User login
Papular Reticulated Rash
The Diagnosis: Prurigo Pigmentosa
Histopathology of the punch biopsy revealed subcorneal collections of neutrophils flanked by a spongiotic epidermis with neutrophil and eosinophil exocytosis. Rare dyskeratotic keratinocytes were identified at the dermoepidermal junction, and grampositive bacterial organisms were seen in a follicular infundibulum with purulent inflammation. The dermis demonstrated a mildly dense superficial perivascular and interstitial infiltrate composed of lymphocytes, histiocytes, scattered neutrophils, and eosinophils (Figure).
.")
Given the combination of clinical and histologic findings, a diagnosis of prurigo pigmentosa (PP) was rendered and a urinalysis was ordered, which confirmed ketonuria. The patient was started on minocycline 100 mg twice daily and was advised to reintroduce carbohydrates into her diet. Resolution of the inflammatory component of the rash was achieved at 3-week follow-up, with residual reticulated postinflammatory hyperpigmentation.
Prurigo pigmentosa is a rare, albeit globally underrecognized, inflammatory dermatosis characterized by pruritic, symmetric, erythematous papules and plaques on the chest, back, neck, and rarely the arms and forehead that subsequently involute, leaving reticular postinflammatory hyperpigmentation.1 Prurigo pigmentosa is predominant in females (2.6:1 ratio). The mean age at presentation is 24.4 years, and it most commonly has been documented among populations in Asian countries, though it is unclear if a genetic predilection exists, as reports of PP are increasing globally with improved clinical awareness.1,2
The etiology of PP remains unknown; however, associations are well documented between PP and a ketogenic state secondary to uncontrolled diabetes, a low-carbohydrate diet, anorexia nervosa, or bariatric surgery.3 It is theorized that high serum ketones lead to perivascular ketone deposition, which induces neutrophil migration and chemotaxis,4 as substantiated by evidence of rash resolution with correction of the ketogenic state and improvement after administration of tetracyclines, a drug class known for neutrophil chemotaxis inhibition.5 Improvement of PP via these treatment mechanisms suggests that ketone bodies may play a role in the pathogenesis of PP.
Interestingly, Kafle et al6 reported that patients with PP commonly have bacterial colonies and associated inflammatory sequelae at the level of the hair follicles, which suggests that follicular involvement plays a role in the pathogenesis of PP. These findings are consistent with our patient’s histopathology consisting of gram-positive organisms and purulent inflammation at the infundibulum. The histopathologic features of PP are stage specific.1 Early stages are characterized by a superficial perivascular infiltrate of neutrophils that then spread to dermal papillae. Neutrophils then quickly sweep through the epidermis, causing spongiosis, ballooning, necrotic keratocytes, and consequent surface epithelium abscess formation. Over time, the dermal infiltrate assumes a lichenoid pattern as eosinophils and lymphocytes invade and predominate over neutrophils. Eventually, melanophages appear in the dermis as the epidermis undergoes hyperplasia, parakeratosis, and hyperpigmentation.1 The histologic differential diagnosis for PP is broad and varies based on the stage-specific progression of clinical and histopathologic findings.
Similar to PP, subacute cutaneous lupus erythematosus has a female predominance and resolves with subsequent dyspigmentation; however, it initially is characterized by annular plaques with central clearing or papulosquamous lesions restricted to sun-exposed skin. Photosensitivity is a prominent feature, and roughly 50% of patients meet diagnostic criteria for systemic lupus erythematosus.7 Histopathology shows interface changes with increased dermal mucin and a perivascular lymphoplasmacytic inflammatory infiltrate.
Papular pityriasis rosea can present as a pruritic papular rash on the back and chest; however, it most commonly is associated with a herald patch and typically follows a flulike prodrome.8 Biopsy reveals mounds of parakeratosis with mild spongiosis, perivascular inflammation, and extravasated erythrocytes.
Galli-Galli disease can present as a pruritic rash with follicular papules under the breasts and other flexural areas but histopathologically shows elongated rete ridges with dermal melanosis and acantholysis.9
Hailey-Hailey disease commonly presents in the third decade of life and can manifest as painful, pruritic, vesicular lesions on erythematous skin distributed on the back, neck, and inframammary region, as seen in our case; however, it is histopathologically associated with widespread epidermal acantholysis unlike the findings seen in our patient.10
First-line treatment of PP includes antibiotics such as minocycline, doxycycline, and dapsone due to their anti-inflammatory properties and ability to inhibit neutrophil chemotaxis. In patients with nutritional deficiencies or ketosis, reintroduction of carbohydrates alone has been effective.5,11
Prurigo pigmentosa is an underrecognized inflammatory dermatosis with a complex stage-dependent clinicopathologic presentation. Clinicians should be aware of the etiologic and histopathologic patterns of this unique dermatosis. Rash presentation in the context of a low-carbohydrate diet should prompt biopsy as well as treatment with antibiotics and dietary reintroduction of carbohydrates.
- Böer A, Misago N, Wolter M, et al. Prurigo pigmentosa: a distinctive inflammatory disease of the skin. Am J Dermatopathol. 2003;25:117-129. doi:10.1097/00000372-200304000-00005
- de Sousa Vargas TJ, Abreu Raposo CM, Lima RB, et al. Prurigo pigmentosa: report of 3 cases from Brazil and literature review. Am J Dermatopathol. 2017;39:267-274. doi:10.1097/DAD.0000000000000643
- Mufti A, Mirali S, Abduelmula A, et al. Clinical manifestations and treatment outcomes in prurigo pigmentosa (Nagashima disease): a systematic review of the literature. JAAD Int. 2021;3:79. doi:10.1016/J .JDIN.2021.03.003
- Beutler BD, Cohen PR, Lee RA. Prurigo pigmentosa: literature review. Am J Clin Dermatol. 2015;16:533-543. doi:10.1007/S40257-015-0154-4
- Chiam LYT, Goh BK, Lim KS, et al. Prurigo pigmentosa: a report of two cases that responded to minocycline. Clin Exp Dermatol. 2009;34. doi:10.1111/J.1365-2230.2009.03253.X
- Kafle SU, Swe SM, Hsiao PF, et al. Folliculitis in prurigo pigmentosa: a proposed pathogenesis based on clinical and pathological observation. J Cutan Pathol. 2017;44:20-27. doi:10.1111/CUP.12829
- Sontheimer RD. Subacute cutaneous lupus erythematosus: 25-year evolution of a prototypic subset (subphenotype) of lupus erythematosus defined by characteristic cutaneous, pathological, immunological, and genetic findings. Autoimmun Rev. 2005;4:253-263. doi:10.1016/J .AUTREV.2004.10.00
- Leung AKC, Lam JM, Leong KF, et al. Pityriasis rosea: an updated review. Curr Pediatr Rev. 2021;17:201-211. doi:10.2174/15733963166662 00923161330
- Sprecher E, Indelman M, Khamaysi Z, et al. Galli-Galli disease is an acantholytic variant of Dowling-Degos disease. Br J Dermatol. 2007;156:572-574. doi:10.1111/J.1365-2133.2006.07703.X
- Burge SM. Hailey-Hailey disease: the clinical features, response to treatment and prognosis. Br J Dermatol. 1992;126:275-282. doi:10.1111/J.1365-2133.1992.TB00658
- Lu L-Y, Chen C-B. Keto rash: ketoacidosis-induced prurigo pigmentosa. Mayo Clin Proc. 2022;97:20-21. doi:10.1016/j.mayocp.2021.11.019
The Diagnosis: Prurigo Pigmentosa
Histopathology of the punch biopsy revealed subcorneal collections of neutrophils flanked by a spongiotic epidermis with neutrophil and eosinophil exocytosis. Rare dyskeratotic keratinocytes were identified at the dermoepidermal junction, and grampositive bacterial organisms were seen in a follicular infundibulum with purulent inflammation. The dermis demonstrated a mildly dense superficial perivascular and interstitial infiltrate composed of lymphocytes, histiocytes, scattered neutrophils, and eosinophils (Figure).
Given the combination of clinical and histologic findings, a diagnosis of prurigo pigmentosa (PP) was rendered and a urinalysis was ordered, which confirmed ketonuria. The patient was started on minocycline 100 mg twice daily and was advised to reintroduce carbohydrates into her diet. Resolution of the inflammatory component of the rash was achieved at 3-week follow-up, with residual reticulated postinflammatory hyperpigmentation.
Prurigo pigmentosa is a rare, albeit globally underrecognized, inflammatory dermatosis characterized by pruritic, symmetric, erythematous papules and plaques on the chest, back, neck, and rarely the arms and forehead that subsequently involute, leaving reticular postinflammatory hyperpigmentation.1 Prurigo pigmentosa is predominant in females (2.6:1 ratio). The mean age at presentation is 24.4 years, and it most commonly has been documented among populations in Asian countries, though it is unclear if a genetic predilection exists, as reports of PP are increasing globally with improved clinical awareness.1,2
The etiology of PP remains unknown; however, associations are well documented between PP and a ketogenic state secondary to uncontrolled diabetes, a low-carbohydrate diet, anorexia nervosa, or bariatric surgery.3 It is theorized that high serum ketones lead to perivascular ketone deposition, which induces neutrophil migration and chemotaxis,4 as substantiated by evidence of rash resolution with correction of the ketogenic state and improvement after administration of tetracyclines, a drug class known for neutrophil chemotaxis inhibition.5 Improvement of PP via these treatment mechanisms suggests that ketone bodies may play a role in the pathogenesis of PP.
Interestingly, Kafle et al6 reported that patients with PP commonly have bacterial colonies and associated inflammatory sequelae at the level of the hair follicles, which suggests that follicular involvement plays a role in the pathogenesis of PP. These findings are consistent with our patient’s histopathology consisting of gram-positive organisms and purulent inflammation at the infundibulum. The histopathologic features of PP are stage specific.1 Early stages are characterized by a superficial perivascular infiltrate of neutrophils that then spread to dermal papillae. Neutrophils then quickly sweep through the epidermis, causing spongiosis, ballooning, necrotic keratocytes, and consequent surface epithelium abscess formation. Over time, the dermal infiltrate assumes a lichenoid pattern as eosinophils and lymphocytes invade and predominate over neutrophils. Eventually, melanophages appear in the dermis as the epidermis undergoes hyperplasia, parakeratosis, and hyperpigmentation.1 The histologic differential diagnosis for PP is broad and varies based on the stage-specific progression of clinical and histopathologic findings.
Similar to PP, subacute cutaneous lupus erythematosus has a female predominance and resolves with subsequent dyspigmentation; however, it initially is characterized by annular plaques with central clearing or papulosquamous lesions restricted to sun-exposed skin. Photosensitivity is a prominent feature, and roughly 50% of patients meet diagnostic criteria for systemic lupus erythematosus.7 Histopathology shows interface changes with increased dermal mucin and a perivascular lymphoplasmacytic inflammatory infiltrate.
Papular pityriasis rosea can present as a pruritic papular rash on the back and chest; however, it most commonly is associated with a herald patch and typically follows a flulike prodrome.8 Biopsy reveals mounds of parakeratosis with mild spongiosis, perivascular inflammation, and extravasated erythrocytes.
Galli-Galli disease can present as a pruritic rash with follicular papules under the breasts and other flexural areas but histopathologically shows elongated rete ridges with dermal melanosis and acantholysis.9
Hailey-Hailey disease commonly presents in the third decade of life and can manifest as painful, pruritic, vesicular lesions on erythematous skin distributed on the back, neck, and inframammary region, as seen in our case; however, it is histopathologically associated with widespread epidermal acantholysis unlike the findings seen in our patient.10
First-line treatment of PP includes antibiotics such as minocycline, doxycycline, and dapsone due to their anti-inflammatory properties and ability to inhibit neutrophil chemotaxis. In patients with nutritional deficiencies or ketosis, reintroduction of carbohydrates alone has been effective.5,11
Prurigo pigmentosa is an underrecognized inflammatory dermatosis with a complex stage-dependent clinicopathologic presentation. Clinicians should be aware of the etiologic and histopathologic patterns of this unique dermatosis. Rash presentation in the context of a low-carbohydrate diet should prompt biopsy as well as treatment with antibiotics and dietary reintroduction of carbohydrates.
The Diagnosis: Prurigo Pigmentosa
Histopathology of the punch biopsy revealed subcorneal collections of neutrophils flanked by a spongiotic epidermis with neutrophil and eosinophil exocytosis. Rare dyskeratotic keratinocytes were identified at the dermoepidermal junction, and grampositive bacterial organisms were seen in a follicular infundibulum with purulent inflammation. The dermis demonstrated a mildly dense superficial perivascular and interstitial infiltrate composed of lymphocytes, histiocytes, scattered neutrophils, and eosinophils (Figure).
Given the combination of clinical and histologic findings, a diagnosis of prurigo pigmentosa (PP) was rendered and a urinalysis was ordered, which confirmed ketonuria. The patient was started on minocycline 100 mg twice daily and was advised to reintroduce carbohydrates into her diet. Resolution of the inflammatory component of the rash was achieved at 3-week follow-up, with residual reticulated postinflammatory hyperpigmentation.
Prurigo pigmentosa is a rare, albeit globally underrecognized, inflammatory dermatosis characterized by pruritic, symmetric, erythematous papules and plaques on the chest, back, neck, and rarely the arms and forehead that subsequently involute, leaving reticular postinflammatory hyperpigmentation.1 Prurigo pigmentosa is predominant in females (2.6:1 ratio). The mean age at presentation is 24.4 years, and it most commonly has been documented among populations in Asian countries, though it is unclear if a genetic predilection exists, as reports of PP are increasing globally with improved clinical awareness.1,2
The etiology of PP remains unknown; however, associations are well documented between PP and a ketogenic state secondary to uncontrolled diabetes, a low-carbohydrate diet, anorexia nervosa, or bariatric surgery.3 It is theorized that high serum ketones lead to perivascular ketone deposition, which induces neutrophil migration and chemotaxis,4 as substantiated by evidence of rash resolution with correction of the ketogenic state and improvement after administration of tetracyclines, a drug class known for neutrophil chemotaxis inhibition.5 Improvement of PP via these treatment mechanisms suggests that ketone bodies may play a role in the pathogenesis of PP.
Interestingly, Kafle et al6 reported that patients with PP commonly have bacterial colonies and associated inflammatory sequelae at the level of the hair follicles, which suggests that follicular involvement plays a role in the pathogenesis of PP. These findings are consistent with our patient’s histopathology consisting of gram-positive organisms and purulent inflammation at the infundibulum. The histopathologic features of PP are stage specific.1 Early stages are characterized by a superficial perivascular infiltrate of neutrophils that then spread to dermal papillae. Neutrophils then quickly sweep through the epidermis, causing spongiosis, ballooning, necrotic keratocytes, and consequent surface epithelium abscess formation. Over time, the dermal infiltrate assumes a lichenoid pattern as eosinophils and lymphocytes invade and predominate over neutrophils. Eventually, melanophages appear in the dermis as the epidermis undergoes hyperplasia, parakeratosis, and hyperpigmentation.1 The histologic differential diagnosis for PP is broad and varies based on the stage-specific progression of clinical and histopathologic findings.
Similar to PP, subacute cutaneous lupus erythematosus has a female predominance and resolves with subsequent dyspigmentation; however, it initially is characterized by annular plaques with central clearing or papulosquamous lesions restricted to sun-exposed skin. Photosensitivity is a prominent feature, and roughly 50% of patients meet diagnostic criteria for systemic lupus erythematosus.7 Histopathology shows interface changes with increased dermal mucin and a perivascular lymphoplasmacytic inflammatory infiltrate.
Papular pityriasis rosea can present as a pruritic papular rash on the back and chest; however, it most commonly is associated with a herald patch and typically follows a flulike prodrome.8 Biopsy reveals mounds of parakeratosis with mild spongiosis, perivascular inflammation, and extravasated erythrocytes.
Galli-Galli disease can present as a pruritic rash with follicular papules under the breasts and other flexural areas but histopathologically shows elongated rete ridges with dermal melanosis and acantholysis.9
Hailey-Hailey disease commonly presents in the third decade of life and can manifest as painful, pruritic, vesicular lesions on erythematous skin distributed on the back, neck, and inframammary region, as seen in our case; however, it is histopathologically associated with widespread epidermal acantholysis unlike the findings seen in our patient.10
First-line treatment of PP includes antibiotics such as minocycline, doxycycline, and dapsone due to their anti-inflammatory properties and ability to inhibit neutrophil chemotaxis. In patients with nutritional deficiencies or ketosis, reintroduction of carbohydrates alone has been effective.5,11
Prurigo pigmentosa is an underrecognized inflammatory dermatosis with a complex stage-dependent clinicopathologic presentation. Clinicians should be aware of the etiologic and histopathologic patterns of this unique dermatosis. Rash presentation in the context of a low-carbohydrate diet should prompt biopsy as well as treatment with antibiotics and dietary reintroduction of carbohydrates.
- Böer A, Misago N, Wolter M, et al. Prurigo pigmentosa: a distinctive inflammatory disease of the skin. Am J Dermatopathol. 2003;25:117-129. doi:10.1097/00000372-200304000-00005
- de Sousa Vargas TJ, Abreu Raposo CM, Lima RB, et al. Prurigo pigmentosa: report of 3 cases from Brazil and literature review. Am J Dermatopathol. 2017;39:267-274. doi:10.1097/DAD.0000000000000643
- Mufti A, Mirali S, Abduelmula A, et al. Clinical manifestations and treatment outcomes in prurigo pigmentosa (Nagashima disease): a systematic review of the literature. JAAD Int. 2021;3:79. doi:10.1016/J .JDIN.2021.03.003
- Beutler BD, Cohen PR, Lee RA. Prurigo pigmentosa: literature review. Am J Clin Dermatol. 2015;16:533-543. doi:10.1007/S40257-015-0154-4
- Chiam LYT, Goh BK, Lim KS, et al. Prurigo pigmentosa: a report of two cases that responded to minocycline. Clin Exp Dermatol. 2009;34. doi:10.1111/J.1365-2230.2009.03253.X
- Kafle SU, Swe SM, Hsiao PF, et al. Folliculitis in prurigo pigmentosa: a proposed pathogenesis based on clinical and pathological observation. J Cutan Pathol. 2017;44:20-27. doi:10.1111/CUP.12829
- Sontheimer RD. Subacute cutaneous lupus erythematosus: 25-year evolution of a prototypic subset (subphenotype) of lupus erythematosus defined by characteristic cutaneous, pathological, immunological, and genetic findings. Autoimmun Rev. 2005;4:253-263. doi:10.1016/J .AUTREV.2004.10.00
- Leung AKC, Lam JM, Leong KF, et al. Pityriasis rosea: an updated review. Curr Pediatr Rev. 2021;17:201-211. doi:10.2174/15733963166662 00923161330
- Sprecher E, Indelman M, Khamaysi Z, et al. Galli-Galli disease is an acantholytic variant of Dowling-Degos disease. Br J Dermatol. 2007;156:572-574. doi:10.1111/J.1365-2133.2006.07703.X
- Burge SM. Hailey-Hailey disease: the clinical features, response to treatment and prognosis. Br J Dermatol. 1992;126:275-282. doi:10.1111/J.1365-2133.1992.TB00658
- Lu L-Y, Chen C-B. Keto rash: ketoacidosis-induced prurigo pigmentosa. Mayo Clin Proc. 2022;97:20-21. doi:10.1016/j.mayocp.2021.11.019
- Böer A, Misago N, Wolter M, et al. Prurigo pigmentosa: a distinctive inflammatory disease of the skin. Am J Dermatopathol. 2003;25:117-129. doi:10.1097/00000372-200304000-00005
- de Sousa Vargas TJ, Abreu Raposo CM, Lima RB, et al. Prurigo pigmentosa: report of 3 cases from Brazil and literature review. Am J Dermatopathol. 2017;39:267-274. doi:10.1097/DAD.0000000000000643
- Mufti A, Mirali S, Abduelmula A, et al. Clinical manifestations and treatment outcomes in prurigo pigmentosa (Nagashima disease): a systematic review of the literature. JAAD Int. 2021;3:79. doi:10.1016/J .JDIN.2021.03.003
- Beutler BD, Cohen PR, Lee RA. Prurigo pigmentosa: literature review. Am J Clin Dermatol. 2015;16:533-543. doi:10.1007/S40257-015-0154-4
- Chiam LYT, Goh BK, Lim KS, et al. Prurigo pigmentosa: a report of two cases that responded to minocycline. Clin Exp Dermatol. 2009;34. doi:10.1111/J.1365-2230.2009.03253.X
- Kafle SU, Swe SM, Hsiao PF, et al. Folliculitis in prurigo pigmentosa: a proposed pathogenesis based on clinical and pathological observation. J Cutan Pathol. 2017;44:20-27. doi:10.1111/CUP.12829
- Sontheimer RD. Subacute cutaneous lupus erythematosus: 25-year evolution of a prototypic subset (subphenotype) of lupus erythematosus defined by characteristic cutaneous, pathological, immunological, and genetic findings. Autoimmun Rev. 2005;4:253-263. doi:10.1016/J .AUTREV.2004.10.00
- Leung AKC, Lam JM, Leong KF, et al. Pityriasis rosea: an updated review. Curr Pediatr Rev. 2021;17:201-211. doi:10.2174/15733963166662 00923161330
- Sprecher E, Indelman M, Khamaysi Z, et al. Galli-Galli disease is an acantholytic variant of Dowling-Degos disease. Br J Dermatol. 2007;156:572-574. doi:10.1111/J.1365-2133.2006.07703.X
- Burge SM. Hailey-Hailey disease: the clinical features, response to treatment and prognosis. Br J Dermatol. 1992;126:275-282. doi:10.1111/J.1365-2133.1992.TB00658
- Lu L-Y, Chen C-B. Keto rash: ketoacidosis-induced prurigo pigmentosa. Mayo Clin Proc. 2022;97:20-21. doi:10.1016/j.mayocp.2021.11.019
An otherwise healthy 22-year-old woman presented with a painful eruption with burning and pruritus that had been slowly worsening as it spread over the last 4 weeks. The rash first appeared on the lower chest and inframammary folds (top) and spread to the upper chest, neck, back (bottom), arms, and lower face. Physical examination revealed multiple illdefined, erythematous papules, patches, and plaques on the chest, back, neck, and upper abdomen. Individual lesions coalesced into plaques that displayed a reticular configuration. There were no lesions in the axillae. The patient had been following a low-carbohydrate diet for 4 months. A punch biopsy was performed.


Miliarial Gout in an Immunocompromised Patient
To the Editor:
Miliarial gout is a rare intradermal manifestation of tophaceous gout. It was first described in 2007 when a patient presented with multiple small papules with a red base containing a white- to cream-colored substance,1 which has rarely been reported,1-6 according to a PubMed search of articles indexed for MEDLINE from 2007 to 2023 using the term miliarial gout. We describe a case of miliarial gout in a patient with a history of gout, uric acid levels within reference range, and immunocompromised status due to a prior orthotopic heart transplant.

A 59-year-old man presented with innumerable subcutaneous, firm, popcornlike clustered papules on the posterior surfaces of the upper arms and thighs of 5 years’ duration (Figure 1). The involved areas were sometimes painful on manipulation, but the patient was otherwise asymptomatic. His medical history was notable for tophaceous gout of more than 10 years’ duration, calcinosis cutis, adrenal insufficiency, essential hypertension, and an orthotopic heart transplant 2 years prior to the current presentation. At the current presentation he was taking tacrolimus, colchicine, febuxostat, and low-dose prednisone. The patient denied any other skin changes such as ulceration or bullae. In addition to the innumerable subcutaneous papules, he had much larger firm deep nodules bilaterally on the elbow (Figure 2). A complete blood cell count with differential and comprehensive metabolic panel results were within reference range. A 4-mm punch biopsy of the right posterior arm revealed dermal deposits consistent with gout on hematoxylin and eosin staining (Figure 3) but no calcium deposits on von Kossa staining, consistent with miliarial gout.

He was treated with 0.6 mg of colchicine daily, 80 mg of febuxostat twice daily, and 2.5 mg of prednisone daily. Unfortunately, the patient had difficulty affording his medications and therefore experienced frequent flares.
.")
Gout is caused by inflammation that occurs from deposition of monosodium urate crystals in tissues, most commonly occurring in the skin and joints. Gout affects8.3 million individuals and is one of the most common rheumatic diseases of adulthood. The classic presentation of the acute form is monoarticular with associated swelling, erythema, and pain. The chronic form (also known as tophaceous gout) affects soft tissue and presents with smooth or multilobulated nodules.2 Miliarial gout is a rare variant of chronic tophaceous gout, and the diagnosis is based on atypical location, size, and distribution of tophi deposition.
In the updated American College of Rheumatology criteria for gout published in 2020, tophi are defined as draining or chalklike subcutaneous nodules that typically are located in joints, ears, olecranon bursae, finger pads, and tendons.3 The term miliarial gout, which is not universally defined, is used to describe the morphology and distribution of tophi deposition in areas outside of the typical locations defined by the American College of Rheumatology criteria. Miliarial refers to the small, multilobulated, and disseminated presentation of tophi. The involvement of atypical locations distinguishes miliarial gout from chronic tophaceous gout.
The cause of tophi deposition in atypical locations is unknown. It is thought that patients with a history of sustained hyperuricemia have a much greater burden of urate crystal deposition, which can lead to involvement of atypical locations. Our patient had innumerable, discrete, 1- to 5-mm, multilobulated tophi located on the posterior upper arms and thighs even though his uric acid levels were within reference range over the last 5 years.
Miliarial gout is a rare entity.1 In 2007, Shukla et al1 coined the term miliarial gout when reporting the first known presentation of a patient with multiple tiny papules containing a white or creamlike substance scattered on an erythematous base. Other cases of miliarial gout have commonly involved the metacarpophalangeal joints of the hands, knees, abdomen, extensor forearms, and thighs.5 Similarly, our patient had disease involvement of the posterior upper arms and thighs. Furthermore, miliarial gout has been associated with carpal tunnel syndrome; monosodium urate crystal deposition in this space can lead to a clinical diagnosis of this condition.6
With a history of orthotopic heart transplant, it is possible that our patient’s immunocompromised status could have increased his susceptibility for the miliarial form of chronic tophaceous gout. Gout reportedly is the most common inflammatory arthritis in transplant recipients, with the highest prevalence following renal and heart transplantation.7 Pretransplant hyperuricemia is correlated with higher probabilities of posttransplant gout.8 In patients with a heart transplant, hyperuricemia may be due to diuretic use. Additionally, the presence of a gout diagnosis before transplant nearly triples the likelihood of posttransplant gout, which often is more severe than de novo gout, as seen in our patient. Calcineurin inhibitors, including tacrolimus, also can predispose patients to hyperuricemia and more severe forms of gout in the posttransplant phase by limiting fractional urate excretion within the first 3 months of therapy.7 Treatment with oral steroids, as in our patient, also has been identified as a potential inciting factor for the development of cutaneous tophaceous gout.9
Treatment with allopurinol and colchicine has been effective in patients with miliarial gout. Obesity and long-term treatment with furosemide (which our patient was not taking) are considered risk factors for the deposition of dermal and hypodermal urates.9 Our patient had a body mass index of 35 (≥30 indicates obesity); therefore, he also should be counseled on lifestyle modifications for optimal disease control.
- Shukla R, Vender RB, Alhabeeb A, et al. Miliarial gout (a new entity). J Cutan Med Surg. 2007;11:31-34.
- Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 2011;63:3136-3141.
- Neogi T, Jansen, TL, Dalbeth N, et al. 2015 gout classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheumatol. 2015;67:2557-2568.
- Hung TL, Wang WM, Chiang CP. Miliarial gout: a rare presentation of extensive cutaneous tophi. QJM. 2016;109:811-812.
- Mireku KA, Burgy JR, Davis LS. Miliarial gout: a rare clinical presentation. J Am Acad Dermatol. 2014;71:E17-E18.
- Sadovici-Bobeica V, Mazur-Nicorici L, Nicorici A, et al. Chronic miliarial gout associated with carpal tunnel syndrome: a very rare clinical presentation. Eur J Case Rep Intern Med. 2018;5:000926.
- Schwab P, Lipton S, Kerr GS. Rheumatologic sequelae and challenges in organ transplantation. Best Pract Res Clin Rheumatol. 2010;24:329-340.
- Hernández-Molina G, Cachafeiro-Vilar A, Villa AR, et al. Gout in renal allograft recipients according to the pretransplant hyperuricemic status. Transplantation. 2008;86:1543-1547.
- Aguayo RS, Baradad M, Soria X, et al. Unilateral milia‐type intradermal tophi associated with underlying urate subcutaneous deposition: an uncommon cutaneous presentation of gout. Clin Exp Dermatol. 2013;38:622-625.
