User login
Molecular Markers and Targeted Therapies in the Management of Non-Small Cell Lung Cancer
INTRODUCTION
Lung cancer is the second most common type of cancer in the United States, with 222,500 estimated new cases in 2017, according to the American Cancer Society.1 However, it is by far the number one cause of death due to cancer, with an estimated 155,870 lung cancer–related deaths occurring in 2017, which is higher than the number of deaths due to breast cancer, prostate cancer, and colorectal cancer combined.1,2 Despite slightly decreasing incidence and mortality over the past decade, largely due to smoking cessation, the 5-year survival rate of lung cancer remains dismal at approximately 18%.2–4
Non-small cell lung cancer (NSCLC) accounts for 80% to 85% of all lung cancer cases.4 Traditionally, it is further divided based on histology: adenocarcinoma, squamous cell carcinoma, large cell carcinoma, and not otherwise specified.5 Chemotherapy had been the cornerstone of treatment for stage IV NSCLC. It is not target-specific and is most effective against rapidly growing cells. Common adverse effects include alopecia, nausea/vomiting, myelosuppression, cardiotoxicity, neuropathy, and nephrotoxicity. However, this paradigm has shifted following the discovery of mutations of the epidermal growth factor receptor (EGFR) gene as an oncogenic driver that confers sensitivity to small molecule tyrosine kinase inhibitors (TKIs) targeting EGFR.6 The EGFR inhibitors are given orally and have a spectrum of toxicities (eg, such as rash, diarrhea, and elevated transaminases) different from that of systemic chemotherapy, which is often administered intravenously. Following the discovery of EGFR mutations, rearrangements of the anaplastic lymphoma kinase (ALK) gene7 and ROS1 gene8 were identified as targetable driver mutations in NSCLC. The frequency of both rearrangements is lower than that of EGFR mutations. Additionally, BRAF V600E mutation has been identified in NSCLC.9–12 This activation mutation is commonly seen in melanoma. Agents that have already been approved for the treatment of melanoma with the BRAF V600E mutation are being tested in NSCLC patients with this mutation.13–16
Given the effectiveness and tolerability of targeted therapy, identifying this distinct molecular subset of NSCLC patients is critical in treatment. Currently, molecular testing is mandatory in all stage IV patients with non-squamous cell carcinoma, as a preponderance of patients with driver mutations have this histology subtype.5,17–19 For patients with squamous cell carcinoma, molecular testing should be considered if the biopsy specimen is small, there is mixed histology, or the patient is a nonsmoker.5,20 Several techniques are commonly utilized in detecting these genetic alterations. EGFR mutation can be detected by polymerase chain reaction (PCR), ALK or ROS1 rearrangement can be detected by fluorescence in-situ hybridization (FISH), and immunohistochemistry (IHC) can also be used to detect ALK rearrangement. The current guideline is to use comprehensive genomic profiling to capture all the potential molecular targets simultaneously instead of running stepwise tests just for EGFR, ALK, and ROS1.5BRAF V600E mutation,13–16 MET exon 14 skipping mutation,21–24 RET rearrangements,25–27 and HER2 mutations28–30 are among the emergent genetic alterations with various responses to targeted therapy.31 Some of these targeted agents have been approved for other types of malignancy, and others are still in the development phase.
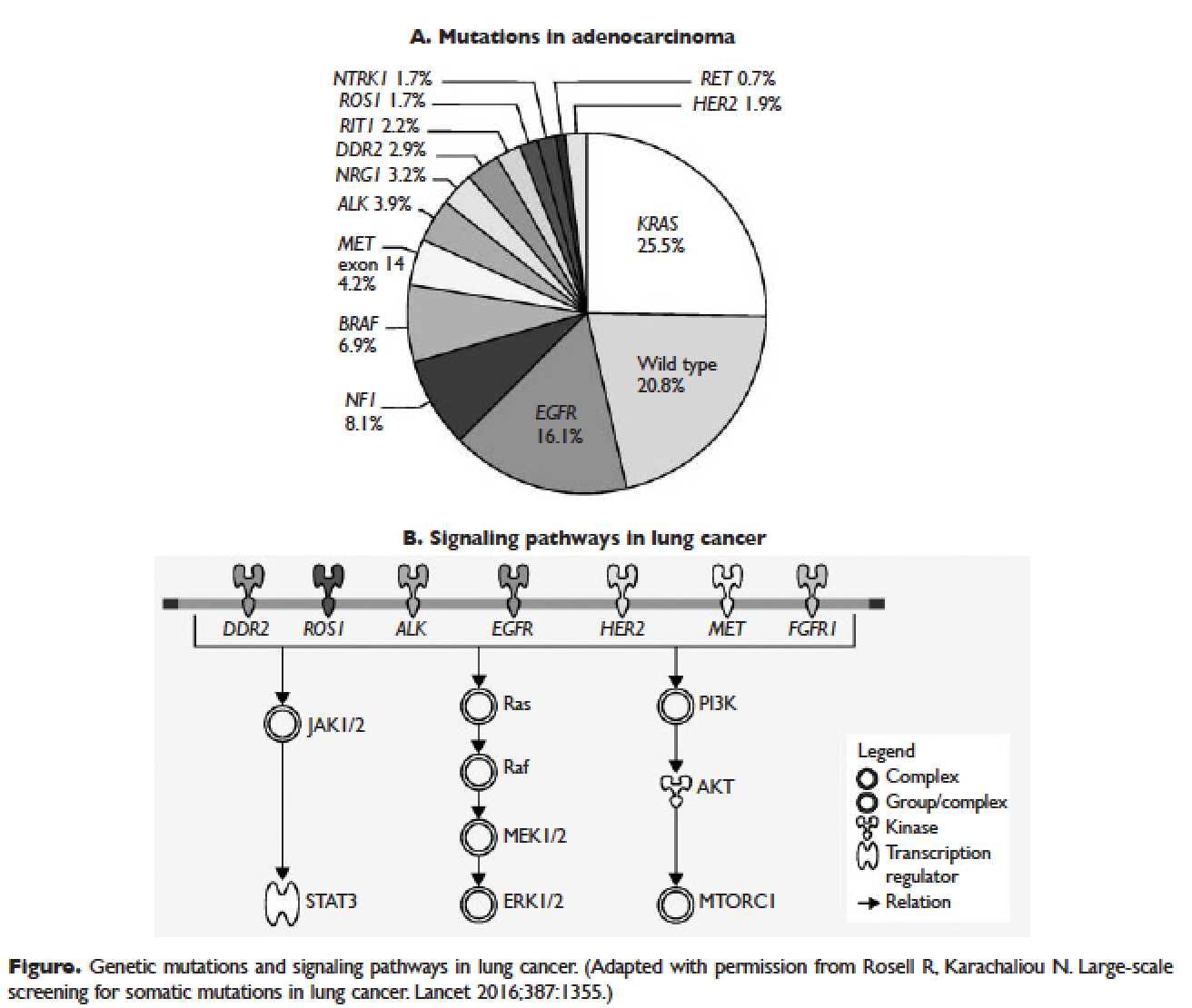
Several initiatives worldwide have reported better outcomes of patients with driver mutations treated with targeted therapy. For instance, the Lung Cancer Mutation Consortium in the United States demonstrated that the median survival of patients without driver mutations, with drivers mutations but not treated with targeted therapy, and with driver mutations and treated with targeted therapy was 2.08 years, 2.38 years, and 3.49 years, respectively.32 The French Cooperative Thoracic Intergroup-French National Cancer Institute demonstrated that the median survival for patients with driver mutations versus those without driver mutations was 16.5 months versus 11.8 months.33 The Spanish Lung Cancer Group demonstrated that the overall survival (OS) for patients with EGFR mutations treated with erlotinib was 27 months.34 The mutations in lung cancer, their frequencies, and the downstream signaling pathways are depicted in the Figure.35
In this article, we discuss targeted therapy for patients with EGFR mutations, ALK rearrangements, ROS1 rearrangements, and BRAF V600E mutation. We also discuss the management of patients with EGFR mutations who develop a secondary mutation after TKI therapy. Almost all of the targeted agents discussed herein have been approved by the US Food and Drug Administration (FDA), so they are considered standard of care. All available phase 3 trials pertinent to these targeted therapies are included in the discussion.
EGFR MUTATIONS
CASE PRESENTATION 1
A 54-year-old Caucasian man who is a former smoker with a 10 pack-year history and past medical history of hypertension and dyslipidemia presents with progressive dyspnea for several weeks. A chest x-ray shows moderate pleural effusion on the left side with possible mass-like opacity on the left upper lung field. An ultrasound-guided thoracentesis is performed and cytology is positive for adenocarcinoma of likely pulmonary origin. Staging workup including positron emission tomography (PET)/computed tomography (CT) and magnetic resonance imaging of the brain with and without contrast is done. PET/CT shows a 5.5-cm mass in the left upper lobe of the lung with high fluorodeoxyglucose (FDG) uptake, several 1- to 2-cm mediastinal lymph nodes with moderate FDG uptake, and small pleural effusion on both sides with moderate FDG uptake. MRI-brain is negative for malignancy. The patient subsequently undergoes a CT-guided biopsy of the lung mass, which shows moderately differentiated adenocarcinoma. Comprehensive molecular profiling reveals EGFR L858R mutation only. The patient now presents for the initial consultation. Of note, his Eastern Cooperative Oncology Group performance status is 1.
What is the next step in the management of this patient?
FIRST-LINE TKI FOR SENSITIZING EGFR MUTATIONS
The 2 most common EGFR mutations are deletions in exon 19 and substitution of arginine for leucine in exon 21 (L858R), found in approximately 45% and 40% of patients with EGFR mutations, respectively.36 Both mutations are sensitive to EGFR TKIs. The benefit may be greater in patients with exon 19 deletions as compared to exon 21 L858R substitution,37,38 but this has not been demonstrated consistently in clinical trials.39-43 In the United States, EGFR mutations are found in approximately 10% of patients with NSCLC, while the incidence can be as high as 50% in Asia.44 Even though the cobas EGFR mutation test is the companion diagnostic approved by the US FDA, a positive test result from any laboratory with the Clinical Laboratory Improvement Amendments (CLIA) certificate should prompt the use of an EGFR TKI as the initial treatment.
Three EGFR TKIs that have been approved as first-line therapy in the United States are available: erlotinib, afatinib, and gefitinib.5 Both erlotinib and gefitinib are considered first-generation TKIs. They have higher binding affinity for the 2 common EGFR mutations than wild-type EGFR. In addition, they reversibly bind to the intracellular tyrosine kinase domain, resulting in inhibition of autophosphorylation of the tyrosine residues. Afatinib, a second-generation and irreversible TKI, targets EGFR (HER1) as well as HER2 and HER4.45
The superior efficacy of the EGFR TKIs over platinum doublet chemotherapy in treatment-naïve patients with EGFR mutations has been demonstrated in 7 randomized trials to date (Table).46 Erlotinib was the TKI arm for the OPTIMAL,41 EURTAC,42 and ENSURE trials;38 afatinib was the TKI arm for LUX-LUNG 337 and 6;43 gefitinib was the TKI arm for NEJ00239,47 and WJTOG3405.40 A meta-analysis of these 7 trials by Lee et al showed that progression-free survival (PFS) was significantly prolonged by EGFR TKIs (hazard ratio [HR] 0.37 [95% confidence interval {CI} 0.32 to 0.42]).46 For instance, in the EURTAC trial, median PFS was 9.7 months for patients treated with erlotinib as compared to 5.2 months for patients treated with platinum/gemcitabine or platinum/docetaxel.42 In this meta-analysis, prespecified subgroups included age, sex, ethnicity, smoking status, performance status, tumor histology, and EGFR mutation subtype. The superior outcome with TKIs was observed in all subgroups. Furthermore, patients with exon 19 deletions, nonsmokers, and women had even better outcomes.46
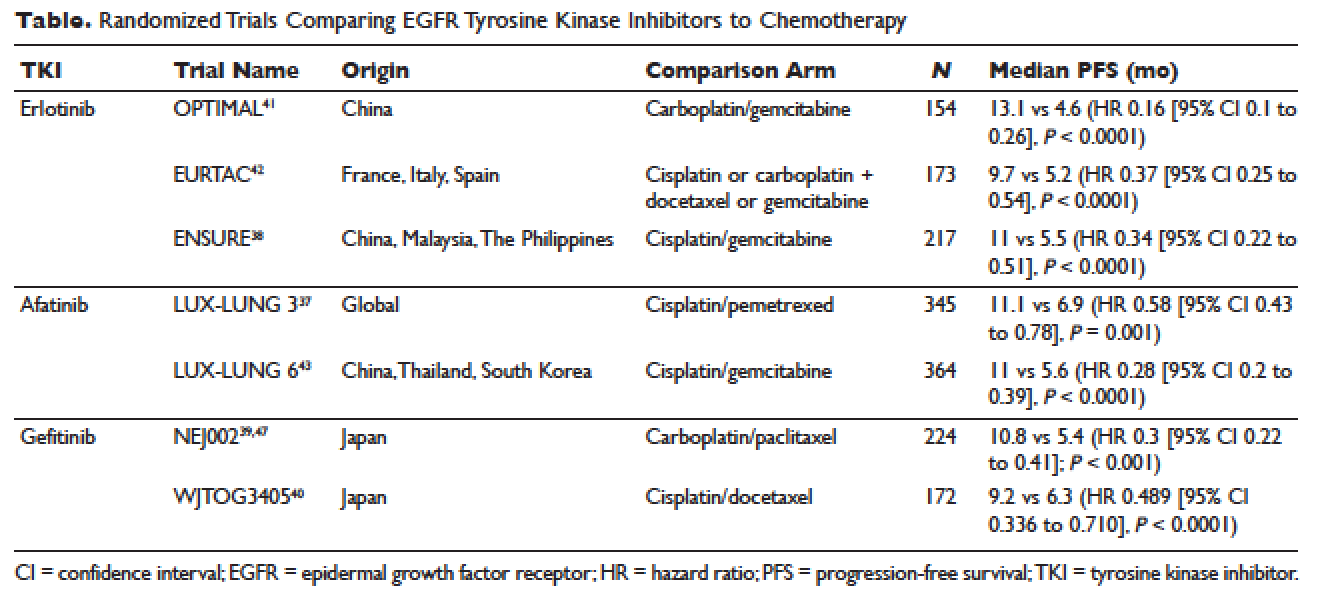
Erlotinib is the most commonly used TKI in the United States largely because gefitinib was off the market for some time until it was re-approved by the FDA in 2015. Interestingly, this “re-approval” was not based on either 1 of the 2 prospective trials (NEJ00239,47 and WJTOG340540), but rather was based on an exploratory analysis of the IPASS trial48,49 as well as a prospective phase 4, single-arm trial in Europe (IFUM).50 The superior efficacy of gefitinib over carboplatin/paclitaxel among patients with EGFR mutations in the IPASS trial was confirmed by blind independent central review, with longer PFS (HR 0.54 [95% CI 0.38 to 0.79] P = 0.0012) and higher objective response rate (ORR; odds ratio 3 [95% CI 1.63 to 5.54], P = 0.0004).49
CASE 1 CONTINUED
Based on the EGFR L858R mutation status, the patient is started on erlotinib. He is quite happy that he does not need intravenous chemotherapy but wants to know what toxicities he might potentially have with erlotinib.
What are the common adverse effects (AEs) of EGFR TKIs? How are AEs of TKIs managed?
Safety Profile
The important toxicities associated with EGFR TKIs are rash, gastrointestinal toxicity, hepatic toxicity, and pulmonary toxicity. Rash is an AE specific to all agents blocking the EGFR pathway, including small molecules and monoclonal antibodies such as cetuximab. The epidermis has a high level of expression of EGFR, which contributes to this toxicity.51 Rash usually presents as dry skin or acneiform eruption. Prophylactic treatment with oral tetracyclines and topical corticosteroids is generally recommended upon initiation of TKI therapy. Diarrhea is the most prevalent gastrointestinal toxicity. All patients starting treatment should be given prescriptions to manage diarrhea such as loperamide and be advised to call when it occurs. Hepatic toxicity is often manifested as elevated transaminases or bilirubin. Interstitial lung disease (ILD) is a rare but potentially fatal pulmonary toxicity.
Rash of any grade was reported in 49.2% of patients treated with erlotinib in clinical trials, while grade 3 rash occurred in 6% of patients and no grade 4 was reported. Diarrhea of any grade was reported in 20.3% of patients, grade 3 diarrhea occurred in 1.8%, and no grade 4 was reported. Grade 2 and 3 alanine aminotransferase (ALT) elevations were seen in 2% and 1% of patients, respectively. Grade 2 and 3 bilirubin elevations were seen in 4% and less than 1% of patients, respectively. The incidence of serious ILD-like events was less than 1%.52
Afatinib is associated with higher incidences of rash and diarrhea. Specifically, diarrhea and rash of all grades were reported in 96% and 90% of patients treated with afatinib, respectively. Paronychia of all grades occurred in 58% of patients. Elevated ALT of all grades was seen in 11% of patients. Approximately 1.5% of patients treated with afatinib across clinical trials had ILD or ILD-like AEs.53
Gefitinib, the most commonly used TKI outside United States, has a toxicity profile similar to erlotinib, except for hepatic toxicity. For instance, rash of all grades occurred in 47% of patients, diarrhea of all grades occurred in 29% of patients, and ILD or ILD-like AEs occurred in 1.3% of patients across clinical trials. In comparison, elevated ALT and aspartate aminotransferase (AST) of all grades was seen in 38% and 40% of patients, respectively.54 Therefore, close monitoring of liver function is clinically warranted. In particular, patients need to be advised to avoid concomitant use of herbal supplements, a common practice in Asian countries.
CASE 1 CONTINUED
The patient does well while on erlotinib at 150 mg orally once daily for about 8 months, until he develops increasing abdominal pain. A CT scan of the abdomen and pelvis with contrast shows a new 8-cm right adrenal mass. Additionally, a repeat CT scan of the chest with contrast shows a stable lung mass but enlarging mediastinal lymphadenopathy.
How would you manage the patient at this point?
MANAGEMENT OF T790M MUTATION AFTER PROGRESSION ON FIRST-LINE EGFR TKIS
As mentioned above, the median PFS of patients with EGFR mutations treated with 1 of the 3 TKIs is around 9 to 13 months.46 Of the various resistance mechanisms that have been described, the T790M mutation is found in approximately 60% of patients who progress after treatment with first-line TKIs.55,56 Other mechanisms, such as HER2 amplification, MET amplification, or rarely small cell transformation, have been reported.56 The first- and second-generation EGFR TKIs function by binding to the ATP-binding domain of mutated EGFR, leading to inhibition of the downstream signaling pathways (Figure, part B) and ultimately cell death.35 The T790M mutation hinders the interaction between the ATP-binding domain of EGFR kinase and TKIs, resulting in treatment resistance and disease progression.57,58
Osimertinib is a third-generation irreversible EGFR TKI with activity against both sensitizing EGFR and resistant T790M mutations. It has low affinity for wide-type EGFR as well as insulin receptor and insulin-like growth factor receptor.59 Osimertinib has been fully approved for NSCLC patients with EGFR mutations who have progressed on first-line EGFR TKIs with the development of T790M mutation. An international phase 3 trial (AURA3) randomly assigned 419 patients in a 2:1 ratio to either osimertinib or platinum/pemetrexed. Eligible patients all had the documented EGFR mutations and disease progression after first-line EGFR TKIs. Central confirmation of the T790M mutation was required. Median PFS by investigator assessment, the trial’s primary end point, was 10.1 months for osimertinib versus 4.4 months for chemotherapy (HR 0.3 [95% CI 0.23 to 0.41]; P < 0.001). ORR was 71% for osimertinib versus 31% for chemotherapy (HR 5.39 [95% CI 3.47 to 8.48], P < 0.001). A total of 144 patients with stable and asymptomatic brain metastases were also eligible. Median PFS for this subset of patients treated with osimertinib and chemotherapy was 8.5 months and 4.2 months, respectively (HR 0.32 [95% CI 0.21 to 0.49]). In the AURA3 trial, osimertinib was better tolerated than chemotherapy, with 23% of patients treated with osimertinib experiencing grade 3 or 4 AEs as compared to 47% of chemotherapy-treated patients. The most common AEs of any grade were diarrhea (41%), rash (34%), dry skin (23%), and paronychia (22%).60
For the case patient, a reasonable approach would be to obtain a tissue biopsy of the adrenal mass and more importantly to check for the T790M mutation. Similar to the companion diagnostic for EGFR mutations, the cobas EGFR mutation test v2 is the FDA-approved test for T790M. However, if this resistance mutation is detected by any CLIA-certified laboratories, osimertinib should be the recommended treatment option. If tissue biopsy is not feasible, plasma-based testing should be considered. A blood-based companion diagnostic also is FDA approved.
ALK REARRANGEMENTS
CASE 2 PRESENTATION
A 42-year-old Korean woman who is a non-smoker with no significant past medical history presents with fatigue, unintentional weight loss of 20 lb in the past 4 months, and vague abdominal pain. A CT can of the abdomen and pelvis without contrast shows multiple foci in the liver and an indeterminate nodule in the right lung base. She subsequently undergoes PET/CT, which confirms multiple liver nodules/masses ranging from 1 to 3 cm with moderate FDG uptake. In addition, there is a 3.5-cm pleura-based lung mass on the right side with moderate FDG uptake. MRI-brain with and without contrast is negative for malignancy. A CT-guided biopsy of 1 of the liver masses is ordered and pathology returns positive for poorly differentiated adenocarcinoma consistent with lung primary. Molecular analysis reveals an echinoderm microtubule-associated protein-like 4 (EML4)-ALK rearrangement. She is placed on crizotinib by an outside oncologist and after about 3 weeks of therapy is doing well. She is now in your clinic for a second opinion. She says that some of her friends told her about another medication called ceritinib and was wondering if she would need to switch her cancer treatment.
How would you respond to this patient’s inquiry?
FIRST-LINE TKIS FOR ALK REARRANGEMENTS
ALK rearrangements are found in 2% to 7% of NSCLC, with EML4-ALK being the most prevalent fusion variant.61 The inversion of chromosome 2p leads to the fusion of the EML4 gene and the ALK gene, which causes the constitutive activation of the fusion protein and ultimately increased transformation and tumorigenicity.7,61 Patients harboring ALK rearrangements tend to be non-smokers. Adenocarcinoma, especially signet ring cell subtype, is the predominant histology. Compared to EGFR mutations, patients with ALK mutations are significantly younger and more likely to be men.62ALK rearrangements can be detected by either FISH or IHC, and most next-generation sequencing (NGS) panels have the ability to identify this driver mutation.
Crizotinib is the first approved ALK inhibitor for the treatment of NSCLC in this molecular subset of patients.63 PROFILE 1014 is a phase 3 randomized trial that compared crizotinib with chemotherapy containing platinum/pemetrexed for up to 6 cycles. Crossover to crizotinib was allowed for patients with disease progression on chemotherapy. The primary end point was PFS by independent radiologic review. The crizotinib arm demonstrated superior PFS (10.9 months versus 7 months; HR 0.45 [95% CI 0.35 to 0.6], P < 0.001) and ORR (74% versus 45%, P < 0.001). Median survival was not reached in either arm (HR 0.82 [95% CI 0.54 to 1.26], P = 0.36).64 Based on this international trial, crizotinib is considered standard of care in the United States for treatment-naïve patients with advanced NSCLC harboring ALK rearrangements. The current recommended dose is 250 mg orally twice daily. Common treatment-related AEs of all grades include vision disorder (62%), nausea (53%), diarrhea (43%), vomiting (40%), edema (28%), and constipation (27%).65 PROFILE 1007 compared crizotinib with pemetrexed or docetaxel in ALK-rearranged NSCLC patients with prior exposure to 1 platinum-based chemotherapy. The median PFS was 7.7 months for crizotinib as compared to 3 months for chemotherapy (HR 0.49 [95% CI 0.37 to 0.64], P < 0.001). The response rates were 65% and 20% for crizotinib and chemotherapy, respectively (P < 0.001).66 In other countries, crizotinib following 1 prior platinum-based regimen may be considered standard of care based on this trial.
Ceritinib is an oral second-generation ALK inhibitor that is 20 times more potent than crizotinib based on enzymatic assays.67 It also targets ROS1 and insulin-like growth factor 1 receptor but not c-MET. It was first approved by the FDA in April 2014 for metastatic ALK-rearranged NSCLC following crizotinib.68 In May 2017, the FDA granted approval of ceritinib for treatment-naïve patients. This decision was based on the results of the ASCEND-4 trial, a randomized phase 3 trial assessing the efficacy and safety of ceritinib over chemotherapy in the first-line setting. The trial assigned 376 patients to either ceritinib at 750 mg once daily or platinum/pemetrexed for 4 cycles followed by maintenance pemetrexed. Median PFS was 16.6 months for ceritinib versus 8.1 months for chemotherapy (HR 0.55 [95% CI 0.42 to 0.73]; P < 0.00001).69 Toxicities of ceritinib are not negligible, with gastrointestinal toxicity being the most prevalent. For instance, diarrhea, nausea, vomiting, abdominal pain, and constipation of all grades were seen in 86%, 80%, 60%, 54%, and 29% of patients, respectively. Furthermore, fatigue and decreased appetite occurred in 52% and 34% of patients, respectively. In terms of laboratory abnormalities, 84% of patients experienced decreased hemoglobin of all grades; 80% increased ALT; 75% increased AST; 58% increased creatinine; 49% increased glucose; 36% decreased phosphate; and 28% increased lipase. Due to these AEs, the incidence of dose reduction was about 58% and the median onset was around 7 weeks.70
Alectinib is another oral second-generation ALK inhibitor that was approved by the FDA in December 2015 for the treatment of NSCLC patients with ALK rearrangements who have progressed on or are intolerant to crizotinib.71 Its indication will soon be broadened to the first-line setting based on the ALEX trial.72 Alectinib is a potent and highly selective TKI of ALK73 with activity against known resistant mutations to crizotinib.74,75 It also inhibits RET but not ROS1 or c-MET.76 ALEX, a randomized phase 3 study, compared alectinib with crizotinib in treatment-naïve patients with NSCLC harboring ALK rearrangements. The trial enrolled 303 patients and the median follow-up was approximately 18 months. The alectinib arm (600 mg twice daily) demonstrated significantly higher PFS by investigator-assessment, the trial’s primary end point. The 12-month event-free survival was 68.4% (95% CI 61% to 75.9%) versus 48.7% (95% CI 40.4% to 56.9%) for alectinib and crizotinib, respectively (HR 0.47 [95% CI 0.34 to 0.65], P < 0.001). The median PFS was not reached in the alectinib arm (95% CI 17.7 months to not estimable) as compared to 11.1 months in the crizotinib arm (95% CI 9.1 to 13.1 months).72 Alectinib is generally well tolerated. Common AEs of all grades include fatigue (41%), constipation (34%), edema (30%), and myalgia (29%). As alectinib can cause anemia, lymphopenia, hepatic toxicity, increased creatine phosphokinase, hyperglycemia, electrolyte abnormalities, and increased creatinine, periodic monitoring of these laboratory values is important, although most of these abnormalities are grade 1 or 2.77
Brigatinib, another oral second-generation ALK inhibitor, was granted accelerated approval by the FDA in April 2017 for ALK-rearranged and crizotinib-resistant NSCLC based on the ALTA trial. This randomized phase 2 study of brigatinib showed an ORR by investigator assessment of 54% (97.5% CI 43% to 65%) in the 180 mg once daily arm with lead-in of 90 mg once daily for 7 days. Median PFS was 12.9 months (95% CI 11.1 months to not reached [NR]).78 Currently, a phase 3 study of brigatinib versus crizotinib in ALK inhibitor–naïve patients is recruiting participants (ALTA-1L). It will be interesting to see if brigatinib can achieve a front-line indication.
Starting the case patient on crizotinib is well within the treatment guidelines. One may consider ceritinib or alectinib in the first-line setting, but both TKIs can be reserved upon disease progression. We would recommend a repeat biopsy at that point to look for resistant mechanisms, as certain secondary ALK mutations may be rescued by certain next-generation ALK inhibitors. For instance, the F1174V mutation has been reported to confer resistance to ceritinib but sensitivity to alectinib, while the opposite is true for I1171T. The G1202R mutation is resistant to ceritinib, alectinib, and brigatinib, but lorlatinib, a third-generation ALK inhibitor, has shown activity against this mutation.79 Furthermore, brain metastasis represents a treatment challenge for patients with ALK rearrangements. It is also an efficacy measure of next-generation ALK inhibitors, all of which have demonstrated better central nervous system activity than crizotinib.69,78,80 If the case patient were found to have brain metastasis at the initial diagnosis, either ceritinib or alectinib would be a reasonable choice since crizotinib has limited penetration of blood-brain barrier.81
ROS1 REARRANGEMENTS
CASE PRESENTATION 3
A 66-year-old Chinese woman who is a non-smoker with a past medical history of hypertension and hypothyroidism presents to the emergency department for worsening lower back pain. Initial workup includes x-ray of the lumbar spine followed by MRI with contrast, which shows a soft tissue mass at L3-4 without cord compression. CT of the chest, abdomen, and pelvis with contrast shows a 7-cm right hilar mass, bilateral small lung nodules, mediastinal lymphadenopathy, and multiple lytic lesions in ribs, lumbar spine, and pelvis. MRI-brain with and without contrast is negative for malignancy. She undergoes endo-bronchial ultrasound and biopsy of the right hilar mass, which shows poorly differentiated adenocarcinoma. While waiting for the result of the molecular analysis, the patient undergoes palliative radiation therapy to L2-5 with good pain relief. She is discharged from the hospital and presents to your clinic for follow up. Molecular analysis now reveals ROS1 rearrangement with CD74-ROS1 fusion.
What treatment plan should be put in place for this patient?
FIRST-LINE THERAPY FOR ROS1 REARRANGEMENTS
Approximately 2.4% of lung adenocarcinomas harbor ROS1 rearrangements.82 This distinct genetic alteration occurs more frequently in NSCLC patients who are younger, female, and never-smokers, and who have adenocarcinomas.8 It has been shown that ROS1 rearrangements rarely overlap with other genetic alterations including KRAS mutations, EGFR mutations, and ALK rearrangements.83 As a receptor tyrosine kinase, ROS1 is similar to ALK and insulin receptor family members.84 Crizotinib, which targets ALK, ROS1, and c-MET, was approved by the FDA on March 11, 2016, for the treatment of metastatic ROS1-rearranged NSCLC.85 The approval was based on a phase 2 expansion cohort of the original phase 1 study. Among 50 US patients enrolled in this expansion cohort, 3 had complete responses and 33 had partial responses with ORR of 72% (95% CI 58% to 84%). Median PFS was 19.2 months (95% CI 14.4 months to NR) and median duration of response (DOR) was 17.6 months (95% CI 14.5 months to NR).86 During longer follow-up, independent radiology review confirmed high ORR of 66% and median DOR of 18.3 months.85
Interestingly, no companion diagnostic assay has been approved for the detection of ROS1 rearrangements with the approval of crizotinib. In the United States, break apart FISH is the most common detection method. In fact, in the above mentioned phase 2 study, ROS1 rearrangements were detected in 49 out of 50 patients by this method.86 FISH can be technically challenging when dealing with high volume and multiple targets. Reverse transcriptase-PCR is another detection method, but it requires knowledge of the fusion partners. To date, at least 14 ROS1 fusion partners have been reported, with CD74 being the most common.87 NGS with appropriate design and validation can also be used to detect ROS1 rearrangements.
For the case patient, the recommendation would be to start her on crizotinib at 250 mg twice daily. Monitoring for vision disturbance, gastrointestinal complaints, and edema is warranted. Because the estimated onset of response is around 7.9 weeks,86 plans should be made to repeat her scans in approximately 2 months.
BRAF V600E MUTATIONS
CASE PRESENTATION 4
A 71-year-old Caucasian man with a past medical history of hypertension, dyslipidemia, and ischemic cerebrovascular accident without residual deficits was diagnosed with stage IV adenocarcinoma of the lung about 8 months ago. He has a 40 pack-year smoking history and quit smoking when he was diagnosed with lung cancer. His disease burden involved a large mediastinal mass, scattered pleural nodules, multiple lymphadenopathy, and several soft tissue masses. His outside oncologist started him on chemotherapy containing carboplatin and pemetrexed for 6 cycles followed by maintenance pemetrexed. The most recent restaging scans show disease progression with enlarging soft tissue masses and several new lytic bone lesions. MRI-brain with and without contrast shows 2 subcentimeter enhancing lesions. He transferred care to you approximately 4 weeks ago. You ordered a repeat biopsy of 1 of the enlarging soft tissue masses. Molecular analysis revealed BRAF V600E mutation. In the interim, he underwent stereotactic radiosurgery for the 2 brain lesions without any complications. The patient is now in your clinic for follow up.
What would be your recommended systemic treatment?
TARGETED THERAPIES FOR BRAF V600E MUTATION
BRAF mutations were first recognized as activating mutations in advanced melanomas, with BRAF V600E, resulting from the substitution of glutamic acid for valine at amino acid 600, being the most common. BRAF plays an important role in the mitogen-activated protein kinase (MAPK) signaling pathway. Briefly, the activation of MAPK pathway occurs upon ligand binding of receptor tyrosine kinases, which then involves RAS/BRAF/MEK/ERK in a stepwise manner, ultimately leading to cell survival. BRAF mutations have been increasingly recognized also as driver mutations in NSCLC.9–12 They can be detected by PCR or NGS method. The characteristics of NSCLC patients harboring BRAF mutations have been described by various groups.9–12 For instance, 1 case series showed that the incidence was 2.2% among patients with advanced lung adenocarcinoma; 50% of mutations were V600E, while G469A and D594G accounted for the remaining 39% and 11% of patients, respectively. All patients were either current or former smokers. The median OS of patients with BRAF mutations in this case series was NR, while it was 37 months for patients with EGFR mutations (P = 0.73) and NR for patients with ALK rearrangements (P = 0.64).9
For patients with BRAF V600E–mutant NSCLC who have progressed on platinum-based chemotherapy, the combination of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) may represent a new treatment paradigm. This was illustrated in a phase 2, nonrandomized, open-label study. A total of 57 patients were enrolled and 36 patients (63.2% [95% CI 49.3% to 75.6%]) achieved an overall response by investigator assessment, the trial’s primary end point. Disease control rate was 78.9% (95% CI 66.1% to 88.6%), with 4% complete response, 60% partial response, and 16% stable disease. PFS was 9.7 months (95% CI [6.9 to 19.6 months]). The safety profile was comparable to what had been observed in patients with melanoma treated with this regimen. More specifically, 56% of patients on this trial reported serious AEs, including pyrexia (16%), anemia (5%), confusional state (4%), decreased appetite (4%), hemoptysis (4%), hypercalcemia (4%), nausea (4%), and cutaneous squamous cell carcinoma (4%). In addition, neutropenia (9%) and hyponatremia (7%) were the most common grade 3-4 AEs.16
The case patient has experienced disease progression after 1 line of platinum-based chemotherapy, so the combination of dabrafenib and trametinib would be a robust systemic treatment option. dabrafenib as a single agent has also been studied in BRAF V600E–mutant NSCLC in a phase 2 trial. The overall response by investigator assessment among 84 patients was 33% (95% CI 23% to 45%).14 Vemurafenib, another oral BRAF TKI, has demonstrated efficacy for NSCLC patients harboring BRAF V600E mutation. In the cohort of 20 patients with NSCLC, the response rate was 42% (95% CI 20% to 67%) and median PFS was 7.3 months (95% CI 3.5 to 10.8 months).13 Patients with non-V600E mutations have shown variable responses to targeted therapies. MEK TKIs may be considered in this setting; however, the details of this discussion are beyond the scope of this review.
CONCLUSION
The management of advanced NSCLC with driver mutations has seen revolutionary changes over the past decade. Tremendous research has been done in order to first understand the molecular pathogenesis of NSCLC and then discover driver mutations that would lead to development of targeted therapies with clinically significant efficacy as well as tolerability. More recently, increasing efforts have focused on how to conquer acquired resistance in patients with disease progression after first-line TKIs. The field of EGFR-mutant NSCLC has set a successful example, but the work is nowhere near finished. The goals are to search for more driver mutations and to design agents that could potentially block cell survival signals once and for all.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
- Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol 2016;893:1–19.
- Alberg AJ, Brock MV, Ford JG, et al. Epidemiology of lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013;143:e1S–29S.
- Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-3013, based on November 2015 SEER data submission, posted to the SEER website, April 2016. Bethesda (MD): National Cancer Institute; 2016.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Non-Small Cell Lung Cancer: 1–190.
- Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–39.
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–6.
- Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancer. J Clin Oncol 2012;30:863–70.
- Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011;29:2046–51.
- Kinno T, Tsuta K, Shiraishi K, et al. Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol 2014;25:138–42.
- Litvak AM, Paik PK, Woo KM, et al. Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J Thorac Oncol 2014;9:1669–74.
- Villaruz LC, Socinski MA, Abberbock S, et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer 2015;121:448–56.
- Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–36.
- Planchard D, Kim TM, Mazieres J, et al. DaBRAFenib in patients with BRAF V600E-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol 2016;17:642–50.
- Gautschi O, Milia J, Cabarrou B, et al. Targeted therapy for patients with BRAF-mutant lung cancer: results from the European EURAF cohort. J Thorac Oncol 2015;10:1451–7.
- Planchard D, Besse B, Groen HJ, et al. DaBRAFenib plus trametinib in patients with previously treated BRAF V600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 2016;17:984–93.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. J Thorac Oncol 2013;8:823–59.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. Arch Pathol Lab Med 2013;137:828–60.
- Leighl NB, Rekhtman N, Biermann WA, et al. Molecular testing for selection of patients with lung cancer for epidermal growth factor receptor and anaplastic lymphoma kinase tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology guideline. J Clin Oncol 2014;32:3673–9.
- Paik PK, Varghese AM, Sima CS, et al. Response to erlotinib in patients with EGFR mutant advanced non-small cell lung cancers with a squamous or squamous-like component. Mol Cancer Ther 2012;11:2535–40.
- Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov 2015;5:842–9.
- Awad MM, Oxnard GR, Jackman DM, et al. MET Exon 14 mutations in non-small-cell lung cancer are associated with advanced age, and stage-dependent MET genomic amplification, and c-MET overexpression. J Clin Oncol 2016;34:721–30.
- Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thorac Oncol 2016;11:1493–502.
- Reungwetwattana T, Liang Y, Zhu V, et al. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The why, the how, the who, the unknown, and the inevitable. Lung Cancer 2017;103:27–37.
- Drilon A, Wang L, Hasanovic A, et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov 2013;3:630–5.
- Lin JJ, Kennedy E, Sequist LV, et al. Clinical activity of alectinib in advanced RET-rearranged non-small cell lung cancer. J Thorac Oncol 2016;11:2027–32.
- Drilon A, Rekhtman N, Arcila M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol 2016;17:1653–60.
- Cappuzzo F, Bemis L, Varella-Garcia M. HER2 mutation and response to trastuzumab therapy in non-small-cell lung cancer. N Engl J Med 2006;354:2619–21.
- Mazieres J. Barlesi F, Filleron T, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targted drugs: results from the European EUHER2 cohort. Annal Oncol 2016;27:281–6.
- Ou SH, Schrock AB, Bocharov EV, et al. HER2 transmembrane (TMD) mutations (V659/G660) that stabilize homo- and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J Thorac Oncol 2017;12:446–57.
- Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emergent therapies. Cancer Discov 2017;7:596–609.
- Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014;311:1998–2006.
- Barlesi F, Mazieres J, Merlio JP, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016;387:1415–26.
- Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361:958–67.
- Rosell R, Karachaliou N, et al. Large-scale screening for somatic mutations in lung cancer. Lancet 2016;387:1354–6.
- Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005;97:339–46.
- Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013;31:3327–34.
- Wu YL, Chou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol 2015;26:1883–9.
- Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Eng J Med 2010;362:2380–8.
- Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121–8.
- Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011;12:735–42.
- Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239–46.
- Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:213–22.
- Hirsch FR, Bunn PA Jr. EGFR testing in lung cancer is ready for prime time. Lancet Oncol 2009;10:432–3.
- Nelson V, Ziehr J, Aqulnik M, et al. Afatinib: emerging next-generation tyrosine kinase inhibitor for NSCLC. Onco Targets Ther 2013;5:135–43.
- Lee CK, Wu YL, Ding PN, et al. Impact of specific epidermal growth factor receptor (EGFR) mutations and clinical characteristics on outcomes after treatment with EGFR tyrosine kinase inhibitors versus chemotherapy in EGFR-mutant lung cancer: a meta-analysis. J Clin Oncol 2015;33:1958–65.
- Inoue A, Kobayashi K, Maemondo M, et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann Oncol 2013;24:54–9.
- Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947–57.
- Wu YL, Saijo N, Thongprasert S, et al. Efficacy according to blind independent central review: post-hoc analyses from the phase III, randomized, multicenter, IPASS study of first-line gefitinib versus carboplatin/paclitaxel in Asian patients with EFGR mutation-positive advanced NSCLC. Lung Cancer 2017;104:119–25.
- Douillard JY, Ostoros G, Cobo M, et al. First-line gefitinib in Caucasian EGFR-mutation positive NSCLC patients: a phase-IV, open-label, single-arm study. Br J Cancer 2014;110:55–62.
- Hu JC, Sadeghi P, Pinter-Brown LC, et al. Cutaneous side effects of epidermal growth factor receptor inhibitors: clinical presentation, pathogenesis, and management. J Am Acad Dermatol 2007;56:317–26.
- Tarceva [package insert]. South San Francisco (CA): Genentech, Inc; 2010. www.accessdata.fda.gov/drugsatfda_docs/label/2010/021743s14s16lbl.pdf. Accessed April 23, 2017.
- Gilotrif [package insert.] Ridgefield (CT): Boehringer Ingelheim, Inc; 2013. www.accessdata.fda.gov/drugsatfda_docs/label/2013/201292s000lbl.pdf. Accessed April 23, 2017.
- Iressa [package insert]. Wilmington (DE): AstraZeneca, Inc; 2015. Error! Hyperlink reference not valid. Accessed April 23, 2017.
- Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res 2011;17:1616–22.
- Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR TKI therapy in 155 patients with EGFR mutant lung cancers. Clin Cancer Res 2013;19:2240–7.
- Yun CH, Mengwasser KE, Tom AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 2008;105:2070–5.
- Sos ML, Rode HB, Heynck S, et al. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Res 2010;70:868–74.
- Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T190M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014;4:1046–61.
- Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 2017;376:629–40.
- Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small cell lung cancer. N Engl J Med 2010;363:1693–703.
- Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009;27:4247–53.
- Kazandjian D, Blumenthal GM, Chen HY, et al. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014;19:e5–11.
- Solomon BJ, Mok T, Kim DW, et al. First-ling crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 2014;371:2167–77.
- Xalkori [package insert]. New York: Pfizer, Inc; 2011. www.accessdata.fda.gov/drugsatfda_docs/label/2012/202570s002lbl.pdf. Accessed April 23, 2017.
- Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013;368:2385–94.
- Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem 2013;56:5675–90.
- Khozin S, Blumenthal GM, Zhang L, et al. FDA approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer. Clin Cancer Res 2015;21:2436–9.
- Soria JC, Tan DS, Chiari R, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet 2017;389:917–29.
- Zykadia [package insert]. East Hanover (NJ): Novartis Pharmaceuticals Corporation, Inc; 2016. www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/zykadia.pdf. Accessed April 23, 2017.
- Larkins E, Blumenthal GM, Chen H, et al. FDA approval: alectinib for the treatment of metastatic, ALK-positive non-small cell lung cancer following crizotinib. Clin Cancer Res 2016;22:5171–6.
- Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. New Engl J Med 2017 June 6 [Epub ahead of print].
- Kinoshita K, Asoh K, Furuichi N, et al. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem 2012;20:1271–80.
- Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011;19:679–90.
- Kodama T, Tsukaguchi T, Yoshida M, et al. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett 2014;351:215–21.
- Kodama T, Tsukaguchi T, Satoh T, et al. Alectinib shows potent antitumor activity against RET-rearranged non-small cell lung cancer. Mol Cancer Ther 2014;13:2910–8.
- Alecensa [package insert]. South San Francisco (CA): Genentech, Inc; 2015. www.accessdata.fda.gov/drugsatfda_docs/label/2015/208434s000lbl.pdf. Accessed April 23, 2017.
- Kim DW, Tiseo M, Ahn MJ, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase positive non-small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol 2017 May 5 [Epub ahead of print].
- Zhu V, Ou SH. Safety of alectinib for the treatment of metastatic ALK-rearranged non-small cell lung cancer. Expert Opin Drug Saf 2017;16:509–14.
- Gadgeel SM, Shaw AT, Govindan R, et al. Pooled analysis of CNS response to alectinib in two studies of pretreated patients with ALK-positive non-small cell lung cancer. J Clin Oncol 2016;34:4079–85.
- Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol 2011;29:e443–5.
- Zhu Q, Zhan P, Zhang X, et al. Clinicopathologic characteristics of patients with ROS1 fusion gene in non-small cell lung cancer: a meta-analysis. Transl Lung Cancer Res 2015;4:300–9.
- Lin JJ, Ritterhouse LL, Ali SM, et al. ROS1 fusions rarely overlap with other oncogenic drivers in non-small cell lung cancer. J Thorac Oncol 2017;12:872–7.
- Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta 2009;1795:37–52.
- Kazandjian D, Blumenthal G, Luo L, et al. Benefit-Risk summary of crizotinib for the treatment of patients with ROS1 alteration-positive metastatic NSCLC. Oncologist 2016;21:974–80.
- Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 2014;371:1963–71.
- Zhu VW, Upadhyay D, Schrock AB, et al. TPD52L1-ROS1, a new ROS1 fusion variant in lung adenosquamous cell carcinoma identified by comprehensive genomic profiling. Lung Cancer 2016;97:48–50.
INTRODUCTION
Lung cancer is the second most common type of cancer in the United States, with 222,500 estimated new cases in 2017, according to the American Cancer Society.1 However, it is by far the number one cause of death due to cancer, with an estimated 155,870 lung cancer–related deaths occurring in 2017, which is higher than the number of deaths due to breast cancer, prostate cancer, and colorectal cancer combined.1,2 Despite slightly decreasing incidence and mortality over the past decade, largely due to smoking cessation, the 5-year survival rate of lung cancer remains dismal at approximately 18%.2–4
Non-small cell lung cancer (NSCLC) accounts for 80% to 85% of all lung cancer cases.4 Traditionally, it is further divided based on histology: adenocarcinoma, squamous cell carcinoma, large cell carcinoma, and not otherwise specified.5 Chemotherapy had been the cornerstone of treatment for stage IV NSCLC. It is not target-specific and is most effective against rapidly growing cells. Common adverse effects include alopecia, nausea/vomiting, myelosuppression, cardiotoxicity, neuropathy, and nephrotoxicity. However, this paradigm has shifted following the discovery of mutations of the epidermal growth factor receptor (EGFR) gene as an oncogenic driver that confers sensitivity to small molecule tyrosine kinase inhibitors (TKIs) targeting EGFR.6 The EGFR inhibitors are given orally and have a spectrum of toxicities (eg, such as rash, diarrhea, and elevated transaminases) different from that of systemic chemotherapy, which is often administered intravenously. Following the discovery of EGFR mutations, rearrangements of the anaplastic lymphoma kinase (ALK) gene7 and ROS1 gene8 were identified as targetable driver mutations in NSCLC. The frequency of both rearrangements is lower than that of EGFR mutations. Additionally, BRAF V600E mutation has been identified in NSCLC.9–12 This activation mutation is commonly seen in melanoma. Agents that have already been approved for the treatment of melanoma with the BRAF V600E mutation are being tested in NSCLC patients with this mutation.13–16
Given the effectiveness and tolerability of targeted therapy, identifying this distinct molecular subset of NSCLC patients is critical in treatment. Currently, molecular testing is mandatory in all stage IV patients with non-squamous cell carcinoma, as a preponderance of patients with driver mutations have this histology subtype.5,17–19 For patients with squamous cell carcinoma, molecular testing should be considered if the biopsy specimen is small, there is mixed histology, or the patient is a nonsmoker.5,20 Several techniques are commonly utilized in detecting these genetic alterations. EGFR mutation can be detected by polymerase chain reaction (PCR), ALK or ROS1 rearrangement can be detected by fluorescence in-situ hybridization (FISH), and immunohistochemistry (IHC) can also be used to detect ALK rearrangement. The current guideline is to use comprehensive genomic profiling to capture all the potential molecular targets simultaneously instead of running stepwise tests just for EGFR, ALK, and ROS1.5BRAF V600E mutation,13–16 MET exon 14 skipping mutation,21–24 RET rearrangements,25–27 and HER2 mutations28–30 are among the emergent genetic alterations with various responses to targeted therapy.31 Some of these targeted agents have been approved for other types of malignancy, and others are still in the development phase.
Several initiatives worldwide have reported better outcomes of patients with driver mutations treated with targeted therapy. For instance, the Lung Cancer Mutation Consortium in the United States demonstrated that the median survival of patients without driver mutations, with drivers mutations but not treated with targeted therapy, and with driver mutations and treated with targeted therapy was 2.08 years, 2.38 years, and 3.49 years, respectively.32 The French Cooperative Thoracic Intergroup-French National Cancer Institute demonstrated that the median survival for patients with driver mutations versus those without driver mutations was 16.5 months versus 11.8 months.33 The Spanish Lung Cancer Group demonstrated that the overall survival (OS) for patients with EGFR mutations treated with erlotinib was 27 months.34 The mutations in lung cancer, their frequencies, and the downstream signaling pathways are depicted in the Figure.35
In this article, we discuss targeted therapy for patients with EGFR mutations, ALK rearrangements, ROS1 rearrangements, and BRAF V600E mutation. We also discuss the management of patients with EGFR mutations who develop a secondary mutation after TKI therapy. Almost all of the targeted agents discussed herein have been approved by the US Food and Drug Administration (FDA), so they are considered standard of care. All available phase 3 trials pertinent to these targeted therapies are included in the discussion.
EGFR MUTATIONS
CASE PRESENTATION 1
A 54-year-old Caucasian man who is a former smoker with a 10 pack-year history and past medical history of hypertension and dyslipidemia presents with progressive dyspnea for several weeks. A chest x-ray shows moderate pleural effusion on the left side with possible mass-like opacity on the left upper lung field. An ultrasound-guided thoracentesis is performed and cytology is positive for adenocarcinoma of likely pulmonary origin. Staging workup including positron emission tomography (PET)/computed tomography (CT) and magnetic resonance imaging of the brain with and without contrast is done. PET/CT shows a 5.5-cm mass in the left upper lobe of the lung with high fluorodeoxyglucose (FDG) uptake, several 1- to 2-cm mediastinal lymph nodes with moderate FDG uptake, and small pleural effusion on both sides with moderate FDG uptake. MRI-brain is negative for malignancy. The patient subsequently undergoes a CT-guided biopsy of the lung mass, which shows moderately differentiated adenocarcinoma. Comprehensive molecular profiling reveals EGFR L858R mutation only. The patient now presents for the initial consultation. Of note, his Eastern Cooperative Oncology Group performance status is 1.
What is the next step in the management of this patient?
FIRST-LINE TKI FOR SENSITIZING EGFR MUTATIONS
The 2 most common EGFR mutations are deletions in exon 19 and substitution of arginine for leucine in exon 21 (L858R), found in approximately 45% and 40% of patients with EGFR mutations, respectively.36 Both mutations are sensitive to EGFR TKIs. The benefit may be greater in patients with exon 19 deletions as compared to exon 21 L858R substitution,37,38 but this has not been demonstrated consistently in clinical trials.39-43 In the United States, EGFR mutations are found in approximately 10% of patients with NSCLC, while the incidence can be as high as 50% in Asia.44 Even though the cobas EGFR mutation test is the companion diagnostic approved by the US FDA, a positive test result from any laboratory with the Clinical Laboratory Improvement Amendments (CLIA) certificate should prompt the use of an EGFR TKI as the initial treatment.
Three EGFR TKIs that have been approved as first-line therapy in the United States are available: erlotinib, afatinib, and gefitinib.5 Both erlotinib and gefitinib are considered first-generation TKIs. They have higher binding affinity for the 2 common EGFR mutations than wild-type EGFR. In addition, they reversibly bind to the intracellular tyrosine kinase domain, resulting in inhibition of autophosphorylation of the tyrosine residues. Afatinib, a second-generation and irreversible TKI, targets EGFR (HER1) as well as HER2 and HER4.45
The superior efficacy of the EGFR TKIs over platinum doublet chemotherapy in treatment-naïve patients with EGFR mutations has been demonstrated in 7 randomized trials to date (Table).46 Erlotinib was the TKI arm for the OPTIMAL,41 EURTAC,42 and ENSURE trials;38 afatinib was the TKI arm for LUX-LUNG 337 and 6;43 gefitinib was the TKI arm for NEJ00239,47 and WJTOG3405.40 A meta-analysis of these 7 trials by Lee et al showed that progression-free survival (PFS) was significantly prolonged by EGFR TKIs (hazard ratio [HR] 0.37 [95% confidence interval {CI} 0.32 to 0.42]).46 For instance, in the EURTAC trial, median PFS was 9.7 months for patients treated with erlotinib as compared to 5.2 months for patients treated with platinum/gemcitabine or platinum/docetaxel.42 In this meta-analysis, prespecified subgroups included age, sex, ethnicity, smoking status, performance status, tumor histology, and EGFR mutation subtype. The superior outcome with TKIs was observed in all subgroups. Furthermore, patients with exon 19 deletions, nonsmokers, and women had even better outcomes.46
Erlotinib is the most commonly used TKI in the United States largely because gefitinib was off the market for some time until it was re-approved by the FDA in 2015. Interestingly, this “re-approval” was not based on either 1 of the 2 prospective trials (NEJ00239,47 and WJTOG340540), but rather was based on an exploratory analysis of the IPASS trial48,49 as well as a prospective phase 4, single-arm trial in Europe (IFUM).50 The superior efficacy of gefitinib over carboplatin/paclitaxel among patients with EGFR mutations in the IPASS trial was confirmed by blind independent central review, with longer PFS (HR 0.54 [95% CI 0.38 to 0.79] P = 0.0012) and higher objective response rate (ORR; odds ratio 3 [95% CI 1.63 to 5.54], P = 0.0004).49
CASE 1 CONTINUED
Based on the EGFR L858R mutation status, the patient is started on erlotinib. He is quite happy that he does not need intravenous chemotherapy but wants to know what toxicities he might potentially have with erlotinib.
What are the common adverse effects (AEs) of EGFR TKIs? How are AEs of TKIs managed?
Safety Profile
The important toxicities associated with EGFR TKIs are rash, gastrointestinal toxicity, hepatic toxicity, and pulmonary toxicity. Rash is an AE specific to all agents blocking the EGFR pathway, including small molecules and monoclonal antibodies such as cetuximab. The epidermis has a high level of expression of EGFR, which contributes to this toxicity.51 Rash usually presents as dry skin or acneiform eruption. Prophylactic treatment with oral tetracyclines and topical corticosteroids is generally recommended upon initiation of TKI therapy. Diarrhea is the most prevalent gastrointestinal toxicity. All patients starting treatment should be given prescriptions to manage diarrhea such as loperamide and be advised to call when it occurs. Hepatic toxicity is often manifested as elevated transaminases or bilirubin. Interstitial lung disease (ILD) is a rare but potentially fatal pulmonary toxicity.
Rash of any grade was reported in 49.2% of patients treated with erlotinib in clinical trials, while grade 3 rash occurred in 6% of patients and no grade 4 was reported. Diarrhea of any grade was reported in 20.3% of patients, grade 3 diarrhea occurred in 1.8%, and no grade 4 was reported. Grade 2 and 3 alanine aminotransferase (ALT) elevations were seen in 2% and 1% of patients, respectively. Grade 2 and 3 bilirubin elevations were seen in 4% and less than 1% of patients, respectively. The incidence of serious ILD-like events was less than 1%.52
Afatinib is associated with higher incidences of rash and diarrhea. Specifically, diarrhea and rash of all grades were reported in 96% and 90% of patients treated with afatinib, respectively. Paronychia of all grades occurred in 58% of patients. Elevated ALT of all grades was seen in 11% of patients. Approximately 1.5% of patients treated with afatinib across clinical trials had ILD or ILD-like AEs.53
Gefitinib, the most commonly used TKI outside United States, has a toxicity profile similar to erlotinib, except for hepatic toxicity. For instance, rash of all grades occurred in 47% of patients, diarrhea of all grades occurred in 29% of patients, and ILD or ILD-like AEs occurred in 1.3% of patients across clinical trials. In comparison, elevated ALT and aspartate aminotransferase (AST) of all grades was seen in 38% and 40% of patients, respectively.54 Therefore, close monitoring of liver function is clinically warranted. In particular, patients need to be advised to avoid concomitant use of herbal supplements, a common practice in Asian countries.
CASE 1 CONTINUED
The patient does well while on erlotinib at 150 mg orally once daily for about 8 months, until he develops increasing abdominal pain. A CT scan of the abdomen and pelvis with contrast shows a new 8-cm right adrenal mass. Additionally, a repeat CT scan of the chest with contrast shows a stable lung mass but enlarging mediastinal lymphadenopathy.
How would you manage the patient at this point?
MANAGEMENT OF T790M MUTATION AFTER PROGRESSION ON FIRST-LINE EGFR TKIS
As mentioned above, the median PFS of patients with EGFR mutations treated with 1 of the 3 TKIs is around 9 to 13 months.46 Of the various resistance mechanisms that have been described, the T790M mutation is found in approximately 60% of patients who progress after treatment with first-line TKIs.55,56 Other mechanisms, such as HER2 amplification, MET amplification, or rarely small cell transformation, have been reported.56 The first- and second-generation EGFR TKIs function by binding to the ATP-binding domain of mutated EGFR, leading to inhibition of the downstream signaling pathways (Figure, part B) and ultimately cell death.35 The T790M mutation hinders the interaction between the ATP-binding domain of EGFR kinase and TKIs, resulting in treatment resistance and disease progression.57,58
Osimertinib is a third-generation irreversible EGFR TKI with activity against both sensitizing EGFR and resistant T790M mutations. It has low affinity for wide-type EGFR as well as insulin receptor and insulin-like growth factor receptor.59 Osimertinib has been fully approved for NSCLC patients with EGFR mutations who have progressed on first-line EGFR TKIs with the development of T790M mutation. An international phase 3 trial (AURA3) randomly assigned 419 patients in a 2:1 ratio to either osimertinib or platinum/pemetrexed. Eligible patients all had the documented EGFR mutations and disease progression after first-line EGFR TKIs. Central confirmation of the T790M mutation was required. Median PFS by investigator assessment, the trial’s primary end point, was 10.1 months for osimertinib versus 4.4 months for chemotherapy (HR 0.3 [95% CI 0.23 to 0.41]; P < 0.001). ORR was 71% for osimertinib versus 31% for chemotherapy (HR 5.39 [95% CI 3.47 to 8.48], P < 0.001). A total of 144 patients with stable and asymptomatic brain metastases were also eligible. Median PFS for this subset of patients treated with osimertinib and chemotherapy was 8.5 months and 4.2 months, respectively (HR 0.32 [95% CI 0.21 to 0.49]). In the AURA3 trial, osimertinib was better tolerated than chemotherapy, with 23% of patients treated with osimertinib experiencing grade 3 or 4 AEs as compared to 47% of chemotherapy-treated patients. The most common AEs of any grade were diarrhea (41%), rash (34%), dry skin (23%), and paronychia (22%).60
For the case patient, a reasonable approach would be to obtain a tissue biopsy of the adrenal mass and more importantly to check for the T790M mutation. Similar to the companion diagnostic for EGFR mutations, the cobas EGFR mutation test v2 is the FDA-approved test for T790M. However, if this resistance mutation is detected by any CLIA-certified laboratories, osimertinib should be the recommended treatment option. If tissue biopsy is not feasible, plasma-based testing should be considered. A blood-based companion diagnostic also is FDA approved.
ALK REARRANGEMENTS
CASE 2 PRESENTATION
A 42-year-old Korean woman who is a non-smoker with no significant past medical history presents with fatigue, unintentional weight loss of 20 lb in the past 4 months, and vague abdominal pain. A CT can of the abdomen and pelvis without contrast shows multiple foci in the liver and an indeterminate nodule in the right lung base. She subsequently undergoes PET/CT, which confirms multiple liver nodules/masses ranging from 1 to 3 cm with moderate FDG uptake. In addition, there is a 3.5-cm pleura-based lung mass on the right side with moderate FDG uptake. MRI-brain with and without contrast is negative for malignancy. A CT-guided biopsy of 1 of the liver masses is ordered and pathology returns positive for poorly differentiated adenocarcinoma consistent with lung primary. Molecular analysis reveals an echinoderm microtubule-associated protein-like 4 (EML4)-ALK rearrangement. She is placed on crizotinib by an outside oncologist and after about 3 weeks of therapy is doing well. She is now in your clinic for a second opinion. She says that some of her friends told her about another medication called ceritinib and was wondering if she would need to switch her cancer treatment.
How would you respond to this patient’s inquiry?
FIRST-LINE TKIS FOR ALK REARRANGEMENTS
ALK rearrangements are found in 2% to 7% of NSCLC, with EML4-ALK being the most prevalent fusion variant.61 The inversion of chromosome 2p leads to the fusion of the EML4 gene and the ALK gene, which causes the constitutive activation of the fusion protein and ultimately increased transformation and tumorigenicity.7,61 Patients harboring ALK rearrangements tend to be non-smokers. Adenocarcinoma, especially signet ring cell subtype, is the predominant histology. Compared to EGFR mutations, patients with ALK mutations are significantly younger and more likely to be men.62ALK rearrangements can be detected by either FISH or IHC, and most next-generation sequencing (NGS) panels have the ability to identify this driver mutation.
Crizotinib is the first approved ALK inhibitor for the treatment of NSCLC in this molecular subset of patients.63 PROFILE 1014 is a phase 3 randomized trial that compared crizotinib with chemotherapy containing platinum/pemetrexed for up to 6 cycles. Crossover to crizotinib was allowed for patients with disease progression on chemotherapy. The primary end point was PFS by independent radiologic review. The crizotinib arm demonstrated superior PFS (10.9 months versus 7 months; HR 0.45 [95% CI 0.35 to 0.6], P < 0.001) and ORR (74% versus 45%, P < 0.001). Median survival was not reached in either arm (HR 0.82 [95% CI 0.54 to 1.26], P = 0.36).64 Based on this international trial, crizotinib is considered standard of care in the United States for treatment-naïve patients with advanced NSCLC harboring ALK rearrangements. The current recommended dose is 250 mg orally twice daily. Common treatment-related AEs of all grades include vision disorder (62%), nausea (53%), diarrhea (43%), vomiting (40%), edema (28%), and constipation (27%).65 PROFILE 1007 compared crizotinib with pemetrexed or docetaxel in ALK-rearranged NSCLC patients with prior exposure to 1 platinum-based chemotherapy. The median PFS was 7.7 months for crizotinib as compared to 3 months for chemotherapy (HR 0.49 [95% CI 0.37 to 0.64], P < 0.001). The response rates were 65% and 20% for crizotinib and chemotherapy, respectively (P < 0.001).66 In other countries, crizotinib following 1 prior platinum-based regimen may be considered standard of care based on this trial.
Ceritinib is an oral second-generation ALK inhibitor that is 20 times more potent than crizotinib based on enzymatic assays.67 It also targets ROS1 and insulin-like growth factor 1 receptor but not c-MET. It was first approved by the FDA in April 2014 for metastatic ALK-rearranged NSCLC following crizotinib.68 In May 2017, the FDA granted approval of ceritinib for treatment-naïve patients. This decision was based on the results of the ASCEND-4 trial, a randomized phase 3 trial assessing the efficacy and safety of ceritinib over chemotherapy in the first-line setting. The trial assigned 376 patients to either ceritinib at 750 mg once daily or platinum/pemetrexed for 4 cycles followed by maintenance pemetrexed. Median PFS was 16.6 months for ceritinib versus 8.1 months for chemotherapy (HR 0.55 [95% CI 0.42 to 0.73]; P < 0.00001).69 Toxicities of ceritinib are not negligible, with gastrointestinal toxicity being the most prevalent. For instance, diarrhea, nausea, vomiting, abdominal pain, and constipation of all grades were seen in 86%, 80%, 60%, 54%, and 29% of patients, respectively. Furthermore, fatigue and decreased appetite occurred in 52% and 34% of patients, respectively. In terms of laboratory abnormalities, 84% of patients experienced decreased hemoglobin of all grades; 80% increased ALT; 75% increased AST; 58% increased creatinine; 49% increased glucose; 36% decreased phosphate; and 28% increased lipase. Due to these AEs, the incidence of dose reduction was about 58% and the median onset was around 7 weeks.70
Alectinib is another oral second-generation ALK inhibitor that was approved by the FDA in December 2015 for the treatment of NSCLC patients with ALK rearrangements who have progressed on or are intolerant to crizotinib.71 Its indication will soon be broadened to the first-line setting based on the ALEX trial.72 Alectinib is a potent and highly selective TKI of ALK73 with activity against known resistant mutations to crizotinib.74,75 It also inhibits RET but not ROS1 or c-MET.76 ALEX, a randomized phase 3 study, compared alectinib with crizotinib in treatment-naïve patients with NSCLC harboring ALK rearrangements. The trial enrolled 303 patients and the median follow-up was approximately 18 months. The alectinib arm (600 mg twice daily) demonstrated significantly higher PFS by investigator-assessment, the trial’s primary end point. The 12-month event-free survival was 68.4% (95% CI 61% to 75.9%) versus 48.7% (95% CI 40.4% to 56.9%) for alectinib and crizotinib, respectively (HR 0.47 [95% CI 0.34 to 0.65], P < 0.001). The median PFS was not reached in the alectinib arm (95% CI 17.7 months to not estimable) as compared to 11.1 months in the crizotinib arm (95% CI 9.1 to 13.1 months).72 Alectinib is generally well tolerated. Common AEs of all grades include fatigue (41%), constipation (34%), edema (30%), and myalgia (29%). As alectinib can cause anemia, lymphopenia, hepatic toxicity, increased creatine phosphokinase, hyperglycemia, electrolyte abnormalities, and increased creatinine, periodic monitoring of these laboratory values is important, although most of these abnormalities are grade 1 or 2.77
Brigatinib, another oral second-generation ALK inhibitor, was granted accelerated approval by the FDA in April 2017 for ALK-rearranged and crizotinib-resistant NSCLC based on the ALTA trial. This randomized phase 2 study of brigatinib showed an ORR by investigator assessment of 54% (97.5% CI 43% to 65%) in the 180 mg once daily arm with lead-in of 90 mg once daily for 7 days. Median PFS was 12.9 months (95% CI 11.1 months to not reached [NR]).78 Currently, a phase 3 study of brigatinib versus crizotinib in ALK inhibitor–naïve patients is recruiting participants (ALTA-1L). It will be interesting to see if brigatinib can achieve a front-line indication.
Starting the case patient on crizotinib is well within the treatment guidelines. One may consider ceritinib or alectinib in the first-line setting, but both TKIs can be reserved upon disease progression. We would recommend a repeat biopsy at that point to look for resistant mechanisms, as certain secondary ALK mutations may be rescued by certain next-generation ALK inhibitors. For instance, the F1174V mutation has been reported to confer resistance to ceritinib but sensitivity to alectinib, while the opposite is true for I1171T. The G1202R mutation is resistant to ceritinib, alectinib, and brigatinib, but lorlatinib, a third-generation ALK inhibitor, has shown activity against this mutation.79 Furthermore, brain metastasis represents a treatment challenge for patients with ALK rearrangements. It is also an efficacy measure of next-generation ALK inhibitors, all of which have demonstrated better central nervous system activity than crizotinib.69,78,80 If the case patient were found to have brain metastasis at the initial diagnosis, either ceritinib or alectinib would be a reasonable choice since crizotinib has limited penetration of blood-brain barrier.81
ROS1 REARRANGEMENTS
CASE PRESENTATION 3
A 66-year-old Chinese woman who is a non-smoker with a past medical history of hypertension and hypothyroidism presents to the emergency department for worsening lower back pain. Initial workup includes x-ray of the lumbar spine followed by MRI with contrast, which shows a soft tissue mass at L3-4 without cord compression. CT of the chest, abdomen, and pelvis with contrast shows a 7-cm right hilar mass, bilateral small lung nodules, mediastinal lymphadenopathy, and multiple lytic lesions in ribs, lumbar spine, and pelvis. MRI-brain with and without contrast is negative for malignancy. She undergoes endo-bronchial ultrasound and biopsy of the right hilar mass, which shows poorly differentiated adenocarcinoma. While waiting for the result of the molecular analysis, the patient undergoes palliative radiation therapy to L2-5 with good pain relief. She is discharged from the hospital and presents to your clinic for follow up. Molecular analysis now reveals ROS1 rearrangement with CD74-ROS1 fusion.
What treatment plan should be put in place for this patient?
FIRST-LINE THERAPY FOR ROS1 REARRANGEMENTS
Approximately 2.4% of lung adenocarcinomas harbor ROS1 rearrangements.82 This distinct genetic alteration occurs more frequently in NSCLC patients who are younger, female, and never-smokers, and who have adenocarcinomas.8 It has been shown that ROS1 rearrangements rarely overlap with other genetic alterations including KRAS mutations, EGFR mutations, and ALK rearrangements.83 As a receptor tyrosine kinase, ROS1 is similar to ALK and insulin receptor family members.84 Crizotinib, which targets ALK, ROS1, and c-MET, was approved by the FDA on March 11, 2016, for the treatment of metastatic ROS1-rearranged NSCLC.85 The approval was based on a phase 2 expansion cohort of the original phase 1 study. Among 50 US patients enrolled in this expansion cohort, 3 had complete responses and 33 had partial responses with ORR of 72% (95% CI 58% to 84%). Median PFS was 19.2 months (95% CI 14.4 months to NR) and median duration of response (DOR) was 17.6 months (95% CI 14.5 months to NR).86 During longer follow-up, independent radiology review confirmed high ORR of 66% and median DOR of 18.3 months.85
Interestingly, no companion diagnostic assay has been approved for the detection of ROS1 rearrangements with the approval of crizotinib. In the United States, break apart FISH is the most common detection method. In fact, in the above mentioned phase 2 study, ROS1 rearrangements were detected in 49 out of 50 patients by this method.86 FISH can be technically challenging when dealing with high volume and multiple targets. Reverse transcriptase-PCR is another detection method, but it requires knowledge of the fusion partners. To date, at least 14 ROS1 fusion partners have been reported, with CD74 being the most common.87 NGS with appropriate design and validation can also be used to detect ROS1 rearrangements.
For the case patient, the recommendation would be to start her on crizotinib at 250 mg twice daily. Monitoring for vision disturbance, gastrointestinal complaints, and edema is warranted. Because the estimated onset of response is around 7.9 weeks,86 plans should be made to repeat her scans in approximately 2 months.
BRAF V600E MUTATIONS
CASE PRESENTATION 4
A 71-year-old Caucasian man with a past medical history of hypertension, dyslipidemia, and ischemic cerebrovascular accident without residual deficits was diagnosed with stage IV adenocarcinoma of the lung about 8 months ago. He has a 40 pack-year smoking history and quit smoking when he was diagnosed with lung cancer. His disease burden involved a large mediastinal mass, scattered pleural nodules, multiple lymphadenopathy, and several soft tissue masses. His outside oncologist started him on chemotherapy containing carboplatin and pemetrexed for 6 cycles followed by maintenance pemetrexed. The most recent restaging scans show disease progression with enlarging soft tissue masses and several new lytic bone lesions. MRI-brain with and without contrast shows 2 subcentimeter enhancing lesions. He transferred care to you approximately 4 weeks ago. You ordered a repeat biopsy of 1 of the enlarging soft tissue masses. Molecular analysis revealed BRAF V600E mutation. In the interim, he underwent stereotactic radiosurgery for the 2 brain lesions without any complications. The patient is now in your clinic for follow up.
What would be your recommended systemic treatment?
TARGETED THERAPIES FOR BRAF V600E MUTATION
BRAF mutations were first recognized as activating mutations in advanced melanomas, with BRAF V600E, resulting from the substitution of glutamic acid for valine at amino acid 600, being the most common. BRAF plays an important role in the mitogen-activated protein kinase (MAPK) signaling pathway. Briefly, the activation of MAPK pathway occurs upon ligand binding of receptor tyrosine kinases, which then involves RAS/BRAF/MEK/ERK in a stepwise manner, ultimately leading to cell survival. BRAF mutations have been increasingly recognized also as driver mutations in NSCLC.9–12 They can be detected by PCR or NGS method. The characteristics of NSCLC patients harboring BRAF mutations have been described by various groups.9–12 For instance, 1 case series showed that the incidence was 2.2% among patients with advanced lung adenocarcinoma; 50% of mutations were V600E, while G469A and D594G accounted for the remaining 39% and 11% of patients, respectively. All patients were either current or former smokers. The median OS of patients with BRAF mutations in this case series was NR, while it was 37 months for patients with EGFR mutations (P = 0.73) and NR for patients with ALK rearrangements (P = 0.64).9
For patients with BRAF V600E–mutant NSCLC who have progressed on platinum-based chemotherapy, the combination of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) may represent a new treatment paradigm. This was illustrated in a phase 2, nonrandomized, open-label study. A total of 57 patients were enrolled and 36 patients (63.2% [95% CI 49.3% to 75.6%]) achieved an overall response by investigator assessment, the trial’s primary end point. Disease control rate was 78.9% (95% CI 66.1% to 88.6%), with 4% complete response, 60% partial response, and 16% stable disease. PFS was 9.7 months (95% CI [6.9 to 19.6 months]). The safety profile was comparable to what had been observed in patients with melanoma treated with this regimen. More specifically, 56% of patients on this trial reported serious AEs, including pyrexia (16%), anemia (5%), confusional state (4%), decreased appetite (4%), hemoptysis (4%), hypercalcemia (4%), nausea (4%), and cutaneous squamous cell carcinoma (4%). In addition, neutropenia (9%) and hyponatremia (7%) were the most common grade 3-4 AEs.16
The case patient has experienced disease progression after 1 line of platinum-based chemotherapy, so the combination of dabrafenib and trametinib would be a robust systemic treatment option. dabrafenib as a single agent has also been studied in BRAF V600E–mutant NSCLC in a phase 2 trial. The overall response by investigator assessment among 84 patients was 33% (95% CI 23% to 45%).14 Vemurafenib, another oral BRAF TKI, has demonstrated efficacy for NSCLC patients harboring BRAF V600E mutation. In the cohort of 20 patients with NSCLC, the response rate was 42% (95% CI 20% to 67%) and median PFS was 7.3 months (95% CI 3.5 to 10.8 months).13 Patients with non-V600E mutations have shown variable responses to targeted therapies. MEK TKIs may be considered in this setting; however, the details of this discussion are beyond the scope of this review.
CONCLUSION
The management of advanced NSCLC with driver mutations has seen revolutionary changes over the past decade. Tremendous research has been done in order to first understand the molecular pathogenesis of NSCLC and then discover driver mutations that would lead to development of targeted therapies with clinically significant efficacy as well as tolerability. More recently, increasing efforts have focused on how to conquer acquired resistance in patients with disease progression after first-line TKIs. The field of EGFR-mutant NSCLC has set a successful example, but the work is nowhere near finished. The goals are to search for more driver mutations and to design agents that could potentially block cell survival signals once and for all.
INTRODUCTION
Lung cancer is the second most common type of cancer in the United States, with 222,500 estimated new cases in 2017, according to the American Cancer Society.1 However, it is by far the number one cause of death due to cancer, with an estimated 155,870 lung cancer–related deaths occurring in 2017, which is higher than the number of deaths due to breast cancer, prostate cancer, and colorectal cancer combined.1,2 Despite slightly decreasing incidence and mortality over the past decade, largely due to smoking cessation, the 5-year survival rate of lung cancer remains dismal at approximately 18%.2–4
Non-small cell lung cancer (NSCLC) accounts for 80% to 85% of all lung cancer cases.4 Traditionally, it is further divided based on histology: adenocarcinoma, squamous cell carcinoma, large cell carcinoma, and not otherwise specified.5 Chemotherapy had been the cornerstone of treatment for stage IV NSCLC. It is not target-specific and is most effective against rapidly growing cells. Common adverse effects include alopecia, nausea/vomiting, myelosuppression, cardiotoxicity, neuropathy, and nephrotoxicity. However, this paradigm has shifted following the discovery of mutations of the epidermal growth factor receptor (EGFR) gene as an oncogenic driver that confers sensitivity to small molecule tyrosine kinase inhibitors (TKIs) targeting EGFR.6 The EGFR inhibitors are given orally and have a spectrum of toxicities (eg, such as rash, diarrhea, and elevated transaminases) different from that of systemic chemotherapy, which is often administered intravenously. Following the discovery of EGFR mutations, rearrangements of the anaplastic lymphoma kinase (ALK) gene7 and ROS1 gene8 were identified as targetable driver mutations in NSCLC. The frequency of both rearrangements is lower than that of EGFR mutations. Additionally, BRAF V600E mutation has been identified in NSCLC.9–12 This activation mutation is commonly seen in melanoma. Agents that have already been approved for the treatment of melanoma with the BRAF V600E mutation are being tested in NSCLC patients with this mutation.13–16
Given the effectiveness and tolerability of targeted therapy, identifying this distinct molecular subset of NSCLC patients is critical in treatment. Currently, molecular testing is mandatory in all stage IV patients with non-squamous cell carcinoma, as a preponderance of patients with driver mutations have this histology subtype.5,17–19 For patients with squamous cell carcinoma, molecular testing should be considered if the biopsy specimen is small, there is mixed histology, or the patient is a nonsmoker.5,20 Several techniques are commonly utilized in detecting these genetic alterations. EGFR mutation can be detected by polymerase chain reaction (PCR), ALK or ROS1 rearrangement can be detected by fluorescence in-situ hybridization (FISH), and immunohistochemistry (IHC) can also be used to detect ALK rearrangement. The current guideline is to use comprehensive genomic profiling to capture all the potential molecular targets simultaneously instead of running stepwise tests just for EGFR, ALK, and ROS1.5BRAF V600E mutation,13–16 MET exon 14 skipping mutation,21–24 RET rearrangements,25–27 and HER2 mutations28–30 are among the emergent genetic alterations with various responses to targeted therapy.31 Some of these targeted agents have been approved for other types of malignancy, and others are still in the development phase.
Several initiatives worldwide have reported better outcomes of patients with driver mutations treated with targeted therapy. For instance, the Lung Cancer Mutation Consortium in the United States demonstrated that the median survival of patients without driver mutations, with drivers mutations but not treated with targeted therapy, and with driver mutations and treated with targeted therapy was 2.08 years, 2.38 years, and 3.49 years, respectively.32 The French Cooperative Thoracic Intergroup-French National Cancer Institute demonstrated that the median survival for patients with driver mutations versus those without driver mutations was 16.5 months versus 11.8 months.33 The Spanish Lung Cancer Group demonstrated that the overall survival (OS) for patients with EGFR mutations treated with erlotinib was 27 months.34 The mutations in lung cancer, their frequencies, and the downstream signaling pathways are depicted in the Figure.35
In this article, we discuss targeted therapy for patients with EGFR mutations, ALK rearrangements, ROS1 rearrangements, and BRAF V600E mutation. We also discuss the management of patients with EGFR mutations who develop a secondary mutation after TKI therapy. Almost all of the targeted agents discussed herein have been approved by the US Food and Drug Administration (FDA), so they are considered standard of care. All available phase 3 trials pertinent to these targeted therapies are included in the discussion.
EGFR MUTATIONS
CASE PRESENTATION 1
A 54-year-old Caucasian man who is a former smoker with a 10 pack-year history and past medical history of hypertension and dyslipidemia presents with progressive dyspnea for several weeks. A chest x-ray shows moderate pleural effusion on the left side with possible mass-like opacity on the left upper lung field. An ultrasound-guided thoracentesis is performed and cytology is positive for adenocarcinoma of likely pulmonary origin. Staging workup including positron emission tomography (PET)/computed tomography (CT) and magnetic resonance imaging of the brain with and without contrast is done. PET/CT shows a 5.5-cm mass in the left upper lobe of the lung with high fluorodeoxyglucose (FDG) uptake, several 1- to 2-cm mediastinal lymph nodes with moderate FDG uptake, and small pleural effusion on both sides with moderate FDG uptake. MRI-brain is negative for malignancy. The patient subsequently undergoes a CT-guided biopsy of the lung mass, which shows moderately differentiated adenocarcinoma. Comprehensive molecular profiling reveals EGFR L858R mutation only. The patient now presents for the initial consultation. Of note, his Eastern Cooperative Oncology Group performance status is 1.
What is the next step in the management of this patient?
FIRST-LINE TKI FOR SENSITIZING EGFR MUTATIONS
The 2 most common EGFR mutations are deletions in exon 19 and substitution of arginine for leucine in exon 21 (L858R), found in approximately 45% and 40% of patients with EGFR mutations, respectively.36 Both mutations are sensitive to EGFR TKIs. The benefit may be greater in patients with exon 19 deletions as compared to exon 21 L858R substitution,37,38 but this has not been demonstrated consistently in clinical trials.39-43 In the United States, EGFR mutations are found in approximately 10% of patients with NSCLC, while the incidence can be as high as 50% in Asia.44 Even though the cobas EGFR mutation test is the companion diagnostic approved by the US FDA, a positive test result from any laboratory with the Clinical Laboratory Improvement Amendments (CLIA) certificate should prompt the use of an EGFR TKI as the initial treatment.
Three EGFR TKIs that have been approved as first-line therapy in the United States are available: erlotinib, afatinib, and gefitinib.5 Both erlotinib and gefitinib are considered first-generation TKIs. They have higher binding affinity for the 2 common EGFR mutations than wild-type EGFR. In addition, they reversibly bind to the intracellular tyrosine kinase domain, resulting in inhibition of autophosphorylation of the tyrosine residues. Afatinib, a second-generation and irreversible TKI, targets EGFR (HER1) as well as HER2 and HER4.45
The superior efficacy of the EGFR TKIs over platinum doublet chemotherapy in treatment-naïve patients with EGFR mutations has been demonstrated in 7 randomized trials to date (Table).46 Erlotinib was the TKI arm for the OPTIMAL,41 EURTAC,42 and ENSURE trials;38 afatinib was the TKI arm for LUX-LUNG 337 and 6;43 gefitinib was the TKI arm for NEJ00239,47 and WJTOG3405.40 A meta-analysis of these 7 trials by Lee et al showed that progression-free survival (PFS) was significantly prolonged by EGFR TKIs (hazard ratio [HR] 0.37 [95% confidence interval {CI} 0.32 to 0.42]).46 For instance, in the EURTAC trial, median PFS was 9.7 months for patients treated with erlotinib as compared to 5.2 months for patients treated with platinum/gemcitabine or platinum/docetaxel.42 In this meta-analysis, prespecified subgroups included age, sex, ethnicity, smoking status, performance status, tumor histology, and EGFR mutation subtype. The superior outcome with TKIs was observed in all subgroups. Furthermore, patients with exon 19 deletions, nonsmokers, and women had even better outcomes.46
Erlotinib is the most commonly used TKI in the United States largely because gefitinib was off the market for some time until it was re-approved by the FDA in 2015. Interestingly, this “re-approval” was not based on either 1 of the 2 prospective trials (NEJ00239,47 and WJTOG340540), but rather was based on an exploratory analysis of the IPASS trial48,49 as well as a prospective phase 4, single-arm trial in Europe (IFUM).50 The superior efficacy of gefitinib over carboplatin/paclitaxel among patients with EGFR mutations in the IPASS trial was confirmed by blind independent central review, with longer PFS (HR 0.54 [95% CI 0.38 to 0.79] P = 0.0012) and higher objective response rate (ORR; odds ratio 3 [95% CI 1.63 to 5.54], P = 0.0004).49
CASE 1 CONTINUED
Based on the EGFR L858R mutation status, the patient is started on erlotinib. He is quite happy that he does not need intravenous chemotherapy but wants to know what toxicities he might potentially have with erlotinib.
What are the common adverse effects (AEs) of EGFR TKIs? How are AEs of TKIs managed?
Safety Profile
The important toxicities associated with EGFR TKIs are rash, gastrointestinal toxicity, hepatic toxicity, and pulmonary toxicity. Rash is an AE specific to all agents blocking the EGFR pathway, including small molecules and monoclonal antibodies such as cetuximab. The epidermis has a high level of expression of EGFR, which contributes to this toxicity.51 Rash usually presents as dry skin or acneiform eruption. Prophylactic treatment with oral tetracyclines and topical corticosteroids is generally recommended upon initiation of TKI therapy. Diarrhea is the most prevalent gastrointestinal toxicity. All patients starting treatment should be given prescriptions to manage diarrhea such as loperamide and be advised to call when it occurs. Hepatic toxicity is often manifested as elevated transaminases or bilirubin. Interstitial lung disease (ILD) is a rare but potentially fatal pulmonary toxicity.
Rash of any grade was reported in 49.2% of patients treated with erlotinib in clinical trials, while grade 3 rash occurred in 6% of patients and no grade 4 was reported. Diarrhea of any grade was reported in 20.3% of patients, grade 3 diarrhea occurred in 1.8%, and no grade 4 was reported. Grade 2 and 3 alanine aminotransferase (ALT) elevations were seen in 2% and 1% of patients, respectively. Grade 2 and 3 bilirubin elevations were seen in 4% and less than 1% of patients, respectively. The incidence of serious ILD-like events was less than 1%.52
Afatinib is associated with higher incidences of rash and diarrhea. Specifically, diarrhea and rash of all grades were reported in 96% and 90% of patients treated with afatinib, respectively. Paronychia of all grades occurred in 58% of patients. Elevated ALT of all grades was seen in 11% of patients. Approximately 1.5% of patients treated with afatinib across clinical trials had ILD or ILD-like AEs.53
Gefitinib, the most commonly used TKI outside United States, has a toxicity profile similar to erlotinib, except for hepatic toxicity. For instance, rash of all grades occurred in 47% of patients, diarrhea of all grades occurred in 29% of patients, and ILD or ILD-like AEs occurred in 1.3% of patients across clinical trials. In comparison, elevated ALT and aspartate aminotransferase (AST) of all grades was seen in 38% and 40% of patients, respectively.54 Therefore, close monitoring of liver function is clinically warranted. In particular, patients need to be advised to avoid concomitant use of herbal supplements, a common practice in Asian countries.
CASE 1 CONTINUED
The patient does well while on erlotinib at 150 mg orally once daily for about 8 months, until he develops increasing abdominal pain. A CT scan of the abdomen and pelvis with contrast shows a new 8-cm right adrenal mass. Additionally, a repeat CT scan of the chest with contrast shows a stable lung mass but enlarging mediastinal lymphadenopathy.
How would you manage the patient at this point?
MANAGEMENT OF T790M MUTATION AFTER PROGRESSION ON FIRST-LINE EGFR TKIS
As mentioned above, the median PFS of patients with EGFR mutations treated with 1 of the 3 TKIs is around 9 to 13 months.46 Of the various resistance mechanisms that have been described, the T790M mutation is found in approximately 60% of patients who progress after treatment with first-line TKIs.55,56 Other mechanisms, such as HER2 amplification, MET amplification, or rarely small cell transformation, have been reported.56 The first- and second-generation EGFR TKIs function by binding to the ATP-binding domain of mutated EGFR, leading to inhibition of the downstream signaling pathways (Figure, part B) and ultimately cell death.35 The T790M mutation hinders the interaction between the ATP-binding domain of EGFR kinase and TKIs, resulting in treatment resistance and disease progression.57,58
Osimertinib is a third-generation irreversible EGFR TKI with activity against both sensitizing EGFR and resistant T790M mutations. It has low affinity for wide-type EGFR as well as insulin receptor and insulin-like growth factor receptor.59 Osimertinib has been fully approved for NSCLC patients with EGFR mutations who have progressed on first-line EGFR TKIs with the development of T790M mutation. An international phase 3 trial (AURA3) randomly assigned 419 patients in a 2:1 ratio to either osimertinib or platinum/pemetrexed. Eligible patients all had the documented EGFR mutations and disease progression after first-line EGFR TKIs. Central confirmation of the T790M mutation was required. Median PFS by investigator assessment, the trial’s primary end point, was 10.1 months for osimertinib versus 4.4 months for chemotherapy (HR 0.3 [95% CI 0.23 to 0.41]; P < 0.001). ORR was 71% for osimertinib versus 31% for chemotherapy (HR 5.39 [95% CI 3.47 to 8.48], P < 0.001). A total of 144 patients with stable and asymptomatic brain metastases were also eligible. Median PFS for this subset of patients treated with osimertinib and chemotherapy was 8.5 months and 4.2 months, respectively (HR 0.32 [95% CI 0.21 to 0.49]). In the AURA3 trial, osimertinib was better tolerated than chemotherapy, with 23% of patients treated with osimertinib experiencing grade 3 or 4 AEs as compared to 47% of chemotherapy-treated patients. The most common AEs of any grade were diarrhea (41%), rash (34%), dry skin (23%), and paronychia (22%).60
For the case patient, a reasonable approach would be to obtain a tissue biopsy of the adrenal mass and more importantly to check for the T790M mutation. Similar to the companion diagnostic for EGFR mutations, the cobas EGFR mutation test v2 is the FDA-approved test for T790M. However, if this resistance mutation is detected by any CLIA-certified laboratories, osimertinib should be the recommended treatment option. If tissue biopsy is not feasible, plasma-based testing should be considered. A blood-based companion diagnostic also is FDA approved.
ALK REARRANGEMENTS
CASE 2 PRESENTATION
A 42-year-old Korean woman who is a non-smoker with no significant past medical history presents with fatigue, unintentional weight loss of 20 lb in the past 4 months, and vague abdominal pain. A CT can of the abdomen and pelvis without contrast shows multiple foci in the liver and an indeterminate nodule in the right lung base. She subsequently undergoes PET/CT, which confirms multiple liver nodules/masses ranging from 1 to 3 cm with moderate FDG uptake. In addition, there is a 3.5-cm pleura-based lung mass on the right side with moderate FDG uptake. MRI-brain with and without contrast is negative for malignancy. A CT-guided biopsy of 1 of the liver masses is ordered and pathology returns positive for poorly differentiated adenocarcinoma consistent with lung primary. Molecular analysis reveals an echinoderm microtubule-associated protein-like 4 (EML4)-ALK rearrangement. She is placed on crizotinib by an outside oncologist and after about 3 weeks of therapy is doing well. She is now in your clinic for a second opinion. She says that some of her friends told her about another medication called ceritinib and was wondering if she would need to switch her cancer treatment.
How would you respond to this patient’s inquiry?
FIRST-LINE TKIS FOR ALK REARRANGEMENTS
ALK rearrangements are found in 2% to 7% of NSCLC, with EML4-ALK being the most prevalent fusion variant.61 The inversion of chromosome 2p leads to the fusion of the EML4 gene and the ALK gene, which causes the constitutive activation of the fusion protein and ultimately increased transformation and tumorigenicity.7,61 Patients harboring ALK rearrangements tend to be non-smokers. Adenocarcinoma, especially signet ring cell subtype, is the predominant histology. Compared to EGFR mutations, patients with ALK mutations are significantly younger and more likely to be men.62ALK rearrangements can be detected by either FISH or IHC, and most next-generation sequencing (NGS) panels have the ability to identify this driver mutation.
Crizotinib is the first approved ALK inhibitor for the treatment of NSCLC in this molecular subset of patients.63 PROFILE 1014 is a phase 3 randomized trial that compared crizotinib with chemotherapy containing platinum/pemetrexed for up to 6 cycles. Crossover to crizotinib was allowed for patients with disease progression on chemotherapy. The primary end point was PFS by independent radiologic review. The crizotinib arm demonstrated superior PFS (10.9 months versus 7 months; HR 0.45 [95% CI 0.35 to 0.6], P < 0.001) and ORR (74% versus 45%, P < 0.001). Median survival was not reached in either arm (HR 0.82 [95% CI 0.54 to 1.26], P = 0.36).64 Based on this international trial, crizotinib is considered standard of care in the United States for treatment-naïve patients with advanced NSCLC harboring ALK rearrangements. The current recommended dose is 250 mg orally twice daily. Common treatment-related AEs of all grades include vision disorder (62%), nausea (53%), diarrhea (43%), vomiting (40%), edema (28%), and constipation (27%).65 PROFILE 1007 compared crizotinib with pemetrexed or docetaxel in ALK-rearranged NSCLC patients with prior exposure to 1 platinum-based chemotherapy. The median PFS was 7.7 months for crizotinib as compared to 3 months for chemotherapy (HR 0.49 [95% CI 0.37 to 0.64], P < 0.001). The response rates were 65% and 20% for crizotinib and chemotherapy, respectively (P < 0.001).66 In other countries, crizotinib following 1 prior platinum-based regimen may be considered standard of care based on this trial.
Ceritinib is an oral second-generation ALK inhibitor that is 20 times more potent than crizotinib based on enzymatic assays.67 It also targets ROS1 and insulin-like growth factor 1 receptor but not c-MET. It was first approved by the FDA in April 2014 for metastatic ALK-rearranged NSCLC following crizotinib.68 In May 2017, the FDA granted approval of ceritinib for treatment-naïve patients. This decision was based on the results of the ASCEND-4 trial, a randomized phase 3 trial assessing the efficacy and safety of ceritinib over chemotherapy in the first-line setting. The trial assigned 376 patients to either ceritinib at 750 mg once daily or platinum/pemetrexed for 4 cycles followed by maintenance pemetrexed. Median PFS was 16.6 months for ceritinib versus 8.1 months for chemotherapy (HR 0.55 [95% CI 0.42 to 0.73]; P < 0.00001).69 Toxicities of ceritinib are not negligible, with gastrointestinal toxicity being the most prevalent. For instance, diarrhea, nausea, vomiting, abdominal pain, and constipation of all grades were seen in 86%, 80%, 60%, 54%, and 29% of patients, respectively. Furthermore, fatigue and decreased appetite occurred in 52% and 34% of patients, respectively. In terms of laboratory abnormalities, 84% of patients experienced decreased hemoglobin of all grades; 80% increased ALT; 75% increased AST; 58% increased creatinine; 49% increased glucose; 36% decreased phosphate; and 28% increased lipase. Due to these AEs, the incidence of dose reduction was about 58% and the median onset was around 7 weeks.70
Alectinib is another oral second-generation ALK inhibitor that was approved by the FDA in December 2015 for the treatment of NSCLC patients with ALK rearrangements who have progressed on or are intolerant to crizotinib.71 Its indication will soon be broadened to the first-line setting based on the ALEX trial.72 Alectinib is a potent and highly selective TKI of ALK73 with activity against known resistant mutations to crizotinib.74,75 It also inhibits RET but not ROS1 or c-MET.76 ALEX, a randomized phase 3 study, compared alectinib with crizotinib in treatment-naïve patients with NSCLC harboring ALK rearrangements. The trial enrolled 303 patients and the median follow-up was approximately 18 months. The alectinib arm (600 mg twice daily) demonstrated significantly higher PFS by investigator-assessment, the trial’s primary end point. The 12-month event-free survival was 68.4% (95% CI 61% to 75.9%) versus 48.7% (95% CI 40.4% to 56.9%) for alectinib and crizotinib, respectively (HR 0.47 [95% CI 0.34 to 0.65], P < 0.001). The median PFS was not reached in the alectinib arm (95% CI 17.7 months to not estimable) as compared to 11.1 months in the crizotinib arm (95% CI 9.1 to 13.1 months).72 Alectinib is generally well tolerated. Common AEs of all grades include fatigue (41%), constipation (34%), edema (30%), and myalgia (29%). As alectinib can cause anemia, lymphopenia, hepatic toxicity, increased creatine phosphokinase, hyperglycemia, electrolyte abnormalities, and increased creatinine, periodic monitoring of these laboratory values is important, although most of these abnormalities are grade 1 or 2.77
Brigatinib, another oral second-generation ALK inhibitor, was granted accelerated approval by the FDA in April 2017 for ALK-rearranged and crizotinib-resistant NSCLC based on the ALTA trial. This randomized phase 2 study of brigatinib showed an ORR by investigator assessment of 54% (97.5% CI 43% to 65%) in the 180 mg once daily arm with lead-in of 90 mg once daily for 7 days. Median PFS was 12.9 months (95% CI 11.1 months to not reached [NR]).78 Currently, a phase 3 study of brigatinib versus crizotinib in ALK inhibitor–naïve patients is recruiting participants (ALTA-1L). It will be interesting to see if brigatinib can achieve a front-line indication.
Starting the case patient on crizotinib is well within the treatment guidelines. One may consider ceritinib or alectinib in the first-line setting, but both TKIs can be reserved upon disease progression. We would recommend a repeat biopsy at that point to look for resistant mechanisms, as certain secondary ALK mutations may be rescued by certain next-generation ALK inhibitors. For instance, the F1174V mutation has been reported to confer resistance to ceritinib but sensitivity to alectinib, while the opposite is true for I1171T. The G1202R mutation is resistant to ceritinib, alectinib, and brigatinib, but lorlatinib, a third-generation ALK inhibitor, has shown activity against this mutation.79 Furthermore, brain metastasis represents a treatment challenge for patients with ALK rearrangements. It is also an efficacy measure of next-generation ALK inhibitors, all of which have demonstrated better central nervous system activity than crizotinib.69,78,80 If the case patient were found to have brain metastasis at the initial diagnosis, either ceritinib or alectinib would be a reasonable choice since crizotinib has limited penetration of blood-brain barrier.81
ROS1 REARRANGEMENTS
CASE PRESENTATION 3
A 66-year-old Chinese woman who is a non-smoker with a past medical history of hypertension and hypothyroidism presents to the emergency department for worsening lower back pain. Initial workup includes x-ray of the lumbar spine followed by MRI with contrast, which shows a soft tissue mass at L3-4 without cord compression. CT of the chest, abdomen, and pelvis with contrast shows a 7-cm right hilar mass, bilateral small lung nodules, mediastinal lymphadenopathy, and multiple lytic lesions in ribs, lumbar spine, and pelvis. MRI-brain with and without contrast is negative for malignancy. She undergoes endo-bronchial ultrasound and biopsy of the right hilar mass, which shows poorly differentiated adenocarcinoma. While waiting for the result of the molecular analysis, the patient undergoes palliative radiation therapy to L2-5 with good pain relief. She is discharged from the hospital and presents to your clinic for follow up. Molecular analysis now reveals ROS1 rearrangement with CD74-ROS1 fusion.
What treatment plan should be put in place for this patient?
FIRST-LINE THERAPY FOR ROS1 REARRANGEMENTS
Approximately 2.4% of lung adenocarcinomas harbor ROS1 rearrangements.82 This distinct genetic alteration occurs more frequently in NSCLC patients who are younger, female, and never-smokers, and who have adenocarcinomas.8 It has been shown that ROS1 rearrangements rarely overlap with other genetic alterations including KRAS mutations, EGFR mutations, and ALK rearrangements.83 As a receptor tyrosine kinase, ROS1 is similar to ALK and insulin receptor family members.84 Crizotinib, which targets ALK, ROS1, and c-MET, was approved by the FDA on March 11, 2016, for the treatment of metastatic ROS1-rearranged NSCLC.85 The approval was based on a phase 2 expansion cohort of the original phase 1 study. Among 50 US patients enrolled in this expansion cohort, 3 had complete responses and 33 had partial responses with ORR of 72% (95% CI 58% to 84%). Median PFS was 19.2 months (95% CI 14.4 months to NR) and median duration of response (DOR) was 17.6 months (95% CI 14.5 months to NR).86 During longer follow-up, independent radiology review confirmed high ORR of 66% and median DOR of 18.3 months.85
Interestingly, no companion diagnostic assay has been approved for the detection of ROS1 rearrangements with the approval of crizotinib. In the United States, break apart FISH is the most common detection method. In fact, in the above mentioned phase 2 study, ROS1 rearrangements were detected in 49 out of 50 patients by this method.86 FISH can be technically challenging when dealing with high volume and multiple targets. Reverse transcriptase-PCR is another detection method, but it requires knowledge of the fusion partners. To date, at least 14 ROS1 fusion partners have been reported, with CD74 being the most common.87 NGS with appropriate design and validation can also be used to detect ROS1 rearrangements.
For the case patient, the recommendation would be to start her on crizotinib at 250 mg twice daily. Monitoring for vision disturbance, gastrointestinal complaints, and edema is warranted. Because the estimated onset of response is around 7.9 weeks,86 plans should be made to repeat her scans in approximately 2 months.
BRAF V600E MUTATIONS
CASE PRESENTATION 4
A 71-year-old Caucasian man with a past medical history of hypertension, dyslipidemia, and ischemic cerebrovascular accident without residual deficits was diagnosed with stage IV adenocarcinoma of the lung about 8 months ago. He has a 40 pack-year smoking history and quit smoking when he was diagnosed with lung cancer. His disease burden involved a large mediastinal mass, scattered pleural nodules, multiple lymphadenopathy, and several soft tissue masses. His outside oncologist started him on chemotherapy containing carboplatin and pemetrexed for 6 cycles followed by maintenance pemetrexed. The most recent restaging scans show disease progression with enlarging soft tissue masses and several new lytic bone lesions. MRI-brain with and without contrast shows 2 subcentimeter enhancing lesions. He transferred care to you approximately 4 weeks ago. You ordered a repeat biopsy of 1 of the enlarging soft tissue masses. Molecular analysis revealed BRAF V600E mutation. In the interim, he underwent stereotactic radiosurgery for the 2 brain lesions without any complications. The patient is now in your clinic for follow up.
What would be your recommended systemic treatment?
TARGETED THERAPIES FOR BRAF V600E MUTATION
BRAF mutations were first recognized as activating mutations in advanced melanomas, with BRAF V600E, resulting from the substitution of glutamic acid for valine at amino acid 600, being the most common. BRAF plays an important role in the mitogen-activated protein kinase (MAPK) signaling pathway. Briefly, the activation of MAPK pathway occurs upon ligand binding of receptor tyrosine kinases, which then involves RAS/BRAF/MEK/ERK in a stepwise manner, ultimately leading to cell survival. BRAF mutations have been increasingly recognized also as driver mutations in NSCLC.9–12 They can be detected by PCR or NGS method. The characteristics of NSCLC patients harboring BRAF mutations have been described by various groups.9–12 For instance, 1 case series showed that the incidence was 2.2% among patients with advanced lung adenocarcinoma; 50% of mutations were V600E, while G469A and D594G accounted for the remaining 39% and 11% of patients, respectively. All patients were either current or former smokers. The median OS of patients with BRAF mutations in this case series was NR, while it was 37 months for patients with EGFR mutations (P = 0.73) and NR for patients with ALK rearrangements (P = 0.64).9
For patients with BRAF V600E–mutant NSCLC who have progressed on platinum-based chemotherapy, the combination of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) may represent a new treatment paradigm. This was illustrated in a phase 2, nonrandomized, open-label study. A total of 57 patients were enrolled and 36 patients (63.2% [95% CI 49.3% to 75.6%]) achieved an overall response by investigator assessment, the trial’s primary end point. Disease control rate was 78.9% (95% CI 66.1% to 88.6%), with 4% complete response, 60% partial response, and 16% stable disease. PFS was 9.7 months (95% CI [6.9 to 19.6 months]). The safety profile was comparable to what had been observed in patients with melanoma treated with this regimen. More specifically, 56% of patients on this trial reported serious AEs, including pyrexia (16%), anemia (5%), confusional state (4%), decreased appetite (4%), hemoptysis (4%), hypercalcemia (4%), nausea (4%), and cutaneous squamous cell carcinoma (4%). In addition, neutropenia (9%) and hyponatremia (7%) were the most common grade 3-4 AEs.16
The case patient has experienced disease progression after 1 line of platinum-based chemotherapy, so the combination of dabrafenib and trametinib would be a robust systemic treatment option. dabrafenib as a single agent has also been studied in BRAF V600E–mutant NSCLC in a phase 2 trial. The overall response by investigator assessment among 84 patients was 33% (95% CI 23% to 45%).14 Vemurafenib, another oral BRAF TKI, has demonstrated efficacy for NSCLC patients harboring BRAF V600E mutation. In the cohort of 20 patients with NSCLC, the response rate was 42% (95% CI 20% to 67%) and median PFS was 7.3 months (95% CI 3.5 to 10.8 months).13 Patients with non-V600E mutations have shown variable responses to targeted therapies. MEK TKIs may be considered in this setting; however, the details of this discussion are beyond the scope of this review.
CONCLUSION
The management of advanced NSCLC with driver mutations has seen revolutionary changes over the past decade. Tremendous research has been done in order to first understand the molecular pathogenesis of NSCLC and then discover driver mutations that would lead to development of targeted therapies with clinically significant efficacy as well as tolerability. More recently, increasing efforts have focused on how to conquer acquired resistance in patients with disease progression after first-line TKIs. The field of EGFR-mutant NSCLC has set a successful example, but the work is nowhere near finished. The goals are to search for more driver mutations and to design agents that could potentially block cell survival signals once and for all.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
- Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol 2016;893:1–19.
- Alberg AJ, Brock MV, Ford JG, et al. Epidemiology of lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013;143:e1S–29S.
- Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-3013, based on November 2015 SEER data submission, posted to the SEER website, April 2016. Bethesda (MD): National Cancer Institute; 2016.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Non-Small Cell Lung Cancer: 1–190.
- Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–39.
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–6.
- Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancer. J Clin Oncol 2012;30:863–70.
- Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011;29:2046–51.
- Kinno T, Tsuta K, Shiraishi K, et al. Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol 2014;25:138–42.
- Litvak AM, Paik PK, Woo KM, et al. Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J Thorac Oncol 2014;9:1669–74.
- Villaruz LC, Socinski MA, Abberbock S, et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer 2015;121:448–56.
- Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–36.
- Planchard D, Kim TM, Mazieres J, et al. DaBRAFenib in patients with BRAF V600E-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol 2016;17:642–50.
- Gautschi O, Milia J, Cabarrou B, et al. Targeted therapy for patients with BRAF-mutant lung cancer: results from the European EURAF cohort. J Thorac Oncol 2015;10:1451–7.
