characterized by malignant endothelial cells dissecting through the dermis. Although the histology can be deceptively bland in some cases, the neoplasm most commonly demonstrates notable atypia with a multilayered endothelium and occasional intravascular atypical cells ("fish in the creek appearance").13,14 There can be frequent mitoses, and the atypical cells may show intracytoplasmic lumina containing red blood cells. The lesional cells are positive for endothelial markers such as erythroblast transformation specific related gene (ERG), CD31, CD34, and friend leukemia integration factor 1 (FLI-1).15,16
Breast cancer also can cause cutaneous metastases in approximately 20% of cases, with the most common presenting site being the anterior chest wall.17 Macroscopically, these lesions appear most commonly as painless nodules but also as telangiectatic, erysipeloid, fibrotic, and alopecic lesions.17-19 The histologic findings from H&E-stained sections of a cutaneous metastasis of breast cancer are variable and depend on the specific tumor subtype (eg, ductal, lobular, mucinous). However, the classic histologic presentation is that of nests and cords of malignant epithelial cells with variable gland formation. Often, tumor cells infiltrate in a single-file fashion (Figure 4).17 Although inflammatory breast carcinoma is a strictly clinical diagnosis, the presence of tumor cells in the lymphovascular spaces is a histologic clue to this diagnosis. Immunohistochemically, GATA binding protein 3 is helpful in identifying both hormone receptor-positive and -negative breast cancer subtypes that have metastasized.20
Within the histologic differential diagnoses, the most useful tool to diagnose metastatic adenocarcinoma of the colon often is a thorough clinical history. In the absence of a clinical history of adenocarcinoma, immunohistochemistry can be a useful adjunct to aid in the correct characterization and classification of a malignant gland-forming tumor.2,3,6
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29:228-236.
- Kumar V, Robbins SL. Robbins Basic Pathology. 8th ed. Philadelphia, PA: Saunders/Elsevier; 2007.
- Taliano RJ, LeGolvan M, Resnick MB. Immunohistochemistry of colorectal carcinoma: current practice and evolving applications. Hum Pathol. 2013;44:151-163.
- Kamalpour L, Brindise RT, Nodzenski M, et al. Primary cutaneous mucinous carcinoma: a systematic review and meta-analysis of outcomes after surgery. JAMA Dermatol. 2014;150:380-384.
- Roshan MH, Tambo A, Pace NP. The role of testosterone in colorectal carcinoma: pathomechanisms and open questions. EPMA J. 2016;7:22.
- Mazoujian G, Pinkus GS, Davis S, et al. Immunohistochemistry of a gross cystic disease fluid protein (GCDFP-15) of the breast. a marker of apocrine epithelium and breast carcinomas with apocrine features. Am J Pathol. 1983;110:105-112.
- Plaza JA, Ortega PF, Stockman DL, et al. Value of p63 and podoplanin (D2-40) immunoreactivity in the distinction between primary cutaneous tumors and adenocarcinomas metastatic to the skin: a clinicopathologic and immunohistochemical study of 79 cases. J Cutan Pathol. 2010;37:403-410.
- Machairiotis N, Stylianaki A, Dryllis G, et al. Extrapelvic endometriosis: a rare entity or an under diagnosed condition? Diagn Pathol. 2013;8:194.
- Chen H, Luo Q, Liu S, et al. Rectal mucosal endometriosis primarily misinterpreted as adenocarcinoma: a case report and review of literature. Int J Clin Exp Pathol. 2015;8:5902-5907.
- Terada S, Miyata Y, Nakazawa H, et al. Immunohistochemical analysis of an ectopic endometriosis in the uterine round ligament. Diagn Pathol. 2006;1:27.
- Yemelyanova A, Gown AM, Wu LS, et al. PAX8 expression in uterine adenocarcinomas and mesonephric proliferations. Int J Gynecol Pathol. 2014;33:492-499.
- Farid M, Ong WS, Lee MJ, et al. Cutaneous versus non-cutaneous angiosarcoma: clinicopathologic features and treatment outcomes in 60 patients at a single Asian cancer centre. Oncology. 2013;85:182-190.
- Requena C, Sendra E, Llombart B, et al. Cutaneous angiosarcoma: clinical and pathology study of 16 cases. Actas Dermosifiliogr. 2017;108:457-465.
- Schmidt AP, Tjarks BJ, Lynch DW. Gone fishing: a unique histologic pattern in cutaneous angiosarcoma. Cutis. 2018;101:270-272.
- Sullivan HC, Edgar MA, Cohen C, et al. The utility of ERG, CD31 and CD34 in the cytological diagnosis of angiosarcoma: an analysis of 25 cases. J Clin Pathol. 2015;68:44-50.
- Rossi S, Orvieto E, Furlanetto A, et al. Utility of the immunohistochemical detection of FLI-1 expression in round cell and vascular neoplasm using a monoclonal antibody. Mod Pathol. 2004;17:547-552.
- Tan AR. Cutaneous manifestations of breast cancer. Semin Oncol. 2016;43:331-334.
- Schwartz RA, Wiederkehr M, Lambert WC. Secondary mucinous carcinoma of the skin: metastatic breast cancer. Dermatol Surg. 2004;30(2, pt 1):234-235.
- Mallon E, Dawber RP. Alopecia neoplastica without alopecia: a unique presentation of breast carcinoma scalp metastasis. J Am Acad Dermatol. 1994;31(2, pt 2):319-321.
- Braxton DR, Cohen C, Siddiqui MT. Utility of GATA3 immunohistochemistry for diagnosis of metastatic breast carcinoma in cytology specimens. Diagn Cytopathol. 2015;43:271-277.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29:228-236.
- Kumar V, Robbins SL. Robbins Basic Pathology. 8th ed. Philadelphia, PA: Saunders/Elsevier; 2007.
- Taliano RJ, LeGolvan M, Resnick MB. Immunohistochemistry of colorectal carcinoma: current practice and evolving applications. Hum Pathol. 2013;44:151-163.
- Kamalpour L, Brindise RT, Nodzenski M, et al. Primary cutaneous mucinous carcinoma: a systematic review and meta-analysis of outcomes after surgery. JAMA Dermatol. 2014;150:380-384.
- Roshan MH, Tambo A, Pace NP. The role of testosterone in colorectal carcinoma: pathomechanisms and open questions. EPMA J. 2016;7:22.
- Mazoujian G, Pinkus GS, Davis S, et al. Immunohistochemistry of a gross cystic disease fluid protein (GCDFP-15) of the breast. a marker of apocrine epithelium and breast carcinomas with apocrine features. Am J Pathol. 1983;110:105-112.
- Plaza JA, Ortega PF, Stockman DL, et al. Value of p63 and podoplanin (D2-40) immunoreactivity in the distinction between primary cutaneous tumors and adenocarcinomas metastatic to the skin: a clinicopathologic and immunohistochemical study of 79 cases. J Cutan Pathol. 2010;37:403-410.
- Machairiotis N, Stylianaki A, Dryllis G, et al. Extrapelvic endometriosis: a rare entity or an under diagnosed condition? Diagn Pathol. 2013;8:194.
- Chen H, Luo Q, Liu S, et al. Rectal mucosal endometriosis primarily misinterpreted as adenocarcinoma: a case report and review of literature. Int J Clin Exp Pathol. 2015;8:5902-5907.
- Terada S, Miyata Y, Nakazawa H, et al. Immunohistochemical analysis of an ectopic endometriosis in the uterine round ligament. Diagn Pathol. 2006;1:27.
- Yemelyanova A, Gown AM, Wu LS, et al. PAX8 expression in uterine adenocarcinomas and mesonephric proliferations. Int J Gynecol Pathol. 2014;33:492-499.
- Farid M, Ong WS, Lee MJ, et al. Cutaneous versus non-cutaneous angiosarcoma: clinicopathologic features and treatment outcomes in 60 patients at a single Asian cancer centre. Oncology. 2013;85:182-190.
- Requena C, Sendra E, Llombart B, et al. Cutaneous angiosarcoma: clinical and pathology study of 16 cases. Actas Dermosifiliogr. 2017;108:457-465.
- Schmidt AP, Tjarks BJ, Lynch DW. Gone fishing: a unique histologic pattern in cutaneous angiosarcoma. Cutis. 2018;101:270-272.
- Sullivan HC, Edgar MA, Cohen C, et al. The utility of ERG, CD31 and CD34 in the cytological diagnosis of angiosarcoma: an analysis of 25 cases. J Clin Pathol. 2015;68:44-50.
- Rossi S, Orvieto E, Furlanetto A, et al. Utility of the immunohistochemical detection of FLI-1 expression in round cell and vascular neoplasm using a monoclonal antibody. Mod Pathol. 2004;17:547-552.
- Tan AR. Cutaneous manifestations of breast cancer. Semin Oncol. 2016;43:331-334.
- Schwartz RA, Wiederkehr M, Lambert WC. Secondary mucinous carcinoma of the skin: metastatic breast cancer. Dermatol Surg. 2004;30(2, pt 1):234-235.
- Mallon E, Dawber RP. Alopecia neoplastica without alopecia: a unique presentation of breast carcinoma scalp metastasis. J Am Acad Dermatol. 1994;31(2, pt 2):319-321.
- Braxton DR, Cohen C, Siddiqui MT. Utility of GATA3 immunohistochemistry for diagnosis of metastatic breast carcinoma in cytology specimens. Diagn Cytopathol. 2015;43:271-277.
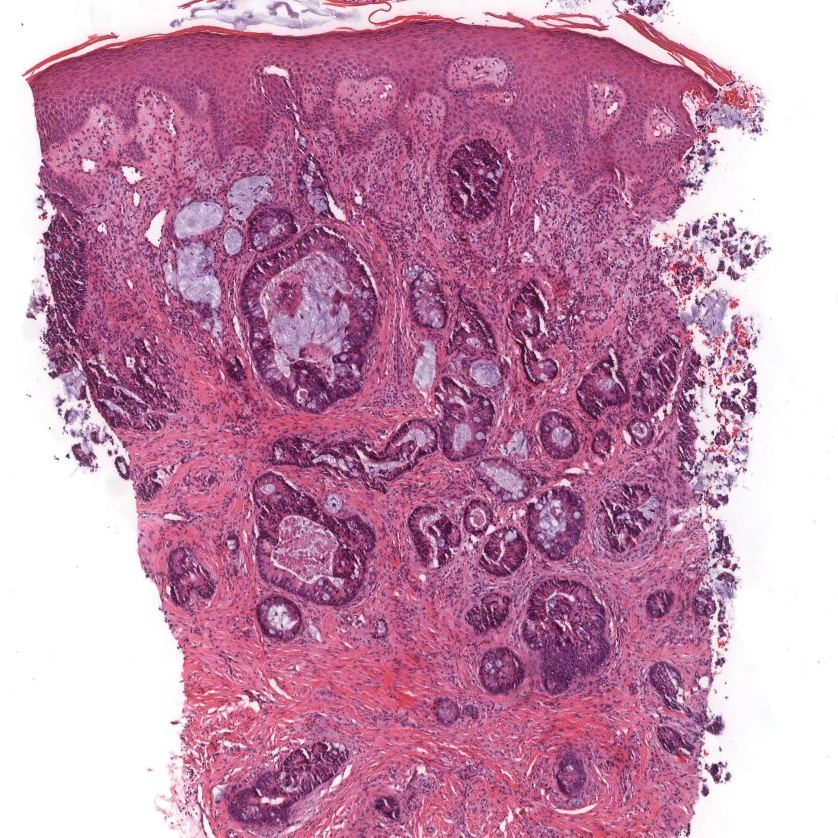
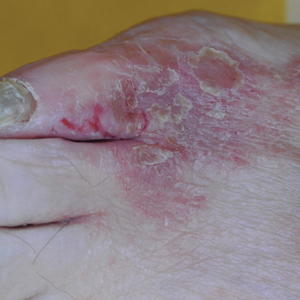
A 68-year-old patient presented with an enlarging flesh-colored nodule on the thigh that was positive for cytokeratin 20 and negative for cytokeratin 7.
Symmetric Lichen Amyloidosis: An Atypical Location on the Bilateral Extensor Surfaces of the Arms
To the Editor:
Lichen amyloidosis (LA) classically presents as a pruritic, hyperkeratotic, papular eruption localized to the pretibial surface of the legs.1 Nonpruritic and generalized variants have been reported but are rare.2 Although it is the most common subtype of primary localized cutaneous amyloidosis, LA is a benign condition but is difficult to eradicate.1 The precise pathophysiology is poorly understood, but chronic frictional irritation is closely associated with the eruption. We present a nongeneralized case of LA in an atypical location.
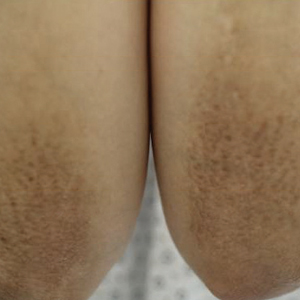
A healthy 30-year-old woman presented with an intermittent itchy rash on the elbows and knees of 2 years’ duration. The patient was first diagnosed with lichen simplex chronicus (LSC) and initially responded well to treatment with fluocinonide ointment 0.05%. Nearly 2 years after the initial presentation, she developed recurrent symptoms and sought further treatment. She reported frequent scratching in association with episodes of anxiety. Examination revealed numerous 1- to 3-mm, flesh-colored to light brown, monomorphic, dome-shaped papules over the extensor surfaces of the bilateral arms and left pretibial surface (Figure 1).
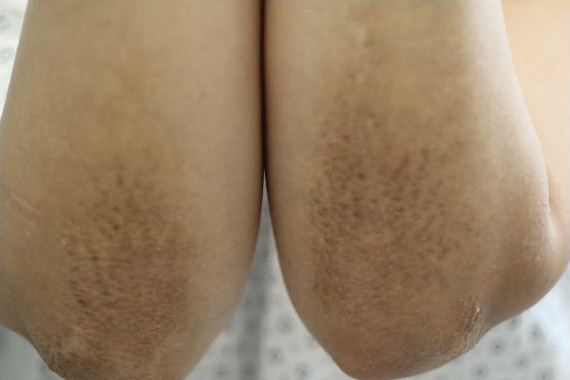
papules (1–3 mm) over the extensor surfaces of the bilateral arms.
Although in an atypical location, LA was clinically suspected due to the morphology, and a biopsy was performed given the evolving nature of the lesions. The differential diagnosis included LSC, hypertrophic lichen planus, papular mucinosis, prurigo nodularis, and pretibial myxedema. Pathology revealed small eosinophilic globules in the papillary dermis (Figure 2), and cytokeratin 5/6 immunostaining showed amorphous papillary dermal deposits consistent with keratin-derived amyloid deposition (Figure 3). The deposits stained positive for Congo red and displayed apple green birefringence under polarized light. Thus, the diagnosis of LA was confirmed. After limited success with triamcinolone ointment 0.1%, the patient was transitioned to clobetasol cream 0.05% with notable physical and symptomatic improvement.
Amyloidosis is histopathologically characterized by extracellular deposits of amyloid, a polypeptide that polymerizes to form cross-β sheets.3 It is believed that the deposits seen in localized amyloidosis result from local production of amyloid, as opposed to the deposition of circulating light chains that is characteristic of systemic amyloidosis.3 Lichen amyloidosis is the most common subtype of primary localized cutaneous amyloidosis.1 The amyloid in this condition has been found to react immunohistochemically with antikeratin antibody, leading to the conclusion that the amyloid is formed by degeneration of keratinocytes locally due to chronic rubbing and scratching.
4-6
The possibility remains that this patient first presented with LSC 2 years prior and secondarily developed LA due to chronic trauma. Indeed, LA has been proposed as a variant of LSC. In both conditions, scratching seems to be the most important factor in the development of lesions. It has been proposed that treatment should primarily focus on the amelioration of pruritus.5
Five percent to 10% of cases of LA have been found to have some form of upper extremity involvement.7 However, these cases typically are associated with a generalized presentation involving the trunk and arms.2,7 Our patient had no evidence of disease elsewhere. When evaluating a localized, pruritic, monomorphic, papular eruption on the extensor surfaces of the arms, LA may be an important consideration.
- Tay CH, Dacosta JL. Lichen amyloidosis. clinical study of 40 cases. Br J Dermatol. 1970;82:129-136.
- Kandhari R, Ramesh V, Singh A. A generalized, non-pruritic variant of lichen amyloidosis: a case report and a brief review. Indian J Dermatol. 2013;58:328.
- Biewend ML, Menke DM, Calamia KT. The spectrum of localized amyloidosis: a case series of 20 patients and review of the literature. Amyloid. 2006;13:135-142.
- Jambrosic J, From L, Hanna W. Lichen amyloidosus. ultrastructure and pathogenesis. Am J Dermatopathol. 1984;6:151-158.
- Weyers W, Weyers I, Bonczkowitz M, et al. Lichen amyloidosis: a consequence of scratching. J Am Acad Dermatol. 1997;37:923-928.
- Kumakiri M, Hashimoto K. Histogenesis of primary localized cutaneous amyloidosis: sequential change of epidermal keratinocytes to amyloid via filamentous degeneration. J Invest Dermatol. 1979;73:150-162.
- Salim T, Shenoi SD, Balachandran C, et al. Lichen amyloidosus: a study of clinical, histopathologic and immunofluorescence findings in 30 cases. Indian J Dermatol Venereol Leprol. 2005;71:166-169.
To the Editor:
Lichen amyloidosis (LA) classically presents as a pruritic, hyperkeratotic, papular eruption localized to the pretibial surface of the legs.1 Nonpruritic and generalized variants have been reported but are rare.2 Although it is the most common subtype of primary localized cutaneous amyloidosis, LA is a benign condition but is difficult to eradicate.1 The precise pathophysiology is poorly understood, but chronic frictional irritation is closely associated with the eruption. We present a nongeneralized case of LA in an atypical location.
A healthy 30-year-old woman presented with an intermittent itchy rash on the elbows and knees of 2 years’ duration. The patient was first diagnosed with lichen simplex chronicus (LSC) and initially responded well to treatment with fluocinonide ointment 0.05%. Nearly 2 years after the initial presentation, she developed recurrent symptoms and sought further treatment. She reported frequent scratching in association with episodes of anxiety. Examination revealed numerous 1- to 3-mm, flesh-colored to light brown, monomorphic, dome-shaped papules over the extensor surfaces of the bilateral arms and left pretibial surface (Figure 1).
papules (1–3 mm) over the extensor surfaces of the bilateral arms.
Although in an atypical location, LA was clinically suspected due to the morphology, and a biopsy was performed given the evolving nature of the lesions. The differential diagnosis included LSC, hypertrophic lichen planus, papular mucinosis, prurigo nodularis, and pretibial myxedema. Pathology revealed small eosinophilic globules in the papillary dermis (Figure 2), and cytokeratin 5/6 immunostaining showed amorphous papillary dermal deposits consistent with keratin-derived amyloid deposition (Figure 3). The deposits stained positive for Congo red and displayed apple green birefringence under polarized light. Thus, the diagnosis of LA was confirmed. After limited success with triamcinolone ointment 0.1%, the patient was transitioned to clobetasol cream 0.05% with notable physical and symptomatic improvement.
Amyloidosis is histopathologically characterized by extracellular deposits of amyloid, a polypeptide that polymerizes to form cross-β sheets.3 It is believed that the deposits seen in localized amyloidosis result from local production of amyloid, as opposed to the deposition of circulating light chains that is characteristic of systemic amyloidosis.3 Lichen amyloidosis is the most common subtype of primary localized cutaneous amyloidosis.1 The amyloid in this condition has been found to react immunohistochemically with antikeratin antibody, leading to the conclusion that the amyloid is formed by degeneration of keratinocytes locally due to chronic rubbing and scratching.
4-6
The possibility remains that this patient first presented with LSC 2 years prior and secondarily developed LA due to chronic trauma. Indeed, LA has been proposed as a variant of LSC. In both conditions, scratching seems to be the most important factor in the development of lesions. It has been proposed that treatment should primarily focus on the amelioration of pruritus.5
Five percent to 10% of cases of LA have been found to have some form of upper extremity involvement.7 However, these cases typically are associated with a generalized presentation involving the trunk and arms.2,7 Our patient had no evidence of disease elsewhere. When evaluating a localized, pruritic, monomorphic, papular eruption on the extensor surfaces of the arms, LA may be an important consideration.
To the Editor:
Lichen amyloidosis (LA) classically presents as a pruritic, hyperkeratotic, papular eruption localized to the pretibial surface of the legs.1 Nonpruritic and generalized variants have been reported but are rare.2 Although it is the most common subtype of primary localized cutaneous amyloidosis, LA is a benign condition but is difficult to eradicate.1 The precise pathophysiology is poorly understood, but chronic frictional irritation is closely associated with the eruption. We present a nongeneralized case of LA in an atypical location.
A healthy 30-year-old woman presented with an intermittent itchy rash on the elbows and knees of 2 years’ duration. The patient was first diagnosed with lichen simplex chronicus (LSC) and initially responded well to treatment with fluocinonide ointment 0.05%. Nearly 2 years after the initial presentation, she developed recurrent symptoms and sought further treatment. She reported frequent scratching in association with episodes of anxiety. Examination revealed numerous 1- to 3-mm, flesh-colored to light brown, monomorphic, dome-shaped papules over the extensor surfaces of the bilateral arms and left pretibial surface (Figure 1).
papules (1–3 mm) over the extensor surfaces of the bilateral arms.
Although in an atypical location, LA was clinically suspected due to the morphology, and a biopsy was performed given the evolving nature of the lesions. The differential diagnosis included LSC, hypertrophic lichen planus, papular mucinosis, prurigo nodularis, and pretibial myxedema. Pathology revealed small eosinophilic globules in the papillary dermis (Figure 2), and cytokeratin 5/6 immunostaining showed amorphous papillary dermal deposits consistent with keratin-derived amyloid deposition (Figure 3). The deposits stained positive for Congo red and displayed apple green birefringence under polarized light. Thus, the diagnosis of LA was confirmed. After limited success with triamcinolone ointment 0.1%, the patient was transitioned to clobetasol cream 0.05% with notable physical and symptomatic improvement.
Amyloidosis is histopathologically characterized by extracellular deposits of amyloid, a polypeptide that polymerizes to form cross-β sheets.3 It is believed that the deposits seen in localized amyloidosis result from local production of amyloid, as opposed to the deposition of circulating light chains that is characteristic of systemic amyloidosis.3 Lichen amyloidosis is the most common subtype of primary localized cutaneous amyloidosis.1 The amyloid in this condition has been found to react immunohistochemically with antikeratin antibody, leading to the conclusion that the amyloid is formed by degeneration of keratinocytes locally due to chronic rubbing and scratching.
4-6
The possibility remains that this patient first presented with LSC 2 years prior and secondarily developed LA due to chronic trauma. Indeed, LA has been proposed as a variant of LSC. In both conditions, scratching seems to be the most important factor in the development of lesions. It has been proposed that treatment should primarily focus on the amelioration of pruritus.5
Five percent to 10% of cases of LA have been found to have some form of upper extremity involvement.7 However, these cases typically are associated with a generalized presentation involving the trunk and arms.2,7 Our patient had no evidence of disease elsewhere. When evaluating a localized, pruritic, monomorphic, papular eruption on the extensor surfaces of the arms, LA may be an important consideration.
- Tay CH, Dacosta JL. Lichen amyloidosis. clinical study of 40 cases. Br J Dermatol. 1970;82:129-136.
- Kandhari R, Ramesh V, Singh A. A generalized, non-pruritic variant of lichen amyloidosis: a case report and a brief review. Indian J Dermatol. 2013;58:328.
- Biewend ML, Menke DM, Calamia KT. The spectrum of localized amyloidosis: a case series of 20 patients and review of the literature. Amyloid. 2006;13:135-142.
- Jambrosic J, From L, Hanna W. Lichen amyloidosus. ultrastructure and pathogenesis. Am J Dermatopathol. 1984;6:151-158.
- Weyers W, Weyers I, Bonczkowitz M, et al. Lichen amyloidosis: a consequence of scratching. J Am Acad Dermatol. 1997;37:923-928.
- Kumakiri M, Hashimoto K. Histogenesis of primary localized cutaneous amyloidosis: sequential change of epidermal keratinocytes to amyloid via filamentous degeneration. J Invest Dermatol. 1979;73:150-162.
- Salim T, Shenoi SD, Balachandran C, et al. Lichen amyloidosus: a study of clinical, histopathologic and immunofluorescence findings in 30 cases. Indian J Dermatol Venereol Leprol. 2005;71:166-169.
- Tay CH, Dacosta JL. Lichen amyloidosis. clinical study of 40 cases. Br J Dermatol. 1970;82:129-136.
- Kandhari R, Ramesh V, Singh A. A generalized, non-pruritic variant of lichen amyloidosis: a case report and a brief review. Indian J Dermatol. 2013;58:328.
- Biewend ML, Menke DM, Calamia KT. The spectrum of localized amyloidosis: a case series of 20 patients and review of the literature. Amyloid. 2006;13:135-142.
- Jambrosic J, From L, Hanna W. Lichen amyloidosus. ultrastructure and pathogenesis. Am J Dermatopathol. 1984;6:151-158.
- Weyers W, Weyers I, Bonczkowitz M, et al. Lichen amyloidosis: a consequence of scratching. J Am Acad Dermatol. 1997;37:923-928.
- Kumakiri M, Hashimoto K. Histogenesis of primary localized cutaneous amyloidosis: sequential change of epidermal keratinocytes to amyloid via filamentous degeneration. J Invest Dermatol. 1979;73:150-162.
- Salim T, Shenoi SD, Balachandran C, et al. Lichen amyloidosus: a study of clinical, histopathologic and immunofluorescence findings in 30 cases. Indian J Dermatol Venereol Leprol. 2005;71:166-169.
Practice Points
- Lichen amyloidosis (LA) classically presents as a pruritic and papular eruption localized to the pretibial surface of the legs.
- Nonpruritic and generalized variants are rare.
- This case represents a pruritic and nongeneralized
case located on the arms; LA should be considered
for any localized and pruritic eruption on the arms.
