User login
What role does asthma medication have in ADHD or depression?
Asthma medications comprise several drug classes, including leukotriene antagonists and steroid-based inhalers. These drugs have been implicated in behavioral changes, such as increased hyperactivity, similar to symptoms of attention-deficit/hyperactivity disorder (ADHD) and oppositional defiant disorder (ODD)1; this scenario is more of a concern in children than adults. This raises the question of whether these medications are physiologically linked to behavioral symptoms because of a suggested association with serotonin.2,3 If this is the case, it is necessary to identify and evaluate possible psychiatric effects of these asthma agents.
How asthma medications work
Some asthma agents, such as montelukast, act as either leukotriene-related enzyme inhibitors (arachidonate 5-lipoxygenase) or leukotriene receptor antagonists. These drugs block production of inflammatory leukotrienes, which cause bronchoconstriction. Leukotrienes also can trigger cytokine synthesis, which can modulate leukotriene receptor function. Therefore, leukotriene antagonists could interfere with cytokine function.3,4
Corticosteroid inhalers suppress inflammatory genes by reversing histone acetylation of inflammatory genes involved in asthma. These inhalers have been shown to reduce cytokine levels in patients with chronic lung disease and those with moderate to
Possible link between asthma and serotonin
Serotonin plays an integral role in observable, dysfunctional behaviors seen in disorders such as ADHD and ODD. In previous studies, serotonin modulated the cytokine network, and patients with asthma had elevated levels of plasma serotonin.2,3 These findings imply that asthma medications could be involved in altering levels of both cytokines and serotonin. Pretorius2 emphasized the importance of monitoring serotonin levels in children who exhibit behavioral dysfunction based on these observations:
- Persons with asthma presenting with medical symptoms have elevated serotonin levels.
- Decreased serotonin levels have been associated with ADHD and ODD; medications for ADHD have been shown to increase serotonin levels.
- Asthma medications have been shown to decrease serotonin levels.2,3
Asthma medications might be partially responsible for behavioral disturbances, and therapeutic management should integrate the role of serotonin with asthma therapy.2,3
Clinical considerations
Therapeutic management of asthma should consider psychiatric conditions and treatments. Future research should investigate the overall predisposition for behavioral dysfunction in persons with respiratory syncytial virus, a precursor for asthma. Once an asthma patient’s risk of a psychiatric disorder has been identified, the clinician can determine the most effective medications for treating the condition. If potential medications or genetic or environmental factors are identified, we might expect a move toward personalized care in the not too distant future.
1. Saricoban HE, Ozen A, Harmanci K, et al. Common behavioral problems among children with asthma: is there a role of asthma treatment? Ann Allergy Asthma Immunol. 2011;106(3):200-204.
2. Pretorius E. Asthma medication may influence the psychological functioning of children. Med Hypotheses. 2004;63(3):409-413.
3. Ménard G, Turmei V, Bissonnette EY. Serotonin modulates the cytokine network in the lung: involvement of prostaglandin E2. Clin Exp Immunol. 2007;150(2):340-348.
4. Rola-Pleszczynski M, Stankova J. Cytokine-leukotriene receptor interactions. Scientific World Journal. 2007;7:1348-1358.
5. Kaur M, Reynolds S, Smyth LJ, et al. The effects of corticosteroids on cytokine production from asthma lung lymphocytes. Int Immunopharmacol. 2014;23(2):581-584.
6. Honda R, Ichiyama T, Sunagawa S, et al. Inhaled corticosteroid therapy reduces cytokine levels in sputum from very preterm infants with chronic lung disease. Acta Paediatr. 2009;98(1):118-122.
7. Pretorius E. Corticosteroids, depression and the role of serotonin. Rev Neurosci. 2004;15(2):109-116.
Asthma medications comprise several drug classes, including leukotriene antagonists and steroid-based inhalers. These drugs have been implicated in behavioral changes, such as increased hyperactivity, similar to symptoms of attention-deficit/hyperactivity disorder (ADHD) and oppositional defiant disorder (ODD)1; this scenario is more of a concern in children than adults. This raises the question of whether these medications are physiologically linked to behavioral symptoms because of a suggested association with serotonin.2,3 If this is the case, it is necessary to identify and evaluate possible psychiatric effects of these asthma agents.
How asthma medications work
Some asthma agents, such as montelukast, act as either leukotriene-related enzyme inhibitors (arachidonate 5-lipoxygenase) or leukotriene receptor antagonists. These drugs block production of inflammatory leukotrienes, which cause bronchoconstriction. Leukotrienes also can trigger cytokine synthesis, which can modulate leukotriene receptor function. Therefore, leukotriene antagonists could interfere with cytokine function.3,4
Corticosteroid inhalers suppress inflammatory genes by reversing histone acetylation of inflammatory genes involved in asthma. These inhalers have been shown to reduce cytokine levels in patients with chronic lung disease and those with moderate to
Possible link between asthma and serotonin
Serotonin plays an integral role in observable, dysfunctional behaviors seen in disorders such as ADHD and ODD. In previous studies, serotonin modulated the cytokine network, and patients with asthma had elevated levels of plasma serotonin.2,3 These findings imply that asthma medications could be involved in altering levels of both cytokines and serotonin. Pretorius2 emphasized the importance of monitoring serotonin levels in children who exhibit behavioral dysfunction based on these observations:
- Persons with asthma presenting with medical symptoms have elevated serotonin levels.
- Decreased serotonin levels have been associated with ADHD and ODD; medications for ADHD have been shown to increase serotonin levels.
- Asthma medications have been shown to decrease serotonin levels.2,3
Asthma medications might be partially responsible for behavioral disturbances, and therapeutic management should integrate the role of serotonin with asthma therapy.2,3
Clinical considerations
Therapeutic management of asthma should consider psychiatric conditions and treatments. Future research should investigate the overall predisposition for behavioral dysfunction in persons with respiratory syncytial virus, a precursor for asthma. Once an asthma patient’s risk of a psychiatric disorder has been identified, the clinician can determine the most effective medications for treating the condition. If potential medications or genetic or environmental factors are identified, we might expect a move toward personalized care in the not too distant future.
Asthma medications comprise several drug classes, including leukotriene antagonists and steroid-based inhalers. These drugs have been implicated in behavioral changes, such as increased hyperactivity, similar to symptoms of attention-deficit/hyperactivity disorder (ADHD) and oppositional defiant disorder (ODD)1; this scenario is more of a concern in children than adults. This raises the question of whether these medications are physiologically linked to behavioral symptoms because of a suggested association with serotonin.2,3 If this is the case, it is necessary to identify and evaluate possible psychiatric effects of these asthma agents.
How asthma medications work
Some asthma agents, such as montelukast, act as either leukotriene-related enzyme inhibitors (arachidonate 5-lipoxygenase) or leukotriene receptor antagonists. These drugs block production of inflammatory leukotrienes, which cause bronchoconstriction. Leukotrienes also can trigger cytokine synthesis, which can modulate leukotriene receptor function. Therefore, leukotriene antagonists could interfere with cytokine function.3,4
Corticosteroid inhalers suppress inflammatory genes by reversing histone acetylation of inflammatory genes involved in asthma. These inhalers have been shown to reduce cytokine levels in patients with chronic lung disease and those with moderate to
Possible link between asthma and serotonin
Serotonin plays an integral role in observable, dysfunctional behaviors seen in disorders such as ADHD and ODD. In previous studies, serotonin modulated the cytokine network, and patients with asthma had elevated levels of plasma serotonin.2,3 These findings imply that asthma medications could be involved in altering levels of both cytokines and serotonin. Pretorius2 emphasized the importance of monitoring serotonin levels in children who exhibit behavioral dysfunction based on these observations:
- Persons with asthma presenting with medical symptoms have elevated serotonin levels.
- Decreased serotonin levels have been associated with ADHD and ODD; medications for ADHD have been shown to increase serotonin levels.
- Asthma medications have been shown to decrease serotonin levels.2,3
Asthma medications might be partially responsible for behavioral disturbances, and therapeutic management should integrate the role of serotonin with asthma therapy.2,3
Clinical considerations
Therapeutic management of asthma should consider psychiatric conditions and treatments. Future research should investigate the overall predisposition for behavioral dysfunction in persons with respiratory syncytial virus, a precursor for asthma. Once an asthma patient’s risk of a psychiatric disorder has been identified, the clinician can determine the most effective medications for treating the condition. If potential medications or genetic or environmental factors are identified, we might expect a move toward personalized care in the not too distant future.
1. Saricoban HE, Ozen A, Harmanci K, et al. Common behavioral problems among children with asthma: is there a role of asthma treatment? Ann Allergy Asthma Immunol. 2011;106(3):200-204.
2. Pretorius E. Asthma medication may influence the psychological functioning of children. Med Hypotheses. 2004;63(3):409-413.
3. Ménard G, Turmei V, Bissonnette EY. Serotonin modulates the cytokine network in the lung: involvement of prostaglandin E2. Clin Exp Immunol. 2007;150(2):340-348.
4. Rola-Pleszczynski M, Stankova J. Cytokine-leukotriene receptor interactions. Scientific World Journal. 2007;7:1348-1358.
5. Kaur M, Reynolds S, Smyth LJ, et al. The effects of corticosteroids on cytokine production from asthma lung lymphocytes. Int Immunopharmacol. 2014;23(2):581-584.
6. Honda R, Ichiyama T, Sunagawa S, et al. Inhaled corticosteroid therapy reduces cytokine levels in sputum from very preterm infants with chronic lung disease. Acta Paediatr. 2009;98(1):118-122.
7. Pretorius E. Corticosteroids, depression and the role of serotonin. Rev Neurosci. 2004;15(2):109-116.
1. Saricoban HE, Ozen A, Harmanci K, et al. Common behavioral problems among children with asthma: is there a role of asthma treatment? Ann Allergy Asthma Immunol. 2011;106(3):200-204.
2. Pretorius E. Asthma medication may influence the psychological functioning of children. Med Hypotheses. 2004;63(3):409-413.
3. Ménard G, Turmei V, Bissonnette EY. Serotonin modulates the cytokine network in the lung: involvement of prostaglandin E2. Clin Exp Immunol. 2007;150(2):340-348.
4. Rola-Pleszczynski M, Stankova J. Cytokine-leukotriene receptor interactions. Scientific World Journal. 2007;7:1348-1358.
5. Kaur M, Reynolds S, Smyth LJ, et al. The effects of corticosteroids on cytokine production from asthma lung lymphocytes. Int Immunopharmacol. 2014;23(2):581-584.
6. Honda R, Ichiyama T, Sunagawa S, et al. Inhaled corticosteroid therapy reduces cytokine levels in sputum from very preterm infants with chronic lung disease. Acta Paediatr. 2009;98(1):118-122.
7. Pretorius E. Corticosteroids, depression and the role of serotonin. Rev Neurosci. 2004;15(2):109-116.
6 Steps to deprescribing: A practical approach
Taking over the care of a patient with a complex medication regimen consisting of multiple psychotropics is a common experience for many practicing psychiatrists. Increasin
We describe a pragmatic approach to deprescribing, outlining 6 steps that we have used successfully in several treatment settings, which can assist prescribers facing similar challenges in their own practices.
1. Obtain a detailed history. First compile a comprehensive list of the patient’s medications, including psychotropics, other drugs, and supplements. If necessary, coordinate with your patient’s primary care provider. Then reassess the patient’s history of illness and efficacy of pharmacologic and non-pharmacologic treatments and how the current regimen has evolved. Understand the patient’s course of illness, coping styles, strengths, and vulnerabilities with an eye toward deprescribing.
2. Investigate underlying meaning. Even the most biologically oriented prescribers can benefit from exploring the underlying meaning the patient ascribes to the medication regimen. Common themes include:
- hesitation to relinquish a complex medication regimen because the patient fears decompensation (which could be either realistic or unrealistic)
- attachment to the “sick role”
- interpreting the complex regimen as evidence of the provider’s care and concern.
A series of sensitive conversations exploring these factors and addressing their underlying meaning can help increase a patient’s trust in the process of deprescribing.
3. Assess risk vs benefit. Weigh and educate the patient on the potential risks and benefits of each medication, as well as drug interactions and additive side effects.
4. Start with:
The most risky. Medications with significant risk for serious adverse effects (eg, high doses of a QTc-prolonging medication in a patient with elevated QTc) should be targeted early.
The least likely to be missed. If there are no high-risk medications that need to take priority, discontinuation of a “redundant” medication, such as a low-dose antihistamine prescribed with multiple other sedating medications, can be an achievable first step. By starting with a medication that the patient is unlikely to miss, the provider can make efficient initial progress while building patient confidence in the deprescribing process.
Medication the patient is most motivated to discontinue. This strategy can enhance the therapeutic alliance and increase the likelihood of successful patient engagement for patients hesitant to decrease medications, so long as there are no significant contraindications to discontinuing the medication.
5. Go slowly. As long as there are no medications that put the patient at risk and require rapid discontinuation, going slowly increases the likelihood of long-term success by:
- permitting careful monitoring for any worsening symptoms
- allowing more time for physiologic readjustment
- enabling the patient and provider to build confidence in the process over time.
With slow discontinuation, normal emotions, such as transient, situationally appropriate anxiety about a life stressor, are less likely to be misinterpreted by the patient or provider as an inability to tolerate medication reduction because there is more opportunity to observe overall trends in symptoms.
6. Replace medications with alternatives. Offering non-pharmacological treatment when possible can greatly facilitate reducing the number of medications. Examples include:
- teaching a patient breathing exercises or mindfulness while preparing to decrease an as needed anxiolytic
- engaging the patient in cognitive-behavioral therapy for insomnia before reducing sleep medications
- working together to identify opportunities for behavioral activation and exercises that are the most achievable for the patient.
This replacement strategy can work in a physiologic sense and address a patient’s fear that medications are “taken away” without alternatives in place.
Although these strategies might not work for every patient and are not recommended for reducing medications that are medically necessary, using this approach will increase the likelihood of long-term success and maintain the patient–provider alliance when reducing unnecessary and potentially risky polypharmacy. An article by Gupta and Cahill1 describes some similar approaches with additional discussion and considerations.
1. Gupta S, Cahill JD. A prescription for “deprescribing” in psychiatry. Psychiatr Serv. 2016;67(8):904-907.
2. Scott IA, Hilmer SN, Reeve E, et al. Reducing inappropriate polypharmacy: the process of deprescribing. JAMA Intern Med. 2015;175(5):827-834.
Taking over the care of a patient with a complex medication regimen consisting of multiple psychotropics is a common experience for many practicing psychiatrists. Increasin
We describe a pragmatic approach to deprescribing, outlining 6 steps that we have used successfully in several treatment settings, which can assist prescribers facing similar challenges in their own practices.
1. Obtain a detailed history. First compile a comprehensive list of the patient’s medications, including psychotropics, other drugs, and supplements. If necessary, coordinate with your patient’s primary care provider. Then reassess the patient’s history of illness and efficacy of pharmacologic and non-pharmacologic treatments and how the current regimen has evolved. Understand the patient’s course of illness, coping styles, strengths, and vulnerabilities with an eye toward deprescribing.
2. Investigate underlying meaning. Even the most biologically oriented prescribers can benefit from exploring the underlying meaning the patient ascribes to the medication regimen. Common themes include:
- hesitation to relinquish a complex medication regimen because the patient fears decompensation (which could be either realistic or unrealistic)
- attachment to the “sick role”
- interpreting the complex regimen as evidence of the provider’s care and concern.
A series of sensitive conversations exploring these factors and addressing their underlying meaning can help increase a patient’s trust in the process of deprescribing.
3. Assess risk vs benefit. Weigh and educate the patient on the potential risks and benefits of each medication, as well as drug interactions and additive side effects.
4. Start with:
The most risky. Medications with significant risk for serious adverse effects (eg, high doses of a QTc-prolonging medication in a patient with elevated QTc) should be targeted early.
The least likely to be missed. If there are no high-risk medications that need to take priority, discontinuation of a “redundant” medication, such as a low-dose antihistamine prescribed with multiple other sedating medications, can be an achievable first step. By starting with a medication that the patient is unlikely to miss, the provider can make efficient initial progress while building patient confidence in the deprescribing process.
Medication the patient is most motivated to discontinue. This strategy can enhance the therapeutic alliance and increase the likelihood of successful patient engagement for patients hesitant to decrease medications, so long as there are no significant contraindications to discontinuing the medication.
5. Go slowly. As long as there are no medications that put the patient at risk and require rapid discontinuation, going slowly increases the likelihood of long-term success by:
- permitting careful monitoring for any worsening symptoms
- allowing more time for physiologic readjustment
- enabling the patient and provider to build confidence in the process over time.
With slow discontinuation, normal emotions, such as transient, situationally appropriate anxiety about a life stressor, are less likely to be misinterpreted by the patient or provider as an inability to tolerate medication reduction because there is more opportunity to observe overall trends in symptoms.
6. Replace medications with alternatives. Offering non-pharmacological treatment when possible can greatly facilitate reducing the number of medications. Examples include:
- teaching a patient breathing exercises or mindfulness while preparing to decrease an as needed anxiolytic
- engaging the patient in cognitive-behavioral therapy for insomnia before reducing sleep medications
- working together to identify opportunities for behavioral activation and exercises that are the most achievable for the patient.
This replacement strategy can work in a physiologic sense and address a patient’s fear that medications are “taken away” without alternatives in place.
Although these strategies might not work for every patient and are not recommended for reducing medications that are medically necessary, using this approach will increase the likelihood of long-term success and maintain the patient–provider alliance when reducing unnecessary and potentially risky polypharmacy. An article by Gupta and Cahill1 describes some similar approaches with additional discussion and considerations.
Taking over the care of a patient with a complex medication regimen consisting of multiple psychotropics is a common experience for many practicing psychiatrists. Increasin
We describe a pragmatic approach to deprescribing, outlining 6 steps that we have used successfully in several treatment settings, which can assist prescribers facing similar challenges in their own practices.
1. Obtain a detailed history. First compile a comprehensive list of the patient’s medications, including psychotropics, other drugs, and supplements. If necessary, coordinate with your patient’s primary care provider. Then reassess the patient’s history of illness and efficacy of pharmacologic and non-pharmacologic treatments and how the current regimen has evolved. Understand the patient’s course of illness, coping styles, strengths, and vulnerabilities with an eye toward deprescribing.
2. Investigate underlying meaning. Even the most biologically oriented prescribers can benefit from exploring the underlying meaning the patient ascribes to the medication regimen. Common themes include:
- hesitation to relinquish a complex medication regimen because the patient fears decompensation (which could be either realistic or unrealistic)
- attachment to the “sick role”
- interpreting the complex regimen as evidence of the provider’s care and concern.
A series of sensitive conversations exploring these factors and addressing their underlying meaning can help increase a patient’s trust in the process of deprescribing.
3. Assess risk vs benefit. Weigh and educate the patient on the potential risks and benefits of each medication, as well as drug interactions and additive side effects.
4. Start with:
The most risky. Medications with significant risk for serious adverse effects (eg, high doses of a QTc-prolonging medication in a patient with elevated QTc) should be targeted early.
The least likely to be missed. If there are no high-risk medications that need to take priority, discontinuation of a “redundant” medication, such as a low-dose antihistamine prescribed with multiple other sedating medications, can be an achievable first step. By starting with a medication that the patient is unlikely to miss, the provider can make efficient initial progress while building patient confidence in the deprescribing process.
Medication the patient is most motivated to discontinue. This strategy can enhance the therapeutic alliance and increase the likelihood of successful patient engagement for patients hesitant to decrease medications, so long as there are no significant contraindications to discontinuing the medication.
5. Go slowly. As long as there are no medications that put the patient at risk and require rapid discontinuation, going slowly increases the likelihood of long-term success by:
- permitting careful monitoring for any worsening symptoms
- allowing more time for physiologic readjustment
- enabling the patient and provider to build confidence in the process over time.
With slow discontinuation, normal emotions, such as transient, situationally appropriate anxiety about a life stressor, are less likely to be misinterpreted by the patient or provider as an inability to tolerate medication reduction because there is more opportunity to observe overall trends in symptoms.
6. Replace medications with alternatives. Offering non-pharmacological treatment when possible can greatly facilitate reducing the number of medications. Examples include:
- teaching a patient breathing exercises or mindfulness while preparing to decrease an as needed anxiolytic
- engaging the patient in cognitive-behavioral therapy for insomnia before reducing sleep medications
- working together to identify opportunities for behavioral activation and exercises that are the most achievable for the patient.
This replacement strategy can work in a physiologic sense and address a patient’s fear that medications are “taken away” without alternatives in place.
Although these strategies might not work for every patient and are not recommended for reducing medications that are medically necessary, using this approach will increase the likelihood of long-term success and maintain the patient–provider alliance when reducing unnecessary and potentially risky polypharmacy. An article by Gupta and Cahill1 describes some similar approaches with additional discussion and considerations.
1. Gupta S, Cahill JD. A prescription for “deprescribing” in psychiatry. Psychiatr Serv. 2016;67(8):904-907.
2. Scott IA, Hilmer SN, Reeve E, et al. Reducing inappropriate polypharmacy: the process of deprescribing. JAMA Intern Med. 2015;175(5):827-834.
1. Gupta S, Cahill JD. A prescription for “deprescribing” in psychiatry. Psychiatr Serv. 2016;67(8):904-907.
2. Scott IA, Hilmer SN, Reeve E, et al. Reducing inappropriate polypharmacy: the process of deprescribing. JAMA Intern Med. 2015;175(5):827-834.
How to diagnose and manage hypertension in a psychiatric patient
Hypertension is a widespread, under-recognized, and undertreated cause of morbidity and mortality in the United States and is associated with several psychiatric illnesses. Left untreated, hypertension can have significant consequences, including increased risk of stroke, coronary heart disease, heart failure, chronic kidney failure, and death. Approximately 70 million adults in the United States have hypertension, but only 60% of them have been diagnosed, and of those only 50% have their blood pressure under control.1 In 2013, 360,000 deaths in the United States were attributed to hypertension.2
Hypertension is associated with major depressive disorder, generalized anxiety disorder, bipolar disorder, and schizophrenia.3-5 Additionally, impulsive eating disorders, substance abuse, anxiety, and depression are associated with a hypertension diagnosis, although patients with panic disorder develop hypertension at a younger age.6 A 2007 study found a 61% prevalence of hypertension in those with bipolar disorder compared with 41% among the general population.7 The strong link between bipolar disorder and hypertension might be because of a common disease mechanism; both are associated with hyperactive cellular calcium signaling and increased platelet intracellular calcium ion concentrations.8
Hypertension not only is common among patients with psychiatric illness, it likely contributes to worse clinical outcomes. Studies across different cultures have found higher mortality rates in individuals with mental illness.9-11 Persons with schizophrenia and other severe mental illnesses may lose ≥25 years of life expectancy, with the primary cause of death being cardiovascular disease, not suicide.12 Patients with depression have a 50% greater risk of cardiovascular disease, which is equivalent to the risk of smoking.13
Schizophrenia is strongly associated with numerous comorbidities and has been linked significantly to an elevated 10-year cardiac risk after controlling for body mass index.5 The high rate of non-treatment of hypertension for patients with schizophrenia (62.4%) is especially concerning.14
Because of the well-documented morbidity and mortality of hypertension and its increased prevalence and undertreatment in the psychiatric population, mental health providers are in an important position to recognize hypertension and evaluate its inherent risks to direct their patients toward proper treatment. This article reviews:
- the signs and symptoms of hypertension
- the mental health provider’s role in the evaluation and diagnosis
- how psychotropic drugs influence blood pressure and drug–drug interactions
- the management of hypertension in psychiatric patients, including strategies for counseling and lifestyle management.
Diagnosing hypertension
Hypertension is defined as a blood pressure >140/90 mm Hg, the average of ≥2 properly measured readings at ≥2 visits in a medical setting.15 The proper equipment, including a well-fitting blood pressure cuff, and technique to measure blood pressure are essential to avoid misdiagnosis. The patient should be at rest for ≥5 minutes, without active pain or emotional distress.
Most cases of hypertension (90% to 95%) are primary, commonly called essential hypertension. However, the differential diagnosis also should consider secondary causes, which may include:
- obesity
- medications
- chronic alcohol use
- methamphetamine or cocaine use
- primary kidney disease
- atherosclerotic renal artery stenosis
- obstructive sleep apnea
- hypothyroidism
- primary hyperaldosteronism
- narrowing of the aorta
- Cushing syndrome
- primary hyperparathyroidism
- polycythemia
- pheochromocytoma.

Medical evaluation. Once the diagnosis of hypertension is made, a medical evaluation is indicated to determine if the patient has end-organ damage from the elevated pressures, such as renal disease or heart disease, to identify other modifiable cardiovascular risk factors, such as hyperlipidemia, and to screen for secondary causes of hypertension. This evaluation includes15:
- a physical exam
- review of medications
- lipid profile
- urinalysis to screen for proteinuria
- serum electrolytes and creatinine
- electrocardiogram to screen for left ventricular hypertrophy or prior infarction
- fasting glucose or hemoglobin A1c to screen for type 2 diabetes mellitus.
Psychotropic drugs. In psychiatric patients, the evaluation must consider the potential impact psychotropic drug effects and drug–drug interactions can have on blood pressure (Table 2). For example, patients taking both diuretics and lithium are at increased risk for dehydration and increased serum lithium levels, which could cause severe neurologic symptoms and renal insufficiency.16 Several antihypertensives when taken with venlafaxine can increase blood pressure, but antihypertensives with α-1 blocking psychotropics can decrease blood pressure. Monoamine oxidase inhibitors can cause hypotension or hypertension with various classes of antihypertensives. Stimulants, such as methylphenidate, atomoxetine, dextroamphetamine, armodafinil, or modafinil, alone or combined with antihypertensives, can cause hypertension.17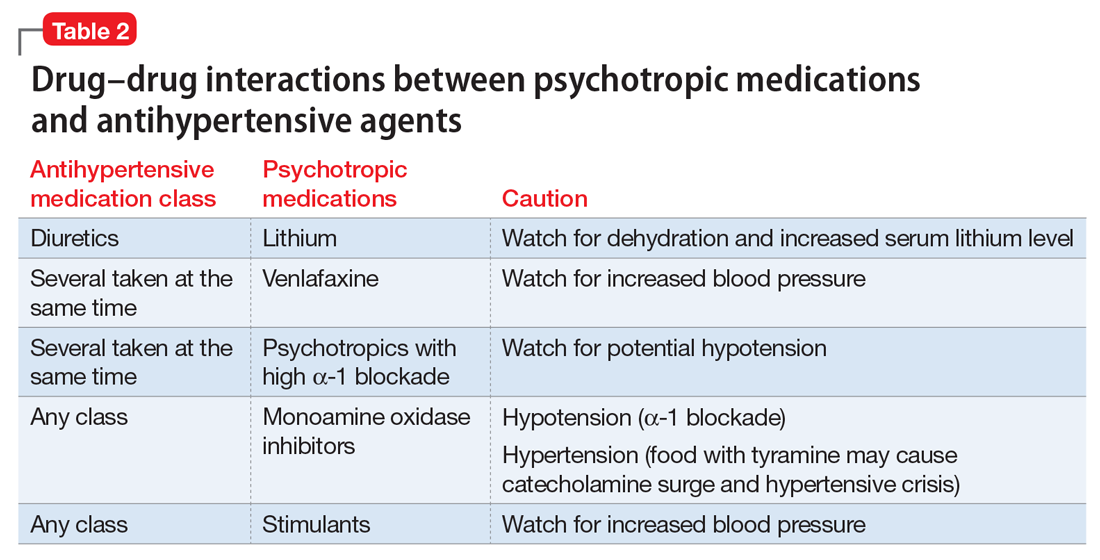
Substance abuse, particularly alcohol, methamphetamine, and cocaine, can cause difficulty controlling blood pressure. Patients with refractory hypertension should have a reassessment of substance abuse as a potential cause.