- Planchard D, Besse B, Groen HJ, et al. DaBRAFenib plus trametinib in patients with previously treated BRAF V600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 2016;17:984–93.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. J Thorac Oncol 2013;8:823–59.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. Arch Pathol Lab Med 2013;137:828–60.
- Leighl NB, Rekhtman N, Biermann WA, et al. Molecular testing for selection of patients with lung cancer for epidermal growth factor receptor and anaplastic lymphoma kinase tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology guideline. J Clin Oncol 2014;32:3673–9.
- Paik PK, Varghese AM, Sima CS, et al. Response to erlotinib in patients with EGFR mutant advanced non-small cell lung cancers with a squamous or squamous-like component. Mol Cancer Ther 2012;11:2535–40.
- Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov 2015;5:842–9.
- Awad MM, Oxnard GR, Jackman DM, et al. MET Exon 14 mutations in non-small-cell lung cancer are associated with advanced age, and stage-dependent MET genomic amplification, and c-MET overexpression. J Clin Oncol 2016;34:721–30.
- Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thorac Oncol 2016;11:1493–502.
- Reungwetwattana T, Liang Y, Zhu V, et al. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The why, the how, the who, the unknown, and the inevitable. Lung Cancer 2017;103:27–37.
- Drilon A, Wang L, Hasanovic A, et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov 2013;3:630–5.
- Lin JJ, Kennedy E, Sequist LV, et al. Clinical activity of alectinib in advanced RET-rearranged non-small cell lung cancer. J Thorac Oncol 2016;11:2027–32.
- Drilon A, Rekhtman N, Arcila M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol 2016;17:1653–60.
- Cappuzzo F, Bemis L, Varella-Garcia M. HER2 mutation and response to trastuzumab therapy in non-small-cell lung cancer. N Engl J Med 2006;354:2619–21.
- Mazieres J. Barlesi F, Filleron T, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targted drugs: results from the European EUHER2 cohort. Annal Oncol 2016;27:281–6.
- Ou SH, Schrock AB, Bocharov EV, et al. HER2 transmembrane (TMD) mutations (V659/G660) that stabilize homo- and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J Thorac Oncol 2017;12:446–57.
- Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emergent therapies. Cancer Discov 2017;7:596–609.
- Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014;311:1998–2006.
- Barlesi F, Mazieres J, Merlio JP, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016;387:1415–26.
- Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361:958–67.
- Rosell R, Karachaliou N, et al. Large-scale screening for somatic mutations in lung cancer. Lancet 2016;387:1354–6.
- Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005;97:339–46.
- Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013;31:3327–34.
- Wu YL, Chou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol 2015;26:1883–9.
- Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Eng J Med 2010;362:2380–8.
- Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121–8.
- Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011;12:735–42.
- Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239–46.
- Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:213–22.
- Hirsch FR, Bunn PA Jr. EGFR testing in lung cancer is ready for prime time. Lancet Oncol 2009;10:432–3.
- Nelson V, Ziehr J, Aqulnik M, et al. Afatinib: emerging next-generation tyrosine kinase inhibitor for NSCLC. Onco Targets Ther 2013;5:135–43.
- Lee CK, Wu YL, Ding PN, et al. Impact of specific epidermal growth factor receptor (EGFR) mutations and clinical characteristics on outcomes after treatment with EGFR tyrosine kinase inhibitors versus chemotherapy in EGFR-mutant lung cancer: a meta-analysis. J Clin Oncol 2015;33:1958–65.
- Inoue A, Kobayashi K, Maemondo M, et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann Oncol 2013;24:54–9.
- Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947–57.
- Wu YL, Saijo N, Thongprasert S, et al. Efficacy according to blind independent central review: post-hoc analyses from the phase III, randomized, multicenter, IPASS study of first-line gefitinib versus carboplatin/paclitaxel in Asian patients with EFGR mutation-positive advanced NSCLC. Lung Cancer 2017;104:119–25.
- Douillard JY, Ostoros G, Cobo M, et al. First-line gefitinib in Caucasian EGFR-mutation positive NSCLC patients: a phase-IV, open-label, single-arm study. Br J Cancer 2014;110:55–62.
- Hu JC, Sadeghi P, Pinter-Brown LC, et al. Cutaneous side effects of epidermal growth factor receptor inhibitors: clinical presentation, pathogenesis, and management. J Am Acad Dermatol 2007;56:317–26.
- Tarceva [package insert]. South San Francisco (CA): Genentech, Inc; 2010. www.accessdata.fda.gov/drugsatfda_docs/label/2010/021743s14s16lbl.pdf. Accessed April 23, 2017.
- Gilotrif [package insert.] Ridgefield (CT): Boehringer Ingelheim, Inc; 2013. www.accessdata.fda.gov/drugsatfda_docs/label/2013/201292s000lbl.pdf. Accessed April 23, 2017.
- Iressa [package insert]. Wilmington (DE): AstraZeneca, Inc; 2015. Error! Hyperlink reference not valid. Accessed April 23, 2017.
- Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res 2011;17:1616–22.
- Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR TKI therapy in 155 patients with EGFR mutant lung cancers. Clin Cancer Res 2013;19:2240–7.
- Yun CH, Mengwasser KE, Tom AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 2008;105:2070–5.
- Sos ML, Rode HB, Heynck S, et al. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Res 2010;70:868–74.
- Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T190M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014;4:1046–61.
- Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 2017;376:629–40.
- Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small cell lung cancer. N Engl J Med 2010;363:1693–703.
- Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009;27:4247–53.
- Kazandjian D, Blumenthal GM, Chen HY, et al. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014;19:e5–11.
- Solomon BJ, Mok T, Kim DW, et al. First-ling crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 2014;371:2167–77.
- Xalkori [package insert]. New York: Pfizer, Inc; 2011. www.accessdata.fda.gov/drugsatfda_docs/label/2012/202570s002lbl.pdf. Accessed April 23, 2017.
- Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013;368:2385–94.
- Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem 2013;56:5675–90.
- Khozin S, Blumenthal GM, Zhang L, et al. FDA approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer. Clin Cancer Res 2015;21:2436–9.
- Soria JC, Tan DS, Chiari R, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet 2017;389:917–29.
- Zykadia [package insert]. East Hanover (NJ): Novartis Pharmaceuticals Corporation, Inc; 2016. www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/zykadia.pdf. Accessed April 23, 2017.
- Larkins E, Blumenthal GM, Chen H, et al. FDA approval: alectinib for the treatment of metastatic, ALK-positive non-small cell lung cancer following crizotinib. Clin Cancer Res 2016;22:5171–6.
- Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. New Engl J Med 2017 June 6 [Epub ahead of print].
- Kinoshita K, Asoh K, Furuichi N, et al. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem 2012;20:1271–80.
- Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011;19:679–90.
- Kodama T, Tsukaguchi T, Yoshida M, et al. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett 2014;351:215–21.
- Kodama T, Tsukaguchi T, Satoh T, et al. Alectinib shows potent antitumor activity against RET-rearranged non-small cell lung cancer. Mol Cancer Ther 2014;13:2910–8.
- Alecensa [package insert]. South San Francisco (CA): Genentech, Inc; 2015. www.accessdata.fda.gov/drugsatfda_docs/label/2015/208434s000lbl.pdf. Accessed April 23, 2017.
- Kim DW, Tiseo M, Ahn MJ, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase positive non-small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol 2017 May 5 [Epub ahead of print].
- Zhu V, Ou SH. Safety of alectinib for the treatment of metastatic ALK-rearranged non-small cell lung cancer. Expert Opin Drug Saf 2017;16:509–14.
- Gadgeel SM, Shaw AT, Govindan R, et al. Pooled analysis of CNS response to alectinib in two studies of pretreated patients with ALK-positive non-small cell lung cancer. J Clin Oncol 2016;34:4079–85.
- Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol 2011;29:e443–5.
- Zhu Q, Zhan P, Zhang X, et al. Clinicopathologic characteristics of patients with ROS1 fusion gene in non-small cell lung cancer: a meta-analysis. Transl Lung Cancer Res 2015;4:300–9.
- Lin JJ, Ritterhouse LL, Ali SM, et al. ROS1 fusions rarely overlap with other oncogenic drivers in non-small cell lung cancer. J Thorac Oncol 2017;12:872–7.
- Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta 2009;1795:37–52.
- Kazandjian D, Blumenthal G, Luo L, et al. Benefit-Risk summary of crizotinib for the treatment of patients with ROS1 alteration-positive metastatic NSCLC. Oncologist 2016;21:974–80.
- Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 2014;371:1963–71.
- Zhu VW, Upadhyay D, Schrock AB, et al. TPD52L1-ROS1, a new ROS1 fusion variant in lung adenosquamous cell carcinoma identified by comprehensive genomic profiling. Lung Cancer 2016;97:48–50.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
- Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol 2016;893:1–19.
- Alberg AJ, Brock MV, Ford JG, et al. Epidemiology of lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013;143:e1S–29S.
- Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-3013, based on November 2015 SEER data submission, posted to the SEER website, April 2016. Bethesda (MD): National Cancer Institute; 2016.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Non-Small Cell Lung Cancer: 1–190.
- Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–39.
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–6.
- Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancer. J Clin Oncol 2012;30:863–70.
- Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011;29:2046–51.
- Kinno T, Tsuta K, Shiraishi K, et al. Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol 2014;25:138–42.
- Litvak AM, Paik PK, Woo KM, et al. Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J Thorac Oncol 2014;9:1669–74.
- Villaruz LC, Socinski MA, Abberbock S, et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer 2015;121:448–56.
- Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–36.
- Planchard D, Kim TM, Mazieres J, et al. DaBRAFenib in patients with BRAF V600E-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol 2016;17:642–50.
- Gautschi O, Milia J, Cabarrou B, et al. Targeted therapy for patients with BRAF-mutant lung cancer: results from the European EURAF cohort. J Thorac Oncol 2015;10:1451–7.
- Planchard D, Besse B, Groen HJ, et al. DaBRAFenib plus trametinib in patients with previously treated BRAF V600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 2016;17:984–93.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. J Thorac Oncol 2013;8:823–59.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. Arch Pathol Lab Med 2013;137:828–60.
- Leighl NB, Rekhtman N, Biermann WA, et al. Molecular testing for selection of patients with lung cancer for epidermal growth factor receptor and anaplastic lymphoma kinase tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology guideline. J Clin Oncol 2014;32:3673–9.
- Paik PK, Varghese AM, Sima CS, et al. Response to erlotinib in patients with EGFR mutant advanced non-small cell lung cancers with a squamous or squamous-like component. Mol Cancer Ther 2012;11:2535–40.
- Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov 2015;5:842–9.
- Awad MM, Oxnard GR, Jackman DM, et al. MET Exon 14 mutations in non-small-cell lung cancer are associated with advanced age, and stage-dependent MET genomic amplification, and c-MET overexpression. J Clin Oncol 2016;34:721–30.
- Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thorac Oncol 2016;11:1493–502.
- Reungwetwattana T, Liang Y, Zhu V, et al. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The why, the how, the who, the unknown, and the inevitable. Lung Cancer 2017;103:27–37.
- Drilon A, Wang L, Hasanovic A, et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov 2013;3:630–5.
- Lin JJ, Kennedy E, Sequist LV, et al. Clinical activity of alectinib in advanced RET-rearranged non-small cell lung cancer. J Thorac Oncol 2016;11:2027–32.
- Drilon A, Rekhtman N, Arcila M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol 2016;17:1653–60.
- Cappuzzo F, Bemis L, Varella-Garcia M. HER2 mutation and response to trastuzumab therapy in non-small-cell lung cancer. N Engl J Med 2006;354:2619–21.
- Mazieres J. Barlesi F, Filleron T, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targted drugs: results from the European EUHER2 cohort. Annal Oncol 2016;27:281–6.
- Ou SH, Schrock AB, Bocharov EV, et al. HER2 transmembrane (TMD) mutations (V659/G660) that stabilize homo- and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J Thorac Oncol 2017;12:446–57.
- Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emergent therapies. Cancer Discov 2017;7:596–609.
- Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014;311:1998–2006.
- Barlesi F, Mazieres J, Merlio JP, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016;387:1415–26.
- Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361:958–67.
- Rosell R, Karachaliou N, et al. Large-scale screening for somatic mutations in lung cancer. Lancet 2016;387:1354–6.
- Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005;97:339–46.
- Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013;31:3327–34.
- Wu YL, Chou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol 2015;26:1883–9.
- Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Eng J Med 2010;362:2380–8.
- Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121–8.
- Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011;12:735–42.
- Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239–46.
- Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:213–22.
- Hirsch FR, Bunn PA Jr. EGFR testing in lung cancer is ready for prime time. Lancet Oncol 2009;10:432–3.
- Nelson V, Ziehr J, Aqulnik M, et al. Afatinib: emerging next-generation tyrosine kinase inhibitor for NSCLC. Onco Targets Ther 2013;5:135–43.
- Lee CK, Wu YL, Ding PN, et al. Impact of specific epidermal growth factor receptor (EGFR) mutations and clinical characteristics on outcomes after treatment with EGFR tyrosine kinase inhibitors versus chemotherapy in EGFR-mutant lung cancer: a meta-analysis. J Clin Oncol 2015;33:1958–65.
- Inoue A, Kobayashi K, Maemondo M, et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann Oncol 2013;24:54–9.
- Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947–57.
- Wu YL, Saijo N, Thongprasert S, et al. Efficacy according to blind independent central review: post-hoc analyses from the phase III, randomized, multicenter, IPASS study of first-line gefitinib versus carboplatin/paclitaxel in Asian patients with EFGR mutation-positive advanced NSCLC. Lung Cancer 2017;104:119–25.
- Douillard JY, Ostoros G, Cobo M, et al. First-line gefitinib in Caucasian EGFR-mutation positive NSCLC patients: a phase-IV, open-label, single-arm study. Br J Cancer 2014;110:55–62.
- Hu JC, Sadeghi P, Pinter-Brown LC, et al. Cutaneous side effects of epidermal growth factor receptor inhibitors: clinical presentation, pathogenesis, and management. J Am Acad Dermatol 2007;56:317–26.
- Tarceva [package insert]. South San Francisco (CA): Genentech, Inc; 2010. www.accessdata.fda.gov/drugsatfda_docs/label/2010/021743s14s16lbl.pdf. Accessed April 23, 2017.
- Gilotrif [package insert.] Ridgefield (CT): Boehringer Ingelheim, Inc; 2013. www.accessdata.fda.gov/drugsatfda_docs/label/2013/201292s000lbl.pdf. Accessed April 23, 2017.
- Iressa [package insert]. Wilmington (DE): AstraZeneca, Inc; 2015. Error! Hyperlink reference not valid. Accessed April 23, 2017.
- Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res 2011;17:1616–22.
- Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR TKI therapy in 155 patients with EGFR mutant lung cancers. Clin Cancer Res 2013;19:2240–7.
- Yun CH, Mengwasser KE, Tom AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 2008;105:2070–5.
- Sos ML, Rode HB, Heynck S, et al. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Res 2010;70:868–74.
- Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T190M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014;4:1046–61.
- Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 2017;376:629–40.
- Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small cell lung cancer. N Engl J Med 2010;363:1693–703.
- Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009;27:4247–53.
- Kazandjian D, Blumenthal GM, Chen HY, et al. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014;19:e5–11.
- Solomon BJ, Mok T, Kim DW, et al. First-ling crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 2014;371:2167–77.
- Xalkori [package insert]. New York: Pfizer, Inc; 2011. www.accessdata.fda.gov/drugsatfda_docs/label/2012/202570s002lbl.pdf. Accessed April 23, 2017.
- Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013;368:2385–94.
- Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem 2013;56:5675–90.
- Khozin S, Blumenthal GM, Zhang L, et al. FDA approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer. Clin Cancer Res 2015;21:2436–9.
- Soria JC, Tan DS, Chiari R, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet 2017;389:917–29.
- Zykadia [package insert]. East Hanover (NJ): Novartis Pharmaceuticals Corporation, Inc; 2016. www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/zykadia.pdf. Accessed April 23, 2017.
- Larkins E, Blumenthal GM, Chen H, et al. FDA approval: alectinib for the treatment of metastatic, ALK-positive non-small cell lung cancer following crizotinib. Clin Cancer Res 2016;22:5171–6.
- Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. New Engl J Med 2017 June 6 [Epub ahead of print].
- Kinoshita K, Asoh K, Furuichi N, et al. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem 2012;20:1271–80.
- Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011;19:679–90.
- Kodama T, Tsukaguchi T, Yoshida M, et al. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett 2014;351:215–21.
- Kodama T, Tsukaguchi T, Satoh T, et al. Alectinib shows potent antitumor activity against RET-rearranged non-small cell lung cancer. Mol Cancer Ther 2014;13:2910–8.
- Alecensa [package insert]. South San Francisco (CA): Genentech, Inc; 2015. www.accessdata.fda.gov/drugsatfda_docs/label/2015/208434s000lbl.pdf. Accessed April 23, 2017.
- Kim DW, Tiseo M, Ahn MJ, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase positive non-small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol 2017 May 5 [Epub ahead of print].
- Zhu V, Ou SH. Safety of alectinib for the treatment of metastatic ALK-rearranged non-small cell lung cancer. Expert Opin Drug Saf 2017;16:509–14.
- Gadgeel SM, Shaw AT, Govindan R, et al. Pooled analysis of CNS response to alectinib in two studies of pretreated patients with ALK-positive non-small cell lung cancer. J Clin Oncol 2016;34:4079–85.
- Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol 2011;29:e443–5.
- Zhu Q, Zhan P, Zhang X, et al. Clinicopathologic characteristics of patients with ROS1 fusion gene in non-small cell lung cancer: a meta-analysis. Transl Lung Cancer Res 2015;4:300–9.
- Lin JJ, Ritterhouse LL, Ali SM, et al. ROS1 fusions rarely overlap with other oncogenic drivers in non-small cell lung cancer. J Thorac Oncol 2017;12:872–7.
- Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta 2009;1795:37–52.
- Kazandjian D, Blumenthal G, Luo L, et al. Benefit-Risk summary of crizotinib for the treatment of patients with ROS1 alteration-positive metastatic NSCLC. Oncologist 2016;21:974–80.
- Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 2014;371:1963–71.
- Zhu VW, Upadhyay D, Schrock AB, et al. TPD52L1-ROS1, a new ROS1 fusion variant in lung adenosquamous cell carcinoma identified by comprehensive genomic profiling. Lung Cancer 2016;97:48–50.
Targeted Therapy and Immunotherapy in the Treatment of Metastatic Cutaneous Melanoma
INTRODUCTION
The incidence of cutaneous melanoma has increased over the past 2 decades, with SEER estimates indicating that the number of new cases of melanoma diagnosed annually rose from 38,300 in 1996 to 76,000 in 2016.1 Among persons younger than 50 years, the incidence is higher in females, and younger women (aged 15–39 years) are especially vulnerable.2 Among persons older than 50, melanoma incidence in men is nearly twice that of women, in whom melanomas are often thicker and often associated with worse outcomes.1,2 Approximately 85% of melanomas are diagnosed at early stages when surgery is curative, but the lifetime probability of developing invasive disease is 3% in men and 2% in women.
Prior to the advent of effective immunotherapies and targeted therapies, melanoma was often managed with chemotherapy, which had dismal response rates and commensurately poor outcomes. Advances in the understanding of the molecular etiopathogenesis and immune escape responses of cutaneous metastatic melanoma have transformed therapeutic approaches. Specifically, improved understanding of the genetic mutations driving melanoma tumorigenesis coupled with insights into mechanisms of tumor-mediated immune evasion resulted in development of inhibitors of mitogen-activated protein kinases (MAPK; BRAF and MEK) along with inhibitors of negative regulatory immune checkpoints (cytotoxic T lymphocyte–associated antigen 4 [CTLA-4] and programmed cell death-1 [PD-1]). In this review, we discuss the role of immune therapy, targeted therapy, and combinations of these in the treatment of metastatic cutaneous melanoma. We limit the immuno-therapy discussion to approved CTLA-4/PD-1 inhibitors and the targeted therapy discussion to approved BRAF/NRAS/MEK inhibitors and do not discuss non-checkpoint immunotherapies including cytokines (HD IL-2), vaccines, or adoptive T-cell approaches. Interested readers are directed to other excellent works covering these important topics.26–29
DEVELOPMENT OF TARGETED AND NOVEL IMMUNE THERAPIES
For many years the degree of ultraviolet (UV) light exposure was considered the sole major risk factor for melanoma oncogenesis, even though its mechanism was largely unknown.3 However, clinical observations regarding the occurrence of melanoma on less exposed areas (trunk and limbs) in individuals with intermittent sun exposure led to the proposition that melanomas that arose in younger patients with intermittent sun exposure were distinct from melanomas that arose in older patients in association with markers of chronic sun exposure—the “divergent pathway” hypothesis.3 Critical to this understanding were whole-exome sequencing data from multiple groups, including The Cancer Genome Atlas, that identified patterns of mutations in oncogenic drivers that were distinct in patients with and without chronically sun-damaged (CSD) skin.4–7 It is now clear that based on its association with CSD skin, melanoma can be subclassified into CSD or non-CSD melanoma. CSD and non-CSD melanoma have distinct clinico-pathological characteristics and are associated with different driver mutations. CSD melanomas typically arise in older patients on sun-exposed areas (head/neck, dorsal surfaces of distal extremities) and are associated with particular driver mutations (BRAF non-V600E, NRAS, NF1, or KIT) and genetic signatures of UV-induced DNA damage (G > T [UVA] or C > T [UVB]) transitions. Conversely, non-CSD melanomas typically arise in younger (< 55 years) patients on intermittently sun-exposed areas (trunk, proximal extremities) and are associated with BRAF V600E/K driver mutations and often lack genetic signatures of UV mutagenesis.
Identification of driver mutations in components of the MAPK pathway, including BRAF and NRAS, facilitated the development of targeted inhibitors. The BRAF inhibitors vemurafenib and dabrafenib have been shown in pivotal phase 3 studies to significantly improve overall and progression-free survival in patients with metastatic melanoma compared with chemotherapy and garnered regulatory approval (vemurafenib, BRIM-3;8,9 dabrafenib, BREAK-310). Concomitant MEK and BRAF inhibition extends the duration of benefit by preventing downstream kinase activation in the MAPK pathway. Notably, concomitant MEK inhibition alters the side-effect profile of BRAF inhibitors, with reduced incidence of keratoacanthomas and cutaneous squamous cell carcinomas that are attributable to on-target, off-tumor effects of BRAF inhibitors. Combined BRAF and MEK inhibition (vemurafenib/cobimetinib and dabrafenib/trametinib) further improved overall and progression-free survival compared to single-agent BRAF inhibition in phase 3 studies (COMBI-d,11 COMBI-v,12 and coBRIM13). Although often deep, the responses seen with the use of targeted kinase inhibitors are not often durable, with the vast majority of patients progressing after 12 to 15 months of therapy.In parallel, work primarily done in murine models of chronic viral infection uncovered the role played by co-inhibitory or co-excitatory immune checkpoints in mediating T-cell immune responses. These efforts clarified that tumor-mediated immune suppression primarily occurs through enhancement of inhibitory signals via the negative T-cell immune checkpoints CTLA-4 or PD-1.14,15 Blockade of negative T-cell immune checkpoints resulted in activation of the adaptive immune system, resulting in durable anti-tumor responses as demonstrated in studies of the CTLA-4 inhibitor ipilimumab (CA184-02016 and CA184-02417) and the PD-1 inhibitors nivolumab (CA209-003,18 CheckMate 037,19 and CheckMate 06620) and pembrolizumab (KEYNOTE-00121 and KEYNOTE-00622). Compared to the deep but short-lived responses seen with targeted kinase inhibitors, patients treated with CTLA-4 or PD-1 immune checkpoint blockade often developed durable responses that persisted even after completion of therapy. Combined CTLA-4 and PD-1 blockade results in greater magnitude of response with proportionately increased toxicity.23–25
IMMUNOTHERAPY
CTLA-4 AND PD-1 IMMUNE CHECKPOINT INHIBITORS
The novel success of immunotherapy in recent decades is largely attributable to improved understanding of adaptive immune physiology, specifically T-cell activation and regulation. T-cell activation requires 2 independent signaling events: it is initiated upon recognition of the antigen-MHC class II-receptor complex on antigen-presenting cells (APC), and requires a secondary co-stimulatory interaction of CD80/CD86 (B7.1/B7.2) on APCs and CD28 molecule on T-cells; without this second event, T-cells enter an anergic state.30–32 Upon successful signaling and co-stimulation, newly activated T-cells upregulate CTLA-4, which can bind to B7 molecules with a nearly 100-fold greater affinity than CD28.33,34 Unlike CD28, CTLA-4 engagement negatively regulates T-cell activation. The opposing signals produced by CD28 and CTLA-4 are integrated by the T-cell to determine eventual response to activation, and provide a means by which T-cell activation is homeostatically regulated to prevent exaggerated physiologic immune responses.35 It was hypothesized that CTLA-4 blockade would permit T-cell activation, which is thwarted in the tumor microenvironment by tumor-mediated CTLA-4 engagement, thereby unleashing an anti-tumor immune response.36
PD-1 is a member of the CD28 and CTLA-4 immunoglobulin super family and, similar to CTLA-4, binds activated T-cells. PD-1 has 2 ligands on activated T-cells: PD-L1 and PD-L2.37 PD-L1 is constitutively expressed by a variety of immune and non-immune cells, particularly in inflammatory environments including tumor microenvironments, in response to the release of inflammatory cytokines such as interferon (IFN)-γ.37,38 Conversely, PD-L2 is only minimally expressed constitutively, although its expression on immune and non-immune cells can be induced by similar cues from inflammatory microenvironments. PD-L1 and PD-L2 cross-compete for binding to PD-1, with PD-L2 exhibiting 2- to 6-fold greater relative affinity than PD-L1.39 PD-L1/PD-1 binding results in phosphorylation of 2 tyrosinases in the intracellular portion of PD-1, which contains immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM). PD-1 ITSM subsequently recruits either of 2 SH2-domain–containing protein tyrosine phosphatases: SHP-1 and SHP-2. SHP-2 signaling suppresses PI3K/Akt activation, down-regulates Bcl-xL, and suppresses expression of multiple transcription factors that mediate T-cell effector function including GATA-3, Eomes, and T-bet.40–42 The net effect of PD-L1/PD-1 engagement is to suppress T-cell proliferation, cytokine production, cytolytic function, and survival. Unlike CTLA-4, which primarily affects the priming phase of naive T-cell activation, PD-1 chiefly regulates the effector phase of T-cell function. Furthermore, because PD-L1/PD-L2 expression is limited to inflammatory microenvironments, the effects of PD-1 are less generalized than those of CTLA-4.
SINGLE AGENT ACTIVITY OF CTLA-4 AND PD-1 INHIBITORS
Ipilimumab (MDX-010) is a human IgG1 monoclonal antibody shown to inhibit CTLA-4.43 Early studies tested different formulations (transfectoma-derived and hybridoma-derived), doses, and schedules of ipilimumab primarily in patients with advanced refractory melanoma.44–46 Although responses were infrequent, responding patients experienced durable remissions at 1- and 2-year time points. Notably, in a foreshadowing of changes to response criteria used to evaluate these agents, several treated patients who initially had radiographically stable disease upon completion of therapy subsequently experienced a gradual decline in tumor burden.
Ipilimumab was subsequently evaluated in 2 phase 3 trials. The first study (MDX010-020/CA184-020), which involved 676 HLA-A*0201–positive patients with advanced melanoma, compared ipilimumab 3 mg/kg every 3 weeks for 4 doses either singly or in combination with gp100 vaccine with a gp100-only control arm.16 Ipilimumab administration resulted in objective responses in 11% of patients and improved progression-free and overall survival compared to gp100 alone. Of note, ipilimumab monotherapy was superior to ipilimumab/gp100 combination, possibly related to timing of vaccine in relation to ipilimumab. A confirmatory study (CA184-024) compared a higher dose of ipilimumab (10 mg/kg) in combination with dacarbazine to dacarbazine monotherapy in previously untreated melanoma and was positive.17 Given the lack of augmented efficacy with the higher (10 mg/kg) dose, ipilimumab received regulatory approval in 2011 for the treatment of melanoma at the lower dose: 3 mg/kg administered every 3 weeks for 4 doses (Table 1). Survival data was strikingly similar to patterns observed in prior phase 2 studies, with survival curves plateauing after 2 years at 23.5% to 28.5% of treated patients. Pooled survival data from prospective and retrospective studies of ipilimumab corroborate the plateau of 22% (26% treated; 20% untreated) reached at year 3 regardless of prior therapy or ipilimumab dose, underscoring the durability of long-term survival in ipilimumab-treated patients.47 Ipilimumab administration resulted in an unusual spectrum of toxicities including diarrhea, rash, hepatitis, and hypophysitis (termed immune-related adverse events, or irAEs) in up to a third of patients.
Pembrolizumab and nivolumab are humanized IgG4 monoclonal antibodies that target the PD-1 receptor found on activated T cells, B cells, and myeloid cells. Pembrolizumab and nivolumab are engineered similarly: by immunizing transgenic mice with recombinant human PD-1-Fc protein and subsequently screening murine splenic cells fused with myeloma cells for hybridomas producing antibodies reactive to PD-1-Fc.48,49 Unlike IgG1, the IgG4 moiety neither engages Fc receptors nor activates complement, avoiding cytotoxic effects of the antibody upon binding to the T cells that it is intended to activate. Both pembrolizumab and nivolumab bind PD-1 with high affinity and specificity, effectively inhibiting the interaction between PD-1 and ligands PD-L1 and PD-L2.
Nivolumab was first studied in a phase 1 study (CA209-003) of 296 patients with advanced cancers who received 1, 3, or 10 mg/kg administered every 2 weeks.18 Histologies tested included melanoma, non–small-cell lung cancer (NSCLC), renal-cell cancer (RCC), castration-resistant prostate cancer (CRPC), and colorectal cancer (CRC). Responses were seen in melanoma and RCC and unusually in NSCLC, including in both squamous and non-squamous tumors. Objective responses were noted in 41% of the 107 melanoma patients treated at 3 mg/kg. Survival was improved, with 1- and 2-year survival rates of 62% and 43% at extended follow up.50
Subsequently, nivolumab was compared to chemotherapy in a pair of phase 3 studies involving both previously untreated (Checkmate 066) and ipilimumab/BRAF inhibitor–refractory (CheckMate 037) patients.19,20 In both studies, nivolumab produced durable responses in 32% to 34% of patients and improved survival over chemotherapy. Compared to ipilimumab, the incidence of irAEs was much lower with nivolumab. The depth and magnitude of responses observed led to regulatory approval for nivolumab in both indications (untreated and ipilimumab/BRAF inhibitor–treated melanoma) in 2014. Data from both studies are summarized in Table 1.
Pembrolizumab was first evaluated in a phase 1 study of 30 patients with a variety of solid organ malignancies in which no dose-limiting toxicities were observed and no defined maximal tolerated dose was reached.51 Per protocol, maximal administered dose was 10 mg/kg every 2 weeks. Following startling responses including 2 complete responses of long duration, pembrolizumab was evaluated in a large phase 1 study (KEYNOTE-001) of 1260 patients that evaluated 3 doses (10 mg/kg every 2 weeks, 10 mg/kg every 3 weeks, and 2 mg/kg every 3 weeks) in separate melanoma and NSCLC substudies.21 Both ipilimumab-naïve and ipilimumab-treated patients were enrolled in the melanoma substudy. Objective responses were seen in 38% ofpatients across all 3 dosing schedules and were similar in both ipilimumab-naïve and ipilimumab-treated patients. Similar to nivolumab, most responders experienced durable remissions.
Pembrolizumab was subsequently compared to ipilimumab in untreated patients (KEYNOTE-006) in which patients were randomly assigned to receive either ipilimumab or pembrolizumab at 1 of 2 doses: 10 mg/kg every 2 weeks and pembrolizumab 10 mg/kg every 3 weeks.22 Response rates were greater with pembrolizumab than ipilimumab, with commensurately greater 1-year survival rates. Rates of treatment-related adverse events requiring discontinuation of study drug were much lower with pembrolizumab than ipilimumab. This trial was instrumental in proving the superior profile of pembrolizumab over ipilimumab. The US Food and Drug Administration (FDA) granted pembrolizumab accelerated approval for second-line treatment of melanoma in 2014, and updated this to include a first-line indication in 2015 (Table 1).
EFFICACY OF COMBINED CTLA-4 AND PD-1 INHIBITION
Preclinical studies demonstrated that PD-1 blockade was more effective than CTLA-4 blockade and combination PD-1/CTLA-4 blockade was synergistic, with complete rejection of tumors in approximately half of the treated animals.14 This hypothesis was evaluated in a phase 1 study that explored both concurrent and sequential combinations of ipilimumab and nivolumab along with increasing doses of both agents in PD-1/CTLA-4–naïve advanced melanoma.23 Responses were greater in the concurrent arm (40%) than in the sequential arm (20%) across dose-levels with a small fraction of patients treated in the concurrent arm experiencing a profound reduction (80%) in tumor burden.
The superiority of ipilimumab/nivolumab combination to ipilimumab monotherapy was demonstrated in a randomized blinded phase 2 study (CheckMate 069).24 Of the 4 different ipilimumab/nivolumab doses explored in the phase 1 study (3 mg/kg and 0.3 mg/kg, 3 mg/kg and 1 mg/kg, 1 mg/kg and 3 mg/kg, 3 mg/kg and 3 mg/kg), ipilimumab 3 mg/kg and nivolumab 1 mg/kg (followed by nivolumab 3 mg/kg) was compared to ipilimumab and nivolumab-matched placebo. Responses were significantly greater with dual PD-1/CTLA-4 blockade compared to CTLA-4 blockade alone (59% versus 11%). Concurrently, a 3-arm randomized phase 3 study compared the same dose of ipilimumab/nivolumab to ipilimumab and nivolumab in previously untreated advanced melanoma (CheckMate 067).25 Similar to CheckMate 069, CheckMate 067 demonstrated that ipilimumab/nivolumab combination resulted in more profound responses (58%) than either ipilimumab (19%) or nivolumab (44%) alone. Toxicity, primarily diarrhea, fatigue, pruritus, and rash, was considerable in the combination arm (55% grade 3/4 adverse events) and resulted in treatment discontinuation in 30% of patients. The profound and durable responses observed led to accelerated approval of ipilimumab/nivolumab combination in 2015 (Table 1).
Efforts to improve the toxicity/benefit ratio of ipilimumab/nivolumab combination have centered around studying lower doses and/or extended dosing schedules of ipilimumab, including ipilimumab 1 mg/kg every 6 or 12 weeks with nivolumab dosed at 3 mg/kg every 2 weeks or 480 mg every 4 weeks. Promising data from a first-line study in NSCLC (CheckMate 012) support the evaluation of nivolumab in combination with lower-dosed ipilimumab (1 mg/kg every 6 or 12 weeks).52 This approach is being tested against platinum doublet chemotherapy in a confirmatory phase 3 study in NSCLC (CheckMate 227).
TARGETED THERAPY
MAPK KINASE PATHWAY IN MELANOMA TUMORIGENESIS
The MAPK pathway mediates cellular responses to growth signals. RAF kinases are central mediators in the MAPK pathway and exert their effect primarily through MEK phosphorylation and activation following dimerization (hetero- or homo-) of RAF molecules. As a result, RAF is integral to multiple cellular processes, including transcriptional regulation, cellular differentiation, and cell proliferation. MAPK pathway activation is a common event in many cancers, primarily due to activating mutations in BRAF or RAS. Alternatively, MAPK pathway activation can occur in the absence of activating mutations in BRAF or NRAS through down-regulation of MAPK pathway inhibitory proteins (RAF-1 inhibitory protein or SPRY-2), C-MET overexpression, or activating mutations in non-BRAF/NRAS kinases including CRAF, HRAS, and NRAS.53,54
Somatic point mutations in BRAF are frequently observed (37%–50%) in malignant melanomas and at lower frequency in a range of human cancers including NSCLC, colorectal cancer, papillary thyroid cancer, ovarian cancer, glioma, and gastrointestinal stromal tumor.6,55,56BRAF mutations in melanoma typically occur within the activation segment of the kinase domain (exon 15). Between 80% and 90% of activating mutations result in an amino acid substitution of glutamate (E) for valine (V) at position 600: V600E.57,58 V600E mutations are true oncogenic drivers, resulting in increased kinase activity with demonstrable transformational capacity in vitro. BRAF mutations are usually mutually exclusive, with tumors typically containing no other driver mutations in NRAS, KIT, NF1, or other genes.
NRAS mutations are less common than BRAF mutations, having a reported frequency of 13% to 25% in melanoma.4NRAS mutations generally occur within the P-loop region of the G domain (exon 2), or less commonly in the switch II region of the G domain (exon 3). Most NRAS exon 2 mutations comprise amino acid substitutions at position 61 from glutamine (Q) to arginine (R; 35%), lysine (K; 34%) and less often to glutamate (E), leucine (L), or proline (P). Preclinical data suggest that NRAS mutations paradoxically stimulate the MAPK pathway and thus enhance tumor growth in vitro.59,60 Several important phenotypic differences distinguish NRAS- from BRAF-mutated melanoma. NRAS-mutated tumors are typically associated with increasing age and CSD skin, while BRAF-mutated tumors arise in younger patients in non-CSD skin. A large population-based study suggested that NRAS-mutated melanomas were associated with mitoses and lower tumor infiltrating lymphocytes (TIL) grade, and arose in anatomic sites other than the head/neck, while BRAF-mutated tumors were associated with mitoses and superficial spreading histology.61 Although the lower TIL grade seen with NRAS-mutated melanomas suggests a more immunosuppressed microenvironment and argues for poorer responses to immune therapies, clinical studies comparing responses to immunotherapies in various categories of driver mutations provide conflicting results for the prognostic role of NRAS mutations in relation to immune checkpoint blockade and other immune therapies.62–64
NF1 represents the third known driver in cutaneous melanoma, with mutations reported in 12% of cases.6,7NF1 encodes neurofibromin, which has GTPase activity and regulates RAS proteins; NF1 loss results in increased RAS.65 Unlike BRAF or NRAS, which are usually mutually exclusive, NF1 mutations in melanoma can occur singly or in combination with either BRAF or NRAS mutations. In these settings, NF1 mutations are associated with RAS activation, MEK-dependence, and resistance to RAF inhibition.66
MAPK PATHWAY INHIBITION SINGLY AND IN COMBINATION
Although multiple MEK 1/2 inhibitors (AS703026, AZD8330/ARRY-704, AZD6244, CH5126766, CI-1040, GSK1120212, PD0325901, RDEA119, and XL518) and RAF inhibitors (ARQ 680, GDC-0879, GSK2118436, PLX4032, RAF265, sorafenib, XL281/BMS-908662) were developed, the initial evaluation of MAPK pathway inhibitors in advanced human cancers began with CI-1040. Preclinical data suggested that CI-1040 potently and selectively inhibited both MEK1 and MEK2, but phase 1 and 2 human trial results were disappointing, likely because these trials were not selectively enriched for NRAS/BRAF–mutated tumors or cancers in which these oncogenic mutations were most commonly detected, such as melanoma.67,68 The subsequent evaluation of selumetinib (AZD6244/ARRY-142886) in a phase 2 study was also negative. Although investigators enrolled a presumably enriched population (cutaneous melanoma), the incidence of NRAS/BRAF–mutated tumors was not ascertained to determine this, but rather assumed, which led to a discrepancy between the assumed (prestudy) and observed (on-study) proportions of BRAF/NRAS mutations that was not accounted for in power calculations.69,70 Lessons learned from these earlier misadventures informed the current paradigm of targeted therapy development: (1) identification of a highly specific and potent inhibitor through high-throughput screening; (2) establishment of maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D) in unselected patients; (3) confirmation of RP2D in selected tumor types enriched for target of interest; and (4) confirmatory study against standard comparator to seek regulatory approval.
Vemurafenib and dabrafenib were evaluated in this tiered fashion in phase 1 dose-finding studies comprising unselected patients, followed by phase 2 studies in advanced BRAF V600E–mutated melanoma. Both were subsequently evaluated in randomized phase 3 trials (vemurafenib, BRIM-38; dabrafenib, BREAK-310) that compared them with dacarbazine (1000 mg/m2 intravenously every 3 weeks) in the treatment of advanced BRAF V600E–mutated melanoma. Response kinetics for both agents were remarkably similar: single-agent BRAF inhibitors resulted in rapid (time to response 2–3 months), profound (approximately 50% objective responses) reductions in tumor burden that lasted 6 to 7 months. Adverse events common to both agents included rash, fatigue, and arthralgia, although clinically significant photosensitivity was more common with vemurafenib and clinically significant pyrexia was more common with dabrafenib. Class-specific adverse events included the development of cutaneous squamous-cell carcinomas and keratoacanthomas secondary to paradoxical activation of MAPK pathway signaling either through activating mutations in HRAS or mutations or amplifications in receptor tyrosine kinases upstream of BRAF, resulting in elevated levels of RAS–guanosine triphosphate complexes.71 Results of these studies resulted in regulatory approval of single-agent BRAF inhibitors for the treatment of BRAF V600E (and later V600K)–mutated melanoma (vemurafenib in 2011; dabrafenib in 2013). Details regarding trial populations, study interventions, efficacy, and adverse events are summarized in Table 2.
Responses to BRAF inhibitors are typically profound but temporary. Mechanisms of acquired resistance are diverse and include reactivation of MAPK pathway–dependent signaling (RAS activation or increased RAF expression), and development of MAPK pathway–independent signaling (COT overexpression; increased PI3K or AKT signaling) that permits bypass of inhibited BRAF signaling within the MAPK pathway.72–76 These findings suggested that upfront inhibition of both MEK and mutant BRAF may produce more durable responses than BRAF inhibition alone. Three pivotal phase 3 studies established the superiority of combination BRAF and MEK inhibition over BRAF inhibition alone: COMBI-d11 (dabrafenib/trametinib versus dabrafenib/placebo), COMBI-v12 (dabrafenib/trametinib versus vemurafenib), and coBRIM13 (vemurafenib/cobimetinib versus vemurafenib/placebo). As expected, compared to BRAF inhibitor monotherapy, combination BRAF and MEK inhibition produced greater responses and improved progression-free and overall survival (Table 2). Interestingly, the rate of cutaneous squamous-cell carcinomas was much lower with combination therapy, reflecting the more profound degree of MAPK pathway inhibition achieved with combination BRAF and MEK inhibition. Based on these results, FDA approval was granted for both dabrafenib/trametinib and vemurafenib/cobimetinib combinations in 2015. Although the dabrafenib/trametinib combination was only approved in 2015, trametinib had independently gained FDA approval in 2013 for the treatment of BRAF V600E/K–mutated melanoma on the basis of the phase 3 METRIC study.77
Encorafenib (LGX818) and binimetinib (MEK162, ARRY-162, ARRY-438162) are new BRAF and MEK inhibitors currently being evaluated in clinical trials. Encorafenib/binimetinib combination was first evaluated in a phase 3 study (COLUMBUS) that compared it with vemurafenib monotherapy in BRAF-mutant melanoma.78 Unsurprisingly, encorafenib/binimetinib combination produced greater and more durable responses compared to vemurafenib monotherapy. The median progression-free survival of the encorafenib/binimetinib combination (14.9 months) was greater than vemurafenib monotherapy (7.3 months) in this study, and intriguingly greater than that seen with vemurafenib/cobimetinib (coBRIM 9.9 months) and dabrafenib/trametinib (COMBI-d 9.3 months; COMBI-v 11.4 months). Of note, although encorafenib has an IC50 midway between dabrafenib and vemurafenib in cell-free assays (0.8 nM dabrafenib, 4 nM encorafenib, and 31 nM vemurafenib), it has an extremely slower off-rate from BRAF V600E, which results in significantly greater target inhibition in cells following drug wash-out.79 This may account for the significantly greater clinical benefit seen with encorafenib/binimetinib in clinical trials. Final study data are eagerly awaited. Regulatory approval has been sought, and is pending at this time.
Binimetinib has been compared to dacarbazine in a phase 3 study (NEMO) of patients with NRAS-mutant melanoma, most of whom had been previously treated with immunotherapy.80 Response rates were low in both arms, although slightly greater with binimetinib than dacarbazine (15% versus 9%), commensurate with a modest improvement in progression-free survival. FDA approval has been sought and remains pending at this time.
KIT INHIBITION SINGLY AND IN COMBINATION
The KIT receptor protein tyrosine kinase is a transmembrane protein consisting of extracellular and intracellular domains. Activating KIT mutations occur in 2% to 8% of all melanoma patients and may be found in all melanoma subtypes but are commonest in acral melanomas (10%–20%) and mucosal melanomas (15%–20%). Activating KIT mutations primarily occur in exons 11 and 13, which code for the juxtamembrane and kinase domains, respectively.5,81–83
Imatinib mesylate is a tyrosine kinase inhibitor of the 2-phenyl amino pyrimidine class that occupies the tyrosine kinase active site with resultant blocking of tyrosine kinase activity. Imatinib mesylate is known to block KIT and has been extensively studied in patients with gastrointestinal stromal tumors (GIST), 80% of whom harbor KIT mutations, in both the adjuvant and the metastatic settings. In melanoma, imatinib mesylate was studied in a Chinese open-label, phase 2 study of imatinib mesylate monotherapy in metastatic melanoma harboring KIT mutation or amplification; 25% of the study patients had mucosal disease and the rest had cutaneous disease, with acral involvement in 50% of all patients.84 Overall response rate was 23%, while 51% of patients remained alive at 1 year with no differences in response rate and/or survival being noted between patients with either KIT mutations or amplifications. In a separate study of imatinib mesylate at 400 mg daily or 400 mg twice daily in Caucasian patients with KIT-mutated/amplified melanoma, similar response and survival rates were reported, although patients with KIT mutations did nonsignificantly better than those with KIT amplifications.85
Other novel studies evaluating KIT inhibitors include KIT inhibition in combination with the VEGF inhibitor bevacizumab and a study of selective BCR-ABL kinase inhibitor nilotinib in imatinib-resistant melanoma. In the former phase 1/2 study, Flaherty and colleagues studied imatinib 800 mg daily and bevacizumab at 10 mg/kg every 2 weeks in 63 patients with advanced tumors, including 23 with metastatic melanoma. Although the combination was relatively nontoxic, no significant efficacy signal was seen and further accrual to the phase 2 portion was halted after the first stage was completed.86 Nilotinib is a BCR-ABL1 tyrosine kinase inhibitor intelligently designed based on the structure of the ABL-imatinib complex that is 10 to 30 times more potent than imatinib in inhibiting BCR-ABL1 tyrosine kinase activity. Nilotinib is approved for the treatment of imatinib-resistant chronic myelogenous leukemia (CML), with reported efficacy in patients with central nervous system (CNS) involvement.87,88 Nilotinib has been studied in a single study of KIT-mutated/amplified melanoma that included patients with imatinib-resistance and those with treated CNS disease. Nilotinib appeared to be active in imatinib-resistant melanoma, although no responses were seen in the CNS disease cohort.89 Overall, the response rates observed with KIT inhibition in melanoma are much lower than those observed in CML and GIST.
CONCLUSION AND FUTURE DIRECTIONS
Prior to 2011, the only approved agents for the treatment of advanced melanoma were dacarbazine and high-dose interleukin-2. Since 2011, drug approvals in melanoma have proceeded at a frenetic pace unmatched in any other disease. The primary events underlying this are advances in our understanding of the gene mutation landscape driving melanoma tumorigenesis, accompanied by insights into the means by which tumors circumvent the induction of effective anti-tumor T-cell responses. These insights have resulted in the development of inhibitors targeting MAPK pathway kinases BRAF, MEK, and NRAS), KIT, and regulatory immune checkpoints (CTLA-4 and PD-1). Although BRAF/MEK inhibition results in profound reductions and even occasional complete responses in patients, these responses are typically short lived, rarely lasting more than 9 to 11 months; the encorafenib/binimetinib combination may improve that duration marginally. However, the signature therapeutic advance in melanoma of the past decade is immunotherapy, particularly the development of inhibitors of CTLA-4 and PD-1 immune checkpoints. With these agents, significant proportions of treated patients remain free of progression off-therapy (ipilimumab 23%; nivolumab 34%; pembrolizumab 35%; ipilimumab/nivolumab 64%), and some patients can be successfully re-induced after delayed progression. Separately, the high response rates observed with the use of KIT inhibitors in CML and GIST have not been observed in KIT mutated/amplified melanoma and development of agents in this space has been limited. The challenges ahead center around identifying predictive biomarkers and circumventing primary or acquired resistance, with the eventual goal of producing durable remissions in the majority of treated patients.
Our improved understanding of the mechanisms of acquired resistance to BRAF/MEK inhibitors suggests that anti-tumor activity may be achieved by targeting multiple pathways, possibly with combination regimens comprising other inhibitors and/or immunotherapy. Preclinical data supports the use of combination strategies targeting both ERK and PI3K/mTOR to circumvent acquired resistance.90 Ongoing studies are evaluating combinations with biguanides (metformin: NCT02143050 and NCT01638676; phenformin: NCT03026517), HSP90 inhibitors (XL888: NCT02721459; AT13387: NCT02097225), and decitabine (NCT01876641).
One complexity affecting management of resistance in the targeted therapy landscape remains tumor heterogeneity, particularly intra- and intertumoral heterogeneity, which may explain the apparent contradiction between continued efficacy of BRAF inhibitors in BRAF-resistant tumors and preclinical data predicting slower progression of resistant tumors on cessation of BRAF inhibitors.91–94 These data provide a rationale to investigate intermittent dosing regimens with BRAF/MEK inhibitors; several studies exploring this approach are ongoing (NCT01894672 and NCT02583516).
Given the specificity, adaptability, and memory response associated with immunotherapy, it is likely that these agents will be used to treat the majority of patients regardless of mutational status. Hence, identifying predictive biomarkers of response to immune checkpoint inhibitors is vital. The presence of CD8+ T-cell infiltrate and IFN-γ gene signature, which indicate an “inflamed” tumor microenvironment, are highly predictive of clinical benefit from PD-1 inhibitors.95,96 However, not all PD-1 responders have “inflamed” tumor microenvironments, and not all patients with an “inflamed” tumor microenvironment respond to immune checkpoint inhibitors. The complexity of the immune system is reflected in the multiple non-redundant immunologic pathways, both positive and negative, with checkpoints and ligands that emerge dynamically in response to treatment. Given the dynamic nature of the immune response, it is unlikely that any single immunologic biomarker identified pre-treatment will be completely predictive. Rather, the complexity of the biomarker approach must match the complexity of the immune response elicited, and will likely incorporate multifarious elements including CD8+ T-cell infiltrate, IFN-γ gene signature, and additional elements including microbiome, genetic polymorphisms, and tumor mutation load. The goal is to use multiple markers to guide development of combinations and then, depending on initial response, to examine tumors for alterations to guide decisions about additional treatment(s) to improve responses, with the eventual goal being durable clinical responses for all patients.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
- Guy GP, Thomas CC, Thompson T, et al. Vital signs: melanoma incidence and mortality trends and projections - United States, 1982-2030. MMWR Morb Mortal Wkly Rep 2015;64:591–6.
- Anderson WF, Pfeiffer RM, Tucker MA, Rosenberg PS. Divergent cancer pathways for early-onset and late-onset cutaneous malignant melanoma. Cancer 2009;115:4176–85.
- Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005;353:2135–47.
- Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 2006;24:4340–6.
- Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell 2012;150:251–63.
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681–96.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol 2014;15:323–32.
- Hauschild A, Grob J-J, Demidov LV, et al. DaBRAFenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380:358–65.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877–88.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined daBRAFenib and trametinib. N Engl J Med 2015;372:30–9.
- Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371:1867–76.
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 2010;107:4275–80.
- Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014;515:577–81.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–23.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517–26.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54.
- Weber JS, Angelo SP D’, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015;16:375–84.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–30.
- Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013;369:134–44.
- Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521–32.
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–33.
- Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015;372:2006–17.
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34.
- Eggermont AMM. Therapeutic vaccines in solid tumours: can they be harmful? Eur J Cancer 2009;45:2087–90.
- Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Transl Med 2012;4:127ps8.
- Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol 2014;192:5451–8.
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015;348:62–8.
- Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 1992;356:607–9.
- Greenfield EA, Nguyen KA, Kuchroo VK. CD28/B7 costimulation: a review. Crit Rev Immunol 1998;18:389–418.
- Sharpe AH, Abbas AK. T-cell costimulation--biology, therapeutic potential, and challenges. N Engl J Med 2006;355:973–5.
- Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol 2001;19:565–94.
- Collins AV, Brodie DW, Gilbert RJC, et al. The interaction properties of costimulatory molecules revisited. Immunity 2002;17:201–10.
- Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995;182:459–65.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271:1734–6.
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704.
- Yamazaki T, Akiba H, Iwai H, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 2002;169:5538–45.
- Youngnak P, Kozono Y, Kozono H, et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem Biophys Res Commun 2003;307:672–7.
- Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 2004;173:945–54.
- Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 2005;25:9543–53.
- Riley JL. PD-1 signaling in primary T cells. Immunol Rev 2009;229:114–25.
- Wolchok JD, Hodi FS, Weber JS, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci 2013;1291:1–13.
- Weber JS, O’Day S, Urba W, et al. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol 2008;26:5950–6.
- Hodi FS, Butler M, Oble DA, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A 2008;105:3005–10.
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol 2010;11:155–64.
- Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015;33:1889–94.
- Wang C, Thudium KB, Han M, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res 2014;2:846–56.
- Poole RM. Pembrolizumab: first global approval. Drugs 2014;74:1973–81.
- Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014;32:1020–30.
- Patnaik A, Kang SP, Rasco D, et al. Phase I study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res 2015;21:4286–93.
- Hellmann MD, Rizvi NA, Goldman JW, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol 2017;18:31–41.
- Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am 2009;23:529–45, ix.
- Satyamoorthy K, Li G, Gerrero MR, et al. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res 2003;63:756–9.
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54.
- Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012;44:1006–14.
- Rubinstein JC, Sznol M, Pavlick AC, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med 2010;8:67.
- Lovly CM, Dahlman KB, Fohn LE, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS ONE 2012;7:e35309.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010;464:431–5.
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010;464:427–30.
- Thomas NE, Edmiston SN, Alexander A, et al. Association between NRAS and BRAF mutational status and melanoma-specific survival among patients with higher-risk primary melanoma. JAMA Oncology 2015;1:359–68.
- Joseph RW, Sullivan RJ, Harrell R, et al. Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother 2012;35:66–72.
- Johnson DB, Lovly CM, Flavin M, et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res 2015;3:288–95.
- Johnson DB, Frampton GM, Rioth MJ, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res 2016;4:959–67.
- Kiuru M, Busam KJ. The NF1 gene in tumor syndromes and melanoma. Lab Invest 2017;97:146–57.
- Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res 2014;74:2340–50.
- Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol 2005;23:5281–93.
- Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 2004;22:4456–62.
- Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res 2012;18:555–67.
- Davar D, Kirkwood JM. CCR 20th anniversary commentary: MAPK/ERK pathway inhibition in melanoma-kinase inhibition redux. Clin Cancer Res 2015;21:5412–4.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 2012;366:207–15.
- Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010;468:968–72.
- Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468:973–7.
- Shi H, Hong A, Kong X, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov 2014;4:69–79.
- Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014;4:80–93.
- Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 2014;4:94–109.
- Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107–14.
- Dummer R, Ascierto PA, Gogas HJ, et al. Results of COLUMBUS Part 1: a phase 3 trial of encorafenib (ENCO) plus binimetinib (BINI) versus vemurafenib (VEM) or ENCO in BRAF-mutant melanoma. Presented at Society for Melanoma Research 2016 Congress. November 6-9, 2016. Boston (MA).
- Adelmann CH, Ching G, Du L, et al. Comparative profiles of BRAF inhibitors: the paradox index as a predictor of clinical toxicity. Oncotarget 2016;7:30453–60.
- Dummer R, Schadendorf D, Ascierto PA, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2017;18:435–45.
- Willmore-Payne C, Holden JA, Tripp S, Layfield LJ. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol 2005;36:486–93.
- Beadling C, Jacobson-Dunlop E, Hodi FS, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res 2008;14:6821–8.
- Handolias D, Salemi R, Murray W, et al. Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment Cell Melanoma Res 2010;23:210–5.
- Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 2011;29:2904–9.
- Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol 2013;31:3182–90.
- Flaherty KT, Hamilton BK, Rosen MA, et al. Phase I/II trial of imatinib and bevacizumab in patients with advanced melanoma and other advanced cancers. Oncologist 2015;20:952–9.
- Giles FJ, le Coutre PD, Pinilla-Ibarz J, et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia 2013;27:107–12.
- Reinwald M, Schleyer E, Kiewe P, et al. Efficacy and pharmacologic data of second-generation tyrosine kinase inhibitor nilotinib in BCR-ABL-positive leukemia patients with central nervous system relapse after allogeneic stem cell transplantation. Biomed Res Int 2014;2014:637059.
- Carvajal RD, Lawrence DP, Weber JS, et al. Phase II study of nilotinib in melanoma harboring KIT alterations following progression to prior KIT inhibition. Clin Cancer Res 2015;21:2289–96.
- Carlino MS, Todd JR, Gowrishankar K, et al. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol Oncol 2014;8:544–54.
- Carlino MS, Gowrishankar K, Saunders CAB, et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther 2013;12:1332–42.
- Chan MMK, Haydu LE, Menzies AM, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer 2014;120:3142–53.
- Thakur M Das, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013;494:251–5.
- Thakur M Das, Stuart DD. Molecular pathways: response and resistance to BRAF and MEK inhibitors in BRAF(V600E) tumors. Clin Cancer Res 2014;20:1074–80.
- Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71.
- Ayers M, Lunceford J, Nebozhyn M, et al. Relationship between immune gene signatures and clinical response to PD-1 blockade with pembrolizumab (MK-3475) in patients with advanced solid tumors. J Immunotherapy Cancer 2015;3(Suppl 2):P80.
INTRODUCTION
The incidence of cutaneous melanoma has increased over the past 2 decades, with SEER estimates indicating that the number of new cases of melanoma diagnosed annually rose from 38,300 in 1996 to 76,000 in 2016.1 Among persons younger than 50 years, the incidence is higher in females, and younger women (aged 15–39 years) are especially vulnerable.2 Among persons older than 50, melanoma incidence in men is nearly twice that of women, in whom melanomas are often thicker and often associated with worse outcomes.1,2 Approximately 85% of melanomas are diagnosed at early stages when surgery is curative, but the lifetime probability of developing invasive disease is 3% in men and 2% in women.
Prior to the advent of effective immunotherapies and targeted therapies, melanoma was often managed with chemotherapy, which had dismal response rates and commensurately poor outcomes. Advances in the understanding of the molecular etiopathogenesis and immune escape responses of cutaneous metastatic melanoma have transformed therapeutic approaches. Specifically, improved understanding of the genetic mutations driving melanoma tumorigenesis coupled with insights into mechanisms of tumor-mediated immune evasion resulted in development of inhibitors of mitogen-activated protein kinases (MAPK; BRAF and MEK) along with inhibitors of negative regulatory immune checkpoints (cytotoxic T lymphocyte–associated antigen 4 [CTLA-4] and programmed cell death-1 [PD-1]). In this review, we discuss the role of immune therapy, targeted therapy, and combinations of these in the treatment of metastatic cutaneous melanoma. We limit the immuno-therapy discussion to approved CTLA-4/PD-1 inhibitors and the targeted therapy discussion to approved BRAF/NRAS/MEK inhibitors and do not discuss non-checkpoint immunotherapies including cytokines (HD IL-2), vaccines, or adoptive T-cell approaches. Interested readers are directed to other excellent works covering these important topics.26–29
DEVELOPMENT OF TARGETED AND NOVEL IMMUNE THERAPIES
For many years the degree of ultraviolet (UV) light exposure was considered the sole major risk factor for melanoma oncogenesis, even though its mechanism was largely unknown.3 However, clinical observations regarding the occurrence of melanoma on less exposed areas (trunk and limbs) in individuals with intermittent sun exposure led to the proposition that melanomas that arose in younger patients with intermittent sun exposure were distinct from melanomas that arose in older patients in association with markers of chronic sun exposure—the “divergent pathway” hypothesis.3 Critical to this understanding were whole-exome sequencing data from multiple groups, including The Cancer Genome Atlas, that identified patterns of mutations in oncogenic drivers that were distinct in patients with and without chronically sun-damaged (CSD) skin.4–7 It is now clear that based on its association with CSD skin, melanoma can be subclassified into CSD or non-CSD melanoma. CSD and non-CSD melanoma have distinct clinico-pathological characteristics and are associated with different driver mutations. CSD melanomas typically arise in older patients on sun-exposed areas (head/neck, dorsal surfaces of distal extremities) and are associated with particular driver mutations (BRAF non-V600E, NRAS, NF1, or KIT) and genetic signatures of UV-induced DNA damage (G > T [UVA] or C > T [UVB]) transitions. Conversely, non-CSD melanomas typically arise in younger (< 55 years) patients on intermittently sun-exposed areas (trunk, proximal extremities) and are associated with BRAF V600E/K driver mutations and often lack genetic signatures of UV mutagenesis.
Identification of driver mutations in components of the MAPK pathway, including BRAF and NRAS, facilitated the development of targeted inhibitors. The BRAF inhibitors vemurafenib and dabrafenib have been shown in pivotal phase 3 studies to significantly improve overall and progression-free survival in patients with metastatic melanoma compared with chemotherapy and garnered regulatory approval (vemurafenib, BRIM-3;8,9 dabrafenib, BREAK-310). Concomitant MEK and BRAF inhibition extends the duration of benefit by preventing downstream kinase activation in the MAPK pathway. Notably, concomitant MEK inhibition alters the side-effect profile of BRAF inhibitors, with reduced incidence of keratoacanthomas and cutaneous squamous cell carcinomas that are attributable to on-target, off-tumor effects of BRAF inhibitors. Combined BRAF and MEK inhibition (vemurafenib/cobimetinib and dabrafenib/trametinib) further improved overall and progression-free survival compared to single-agent BRAF inhibition in phase 3 studies (COMBI-d,11 COMBI-v,12 and coBRIM13). Although often deep, the responses seen with the use of targeted kinase inhibitors are not often durable, with the vast majority of patients progressing after 12 to 15 months of therapy.In parallel, work primarily done in murine models of chronic viral infection uncovered the role played by co-inhibitory or co-excitatory immune checkpoints in mediating T-cell immune responses. These efforts clarified that tumor-mediated immune suppression primarily occurs through enhancement of inhibitory signals via the negative T-cell immune checkpoints CTLA-4 or PD-1.14,15 Blockade of negative T-cell immune checkpoints resulted in activation of the adaptive immune system, resulting in durable anti-tumor responses as demonstrated in studies of the CTLA-4 inhibitor ipilimumab (CA184-02016 and CA184-02417) and the PD-1 inhibitors nivolumab (CA209-003,18 CheckMate 037,19 and CheckMate 06620) and pembrolizumab (KEYNOTE-00121 and KEYNOTE-00622). Compared to the deep but short-lived responses seen with targeted kinase inhibitors, patients treated with CTLA-4 or PD-1 immune checkpoint blockade often developed durable responses that persisted even after completion of therapy. Combined CTLA-4 and PD-1 blockade results in greater magnitude of response with proportionately increased toxicity.23–25
IMMUNOTHERAPY
CTLA-4 AND PD-1 IMMUNE CHECKPOINT INHIBITORS
The novel success of immunotherapy in recent decades is largely attributable to improved understanding of adaptive immune physiology, specifically T-cell activation and regulation. T-cell activation requires 2 independent signaling events: it is initiated upon recognition of the antigen-MHC class II-receptor complex on antigen-presenting cells (APC), and requires a secondary co-stimulatory interaction of CD80/CD86 (B7.1/B7.2) on APCs and CD28 molecule on T-cells; without this second event, T-cells enter an anergic state.30–32 Upon successful signaling and co-stimulation, newly activated T-cells upregulate CTLA-4, which can bind to B7 molecules with a nearly 100-fold greater affinity than CD28.33,34 Unlike CD28, CTLA-4 engagement negatively regulates T-cell activation. The opposing signals produced by CD28 and CTLA-4 are integrated by the T-cell to determine eventual response to activation, and provide a means by which T-cell activation is homeostatically regulated to prevent exaggerated physiologic immune responses.35 It was hypothesized that CTLA-4 blockade would permit T-cell activation, which is thwarted in the tumor microenvironment by tumor-mediated CTLA-4 engagement, thereby unleashing an anti-tumor immune response.36
PD-1 is a member of the CD28 and CTLA-4 immunoglobulin super family and, similar to CTLA-4, binds activated T-cells. PD-1 has 2 ligands on activated T-cells: PD-L1 and PD-L2.37 PD-L1 is constitutively expressed by a variety of immune and non-immune cells, particularly in inflammatory environments including tumor microenvironments, in response to the release of inflammatory cytokines such as interferon (IFN)-γ.37,38 Conversely, PD-L2 is only minimally expressed constitutively, although its expression on immune and non-immune cells can be induced by similar cues from inflammatory microenvironments. PD-L1 and PD-L2 cross-compete for binding to PD-1, with PD-L2 exhibiting 2- to 6-fold greater relative affinity than PD-L1.39 PD-L1/PD-1 binding results in phosphorylation of 2 tyrosinases in the intracellular portion of PD-1, which contains immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM). PD-1 ITSM subsequently recruits either of 2 SH2-domain–containing protein tyrosine phosphatases: SHP-1 and SHP-2. SHP-2 signaling suppresses PI3K/Akt activation, down-regulates Bcl-xL, and suppresses expression of multiple transcription factors that mediate T-cell effector function including GATA-3, Eomes, and T-bet.40–42 The net effect of PD-L1/PD-1 engagement is to suppress T-cell proliferation, cytokine production, cytolytic function, and survival. Unlike CTLA-4, which primarily affects the priming phase of naive T-cell activation, PD-1 chiefly regulates the effector phase of T-cell function. Furthermore, because PD-L1/PD-L2 expression is limited to inflammatory microenvironments, the effects of PD-1 are less generalized than those of CTLA-4.
SINGLE AGENT ACTIVITY OF CTLA-4 AND PD-1 INHIBITORS
Ipilimumab (MDX-010) is a human IgG1 monoclonal antibody shown to inhibit CTLA-4.43 Early studies tested different formulations (transfectoma-derived and hybridoma-derived), doses, and schedules of ipilimumab primarily in patients with advanced refractory melanoma.44–46 Although responses were infrequent, responding patients experienced durable remissions at 1- and 2-year time points. Notably, in a foreshadowing of changes to response criteria used to evaluate these agents, several treated patients who initially had radiographically stable disease upon completion of therapy subsequently experienced a gradual decline in tumor burden.
Ipilimumab was subsequently evaluated in 2 phase 3 trials. The first study (MDX010-020/CA184-020), which involved 676 HLA-A*0201–positive patients with advanced melanoma, compared ipilimumab 3 mg/kg every 3 weeks for 4 doses either singly or in combination with gp100 vaccine with a gp100-only control arm.16 Ipilimumab administration resulted in objective responses in 11% of patients and improved progression-free and overall survival compared to gp100 alone. Of note, ipilimumab monotherapy was superior to ipilimumab/gp100 combination, possibly related to timing of vaccine in relation to ipilimumab. A confirmatory study (CA184-024) compared a higher dose of ipilimumab (10 mg/kg) in combination with dacarbazine to dacarbazine monotherapy in previously untreated melanoma and was positive.17 Given the lack of augmented efficacy with the higher (10 mg/kg) dose, ipilimumab received regulatory approval in 2011 for the treatment of melanoma at the lower dose: 3 mg/kg administered every 3 weeks for 4 doses (Table 1). Survival data was strikingly similar to patterns observed in prior phase 2 studies, with survival curves plateauing after 2 years at 23.5% to 28.5% of treated patients. Pooled survival data from prospective and retrospective studies of ipilimumab corroborate the plateau of 22% (26% treated; 20% untreated) reached at year 3 regardless of prior therapy or ipilimumab dose, underscoring the durability of long-term survival in ipilimumab-treated patients.47 Ipilimumab administration resulted in an unusual spectrum of toxicities including diarrhea, rash, hepatitis, and hypophysitis (termed immune-related adverse events, or irAEs) in up to a third of patients.
Pembrolizumab and nivolumab are humanized IgG4 monoclonal antibodies that target the PD-1 receptor found on activated T cells, B cells, and myeloid cells. Pembrolizumab and nivolumab are engineered similarly: by immunizing transgenic mice with recombinant human PD-1-Fc protein and subsequently screening murine splenic cells fused with myeloma cells for hybridomas producing antibodies reactive to PD-1-Fc.48,49 Unlike IgG1, the IgG4 moiety neither engages Fc receptors nor activates complement, avoiding cytotoxic effects of the antibody upon binding to the T cells that it is intended to activate. Both pembrolizumab and nivolumab bind PD-1 with high affinity and specificity, effectively inhibiting the interaction between PD-1 and ligands PD-L1 and PD-L2.
Nivolumab was first studied in a phase 1 study (CA209-003) of 296 patients with advanced cancers who received 1, 3, or 10 mg/kg administered every 2 weeks.18 Histologies tested included melanoma, non–small-cell lung cancer (NSCLC), renal-cell cancer (RCC), castration-resistant prostate cancer (CRPC), and colorectal cancer (CRC). Responses were seen in melanoma and RCC and unusually in NSCLC, including in both squamous and non-squamous tumors. Objective responses were noted in 41% of the 107 melanoma patients treated at 3 mg/kg. Survival was improved, with 1- and 2-year survival rates of 62% and 43% at extended follow up.50
Subsequently, nivolumab was compared to chemotherapy in a pair of phase 3 studies involving both previously untreated (Checkmate 066) and ipilimumab/BRAF inhibitor–refractory (CheckMate 037) patients.19,20 In both studies, nivolumab produced durable responses in 32% to 34% of patients and improved survival over chemotherapy. Compared to ipilimumab, the incidence of irAEs was much lower with nivolumab. The depth and magnitude of responses observed led to regulatory approval for nivolumab in both indications (untreated and ipilimumab/BRAF inhibitor–treated melanoma) in 2014. Data from both studies are summarized in Table 1.
Pembrolizumab was first evaluated in a phase 1 study of 30 patients with a variety of solid organ malignancies in which no dose-limiting toxicities were observed and no defined maximal tolerated dose was reached.51 Per protocol, maximal administered dose was 10 mg/kg every 2 weeks. Following startling responses including 2 complete responses of long duration, pembrolizumab was evaluated in a large phase 1 study (KEYNOTE-001) of 1260 patients that evaluated 3 doses (10 mg/kg every 2 weeks, 10 mg/kg every 3 weeks, and 2 mg/kg every 3 weeks) in separate melanoma and NSCLC substudies.21 Both ipilimumab-naïve and ipilimumab-treated patients were enrolled in the melanoma substudy. Objective responses were seen in 38% ofpatients across all 3 dosing schedules and were similar in both ipilimumab-naïve and ipilimumab-treated patients. Similar to nivolumab, most responders experienced durable remissions.
Pembrolizumab was subsequently compared to ipilimumab in untreated patients (KEYNOTE-006) in which patients were randomly assigned to receive either ipilimumab or pembrolizumab at 1 of 2 doses: 10 mg/kg every 2 weeks and pembrolizumab 10 mg/kg every 3 weeks.22 Response rates were greater with pembrolizumab than ipilimumab, with commensurately greater 1-year survival rates. Rates of treatment-related adverse events requiring discontinuation of study drug were much lower with pembrolizumab than ipilimumab. This trial was instrumental in proving the superior profile of pembrolizumab over ipilimumab. The US Food and Drug Administration (FDA) granted pembrolizumab accelerated approval for second-line treatment of melanoma in 2014, and updated this to include a first-line indication in 2015 (Table 1).
EFFICACY OF COMBINED CTLA-4 AND PD-1 INHIBITION
Preclinical studies demonstrated that PD-1 blockade was more effective than CTLA-4 blockade and combination PD-1/CTLA-4 blockade was synergistic, with complete rejection of tumors in approximately half of the treated animals.14 This hypothesis was evaluated in a phase 1 study that explored both concurrent and sequential combinations of ipilimumab and nivolumab along with increasing doses of both agents in PD-1/CTLA-4–naïve advanced melanoma.23 Responses were greater in the concurrent arm (40%) than in the sequential arm (20%) across dose-levels with a small fraction of patients treated in the concurrent arm experiencing a profound reduction (80%) in tumor burden.
The superiority of ipilimumab/nivolumab combination to ipilimumab monotherapy was demonstrated in a randomized blinded phase 2 study (CheckMate 069).24 Of the 4 different ipilimumab/nivolumab doses explored in the phase 1 study (3 mg/kg and 0.3 mg/kg, 3 mg/kg and 1 mg/kg, 1 mg/kg and 3 mg/kg, 3 mg/kg and 3 mg/kg), ipilimumab 3 mg/kg and nivolumab 1 mg/kg (followed by nivolumab 3 mg/kg) was compared to ipilimumab and nivolumab-matched placebo. Responses were significantly greater with dual PD-1/CTLA-4 blockade compared to CTLA-4 blockade alone (59% versus 11%). Concurrently, a 3-arm randomized phase 3 study compared the same dose of ipilimumab/nivolumab to ipilimumab and nivolumab in previously untreated advanced melanoma (CheckMate 067).25 Similar to CheckMate 069, CheckMate 067 demonstrated that ipilimumab/nivolumab combination resulted in more profound responses (58%) than either ipilimumab (19%) or nivolumab (44%) alone. Toxicity, primarily diarrhea, fatigue, pruritus, and rash, was considerable in the combination arm (55% grade 3/4 adverse events) and resulted in treatment discontinuation in 30% of patients. The profound and durable responses observed led to accelerated approval of ipilimumab/nivolumab combination in 2015 (Table 1).
Efforts to improve the toxicity/benefit ratio of ipilimumab/nivolumab combination have centered around studying lower doses and/or extended dosing schedules of ipilimumab, including ipilimumab 1 mg/kg every 6 or 12 weeks with nivolumab dosed at 3 mg/kg every 2 weeks or 480 mg every 4 weeks. Promising data from a first-line study in NSCLC (CheckMate 012) support the evaluation of nivolumab in combination with lower-dosed ipilimumab (1 mg/kg every 6 or 12 weeks).52 This approach is being tested against platinum doublet chemotherapy in a confirmatory phase 3 study in NSCLC (CheckMate 227).
TARGETED THERAPY
MAPK KINASE PATHWAY IN MELANOMA TUMORIGENESIS
The MAPK pathway mediates cellular responses to growth signals. RAF kinases are central mediators in the MAPK pathway and exert their effect primarily through MEK phosphorylation and activation following dimerization (hetero- or homo-) of RAF molecules. As a result, RAF is integral to multiple cellular processes, including transcriptional regulation, cellular differentiation, and cell proliferation. MAPK pathway activation is a common event in many cancers, primarily due to activating mutations in BRAF or RAS. Alternatively, MAPK pathway activation can occur in the absence of activating mutations in BRAF or NRAS through down-regulation of MAPK pathway inhibitory proteins (RAF-1 inhibitory protein or SPRY-2), C-MET overexpression, or activating mutations in non-BRAF/NRAS kinases including CRAF, HRAS, and NRAS.53,54
Somatic point mutations in BRAF are frequently observed (37%–50%) in malignant melanomas and at lower frequency in a range of human cancers including NSCLC, colorectal cancer, papillary thyroid cancer, ovarian cancer, glioma, and gastrointestinal stromal tumor.6,55,56BRAF mutations in melanoma typically occur within the activation segment of the kinase domain (exon 15). Between 80% and 90% of activating mutations result in an amino acid substitution of glutamate (E) for valine (V) at position 600: V600E.57,58 V600E mutations are true oncogenic drivers, resulting in increased kinase activity with demonstrable transformational capacity in vitro. BRAF mutations are usually mutually exclusive, with tumors typically containing no other driver mutations in NRAS, KIT, NF1, or other genes.
NRAS mutations are less common than BRAF mutations, having a reported frequency of 13% to 25% in melanoma.4NRAS mutations generally occur within the P-loop region of the G domain (exon 2), or less commonly in the switch II region of the G domain (exon 3). Most NRAS exon 2 mutations comprise amino acid substitutions at position 61 from glutamine (Q) to arginine (R; 35%), lysine (K; 34%) and less often to glutamate (E), leucine (L), or proline (P). Preclinical data suggest that NRAS mutations paradoxically stimulate the MAPK pathway and thus enhance tumor growth in vitro.59,60 Several important phenotypic differences distinguish NRAS- from BRAF-mutated melanoma. NRAS-mutated tumors are typically associated with increasing age and CSD skin, while BRAF-mutated tumors arise in younger patients in non-CSD skin. A large population-based study suggested that NRAS-mutated melanomas were associated with mitoses and lower tumor infiltrating lymphocytes (TIL) grade, and arose in anatomic sites other than the head/neck, while BRAF-mutated tumors were associated with mitoses and superficial spreading histology.61 Although the lower TIL grade seen with NRAS-mutated melanomas suggests a more immunosuppressed microenvironment and argues for poorer responses to immune therapies, clinical studies comparing responses to immunotherapies in various categories of driver mutations provide conflicting results for the prognostic role of NRAS mutations in relation to immune checkpoint blockade and other immune therapies.62–64
NF1 represents the third known driver in cutaneous melanoma, with mutations reported in 12% of cases.6,7NF1 encodes neurofibromin, which has GTPase activity and regulates RAS proteins; NF1 loss results in increased RAS.65 Unlike BRAF or NRAS, which are usually mutually exclusive, NF1 mutations in melanoma can occur singly or in combination with either BRAF or NRAS mutations. In these settings, NF1 mutations are associated with RAS activation, MEK-dependence, and resistance to RAF inhibition.66
MAPK PATHWAY INHIBITION SINGLY AND IN COMBINATION
Although multiple MEK 1/2 inhibitors (AS703026, AZD8330/ARRY-704, AZD6244, CH5126766, CI-1040, GSK1120212, PD0325901, RDEA119, and XL518) and RAF inhibitors (ARQ 680, GDC-0879, GSK2118436, PLX4032, RAF265, sorafenib, XL281/BMS-908662) were developed, the initial evaluation of MAPK pathway inhibitors in advanced human cancers began with CI-1040. Preclinical data suggested that CI-1040 potently and selectively inhibited both MEK1 and MEK2, but phase 1 and 2 human trial results were disappointing, likely because these trials were not selectively enriched for NRAS/BRAF–mutated tumors or cancers in which these oncogenic mutations were most commonly detected, such as melanoma.67,68 The subsequent evaluation of selumetinib (AZD6244/ARRY-142886) in a phase 2 study was also negative. Although investigators enrolled a presumably enriched population (cutaneous melanoma), the incidence of NRAS/BRAF–mutated tumors was not ascertained to determine this, but rather assumed, which led to a discrepancy between the assumed (prestudy) and observed (on-study) proportions of BRAF/NRAS mutations that was not accounted for in power calculations.69,70 Lessons learned from these earlier misadventures informed the current paradigm of targeted therapy development: (1) identification of a highly specific and potent inhibitor through high-throughput screening; (2) establishment of maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D) in unselected patients; (3) confirmation of RP2D in selected tumor types enriched for target of interest; and (4) confirmatory study against standard comparator to seek regulatory approval.
Vemurafenib and dabrafenib were evaluated in this tiered fashion in phase 1 dose-finding studies comprising unselected patients, followed by phase 2 studies in advanced BRAF V600E–mutated melanoma. Both were subsequently evaluated in randomized phase 3 trials (vemurafenib, BRIM-38; dabrafenib, BREAK-310) that compared them with dacarbazine (1000 mg/m2 intravenously every 3 weeks) in the treatment of advanced BRAF V600E–mutated melanoma. Response kinetics for both agents were remarkably similar: single-agent BRAF inhibitors resulted in rapid (time to response 2–3 months), profound (approximately 50% objective responses) reductions in tumor burden that lasted 6 to 7 months. Adverse events common to both agents included rash, fatigue, and arthralgia, although clinically significant photosensitivity was more common with vemurafenib and clinically significant pyrexia was more common with dabrafenib. Class-specific adverse events included the development of cutaneous squamous-cell carcinomas and keratoacanthomas secondary to paradoxical activation of MAPK pathway signaling either through activating mutations in HRAS or mutations or amplifications in receptor tyrosine kinases upstream of BRAF, resulting in elevated levels of RAS–guanosine triphosphate complexes.71 Results of these studies resulted in regulatory approval of single-agent BRAF inhibitors for the treatment of BRAF V600E (and later V600K)–mutated melanoma (vemurafenib in 2011; dabrafenib in 2013). Details regarding trial populations, study interventions, efficacy, and adverse events are summarized in Table 2.
Responses to BRAF inhibitors are typically profound but temporary. Mechanisms of acquired resistance are diverse and include reactivation of MAPK pathway–dependent signaling (RAS activation or increased RAF expression), and development of MAPK pathway–independent signaling (COT overexpression; increased PI3K or AKT signaling) that permits bypass of inhibited BRAF signaling within the MAPK pathway.72–76 These findings suggested that upfront inhibition of both MEK and mutant BRAF may produce more durable responses than BRAF inhibition alone. Three pivotal phase 3 studies established the superiority of combination BRAF and MEK inhibition over BRAF inhibition alone: COMBI-d11 (dabrafenib/trametinib versus dabrafenib/placebo), COMBI-v12 (dabrafenib/trametinib versus vemurafenib), and coBRIM13 (vemurafenib/cobimetinib versus vemurafenib/placebo). As expected, compared to BRAF inhibitor monotherapy, combination BRAF and MEK inhibition produced greater responses and improved progression-free and overall survival (Table 2). Interestingly, the rate of cutaneous squamous-cell carcinomas was much lower with combination therapy, reflecting the more profound degree of MAPK pathway inhibition achieved with combination BRAF and MEK inhibition. Based on these results, FDA approval was granted for both dabrafenib/trametinib and vemurafenib/cobimetinib combinations in 2015. Although the dabrafenib/trametinib combination was only approved in 2015, trametinib had independently gained FDA approval in 2013 for the treatment of BRAF V600E/K–mutated melanoma on the basis of the phase 3 METRIC study.77
Encorafenib (LGX818) and binimetinib (MEK162, ARRY-162, ARRY-438162) are new BRAF and MEK inhibitors currently being evaluated in clinical trials. Encorafenib/binimetinib combination was first evaluated in a phase 3 study (COLUMBUS) that compared it with vemurafenib monotherapy in BRAF-mutant melanoma.78 Unsurprisingly, encorafenib/binimetinib combination produced greater and more durable responses compared to vemurafenib monotherapy. The median progression-free survival of the encorafenib/binimetinib combination (14.9 months) was greater than vemurafenib monotherapy (7.3 months) in this study, and intriguingly greater than that seen with vemurafenib/cobimetinib (coBRIM 9.9 months) and dabrafenib/trametinib (COMBI-d 9.3 months; COMBI-v 11.4 months). Of note, although encorafenib has an IC50 midway between dabrafenib and vemurafenib in cell-free assays (0.8 nM dabrafenib, 4 nM encorafenib, and 31 nM vemurafenib), it has an extremely slower off-rate from BRAF V600E, which results in significantly greater target inhibition in cells following drug wash-out.79 This may account for the significantly greater clinical benefit seen with encorafenib/binimetinib in clinical trials. Final study data are eagerly awaited. Regulatory approval has been sought, and is pending at this time.
Binimetinib has been compared to dacarbazine in a phase 3 study (NEMO) of patients with NRAS-mutant melanoma, most of whom had been previously treated with immunotherapy.80 Response rates were low in both arms, although slightly greater with binimetinib than dacarbazine (15% versus 9%), commensurate with a modest improvement in progression-free survival. FDA approval has been sought and remains pending at this time.
KIT INHIBITION SINGLY AND IN COMBINATION
The KIT receptor protein tyrosine kinase is a transmembrane protein consisting of extracellular and intracellular domains. Activating KIT mutations occur in 2% to 8% of all melanoma patients and may be found in all melanoma subtypes but are commonest in acral melanomas (10%–20%) and mucosal melanomas (15%–20%). Activating KIT mutations primarily occur in exons 11 and 13, which code for the juxtamembrane and kinase domains, respectively.5,81–83
Imatinib mesylate is a tyrosine kinase inhibitor of the 2-phenyl amino pyrimidine class that occupies the tyrosine kinase active site with resultant blocking of tyrosine kinase activity. Imatinib mesylate is known to block KIT and has been extensively studied in patients with gastrointestinal stromal tumors (GIST), 80% of whom harbor KIT mutations, in both the adjuvant and the metastatic settings. In melanoma, imatinib mesylate was studied in a Chinese open-label, phase 2 study of imatinib mesylate monotherapy in metastatic melanoma harboring KIT mutation or amplification; 25% of the study patients had mucosal disease and the rest had cutaneous disease, with acral involvement in 50% of all patients.84 Overall response rate was 23%, while 51% of patients remained alive at 1 year with no differences in response rate and/or survival being noted between patients with either KIT mutations or amplifications. In a separate study of imatinib mesylate at 400 mg daily or 400 mg twice daily in Caucasian patients with KIT-mutated/amplified melanoma, similar response and survival rates were reported, although patients with KIT mutations did nonsignificantly better than those with KIT amplifications.85
Other novel studies evaluating KIT inhibitors include KIT inhibition in combination with the VEGF inhibitor bevacizumab and a study of selective BCR-ABL kinase inhibitor nilotinib in imatinib-resistant melanoma. In the former phase 1/2 study, Flaherty and colleagues studied imatinib 800 mg daily and bevacizumab at 10 mg/kg every 2 weeks in 63 patients with advanced tumors, including 23 with metastatic melanoma. Although the combination was relatively nontoxic, no significant efficacy signal was seen and further accrual to the phase 2 portion was halted after the first stage was completed.86 Nilotinib is a BCR-ABL1 tyrosine kinase inhibitor intelligently designed based on the structure of the ABL-imatinib complex that is 10 to 30 times more potent than imatinib in inhibiting BCR-ABL1 tyrosine kinase activity. Nilotinib is approved for the treatment of imatinib-resistant chronic myelogenous leukemia (CML), with reported efficacy in patients with central nervous system (CNS) involvement.87,88 Nilotinib has been studied in a single study of KIT-mutated/amplified melanoma that included patients with imatinib-resistance and those with treated CNS disease. Nilotinib appeared to be active in imatinib-resistant melanoma, although no responses were seen in the CNS disease cohort.89 Overall, the response rates observed with KIT inhibition in melanoma are much lower than those observed in CML and GIST.
CONCLUSION AND FUTURE DIRECTIONS
Prior to 2011, the only approved agents for the treatment of advanced melanoma were dacarbazine and high-dose interleukin-2. Since 2011, drug approvals in melanoma have proceeded at a frenetic pace unmatched in any other disease. The primary events underlying this are advances in our understanding of the gene mutation landscape driving melanoma tumorigenesis, accompanied by insights into the means by which tumors circumvent the induction of effective anti-tumor T-cell responses. These insights have resulted in the development of inhibitors targeting MAPK pathway kinases BRAF, MEK, and NRAS), KIT, and regulatory immune checkpoints (CTLA-4 and PD-1). Although BRAF/MEK inhibition results in profound reductions and even occasional complete responses in patients, these responses are typically short lived, rarely lasting more than 9 to 11 months; the encorafenib/binimetinib combination may improve that duration marginally. However, the signature therapeutic advance in melanoma of the past decade is immunotherapy, particularly the development of inhibitors of CTLA-4 and PD-1 immune checkpoints. With these agents, significant proportions of treated patients remain free of progression off-therapy (ipilimumab 23%; nivolumab 34%; pembrolizumab 35%; ipilimumab/nivolumab 64%), and some patients can be successfully re-induced after delayed progression. Separately, the high response rates observed with the use of KIT inhibitors in CML and GIST have not been observed in KIT mutated/amplified melanoma and development of agents in this space has been limited. The challenges ahead center around identifying predictive biomarkers and circumventing primary or acquired resistance, with the eventual goal of producing durable remissions in the majority of treated patients.
Our improved understanding of the mechanisms of acquired resistance to BRAF/MEK inhibitors suggests that anti-tumor activity may be achieved by targeting multiple pathways, possibly with combination regimens comprising other inhibitors and/or immunotherapy. Preclinical data supports the use of combination strategies targeting both ERK and PI3K/mTOR to circumvent acquired resistance.90 Ongoing studies are evaluating combinations with biguanides (metformin: NCT02143050 and NCT01638676; phenformin: NCT03026517), HSP90 inhibitors (XL888: NCT02721459; AT13387: NCT02097225), and decitabine (NCT01876641).
One complexity affecting management of resistance in the targeted therapy landscape remains tumor heterogeneity, particularly intra- and intertumoral heterogeneity, which may explain the apparent contradiction between continued efficacy of BRAF inhibitors in BRAF-resistant tumors and preclinical data predicting slower progression of resistant tumors on cessation of BRAF inhibitors.91–94 These data provide a rationale to investigate intermittent dosing regimens with BRAF/MEK inhibitors; several studies exploring this approach are ongoing (NCT01894672 and NCT02583516).
Given the specificity, adaptability, and memory response associated with immunotherapy, it is likely that these agents will be used to treat the majority of patients regardless of mutational status. Hence, identifying predictive biomarkers of response to immune checkpoint inhibitors is vital. The presence of CD8+ T-cell infiltrate and IFN-γ gene signature, which indicate an “inflamed” tumor microenvironment, are highly predictive of clinical benefit from PD-1 inhibitors.95,96 However, not all PD-1 responders have “inflamed” tumor microenvironments, and not all patients with an “inflamed” tumor microenvironment respond to immune checkpoint inhibitors. The complexity of the immune system is reflected in the multiple non-redundant immunologic pathways, both positive and negative, with checkpoints and ligands that emerge dynamically in response to treatment. Given the dynamic nature of the immune response, it is unlikely that any single immunologic biomarker identified pre-treatment will be completely predictive. Rather, the complexity of the biomarker approach must match the complexity of the immune response elicited, and will likely incorporate multifarious elements including CD8+ T-cell infiltrate, IFN-γ gene signature, and additional elements including microbiome, genetic polymorphisms, and tumor mutation load. The goal is to use multiple markers to guide development of combinations and then, depending on initial response, to examine tumors for alterations to guide decisions about additional treatment(s) to improve responses, with the eventual goal being durable clinical responses for all patients.
INTRODUCTION
The incidence of cutaneous melanoma has increased over the past 2 decades, with SEER estimates indicating that the number of new cases of melanoma diagnosed annually rose from 38,300 in 1996 to 76,000 in 2016.1 Among persons younger than 50 years, the incidence is higher in females, and younger women (aged 15–39 years) are especially vulnerable.2 Among persons older than 50, melanoma incidence in men is nearly twice that of women, in whom melanomas are often thicker and often associated with worse outcomes.1,2 Approximately 85% of melanomas are diagnosed at early stages when surgery is curative, but the lifetime probability of developing invasive disease is 3% in men and 2% in women.
Prior to the advent of effective immunotherapies and targeted therapies, melanoma was often managed with chemotherapy, which had dismal response rates and commensurately poor outcomes. Advances in the understanding of the molecular etiopathogenesis and immune escape responses of cutaneous metastatic melanoma have transformed therapeutic approaches. Specifically, improved understanding of the genetic mutations driving melanoma tumorigenesis coupled with insights into mechanisms of tumor-mediated immune evasion resulted in development of inhibitors of mitogen-activated protein kinases (MAPK; BRAF and MEK) along with inhibitors of negative regulatory immune checkpoints (cytotoxic T lymphocyte–associated antigen 4 [CTLA-4] and programmed cell death-1 [PD-1]). In this review, we discuss the role of immune therapy, targeted therapy, and combinations of these in the treatment of metastatic cutaneous melanoma. We limit the immuno-therapy discussion to approved CTLA-4/PD-1 inhibitors and the targeted therapy discussion to approved BRAF/NRAS/MEK inhibitors and do not discuss non-checkpoint immunotherapies including cytokines (HD IL-2), vaccines, or adoptive T-cell approaches. Interested readers are directed to other excellent works covering these important topics.26–29
DEVELOPMENT OF TARGETED AND NOVEL IMMUNE THERAPIES
For many years the degree of ultraviolet (UV) light exposure was considered the sole major risk factor for melanoma oncogenesis, even though its mechanism was largely unknown.3 However, clinical observations regarding the occurrence of melanoma on less exposed areas (trunk and limbs) in individuals with intermittent sun exposure led to the proposition that melanomas that arose in younger patients with intermittent sun exposure were distinct from melanomas that arose in older patients in association with markers of chronic sun exposure—the “divergent pathway” hypothesis.3 Critical to this understanding were whole-exome sequencing data from multiple groups, including The Cancer Genome Atlas, that identified patterns of mutations in oncogenic drivers that were distinct in patients with and without chronically sun-damaged (CSD) skin.4–7 It is now clear that based on its association with CSD skin, melanoma can be subclassified into CSD or non-CSD melanoma. CSD and non-CSD melanoma have distinct clinico-pathological characteristics and are associated with different driver mutations. CSD melanomas typically arise in older patients on sun-exposed areas (head/neck, dorsal surfaces of distal extremities) and are associated with particular driver mutations (BRAF non-V600E, NRAS, NF1, or KIT) and genetic signatures of UV-induced DNA damage (G > T [UVA] or C > T [UVB]) transitions. Conversely, non-CSD melanomas typically arise in younger (< 55 years) patients on intermittently sun-exposed areas (trunk, proximal extremities) and are associated with BRAF V600E/K driver mutations and often lack genetic signatures of UV mutagenesis.
Identification of driver mutations in components of the MAPK pathway, including BRAF and NRAS, facilitated the development of targeted inhibitors. The BRAF inhibitors vemurafenib and dabrafenib have been shown in pivotal phase 3 studies to significantly improve overall and progression-free survival in patients with metastatic melanoma compared with chemotherapy and garnered regulatory approval (vemurafenib, BRIM-3;8,9 dabrafenib, BREAK-310). Concomitant MEK and BRAF inhibition extends the duration of benefit by preventing downstream kinase activation in the MAPK pathway. Notably, concomitant MEK inhibition alters the side-effect profile of BRAF inhibitors, with reduced incidence of keratoacanthomas and cutaneous squamous cell carcinomas that are attributable to on-target, off-tumor effects of BRAF inhibitors. Combined BRAF and MEK inhibition (vemurafenib/cobimetinib and dabrafenib/trametinib) further improved overall and progression-free survival compared to single-agent BRAF inhibition in phase 3 studies (COMBI-d,11 COMBI-v,12 and coBRIM13). Although often deep, the responses seen with the use of targeted kinase inhibitors are not often durable, with the vast majority of patients progressing after 12 to 15 months of therapy.In parallel, work primarily done in murine models of chronic viral infection uncovered the role played by co-inhibitory or co-excitatory immune checkpoints in mediating T-cell immune responses. These efforts clarified that tumor-mediated immune suppression primarily occurs through enhancement of inhibitory signals via the negative T-cell immune checkpoints CTLA-4 or PD-1.14,15 Blockade of negative T-cell immune checkpoints resulted in activation of the adaptive immune system, resulting in durable anti-tumor responses as demonstrated in studies of the CTLA-4 inhibitor ipilimumab (CA184-02016 and CA184-02417) and the PD-1 inhibitors nivolumab (CA209-003,18 CheckMate 037,19 and CheckMate 06620) and pembrolizumab (KEYNOTE-00121 and KEYNOTE-00622). Compared to the deep but short-lived responses seen with targeted kinase inhibitors, patients treated with CTLA-4 or PD-1 immune checkpoint blockade often developed durable responses that persisted even after completion of therapy. Combined CTLA-4 and PD-1 blockade results in greater magnitude of response with proportionately increased toxicity.23–25
IMMUNOTHERAPY
CTLA-4 AND PD-1 IMMUNE CHECKPOINT INHIBITORS
The novel success of immunotherapy in recent decades is largely attributable to improved understanding of adaptive immune physiology, specifically T-cell activation and regulation. T-cell activation requires 2 independent signaling events: it is initiated upon recognition of the antigen-MHC class II-receptor complex on antigen-presenting cells (APC), and requires a secondary co-stimulatory interaction of CD80/CD86 (B7.1/B7.2) on APCs and CD28 molecule on T-cells; without this second event, T-cells enter an anergic state.30–32 Upon successful signaling and co-stimulation, newly activated T-cells upregulate CTLA-4, which can bind to B7 molecules with a nearly 100-fold greater affinity than CD28.33,34 Unlike CD28, CTLA-4 engagement negatively regulates T-cell activation. The opposing signals produced by CD28 and CTLA-4 are integrated by the T-cell to determine eventual response to activation, and provide a means by which T-cell activation is homeostatically regulated to prevent exaggerated physiologic immune responses.35 It was hypothesized that CTLA-4 blockade would permit T-cell activation, which is thwarted in the tumor microenvironment by tumor-mediated CTLA-4 engagement, thereby unleashing an anti-tumor immune response.36
PD-1 is a member of the CD28 and CTLA-4 immunoglobulin super family and, similar to CTLA-4, binds activated T-cells. PD-1 has 2 ligands on activated T-cells: PD-L1 and PD-L2.37 PD-L1 is constitutively expressed by a variety of immune and non-immune cells, particularly in inflammatory environments including tumor microenvironments, in response to the release of inflammatory cytokines such as interferon (IFN)-γ.37,38 Conversely, PD-L2 is only minimally expressed constitutively, although its expression on immune and non-immune cells can be induced by similar cues from inflammatory microenvironments. PD-L1 and PD-L2 cross-compete for binding to PD-1, with PD-L2 exhibiting 2- to 6-fold greater relative affinity than PD-L1.39 PD-L1/PD-1 binding results in phosphorylation of 2 tyrosinases in the intracellular portion of PD-1, which contains immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM). PD-1 ITSM subsequently recruits either of 2 SH2-domain–containing protein tyrosine phosphatases: SHP-1 and SHP-2. SHP-2 signaling suppresses PI3K/Akt activation, down-regulates Bcl-xL, and suppresses expression of multiple transcription factors that mediate T-cell effector function including GATA-3, Eomes, and T-bet.40–42 The net effect of PD-L1/PD-1 engagement is to suppress T-cell proliferation, cytokine production, cytolytic function, and survival. Unlike CTLA-4, which primarily affects the priming phase of naive T-cell activation, PD-1 chiefly regulates the effector phase of T-cell function. Furthermore, because PD-L1/PD-L2 expression is limited to inflammatory microenvironments, the effects of PD-1 are less generalized than those of CTLA-4.
SINGLE AGENT ACTIVITY OF CTLA-4 AND PD-1 INHIBITORS
Ipilimumab (MDX-010) is a human IgG1 monoclonal antibody shown to inhibit CTLA-4.43 Early studies tested different formulations (transfectoma-derived and hybridoma-derived), doses, and schedules of ipilimumab primarily in patients with advanced refractory melanoma.44–46 Although responses were infrequent, responding patients experienced durable remissions at 1- and 2-year time points. Notably, in a foreshadowing of changes to response criteria used to evaluate these agents, several treated patients who initially had radiographically stable disease upon completion of therapy subsequently experienced a gradual decline in tumor burden.
Ipilimumab was subsequently evaluated in 2 phase 3 trials. The first study (MDX010-020/CA184-020), which involved 676 HLA-A*0201–positive patients with advanced melanoma, compared ipilimumab 3 mg/kg every 3 weeks for 4 doses either singly or in combination with gp100 vaccine with a gp100-only control arm.16 Ipilimumab administration resulted in objective responses in 11% of patients and improved progression-free and overall survival compared to gp100 alone. Of note, ipilimumab monotherapy was superior to ipilimumab/gp100 combination, possibly related to timing of vaccine in relation to ipilimumab. A confirmatory study (CA184-024) compared a higher dose of ipilimumab (10 mg/kg) in combination with dacarbazine to dacarbazine monotherapy in previously untreated melanoma and was positive.17 Given the lack of augmented efficacy with the higher (10 mg/kg) dose, ipilimumab received regulatory approval in 2011 for the treatment of melanoma at the lower dose: 3 mg/kg administered every 3 weeks for 4 doses (Table 1). Survival data was strikingly similar to patterns observed in prior phase 2 studies, with survival curves plateauing after 2 years at 23.5% to 28.5% of treated patients. Pooled survival data from prospective and retrospective studies of ipilimumab corroborate the plateau of 22% (26% treated; 20% untreated) reached at year 3 regardless of prior therapy or ipilimumab dose, underscoring the durability of long-term survival in ipilimumab-treated patients.47 Ipilimumab administration resulted in an unusual spectrum of toxicities including diarrhea, rash, hepatitis, and hypophysitis (termed immune-related adverse events, or irAEs) in up to a third of patients.
Pembrolizumab and nivolumab are humanized IgG4 monoclonal antibodies that target the PD-1 receptor found on activated T cells, B cells, and myeloid cells. Pembrolizumab and nivolumab are engineered similarly: by immunizing transgenic mice with recombinant human PD-1-Fc protein and subsequently screening murine splenic cells fused with myeloma cells for hybridomas producing antibodies reactive to PD-1-Fc.48,49 Unlike IgG1, the IgG4 moiety neither engages Fc receptors nor activates complement, avoiding cytotoxic effects of the antibody upon binding to the T cells that it is intended to activate. Both pembrolizumab and nivolumab bind PD-1 with high affinity and specificity, effectively inhibiting the interaction between PD-1 and ligands PD-L1 and PD-L2.
Nivolumab was first studied in a phase 1 study (CA209-003) of 296 patients with advanced cancers who received 1, 3, or 10 mg/kg administered every 2 weeks.18 Histologies tested included melanoma, non–small-cell lung cancer (NSCLC), renal-cell cancer (RCC), castration-resistant prostate cancer (CRPC), and colorectal cancer (CRC). Responses were seen in melanoma and RCC and unusually in NSCLC, including in both squamous and non-squamous tumors. Objective responses were noted in 41% of the 107 melanoma patients treated at 3 mg/kg. Survival was improved, with 1- and 2-year survival rates of 62% and 43% at extended follow up.50
Subsequently, nivolumab was compared to chemotherapy in a pair of phase 3 studies involving both previously untreated (Checkmate 066) and ipilimumab/BRAF inhibitor–refractory (CheckMate 037) patients.19,20 In both studies, nivolumab produced durable responses in 32% to 34% of patients and improved survival over chemotherapy. Compared to ipilimumab, the incidence of irAEs was much lower with nivolumab. The depth and magnitude of responses observed led to regulatory approval for nivolumab in both indications (untreated and ipilimumab/BRAF inhibitor–treated melanoma) in 2014. Data from both studies are summarized in Table 1.
Pembrolizumab was first evaluated in a phase 1 study of 30 patients with a variety of solid organ malignancies in which no dose-limiting toxicities were observed and no defined maximal tolerated dose was reached.51 Per protocol, maximal administered dose was 10 mg/kg every 2 weeks. Following startling responses including 2 complete responses of long duration, pembrolizumab was evaluated in a large phase 1 study (KEYNOTE-001) of 1260 patients that evaluated 3 doses (10 mg/kg every 2 weeks, 10 mg/kg every 3 weeks, and 2 mg/kg every 3 weeks) in separate melanoma and NSCLC substudies.21 Both ipilimumab-naïve and ipilimumab-treated patients were enrolled in the melanoma substudy. Objective responses were seen in 38% ofpatients across all 3 dosing schedules and were similar in both ipilimumab-naïve and ipilimumab-treated patients. Similar to nivolumab, most responders experienced durable remissions.
Pembrolizumab was subsequently compared to ipilimumab in untreated patients (KEYNOTE-006) in which patients were randomly assigned to receive either ipilimumab or pembrolizumab at 1 of 2 doses: 10 mg/kg every 2 weeks and pembrolizumab 10 mg/kg every 3 weeks.22 Response rates were greater with pembrolizumab than ipilimumab, with commensurately greater 1-year survival rates. Rates of treatment-related adverse events requiring discontinuation of study drug were much lower with pembrolizumab than ipilimumab. This trial was instrumental in proving the superior profile of pembrolizumab over ipilimumab. The US Food and Drug Administration (FDA) granted pembrolizumab accelerated approval for second-line treatment of melanoma in 2014, and updated this to include a first-line indication in 2015 (Table 1).
EFFICACY OF COMBINED CTLA-4 AND PD-1 INHIBITION
Preclinical studies demonstrated that PD-1 blockade was more effective than CTLA-4 blockade and combination PD-1/CTLA-4 blockade was synergistic, with complete rejection of tumors in approximately half of the treated animals.14 This hypothesis was evaluated in a phase 1 study that explored both concurrent and sequential combinations of ipilimumab and nivolumab along with increasing doses of both agents in PD-1/CTLA-4–naïve advanced melanoma.23 Responses were greater in the concurrent arm (40%) than in the sequential arm (20%) across dose-levels with a small fraction of patients treated in the concurrent arm experiencing a profound reduction (80%) in tumor burden.
The superiority of ipilimumab/nivolumab combination to ipilimumab monotherapy was demonstrated in a randomized blinded phase 2 study (CheckMate 069).24 Of the 4 different ipilimumab/nivolumab doses explored in the phase 1 study (3 mg/kg and 0.3 mg/kg, 3 mg/kg and 1 mg/kg, 1 mg/kg and 3 mg/kg, 3 mg/kg and 3 mg/kg), ipilimumab 3 mg/kg and nivolumab 1 mg/kg (followed by nivolumab 3 mg/kg) was compared to ipilimumab and nivolumab-matched placebo. Responses were significantly greater with dual PD-1/CTLA-4 blockade compared to CTLA-4 blockade alone (59% versus 11%). Concurrently, a 3-arm randomized phase 3 study compared the same dose of ipilimumab/nivolumab to ipilimumab and nivolumab in previously untreated advanced melanoma (CheckMate 067).25 Similar to CheckMate 069, CheckMate 067 demonstrated that ipilimumab/nivolumab combination resulted in more profound responses (58%) than either ipilimumab (19%) or nivolumab (44%) alone. Toxicity, primarily diarrhea, fatigue, pruritus, and rash, was considerable in the combination arm (55% grade 3/4 adverse events) and resulted in treatment discontinuation in 30% of patients. The profound and durable responses observed led to accelerated approval of ipilimumab/nivolumab combination in 2015 (Table 1).