Acral Flesh-Colored Papules on the Fingers
The Diagnosis: Lichen Nitidus
Our patient represents a case of lichen nitidus (LN) that was diagnosed through clinicopathologic correlation, with the pathology results showing a lymphohistiocytic infiltrate in the papillary dermis enclosed by acanthotic rete ridges on either side. Lichen nitidus was first described by Pinkus in 1901 as a variant of lichen planus.1 It is a rare chronic inflammatory disease that is most prevalent in children and adolescents.2 Clinically, the lesions appear as 1- to 2-mm, shiny, flesh-colored papules with central umbilication.3 Typically, lesions are localized and discrete; however, vesicular, hemorrhagic, perforating, spinous follicular, linear, generalized, and actinic variants all have been reported in the literature. Lichen nitidus has a predilection for the lower abdomen, medial thighs, penis, forearms, ventral wrists, and hands.4 Cases of LN have been reported on the palms, soles, nails, and mucosa, presenting a diagnostic challenge.5 The pathogenesis of LN is unknown, and all races and sexes are affected equally.6
Histopathologically, LN has distinct findings including a well-circumscribed lymphohistiocytic infiltrate in the papillary dermis embraced by elongated and acanthotic rete ridges.2 These histopathologic characteristics were seen in our patient's biopsy specimen (Figure) and have been described as the ball-and-claw configuration. Lichen nitidus may be pruritic but typically is asymptomatic.7 It often spontaneously regresses within months to years without any treatment7; however, successful outcomes have been seen with topical steroids, UVA/UVB phototherapy, and retinoids.2 Our patient was treated with topical steroids.

The differential diagnosis for LN includes verruca plana, dyshidrotic eczema, acral persistent papular mucinosis (APPM), and molluscum contagiosum. Verruca plana can occur as 1- to 5-mm, grouped, flesh-colored papules on the face, neck, dorsal hands, wrists, or knees.8 Most commonly, verruca plana occurs due to human papillomavirus type 3 and less commonly human papillomavirus types 10, 27, and 41. Verruca plana is easily differentiated from LN on pathology with findings of epidermal hyperkeratosis, irregular acanthosis, and koilocytic changes.8
Dyshidrotic eczema is a pruritic vesicular rash that is classically distributed symmetrically on the palmar aspects of the hands and lateral fingers.9 Histopathology of the lesions reveals spongiosis with an epidermal lymphocytic infiltrate. Exacerbating factors include exposure to allergens, stress, fungal infections, and genetic predisposition.9
Acral persistent papular mucinosis can present as multiple, 2- to 5-mm, flesh-colored papules on the dorsal aspects of the hands.10 However, the demographic is different from LN, as APPM most commonly affects middle-aged females versus adolescents. Lesions of APPM may multiply or spontaneously remit over time. Acral persistent papular mucinosis generally is asymptomatic but can be treated with cryotherapy, topical corticosteroids, electrodesiccation, or CO2 lasers for cosmetic purposes. Acral persistent papular mucinosis can be easily distinguished from LN on histology, as it will show areas of focal, well-circumscribed mucin in the papillary dermis and a spared Grenz zone.10
Molluscum contagiosum is a common viral skin infection caused by the poxvirus that affects children and adults.11 The skin lesions appear as 2- to 4-mm, dome-shaped, flesh-colored papules with central umbilication on the limbs, trunk, or face. Clinicians may choose to monitor lesions of molluscum contagiosum, as it is a self-limited condition, or it may be treated with cryotherapy, salicylic acid, imiquimod, curettage, laser, or cimetidine.11 On histology, epidermal budlike proliferations can be appreciated in the dermis, and characteristic large, eosinophilic, intracytoplasmic inclusion or molluscum bodies are found in the epidermis.12
- Barber HW. Case of lichen nitidus (Pinkus) or tuberculide lichéniforme et nitida (Chatellier). Proc R Soc Med. 1924;17:39.
- Frey MN, Luzzatto L, Seidel GB, et al. Case for diagnosis. An Bras Dermatol. 2010;85:561-563.
- Pielop JA, Hsu S. Tiny, skin-colored papules on the arms and hands. Am Fam Physician. 2005;72:343-344.
- Cho EB, Kim HY, Park EJ, et al. Three cases of lichen nitidus associated with various cutaneous diseases. Ann Dermatol. 2014;26:505-509.
- Podder I, Mohanty S, Chandra S, et al. Isolated palmar lichen nitidus--a diagnostic challenge: first case from Eastern India. Indian J Dermatol. 2015;60:308-309.
- Chen W, Schramm M, Zouboulis C. Generalized lichen nitidus. J Am Acad Dermatol. 1997;36:630-631.
- Rallis E, Verros C, Moussatou V, et al. Generalized purpuric lichen nitidus: a case report and review of the literature. Dermatol Online J. 2007;13:5.
- Pavithra S, Mallya H, Pai GS. Extensive presentation of verruca plana in a healthy individual. Indian J Dermatol. 2011;56:324-325.
- Paulsen L, Geller D, Guggenbiller M. Symmetrical vesicular eruption on the palms. Am Fam Physician. 2012;15:811-812.
- Alvarez-Garrido H, Najera L, Garrido-Rios A, et al. Acral persistent papular mucinosis: is it an under-diagnosed disease? Dermatol Online J. 2014;20:10
- Diaconu R, Oprea B, Vasilescu M, et al. Inflamed molluscum contagiosum in a 6-year-old boy: a case report. Rom J Morphol Embryol. 2015;56:843-845.
- Krishnamurthy J, Nagappa D. The cytology of molluscum contagiosum mimicking skin adnexal tumor. J Cytol. 2010;27:74.
The Diagnosis: Lichen Nitidus
Our patient represents a case of lichen nitidus (LN) that was diagnosed through clinicopathologic correlation, with the pathology results showing a lymphohistiocytic infiltrate in the papillary dermis enclosed by acanthotic rete ridges on either side. Lichen nitidus was first described by Pinkus in 1901 as a variant of lichen planus.1 It is a rare chronic inflammatory disease that is most prevalent in children and adolescents.2 Clinically, the lesions appear as 1- to 2-mm, shiny, flesh-colored papules with central umbilication.3 Typically, lesions are localized and discrete; however, vesicular, hemorrhagic, perforating, spinous follicular, linear, generalized, and actinic variants all have been reported in the literature. Lichen nitidus has a predilection for the lower abdomen, medial thighs, penis, forearms, ventral wrists, and hands.4 Cases of LN have been reported on the palms, soles, nails, and mucosa, presenting a diagnostic challenge.5 The pathogenesis of LN is unknown, and all races and sexes are affected equally.6
Histopathologically, LN has distinct findings including a well-circumscribed lymphohistiocytic infiltrate in the papillary dermis embraced by elongated and acanthotic rete ridges.2 These histopathologic characteristics were seen in our patient's biopsy specimen (Figure) and have been described as the ball-and-claw configuration. Lichen nitidus may be pruritic but typically is asymptomatic.7 It often spontaneously regresses within months to years without any treatment7; however, successful outcomes have been seen with topical steroids, UVA/UVB phototherapy, and retinoids.2 Our patient was treated with topical steroids.
The differential diagnosis for LN includes verruca plana, dyshidrotic eczema, acral persistent papular mucinosis (APPM), and molluscum contagiosum. Verruca plana can occur as 1- to 5-mm, grouped, flesh-colored papules on the face, neck, dorsal hands, wrists, or knees.8 Most commonly, verruca plana occurs due to human papillomavirus type 3 and less commonly human papillomavirus types 10, 27, and 41. Verruca plana is easily differentiated from LN on pathology with findings of epidermal hyperkeratosis, irregular acanthosis, and koilocytic changes.8
Dyshidrotic eczema is a pruritic vesicular rash that is classically distributed symmetrically on the palmar aspects of the hands and lateral fingers.9 Histopathology of the lesions reveals spongiosis with an epidermal lymphocytic infiltrate. Exacerbating factors include exposure to allergens, stress, fungal infections, and genetic predisposition.9
Acral persistent papular mucinosis can present as multiple, 2- to 5-mm, flesh-colored papules on the dorsal aspects of the hands.10 However, the demographic is different from LN, as APPM most commonly affects middle-aged females versus adolescents. Lesions of APPM may multiply or spontaneously remit over time. Acral persistent papular mucinosis generally is asymptomatic but can be treated with cryotherapy, topical corticosteroids, electrodesiccation, or CO2 lasers for cosmetic purposes. Acral persistent papular mucinosis can be easily distinguished from LN on histology, as it will show areas of focal, well-circumscribed mucin in the papillary dermis and a spared Grenz zone.10
Molluscum contagiosum is a common viral skin infection caused by the poxvirus that affects children and adults.11 The skin lesions appear as 2- to 4-mm, dome-shaped, flesh-colored papules with central umbilication on the limbs, trunk, or face. Clinicians may choose to monitor lesions of molluscum contagiosum, as it is a self-limited condition, or it may be treated with cryotherapy, salicylic acid, imiquimod, curettage, laser, or cimetidine.11 On histology, epidermal budlike proliferations can be appreciated in the dermis, and characteristic large, eosinophilic, intracytoplasmic inclusion or molluscum bodies are found in the epidermis.12
The Diagnosis: Lichen Nitidus
Our patient represents a case of lichen nitidus (LN) that was diagnosed through clinicopathologic correlation, with the pathology results showing a lymphohistiocytic infiltrate in the papillary dermis enclosed by acanthotic rete ridges on either side. Lichen nitidus was first described by Pinkus in 1901 as a variant of lichen planus.1 It is a rare chronic inflammatory disease that is most prevalent in children and adolescents.2 Clinically, the lesions appear as 1- to 2-mm, shiny, flesh-colored papules with central umbilication.3 Typically, lesions are localized and discrete; however, vesicular, hemorrhagic, perforating, spinous follicular, linear, generalized, and actinic variants all have been reported in the literature. Lichen nitidus has a predilection for the lower abdomen, medial thighs, penis, forearms, ventral wrists, and hands.4 Cases of LN have been reported on the palms, soles, nails, and mucosa, presenting a diagnostic challenge.5 The pathogenesis of LN is unknown, and all races and sexes are affected equally.6
Histopathologically, LN has distinct findings including a well-circumscribed lymphohistiocytic infiltrate in the papillary dermis embraced by elongated and acanthotic rete ridges.2 These histopathologic characteristics were seen in our patient's biopsy specimen (Figure) and have been described as the ball-and-claw configuration. Lichen nitidus may be pruritic but typically is asymptomatic.7 It often spontaneously regresses within months to years without any treatment7; however, successful outcomes have been seen with topical steroids, UVA/UVB phototherapy, and retinoids.2 Our patient was treated with topical steroids.
The differential diagnosis for LN includes verruca plana, dyshidrotic eczema, acral persistent papular mucinosis (APPM), and molluscum contagiosum. Verruca plana can occur as 1- to 5-mm, grouped, flesh-colored papules on the face, neck, dorsal hands, wrists, or knees.8 Most commonly, verruca plana occurs due to human papillomavirus type 3 and less commonly human papillomavirus types 10, 27, and 41. Verruca plana is easily differentiated from LN on pathology with findings of epidermal hyperkeratosis, irregular acanthosis, and koilocytic changes.8
Dyshidrotic eczema is a pruritic vesicular rash that is classically distributed symmetrically on the palmar aspects of the hands and lateral fingers.9 Histopathology of the lesions reveals spongiosis with an epidermal lymphocytic infiltrate. Exacerbating factors include exposure to allergens, stress, fungal infections, and genetic predisposition.9
Acral persistent papular mucinosis can present as multiple, 2- to 5-mm, flesh-colored papules on the dorsal aspects of the hands.10 However, the demographic is different from LN, as APPM most commonly affects middle-aged females versus adolescents. Lesions of APPM may multiply or spontaneously remit over time. Acral persistent papular mucinosis generally is asymptomatic but can be treated with cryotherapy, topical corticosteroids, electrodesiccation, or CO2 lasers for cosmetic purposes. Acral persistent papular mucinosis can be easily distinguished from LN on histology, as it will show areas of focal, well-circumscribed mucin in the papillary dermis and a spared Grenz zone.10
Molluscum contagiosum is a common viral skin infection caused by the poxvirus that affects children and adults.11 The skin lesions appear as 2- to 4-mm, dome-shaped, flesh-colored papules with central umbilication on the limbs, trunk, or face. Clinicians may choose to monitor lesions of molluscum contagiosum, as it is a self-limited condition, or it may be treated with cryotherapy, salicylic acid, imiquimod, curettage, laser, or cimetidine.11 On histology, epidermal budlike proliferations can be appreciated in the dermis, and characteristic large, eosinophilic, intracytoplasmic inclusion or molluscum bodies are found in the epidermis.12
- Barber HW. Case of lichen nitidus (Pinkus) or tuberculide lichéniforme et nitida (Chatellier). Proc R Soc Med. 1924;17:39.
- Frey MN, Luzzatto L, Seidel GB, et al. Case for diagnosis. An Bras Dermatol. 2010;85:561-563.
- Pielop JA, Hsu S. Tiny, skin-colored papules on the arms and hands. Am Fam Physician. 2005;72:343-344.
- Cho EB, Kim HY, Park EJ, et al. Three cases of lichen nitidus associated with various cutaneous diseases. Ann Dermatol. 2014;26:505-509.
- Podder I, Mohanty S, Chandra S, et al. Isolated palmar lichen nitidus--a diagnostic challenge: first case from Eastern India. Indian J Dermatol. 2015;60:308-309.
- Chen W, Schramm M, Zouboulis C. Generalized lichen nitidus. J Am Acad Dermatol. 1997;36:630-631.
- Rallis E, Verros C, Moussatou V, et al. Generalized purpuric lichen nitidus: a case report and review of the literature. Dermatol Online J. 2007;13:5.
- Pavithra S, Mallya H, Pai GS. Extensive presentation of verruca plana in a healthy individual. Indian J Dermatol. 2011;56:324-325.
- Paulsen L, Geller D, Guggenbiller M. Symmetrical vesicular eruption on the palms. Am Fam Physician. 2012;15:811-812.
- Alvarez-Garrido H, Najera L, Garrido-Rios A, et al. Acral persistent papular mucinosis: is it an under-diagnosed disease? Dermatol Online J. 2014;20:10
- Diaconu R, Oprea B, Vasilescu M, et al. Inflamed molluscum contagiosum in a 6-year-old boy: a case report. Rom J Morphol Embryol. 2015;56:843-845.
- Krishnamurthy J, Nagappa D. The cytology of molluscum contagiosum mimicking skin adnexal tumor. J Cytol. 2010;27:74.
- Barber HW. Case of lichen nitidus (Pinkus) or tuberculide lichéniforme et nitida (Chatellier). Proc R Soc Med. 1924;17:39.
- Frey MN, Luzzatto L, Seidel GB, et al. Case for diagnosis. An Bras Dermatol. 2010;85:561-563.
- Pielop JA, Hsu S. Tiny, skin-colored papules on the arms and hands. Am Fam Physician. 2005;72:343-344.
- Cho EB, Kim HY, Park EJ, et al. Three cases of lichen nitidus associated with various cutaneous diseases. Ann Dermatol. 2014;26:505-509.
- Podder I, Mohanty S, Chandra S, et al. Isolated palmar lichen nitidus--a diagnostic challenge: first case from Eastern India. Indian J Dermatol. 2015;60:308-309.
- Chen W, Schramm M, Zouboulis C. Generalized lichen nitidus. J Am Acad Dermatol. 1997;36:630-631.
- Rallis E, Verros C, Moussatou V, et al. Generalized purpuric lichen nitidus: a case report and review of the literature. Dermatol Online J. 2007;13:5.
- Pavithra S, Mallya H, Pai GS. Extensive presentation of verruca plana in a healthy individual. Indian J Dermatol. 2011;56:324-325.
- Paulsen L, Geller D, Guggenbiller M. Symmetrical vesicular eruption on the palms. Am Fam Physician. 2012;15:811-812.
- Alvarez-Garrido H, Najera L, Garrido-Rios A, et al. Acral persistent papular mucinosis: is it an under-diagnosed disease? Dermatol Online J. 2014;20:10
- Diaconu R, Oprea B, Vasilescu M, et al. Inflamed molluscum contagiosum in a 6-year-old boy: a case report. Rom J Morphol Embryol. 2015;56:843-845.
- Krishnamurthy J, Nagappa D. The cytology of molluscum contagiosum mimicking skin adnexal tumor. J Cytol. 2010;27:74.
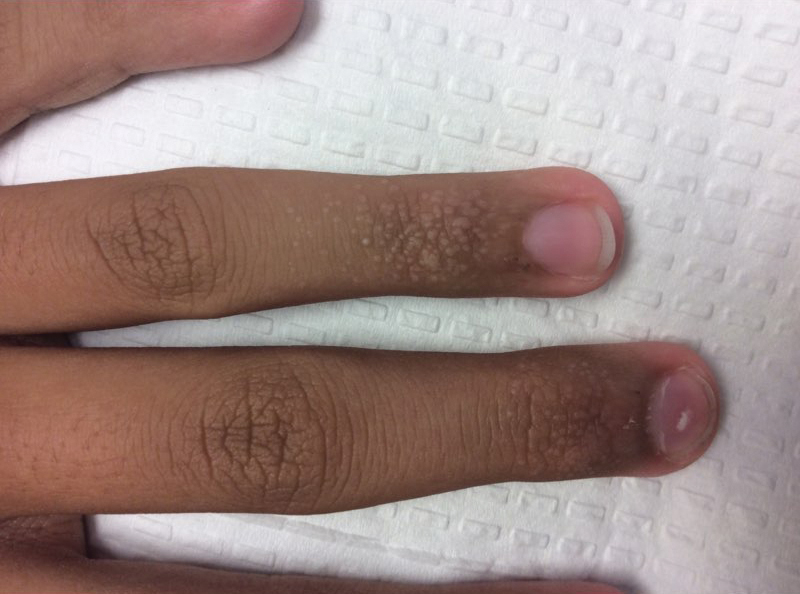
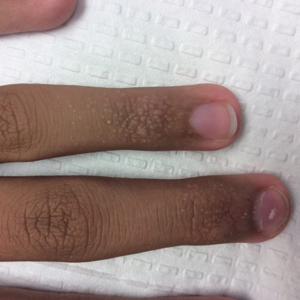
A 13-year-old otherwise healthy adolescent boy presented to the dermatology clinic for a rash on the bilateral dorsal hands of approximately 1 year’s duration. The rash was asymptomatic with no pain or pruritus reported. Physical examination revealed a well-nourished adolescent boy in no acute distress with 1- to 2-mm flesh-colored papules clustered on the bilateral dorsal fingers.
Unilateral Facial Papules and Plaques
The Diagnosis: Unilateral Dermatomal Trichoepithelioma
Adnexal lesions presenting with a linear and/or dermatomal pattern rarely have been reported. Bolognia et al1 performed a comprehensive review of Blaschko lines and skin conditions that follow such a pattern. The authors found that adnexal-related lesions included linear nevus comedonicus, linear basal cell nevus with comedones (linear basaloid follicular hamartoma), unilateral nevoid basal cell carcinoma (BCC), linear trichoepithelioma, linear trichodiscoma, linear hamartoma of the follicular infundibulum, nevus sebaceous, syringocystadenoma papilliferum, porokeratotic eccrine ostial and dermal duct nevus, linear eccrine poroma, linear spiradenoma, linear syringoma, and linear eccrine syringofibroadenoma.1
Trichoepithelioma is a hair follicle-related neoplastic lesion presenting most commonly as the autosomal-dominant multiple familial type with lesions mainly centered on the face. Initial genetic studies associated the disease with loss of heterozygosity in the 9p21 region and further studies identified mutations in the CYLD (cylindromatosis [turban tumor syndrome]) gene on chromosome 16q12-q13.2,3 Unilateral, linear, and dermatomal forms of trichoepithelioma rarely are reported. In 1986, Geffner et al4 reported a case of linear and dermatomal trichoepithelioma in a 10-year-old girl. In addition to discrete solitary lesions affecting the face, she developed lesions on the left shoulder, left side of the trunk, and left lower leg following dermatomal distribution. In 2006, 2 cases of dermatomal trichoepitheliomas affecting the face in children, as in our case, were reported.5,6 Another case involving the neck was reported in 2016.7 Although classic multiple familial trichoepithelioma can be part of conditions such as Brooke-Spiegler8 and Rombo syndromes,9 no syndromal association has been reported thus far with the unilateral, linear, or dermatomal variants.
Our case showed typical histopathologic features of trichoepithelioma, including discrete islands of basaloid cells in the dermis set in a conspicuous fibroblastic stroma. Focal connection with the epidermis was present. Most of the islands showed peripheral palisading and horn cysts lined by eosinophilic cells. The fibroblastic component was tightly adherent to the epithelial component, and only stromal clefts were detected. Papillary mesenchymal bodies also were detected as oval aggregates of fibroblastic cells invaginating into epithelial islands to form hair papillae.
Histopathologically, the 2 most important differential diagnoses of trichoepithelioma include BCC and basaloid follicular hamartoma. In differentiating BCC from trichoepithelioma, the presence of dense fibroblastic stroma and papillary mesenchymal bodies characterize trichoepithelioma, while a fibromucinous stroma with mucinous retraction artifacts and clefting between the basaloid islands and the stroma characterize BCC (Figure 1).10 Immunohistochemical studies using antibodies against Bcl-2, CD34, CD10, androgen receptor, Ki-67, cytokeratin 19, and PHLDA1 (pleckstrin homologylike domain family A member 1) have reportedly been utilized to differentiate trichoepithelioma from BCC.11,12 Basaloid follicular hamartoma is characterized by thin anastomosing strands and branching cords of undifferentiated basaloid cells that replace or associate hair follicles in a latticelike pattern (Figure 2). The strands usually are vertically oriented perpendicular to the epidermis. Peripheral palisading is possible, and the basaloid strands are surrounded with cellular connective tissue stroma.13 Tumor islands in eccrine poroma show broad connections with the epidermis and are composed of poroid cells that show evident ductal differentiation with eosinophilic cuticles (Figure 3).14 Spiradenoma is characterized by capsulated deep-seated tumorous nodules not connected with the epidermis and composed of light and dark cells with ductal differentiation and vascular stroma (Figure 4). Scattered lymphocytes within the tumor lobules and in the stroma also are seen. Eosinophilic hyaline globules rarely can be present.15


Many pathologists consider trichoepithelioma as the superficial variant of trichoblastoma. According to the recent World Health Organization classification of benign tumors with follicular differentiation, trichoepithelioma is considered synonymous with trichoblastoma.16
Trichoepitheliomas are benign tumors, and therapy is mainly directed at removal for cosmetic purposes. Several methods of removal are available including electrocautery, laser therapy, and surgery. Awareness of the possible dermatomal distribution of hair follicle and other adnexal-related conditions is important, and such lesions should be thought of in the differential diagnosis of unilateral and/or dermatomal lesions.
- Bolognia JL, Orlow SJ, Glick SA. Lines of Blaschko. J Am Acad Dermatol. 1994;31(2, pt 1):157-190.
- Harada H, Hashimoto K, Ko MS. The gene for multiple familial trichoepithelioma maps to chromosome 9p21. J Invest Dermatol. 1996;107:41-43.
- Zheng G, Hu L, Huang W, et al. CYLD mutation causes multiple familial trichoepithelioma in three Chinese families. Hum Mutat. 2004;23:400.
- Geffner RE, Goslen JB, Santa Cruz DJ. Linear and dermatomal trichoepitheliomas. J Am Acad Dermatol. 1986;14(5, pt 2):927-930.
- Chang YC, Colome-Grimmer M, Kelly E. Multiple trichoepitheliomas in the lines of Blaschko. Pediatr Dermatol. 2006;23:149-151.
- Strauss RM, Merchant WJ, Stainforth JM, et al. Unilateral naevoid trichoepitheliomas on the face of a child. Clin Exp Dermatol. 2006;6:778-780.
- Laska AJ, Belli RA, Kobayashi TT. Linear trichoepithelioma on the neck of a 15-year-old girl. Dermatol Online J. 2016;22. pii:13030/qt87b6h4q8.
- Rasmussen JE. A syndrome of trichoepitheliomas, milia and cylindroma. Arch Dermatol. 1975;111:610-614.
- Michaelson G, Olsson E, Westermark P. The Rombo syndrome. Acta Derm Venereol. 1981;61:497-503.
- Brooke JD, Fitzpatrick JE, Golitz LE. Papillary mesenchymal bodies: a histologic finding useful in differentiating trichoepitheliomas from basal cell carcinomas. J Am Acad Dermatol. 1989;21(3, pt 1):523-528.
- Mostafa NA, Assaf M, Elhakim S, et al. Diagnostic accuracy of immunohistochemical markers in differentiation between basal cell carcinoma and trichoepithelioma in small biopsy specimens. J Cutan Pathol. 2018;45:807-816.
- Poniecka AW, Alexis JB. An immunohistochemical study of basal cell carcinoma and trichoepithelioma. Am J Dermatopathol. 1999;21:332-336.
- Abdel-Halim MRE, Fawzy M, Saleh M, et al. Linear unilateral basal cell nevus with comedones (linear nevoid basaloid follicular hamartoma): a case report. J Egypt Womens Dermatol Soc. 2016;13:46-48.
- Hyman AB, Brownstein MH. Eccrine poroma: analysis of 45 new cases. Dermatologica. 1969;138:28-38.
- Mambo NC. Eccrine spiradenoma: clinical and pathologic study of 49 tumors. J Cutan Pathol. 1983;10:312-320.
- Kutzner H, Kaddu S, Kanitakis J, et al. Trichoblastoma. In: Elder D, Massi D, Scolyer RA, et al, eds. WHO Classification of Skin Tumours. 4th ed. Lyon, France: IARC; 2018.
The Diagnosis: Unilateral Dermatomal Trichoepithelioma
Adnexal lesions presenting with a linear and/or dermatomal pattern rarely have been reported. Bolognia et al1 performed a comprehensive review of Blaschko lines and skin conditions that follow such a pattern. The authors found that adnexal-related lesions included linear nevus comedonicus, linear basal cell nevus with comedones (linear basaloid follicular hamartoma), unilateral nevoid basal cell carcinoma (BCC), linear trichoepithelioma, linear trichodiscoma, linear hamartoma of the follicular infundibulum, nevus sebaceous, syringocystadenoma papilliferum, porokeratotic eccrine ostial and dermal duct nevus, linear eccrine poroma, linear spiradenoma, linear syringoma, and linear eccrine syringofibroadenoma.1
Trichoepithelioma is a hair follicle-related neoplastic lesion presenting most commonly as the autosomal-dominant multiple familial type with lesions mainly centered on the face. Initial genetic studies associated the disease with loss of heterozygosity in the 9p21 region and further studies identified mutations in the CYLD (cylindromatosis [turban tumor syndrome]) gene on chromosome 16q12-q13.2,3 Unilateral, linear, and dermatomal forms of trichoepithelioma rarely are reported. In 1986, Geffner et al4 reported a case of linear and dermatomal trichoepithelioma in a 10-year-old girl. In addition to discrete solitary lesions affecting the face, she developed lesions on the left shoulder, left side of the trunk, and left lower leg following dermatomal distribution. In 2006, 2 cases of dermatomal trichoepitheliomas affecting the face in children, as in our case, were reported.5,6 Another case involving the neck was reported in 2016.7 Although classic multiple familial trichoepithelioma can be part of conditions such as Brooke-Spiegler8 and Rombo syndromes,9 no syndromal association has been reported thus far with the unilateral, linear, or dermatomal variants.