Screening guidelines for mental health providers
For many patients with severe mental illness, visits to their mental health providers might be their only contact with the medical system. Therefore, screening in the mental health settings could detect cases that otherwise would be missed.
Screening recommendations. The U.S. Preventive Services Task Force recommends screening for hypertension in the general population beginning at age 18.18 Adults age 18 to 39 with normal blood pressure (<130/85 mm Hg) and no other risk factors (eg, overweight, obese, or African American) can be screened every 3 years. Those with risk factors or a blood pressure of 130/85 to 139/89 mm Hg and adults age ≥40 should have annual screenings.
Ideally, psychiatrists and other mental health providers should monitor blood pressure at each visit, especially in patients taking psychotropics because of their higher risk for hypertension.
Optimizing treatment. Once the diagnosis of essential hypertension is established, identifying psychiatric comorbidities and the severity of psychiatric symptoms are important to optimize treatment adherence. Patients with increased depressive symptoms are less likely to comply with antihypertensive medication,19 and patients with confirmed depression are 3 times more likely to not adhere to medical treatment recommendations than non-depressed patients.20
Physicians’ attitudes toward hypertension also can affect patients’ compliance and blood pressure control.21 Psychiatrists should be empathetic and motivational toward patients attempting to control their blood pressure. The Seventh Joint National Committee on the Prevention, Detection, Evaluation, and Treatment of High Blood Pressure states, “Motivation improves when patients have positive experiences with, and trust in, the clinician. Empathy builds trust and is a potent motivator.”22
Treatment and management
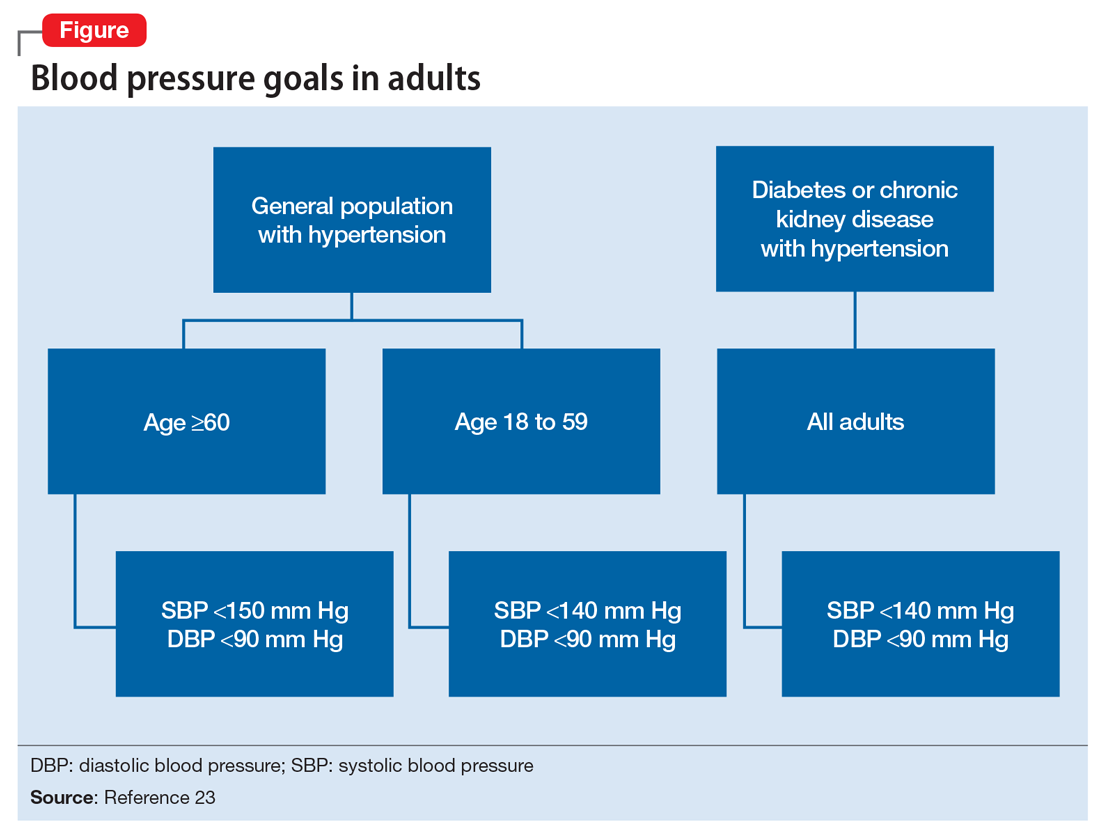
Treatment of hypertension significantly reduces the risk of stroke, myocardial infarction, renal injury, heart failure, and premature death. Studies show that treatment that reduces systolic blood pressure by 12 mm Hg over 10 years will prevent 1 death for every 11 patients with essential hypertension. In those with concomitant cardiovascular disease or target organ damage, such a reduction would prevent death in 1 of every 9 patients treated.15Blood pressure goals. The 2014 Eighth Joint National Committee Guideline for Management of High Blood Pressure in Adults provides guidance on blood pressure goals depending on patients’ underlying medical history (Figure).23 Based on expert opinion and randomized controlled studies, blood pressure goals for patients without diabetes or chronic kidney disease (CKD)—an estimated or measured glomerular filtration rate (GFR) of ≤60 mL/min/1.73 m2—depend on age: <140/90 mm Hg for age 18 to 59 and <150/90 mm Hg for age ≥60. For patients with diabetes or CKD, the blood pressure goal is <140/90 mm Hg, regardless of age.
However, not all experts agree on these specific blood pressure goals. A major trial (SPRINT) published in 2015 found that intensive blood pressure goals do benefit higher-risk, non-diabetic patients.24 Specifically, the study randomized patients age ≥50 with systolic blood pressure of 130 to 180 mm Hg and increased cardiovascular risk to systolic blood pressure targets of <140 mm Hg (standard) or <120 mm Hg (intensive). Characteristics of increased cardiovascular risk were clinical or subclinical cardiovascular disease other than stroke, CKD with GFR of 20 to 60 mL/min/1.73 m2, age ≥75, or Framingham 10-year coronary heart disease risk score ≥15%. Intensive treatment significantly reduced overall mortality and the rate of acute coronary syndrome, myocardial infarction, heart failure, stroke, or cardiovascular death. However, the results of this study have not been assimilated into any recent guidelines. Therefore, consider a goal of <120 mm Hg for non-diabetic patients age ≥50 with any of these factors.
Lifestyle modifications. Psychiatrists are well equipped to motivate and encourage behavioral modification in patients with hypertension. Counseling and structured training courses could help to effectively lower blood pressure.25 Patients should receive education on lifestyle modifications including:
- weight reduction
- physical activity
- moderate alcohol consumption
- decreased sodium consumption
- implementation of the Dietary Approaches to Stop Hypertension (DASH) or Mediterranean diets.15
Maintaining a normal body weight is ideal, but weight reduction of 10 lb can reduce blood pressure in overweight patients. The DASH diet, consisting of fruits, vegetables, low-fat dairy products, high calcium and potassium intake, and reduced saturated and total fat intake can decrease systolic blood pressure from 8 to 14 mm Hg. Reduction of sodium intake to ≤2,400 mg/d can reduce systolic blood pressure from 2 to 8 mm Hg. Regular aerobic exercise of 30 minutes a day most days of the week can reduce systolic blood pressure up to 9 mm Hg. Patients also should be encouraged to quit smoking. Patients who implement ≥2 these modifications get better results.
Antihypertensive medications. Patients who do not reach their goals with lifestyle measures alone should receive antihypertensive medications. Most patients will require ≥2 agents to control their blood pressure. Clinical trials show that some patient subgroups have better outcomes with different first-line agents.
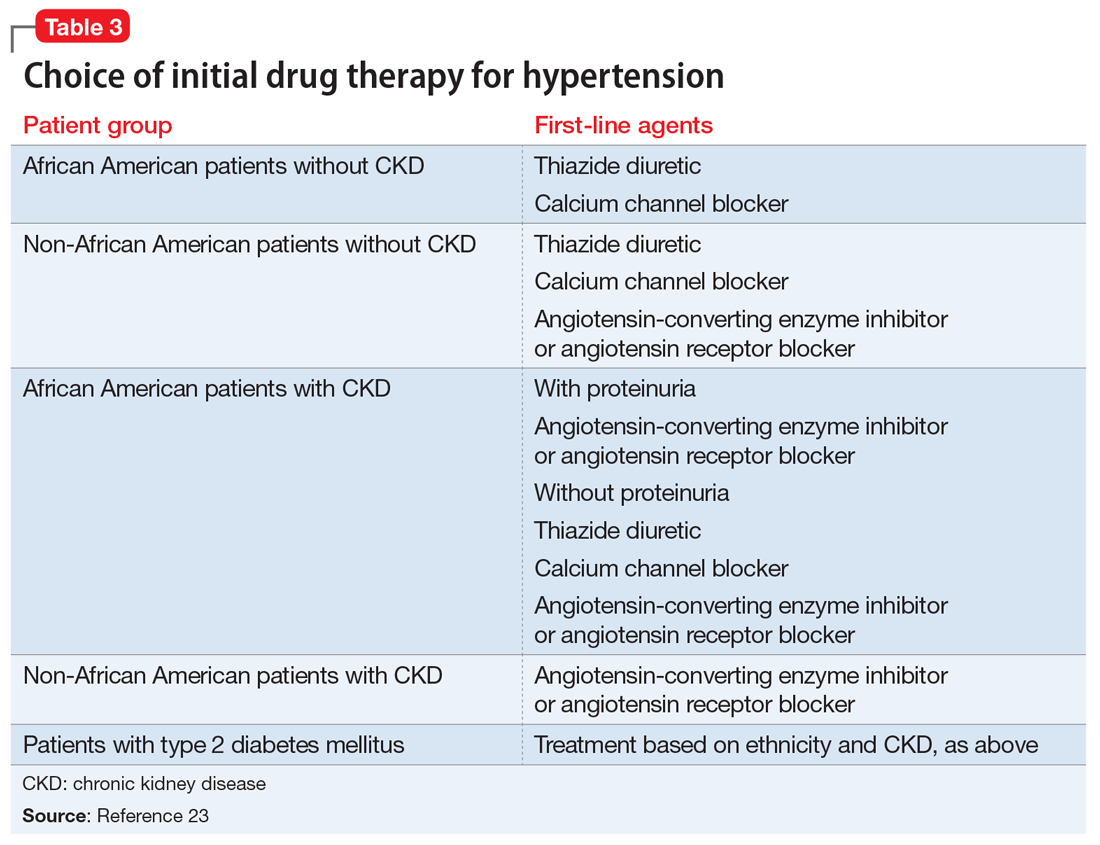
For example, in non-African American patients, thiazide diuretics, calcium channel blockers, angiotensin receptor blockers, and angiotensin-converting enzyme inhibitors are first-line treatments (Table 3). For African American patients without CKD, first-line treatments should be thiazide diuretics and calcium channel blockers, because angiotensin-converting enzyme inhibitors and angiotensin receptor blockers do not reduce cardiovascular events as effectively. African American patients with CKD and proteinuria, however, benefit from angiotensin-converting enzyme inhibitors or angiotensin receptor blockers and are preferred first-line agents. However, blood pressure control is a more important factor in improving outcomes than the choice of medication.
Psychiatrists’ role. Psychiatrists should aim to collaborate with the primary care provider when treating hypertension. However, when integrative care is not possible, they should start a first-line medication with follow-up in 1 month or sooner for patients with severe hypertension (>160/100 mm Hg) or significant comorbidities (eg, CKD, congestive heart failure, coronary disease). Patients with blood pressure >160/100 mm Hg often are started on a thiazide diuretic with one other medication because a single agent usually does not achieve goal blood pressure. Patients with CKD need close monitoring of potassium and creatinine when starting angiotensin-converting enzyme inhibitor or angiotensin receptor blocker therapy, usually within 1 to 2 days of starting or adjusting their medication. Adjust or add medication dosages monthly until blood pressure goals are reached.
A general internist, cardiologist, or nephrologist who has expertise in managing complex cases should oversee care of a psychiatric patient in any of the following scenarios:
- suspected secondary cause of hypertension
- adverse reaction to antihypertensive medications
- complicated comorbid conditions (ie, creatinine >1.8 mg/dL, worsening renal failure, hyperkalemia, heart failure, coronary disease)
- blood pressure >180/120 mm Hg
- requires ≥3 antihypertensive medications.
Summing up
Hypertension is a significant comorbidity in many psychiatric patients, but usually is asymptomatic. Often the psychiatrist or other mental health provider will diagnose hypertension because of their frequent contact with these patients. Once the diagnosis is made, an initial evaluation can direct lifestyle modifications. Patients who continue to have significant elevation of blood pressure should start pharmacotherapy, either by the psychiatrist or by ensuring follow-up with a primary care physician. The psychiatrist may be able to manage cases of essential hypertension, but always must be vigilant for potential drug–disease or drug–drug interactions during treatment. A team-based approach may improve health outcomes in psychiatric patients.
1. Centers for Disease Control and Prevention (CDC). Vital signs: awareness and treatment of uncontrolled hypertension among adults—United States, 2003-2010. MMWR Morb Mortal Wkly Rep. 2012;61:703-709.
2. Mozzafarian D, Benjamin EJ, Go AS, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics—2015 update: a report from the American Heart Association. Circulation. 2015;131(4):e29-e322.
3. Carroll D, Phillips AC, Gale CR, et al. Generalized anxiety and major depressive disorders, their comorbidity and hypertension in middle-aged men. Psychosom Med. 2010;72(1):16-19.
4. Leboyer M, Soreca I, Scott J, et al. Can bipolar disorder be viewed as a multi-system inflammatory disease? J Affect Disord. 2012;141(1):1-10.
5. Goff DC, Sullivan LM, McEvoy JP, et al. A comparison of ten-year cardiac risk estimates in schizophrenia patients from the CATIE study and matched controls. Schizophr Res. 2005;80(1):45-53.
6. Stein DJ, Aguilar-Gaxiola S, Alonso J, et al. Associations between mental disorders and subsequent onset of hypertension. Gen Hosp Psychiatry. 2014;36(2):142-149.
7. Birkenaes AB, Opjordsmoen S, Brunborg C, et al. The level of cardiovascular risk factors in bipolar disorder equals that of schizophrenia: a comparative study. J Clin Psychiatry. 2007;68(6):917-923.
8. Izzo JL, Black HR, Goodfriend TL. Hypertension primer: the essentials of high blood pressure. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008.
9. Osby U, Correia N, Brandt L, et al. Mortality and causes of death in schizophrenia in Stockholm County, Sweden. Schizophr Res. 2000;45(1-2):21-28.
10. Brown S, Inskip H, Barraclough B. Causes of the excess mortality of schizophrenia. Br J Psychiatry. 2000;177:212-217.
11. Auquier P, Lançon C, Rouillon F, et al. Mortality in schizophrenia. Pharmacoepidemiol Drug Saf. 2007;16(12):1308-1312.
12. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
13. Bowis J, Parvanova A, McDaid D, et al. Mental and Physical Health Charter: bridging the gap between mental and physical health. https://www.idf.org/sites/default/files/Mental%2520and%2520Physical%2520Health%2520Charter%2520-%2520FINAL.pdf. Published October 7, 2009. Accessed March 6, 2017.
14. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
15. Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289(19):2560-2571.
16. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
17. National Collaborating Centre for Mental Health (UK). Depression in adults with a chronic physical health problem: treatment and Management. Appendix 16: table of drug interactions. http://www.ncbi.nlm.nih.gov/books/NBK82914. Published 2010. Accessed March 6, 2017.
18. Siu AL; U.S. Preventive Services Task Force. Screening for high blood pressure in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2015:163(10):778-786.
19. Wang PS, Bohn RL, Knight E, et al. Noncompliance with antihypertensive medications: the impact of depressive symptoms and psychosocial factors. J Gen Intern Med. 2002;17(7):504-511.
20. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107.
21. Consoli SM, Lemogne C, Levy A, et al. Physicians’ degree of motivation regarding their perception of hypertension, and blood pressure control. J Hypertens. 2010;28(6):1330-1339.
22. National High Blood Pressure Education Program. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Improving Hypertension Control. Bethesda, MD: U.S. Department of Health and Human Services; 2004:61-64.
23. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-520.
24. The SPRINT Research Group; Wright JT Jr, Williamson JD, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373(22):2103-2016.
25. Boulware LE, Daumit GL, Frick KD, et al. An evidence-based review of patient-centered behavioral interventions for hypertension. Am J Prev Med. 2001;21(3):221-232.
Hypertension is a widespread, under-recognized, and undertreated cause of morbidity and mortality in the United States and is associated with several psychiatric illnesses. Left untreated, hypertension can have significant consequences, including increased risk of stroke, coronary heart disease, heart failure, chronic kidney failure, and death. Approximately 70 million adults in the United States have hypertension, but only 60% of them have been diagnosed, and of those only 50% have their blood pressure under control.1 In 2013, 360,000 deaths in the United States were attributed to hypertension.2
Hypertension is associated with major depressive disorder, generalized anxiety disorder, bipolar disorder, and schizophrenia.3-5 Additionally, impulsive eating disorders, substance abuse, anxiety, and depression are associated with a hypertension diagnosis, although patients with panic disorder develop hypertension at a younger age.6 A 2007 study found a 61% prevalence of hypertension in those with bipolar disorder compared with 41% among the general population.7 The strong link between bipolar disorder and hypertension might be because of a common disease mechanism; both are associated with hyperactive cellular calcium signaling and increased platelet intracellular calcium ion concentrations.8
Hypertension not only is common among patients with psychiatric illness, it likely contributes to worse clinical outcomes. Studies across different cultures have found higher mortality rates in individuals with mental illness.9-11 Persons with schizophrenia and other severe mental illnesses may lose ≥25 years of life expectancy, with the primary cause of death being cardiovascular disease, not suicide.12 Patients with depression have a 50% greater risk of cardiovascular disease, which is equivalent to the risk of smoking.13
Schizophrenia is strongly associated with numerous comorbidities and has been linked significantly to an elevated 10-year cardiac risk after controlling for body mass index.5 The high rate of non-treatment of hypertension for patients with schizophrenia (62.4%) is especially concerning.14
Because of the well-documented morbidity and mortality of hypertension and its increased prevalence and undertreatment in the psychiatric population, mental health providers are in an important position to recognize hypertension and evaluate its inherent risks to direct their patients toward proper treatment. This article reviews:
- the signs and symptoms of hypertension
- the mental health provider’s role in the evaluation and diagnosis
- how psychotropic drugs influence blood pressure and drug–drug interactions
- the management of hypertension in psychiatric patients, including strategies for counseling and lifestyle management.
Diagnosing hypertension
Hypertension is defined as a blood pressure >140/90 mm Hg, the average of ≥2 properly measured readings at ≥2 visits in a medical setting.15 The proper equipment, including a well-fitting blood pressure cuff, and technique to measure blood pressure are essential to avoid misdiagnosis. The patient should be at rest for ≥5 minutes, without active pain or emotional distress.
Most cases of hypertension (90% to 95%) are primary, commonly called essential hypertension. However, the differential diagnosis also should consider secondary causes, which may include:
- obesity
- medications
- chronic alcohol use
- methamphetamine or cocaine use
- primary kidney disease
- atherosclerotic renal artery stenosis
- obstructive sleep apnea
- hypothyroidism
- primary hyperaldosteronism
- narrowing of the aorta
- Cushing syndrome
- primary hyperparathyroidism
- polycythemia
- pheochromocytoma.
Medical evaluation. Once the diagnosis of hypertension is made, a medical evaluation is indicated to determine if the patient has end-organ damage from the elevated pressures, such as renal disease or heart disease, to identify other modifiable cardiovascular risk factors, such as hyperlipidemia, and to screen for secondary causes of hypertension. This evaluation includes15:
- a physical exam
- review of medications
- lipid profile
- urinalysis to screen for proteinuria
- serum electrolytes and creatinine
- electrocardiogram to screen for left ventricular hypertrophy or prior infarction
- fasting glucose or hemoglobin A1c to screen for type 2 diabetes mellitus.
Psychotropic drugs. In psychiatric patients, the evaluation must consider the potential impact psychotropic drug effects and drug–drug interactions can have on blood pressure (Table 2). For example, patients taking both diuretics and lithium are at increased risk for dehydration and increased serum lithium levels, which could cause severe neurologic symptoms and renal insufficiency.16 Several antihypertensives when taken with venlafaxine can increase blood pressure, but antihypertensives with α-1 blocking psychotropics can decrease blood pressure. Monoamine oxidase inhibitors can cause hypotension or hypertension with various classes of antihypertensives. Stimulants, such as methylphenidate, atomoxetine, dextroamphetamine, armodafinil, or modafinil, alone or combined with antihypertensives, can cause hypertension.17
Substance abuse, particularly alcohol, methamphetamine, and cocaine, can cause difficulty controlling blood pressure. Patients with refractory hypertension should have a reassessment of substance abuse as a potential cause.
Screening guidelines for mental health providers
For many patients with severe mental illness, visits to their mental health providers might be their only contact with the medical system. Therefore, screening in the mental health settings could detect cases that otherwise would be missed.
Screening recommendations. The U.S. Preventive Services Task Force recommends screening for hypertension in the general population beginning at age 18.18 Adults age 18 to 39 with normal blood pressure (<130/85 mm Hg) and no other risk factors (eg, overweight, obese, or African American) can be screened every 3 years. Those with risk factors or a blood pressure of 130/85 to 139/89 mm Hg and adults age ≥40 should have annual screenings.
Ideally, psychiatrists and other mental health providers should monitor blood pressure at each visit, especially in patients taking psychotropics because of their higher risk for hypertension.
Optimizing treatment. Once the diagnosis of essential hypertension is established, identifying psychiatric comorbidities and the severity of psychiatric symptoms are important to optimize treatment adherence. Patients with increased depressive symptoms are less likely to comply with antihypertensive medication,19 and patients with confirmed depression are 3 times more likely to not adhere to medical treatment recommendations than non-depressed patients.20
Physicians’ attitudes toward hypertension also can affect patients’ compliance and blood pressure control.21 Psychiatrists should be empathetic and motivational toward patients attempting to control their blood pressure. The Seventh Joint National Committee on the Prevention, Detection, Evaluation, and Treatment of High Blood Pressure states, “Motivation improves when patients have positive experiences with, and trust in, the clinician. Empathy builds trust and is a potent motivator.”22
Treatment and management
Treatment of hypertension significantly reduces the risk of stroke, myocardial infarction, renal injury, heart failure, and premature death. Studies show that treatment that reduces systolic blood pressure by 12 mm Hg over 10 years will prevent 1 death for every 11 patients with essential hypertension. In those with concomitant cardiovascular disease or target organ damage, such a reduction would prevent death in 1 of every 9 patients treated.15Blood pressure goals. The 2014 Eighth Joint National Committee Guideline for Management of High Blood Pressure in Adults provides guidance on blood pressure goals depending on patients’ underlying medical history (Figure).23 Based on expert opinion and randomized controlled studies, blood pressure goals for patients without diabetes or chronic kidney disease (CKD)—an estimated or measured glomerular filtration rate (GFR) of ≤60 mL/min/1.73 m2—depend on age: <140/90 mm Hg for age 18 to 59 and <150/90 mm Hg for age ≥60. For patients with diabetes or CKD, the blood pressure goal is <140/90 mm Hg, regardless of age.
However, not all experts agree on these specific blood pressure goals. A major trial (SPRINT) published in 2015 found that intensive blood pressure goals do benefit higher-risk, non-diabetic patients.24 Specifically, the study randomized patients age ≥50 with systolic blood pressure of 130 to 180 mm Hg and increased cardiovascular risk to systolic blood pressure targets of <140 mm Hg (standard) or <120 mm Hg (intensive). Characteristics of increased cardiovascular risk were clinical or subclinical cardiovascular disease other than stroke, CKD with GFR of 20 to 60 mL/min/1.73 m2, age ≥75, or Framingham 10-year coronary heart disease risk score ≥15%. Intensive treatment significantly reduced overall mortality and the rate of acute coronary syndrome, myocardial infarction, heart failure, stroke, or cardiovascular death. However, the results of this study have not been assimilated into any recent guidelines. Therefore, consider a goal of <120 mm Hg for non-diabetic patients age ≥50 with any of these factors.
Lifestyle modifications. Psychiatrists are well equipped to motivate and encourage behavioral modification in patients with hypertension. Counseling and structured training courses could help to effectively lower blood pressure.25 Patients should receive education on lifestyle modifications including:
- weight reduction
- physical activity
- moderate alcohol consumption
- decreased sodium consumption
- implementation of the Dietary Approaches to Stop Hypertension (DASH) or Mediterranean diets.15
Maintaining a normal body weight is ideal, but weight reduction of 10 lb can reduce blood pressure in overweight patients. The DASH diet, consisting of fruits, vegetables, low-fat dairy products, high calcium and potassium intake, and reduced saturated and total fat intake can decrease systolic blood pressure from 8 to 14 mm Hg. Reduction of sodium intake to ≤2,400 mg/d can reduce systolic blood pressure from 2 to 8 mm Hg. Regular aerobic exercise of 30 minutes a day most days of the week can reduce systolic blood pressure up to 9 mm Hg. Patients also should be encouraged to quit smoking. Patients who implement ≥2 these modifications get better results.
Antihypertensive medications. Patients who do not reach their goals with lifestyle measures alone should receive antihypertensive medications. Most patients will require ≥2 agents to control their blood pressure. Clinical trials show that some patient subgroups have better outcomes with different first-line agents.
For example, in non-African American patients, thiazide diuretics, calcium channel blockers, angiotensin receptor blockers, and angiotensin-converting enzyme inhibitors are first-line treatments (Table 3). For African American patients without CKD, first-line treatments should be thiazide diuretics and calcium channel blockers, because angiotensin-converting enzyme inhibitors and angiotensin receptor blockers do not reduce cardiovascular events as effectively. African American patients with CKD and proteinuria, however, benefit from angiotensin-converting enzyme inhibitors or angiotensin receptor blockers and are preferred first-line agents. However, blood pressure control is a more important factor in improving outcomes than the choice of medication.
Psychiatrists’ role. Psychiatrists should aim to collaborate with the primary care provider when treating hypertension. However, when integrative care is not possible, they should start a first-line medication with follow-up in 1 month or sooner for patients with severe hypertension (>160/100 mm Hg) or significant comorbidities (eg, CKD, congestive heart failure, coronary disease). Patients with blood pressure >160/100 mm Hg often are started on a thiazide diuretic with one other medication because a single agent usually does not achieve goal blood pressure. Patients with CKD need close monitoring of potassium and creatinine when starting angiotensin-converting enzyme inhibitor or angiotensin receptor blocker therapy, usually within 1 to 2 days of starting or adjusting their medication. Adjust or add medication dosages monthly until blood pressure goals are reached.
A general internist, cardiologist, or nephrologist who has expertise in managing complex cases should oversee care of a psychiatric patient in any of the following scenarios:
- suspected secondary cause of hypertension
- adverse reaction to antihypertensive medications
- complicated comorbid conditions (ie, creatinine >1.8 mg/dL, worsening renal failure, hyperkalemia, heart failure, coronary disease)
- blood pressure >180/120 mm Hg
- requires ≥3 antihypertensive medications.