To the Editor:
Miliarial gout is a rare intradermal manifestation of tophaceous gout. It was first described in 2007 when a patient presented with multiple small papules with a red base containing a white- to cream-colored substance,1 which has rarely been reported,1-6 according to a PubMed search of articles indexed for MEDLINE from 2007 to 2023 using the term miliarial gout. We describe a case of miliarial gout in a patient with a history of gout, uric acid levels within reference range, and immunocompromised status due to a prior orthotopic heart transplant.
A 59-year-old man presented with innumerable subcutaneous, firm, popcornlike clustered papules on the posterior surfaces of the upper arms and thighs of 5 years’ duration (Figure 1). The involved areas were sometimes painful on manipulation, but the patient was otherwise asymptomatic. His medical history was notable for tophaceous gout of more than 10 years’ duration, calcinosis cutis, adrenal insufficiency, essential hypertension, and an orthotopic heart transplant 2 years prior to the current presentation. At the current presentation he was taking tacrolimus, colchicine, febuxostat, and low-dose prednisone. The patient denied any other skin changes such as ulceration or bullae. In addition to the innumerable subcutaneous papules, he had much larger firm deep nodules bilaterally on the elbow (Figure 2). A complete blood cell count with differential and comprehensive metabolic panel results were within reference range. A 4-mm punch biopsy of the right posterior arm revealed dermal deposits consistent with gout on hematoxylin and eosin staining (Figure 3) but no calcium deposits on von Kossa staining, consistent with miliarial gout.
He was treated with 0.6 mg of colchicine daily, 80 mg of febuxostat twice daily, and 2.5 mg of prednisone daily. Unfortunately, the patient had difficulty affording his medications and therefore experienced frequent flares.
Gout is caused by inflammation that occurs from deposition of monosodium urate crystals in tissues, most commonly occurring in the skin and joints. Gout affects8.3 million individuals and is one of the most common rheumatic diseases of adulthood. The classic presentation of the acute form is monoarticular with associated swelling, erythema, and pain. The chronic form (also known as tophaceous gout) affects soft tissue and presents with smooth or multilobulated nodules.2 Miliarial gout is a rare variant of chronic tophaceous gout, and the diagnosis is based on atypical location, size, and distribution of tophi deposition.
In the updated American College of Rheumatology criteria for gout published in 2020, tophi are defined as draining or chalklike subcutaneous nodules that typically are located in joints, ears, olecranon bursae, finger pads, and tendons.3 The term miliarial gout, which is not universally defined, is used to describe the morphology and distribution of tophi deposition in areas outside of the typical locations defined by the American College of Rheumatology criteria. Miliarial refers to the small, multilobulated, and disseminated presentation of tophi. The involvement of atypical locations distinguishes miliarial gout from chronic tophaceous gout.
The cause of tophi deposition in atypical locations is unknown. It is thought that patients with a history of sustained hyperuricemia have a much greater burden of urate crystal deposition, which can lead to involvement of atypical locations. Our patient had innumerable, discrete, 1- to 5-mm, multilobulated tophi located on the posterior upper arms and thighs even though his uric acid levels were within reference range over the last 5 years.
Miliarial gout is a rare entity.1 In 2007, Shukla et al1 coined the term miliarial gout when reporting the first known presentation of a patient with multiple tiny papules containing a white or creamlike substance scattered on an erythematous base. Other cases of miliarial gout have commonly involved the metacarpophalangeal joints of the hands, knees, abdomen, extensor forearms, and thighs.5 Similarly, our patient had disease involvement of the posterior upper arms and thighs. Furthermore, miliarial gout has been associated with carpal tunnel syndrome; monosodium urate crystal deposition in this space can lead to a clinical diagnosis of this condition.6
With a history of orthotopic heart transplant, it is possible that our patient’s immunocompromised status could have increased his susceptibility for the miliarial form of chronic tophaceous gout. Gout reportedly is the most common inflammatory arthritis in transplant recipients, with the highest prevalence following renal and heart transplantation.7 Pretransplant hyperuricemia is correlated with higher probabilities of posttransplant gout.8 In patients with a heart transplant, hyperuricemia may be due to diuretic use. Additionally, the presence of a gout diagnosis before transplant nearly triples the likelihood of posttransplant gout, which often is more severe than de novo gout, as seen in our patient. Calcineurin inhibitors, including tacrolimus, also can predispose patients to hyperuricemia and more severe forms of gout in the posttransplant phase by limiting fractional urate excretion within the first 3 months of therapy.7 Treatment with oral steroids, as in our patient, also has been identified as a potential inciting factor for the development of cutaneous tophaceous gout.9
Treatment with allopurinol and colchicine has been effective in patients with miliarial gout. Obesity and long-term treatment with furosemide (which our patient was not taking) are considered risk factors for the deposition of dermal and hypodermal urates.9 Our patient had a body mass index of 35 (≥30 indicates obesity); therefore, he also should be counseled on lifestyle modifications for optimal disease control.
To the Editor:
Miliarial gout is a rare intradermal manifestation of tophaceous gout. It was first described in 2007 when a patient presented with multiple small papules with a red base containing a white- to cream-colored substance,1 which has rarely been reported,1-6 according to a PubMed search of articles indexed for MEDLINE from 2007 to 2023 using the term miliarial gout. We describe a case of miliarial gout in a patient with a history of gout, uric acid levels within reference range, and immunocompromised status due to a prior orthotopic heart transplant.
A 59-year-old man presented with innumerable subcutaneous, firm, popcornlike clustered papules on the posterior surfaces of the upper arms and thighs of 5 years’ duration (Figure 1). The involved areas were sometimes painful on manipulation, but the patient was otherwise asymptomatic. His medical history was notable for tophaceous gout of more than 10 years’ duration, calcinosis cutis, adrenal insufficiency, essential hypertension, and an orthotopic heart transplant 2 years prior to the current presentation. At the current presentation he was taking tacrolimus, colchicine, febuxostat, and low-dose prednisone. The patient denied any other skin changes such as ulceration or bullae. In addition to the innumerable subcutaneous papules, he had much larger firm deep nodules bilaterally on the elbow (Figure 2). A complete blood cell count with differential and comprehensive metabolic panel results were within reference range. A 4-mm punch biopsy of the right posterior arm revealed dermal deposits consistent with gout on hematoxylin and eosin staining (Figure 3) but no calcium deposits on von Kossa staining, consistent with miliarial gout.
He was treated with 0.6 mg of colchicine daily, 80 mg of febuxostat twice daily, and 2.5 mg of prednisone daily. Unfortunately, the patient had difficulty affording his medications and therefore experienced frequent flares.
Gout is caused by inflammation that occurs from deposition of monosodium urate crystals in tissues, most commonly occurring in the skin and joints. Gout affects8.3 million individuals and is one of the most common rheumatic diseases of adulthood. The classic presentation of the acute form is monoarticular with associated swelling, erythema, and pain. The chronic form (also known as tophaceous gout) affects soft tissue and presents with smooth or multilobulated nodules.2 Miliarial gout is a rare variant of chronic tophaceous gout, and the diagnosis is based on atypical location, size, and distribution of tophi deposition.
In the updated American College of Rheumatology criteria for gout published in 2020, tophi are defined as draining or chalklike subcutaneous nodules that typically are located in joints, ears, olecranon bursae, finger pads, and tendons.3 The term miliarial gout, which is not universally defined, is used to describe the morphology and distribution of tophi deposition in areas outside of the typical locations defined by the American College of Rheumatology criteria. Miliarial refers to the small, multilobulated, and disseminated presentation of tophi. The involvement of atypical locations distinguishes miliarial gout from chronic tophaceous gout.
The cause of tophi deposition in atypical locations is unknown. It is thought that patients with a history of sustained hyperuricemia have a much greater burden of urate crystal deposition, which can lead to involvement of atypical locations. Our patient had innumerable, discrete, 1- to 5-mm, multilobulated tophi located on the posterior upper arms and thighs even though his uric acid levels were within reference range over the last 5 years.
Miliarial gout is a rare entity.1 In 2007, Shukla et al1 coined the term miliarial gout when reporting the first known presentation of a patient with multiple tiny papules containing a white or creamlike substance scattered on an erythematous base. Other cases of miliarial gout have commonly involved the metacarpophalangeal joints of the hands, knees, abdomen, extensor forearms, and thighs.5 Similarly, our patient had disease involvement of the posterior upper arms and thighs. Furthermore, miliarial gout has been associated with carpal tunnel syndrome; monosodium urate crystal deposition in this space can lead to a clinical diagnosis of this condition.6
With a history of orthotopic heart transplant, it is possible that our patient’s immunocompromised status could have increased his susceptibility for the miliarial form of chronic tophaceous gout. Gout reportedly is the most common inflammatory arthritis in transplant recipients, with the highest prevalence following renal and heart transplantation.7 Pretransplant hyperuricemia is correlated with higher probabilities of posttransplant gout.8 In patients with a heart transplant, hyperuricemia may be due to diuretic use. Additionally, the presence of a gout diagnosis before transplant nearly triples the likelihood of posttransplant gout, which often is more severe than de novo gout, as seen in our patient. Calcineurin inhibitors, including tacrolimus, also can predispose patients to hyperuricemia and more severe forms of gout in the posttransplant phase by limiting fractional urate excretion within the first 3 months of therapy.7 Treatment with oral steroids, as in our patient, also has been identified as a potential inciting factor for the development of cutaneous tophaceous gout.9
Treatment with allopurinol and colchicine has been effective in patients with miliarial gout. Obesity and long-term treatment with furosemide (which our patient was not taking) are considered risk factors for the deposition of dermal and hypodermal urates.9 Our patient had a body mass index of 35 (≥30 indicates obesity); therefore, he also should be counseled on lifestyle modifications for optimal disease control.
- Shukla R, Vender RB, Alhabeeb A, et al. Miliarial gout (a new entity). J Cutan Med Surg. 2007;11:31-34.
- Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 2011;63:3136-3141.
- Neogi T, Jansen, TL, Dalbeth N, et al. 2015 gout classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheumatol. 2015;67:2557-2568.
- Hung TL, Wang WM, Chiang CP. Miliarial gout: a rare presentation of extensive cutaneous tophi. QJM. 2016;109:811-812.
- Mireku KA, Burgy JR, Davis LS. Miliarial gout: a rare clinical presentation. J Am Acad Dermatol. 2014;71:E17-E18.
- Sadovici-Bobeica V, Mazur-Nicorici L, Nicorici A, et al. Chronic miliarial gout associated with carpal tunnel syndrome: a very rare clinical presentation. Eur J Case Rep Intern Med. 2018;5:000926.
- Schwab P, Lipton S, Kerr GS. Rheumatologic sequelae and challenges in organ transplantation. Best Pract Res Clin Rheumatol. 2010;24:329-340.
- Hernández-Molina G, Cachafeiro-Vilar A, Villa AR, et al. Gout in renal allograft recipients according to the pretransplant hyperuricemic status. Transplantation. 2008;86:1543-1547.
- Aguayo RS, Baradad M, Soria X, et al. Unilateral milia‐type intradermal tophi associated with underlying urate subcutaneous deposition: an uncommon cutaneous presentation of gout. Clin Exp Dermatol. 2013;38:622-625.
- Shukla R, Vender RB, Alhabeeb A, et al. Miliarial gout (a new entity). J Cutan Med Surg. 2007;11:31-34.
- Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 2011;63:3136-3141.
- Neogi T, Jansen, TL, Dalbeth N, et al. 2015 gout classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheumatol. 2015;67:2557-2568.
- Hung TL, Wang WM, Chiang CP. Miliarial gout: a rare presentation of extensive cutaneous tophi. QJM. 2016;109:811-812.
- Mireku KA, Burgy JR, Davis LS. Miliarial gout: a rare clinical presentation. J Am Acad Dermatol. 2014;71:E17-E18.
- Sadovici-Bobeica V, Mazur-Nicorici L, Nicorici A, et al. Chronic miliarial gout associated with carpal tunnel syndrome: a very rare clinical presentation. Eur J Case Rep Intern Med. 2018;5:000926.
- Schwab P, Lipton S, Kerr GS. Rheumatologic sequelae and challenges in organ transplantation. Best Pract Res Clin Rheumatol. 2010;24:329-340.
- Hernández-Molina G, Cachafeiro-Vilar A, Villa AR, et al. Gout in renal allograft recipients according to the pretransplant hyperuricemic status. Transplantation. 2008;86:1543-1547.
- Aguayo RS, Baradad M, Soria X, et al. Unilateral milia‐type intradermal tophi associated with underlying urate subcutaneous deposition: an uncommon cutaneous presentation of gout. Clin Exp Dermatol. 2013;38:622-625.
Practice Points
- Miliarial gout is a rare intradermal manifestation of tophaceous gout and often presents as multiple small papules containing a white- to cream-colored substance.
- Immunocompromised status may be a risk factor for miliarial gout, especially in patients with a history of gout or hyperuricemia.
- Effective treatments for miliarial gout include allopurinol and colchicine.

Subcutaneous Nodule on the Postauricular Neck
The Diagnosis: Pleomorphic Lipoma
Pleomorphic lipoma is a rare, benign, adipocytic neoplasm that presents in the subcutaneous tissues of the upper shoulder, back, or neck. It predominantly affects men aged 50 to 70 years. Most lesions are situated in the subcutaneous tissues; few cases of intramuscular and retroperitoneal tumors have been reported.1 Clinically, pleomorphic lipomas present as painless, well-circumscribed lesions of the subcutaneous tissue that often resemble a lipoma or occasionally may be mistaken for liposarcoma. Histopathologic examination of ordinary lipomas reveals uniform mature adipocytes. However, pleomorphic lipomas consist of a mixture of multinucleated floretlike giant cells, variable-sized adipocytes, and fibrous tissue (ropy collagen bundles) with some myxoid and spindled areas.1,2 The most characteristic histologic feature of pleomorphic lipoma is multinucleated floretlike giant cells. The nuclei of these giant cells appear hyperchromatic, enlarged, and disposed to the periphery of the cell in a circular pattern. Additionally, tumors frequently contain excess mature dense collagen bundles that are strongly refractile in polarized light. Numerous mast cells are present. Atypical lipoblasts and capillary networks commonly are not visible in pleomorphic lipoma.3 The spindle cells express CD34 on immunohistochemistry. Loss of Rb-1 expression is typical.4
Dermatofibrosarcoma protuberans is a slow-growing soft tissue sarcoma that commonly begins as a pink or violet plaque on the trunk or upper limbs. Involvement of the head or neck accounts for only 10% to 15% of cases.5 This tumor has low metastatic potential but is highly infiltrative of surrounding tissues. It is associated with a translocation between chromosomes 22 and 17, leading to the fusion of the platelet-derived growth factor subunit β, PDGFB, and collagen type 1α1, COL1A1, genes.5 Clinically, patients often report that the lesion was present for several years prior to presentation with general stability in size and shape. Eventually, untreated lesions progress to become nodules or tumors and may even bleed or ulcerate. Histology reveals a storiform spindle cell proliferation throughout the dermis with infiltration into subcutaneous fat, commonly appearing in a honeycomblike pattern (Figure 1). Numerous histologic variants exist, including myxoid, sclerosing, pigmented (Bednar tumor), myoid, atrophic, or fibrosarcomatous dermatofibrosarcoma protuberans, as well as a giant cell fibroblastoma variant.6 These tumor subtypes can exist independently or in association with one another, creating hybrid lesions that can closely mimic other entities such as pleomorphic lipoma. The spindle cells stain positively for CD34. Treatment of these tumors involves complete surgical excision or Mohs micrographic surgery; however, recurrence is common for tumors involving the head or neck.5
.")
Superficial angiomyxoma is a slow-growing papule that most commonly appears on the trunk, head, or neck in middle-aged adults. Occasionally, patients with Carney complex also can develop lesions on the external ear or breast.7 Histologically, superficial angiomyxoma is a hypocellular tumor characterized by abundant myxoid stroma, thin blood vessels, and small spindled and stellate cells with minimal cytoplasm (Figure 2).8 Superficial angiomyxoma and pleomorphic lipoma present differently on histology; superficial angiomyxoma is not associated with nuclear atypia or pleomorphism, whereas pleomorphic lipoma characteristically contains multinucleated floretlike giant cells and pleomorphism. Frequently, there also is loss of normal PRKAR1A gene expression, which is responsible for protein kinase A regulatory subunit 1-alpha expression.8
.")
Multinucleate cell angiohistiocytoma is a rare benign proliferation that presents with numerous red-violet asymptomatic papules that commonly appear on the upper and lower extremities of women aged 40 to 70 years. Lesions feature both a fibrohistiocytic and vascular component.9 Histologic examination commonly shows multinucleated cells with angular outlining in the superficial dermis accompanied by fibrosis and ectatic small-caliber vessels (Figure 3). Although both pleomorphic lipoma and multinucleate cell angiohistiocytoma have similar-appearing multinucleated giant cells, the latter has a proliferation of narrow vessels in thick collagen bundles and lacks an adipocytic component, which distinguishes it from the former.10 Multinucleate cell angiohistiocytoma also is characterized by a substantial number of factor XIIIa–positive fibrohistiocytic interstitial cells and vascular hyperplasia.9
.")
Nodular fasciitis is a benign lesion involving the rapid proliferation of myofibroblasts and fibroblasts in the subcutaneous tissue and most commonly is encountered on the extremities or head and neck regions. Many cases appear at sites of prior trauma, especially in patients aged 20 to 40 years. However, in infants and children the lesions typically are found in the head and neck regions.11 Clinically, lesions present as subcutaneous nodules. Histology reveals an infiltrative and poorly circumscribed proliferation of spindled myofibroblasts associated with myxoid stroma and dense collagen depositions. The spindled cells are loosely associated, rendering a tissue culture–like appearance (Figure 4). It also is common to see erythrocyte extravasation adjacent to myxoid stroma.11 Positive stains include vimentin, smooth muscle actin, and CD68, though immunohistochemistry often is not necessary for diagnosis.12 There often is abundant mitotic activity in nodular fasciitis, especially in early lesions, and the differential diagnosis includes sarcoma. Although nodular fasciitis is mitotically active, it does not show atypical mitotic figures. Nodular fasciitis commonly harbors a gene translocation of the MYH9 gene’s promoter region to the USP6 gene’s coding region.13
.")
- Sakhadeo U, Mundhe R, DeSouza MA, et al. Pleomorphic lipoma: a gentle giant of pathology. J Cytol. 2015;32:201-203. doi:10.4103 /0970-9371.168904
- Shmookler BM, Enzinger FM. Pleomorphic lipoma: a benign tumor simulating liposarcoma. a clinicopathologic analysis of 48 cases. Cancer. 1981;47:126-133.
- Azzopardi JG, Iocco J, Salm R. Pleomorphic lipoma: a tumour simulating liposarcoma. Histopathology. 1983;7:511-523. doi:10.1111/j.1365-2559.1983.tb02264.x
- Jäger M, Winkelmann R, Eichler K, et al. Pleomorphic lipoma. J Dtsch Dermatol Ges. 2018;16:208-210. doi:10.1111/ddg.13422
- Allen A, Ahn C, Sangüeza OP. Dermatofibrosarcoma protuberans. Dermatol Clin. 2019;37:483-488. doi:10.1016/j.det.2019.05.006
- Socoliuc C, Zurac S, Andrei R, et al. Multiple histological subtypes of dermatofibrosarcoma protuberans occurring in the same tumor. Rom J Intern Med. 2015;53:79-88. doi:10.1515/rjim-2015-0011
- Abarzúa-Araya A, Lallas A, Piana S, et al. Superficial angiomyxoma of the skin. Dermatol Pract Concept. 2016;6:47-49. doi:10.5826 /dpc.0603a09
- Hornick J. Practical Soft Tissue Pathology A Diagnostic Approach. 2nd ed. Elsevier Health Sciences; 2017.
- Rato M, Monteiro AF, Parente J, et al. Case for diagnosis. multinucleated cell angiohistiocytoma. An Bras Dermatol. 2018;93:291-293. doi:10.1590 /abd1806-4841.20186821
- Grgurich E, Quinn K, Oram C, et al. Multinucleate cell angiohistiocytoma: case report and literature review. J Cutan Pathol. 2019;46:59-61. doi:10.1111/cup.13361
- Zuber TJ, Finley JL. Nodular fasciitis. South Med J. 1994;87:842-844. doi:10.1097/00007611-199408000-00020
- Yver CM, Husson MA, Friedman O. Pathology clinic: nodular fasciitis involving the external ear [published online March 18, 2021]. Ear Nose Throat J. doi:10.1177/01455613211001958
- Erickson-Johnson M, Chou M, Evers B, et al. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab Invest. 2011;91:1427-1433. https://doi.org/10.1038 /labinvest.2011.118
The Diagnosis: Pleomorphic Lipoma
Pleomorphic lipoma is a rare, benign, adipocytic neoplasm that presents in the subcutaneous tissues of the upper shoulder, back, or neck. It predominantly affects men aged 50 to 70 years. Most lesions are situated in the subcutaneous tissues; few cases of intramuscular and retroperitoneal tumors have been reported.1 Clinically, pleomorphic lipomas present as painless, well-circumscribed lesions of the subcutaneous tissue that often resemble a lipoma or occasionally may be mistaken for liposarcoma. Histopathologic examination of ordinary lipomas reveals uniform mature adipocytes. However, pleomorphic lipomas consist of a mixture of multinucleated floretlike giant cells, variable-sized adipocytes, and fibrous tissue (ropy collagen bundles) with some myxoid and spindled areas.1,2 The most characteristic histologic feature of pleomorphic lipoma is multinucleated floretlike giant cells. The nuclei of these giant cells appear hyperchromatic, enlarged, and disposed to the periphery of the cell in a circular pattern. Additionally, tumors frequently contain excess mature dense collagen bundles that are strongly refractile in polarized light. Numerous mast cells are present. Atypical lipoblasts and capillary networks commonly are not visible in pleomorphic lipoma.3 The spindle cells express CD34 on immunohistochemistry. Loss of Rb-1 expression is typical.4
Dermatofibrosarcoma protuberans is a slow-growing soft tissue sarcoma that commonly begins as a pink or violet plaque on the trunk or upper limbs. Involvement of the head or neck accounts for only 10% to 15% of cases.5 This tumor has low metastatic potential but is highly infiltrative of surrounding tissues. It is associated with a translocation between chromosomes 22 and 17, leading to the fusion of the platelet-derived growth factor subunit β, PDGFB, and collagen type 1α1, COL1A1, genes.5 Clinically, patients often report that the lesion was present for several years prior to presentation with general stability in size and shape. Eventually, untreated lesions progress to become nodules or tumors and may even bleed or ulcerate. Histology reveals a storiform spindle cell proliferation throughout the dermis with infiltration into subcutaneous fat, commonly appearing in a honeycomblike pattern (Figure 1). Numerous histologic variants exist, including myxoid, sclerosing, pigmented (Bednar tumor), myoid, atrophic, or fibrosarcomatous dermatofibrosarcoma protuberans, as well as a giant cell fibroblastoma variant.6 These tumor subtypes can exist independently or in association with one another, creating hybrid lesions that can closely mimic other entities such as pleomorphic lipoma. The spindle cells stain positively for CD34. Treatment of these tumors involves complete surgical excision or Mohs micrographic surgery; however, recurrence is common for tumors involving the head or neck.5
Superficial angiomyxoma is a slow-growing papule that most commonly appears on the trunk, head, or neck in middle-aged adults. Occasionally, patients with Carney complex also can develop lesions on the external ear or breast.7 Histologically, superficial angiomyxoma is a hypocellular tumor characterized by abundant myxoid stroma, thin blood vessels, and small spindled and stellate cells with minimal cytoplasm (Figure 2).8 Superficial angiomyxoma and pleomorphic lipoma present differently on histology; superficial angiomyxoma is not associated with nuclear atypia or pleomorphism, whereas pleomorphic lipoma characteristically contains multinucleated floretlike giant cells and pleomorphism. Frequently, there also is loss of normal PRKAR1A gene expression, which is responsible for protein kinase A regulatory subunit 1-alpha expression.8
Multinucleate cell angiohistiocytoma is a rare benign proliferation that presents with numerous red-violet asymptomatic papules that commonly appear on the upper and lower extremities of women aged 40 to 70 years. Lesions feature both a fibrohistiocytic and vascular component.9 Histologic examination commonly shows multinucleated cells with angular outlining in the superficial dermis accompanied by fibrosis and ectatic small-caliber vessels (Figure 3). Although both pleomorphic lipoma and multinucleate cell angiohistiocytoma have similar-appearing multinucleated giant cells, the latter has a proliferation of narrow vessels in thick collagen bundles and lacks an adipocytic component, which distinguishes it from the former.10 Multinucleate cell angiohistiocytoma also is characterized by a substantial number of factor XIIIa–positive fibrohistiocytic interstitial cells and vascular hyperplasia.9
Nodular fasciitis is a benign lesion involving the rapid proliferation of myofibroblasts and fibroblasts in the subcutaneous tissue and most commonly is encountered on the extremities or head and neck regions. Many cases appear at sites of prior trauma, especially in patients aged 20 to 40 years. However, in infants and children the lesions typically are found in the head and neck regions.11 Clinically, lesions present as subcutaneous nodules. Histology reveals an infiltrative and poorly circumscribed proliferation of spindled myofibroblasts associated with myxoid stroma and dense collagen depositions. The spindled cells are loosely associated, rendering a tissue culture–like appearance (Figure 4). It also is common to see erythrocyte extravasation adjacent to myxoid stroma.11 Positive stains include vimentin, smooth muscle actin, and CD68, though immunohistochemistry often is not necessary for diagnosis.12 There often is abundant mitotic activity in nodular fasciitis, especially in early lesions, and the differential diagnosis includes sarcoma. Although nodular fasciitis is mitotically active, it does not show atypical mitotic figures. Nodular fasciitis commonly harbors a gene translocation of the MYH9 gene’s promoter region to the USP6 gene’s coding region.13
The Diagnosis: Pleomorphic Lipoma
Pleomorphic lipoma is a rare, benign, adipocytic neoplasm that presents in the subcutaneous tissues of the upper shoulder, back, or neck. It predominantly affects men aged 50 to 70 years. Most lesions are situated in the subcutaneous tissues; few cases of intramuscular and retroperitoneal tumors have been reported.1 Clinically, pleomorphic lipomas present as painless, well-circumscribed lesions of the subcutaneous tissue that often resemble a lipoma or occasionally may be mistaken for liposarcoma. Histopathologic examination of ordinary lipomas reveals uniform mature adipocytes. However, pleomorphic lipomas consist of a mixture of multinucleated floretlike giant cells, variable-sized adipocytes, and fibrous tissue (ropy collagen bundles) with some myxoid and spindled areas.1,2 The most characteristic histologic feature of pleomorphic lipoma is multinucleated floretlike giant cells. The nuclei of these giant cells appear hyperchromatic, enlarged, and disposed to the periphery of the cell in a circular pattern. Additionally, tumors frequently contain excess mature dense collagen bundles that are strongly refractile in polarized light. Numerous mast cells are present. Atypical lipoblasts and capillary networks commonly are not visible in pleomorphic lipoma.3 The spindle cells express CD34 on immunohistochemistry. Loss of Rb-1 expression is typical.4
Dermatofibrosarcoma protuberans is a slow-growing soft tissue sarcoma that commonly begins as a pink or violet plaque on the trunk or upper limbs. Involvement of the head or neck accounts for only 10% to 15% of cases.5 This tumor has low metastatic potential but is highly infiltrative of surrounding tissues. It is associated with a translocation between chromosomes 22 and 17, leading to the fusion of the platelet-derived growth factor subunit β, PDGFB, and collagen type 1α1, COL1A1, genes.5 Clinically, patients often report that the lesion was present for several years prior to presentation with general stability in size and shape. Eventually, untreated lesions progress to become nodules or tumors and may even bleed or ulcerate. Histology reveals a storiform spindle cell proliferation throughout the dermis with infiltration into subcutaneous fat, commonly appearing in a honeycomblike pattern (Figure 1). Numerous histologic variants exist, including myxoid, sclerosing, pigmented (Bednar tumor), myoid, atrophic, or fibrosarcomatous dermatofibrosarcoma protuberans, as well as a giant cell fibroblastoma variant.6 These tumor subtypes can exist independently or in association with one another, creating hybrid lesions that can closely mimic other entities such as pleomorphic lipoma. The spindle cells stain positively for CD34. Treatment of these tumors involves complete surgical excision or Mohs micrographic surgery; however, recurrence is common for tumors involving the head or neck.5
Superficial angiomyxoma is a slow-growing papule that most commonly appears on the trunk, head, or neck in middle-aged adults. Occasionally, patients with Carney complex also can develop lesions on the external ear or breast.7 Histologically, superficial angiomyxoma is a hypocellular tumor characterized by abundant myxoid stroma, thin blood vessels, and small spindled and stellate cells with minimal cytoplasm (Figure 2).8 Superficial angiomyxoma and pleomorphic lipoma present differently on histology; superficial angiomyxoma is not associated with nuclear atypia or pleomorphism, whereas pleomorphic lipoma characteristically contains multinucleated floretlike giant cells and pleomorphism. Frequently, there also is loss of normal PRKAR1A gene expression, which is responsible for protein kinase A regulatory subunit 1-alpha expression.8
Multinucleate cell angiohistiocytoma is a rare benign proliferation that presents with numerous red-violet asymptomatic papules that commonly appear on the upper and lower extremities of women aged 40 to 70 years. Lesions feature both a fibrohistiocytic and vascular component.9 Histologic examination commonly shows multinucleated cells with angular outlining in the superficial dermis accompanied by fibrosis and ectatic small-caliber vessels (Figure 3). Although both pleomorphic lipoma and multinucleate cell angiohistiocytoma have similar-appearing multinucleated giant cells, the latter has a proliferation of narrow vessels in thick collagen bundles and lacks an adipocytic component, which distinguishes it from the former.10 Multinucleate cell angiohistiocytoma also is characterized by a substantial number of factor XIIIa–positive fibrohistiocytic interstitial cells and vascular hyperplasia.9
Nodular fasciitis is a benign lesion involving the rapid proliferation of myofibroblasts and fibroblasts in the subcutaneous tissue and most commonly is encountered on the extremities or head and neck regions. Many cases appear at sites of prior trauma, especially in patients aged 20 to 40 years. However, in infants and children the lesions typically are found in the head and neck regions.11 Clinically, lesions present as subcutaneous nodules. Histology reveals an infiltrative and poorly circumscribed proliferation of spindled myofibroblasts associated with myxoid stroma and dense collagen depositions. The spindled cells are loosely associated, rendering a tissue culture–like appearance (Figure 4). It also is common to see erythrocyte extravasation adjacent to myxoid stroma.11 Positive stains include vimentin, smooth muscle actin, and CD68, though immunohistochemistry often is not necessary for diagnosis.12 There often is abundant mitotic activity in nodular fasciitis, especially in early lesions, and the differential diagnosis includes sarcoma. Although nodular fasciitis is mitotically active, it does not show atypical mitotic figures. Nodular fasciitis commonly harbors a gene translocation of the MYH9 gene’s promoter region to the USP6 gene’s coding region.13
- Sakhadeo U, Mundhe R, DeSouza MA, et al. Pleomorphic lipoma: a gentle giant of pathology. J Cytol. 2015;32:201-203. doi:10.4103 /0970-9371.168904
- Shmookler BM, Enzinger FM. Pleomorphic lipoma: a benign tumor simulating liposarcoma. a clinicopathologic analysis of 48 cases. Cancer. 1981;47:126-133.