Our case showed typical histopathologic features of trichoepithelioma, including discrete islands of basaloid cells in the dermis set in a conspicuous fibroblastic stroma. Focal connection with the epidermis was present. Most of the islands showed peripheral palisading and horn cysts lined by eosinophilic cells. The fibroblastic component was tightly adherent to the epithelial component, and only stromal clefts were detected. Papillary mesenchymal bodies also were detected as oval aggregates of fibroblastic cells invaginating into epithelial islands to form hair papillae.
Histopathologically, the 2 most important differential diagnoses of trichoepithelioma include BCC and basaloid follicular hamartoma. In differentiating BCC from trichoepithelioma, the presence of dense fibroblastic stroma and papillary mesenchymal bodies characterize trichoepithelioma, while a fibromucinous stroma with mucinous retraction artifacts and clefting between the basaloid islands and the stroma characterize BCC (Figure 1).10 Immunohistochemical studies using antibodies against Bcl-2, CD34, CD10, androgen receptor, Ki-67, cytokeratin 19, and PHLDA1 (pleckstrin homologylike domain family A member 1) have reportedly been utilized to differentiate trichoepithelioma from BCC.11,12 Basaloid follicular hamartoma is characterized by thin anastomosing strands and branching cords of undifferentiated basaloid cells that replace or associate hair follicles in a latticelike pattern (Figure 2). The strands usually are vertically oriented perpendicular to the epidermis. Peripheral palisading is possible, and the basaloid strands are surrounded with cellular connective tissue stroma.13 Tumor islands in eccrine poroma show broad connections with the epidermis and are composed of poroid cells that show evident ductal differentiation with eosinophilic cuticles (Figure 3).14 Spiradenoma is characterized by capsulated deep-seated tumorous nodules not connected with the epidermis and composed of light and dark cells with ductal differentiation and vascular stroma (Figure 4). Scattered lymphocytes within the tumor lobules and in the stroma also are seen. Eosinophilic hyaline globules rarely can be present.15
Many pathologists consider trichoepithelioma as the superficial variant of trichoblastoma. According to the recent World Health Organization classification of benign tumors with follicular differentiation, trichoepithelioma is considered synonymous with trichoblastoma.16
Trichoepitheliomas are benign tumors, and therapy is mainly directed at removal for cosmetic purposes. Several methods of removal are available including electrocautery, laser therapy, and surgery. Awareness of the possible dermatomal distribution of hair follicle and other adnexal-related conditions is important, and such lesions should be thought of in the differential diagnosis of unilateral and/or dermatomal lesions.
The Diagnosis: Unilateral Dermatomal Trichoepithelioma
Adnexal lesions presenting with a linear and/or dermatomal pattern rarely have been reported. Bolognia et al1 performed a comprehensive review of Blaschko lines and skin conditions that follow such a pattern. The authors found that adnexal-related lesions included linear nevus comedonicus, linear basal cell nevus with comedones (linear basaloid follicular hamartoma), unilateral nevoid basal cell carcinoma (BCC), linear trichoepithelioma, linear trichodiscoma, linear hamartoma of the follicular infundibulum, nevus sebaceous, syringocystadenoma papilliferum, porokeratotic eccrine ostial and dermal duct nevus, linear eccrine poroma, linear spiradenoma, linear syringoma, and linear eccrine syringofibroadenoma.1
Trichoepithelioma is a hair follicle-related neoplastic lesion presenting most commonly as the autosomal-dominant multiple familial type with lesions mainly centered on the face. Initial genetic studies associated the disease with loss of heterozygosity in the 9p21 region and further studies identified mutations in the CYLD (cylindromatosis [turban tumor syndrome]) gene on chromosome 16q12-q13.2,3 Unilateral, linear, and dermatomal forms of trichoepithelioma rarely are reported. In 1986, Geffner et al4 reported a case of linear and dermatomal trichoepithelioma in a 10-year-old girl. In addition to discrete solitary lesions affecting the face, she developed lesions on the left shoulder, left side of the trunk, and left lower leg following dermatomal distribution. In 2006, 2 cases of dermatomal trichoepitheliomas affecting the face in children, as in our case, were reported.5,6 Another case involving the neck was reported in 2016.7 Although classic multiple familial trichoepithelioma can be part of conditions such as Brooke-Spiegler8 and Rombo syndromes,9 no syndromal association has been reported thus far with the unilateral, linear, or dermatomal variants.
Our case showed typical histopathologic features of trichoepithelioma, including discrete islands of basaloid cells in the dermis set in a conspicuous fibroblastic stroma. Focal connection with the epidermis was present. Most of the islands showed peripheral palisading and horn cysts lined by eosinophilic cells. The fibroblastic component was tightly adherent to the epithelial component, and only stromal clefts were detected. Papillary mesenchymal bodies also were detected as oval aggregates of fibroblastic cells invaginating into epithelial islands to form hair papillae.
Histopathologically, the 2 most important differential diagnoses of trichoepithelioma include BCC and basaloid follicular hamartoma. In differentiating BCC from trichoepithelioma, the presence of dense fibroblastic stroma and papillary mesenchymal bodies characterize trichoepithelioma, while a fibromucinous stroma with mucinous retraction artifacts and clefting between the basaloid islands and the stroma characterize BCC (Figure 1).10 Immunohistochemical studies using antibodies against Bcl-2, CD34, CD10, androgen receptor, Ki-67, cytokeratin 19, and PHLDA1 (pleckstrin homologylike domain family A member 1) have reportedly been utilized to differentiate trichoepithelioma from BCC.11,12 Basaloid follicular hamartoma is characterized by thin anastomosing strands and branching cords of undifferentiated basaloid cells that replace or associate hair follicles in a latticelike pattern (Figure 2). The strands usually are vertically oriented perpendicular to the epidermis. Peripheral palisading is possible, and the basaloid strands are surrounded with cellular connective tissue stroma.13 Tumor islands in eccrine poroma show broad connections with the epidermis and are composed of poroid cells that show evident ductal differentiation with eosinophilic cuticles (Figure 3).14 Spiradenoma is characterized by capsulated deep-seated tumorous nodules not connected with the epidermis and composed of light and dark cells with ductal differentiation and vascular stroma (Figure 4). Scattered lymphocytes within the tumor lobules and in the stroma also are seen. Eosinophilic hyaline globules rarely can be present.15
Many pathologists consider trichoepithelioma as the superficial variant of trichoblastoma. According to the recent World Health Organization classification of benign tumors with follicular differentiation, trichoepithelioma is considered synonymous with trichoblastoma.16
Trichoepitheliomas are benign tumors, and therapy is mainly directed at removal for cosmetic purposes. Several methods of removal are available including electrocautery, laser therapy, and surgery. Awareness of the possible dermatomal distribution of hair follicle and other adnexal-related conditions is important, and such lesions should be thought of in the differential diagnosis of unilateral and/or dermatomal lesions.
- Bolognia JL, Orlow SJ, Glick SA. Lines of Blaschko. J Am Acad Dermatol. 1994;31(2, pt 1):157-190.
- Harada H, Hashimoto K, Ko MS. The gene for multiple familial trichoepithelioma maps to chromosome 9p21. J Invest Dermatol. 1996;107:41-43.
- Zheng G, Hu L, Huang W, et al. CYLD mutation causes multiple familial trichoepithelioma in three Chinese families. Hum Mutat. 2004;23:400.
- Geffner RE, Goslen JB, Santa Cruz DJ. Linear and dermatomal trichoepitheliomas. J Am Acad Dermatol. 1986;14(5, pt 2):927-930.
- Chang YC, Colome-Grimmer M, Kelly E. Multiple trichoepitheliomas in the lines of Blaschko. Pediatr Dermatol. 2006;23:149-151.
- Strauss RM, Merchant WJ, Stainforth JM, et al. Unilateral naevoid trichoepitheliomas on the face of a child. Clin Exp Dermatol. 2006;6:778-780.
- Laska AJ, Belli RA, Kobayashi TT. Linear trichoepithelioma on the neck of a 15-year-old girl. Dermatol Online J. 2016;22. pii:13030/qt87b6h4q8.
- Rasmussen JE. A syndrome of trichoepitheliomas, milia and cylindroma. Arch Dermatol. 1975;111:610-614.
- Michaelson G, Olsson E, Westermark P. The Rombo syndrome. Acta Derm Venereol. 1981;61:497-503.
- Brooke JD, Fitzpatrick JE, Golitz LE. Papillary mesenchymal bodies: a histologic finding useful in differentiating trichoepitheliomas from basal cell carcinomas. J Am Acad Dermatol. 1989;21(3, pt 1):523-528.
- Mostafa NA, Assaf M, Elhakim S, et al. Diagnostic accuracy of immunohistochemical markers in differentiation between basal cell carcinoma and trichoepithelioma in small biopsy specimens. J Cutan Pathol. 2018;45:807-816.
- Poniecka AW, Alexis JB. An immunohistochemical study of basal cell carcinoma and trichoepithelioma. Am J Dermatopathol. 1999;21:332-336.
- Abdel-Halim MRE, Fawzy M, Saleh M, et al. Linear unilateral basal cell nevus with comedones (linear nevoid basaloid follicular hamartoma): a case report. J Egypt Womens Dermatol Soc. 2016;13:46-48.
- Hyman AB, Brownstein MH. Eccrine poroma: analysis of 45 new cases. Dermatologica. 1969;138:28-38.
- Mambo NC. Eccrine spiradenoma: clinical and pathologic study of 49 tumors. J Cutan Pathol. 1983;10:312-320.
- Kutzner H, Kaddu S, Kanitakis J, et al. Trichoblastoma. In: Elder D, Massi D, Scolyer RA, et al, eds. WHO Classification of Skin Tumours. 4th ed. Lyon, France: IARC; 2018.
- Bolognia JL, Orlow SJ, Glick SA. Lines of Blaschko. J Am Acad Dermatol. 1994;31(2, pt 1):157-190.
- Harada H, Hashimoto K, Ko MS. The gene for multiple familial trichoepithelioma maps to chromosome 9p21. J Invest Dermatol. 1996;107:41-43.
- Zheng G, Hu L, Huang W, et al. CYLD mutation causes multiple familial trichoepithelioma in three Chinese families. Hum Mutat. 2004;23:400.
- Geffner RE, Goslen JB, Santa Cruz DJ. Linear and dermatomal trichoepitheliomas. J Am Acad Dermatol. 1986;14(5, pt 2):927-930.
- Chang YC, Colome-Grimmer M, Kelly E. Multiple trichoepitheliomas in the lines of Blaschko. Pediatr Dermatol. 2006;23:149-151.
- Strauss RM, Merchant WJ, Stainforth JM, et al. Unilateral naevoid trichoepitheliomas on the face of a child. Clin Exp Dermatol. 2006;6:778-780.
- Laska AJ, Belli RA, Kobayashi TT. Linear trichoepithelioma on the neck of a 15-year-old girl. Dermatol Online J. 2016;22. pii:13030/qt87b6h4q8.
- Rasmussen JE. A syndrome of trichoepitheliomas, milia and cylindroma. Arch Dermatol. 1975;111:610-614.
- Michaelson G, Olsson E, Westermark P. The Rombo syndrome. Acta Derm Venereol. 1981;61:497-503.
- Brooke JD, Fitzpatrick JE, Golitz LE. Papillary mesenchymal bodies: a histologic finding useful in differentiating trichoepitheliomas from basal cell carcinomas. J Am Acad Dermatol. 1989;21(3, pt 1):523-528.
- Mostafa NA, Assaf M, Elhakim S, et al. Diagnostic accuracy of immunohistochemical markers in differentiation between basal cell carcinoma and trichoepithelioma in small biopsy specimens. J Cutan Pathol. 2018;45:807-816.
- Poniecka AW, Alexis JB. An immunohistochemical study of basal cell carcinoma and trichoepithelioma. Am J Dermatopathol. 1999;21:332-336.
- Abdel-Halim MRE, Fawzy M, Saleh M, et al. Linear unilateral basal cell nevus with comedones (linear nevoid basaloid follicular hamartoma): a case report. J Egypt Womens Dermatol Soc. 2016;13:46-48.
- Hyman AB, Brownstein MH. Eccrine poroma: analysis of 45 new cases. Dermatologica. 1969;138:28-38.
- Mambo NC. Eccrine spiradenoma: clinical and pathologic study of 49 tumors. J Cutan Pathol. 1983;10:312-320.
- Kutzner H, Kaddu S, Kanitakis J, et al. Trichoblastoma. In: Elder D, Massi D, Scolyer RA, et al, eds. WHO Classification of Skin Tumours. 4th ed. Lyon, France: IARC; 2018.

A 9-year-old boy presented with a slowly progressive lesion of 5 years’ duration affecting only the left side of the face in a dermatomal pattern. The patient denied any symptoms and had no other anomalies or family history of similar lesions. On physical examination the lesion was found to span a 12×7-cm area of the lateral half of the left cheek and was composed of multiple variable-sized, pinkish to flesh-colored papules that coalesced in some areas to form small plaques. Few milialike cysts were present. One papule was biopsied.
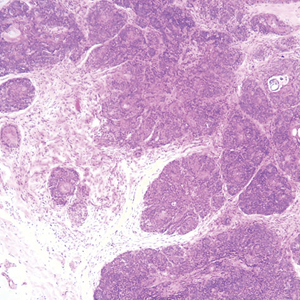
Bothersome Blisters: Localized Epidermolysis Bullosa Simplex
To the Editor:
Epidermolysis bullosa (EB) was first described in 1886, with the first classification scheme proposed in 1962 utilizing transmission electron microscopy (TEM) findings to delineate categories: epidermolytic (EB simplex [EBS]), lucidolytic (junctional EB), and dermolytic (dystrophic EB).1 Localized EBS (EBS-loc) is an autosomal-dominant disorder caused by negative mutations in keratin-5 and keratin-14, proteins expressed in the intermediate filaments of basal keratinocytes, which result in fragility of the skin in response to minor trauma.2 The incidence of EBS-loc is approximately 10 to 30 cases per million live births, with the age of presentation typically between the first and third decades of life.3,4 Because EBS-loc is the most common and often mildest form of EB, not all patients present for medical evaluation and true prevalence may be underestimated.4 We report a case of EBS-loc.
A 26-year-old woman with no notable medical history presented to the dermatology clinic for evaluation of skin blisters that had been intermittently present since infancy. The blisters primarily occurred on the feet, but she did occasionally develop blisters on the hands, knees, and elbows and at sites of friction or trauma (eg, bra line, medial thighs) following exercise. The blisters were worsened by heat and tight-fitting shoes. Because of the painful nature of the blisters, she would lance them with a needle. On the medial thighs, she utilized nonstick and gauze bandage roll dressings to minimize friction. A review of systems was positive for hyperhidrosis. Her family history revealed multiple family members with blisters involving the feet and areas of friction or trauma for 4 generations with no known diagnosis.
Physical examination revealed multiple tense bullae and calluses scattered over the bilateral plantar and distal dorsal feet with a few healing, superficially eroded, erythematous papules and plaques on the bilateral medial thighs (Figure 1). A biopsy from an induced blister on the right dorsal second toe was performed and sent in glutaraldehyde to the Epidermolysis Bullosa Clinic at Stanford University (Redwood City, California) for electron microscopy, which revealed lysis within the basal keratinocytes through the tonofilaments with continuous and intact lamina densa and lamina lucida (Figure 2). In this clinical context with the relevant family history, the findings were consistent with the diagnosis of EBS-loc (formerly Weber-Cockayne syndrome).2

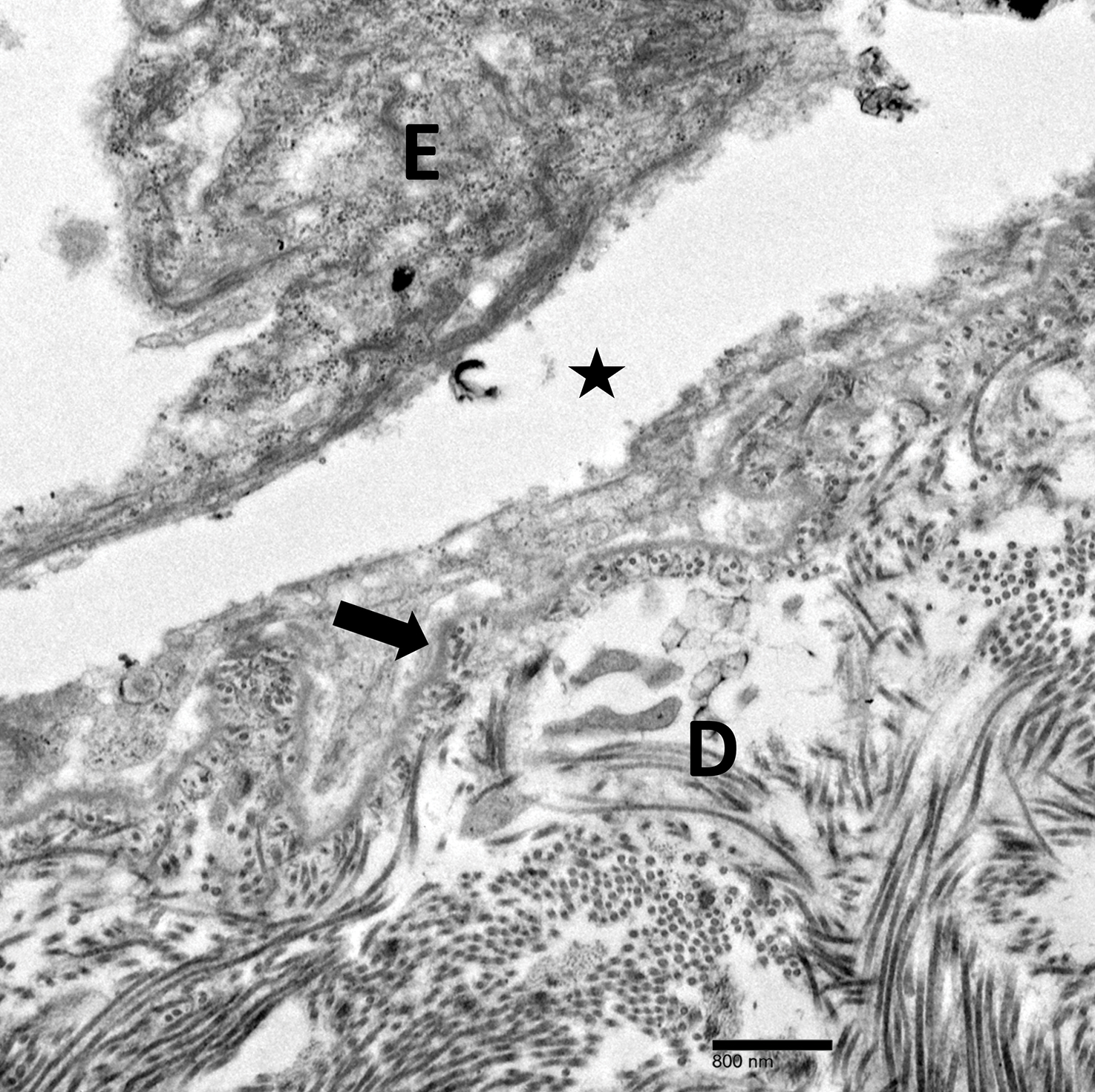
Skin manifestations of EBS-loc typically consist of friction-induced blisters, erosions, and calluses primarily on the palms and soles, often associated with hyperhidrosis and worsening of symptoms in summer months and hot temperatures.3 Milia, atrophic scarring, and dystrophic nails are uncommon.1 Extracutaneous involvement is rare with the exception of oral cavity erosions, which typically are asymptomatic and usually are only seen during infancy.1
Light microscopy does not have a notable role in diagnosis of classic forms of inherited EB unless another autoimmune blistering disorder is suspected.2,5 Both TEM and immunofluorescence mapping are used to diagnose EB.1 DNA mutational analysis is not considered a first-line diagnostic test for EB given it is a costly labor-intensive technique with limited access at present, but it may be considered in settings of prenatal diagnosis or in vitro fertilization.1 Biopsy of a freshly induced blister should be performed, as early reepithelialization of an existing blister makes it difficult to establish the level of cleavage.5 Applying firm pressure using a pencil eraser and rotating it on intact skin induces a subclinical blister. Two punch biopsies (4 mm) at the edge of the blister with one-third lesional and two-thirds perilesional skin should be obtained, with one biopsy sent for immunofluorescence mapping in Michel fixative and the other for TEM in glutaraldehyde.3,5 Transmission electron microscopy of an induced blister in EBS-loc shows cleavage within the most inferior portion of the basilar keratinocyte.2 Immunofluorescence mapping with anti–epidermal basement membrane monoclonal antibodies can distinguish between EB subtypes and assess expression of specific skin-associated proteins on both a qualitative or semiquantitative basis, providing insight on which structural protein is mutated.1,5
No specific treatments are available for EBS-loc. Mainstays of treatment include prevention of mechanical trauma and secondary infection. Hyperhidrosis of thepalms and soles may be treated with topical aluminum chloride hexahydrate or injections of botulinum toxin type A.2,6 Patients have normal life expectancy, though some cases may have complications with substantial morbidity.1 Awareness of this disease, its clinical course, and therapeutic options will allow physicians to more appropriately counsel patients on the disease process.
Localized EBS may be more common than previously thought, as not all patients seek medical care. Given its impact on patient quality of life, it is important for clinicians to recognize EBS-loc. Although no specific treatments are available, wound care counseling and explanation of the genetics of the disease should be provided to patients.
- Fine JD, Eady RA, Bauer EA, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58:931-950.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Limited; 2012.
- Eichenfield LF, Frieden IJ, Mathes EF, et al, eds. Neonatal and Infant Dermatology. 3rd ed. New York, NY: Elsevier Health Sciences; 2015.
- Spitz JL. Genodermatoses: A Clinical Guide to Genetic Skin Disorders. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005.
- Epidermolysis bullosa. Stanford Medicine website. http://med.stanford.edu/dermatopathology/dermpath-services/epiderm.html. Accessed April 3, 2019.
- Abitbol RJ, Zhou LH. Treatment of epidermolysis bullosa simplex, Weber-Cockayne type, with botulinum toxin type A. Arch Dermatol. 2009;145:13-15.
To the Editor:
Epidermolysis bullosa (EB) was first described in 1886, with the first classification scheme proposed in 1962 utilizing transmission electron microscopy (TEM) findings to delineate categories: epidermolytic (EB simplex [EBS]), lucidolytic (junctional EB), and dermolytic (dystrophic EB).1 Localized EBS (EBS-loc) is an autosomal-dominant disorder caused by negative mutations in keratin-5 and keratin-14, proteins expressed in the intermediate filaments of basal keratinocytes, which result in fragility of the skin in response to minor trauma.2 The incidence of EBS-loc is approximately 10 to 30 cases per million live births, with the age of presentation typically between the first and third decades of life.3,4 Because EBS-loc is the most common and often mildest form of EB, not all patients present for medical evaluation and true prevalence may be underestimated.4 We report a case of EBS-loc.
A 26-year-old woman with no notable medical history presented to the dermatology clinic for evaluation of skin blisters that had been intermittently present since infancy. The blisters primarily occurred on the feet, but she did occasionally develop blisters on the hands, knees, and elbows and at sites of friction or trauma (eg, bra line, medial thighs) following exercise. The blisters were worsened by heat and tight-fitting shoes. Because of the painful nature of the blisters, she would lance them with a needle. On the medial thighs, she utilized nonstick and gauze bandage roll dressings to minimize friction. A review of systems was positive for hyperhidrosis. Her family history revealed multiple family members with blisters involving the feet and areas of friction or trauma for 4 generations with no known diagnosis.
Physical examination revealed multiple tense bullae and calluses scattered over the bilateral plantar and distal dorsal feet with a few healing, superficially eroded, erythematous papules and plaques on the bilateral medial thighs (Figure 1). A biopsy from an induced blister on the right dorsal second toe was performed and sent in glutaraldehyde to the Epidermolysis Bullosa Clinic at Stanford University (Redwood City, California) for electron microscopy, which revealed lysis within the basal keratinocytes through the tonofilaments with continuous and intact lamina densa and lamina lucida (Figure 2). In this clinical context with the relevant family history, the findings were consistent with the diagnosis of EBS-loc (formerly Weber-Cockayne syndrome).2
Skin manifestations of EBS-loc typically consist of friction-induced blisters, erosions, and calluses primarily on the palms and soles, often associated with hyperhidrosis and worsening of symptoms in summer months and hot temperatures.3 Milia, atrophic scarring, and dystrophic nails are uncommon.1 Extracutaneous involvement is rare with the exception of oral cavity erosions, which typically are asymptomatic and usually are only seen during infancy.1
Light microscopy does not have a notable role in diagnosis of classic forms of inherited EB unless another autoimmune blistering disorder is suspected.2,5 Both TEM and immunofluorescence mapping are used to diagnose EB.1 DNA mutational analysis is not considered a first-line diagnostic test for EB given it is a costly labor-intensive technique with limited access at present, but it may be considered in settings of prenatal diagnosis or in vitro fertilization.1 Biopsy of a freshly induced blister should be performed, as early reepithelialization of an existing blister makes it difficult to establish the level of cleavage.5 Applying firm pressure using a pencil eraser and rotating it on intact skin induces a subclinical blister. Two punch biopsies (4 mm) at the edge of the blister with one-third lesional and two-thirds perilesional skin should be obtained, with one biopsy sent for immunofluorescence mapping in Michel fixative and the other for TEM in glutaraldehyde.3,5 Transmission electron microscopy of an induced blister in EBS-loc shows cleavage within the most inferior portion of the basilar keratinocyte.2 Immunofluorescence mapping with anti–epidermal basement membrane monoclonal antibodies can distinguish between EB subtypes and assess expression of specific skin-associated proteins on both a qualitative or semiquantitative basis, providing insight on which structural protein is mutated.1,5
No specific treatments are available for EBS-loc. Mainstays of treatment include prevention of mechanical trauma and secondary infection. Hyperhidrosis of thepalms and soles may be treated with topical aluminum chloride hexahydrate or injections of botulinum toxin type A.2,6 Patients have normal life expectancy, though some cases may have complications with substantial morbidity.1 Awareness of this disease, its clinical course, and therapeutic options will allow physicians to more appropriately counsel patients on the disease process.
Localized EBS may be more common than previously thought, as not all patients seek medical care. Given its impact on patient quality of life, it is important for clinicians to recognize EBS-loc. Although no specific treatments are available, wound care counseling and explanation of the genetics of the disease should be provided to patients.