Summing up
Hypertension is a significant comorbidity in many psychiatric patients, but usually is asymptomatic. Often the psychiatrist or other mental health provider will diagnose hypertension because of their frequent contact with these patients. Once the diagnosis is made, an initial evaluation can direct lifestyle modifications. Patients who continue to have significant elevation of blood pressure should start pharmacotherapy, either by the psychiatrist or by ensuring follow-up with a primary care physician. The psychiatrist may be able to manage cases of essential hypertension, but always must be vigilant for potential drug–disease or drug–drug interactions during treatment. A team-based approach may improve health outcomes in psychiatric patients.
Hypertension is a widespread, under-recognized, and undertreated cause of morbidity and mortality in the United States and is associated with several psychiatric illnesses. Left untreated, hypertension can have significant consequences, including increased risk of stroke, coronary heart disease, heart failure, chronic kidney failure, and death. Approximately 70 million adults in the United States have hypertension, but only 60% of them have been diagnosed, and of those only 50% have their blood pressure under control.1 In 2013, 360,000 deaths in the United States were attributed to hypertension.2
Hypertension is associated with major depressive disorder, generalized anxiety disorder, bipolar disorder, and schizophrenia.3-5 Additionally, impulsive eating disorders, substance abuse, anxiety, and depression are associated with a hypertension diagnosis, although patients with panic disorder develop hypertension at a younger age.6 A 2007 study found a 61% prevalence of hypertension in those with bipolar disorder compared with 41% among the general population.7 The strong link between bipolar disorder and hypertension might be because of a common disease mechanism; both are associated with hyperactive cellular calcium signaling and increased platelet intracellular calcium ion concentrations.8
Hypertension not only is common among patients with psychiatric illness, it likely contributes to worse clinical outcomes. Studies across different cultures have found higher mortality rates in individuals with mental illness.9-11 Persons with schizophrenia and other severe mental illnesses may lose ≥25 years of life expectancy, with the primary cause of death being cardiovascular disease, not suicide.12 Patients with depression have a 50% greater risk of cardiovascular disease, which is equivalent to the risk of smoking.13
Schizophrenia is strongly associated with numerous comorbidities and has been linked significantly to an elevated 10-year cardiac risk after controlling for body mass index.5 The high rate of non-treatment of hypertension for patients with schizophrenia (62.4%) is especially concerning.14
Because of the well-documented morbidity and mortality of hypertension and its increased prevalence and undertreatment in the psychiatric population, mental health providers are in an important position to recognize hypertension and evaluate its inherent risks to direct their patients toward proper treatment. This article reviews:
- the signs and symptoms of hypertension
- the mental health provider’s role in the evaluation and diagnosis
- how psychotropic drugs influence blood pressure and drug–drug interactions
- the management of hypertension in psychiatric patients, including strategies for counseling and lifestyle management.
Diagnosing hypertension
Hypertension is defined as a blood pressure >140/90 mm Hg, the average of ≥2 properly measured readings at ≥2 visits in a medical setting.15 The proper equipment, including a well-fitting blood pressure cuff, and technique to measure blood pressure are essential to avoid misdiagnosis. The patient should be at rest for ≥5 minutes, without active pain or emotional distress.
Most cases of hypertension (90% to 95%) are primary, commonly called essential hypertension. However, the differential diagnosis also should consider secondary causes, which may include:
- obesity
- medications
- chronic alcohol use
- methamphetamine or cocaine use
- primary kidney disease
- atherosclerotic renal artery stenosis
- obstructive sleep apnea
- hypothyroidism
- primary hyperaldosteronism
- narrowing of the aorta
- Cushing syndrome
- primary hyperparathyroidism
- polycythemia
- pheochromocytoma.
Medical evaluation. Once the diagnosis of hypertension is made, a medical evaluation is indicated to determine if the patient has end-organ damage from the elevated pressures, such as renal disease or heart disease, to identify other modifiable cardiovascular risk factors, such as hyperlipidemia, and to screen for secondary causes of hypertension. This evaluation includes15:
- a physical exam
- review of medications
- lipid profile
- urinalysis to screen for proteinuria
- serum electrolytes and creatinine
- electrocardiogram to screen for left ventricular hypertrophy or prior infarction
- fasting glucose or hemoglobin A1c to screen for type 2 diabetes mellitus.
Psychotropic drugs. In psychiatric patients, the evaluation must consider the potential impact psychotropic drug effects and drug–drug interactions can have on blood pressure (Table 2). For example, patients taking both diuretics and lithium are at increased risk for dehydration and increased serum lithium levels, which could cause severe neurologic symptoms and renal insufficiency.16 Several antihypertensives when taken with venlafaxine can increase blood pressure, but antihypertensives with α-1 blocking psychotropics can decrease blood pressure. Monoamine oxidase inhibitors can cause hypotension or hypertension with various classes of antihypertensives. Stimulants, such as methylphenidate, atomoxetine, dextroamphetamine, armodafinil, or modafinil, alone or combined with antihypertensives, can cause hypertension.17
Substance abuse, particularly alcohol, methamphetamine, and cocaine, can cause difficulty controlling blood pressure. Patients with refractory hypertension should have a reassessment of substance abuse as a potential cause.
Screening guidelines for mental health providers
For many patients with severe mental illness, visits to their mental health providers might be their only contact with the medical system. Therefore, screening in the mental health settings could detect cases that otherwise would be missed.
Screening recommendations. The U.S. Preventive Services Task Force recommends screening for hypertension in the general population beginning at age 18.18 Adults age 18 to 39 with normal blood pressure (<130/85 mm Hg) and no other risk factors (eg, overweight, obese, or African American) can be screened every 3 years. Those with risk factors or a blood pressure of 130/85 to 139/89 mm Hg and adults age ≥40 should have annual screenings.
Ideally, psychiatrists and other mental health providers should monitor blood pressure at each visit, especially in patients taking psychotropics because of their higher risk for hypertension.
Optimizing treatment. Once the diagnosis of essential hypertension is established, identifying psychiatric comorbidities and the severity of psychiatric symptoms are important to optimize treatment adherence. Patients with increased depressive symptoms are less likely to comply with antihypertensive medication,19 and patients with confirmed depression are 3 times more likely to not adhere to medical treatment recommendations than non-depressed patients.20
Physicians’ attitudes toward hypertension also can affect patients’ compliance and blood pressure control.21 Psychiatrists should be empathetic and motivational toward patients attempting to control their blood pressure. The Seventh Joint National Committee on the Prevention, Detection, Evaluation, and Treatment of High Blood Pressure states, “Motivation improves when patients have positive experiences with, and trust in, the clinician. Empathy builds trust and is a potent motivator.”22
Treatment and management
Treatment of hypertension significantly reduces the risk of stroke, myocardial infarction, renal injury, heart failure, and premature death. Studies show that treatment that reduces systolic blood pressure by 12 mm Hg over 10 years will prevent 1 death for every 11 patients with essential hypertension. In those with concomitant cardiovascular disease or target organ damage, such a reduction would prevent death in 1 of every 9 patients treated.15Blood pressure goals. The 2014 Eighth Joint National Committee Guideline for Management of High Blood Pressure in Adults provides guidance on blood pressure goals depending on patients’ underlying medical history (Figure).23 Based on expert opinion and randomized controlled studies, blood pressure goals for patients without diabetes or chronic kidney disease (CKD)—an estimated or measured glomerular filtration rate (GFR) of ≤60 mL/min/1.73 m2—depend on age: <140/90 mm Hg for age 18 to 59 and <150/90 mm Hg for age ≥60. For patients with diabetes or CKD, the blood pressure goal is <140/90 mm Hg, regardless of age.
However, not all experts agree on these specific blood pressure goals. A major trial (SPRINT) published in 2015 found that intensive blood pressure goals do benefit higher-risk, non-diabetic patients.24 Specifically, the study randomized patients age ≥50 with systolic blood pressure of 130 to 180 mm Hg and increased cardiovascular risk to systolic blood pressure targets of <140 mm Hg (standard) or <120 mm Hg (intensive). Characteristics of increased cardiovascular risk were clinical or subclinical cardiovascular disease other than stroke, CKD with GFR of 20 to 60 mL/min/1.73 m2, age ≥75, or Framingham 10-year coronary heart disease risk score ≥15%. Intensive treatment significantly reduced overall mortality and the rate of acute coronary syndrome, myocardial infarction, heart failure, stroke, or cardiovascular death. However, the results of this study have not been assimilated into any recent guidelines. Therefore, consider a goal of <120 mm Hg for non-diabetic patients age ≥50 with any of these factors.
Lifestyle modifications. Psychiatrists are well equipped to motivate and encourage behavioral modification in patients with hypertension. Counseling and structured training courses could help to effectively lower blood pressure.25 Patients should receive education on lifestyle modifications including:
- weight reduction
- physical activity
- moderate alcohol consumption
- decreased sodium consumption
- implementation of the Dietary Approaches to Stop Hypertension (DASH) or Mediterranean diets.15
Maintaining a normal body weight is ideal, but weight reduction of 10 lb can reduce blood pressure in overweight patients. The DASH diet, consisting of fruits, vegetables, low-fat dairy products, high calcium and potassium intake, and reduced saturated and total fat intake can decrease systolic blood pressure from 8 to 14 mm Hg. Reduction of sodium intake to ≤2,400 mg/d can reduce systolic blood pressure from 2 to 8 mm Hg. Regular aerobic exercise of 30 minutes a day most days of the week can reduce systolic blood pressure up to 9 mm Hg. Patients also should be encouraged to quit smoking. Patients who implement ≥2 these modifications get better results.
Antihypertensive medications. Patients who do not reach their goals with lifestyle measures alone should receive antihypertensive medications. Most patients will require ≥2 agents to control their blood pressure. Clinical trials show that some patient subgroups have better outcomes with different first-line agents.
For example, in non-African American patients, thiazide diuretics, calcium channel blockers, angiotensin receptor blockers, and angiotensin-converting enzyme inhibitors are first-line treatments (Table 3). For African American patients without CKD, first-line treatments should be thiazide diuretics and calcium channel blockers, because angiotensin-converting enzyme inhibitors and angiotensin receptor blockers do not reduce cardiovascular events as effectively. African American patients with CKD and proteinuria, however, benefit from angiotensin-converting enzyme inhibitors or angiotensin receptor blockers and are preferred first-line agents. However, blood pressure control is a more important factor in improving outcomes than the choice of medication.
Psychiatrists’ role. Psychiatrists should aim to collaborate with the primary care provider when treating hypertension. However, when integrative care is not possible, they should start a first-line medication with follow-up in 1 month or sooner for patients with severe hypertension (>160/100 mm Hg) or significant comorbidities (eg, CKD, congestive heart failure, coronary disease). Patients with blood pressure >160/100 mm Hg often are started on a thiazide diuretic with one other medication because a single agent usually does not achieve goal blood pressure. Patients with CKD need close monitoring of potassium and creatinine when starting angiotensin-converting enzyme inhibitor or angiotensin receptor blocker therapy, usually within 1 to 2 days of starting or adjusting their medication. Adjust or add medication dosages monthly until blood pressure goals are reached.
A general internist, cardiologist, or nephrologist who has expertise in managing complex cases should oversee care of a psychiatric patient in any of the following scenarios:
- suspected secondary cause of hypertension
- adverse reaction to antihypertensive medications
- complicated comorbid conditions (ie, creatinine >1.8 mg/dL, worsening renal failure, hyperkalemia, heart failure, coronary disease)
- blood pressure >180/120 mm Hg
- requires ≥3 antihypertensive medications.
Summing up
Hypertension is a significant comorbidity in many psychiatric patients, but usually is asymptomatic. Often the psychiatrist or other mental health provider will diagnose hypertension because of their frequent contact with these patients. Once the diagnosis is made, an initial evaluation can direct lifestyle modifications. Patients who continue to have significant elevation of blood pressure should start pharmacotherapy, either by the psychiatrist or by ensuring follow-up with a primary care physician. The psychiatrist may be able to manage cases of essential hypertension, but always must be vigilant for potential drug–disease or drug–drug interactions during treatment. A team-based approach may improve health outcomes in psychiatric patients.
1. Centers for Disease Control and Prevention (CDC). Vital signs: awareness and treatment of uncontrolled hypertension among adults—United States, 2003-2010. MMWR Morb Mortal Wkly Rep. 2012;61:703-709.
2. Mozzafarian D, Benjamin EJ, Go AS, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics—2015 update: a report from the American Heart Association. Circulation. 2015;131(4):e29-e322.
3. Carroll D, Phillips AC, Gale CR, et al. Generalized anxiety and major depressive disorders, their comorbidity and hypertension in middle-aged men. Psychosom Med. 2010;72(1):16-19.
4. Leboyer M, Soreca I, Scott J, et al. Can bipolar disorder be viewed as a multi-system inflammatory disease? J Affect Disord. 2012;141(1):1-10.
5. Goff DC, Sullivan LM, McEvoy JP, et al. A comparison of ten-year cardiac risk estimates in schizophrenia patients from the CATIE study and matched controls. Schizophr Res. 2005;80(1):45-53.
6. Stein DJ, Aguilar-Gaxiola S, Alonso J, et al. Associations between mental disorders and subsequent onset of hypertension. Gen Hosp Psychiatry. 2014;36(2):142-149.
7. Birkenaes AB, Opjordsmoen S, Brunborg C, et al. The level of cardiovascular risk factors in bipolar disorder equals that of schizophrenia: a comparative study. J Clin Psychiatry. 2007;68(6):917-923.
8. Izzo JL, Black HR, Goodfriend TL. Hypertension primer: the essentials of high blood pressure. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008.
9. Osby U, Correia N, Brandt L, et al. Mortality and causes of death in schizophrenia in Stockholm County, Sweden. Schizophr Res. 2000;45(1-2):21-28.
10. Brown S, Inskip H, Barraclough B. Causes of the excess mortality of schizophrenia. Br J Psychiatry. 2000;177:212-217.
11. Auquier P, Lançon C, Rouillon F, et al. Mortality in schizophrenia. Pharmacoepidemiol Drug Saf. 2007;16(12):1308-1312.
12. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
13. Bowis J, Parvanova A, McDaid D, et al. Mental and Physical Health Charter: bridging the gap between mental and physical health. https://www.idf.org/sites/default/files/Mental%2520and%2520Physical%2520Health%2520Charter%2520-%2520FINAL.pdf. Published October 7, 2009. Accessed March 6, 2017.
14. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
15. Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289(19):2560-2571.
16. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
17. National Collaborating Centre for Mental Health (UK). Depression in adults with a chronic physical health problem: treatment and Management. Appendix 16: table of drug interactions. http://www.ncbi.nlm.nih.gov/books/NBK82914. Published 2010. Accessed March 6, 2017.
18. Siu AL; U.S. Preventive Services Task Force. Screening for high blood pressure in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2015:163(10):778-786.
19. Wang PS, Bohn RL, Knight E, et al. Noncompliance with antihypertensive medications: the impact of depressive symptoms and psychosocial factors. J Gen Intern Med. 2002;17(7):504-511.
20. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107.
21. Consoli SM, Lemogne C, Levy A, et al. Physicians’ degree of motivation regarding their perception of hypertension, and blood pressure control. J Hypertens. 2010;28(6):1330-1339.
22. National High Blood Pressure Education Program. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Improving Hypertension Control. Bethesda, MD: U.S. Department of Health and Human Services; 2004:61-64.
23. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-520.
24. The SPRINT Research Group; Wright JT Jr, Williamson JD, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373(22):2103-2016.
25. Boulware LE, Daumit GL, Frick KD, et al. An evidence-based review of patient-centered behavioral interventions for hypertension. Am J Prev Med. 2001;21(3):221-232.
1. Centers for Disease Control and Prevention (CDC). Vital signs: awareness and treatment of uncontrolled hypertension among adults—United States, 2003-2010. MMWR Morb Mortal Wkly Rep. 2012;61:703-709.
2. Mozzafarian D, Benjamin EJ, Go AS, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics—2015 update: a report from the American Heart Association. Circulation. 2015;131(4):e29-e322.
3. Carroll D, Phillips AC, Gale CR, et al. Generalized anxiety and major depressive disorders, their comorbidity and hypertension in middle-aged men. Psychosom Med. 2010;72(1):16-19.
4. Leboyer M, Soreca I, Scott J, et al. Can bipolar disorder be viewed as a multi-system inflammatory disease? J Affect Disord. 2012;141(1):1-10.
5. Goff DC, Sullivan LM, McEvoy JP, et al. A comparison of ten-year cardiac risk estimates in schizophrenia patients from the CATIE study and matched controls. Schizophr Res. 2005;80(1):45-53.
6. Stein DJ, Aguilar-Gaxiola S, Alonso J, et al. Associations between mental disorders and subsequent onset of hypertension. Gen Hosp Psychiatry. 2014;36(2):142-149.
7. Birkenaes AB, Opjordsmoen S, Brunborg C, et al. The level of cardiovascular risk factors in bipolar disorder equals that of schizophrenia: a comparative study. J Clin Psychiatry. 2007;68(6):917-923.
8. Izzo JL, Black HR, Goodfriend TL. Hypertension primer: the essentials of high blood pressure. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008.
9. Osby U, Correia N, Brandt L, et al. Mortality and causes of death in schizophrenia in Stockholm County, Sweden. Schizophr Res. 2000;45(1-2):21-28.
10. Brown S, Inskip H, Barraclough B. Causes of the excess mortality of schizophrenia. Br J Psychiatry. 2000;177:212-217.
11. Auquier P, Lançon C, Rouillon F, et al. Mortality in schizophrenia. Pharmacoepidemiol Drug Saf. 2007;16(12):1308-1312.
12. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
13. Bowis J, Parvanova A, McDaid D, et al. Mental and Physical Health Charter: bridging the gap between mental and physical health. https://www.idf.org/sites/default/files/Mental%2520and%2520Physical%2520Health%2520Charter%2520-%2520FINAL.pdf. Published October 7, 2009. Accessed March 6, 2017.
14. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
15. Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289(19):2560-2571.
16. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
17. National Collaborating Centre for Mental Health (UK). Depression in adults with a chronic physical health problem: treatment and Management. Appendix 16: table of drug interactions. http://www.ncbi.nlm.nih.gov/books/NBK82914. Published 2010. Accessed March 6, 2017.
18. Siu AL; U.S. Preventive Services Task Force. Screening for high blood pressure in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2015:163(10):778-786.
19. Wang PS, Bohn RL, Knight E, et al. Noncompliance with antihypertensive medications: the impact of depressive symptoms and psychosocial factors. J Gen Intern Med. 2002;17(7):504-511.
20. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107.
21. Consoli SM, Lemogne C, Levy A, et al. Physicians’ degree of motivation regarding their perception of hypertension, and blood pressure control. J Hypertens. 2010;28(6):1330-1339.
22. National High Blood Pressure Education Program. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Improving Hypertension Control. Bethesda, MD: U.S. Department of Health and Human Services; 2004:61-64.
23. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-520.
24. The SPRINT Research Group; Wright JT Jr, Williamson JD, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373(22):2103-2016.
25. Boulware LE, Daumit GL, Frick KD, et al. An evidence-based review of patient-centered behavioral interventions for hypertension. Am J Prev Med. 2001;21(3):221-232.

Prevention and treatment options for mTOR inhibitor-associated stomatitis
Mammalian target of rapamycin (mTOR), a serine–threonine protein kinase, operates in the phosphoinositide 3-kinase (PI3K)–protein kinase B (AKT)–mTOR signal transduction pathway regulating both normal and cancer cellular processes, including cell growth, proliferation, motility, survival, and protein and lipid synthesis.1 Genetic alterations affecting this pathway, including mutations in receptor tyrosine kinases PI3K and AKT, occur frequently in human cancers,2 supporting the rationale to develop drugs that target pathway components, such as mTOR inhibitors.
Two mTOR inhibitors are currently approved by the US Food and Drug Administration for cancer treatment: temsirolimus, for advanced renal cell carcinoma (RCC; approved 2007)3 and everolimus, for advanced RCC (approved 2009), advanced pancreatic neuroendocrine tumors (pNET; approved 2011), and hormone receptor-positive (HR-positive), human epidermal growth factor receptor-2 (HER2)-negative advanced breast cancer (approved 2012).4 Another mTOR inhibitor, sirolimus, is approved for use as an immunosuppressive agent and prophylactic against organ rejection after kidney transplant.5
Stomatitis, inflammation of the oral mucosa with contributing factors of genetic predisposition, nutritional deficiencies, infections, and immunological or hematologic dysfunction,6 occurs frequently as a side effect associated with mTOR inhibitor treatment.7-9 Left untreated or managed unsatisfactorily, mTOR inhibitor-associated stomatitis (mIAS) may cause patients discomfort and trouble with maintaining adequate nutritional intake and proper oral hygiene, as well as strict adherence to cancer treatment. It is therefore important for health care providers of cancer patients receiving mTOR inhibitor treatment to be knowledgeable about this side effect. The purpose of the present systematic review of published literature is to provide a better understanding of the differential diagnosis of mIAS, the pathophysiology of mIAS, preventive strategies for patients initiating mTOR inhibitor treatment, and treatment options available to manage mIAS.
Method
The PubMed database was searched with the terms mTOR inhibitor and stomatitis (no date restriction); 79 articles were retrieved, and all abstracts were reviewed to select those relevant to the aims of this review article. To understand future directions for management and prevention of mIAS, a search of clinicaltrials.gov was performed with the terms temsirolimus everolimus stomatitis yielding 12 clinical trials, of which 4 were excluded: 1 trial was terminated due to slow accrual, the status of 1 trial had not been verified in >2 years, and 2 studies focused on efficacy outcomes. A search of the American Society of Clinical Oncology (ASCO) meeting abstracts database was performed to assess the availability of clinical trial data; the search was limited to 2011-2016 and terms were stomatitis in the title and mTOR in the abstract or title. Seven abstracts were retrieved; 2 discussed stomatitis prevention (1 as a “trial-in-progress” and 1 presented results of the trial); the other 5 abstracts presented meta-analyses or reviews of previous clinical studies to assess the risk, incidence, management, and resolution of mIAS.
Review findings
Incidence of mIAS in patients treated for cancer
Two recent meta-analyses quantified the rate of mIAS in patients receiving mTOR inhibitors. Shameem and colleagues10 identified 9 randomized studies of everolimus (8 phase 3, 1 phase 2) and 2 of temsirolimus (1 each phase 2 and 3) involving a total of 4752 patients with a variety of tumor types including angiomyolipoma, breast, gastric, giant cell astrocytoma, pNET, and RCC. Patients received everolimus monotherapy (n = 1,075) or in combination with exemestane (n = 485), tamoxifen (n = 54), letrozole (n = 137), or octreotide (n = 216). Temsirolimus was administered as monotherapy (n = 208) or in combination with interferon
(n = 210) or letrozole (n = 550). The incidence of all-grade stomatitis in the 11 studies ranged from 11%-63%, and the overall incidence of any grade stomatitis was 33.5% (95% confidence interval [CI], 21.9%-47.6%). The concurrent use of a second agent may have confounded these findings because, for example, stomatitis has been reported in pooled analyses and in postmarketing experience with letrozole.11
Rugo and colleagues12 evaluated the incidence of stomatitis in 1455 patients participating in 5 phase 3 randomized clinical trials of everolimus in breast cancer, carcinoid tumor, pNET, and RCC. Patients received everolimus monotherapy
(n = 478) or in combination with exemestane (n = 482), trastuzumab plus vinorelbine (n = 280), or octreotide
(n = 215). The incidence of stomatitis in patients receiving everolimus was 59%-71%, compared with 19%-29% in 1,071 patients of the comparator arms (placebo, and placebo–trastuzumab–vinorelbine). The overall incidence of any grade stomatitis was 67%; most events were mild (grade 1/2); 9% of stomatitis events were moderate to severe (grade 3/4).
Differential clinical presentation of mIAS and severity
Oral mucositis is a common significant adverse event (AE) that occurs in patients with cancer who receive standard chemotherapy regimens and/or radiation therapy,13 so it is important to recognize that the clinical presentation of mIAS differs from that of oral mucositis (Table 1, Figure 14,15).16 mIAS shares some similarities with aphthous ulcers (also referred to as canker sores), a common oral condition with varied causes related to systemic disorders, gastrointestinal disorders, and infections, among others .17 In general, mIAS ulcers develop with a median onset of 10 days (range, 4-25 days) after initiation of mTOR inhibitor treatment and resolve in about 1-3 weeks after dose interruption/reduction of everolimus.16,18,19 mIAS ulcers appear as distinct, oval lesions with a central gray area surrounded by peripheral erythema. They are usually localized to the movable mucosa of the mouth and oropharynx. Although mIAS lesions are usually small, they are quite painful and may cluster.
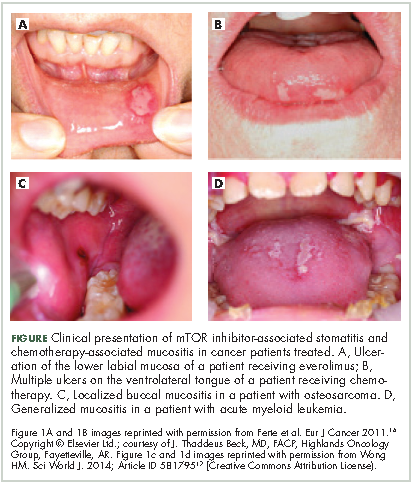
Differential diagnosis of mIAS should be made based on physical examination and medical history, with consideration given to appearance of lesions (number, size, and location), current infection status, and current medications. Specific diagnostic testing should be conducted to confirm a coexisting or alternative cause of oral lesions.17
Although there are many different scales for grading mIAS severity, the most commonly used are the National Cancer Institute Common Terminology Criteria for Adverse Events (based on patient function, symptoms, and intervention needs) and the World Health Organization oral mucositis scales (based on symptoms, clinical presentation, and interference with patient function).20-22 These scales distinguish between mild lesions (grade 1/2) and moderate to severe lesions (grade 3/4) that cause significant pain or interfere with oral intake.
Pathophysiology of mIAS
The pathophysiology mIAS is incompletely understood. The ubiquitous role of the PI3K-AKT-mTOR pathway in regulating broad cellular functions suggests that mTOR inhibition is likely to have wide-ranging effects on many biological processes. It is not known whether disruption of one or more processes – or upsetting the balance of mTOR activities – underlies the formation of mIAS.
Differences between mIAS and oral mucositis, including clinical presentation and concomitant toxicities,16,23 suggest that the two types of oral lesions are fundamentally distinct. This distinction is supported by animal studies in which mTOR inhibition was found to almost completely prevent the appearance of oral mucositis in irradiated mice. The protective effect of mTOR inhibition is mediated through suppression of oxidative stress generated by radiation therapy.24
Although mIAS and recurrent aphthous ulcers share some similarities, it is not clear whether they also share a common pathophysiology. Recent studies suggest that patients with recurrent aphthous ulcers have immune dysfunction that leads to excessive immune response to normally innocuous substrates in the oral mucosa.25 mTOR inhibition can have proinflammatory activity by promoting autophagy, a process that stimulates antigen presentation and activation of T cells that produce proinflammatory cytokines.26 It is interesting to note that the incidence of stomatitis in patients receiving sirolimus after kidney transplant is relatively low, 3%-20%.5 Sirolimus is administered in combination with other immunosuppressants, namely cyclosporine and corticosteroids, so it suggests that concomitant use of a steroid-based regimen may have a preventive or therapeutic effect. However, posttransplant sirolimus is typically administered at relatively low doses, which might account in part for the lower incidence of mIAS observed. Ongoing clinical studies of steroid-based mouthwashes in patients receiving everolimus should shed light on this.
Other study findings have shown that inhibition of the PI3K-AKT-mTOR signaling pathway affects skin wound healing,27,28 which raises the possibility that mIAS may stem from a diminished capacity to repair physical injuries to the oral mucosa. More research is needed to elucidate the pathophysiology of mIAS.