- Azzopardi JG, Iocco J, Salm R. Pleomorphic lipoma: a tumour simulating liposarcoma. Histopathology. 1983;7:511-523. doi:10.1111/j.1365-2559.1983.tb02264.x
- Jäger M, Winkelmann R, Eichler K, et al. Pleomorphic lipoma. J Dtsch Dermatol Ges. 2018;16:208-210. doi:10.1111/ddg.13422
- Allen A, Ahn C, Sangüeza OP. Dermatofibrosarcoma protuberans. Dermatol Clin. 2019;37:483-488. doi:10.1016/j.det.2019.05.006
- Socoliuc C, Zurac S, Andrei R, et al. Multiple histological subtypes of dermatofibrosarcoma protuberans occurring in the same tumor. Rom J Intern Med. 2015;53:79-88. doi:10.1515/rjim-2015-0011
- Abarzúa-Araya A, Lallas A, Piana S, et al. Superficial angiomyxoma of the skin. Dermatol Pract Concept. 2016;6:47-49. doi:10.5826 /dpc.0603a09
- Hornick J. Practical Soft Tissue Pathology A Diagnostic Approach. 2nd ed. Elsevier Health Sciences; 2017.
- Rato M, Monteiro AF, Parente J, et al. Case for diagnosis. multinucleated cell angiohistiocytoma. An Bras Dermatol. 2018;93:291-293. doi:10.1590 /abd1806-4841.20186821
- Grgurich E, Quinn K, Oram C, et al. Multinucleate cell angiohistiocytoma: case report and literature review. J Cutan Pathol. 2019;46:59-61. doi:10.1111/cup.13361
- Zuber TJ, Finley JL. Nodular fasciitis. South Med J. 1994;87:842-844. doi:10.1097/00007611-199408000-00020
- Yver CM, Husson MA, Friedman O. Pathology clinic: nodular fasciitis involving the external ear [published online March 18, 2021]. Ear Nose Throat J. doi:10.1177/01455613211001958
- Erickson-Johnson M, Chou M, Evers B, et al. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab Invest. 2011;91:1427-1433. https://doi.org/10.1038 /labinvest.2011.118
- Sakhadeo U, Mundhe R, DeSouza MA, et al. Pleomorphic lipoma: a gentle giant of pathology. J Cytol. 2015;32:201-203. doi:10.4103 /0970-9371.168904
- Shmookler BM, Enzinger FM. Pleomorphic lipoma: a benign tumor simulating liposarcoma. a clinicopathologic analysis of 48 cases. Cancer. 1981;47:126-133.
- Azzopardi JG, Iocco J, Salm R. Pleomorphic lipoma: a tumour simulating liposarcoma. Histopathology. 1983;7:511-523. doi:10.1111/j.1365-2559.1983.tb02264.x
- Jäger M, Winkelmann R, Eichler K, et al. Pleomorphic lipoma. J Dtsch Dermatol Ges. 2018;16:208-210. doi:10.1111/ddg.13422
- Allen A, Ahn C, Sangüeza OP. Dermatofibrosarcoma protuberans. Dermatol Clin. 2019;37:483-488. doi:10.1016/j.det.2019.05.006
- Socoliuc C, Zurac S, Andrei R, et al. Multiple histological subtypes of dermatofibrosarcoma protuberans occurring in the same tumor. Rom J Intern Med. 2015;53:79-88. doi:10.1515/rjim-2015-0011
- Abarzúa-Araya A, Lallas A, Piana S, et al. Superficial angiomyxoma of the skin. Dermatol Pract Concept. 2016;6:47-49. doi:10.5826 /dpc.0603a09
- Hornick J. Practical Soft Tissue Pathology A Diagnostic Approach. 2nd ed. Elsevier Health Sciences; 2017.
- Rato M, Monteiro AF, Parente J, et al. Case for diagnosis. multinucleated cell angiohistiocytoma. An Bras Dermatol. 2018;93:291-293. doi:10.1590 /abd1806-4841.20186821
- Grgurich E, Quinn K, Oram C, et al. Multinucleate cell angiohistiocytoma: case report and literature review. J Cutan Pathol. 2019;46:59-61. doi:10.1111/cup.13361
- Zuber TJ, Finley JL. Nodular fasciitis. South Med J. 1994;87:842-844. doi:10.1097/00007611-199408000-00020
- Yver CM, Husson MA, Friedman O. Pathology clinic: nodular fasciitis involving the external ear [published online March 18, 2021]. Ear Nose Throat J. doi:10.1177/01455613211001958
- Erickson-Johnson M, Chou M, Evers B, et al. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab Invest. 2011;91:1427-1433. https://doi.org/10.1038 /labinvest.2011.118
An otherwise healthy 56-year-old man with a family history of lymphoma presented with a raised lesion on the postauricular neck. He first noticed the nodule 3 months prior and was unsure if it was still getting larger. It was predominantly asymptomatic. Physical examination revealed a 1.5×1.5-cm, mobile, subcutaneous nodule. An incisional biopsy was performed and submitted for histologic evaluation.



Collision Course of a Basal Cell Carcinoma and Apocrine Hidrocystoma on the Scalp
To the Editor:
A collision tumor is the coexistence of 2 discrete tumors in the same neoplasm, possibly comprising a malignant tumor and a benign tumor, and thereby complicating appropriate diagnosis and treatment. We present a case of a basal cell carcinoma (BCC) of the scalp that was later found to be in collision with an apocrine hidrocystoma that might have arisen from a nevus sebaceus. Although rare, BCC can coexist with apocrine hidrocystoma. Jayaprakasam and Rene1 reported a case of a collision tumor containing BCC and hidrocystoma on the eyelid.1 We present a case of a BCC on the scalp that was later found to be in collision with an apocrine hidrocystoma that possibly arose from a nevus sebaceus.
.")
A 92-year-old Black woman with a biopsy-confirmed primary BCC of the left parietal scalp presented for Mohs micrographic surgery. The pathology report from an outside facility was reviewed. The initial diagnosis had been made with 2 punch biopsies from separate areas of the large nodule—one consistent with nodular and pigmented BCC (Figure 1), and the other revealed nodular ulcerated BCC. Physical examination prior to Mohs surgery revealed a mobile, flesh-colored, 6.2×6.0-cm nodule with minimal overlying hair on the left parietal scalp (Figure 2). During stage-I processing by the histopathology laboratory, large cystic structures were encountered; en face frozen sections showed a cystic tumor. Excised tissue was submitted for permanent processing to aid in diagnosis; the initial diagnostic biopsy slides were requested from the outside facility for review.

The initial diagnostic biopsy slides were reviewed and found to be consistent with nodular and pigmented BCC, as previously reported. Findings from hematoxylin and eosin staining of tissue obtained from Mohs sections were consistent with a combined neoplasm comprising BCC (Figure 3A) and apocrine hidrocystoma (Figure 3B). In addition, one section was characterized by acanthosis, papillomatosis, and sebaceous glands—similar to findings that are seen in a nevus sebaceus (Figure 3C).
")
The BCC was cleared after stage I; the final wound size was 7×6.6 cm. Although benign apocrine hidrocystoma was still evident at the margin, further excision was not performed at the request of the patient and her family. Partial primary wound closure was performed with pulley sutures. A xenograft was placed over the unclosed central portion. The wound was permitted to heal by second intention.
The clinical differential diagnosis of a scalp nodule includes a pilar cyst, BCC, squamous cell carcinoma, melanoma, cutaneous metastasis, adnexal tumor, atypical fibroxanthoma, and collision tumor. A collision tumor—the association of 2 or more benign or malignant neoplasms—represents a well-known pitfall in making a correct clinical and pathologic diagnosis.2 Many theories have been proposed to explain the pathophysiology of collision tumors. Some authors have speculated that they arise from involvement of related cell types.1 Other theories include induction by cytokines and growth factors secreted from one tumor that provides an ideal environment for proliferation of other cell types, a field cancerization effect of sun-damaged skin, or a coincidence.2
In our case, it is possible that the 2 tumors arose from a nevus sebaceus. One retrospective study of 706 cases of nevus sebaceus (707 specimens) found that 22.5% of cases developed secondary proliferation; of those cases, 18.9% were benign.3 Additionally, in 4.2% of cases of nevus sebaceus, proliferation of 2 or more tumors developed. The most common malignant neoplasm to develop from nevus sebaceus was BCC, followed by squamous cell carcinoma and sebaceous carcinoma. The most common benign neoplasm to develop from nevus sebaceus was trichoblastoma, followed by syringocystadenoma papilliferum.3
Our case highlights the possibility of a sampling error when performing a biopsy of any large neoplasm. Additionally, Mohs surgeons should maintain high clinical suspicion for collision tumors when encountering a large tumor with pathology inconsistent with the original biopsy. Apocrine hidrocystoma should be considered in the differential diagnosis of a large cystic mass of the scalp. Also, it is important to recognize that malignant lesions, such as BCC, can coexist with another benign tumor. Basal cell carcinoma is rare in Black patients, supporting our belief that our patient’s tumors arose from a nevus sebaceus.
It also is important for Mohs surgeons to consider any potential discrepancy between the initial pathology report and Mohs intraoperative pathology that can impact diagnosis, the aggressiveness of the tumors identified, and how such aggressiveness may affect management options.4,5 Some dermatology practices request biopsy slides from patients who are referred for Mohs micrographic surgery for internal review by a dermatopathologist before surgery is performed; however, this protocol requires additional time and adds costs for the overall health care system.4 One study found that internal review of outside biopsy slides resulted in a change in diagnosis in 2.2% of patients (N=3345)—affecting management in 61% of cases in which the diagnosis was changed.4 Another study (N=163) found that the reported aggressiveness of 50.5% of nonmelanoma cases in an initial biopsy report was changed during Mohs micrographic surgery.5 Mohs surgeons should be aware that discrepancies can occur, and if a discrepancy is discovered, the procedure may be paused until the initial biopsy slide is reviewed and further information is collected.
- Jayaprakasam A, Rene C. A benign or malignant eyelid lump—can you tell? an unusual collision tumour highlighting the difficulty differentiating a hidrocystoma from a basal cell carcinoma. BMJ Case Reports. 2012;2012:bcr1220115307. doi:10.1136/bcr.12.2011.5307
- Miteva M, Herschthal D, Ricotti C, et al. A rare case of a cutaneous squamomelanocytic tumor: revisiting the histogenesis of combined neoplasms. Am J Dermatopathol. 2009;31:599-603. doi:10.1097/DAD.0b013e3181a88116
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337. doi:10.1016/j.jaad.2013.10.004
- Butler ST, Youker SR, Mandrell J, et al. The importance of reviewing pathology specimens before Mohs surgery. Dermatol Surg. 2009;35:407-412. doi:10.1111/j.1524-4725.2008.01056.x
- Stiegel E, Lam C, Schowalter M, et al. Correlation between original biopsy pathology and Mohs intraoperative pathology. Dermatol Surg. 2018;44:193-197. doi:10.1097/DSS.0000000000001276
To the Editor:
A collision tumor is the coexistence of 2 discrete tumors in the same neoplasm, possibly comprising a malignant tumor and a benign tumor, and thereby complicating appropriate diagnosis and treatment. We present a case of a basal cell carcinoma (BCC) of the scalp that was later found to be in collision with an apocrine hidrocystoma that might have arisen from a nevus sebaceus. Although rare, BCC can coexist with apocrine hidrocystoma. Jayaprakasam and Rene1 reported a case of a collision tumor containing BCC and hidrocystoma on the eyelid.1 We present a case of a BCC on the scalp that was later found to be in collision with an apocrine hidrocystoma that possibly arose from a nevus sebaceus.
A 92-year-old Black woman with a biopsy-confirmed primary BCC of the left parietal scalp presented for Mohs micrographic surgery. The pathology report from an outside facility was reviewed. The initial diagnosis had been made with 2 punch biopsies from separate areas of the large nodule—one consistent with nodular and pigmented BCC (Figure 1), and the other revealed nodular ulcerated BCC. Physical examination prior to Mohs surgery revealed a mobile, flesh-colored, 6.2×6.0-cm nodule with minimal overlying hair on the left parietal scalp (Figure 2). During stage-I processing by the histopathology laboratory, large cystic structures were encountered; en face frozen sections showed a cystic tumor. Excised tissue was submitted for permanent processing to aid in diagnosis; the initial diagnostic biopsy slides were requested from the outside facility for review.
The initial diagnostic biopsy slides were reviewed and found to be consistent with nodular and pigmented BCC, as previously reported. Findings from hematoxylin and eosin staining of tissue obtained from Mohs sections were consistent with a combined neoplasm comprising BCC (Figure 3A) and apocrine hidrocystoma (Figure 3B). In addition, one section was characterized by acanthosis, papillomatosis, and sebaceous glands—similar to findings that are seen in a nevus sebaceus (Figure 3C).
The BCC was cleared after stage I; the final wound size was 7×6.6 cm. Although benign apocrine hidrocystoma was still evident at the margin, further excision was not performed at the request of the patient and her family. Partial primary wound closure was performed with pulley sutures. A xenograft was placed over the unclosed central portion. The wound was permitted to heal by second intention.
The clinical differential diagnosis of a scalp nodule includes a pilar cyst, BCC, squamous cell carcinoma, melanoma, cutaneous metastasis, adnexal tumor, atypical fibroxanthoma, and collision tumor. A collision tumor—the association of 2 or more benign or malignant neoplasms—represents a well-known pitfall in making a correct clinical and pathologic diagnosis.2 Many theories have been proposed to explain the pathophysiology of collision tumors. Some authors have speculated that they arise from involvement of related cell types.1 Other theories include induction by cytokines and growth factors secreted from one tumor that provides an ideal environment for proliferation of other cell types, a field cancerization effect of sun-damaged skin, or a coincidence.2
In our case, it is possible that the 2 tumors arose from a nevus sebaceus. One retrospective study of 706 cases of nevus sebaceus (707 specimens) found that 22.5% of cases developed secondary proliferation; of those cases, 18.9% were benign.3 Additionally, in 4.2% of cases of nevus sebaceus, proliferation of 2 or more tumors developed. The most common malignant neoplasm to develop from nevus sebaceus was BCC, followed by squamous cell carcinoma and sebaceous carcinoma. The most common benign neoplasm to develop from nevus sebaceus was trichoblastoma, followed by syringocystadenoma papilliferum.3
Our case highlights the possibility of a sampling error when performing a biopsy of any large neoplasm. Additionally, Mohs surgeons should maintain high clinical suspicion for collision tumors when encountering a large tumor with pathology inconsistent with the original biopsy. Apocrine hidrocystoma should be considered in the differential diagnosis of a large cystic mass of the scalp. Also, it is important to recognize that malignant lesions, such as BCC, can coexist with another benign tumor. Basal cell carcinoma is rare in Black patients, supporting our belief that our patient’s tumors arose from a nevus sebaceus.
It also is important for Mohs surgeons to consider any potential discrepancy between the initial pathology report and Mohs intraoperative pathology that can impact diagnosis, the aggressiveness of the tumors identified, and how such aggressiveness may affect management options.4,5 Some dermatology practices request biopsy slides from patients who are referred for Mohs micrographic surgery for internal review by a dermatopathologist before surgery is performed; however, this protocol requires additional time and adds costs for the overall health care system.4 One study found that internal review of outside biopsy slides resulted in a change in diagnosis in 2.2% of patients (N=3345)—affecting management in 61% of cases in which the diagnosis was changed.4 Another study (N=163) found that the reported aggressiveness of 50.5% of nonmelanoma cases in an initial biopsy report was changed during Mohs micrographic surgery.5 Mohs surgeons should be aware that discrepancies can occur, and if a discrepancy is discovered, the procedure may be paused until the initial biopsy slide is reviewed and further information is collected.
To the Editor:
A collision tumor is the coexistence of 2 discrete tumors in the same neoplasm, possibly comprising a malignant tumor and a benign tumor, and thereby complicating appropriate diagnosis and treatment. We present a case of a basal cell carcinoma (BCC) of the scalp that was later found to be in collision with an apocrine hidrocystoma that might have arisen from a nevus sebaceus. Although rare, BCC can coexist with apocrine hidrocystoma. Jayaprakasam and Rene1 reported a case of a collision tumor containing BCC and hidrocystoma on the eyelid.1 We present a case of a BCC on the scalp that was later found to be in collision with an apocrine hidrocystoma that possibly arose from a nevus sebaceus.
A 92-year-old Black woman with a biopsy-confirmed primary BCC of the left parietal scalp presented for Mohs micrographic surgery. The pathology report from an outside facility was reviewed. The initial diagnosis had been made with 2 punch biopsies from separate areas of the large nodule—one consistent with nodular and pigmented BCC (Figure 1), and the other revealed nodular ulcerated BCC. Physical examination prior to Mohs surgery revealed a mobile, flesh-colored, 6.2×6.0-cm nodule with minimal overlying hair on the left parietal scalp (Figure 2). During stage-I processing by the histopathology laboratory, large cystic structures were encountered; en face frozen sections showed a cystic tumor. Excised tissue was submitted for permanent processing to aid in diagnosis; the initial diagnostic biopsy slides were requested from the outside facility for review.
The initial diagnostic biopsy slides were reviewed and found to be consistent with nodular and pigmented BCC, as previously reported. Findings from hematoxylin and eosin staining of tissue obtained from Mohs sections were consistent with a combined neoplasm comprising BCC (Figure 3A) and apocrine hidrocystoma (Figure 3B). In addition, one section was characterized by acanthosis, papillomatosis, and sebaceous glands—similar to findings that are seen in a nevus sebaceus (Figure 3C).
The BCC was cleared after stage I; the final wound size was 7×6.6 cm. Although benign apocrine hidrocystoma was still evident at the margin, further excision was not performed at the request of the patient and her family. Partial primary wound closure was performed with pulley sutures. A xenograft was placed over the unclosed central portion. The wound was permitted to heal by second intention.
The clinical differential diagnosis of a scalp nodule includes a pilar cyst, BCC, squamous cell carcinoma, melanoma, cutaneous metastasis, adnexal tumor, atypical fibroxanthoma, and collision tumor. A collision tumor—the association of 2 or more benign or malignant neoplasms—represents a well-known pitfall in making a correct clinical and pathologic diagnosis.2 Many theories have been proposed to explain the pathophysiology of collision tumors. Some authors have speculated that they arise from involvement of related cell types.1 Other theories include induction by cytokines and growth factors secreted from one tumor that provides an ideal environment for proliferation of other cell types, a field cancerization effect of sun-damaged skin, or a coincidence.2
In our case, it is possible that the 2 tumors arose from a nevus sebaceus. One retrospective study of 706 cases of nevus sebaceus (707 specimens) found that 22.5% of cases developed secondary proliferation; of those cases, 18.9% were benign.3 Additionally, in 4.2% of cases of nevus sebaceus, proliferation of 2 or more tumors developed. The most common malignant neoplasm to develop from nevus sebaceus was BCC, followed by squamous cell carcinoma and sebaceous carcinoma. The most common benign neoplasm to develop from nevus sebaceus was trichoblastoma, followed by syringocystadenoma papilliferum.3
Our case highlights the possibility of a sampling error when performing a biopsy of any large neoplasm. Additionally, Mohs surgeons should maintain high clinical suspicion for collision tumors when encountering a large tumor with pathology inconsistent with the original biopsy. Apocrine hidrocystoma should be considered in the differential diagnosis of a large cystic mass of the scalp. Also, it is important to recognize that malignant lesions, such as BCC, can coexist with another benign tumor. Basal cell carcinoma is rare in Black patients, supporting our belief that our patient’s tumors arose from a nevus sebaceus.
It also is important for Mohs surgeons to consider any potential discrepancy between the initial pathology report and Mohs intraoperative pathology that can impact diagnosis, the aggressiveness of the tumors identified, and how such aggressiveness may affect management options.4,5 Some dermatology practices request biopsy slides from patients who are referred for Mohs micrographic surgery for internal review by a dermatopathologist before surgery is performed; however, this protocol requires additional time and adds costs for the overall health care system.4 One study found that internal review of outside biopsy slides resulted in a change in diagnosis in 2.2% of patients (N=3345)—affecting management in 61% of cases in which the diagnosis was changed.4 Another study (N=163) found that the reported aggressiveness of 50.5% of nonmelanoma cases in an initial biopsy report was changed during Mohs micrographic surgery.5 Mohs surgeons should be aware that discrepancies can occur, and if a discrepancy is discovered, the procedure may be paused until the initial biopsy slide is reviewed and further information is collected.
- Jayaprakasam A, Rene C. A benign or malignant eyelid lump—can you tell? an unusual collision tumour highlighting the difficulty differentiating a hidrocystoma from a basal cell carcinoma. BMJ Case Reports. 2012;2012:bcr1220115307. doi:10.1136/bcr.12.2011.5307
- Miteva M, Herschthal D, Ricotti C, et al. A rare case of a cutaneous squamomelanocytic tumor: revisiting the histogenesis of combined neoplasms. Am J Dermatopathol. 2009;31:599-603. doi:10.1097/DAD.0b013e3181a88116
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337. doi:10.1016/j.jaad.2013.10.004
- Butler ST, Youker SR, Mandrell J, et al. The importance of reviewing pathology specimens before Mohs surgery. Dermatol Surg. 2009;35:407-412. doi:10.1111/j.1524-4725.2008.01056.x
- Stiegel E, Lam C, Schowalter M, et al. Correlation between original biopsy pathology and Mohs intraoperative pathology. Dermatol Surg. 2018;44:193-197. doi:10.1097/DSS.0000000000001276
- Jayaprakasam A, Rene C. A benign or malignant eyelid lump—can you tell? an unusual collision tumour highlighting the difficulty differentiating a hidrocystoma from a basal cell carcinoma. BMJ Case Reports. 2012;2012:bcr1220115307. doi:10.1136/bcr.12.2011.5307
- Miteva M, Herschthal D, Ricotti C, et al. A rare case of a cutaneous squamomelanocytic tumor: revisiting the histogenesis of combined neoplasms. Am J Dermatopathol. 2009;31:599-603. doi:10.1097/DAD.0b013e3181a88116
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337. doi:10.1016/j.jaad.2013.10.004
- Butler ST, Youker SR, Mandrell J, et al. The importance of reviewing pathology specimens before Mohs surgery. Dermatol Surg. 2009;35:407-412. doi:10.1111/j.1524-4725.2008.01056.x
- Stiegel E, Lam C, Schowalter M, et al. Correlation between original biopsy pathology and Mohs intraoperative pathology. Dermatol Surg. 2018;44:193-197. doi:10.1097/DSS.0000000000001276
PRACTICE POINTS
- When collision tumors are encountered during Mohs micrographic surgery, review of the initial diagnostic material is recommended.
- Permanent processing of Mohs excisions may be helpful in determining the diagnosis of the occult second tumor diagnosis.

Scattered Red-Brown, Centrally Violaceous, Blanching Papules on an Infant
The Diagnosis: Neonatal-Onset Multisystem Inflammatory Disorder (NOMID)
The punch biopsy demonstrated a predominantly deep but somewhat superficial, periadnexal, neutrophilic and eosinophilic infiltrate (Figure). The eruption resolved 3 days later with supportive treatment, including appropriate wound care. Genetic analysis revealed an autosomal-dominant NLR family pyrin domain containing 3 gene, NLRP3, de novo variant associated with neonatal-onset multisystem inflammatory disorder (NOMID). Additional workup to characterize our patient’s inflammatory profile revealed elevated IL-18, CD3, CD4, S100A12, and S100A8/A9 levels. On day 48 of life, she was started on anakinra, an IL-1 inhibitor, at a dose of 1 mg/kg subcutaneously, which eventually was titrated to 10 mg/kg at hospital discharge. Hearing screenings were within normal limits.
, neutrophils, and macrophages in the deep dermis (H&E, original magnification ×200).")
Cryopyrin-associated periodic syndromes (CAPS) consist of 3 rare, IL-1–associated, autoinflammatory disorders, including familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), and NOMID (also known as chronic infantile neurologic cutaneous and articular syndrome). These conditions result from a sporadic or autosomal-dominant gain-of-function mutations in a single gene, NLRP3, on chromosome 1q44. NLRP3 encodes for cryopyrin, an important component of an IL-1 and IL-18 activating inflammasome.1 The most severe manifestation of CAPS is NOMID, which typically presents at birth as a migratory urticarial eruption, growth failure, myalgia, fever, and abnormal facial features, including frontal bossing, saddle-shaped nose, and protruding eyes.2 The illness also can manifest with hepatosplenomegaly, lymphadenopathy, uveitis, sensorineural hearing loss, cerebral atrophy, and other neurologic manifestations.3 A diagnosis of chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome was less likely given that our patient remained afebrile and did not show signs of lipodystrophy and persistent violaceous eyelid swelling. Both FCAS and MWS are less severe forms of CAPS when compared to NOMID. Familial cold autoinflammatory syndrome was less likely given the absence of the typical periodic fever pattern associated with the condition and severity of our patient’s symptoms. Muckle-Wells syndrome typically presents in adolescence with symptoms of FCAS, painful urticarial plaques, and progressive sensorinueral hearing loss. Tumor necrosis factor receptor–associated periodic fever (TRAPS) usually is associated with episodic fevers, abdominal pain, periorbital edema, migratory erythema, and arthralgia.1,3,4
Diagnostic criteria for CAPS include elevated inflammatory markers and serum amyloid, plus at least 2 of the typical CAPS symptoms: urticarial rash, cold-triggered episodes, sensorineural hearing loss, musculoskeletal symptoms, chronic aseptic meningitis, and skeletal abnormalities.4 The sensitivity and specificity of these diagnostic criteria are 84% and 91%, respectively. Additional findings that can be seen but are not part of the diagnostic criteria include intermittent fever, transient joint swelling, bony overgrowths, uveitis, optic disc edema, impaired growth, and hepatosplenomegaly.5 Laboratory findings may reveal leukocytosis, eosinophilia, anemia, and/or thrombocytopenia.3,5
Genetic testing, skin biopsies, ophthalmic examinations, neuroimaging, joint radiography, cerebrospinal fluid tests, and hearing examinations can be performed for confirmation of diagnosis and evaluation of systemic complications.4 A skin biopsy may reveal a neutrophilic infiltrate. Ophthalmic examination can demonstrate uveitis and optic disk edema. Neuroimaging may reveal cerebral atrophy or ventricular dilation. Lastly, joint radiography can be used to evaluate for the presence of premature long bone ossification or osseous overgrowth.4
In summary, NOMID is a multisystemic disorder with cutaneous manifestations. Early recognition of this entity is important given the severe sequelae and available efficacious therapy. Dermatologists should be aware of these manifestations, as dermatologic consultation and a skin biopsy may aid in diagnosis.