To the Editor:
Epidermolysis bullosa (EB) was first described in 1886, with the first classification scheme proposed in 1962 utilizing transmission electron microscopy (TEM) findings to delineate categories: epidermolytic (EB simplex [EBS]), lucidolytic (junctional EB), and dermolytic (dystrophic EB).1 Localized EBS (EBS-loc) is an autosomal-dominant disorder caused by negative mutations in keratin-5 and keratin-14, proteins expressed in the intermediate filaments of basal keratinocytes, which result in fragility of the skin in response to minor trauma.2 The incidence of EBS-loc is approximately 10 to 30 cases per million live births, with the age of presentation typically between the first and third decades of life.3,4 Because EBS-loc is the most common and often mildest form of EB, not all patients present for medical evaluation and true prevalence may be underestimated.4 We report a case of EBS-loc.
A 26-year-old woman with no notable medical history presented to the dermatology clinic for evaluation of skin blisters that had been intermittently present since infancy. The blisters primarily occurred on the feet, but she did occasionally develop blisters on the hands, knees, and elbows and at sites of friction or trauma (eg, bra line, medial thighs) following exercise. The blisters were worsened by heat and tight-fitting shoes. Because of the painful nature of the blisters, she would lance them with a needle. On the medial thighs, she utilized nonstick and gauze bandage roll dressings to minimize friction. A review of systems was positive for hyperhidrosis. Her family history revealed multiple family members with blisters involving the feet and areas of friction or trauma for 4 generations with no known diagnosis.
Physical examination revealed multiple tense bullae and calluses scattered over the bilateral plantar and distal dorsal feet with a few healing, superficially eroded, erythematous papules and plaques on the bilateral medial thighs (Figure 1). A biopsy from an induced blister on the right dorsal second toe was performed and sent in glutaraldehyde to the Epidermolysis Bullosa Clinic at Stanford University (Redwood City, California) for electron microscopy, which revealed lysis within the basal keratinocytes through the tonofilaments with continuous and intact lamina densa and lamina lucida (Figure 2). In this clinical context with the relevant family history, the findings were consistent with the diagnosis of EBS-loc (formerly Weber-Cockayne syndrome).2
Skin manifestations of EBS-loc typically consist of friction-induced blisters, erosions, and calluses primarily on the palms and soles, often associated with hyperhidrosis and worsening of symptoms in summer months and hot temperatures.3 Milia, atrophic scarring, and dystrophic nails are uncommon.1 Extracutaneous involvement is rare with the exception of oral cavity erosions, which typically are asymptomatic and usually are only seen during infancy.1
Light microscopy does not have a notable role in diagnosis of classic forms of inherited EB unless another autoimmune blistering disorder is suspected.2,5 Both TEM and immunofluorescence mapping are used to diagnose EB.1 DNA mutational analysis is not considered a first-line diagnostic test for EB given it is a costly labor-intensive technique with limited access at present, but it may be considered in settings of prenatal diagnosis or in vitro fertilization.1 Biopsy of a freshly induced blister should be performed, as early reepithelialization of an existing blister makes it difficult to establish the level of cleavage.5 Applying firm pressure using a pencil eraser and rotating it on intact skin induces a subclinical blister. Two punch biopsies (4 mm) at the edge of the blister with one-third lesional and two-thirds perilesional skin should be obtained, with one biopsy sent for immunofluorescence mapping in Michel fixative and the other for TEM in glutaraldehyde.3,5 Transmission electron microscopy of an induced blister in EBS-loc shows cleavage within the most inferior portion of the basilar keratinocyte.2 Immunofluorescence mapping with anti–epidermal basement membrane monoclonal antibodies can distinguish between EB subtypes and assess expression of specific skin-associated proteins on both a qualitative or semiquantitative basis, providing insight on which structural protein is mutated.1,5
No specific treatments are available for EBS-loc. Mainstays of treatment include prevention of mechanical trauma and secondary infection. Hyperhidrosis of thepalms and soles may be treated with topical aluminum chloride hexahydrate or injections of botulinum toxin type A.2,6 Patients have normal life expectancy, though some cases may have complications with substantial morbidity.1 Awareness of this disease, its clinical course, and therapeutic options will allow physicians to more appropriately counsel patients on the disease process.
Localized EBS may be more common than previously thought, as not all patients seek medical care. Given its impact on patient quality of life, it is important for clinicians to recognize EBS-loc. Although no specific treatments are available, wound care counseling and explanation of the genetics of the disease should be provided to patients.
- Fine JD, Eady RA, Bauer EA, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58:931-950.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Limited; 2012.
- Eichenfield LF, Frieden IJ, Mathes EF, et al, eds. Neonatal and Infant Dermatology. 3rd ed. New York, NY: Elsevier Health Sciences; 2015.
- Spitz JL. Genodermatoses: A Clinical Guide to Genetic Skin Disorders. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005.
- Epidermolysis bullosa. Stanford Medicine website. http://med.stanford.edu/dermatopathology/dermpath-services/epiderm.html. Accessed April 3, 2019.
- Abitbol RJ, Zhou LH. Treatment of epidermolysis bullosa simplex, Weber-Cockayne type, with botulinum toxin type A. Arch Dermatol. 2009;145:13-15.
- Fine JD, Eady RA, Bauer EA, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58:931-950.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Limited; 2012.
- Eichenfield LF, Frieden IJ, Mathes EF, et al, eds. Neonatal and Infant Dermatology. 3rd ed. New York, NY: Elsevier Health Sciences; 2015.
- Spitz JL. Genodermatoses: A Clinical Guide to Genetic Skin Disorders. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005.
- Epidermolysis bullosa. Stanford Medicine website. http://med.stanford.edu/dermatopathology/dermpath-services/epiderm.html. Accessed April 3, 2019.
- Abitbol RJ, Zhou LH. Treatment of epidermolysis bullosa simplex, Weber-Cockayne type, with botulinum toxin type A. Arch Dermatol. 2009;145:13-15.
Practice Points
- Localized epidermolysis bullosa simplex (formerly Weber-Cockayne syndrome) presents with flaccid bullae and erosions predominantly on the hands and feet, most commonly related to mechanical friction and heat.
- It is inherited in an autosomal-dominant fashion with defects in keratin-5 and keratin-14.
- Biopsy of a freshly induced blister should be examined by transmission electron microscopy or immunofluorescence mapping.
- Treatment is focused on wound management and infection control of the blisters.
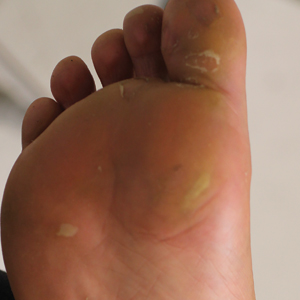
Relapsing Polychondritis in Human Immunodeficiency Virus
Relapsing polychondritis (RP) is a recurrent inflammatory condition involving primarily cartilaginous structures. The disease, first described as a clinical entity in 1960 by Pearson et al,1 is rare with an estimated incidence of 3.5 cases per 1 million individuals.2 The pathogenesis of RP is widely accepted as being autoimmune in nature, largely due to the identification of circulating autoantibodies seen in the sera of patients with similar clinical pictures.3
Although in most patients the primary process involves inflammation of cartilage, a subset of patients experience involvement of noncartilaginous sites.4 The degree of systemic involvement varies from none to notable, affecting the cardiovascular and respiratory systems and potentially leading to life-threatening complications, including cardiac valve compromise and airway collapse. Relapsing polychondritis is considered to be a progressive disease with the ultimate potential to be life-threatening.5
Human immunodeficiency virus (HIV) infection leads to a profound state of immune dysregulation, affecting innate, adaptive, and natural killer components of the immune system.6 There is variability in the development of autoimmune disease in HIV patients depending on the stage of infection. The frequency of rheumatologic disease in HIV patients might be as high as 60%.6 Relapsing polychondritis is rare in patients with HIV.7-9 Of 4 reported cases, 2 patients had other coexisting autoimmune disease—sarcoidosis and Behçet disease.8,9
Case Report
A 36-year-old man presented to the clinic with a concern of recurrent ear pain and swelling of approximately 2 years’ duration. Onset was sudden without inciting event. Symptoms initially involved the right ear with eventual progression to both ears. Additional symptoms included an auditory perception of underwater submersion, intermittent vertigo, and 3 episodes of throat closure sensation.
The patient’s medical history was notable for asthma; gastritis; depression; and HIV infection, which was diagnosed 4 years earlier and adequately managed with highly active antiretroviral therapy. His family history was notable for systemic lupus erythematosus in his mother, maternal aunt, and maternal cousin.
At presentation, the patient’s CD4 count was 799 cells/mm3 with an undetectable viral load. Medications included abacavir-dolutegravir-lamivudine, hydroxyzine, meclizine, mometasone, and quetiapine. Physical examination showed erythema, swelling, and tenderness of the left and right auricles with sparing of the earlobe that was more noticeable on the left ear (Figure 1). Bacterial culture from the external auditory meatus was positive for methicillin-resistant Staphylococcus aureus. Biopsy revealed chronic inflammatory perichondritis with mild to moderate fibrosis and chronic lymphocytic inflammation at the dermal cartilaginous junction (Figure 2). A direct immunofluorescent biopsy was unremarkable, but subsequent type II collagen antibodies were positive (35.5 endotoxin units/mL [reference range, <20 endotoxin units/mL]).
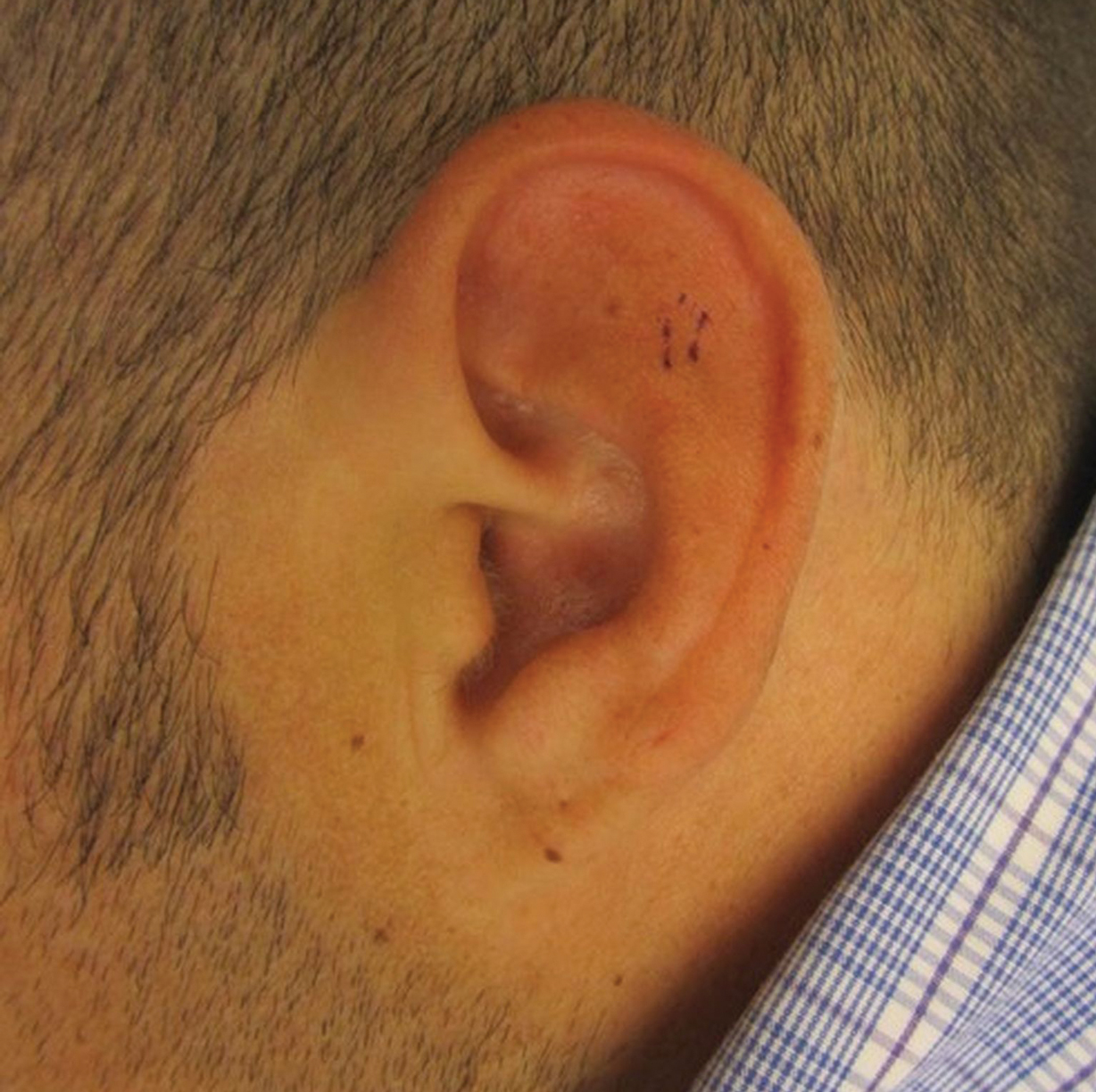
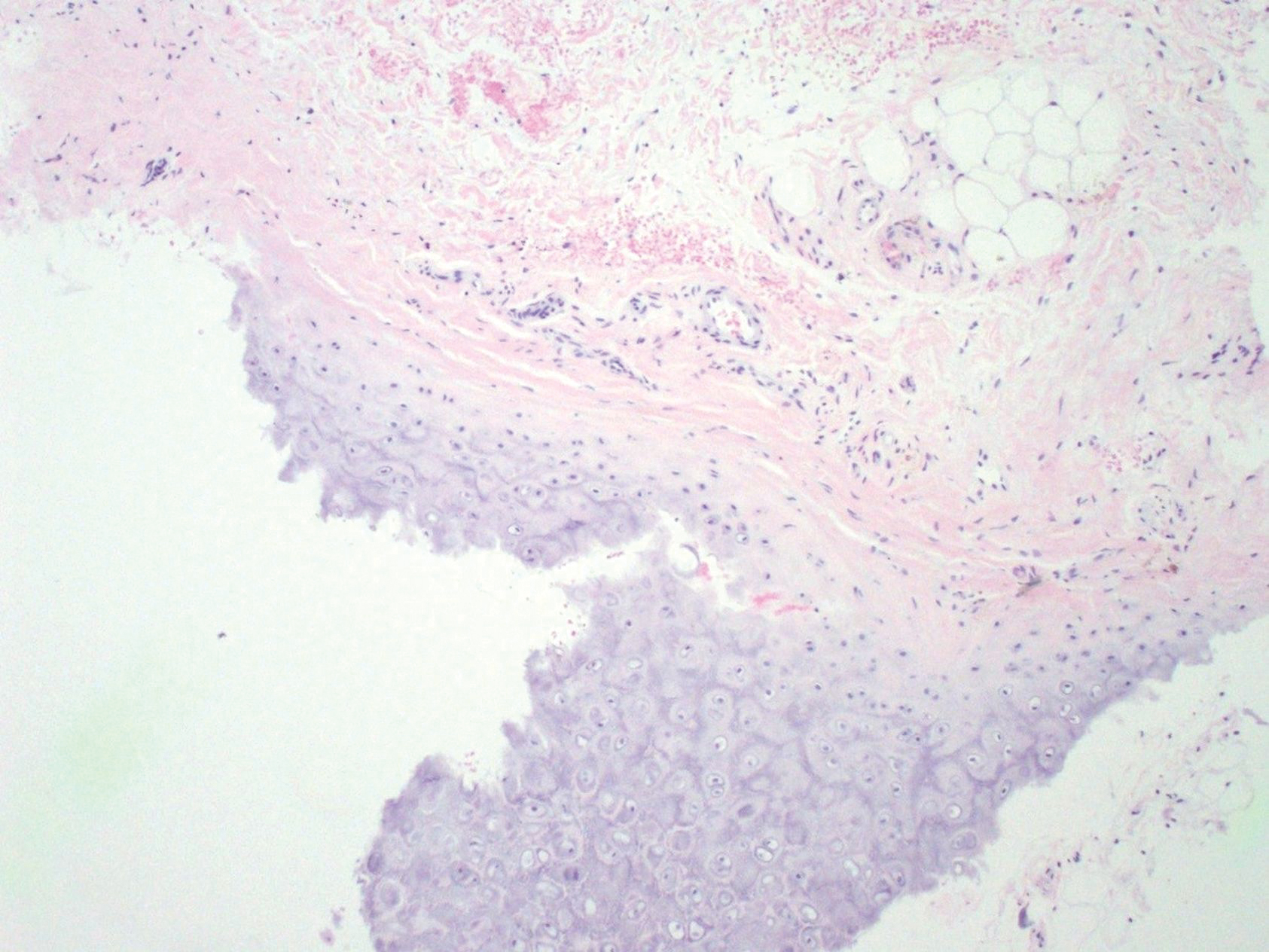
Comment
Relapsing polychondritis is an uncommon progressive disease characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures.4 The most consistent clinical features of RP are ear inflammation that involves the auricle and spares the lobe, nasal chondritis, and arthralgia.10 Laryngotracheal compromise may occur from tracheal cartilage inflammation. The involvement of these specific structures is due to commonality between their component collagens.5 Although any organ system can be affected, as many as 50% of patients have respiratory tract involvement, which may affect any portion of the respiratory tree.11 If involving the larynx, this inflammation can lead to severe edema warranting intubation. Cardiovascular involvement is present in 24% to 52% of patients,10 which most commonly manifests as valvular impairment affecting the aortic valve more frequently than the mitral valve.5
Pathogenesis
Although the etiology of RP remains undetermined, multiple hypotheses have been proposed. One is that a certain subset of patients is predisposed to autoimmunity, and a secondary triggering event in the form of infection, malignancy, or medication catalyzes development of RP. A second hypothesis is that mechanical trauma to cartilage exposes the immune system to certain antigens that would have otherwise remained hidden, prompting autosensitization.12,13
Regardless of cause, an autoimmune pathogenesis is favored based on the following observations: RP is frequently associated with other autoimmune diseases in the same patient, glucocorticosteroids and other immunosuppressive therapies are effective for treatment, and histopathologic findings include an infiltrate of CD4+ T lymphocytes with detection of immunoglobulins and plasma cells in different lesions.5 The detection of autoantibodies against collagen in the serum of patients with RP further supports an autoimmune pathogenesis.3 The earliest identified autoantibodies in patients with RP were against type II collagen. Subsequent studies have identified autoantibodies against type IV and type XI collagens as well as other cartilage-related proteins such as matrilin 114 and cartilage oligomeric matrix proteins.15 Although circulating antibodies to type II collagen are present in a variable number of patients with the disease (30%–70%), levels likely correlate with disease activity and are highest at times of acute inflammation.3 Additionally, titers of type II collagen antibodies have been shown to decrease upon institution of immunosuppressive therapy.16
Although a humoral response dominates the picture of RP, there also is an associated T cell–mediated response.13 Histopathologically, biopsy of an active lesion of auricular cartilage shows a mixed inflammatory infiltrate composed primarily of lymphocytes, with variable numbers of polymorphonuclear cells, monocytes, and plasma cells. Loss of basophilia of the cartilage matrix can be observed, thought to be the result of proteoglycan depletion.13 Later, lesions classically display apoptosis of chondrocytes, focal calcification, or fibrosis.5
Diagnosis
Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge. Many sets of diagnostic criteria have been proposed. The most referenced remains the original criteria described by McAdam et al.17 In 2012, the Relapsing Polychondritis Disease Activity Index modified criteria set forth by Michet et al18 and might serve as the standard for diagnosis going forward.19
McAdam et al17 proposed that 3 of 6 clinical features are necessary for diagnosis: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation, respiratory tract chondritis, and audiovestibular damage. Michet et al18 proposed that 1 of 2 conditions are necessary for diagnosis of RP: (1) proven inflammation in 2 of 3 of the auricular, nasal, or laryngotracheal cartilages; or (2) proven inflammation in 1 of 3 of the auricular, nasal, or laryngotracheal cartilages, plus 2 other signs, including ocular inflammation, vestibular dysfunction, seronegative inflammatory arthritis, and hearing loss.
These criteria were proposed originally in 197617 and modified in 1986.18 No further updates have been offered since then. As such, serologic findings, such as antibodies against type II collagen, are not included in the diagnostic criteria. Additionally, these antibodies are not specific for RP and can be seen in other conditions such as rheumatoid arthritis.20
More recently, imaging analysis has been employed in conjunction with clinical and serologic data to diagnose the disease and evaluate its severity. The use of imaging modalities for these purposes is most beneficial in patients with notable disease and respiratory involvement.21
Although the clinical picture is typified by the classic findings described above, the clinician must be aware of more subtle clues to diagnosis,11 which is of particular importance to the dermatologist because 35% of patients with RP alone will have skin manifestations that can precede onset of chondritis.10 Most commonly, dermatologic manifestations are nonspecific and can include nodules on the limbs, purpura, and urticarial lesions.22 Individual case reports have noted the coexistence of RP with erythema multiforme,18 erythema annulare centrifugum,23 pyoderma gangrenosum,24 and panniculitis,18 among other disorders.
Treatment
Standardized guidelines for treatment do not exist. Treatments should be chosen based on severity of disease. Mild disease, presenting with recurrent chondritis and arthritis without evidence of systemic involvement, can be treated with nonsteroidal anti-inflammatory drugs, dapsone, or colchicine. Refractory disease often requires high-dose systemic corticosteroids.5
Severe systemic involvement leads to increased mortality and warrants more aggressive treatment.22 Commonly used agents include the immunosuppressants cyclophosphamide, cyclosporine, and methotrexate. Tumor necrosis factor α inhibitors have been the most widely utilized immunomodulatory agent for treatment of RP.25,26 Abatacept and rituximab also have been used with variable efficacy in patients with severe disease. Recently, the IL-6 receptor blocker tocilizumab has been used with some success.27
Prognosis
The prognosis for patients with RP largely depends on the severity of disease and degree of internal involvement. With improved management, largely due to awareness and recognition of disease, the survival rate among RP patients has increased from 55% at 10 years to 94% at the end of 8 years.18 The main cause of death in RP patients is airway complications related to laryngotracheal involvement.10 The second most common cause of death is cardiovascular complications in which valvular disease predominates.5
Concomitant Illness
Thirty-five percent of RP patients have coexisting autoimmune disease, the most common being antineutrophil cytoplasmic antibody–associated vasculitis.5,28 Although this association with autoimmune disease is well described, reports of RP occurring in other states of immune dysfunction are sparse. One case of RP has been reported in a child with common variable immunodeficiency thought to be related to underlying abnormal immune regulation and immunodeficiency.29 Relapsing polychondritis has been described in 4 patients with HIV, 2 of whom had concomitant autoimmune disease.7-9
Human immunodeficiency virus infection is a well-established cause of immune dysregulation and has variable association with autoimmunity. This variability depends largely on the stage of infection. When divided into stages, autoimmune diseases develop predominantly in stage I, during acute infection with an intact immune system; in stage III, with immunosuppression, a low CD4 count, and development of AIDS; and in stage IV, when the immune system is restored with the institution of highly active antiretroviral therapy.6 The interplay between HIV infection and development of autoimmune disease is complex, and pathogenesis remains speculative.
Conclusion
Our patient represents a case of RP in an HIV-positive patient. Additionally, our patient had no other identifiable autoimmune conditions but did have a strong family history of them. It is important for providers to be aware of the potential for development of RP as well as other autoimmune disease in the setting of HIV infection. The implications of a missed diagnosis could be dire because the disease course of RP is progressive and has the potential to decrease survival.
- Pearson CM, Kline HM, Newcomer VD. Relapsing polychondritis. N Engl J Med. 1960;263:51-58.
- Kent PD, Michet CJ Jr, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol. 2004;16:56-61.
- Ebringer R, Rook G, Swana GT, et al. Autoantibodies to cartilage and type II collagen in relapsing polychondritis and other rheumatic diseases. Ann Rheum Dis. 1981;40:473-479.
- Sharma A, Law AD, Bambery P, et al. Relapsing polychondritis: clinical presentations, disease activity and outcomes. Orphanet J Rare Dis. 2014;9:198.
- Vitale A, Sota J, Rigante D, et al. Relapsing polychondritis: an update on pathogenesis, clinical features, diagnostic tools, and therapeutic perspectives. Curr Rheumatol Rep. 2016;18:3.
- Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329-337.
- Dolev JC, Maurer TA, Reddy SG, et al. Relapsing polychondritis in HIV-infected patients: a report of two cases. J Am Acad Dermatol. 2004;51:1023-1025.
- Zandman-Goddard G, Peeva E, Barland P. Combined autoimmune disease in a patient with AIDS. Clin Rheumatol. 2002;21:70-72.
- Belzunegui J, Cancio J, Pego JM, et al. Relapsing polychondritis and Behc¸et’s syndrome in a patient with HIV infection. Ann Rheum Dis. 1995;54:780.
- Sharma A, Gnanapandithan K, Sharma K, et al. Relapsing polychondritis: a review. Clin Rheumatol. 2013;32:1575-1583.
- Cantarini L, Vitale A, Brizi MG, et al. Diagnosis and classification of relapsing polychondritis. J Autoimmun. 2014;48-49:53-59.
- Cañas CA, Bonilla Abadía F. Local cartilage trauma as a pathogenic factor in autoimmunity (one hypothesis based on patients with relapsing polychondritis triggered by cartilage trauma). Autoimmune Dis. 2012;2012:453698.
- Ouchi N, Uzuki M, Kamataki A, et al. Cartilage destruction is partly induced by the internal proteolytic enzymes and apoptotic phenomenon of chondrocytes in relapsing polychondritis. J Rheumatol. 2011;38:730-737.
- Buckner JH, Wu JJ, Reife RA, et al. Autoreactivity against matrilin-1 in a patient with relapsing polychondritis. Arthritis Rheum. 2000;43:939-943.
- Kempta Lekpa F, Piette JC, Bastuji-Garin S, et al. Serum cartilage oligomeric matrix protein (COMP) is a marker of disease activity in relapsing polychondritis. Clin Exp Rheumatol. 2010;28:553-555.
- Foidart JM, Abe S, Martin GR, et al. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med. 1978;299:1203-1207.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet CJ, McKenna CH, Luthra HS, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Arnaud L, Devilliers H, Peng SL, et al. The Relapsing Polychondritis Disease Activity Index: development of a disease activity score for relapsing polychondritis. Autoimmun Rev. 2012;12:204-209.
- Brand DD, Kang AH, Rosloniec EF. Immunopathogenesis of collagen arthritis. Springer Semin Immunopathol. 2003;25:3-18.