Preventive measures for patients initiating mTOR inhibitor treatment
There are preventive measures for mIAS that have not yet been backed up with evidence-based findings, although several clinical studies that are underway aim to address this gap (Table 2). The hypotheses about the pathophysiology of mIAS suggest that certain preventive and therapeutic interventions might be effective against mIAS. For example, two studies are evaluating the use of steroid-based mouthwashes in patients receiving everolimus, based on the hypothesis that mIAS may arise from an inflammatory process; another study will evaluate a mucoadhesive oral wound rinse, based on the hypothesis that wound protection might prevent mIAS. Glutamine suspension is also under evaluation as it is understood to have wound-preventative and tissue-repair properties, and another study is focused on dentist-guided oral management. Recent results of one of these trials (SWISH),29 reported that preventative care with a dexamethasone mouthwash 3-4 times a day significantly minimized or prevented the incidence of all grades of stomatitis in women receiving everolimus plus exemestane therapy for advanced/metastatic breast cancer compared with the incidence of stomatitis observed in a previously published phase 3 trial (BOLERO-2)30,31 of everolimus plus exemestane in the same patient population. Results from several other studies are expected soon.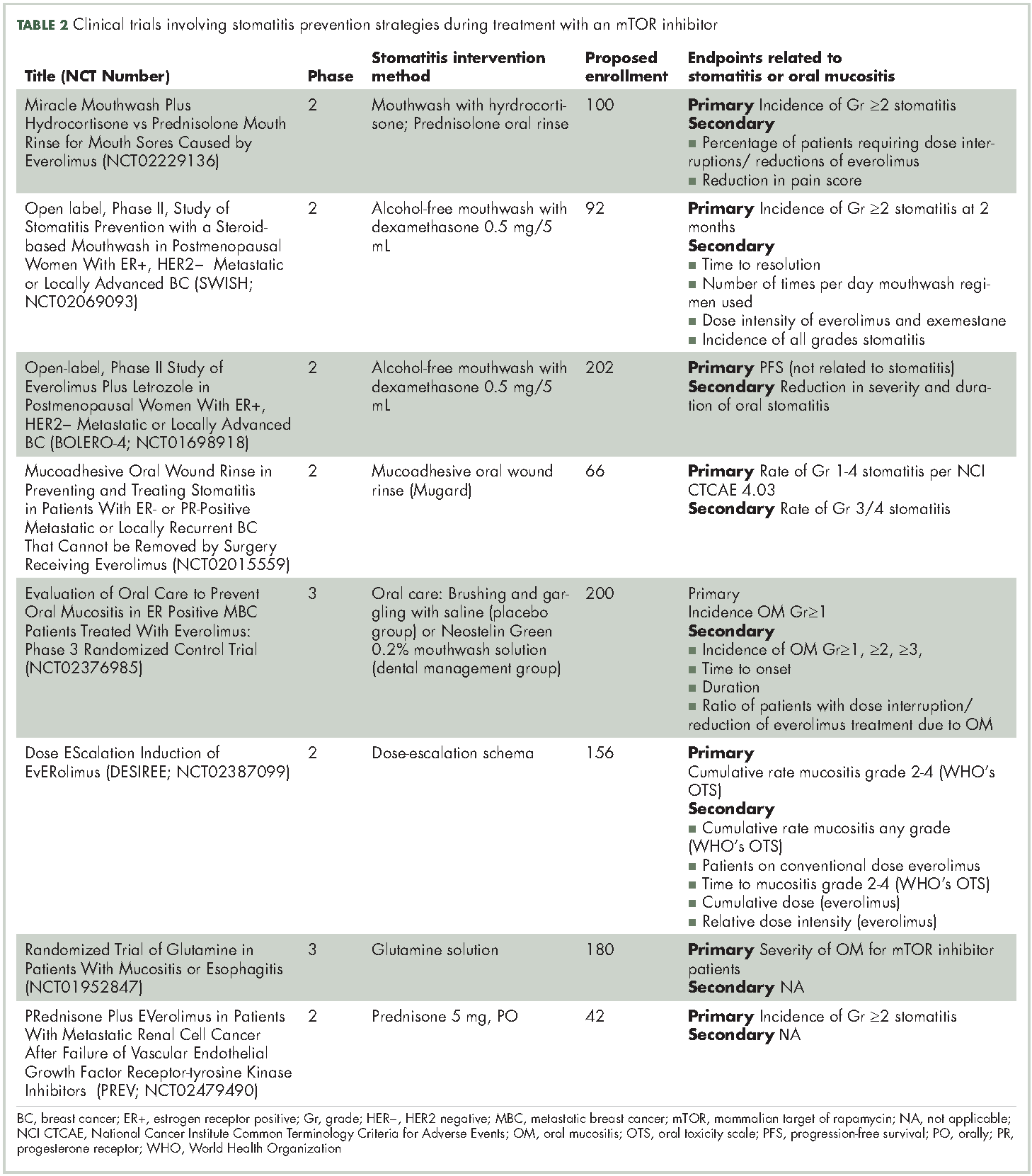
Current approaches to mIAS prevention are based largely on clinical experience with chemotherapy- or radiation-induced oral mucositis (Table 3).13,32 Preventive measures use three main strategies: establish and maintain good routine oral care; modify diet to avoid potentially damaging foods; and improve patient education about mIAS. In regard to patient education, numerous studies have reported that establishing an institutional protocol for oral care helped reduce the incidence of chemotherapy- or radiation-induced oral mucositis.33-40 An ongoing clinical study that will randomize patients to receive oral care education from oral surgeons or instruction on brushing only (NCT02376985) is investigating whether having an oral care protocol holds for patients with mIAS. The hypothesis is that focusing attention on oral care and educating patients to recognize the onset of mIAS facilitates early detection and promotes early intervention.
Therapeutic measures for patients with mIAS
Therapeutic measures for mIAS are based largely on experience with chemotherapy- or radiation-induced oral mucositis or recurrent aphthous ulcers (Table 3) and vary in part by the severity of lesions. Treatments for mild mIAS aim to ameliorate symptoms (eg, topical analgesics for pain), protect the oral mucosa (eg, mucoadhesive gels or viscous solutions that coat the oral cavity), prevent potential sequelae (eg, prophylactic antibiotics to avoid secondary infections), and reduce inflammation/immune response (eg, steroid-based mouth rinses, topical steroids, or topical anti-inflammatory agents). Treatments for mild mIAS are generally local rather than systemic.
Treatment options for moderate to severe mIAS include systemic approaches that generally carry increased risk of AEs and, therefore, should be reserved for patients with multiple lesions, uncontrolled or poorly controlled pain, or greatly diminished oral food intake (Table 3).41 When mIAS cannot be controlled with the interventions described, the dose of the mTOR inhibitor can be reduced with the recognition that dose modification of anticancer therapy may affect disease outcomes.29 The experience of reduction or interruption of treatment with everolimus in the BOLERO-2 trial as a strategy for management of AEs is discussed in a recent review.29 Prescribing information for both temsirolimus and everolimus specify that grade 3 AEs be treated with temporary dose interruption, and with resolution (temsirolimus: grade ≤2; everolimus: grade ≤1), treatment may be resumed at lower doses (temsirolimus: reduce by 5 mg/week; no lower than 15 mg/week; everolimus: reduce by half the previously administered dose).3,4 Grade 4 events due to treatment with temsirolimus may also be treated with dose interruption/reduction; the everolimus prescribing information advises treatment discontinuation for grade 4 stomatitis.
Summary and discussion
mTOR inhibitors can be effective treatments for patients with advanced cancer, specifically for advanced RCC, advanced pNET, and HR+, HER2-negative advanced breast cancer. Although mIAS may occur in many patients, it is usually grade 1 or 2 in severity. mIAS has an early onset, usually within the first 2 weeks of treatment16,19,42 and a relatively rapid resolution, usually within 3 weeks.16,19 Thus, most cases of mIAS are self-limiting.
The relatively recent emergence of mIAS poses short-term challenges regarding diagnosis, assessment, prevention, and treatment. Several clinical studies are underway to evaluate a range of interventions for their preventive and therapeutic efficacy in mIAS. Furthermore, our growing understanding of the underlying pathophysiology of mIAS can guide how mIAS is managed and what interventions patients receive.
Although mIAS is believed to differ from chemotherapy- or radiation-induced oral mucositis and aphthous ulcers, much can be learned from the treatment of both of these. Several strategies have been proposed to limit the occurrence of mIAS (Table 3). First, establish an oral care protocol. Educate patients who are initiating treatment with an mTOR inhibitor on implementation of the oral care protocol and emphasize adherence. Second, educate patients on the symptoms and timing of mIAS. Patients may hesitate to report mild symptoms or assume they are innocuous, so be clear that reporting all symptoms is important to allow timely clinical evaluation. Early recognition of mIAS facilitates early intervention and can prevent dose modification and interruption. Third, implement the preventive and treatment measures described. Many of the preventive measures can be incorporated into an oral care protocol.
The advent of mTOR inhibitors has clinically benefited many patients with cancer. Although side effects, like mIAS, may develop during treatment, they should not be considered insurmountable. Through education, vigilance, and aggressive management, health care providers and patients can work together to help patients maintain their quality of life while continuing to optimally address their disease.
Acknowledgment
The authors thank Anna Lau, PhD, and Patricia Segarini, PhD, of Percolation Communications LLC, for their editorial assistance. Funding for manuscript development was provided by Novartis Pharmaceuticals Corp.
1. Lauring J, Park BH, Wolff AC. The phosphoinositide-3-kinase-Akt-mTOR pathway as a therapeutic target in breast cancer. J Natl Compr Canc Netw. 2013;11:670-678.
2. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140-156.
3. Torisel (temsirolimus) [prescribing information]. Philadelphia, PA: Wyeth Pharmaceuticals; 2014.
4. Afinitor (everolimus) [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2015.
5. Rapamune (sirolimus) [prescribing information]. Philadelphia, PA: Wyeth Pharmaceuticals; 2012.
6. Peterson DE, Boers-Doets CB, Bensadoun RJ, Herrstedt J, ESMO Guidelines Committee. Management of oral and gastrointestinal mucosal injury: ESMO Clinical Practice Guidelines for diagnosis, treatment, and follow-up. Ann Oncol. 2015;26 Suppl 5:v139-151.
7. Hidalgo M, Buckner JC, Erlichman C, et al. A phase I and pharmacokinetic study of temsirolimus (CCI-779) administered intravenously daily for 5 days every 2 weeks to patients with advanced cancer. Clin Cancer Res. 2006;12:5755-5763.
8. Martins F, de Oliveira MA, Wang Q, et al. A review of oral toxicity associated with mTOR inhibitor therapy in cancer patients. Oral Oncol. 2013;49:293-298.
9. O’Donnell A, Faivre S, Burris HA, 3rd, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol. 2008;26:1588-1595.
10. Shameem R, Lacouture M, Wu S. Incidence and risk of high-grade stomatitis with mTOR inhibitors in cancer patients. Cancer Invest. 2015;33:70-77.
11. Femara (letrozole) [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
12. Rugo HS, Hortobagyi GN, Yao J, et al. Meta-analysis of stomatitis in clinical studies of everolimus: incidence and relationship with efficacy. Ann Oncol. 2016;27:519-525.
13. Keefe DM, Schubert MM, Elting LS, et al. Updated clinical practice guidelines for the prevention and treatment of mucositis. Cancer. 2007;109:820-831.
14. Sonis S, Treister N, Chawla S, Demetri G, Haluska F. Preliminary characterization of oral lesions associated with inhibitors of mammalian target of rapamycin in cancer patients. Cancer. 2010;116:210-215.
15. Scully C. Clinical practice. Aphthous ulceration. N Engl J Med. 2006;355:165-172.
16. Ferte C, Paci A, Zizi M, et al. Natural history, management and pharmacokinetics of everolimus-induced-oral ulcers: insights into compliance issues. Eur J Cancer. 2011;47:2249-2255.
17. Wong HM. Oral complications and management strategies for patients undergoing cancer therapy ScienceWorldJournal. 2014;581795.
18. de Oliveira MA, Martins EMF, Wang Q, et al. Clinical presentation and management of mTOR inhibitor-associated stomatitis. Oral Oncol. 2011;47:998-1003.
19. Rugo HS, Pritchard KI, Gnant M, et al. Incidence and time course of everolimus-related adverse events in postmenopausal women with hormone receptor-positive advanced breast cancer: insights from BOLERO-2. Ann Oncol. 2014;25:808-815.
20. National Cancer Institute. Cancer Therapy Evaluation Program. Common Terminology Criteria for Adverse Events v3.0 (CTCAE). http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed February 13, 2017.
21. National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) v4.03. http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf. Accessed February 13, 2017.
22. World Health Organization. WHO Handbook for Reporting Results of Cancer Treatment. Geneva, Switzerland: World Health Organization (WHO Offset Publication No. 48); 1979.
23. Epstein JB, Thariat J, Bensadoun RJ, et al. Oral complications of cancer and cancer therapy: from cancer treatment to survivorship. CA Cancer J Clin. 2012;62:400-422.
24. Iglesias-Bartolome R, Patel V, Cotrim A, et al. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell. 2012;11:401-414.
25. Lewkowicz N, Lewkowicz P, Dzitko K, et al. Dysfunction of CD4+CD25high T regulatory cells in patients with recurrent aphthous stomatitis. J Oral Pathol Med. 2008;37:454-461.
26. Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767-777.
27. Jin Y, Tymen SD, Chen D, et al. MicroRNA-99 family targets AKT/mTOR signaling pathway in dermal wound healing. PLoS One. 2013;8:e64434.
28. Rosselli-Murai LK, Almeida LO, Zagni C, et al. Periostin responds to mechanical stress and tension by activating the MTOR signaling pathway. PLoS One. 2013;8:e83580.
29. Rugo HS. Dosing and safety implications for oncologists when administering everolimus to patients with hormone receptor-positive breast cancer. Clin Breast Cancer. 2016;16:18-22.
30. Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520-529.
31. Yardley DA, Noguchi S, Pritchard KI, et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther. 2013;30:870-884.
32. Rubenstein EB, Peterson DE, Schubert M, et al. Clinical practice guidelines for the prevention and treatment of cancer therapy-induced oral and gastrointestinal mucositis. Cancer. 2004;100:2026-2046.
33. Borowski B, Benhamou E, Pico JL, Laplanche A, Margainaud JP, Hayat M. Prevention of oral mucositis in patients treated with high-dose chemotherapy and bone marrow transplantation: a randomised controlled trial comparing two protocols of dental care. Eur J Cancer B Oral Oncol. 1994;30B:93-97.
34. Cheng KK, Molassiotis A, Chang AM, Wai WC, Cheung SS. Evaluation of an oral care protocol intervention in the prevention of chemotherapy-induced oral mucositis in paediatric cancer patients. Eur J Cancer. 2001;37:2056-2063.
35. Dudjak LA. Mouth care for mucositis due to radiation therapy. Cancer Nurs. 1987;10:131-140.
36. Graham KM, Pecoraro DA, Ventura M, Meyer CC. Reducing the incidence of stomatitis using a quality assessment and improvement approach. Cancer Nurs. 1993;16:117-122.
37. Kenny SA. Effect of two oral care protocols on the incidence of stomatitis in hematology patients. Cancer Nurs. 1990;13:345-353.
38. Larson PJ, Miaskowski C, MacPhail L, et al. The PRO-SELF Mouth Aware program: an effective approach for reducing chemotherapy-induced mucositis. Cancer Nurs. 1998;21:263-268.
39. Levy-Polack MP, Sebelli P, Polack NL. Incidence of oral complications and application of a preventive protocol in children with acute leukemia. Spec Care Dentist. 1998;18:189-193.
40. Yeager KA, Webster J, Crain M, Kasow J, McGuire DB. Implementation of an oral care standard for leukemia and transplantation patients. Cancer Nurs. 2000;23:40-47; quiz 47-48.
41. Pilotte AP, Hohos MB, Polson KM, Huftalen TM, Treister N. Managing stomatitis in patients treated with mammalian target of rapamycin inhibitors. Clin J Oncol Nurs. 2011;15:E83-89.
42. Gomez-Fernandez C, Garden BC, Wu S, Feldman DR, Lacouture ME. The risk of skin rash and stomatitis with the mammalian target of rapamycin inhibitor temsirolimus: a systematic review of the literature and meta-analysis. Eur J Cancer. 2012;48:340-346.
43. Bonnaure-Mallet M, Bunetel L, Tricot-Doleux S, Guerin J, Bergeron C, LeGall E. Oral complications during treatment of malignant diseases in childhood: effects of tooth brushing. Eur J Cancer. 1998;34:1588-1591.
44. Chuang P, Langone AJ. Clobetasol ameliorates aphthous ulceration in renal transplant patients on sirolimus. Am J Transplant. 2007;7:714-717.
45. Femiano F, Buonaiuto C, Gombos F, Lanza A, Cirillo N. Pilot study on recurrent aphthous stomatitis (RAS): a randomized placebo-controlled trial for the comparative therapeutic effects of systemic prednisone and systemic montelukast in subjects unresponsive to topical therapy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010;109:402-407.
Mammalian target of rapamycin (mTOR), a serine–threonine protein kinase, operates in the phosphoinositide 3-kinase (PI3K)–protein kinase B (AKT)–mTOR signal transduction pathway regulating both normal and cancer cellular processes, including cell growth, proliferation, motility, survival, and protein and lipid synthesis.1 Genetic alterations affecting this pathway, including mutations in receptor tyrosine kinases PI3K and AKT, occur frequently in human cancers,2 supporting the rationale to develop drugs that target pathway components, such as mTOR inhibitors.
Two mTOR inhibitors are currently approved by the US Food and Drug Administration for cancer treatment: temsirolimus, for advanced renal cell carcinoma (RCC; approved 2007)3 and everolimus, for advanced RCC (approved 2009), advanced pancreatic neuroendocrine tumors (pNET; approved 2011), and hormone receptor-positive (HR-positive), human epidermal growth factor receptor-2 (HER2)-negative advanced breast cancer (approved 2012).4 Another mTOR inhibitor, sirolimus, is approved for use as an immunosuppressive agent and prophylactic against organ rejection after kidney transplant.5
Stomatitis, inflammation of the oral mucosa with contributing factors of genetic predisposition, nutritional deficiencies, infections, and immunological or hematologic dysfunction,6 occurs frequently as a side effect associated with mTOR inhibitor treatment.7-9 Left untreated or managed unsatisfactorily, mTOR inhibitor-associated stomatitis (mIAS) may cause patients discomfort and trouble with maintaining adequate nutritional intake and proper oral hygiene, as well as strict adherence to cancer treatment. It is therefore important for health care providers of cancer patients receiving mTOR inhibitor treatment to be knowledgeable about this side effect. The purpose of the present systematic review of published literature is to provide a better understanding of the differential diagnosis of mIAS, the pathophysiology of mIAS, preventive strategies for patients initiating mTOR inhibitor treatment, and treatment options available to manage mIAS.
Method
The PubMed database was searched with the terms mTOR inhibitor and stomatitis (no date restriction); 79 articles were retrieved, and all abstracts were reviewed to select those relevant to the aims of this review article. To understand future directions for management and prevention of mIAS, a search of clinicaltrials.gov was performed with the terms temsirolimus everolimus stomatitis yielding 12 clinical trials, of which 4 were excluded: 1 trial was terminated due to slow accrual, the status of 1 trial had not been verified in >2 years, and 2 studies focused on efficacy outcomes. A search of the American Society of Clinical Oncology (ASCO) meeting abstracts database was performed to assess the availability of clinical trial data; the search was limited to 2011-2016 and terms were stomatitis in the title and mTOR in the abstract or title. Seven abstracts were retrieved; 2 discussed stomatitis prevention (1 as a “trial-in-progress” and 1 presented results of the trial); the other 5 abstracts presented meta-analyses or reviews of previous clinical studies to assess the risk, incidence, management, and resolution of mIAS.
Review findings
Incidence of mIAS in patients treated for cancer
Two recent meta-analyses quantified the rate of mIAS in patients receiving mTOR inhibitors. Shameem and colleagues10 identified 9 randomized studies of everolimus (8 phase 3, 1 phase 2) and 2 of temsirolimus (1 each phase 2 and 3) involving a total of 4752 patients with a variety of tumor types including angiomyolipoma, breast, gastric, giant cell astrocytoma, pNET, and RCC. Patients received everolimus monotherapy (n = 1,075) or in combination with exemestane (n = 485), tamoxifen (n = 54), letrozole (n = 137), or octreotide (n = 216). Temsirolimus was administered as monotherapy (n = 208) or in combination with interferon
(n = 210) or letrozole (n = 550). The incidence of all-grade stomatitis in the 11 studies ranged from 11%-63%, and the overall incidence of any grade stomatitis was 33.5% (95% confidence interval [CI], 21.9%-47.6%). The concurrent use of a second agent may have confounded these findings because, for example, stomatitis has been reported in pooled analyses and in postmarketing experience with letrozole.11
Rugo and colleagues12 evaluated the incidence of stomatitis in 1455 patients participating in 5 phase 3 randomized clinical trials of everolimus in breast cancer, carcinoid tumor, pNET, and RCC. Patients received everolimus monotherapy
(n = 478) or in combination with exemestane (n = 482), trastuzumab plus vinorelbine (n = 280), or octreotide
(n = 215). The incidence of stomatitis in patients receiving everolimus was 59%-71%, compared with 19%-29% in 1,071 patients of the comparator arms (placebo, and placebo–trastuzumab–vinorelbine). The overall incidence of any grade stomatitis was 67%; most events were mild (grade 1/2); 9% of stomatitis events were moderate to severe (grade 3/4).
Differential clinical presentation of mIAS and severity
Oral mucositis is a common significant adverse event (AE) that occurs in patients with cancer who receive standard chemotherapy regimens and/or radiation therapy,13 so it is important to recognize that the clinical presentation of mIAS differs from that of oral mucositis (Table 1, Figure 14,15).16 mIAS shares some similarities with aphthous ulcers (also referred to as canker sores), a common oral condition with varied causes related to systemic disorders, gastrointestinal disorders, and infections, among others .17 In general, mIAS ulcers develop with a median onset of 10 days (range, 4-25 days) after initiation of mTOR inhibitor treatment and resolve in about 1-3 weeks after dose interruption/reduction of everolimus.16,18,19 mIAS ulcers appear as distinct, oval lesions with a central gray area surrounded by peripheral erythema. They are usually localized to the movable mucosa of the mouth and oropharynx. Although mIAS lesions are usually small, they are quite painful and may cluster.
Differential diagnosis of mIAS should be made based on physical examination and medical history, with consideration given to appearance of lesions (number, size, and location), current infection status, and current medications. Specific diagnostic testing should be conducted to confirm a coexisting or alternative cause of oral lesions.17
Although there are many different scales for grading mIAS severity, the most commonly used are the National Cancer Institute Common Terminology Criteria for Adverse Events (based on patient function, symptoms, and intervention needs) and the World Health Organization oral mucositis scales (based on symptoms, clinical presentation, and interference with patient function).20-22 These scales distinguish between mild lesions (grade 1/2) and moderate to severe lesions (grade 3/4) that cause significant pain or interfere with oral intake.
Pathophysiology of mIAS
The pathophysiology mIAS is incompletely understood. The ubiquitous role of the PI3K-AKT-mTOR pathway in regulating broad cellular functions suggests that mTOR inhibition is likely to have wide-ranging effects on many biological processes. It is not known whether disruption of one or more processes – or upsetting the balance of mTOR activities – underlies the formation of mIAS.
Differences between mIAS and oral mucositis, including clinical presentation and concomitant toxicities,16,23 suggest that the two types of oral lesions are fundamentally distinct. This distinction is supported by animal studies in which mTOR inhibition was found to almost completely prevent the appearance of oral mucositis in irradiated mice. The protective effect of mTOR inhibition is mediated through suppression of oxidative stress generated by radiation therapy.24
Although mIAS and recurrent aphthous ulcers share some similarities, it is not clear whether they also share a common pathophysiology. Recent studies suggest that patients with recurrent aphthous ulcers have immune dysfunction that leads to excessive immune response to normally innocuous substrates in the oral mucosa.25 mTOR inhibition can have proinflammatory activity by promoting autophagy, a process that stimulates antigen presentation and activation of T cells that produce proinflammatory cytokines.26 It is interesting to note that the incidence of stomatitis in patients receiving sirolimus after kidney transplant is relatively low, 3%-20%.5 Sirolimus is administered in combination with other immunosuppressants, namely cyclosporine and corticosteroids, so it suggests that concomitant use of a steroid-based regimen may have a preventive or therapeutic effect. However, posttransplant sirolimus is typically administered at relatively low doses, which might account in part for the lower incidence of mIAS observed. Ongoing clinical studies of steroid-based mouthwashes in patients receiving everolimus should shed light on this.
Other study findings have shown that inhibition of the PI3K-AKT-mTOR signaling pathway affects skin wound healing,27,28 which raises the possibility that mIAS may stem from a diminished capacity to repair physical injuries to the oral mucosa. More research is needed to elucidate the pathophysiology of mIAS.
Preventive measures for patients initiating mTOR inhibitor treatment
There are preventive measures for mIAS that have not yet been backed up with evidence-based findings, although several clinical studies that are underway aim to address this gap (Table 2). The hypotheses about the pathophysiology of mIAS suggest that certain preventive and therapeutic interventions might be effective against mIAS. For example, two studies are evaluating the use of steroid-based mouthwashes in patients receiving everolimus, based on the hypothesis that mIAS may arise from an inflammatory process; another study will evaluate a mucoadhesive oral wound rinse, based on the hypothesis that wound protection might prevent mIAS. Glutamine suspension is also under evaluation as it is understood to have wound-preventative and tissue-repair properties, and another study is focused on dentist-guided oral management. Recent results of one of these trials (SWISH),29 reported that preventative care with a dexamethasone mouthwash 3-4 times a day significantly minimized or prevented the incidence of all grades of stomatitis in women receiving everolimus plus exemestane therapy for advanced/metastatic breast cancer compared with the incidence of stomatitis observed in a previously published phase 3 trial (BOLERO-2)30,31 of everolimus plus exemestane in the same patient population. Results from several other studies are expected soon.
Current approaches to mIAS prevention are based largely on clinical experience with chemotherapy- or radiation-induced oral mucositis (Table 3).13,32 Preventive measures use three main strategies: establish and maintain good routine oral care; modify diet to avoid potentially damaging foods; and improve patient education about mIAS. In regard to patient education, numerous studies have reported that establishing an institutional protocol for oral care helped reduce the incidence of chemotherapy- or radiation-induced oral mucositis.33-40 An ongoing clinical study that will randomize patients to receive oral care education from oral surgeons or instruction on brushing only (NCT02376985) is investigating whether having an oral care protocol holds for patients with mIAS. The hypothesis is that focusing attention on oral care and educating patients to recognize the onset of mIAS facilitates early detection and promotes early intervention.
Therapeutic measures for patients with mIAS
Therapeutic measures for mIAS are based largely on experience with chemotherapy- or radiation-induced oral mucositis or recurrent aphthous ulcers (Table 3) and vary in part by the severity of lesions. Treatments for mild mIAS aim to ameliorate symptoms (eg, topical analgesics for pain), protect the oral mucosa (eg, mucoadhesive gels or viscous solutions that coat the oral cavity), prevent potential sequelae (eg, prophylactic antibiotics to avoid secondary infections), and reduce inflammation/immune response (eg, steroid-based mouth rinses, topical steroids, or topical anti-inflammatory agents). Treatments for mild mIAS are generally local rather than systemic.