Efforts to improve the toxicity/benefit ratio of ipilimumab/nivolumab combination have centered around studying lower doses and/or extended dosing schedules of ipilimumab, including ipilimumab 1 mg/kg every 6 or 12 weeks with nivolumab dosed at 3 mg/kg every 2 weeks or 480 mg every 4 weeks. Promising data from a first-line study in NSCLC (CheckMate 012) support the evaluation of nivolumab in combination with lower-dosed ipilimumab (1 mg/kg every 6 or 12 weeks).52 This approach is being tested against platinum doublet chemotherapy in a confirmatory phase 3 study in NSCLC (CheckMate 227).
TARGETED THERAPY
MAPK KINASE PATHWAY IN MELANOMA TUMORIGENESIS
The MAPK pathway mediates cellular responses to growth signals. RAF kinases are central mediators in the MAPK pathway and exert their effect primarily through MEK phosphorylation and activation following dimerization (hetero- or homo-) of RAF molecules. As a result, RAF is integral to multiple cellular processes, including transcriptional regulation, cellular differentiation, and cell proliferation. MAPK pathway activation is a common event in many cancers, primarily due to activating mutations in BRAF or RAS. Alternatively, MAPK pathway activation can occur in the absence of activating mutations in BRAF or NRAS through down-regulation of MAPK pathway inhibitory proteins (RAF-1 inhibitory protein or SPRY-2), C-MET overexpression, or activating mutations in non-BRAF/NRAS kinases including CRAF, HRAS, and NRAS.53,54
Somatic point mutations in BRAF are frequently observed (37%–50%) in malignant melanomas and at lower frequency in a range of human cancers including NSCLC, colorectal cancer, papillary thyroid cancer, ovarian cancer, glioma, and gastrointestinal stromal tumor.6,55,56BRAF mutations in melanoma typically occur within the activation segment of the kinase domain (exon 15). Between 80% and 90% of activating mutations result in an amino acid substitution of glutamate (E) for valine (V) at position 600: V600E.57,58 V600E mutations are true oncogenic drivers, resulting in increased kinase activity with demonstrable transformational capacity in vitro. BRAF mutations are usually mutually exclusive, with tumors typically containing no other driver mutations in NRAS, KIT, NF1, or other genes.
NRAS mutations are less common than BRAF mutations, having a reported frequency of 13% to 25% in melanoma.4NRAS mutations generally occur within the P-loop region of the G domain (exon 2), or less commonly in the switch II region of the G domain (exon 3). Most NRAS exon 2 mutations comprise amino acid substitutions at position 61 from glutamine (Q) to arginine (R; 35%), lysine (K; 34%) and less often to glutamate (E), leucine (L), or proline (P). Preclinical data suggest that NRAS mutations paradoxically stimulate the MAPK pathway and thus enhance tumor growth in vitro.59,60 Several important phenotypic differences distinguish NRAS- from BRAF-mutated melanoma. NRAS-mutated tumors are typically associated with increasing age and CSD skin, while BRAF-mutated tumors arise in younger patients in non-CSD skin. A large population-based study suggested that NRAS-mutated melanomas were associated with mitoses and lower tumor infiltrating lymphocytes (TIL) grade, and arose in anatomic sites other than the head/neck, while BRAF-mutated tumors were associated with mitoses and superficial spreading histology.61 Although the lower TIL grade seen with NRAS-mutated melanomas suggests a more immunosuppressed microenvironment and argues for poorer responses to immune therapies, clinical studies comparing responses to immunotherapies in various categories of driver mutations provide conflicting results for the prognostic role of NRAS mutations in relation to immune checkpoint blockade and other immune therapies.62–64
NF1 represents the third known driver in cutaneous melanoma, with mutations reported in 12% of cases.6,7NF1 encodes neurofibromin, which has GTPase activity and regulates RAS proteins; NF1 loss results in increased RAS.65 Unlike BRAF or NRAS, which are usually mutually exclusive, NF1 mutations in melanoma can occur singly or in combination with either BRAF or NRAS mutations. In these settings, NF1 mutations are associated with RAS activation, MEK-dependence, and resistance to RAF inhibition.66
MAPK PATHWAY INHIBITION SINGLY AND IN COMBINATION
Although multiple MEK 1/2 inhibitors (AS703026, AZD8330/ARRY-704, AZD6244, CH5126766, CI-1040, GSK1120212, PD0325901, RDEA119, and XL518) and RAF inhibitors (ARQ 680, GDC-0879, GSK2118436, PLX4032, RAF265, sorafenib, XL281/BMS-908662) were developed, the initial evaluation of MAPK pathway inhibitors in advanced human cancers began with CI-1040. Preclinical data suggested that CI-1040 potently and selectively inhibited both MEK1 and MEK2, but phase 1 and 2 human trial results were disappointing, likely because these trials were not selectively enriched for NRAS/BRAF–mutated tumors or cancers in which these oncogenic mutations were most commonly detected, such as melanoma.67,68 The subsequent evaluation of selumetinib (AZD6244/ARRY-142886) in a phase 2 study was also negative. Although investigators enrolled a presumably enriched population (cutaneous melanoma), the incidence of NRAS/BRAF–mutated tumors was not ascertained to determine this, but rather assumed, which led to a discrepancy between the assumed (prestudy) and observed (on-study) proportions of BRAF/NRAS mutations that was not accounted for in power calculations.69,70 Lessons learned from these earlier misadventures informed the current paradigm of targeted therapy development: (1) identification of a highly specific and potent inhibitor through high-throughput screening; (2) establishment of maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D) in unselected patients; (3) confirmation of RP2D in selected tumor types enriched for target of interest; and (4) confirmatory study against standard comparator to seek regulatory approval.
Vemurafenib and dabrafenib were evaluated in this tiered fashion in phase 1 dose-finding studies comprising unselected patients, followed by phase 2 studies in advanced BRAF V600E–mutated melanoma. Both were subsequently evaluated in randomized phase 3 trials (vemurafenib, BRIM-38; dabrafenib, BREAK-310) that compared them with dacarbazine (1000 mg/m2 intravenously every 3 weeks) in the treatment of advanced BRAF V600E–mutated melanoma. Response kinetics for both agents were remarkably similar: single-agent BRAF inhibitors resulted in rapid (time to response 2–3 months), profound (approximately 50% objective responses) reductions in tumor burden that lasted 6 to 7 months. Adverse events common to both agents included rash, fatigue, and arthralgia, although clinically significant photosensitivity was more common with vemurafenib and clinically significant pyrexia was more common with dabrafenib. Class-specific adverse events included the development of cutaneous squamous-cell carcinomas and keratoacanthomas secondary to paradoxical activation of MAPK pathway signaling either through activating mutations in HRAS or mutations or amplifications in receptor tyrosine kinases upstream of BRAF, resulting in elevated levels of RAS–guanosine triphosphate complexes.71 Results of these studies resulted in regulatory approval of single-agent BRAF inhibitors for the treatment of BRAF V600E (and later V600K)–mutated melanoma (vemurafenib in 2011; dabrafenib in 2013). Details regarding trial populations, study interventions, efficacy, and adverse events are summarized in Table 2.
Responses to BRAF inhibitors are typically profound but temporary. Mechanisms of acquired resistance are diverse and include reactivation of MAPK pathway–dependent signaling (RAS activation or increased RAF expression), and development of MAPK pathway–independent signaling (COT overexpression; increased PI3K or AKT signaling) that permits bypass of inhibited BRAF signaling within the MAPK pathway.72–76 These findings suggested that upfront inhibition of both MEK and mutant BRAF may produce more durable responses than BRAF inhibition alone. Three pivotal phase 3 studies established the superiority of combination BRAF and MEK inhibition over BRAF inhibition alone: COMBI-d11 (dabrafenib/trametinib versus dabrafenib/placebo), COMBI-v12 (dabrafenib/trametinib versus vemurafenib), and coBRIM13 (vemurafenib/cobimetinib versus vemurafenib/placebo). As expected, compared to BRAF inhibitor monotherapy, combination BRAF and MEK inhibition produced greater responses and improved progression-free and overall survival (Table 2). Interestingly, the rate of cutaneous squamous-cell carcinomas was much lower with combination therapy, reflecting the more profound degree of MAPK pathway inhibition achieved with combination BRAF and MEK inhibition. Based on these results, FDA approval was granted for both dabrafenib/trametinib and vemurafenib/cobimetinib combinations in 2015. Although the dabrafenib/trametinib combination was only approved in 2015, trametinib had independently gained FDA approval in 2013 for the treatment of BRAF V600E/K–mutated melanoma on the basis of the phase 3 METRIC study.77
Encorafenib (LGX818) and binimetinib (MEK162, ARRY-162, ARRY-438162) are new BRAF and MEK inhibitors currently being evaluated in clinical trials. Encorafenib/binimetinib combination was first evaluated in a phase 3 study (COLUMBUS) that compared it with vemurafenib monotherapy in BRAF-mutant melanoma.78 Unsurprisingly, encorafenib/binimetinib combination produced greater and more durable responses compared to vemurafenib monotherapy. The median progression-free survival of the encorafenib/binimetinib combination (14.9 months) was greater than vemurafenib monotherapy (7.3 months) in this study, and intriguingly greater than that seen with vemurafenib/cobimetinib (coBRIM 9.9 months) and dabrafenib/trametinib (COMBI-d 9.3 months; COMBI-v 11.4 months). Of note, although encorafenib has an IC50 midway between dabrafenib and vemurafenib in cell-free assays (0.8 nM dabrafenib, 4 nM encorafenib, and 31 nM vemurafenib), it has an extremely slower off-rate from BRAF V600E, which results in significantly greater target inhibition in cells following drug wash-out.79 This may account for the significantly greater clinical benefit seen with encorafenib/binimetinib in clinical trials. Final study data are eagerly awaited. Regulatory approval has been sought, and is pending at this time.
Binimetinib has been compared to dacarbazine in a phase 3 study (NEMO) of patients with NRAS-mutant melanoma, most of whom had been previously treated with immunotherapy.80 Response rates were low in both arms, although slightly greater with binimetinib than dacarbazine (15% versus 9%), commensurate with a modest improvement in progression-free survival. FDA approval has been sought and remains pending at this time.
KIT INHIBITION SINGLY AND IN COMBINATION
The KIT receptor protein tyrosine kinase is a transmembrane protein consisting of extracellular and intracellular domains. Activating KIT mutations occur in 2% to 8% of all melanoma patients and may be found in all melanoma subtypes but are commonest in acral melanomas (10%–20%) and mucosal melanomas (15%–20%). Activating KIT mutations primarily occur in exons 11 and 13, which code for the juxtamembrane and kinase domains, respectively.5,81–83
Imatinib mesylate is a tyrosine kinase inhibitor of the 2-phenyl amino pyrimidine class that occupies the tyrosine kinase active site with resultant blocking of tyrosine kinase activity. Imatinib mesylate is known to block KIT and has been extensively studied in patients with gastrointestinal stromal tumors (GIST), 80% of whom harbor KIT mutations, in both the adjuvant and the metastatic settings. In melanoma, imatinib mesylate was studied in a Chinese open-label, phase 2 study of imatinib mesylate monotherapy in metastatic melanoma harboring KIT mutation or amplification; 25% of the study patients had mucosal disease and the rest had cutaneous disease, with acral involvement in 50% of all patients.84 Overall response rate was 23%, while 51% of patients remained alive at 1 year with no differences in response rate and/or survival being noted between patients with either KIT mutations or amplifications. In a separate study of imatinib mesylate at 400 mg daily or 400 mg twice daily in Caucasian patients with KIT-mutated/amplified melanoma, similar response and survival rates were reported, although patients with KIT mutations did nonsignificantly better than those with KIT amplifications.85
Other novel studies evaluating KIT inhibitors include KIT inhibition in combination with the VEGF inhibitor bevacizumab and a study of selective BCR-ABL kinase inhibitor nilotinib in imatinib-resistant melanoma. In the former phase 1/2 study, Flaherty and colleagues studied imatinib 800 mg daily and bevacizumab at 10 mg/kg every 2 weeks in 63 patients with advanced tumors, including 23 with metastatic melanoma. Although the combination was relatively nontoxic, no significant efficacy signal was seen and further accrual to the phase 2 portion was halted after the first stage was completed.86 Nilotinib is a BCR-ABL1 tyrosine kinase inhibitor intelligently designed based on the structure of the ABL-imatinib complex that is 10 to 30 times more potent than imatinib in inhibiting BCR-ABL1 tyrosine kinase activity. Nilotinib is approved for the treatment of imatinib-resistant chronic myelogenous leukemia (CML), with reported efficacy in patients with central nervous system (CNS) involvement.87,88 Nilotinib has been studied in a single study of KIT-mutated/amplified melanoma that included patients with imatinib-resistance and those with treated CNS disease. Nilotinib appeared to be active in imatinib-resistant melanoma, although no responses were seen in the CNS disease cohort.89 Overall, the response rates observed with KIT inhibition in melanoma are much lower than those observed in CML and GIST.
CONCLUSION AND FUTURE DIRECTIONS
Prior to 2011, the only approved agents for the treatment of advanced melanoma were dacarbazine and high-dose interleukin-2. Since 2011, drug approvals in melanoma have proceeded at a frenetic pace unmatched in any other disease. The primary events underlying this are advances in our understanding of the gene mutation landscape driving melanoma tumorigenesis, accompanied by insights into the means by which tumors circumvent the induction of effective anti-tumor T-cell responses. These insights have resulted in the development of inhibitors targeting MAPK pathway kinases BRAF, MEK, and NRAS), KIT, and regulatory immune checkpoints (CTLA-4 and PD-1). Although BRAF/MEK inhibition results in profound reductions and even occasional complete responses in patients, these responses are typically short lived, rarely lasting more than 9 to 11 months; the encorafenib/binimetinib combination may improve that duration marginally. However, the signature therapeutic advance in melanoma of the past decade is immunotherapy, particularly the development of inhibitors of CTLA-4 and PD-1 immune checkpoints. With these agents, significant proportions of treated patients remain free of progression off-therapy (ipilimumab 23%; nivolumab 34%; pembrolizumab 35%; ipilimumab/nivolumab 64%), and some patients can be successfully re-induced after delayed progression. Separately, the high response rates observed with the use of KIT inhibitors in CML and GIST have not been observed in KIT mutated/amplified melanoma and development of agents in this space has been limited. The challenges ahead center around identifying predictive biomarkers and circumventing primary or acquired resistance, with the eventual goal of producing durable remissions in the majority of treated patients.
Our improved understanding of the mechanisms of acquired resistance to BRAF/MEK inhibitors suggests that anti-tumor activity may be achieved by targeting multiple pathways, possibly with combination regimens comprising other inhibitors and/or immunotherapy. Preclinical data supports the use of combination strategies targeting both ERK and PI3K/mTOR to circumvent acquired resistance.90 Ongoing studies are evaluating combinations with biguanides (metformin: NCT02143050 and NCT01638676; phenformin: NCT03026517), HSP90 inhibitors (XL888: NCT02721459; AT13387: NCT02097225), and decitabine (NCT01876641).
One complexity affecting management of resistance in the targeted therapy landscape remains tumor heterogeneity, particularly intra- and intertumoral heterogeneity, which may explain the apparent contradiction between continued efficacy of BRAF inhibitors in BRAF-resistant tumors and preclinical data predicting slower progression of resistant tumors on cessation of BRAF inhibitors.91–94 These data provide a rationale to investigate intermittent dosing regimens with BRAF/MEK inhibitors; several studies exploring this approach are ongoing (NCT01894672 and NCT02583516).
Given the specificity, adaptability, and memory response associated with immunotherapy, it is likely that these agents will be used to treat the majority of patients regardless of mutational status. Hence, identifying predictive biomarkers of response to immune checkpoint inhibitors is vital. The presence of CD8+ T-cell infiltrate and IFN-γ gene signature, which indicate an “inflamed” tumor microenvironment, are highly predictive of clinical benefit from PD-1 inhibitors.95,96 However, not all PD-1 responders have “inflamed” tumor microenvironments, and not all patients with an “inflamed” tumor microenvironment respond to immune checkpoint inhibitors. The complexity of the immune system is reflected in the multiple non-redundant immunologic pathways, both positive and negative, with checkpoints and ligands that emerge dynamically in response to treatment. Given the dynamic nature of the immune response, it is unlikely that any single immunologic biomarker identified pre-treatment will be completely predictive. Rather, the complexity of the biomarker approach must match the complexity of the immune response elicited, and will likely incorporate multifarious elements including CD8+ T-cell infiltrate, IFN-γ gene signature, and additional elements including microbiome, genetic polymorphisms, and tumor mutation load. The goal is to use multiple markers to guide development of combinations and then, depending on initial response, to examine tumors for alterations to guide decisions about additional treatment(s) to improve responses, with the eventual goal being durable clinical responses for all patients.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
- Guy GP, Thomas CC, Thompson T, et al. Vital signs: melanoma incidence and mortality trends and projections - United States, 1982-2030. MMWR Morb Mortal Wkly Rep 2015;64:591–6.
- Anderson WF, Pfeiffer RM, Tucker MA, Rosenberg PS. Divergent cancer pathways for early-onset and late-onset cutaneous malignant melanoma. Cancer 2009;115:4176–85.
- Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005;353:2135–47.
- Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 2006;24:4340–6.
- Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell 2012;150:251–63.
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681–96.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol 2014;15:323–32.
- Hauschild A, Grob J-J, Demidov LV, et al. DaBRAFenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380:358–65.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877–88.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined daBRAFenib and trametinib. N Engl J Med 2015;372:30–9.
- Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371:1867–76.
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 2010;107:4275–80.
- Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014;515:577–81.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–23.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517–26.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54.
- Weber JS, Angelo SP D’, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015;16:375–84.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–30.
- Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013;369:134–44.
- Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521–32.
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–33.
- Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015;372:2006–17.
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34.
- Eggermont AMM. Therapeutic vaccines in solid tumours: can they be harmful? Eur J Cancer 2009;45:2087–90.
- Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Transl Med 2012;4:127ps8.
- Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol 2014;192:5451–8.
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015;348:62–8.
- Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 1992;356:607–9.
- Greenfield EA, Nguyen KA, Kuchroo VK. CD28/B7 costimulation: a review. Crit Rev Immunol 1998;18:389–418.
- Sharpe AH, Abbas AK. T-cell costimulation--biology, therapeutic potential, and challenges. N Engl J Med 2006;355:973–5.
- Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol 2001;19:565–94.
- Collins AV, Brodie DW, Gilbert RJC, et al. The interaction properties of costimulatory molecules revisited. Immunity 2002;17:201–10.
- Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995;182:459–65.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271:1734–6.
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704.
- Yamazaki T, Akiba H, Iwai H, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 2002;169:5538–45.
- Youngnak P, Kozono Y, Kozono H, et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem Biophys Res Commun 2003;307:672–7.
- Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 2004;173:945–54.
- Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 2005;25:9543–53.
- Riley JL. PD-1 signaling in primary T cells. Immunol Rev 2009;229:114–25.
- Wolchok JD, Hodi FS, Weber JS, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci 2013;1291:1–13.
- Weber JS, O’Day S, Urba W, et al. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol 2008;26:5950–6.
- Hodi FS, Butler M, Oble DA, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A 2008;105:3005–10.
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol 2010;11:155–64.
- Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015;33:1889–94.
- Wang C, Thudium KB, Han M, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res 2014;2:846–56.
- Poole RM. Pembrolizumab: first global approval. Drugs 2014;74:1973–81.
- Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014;32:1020–30.
- Patnaik A, Kang SP, Rasco D, et al. Phase I study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res 2015;21:4286–93.
- Hellmann MD, Rizvi NA, Goldman JW, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol 2017;18:31–41.
- Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am 2009;23:529–45, ix.
- Satyamoorthy K, Li G, Gerrero MR, et al. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res 2003;63:756–9.
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54.
- Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012;44:1006–14.
- Rubinstein JC, Sznol M, Pavlick AC, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med 2010;8:67.
- Lovly CM, Dahlman KB, Fohn LE, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS ONE 2012;7:e35309.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010;464:431–5.
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010;464:427–30.
- Thomas NE, Edmiston SN, Alexander A, et al. Association between NRAS and BRAF mutational status and melanoma-specific survival among patients with higher-risk primary melanoma. JAMA Oncology 2015;1:359–68.
- Joseph RW, Sullivan RJ, Harrell R, et al. Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother 2012;35:66–72.
- Johnson DB, Lovly CM, Flavin M, et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res 2015;3:288–95.
- Johnson DB, Frampton GM, Rioth MJ, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res 2016;4:959–67.
- Kiuru M, Busam KJ. The NF1 gene in tumor syndromes and melanoma. Lab Invest 2017;97:146–57.
- Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res 2014;74:2340–50.
- Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol 2005;23:5281–93.
- Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 2004;22:4456–62.
- Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res 2012;18:555–67.
- Davar D, Kirkwood JM. CCR 20th anniversary commentary: MAPK/ERK pathway inhibition in melanoma-kinase inhibition redux. Clin Cancer Res 2015;21:5412–4.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 2012;366:207–15.
- Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010;468:968–72.
- Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468:973–7.
- Shi H, Hong A, Kong X, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov 2014;4:69–79.
- Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014;4:80–93.
- Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 2014;4:94–109.
- Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107–14.
- Dummer R, Ascierto PA, Gogas HJ, et al. Results of COLUMBUS Part 1: a phase 3 trial of encorafenib (ENCO) plus binimetinib (BINI) versus vemurafenib (VEM) or ENCO in BRAF-mutant melanoma. Presented at Society for Melanoma Research 2016 Congress. November 6-9, 2016. Boston (MA).
- Adelmann CH, Ching G, Du L, et al. Comparative profiles of BRAF inhibitors: the paradox index as a predictor of clinical toxicity. Oncotarget 2016;7:30453–60.
- Dummer R, Schadendorf D, Ascierto PA, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2017;18:435–45.
- Willmore-Payne C, Holden JA, Tripp S, Layfield LJ. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol 2005;36:486–93.
- Beadling C, Jacobson-Dunlop E, Hodi FS, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res 2008;14:6821–8.
- Handolias D, Salemi R, Murray W, et al. Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment Cell Melanoma Res 2010;23:210–5.
- Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 2011;29:2904–9.
- Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol 2013;31:3182–90.
- Flaherty KT, Hamilton BK, Rosen MA, et al. Phase I/II trial of imatinib and bevacizumab in patients with advanced melanoma and other advanced cancers. Oncologist 2015;20:952–9.
- Giles FJ, le Coutre PD, Pinilla-Ibarz J, et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia 2013;27:107–12.
- Reinwald M, Schleyer E, Kiewe P, et al. Efficacy and pharmacologic data of second-generation tyrosine kinase inhibitor nilotinib in BCR-ABL-positive leukemia patients with central nervous system relapse after allogeneic stem cell transplantation. Biomed Res Int 2014;2014:637059.
- Carvajal RD, Lawrence DP, Weber JS, et al. Phase II study of nilotinib in melanoma harboring KIT alterations following progression to prior KIT inhibition. Clin Cancer Res 2015;21:2289–96.
- Carlino MS, Todd JR, Gowrishankar K, et al. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol Oncol 2014;8:544–54.
- Carlino MS, Gowrishankar K, Saunders CAB, et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther 2013;12:1332–42.
- Chan MMK, Haydu LE, Menzies AM, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer 2014;120:3142–53.
- Thakur M Das, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013;494:251–5.
- Thakur M Das, Stuart DD. Molecular pathways: response and resistance to BRAF and MEK inhibitors in BRAF(V600E) tumors. Clin Cancer Res 2014;20:1074–80.
- Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71.
- Ayers M, Lunceford J, Nebozhyn M, et al. Relationship between immune gene signatures and clinical response to PD-1 blockade with pembrolizumab (MK-3475) in patients with advanced solid tumors. J Immunotherapy Cancer 2015;3(Suppl 2):P80.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
- Guy GP, Thomas CC, Thompson T, et al. Vital signs: melanoma incidence and mortality trends and projections - United States, 1982-2030. MMWR Morb Mortal Wkly Rep 2015;64:591–6.
- Anderson WF, Pfeiffer RM, Tucker MA, Rosenberg PS. Divergent cancer pathways for early-onset and late-onset cutaneous malignant melanoma. Cancer 2009;115:4176–85.
- Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005;353:2135–47.
- Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 2006;24:4340–6.
- Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell 2012;150:251–63.
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681–96.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol 2014;15:323–32.
- Hauschild A, Grob J-J, Demidov LV, et al. DaBRAFenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380:358–65.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877–88.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined daBRAFenib and trametinib. N Engl J Med 2015;372:30–9.
- Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371:1867–76.
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 2010;107:4275–80.
- Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014;515:577–81.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–23.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517–26.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54.
- Weber JS, Angelo SP D’, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015;16:375–84.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–30.
- Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013;369:134–44.
- Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521–32.
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–33.
- Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015;372:2006–17.
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34.
- Eggermont AMM. Therapeutic vaccines in solid tumours: can they be harmful? Eur J Cancer 2009;45:2087–90.
- Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Transl Med 2012;4:127ps8.
- Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol 2014;192:5451–8.
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015;348:62–8.
- Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 1992;356:607–9.
- Greenfield EA, Nguyen KA, Kuchroo VK. CD28/B7 costimulation: a review. Crit Rev Immunol 1998;18:389–418.
- Sharpe AH, Abbas AK. T-cell costimulation--biology, therapeutic potential, and challenges. N Engl J Med 2006;355:973–5.
- Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol 2001;19:565–94.
- Collins AV, Brodie DW, Gilbert RJC, et al. The interaction properties of costimulatory molecules revisited. Immunity 2002;17:201–10.
- Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995;182:459–65.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271:1734–6.
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704.
- Yamazaki T, Akiba H, Iwai H, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 2002;169:5538–45.
- Youngnak P, Kozono Y, Kozono H, et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem Biophys Res Commun 2003;307:672–7.
- Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 2004;173:945–54.
- Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 2005;25:9543–53.
- Riley JL. PD-1 signaling in primary T cells. Immunol Rev 2009;229:114–25.
- Wolchok JD, Hodi FS, Weber JS, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci 2013;1291:1–13.
- Weber JS, O’Day S, Urba W, et al. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol 2008;26:5950–6.
- Hodi FS, Butler M, Oble DA, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A 2008;105:3005–10.
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol 2010;11:155–64.
- Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015;33:1889–94.
- Wang C, Thudium KB, Han M, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res 2014;2:846–56.
- Poole RM. Pembrolizumab: first global approval. Drugs 2014;74:1973–81.
- Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014;32:1020–30.
- Patnaik A, Kang SP, Rasco D, et al. Phase I study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res 2015;21:4286–93.
- Hellmann MD, Rizvi NA, Goldman JW, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol 2017;18:31–41.
- Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am 2009;23:529–45, ix.
- Satyamoorthy K, Li G, Gerrero MR, et al. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res 2003;63:756–9.
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54.
- Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012;44:1006–14.
- Rubinstein JC, Sznol M, Pavlick AC, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med 2010;8:67.
- Lovly CM, Dahlman KB, Fohn LE, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS ONE 2012;7:e35309.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010;464:431–5.
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010;464:427–30.
- Thomas NE, Edmiston SN, Alexander A, et al. Association between NRAS and BRAF mutational status and melanoma-specific survival among patients with higher-risk primary melanoma. JAMA Oncology 2015;1:359–68.
- Joseph RW, Sullivan RJ, Harrell R, et al. Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother 2012;35:66–72.
- Johnson DB, Lovly CM, Flavin M, et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res 2015;3:288–95.
- Johnson DB, Frampton GM, Rioth MJ, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res 2016;4:959–67.
- Kiuru M, Busam KJ. The NF1 gene in tumor syndromes and melanoma. Lab Invest 2017;97:146–57.
- Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res 2014;74:2340–50.
- Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol 2005;23:5281–93.
- Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 2004;22:4456–62.
- Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res 2012;18:555–67.
- Davar D, Kirkwood JM. CCR 20th anniversary commentary: MAPK/ERK pathway inhibition in melanoma-kinase inhibition redux. Clin Cancer Res 2015;21:5412–4.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 2012;366:207–15.
- Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010;468:968–72.
- Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468:973–7.
- Shi H, Hong A, Kong X, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov 2014;4:69–79.
- Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014;4:80–93.
- Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 2014;4:94–109.
- Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107–14.
- Dummer R, Ascierto PA, Gogas HJ, et al. Results of COLUMBUS Part 1: a phase 3 trial of encorafenib (ENCO) plus binimetinib (BINI) versus vemurafenib (VEM) or ENCO in BRAF-mutant melanoma. Presented at Society for Melanoma Research 2016 Congress. November 6-9, 2016. Boston (MA).
- Adelmann CH, Ching G, Du L, et al. Comparative profiles of BRAF inhibitors: the paradox index as a predictor of clinical toxicity. Oncotarget 2016;7:30453–60.
- Dummer R, Schadendorf D, Ascierto PA, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2017;18:435–45.
- Willmore-Payne C, Holden JA, Tripp S, Layfield LJ. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol 2005;36:486–93.
- Beadling C, Jacobson-Dunlop E, Hodi FS, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res 2008;14:6821–8.
- Handolias D, Salemi R, Murray W, et al. Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment Cell Melanoma Res 2010;23:210–5.
- Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 2011;29:2904–9.
- Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol 2013;31:3182–90.
- Flaherty KT, Hamilton BK, Rosen MA, et al. Phase I/II trial of imatinib and bevacizumab in patients with advanced melanoma and other advanced cancers. Oncologist 2015;20:952–9.
- Giles FJ, le Coutre PD, Pinilla-Ibarz J, et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia 2013;27:107–12.
- Reinwald M, Schleyer E, Kiewe P, et al. Efficacy and pharmacologic data of second-generation tyrosine kinase inhibitor nilotinib in BCR-ABL-positive leukemia patients with central nervous system relapse after allogeneic stem cell transplantation. Biomed Res Int 2014;2014:637059.
- Carvajal RD, Lawrence DP, Weber JS, et al. Phase II study of nilotinib in melanoma harboring KIT alterations following progression to prior KIT inhibition. Clin Cancer Res 2015;21:2289–96.
- Carlino MS, Todd JR, Gowrishankar K, et al. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol Oncol 2014;8:544–54.
- Carlino MS, Gowrishankar K, Saunders CAB, et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther 2013;12:1332–42.
- Chan MMK, Haydu LE, Menzies AM, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer 2014;120:3142–53.
- Thakur M Das, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013;494:251–5.
- Thakur M Das, Stuart DD. Molecular pathways: response and resistance to BRAF and MEK inhibitors in BRAF(V600E) tumors. Clin Cancer Res 2014;20:1074–80.
- Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71.
- Ayers M, Lunceford J, Nebozhyn M, et al. Relationship between immune gene signatures and clinical response to PD-1 blockade with pembrolizumab (MK-3475) in patients with advanced solid tumors. J Immunotherapy Cancer 2015;3(Suppl 2):P80.
Adjuvant Chemotherapy in the Treatment of Colon Cancer
INTRODUCTION
Colorectal cancer (CRC) is one of the most prevalent malignancies and is the fourth most common cancer in the United States, with an estimated 133,490 new cases diagnosed in 2016. Of these, approximately 95,520 are located in the colon and 39,970 are in the rectum.1 CRC is the third leading cause of cancer death in women and the second leading cause of cancer death in men, with an estimated 49,190 total deaths in 2016.2 The incidence appears to be increasing,3 especially in patients younger than 55 years of age;4 the reason for this increase remains uncertain.
A number of risk factors for the development of CRC have been identified. Numerous hered-itary CRC syndromes have been described, including familial adenomatous polyposis,5 hereditary non-polyposis colorectal cancer (HNPCC) or Lynch syndrome,6 and MUTYH-associated polyposis.7,8 A family history of CRC doubles the risk of developing CRC,9 and current guidelines support lowering the age of screening in individuals with a family history of CRC to 10 years younger than the age of diagnosis of the family member or 40 years of age, whichever is lower.10 Patients with a personal history of adenomatous polyps are at increased risk for developing CRC, as are patients with a personal history of CRC, with a relative risk ranging from 3 to 6.11 Ulcerative colitis and Crohn’s disease are associated with the development of CRC and also influence screening, though evidence suggests good control of these diseases may mitigate risk.12 Finally, modifiable risk factors for the development of CRC include high red meat consumption,13 diets low in fiber,14 obesity,13 smoking, alcohol use,15 and physical inactivity16; lifestyle modification targeting these factors has been shown to decrease rates of CRC.17 The majority of colon cancers present with clinical symptoms, often with rectal bleeding, abdominal pain, change in bowel habits, or obstructive symptoms. More rarely, these tumors are detected during screening colonoscopy, in which case they tend to be at an early stage.
SURGICAL MANAGEMENT
A critical goal in the resection of early-stage colon cancer is attaining R0 resection. Patients who achieve R0 resection as compared to R1 (microscopic residual tumor) and R2 (macroscopic residual tumor)18 have significantly improved long-term overall survival.19 Traditionally, open resection of the involved colonic segment was employed, with end-end anastomosis of the uninvolved free margins. Laparoscopic resection for early-stage disease has been utilized in attempts to decrease morbidity of open procedures, with similar outcomes and node sampling.20 Laparoscopic resection appears to provide similar outcomes even in locally advanced disease.21 Right-sided lesions are treated with right colectomy and primary ileocolic anastomosis.22 For patients presenting with obstructing masses, the Hartmann procedure is the most commonly performed operation. This involves creation of an ostomy with subtotal colectomy and subsequent ostomy reversal in a 2- or 3-stage protocol.23 Patients with locally advanced disease and invasion into surrounding structures require multivisceral resection, which involves resection en bloc with secondarily involved organs.24 Intestinal perforation presents a unique challenge and is associated with surgical complications, infection, and lower overall survival (OS) and 5-year disease-free survival (DFS). Complete mesocolic excision is a newer technique that has been performed with reports of better oncologic outcome at some centers; however, this approach is not currently considered standard of care.25
STAGING
According to a report by the National Cancer Institute, the estimated 5-year relative survival rates for localized colon cancer (lymph node negative), regional (lymph node positive) disease, and distant (metastatic) disease are 89.9%, 71.3%, and 13.9%, respectively.1 However, efforts have been made to further classify patients into distinct categories to allow fine-tuning of prognostication. In the current system, staging of colon cancer utilizes the American Joint Committee on Cancer tumor/node/metastasis (TNM) system.20 Clinical and pathologic features include depth of invasion, local invasion of other organs, nodal involvement, and presence of distant metastasis (Table 1). Studies completed prior to the adoption of the TNM system used the Dukes criteria, which divided colon cancer into A, B, and C, corresponding to TNM stage I, stage IIA–IIC, and stage IIIA-IIIC. This classification is rarely used in more contemporary studies.
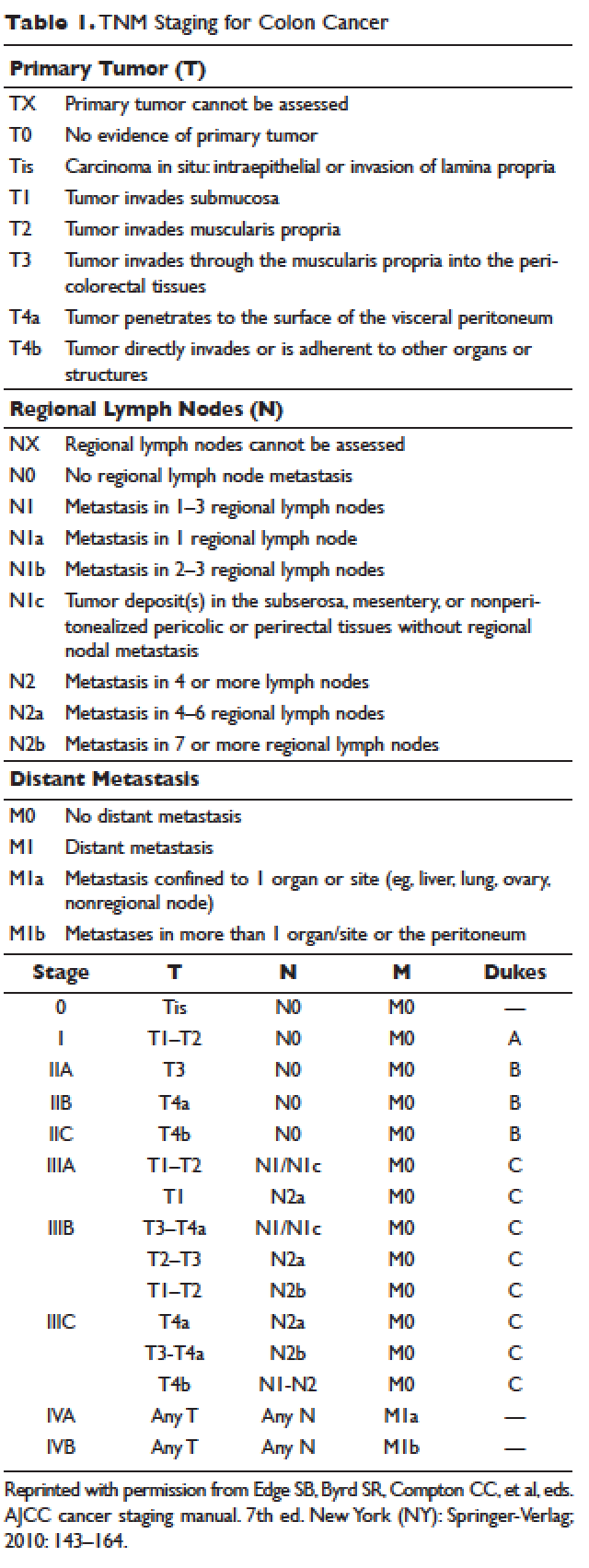
APPROACH TO ADJUVANT CHEMOTHERAPY
Adjuvant chemotherapy seeks to eliminate micrometastatic disease present following curative surgical resection. When stage 0 cancer is discovered incidentally during colonoscopy, endoscopic resection alone is the management of choice, as presence of micrometastatic disease is exceedingly unlikely.26 Stage I–III CRCs are treated with surgical resection withcurative intent. The 5-year survival rate for stage I and early-stage II CRC is estimated at 97% with surgery alone.27,28 The survival rate drops to about 60% for high-risk stage II tumors (T4aN0), and down to 50% or less for stage II-T4N0 or stage III cancers. Adjuvant chemotherapy is generally recommended to further decrease the rates of distant recurrence in certain cases of stage II and in all stage III tumors.
DETERMINATION OF BENEFIT FROM CHEMOTHERAPY: PROGNOSTIC MARKERS
Prior to administration of adjuvant chemotherapy, a clinical evaluation by the medical oncologist to determine appropriateness and safety of treatment is paramount. Poor performance status and comorbid conditions may indicate risk for excessive toxicity and minimal benefit from chemotherapy. CRC commonly presents in older individuals, with the median age at diagnosis of 69 years for men and 73 years for women.29 In this patient population, comorbidities such as cardiovascular disease, diabetes, and renal dysfunction are more prevalent.30 Decisions regarding adjuvant chemotherapy in this patient population have to take into consideration the fact that older patients may experience higher rates of toxicity with chemotherapy, including gastrointestinal toxicities and marrow suppression.31 Though some reports indicate patients older than 70 years derive similar benefit from adjuvant chemotherapy,32,33 a large pooled analysis of the ACCENT database, which included 7 adjuvant therapy trials and 14,528 patients, suggested limited benefit from the addition of oxaliplatin to fluorouracil in elderly patients.32 Other factors that weigh on the decision include stage, pathology, and presence of high-risk features. A common concern in the postoperative setting is delaying initiation of chemotherapy to allow adequate wound healing; however, evidence suggests that delays longer than 8 weeks leads to worse overall survival, with hazard ratios (HR) ranging from 1.4 to 1.7.34,35 Thus, the start of adjuvant therapy should ideally be within this time frame.
HIGH-RISK FEATURES
Multiple factors have been found to predict worse outcome and are classified as high-risk features (Table 2). Histologically, high-grade or poorly differentiated tumors are associated with higher recurrence rate and worse outcome.36 Certain histological subtypes, including mucinous and signet-ring, both appear to have more aggressive biology.37 Presence of microscopic invasion into surrounding blood vessels (vascular invasion) and nerves (perineural invasion) is associated with lower survival.38 Penetration of the cancer through the visceral peritoneum (T4a) or into surrounding structures (T4b) is associated with lower survival.36 During surgical resection, multiple lymph nodes are removed along with the primary tumor to evaluate for metastasis to the regional nodes. Multiple analyses have demonstrated that removal and pathologic assessment of fewer than 12 lymph nodes is associated with high risk of missing a positive node, and is thus equated with high risk.39–41 In addition, extension of tumor beyond the capsules of any single lymph node, termed extracapsular extension, is associated with an increased risk of all-cause mortality.42 Tumor deposits, or focal aggregates of adenocarcinoma in the pericolic fat that are not contiguous with the primary tumor and are not associated with lymph nodes, are currently classified as lymph nodes as N1c in the current TNM staging system. Presence of these deposits has been found to predict poor outcome stage for stage.43 Obstruction and/or perforation secondary to the tumor are also considered high-risk features that predict poor outcome.

SIDEDNESS
As reported at the 2016 American Society of Clinical Oncology annual meeting, tumor location predicts outcome in the metastatic setting. A report by Venook and colleagues based on a post-hoc analysis found that in the metastatic setting, location of the tumor primary in the left side is associated with longer OS (33.3 months) when compared to the right side of the colon (19.4 months).44 A retrospective analysis of multiple databases presented by Schrag and colleagues similarly reported inferior outcomes in patients with stage III and IV disease who had right-sided primary tumors.45 However, the prognostic implications for stage II disease remain uncertain.
BIOMARKERS
Given the controversy regarding adjuvant therapy of patients with stage II colon cancer, multiple biomarkers have been evaluated as possible predictive markers that can assist in this decision. The mismatch repair (MMR) system is a complex cellular enzymatic mechanism that identifies and corrects DNA errors during cell division and prevents mutagenesis.46 The familial cancer syndrome HNPCC is linked to alteration in a variety of MMR genes, leading to deficient mismatch repair (dMMR), also termed microsatellite instability-high (MSI-high).47,48 Epigenetic modification can also lead to silencing of the same implicated genes and accounts for 15% to 20% of sporadic colorectal cancer.49 These epigenetic modifications lead to hypermethylation of the promotor region of MLH1 in 70% of cases.50 The 4 MMR genes most commonly tested are MLH-1, MSH2, MSH6, and PMS2. Testing can be performed by immunohistochemistry or polymerase chain reaction.51 Across tumor histology and stage, MSI status is prognostic. Patients with MSI-high tumors have been shown to have improved prognosis and longer OS both in stage II and III disease52–54 and in the metastatic setting.55 However, despite this survival benefit, there is conflicting data as to whether patients with stage II, MSI-high colon cancer may benefit less from adjuvant chemotherapy. One early retrospective study compared outcomes of 70 patients with stage II and III disease and dMMR to those of 387 patients with stage II and III disease and proficient mismatch repair (pMMR). Adjuvant fluorouracil with leucovorin improved DFS for patients with pMMR (HR 0.67) but not for those with dMMR (HR 1.10). In addition, for patients with stage II disease and dMMR, the HR for OS was inferior at 2.95.56 Data collected from randomized clinical trials using fluorouracil-based adjuvant chemotherapy were analyzed in an attempt to predict benefit based on MSI status. Benefit was only seen in pMMR patients, with a HR of 0.72; this was not seen in the dMMR patients.57 Subsequent studies have had different findings and did not demonstrate a detrimental effect of fluorouracil in dMMR.58,59 For stage III patients, MSI status does not appear to affect benefit from chemotherapy, as analysis of data from the NSABP C-07 trial (Table 3) demonstrated benefit of FOLFOX (leucovorin, fluorouracil, oxaliplatin) in patients with dMMR status and stage III disease.59
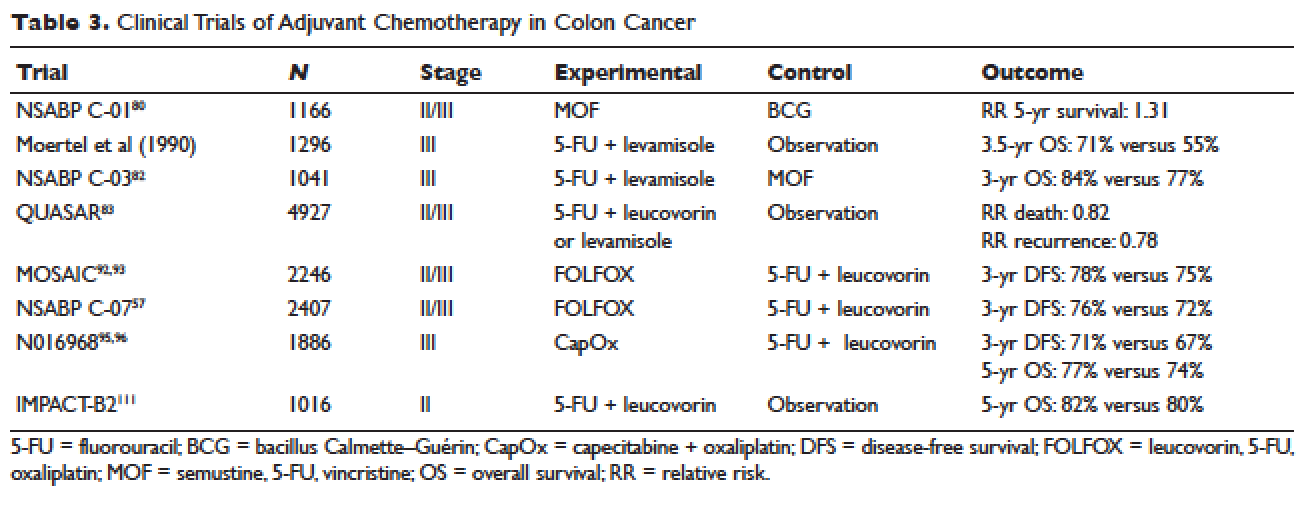
Another genetic abnormality identified in colon cancers is chromosome 18q loss of heterozygosity (LOH). The presence of 18q LOH appears to be inversely associated with MSI-high status. Some reports have linked presence of 18q with worse outcome,60 but others question this, arguing the finding may simply be related to MSI status.61,62 This biomarker has not been established as a clear prognostic marker that can aid clinical decisions.
Most recently, expression of caudal-type homeobox transcription factor 2 (CDX2) has been reported as a novel prognostic and predictive tool. A 2015 report linked lack of expression of CDX2 to worse outcome; in this study, 5-year DFS was 41% in patients with CDX2-negative tumors versus 74% in the CDX2-positive tumors, with a HR of disease recurrence of 2.73 for CDX2-negative tumors.63 Similar numbers were observed in patients with stage II disease, with 5-year OS of 40% in patients with CDX2-negative tumors versus 70% in those with CDX2-positive tumors. Treatment of CDX2-negative patients with adjuvant chemotherapy improved outcomes: 5-year DFS in the stage II subgroup was 91% with chemotherapy versus 56% without, and in the stage III subgroup, 74% with chemotherapy versus 37% without. The authors concluded that patients with stage II and III colon cancer that is CDX2-negative may benefit from adjuvant chemotherapy. Importantly, CDX2-negativity is a rare event, occurring in only 6.9% of evaluable tumors.
RISK ASSESSMENT TOOLS
Several risk assessment tools have been developed in an attempt to aid clinical decision making regarding adjuvant chemotherapy for patients with stage II colon cancer. The Oncotype DX Colon Assay analyses a 12-gene signature in the pathologic sample and was developed with the goal to improve prognostication and aid in treatment decision making. The test utilizes reverse transcription-PCR on RNA extracted from the tumor.64 After evaluating 12 genes, a recurrence score is generated that predicts the risk of disease recurrence. This score was validated using data from 3 large clinical trials.65–67 Unlike the Oncotype Dx score used in breast cancer, the test in colon cancer has not been found to predict the benefit from chemotherapy and has not been incorporated widely into clinical practice.
Adjuvant! Online (available at www.adjuvantonline.com) is a web-based tool that combines clinical and histological features to estimate outcome. Calculations are based on US SEER tumor registry-reported outcomes.68 A second web-based tool, Numeracy (available at www.mayoclinic.com/calcs), was developed by the Mayo Clinic using pooled data from 7 randomized clinical trials including 3341 patients.68 Both tools seek to predict absolute benefit for patients treated with fluorouracil, though data suggests Adjuvant! Online may be more reliable in its predictive ability.69 Adjuvant! Online has also been validated in an Asian population70 and patients older than 70 years.71
MUTATIONAL ANALYSIS
Multiple mutations in proto-oncogenes have been found in colon cancer cells. One such proto-oncogene is BRAF, which encodes a serine-threonine kinase in the rapidly accelerated fibrosarcoma (RAF). Mutations in BRAF have been found in 5% to 10% of colon cancers and are associated with right-sided tumors.72 As a prognostic marker, some studies have associated BRAF mutations with worse prognosis, including shorter time to relapse and shorter OS.73,74 Two other proto-oncogenes are Kristen rat sarcoma viral oncogene homolog (KRAS) and neuroblastoma rat sarcoma viral oncogene homolog (NRAS), both of which encode proteins downstream of epidermal growth factor receptor (EGFR). KRAS and NRAS mutations have been shown to be predictive in the metastatic setting where they predict resistance to the EGFR inhibitors cetuximab and panitumumab.75,76 The effect of KRAS and NRAS mutations on outcome in stage II and III colon cancer is uncertain. Some studies suggest worse outcome in KRAS-mutated cancers,77 while others failed to demonstrate this finding.73
CASE PRESENTATION 1
A 53-year-old man with no past medical history presents to the emergency department with early satiety and generalized abdominal pain. Laboratory evaluation shows a microcytic anemia with normal white blood cell count, platelet count, renal function, and liver function tests. Computed tomography (CT) scan of the abdomen and pelvis show a 4-cm mass in the transverse colon without obstruction and without abnormality in the liver. CT scan of the chest does not demonstrate pathologic lymphadenopathy or other findings. He undergoes robotic laparoscopic transverse colon resection and appendectomy. Pathology confirms a 3.5-cm focus of adenocarcinoma of the colon with invasion through the muscularis propria and 5 of 27 regional lymph nodes positive for adenocarcinoma and uninvolved proximal, distal, and radial margins. He is given a stage of IIIB pT3 pN2a M0 and referred to medical oncology for further management, where 6 months of adjuvant FOLFOX chemotherapy is recommended.
ADJUVANT CHEMOTHERAPY IN STAGE III COLON CANCER
Postoperative adjuvant chemotherapy is the standard of care for patients with stage III disease. In the 1960s, infusional fluorouracil was first used to treat inoperable colon cancer.78,79 After encouraging results, the agent was used both intraluminally and intravenously as an adjuvant therapy for patients undergoing resection with curative intent; however, only modest benefits were described.80,81 The National Surgical Adjuvant Breast and Bowel Project (NSABP) C-01 trial (Table 3) was the first study to demonstrate a benefit from adjuvant chemotherapy in colon cancer. This study randomly assigned patients with stage II and III colon cancer to surgery alone, postoperative chemotherapy with fluorouracil, semustine, and vincristine (MOF), or postoperative bacillus Calmette-Guérin (BCG). DFS and OS were significantly improved with MOF chemotherapy.82 In 1990, a landmark study reported on outcomes after treatment of 1296 patients with stage III colon cancer with adjuvant fluorouracil and levamisole for 12 months. The combination was associated with a 41% reduction in risk of cancer recurrence and a 33% reduction in risk of death.83 The NSABP C-03 trial (Table 3) compared MOF to the combination of fluorouracil and leucovorin and demonstrated improved 3-year DFS (69% versus 73%) and 3-year OS (77% versus 84%) in patients with stage III disease.84 Building on these outcomes, the QUASAR study (Table 3) compared fluorouracil in combination with one of levamisole, low-dose leucovorin, or high-dose leucovorin. The study enrolled 4927 patients and found worse outcomes with fluorouracil plus levamisole and no difference in low-doseversus high-dose leucovorin.85 Levamisole fell out of use after associations with development of multifocal leukoencephalopathy,86 and was later shown to have inferior outcomes versus leucovorin when combined with fluorouracil.87,88 Intravenous fluorouracil has shown similar benefit when administered by bolus or infusion,89 although continuous infusion has been associated with lower incidence of severe toxicity.90 The efficacy of the oral fluoropyrimidine capecitabine has been shown to be equivalent to that of fluorouracil.91
Fluorouracil-based treatment remained the standard of care until the introduction of oxaliplatin in the mid-1990s. After encouraging results in the metastatic setting,92,93 the agent was moved to the adjuvant setting. The MOSAIC trial (Table 3) randomly assigned patients with stage II and III colon cancer to fluorouracil with leucovorin (FULV) versus FOLFOX given once every 2 weeks for 12 cycles. Analysis with respect to stage III patients showed a clear survival benefit, with a 10-year OS of 67.1% with FOLFOX chemotherapy versus 59% with fluorouracil and leucovorin.94,95 The NSABP C-07 (Table 3) trial used a similar trial design but employed bolus fluorouracil. More than 2400 patients with stage II and III colon cancer were randomly assigned to bolus FULV or bolus fluorouracil, leucovorin, and oxaliplatin (FLOX). The addition of oxaliplatin significantly improved outcomes, with 4-year DFS of 67% versus 71.8% for FULV and FLOX, respectively, and a HR of death of 0.80 with FLOX.59,96 The multicenter N016968 trial (Table 3) randomly assigned 1886 patients with stage III colon cancer to adjuvant capecitabine plus oxaliplatin (XELOX) or bolus fluorouracil plus leucovorin (FU/FA). The 3-year DFS was 70.9% versus 66.5% with XELOX and FU/FA, respectively, and 5-year OS was 77.6% versus 74.2%, respectively.97,98
In the metastatic setting, additional agents have shown efficacy, including irinotecan,99,100 bevacizumab,101,102 cetuximab,103,104 and regorafenib.105 This observation led to testing of these agents in earlier stage disease. The CALGB 89803 trial compared fluorouracil, leucovorin, and irinotecan to fluorouracil with leucovorin alone. No benefit in 5-year DFS or OS was seen.106 Similarly, infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) was not found to improve 5-year DFS as compared to fluorouracil with leucovorin alone in the PETACC-3 trial.107 The NSABP C-08 trial considered the addition of bevacizumab to FOLFOX. When compared to FOLFOX alone, the combination of bevacizumab to FOLFOX had similar 3-year DFS (77.9% versus 75.1%) and 5-year OS (82.5% versus 80.7%).108 This finding was confirmed in the Avant trial.109 The addition of cetuximab to FOLFOX was equally disappointing, as shown in the N0147 trial110 and PETACC-8 trial.111 Data on regorafenib in the adjuvant setting for stage III colon cancer is lacking; however, 2 ongoing clinical trials, NCT02425683 and NCT02664077, are each studying the use of regorafenib following completion of FOLFOX for patients with stage III disease.
Thus, after multiple trials comparing various regimens and despite attempts to improve outcomes by the addition of a third agent, the standard of care per National Comprehensive Cancer Network (NCCN) guidelines for management of stage III colon cancer remains 12 cycles of FOLFOX chemotherapy. Therapy should be initiated within 8 weeks of surgery. Data are emerging to support a short duration of therapy for patients with low-risk stage III tumors, as shown in an abstract presented at the 2017 American Society of Clinical Oncology annual meeting. The IDEA trial was a pooled analysis of 6 randomized clinical trials across multiple countries, all of which evaluated 3 versus 6 months of FOLFOX or capecitabine and oxaliplatin in the treatment of stage III colon cancer. The analysis was designed to test non-inferiority of 3 months of therapy as compared to 6 months. The analysis included 6088 patients across 244 centers in 6 countries. The overall analysis failed to establish noninferiority. The 3-year DFS rate was 74.6% for 3 months and 75.5% for 6 months, with a DFS HR of 1.07 and a confidence interval that did not meet the prespecified endpoint. Subgroup analysis suggested noninferiority for lower stage disease (T1–3 or N1) but not for higher stage disease (T4 or N2). Given the high rates of neuropathy with 6 months of oxaliplatin, these results suggest that 3 months of adjuvant therapy can be considered for patients with T1–3 or N1 disease in an attempt to limit toxicity.112
CASE PRESENTATION 2
A 57-year-old woman presents to the emergency department with fever and abdominal pain. CT of the abdomen and pelvis demonstrates a left-sided colonic mass with surrounding fat stranding and pelvic abscess. She is taken emergently for left hemicolectomy, cholecystectomy, and evacuation of pelvic abscess. Pathology reveals a 5-cm adenocarcinoma with invasion through the visceral peritoneum; 0/22 lymph nodes are involved. She is given a diagnosis of stage IIC and referred to medical oncology for further management. Due to her young age and presence of high-risk features, she is recommended adjuvant therapy with FOLFOX for 6 months.
ADJUVANT CHEMOTHERAPY IN STAGE II COLON CANCER
Because of excellent outcomes with surgical resection alone for stage II cancers, the use of adjuvant chemotherapy for patients with stage II disease is controversial. Limited prospective data is available to guide adjuvant treatment decisions for stage II patients. The QUASAR trial, which compared observation to adjuvant fluorouracil and leucovorin in patients with early-stage colon cancer, included 2963 patients with stage II disease and found a relative risk (RR) of death or recurrence of 0.82 and 0.78, respectively. Importantly, the absolute benefit of therapy was less than 5%.113 The IMPACT-B2 trial (Table 3) combined data from 5 separate trials and analyzed 1016 patients with stage II colon cancer who received fluorouracil with leucovorin or observation. Event-free survival was 0.86 versus 0.83 and 5-year OS was 82% versus 80%, suggesting no benefit.114 The benefit of addition of oxaliplatin to fluorouracil in stage II disease appears to be less than the benefit of adding this agent in the treatment of stage III CRC. As noted above, the MOSAIC trial randomly assigned patients with stage II and III colon cancer to receive adjuvant fluorouracil and leucovorin with or without oxaliplatin for 12 cycles. After a median follow-up of 9.5 years, 10-year OS rates for patients with stage II disease were 78.4% versus 79.5%. For patients with high-risk stage II disease (defined as T4, bowel perforation, or fewer than 10 lymph nodes examined), 10-year OS was 71.7% and 75.4% respectively, but these differences were not statistically significant.94
Because of conflicting data as to the benefit of adding oxaliplatin in stage II disease, oxaliplatin is not recommended for standard-risk stage II patients. The use of oxaliplatin in high-risk stage II tumors should be weighed carefully given the toxicity risk. Oxaliplatin is recognized to cause sensory neuropathy in many patients, which can become painful and debilitating.115 Two types of neuropathy are associated with oxaliplatin: acute and chronic. Acute neuropathy manifests most often as cold-induced paresthesias in the fingers and toes and is quite common, affecting up to 90% of patients. These symptoms are self-limited and resolve usually within 1 week of each treatment.116 Some patients, with reports ranging from 10% to 79%, develop chronic neuropathy that persists for 1 year or more and causes significant decrements in quality of life.117 Patients older than age 70 may be at greater risk for oxaliplatin-induced neuropathy, which would increase risk of falls in this population.118 In addition to neuropathy, oxaliplatin is associated with hypersensitivity reactions that can be severe and even fatal.119 In a single institution series, the incidence of severe reactions was 2%.120 Desensitization following hypersensitivity reactions is possible but requires a time-intensive protocol.121
Based on the inconclusive efficacy findings and due to concerns over toxicity, each decision must be individualized to fit patient characteristics and preferences. In general, for patients with stage II disease without high-risk features, an individualized discussion should be held as to the risks and benefits of single-agent fluorouracil, and this treatment should be offered in cases where the patient or provider would like to be aggressive. Patients with stage II cancer who have 1 or more high-risk features are often recommended adjuvant chemotherapy. Whether treatment with fluorouracil plus leucovorin or FOLFOX is preferred remains uncertain, and thus the risks and the potential gains of oxaliplatin must be discussed with the individual patient. MMR status can also influence the treatment recommendation for patients with stage II disease. In general, patients with standard-risk stage II tumors that are pMMR are offered MMR with leucovorin or oral capecitabine for 12 cycles. FOLFOX is considered for patients with MSI-high disease and those with multiple high-risk features.
MONITORING AFTER THERAPY
After completion of adjuvant chemotherapy, patients enter a period of survivorship. Patients are seen in clinic for symptom and laboratory monitoring of the complete blood count, liver function tests, and carcinoembryonic antigen (CEA). NCCN guidelines support history and physical examination with CEA testing every 3 to 6 months for the first 2 years, then every 6 months for the next 3 years, after which many patients continue to be seen annually. CT imaging of the chest, abdomen, and pelvis for monitoring of disease recurrence is recommended every 6 to 12 months for a total of 5 years. New elevations in CEA or liver function tests should prompt early imaging. Colonoscopy should be performed 1 year after completion of therapy; however, if no preoperative colonoscopy was performed, this should be done 3 to 6 months after completion. Colonoscopy is then repeated in 3 years and then every 5 years unless advanced adenomas are present.122
SUMMARY
The addition of chemotherapy to surgical management of colon cancer has lowered the rate of disease recurrence and improved long-term survival. Adjuvant FOLFOX for 12 cycles is the standard of care for patients with stage III colon cancer and for patients with stage II disease with certain high-risk features. Use of adjuvant chemotherapy in stage II disease without high-risk features is controversial, and treatment decisions should be individualized. Biologic markers such as MSI and CDX2 status as well as patient-related factors including age, overall health, and personal preferences can inform treatment decisions. If chemotherapy is recommended in this setting, it would be with single-agent fluorouracil in an infusional or oral formulation, unless the tumor has the MSI-high feature. Following completion of adjuvant therapy, patients should be followed with clinical evaluation, laboratory testing, and imaging for a total of 5 years as per recommended guidelines.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67(1):7–30.
- United States Cancer Statistics. 1999–2013 incidence and mortality web-based report. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute, 2016. www.cdc.gov/uscs. Accessed June 12, 2017.
- Ahnen DJ, Wade SW, Jones WF, et al. The increasing incidence of young-onset colorectal cancer: a call to action. Mayo Clin Proc 2014;89:216–24.
- Jemal A, Fedewa SA, Anderson WF, et al. Colorectal cancer incidence patterns in the United States, 1974–2013. J Natl Cancer Inst 2017;109(8).
- Boursi B, Sella T, Liberman E, et al. The APC p.I1307K polymorphism is a significant risk factor for CRC in average risk Ashkenazi Jews. Eur J Cancer 2013;49:3680–5.
- Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011;60: 950–7.
- van Puijenbroek M, Nielsen M, Tops CM, et al. Identification of patients with (atypical) MUTYH-associated polyposis by KRAS2 c.34G > T prescreening followed by MUTYH hotspot analysis in formalin-fixed paraffin-embedded tissue. Clin Cancer Res 2008;14:139–42.
- Aretz S, Uhlhaas S, Goergens H, et al. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006;119:807–14.
- Tuohy TM, Rowe KG, Mineau GP, et al. Risk of colorectal cancer and adenomas in the families of patients with adenomas: a population-based study in Utah. Cancer 2014;120:35–42.
- Choi Y, Sateia HF, Peairs KS, Stewart RW. Screening for colorectal cancer. Semin Oncol 2017; 44:34–44.
- Atkin WS, Morson BC, Cuzick J. Long-term risk of colorectal cancer after excision of rectosigmoid adenomas. N Engl J Med 1992;326:658–62.
- Rutter MD. Surveillance programmes for neoplasia in colitis. J Gastroenterol 2011;46 Suppl 1:1–5.
- Giovannucci E. Modifiable risk factors for colon cancer. Gastroenterol Clin North Am 2002;31:925–43.
- Michels KB, Fuchs GS, Giovannucci E, et al. Fiber intake and incidence of colorectal cancer among 76,947 women and 47,279 men. Cancer Epidemiol Biomarkers Prev 2005;14:842–9.
- Omata F, Brown WR, Tokuda Y, et al. Modifiable risk factors for colorectal neoplasms and hyperplastic polyps. Intern Med 2009;48:123–8.
- Friedenreich CM, Neilson HK, Lynch BM. State of the epidemiological evidence on physical activity and cancer prevention. Eur J Cancer 2010;46:2593–604.
- Aleksandrova K, Pischon T, Jenab M, et al. Combined impact of healthy lifestyle factors on colorectal cancer: a large European cohort study. BMC Med 2014;12:168.
- Hermanek P, Wittekind C. The pathologist and the residual tumor (R) classification. Pathol Res Pract 1994;190:115–23.
- Lehnert T, Methner M, Pollok A, et al. Multivisceral resection for locally advanced primary colon and rectal cancer: an analysis of prognostic factors in 201 patients. Ann Surg 2002;235:217–25.
- Feinberg AE, et al. Oncologic outcomes following laparoscopic versus open resection of pT4 colon cancer: a systematic review and meta-analysis. Dis Colon Rectum 2017;60:116–125.
- Vignali A, et al. Laparoscopic treatment of advanced colonic cancer: a case-matched control with open surgery. Colorectal Dis 2013;15:944–8.
- Gainant A. Emergency management of acute colonic cancer obstruction. J Visc Surg 2012;149: e3–e10.
- Rosenman LD. Hartmann’s operation. Am J Surg 1994;168:283–4.
- Lee-Kong S, Lisle D. Surgical management of complicated colon cancer. Clin Colon Rectal Surg 2015;28:228–33.
- Bertelsen CA. Complete mesocolic excision an assessment of feasibility and outcome. Dan Med J 2017;64(2).
- Wolff WI SH. Definitive treatment of “malignant” polyps of the colon. Ann Surg 1975;182:516–25.
- Clinical Outcomes of Surgical Therapy Study Group, Nelson H, Sargent DJ, Wieand HS, et al. A comparison of laparoscopically assisted and open colectomy for colon cancer. N Engl J Med 2004;350:2050–9.
- Gunderson LL, Jessup JM, Sarjent DJ, et al. Revised tumor and node categorization for rectal cancer based on surveillance, epidemiology, and end results and rectal pooled analysis outcomes. J Clin Oncol 2010;28:256–63.
- Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41.
- Alves A, Panis Y, Mathieu P, et al. Postoperative mortality and morbidity in French patients undergoing colorectal surgery: results of a prospective multicenter study. Arch Surg 2005;140:278–83.
- Popescu RA, Norman A, Ross PJ, et al, Adjuvant or palliative chemotherapy for colorectal cancer in patients 70 years or older. J Clin Oncol 1999;17:2412–8.
- McCleary NJ, Meyerhardt JA, Green E, et al. Impact of age on the efficacy of newer adjuvant therapies in patients with stage II/III colon cancer: findings from the ACCENT database. J Clin Oncol 2013;31:2600–6.
- Tominaga T, Nonaka T, Sumida Y, et al. Effectiveness of adjuvant chemotherapy for elderly patients with lymph node-positive colorectal cancer. World J Surg Oncol 2016;14:197.
- Bos AC, van Erning FN, van Gestel YR, et al. Timing of adjuvant chemotherapy and its relation to survival among patients with stage III colon cancer. Eur J Cancer 2015;51:2553–61.
- Peixoto RD, Kumar A, Speers C, et al. Effect of delay in adjuvant oxaliplatin-based chemotherapy for stage III colon cancer. Clin Colorectal Cancer 2015;14:25–30.
- Compton CC, Fielding LP, Burgart LJ, et al. Prognostic factors in colorectal cancer. College of American Pathologists Consensus Statement 1999. Arch Pathol Lab Med 2000;124:979–94.
- Lieu CH, Lambert LA, Wolff RA, et al. Systemic chemotherapy and surgical cytoreduction for poorly differentiated and signet ring cell adenocarcinomas of the appendix. Ann Oncol 2012;23:652–8.
- Krasna MJ, Flancbaum L, Cody RP, et al. Vascular and neural invasion in colorectal carcinoma. Incidence and prognostic significance. Cancer 1988;61:1018–23.
- Cianchi F, Palomba A, Boddi V, et al. Lymph node recovery from colorectal tumor specimens: recommendation for a minimum number of lymph nodes to be examined. World J Surg 2002;26:384–9.
- Yoshimatsu K, et al. How many lymph nodes should be examined in Dukes’ B colorectal cancer? Determination on the basis of cumulative survival rate. Hepatogastroenterology 2005;52:1703–6.
- Caplin S, Cerottini JP, Bosman FT, et al. For patients with Dukes’ B (TNM Stage II) colorectal carcinoma, examination of six or fewer lymph nodes is related to poor prognosis. Cancer 1998;83:666–72.
- Veronese N, Nottegar A, Pea A, et al. Prognostic impact and implications of extracapsular lymph node involvement in colorectal cancer: a systematic review with meta-analysis. Ann Oncol 2016;27:42–8.
- Li J, Yang S, Hu J, et al. Tumor deposits counted as positive lymph nodes in TNM staging for advanced colorectal cancer: a retrospective multicenter study. Oncotarget 2016;7:18269–79.
- Venook A, Niedzwiecki D, Innocenti Fet al. Impact of primary (1º) tumor location on overall survival (OS) and progression-free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). J Clin Oncol 2016;34 no. 15 suppl. Abstract 3504.
- Schrag D, Brooks G, Meyerhardt JA ,et al. The relationship between primary tumor sidedness and prognosis in colorectal cancer. J Clin Oncol 2016;34 no. 15 suppl. Abstract 3505.
- Larrea AA, Lujan SA, Kunkel TA. SnapShot: DNA mismatch repair. Cell 2010;141:730 e1.
- Jass JR. Pathology of hereditary nonpolyposis colorectal cancer. Ann N Y Acad Sci 2000;910:62–73.
- Lynch HT, Smyrk T. Hereditary nonpolyposis colorectal cancer (Lynch syndrome). An updated review. Cancer 1996;78:1149–67.
- Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993;260:812–6.
- Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol 2017;12:24.
- Bupathi M, Wu C. Biomarkers for immune therapy in colorectal cancer: mismatch-repair deficiency and others. J Gastrointest Oncol 2016;7:713–20.
- Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 2005;23:609–18.
- Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med 2000;342:69–77.
- Ogino S, Kuchiba A, Qian ZR, et al. Prognostic significance and molecular associations of 18q loss of heterozygosity: a cohort study of microsatellite stable colorectal cancers. J Clin Oncol 2009; 27:4591–8.
- Kim ST, Lee J, Park SH, et al. The effect of DNA mismatch repair (MMR) status on oxaliplatin-based first-line chemotherapy as in recurrent or metastatic colon cancer. Med Oncol 2010;27:1277–85.
- Sargent DJ, Monges G, Thibodeau SN, et al. Therapy in colon cancer. J Clin Oncol 2010;28:4664.
- Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003;349:247–57.
- Hutchins G, Southward K, Handley K, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol 2011;29:1261–270.
- Yothers G, O’Connell MJ, Allegra CJ, et al. Oxaliplatin as adjuvant therapy for colon cancer: updated results of NSABP C-07 trial, including survival and subset analyses J Clin Oncol 2011;29:3768–74.
- Chang SC, Lin JK, Lin TC, Liang WY. Loss of heterozygosity: an independent prognostic factor of colorectal cancer. World J Gastroenterol 2005;11:778–84.
- Bertagnolli MM, Niedzwiecki D, Compton CC, et al. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: Cancer and Leukemia Group B Protocol 89803. J Clin Oncol 2009;27:1814–21.
- Bertagnolli MM, Redston M, Compton CC, et al. Microsatellite instability and loss of heterozygosity at chromosomal location 18q: prospective evaluation of biomarkers for stages II and III colon cancer--a study of CALGB 9581 and 89803. J Clin Oncol 2011;29:3153–62.
- Dalerba P, et al. CDX2 as a prognostic biomarker in stage II and stage III colon cancer. N Engl J Med 2016;374: 211–22.
- Clark-Langone KM, Wu JY, Sangli C, et al. Biomarker discovery for colon cancer using a 761 gene RT-PCR assay. BMC Genomics 2007;8:279.
- Gray RG, Quirke P, Handley K, et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol 2011;29:4611–9.
- Niedzwiecki D, Bertagnolli MM, Warren RS, et al. Documenting the natural history of patients with resected stage II adenocarcinoma of the colon after random assignment to adjuvant treatment with edrecolomab or observation: results from CALGB 9581. J Clin Oncol 2011;29:3146–52.
- Yothers G, O’Connell MJ, Lee M, et al. Validation of the 12-gene colon cancer recurrence score in NSABP C-07 as a predictor of recurrence in patients with stage II and III colon cancer treated with fluorouracil and leucovorin (FU/LV) and FU/LV plus oxaliplatin. J Clin Oncol 2013;31:4512–9.
- Gill S, Loprinzi CL, Sargent DJ, et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much? J Clin Oncol 2004;22:1797–806.
- Gill S, Loprinzi C, Kennecke H, et al. Prognostic web-based models for stage II and III colon cancer: A population and clinical trials-based validation of numeracy and adjuvant! online. Cancer 2011;117:4155–65.
- Jung M, Kim GW, Jung I, et al. Application of the Western-based adjuvant online model to Korean colon cancer patients; a single institution experience. BMC Cancer 2012;12:471.
- Papamichael D, Renfro LA, Matthaiou C, et al. Validity of Adjuvant! Online in older patients with stage III colon cancer based on 2967 patients from the ACCENT database. J Geriatr Oncol 2016;7:422–9.
- Tran B, Kopetz S, Tie J, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 2011;117:4623–32.
- Roth AD, Tejpar S, Delorenzi M, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol 2010;28:466–74.
- Lochhead P, Kuchiba A, Imamura Y, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst 2013;105:1151–6.
- Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 2007;67:2643–8.
- Therkildsen C, Bergmann TK, Henrichsen-Schnack T, et al. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol 2014;53:852–64.
- Taieb J, Le Malicot K, Shi Q, et al. Prognostic value of BRAF and KRAS mutations in MSI and MSS stage III colon cancer. J Natl Cancer Inst 2017;109(5).
- Palumbo LT, Sharpe WS, Henry JS. Cancer of the colon and rectum; analysis of 300 cases. Am J Surg 1965;109:439–44.
- Sharp GS, Benefiel WW. 5-Fluorouracil in the treatment of inoperable carcinoma of the colon and rectum. Cancer Chemother Rep 1962;20:97–101.
- Lawrence W Jr, Terz JJ, Horsley JS 3rd, et al. Chemotherapy as an adjuvant to surgery for colorectal cancer. Ann Surg 1975;181:616–23.
- Grage TD, et al. Adjuvant chemotherapy with 5-fluorouracil after surgical resection of colorectal carcinoma (COG protocol 7041). A preliminary report. Am J Surg 1977;133:59–66.
- Wolmark N, Fisher B, Rockette H, et al. Postoperative adjuvant chemotherapy or BCG for colon cancer: results from NSABP protocol C-01. J Natl Cancer Inst 1988;80:30–6.
- Moertel CG, Fleming TR, Macdonald JS, et al. Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma. N Engl J Med 1990;322:352–8.
- Wolmark N, Rockette H, Fisher B, et al. The benefit of leucovorin-modulated fluorouracil as postoperative adjuvant therapy for primary colon cancer: results from National Surgical Adjuvant Breast and Bowel Project protocol C-03. J Clin Oncol 1993;11:1879–87.
- Comparison of fluorouracil with additional levamisole, higher-dose folinic acid, or both, as adjuvant chemotherapy for colorectal cancer: a randomised trial. QUASAR Collaborative Group. Lancet 2000;355(9215):1588–96.
- Chen TC, Hinton DR, Leichman L, et al. Multifocal inflammatory leukoencephalopathy associated with levamisole and 5-fluorouracil: case report. Neurosurgery 1994;35:1138-42.
- Porschen R, Bermann A, Löffler T, et al. Fluorouracil plus leucovorin as effective adjuvant chemotherapy in curatively resected stage III colon cancer: results of the trial adjCCA-01. J Clin Oncol 2001;19:1787–94.
- Arkenau HT, Bermann A, Rettig K, et al. 5-Fluorouracil plus leucovorin is an effective adjuvant chemotherapy in curatively resected stage III colon cancer: long-term follow-up results of the adjCCA-01 trial. Ann Oncol 2003;14:395–9.
- Weinerman B, Shah A, Fields A, et al. Systemic infusion versus bolus chemotherapy with 5-fluorouracil in measurable metastatic colorectal cancer. Am J Clin Oncol 1992;15:518–23.
- Poplin EA, Benedetti JK, Estes NC, et al. Phase III Southwest Oncology Group 9415/Intergroup 0153 randomized trial of fluorouracil, leucovorin, and levamisole versus fluorouracil continuous infusion and levamisole for adjuvant treatment of stage III and high-risk stage II colon cancer. J Clin Oncol 2005;23:1819–25.
- Twelves C, Wong A, Nowacki MP, et al. Capecitabine as adjuvant treatment for stage III colon cancer. N Engl J Med 2005;352:2696–704.
- de Gramont A, Vignoud J, Tournigand C, et al. Oxaliplatin with high-dose leucovorin and 5-fluorouracil 48-hour continuous infusion in pretreated metastatic colorectal cancer. Eur J Cancer 1997;33:214–9.
- Diaz-Rubio E, Sastre J, Zaniboni A, et al. Oxaliplatin as single agent in previously untreated colorectal carcinoma patients: a phase II multicentric study. Ann Oncol 1998;9:105–8.
- André T, de Gramont A, Vernerey D, et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF mutation and mismatch repair status of the MOSAIC Study. J Clin Oncol 2015;33:4176–87.
- Andre T, Boni C, Mounedji-Boudiaf L, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med 2004;350:2343–51.
- Kuebler JP, Wieand HS, O’Connell MJ, et al. Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C-07. J Clin Oncol 2007;25:2198–204.
- Haller DG, Tabernero J, Maroun J, et al. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. J Clin Oncol 2011;29:1465–71.
- Schmoll HJ, et al. Capecitabine plus oxaliplatin compared with fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results of the NO16968 randomized controlled phase III trial. J Clin Oncol 2015;33:3733–40.
- Colucci G, Gebbia V, Paoletti G, et al. Phase III randomized trial of FOLFIRI versus FOLFOX4 in the treatment of advanced colorectal cancer: a multicenter study of the Gruppo Oncologico Dell’Italia Meridionale. J Clin Oncol 2005;23:4866–75.
- Tournigand C, André T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol 2004;22:229–37.
- Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004;350:2335–42.
- Saltz LB, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 2008;26:2013–9.
- Cremolini C, Loupakis F, Ruzzo A, et al. Predictors of benefit in colorectal cancer treated with cetuximab: are we getting “Lost in TranslationAL”? J Clin Oncol 2010;28:e173–4.
- Sorich MJ, Wiese MD, Rowland D, et al. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol 2015;26:13–21.
- Grothey A, van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381(9863):303–12.
- Saltz LB, Niedzwiecki D, Hollis D, et al. Irinotecan fluorouracil plus leucovorin is not superior to fluorouracil plus leucovorin alone as adjuvant treatment for stage III colon cancer: results of CALGB 89803. J Clin Oncol 2007;25:3456–61.
- Van Cutsem E, et al. Randomized phase III trial comparing biweekly infusional fluorouracil/leucovorin alone or with irinotecan in the adjuvant treatment of stage III colon cancer: PETACC-3. J Clin Oncol 2009;27:3117–25.
- Allegra CJ, et al. Bevacizumab in stage II-III colon cancer: 5-year update of the National Surgical Adjuvant Breast and Bowel Project C-08 trial. J Clin Oncol 2013;31:359–64.
- de Gramont A, et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): a phase 3 randomised controlled trial. Lancet Oncol 2012;13:1225–33.
- Alberts SR, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA 2012;307:1383–93.
- Taieb J, et al. Oxaliplatin, fluorouracil, and leucovorin with or without cetuximab in patients with resected stage III colon cancer (PETACC-8): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:862–73.
- Shi Q, Sobrero AF, Shields AF, et al. Prospective pooled analysis of six phase III trials investigating duration of adjuvant (adjuvant) oxaliplatin-based therapy (3 vs 6 months) for patients (pts) with stage III colon cancer (CC): The IDEA (International Duration Evaluation of Adjuvant chemotherapy) collaboration. In: Proceedings from the American Society of Clinical Oncology; June 1–5, 2017; Chicago. Abstract LBA1.
- Quasar Collaborative Group; Gray R, Barnwell J, McConkey C, et al. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet 2007;370(9604):2020–9.
- Efficacy of adjuvant fluorouracil and folinic acid in B2 colon cancer. International Multicentre Pooled Analysis of B2 Colon Cancer Trials (IMPACT B2) Investigators. J Clin Oncol 1999;17:1356–63.
- Kidwell KM, et al. Long-term neurotoxicity effects of oxaliplatin added to fluorouracil and leucovorin as adjuvant therapy for colon cancer: results from National Surgical Adjuvant Breast and Bowel Project trials C-07 and LTS-01. Cancer 2012;118:5614–22.
- Beijers AJ, Mols F, Vreugdenhil G. A systematic review on chronic oxaliplatin-induced peripheral neuropathy and the relation with oxaliplatin administration. Support Care Cancer 2014;22:1999–2007.
- Mols F, Beijers T, Lemmens V, et al. Chemotherapy-induced neuropathy and its association with quality of life among 2- to 11-year colorectal cancer survivors: results from the population-based PROFILES registry. J Clin Oncol 2013;31:2699–707.
- Raphael MJ, Fischer HD, Fung K, et al. Neurotoxicity outcomes in a population-based cohort of elderly patients treated with adjuvant oxaliplatin for colorectal cancer. Clin Colorectal Cancer 2017 March 24.
- Toki MI, Saif MW, Syrigos KN. Hypersensitivity reactions associated with oxaliplatin and their clinical management. Expert Opin Drug Saf 2014;13:1545–54.
- Siu SW, Chan RT, Au GK. Hypersensitivity reactions to oxaliplatin: experience in a single institute. Ann Oncol 2006;17:259–61.
- Wong JT, Ling M, Patil S, et al. Oxaliplatin hypersensitivity: evaluation, implications of skin testing, and desensitization. J Allergy Clin Immunol Pract 2014;2:40–5.
- Benson AB 3rd, Venook AP, Cederquist L, et al. NCCN Guidelines Colon Cancer Version 2.2017. www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed May 8, 2017.
- Wolmark N, Rockette H, Mamounas E, et al. Clinical trial to assess the relative efficacy of fluorouracil and leucovorin, fluorouracil and levamisole, and fluorouracil, leucovorin, and levamisole in patients with Dukes’ B and C carcinoma of the colon: results from National Surgical Adjuvant Breast and Bowel Project C-04. J Clin Oncol 1999;17:3553–9.
INTRODUCTION
Colorectal cancer (CRC) is one of the most prevalent malignancies and is the fourth most common cancer in the United States, with an estimated 133,490 new cases diagnosed in 2016. Of these, approximately 95,520 are located in the colon and 39,970 are in the rectum.1 CRC is the third leading cause of cancer death in women and the second leading cause of cancer death in men, with an estimated 49,190 total deaths in 2016.2 The incidence appears to be increasing,3 especially in patients younger than 55 years of age;4 the reason for this increase remains uncertain.
A number of risk factors for the development of CRC have been identified. Numerous hered-itary CRC syndromes have been described, including familial adenomatous polyposis,5 hereditary non-polyposis colorectal cancer (HNPCC) or Lynch syndrome,6 and MUTYH-associated polyposis.7,8 A family history of CRC doubles the risk of developing CRC,9 and current guidelines support lowering the age of screening in individuals with a family history of CRC to 10 years younger than the age of diagnosis of the family member or 40 years of age, whichever is lower.10 Patients with a personal history of adenomatous polyps are at increased risk for developing CRC, as are patients with a personal history of CRC, with a relative risk ranging from 3 to 6.11 Ulcerative colitis and Crohn’s disease are associated with the development of CRC and also influence screening, though evidence suggests good control of these diseases may mitigate risk.12 Finally, modifiable risk factors for the development of CRC include high red meat consumption,13 diets low in fiber,14 obesity,13 smoking, alcohol use,15 and physical inactivity16; lifestyle modification targeting these factors has been shown to decrease rates of CRC.17 The majority of colon cancers present with clinical symptoms, often with rectal bleeding, abdominal pain, change in bowel habits, or obstructive symptoms. More rarely, these tumors are detected during screening colonoscopy, in which case they tend to be at an early stage.
SURGICAL MANAGEMENT
A critical goal in the resection of early-stage colon cancer is attaining R0 resection. Patients who achieve R0 resection as compared to R1 (microscopic residual tumor) and R2 (macroscopic residual tumor)18 have significantly improved long-term overall survival.19 Traditionally, open resection of the involved colonic segment was employed, with end-end anastomosis of the uninvolved free margins. Laparoscopic resection for early-stage disease has been utilized in attempts to decrease morbidity of open procedures, with similar outcomes and node sampling.20 Laparoscopic resection appears to provide similar outcomes even in locally advanced disease.21 Right-sided lesions are treated with right colectomy and primary ileocolic anastomosis.22 For patients presenting with obstructing masses, the Hartmann procedure is the most commonly performed operation. This involves creation of an ostomy with subtotal colectomy and subsequent ostomy reversal in a 2- or 3-stage protocol.23 Patients with locally advanced disease and invasion into surrounding structures require multivisceral resection, which involves resection en bloc with secondarily involved organs.24 Intestinal perforation presents a unique challenge and is associated with surgical complications, infection, and lower overall survival (OS) and 5-year disease-free survival (DFS). Complete mesocolic excision is a newer technique that has been performed with reports of better oncologic outcome at some centers; however, this approach is not currently considered standard of care.25
STAGING
According to a report by the National Cancer Institute, the estimated 5-year relative survival rates for localized colon cancer (lymph node negative), regional (lymph node positive) disease, and distant (metastatic) disease are 89.9%, 71.3%, and 13.9%, respectively.1 However, efforts have been made to further classify patients into distinct categories to allow fine-tuning of prognostication. In the current system, staging of colon cancer utilizes the American Joint Committee on Cancer tumor/node/metastasis (TNM) system.20 Clinical and pathologic features include depth of invasion, local invasion of other organs, nodal involvement, and presence of distant metastasis (Table 1). Studies completed prior to the adoption of the TNM system used the Dukes criteria, which divided colon cancer into A, B, and C, corresponding to TNM stage I, stage IIA–IIC, and stage IIIA-IIIC. This classification is rarely used in more contemporary studies.
APPROACH TO ADJUVANT CHEMOTHERAPY
Adjuvant chemotherapy seeks to eliminate micrometastatic disease present following curative surgical resection. When stage 0 cancer is discovered incidentally during colonoscopy, endoscopic resection alone is the management of choice, as presence of micrometastatic disease is exceedingly unlikely.26 Stage I–III CRCs are treated with surgical resection withcurative intent. The 5-year survival rate for stage I and early-stage II CRC is estimated at 97% with surgery alone.27,28 The survival rate drops to about 60% for high-risk stage II tumors (T4aN0), and down to 50% or less for stage II-T4N0 or stage III cancers. Adjuvant chemotherapy is generally recommended to further decrease the rates of distant recurrence in certain cases of stage II and in all stage III tumors.
DETERMINATION OF BENEFIT FROM CHEMOTHERAPY: PROGNOSTIC MARKERS
Prior to administration of adjuvant chemotherapy, a clinical evaluation by the medical oncologist to determine appropriateness and safety of treatment is paramount. Poor performance status and comorbid conditions may indicate risk for excessive toxicity and minimal benefit from chemotherapy. CRC commonly presents in older individuals, with the median age at diagnosis of 69 years for men and 73 years for women.29 In this patient population, comorbidities such as cardiovascular disease, diabetes, and renal dysfunction are more prevalent.30 Decisions regarding adjuvant chemotherapy in this patient population have to take into consideration the fact that older patients may experience higher rates of toxicity with chemotherapy, including gastrointestinal toxicities and marrow suppression.31 Though some reports indicate patients older than 70 years derive similar benefit from adjuvant chemotherapy,32,33 a large pooled analysis of the ACCENT database, which included 7 adjuvant therapy trials and 14,528 patients, suggested limited benefit from the addition of oxaliplatin to fluorouracil in elderly patients.32 Other factors that weigh on the decision include stage, pathology, and presence of high-risk features. A common concern in the postoperative setting is delaying initiation of chemotherapy to allow adequate wound healing; however, evidence suggests that delays longer than 8 weeks leads to worse overall survival, with hazard ratios (HR) ranging from 1.4 to 1.7.34,35 Thus, the start of adjuvant therapy should ideally be within this time frame.
HIGH-RISK FEATURES
Multiple factors have been found to predict worse outcome and are classified as high-risk features (Table 2). Histologically, high-grade or poorly differentiated tumors are associated with higher recurrence rate and worse outcome.36 Certain histological subtypes, including mucinous and signet-ring, both appear to have more aggressive biology.37 Presence of microscopic invasion into surrounding blood vessels (vascular invasion) and nerves (perineural invasion) is associated with lower survival.38 Penetration of the cancer through the visceral peritoneum (T4a) or into surrounding structures (T4b) is associated with lower survival.36 During surgical resection, multiple lymph nodes are removed along with the primary tumor to evaluate for metastasis to the regional nodes. Multiple analyses have demonstrated that removal and pathologic assessment of fewer than 12 lymph nodes is associated with high risk of missing a positive node, and is thus equated with high risk.39–41 In addition, extension of tumor beyond the capsules of any single lymph node, termed extracapsular extension, is associated with an increased risk of all-cause mortality.42 Tumor deposits, or focal aggregates of adenocarcinoma in the pericolic fat that are not contiguous with the primary tumor and are not associated with lymph nodes, are currently classified as lymph nodes as N1c in the current TNM staging system. Presence of these deposits has been found to predict poor outcome stage for stage.43 Obstruction and/or perforation secondary to the tumor are also considered high-risk features that predict poor outcome.
SIDEDNESS
As reported at the 2016 American Society of Clinical Oncology annual meeting, tumor location predicts outcome in the metastatic setting. A report by Venook and colleagues based on a post-hoc analysis found that in the metastatic setting, location of the tumor primary in the left side is associated with longer OS (33.3 months) when compared to the right side of the colon (19.4 months).44 A retrospective analysis of multiple databases presented by Schrag and colleagues similarly reported inferior outcomes in patients with stage III and IV disease who had right-sided primary tumors.45 However, the prognostic implications for stage II disease remain uncertain.
BIOMARKERS
Given the controversy regarding adjuvant therapy of patients with stage II colon cancer, multiple biomarkers have been evaluated as possible predictive markers that can assist in this decision. The mismatch repair (MMR) system is a complex cellular enzymatic mechanism that identifies and corrects DNA errors during cell division and prevents mutagenesis.46 The familial cancer syndrome HNPCC is linked to alteration in a variety of MMR genes, leading to deficient mismatch repair (dMMR), also termed microsatellite instability-high (MSI-high).47,48 Epigenetic modification can also lead to silencing of the same implicated genes and accounts for 15% to 20% of sporadic colorectal cancer.49 These epigenetic modifications lead to hypermethylation of the promotor region of MLH1 in 70% of cases.50 The 4 MMR genes most commonly tested are MLH-1, MSH2, MSH6, and PMS2. Testing can be performed by immunohistochemistry or polymerase chain reaction.51 Across tumor histology and stage, MSI status is prognostic. Patients with MSI-high tumors have been shown to have improved prognosis and longer OS both in stage II and III disease52–54 and in the metastatic setting.55 However, despite this survival benefit, there is conflicting data as to whether patients with stage II, MSI-high colon cancer may benefit less from adjuvant chemotherapy. One early retrospective study compared outcomes of 70 patients with stage II and III disease and dMMR to those of 387 patients with stage II and III disease and proficient mismatch repair (pMMR). Adjuvant fluorouracil with leucovorin improved DFS for patients with pMMR (HR 0.67) but not for those with dMMR (HR 1.10). In addition, for patients with stage II disease and dMMR, the HR for OS was inferior at 2.95.56 Data collected from randomized clinical trials using fluorouracil-based adjuvant chemotherapy were analyzed in an attempt to predict benefit based on MSI status. Benefit was only seen in pMMR patients, with a HR of 0.72; this was not seen in the dMMR patients.57 Subsequent studies have had different findings and did not demonstrate a detrimental effect of fluorouracil in dMMR.58,59 For stage III patients, MSI status does not appear to affect benefit from chemotherapy, as analysis of data from the NSABP C-07 trial (Table 3) demonstrated benefit of FOLFOX (leucovorin, fluorouracil, oxaliplatin) in patients with dMMR status and stage III disease.59
Another genetic abnormality identified in colon cancers is chromosome 18q loss of heterozygosity (LOH). The presence of 18q LOH appears to be inversely associated with MSI-high status. Some reports have linked presence of 18q with worse outcome,60 but others question this, arguing the finding may simply be related to MSI status.61,62 This biomarker has not been established as a clear prognostic marker that can aid clinical decisions.
Most recently, expression of caudal-type homeobox transcription factor 2 (CDX2) has been reported as a novel prognostic and predictive tool. A 2015 report linked lack of expression of CDX2 to worse outcome; in this study, 5-year DFS was 41% in patients with CDX2-negative tumors versus 74% in the CDX2-positive tumors, with a HR of disease recurrence of 2.73 for CDX2-negative tumors.63 Similar numbers were observed in patients with stage II disease, with 5-year OS of 40% in patients with CDX2-negative tumors versus 70% in those with CDX2-positive tumors. Treatment of CDX2-negative patients with adjuvant chemotherapy improved outcomes: 5-year DFS in the stage II subgroup was 91% with chemotherapy versus 56% without, and in the stage III subgroup, 74% with chemotherapy versus 37% without. The authors concluded that patients with stage II and III colon cancer that is CDX2-negative may benefit from adjuvant chemotherapy. Importantly, CDX2-negativity is a rare event, occurring in only 6.9% of evaluable tumors.
RISK ASSESSMENT TOOLS
Several risk assessment tools have been developed in an attempt to aid clinical decision making regarding adjuvant chemotherapy for patients with stage II colon cancer. The Oncotype DX Colon Assay analyses a 12-gene signature in the pathologic sample and was developed with the goal to improve prognostication and aid in treatment decision making. The test utilizes reverse transcription-PCR on RNA extracted from the tumor.64 After evaluating 12 genes, a recurrence score is generated that predicts the risk of disease recurrence. This score was validated using data from 3 large clinical trials.65–67 Unlike the Oncotype Dx score used in breast cancer, the test in colon cancer has not been found to predict the benefit from chemotherapy and has not been incorporated widely into clinical practice.
Adjuvant! Online (available at www.adjuvantonline.com) is a web-based tool that combines clinical and histological features to estimate outcome. Calculations are based on US SEER tumor registry-reported outcomes.68 A second web-based tool, Numeracy (available at www.mayoclinic.com/calcs), was developed by the Mayo Clinic using pooled data from 7 randomized clinical trials including 3341 patients.68 Both tools seek to predict absolute benefit for patients treated with fluorouracil, though data suggests Adjuvant! Online may be more reliable in its predictive ability.69 Adjuvant! Online has also been validated in an Asian population70 and patients older than 70 years.71
MUTATIONAL ANALYSIS
Multiple mutations in proto-oncogenes have been found in colon cancer cells. One such proto-oncogene is BRAF, which encodes a serine-threonine kinase in the rapidly accelerated fibrosarcoma (RAF). Mutations in BRAF have been found in 5% to 10% of colon cancers and are associated with right-sided tumors.72 As a prognostic marker, some studies have associated BRAF mutations with worse prognosis, including shorter time to relapse and shorter OS.73,74 Two other proto-oncogenes are Kristen rat sarcoma viral oncogene homolog (KRAS) and neuroblastoma rat sarcoma viral oncogene homolog (NRAS), both of which encode proteins downstream of epidermal growth factor receptor (EGFR). KRAS and NRAS mutations have been shown to be predictive in the metastatic setting where they predict resistance to the EGFR inhibitors cetuximab and panitumumab.75,76 The effect of KRAS and NRAS mutations on outcome in stage II and III colon cancer is uncertain. Some studies suggest worse outcome in KRAS-mutated cancers,77 while others failed to demonstrate this finding.73
CASE PRESENTATION 1
A 53-year-old man with no past medical history presents to the emergency department with early satiety and generalized abdominal pain. Laboratory evaluation shows a microcytic anemia with normal white blood cell count, platelet count, renal function, and liver function tests. Computed tomography (CT) scan of the abdomen and pelvis show a 4-cm mass in the transverse colon without obstruction and without abnormality in the liver. CT scan of the chest does not demonstrate pathologic lymphadenopathy or other findings. He undergoes robotic laparoscopic transverse colon resection and appendectomy. Pathology confirms a 3.5-cm focus of adenocarcinoma of the colon with invasion through the muscularis propria and 5 of 27 regional lymph nodes positive for adenocarcinoma and uninvolved proximal, distal, and radial margins. He is given a stage of IIIB pT3 pN2a M0 and referred to medical oncology for further management, where 6 months of adjuvant FOLFOX chemotherapy is recommended.
ADJUVANT CHEMOTHERAPY IN STAGE III COLON CANCER
Postoperative adjuvant chemotherapy is the standard of care for patients with stage III disease. In the 1960s, infusional fluorouracil was first used to treat inoperable colon cancer.78,79 After encouraging results, the agent was used both intraluminally and intravenously as an adjuvant therapy for patients undergoing resection with curative intent; however, only modest benefits were described.80,81 The National Surgical Adjuvant Breast and Bowel Project (NSABP) C-01 trial (Table 3) was the first study to demonstrate a benefit from adjuvant chemotherapy in colon cancer. This study randomly assigned patients with stage II and III colon cancer to surgery alone, postoperative chemotherapy with fluorouracil, semustine, and vincristine (MOF), or postoperative bacillus Calmette-Guérin (BCG). DFS and OS were significantly improved with MOF chemotherapy.82 In 1990, a landmark study reported on outcomes after treatment of 1296 patients with stage III colon cancer with adjuvant fluorouracil and levamisole for 12 months. The combination was associated with a 41% reduction in risk of cancer recurrence and a 33% reduction in risk of death.83 The NSABP C-03 trial (Table 3) compared MOF to the combination of fluorouracil and leucovorin and demonstrated improved 3-year DFS (69% versus 73%) and 3-year OS (77% versus 84%) in patients with stage III disease.84 Building on these outcomes, the QUASAR study (Table 3) compared fluorouracil in combination with one of levamisole, low-dose leucovorin, or high-dose leucovorin. The study enrolled 4927 patients and found worse outcomes with fluorouracil plus levamisole and no difference in low-doseversus high-dose leucovorin.85 Levamisole fell out of use after associations with development of multifocal leukoencephalopathy,86 and was later shown to have inferior outcomes versus leucovorin when combined with fluorouracil.87,88 Intravenous fluorouracil has shown similar benefit when administered by bolus or infusion,89 although continuous infusion has been associated with lower incidence of severe toxicity.90 The efficacy of the oral fluoropyrimidine capecitabine has been shown to be equivalent to that of fluorouracil.91
Fluorouracil-based treatment remained the standard of care until the introduction of oxaliplatin in the mid-1990s. After encouraging results in the metastatic setting,92,93 the agent was moved to the adjuvant setting. The MOSAIC trial (Table 3) randomly assigned patients with stage II and III colon cancer to fluorouracil with leucovorin (FULV) versus FOLFOX given once every 2 weeks for 12 cycles. Analysis with respect to stage III patients showed a clear survival benefit, with a 10-year OS of 67.1% with FOLFOX chemotherapy versus 59% with fluorouracil and leucovorin.94,95 The NSABP C-07 (Table 3) trial used a similar trial design but employed bolus fluorouracil. More than 2400 patients with stage II and III colon cancer were randomly assigned to bolus FULV or bolus fluorouracil, leucovorin, and oxaliplatin (FLOX). The addition of oxaliplatin significantly improved outcomes, with 4-year DFS of 67% versus 71.8% for FULV and FLOX, respectively, and a HR of death of 0.80 with FLOX.59,96 The multicenter N016968 trial (Table 3) randomly assigned 1886 patients with stage III colon cancer to adjuvant capecitabine plus oxaliplatin (XELOX) or bolus fluorouracil plus leucovorin (FU/FA). The 3-year DFS was 70.9% versus 66.5% with XELOX and FU/FA, respectively, and 5-year OS was 77.6% versus 74.2%, respectively.97,98
In the metastatic setting, additional agents have shown efficacy, including irinotecan,99,100 bevacizumab,101,102 cetuximab,103,104 and regorafenib.105 This observation led to testing of these agents in earlier stage disease. The CALGB 89803 trial compared fluorouracil, leucovorin, and irinotecan to fluorouracil with leucovorin alone. No benefit in 5-year DFS or OS was seen.106 Similarly, infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) was not found to improve 5-year DFS as compared to fluorouracil with leucovorin alone in the PETACC-3 trial.107 The NSABP C-08 trial considered the addition of bevacizumab to FOLFOX. When compared to FOLFOX alone, the combination of bevacizumab to FOLFOX had similar 3-year DFS (77.9% versus 75.1%) and 5-year OS (82.5% versus 80.7%).108 This finding was confirmed in the Avant trial.109 The addition of cetuximab to FOLFOX was equally disappointing, as shown in the N0147 trial110 and PETACC-8 trial.111 Data on regorafenib in the adjuvant setting for stage III colon cancer is lacking; however, 2 ongoing clinical trials, NCT02425683 and NCT02664077, are each studying the use of regorafenib following completion of FOLFOX for patients with stage III disease.
Thus, after multiple trials comparing various regimens and despite attempts to improve outcomes by the addition of a third agent, the standard of care per National Comprehensive Cancer Network (NCCN) guidelines for management of stage III colon cancer remains 12 cycles of FOLFOX chemotherapy. Therapy should be initiated within 8 weeks of surgery. Data are emerging to support a short duration of therapy for patients with low-risk stage III tumors, as shown in an abstract presented at the 2017 American Society of Clinical Oncology annual meeting. The IDEA trial was a pooled analysis of 6 randomized clinical trials across multiple countries, all of which evaluated 3 versus 6 months of FOLFOX or capecitabine and oxaliplatin in the treatment of stage III colon cancer. The analysis was designed to test non-inferiority of 3 months of therapy as compared to 6 months. The analysis included 6088 patients across 244 centers in 6 countries. The overall analysis failed to establish noninferiority. The 3-year DFS rate was 74.6% for 3 months and 75.5% for 6 months, with a DFS HR of 1.07 and a confidence interval that did not meet the prespecified endpoint. Subgroup analysis suggested noninferiority for lower stage disease (T1–3 or N1) but not for higher stage disease (T4 or N2). Given the high rates of neuropathy with 6 months of oxaliplatin, these results suggest that 3 months of adjuvant therapy can be considered for patients with T1–3 or N1 disease in an attempt to limit toxicity.112
CASE PRESENTATION 2
A 57-year-old woman presents to the emergency department with fever and abdominal pain. CT of the abdomen and pelvis demonstrates a left-sided colonic mass with surrounding fat stranding and pelvic abscess. She is taken emergently for left hemicolectomy, cholecystectomy, and evacuation of pelvic abscess. Pathology reveals a 5-cm adenocarcinoma with invasion through the visceral peritoneum; 0/22 lymph nodes are involved. She is given a diagnosis of stage IIC and referred to medical oncology for further management. Due to her young age and presence of high-risk features, she is recommended adjuvant therapy with FOLFOX for 6 months.
ADJUVANT CHEMOTHERAPY IN STAGE II COLON CANCER
Because of excellent outcomes with surgical resection alone for stage II cancers, the use of adjuvant chemotherapy for patients with stage II disease is controversial. Limited prospective data is available to guide adjuvant treatment decisions for stage II patients. The QUASAR trial, which compared observation to adjuvant fluorouracil and leucovorin in patients with early-stage colon cancer, included 2963 patients with stage II disease and found a relative risk (RR) of death or recurrence of 0.82 and 0.78, respectively. Importantly, the absolute benefit of therapy was less than 5%.113 The IMPACT-B2 trial (Table 3) combined data from 5 separate trials and analyzed 1016 patients with stage II colon cancer who received fluorouracil with leucovorin or observation. Event-free survival was 0.86 versus 0.83 and 5-year OS was 82% versus 80%, suggesting no benefit.114 The benefit of addition of oxaliplatin to fluorouracil in stage II disease appears to be less than the benefit of adding this agent in the treatment of stage III CRC. As noted above, the MOSAIC trial randomly assigned patients with stage II and III colon cancer to receive adjuvant fluorouracil and leucovorin with or without oxaliplatin for 12 cycles. After a median follow-up of 9.5 years, 10-year OS rates for patients with stage II disease were 78.4% versus 79.5%. For patients with high-risk stage II disease (defined as T4, bowel perforation, or fewer than 10 lymph nodes examined), 10-year OS was 71.7% and 75.4% respectively, but these differences were not statistically significant.94
Because of conflicting data as to the benefit of adding oxaliplatin in stage II disease, oxaliplatin is not recommended for standard-risk stage II patients. The use of oxaliplatin in high-risk stage II tumors should be weighed carefully given the toxicity risk. Oxaliplatin is recognized to cause sensory neuropathy in many patients, which can become painful and debilitating.115 Two types of neuropathy are associated with oxaliplatin: acute and chronic. Acute neuropathy manifests most often as cold-induced paresthesias in the fingers and toes and is quite common, affecting up to 90% of patients. These symptoms are self-limited and resolve usually within 1 week of each treatment.116 Some patients, with reports ranging from 10% to 79%, develop chronic neuropathy that persists for 1 year or more and causes significant decrements in quality of life.117 Patients older than age 70 may be at greater risk for oxaliplatin-induced neuropathy, which would increase risk of falls in this population.118 In addition to neuropathy, oxaliplatin is associated with hypersensitivity reactions that can be severe and even fatal.119 In a single institution series, the incidence of severe reactions was 2%.120 Desensitization following hypersensitivity reactions is possible but requires a time-intensive protocol.121
Based on the inconclusive efficacy findings and due to concerns over toxicity, each decision must be individualized to fit patient characteristics and preferences. In general, for patients with stage II disease without high-risk features, an individualized discussion should be held as to the risks and benefits of single-agent fluorouracil, and this treatment should be offered in cases where the patient or provider would like to be aggressive. Patients with stage II cancer who have 1 or more high-risk features are often recommended adjuvant chemotherapy. Whether treatment with fluorouracil plus leucovorin or FOLFOX is preferred remains uncertain, and thus the risks and the potential gains of oxaliplatin must be discussed with the individual patient. MMR status can also influence the treatment recommendation for patients with stage II disease. In general, patients with standard-risk stage II tumors that are pMMR are offered MMR with leucovorin or oral capecitabine for 12 cycles. FOLFOX is considered for patients with MSI-high disease and those with multiple high-risk features.