- Thaiss WM, Nikolaou K, Spengler W, et al. Imaging diagnosis in relapsing polychondritis and correlation with clinical and serological data. Skeletal Radiol. 2015;5:339-346.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Francès C, el Rassi R, Laporte JL, et al. Dermatologic manifestations of relapsing polychondritis. A study of 200 cases at a single center. Medicine (Baltimore). 2001;80:173-179.
- Chopra R, Chaudhary N, Kay J. Relapsing polychondritis. Rheum Dis Clin North Am. 2013;39:263-276.
- Moulis G, Sailler L, Pugnet G, et al. Biologics in relapsing polychondritis: a case series. Clin Exp Rheumatol. 2013;31:937-939.
- Henes CJ, Xenitidis T, Horger M. Tocilizumab for refractory relapsing polychondritis—long-term response monitoring by magnetic resonance imaging. Joint Bone Spine. 2016;83:365-366.
- Weinberger A, Myers AR. Relapsing polychondritis associated with cutaneous vasculitis. Arch Dermatol. 1979;115:980-981.
- Karaca NE, Aksu G, Yildiz B, et al. Relapsing polychondritis in a child with common variable immunodeficiency. Int J Dermatol. 2009;48:525-528.
Relapsing polychondritis (RP) is a recurrent inflammatory condition involving primarily cartilaginous structures. The disease, first described as a clinical entity in 1960 by Pearson et al,1 is rare with an estimated incidence of 3.5 cases per 1 million individuals.2 The pathogenesis of RP is widely accepted as being autoimmune in nature, largely due to the identification of circulating autoantibodies seen in the sera of patients with similar clinical pictures.3
Although in most patients the primary process involves inflammation of cartilage, a subset of patients experience involvement of noncartilaginous sites.4 The degree of systemic involvement varies from none to notable, affecting the cardiovascular and respiratory systems and potentially leading to life-threatening complications, including cardiac valve compromise and airway collapse. Relapsing polychondritis is considered to be a progressive disease with the ultimate potential to be life-threatening.5
Human immunodeficiency virus (HIV) infection leads to a profound state of immune dysregulation, affecting innate, adaptive, and natural killer components of the immune system.6 There is variability in the development of autoimmune disease in HIV patients depending on the stage of infection. The frequency of rheumatologic disease in HIV patients might be as high as 60%.6 Relapsing polychondritis is rare in patients with HIV.7-9 Of 4 reported cases, 2 patients had other coexisting autoimmune disease—sarcoidosis and Behçet disease.8,9
Case Report
A 36-year-old man presented to the clinic with a concern of recurrent ear pain and swelling of approximately 2 years’ duration. Onset was sudden without inciting event. Symptoms initially involved the right ear with eventual progression to both ears. Additional symptoms included an auditory perception of underwater submersion, intermittent vertigo, and 3 episodes of throat closure sensation.
The patient’s medical history was notable for asthma; gastritis; depression; and HIV infection, which was diagnosed 4 years earlier and adequately managed with highly active antiretroviral therapy. His family history was notable for systemic lupus erythematosus in his mother, maternal aunt, and maternal cousin.
At presentation, the patient’s CD4 count was 799 cells/mm3 with an undetectable viral load. Medications included abacavir-dolutegravir-lamivudine, hydroxyzine, meclizine, mometasone, and quetiapine. Physical examination showed erythema, swelling, and tenderness of the left and right auricles with sparing of the earlobe that was more noticeable on the left ear (Figure 1). Bacterial culture from the external auditory meatus was positive for methicillin-resistant Staphylococcus aureus. Biopsy revealed chronic inflammatory perichondritis with mild to moderate fibrosis and chronic lymphocytic inflammation at the dermal cartilaginous junction (Figure 2). A direct immunofluorescent biopsy was unremarkable, but subsequent type II collagen antibodies were positive (35.5 endotoxin units/mL [reference range, <20 endotoxin units/mL]).
Comment
Relapsing polychondritis is an uncommon progressive disease characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures.4 The most consistent clinical features of RP are ear inflammation that involves the auricle and spares the lobe, nasal chondritis, and arthralgia.10 Laryngotracheal compromise may occur from tracheal cartilage inflammation. The involvement of these specific structures is due to commonality between their component collagens.5 Although any organ system can be affected, as many as 50% of patients have respiratory tract involvement, which may affect any portion of the respiratory tree.11 If involving the larynx, this inflammation can lead to severe edema warranting intubation. Cardiovascular involvement is present in 24% to 52% of patients,10 which most commonly manifests as valvular impairment affecting the aortic valve more frequently than the mitral valve.5
Pathogenesis
Although the etiology of RP remains undetermined, multiple hypotheses have been proposed. One is that a certain subset of patients is predisposed to autoimmunity, and a secondary triggering event in the form of infection, malignancy, or medication catalyzes development of RP. A second hypothesis is that mechanical trauma to cartilage exposes the immune system to certain antigens that would have otherwise remained hidden, prompting autosensitization.12,13
Regardless of cause, an autoimmune pathogenesis is favored based on the following observations: RP is frequently associated with other autoimmune diseases in the same patient, glucocorticosteroids and other immunosuppressive therapies are effective for treatment, and histopathologic findings include an infiltrate of CD4+ T lymphocytes with detection of immunoglobulins and plasma cells in different lesions.5 The detection of autoantibodies against collagen in the serum of patients with RP further supports an autoimmune pathogenesis.3 The earliest identified autoantibodies in patients with RP were against type II collagen. Subsequent studies have identified autoantibodies against type IV and type XI collagens as well as other cartilage-related proteins such as matrilin 114 and cartilage oligomeric matrix proteins.15 Although circulating antibodies to type II collagen are present in a variable number of patients with the disease (30%–70%), levels likely correlate with disease activity and are highest at times of acute inflammation.3 Additionally, titers of type II collagen antibodies have been shown to decrease upon institution of immunosuppressive therapy.16
Although a humoral response dominates the picture of RP, there also is an associated T cell–mediated response.13 Histopathologically, biopsy of an active lesion of auricular cartilage shows a mixed inflammatory infiltrate composed primarily of lymphocytes, with variable numbers of polymorphonuclear cells, monocytes, and plasma cells. Loss of basophilia of the cartilage matrix can be observed, thought to be the result of proteoglycan depletion.13 Later, lesions classically display apoptosis of chondrocytes, focal calcification, or fibrosis.5
Diagnosis
Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge. Many sets of diagnostic criteria have been proposed. The most referenced remains the original criteria described by McAdam et al.17 In 2012, the Relapsing Polychondritis Disease Activity Index modified criteria set forth by Michet et al18 and might serve as the standard for diagnosis going forward.19
McAdam et al17 proposed that 3 of 6 clinical features are necessary for diagnosis: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation, respiratory tract chondritis, and audiovestibular damage. Michet et al18 proposed that 1 of 2 conditions are necessary for diagnosis of RP: (1) proven inflammation in 2 of 3 of the auricular, nasal, or laryngotracheal cartilages; or (2) proven inflammation in 1 of 3 of the auricular, nasal, or laryngotracheal cartilages, plus 2 other signs, including ocular inflammation, vestibular dysfunction, seronegative inflammatory arthritis, and hearing loss.
These criteria were proposed originally in 197617 and modified in 1986.18 No further updates have been offered since then. As such, serologic findings, such as antibodies against type II collagen, are not included in the diagnostic criteria. Additionally, these antibodies are not specific for RP and can be seen in other conditions such as rheumatoid arthritis.20
More recently, imaging analysis has been employed in conjunction with clinical and serologic data to diagnose the disease and evaluate its severity. The use of imaging modalities for these purposes is most beneficial in patients with notable disease and respiratory involvement.21
Although the clinical picture is typified by the classic findings described above, the clinician must be aware of more subtle clues to diagnosis,11 which is of particular importance to the dermatologist because 35% of patients with RP alone will have skin manifestations that can precede onset of chondritis.10 Most commonly, dermatologic manifestations are nonspecific and can include nodules on the limbs, purpura, and urticarial lesions.22 Individual case reports have noted the coexistence of RP with erythema multiforme,18 erythema annulare centrifugum,23 pyoderma gangrenosum,24 and panniculitis,18 among other disorders.
Treatment
Standardized guidelines for treatment do not exist. Treatments should be chosen based on severity of disease. Mild disease, presenting with recurrent chondritis and arthritis without evidence of systemic involvement, can be treated with nonsteroidal anti-inflammatory drugs, dapsone, or colchicine. Refractory disease often requires high-dose systemic corticosteroids.5
Severe systemic involvement leads to increased mortality and warrants more aggressive treatment.22 Commonly used agents include the immunosuppressants cyclophosphamide, cyclosporine, and methotrexate. Tumor necrosis factor α inhibitors have been the most widely utilized immunomodulatory agent for treatment of RP.25,26 Abatacept and rituximab also have been used with variable efficacy in patients with severe disease. Recently, the IL-6 receptor blocker tocilizumab has been used with some success.27
Prognosis
The prognosis for patients with RP largely depends on the severity of disease and degree of internal involvement. With improved management, largely due to awareness and recognition of disease, the survival rate among RP patients has increased from 55% at 10 years to 94% at the end of 8 years.18 The main cause of death in RP patients is airway complications related to laryngotracheal involvement.10 The second most common cause of death is cardiovascular complications in which valvular disease predominates.5
Concomitant Illness
Thirty-five percent of RP patients have coexisting autoimmune disease, the most common being antineutrophil cytoplasmic antibody–associated vasculitis.5,28 Although this association with autoimmune disease is well described, reports of RP occurring in other states of immune dysfunction are sparse. One case of RP has been reported in a child with common variable immunodeficiency thought to be related to underlying abnormal immune regulation and immunodeficiency.29 Relapsing polychondritis has been described in 4 patients with HIV, 2 of whom had concomitant autoimmune disease.7-9
Human immunodeficiency virus infection is a well-established cause of immune dysregulation and has variable association with autoimmunity. This variability depends largely on the stage of infection. When divided into stages, autoimmune diseases develop predominantly in stage I, during acute infection with an intact immune system; in stage III, with immunosuppression, a low CD4 count, and development of AIDS; and in stage IV, when the immune system is restored with the institution of highly active antiretroviral therapy.6 The interplay between HIV infection and development of autoimmune disease is complex, and pathogenesis remains speculative.
Conclusion
Our patient represents a case of RP in an HIV-positive patient. Additionally, our patient had no other identifiable autoimmune conditions but did have a strong family history of them. It is important for providers to be aware of the potential for development of RP as well as other autoimmune disease in the setting of HIV infection. The implications of a missed diagnosis could be dire because the disease course of RP is progressive and has the potential to decrease survival.
Relapsing polychondritis (RP) is a recurrent inflammatory condition involving primarily cartilaginous structures. The disease, first described as a clinical entity in 1960 by Pearson et al,1 is rare with an estimated incidence of 3.5 cases per 1 million individuals.2 The pathogenesis of RP is widely accepted as being autoimmune in nature, largely due to the identification of circulating autoantibodies seen in the sera of patients with similar clinical pictures.3
Although in most patients the primary process involves inflammation of cartilage, a subset of patients experience involvement of noncartilaginous sites.4 The degree of systemic involvement varies from none to notable, affecting the cardiovascular and respiratory systems and potentially leading to life-threatening complications, including cardiac valve compromise and airway collapse. Relapsing polychondritis is considered to be a progressive disease with the ultimate potential to be life-threatening.5
Human immunodeficiency virus (HIV) infection leads to a profound state of immune dysregulation, affecting innate, adaptive, and natural killer components of the immune system.6 There is variability in the development of autoimmune disease in HIV patients depending on the stage of infection. The frequency of rheumatologic disease in HIV patients might be as high as 60%.6 Relapsing polychondritis is rare in patients with HIV.7-9 Of 4 reported cases, 2 patients had other coexisting autoimmune disease—sarcoidosis and Behçet disease.8,9
Case Report
A 36-year-old man presented to the clinic with a concern of recurrent ear pain and swelling of approximately 2 years’ duration. Onset was sudden without inciting event. Symptoms initially involved the right ear with eventual progression to both ears. Additional symptoms included an auditory perception of underwater submersion, intermittent vertigo, and 3 episodes of throat closure sensation.
The patient’s medical history was notable for asthma; gastritis; depression; and HIV infection, which was diagnosed 4 years earlier and adequately managed with highly active antiretroviral therapy. His family history was notable for systemic lupus erythematosus in his mother, maternal aunt, and maternal cousin.
At presentation, the patient’s CD4 count was 799 cells/mm3 with an undetectable viral load. Medications included abacavir-dolutegravir-lamivudine, hydroxyzine, meclizine, mometasone, and quetiapine. Physical examination showed erythema, swelling, and tenderness of the left and right auricles with sparing of the earlobe that was more noticeable on the left ear (Figure 1). Bacterial culture from the external auditory meatus was positive for methicillin-resistant Staphylococcus aureus. Biopsy revealed chronic inflammatory perichondritis with mild to moderate fibrosis and chronic lymphocytic inflammation at the dermal cartilaginous junction (Figure 2). A direct immunofluorescent biopsy was unremarkable, but subsequent type II collagen antibodies were positive (35.5 endotoxin units/mL [reference range, <20 endotoxin units/mL]).
Comment
Relapsing polychondritis is an uncommon progressive disease characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures.4 The most consistent clinical features of RP are ear inflammation that involves the auricle and spares the lobe, nasal chondritis, and arthralgia.10 Laryngotracheal compromise may occur from tracheal cartilage inflammation. The involvement of these specific structures is due to commonality between their component collagens.5 Although any organ system can be affected, as many as 50% of patients have respiratory tract involvement, which may affect any portion of the respiratory tree.11 If involving the larynx, this inflammation can lead to severe edema warranting intubation. Cardiovascular involvement is present in 24% to 52% of patients,10 which most commonly manifests as valvular impairment affecting the aortic valve more frequently than the mitral valve.5
Pathogenesis
Although the etiology of RP remains undetermined, multiple hypotheses have been proposed. One is that a certain subset of patients is predisposed to autoimmunity, and a secondary triggering event in the form of infection, malignancy, or medication catalyzes development of RP. A second hypothesis is that mechanical trauma to cartilage exposes the immune system to certain antigens that would have otherwise remained hidden, prompting autosensitization.12,13
Regardless of cause, an autoimmune pathogenesis is favored based on the following observations: RP is frequently associated with other autoimmune diseases in the same patient, glucocorticosteroids and other immunosuppressive therapies are effective for treatment, and histopathologic findings include an infiltrate of CD4+ T lymphocytes with detection of immunoglobulins and plasma cells in different lesions.5 The detection of autoantibodies against collagen in the serum of patients with RP further supports an autoimmune pathogenesis.3 The earliest identified autoantibodies in patients with RP were against type II collagen. Subsequent studies have identified autoantibodies against type IV and type XI collagens as well as other cartilage-related proteins such as matrilin 114 and cartilage oligomeric matrix proteins.15 Although circulating antibodies to type II collagen are present in a variable number of patients with the disease (30%–70%), levels likely correlate with disease activity and are highest at times of acute inflammation.3 Additionally, titers of type II collagen antibodies have been shown to decrease upon institution of immunosuppressive therapy.16
Although a humoral response dominates the picture of RP, there also is an associated T cell–mediated response.13 Histopathologically, biopsy of an active lesion of auricular cartilage shows a mixed inflammatory infiltrate composed primarily of lymphocytes, with variable numbers of polymorphonuclear cells, monocytes, and plasma cells. Loss of basophilia of the cartilage matrix can be observed, thought to be the result of proteoglycan depletion.13 Later, lesions classically display apoptosis of chondrocytes, focal calcification, or fibrosis.5
Diagnosis
Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge. Many sets of diagnostic criteria have been proposed. The most referenced remains the original criteria described by McAdam et al.17 In 2012, the Relapsing Polychondritis Disease Activity Index modified criteria set forth by Michet et al18 and might serve as the standard for diagnosis going forward.19
McAdam et al17 proposed that 3 of 6 clinical features are necessary for diagnosis: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation, respiratory tract chondritis, and audiovestibular damage. Michet et al18 proposed that 1 of 2 conditions are necessary for diagnosis of RP: (1) proven inflammation in 2 of 3 of the auricular, nasal, or laryngotracheal cartilages; or (2) proven inflammation in 1 of 3 of the auricular, nasal, or laryngotracheal cartilages, plus 2 other signs, including ocular inflammation, vestibular dysfunction, seronegative inflammatory arthritis, and hearing loss.
These criteria were proposed originally in 197617 and modified in 1986.18 No further updates have been offered since then. As such, serologic findings, such as antibodies against type II collagen, are not included in the diagnostic criteria. Additionally, these antibodies are not specific for RP and can be seen in other conditions such as rheumatoid arthritis.20
More recently, imaging analysis has been employed in conjunction with clinical and serologic data to diagnose the disease and evaluate its severity. The use of imaging modalities for these purposes is most beneficial in patients with notable disease and respiratory involvement.21
Although the clinical picture is typified by the classic findings described above, the clinician must be aware of more subtle clues to diagnosis,11 which is of particular importance to the dermatologist because 35% of patients with RP alone will have skin manifestations that can precede onset of chondritis.10 Most commonly, dermatologic manifestations are nonspecific and can include nodules on the limbs, purpura, and urticarial lesions.22 Individual case reports have noted the coexistence of RP with erythema multiforme,18 erythema annulare centrifugum,23 pyoderma gangrenosum,24 and panniculitis,18 among other disorders.
Treatment
Standardized guidelines for treatment do not exist. Treatments should be chosen based on severity of disease. Mild disease, presenting with recurrent chondritis and arthritis without evidence of systemic involvement, can be treated with nonsteroidal anti-inflammatory drugs, dapsone, or colchicine. Refractory disease often requires high-dose systemic corticosteroids.5
Severe systemic involvement leads to increased mortality and warrants more aggressive treatment.22 Commonly used agents include the immunosuppressants cyclophosphamide, cyclosporine, and methotrexate. Tumor necrosis factor α inhibitors have been the most widely utilized immunomodulatory agent for treatment of RP.25,26 Abatacept and rituximab also have been used with variable efficacy in patients with severe disease. Recently, the IL-6 receptor blocker tocilizumab has been used with some success.27
Prognosis
The prognosis for patients with RP largely depends on the severity of disease and degree of internal involvement. With improved management, largely due to awareness and recognition of disease, the survival rate among RP patients has increased from 55% at 10 years to 94% at the end of 8 years.18 The main cause of death in RP patients is airway complications related to laryngotracheal involvement.10 The second most common cause of death is cardiovascular complications in which valvular disease predominates.5
Concomitant Illness
Thirty-five percent of RP patients have coexisting autoimmune disease, the most common being antineutrophil cytoplasmic antibody–associated vasculitis.5,28 Although this association with autoimmune disease is well described, reports of RP occurring in other states of immune dysfunction are sparse. One case of RP has been reported in a child with common variable immunodeficiency thought to be related to underlying abnormal immune regulation and immunodeficiency.29 Relapsing polychondritis has been described in 4 patients with HIV, 2 of whom had concomitant autoimmune disease.7-9
Human immunodeficiency virus infection is a well-established cause of immune dysregulation and has variable association with autoimmunity. This variability depends largely on the stage of infection. When divided into stages, autoimmune diseases develop predominantly in stage I, during acute infection with an intact immune system; in stage III, with immunosuppression, a low CD4 count, and development of AIDS; and in stage IV, when the immune system is restored with the institution of highly active antiretroviral therapy.6 The interplay between HIV infection and development of autoimmune disease is complex, and pathogenesis remains speculative.
Conclusion
Our patient represents a case of RP in an HIV-positive patient. Additionally, our patient had no other identifiable autoimmune conditions but did have a strong family history of them. It is important for providers to be aware of the potential for development of RP as well as other autoimmune disease in the setting of HIV infection. The implications of a missed diagnosis could be dire because the disease course of RP is progressive and has the potential to decrease survival.
- Pearson CM, Kline HM, Newcomer VD. Relapsing polychondritis. N Engl J Med. 1960;263:51-58.
- Kent PD, Michet CJ Jr, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol. 2004;16:56-61.
- Ebringer R, Rook G, Swana GT, et al. Autoantibodies to cartilage and type II collagen in relapsing polychondritis and other rheumatic diseases. Ann Rheum Dis. 1981;40:473-479.
- Sharma A, Law AD, Bambery P, et al. Relapsing polychondritis: clinical presentations, disease activity and outcomes. Orphanet J Rare Dis. 2014;9:198.
- Vitale A, Sota J, Rigante D, et al. Relapsing polychondritis: an update on pathogenesis, clinical features, diagnostic tools, and therapeutic perspectives. Curr Rheumatol Rep. 2016;18:3.
- Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329-337.
- Dolev JC, Maurer TA, Reddy SG, et al. Relapsing polychondritis in HIV-infected patients: a report of two cases. J Am Acad Dermatol. 2004;51:1023-1025.
- Zandman-Goddard G, Peeva E, Barland P. Combined autoimmune disease in a patient with AIDS. Clin Rheumatol. 2002;21:70-72.
- Belzunegui J, Cancio J, Pego JM, et al. Relapsing polychondritis and Behc¸et’s syndrome in a patient with HIV infection. Ann Rheum Dis. 1995;54:780.
- Sharma A, Gnanapandithan K, Sharma K, et al. Relapsing polychondritis: a review. Clin Rheumatol. 2013;32:1575-1583.
- Cantarini L, Vitale A, Brizi MG, et al. Diagnosis and classification of relapsing polychondritis. J Autoimmun. 2014;48-49:53-59.
- Cañas CA, Bonilla Abadía F. Local cartilage trauma as a pathogenic factor in autoimmunity (one hypothesis based on patients with relapsing polychondritis triggered by cartilage trauma). Autoimmune Dis. 2012;2012:453698.
- Ouchi N, Uzuki M, Kamataki A, et al. Cartilage destruction is partly induced by the internal proteolytic enzymes and apoptotic phenomenon of chondrocytes in relapsing polychondritis. J Rheumatol. 2011;38:730-737.
- Buckner JH, Wu JJ, Reife RA, et al. Autoreactivity against matrilin-1 in a patient with relapsing polychondritis. Arthritis Rheum. 2000;43:939-943.
- Kempta Lekpa F, Piette JC, Bastuji-Garin S, et al. Serum cartilage oligomeric matrix protein (COMP) is a marker of disease activity in relapsing polychondritis. Clin Exp Rheumatol. 2010;28:553-555.
- Foidart JM, Abe S, Martin GR, et al. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med. 1978;299:1203-1207.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet CJ, McKenna CH, Luthra HS, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Arnaud L, Devilliers H, Peng SL, et al. The Relapsing Polychondritis Disease Activity Index: development of a disease activity score for relapsing polychondritis. Autoimmun Rev. 2012;12:204-209.
- Brand DD, Kang AH, Rosloniec EF. Immunopathogenesis of collagen arthritis. Springer Semin Immunopathol. 2003;25:3-18.
- Thaiss WM, Nikolaou K, Spengler W, et al. Imaging diagnosis in relapsing polychondritis and correlation with clinical and serological data. Skeletal Radiol. 2015;5:339-346.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Francès C, el Rassi R, Laporte JL, et al. Dermatologic manifestations of relapsing polychondritis. A study of 200 cases at a single center. Medicine (Baltimore). 2001;80:173-179.
- Chopra R, Chaudhary N, Kay J. Relapsing polychondritis. Rheum Dis Clin North Am. 2013;39:263-276.
- Moulis G, Sailler L, Pugnet G, et al. Biologics in relapsing polychondritis: a case series. Clin Exp Rheumatol. 2013;31:937-939.
- Henes CJ, Xenitidis T, Horger M. Tocilizumab for refractory relapsing polychondritis—long-term response monitoring by magnetic resonance imaging. Joint Bone Spine. 2016;83:365-366.
- Weinberger A, Myers AR. Relapsing polychondritis associated with cutaneous vasculitis. Arch Dermatol. 1979;115:980-981.
- Karaca NE, Aksu G, Yildiz B, et al. Relapsing polychondritis in a child with common variable immunodeficiency. Int J Dermatol. 2009;48:525-528.
- Pearson CM, Kline HM, Newcomer VD. Relapsing polychondritis. N Engl J Med. 1960;263:51-58.
- Kent PD, Michet CJ Jr, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol. 2004;16:56-61.
- Ebringer R, Rook G, Swana GT, et al. Autoantibodies to cartilage and type II collagen in relapsing polychondritis and other rheumatic diseases. Ann Rheum Dis. 1981;40:473-479.
- Sharma A, Law AD, Bambery P, et al. Relapsing polychondritis: clinical presentations, disease activity and outcomes. Orphanet J Rare Dis. 2014;9:198.
- Vitale A, Sota J, Rigante D, et al. Relapsing polychondritis: an update on pathogenesis, clinical features, diagnostic tools, and therapeutic perspectives. Curr Rheumatol Rep. 2016;18:3.
- Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329-337.
- Dolev JC, Maurer TA, Reddy SG, et al. Relapsing polychondritis in HIV-infected patients: a report of two cases. J Am Acad Dermatol. 2004;51:1023-1025.
- Zandman-Goddard G, Peeva E, Barland P. Combined autoimmune disease in a patient with AIDS. Clin Rheumatol. 2002;21:70-72.
- Belzunegui J, Cancio J, Pego JM, et al. Relapsing polychondritis and Behc¸et’s syndrome in a patient with HIV infection. Ann Rheum Dis. 1995;54:780.
- Sharma A, Gnanapandithan K, Sharma K, et al. Relapsing polychondritis: a review. Clin Rheumatol. 2013;32:1575-1583.
- Cantarini L, Vitale A, Brizi MG, et al. Diagnosis and classification of relapsing polychondritis. J Autoimmun. 2014;48-49:53-59.
- Cañas CA, Bonilla Abadía F. Local cartilage trauma as a pathogenic factor in autoimmunity (one hypothesis based on patients with relapsing polychondritis triggered by cartilage trauma). Autoimmune Dis. 2012;2012:453698.
- Ouchi N, Uzuki M, Kamataki A, et al. Cartilage destruction is partly induced by the internal proteolytic enzymes and apoptotic phenomenon of chondrocytes in relapsing polychondritis. J Rheumatol. 2011;38:730-737.
- Buckner JH, Wu JJ, Reife RA, et al. Autoreactivity against matrilin-1 in a patient with relapsing polychondritis. Arthritis Rheum. 2000;43:939-943.
- Kempta Lekpa F, Piette JC, Bastuji-Garin S, et al. Serum cartilage oligomeric matrix protein (COMP) is a marker of disease activity in relapsing polychondritis. Clin Exp Rheumatol. 2010;28:553-555.
- Foidart JM, Abe S, Martin GR, et al. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med. 1978;299:1203-1207.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet CJ, McKenna CH, Luthra HS, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Arnaud L, Devilliers H, Peng SL, et al. The Relapsing Polychondritis Disease Activity Index: development of a disease activity score for relapsing polychondritis. Autoimmun Rev. 2012;12:204-209.