Treatment options for moderate to severe mIAS include systemic approaches that generally carry increased risk of AEs and, therefore, should be reserved for patients with multiple lesions, uncontrolled or poorly controlled pain, or greatly diminished oral food intake (Table 3).41 When mIAS cannot be controlled with the interventions described, the dose of the mTOR inhibitor can be reduced with the recognition that dose modification of anticancer therapy may affect disease outcomes.29 The experience of reduction or interruption of treatment with everolimus in the BOLERO-2 trial as a strategy for management of AEs is discussed in a recent review.29 Prescribing information for both temsirolimus and everolimus specify that grade 3 AEs be treated with temporary dose interruption, and with resolution (temsirolimus: grade ≤2; everolimus: grade ≤1), treatment may be resumed at lower doses (temsirolimus: reduce by 5 mg/week; no lower than 15 mg/week; everolimus: reduce by half the previously administered dose).3,4 Grade 4 events due to treatment with temsirolimus may also be treated with dose interruption/reduction; the everolimus prescribing information advises treatment discontinuation for grade 4 stomatitis.
Summary and discussion
mTOR inhibitors can be effective treatments for patients with advanced cancer, specifically for advanced RCC, advanced pNET, and HR+, HER2-negative advanced breast cancer. Although mIAS may occur in many patients, it is usually grade 1 or 2 in severity. mIAS has an early onset, usually within the first 2 weeks of treatment16,19,42 and a relatively rapid resolution, usually within 3 weeks.16,19 Thus, most cases of mIAS are self-limiting.
The relatively recent emergence of mIAS poses short-term challenges regarding diagnosis, assessment, prevention, and treatment. Several clinical studies are underway to evaluate a range of interventions for their preventive and therapeutic efficacy in mIAS. Furthermore, our growing understanding of the underlying pathophysiology of mIAS can guide how mIAS is managed and what interventions patients receive.
Although mIAS is believed to differ from chemotherapy- or radiation-induced oral mucositis and aphthous ulcers, much can be learned from the treatment of both of these. Several strategies have been proposed to limit the occurrence of mIAS (Table 3). First, establish an oral care protocol. Educate patients who are initiating treatment with an mTOR inhibitor on implementation of the oral care protocol and emphasize adherence. Second, educate patients on the symptoms and timing of mIAS. Patients may hesitate to report mild symptoms or assume they are innocuous, so be clear that reporting all symptoms is important to allow timely clinical evaluation. Early recognition of mIAS facilitates early intervention and can prevent dose modification and interruption. Third, implement the preventive and treatment measures described. Many of the preventive measures can be incorporated into an oral care protocol.
The advent of mTOR inhibitors has clinically benefited many patients with cancer. Although side effects, like mIAS, may develop during treatment, they should not be considered insurmountable. Through education, vigilance, and aggressive management, health care providers and patients can work together to help patients maintain their quality of life while continuing to optimally address their disease.
Acknowledgment
The authors thank Anna Lau, PhD, and Patricia Segarini, PhD, of Percolation Communications LLC, for their editorial assistance. Funding for manuscript development was provided by Novartis Pharmaceuticals Corp.
Mammalian target of rapamycin (mTOR), a serine–threonine protein kinase, operates in the phosphoinositide 3-kinase (PI3K)–protein kinase B (AKT)–mTOR signal transduction pathway regulating both normal and cancer cellular processes, including cell growth, proliferation, motility, survival, and protein and lipid synthesis.1 Genetic alterations affecting this pathway, including mutations in receptor tyrosine kinases PI3K and AKT, occur frequently in human cancers,2 supporting the rationale to develop drugs that target pathway components, such as mTOR inhibitors.
Two mTOR inhibitors are currently approved by the US Food and Drug Administration for cancer treatment: temsirolimus, for advanced renal cell carcinoma (RCC; approved 2007)3 and everolimus, for advanced RCC (approved 2009), advanced pancreatic neuroendocrine tumors (pNET; approved 2011), and hormone receptor-positive (HR-positive), human epidermal growth factor receptor-2 (HER2)-negative advanced breast cancer (approved 2012).4 Another mTOR inhibitor, sirolimus, is approved for use as an immunosuppressive agent and prophylactic against organ rejection after kidney transplant.5
Stomatitis, inflammation of the oral mucosa with contributing factors of genetic predisposition, nutritional deficiencies, infections, and immunological or hematologic dysfunction,6 occurs frequently as a side effect associated with mTOR inhibitor treatment.7-9 Left untreated or managed unsatisfactorily, mTOR inhibitor-associated stomatitis (mIAS) may cause patients discomfort and trouble with maintaining adequate nutritional intake and proper oral hygiene, as well as strict adherence to cancer treatment. It is therefore important for health care providers of cancer patients receiving mTOR inhibitor treatment to be knowledgeable about this side effect. The purpose of the present systematic review of published literature is to provide a better understanding of the differential diagnosis of mIAS, the pathophysiology of mIAS, preventive strategies for patients initiating mTOR inhibitor treatment, and treatment options available to manage mIAS.
Method
The PubMed database was searched with the terms mTOR inhibitor and stomatitis (no date restriction); 79 articles were retrieved, and all abstracts were reviewed to select those relevant to the aims of this review article. To understand future directions for management and prevention of mIAS, a search of clinicaltrials.gov was performed with the terms temsirolimus everolimus stomatitis yielding 12 clinical trials, of which 4 were excluded: 1 trial was terminated due to slow accrual, the status of 1 trial had not been verified in >2 years, and 2 studies focused on efficacy outcomes. A search of the American Society of Clinical Oncology (ASCO) meeting abstracts database was performed to assess the availability of clinical trial data; the search was limited to 2011-2016 and terms were stomatitis in the title and mTOR in the abstract or title. Seven abstracts were retrieved; 2 discussed stomatitis prevention (1 as a “trial-in-progress” and 1 presented results of the trial); the other 5 abstracts presented meta-analyses or reviews of previous clinical studies to assess the risk, incidence, management, and resolution of mIAS.
Review findings
Incidence of mIAS in patients treated for cancer
Two recent meta-analyses quantified the rate of mIAS in patients receiving mTOR inhibitors. Shameem and colleagues10 identified 9 randomized studies of everolimus (8 phase 3, 1 phase 2) and 2 of temsirolimus (1 each phase 2 and 3) involving a total of 4752 patients with a variety of tumor types including angiomyolipoma, breast, gastric, giant cell astrocytoma, pNET, and RCC. Patients received everolimus monotherapy (n = 1,075) or in combination with exemestane (n = 485), tamoxifen (n = 54), letrozole (n = 137), or octreotide (n = 216). Temsirolimus was administered as monotherapy (n = 208) or in combination with interferon
(n = 210) or letrozole (n = 550). The incidence of all-grade stomatitis in the 11 studies ranged from 11%-63%, and the overall incidence of any grade stomatitis was 33.5% (95% confidence interval [CI], 21.9%-47.6%). The concurrent use of a second agent may have confounded these findings because, for example, stomatitis has been reported in pooled analyses and in postmarketing experience with letrozole.11
Rugo and colleagues12 evaluated the incidence of stomatitis in 1455 patients participating in 5 phase 3 randomized clinical trials of everolimus in breast cancer, carcinoid tumor, pNET, and RCC. Patients received everolimus monotherapy
(n = 478) or in combination with exemestane (n = 482), trastuzumab plus vinorelbine (n = 280), or octreotide
(n = 215). The incidence of stomatitis in patients receiving everolimus was 59%-71%, compared with 19%-29% in 1,071 patients of the comparator arms (placebo, and placebo–trastuzumab–vinorelbine). The overall incidence of any grade stomatitis was 67%; most events were mild (grade 1/2); 9% of stomatitis events were moderate to severe (grade 3/4).
Differential clinical presentation of mIAS and severity
Oral mucositis is a common significant adverse event (AE) that occurs in patients with cancer who receive standard chemotherapy regimens and/or radiation therapy,13 so it is important to recognize that the clinical presentation of mIAS differs from that of oral mucositis (Table 1, Figure 14,15).16 mIAS shares some similarities with aphthous ulcers (also referred to as canker sores), a common oral condition with varied causes related to systemic disorders, gastrointestinal disorders, and infections, among others .17 In general, mIAS ulcers develop with a median onset of 10 days (range, 4-25 days) after initiation of mTOR inhibitor treatment and resolve in about 1-3 weeks after dose interruption/reduction of everolimus.16,18,19 mIAS ulcers appear as distinct, oval lesions with a central gray area surrounded by peripheral erythema. They are usually localized to the movable mucosa of the mouth and oropharynx. Although mIAS lesions are usually small, they are quite painful and may cluster.
Differential diagnosis of mIAS should be made based on physical examination and medical history, with consideration given to appearance of lesions (number, size, and location), current infection status, and current medications. Specific diagnostic testing should be conducted to confirm a coexisting or alternative cause of oral lesions.17
Although there are many different scales for grading mIAS severity, the most commonly used are the National Cancer Institute Common Terminology Criteria for Adverse Events (based on patient function, symptoms, and intervention needs) and the World Health Organization oral mucositis scales (based on symptoms, clinical presentation, and interference with patient function).20-22 These scales distinguish between mild lesions (grade 1/2) and moderate to severe lesions (grade 3/4) that cause significant pain or interfere with oral intake.
Pathophysiology of mIAS
The pathophysiology mIAS is incompletely understood. The ubiquitous role of the PI3K-AKT-mTOR pathway in regulating broad cellular functions suggests that mTOR inhibition is likely to have wide-ranging effects on many biological processes. It is not known whether disruption of one or more processes – or upsetting the balance of mTOR activities – underlies the formation of mIAS.
Differences between mIAS and oral mucositis, including clinical presentation and concomitant toxicities,16,23 suggest that the two types of oral lesions are fundamentally distinct. This distinction is supported by animal studies in which mTOR inhibition was found to almost completely prevent the appearance of oral mucositis in irradiated mice. The protective effect of mTOR inhibition is mediated through suppression of oxidative stress generated by radiation therapy.24
Although mIAS and recurrent aphthous ulcers share some similarities, it is not clear whether they also share a common pathophysiology. Recent studies suggest that patients with recurrent aphthous ulcers have immune dysfunction that leads to excessive immune response to normally innocuous substrates in the oral mucosa.25 mTOR inhibition can have proinflammatory activity by promoting autophagy, a process that stimulates antigen presentation and activation of T cells that produce proinflammatory cytokines.26 It is interesting to note that the incidence of stomatitis in patients receiving sirolimus after kidney transplant is relatively low, 3%-20%.5 Sirolimus is administered in combination with other immunosuppressants, namely cyclosporine and corticosteroids, so it suggests that concomitant use of a steroid-based regimen may have a preventive or therapeutic effect. However, posttransplant sirolimus is typically administered at relatively low doses, which might account in part for the lower incidence of mIAS observed. Ongoing clinical studies of steroid-based mouthwashes in patients receiving everolimus should shed light on this.
Other study findings have shown that inhibition of the PI3K-AKT-mTOR signaling pathway affects skin wound healing,27,28 which raises the possibility that mIAS may stem from a diminished capacity to repair physical injuries to the oral mucosa. More research is needed to elucidate the pathophysiology of mIAS.
Preventive measures for patients initiating mTOR inhibitor treatment
There are preventive measures for mIAS that have not yet been backed up with evidence-based findings, although several clinical studies that are underway aim to address this gap (Table 2). The hypotheses about the pathophysiology of mIAS suggest that certain preventive and therapeutic interventions might be effective against mIAS. For example, two studies are evaluating the use of steroid-based mouthwashes in patients receiving everolimus, based on the hypothesis that mIAS may arise from an inflammatory process; another study will evaluate a mucoadhesive oral wound rinse, based on the hypothesis that wound protection might prevent mIAS. Glutamine suspension is also under evaluation as it is understood to have wound-preventative and tissue-repair properties, and another study is focused on dentist-guided oral management. Recent results of one of these trials (SWISH),29 reported that preventative care with a dexamethasone mouthwash 3-4 times a day significantly minimized or prevented the incidence of all grades of stomatitis in women receiving everolimus plus exemestane therapy for advanced/metastatic breast cancer compared with the incidence of stomatitis observed in a previously published phase 3 trial (BOLERO-2)30,31 of everolimus plus exemestane in the same patient population. Results from several other studies are expected soon.
Current approaches to mIAS prevention are based largely on clinical experience with chemotherapy- or radiation-induced oral mucositis (Table 3).13,32 Preventive measures use three main strategies: establish and maintain good routine oral care; modify diet to avoid potentially damaging foods; and improve patient education about mIAS. In regard to patient education, numerous studies have reported that establishing an institutional protocol for oral care helped reduce the incidence of chemotherapy- or radiation-induced oral mucositis.33-40 An ongoing clinical study that will randomize patients to receive oral care education from oral surgeons or instruction on brushing only (NCT02376985) is investigating whether having an oral care protocol holds for patients with mIAS. The hypothesis is that focusing attention on oral care and educating patients to recognize the onset of mIAS facilitates early detection and promotes early intervention.
Therapeutic measures for patients with mIAS
Therapeutic measures for mIAS are based largely on experience with chemotherapy- or radiation-induced oral mucositis or recurrent aphthous ulcers (Table 3) and vary in part by the severity of lesions. Treatments for mild mIAS aim to ameliorate symptoms (eg, topical analgesics for pain), protect the oral mucosa (eg, mucoadhesive gels or viscous solutions that coat the oral cavity), prevent potential sequelae (eg, prophylactic antibiotics to avoid secondary infections), and reduce inflammation/immune response (eg, steroid-based mouth rinses, topical steroids, or topical anti-inflammatory agents). Treatments for mild mIAS are generally local rather than systemic.
Treatment options for moderate to severe mIAS include systemic approaches that generally carry increased risk of AEs and, therefore, should be reserved for patients with multiple lesions, uncontrolled or poorly controlled pain, or greatly diminished oral food intake (Table 3).41 When mIAS cannot be controlled with the interventions described, the dose of the mTOR inhibitor can be reduced with the recognition that dose modification of anticancer therapy may affect disease outcomes.29 The experience of reduction or interruption of treatment with everolimus in the BOLERO-2 trial as a strategy for management of AEs is discussed in a recent review.29 Prescribing information for both temsirolimus and everolimus specify that grade 3 AEs be treated with temporary dose interruption, and with resolution (temsirolimus: grade ≤2; everolimus: grade ≤1), treatment may be resumed at lower doses (temsirolimus: reduce by 5 mg/week; no lower than 15 mg/week; everolimus: reduce by half the previously administered dose).3,4 Grade 4 events due to treatment with temsirolimus may also be treated with dose interruption/reduction; the everolimus prescribing information advises treatment discontinuation for grade 4 stomatitis.
Summary and discussion
mTOR inhibitors can be effective treatments for patients with advanced cancer, specifically for advanced RCC, advanced pNET, and HR+, HER2-negative advanced breast cancer. Although mIAS may occur in many patients, it is usually grade 1 or 2 in severity. mIAS has an early onset, usually within the first 2 weeks of treatment16,19,42 and a relatively rapid resolution, usually within 3 weeks.16,19 Thus, most cases of mIAS are self-limiting.
The relatively recent emergence of mIAS poses short-term challenges regarding diagnosis, assessment, prevention, and treatment. Several clinical studies are underway to evaluate a range of interventions for their preventive and therapeutic efficacy in mIAS. Furthermore, our growing understanding of the underlying pathophysiology of mIAS can guide how mIAS is managed and what interventions patients receive.
Although mIAS is believed to differ from chemotherapy- or radiation-induced oral mucositis and aphthous ulcers, much can be learned from the treatment of both of these. Several strategies have been proposed to limit the occurrence of mIAS (Table 3). First, establish an oral care protocol. Educate patients who are initiating treatment with an mTOR inhibitor on implementation of the oral care protocol and emphasize adherence. Second, educate patients on the symptoms and timing of mIAS. Patients may hesitate to report mild symptoms or assume they are innocuous, so be clear that reporting all symptoms is important to allow timely clinical evaluation. Early recognition of mIAS facilitates early intervention and can prevent dose modification and interruption. Third, implement the preventive and treatment measures described. Many of the preventive measures can be incorporated into an oral care protocol.
The advent of mTOR inhibitors has clinically benefited many patients with cancer. Although side effects, like mIAS, may develop during treatment, they should not be considered insurmountable. Through education, vigilance, and aggressive management, health care providers and patients can work together to help patients maintain their quality of life while continuing to optimally address their disease.
Acknowledgment
The authors thank Anna Lau, PhD, and Patricia Segarini, PhD, of Percolation Communications LLC, for their editorial assistance. Funding for manuscript development was provided by Novartis Pharmaceuticals Corp.
1. Lauring J, Park BH, Wolff AC. The phosphoinositide-3-kinase-Akt-mTOR pathway as a therapeutic target in breast cancer. J Natl Compr Canc Netw. 2013;11:670-678.
2. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140-156.
3. Torisel (temsirolimus) [prescribing information]. Philadelphia, PA: Wyeth Pharmaceuticals; 2014.
4. Afinitor (everolimus) [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2015.
5. Rapamune (sirolimus) [prescribing information]. Philadelphia, PA: Wyeth Pharmaceuticals; 2012.
6. Peterson DE, Boers-Doets CB, Bensadoun RJ, Herrstedt J, ESMO Guidelines Committee. Management of oral and gastrointestinal mucosal injury: ESMO Clinical Practice Guidelines for diagnosis, treatment, and follow-up. Ann Oncol. 2015;26 Suppl 5:v139-151.
7. Hidalgo M, Buckner JC, Erlichman C, et al. A phase I and pharmacokinetic study of temsirolimus (CCI-779) administered intravenously daily for 5 days every 2 weeks to patients with advanced cancer. Clin Cancer Res. 2006;12:5755-5763.
8. Martins F, de Oliveira MA, Wang Q, et al. A review of oral toxicity associated with mTOR inhibitor therapy in cancer patients. Oral Oncol. 2013;49:293-298.
9. O’Donnell A, Faivre S, Burris HA, 3rd, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol. 2008;26:1588-1595.
10. Shameem R, Lacouture M, Wu S. Incidence and risk of high-grade stomatitis with mTOR inhibitors in cancer patients. Cancer Invest. 2015;33:70-77.
11. Femara (letrozole) [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
12. Rugo HS, Hortobagyi GN, Yao J, et al. Meta-analysis of stomatitis in clinical studies of everolimus: incidence and relationship with efficacy. Ann Oncol. 2016;27:519-525.
13. Keefe DM, Schubert MM, Elting LS, et al. Updated clinical practice guidelines for the prevention and treatment of mucositis. Cancer. 2007;109:820-831.
14. Sonis S, Treister N, Chawla S, Demetri G, Haluska F. Preliminary characterization of oral lesions associated with inhibitors of mammalian target of rapamycin in cancer patients. Cancer. 2010;116:210-215.
15. Scully C. Clinical practice. Aphthous ulceration. N Engl J Med. 2006;355:165-172.
16. Ferte C, Paci A, Zizi M, et al. Natural history, management and pharmacokinetics of everolimus-induced-oral ulcers: insights into compliance issues. Eur J Cancer. 2011;47:2249-2255.
17. Wong HM. Oral complications and management strategies for patients undergoing cancer therapy ScienceWorldJournal. 2014;581795.
18. de Oliveira MA, Martins EMF, Wang Q, et al. Clinical presentation and management of mTOR inhibitor-associated stomatitis. Oral Oncol. 2011;47:998-1003.
19. Rugo HS, Pritchard KI, Gnant M, et al. Incidence and time course of everolimus-related adverse events in postmenopausal women with hormone receptor-positive advanced breast cancer: insights from BOLERO-2. Ann Oncol. 2014;25:808-815.
20. National Cancer Institute. Cancer Therapy Evaluation Program. Common Terminology Criteria for Adverse Events v3.0 (CTCAE). http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed February 13, 2017.
21. National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) v4.03. http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf. Accessed February 13, 2017.
22. World Health Organization. WHO Handbook for Reporting Results of Cancer Treatment. Geneva, Switzerland: World Health Organization (WHO Offset Publication No. 48); 1979.
23. Epstein JB, Thariat J, Bensadoun RJ, et al. Oral complications of cancer and cancer therapy: from cancer treatment to survivorship. CA Cancer J Clin. 2012;62:400-422.
24. Iglesias-Bartolome R, Patel V, Cotrim A, et al. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell. 2012;11:401-414.
25. Lewkowicz N, Lewkowicz P, Dzitko K, et al. Dysfunction of CD4+CD25high T regulatory cells in patients with recurrent aphthous stomatitis. J Oral Pathol Med. 2008;37:454-461.
26. Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767-777.
27. Jin Y, Tymen SD, Chen D, et al. MicroRNA-99 family targets AKT/mTOR signaling pathway in dermal wound healing. PLoS One. 2013;8:e64434.
28. Rosselli-Murai LK, Almeida LO, Zagni C, et al. Periostin responds to mechanical stress and tension by activating the MTOR signaling pathway. PLoS One. 2013;8:e83580.
29. Rugo HS. Dosing and safety implications for oncologists when administering everolimus to patients with hormone receptor-positive breast cancer. Clin Breast Cancer. 2016;16:18-22.
30. Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520-529.
31. Yardley DA, Noguchi S, Pritchard KI, et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther. 2013;30:870-884.
32. Rubenstein EB, Peterson DE, Schubert M, et al. Clinical practice guidelines for the prevention and treatment of cancer therapy-induced oral and gastrointestinal mucositis. Cancer. 2004;100:2026-2046.
33. Borowski B, Benhamou E, Pico JL, Laplanche A, Margainaud JP, Hayat M. Prevention of oral mucositis in patients treated with high-dose chemotherapy and bone marrow transplantation: a randomised controlled trial comparing two protocols of dental care. Eur J Cancer B Oral Oncol. 1994;30B:93-97.
34. Cheng KK, Molassiotis A, Chang AM, Wai WC, Cheung SS. Evaluation of an oral care protocol intervention in the prevention of chemotherapy-induced oral mucositis in paediatric cancer patients. Eur J Cancer. 2001;37:2056-2063.
35. Dudjak LA. Mouth care for mucositis due to radiation therapy. Cancer Nurs. 1987;10:131-140.
36. Graham KM, Pecoraro DA, Ventura M, Meyer CC. Reducing the incidence of stomatitis using a quality assessment and improvement approach. Cancer Nurs. 1993;16:117-122.
37. Kenny SA. Effect of two oral care protocols on the incidence of stomatitis in hematology patients. Cancer Nurs. 1990;13:345-353.
38. Larson PJ, Miaskowski C, MacPhail L, et al. The PRO-SELF Mouth Aware program: an effective approach for reducing chemotherapy-induced mucositis. Cancer Nurs. 1998;21:263-268.
39. Levy-Polack MP, Sebelli P, Polack NL. Incidence of oral complications and application of a preventive protocol in children with acute leukemia. Spec Care Dentist. 1998;18:189-193.
40. Yeager KA, Webster J, Crain M, Kasow J, McGuire DB. Implementation of an oral care standard for leukemia and transplantation patients. Cancer Nurs. 2000;23:40-47; quiz 47-48.
41. Pilotte AP, Hohos MB, Polson KM, Huftalen TM, Treister N. Managing stomatitis in patients treated with mammalian target of rapamycin inhibitors. Clin J Oncol Nurs. 2011;15:E83-89.
42. Gomez-Fernandez C, Garden BC, Wu S, Feldman DR, Lacouture ME. The risk of skin rash and stomatitis with the mammalian target of rapamycin inhibitor temsirolimus: a systematic review of the literature and meta-analysis. Eur J Cancer. 2012;48:340-346.
43. Bonnaure-Mallet M, Bunetel L, Tricot-Doleux S, Guerin J, Bergeron C, LeGall E. Oral complications during treatment of malignant diseases in childhood: effects of tooth brushing. Eur J Cancer. 1998;34:1588-1591.
44. Chuang P, Langone AJ. Clobetasol ameliorates aphthous ulceration in renal transplant patients on sirolimus. Am J Transplant. 2007;7:714-717.
45. Femiano F, Buonaiuto C, Gombos F, Lanza A, Cirillo N. Pilot study on recurrent aphthous stomatitis (RAS): a randomized placebo-controlled trial for the comparative therapeutic effects of systemic prednisone and systemic montelukast in subjects unresponsive to topical therapy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010;109:402-407.
1. Lauring J, Park BH, Wolff AC. The phosphoinositide-3-kinase-Akt-mTOR pathway as a therapeutic target in breast cancer. J Natl Compr Canc Netw. 2013;11:670-678.
2. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140-156.
3. Torisel (temsirolimus) [prescribing information]. Philadelphia, PA: Wyeth Pharmaceuticals; 2014.
4. Afinitor (everolimus) [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2015.
5. Rapamune (sirolimus) [prescribing information]. Philadelphia, PA: Wyeth Pharmaceuticals; 2012.
6. Peterson DE, Boers-Doets CB, Bensadoun RJ, Herrstedt J, ESMO Guidelines Committee. Management of oral and gastrointestinal mucosal injury: ESMO Clinical Practice Guidelines for diagnosis, treatment, and follow-up. Ann Oncol. 2015;26 Suppl 5:v139-151.
7. Hidalgo M, Buckner JC, Erlichman C, et al. A phase I and pharmacokinetic study of temsirolimus (CCI-779) administered intravenously daily for 5 days every 2 weeks to patients with advanced cancer. Clin Cancer Res. 2006;12:5755-5763.
8. Martins F, de Oliveira MA, Wang Q, et al. A review of oral toxicity associated with mTOR inhibitor therapy in cancer patients. Oral Oncol. 2013;49:293-298.
9. O’Donnell A, Faivre S, Burris HA, 3rd, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol. 2008;26:1588-1595.
10. Shameem R, Lacouture M, Wu S. Incidence and risk of high-grade stomatitis with mTOR inhibitors in cancer patients. Cancer Invest. 2015;33:70-77.
11. Femara (letrozole) [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
12. Rugo HS, Hortobagyi GN, Yao J, et al. Meta-analysis of stomatitis in clinical studies of everolimus: incidence and relationship with efficacy. Ann Oncol. 2016;27:519-525.
13. Keefe DM, Schubert MM, Elting LS, et al. Updated clinical practice guidelines for the prevention and treatment of mucositis. Cancer. 2007;109:820-831.
14. Sonis S, Treister N, Chawla S, Demetri G, Haluska F. Preliminary characterization of oral lesions associated with inhibitors of mammalian target of rapamycin in cancer patients. Cancer. 2010;116:210-215.
15. Scully C. Clinical practice. Aphthous ulceration. N Engl J Med. 2006;355:165-172.
16. Ferte C, Paci A, Zizi M, et al. Natural history, management and pharmacokinetics of everolimus-induced-oral ulcers: insights into compliance issues. Eur J Cancer. 2011;47:2249-2255.
17. Wong HM. Oral complications and management strategies for patients undergoing cancer therapy ScienceWorldJournal. 2014;581795.
18. de Oliveira MA, Martins EMF, Wang Q, et al. Clinical presentation and management of mTOR inhibitor-associated stomatitis. Oral Oncol. 2011;47:998-1003.
19. Rugo HS, Pritchard KI, Gnant M, et al. Incidence and time course of everolimus-related adverse events in postmenopausal women with hormone receptor-positive advanced breast cancer: insights from BOLERO-2. Ann Oncol. 2014;25:808-815.
20. National Cancer Institute. Cancer Therapy Evaluation Program. Common Terminology Criteria for Adverse Events v3.0 (CTCAE). http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed February 13, 2017.
21. National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) v4.03. http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf. Accessed February 13, 2017.
22. World Health Organization. WHO Handbook for Reporting Results of Cancer Treatment. Geneva, Switzerland: World Health Organization (WHO Offset Publication No. 48); 1979.
23. Epstein JB, Thariat J, Bensadoun RJ, et al. Oral complications of cancer and cancer therapy: from cancer treatment to survivorship. CA Cancer J Clin. 2012;62:400-422.