- Lachmann HJ. Periodic fever syndromes. Best Pract Res Clin Rheumatol. 2017;31:596-609. doi:10.1016/j.berh.2017.12.001
- Hull KM, Shoham N, Jin Chae J, et al. The expanding spectrum of systemic autoinflammatory disorders and their rheumatic manifestations. Curr Opin Rheumatol. 2003;15:61-69. doi:10.1097/00002281-200301000-00011
- Ahmadi N, Brewer CC, Zalewski C, et al. Cryopyrin-associated periodic syndromes: otolaryngologic and audiologic manifestations. Otolaryngol Head Neck Surg. 2011;145:295-302. doi:10.1177/0194599811402296
- Kuemmerle-Deschner JB, Ozen S, Tyrrell PN, et al. Diagnostic criteria for cryopyrin-associated periodic syndrome (CAPS). Ann Rheum Dis. 2017;76:942-947. doi:10.1136/annrheumdis-2016-209686
- Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrinassociated autoinflammatory diseases. Arthritis Rheum. 2002; 46:3340-3348. doi:10.1002/art.10688
The Diagnosis: Neonatal-Onset Multisystem Inflammatory Disorder (NOMID)
The punch biopsy demonstrated a predominantly deep but somewhat superficial, periadnexal, neutrophilic and eosinophilic infiltrate (Figure). The eruption resolved 3 days later with supportive treatment, including appropriate wound care. Genetic analysis revealed an autosomal-dominant NLR family pyrin domain containing 3 gene, NLRP3, de novo variant associated with neonatal-onset multisystem inflammatory disorder (NOMID). Additional workup to characterize our patient’s inflammatory profile revealed elevated IL-18, CD3, CD4, S100A12, and S100A8/A9 levels. On day 48 of life, she was started on anakinra, an IL-1 inhibitor, at a dose of 1 mg/kg subcutaneously, which eventually was titrated to 10 mg/kg at hospital discharge. Hearing screenings were within normal limits.
Cryopyrin-associated periodic syndromes (CAPS) consist of 3 rare, IL-1–associated, autoinflammatory disorders, including familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), and NOMID (also known as chronic infantile neurologic cutaneous and articular syndrome). These conditions result from a sporadic or autosomal-dominant gain-of-function mutations in a single gene, NLRP3, on chromosome 1q44. NLRP3 encodes for cryopyrin, an important component of an IL-1 and IL-18 activating inflammasome.1 The most severe manifestation of CAPS is NOMID, which typically presents at birth as a migratory urticarial eruption, growth failure, myalgia, fever, and abnormal facial features, including frontal bossing, saddle-shaped nose, and protruding eyes.2 The illness also can manifest with hepatosplenomegaly, lymphadenopathy, uveitis, sensorineural hearing loss, cerebral atrophy, and other neurologic manifestations.3 A diagnosis of chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome was less likely given that our patient remained afebrile and did not show signs of lipodystrophy and persistent violaceous eyelid swelling. Both FCAS and MWS are less severe forms of CAPS when compared to NOMID. Familial cold autoinflammatory syndrome was less likely given the absence of the typical periodic fever pattern associated with the condition and severity of our patient’s symptoms. Muckle-Wells syndrome typically presents in adolescence with symptoms of FCAS, painful urticarial plaques, and progressive sensorinueral hearing loss. Tumor necrosis factor receptor–associated periodic fever (TRAPS) usually is associated with episodic fevers, abdominal pain, periorbital edema, migratory erythema, and arthralgia.1,3,4
Diagnostic criteria for CAPS include elevated inflammatory markers and serum amyloid, plus at least 2 of the typical CAPS symptoms: urticarial rash, cold-triggered episodes, sensorineural hearing loss, musculoskeletal symptoms, chronic aseptic meningitis, and skeletal abnormalities.4 The sensitivity and specificity of these diagnostic criteria are 84% and 91%, respectively. Additional findings that can be seen but are not part of the diagnostic criteria include intermittent fever, transient joint swelling, bony overgrowths, uveitis, optic disc edema, impaired growth, and hepatosplenomegaly.5 Laboratory findings may reveal leukocytosis, eosinophilia, anemia, and/or thrombocytopenia.3,5
Genetic testing, skin biopsies, ophthalmic examinations, neuroimaging, joint radiography, cerebrospinal fluid tests, and hearing examinations can be performed for confirmation of diagnosis and evaluation of systemic complications.4 A skin biopsy may reveal a neutrophilic infiltrate. Ophthalmic examination can demonstrate uveitis and optic disk edema. Neuroimaging may reveal cerebral atrophy or ventricular dilation. Lastly, joint radiography can be used to evaluate for the presence of premature long bone ossification or osseous overgrowth.4
In summary, NOMID is a multisystemic disorder with cutaneous manifestations. Early recognition of this entity is important given the severe sequelae and available efficacious therapy. Dermatologists should be aware of these manifestations, as dermatologic consultation and a skin biopsy may aid in diagnosis.
The Diagnosis: Neonatal-Onset Multisystem Inflammatory Disorder (NOMID)
The punch biopsy demonstrated a predominantly deep but somewhat superficial, periadnexal, neutrophilic and eosinophilic infiltrate (Figure). The eruption resolved 3 days later with supportive treatment, including appropriate wound care. Genetic analysis revealed an autosomal-dominant NLR family pyrin domain containing 3 gene, NLRP3, de novo variant associated with neonatal-onset multisystem inflammatory disorder (NOMID). Additional workup to characterize our patient’s inflammatory profile revealed elevated IL-18, CD3, CD4, S100A12, and S100A8/A9 levels. On day 48 of life, she was started on anakinra, an IL-1 inhibitor, at a dose of 1 mg/kg subcutaneously, which eventually was titrated to 10 mg/kg at hospital discharge. Hearing screenings were within normal limits.
Cryopyrin-associated periodic syndromes (CAPS) consist of 3 rare, IL-1–associated, autoinflammatory disorders, including familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), and NOMID (also known as chronic infantile neurologic cutaneous and articular syndrome). These conditions result from a sporadic or autosomal-dominant gain-of-function mutations in a single gene, NLRP3, on chromosome 1q44. NLRP3 encodes for cryopyrin, an important component of an IL-1 and IL-18 activating inflammasome.1 The most severe manifestation of CAPS is NOMID, which typically presents at birth as a migratory urticarial eruption, growth failure, myalgia, fever, and abnormal facial features, including frontal bossing, saddle-shaped nose, and protruding eyes.2 The illness also can manifest with hepatosplenomegaly, lymphadenopathy, uveitis, sensorineural hearing loss, cerebral atrophy, and other neurologic manifestations.3 A diagnosis of chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome was less likely given that our patient remained afebrile and did not show signs of lipodystrophy and persistent violaceous eyelid swelling. Both FCAS and MWS are less severe forms of CAPS when compared to NOMID. Familial cold autoinflammatory syndrome was less likely given the absence of the typical periodic fever pattern associated with the condition and severity of our patient’s symptoms. Muckle-Wells syndrome typically presents in adolescence with symptoms of FCAS, painful urticarial plaques, and progressive sensorinueral hearing loss. Tumor necrosis factor receptor–associated periodic fever (TRAPS) usually is associated with episodic fevers, abdominal pain, periorbital edema, migratory erythema, and arthralgia.1,3,4
Diagnostic criteria for CAPS include elevated inflammatory markers and serum amyloid, plus at least 2 of the typical CAPS symptoms: urticarial rash, cold-triggered episodes, sensorineural hearing loss, musculoskeletal symptoms, chronic aseptic meningitis, and skeletal abnormalities.4 The sensitivity and specificity of these diagnostic criteria are 84% and 91%, respectively. Additional findings that can be seen but are not part of the diagnostic criteria include intermittent fever, transient joint swelling, bony overgrowths, uveitis, optic disc edema, impaired growth, and hepatosplenomegaly.5 Laboratory findings may reveal leukocytosis, eosinophilia, anemia, and/or thrombocytopenia.3,5
Genetic testing, skin biopsies, ophthalmic examinations, neuroimaging, joint radiography, cerebrospinal fluid tests, and hearing examinations can be performed for confirmation of diagnosis and evaluation of systemic complications.4 A skin biopsy may reveal a neutrophilic infiltrate. Ophthalmic examination can demonstrate uveitis and optic disk edema. Neuroimaging may reveal cerebral atrophy or ventricular dilation. Lastly, joint radiography can be used to evaluate for the presence of premature long bone ossification or osseous overgrowth.4
In summary, NOMID is a multisystemic disorder with cutaneous manifestations. Early recognition of this entity is important given the severe sequelae and available efficacious therapy. Dermatologists should be aware of these manifestations, as dermatologic consultation and a skin biopsy may aid in diagnosis.
- Lachmann HJ. Periodic fever syndromes. Best Pract Res Clin Rheumatol. 2017;31:596-609. doi:10.1016/j.berh.2017.12.001
- Hull KM, Shoham N, Jin Chae J, et al. The expanding spectrum of systemic autoinflammatory disorders and their rheumatic manifestations. Curr Opin Rheumatol. 2003;15:61-69. doi:10.1097/00002281-200301000-00011
- Ahmadi N, Brewer CC, Zalewski C, et al. Cryopyrin-associated periodic syndromes: otolaryngologic and audiologic manifestations. Otolaryngol Head Neck Surg. 2011;145:295-302. doi:10.1177/0194599811402296
- Kuemmerle-Deschner JB, Ozen S, Tyrrell PN, et al. Diagnostic criteria for cryopyrin-associated periodic syndrome (CAPS). Ann Rheum Dis. 2017;76:942-947. doi:10.1136/annrheumdis-2016-209686
- Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrinassociated autoinflammatory diseases. Arthritis Rheum. 2002; 46:3340-3348. doi:10.1002/art.10688
- Lachmann HJ. Periodic fever syndromes. Best Pract Res Clin Rheumatol. 2017;31:596-609. doi:10.1016/j.berh.2017.12.001
- Hull KM, Shoham N, Jin Chae J, et al. The expanding spectrum of systemic autoinflammatory disorders and their rheumatic manifestations. Curr Opin Rheumatol. 2003;15:61-69. doi:10.1097/00002281-200301000-00011
- Ahmadi N, Brewer CC, Zalewski C, et al. Cryopyrin-associated periodic syndromes: otolaryngologic and audiologic manifestations. Otolaryngol Head Neck Surg. 2011;145:295-302. doi:10.1177/0194599811402296
- Kuemmerle-Deschner JB, Ozen S, Tyrrell PN, et al. Diagnostic criteria for cryopyrin-associated periodic syndrome (CAPS). Ann Rheum Dis. 2017;76:942-947. doi:10.1136/annrheumdis-2016-209686
- Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrinassociated autoinflammatory diseases. Arthritis Rheum. 2002; 46:3340-3348. doi:10.1002/art.10688
A 2-week-old infant girl was transferred to a specialty pediatric hospital where dermatology was consulted for evaluation of a diffuse eruption triggered by cold that was similar to an eruption present at birth. She was born at 31 weeks and 2 days’ gestation at an outside hospital via caesarean delivery. Early delivery was prompted by superimposed pre-eclampsia with severe hypertension after administration of antenatal steroids. At birth, the infant was cyanotic and apneic and had a documented skin eruption, according to the medical record. She had thrombocytopenia, elevated C-reactive protein, and an elevated temperature without fever. Extensive septic workup, including blood, urine, and cerebrospinal fluid cultures; herpes simplex virus and cytomegalovirus screening; and Toxoplasma polymerase chain reaction were negative. Magnetic resonance imaging of the brain revealed no evidence of intracranial congenital infection. Ampicillinsulbactam was initiated for presumed culture-negative sepsis. On day 2 of hospitalization, she developed conjunctival icterus, hepatomegaly, and jaundice. Direct hyperbilirubinemia; anemia; and elevated triglycerides, ferritin, and ammonia all were present. Coagulation studies were normal. Subsequent workup, including abdominal ultrasonography and hepatobiliary iminodiacetic acid scan, was concerning for biliary atresia. Despite appropriate treatment, her condition did not improve and she was transferred. Repeat abdominal ultrasonography on day 24 of life confirmed hepatomegaly but did not demonstrate other findings of biliary atresia. At the current presentation, physical examination revealed many scattered, redbrown and centrally violaceous, blanching papules measuring a few millimeters involving the trunk, arms, buttocks, and legs. A punch biopsy was obtained.


A 7-month-old male presents with pustules and inflamed papules on the scalp and extremities
The bacterial, fungal, and atypical mycobacterial cultures from the lesions performed at the emergency department were all negative.
Pediatric dermatology was consulted and a punch biopsy of one of the lesions was done. Histopathologic examination showed a mixed perifollicular infiltrate of predominantly eosinophils with some neutrophils and associated microabscesses. Periodic acid Schiff and Fite stains failed to reveal any organisms. CD1 immunostain was negative. Fresh tissue cultures for bacteria, fungi, and atypical mycobacteria were negative.
Given the clinical presentation of chronic recurrent sterile pustules on an infant with associated eosinophilia and the reported histopathologic findings, the patient was diagnosed with eosinophilic pustular folliculitis of infancy (EPFI).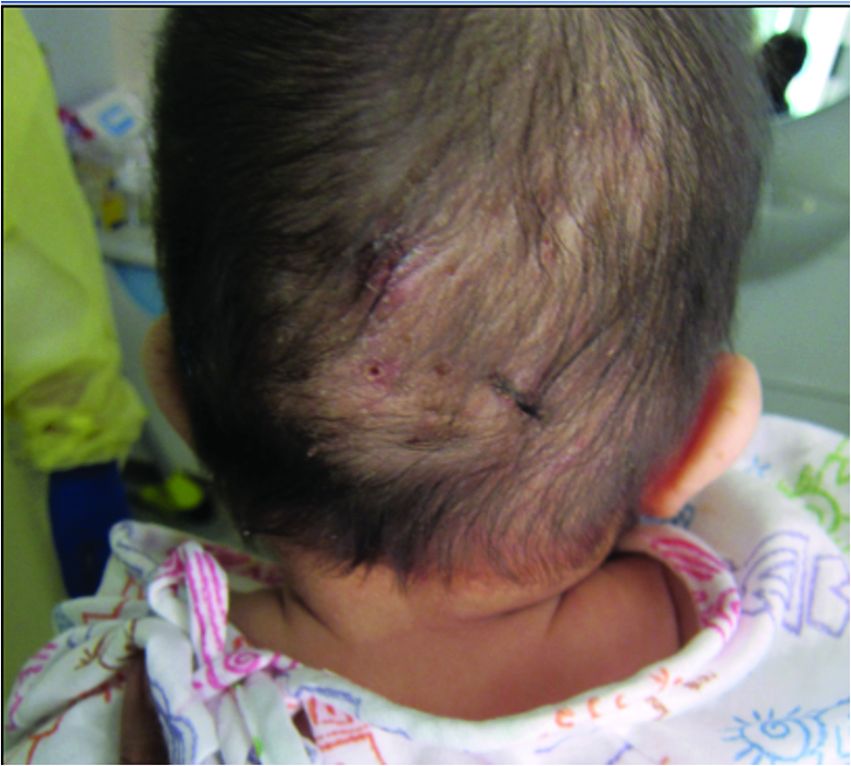
EPFI is a rare and idiopathic cutaneous disorder present in children. About 70% of the cases reported occur in the first 6 month of life and rarely present past 3 years of age. EPF encompasses a group of conditions including the classic adult form, or Ofuji disease. EPF is seen in immunosuppressed patients, mainly HIV positive, and EPF is also seen in infants and children.
In EPFI, males are most commonly affected. The condition presents, as it did in our patient, with recurrent crops of sterile papules and pustules mainly on the scalp, but they can occur in other parts of the body. The lesions go away within a few weeks to months without leaving any scars but it can take months to years to resolve. Histopathologic analysis of the lesions show an eosinophilic infiltrate which can be follicular, perifollicular, or periadnexal with associated flame figures in about 26% of cases.
Aggressive treatment is usually not needed as lesions are self-limited. Lesions can be treated with topical corticosteroids and oral antihistamine medications like cetirizine if symptomatic.
If the lesions start to present during the neonatal period, one may consider in the differential diagnosis, neonatal rashes like transient neonatal pustular melanosis and erythema toxicum neonatorum. Both of these neonatal conditions tend to resolve in the first month of life, compared with EPFI where lesions can come and go for months to years. EPFI lesions can be described as pustules and inflammatory papules, as well as furuncles and vesicles. All of the lesions may be seen in one patient at one time, which will not be typical for transient neonatal pustular melanosis or erythema toxicum. Eosinophils can be seen in erythema toxicum but folliculitis is not present. The inflammatory infiltrate seen in transient neonatal pustular melanosis is polymorphonuclear, not eosinophilic.
Early in the presentation, infectious conditions like staphylococcal or streptococcal folliculitis, cellulitis and furunculosis, tinea capitis, atypical mycobacterial infections, herpes simplex, and parasitic infections like scabies should be considered. In young infants, empiric antibiotic treatment may be started until cultures are finalized. If there is a family history of pruritic papules and pustules, scabies should be considered. A scabies prep can be done to rule out this entity.
Langerhans cell histiocytosis can also present with pustules and papules in early infancy and also has a predilection for the scalp. When this condition is in question, a skin biopsy should be performed which shows a CD1 positive histiocytic infiltrate.
In conclusion, EPFI is a benign rare condition that can present in infants as recurrent pustules and papules, mainly on the scalp, which are self-limited and if symptomatic can be treated with topical corticosteroids and antihistamines.
References
Alonso-Castro L et al. Dermatol Online J. 2012 Oct 15;18(10):6.
Frølunde AS et al. Clin Case Rep. 2021 May 11;9(5):e04167.
Hernández-Martín Á et al. J Am Acad Dermatol. 2013 Jan;68(1):150-5.
The bacterial, fungal, and atypical mycobacterial cultures from the lesions performed at the emergency department were all negative.
Pediatric dermatology was consulted and a punch biopsy of one of the lesions was done. Histopathologic examination showed a mixed perifollicular infiltrate of predominantly eosinophils with some neutrophils and associated microabscesses. Periodic acid Schiff and Fite stains failed to reveal any organisms. CD1 immunostain was negative. Fresh tissue cultures for bacteria, fungi, and atypical mycobacteria were negative.
Given the clinical presentation of chronic recurrent sterile pustules on an infant with associated eosinophilia and the reported histopathologic findings, the patient was diagnosed with eosinophilic pustular folliculitis of infancy (EPFI).
EPFI is a rare and idiopathic cutaneous disorder present in children. About 70% of the cases reported occur in the first 6 month of life and rarely present past 3 years of age. EPF encompasses a group of conditions including the classic adult form, or Ofuji disease. EPF is seen in immunosuppressed patients, mainly HIV positive, and EPF is also seen in infants and children.
In EPFI, males are most commonly affected. The condition presents, as it did in our patient, with recurrent crops of sterile papules and pustules mainly on the scalp, but they can occur in other parts of the body. The lesions go away within a few weeks to months without leaving any scars but it can take months to years to resolve. Histopathologic analysis of the lesions show an eosinophilic infiltrate which can be follicular, perifollicular, or periadnexal with associated flame figures in about 26% of cases.
Aggressive treatment is usually not needed as lesions are self-limited. Lesions can be treated with topical corticosteroids and oral antihistamine medications like cetirizine if symptomatic.
If the lesions start to present during the neonatal period, one may consider in the differential diagnosis, neonatal rashes like transient neonatal pustular melanosis and erythema toxicum neonatorum. Both of these neonatal conditions tend to resolve in the first month of life, compared with EPFI where lesions can come and go for months to years. EPFI lesions can be described as pustules and inflammatory papules, as well as furuncles and vesicles. All of the lesions may be seen in one patient at one time, which will not be typical for transient neonatal pustular melanosis or erythema toxicum. Eosinophils can be seen in erythema toxicum but folliculitis is not present. The inflammatory infiltrate seen in transient neonatal pustular melanosis is polymorphonuclear, not eosinophilic.
Early in the presentation, infectious conditions like staphylococcal or streptococcal folliculitis, cellulitis and furunculosis, tinea capitis, atypical mycobacterial infections, herpes simplex, and parasitic infections like scabies should be considered. In young infants, empiric antibiotic treatment may be started until cultures are finalized. If there is a family history of pruritic papules and pustules, scabies should be considered. A scabies prep can be done to rule out this entity.
Langerhans cell histiocytosis can also present with pustules and papules in early infancy and also has a predilection for the scalp. When this condition is in question, a skin biopsy should be performed which shows a CD1 positive histiocytic infiltrate.
In conclusion, EPFI is a benign rare condition that can present in infants as recurrent pustules and papules, mainly on the scalp, which are self-limited and if symptomatic can be treated with topical corticosteroids and antihistamines.
References
Alonso-Castro L et al. Dermatol Online J. 2012 Oct 15;18(10):6.
Frølunde AS et al. Clin Case Rep. 2021 May 11;9(5):e04167.
Hernández-Martín Á et al. J Am Acad Dermatol. 2013 Jan;68(1):150-5.
The bacterial, fungal, and atypical mycobacterial cultures from the lesions performed at the emergency department were all negative.
Pediatric dermatology was consulted and a punch biopsy of one of the lesions was done. Histopathologic examination showed a mixed perifollicular infiltrate of predominantly eosinophils with some neutrophils and associated microabscesses. Periodic acid Schiff and Fite stains failed to reveal any organisms. CD1 immunostain was negative. Fresh tissue cultures for bacteria, fungi, and atypical mycobacteria were negative.
Given the clinical presentation of chronic recurrent sterile pustules on an infant with associated eosinophilia and the reported histopathologic findings, the patient was diagnosed with eosinophilic pustular folliculitis of infancy (EPFI).
EPFI is a rare and idiopathic cutaneous disorder present in children. About 70% of the cases reported occur in the first 6 month of life and rarely present past 3 years of age. EPF encompasses a group of conditions including the classic adult form, or Ofuji disease. EPF is seen in immunosuppressed patients, mainly HIV positive, and EPF is also seen in infants and children.
In EPFI, males are most commonly affected. The condition presents, as it did in our patient, with recurrent crops of sterile papules and pustules mainly on the scalp, but they can occur in other parts of the body. The lesions go away within a few weeks to months without leaving any scars but it can take months to years to resolve. Histopathologic analysis of the lesions show an eosinophilic infiltrate which can be follicular, perifollicular, or periadnexal with associated flame figures in about 26% of cases.
Aggressive treatment is usually not needed as lesions are self-limited. Lesions can be treated with topical corticosteroids and oral antihistamine medications like cetirizine if symptomatic.
If the lesions start to present during the neonatal period, one may consider in the differential diagnosis, neonatal rashes like transient neonatal pustular melanosis and erythema toxicum neonatorum. Both of these neonatal conditions tend to resolve in the first month of life, compared with EPFI where lesions can come and go for months to years. EPFI lesions can be described as pustules and inflammatory papules, as well as furuncles and vesicles. All of the lesions may be seen in one patient at one time, which will not be typical for transient neonatal pustular melanosis or erythema toxicum. Eosinophils can be seen in erythema toxicum but folliculitis is not present. The inflammatory infiltrate seen in transient neonatal pustular melanosis is polymorphonuclear, not eosinophilic.
Early in the presentation, infectious conditions like staphylococcal or streptococcal folliculitis, cellulitis and furunculosis, tinea capitis, atypical mycobacterial infections, herpes simplex, and parasitic infections like scabies should be considered. In young infants, empiric antibiotic treatment may be started until cultures are finalized. If there is a family history of pruritic papules and pustules, scabies should be considered. A scabies prep can be done to rule out this entity.
Langerhans cell histiocytosis can also present with pustules and papules in early infancy and also has a predilection for the scalp. When this condition is in question, a skin biopsy should be performed which shows a CD1 positive histiocytic infiltrate.
In conclusion, EPFI is a benign rare condition that can present in infants as recurrent pustules and papules, mainly on the scalp, which are self-limited and if symptomatic can be treated with topical corticosteroids and antihistamines.
References
Alonso-Castro L et al. Dermatol Online J. 2012 Oct 15;18(10):6.
Frølunde AS et al. Clin Case Rep. 2021 May 11;9(5):e04167.
Hernández-Martín Á et al. J Am Acad Dermatol. 2013 Jan;68(1):150-5.
A 7-month-old male is brought to the emergency department for evaluation of pustules and inflamed papules on the scalp and extremities for several weeks of duration. The parents report the lesions started about a month prior and he has already been treated with cephalexin, clindamycin, and sulfamethoxazole without any improvement. Cultures sent prior by the child's pediatrician did not reveal any fungus or bacteria. The parents report a low-grade fever for about 3 days.
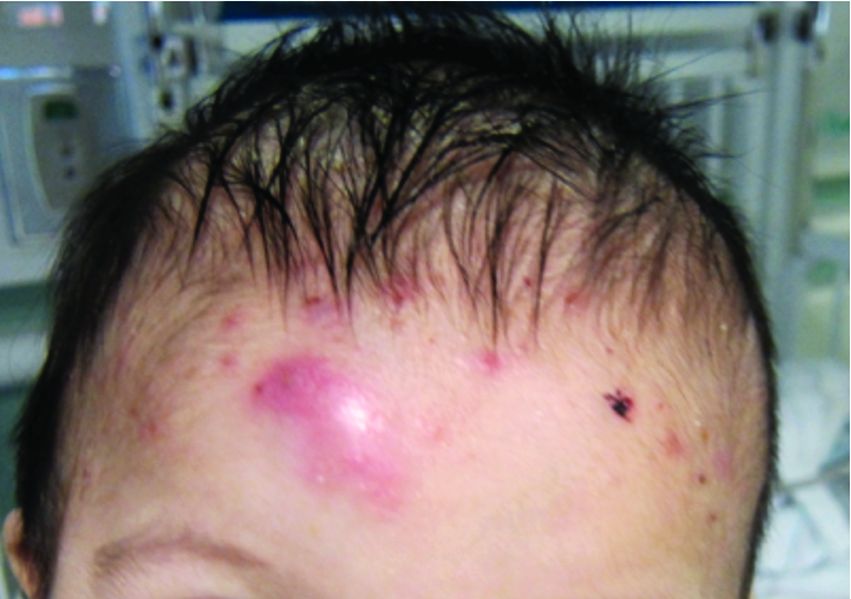
He was born via natural vaginal delivery with no instrumentation or external monitoring. Mom had prenatal care. Besides the skin lesions, the baby has been healthy and growing well. He has no history of eczema or severe infections. He has not been hospitalized before.
On physical examination the baby was not febrile. On the scalp and forehead, he had diffusely distributed pustules, erythematous papules, and nodules. He also presented with scattered, fine, small, crusted 1-2-mm pink papules on the trunk and extremities. He had no adenopathy or hepatosplenomegaly.
At the emergency department, samples from one of the pustules were sent for bacterial, fungal, and atypical mycobacteria cultures. Laboratory test showed a normal blood count with associated eosinophilia (2.8 x 109 L), and normal liver and kidney function. A head ultrasound showed three ill-defined hypoechoic foci within the scalp.
The patient was admitted for treatment with broad-spectrum antibiotics and dermatology was consulted.