- Brand DD, Kang AH, Rosloniec EF. Immunopathogenesis of collagen arthritis. Springer Semin Immunopathol. 2003;25:3-18.
- Thaiss WM, Nikolaou K, Spengler W, et al. Imaging diagnosis in relapsing polychondritis and correlation with clinical and serological data. Skeletal Radiol. 2015;5:339-346.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Francès C, el Rassi R, Laporte JL, et al. Dermatologic manifestations of relapsing polychondritis. A study of 200 cases at a single center. Medicine (Baltimore). 2001;80:173-179.
- Chopra R, Chaudhary N, Kay J. Relapsing polychondritis. Rheum Dis Clin North Am. 2013;39:263-276.
- Moulis G, Sailler L, Pugnet G, et al. Biologics in relapsing polychondritis: a case series. Clin Exp Rheumatol. 2013;31:937-939.
- Henes CJ, Xenitidis T, Horger M. Tocilizumab for refractory relapsing polychondritis—long-term response monitoring by magnetic resonance imaging. Joint Bone Spine. 2016;83:365-366.
- Weinberger A, Myers AR. Relapsing polychondritis associated with cutaneous vasculitis. Arch Dermatol. 1979;115:980-981.
- Karaca NE, Aksu G, Yildiz B, et al. Relapsing polychondritis in a child with common variable immunodeficiency. Int J Dermatol. 2009;48:525-528.
Practice Points
- Relapsing polychondritis (RP) is characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures, most often manifesting as ear inflammation that involves the auricle but spares the lobe, nasal chondritis, and arthralgia.
- Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge.
- One-third of RP patients have coexisting autoimmune disease.
- Treatment of RP depends on severity of disease.
- Dermatologists must be aware of the potential for development of RP in the setting of human immunodeficiency virus infection; a missed diagnosis of this progressive disease has the potential to be life-threatening.
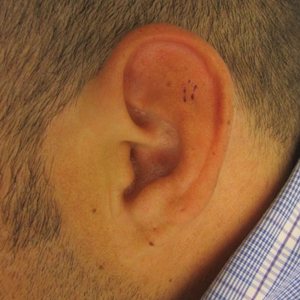
Indurated Plaque on the Shoulder
Herpes zoster (HZ) is a painful skin condition caused by reactivation of latent varicella-zoster virus (VZV) in dorsal root ganglion cells.1 Upon reactivation, VZV replicates in the dorsal root ganglion, which ultimately results in inflammation and necrosis of the neuron and intense neuralgia. Reactivation of latent VZV may occur spontaneously or may be induced by various factors including immunosuppression, stress, illness, and trauma. Prior to the development of skin lesions, many patients experience a prodrome of tingling, pain, or pruritus. Herpes zoster classically presents with grouped vesicles on an erythematous base in a unilateral dermatomal distribution; however, more than one adjacent dermatome may be involved, and the lesions can cross the midline. Furthermore, the development of vesicles may be preceded by the development of edematous papules or plaques.1
On histology, VZV closely resembles herpes simplex virus type 1 and herpes simplex virus type 2 infections.2 Classic histologic findings include ballooning degeneration of keratinocytes, acantholysis, nuclear molding, ground-glass nuclear inclusions, marginated chromatin, and multinucleated keratinocytes, as well as necrosis of follicles and sebaceous glands.2 Varicella-zoster virus polymerase chain reaction or immunostaining can be used to confirm the diagnosis.2
Classic mycosis fungoides (MF) presents with well-circumscribed erythematous patches in non–sun-exposed areas and eventually may progress to plaques and tumors.3 Patients with cutaneous T-cell lymphomas, such as MF, are at a higher risk for skin infections including HZ4,5; however, immunocompromised patients, such as those with cutaneous lymphomas, can have atypical clinical presentations of HZ that may be concerning for cutaneous lymphoma.6 Furthermore, cutaneous malignancies can occur in dermatomal distributions that may mimic HZ.7 Therefore, the threshold for biopsy should be lowered in those patients with dermatomal lesions and history concerning for possible malignancy.
Classically, histologic examination of MF demonstrates an infiltrate of haloed cells at the dermoepidermal junction, which are atypical T cells with hyperchromatic cerebriform nuclei that are larger, darker, and more angulated than the benign recruited lymphocytes in the perivascular infiltrate seen in VZV infection (Figure 1).3 Papillary dermal fibrosis typically is present, and the perivascular infiltrate is denser above the postcapillary venule rather than being symmetrical around the vessel (bare underbelly sign). Clusters of these cells may form within the epidermis, which are called Pautrier microabscesses.3 Mycosis fungoides also can exhibit large cell transformation in which small lymphocytes transform into larger cells, thereby associated with a poorer prognosis.8
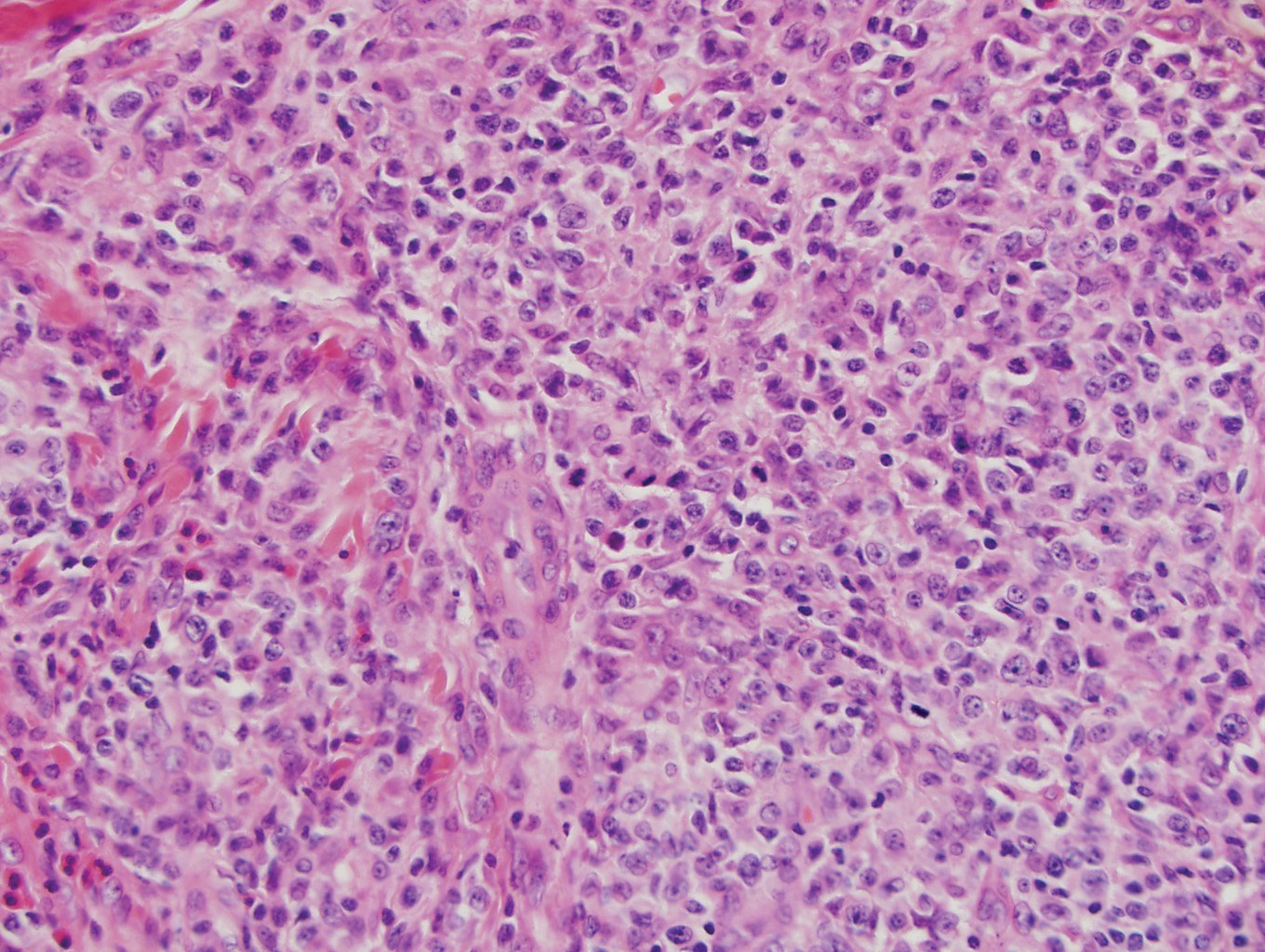
Lymphomatoid papulosis is a CD30+-predominant form of cutaneous T-cell lymphoma characterized by papules and nodules that spontaneously involute.9 This condition is most commonly associated with MF but can be associated with other lymphomas. This condition may be mistaken for HZ clinically, but histology classically demonstrates large atypical lymphocytes resembling Reed-Sternberg cells in small clusters rather than follicular necrosis (Figure 2).9
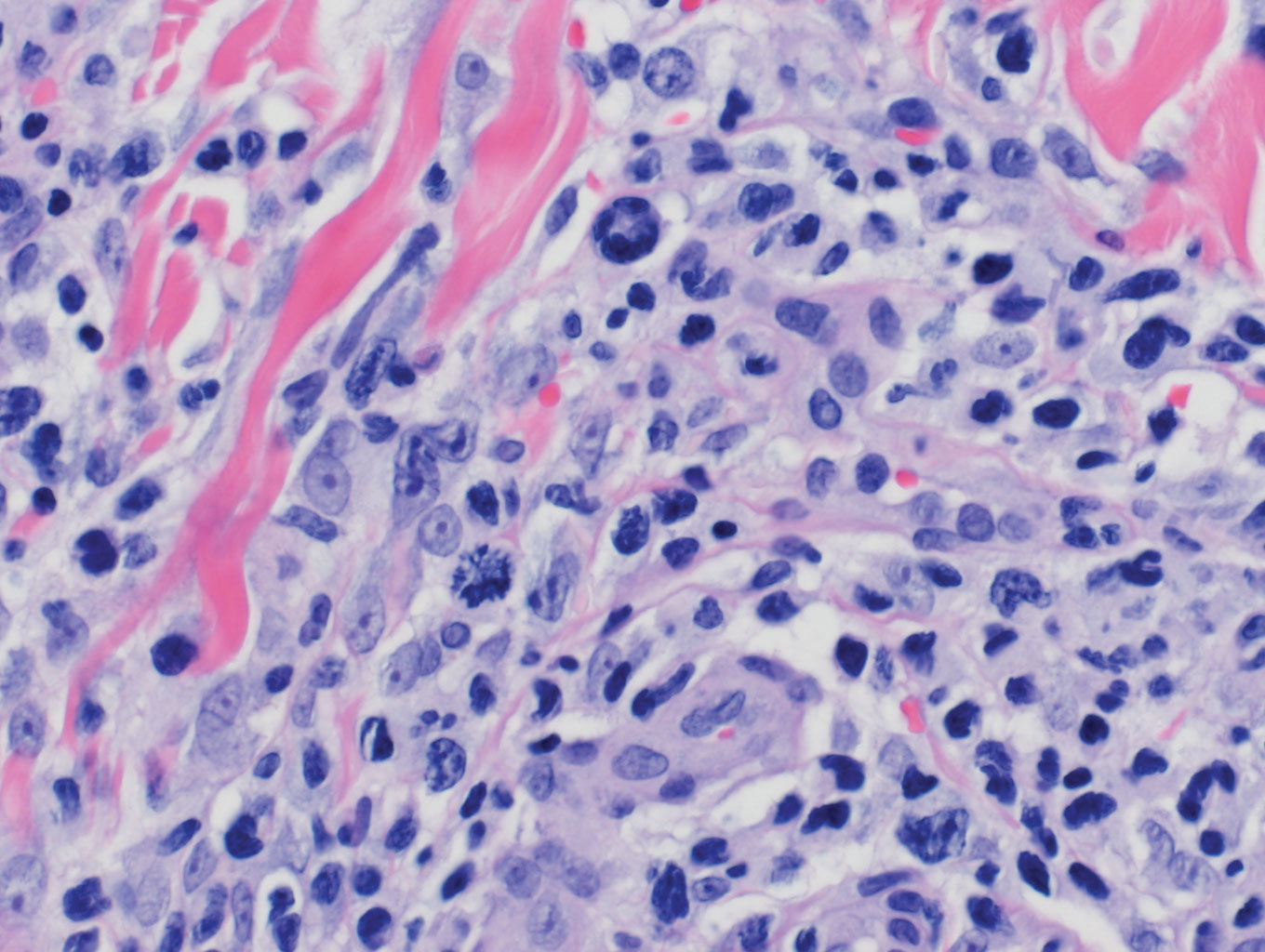
Patients with lymphoma may sequentially develop a secondary lymphoma. There have been reports of secondary B-cell lymphomas associated with MF, but this phenomenon is rare.10 The histology depends on the type of B-cell lymphoma present, but follicular necrosis would not be expected (Figure 3).
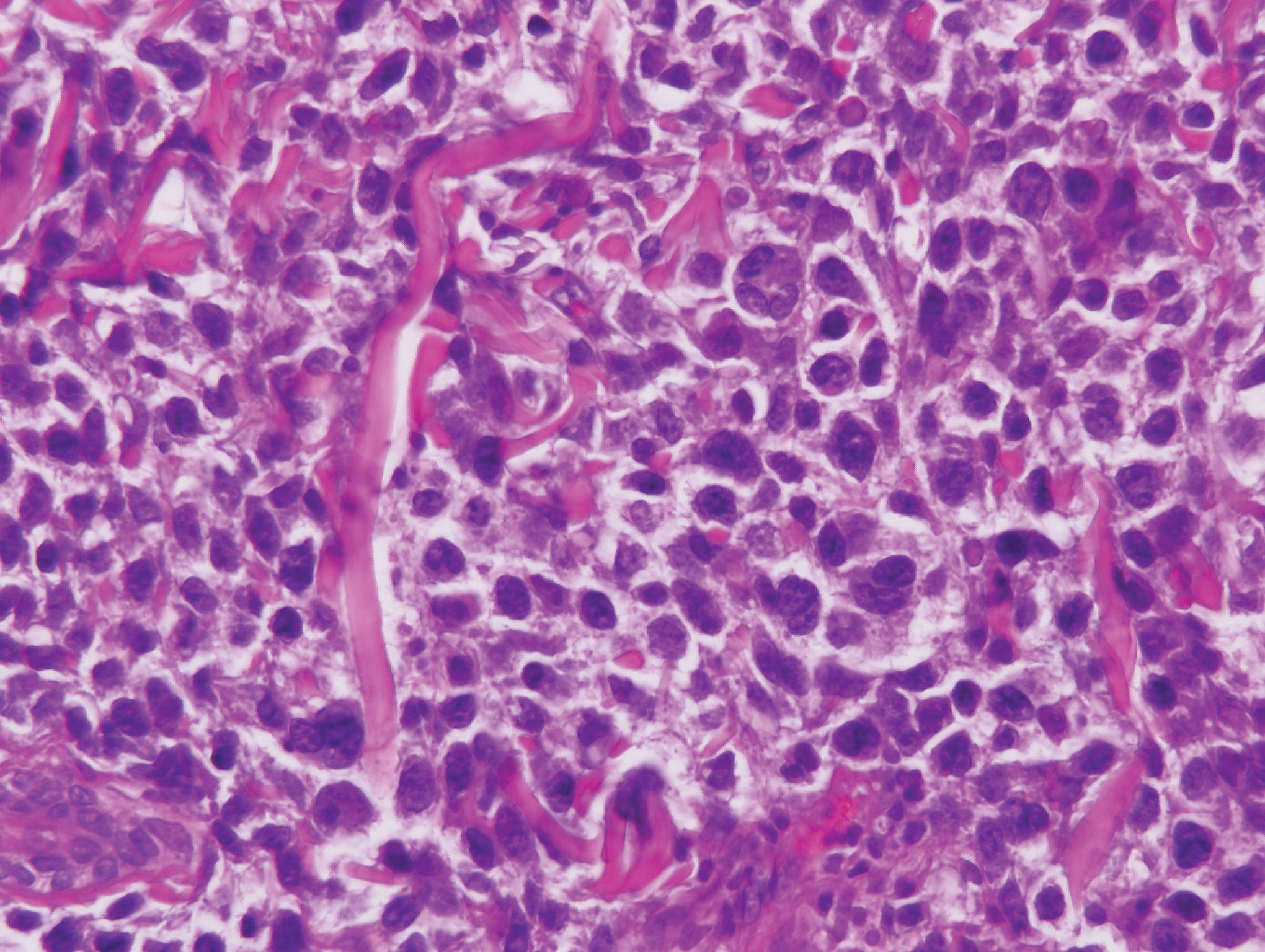
Unusual hypersensitivity reactions to arthropod attacks have been described in patients with lymphoproliferative disorders and could be mistaken for HZ. Histology may demonstrate a wedge-shaped perivascular and/or interstitial infiltrate containing eosinophils with endothelial swelling (Figure 4), but these findings may vary depending on the type of arthropod involved.11
Our case provided a unique example of HZ in a patient with a known history of MF. Clinically, there was concern for progression of the patient’s underlying disease; however, histology demonstrated ballooning keratinocytes and follicular necrosis, which are classically seen in HZ infection.
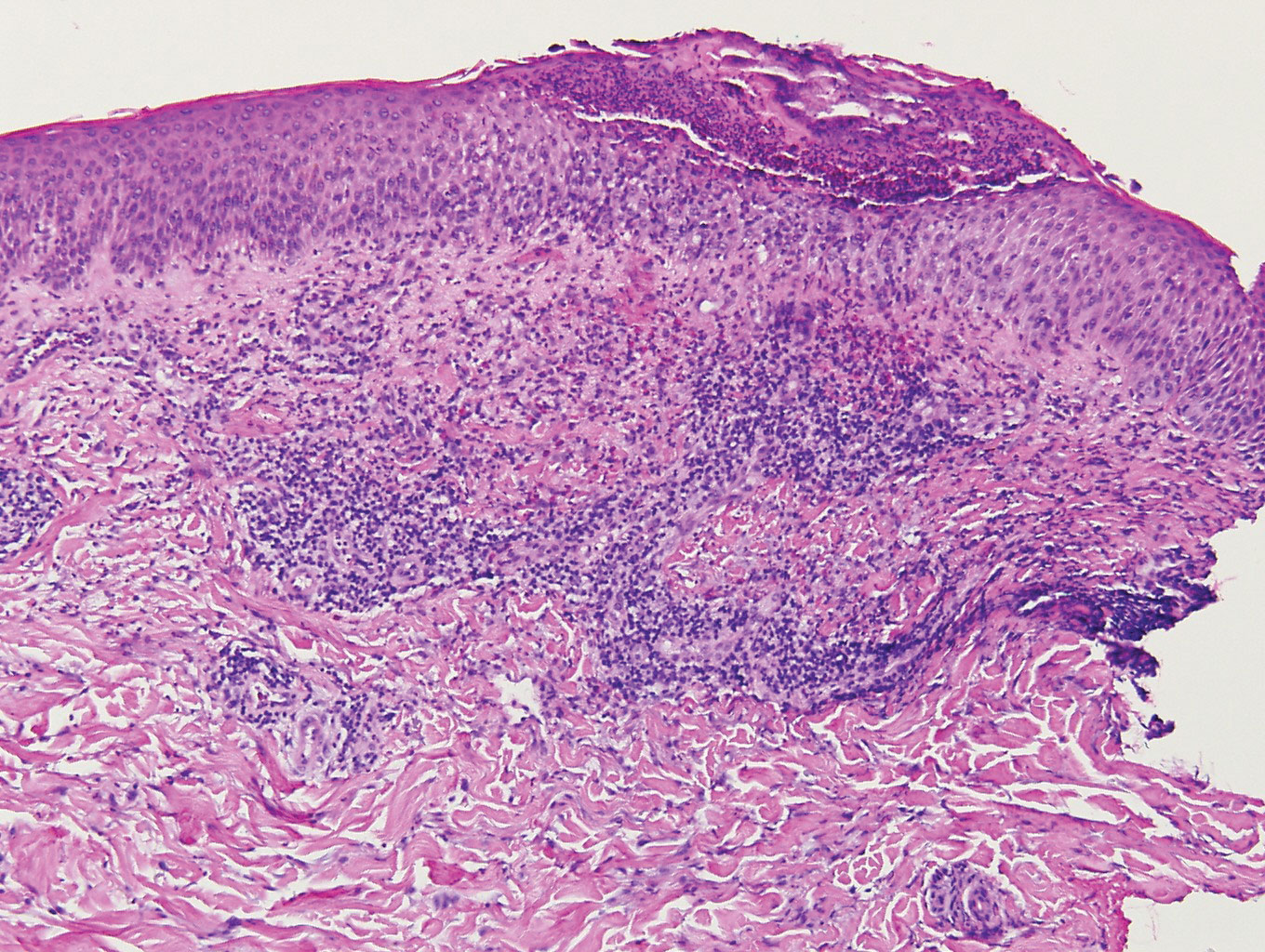
- Downing C, Medoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- Chisholm C, Lopez L. Cutaneous infections caused by Herpesviridae: a review. Arch Pathol Lab Med. 2011;135:1357-1362.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70: 205.e1-205.e16.
- Vonderheid EC, van Voorst Vader PC. Herpes zoster-varicella in cutaneous T-cell lymphomas. Arch Dermatol. 1980;116:408-412.
- Lebas E, Arrese JE, Nikkels AF. Risk factors for skin infections in mycosis fungoides. Dermatology. 2016;232:731-737.
- Leinweber B, Kerl H, Cerroni L. Histopathologic features of cutaneous herpes virus infections (herpes simplex, herpes varicella/zoster): a broad spectrum of presentations with common pseudolymphomatous aspects. Am J Surg Pathol. 2006;30:50-58.
- Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
- Pulitzer M, Myskowski PL, Horwitz SM, et al. Mycosis fungoides with large cell transformation:clinicopathological features and prognostic factors. Pathology. 2014;46:610-616.
- Zackheim HS, Jones C, Leboit PE, et al. Lymphomatoid papulosis associated with mycosis fungoides: a study of 21 patients including analyses for clonality. J Am Acad Dermatol. 2003;49:620-623.
- Barzilai A, Trau H, David M, et al. Mycosis fungoides associated with B-cell malignancies. Br J Dermatol. 2006;155:379-386.
- Vassallo C, Passamonti F, Cananzi R, et al. Exaggerated insect bite-like reaction in patients affected by oncohaematological diseases. Acta Derm Venereol. 2005;85:76-77.
Herpes zoster (HZ) is a painful skin condition caused by reactivation of latent varicella-zoster virus (VZV) in dorsal root ganglion cells.1 Upon reactivation, VZV replicates in the dorsal root ganglion, which ultimately results in inflammation and necrosis of the neuron and intense neuralgia. Reactivation of latent VZV may occur spontaneously or may be induced by various factors including immunosuppression, stress, illness, and trauma. Prior to the development of skin lesions, many patients experience a prodrome of tingling, pain, or pruritus. Herpes zoster classically presents with grouped vesicles on an erythematous base in a unilateral dermatomal distribution; however, more than one adjacent dermatome may be involved, and the lesions can cross the midline. Furthermore, the development of vesicles may be preceded by the development of edematous papules or plaques.1
On histology, VZV closely resembles herpes simplex virus type 1 and herpes simplex virus type 2 infections.2 Classic histologic findings include ballooning degeneration of keratinocytes, acantholysis, nuclear molding, ground-glass nuclear inclusions, marginated chromatin, and multinucleated keratinocytes, as well as necrosis of follicles and sebaceous glands.2 Varicella-zoster virus polymerase chain reaction or immunostaining can be used to confirm the diagnosis.2
Classic mycosis fungoides (MF) presents with well-circumscribed erythematous patches in non–sun-exposed areas and eventually may progress to plaques and tumors.3 Patients with cutaneous T-cell lymphomas, such as MF, are at a higher risk for skin infections including HZ4,5; however, immunocompromised patients, such as those with cutaneous lymphomas, can have atypical clinical presentations of HZ that may be concerning for cutaneous lymphoma.6 Furthermore, cutaneous malignancies can occur in dermatomal distributions that may mimic HZ.7 Therefore, the threshold for biopsy should be lowered in those patients with dermatomal lesions and history concerning for possible malignancy.
Classically, histologic examination of MF demonstrates an infiltrate of haloed cells at the dermoepidermal junction, which are atypical T cells with hyperchromatic cerebriform nuclei that are larger, darker, and more angulated than the benign recruited lymphocytes in the perivascular infiltrate seen in VZV infection (Figure 1).3 Papillary dermal fibrosis typically is present, and the perivascular infiltrate is denser above the postcapillary venule rather than being symmetrical around the vessel (bare underbelly sign). Clusters of these cells may form within the epidermis, which are called Pautrier microabscesses.3 Mycosis fungoides also can exhibit large cell transformation in which small lymphocytes transform into larger cells, thereby associated with a poorer prognosis.8
Lymphomatoid papulosis is a CD30+-predominant form of cutaneous T-cell lymphoma characterized by papules and nodules that spontaneously involute.9 This condition is most commonly associated with MF but can be associated with other lymphomas. This condition may be mistaken for HZ clinically, but histology classically demonstrates large atypical lymphocytes resembling Reed-Sternberg cells in small clusters rather than follicular necrosis (Figure 2).9
Patients with lymphoma may sequentially develop a secondary lymphoma. There have been reports of secondary B-cell lymphomas associated with MF, but this phenomenon is rare.10 The histology depends on the type of B-cell lymphoma present, but follicular necrosis would not be expected (Figure 3).
Unusual hypersensitivity reactions to arthropod attacks have been described in patients with lymphoproliferative disorders and could be mistaken for HZ. Histology may demonstrate a wedge-shaped perivascular and/or interstitial infiltrate containing eosinophils with endothelial swelling (Figure 4), but these findings may vary depending on the type of arthropod involved.11
Our case provided a unique example of HZ in a patient with a known history of MF. Clinically, there was concern for progression of the patient’s underlying disease; however, histology demonstrated ballooning keratinocytes and follicular necrosis, which are classically seen in HZ infection.