24. Iglesias-Bartolome R, Patel V, Cotrim A, et al. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell. 2012;11:401-414.
25. Lewkowicz N, Lewkowicz P, Dzitko K, et al. Dysfunction of CD4+CD25high T regulatory cells in patients with recurrent aphthous stomatitis. J Oral Pathol Med. 2008;37:454-461.
26. Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767-777.
27. Jin Y, Tymen SD, Chen D, et al. MicroRNA-99 family targets AKT/mTOR signaling pathway in dermal wound healing. PLoS One. 2013;8:e64434.
28. Rosselli-Murai LK, Almeida LO, Zagni C, et al. Periostin responds to mechanical stress and tension by activating the MTOR signaling pathway. PLoS One. 2013;8:e83580.
29. Rugo HS. Dosing and safety implications for oncologists when administering everolimus to patients with hormone receptor-positive breast cancer. Clin Breast Cancer. 2016;16:18-22.
30. Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520-529.
31. Yardley DA, Noguchi S, Pritchard KI, et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther. 2013;30:870-884.
32. Rubenstein EB, Peterson DE, Schubert M, et al. Clinical practice guidelines for the prevention and treatment of cancer therapy-induced oral and gastrointestinal mucositis. Cancer. 2004;100:2026-2046.
33. Borowski B, Benhamou E, Pico JL, Laplanche A, Margainaud JP, Hayat M. Prevention of oral mucositis in patients treated with high-dose chemotherapy and bone marrow transplantation: a randomised controlled trial comparing two protocols of dental care. Eur J Cancer B Oral Oncol. 1994;30B:93-97.
34. Cheng KK, Molassiotis A, Chang AM, Wai WC, Cheung SS. Evaluation of an oral care protocol intervention in the prevention of chemotherapy-induced oral mucositis in paediatric cancer patients. Eur J Cancer. 2001;37:2056-2063.
35. Dudjak LA. Mouth care for mucositis due to radiation therapy. Cancer Nurs. 1987;10:131-140.
36. Graham KM, Pecoraro DA, Ventura M, Meyer CC. Reducing the incidence of stomatitis using a quality assessment and improvement approach. Cancer Nurs. 1993;16:117-122.
37. Kenny SA. Effect of two oral care protocols on the incidence of stomatitis in hematology patients. Cancer Nurs. 1990;13:345-353.
38. Larson PJ, Miaskowski C, MacPhail L, et al. The PRO-SELF Mouth Aware program: an effective approach for reducing chemotherapy-induced mucositis. Cancer Nurs. 1998;21:263-268.
39. Levy-Polack MP, Sebelli P, Polack NL. Incidence of oral complications and application of a preventive protocol in children with acute leukemia. Spec Care Dentist. 1998;18:189-193.
40. Yeager KA, Webster J, Crain M, Kasow J, McGuire DB. Implementation of an oral care standard for leukemia and transplantation patients. Cancer Nurs. 2000;23:40-47; quiz 47-48.
41. Pilotte AP, Hohos MB, Polson KM, Huftalen TM, Treister N. Managing stomatitis in patients treated with mammalian target of rapamycin inhibitors. Clin J Oncol Nurs. 2011;15:E83-89.
42. Gomez-Fernandez C, Garden BC, Wu S, Feldman DR, Lacouture ME. The risk of skin rash and stomatitis with the mammalian target of rapamycin inhibitor temsirolimus: a systematic review of the literature and meta-analysis. Eur J Cancer. 2012;48:340-346.
43. Bonnaure-Mallet M, Bunetel L, Tricot-Doleux S, Guerin J, Bergeron C, LeGall E. Oral complications during treatment of malignant diseases in childhood: effects of tooth brushing. Eur J Cancer. 1998;34:1588-1591.
44. Chuang P, Langone AJ. Clobetasol ameliorates aphthous ulceration in renal transplant patients on sirolimus. Am J Transplant. 2007;7:714-717.
45. Femiano F, Buonaiuto C, Gombos F, Lanza A, Cirillo N. Pilot study on recurrent aphthous stomatitis (RAS): a randomized placebo-controlled trial for the comparative therapeutic effects of systemic prednisone and systemic montelukast in subjects unresponsive to topical therapy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010;109:402-407.
E-cigarettes and vapes: Do they work for smoking cessation and should we be recommending their use?
The popularity of electronic cigarettes (E-cigs) and “vapes” has grown dramatically, spawning a new industry of electronic nicotine delivery systems (ENDS). With the increasing use of E-cigs not only for smoking cessation, but also as a primary nicotine source, it is important for mental health professionals to be prepared to discuss use of these devices with patients. In this article, we will describe:
- the composition of E-cigs and their current use
- evidence for their use for smoking cessation
- adverse health effects
- recommendations of major regulatory agencies.
Finally, we will provide recommendations for E-cig use in clinical populations.
What is an electronic nicotine delivery system?
ENDS produce an aerosol with or without nicotine that is inhaled and is thought to mimic the use of combustible cigarettes. ENDS evolved from basic E-cigs into a less “cigarette-like” and more customizable product (Figure 1). ENDS include a range of designs and go by various names, including “personal vaporizers,” “e-cigars,” and “e-hookahs” (in this article, we will use the term “ENDS” to refer to these devices).
The general design of ENDS is a plastic tubing system that contains a mouthpiece, battery, electronic heating element (“vaporizer”), and a cartridge with liquid solvent with or without nicotine or flavoring (Figure 2). One draw on the mouthpiece or press of a button activates the device, heats the solution, and delivers a vapor in a similar manner to taking a puff of a cigarette. Although studies have shown that ENDS result in significant increases in plasma nicotine concentrations in 5 minutes,1 the plasma nicotine levels obtained with the first-generation “cigarette-like” ENDS are much lower than those caused by inhaling tobacco smoke.2 Over time nicotine delivery capability has improved as ENDS have evolved such that the rate of nicotine delivery and peak concentration obtained with newer models more closely mirror tobacco cigarettes.3 Whether the rapid delivery of larger amounts of nicotine helps or hinders one’s efforts to break nicotine addiction remains to be determined because of the reinforcing properties of the drug.
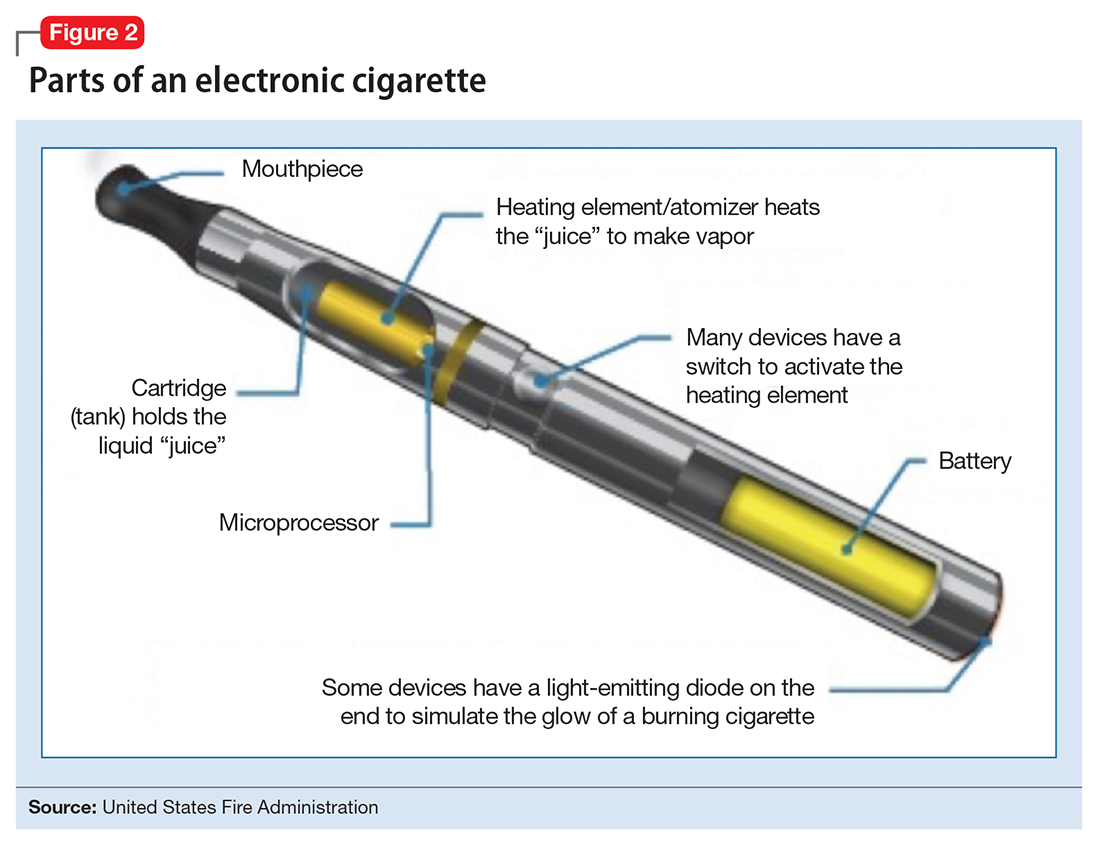
The liquid in the E-cig cartridge typically contains not only nicotine but a number of chemical compounds with potentially deleterious or unknown health risks. The 3 main ingredients include:
- a solvent of glycerin and/or propylene glycol
- nicotine in various concentrations
- flavorings.
The glycerin or propylene glycol forms the basis for the aerosol. Nicotine concentrations vary from 0 (denicotinized) to 35 mcg per puff.4 A study reported 7,700 unique flavors available for vaping liquid.5 The liquid also contains impurities, such as anabasine, which has effects on the α-7 nicotinic acetylcholine receptor and its principal use is as an insecticide and β-nicotyrine, which inhibits cytochrome P450 2A.
Epidemiology and end-user perspectives
In 2014, 12.4% of U.S. adults classified themselves as “ever users” of ENDS (used at least once) and 3.7% of adults classified themselves as current users, according to the National Health Interview Study.6 Importantly, among E-cig users who had not used combustible cigarettes, young adults (age 18 to 24) were more likely to have tried ENDS than older adults. ENDS are becoming more popular across the globe. A study in the European Union found that ever users of ENDS most commonly were current cigarette smokers (31%) followed by former (10.8%) and never smokers (2.3%).7
ENDS use is relevant for mental health professionals because of the high rate of comorbid tobacco use disorder in individuals with psychiatric conditions. For example, 2 U.S. population surveys8,9 revealed those with mental health conditions were 1.5 to 2 times more likely to have tried ENDS and 2 to 3 times more likely to be current users. Those with psychiatric illness reported similar reasons for ENDS use as other individuals, including “just because,” use as a smoking cessation aid, ease of use, and perceived safety vs combustible cigarettes.
A recent review that included 9 studies focusing on ENDS use in those with mental illness reported mixed findings on the utility of these devices to reduce or stop use of combustible cigarettes.10 Additionally, it is important to monitor the use of cigarettes and ENDS in patients with psychiatric illness because the byproducts of tobacco smoke can affect the metabolism of some psychotropic medications.11 Although reduced use of combustible cigarettes could lead to lower dosing of some psychotropics, an unreported decrease in combustible cigarette use could lead to supratherapeutic drug levels. There are no data on the effect of ENDS on the metabolism of psychotropics.
ENDS are increasingly popular among adolescents. In 2015, there were an estimated 4.6 million current tobacco users among middle/high school youths in the United States and 3 million current ENDS users, according to the National Youth Tobacco Surveys.12 The shift from combustible cigarettes to ENDS is notable, with an increase in the percentage of current E-cig users and a decrease in the percentage of exclusive combustible cigarette users. In addition, there has been no change in the prevalence of lifetime tobacco users.12 This is a global issue, as reports of ever use of ENDS by adolescents range from 6.5% to 31% in the United States, 14.6% in Canada, and 4.7% to 38.5% in Europe.13 Based on these trends, the U.S. Surgeon General released a statement warning against the use of ENDS in youth because of the lack of safety data and strong association with use of tobacco products.14
There are a number of possible reasons for the increasing popularity of ENDS, including the product’s novelty, lack of regulations regarding their sale, availability of flavorings, and the perception that ENDS are safe alternatives to cigarettes. E-cig–using youths have described ENDS as “not at all harmful” and “not at all addictive” and believe that ENDS with flavoring are less harmful than those without.15 Although studies in adults show some users reporting that ENDS are less satisfying, they are seen as useful in decreasing craving and a safer alternative to cigarettes.16,17
Are ENDS effective for smoking cessation?
The evidence for ENDS as aids to smoking cessation remains murky (Table 118-22). There is a paucity of randomized controlled clinical trials (RCTs) investigating ENDS for smoking cessation or reduction, and it is difficult to quantify the amount of nicotine used in ENDS because of the variety of delivery systems and cartridges. In a recent Cochrane review, those using ENDS to quit smoking were more likely to be abstinent from combustible cigarettes at 6 months vs those using nicotine-free ENDS (relative risk = 2.29; 95% CI, 1.05 to 4.96), but there was no significant difference in quit rates compared with nicotine patches.23 However, the confidence in this finding was rated as low because of the limited number of RCTs. Of note, the authors found 15 ongoing RCTs at the time of publication that might be eligible for later evaluation.
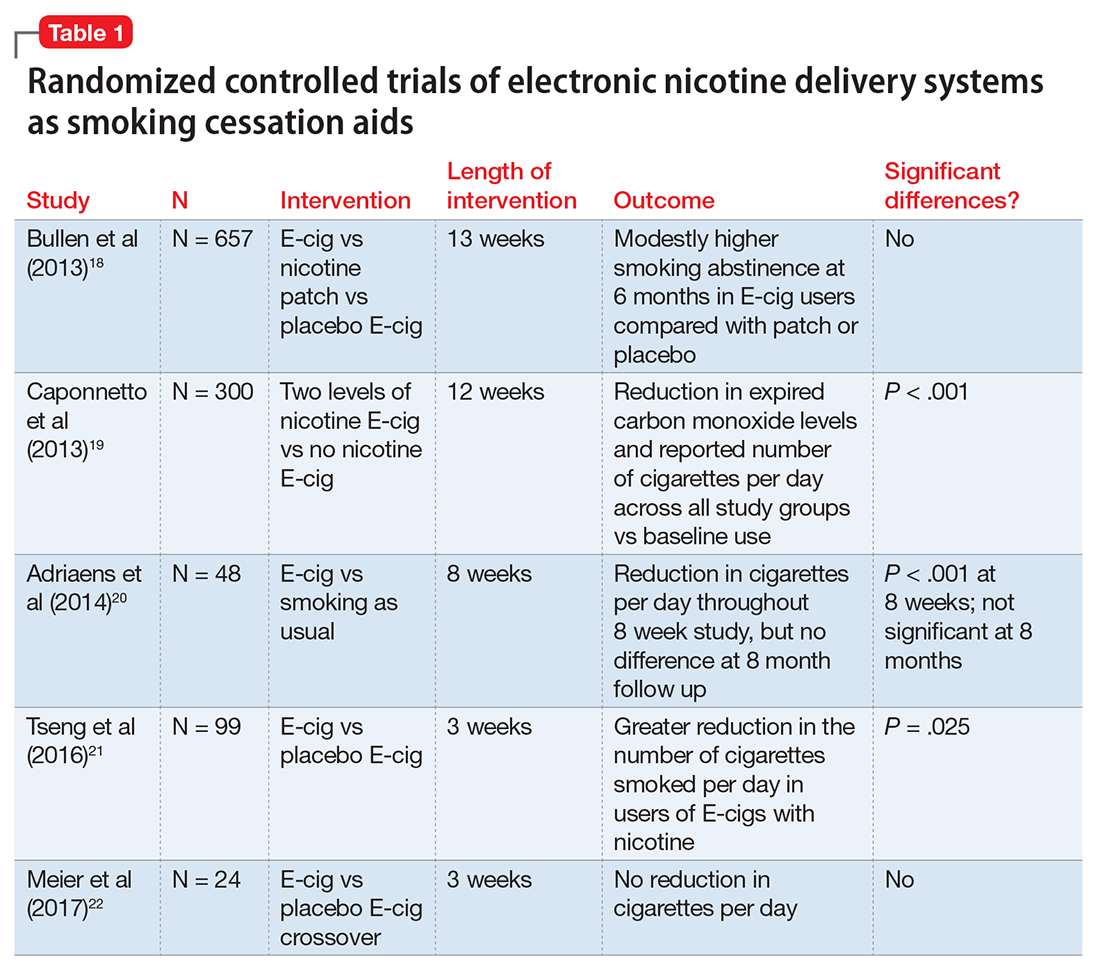
Non-RCTs reveal mixed data. Positive results include 1 study with an odds ratio of 6.07 to quit for intensive ENDS users vs non-users,24 and another with dual users of combustible and electronic cigarettes having a 46% quit rate at 1 year.25 Additionally, in a pilot study providing ENDS to 14 patients with schizophrenia who had no previous desire to quit smoking, authors noted a reduction in the number of cigarettes smoked per day by 50% in one-half of participants and abstinence in 14% of participants at 52 weeks.26 Studies with neutral or negative results include those showing ENDS users to be current combustible tobacco smokers, and use of ENDS not predicting smoking cessation.4,27 Data also are mixed regarding the use of ENDS as a harm reduction strategy. One study found that ENDS decreased cigarette consumption, but did not increase the likelihood of quitting,28 while another reported that daily use of ENDS increased the odds of reducing smoking by as much as 2.5 times compared with non-use of such aids.29 In a 24-month prospective cohort study following tobacco users, there was no difference in the number of cigarettes smoked per day in those who started the trial as users of combustible cigarettes alone vs combustible cigarettes plus ENDS users.30 Interestingly, those who started the study as combustible cigarette users and switched to ENDS and those who had continued dual use throughout the 24 months smoked fewer combustible cigarettes per day than those who never tried ENDS or quit during the study period.
Health effects

To better understand the adverse health effects of ENDS, one must consider potential short- and long-term consequences (Table 2). In the short-term, ENDS have been found to increase markers of inflammation and oxidative stress acutely as evidenced by in vivo laboratory studies.31,32 ENDS also have been linked to upper respiratory irritation, in part, because of the transformation of glycerin in the nicotine cartridge to acrolein upon combustion.33 Even 5 minutes of ad lib E-cig use has been found to significantly increase airflow resistance during pulmonary function tests34—changes that have been shown to precede more persistent alterations in peak expiratory flow, such as those seen in chronic obstructive pulmonary disease. The more common patient-reported side effects include:
- daytime cough (27%)
- phlegm production (25%)
- headache (21%)
- dry mouth/throat (20%)
- vertigo, headache, or nausea (9%).35,36
A RCT investigating efficacy of E-cigs vs nicotine patches vs denicotinized E-cigs found no difference among the groups in the number of reported adverse events.18 Interestingly, another RCT found a decrease in adverse events, such as dry cough, mouth irritation, throat irritation, shortness of breath, and headache, compared with baseline in combustible cigarette smokers who used regular or denicotinized E-cigs.19
Although no studies have directly investigated long-term health consequences of ENDS because of their relative novelty, one can extrapolate potential harmful long-term effects based on knowledge of the products’ chemical constituents. For example, propylene glycol can degrade into propylene oxide, a class 2B carcinogen.37 Other potential carcinogens in the aerosol include formaldehyde and acetaldehyde. On a broader scale, many of the particulates have been shown to cause systemic inflammation, which is thought to increase cardiovascular and respiratory disease and death.38 Flavorings in ENDS include a variety of components including, but not limited to, aldehydes, which are irritants, and other additives that have been associated with respiratory disease.39
Second-hand exposure. There are no long-term studies of second-hand vapor exposure, but similar to long-term health on primary users, one can glean some observations from the literature. It is promising that compared with cigarettes, ENDS lack sidestream smoke and the vapor has not been found to contain carbon monoxide.40 Some research has demonstrated that the size and spray of fine particles in the aerosol is as large or larger than combustible cigarettes.41 Formaldehyde, acetaldehyde, isoprene, and acetic acid have been found in ENDS vapor.40 Interestingly, a simulated café study found elevated nicotine, glycerine, hydrocarbon, and other materials classified as carcinogens in the air.42
Although it is popularly thought that ENDS are less toxic than tobacco cigarettes, there is not enough evidence to estimate precisely as to how much less toxic or the consequences of use. ENDS are increasingly popular and are being used by never smokers who should be educated on the potential harm that ENDS pose.
Recommendations from agencies and medical organizations
The World Health Organization (WHO) recommended prohibiting the use of ENDS in indoor spaces to minimize potential health risks to users and non-users. The WHO also aims to prevent dissemination of unproven health claims, including claims that ENDS are effective—or not—or that the devices are innocuous.36 In the United States, the FDA has stated that ENDS are not recommended for safe quitting (2009). In August 2016, the FDA introduced regulations banning the sale of ENDS to individuals age <18 and required manufacturers to submit documents detailing all ingredients for review and possible approval.
The American Lung Association has stated its concerns about the use of ENDS but has not made any direct recommendations. The American Heart Association reports a potential negative public health impact and provides clinical guideline recommendations.43 Prominent psychiatric organizations such as the American Psychiatric Association, American Academy of Addiction Psychiatry (AAAP), the Substance Abuse and Mental Health Services Administration (SAMHSA), and the National Institute of Drug Abuse do not have official statements supporting or rejecting the use of ENDS. However, they do note the potential harm and lack of substantial evidence for efficacy of ENDS as a smoking cessation tool, and the AAAP and SAMHSA state that they will work with regulatory agencies to reduce the use of toxic products with addictive potential including ENDS.44-46
Clinical recommendations
We do not recommend ENDS as a first-line treatment for smoking cessation because there is no evidence they are superior to the FDA-approved nicotine replacement therapies (NRTs), the paucity of research into the potential short- and long-term health risks of ENDS, and the fact that these products are not regulated for use as smoking cessation aids. It is, however, advisable to discuss ENDS use with patients by:
- asking if they are using the products
- assessing whether the user also is a smoker
- advising the patient to quit.
It also is important to assess the patient’s knowledge and attitudes regarding ENDS use and provide education about the products. Some patients firmly believe that ENDS are the lesser of 2 evils, and they are decreasing the harms of smoking by using these devices. While the debate over a potential harm reduction strategy unfolds,47 we think that because of the state of the evidence it is prudent to adopt a more precautionary stance and recommend that patients work toward abstinence from nicotine in any form.
For dual tobacco/ENDS users and for patients using ENDS who want to quit smoking, we recommend treatment with an approved pharmacotherapy (ie, NRTs, bupropion, and varenicline) combined with counseling. A 2013 Cochrane Review found that all pharamacotherapy options are more effective than placebo, and combination NRT and varenicline are superior to single NRT or bupropion (Box).23,48

1. Hajek P, Goniewicz ML, Phillips A, et al. Nicotine intake from electronic cigarettes on initial use and after 4 weeks of regular use. Nicotine Tob Res. 2015;17(2):175-179.
2. Farsalinos KE, Polosa R. Safety evaluation and risk assessment of electronic cigarettes as tobacco cigarette substitutes: a systematic review. Ther Adv Drug Saf. 2014;5(2):67-86.
3. St Helen G, Havel C, Dempsey DA, et al. Nicotine delivery, retention and pharmacokinetics from various electronic cigarettes. Addiction. 2016;111(3):535-544.
4. Grana R, Benowitz N, Glantz SA. E-cigarettes: a scientific review. Circulation. 2014;129(19):1972-1986.
5. Zhu SH, Sun JY, Bonnevie E, et al. Four hundred and sixty brands of e-cigarettes and counting: implications for product regulation. Tob Control. 2014;23(suppl 3):iii3-iii9. doi: 10.1136/tobaccocontrol-2014-051670.
6. Schoenborn CA, Gindi RM. Electronic cigarette use among adults: United States, 2014. NCHS Data Brief. 2015;(217):1-8.
7. Farsalinos KE, Poulas K, Voudris V, et al. Electronic cigarette use in the European Union: analysis of a representative sample of 27 460 Europeans from 28 countries. Addiction. 2016;111(11):2032-2040.
8. Cummins SE, Zhu SH, Tedeschi GJ, et al. Use of e-cigarettes by individuals with mental health conditions. Tob Control. 2015;23(suppl 3):iii48-iii53. doi: 10.1136/tobaccocontrol-2013-051511.
9. Spears CA, Jones DM, Weaver SR, et al. Use of electronic nicotine delivery systems among adults with mental health conditions, 2015. Int J Environ Res Public Heal. 2017;14(1):10.
10. Hefner K, Valentine G, Sofuoglu M. Electronic cigarettes and mental illness: reviewing the evidence for help and harm among those with psychiatric and substance use disorders [published online February 2, 2017]. Am J Addict. doi: 10.1111/ajad.12504.
11. Anthenelli R. How—and why—to help psychiatric patients stop smoking. Current Psychiatry. 2005;4(1):77-87.
12. Singh T, Arrazola RA, Corey CG, et al. Tobacco use among middle and high school students—United States, 2011-2015. MMWR Morb Mortal Wkly Rep. 2016;65(14):361-367.
13. Greenhill R, Dawkins L, Notley C, et al. Adolescent awareness and use of electronic cigarettes: a review of emerging trends and findings. J Adolesc Heal. 2016;59(6):612-619.
14. U.S. Department of Health and Human Services. E-cigarette use among youth and young adults: a report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention; 2016.
15. Cooper M, Harrell MB, Pérez A, et al. Flavorings and perceived harm and addictiveness of e-cigarettes among youth. Tob Regul Sci. 2016;2(3):278-289.
16. Kim H, Davis AH, Dohack JL, et al. E-cigarettes use behavior and experience of adults: qualitative research findings to inform e-cigarette use measure development. Nicotine Tob Res. 2017;19(2):190-196.
17. Czoli CD, Fong GT, Mays D, et al. How do consumers perceive differences in risk across nicotine products? A review of relative risk perceptions across smokeless tobacco, e-cigarettes, nicotine replacement therapy and combustible cigarettes. Tob Control. 2017;26(e1):e49-e58.
18. Bullen C, Howe C, Laugesen M, et al. Electronic cigarettes for smoking cessation: a randomised controlled trial. Lancet. 2013;382(9905):1629-1637.
19. Caponnetto P, Campagna D, Cibella F, et al. EffiCiency and safety of an eLectronic cigAreTte (ECLAT) as tobacco cigarettes substitute: a prospective 12-month randomized control design study. PLoS One. 2013;8(6):e66317. doi: 10.1371/journal.pone.0066317.
20. Adriaens K, Van Gucht D, Declerck P, et al. Effectiveness of the electronic cigarette: an eight-week Flemish study with six-month follow-up on smoking reduction, craving and experienced benefits and complaints. Int J Environ Res Public Health. 2014;11(11):11220-11248.
21. Tseng TY, Ostroff JS, Campo A, et al. A randomized trial comparing the effect of nicotine versus placebo electronic cigarettes on smoking reduction among young adult smokers. Nicotine Tob Res. 2016;18(10):1937-1943.
22. Meier E, Wahlquist AE, Heckman BW, et al. A pilot randomized crossover trial of electronic cigarette sampling among smokers. Nicotine Tob Res. 2017;19(2):176-182.
23. Hartmann-Boyce J, McRobbie H, Bullen C, et al. Electronic cigarettes for smoking cessation [published online September 14, 2016]. Cochrane Database Syst Rev. 2016;9:CD010216.
24. Biener L, Hargraves JL. A longitudinal study of electronic cigarette use among a population-based sample of adult smokers: association with smoking cessation and motivation to quit. Nicotine Tob Res. 2014;17(2):127-133.
25. Etter JF, Bullen C. A longitudinal study of electronic cigarette users. Addict Behav. 2014;39(2):491-494.
26. Caponnetto P, Auditore R, Russo C, et al. Impact of an electronic cigarette on smoking reduction and cessation in schizophrenic smokers: a prospective 12-month pilot study. Int J Environ Res Public Health. 2013;10(2):446-461.