Retiform Purpura on the Lower Legs
The Diagnosis: Type I Cryoglobulinemia
Retiform purpura with overlying necrosis subsequently developed over the course of a week following presentation (Figure 1). A skin biopsy showed fibrin thrombi and congestion of small- and medium-sized blood vessels, consistent with vasculopathy (Figure 2). Urinalysis revealed hematuria and proteinuria. A renal biopsy performed due to a continually elevated serum creatinine level revealed glomerulonephritis with numerous IgG1 lambda–restricted glomerular capillary hyaline thrombi, compatible with a lymphoproliferative disorder–associated type I cryoglobulinemia. A serum cryoglobulin immunofixation test confirmed type I cryoglobulinemia involving monoclonal IgG lambda. The combination of cutaneous, renal, and hematologic findings was consistent with type I cryoglobulinemia. A subsequent bone marrow biopsy demonstrated a CD20+ lambda–restricted plasma cell neoplasm. Initial treatment with high-dose corticosteroids followed by targeted treatment of the underlying hematologic condition with bortezomib, rituximab, and dexamethasone improved the skin disease.

Cryoglobulins are abnormal immunoglobulins that precipitate at temperatures below 37 °C. The persistent presence of cryoglobulins in the serum is termed cryoglobulinemia.1 Type I cryoglobulinemia is distinguished from mixed cryoglobulinemia—types II and III—by the presence of a single monoclonal immunoglobulin, typically IgM or IgG. It is associated with lymphoproliferative disorders, most commonly monoclonal gammopathy of undetermined significance and B-cell malignancies such as Waldenström macroglobulinemia, multiple myeloma, or chronic lymphocytic leukemia. Histopathology shows occlusion of small vessel lumina with homogenous eosinophilic material containing the monoclonal cryoprecipitate.2 Disease manifestations are caused by small vessel occlusion, which leads to ischemia and tissue damage.
.")
Retiform purpura, livedo reticularis/racemosa, and necrosis leading to ulcers are the most common cutaneous clinical findings. Extracutaneous signs include peripheral neuropathy, arthralgia, Raynaud phenomenon, and acrocyanosis. Renal involvement, most commonly glomerulonephritis with associated proteinuria, is noted in 14% to 20% of cases.3,4 An elevated cryocrit can lead to symptoms of hyperviscosity syndrome.2
Treatment is difficult and primarily is focused on addressing the underlying hematologic condition, which is responsible for synthesis of the cryoglobulin. Decreasing cryoglobulin production leads to decreased occlusion of blood vessels, thus alleviating the ischemia and skin damage. Monoclonal gammopathy of undetermined significance–related type I cryoglobulinemia initially is treated with corticosteroids followed by rituximab if a CD20+ B-cell clone is identified.2 Bortezomib is recommended for cases associated with Waldenström macroglobulinemia and cases associated with multiple myeloma with concurrent renal failure. In patients with neuropathy, a lenalidomide-based treatment can be employed. Patients should be instructed to keep extremities warm.2 Diabetic foot care guidelines should be followed to prevent wound complications. The differential diagnosis for type I cryoglobulinemia includes other causes of retiform purpura–like angioinvasive fungal infection, antiphospholipid antibody syndrome, calciphylaxis, and livedoid vasculopathy.5 Angioinvasive fungal infections are caused by Candida, Aspergillus, and Mucorales species, as well as other hyaline molds. They typically occur in immunocompromised patients and invade the blood vessels via direct inoculation or dissemination.6 Patients present with retiform purpura but typically will be acutely ill with fevers and vital sign abnormalities. Histopathology with special stains often will identify the fungal organisms in the dermis or inside blood vessel walls with vessel wall destruction and hemorrhage.7 Accurate diagnosis is essential to selecting appropriate antifungal agents. If angioinvasive fungal infection is clinically suspected, treatment should begin before culture and histopathologic data are available.7
Antiphospholipid antibody syndrome is an autoimmune thrombophilia that can occur as primary disease or in association with other autoimmune conditions, most commonly systemic lupus erythematosus. Diagnosis requires the presence of antiphospholipid antibodies, such as lupus anticoagulant, anticardiolipin antibody, anti–β2-glycoprotein-1 antibody, with arterial or venous thrombosis and/or recurrent pregnancy loss. Paraproteinemia is not seen. The most common cutaneous finding is livedo reticularis, with livedo racemosa being a more distinctive finding.8 Small vessel thrombosis is seen histopathologically. Treatment includes antiplatelet and anticoagulant medications. Patients with refractory disease may benefit from additional therapy with hydroxychloroquine or intravenous immunoglobulins.8
Calciphylaxis is a rare depositional vasculopathy that often occurs in patients with end-stage renal disease on dialysis. Patients present with painful and poor-healing skin lesions including indurated nodules, violaceous plaques, and retiform purpura that typically affect areas of high adiposity such as the thighs, abdomen, and buttocks.9 Ulceration and superimposed infections are common complications. Histopathologically, small dermal and subcutaneous vessels demonstrate calcification, microthrombosis, and fibrointimal hyperplasia.9 Wound management is critically important in patients with calciphylaxis. Treatment with intravenous sodium thiosulfate is typical, but prognosis remains poor. Although livedoid vasculopathy may present with retiform purpura in the ankles, paraproteinemia is not seen and patients frequently present with punched-out ulcerations that tend to heal into atrophie blanche.10 Livedoid vasculopathy has been associated with underlying hypercoagulable states, connective tissue diseases, and chronic venous hypertension. Hypercoagulability and endothelial cell damage contribute to the formation of fibrin thrombi in the superficial dermal blood vessels. Histopathology demonstrates thickening of vessel walls and intraluminal hyaline thrombi. Successful treatment in most cases is achieved with anticoagulation therapy, typically rivaroxaban, especially in patients with underlying hypercoagulability. Antiplatelet therapy also may be considered, while anabolic agents have been shown to be helpful in patients with connective tissue disease.10
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j .jbspin.2019.01.016
- Muchtar E, Magen H, Gertz MA. How I treat cryoglobulinemia. Blood. 2017;129:289-298. doi:10.1182/blood-2016-09-719773
- Sidana S, Rajkumar SV, Dispenzieri A, et al. Clinical presentation and outcomes of patients with type 1 monoclonal cryoglobulinemia. Am J Hematol. 2017;92:668-673. doi:10.1002/ajh.24745
- Harel S, Mohr M, Jahn I, et al. Clinico-biological characteristics and treatment of type I monoclonal cryoglobulinaemia: a study of 64 cases. Br J Haematol. 2015;168:671-678. doi:10.1111/bjh.13196
- Georgesen C, Fox LP, Harp J. Retiform purpura: a diagnostic approach. J Am Acad Dermatol. 2020;82:783-796. doi:10.1016/j.jaad.2019.07.112
- Shields BE, Rosenbach M, Brown-Joel Z, et al. Angioinvasive fungal infections impacting the skin: background, epidemiology, and clinical presentation. J Am Acad Dermatol. 2019;80:869-880.e5. doi:10.1016/j.jaad.2018.04.059
- Berger AP, Ford BA, Brown-Joel Z, et al. Angioinvasive fungal infections impacting the skin: diagnosis, management, and complications. J Am Acad Dermatol. 2019;80:883-898.e2. doi:10.1016/j.jaad.2018.04.058
- Negrini S, Pappalardo F, Murdaca G, et al. The antiphospholipid syndrome: from pathophysiology to treatment. Clin Exp Med. 2017;17:257-267. doi:10.1007/s10238-016-0430-5
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Georgesen C, Fox LP, Harp J. Retiform purpura: workup and therapeutic considerations in select conditions. J Am Acad Dermatol. 2020;82:799-816. doi:10.1016/j.jaad.2019.07.113
The Diagnosis: Type I Cryoglobulinemia
Retiform purpura with overlying necrosis subsequently developed over the course of a week following presentation (Figure 1). A skin biopsy showed fibrin thrombi and congestion of small- and medium-sized blood vessels, consistent with vasculopathy (Figure 2). Urinalysis revealed hematuria and proteinuria. A renal biopsy performed due to a continually elevated serum creatinine level revealed glomerulonephritis with numerous IgG1 lambda–restricted glomerular capillary hyaline thrombi, compatible with a lymphoproliferative disorder–associated type I cryoglobulinemia. A serum cryoglobulin immunofixation test confirmed type I cryoglobulinemia involving monoclonal IgG lambda. The combination of cutaneous, renal, and hematologic findings was consistent with type I cryoglobulinemia. A subsequent bone marrow biopsy demonstrated a CD20+ lambda–restricted plasma cell neoplasm. Initial treatment with high-dose corticosteroids followed by targeted treatment of the underlying hematologic condition with bortezomib, rituximab, and dexamethasone improved the skin disease.
Cryoglobulins are abnormal immunoglobulins that precipitate at temperatures below 37 °C. The persistent presence of cryoglobulins in the serum is termed cryoglobulinemia.1 Type I cryoglobulinemia is distinguished from mixed cryoglobulinemia—types II and III—by the presence of a single monoclonal immunoglobulin, typically IgM or IgG. It is associated with lymphoproliferative disorders, most commonly monoclonal gammopathy of undetermined significance and B-cell malignancies such as Waldenström macroglobulinemia, multiple myeloma, or chronic lymphocytic leukemia. Histopathology shows occlusion of small vessel lumina with homogenous eosinophilic material containing the monoclonal cryoprecipitate.2 Disease manifestations are caused by small vessel occlusion, which leads to ischemia and tissue damage.
Retiform purpura, livedo reticularis/racemosa, and necrosis leading to ulcers are the most common cutaneous clinical findings. Extracutaneous signs include peripheral neuropathy, arthralgia, Raynaud phenomenon, and acrocyanosis. Renal involvement, most commonly glomerulonephritis with associated proteinuria, is noted in 14% to 20% of cases.3,4 An elevated cryocrit can lead to symptoms of hyperviscosity syndrome.2
Treatment is difficult and primarily is focused on addressing the underlying hematologic condition, which is responsible for synthesis of the cryoglobulin. Decreasing cryoglobulin production leads to decreased occlusion of blood vessels, thus alleviating the ischemia and skin damage. Monoclonal gammopathy of undetermined significance–related type I cryoglobulinemia initially is treated with corticosteroids followed by rituximab if a CD20+ B-cell clone is identified.2 Bortezomib is recommended for cases associated with Waldenström macroglobulinemia and cases associated with multiple myeloma with concurrent renal failure. In patients with neuropathy, a lenalidomide-based treatment can be employed. Patients should be instructed to keep extremities warm.2 Diabetic foot care guidelines should be followed to prevent wound complications. The differential diagnosis for type I cryoglobulinemia includes other causes of retiform purpura–like angioinvasive fungal infection, antiphospholipid antibody syndrome, calciphylaxis, and livedoid vasculopathy.5 Angioinvasive fungal infections are caused by Candida, Aspergillus, and Mucorales species, as well as other hyaline molds. They typically occur in immunocompromised patients and invade the blood vessels via direct inoculation or dissemination.6 Patients present with retiform purpura but typically will be acutely ill with fevers and vital sign abnormalities. Histopathology with special stains often will identify the fungal organisms in the dermis or inside blood vessel walls with vessel wall destruction and hemorrhage.7 Accurate diagnosis is essential to selecting appropriate antifungal agents. If angioinvasive fungal infection is clinically suspected, treatment should begin before culture and histopathologic data are available.7
Antiphospholipid antibody syndrome is an autoimmune thrombophilia that can occur as primary disease or in association with other autoimmune conditions, most commonly systemic lupus erythematosus. Diagnosis requires the presence of antiphospholipid antibodies, such as lupus anticoagulant, anticardiolipin antibody, anti–β2-glycoprotein-1 antibody, with arterial or venous thrombosis and/or recurrent pregnancy loss. Paraproteinemia is not seen. The most common cutaneous finding is livedo reticularis, with livedo racemosa being a more distinctive finding.8 Small vessel thrombosis is seen histopathologically. Treatment includes antiplatelet and anticoagulant medications. Patients with refractory disease may benefit from additional therapy with hydroxychloroquine or intravenous immunoglobulins.8
Calciphylaxis is a rare depositional vasculopathy that often occurs in patients with end-stage renal disease on dialysis. Patients present with painful and poor-healing skin lesions including indurated nodules, violaceous plaques, and retiform purpura that typically affect areas of high adiposity such as the thighs, abdomen, and buttocks.9 Ulceration and superimposed infections are common complications. Histopathologically, small dermal and subcutaneous vessels demonstrate calcification, microthrombosis, and fibrointimal hyperplasia.9 Wound management is critically important in patients with calciphylaxis. Treatment with intravenous sodium thiosulfate is typical, but prognosis remains poor. Although livedoid vasculopathy may present with retiform purpura in the ankles, paraproteinemia is not seen and patients frequently present with punched-out ulcerations that tend to heal into atrophie blanche.10 Livedoid vasculopathy has been associated with underlying hypercoagulable states, connective tissue diseases, and chronic venous hypertension. Hypercoagulability and endothelial cell damage contribute to the formation of fibrin thrombi in the superficial dermal blood vessels. Histopathology demonstrates thickening of vessel walls and intraluminal hyaline thrombi. Successful treatment in most cases is achieved with anticoagulation therapy, typically rivaroxaban, especially in patients with underlying hypercoagulability. Antiplatelet therapy also may be considered, while anabolic agents have been shown to be helpful in patients with connective tissue disease.10
The Diagnosis: Type I Cryoglobulinemia
Retiform purpura with overlying necrosis subsequently developed over the course of a week following presentation (Figure 1). A skin biopsy showed fibrin thrombi and congestion of small- and medium-sized blood vessels, consistent with vasculopathy (Figure 2). Urinalysis revealed hematuria and proteinuria. A renal biopsy performed due to a continually elevated serum creatinine level revealed glomerulonephritis with numerous IgG1 lambda–restricted glomerular capillary hyaline thrombi, compatible with a lymphoproliferative disorder–associated type I cryoglobulinemia. A serum cryoglobulin immunofixation test confirmed type I cryoglobulinemia involving monoclonal IgG lambda. The combination of cutaneous, renal, and hematologic findings was consistent with type I cryoglobulinemia. A subsequent bone marrow biopsy demonstrated a CD20+ lambda–restricted plasma cell neoplasm. Initial treatment with high-dose corticosteroids followed by targeted treatment of the underlying hematologic condition with bortezomib, rituximab, and dexamethasone improved the skin disease.
Cryoglobulins are abnormal immunoglobulins that precipitate at temperatures below 37 °C. The persistent presence of cryoglobulins in the serum is termed cryoglobulinemia.1 Type I cryoglobulinemia is distinguished from mixed cryoglobulinemia—types II and III—by the presence of a single monoclonal immunoglobulin, typically IgM or IgG. It is associated with lymphoproliferative disorders, most commonly monoclonal gammopathy of undetermined significance and B-cell malignancies such as Waldenström macroglobulinemia, multiple myeloma, or chronic lymphocytic leukemia. Histopathology shows occlusion of small vessel lumina with homogenous eosinophilic material containing the monoclonal cryoprecipitate.2 Disease manifestations are caused by small vessel occlusion, which leads to ischemia and tissue damage.
Retiform purpura, livedo reticularis/racemosa, and necrosis leading to ulcers are the most common cutaneous clinical findings. Extracutaneous signs include peripheral neuropathy, arthralgia, Raynaud phenomenon, and acrocyanosis. Renal involvement, most commonly glomerulonephritis with associated proteinuria, is noted in 14% to 20% of cases.3,4 An elevated cryocrit can lead to symptoms of hyperviscosity syndrome.2
Treatment is difficult and primarily is focused on addressing the underlying hematologic condition, which is responsible for synthesis of the cryoglobulin. Decreasing cryoglobulin production leads to decreased occlusion of blood vessels, thus alleviating the ischemia and skin damage. Monoclonal gammopathy of undetermined significance–related type I cryoglobulinemia initially is treated with corticosteroids followed by rituximab if a CD20+ B-cell clone is identified.2 Bortezomib is recommended for cases associated with Waldenström macroglobulinemia and cases associated with multiple myeloma with concurrent renal failure. In patients with neuropathy, a lenalidomide-based treatment can be employed. Patients should be instructed to keep extremities warm.2 Diabetic foot care guidelines should be followed to prevent wound complications. The differential diagnosis for type I cryoglobulinemia includes other causes of retiform purpura–like angioinvasive fungal infection, antiphospholipid antibody syndrome, calciphylaxis, and livedoid vasculopathy.5 Angioinvasive fungal infections are caused by Candida, Aspergillus, and Mucorales species, as well as other hyaline molds. They typically occur in immunocompromised patients and invade the blood vessels via direct inoculation or dissemination.6 Patients present with retiform purpura but typically will be acutely ill with fevers and vital sign abnormalities. Histopathology with special stains often will identify the fungal organisms in the dermis or inside blood vessel walls with vessel wall destruction and hemorrhage.7 Accurate diagnosis is essential to selecting appropriate antifungal agents. If angioinvasive fungal infection is clinically suspected, treatment should begin before culture and histopathologic data are available.7
Antiphospholipid antibody syndrome is an autoimmune thrombophilia that can occur as primary disease or in association with other autoimmune conditions, most commonly systemic lupus erythematosus. Diagnosis requires the presence of antiphospholipid antibodies, such as lupus anticoagulant, anticardiolipin antibody, anti–β2-glycoprotein-1 antibody, with arterial or venous thrombosis and/or recurrent pregnancy loss. Paraproteinemia is not seen. The most common cutaneous finding is livedo reticularis, with livedo racemosa being a more distinctive finding.8 Small vessel thrombosis is seen histopathologically. Treatment includes antiplatelet and anticoagulant medications. Patients with refractory disease may benefit from additional therapy with hydroxychloroquine or intravenous immunoglobulins.8
Calciphylaxis is a rare depositional vasculopathy that often occurs in patients with end-stage renal disease on dialysis. Patients present with painful and poor-healing skin lesions including indurated nodules, violaceous plaques, and retiform purpura that typically affect areas of high adiposity such as the thighs, abdomen, and buttocks.9 Ulceration and superimposed infections are common complications. Histopathologically, small dermal and subcutaneous vessels demonstrate calcification, microthrombosis, and fibrointimal hyperplasia.9 Wound management is critically important in patients with calciphylaxis. Treatment with intravenous sodium thiosulfate is typical, but prognosis remains poor. Although livedoid vasculopathy may present with retiform purpura in the ankles, paraproteinemia is not seen and patients frequently present with punched-out ulcerations that tend to heal into atrophie blanche.10 Livedoid vasculopathy has been associated with underlying hypercoagulable states, connective tissue diseases, and chronic venous hypertension. Hypercoagulability and endothelial cell damage contribute to the formation of fibrin thrombi in the superficial dermal blood vessels. Histopathology demonstrates thickening of vessel walls and intraluminal hyaline thrombi. Successful treatment in most cases is achieved with anticoagulation therapy, typically rivaroxaban, especially in patients with underlying hypercoagulability. Antiplatelet therapy also may be considered, while anabolic agents have been shown to be helpful in patients with connective tissue disease.10
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j .jbspin.2019.01.016
- Muchtar E, Magen H, Gertz MA. How I treat cryoglobulinemia. Blood. 2017;129:289-298. doi:10.1182/blood-2016-09-719773
- Sidana S, Rajkumar SV, Dispenzieri A, et al. Clinical presentation and outcomes of patients with type 1 monoclonal cryoglobulinemia. Am J Hematol. 2017;92:668-673. doi:10.1002/ajh.24745
- Harel S, Mohr M, Jahn I, et al. Clinico-biological characteristics and treatment of type I monoclonal cryoglobulinaemia: a study of 64 cases. Br J Haematol. 2015;168:671-678. doi:10.1111/bjh.13196
- Georgesen C, Fox LP, Harp J. Retiform purpura: a diagnostic approach. J Am Acad Dermatol. 2020;82:783-796. doi:10.1016/j.jaad.2019.07.112
- Shields BE, Rosenbach M, Brown-Joel Z, et al. Angioinvasive fungal infections impacting the skin: background, epidemiology, and clinical presentation. J Am Acad Dermatol. 2019;80:869-880.e5. doi:10.1016/j.jaad.2018.04.059
- Berger AP, Ford BA, Brown-Joel Z, et al. Angioinvasive fungal infections impacting the skin: diagnosis, management, and complications. J Am Acad Dermatol. 2019;80:883-898.e2. doi:10.1016/j.jaad.2018.04.058
- Negrini S, Pappalardo F, Murdaca G, et al. The antiphospholipid syndrome: from pathophysiology to treatment. Clin Exp Med. 2017;17:257-267. doi:10.1007/s10238-016-0430-5
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Georgesen C, Fox LP, Harp J. Retiform purpura: workup and therapeutic considerations in select conditions. J Am Acad Dermatol. 2020;82:799-816. doi:10.1016/j.jaad.2019.07.113
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j .jbspin.2019.01.016
- Muchtar E, Magen H, Gertz MA. How I treat cryoglobulinemia. Blood. 2017;129:289-298. doi:10.1182/blood-2016-09-719773
- Sidana S, Rajkumar SV, Dispenzieri A, et al. Clinical presentation and outcomes of patients with type 1 monoclonal cryoglobulinemia. Am J Hematol. 2017;92:668-673. doi:10.1002/ajh.24745
- Harel S, Mohr M, Jahn I, et al. Clinico-biological characteristics and treatment of type I monoclonal cryoglobulinaemia: a study of 64 cases. Br J Haematol. 2015;168:671-678. doi:10.1111/bjh.13196
- Georgesen C, Fox LP, Harp J. Retiform purpura: a diagnostic approach. J Am Acad Dermatol. 2020;82:783-796. doi:10.1016/j.jaad.2019.07.112
- Shields BE, Rosenbach M, Brown-Joel Z, et al. Angioinvasive fungal infections impacting the skin: background, epidemiology, and clinical presentation. J Am Acad Dermatol. 2019;80:869-880.e5. doi:10.1016/j.jaad.2018.04.059
- Berger AP, Ford BA, Brown-Joel Z, et al. Angioinvasive fungal infections impacting the skin: diagnosis, management, and complications. J Am Acad Dermatol. 2019;80:883-898.e2. doi:10.1016/j.jaad.2018.04.058
- Negrini S, Pappalardo F, Murdaca G, et al. The antiphospholipid syndrome: from pathophysiology to treatment. Clin Exp Med. 2017;17:257-267. doi:10.1007/s10238-016-0430-5
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Georgesen C, Fox LP, Harp J. Retiform purpura: workup and therapeutic considerations in select conditions. J Am Acad Dermatol. 2020;82:799-816. doi:10.1016/j.jaad.2019.07.113
A 58-year-old man presented with a petechial and purpuric rash limited to the lower extremities. He reported that the rash had been present for months but worsened acutely over the last 3 days with new-onset dark urine, joint pain, and edema limiting his ability to walk. Physical examination showed areas of violaceous macules and papules on the legs and dorsal feet in a reticular distribution. Laboratory findings were remarkable for an elevated serum creatinine level of 2.75 mg/dL (reference range, 0.70–1.30 mg/dL), and serum immunofixation revealed the presence of markedly elevated IgG lambda monoclonal proteins. He was afebrile and his vital signs were stable. Dermatology, nephrology, and rheumatology services were consulted.


Recurrent Oral and Gluteal Cleft Erosions
The Diagnosis: Lichen Planus Pemphigoides
Lichen planus pemphigoides (LPP) is a rare acquired autoimmune blistering disorder with an estimated worldwide prevalence of approximately 1 in 1,000,000 individuals.1 It often manifests with overlapping features of both LP and bullous pemphigoid (BP). The condition usually presents in the fifth decade of life and has a slight female predominance.2 Although primarily idiopathic, it has been associated with certain medications and treatments, such as angiotensin-converting enzyme inhibitors, programmed cell death protein 1 inhibitors, programmed cell death ligand 1 inhibitors, labetalol, narrowband UVB, and psoralen plus UVA.3,4
Patients initially present with lesions of classic lichen planus (LP) with pink-purple, flat-topped, pruritic, polygonal papules and plaques.5 After weeks to months, tense vesicles and bullae usually develop on the sites of LP as well as on uninvolved skin. One study found a mean lag time of about 8.3 months for blistering to present after LP,5 but concurrent presentations of both have been reported.1 In addition, oral mucosal involvement has been seen in 36% of cases. The most commonly affected sites are the extremities; however, involvement can be widespread.2
The pathogenesis of LPP currently is unknown. It has been proposed that in LP, injury of basal keratinocytes exposes hidden basement membrane and hemidesmosome antigens including BP180, a 180 kDa transmembrane protein of the basement membrane zone (BMZ),6 which triggers an immune response where T cells recognize the extracellular portion of BP180 and antibodies are formed against the likely autoantigen.1 One study has suggested that the autoantigen in LPP is the MCW-4 epitope within the C-terminal end of the NC16A domain of BP180.7
Histopathology of LPP reveals characteristics of both LP as well as BP. Typical features of LP on hematoxylin and eosin (H&E) staining include lichenoid lymphocytic interface dermatitis, sawtooth rete ridges, wedge-shaped hypergranulosis, and colloid bodies, as demonstrated from the biopsy of our patient’s gluteal cleft lesion (quiz image 1), while the predominant feature of BP on H&E staining includes a subepidermal bulla with eosinophils.2 Typically, direct immunofluorescence (DIF) shows linear deposits of IgG and/or C3 along the BMZ. Indirect immunofluorescence (IIF) often reveals IgG against the roof of the BMZ in a human split-skin substrate.1 Antibodies against BP180 or uncommonly BP230 often are detected on enzyme-linked immunosorbent assay (ELISA). For our patient, IIF and ELISA tests were positive. Given the clinical presentation with recurrent oral and gluteal cleft erosions, histologic findings, and the results of our patient’s immunological testing, the diagnosis of LPP was made.
Topical steroids often are used to treat localized disease of LPP.8 Oral prednisone also may be given for widespread or unresponsive disease.9 Other treatments include azathioprine, mycophenolate mofetil, hydroxychloroquine, dapsone, tetracycline in combination with nicotinamide, acitretin, ustekinumab, baricitinib, and rituximab with intravenous immunoglobulin.3,8,10-12 Any potential medication culprits should be discontinued.9 Patients with oral involvement may require a soft diet to avoid further mucosal insult.10 Additionally, providers should consider dentistry, ophthalmology, and/or otolaryngology referrals depending on disease severity.
Bullous pemphigoid, the most common autoimmune blistering disease, has an estimated incidence of 10 to 43 per million individuals per year.2 Classically, it presents with tense bullae on the skin of the lower abdomen, thighs, groin, forearms, and axillae. Circulating antibodies against 2 BMZ proteins—BP180 and BP230—are important factors in BP pathogenesis.2 Diagnosis of BP is based on clinical features, histologic findings, and immunological studies including DIF, IIF, and ELISA. An eosinophil-rich subepidermal split typically is seen on H&E staining (Figure 1).
.")
Direct immunofluorescence displays linear IgG and/ or C3 staining at the BMZ. Indirect immunofluorescence on a human salt-split skin substrate commonly shows linear BMZ deposition on the roof of the blister.2 Indirect immunofluorescence for IgG deposition on monkey esophagus substrate shows linear BMZ deposition. Antibodies against the NC16A domain of BP180 (NC16A-BP180) are dominant, but BP230 antibodies against BP230 also are detected with ELISA.2 Further studies have indicated that the NC16A epitopes of BP180 that are targeted in BP are MCW-0-3,2 different from the autoantigen MCW-4 that is targeted in LPP.7
Paraneoplastic pemphigus (PNP) is another diagnosis to consider. Patients with PNP initially present with oral findings—most commonly chronic, erosive, and painful mucositis—followed by cutaneous involvement, which varies from the development of bullae to the formation of plaques similar to those of LP.13 The latter, in combination with oral erosions, may appear clinically similar to LPP. The results of DIF in conjugation with IIF and ELISA may help to further differentiate these disorders. Direct immunofluorescence in PNP typically reveals positive intercellular and/or BMZ IgG and C3, while DIF in LPP reveals depositions along the BMZ alone. Indirect immunofluorescence performed on rat bladder epithelium is particularly useful, as binding of IgG to rat bladder epithelium is characteristic of PNP and not seen in other disorders.14 Lastly, patients with PNP may develop IgG antibodies to various antigens such as desmoplakin I, desmoplakin II, envoplakin, periplakin, BP230, desmoglein 1, and desmoglein 3, which would not be expected in LPP patients.15 Hematoxylin and eosin staining differs from LPP, primarily with the location of the blister being intraepidermal. Acantholysis with hemorrhagic bullae can be seen (Figure 2).
.")
Classic LP is an inflammatory disorder that mainly affects adults, with an estimated incidence of less than 1%.16 The classic form presents with purple, flat-topped, pruritic, polygonal papules and plaques of varying size that often are characterized by Wickham striae. Lichen planus possesses a broad spectrum of subtypes involving different locations, though skin lesions usually are localized to the extremities. Despite an unknown etiology, activated T cells and T helper type 1 cytokines are considered key in keratinocyte injury. Compact orthokeratosis, wedge-shaped hypergranulosis, focal dyskeratosis, and colloid bodies typically are found on H&E staining, along with a dense bandlike lymphohistiocytic infiltrate at the dermoepidermal junction (DEJ)(Figure 3). Direct immunofluorescence typically shows a shaggy band of fibrinogen along the DEJ in addition to colloid bodies that stain with various autoantibodies including IgM, IgG, IgA, and C3.16
.")