Herpes zoster (HZ) is a painful skin condition caused by reactivation of latent varicella-zoster virus (VZV) in dorsal root ganglion cells.1 Upon reactivation, VZV replicates in the dorsal root ganglion, which ultimately results in inflammation and necrosis of the neuron and intense neuralgia. Reactivation of latent VZV may occur spontaneously or may be induced by various factors including immunosuppression, stress, illness, and trauma. Prior to the development of skin lesions, many patients experience a prodrome of tingling, pain, or pruritus. Herpes zoster classically presents with grouped vesicles on an erythematous base in a unilateral dermatomal distribution; however, more than one adjacent dermatome may be involved, and the lesions can cross the midline. Furthermore, the development of vesicles may be preceded by the development of edematous papules or plaques.1
On histology, VZV closely resembles herpes simplex virus type 1 and herpes simplex virus type 2 infections.2 Classic histologic findings include ballooning degeneration of keratinocytes, acantholysis, nuclear molding, ground-glass nuclear inclusions, marginated chromatin, and multinucleated keratinocytes, as well as necrosis of follicles and sebaceous glands.2 Varicella-zoster virus polymerase chain reaction or immunostaining can be used to confirm the diagnosis.2
Classic mycosis fungoides (MF) presents with well-circumscribed erythematous patches in non–sun-exposed areas and eventually may progress to plaques and tumors.3 Patients with cutaneous T-cell lymphomas, such as MF, are at a higher risk for skin infections including HZ4,5; however, immunocompromised patients, such as those with cutaneous lymphomas, can have atypical clinical presentations of HZ that may be concerning for cutaneous lymphoma.6 Furthermore, cutaneous malignancies can occur in dermatomal distributions that may mimic HZ.7 Therefore, the threshold for biopsy should be lowered in those patients with dermatomal lesions and history concerning for possible malignancy.
Classically, histologic examination of MF demonstrates an infiltrate of haloed cells at the dermoepidermal junction, which are atypical T cells with hyperchromatic cerebriform nuclei that are larger, darker, and more angulated than the benign recruited lymphocytes in the perivascular infiltrate seen in VZV infection (Figure 1).3 Papillary dermal fibrosis typically is present, and the perivascular infiltrate is denser above the postcapillary venule rather than being symmetrical around the vessel (bare underbelly sign). Clusters of these cells may form within the epidermis, which are called Pautrier microabscesses.3 Mycosis fungoides also can exhibit large cell transformation in which small lymphocytes transform into larger cells, thereby associated with a poorer prognosis.8
Lymphomatoid papulosis is a CD30+-predominant form of cutaneous T-cell lymphoma characterized by papules and nodules that spontaneously involute.9 This condition is most commonly associated with MF but can be associated with other lymphomas. This condition may be mistaken for HZ clinically, but histology classically demonstrates large atypical lymphocytes resembling Reed-Sternberg cells in small clusters rather than follicular necrosis (Figure 2).9
Patients with lymphoma may sequentially develop a secondary lymphoma. There have been reports of secondary B-cell lymphomas associated with MF, but this phenomenon is rare.10 The histology depends on the type of B-cell lymphoma present, but follicular necrosis would not be expected (Figure 3).
Unusual hypersensitivity reactions to arthropod attacks have been described in patients with lymphoproliferative disorders and could be mistaken for HZ. Histology may demonstrate a wedge-shaped perivascular and/or interstitial infiltrate containing eosinophils with endothelial swelling (Figure 4), but these findings may vary depending on the type of arthropod involved.11
Our case provided a unique example of HZ in a patient with a known history of MF. Clinically, there was concern for progression of the patient’s underlying disease; however, histology demonstrated ballooning keratinocytes and follicular necrosis, which are classically seen in HZ infection.
- Downing C, Medoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- Chisholm C, Lopez L. Cutaneous infections caused by Herpesviridae: a review. Arch Pathol Lab Med. 2011;135:1357-1362.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70: 205.e1-205.e16.
- Vonderheid EC, van Voorst Vader PC. Herpes zoster-varicella in cutaneous T-cell lymphomas. Arch Dermatol. 1980;116:408-412.
- Lebas E, Arrese JE, Nikkels AF. Risk factors for skin infections in mycosis fungoides. Dermatology. 2016;232:731-737.
- Leinweber B, Kerl H, Cerroni L. Histopathologic features of cutaneous herpes virus infections (herpes simplex, herpes varicella/zoster): a broad spectrum of presentations with common pseudolymphomatous aspects. Am J Surg Pathol. 2006;30:50-58.
- Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
- Pulitzer M, Myskowski PL, Horwitz SM, et al. Mycosis fungoides with large cell transformation:clinicopathological features and prognostic factors. Pathology. 2014;46:610-616.
- Zackheim HS, Jones C, Leboit PE, et al. Lymphomatoid papulosis associated with mycosis fungoides: a study of 21 patients including analyses for clonality. J Am Acad Dermatol. 2003;49:620-623.
- Barzilai A, Trau H, David M, et al. Mycosis fungoides associated with B-cell malignancies. Br J Dermatol. 2006;155:379-386.
- Vassallo C, Passamonti F, Cananzi R, et al. Exaggerated insect bite-like reaction in patients affected by oncohaematological diseases. Acta Derm Venereol. 2005;85:76-77.
- Downing C, Medoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- Chisholm C, Lopez L. Cutaneous infections caused by Herpesviridae: a review. Arch Pathol Lab Med. 2011;135:1357-1362.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70: 205.e1-205.e16.
- Vonderheid EC, van Voorst Vader PC. Herpes zoster-varicella in cutaneous T-cell lymphomas. Arch Dermatol. 1980;116:408-412.
- Lebas E, Arrese JE, Nikkels AF. Risk factors for skin infections in mycosis fungoides. Dermatology. 2016;232:731-737.
- Leinweber B, Kerl H, Cerroni L. Histopathologic features of cutaneous herpes virus infections (herpes simplex, herpes varicella/zoster): a broad spectrum of presentations with common pseudolymphomatous aspects. Am J Surg Pathol. 2006;30:50-58.
- Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
- Pulitzer M, Myskowski PL, Horwitz SM, et al. Mycosis fungoides with large cell transformation:clinicopathological features and prognostic factors. Pathology. 2014;46:610-616.
- Zackheim HS, Jones C, Leboit PE, et al. Lymphomatoid papulosis associated with mycosis fungoides: a study of 21 patients including analyses for clonality. J Am Acad Dermatol. 2003;49:620-623.
- Barzilai A, Trau H, David M, et al. Mycosis fungoides associated with B-cell malignancies. Br J Dermatol. 2006;155:379-386.
- Vassallo C, Passamonti F, Cananzi R, et al. Exaggerated insect bite-like reaction in patients affected by oncohaematological diseases. Acta Derm Venereol. 2005;85:76-77.
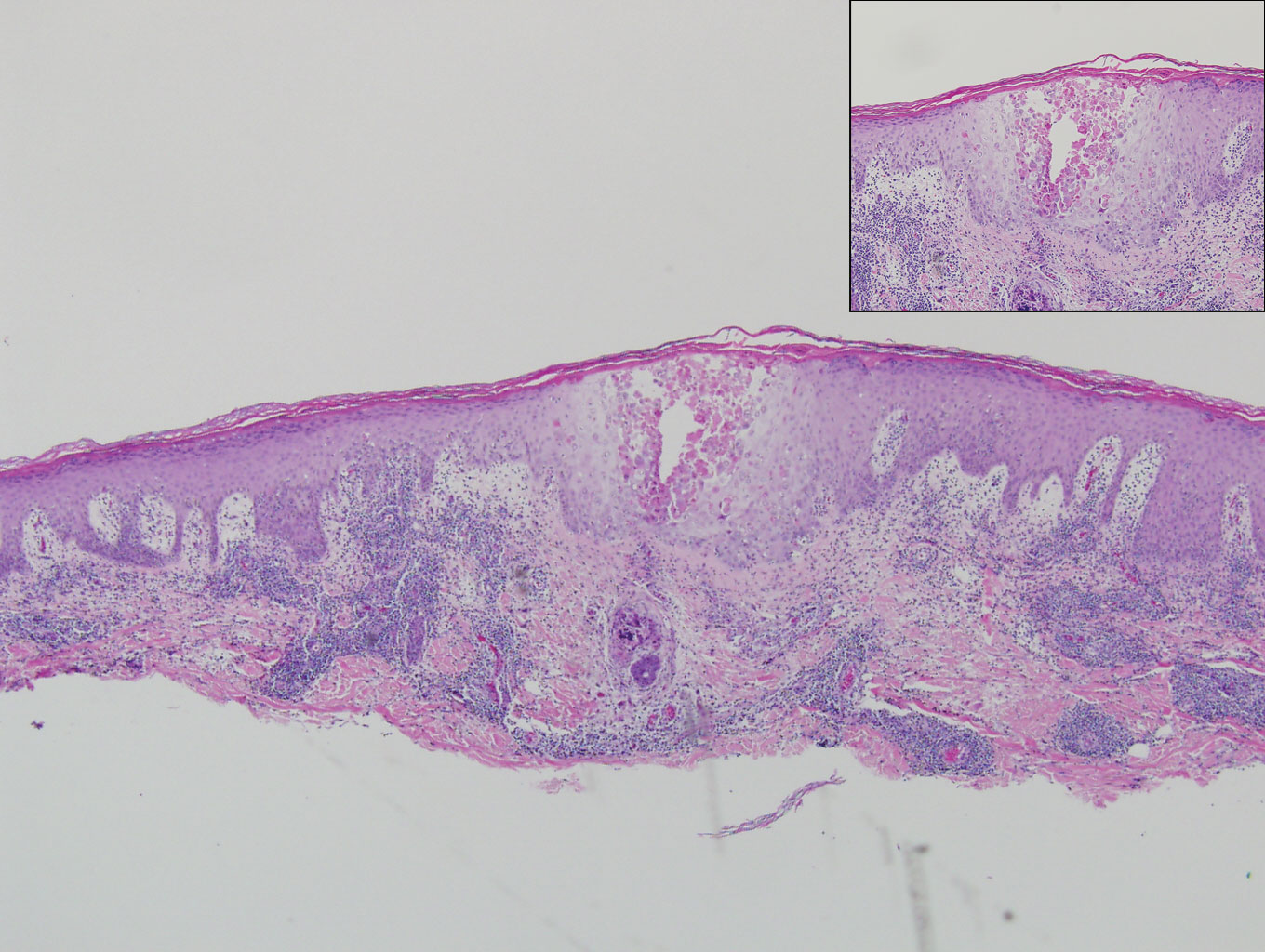
A 66-year-old man with mycosis fungoides presented with a new indurated plaque on the left shoulder. Biopsies of the left shoulder and back lesions were obtained.
31-GEP test predicts likelihood of metastasis for cutaneous melanoma
WASHINGTON – The for accurately predicting recurrence-free survival and distant metastasis-free survival and melanoma-specific survival, according to results presented by Bradley N. Greenhaw, MD, at a late-breaking research session at the annual meeting of the American Academy of Dermatology.
Dr. Greenhaw, a dermatologist affiliated with the North Mississippi Medical Center-Tupelo, and his colleagues pooled together 1,268 patients from the following studies that analyzed results from melanoma patients who had their disease classified with the 31-gene expression profile (31-GEP) test.
- A single-center study, conducted by Dr. Greenhaw and his associates (Greenhaw BN et al. Dermatol Surg. 2018 Dec. doi: 10.1097/DSS.0000000000001588.
- A multicenter prospective study (J Hematol Oncol. 2017 Aug. doi: 10.1186/s13045-017-0520-1.
- A retrospective archival study (J Am Acad Dermatol. 2019 Jan. doi: 10.1016/j.jaad.2018.07.028.
The 31-GEP test stratifies an individual’s likelihood of developing metastasis within 5 years as low and high risk. In the three studies, the test was used to identify tumors with low-risk (class 1A, class 1B), higher-risk (class 2A), and highest-risk (class 2B) melanoma based on tumor gene expression. In these individual studies, class 2B melanoma independently predicted recurrence-free survival (RFS), distant metastasis–free, and melanoma-specific survival.
Dr. Greenhaw and colleagues performed a meta-analysis of 1,268 patients with stage I through stage III melanoma from those three studies, using fixed and random effects weighting to account for study differences and heterogeneity, respectively. For class 2B tumors, they found a 2.96 increased risk for recurrent metastases and a 2.88 increased risk for distant metastases. The researchers also found no heterogeneity across the studies.
Melanoma-specific survival was not included in the meta-analysis because one paper did not contain any mortality events in class 1A melanoma patients.
“The meta-analysis demonstrated that the GEP test was able to accurately identify those melanoma patients who were at higher risk of metastasis, and we saw a consistent effect across multiple studies,” Dr. Greenhaw said.
Since publication of the 2019 JAAD paper, there were an additional 211 patients who met inclusion criteria and were included in an additional meta-analysis to determine whether inclusion of these patients affected the results. Dr. Greenhaw and colleagues found a 91.4% recurrence-free survival rate and a 94.1% distant metastasis–free survival rate for class 1A melanomas, compared with 45.7% and 55.5% , respectively, for class 2B tumors.
“You can see a big divergence,” Dr. Greenhaw said at the meeting. “Just by using this one test, it’s able to separate out melanomas that otherwise may be grouped in together under current AJCC [American Joint Committee on Cancer] staging,” he added. “The class 2B designation really did confirm a higher risk for recurrence in distant metastasis.”
The researchers used the SORT method to rate the quality of the data across all three studies. Level 1 evidence under the SORT method represents a systematic review or meta-analysis of good-quality studies and/or a prospective study with good follow-up, while an A-level recommendation represents good, quality evidence. Based on the meta-analysis results, the 31-GEP test meets level 1A evidence under the SORT method, Dr. Greenhaw said.
As a prognostic tool, 31-GEP has the potential to change how dermatologists manage their patients with regard to follow-up and adjuvant therapy. “It is being used not just as this novel test that gives us more information, it’s being used clinically,” said Dr. Greenhaw, who noted he regularly uses the 31-GEP test in his practice.
This is the first time that a meta-analysis has been performed for this test, he noted.
Dr. Greenhaw reports a pending relationship with Castle Biosciences.
SOURCE: Greenhaw BN et al. AAD 19. Session F055, Abstract 11370.
WASHINGTON – The for accurately predicting recurrence-free survival and distant metastasis-free survival and melanoma-specific survival, according to results presented by Bradley N. Greenhaw, MD, at a late-breaking research session at the annual meeting of the American Academy of Dermatology.
Dr. Greenhaw, a dermatologist affiliated with the North Mississippi Medical Center-Tupelo, and his colleagues pooled together 1,268 patients from the following studies that analyzed results from melanoma patients who had their disease classified with the 31-gene expression profile (31-GEP) test.
- A single-center study, conducted by Dr. Greenhaw and his associates (Greenhaw BN et al. Dermatol Surg. 2018 Dec. doi: 10.1097/DSS.0000000000001588.
- A multicenter prospective study (J Hematol Oncol. 2017 Aug. doi: 10.1186/s13045-017-0520-1.
- A retrospective archival study (J Am Acad Dermatol. 2019 Jan. doi: 10.1016/j.jaad.2018.07.028.
The 31-GEP test stratifies an individual’s likelihood of developing metastasis within 5 years as low and high risk. In the three studies, the test was used to identify tumors with low-risk (class 1A, class 1B), higher-risk (class 2A), and highest-risk (class 2B) melanoma based on tumor gene expression. In these individual studies, class 2B melanoma independently predicted recurrence-free survival (RFS), distant metastasis–free, and melanoma-specific survival.
Dr. Greenhaw and colleagues performed a meta-analysis of 1,268 patients with stage I through stage III melanoma from those three studies, using fixed and random effects weighting to account for study differences and heterogeneity, respectively. For class 2B tumors, they found a 2.96 increased risk for recurrent metastases and a 2.88 increased risk for distant metastases. The researchers also found no heterogeneity across the studies.
Melanoma-specific survival was not included in the meta-analysis because one paper did not contain any mortality events in class 1A melanoma patients.
“The meta-analysis demonstrated that the GEP test was able to accurately identify those melanoma patients who were at higher risk of metastasis, and we saw a consistent effect across multiple studies,” Dr. Greenhaw said.
Since publication of the 2019 JAAD paper, there were an additional 211 patients who met inclusion criteria and were included in an additional meta-analysis to determine whether inclusion of these patients affected the results. Dr. Greenhaw and colleagues found a 91.4% recurrence-free survival rate and a 94.1% distant metastasis–free survival rate for class 1A melanomas, compared with 45.7% and 55.5% , respectively, for class 2B tumors.
“You can see a big divergence,” Dr. Greenhaw said at the meeting. “Just by using this one test, it’s able to separate out melanomas that otherwise may be grouped in together under current AJCC [American Joint Committee on Cancer] staging,” he added. “The class 2B designation really did confirm a higher risk for recurrence in distant metastasis.”
The researchers used the SORT method to rate the quality of the data across all three studies. Level 1 evidence under the SORT method represents a systematic review or meta-analysis of good-quality studies and/or a prospective study with good follow-up, while an A-level recommendation represents good, quality evidence. Based on the meta-analysis results, the 31-GEP test meets level 1A evidence under the SORT method, Dr. Greenhaw said.
As a prognostic tool, 31-GEP has the potential to change how dermatologists manage their patients with regard to follow-up and adjuvant therapy. “It is being used not just as this novel test that gives us more information, it’s being used clinically,” said Dr. Greenhaw, who noted he regularly uses the 31-GEP test in his practice.
This is the first time that a meta-analysis has been performed for this test, he noted.
Dr. Greenhaw reports a pending relationship with Castle Biosciences.
SOURCE: Greenhaw BN et al. AAD 19. Session F055, Abstract 11370.
WASHINGTON – The for accurately predicting recurrence-free survival and distant metastasis-free survival and melanoma-specific survival, according to results presented by Bradley N. Greenhaw, MD, at a late-breaking research session at the annual meeting of the American Academy of Dermatology.
Dr. Greenhaw, a dermatologist affiliated with the North Mississippi Medical Center-Tupelo, and his colleagues pooled together 1,268 patients from the following studies that analyzed results from melanoma patients who had their disease classified with the 31-gene expression profile (31-GEP) test.
- A single-center study, conducted by Dr. Greenhaw and his associates (Greenhaw BN et al. Dermatol Surg. 2018 Dec. doi: 10.1097/DSS.0000000000001588.
- A multicenter prospective study (J Hematol Oncol. 2017 Aug. doi: 10.1186/s13045-017-0520-1.
- A retrospective archival study (J Am Acad Dermatol. 2019 Jan. doi: 10.1016/j.jaad.2018.07.028.
The 31-GEP test stratifies an individual’s likelihood of developing metastasis within 5 years as low and high risk. In the three studies, the test was used to identify tumors with low-risk (class 1A, class 1B), higher-risk (class 2A), and highest-risk (class 2B) melanoma based on tumor gene expression. In these individual studies, class 2B melanoma independently predicted recurrence-free survival (RFS), distant metastasis–free, and melanoma-specific survival.
Dr. Greenhaw and colleagues performed a meta-analysis of 1,268 patients with stage I through stage III melanoma from those three studies, using fixed and random effects weighting to account for study differences and heterogeneity, respectively. For class 2B tumors, they found a 2.96 increased risk for recurrent metastases and a 2.88 increased risk for distant metastases. The researchers also found no heterogeneity across the studies.
Melanoma-specific survival was not included in the meta-analysis because one paper did not contain any mortality events in class 1A melanoma patients.
“The meta-analysis demonstrated that the GEP test was able to accurately identify those melanoma patients who were at higher risk of metastasis, and we saw a consistent effect across multiple studies,” Dr. Greenhaw said.
Since publication of the 2019 JAAD paper, there were an additional 211 patients who met inclusion criteria and were included in an additional meta-analysis to determine whether inclusion of these patients affected the results. Dr. Greenhaw and colleagues found a 91.4% recurrence-free survival rate and a 94.1% distant metastasis–free survival rate for class 1A melanomas, compared with 45.7% and 55.5% , respectively, for class 2B tumors.
“You can see a big divergence,” Dr. Greenhaw said at the meeting. “Just by using this one test, it’s able to separate out melanomas that otherwise may be grouped in together under current AJCC [American Joint Committee on Cancer] staging,” he added. “The class 2B designation really did confirm a higher risk for recurrence in distant metastasis.”
The researchers used the SORT method to rate the quality of the data across all three studies. Level 1 evidence under the SORT method represents a systematic review or meta-analysis of good-quality studies and/or a prospective study with good follow-up, while an A-level recommendation represents good, quality evidence. Based on the meta-analysis results, the 31-GEP test meets level 1A evidence under the SORT method, Dr. Greenhaw said.
As a prognostic tool, 31-GEP has the potential to change how dermatologists manage their patients with regard to follow-up and adjuvant therapy. “It is being used not just as this novel test that gives us more information, it’s being used clinically,” said Dr. Greenhaw, who noted he regularly uses the 31-GEP test in his practice.
This is the first time that a meta-analysis has been performed for this test, he noted.
Dr. Greenhaw reports a pending relationship with Castle Biosciences.
SOURCE: Greenhaw BN et al. AAD 19. Session F055, Abstract 11370.
REPORTING FROM AAD 19
Hailey-Hailey Disease: A Diagnostic Challenge
Hailey-Hailey disease (HHD), also known as benign familial chronic pemphigus, is an autosomal-dominant genodermatosis caused by mutations of the ATPase secretory pathway Ca2+ transporting 1 gene, ATP2C1.1 It is characterized by crusted macerated erosions and velvety, fissured, hypertrophic plaques classically involving the intertriginous areas. The diagnosis is suggested by characteristic clinical morphology, involvement of the intertriginous areas, and a positive family history. Histology often confirms the diagnosis and demonstrates a characteristic dilapidated brick wall appearance. If there is a need to distinguish HHD from pemphigus, direct immunofluorescence studies also should be performed, which would be negative.2,3 However, HHD often is misdiagnosed due to lack of knowledge of this uncommon disorder and its resemblance to other dermatoses of the intertriginous areas.4 We present an unusual presentation of HHD with late onset and involvement of the skin of the abdomen and foot.
Case Report
A 61-year-old woman presented with a 3×4-cm fissured plaque with erosions and a peripheral yellow crust on the left side of the anterior abdomen (Figure 1A). There was another fissured plaque with surrounding erythema and scaling on the fifth digit of the right foot (Figure 1B). For the last 11 years, she periodically experienced erosive and scabbing skin plaques under the breasts and on the axillae and groin. Her mother and maternal grandfather had a history of similar skin lesions. Due to a suspicion of HHD, a skin biopsy specimen of the abdominal plaque was performed, which demonstrated epidermal acanthosis and suprabasal acantholysis with lacunae formation (Figure 2). There was uneven thickening of the epidermal keratin layer with parakeratotic nests. The upper layer of the dermis demonstrated edema and focal fibrosis, enlarged capillaries, and pericapillary lymphohistiocytic infiltration with eosinophils and neutrophils. Accordingly, a diagnosis of HHD was established.