MONITORING AFTER THERAPY
After completion of adjuvant chemotherapy, patients enter a period of survivorship. Patients are seen in clinic for symptom and laboratory monitoring of the complete blood count, liver function tests, and carcinoembryonic antigen (CEA). NCCN guidelines support history and physical examination with CEA testing every 3 to 6 months for the first 2 years, then every 6 months for the next 3 years, after which many patients continue to be seen annually. CT imaging of the chest, abdomen, and pelvis for monitoring of disease recurrence is recommended every 6 to 12 months for a total of 5 years. New elevations in CEA or liver function tests should prompt early imaging. Colonoscopy should be performed 1 year after completion of therapy; however, if no preoperative colonoscopy was performed, this should be done 3 to 6 months after completion. Colonoscopy is then repeated in 3 years and then every 5 years unless advanced adenomas are present.122
SUMMARY
The addition of chemotherapy to surgical management of colon cancer has lowered the rate of disease recurrence and improved long-term survival. Adjuvant FOLFOX for 12 cycles is the standard of care for patients with stage III colon cancer and for patients with stage II disease with certain high-risk features. Use of adjuvant chemotherapy in stage II disease without high-risk features is controversial, and treatment decisions should be individualized. Biologic markers such as MSI and CDX2 status as well as patient-related factors including age, overall health, and personal preferences can inform treatment decisions. If chemotherapy is recommended in this setting, it would be with single-agent fluorouracil in an infusional or oral formulation, unless the tumor has the MSI-high feature. Following completion of adjuvant therapy, patients should be followed with clinical evaluation, laboratory testing, and imaging for a total of 5 years as per recommended guidelines.
INTRODUCTION
Colorectal cancer (CRC) is one of the most prevalent malignancies and is the fourth most common cancer in the United States, with an estimated 133,490 new cases diagnosed in 2016. Of these, approximately 95,520 are located in the colon and 39,970 are in the rectum.1 CRC is the third leading cause of cancer death in women and the second leading cause of cancer death in men, with an estimated 49,190 total deaths in 2016.2 The incidence appears to be increasing,3 especially in patients younger than 55 years of age;4 the reason for this increase remains uncertain.
A number of risk factors for the development of CRC have been identified. Numerous hered-itary CRC syndromes have been described, including familial adenomatous polyposis,5 hereditary non-polyposis colorectal cancer (HNPCC) or Lynch syndrome,6 and MUTYH-associated polyposis.7,8 A family history of CRC doubles the risk of developing CRC,9 and current guidelines support lowering the age of screening in individuals with a family history of CRC to 10 years younger than the age of diagnosis of the family member or 40 years of age, whichever is lower.10 Patients with a personal history of adenomatous polyps are at increased risk for developing CRC, as are patients with a personal history of CRC, with a relative risk ranging from 3 to 6.11 Ulcerative colitis and Crohn’s disease are associated with the development of CRC and also influence screening, though evidence suggests good control of these diseases may mitigate risk.12 Finally, modifiable risk factors for the development of CRC include high red meat consumption,13 diets low in fiber,14 obesity,13 smoking, alcohol use,15 and physical inactivity16; lifestyle modification targeting these factors has been shown to decrease rates of CRC.17 The majority of colon cancers present with clinical symptoms, often with rectal bleeding, abdominal pain, change in bowel habits, or obstructive symptoms. More rarely, these tumors are detected during screening colonoscopy, in which case they tend to be at an early stage.
SURGICAL MANAGEMENT
A critical goal in the resection of early-stage colon cancer is attaining R0 resection. Patients who achieve R0 resection as compared to R1 (microscopic residual tumor) and R2 (macroscopic residual tumor)18 have significantly improved long-term overall survival.19 Traditionally, open resection of the involved colonic segment was employed, with end-end anastomosis of the uninvolved free margins. Laparoscopic resection for early-stage disease has been utilized in attempts to decrease morbidity of open procedures, with similar outcomes and node sampling.20 Laparoscopic resection appears to provide similar outcomes even in locally advanced disease.21 Right-sided lesions are treated with right colectomy and primary ileocolic anastomosis.22 For patients presenting with obstructing masses, the Hartmann procedure is the most commonly performed operation. This involves creation of an ostomy with subtotal colectomy and subsequent ostomy reversal in a 2- or 3-stage protocol.23 Patients with locally advanced disease and invasion into surrounding structures require multivisceral resection, which involves resection en bloc with secondarily involved organs.24 Intestinal perforation presents a unique challenge and is associated with surgical complications, infection, and lower overall survival (OS) and 5-year disease-free survival (DFS). Complete mesocolic excision is a newer technique that has been performed with reports of better oncologic outcome at some centers; however, this approach is not currently considered standard of care.25
STAGING
According to a report by the National Cancer Institute, the estimated 5-year relative survival rates for localized colon cancer (lymph node negative), regional (lymph node positive) disease, and distant (metastatic) disease are 89.9%, 71.3%, and 13.9%, respectively.1 However, efforts have been made to further classify patients into distinct categories to allow fine-tuning of prognostication. In the current system, staging of colon cancer utilizes the American Joint Committee on Cancer tumor/node/metastasis (TNM) system.20 Clinical and pathologic features include depth of invasion, local invasion of other organs, nodal involvement, and presence of distant metastasis (Table 1). Studies completed prior to the adoption of the TNM system used the Dukes criteria, which divided colon cancer into A, B, and C, corresponding to TNM stage I, stage IIA–IIC, and stage IIIA-IIIC. This classification is rarely used in more contemporary studies.
APPROACH TO ADJUVANT CHEMOTHERAPY
Adjuvant chemotherapy seeks to eliminate micrometastatic disease present following curative surgical resection. When stage 0 cancer is discovered incidentally during colonoscopy, endoscopic resection alone is the management of choice, as presence of micrometastatic disease is exceedingly unlikely.26 Stage I–III CRCs are treated with surgical resection withcurative intent. The 5-year survival rate for stage I and early-stage II CRC is estimated at 97% with surgery alone.27,28 The survival rate drops to about 60% for high-risk stage II tumors (T4aN0), and down to 50% or less for stage II-T4N0 or stage III cancers. Adjuvant chemotherapy is generally recommended to further decrease the rates of distant recurrence in certain cases of stage II and in all stage III tumors.
DETERMINATION OF BENEFIT FROM CHEMOTHERAPY: PROGNOSTIC MARKERS
Prior to administration of adjuvant chemotherapy, a clinical evaluation by the medical oncologist to determine appropriateness and safety of treatment is paramount. Poor performance status and comorbid conditions may indicate risk for excessive toxicity and minimal benefit from chemotherapy. CRC commonly presents in older individuals, with the median age at diagnosis of 69 years for men and 73 years for women.29 In this patient population, comorbidities such as cardiovascular disease, diabetes, and renal dysfunction are more prevalent.30 Decisions regarding adjuvant chemotherapy in this patient population have to take into consideration the fact that older patients may experience higher rates of toxicity with chemotherapy, including gastrointestinal toxicities and marrow suppression.31 Though some reports indicate patients older than 70 years derive similar benefit from adjuvant chemotherapy,32,33 a large pooled analysis of the ACCENT database, which included 7 adjuvant therapy trials and 14,528 patients, suggested limited benefit from the addition of oxaliplatin to fluorouracil in elderly patients.32 Other factors that weigh on the decision include stage, pathology, and presence of high-risk features. A common concern in the postoperative setting is delaying initiation of chemotherapy to allow adequate wound healing; however, evidence suggests that delays longer than 8 weeks leads to worse overall survival, with hazard ratios (HR) ranging from 1.4 to 1.7.34,35 Thus, the start of adjuvant therapy should ideally be within this time frame.
HIGH-RISK FEATURES
Multiple factors have been found to predict worse outcome and are classified as high-risk features (Table 2). Histologically, high-grade or poorly differentiated tumors are associated with higher recurrence rate and worse outcome.36 Certain histological subtypes, including mucinous and signet-ring, both appear to have more aggressive biology.37 Presence of microscopic invasion into surrounding blood vessels (vascular invasion) and nerves (perineural invasion) is associated with lower survival.38 Penetration of the cancer through the visceral peritoneum (T4a) or into surrounding structures (T4b) is associated with lower survival.36 During surgical resection, multiple lymph nodes are removed along with the primary tumor to evaluate for metastasis to the regional nodes. Multiple analyses have demonstrated that removal and pathologic assessment of fewer than 12 lymph nodes is associated with high risk of missing a positive node, and is thus equated with high risk.39–41 In addition, extension of tumor beyond the capsules of any single lymph node, termed extracapsular extension, is associated with an increased risk of all-cause mortality.42 Tumor deposits, or focal aggregates of adenocarcinoma in the pericolic fat that are not contiguous with the primary tumor and are not associated with lymph nodes, are currently classified as lymph nodes as N1c in the current TNM staging system. Presence of these deposits has been found to predict poor outcome stage for stage.43 Obstruction and/or perforation secondary to the tumor are also considered high-risk features that predict poor outcome.
SIDEDNESS
As reported at the 2016 American Society of Clinical Oncology annual meeting, tumor location predicts outcome in the metastatic setting. A report by Venook and colleagues based on a post-hoc analysis found that in the metastatic setting, location of the tumor primary in the left side is associated with longer OS (33.3 months) when compared to the right side of the colon (19.4 months).44 A retrospective analysis of multiple databases presented by Schrag and colleagues similarly reported inferior outcomes in patients with stage III and IV disease who had right-sided primary tumors.45 However, the prognostic implications for stage II disease remain uncertain.
BIOMARKERS
Given the controversy regarding adjuvant therapy of patients with stage II colon cancer, multiple biomarkers have been evaluated as possible predictive markers that can assist in this decision. The mismatch repair (MMR) system is a complex cellular enzymatic mechanism that identifies and corrects DNA errors during cell division and prevents mutagenesis.46 The familial cancer syndrome HNPCC is linked to alteration in a variety of MMR genes, leading to deficient mismatch repair (dMMR), also termed microsatellite instability-high (MSI-high).47,48 Epigenetic modification can also lead to silencing of the same implicated genes and accounts for 15% to 20% of sporadic colorectal cancer.49 These epigenetic modifications lead to hypermethylation of the promotor region of MLH1 in 70% of cases.50 The 4 MMR genes most commonly tested are MLH-1, MSH2, MSH6, and PMS2. Testing can be performed by immunohistochemistry or polymerase chain reaction.51 Across tumor histology and stage, MSI status is prognostic. Patients with MSI-high tumors have been shown to have improved prognosis and longer OS both in stage II and III disease52–54 and in the metastatic setting.55 However, despite this survival benefit, there is conflicting data as to whether patients with stage II, MSI-high colon cancer may benefit less from adjuvant chemotherapy. One early retrospective study compared outcomes of 70 patients with stage II and III disease and dMMR to those of 387 patients with stage II and III disease and proficient mismatch repair (pMMR). Adjuvant fluorouracil with leucovorin improved DFS for patients with pMMR (HR 0.67) but not for those with dMMR (HR 1.10). In addition, for patients with stage II disease and dMMR, the HR for OS was inferior at 2.95.56 Data collected from randomized clinical trials using fluorouracil-based adjuvant chemotherapy were analyzed in an attempt to predict benefit based on MSI status. Benefit was only seen in pMMR patients, with a HR of 0.72; this was not seen in the dMMR patients.57 Subsequent studies have had different findings and did not demonstrate a detrimental effect of fluorouracil in dMMR.58,59 For stage III patients, MSI status does not appear to affect benefit from chemotherapy, as analysis of data from the NSABP C-07 trial (Table 3) demonstrated benefit of FOLFOX (leucovorin, fluorouracil, oxaliplatin) in patients with dMMR status and stage III disease.59
Another genetic abnormality identified in colon cancers is chromosome 18q loss of heterozygosity (LOH). The presence of 18q LOH appears to be inversely associated with MSI-high status. Some reports have linked presence of 18q with worse outcome,60 but others question this, arguing the finding may simply be related to MSI status.61,62 This biomarker has not been established as a clear prognostic marker that can aid clinical decisions.
Most recently, expression of caudal-type homeobox transcription factor 2 (CDX2) has been reported as a novel prognostic and predictive tool. A 2015 report linked lack of expression of CDX2 to worse outcome; in this study, 5-year DFS was 41% in patients with CDX2-negative tumors versus 74% in the CDX2-positive tumors, with a HR of disease recurrence of 2.73 for CDX2-negative tumors.63 Similar numbers were observed in patients with stage II disease, with 5-year OS of 40% in patients with CDX2-negative tumors versus 70% in those with CDX2-positive tumors. Treatment of CDX2-negative patients with adjuvant chemotherapy improved outcomes: 5-year DFS in the stage II subgroup was 91% with chemotherapy versus 56% without, and in the stage III subgroup, 74% with chemotherapy versus 37% without. The authors concluded that patients with stage II and III colon cancer that is CDX2-negative may benefit from adjuvant chemotherapy. Importantly, CDX2-negativity is a rare event, occurring in only 6.9% of evaluable tumors.
RISK ASSESSMENT TOOLS
Several risk assessment tools have been developed in an attempt to aid clinical decision making regarding adjuvant chemotherapy for patients with stage II colon cancer. The Oncotype DX Colon Assay analyses a 12-gene signature in the pathologic sample and was developed with the goal to improve prognostication and aid in treatment decision making. The test utilizes reverse transcription-PCR on RNA extracted from the tumor.64 After evaluating 12 genes, a recurrence score is generated that predicts the risk of disease recurrence. This score was validated using data from 3 large clinical trials.65–67 Unlike the Oncotype Dx score used in breast cancer, the test in colon cancer has not been found to predict the benefit from chemotherapy and has not been incorporated widely into clinical practice.
Adjuvant! Online (available at www.adjuvantonline.com) is a web-based tool that combines clinical and histological features to estimate outcome. Calculations are based on US SEER tumor registry-reported outcomes.68 A second web-based tool, Numeracy (available at www.mayoclinic.com/calcs), was developed by the Mayo Clinic using pooled data from 7 randomized clinical trials including 3341 patients.68 Both tools seek to predict absolute benefit for patients treated with fluorouracil, though data suggests Adjuvant! Online may be more reliable in its predictive ability.69 Adjuvant! Online has also been validated in an Asian population70 and patients older than 70 years.71
MUTATIONAL ANALYSIS
Multiple mutations in proto-oncogenes have been found in colon cancer cells. One such proto-oncogene is BRAF, which encodes a serine-threonine kinase in the rapidly accelerated fibrosarcoma (RAF). Mutations in BRAF have been found in 5% to 10% of colon cancers and are associated with right-sided tumors.72 As a prognostic marker, some studies have associated BRAF mutations with worse prognosis, including shorter time to relapse and shorter OS.73,74 Two other proto-oncogenes are Kristen rat sarcoma viral oncogene homolog (KRAS) and neuroblastoma rat sarcoma viral oncogene homolog (NRAS), both of which encode proteins downstream of epidermal growth factor receptor (EGFR). KRAS and NRAS mutations have been shown to be predictive in the metastatic setting where they predict resistance to the EGFR inhibitors cetuximab and panitumumab.75,76 The effect of KRAS and NRAS mutations on outcome in stage II and III colon cancer is uncertain. Some studies suggest worse outcome in KRAS-mutated cancers,77 while others failed to demonstrate this finding.73
CASE PRESENTATION 1
A 53-year-old man with no past medical history presents to the emergency department with early satiety and generalized abdominal pain. Laboratory evaluation shows a microcytic anemia with normal white blood cell count, platelet count, renal function, and liver function tests. Computed tomography (CT) scan of the abdomen and pelvis show a 4-cm mass in the transverse colon without obstruction and without abnormality in the liver. CT scan of the chest does not demonstrate pathologic lymphadenopathy or other findings. He undergoes robotic laparoscopic transverse colon resection and appendectomy. Pathology confirms a 3.5-cm focus of adenocarcinoma of the colon with invasion through the muscularis propria and 5 of 27 regional lymph nodes positive for adenocarcinoma and uninvolved proximal, distal, and radial margins. He is given a stage of IIIB pT3 pN2a M0 and referred to medical oncology for further management, where 6 months of adjuvant FOLFOX chemotherapy is recommended.
ADJUVANT CHEMOTHERAPY IN STAGE III COLON CANCER
Postoperative adjuvant chemotherapy is the standard of care for patients with stage III disease. In the 1960s, infusional fluorouracil was first used to treat inoperable colon cancer.78,79 After encouraging results, the agent was used both intraluminally and intravenously as an adjuvant therapy for patients undergoing resection with curative intent; however, only modest benefits were described.80,81 The National Surgical Adjuvant Breast and Bowel Project (NSABP) C-01 trial (Table 3) was the first study to demonstrate a benefit from adjuvant chemotherapy in colon cancer. This study randomly assigned patients with stage II and III colon cancer to surgery alone, postoperative chemotherapy with fluorouracil, semustine, and vincristine (MOF), or postoperative bacillus Calmette-Guérin (BCG). DFS and OS were significantly improved with MOF chemotherapy.82 In 1990, a landmark study reported on outcomes after treatment of 1296 patients with stage III colon cancer with adjuvant fluorouracil and levamisole for 12 months. The combination was associated with a 41% reduction in risk of cancer recurrence and a 33% reduction in risk of death.83 The NSABP C-03 trial (Table 3) compared MOF to the combination of fluorouracil and leucovorin and demonstrated improved 3-year DFS (69% versus 73%) and 3-year OS (77% versus 84%) in patients with stage III disease.84 Building on these outcomes, the QUASAR study (Table 3) compared fluorouracil in combination with one of levamisole, low-dose leucovorin, or high-dose leucovorin. The study enrolled 4927 patients and found worse outcomes with fluorouracil plus levamisole and no difference in low-doseversus high-dose leucovorin.85 Levamisole fell out of use after associations with development of multifocal leukoencephalopathy,86 and was later shown to have inferior outcomes versus leucovorin when combined with fluorouracil.87,88 Intravenous fluorouracil has shown similar benefit when administered by bolus or infusion,89 although continuous infusion has been associated with lower incidence of severe toxicity.90 The efficacy of the oral fluoropyrimidine capecitabine has been shown to be equivalent to that of fluorouracil.91
Fluorouracil-based treatment remained the standard of care until the introduction of oxaliplatin in the mid-1990s. After encouraging results in the metastatic setting,92,93 the agent was moved to the adjuvant setting. The MOSAIC trial (Table 3) randomly assigned patients with stage II and III colon cancer to fluorouracil with leucovorin (FULV) versus FOLFOX given once every 2 weeks for 12 cycles. Analysis with respect to stage III patients showed a clear survival benefit, with a 10-year OS of 67.1% with FOLFOX chemotherapy versus 59% with fluorouracil and leucovorin.94,95 The NSABP C-07 (Table 3) trial used a similar trial design but employed bolus fluorouracil. More than 2400 patients with stage II and III colon cancer were randomly assigned to bolus FULV or bolus fluorouracil, leucovorin, and oxaliplatin (FLOX). The addition of oxaliplatin significantly improved outcomes, with 4-year DFS of 67% versus 71.8% for FULV and FLOX, respectively, and a HR of death of 0.80 with FLOX.59,96 The multicenter N016968 trial (Table 3) randomly assigned 1886 patients with stage III colon cancer to adjuvant capecitabine plus oxaliplatin (XELOX) or bolus fluorouracil plus leucovorin (FU/FA). The 3-year DFS was 70.9% versus 66.5% with XELOX and FU/FA, respectively, and 5-year OS was 77.6% versus 74.2%, respectively.97,98
In the metastatic setting, additional agents have shown efficacy, including irinotecan,99,100 bevacizumab,101,102 cetuximab,103,104 and regorafenib.105 This observation led to testing of these agents in earlier stage disease. The CALGB 89803 trial compared fluorouracil, leucovorin, and irinotecan to fluorouracil with leucovorin alone. No benefit in 5-year DFS or OS was seen.106 Similarly, infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) was not found to improve 5-year DFS as compared to fluorouracil with leucovorin alone in the PETACC-3 trial.107 The NSABP C-08 trial considered the addition of bevacizumab to FOLFOX. When compared to FOLFOX alone, the combination of bevacizumab to FOLFOX had similar 3-year DFS (77.9% versus 75.1%) and 5-year OS (82.5% versus 80.7%).108 This finding was confirmed in the Avant trial.109 The addition of cetuximab to FOLFOX was equally disappointing, as shown in the N0147 trial110 and PETACC-8 trial.111 Data on regorafenib in the adjuvant setting for stage III colon cancer is lacking; however, 2 ongoing clinical trials, NCT02425683 and NCT02664077, are each studying the use of regorafenib following completion of FOLFOX for patients with stage III disease.
Thus, after multiple trials comparing various regimens and despite attempts to improve outcomes by the addition of a third agent, the standard of care per National Comprehensive Cancer Network (NCCN) guidelines for management of stage III colon cancer remains 12 cycles of FOLFOX chemotherapy. Therapy should be initiated within 8 weeks of surgery. Data are emerging to support a short duration of therapy for patients with low-risk stage III tumors, as shown in an abstract presented at the 2017 American Society of Clinical Oncology annual meeting. The IDEA trial was a pooled analysis of 6 randomized clinical trials across multiple countries, all of which evaluated 3 versus 6 months of FOLFOX or capecitabine and oxaliplatin in the treatment of stage III colon cancer. The analysis was designed to test non-inferiority of 3 months of therapy as compared to 6 months. The analysis included 6088 patients across 244 centers in 6 countries. The overall analysis failed to establish noninferiority. The 3-year DFS rate was 74.6% for 3 months and 75.5% for 6 months, with a DFS HR of 1.07 and a confidence interval that did not meet the prespecified endpoint. Subgroup analysis suggested noninferiority for lower stage disease (T1–3 or N1) but not for higher stage disease (T4 or N2). Given the high rates of neuropathy with 6 months of oxaliplatin, these results suggest that 3 months of adjuvant therapy can be considered for patients with T1–3 or N1 disease in an attempt to limit toxicity.112
CASE PRESENTATION 2
A 57-year-old woman presents to the emergency department with fever and abdominal pain. CT of the abdomen and pelvis demonstrates a left-sided colonic mass with surrounding fat stranding and pelvic abscess. She is taken emergently for left hemicolectomy, cholecystectomy, and evacuation of pelvic abscess. Pathology reveals a 5-cm adenocarcinoma with invasion through the visceral peritoneum; 0/22 lymph nodes are involved. She is given a diagnosis of stage IIC and referred to medical oncology for further management. Due to her young age and presence of high-risk features, she is recommended adjuvant therapy with FOLFOX for 6 months.
ADJUVANT CHEMOTHERAPY IN STAGE II COLON CANCER
Because of excellent outcomes with surgical resection alone for stage II cancers, the use of adjuvant chemotherapy for patients with stage II disease is controversial. Limited prospective data is available to guide adjuvant treatment decisions for stage II patients. The QUASAR trial, which compared observation to adjuvant fluorouracil and leucovorin in patients with early-stage colon cancer, included 2963 patients with stage II disease and found a relative risk (RR) of death or recurrence of 0.82 and 0.78, respectively. Importantly, the absolute benefit of therapy was less than 5%.113 The IMPACT-B2 trial (Table 3) combined data from 5 separate trials and analyzed 1016 patients with stage II colon cancer who received fluorouracil with leucovorin or observation. Event-free survival was 0.86 versus 0.83 and 5-year OS was 82% versus 80%, suggesting no benefit.114 The benefit of addition of oxaliplatin to fluorouracil in stage II disease appears to be less than the benefit of adding this agent in the treatment of stage III CRC. As noted above, the MOSAIC trial randomly assigned patients with stage II and III colon cancer to receive adjuvant fluorouracil and leucovorin with or without oxaliplatin for 12 cycles. After a median follow-up of 9.5 years, 10-year OS rates for patients with stage II disease were 78.4% versus 79.5%. For patients with high-risk stage II disease (defined as T4, bowel perforation, or fewer than 10 lymph nodes examined), 10-year OS was 71.7% and 75.4% respectively, but these differences were not statistically significant.94
Because of conflicting data as to the benefit of adding oxaliplatin in stage II disease, oxaliplatin is not recommended for standard-risk stage II patients. The use of oxaliplatin in high-risk stage II tumors should be weighed carefully given the toxicity risk. Oxaliplatin is recognized to cause sensory neuropathy in many patients, which can become painful and debilitating.115 Two types of neuropathy are associated with oxaliplatin: acute and chronic. Acute neuropathy manifests most often as cold-induced paresthesias in the fingers and toes and is quite common, affecting up to 90% of patients. These symptoms are self-limited and resolve usually within 1 week of each treatment.116 Some patients, with reports ranging from 10% to 79%, develop chronic neuropathy that persists for 1 year or more and causes significant decrements in quality of life.117 Patients older than age 70 may be at greater risk for oxaliplatin-induced neuropathy, which would increase risk of falls in this population.118 In addition to neuropathy, oxaliplatin is associated with hypersensitivity reactions that can be severe and even fatal.119 In a single institution series, the incidence of severe reactions was 2%.120 Desensitization following hypersensitivity reactions is possible but requires a time-intensive protocol.121
Based on the inconclusive efficacy findings and due to concerns over toxicity, each decision must be individualized to fit patient characteristics and preferences. In general, for patients with stage II disease without high-risk features, an individualized discussion should be held as to the risks and benefits of single-agent fluorouracil, and this treatment should be offered in cases where the patient or provider would like to be aggressive. Patients with stage II cancer who have 1 or more high-risk features are often recommended adjuvant chemotherapy. Whether treatment with fluorouracil plus leucovorin or FOLFOX is preferred remains uncertain, and thus the risks and the potential gains of oxaliplatin must be discussed with the individual patient. MMR status can also influence the treatment recommendation for patients with stage II disease. In general, patients with standard-risk stage II tumors that are pMMR are offered MMR with leucovorin or oral capecitabine for 12 cycles. FOLFOX is considered for patients with MSI-high disease and those with multiple high-risk features.
MONITORING AFTER THERAPY
After completion of adjuvant chemotherapy, patients enter a period of survivorship. Patients are seen in clinic for symptom and laboratory monitoring of the complete blood count, liver function tests, and carcinoembryonic antigen (CEA). NCCN guidelines support history and physical examination with CEA testing every 3 to 6 months for the first 2 years, then every 6 months for the next 3 years, after which many patients continue to be seen annually. CT imaging of the chest, abdomen, and pelvis for monitoring of disease recurrence is recommended every 6 to 12 months for a total of 5 years. New elevations in CEA or liver function tests should prompt early imaging. Colonoscopy should be performed 1 year after completion of therapy; however, if no preoperative colonoscopy was performed, this should be done 3 to 6 months after completion. Colonoscopy is then repeated in 3 years and then every 5 years unless advanced adenomas are present.122
SUMMARY
The addition of chemotherapy to surgical management of colon cancer has lowered the rate of disease recurrence and improved long-term survival. Adjuvant FOLFOX for 12 cycles is the standard of care for patients with stage III colon cancer and for patients with stage II disease with certain high-risk features. Use of adjuvant chemotherapy in stage II disease without high-risk features is controversial, and treatment decisions should be individualized. Biologic markers such as MSI and CDX2 status as well as patient-related factors including age, overall health, and personal preferences can inform treatment decisions. If chemotherapy is recommended in this setting, it would be with single-agent fluorouracil in an infusional or oral formulation, unless the tumor has the MSI-high feature. Following completion of adjuvant therapy, patients should be followed with clinical evaluation, laboratory testing, and imaging for a total of 5 years as per recommended guidelines.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67(1):7–30.
- United States Cancer Statistics. 1999–2013 incidence and mortality web-based report. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute, 2016. www.cdc.gov/uscs. Accessed June 12, 2017.
- Ahnen DJ, Wade SW, Jones WF, et al. The increasing incidence of young-onset colorectal cancer: a call to action. Mayo Clin Proc 2014;89:216–24.
- Jemal A, Fedewa SA, Anderson WF, et al. Colorectal cancer incidence patterns in the United States, 1974–2013. J Natl Cancer Inst 2017;109(8).
- Boursi B, Sella T, Liberman E, et al. The APC p.I1307K polymorphism is a significant risk factor for CRC in average risk Ashkenazi Jews. Eur J Cancer 2013;49:3680–5.
- Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011;60: 950–7.
- van Puijenbroek M, Nielsen M, Tops CM, et al. Identification of patients with (atypical) MUTYH-associated polyposis by KRAS2 c.34G > T prescreening followed by MUTYH hotspot analysis in formalin-fixed paraffin-embedded tissue. Clin Cancer Res 2008;14:139–42.
- Aretz S, Uhlhaas S, Goergens H, et al. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006;119:807–14.
- Tuohy TM, Rowe KG, Mineau GP, et al. Risk of colorectal cancer and adenomas in the families of patients with adenomas: a population-based study in Utah. Cancer 2014;120:35–42.
- Choi Y, Sateia HF, Peairs KS, Stewart RW. Screening for colorectal cancer. Semin Oncol 2017; 44:34–44.
- Atkin WS, Morson BC, Cuzick J. Long-term risk of colorectal cancer after excision of rectosigmoid adenomas. N Engl J Med 1992;326:658–62.
- Rutter MD. Surveillance programmes for neoplasia in colitis. J Gastroenterol 2011;46 Suppl 1:1–5.
- Giovannucci E. Modifiable risk factors for colon cancer. Gastroenterol Clin North Am 2002;31:925–43.
- Michels KB, Fuchs GS, Giovannucci E, et al. Fiber intake and incidence of colorectal cancer among 76,947 women and 47,279 men. Cancer Epidemiol Biomarkers Prev 2005;14:842–9.
- Omata F, Brown WR, Tokuda Y, et al. Modifiable risk factors for colorectal neoplasms and hyperplastic polyps. Intern Med 2009;48:123–8.
- Friedenreich CM, Neilson HK, Lynch BM. State of the epidemiological evidence on physical activity and cancer prevention. Eur J Cancer 2010;46:2593–604.
- Aleksandrova K, Pischon T, Jenab M, et al. Combined impact of healthy lifestyle factors on colorectal cancer: a large European cohort study. BMC Med 2014;12:168.
- Hermanek P, Wittekind C. The pathologist and the residual tumor (R) classification. Pathol Res Pract 1994;190:115–23.
- Lehnert T, Methner M, Pollok A, et al. Multivisceral resection for locally advanced primary colon and rectal cancer: an analysis of prognostic factors in 201 patients. Ann Surg 2002;235:217–25.
- Feinberg AE, et al. Oncologic outcomes following laparoscopic versus open resection of pT4 colon cancer: a systematic review and meta-analysis. Dis Colon Rectum 2017;60:116–125.
- Vignali A, et al. Laparoscopic treatment of advanced colonic cancer: a case-matched control with open surgery. Colorectal Dis 2013;15:944–8.
- Gainant A. Emergency management of acute colonic cancer obstruction. J Visc Surg 2012;149: e3–e10.
- Rosenman LD. Hartmann’s operation. Am J Surg 1994;168:283–4.
- Lee-Kong S, Lisle D. Surgical management of complicated colon cancer. Clin Colon Rectal Surg 2015;28:228–33.
- Bertelsen CA. Complete mesocolic excision an assessment of feasibility and outcome. Dan Med J 2017;64(2).
- Wolff WI SH. Definitive treatment of “malignant” polyps of the colon. Ann Surg 1975;182:516–25.
- Clinical Outcomes of Surgical Therapy Study Group, Nelson H, Sargent DJ, Wieand HS, et al. A comparison of laparoscopically assisted and open colectomy for colon cancer. N Engl J Med 2004;350:2050–9.
- Gunderson LL, Jessup JM, Sarjent DJ, et al. Revised tumor and node categorization for rectal cancer based on surveillance, epidemiology, and end results and rectal pooled analysis outcomes. J Clin Oncol 2010;28:256–63.
- Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41.
- Alves A, Panis Y, Mathieu P, et al. Postoperative mortality and morbidity in French patients undergoing colorectal surgery: results of a prospective multicenter study. Arch Surg 2005;140:278–83.
- Popescu RA, Norman A, Ross PJ, et al, Adjuvant or palliative chemotherapy for colorectal cancer in patients 70 years or older. J Clin Oncol 1999;17:2412–8.
- McCleary NJ, Meyerhardt JA, Green E, et al. Impact of age on the efficacy of newer adjuvant therapies in patients with stage II/III colon cancer: findings from the ACCENT database. J Clin Oncol 2013;31:2600–6.
- Tominaga T, Nonaka T, Sumida Y, et al. Effectiveness of adjuvant chemotherapy for elderly patients with lymph node-positive colorectal cancer. World J Surg Oncol 2016;14:197.
- Bos AC, van Erning FN, van Gestel YR, et al. Timing of adjuvant chemotherapy and its relation to survival among patients with stage III colon cancer. Eur J Cancer 2015;51:2553–61.
- Peixoto RD, Kumar A, Speers C, et al. Effect of delay in adjuvant oxaliplatin-based chemotherapy for stage III colon cancer. Clin Colorectal Cancer 2015;14:25–30.
- Compton CC, Fielding LP, Burgart LJ, et al. Prognostic factors in colorectal cancer. College of American Pathologists Consensus Statement 1999. Arch Pathol Lab Med 2000;124:979–94.
- Lieu CH, Lambert LA, Wolff RA, et al. Systemic chemotherapy and surgical cytoreduction for poorly differentiated and signet ring cell adenocarcinomas of the appendix. Ann Oncol 2012;23:652–8.
- Krasna MJ, Flancbaum L, Cody RP, et al. Vascular and neural invasion in colorectal carcinoma. Incidence and prognostic significance. Cancer 1988;61:1018–23.
- Cianchi F, Palomba A, Boddi V, et al. Lymph node recovery from colorectal tumor specimens: recommendation for a minimum number of lymph nodes to be examined. World J Surg 2002;26:384–9.
- Yoshimatsu K, et al. How many lymph nodes should be examined in Dukes’ B colorectal cancer? Determination on the basis of cumulative survival rate. Hepatogastroenterology 2005;52:1703–6.
- Caplin S, Cerottini JP, Bosman FT, et al. For patients with Dukes’ B (TNM Stage II) colorectal carcinoma, examination of six or fewer lymph nodes is related to poor prognosis. Cancer 1998;83:666–72.
- Veronese N, Nottegar A, Pea A, et al. Prognostic impact and implications of extracapsular lymph node involvement in colorectal cancer: a systematic review with meta-analysis. Ann Oncol 2016;27:42–8.
- Li J, Yang S, Hu J, et al. Tumor deposits counted as positive lymph nodes in TNM staging for advanced colorectal cancer: a retrospective multicenter study. Oncotarget 2016;7:18269–79.
- Venook A, Niedzwiecki D, Innocenti Fet al. Impact of primary (1º) tumor location on overall survival (OS) and progression-free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). J Clin Oncol 2016;34 no. 15 suppl. Abstract 3504.
- Schrag D, Brooks G, Meyerhardt JA ,et al. The relationship between primary tumor sidedness and prognosis in colorectal cancer. J Clin Oncol 2016;34 no. 15 suppl. Abstract 3505.
- Larrea AA, Lujan SA, Kunkel TA. SnapShot: DNA mismatch repair. Cell 2010;141:730 e1.
- Jass JR. Pathology of hereditary nonpolyposis colorectal cancer. Ann N Y Acad Sci 2000;910:62–73.
- Lynch HT, Smyrk T. Hereditary nonpolyposis colorectal cancer (Lynch syndrome). An updated review. Cancer 1996;78:1149–67.
- Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993;260:812–6.
- Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol 2017;12:24.
- Bupathi M, Wu C. Biomarkers for immune therapy in colorectal cancer: mismatch-repair deficiency and others. J Gastrointest Oncol 2016;7:713–20.
- Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 2005;23:609–18.
- Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med 2000;342:69–77.
- Ogino S, Kuchiba A, Qian ZR, et al. Prognostic significance and molecular associations of 18q loss of heterozygosity: a cohort study of microsatellite stable colorectal cancers. J Clin Oncol 2009; 27:4591–8.
- Kim ST, Lee J, Park SH, et al. The effect of DNA mismatch repair (MMR) status on oxaliplatin-based first-line chemotherapy as in recurrent or metastatic colon cancer. Med Oncol 2010;27:1277–85.
- Sargent DJ, Monges G, Thibodeau SN, et al. Therapy in colon cancer. J Clin Oncol 2010;28:4664.
- Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003;349:247–57.
- Hutchins G, Southward K, Handley K, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol 2011;29:1261–270.
- Yothers G, O’Connell MJ, Allegra CJ, et al. Oxaliplatin as adjuvant therapy for colon cancer: updated results of NSABP C-07 trial, including survival and subset analyses J Clin Oncol 2011;29:3768–74.
- Chang SC, Lin JK, Lin TC, Liang WY. Loss of heterozygosity: an independent prognostic factor of colorectal cancer. World J Gastroenterol 2005;11:778–84.
- Bertagnolli MM, Niedzwiecki D, Compton CC, et al. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: Cancer and Leukemia Group B Protocol 89803. J Clin Oncol 2009;27:1814–21.
- Bertagnolli MM, Redston M, Compton CC, et al. Microsatellite instability and loss of heterozygosity at chromosomal location 18q: prospective evaluation of biomarkers for stages II and III colon cancer--a study of CALGB 9581 and 89803. J Clin Oncol 2011;29:3153–62.
- Dalerba P, et al. CDX2 as a prognostic biomarker in stage II and stage III colon cancer. N Engl J Med 2016;374: 211–22.
- Clark-Langone KM, Wu JY, Sangli C, et al. Biomarker discovery for colon cancer using a 761 gene RT-PCR assay. BMC Genomics 2007;8:279.
- Gray RG, Quirke P, Handley K, et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol 2011;29:4611–9.
- Niedzwiecki D, Bertagnolli MM, Warren RS, et al. Documenting the natural history of patients with resected stage II adenocarcinoma of the colon after random assignment to adjuvant treatment with edrecolomab or observation: results from CALGB 9581. J Clin Oncol 2011;29:3146–52.
- Yothers G, O’Connell MJ, Lee M, et al. Validation of the 12-gene colon cancer recurrence score in NSABP C-07 as a predictor of recurrence in patients with stage II and III colon cancer treated with fluorouracil and leucovorin (FU/LV) and FU/LV plus oxaliplatin. J Clin Oncol 2013;31:4512–9.
- Gill S, Loprinzi CL, Sargent DJ, et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much? J Clin Oncol 2004;22:1797–806.
- Gill S, Loprinzi C, Kennecke H, et al. Prognostic web-based models for stage II and III colon cancer: A population and clinical trials-based validation of numeracy and adjuvant! online. Cancer 2011;117:4155–65.
- Jung M, Kim GW, Jung I, et al. Application of the Western-based adjuvant online model to Korean colon cancer patients; a single institution experience. BMC Cancer 2012;12:471.
- Papamichael D, Renfro LA, Matthaiou C, et al. Validity of Adjuvant! Online in older patients with stage III colon cancer based on 2967 patients from the ACCENT database. J Geriatr Oncol 2016;7:422–9.
- Tran B, Kopetz S, Tie J, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 2011;117:4623–32.
- Roth AD, Tejpar S, Delorenzi M, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol 2010;28:466–74.
- Lochhead P, Kuchiba A, Imamura Y, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst 2013;105:1151–6.
- Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 2007;67:2643–8.
- Therkildsen C, Bergmann TK, Henrichsen-Schnack T, et al. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol 2014;53:852–64.
- Taieb J, Le Malicot K, Shi Q, et al. Prognostic value of BRAF and KRAS mutations in MSI and MSS stage III colon cancer. J Natl Cancer Inst 2017;109(5).
- Palumbo LT, Sharpe WS, Henry JS. Cancer of the colon and rectum; analysis of 300 cases. Am J Surg 1965;109:439–44.
- Sharp GS, Benefiel WW. 5-Fluorouracil in the treatment of inoperable carcinoma of the colon and rectum. Cancer Chemother Rep 1962;20:97–101.
- Lawrence W Jr, Terz JJ, Horsley JS 3rd, et al. Chemotherapy as an adjuvant to surgery for colorectal cancer. Ann Surg 1975;181:616–23.
- Grage TD, et al. Adjuvant chemotherapy with 5-fluorouracil after surgical resection of colorectal carcinoma (COG protocol 7041). A preliminary report. Am J Surg 1977;133:59–66.
- Wolmark N, Fisher B, Rockette H, et al. Postoperative adjuvant chemotherapy or BCG for colon cancer: results from NSABP protocol C-01. J Natl Cancer Inst 1988;80:30–6.
- Moertel CG, Fleming TR, Macdonald JS, et al. Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma. N Engl J Med 1990;322:352–8.
- Wolmark N, Rockette H, Fisher B, et al. The benefit of leucovorin-modulated fluorouracil as postoperative adjuvant therapy for primary colon cancer: results from National Surgical Adjuvant Breast and Bowel Project protocol C-03. J Clin Oncol 1993;11:1879–87.
- Comparison of fluorouracil with additional levamisole, higher-dose folinic acid, or both, as adjuvant chemotherapy for colorectal cancer: a randomised trial. QUASAR Collaborative Group. Lancet 2000;355(9215):1588–96.
- Chen TC, Hinton DR, Leichman L, et al. Multifocal inflammatory leukoencephalopathy associated with levamisole and 5-fluorouracil: case report. Neurosurgery 1994;35:1138-42.
- Porschen R, Bermann A, Löffler T, et al. Fluorouracil plus leucovorin as effective adjuvant chemotherapy in curatively resected stage III colon cancer: results of the trial adjCCA-01. J Clin Oncol 2001;19:1787–94.
- Arkenau HT, Bermann A, Rettig K, et al. 5-Fluorouracil plus leucovorin is an effective adjuvant chemotherapy in curatively resected stage III colon cancer: long-term follow-up results of the adjCCA-01 trial. Ann Oncol 2003;14:395–9.
- Weinerman B, Shah A, Fields A, et al. Systemic infusion versus bolus chemotherapy with 5-fluorouracil in measurable metastatic colorectal cancer. Am J Clin Oncol 1992;15:518–23.
- Poplin EA, Benedetti JK, Estes NC, et al. Phase III Southwest Oncology Group 9415/Intergroup 0153 randomized trial of fluorouracil, leucovorin, and levamisole versus fluorouracil continuous infusion and levamisole for adjuvant treatment of stage III and high-risk stage II colon cancer. J Clin Oncol 2005;23:1819–25.
- Twelves C, Wong A, Nowacki MP, et al. Capecitabine as adjuvant treatment for stage III colon cancer. N Engl J Med 2005;352:2696–704.
- de Gramont A, Vignoud J, Tournigand C, et al. Oxaliplatin with high-dose leucovorin and 5-fluorouracil 48-hour continuous infusion in pretreated metastatic colorectal cancer. Eur J Cancer 1997;33:214–9.
- Diaz-Rubio E, Sastre J, Zaniboni A, et al. Oxaliplatin as single agent in previously untreated colorectal carcinoma patients: a phase II multicentric study. Ann Oncol 1998;9:105–8.
- André T, de Gramont A, Vernerey D, et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF mutation and mismatch repair status of the MOSAIC Study. J Clin Oncol 2015;33:4176–87.
- Andre T, Boni C, Mounedji-Boudiaf L, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med 2004;350:2343–51.
- Kuebler JP, Wieand HS, O’Connell MJ, et al. Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C-07. J Clin Oncol 2007;25:2198–204.
- Haller DG, Tabernero J, Maroun J, et al. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. J Clin Oncol 2011;29:1465–71.
- Schmoll HJ, et al. Capecitabine plus oxaliplatin compared with fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results of the NO16968 randomized controlled phase III trial. J Clin Oncol 2015;33:3733–40.
- Colucci G, Gebbia V, Paoletti G, et al. Phase III randomized trial of FOLFIRI versus FOLFOX4 in the treatment of advanced colorectal cancer: a multicenter study of the Gruppo Oncologico Dell’Italia Meridionale. J Clin Oncol 2005;23:4866–75.
- Tournigand C, André T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol 2004;22:229–37.
- Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004;350:2335–42.
- Saltz LB, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 2008;26:2013–9.
- Cremolini C, Loupakis F, Ruzzo A, et al. Predictors of benefit in colorectal cancer treated with cetuximab: are we getting “Lost in TranslationAL”? J Clin Oncol 2010;28:e173–4.
- Sorich MJ, Wiese MD, Rowland D, et al. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol 2015;26:13–21.
- Grothey A, van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381(9863):303–12.
- Saltz LB, Niedzwiecki D, Hollis D, et al. Irinotecan fluorouracil plus leucovorin is not superior to fluorouracil plus leucovorin alone as adjuvant treatment for stage III colon cancer: results of CALGB 89803. J Clin Oncol 2007;25:3456–61.
- Van Cutsem E, et al. Randomized phase III trial comparing biweekly infusional fluorouracil/leucovorin alone or with irinotecan in the adjuvant treatment of stage III colon cancer: PETACC-3. J Clin Oncol 2009;27:3117–25.
- Allegra CJ, et al. Bevacizumab in stage II-III colon cancer: 5-year update of the National Surgical Adjuvant Breast and Bowel Project C-08 trial. J Clin Oncol 2013;31:359–64.
- de Gramont A, et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): a phase 3 randomised controlled trial. Lancet Oncol 2012;13:1225–33.
- Alberts SR, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA 2012;307:1383–93.
- Taieb J, et al. Oxaliplatin, fluorouracil, and leucovorin with or without cetuximab in patients with resected stage III colon cancer (PETACC-8): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:862–73.
- Shi Q, Sobrero AF, Shields AF, et al. Prospective pooled analysis of six phase III trials investigating duration of adjuvant (adjuvant) oxaliplatin-based therapy (3 vs 6 months) for patients (pts) with stage III colon cancer (CC): The IDEA (International Duration Evaluation of Adjuvant chemotherapy) collaboration. In: Proceedings from the American Society of Clinical Oncology; June 1–5, 2017; Chicago. Abstract LBA1.
- Quasar Collaborative Group; Gray R, Barnwell J, McConkey C, et al. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet 2007;370(9604):2020–9.
- Efficacy of adjuvant fluorouracil and folinic acid in B2 colon cancer. International Multicentre Pooled Analysis of B2 Colon Cancer Trials (IMPACT B2) Investigators. J Clin Oncol 1999;17:1356–63.
- Kidwell KM, et al. Long-term neurotoxicity effects of oxaliplatin added to fluorouracil and leucovorin as adjuvant therapy for colon cancer: results from National Surgical Adjuvant Breast and Bowel Project trials C-07 and LTS-01. Cancer 2012;118:5614–22.
- Beijers AJ, Mols F, Vreugdenhil G. A systematic review on chronic oxaliplatin-induced peripheral neuropathy and the relation with oxaliplatin administration. Support Care Cancer 2014;22:1999–2007.
- Mols F, Beijers T, Lemmens V, et al. Chemotherapy-induced neuropathy and its association with quality of life among 2- to 11-year colorectal cancer survivors: results from the population-based PROFILES registry. J Clin Oncol 2013;31:2699–707.
- Raphael MJ, Fischer HD, Fung K, et al. Neurotoxicity outcomes in a population-based cohort of elderly patients treated with adjuvant oxaliplatin for colorectal cancer. Clin Colorectal Cancer 2017 March 24.
- Toki MI, Saif MW, Syrigos KN. Hypersensitivity reactions associated with oxaliplatin and their clinical management. Expert Opin Drug Saf 2014;13:1545–54.
- Siu SW, Chan RT, Au GK. Hypersensitivity reactions to oxaliplatin: experience in a single institute. Ann Oncol 2006;17:259–61.
- Wong JT, Ling M, Patil S, et al. Oxaliplatin hypersensitivity: evaluation, implications of skin testing, and desensitization. J Allergy Clin Immunol Pract 2014;2:40–5.
- Benson AB 3rd, Venook AP, Cederquist L, et al. NCCN Guidelines Colon Cancer Version 2.2017. www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed May 8, 2017.
- Wolmark N, Rockette H, Mamounas E, et al. Clinical trial to assess the relative efficacy of fluorouracil and leucovorin, fluorouracil and levamisole, and fluorouracil, leucovorin, and levamisole in patients with Dukes’ B and C carcinoma of the colon: results from National Surgical Adjuvant Breast and Bowel Project C-04. J Clin Oncol 1999;17:3553–9.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67(1):7–30.
- United States Cancer Statistics. 1999–2013 incidence and mortality web-based report. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute, 2016. www.cdc.gov/uscs. Accessed June 12, 2017.
- Ahnen DJ, Wade SW, Jones WF, et al. The increasing incidence of young-onset colorectal cancer: a call to action. Mayo Clin Proc 2014;89:216–24.
- Jemal A, Fedewa SA, Anderson WF, et al. Colorectal cancer incidence patterns in the United States, 1974–2013. J Natl Cancer Inst 2017;109(8).
- Boursi B, Sella T, Liberman E, et al. The APC p.I1307K polymorphism is a significant risk factor for CRC in average risk Ashkenazi Jews. Eur J Cancer 2013;49:3680–5.
- Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011;60: 950–7.
- van Puijenbroek M, Nielsen M, Tops CM, et al. Identification of patients with (atypical) MUTYH-associated polyposis by KRAS2 c.34G > T prescreening followed by MUTYH hotspot analysis in formalin-fixed paraffin-embedded tissue. Clin Cancer Res 2008;14:139–42.
- Aretz S, Uhlhaas S, Goergens H, et al. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006;119:807–14.
- Tuohy TM, Rowe KG, Mineau GP, et al. Risk of colorectal cancer and adenomas in the families of patients with adenomas: a population-based study in Utah. Cancer 2014;120:35–42.
- Choi Y, Sateia HF, Peairs KS, Stewart RW. Screening for colorectal cancer. Semin Oncol 2017; 44:34–44.
- Atkin WS, Morson BC, Cuzick J. Long-term risk of colorectal cancer after excision of rectosigmoid adenomas. N Engl J Med 1992;326:658–62.
- Rutter MD. Surveillance programmes for neoplasia in colitis. J Gastroenterol 2011;46 Suppl 1:1–5.
- Giovannucci E. Modifiable risk factors for colon cancer. Gastroenterol Clin North Am 2002;31:925–43.
- Michels KB, Fuchs GS, Giovannucci E, et al. Fiber intake and incidence of colorectal cancer among 76,947 women and 47,279 men. Cancer Epidemiol Biomarkers Prev 2005;14:842–9.
- Omata F, Brown WR, Tokuda Y, et al. Modifiable risk factors for colorectal neoplasms and hyperplastic polyps. Intern Med 2009;48:123–8.
- Friedenreich CM, Neilson HK, Lynch BM. State of the epidemiological evidence on physical activity and cancer prevention. Eur J Cancer 2010;46:2593–604.
- Aleksandrova K, Pischon T, Jenab M, et al. Combined impact of healthy lifestyle factors on colorectal cancer: a large European cohort study. BMC Med 2014;12:168.
- Hermanek P, Wittekind C. The pathologist and the residual tumor (R) classification. Pathol Res Pract 1994;190:115–23.
- Lehnert T, Methner M, Pollok A, et al. Multivisceral resection for locally advanced primary colon and rectal cancer: an analysis of prognostic factors in 201 patients. Ann Surg 2002;235:217–25.
- Feinberg AE, et al. Oncologic outcomes following laparoscopic versus open resection of pT4 colon cancer: a systematic review and meta-analysis. Dis Colon Rectum 2017;60:116–125.
- Vignali A, et al. Laparoscopic treatment of advanced colonic cancer: a case-matched control with open surgery. Colorectal Dis 2013;15:944–8.
- Gainant A. Emergency management of acute colonic cancer obstruction. J Visc Surg 2012;149: e3–e10.
- Rosenman LD. Hartmann’s operation. Am J Surg 1994;168:283–4.
- Lee-Kong S, Lisle D. Surgical management of complicated colon cancer. Clin Colon Rectal Surg 2015;28:228–33.
- Bertelsen CA. Complete mesocolic excision an assessment of feasibility and outcome. Dan Med J 2017;64(2).
- Wolff WI SH. Definitive treatment of “malignant” polyps of the colon. Ann Surg 1975;182:516–25.
- Clinical Outcomes of Surgical Therapy Study Group, Nelson H, Sargent DJ, Wieand HS, et al. A comparison of laparoscopically assisted and open colectomy for colon cancer. N Engl J Med 2004;350:2050–9.
- Gunderson LL, Jessup JM, Sarjent DJ, et al. Revised tumor and node categorization for rectal cancer based on surveillance, epidemiology, and end results and rectal pooled analysis outcomes. J Clin Oncol 2010;28:256–63.
- Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41.
- Alves A, Panis Y, Mathieu P, et al. Postoperative mortality and morbidity in French patients undergoing colorectal surgery: results of a prospective multicenter study. Arch Surg 2005;140:278–83.
- Popescu RA, Norman A, Ross PJ, et al, Adjuvant or palliative chemotherapy for colorectal cancer in patients 70 years or older. J Clin Oncol 1999;17:2412–8.
- McCleary NJ, Meyerhardt JA, Green E, et al. Impact of age on the efficacy of newer adjuvant therapies in patients with stage II/III colon cancer: findings from the ACCENT database. J Clin Oncol 2013;31:2600–6.
- Tominaga T, Nonaka T, Sumida Y, et al. Effectiveness of adjuvant chemotherapy for elderly patients with lymph node-positive colorectal cancer. World J Surg Oncol 2016;14:197.
- Bos AC, van Erning FN, van Gestel YR, et al. Timing of adjuvant chemotherapy and its relation to survival among patients with stage III colon cancer. Eur J Cancer 2015;51:2553–61.
- Peixoto RD, Kumar A, Speers C, et al. Effect of delay in adjuvant oxaliplatin-based chemotherapy for stage III colon cancer. Clin Colorectal Cancer 2015;14:25–30.
- Compton CC, Fielding LP, Burgart LJ, et al. Prognostic factors in colorectal cancer. College of American Pathologists Consensus Statement 1999. Arch Pathol Lab Med 2000;124:979–94.
- Lieu CH, Lambert LA, Wolff RA, et al. Systemic chemotherapy and surgical cytoreduction for poorly differentiated and signet ring cell adenocarcinomas of the appendix. Ann Oncol 2012;23:652–8.
- Krasna MJ, Flancbaum L, Cody RP, et al. Vascular and neural invasion in colorectal carcinoma. Incidence and prognostic significance. Cancer 1988;61:1018–23.
- Cianchi F, Palomba A, Boddi V, et al. Lymph node recovery from colorectal tumor specimens: recommendation for a minimum number of lymph nodes to be examined. World J Surg 2002;26:384–9.
- Yoshimatsu K, et al. How many lymph nodes should be examined in Dukes’ B colorectal cancer? Determination on the basis of cumulative survival rate. Hepatogastroenterology 2005;52:1703–6.
- Caplin S, Cerottini JP, Bosman FT, et al. For patients with Dukes’ B (TNM Stage II) colorectal carcinoma, examination of six or fewer lymph nodes is related to poor prognosis. Cancer 1998;83:666–72.
- Veronese N, Nottegar A, Pea A, et al. Prognostic impact and implications of extracapsular lymph node involvement in colorectal cancer: a systematic review with meta-analysis. Ann Oncol 2016;27:42–8.
- Li J, Yang S, Hu J, et al. Tumor deposits counted as positive lymph nodes in TNM staging for advanced colorectal cancer: a retrospective multicenter study. Oncotarget 2016;7:18269–79.
- Venook A, Niedzwiecki D, Innocenti Fet al. Impact of primary (1º) tumor location on overall survival (OS) and progression-free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). J Clin Oncol 2016;34 no. 15 suppl. Abstract 3504.
- Schrag D, Brooks G, Meyerhardt JA ,et al. The relationship between primary tumor sidedness and prognosis in colorectal cancer. J Clin Oncol 2016;34 no. 15 suppl. Abstract 3505.
- Larrea AA, Lujan SA, Kunkel TA. SnapShot: DNA mismatch repair. Cell 2010;141:730 e1.
- Jass JR. Pathology of hereditary nonpolyposis colorectal cancer. Ann N Y Acad Sci 2000;910:62–73.
- Lynch HT, Smyrk T. Hereditary nonpolyposis colorectal cancer (Lynch syndrome). An updated review. Cancer 1996;78:1149–67.
- Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993;260:812–6.
- Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol 2017;12:24.
- Bupathi M, Wu C. Biomarkers for immune therapy in colorectal cancer: mismatch-repair deficiency and others. J Gastrointest Oncol 2016;7:713–20.
- Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 2005;23:609–18.
- Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med 2000;342:69–77.
- Ogino S, Kuchiba A, Qian ZR, et al. Prognostic significance and molecular associations of 18q loss of heterozygosity: a cohort study of microsatellite stable colorectal cancers. J Clin Oncol 2009; 27:4591–8.
- Kim ST, Lee J, Park SH, et al. The effect of DNA mismatch repair (MMR) status on oxaliplatin-based first-line chemotherapy as in recurrent or metastatic colon cancer. Med Oncol 2010;27:1277–85.
- Sargent DJ, Monges G, Thibodeau SN, et al. Therapy in colon cancer. J Clin Oncol 2010;28:4664.
- Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003;349:247–57.
- Hutchins G, Southward K, Handley K, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol 2011;29:1261–270.
- Yothers G, O’Connell MJ, Allegra CJ, et al. Oxaliplatin as adjuvant therapy for colon cancer: updated results of NSABP C-07 trial, including survival and subset analyses J Clin Oncol 2011;29:3768–74.
- Chang SC, Lin JK, Lin TC, Liang WY. Loss of heterozygosity: an independent prognostic factor of colorectal cancer. World J Gastroenterol 2005;11:778–84.
- Bertagnolli MM, Niedzwiecki D, Compton CC, et al. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: Cancer and Leukemia Group B Protocol 89803. J Clin Oncol 2009;27:1814–21.
- Bertagnolli MM, Redston M, Compton CC, et al. Microsatellite instability and loss of heterozygosity at chromosomal location 18q: prospective evaluation of biomarkers for stages II and III colon cancer--a study of CALGB 9581 and 89803. J Clin Oncol 2011;29:3153–62.
- Dalerba P, et al. CDX2 as a prognostic biomarker in stage II and stage III colon cancer. N Engl J Med 2016;374: 211–22.
- Clark-Langone KM, Wu JY, Sangli C, et al. Biomarker discovery for colon cancer using a 761 gene RT-PCR assay. BMC Genomics 2007;8:279.
- Gray RG, Quirke P, Handley K, et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol 2011;29:4611–9.
- Niedzwiecki D, Bertagnolli MM, Warren RS, et al. Documenting the natural history of patients with resected stage II adenocarcinoma of the colon after random assignment to adjuvant treatment with edrecolomab or observation: results from CALGB 9581. J Clin Oncol 2011;29:3146–52.
- Yothers G, O’Connell MJ, Lee M, et al. Validation of the 12-gene colon cancer recurrence score in NSABP C-07 as a predictor of recurrence in patients with stage II and III colon cancer treated with fluorouracil and leucovorin (FU/LV) and FU/LV plus oxaliplatin. J Clin Oncol 2013;31:4512–9.
- Gill S, Loprinzi CL, Sargent DJ, et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much? J Clin Oncol 2004;22:1797–806.
- Gill S, Loprinzi C, Kennecke H, et al. Prognostic web-based models for stage II and III colon cancer: A population and clinical trials-based validation of numeracy and adjuvant! online. Cancer 2011;117:4155–65.
- Jung M, Kim GW, Jung I, et al. Application of the Western-based adjuvant online model to Korean colon cancer patients; a single institution experience. BMC Cancer 2012;12:471.
- Papamichael D, Renfro LA, Matthaiou C, et al. Validity of Adjuvant! Online in older patients with stage III colon cancer based on 2967 patients from the ACCENT database. J Geriatr Oncol 2016;7:422–9.
- Tran B, Kopetz S, Tie J, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 2011;117:4623–32.
- Roth AD, Tejpar S, Delorenzi M, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol 2010;28:466–74.
- Lochhead P, Kuchiba A, Imamura Y, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst 2013;105:1151–6.
- Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 2007;67:2643–8.
- Therkildsen C, Bergmann TK, Henrichsen-Schnack T, et al. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol 2014;53:852–64.
- Taieb J, Le Malicot K, Shi Q, et al. Prognostic value of BRAF and KRAS mutations in MSI and MSS stage III colon cancer. J Natl Cancer Inst 2017;109(5).
- Palumbo LT, Sharpe WS, Henry JS. Cancer of the colon and rectum; analysis of 300 cases. Am J Surg 1965;109:439–44.
- Sharp GS, Benefiel WW. 5-Fluorouracil in the treatment of inoperable carcinoma of the colon and rectum. Cancer Chemother Rep 1962;20:97–101.
- Lawrence W Jr, Terz JJ, Horsley JS 3rd, et al. Chemotherapy as an adjuvant to surgery for colorectal cancer. Ann Surg 1975;181:616–23.
- Grage TD, et al. Adjuvant chemotherapy with 5-fluorouracil after surgical resection of colorectal carcinoma (COG protocol 7041). A preliminary report. Am J Surg 1977;133:59–66.
- Wolmark N, Fisher B, Rockette H, et al. Postoperative adjuvant chemotherapy or BCG for colon cancer: results from NSABP protocol C-01. J Natl Cancer Inst 1988;80:30–6.
- Moertel CG, Fleming TR, Macdonald JS, et al. Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma. N Engl J Med 1990;322:352–8.
- Wolmark N, Rockette H, Fisher B, et al. The benefit of leucovorin-modulated fluorouracil as postoperative adjuvant therapy for primary colon cancer: results from National Surgical Adjuvant Breast and Bowel Project protocol C-03. J Clin Oncol 1993;11:1879–87.
- Comparison of fluorouracil with additional levamisole, higher-dose folinic acid, or both, as adjuvant chemotherapy for colorectal cancer: a randomised trial. QUASAR Collaborative Group. Lancet 2000;355(9215):1588–96.
- Chen TC, Hinton DR, Leichman L, et al. Multifocal inflammatory leukoencephalopathy associated with levamisole and 5-fluorouracil: case report. Neurosurgery 1994;35:1138-42.
- Porschen R, Bermann A, Löffler T, et al. Fluorouracil plus leucovorin as effective adjuvant chemotherapy in curatively resected stage III colon cancer: results of the trial adjCCA-01. J Clin Oncol 2001;19:1787–94.
- Arkenau HT, Bermann A, Rettig K, et al. 5-Fluorouracil plus leucovorin is an effective adjuvant chemotherapy in curatively resected stage III colon cancer: long-term follow-up results of the adjCCA-01 trial. Ann Oncol 2003;14:395–9.
- Weinerman B, Shah A, Fields A, et al. Systemic infusion versus bolus chemotherapy with 5-fluorouracil in measurable metastatic colorectal cancer. Am J Clin Oncol 1992;15:518–23.
- Poplin EA, Benedetti JK, Estes NC, et al. Phase III Southwest Oncology Group 9415/Intergroup 0153 randomized trial of fluorouracil, leucovorin, and levamisole versus fluorouracil continuous infusion and levamisole for adjuvant treatment of stage III and high-risk stage II colon cancer. J Clin Oncol 2005;23:1819–25.
- Twelves C, Wong A, Nowacki MP, et al. Capecitabine as adjuvant treatment for stage III colon cancer. N Engl J Med 2005;352:2696–704.
- de Gramont A, Vignoud J, Tournigand C, et al. Oxaliplatin with high-dose leucovorin and 5-fluorouracil 48-hour continuous infusion in pretreated metastatic colorectal cancer. Eur J Cancer 1997;33:214–9.
- Diaz-Rubio E, Sastre J, Zaniboni A, et al. Oxaliplatin as single agent in previously untreated colorectal carcinoma patients: a phase II multicentric study. Ann Oncol 1998;9:105–8.
- André T, de Gramont A, Vernerey D, et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF mutation and mismatch repair status of the MOSAIC Study. J Clin Oncol 2015;33:4176–87.
- Andre T, Boni C, Mounedji-Boudiaf L, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med 2004;350:2343–51.
- Kuebler JP, Wieand HS, O’Connell MJ, et al. Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C-07. J Clin Oncol 2007;25:2198–204.
- Haller DG, Tabernero J, Maroun J, et al. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. J Clin Oncol 2011;29:1465–71.
- Schmoll HJ, et al. Capecitabine plus oxaliplatin compared with fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results of the NO16968 randomized controlled phase III trial. J Clin Oncol 2015;33:3733–40.
- Colucci G, Gebbia V, Paoletti G, et al. Phase III randomized trial of FOLFIRI versus FOLFOX4 in the treatment of advanced colorectal cancer: a multicenter study of the Gruppo Oncologico Dell’Italia Meridionale. J Clin Oncol 2005;23:4866–75.
- Tournigand C, André T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol 2004;22:229–37.
- Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004;350:2335–42.
- Saltz LB, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 2008;26:2013–9.
- Cremolini C, Loupakis F, Ruzzo A, et al. Predictors of benefit in colorectal cancer treated with cetuximab: are we getting “Lost in TranslationAL”? J Clin Oncol 2010;28:e173–4.
- Sorich MJ, Wiese MD, Rowland D, et al. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol 2015;26:13–21.
- Grothey A, van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381(9863):303–12.
- Saltz LB, Niedzwiecki D, Hollis D, et al. Irinotecan fluorouracil plus leucovorin is not superior to fluorouracil plus leucovorin alone as adjuvant treatment for stage III colon cancer: results of CALGB 89803. J Clin Oncol 2007;25:3456–61.
- Van Cutsem E, et al. Randomized phase III trial comparing biweekly infusional fluorouracil/leucovorin alone or with irinotecan in the adjuvant treatment of stage III colon cancer: PETACC-3. J Clin Oncol 2009;27:3117–25.
- Allegra CJ, et al. Bevacizumab in stage II-III colon cancer: 5-year update of the National Surgical Adjuvant Breast and Bowel Project C-08 trial. J Clin Oncol 2013;31:359–64.
- de Gramont A, et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): a phase 3 randomised controlled trial. Lancet Oncol 2012;13:1225–33.
- Alberts SR, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA 2012;307:1383–93.
- Taieb J, et al. Oxaliplatin, fluorouracil, and leucovorin with or without cetuximab in patients with resected stage III colon cancer (PETACC-8): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:862–73.
- Shi Q, Sobrero AF, Shields AF, et al. Prospective pooled analysis of six phase III trials investigating duration of adjuvant (adjuvant) oxaliplatin-based therapy (3 vs 6 months) for patients (pts) with stage III colon cancer (CC): The IDEA (International Duration Evaluation of Adjuvant chemotherapy) collaboration. In: Proceedings from the American Society of Clinical Oncology; June 1–5, 2017; Chicago. Abstract LBA1.
- Quasar Collaborative Group; Gray R, Barnwell J, McConkey C, et al. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet 2007;370(9604):2020–9.
- Efficacy of adjuvant fluorouracil and folinic acid in B2 colon cancer. International Multicentre Pooled Analysis of B2 Colon Cancer Trials (IMPACT B2) Investigators. J Clin Oncol 1999;17:1356–63.
- Kidwell KM, et al. Long-term neurotoxicity effects of oxaliplatin added to fluorouracil and leucovorin as adjuvant therapy for colon cancer: results from National Surgical Adjuvant Breast and Bowel Project trials C-07 and LTS-01. Cancer 2012;118:5614–22.
- Beijers AJ, Mols F, Vreugdenhil G. A systematic review on chronic oxaliplatin-induced peripheral neuropathy and the relation with oxaliplatin administration. Support Care Cancer 2014;22:1999–2007.
- Mols F, Beijers T, Lemmens V, et al. Chemotherapy-induced neuropathy and its association with quality of life among 2- to 11-year colorectal cancer survivors: results from the population-based PROFILES registry. J Clin Oncol 2013;31:2699–707.
- Raphael MJ, Fischer HD, Fung K, et al. Neurotoxicity outcomes in a population-based cohort of elderly patients treated with adjuvant oxaliplatin for colorectal cancer. Clin Colorectal Cancer 2017 March 24.
- Toki MI, Saif MW, Syrigos KN. Hypersensitivity reactions associated with oxaliplatin and their clinical management. Expert Opin Drug Saf 2014;13:1545–54.
- Siu SW, Chan RT, Au GK. Hypersensitivity reactions to oxaliplatin: experience in a single institute. Ann Oncol 2006;17:259–61.
- Wong JT, Ling M, Patil S, et al. Oxaliplatin hypersensitivity: evaluation, implications of skin testing, and desensitization. J Allergy Clin Immunol Pract 2014;2:40–5.
- Benson AB 3rd, Venook AP, Cederquist L, et al. NCCN Guidelines Colon Cancer Version 2.2017. www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed May 8, 2017.
- Wolmark N, Rockette H, Mamounas E, et al. Clinical trial to assess the relative efficacy of fluorouracil and leucovorin, fluorouracil and levamisole, and fluorouracil, leucovorin, and levamisole in patients with Dukes’ B and C carcinoma of the colon: results from National Surgical Adjuvant Breast and Bowel Project C-04. J Clin Oncol 1999;17:3553–9.
Is there a doctor on board? In-flight medical emergencies
It could happen. You are on a plane, perhaps on your way to a medical conference or a well-deserved vacation, when the flight attendant asks you to help a passenger experiencing an in-flight medical emergency. What is your role in this situation?
FLIGHT ATTENDANTS USED TO BE NURSES
Before World War II, nearly all American flight attendants were nurses, who could address most medical issues that arose during flights.1 Airlines eliminated this preferential hiring practice to support the war effort. Traveling healthcare providers thereafter often volunteered to assist when in-flight medical issues arose, but aircraft carried minimal medical equipment and volunteers’ liability was uncertain.
In 1998, Congress passed the Aviation Medical Assistance Act (AMAA), which provides liability protection for on-board healthcare providers who render medical assistance. It also required the Federal Aviation Administration (FAA) to improve its standards for in-flight medical equipment.2,3
HOW OFTEN DO EMERGENCIES ARISE?
How often medical events occur during flight is difficult to estimate because airlines are not mandated to report such issues.4 Based on data from a ground-based communications center that provides medical consultation service to airlines, medical events occur in approximately 1 in every 604 flights.5 This is likely an underestimate, as many medical events may be handled on board without involving a ground-based consultation center.
The most common emergencies are syncope or presyncope, representing 37.4% of consultations, followed by respiratory symptoms (12.1%), nausea or vomiting (9.5%), cardiac symptoms (7.7%), seizures (5.8%), and abdominal pain (4.1%).5 Very few in-flight medical emergencies progress to death; the reported mortality rate is 0.3%.5
CABIN PRESSURES ARE RELATIVELY LOW
The cabins of commercial airliners are pressurized, but the pressure is still lower than on the ground. The cabin pressure in flight is equivalent to that at an altitude of 6,000 to 8,000 feet,6,7 ie, about 23 or 24 mm Hg, compared with about 30 mm Hg at sea level. At this pressure, passengers have a partial pressure of arterial oxygen (Pao2) of 60 mm Hg (normal at sea level is > 80).8
This reduced oxygen pressure is typically not clinically meaningful in healthy people. However, people with underlying pulmonary or cardiac illness may be starting further to the left on the oxygen dissociation curve before gaining altitude, putting them at risk for acute exacerbations of underlying medical conditions. Many patients who rely on supplemental oxygen, such as those with chronic obstructive pulmonary disease, are advised to increase their oxygen support during flight.9
Boyle’s law says that the volume of a gas is inversely proportional to its pressure. As the pressure drops in the cabin after takeoff, air trapped in an enclosed space—eg, in some patient’s bodies—can increase in volume up to 30%,10 which can have medical ramifications. Clinically significant pneumothorax during flight has been reported.11–13 Partially because of these volumetric changes, patients who have undergone abdominal surgery are advised to avoid flying for at least 2 weeks after their procedure.10,14 Patients who have had recent ocular or intracranial surgery may also be at risk of in-flight complications.15
IN-FLIGHT MEDICAL RESOURCES
The limited medical supplies available on aircraft often challenge healthcare providers who offer to respond to in-flight medical events. However, several important medical resources are available.
Medical kits and defibrillators

FAA regulations require airlines based in the United States to carry basic first aid supplies such as bandages and splints.3 Airlines are also required to carry a medical kit containing the items listed in Table 1.
The FAA-mandated kit does not cover every circumstance that may arise. Although in-flight pediatric events occasionally occur,16 many of the available medications are inappropriate for young children. The FAA does not require sedative or antipsychotic agents, which could be useful for passengers who have acute psychiatric episodes. Obstetric supplies are absent. On international carriers, the contents of medical kits are highly variable,17 as are the names used for some medications.
The FAA requires at least 1 automated external defibrillator (AED) to be available on each commercial aircraft.3 The timely use of AEDs greatly improves survivability after out-of-hospital cardiac arrest.18,19 One study involving a major US airline found a 40% survival rate to hospital discharge in patients who received in-flight defibrillation.20 Without this intervention, very few of the patients would have been expected to survive. In addition to being clinically effective, placing AEDs aboard commercial aircraft is a cost-effective public health intervention.21
Consultation services
Most major airlines can contact ground-based medical consultation services during flight.10 These centers are staffed with healthcare providers who can provide flight crews with advice on how to handle medical events in real time. Healthcare providers can likewise discuss specific medical issues with these services if they respond to an in-flight medical event. Ground-based call centers can also communicate with prehospital providers should a flight need to be diverted.
Other on-board providers
Some medical events require the involvement of more than one medical provider. Other physicians, nurses, and prehospital providers are often also on board.22 Responding physicians can also request the assistance of these other healthcare providers. Flight attendants in the United States are required to be trained in cardiopulmonary resuscitation (CPR).23
Flight diversion
Critically ill patients or those with time-sensitive medical emergencies may require the aircraft to divert from its intended destination. As may be expected, medical emergencies suspected to involve the cardiovascular, neurologic, or respiratory system have been shown to most likely result in aircraft diversion.5,24 Approximately 7% of in-flight medical events in which a ground-based medical consultation service is contacted result in diversion.5
While an on-board responding physician can make a recommendation to divert based on the patient’s acute medical status, only the captain can make the ultimate decision.4 On-board healthcare providers should clearly state that a patient might benefit from an unscheduled landing if that is truly their assessment. In addition to communicating their clinical concerns with the flight crew, the responding physician may also be able to discuss the situation with the airline’s ground-based consultation service. On-board physicians can make important contributions to the assessment of illness severity and triage decisions.
MEDICOLEGAL ISSUES
No legal duty to assist
US healthcare providers are not legally required to respond to on-board medical emergencies on US-based airlines. Canada and the United Kingdom also do not require providers to render assistance. But the General Medical Council (the regulatory body for UK doctors) states that doctors have an ethical duty to respond in the event of a medical emergency, including one on board an aircraft. Other countries, notably Australia and some in the European Union, require healthcare professionals to respond to on-board medical emergencies.10
Regardless of potential legal duties to assist, healthcare providers are arguably ethically obliged to render assistance if they can.
Aviation Medical Assistance Act
The extent of an American healthcare provider’s liability risk for assisting in a medical emergency on a plane registered in the United States is limited by statute. The 1998 AMAA provides liability protection for on-board medical providers who are asked to assist during an in-flight medical emergency. This statute covers all US-certified air carriers on domestic flights and would likely be held to apply to US aircraft in foreign airspace because of the general rule that the law of the country where the air carrier is registered applies to in-flight events.
Under the AMAA, providers asked to assist with in-flight medical emergencies are not liable for malpractice as long as their actions are not “grossly” negligent or intended to cause the patient harm.25 This is distinguishable from a standard malpractice liability scenario, in which the plaintiff only needs to show ordinary negligence. In a traditional healthcare setting, a provider has to act within the “standard of care” when assessing and treating a patient. If the provider deviates from the standard of care, such as by making an error in judgment or diagnosis, the provider is legally negligent. Under traditional malpractice law, even if a provider is minimally negligent, he or she is liable for any damages resulting from that negligence. Under a gross negligence standard, providers are protected from liability unless they demonstrate flagrant disregard for the patient’s health and safety.
Postflight issues
A provider who undertakes care should continue to provide care until it is no longer necessary, either because the patient recovers or the responsibility has been transferred to another provider. At the point of transfer, the healthcare provider’s relationship with the patient terminates.
The provider should document the encounter, typically using airline-specific documentation. The responding physician needs to be mindful of the patient’s privacy, refraining from discussing the event with others without the patient’s authorization.26
SUGGESTED RESPONSE
Healthcare providers who wish to respond to in-flight medical emergencies must first determine if they are sufficiently capable of providing care. During a flight, providers do not expect to be on duty and so may have consumed alcoholic beverages to an extent that would potentially render them unsuitable to respond. When it is appropriate to become involved in a medical emergency during flight, the healthcare provider should state his or her qualifications to the passenger and to flight personnel.
If circumstances allow, the volunteer provider should obtain the patient’s consent for evaluation and treatment.10 Additionally, with the multilingual nature of commercial air travel, especially on international flights, the provider may need to enlist a translator’s assistance.
Providers may find it preferable to treat passengers in their seats.27 Given the confined space in an aircraft, keeping ill passengers out of the aisle allows others to move about the cabin. If it becomes necessary to move the patient, a location should be sought that minimally interferes with other passengers’ needs.
If a passenger has critical medical needs, in-flight medical volunteers can recommend flight diversion, which should also be discussed with ground-based medical staff. However, as emphasized earlier, the captain makes the ultimate decision to divert, taking into account other operational factors that affect the safety of the aircraft and its occupants. In-flight medical care providers should perform only the treatments they are qualified to provide and should operate within their scope of training.
After the aircraft lands, if the passenger must be transported to a hospital, providers should supply prehospital personnel with a requisite transfer-of-care communication. In-flight medical providers who have performed a significant medical intervention might find it appropriate to accompany the patient to the hospital.
SPECIFIC CONDITIONS
The list of possible acute medical issues that occur aboard aircraft is extensive. Here are a few of them.
Trauma
Passengers may experience injuries during flight, for example during periods of heavy air turbulence. Responding physicians should assess for potential life-threatening injuries, keeping in mind that some passengers may be at higher risk. For example, if a passenger on anticoagulation experiences a blunt head injury, this would raise suspicion for possible intracranial hemorrhage, and frequent reassessment of neurologic status may be necessary. If an extremity fracture is suspected, the physician should splint the affected limb. Analgesia may be provided from the medical kit, if appropriate.
Gastrointestinal issues
Acute gastrointestinal issues such as nausea and vomiting are often reported to ground-based medical consultation services.5 Responding on-board providers must consider if the passenger is simply experiencing gastrointestinal upset from a benign condition such as gastroenteritis or has a more serious condition. For some patients, vomiting may be a symptom of a myocardial infarction.28 Bilious emesis with abdominal distention may be associated with small-bowel obstruction. While antiemetics are not included in the FAA-mandated medical kit, providers can initiate intravenous fluid therapy for passengers who show signs of hypovolemia.
Cardiac arrest
Although cardiac arrest during flight is rare,5 medical providers should nonetheless be prepared to handle it. Upon recognition of cardiac arrest, the provider should immediately begin cardiopulmonary resuscitation and use the on-board AED to defibrillate a potentially shockable rhythm. Flight attendants are trained in cardiopulmonary resuscitation and therefore may assist with resuscitation efforts. If the patient is resuscitated, the responding physician should recommend diversion of the flight.
Anaphylaxis
In the event of a severe life-threatening allergic reaction, the FAA-mandated emergency medical kit contains both diphenhydramine and epinephrine. For an adult experiencing anaphylaxis, a responding on-board physician can administer diphenhydramine 50 mg and epinephrine 0.3 mg (using the 1:1000 formulation), both intramuscularly. For patients with bronchospasm, a metered-dose inhaler of albuterol can be given. As anaphylaxis is an acute and potentially lethal condition, diversion of the aircraft would also be appropriate.29
Myocardial infarction
When acute myocardial infarction is suspected, it is appropriate for the provider to give aspirin, with important exceptions for patients who are experiencing an acute hemorrhage or who have an aspirin allergy.30 Supplemental oxygen should likewise be provided if the responding physician suspects compromised oxygenation. As acute myocardial infarction is also a time-sensitive condition, the clinician who suspects this diagnosis should recommend diversion of the aircraft.
Acute psychiatric issues
While approximately 2.4% of on-board medical events are attributed to psychiatric issues,5 there are few tools at the clinician’s disposal in the FAA-mandated emergency medical kit. Antipsychotics and sedatives are not included. The responding physician may need to attempt verbal de-escalation of aggressive behavior. If the safety of the flight is compromised, the application of improvised physical restraints may be appropriate.
Altered mental status
The differential diagnosis for altered mental status is extensive. The on-board physician should try to identify reversible and potentially lethal conditions and determine the potential need for aircraft diversion.
If possible, a blood sugar level should be measured (although the FAA-mandated kit does not contain a glucometer). It may be appropriate to empirically give intravenous dextrose to patients strongly suspected of having hypoglycemia.
If respiratory or cerebrovascular compromise is suspected, supplemental oxygen should be provided.
Unless a reversible cause of altered mental status is identified and treated successfully, it will likely be appropriate to recommend diversion of the aircraft.
Acknowledgment: The authors acknowledge Linda J. Kesselring, MS, ELS, the technical editor/writer in the Department of Emergency Medicine University of Maryland School of Medicine, for her contributions as copy editor of a previous version of this manuscript.
- Gazdik M. Vault guide to flight attendant careers. New York, NY: Vault, Inc.; 2005.
- Stewart PH, Agin WS, Douglas SP. What does the law say to Good Samaritans? A review of Good Samaritan statutes in 50 states and on US airlines. Chest 2013; 143:1774–1783.
- Federal Aviation Administration (FAA), DOT. Emergency medical equipment. Final rule. Fed Regist 2001; 66:19028–19046.
- Goodwin T. In-flight medical emergencies: an overview. BMJ 2000; 321:1338–1341.
- Peterson DC, Martin-Gill C, Guyette FX, et al. Outcomes of medical emergencies on commercial airline flights. N Engl J Med 2013; 368:2075–2083.
- Aerospace Medical Association, Aviation Safety Committee, Civil Aviation Subcommittee. Cabin cruising altitudes for regular transport aircraft. Aviat Space Environ Med 2008; 79:433–439.
- Cottrell JJ. Altitude exposures during aircraft flight. Flying higher. Chest 1988; 93:81–84.
- Humphreys S, Deyermond R, Bali I, Stevenson M, Fee JP. The effect of high altitude commercial air travel on oxygen saturation. Anaesthesia 2005; 60:458–460.
- Shrikrishna D, Coker RK; Air Travel Working Party of the British Thoracic Society Standards of Care Committee. Managing passengers with stable respiratory disease planning air travel: British Thoracic Society recommendations. Thorax 2011; 66:831–833.
- Gendreau MA, DeJohn C. Responding to medical events during commercial airline flights. N Engl J Med 2002; 346:1067–1073.
- Hu X, Cowl CT, Baqir M, Ryu JH. Air travel and pneumothorax. Chest 2014; 145:688–694.
- Madan K, Vishwanath G, Singh N. In-flight spontaneous pneumothorax: congenital cystic adenomatoid malformation of the lung. Respiration 2012; 83:554–558.
- Wallace TW, Wong T, O’Bichere A, Ellis BW. Managing in flight emergencies. BMJ 1995; 311:374–376.
- Medical aspects of transportation aboard commercial aircraft. AMA commission on emergency medical services. JAMA 1982; 247:1007–1011.
- Mills MD, Devenyi RG, Lam WC, Berger AR, Beijer CD, Lam SR. An assessment of intraocular pressure rise in patients with gas-filled eyes during simulated air flight. Ophthalmology 2001; 108:40–44.
- Moore BR, Ping JM, Claypool DW. Pediatric emergencies on a US-based commercial airline. Pediatr Emerg Care 2005; 21:725–729.
- Sand M, Gambichler T, Sand D, Thrandorf C, Altmeyer P, Bechara FG. Emergency medical kits on board commercial aircraft: a comparative study. Travel Med Infect Dis 2010; 8:388–394.
- Auble TE, Menegazzi JJ, Paris PM. Effect of out-of-hospital defibrillation by basic life support providers on cardiac arrest mortality: a metaanalysis. Ann Emerg Med 1995; 25:642–648.
- Marenco JP, Wang PJ, Link MS, Homoud MK, Estes NA. Improving survival from sudden cardiac arrest: the role of the automated external defibrillator. JAMA 2001; 285:1193–1200.
- Page RL, Joglar JA, Kowal RC, et al. Use of automated external defibrillators by a US airline. N Engl J Med 2000; 343:1210–1216.
- Groeneveld PW, Kwong JL, Liu Y, et al. Cost-effectiveness of automated external defibrillators on airlines. JAMA 2001; 286:1482–1489.
- Baltsezak S. Clinic in the air? A retrospective study of medical emergency calls from a major international airline. J Travel Med 2008; 15:391–394.
- Federal Aviation Administration (FAA). Advisory circular: emergency medical equipment training AC 121-34B. www.faa.gov/documentLibrary/media/Advisory_Circular/AC121-34B.pdf. Accessed April 6, 2017.
- Cummins RO, Schubach JA. Medical emergencies among commercial air travelers. JAMA 1989; 261:1295–1299.
- US Government Publishing Office. Public Law 105-170. Aviation Medical Assistance Act of 1998.
- US Government Publishing Office. Public Law 104-191. Health Insurance Portability and Accountability Act of 1996.
- Chandra A, Conry S. In-flight medical emergencies. West J Emerg Med 2013; 14:499–504.
- Kirchberger I, Meisinger C, Heier M, et al. Patient-reported symptoms in acute myocardial infarction: differences related to ST-segment elevation: the MONICA/KORA Myocardial Infarction Registry. J Intern Med 2011; 270:58–64.
- Brady WJ Jr, Bright HL. Occurrence of multiphasic anaphylaxis during a transcontinental air flight. Am J Emerg Med 1999; 17:695–696.
- O’Connor RE, Brady W, Brooks SC, et al. Part 10: acute coronary syndromes: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2010; 122:(suppl 3):S787–S817.
It could happen. You are on a plane, perhaps on your way to a medical conference or a well-deserved vacation, when the flight attendant asks you to help a passenger experiencing an in-flight medical emergency. What is your role in this situation?
FLIGHT ATTENDANTS USED TO BE NURSES
Before World War II, nearly all American flight attendants were nurses, who could address most medical issues that arose during flights.1 Airlines eliminated this preferential hiring practice to support the war effort. Traveling healthcare providers thereafter often volunteered to assist when in-flight medical issues arose, but aircraft carried minimal medical equipment and volunteers’ liability was uncertain.
In 1998, Congress passed the Aviation Medical Assistance Act (AMAA), which provides liability protection for on-board healthcare providers who render medical assistance. It also required the Federal Aviation Administration (FAA) to improve its standards for in-flight medical equipment.2,3
HOW OFTEN DO EMERGENCIES ARISE?
How often medical events occur during flight is difficult to estimate because airlines are not mandated to report such issues.4 Based on data from a ground-based communications center that provides medical consultation service to airlines, medical events occur in approximately 1 in every 604 flights.5 This is likely an underestimate, as many medical events may be handled on board without involving a ground-based consultation center.
The most common emergencies are syncope or presyncope, representing 37.4% of consultations, followed by respiratory symptoms (12.1%), nausea or vomiting (9.5%), cardiac symptoms (7.7%), seizures (5.8%), and abdominal pain (4.1%).5 Very few in-flight medical emergencies progress to death; the reported mortality rate is 0.3%.5
CABIN PRESSURES ARE RELATIVELY LOW
The cabins of commercial airliners are pressurized, but the pressure is still lower than on the ground. The cabin pressure in flight is equivalent to that at an altitude of 6,000 to 8,000 feet,6,7 ie, about 23 or 24 mm Hg, compared with about 30 mm Hg at sea level. At this pressure, passengers have a partial pressure of arterial oxygen (Pao2) of 60 mm Hg (normal at sea level is > 80).8
This reduced oxygen pressure is typically not clinically meaningful in healthy people. However, people with underlying pulmonary or cardiac illness may be starting further to the left on the oxygen dissociation curve before gaining altitude, putting them at risk for acute exacerbations of underlying medical conditions. Many patients who rely on supplemental oxygen, such as those with chronic obstructive pulmonary disease, are advised to increase their oxygen support during flight.9
Boyle’s law says that the volume of a gas is inversely proportional to its pressure. As the pressure drops in the cabin after takeoff, air trapped in an enclosed space—eg, in some patient’s bodies—can increase in volume up to 30%,10 which can have medical ramifications. Clinically significant pneumothorax during flight has been reported.11–13 Partially because of these volumetric changes, patients who have undergone abdominal surgery are advised to avoid flying for at least 2 weeks after their procedure.10,14 Patients who have had recent ocular or intracranial surgery may also be at risk of in-flight complications.15
IN-FLIGHT MEDICAL RESOURCES
The limited medical supplies available on aircraft often challenge healthcare providers who offer to respond to in-flight medical events. However, several important medical resources are available.
Medical kits and defibrillators
FAA regulations require airlines based in the United States to carry basic first aid supplies such as bandages and splints.3 Airlines are also required to carry a medical kit containing the items listed in Table 1.
The FAA-mandated kit does not cover every circumstance that may arise. Although in-flight pediatric events occasionally occur,16 many of the available medications are inappropriate for young children. The FAA does not require sedative or antipsychotic agents, which could be useful for passengers who have acute psychiatric episodes. Obstetric supplies are absent. On international carriers, the contents of medical kits are highly variable,17 as are the names used for some medications.
The FAA requires at least 1 automated external defibrillator (AED) to be available on each commercial aircraft.3 The timely use of AEDs greatly improves survivability after out-of-hospital cardiac arrest.18,19 One study involving a major US airline found a 40% survival rate to hospital discharge in patients who received in-flight defibrillation.20 Without this intervention, very few of the patients would have been expected to survive. In addition to being clinically effective, placing AEDs aboard commercial aircraft is a cost-effective public health intervention.21
Consultation services
Most major airlines can contact ground-based medical consultation services during flight.10 These centers are staffed with healthcare providers who can provide flight crews with advice on how to handle medical events in real time. Healthcare providers can likewise discuss specific medical issues with these services if they respond to an in-flight medical event. Ground-based call centers can also communicate with prehospital providers should a flight need to be diverted.
Other on-board providers
Some medical events require the involvement of more than one medical provider. Other physicians, nurses, and prehospital providers are often also on board.22 Responding physicians can also request the assistance of these other healthcare providers. Flight attendants in the United States are required to be trained in cardiopulmonary resuscitation (CPR).23
Flight diversion
Critically ill patients or those with time-sensitive medical emergencies may require the aircraft to divert from its intended destination. As may be expected, medical emergencies suspected to involve the cardiovascular, neurologic, or respiratory system have been shown to most likely result in aircraft diversion.5,24 Approximately 7% of in-flight medical events in which a ground-based medical consultation service is contacted result in diversion.5
While an on-board responding physician can make a recommendation to divert based on the patient’s acute medical status, only the captain can make the ultimate decision.4 On-board healthcare providers should clearly state that a patient might benefit from an unscheduled landing if that is truly their assessment. In addition to communicating their clinical concerns with the flight crew, the responding physician may also be able to discuss the situation with the airline’s ground-based consultation service. On-board physicians can make important contributions to the assessment of illness severity and triage decisions.
MEDICOLEGAL ISSUES
No legal duty to assist
US healthcare providers are not legally required to respond to on-board medical emergencies on US-based airlines. Canada and the United Kingdom also do not require providers to render assistance. But the General Medical Council (the regulatory body for UK doctors) states that doctors have an ethical duty to respond in the event of a medical emergency, including one on board an aircraft. Other countries, notably Australia and some in the European Union, require healthcare professionals to respond to on-board medical emergencies.10
Regardless of potential legal duties to assist, healthcare providers are arguably ethically obliged to render assistance if they can.
Aviation Medical Assistance Act
The extent of an American healthcare provider’s liability risk for assisting in a medical emergency on a plane registered in the United States is limited by statute. The 1998 AMAA provides liability protection for on-board medical providers who are asked to assist during an in-flight medical emergency. This statute covers all US-certified air carriers on domestic flights and would likely be held to apply to US aircraft in foreign airspace because of the general rule that the law of the country where the air carrier is registered applies to in-flight events.
Under the AMAA, providers asked to assist with in-flight medical emergencies are not liable for malpractice as long as their actions are not “grossly” negligent or intended to cause the patient harm.25 This is distinguishable from a standard malpractice liability scenario, in which the plaintiff only needs to show ordinary negligence. In a traditional healthcare setting, a provider has to act within the “standard of care” when assessing and treating a patient. If the provider deviates from the standard of care, such as by making an error in judgment or diagnosis, the provider is legally negligent. Under traditional malpractice law, even if a provider is minimally negligent, he or she is liable for any damages resulting from that negligence. Under a gross negligence standard, providers are protected from liability unless they demonstrate flagrant disregard for the patient’s health and safety.
Postflight issues
A provider who undertakes care should continue to provide care until it is no longer necessary, either because the patient recovers or the responsibility has been transferred to another provider. At the point of transfer, the healthcare provider’s relationship with the patient terminates.
The provider should document the encounter, typically using airline-specific documentation. The responding physician needs to be mindful of the patient’s privacy, refraining from discussing the event with others without the patient’s authorization.26
SUGGESTED RESPONSE
Healthcare providers who wish to respond to in-flight medical emergencies must first determine if they are sufficiently capable of providing care. During a flight, providers do not expect to be on duty and so may have consumed alcoholic beverages to an extent that would potentially render them unsuitable to respond. When it is appropriate to become involved in a medical emergency during flight, the healthcare provider should state his or her qualifications to the passenger and to flight personnel.
If circumstances allow, the volunteer provider should obtain the patient’s consent for evaluation and treatment.10 Additionally, with the multilingual nature of commercial air travel, especially on international flights, the provider may need to enlist a translator’s assistance.
Providers may find it preferable to treat passengers in their seats.27 Given the confined space in an aircraft, keeping ill passengers out of the aisle allows others to move about the cabin. If it becomes necessary to move the patient, a location should be sought that minimally interferes with other passengers’ needs.
If a passenger has critical medical needs, in-flight medical volunteers can recommend flight diversion, which should also be discussed with ground-based medical staff. However, as emphasized earlier, the captain makes the ultimate decision to divert, taking into account other operational factors that affect the safety of the aircraft and its occupants. In-flight medical care providers should perform only the treatments they are qualified to provide and should operate within their scope of training.
After the aircraft lands, if the passenger must be transported to a hospital, providers should supply prehospital personnel with a requisite transfer-of-care communication. In-flight medical providers who have performed a significant medical intervention might find it appropriate to accompany the patient to the hospital.
SPECIFIC CONDITIONS
The list of possible acute medical issues that occur aboard aircraft is extensive. Here are a few of them.
Trauma
Passengers may experience injuries during flight, for example during periods of heavy air turbulence. Responding physicians should assess for potential life-threatening injuries, keeping in mind that some passengers may be at higher risk. For example, if a passenger on anticoagulation experiences a blunt head injury, this would raise suspicion for possible intracranial hemorrhage, and frequent reassessment of neurologic status may be necessary. If an extremity fracture is suspected, the physician should splint the affected limb. Analgesia may be provided from the medical kit, if appropriate.
Gastrointestinal issues
Acute gastrointestinal issues such as nausea and vomiting are often reported to ground-based medical consultation services.5 Responding on-board providers must consider if the passenger is simply experiencing gastrointestinal upset from a benign condition such as gastroenteritis or has a more serious condition. For some patients, vomiting may be a symptom of a myocardial infarction.28 Bilious emesis with abdominal distention may be associated with small-bowel obstruction. While antiemetics are not included in the FAA-mandated medical kit, providers can initiate intravenous fluid therapy for passengers who show signs of hypovolemia.
Cardiac arrest
Although cardiac arrest during flight is rare,5 medical providers should nonetheless be prepared to handle it. Upon recognition of cardiac arrest, the provider should immediately begin cardiopulmonary resuscitation and use the on-board AED to defibrillate a potentially shockable rhythm. Flight attendants are trained in cardiopulmonary resuscitation and therefore may assist with resuscitation efforts. If the patient is resuscitated, the responding physician should recommend diversion of the flight.
Anaphylaxis
In the event of a severe life-threatening allergic reaction, the FAA-mandated emergency medical kit contains both diphenhydramine and epinephrine. For an adult experiencing anaphylaxis, a responding on-board physician can administer diphenhydramine 50 mg and epinephrine 0.3 mg (using the 1:1000 formulation), both intramuscularly. For patients with bronchospasm, a metered-dose inhaler of albuterol can be given. As anaphylaxis is an acute and potentially lethal condition, diversion of the aircraft would also be appropriate.29
Myocardial infarction
When acute myocardial infarction is suspected, it is appropriate for the provider to give aspirin, with important exceptions for patients who are experiencing an acute hemorrhage or who have an aspirin allergy.30 Supplemental oxygen should likewise be provided if the responding physician suspects compromised oxygenation. As acute myocardial infarction is also a time-sensitive condition, the clinician who suspects this diagnosis should recommend diversion of the aircraft.
Acute psychiatric issues
While approximately 2.4% of on-board medical events are attributed to psychiatric issues,5 there are few tools at the clinician’s disposal in the FAA-mandated emergency medical kit. Antipsychotics and sedatives are not included. The responding physician may need to attempt verbal de-escalation of aggressive behavior. If the safety of the flight is compromised, the application of improvised physical restraints may be appropriate.
Altered mental status
The differential diagnosis for altered mental status is extensive. The on-board physician should try to identify reversible and potentially lethal conditions and determine the potential need for aircraft diversion.
If possible, a blood sugar level should be measured (although the FAA-mandated kit does not contain a glucometer). It may be appropriate to empirically give intravenous dextrose to patients strongly suspected of having hypoglycemia.
If respiratory or cerebrovascular compromise is suspected, supplemental oxygen should be provided.
Unless a reversible cause of altered mental status is identified and treated successfully, it will likely be appropriate to recommend diversion of the aircraft.
Acknowledgment: The authors acknowledge Linda J. Kesselring, MS, ELS, the technical editor/writer in the Department of Emergency Medicine University of Maryland School of Medicine, for her contributions as copy editor of a previous version of this manuscript.
It could happen. You are on a plane, perhaps on your way to a medical conference or a well-deserved vacation, when the flight attendant asks you to help a passenger experiencing an in-flight medical emergency. What is your role in this situation?
FLIGHT ATTENDANTS USED TO BE NURSES
Before World War II, nearly all American flight attendants were nurses, who could address most medical issues that arose during flights.1 Airlines eliminated this preferential hiring practice to support the war effort. Traveling healthcare providers thereafter often volunteered to assist when in-flight medical issues arose, but aircraft carried minimal medical equipment and volunteers’ liability was uncertain.
In 1998, Congress passed the Aviation Medical Assistance Act (AMAA), which provides liability protection for on-board healthcare providers who render medical assistance. It also required the Federal Aviation Administration (FAA) to improve its standards for in-flight medical equipment.2,3
HOW OFTEN DO EMERGENCIES ARISE?
How often medical events occur during flight is difficult to estimate because airlines are not mandated to report such issues.4 Based on data from a ground-based communications center that provides medical consultation service to airlines, medical events occur in approximately 1 in every 604 flights.5 This is likely an underestimate, as many medical events may be handled on board without involving a ground-based consultation center.
The most common emergencies are syncope or presyncope, representing 37.4% of consultations, followed by respiratory symptoms (12.1%), nausea or vomiting (9.5%), cardiac symptoms (7.7%), seizures (5.8%), and abdominal pain (4.1%).5 Very few in-flight medical emergencies progress to death; the reported mortality rate is 0.3%.5
CABIN PRESSURES ARE RELATIVELY LOW
The cabins of commercial airliners are pressurized, but the pressure is still lower than on the ground. The cabin pressure in flight is equivalent to that at an altitude of 6,000 to 8,000 feet,6,7 ie, about 23 or 24 mm Hg, compared with about 30 mm Hg at sea level. At this pressure, passengers have a partial pressure of arterial oxygen (Pao2) of 60 mm Hg (normal at sea level is > 80).8
This reduced oxygen pressure is typically not clinically meaningful in healthy people. However, people with underlying pulmonary or cardiac illness may be starting further to the left on the oxygen dissociation curve before gaining altitude, putting them at risk for acute exacerbations of underlying medical conditions. Many patients who rely on supplemental oxygen, such as those with chronic obstructive pulmonary disease, are advised to increase their oxygen support during flight.9
Boyle’s law says that the volume of a gas is inversely proportional to its pressure. As the pressure drops in the cabin after takeoff, air trapped in an enclosed space—eg, in some patient’s bodies—can increase in volume up to 30%,10 which can have medical ramifications. Clinically significant pneumothorax during flight has been reported.11–13 Partially because of these volumetric changes, patients who have undergone abdominal surgery are advised to avoid flying for at least 2 weeks after their procedure.10,14 Patients who have had recent ocular or intracranial surgery may also be at risk of in-flight complications.15
IN-FLIGHT MEDICAL RESOURCES
The limited medical supplies available on aircraft often challenge healthcare providers who offer to respond to in-flight medical events. However, several important medical resources are available.
Medical kits and defibrillators
FAA regulations require airlines based in the United States to carry basic first aid supplies such as bandages and splints.3 Airlines are also required to carry a medical kit containing the items listed in Table 1.
The FAA-mandated kit does not cover every circumstance that may arise. Although in-flight pediatric events occasionally occur,16 many of the available medications are inappropriate for young children. The FAA does not require sedative or antipsychotic agents, which could be useful for passengers who have acute psychiatric episodes. Obstetric supplies are absent. On international carriers, the contents of medical kits are highly variable,17 as are the names used for some medications.
The FAA requires at least 1 automated external defibrillator (AED) to be available on each commercial aircraft.3 The timely use of AEDs greatly improves survivability after out-of-hospital cardiac arrest.18,19 One study involving a major US airline found a 40% survival rate to hospital discharge in patients who received in-flight defibrillation.20 Without this intervention, very few of the patients would have been expected to survive. In addition to being clinically effective, placing AEDs aboard commercial aircraft is a cost-effective public health intervention.21
Consultation services
Most major airlines can contact ground-based medical consultation services during flight.10 These centers are staffed with healthcare providers who can provide flight crews with advice on how to handle medical events in real time. Healthcare providers can likewise discuss specific medical issues with these services if they respond to an in-flight medical event. Ground-based call centers can also communicate with prehospital providers should a flight need to be diverted.
Other on-board providers
Some medical events require the involvement of more than one medical provider. Other physicians, nurses, and prehospital providers are often also on board.22 Responding physicians can also request the assistance of these other healthcare providers. Flight attendants in the United States are required to be trained in cardiopulmonary resuscitation (CPR).23
Flight diversion
Critically ill patients or those with time-sensitive medical emergencies may require the aircraft to divert from its intended destination. As may be expected, medical emergencies suspected to involve the cardiovascular, neurologic, or respiratory system have been shown to most likely result in aircraft diversion.5,24 Approximately 7% of in-flight medical events in which a ground-based medical consultation service is contacted result in diversion.5
While an on-board responding physician can make a recommendation to divert based on the patient’s acute medical status, only the captain can make the ultimate decision.4 On-board healthcare providers should clearly state that a patient might benefit from an unscheduled landing if that is truly their assessment. In addition to communicating their clinical concerns with the flight crew, the responding physician may also be able to discuss the situation with the airline’s ground-based consultation service. On-board physicians can make important contributions to the assessment of illness severity and triage decisions.
MEDICOLEGAL ISSUES
No legal duty to assist
US healthcare providers are not legally required to respond to on-board medical emergencies on US-based airlines. Canada and the United Kingdom also do not require providers to render assistance. But the General Medical Council (the regulatory body for UK doctors) states that doctors have an ethical duty to respond in the event of a medical emergency, including one on board an aircraft. Other countries, notably Australia and some in the European Union, require healthcare professionals to respond to on-board medical emergencies.10
Regardless of potential legal duties to assist, healthcare providers are arguably ethically obliged to render assistance if they can.
Aviation Medical Assistance Act
The extent of an American healthcare provider’s liability risk for assisting in a medical emergency on a plane registered in the United States is limited by statute. The 1998 AMAA provides liability protection for on-board medical providers who are asked to assist during an in-flight medical emergency. This statute covers all US-certified air carriers on domestic flights and would likely be held to apply to US aircraft in foreign airspace because of the general rule that the law of the country where the air carrier is registered applies to in-flight events.
Under the AMAA, providers asked to assist with in-flight medical emergencies are not liable for malpractice as long as their actions are not “grossly” negligent or intended to cause the patient harm.25 This is distinguishable from a standard malpractice liability scenario, in which the plaintiff only needs to show ordinary negligence. In a traditional healthcare setting, a provider has to act within the “standard of care” when assessing and treating a patient. If the provider deviates from the standard of care, such as by making an error in judgment or diagnosis, the provider is legally negligent. Under traditional malpractice law, even if a provider is minimally negligent, he or she is liable for any damages resulting from that negligence. Under a gross negligence standard, providers are protected from liability unless they demonstrate flagrant disregard for the patient’s health and safety.
Postflight issues
A provider who undertakes care should continue to provide care until it is no longer necessary, either because the patient recovers or the responsibility has been transferred to another provider. At the point of transfer, the healthcare provider’s relationship with the patient terminates.
The provider should document the encounter, typically using airline-specific documentation. The responding physician needs to be mindful of the patient’s privacy, refraining from discussing the event with others without the patient’s authorization.26
SUGGESTED RESPONSE
Healthcare providers who wish to respond to in-flight medical emergencies must first determine if they are sufficiently capable of providing care. During a flight, providers do not expect to be on duty and so may have consumed alcoholic beverages to an extent that would potentially render them unsuitable to respond. When it is appropriate to become involved in a medical emergency during flight, the healthcare provider should state his or her qualifications to the passenger and to flight personnel.
If circumstances allow, the volunteer provider should obtain the patient’s consent for evaluation and treatment.10 Additionally, with the multilingual nature of commercial air travel, especially on international flights, the provider may need to enlist a translator’s assistance.
Providers may find it preferable to treat passengers in their seats.27 Given the confined space in an aircraft, keeping ill passengers out of the aisle allows others to move about the cabin. If it becomes necessary to move the patient, a location should be sought that minimally interferes with other passengers’ needs.