Bullous LP is a rare variant of LP that commonly develops on the oral mucosa and the legs, with blisters confined on pre-existing LP lesions.9 The pathogenesis is related to an epidermal inflammatory infiltrate that leads to basal layer destruction followed by dermal-epidermal separations that cause blistering.17 Bullous LP does not have positive DIF, IIF, or ELISA because the pathophysiology does not involve autoantibody production. Histopathology typically displays an extensive inflammatory infiltrate and degeneration of the basal keratinocytes, resulting in large dermal-epidermal separations called Max-Joseph spaces (Figure 4).17 Colloid bodies are prominent in bullous LP but rarely are seen in LPP; eosinophils also are much more prominent in LPP compared to bullous LP.18 Unlike in LPP, DIF usually is negative in bullous LP, though lichenoid lesions may exhibit globular deposition of IgM, IgG, and IgA in the colloid bodies of the lower epidermis and/or papillary dermis. Similar to LP, DIF of the biopsy specimen shows linear or shaggy deposits of fibrinogen at the DEJ.17
.")
- Hübner F, Langan EA, Recke A. Lichen planus pemphigoides: from lichenoid inflammation to autoantibody-mediated blistering. Front Immunol. 2019;10:1389.
- Montagnon CM, Tolkachjov SN, Murrell DF, et al. Subepithelial autoimmune blistering dermatoses: clinical features and diagnosis. J Am Acad Dermatol. 2021;85:1-14.
- Hackländer K, Lehmann P, Hofmann SC. Successful treatment of lichen planus pemphigoides using acitretin as monotherapy. J Dtsch Dermatol Ges. 2014;12:818-819.
- Boyle M, Ashi S, Puiu T, et al. Lichen planus pemphigoides associated with PD-1 and PD-L1 inhibitors: a case series and review of the literature. Am J Dermatopathol. 2022;44:360-367.
- Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol. 2013;52:406-412.
- Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018.
- Zillikens D, Caux F, Mascaru JM Jr, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Knisley RR, Petropolis AA, Mackey VT. Lichen planus pemphigoides treated with ustekinumab. Cutis. 2017;100:415-418.
- Liakopoulou A, Rallis E. Bullous lichen planus—a review. J Dermatol Case Rep. 2017;11:1-4.
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Womens Dermatol. 2015;1:140-149.
- Moussa A, Colla TG, Asfour L, et al. Effective treatment of refractory lichen planus pemphigoides with a Janus kinase-1/2 inhibitor. Clin Exp Dermatol. 2022;47:2040-2041.
- Brennan M, Baldissano M, King L, et al. Successful use of rituximab and intravenous gamma globulin to treat checkpoint inhibitor-induced severe lichen planus pemphigoides. Skinmed. 2020;18:246-249.
- Kim JH, Kim SC. Paraneoplastic pemphigus: paraneoplastic autoimmune disease of the skin and mucosa. Front Immunol. 2019;10:1259.
- Stevens SR, Griffiths CE, Anhalt GJ, et al. Paraneoplastic pemphigus presenting as a lichen planus pemphigoides-like eruption. Arch Dermatol. 1993;129:866-869.
- Ohzono A, Sogame R, Li X, et al. Clinical and immunological findings in 104 cases of paraneoplastic pemphigus. Br J Dermatol. 2015;173:1447-1452.
- Tziotzios C, Lee JYW, Brier T, et al. Lichen planus and lichenoid dermatoses: clinical overview and molecular basis. J Am Acad Dermatol. 2018;79:789-804.
- Papara C, Danescu S, Sitaru C, et al. Challenges and pitfalls between lichen planus pemphigoides and bullous lichen planus. Australas J Dermatol. 2022;63:165-171.
- Tripathy DM, Vashisht D, Rathore G, et al. Bullous lichen planus vs lichen planus pemphigoides: a diagnostic dilemma. Indian Dermatol Online J. 2022;13:282-284.
The Diagnosis: Lichen Planus Pemphigoides
Lichen planus pemphigoides (LPP) is a rare acquired autoimmune blistering disorder with an estimated worldwide prevalence of approximately 1 in 1,000,000 individuals.1 It often manifests with overlapping features of both LP and bullous pemphigoid (BP). The condition usually presents in the fifth decade of life and has a slight female predominance.2 Although primarily idiopathic, it has been associated with certain medications and treatments, such as angiotensin-converting enzyme inhibitors, programmed cell death protein 1 inhibitors, programmed cell death ligand 1 inhibitors, labetalol, narrowband UVB, and psoralen plus UVA.3,4
Patients initially present with lesions of classic lichen planus (LP) with pink-purple, flat-topped, pruritic, polygonal papules and plaques.5 After weeks to months, tense vesicles and bullae usually develop on the sites of LP as well as on uninvolved skin. One study found a mean lag time of about 8.3 months for blistering to present after LP,5 but concurrent presentations of both have been reported.1 In addition, oral mucosal involvement has been seen in 36% of cases. The most commonly affected sites are the extremities; however, involvement can be widespread.2
The pathogenesis of LPP currently is unknown. It has been proposed that in LP, injury of basal keratinocytes exposes hidden basement membrane and hemidesmosome antigens including BP180, a 180 kDa transmembrane protein of the basement membrane zone (BMZ),6 which triggers an immune response where T cells recognize the extracellular portion of BP180 and antibodies are formed against the likely autoantigen.1 One study has suggested that the autoantigen in LPP is the MCW-4 epitope within the C-terminal end of the NC16A domain of BP180.7
Histopathology of LPP reveals characteristics of both LP as well as BP. Typical features of LP on hematoxylin and eosin (H&E) staining include lichenoid lymphocytic interface dermatitis, sawtooth rete ridges, wedge-shaped hypergranulosis, and colloid bodies, as demonstrated from the biopsy of our patient’s gluteal cleft lesion (quiz image 1), while the predominant feature of BP on H&E staining includes a subepidermal bulla with eosinophils.2 Typically, direct immunofluorescence (DIF) shows linear deposits of IgG and/or C3 along the BMZ. Indirect immunofluorescence (IIF) often reveals IgG against the roof of the BMZ in a human split-skin substrate.1 Antibodies against BP180 or uncommonly BP230 often are detected on enzyme-linked immunosorbent assay (ELISA). For our patient, IIF and ELISA tests were positive. Given the clinical presentation with recurrent oral and gluteal cleft erosions, histologic findings, and the results of our patient’s immunological testing, the diagnosis of LPP was made.
Topical steroids often are used to treat localized disease of LPP.8 Oral prednisone also may be given for widespread or unresponsive disease.9 Other treatments include azathioprine, mycophenolate mofetil, hydroxychloroquine, dapsone, tetracycline in combination with nicotinamide, acitretin, ustekinumab, baricitinib, and rituximab with intravenous immunoglobulin.3,8,10-12 Any potential medication culprits should be discontinued.9 Patients with oral involvement may require a soft diet to avoid further mucosal insult.10 Additionally, providers should consider dentistry, ophthalmology, and/or otolaryngology referrals depending on disease severity.
Bullous pemphigoid, the most common autoimmune blistering disease, has an estimated incidence of 10 to 43 per million individuals per year.2 Classically, it presents with tense bullae on the skin of the lower abdomen, thighs, groin, forearms, and axillae. Circulating antibodies against 2 BMZ proteins—BP180 and BP230—are important factors in BP pathogenesis.2 Diagnosis of BP is based on clinical features, histologic findings, and immunological studies including DIF, IIF, and ELISA. An eosinophil-rich subepidermal split typically is seen on H&E staining (Figure 1).
Direct immunofluorescence displays linear IgG and/ or C3 staining at the BMZ. Indirect immunofluorescence on a human salt-split skin substrate commonly shows linear BMZ deposition on the roof of the blister.2 Indirect immunofluorescence for IgG deposition on monkey esophagus substrate shows linear BMZ deposition. Antibodies against the NC16A domain of BP180 (NC16A-BP180) are dominant, but BP230 antibodies against BP230 also are detected with ELISA.2 Further studies have indicated that the NC16A epitopes of BP180 that are targeted in BP are MCW-0-3,2 different from the autoantigen MCW-4 that is targeted in LPP.7
Paraneoplastic pemphigus (PNP) is another diagnosis to consider. Patients with PNP initially present with oral findings—most commonly chronic, erosive, and painful mucositis—followed by cutaneous involvement, which varies from the development of bullae to the formation of plaques similar to those of LP.13 The latter, in combination with oral erosions, may appear clinically similar to LPP. The results of DIF in conjugation with IIF and ELISA may help to further differentiate these disorders. Direct immunofluorescence in PNP typically reveals positive intercellular and/or BMZ IgG and C3, while DIF in LPP reveals depositions along the BMZ alone. Indirect immunofluorescence performed on rat bladder epithelium is particularly useful, as binding of IgG to rat bladder epithelium is characteristic of PNP and not seen in other disorders.14 Lastly, patients with PNP may develop IgG antibodies to various antigens such as desmoplakin I, desmoplakin II, envoplakin, periplakin, BP230, desmoglein 1, and desmoglein 3, which would not be expected in LPP patients.15 Hematoxylin and eosin staining differs from LPP, primarily with the location of the blister being intraepidermal. Acantholysis with hemorrhagic bullae can be seen (Figure 2).
Classic LP is an inflammatory disorder that mainly affects adults, with an estimated incidence of less than 1%.16 The classic form presents with purple, flat-topped, pruritic, polygonal papules and plaques of varying size that often are characterized by Wickham striae. Lichen planus possesses a broad spectrum of subtypes involving different locations, though skin lesions usually are localized to the extremities. Despite an unknown etiology, activated T cells and T helper type 1 cytokines are considered key in keratinocyte injury. Compact orthokeratosis, wedge-shaped hypergranulosis, focal dyskeratosis, and colloid bodies typically are found on H&E staining, along with a dense bandlike lymphohistiocytic infiltrate at the dermoepidermal junction (DEJ)(Figure 3). Direct immunofluorescence typically shows a shaggy band of fibrinogen along the DEJ in addition to colloid bodies that stain with various autoantibodies including IgM, IgG, IgA, and C3.16
Bullous LP is a rare variant of LP that commonly develops on the oral mucosa and the legs, with blisters confined on pre-existing LP lesions.9 The pathogenesis is related to an epidermal inflammatory infiltrate that leads to basal layer destruction followed by dermal-epidermal separations that cause blistering.17 Bullous LP does not have positive DIF, IIF, or ELISA because the pathophysiology does not involve autoantibody production. Histopathology typically displays an extensive inflammatory infiltrate and degeneration of the basal keratinocytes, resulting in large dermal-epidermal separations called Max-Joseph spaces (Figure 4).17 Colloid bodies are prominent in bullous LP but rarely are seen in LPP; eosinophils also are much more prominent in LPP compared to bullous LP.18 Unlike in LPP, DIF usually is negative in bullous LP, though lichenoid lesions may exhibit globular deposition of IgM, IgG, and IgA in the colloid bodies of the lower epidermis and/or papillary dermis. Similar to LP, DIF of the biopsy specimen shows linear or shaggy deposits of fibrinogen at the DEJ.17
The Diagnosis: Lichen Planus Pemphigoides
Lichen planus pemphigoides (LPP) is a rare acquired autoimmune blistering disorder with an estimated worldwide prevalence of approximately 1 in 1,000,000 individuals.1 It often manifests with overlapping features of both LP and bullous pemphigoid (BP). The condition usually presents in the fifth decade of life and has a slight female predominance.2 Although primarily idiopathic, it has been associated with certain medications and treatments, such as angiotensin-converting enzyme inhibitors, programmed cell death protein 1 inhibitors, programmed cell death ligand 1 inhibitors, labetalol, narrowband UVB, and psoralen plus UVA.3,4
Patients initially present with lesions of classic lichen planus (LP) with pink-purple, flat-topped, pruritic, polygonal papules and plaques.5 After weeks to months, tense vesicles and bullae usually develop on the sites of LP as well as on uninvolved skin. One study found a mean lag time of about 8.3 months for blistering to present after LP,5 but concurrent presentations of both have been reported.1 In addition, oral mucosal involvement has been seen in 36% of cases. The most commonly affected sites are the extremities; however, involvement can be widespread.2
The pathogenesis of LPP currently is unknown. It has been proposed that in LP, injury of basal keratinocytes exposes hidden basement membrane and hemidesmosome antigens including BP180, a 180 kDa transmembrane protein of the basement membrane zone (BMZ),6 which triggers an immune response where T cells recognize the extracellular portion of BP180 and antibodies are formed against the likely autoantigen.1 One study has suggested that the autoantigen in LPP is the MCW-4 epitope within the C-terminal end of the NC16A domain of BP180.7
Histopathology of LPP reveals characteristics of both LP as well as BP. Typical features of LP on hematoxylin and eosin (H&E) staining include lichenoid lymphocytic interface dermatitis, sawtooth rete ridges, wedge-shaped hypergranulosis, and colloid bodies, as demonstrated from the biopsy of our patient’s gluteal cleft lesion (quiz image 1), while the predominant feature of BP on H&E staining includes a subepidermal bulla with eosinophils.2 Typically, direct immunofluorescence (DIF) shows linear deposits of IgG and/or C3 along the BMZ. Indirect immunofluorescence (IIF) often reveals IgG against the roof of the BMZ in a human split-skin substrate.1 Antibodies against BP180 or uncommonly BP230 often are detected on enzyme-linked immunosorbent assay (ELISA). For our patient, IIF and ELISA tests were positive. Given the clinical presentation with recurrent oral and gluteal cleft erosions, histologic findings, and the results of our patient’s immunological testing, the diagnosis of LPP was made.
Topical steroids often are used to treat localized disease of LPP.8 Oral prednisone also may be given for widespread or unresponsive disease.9 Other treatments include azathioprine, mycophenolate mofetil, hydroxychloroquine, dapsone, tetracycline in combination with nicotinamide, acitretin, ustekinumab, baricitinib, and rituximab with intravenous immunoglobulin.3,8,10-12 Any potential medication culprits should be discontinued.9 Patients with oral involvement may require a soft diet to avoid further mucosal insult.10 Additionally, providers should consider dentistry, ophthalmology, and/or otolaryngology referrals depending on disease severity.
Bullous pemphigoid, the most common autoimmune blistering disease, has an estimated incidence of 10 to 43 per million individuals per year.2 Classically, it presents with tense bullae on the skin of the lower abdomen, thighs, groin, forearms, and axillae. Circulating antibodies against 2 BMZ proteins—BP180 and BP230—are important factors in BP pathogenesis.2 Diagnosis of BP is based on clinical features, histologic findings, and immunological studies including DIF, IIF, and ELISA. An eosinophil-rich subepidermal split typically is seen on H&E staining (Figure 1).
Direct immunofluorescence displays linear IgG and/ or C3 staining at the BMZ. Indirect immunofluorescence on a human salt-split skin substrate commonly shows linear BMZ deposition on the roof of the blister.2 Indirect immunofluorescence for IgG deposition on monkey esophagus substrate shows linear BMZ deposition. Antibodies against the NC16A domain of BP180 (NC16A-BP180) are dominant, but BP230 antibodies against BP230 also are detected with ELISA.2 Further studies have indicated that the NC16A epitopes of BP180 that are targeted in BP are MCW-0-3,2 different from the autoantigen MCW-4 that is targeted in LPP.7
Paraneoplastic pemphigus (PNP) is another diagnosis to consider. Patients with PNP initially present with oral findings—most commonly chronic, erosive, and painful mucositis—followed by cutaneous involvement, which varies from the development of bullae to the formation of plaques similar to those of LP.13 The latter, in combination with oral erosions, may appear clinically similar to LPP. The results of DIF in conjugation with IIF and ELISA may help to further differentiate these disorders. Direct immunofluorescence in PNP typically reveals positive intercellular and/or BMZ IgG and C3, while DIF in LPP reveals depositions along the BMZ alone. Indirect immunofluorescence performed on rat bladder epithelium is particularly useful, as binding of IgG to rat bladder epithelium is characteristic of PNP and not seen in other disorders.14 Lastly, patients with PNP may develop IgG antibodies to various antigens such as desmoplakin I, desmoplakin II, envoplakin, periplakin, BP230, desmoglein 1, and desmoglein 3, which would not be expected in LPP patients.15 Hematoxylin and eosin staining differs from LPP, primarily with the location of the blister being intraepidermal. Acantholysis with hemorrhagic bullae can be seen (Figure 2).
Classic LP is an inflammatory disorder that mainly affects adults, with an estimated incidence of less than 1%.16 The classic form presents with purple, flat-topped, pruritic, polygonal papules and plaques of varying size that often are characterized by Wickham striae. Lichen planus possesses a broad spectrum of subtypes involving different locations, though skin lesions usually are localized to the extremities. Despite an unknown etiology, activated T cells and T helper type 1 cytokines are considered key in keratinocyte injury. Compact orthokeratosis, wedge-shaped hypergranulosis, focal dyskeratosis, and colloid bodies typically are found on H&E staining, along with a dense bandlike lymphohistiocytic infiltrate at the dermoepidermal junction (DEJ)(Figure 3). Direct immunofluorescence typically shows a shaggy band of fibrinogen along the DEJ in addition to colloid bodies that stain with various autoantibodies including IgM, IgG, IgA, and C3.16
Bullous LP is a rare variant of LP that commonly develops on the oral mucosa and the legs, with blisters confined on pre-existing LP lesions.9 The pathogenesis is related to an epidermal inflammatory infiltrate that leads to basal layer destruction followed by dermal-epidermal separations that cause blistering.17 Bullous LP does not have positive DIF, IIF, or ELISA because the pathophysiology does not involve autoantibody production. Histopathology typically displays an extensive inflammatory infiltrate and degeneration of the basal keratinocytes, resulting in large dermal-epidermal separations called Max-Joseph spaces (Figure 4).17 Colloid bodies are prominent in bullous LP but rarely are seen in LPP; eosinophils also are much more prominent in LPP compared to bullous LP.18 Unlike in LPP, DIF usually is negative in bullous LP, though lichenoid lesions may exhibit globular deposition of IgM, IgG, and IgA in the colloid bodies of the lower epidermis and/or papillary dermis. Similar to LP, DIF of the biopsy specimen shows linear or shaggy deposits of fibrinogen at the DEJ.17
- Hübner F, Langan EA, Recke A. Lichen planus pemphigoides: from lichenoid inflammation to autoantibody-mediated blistering. Front Immunol. 2019;10:1389.
- Montagnon CM, Tolkachjov SN, Murrell DF, et al. Subepithelial autoimmune blistering dermatoses: clinical features and diagnosis. J Am Acad Dermatol. 2021;85:1-14.
- Hackländer K, Lehmann P, Hofmann SC. Successful treatment of lichen planus pemphigoides using acitretin as monotherapy. J Dtsch Dermatol Ges. 2014;12:818-819.
- Boyle M, Ashi S, Puiu T, et al. Lichen planus pemphigoides associated with PD-1 and PD-L1 inhibitors: a case series and review of the literature. Am J Dermatopathol. 2022;44:360-367.
- Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol. 2013;52:406-412.
- Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018.
- Zillikens D, Caux F, Mascaru JM Jr, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Knisley RR, Petropolis AA, Mackey VT. Lichen planus pemphigoides treated with ustekinumab. Cutis. 2017;100:415-418.
- Liakopoulou A, Rallis E. Bullous lichen planus—a review. J Dermatol Case Rep. 2017;11:1-4.
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Womens Dermatol. 2015;1:140-149.
- Moussa A, Colla TG, Asfour L, et al. Effective treatment of refractory lichen planus pemphigoides with a Janus kinase-1/2 inhibitor. Clin Exp Dermatol. 2022;47:2040-2041.
- Brennan M, Baldissano M, King L, et al. Successful use of rituximab and intravenous gamma globulin to treat checkpoint inhibitor-induced severe lichen planus pemphigoides. Skinmed. 2020;18:246-249.
- Kim JH, Kim SC. Paraneoplastic pemphigus: paraneoplastic autoimmune disease of the skin and mucosa. Front Immunol. 2019;10:1259.
- Stevens SR, Griffiths CE, Anhalt GJ, et al. Paraneoplastic pemphigus presenting as a lichen planus pemphigoides-like eruption. Arch Dermatol. 1993;129:866-869.
- Ohzono A, Sogame R, Li X, et al. Clinical and immunological findings in 104 cases of paraneoplastic pemphigus. Br J Dermatol. 2015;173:1447-1452.
- Tziotzios C, Lee JYW, Brier T, et al. Lichen planus and lichenoid dermatoses: clinical overview and molecular basis. J Am Acad Dermatol. 2018;79:789-804.
- Papara C, Danescu S, Sitaru C, et al. Challenges and pitfalls between lichen planus pemphigoides and bullous lichen planus. Australas J Dermatol. 2022;63:165-171.
- Tripathy DM, Vashisht D, Rathore G, et al. Bullous lichen planus vs lichen planus pemphigoides: a diagnostic dilemma. Indian Dermatol Online J. 2022;13:282-284.
- Hübner F, Langan EA, Recke A. Lichen planus pemphigoides: from lichenoid inflammation to autoantibody-mediated blistering. Front Immunol. 2019;10:1389.
- Montagnon CM, Tolkachjov SN, Murrell DF, et al. Subepithelial autoimmune blistering dermatoses: clinical features and diagnosis. J Am Acad Dermatol. 2021;85:1-14.
- Hackländer K, Lehmann P, Hofmann SC. Successful treatment of lichen planus pemphigoides using acitretin as monotherapy. J Dtsch Dermatol Ges. 2014;12:818-819.
- Boyle M, Ashi S, Puiu T, et al. Lichen planus pemphigoides associated with PD-1 and PD-L1 inhibitors: a case series and review of the literature. Am J Dermatopathol. 2022;44:360-367.
- Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol. 2013;52:406-412.
- Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018.
- Zillikens D, Caux F, Mascaru JM Jr, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Knisley RR, Petropolis AA, Mackey VT. Lichen planus pemphigoides treated with ustekinumab. Cutis. 2017;100:415-418.
- Liakopoulou A, Rallis E. Bullous lichen planus—a review. J Dermatol Case Rep. 2017;11:1-4.
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Womens Dermatol. 2015;1:140-149.
- Moussa A, Colla TG, Asfour L, et al. Effective treatment of refractory lichen planus pemphigoides with a Janus kinase-1/2 inhibitor. Clin Exp Dermatol. 2022;47:2040-2041.
- Brennan M, Baldissano M, King L, et al. Successful use of rituximab and intravenous gamma globulin to treat checkpoint inhibitor-induced severe lichen planus pemphigoides. Skinmed. 2020;18:246-249.
- Kim JH, Kim SC. Paraneoplastic pemphigus: paraneoplastic autoimmune disease of the skin and mucosa. Front Immunol. 2019;10:1259.
- Stevens SR, Griffiths CE, Anhalt GJ, et al. Paraneoplastic pemphigus presenting as a lichen planus pemphigoides-like eruption. Arch Dermatol. 1993;129:866-869.
- Ohzono A, Sogame R, Li X, et al. Clinical and immunological findings in 104 cases of paraneoplastic pemphigus. Br J Dermatol. 2015;173:1447-1452.
- Tziotzios C, Lee JYW, Brier T, et al. Lichen planus and lichenoid dermatoses: clinical overview and molecular basis. J Am Acad Dermatol. 2018;79:789-804.
- Papara C, Danescu S, Sitaru C, et al. Challenges and pitfalls between lichen planus pemphigoides and bullous lichen planus. Australas J Dermatol. 2022;63:165-171.
- Tripathy DM, Vashisht D, Rathore G, et al. Bullous lichen planus vs lichen planus pemphigoides: a diagnostic dilemma. Indian Dermatol Online J. 2022;13:282-284.
A 71-year-old woman with no relevant medical history presented with recurrent painful erosions on the gingivae and gluteal cleft of 1 year’s duration. She previously was diagnosed by her periodontist with erosive lichen planus and was prescribed topical and oral steroids with minimal improvement. She denied fever, chills, weakness, fatigue, vision changes, eye pain, and sore throat. Dermatologic examination revealed edematous and erythematous upper and lower gingivae with mild erosions, as well as thin, eroded, erythematous plaques within the gluteal cleft. Indirect immunofluorescence revealed IgG with epidermal localization in a human split-skin substrate, and an enzyme-linked immunosorbent assay revealed positive IgG to bullous pemphigoid (BP) 180 and negative IgG to BP230. A 4-mm punch biopsy of the gluteal cleft was performed.



Symmetric Palmoplantar Papules With a Keratotic Border
The Diagnosis: Porokeratosis Plantaris Palmaris et Disseminata
A 3-mm punch biopsy of the right upper arm showed incipient cornoid lamellae formation, pigment incontinence, and sparse dermal lymphocytic inflammation (Figure), suggestive of porokeratosis plantaris palmaris et disseminata (PPPD). The dermatopathologist recommended a second biopsy to confirm the diagnosis and to confirm that the lesions on the palms and soles also were suggestive of porokeratosis. A second 4-mm punch biopsy of the left palm was consistent with PPPD.

The risks of PPPD as a precancerous entity along with the benefits and side effects of the various management options were discussed with our patient. We recommended that he start low-dose isotretinoin (20 mg/d) due to the large body surface area affected, making focal and field treatments likely insufficient. However, our patient opted not to treat and did not return for follow-up.
Subtypes of porokeratosis, including disseminated superficial actinic porokeratosis (DSAP) and PPPD, are conditions that disrupt the normal maturation of keratin and present clinically with symmetric, crusted, annular papules.1 The signature but nonspecific histopathologic feature shared among the subtypes is the presence of a cornoid lamellae.2 Several triggers of porokeratosis have been proposed, including trauma and exposure to UV and ionizing radiation.2,3 The clinical variants of porokeratosis are important conditions to diagnose correctly because they portend a risk for Bowen disease and invasive squamous cell carcinoma and may indicate the presence of an underlying hematologic and/or solid organ malignancy.4 Management of porokeratosis is difficult, as treatments have shown limited efficacy and variable recurrence rates. Treatment options include focal, field, and systemic options, such as 5-fluorouracil, topical compound of cholesterol and lovastatin, isotretinoin, and acitretin.1,2
Porokeratoses may arise from gene mutations in the mevalonate pathway,5 which is essential for the production of cholesterol.6 Topical cholesterol alone has not been shown to improve porokeratosis, but the combination topical therapy of cholesterol and lovastatin is promising. It is theorized to deliver benefit by both providing the essential end product of the pathway and simultaneously reducing the number of potentially toxic intermediates.6
Porokeratosis plantaris palmaris et disseminata (also known as porokeratosis plantaris) is unique among the subtypes of porokeratosis in that its annular, red-pink, papular rash with scaling and a keratotic border tends to start distally, involving the palms and soles, and progresses proximally to the trunk with smaller lesions.1,7 This centripetal progression can take years, as was seen in our patient.1 The disease is uncommon, with a dearth of published reports on PPPD.2 However, case reports have shown that PPPD is strongly linked to family history and may have an autosomal-dominant inheritance pattern. Penetrance is greater in men than in women, as PPPD is twice as common in men.8 Most cases of PPPD have been diagnosed in patients in their 20s and 30s, but Hartman et al9 reported a case wherein a patient was diagnosed with PPPD after 65 years of age, similar to our patient.
Although the lesions in DSAP can appear similar to those in PPPD, DSAP is more common among the family of porokeratotic conditions, affecting women twice as often as men, with a sporadic pattern of inheritance.2 These same features are present in some other types of porokeratosis but not PPPD. Furthermore, DSAP progresses proximally to distally but often with truncal sparing.2
Akin to PPPD, pityriasis rubra pilaris (PRP) often presents with palmoplantar keratoderma.10 There are at least 6 types of PRP with varying degrees of similarity to PPPD. However, in many cases PRP is associated with a background of diffuse erythema on the body with islands of spared skin. In addition, cases of PRP have been linked to extracutaneous findings such as ectropion and joint pain.11
Darier disease, especially the acrokeratosis verruciformis of Hopf variant, is more common in men and involves younger populations, as in PPPD.11 However, the crusted lesions seen in Darier disease frequently involve the skin folds. These intertriginous lesions may coalesce, mimicking warts in appearance, and are at risk for secondary infection. Nail findings in Darier disease also are distinct and include longitudinal white or red stripes running along the nail bed, in addition to V-shaped nicks at the nail tips.