27. Popova L, Ling PM. Alternative tobacco product use and smoking cessation: a national study. Am J Public Health. 2013;103(5):923-930.
28. Adkison SE, O’Connor RJ, Bansal-Travers M, et al. Electronic nicotine delivery systems: International Tobacco Control Four-Country Survey. Am J Prev Med. 2013;44(3):207-215.
29. Brose LS, Hitchman SC, Brown J, et al. Is the use of electronic cigarettes while smoking associated with smoking cessation attempts, cessation and reduced cigarette consumption? A survey with a 1-year follow-up. Addiction. 2015;110(7):1160-1168.
30. Manzoli L, Flacco ME, Ferrante M, et al; ISLESE Working Group. Cohort study of electronic cigarette use: effectiveness and safety at 24 months [published online June 6, 2016]. Tob Control. doi: 10.1136/tobaccocontrol-2015-052822.
31. Lerner CA, Sundar IK, Yao H, et al. Vapors produced by electronic cigarettes and E-juices with flavorings induce toxicity, oxidative stress, and inflammatory response in lung epithelial cells and in mouse lung. PLoS One. 2015;10(2):e0116732. doi: 10.1371/journal.pone.0116732.
32. Sussan TE, Gajghate S, Thimmulappa RK, et al. Exposure to electronic cigarettes impairs pulmonary anti-bacterial and anti-viral defenses in a mouse model. PLoS One. 2015;10(2):e0116861. doi: 10.1371/journal.pone.0116861.
33. US Environmental Protection Agency. Acrolein. https://www.epa.gov/sites/production/files/2016-08/documents/acrolein.pdf. Updated September 2009. Accessed April 7, 2017.
34. Vardavas CI, Anagnostopoulos N, Kougias M, et al. Short-term pulmonary effects of using an electronic cigarette: impact on respiratory flow resistance, impedance, and exhaled nitric oxide. Chest. 2012;141(6):1400-1406.
35. Etter JF. Electronic cigarettes: a survey of users. BMC Public Health. 2010;10:231.
36. Goniewicz ML, Lingas EO, Hajek P. Patterns of electronic cigarette use and user beliefs about their safety and benefits: an internet survey. Drug Alcohol Rev. 2013;32(2):133-140.
37. Laino T, Tuma C, Moor P, et al. Mechanisms of propylene glycol and triacetin pyrolysis. J Phys Chem A. 2012;116(18):4602-4609.
38. Brook RD, Rajagopalan S, Pope CA 3rd, et al; American Heart Association Council on Epidemiology and Prevention; Council on the Kidney in Cardiovascular Disease; Council on Nutrition, Physical Activity and Metabolism. Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation. 2010;121(21):2331-2378.
39. Barrington-Trimis JL, Samet JM, McConnell R. Flavorings in electronic cigarettes: an unrecognized respiratory health hazard? JAMA. 2014;312(23):2493-2494.
40. Schripp T, Markewitz D, Uhde E, et al. Does e-cigarette consumption cause passive vaping? Indoor Air. 2013;23(1):25-31.
41. Fuoco FC, Buonanno G, Stabile L, et al. Influential parameters on particle concentration and size distribution in the mainstream of e-cigarettes. Environ Pollut. 2014;184:523-529.
42. Schober W, Szendrei K, Matzen W, et al. Use of electronic cigarettes (e-cigarettes) impairs indoor air quality and increases FeNO levels of e-cigarette consumers. Int J Hyg Environ Health. 2014;217(6):628-637.
43. Bhatnagar A, Whitsel L, Ribisl K, et al; American Heart Association Advocacy Coordinating Committee; Council on Cardiovascular and Stroke Nursing; Council on Clinical Cardiology; Council on Quality of Care and Outcomes Research. Electronic cigarettes: a policy statement from the American Heart Association. Circulation. 2014;130(16):1418-1436.
44. E-cigarettes pose risks. SAMHSA News. https://www.samhsa.gov/samhsaNewsLetter/Volume_22_Number_3/e_cigarettes. Published 2014. Accessed April 7, 2017.
45. National Institute on Drug Abuse. Electronic cigarettes (e-cigarettes). https://www.drugabuse.gov/publications/drugfacts/electronic-cigarettes-e-cigarettes. Revised May 2016. Accessed April 7, 2017.
46. American Academy of Addiction Psychiatry. Nicotine dependence. East Providence, RI: American Academy of Addition Psychiatry; 2015.
47. Green SH, Bayer R, Fairchild AL. Evidence, policy, and e-cigarettes — will England reframe the debate. N Engl J Med. 2016;374(14):1301-1303.
48. Cahill K, Stevens S, Lancaster T. Pharmacological treatments for smoking cessation. JAMA. 2014;311(2):193-194.
The popularity of electronic cigarettes (E-cigs) and “vapes” has grown dramatically, spawning a new industry of electronic nicotine delivery systems (ENDS). With the increasing use of E-cigs not only for smoking cessation, but also as a primary nicotine source, it is important for mental health professionals to be prepared to discuss use of these devices with patients. In this article, we will describe:
- the composition of E-cigs and their current use
- evidence for their use for smoking cessation
- adverse health effects
- recommendations of major regulatory agencies.
Finally, we will provide recommendations for E-cig use in clinical populations.
What is an electronic nicotine delivery system?
ENDS produce an aerosol with or without nicotine that is inhaled and is thought to mimic the use of combustible cigarettes. ENDS evolved from basic E-cigs into a less “cigarette-like” and more customizable product (Figure 1). ENDS include a range of designs and go by various names, including “personal vaporizers,” “e-cigars,” and “e-hookahs” (in this article, we will use the term “ENDS” to refer to these devices).
The general design of ENDS is a plastic tubing system that contains a mouthpiece, battery, electronic heating element (“vaporizer”), and a cartridge with liquid solvent with or without nicotine or flavoring (Figure 2). One draw on the mouthpiece or press of a button activates the device, heats the solution, and delivers a vapor in a similar manner to taking a puff of a cigarette. Although studies have shown that ENDS result in significant increases in plasma nicotine concentrations in 5 minutes,1 the plasma nicotine levels obtained with the first-generation “cigarette-like” ENDS are much lower than those caused by inhaling tobacco smoke.2 Over time nicotine delivery capability has improved as ENDS have evolved such that the rate of nicotine delivery and peak concentration obtained with newer models more closely mirror tobacco cigarettes.3 Whether the rapid delivery of larger amounts of nicotine helps or hinders one’s efforts to break nicotine addiction remains to be determined because of the reinforcing properties of the drug.
The liquid in the E-cig cartridge typically contains not only nicotine but a number of chemical compounds with potentially deleterious or unknown health risks. The 3 main ingredients include:
- a solvent of glycerin and/or propylene glycol
- nicotine in various concentrations
- flavorings.
The glycerin or propylene glycol forms the basis for the aerosol. Nicotine concentrations vary from 0 (denicotinized) to 35 mcg per puff.4 A study reported 7,700 unique flavors available for vaping liquid.5 The liquid also contains impurities, such as anabasine, which has effects on the α-7 nicotinic acetylcholine receptor and its principal use is as an insecticide and β-nicotyrine, which inhibits cytochrome P450 2A.
Epidemiology and end-user perspectives
In 2014, 12.4% of U.S. adults classified themselves as “ever users” of ENDS (used at least once) and 3.7% of adults classified themselves as current users, according to the National Health Interview Study.6 Importantly, among E-cig users who had not used combustible cigarettes, young adults (age 18 to 24) were more likely to have tried ENDS than older adults. ENDS are becoming more popular across the globe. A study in the European Union found that ever users of ENDS most commonly were current cigarette smokers (31%) followed by former (10.8%) and never smokers (2.3%).7
ENDS use is relevant for mental health professionals because of the high rate of comorbid tobacco use disorder in individuals with psychiatric conditions. For example, 2 U.S. population surveys8,9 revealed those with mental health conditions were 1.5 to 2 times more likely to have tried ENDS and 2 to 3 times more likely to be current users. Those with psychiatric illness reported similar reasons for ENDS use as other individuals, including “just because,” use as a smoking cessation aid, ease of use, and perceived safety vs combustible cigarettes.
A recent review that included 9 studies focusing on ENDS use in those with mental illness reported mixed findings on the utility of these devices to reduce or stop use of combustible cigarettes.10 Additionally, it is important to monitor the use of cigarettes and ENDS in patients with psychiatric illness because the byproducts of tobacco smoke can affect the metabolism of some psychotropic medications.11 Although reduced use of combustible cigarettes could lead to lower dosing of some psychotropics, an unreported decrease in combustible cigarette use could lead to supratherapeutic drug levels. There are no data on the effect of ENDS on the metabolism of psychotropics.
ENDS are increasingly popular among adolescents. In 2015, there were an estimated 4.6 million current tobacco users among middle/high school youths in the United States and 3 million current ENDS users, according to the National Youth Tobacco Surveys.12 The shift from combustible cigarettes to ENDS is notable, with an increase in the percentage of current E-cig users and a decrease in the percentage of exclusive combustible cigarette users. In addition, there has been no change in the prevalence of lifetime tobacco users.12 This is a global issue, as reports of ever use of ENDS by adolescents range from 6.5% to 31% in the United States, 14.6% in Canada, and 4.7% to 38.5% in Europe.13 Based on these trends, the U.S. Surgeon General released a statement warning against the use of ENDS in youth because of the lack of safety data and strong association with use of tobacco products.14
There are a number of possible reasons for the increasing popularity of ENDS, including the product’s novelty, lack of regulations regarding their sale, availability of flavorings, and the perception that ENDS are safe alternatives to cigarettes. E-cig–using youths have described ENDS as “not at all harmful” and “not at all addictive” and believe that ENDS with flavoring are less harmful than those without.15 Although studies in adults show some users reporting that ENDS are less satisfying, they are seen as useful in decreasing craving and a safer alternative to cigarettes.16,17
Are ENDS effective for smoking cessation?
The evidence for ENDS as aids to smoking cessation remains murky (Table 118-22). There is a paucity of randomized controlled clinical trials (RCTs) investigating ENDS for smoking cessation or reduction, and it is difficult to quantify the amount of nicotine used in ENDS because of the variety of delivery systems and cartridges. In a recent Cochrane review, those using ENDS to quit smoking were more likely to be abstinent from combustible cigarettes at 6 months vs those using nicotine-free ENDS (relative risk = 2.29; 95% CI, 1.05 to 4.96), but there was no significant difference in quit rates compared with nicotine patches.23 However, the confidence in this finding was rated as low because of the limited number of RCTs. Of note, the authors found 15 ongoing RCTs at the time of publication that might be eligible for later evaluation.
Non-RCTs reveal mixed data. Positive results include 1 study with an odds ratio of 6.07 to quit for intensive ENDS users vs non-users,24 and another with dual users of combustible and electronic cigarettes having a 46% quit rate at 1 year.25 Additionally, in a pilot study providing ENDS to 14 patients with schizophrenia who had no previous desire to quit smoking, authors noted a reduction in the number of cigarettes smoked per day by 50% in one-half of participants and abstinence in 14% of participants at 52 weeks.26 Studies with neutral or negative results include those showing ENDS users to be current combustible tobacco smokers, and use of ENDS not predicting smoking cessation.4,27 Data also are mixed regarding the use of ENDS as a harm reduction strategy. One study found that ENDS decreased cigarette consumption, but did not increase the likelihood of quitting,28 while another reported that daily use of ENDS increased the odds of reducing smoking by as much as 2.5 times compared with non-use of such aids.29 In a 24-month prospective cohort study following tobacco users, there was no difference in the number of cigarettes smoked per day in those who started the trial as users of combustible cigarettes alone vs combustible cigarettes plus ENDS users.30 Interestingly, those who started the study as combustible cigarette users and switched to ENDS and those who had continued dual use throughout the 24 months smoked fewer combustible cigarettes per day than those who never tried ENDS or quit during the study period.
Health effects
To better understand the adverse health effects of ENDS, one must consider potential short- and long-term consequences (Table 2). In the short-term, ENDS have been found to increase markers of inflammation and oxidative stress acutely as evidenced by in vivo laboratory studies.31,32 ENDS also have been linked to upper respiratory irritation, in part, because of the transformation of glycerin in the nicotine cartridge to acrolein upon combustion.33 Even 5 minutes of ad lib E-cig use has been found to significantly increase airflow resistance during pulmonary function tests34—changes that have been shown to precede more persistent alterations in peak expiratory flow, such as those seen in chronic obstructive pulmonary disease. The more common patient-reported side effects include:
- daytime cough (27%)
- phlegm production (25%)
- headache (21%)
- dry mouth/throat (20%)
- vertigo, headache, or nausea (9%).35,36
A RCT investigating efficacy of E-cigs vs nicotine patches vs denicotinized E-cigs found no difference among the groups in the number of reported adverse events.18 Interestingly, another RCT found a decrease in adverse events, such as dry cough, mouth irritation, throat irritation, shortness of breath, and headache, compared with baseline in combustible cigarette smokers who used regular or denicotinized E-cigs.19
Although no studies have directly investigated long-term health consequences of ENDS because of their relative novelty, one can extrapolate potential harmful long-term effects based on knowledge of the products’ chemical constituents. For example, propylene glycol can degrade into propylene oxide, a class 2B carcinogen.37 Other potential carcinogens in the aerosol include formaldehyde and acetaldehyde. On a broader scale, many of the particulates have been shown to cause systemic inflammation, which is thought to increase cardiovascular and respiratory disease and death.38 Flavorings in ENDS include a variety of components including, but not limited to, aldehydes, which are irritants, and other additives that have been associated with respiratory disease.39
Second-hand exposure. There are no long-term studies of second-hand vapor exposure, but similar to long-term health on primary users, one can glean some observations from the literature. It is promising that compared with cigarettes, ENDS lack sidestream smoke and the vapor has not been found to contain carbon monoxide.40 Some research has demonstrated that the size and spray of fine particles in the aerosol is as large or larger than combustible cigarettes.41 Formaldehyde, acetaldehyde, isoprene, and acetic acid have been found in ENDS vapor.40 Interestingly, a simulated café study found elevated nicotine, glycerine, hydrocarbon, and other materials classified as carcinogens in the air.42
Although it is popularly thought that ENDS are less toxic than tobacco cigarettes, there is not enough evidence to estimate precisely as to how much less toxic or the consequences of use. ENDS are increasingly popular and are being used by never smokers who should be educated on the potential harm that ENDS pose.
Recommendations from agencies and medical organizations
The World Health Organization (WHO) recommended prohibiting the use of ENDS in indoor spaces to minimize potential health risks to users and non-users. The WHO also aims to prevent dissemination of unproven health claims, including claims that ENDS are effective—or not—or that the devices are innocuous.36 In the United States, the FDA has stated that ENDS are not recommended for safe quitting (2009). In August 2016, the FDA introduced regulations banning the sale of ENDS to individuals age <18 and required manufacturers to submit documents detailing all ingredients for review and possible approval.
The American Lung Association has stated its concerns about the use of ENDS but has not made any direct recommendations. The American Heart Association reports a potential negative public health impact and provides clinical guideline recommendations.43 Prominent psychiatric organizations such as the American Psychiatric Association, American Academy of Addiction Psychiatry (AAAP), the Substance Abuse and Mental Health Services Administration (SAMHSA), and the National Institute of Drug Abuse do not have official statements supporting or rejecting the use of ENDS. However, they do note the potential harm and lack of substantial evidence for efficacy of ENDS as a smoking cessation tool, and the AAAP and SAMHSA state that they will work with regulatory agencies to reduce the use of toxic products with addictive potential including ENDS.44-46
Clinical recommendations
We do not recommend ENDS as a first-line treatment for smoking cessation because there is no evidence they are superior to the FDA-approved nicotine replacement therapies (NRTs), the paucity of research into the potential short- and long-term health risks of ENDS, and the fact that these products are not regulated for use as smoking cessation aids. It is, however, advisable to discuss ENDS use with patients by:
- asking if they are using the products
- assessing whether the user also is a smoker
- advising the patient to quit.
It also is important to assess the patient’s knowledge and attitudes regarding ENDS use and provide education about the products. Some patients firmly believe that ENDS are the lesser of 2 evils, and they are decreasing the harms of smoking by using these devices. While the debate over a potential harm reduction strategy unfolds,47 we think that because of the state of the evidence it is prudent to adopt a more precautionary stance and recommend that patients work toward abstinence from nicotine in any form.
For dual tobacco/ENDS users and for patients using ENDS who want to quit smoking, we recommend treatment with an approved pharmacotherapy (ie, NRTs, bupropion, and varenicline) combined with counseling. A 2013 Cochrane Review found that all pharamacotherapy options are more effective than placebo, and combination NRT and varenicline are superior to single NRT or bupropion (Box).23,48
The popularity of electronic cigarettes (E-cigs) and “vapes” has grown dramatically, spawning a new industry of electronic nicotine delivery systems (ENDS). With the increasing use of E-cigs not only for smoking cessation, but also as a primary nicotine source, it is important for mental health professionals to be prepared to discuss use of these devices with patients. In this article, we will describe:
- the composition of E-cigs and their current use
- evidence for their use for smoking cessation
- adverse health effects
- recommendations of major regulatory agencies.
Finally, we will provide recommendations for E-cig use in clinical populations.
What is an electronic nicotine delivery system?
ENDS produce an aerosol with or without nicotine that is inhaled and is thought to mimic the use of combustible cigarettes. ENDS evolved from basic E-cigs into a less “cigarette-like” and more customizable product (Figure 1). ENDS include a range of designs and go by various names, including “personal vaporizers,” “e-cigars,” and “e-hookahs” (in this article, we will use the term “ENDS” to refer to these devices).
The general design of ENDS is a plastic tubing system that contains a mouthpiece, battery, electronic heating element (“vaporizer”), and a cartridge with liquid solvent with or without nicotine or flavoring (Figure 2). One draw on the mouthpiece or press of a button activates the device, heats the solution, and delivers a vapor in a similar manner to taking a puff of a cigarette. Although studies have shown that ENDS result in significant increases in plasma nicotine concentrations in 5 minutes,1 the plasma nicotine levels obtained with the first-generation “cigarette-like” ENDS are much lower than those caused by inhaling tobacco smoke.2 Over time nicotine delivery capability has improved as ENDS have evolved such that the rate of nicotine delivery and peak concentration obtained with newer models more closely mirror tobacco cigarettes.3 Whether the rapid delivery of larger amounts of nicotine helps or hinders one’s efforts to break nicotine addiction remains to be determined because of the reinforcing properties of the drug.
The liquid in the E-cig cartridge typically contains not only nicotine but a number of chemical compounds with potentially deleterious or unknown health risks. The 3 main ingredients include:
- a solvent of glycerin and/or propylene glycol
- nicotine in various concentrations
- flavorings.
The glycerin or propylene glycol forms the basis for the aerosol. Nicotine concentrations vary from 0 (denicotinized) to 35 mcg per puff.4 A study reported 7,700 unique flavors available for vaping liquid.5 The liquid also contains impurities, such as anabasine, which has effects on the α-7 nicotinic acetylcholine receptor and its principal use is as an insecticide and β-nicotyrine, which inhibits cytochrome P450 2A.
Epidemiology and end-user perspectives
In 2014, 12.4% of U.S. adults classified themselves as “ever users” of ENDS (used at least once) and 3.7% of adults classified themselves as current users, according to the National Health Interview Study.6 Importantly, among E-cig users who had not used combustible cigarettes, young adults (age 18 to 24) were more likely to have tried ENDS than older adults. ENDS are becoming more popular across the globe. A study in the European Union found that ever users of ENDS most commonly were current cigarette smokers (31%) followed by former (10.8%) and never smokers (2.3%).7
ENDS use is relevant for mental health professionals because of the high rate of comorbid tobacco use disorder in individuals with psychiatric conditions. For example, 2 U.S. population surveys8,9 revealed those with mental health conditions were 1.5 to 2 times more likely to have tried ENDS and 2 to 3 times more likely to be current users. Those with psychiatric illness reported similar reasons for ENDS use as other individuals, including “just because,” use as a smoking cessation aid, ease of use, and perceived safety vs combustible cigarettes.
A recent review that included 9 studies focusing on ENDS use in those with mental illness reported mixed findings on the utility of these devices to reduce or stop use of combustible cigarettes.10 Additionally, it is important to monitor the use of cigarettes and ENDS in patients with psychiatric illness because the byproducts of tobacco smoke can affect the metabolism of some psychotropic medications.11 Although reduced use of combustible cigarettes could lead to lower dosing of some psychotropics, an unreported decrease in combustible cigarette use could lead to supratherapeutic drug levels. There are no data on the effect of ENDS on the metabolism of psychotropics.
ENDS are increasingly popular among adolescents. In 2015, there were an estimated 4.6 million current tobacco users among middle/high school youths in the United States and 3 million current ENDS users, according to the National Youth Tobacco Surveys.12 The shift from combustible cigarettes to ENDS is notable, with an increase in the percentage of current E-cig users and a decrease in the percentage of exclusive combustible cigarette users. In addition, there has been no change in the prevalence of lifetime tobacco users.12 This is a global issue, as reports of ever use of ENDS by adolescents range from 6.5% to 31% in the United States, 14.6% in Canada, and 4.7% to 38.5% in Europe.13 Based on these trends, the U.S. Surgeon General released a statement warning against the use of ENDS in youth because of the lack of safety data and strong association with use of tobacco products.14
There are a number of possible reasons for the increasing popularity of ENDS, including the product’s novelty, lack of regulations regarding their sale, availability of flavorings, and the perception that ENDS are safe alternatives to cigarettes. E-cig–using youths have described ENDS as “not at all harmful” and “not at all addictive” and believe that ENDS with flavoring are less harmful than those without.15 Although studies in adults show some users reporting that ENDS are less satisfying, they are seen as useful in decreasing craving and a safer alternative to cigarettes.16,17
Are ENDS effective for smoking cessation?
The evidence for ENDS as aids to smoking cessation remains murky (Table 118-22). There is a paucity of randomized controlled clinical trials (RCTs) investigating ENDS for smoking cessation or reduction, and it is difficult to quantify the amount of nicotine used in ENDS because of the variety of delivery systems and cartridges. In a recent Cochrane review, those using ENDS to quit smoking were more likely to be abstinent from combustible cigarettes at 6 months vs those using nicotine-free ENDS (relative risk = 2.29; 95% CI, 1.05 to 4.96), but there was no significant difference in quit rates compared with nicotine patches.23 However, the confidence in this finding was rated as low because of the limited number of RCTs. Of note, the authors found 15 ongoing RCTs at the time of publication that might be eligible for later evaluation.
Non-RCTs reveal mixed data. Positive results include 1 study with an odds ratio of 6.07 to quit for intensive ENDS users vs non-users,24 and another with dual users of combustible and electronic cigarettes having a 46% quit rate at 1 year.25 Additionally, in a pilot study providing ENDS to 14 patients with schizophrenia who had no previous desire to quit smoking, authors noted a reduction in the number of cigarettes smoked per day by 50% in one-half of participants and abstinence in 14% of participants at 52 weeks.26 Studies with neutral or negative results include those showing ENDS users to be current combustible tobacco smokers, and use of ENDS not predicting smoking cessation.4,27 Data also are mixed regarding the use of ENDS as a harm reduction strategy. One study found that ENDS decreased cigarette consumption, but did not increase the likelihood of quitting,28 while another reported that daily use of ENDS increased the odds of reducing smoking by as much as 2.5 times compared with non-use of such aids.29 In a 24-month prospective cohort study following tobacco users, there was no difference in the number of cigarettes smoked per day in those who started the trial as users of combustible cigarettes alone vs combustible cigarettes plus ENDS users.30 Interestingly, those who started the study as combustible cigarette users and switched to ENDS and those who had continued dual use throughout the 24 months smoked fewer combustible cigarettes per day than those who never tried ENDS or quit during the study period.
Health effects
To better understand the adverse health effects of ENDS, one must consider potential short- and long-term consequences (Table 2). In the short-term, ENDS have been found to increase markers of inflammation and oxidative stress acutely as evidenced by in vivo laboratory studies.31,32 ENDS also have been linked to upper respiratory irritation, in part, because of the transformation of glycerin in the nicotine cartridge to acrolein upon combustion.33 Even 5 minutes of ad lib E-cig use has been found to significantly increase airflow resistance during pulmonary function tests34—changes that have been shown to precede more persistent alterations in peak expiratory flow, such as those seen in chronic obstructive pulmonary disease. The more common patient-reported side effects include:
- daytime cough (27%)
- phlegm production (25%)
- headache (21%)
- dry mouth/throat (20%)
- vertigo, headache, or nausea (9%).35,36
A RCT investigating efficacy of E-cigs vs nicotine patches vs denicotinized E-cigs found no difference among the groups in the number of reported adverse events.18 Interestingly, another RCT found a decrease in adverse events, such as dry cough, mouth irritation, throat irritation, shortness of breath, and headache, compared with baseline in combustible cigarette smokers who used regular or denicotinized E-cigs.19
Although no studies have directly investigated long-term health consequences of ENDS because of their relative novelty, one can extrapolate potential harmful long-term effects based on knowledge of the products’ chemical constituents. For example, propylene glycol can degrade into propylene oxide, a class 2B carcinogen.37 Other potential carcinogens in the aerosol include formaldehyde and acetaldehyde. On a broader scale, many of the particulates have been shown to cause systemic inflammation, which is thought to increase cardiovascular and respiratory disease and death.38 Flavorings in ENDS include a variety of components including, but not limited to, aldehydes, which are irritants, and other additives that have been associated with respiratory disease.39
Second-hand exposure. There are no long-term studies of second-hand vapor exposure, but similar to long-term health on primary users, one can glean some observations from the literature. It is promising that compared with cigarettes, ENDS lack sidestream smoke and the vapor has not been found to contain carbon monoxide.40 Some research has demonstrated that the size and spray of fine particles in the aerosol is as large or larger than combustible cigarettes.41 Formaldehyde, acetaldehyde, isoprene, and acetic acid have been found in ENDS vapor.40 Interestingly, a simulated café study found elevated nicotine, glycerine, hydrocarbon, and other materials classified as carcinogens in the air.42
Although it is popularly thought that ENDS are less toxic than tobacco cigarettes, there is not enough evidence to estimate precisely as to how much less toxic or the consequences of use. ENDS are increasingly popular and are being used by never smokers who should be educated on the potential harm that ENDS pose.
Recommendations from agencies and medical organizations
The World Health Organization (WHO) recommended prohibiting the use of ENDS in indoor spaces to minimize potential health risks to users and non-users. The WHO also aims to prevent dissemination of unproven health claims, including claims that ENDS are effective—or not—or that the devices are innocuous.36 In the United States, the FDA has stated that ENDS are not recommended for safe quitting (2009). In August 2016, the FDA introduced regulations banning the sale of ENDS to individuals age <18 and required manufacturers to submit documents detailing all ingredients for review and possible approval.
The American Lung Association has stated its concerns about the use of ENDS but has not made any direct recommendations. The American Heart Association reports a potential negative public health impact and provides clinical guideline recommendations.43 Prominent psychiatric organizations such as the American Psychiatric Association, American Academy of Addiction Psychiatry (AAAP), the Substance Abuse and Mental Health Services Administration (SAMHSA), and the National Institute of Drug Abuse do not have official statements supporting or rejecting the use of ENDS. However, they do note the potential harm and lack of substantial evidence for efficacy of ENDS as a smoking cessation tool, and the AAAP and SAMHSA state that they will work with regulatory agencies to reduce the use of toxic products with addictive potential including ENDS.44-46
Clinical recommendations
We do not recommend ENDS as a first-line treatment for smoking cessation because there is no evidence they are superior to the FDA-approved nicotine replacement therapies (NRTs), the paucity of research into the potential short- and long-term health risks of ENDS, and the fact that these products are not regulated for use as smoking cessation aids. It is, however, advisable to discuss ENDS use with patients by:
- asking if they are using the products
- assessing whether the user also is a smoker
- advising the patient to quit.