If a passenger has critical medical needs, in-flight medical volunteers can recommend flight diversion, which should also be discussed with ground-based medical staff. However, as emphasized earlier, the captain makes the ultimate decision to divert, taking into account other operational factors that affect the safety of the aircraft and its occupants. In-flight medical care providers should perform only the treatments they are qualified to provide and should operate within their scope of training.
After the aircraft lands, if the passenger must be transported to a hospital, providers should supply prehospital personnel with a requisite transfer-of-care communication. In-flight medical providers who have performed a significant medical intervention might find it appropriate to accompany the patient to the hospital.
SPECIFIC CONDITIONS
The list of possible acute medical issues that occur aboard aircraft is extensive. Here are a few of them.
Trauma
Passengers may experience injuries during flight, for example during periods of heavy air turbulence. Responding physicians should assess for potential life-threatening injuries, keeping in mind that some passengers may be at higher risk. For example, if a passenger on anticoagulation experiences a blunt head injury, this would raise suspicion for possible intracranial hemorrhage, and frequent reassessment of neurologic status may be necessary. If an extremity fracture is suspected, the physician should splint the affected limb. Analgesia may be provided from the medical kit, if appropriate.
Gastrointestinal issues
Acute gastrointestinal issues such as nausea and vomiting are often reported to ground-based medical consultation services.5 Responding on-board providers must consider if the passenger is simply experiencing gastrointestinal upset from a benign condition such as gastroenteritis or has a more serious condition. For some patients, vomiting may be a symptom of a myocardial infarction.28 Bilious emesis with abdominal distention may be associated with small-bowel obstruction. While antiemetics are not included in the FAA-mandated medical kit, providers can initiate intravenous fluid therapy for passengers who show signs of hypovolemia.
Cardiac arrest
Although cardiac arrest during flight is rare,5 medical providers should nonetheless be prepared to handle it. Upon recognition of cardiac arrest, the provider should immediately begin cardiopulmonary resuscitation and use the on-board AED to defibrillate a potentially shockable rhythm. Flight attendants are trained in cardiopulmonary resuscitation and therefore may assist with resuscitation efforts. If the patient is resuscitated, the responding physician should recommend diversion of the flight.
Anaphylaxis
In the event of a severe life-threatening allergic reaction, the FAA-mandated emergency medical kit contains both diphenhydramine and epinephrine. For an adult experiencing anaphylaxis, a responding on-board physician can administer diphenhydramine 50 mg and epinephrine 0.3 mg (using the 1:1000 formulation), both intramuscularly. For patients with bronchospasm, a metered-dose inhaler of albuterol can be given. As anaphylaxis is an acute and potentially lethal condition, diversion of the aircraft would also be appropriate.29
Myocardial infarction
When acute myocardial infarction is suspected, it is appropriate for the provider to give aspirin, with important exceptions for patients who are experiencing an acute hemorrhage or who have an aspirin allergy.30 Supplemental oxygen should likewise be provided if the responding physician suspects compromised oxygenation. As acute myocardial infarction is also a time-sensitive condition, the clinician who suspects this diagnosis should recommend diversion of the aircraft.
Acute psychiatric issues
While approximately 2.4% of on-board medical events are attributed to psychiatric issues,5 there are few tools at the clinician’s disposal in the FAA-mandated emergency medical kit. Antipsychotics and sedatives are not included. The responding physician may need to attempt verbal de-escalation of aggressive behavior. If the safety of the flight is compromised, the application of improvised physical restraints may be appropriate.
Altered mental status
The differential diagnosis for altered mental status is extensive. The on-board physician should try to identify reversible and potentially lethal conditions and determine the potential need for aircraft diversion.
If possible, a blood sugar level should be measured (although the FAA-mandated kit does not contain a glucometer). It may be appropriate to empirically give intravenous dextrose to patients strongly suspected of having hypoglycemia.
If respiratory or cerebrovascular compromise is suspected, supplemental oxygen should be provided.
Unless a reversible cause of altered mental status is identified and treated successfully, it will likely be appropriate to recommend diversion of the aircraft.
Acknowledgment: The authors acknowledge Linda J. Kesselring, MS, ELS, the technical editor/writer in the Department of Emergency Medicine University of Maryland School of Medicine, for her contributions as copy editor of a previous version of this manuscript.
- Gazdik M. Vault guide to flight attendant careers. New York, NY: Vault, Inc.; 2005.
- Stewart PH, Agin WS, Douglas SP. What does the law say to Good Samaritans? A review of Good Samaritan statutes in 50 states and on US airlines. Chest 2013; 143:1774–1783.
- Federal Aviation Administration (FAA), DOT. Emergency medical equipment. Final rule. Fed Regist 2001; 66:19028–19046.
- Goodwin T. In-flight medical emergencies: an overview. BMJ 2000; 321:1338–1341.
- Peterson DC, Martin-Gill C, Guyette FX, et al. Outcomes of medical emergencies on commercial airline flights. N Engl J Med 2013; 368:2075–2083.
- Aerospace Medical Association, Aviation Safety Committee, Civil Aviation Subcommittee. Cabin cruising altitudes for regular transport aircraft. Aviat Space Environ Med 2008; 79:433–439.
- Cottrell JJ. Altitude exposures during aircraft flight. Flying higher. Chest 1988; 93:81–84.
- Humphreys S, Deyermond R, Bali I, Stevenson M, Fee JP. The effect of high altitude commercial air travel on oxygen saturation. Anaesthesia 2005; 60:458–460.
- Shrikrishna D, Coker RK; Air Travel Working Party of the British Thoracic Society Standards of Care Committee. Managing passengers with stable respiratory disease planning air travel: British Thoracic Society recommendations. Thorax 2011; 66:831–833.
- Gendreau MA, DeJohn C. Responding to medical events during commercial airline flights. N Engl J Med 2002; 346:1067–1073.
- Hu X, Cowl CT, Baqir M, Ryu JH. Air travel and pneumothorax. Chest 2014; 145:688–694.
- Madan K, Vishwanath G, Singh N. In-flight spontaneous pneumothorax: congenital cystic adenomatoid malformation of the lung. Respiration 2012; 83:554–558.
- Wallace TW, Wong T, O’Bichere A, Ellis BW. Managing in flight emergencies. BMJ 1995; 311:374–376.
- Medical aspects of transportation aboard commercial aircraft. AMA commission on emergency medical services. JAMA 1982; 247:1007–1011.
- Mills MD, Devenyi RG, Lam WC, Berger AR, Beijer CD, Lam SR. An assessment of intraocular pressure rise in patients with gas-filled eyes during simulated air flight. Ophthalmology 2001; 108:40–44.
- Moore BR, Ping JM, Claypool DW. Pediatric emergencies on a US-based commercial airline. Pediatr Emerg Care 2005; 21:725–729.
- Sand M, Gambichler T, Sand D, Thrandorf C, Altmeyer P, Bechara FG. Emergency medical kits on board commercial aircraft: a comparative study. Travel Med Infect Dis 2010; 8:388–394.
- Auble TE, Menegazzi JJ, Paris PM. Effect of out-of-hospital defibrillation by basic life support providers on cardiac arrest mortality: a metaanalysis. Ann Emerg Med 1995; 25:642–648.
- Marenco JP, Wang PJ, Link MS, Homoud MK, Estes NA. Improving survival from sudden cardiac arrest: the role of the automated external defibrillator. JAMA 2001; 285:1193–1200.
- Page RL, Joglar JA, Kowal RC, et al. Use of automated external defibrillators by a US airline. N Engl J Med 2000; 343:1210–1216.
- Groeneveld PW, Kwong JL, Liu Y, et al. Cost-effectiveness of automated external defibrillators on airlines. JAMA 2001; 286:1482–1489.
- Baltsezak S. Clinic in the air? A retrospective study of medical emergency calls from a major international airline. J Travel Med 2008; 15:391–394.
- Federal Aviation Administration (FAA). Advisory circular: emergency medical equipment training AC 121-34B. www.faa.gov/documentLibrary/media/Advisory_Circular/AC121-34B.pdf. Accessed April 6, 2017.
- Cummins RO, Schubach JA. Medical emergencies among commercial air travelers. JAMA 1989; 261:1295–1299.
- US Government Publishing Office. Public Law 105-170. Aviation Medical Assistance Act of 1998.
- US Government Publishing Office. Public Law 104-191. Health Insurance Portability and Accountability Act of 1996.
- Chandra A, Conry S. In-flight medical emergencies. West J Emerg Med 2013; 14:499–504.
- Kirchberger I, Meisinger C, Heier M, et al. Patient-reported symptoms in acute myocardial infarction: differences related to ST-segment elevation: the MONICA/KORA Myocardial Infarction Registry. J Intern Med 2011; 270:58–64.
- Brady WJ Jr, Bright HL. Occurrence of multiphasic anaphylaxis during a transcontinental air flight. Am J Emerg Med 1999; 17:695–696.
- O’Connor RE, Brady W, Brooks SC, et al. Part 10: acute coronary syndromes: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2010; 122:(suppl 3):S787–S817.
- Gazdik M. Vault guide to flight attendant careers. New York, NY: Vault, Inc.; 2005.
- Stewart PH, Agin WS, Douglas SP. What does the law say to Good Samaritans? A review of Good Samaritan statutes in 50 states and on US airlines. Chest 2013; 143:1774–1783.
- Federal Aviation Administration (FAA), DOT. Emergency medical equipment. Final rule. Fed Regist 2001; 66:19028–19046.
- Goodwin T. In-flight medical emergencies: an overview. BMJ 2000; 321:1338–1341.
- Peterson DC, Martin-Gill C, Guyette FX, et al. Outcomes of medical emergencies on commercial airline flights. N Engl J Med 2013; 368:2075–2083.
- Aerospace Medical Association, Aviation Safety Committee, Civil Aviation Subcommittee. Cabin cruising altitudes for regular transport aircraft. Aviat Space Environ Med 2008; 79:433–439.
- Cottrell JJ. Altitude exposures during aircraft flight. Flying higher. Chest 1988; 93:81–84.
- Humphreys S, Deyermond R, Bali I, Stevenson M, Fee JP. The effect of high altitude commercial air travel on oxygen saturation. Anaesthesia 2005; 60:458–460.
- Shrikrishna D, Coker RK; Air Travel Working Party of the British Thoracic Society Standards of Care Committee. Managing passengers with stable respiratory disease planning air travel: British Thoracic Society recommendations. Thorax 2011; 66:831–833.
- Gendreau MA, DeJohn C. Responding to medical events during commercial airline flights. N Engl J Med 2002; 346:1067–1073.
- Hu X, Cowl CT, Baqir M, Ryu JH. Air travel and pneumothorax. Chest 2014; 145:688–694.
- Madan K, Vishwanath G, Singh N. In-flight spontaneous pneumothorax: congenital cystic adenomatoid malformation of the lung. Respiration 2012; 83:554–558.
- Wallace TW, Wong T, O’Bichere A, Ellis BW. Managing in flight emergencies. BMJ 1995; 311:374–376.
- Medical aspects of transportation aboard commercial aircraft. AMA commission on emergency medical services. JAMA 1982; 247:1007–1011.
- Mills MD, Devenyi RG, Lam WC, Berger AR, Beijer CD, Lam SR. An assessment of intraocular pressure rise in patients with gas-filled eyes during simulated air flight. Ophthalmology 2001; 108:40–44.
- Moore BR, Ping JM, Claypool DW. Pediatric emergencies on a US-based commercial airline. Pediatr Emerg Care 2005; 21:725–729.
- Sand M, Gambichler T, Sand D, Thrandorf C, Altmeyer P, Bechara FG. Emergency medical kits on board commercial aircraft: a comparative study. Travel Med Infect Dis 2010; 8:388–394.
- Auble TE, Menegazzi JJ, Paris PM. Effect of out-of-hospital defibrillation by basic life support providers on cardiac arrest mortality: a metaanalysis. Ann Emerg Med 1995; 25:642–648.
- Marenco JP, Wang PJ, Link MS, Homoud MK, Estes NA. Improving survival from sudden cardiac arrest: the role of the automated external defibrillator. JAMA 2001; 285:1193–1200.
- Page RL, Joglar JA, Kowal RC, et al. Use of automated external defibrillators by a US airline. N Engl J Med 2000; 343:1210–1216.
- Groeneveld PW, Kwong JL, Liu Y, et al. Cost-effectiveness of automated external defibrillators on airlines. JAMA 2001; 286:1482–1489.
- Baltsezak S. Clinic in the air? A retrospective study of medical emergency calls from a major international airline. J Travel Med 2008; 15:391–394.
- Federal Aviation Administration (FAA). Advisory circular: emergency medical equipment training AC 121-34B. www.faa.gov/documentLibrary/media/Advisory_Circular/AC121-34B.pdf. Accessed April 6, 2017.
- Cummins RO, Schubach JA. Medical emergencies among commercial air travelers. JAMA 1989; 261:1295–1299.
- US Government Publishing Office. Public Law 105-170. Aviation Medical Assistance Act of 1998.
- US Government Publishing Office. Public Law 104-191. Health Insurance Portability and Accountability Act of 1996.
- Chandra A, Conry S. In-flight medical emergencies. West J Emerg Med 2013; 14:499–504.
- Kirchberger I, Meisinger C, Heier M, et al. Patient-reported symptoms in acute myocardial infarction: differences related to ST-segment elevation: the MONICA/KORA Myocardial Infarction Registry. J Intern Med 2011; 270:58–64.
- Brady WJ Jr, Bright HL. Occurrence of multiphasic anaphylaxis during a transcontinental air flight. Am J Emerg Med 1999; 17:695–696.
- O’Connor RE, Brady W, Brooks SC, et al. Part 10: acute coronary syndromes: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2010; 122:(suppl 3):S787–S817.
KEY POINTS
- The exact incidence of medical emergencies aboard airplanes is unknown, but they occurred in 1 in 604 flights in 1 study, which is likely an underestimate.
- The relatively low air pressure in the cabin can contribute to the development of acute medical issues.
- In the United States, the Federal Aviation Administration mandates that airlines carry a limited set of medical resources.
- The Aviation Medical Assistance Act protects responding providers against liability except in cases of “gross negligence.”
- You the physician can recommend that the flight be diverted to the closest airport, but only the captain can make the actual decision.

ERAAs for menopause treatment: Welcome the ‘designer estrogens’
Estrogen receptor agonist-antagonists (ERAAs), previously called selective estrogen receptor modulators (SERMs), have extended the options for treating the various conditions that menopausal women suffer from. These drugs act differently on estrogen receptors in different tissues, stimulating receptors in some tissues but inhibiting them in others. This allows selective inhibition or stimulation of estrogen-like action in various target tissues.1
This article highlights the use of ERAAs to treat menopausal vasomotor symptoms (eg, hot flashes, night sweats), genitourinary syndrome of menopause, osteoporosis, breast cancer (and the risk of breast cancer), and other health concerns unique to women at midlife.
SYMPTOMS OF MENOPAUSE: COMMON AND TROUBLESOME
Vasomotor symptoms such as hot flashes and night sweats are common during perimenopause—most women experience them. They are most frequent during the menopause transition but can persist for 10 years or more afterward.2
Genitourinary syndrome of menopause is also common and often worsens with years after menopause.3 It can lead to dyspareunia and vaginal dryness, which may in turn result in lower libido, vaginismus, and hypoactive sexual desire disorder, problems that often arise at the same time as vaginal dryness and atrophy.4
Osteopenia and osteoporosis. A drop in systemic estrogen leads to a decline in bone mineral density, increasing the risk of fractures.5
ESTROGEN-PROGESTIN TREATMENT: THE GOLD STANDARD, BUT NOT IDEAL
The current gold standard for treating moderate to severe hot flashes is estrogen, available in oral, transdermal, and vaginal formulations.6 Estrogen also has antiresorptive effects on bone and is approved for preventing osteoporosis. Systemic estrogen may also be prescribed for genitourinary syndrome of menopause if local vaginal treatment alone is insufficient.
If women who have an intact uterus receive estrogen, they should also receive a progestin to protect against endometrial hyperplasia and reduce the risk of endometrial cancer.
Despite its status as the gold standard, estrogen-progestin therapy presents challenges. In some women, progestins cause side effects such as breast tenderness, bloating, fatigue, and depression.7 Estrogen-progestin therapy often causes vaginal bleeding, which for some women is troublesome or distressing; bleeding may be the reason for repeated evaluations, can increase anxiety, and can lead to poor adherence with hormonal treatment. Women who carry a higher-than-normal risk of developing breast cancer or fear that taking hormones will lead to breast cancer may show decreased adherence to therapy. Women who have estrogen receptor-positive breast cancer cannot take estrogen.
Individualized options are needed for women who have progestin-related side effects, unwanted vaginal bleeding, or a higher risk of breast cancer.
WELCOME THE ERAAs
An ideal treatment for menopause would relieve vasomotor symptoms and genitourinary syndrome of menopause and increase bone mineral density without causing breast tenderness, vaginal bleeding, or endometrial proliferation.
The “designer estrogens,” or ERAAs, have specific positive effects on the bone, heart, and brain with neutral or antagonist effects on estrogen receptors in other tissues such as the breasts and endometrium.8 While not entirely free of adverse effects, these agents have been developed with the aim of minimizing the most common ones related to estrogen and progestin.

Several ERAAs are currently approved by the US Food and Drug Administration (FDA)for various indications, each having a unique profile. Clomifene was the first agent of this class, and it is still used clinically to induce ovulation. This article highlights subsequently approved agents, ie, tamoxifen, raloxifene, ospemifene, and the combination of conjugated estrogens and bazedoxifene (Table 1).
All ERAAs increase the risk of venous thromboembolism, and therefore none of them should be used in women with known venous thromboembolism or at high risk of it.
TAMOXIFEN: CANCER TREATMENT AND PREVENTION
After clomiphene, tamoxifen was the second ERAA on the market. Although researchers were looking for a new contraceptive drug, they found tamoxifen to be useful as a chemotherapeutic agent for breast cancer. First used in 1971, tamoxifen continues to be one of the most commonly prescribed chemotherapeutic medications today.
The FDA has approved tamoxifen to treat breast cancer as well as to prevent breast cancer in pre- and postmenopausal women at risk. It may also have beneficial effects on bone and on cardiovascular risk factors, but these are not approved uses for it.
Trials of tamoxifen for cancer treatment
The Early Breast Cancer Trialists’ Collaborative Group9 performed a meta-analysis and found that 5 years of adjuvant treatment with tamoxifen is associated with a 26% reduction in mortality and a 47% reduction in breast cancer recurrence at 10 years. In absolute terms, we estimate that 21 women would need to be treated to prevent 1 death and 8 would need to be treated to prevent 1 recurrence.
The ATLAS Trial (Adjuvant Tamoxifen Longer Against Shorter)10 and later the UK ATTOM (Adjuvant Tamoxifen Treatment to Offer More)11 trial confirmed an even greater reduction in recurrence and mortality after a total of 10 years of treatment.
Trials of tamoxifen for cancer prevention
Cuzik et al12 performed a meta-analysis of 4 trials of tamoxifen’s effectiveness in preventing breast cancer for women at elevated risk. The incidence of estrogen receptor-positive breast cancer was 48% lower with tamoxifen use, but there was no effect on estrogen-negative breast cancer. From their data, we estimate that 77 women would need to be treated to prevent 1 case of breast cancer.
The IBIS-I trial (International Breast Cancer Intervention Study I)13 found that, in healthy women at high risk of breast cancer, the benefit of taking tamoxifen for 5 years as preventive treatment persisted long afterward. The investigators estimated that at 20 years of follow-up the risk of breast cancer would be 12.3% in placebo recipients and 7.8% in tamoxifen recipients, a 4.5% absolute risk reduction; number needed to treat (NNT) 22.
Data on tamoxifen and osteoporosis
The Breast Cancer Prevention Trial revealed a 19% reduction in the incidence of osteoporotic fractures with tamoxifen, but the difference was not statistically significant.14 The 1-year rates of fracture in women age 50 and older were 0.727% with placebo and 0.567% with tamoxifen, an absolute difference of 0.151%; therefore, if the effect is real, 662 women age 50 or older would need to be treated for 1 year to prevent 1 fracture. Tamoxifen is not FDA-approved to treat osteoporosis.
Data on tamoxifen and cardiovascular risk reduction
Chang et al,15 in a study in women at risk of breast cancer, incidentally found that tamoxifen was associated with a 13% reduction in total cholesterol compared with placebo.
Herrington and Klein,16 in a systematic review, noted similar findings in multiple studies of tamoxifen, with decreases in total cholesterol ranging from 7% to 17% and decreases in low-density lipoprotein cholesterol ranging from 10% to 28%. However, they found no change in high-density lipoprotein cholesterol concentrations or in the cardiovascular mortality rate.
The ATLAS trial10 revealed a relative risk reduction of 0.76 (95% confidence interval [CI] 0.60–0.95, P = .02) in ischemic heart disease for women who took tamoxifen for 10 years compared with 5 years. We calculate that ischemic heart disease occurred in 163 (2.5%) of 6,440 women who took tamoxifen for 5 years compared with 127 (1.9%) of 6,454 women who took it for 10 years, a 0.6% absolute risk reduction, NNT = 167.
Adverse effects of tamoxifen
Uterine neoplasia. Women taking tamoxifen have a 2.5-fold increased risk of endometrial cancer.14 Tamoxifen also increases the risk of benign uterine disease such as endometrial hyperplasia and polyps. As many as 39% of women taking tamoxifen will have evidence of benign uterine changes on pathology.17 Other adverse effects:
Venous thromboembolism (the risk of pulmonary embolism is increased approximately threefold14)
Cataracts (there is a slight increase in cataract diagnosis in tamoxifen users)
Vasomotor symptoms, which limit the use of tamoxifen in many women.
Ideal candidate for tamoxifen
The ideal candidate for tamoxifen is a woman with breast cancer that is estrogen receptor-positive and who has a history of osteopenia or osteoporosis and no risk factors for venous thromboembolism.
RALOXIFENE: FOR OSTEOPOROSIS AND FOR CANCER PREVENTION
Raloxifene, a second-generation ERAA, was first approved for preventing and treating osteoporosis and later for reducing the risk of invasive estrogen receptor-positive breast cancer in postmenopausal women.
Trials of raloxifene for osteoporosis
The MORE trial (Multiple Outcomes of Raloxifene)18 was a large multicenter randomized double-blind study. Raloxifene recipients showed a significant increase in bone mineral density in the lumbar spine and femoral neck at year 3 (P < .001) compared with those receiving placebo. Even after only 1 year of treatment, raloxifene significantly reduced the risk of new fractures, despite only modest gains in bone mineral density. After 3 years of treatment, new clinical vertebral fractures had occurred in 3.5% of the placebo group compared with 2.1% of the group receiving raloxifene 60 mg.19 Relative risk reductions were similar in women who had already had a clinical vertebral fracture at baseline, whose absolute risk is higher. However, no significant effect was seen on the incidence of hip or nonvertebral fractures.
The CORE trial (Continuing Outcomes Relevant to Raloxifene)20 extended the treatment of the women enrolled in the MORE trial another 4 years and found that the benefit of raloxifene with regard to bone mineral density persisted with continued use.
Trials of raloxifene for breast cancer prevention
The MORE trial,21 in postmenopausal women with osteoporosis included breast cancer as a secondary end point, and raloxifene was shown to decrease the incidence of invasive breast cancer. At a median of 40 months, invasive breast cancer had arisen in 13 (0.25%) of the 5,129 women assigned to raloxifene and 27 (1.0%) of the 2,576 women assigned to placebo. The authors calculated that 126 women would need to be treated to prevent 1 case of breast cancer.
The CORE trial,22 as noted, extended the treatment of the women enrolled in the MORE trial another 4 years. The risk of any invasive breast cancer in postmenopausal women with osteoporosis was significantly reduced by 59% after 8 years, and the risk of estrogen receptor-positive invasive breast cancer was reduced by 66%.
There is evidence that raloxifene’s effect on breast cancer risk persists after discontinuation of use.23
Does raloxifene reduce mortality?
Grady et al24 studied the effect of raloxifene on all-cause mortality in a pooled analysis of mortality data from the MORE, CORE, and Raloxifene Use for the Heart (RUTH)25 trials. In older postmenopausal women, the rate of all-cause mortality was 8.65% in those taking placebo compared with 7.88% in those taking raloxifene 60 mg daily—10% lower. The mechanism behind the lower mortality rate is unclear, and Grady et al recommend that the finding be interpreted with caution.
Trials of raloxifene for heart protection
The RUTH trial25 was a 5.6-year study undertaken to study the effects of raloxifene on coronary outcomes and invasive breast cancer in postmenopausal women. Results were mixed. Active treatment:
- Did not significantly affect the risk of coronary artery disease compared with placebo
- Significantly decreased the risk of invasive breast cancer
- Significantly decreased the risk of clinical vertebral fractures
- Increased the risk of fatal stroke (59 vs 39 events, hazard ratio 1.49, 95% CI 1.00–2.24) and venous thromboembolism (103 vs 71 events, hazard ratio 1.44, 95% CI 1.06–1.95).
The STAR trial (Study of Tamoxifen and Raloxifene)26,27 compared raloxifene and tamoxifen in postmenopausal women at increased risk of breast cancer. Women were randomized to receive either tamoxifen 20 mg or raloxifene 60 mg for 5 years. Results:
- No difference in the number of new cases of invasive breast cancer between the groups
- Fewer cases of noninvasive breast cancer in the tamoxifen group, but the difference was not statistically significant
- Fewer cases of uterine cancer in the raloxifene group, annual incidence rates 0.125% vs 0.199%, absolute risk reduction 0.74%, NNT 1,351, relative risk with raloxifene 0.62, 95% CI 0.30–0.50
- Fewer thromboembolic events with raloxifene
- Fewer cataracts with raloxifene.
Adverse effects of raloxifene
Raloxifene increases the risk of venous thromboembolism and stroke in women at high risk of coronary artery disease.19
Ideal candidates for raloxifene
Postmenopausal women with osteopenia or osteoporosis and a higher risk of breast cancer who have minimal to no vasomotor symptoms or genitourinary syndrome of menopause are good candidates for raloxifene. Raloxifene is also a good choice for women who have genitourinary syndrome of menopause treated with local vaginal estrogen. Raloxifene has no effect on vasomotor symptoms or genitourinary syndrome of menopause.
OSPEMIFENE: FOR GENITOURINARY SYNDROME OF MENOPAUSE
Although ospemifene does not have the steroid structure of estrogen, it acts as an estrogen agonist specifically in the vaginal mucosa and an antagonist in other tissues.28 It has been shown on Papanicolaou smears to reduce the number of parabasal cells and increase the number of intermediate and superficial cells after 3 months of treatment.29
Ospemifene 60 mg taken orally with food is approved by the FDA to treat genitourinary syndrome of menopause.
Why ospemifene is needed
First-line treatment options for genitourinary syndrome of menopause include over-the-counter lubricants. However, there is no evidence that these products reverse vaginal atrophy,30 and many women report no relief of symptoms with them.
While various local estrogen preparations positively affect genitourinary syndrome of menopause, some of them can be messy, which can limit-long term adherence.
In one of the largest surveys on genitourinary syndrome of menopause (the REVIVE survey—the Real Women’s View of Treatment Options for Menopausal Vaginal Changes29), 59% of women reported that their vaginal symptoms negatively affected sexual activity. The problem affects not only the patient but also her sexual partner.31 Another large study showed that 38% of women and 39% of male partners reported that it had a worse-than-expected impact on their intimate relationships.31
Genitourinary syndrome of menopause also makes pelvic examinations difficult, may worsen or exacerbate cystitis, and may increase urinary tract infections.
Trials of ospemifene for genitourinary syndrome of menopause
To date, 3 randomized, double-blind clinical trials have demonstrated ospemifene 60 mg to be superior to placebo in treating genitourinary syndrome of menopause. Two were short-term (12-week) and showed significant positive changes in the percent of superficial cells, vaginal pH (lower is better), and number of parabasal cells, along with improvements in the Likert rating of both vaginal dryness and dyspareunia.32,33
A long-term (52-week) randomized placebo-controlled trial compared ospemifene and placebo and showed significant improvement in vaginal maturation index and pH at weeks 12 and 52.34 Other outcome measures included petechiae, pallor, friability, erythema, and dryness, all of which improved from baseline (P < .001). At the end of the trial, 80% of the patients who received ospemifene had no vaginal atrophy.
No serious adverse events were noted in any of the clinical trials to date, and a systemic review and meta-analysis demonstrated ospemifene to be safe and efficacious.35 The most frequently reported reasons for discontinuation were hot flashes, vaginal discharge, muscle spasms, and hyperhidrosis, but the rates of these effects were similar to those with placebo.
Trial of ospemifene’s effect on bone turnover
As an estrogen receptor agonist in bone, ospemifene decreases the levels of bone turnover markers in postmenopausal women.36 A study found ospemifene to be about as effective as raloxifene in suppressing bone turnover,37 but ospemifene does not carry FDA approval for preventing or treating osteoporosis.
Other effects
In experiments in rats, the incidence of breast cancer appears to be lower with ospemifene, and the higher the dose, the lower the incidence.38
Ospemifene also has antagonistic effects on uterine tissue, and no cases of endometrial hyperplasia or carcinoma have been reported in short-term or long-term studies.35
Ospemifene has no effect however on vasomotor symptoms and may in fact worsen vasomotor symptoms in women suffering with hot flashes and night sweats. Further investigation into its long-term safety and effects on breast tissue and bone would provide more insight.
Ideal candidates for ospemifene
Ospemifene could help postmenopausal women with genitourinary syndrome of menopause for whom over-the-counter lubricants fail, who dislike local vaginal estrogen, or who decline systemic hormone therapy, and who do not meet the criteria for treatment with systemic hormone therapy.
CONJUGATED ESTROGENS AND BAZEDOXIFENE COMBINATION
A combination agent consisting of conjugated estrogens 0.45 mg plus bazedoxifene 20 mg has been approved by the FDA for treating moderate to severe vasomotor symptoms associated with menopause and also for preventing postmenopausal osteoporosis in women who have an intact uterus.
Trials of estrogen-bazedoxifene for vasomotor symptoms
The Selective Estrogen Menopause and Response to Therapy (SMART) trials39,40 were a series of randomized, double-blind, placebo-controlled phase 3 studies evaluating the efficacy and safety of the estrogen-bazedoxifene combination in postmenopausal women.
The SMART-2 trial39 evaluated the combination of conjugated estrogens (either 0.45 mg or 0.625) plus bazedoxifene 20 mg and found both dosages significantly reduced the number and severity of hot flashes at weeks 4 and 12 (P < .001). At week 12, the combination with 0.45 mg of estrogen reduced vasomotor symptoms from baseline by 74% (10.3 hot flashes per week at baseline vs 2.8 at week 12); the combination with 0.625 mg of estrogen reduced vasomotor symptoms by 80% (10.4 vs 2.4 flashes); and placebo reduced them by 51% (10.5 vs 5.4 flashes).
For bone density. The SMART-1 trial40 showed that the estrogen-bazedoxifene combination in both estrogen dosages significantly increased mean lumbar spine bone mineral density (P < .001) and total hip bone mineral density (P < .05) from baseline at 12 and 24 months compared with placebo. Increases in density tended to be higher with the higher estrogen dose (0.625 mg), but less with higher doses of bazedoxifene.41 At 24 months, the increase in bone mineral density was even greater than in women treated with raloxifene.42 However, the effect of estrogen-bazedoxifene on the incidence of fractures remains to be studied.
For genitourinary syndrome of menopause. The SMART-3 trial showed that treatment with conjugated estrogens plus bazedoxifene (0.45/20 mg or 0.625/20 mg) was more effective than placebo in increasing the percent of superficial and intermediate cells and decreased the number of parabasal cells at 12 weeks compared with placebo (P < .01).43 Both doses also significantly decreased the mean vaginal pH and improved vaginal dryness.
Patients treated with estrogen-bazedoxifene for a minimum of 12 weeks in a double-blind placebo-controlled study also showed a significant improvement in sexual function and quality-of-life measurements based on 3 well-defined scales, which included ease of lubrication, satisfaction with treatment, control of hot flashes, and sleep parameters.43
Low rates of side effects
To evaluate this regimen’s antagonistic effects on uterine tissue, endometrial hyperplasia was diagnosed by blinded pathologists using endometrial biopsies taken at 6, 12, and 24 months or more if cancer was a suspected diagnosis. At 12 and 24 months of treatment, the incidence of hyperplasia with bazedoxifene 20 or 40 mg at doses of either 0.45 or 0.625 mg of conjugated estrogens was less than 1%, which was similar to placebo rates over the 24 months.44 The lowest dose studied, bazedoxifene 10 mg, did not prevent hyperplasia with conjugated estrogens 0.45 or 0.625 mg, and its use was discontinued.
Rates of amenorrhea with bazedoxifene 20 or 40 mg and conjugated estrogens 0.45 or 0.625 mg were very favorable (83%–93%) and similar to those with placebo.45 For women with continued bleeding on hormone therapy requiring multiple evaluations, or for women who won’t accept the risk of bleeding on hormone therapy, conjugated estrogens and bazedoxifene may be a sustainable option. However, any woman with abnormal bleeding should undergo prompt immediate evaluation.
A typical side effect of estrogen replacement therapy is breast tenderness. For women seeking vasomotor symptom treatment but who experience breast tenderness, this may be a deterrent from continuing hormone therapy. As shown in the SMART-1 and SMART-2 trials,46 conjugated estrogens and bazedoxifene did not cause an increase in breast tenderness, which may enhance medication adherence.
Ideal candidates for conjugated estrogens plus bazedoxifene
This product could help postmenopausal women who have an intact uterus and are suffering with moderate to severe vasomotor symptoms and genitourinary syndrome of menopause who cannot tolerate the side effects of hormone therapy such as bleeding, bloating, or breast tenderness, or who prefer to take an estrogen but without a progestin. It is also ideal for women at higher risk of osteoporosis.
WHO SHOULD GET WHAT?
Not all postmenopausal women have vasomotor symptoms, genitourinary syndrome of menopause, or bone loss. For those who do, standard hormone therapy is an option.
For those who have symptoms and a lower threshold of side effects such as breast tenderness and vaginal bleeding, a combination of an estrogen plus an ERAA (eg, bazedoxifene) is an option.
For women who have no vasomotor symptoms but do have genitourinary syndrome of menopause and don’t want local vaginal treatment, ospemifene is an option.
For women with no vasomotor symptoms but who have bone loss and increased risk of estrogen receptor-positive breast cancer, raloxifene is a good option.
Both premenopausal and postmenopausal women who are at increased risk for breast cancer should be considered for tamoxifen chemoprevention. Postmenopausal women with a uterus at increased risk for breast cancer should be considered for raloxifene, as it has no uterine effect. Raloxifene is not indicated in premenopausal women.
No woman at increased risk of venous thromboembolism is a candidate for ERAA treatment or for oral estrogen. However, the clinician has multiple options to improve quality of life and work productivity and reduce office visits of women at midlife, especially when they are individually assessed and treated.
- Giannini A, Russo E, Mannella P, Simoncini T. Selective steroid receptor modulators in reproductive medicine. Minerva Ginecol 2015; 67:431–455.
- Feldman BM, Voda A, Gronseth E. The prevalence of hot flash and associated variables among perimenopausal women. Res Nurs Health 1985; 8:261–268.
- Versi E, Harvey MA, Cardozo L, Brincat M, Studd JW. Urogenital prolapse and atrophy at menopause: a prevalence study. Int Urogynecol J Pelvic Floor Dysfunct 2001; 12:107–110.
- Hess R, Chang CC, Conigliaro J, McNeil M. Understanding physicians’ attitudes towards hormone therapy. Womens Health Issues 2005; 15:31–38.
- Melton LJ 3rd, Khosla S, Atkinson EJ, O’Fallon WM, Riggs BL. Relationship of bone turnover to bone density and fractures. J Bone Miner Res 1997; 12:1083–1091.
- Sikon A, Thacker HL. Treatment options for menopausal hot flashes. Cleve Clin J Med 2004; 71:578–582.
- Levine JP. Treating menopausal symptoms with a tissue-selective estrogen complex. Gend Med 2011; 8:57–68.
- Pinkerton JV, Thomas S. Use of SERMs for treatment in postmenopausal women. J Steroid Biochem Mol Biol 2014; 142:142–154.
- Tamoxifen for early breast cancer: an overview of the randomised trials. Early Breast Cancer Trialists’ Collaborative Group. Lancet 1998; 351:1451–1467.
- Davies C, Pan H, Godwin J, et al; Adjuvant Tamoxifen: Longer Against Shorter (ATLAS) Collaborative Group. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013; 381:805–816.
- Gray RG, Rea D, Handley K, et al. aTTom: Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years in 6,953 women with early breast cancer. J Clin Oncol 2013; (suppl): abstract 5.
- Cuzick J, Powles T, Veronesi U, et al. Overview of the main outcomes in breast-cancer prevention trials. Lancet 2003; 361:296–300.
- Cuzick J, Sestak I, Cawthorn S, et al. Tamoxifen for prevention of breast cancer: extended long-term follow-up of the IBIS-I breast cancer prevention trial. Lancet Oncol 2015; 16:67–75.
- Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst 1998; 90:1371–1388.
- Chang J, Powles TJ, Ashley SE, et al. The effect of tamoxifen and hormone replacement therapy on serum cholesterol, bone mineral density and coagulation factors in healthy postmenopausal women participating in a randomised, controlled tamoxifen prevention study. Ann Oncol 1996; 7:671–675.
- Herrington DM, Klein KP. Effects of SERMs on important indicators of cardiovascular health: lipoproteins, hemostatic factors and endothelial function. Womens Health Issues 2001; 11:95–102.
- Kedar RP, Bourne TH, Powles TJ, et al. Effects of tamoxifen on uterus and ovaries of postmenopausal women in a randomized breast cancer prevention trial. Lancet 1994; 343:1318–1321.
- Ettinger B, Black DM, Mitlak BH, et al. Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene: results from a 3-year randomized clinical trial. Multiple Outcomes of Raloxifene Evaluation (MORE) Investigators. JAMA 1999; 282:637–645.
- Maricic M, Adachi JD, Sarkar S, Wu W, Wong M, Harper KD. Early effects of raloxifene on clinical vertebral fractures at 12 months in postmenopausal women with osteoporosis. Arch Intern Med 2002; 162:1140–1143.
- Recker RR, Mitlak BH, Ni X, Krege JH. Long-term raloxifene for postmenopausal osteoporosis. Curr Med Res Opin 2011; 27:1755–1761.
- Cummings SR, Eckert S, Krueger KA, et al. The effect of raloxifene on risk of breast cancer in postmenopausal women: results from the MORE randomized trial. Multiple Outcomes of Raloxifene Evaluation. JAMA 1999; 281:2189–2197.
- Martino S, Cauley JA, Barrett-Connor E, et al; CORE Investigators. Continuing outcomes relevant to Evista: breast cancer incidence in postmenopausal osteoporotic women in a randomized trial of raloxifene. J Natl Cancer Inst 2004; 96:1751–1761.
- Vogel VG, Qu Y, Wong M, Mitchell B, Mershon JL. Incidence of invasive breast cancer in postmenopausal women after discontinuation of long-term raloxifene administration. Clin Breast Cancer 2009; 9:45–50.
- Grady D, Cauley JA, Stock JL, et al. Effect of raloxifene on all-cause mortality. Am J Med 2010; 123:469.e1–e7.
- Barrett-Connor E, Mosca L, Collins P, et al; Raloxifene Use for The Heart (RUTH) Trial Investigators. Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. N Engl J Med 2006; 355:125–137.
- Vogel VG. The NSABP Study of Tamoxifen and Raloxifene (STAR) trial. Expert Rev Anticancer Ther 2009; 9:51–60.
- Vogel VG, Costantino JP, Wickerham DL, et al; National Surgical Adjuvant Breast and Bowel Project (NSABP). Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA 2006; 295:2727–2741.
- Barnes KN, Pearce EF, Yancey AM, Forinash AB. Ospemifene in the treatment of vulvovaginal atrophy. Ann Pharmacother 2014; 48:752–757.
- Rutanen EM, Heikkinen J, Halonen K, Komi J, Lammintausta R, Ylikorkala O. Effects of ospemifene, a novel SERM, on hormones, genital tract, climacteric symptoms, and quality of life in postmenopausal women: a double-blind, randomized trial. Menopause 2003; 10:433–439.
- Constantine G, Graham S, Koltun WD, Kingsberg SA. Assessment of ospemifene or lubricants on clinical signs of VVA. J Sex Med 2014; 11:1033–1041.
- Kingsberg SA, Wysocki S, Magnus L, Krychman ML. Vulvar and vaginal atrophy in postmenopausal women: findings from the REVIVE survey. J Sex Med 2013; 10:1790–1799.
- Portman DJ, Bachmann GA, Simon JA; Ospemifene Study Group. Ospemifene, a novel selective estrogen receptor modulator for treating dyspareunia associated with postmenopausal vulvar and vaginal atrophy. Menopause 2013; 20:623–630.
- Bachmann GA, Komi JO; Ospemifene Study Group. Ospemifene effectively treats vulvovaginal atrophy in postmenopausal women: results from a pivotal phase 3 study. Menopause 2010; 17:480–486.
- Goldstein SR, Bachmann GA, Koninckx PR, Lin VH, Portman DJ, Ylikorkala O; Ospemifene Study Group. Ospemifene 12-month safety and efficacy in postmenopausal women with vulvar and vaginal atrophy. Climacteric 2014; 17:173–182.
- Cui Y, Zong H, Yan H, Li N, Zhang Y. The efficacy and safety of ospemifene in treating dyspareunia associated with postmenopausal vulvar and vaginal atrophy: a systematic review and meta-analysis. J Sex Med 2014; 11:487–497.
- Komi J, Heikkinen J, Rutanen EM, Halonen K, Lammintausta R, Ylikorkala O. Effects of ospemifene, a novel SERM, on biochemical markers of bone turnover in healthy postmenopausal women. Gynecal Endocrinol 2004; 18:152–158.
- Komi J, Lankinen KS, DeGregorio M, et al. Effects of ospemifene and raloxifene on biochemical markers of bone turnover in postmenopausal women. J Bone Miner Metab 2006; 24:314–318.
- Wurz GT, Read KC, Marchisano-Karpman C, et al. Ospemifene inhibits the growth of dimethylbenzanthracene-induced mammary tumors in Sencar mice. J Steroid Biochem Mol Biol 2005; 97:230–240.
- Pinkerton JV, Utian WH, Constantine GD, Olivier S, Pickar JH. Relief of vasomotor symptoms with the tissue-selective estrogen complex containing bazedoxifene/conjugated estrogens: a randomized, controlled trial. Menopause 2009; 16:1116–1124.
- Pickar JH, Mirkin S. Tissue-selective agents: selective estrogen receptor modulators and the tissue-selective estrogen complex. Menopause Int 2010; 16:121–128.
- Levine JP. Treating menopausal symptoms with a tissue-selective estrogen complex. Gend Med 2011; 8:57–68.
- Lindsay R, Gallagher JC, Kagan R, Pickar JH, Constantine G. Efficacy of tissue-selective estrange complex of bazedoxifene/conjugated estrogens for osteoporosis prevention in at-risk postmenopausal women. Fertil Steril 2009; 92:1045–1052.
- Bachmann G, Bobula J, Mirkin S. Effects of bazedoxifene/conjugated estrogens on quality of life in postmenopausal women with symptoms of vulvar/vaginal atrophy. Climacteric 2010; 13:132–140.
- Pickar JH, Yeh IT, Bachmann G, Speroff L. Endometrial effects of a tissue selective estrogen complex containing bazedoxifene/conjugated estrogens as a menopausal therapy. Fertil Steril 2009; 92:1018–1024.
- Archer DF, Lewis V, Carr BR, Olivier S, Pickar JH. Bazedoxifene/conjugated estrogens (BZA/CE): incidence of uterine bleeding in postmenopausal women. Fertil Steril 2009: 92:1039–1044.
- Pinkerton JV, Abraham L, Bushmakin AG, et al. Evaluation of the efficacy and safety of bazedoxifene/conjugated estrogens for secondary outcomes including vasomotor symptoms in postmenopausal women by years since menopause in the Selective estrogens, Menopause and Response to Therapy (SMART) trials. J Womens Health (Larchmt) 2014; 23:18–28.
Estrogen receptor agonist-antagonists (ERAAs), previously called selective estrogen receptor modulators (SERMs), have extended the options for treating the various conditions that menopausal women suffer from. These drugs act differently on estrogen receptors in different tissues, stimulating receptors in some tissues but inhibiting them in others. This allows selective inhibition or stimulation of estrogen-like action in various target tissues.1
This article highlights the use of ERAAs to treat menopausal vasomotor symptoms (eg, hot flashes, night sweats), genitourinary syndrome of menopause, osteoporosis, breast cancer (and the risk of breast cancer), and other health concerns unique to women at midlife.
SYMPTOMS OF MENOPAUSE: COMMON AND TROUBLESOME
Vasomotor symptoms such as hot flashes and night sweats are common during perimenopause—most women experience them. They are most frequent during the menopause transition but can persist for 10 years or more afterward.2
Genitourinary syndrome of menopause is also common and often worsens with years after menopause.3 It can lead to dyspareunia and vaginal dryness, which may in turn result in lower libido, vaginismus, and hypoactive sexual desire disorder, problems that often arise at the same time as vaginal dryness and atrophy.4
Osteopenia and osteoporosis. A drop in systemic estrogen leads to a decline in bone mineral density, increasing the risk of fractures.5
ESTROGEN-PROGESTIN TREATMENT: THE GOLD STANDARD, BUT NOT IDEAL
The current gold standard for treating moderate to severe hot flashes is estrogen, available in oral, transdermal, and vaginal formulations.6 Estrogen also has antiresorptive effects on bone and is approved for preventing osteoporosis. Systemic estrogen may also be prescribed for genitourinary syndrome of menopause if local vaginal treatment alone is insufficient.
If women who have an intact uterus receive estrogen, they should also receive a progestin to protect against endometrial hyperplasia and reduce the risk of endometrial cancer.
Despite its status as the gold standard, estrogen-progestin therapy presents challenges. In some women, progestins cause side effects such as breast tenderness, bloating, fatigue, and depression.7 Estrogen-progestin therapy often causes vaginal bleeding, which for some women is troublesome or distressing; bleeding may be the reason for repeated evaluations, can increase anxiety, and can lead to poor adherence with hormonal treatment. Women who carry a higher-than-normal risk of developing breast cancer or fear that taking hormones will lead to breast cancer may show decreased adherence to therapy. Women who have estrogen receptor-positive breast cancer cannot take estrogen.
Individualized options are needed for women who have progestin-related side effects, unwanted vaginal bleeding, or a higher risk of breast cancer.
WELCOME THE ERAAs
An ideal treatment for menopause would relieve vasomotor symptoms and genitourinary syndrome of menopause and increase bone mineral density without causing breast tenderness, vaginal bleeding, or endometrial proliferation.
The “designer estrogens,” or ERAAs, have specific positive effects on the bone, heart, and brain with neutral or antagonist effects on estrogen receptors in other tissues such as the breasts and endometrium.8 While not entirely free of adverse effects, these agents have been developed with the aim of minimizing the most common ones related to estrogen and progestin.
Several ERAAs are currently approved by the US Food and Drug Administration (FDA)for various indications, each having a unique profile. Clomifene was the first agent of this class, and it is still used clinically to induce ovulation. This article highlights subsequently approved agents, ie, tamoxifen, raloxifene, ospemifene, and the combination of conjugated estrogens and bazedoxifene (Table 1).
All ERAAs increase the risk of venous thromboembolism, and therefore none of them should be used in women with known venous thromboembolism or at high risk of it.
TAMOXIFEN: CANCER TREATMENT AND PREVENTION
After clomiphene, tamoxifen was the second ERAA on the market. Although researchers were looking for a new contraceptive drug, they found tamoxifen to be useful as a chemotherapeutic agent for breast cancer. First used in 1971, tamoxifen continues to be one of the most commonly prescribed chemotherapeutic medications today.
The FDA has approved tamoxifen to treat breast cancer as well as to prevent breast cancer in pre- and postmenopausal women at risk. It may also have beneficial effects on bone and on cardiovascular risk factors, but these are not approved uses for it.
Trials of tamoxifen for cancer treatment
The Early Breast Cancer Trialists’ Collaborative Group9 performed a meta-analysis and found that 5 years of adjuvant treatment with tamoxifen is associated with a 26% reduction in mortality and a 47% reduction in breast cancer recurrence at 10 years. In absolute terms, we estimate that 21 women would need to be treated to prevent 1 death and 8 would need to be treated to prevent 1 recurrence.
The ATLAS Trial (Adjuvant Tamoxifen Longer Against Shorter)10 and later the UK ATTOM (Adjuvant Tamoxifen Treatment to Offer More)11 trial confirmed an even greater reduction in recurrence and mortality after a total of 10 years of treatment.
Trials of tamoxifen for cancer prevention
Cuzik et al12 performed a meta-analysis of 4 trials of tamoxifen’s effectiveness in preventing breast cancer for women at elevated risk. The incidence of estrogen receptor-positive breast cancer was 48% lower with tamoxifen use, but there was no effect on estrogen-negative breast cancer. From their data, we estimate that 77 women would need to be treated to prevent 1 case of breast cancer.
The IBIS-I trial (International Breast Cancer Intervention Study I)13 found that, in healthy women at high risk of breast cancer, the benefit of taking tamoxifen for 5 years as preventive treatment persisted long afterward. The investigators estimated that at 20 years of follow-up the risk of breast cancer would be 12.3% in placebo recipients and 7.8% in tamoxifen recipients, a 4.5% absolute risk reduction; number needed to treat (NNT) 22.
Data on tamoxifen and osteoporosis
The Breast Cancer Prevention Trial revealed a 19% reduction in the incidence of osteoporotic fractures with tamoxifen, but the difference was not statistically significant.14 The 1-year rates of fracture in women age 50 and older were 0.727% with placebo and 0.567% with tamoxifen, an absolute difference of 0.151%; therefore, if the effect is real, 662 women age 50 or older would need to be treated for 1 year to prevent 1 fracture. Tamoxifen is not FDA-approved to treat osteoporosis.
Data on tamoxifen and cardiovascular risk reduction
Chang et al,15 in a study in women at risk of breast cancer, incidentally found that tamoxifen was associated with a 13% reduction in total cholesterol compared with placebo.
Herrington and Klein,16 in a systematic review, noted similar findings in multiple studies of tamoxifen, with decreases in total cholesterol ranging from 7% to 17% and decreases in low-density lipoprotein cholesterol ranging from 10% to 28%. However, they found no change in high-density lipoprotein cholesterol concentrations or in the cardiovascular mortality rate.
The ATLAS trial10 revealed a relative risk reduction of 0.76 (95% confidence interval [CI] 0.60–0.95, P = .02) in ischemic heart disease for women who took tamoxifen for 10 years compared with 5 years. We calculate that ischemic heart disease occurred in 163 (2.5%) of 6,440 women who took tamoxifen for 5 years compared with 127 (1.9%) of 6,454 women who took it for 10 years, a 0.6% absolute risk reduction, NNT = 167.
Adverse effects of tamoxifen
Uterine neoplasia. Women taking tamoxifen have a 2.5-fold increased risk of endometrial cancer.14 Tamoxifen also increases the risk of benign uterine disease such as endometrial hyperplasia and polyps. As many as 39% of women taking tamoxifen will have evidence of benign uterine changes on pathology.17 Other adverse effects:
Venous thromboembolism (the risk of pulmonary embolism is increased approximately threefold14)
Cataracts (there is a slight increase in cataract diagnosis in tamoxifen users)
Vasomotor symptoms, which limit the use of tamoxifen in many women.
Ideal candidate for tamoxifen
The ideal candidate for tamoxifen is a woman with breast cancer that is estrogen receptor-positive and who has a history of osteopenia or osteoporosis and no risk factors for venous thromboembolism.
RALOXIFENE: FOR OSTEOPOROSIS AND FOR CANCER PREVENTION
Raloxifene, a second-generation ERAA, was first approved for preventing and treating osteoporosis and later for reducing the risk of invasive estrogen receptor-positive breast cancer in postmenopausal women.
Trials of raloxifene for osteoporosis
The MORE trial (Multiple Outcomes of Raloxifene)18 was a large multicenter randomized double-blind study. Raloxifene recipients showed a significant increase in bone mineral density in the lumbar spine and femoral neck at year 3 (P < .001) compared with those receiving placebo. Even after only 1 year of treatment, raloxifene significantly reduced the risk of new fractures, despite only modest gains in bone mineral density. After 3 years of treatment, new clinical vertebral fractures had occurred in 3.5% of the placebo group compared with 2.1% of the group receiving raloxifene 60 mg.19 Relative risk reductions were similar in women who had already had a clinical vertebral fracture at baseline, whose absolute risk is higher. However, no significant effect was seen on the incidence of hip or nonvertebral fractures.
The CORE trial (Continuing Outcomes Relevant to Raloxifene)20 extended the treatment of the women enrolled in the MORE trial another 4 years and found that the benefit of raloxifene with regard to bone mineral density persisted with continued use.
Trials of raloxifene for breast cancer prevention
The MORE trial,21 in postmenopausal women with osteoporosis included breast cancer as a secondary end point, and raloxifene was shown to decrease the incidence of invasive breast cancer. At a median of 40 months, invasive breast cancer had arisen in 13 (0.25%) of the 5,129 women assigned to raloxifene and 27 (1.0%) of the 2,576 women assigned to placebo. The authors calculated that 126 women would need to be treated to prevent 1 case of breast cancer.
The CORE trial,22 as noted, extended the treatment of the women enrolled in the MORE trial another 4 years. The risk of any invasive breast cancer in postmenopausal women with osteoporosis was significantly reduced by 59% after 8 years, and the risk of estrogen receptor-positive invasive breast cancer was reduced by 66%.
There is evidence that raloxifene’s effect on breast cancer risk persists after discontinuation of use.23
Does raloxifene reduce mortality?
Grady et al24 studied the effect of raloxifene on all-cause mortality in a pooled analysis of mortality data from the MORE, CORE, and Raloxifene Use for the Heart (RUTH)25 trials. In older postmenopausal women, the rate of all-cause mortality was 8.65% in those taking placebo compared with 7.88% in those taking raloxifene 60 mg daily—10% lower. The mechanism behind the lower mortality rate is unclear, and Grady et al recommend that the finding be interpreted with caution.
Trials of raloxifene for heart protection
The RUTH trial25 was a 5.6-year study undertaken to study the effects of raloxifene on coronary outcomes and invasive breast cancer in postmenopausal women. Results were mixed. Active treatment:
- Did not significantly affect the risk of coronary artery disease compared with placebo
- Significantly decreased the risk of invasive breast cancer
- Significantly decreased the risk of clinical vertebral fractures
- Increased the risk of fatal stroke (59 vs 39 events, hazard ratio 1.49, 95% CI 1.00–2.24) and venous thromboembolism (103 vs 71 events, hazard ratio 1.44, 95% CI 1.06–1.95).
The STAR trial (Study of Tamoxifen and Raloxifene)26,27 compared raloxifene and tamoxifen in postmenopausal women at increased risk of breast cancer. Women were randomized to receive either tamoxifen 20 mg or raloxifene 60 mg for 5 years. Results:
- No difference in the number of new cases of invasive breast cancer between the groups
- Fewer cases of noninvasive breast cancer in the tamoxifen group, but the difference was not statistically significant
- Fewer cases of uterine cancer in the raloxifene group, annual incidence rates 0.125% vs 0.199%, absolute risk reduction 0.74%, NNT 1,351, relative risk with raloxifene 0.62, 95% CI 0.30–0.50
- Fewer thromboembolic events with raloxifene
- Fewer cataracts with raloxifene.
Adverse effects of raloxifene
Raloxifene increases the risk of venous thromboembolism and stroke in women at high risk of coronary artery disease.19
Ideal candidates for raloxifene
Postmenopausal women with osteopenia or osteoporosis and a higher risk of breast cancer who have minimal to no vasomotor symptoms or genitourinary syndrome of menopause are good candidates for raloxifene. Raloxifene is also a good choice for women who have genitourinary syndrome of menopause treated with local vaginal estrogen. Raloxifene has no effect on vasomotor symptoms or genitourinary syndrome of menopause.
OSPEMIFENE: FOR GENITOURINARY SYNDROME OF MENOPAUSE
Although ospemifene does not have the steroid structure of estrogen, it acts as an estrogen agonist specifically in the vaginal mucosa and an antagonist in other tissues.28 It has been shown on Papanicolaou smears to reduce the number of parabasal cells and increase the number of intermediate and superficial cells after 3 months of treatment.29
Ospemifene 60 mg taken orally with food is approved by the FDA to treat genitourinary syndrome of menopause.
Why ospemifene is needed
First-line treatment options for genitourinary syndrome of menopause include over-the-counter lubricants. However, there is no evidence that these products reverse vaginal atrophy,30 and many women report no relief of symptoms with them.
While various local estrogen preparations positively affect genitourinary syndrome of menopause, some of them can be messy, which can limit-long term adherence.
In one of the largest surveys on genitourinary syndrome of menopause (the REVIVE survey—the Real Women’s View of Treatment Options for Menopausal Vaginal Changes29), 59% of women reported that their vaginal symptoms negatively affected sexual activity. The problem affects not only the patient but also her sexual partner.31 Another large study showed that 38% of women and 39% of male partners reported that it had a worse-than-expected impact on their intimate relationships.31
Genitourinary syndrome of menopause also makes pelvic examinations difficult, may worsen or exacerbate cystitis, and may increase urinary tract infections.
Trials of ospemifene for genitourinary syndrome of menopause
To date, 3 randomized, double-blind clinical trials have demonstrated ospemifene 60 mg to be superior to placebo in treating genitourinary syndrome of menopause. Two were short-term (12-week) and showed significant positive changes in the percent of superficial cells, vaginal pH (lower is better), and number of parabasal cells, along with improvements in the Likert rating of both vaginal dryness and dyspareunia.32,33
A long-term (52-week) randomized placebo-controlled trial compared ospemifene and placebo and showed significant improvement in vaginal maturation index and pH at weeks 12 and 52.34 Other outcome measures included petechiae, pallor, friability, erythema, and dryness, all of which improved from baseline (P < .001). At the end of the trial, 80% of the patients who received ospemifene had no vaginal atrophy.
No serious adverse events were noted in any of the clinical trials to date, and a systemic review and meta-analysis demonstrated ospemifene to be safe and efficacious.35 The most frequently reported reasons for discontinuation were hot flashes, vaginal discharge, muscle spasms, and hyperhidrosis, but the rates of these effects were similar to those with placebo.
Trial of ospemifene’s effect on bone turnover
As an estrogen receptor agonist in bone, ospemifene decreases the levels of bone turnover markers in postmenopausal women.36 A study found ospemifene to be about as effective as raloxifene in suppressing bone turnover,37 but ospemifene does not carry FDA approval for preventing or treating osteoporosis.
Other effects
In experiments in rats, the incidence of breast cancer appears to be lower with ospemifene, and the higher the dose, the lower the incidence.38
Ospemifene also has antagonistic effects on uterine tissue, and no cases of endometrial hyperplasia or carcinoma have been reported in short-term or long-term studies.35
Ospemifene has no effect however on vasomotor symptoms and may in fact worsen vasomotor symptoms in women suffering with hot flashes and night sweats. Further investigation into its long-term safety and effects on breast tissue and bone would provide more insight.
Ideal candidates for ospemifene
Ospemifene could help postmenopausal women with genitourinary syndrome of menopause for whom over-the-counter lubricants fail, who dislike local vaginal estrogen, or who decline systemic hormone therapy, and who do not meet the criteria for treatment with systemic hormone therapy.
CONJUGATED ESTROGENS AND BAZEDOXIFENE COMBINATION
A combination agent consisting of conjugated estrogens 0.45 mg plus bazedoxifene 20 mg has been approved by the FDA for treating moderate to severe vasomotor symptoms associated with menopause and also for preventing postmenopausal osteoporosis in women who have an intact uterus.
Trials of estrogen-bazedoxifene for vasomotor symptoms
The Selective Estrogen Menopause and Response to Therapy (SMART) trials39,40 were a series of randomized, double-blind, placebo-controlled phase 3 studies evaluating the efficacy and safety of the estrogen-bazedoxifene combination in postmenopausal women.
The SMART-2 trial39 evaluated the combination of conjugated estrogens (either 0.45 mg or 0.625) plus bazedoxifene 20 mg and found both dosages significantly reduced the number and severity of hot flashes at weeks 4 and 12 (P < .001). At week 12, the combination with 0.45 mg of estrogen reduced vasomotor symptoms from baseline by 74% (10.3 hot flashes per week at baseline vs 2.8 at week 12); the combination with 0.625 mg of estrogen reduced vasomotor symptoms by 80% (10.4 vs 2.4 flashes); and placebo reduced them by 51% (10.5 vs 5.4 flashes).
For bone density. The SMART-1 trial40 showed that the estrogen-bazedoxifene combination in both estrogen dosages significantly increased mean lumbar spine bone mineral density (P < .001) and total hip bone mineral density (P < .05) from baseline at 12 and 24 months compared with placebo. Increases in density tended to be higher with the higher estrogen dose (0.625 mg), but less with higher doses of bazedoxifene.41 At 24 months, the increase in bone mineral density was even greater than in women treated with raloxifene.42 However, the effect of estrogen-bazedoxifene on the incidence of fractures remains to be studied.
For genitourinary syndrome of menopause. The SMART-3 trial showed that treatment with conjugated estrogens plus bazedoxifene (0.45/20 mg or 0.625/20 mg) was more effective than placebo in increasing the percent of superficial and intermediate cells and decreased the number of parabasal cells at 12 weeks compared with placebo (P < .01).43 Both doses also significantly decreased the mean vaginal pH and improved vaginal dryness.
Patients treated with estrogen-bazedoxifene for a minimum of 12 weeks in a double-blind placebo-controlled study also showed a significant improvement in sexual function and quality-of-life measurements based on 3 well-defined scales, which included ease of lubrication, satisfaction with treatment, control of hot flashes, and sleep parameters.43
Low rates of side effects
To evaluate this regimen’s antagonistic effects on uterine tissue, endometrial hyperplasia was diagnosed by blinded pathologists using endometrial biopsies taken at 6, 12, and 24 months or more if cancer was a suspected diagnosis. At 12 and 24 months of treatment, the incidence of hyperplasia with bazedoxifene 20 or 40 mg at doses of either 0.45 or 0.625 mg of conjugated estrogens was less than 1%, which was similar to placebo rates over the 24 months.44 The lowest dose studied, bazedoxifene 10 mg, did not prevent hyperplasia with conjugated estrogens 0.45 or 0.625 mg, and its use was discontinued.
Rates of amenorrhea with bazedoxifene 20 or 40 mg and conjugated estrogens 0.45 or 0.625 mg were very favorable (83%–93%) and similar to those with placebo.45 For women with continued bleeding on hormone therapy requiring multiple evaluations, or for women who won’t accept the risk of bleeding on hormone therapy, conjugated estrogens and bazedoxifene may be a sustainable option. However, any woman with abnormal bleeding should undergo prompt immediate evaluation.
A typical side effect of estrogen replacement therapy is breast tenderness. For women seeking vasomotor symptom treatment but who experience breast tenderness, this may be a deterrent from continuing hormone therapy. As shown in the SMART-1 and SMART-2 trials,46 conjugated estrogens and bazedoxifene did not cause an increase in breast tenderness, which may enhance medication adherence.
Ideal candidates for conjugated estrogens plus bazedoxifene
This product could help postmenopausal women who have an intact uterus and are suffering with moderate to severe vasomotor symptoms and genitourinary syndrome of menopause who cannot tolerate the side effects of hormone therapy such as bleeding, bloating, or breast tenderness, or who prefer to take an estrogen but without a progestin. It is also ideal for women at higher risk of osteoporosis.
WHO SHOULD GET WHAT?
Not all postmenopausal women have vasomotor symptoms, genitourinary syndrome of menopause, or bone loss. For those who do, standard hormone therapy is an option.
For those who have symptoms and a lower threshold of side effects such as breast tenderness and vaginal bleeding, a combination of an estrogen plus an ERAA (eg, bazedoxifene) is an option.
For women who have no vasomotor symptoms but do have genitourinary syndrome of menopause and don’t want local vaginal treatment, ospemifene is an option.
For women with no vasomotor symptoms but who have bone loss and increased risk of estrogen receptor-positive breast cancer, raloxifene is a good option.
Both premenopausal and postmenopausal women who are at increased risk for breast cancer should be considered for tamoxifen chemoprevention. Postmenopausal women with a uterus at increased risk for breast cancer should be considered for raloxifene, as it has no uterine effect. Raloxifene is not indicated in premenopausal women.
No woman at increased risk of venous thromboembolism is a candidate for ERAA treatment or for oral estrogen. However, the clinician has multiple options to improve quality of life and work productivity and reduce office visits of women at midlife, especially when they are individually assessed and treated.
Estrogen receptor agonist-antagonists (ERAAs), previously called selective estrogen receptor modulators (SERMs), have extended the options for treating the various conditions that menopausal women suffer from. These drugs act differently on estrogen receptors in different tissues, stimulating receptors in some tissues but inhibiting them in others. This allows selective inhibition or stimulation of estrogen-like action in various target tissues.1
This article highlights the use of ERAAs to treat menopausal vasomotor symptoms (eg, hot flashes, night sweats), genitourinary syndrome of menopause, osteoporosis, breast cancer (and the risk of breast cancer), and other health concerns unique to women at midlife.
SYMPTOMS OF MENOPAUSE: COMMON AND TROUBLESOME
Vasomotor symptoms such as hot flashes and night sweats are common during perimenopause—most women experience them. They are most frequent during the menopause transition but can persist for 10 years or more afterward.2
Genitourinary syndrome of menopause is also common and often worsens with years after menopause.3 It can lead to dyspareunia and vaginal dryness, which may in turn result in lower libido, vaginismus, and hypoactive sexual desire disorder, problems that often arise at the same time as vaginal dryness and atrophy.4
Osteopenia and osteoporosis. A drop in systemic estrogen leads to a decline in bone mineral density, increasing the risk of fractures.5
ESTROGEN-PROGESTIN TREATMENT: THE GOLD STANDARD, BUT NOT IDEAL
The current gold standard for treating moderate to severe hot flashes is estrogen, available in oral, transdermal, and vaginal formulations.6 Estrogen also has antiresorptive effects on bone and is approved for preventing osteoporosis. Systemic estrogen may also be prescribed for genitourinary syndrome of menopause if local vaginal treatment alone is insufficient.
If women who have an intact uterus receive estrogen, they should also receive a progestin to protect against endometrial hyperplasia and reduce the risk of endometrial cancer.
Despite its status as the gold standard, estrogen-progestin therapy presents challenges. In some women, progestins cause side effects such as breast tenderness, bloating, fatigue, and depression.7 Estrogen-progestin therapy often causes vaginal bleeding, which for some women is troublesome or distressing; bleeding may be the reason for repeated evaluations, can increase anxiety, and can lead to poor adherence with hormonal treatment. Women who carry a higher-than-normal risk of developing breast cancer or fear that taking hormones will lead to breast cancer may show decreased adherence to therapy. Women who have estrogen receptor-positive breast cancer cannot take estrogen.
Individualized options are needed for women who have progestin-related side effects, unwanted vaginal bleeding, or a higher risk of breast cancer.
WELCOME THE ERAAs
An ideal treatment for menopause would relieve vasomotor symptoms and genitourinary syndrome of menopause and increase bone mineral density without causing breast tenderness, vaginal bleeding, or endometrial proliferation.
The “designer estrogens,” or ERAAs, have specific positive effects on the bone, heart, and brain with neutral or antagonist effects on estrogen receptors in other tissues such as the breasts and endometrium.8 While not entirely free of adverse effects, these agents have been developed with the aim of minimizing the most common ones related to estrogen and progestin.
Several ERAAs are currently approved by the US Food and Drug Administration (FDA)for various indications, each having a unique profile. Clomifene was the first agent of this class, and it is still used clinically to induce ovulation. This article highlights subsequently approved agents, ie, tamoxifen, raloxifene, ospemifene, and the combination of conjugated estrogens and bazedoxifene (Table 1).
All ERAAs increase the risk of venous thromboembolism, and therefore none of them should be used in women with known venous thromboembolism or at high risk of it.
TAMOXIFEN: CANCER TREATMENT AND PREVENTION
After clomiphene, tamoxifen was the second ERAA on the market. Although researchers were looking for a new contraceptive drug, they found tamoxifen to be useful as a chemotherapeutic agent for breast cancer. First used in 1971, tamoxifen continues to be one of the most commonly prescribed chemotherapeutic medications today.
The FDA has approved tamoxifen to treat breast cancer as well as to prevent breast cancer in pre- and postmenopausal women at risk. It may also have beneficial effects on bone and on cardiovascular risk factors, but these are not approved uses for it.
Trials of tamoxifen for cancer treatment
The Early Breast Cancer Trialists’ Collaborative Group9 performed a meta-analysis and found that 5 years of adjuvant treatment with tamoxifen is associated with a 26% reduction in mortality and a 47% reduction in breast cancer recurrence at 10 years. In absolute terms, we estimate that 21 women would need to be treated to prevent 1 death and 8 would need to be treated to prevent 1 recurrence.
The ATLAS Trial (Adjuvant Tamoxifen Longer Against Shorter)10 and later the UK ATTOM (Adjuvant Tamoxifen Treatment to Offer More)11 trial confirmed an even greater reduction in recurrence and mortality after a total of 10 years of treatment.
Trials of tamoxifen for cancer prevention
Cuzik et al12 performed a meta-analysis of 4 trials of tamoxifen’s effectiveness in preventing breast cancer for women at elevated risk. The incidence of estrogen receptor-positive breast cancer was 48% lower with tamoxifen use, but there was no effect on estrogen-negative breast cancer. From their data, we estimate that 77 women would need to be treated to prevent 1 case of breast cancer.
The IBIS-I trial (International Breast Cancer Intervention Study I)13 found that, in healthy women at high risk of breast cancer, the benefit of taking tamoxifen for 5 years as preventive treatment persisted long afterward. The investigators estimated that at 20 years of follow-up the risk of breast cancer would be 12.3% in placebo recipients and 7.8% in tamoxifen recipients, a 4.5% absolute risk reduction; number needed to treat (NNT) 22.
Data on tamoxifen and osteoporosis
The Breast Cancer Prevention Trial revealed a 19% reduction in the incidence of osteoporotic fractures with tamoxifen, but the difference was not statistically significant.14 The 1-year rates of fracture in women age 50 and older were 0.727% with placebo and 0.567% with tamoxifen, an absolute difference of 0.151%; therefore, if the effect is real, 662 women age 50 or older would need to be treated for 1 year to prevent 1 fracture. Tamoxifen is not FDA-approved to treat osteoporosis.
Data on tamoxifen and cardiovascular risk reduction
Chang et al,15 in a study in women at risk of breast cancer, incidentally found that tamoxifen was associated with a 13% reduction in total cholesterol compared with placebo.
Herrington and Klein,16 in a systematic review, noted similar findings in multiple studies of tamoxifen, with decreases in total cholesterol ranging from 7% to 17% and decreases in low-density lipoprotein cholesterol ranging from 10% to 28%. However, they found no change in high-density lipoprotein cholesterol concentrations or in the cardiovascular mortality rate.
The ATLAS trial10 revealed a relative risk reduction of 0.76 (95% confidence interval [CI] 0.60–0.95, P = .02) in ischemic heart disease for women who took tamoxifen for 10 years compared with 5 years. We calculate that ischemic heart disease occurred in 163 (2.5%) of 6,440 women who took tamoxifen for 5 years compared with 127 (1.9%) of 6,454 women who took it for 10 years, a 0.6% absolute risk reduction, NNT = 167.
Adverse effects of tamoxifen
Uterine neoplasia. Women taking tamoxifen have a 2.5-fold increased risk of endometrial cancer.14 Tamoxifen also increases the risk of benign uterine disease such as endometrial hyperplasia and polyps. As many as 39% of women taking tamoxifen will have evidence of benign uterine changes on pathology.17 Other adverse effects:
Venous thromboembolism (the risk of pulmonary embolism is increased approximately threefold14)
Cataracts (there is a slight increase in cataract diagnosis in tamoxifen users)
Vasomotor symptoms, which limit the use of tamoxifen in many women.
Ideal candidate for tamoxifen
The ideal candidate for tamoxifen is a woman with breast cancer that is estrogen receptor-positive and who has a history of osteopenia or osteoporosis and no risk factors for venous thromboembolism.
RALOXIFENE: FOR OSTEOPOROSIS AND FOR CANCER PREVENTION
Raloxifene, a second-generation ERAA, was first approved for preventing and treating osteoporosis and later for reducing the risk of invasive estrogen receptor-positive breast cancer in postmenopausal women.
Trials of raloxifene for osteoporosis
The MORE trial (Multiple Outcomes of Raloxifene)18 was a large multicenter randomized double-blind study. Raloxifene recipients showed a significant increase in bone mineral density in the lumbar spine and femoral neck at year 3 (P < .001) compared with those receiving placebo. Even after only 1 year of treatment, raloxifene significantly reduced the risk of new fractures, despite only modest gains in bone mineral density. After 3 years of treatment, new clinical vertebral fractures had occurred in 3.5% of the placebo group compared with 2.1% of the group receiving raloxifene 60 mg.19 Relative risk reductions were similar in women who had already had a clinical vertebral fracture at baseline, whose absolute risk is higher. However, no significant effect was seen on the incidence of hip or nonvertebral fractures.
The CORE trial (Continuing Outcomes Relevant to Raloxifene)20 extended the treatment of the women enrolled in the MORE trial another 4 years and found that the benefit of raloxifene with regard to bone mineral density persisted with continued use.
Trials of raloxifene for breast cancer prevention
The MORE trial,21 in postmenopausal women with osteoporosis included breast cancer as a secondary end point, and raloxifene was shown to decrease the incidence of invasive breast cancer. At a median of 40 months, invasive breast cancer had arisen in 13 (0.25%) of the 5,129 women assigned to raloxifene and 27 (1.0%) of the 2,576 women assigned to placebo. The authors calculated that 126 women would need to be treated to prevent 1 case of breast cancer.
The CORE trial,22 as noted, extended the treatment of the women enrolled in the MORE trial another 4 years. The risk of any invasive breast cancer in postmenopausal women with osteoporosis was significantly reduced by 59% after 8 years, and the risk of estrogen receptor-positive invasive breast cancer was reduced by 66%.
There is evidence that raloxifene’s effect on breast cancer risk persists after discontinuation of use.23
Does raloxifene reduce mortality?
Grady et al24 studied the effect of raloxifene on all-cause mortality in a pooled analysis of mortality data from the MORE, CORE, and Raloxifene Use for the Heart (RUTH)25 trials. In older postmenopausal women, the rate of all-cause mortality was 8.65% in those taking placebo compared with 7.88% in those taking raloxifene 60 mg daily—10% lower. The mechanism behind the lower mortality rate is unclear, and Grady et al recommend that the finding be interpreted with caution.
Trials of raloxifene for heart protection
The RUTH trial25 was a 5.6-year study undertaken to study the effects of raloxifene on coronary outcomes and invasive breast cancer in postmenopausal women. Results were mixed. Active treatment:
- Did not significantly affect the risk of coronary artery disease compared with placebo
- Significantly decreased the risk of invasive breast cancer
- Significantly decreased the risk of clinical vertebral fractures
- Increased the risk of fatal stroke (59 vs 39 events, hazard ratio 1.49, 95% CI 1.00–2.24) and venous thromboembolism (103 vs 71 events, hazard ratio 1.44, 95% CI 1.06–1.95).
The STAR trial (Study of Tamoxifen and Raloxifene)26,27 compared raloxifene and tamoxifen in postmenopausal women at increased risk of breast cancer. Women were randomized to receive either tamoxifen 20 mg or raloxifene 60 mg for 5 years. Results:
- No difference in the number of new cases of invasive breast cancer between the groups
- Fewer cases of noninvasive breast cancer in the tamoxifen group, but the difference was not statistically significant
- Fewer cases of uterine cancer in the raloxifene group, annual incidence rates 0.125% vs 0.199%, absolute risk reduction 0.74%, NNT 1,351, relative risk with raloxifene 0.62, 95% CI 0.30–0.50
- Fewer thromboembolic events with raloxifene
- Fewer cataracts with raloxifene.
Adverse effects of raloxifene
Raloxifene increases the risk of venous thromboembolism and stroke in women at high risk of coronary artery disease.19
Ideal candidates for raloxifene
Postmenopausal women with osteopenia or osteoporosis and a higher risk of breast cancer who have minimal to no vasomotor symptoms or genitourinary syndrome of menopause are good candidates for raloxifene. Raloxifene is also a good choice for women who have genitourinary syndrome of menopause treated with local vaginal estrogen. Raloxifene has no effect on vasomotor symptoms or genitourinary syndrome of menopause.
OSPEMIFENE: FOR GENITOURINARY SYNDROME OF MENOPAUSE
Although ospemifene does not have the steroid structure of estrogen, it acts as an estrogen agonist specifically in the vaginal mucosa and an antagonist in other tissues.28 It has been shown on Papanicolaou smears to reduce the number of parabasal cells and increase the number of intermediate and superficial cells after 3 months of treatment.29
Ospemifene 60 mg taken orally with food is approved by the FDA to treat genitourinary syndrome of menopause.
Why ospemifene is needed
First-line treatment options for genitourinary syndrome of menopause include over-the-counter lubricants. However, there is no evidence that these products reverse vaginal atrophy,30 and many women report no relief of symptoms with them.
While various local estrogen preparations positively affect genitourinary syndrome of menopause, some of them can be messy, which can limit-long term adherence.
In one of the largest surveys on genitourinary syndrome of menopause (the REVIVE survey—the Real Women’s View of Treatment Options for Menopausal Vaginal Changes29), 59% of women reported that their vaginal symptoms negatively affected sexual activity. The problem affects not only the patient but also her sexual partner.31 Another large study showed that 38% of women and 39% of male partners reported that it had a worse-than-expected impact on their intimate relationships.31
Genitourinary syndrome of menopause also makes pelvic examinations difficult, may worsen or exacerbate cystitis, and may increase urinary tract infections.
Trials of ospemifene for genitourinary syndrome of menopause
To date, 3 randomized, double-blind clinical trials have demonstrated ospemifene 60 mg to be superior to placebo in treating genitourinary syndrome of menopause. Two were short-term (12-week) and showed significant positive changes in the percent of superficial cells, vaginal pH (lower is better), and number of parabasal cells, along with improvements in the Likert rating of both vaginal dryness and dyspareunia.32,33
A long-term (52-week) randomized placebo-controlled trial compared ospemifene and placebo and showed significant improvement in vaginal maturation index and pH at weeks 12 and 52.34 Other outcome measures included petechiae, pallor, friability, erythema, and dryness, all of which improved from baseline (P < .001). At the end of the trial, 80% of the patients who received ospemifene had no vaginal atrophy.
No serious adverse events were noted in any of the clinical trials to date, and a systemic review and meta-analysis demonstrated ospemifene to be safe and efficacious.35 The most frequently reported reasons for discontinuation were hot flashes, vaginal discharge, muscle spasms, and hyperhidrosis, but the rates of these effects were similar to those with placebo.
Trial of ospemifene’s effect on bone turnover
As an estrogen receptor agonist in bone, ospemifene decreases the levels of bone turnover markers in postmenopausal women.36 A study found ospemifene to be about as effective as raloxifene in suppressing bone turnover,37 but ospemifene does not carry FDA approval for preventing or treating osteoporosis.
Other effects
In experiments in rats, the incidence of breast cancer appears to be lower with ospemifene, and the higher the dose, the lower the incidence.38
Ospemifene also has antagonistic effects on uterine tissue, and no cases of endometrial hyperplasia or carcinoma have been reported in short-term or long-term studies.35
Ospemifene has no effect however on vasomotor symptoms and may in fact worsen vasomotor symptoms in women suffering with hot flashes and night sweats. Further investigation into its long-term safety and effects on breast tissue and bone would provide more insight.
Ideal candidates for ospemifene
Ospemifene could help postmenopausal women with genitourinary syndrome of menopause for whom over-the-counter lubricants fail, who dislike local vaginal estrogen, or who decline systemic hormone therapy, and who do not meet the criteria for treatment with systemic hormone therapy.
CONJUGATED ESTROGENS AND BAZEDOXIFENE COMBINATION
A combination agent consisting of conjugated estrogens 0.45 mg plus bazedoxifene 20 mg has been approved by the FDA for treating moderate to severe vasomotor symptoms associated with menopause and also for preventing postmenopausal osteoporosis in women who have an intact uterus.
Trials of estrogen-bazedoxifene for vasomotor symptoms
The Selective Estrogen Menopause and Response to Therapy (SMART) trials39,40 were a series of randomized, double-blind, placebo-controlled phase 3 studies evaluating the efficacy and safety of the estrogen-bazedoxifene combination in postmenopausal women.
The SMART-2 trial39 evaluated the combination of conjugated estrogens (either 0.45 mg or 0.625) plus bazedoxifene 20 mg and found both dosages significantly reduced the number and severity of hot flashes at weeks 4 and 12 (P < .001). At week 12, the combination with 0.45 mg of estrogen reduced vasomotor symptoms from baseline by 74% (10.3 hot flashes per week at baseline vs 2.8 at week 12); the combination with 0.625 mg of estrogen reduced vasomotor symptoms by 80% (10.4 vs 2.4 flashes); and placebo reduced them by 51% (10.5 vs 5.4 flashes).
For bone density. The SMART-1 trial40 showed that the estrogen-bazedoxifene combination in both estrogen dosages significantly increased mean lumbar spine bone mineral density (P < .001) and total hip bone mineral density (P < .05) from baseline at 12 and 24 months compared with placebo. Increases in density tended to be higher with the higher estrogen dose (0.625 mg), but less with higher doses of bazedoxifene.41 At 24 months, the increase in bone mineral density was even greater than in women treated with raloxifene.42 However, the effect of estrogen-bazedoxifene on the incidence of fractures remains to be studied.
For genitourinary syndrome of menopause. The SMART-3 trial showed that treatment with conjugated estrogens plus bazedoxifene (0.45/20 mg or 0.625/20 mg) was more effective than placebo in increasing the percent of superficial and intermediate cells and decreased the number of parabasal cells at 12 weeks compared with placebo (P < .01).43 Both doses also significantly decreased the mean vaginal pH and improved vaginal dryness.
Patients treated with estrogen-bazedoxifene for a minimum of 12 weeks in a double-blind placebo-controlled study also showed a significant improvement in sexual function and quality-of-life measurements based on 3 well-defined scales, which included ease of lubrication, satisfaction with treatment, control of hot flashes, and sleep parameters.43
Low rates of side effects
To evaluate this regimen’s antagonistic effects on uterine tissue, endometrial hyperplasia was diagnosed by blinded pathologists using endometrial biopsies taken at 6, 12, and 24 months or more if cancer was a suspected diagnosis. At 12 and 24 months of treatment, the incidence of hyperplasia with bazedoxifene 20 or 40 mg at doses of either 0.45 or 0.625 mg of conjugated estrogens was less than 1%, which was similar to placebo rates over the 24 months.44 The lowest dose studied, bazedoxifene 10 mg, did not prevent hyperplasia with conjugated estrogens 0.45 or 0.625 mg, and its use was discontinued.
Rates of amenorrhea with bazedoxifene 20 or 40 mg and conjugated estrogens 0.45 or 0.625 mg were very favorable (83%–93%) and similar to those with placebo.45 For women with continued bleeding on hormone therapy requiring multiple evaluations, or for women who won’t accept the risk of bleeding on hormone therapy, conjugated estrogens and bazedoxifene may be a sustainable option. However, any woman with abnormal bleeding should undergo prompt immediate evaluation.
A typical side effect of estrogen replacement therapy is breast tenderness. For women seeking vasomotor symptom treatment but who experience breast tenderness, this may be a deterrent from continuing hormone therapy. As shown in the SMART-1 and SMART-2 trials,46 conjugated estrogens and bazedoxifene did not cause an increase in breast tenderness, which may enhance medication adherence.
Ideal candidates for conjugated estrogens plus bazedoxifene
This product could help postmenopausal women who have an intact uterus and are suffering with moderate to severe vasomotor symptoms and genitourinary syndrome of menopause who cannot tolerate the side effects of hormone therapy such as bleeding, bloating, or breast tenderness, or who prefer to take an estrogen but without a progestin. It is also ideal for women at higher risk of osteoporosis.
WHO SHOULD GET WHAT?
Not all postmenopausal women have vasomotor symptoms, genitourinary syndrome of menopause, or bone loss. For those who do, standard hormone therapy is an option.
For those who have symptoms and a lower threshold of side effects such as breast tenderness and vaginal bleeding, a combination of an estrogen plus an ERAA (eg, bazedoxifene) is an option.
For women who have no vasomotor symptoms but do have genitourinary syndrome of menopause and don’t want local vaginal treatment, ospemifene is an option.
For women with no vasomotor symptoms but who have bone loss and increased risk of estrogen receptor-positive breast cancer, raloxifene is a good option.
Both premenopausal and postmenopausal women who are at increased risk for breast cancer should be considered for tamoxifen chemoprevention. Postmenopausal women with a uterus at increased risk for breast cancer should be considered for raloxifene, as it has no uterine effect. Raloxifene is not indicated in premenopausal women.
No woman at increased risk of venous thromboembolism is a candidate for ERAA treatment or for oral estrogen. However, the clinician has multiple options to improve quality of life and work productivity and reduce office visits of women at midlife, especially when they are individually assessed and treated.
- Giannini A, Russo E, Mannella P, Simoncini T. Selective steroid receptor modulators in reproductive medicine. Minerva Ginecol 2015; 67:431–455.
- Feldman BM, Voda A, Gronseth E. The prevalence of hot flash and associated variables among perimenopausal women. Res Nurs Health 1985; 8:261–268.
- Versi E, Harvey MA, Cardozo L, Brincat M, Studd JW. Urogenital prolapse and atrophy at menopause: a prevalence study. Int Urogynecol J Pelvic Floor Dysfunct 2001; 12:107–110.
- Hess R, Chang CC, Conigliaro J, McNeil M. Understanding physicians’ attitudes towards hormone therapy. Womens Health Issues 2005; 15:31–38.
- Melton LJ 3rd, Khosla S, Atkinson EJ, O’Fallon WM, Riggs BL. Relationship of bone turnover to bone density and fractures. J Bone Miner Res 1997; 12:1083–1091.
- Sikon A, Thacker HL. Treatment options for menopausal hot flashes. Cleve Clin J Med 2004; 71:578–582.
- Levine JP. Treating menopausal symptoms with a tissue-selective estrogen complex. Gend Med 2011; 8:57–68.
- Pinkerton JV, Thomas S. Use of SERMs for treatment in postmenopausal women. J Steroid Biochem Mol Biol 2014; 142:142–154.
- Tamoxifen for early breast cancer: an overview of the randomised trials. Early Breast Cancer Trialists’ Collaborative Group. Lancet 1998; 351:1451–1467.
- Davies C, Pan H, Godwin J, et al; Adjuvant Tamoxifen: Longer Against Shorter (ATLAS) Collaborative Group. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013; 381:805–816.
- Gray RG, Rea D, Handley K, et al. aTTom: Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years in 6,953 women with early breast cancer. J Clin Oncol 2013; (suppl): abstract 5.
- Cuzick J, Powles T, Veronesi U, et al. Overview of the main outcomes in breast-cancer prevention trials. Lancet 2003; 361:296–300.
- Cuzick J, Sestak I, Cawthorn S, et al. Tamoxifen for prevention of breast cancer: extended long-term follow-up of the IBIS-I breast cancer prevention trial. Lancet Oncol 2015; 16:67–75.
- Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst 1998; 90:1371–1388.
- Chang J, Powles TJ, Ashley SE, et al. The effect of tamoxifen and hormone replacement therapy on serum cholesterol, bone mineral density and coagulation factors in healthy postmenopausal women participating in a randomised, controlled tamoxifen prevention study. Ann Oncol 1996; 7:671–675.
- Herrington DM, Klein KP. Effects of SERMs on important indicators of cardiovascular health: lipoproteins, hemostatic factors and endothelial function. Womens Health Issues 2001; 11:95–102.
- Kedar RP, Bourne TH, Powles TJ, et al. Effects of tamoxifen on uterus and ovaries of postmenopausal women in a randomized breast cancer prevention trial. Lancet 1994; 343:1318–1321.
- Ettinger B, Black DM, Mitlak BH, et al. Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene: results from a 3-year randomized clinical trial. Multiple Outcomes of Raloxifene Evaluation (MORE) Investigators. JAMA 1999; 282:637–645.
- Maricic M, Adachi JD, Sarkar S, Wu W, Wong M, Harper KD. Early effects of raloxifene on clinical vertebral fractures at 12 months in postmenopausal women with osteoporosis. Arch Intern Med 2002; 162:1140–1143.
- Recker RR, Mitlak BH, Ni X, Krege JH. Long-term raloxifene for postmenopausal osteoporosis. Curr Med Res Opin 2011; 27:1755–1761.
- Cummings SR, Eckert S, Krueger KA, et al. The effect of raloxifene on risk of breast cancer in postmenopausal women: results from the MORE randomized trial. Multiple Outcomes of Raloxifene Evaluation. JAMA 1999; 281:2189–2197.
- Martino S, Cauley JA, Barrett-Connor E, et al; CORE Investigators. Continuing outcomes relevant to Evista: breast cancer incidence in postmenopausal osteoporotic women in a randomized trial of raloxifene. J Natl Cancer Inst 2004; 96:1751–1761.
- Vogel VG, Qu Y, Wong M, Mitchell B, Mershon JL. Incidence of invasive breast cancer in postmenopausal women after discontinuation of long-term raloxifene administration. Clin Breast Cancer 2009; 9:45–50.
- Grady D, Cauley JA, Stock JL, et al. Effect of raloxifene on all-cause mortality. Am J Med 2010; 123:469.e1–e7.
- Barrett-Connor E, Mosca L, Collins P, et al; Raloxifene Use for The Heart (RUTH) Trial Investigators. Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. N Engl J Med 2006; 355:125–137.
- Vogel VG. The NSABP Study of Tamoxifen and Raloxifene (STAR) trial. Expert Rev Anticancer Ther 2009; 9:51–60.
- Vogel VG, Costantino JP, Wickerham DL, et al; National Surgical Adjuvant Breast and Bowel Project (NSABP). Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA 2006; 295:2727–2741.
- Barnes KN, Pearce EF, Yancey AM, Forinash AB. Ospemifene in the treatment of vulvovaginal atrophy. Ann Pharmacother 2014; 48:752–757.
- Rutanen EM, Heikkinen J, Halonen K, Komi J, Lammintausta R, Ylikorkala O. Effects of ospemifene, a novel SERM, on hormones, genital tract, climacteric symptoms, and quality of life in postmenopausal women: a double-blind, randomized trial. Menopause 2003; 10:433–439.
- Constantine G, Graham S, Koltun WD, Kingsberg SA. Assessment of ospemifene or lubricants on clinical signs of VVA. J Sex Med 2014; 11:1033–1041.
- Kingsberg SA, Wysocki S, Magnus L, Krychman ML. Vulvar and vaginal atrophy in postmenopausal women: findings from the REVIVE survey. J Sex Med 2013; 10:1790–1799.
- Portman DJ, Bachmann GA, Simon JA; Ospemifene Study Group. Ospemifene, a novel selective estrogen receptor modulator for treating dyspareunia associated with postmenopausal vulvar and vaginal atrophy. Menopause 2013; 20:623–630.
- Bachmann GA, Komi JO; Ospemifene Study Group. Ospemifene effectively treats vulvovaginal atrophy in postmenopausal women: results from a pivotal phase 3 study. Menopause 2010; 17:480–486.
- Goldstein SR, Bachmann GA, Koninckx PR, Lin VH, Portman DJ, Ylikorkala O; Ospemifene Study Group. Ospemifene 12-month safety and efficacy in postmenopausal women with vulvar and vaginal atrophy. Climacteric 2014; 17:173–182.
- Cui Y, Zong H, Yan H, Li N, Zhang Y. The efficacy and safety of ospemifene in treating dyspareunia associated with postmenopausal vulvar and vaginal atrophy: a systematic review and meta-analysis. J Sex Med 2014; 11:487–497.
- Komi J, Heikkinen J, Rutanen EM, Halonen K, Lammintausta R, Ylikorkala O. Effects of ospemifene, a novel SERM, on biochemical markers of bone turnover in healthy postmenopausal women. Gynecal Endocrinol 2004; 18:152–158.
- Komi J, Lankinen KS, DeGregorio M, et al. Effects of ospemifene and raloxifene on biochemical markers of bone turnover in postmenopausal women. J Bone Miner Metab 2006; 24:314–318.
- Wurz GT, Read KC, Marchisano-Karpman C, et al. Ospemifene inhibits the growth of dimethylbenzanthracene-induced mammary tumors in Sencar mice. J Steroid Biochem Mol Biol 2005; 97:230–240.
- Pinkerton JV, Utian WH, Constantine GD, Olivier S, Pickar JH. Relief of vasomotor symptoms with the tissue-selective estrogen complex containing bazedoxifene/conjugated estrogens: a randomized, controlled trial. Menopause 2009; 16:1116–1124.
- Pickar JH, Mirkin S. Tissue-selective agents: selective estrogen receptor modulators and the tissue-selective estrogen complex. Menopause Int 2010; 16:121–128.
- Levine JP. Treating menopausal symptoms with a tissue-selective estrogen complex. Gend Med 2011; 8:57–68.
- Lindsay R, Gallagher JC, Kagan R, Pickar JH, Constantine G. Efficacy of tissue-selective estrange complex of bazedoxifene/conjugated estrogens for osteoporosis prevention in at-risk postmenopausal women. Fertil Steril 2009; 92:1045–1052.
- Bachmann G, Bobula J, Mirkin S. Effects of bazedoxifene/conjugated estrogens on quality of life in postmenopausal women with symptoms of vulvar/vaginal atrophy. Climacteric 2010; 13:132–140.
- Pickar JH, Yeh IT, Bachmann G, Speroff L. Endometrial effects of a tissue selective estrogen complex containing bazedoxifene/conjugated estrogens as a menopausal therapy. Fertil Steril 2009; 92:1018–1024.
- Archer DF, Lewis V, Carr BR, Olivier S, Pickar JH. Bazedoxifene/conjugated estrogens (BZA/CE): incidence of uterine bleeding in postmenopausal women. Fertil Steril 2009: 92:1039–1044.
- Pinkerton JV, Abraham L, Bushmakin AG, et al. Evaluation of the efficacy and safety of bazedoxifene/conjugated estrogens for secondary outcomes including vasomotor symptoms in postmenopausal women by years since menopause in the Selective estrogens, Menopause and Response to Therapy (SMART) trials. J Womens Health (Larchmt) 2014; 23:18–28.
- Giannini A, Russo E, Mannella P, Simoncini T. Selective steroid receptor modulators in reproductive medicine. Minerva Ginecol 2015; 67:431–455.
- Feldman BM, Voda A, Gronseth E. The prevalence of hot flash and associated variables among perimenopausal women. Res Nurs Health 1985; 8:261–268.
- Versi E, Harvey MA, Cardozo L, Brincat M, Studd JW. Urogenital prolapse and atrophy at menopause: a prevalence study. Int Urogynecol J Pelvic Floor Dysfunct 2001; 12:107–110.
- Hess R, Chang CC, Conigliaro J, McNeil M. Understanding physicians’ attitudes towards hormone therapy. Womens Health Issues 2005; 15:31–38.
- Melton LJ 3rd, Khosla S, Atkinson EJ, O’Fallon WM, Riggs BL. Relationship of bone turnover to bone density and fractures. J Bone Miner Res 1997; 12:1083–1091.
- Sikon A, Thacker HL. Treatment options for menopausal hot flashes. Cleve Clin J Med 2004; 71:578–582.
- Levine JP. Treating menopausal symptoms with a tissue-selective estrogen complex. Gend Med 2011; 8:57–68.
- Pinkerton JV, Thomas S. Use of SERMs for treatment in postmenopausal women. J Steroid Biochem Mol Biol 2014; 142:142–154.
- Tamoxifen for early breast cancer: an overview of the randomised trials. Early Breast Cancer Trialists’ Collaborative Group. Lancet 1998; 351:1451–1467.
- Davies C, Pan H, Godwin J, et al; Adjuvant Tamoxifen: Longer Against Shorter (ATLAS) Collaborative Group. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013; 381:805–816.
- Gray RG, Rea D, Handley K, et al. aTTom: Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years in 6,953 women with early breast cancer. J Clin Oncol 2013; (suppl): abstract 5.
- Cuzick J, Powles T, Veronesi U, et al. Overview of the main outcomes in breast-cancer prevention trials. Lancet 2003; 361:296–300.
- Cuzick J, Sestak I, Cawthorn S, et al. Tamoxifen for prevention of breast cancer: extended long-term follow-up of the IBIS-I breast cancer prevention trial. Lancet Oncol 2015; 16:67–75.
- Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst 1998; 90:1371–1388.
- Chang J, Powles TJ, Ashley SE, et al. The effect of tamoxifen and hormone replacement therapy on serum cholesterol, bone mineral density and coagulation factors in healthy postmenopausal women participating in a randomised, controlled tamoxifen prevention study. Ann Oncol 1996; 7:671–675.
- Herrington DM, Klein KP. Effects of SERMs on important indicators of cardiovascular health: lipoproteins, hemostatic factors and endothelial function. Womens Health Issues 2001; 11:95–102.
- Kedar RP, Bourne TH, Powles TJ, et al. Effects of tamoxifen on uterus and ovaries of postmenopausal women in a randomized breast cancer prevention trial. Lancet 1994; 343:1318–1321.
- Ettinger B, Black DM, Mitlak BH, et al. Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene: results from a 3-year randomized clinical trial. Multiple Outcomes of Raloxifene Evaluation (MORE) Investigators. JAMA 1999; 282:637–645.
- Maricic M, Adachi JD, Sarkar S, Wu W, Wong M, Harper KD. Early effects of raloxifene on clinical vertebral fractures at 12 months in postmenopausal women with osteoporosis. Arch Intern Med 2002; 162:1140–1143.
- Recker RR, Mitlak BH, Ni X, Krege JH. Long-term raloxifene for postmenopausal osteoporosis. Curr Med Res Opin 2011; 27:1755–1761.
- Cummings SR, Eckert S, Krueger KA, et al. The effect of raloxifene on risk of breast cancer in postmenopausal women: results from the MORE randomized trial. Multiple Outcomes of Raloxifene Evaluation. JAMA 1999; 281:2189–2197.
- Martino S, Cauley JA, Barrett-Connor E, et al; CORE Investigators. Continuing outcomes relevant to Evista: breast cancer incidence in postmenopausal osteoporotic women in a randomized trial of raloxifene. J Natl Cancer Inst 2004; 96:1751–1761.
- Vogel VG, Qu Y, Wong M, Mitchell B, Mershon JL. Incidence of invasive breast cancer in postmenopausal women after discontinuation of long-term raloxifene administration. Clin Breast Cancer 2009; 9:45–50.
- Grady D, Cauley JA, Stock JL, et al. Effect of raloxifene on all-cause mortality. Am J Med 2010; 123:469.e1–e7.
- Barrett-Connor E, Mosca L, Collins P, et al; Raloxifene Use for The Heart (RUTH) Trial Investigators. Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. N Engl J Med 2006; 355:125–137.
- Vogel VG. The NSABP Study of Tamoxifen and Raloxifene (STAR) trial. Expert Rev Anticancer Ther 2009; 9:51–60.
- Vogel VG, Costantino JP, Wickerham DL, et al; National Surgical Adjuvant Breast and Bowel Project (NSABP). Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA 2006; 295:2727–2741.
- Barnes KN, Pearce EF, Yancey AM, Forinash AB. Ospemifene in the treatment of vulvovaginal atrophy. Ann Pharmacother 2014; 48:752–757.
- Rutanen EM, Heikkinen J, Halonen K, Komi J, Lammintausta R, Ylikorkala O. Effects of ospemifene, a novel SERM, on hormones, genital tract, climacteric symptoms, and quality of life in postmenopausal women: a double-blind, randomized trial. Menopause 2003; 10:433–439.
- Constantine G, Graham S, Koltun WD, Kingsberg SA. Assessment of ospemifene or lubricants on clinical signs of VVA. J Sex Med 2014; 11:1033–1041.
- Kingsberg SA, Wysocki S, Magnus L, Krychman ML. Vulvar and vaginal atrophy in postmenopausal women: findings from the REVIVE survey. J Sex Med 2013; 10:1790–1799.
- Portman DJ, Bachmann GA, Simon JA; Ospemifene Study Group. Ospemifene, a novel selective estrogen receptor modulator for treating dyspareunia associated with postmenopausal vulvar and vaginal atrophy. Menopause 2013; 20:623–630.
- Bachmann GA, Komi JO; Ospemifene Study Group. Ospemifene effectively treats vulvovaginal atrophy in postmenopausal women: results from a pivotal phase 3 study. Menopause 2010; 17:480–486.
- Goldstein SR, Bachmann GA, Koninckx PR, Lin VH, Portman DJ, Ylikorkala O; Ospemifene Study Group. Ospemifene 12-month safety and efficacy in postmenopausal women with vulvar and vaginal atrophy. Climacteric 2014; 17:173–182.
- Cui Y, Zong H, Yan H, Li N, Zhang Y. The efficacy and safety of ospemifene in treating dyspareunia associated with postmenopausal vulvar and vaginal atrophy: a systematic review and meta-analysis. J Sex Med 2014; 11:487–497.
- Komi J, Heikkinen J, Rutanen EM, Halonen K, Lammintausta R, Ylikorkala O. Effects of ospemifene, a novel SERM, on biochemical markers of bone turnover in healthy postmenopausal women. Gynecal Endocrinol 2004; 18:152–158.
- Komi J, Lankinen KS, DeGregorio M, et al. Effects of ospemifene and raloxifene on biochemical markers of bone turnover in postmenopausal women. J Bone Miner Metab 2006; 24:314–318.
- Wurz GT, Read KC, Marchisano-Karpman C, et al. Ospemifene inhibits the growth of dimethylbenzanthracene-induced mammary tumors in Sencar mice. J Steroid Biochem Mol Biol 2005; 97:230–240.
- Pinkerton JV, Utian WH, Constantine GD, Olivier S, Pickar JH. Relief of vasomotor symptoms with the tissue-selective estrogen complex containing bazedoxifene/conjugated estrogens: a randomized, controlled trial. Menopause 2009; 16:1116–1124.
- Pickar JH, Mirkin S. Tissue-selective agents: selective estrogen receptor modulators and the tissue-selective estrogen complex. Menopause Int 2010; 16:121–128.
- Levine JP. Treating menopausal symptoms with a tissue-selective estrogen complex. Gend Med 2011; 8:57–68.
- Lindsay R, Gallagher JC, Kagan R, Pickar JH, Constantine G. Efficacy of tissue-selective estrange complex of bazedoxifene/conjugated estrogens for osteoporosis prevention in at-risk postmenopausal women. Fertil Steril 2009; 92:1045–1052.
- Bachmann G, Bobula J, Mirkin S. Effects of bazedoxifene/conjugated estrogens on quality of life in postmenopausal women with symptoms of vulvar/vaginal atrophy. Climacteric 2010; 13:132–140.
- Pickar JH, Yeh IT, Bachmann G, Speroff L. Endometrial effects of a tissue selective estrogen complex containing bazedoxifene/conjugated estrogens as a menopausal therapy. Fertil Steril 2009; 92:1018–1024.
- Archer DF, Lewis V, Carr BR, Olivier S, Pickar JH. Bazedoxifene/conjugated estrogens (BZA/CE): incidence of uterine bleeding in postmenopausal women. Fertil Steril 2009: 92:1039–1044.
- Pinkerton JV, Abraham L, Bushmakin AG, et al. Evaluation of the efficacy and safety of bazedoxifene/conjugated estrogens for secondary outcomes including vasomotor symptoms in postmenopausal women by years since menopause in the Selective estrogens, Menopause and Response to Therapy (SMART) trials. J Womens Health (Larchmt) 2014; 23:18–28.
KEY POINTS
- Tamoxifen is approved to prevent and treat breast cancer. It may also have beneficial effects on bone and on cardiovascular risk factors, but these are not approved uses.
- Raloxifene, a second-generation ERAA, was initially approved for preventing and treating osteoporosis and later received approval to reduce the risk of invasive estrogen receptor-positive breast cancer in postmenopausal women.
- Ospemifene is approved for treatment of genitourinary syndrome of menopause.
- The combination of conjugated estrogen and bazedoxifene is approved for treating moderate to severe vasomotor symptoms associated with menopause and also for preventing postmenopausal osteoporosis in women with an intact uterus.

Apps and fitness trackers that measure sleep: Are they useful?
More and more consumers are using wearable devices and smartphones to monitor and measure various body functions, including sleep. Many patients now present their providers with sleep data obtained from their phones and other devices. But can these devices provide valid, useful clinical information?
This article describes common sleep tracking devices available to consumers and the mechanisms the devices probably use to distinguish sleep from wakefulness (their algorithms are secret), the studies evaluating the validity of device manufacturers’ claims, and their clinical utility and limitations.
DEVICES ARE COMMON
Close to 1 in 10 adults over age 18 owns an activity tracker, and sales are projected to reach $50 billion by 2018.1 Even more impressive, close to 69% of Americans own a smartphone,1 and more than half use it as an alarm clock.2
At the same time that these devices have become so popular, sleep medicine has come of age, and experts have been pushing to improve people’s sleep and increase awareness of sleep disorders.3,4 While the technology has significantly advanced, adoption of data from these devices for clinical evaluation has been limited. Studies examining the validity of these devices have only recently been conducted, and companies that make the devices have not been forthcoming with details of the specific algorithms they use to tell if the patient is asleep or awake or what stage of sleep the patient is in.
WHAT ARE THESE DEVICES?
Consumer tracking devices that claim to measure sleep are easily available for purchase and include wearable fitness trackers such as Fitbit, Jawbone UP, and Nike+ Fuelband. Other sleep tracking devices are catalogued by Ko et al.5 Various smartphone applications (apps) are also available.