Psoriasis can occur anywhere on the body and is associated with silver scaling atop a salmon-colored dermatitis.12 It results from aberrant proliferation of keratinocytes. Some distinguishing features of psoriasis include a disease course that waxes and wanes as well as pitting of the nails.
Although PPPD typically affects young adults, we presented a case of PPPD in an older man. Porokeratosis plantaris palmaris et disseminata in older adults may represent a delayed diagnosis, imply a broader range for the age of onset, or suggest its manifestation secondary to radiation treatment or another phenomenon. For example, our patient received 35 radiotherapy cycles for tongue cancer more than 5 years prior to the onset of PPPD.
- Irisawa R, Yamazaki M, Yamamoto T, et al. A case of porokeratosis plantaris palmaris et disseminata and literature review. Dermatol Online J. 2012;18:5.
- Vargas-Mora P, Morgado-Carrasco D, Fusta-Novell X. Porokeratosis: a review of its pathophysiology, clinical manifestations, diagnosis, and treatment. Actas Dermosifiliogr. 2020;111:545-560.
- James AJ, Clarke LE, Elenitsas R, et al. Segmental porokeratosis after radiation therapy for follicular lymphoma. J Am Acad Dermatol. 2008;58(2 suppl):S49-S50.
- Schena D, Papagrigoraki A, Frigo A, et al. Eruptive disseminated porokeratosis associated with internal malignancies: a case report. Cutis. 2010;85:156-159.
- Zhang Z, Li C, Wu F, et al. Genomic variations of the mevalonate pathway in porokeratosis. Elife. 2015;4:E06322. doi:10.7554/eLife.06322
- Atzmony L, Lim YH, Hamilton C, et al. Topical cholesterol/lovastatin for the treatment of porokeratosis: a pathogenesis-directed therapy. J Am Acad Dermatol. 2020;82:123-131. doi:10.1016/j.jaad.2019.08.043
- Guss SB, Osbourn RA, Lutzner MA. Porokeratosis plantaris, palmaris, et disseminata. a third type of porokeratosis. Arch Dermatol. 1971;104:366-373.
- Kanitakis J. Porokeratoses: an update of clinical, aetiopathogenic and therapeutic features. Eur J Dermatol. 2014;24:533-544.
- Hartman R, Mandal R, Sanchez M, et al. Porokeratosis plantaris, palmaris, et disseminata. Dermatol Online J. 2010;16:22.
- Suryawanshi H, Dhobley A, Sharma A, et al. Darier disease: a rare genodermatosis. J Oral Maxillofac Pathol. 2017;21:321. doi:10.4103/jomfp.JOMFP_170_16
- Eastham AB. Pityriasis rubra pilaris. JAMA Dermatol. 2019;155:404. doi:10.1001/jamadermatol.2018.5030
- Nair PA, Badri T. Psoriasis. StatPearls Publishing; 2022. Updated April 6, 2022. Accessed March 13, 2023. https://www.ncbi.nlm.nih.gov/books/NBK448194/
The Diagnosis: Porokeratosis Plantaris Palmaris et Disseminata
A 3-mm punch biopsy of the right upper arm showed incipient cornoid lamellae formation, pigment incontinence, and sparse dermal lymphocytic inflammation (Figure), suggestive of porokeratosis plantaris palmaris et disseminata (PPPD). The dermatopathologist recommended a second biopsy to confirm the diagnosis and to confirm that the lesions on the palms and soles also were suggestive of porokeratosis. A second 4-mm punch biopsy of the left palm was consistent with PPPD.
The risks of PPPD as a precancerous entity along with the benefits and side effects of the various management options were discussed with our patient. We recommended that he start low-dose isotretinoin (20 mg/d) due to the large body surface area affected, making focal and field treatments likely insufficient. However, our patient opted not to treat and did not return for follow-up.
Subtypes of porokeratosis, including disseminated superficial actinic porokeratosis (DSAP) and PPPD, are conditions that disrupt the normal maturation of keratin and present clinically with symmetric, crusted, annular papules.1 The signature but nonspecific histopathologic feature shared among the subtypes is the presence of a cornoid lamellae.2 Several triggers of porokeratosis have been proposed, including trauma and exposure to UV and ionizing radiation.2,3 The clinical variants of porokeratosis are important conditions to diagnose correctly because they portend a risk for Bowen disease and invasive squamous cell carcinoma and may indicate the presence of an underlying hematologic and/or solid organ malignancy.4 Management of porokeratosis is difficult, as treatments have shown limited efficacy and variable recurrence rates. Treatment options include focal, field, and systemic options, such as 5-fluorouracil, topical compound of cholesterol and lovastatin, isotretinoin, and acitretin.1,2
Porokeratoses may arise from gene mutations in the mevalonate pathway,5 which is essential for the production of cholesterol.6 Topical cholesterol alone has not been shown to improve porokeratosis, but the combination topical therapy of cholesterol and lovastatin is promising. It is theorized to deliver benefit by both providing the essential end product of the pathway and simultaneously reducing the number of potentially toxic intermediates.6
Porokeratosis plantaris palmaris et disseminata (also known as porokeratosis plantaris) is unique among the subtypes of porokeratosis in that its annular, red-pink, papular rash with scaling and a keratotic border tends to start distally, involving the palms and soles, and progresses proximally to the trunk with smaller lesions.1,7 This centripetal progression can take years, as was seen in our patient.1 The disease is uncommon, with a dearth of published reports on PPPD.2 However, case reports have shown that PPPD is strongly linked to family history and may have an autosomal-dominant inheritance pattern. Penetrance is greater in men than in women, as PPPD is twice as common in men.8 Most cases of PPPD have been diagnosed in patients in their 20s and 30s, but Hartman et al9 reported a case wherein a patient was diagnosed with PPPD after 65 years of age, similar to our patient.
Although the lesions in DSAP can appear similar to those in PPPD, DSAP is more common among the family of porokeratotic conditions, affecting women twice as often as men, with a sporadic pattern of inheritance.2 These same features are present in some other types of porokeratosis but not PPPD. Furthermore, DSAP progresses proximally to distally but often with truncal sparing.2
Akin to PPPD, pityriasis rubra pilaris (PRP) often presents with palmoplantar keratoderma.10 There are at least 6 types of PRP with varying degrees of similarity to PPPD. However, in many cases PRP is associated with a background of diffuse erythema on the body with islands of spared skin. In addition, cases of PRP have been linked to extracutaneous findings such as ectropion and joint pain.11
Darier disease, especially the acrokeratosis verruciformis of Hopf variant, is more common in men and involves younger populations, as in PPPD.11 However, the crusted lesions seen in Darier disease frequently involve the skin folds. These intertriginous lesions may coalesce, mimicking warts in appearance, and are at risk for secondary infection. Nail findings in Darier disease also are distinct and include longitudinal white or red stripes running along the nail bed, in addition to V-shaped nicks at the nail tips.
Psoriasis can occur anywhere on the body and is associated with silver scaling atop a salmon-colored dermatitis.12 It results from aberrant proliferation of keratinocytes. Some distinguishing features of psoriasis include a disease course that waxes and wanes as well as pitting of the nails.
Although PPPD typically affects young adults, we presented a case of PPPD in an older man. Porokeratosis plantaris palmaris et disseminata in older adults may represent a delayed diagnosis, imply a broader range for the age of onset, or suggest its manifestation secondary to radiation treatment or another phenomenon. For example, our patient received 35 radiotherapy cycles for tongue cancer more than 5 years prior to the onset of PPPD.
The Diagnosis: Porokeratosis Plantaris Palmaris et Disseminata
A 3-mm punch biopsy of the right upper arm showed incipient cornoid lamellae formation, pigment incontinence, and sparse dermal lymphocytic inflammation (Figure), suggestive of porokeratosis plantaris palmaris et disseminata (PPPD). The dermatopathologist recommended a second biopsy to confirm the diagnosis and to confirm that the lesions on the palms and soles also were suggestive of porokeratosis. A second 4-mm punch biopsy of the left palm was consistent with PPPD.
The risks of PPPD as a precancerous entity along with the benefits and side effects of the various management options were discussed with our patient. We recommended that he start low-dose isotretinoin (20 mg/d) due to the large body surface area affected, making focal and field treatments likely insufficient. However, our patient opted not to treat and did not return for follow-up.
Subtypes of porokeratosis, including disseminated superficial actinic porokeratosis (DSAP) and PPPD, are conditions that disrupt the normal maturation of keratin and present clinically with symmetric, crusted, annular papules.1 The signature but nonspecific histopathologic feature shared among the subtypes is the presence of a cornoid lamellae.2 Several triggers of porokeratosis have been proposed, including trauma and exposure to UV and ionizing radiation.2,3 The clinical variants of porokeratosis are important conditions to diagnose correctly because they portend a risk for Bowen disease and invasive squamous cell carcinoma and may indicate the presence of an underlying hematologic and/or solid organ malignancy.4 Management of porokeratosis is difficult, as treatments have shown limited efficacy and variable recurrence rates. Treatment options include focal, field, and systemic options, such as 5-fluorouracil, topical compound of cholesterol and lovastatin, isotretinoin, and acitretin.1,2
Porokeratoses may arise from gene mutations in the mevalonate pathway,5 which is essential for the production of cholesterol.6 Topical cholesterol alone has not been shown to improve porokeratosis, but the combination topical therapy of cholesterol and lovastatin is promising. It is theorized to deliver benefit by both providing the essential end product of the pathway and simultaneously reducing the number of potentially toxic intermediates.6
Porokeratosis plantaris palmaris et disseminata (also known as porokeratosis plantaris) is unique among the subtypes of porokeratosis in that its annular, red-pink, papular rash with scaling and a keratotic border tends to start distally, involving the palms and soles, and progresses proximally to the trunk with smaller lesions.1,7 This centripetal progression can take years, as was seen in our patient.1 The disease is uncommon, with a dearth of published reports on PPPD.2 However, case reports have shown that PPPD is strongly linked to family history and may have an autosomal-dominant inheritance pattern. Penetrance is greater in men than in women, as PPPD is twice as common in men.8 Most cases of PPPD have been diagnosed in patients in their 20s and 30s, but Hartman et al9 reported a case wherein a patient was diagnosed with PPPD after 65 years of age, similar to our patient.
Although the lesions in DSAP can appear similar to those in PPPD, DSAP is more common among the family of porokeratotic conditions, affecting women twice as often as men, with a sporadic pattern of inheritance.2 These same features are present in some other types of porokeratosis but not PPPD. Furthermore, DSAP progresses proximally to distally but often with truncal sparing.2
Akin to PPPD, pityriasis rubra pilaris (PRP) often presents with palmoplantar keratoderma.10 There are at least 6 types of PRP with varying degrees of similarity to PPPD. However, in many cases PRP is associated with a background of diffuse erythema on the body with islands of spared skin. In addition, cases of PRP have been linked to extracutaneous findings such as ectropion and joint pain.11
Darier disease, especially the acrokeratosis verruciformis of Hopf variant, is more common in men and involves younger populations, as in PPPD.11 However, the crusted lesions seen in Darier disease frequently involve the skin folds. These intertriginous lesions may coalesce, mimicking warts in appearance, and are at risk for secondary infection. Nail findings in Darier disease also are distinct and include longitudinal white or red stripes running along the nail bed, in addition to V-shaped nicks at the nail tips.
Psoriasis can occur anywhere on the body and is associated with silver scaling atop a salmon-colored dermatitis.12 It results from aberrant proliferation of keratinocytes. Some distinguishing features of psoriasis include a disease course that waxes and wanes as well as pitting of the nails.
Although PPPD typically affects young adults, we presented a case of PPPD in an older man. Porokeratosis plantaris palmaris et disseminata in older adults may represent a delayed diagnosis, imply a broader range for the age of onset, or suggest its manifestation secondary to radiation treatment or another phenomenon. For example, our patient received 35 radiotherapy cycles for tongue cancer more than 5 years prior to the onset of PPPD.
- Irisawa R, Yamazaki M, Yamamoto T, et al. A case of porokeratosis plantaris palmaris et disseminata and literature review. Dermatol Online J. 2012;18:5.
- Vargas-Mora P, Morgado-Carrasco D, Fusta-Novell X. Porokeratosis: a review of its pathophysiology, clinical manifestations, diagnosis, and treatment. Actas Dermosifiliogr. 2020;111:545-560.
- James AJ, Clarke LE, Elenitsas R, et al. Segmental porokeratosis after radiation therapy for follicular lymphoma. J Am Acad Dermatol. 2008;58(2 suppl):S49-S50.
- Schena D, Papagrigoraki A, Frigo A, et al. Eruptive disseminated porokeratosis associated with internal malignancies: a case report. Cutis. 2010;85:156-159.
- Zhang Z, Li C, Wu F, et al. Genomic variations of the mevalonate pathway in porokeratosis. Elife. 2015;4:E06322. doi:10.7554/eLife.06322
- Atzmony L, Lim YH, Hamilton C, et al. Topical cholesterol/lovastatin for the treatment of porokeratosis: a pathogenesis-directed therapy. J Am Acad Dermatol. 2020;82:123-131. doi:10.1016/j.jaad.2019.08.043
- Guss SB, Osbourn RA, Lutzner MA. Porokeratosis plantaris, palmaris, et disseminata. a third type of porokeratosis. Arch Dermatol. 1971;104:366-373.
- Kanitakis J. Porokeratoses: an update of clinical, aetiopathogenic and therapeutic features. Eur J Dermatol. 2014;24:533-544.
- Hartman R, Mandal R, Sanchez M, et al. Porokeratosis plantaris, palmaris, et disseminata. Dermatol Online J. 2010;16:22.
- Suryawanshi H, Dhobley A, Sharma A, et al. Darier disease: a rare genodermatosis. J Oral Maxillofac Pathol. 2017;21:321. doi:10.4103/jomfp.JOMFP_170_16
- Eastham AB. Pityriasis rubra pilaris. JAMA Dermatol. 2019;155:404. doi:10.1001/jamadermatol.2018.5030
- Nair PA, Badri T. Psoriasis. StatPearls Publishing; 2022. Updated April 6, 2022. Accessed March 13, 2023. https://www.ncbi.nlm.nih.gov/books/NBK448194/
- Irisawa R, Yamazaki M, Yamamoto T, et al. A case of porokeratosis plantaris palmaris et disseminata and literature review. Dermatol Online J. 2012;18:5.
- Vargas-Mora P, Morgado-Carrasco D, Fusta-Novell X. Porokeratosis: a review of its pathophysiology, clinical manifestations, diagnosis, and treatment. Actas Dermosifiliogr. 2020;111:545-560.
- James AJ, Clarke LE, Elenitsas R, et al. Segmental porokeratosis after radiation therapy for follicular lymphoma. J Am Acad Dermatol. 2008;58(2 suppl):S49-S50.
- Schena D, Papagrigoraki A, Frigo A, et al. Eruptive disseminated porokeratosis associated with internal malignancies: a case report. Cutis. 2010;85:156-159.
- Zhang Z, Li C, Wu F, et al. Genomic variations of the mevalonate pathway in porokeratosis. Elife. 2015;4:E06322. doi:10.7554/eLife.06322
- Atzmony L, Lim YH, Hamilton C, et al. Topical cholesterol/lovastatin for the treatment of porokeratosis: a pathogenesis-directed therapy. J Am Acad Dermatol. 2020;82:123-131. doi:10.1016/j.jaad.2019.08.043
- Guss SB, Osbourn RA, Lutzner MA. Porokeratosis plantaris, palmaris, et disseminata. a third type of porokeratosis. Arch Dermatol. 1971;104:366-373.
- Kanitakis J. Porokeratoses: an update of clinical, aetiopathogenic and therapeutic features. Eur J Dermatol. 2014;24:533-544.
- Hartman R, Mandal R, Sanchez M, et al. Porokeratosis plantaris, palmaris, et disseminata. Dermatol Online J. 2010;16:22.
- Suryawanshi H, Dhobley A, Sharma A, et al. Darier disease: a rare genodermatosis. J Oral Maxillofac Pathol. 2017;21:321. doi:10.4103/jomfp.JOMFP_170_16
- Eastham AB. Pityriasis rubra pilaris. JAMA Dermatol. 2019;155:404. doi:10.1001/jamadermatol.2018.5030
- Nair PA, Badri T. Psoriasis. StatPearls Publishing; 2022. Updated April 6, 2022. Accessed March 13, 2023. https://www.ncbi.nlm.nih.gov/books/NBK448194/
A 67-year-old man presented to our office with a rash on the hands, feet, and periungual skin that began with wartlike growths many years prior and recently had started to involve the proximal arms and legs up to the thighs as well as the trunk. He had a medical history of essential hypertension and chronic obstructive pulmonary disease. He had an 18-year smoking history and had quit more than 25 years prior, with tongue cancer diagnosed more than 5 years prior that was treated with surgery, chemotherapy, and radiation. The lesions occasionally were itchy but not painful. He also reported that his nails frequently split down the middle. He denied any oral lesions and was not using any treatments for the rash. He had no history of skin cancer or other skin conditions. His family history was unclear. Physical examination revealed annular red-pink scaling with a keratotic border on the soles of the feet, palms, and periungual skin. There also were small hyperpigmented papules on the arms, legs, thighs, and trunk over a background of dry and discolored skin, as well as dystrophy of all nails.


Subcutaneous Panniculitic T-cell Lymphoma Presenting With Anasarca in a Patient With Known Chronic Lymphocytic Leukemia
To the Editor:
Subcutaneous panniculitic T-cell lymphoma (SPTCL) is a rare cutaneous T-cell lymphoma that was first described in 19911 and comprises less than 1% of all non-Hodgkin lymphomas (NHLs). It most commonly occurs in young adults, with a median patient age of 36 years and a slight female predominance.2 Patients typically present with skin nodules or deep-seated plaques involving the legs, arms, and/or trunk. Presentation on the face is less common.2,3 Paraneoplastic edema has been reported in several cases of SPTCL with facial and periorbital swelling.4-9
Diagnosis of SPTCL is achieved via analysis of a deep tissue skin biopsy and close clinicopathologic correlation. Histopathology demonstrates lobular panniculitis with an atypical lymphoid infiltrate in the subcutaneous tissue with predominantly CD8+ T cells without overlying epidermotropism or interface dermatitis.3 The degree of cellular atypia, fat necrosis, karyorrhexis, cytophagia, and lack of angioinvasion can help to distinguish SPTCL from other panniculitides.2,3
The prognosis of SPTCL is good, with a 5-year survival rate of 82%, and many patients are able to achieve remission.2 However, SPTCL can progress to a fatal hemophagocytic syndrome, which has been reported in 17% of cases, making early diagnosis and treatment of this malignancy imperative.1,2 Treatment varies depending on the progression and extent of disease and can include the use of steroids, multidrug chemotherapy regimens, radiotherapy, and stem cell transplant in refractory cases.2-4,10,11
Subcutaneous panniculitic T-cell lymphoma with edema has been reported in a 2-year-old child.12 We present a case of SPTCL in an adult patient with known stage IV chronic lymphocytic leukemia (CLL) who also had full-body edema.
A 60-year-old woman with a 7-year history of stage IV CLL presented with anasarca of 3 months’ duration. At the time of presentation to dermatology, physical examination revealed erythematous tender nodules on the arms and legs. She had no other medical conditions and was undergoing treatment with ibrutinib for the CLL. The patient reported profound fatigue but no fever, chills, night sweats, cough, or dyspnea. The swelling had begun initially in the legs and progressively worsened to involve the arms, face, and body. She was hospitalized and treated with intravenous steroids and antihistamines, which led to minor improvement in the swelling. The patient’s preliminary diagnosis of erythema nodosum was thought to be related to the CLL or ibrutinib; therefore, treatment subsequently was discontinued and she was discharged from the hospital.
The swelling continued to worsen over the following 3 months, and the patient gained approximately 25 pounds. She presented to our office again with severe periorbital, facial, and lip edema as well as diffuse edema of the torso, arms, and legs (Figure 1). Erythematous tender subcutaneous nodules were noted on the right proximal thigh, left lateral calf, and forearms. She was again hospitalized, and extensive evaluation was performed to exclude other causes of anasarca, including a complete blood cell count; comprehensive metabolic profile; hepatitis panels; HIV test; C3 and C4, complement CH50, C1 esterase inhibitor, IgE, and angiotensin-converting enzyme levels; urine protein to creatinine ratio; computed tomography of the chest, abdomen, and pelvis; and allergy evaluation. The analyses failed to reveal the cause of the anasarca.
.")
During hospitalization, the patient underwent a lymph node biopsy, bone marrow biopsy, and a 6-mm punch biopsy of the right thigh nodule. The lymph node and bone marrow biopsy results were consistent with the known diagnosis of CLL, and the patient was started on intravenous chemotherapy with bendamustine. The skin biopsy demonstrated a predominant T-cell infiltrate consistent with a lobular panniculitis with variable amounts of adipocytes rimmed by lymphocytes, nuclear debris, and karyorrhexis (Figure 2). CD3+, CD8+, and CD4− T cells were positive for T-cell receptor (TCR) βF1 and negative for TCR-γ with strong expression of cytotoxic markers including granzyme B, perforin, and T-cell intracytoplasmic antigen 1. Rare CD56+ cells also were noted. The biopsy did not demonstrate any notable interface dermatitis, epidermotropism, or angioinvasion. T-cell receptor gene rearrangement studies did not show clonality for γ- or β-chain probes. Subcutaneous panniculitic T-cell lymphoma was diagnosed, making this case unique with the presentation of anasarca. This case also is noteworthy due to the rare diagnosis of the secondary malignancy of SPTCL in a patient with known CLL. The patient opted to pursue hospice and comfort measures due to the effects of persistent pancytopenia and the progression of CLL. She died 2 months later.
. B, Variable amounts of adipocytes rimmed by lymphocytes, nuclear debris, and karyorrhexis were shown on high power")
Clinical courses of SPTCL vary based on the TCR phenotype and immunophenotypic characteristics of the tumor cells. The TCR-αβ phenotype, as described in this case, typically is CD4−, CD8+, and CD56– and leads to a more indolent disease course. Lymphomas with the TCR-γδ phenotype typically are CD4−, CD8−, and CD56+; they often are associated with hemophagocytic syndrome and thus a worse prognosis. In 2009, the World Health Organization–European Organization for Research and Treatment of Cancer classification of primary cutaneous lymphomas restricted the category of SPTCL to the TCR-αβ phenotype due to the stark differences between the 2 types. The TCR-γδ phenotype was given its own diagnostic category—primary cutaneous γδ T-cell lymphoma.3
Patients with SPTCL commonly present with nodular skin lesions or deep-seated plaques on the legs, arms, and/or trunk; presentation on the face is rare.2,3 Fever, chills, night sweats, and/or weight loss were present in approximately 50% of recorded cases. Underlying autoimmune disease was present in 12 of 63 (19%) patients in a 2008 study.2 Facial and periorbital swelling with SPTCL has been reported.4-9 The presentation of anasarca, as seen in our adult patient, has been reported in a 2-year-old child.12 Anasarca as a presenting symptom of NHL is a rare phenomenon proposed to be induced by malignant cells secreting a cytokine that causes a vascular leak syndrome.13 Specifically, tumor necrosis factor α was found to be elevated in at least 2 patients with NHL presenting with anasarca in a prior study. Tumor necrosis factor α is known to cause increased capillary permeability, vascular leakage, and development of edema.13 In retrospect, obtaining cytokine levels in our patient would have been useful to support or refute tumor necrosis factor α as a possible cause of anasarca in the setting of NHL. This case continues to highlight that a diagnosis of SPTCL and analysis of a skin biopsy should be considered in cases of sudden unremitting facial and/or body swelling that cannot be explained by other more common causes.
Subcutaneous panniculitic T-cell lymphoma can be diagnosed and distinguished from other panniculitides via analysis of a deep tissue skin biopsy. Multiple biopsies may be required to ensure an adequate sample is obtained.4 Histopathology displays an atypical lymphoid infiltrate with a predominant presence of T cells. Neoplastic cells show CD3+, CD8+, and CD4− T cells, which strongly express cytotoxic proteins such as granzyme B, T-cell intracellular antigen 1, and perforin.3 The degree of cellular atypia, fat necrosis, karyorrhexis, and cytophagia, as well as the lack of angioinvasion, interface dermatitis, and epidermotropism help to distinguish SPTCL from other panniculitides.2,3 According to a previous study, clonal TCR gene rearrangement was identified in 50% to 80% of cases, but the absence of this clonal rearrangement does not exclude the diagnosis.14
This case also highlights the occurrence of secondary malignancies in patients with CLL, an NHL that is classified as a low-grade lymphoproliferative malignancy with clonal expansion of B cells.15 Secondary CTCLs in patients with CLL are rare, but they have been previously described. In 2017, Chang et al16 identified 12 patients with CLL who subsequently developed CTCL between 1992 and 2008. Of the 12 patients, 7 developed mycosis fungoides, 3 had CTCL not otherwise specified, 1 had mature T-cell lymphoma not otherwise specified, and 1 had primary cutaneous CD30+ T-cell lymphoma.16 The proliferation of 2 separate lymphocytic lineages is rare, but this study demonstrated an increased risk for CTCL to develop in patients with CLL. One possible explanation is that malignant cells come from a common stem cell progenitor or from genetic events. They occur secondary to carcinogens, viruses, or cytokines from T-cell or B-cell clones; they evolve due to treatment of the preexisting lymphoproliferative disease; or they occur simply by coincidence. The behavior of the CTCL may be more aggressive in patients with CLL due to immunosuppression, which may have contributed to the extreme presentation in our patient.16 Subcutaneous panniculitic T-cell lymphoma also has been reported in a patient with CLL that was thought to be associated with prior rituximab treatment.17
Treatment of SPTCL depends on the severity and course of the disease. In patients with more indolent disease, systemic steroids have been the most frequently used initial treatment.2,3,10 However, the disease often will progress after steroid tapering and require further intervention. Localized lesions may be treated with radiation alone or in combination with other systemic therapies.3,10 In refractory, aggressive, or relapsing cases, polychemotherapeutic regimens have proven to produce long-term remission in 30% of patients, with an overall response rate of 50%.10 These regimens most commonly have included cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or CHOP-like treatment (EPOCH regimen [etoposide, prednisone, oncovin, cyclophosphamide, and doxorubicin hydrochloride]).3,10 A stem cell transplant can be considered in patients with recurrent and refractory disease, and it also has been shown to induce remission.4,17 In patients with a good response to therapy, the disease often can be controlled for long periods of time, with an estimated 5-year survival rate of 80%.15
This case highlights the diagnostic challenges and variable presentations of SPTCL. Dermatologists, oncologists, and dermatopathologists should be aware of this condition and consider it in the differential diagnosis of a patient with a hematologic malignancy and unremitting facial and/or body swelling without any other cause. The possibility of a secondary hematologic cancer in a patient with CLL also must be taken into consideration. Early diagnosis and treatment can minimize morbidity and induce remission in most patients.
- Gonzalez CL, Medeiros LJ, Braziel RM, et al. T-cell lymphoma involving subcutaneous tissue. a clinicopathologic entity commonly associated with hemophagocytic syndrome. Am J Surg Pathol. 1991;15:17-27.
- Willemze R, Jansen P, Cerroni L, et al.
Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood. 2008;111:38-45. - Parveen Z, Thompson K. Subcutaneous panniculitis-like T-cell lymphoma: redefinition of diagnostic criteria in the recent World Health Organization–European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas. Arch Pathol Lab Med. 2009;133:303-308.
- Velez N, Ishizawar R, Dellaripa P, et al. Full facial edema: a novel presentation of subcutaneous panniculitis-like T-cell lymphoma. J Clin Oncol. 2012;30:e233-236.
- Asati D, Ingle V, Joshi D, et al. Subcutaneous panniculitis-like T-cell lymphoma with macrophage activation syndrome treated by cyclosporine and prednisolone. Indian Dermatol Online J. 2016;7:529-532.
- Fricker M, Dubach P, Helbing A, et al. Not all facial swellings are angioedemas! J Investig Allergol Clin Immunol. 2015;25:146-147.
- Kosari F, Akbarzadeh H. Local facial edema: a novel presentation of subcutaneous panniculitis-like T-cell lymphoma in a 30-year-old Iranian woman. Acta Med Iran. 2014;52:950-953.
- Bhojaraja M, Kistampally P, Udupa K, et al. Subcutaneous panniculitis-like T-cell lymphoma: a rare tumour. J Clin Diagn Res. 2016;10:OD29-OD30.
- Hashimoto R, Uchiyama M, Maeno T. Case report of subcutaneous panniculitis-like T-cell lymphoma complicated by eyelid swelling. BMC Ophthalmol. 2016;16:117.