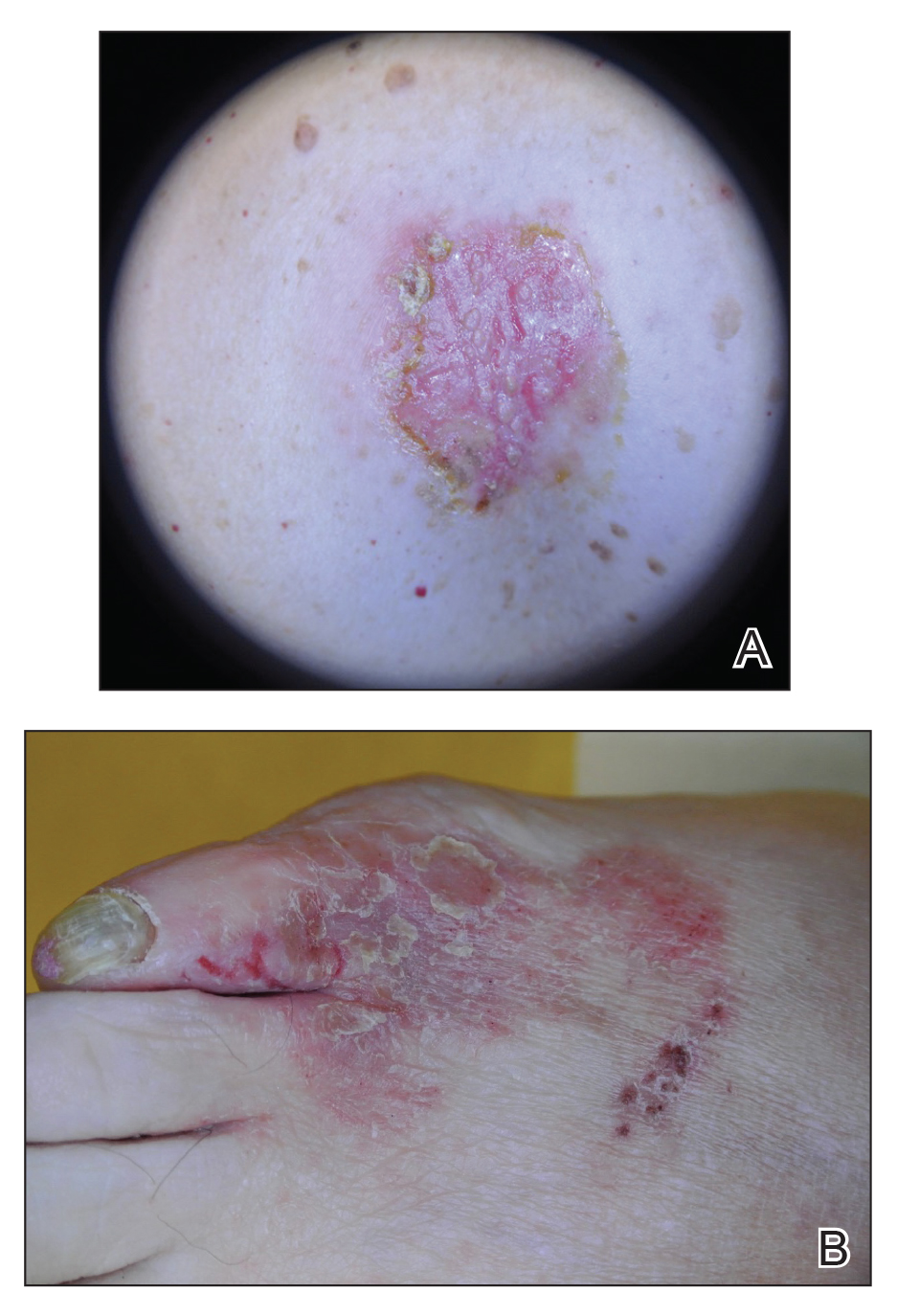
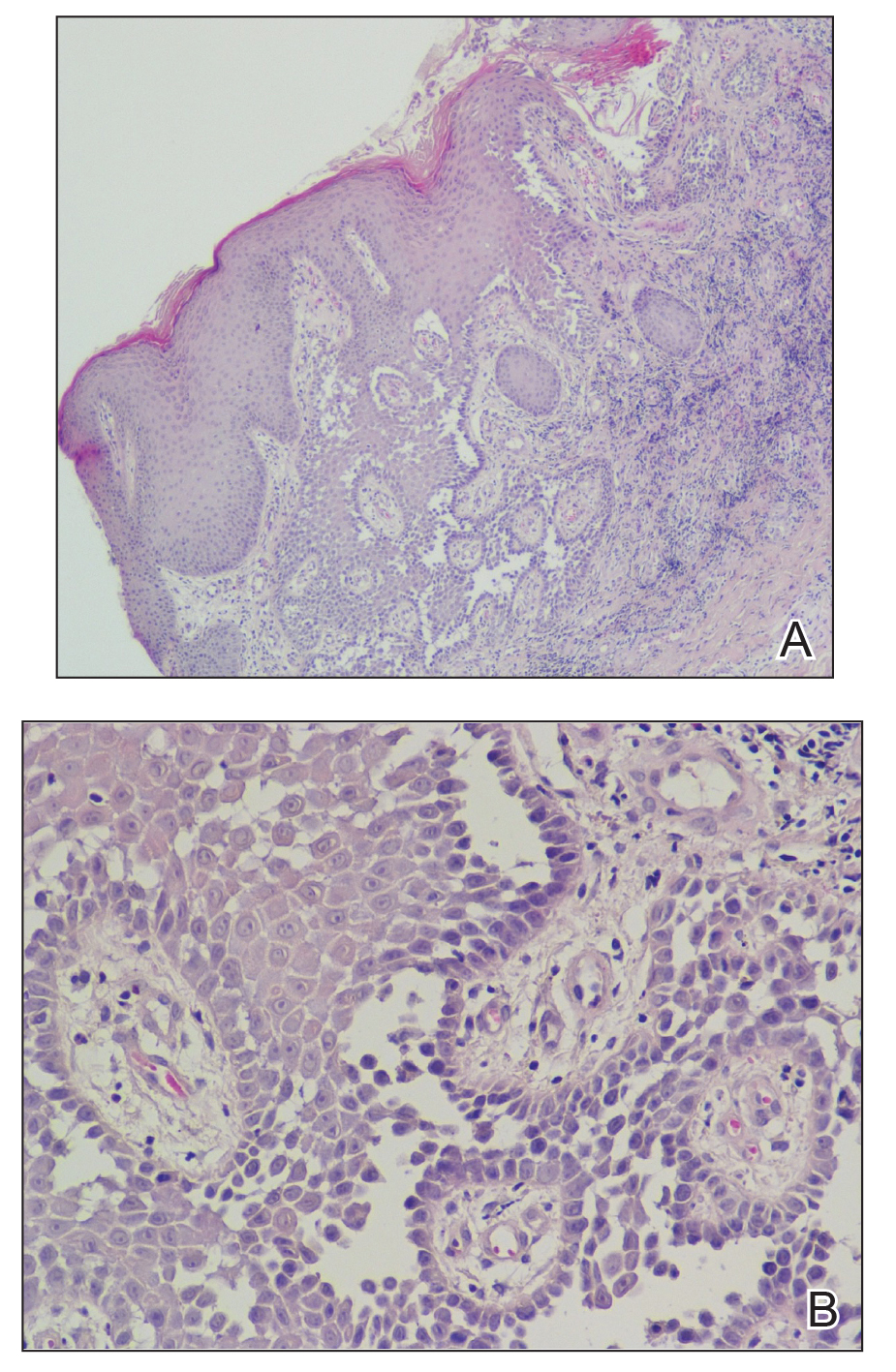
Comment
Hailey-Hailey disease occurs in 1 to 4 per 100,000 individuals without predilection for sex or ethnic group.5-9 Onset usually occurs after puberty, most commonly in the third decade of life.8,10-12 Mutations of the ATP2C1 gene on band 3q22.1 cause haploinsufficiency of Ca2+/Mn2+−ATPase protein 1 (hSPCA1) that alters the intracellular calcium gradient, leading to disruptions in assembly and trafficking of desmosomal proteins to the cell membrane. Consequently, altered intercellular connections and acantholysis of the epidermis occur.1,13-16
Hailey-Hailey disease initially manifests as grouped flaccid vesicles that rupture easily, leaving behind crusted erosions and dry, scaly, eczematous patches.17,18 Over time, velvety, fissured, and hypertrophic plaques develop. Up to 80% of patients experience secondary bacterial and fungal superinfections that may cause vegetative or malodorous plaques.9 Although HHD has no specific treatment, symptoms are managed with topical corticosteroids and antimicrobial agents. Patients should be advised to avoid irritants such as friction, sunlight, or sweat. For severe cases, botulinum toxin type A, laser therapy, dermabrasion, and surgery have been utilized with variable success.19-22 The responsiveness of HHD to corticosteroids and antimicrobial agents facilitates misdiagnosis as intertrigo, erythrasma, or dermatophytosis.
Our patient presented with late-onset HHD (age, 50 years) compared to the typical age of onset in the third decade of life.8 Furthermore, her presentation was atypical for HHD, which characteristically affects intertriginous areas due to sweat, heat, friction, and microorganisms. Hailey-Hailey disease involving the abdominal skin is unusual, as it typically occurs in regions of friction such as the belt area.23 Our patient lacked a history of friction or trauma at the site of the abdominal plaque. In addition, HHD involving the feet is exceedingly rare. It is plausible that friction and heat caused by footwear may have predisposed her to these skin changes.
Conclusion
This case highlights the difficulties of diagnosing HHD, especially if it appears in atypical locations.24 Obtaining a thorough family history and detailed dermatologic examination as well as maintaining a high level of suspicion can assist in diagnosing this uncommon disorder.
- Hu Z, Bonifas JM, Beech J, et al. Mutations in ATP2C1, encoding a alcium pump, cause Hailey-Hailey disease. Nat Genet. 2000;24:61-65.
- Ohata C. Hailey-Hailey disease. Cutis. 2014;94:33-34.
- Abdullah L, Abbas O. Dermacase. can you identify this condition? benign familial chronic pemphigus. Can Fam Physician. 2011;57:1157-1158.
- Le Donne M, Lentini M, Moretti G, et al. Chronic vulvocrural dermatitis with burning and itching. CMAJ. 2008;179:555-556.
- Hohl D. Darier disease and Hailey-Hailey disease. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. Philadelphia, PA: Saunders; 2012:887-897.
- Cooper SM, Burge SM. Darier’s disease: epidemiology, pathophysiology, and management. Am J Clin Dermatol. 2003;4:97-105.
- Godic A, Miljkovic J, Kansky A, et al. Epidemiology of Darier’s disease in Slovenia. Acta Dermatovenerol Alp Pannonica Adriat. 2005;14:43-48.
- Burge SM. Hailey-Hailey disease: the clinical features, response to treatment and prognosis. Br J Dermatol. 1992;126:275-282.
- Benmously-Mlika R, Bchetnia M, Deghais S, et al. Hailey-Hailey disease in Tunisia. Int J Dermatol. 2010;49:396-401.
- Bessa GR, Grazziotin TC, Manzoni AP, et al. Hailey-Hailey disease treatment with botulinum toxin type A. An Bras Dermatol. 2010;85:717-722.
- Gu H, Chang B, Chen W, et al. Clinical analysis of 69 patients with familial benign chronic pemphigus. Chin Med J (Engl). 1999;112:761-763.
- Dobson-Stone C, Fairclough R, Dunne E, et al. Hailey-Hailey disease: molecular and clinical characterization of novel mutations in the ATP2C1 gene. J Invest Dermatol. 2002;118:338-343.
- Fairclough RJ, Lonie L, Van Baelen K, et al. Hailey-Hailey disease: identification of novel mutations in ATP2C1 and effect of missense mutation A528P on protein expression levels. J Invest Dermatol. 2004;123:6771.
- Shibata A, Sugiura K, Kimura U, et al. A novel ATP2C1 early truncation mutation suggests haploinsufficiency as a pathogenic mechanism in a patient with Hailey-Hailey disease. Acta Derm Venereol. 2013;93:719-720.
- Dhitavat J, Fairclough RJ, Hovnanian A, et al. Calcium pumps and keratinocytes: lessons from Darier’s disease and Hailey-Hailey disease. Br J Dermatol. 2004;150:821-828.
- Raiko L, Siljamaki E, Mahoney MG, et al. Hailey-Hailey disease and tight junctions: claudins 1 and 4 are regulated by ATP2C1 gene encoding Ca(2+)/Mn(2+) ATPase SPCA1 in cultured keratinocytes. Exp Dermatol. 2012;21:586-591.
- Yadav N, Madke B, Kar S, et al. Hailey-Hailey disease. Indian Dermatol Online J. 2016;7:147-148.
- Vasudevan B, Verma R, Badwal S, et al. Hailey-Hailey disease with skin lesions at unusual sites and a good response to acitretin. Indian J Dermatol Venereol Leprol. 2015;81:88-91.
- Bagherani N, Smoller BR. The efficacy of botulinum toxin type A in the treatment of Hailey Hailey disease. Dermatol Ther. 2016;29:394-395.
- Hochwalt PC, Christensen KN, Cantwell SR, et al. Carbon dioxide laser treatment for Hailey-Hailey disease: a retrospective chart review with patient-reported outcomes. Int J Dermatol. 2015;54:1309-1314.
- Falto-Aizpurua LA, Griffith RD, Yazdani Abyaneh MA, et al. Laser therapy for the treatment of Hailey-Hailey disease: a systematic review with focus on carbon dioxide laser resurfacing. J Eur Acad Dermatol Venereol. 2015;29:1045-1052.
- Arora H, Bray FN, Cervantes J, et al. Management of familial benign chronic pemphigus. Clin Cosmet Investig Dermatol. 2016;9:281-290.
- Iijima S, Hamada T, Kanzaki M, et al. Sibling cases of Hailey-Hailey disease showing atypical clinical features and unique disease course. JAMA Dermatol. 2014;150:97-99.
- Saied NK, Schwartz RA, Hansen RC, et al. Atypical familial benign chronic pemphigus. Cutis. 1981;27:666-669.
Hailey-Hailey disease (HHD), also known as benign familial chronic pemphigus, is an autosomal-dominant genodermatosis caused by mutations of the ATPase secretory pathway Ca2+ transporting 1 gene, ATP2C1.1 It is characterized by crusted macerated erosions and velvety, fissured, hypertrophic plaques classically involving the intertriginous areas. The diagnosis is suggested by characteristic clinical morphology, involvement of the intertriginous areas, and a positive family history. Histology often confirms the diagnosis and demonstrates a characteristic dilapidated brick wall appearance. If there is a need to distinguish HHD from pemphigus, direct immunofluorescence studies also should be performed, which would be negative.2,3 However, HHD often is misdiagnosed due to lack of knowledge of this uncommon disorder and its resemblance to other dermatoses of the intertriginous areas.4 We present an unusual presentation of HHD with late onset and involvement of the skin of the abdomen and foot.
Case Report
A 61-year-old woman presented with a 3×4-cm fissured plaque with erosions and a peripheral yellow crust on the left side of the anterior abdomen (Figure 1A). There was another fissured plaque with surrounding erythema and scaling on the fifth digit of the right foot (Figure 1B). For the last 11 years, she periodically experienced erosive and scabbing skin plaques under the breasts and on the axillae and groin. Her mother and maternal grandfather had a history of similar skin lesions. Due to a suspicion of HHD, a skin biopsy specimen of the abdominal plaque was performed, which demonstrated epidermal acanthosis and suprabasal acantholysis with lacunae formation (Figure 2). There was uneven thickening of the epidermal keratin layer with parakeratotic nests. The upper layer of the dermis demonstrated edema and focal fibrosis, enlarged capillaries, and pericapillary lymphohistiocytic infiltration with eosinophils and neutrophils. Accordingly, a diagnosis of HHD was established.
Comment
Hailey-Hailey disease occurs in 1 to 4 per 100,000 individuals without predilection for sex or ethnic group.5-9 Onset usually occurs after puberty, most commonly in the third decade of life.8,10-12 Mutations of the ATP2C1 gene on band 3q22.1 cause haploinsufficiency of Ca2+/Mn2+−ATPase protein 1 (hSPCA1) that alters the intracellular calcium gradient, leading to disruptions in assembly and trafficking of desmosomal proteins to the cell membrane. Consequently, altered intercellular connections and acantholysis of the epidermis occur.1,13-16
Hailey-Hailey disease initially manifests as grouped flaccid vesicles that rupture easily, leaving behind crusted erosions and dry, scaly, eczematous patches.17,18 Over time, velvety, fissured, and hypertrophic plaques develop. Up to 80% of patients experience secondary bacterial and fungal superinfections that may cause vegetative or malodorous plaques.9 Although HHD has no specific treatment, symptoms are managed with topical corticosteroids and antimicrobial agents. Patients should be advised to avoid irritants such as friction, sunlight, or sweat. For severe cases, botulinum toxin type A, laser therapy, dermabrasion, and surgery have been utilized with variable success.19-22 The responsiveness of HHD to corticosteroids and antimicrobial agents facilitates misdiagnosis as intertrigo, erythrasma, or dermatophytosis.
Our patient presented with late-onset HHD (age, 50 years) compared to the typical age of onset in the third decade of life.8 Furthermore, her presentation was atypical for HHD, which characteristically affects intertriginous areas due to sweat, heat, friction, and microorganisms. Hailey-Hailey disease involving the abdominal skin is unusual, as it typically occurs in regions of friction such as the belt area.23 Our patient lacked a history of friction or trauma at the site of the abdominal plaque. In addition, HHD involving the feet is exceedingly rare. It is plausible that friction and heat caused by footwear may have predisposed her to these skin changes.
Conclusion
This case highlights the difficulties of diagnosing HHD, especially if it appears in atypical locations.24 Obtaining a thorough family history and detailed dermatologic examination as well as maintaining a high level of suspicion can assist in diagnosing this uncommon disorder.
Hailey-Hailey disease (HHD), also known as benign familial chronic pemphigus, is an autosomal-dominant genodermatosis caused by mutations of the ATPase secretory pathway Ca2+ transporting 1 gene, ATP2C1.1 It is characterized by crusted macerated erosions and velvety, fissured, hypertrophic plaques classically involving the intertriginous areas. The diagnosis is suggested by characteristic clinical morphology, involvement of the intertriginous areas, and a positive family history. Histology often confirms the diagnosis and demonstrates a characteristic dilapidated brick wall appearance. If there is a need to distinguish HHD from pemphigus, direct immunofluorescence studies also should be performed, which would be negative.2,3 However, HHD often is misdiagnosed due to lack of knowledge of this uncommon disorder and its resemblance to other dermatoses of the intertriginous areas.4 We present an unusual presentation of HHD with late onset and involvement of the skin of the abdomen and foot.
Case Report
A 61-year-old woman presented with a 3×4-cm fissured plaque with erosions and a peripheral yellow crust on the left side of the anterior abdomen (Figure 1A). There was another fissured plaque with surrounding erythema and scaling on the fifth digit of the right foot (Figure 1B). For the last 11 years, she periodically experienced erosive and scabbing skin plaques under the breasts and on the axillae and groin. Her mother and maternal grandfather had a history of similar skin lesions. Due to a suspicion of HHD, a skin biopsy specimen of the abdominal plaque was performed, which demonstrated epidermal acanthosis and suprabasal acantholysis with lacunae formation (Figure 2). There was uneven thickening of the epidermal keratin layer with parakeratotic nests. The upper layer of the dermis demonstrated edema and focal fibrosis, enlarged capillaries, and pericapillary lymphohistiocytic infiltration with eosinophils and neutrophils. Accordingly, a diagnosis of HHD was established.
Comment
Hailey-Hailey disease occurs in 1 to 4 per 100,000 individuals without predilection for sex or ethnic group.5-9 Onset usually occurs after puberty, most commonly in the third decade of life.8,10-12 Mutations of the ATP2C1 gene on band 3q22.1 cause haploinsufficiency of Ca2+/Mn2+−ATPase protein 1 (hSPCA1) that alters the intracellular calcium gradient, leading to disruptions in assembly and trafficking of desmosomal proteins to the cell membrane. Consequently, altered intercellular connections and acantholysis of the epidermis occur.1,13-16
Hailey-Hailey disease initially manifests as grouped flaccid vesicles that rupture easily, leaving behind crusted erosions and dry, scaly, eczematous patches.17,18 Over time, velvety, fissured, and hypertrophic plaques develop. Up to 80% of patients experience secondary bacterial and fungal superinfections that may cause vegetative or malodorous plaques.9 Although HHD has no specific treatment, symptoms are managed with topical corticosteroids and antimicrobial agents. Patients should be advised to avoid irritants such as friction, sunlight, or sweat. For severe cases, botulinum toxin type A, laser therapy, dermabrasion, and surgery have been utilized with variable success.19-22 The responsiveness of HHD to corticosteroids and antimicrobial agents facilitates misdiagnosis as intertrigo, erythrasma, or dermatophytosis.
Our patient presented with late-onset HHD (age, 50 years) compared to the typical age of onset in the third decade of life.8 Furthermore, her presentation was atypical for HHD, which characteristically affects intertriginous areas due to sweat, heat, friction, and microorganisms. Hailey-Hailey disease involving the abdominal skin is unusual, as it typically occurs in regions of friction such as the belt area.23 Our patient lacked a history of friction or trauma at the site of the abdominal plaque. In addition, HHD involving the feet is exceedingly rare. It is plausible that friction and heat caused by footwear may have predisposed her to these skin changes.
Conclusion
This case highlights the difficulties of diagnosing HHD, especially if it appears in atypical locations.24 Obtaining a thorough family history and detailed dermatologic examination as well as maintaining a high level of suspicion can assist in diagnosing this uncommon disorder.
- Hu Z, Bonifas JM, Beech J, et al. Mutations in ATP2C1, encoding a alcium pump, cause Hailey-Hailey disease. Nat Genet. 2000;24:61-65.
- Ohata C. Hailey-Hailey disease. Cutis. 2014;94:33-34.
- Abdullah L, Abbas O. Dermacase. can you identify this condition? benign familial chronic pemphigus. Can Fam Physician. 2011;57:1157-1158.
- Le Donne M, Lentini M, Moretti G, et al. Chronic vulvocrural dermatitis with burning and itching. CMAJ. 2008;179:555-556.
- Hohl D. Darier disease and Hailey-Hailey disease. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. Philadelphia, PA: Saunders; 2012:887-897.
- Cooper SM, Burge SM. Darier’s disease: epidemiology, pathophysiology, and management. Am J Clin Dermatol. 2003;4:97-105.
- Godic A, Miljkovic J, Kansky A, et al. Epidemiology of Darier’s disease in Slovenia. Acta Dermatovenerol Alp Pannonica Adriat. 2005;14:43-48.
- Burge SM. Hailey-Hailey disease: the clinical features, response to treatment and prognosis. Br J Dermatol. 1992;126:275-282.
- Benmously-Mlika R, Bchetnia M, Deghais S, et al. Hailey-Hailey disease in Tunisia. Int J Dermatol. 2010;49:396-401.
- Bessa GR, Grazziotin TC, Manzoni AP, et al. Hailey-Hailey disease treatment with botulinum toxin type A. An Bras Dermatol. 2010;85:717-722.
- Gu H, Chang B, Chen W, et al. Clinical analysis of 69 patients with familial benign chronic pemphigus. Chin Med J (Engl). 1999;112:761-763.
- Dobson-Stone C, Fairclough R, Dunne E, et al. Hailey-Hailey disease: molecular and clinical characterization of novel mutations in the ATP2C1 gene. J Invest Dermatol. 2002;118:338-343.
- Fairclough RJ, Lonie L, Van Baelen K, et al. Hailey-Hailey disease: identification of novel mutations in ATP2C1 and effect of missense mutation A528P on protein expression levels. J Invest Dermatol. 2004;123:6771.
- Shibata A, Sugiura K, Kimura U, et al. A novel ATP2C1 early truncation mutation suggests haploinsufficiency as a pathogenic mechanism in a patient with Hailey-Hailey disease. Acta Derm Venereol. 2013;93:719-720.
- Dhitavat J, Fairclough RJ, Hovnanian A, et al. Calcium pumps and keratinocytes: lessons from Darier’s disease and Hailey-Hailey disease. Br J Dermatol. 2004;150:821-828.
- Raiko L, Siljamaki E, Mahoney MG, et al. Hailey-Hailey disease and tight junctions: claudins 1 and 4 are regulated by ATP2C1 gene encoding Ca(2+)/Mn(2+) ATPase SPCA1 in cultured keratinocytes. Exp Dermatol. 2012;21:586-591.
- Yadav N, Madke B, Kar S, et al. Hailey-Hailey disease. Indian Dermatol Online J. 2016;7:147-148.
- Vasudevan B, Verma R, Badwal S, et al. Hailey-Hailey disease with skin lesions at unusual sites and a good response to acitretin. Indian J Dermatol Venereol Leprol. 2015;81:88-91.
- Bagherani N, Smoller BR. The efficacy of botulinum toxin type A in the treatment of Hailey Hailey disease. Dermatol Ther. 2016;29:394-395.
- Hochwalt PC, Christensen KN, Cantwell SR, et al. Carbon dioxide laser treatment for Hailey-Hailey disease: a retrospective chart review with patient-reported outcomes. Int J Dermatol. 2015;54:1309-1314.
- Falto-Aizpurua LA, Griffith RD, Yazdani Abyaneh MA, et al. Laser therapy for the treatment of Hailey-Hailey disease: a systematic review with focus on carbon dioxide laser resurfacing. J Eur Acad Dermatol Venereol. 2015;29:1045-1052.
- Arora H, Bray FN, Cervantes J, et al. Management of familial benign chronic pemphigus. Clin Cosmet Investig Dermatol. 2016;9:281-290.
- Iijima S, Hamada T, Kanzaki M, et al. Sibling cases of Hailey-Hailey disease showing atypical clinical features and unique disease course. JAMA Dermatol. 2014;150:97-99.
- Saied NK, Schwartz RA, Hansen RC, et al. Atypical familial benign chronic pemphigus. Cutis. 1981;27:666-669.
- Hu Z, Bonifas JM, Beech J, et al. Mutations in ATP2C1, encoding a alcium pump, cause Hailey-Hailey disease. Nat Genet. 2000;24:61-65.
- Ohata C. Hailey-Hailey disease. Cutis. 2014;94:33-34.
- Abdullah L, Abbas O. Dermacase. can you identify this condition? benign familial chronic pemphigus. Can Fam Physician. 2011;57:1157-1158.
- Le Donne M, Lentini M, Moretti G, et al. Chronic vulvocrural dermatitis with burning and itching. CMAJ. 2008;179:555-556.
- Hohl D. Darier disease and Hailey-Hailey disease. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. Philadelphia, PA: Saunders; 2012:887-897.
- Cooper SM, Burge SM. Darier’s disease: epidemiology, pathophysiology, and management. Am J Clin Dermatol. 2003;4:97-105.
- Godic A, Miljkovic J, Kansky A, et al. Epidemiology of Darier’s disease in Slovenia. Acta Dermatovenerol Alp Pannonica Adriat. 2005;14:43-48.
- Burge SM. Hailey-Hailey disease: the clinical features, response to treatment and prognosis. Br J Dermatol. 1992;126:275-282.
- Benmously-Mlika R, Bchetnia M, Deghais S, et al. Hailey-Hailey disease in Tunisia. Int J Dermatol. 2010;49:396-401.
- Bessa GR, Grazziotin TC, Manzoni AP, et al. Hailey-Hailey disease treatment with botulinum toxin type A. An Bras Dermatol. 2010;85:717-722.
- Gu H, Chang B, Chen W, et al. Clinical analysis of 69 patients with familial benign chronic pemphigus. Chin Med J (Engl). 1999;112:761-763.
- Dobson-Stone C, Fairclough R, Dunne E, et al. Hailey-Hailey disease: molecular and clinical characterization of novel mutations in the ATP2C1 gene. J Invest Dermatol. 2002;118:338-343.
- Fairclough RJ, Lonie L, Van Baelen K, et al. Hailey-Hailey disease: identification of novel mutations in ATP2C1 and effect of missense mutation A528P on protein expression levels. J Invest Dermatol. 2004;123:6771.
- Shibata A, Sugiura K, Kimura U, et al. A novel ATP2C1 early truncation mutation suggests haploinsufficiency as a pathogenic mechanism in a patient with Hailey-Hailey disease. Acta Derm Venereol. 2013;93:719-720.
- Dhitavat J, Fairclough RJ, Hovnanian A, et al. Calcium pumps and keratinocytes: lessons from Darier’s disease and Hailey-Hailey disease. Br J Dermatol. 2004;150:821-828.
- Raiko L, Siljamaki E, Mahoney MG, et al. Hailey-Hailey disease and tight junctions: claudins 1 and 4 are regulated by ATP2C1 gene encoding Ca(2+)/Mn(2+) ATPase SPCA1 in cultured keratinocytes. Exp Dermatol. 2012;21:586-591.
- Yadav N, Madke B, Kar S, et al. Hailey-Hailey disease. Indian Dermatol Online J. 2016;7:147-148.
- Vasudevan B, Verma R, Badwal S, et al. Hailey-Hailey disease with skin lesions at unusual sites and a good response to acitretin. Indian J Dermatol Venereol Leprol. 2015;81:88-91.
- Bagherani N, Smoller BR. The efficacy of botulinum toxin type A in the treatment of Hailey Hailey disease. Dermatol Ther. 2016;29:394-395.
- Hochwalt PC, Christensen KN, Cantwell SR, et al. Carbon dioxide laser treatment for Hailey-Hailey disease: a retrospective chart review with patient-reported outcomes. Int J Dermatol. 2015;54:1309-1314.
- Falto-Aizpurua LA, Griffith RD, Yazdani Abyaneh MA, et al. Laser therapy for the treatment of Hailey-Hailey disease: a systematic review with focus on carbon dioxide laser resurfacing. J Eur Acad Dermatol Venereol. 2015;29:1045-1052.
- Arora H, Bray FN, Cervantes J, et al. Management of familial benign chronic pemphigus. Clin Cosmet Investig Dermatol. 2016;9:281-290.
- Iijima S, Hamada T, Kanzaki M, et al. Sibling cases of Hailey-Hailey disease showing atypical clinical features and unique disease course. JAMA Dermatol. 2014;150:97-99.
- Saied NK, Schwartz RA, Hansen RC, et al. Atypical familial benign chronic pemphigus. Cutis. 1981;27:666-669.
Practice Points
- Hailey-Hailey disease may present atypically with a late age of onset, involvement of nonintertriginous areas, and lack of clear exacerbating factors such as friction.
- A detailed history and physical examination as well as a high degree of suspicion can aid in diagnosing this uncommon disorder.