Fitness trackers, usually worn as a wrist band, are primarily designed to measure movement and activity, but manufacturers now claim the trackers can also measure sleep. Collected data are available for the user to review the following day. In most cases, these trackers display sleep and wake times; others also claim to record sound sleep, light sleep, and the number and duration of awakenings. Most fitness trackers have complementary apps available for download that graphically display the data on smartphones and interact with social media to allow users to post their sleep and activity data.
More than 500 sleep-related apps are available for download to smartphones in the iTunes app store5; the Sleep Cycle alarm clock app was among the top 5 sleep-tracking apps downloaded in 2014.6 Because sleep data collection relies on the smartphone being placed on the user’s mattress, movements of bed partners, pets, and bedding may interfere with results. In most cases, the apps display data in a format similar to that of fitness trackers. Some claim to determine the optimal sleep phase for the alarm to wake the user.
HOW DOES THE TECHNOLOGY WORK?
Older activity-tracking devices used single-channel electroencephalographic recordings or multiple physiologic channels such as galvanic skin response, skin temperature, and heat flux to measure activity to determine transitions between periods of sleep and wakefulness.7,8
None of the currently available consumer sleep tracking devices discloses the exact mechanisms used to measure sleep and wakefulness, but most appear to rely on 3-axis accelerometers,9 ie, microelectromechanical devices that measure front-to-back, side-to-side, and up-and-down motion and convert the data into an activity count. Activity counts are acquired over 30- or 60-second intervals and are entered into algorithms that determine if the pattern indicates that the patient is awake or asleep. This is the same method that actigraphy uses to evaluate sleep, but most actigraphs used in medicine disclose their mechanisms and provide clinicians with the option of using various validated algorithms to classify the activity counts into sleep or awake periods.9–11
ARE THE MEASURES VALID?
Only a few studies have examined the validity and accuracy of current fitness trackers and apps for measuring sleep.
The available studies are difficult to compare; most have been small and used different actigraphy devices for comparison. Some tested healthy volunteers, others included people with suspected or confirmed sleep disorders, and some had both types of participants. In many studies, the device was compared with polysomnography for only 1 night, making the “first-night effect” likely to be a confounding factor, as people tend to sleep worse during the first night of testing. Technical failures for the devices were noted in some studies.12 In addition, some currently used apps may use different platforms than the devices used in these studies, limiting the extrapolation of results.
![]()
As with fitness trackers, few studies have been done to examine the validity of smartphone apps.5 Findings of 3 studies are summarized in Table 2.17–19 In addition to tracking the duration and depth of sleep, some apps purport to detect snoring, sleep apnea, and periodic limb movements of sleep. Discussion of these apps is beyond the scope of this review.
![]()
ARE THE DEVICES CLINICALLY USEFUL?
Although a thorough history remains the cornerstone of a good evaluation of sleep problems, testing is sometimes essential, and in certain situations, objective data can complement the history and clarify the diagnosis.
Polysomnography remains the gold standard for telling when the patient is asleep vs awake, diagnosing sleep-disordered breathing, detecting periodic limb movements and parasomnias, and aiding in the diagnosis of narcolepsy.
Actigraphy, which uses technology similar to fitness trackers, can help distinguish sleep from wakefulness, reveal erratic sleep schedules, and help diagnose circadian rhythm sleep disorders. In patients with insomnia, actigraphy can help determine daily sleep patterns and response to treatment.20 It can be especially useful for patients who cannot provide a clear history, eg, children and those with developmental disabilities or cognitive dysfunction.
Consumer sleep tracking devices, like actigraphy, are portable and unobtrusive, providing a way to measure sleep duration and demonstrate sleep patterns in a patient’s natural environment. Being more accessible, cheaper, and less time-consuming than clinical tests, the commercially available devices could be clinically useful in some situations, eg, for monitoring overall sleep patterns to look for circadian sleep-wake disorders, commonly seen in shift workers (shift work disorder) or adolescents (delayed sleep-wake phase disorder); or in patients with poor motivation to maintain a sleep diary. Because of their poor performance in clinical trials, they should not be relied upon to distinguish sleep from wakefulness, quantify the amount of sleep, determine sleep stages, and awaken patients exclusively from light sleep.
Discerning poor sleep hygiene from insomnia
Patients with insomnia tend to take longer to go to sleep (have longer sleep latencies), wake up more (have more disturbed sleep with increased awakenings), and have shorter sleep times with reduced sleep efficiencies.21 Sleep tracking devices tend to be less accurate for patients with short sleep duration and disturbed sleep, limiting their usefulness in this group. Furthermore, patients with insomnia tend to underestimate their sleep time and overestimate sleep latency; some devices also tend to overestimate the time to fall asleep, reinforcing this common error made by patients.22,23
On the other hand, data from sleep tracking devices could help distinguish poor sleep hygiene from an insomnia disorder. For example, the data may indicate that a patient has poor sleep habits, such as taking long daytime naps or having significantly variable time in and out of bed from day to day. The total times asleep and awake in the middle of the night may also be substantially different on each night, which would also possibly indicate poor sleep hygiene.
Detecting circadian rhythms
A device may show that a patient has a clear circadian preference that is not in line with his or her daily routines, suggesting an underlying circadian rhythm sleep-wake disorder. This may be evident by bedtimes and wake times that are consistent but substantially out of sync with one’s social or occupational needs.
Measuring overall sleep duration
In people with normal sleep, fitness trackers perform reasonably well for measuring overall sleep duration. This information could be used to assess a patient with daytime sleepiness and fatigue to evaluate insufficient sleep as an etiologic factor.
![]()
Table 3 summarizes how to evaluate the data from sleep apps and fitness tracking devices for clinical use. While these features of consumer sleep tracking devices could conceivably help in the above clinical scenarios, further validation of devices in clinical populations is necessary before their use can be recommended without reservation.
ADVISING PATIENTS
Patients sometimes present to clinicians with concerns about the duration of sleep time and time spent in various sleep stages as delineated by their sleep tracking devices. Currently, these devices do not appear to be able to adequately distinguish various sleep stages, and in many users, they can substantially underestimate or overestimate sleep parameters such as time taken to fall asleep or duration of awakenings in the middle of the night. Patients can be reassured about this lack of evidence and should be advised to not place too much weight on such data alone.
Sleep “goals” set by many devices have not been scientifically validated. People without sleep problems should be discouraged from making substantial changes to their routines to accommodate sleep targets set by the devices. Patients should be counseled about the pitfalls of the data and can be reassured that little evidence suggests that time spent in various sleep stages correlates with adverse daytime consequences or with poor health outcomes.
Some of the apps used as alarm clocks claim to be able to tell what stage of sleep people are in and wait to awaken them until they are in a light stage, which is less jarring than being awakened from a deep stage, but the evidence for this is unclear. In the one study that tested this claim, the app did not awaken participants from light sleep more often than is likely to occur by chance.17 The utility of these apps as personalized alarm clocks is still extremely limited, and patients should be counseled to obtain an adequate amount of sleep rather than rely on devices to awaken them during specific sleep stages.
The rates for discontinuing the use of these devices are high, which could limit their utility. Some surveys have shown that close to 50% of users stop using fitness trackers; 33% stop using them within 6 months of obtaining the device.24 Also, there is little evidence that close monitoring of sleep results in behavior changes or improved sleep duration. Conversely, the potential harms of excessive monitoring of one’s sleep are currently unknown.
- Rock Health. The future of biosensing wearables. http://rockhealth.com/reports/the-future-of-biosensing-wearables/. Accessed March 16, 2017.
- Time, Inc. Your wireless life: results of Time’s mobility poll. http://content.time.com/time/interactive/0,31813,2122187,00.html. Accessed March 16, 2017.
- Office of Disease Prevention and Health Promotion (ODPHP). Healthy people 2020. Sleep health. www.healthypeople.gov/2020/topics-objectives/topic/sleep-health. Accessed March 16, 2017.
- Consensus Conference Panel; Watson NF, Badr MS, Belenky G, et al. Joint consensus statement of the American Academy of Sleep Medicine and Sleep Research Society on the recommended amount of sleep for a healthy adult: methodology and discussion. J Clin Sleep Med 2015; 11:931–952.
- Ko PT, Kientz JA, Choe EK, Kay M, Landis CA, Watson NF. Consumer sleep technologies: a review of the landscape. J Clin Sleep Med 2015; 11:1455–1461.
- Investor Place Media, LLC. Top iTunes picks: Apple names best apps of 2014. http://investorplace.com/2014/12/apple-best-apps-of-2014-aapl/#.VIYeE9LF98E/. Accessed April 13, 2017.
- Sunseri M, Liden CB, Farringdon J, et al. The SenseWear armband as a sleep detection device. Internal publication.
- Shambroom JR, Fábregas SE, Johnstone J. Validation of an automated wireless system to monitor sleep in healthy adults. J Sleep Res 2012; 21:221–230.
- John D, Freedson P. ActiGraph and Actical physical activity monitors: a peek under the hood. Med Sci Sports Exerc 2012; 44(suppl 1):S86–S89.
- Sadeh A, Sharkey KM, Carskadon MA. Activity-based sleep-wake identification: an empirical test of methodological issues. Sleep 1994; 17:201–207.
- Kripke DF, Hahn EK, Grizas AP, et al. Wrist actigraphic scoring for sleep laboratory patients: algorithm development. J Sleep Res 2010; 19:612–619.
- Meltzer LJ, Marcus CL. Reply: caffeine therapy for apnea of prematurity: long-term effect on sleep by actigraphy and polysomnography. Am J Respir Crit Care Med 2014; 190:1457–1458.
- Montgomery-Downs HE, Insana SP, Bond JA. Movement toward a novel activity monitoring device. Sleep Breath 2012; 16:913–917.
- Meltzer LJ, Hiruma LS, Avis K, Montgomery-Downs H, Valentin J. Comparison of a commercial accelerometer with polysomnography and actigraphy in children and adolescents. Sleep 2015; 38:1323–1330.
- de Zambotti M, Baker FC, Colrain IM. Validation of sleep-tracking technology compared with polysomnography in adolescents. Sleep 2015; 38:1461–1468.
- de Zambotti M, Claudatos S, Inkelis S, Colrain IM, Baker FC. Evaluation of a consumer fitness-tracking device to assess sleep in adults. Chronobiol Int 2015; 32:1024–1028.
- Toon E, Davey MJ, Hollis SL, Nixon GM, Horne RS, Biggs SN. Comparison of commercial wrist-based and smartphone accelerometers, actigraphy, and PSG in a clinical cohort of children and adolescents. J Clin Sleep Med 2016; 12:343–350.
- Bhat S, Ferraris A, Gupta D, et al. Is there a clinical role for smartphone sleep apps? Comparison of sleep cycle detection by a smartphone application to polysomnography. J Clin Sleep Med 2015; 11:709–715.
- Min JK, Doryab A, Wiese J, Amini S, Zimmerman J, Hong JI. Toss ‘n’ turn: smartphone as sleep and sleep quality detector. Proceedings of the SIGCHI Conference on Human Factors in Computing Systems. Toronto, Ontario, Canada: ACM; 2014:477-486.
- Morgenthaler T, Alessi C, Friedman L, et al; Standards of Practice Committee; American Academy of Sleep Medicine. Practice parameters for the use of actigraphy in the assessment of sleep and sleep disorders: an update for 2007. Sleep 2007; 30:519-529.
- Lichstein KL, Durrence HH, Taylor DJ, Bush AJ, Riedel BW. Quantitative criteria for insomnia. Behav Res Ther 2003; 41:427–445.
- Carskadon MA, Dement WC, Mitler MM, Guilleminault C, Zarcone VP, Spiegel R. Self-reports versus sleep laboratory findings in 122 drug-free subjects with complaints of chronic insomnia. Am J Psychiatry 1976; 133:1382–1388.
- Perlis ML, Giles DE, Mendelson WB, Bootzin RR, Wyatt JK. Psychophysiological insomnia: the behavioural model and a neurocognitive perspective. J Sleep Res 1997; 6:179–188.
- Endeavour Partners LLC. Inside wearables: how the science of human behavior change offers the secret to long-term engagement. http://endeavourpartners.net/assets/Wearables-and-the-Science-of-Human-Behavior-Change-EP4.pdf. Accessed March 16, 2017.
More and more consumers are using wearable devices and smartphones to monitor and measure various body functions, including sleep. Many patients now present their providers with sleep data obtained from their phones and other devices. But can these devices provide valid, useful clinical information?
This article describes common sleep tracking devices available to consumers and the mechanisms the devices probably use to distinguish sleep from wakefulness (their algorithms are secret), the studies evaluating the validity of device manufacturers’ claims, and their clinical utility and limitations.
DEVICES ARE COMMON
Close to 1 in 10 adults over age 18 owns an activity tracker, and sales are projected to reach $50 billion by 2018.1 Even more impressive, close to 69% of Americans own a smartphone,1 and more than half use it as an alarm clock.2
At the same time that these devices have become so popular, sleep medicine has come of age, and experts have been pushing to improve people’s sleep and increase awareness of sleep disorders.3,4 While the technology has significantly advanced, adoption of data from these devices for clinical evaluation has been limited. Studies examining the validity of these devices have only recently been conducted, and companies that make the devices have not been forthcoming with details of the specific algorithms they use to tell if the patient is asleep or awake or what stage of sleep the patient is in.
WHAT ARE THESE DEVICES?
Consumer tracking devices that claim to measure sleep are easily available for purchase and include wearable fitness trackers such as Fitbit, Jawbone UP, and Nike+ Fuelband. Other sleep tracking devices are catalogued by Ko et al.5 Various smartphone applications (apps) are also available.
Fitness trackers, usually worn as a wrist band, are primarily designed to measure movement and activity, but manufacturers now claim the trackers can also measure sleep. Collected data are available for the user to review the following day. In most cases, these trackers display sleep and wake times; others also claim to record sound sleep, light sleep, and the number and duration of awakenings. Most fitness trackers have complementary apps available for download that graphically display the data on smartphones and interact with social media to allow users to post their sleep and activity data.
More than 500 sleep-related apps are available for download to smartphones in the iTunes app store5; the Sleep Cycle alarm clock app was among the top 5 sleep-tracking apps downloaded in 2014.6 Because sleep data collection relies on the smartphone being placed on the user’s mattress, movements of bed partners, pets, and bedding may interfere with results. In most cases, the apps display data in a format similar to that of fitness trackers. Some claim to determine the optimal sleep phase for the alarm to wake the user.
HOW DOES THE TECHNOLOGY WORK?
Older activity-tracking devices used single-channel electroencephalographic recordings or multiple physiologic channels such as galvanic skin response, skin temperature, and heat flux to measure activity to determine transitions between periods of sleep and wakefulness.7,8
None of the currently available consumer sleep tracking devices discloses the exact mechanisms used to measure sleep and wakefulness, but most appear to rely on 3-axis accelerometers,9 ie, microelectromechanical devices that measure front-to-back, side-to-side, and up-and-down motion and convert the data into an activity count. Activity counts are acquired over 30- or 60-second intervals and are entered into algorithms that determine if the pattern indicates that the patient is awake or asleep. This is the same method that actigraphy uses to evaluate sleep, but most actigraphs used in medicine disclose their mechanisms and provide clinicians with the option of using various validated algorithms to classify the activity counts into sleep or awake periods.9–11
ARE THE MEASURES VALID?
Only a few studies have examined the validity and accuracy of current fitness trackers and apps for measuring sleep.
The available studies are difficult to compare; most have been small and used different actigraphy devices for comparison. Some tested healthy volunteers, others included people with suspected or confirmed sleep disorders, and some had both types of participants. In many studies, the device was compared with polysomnography for only 1 night, making the “first-night effect” likely to be a confounding factor, as people tend to sleep worse during the first night of testing. Technical failures for the devices were noted in some studies.12 In addition, some currently used apps may use different platforms than the devices used in these studies, limiting the extrapolation of results.
![]()
As with fitness trackers, few studies have been done to examine the validity of smartphone apps.5 Findings of 3 studies are summarized in Table 2.17–19 In addition to tracking the duration and depth of sleep, some apps purport to detect snoring, sleep apnea, and periodic limb movements of sleep. Discussion of these apps is beyond the scope of this review.
![]()
ARE THE DEVICES CLINICALLY USEFUL?
Although a thorough history remains the cornerstone of a good evaluation of sleep problems, testing is sometimes essential, and in certain situations, objective data can complement the history and clarify the diagnosis.
Polysomnography remains the gold standard for telling when the patient is asleep vs awake, diagnosing sleep-disordered breathing, detecting periodic limb movements and parasomnias, and aiding in the diagnosis of narcolepsy.
Actigraphy, which uses technology similar to fitness trackers, can help distinguish sleep from wakefulness, reveal erratic sleep schedules, and help diagnose circadian rhythm sleep disorders. In patients with insomnia, actigraphy can help determine daily sleep patterns and response to treatment.20 It can be especially useful for patients who cannot provide a clear history, eg, children and those with developmental disabilities or cognitive dysfunction.
Consumer sleep tracking devices, like actigraphy, are portable and unobtrusive, providing a way to measure sleep duration and demonstrate sleep patterns in a patient’s natural environment. Being more accessible, cheaper, and less time-consuming than clinical tests, the commercially available devices could be clinically useful in some situations, eg, for monitoring overall sleep patterns to look for circadian sleep-wake disorders, commonly seen in shift workers (shift work disorder) or adolescents (delayed sleep-wake phase disorder); or in patients with poor motivation to maintain a sleep diary. Because of their poor performance in clinical trials, they should not be relied upon to distinguish sleep from wakefulness, quantify the amount of sleep, determine sleep stages, and awaken patients exclusively from light sleep.
Discerning poor sleep hygiene from insomnia
Patients with insomnia tend to take longer to go to sleep (have longer sleep latencies), wake up more (have more disturbed sleep with increased awakenings), and have shorter sleep times with reduced sleep efficiencies.21 Sleep tracking devices tend to be less accurate for patients with short sleep duration and disturbed sleep, limiting their usefulness in this group. Furthermore, patients with insomnia tend to underestimate their sleep time and overestimate sleep latency; some devices also tend to overestimate the time to fall asleep, reinforcing this common error made by patients.22,23
On the other hand, data from sleep tracking devices could help distinguish poor sleep hygiene from an insomnia disorder. For example, the data may indicate that a patient has poor sleep habits, such as taking long daytime naps or having significantly variable time in and out of bed from day to day. The total times asleep and awake in the middle of the night may also be substantially different on each night, which would also possibly indicate poor sleep hygiene.
Detecting circadian rhythms
A device may show that a patient has a clear circadian preference that is not in line with his or her daily routines, suggesting an underlying circadian rhythm sleep-wake disorder. This may be evident by bedtimes and wake times that are consistent but substantially out of sync with one’s social or occupational needs.
Measuring overall sleep duration
In people with normal sleep, fitness trackers perform reasonably well for measuring overall sleep duration. This information could be used to assess a patient with daytime sleepiness and fatigue to evaluate insufficient sleep as an etiologic factor.
![]()
Table 3 summarizes how to evaluate the data from sleep apps and fitness tracking devices for clinical use. While these features of consumer sleep tracking devices could conceivably help in the above clinical scenarios, further validation of devices in clinical populations is necessary before their use can be recommended without reservation.
ADVISING PATIENTS
Patients sometimes present to clinicians with concerns about the duration of sleep time and time spent in various sleep stages as delineated by their sleep tracking devices. Currently, these devices do not appear to be able to adequately distinguish various sleep stages, and in many users, they can substantially underestimate or overestimate sleep parameters such as time taken to fall asleep or duration of awakenings in the middle of the night. Patients can be reassured about this lack of evidence and should be advised to not place too much weight on such data alone.
Sleep “goals” set by many devices have not been scientifically validated. People without sleep problems should be discouraged from making substantial changes to their routines to accommodate sleep targets set by the devices. Patients should be counseled about the pitfalls of the data and can be reassured that little evidence suggests that time spent in various sleep stages correlates with adverse daytime consequences or with poor health outcomes.
Some of the apps used as alarm clocks claim to be able to tell what stage of sleep people are in and wait to awaken them until they are in a light stage, which is less jarring than being awakened from a deep stage, but the evidence for this is unclear. In the one study that tested this claim, the app did not awaken participants from light sleep more often than is likely to occur by chance.17 The utility of these apps as personalized alarm clocks is still extremely limited, and patients should be counseled to obtain an adequate amount of sleep rather than rely on devices to awaken them during specific sleep stages.
The rates for discontinuing the use of these devices are high, which could limit their utility. Some surveys have shown that close to 50% of users stop using fitness trackers; 33% stop using them within 6 months of obtaining the device.24 Also, there is little evidence that close monitoring of sleep results in behavior changes or improved sleep duration. Conversely, the potential harms of excessive monitoring of one’s sleep are currently unknown.
More and more consumers are using wearable devices and smartphones to monitor and measure various body functions, including sleep. Many patients now present their providers with sleep data obtained from their phones and other devices. But can these devices provide valid, useful clinical information?
This article describes common sleep tracking devices available to consumers and the mechanisms the devices probably use to distinguish sleep from wakefulness (their algorithms are secret), the studies evaluating the validity of device manufacturers’ claims, and their clinical utility and limitations.
DEVICES ARE COMMON
Close to 1 in 10 adults over age 18 owns an activity tracker, and sales are projected to reach $50 billion by 2018.1 Even more impressive, close to 69% of Americans own a smartphone,1 and more than half use it as an alarm clock.2
At the same time that these devices have become so popular, sleep medicine has come of age, and experts have been pushing to improve people’s sleep and increase awareness of sleep disorders.3,4 While the technology has significantly advanced, adoption of data from these devices for clinical evaluation has been limited. Studies examining the validity of these devices have only recently been conducted, and companies that make the devices have not been forthcoming with details of the specific algorithms they use to tell if the patient is asleep or awake or what stage of sleep the patient is in.
WHAT ARE THESE DEVICES?
Consumer tracking devices that claim to measure sleep are easily available for purchase and include wearable fitness trackers such as Fitbit, Jawbone UP, and Nike+ Fuelband. Other sleep tracking devices are catalogued by Ko et al.5 Various smartphone applications (apps) are also available.
Fitness trackers, usually worn as a wrist band, are primarily designed to measure movement and activity, but manufacturers now claim the trackers can also measure sleep. Collected data are available for the user to review the following day. In most cases, these trackers display sleep and wake times; others also claim to record sound sleep, light sleep, and the number and duration of awakenings. Most fitness trackers have complementary apps available for download that graphically display the data on smartphones and interact with social media to allow users to post their sleep and activity data.
More than 500 sleep-related apps are available for download to smartphones in the iTunes app store5; the Sleep Cycle alarm clock app was among the top 5 sleep-tracking apps downloaded in 2014.6 Because sleep data collection relies on the smartphone being placed on the user’s mattress, movements of bed partners, pets, and bedding may interfere with results. In most cases, the apps display data in a format similar to that of fitness trackers. Some claim to determine the optimal sleep phase for the alarm to wake the user.
HOW DOES THE TECHNOLOGY WORK?
Older activity-tracking devices used single-channel electroencephalographic recordings or multiple physiologic channels such as galvanic skin response, skin temperature, and heat flux to measure activity to determine transitions between periods of sleep and wakefulness.7,8
None of the currently available consumer sleep tracking devices discloses the exact mechanisms used to measure sleep and wakefulness, but most appear to rely on 3-axis accelerometers,9 ie, microelectromechanical devices that measure front-to-back, side-to-side, and up-and-down motion and convert the data into an activity count. Activity counts are acquired over 30- or 60-second intervals and are entered into algorithms that determine if the pattern indicates that the patient is awake or asleep. This is the same method that actigraphy uses to evaluate sleep, but most actigraphs used in medicine disclose their mechanisms and provide clinicians with the option of using various validated algorithms to classify the activity counts into sleep or awake periods.9–11
ARE THE MEASURES VALID?
Only a few studies have examined the validity and accuracy of current fitness trackers and apps for measuring sleep.
The available studies are difficult to compare; most have been small and used different actigraphy devices for comparison. Some tested healthy volunteers, others included people with suspected or confirmed sleep disorders, and some had both types of participants. In many studies, the device was compared with polysomnography for only 1 night, making the “first-night effect” likely to be a confounding factor, as people tend to sleep worse during the first night of testing. Technical failures for the devices were noted in some studies.12 In addition, some currently used apps may use different platforms than the devices used in these studies, limiting the extrapolation of results.
![]()
As with fitness trackers, few studies have been done to examine the validity of smartphone apps.5 Findings of 3 studies are summarized in Table 2.17–19 In addition to tracking the duration and depth of sleep, some apps purport to detect snoring, sleep apnea, and periodic limb movements of sleep. Discussion of these apps is beyond the scope of this review.
![]()
ARE THE DEVICES CLINICALLY USEFUL?
Although a thorough history remains the cornerstone of a good evaluation of sleep problems, testing is sometimes essential, and in certain situations, objective data can complement the history and clarify the diagnosis.
Polysomnography remains the gold standard for telling when the patient is asleep vs awake, diagnosing sleep-disordered breathing, detecting periodic limb movements and parasomnias, and aiding in the diagnosis of narcolepsy.
Actigraphy, which uses technology similar to fitness trackers, can help distinguish sleep from wakefulness, reveal erratic sleep schedules, and help diagnose circadian rhythm sleep disorders. In patients with insomnia, actigraphy can help determine daily sleep patterns and response to treatment.20 It can be especially useful for patients who cannot provide a clear history, eg, children and those with developmental disabilities or cognitive dysfunction.
Consumer sleep tracking devices, like actigraphy, are portable and unobtrusive, providing a way to measure sleep duration and demonstrate sleep patterns in a patient’s natural environment. Being more accessible, cheaper, and less time-consuming than clinical tests, the commercially available devices could be clinically useful in some situations, eg, for monitoring overall sleep patterns to look for circadian sleep-wake disorders, commonly seen in shift workers (shift work disorder) or adolescents (delayed sleep-wake phase disorder); or in patients with poor motivation to maintain a sleep diary. Because of their poor performance in clinical trials, they should not be relied upon to distinguish sleep from wakefulness, quantify the amount of sleep, determine sleep stages, and awaken patients exclusively from light sleep.
Discerning poor sleep hygiene from insomnia
Patients with insomnia tend to take longer to go to sleep (have longer sleep latencies), wake up more (have more disturbed sleep with increased awakenings), and have shorter sleep times with reduced sleep efficiencies.21 Sleep tracking devices tend to be less accurate for patients with short sleep duration and disturbed sleep, limiting their usefulness in this group. Furthermore, patients with insomnia tend to underestimate their sleep time and overestimate sleep latency; some devices also tend to overestimate the time to fall asleep, reinforcing this common error made by patients.22,23
On the other hand, data from sleep tracking devices could help distinguish poor sleep hygiene from an insomnia disorder. For example, the data may indicate that a patient has poor sleep habits, such as taking long daytime naps or having significantly variable time in and out of bed from day to day. The total times asleep and awake in the middle of the night may also be substantially different on each night, which would also possibly indicate poor sleep hygiene.
Detecting circadian rhythms
A device may show that a patient has a clear circadian preference that is not in line with his or her daily routines, suggesting an underlying circadian rhythm sleep-wake disorder. This may be evident by bedtimes and wake times that are consistent but substantially out of sync with one’s social or occupational needs.
Measuring overall sleep duration
In people with normal sleep, fitness trackers perform reasonably well for measuring overall sleep duration. This information could be used to assess a patient with daytime sleepiness and fatigue to evaluate insufficient sleep as an etiologic factor.
![]()
Table 3 summarizes how to evaluate the data from sleep apps and fitness tracking devices for clinical use. While these features of consumer sleep tracking devices could conceivably help in the above clinical scenarios, further validation of devices in clinical populations is necessary before their use can be recommended without reservation.
ADVISING PATIENTS
Patients sometimes present to clinicians with concerns about the duration of sleep time and time spent in various sleep stages as delineated by their sleep tracking devices. Currently, these devices do not appear to be able to adequately distinguish various sleep stages, and in many users, they can substantially underestimate or overestimate sleep parameters such as time taken to fall asleep or duration of awakenings in the middle of the night. Patients can be reassured about this lack of evidence and should be advised to not place too much weight on such data alone.
Sleep “goals” set by many devices have not been scientifically validated. People without sleep problems should be discouraged from making substantial changes to their routines to accommodate sleep targets set by the devices. Patients should be counseled about the pitfalls of the data and can be reassured that little evidence suggests that time spent in various sleep stages correlates with adverse daytime consequences or with poor health outcomes.
Some of the apps used as alarm clocks claim to be able to tell what stage of sleep people are in and wait to awaken them until they are in a light stage, which is less jarring than being awakened from a deep stage, but the evidence for this is unclear. In the one study that tested this claim, the app did not awaken participants from light sleep more often than is likely to occur by chance.17 The utility of these apps as personalized alarm clocks is still extremely limited, and patients should be counseled to obtain an adequate amount of sleep rather than rely on devices to awaken them during specific sleep stages.
The rates for discontinuing the use of these devices are high, which could limit their utility. Some surveys have shown that close to 50% of users stop using fitness trackers; 33% stop using them within 6 months of obtaining the device.24 Also, there is little evidence that close monitoring of sleep results in behavior changes or improved sleep duration. Conversely, the potential harms of excessive monitoring of one’s sleep are currently unknown.
- Rock Health. The future of biosensing wearables. http://rockhealth.com/reports/the-future-of-biosensing-wearables/. Accessed March 16, 2017.
- Time, Inc. Your wireless life: results of Time’s mobility poll. http://content.time.com/time/interactive/0,31813,2122187,00.html. Accessed March 16, 2017.
- Office of Disease Prevention and Health Promotion (ODPHP). Healthy people 2020. Sleep health. www.healthypeople.gov/2020/topics-objectives/topic/sleep-health. Accessed March 16, 2017.
- Consensus Conference Panel; Watson NF, Badr MS, Belenky G, et al. Joint consensus statement of the American Academy of Sleep Medicine and Sleep Research Society on the recommended amount of sleep for a healthy adult: methodology and discussion. J Clin Sleep Med 2015; 11:931–952.
- Ko PT, Kientz JA, Choe EK, Kay M, Landis CA, Watson NF. Consumer sleep technologies: a review of the landscape. J Clin Sleep Med 2015; 11:1455–1461.
- Investor Place Media, LLC. Top iTunes picks: Apple names best apps of 2014. http://investorplace.com/2014/12/apple-best-apps-of-2014-aapl/#.VIYeE9LF98E/. Accessed April 13, 2017.
- Sunseri M, Liden CB, Farringdon J, et al. The SenseWear armband as a sleep detection device. Internal publication.
- Shambroom JR, Fábregas SE, Johnstone J. Validation of an automated wireless system to monitor sleep in healthy adults. J Sleep Res 2012; 21:221–230.
- John D, Freedson P. ActiGraph and Actical physical activity monitors: a peek under the hood. Med Sci Sports Exerc 2012; 44(suppl 1):S86–S89.
- Sadeh A, Sharkey KM, Carskadon MA. Activity-based sleep-wake identification: an empirical test of methodological issues. Sleep 1994; 17:201–207.
- Kripke DF, Hahn EK, Grizas AP, et al. Wrist actigraphic scoring for sleep laboratory patients: algorithm development. J Sleep Res 2010; 19:612–619.
- Meltzer LJ, Marcus CL. Reply: caffeine therapy for apnea of prematurity: long-term effect on sleep by actigraphy and polysomnography. Am J Respir Crit Care Med 2014; 190:1457–1458.
- Montgomery-Downs HE, Insana SP, Bond JA. Movement toward a novel activity monitoring device. Sleep Breath 2012; 16:913–917.
- Meltzer LJ, Hiruma LS, Avis K, Montgomery-Downs H, Valentin J. Comparison of a commercial accelerometer with polysomnography and actigraphy in children and adolescents. Sleep 2015; 38:1323–1330.
- de Zambotti M, Baker FC, Colrain IM. Validation of sleep-tracking technology compared with polysomnography in adolescents. Sleep 2015; 38:1461–1468.
- de Zambotti M, Claudatos S, Inkelis S, Colrain IM, Baker FC. Evaluation of a consumer fitness-tracking device to assess sleep in adults. Chronobiol Int 2015; 32:1024–1028.
- Toon E, Davey MJ, Hollis SL, Nixon GM, Horne RS, Biggs SN. Comparison of commercial wrist-based and smartphone accelerometers, actigraphy, and PSG in a clinical cohort of children and adolescents. J Clin Sleep Med 2016; 12:343–350.
- Bhat S, Ferraris A, Gupta D, et al. Is there a clinical role for smartphone sleep apps? Comparison of sleep cycle detection by a smartphone application to polysomnography. J Clin Sleep Med 2015; 11:709–715.
- Min JK, Doryab A, Wiese J, Amini S, Zimmerman J, Hong JI. Toss ‘n’ turn: smartphone as sleep and sleep quality detector. Proceedings of the SIGCHI Conference on Human Factors in Computing Systems. Toronto, Ontario, Canada: ACM; 2014:477-486.
- Morgenthaler T, Alessi C, Friedman L, et al; Standards of Practice Committee; American Academy of Sleep Medicine. Practice parameters for the use of actigraphy in the assessment of sleep and sleep disorders: an update for 2007. Sleep 2007; 30:519-529.
- Lichstein KL, Durrence HH, Taylor DJ, Bush AJ, Riedel BW. Quantitative criteria for insomnia. Behav Res Ther 2003; 41:427–445.
- Carskadon MA, Dement WC, Mitler MM, Guilleminault C, Zarcone VP, Spiegel R. Self-reports versus sleep laboratory findings in 122 drug-free subjects with complaints of chronic insomnia. Am J Psychiatry 1976; 133:1382–1388.
- Perlis ML, Giles DE, Mendelson WB, Bootzin RR, Wyatt JK. Psychophysiological insomnia: the behavioural model and a neurocognitive perspective. J Sleep Res 1997; 6:179–188.
- Endeavour Partners LLC. Inside wearables: how the science of human behavior change offers the secret to long-term engagement. http://endeavourpartners.net/assets/Wearables-and-the-Science-of-Human-Behavior-Change-EP4.pdf. Accessed March 16, 2017.
- Rock Health. The future of biosensing wearables. http://rockhealth.com/reports/the-future-of-biosensing-wearables/. Accessed March 16, 2017.
- Time, Inc. Your wireless life: results of Time’s mobility poll. http://content.time.com/time/interactive/0,31813,2122187,00.html. Accessed March 16, 2017.
- Office of Disease Prevention and Health Promotion (ODPHP). Healthy people 2020. Sleep health. www.healthypeople.gov/2020/topics-objectives/topic/sleep-health. Accessed March 16, 2017.
- Consensus Conference Panel; Watson NF, Badr MS, Belenky G, et al. Joint consensus statement of the American Academy of Sleep Medicine and Sleep Research Society on the recommended amount of sleep for a healthy adult: methodology and discussion. J Clin Sleep Med 2015; 11:931–952.
- Ko PT, Kientz JA, Choe EK, Kay M, Landis CA, Watson NF. Consumer sleep technologies: a review of the landscape. J Clin Sleep Med 2015; 11:1455–1461.
- Investor Place Media, LLC. Top iTunes picks: Apple names best apps of 2014. http://investorplace.com/2014/12/apple-best-apps-of-2014-aapl/#.VIYeE9LF98E/. Accessed April 13, 2017.
- Sunseri M, Liden CB, Farringdon J, et al. The SenseWear armband as a sleep detection device. Internal publication.
- Shambroom JR, Fábregas SE, Johnstone J. Validation of an automated wireless system to monitor sleep in healthy adults. J Sleep Res 2012; 21:221–230.
- John D, Freedson P. ActiGraph and Actical physical activity monitors: a peek under the hood. Med Sci Sports Exerc 2012; 44(suppl 1):S86–S89.
- Sadeh A, Sharkey KM, Carskadon MA. Activity-based sleep-wake identification: an empirical test of methodological issues. Sleep 1994; 17:201–207.
- Kripke DF, Hahn EK, Grizas AP, et al. Wrist actigraphic scoring for sleep laboratory patients: algorithm development. J Sleep Res 2010; 19:612–619.
- Meltzer LJ, Marcus CL. Reply: caffeine therapy for apnea of prematurity: long-term effect on sleep by actigraphy and polysomnography. Am J Respir Crit Care Med 2014; 190:1457–1458.
- Montgomery-Downs HE, Insana SP, Bond JA. Movement toward a novel activity monitoring device. Sleep Breath 2012; 16:913–917.
- Meltzer LJ, Hiruma LS, Avis K, Montgomery-Downs H, Valentin J. Comparison of a commercial accelerometer with polysomnography and actigraphy in children and adolescents. Sleep 2015; 38:1323–1330.
- de Zambotti M, Baker FC, Colrain IM. Validation of sleep-tracking technology compared with polysomnography in adolescents. Sleep 2015; 38:1461–1468.
- de Zambotti M, Claudatos S, Inkelis S, Colrain IM, Baker FC. Evaluation of a consumer fitness-tracking device to assess sleep in adults. Chronobiol Int 2015; 32:1024–1028.
- Toon E, Davey MJ, Hollis SL, Nixon GM, Horne RS, Biggs SN. Comparison of commercial wrist-based and smartphone accelerometers, actigraphy, and PSG in a clinical cohort of children and adolescents. J Clin Sleep Med 2016; 12:343–350.
- Bhat S, Ferraris A, Gupta D, et al. Is there a clinical role for smartphone sleep apps? Comparison of sleep cycle detection by a smartphone application to polysomnography. J Clin Sleep Med 2015; 11:709–715.
- Min JK, Doryab A, Wiese J, Amini S, Zimmerman J, Hong JI. Toss ‘n’ turn: smartphone as sleep and sleep quality detector. Proceedings of the SIGCHI Conference on Human Factors in Computing Systems. Toronto, Ontario, Canada: ACM; 2014:477-486.
- Morgenthaler T, Alessi C, Friedman L, et al; Standards of Practice Committee; American Academy of Sleep Medicine. Practice parameters for the use of actigraphy in the assessment of sleep and sleep disorders: an update for 2007. Sleep 2007; 30:519-529.
- Lichstein KL, Durrence HH, Taylor DJ, Bush AJ, Riedel BW. Quantitative criteria for insomnia. Behav Res Ther 2003; 41:427–445.
- Carskadon MA, Dement WC, Mitler MM, Guilleminault C, Zarcone VP, Spiegel R. Self-reports versus sleep laboratory findings in 122 drug-free subjects with complaints of chronic insomnia. Am J Psychiatry 1976; 133:1382–1388.
- Perlis ML, Giles DE, Mendelson WB, Bootzin RR, Wyatt JK. Psychophysiological insomnia: the behavioural model and a neurocognitive perspective. J Sleep Res 1997; 6:179–188.
- Endeavour Partners LLC. Inside wearables: how the science of human behavior change offers the secret to long-term engagement. http://endeavourpartners.net/assets/Wearables-and-the-Science-of-Human-Behavior-Change-EP4.pdf. Accessed March 16, 2017.
KEY POINTS
- Wearable fitness trackers tend to perform better than smartphone applications, which are more prone to interference from bed partners and pets.
- Sleep data from tracking devices are less reliable in patients with fragmented sleep and insomnia.
- In normal sleepers, devices tend to measure sleep duration with reasonable accuracy, so that one can tell if a patient is getting too little sleep or reassure someone who is getting enough sleep.
- Devices may help identify patients with poor sleep hygiene or atypical circadian rhythms.
Autosomal dominant polycystic kidney disease and the heart and brain
Autosomal dominant polycystic kidney disease (ADPKD) has significant extrarenal manifestations. Hypertension is a common complication, arises early in the course of the disease, and is implicated in the development of left ventricular hypertrophy. Patients with ADPKD are also at risk of other cardiovascular complications (Table 1).
This article reviews the timely diagnosis of these common ADPKD complications and how to manage them.
ADPKD ACCOUNTS FOR 10% OF END-STAGE RENAL DISEASE

ADPKD is a genetic condition characterized by multiple renal cysts.1 Progressive enlargement of these cysts leads to a gradual decline in kidney function and eventually end-stage renal disease by the fifth or sixth decade of life.2 Worldwide, about 12.5 million people have ADPKD, and it accounts for about 10% of cases of end-stage renal disease.1,3,4
ADPKD has a variety of clinical presentations, including (in decreasing order of frequency) hypertension, flank pain, abdominal masses, urinary tract infection, renal failure, renal stones, and cerebrovascular accidents.2
Extrarenal complications are common and include hepatic cysts, hypertension, left ventricular hypertrophy, valvular heart disease, intracranial and extracranial aneurysms, pancreatic cysts, and diverticulosis.1–5
Less-common complications are dissection of the aorta and the internal carotid, vertebral, and iliac arteries6–10; aneurysm of the coronary, popliteal, and splenic arteries11–14; atrial myxoma15; cardiomyopathy16; pericardial effusion17; intracranial arterial dolichoectasia18; arachnoid cysts2; and intraoperative inferior vena cava syndrome (normally in ADPKD patients, pressure on the inferior vena cava results in compensatory sympathetic overactivity to maintain blood pressure), which occurs due to reduced sympathetic output under the influence of epidural or general anesthesia.19
Cardiovascular complications, especially cardiac hypertrophy and coronary artery disease, are now the leading cause of death in patients with ADPKD, as renal replacement therapy has improved and made death from end-stage renal disease less common.20,21
HYPERTENSION IN ADPKD
Hypertension is the most frequent initial presentation of ADPKD, occurring in 50% to 75% of cases and usually preceding the onset of renal failure.2,22 Hypertension is more common in male ADPKD patients, begins early in the course of the disease, and is diagnosed around the fourth decade of life.21
In a study in 2007, de Almeida et al23 used 24-hour ambulatory blood pressure monitoring early in the course of ADPKD and found significantly higher systolic, diastolic, and mean 24-hour blood pressures in ADPKD patients who had normal in-office blood pressure than in normotensive controls. In addition, nighttime systolic, nighttime diastolic, and nighttime mean blood pressures were significantly higher in the ADPKD group.
Hypertension is strongly associated with an accelerated decline in renal function to end-stage renal disease, development of left ventricular hypertrophy, and cardiovascular death.20,24
Although a prospective study25 showed a strong association between renal stones and hypertension in ADPKD, the relation between them is not clear. The incidence of renal stones is higher in hypertensive than in normotensive ADPKD patients, although evidence has to be established whether nephrolithiasis is a risk factor for hypertension or the other way around.25

Hypertension in ADPKD is multifactorial (Figure 1). The major factors associated with its development are increased activation of the renin-angiotensin-aldosterone system (RAAS); overexpression of endothelin receptor subtype A (ET-A) in cystic kidneys; increased production of endothelin 1 (ET-1); and sodium retention.26–31
The renin-angiotensin-aldosterone system
Activation of the RAAS plays a major role in the development and maintenance of hypertension in ADPKD. This is thought to be mainly due to progressive enlargement of renal cysts, which causes renal arteriolar attenuation and ischemia secondary to pressure effects, which in turn activates the RAAS.26,30,32–34 Two studies in patients with normal renal function found that cyst growth and increasing kidney volume have a strong relationship with the development of hypertension and declining kidney function.35,36
Ectopic secretion of RAAS components in polycystic kidneys has also been implicated in the development of hypertension, whereby renin, angiotensinogen, angiotensin-converting enzyme (ACE), angiotensin II, and angiotensin II receptors are produced in the epithelium of cysts and dilated tubules in polycystic kidneys.37–39 Proximal renal cysts and tubules produce ectopic angiotensinogen, which is converted to angiotensin I by renin in distal renal cysts. Angiotensin I is converted to angiotensin II by ACE in distal tubules, which in turn stimulates angiotensin II receptors, causing sodium and water retention in distal tubules.37 This may be responsible for hypertension in the initial stages; however, RAAS hyperactivity due to renal injury may predominate during later stages.37
Increased RAAS activity also increases sympathetic output, which in turn raises catecholamine levels and blood pressure.34 A study showed higher levels of plasma catecholamines in ADPKD hypertensive patients irrespective of renal function than in patients with essential hypertension.40
ET-A receptor and ET-1
A few studies have shown that in ADPKD patients, increased density of ET-A receptors and overproduction of ET-1, a potent vasoconstrictor, play a significant role in the development of hypertension and gradual loss of kidney function due to cyst enlargement and interstitial scarring.28,29 Ong et al29 found that expression of ET-A receptors is increased in smooth muscle cells of renal arteries, glomerular mesangial cells, and cyst epithelia in ADPKD.
Sodium retention
Studies in ADPKD patients with preserved renal function have linked high blood pressure to sodium retention and volume expansion.30,31,41 However, this phenomenon reverses when there is significant renal impairment in ADPKD.
As evidence of this, a study demonstrated significantly more natriuresis in patients with renal failure due to ADPKD than in patients with a similar degree of renal failure due to chronic glomerulonephritis.31 Moreover, another study found that the prevalence of hypertension is higher in ADPKD patients than in those with other nephropathies with preserved renal function, but this association reverses with significant decline in kidney function.22
MANAGING HYPERTENSION IN ADPKD
Early diagnosis of hypertension and effective control of it, even before ADPKD is diagnosed, is crucial to reduce cardiovascular mortality. Aggressive blood pressure control in the prehypertensive phase of ADPKD will also help reduce the incidence of left ventricular hypertrophy and mitral regurgitation and slow the progression of renal failure (Figure 2).

A meta-analysis42 revealed hypertension to be present in 20% of ADPKD patients younger than 21, and many of them were undiagnosed. This study also suggests that patients at risk of hypertension (ie, all patients with ADPKD) should be routinely screened for it.
Ambulatory blood pressure monitoring may play an important role in diagnosing hypertension early in the prehypertensive stage of ADPKD.23
Target blood pressures: No consensus
Two well-powered double-blind, placebo-controlled trials, known as HALT-PKD Study A and HALT-PKD Study B, tested the effects of 2 different blood pressure targets and of monotherapy with an ACE inhibitor vs combination therapy with an ACE inhibitor plus an angiotensin II receptor blocker (ARB) on renal function, total kidney volume, left ventricular mass index, and urinary albumin excretion in the early (estimated glomerular filtration rate [eGFR] > 60 mL/min) and late (eGFR 25–60 mL/min) stages of ADPKD, respectively.43,44
HALT-PKD Study A43 found that, in the early stages of ADPKD with preserved renal function, meticulous control of blood pressure (95–110/60–75 mm Hg) was strongly correlated with significant reductions in left ventricular mass index, albuminuria, and rate of total kidney volume growth without remarkable alteration in renal function compared with standard blood pressure control (120–130/70–80 mm Hg). However, no notable differences were observed between the ACE inhibitor and ACE inhibitor-plus-ARB groups.43
Despite the evidence, universal consensus guidelines are lacking, and the available guidelines on hypertension management have different blood pressure goals in patients with chronic kidney disease.
The eighth Joint National Committee guideline of 2014 recommends a blood pressure goal of less than 140/90 mm Hg in patients with diabetic and nondiabetic chronic kidney disease.45
The National Institute for Health and Care Excellence 2011 guideline recommends a blood pressure goal of less than 130/80 mm Hg in chronic kidney disease patients.46
The European Society of Hypertension and European Society of Cardiology joint 2013 guideline recommends a systolic blood pressure goal of less than 140 mm Hg in diabetic and nondiabetic patients with chronic kidney disease.47
The 2016 Kidney Health Australia-Caring for Australians With Renal Impairment guideline for diagnosis and management of ADPKD48 recommends a lower blood pressure goal of 96–110/60–75 mm Hg in patients with an eGFR greater than 60 mL/min/1.73 m2 who can tolerate it without side effects, which is based on the findings of HALT-PKD Study A.43
Helal et al recommend that blood pressure be controlled to less than 130/80 mm Hg, until there is more evidence for a safe and effective target blood pressure goal in ADPKD patients.49
We recommend a target blood pressure less than 110/75 mm Hg in hypertensive ADPKD patients with preserved renal function who can tolerate this level, and less than 130/80 mm Hg in ADPKD patients with stage 3 chronic kidney disease. These targets can be achieved with ACE inhibitor or ARB monotherapy.43,44 However, no studies have established the safest lower limit of target blood pressure in ADPKD.
ACE inhibitors, ARBs are mainstays
Mainstays of antihypertensive drug therapy in ADPKD are ACE inhibitors and ARBs.
HALT-PKD Study B44 demonstrated that, in the late stages of ADPKD, target blood pressure control (110–130/70–80 mm Hg) can be attained with ACE inhibitor monotherapy or with an ACE inhibitor plus an ARB, but the latter produced no additive benefit.
Patch et al,50 in a retrospective cohort study, showed that broadening the spectrum of antihypertensive therapy decreases mortality in ADPKD patients. Evaluating ADPKD patients from the UK General Practice Research Database between 1991 and 2008, they found a trend toward lower mortality rates as the number of antihypertensive drugs prescribed within 1 year increased. They also observed that the prescription of RAAS-blocking agents increased from 7% in 1991 to 46% in 2008.50
However, a 3-year prospective randomized double-blind study compared the effects of the ACE inhibitor ramipril and the beta-blocker metoprolol in hypertensive ADPKD patients.51 The results showed that effective blood pressure control could be achieved in both groups with no significant differences in left ventricular mass index, albuminuria, or kidney function.51
Treatment strategies
Lifestyle modification is the initial approach to the management of hypertension before starting drug therapy. Lifestyle changes include dietary salt restriction to less than 6 g/day, weight reduction, regular exercise, increased fluid intake (up to 3 L/day or to satisfy thirst), smoking cessation, and avoidance of caffeine.47–49
ACE inhibitors are first-line drugs in hypertensive ADPKD patients.
ARBs can also be considered, but there is no role for dual ACE inhibitor and ARB therapy.43,48 A study found ACE inhibitors to be more cost-effective and to decrease mortality rates to a greater extent than ARBs.52
Beta-blockers or calcium channel blockers should be considered instead if ACE inhibitors and ARBs are contraindicated, or as add-on drugs if ACE inhibitors and ARBs fail to reduce blood pressure adequately.48,49
Diuretics are third-line agents. Thiazides are preferred in ADPKD patients with normal renal function and loop diuretics in those with impaired renal function.49
LEFT VENTRICULAR HYPERTROPHY IN ADPKD
Increased left ventricular mass is an indirect indicator of untreated hypertension, and it often goes unnoticed in patients with undiagnosed ADPKD. Left ventricular hypertrophy is associated with arrhythmias and heart failure, which contribute significantly to cardiovascular mortality and adverse renal outcomes.20,24
A 5-year randomized clinical trial by Cadnapaphornchai et al36 in ADPKD patients between 4 and 21 years of age showed strong correlations between hypertension, left ventricular mass index, and kidney volume and a negative correlation between left ventricular mass index and renal function.
Several factors are thought to contribute to left ventricular hypertrophy in ADPKD (Figure 1).
Hypertension. Two studies of 24-hour ambulatory blood pressure monitoring showed that nocturnal blood pressures decreased less in normotensive and hypertensive ADPKD patients than in normotensive and hypertensive controls.23,53 This persistent elevation of nocturnal blood pressure may contribute to the development and progression of left ventricular hypertrophy.
On the other hand, Valero et al54 reported that the left ventricular mass index was strongly associated with ambulatory systolic blood pressure rather than elevated nocturnal blood pressure in ADPKD patients compared with healthy controls.
FGF23. High levels of fibroblast growth factor 23 (FGF23) have been shown to be strongly associated with left ventricular hypertrophy in ADPKD. Experimental studies have shown that FGF23 is directly involved in the pathogenesis of left ventricular hypertrophy through stimulation of the calcineurin-nuclear factor of activated T cells pathway.
Faul et al55 induced cardiac hypertrophy in mice that were deficient in klotho (a transmembrane protein that increases FGF23 affinity for FGF receptors) by injecting FGF23 intravenously.
Yildiz et al56 observed higher levels of FGF23 in hypertensive and normotensive ADPKD patients with normal renal function than in healthy controls. They also found a lower elasticity index in the large and small arteries in normotensive and hypertensive ADPKD patients, which accounts for vascular dysfunction. High FGF23 levels may be responsible for the left ventricular hypertrophy seen in normotensive ADPKD patients with preserved renal function.
Polymorphisms in the ACE gene have been implicated in the development of cardiac hypertrophy in ADPKD.
Wanic-Kossowska et al57 studied the association between ACE gene polymorphisms and cardiovascular complications in ADPKD patients. They found a higher prevalence of the homozygous DD genotype among ADPKD patients with end-stage renal disease than in those in the early stages of chronic kidney disease in ADPKD. Also, the DD genotype has been shown to be more strongly associated with left ventricular hypertrophy and left ventricular dysfunction than other (II or ID) genotypes. These findings suggest that the DD genotype carries higher risk for the development of end-stage renal disease, left ventricular hypertrophy, and other cardiovascular complications.
MANAGING LEFT VENTRICULAR HYPERTROPHY IN ADPKD
Preventing and halting progression of left ventricular hypertrophy primarily involves effective blood pressure control, especially in the early stages of ADPKD (Figure 2).
A 7-year prospective randomized trial in ADPKD patients with established hypertension and left ventricular hypertrophy proved that aggressive (< 120/80 mm Hg) compared with standard blood pressure control (135–140/85–90 mm Hg) significantly reduces left ventricular mass index. ACE inhibitors were preferred over calcium channel blockers.58
HALT-PKD Study A showed that a significant decrease in left ventricular mass index can be achieved by aggressive blood pressure control (95–110/60–75 mm Hg) with an ACE inhibitor alone or in combination with an ARB in the early stages of ADPKD with preserved renal function.43
A 5-year randomized clinical trial in children with borderline hypertension treated with an ACE inhibitor for effective control of blood pressure showed no change in left ventricular mass index or renal function.36,59
These results support starting ACE inhibitor therapy early in the disease process when blood pressure is still normal or borderline to prevent the progression of left ventricular hypertrophy or worsening kidney function.
Since FGF23 is directly involved in the causation of left ventricular hypertrophy, FGF receptors may be potential therapeutic targets to prevent left ventricular hypertrophy in ADPKD. An FGF receptor blocker was shown to decrease left ventricular hypertrophy in rats with chronic kidney disease without affecting blood pressure.55
INTRACRANIAL ANEURYSM IN ADPKD
Intracranial aneurysm is the most dangerous complication of ADPKD. When an aneurysm ruptures, the mortality rate is 4% to 7%, and 50% of survivors are left with residual neurologic deficits.5,60,61
In various studies, the prevalence of intracranial aneurysm in ADPKD ranged from 4% to 41.2%, compared with 1% in the general population.5,62,63 On follow-up ranging from 18 months to about 10 years, the incidence of new intracranial aneurysm was 2.6% to 13.3% in patients with previously normal findings on magnetic resonance angiography and 25% in patients with a history of intracranial aneurysm.62,64,65
The most common sites are the middle cerebral artery (45%), internal carotid artery (40.5%), and anterior communicating artery (35.1%).66 (The numbers add up to more than 100% because some patients have aneurysms in more than 1 site.) The mean size of a ruptured aneurysm was 6 mm per a recent systematic review.66 Intracranial aneurysms 6 mm or larger are at highest risk of rupture.66
SCREENING FOR INTRACRANIAL ANEURYSM
Timely screening and intervention for intracranial aneurysm is crucial to prevent death from intracranial hemorrhage.
Currently, there are no standard guidelines for screening and follow-up of intracranial aneurysm in ADPKD patients. However, some recommendations are available from the ADPKD Kidney Disease Improving Global Outcomes Controversies Conference3 and Kidney Health Australia—Caring for Australasians With Renal Impairment ADPKD guidelines67 (Figure 3).
Imaging tests
Magnetic resonance angiography (MRA) with gadolinium enhancement and computed tomographic angiography (CTA) are recommended for screening in ADPKD patients with normal renal function,67 but time-of-flight MRA without gadolinium is the imaging test of choice because it is noninvasive and poses no risk of nephrotoxicity or contrast allergy.3,68 Further, gadolinium should be avoided in patients whose eGFR is 30 mL/min/1.73 m2 or less because of risk of nephrogenic systemic sclerosis and fibrosis.67,68
The sensitivity of time-of-flight MRA screening for intracranial aneurysm varies depending on the size of aneurysm; 67% for those less than 3 mm, 79% for those 3 to 5 mm, and 95% for those larger than 5 mm.69 The sensitivity of CTA screening is 95% for aneurysms larger than 7 mm and 53% for those measuring 2 mm.70,71 The specificity of CTA screening was reported to be 98.9% overall.71
When to screen
Screening for intracranial aneurysm is recommended at the time of ADPKD diagnosis for all high-risk patients, ie, those who have a family history of intracranial hemorrhage or aneurysm in an affected first-degree relative.67 It is also recommended for ADPKD patients with a history of sudden-onset severe headache or neurologic symptoms.67 A third group for whom screening is recommended is ADPKD patients who have no family history of intracranial aneurysm or hemorrhage but who are at risk of poor outcome if an intracranial aneurysm ruptures (eg, those undergoing major elective surgery, with uncontrolled blood pressure, on anticoagulation, with a history of or current smoking, and airline pilots).67
Patients found to have an intracranial aneurysm on screening should be referred to a neurosurgeon and should undergo repeat MRA or CTA imaging every 6 to 24 months.3 High-risk ADPKD patients with normal findings on initial screening should have repeat MRA or CTA screening in 5 to 10 years unless they suffer from sudden-onset severe headache or neurologic symptoms.65,67
Both smoking and high blood pressure increase the risk of formation and growth of intracranial aneurysm. Hence, meticulous control of blood pressure and smoking cessation are recommended in ADPKD patients.3,67
CARDIAC VALVULAR ABNORMALITIES IN ADPKD
Of the valvular abnormalities that complicate ADPKD, the more common ones are mitral valve prolapse and mitral and aortic regurgitation. The less common ones are tricuspid valve prolapse and tricuspid regurgitation.72–74
The pathophysiology underlying these valvular abnormalities is unclear. However, defective collagen synthesis and myxomatous degeneration have been demonstrated in histopathologic examination of affected valvular tissue.75 Also, ACE gene polymorphism, especially the DD genotype, has been shown to be associated with cardiac valvular calcifications and valvular insufficiency.57
Lumiaho et al72 found a higher prevalence of mitral valve prolapse, mitral regurgitation, and left ventricular hypertrophy in patients with ADPKD type 1 (due to abnormalities in PDK1) than in unaffected family members and healthy controls. The investigators speculated that mitral regurgitation is caused by the high blood pressure observed in ADPKD type 1 patients, since hypertension causes left ventricular hypertrophy and left ventricular dilatation. The severity of renal failure was related to mitral regurgitation but not mitral valve prolapse.
Similarly, Gabow et al24 showed that there is no significant relationship between mitral valve prolapse and progression of renal disease in ADPKD.
Interestingly, Fick et al20 found that mitral valve prolapse has no significant effect on cardiovascular mortality.
CORONARY ARTERY DISEASE AND ANEURYSM IN ADPKD
Atherosclerosis sets in early in ADPKD, resulting in coronary artery disease and adverse cardiovascular outcomes.
Coronary flow velocity reserve is the ability of coronary arteries to dilate in response to myocardial oxygen demand. Atherosclerosis decreases this reserve in ADPKD patients, as shown in several studies.
Turkmen et al, in a series of studies,76–78 found that ADPKD patients had significantly less coronary flow velocity reserve, thicker carotid intima media (a surrogate marker of atherosclerosis), and greater insulin resistance than healthy controls. These findings imply that atherosclerosis begins very early in the course of ADPKD and has remarkable effects on cardiovascular morbidity and mortality.76
Aneurysm. Although the risk of extracranial aneurysm is higher with ADPKD, coronary artery aneurysm is uncommon. The pathogenesis of coronary aneurysm has been linked to abnormal expression of the proteins polycystin 1 and polycystin 2 in vascular smooth muscle.11,79 The PKD1 and PKD2 genes encode polycystin 1 and 2, respectively, in ADPKD. These polycystins are also expressed in the liver, kidneys, and myocardium and are involved in the regulation of intracellular calcium, stretch-activated ion channels, and vascular smooth muscle cell proliferation and apoptosis.11,16 Abnormally expressed polycystin in ADPKD therefore has an impact on arterial wall integrity, resulting in focal medial defects in the vasculature that eventually develop into micro- and macroaneurysms.11
Hadimeri et al79 found a higher prevalence of coronary aneurysm and ectasia in ADPKD patients than in controls. Most coronary aneurysms are smaller than 1 cm; however, a coronary aneurysm measuring 4 cm in diameter was found at autopsy of an ADPKD patient.11
Spontaneous coronary artery dissection is very rare in the general population, but Bobrie et al reported a case of it in an ADPKD patient.9
ATRIAL MYXOMA, CARDIOMYOPATHY, AND PERICARDIAL EFFUSION IN ADPKD
Atrial myxoma in ADPKD patients has been described in 2 case reports.15,80 However, the association of atrial myxoma with ADPKD is poorly understood and may be a coincidental finding.
Cardiomyopathy in ADPKD has been linked to abnormalities in the intracellular calcium pathway, although a clear picture of its involvement has yet to be established.
Paavola et al16 described the pathophysiology of ADPKD-associated cardiomyopathy in PKD2 mutant zebrafish lacking polycystin 2. These mutants showed decreased cardiac output and had atrioventricular blocks. The findings were attributed to abnormal intracellular calcium cycling. These findings correlated well with the frequent finding of idiopathic dilated cardiomyopathy in ADPKD patients, especially with PKD2 mutations.16 Also, 2 cases of dilated cardiomyopathy in ADPKD have been reported and thought to be related to PKD2 mutations.81,82
Pericardial effusion. Even though the exact pathophysiology of pericardial effusion in ADPKD is unknown, it has been theorized to be related to defects in connective tissue and extracellular matrix due to PKD1 and PKD2 mutations. These abnormalities increase the compliance and impair the recoil capacity of connective tissue, which results in unusual distention of the parietal pericardium. This abnormal distention of the parietal pericardium together with increased extracellular volume may lead to pericardial effusion.17
Qian et al17 found a higher prevalence of pericardial effusion in ADPKD patients. It was generally asymptomatic, and the cause was attributed to these connective tissue and extracellular matrix abnormalities.
EMERGING THERAPIES AND TESTS
Recent trials have investigated the effects of vasopressin receptor antagonists, specifically V2 receptor blockers in ADPKD and its complications.
Tolvaptan has been shown to slow the rate of increase in cyst size and total kidney volume.83,84 Also, a correlation between kidney size and diastolic blood pressure has been observed in ADPKD patients.85 Reducing cyst volume may reduce pressure effects and decrease renal ischemia, which in turn may reduce RAAS activation; however, the evidence to support this hypothesis is poor. A major clinical trial of tolvaptan in ADPKD patients showed no effect on blood pressure control, but the drug slowed the rate of increase in total kidney volume and fall in eGFR.83
Endothelin receptor antagonists are also in the preliminary stage of development for use in ADPKD. The effects of acute blockade of the renal endothelial system with bosentan were investigated in animal models by Hocher et al.28 This study showed a greater reduction in mean arterial pressure after bosentan administration, resulting in significantly decreased GFR and renal blood flow. Nonetheless, the mean arterial pressure-lowering effect of bosentan was more marked than the reductions in GFR and renal blood flow.
Raina et al,86 in a pilot cross-sectional analysis, showed that urinary excretion of ET-1 is increased in ADPKD patients, and may serve as a surrogate marker for ET-1 in renal tissue and a noninvasive marker of early kidney injury.
- Ha SK, Park CH, Kna JS, et al. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Yonsei Med J 1997; 38:111–116.
- Romao EA, Moyses Neto M, Teixeira SR, Muglia VF, Vieira-Neto OM, Dantas M. Renal and extrarenal manifestations of autosomal dominant polycystic kidney disease. Brazilian J Med Biol Res 2006; 39:533–538.
- Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 2015; 88:17–27.
- Perrone RD, Ruthazer R, Terrin NC. Survival after end-stage renal disease in autosomal dominant polycystic kidney disease: contribution of extrarenal complications to mortality. Am J Kidney Dis 2001; 38:777–784.
- Ecder T. Cardiovascular complications in autosomal dominant polycystic kidney disease. Curr Hypertens Rev 2013; 9:2–11.
- Silverio A, Prota C, Di Maio M, et al. Aortic dissection in patients with autosomal dominant polycystic kidney disease: a series of two cases and a review of the literature. Nephrology 2015; 20:229–235.
- Ramineni R, Daniel GK. Use of endovascular stent-graft repair for type B aortic dissection in polycystic kidney disease. J Invas Cardiol 2010; 22:E171–E174.
- Courtois A, Nusgens BV, Delvenne P, et al. Dissection of iliac artery in a patient with autosomal dominant polycystic kidney disease: a case report. Aorta 2013; 1:123–125.
- Bobrie G, Brunet-Bourgin F, Alamowitch S, et al. Spontaneous artery dissection: is it part of the spectrum of autosomal dominant polycystic kidney disease? Nephrol Dial Transplant 1998; 13:2138–2141.
- Minami T, Karube N, Sakamoto A. [Thoracic aortic dissection complicating autosomal dominant polycystic kidney disease; report of a case]. Kyobu Geka 2009; 62:924–927.
- Ohara K, Kimura T, Karasawa T, et al. A large coronary aneurysm and its probable precursor lesions in a patient with autosomal dominant polycystic kidney disease: an implication for the process of aneurysmogenesis. Pathol Int 2012; 62:758–762.
- Al-Hakim W, Goldsmith DJ. Bilateral popliteal aneurysms complicating adult polycystic kidney disease in a patient with a marfanoid habitus. Postgrad Med J 2003; 79:474–475.
- Kanagasundaram NS, Perry EP, Turney JH. Aneurysm of the splenic artery in a patient with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1999; 14:183–184.
- Kang YR, Ahn JH, Kim KH, Choi YM, Choi J, Park JR. Multiple cardiovascular manifestations in a patient with autosomal dominant polycystic kidney disease. J Cardiovasc Ultrasound 2014; 22:144–147.
- Iglesias D, Fraga AR, Arrizurieta E, et al. Atrial myxoma in a woman with autosomal dominant polycystic kidney disease type 2. Am J Kidney Dis 1997; 29:164–165.
- Paavola J, Schliffke S, Rossetti S, et al. Polycystin-2 mutations lead to impaired calcium cycling in the heart and predispose to dilated cardiomyopathy. J Mol Cell Cardiol 2013; 58:199–208.
- Qian Q, Hartman RP, King BF, Torres VE. Increased occurrence of pericardial effusion in patients with autosomal dominant polycystic kidney disease.
- Schievink WI, Torres VE, Wiebers DO, Huston J 3rd. Intracranial arterial dolichoectasia in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1997; 8:1298–1303.
- Pierre SA, Jaeger MT, Siemens DR. Intra-operative inferior vena cava syndrome in a patient with autosomal dominant polycystic kidney disease. World J Urol 2006; 24:110–112.
- Fick GM, Johnson AM, Hammond WS, Gabow PA. Causes of death in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1995; 5:2048–2056.
- Helal I, Reed B, Mettler P, et al. Prevalence of cardiovascular events in patients with autosomal dominant polycystic kidney disease. Am J Nephrol 2012; 36:362–370.
- Calabrese G, Vagelli G, Cristofano C, Barsotti G. Behaviour of arterial pressure in different stages of polycystic kidney disease. Nephron 1982; 32:207–208.
- de Almeida EA, de Oliveira EI, Lopes JA, Almeida AG, Lopes MG, Prata MM. Ambulatory blood pressure measurement in young normotensive patients with autosomal dominant polycystic kidney disease. Rev Port Cardiol 2007; 26:235–243.
- Gabow PA, Johnson AM, Kaehny WD, et al. Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 41:1311–1319.
- Bajrami V, Idrizi A, Roshi E, Barbullushi M. Association between nephrolithiasis, hypertension and obesity in polycystic kidney disease. Open Access Maced J Med Sci 2016; 4:43–46.
- Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med 1990; 323:1091–1096.
- Nakamura T, Ebihara I, Fukui M, et al. Increased endothelin and endothelin receptor mRNA expression in polycystic kidneys of cpk mice. J Am Soc Nephrol 1993; 4:1064–1072.
- Hocher B, Zart R, Schwarz A, et al. Renal endothelin system in polycystic kidney disease. J Am Soc Nephrol 1998; 9:1169–1177.
- Ong AC, Newby LJ, Dashwood MR. Expression and cellular localisation of renal endothelin-1 and endothelin receptor subtypes in autosomal-dominant polycystic kidney disease. Nephron Exper Nephrol 2003; 93:e80.
- Nash DA Jr. Hypertension in polycystic kidney disease without renal failure. Arch Intern Med 1977; 137:1571–1575.
- D’Angelo A, Mioni G, Ossi E, Lupo A, Valvo E, Maschio G. Alterations in renal tubular sodium and water transport in polycystic kidney disease. Clin Nephrol 1975; 3:99–105.
- Ettinger A, Kahn PC, Wise HM Jr. The importance of selective renal angiography in the diagnosis of polycystic disease. J Urol 1969; 102:156–161.
- Cornell SH. Angiography in polycystic disease of the kidneys. J Urol 1970; 103:24–26.
- Schrier RW. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2009; 20:1888–1893.
- Chapman AB, Guay-Woodford LM, Grantham JJ, et al. Renal structure in early autosomal-dominant polycystic kidney disease (ADPKD): The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) cohort. Kidney Int 2003; 64:1035–1045.
- Cadnapaphornchai MA, McFann K, Strain JD, Masoumi A, Schrier RW. Prospective change in renal volume and function in children with ADPKD. Clin J Am Soc Nephrol 2009; 4:820–829.
- Loghman-Adham M, Soto CE, Inagami T, Cassis L. The intrarenal renin-angiotensin system in autosomal dominant polycystic kidney disease. Am J Physiol Renal Physiol 2004; 287:F775–F788.
- Torres VE, Donovan KA, Scicli G, et al. Synthesis of renin by tubulocystic epithelium in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 42:364–373.
- Graham PC, Lindop GB. The anatomy of the renin-secreting cell in adult polycystic kidney disease. Kidney Int 1988; 33:1084–1090.
- Cerasola G, Vecchi M, Mule G, et al. Sympathetic activity and blood pressure pattern in autosomal dominant polycystic kidney disease hypertensives. Am J Nephrol 1998; 18:391–398.
- Valvo E, Gammaro L, Tessitore N, et al. Hypertension of polycystic kidney disease: mechanisms and hemodynamic alterations. Am J Nephrol 1985; 5:176–181.
- Marlais M, Cuthell O, Langan D, Dudley J, Sinha MD, Winyard PJ. Hypertension in autosomal dominant polycystic kidney disease: a meta-analysis. Arch Dis Child 2016; 101:1142–1147.
- Schrier RW, Abebe KZ, Perrone RD, et al. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371:2255–2266.
- Torres VE, Abebe KZ, Chapman AB, et al. Angiotensin blockade in late autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371:2267–2276.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Ritchie LD, Campbell NC, Murchie P. New NICE guidelines for hypertension. BMJ 2011; 343:d5644.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34:2159–2219.
- Rangan GK, Alexander SI, Campbell KL, et al. KHA-CARI guideline recommendations for the diagnosis and management of autosomal dominant polycystic kidney disease. Nephrology 2016; 21:705–716.
- Helal I, Al-Rowaie F, Abderrahim E, Kheder A. Update on pathogenesis, management, and treatment of hypertension in autosomal dominant polycystic kidney disease. Saudi J Kidney Dis Transpl 2017; 28:253–260.
- Patch C, Charlton J, Roderick PJ, Gulliford MC. Use of antihypertensive medications and mortality of patients with autosomal dominant polycystic kidney disease: a population-based study. Am J Kidney Dis 2011; 57:856–862.
- Zeltner R, Poliak R, Stiasny B, Schmieder RE, Schulze BD. Renal and cardiac effects of antihypertensive treatment with ramipril vs metoprolol in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2008; 23:573–579.
- Clark LA, Whitmire S, Patton S, Clark C, Blanchette CM, Howden R. Cost-effectiveness of angiotensin-converting enzyme inhibitors versus angiotensin II receptor blockers as first-line treatment in autosomal dominant polycystic kidney disease. J Med Econ 2017:1–17.
- Li Kam Wa TC, Macnicol AM, Watson ML. Ambulatory blood pressure in hypertensive patients with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1997; 12:2075–2080.
- Valero FA, Martinez-Vea A, Bardaji A, et al. Ambulatory blood pressure and left ventricular mass in normotensive patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1999; 10:1020–1026.
- Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest 2011; 121:4393–4408.
- Yildiz A, Gul CB, Ersoy A, Asiltas B, Ermurat S, Dogan S, et al. Arterial dysfunction in early autosomal dominant polycystic kidney disease independent of fibroblast growth factor 23. Iranian J Kidney Dis 2014; 8:443–449.
- Wanic-Kossowska M, Posnik B, Kobelski M, et al. The polymorphism of the ACE gene affects left ventricular hypertrophy and causes disturbances in left ventricular systolic/diastolic function in patients with autosomal dominant polycystic kidney disease. ScientificWorldJournal 2014; 2014:707658.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Cadnapaphornchai MA. Hypertension in children with autosomal dominant polycystic kidney disease (ADPKD). Curr Hypertens Rev 2013; 9:21–26.
- Schievink WI, Prendergast V, Zabramski JM. Rupture of a previously documented small asymptomatic intracranial aneurysm in a patient with autosomal dominant polycystic kidney disease. Case report. J Neurosurg 1998; 89:479–482.
- Graf S, Schischma A, Eberhardt KE, Istel R, Stiasny B, Schulze BD. Intracranial aneurysms and dolichoectasia in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2002; 17:819–823.
- Nakajima F, Shibahara N, Arai M, Gohji K, Ueda H, Katsuoka Y. Intracranial aneurysms and autosomal dominant polycystic kidney disease: followup study by magnetic resonance angiography. J Urol 2000; 164:311–313.
- Wakabayashi T, Fujita S, Ohbora Y, Suyama T, Tamaki N, Matsumoto S. Polycystic kidney disease and intracranial aneurysms. Early angiographic diagnosis and early operation for the unruptured aneurysm. J Neurosurg 1983; 58:488–491.
- Belz MM, Fick-Brosnahan GM, Hughes RL, et al. Recurrence of intracranial aneurysms in autosomal-dominant polycystic kidney disease. Kidney Int 2003; 63:1824–1830.
- Schrier RW, Belz MM, Johnson AM, et al. Repeat imaging for intracranial aneurysms in patients with autosomal dominant polycystic kidney disease with initially negative studies: a prospective ten-year follow-up. J Am Soc Nephrol 2004; 15:1023–1028.
- Cagnazzo F, Gambacciani C, Morganti R, Perrini P. Intracranial aneurysms in patients with autosomal dominant polycystic kidney disease: prevalence, risk of rupture, and management. A systematic review. Acta Neurochirurgica 2017; 5:811–821.
- Lee VW, Dexter MA, Mai J, Vladica P, Lopez-Vargas P, Rangan GK. KHA-CARI autosomal dominant polycystic kidney disease guideline: management of intracranial aneurysms. Semin Nephrol 2015; 35:612–617.
- Rozenfeld MN, Ansari SA, Shaibani A, Russell EJ, Mohan P, Hurley MC. Should patients with autosomal dominant polycystic kidney disease be screened for cerebral aneurysms? AJNR Am J Neuroradiol 2014; 35:3–9.
- Hiratsuka Y, Miki H, Kiriyama I, et al. Diagnosis of unruptured intracranial aneurysms: 3T MR angiography versus 64-channel multi-detector row CT angiography. Magn Reson Med Sci 2008; 7:169–178.
- van Gelder JM. Computed tomographic angiography for detecting cerebral aneurysms: implications of aneurysm size distribution for the sensitivity, specificity, and likelihood ratios. Neurosurgery 2003; 53:597–605.
- Villablanca JP, Jahan R, Hooshi P, et al. Detection and characterization of very small cerebral aneurysms by using 2D and 3D helical CT angiography. AJNR Am J Neuroradiol 2002; 23:1187–1198.
- Lumiaho A, Ikäheimo R, Miettinen R, et al. Mitral valve prolapse and mitral regurgitation are common in patients with polycystic kidney disease type 1. Am J Kidney Dis 2001; 38:1208–1216.
- Timio M, Monarca C, Pede S, Gentili S, Verdura C, Lolli S. The spectrum of cardiovascular abnormalities in autosomal dominant polycystic kidney disease: a 10-year follow-up in a five-generation kindred. Clin Nephrol 1992; 37:245–251.
- Hossack KF, Leddy CL, Johnson AM, Schrier RW, Gabow PA. Echocardiographic findings in autosomal dominant polycystic kidney disease. N Engl J Med 1988; 319:907–912.
- Leier CV, Baker PB, Kilman JW, Wooley CF. Cardiovascular abnormalities associated with adult polycystic kidney disease. Ann Intern Med 1984; 100:683–688.
- Turkmen K, Oflaz H, Uslu B, et al. Coronary flow velocity reserve and carotid intima media thickness in patients with autosomal dominant polycystic kidney disease: from impaired tubules to impaired carotid and coronary arteries. Clin J Am Soc Nephrol 2008; 3:986–991.
- Turkmen K, Tufan F, Alpay N, et al. Insulin resistance and coronary flow velocity reserve in patients with autosomal dominant polycystic kidney disease. Intern Med J 2012; 42:146–153.
- Turkmen K, Tufan F, Selcuk E, Akpinar T, Oflaz H, Ecder T. Neutrophil-to-lymphocyte ratio, insulin resistance, and endothelial dysfunction in patients with autosomal dominant polycystic kidney disease. Indian J Nephrol 2013; 23:34–40.
- Hadimeri H, Lamm C, Nyberg G. Coronary aneurysms in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1998; 9:837–841.
- Earle K, Hoffbrand BI. Adult dominant polycystic kidney disease and atrial myxoma. Nephron 1989; 52:197.
- Chung BM, Chong S, Lee W-S, Hwang S-N. Autosomal dominant polycystic kidney disease combined with intracranial aneurysm and dilated cardiomyopathy: a case report. J Korean Soc Radiol 2014; 71:84–88.
- Mariathasan DAL, Kumanan T. Adult polycystic kidney disease and idiopathic dilated cardiomyopathy: a rare genetic association. J Ceylon Coll Physicians 2016: 46:42–44.
- Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2012; 367:2407–2018.
- Higashihara E, Torres VE, Chapman AB, et al. Tolvaptan in autosomal dominant polycystic kidney disease: three years’ experience. Clin J Am Soc Nephrol 2011; 6:2499–2507.
- Sans Atxer L, Roca-Cusachs A, Torra R, et al. Relationship between renal size and blood pressure profile in patients with autosomal dominant polycystic kidney disease without renal failure. Nefrologia 2010; 30:567–572. In Spanish.
- Raina R, Lou L, Berger B, et al. Relationship of urinary endothelin-1 with estimated glomerular filtration rate in autosomal dominant polycystic kidney disease: a pilot cross-sectional analysis. BMC Nephrol 2016; 17:22.
Autosomal dominant polycystic kidney disease (ADPKD) has significant extrarenal manifestations. Hypertension is a common complication, arises early in the course of the disease, and is implicated in the development of left ventricular hypertrophy. Patients with ADPKD are also at risk of other cardiovascular complications (Table 1).
This article reviews the timely diagnosis of these common ADPKD complications and how to manage them.
ADPKD ACCOUNTS FOR 10% OF END-STAGE RENAL DISEASE
ADPKD is a genetic condition characterized by multiple renal cysts.1 Progressive enlargement of these cysts leads to a gradual decline in kidney function and eventually end-stage renal disease by the fifth or sixth decade of life.2 Worldwide, about 12.5 million people have ADPKD, and it accounts for about 10% of cases of end-stage renal disease.1,3,4
ADPKD has a variety of clinical presentations, including (in decreasing order of frequency) hypertension, flank pain, abdominal masses, urinary tract infection, renal failure, renal stones, and cerebrovascular accidents.2
Extrarenal complications are common and include hepatic cysts, hypertension, left ventricular hypertrophy, valvular heart disease, intracranial and extracranial aneurysms, pancreatic cysts, and diverticulosis.1–5
Less-common complications are dissection of the aorta and the internal carotid, vertebral, and iliac arteries6–10; aneurysm of the coronary, popliteal, and splenic arteries11–14; atrial myxoma15; cardiomyopathy16; pericardial effusion17; intracranial arterial dolichoectasia18; arachnoid cysts2; and intraoperative inferior vena cava syndrome (normally in ADPKD patients, pressure on the inferior vena cava results in compensatory sympathetic overactivity to maintain blood pressure), which occurs due to reduced sympathetic output under the influence of epidural or general anesthesia.19
Cardiovascular complications, especially cardiac hypertrophy and coronary artery disease, are now the leading cause of death in patients with ADPKD, as renal replacement therapy has improved and made death from end-stage renal disease less common.20,21
HYPERTENSION IN ADPKD
Hypertension is the most frequent initial presentation of ADPKD, occurring in 50% to 75% of cases and usually preceding the onset of renal failure.2,22 Hypertension is more common in male ADPKD patients, begins early in the course of the disease, and is diagnosed around the fourth decade of life.21
In a study in 2007, de Almeida et al23 used 24-hour ambulatory blood pressure monitoring early in the course of ADPKD and found significantly higher systolic, diastolic, and mean 24-hour blood pressures in ADPKD patients who had normal in-office blood pressure than in normotensive controls. In addition, nighttime systolic, nighttime diastolic, and nighttime mean blood pressures were significantly higher in the ADPKD group.
Hypertension is strongly associated with an accelerated decline in renal function to end-stage renal disease, development of left ventricular hypertrophy, and cardiovascular death.20,24
Although a prospective study25 showed a strong association between renal stones and hypertension in ADPKD, the relation between them is not clear. The incidence of renal stones is higher in hypertensive than in normotensive ADPKD patients, although evidence has to be established whether nephrolithiasis is a risk factor for hypertension or the other way around.25
Hypertension in ADPKD is multifactorial (Figure 1). The major factors associated with its development are increased activation of the renin-angiotensin-aldosterone system (RAAS); overexpression of endothelin receptor subtype A (ET-A) in cystic kidneys; increased production of endothelin 1 (ET-1); and sodium retention.26–31
The renin-angiotensin-aldosterone system
Activation of the RAAS plays a major role in the development and maintenance of hypertension in ADPKD. This is thought to be mainly due to progressive enlargement of renal cysts, which causes renal arteriolar attenuation and ischemia secondary to pressure effects, which in turn activates the RAAS.26,30,32–34 Two studies in patients with normal renal function found that cyst growth and increasing kidney volume have a strong relationship with the development of hypertension and declining kidney function.35,36
Ectopic secretion of RAAS components in polycystic kidneys has also been implicated in the development of hypertension, whereby renin, angiotensinogen, angiotensin-converting enzyme (ACE), angiotensin II, and angiotensin II receptors are produced in the epithelium of cysts and dilated tubules in polycystic kidneys.37–39 Proximal renal cysts and tubules produce ectopic angiotensinogen, which is converted to angiotensin I by renin in distal renal cysts. Angiotensin I is converted to angiotensin II by ACE in distal tubules, which in turn stimulates angiotensin II receptors, causing sodium and water retention in distal tubules.37 This may be responsible for hypertension in the initial stages; however, RAAS hyperactivity due to renal injury may predominate during later stages.37
Increased RAAS activity also increases sympathetic output, which in turn raises catecholamine levels and blood pressure.34 A study showed higher levels of plasma catecholamines in ADPKD hypertensive patients irrespective of renal function than in patients with essential hypertension.40
ET-A receptor and ET-1
A few studies have shown that in ADPKD patients, increased density of ET-A receptors and overproduction of ET-1, a potent vasoconstrictor, play a significant role in the development of hypertension and gradual loss of kidney function due to cyst enlargement and interstitial scarring.28,29 Ong et al29 found that expression of ET-A receptors is increased in smooth muscle cells of renal arteries, glomerular mesangial cells, and cyst epithelia in ADPKD.
Sodium retention
Studies in ADPKD patients with preserved renal function have linked high blood pressure to sodium retention and volume expansion.30,31,41 However, this phenomenon reverses when there is significant renal impairment in ADPKD.
As evidence of this, a study demonstrated significantly more natriuresis in patients with renal failure due to ADPKD than in patients with a similar degree of renal failure due to chronic glomerulonephritis.31 Moreover, another study found that the prevalence of hypertension is higher in ADPKD patients than in those with other nephropathies with preserved renal function, but this association reverses with significant decline in kidney function.22
MANAGING HYPERTENSION IN ADPKD
Early diagnosis of hypertension and effective control of it, even before ADPKD is diagnosed, is crucial to reduce cardiovascular mortality. Aggressive blood pressure control in the prehypertensive phase of ADPKD will also help reduce the incidence of left ventricular hypertrophy and mitral regurgitation and slow the progression of renal failure (Figure 2).
A meta-analysis42 revealed hypertension to be present in 20% of ADPKD patients younger than 21, and many of them were undiagnosed. This study also suggests that patients at risk of hypertension (ie, all patients with ADPKD) should be routinely screened for it.
Ambulatory blood pressure monitoring may play an important role in diagnosing hypertension early in the prehypertensive stage of ADPKD.23
Target blood pressures: No consensus
Two well-powered double-blind, placebo-controlled trials, known as HALT-PKD Study A and HALT-PKD Study B, tested the effects of 2 different blood pressure targets and of monotherapy with an ACE inhibitor vs combination therapy with an ACE inhibitor plus an angiotensin II receptor blocker (ARB) on renal function, total kidney volume, left ventricular mass index, and urinary albumin excretion in the early (estimated glomerular filtration rate [eGFR] > 60 mL/min) and late (eGFR 25–60 mL/min) stages of ADPKD, respectively.43,44
HALT-PKD Study A43 found that, in the early stages of ADPKD with preserved renal function, meticulous control of blood pressure (95–110/60–75 mm Hg) was strongly correlated with significant reductions in left ventricular mass index, albuminuria, and rate of total kidney volume growth without remarkable alteration in renal function compared with standard blood pressure control (120–130/70–80 mm Hg). However, no notable differences were observed between the ACE inhibitor and ACE inhibitor-plus-ARB groups.43
Despite the evidence, universal consensus guidelines are lacking, and the available guidelines on hypertension management have different blood pressure goals in patients with chronic kidney disease.
The eighth Joint National Committee guideline of 2014 recommends a blood pressure goal of less than 140/90 mm Hg in patients with diabetic and nondiabetic chronic kidney disease.45
The National Institute for Health and Care Excellence 2011 guideline recommends a blood pressure goal of less than 130/80 mm Hg in chronic kidney disease patients.46
The European Society of Hypertension and European Society of Cardiology joint 2013 guideline recommends a systolic blood pressure goal of less than 140 mm Hg in diabetic and nondiabetic patients with chronic kidney disease.47
The 2016 Kidney Health Australia-Caring for Australians With Renal Impairment guideline for diagnosis and management of ADPKD48 recommends a lower blood pressure goal of 96–110/60–75 mm Hg in patients with an eGFR greater than 60 mL/min/1.73 m2 who can tolerate it without side effects, which is based on the findings of HALT-PKD Study A.43
Helal et al recommend that blood pressure be controlled to less than 130/80 mm Hg, until there is more evidence for a safe and effective target blood pressure goal in ADPKD patients.49
We recommend a target blood pressure less than 110/75 mm Hg in hypertensive ADPKD patients with preserved renal function who can tolerate this level, and less than 130/80 mm Hg in ADPKD patients with stage 3 chronic kidney disease. These targets can be achieved with ACE inhibitor or ARB monotherapy.43,44 However, no studies have established the safest lower limit of target blood pressure in ADPKD.
ACE inhibitors, ARBs are mainstays
Mainstays of antihypertensive drug therapy in ADPKD are ACE inhibitors and ARBs.
HALT-PKD Study B44 demonstrated that, in the late stages of ADPKD, target blood pressure control (110–130/70–80 mm Hg) can be attained with ACE inhibitor monotherapy or with an ACE inhibitor plus an ARB, but the latter produced no additive benefit.
Patch et al,50 in a retrospective cohort study, showed that broadening the spectrum of antihypertensive therapy decreases mortality in ADPKD patients. Evaluating ADPKD patients from the UK General Practice Research Database between 1991 and 2008, they found a trend toward lower mortality rates as the number of antihypertensive drugs prescribed within 1 year increased. They also observed that the prescription of RAAS-blocking agents increased from 7% in 1991 to 46% in 2008.50
However, a 3-year prospective randomized double-blind study compared the effects of the ACE inhibitor ramipril and the beta-blocker metoprolol in hypertensive ADPKD patients.51 The results showed that effective blood pressure control could be achieved in both groups with no significant differences in left ventricular mass index, albuminuria, or kidney function.51
Treatment strategies
Lifestyle modification is the initial approach to the management of hypertension before starting drug therapy. Lifestyle changes include dietary salt restriction to less than 6 g/day, weight reduction, regular exercise, increased fluid intake (up to 3 L/day or to satisfy thirst), smoking cessation, and avoidance of caffeine.47–49
ACE inhibitors are first-line drugs in hypertensive ADPKD patients.
ARBs can also be considered, but there is no role for dual ACE inhibitor and ARB therapy.43,48 A study found ACE inhibitors to be more cost-effective and to decrease mortality rates to a greater extent than ARBs.52
Beta-blockers or calcium channel blockers should be considered instead if ACE inhibitors and ARBs are contraindicated, or as add-on drugs if ACE inhibitors and ARBs fail to reduce blood pressure adequately.48,49
Diuretics are third-line agents. Thiazides are preferred in ADPKD patients with normal renal function and loop diuretics in those with impaired renal function.49
LEFT VENTRICULAR HYPERTROPHY IN ADPKD
Increased left ventricular mass is an indirect indicator of untreated hypertension, and it often goes unnoticed in patients with undiagnosed ADPKD. Left ventricular hypertrophy is associated with arrhythmias and heart failure, which contribute significantly to cardiovascular mortality and adverse renal outcomes.20,24
A 5-year randomized clinical trial by Cadnapaphornchai et al36 in ADPKD patients between 4 and 21 years of age showed strong correlations between hypertension, left ventricular mass index, and kidney volume and a negative correlation between left ventricular mass index and renal function.
Several factors are thought to contribute to left ventricular hypertrophy in ADPKD (Figure 1).
Hypertension. Two studies of 24-hour ambulatory blood pressure monitoring showed that nocturnal blood pressures decreased less in normotensive and hypertensive ADPKD patients than in normotensive and hypertensive controls.23,53 This persistent elevation of nocturnal blood pressure may contribute to the development and progression of left ventricular hypertrophy.
On the other hand, Valero et al54 reported that the left ventricular mass index was strongly associated with ambulatory systolic blood pressure rather than elevated nocturnal blood pressure in ADPKD patients compared with healthy controls.
FGF23. High levels of fibroblast growth factor 23 (FGF23) have been shown to be strongly associated with left ventricular hypertrophy in ADPKD. Experimental studies have shown that FGF23 is directly involved in the pathogenesis of left ventricular hypertrophy through stimulation of the calcineurin-nuclear factor of activated T cells pathway.
Faul et al55 induced cardiac hypertrophy in mice that were deficient in klotho (a transmembrane protein that increases FGF23 affinity for FGF receptors) by injecting FGF23 intravenously.
Yildiz et al56 observed higher levels of FGF23 in hypertensive and normotensive ADPKD patients with normal renal function than in healthy controls. They also found a lower elasticity index in the large and small arteries in normotensive and hypertensive ADPKD patients, which accounts for vascular dysfunction. High FGF23 levels may be responsible for the left ventricular hypertrophy seen in normotensive ADPKD patients with preserved renal function.
Polymorphisms in the ACE gene have been implicated in the development of cardiac hypertrophy in ADPKD.
Wanic-Kossowska et al57 studied the association between ACE gene polymorphisms and cardiovascular complications in ADPKD patients. They found a higher prevalence of the homozygous DD genotype among ADPKD patients with end-stage renal disease than in those in the early stages of chronic kidney disease in ADPKD. Also, the DD genotype has been shown to be more strongly associated with left ventricular hypertrophy and left ventricular dysfunction than other (II or ID) genotypes. These findings suggest that the DD genotype carries higher risk for the development of end-stage renal disease, left ventricular hypertrophy, and other cardiovascular complications.
MANAGING LEFT VENTRICULAR HYPERTROPHY IN ADPKD
Preventing and halting progression of left ventricular hypertrophy primarily involves effective blood pressure control, especially in the early stages of ADPKD (Figure 2).
A 7-year prospective randomized trial in ADPKD patients with established hypertension and left ventricular hypertrophy proved that aggressive (< 120/80 mm Hg) compared with standard blood pressure control (135–140/85–90 mm Hg) significantly reduces left ventricular mass index. ACE inhibitors were preferred over calcium channel blockers.58
HALT-PKD Study A showed that a significant decrease in left ventricular mass index can be achieved by aggressive blood pressure control (95–110/60–75 mm Hg) with an ACE inhibitor alone or in combination with an ARB in the early stages of ADPKD with preserved renal function.43
A 5-year randomized clinical trial in children with borderline hypertension treated with an ACE inhibitor for effective control of blood pressure showed no change in left ventricular mass index or renal function.36,59
These results support starting ACE inhibitor therapy early in the disease process when blood pressure is still normal or borderline to prevent the progression of left ventricular hypertrophy or worsening kidney function.
Since FGF23 is directly involved in the causation of left ventricular hypertrophy, FGF receptors may be potential therapeutic targets to prevent left ventricular hypertrophy in ADPKD. An FGF receptor blocker was shown to decrease left ventricular hypertrophy in rats with chronic kidney disease without affecting blood pressure.55
INTRACRANIAL ANEURYSM IN ADPKD
Intracranial aneurysm is the most dangerous complication of ADPKD. When an aneurysm ruptures, the mortality rate is 4% to 7%, and 50% of survivors are left with residual neurologic deficits.5,60,61
In various studies, the prevalence of intracranial aneurysm in ADPKD ranged from 4% to 41.2%, compared with 1% in the general population.5,62,63 On follow-up ranging from 18 months to about 10 years, the incidence of new intracranial aneurysm was 2.6% to 13.3% in patients with previously normal findings on magnetic resonance angiography and 25% in patients with a history of intracranial aneurysm.62,64,65
The most common sites are the middle cerebral artery (45%), internal carotid artery (40.5%), and anterior communicating artery (35.1%).66 (The numbers add up to more than 100% because some patients have aneurysms in more than 1 site.) The mean size of a ruptured aneurysm was 6 mm per a recent systematic review.66 Intracranial aneurysms 6 mm or larger are at highest risk of rupture.66
SCREENING FOR INTRACRANIAL ANEURYSM
Timely screening and intervention for intracranial aneurysm is crucial to prevent death from intracranial hemorrhage.
Currently, there are no standard guidelines for screening and follow-up of intracranial aneurysm in ADPKD patients. However, some recommendations are available from the ADPKD Kidney Disease Improving Global Outcomes Controversies Conference3 and Kidney Health Australia—Caring for Australasians With Renal Impairment ADPKD guidelines67 (Figure 3).
Imaging tests
Magnetic resonance angiography (MRA) with gadolinium enhancement and computed tomographic angiography (CTA) are recommended for screening in ADPKD patients with normal renal function,67 but time-of-flight MRA without gadolinium is the imaging test of choice because it is noninvasive and poses no risk of nephrotoxicity or contrast allergy.3,68 Further, gadolinium should be avoided in patients whose eGFR is 30 mL/min/1.73 m2 or less because of risk of nephrogenic systemic sclerosis and fibrosis.67,68
The sensitivity of time-of-flight MRA screening for intracranial aneurysm varies depending on the size of aneurysm; 67% for those less than 3 mm, 79% for those 3 to 5 mm, and 95% for those larger than 5 mm.69 The sensitivity of CTA screening is 95% for aneurysms larger than 7 mm and 53% for those measuring 2 mm.70,71 The specificity of CTA screening was reported to be 98.9% overall.71
When to screen
Screening for intracranial aneurysm is recommended at the time of ADPKD diagnosis for all high-risk patients, ie, those who have a family history of intracranial hemorrhage or aneurysm in an affected first-degree relative.67 It is also recommended for ADPKD patients with a history of sudden-onset severe headache or neurologic symptoms.67 A third group for whom screening is recommended is ADPKD patients who have no family history of intracranial aneurysm or hemorrhage but who are at risk of poor outcome if an intracranial aneurysm ruptures (eg, those undergoing major elective surgery, with uncontrolled blood pressure, on anticoagulation, with a history of or current smoking, and airline pilots).67
Patients found to have an intracranial aneurysm on screening should be referred to a neurosurgeon and should undergo repeat MRA or CTA imaging every 6 to 24 months.3 High-risk ADPKD patients with normal findings on initial screening should have repeat MRA or CTA screening in 5 to 10 years unless they suffer from sudden-onset severe headache or neurologic symptoms.65,67
Both smoking and high blood pressure increase the risk of formation and growth of intracranial aneurysm. Hence, meticulous control of blood pressure and smoking cessation are recommended in ADPKD patients.3,67
CARDIAC VALVULAR ABNORMALITIES IN ADPKD
Of the valvular abnormalities that complicate ADPKD, the more common ones are mitral valve prolapse and mitral and aortic regurgitation. The less common ones are tricuspid valve prolapse and tricuspid regurgitation.72–74
The pathophysiology underlying these valvular abnormalities is unclear. However, defective collagen synthesis and myxomatous degeneration have been demonstrated in histopathologic examination of affected valvular tissue.75 Also, ACE gene polymorphism, especially the DD genotype, has been shown to be associated with cardiac valvular calcifications and valvular insufficiency.57
Lumiaho et al72 found a higher prevalence of mitral valve prolapse, mitral regurgitation, and left ventricular hypertrophy in patients with ADPKD type 1 (due to abnormalities in PDK1) than in unaffected family members and healthy controls. The investigators speculated that mitral regurgitation is caused by the high blood pressure observed in ADPKD type 1 patients, since hypertension causes left ventricular hypertrophy and left ventricular dilatation. The severity of renal failure was related to mitral regurgitation but not mitral valve prolapse.
Similarly, Gabow et al24 showed that there is no significant relationship between mitral valve prolapse and progression of renal disease in ADPKD.
Interestingly, Fick et al20 found that mitral valve prolapse has no significant effect on cardiovascular mortality.
CORONARY ARTERY DISEASE AND ANEURYSM IN ADPKD
Atherosclerosis sets in early in ADPKD, resulting in coronary artery disease and adverse cardiovascular outcomes.
Coronary flow velocity reserve is the ability of coronary arteries to dilate in response to myocardial oxygen demand. Atherosclerosis decreases this reserve in ADPKD patients, as shown in several studies.
Turkmen et al, in a series of studies,76–78 found that ADPKD patients had significantly less coronary flow velocity reserve, thicker carotid intima media (a surrogate marker of atherosclerosis), and greater insulin resistance than healthy controls. These findings imply that atherosclerosis begins very early in the course of ADPKD and has remarkable effects on cardiovascular morbidity and mortality.76
Aneurysm. Although the risk of extracranial aneurysm is higher with ADPKD, coronary artery aneurysm is uncommon. The pathogenesis of coronary aneurysm has been linked to abnormal expression of the proteins polycystin 1 and polycystin 2 in vascular smooth muscle.11,79 The PKD1 and PKD2 genes encode polycystin 1 and 2, respectively, in ADPKD. These polycystins are also expressed in the liver, kidneys, and myocardium and are involved in the regulation of intracellular calcium, stretch-activated ion channels, and vascular smooth muscle cell proliferation and apoptosis.11,16 Abnormally expressed polycystin in ADPKD therefore has an impact on arterial wall integrity, resulting in focal medial defects in the vasculature that eventually develop into micro- and macroaneurysms.11
Hadimeri et al79 found a higher prevalence of coronary aneurysm and ectasia in ADPKD patients than in controls. Most coronary aneurysms are smaller than 1 cm; however, a coronary aneurysm measuring 4 cm in diameter was found at autopsy of an ADPKD patient.11
Spontaneous coronary artery dissection is very rare in the general population, but Bobrie et al reported a case of it in an ADPKD patient.9
ATRIAL MYXOMA, CARDIOMYOPATHY, AND PERICARDIAL EFFUSION IN ADPKD
Atrial myxoma in ADPKD patients has been described in 2 case reports.15,80 However, the association of atrial myxoma with ADPKD is poorly understood and may be a coincidental finding.
Cardiomyopathy in ADPKD has been linked to abnormalities in the intracellular calcium pathway, although a clear picture of its involvement has yet to be established.
Paavola et al16 described the pathophysiology of ADPKD-associated cardiomyopathy in PKD2 mutant zebrafish lacking polycystin 2. These mutants showed decreased cardiac output and had atrioventricular blocks. The findings were attributed to abnormal intracellular calcium cycling. These findings correlated well with the frequent finding of idiopathic dilated cardiomyopathy in ADPKD patients, especially with PKD2 mutations.16 Also, 2 cases of dilated cardiomyopathy in ADPKD have been reported and thought to be related to PKD2 mutations.81,82
Pericardial effusion. Even though the exact pathophysiology of pericardial effusion in ADPKD is unknown, it has been theorized to be related to defects in connective tissue and extracellular matrix due to PKD1 and PKD2 mutations. These abnormalities increase the compliance and impair the recoil capacity of connective tissue, which results in unusual distention of the parietal pericardium. This abnormal distention of the parietal pericardium together with increased extracellular volume may lead to pericardial effusion.17
Qian et al17 found a higher prevalence of pericardial effusion in ADPKD patients. It was generally asymptomatic, and the cause was attributed to these connective tissue and extracellular matrix abnormalities.
EMERGING THERAPIES AND TESTS
Recent trials have investigated the effects of vasopressin receptor antagonists, specifically V2 receptor blockers in ADPKD and its complications.
Tolvaptan has been shown to slow the rate of increase in cyst size and total kidney volume.83,84 Also, a correlation between kidney size and diastolic blood pressure has been observed in ADPKD patients.85 Reducing cyst volume may reduce pressure effects and decrease renal ischemia, which in turn may reduce RAAS activation; however, the evidence to support this hypothesis is poor. A major clinical trial of tolvaptan in ADPKD patients showed no effect on blood pressure control, but the drug slowed the rate of increase in total kidney volume and fall in eGFR.83
Endothelin receptor antagonists are also in the preliminary stage of development for use in ADPKD. The effects of acute blockade of the renal endothelial system with bosentan were investigated in animal models by Hocher et al.28 This study showed a greater reduction in mean arterial pressure after bosentan administration, resulting in significantly decreased GFR and renal blood flow. Nonetheless, the mean arterial pressure-lowering effect of bosentan was more marked than the reductions in GFR and renal blood flow.
Raina et al,86 in a pilot cross-sectional analysis, showed that urinary excretion of ET-1 is increased in ADPKD patients, and may serve as a surrogate marker for ET-1 in renal tissue and a noninvasive marker of early kidney injury.
Autosomal dominant polycystic kidney disease (ADPKD) has significant extrarenal manifestations. Hypertension is a common complication, arises early in the course of the disease, and is implicated in the development of left ventricular hypertrophy. Patients with ADPKD are also at risk of other cardiovascular complications (Table 1).
This article reviews the timely diagnosis of these common ADPKD complications and how to manage them.
ADPKD ACCOUNTS FOR 10% OF END-STAGE RENAL DISEASE
ADPKD is a genetic condition characterized by multiple renal cysts.1 Progressive enlargement of these cysts leads to a gradual decline in kidney function and eventually end-stage renal disease by the fifth or sixth decade of life.2 Worldwide, about 12.5 million people have ADPKD, and it accounts for about 10% of cases of end-stage renal disease.1,3,4
ADPKD has a variety of clinical presentations, including (in decreasing order of frequency) hypertension, flank pain, abdominal masses, urinary tract infection, renal failure, renal stones, and cerebrovascular accidents.2
Extrarenal complications are common and include hepatic cysts, hypertension, left ventricular hypertrophy, valvular heart disease, intracranial and extracranial aneurysms, pancreatic cysts, and diverticulosis.1–5
Less-common complications are dissection of the aorta and the internal carotid, vertebral, and iliac arteries6–10; aneurysm of the coronary, popliteal, and splenic arteries11–14; atrial myxoma15; cardiomyopathy16; pericardial effusion17; intracranial arterial dolichoectasia18; arachnoid cysts2; and intraoperative inferior vena cava syndrome (normally in ADPKD patients, pressure on the inferior vena cava results in compensatory sympathetic overactivity to maintain blood pressure), which occurs due to reduced sympathetic output under the influence of epidural or general anesthesia.19
Cardiovascular complications, especially cardiac hypertrophy and coronary artery disease, are now the leading cause of death in patients with ADPKD, as renal replacement therapy has improved and made death from end-stage renal disease less common.20,21
HYPERTENSION IN ADPKD
Hypertension is the most frequent initial presentation of ADPKD, occurring in 50% to 75% of cases and usually preceding the onset of renal failure.2,22 Hypertension is more common in male ADPKD patients, begins early in the course of the disease, and is diagnosed around the fourth decade of life.21
In a study in 2007, de Almeida et al23 used 24-hour ambulatory blood pressure monitoring early in the course of ADPKD and found significantly higher systolic, diastolic, and mean 24-hour blood pressures in ADPKD patients who had normal in-office blood pressure than in normotensive controls. In addition, nighttime systolic, nighttime diastolic, and nighttime mean blood pressures were significantly higher in the ADPKD group.