- Chinello MN, Naviglio S, Remotti D, et al. Subcutaneous panniculitis-like T-cell lymphoma presenting with diffuse cutaneous edema in a 2-year-old child. J Pediatr Hematol Oncol. 2015;37:329-330.
- Chang TW, Weaver AL, Shanafelt TD, et al. Risk of cutaneous T-cell lymphoma in patients with chronic lymphocytic leukemia and other subtypes of non-Hodgkin lymphoma. Int J Dermatol. 2017;56:1125-1129.
- Chinello MN, Naviglio S, Remotti D, et al. Subcutaneous panniculitis-like T-cell lymphoma presenting with diffuse cutaneous edema in a 2-year-old child. J Pediatr Hematol Oncol. 2015;37:329-330.
- Jillella A, Day D, Severson K, et al. Non-Hodgkin’s lymphoma presenting as anasarca: probably mediated by tumor necrosis factor alpha (TNF-α). Leuk Lymphoma. 2000;38:419-422.
- Lee D-W, Yang J-H, Lee S-M, et al. Subcutaneous panniculitis-like T-cell lymphoma: a clinical and pathologic study of 14 Korean patients. Ann Dermatol. 2011;23:329-337.
- Jaffe ES. The 2008 WHO classification of lymphomas: implications for clinical practice and translational research [published online January 1, 2009]. Hematology Am Soc Hematol Educ Program. https://doi.org/10.1182/asheducation-2009.1.523
- Chang TW, Weaver AL, Shanafelt TD, et al. Risk of cutaneous T-cell lymphoma in patients with chronic lymphocytic leukemia and other subtypes of non-Hodgkin lymphoma. Int J Dermatol. 2017;56:1125-1129.
- Hall M, Sluzevich J, Snow J. Generalized subcutaneous panniculitis-like T-cell lymphoma following rituximab for hemolytic anemia in a patient with chronic lymphocytic leukemia. J Am Acad Dermatol. 2010;62(suppl 1):AB96.
To the Editor:
Subcutaneous panniculitic T-cell lymphoma (SPTCL) is a rare cutaneous T-cell lymphoma that was first described in 19911 and comprises less than 1% of all non-Hodgkin lymphomas (NHLs). It most commonly occurs in young adults, with a median patient age of 36 years and a slight female predominance.2 Patients typically present with skin nodules or deep-seated plaques involving the legs, arms, and/or trunk. Presentation on the face is less common.2,3 Paraneoplastic edema has been reported in several cases of SPTCL with facial and periorbital swelling.4-9
Diagnosis of SPTCL is achieved via analysis of a deep tissue skin biopsy and close clinicopathologic correlation. Histopathology demonstrates lobular panniculitis with an atypical lymphoid infiltrate in the subcutaneous tissue with predominantly CD8+ T cells without overlying epidermotropism or interface dermatitis.3 The degree of cellular atypia, fat necrosis, karyorrhexis, cytophagia, and lack of angioinvasion can help to distinguish SPTCL from other panniculitides.2,3
The prognosis of SPTCL is good, with a 5-year survival rate of 82%, and many patients are able to achieve remission.2 However, SPTCL can progress to a fatal hemophagocytic syndrome, which has been reported in 17% of cases, making early diagnosis and treatment of this malignancy imperative.1,2 Treatment varies depending on the progression and extent of disease and can include the use of steroids, multidrug chemotherapy regimens, radiotherapy, and stem cell transplant in refractory cases.2-4,10,11
Subcutaneous panniculitic T-cell lymphoma with edema has been reported in a 2-year-old child.12 We present a case of SPTCL in an adult patient with known stage IV chronic lymphocytic leukemia (CLL) who also had full-body edema.
A 60-year-old woman with a 7-year history of stage IV CLL presented with anasarca of 3 months’ duration. At the time of presentation to dermatology, physical examination revealed erythematous tender nodules on the arms and legs. She had no other medical conditions and was undergoing treatment with ibrutinib for the CLL. The patient reported profound fatigue but no fever, chills, night sweats, cough, or dyspnea. The swelling had begun initially in the legs and progressively worsened to involve the arms, face, and body. She was hospitalized and treated with intravenous steroids and antihistamines, which led to minor improvement in the swelling. The patient’s preliminary diagnosis of erythema nodosum was thought to be related to the CLL or ibrutinib; therefore, treatment subsequently was discontinued and she was discharged from the hospital.
The swelling continued to worsen over the following 3 months, and the patient gained approximately 25 pounds. She presented to our office again with severe periorbital, facial, and lip edema as well as diffuse edema of the torso, arms, and legs (Figure 1). Erythematous tender subcutaneous nodules were noted on the right proximal thigh, left lateral calf, and forearms. She was again hospitalized, and extensive evaluation was performed to exclude other causes of anasarca, including a complete blood cell count; comprehensive metabolic profile; hepatitis panels; HIV test; C3 and C4, complement CH50, C1 esterase inhibitor, IgE, and angiotensin-converting enzyme levels; urine protein to creatinine ratio; computed tomography of the chest, abdomen, and pelvis; and allergy evaluation. The analyses failed to reveal the cause of the anasarca.
During hospitalization, the patient underwent a lymph node biopsy, bone marrow biopsy, and a 6-mm punch biopsy of the right thigh nodule. The lymph node and bone marrow biopsy results were consistent with the known diagnosis of CLL, and the patient was started on intravenous chemotherapy with bendamustine. The skin biopsy demonstrated a predominant T-cell infiltrate consistent with a lobular panniculitis with variable amounts of adipocytes rimmed by lymphocytes, nuclear debris, and karyorrhexis (Figure 2). CD3+, CD8+, and CD4− T cells were positive for T-cell receptor (TCR) βF1 and negative for TCR-γ with strong expression of cytotoxic markers including granzyme B, perforin, and T-cell intracytoplasmic antigen 1. Rare CD56+ cells also were noted. The biopsy did not demonstrate any notable interface dermatitis, epidermotropism, or angioinvasion. T-cell receptor gene rearrangement studies did not show clonality for γ- or β-chain probes. Subcutaneous panniculitic T-cell lymphoma was diagnosed, making this case unique with the presentation of anasarca. This case also is noteworthy due to the rare diagnosis of the secondary malignancy of SPTCL in a patient with known CLL. The patient opted to pursue hospice and comfort measures due to the effects of persistent pancytopenia and the progression of CLL. She died 2 months later.
Clinical courses of SPTCL vary based on the TCR phenotype and immunophenotypic characteristics of the tumor cells. The TCR-αβ phenotype, as described in this case, typically is CD4−, CD8+, and CD56– and leads to a more indolent disease course. Lymphomas with the TCR-γδ phenotype typically are CD4−, CD8−, and CD56+; they often are associated with hemophagocytic syndrome and thus a worse prognosis. In 2009, the World Health Organization–European Organization for Research and Treatment of Cancer classification of primary cutaneous lymphomas restricted the category of SPTCL to the TCR-αβ phenotype due to the stark differences between the 2 types. The TCR-γδ phenotype was given its own diagnostic category—primary cutaneous γδ T-cell lymphoma.3
Patients with SPTCL commonly present with nodular skin lesions or deep-seated plaques on the legs, arms, and/or trunk; presentation on the face is rare.2,3 Fever, chills, night sweats, and/or weight loss were present in approximately 50% of recorded cases. Underlying autoimmune disease was present in 12 of 63 (19%) patients in a 2008 study.2 Facial and periorbital swelling with SPTCL has been reported.4-9 The presentation of anasarca, as seen in our adult patient, has been reported in a 2-year-old child.12 Anasarca as a presenting symptom of NHL is a rare phenomenon proposed to be induced by malignant cells secreting a cytokine that causes a vascular leak syndrome.13 Specifically, tumor necrosis factor α was found to be elevated in at least 2 patients with NHL presenting with anasarca in a prior study. Tumor necrosis factor α is known to cause increased capillary permeability, vascular leakage, and development of edema.13 In retrospect, obtaining cytokine levels in our patient would have been useful to support or refute tumor necrosis factor α as a possible cause of anasarca in the setting of NHL. This case continues to highlight that a diagnosis of SPTCL and analysis of a skin biopsy should be considered in cases of sudden unremitting facial and/or body swelling that cannot be explained by other more common causes.
Subcutaneous panniculitic T-cell lymphoma can be diagnosed and distinguished from other panniculitides via analysis of a deep tissue skin biopsy. Multiple biopsies may be required to ensure an adequate sample is obtained.4 Histopathology displays an atypical lymphoid infiltrate with a predominant presence of T cells. Neoplastic cells show CD3+, CD8+, and CD4− T cells, which strongly express cytotoxic proteins such as granzyme B, T-cell intracellular antigen 1, and perforin.3 The degree of cellular atypia, fat necrosis, karyorrhexis, and cytophagia, as well as the lack of angioinvasion, interface dermatitis, and epidermotropism help to distinguish SPTCL from other panniculitides.2,3 According to a previous study, clonal TCR gene rearrangement was identified in 50% to 80% of cases, but the absence of this clonal rearrangement does not exclude the diagnosis.14
This case also highlights the occurrence of secondary malignancies in patients with CLL, an NHL that is classified as a low-grade lymphoproliferative malignancy with clonal expansion of B cells.15 Secondary CTCLs in patients with CLL are rare, but they have been previously described. In 2017, Chang et al16 identified 12 patients with CLL who subsequently developed CTCL between 1992 and 2008. Of the 12 patients, 7 developed mycosis fungoides, 3 had CTCL not otherwise specified, 1 had mature T-cell lymphoma not otherwise specified, and 1 had primary cutaneous CD30+ T-cell lymphoma.16 The proliferation of 2 separate lymphocytic lineages is rare, but this study demonstrated an increased risk for CTCL to develop in patients with CLL. One possible explanation is that malignant cells come from a common stem cell progenitor or from genetic events. They occur secondary to carcinogens, viruses, or cytokines from T-cell or B-cell clones; they evolve due to treatment of the preexisting lymphoproliferative disease; or they occur simply by coincidence. The behavior of the CTCL may be more aggressive in patients with CLL due to immunosuppression, which may have contributed to the extreme presentation in our patient.16 Subcutaneous panniculitic T-cell lymphoma also has been reported in a patient with CLL that was thought to be associated with prior rituximab treatment.17
Treatment of SPTCL depends on the severity and course of the disease. In patients with more indolent disease, systemic steroids have been the most frequently used initial treatment.2,3,10 However, the disease often will progress after steroid tapering and require further intervention. Localized lesions may be treated with radiation alone or in combination with other systemic therapies.3,10 In refractory, aggressive, or relapsing cases, polychemotherapeutic regimens have proven to produce long-term remission in 30% of patients, with an overall response rate of 50%.10 These regimens most commonly have included cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or CHOP-like treatment (EPOCH regimen [etoposide, prednisone, oncovin, cyclophosphamide, and doxorubicin hydrochloride]).3,10 A stem cell transplant can be considered in patients with recurrent and refractory disease, and it also has been shown to induce remission.4,17 In patients with a good response to therapy, the disease often can be controlled for long periods of time, with an estimated 5-year survival rate of 80%.15
This case highlights the diagnostic challenges and variable presentations of SPTCL. Dermatologists, oncologists, and dermatopathologists should be aware of this condition and consider it in the differential diagnosis of a patient with a hematologic malignancy and unremitting facial and/or body swelling without any other cause. The possibility of a secondary hematologic cancer in a patient with CLL also must be taken into consideration. Early diagnosis and treatment can minimize morbidity and induce remission in most patients.
To the Editor:
Subcutaneous panniculitic T-cell lymphoma (SPTCL) is a rare cutaneous T-cell lymphoma that was first described in 19911 and comprises less than 1% of all non-Hodgkin lymphomas (NHLs). It most commonly occurs in young adults, with a median patient age of 36 years and a slight female predominance.2 Patients typically present with skin nodules or deep-seated plaques involving the legs, arms, and/or trunk. Presentation on the face is less common.2,3 Paraneoplastic edema has been reported in several cases of SPTCL with facial and periorbital swelling.4-9
Diagnosis of SPTCL is achieved via analysis of a deep tissue skin biopsy and close clinicopathologic correlation. Histopathology demonstrates lobular panniculitis with an atypical lymphoid infiltrate in the subcutaneous tissue with predominantly CD8+ T cells without overlying epidermotropism or interface dermatitis.3 The degree of cellular atypia, fat necrosis, karyorrhexis, cytophagia, and lack of angioinvasion can help to distinguish SPTCL from other panniculitides.2,3
The prognosis of SPTCL is good, with a 5-year survival rate of 82%, and many patients are able to achieve remission.2 However, SPTCL can progress to a fatal hemophagocytic syndrome, which has been reported in 17% of cases, making early diagnosis and treatment of this malignancy imperative.1,2 Treatment varies depending on the progression and extent of disease and can include the use of steroids, multidrug chemotherapy regimens, radiotherapy, and stem cell transplant in refractory cases.2-4,10,11
Subcutaneous panniculitic T-cell lymphoma with edema has been reported in a 2-year-old child.12 We present a case of SPTCL in an adult patient with known stage IV chronic lymphocytic leukemia (CLL) who also had full-body edema.
A 60-year-old woman with a 7-year history of stage IV CLL presented with anasarca of 3 months’ duration. At the time of presentation to dermatology, physical examination revealed erythematous tender nodules on the arms and legs. She had no other medical conditions and was undergoing treatment with ibrutinib for the CLL. The patient reported profound fatigue but no fever, chills, night sweats, cough, or dyspnea. The swelling had begun initially in the legs and progressively worsened to involve the arms, face, and body. She was hospitalized and treated with intravenous steroids and antihistamines, which led to minor improvement in the swelling. The patient’s preliminary diagnosis of erythema nodosum was thought to be related to the CLL or ibrutinib; therefore, treatment subsequently was discontinued and she was discharged from the hospital.
The swelling continued to worsen over the following 3 months, and the patient gained approximately 25 pounds. She presented to our office again with severe periorbital, facial, and lip edema as well as diffuse edema of the torso, arms, and legs (Figure 1). Erythematous tender subcutaneous nodules were noted on the right proximal thigh, left lateral calf, and forearms. She was again hospitalized, and extensive evaluation was performed to exclude other causes of anasarca, including a complete blood cell count; comprehensive metabolic profile; hepatitis panels; HIV test; C3 and C4, complement CH50, C1 esterase inhibitor, IgE, and angiotensin-converting enzyme levels; urine protein to creatinine ratio; computed tomography of the chest, abdomen, and pelvis; and allergy evaluation. The analyses failed to reveal the cause of the anasarca.
During hospitalization, the patient underwent a lymph node biopsy, bone marrow biopsy, and a 6-mm punch biopsy of the right thigh nodule. The lymph node and bone marrow biopsy results were consistent with the known diagnosis of CLL, and the patient was started on intravenous chemotherapy with bendamustine. The skin biopsy demonstrated a predominant T-cell infiltrate consistent with a lobular panniculitis with variable amounts of adipocytes rimmed by lymphocytes, nuclear debris, and karyorrhexis (Figure 2). CD3+, CD8+, and CD4− T cells were positive for T-cell receptor (TCR) βF1 and negative for TCR-γ with strong expression of cytotoxic markers including granzyme B, perforin, and T-cell intracytoplasmic antigen 1. Rare CD56+ cells also were noted. The biopsy did not demonstrate any notable interface dermatitis, epidermotropism, or angioinvasion. T-cell receptor gene rearrangement studies did not show clonality for γ- or β-chain probes. Subcutaneous panniculitic T-cell lymphoma was diagnosed, making this case unique with the presentation of anasarca. This case also is noteworthy due to the rare diagnosis of the secondary malignancy of SPTCL in a patient with known CLL. The patient opted to pursue hospice and comfort measures due to the effects of persistent pancytopenia and the progression of CLL. She died 2 months later.
Clinical courses of SPTCL vary based on the TCR phenotype and immunophenotypic characteristics of the tumor cells. The TCR-αβ phenotype, as described in this case, typically is CD4−, CD8+, and CD56– and leads to a more indolent disease course. Lymphomas with the TCR-γδ phenotype typically are CD4−, CD8−, and CD56+; they often are associated with hemophagocytic syndrome and thus a worse prognosis. In 2009, the World Health Organization–European Organization for Research and Treatment of Cancer classification of primary cutaneous lymphomas restricted the category of SPTCL to the TCR-αβ phenotype due to the stark differences between the 2 types. The TCR-γδ phenotype was given its own diagnostic category—primary cutaneous γδ T-cell lymphoma.3
Patients with SPTCL commonly present with nodular skin lesions or deep-seated plaques on the legs, arms, and/or trunk; presentation on the face is rare.2,3 Fever, chills, night sweats, and/or weight loss were present in approximately 50% of recorded cases. Underlying autoimmune disease was present in 12 of 63 (19%) patients in a 2008 study.2 Facial and periorbital swelling with SPTCL has been reported.4-9 The presentation of anasarca, as seen in our adult patient, has been reported in a 2-year-old child.12 Anasarca as a presenting symptom of NHL is a rare phenomenon proposed to be induced by malignant cells secreting a cytokine that causes a vascular leak syndrome.13 Specifically, tumor necrosis factor α was found to be elevated in at least 2 patients with NHL presenting with anasarca in a prior study. Tumor necrosis factor α is known to cause increased capillary permeability, vascular leakage, and development of edema.13 In retrospect, obtaining cytokine levels in our patient would have been useful to support or refute tumor necrosis factor α as a possible cause of anasarca in the setting of NHL. This case continues to highlight that a diagnosis of SPTCL and analysis of a skin biopsy should be considered in cases of sudden unremitting facial and/or body swelling that cannot be explained by other more common causes.
Subcutaneous panniculitic T-cell lymphoma can be diagnosed and distinguished from other panniculitides via analysis of a deep tissue skin biopsy. Multiple biopsies may be required to ensure an adequate sample is obtained.4 Histopathology displays an atypical lymphoid infiltrate with a predominant presence of T cells. Neoplastic cells show CD3+, CD8+, and CD4− T cells, which strongly express cytotoxic proteins such as granzyme B, T-cell intracellular antigen 1, and perforin.3 The degree of cellular atypia, fat necrosis, karyorrhexis, and cytophagia, as well as the lack of angioinvasion, interface dermatitis, and epidermotropism help to distinguish SPTCL from other panniculitides.2,3 According to a previous study, clonal TCR gene rearrangement was identified in 50% to 80% of cases, but the absence of this clonal rearrangement does not exclude the diagnosis.14
This case also highlights the occurrence of secondary malignancies in patients with CLL, an NHL that is classified as a low-grade lymphoproliferative malignancy with clonal expansion of B cells.15 Secondary CTCLs in patients with CLL are rare, but they have been previously described. In 2017, Chang et al16 identified 12 patients with CLL who subsequently developed CTCL between 1992 and 2008. Of the 12 patients, 7 developed mycosis fungoides, 3 had CTCL not otherwise specified, 1 had mature T-cell lymphoma not otherwise specified, and 1 had primary cutaneous CD30+ T-cell lymphoma.16 The proliferation of 2 separate lymphocytic lineages is rare, but this study demonstrated an increased risk for CTCL to develop in patients with CLL. One possible explanation is that malignant cells come from a common stem cell progenitor or from genetic events. They occur secondary to carcinogens, viruses, or cytokines from T-cell or B-cell clones; they evolve due to treatment of the preexisting lymphoproliferative disease; or they occur simply by coincidence. The behavior of the CTCL may be more aggressive in patients with CLL due to immunosuppression, which may have contributed to the extreme presentation in our patient.16 Subcutaneous panniculitic T-cell lymphoma also has been reported in a patient with CLL that was thought to be associated with prior rituximab treatment.17
Treatment of SPTCL depends on the severity and course of the disease. In patients with more indolent disease, systemic steroids have been the most frequently used initial treatment.2,3,10 However, the disease often will progress after steroid tapering and require further intervention. Localized lesions may be treated with radiation alone or in combination with other systemic therapies.3,10 In refractory, aggressive, or relapsing cases, polychemotherapeutic regimens have proven to produce long-term remission in 30% of patients, with an overall response rate of 50%.10 These regimens most commonly have included cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or CHOP-like treatment (EPOCH regimen [etoposide, prednisone, oncovin, cyclophosphamide, and doxorubicin hydrochloride]).3,10 A stem cell transplant can be considered in patients with recurrent and refractory disease, and it also has been shown to induce remission.4,17 In patients with a good response to therapy, the disease often can be controlled for long periods of time, with an estimated 5-year survival rate of 80%.15
This case highlights the diagnostic challenges and variable presentations of SPTCL. Dermatologists, oncologists, and dermatopathologists should be aware of this condition and consider it in the differential diagnosis of a patient with a hematologic malignancy and unremitting facial and/or body swelling without any other cause. The possibility of a secondary hematologic cancer in a patient with CLL also must be taken into consideration. Early diagnosis and treatment can minimize morbidity and induce remission in most patients.
- Gonzalez CL, Medeiros LJ, Braziel RM, et al. T-cell lymphoma involving subcutaneous tissue. a clinicopathologic entity commonly associated with hemophagocytic syndrome. Am J Surg Pathol. 1991;15:17-27.
- Willemze R, Jansen P, Cerroni L, et al.
Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood. 2008;111:38-45. - Parveen Z, Thompson K. Subcutaneous panniculitis-like T-cell lymphoma: redefinition of diagnostic criteria in the recent World Health Organization–European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas. Arch Pathol Lab Med. 2009;133:303-308.
- Velez N, Ishizawar R, Dellaripa P, et al. Full facial edema: a novel presentation of subcutaneous panniculitis-like T-cell lymphoma. J Clin Oncol. 2012;30:e233-236.
- Asati D, Ingle V, Joshi D, et al. Subcutaneous panniculitis-like T-cell lymphoma with macrophage activation syndrome treated by cyclosporine and prednisolone. Indian Dermatol Online J. 2016;7:529-532.
- Fricker M, Dubach P, Helbing A, et al. Not all facial swellings are angioedemas! J Investig Allergol Clin Immunol. 2015;25:146-147.
- Kosari F, Akbarzadeh H. Local facial edema: a novel presentation of subcutaneous panniculitis-like T-cell lymphoma in a 30-year-old Iranian woman. Acta Med Iran. 2014;52:950-953.
- Bhojaraja M, Kistampally P, Udupa K, et al. Subcutaneous panniculitis-like T-cell lymphoma: a rare tumour. J Clin Diagn Res. 2016;10:OD29-OD30.
- Hashimoto R, Uchiyama M, Maeno T. Case report of subcutaneous panniculitis-like T-cell lymphoma complicated by eyelid swelling. BMC Ophthalmol. 2016;16:117.
- Chinello MN, Naviglio S, Remotti D, et al. Subcutaneous panniculitis-like T-cell lymphoma presenting with diffuse cutaneous edema in a 2-year-old child. J Pediatr Hematol Oncol. 2015;37:329-330.
- Chang TW, Weaver AL, Shanafelt TD, et al. Risk of cutaneous T-cell lymphoma in patients with chronic lymphocytic leukemia and other subtypes of non-Hodgkin lymphoma. Int J Dermatol. 2017;56:1125-1129.
- Chinello MN, Naviglio S, Remotti D, et al. Subcutaneous panniculitis-like T-cell lymphoma presenting with diffuse cutaneous edema in a 2-year-old child. J Pediatr Hematol Oncol. 2015;37:329-330.
- Jillella A, Day D, Severson K, et al. Non-Hodgkin’s lymphoma presenting as anasarca: probably mediated by tumor necrosis factor alpha (TNF-α). Leuk Lymphoma. 2000;38:419-422.
- Lee D-W, Yang J-H, Lee S-M, et al. Subcutaneous panniculitis-like T-cell lymphoma: a clinical and pathologic study of 14 Korean patients. Ann Dermatol. 2011;23:329-337.
- Jaffe ES. The 2008 WHO classification of lymphomas: implications for clinical practice and translational research [published online January 1, 2009]. Hematology Am Soc Hematol Educ Program. https://doi.org/10.1182/asheducation-2009.1.523
- Chang TW, Weaver AL, Shanafelt TD, et al. Risk of cutaneous T-cell lymphoma in patients with chronic lymphocytic leukemia and other subtypes of non-Hodgkin lymphoma. Int J Dermatol. 2017;56:1125-1129.
- Hall M, Sluzevich J, Snow J. Generalized subcutaneous panniculitis-like T-cell lymphoma following rituximab for hemolytic anemia in a patient with chronic lymphocytic leukemia. J Am Acad Dermatol. 2010;62(suppl 1):AB96.
- Gonzalez CL, Medeiros LJ, Braziel RM, et al. T-cell lymphoma involving subcutaneous tissue. a clinicopathologic entity commonly associated with hemophagocytic syndrome. Am J Surg Pathol. 1991;15:17-27.
- Willemze R, Jansen P, Cerroni L, et al.
Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood. 2008;111:38-45. - Parveen Z, Thompson K. Subcutaneous panniculitis-like T-cell lymphoma: redefinition of diagnostic criteria in the recent World Health Organization–European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas. Arch Pathol Lab Med. 2009;133:303-308.
- Velez N, Ishizawar R, Dellaripa P, et al. Full facial edema: a novel presentation of subcutaneous panniculitis-like T-cell lymphoma. J Clin Oncol. 2012;30:e233-236.
- Asati D, Ingle V, Joshi D, et al. Subcutaneous panniculitis-like T-cell lymphoma with macrophage activation syndrome treated by cyclosporine and prednisolone. Indian Dermatol Online J. 2016;7:529-532.
- Fricker M, Dubach P, Helbing A, et al. Not all facial swellings are angioedemas! J Investig Allergol Clin Immunol. 2015;25:146-147.
- Kosari F, Akbarzadeh H. Local facial edema: a novel presentation of subcutaneous panniculitis-like T-cell lymphoma in a 30-year-old Iranian woman. Acta Med Iran. 2014;52:950-953.
- Bhojaraja M, Kistampally P, Udupa K, et al. Subcutaneous panniculitis-like T-cell lymphoma: a rare tumour. J Clin Diagn Res. 2016;10:OD29-OD30.
- Hashimoto R, Uchiyama M, Maeno T. Case report of subcutaneous panniculitis-like T-cell lymphoma complicated by eyelid swelling. BMC Ophthalmol. 2016;16:117.
- Chinello MN, Naviglio S, Remotti D, et al. Subcutaneous panniculitis-like T-cell lymphoma presenting with diffuse cutaneous edema in a 2-year-old child. J Pediatr Hematol Oncol. 2015;37:329-330.
- Chang TW, Weaver AL, Shanafelt TD, et al. Risk of cutaneous T-cell lymphoma in patients with chronic lymphocytic leukemia and other subtypes of non-Hodgkin lymphoma. Int J Dermatol. 2017;56:1125-1129.
- Chinello MN, Naviglio S, Remotti D, et al. Subcutaneous panniculitis-like T-cell lymphoma presenting with diffuse cutaneous edema in a 2-year-old child. J Pediatr Hematol Oncol. 2015;37:329-330.
- Jillella A, Day D, Severson K, et al. Non-Hodgkin’s lymphoma presenting as anasarca: probably mediated by tumor necrosis factor alpha (TNF-α). Leuk Lymphoma. 2000;38:419-422.
- Lee D-W, Yang J-H, Lee S-M, et al. Subcutaneous panniculitis-like T-cell lymphoma: a clinical and pathologic study of 14 Korean patients. Ann Dermatol. 2011;23:329-337.
- Jaffe ES. The 2008 WHO classification of lymphomas: implications for clinical practice and translational research [published online January 1, 2009]. Hematology Am Soc Hematol Educ Program. https://doi.org/10.1182/asheducation-2009.1.523
- Chang TW, Weaver AL, Shanafelt TD, et al. Risk of cutaneous T-cell lymphoma in patients with chronic lymphocytic leukemia and other subtypes of non-Hodgkin lymphoma. Int J Dermatol. 2017;56:1125-1129.
- Hall M, Sluzevich J, Snow J. Generalized subcutaneous panniculitis-like T-cell lymphoma following rituximab for hemolytic anemia in a patient with chronic lymphocytic leukemia. J Am Acad Dermatol. 2010;62(suppl 1):AB96.
Practice Points
- Subcutaneous panniculitic T-cell lymphoma (SPTCL) is a rare type of cutaneous T-cell lymphoma that may be complicated by fatal hemophagocytic syndrome.
- Patients typically present with deep-seated plaques or nodules that may be masked by localized edema.
- A biopsy is necessary to diagnose SPTCL, as well as to assess the degree of cellular atypia, fat necrosis, karyorrhexis, cytophagia, and angioinvasion to distinguish it from other panniculitides.
- In patients with a known hematologic malignancy, a secondary malignancy must be considered in the differential diagnosis of paraneoplastic edema.