Hypertension is strongly associated with an accelerated decline in renal function to end-stage renal disease, development of left ventricular hypertrophy, and cardiovascular death.20,24
Although a prospective study25 showed a strong association between renal stones and hypertension in ADPKD, the relation between them is not clear. The incidence of renal stones is higher in hypertensive than in normotensive ADPKD patients, although evidence has to be established whether nephrolithiasis is a risk factor for hypertension or the other way around.25
Hypertension in ADPKD is multifactorial (Figure 1). The major factors associated with its development are increased activation of the renin-angiotensin-aldosterone system (RAAS); overexpression of endothelin receptor subtype A (ET-A) in cystic kidneys; increased production of endothelin 1 (ET-1); and sodium retention.26–31
The renin-angiotensin-aldosterone system
Activation of the RAAS plays a major role in the development and maintenance of hypertension in ADPKD. This is thought to be mainly due to progressive enlargement of renal cysts, which causes renal arteriolar attenuation and ischemia secondary to pressure effects, which in turn activates the RAAS.26,30,32–34 Two studies in patients with normal renal function found that cyst growth and increasing kidney volume have a strong relationship with the development of hypertension and declining kidney function.35,36
Ectopic secretion of RAAS components in polycystic kidneys has also been implicated in the development of hypertension, whereby renin, angiotensinogen, angiotensin-converting enzyme (ACE), angiotensin II, and angiotensin II receptors are produced in the epithelium of cysts and dilated tubules in polycystic kidneys.37–39 Proximal renal cysts and tubules produce ectopic angiotensinogen, which is converted to angiotensin I by renin in distal renal cysts. Angiotensin I is converted to angiotensin II by ACE in distal tubules, which in turn stimulates angiotensin II receptors, causing sodium and water retention in distal tubules.37 This may be responsible for hypertension in the initial stages; however, RAAS hyperactivity due to renal injury may predominate during later stages.37
Increased RAAS activity also increases sympathetic output, which in turn raises catecholamine levels and blood pressure.34 A study showed higher levels of plasma catecholamines in ADPKD hypertensive patients irrespective of renal function than in patients with essential hypertension.40
ET-A receptor and ET-1
A few studies have shown that in ADPKD patients, increased density of ET-A receptors and overproduction of ET-1, a potent vasoconstrictor, play a significant role in the development of hypertension and gradual loss of kidney function due to cyst enlargement and interstitial scarring.28,29 Ong et al29 found that expression of ET-A receptors is increased in smooth muscle cells of renal arteries, glomerular mesangial cells, and cyst epithelia in ADPKD.
Sodium retention
Studies in ADPKD patients with preserved renal function have linked high blood pressure to sodium retention and volume expansion.30,31,41 However, this phenomenon reverses when there is significant renal impairment in ADPKD.
As evidence of this, a study demonstrated significantly more natriuresis in patients with renal failure due to ADPKD than in patients with a similar degree of renal failure due to chronic glomerulonephritis.31 Moreover, another study found that the prevalence of hypertension is higher in ADPKD patients than in those with other nephropathies with preserved renal function, but this association reverses with significant decline in kidney function.22
MANAGING HYPERTENSION IN ADPKD
Early diagnosis of hypertension and effective control of it, even before ADPKD is diagnosed, is crucial to reduce cardiovascular mortality. Aggressive blood pressure control in the prehypertensive phase of ADPKD will also help reduce the incidence of left ventricular hypertrophy and mitral regurgitation and slow the progression of renal failure (Figure 2).
A meta-analysis42 revealed hypertension to be present in 20% of ADPKD patients younger than 21, and many of them were undiagnosed. This study also suggests that patients at risk of hypertension (ie, all patients with ADPKD) should be routinely screened for it.
Ambulatory blood pressure monitoring may play an important role in diagnosing hypertension early in the prehypertensive stage of ADPKD.23
Target blood pressures: No consensus
Two well-powered double-blind, placebo-controlled trials, known as HALT-PKD Study A and HALT-PKD Study B, tested the effects of 2 different blood pressure targets and of monotherapy with an ACE inhibitor vs combination therapy with an ACE inhibitor plus an angiotensin II receptor blocker (ARB) on renal function, total kidney volume, left ventricular mass index, and urinary albumin excretion in the early (estimated glomerular filtration rate [eGFR] > 60 mL/min) and late (eGFR 25–60 mL/min) stages of ADPKD, respectively.43,44
HALT-PKD Study A43 found that, in the early stages of ADPKD with preserved renal function, meticulous control of blood pressure (95–110/60–75 mm Hg) was strongly correlated with significant reductions in left ventricular mass index, albuminuria, and rate of total kidney volume growth without remarkable alteration in renal function compared with standard blood pressure control (120–130/70–80 mm Hg). However, no notable differences were observed between the ACE inhibitor and ACE inhibitor-plus-ARB groups.43
Despite the evidence, universal consensus guidelines are lacking, and the available guidelines on hypertension management have different blood pressure goals in patients with chronic kidney disease.
The eighth Joint National Committee guideline of 2014 recommends a blood pressure goal of less than 140/90 mm Hg in patients with diabetic and nondiabetic chronic kidney disease.45
The National Institute for Health and Care Excellence 2011 guideline recommends a blood pressure goal of less than 130/80 mm Hg in chronic kidney disease patients.46
The European Society of Hypertension and European Society of Cardiology joint 2013 guideline recommends a systolic blood pressure goal of less than 140 mm Hg in diabetic and nondiabetic patients with chronic kidney disease.47
The 2016 Kidney Health Australia-Caring for Australians With Renal Impairment guideline for diagnosis and management of ADPKD48 recommends a lower blood pressure goal of 96–110/60–75 mm Hg in patients with an eGFR greater than 60 mL/min/1.73 m2 who can tolerate it without side effects, which is based on the findings of HALT-PKD Study A.43
Helal et al recommend that blood pressure be controlled to less than 130/80 mm Hg, until there is more evidence for a safe and effective target blood pressure goal in ADPKD patients.49
We recommend a target blood pressure less than 110/75 mm Hg in hypertensive ADPKD patients with preserved renal function who can tolerate this level, and less than 130/80 mm Hg in ADPKD patients with stage 3 chronic kidney disease. These targets can be achieved with ACE inhibitor or ARB monotherapy.43,44 However, no studies have established the safest lower limit of target blood pressure in ADPKD.
ACE inhibitors, ARBs are mainstays
Mainstays of antihypertensive drug therapy in ADPKD are ACE inhibitors and ARBs.
HALT-PKD Study B44 demonstrated that, in the late stages of ADPKD, target blood pressure control (110–130/70–80 mm Hg) can be attained with ACE inhibitor monotherapy or with an ACE inhibitor plus an ARB, but the latter produced no additive benefit.
Patch et al,50 in a retrospective cohort study, showed that broadening the spectrum of antihypertensive therapy decreases mortality in ADPKD patients. Evaluating ADPKD patients from the UK General Practice Research Database between 1991 and 2008, they found a trend toward lower mortality rates as the number of antihypertensive drugs prescribed within 1 year increased. They also observed that the prescription of RAAS-blocking agents increased from 7% in 1991 to 46% in 2008.50
However, a 3-year prospective randomized double-blind study compared the effects of the ACE inhibitor ramipril and the beta-blocker metoprolol in hypertensive ADPKD patients.51 The results showed that effective blood pressure control could be achieved in both groups with no significant differences in left ventricular mass index, albuminuria, or kidney function.51
Treatment strategies
Lifestyle modification is the initial approach to the management of hypertension before starting drug therapy. Lifestyle changes include dietary salt restriction to less than 6 g/day, weight reduction, regular exercise, increased fluid intake (up to 3 L/day or to satisfy thirst), smoking cessation, and avoidance of caffeine.47–49
ACE inhibitors are first-line drugs in hypertensive ADPKD patients.
ARBs can also be considered, but there is no role for dual ACE inhibitor and ARB therapy.43,48 A study found ACE inhibitors to be more cost-effective and to decrease mortality rates to a greater extent than ARBs.52
Beta-blockers or calcium channel blockers should be considered instead if ACE inhibitors and ARBs are contraindicated, or as add-on drugs if ACE inhibitors and ARBs fail to reduce blood pressure adequately.48,49
Diuretics are third-line agents. Thiazides are preferred in ADPKD patients with normal renal function and loop diuretics in those with impaired renal function.49
LEFT VENTRICULAR HYPERTROPHY IN ADPKD
Increased left ventricular mass is an indirect indicator of untreated hypertension, and it often goes unnoticed in patients with undiagnosed ADPKD. Left ventricular hypertrophy is associated with arrhythmias and heart failure, which contribute significantly to cardiovascular mortality and adverse renal outcomes.20,24
A 5-year randomized clinical trial by Cadnapaphornchai et al36 in ADPKD patients between 4 and 21 years of age showed strong correlations between hypertension, left ventricular mass index, and kidney volume and a negative correlation between left ventricular mass index and renal function.
Several factors are thought to contribute to left ventricular hypertrophy in ADPKD (Figure 1).
Hypertension. Two studies of 24-hour ambulatory blood pressure monitoring showed that nocturnal blood pressures decreased less in normotensive and hypertensive ADPKD patients than in normotensive and hypertensive controls.23,53 This persistent elevation of nocturnal blood pressure may contribute to the development and progression of left ventricular hypertrophy.
On the other hand, Valero et al54 reported that the left ventricular mass index was strongly associated with ambulatory systolic blood pressure rather than elevated nocturnal blood pressure in ADPKD patients compared with healthy controls.
FGF23. High levels of fibroblast growth factor 23 (FGF23) have been shown to be strongly associated with left ventricular hypertrophy in ADPKD. Experimental studies have shown that FGF23 is directly involved in the pathogenesis of left ventricular hypertrophy through stimulation of the calcineurin-nuclear factor of activated T cells pathway.
Faul et al55 induced cardiac hypertrophy in mice that were deficient in klotho (a transmembrane protein that increases FGF23 affinity for FGF receptors) by injecting FGF23 intravenously.
Yildiz et al56 observed higher levels of FGF23 in hypertensive and normotensive ADPKD patients with normal renal function than in healthy controls. They also found a lower elasticity index in the large and small arteries in normotensive and hypertensive ADPKD patients, which accounts for vascular dysfunction. High FGF23 levels may be responsible for the left ventricular hypertrophy seen in normotensive ADPKD patients with preserved renal function.
Polymorphisms in the ACE gene have been implicated in the development of cardiac hypertrophy in ADPKD.
Wanic-Kossowska et al57 studied the association between ACE gene polymorphisms and cardiovascular complications in ADPKD patients. They found a higher prevalence of the homozygous DD genotype among ADPKD patients with end-stage renal disease than in those in the early stages of chronic kidney disease in ADPKD. Also, the DD genotype has been shown to be more strongly associated with left ventricular hypertrophy and left ventricular dysfunction than other (II or ID) genotypes. These findings suggest that the DD genotype carries higher risk for the development of end-stage renal disease, left ventricular hypertrophy, and other cardiovascular complications.
MANAGING LEFT VENTRICULAR HYPERTROPHY IN ADPKD
Preventing and halting progression of left ventricular hypertrophy primarily involves effective blood pressure control, especially in the early stages of ADPKD (Figure 2).
A 7-year prospective randomized trial in ADPKD patients with established hypertension and left ventricular hypertrophy proved that aggressive (< 120/80 mm Hg) compared with standard blood pressure control (135–140/85–90 mm Hg) significantly reduces left ventricular mass index. ACE inhibitors were preferred over calcium channel blockers.58
HALT-PKD Study A showed that a significant decrease in left ventricular mass index can be achieved by aggressive blood pressure control (95–110/60–75 mm Hg) with an ACE inhibitor alone or in combination with an ARB in the early stages of ADPKD with preserved renal function.43
A 5-year randomized clinical trial in children with borderline hypertension treated with an ACE inhibitor for effective control of blood pressure showed no change in left ventricular mass index or renal function.36,59
These results support starting ACE inhibitor therapy early in the disease process when blood pressure is still normal or borderline to prevent the progression of left ventricular hypertrophy or worsening kidney function.
Since FGF23 is directly involved in the causation of left ventricular hypertrophy, FGF receptors may be potential therapeutic targets to prevent left ventricular hypertrophy in ADPKD. An FGF receptor blocker was shown to decrease left ventricular hypertrophy in rats with chronic kidney disease without affecting blood pressure.55
INTRACRANIAL ANEURYSM IN ADPKD
Intracranial aneurysm is the most dangerous complication of ADPKD. When an aneurysm ruptures, the mortality rate is 4% to 7%, and 50% of survivors are left with residual neurologic deficits.5,60,61
In various studies, the prevalence of intracranial aneurysm in ADPKD ranged from 4% to 41.2%, compared with 1% in the general population.5,62,63 On follow-up ranging from 18 months to about 10 years, the incidence of new intracranial aneurysm was 2.6% to 13.3% in patients with previously normal findings on magnetic resonance angiography and 25% in patients with a history of intracranial aneurysm.62,64,65
The most common sites are the middle cerebral artery (45%), internal carotid artery (40.5%), and anterior communicating artery (35.1%).66 (The numbers add up to more than 100% because some patients have aneurysms in more than 1 site.) The mean size of a ruptured aneurysm was 6 mm per a recent systematic review.66 Intracranial aneurysms 6 mm or larger are at highest risk of rupture.66
SCREENING FOR INTRACRANIAL ANEURYSM
Timely screening and intervention for intracranial aneurysm is crucial to prevent death from intracranial hemorrhage.
Currently, there are no standard guidelines for screening and follow-up of intracranial aneurysm in ADPKD patients. However, some recommendations are available from the ADPKD Kidney Disease Improving Global Outcomes Controversies Conference3 and Kidney Health Australia—Caring for Australasians With Renal Impairment ADPKD guidelines67 (Figure 3).
Imaging tests
Magnetic resonance angiography (MRA) with gadolinium enhancement and computed tomographic angiography (CTA) are recommended for screening in ADPKD patients with normal renal function,67 but time-of-flight MRA without gadolinium is the imaging test of choice because it is noninvasive and poses no risk of nephrotoxicity or contrast allergy.3,68 Further, gadolinium should be avoided in patients whose eGFR is 30 mL/min/1.73 m2 or less because of risk of nephrogenic systemic sclerosis and fibrosis.67,68
The sensitivity of time-of-flight MRA screening for intracranial aneurysm varies depending on the size of aneurysm; 67% for those less than 3 mm, 79% for those 3 to 5 mm, and 95% for those larger than 5 mm.69 The sensitivity of CTA screening is 95% for aneurysms larger than 7 mm and 53% for those measuring 2 mm.70,71 The specificity of CTA screening was reported to be 98.9% overall.71
When to screen
Screening for intracranial aneurysm is recommended at the time of ADPKD diagnosis for all high-risk patients, ie, those who have a family history of intracranial hemorrhage or aneurysm in an affected first-degree relative.67 It is also recommended for ADPKD patients with a history of sudden-onset severe headache or neurologic symptoms.67 A third group for whom screening is recommended is ADPKD patients who have no family history of intracranial aneurysm or hemorrhage but who are at risk of poor outcome if an intracranial aneurysm ruptures (eg, those undergoing major elective surgery, with uncontrolled blood pressure, on anticoagulation, with a history of or current smoking, and airline pilots).67
Patients found to have an intracranial aneurysm on screening should be referred to a neurosurgeon and should undergo repeat MRA or CTA imaging every 6 to 24 months.3 High-risk ADPKD patients with normal findings on initial screening should have repeat MRA or CTA screening in 5 to 10 years unless they suffer from sudden-onset severe headache or neurologic symptoms.65,67
Both smoking and high blood pressure increase the risk of formation and growth of intracranial aneurysm. Hence, meticulous control of blood pressure and smoking cessation are recommended in ADPKD patients.3,67
CARDIAC VALVULAR ABNORMALITIES IN ADPKD
Of the valvular abnormalities that complicate ADPKD, the more common ones are mitral valve prolapse and mitral and aortic regurgitation. The less common ones are tricuspid valve prolapse and tricuspid regurgitation.72–74
The pathophysiology underlying these valvular abnormalities is unclear. However, defective collagen synthesis and myxomatous degeneration have been demonstrated in histopathologic examination of affected valvular tissue.75 Also, ACE gene polymorphism, especially the DD genotype, has been shown to be associated with cardiac valvular calcifications and valvular insufficiency.57
Lumiaho et al72 found a higher prevalence of mitral valve prolapse, mitral regurgitation, and left ventricular hypertrophy in patients with ADPKD type 1 (due to abnormalities in PDK1) than in unaffected family members and healthy controls. The investigators speculated that mitral regurgitation is caused by the high blood pressure observed in ADPKD type 1 patients, since hypertension causes left ventricular hypertrophy and left ventricular dilatation. The severity of renal failure was related to mitral regurgitation but not mitral valve prolapse.
Similarly, Gabow et al24 showed that there is no significant relationship between mitral valve prolapse and progression of renal disease in ADPKD.
Interestingly, Fick et al20 found that mitral valve prolapse has no significant effect on cardiovascular mortality.
CORONARY ARTERY DISEASE AND ANEURYSM IN ADPKD
Atherosclerosis sets in early in ADPKD, resulting in coronary artery disease and adverse cardiovascular outcomes.
Coronary flow velocity reserve is the ability of coronary arteries to dilate in response to myocardial oxygen demand. Atherosclerosis decreases this reserve in ADPKD patients, as shown in several studies.
Turkmen et al, in a series of studies,76–78 found that ADPKD patients had significantly less coronary flow velocity reserve, thicker carotid intima media (a surrogate marker of atherosclerosis), and greater insulin resistance than healthy controls. These findings imply that atherosclerosis begins very early in the course of ADPKD and has remarkable effects on cardiovascular morbidity and mortality.76
Aneurysm. Although the risk of extracranial aneurysm is higher with ADPKD, coronary artery aneurysm is uncommon. The pathogenesis of coronary aneurysm has been linked to abnormal expression of the proteins polycystin 1 and polycystin 2 in vascular smooth muscle.11,79 The PKD1 and PKD2 genes encode polycystin 1 and 2, respectively, in ADPKD. These polycystins are also expressed in the liver, kidneys, and myocardium and are involved in the regulation of intracellular calcium, stretch-activated ion channels, and vascular smooth muscle cell proliferation and apoptosis.11,16 Abnormally expressed polycystin in ADPKD therefore has an impact on arterial wall integrity, resulting in focal medial defects in the vasculature that eventually develop into micro- and macroaneurysms.11
Hadimeri et al79 found a higher prevalence of coronary aneurysm and ectasia in ADPKD patients than in controls. Most coronary aneurysms are smaller than 1 cm; however, a coronary aneurysm measuring 4 cm in diameter was found at autopsy of an ADPKD patient.11
Spontaneous coronary artery dissection is very rare in the general population, but Bobrie et al reported a case of it in an ADPKD patient.9
ATRIAL MYXOMA, CARDIOMYOPATHY, AND PERICARDIAL EFFUSION IN ADPKD
Atrial myxoma in ADPKD patients has been described in 2 case reports.15,80 However, the association of atrial myxoma with ADPKD is poorly understood and may be a coincidental finding.
Cardiomyopathy in ADPKD has been linked to abnormalities in the intracellular calcium pathway, although a clear picture of its involvement has yet to be established.
Paavola et al16 described the pathophysiology of ADPKD-associated cardiomyopathy in PKD2 mutant zebrafish lacking polycystin 2. These mutants showed decreased cardiac output and had atrioventricular blocks. The findings were attributed to abnormal intracellular calcium cycling. These findings correlated well with the frequent finding of idiopathic dilated cardiomyopathy in ADPKD patients, especially with PKD2 mutations.16 Also, 2 cases of dilated cardiomyopathy in ADPKD have been reported and thought to be related to PKD2 mutations.81,82
Pericardial effusion. Even though the exact pathophysiology of pericardial effusion in ADPKD is unknown, it has been theorized to be related to defects in connective tissue and extracellular matrix due to PKD1 and PKD2 mutations. These abnormalities increase the compliance and impair the recoil capacity of connective tissue, which results in unusual distention of the parietal pericardium. This abnormal distention of the parietal pericardium together with increased extracellular volume may lead to pericardial effusion.17
Qian et al17 found a higher prevalence of pericardial effusion in ADPKD patients. It was generally asymptomatic, and the cause was attributed to these connective tissue and extracellular matrix abnormalities.
EMERGING THERAPIES AND TESTS
Recent trials have investigated the effects of vasopressin receptor antagonists, specifically V2 receptor blockers in ADPKD and its complications.
Tolvaptan has been shown to slow the rate of increase in cyst size and total kidney volume.83,84 Also, a correlation between kidney size and diastolic blood pressure has been observed in ADPKD patients.85 Reducing cyst volume may reduce pressure effects and decrease renal ischemia, which in turn may reduce RAAS activation; however, the evidence to support this hypothesis is poor. A major clinical trial of tolvaptan in ADPKD patients showed no effect on blood pressure control, but the drug slowed the rate of increase in total kidney volume and fall in eGFR.83
Endothelin receptor antagonists are also in the preliminary stage of development for use in ADPKD. The effects of acute blockade of the renal endothelial system with bosentan were investigated in animal models by Hocher et al.28 This study showed a greater reduction in mean arterial pressure after bosentan administration, resulting in significantly decreased GFR and renal blood flow. Nonetheless, the mean arterial pressure-lowering effect of bosentan was more marked than the reductions in GFR and renal blood flow.
Raina et al,86 in a pilot cross-sectional analysis, showed that urinary excretion of ET-1 is increased in ADPKD patients, and may serve as a surrogate marker for ET-1 in renal tissue and a noninvasive marker of early kidney injury.
- Ha SK, Park CH, Kna JS, et al. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Yonsei Med J 1997; 38:111–116.
- Romao EA, Moyses Neto M, Teixeira SR, Muglia VF, Vieira-Neto OM, Dantas M. Renal and extrarenal manifestations of autosomal dominant polycystic kidney disease. Brazilian J Med Biol Res 2006; 39:533–538.
- Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 2015; 88:17–27.
- Perrone RD, Ruthazer R, Terrin NC. Survival after end-stage renal disease in autosomal dominant polycystic kidney disease: contribution of extrarenal complications to mortality. Am J Kidney Dis 2001; 38:777–784.
- Ecder T. Cardiovascular complications in autosomal dominant polycystic kidney disease. Curr Hypertens Rev 2013; 9:2–11.
- Silverio A, Prota C, Di Maio M, et al. Aortic dissection in patients with autosomal dominant polycystic kidney disease: a series of two cases and a review of the literature. Nephrology 2015; 20:229–235.
- Ramineni R, Daniel GK. Use of endovascular stent-graft repair for type B aortic dissection in polycystic kidney disease. J Invas Cardiol 2010; 22:E171–E174.
- Courtois A, Nusgens BV, Delvenne P, et al. Dissection of iliac artery in a patient with autosomal dominant polycystic kidney disease: a case report. Aorta 2013; 1:123–125.
- Bobrie G, Brunet-Bourgin F, Alamowitch S, et al. Spontaneous artery dissection: is it part of the spectrum of autosomal dominant polycystic kidney disease? Nephrol Dial Transplant 1998; 13:2138–2141.
- Minami T, Karube N, Sakamoto A. [Thoracic aortic dissection complicating autosomal dominant polycystic kidney disease; report of a case]. Kyobu Geka 2009; 62:924–927.
- Ohara K, Kimura T, Karasawa T, et al. A large coronary aneurysm and its probable precursor lesions in a patient with autosomal dominant polycystic kidney disease: an implication for the process of aneurysmogenesis. Pathol Int 2012; 62:758–762.
- Al-Hakim W, Goldsmith DJ. Bilateral popliteal aneurysms complicating adult polycystic kidney disease in a patient with a marfanoid habitus. Postgrad Med J 2003; 79:474–475.
- Kanagasundaram NS, Perry EP, Turney JH. Aneurysm of the splenic artery in a patient with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1999; 14:183–184.
- Kang YR, Ahn JH, Kim KH, Choi YM, Choi J, Park JR. Multiple cardiovascular manifestations in a patient with autosomal dominant polycystic kidney disease. J Cardiovasc Ultrasound 2014; 22:144–147.
- Iglesias D, Fraga AR, Arrizurieta E, et al. Atrial myxoma in a woman with autosomal dominant polycystic kidney disease type 2. Am J Kidney Dis 1997; 29:164–165.
- Paavola J, Schliffke S, Rossetti S, et al. Polycystin-2 mutations lead to impaired calcium cycling in the heart and predispose to dilated cardiomyopathy. J Mol Cell Cardiol 2013; 58:199–208.
- Qian Q, Hartman RP, King BF, Torres VE. Increased occurrence of pericardial effusion in patients with autosomal dominant polycystic kidney disease.
- Schievink WI, Torres VE, Wiebers DO, Huston J 3rd. Intracranial arterial dolichoectasia in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1997; 8:1298–1303.
- Pierre SA, Jaeger MT, Siemens DR. Intra-operative inferior vena cava syndrome in a patient with autosomal dominant polycystic kidney disease. World J Urol 2006; 24:110–112.
- Fick GM, Johnson AM, Hammond WS, Gabow PA. Causes of death in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1995; 5:2048–2056.
- Helal I, Reed B, Mettler P, et al. Prevalence of cardiovascular events in patients with autosomal dominant polycystic kidney disease. Am J Nephrol 2012; 36:362–370.
- Calabrese G, Vagelli G, Cristofano C, Barsotti G. Behaviour of arterial pressure in different stages of polycystic kidney disease. Nephron 1982; 32:207–208.
- de Almeida EA, de Oliveira EI, Lopes JA, Almeida AG, Lopes MG, Prata MM. Ambulatory blood pressure measurement in young normotensive patients with autosomal dominant polycystic kidney disease. Rev Port Cardiol 2007; 26:235–243.
- Gabow PA, Johnson AM, Kaehny WD, et al. Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 41:1311–1319.
- Bajrami V, Idrizi A, Roshi E, Barbullushi M. Association between nephrolithiasis, hypertension and obesity in polycystic kidney disease. Open Access Maced J Med Sci 2016; 4:43–46.
- Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med 1990; 323:1091–1096.
- Nakamura T, Ebihara I, Fukui M, et al. Increased endothelin and endothelin receptor mRNA expression in polycystic kidneys of cpk mice. J Am Soc Nephrol 1993; 4:1064–1072.
- Hocher B, Zart R, Schwarz A, et al. Renal endothelin system in polycystic kidney disease. J Am Soc Nephrol 1998; 9:1169–1177.
- Ong AC, Newby LJ, Dashwood MR. Expression and cellular localisation of renal endothelin-1 and endothelin receptor subtypes in autosomal-dominant polycystic kidney disease. Nephron Exper Nephrol 2003; 93:e80.
- Nash DA Jr. Hypertension in polycystic kidney disease without renal failure. Arch Intern Med 1977; 137:1571–1575.
- D’Angelo A, Mioni G, Ossi E, Lupo A, Valvo E, Maschio G. Alterations in renal tubular sodium and water transport in polycystic kidney disease. Clin Nephrol 1975; 3:99–105.
- Ettinger A, Kahn PC, Wise HM Jr. The importance of selective renal angiography in the diagnosis of polycystic disease. J Urol 1969; 102:156–161.
- Cornell SH. Angiography in polycystic disease of the kidneys. J Urol 1970; 103:24–26.
- Schrier RW. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2009; 20:1888–1893.
- Chapman AB, Guay-Woodford LM, Grantham JJ, et al. Renal structure in early autosomal-dominant polycystic kidney disease (ADPKD): The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) cohort. Kidney Int 2003; 64:1035–1045.
- Cadnapaphornchai MA, McFann K, Strain JD, Masoumi A, Schrier RW. Prospective change in renal volume and function in children with ADPKD. Clin J Am Soc Nephrol 2009; 4:820–829.
- Loghman-Adham M, Soto CE, Inagami T, Cassis L. The intrarenal renin-angiotensin system in autosomal dominant polycystic kidney disease. Am J Physiol Renal Physiol 2004; 287:F775–F788.
- Torres VE, Donovan KA, Scicli G, et al. Synthesis of renin by tubulocystic epithelium in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 42:364–373.
- Graham PC, Lindop GB. The anatomy of the renin-secreting cell in adult polycystic kidney disease. Kidney Int 1988; 33:1084–1090.
- Cerasola G, Vecchi M, Mule G, et al. Sympathetic activity and blood pressure pattern in autosomal dominant polycystic kidney disease hypertensives. Am J Nephrol 1998; 18:391–398.
- Valvo E, Gammaro L, Tessitore N, et al. Hypertension of polycystic kidney disease: mechanisms and hemodynamic alterations. Am J Nephrol 1985; 5:176–181.
- Marlais M, Cuthell O, Langan D, Dudley J, Sinha MD, Winyard PJ. Hypertension in autosomal dominant polycystic kidney disease: a meta-analysis. Arch Dis Child 2016; 101:1142–1147.
- Schrier RW, Abebe KZ, Perrone RD, et al. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371:2255–2266.
- Torres VE, Abebe KZ, Chapman AB, et al. Angiotensin blockade in late autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371:2267–2276.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Ritchie LD, Campbell NC, Murchie P. New NICE guidelines for hypertension. BMJ 2011; 343:d5644.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34:2159–2219.
- Rangan GK, Alexander SI, Campbell KL, et al. KHA-CARI guideline recommendations for the diagnosis and management of autosomal dominant polycystic kidney disease. Nephrology 2016; 21:705–716.
- Helal I, Al-Rowaie F, Abderrahim E, Kheder A. Update on pathogenesis, management, and treatment of hypertension in autosomal dominant polycystic kidney disease. Saudi J Kidney Dis Transpl 2017; 28:253–260.
- Patch C, Charlton J, Roderick PJ, Gulliford MC. Use of antihypertensive medications and mortality of patients with autosomal dominant polycystic kidney disease: a population-based study. Am J Kidney Dis 2011; 57:856–862.
- Zeltner R, Poliak R, Stiasny B, Schmieder RE, Schulze BD. Renal and cardiac effects of antihypertensive treatment with ramipril vs metoprolol in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2008; 23:573–579.
- Clark LA, Whitmire S, Patton S, Clark C, Blanchette CM, Howden R. Cost-effectiveness of angiotensin-converting enzyme inhibitors versus angiotensin II receptor blockers as first-line treatment in autosomal dominant polycystic kidney disease. J Med Econ 2017:1–17.
- Li Kam Wa TC, Macnicol AM, Watson ML. Ambulatory blood pressure in hypertensive patients with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1997; 12:2075–2080.
- Valero FA, Martinez-Vea A, Bardaji A, et al. Ambulatory blood pressure and left ventricular mass in normotensive patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1999; 10:1020–1026.
- Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest 2011; 121:4393–4408.
- Yildiz A, Gul CB, Ersoy A, Asiltas B, Ermurat S, Dogan S, et al. Arterial dysfunction in early autosomal dominant polycystic kidney disease independent of fibroblast growth factor 23. Iranian J Kidney Dis 2014; 8:443–449.
- Wanic-Kossowska M, Posnik B, Kobelski M, et al. The polymorphism of the ACE gene affects left ventricular hypertrophy and causes disturbances in left ventricular systolic/diastolic function in patients with autosomal dominant polycystic kidney disease. ScientificWorldJournal 2014; 2014:707658.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Cadnapaphornchai MA. Hypertension in children with autosomal dominant polycystic kidney disease (ADPKD). Curr Hypertens Rev 2013; 9:21–26.
- Schievink WI, Prendergast V, Zabramski JM. Rupture of a previously documented small asymptomatic intracranial aneurysm in a patient with autosomal dominant polycystic kidney disease. Case report. J Neurosurg 1998; 89:479–482.
- Graf S, Schischma A, Eberhardt KE, Istel R, Stiasny B, Schulze BD. Intracranial aneurysms and dolichoectasia in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2002; 17:819–823.
- Nakajima F, Shibahara N, Arai M, Gohji K, Ueda H, Katsuoka Y. Intracranial aneurysms and autosomal dominant polycystic kidney disease: followup study by magnetic resonance angiography. J Urol 2000; 164:311–313.
- Wakabayashi T, Fujita S, Ohbora Y, Suyama T, Tamaki N, Matsumoto S. Polycystic kidney disease and intracranial aneurysms. Early angiographic diagnosis and early operation for the unruptured aneurysm. J Neurosurg 1983; 58:488–491.
- Belz MM, Fick-Brosnahan GM, Hughes RL, et al. Recurrence of intracranial aneurysms in autosomal-dominant polycystic kidney disease. Kidney Int 2003; 63:1824–1830.
- Schrier RW, Belz MM, Johnson AM, et al. Repeat imaging for intracranial aneurysms in patients with autosomal dominant polycystic kidney disease with initially negative studies: a prospective ten-year follow-up. J Am Soc Nephrol 2004; 15:1023–1028.
- Cagnazzo F, Gambacciani C, Morganti R, Perrini P. Intracranial aneurysms in patients with autosomal dominant polycystic kidney disease: prevalence, risk of rupture, and management. A systematic review. Acta Neurochirurgica 2017; 5:811–821.
- Lee VW, Dexter MA, Mai J, Vladica P, Lopez-Vargas P, Rangan GK. KHA-CARI autosomal dominant polycystic kidney disease guideline: management of intracranial aneurysms. Semin Nephrol 2015; 35:612–617.
- Rozenfeld MN, Ansari SA, Shaibani A, Russell EJ, Mohan P, Hurley MC. Should patients with autosomal dominant polycystic kidney disease be screened for cerebral aneurysms? AJNR Am J Neuroradiol 2014; 35:3–9.
- Hiratsuka Y, Miki H, Kiriyama I, et al. Diagnosis of unruptured intracranial aneurysms: 3T MR angiography versus 64-channel multi-detector row CT angiography. Magn Reson Med Sci 2008; 7:169–178.
- van Gelder JM. Computed tomographic angiography for detecting cerebral aneurysms: implications of aneurysm size distribution for the sensitivity, specificity, and likelihood ratios. Neurosurgery 2003; 53:597–605.
- Villablanca JP, Jahan R, Hooshi P, et al. Detection and characterization of very small cerebral aneurysms by using 2D and 3D helical CT angiography. AJNR Am J Neuroradiol 2002; 23:1187–1198.
- Lumiaho A, Ikäheimo R, Miettinen R, et al. Mitral valve prolapse and mitral regurgitation are common in patients with polycystic kidney disease type 1. Am J Kidney Dis 2001; 38:1208–1216.
- Timio M, Monarca C, Pede S, Gentili S, Verdura C, Lolli S. The spectrum of cardiovascular abnormalities in autosomal dominant polycystic kidney disease: a 10-year follow-up in a five-generation kindred. Clin Nephrol 1992; 37:245–251.
- Hossack KF, Leddy CL, Johnson AM, Schrier RW, Gabow PA. Echocardiographic findings in autosomal dominant polycystic kidney disease. N Engl J Med 1988; 319:907–912.
- Leier CV, Baker PB, Kilman JW, Wooley CF. Cardiovascular abnormalities associated with adult polycystic kidney disease. Ann Intern Med 1984; 100:683–688.
- Turkmen K, Oflaz H, Uslu B, et al. Coronary flow velocity reserve and carotid intima media thickness in patients with autosomal dominant polycystic kidney disease: from impaired tubules to impaired carotid and coronary arteries. Clin J Am Soc Nephrol 2008; 3:986–991.
- Turkmen K, Tufan F, Alpay N, et al. Insulin resistance and coronary flow velocity reserve in patients with autosomal dominant polycystic kidney disease. Intern Med J 2012; 42:146–153.
- Turkmen K, Tufan F, Selcuk E, Akpinar T, Oflaz H, Ecder T. Neutrophil-to-lymphocyte ratio, insulin resistance, and endothelial dysfunction in patients with autosomal dominant polycystic kidney disease. Indian J Nephrol 2013; 23:34–40.
- Hadimeri H, Lamm C, Nyberg G. Coronary aneurysms in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1998; 9:837–841.
- Earle K, Hoffbrand BI. Adult dominant polycystic kidney disease and atrial myxoma. Nephron 1989; 52:197.
- Chung BM, Chong S, Lee W-S, Hwang S-N. Autosomal dominant polycystic kidney disease combined with intracranial aneurysm and dilated cardiomyopathy: a case report. J Korean Soc Radiol 2014; 71:84–88.
- Mariathasan DAL, Kumanan T. Adult polycystic kidney disease and idiopathic dilated cardiomyopathy: a rare genetic association. J Ceylon Coll Physicians 2016: 46:42–44.
- Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2012; 367:2407–2018.
- Higashihara E, Torres VE, Chapman AB, et al. Tolvaptan in autosomal dominant polycystic kidney disease: three years’ experience. Clin J Am Soc Nephrol 2011; 6:2499–2507.
- Sans Atxer L, Roca-Cusachs A, Torra R, et al. Relationship between renal size and blood pressure profile in patients with autosomal dominant polycystic kidney disease without renal failure. Nefrologia 2010; 30:567–572. In Spanish.
- Raina R, Lou L, Berger B, et al. Relationship of urinary endothelin-1 with estimated glomerular filtration rate in autosomal dominant polycystic kidney disease: a pilot cross-sectional analysis. BMC Nephrol 2016; 17:22.
- Ha SK, Park CH, Kna JS, et al. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Yonsei Med J 1997; 38:111–116.
- Romao EA, Moyses Neto M, Teixeira SR, Muglia VF, Vieira-Neto OM, Dantas M. Renal and extrarenal manifestations of autosomal dominant polycystic kidney disease. Brazilian J Med Biol Res 2006; 39:533–538.
- Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 2015; 88:17–27.
- Perrone RD, Ruthazer R, Terrin NC. Survival after end-stage renal disease in autosomal dominant polycystic kidney disease: contribution of extrarenal complications to mortality. Am J Kidney Dis 2001; 38:777–784.
- Ecder T. Cardiovascular complications in autosomal dominant polycystic kidney disease. Curr Hypertens Rev 2013; 9:2–11.
- Silverio A, Prota C, Di Maio M, et al. Aortic dissection in patients with autosomal dominant polycystic kidney disease: a series of two cases and a review of the literature. Nephrology 2015; 20:229–235.
- Ramineni R, Daniel GK. Use of endovascular stent-graft repair for type B aortic dissection in polycystic kidney disease. J Invas Cardiol 2010; 22:E171–E174.
- Courtois A, Nusgens BV, Delvenne P, et al. Dissection of iliac artery in a patient with autosomal dominant polycystic kidney disease: a case report. Aorta 2013; 1:123–125.
- Bobrie G, Brunet-Bourgin F, Alamowitch S, et al. Spontaneous artery dissection: is it part of the spectrum of autosomal dominant polycystic kidney disease? Nephrol Dial Transplant 1998; 13:2138–2141.
- Minami T, Karube N, Sakamoto A. [Thoracic aortic dissection complicating autosomal dominant polycystic kidney disease; report of a case]. Kyobu Geka 2009; 62:924–927.
- Ohara K, Kimura T, Karasawa T, et al. A large coronary aneurysm and its probable precursor lesions in a patient with autosomal dominant polycystic kidney disease: an implication for the process of aneurysmogenesis. Pathol Int 2012; 62:758–762.
- Al-Hakim W, Goldsmith DJ. Bilateral popliteal aneurysms complicating adult polycystic kidney disease in a patient with a marfanoid habitus. Postgrad Med J 2003; 79:474–475.
- Kanagasundaram NS, Perry EP, Turney JH. Aneurysm of the splenic artery in a patient with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1999; 14:183–184.
- Kang YR, Ahn JH, Kim KH, Choi YM, Choi J, Park JR. Multiple cardiovascular manifestations in a patient with autosomal dominant polycystic kidney disease. J Cardiovasc Ultrasound 2014; 22:144–147.
- Iglesias D, Fraga AR, Arrizurieta E, et al. Atrial myxoma in a woman with autosomal dominant polycystic kidney disease type 2. Am J Kidney Dis 1997; 29:164–165.
- Paavola J, Schliffke S, Rossetti S, et al. Polycystin-2 mutations lead to impaired calcium cycling in the heart and predispose to dilated cardiomyopathy. J Mol Cell Cardiol 2013; 58:199–208.
- Qian Q, Hartman RP, King BF, Torres VE. Increased occurrence of pericardial effusion in patients with autosomal dominant polycystic kidney disease.
- Schievink WI, Torres VE, Wiebers DO, Huston J 3rd. Intracranial arterial dolichoectasia in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1997; 8:1298–1303.
- Pierre SA, Jaeger MT, Siemens DR. Intra-operative inferior vena cava syndrome in a patient with autosomal dominant polycystic kidney disease. World J Urol 2006; 24:110–112.
- Fick GM, Johnson AM, Hammond WS, Gabow PA. Causes of death in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1995; 5:2048–2056.
- Helal I, Reed B, Mettler P, et al. Prevalence of cardiovascular events in patients with autosomal dominant polycystic kidney disease. Am J Nephrol 2012; 36:362–370.
- Calabrese G, Vagelli G, Cristofano C, Barsotti G. Behaviour of arterial pressure in different stages of polycystic kidney disease. Nephron 1982; 32:207–208.
- de Almeida EA, de Oliveira EI, Lopes JA, Almeida AG, Lopes MG, Prata MM. Ambulatory blood pressure measurement in young normotensive patients with autosomal dominant polycystic kidney disease. Rev Port Cardiol 2007; 26:235–243.
- Gabow PA, Johnson AM, Kaehny WD, et al. Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 41:1311–1319.
- Bajrami V, Idrizi A, Roshi E, Barbullushi M. Association between nephrolithiasis, hypertension and obesity in polycystic kidney disease. Open Access Maced J Med Sci 2016; 4:43–46.
- Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med 1990; 323:1091–1096.
- Nakamura T, Ebihara I, Fukui M, et al. Increased endothelin and endothelin receptor mRNA expression in polycystic kidneys of cpk mice. J Am Soc Nephrol 1993; 4:1064–1072.
- Hocher B, Zart R, Schwarz A, et al. Renal endothelin system in polycystic kidney disease. J Am Soc Nephrol 1998; 9:1169–1177.
- Ong AC, Newby LJ, Dashwood MR. Expression and cellular localisation of renal endothelin-1 and endothelin receptor subtypes in autosomal-dominant polycystic kidney disease. Nephron Exper Nephrol 2003; 93:e80.
- Nash DA Jr. Hypertension in polycystic kidney disease without renal failure. Arch Intern Med 1977; 137:1571–1575.
- D’Angelo A, Mioni G, Ossi E, Lupo A, Valvo E, Maschio G. Alterations in renal tubular sodium and water transport in polycystic kidney disease. Clin Nephrol 1975; 3:99–105.
- Ettinger A, Kahn PC, Wise HM Jr. The importance of selective renal angiography in the diagnosis of polycystic disease. J Urol 1969; 102:156–161.
- Cornell SH. Angiography in polycystic disease of the kidneys. J Urol 1970; 103:24–26.
- Schrier RW. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2009; 20:1888–1893.
- Chapman AB, Guay-Woodford LM, Grantham JJ, et al. Renal structure in early autosomal-dominant polycystic kidney disease (ADPKD): The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) cohort. Kidney Int 2003; 64:1035–1045.
- Cadnapaphornchai MA, McFann K, Strain JD, Masoumi A, Schrier RW. Prospective change in renal volume and function in children with ADPKD. Clin J Am Soc Nephrol 2009; 4:820–829.
- Loghman-Adham M, Soto CE, Inagami T, Cassis L. The intrarenal renin-angiotensin system in autosomal dominant polycystic kidney disease. Am J Physiol Renal Physiol 2004; 287:F775–F788.
- Torres VE, Donovan KA, Scicli G, et al. Synthesis of renin by tubulocystic epithelium in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 42:364–373.
- Graham PC, Lindop GB. The anatomy of the renin-secreting cell in adult polycystic kidney disease. Kidney Int 1988; 33:1084–1090.
- Cerasola G, Vecchi M, Mule G, et al. Sympathetic activity and blood pressure pattern in autosomal dominant polycystic kidney disease hypertensives. Am J Nephrol 1998; 18:391–398.
- Valvo E, Gammaro L, Tessitore N, et al. Hypertension of polycystic kidney disease: mechanisms and hemodynamic alterations. Am J Nephrol 1985; 5:176–181.
- Marlais M, Cuthell O, Langan D, Dudley J, Sinha MD, Winyard PJ. Hypertension in autosomal dominant polycystic kidney disease: a meta-analysis. Arch Dis Child 2016; 101:1142–1147.
- Schrier RW, Abebe KZ, Perrone RD, et al. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371:2255–2266.
- Torres VE, Abebe KZ, Chapman AB, et al. Angiotensin blockade in late autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371:2267–2276.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Ritchie LD, Campbell NC, Murchie P. New NICE guidelines for hypertension. BMJ 2011; 343:d5644.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34:2159–2219.
- Rangan GK, Alexander SI, Campbell KL, et al. KHA-CARI guideline recommendations for the diagnosis and management of autosomal dominant polycystic kidney disease. Nephrology 2016; 21:705–716.
- Helal I, Al-Rowaie F, Abderrahim E, Kheder A. Update on pathogenesis, management, and treatment of hypertension in autosomal dominant polycystic kidney disease. Saudi J Kidney Dis Transpl 2017; 28:253–260.
- Patch C, Charlton J, Roderick PJ, Gulliford MC. Use of antihypertensive medications and mortality of patients with autosomal dominant polycystic kidney disease: a population-based study. Am J Kidney Dis 2011; 57:856–862.
- Zeltner R, Poliak R, Stiasny B, Schmieder RE, Schulze BD. Renal and cardiac effects of antihypertensive treatment with ramipril vs metoprolol in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2008; 23:573–579.
- Clark LA, Whitmire S, Patton S, Clark C, Blanchette CM, Howden R. Cost-effectiveness of angiotensin-converting enzyme inhibitors versus angiotensin II receptor blockers as first-line treatment in autosomal dominant polycystic kidney disease. J Med Econ 2017:1–17.
- Li Kam Wa TC, Macnicol AM, Watson ML. Ambulatory blood pressure in hypertensive patients with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1997; 12:2075–2080.
- Valero FA, Martinez-Vea A, Bardaji A, et al. Ambulatory blood pressure and left ventricular mass in normotensive patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1999; 10:1020–1026.
- Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest 2011; 121:4393–4408.
- Yildiz A, Gul CB, Ersoy A, Asiltas B, Ermurat S, Dogan S, et al. Arterial dysfunction in early autosomal dominant polycystic kidney disease independent of fibroblast growth factor 23. Iranian J Kidney Dis 2014; 8:443–449.
- Wanic-Kossowska M, Posnik B, Kobelski M, et al. The polymorphism of the ACE gene affects left ventricular hypertrophy and causes disturbances in left ventricular systolic/diastolic function in patients with autosomal dominant polycystic kidney disease. ScientificWorldJournal 2014; 2014:707658.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Cadnapaphornchai MA. Hypertension in children with autosomal dominant polycystic kidney disease (ADPKD). Curr Hypertens Rev 2013; 9:21–26.
- Schievink WI, Prendergast V, Zabramski JM. Rupture of a previously documented small asymptomatic intracranial aneurysm in a patient with autosomal dominant polycystic kidney disease. Case report. J Neurosurg 1998; 89:479–482.
- Graf S, Schischma A, Eberhardt KE, Istel R, Stiasny B, Schulze BD. Intracranial aneurysms and dolichoectasia in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2002; 17:819–823.
- Nakajima F, Shibahara N, Arai M, Gohji K, Ueda H, Katsuoka Y. Intracranial aneurysms and autosomal dominant polycystic kidney disease: followup study by magnetic resonance angiography. J Urol 2000; 164:311–313.
- Wakabayashi T, Fujita S, Ohbora Y, Suyama T, Tamaki N, Matsumoto S. Polycystic kidney disease and intracranial aneurysms. Early angiographic diagnosis and early operation for the unruptured aneurysm. J Neurosurg 1983; 58:488–491.
- Belz MM, Fick-Brosnahan GM, Hughes RL, et al. Recurrence of intracranial aneurysms in autosomal-dominant polycystic kidney disease. Kidney Int 2003; 63:1824–1830.
- Schrier RW, Belz MM, Johnson AM, et al. Repeat imaging for intracranial aneurysms in patients with autosomal dominant polycystic kidney disease with initially negative studies: a prospective ten-year follow-up. J Am Soc Nephrol 2004; 15:1023–1028.
- Cagnazzo F, Gambacciani C, Morganti R, Perrini P. Intracranial aneurysms in patients with autosomal dominant polycystic kidney disease: prevalence, risk of rupture, and management. A systematic review. Acta Neurochirurgica 2017; 5:811–821.
- Lee VW, Dexter MA, Mai J, Vladica P, Lopez-Vargas P, Rangan GK. KHA-CARI autosomal dominant polycystic kidney disease guideline: management of intracranial aneurysms. Semin Nephrol 2015; 35:612–617.
- Rozenfeld MN, Ansari SA, Shaibani A, Russell EJ, Mohan P, Hurley MC. Should patients with autosomal dominant polycystic kidney disease be screened for cerebral aneurysms? AJNR Am J Neuroradiol 2014; 35:3–9.
- Hiratsuka Y, Miki H, Kiriyama I, et al. Diagnosis of unruptured intracranial aneurysms: 3T MR angiography versus 64-channel multi-detector row CT angiography. Magn Reson Med Sci 2008; 7:169–178.
- van Gelder JM. Computed tomographic angiography for detecting cerebral aneurysms: implications of aneurysm size distribution for the sensitivity, specificity, and likelihood ratios. Neurosurgery 2003; 53:597–605.
- Villablanca JP, Jahan R, Hooshi P, et al. Detection and characterization of very small cerebral aneurysms by using 2D and 3D helical CT angiography. AJNR Am J Neuroradiol 2002; 23:1187–1198.
- Lumiaho A, Ikäheimo R, Miettinen R, et al. Mitral valve prolapse and mitral regurgitation are common in patients with polycystic kidney disease type 1. Am J Kidney Dis 2001; 38:1208–1216.
- Timio M, Monarca C, Pede S, Gentili S, Verdura C, Lolli S. The spectrum of cardiovascular abnormalities in autosomal dominant polycystic kidney disease: a 10-year follow-up in a five-generation kindred. Clin Nephrol 1992; 37:245–251.
- Hossack KF, Leddy CL, Johnson AM, Schrier RW, Gabow PA. Echocardiographic findings in autosomal dominant polycystic kidney disease. N Engl J Med 1988; 319:907–912.
- Leier CV, Baker PB, Kilman JW, Wooley CF. Cardiovascular abnormalities associated with adult polycystic kidney disease. Ann Intern Med 1984; 100:683–688.
- Turkmen K, Oflaz H, Uslu B, et al. Coronary flow velocity reserve and carotid intima media thickness in patients with autosomal dominant polycystic kidney disease: from impaired tubules to impaired carotid and coronary arteries. Clin J Am Soc Nephrol 2008; 3:986–991.
- Turkmen K, Tufan F, Alpay N, et al. Insulin resistance and coronary flow velocity reserve in patients with autosomal dominant polycystic kidney disease. Intern Med J 2012; 42:146–153.
- Turkmen K, Tufan F, Selcuk E, Akpinar T, Oflaz H, Ecder T. Neutrophil-to-lymphocyte ratio, insulin resistance, and endothelial dysfunction in patients with autosomal dominant polycystic kidney disease. Indian J Nephrol 2013; 23:34–40.
- Hadimeri H, Lamm C, Nyberg G. Coronary aneurysms in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1998; 9:837–841.
- Earle K, Hoffbrand BI. Adult dominant polycystic kidney disease and atrial myxoma. Nephron 1989; 52:197.
- Chung BM, Chong S, Lee W-S, Hwang S-N. Autosomal dominant polycystic kidney disease combined with intracranial aneurysm and dilated cardiomyopathy: a case report. J Korean Soc Radiol 2014; 71:84–88.
- Mariathasan DAL, Kumanan T. Adult polycystic kidney disease and idiopathic dilated cardiomyopathy: a rare genetic association. J Ceylon Coll Physicians 2016: 46:42–44.
- Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2012; 367:2407–2018.
- Higashihara E, Torres VE, Chapman AB, et al. Tolvaptan in autosomal dominant polycystic kidney disease: three years’ experience. Clin J Am Soc Nephrol 2011; 6:2499–2507.
- Sans Atxer L, Roca-Cusachs A, Torra R, et al. Relationship between renal size and blood pressure profile in patients with autosomal dominant polycystic kidney disease without renal failure. Nefrologia 2010; 30:567–572. In Spanish.
- Raina R, Lou L, Berger B, et al. Relationship of urinary endothelin-1 with estimated glomerular filtration rate in autosomal dominant polycystic kidney disease: a pilot cross-sectional analysis. BMC Nephrol 2016; 17:22.
KEY POINTS
- Hypertension and left ventricular hypertrophy are common complications of ADPKD.
- Cardiovascular disease is a major cause of morbidity and death in ADPKD.
- Early diagnosis and aggressive management of high blood pressure, specifically with agents that block the renin-angiotensin-aldosterone system, are necessary to prevent left ventricular hypertrophy and progression of renal failure in ADPKD.
- Timely screening and intervention for intracranial aneurysm would lessen the rates of morbidity and death from intracranial hemorrhage.
Postdischarge clinics and hospitalists: A review of the evidence and existing models
Readmission prevention is paramount for hospitals and, by extension, hospitalist programs. Hospitalists see early and reliable outpatient follow-up as a safe landing for their most complicated patient cases. The option of a postdischarge clinic arises from the challenge to arrange adequate postdischarge care for patients who lack easy access because of insurance or provider availability. Guaranteeing postdischarge access by opening a dedicated, hospitalist-led postdischarge clinic appears to be an easy solution, but it is a solution that requires significant investment (including investment in physician and staff training and administrative support) and careful navigation of existing primary care relationships. In addition, a clinic staffed only with physicians may not be well equipped to address the complex social factors in healthcare utilization and readmission. Better understanding of the evidence supporting post discharge physician visits, several models of clinics, and the key operational questions are essential to address before crossing the inpatient-outpatient divide.
POSTDISCHARGE PHYSICIAN VISITS AND READMISSIONS
A postdischarge outpatient provider visit is often seen as a key factor in reducing readmissions. In 2013, Medicare added strength to this association by establishing transitional care management codes, which provide enhanced reimbursement to providers for a visit within 7 or 14 days of discharge, with focused attention on transitional issues.1 However, whether a postdischarge visit reduces readmissions remains unclear. Given evidence that higher primary care density is associated with lower healthcare utilization,2 CMS’s financial investment in incentivizing post discharge physician visits may be a good bet. On the other hand, simply having a primary care physician (PCP) may be a risk factor for readmission. This association suggests that postdischarge vigilance leads to identification of medical problems that lead to rehospitalization.3 This uncertainty is not resolved in systematic reviews of readmission reduction initiatives, which were not focused solely on the impact of a physician visit.4,5
The earliest study of postdischarge visits in a general medical population found an association between intensive outpatient follow-up by new providers in a Veterans Affairs population and an increase in hospital readmissions.6 This model is similar to some hospitalist models for postdischarge clinics, as the visit was with a noncontinuity provider. The largest recent study, of patients hospitalized with acute myocardial infarction, community-acquired pneumonia, or congestive heart failure (CHF) between 2009 and 2012, found increased frequency of postdischarge follow-up but no concomitant reduction in readmissions.7 Although small observational studies8 have found a postdischarge primary care visit may reduce the risk for readmission in general medical patients, the bulk of the recent data is negative.
In high-risk patients, however, there may be a clear benefit to postdischarge follow-up. In a North Carolina Medicaid population, a physician visit after discharge was associated with fewer readmissions among high-risk patients, but not among lower risk patients, whose readmission rates were low to start.9 The results of that study support the idea that risk stratification may identify patients who can benefit from more intensive outpatient follow-up. In general medical populations, existing studies may suffer from an absence of adequate risk assessment.
The evidence in specific disease states may show a clearer association between a postdischarge physician visit and reduced risk for readmission. One quarter of patients with CHF are rehospitalized within 30 days of discharge.10 In this disease with frequent exacerbations, a clinic visit to monitor volume status, weight, and medication adherence might reduce the frequency of readmissions or prolong the interval between rehospitalizations. A large observational study observed that earlier post discharge follow up by a cardiologist or a PCP was associated with lower risk of readmission, but only in the quintile with the closest follow-up. In addition, fewer than 40% of patients in this group had a visit within 7 days.11 In another heart failure population, follow-up with either a PCP or cardiologist within 7 days of discharge was again associated with lower risk for readmission.12 Thus, data suggest a protective effect of postdischarge visits in CHF patients, in contrast to a general medical population. Patients with end-stage renal disease may also fit in this group protected by a postdischarge physician visit, as 1 additional visit within the month after discharge was estimated to reduce rehospitalizations and produce significant cost savings.13
With other specific discharge diagnoses, results are varied. Two small observational studies in chronic obstructive pulmonary disease had conflicting results—one found a modest reduction in readmission and emergency department (ED) visits for patients seen by a PCP or pulmonologist within 30 days of discharge,14 and the other found no effect on readmissions but an associated reduction in mortality.15 More data are needed to clarify further the interaction of postdischarge visits with mortality, but the association between postdischarge physician visits and readmission reduction is controversial for patients with chronic obstructive pulmonary disease.
Finally, the evidence for dedicated postdischarge clinics is even more limited. A study of a hospitalist-led postdischarge clinic in a Veterans Affairs hospital found reduced length of stay and earlier postdischarge follow-up in a postdischarge clinic, but no effect on readmissions.16 Other studies have found earlier postdischarge follow-up with dedicated discharge clinics but have not evaluated readmission rates specifically.17In summary, the effect of postdischarge visits on risk for readmission is an area of active research, but remains unclear. The data reviewed suggest a benefit for the highest risk patients, specifically those with severe chronic illness, or those deemed high-risk with a readmission tool.9 At present, because physicians cannot accurately predict which patients will be readmitted,18 discharging physicians often take a broad approach and schedule outpatient visits for all patients. As readmission tools are further refined, the group of patients who will benefit from postdischarge care will be easier to identify, and a benefit to postdischarge visits may be seen
It is also important to note that this review emphasizes the physician visit and its potential impact on readmissions. Socioeconomic causes are increasingly being recognized as driving readmissions and other utilization.19 Whether an isolated physician visit is sufficient to prevent readmissions for patients with nonmedical drivers of healthcare utilization is unclear. For those patients, a discharge visit likely is a necessary component of a readmission reduction strategy for high-risk patients, but may be insufficient for patients who require not just an isolated visit but rather a more integrated and comprehensive care program.8,20,21
POSTDISCHARGE CLINIC MODELS
Despite the unclear relationship between postdischarge physician care and readmissions, dedicated postdischarge clinics, some staffed by hospitalists, have been adopted over the past 10 years. The three primary types of clinics arise in safety net environments, in academic medical centers, and as comprehensive high-risk patient solutions. Reviewing several types of clinics further clarifies the nature of this structural innovation.
Safety Net Hospital Models
Safety net hospitals and their hospitalists struggle with securing adequate postdischarge access for their population, which has inadequate insurance and poor access to primary care. Patient characteristics also play a role in the complex postdischarge care for this population, given its high rate of ED use (owing to perceived convenience and capabilities) for ambulatory-sensitive conditions.22 In addition, immigrants, particularly those with low English-language proficiency, underuse and have poor access to primary care.23,24 Postdischarge clinics in this environment focus first on providing a reliable postdischarge plan and then on linking to primary care. Examples of two clinics are at Harborview Medical Center in Seattle, Washington25 and Texas Health in Fort Worth.
Harborview is a 400-bed hospital affiliated with the University of Washington. More than 50% of its patients are considered indigent. The clinic was established in 2007 to provide a postdischarge option for uninsured patients, and a link to primary care in federally qualified health centers. The clinic was staffed 5 days a week with one or two hospitalists or advanced practice nurses. Visit duration was 20 minutes, 270 visits occurred per month, and the no-show rate was 30%. A small subgroup of the hospitalist group staffed the clinic. Particular clinical foci included CHF patients, patients with wound-care needs, and homeless, immigrant, and recently incarcerated patients. A key goal was connecting to longitudinal primary care, and the clinic successfully connected more than 70% of patients to primary care in community health centers. This clinic ultimately transitioned from a hospitalist practice to a primary care practice with a primary focus on post-ED follow-up for unaffiliated patients.26
In 2010, Texas Health faced a similar challenge with unaffiliated patients, and established a nurse practitioner–based clinic with hospitalist oversight to provide care primarily for patients without insurance or without an existing primary care relationship.
Academic Medical Center Models
Another clinical model is designed for patients who receive primary care at practices affiliated with academic medical centers. Although many of these patients have insurance and a PCP, there is often no availability with their continuity provider, because of the resident’s inpatient schedule or the faculty member’s conflicting priorities.27,28 Academic medical centers, including the University of California at San Francisco, the University of New Mexico, and the Beth Israel Deaconess Medical Center, have established discharge clinics within their faculty primary care practices. A model of this type of clinic was set up at Beth Israel Deaconess in 2010. Staffed by four hospitalists and using 40-minute appointments, this clinic was physically based in the primary care practice. As such, it took advantage of the existing clinic’s administrative and clinical functions, including triage, billing, and scheduling. A visit was scheduled in that clinic by the discharging physician team if a primary care appointment was not available with the patient’s continuity provider. Visits were standardized and focused on outstanding issues at discharge, medication reconciliation, and symptom trajectory. The hospitalists used the clinic’s clinical resources, including nurses, social workers, and pharmacists, but had no other dedicated staff. That there were only four hospitalists meant they were able to gain sufficient exposure to the outpatient setting, provide consistent high-quality care, and gain credibility with the PCPs. As the patients who were seen had PCPs of their own, during the visit significant attention was focused first on the postdischarge concerns, and then on promptly returning the patients to routine primary care. Significant patient outreach was used to address the clinic’s no-show rate, which was almost 50% in the early months. Within a year, the rate was down, closer to 20%. This clinic closed in 2015 after the primary care practice, in which it was based, transitioned to a patient-centered medical home. Since that time, this type of initiative has spread further, with neurohospitalist discharge clinics established, and postdischarge neurology follow-up becoming faster and more reliable.29
Academic medical centers and safety net hospitals substitute for routine primary care to address the basic challenge of primary care access, often without significant enhancements or additional resources, such as dedicated care management and pharmacy, social work, and nursing support. Commonalities of these clinics include dedicated physician staff, appointments generally longer than average outpatient appointments, and visit content concentrated on the key issues at transition (medication reconciliation, outstanding tests, symptom trajectory). As possible, clinics adopted a multidisciplinary approach, with social workers, community health workers, and nurses, to respond to the breadth of patients’ postdischarge needs, which often extend beyond pure medical need. The most frequent barriers encountered included the knowledge gap for hospitalist providers in the outpatient setting (a gap mitigated by using dedicated providers) and the patients’ high no-show rate (not surprising given that the providers are generally new to them). Few clinics have attempted to create continuity across inpatient and outpatient providers, though continuity might reduce no-shows as well as eliminate at least 1 transition.
Comprehensive High-Risk Patient Solutions
At the other end of the clinic spectrum are more integrated postdischarge approaches, which also evolved from the hospitalist model with hospitalist staffing. However, these approaches were introduced in response to the clinical needs of the highest risk patients (who are most vulnerable to frequent provider transitions), not to a systemic inability to provide routine postdischarge care.30
The most long-standing model for this type of clinic is represented by CareMore Health System, a subsidiary of Anthem.30-32 The extensivist, an expanded-scope hospitalist, acts as primary care coordinator, coordinating a multidisciplinary team for a panel of about 100 patients, representing the sickest 5% of the Medicare Advantage–insured population. Unlike the traditional hospitalist, the extensivist follows patients across all care sites, including hospital, rehabilitation sites, and outpatient clinic. For the most part, this relationship is not designed to evolve into a longitudinal relationship, but rather is an intervention only for the several-months period of acute need. Internal data have shown effects on hospital readmissions as well as length of stay.30
Another integrated clinic was established in 2013, at the University of Chicago. This was an effort to redesign care for patients at highest risk for hospitalization.33 Similar to the CareMore process, a high-risk population is identified by prior hospitalization and expected high Medicare costs. A comprehensive care physician cares for these patients across care settings. The clinic takes a team-based approach to patient care, with team members selected on the basis of patient need. Physicians have panels limited to only 200 patients, and generally spend part of the day in clinic, and part in seeing their hospitalized patients. Although reminiscent of a traditional primary care setting, this clinic is designed specifically for a high-risk, frequently hospitalized population, and therefore requires physicians with both a skill set akin to that of hospitalists, and an approach of palliative care and holistic patient care. Outcomes from this trial clinic are expected in 2017 or 2018.

LOGISTICAL CONSIDERATIONS FOR DISCHARGE CLINICS
Considering some key operational questions (Table) can help guide hospitals, hospitalists, and healthcare systems as they venture into the postdischarge clinic space. Return on investment and sustainability are two key questions for postdischarge clinics.
Return on investment varies by payment structure. In capitated environments with a strong emphasis on readmissions and total medical expenditure, a successful postdischarge clinic would recoup the investment through readmission reduction. However, maintaining adequate patient volume against high no-show rates may strain the group financially. In addition, although a hospitalist group may reap few measurable benefits from this clinical exposure, the unique view of the outpatient world afforded to hospitalists working in this environment could enrich the group as a whole by providing a more well-rounded vantage point.
Another key question surrounds sustainability. The clinic at the Beth Israel Deaconess Medical Center in Boston temporarily closed due to high inpatient volume and corresponding need for those hospitalists in the inpatient setting, early in its inception. It subsequently closed due to evolution in the clinic where it was based, rendering it unnecessary. Clinics that are contingent on other clinics will be vulnerable to external forces. Finally, staffing these clinics may be a stretch for a hospitalist group, as a partly different skill set is required for patient care in the outpatient setting. Hospitalists interested in care transitions are well suited for this role. In addition, hospitalists interested in more clinical variety, or in more schedule variety than that provided in a traditional hospitalist schedule, often enjoy the work. A vast majority of hospitalists think PCPs are responsible for postdischarge problems, and would not be interested in working in the postdischarge world.34 A poor fit for providers may lead to clinic failure.
As evident from this review, gaps in understanding the benefits of postdischarge care have persisted for 10 years. Discharge clinics have been scantly described in the literature. The primary unanswered question remains the effect on readmissions, but this has been the sole research focus to date. Other key research areas are the impact on other patient-centered clinical and system outcomes (eg, patient satisfaction, particularly for patients seeing new providers), postdischarge mortality, the effect on other adverse events, and total medical expenditure.
The healthcare system is evolving in the context of a focus on readmissions, primary care access challenges, and high-risk patients’ specific needs. These forces are spurring innovation in the realm of postdischarge physician clinics, as even the basic need for an appointment may not be met by the existing outpatient primary care system. In this context, multiple new outpatient care structures have arisen, many staffed by hospitalists. Some, such as clinics based in safety net hospitals and academic medical centers, address the simple requirement that patients who lack easy access, because of insurance status or provider availability, can see a doctor after discharge. This type of clinic may be an essential step in alleviating a strained system but may not represent a sustainable long-term solution. More comprehensive solutions for improving patient care and clinical outcomes may be offered by integrated systems, such as CareMore, which also emerged from the hospitalist model. A lasting question is whether these clinics, both the narrowly focused and the comprehensive, will have longevity in the evolving healthcare market. Inevitably, though, hospitalist directors will continue to raise such questions, and should stand to benefit from the experiences of others described in this review.
Disclosure
Nothing to report.
1. US Department of Health and Human Services, Centers for Medicare & Medicaid Services. Transitional Care Management Services. https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNProducts/Downloads/Transitional-Care-Management-Services-Fact-Sheet-ICN908628.pdf. Fact sheet ICN 908628.. Accessed June 29, 2016.
2. Kravet SJ, Shore AD, Miller R, Green GB, Kolodner K, Wright SM. Health care utilization and the proportion of primary care physicians. Am J Med. 2008;121(2):142-148. PubMed
3. Hasan O, Meltzer DO, Shaykevich SA, et al. Hospital readmission in general medicine patients: a prediction model. J Gen Intern Med. 2010;25(3):211-219. PubMed
4. Hansen LO, Young RS, Hinami K, Leung A, Williams MV. Interventions to reduce 30-day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520-528. PubMed
5. Leppin AL, Gionfriddo MR, Kessler M, et al. Preventing 30-day hospital readmissions: a systematic review and meta-analysis of randomized trials. JAMA Intern Med. 2014;174(7):1095-1107. PubMed
6. Weinberger M, Oddone EZ, Henderson WG. Does increased access to primary care reduce hospital readmissions? Veterans Affairs Cooperative Study Group on Primary Care and Hospital Readmission. N Engl J Med. 1996;334(22):1441-1447. PubMed
7. DeLia D, Tong J, Gaboda D, Casalino LP. Post-discharge follow-up visits and hospital utilization by Medicare patients, 2007-2010. Medicare Medicaid Res Rev. 2014;4(2). PubMed
8. Dedhia P, Kravet S, Bulger J, et al. A quality improvement intervention to facilitate the transition of older adults from three hospitals back to their homes. J Am Geriatr Soc. 2009;57(9):1540-1546. PubMed
9. Jackson C, Shahsahebi M, Wedlake T, DuBard CA. Timeliness of outpatient follow-up: an evidence-based approach for planning after hospital discharge. Ann Fam Med. 2015;13(2):115-122. PubMed
10. Dharmarajan K, Hsieh AF, Lin Z, et al. Diagnoses and timing of 30-day readmissions after hospitalization for heart failure, acute myocardial infarction, or pneumonia. JAMA. 2013;309(4):355-363. PubMed
11. Hernandez AF, Greiner MA, Fonarow GC, et al. Relationship between early physician follow-up and 30-day readmission among Medicare beneficiaries hospitalized for heart failure. JAMA. 2010;303(17):1716-1722. PubMed
12. Lee KK, Yang J, Hernandez AF, Steimle AE, Go AS. Post-discharge follow-up characteristics associated with 30-day readmission after heart failure hospitalization. Med Care. 2016;54(4):365-372. PubMed
13. Erickson KF, Winkelmayer WC, Chertow GM, Bhattacharya J. Physician visits and 30-day hospital readmissions in patients receiving hemodialysis. J Am Soc Nephrol. 2014;25(9):2079-2087. PubMed
14. Sharma G, Kuo YF, Freeman JL, Zhang DD, Goodwin JS. Outpatient follow-up visit and 30-day emergency department visit and readmission in patients hospitalized for chronic obstructive pulmonary disease. Arch Intern Med. 2010;170(18):1664-1670. PubMed
15. Fidahussein SS, Croghan IT, Cha SS, Klocke DL. Posthospital follow-up visits and 30-day readmission rates in chronic obstructive pulmonary disease. Risk Manag Healthc Policy. 2014;7:105-112. PubMed
16. Burke RE, Whitfield E, Prochazka AV. Effect of a hospitalist-run postdischarge clinic on outcomes. J Hosp Med. 2014;9(1):7-12. PubMed
17. Doctoroff L, Nijhawan A, McNally D, Vanka A, Yu R, Mukamal KJ. The characteristics and impact of a hospitalist-staffed post-discharge clinic. Am J Med. 2013;126(11):1016.e9-e15. PubMed
18. Allaudeen N, Schnipper JL, Orav EJ, Wachter RM, Vidyarthi AR. Inability of providers to predict unplanned readmissions. J Gen Intern Med. 2011;26(7):771-776. PubMed
19. Barnett ML, Hsu J, McWilliams J. Patient characteristics and differences in hospital readmission rates. JAMA Intern Med. 2015;175(11):1803-1812. PubMed
20. Jack BW, Chetty VK, Anthony D, et al. A reengineered hospital discharge program to decrease rehospitalization: a randomized trial. Ann Intern Med. 2009;150(3):178-187. PubMed
21. Naylor M, Brooten D, Jones R, Lavizzo-Mourey R, Mezey M, Pauly M. Comprehensive discharge planning for the hospitalized elderly. A randomized clinical trial. Ann Intern Med. 1994;120(12):999-1006. PubMed
22. Capp R, Camp-Binford M, Sobolewski S, Bulmer S, Kelley L. Do adult Medicaid enrollees prefer going to their primary care provider’s clinic rather than emergency department (ED) for low acuity conditions? Med Care. 2015;53(6):530-533. PubMed
23. Vargas Bustamante A, Fang H, Garza J, et al. Variations in healthcare access and utilization among Mexican immigrants: the role of documentation status. J Immigr Minor Health. 2012;14(1):146-155. PubMed
24. Chi JT, Handcock MS. Identifying sources of health care underutilization among California’s immigrants. J Racial Ethn Health Disparities. 2014;1(3):207-218. PubMed
25. Martinez S. Bridging the Gap: Discharge Clinics Providing Safe Transitions for High Risk Patients. Workshop presented at: Northwest Patient Safety Conference; May 15, 2012; Seattle, WA. http://www.wapatientsafety.org/downloads/Martinez.pdf. Published 2011. Accessed April 26, 2017.
26. Elliott K, W Klein J, Basu A, Sabbatini AK. Transitional care clinics for follow-up and primary care linkage for patients discharged from the ED. Am J Emerg Med. 2016;34(7):1230-1235. PubMed
27. Baxley EG, Weir S. Advanced access in academic settings: definitional challenges. Ann Fam Med. 2009;7(1):90-91. PubMed
28. Doctoroff L, McNally D, Vanka A, Nall R, Mukamal KJ. Inpatient–outpatient transitions for patients with resident primary care physicians: access and readmission. Am J Med. 2014;127(9):886.e15-e20. PubMed
29. Shah M, Douglas V, Scott B, Josephson SA. A neurohospitalist discharge clinic shortens the transition from inpatient to outpatient care. Neurohospitalist. 2016;6(2):64-69. PubMed
30. Powers BW, Milstein A, Jain SH. Delivery models for high-risk older patients: back to the future? JAMA. 2016;315(1):23-24. PubMed
31. Milstein A, Gilbertson E. American medical home runs. Health Aff (Millwood). 2009;28(5):1317-1326. PubMed
32. Reuben DB. Physicians in supporting roles in chronic disease care: the CareMore model. J Am Geriatr Soc. 2011;59(1):158-160. PubMed
33. Meltzer DO, Ruhnke GW. Redesigning care for patients at increased hospitalization risk: the comprehensive care physician model. Health Aff (Millwood). 2014;33(5):770-777. PubMed
34. Burke RE, Ryan P. Postdischarge clinics: hospitalist attitudes and experiences. J Hosp Med. 2013;8(10):578-581. PubMed
Readmission prevention is paramount for hospitals and, by extension, hospitalist programs. Hospitalists see early and reliable outpatient follow-up as a safe landing for their most complicated patient cases. The option of a postdischarge clinic arises from the challenge to arrange adequate postdischarge care for patients who lack easy access because of insurance or provider availability. Guaranteeing postdischarge access by opening a dedicated, hospitalist-led postdischarge clinic appears to be an easy solution, but it is a solution that requires significant investment (including investment in physician and staff training and administrative support) and careful navigation of existing primary care relationships. In addition, a clinic staffed only with physicians may not be well equipped to address the complex social factors in healthcare utilization and readmission. Better understanding of the evidence supporting post discharge physician visits, several models of clinics, and the key operational questions are essential to address before crossing the inpatient-outpatient divide.
POSTDISCHARGE PHYSICIAN VISITS AND READMISSIONS
A postdischarge outpatient provider visit is often seen as a key factor in reducing readmissions. In 2013, Medicare added strength to this association by establishing transitional care management codes, which provide enhanced reimbursement to providers for a visit within 7 or 14 days of discharge, with focused attention on transitional issues.1 However, whether a postdischarge visit reduces readmissions remains unclear. Given evidence that higher primary care density is associated with lower healthcare utilization,2 CMS’s financial investment in incentivizing post discharge physician visits may be a good bet. On the other hand, simply having a primary care physician (PCP) may be a risk factor for readmission. This association suggests that postdischarge vigilance leads to identification of medical problems that lead to rehospitalization.3 This uncertainty is not resolved in systematic reviews of readmission reduction initiatives, which were not focused solely on the impact of a physician visit.4,5
The earliest study of postdischarge visits in a general medical population found an association between intensive outpatient follow-up by new providers in a Veterans Affairs population and an increase in hospital readmissions.6 This model is similar to some hospitalist models for postdischarge clinics, as the visit was with a noncontinuity provider. The largest recent study, of patients hospitalized with acute myocardial infarction, community-acquired pneumonia, or congestive heart failure (CHF) between 2009 and 2012, found increased frequency of postdischarge follow-up but no concomitant reduction in readmissions.7 Although small observational studies8 have found a postdischarge primary care visit may reduce the risk for readmission in general medical patients, the bulk of the recent data is negative.
In high-risk patients, however, there may be a clear benefit to postdischarge follow-up. In a North Carolina Medicaid population, a physician visit after discharge was associated with fewer readmissions among high-risk patients, but not among lower risk patients, whose readmission rates were low to start.9 The results of that study support the idea that risk stratification may identify patients who can benefit from more intensive outpatient follow-up. In general medical populations, existing studies may suffer from an absence of adequate risk assessment.
The evidence in specific disease states may show a clearer association between a postdischarge physician visit and reduced risk for readmission. One quarter of patients with CHF are rehospitalized within 30 days of discharge.10 In this disease with frequent exacerbations, a clinic visit to monitor volume status, weight, and medication adherence might reduce the frequency of readmissions or prolong the interval between rehospitalizations. A large observational study observed that earlier post discharge follow up by a cardiologist or a PCP was associated with lower risk of readmission, but only in the quintile with the closest follow-up. In addition, fewer than 40% of patients in this group had a visit within 7 days.11 In another heart failure population, follow-up with either a PCP or cardiologist within 7 days of discharge was again associated with lower risk for readmission.12 Thus, data suggest a protective effect of postdischarge visits in CHF patients, in contrast to a general medical population. Patients with end-stage renal disease may also fit in this group protected by a postdischarge physician visit, as 1 additional visit within the month after discharge was estimated to reduce rehospitalizations and produce significant cost savings.13
With other specific discharge diagnoses, results are varied. Two small observational studies in chronic obstructive pulmonary disease had conflicting results—one found a modest reduction in readmission and emergency department (ED) visits for patients seen by a PCP or pulmonologist within 30 days of discharge,14 and the other found no effect on readmissions but an associated reduction in mortality.15 More data are needed to clarify further the interaction of postdischarge visits with mortality, but the association between postdischarge physician visits and readmission reduction is controversial for patients with chronic obstructive pulmonary disease.
Finally, the evidence for dedicated postdischarge clinics is even more limited. A study of a hospitalist-led postdischarge clinic in a Veterans Affairs hospital found reduced length of stay and earlier postdischarge follow-up in a postdischarge clinic, but no effect on readmissions.16 Other studies have found earlier postdischarge follow-up with dedicated discharge clinics but have not evaluated readmission rates specifically.17In summary, the effect of postdischarge visits on risk for readmission is an area of active research, but remains unclear. The data reviewed suggest a benefit for the highest risk patients, specifically those with severe chronic illness, or those deemed high-risk with a readmission tool.9 At present, because physicians cannot accurately predict which patients will be readmitted,18 discharging physicians often take a broad approach and schedule outpatient visits for all patients. As readmission tools are further refined, the group of patients who will benefit from postdischarge care will be easier to identify, and a benefit to postdischarge visits may be seen
It is also important to note that this review emphasizes the physician visit and its potential impact on readmissions. Socioeconomic causes are increasingly being recognized as driving readmissions and other utilization.19 Whether an isolated physician visit is sufficient to prevent readmissions for patients with nonmedical drivers of healthcare utilization is unclear. For those patients, a discharge visit likely is a necessary component of a readmission reduction strategy for high-risk patients, but may be insufficient for patients who require not just an isolated visit but rather a more integrated and comprehensive care program.8,20,21
POSTDISCHARGE CLINIC MODELS
Despite the unclear relationship between postdischarge physician care and readmissions, dedicated postdischarge clinics, some staffed by hospitalists, have been adopted over the past 10 years. The three primary types of clinics arise in safety net environments, in academic medical centers, and as comprehensive high-risk patient solutions. Reviewing several types of clinics further clarifies the nature of this structural innovation.
Safety Net Hospital Models
Safety net hospitals and their hospitalists struggle with securing adequate postdischarge access for their population, which has inadequate insurance and poor access to primary care. Patient characteristics also play a role in the complex postdischarge care for this population, given its high rate of ED use (owing to perceived convenience and capabilities) for ambulatory-sensitive conditions.22 In addition, immigrants, particularly those with low English-language proficiency, underuse and have poor access to primary care.23,24 Postdischarge clinics in this environment focus first on providing a reliable postdischarge plan and then on linking to primary care. Examples of two clinics are at Harborview Medical Center in Seattle, Washington25 and Texas Health in Fort Worth.
Harborview is a 400-bed hospital affiliated with the University of Washington. More than 50% of its patients are considered indigent. The clinic was established in 2007 to provide a postdischarge option for uninsured patients, and a link to primary care in federally qualified health centers. The clinic was staffed 5 days a week with one or two hospitalists or advanced practice nurses. Visit duration was 20 minutes, 270 visits occurred per month, and the no-show rate was 30%. A small subgroup of the hospitalist group staffed the clinic. Particular clinical foci included CHF patients, patients with wound-care needs, and homeless, immigrant, and recently incarcerated patients. A key goal was connecting to longitudinal primary care, and the clinic successfully connected more than 70% of patients to primary care in community health centers. This clinic ultimately transitioned from a hospitalist practice to a primary care practice with a primary focus on post-ED follow-up for unaffiliated patients.26
In 2010, Texas Health faced a similar challenge with unaffiliated patients, and established a nurse practitioner–based clinic with hospitalist oversight to provide care primarily for patients without insurance or without an existing primary care relationship.
Academic Medical Center Models
Another clinical model is designed for patients who receive primary care at practices affiliated with academic medical centers. Although many of these patients have insurance and a PCP, there is often no availability with their continuity provider, because of the resident’s inpatient schedule or the faculty member’s conflicting priorities.27,28 Academic medical centers, including the University of California at San Francisco, the University of New Mexico, and the Beth Israel Deaconess Medical Center, have established discharge clinics within their faculty primary care practices. A model of this type of clinic was set up at Beth Israel Deaconess in 2010. Staffed by four hospitalists and using 40-minute appointments, this clinic was physically based in the primary care practice. As such, it took advantage of the existing clinic’s administrative and clinical functions, including triage, billing, and scheduling. A visit was scheduled in that clinic by the discharging physician team if a primary care appointment was not available with the patient’s continuity provider. Visits were standardized and focused on outstanding issues at discharge, medication reconciliation, and symptom trajectory. The hospitalists used the clinic’s clinical resources, including nurses, social workers, and pharmacists, but had no other dedicated staff. That there were only four hospitalists meant they were able to gain sufficient exposure to the outpatient setting, provide consistent high-quality care, and gain credibility with the PCPs. As the patients who were seen had PCPs of their own, during the visit significant attention was focused first on the postdischarge concerns, and then on promptly returning the patients to routine primary care. Significant patient outreach was used to address the clinic’s no-show rate, which was almost 50% in the early months. Within a year, the rate was down, closer to 20%. This clinic closed in 2015 after the primary care practice, in which it was based, transitioned to a patient-centered medical home. Since that time, this type of initiative has spread further, with neurohospitalist discharge clinics established, and postdischarge neurology follow-up becoming faster and more reliable.29
Academic medical centers and safety net hospitals substitute for routine primary care to address the basic challenge of primary care access, often without significant enhancements or additional resources, such as dedicated care management and pharmacy, social work, and nursing support. Commonalities of these clinics include dedicated physician staff, appointments generally longer than average outpatient appointments, and visit content concentrated on the key issues at transition (medication reconciliation, outstanding tests, symptom trajectory). As possible, clinics adopted a multidisciplinary approach, with social workers, community health workers, and nurses, to respond to the breadth of patients’ postdischarge needs, which often extend beyond pure medical need. The most frequent barriers encountered included the knowledge gap for hospitalist providers in the outpatient setting (a gap mitigated by using dedicated providers) and the patients’ high no-show rate (not surprising given that the providers are generally new to them). Few clinics have attempted to create continuity across inpatient and outpatient providers, though continuity might reduce no-shows as well as eliminate at least 1 transition.
Comprehensive High-Risk Patient Solutions
At the other end of the clinic spectrum are more integrated postdischarge approaches, which also evolved from the hospitalist model with hospitalist staffing. However, these approaches were introduced in response to the clinical needs of the highest risk patients (who are most vulnerable to frequent provider transitions), not to a systemic inability to provide routine postdischarge care.30
The most long-standing model for this type of clinic is represented by CareMore Health System, a subsidiary of Anthem.30-32 The extensivist, an expanded-scope hospitalist, acts as primary care coordinator, coordinating a multidisciplinary team for a panel of about 100 patients, representing the sickest 5% of the Medicare Advantage–insured population. Unlike the traditional hospitalist, the extensivist follows patients across all care sites, including hospital, rehabilitation sites, and outpatient clinic. For the most part, this relationship is not designed to evolve into a longitudinal relationship, but rather is an intervention only for the several-months period of acute need. Internal data have shown effects on hospital readmissions as well as length of stay.30
Another integrated clinic was established in 2013, at the University of Chicago. This was an effort to redesign care for patients at highest risk for hospitalization.33 Similar to the CareMore process, a high-risk population is identified by prior hospitalization and expected high Medicare costs. A comprehensive care physician cares for these patients across care settings. The clinic takes a team-based approach to patient care, with team members selected on the basis of patient need. Physicians have panels limited to only 200 patients, and generally spend part of the day in clinic, and part in seeing their hospitalized patients. Although reminiscent of a traditional primary care setting, this clinic is designed specifically for a high-risk, frequently hospitalized population, and therefore requires physicians with both a skill set akin to that of hospitalists, and an approach of palliative care and holistic patient care. Outcomes from this trial clinic are expected in 2017 or 2018.
LOGISTICAL CONSIDERATIONS FOR DISCHARGE CLINICS
Considering some key operational questions (Table) can help guide hospitals, hospitalists, and healthcare systems as they venture into the postdischarge clinic space. Return on investment and sustainability are two key questions for postdischarge clinics.
Return on investment varies by payment structure. In capitated environments with a strong emphasis on readmissions and total medical expenditure, a successful postdischarge clinic would recoup the investment through readmission reduction. However, maintaining adequate patient volume against high no-show rates may strain the group financially. In addition, although a hospitalist group may reap few measurable benefits from this clinical exposure, the unique view of the outpatient world afforded to hospitalists working in this environment could enrich the group as a whole by providing a more well-rounded vantage point.
Another key question surrounds sustainability. The clinic at the Beth Israel Deaconess Medical Center in Boston temporarily closed due to high inpatient volume and corresponding need for those hospitalists in the inpatient setting, early in its inception. It subsequently closed due to evolution in the clinic where it was based, rendering it unnecessary. Clinics that are contingent on other clinics will be vulnerable to external forces. Finally, staffing these clinics may be a stretch for a hospitalist group, as a partly different skill set is required for patient care in the outpatient setting. Hospitalists interested in care transitions are well suited for this role. In addition, hospitalists interested in more clinical variety, or in more schedule variety than that provided in a traditional hospitalist schedule, often enjoy the work. A vast majority of hospitalists think PCPs are responsible for postdischarge problems, and would not be interested in working in the postdischarge world.34 A poor fit for providers may lead to clinic failure.
As evident from this review, gaps in understanding the benefits of postdischarge care have persisted for 10 years. Discharge clinics have been scantly described in the literature. The primary unanswered question remains the effect on readmissions, but this has been the sole research focus to date. Other key research areas are the impact on other patient-centered clinical and system outcomes (eg, patient satisfaction, particularly for patients seeing new providers), postdischarge mortality, the effect on other adverse events, and total medical expenditure.
The healthcare system is evolving in the context of a focus on readmissions, primary care access challenges, and high-risk patients’ specific needs. These forces are spurring innovation in the realm of postdischarge physician clinics, as even the basic need for an appointment may not be met by the existing outpatient primary care system. In this context, multiple new outpatient care structures have arisen, many staffed by hospitalists. Some, such as clinics based in safety net hospitals and academic medical centers, address the simple requirement that patients who lack easy access, because of insurance status or provider availability, can see a doctor after discharge. This type of clinic may be an essential step in alleviating a strained system but may not represent a sustainable long-term solution. More comprehensive solutions for improving patient care and clinical outcomes may be offered by integrated systems, such as CareMore, which also emerged from the hospitalist model. A lasting question is whether these clinics, both the narrowly focused and the comprehensive, will have longevity in the evolving healthcare market. Inevitably, though, hospitalist directors will continue to raise such questions, and should stand to benefit from the experiences of others described in this review.
Disclosure
Nothing to report.
Readmission prevention is paramount for hospitals and, by extension, hospitalist programs. Hospitalists see early and reliable outpatient follow-up as a safe landing for their most complicated patient cases. The option of a postdischarge clinic arises from the challenge to arrange adequate postdischarge care for patients who lack easy access because of insurance or provider availability. Guaranteeing postdischarge access by opening a dedicated, hospitalist-led postdischarge clinic appears to be an easy solution, but it is a solution that requires significant investment (including investment in physician and staff training and administrative support) and careful navigation of existing primary care relationships. In addition, a clinic staffed only with physicians may not be well equipped to address the complex social factors in healthcare utilization and readmission. Better understanding of the evidence supporting post discharge physician visits, several models of clinics, and the key operational questions are essential to address before crossing the inpatient-outpatient divide.
POSTDISCHARGE PHYSICIAN VISITS AND READMISSIONS
A postdischarge outpatient provider visit is often seen as a key factor in reducing readmissions. In 2013, Medicare added strength to this association by establishing transitional care management codes, which provide enhanced reimbursement to providers for a visit within 7 or 14 days of discharge, with focused attention on transitional issues.1 However, whether a postdischarge visit reduces readmissions remains unclear. Given evidence that higher primary care density is associated with lower healthcare utilization,2 CMS’s financial investment in incentivizing post discharge physician visits may be a good bet. On the other hand, simply having a primary care physician (PCP) may be a risk factor for readmission. This association suggests that postdischarge vigilance leads to identification of medical problems that lead to rehospitalization.3 This uncertainty is not resolved in systematic reviews of readmission reduction initiatives, which were not focused solely on the impact of a physician visit.4,5
The earliest study of postdischarge visits in a general medical population found an association between intensive outpatient follow-up by new providers in a Veterans Affairs population and an increase in hospital readmissions.6 This model is similar to some hospitalist models for postdischarge clinics, as the visit was with a noncontinuity provider. The largest recent study, of patients hospitalized with acute myocardial infarction, community-acquired pneumonia, or congestive heart failure (CHF) between 2009 and 2012, found increased frequency of postdischarge follow-up but no concomitant reduction in readmissions.7 Although small observational studies8 have found a postdischarge primary care visit may reduce the risk for readmission in general medical patients, the bulk of the recent data is negative.
In high-risk patients, however, there may be a clear benefit to postdischarge follow-up. In a North Carolina Medicaid population, a physician visit after discharge was associated with fewer readmissions among high-risk patients, but not among lower risk patients, whose readmission rates were low to start.9 The results of that study support the idea that risk stratification may identify patients who can benefit from more intensive outpatient follow-up. In general medical populations, existing studies may suffer from an absence of adequate risk assessment.
The evidence in specific disease states may show a clearer association between a postdischarge physician visit and reduced risk for readmission. One quarter of patients with CHF are rehospitalized within 30 days of discharge.10 In this disease with frequent exacerbations, a clinic visit to monitor volume status, weight, and medication adherence might reduce the frequency of readmissions or prolong the interval between rehospitalizations. A large observational study observed that earlier post discharge follow up by a cardiologist or a PCP was associated with lower risk of readmission, but only in the quintile with the closest follow-up. In addition, fewer than 40% of patients in this group had a visit within 7 days.11 In another heart failure population, follow-up with either a PCP or cardiologist within 7 days of discharge was again associated with lower risk for readmission.12 Thus, data suggest a protective effect of postdischarge visits in CHF patients, in contrast to a general medical population. Patients with end-stage renal disease may also fit in this group protected by a postdischarge physician visit, as 1 additional visit within the month after discharge was estimated to reduce rehospitalizations and produce significant cost savings.13
With other specific discharge diagnoses, results are varied. Two small observational studies in chronic obstructive pulmonary disease had conflicting results—one found a modest reduction in readmission and emergency department (ED) visits for patients seen by a PCP or pulmonologist within 30 days of discharge,14 and the other found no effect on readmissions but an associated reduction in mortality.15 More data are needed to clarify further the interaction of postdischarge visits with mortality, but the association between postdischarge physician visits and readmission reduction is controversial for patients with chronic obstructive pulmonary disease.
Finally, the evidence for dedicated postdischarge clinics is even more limited. A study of a hospitalist-led postdischarge clinic in a Veterans Affairs hospital found reduced length of stay and earlier postdischarge follow-up in a postdischarge clinic, but no effect on readmissions.16 Other studies have found earlier postdischarge follow-up with dedicated discharge clinics but have not evaluated readmission rates specifically.17In summary, the effect of postdischarge visits on risk for readmission is an area of active research, but remains unclear. The data reviewed suggest a benefit for the highest risk patients, specifically those with severe chronic illness, or those deemed high-risk with a readmission tool.9 At present, because physicians cannot accurately predict which patients will be readmitted,18 discharging physicians often take a broad approach and schedule outpatient visits for all patients. As readmission tools are further refined, the group of patients who will benefit from postdischarge care will be easier to identify, and a benefit to postdischarge visits may be seen
It is also important to note that this review emphasizes the physician visit and its potential impact on readmissions. Socioeconomic causes are increasingly being recognized as driving readmissions and other utilization.19 Whether an isolated physician visit is sufficient to prevent readmissions for patients with nonmedical drivers of healthcare utilization is unclear. For those patients, a discharge visit likely is a necessary component of a readmission reduction strategy for high-risk patients, but may be insufficient for patients who require not just an isolated visit but rather a more integrated and comprehensive care program.8,20,21
POSTDISCHARGE CLINIC MODELS
Despite the unclear relationship between postdischarge physician care and readmissions, dedicated postdischarge clinics, some staffed by hospitalists, have been adopted over the past 10 years. The three primary types of clinics arise in safety net environments, in academic medical centers, and as comprehensive high-risk patient solutions. Reviewing several types of clinics further clarifies the nature of this structural innovation.
Safety Net Hospital Models
Safety net hospitals and their hospitalists struggle with securing adequate postdischarge access for their population, which has inadequate insurance and poor access to primary care. Patient characteristics also play a role in the complex postdischarge care for this population, given its high rate of ED use (owing to perceived convenience and capabilities) for ambulatory-sensitive conditions.22 In addition, immigrants, particularly those with low English-language proficiency, underuse and have poor access to primary care.23,24 Postdischarge clinics in this environment focus first on providing a reliable postdischarge plan and then on linking to primary care. Examples of two clinics are at Harborview Medical Center in Seattle, Washington25 and Texas Health in Fort Worth.
Harborview is a 400-bed hospital affiliated with the University of Washington. More than 50% of its patients are considered indigent. The clinic was established in 2007 to provide a postdischarge option for uninsured patients, and a link to primary care in federally qualified health centers. The clinic was staffed 5 days a week with one or two hospitalists or advanced practice nurses. Visit duration was 20 minutes, 270 visits occurred per month, and the no-show rate was 30%. A small subgroup of the hospitalist group staffed the clinic. Particular clinical foci included CHF patients, patients with wound-care needs, and homeless, immigrant, and recently incarcerated patients. A key goal was connecting to longitudinal primary care, and the clinic successfully connected more than 70% of patients to primary care in community health centers. This clinic ultimately transitioned from a hospitalist practice to a primary care practice with a primary focus on post-ED follow-up for unaffiliated patients.26
In 2010, Texas Health faced a similar challenge with unaffiliated patients, and established a nurse practitioner–based clinic with hospitalist oversight to provide care primarily for patients without insurance or without an existing primary care relationship.
Academic Medical Center Models
Another clinical model is designed for patients who receive primary care at practices affiliated with academic medical centers. Although many of these patients have insurance and a PCP, there is often no availability with their continuity provider, because of the resident’s inpatient schedule or the faculty member’s conflicting priorities.27,28 Academic medical centers, including the University of California at San Francisco, the University of New Mexico, and the Beth Israel Deaconess Medical Center, have established discharge clinics within their faculty primary care practices. A model of this type of clinic was set up at Beth Israel Deaconess in 2010. Staffed by four hospitalists and using 40-minute appointments, this clinic was physically based in the primary care practice. As such, it took advantage of the existing clinic’s administrative and clinical functions, including triage, billing, and scheduling. A visit was scheduled in that clinic by the discharging physician team if a primary care appointment was not available with the patient’s continuity provider. Visits were standardized and focused on outstanding issues at discharge, medication reconciliation, and symptom trajectory. The hospitalists used the clinic’s clinical resources, including nurses, social workers, and pharmacists, but had no other dedicated staff. That there were only four hospitalists meant they were able to gain sufficient exposure to the outpatient setting, provide consistent high-quality care, and gain credibility with the PCPs. As the patients who were seen had PCPs of their own, during the visit significant attention was focused first on the postdischarge concerns, and then on promptly returning the patients to routine primary care. Significant patient outreach was used to address the clinic’s no-show rate, which was almost 50% in the early months. Within a year, the rate was down, closer to 20%. This clinic closed in 2015 after the primary care practice, in which it was based, transitioned to a patient-centered medical home. Since that time, this type of initiative has spread further, with neurohospitalist discharge clinics established, and postdischarge neurology follow-up becoming faster and more reliable.29
Academic medical centers and safety net hospitals substitute for routine primary care to address the basic challenge of primary care access, often without significant enhancements or additional resources, such as dedicated care management and pharmacy, social work, and nursing support. Commonalities of these clinics include dedicated physician staff, appointments generally longer than average outpatient appointments, and visit content concentrated on the key issues at transition (medication reconciliation, outstanding tests, symptom trajectory). As possible, clinics adopted a multidisciplinary approach, with social workers, community health workers, and nurses, to respond to the breadth of patients’ postdischarge needs, which often extend beyond pure medical need. The most frequent barriers encountered included the knowledge gap for hospitalist providers in the outpatient setting (a gap mitigated by using dedicated providers) and the patients’ high no-show rate (not surprising given that the providers are generally new to them). Few clinics have attempted to create continuity across inpatient and outpatient providers, though continuity might reduce no-shows as well as eliminate at least 1 transition.
Comprehensive High-Risk Patient Solutions
At the other end of the clinic spectrum are more integrated postdischarge approaches, which also evolved from the hospitalist model with hospitalist staffing. However, these approaches were introduced in response to the clinical needs of the highest risk patients (who are most vulnerable to frequent provider transitions), not to a systemic inability to provide routine postdischarge care.30
The most long-standing model for this type of clinic is represented by CareMore Health System, a subsidiary of Anthem.30-32 The extensivist, an expanded-scope hospitalist, acts as primary care coordinator, coordinating a multidisciplinary team for a panel of about 100 patients, representing the sickest 5% of the Medicare Advantage–insured population. Unlike the traditional hospitalist, the extensivist follows patients across all care sites, including hospital, rehabilitation sites, and outpatient clinic. For the most part, this relationship is not designed to evolve into a longitudinal relationship, but rather is an intervention only for the several-months period of acute need. Internal data have shown effects on hospital readmissions as well as length of stay.30
Another integrated clinic was established in 2013, at the University of Chicago. This was an effort to redesign care for patients at highest risk for hospitalization.33 Similar to the CareMore process, a high-risk population is identified by prior hospitalization and expected high Medicare costs. A comprehensive care physician cares for these patients across care settings. The clinic takes a team-based approach to patient care, with team members selected on the basis of patient need. Physicians have panels limited to only 200 patients, and generally spend part of the day in clinic, and part in seeing their hospitalized patients. Although reminiscent of a traditional primary care setting, this clinic is designed specifically for a high-risk, frequently hospitalized population, and therefore requires physicians with both a skill set akin to that of hospitalists, and an approach of palliative care and holistic patient care. Outcomes from this trial clinic are expected in 2017 or 2018.
LOGISTICAL CONSIDERATIONS FOR DISCHARGE CLINICS
Considering some key operational questions (Table) can help guide hospitals, hospitalists, and healthcare systems as they venture into the postdischarge clinic space. Return on investment and sustainability are two key questions for postdischarge clinics.
Return on investment varies by payment structure. In capitated environments with a strong emphasis on readmissions and total medical expenditure, a successful postdischarge clinic would recoup the investment through readmission reduction. However, maintaining adequate patient volume against high no-show rates may strain the group financially. In addition, although a hospitalist group may reap few measurable benefits from this clinical exposure, the unique view of the outpatient world afforded to hospitalists working in this environment could enrich the group as a whole by providing a more well-rounded vantage point.
Another key question surrounds sustainability. The clinic at the Beth Israel Deaconess Medical Center in Boston temporarily closed due to high inpatient volume and corresponding need for those hospitalists in the inpatient setting, early in its inception. It subsequently closed due to evolution in the clinic where it was based, rendering it unnecessary. Clinics that are contingent on other clinics will be vulnerable to external forces. Finally, staffing these clinics may be a stretch for a hospitalist group, as a partly different skill set is required for patient care in the outpatient setting. Hospitalists interested in care transitions are well suited for this role. In addition, hospitalists interested in more clinical variety, or in more schedule variety than that provided in a traditional hospitalist schedule, often enjoy the work. A vast majority of hospitalists think PCPs are responsible for postdischarge problems, and would not be interested in working in the postdischarge world.34 A poor fit for providers may lead to clinic failure.
As evident from this review, gaps in understanding the benefits of postdischarge care have persisted for 10 years. Discharge clinics have been scantly described in the literature. The primary unanswered question remains the effect on readmissions, but this has been the sole research focus to date. Other key research areas are the impact on other patient-centered clinical and system outcomes (eg, patient satisfaction, particularly for patients seeing new providers), postdischarge mortality, the effect on other adverse events, and total medical expenditure.
The healthcare system is evolving in the context of a focus on readmissions, primary care access challenges, and high-risk patients’ specific needs. These forces are spurring innovation in the realm of postdischarge physician clinics, as even the basic need for an appointment may not be met by the existing outpatient primary care system. In this context, multiple new outpatient care structures have arisen, many staffed by hospitalists. Some, such as clinics based in safety net hospitals and academic medical centers, address the simple requirement that patients who lack easy access, because of insurance status or provider availability, can see a doctor after discharge. This type of clinic may be an essential step in alleviating a strained system but may not represent a sustainable long-term solution. More comprehensive solutions for improving patient care and clinical outcomes may be offered by integrated systems, such as CareMore, which also emerged from the hospitalist model. A lasting question is whether these clinics, both the narrowly focused and the comprehensive, will have longevity in the evolving healthcare market. Inevitably, though, hospitalist directors will continue to raise such questions, and should stand to benefit from the experiences of others described in this review.
Disclosure
Nothing to report.
1. US Department of Health and Human Services, Centers for Medicare & Medicaid Services. Transitional Care Management Services. https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNProducts/Downloads/Transitional-Care-Management-Services-Fact-Sheet-ICN908628.pdf. Fact sheet ICN 908628.. Accessed June 29, 2016.
2. Kravet SJ, Shore AD, Miller R, Green GB, Kolodner K, Wright SM. Health care utilization and the proportion of primary care physicians. Am J Med. 2008;121(2):142-148. PubMed
3. Hasan O, Meltzer DO, Shaykevich SA, et al. Hospital readmission in general medicine patients: a prediction model. J Gen Intern Med. 2010;25(3):211-219. PubMed
4. Hansen LO, Young RS, Hinami K, Leung A, Williams MV. Interventions to reduce 30-day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520-528. PubMed
5. Leppin AL, Gionfriddo MR, Kessler M, et al. Preventing 30-day hospital readmissions: a systematic review and meta-analysis of randomized trials. JAMA Intern Med. 2014;174(7):1095-1107. PubMed
6. Weinberger M, Oddone EZ, Henderson WG. Does increased access to primary care reduce hospital readmissions? Veterans Affairs Cooperative Study Group on Primary Care and Hospital Readmission. N Engl J Med. 1996;334(22):1441-1447. PubMed
7. DeLia D, Tong J, Gaboda D, Casalino LP. Post-discharge follow-up visits and hospital utilization by Medicare patients, 2007-2010. Medicare Medicaid Res Rev. 2014;4(2). PubMed
8. Dedhia P, Kravet S, Bulger J, et al. A quality improvement intervention to facilitate the transition of older adults from three hospitals back to their homes. J Am Geriatr Soc. 2009;57(9):1540-1546. PubMed
9. Jackson C, Shahsahebi M, Wedlake T, DuBard CA. Timeliness of outpatient follow-up: an evidence-based approach for planning after hospital discharge. Ann Fam Med. 2015;13(2):115-122. PubMed
10. Dharmarajan K, Hsieh AF, Lin Z, et al. Diagnoses and timing of 30-day readmissions after hospitalization for heart failure, acute myocardial infarction, or pneumonia. JAMA. 2013;309(4):355-363. PubMed
11. Hernandez AF, Greiner MA, Fonarow GC, et al. Relationship between early physician follow-up and 30-day readmission among Medicare beneficiaries hospitalized for heart failure. JAMA. 2010;303(17):1716-1722. PubMed
12. Lee KK, Yang J, Hernandez AF, Steimle AE, Go AS. Post-discharge follow-up characteristics associated with 30-day readmission after heart failure hospitalization. Med Care. 2016;54(4):365-372. PubMed
13. Erickson KF, Winkelmayer WC, Chertow GM, Bhattacharya J. Physician visits and 30-day hospital readmissions in patients receiving hemodialysis. J Am Soc Nephrol. 2014;25(9):2079-2087. PubMed
14. Sharma G, Kuo YF, Freeman JL, Zhang DD, Goodwin JS. Outpatient follow-up visit and 30-day emergency department visit and readmission in patients hospitalized for chronic obstructive pulmonary disease. Arch Intern Med. 2010;170(18):1664-1670. PubMed
15. Fidahussein SS, Croghan IT, Cha SS, Klocke DL. Posthospital follow-up visits and 30-day readmission rates in chronic obstructive pulmonary disease. Risk Manag Healthc Policy. 2014;7:105-112. PubMed
16. Burke RE, Whitfield E, Prochazka AV. Effect of a hospitalist-run postdischarge clinic on outcomes. J Hosp Med. 2014;9(1):7-12. PubMed
17. Doctoroff L, Nijhawan A, McNally D, Vanka A, Yu R, Mukamal KJ. The characteristics and impact of a hospitalist-staffed post-discharge clinic. Am J Med. 2013;126(11):1016.e9-e15. PubMed
18. Allaudeen N, Schnipper JL, Orav EJ, Wachter RM, Vidyarthi AR. Inability of providers to predict unplanned readmissions. J Gen Intern Med. 2011;26(7):771-776. PubMed
19. Barnett ML, Hsu J, McWilliams J. Patient characteristics and differences in hospital readmission rates. JAMA Intern Med. 2015;175(11):1803-1812. PubMed
20. Jack BW, Chetty VK, Anthony D, et al. A reengineered hospital discharge program to decrease rehospitalization: a randomized trial. Ann Intern Med. 2009;150(3):178-187. PubMed
21. Naylor M, Brooten D, Jones R, Lavizzo-Mourey R, Mezey M, Pauly M. Comprehensive discharge planning for the hospitalized elderly. A randomized clinical trial. Ann Intern Med. 1994;120(12):999-1006. PubMed
22. Capp R, Camp-Binford M, Sobolewski S, Bulmer S, Kelley L. Do adult Medicaid enrollees prefer going to their primary care provider’s clinic rather than emergency department (ED) for low acuity conditions? Med Care. 2015;53(6):530-533. PubMed
23. Vargas Bustamante A, Fang H, Garza J, et al. Variations in healthcare access and utilization among Mexican immigrants: the role of documentation status. J Immigr Minor Health. 2012;14(1):146-155. PubMed
24. Chi JT, Handcock MS. Identifying sources of health care underutilization among California’s immigrants. J Racial Ethn Health Disparities. 2014;1(3):207-218. PubMed
25. Martinez S. Bridging the Gap: Discharge Clinics Providing Safe Transitions for High Risk Patients. Workshop presented at: Northwest Patient Safety Conference; May 15, 2012; Seattle, WA. http://www.wapatientsafety.org/downloads/Martinez.pdf. Published 2011. Accessed April 26, 2017.
26. Elliott K, W Klein J, Basu A, Sabbatini AK. Transitional care clinics for follow-up and primary care linkage for patients discharged from the ED. Am J Emerg Med. 2016;34(7):1230-1235. PubMed
27. Baxley EG, Weir S. Advanced access in academic settings: definitional challenges. Ann Fam Med. 2009;7(1):90-91. PubMed
28. Doctoroff L, McNally D, Vanka A, Nall R, Mukamal KJ. Inpatient–outpatient transitions for patients with resident primary care physicians: access and readmission. Am J Med. 2014;127(9):886.e15-e20. PubMed
29. Shah M, Douglas V, Scott B, Josephson SA. A neurohospitalist discharge clinic shortens the transition from inpatient to outpatient care. Neurohospitalist. 2016;6(2):64-69. PubMed
30. Powers BW, Milstein A, Jain SH. Delivery models for high-risk older patients: back to the future? JAMA. 2016;315(1):23-24. PubMed
31. Milstein A, Gilbertson E. American medical home runs. Health Aff (Millwood). 2009;28(5):1317-1326. PubMed
32. Reuben DB. Physicians in supporting roles in chronic disease care: the CareMore model. J Am Geriatr Soc. 2011;59(1):158-160. PubMed
33. Meltzer DO, Ruhnke GW. Redesigning care for patients at increased hospitalization risk: the comprehensive care physician model. Health Aff (Millwood). 2014;33(5):770-777. PubMed
34. Burke RE, Ryan P. Postdischarge clinics: hospitalist attitudes and experiences. J Hosp Med. 2013;8(10):578-581. PubMed
1. US Department of Health and Human Services, Centers for Medicare & Medicaid Services. Transitional Care Management Services. https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNProducts/Downloads/Transitional-Care-Management-Services-Fact-Sheet-ICN908628.pdf. Fact sheet ICN 908628.. Accessed June 29, 2016.
2. Kravet SJ, Shore AD, Miller R, Green GB, Kolodner K, Wright SM. Health care utilization and the proportion of primary care physicians. Am J Med. 2008;121(2):142-148. PubMed
3. Hasan O, Meltzer DO, Shaykevich SA, et al. Hospital readmission in general medicine patients: a prediction model. J Gen Intern Med. 2010;25(3):211-219. PubMed
4. Hansen LO, Young RS, Hinami K, Leung A, Williams MV. Interventions to reduce 30-day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520-528. PubMed
5. Leppin AL, Gionfriddo MR, Kessler M, et al. Preventing 30-day hospital readmissions: a systematic review and meta-analysis of randomized trials. JAMA Intern Med. 2014;174(7):1095-1107. PubMed
6. Weinberger M, Oddone EZ, Henderson WG. Does increased access to primary care reduce hospital readmissions? Veterans Affairs Cooperative Study Group on Primary Care and Hospital Readmission. N Engl J Med. 1996;334(22):1441-1447. PubMed
7. DeLia D, Tong J, Gaboda D, Casalino LP. Post-discharge follow-up visits and hospital utilization by Medicare patients, 2007-2010. Medicare Medicaid Res Rev. 2014;4(2). PubMed
8. Dedhia P, Kravet S, Bulger J, et al. A quality improvement intervention to facilitate the transition of older adults from three hospitals back to their homes. J Am Geriatr Soc. 2009;57(9):1540-1546. PubMed
9. Jackson C, Shahsahebi M, Wedlake T, DuBard CA. Timeliness of outpatient follow-up: an evidence-based approach for planning after hospital discharge. Ann Fam Med. 2015;13(2):115-122. PubMed
10. Dharmarajan K, Hsieh AF, Lin Z, et al. Diagnoses and timing of 30-day readmissions after hospitalization for heart failure, acute myocardial infarction, or pneumonia. JAMA. 2013;309(4):355-363. PubMed
11. Hernandez AF, Greiner MA, Fonarow GC, et al. Relationship between early physician follow-up and 30-day readmission among Medicare beneficiaries hospitalized for heart failure. JAMA. 2010;303(17):1716-1722. PubMed
12. Lee KK, Yang J, Hernandez AF, Steimle AE, Go AS. Post-discharge follow-up characteristics associated with 30-day readmission after heart failure hospitalization. Med Care. 2016;54(4):365-372. PubMed
13. Erickson KF, Winkelmayer WC, Chertow GM, Bhattacharya J. Physician visits and 30-day hospital readmissions in patients receiving hemodialysis. J Am Soc Nephrol. 2014;25(9):2079-2087. PubMed
14. Sharma G, Kuo YF, Freeman JL, Zhang DD, Goodwin JS. Outpatient follow-up visit and 30-day emergency department visit and readmission in patients hospitalized for chronic obstructive pulmonary disease. Arch Intern Med. 2010;170(18):1664-1670. PubMed
15. Fidahussein SS, Croghan IT, Cha SS, Klocke DL. Posthospital follow-up visits and 30-day readmission rates in chronic obstructive pulmonary disease. Risk Manag Healthc Policy. 2014;7:105-112. PubMed
16. Burke RE, Whitfield E, Prochazka AV. Effect of a hospitalist-run postdischarge clinic on outcomes. J Hosp Med. 2014;9(1):7-12. PubMed
17. Doctoroff L, Nijhawan A, McNally D, Vanka A, Yu R, Mukamal KJ. The characteristics and impact of a hospitalist-staffed post-discharge clinic. Am J Med. 2013;126(11):1016.e9-e15. PubMed
18. Allaudeen N, Schnipper JL, Orav EJ, Wachter RM, Vidyarthi AR. Inability of providers to predict unplanned readmissions. J Gen Intern Med. 2011;26(7):771-776. PubMed
19. Barnett ML, Hsu J, McWilliams J. Patient characteristics and differences in hospital readmission rates. JAMA Intern Med. 2015;175(11):1803-1812. PubMed
20. Jack BW, Chetty VK, Anthony D, et al. A reengineered hospital discharge program to decrease rehospitalization: a randomized trial. Ann Intern Med. 2009;150(3):178-187. PubMed
21. Naylor M, Brooten D, Jones R, Lavizzo-Mourey R, Mezey M, Pauly M. Comprehensive discharge planning for the hospitalized elderly. A randomized clinical trial. Ann Intern Med. 1994;120(12):999-1006. PubMed
22. Capp R, Camp-Binford M, Sobolewski S, Bulmer S, Kelley L. Do adult Medicaid enrollees prefer going to their primary care provider’s clinic rather than emergency department (ED) for low acuity conditions? Med Care. 2015;53(6):530-533. PubMed
23. Vargas Bustamante A, Fang H, Garza J, et al. Variations in healthcare access and utilization among Mexican immigrants: the role of documentation status. J Immigr Minor Health. 2012;14(1):146-155. PubMed
24. Chi JT, Handcock MS. Identifying sources of health care underutilization among California’s immigrants. J Racial Ethn Health Disparities. 2014;1(3):207-218. PubMed
25. Martinez S. Bridging the Gap: Discharge Clinics Providing Safe Transitions for High Risk Patients. Workshop presented at: Northwest Patient Safety Conference; May 15, 2012; Seattle, WA. http://www.wapatientsafety.org/downloads/Martinez.pdf. Published 2011. Accessed April 26, 2017.
26. Elliott K, W Klein J, Basu A, Sabbatini AK. Transitional care clinics for follow-up and primary care linkage for patients discharged from the ED. Am J Emerg Med. 2016;34(7):1230-1235. PubMed
27. Baxley EG, Weir S. Advanced access in academic settings: definitional challenges. Ann Fam Med. 2009;7(1):90-91. PubMed
28. Doctoroff L, McNally D, Vanka A, Nall R, Mukamal KJ. Inpatient–outpatient transitions for patients with resident primary care physicians: access and readmission. Am J Med. 2014;127(9):886.e15-e20. PubMed
29. Shah M, Douglas V, Scott B, Josephson SA. A neurohospitalist discharge clinic shortens the transition from inpatient to outpatient care. Neurohospitalist. 2016;6(2):64-69. PubMed
30. Powers BW, Milstein A, Jain SH. Delivery models for high-risk older patients: back to the future? JAMA. 2016;315(1):23-24. PubMed
31. Milstein A, Gilbertson E. American medical home runs. Health Aff (Millwood). 2009;28(5):1317-1326. PubMed
32. Reuben DB. Physicians in supporting roles in chronic disease care: the CareMore model. J Am Geriatr Soc. 2011;59(1):158-160. PubMed
33. Meltzer DO, Ruhnke GW. Redesigning care for patients at increased hospitalization risk: the comprehensive care physician model. Health Aff (Millwood). 2014;33(5):770-777. PubMed
34. Burke RE, Ryan P. Postdischarge clinics: hospitalist attitudes and experiences. J Hosp Med. 2013;8(10):578-581. PubMed
© 2017 Society of Hospital Medicine
Forgotten but not gone: Update on measles infection for hospitalists
Measles is a highly contagious acute respiratory illness that includes a characteristic rash. After exposure, up to 90% of susceptible persons develop measles.1 Even though it is considered a childhood illness, measles can affect people of all age groups. Measles continues to be major health problem around the world, despite the availability of a safe and effective vaccine, and it remains one of the leading causes of childhood mortality, with nearly 115,000 deaths reported by the World Health Organization2 in 2014. In 2000, measles was declared eliminated from the United States, but outbreaks still occasionally occur.3-6
The disease is self-limited, but some patients develop complications that may require hospitalization for treatment. People at highest risk for complications are children younger than 5 years, adults older than 20 years, pregnant women, and immunocompromised individuals.7
HISTORY AND EPIDEMIOLOGY
During the licensure of live measles vaccine in 1963, an average of 549,000 measles cases and 495 measles deaths, as well as 48,000 hospitalizations and 4000 encephalitis cases, were reported annually in the United States. Almost all Americans were affected by measles by adolescence.
Implementation of the 1-dose vaccine program substantially reduced reported incidence in the United States by 1988, and led to a dramatic decline in measles-related hospitalizations and deaths.3-6 The 2-dose MMR (measles, mumps, rubella) vaccination was introduced in 1989, and measles was declared eliminated in the United States in 2000.3-6
National–level one-dose MMR coverage among children 19-35 months has remained above 90% during the last two decades.8 NIS-Teen vaccination coverage data for 13- to 17-year-olds since 2008 has been near or above 90%,9 and 94% of children enrolled in kindergarten had evidence of 2 MMR doses in the 2014-2015 school year.10
A large multistate measles outbreak was reported in the United States in 2014-2015.4,11 One hundred fifty-nine cases were reported in the United States between January 4 and April 5, 2015. The majority of patients either were unvaccinated (45%) or had an unknown vaccination status (38%). Age ranged from 6 weeks to 70 years, and 22 patients (14%) were hospitalized.4

CLINICAL PRESENTATION AND PATHOPHYSIOLOGY
Measles is caused by an RNA-containing paramyxovirus that is spread by the respiratory route. Average incubation period from exposure to rash onset is 14 days (range, 7-21 days).12,13 Peak infectivity occurs during the prodromal phase, before rash onset (Figure 1), but patients are infectious from 4 days before rash onset through 4 days after rash onset.7,12,13
The disease prodrome consists of a high fever (39°C-40.5°C), coryza, cough, and conjunctivitis followed by Koplik spots (Figure 2A). Koplik spots are pathognomonic for measles but rarely discovered. They appear before the skin rash alongside second molars on the buccal surface of the cheeks. The spots usually disappear when the characteristic maculopapular, nonpruritic rash erupts initially at the hairline and behind the ears, and within four days progresses toward the trunk and limbs, including the palms and soles (Figures 2B, 2C).
 Pathognomonic buccal exanthem, Koplik spots. (B) Typical small, reddish, flat, macular and papular exanthemous rash on head and neck of patient with measles infection. (C) Rash spreads to arms, back, upper trunk, and legs.")
The patient remains febrile while the rash spreads.12,13 Usually the fever resolves while the rash fades in the same order in which it appeared. Fever that persists for more than 5 days usually indicates complications.13
Cellular immunity plays an important role in host defense; the virus invades T lymphocytes and triggers suppressive cytokine (interleukin 4) production. Leukopenia, expansion of mainly measles-specific T and B lymphocytes, and replacement of lymphocyte memory cell population results in further depression of cellular immunity, and predisposes patients to secondary bacterial infections for up to 2 years after measles infection.14,15
Patients immunocompromised by congenital cellular immunity deficiency, cancer, human immunodeficiency virus (HIV) infection without effective antiretroviral therapy, or immunosuppression treatment are at higher risk for developing severe complications or dying from measles. As the rash may fail to develop in these patients, diagnosis can be challenging.16
Modified measles is milder and may occur in patients with preexisting partial immunity: those with an immunization history (2-dose vaccine effectiveness is ∼97%), and infants with minimal immunity from their mothers.1,7 Patients may have mild respiratory symptoms with rash but little or no fever.7
Atypical measles is now extremely rare. It was described only among people who were vaccinated with the killed vaccine in the United States between 1963 and 1968 and subsequently exposed to measles. The disease is characterized by high fever, edema of extremities, and a rash that develops on the palms and soles and spreads centerward. It is considered noncommunicable.17
Measles infection during pregnancy is associated with increased maternal and fetal morbidity. The virus can induce neonatal low birth weight, spontaneous abortion, intrauterine fetal death, and maternal death. Pregnant women with measles are more likely to be hospitalized.18,19
DIFFERENTIAL DIAGNOSIS
The presenting symptoms of primary measles infection are nonspecific, particularly if Koplik spots are not identified. The differential diagnosis for a patient who presents with high fever and rash include Kawasaki disease, dengue, parvovirus B19, serum sickness, syphilis, systemic lupus erythematous, toxic shock syndrome, enterovirus infection, human herpes virus 6 (roseola), viral hemorrhagic fever, drug eruption, infectious mononucleosis, Rocky Mountain spotted fever, rubella, scarlet fever, chikungunya, and Zika virus infection.
COMPLICATIONS
Measles complications can affect nearly every organ system (Table). Rates of complications from measles infection depend on age and underlying condition. Coexisting vitamin A deficiency increases complication rates.20

Bacterial infections in the setting of measles infection are more common in adults than in children, and are more severe among people who are malnourished or have an immunodeficiency disorder. The most common infectious complications, which involve the respiratory tract, include pneumonia, laryngotracheitis (“measles croup”), bronchitis, otitis media (most common complication among children in the United States), and sinusitis.7,13,21
Indications for hospitalizing children include respiratory distress, laryngeal obstruction, dehydration that requires intravenous fluids, diarrhea with more than 10 stools a day or bloody stool, severe anemia, altered mental status, convulsion, severe rash with developing hemorrhagic areas, extensive mouth ulcers, corneal clouding or ulcers, visual disturbance, and mastoiditis.22
Pneumonia is a common indication for hospitalizing adults.23,24 Measles-associated interstitial giant cell (Hecht) pneumonia is most often recognized among immunocompromised and malnourished patients.13 Primary pneumonia is caused by the measles virus, but bacterial superinfection can occur. The most common bacterial pathogens include Streptococcus, Pneumococcus, and Staphylococcus,13,24 and less commonly isolated organisms include gram-negative bacteria, such as Haemophilus influenzae, Pseudomonas aeruginosa, Neisseria meningitides, and Enterobacter cloacae.23
Uncommon complications of measles are myocarditis, glomerulonephritis, acute renal failure, and thrombocytopenic purpura.25,26
Neurologic complications in measles are an important concern. Measles-associated central nervous system complications are considered a result of an immune-mediated reaction to myelin protein and not from direct viral insult.26-28 Immunocompromised patients are at risk for developing fatal encephalitis, and those who survive often experience cognitive decline or seizures.
Measles is associated with four different encephalitic diseases: primary measles encephalitis, acute post-measles encephalomyelitis, measles inclusion body encephalitis, and subacute sclerosing panencephalitis.
Primary measles encephalitis is characterized by fever, headache, stiff neck, and meningeal signs. Onset occurs between 1 and 15 days after rash onset, and the disease affects 1/1000 patients. Seizure, altered mental status, and coma can also develop. Viral RNA detection in the cerebrospinal fluid (CSF) confirms the diagnosis.29Acute post-measles encephalomyelitis is more common in adults than in children.12 It typically develops after the rash fades and the other symptoms subside. Patients suddenly experience a recurrence of fevers or seizures. Deafness, intellectual decline, epilepsy, postencephalitic hyperkinesia, hemiplegia, and/or paraplegia also can develop.27-29
Measles inclusion body encephalitis is described only in immunocompromised patients, and onset occurs within 1 year of infection. Seizures are an initial and common symptom, and some patients also experience hemiplegia, stupor, hypertonia, and dysarthria.29 Diagnostic findings include seroconversion during the disease course, improvement after withholding of the immunosuppressive regimen, and normal CSF. Brain biopsy confirms the diagnosis.
Subacute sclerosing panencephalitis (SSPE) is a slowly progressing and untreatable degenerative neurologic disorder characterized by demyelination of multiple brain areas. SSPE develops 7 to 10 years after natural measles infection, and usually affects children or adolescents. Clinical presentation includes intellectual decline, frequent rhythmic myoclonic jerks, seizure, and dementia. As the disease progresses, coma, quadriplegia, vegetative state, and autonomic instability develop. Death usually occurs within 2 years of onset.30,31 In children, the risk for SSPE after measles infection is estimated to be 4 to 11 per 100,000 infections. After the 1989-1991 resurgence of measles in the United States, however, the risk for SSPE was estimated to be 22 per 100,000 infections.30-32 The pathogenesis of SSPE is not fully understood but is thought to result from persistent aberrant measles virus infection.32
The SSPE diagnosis is based on clinical presentation, presence of anti-measles antibodies in CSF, typical electroencephalography pattern (periodic paroxysmal bursts) with accompanying myoclonus, tissue analysis, and magnetic resonance imaging.30
LABORATORY DIAGNOSIS
Suspicion for measles should prompt immediate consultation with local or state public health officials. Laboratory testing can be carefully considered after consultation, and care is needed in interpreting serologic studies.
The mainstays of measles infection diagnosis are detection of viral RNA by reverse transcriptase–polymerase chain reaction, or isolation of the virus in the clinical specimen, and detection of measles-specific IgM (immunoglobulin M) antibodies. A detailed protocol for collecting specimens for viral isolation appears on the Centers for Disease Control and Prevention website (http://www.cdc.gov/measles/lab-tools/rt-pcr.html).
IgM antibodies are detectable over the 15 weeks after rash onset, but the recommendation is to collect serum between 72 hours and 4 weeks after rash onset.33 Clinicians should be aware that false-positive IgM results may occur with rheumatologic diseases, parvovirus B19 infection, rubella, and infectious mononucleosis.
IgG (immunoglobulin G) antibodies are usually detectable a week after rash onset. The laboratory can confirm measles by detecting more than a 4-fold increase in IgG titers between the acute phase and the convalescent phase. After measles infection, most adults develop lifelong immunity with positive IgG serology.34
Additional tests, such as IgG avidity and plaque reduction neutralization assay, can be used to confirm suspected cases in previously vaccinated individuals.34
MANAGEMENT
General Principles
Uncomplicated measles treatment is supportive and includes oral fluids and antipyretics.7,22 Severe bacterial infections, encephalitis, or dehydration may require hospitalization, and in these cases infectious disease consultation is recommended. Patients with pneumonia, purulent otitis media, or tonsillitis should be treated with antibiotics.35 Observational data suggest antibiotics may reduce the occurrence of bacterial infection in children, but there are no usage guidelines.35 Vitamin A supplementation has been associated with a 50% decrease in morbidity and mortality and with blindness prevention.22 This supplementation should be considered in severe measles cases (all hospitalized patients), especially for children, regardless of country of residence, and for adult patients who exhibit clinical signs of vitamin A deficiency.22,24
Antiviral Treatment
No specific treatment is available.36 Ribavirin demonstrates in vitro activity against the virus, but the Food and Drug Administration has not approved the drug for treatment of measles. Ribavirin has been used for cases of severe measles, and for patients with SSPE along with intrathecal interferon alpha. This antiviral treatment is considered experimental.37
All patients hospitalized with measles infection should be cautioned about the potential downstream complications of the disease and should follow up with their primary care physician for surveillance after discharge.38
If measles symptoms develop, patients should self-quarantine and contact their primary care physician or public health department as soon as possible. Regardless of immune status, family members and other exposed persons should be educated about the measles symptoms that may occur during the 21 days after exposure.38
Both suspected and confirmed cases of measles should be reported immediately to local public health authorities.
Infection Control and Prophylaxis
Current guidelines recommend 2 doses of measles-containing vaccine to all adults at higher risk for contracting measles: international travelers, healthcare personnel, and high school and college students. Infants 6 or 11 months old should receive 1 MMR dose before international travel.1,38
Strict airborne isolation—use of N95 respirator or respirator with similar effectiveness in preventing airborne transmission—is mandatory from 3 to 5 days before rash onset to 4 days after rash onset (immunocompetent patients) or for the duration of the disease (immunocompromised patients).38
Healthcare workers should have documented presumptive evidence of immunity to measles.39 Healthcare providers without evidence of immunity should be excused from work from day 5 to day 21 of exposure, even if they have received postexposure vaccine or intramuscular immunoglobulin. They should be offered the first MMR dose within 72 hours of measles exposure to prevent or modify the disease. Susceptible family members or visitors should not be allowed in the patient’s room.1
Postexposure Prophylaxis
Standard MMR vaccination within 72 hours after exposure may protect against disease in people without a contraindication to measles vaccine. The public health department usually identifies these individuals and provides postexposure prophylaxis recommendations.38,39
People with HIV, patients receiving immunosuppressive therapy, and pregnant women and infants who have been exposed to measles and who are at risk for developing morbid disease can be treated with immunoglobulin (IG). If administered within 6 days of exposure, IG can prevent or modify disease in people who are unvaccinated or severely immunocompromised (ie, not immune). The recommended dose of IG administered intramuscularly is 0.5 mL/kg of body weight (maximum, 15 mL), and the recommended dose of IG given intravenously is 400 mg/kg. Anyone heavier than 30 kg would require intravenous IG to achieve adequate antibody levels.
Physicians should not vaccinate pregnant women, patients with severe immunosuppression from disease or therapy, patients with moderate or severe illness, and people with a history of severe allergic reaction to the vaccine.1,40 The measles vaccine should be deferred for 6 months after IG administration.36 More details are available in the recommendations made by the Advisory Committee on Immunization Practices.1
CONCLUSION
Although rare in the United States, measles remains a common and potentially devastating infection among patients who have not been vaccinated. Diagnosis requires clinical suspicion, engagement of public health authorities, and judicious use of laboratory testing. Hospitalists may encounter infectious and neurologic complications of measles long after the initial infection and should be aware of these associations.
Disclosure
Nothing to report.
1. McLean HQ, Fiebelkorn AP, Temte JL, Wallace, GS; Centers for Disease Control and Prevention. Prevention of measles, rubella, congenital rubella syndrome, and mumps, 2013: summary recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2013;62(RR-04):1-34.
2. World Health Organization. Measles [fact sheet]. http://www.who.int/mediacentre/factsheets/fs286/en/. Accessed April 27, 2017.
3. Kutty P, Rota J, Bellini W, Redd SB, Barskey A, Wallace G. Chapter 7: measles. In: Manual for the Surveillance of Vaccine-Preventable Disease. 6th ed. https://www.cdc.gov/vaccines/pubs/surv-manual/chpt07-measles.html. Published 2013. Accessed April 27, 2017.
4. Clemmons NS, Gastanaduy PA, Fiebelkorn AP, Redd SB, Wallace GS; Centers for Disease Control and Prevention (CDC). Measles—United States, January 4-April 2, 2015. MMWR Morb Mortal Wkly Rep. 2015;64(14):373-376.
5. Fiebelkorn AP, Redd SB, Gallagher K, et al. Measles in the United States during the postelimination era. J Infect Dis. 2010;202(10):1520-1528.
6. Fiebelkorn AP, Redd SB, Gastañaduy PA, et al. A comparison of postelimination measles epidemiology in the United States, 2009-2014 versus 2001-2008. J Pediatric Infect Dis Soc. 2017;6(1):40-48.
7. Gershon A. Measles (rubeola). In: Braunwald E, Fauci AS, Kasper DL, Hauser SL, Longo DL, Jameson JL, eds. Harrison’s Principles of Internal Medicine. 15th ed. New York, NY: McGraw-Hill; 2001:1143-1145.
8. Hill HA, Elam-Evans LD, Yankey D, Singleton JA, Kolasa M. National, state, and selected local area vaccination coverage among children aged 19-35 months—United States, 2014. MMWR Morb Mortal Wkly Rep. 2015;64(33):889-896.
9. Reagan-Steiner S, Yankey D, Jayarajah J, et al. National, state and selected local area vaccination coverage among children aged 13-17 years—United States, 2014. MMWR Morb Mortal Wkly Rep. 2015;64(29):784-792.
10. Seither R, Calhoun K, Knighton CL, et al. Vaccination coverage among children in kindergarten—United States, 2014-15 school year. MMWR Morb Mortal Wkly Rep. 2015;64(33):897-904.
11. Zipprich J, Winter K, Hacker J, Xia D, Watt J, Harriman K; Centers for Disease Control and Prevention (CDC). Measles outbreak—California, December 2014-February 2015. MMWR Morb Mortal Wkly Rep. 2015;64(6):153-154.
12. Perry RT, Halsey NA. The clinical significance of measles: a review. J Infect Dis. 2004;189(suppl 1):S4-S6.
13. Bernstein DI, Schiff GM. Measles. In: Gorbach SL, Bartlett JG, Blacklow NR, eds. Infectious Diseases. Philadelphia, PA: Saunders; 1998:1296.
14. Scheider-Schaulies S, Schneider-Schaulies J. Measles virus induced immunosuppression. Curr Top Microbiol Immunol. 2009;330:243-69
15. Mina MJ, Metcalf JE, de Swart RL, Osterhaus AD, Grenfell BT. Vaccines. Long-term measles-induced immunomodulation increases overall childhood infectious disease mortality. Science. 2015;348(6235):694-699.
16. Kaplan LJ, Daum RS, Smaron M, McCarthy CA. Severe measles may occur in immunocompromised patients. JAMA. 1992;267(9):1237-1241.
17. Melenotte C, Cassir N, Tessonnier L, Brouqui P. Atypical measles syndrome in adults: still around [published online September 23, 2015]. BMJ Case Rep. doi:10.1136/bcr-2015-211054.
18. Ogbuano IU, Zeko S, Chu SY, et al. Maternal, fetal and neonatal outcomes associated with measles during pregnancy: Namibia, 2009-2010. Clin Infect Dis. 2014;58(8):1086-1092.
19. Rasmussen SA, Jameson DJ. What obstetric healthcare providers need to know about measles and pregnancy. Obstet Gynecol. 2015;126(1):163-170.
20. Davis AT. Exanthematous diseases. In: Shulman ST, Phair JP, Peterson LR, Warren JR, eds. The Biologic and Clinical Basis of Infectious Diseases. 5th ed. Philadelphia, PA: Saunders; 1997:467-469.
21. Fortenberry JD, Mariscalco MM, Louis PT, Stein F, Jones JK, Jefferson LS. Severe laryngotracheobronchitis complicating measles. Am J Dis Child. 1992;146(9):1040-1043.
22. World Health Organization, Department of Immunization, Vaccines and Biologicals. Treating Measles in Children. http://www.who.int/immunization/programmes_systems/interventions/TreatingMeaslesENG300.pdf. Published 1997. Updated 2004. Accessed April 27, 2017.
23. Rafat C, Klouche K, Ricard JD, et al. Severe measles infection: the spectrum of disease in 36 critically ill adult patients. Medicine (Baltimore). 2013;92(5):257-272.
24. Ortac Ersoy E, Tanriover MD, Ocal S, Ozisik L, Inkaya C, Topeli A. Severe measles pneumonia in adults with respiratory failure: role of ribavirin and high-dose vitamin A. Clin Respir J. 2016;10(5):673-675.
25. Chassort A, Coutherut J, Moreau-Klein A, et al. Renal dysfunction in adults during measles. Med Mal Infect. 2015;45(5):165-168.
26. Sunnetcioglu M, Baran A, Sunnetcioglu A, Mentes O, Karadas S, Aypak A. Clinical and laboratory features of adult measles cases detected in Van, Turkey. J Pak Med Assoc. 2015;65(3):273-276.
27. Honarmand S, Glaser CA, Chow E, et al. Subacute sclerosing panencephalitis in the differential diagnosis of encephalitis. Neurology. 2004;63(8):1489-1493.
28. Liko J, Guzman-Cottrill JA, Cieslak PR. Notes from the field: subacute sclerosing panencephalitis death—Oregon, 2015. MMWR Morb Mortal Wkly Rep. 2016;65(1):10-11.
29. Fisher DL, Defres S, Solomon T. Measles-induced encephalitis. QJM. 2015;108(3):177-182.
30. Rodriguez D, Fishman D. Measles and subacute sclerosing panencephalitis. In: Samuels MA, Feske SK, eds. Office Practice of Neurology. Philadelphia, PA: Churchill Livingstone; 2003:419-420.
31. Gutierrez J, Issacson RS, Koppel BS. Subacute sclerosing panencephalitis: an update. Dev Med Child Neurol. 2010;52(10):901-907.
32. Bellini WJ, Rota JS, Lowe LE, et al. Subacute sclerosing panencephalitis: more cases
of this fatal disease are prevented by measles immunization than was previously
recognized. J Infect Dis. 2005;192(10);1686-1693.
33. Helfand RF, Heath JL, Anderson LJ, Maes EF, Guris D, Bellini WJ. Diagnosis of
measles with an IgM capture EIA: the optimal timing of specimen collection after
rash onset. J Infect Dis. 1997;175(1):195-199.
34. Hickman CJ, Hyde TB, Sowers SB, et al. Laboratory characterization of measles
virus infection in previously vaccinated and unvaccinated individuals. J Infect Dis.
2011;204(suppl 1):S549-S558.
35. Kabra SK, Lodha R. Antibiotics for preventing complications in children with
measles. Cochrane Database Syst Rev. 2013;(8):CD001477.
36. Sabella C. Measles: not just a childhood rash. Cleve Clin J Med. 2010;77(3):
207-213.
37. Hosoya M, Shigeta S, Mori S, et al. High-dose intravenous ribavirin therapy
for subacute sclerosing panencephalitis. Antimicrob Agents Chemother.
2001;45(3):943-945.
38. Siegel JD, Rhinehart E, Jackson M, Chiarello L; Healthcare Infection Control
Practices Advisory Committee. 2007 Guideline for Isolation Precautions: Preventing
Transmission of Infectious Agents in Healthcare Settings. Centers for Disease Control
and Prevention website. https://www.cdc.gov/hicpac/pdf/isolation/isolation2007.
pdf. Accessed April 27, 2017.
39. Houck P, Scott-Johnson G, Krebs L. Measles immunity among community hospital
employees. Infect Control Hosp Epidemiol. 1991;12(11):663-668.
40. Kumar D, Sabella C. Measles: back again. Cleve Clin J Med. 2016;83(5):340-344.
Measles is a highly contagious acute respiratory illness that includes a characteristic rash. After exposure, up to 90% of susceptible persons develop measles.1 Even though it is considered a childhood illness, measles can affect people of all age groups. Measles continues to be major health problem around the world, despite the availability of a safe and effective vaccine, and it remains one of the leading causes of childhood mortality, with nearly 115,000 deaths reported by the World Health Organization2 in 2014. In 2000, measles was declared eliminated from the United States, but outbreaks still occasionally occur.3-6
The disease is self-limited, but some patients develop complications that may require hospitalization for treatment. People at highest risk for complications are children younger than 5 years, adults older than 20 years, pregnant women, and immunocompromised individuals.7
HISTORY AND EPIDEMIOLOGY
During the licensure of live measles vaccine in 1963, an average of 549,000 measles cases and 495 measles deaths, as well as 48,000 hospitalizations and 4000 encephalitis cases, were reported annually in the United States. Almost all Americans were affected by measles by adolescence.
Implementation of the 1-dose vaccine program substantially reduced reported incidence in the United States by 1988, and led to a dramatic decline in measles-related hospitalizations and deaths.3-6 The 2-dose MMR (measles, mumps, rubella) vaccination was introduced in 1989, and measles was declared eliminated in the United States in 2000.3-6
National–level one-dose MMR coverage among children 19-35 months has remained above 90% during the last two decades.8 NIS-Teen vaccination coverage data for 13- to 17-year-olds since 2008 has been near or above 90%,9 and 94% of children enrolled in kindergarten had evidence of 2 MMR doses in the 2014-2015 school year.10
A large multistate measles outbreak was reported in the United States in 2014-2015.4,11 One hundred fifty-nine cases were reported in the United States between January 4 and April 5, 2015. The majority of patients either were unvaccinated (45%) or had an unknown vaccination status (38%). Age ranged from 6 weeks to 70 years, and 22 patients (14%) were hospitalized.4
CLINICAL PRESENTATION AND PATHOPHYSIOLOGY
Measles is caused by an RNA-containing paramyxovirus that is spread by the respiratory route. Average incubation period from exposure to rash onset is 14 days (range, 7-21 days).12,13 Peak infectivity occurs during the prodromal phase, before rash onset (Figure 1), but patients are infectious from 4 days before rash onset through 4 days after rash onset.7,12,13
The disease prodrome consists of a high fever (39°C-40.5°C), coryza, cough, and conjunctivitis followed by Koplik spots (Figure 2A). Koplik spots are pathognomonic for measles but rarely discovered. They appear before the skin rash alongside second molars on the buccal surface of the cheeks. The spots usually disappear when the characteristic maculopapular, nonpruritic rash erupts initially at the hairline and behind the ears, and within four days progresses toward the trunk and limbs, including the palms and soles (Figures 2B, 2C).
The patient remains febrile while the rash spreads.12,13 Usually the fever resolves while the rash fades in the same order in which it appeared. Fever that persists for more than 5 days usually indicates complications.13
Cellular immunity plays an important role in host defense; the virus invades T lymphocytes and triggers suppressive cytokine (interleukin 4) production. Leukopenia, expansion of mainly measles-specific T and B lymphocytes, and replacement of lymphocyte memory cell population results in further depression of cellular immunity, and predisposes patients to secondary bacterial infections for up to 2 years after measles infection.14,15
Patients immunocompromised by congenital cellular immunity deficiency, cancer, human immunodeficiency virus (HIV) infection without effective antiretroviral therapy, or immunosuppression treatment are at higher risk for developing severe complications or dying from measles. As the rash may fail to develop in these patients, diagnosis can be challenging.16
Modified measles is milder and may occur in patients with preexisting partial immunity: those with an immunization history (2-dose vaccine effectiveness is ∼97%), and infants with minimal immunity from their mothers.1,7 Patients may have mild respiratory symptoms with rash but little or no fever.7
Atypical measles is now extremely rare. It was described only among people who were vaccinated with the killed vaccine in the United States between 1963 and 1968 and subsequently exposed to measles. The disease is characterized by high fever, edema of extremities, and a rash that develops on the palms and soles and spreads centerward. It is considered noncommunicable.17
Measles infection during pregnancy is associated with increased maternal and fetal morbidity. The virus can induce neonatal low birth weight, spontaneous abortion, intrauterine fetal death, and maternal death. Pregnant women with measles are more likely to be hospitalized.18,19
DIFFERENTIAL DIAGNOSIS
The presenting symptoms of primary measles infection are nonspecific, particularly if Koplik spots are not identified. The differential diagnosis for a patient who presents with high fever and rash include Kawasaki disease, dengue, parvovirus B19, serum sickness, syphilis, systemic lupus erythematous, toxic shock syndrome, enterovirus infection, human herpes virus 6 (roseola), viral hemorrhagic fever, drug eruption, infectious mononucleosis, Rocky Mountain spotted fever, rubella, scarlet fever, chikungunya, and Zika virus infection.
COMPLICATIONS
Measles complications can affect nearly every organ system (Table). Rates of complications from measles infection depend on age and underlying condition. Coexisting vitamin A deficiency increases complication rates.20
Bacterial infections in the setting of measles infection are more common in adults than in children, and are more severe among people who are malnourished or have an immunodeficiency disorder. The most common infectious complications, which involve the respiratory tract, include pneumonia, laryngotracheitis (“measles croup”), bronchitis, otitis media (most common complication among children in the United States), and sinusitis.7,13,21
Indications for hospitalizing children include respiratory distress, laryngeal obstruction, dehydration that requires intravenous fluids, diarrhea with more than 10 stools a day or bloody stool, severe anemia, altered mental status, convulsion, severe rash with developing hemorrhagic areas, extensive mouth ulcers, corneal clouding or ulcers, visual disturbance, and mastoiditis.22
Pneumonia is a common indication for hospitalizing adults.23,24 Measles-associated interstitial giant cell (Hecht) pneumonia is most often recognized among immunocompromised and malnourished patients.13 Primary pneumonia is caused by the measles virus, but bacterial superinfection can occur. The most common bacterial pathogens include Streptococcus, Pneumococcus, and Staphylococcus,13,24 and less commonly isolated organisms include gram-negative bacteria, such as Haemophilus influenzae, Pseudomonas aeruginosa, Neisseria meningitides, and Enterobacter cloacae.23
Uncommon complications of measles are myocarditis, glomerulonephritis, acute renal failure, and thrombocytopenic purpura.25,26
Neurologic complications in measles are an important concern. Measles-associated central nervous system complications are considered a result of an immune-mediated reaction to myelin protein and not from direct viral insult.26-28 Immunocompromised patients are at risk for developing fatal encephalitis, and those who survive often experience cognitive decline or seizures.
Measles is associated with four different encephalitic diseases: primary measles encephalitis, acute post-measles encephalomyelitis, measles inclusion body encephalitis, and subacute sclerosing panencephalitis.
Primary measles encephalitis is characterized by fever, headache, stiff neck, and meningeal signs. Onset occurs between 1 and 15 days after rash onset, and the disease affects 1/1000 patients. Seizure, altered mental status, and coma can also develop. Viral RNA detection in the cerebrospinal fluid (CSF) confirms the diagnosis.29Acute post-measles encephalomyelitis is more common in adults than in children.12 It typically develops after the rash fades and the other symptoms subside. Patients suddenly experience a recurrence of fevers or seizures. Deafness, intellectual decline, epilepsy, postencephalitic hyperkinesia, hemiplegia, and/or paraplegia also can develop.27-29
Measles inclusion body encephalitis is described only in immunocompromised patients, and onset occurs within 1 year of infection. Seizures are an initial and common symptom, and some patients also experience hemiplegia, stupor, hypertonia, and dysarthria.29 Diagnostic findings include seroconversion during the disease course, improvement after withholding of the immunosuppressive regimen, and normal CSF. Brain biopsy confirms the diagnosis.
Subacute sclerosing panencephalitis (SSPE) is a slowly progressing and untreatable degenerative neurologic disorder characterized by demyelination of multiple brain areas. SSPE develops 7 to 10 years after natural measles infection, and usually affects children or adolescents. Clinical presentation includes intellectual decline, frequent rhythmic myoclonic jerks, seizure, and dementia. As the disease progresses, coma, quadriplegia, vegetative state, and autonomic instability develop. Death usually occurs within 2 years of onset.30,31 In children, the risk for SSPE after measles infection is estimated to be 4 to 11 per 100,000 infections. After the 1989-1991 resurgence of measles in the United States, however, the risk for SSPE was estimated to be 22 per 100,000 infections.30-32 The pathogenesis of SSPE is not fully understood but is thought to result from persistent aberrant measles virus infection.32
The SSPE diagnosis is based on clinical presentation, presence of anti-measles antibodies in CSF, typical electroencephalography pattern (periodic paroxysmal bursts) with accompanying myoclonus, tissue analysis, and magnetic resonance imaging.30
LABORATORY DIAGNOSIS
Suspicion for measles should prompt immediate consultation with local or state public health officials. Laboratory testing can be carefully considered after consultation, and care is needed in interpreting serologic studies.
The mainstays of measles infection diagnosis are detection of viral RNA by reverse transcriptase–polymerase chain reaction, or isolation of the virus in the clinical specimen, and detection of measles-specific IgM (immunoglobulin M) antibodies. A detailed protocol for collecting specimens for viral isolation appears on the Centers for Disease Control and Prevention website (http://www.cdc.gov/measles/lab-tools/rt-pcr.html).
IgM antibodies are detectable over the 15 weeks after rash onset, but the recommendation is to collect serum between 72 hours and 4 weeks after rash onset.33 Clinicians should be aware that false-positive IgM results may occur with rheumatologic diseases, parvovirus B19 infection, rubella, and infectious mononucleosis.
IgG (immunoglobulin G) antibodies are usually detectable a week after rash onset. The laboratory can confirm measles by detecting more than a 4-fold increase in IgG titers between the acute phase and the convalescent phase. After measles infection, most adults develop lifelong immunity with positive IgG serology.34
Additional tests, such as IgG avidity and plaque reduction neutralization assay, can be used to confirm suspected cases in previously vaccinated individuals.34
MANAGEMENT
General Principles
Uncomplicated measles treatment is supportive and includes oral fluids and antipyretics.7,22 Severe bacterial infections, encephalitis, or dehydration may require hospitalization, and in these cases infectious disease consultation is recommended. Patients with pneumonia, purulent otitis media, or tonsillitis should be treated with antibiotics.35 Observational data suggest antibiotics may reduce the occurrence of bacterial infection in children, but there are no usage guidelines.35 Vitamin A supplementation has been associated with a 50% decrease in morbidity and mortality and with blindness prevention.22 This supplementation should be considered in severe measles cases (all hospitalized patients), especially for children, regardless of country of residence, and for adult patients who exhibit clinical signs of vitamin A deficiency.22,24
Antiviral Treatment
No specific treatment is available.36 Ribavirin demonstrates in vitro activity against the virus, but the Food and Drug Administration has not approved the drug for treatment of measles. Ribavirin has been used for cases of severe measles, and for patients with SSPE along with intrathecal interferon alpha. This antiviral treatment is considered experimental.37
All patients hospitalized with measles infection should be cautioned about the potential downstream complications of the disease and should follow up with their primary care physician for surveillance after discharge.38
If measles symptoms develop, patients should self-quarantine and contact their primary care physician or public health department as soon as possible. Regardless of immune status, family members and other exposed persons should be educated about the measles symptoms that may occur during the 21 days after exposure.38
Both suspected and confirmed cases of measles should be reported immediately to local public health authorities.
Infection Control and Prophylaxis
Current guidelines recommend 2 doses of measles-containing vaccine to all adults at higher risk for contracting measles: international travelers, healthcare personnel, and high school and college students. Infants 6 or 11 months old should receive 1 MMR dose before international travel.1,38
Strict airborne isolation—use of N95 respirator or respirator with similar effectiveness in preventing airborne transmission—is mandatory from 3 to 5 days before rash onset to 4 days after rash onset (immunocompetent patients) or for the duration of the disease (immunocompromised patients).38
Healthcare workers should have documented presumptive evidence of immunity to measles.39 Healthcare providers without evidence of immunity should be excused from work from day 5 to day 21 of exposure, even if they have received postexposure vaccine or intramuscular immunoglobulin. They should be offered the first MMR dose within 72 hours of measles exposure to prevent or modify the disease. Susceptible family members or visitors should not be allowed in the patient’s room.1
Postexposure Prophylaxis
Standard MMR vaccination within 72 hours after exposure may protect against disease in people without a contraindication to measles vaccine. The public health department usually identifies these individuals and provides postexposure prophylaxis recommendations.38,39
People with HIV, patients receiving immunosuppressive therapy, and pregnant women and infants who have been exposed to measles and who are at risk for developing morbid disease can be treated with immunoglobulin (IG). If administered within 6 days of exposure, IG can prevent or modify disease in people who are unvaccinated or severely immunocompromised (ie, not immune). The recommended dose of IG administered intramuscularly is 0.5 mL/kg of body weight (maximum, 15 mL), and the recommended dose of IG given intravenously is 400 mg/kg. Anyone heavier than 30 kg would require intravenous IG to achieve adequate antibody levels.
Physicians should not vaccinate pregnant women, patients with severe immunosuppression from disease or therapy, patients with moderate or severe illness, and people with a history of severe allergic reaction to the vaccine.1,40 The measles vaccine should be deferred for 6 months after IG administration.36 More details are available in the recommendations made by the Advisory Committee on Immunization Practices.1
CONCLUSION
Although rare in the United States, measles remains a common and potentially devastating infection among patients who have not been vaccinated. Diagnosis requires clinical suspicion, engagement of public health authorities, and judicious use of laboratory testing. Hospitalists may encounter infectious and neurologic complications of measles long after the initial infection and should be aware of these associations.
Disclosure
Nothing to report.
Measles is a highly contagious acute respiratory illness that includes a characteristic rash. After exposure, up to 90% of susceptible persons develop measles.1 Even though it is considered a childhood illness, measles can affect people of all age groups. Measles continues to be major health problem around the world, despite the availability of a safe and effective vaccine, and it remains one of the leading causes of childhood mortality, with nearly 115,000 deaths reported by the World Health Organization2 in 2014. In 2000, measles was declared eliminated from the United States, but outbreaks still occasionally occur.3-6
The disease is self-limited, but some patients develop complications that may require hospitalization for treatment. People at highest risk for complications are children younger than 5 years, adults older than 20 years, pregnant women, and immunocompromised individuals.7
HISTORY AND EPIDEMIOLOGY
During the licensure of live measles vaccine in 1963, an average of 549,000 measles cases and 495 measles deaths, as well as 48,000 hospitalizations and 4000 encephalitis cases, were reported annually in the United States. Almost all Americans were affected by measles by adolescence.
Implementation of the 1-dose vaccine program substantially reduced reported incidence in the United States by 1988, and led to a dramatic decline in measles-related hospitalizations and deaths.3-6 The 2-dose MMR (measles, mumps, rubella) vaccination was introduced in 1989, and measles was declared eliminated in the United States in 2000.3-6
National–level one-dose MMR coverage among children 19-35 months has remained above 90% during the last two decades.8 NIS-Teen vaccination coverage data for 13- to 17-year-olds since 2008 has been near or above 90%,9 and 94% of children enrolled in kindergarten had evidence of 2 MMR doses in the 2014-2015 school year.10
A large multistate measles outbreak was reported in the United States in 2014-2015.4,11 One hundred fifty-nine cases were reported in the United States between January 4 and April 5, 2015. The majority of patients either were unvaccinated (45%) or had an unknown vaccination status (38%). Age ranged from 6 weeks to 70 years, and 22 patients (14%) were hospitalized.4
CLINICAL PRESENTATION AND PATHOPHYSIOLOGY
Measles is caused by an RNA-containing paramyxovirus that is spread by the respiratory route. Average incubation period from exposure to rash onset is 14 days (range, 7-21 days).12,13 Peak infectivity occurs during the prodromal phase, before rash onset (Figure 1), but patients are infectious from 4 days before rash onset through 4 days after rash onset.7,12,13
The disease prodrome consists of a high fever (39°C-40.5°C), coryza, cough, and conjunctivitis followed by Koplik spots (Figure 2A). Koplik spots are pathognomonic for measles but rarely discovered. They appear before the skin rash alongside second molars on the buccal surface of the cheeks. The spots usually disappear when the characteristic maculopapular, nonpruritic rash erupts initially at the hairline and behind the ears, and within four days progresses toward the trunk and limbs, including the palms and soles (Figures 2B, 2C).
The patient remains febrile while the rash spreads.12,13 Usually the fever resolves while the rash fades in the same order in which it appeared. Fever that persists for more than 5 days usually indicates complications.13
Cellular immunity plays an important role in host defense; the virus invades T lymphocytes and triggers suppressive cytokine (interleukin 4) production. Leukopenia, expansion of mainly measles-specific T and B lymphocytes, and replacement of lymphocyte memory cell population results in further depression of cellular immunity, and predisposes patients to secondary bacterial infections for up to 2 years after measles infection.14,15
Patients immunocompromised by congenital cellular immunity deficiency, cancer, human immunodeficiency virus (HIV) infection without effective antiretroviral therapy, or immunosuppression treatment are at higher risk for developing severe complications or dying from measles. As the rash may fail to develop in these patients, diagnosis can be challenging.16
Modified measles is milder and may occur in patients with preexisting partial immunity: those with an immunization history (2-dose vaccine effectiveness is ∼97%), and infants with minimal immunity from their mothers.1,7 Patients may have mild respiratory symptoms with rash but little or no fever.7
Atypical measles is now extremely rare. It was described only among people who were vaccinated with the killed vaccine in the United States between 1963 and 1968 and subsequently exposed to measles. The disease is characterized by high fever, edema of extremities, and a rash that develops on the palms and soles and spreads centerward. It is considered noncommunicable.17
Measles infection during pregnancy is associated with increased maternal and fetal morbidity. The virus can induce neonatal low birth weight, spontaneous abortion, intrauterine fetal death, and maternal death. Pregnant women with measles are more likely to be hospitalized.18,19
DIFFERENTIAL DIAGNOSIS
The presenting symptoms of primary measles infection are nonspecific, particularly if Koplik spots are not identified. The differential diagnosis for a patient who presents with high fever and rash include Kawasaki disease, dengue, parvovirus B19, serum sickness, syphilis, systemic lupus erythematous, toxic shock syndrome, enterovirus infection, human herpes virus 6 (roseola), viral hemorrhagic fever, drug eruption, infectious mononucleosis, Rocky Mountain spotted fever, rubella, scarlet fever, chikungunya, and Zika virus infection.
COMPLICATIONS
Measles complications can affect nearly every organ system (Table). Rates of complications from measles infection depend on age and underlying condition. Coexisting vitamin A deficiency increases complication rates.20
Bacterial infections in the setting of measles infection are more common in adults than in children, and are more severe among people who are malnourished or have an immunodeficiency disorder. The most common infectious complications, which involve the respiratory tract, include pneumonia, laryngotracheitis (“measles croup”), bronchitis, otitis media (most common complication among children in the United States), and sinusitis.7,13,21
Indications for hospitalizing children include respiratory distress, laryngeal obstruction, dehydration that requires intravenous fluids, diarrhea with more than 10 stools a day or bloody stool, severe anemia, altered mental status, convulsion, severe rash with developing hemorrhagic areas, extensive mouth ulcers, corneal clouding or ulcers, visual disturbance, and mastoiditis.22
Pneumonia is a common indication for hospitalizing adults.23,24 Measles-associated interstitial giant cell (Hecht) pneumonia is most often recognized among immunocompromised and malnourished patients.13 Primary pneumonia is caused by the measles virus, but bacterial superinfection can occur. The most common bacterial pathogens include Streptococcus, Pneumococcus, and Staphylococcus,13,24 and less commonly isolated organisms include gram-negative bacteria, such as Haemophilus influenzae, Pseudomonas aeruginosa, Neisseria meningitides, and Enterobacter cloacae.23
Uncommon complications of measles are myocarditis, glomerulonephritis, acute renal failure, and thrombocytopenic purpura.25,26
Neurologic complications in measles are an important concern. Measles-associated central nervous system complications are considered a result of an immune-mediated reaction to myelin protein and not from direct viral insult.26-28 Immunocompromised patients are at risk for developing fatal encephalitis, and those who survive often experience cognitive decline or seizures.
Measles is associated with four different encephalitic diseases: primary measles encephalitis, acute post-measles encephalomyelitis, measles inclusion body encephalitis, and subacute sclerosing panencephalitis.
Primary measles encephalitis is characterized by fever, headache, stiff neck, and meningeal signs. Onset occurs between 1 and 15 days after rash onset, and the disease affects 1/1000 patients. Seizure, altered mental status, and coma can also develop. Viral RNA detection in the cerebrospinal fluid (CSF) confirms the diagnosis.29Acute post-measles encephalomyelitis is more common in adults than in children.12 It typically develops after the rash fades and the other symptoms subside. Patients suddenly experience a recurrence of fevers or seizures. Deafness, intellectual decline, epilepsy, postencephalitic hyperkinesia, hemiplegia, and/or paraplegia also can develop.27-29
Measles inclusion body encephalitis is described only in immunocompromised patients, and onset occurs within 1 year of infection. Seizures are an initial and common symptom, and some patients also experience hemiplegia, stupor, hypertonia, and dysarthria.29 Diagnostic findings include seroconversion during the disease course, improvement after withholding of the immunosuppressive regimen, and normal CSF. Brain biopsy confirms the diagnosis.
Subacute sclerosing panencephalitis (SSPE) is a slowly progressing and untreatable degenerative neurologic disorder characterized by demyelination of multiple brain areas. SSPE develops 7 to 10 years after natural measles infection, and usually affects children or adolescents. Clinical presentation includes intellectual decline, frequent rhythmic myoclonic jerks, seizure, and dementia. As the disease progresses, coma, quadriplegia, vegetative state, and autonomic instability develop. Death usually occurs within 2 years of onset.30,31 In children, the risk for SSPE after measles infection is estimated to be 4 to 11 per 100,000 infections. After the 1989-1991 resurgence of measles in the United States, however, the risk for SSPE was estimated to be 22 per 100,000 infections.30-32 The pathogenesis of SSPE is not fully understood but is thought to result from persistent aberrant measles virus infection.32
The SSPE diagnosis is based on clinical presentation, presence of anti-measles antibodies in CSF, typical electroencephalography pattern (periodic paroxysmal bursts) with accompanying myoclonus, tissue analysis, and magnetic resonance imaging.30
LABORATORY DIAGNOSIS
Suspicion for measles should prompt immediate consultation with local or state public health officials. Laboratory testing can be carefully considered after consultation, and care is needed in interpreting serologic studies.
The mainstays of measles infection diagnosis are detection of viral RNA by reverse transcriptase–polymerase chain reaction, or isolation of the virus in the clinical specimen, and detection of measles-specific IgM (immunoglobulin M) antibodies. A detailed protocol for collecting specimens for viral isolation appears on the Centers for Disease Control and Prevention website (http://www.cdc.gov/measles/lab-tools/rt-pcr.html).
IgM antibodies are detectable over the 15 weeks after rash onset, but the recommendation is to collect serum between 72 hours and 4 weeks after rash onset.33 Clinicians should be aware that false-positive IgM results may occur with rheumatologic diseases, parvovirus B19 infection, rubella, and infectious mononucleosis.
IgG (immunoglobulin G) antibodies are usually detectable a week after rash onset. The laboratory can confirm measles by detecting more than a 4-fold increase in IgG titers between the acute phase and the convalescent phase. After measles infection, most adults develop lifelong immunity with positive IgG serology.34
Additional tests, such as IgG avidity and plaque reduction neutralization assay, can be used to confirm suspected cases in previously vaccinated individuals.34
MANAGEMENT
General Principles
Uncomplicated measles treatment is supportive and includes oral fluids and antipyretics.7,22 Severe bacterial infections, encephalitis, or dehydration may require hospitalization, and in these cases infectious disease consultation is recommended. Patients with pneumonia, purulent otitis media, or tonsillitis should be treated with antibiotics.35 Observational data suggest antibiotics may reduce the occurrence of bacterial infection in children, but there are no usage guidelines.35 Vitamin A supplementation has been associated with a 50% decrease in morbidity and mortality and with blindness prevention.22 This supplementation should be considered in severe measles cases (all hospitalized patients), especially for children, regardless of country of residence, and for adult patients who exhibit clinical signs of vitamin A deficiency.22,24
Antiviral Treatment
No specific treatment is available.36 Ribavirin demonstrates in vitro activity against the virus, but the Food and Drug Administration has not approved the drug for treatment of measles. Ribavirin has been used for cases of severe measles, and for patients with SSPE along with intrathecal interferon alpha. This antiviral treatment is considered experimental.37
All patients hospitalized with measles infection should be cautioned about the potential downstream complications of the disease and should follow up with their primary care physician for surveillance after discharge.38
If measles symptoms develop, patients should self-quarantine and contact their primary care physician or public health department as soon as possible. Regardless of immune status, family members and other exposed persons should be educated about the measles symptoms that may occur during the 21 days after exposure.38
Both suspected and confirmed cases of measles should be reported immediately to local public health authorities.
Infection Control and Prophylaxis
Current guidelines recommend 2 doses of measles-containing vaccine to all adults at higher risk for contracting measles: international travelers, healthcare personnel, and high school and college students. Infants 6 or 11 months old should receive 1 MMR dose before international travel.1,38
Strict airborne isolation—use of N95 respirator or respirator with similar effectiveness in preventing airborne transmission—is mandatory from 3 to 5 days before rash onset to 4 days after rash onset (immunocompetent patients) or for the duration of the disease (immunocompromised patients).38
Healthcare workers should have documented presumptive evidence of immunity to measles.39 Healthcare providers without evidence of immunity should be excused from work from day 5 to day 21 of exposure, even if they have received postexposure vaccine or intramuscular immunoglobulin. They should be offered the first MMR dose within 72 hours of measles exposure to prevent or modify the disease. Susceptible family members or visitors should not be allowed in the patient’s room.1
Postexposure Prophylaxis
Standard MMR vaccination within 72 hours after exposure may protect against disease in people without a contraindication to measles vaccine. The public health department usually identifies these individuals and provides postexposure prophylaxis recommendations.38,39
People with HIV, patients receiving immunosuppressive therapy, and pregnant women and infants who have been exposed to measles and who are at risk for developing morbid disease can be treated with immunoglobulin (IG). If administered within 6 days of exposure, IG can prevent or modify disease in people who are unvaccinated or severely immunocompromised (ie, not immune). The recommended dose of IG administered intramuscularly is 0.5 mL/kg of body weight (maximum, 15 mL), and the recommended dose of IG given intravenously is 400 mg/kg. Anyone heavier than 30 kg would require intravenous IG to achieve adequate antibody levels.
Physicians should not vaccinate pregnant women, patients with severe immunosuppression from disease or therapy, patients with moderate or severe illness, and people with a history of severe allergic reaction to the vaccine.1,40 The measles vaccine should be deferred for 6 months after IG administration.36 More details are available in the recommendations made by the Advisory Committee on Immunization Practices.1
CONCLUSION
Although rare in the United States, measles remains a common and potentially devastating infection among patients who have not been vaccinated. Diagnosis requires clinical suspicion, engagement of public health authorities, and judicious use of laboratory testing. Hospitalists may encounter infectious and neurologic complications of measles long after the initial infection and should be aware of these associations.
Disclosure
Nothing to report.
1. McLean HQ, Fiebelkorn AP, Temte JL, Wallace, GS; Centers for Disease Control and Prevention. Prevention of measles, rubella, congenital rubella syndrome, and mumps, 2013: summary recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2013;62(RR-04):1-34.
2. World Health Organization. Measles [fact sheet]. http://www.who.int/mediacentre/factsheets/fs286/en/. Accessed April 27, 2017.
3. Kutty P, Rota J, Bellini W, Redd SB, Barskey A, Wallace G. Chapter 7: measles. In: Manual for the Surveillance of Vaccine-Preventable Disease. 6th ed. https://www.cdc.gov/vaccines/pubs/surv-manual/chpt07-measles.html. Published 2013. Accessed April 27, 2017.
4. Clemmons NS, Gastanaduy PA, Fiebelkorn AP, Redd SB, Wallace GS; Centers for Disease Control and Prevention (CDC). Measles—United States, January 4-April 2, 2015. MMWR Morb Mortal Wkly Rep. 2015;64(14):373-376.
5. Fiebelkorn AP, Redd SB, Gallagher K, et al. Measles in the United States during the postelimination era. J Infect Dis. 2010;202(10):1520-1528.
6. Fiebelkorn AP, Redd SB, Gastañaduy PA, et al. A comparison of postelimination measles epidemiology in the United States, 2009-2014 versus 2001-2008. J Pediatric Infect Dis Soc. 2017;6(1):40-48.
7. Gershon A. Measles (rubeola). In: Braunwald E, Fauci AS, Kasper DL, Hauser SL, Longo DL, Jameson JL, eds. Harrison’s Principles of Internal Medicine. 15th ed. New York, NY: McGraw-Hill; 2001:1143-1145.
8. Hill HA, Elam-Evans LD, Yankey D, Singleton JA, Kolasa M. National, state, and selected local area vaccination coverage among children aged 19-35 months—United States, 2014. MMWR Morb Mortal Wkly Rep. 2015;64(33):889-896.
9. Reagan-Steiner S, Yankey D, Jayarajah J, et al. National, state and selected local area vaccination coverage among children aged 13-17 years—United States, 2014. MMWR Morb Mortal Wkly Rep. 2015;64(29):784-792.
10. Seither R, Calhoun K, Knighton CL, et al. Vaccination coverage among children in kindergarten—United States, 2014-15 school year. MMWR Morb Mortal Wkly Rep. 2015;64(33):897-904.
11. Zipprich J, Winter K, Hacker J, Xia D, Watt J, Harriman K; Centers for Disease Control and Prevention (CDC). Measles outbreak—California, December 2014-February 2015. MMWR Morb Mortal Wkly Rep. 2015;64(6):153-154.
12. Perry RT, Halsey NA. The clinical significance of measles: a review. J Infect Dis. 2004;189(suppl 1):S4-S6.
13. Bernstein DI, Schiff GM. Measles. In: Gorbach SL, Bartlett JG, Blacklow NR, eds. Infectious Diseases. Philadelphia, PA: Saunders; 1998:1296.
14. Scheider-Schaulies S, Schneider-Schaulies J. Measles virus induced immunosuppression. Curr Top Microbiol Immunol. 2009;330:243-69
15. Mina MJ, Metcalf JE, de Swart RL, Osterhaus AD, Grenfell BT. Vaccines. Long-term measles-induced immunomodulation increases overall childhood infectious disease mortality. Science. 2015;348(6235):694-699.
16. Kaplan LJ, Daum RS, Smaron M, McCarthy CA. Severe measles may occur in immunocompromised patients. JAMA. 1992;267(9):1237-1241.
17. Melenotte C, Cassir N, Tessonnier L, Brouqui P. Atypical measles syndrome in adults: still around [published online September 23, 2015]. BMJ Case Rep. doi:10.1136/bcr-2015-211054.
18. Ogbuano IU, Zeko S, Chu SY, et al. Maternal, fetal and neonatal outcomes associated with measles during pregnancy: Namibia, 2009-2010. Clin Infect Dis. 2014;58(8):1086-1092.
19. Rasmussen SA, Jameson DJ. What obstetric healthcare providers need to know about measles and pregnancy. Obstet Gynecol. 2015;126(1):163-170.
20. Davis AT. Exanthematous diseases. In: Shulman ST, Phair JP, Peterson LR, Warren JR, eds. The Biologic and Clinical Basis of Infectious Diseases. 5th ed. Philadelphia, PA: Saunders; 1997:467-469.
21. Fortenberry JD, Mariscalco MM, Louis PT, Stein F, Jones JK, Jefferson LS. Severe laryngotracheobronchitis complicating measles. Am J Dis Child. 1992;146(9):1040-1043.
22. World Health Organization, Department of Immunization, Vaccines and Biologicals. Treating Measles in Children. http://www.who.int/immunization/programmes_systems/interventions/TreatingMeaslesENG300.pdf. Published 1997. Updated 2004. Accessed April 27, 2017.
23. Rafat C, Klouche K, Ricard JD, et al. Severe measles infection: the spectrum of disease in 36 critically ill adult patients. Medicine (Baltimore). 2013;92(5):257-272.
24. Ortac Ersoy E, Tanriover MD, Ocal S, Ozisik L, Inkaya C, Topeli A. Severe measles pneumonia in adults with respiratory failure: role of ribavirin and high-dose vitamin A. Clin Respir J. 2016;10(5):673-675.
25. Chassort A, Coutherut J, Moreau-Klein A, et al. Renal dysfunction in adults during measles. Med Mal Infect. 2015;45(5):165-168.
26. Sunnetcioglu M, Baran A, Sunnetcioglu A, Mentes O, Karadas S, Aypak A. Clinical and laboratory features of adult measles cases detected in Van, Turkey. J Pak Med Assoc. 2015;65(3):273-276.
27. Honarmand S, Glaser CA, Chow E, et al. Subacute sclerosing panencephalitis in the differential diagnosis of encephalitis. Neurology. 2004;63(8):1489-1493.
28. Liko J, Guzman-Cottrill JA, Cieslak PR. Notes from the field: subacute sclerosing panencephalitis death—Oregon, 2015. MMWR Morb Mortal Wkly Rep. 2016;65(1):10-11.
29. Fisher DL, Defres S, Solomon T. Measles-induced encephalitis. QJM. 2015;108(3):177-182.
30. Rodriguez D, Fishman D. Measles and subacute sclerosing panencephalitis. In: Samuels MA, Feske SK, eds. Office Practice of Neurology. Philadelphia, PA: Churchill Livingstone; 2003:419-420.
31. Gutierrez J, Issacson RS, Koppel BS. Subacute sclerosing panencephalitis: an update. Dev Med Child Neurol. 2010;52(10):901-907.
32. Bellini WJ, Rota JS, Lowe LE, et al. Subacute sclerosing panencephalitis: more cases
of this fatal disease are prevented by measles immunization than was previously
recognized. J Infect Dis. 2005;192(10);1686-1693.
33. Helfand RF, Heath JL, Anderson LJ, Maes EF, Guris D, Bellini WJ. Diagnosis of
measles with an IgM capture EIA: the optimal timing of specimen collection after
rash onset. J Infect Dis. 1997;175(1):195-199.
34. Hickman CJ, Hyde TB, Sowers SB, et al. Laboratory characterization of measles
virus infection in previously vaccinated and unvaccinated individuals. J Infect Dis.
2011;204(suppl 1):S549-S558.
35. Kabra SK, Lodha R. Antibiotics for preventing complications in children with
measles. Cochrane Database Syst Rev. 2013;(8):CD001477.
36. Sabella C. Measles: not just a childhood rash. Cleve Clin J Med. 2010;77(3):
207-213.
37. Hosoya M, Shigeta S, Mori S, et al. High-dose intravenous ribavirin therapy
for subacute sclerosing panencephalitis. Antimicrob Agents Chemother.
2001;45(3):943-945.
38. Siegel JD, Rhinehart E, Jackson M, Chiarello L; Healthcare Infection Control
Practices Advisory Committee. 2007 Guideline for Isolation Precautions: Preventing
Transmission of Infectious Agents in Healthcare Settings. Centers for Disease Control
and Prevention website. https://www.cdc.gov/hicpac/pdf/isolation/isolation2007.
pdf. Accessed April 27, 2017.
39. Houck P, Scott-Johnson G, Krebs L. Measles immunity among community hospital
employees. Infect Control Hosp Epidemiol. 1991;12(11):663-668.
40. Kumar D, Sabella C. Measles: back again. Cleve Clin J Med. 2016;83(5):340-344.
1. McLean HQ, Fiebelkorn AP, Temte JL, Wallace, GS; Centers for Disease Control and Prevention. Prevention of measles, rubella, congenital rubella syndrome, and mumps, 2013: summary recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2013;62(RR-04):1-34.
2. World Health Organization. Measles [fact sheet]. http://www.who.int/mediacentre/factsheets/fs286/en/. Accessed April 27, 2017.
3. Kutty P, Rota J, Bellini W, Redd SB, Barskey A, Wallace G. Chapter 7: measles. In: Manual for the Surveillance of Vaccine-Preventable Disease. 6th ed. https://www.cdc.gov/vaccines/pubs/surv-manual/chpt07-measles.html. Published 2013. Accessed April 27, 2017.
4. Clemmons NS, Gastanaduy PA, Fiebelkorn AP, Redd SB, Wallace GS; Centers for Disease Control and Prevention (CDC). Measles—United States, January 4-April 2, 2015. MMWR Morb Mortal Wkly Rep. 2015;64(14):373-376.
5. Fiebelkorn AP, Redd SB, Gallagher K, et al. Measles in the United States during the postelimination era. J Infect Dis. 2010;202(10):1520-1528.
6. Fiebelkorn AP, Redd SB, Gastañaduy PA, et al. A comparison of postelimination measles epidemiology in the United States, 2009-2014 versus 2001-2008. J Pediatric Infect Dis Soc. 2017;6(1):40-48.
7. Gershon A. Measles (rubeola). In: Braunwald E, Fauci AS, Kasper DL, Hauser SL, Longo DL, Jameson JL, eds. Harrison’s Principles of Internal Medicine. 15th ed. New York, NY: McGraw-Hill; 2001:1143-1145.
8. Hill HA, Elam-Evans LD, Yankey D, Singleton JA, Kolasa M. National, state, and selected local area vaccination coverage among children aged 19-35 months—United States, 2014. MMWR Morb Mortal Wkly Rep. 2015;64(33):889-896.
9. Reagan-Steiner S, Yankey D, Jayarajah J, et al. National, state and selected local area vaccination coverage among children aged 13-17 years—United States, 2014. MMWR Morb Mortal Wkly Rep. 2015;64(29):784-792.
10. Seither R, Calhoun K, Knighton CL, et al. Vaccination coverage among children in kindergarten—United States, 2014-15 school year. MMWR Morb Mortal Wkly Rep. 2015;64(33):897-904.
11. Zipprich J, Winter K, Hacker J, Xia D, Watt J, Harriman K; Centers for Disease Control and Prevention (CDC). Measles outbreak—California, December 2014-February 2015. MMWR Morb Mortal Wkly Rep. 2015;64(6):153-154.
12. Perry RT, Halsey NA. The clinical significance of measles: a review. J Infect Dis. 2004;189(suppl 1):S4-S6.
13. Bernstein DI, Schiff GM. Measles. In: Gorbach SL, Bartlett JG, Blacklow NR, eds. Infectious Diseases. Philadelphia, PA: Saunders; 1998:1296.
14. Scheider-Schaulies S, Schneider-Schaulies J. Measles virus induced immunosuppression. Curr Top Microbiol Immunol. 2009;330:243-69
15. Mina MJ, Metcalf JE, de Swart RL, Osterhaus AD, Grenfell BT. Vaccines. Long-term measles-induced immunomodulation increases overall childhood infectious disease mortality. Science. 2015;348(6235):694-699.
16. Kaplan LJ, Daum RS, Smaron M, McCarthy CA. Severe measles may occur in immunocompromised patients. JAMA. 1992;267(9):1237-1241.
17. Melenotte C, Cassir N, Tessonnier L, Brouqui P. Atypical measles syndrome in adults: still around [published online September 23, 2015]. BMJ Case Rep. doi:10.1136/bcr-2015-211054.
18. Ogbuano IU, Zeko S, Chu SY, et al. Maternal, fetal and neonatal outcomes associated with measles during pregnancy: Namibia, 2009-2010. Clin Infect Dis. 2014;58(8):1086-1092.
19. Rasmussen SA, Jameson DJ. What obstetric healthcare providers need to know about measles and pregnancy. Obstet Gynecol. 2015;126(1):163-170.
20. Davis AT. Exanthematous diseases. In: Shulman ST, Phair JP, Peterson LR, Warren JR, eds. The Biologic and Clinical Basis of Infectious Diseases. 5th ed. Philadelphia, PA: Saunders; 1997:467-469.
21. Fortenberry JD, Mariscalco MM, Louis PT, Stein F, Jones JK, Jefferson LS. Severe laryngotracheobronchitis complicating measles. Am J Dis Child. 1992;146(9):1040-1043.
22. World Health Organization, Department of Immunization, Vaccines and Biologicals. Treating Measles in Children. http://www.who.int/immunization/programmes_systems/interventions/TreatingMeaslesENG300.pdf. Published 1997. Updated 2004. Accessed April 27, 2017.
23. Rafat C, Klouche K, Ricard JD, et al. Severe measles infection: the spectrum of disease in 36 critically ill adult patients. Medicine (Baltimore). 2013;92(5):257-272.
24. Ortac Ersoy E, Tanriover MD, Ocal S, Ozisik L, Inkaya C, Topeli A. Severe measles pneumonia in adults with respiratory failure: role of ribavirin and high-dose vitamin A. Clin Respir J. 2016;10(5):673-675.
25. Chassort A, Coutherut J, Moreau-Klein A, et al. Renal dysfunction in adults during measles. Med Mal Infect. 2015;45(5):165-168.
26. Sunnetcioglu M, Baran A, Sunnetcioglu A, Mentes O, Karadas S, Aypak A. Clinical and laboratory features of adult measles cases detected in Van, Turkey. J Pak Med Assoc. 2015;65(3):273-276.
27. Honarmand S, Glaser CA, Chow E, et al. Subacute sclerosing panencephalitis in the differential diagnosis of encephalitis. Neurology. 2004;63(8):1489-1493.
28. Liko J, Guzman-Cottrill JA, Cieslak PR. Notes from the field: subacute sclerosing panencephalitis death—Oregon, 2015. MMWR Morb Mortal Wkly Rep. 2016;65(1):10-11.
29. Fisher DL, Defres S, Solomon T. Measles-induced encephalitis. QJM. 2015;108(3):177-182.
30. Rodriguez D, Fishman D. Measles and subacute sclerosing panencephalitis. In: Samuels MA, Feske SK, eds. Office Practice of Neurology. Philadelphia, PA: Churchill Livingstone; 2003:419-420.
31. Gutierrez J, Issacson RS, Koppel BS. Subacute sclerosing panencephalitis: an update. Dev Med Child Neurol. 2010;52(10):901-907.
32. Bellini WJ, Rota JS, Lowe LE, et al. Subacute sclerosing panencephalitis: more cases
of this fatal disease are prevented by measles immunization than was previously
recognized. J Infect Dis. 2005;192(10);1686-1693.
33. Helfand RF, Heath JL, Anderson LJ, Maes EF, Guris D, Bellini WJ. Diagnosis of
measles with an IgM capture EIA: the optimal timing of specimen collection after
rash onset. J Infect Dis. 1997;175(1):195-199.
34. Hickman CJ, Hyde TB, Sowers SB, et al. Laboratory characterization of measles
virus infection in previously vaccinated and unvaccinated individuals. J Infect Dis.
2011;204(suppl 1):S549-S558.
35. Kabra SK, Lodha R. Antibiotics for preventing complications in children with
measles. Cochrane Database Syst Rev. 2013;(8):CD001477.
36. Sabella C. Measles: not just a childhood rash. Cleve Clin J Med. 2010;77(3):
207-213.
37. Hosoya M, Shigeta S, Mori S, et al. High-dose intravenous ribavirin therapy
for subacute sclerosing panencephalitis. Antimicrob Agents Chemother.
2001;45(3):943-945.
38. Siegel JD, Rhinehart E, Jackson M, Chiarello L; Healthcare Infection Control
Practices Advisory Committee. 2007 Guideline for Isolation Precautions: Preventing
Transmission of Infectious Agents in Healthcare Settings. Centers for Disease Control
and Prevention website. https://www.cdc.gov/hicpac/pdf/isolation/isolation2007.
pdf. Accessed April 27, 2017.
39. Houck P, Scott-Johnson G, Krebs L. Measles immunity among community hospital
employees. Infect Control Hosp Epidemiol. 1991;12(11):663-668.
40. Kumar D, Sabella C. Measles: back again. Cleve Clin J Med. 2016;83(5):340-344.
© 2017 Society of Hospital Medicine
Malingering in apparently psychotic patients: Detecting it and dealing with it
Imagine you’re on call in a busy emergency department (ED) overnight. Things are tough. The consults are piling up, no one is returning your calls for collateral information, and you’re dealing with a myriad of emergencies.
In walks Mr. D, age 45, complaining of hearing voices, feeling unsafe, and asking for admission. It’s now 2
Of course, like all qualified psychiatrists, you will dig a little deeper, and in doing so you learn that Mr. D has visited this hospital before and has been admitted to the psychiatry unit. Now you go from having a dearth of information to having more records than you can count.
You discover that Mr. D has a history of coming to the ED during precarious hours, with similar complaints, demanding admission.
Mr. D, you learn, is unemployed, single, and homeless. Your meticulous search through his hospital records and previous admission and discharge notes reveal that once he has slept for a night, eaten a hot meal, and received narcotics for his back pain and benzodiazepines for his “symptoms” he demands to leave the hospital. His psychotic symptoms disappear despite his consistent refusal to take antipsychotics throughout his stay.
Now, what would you do?
As earnest medical students and psychiatrists, we enjoy helping patients on their path toward recovery. We want to advocate for our patients and give them the benefit of the doubt. We’re taught in medical school to be non-judgmental and invite patients to share their narrative. But through experience, we start to become aware of malingering.
Suspecting malingering, diagnosed as a condition, often is avoided by psychiatrists.1 This makes sense—it goes against the essence of our training and imposes a pejorative label on someone who has reached out for help.
Often persons with mental illness will suffer for years until they to receive help.2 That’s exactly why, when patients like Mr. D come to the ED and report hearing voices, we’re not likely to shout, “Liar!” and invite them to leave.
However, malingering is a real problem, especially because the number of psychiatric hospital beds have dwindled to record lows, thereby overcrowding EDs. Resources are skimpy, and clinicians want to help those who need it the most and not waste resources on someone who is “faking it” for secondary gain.
To navigate this diagnostic challenge, psychiatrists need the skills to detect malingering and the confidence to deal with it appropriately. This article aims to:
- define psychosis and malingering
- review the prevalence and historical considerations of malingering
- offer practical strategies to deal with malingering.
Know the real thing
Clinicians first must have the clinical acumen and expertise to identify a true mental illness such as psychosis2 (Table 1). The differential diagnosis for psychotic symptoms is broad. The astute clinician might suspect that untreated bipolar disorder or depression led to the emergence of perceptual disturbances or disordered thinking. Transient psychotic symptoms can be associated with trauma disorders, borderline personality disorder, and acute intoxication. Psychotic spectrum disorders range from brief psychosis to schizophreniform to schizoaffective disorder or schizophrenia.
Malingering—which is a condition, not a diagnosis—is characterized by the intentional production of false or grossly exaggerated physical or psychological symptoms motivated by external incentives.3,4 The presence of external incentives differentiates malingering from true psychiatric disorders, including factitious disorder, somatoform disorder, and dissociative disorder, and specific medical conditions.1 In those disorders, there is no external incentive.
It is important to remember that malingering can coexist with a serious mental illness. For example, a truly psychotic person might malinger, feign, or exaggerate symptoms to try to receive much needed help. Individuals with true psychosis might have become disenchanted with the mental health system, and thereby have a tendency to over-report or exaggerate symptoms in an effort to obtain treatment. This also could explain why many clinicians intuitively are reluctant to make the determination that someone is malingering. Malingering also can be present in an individual who has antisocial personality disorder, factitious disorder, Ganser syndrome, and Munchausen syndrome.4 When symptoms or diseases that either are thought to be exaggerated or do not exist, consider a diagnosis of malingering.
A key challenge in any discussion of abnormal health care–seeking behavior is the extent to which a person’s reported symptoms are considered to be a product of choice, psychopathology beyond volitional control, or perhaps both. Clinical skills alone typically are not sufficient for diagnosing or detecting malingering. Medical education needs to provide doctors with the conceptual, developmental, and management frameworks to understand and manage patients whose symptoms appear to be simulated. Central to understanding factitious disorders and malingering are the explanatory models and beliefs used to provide meaning for both patients and doctors.7
When considering malingered psychosis, the suspecting physician must stay alert to possible motives. Also, the patient’s presentation might provide some clues when there is marked variability, such as discrepancies in the history, gross inconsistencies, or blatant contradictions. Hallucinations are a heterogeneous experience, and discerning between true vs feigned symptoms can be challenging for even the seasoned clinician. It can be helpful to study the phenomenology of typical vs atypical hallucinatory symptoms.8 Examples of atypical symptoms include:
- vague hallucinations
- experiencing hallucinations of only 1 sensory modality (such as voices alone, visual images in black and white only)
- delusions that have an abrupt onset
- bizarre content without disordered thinking.2,6,9,10

The truth about an untruthful condition
Although the exact prevalence of malingering varies by circumstance, Rissmiller et al12,13 demonstrated—and later replicated—a prevalence of approximately 10% among patients hospitalized for suicidal ideation or suicide attempts. Studies have demonstrated even higher prevalence within forensic populations, which seems reasonable because evading criminal responsibility is a large incentive to feign symptoms. Studies also have shown that 5% of military recruits will feign symptoms to avoid service. Moreover, 1% of psychiatric patients, such as Mr. D, feign symptoms for secondary gain.13
Although there are no psychometrically validated assessment tools to distinguish between real vs feigned hallucinations, several standardized tests can help tease out the truth.9 The preferred personality test used in forensic settings is the Minnesota Multiphasic Personality Inventory,14 which consists of 567 items, with 10 clinical scales and several validity scales. The F scale, “faking good” or “faking bad,” detects people who are answering questions with the goal of appearing better or worse than they actually are. In studies of patients hospitalized for being at risk for suicide who were administered tests of self-reported malingering, approximately 10% of people admitted to psychiatric units were “faking” their symptoms.14
It is important to identify malingering from a professional and public health standpoint. Society incurs incremental costs when a person uses dwindling mental health resources for their own reward, leaving others to suffer without treatment. The number of psychiatric hospital beds has fallen from half a million in the 1950s to approximately 100,000 today.15
Practical guidelines
Malingering presents specific challenges to clinicians, such as:
- diagnostic uncertainty
- inaccurately branding one a liar
- countertransference
- personal reactions.
Our ethical and fiduciary responsibility is to our patient. In examining the art in medicine, it has been suggested that malingering could be viewed as an immature or primitive defense.16
Although there often is suspicion that a person is malingering, a definitive statement of such must be confirmed. Without clarity, labeling an individual as a malingerer could have detrimental effects to his (her) future care, defames his character, and places a thoughtless examiner at risk of a lawsuit. Confirmation can be achieved by observation or psychological testing methods.
Observation. When in doubt of what to do with someone such as Mr. D, there is little harm in acting prudently by holding him in a controlled setting—whether keeping him overnight in an ED or admitting him for a brief psychiatric stay. By observing someone in a controlled environment, where there are multiple professional watchful eyes, inferences will be more accurate.1
Structured assessments have been developed to help detect malingering—one example is the Test of Memory Malingering—however, in daily practice, the physician generally should suspect malingering when there are tangible incentives and when reported symptoms do not match the physical examination or there is no organic basis for the physical complaints.17 Detecting illness deception relies on converging evidence sources, including detailed interview assessments, clinical notes, and consultations.7
When you feel certain that you are encountering someone who is malingering, the final step is to get a consult. Malingering is a serious label and warrants due diligence by the provider, rather than a haphazard guess that a patient is lying. Once you receive confirmatory opinions, great care should be taken in documenting a clear and accurate note that will benefit your clinical counterpart who might encounter a patient such as Mr. D when he (she) shows up again, and will go a long way toward appropriately directing his care.
1. LoPiccolo CJ, Goodkin K, Baldewicz TT. Current issues in the diagnosis and management of malingering. Ann Med. 1999;31(3):166-174.
2. Resnick PJ, Knoll J. Faking it: how to detect malingered psychosis. Current Psychiatry. 2005;4(11):12-25.
3. Sadock VA. Kaplan and Sadock’s synopsis of psychiatry: behavioral sciences/clinical psychiatry. 10th ed. Philadelphia, PA: Lippincott, Williams & Wilkins; 2007:887.
4. Gorman WF. Defining malingering. J Forensic Sci. 1982;27(2):401-407.
5. Mendelson G, Mendelson D. Malingering pain in the medicolegal context. Clin J Pain. 2004;20(6):423-432.
6. Resnick PJ. Malingered psychosis. In: Rogers R, ed. Clinical assessment of malingering and deception. 2nd ed. New York, NY: The Guilford Press; 1997:47-67.
7. Bass C, Halligan P. Factitious disorders and malingering: challenges for clinical assessment and management. Lancet. 2014;383(9926):1422-1432.
8. McCarthy-Jones S, Resnick PJ. Listening to the voices: the use of phenomenology to differentiate malingered from genuine auditory verbal hallucinations. Int J Law Psychiatry. 2014;37(2):183-189.
9. Resnick PJ. Defrocking the fraud: the detection of malingering. Isr J Psychiatry Relat Sci. 1993;30(2):93-101.
10. Nayani TH, David AS. The auditory hallucination: a phenomenological survey. Psychol Med. 1996;26(1):177-189.
11. Pollock P. Feigning auditory hallucinations by offenders. Journal of Forensic Psychiatry. 1998;9(2)305-327.
12. Rissmiller DJ, Wayslow A, Madison H, et al. Prevalence of malingering in inpatient suicide ideators and attempters. Crisis. 1998;19(2):62-66.
13. Rissmiller DA, Steer RA, Friedman M, et al. Prevalence of malingering in suicidal psychiatric patients: a replication. Psychol Rep. 1999;84(3 pt 1):726-730.
14. Hathaway SR, McKinley JC. The Minnesota Multiphasic Personality Inventory-2. Minneapolis, MN: University of Minnesota Press; 1989.
15. Szabo L. Cost of not caring: Stigma set in stone. USA Today. http://www.usatoday.com/story/news/nation/2014/06/25/stigma-of-mental-illness/9875351. Published June 25, 2014. Accessed May 5, 2017.
16. Malone RD, Lange CL. A clinical approach to the malingering patient. J Am Acad Psychoanal Dyn Psychiatry. 2007;35(1):13-21.
17. McDermott BE, Feldman MD. Malingering in the medical setting. Psychiatr Clin North Am. 2007;30(4):645-662.
Imagine you’re on call in a busy emergency department (ED) overnight. Things are tough. The consults are piling up, no one is returning your calls for collateral information, and you’re dealing with a myriad of emergencies.
In walks Mr. D, age 45, complaining of hearing voices, feeling unsafe, and asking for admission. It’s now 2
Of course, like all qualified psychiatrists, you will dig a little deeper, and in doing so you learn that Mr. D has visited this hospital before and has been admitted to the psychiatry unit. Now you go from having a dearth of information to having more records than you can count.
You discover that Mr. D has a history of coming to the ED during precarious hours, with similar complaints, demanding admission.
Mr. D, you learn, is unemployed, single, and homeless. Your meticulous search through his hospital records and previous admission and discharge notes reveal that once he has slept for a night, eaten a hot meal, and received narcotics for his back pain and benzodiazepines for his “symptoms” he demands to leave the hospital. His psychotic symptoms disappear despite his consistent refusal to take antipsychotics throughout his stay.
Now, what would you do?
As earnest medical students and psychiatrists, we enjoy helping patients on their path toward recovery. We want to advocate for our patients and give them the benefit of the doubt. We’re taught in medical school to be non-judgmental and invite patients to share their narrative. But through experience, we start to become aware of malingering.
Suspecting malingering, diagnosed as a condition, often is avoided by psychiatrists.1 This makes sense—it goes against the essence of our training and imposes a pejorative label on someone who has reached out for help.
Often persons with mental illness will suffer for years until they to receive help.2 That’s exactly why, when patients like Mr. D come to the ED and report hearing voices, we’re not likely to shout, “Liar!” and invite them to leave.
However, malingering is a real problem, especially because the number of psychiatric hospital beds have dwindled to record lows, thereby overcrowding EDs. Resources are skimpy, and clinicians want to help those who need it the most and not waste resources on someone who is “faking it” for secondary gain.
To navigate this diagnostic challenge, psychiatrists need the skills to detect malingering and the confidence to deal with it appropriately. This article aims to:
- define psychosis and malingering
- review the prevalence and historical considerations of malingering
- offer practical strategies to deal with malingering.
Know the real thing
Clinicians first must have the clinical acumen and expertise to identify a true mental illness such as psychosis2 (Table 1). The differential diagnosis for psychotic symptoms is broad. The astute clinician might suspect that untreated bipolar disorder or depression led to the emergence of perceptual disturbances or disordered thinking. Transient psychotic symptoms can be associated with trauma disorders, borderline personality disorder, and acute intoxication. Psychotic spectrum disorders range from brief psychosis to schizophreniform to schizoaffective disorder or schizophrenia.
Malingering—which is a condition, not a diagnosis—is characterized by the intentional production of false or grossly exaggerated physical or psychological symptoms motivated by external incentives.3,4 The presence of external incentives differentiates malingering from true psychiatric disorders, including factitious disorder, somatoform disorder, and dissociative disorder, and specific medical conditions.1 In those disorders, there is no external incentive.
It is important to remember that malingering can coexist with a serious mental illness. For example, a truly psychotic person might malinger, feign, or exaggerate symptoms to try to receive much needed help. Individuals with true psychosis might have become disenchanted with the mental health system, and thereby have a tendency to over-report or exaggerate symptoms in an effort to obtain treatment. This also could explain why many clinicians intuitively are reluctant to make the determination that someone is malingering. Malingering also can be present in an individual who has antisocial personality disorder, factitious disorder, Ganser syndrome, and Munchausen syndrome.4 When symptoms or diseases that either are thought to be exaggerated or do not exist, consider a diagnosis of malingering.
A key challenge in any discussion of abnormal health care–seeking behavior is the extent to which a person’s reported symptoms are considered to be a product of choice, psychopathology beyond volitional control, or perhaps both. Clinical skills alone typically are not sufficient for diagnosing or detecting malingering. Medical education needs to provide doctors with the conceptual, developmental, and management frameworks to understand and manage patients whose symptoms appear to be simulated. Central to understanding factitious disorders and malingering are the explanatory models and beliefs used to provide meaning for both patients and doctors.7
When considering malingered psychosis, the suspecting physician must stay alert to possible motives. Also, the patient’s presentation might provide some clues when there is marked variability, such as discrepancies in the history, gross inconsistencies, or blatant contradictions. Hallucinations are a heterogeneous experience, and discerning between true vs feigned symptoms can be challenging for even the seasoned clinician. It can be helpful to study the phenomenology of typical vs atypical hallucinatory symptoms.8 Examples of atypical symptoms include:
- vague hallucinations
- experiencing hallucinations of only 1 sensory modality (such as voices alone, visual images in black and white only)
- delusions that have an abrupt onset
- bizarre content without disordered thinking.2,6,9,10
The truth about an untruthful condition
Although the exact prevalence of malingering varies by circumstance, Rissmiller et al12,13 demonstrated—and later replicated—a prevalence of approximately 10% among patients hospitalized for suicidal ideation or suicide attempts. Studies have demonstrated even higher prevalence within forensic populations, which seems reasonable because evading criminal responsibility is a large incentive to feign symptoms. Studies also have shown that 5% of military recruits will feign symptoms to avoid service. Moreover, 1% of psychiatric patients, such as Mr. D, feign symptoms for secondary gain.13
Although there are no psychometrically validated assessment tools to distinguish between real vs feigned hallucinations, several standardized tests can help tease out the truth.9 The preferred personality test used in forensic settings is the Minnesota Multiphasic Personality Inventory,14 which consists of 567 items, with 10 clinical scales and several validity scales. The F scale, “faking good” or “faking bad,” detects people who are answering questions with the goal of appearing better or worse than they actually are. In studies of patients hospitalized for being at risk for suicide who were administered tests of self-reported malingering, approximately 10% of people admitted to psychiatric units were “faking” their symptoms.14
It is important to identify malingering from a professional and public health standpoint. Society incurs incremental costs when a person uses dwindling mental health resources for their own reward, leaving others to suffer without treatment. The number of psychiatric hospital beds has fallen from half a million in the 1950s to approximately 100,000 today.15
Practical guidelines
Malingering presents specific challenges to clinicians, such as:
- diagnostic uncertainty
- inaccurately branding one a liar
- countertransference
- personal reactions.
Our ethical and fiduciary responsibility is to our patient. In examining the art in medicine, it has been suggested that malingering could be viewed as an immature or primitive defense.16
Although there often is suspicion that a person is malingering, a definitive statement of such must be confirmed. Without clarity, labeling an individual as a malingerer could have detrimental effects to his (her) future care, defames his character, and places a thoughtless examiner at risk of a lawsuit. Confirmation can be achieved by observation or psychological testing methods.
Observation. When in doubt of what to do with someone such as Mr. D, there is little harm in acting prudently by holding him in a controlled setting—whether keeping him overnight in an ED or admitting him for a brief psychiatric stay. By observing someone in a controlled environment, where there are multiple professional watchful eyes, inferences will be more accurate.1
Structured assessments have been developed to help detect malingering—one example is the Test of Memory Malingering—however, in daily practice, the physician generally should suspect malingering when there are tangible incentives and when reported symptoms do not match the physical examination or there is no organic basis for the physical complaints.17 Detecting illness deception relies on converging evidence sources, including detailed interview assessments, clinical notes, and consultations.7
When you feel certain that you are encountering someone who is malingering, the final step is to get a consult. Malingering is a serious label and warrants due diligence by the provider, rather than a haphazard guess that a patient is lying. Once you receive confirmatory opinions, great care should be taken in documenting a clear and accurate note that will benefit your clinical counterpart who might encounter a patient such as Mr. D when he (she) shows up again, and will go a long way toward appropriately directing his care.
Imagine you’re on call in a busy emergency department (ED) overnight. Things are tough. The consults are piling up, no one is returning your calls for collateral information, and you’re dealing with a myriad of emergencies.
In walks Mr. D, age 45, complaining of hearing voices, feeling unsafe, and asking for admission. It’s now 2
Of course, like all qualified psychiatrists, you will dig a little deeper, and in doing so you learn that Mr. D has visited this hospital before and has been admitted to the psychiatry unit. Now you go from having a dearth of information to having more records than you can count.
You discover that Mr. D has a history of coming to the ED during precarious hours, with similar complaints, demanding admission.
Mr. D, you learn, is unemployed, single, and homeless. Your meticulous search through his hospital records and previous admission and discharge notes reveal that once he has slept for a night, eaten a hot meal, and received narcotics for his back pain and benzodiazepines for his “symptoms” he demands to leave the hospital. His psychotic symptoms disappear despite his consistent refusal to take antipsychotics throughout his stay.
Now, what would you do?
As earnest medical students and psychiatrists, we enjoy helping patients on their path toward recovery. We want to advocate for our patients and give them the benefit of the doubt. We’re taught in medical school to be non-judgmental and invite patients to share their narrative. But through experience, we start to become aware of malingering.
Suspecting malingering, diagnosed as a condition, often is avoided by psychiatrists.1 This makes sense—it goes against the essence of our training and imposes a pejorative label on someone who has reached out for help.
Often persons with mental illness will suffer for years until they to receive help.2 That’s exactly why, when patients like Mr. D come to the ED and report hearing voices, we’re not likely to shout, “Liar!” and invite them to leave.
However, malingering is a real problem, especially because the number of psychiatric hospital beds have dwindled to record lows, thereby overcrowding EDs. Resources are skimpy, and clinicians want to help those who need it the most and not waste resources on someone who is “faking it” for secondary gain.
To navigate this diagnostic challenge, psychiatrists need the skills to detect malingering and the confidence to deal with it appropriately. This article aims to:
- define psychosis and malingering
- review the prevalence and historical considerations of malingering
- offer practical strategies to deal with malingering.
Know the real thing
Clinicians first must have the clinical acumen and expertise to identify a true mental illness such as psychosis2 (Table 1). The differential diagnosis for psychotic symptoms is broad. The astute clinician might suspect that untreated bipolar disorder or depression led to the emergence of perceptual disturbances or disordered thinking. Transient psychotic symptoms can be associated with trauma disorders, borderline personality disorder, and acute intoxication. Psychotic spectrum disorders range from brief psychosis to schizophreniform to schizoaffective disorder or schizophrenia.
Malingering—which is a condition, not a diagnosis—is characterized by the intentional production of false or grossly exaggerated physical or psychological symptoms motivated by external incentives.3,4 The presence of external incentives differentiates malingering from true psychiatric disorders, including factitious disorder, somatoform disorder, and dissociative disorder, and specific medical conditions.1 In those disorders, there is no external incentive.
It is important to remember that malingering can coexist with a serious mental illness. For example, a truly psychotic person might malinger, feign, or exaggerate symptoms to try to receive much needed help. Individuals with true psychosis might have become disenchanted with the mental health system, and thereby have a tendency to over-report or exaggerate symptoms in an effort to obtain treatment. This also could explain why many clinicians intuitively are reluctant to make the determination that someone is malingering. Malingering also can be present in an individual who has antisocial personality disorder, factitious disorder, Ganser syndrome, and Munchausen syndrome.4 When symptoms or diseases that either are thought to be exaggerated or do not exist, consider a diagnosis of malingering.
A key challenge in any discussion of abnormal health care–seeking behavior is the extent to which a person’s reported symptoms are considered to be a product of choice, psychopathology beyond volitional control, or perhaps both. Clinical skills alone typically are not sufficient for diagnosing or detecting malingering. Medical education needs to provide doctors with the conceptual, developmental, and management frameworks to understand and manage patients whose symptoms appear to be simulated. Central to understanding factitious disorders and malingering are the explanatory models and beliefs used to provide meaning for both patients and doctors.7
When considering malingered psychosis, the suspecting physician must stay alert to possible motives. Also, the patient’s presentation might provide some clues when there is marked variability, such as discrepancies in the history, gross inconsistencies, or blatant contradictions. Hallucinations are a heterogeneous experience, and discerning between true vs feigned symptoms can be challenging for even the seasoned clinician. It can be helpful to study the phenomenology of typical vs atypical hallucinatory symptoms.8 Examples of atypical symptoms include:
- vague hallucinations
- experiencing hallucinations of only 1 sensory modality (such as voices alone, visual images in black and white only)
- delusions that have an abrupt onset
- bizarre content without disordered thinking.2,6,9,10
The truth about an untruthful condition
Although the exact prevalence of malingering varies by circumstance, Rissmiller et al12,13 demonstrated—and later replicated—a prevalence of approximately 10% among patients hospitalized for suicidal ideation or suicide attempts. Studies have demonstrated even higher prevalence within forensic populations, which seems reasonable because evading criminal responsibility is a large incentive to feign symptoms. Studies also have shown that 5% of military recruits will feign symptoms to avoid service. Moreover, 1% of psychiatric patients, such as Mr. D, feign symptoms for secondary gain.13
Although there are no psychometrically validated assessment tools to distinguish between real vs feigned hallucinations, several standardized tests can help tease out the truth.9 The preferred personality test used in forensic settings is the Minnesota Multiphasic Personality Inventory,14 which consists of 567 items, with 10 clinical scales and several validity scales. The F scale, “faking good” or “faking bad,” detects people who are answering questions with the goal of appearing better or worse than they actually are. In studies of patients hospitalized for being at risk for suicide who were administered tests of self-reported malingering, approximately 10% of people admitted to psychiatric units were “faking” their symptoms.14
It is important to identify malingering from a professional and public health standpoint. Society incurs incremental costs when a person uses dwindling mental health resources for their own reward, leaving others to suffer without treatment. The number of psychiatric hospital beds has fallen from half a million in the 1950s to approximately 100,000 today.15
Practical guidelines
Malingering presents specific challenges to clinicians, such as:
- diagnostic uncertainty
- inaccurately branding one a liar
- countertransference
- personal reactions.
Our ethical and fiduciary responsibility is to our patient. In examining the art in medicine, it has been suggested that malingering could be viewed as an immature or primitive defense.16
Although there often is suspicion that a person is malingering, a definitive statement of such must be confirmed. Without clarity, labeling an individual as a malingerer could have detrimental effects to his (her) future care, defames his character, and places a thoughtless examiner at risk of a lawsuit. Confirmation can be achieved by observation or psychological testing methods.
Observation. When in doubt of what to do with someone such as Mr. D, there is little harm in acting prudently by holding him in a controlled setting—whether keeping him overnight in an ED or admitting him for a brief psychiatric stay. By observing someone in a controlled environment, where there are multiple professional watchful eyes, inferences will be more accurate.1
Structured assessments have been developed to help detect malingering—one example is the Test of Memory Malingering—however, in daily practice, the physician generally should suspect malingering when there are tangible incentives and when reported symptoms do not match the physical examination or there is no organic basis for the physical complaints.17 Detecting illness deception relies on converging evidence sources, including detailed interview assessments, clinical notes, and consultations.7
When you feel certain that you are encountering someone who is malingering, the final step is to get a consult. Malingering is a serious label and warrants due diligence by the provider, rather than a haphazard guess that a patient is lying. Once you receive confirmatory opinions, great care should be taken in documenting a clear and accurate note that will benefit your clinical counterpart who might encounter a patient such as Mr. D when he (she) shows up again, and will go a long way toward appropriately directing his care.
1. LoPiccolo CJ, Goodkin K, Baldewicz TT. Current issues in the diagnosis and management of malingering. Ann Med. 1999;31(3):166-174.
2. Resnick PJ, Knoll J. Faking it: how to detect malingered psychosis. Current Psychiatry. 2005;4(11):12-25.
3. Sadock VA. Kaplan and Sadock’s synopsis of psychiatry: behavioral sciences/clinical psychiatry. 10th ed. Philadelphia, PA: Lippincott, Williams & Wilkins; 2007:887.
4. Gorman WF. Defining malingering. J Forensic Sci. 1982;27(2):401-407.
5. Mendelson G, Mendelson D. Malingering pain in the medicolegal context. Clin J Pain. 2004;20(6):423-432.
6. Resnick PJ. Malingered psychosis. In: Rogers R, ed. Clinical assessment of malingering and deception. 2nd ed. New York, NY: The Guilford Press; 1997:47-67.
7. Bass C, Halligan P. Factitious disorders and malingering: challenges for clinical assessment and management. Lancet. 2014;383(9926):1422-1432.
8. McCarthy-Jones S, Resnick PJ. Listening to the voices: the use of phenomenology to differentiate malingered from genuine auditory verbal hallucinations. Int J Law Psychiatry. 2014;37(2):183-189.
9. Resnick PJ. Defrocking the fraud: the detection of malingering. Isr J Psychiatry Relat Sci. 1993;30(2):93-101.
10. Nayani TH, David AS. The auditory hallucination: a phenomenological survey. Psychol Med. 1996;26(1):177-189.
11. Pollock P. Feigning auditory hallucinations by offenders. Journal of Forensic Psychiatry. 1998;9(2)305-327.
12. Rissmiller DJ, Wayslow A, Madison H, et al. Prevalence of malingering in inpatient suicide ideators and attempters. Crisis. 1998;19(2):62-66.
13. Rissmiller DA, Steer RA, Friedman M, et al. Prevalence of malingering in suicidal psychiatric patients: a replication. Psychol Rep. 1999;84(3 pt 1):726-730.
14. Hathaway SR, McKinley JC. The Minnesota Multiphasic Personality Inventory-2. Minneapolis, MN: University of Minnesota Press; 1989.
15. Szabo L. Cost of not caring: Stigma set in stone. USA Today. http://www.usatoday.com/story/news/nation/2014/06/25/stigma-of-mental-illness/9875351. Published June 25, 2014. Accessed May 5, 2017.
16. Malone RD, Lange CL. A clinical approach to the malingering patient. J Am Acad Psychoanal Dyn Psychiatry. 2007;35(1):13-21.
17. McDermott BE, Feldman MD. Malingering in the medical setting. Psychiatr Clin North Am. 2007;30(4):645-662.
1. LoPiccolo CJ, Goodkin K, Baldewicz TT. Current issues in the diagnosis and management of malingering. Ann Med. 1999;31(3):166-174.
2. Resnick PJ, Knoll J. Faking it: how to detect malingered psychosis. Current Psychiatry. 2005;4(11):12-25.
3. Sadock VA. Kaplan and Sadock’s synopsis of psychiatry: behavioral sciences/clinical psychiatry. 10th ed. Philadelphia, PA: Lippincott, Williams & Wilkins; 2007:887.
4. Gorman WF. Defining malingering. J Forensic Sci. 1982;27(2):401-407.
5. Mendelson G, Mendelson D. Malingering pain in the medicolegal context. Clin J Pain. 2004;20(6):423-432.
6. Resnick PJ. Malingered psychosis. In: Rogers R, ed. Clinical assessment of malingering and deception. 2nd ed. New York, NY: The Guilford Press; 1997:47-67.
7. Bass C, Halligan P. Factitious disorders and malingering: challenges for clinical assessment and management. Lancet. 2014;383(9926):1422-1432.
8. McCarthy-Jones S, Resnick PJ. Listening to the voices: the use of phenomenology to differentiate malingered from genuine auditory verbal hallucinations. Int J Law Psychiatry. 2014;37(2):183-189.
9. Resnick PJ. Defrocking the fraud: the detection of malingering. Isr J Psychiatry Relat Sci. 1993;30(2):93-101.
10. Nayani TH, David AS. The auditory hallucination: a phenomenological survey. Psychol Med. 1996;26(1):177-189.
11. Pollock P. Feigning auditory hallucinations by offenders. Journal of Forensic Psychiatry. 1998;9(2)305-327.
12. Rissmiller DJ, Wayslow A, Madison H, et al. Prevalence of malingering in inpatient suicide ideators and attempters. Crisis. 1998;19(2):62-66.
13. Rissmiller DA, Steer RA, Friedman M, et al. Prevalence of malingering in suicidal psychiatric patients: a replication. Psychol Rep. 1999;84(3 pt 1):726-730.
14. Hathaway SR, McKinley JC. The Minnesota Multiphasic Personality Inventory-2. Minneapolis, MN: University of Minnesota Press; 1989.
15. Szabo L. Cost of not caring: Stigma set in stone. USA Today. http://www.usatoday.com/story/news/nation/2014/06/25/stigma-of-mental-illness/9875351. Published June 25, 2014. Accessed May 5, 2017.
16. Malone RD, Lange CL. A clinical approach to the malingering patient. J Am Acad Psychoanal Dyn Psychiatry. 2007;35(1):13-21.
17. McDermott BE, Feldman MD. Malingering in the medical setting. Psychiatr Clin North Am. 2007;30(4):645-662.