It also is important to assess the patient’s knowledge and attitudes regarding ENDS use and provide education about the products. Some patients firmly believe that ENDS are the lesser of 2 evils, and they are decreasing the harms of smoking by using these devices. While the debate over a potential harm reduction strategy unfolds,47 we think that because of the state of the evidence it is prudent to adopt a more precautionary stance and recommend that patients work toward abstinence from nicotine in any form.
For dual tobacco/ENDS users and for patients using ENDS who want to quit smoking, we recommend treatment with an approved pharmacotherapy (ie, NRTs, bupropion, and varenicline) combined with counseling. A 2013 Cochrane Review found that all pharamacotherapy options are more effective than placebo, and combination NRT and varenicline are superior to single NRT or bupropion (Box).23,48
1. Hajek P, Goniewicz ML, Phillips A, et al. Nicotine intake from electronic cigarettes on initial use and after 4 weeks of regular use. Nicotine Tob Res. 2015;17(2):175-179.
2. Farsalinos KE, Polosa R. Safety evaluation and risk assessment of electronic cigarettes as tobacco cigarette substitutes: a systematic review. Ther Adv Drug Saf. 2014;5(2):67-86.
3. St Helen G, Havel C, Dempsey DA, et al. Nicotine delivery, retention and pharmacokinetics from various electronic cigarettes. Addiction. 2016;111(3):535-544.
4. Grana R, Benowitz N, Glantz SA. E-cigarettes: a scientific review. Circulation. 2014;129(19):1972-1986.
5. Zhu SH, Sun JY, Bonnevie E, et al. Four hundred and sixty brands of e-cigarettes and counting: implications for product regulation. Tob Control. 2014;23(suppl 3):iii3-iii9. doi: 10.1136/tobaccocontrol-2014-051670.
6. Schoenborn CA, Gindi RM. Electronic cigarette use among adults: United States, 2014. NCHS Data Brief. 2015;(217):1-8.
7. Farsalinos KE, Poulas K, Voudris V, et al. Electronic cigarette use in the European Union: analysis of a representative sample of 27 460 Europeans from 28 countries. Addiction. 2016;111(11):2032-2040.
8. Cummins SE, Zhu SH, Tedeschi GJ, et al. Use of e-cigarettes by individuals with mental health conditions. Tob Control. 2015;23(suppl 3):iii48-iii53. doi: 10.1136/tobaccocontrol-2013-051511.
9. Spears CA, Jones DM, Weaver SR, et al. Use of electronic nicotine delivery systems among adults with mental health conditions, 2015. Int J Environ Res Public Heal. 2017;14(1):10.
10. Hefner K, Valentine G, Sofuoglu M. Electronic cigarettes and mental illness: reviewing the evidence for help and harm among those with psychiatric and substance use disorders [published online February 2, 2017]. Am J Addict. doi: 10.1111/ajad.12504.
11. Anthenelli R. How—and why—to help psychiatric patients stop smoking. Current Psychiatry. 2005;4(1):77-87.
12. Singh T, Arrazola RA, Corey CG, et al. Tobacco use among middle and high school students—United States, 2011-2015. MMWR Morb Mortal Wkly Rep. 2016;65(14):361-367.
13. Greenhill R, Dawkins L, Notley C, et al. Adolescent awareness and use of electronic cigarettes: a review of emerging trends and findings. J Adolesc Heal. 2016;59(6):612-619.
14. U.S. Department of Health and Human Services. E-cigarette use among youth and young adults: a report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention; 2016.
15. Cooper M, Harrell MB, Pérez A, et al. Flavorings and perceived harm and addictiveness of e-cigarettes among youth. Tob Regul Sci. 2016;2(3):278-289.
16. Kim H, Davis AH, Dohack JL, et al. E-cigarettes use behavior and experience of adults: qualitative research findings to inform e-cigarette use measure development. Nicotine Tob Res. 2017;19(2):190-196.
17. Czoli CD, Fong GT, Mays D, et al. How do consumers perceive differences in risk across nicotine products? A review of relative risk perceptions across smokeless tobacco, e-cigarettes, nicotine replacement therapy and combustible cigarettes. Tob Control. 2017;26(e1):e49-e58.
18. Bullen C, Howe C, Laugesen M, et al. Electronic cigarettes for smoking cessation: a randomised controlled trial. Lancet. 2013;382(9905):1629-1637.
19. Caponnetto P, Campagna D, Cibella F, et al. EffiCiency and safety of an eLectronic cigAreTte (ECLAT) as tobacco cigarettes substitute: a prospective 12-month randomized control design study. PLoS One. 2013;8(6):e66317. doi: 10.1371/journal.pone.0066317.
20. Adriaens K, Van Gucht D, Declerck P, et al. Effectiveness of the electronic cigarette: an eight-week Flemish study with six-month follow-up on smoking reduction, craving and experienced benefits and complaints. Int J Environ Res Public Health. 2014;11(11):11220-11248.
21. Tseng TY, Ostroff JS, Campo A, et al. A randomized trial comparing the effect of nicotine versus placebo electronic cigarettes on smoking reduction among young adult smokers. Nicotine Tob Res. 2016;18(10):1937-1943.
22. Meier E, Wahlquist AE, Heckman BW, et al. A pilot randomized crossover trial of electronic cigarette sampling among smokers. Nicotine Tob Res. 2017;19(2):176-182.
23. Hartmann-Boyce J, McRobbie H, Bullen C, et al. Electronic cigarettes for smoking cessation [published online September 14, 2016]. Cochrane Database Syst Rev. 2016;9:CD010216.
24. Biener L, Hargraves JL. A longitudinal study of electronic cigarette use among a population-based sample of adult smokers: association with smoking cessation and motivation to quit. Nicotine Tob Res. 2014;17(2):127-133.
25. Etter JF, Bullen C. A longitudinal study of electronic cigarette users. Addict Behav. 2014;39(2):491-494.
26. Caponnetto P, Auditore R, Russo C, et al. Impact of an electronic cigarette on smoking reduction and cessation in schizophrenic smokers: a prospective 12-month pilot study. Int J Environ Res Public Health. 2013;10(2):446-461.
27. Popova L, Ling PM. Alternative tobacco product use and smoking cessation: a national study. Am J Public Health. 2013;103(5):923-930.
28. Adkison SE, O’Connor RJ, Bansal-Travers M, et al. Electronic nicotine delivery systems: International Tobacco Control Four-Country Survey. Am J Prev Med. 2013;44(3):207-215.
29. Brose LS, Hitchman SC, Brown J, et al. Is the use of electronic cigarettes while smoking associated with smoking cessation attempts, cessation and reduced cigarette consumption? A survey with a 1-year follow-up. Addiction. 2015;110(7):1160-1168.
30. Manzoli L, Flacco ME, Ferrante M, et al; ISLESE Working Group. Cohort study of electronic cigarette use: effectiveness and safety at 24 months [published online June 6, 2016]. Tob Control. doi: 10.1136/tobaccocontrol-2015-052822.
31. Lerner CA, Sundar IK, Yao H, et al. Vapors produced by electronic cigarettes and E-juices with flavorings induce toxicity, oxidative stress, and inflammatory response in lung epithelial cells and in mouse lung. PLoS One. 2015;10(2):e0116732. doi: 10.1371/journal.pone.0116732.
32. Sussan TE, Gajghate S, Thimmulappa RK, et al. Exposure to electronic cigarettes impairs pulmonary anti-bacterial and anti-viral defenses in a mouse model. PLoS One. 2015;10(2):e0116861. doi: 10.1371/journal.pone.0116861.
33. US Environmental Protection Agency. Acrolein. https://www.epa.gov/sites/production/files/2016-08/documents/acrolein.pdf. Updated September 2009. Accessed April 7, 2017.
34. Vardavas CI, Anagnostopoulos N, Kougias M, et al. Short-term pulmonary effects of using an electronic cigarette: impact on respiratory flow resistance, impedance, and exhaled nitric oxide. Chest. 2012;141(6):1400-1406.
35. Etter JF. Electronic cigarettes: a survey of users. BMC Public Health. 2010;10:231.
36. Goniewicz ML, Lingas EO, Hajek P. Patterns of electronic cigarette use and user beliefs about their safety and benefits: an internet survey. Drug Alcohol Rev. 2013;32(2):133-140.
37. Laino T, Tuma C, Moor P, et al. Mechanisms of propylene glycol and triacetin pyrolysis. J Phys Chem A. 2012;116(18):4602-4609.
38. Brook RD, Rajagopalan S, Pope CA 3rd, et al; American Heart Association Council on Epidemiology and Prevention; Council on the Kidney in Cardiovascular Disease; Council on Nutrition, Physical Activity and Metabolism. Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation. 2010;121(21):2331-2378.
39. Barrington-Trimis JL, Samet JM, McConnell R. Flavorings in electronic cigarettes: an unrecognized respiratory health hazard? JAMA. 2014;312(23):2493-2494.
40. Schripp T, Markewitz D, Uhde E, et al. Does e-cigarette consumption cause passive vaping? Indoor Air. 2013;23(1):25-31.
41. Fuoco FC, Buonanno G, Stabile L, et al. Influential parameters on particle concentration and size distribution in the mainstream of e-cigarettes. Environ Pollut. 2014;184:523-529.
42. Schober W, Szendrei K, Matzen W, et al. Use of electronic cigarettes (e-cigarettes) impairs indoor air quality and increases FeNO levels of e-cigarette consumers. Int J Hyg Environ Health. 2014;217(6):628-637.
43. Bhatnagar A, Whitsel L, Ribisl K, et al; American Heart Association Advocacy Coordinating Committee; Council on Cardiovascular and Stroke Nursing; Council on Clinical Cardiology; Council on Quality of Care and Outcomes Research. Electronic cigarettes: a policy statement from the American Heart Association. Circulation. 2014;130(16):1418-1436.
44. E-cigarettes pose risks. SAMHSA News. https://www.samhsa.gov/samhsaNewsLetter/Volume_22_Number_3/e_cigarettes. Published 2014. Accessed April 7, 2017.
45. National Institute on Drug Abuse. Electronic cigarettes (e-cigarettes). https://www.drugabuse.gov/publications/drugfacts/electronic-cigarettes-e-cigarettes. Revised May 2016. Accessed April 7, 2017.
46. American Academy of Addiction Psychiatry. Nicotine dependence. East Providence, RI: American Academy of Addition Psychiatry; 2015.
47. Green SH, Bayer R, Fairchild AL. Evidence, policy, and e-cigarettes — will England reframe the debate. N Engl J Med. 2016;374(14):1301-1303.
48. Cahill K, Stevens S, Lancaster T. Pharmacological treatments for smoking cessation. JAMA. 2014;311(2):193-194.
1. Hajek P, Goniewicz ML, Phillips A, et al. Nicotine intake from electronic cigarettes on initial use and after 4 weeks of regular use. Nicotine Tob Res. 2015;17(2):175-179.
2. Farsalinos KE, Polosa R. Safety evaluation and risk assessment of electronic cigarettes as tobacco cigarette substitutes: a systematic review. Ther Adv Drug Saf. 2014;5(2):67-86.
3. St Helen G, Havel C, Dempsey DA, et al. Nicotine delivery, retention and pharmacokinetics from various electronic cigarettes. Addiction. 2016;111(3):535-544.
4. Grana R, Benowitz N, Glantz SA. E-cigarettes: a scientific review. Circulation. 2014;129(19):1972-1986.
5. Zhu SH, Sun JY, Bonnevie E, et al. Four hundred and sixty brands of e-cigarettes and counting: implications for product regulation. Tob Control. 2014;23(suppl 3):iii3-iii9. doi: 10.1136/tobaccocontrol-2014-051670.
6. Schoenborn CA, Gindi RM. Electronic cigarette use among adults: United States, 2014. NCHS Data Brief. 2015;(217):1-8.
7. Farsalinos KE, Poulas K, Voudris V, et al. Electronic cigarette use in the European Union: analysis of a representative sample of 27 460 Europeans from 28 countries. Addiction. 2016;111(11):2032-2040.
8. Cummins SE, Zhu SH, Tedeschi GJ, et al. Use of e-cigarettes by individuals with mental health conditions. Tob Control. 2015;23(suppl 3):iii48-iii53. doi: 10.1136/tobaccocontrol-2013-051511.
9. Spears CA, Jones DM, Weaver SR, et al. Use of electronic nicotine delivery systems among adults with mental health conditions, 2015. Int J Environ Res Public Heal. 2017;14(1):10.
10. Hefner K, Valentine G, Sofuoglu M. Electronic cigarettes and mental illness: reviewing the evidence for help and harm among those with psychiatric and substance use disorders [published online February 2, 2017]. Am J Addict. doi: 10.1111/ajad.12504.
11. Anthenelli R. How—and why—to help psychiatric patients stop smoking. Current Psychiatry. 2005;4(1):77-87.
12. Singh T, Arrazola RA, Corey CG, et al. Tobacco use among middle and high school students—United States, 2011-2015. MMWR Morb Mortal Wkly Rep. 2016;65(14):361-367.
13. Greenhill R, Dawkins L, Notley C, et al. Adolescent awareness and use of electronic cigarettes: a review of emerging trends and findings. J Adolesc Heal. 2016;59(6):612-619.
14. U.S. Department of Health and Human Services. E-cigarette use among youth and young adults: a report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention; 2016.
15. Cooper M, Harrell MB, Pérez A, et al. Flavorings and perceived harm and addictiveness of e-cigarettes among youth. Tob Regul Sci. 2016;2(3):278-289.
16. Kim H, Davis AH, Dohack JL, et al. E-cigarettes use behavior and experience of adults: qualitative research findings to inform e-cigarette use measure development. Nicotine Tob Res. 2017;19(2):190-196.
17. Czoli CD, Fong GT, Mays D, et al. How do consumers perceive differences in risk across nicotine products? A review of relative risk perceptions across smokeless tobacco, e-cigarettes, nicotine replacement therapy and combustible cigarettes. Tob Control. 2017;26(e1):e49-e58.
18. Bullen C, Howe C, Laugesen M, et al. Electronic cigarettes for smoking cessation: a randomised controlled trial. Lancet. 2013;382(9905):1629-1637.
19. Caponnetto P, Campagna D, Cibella F, et al. EffiCiency and safety of an eLectronic cigAreTte (ECLAT) as tobacco cigarettes substitute: a prospective 12-month randomized control design study. PLoS One. 2013;8(6):e66317. doi: 10.1371/journal.pone.0066317.
20. Adriaens K, Van Gucht D, Declerck P, et al. Effectiveness of the electronic cigarette: an eight-week Flemish study with six-month follow-up on smoking reduction, craving and experienced benefits and complaints. Int J Environ Res Public Health. 2014;11(11):11220-11248.
21. Tseng TY, Ostroff JS, Campo A, et al. A randomized trial comparing the effect of nicotine versus placebo electronic cigarettes on smoking reduction among young adult smokers. Nicotine Tob Res. 2016;18(10):1937-1943.
22. Meier E, Wahlquist AE, Heckman BW, et al. A pilot randomized crossover trial of electronic cigarette sampling among smokers. Nicotine Tob Res. 2017;19(2):176-182.
23. Hartmann-Boyce J, McRobbie H, Bullen C, et al. Electronic cigarettes for smoking cessation [published online September 14, 2016]. Cochrane Database Syst Rev. 2016;9:CD010216.
24. Biener L, Hargraves JL. A longitudinal study of electronic cigarette use among a population-based sample of adult smokers: association with smoking cessation and motivation to quit. Nicotine Tob Res. 2014;17(2):127-133.
25. Etter JF, Bullen C. A longitudinal study of electronic cigarette users. Addict Behav. 2014;39(2):491-494.
26. Caponnetto P, Auditore R, Russo C, et al. Impact of an electronic cigarette on smoking reduction and cessation in schizophrenic smokers: a prospective 12-month pilot study. Int J Environ Res Public Health. 2013;10(2):446-461.
27. Popova L, Ling PM. Alternative tobacco product use and smoking cessation: a national study. Am J Public Health. 2013;103(5):923-930.
28. Adkison SE, O’Connor RJ, Bansal-Travers M, et al. Electronic nicotine delivery systems: International Tobacco Control Four-Country Survey. Am J Prev Med. 2013;44(3):207-215.
29. Brose LS, Hitchman SC, Brown J, et al. Is the use of electronic cigarettes while smoking associated with smoking cessation attempts, cessation and reduced cigarette consumption? A survey with a 1-year follow-up. Addiction. 2015;110(7):1160-1168.
30. Manzoli L, Flacco ME, Ferrante M, et al; ISLESE Working Group. Cohort study of electronic cigarette use: effectiveness and safety at 24 months [published online June 6, 2016]. Tob Control. doi: 10.1136/tobaccocontrol-2015-052822.
31. Lerner CA, Sundar IK, Yao H, et al. Vapors produced by electronic cigarettes and E-juices with flavorings induce toxicity, oxidative stress, and inflammatory response in lung epithelial cells and in mouse lung. PLoS One. 2015;10(2):e0116732. doi: 10.1371/journal.pone.0116732.
32. Sussan TE, Gajghate S, Thimmulappa RK, et al. Exposure to electronic cigarettes impairs pulmonary anti-bacterial and anti-viral defenses in a mouse model. PLoS One. 2015;10(2):e0116861. doi: 10.1371/journal.pone.0116861.
33. US Environmental Protection Agency. Acrolein. https://www.epa.gov/sites/production/files/2016-08/documents/acrolein.pdf. Updated September 2009. Accessed April 7, 2017.
34. Vardavas CI, Anagnostopoulos N, Kougias M, et al. Short-term pulmonary effects of using an electronic cigarette: impact on respiratory flow resistance, impedance, and exhaled nitric oxide. Chest. 2012;141(6):1400-1406.
35. Etter JF. Electronic cigarettes: a survey of users. BMC Public Health. 2010;10:231.
36. Goniewicz ML, Lingas EO, Hajek P. Patterns of electronic cigarette use and user beliefs about their safety and benefits: an internet survey. Drug Alcohol Rev. 2013;32(2):133-140.
37. Laino T, Tuma C, Moor P, et al. Mechanisms of propylene glycol and triacetin pyrolysis. J Phys Chem A. 2012;116(18):4602-4609.
38. Brook RD, Rajagopalan S, Pope CA 3rd, et al; American Heart Association Council on Epidemiology and Prevention; Council on the Kidney in Cardiovascular Disease; Council on Nutrition, Physical Activity and Metabolism. Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation. 2010;121(21):2331-2378.
39. Barrington-Trimis JL, Samet JM, McConnell R. Flavorings in electronic cigarettes: an unrecognized respiratory health hazard? JAMA. 2014;312(23):2493-2494.
40. Schripp T, Markewitz D, Uhde E, et al. Does e-cigarette consumption cause passive vaping? Indoor Air. 2013;23(1):25-31.
41. Fuoco FC, Buonanno G, Stabile L, et al. Influential parameters on particle concentration and size distribution in the mainstream of e-cigarettes. Environ Pollut. 2014;184:523-529.
42. Schober W, Szendrei K, Matzen W, et al. Use of electronic cigarettes (e-cigarettes) impairs indoor air quality and increases FeNO levels of e-cigarette consumers. Int J Hyg Environ Health. 2014;217(6):628-637.
43. Bhatnagar A, Whitsel L, Ribisl K, et al; American Heart Association Advocacy Coordinating Committee; Council on Cardiovascular and Stroke Nursing; Council on Clinical Cardiology; Council on Quality of Care and Outcomes Research. Electronic cigarettes: a policy statement from the American Heart Association. Circulation. 2014;130(16):1418-1436.
44. E-cigarettes pose risks. SAMHSA News. https://www.samhsa.gov/samhsaNewsLetter/Volume_22_Number_3/e_cigarettes. Published 2014. Accessed April 7, 2017.
45. National Institute on Drug Abuse. Electronic cigarettes (e-cigarettes). https://www.drugabuse.gov/publications/drugfacts/electronic-cigarettes-e-cigarettes. Revised May 2016. Accessed April 7, 2017.
46. American Academy of Addiction Psychiatry. Nicotine dependence. East Providence, RI: American Academy of Addition Psychiatry; 2015.
47. Green SH, Bayer R, Fairchild AL. Evidence, policy, and e-cigarettes — will England reframe the debate. N Engl J Med. 2016;374(14):1301-1303.
48. Cahill K, Stevens S, Lancaster T. Pharmacological treatments for smoking cessation. JAMA. 2014;311(2):193-194.

Use and misuse of opioid agonists in opioid addiction
For a patient struggling with opioid addiction, opioid agonist therapy with methadone or buprenorphine can reduce craving and opioid use and may even save his or her life. But many clinicians are unfamiliar with this evidence-based treatment,1,2 which is best started early in the course of addiction.3
This article outlines the pharmacology of these drugs, their clinical uses, and the challenges of using them to treat opioid addiction.
DIAGNOSTIC CRITERIA
Opioid addiction, formally known as opioid use disorder, is a pattern of opioid misuse leading to clinically significant impairment in multiple areas of life. The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, lists 11 diagnostic criteria, but only 2 need to be present within the past year to make the diagnosis4:
- Taking opioids longer or in higher doses than was intended
- A persistent desire or unsuccessful efforts to cut down or control opioid use
- Spending a great deal of time obtaining, using, or recovering from using opioids
- Craving opioids
- Repeatedly failing to fulfill obligations at work, school, or home due to opioid use
- Continuing to use opioids even though it causes or exacerbates social or interpersonal problems
- Giving up or curtailing important social, occupational, or recreational activities because of opioid use
- Repeatedly using opioids in situations in which it is physically hazardous
- Continuing to use opioids despite knowledge of having a persistent or recurrent physical or psychological problem that is likely to have been caused or exacerbated by the substance
- Tolerance
- Withdrawal.
Recent estimates indicate that 2.23 million people in the United States have opioid use disorder (426,000 with heroin and 1.8 million with prescription opioids).5
Progression from prescription opioids to heroin
We have observed that many patients with opioid use disorder start by misusing prescription opioids. Over time, tolerance can develop, which drives patients to use higher and higher doses.6
As the addiction progresses, a subset of prescription opioid users advances to using heroin, which is typically less expensive and easier to obtain.7 Most patients start with the intranasal route but eventually inject it intravenously.6,7
For many addicts, heroin use has medical consequences such as hepatitis C virus (HCV) and human immunodeficiency virus (HIV) infection, psychiatric problems such as depression and anxiety, and illegal activities such as theft and sex work.8 People who use heroin appear to have more severe addiction and a lower socioeconomic status than prescription opioid users.9–11 But recently, a growing number of middle class individuals are becoming addicted to heroin.12
METHADONE
Methadone is a long-acting synthetic opioid that functions as a full agonist on the mu-opioid receptor. The drug binds, occupies, and stimulates the receptor, preventing withdrawal symptoms and reducing opioid cravings for at least 24 hours.13
Adverse effects of methadone
The most common adverse effects include lightheadedness, dizziness, sedation, nausea, vomiting, and sweating.14 Other adverse effects:
Unintentional overdose. The risk is serious, as a single 30-mg dose can be fatal in people who are opioid-naïve.13
QTc prolongation, which can lead to torsade de pointes. This risk, which is dose-related, must be taken into consideration in patients who have any cardiac symptoms (eg, syncope, arrhythmia), pathology (familial QT prolongation), or other risk factors for QTc prolongation (eg, hypokalemia, QTc-prolonging medications).15
Respiratory depression, which can be fatal. This dose-related risk is heightened during the first 4 weeks of treatment if titration is too rapid or if methadone is used in combination with other drugs that cause central nervous system or respiratory depression.13,14
Starting methadone
To prevent respiratory depression and death related to rapid induction, the general rule is to start methadone at a low daily dose (20–30 mg) depending on the patient’s withdrawal symptoms.14 During this period, patients need to be closely monitored and educated on the perils of concomitant use of central nervous system depressants.14
In most patients, the dose is titrated up until their withdrawal symptoms and cravings are eliminated, which generally requires 60 to 120 mg daily.14 Hepatic and renal impairment, pregnancy, and advanced age can alter methadone pharmacokinetics and may therefore necessitate dose adjustment.
BUPRENORPHINE
Buprenorphine is an alkaloid thebaine opioid derivative that acts as a partial mu-opioid agonist and a kappa antagonist.16 Like methadone, buprenorphine is used to manage cravings and withdrawal symptoms.16 Dosages of 4 to 16 mg (up to 32 mg) per day of buprenorphine are usually required to adequately control opioid cravings.16
Sublingual and subdermal products
Buprenorphine is currently available in the United States in sublingual and subdermal formulations.16,17
Sublingual buprenorphine is usually combined with naloxone in a 4:1 ratio to deter intravenous use. Intravenous injection of the combination product can precipitate withdrawal due to the antagonist action of naloxone. (Taken orally or sublingually, naloxone is poorly absorbed and has little or no clinical effect.) Buprenorphine-naloxone is available in tablets, a sublingual film strip, and a buccal film strip. Buprenorphine is also available by itself in a sublingual formulation.
The US Food and Drug Administration has approved a buprenorphine subdermal implant, Probuphine. Four rods, about 1 inch long, are placed under the skin in the inner aspect of the upper arm and provide the equivalent of 8 mg of buprenorphine daily for 6 months.17 However, this method is formulated only for maintenance treatment and cannot be used for induction. Additionally, it is recommended that the implants be surgically removed at the end of 6 months, after which another set of implants can be inserted in the other arm or the patient can switch to sublingual therapy, depending on the clinical situation and patient preference.17
Generally safer than methadone
Buprenorphine works on the same receptor as methadone and therefore has a similar side effect profile. However, buprenorphine has a ceiling effect, which greatly reduces the risk of fatal respiratory depression.18 It also does not cause clinically significant QTc prolongation and is preferable in patients who have cardiac risk factors.18
Another advantage is that buprenorphine has fewer identified medication interactions than methadone.18 Further, induction of buprenorphine in patients with opioid use disorder has been shown to be safer than methadone.19
Although buprenorphine has been found to be 6 times safer than methadone with regard to overdose among the general population,20 it can still cause fatal intoxication if used in combination with central nervous system depressants.21
Buprenorphine has been also associated with hepatotoxicity, though the risk of new-onset liver disease appears to be low.22
NALTREXONE IS LESS EFFECTIVE THAN METHADONE, BUPRENORPHINE
Besides methadone and buprenorphine, the only other approved option for treating opioid use disorder is the opioid antagonist naltrexone.
Naltrexone has significantly less abuse potential, as it provides no euphoria, but patients do not like it. Even with the long-acting formulation (Vivitrol), naltrexone treatment is significantly less effective than methadone or buprenorphine.23–25 Further, although naltrexone is not a controlled substance and so does not face the same scrutiny as the agonist therapies, there are other significant barriers. Additional information on naltrexone is presented in reviews by Modesto-Lowe and Van Kirk24 and Woody.25
OBSTACLES TO TREATMENT
People hold conflicting views about opioid agonist therapy. Some believe that “trading one drug for another” is not a legitimate therapeutic strategy, and they may feel ashamed of being on maintenance therapy.26 Similarly, some argue that the answer to establishing stable abstinence does not lie simply in prescribing medications.
The contrary argument is that these medications, if used appropriately, confer many benefits such as reducing the medical and psychosocial sequelae of opioid addiction.18 In fact, properly treated patients no longer meet the diagnostic criteria of opioid use disorder, and both methadone and buprenorphine are on the World Health Organization’s (WHO) list of essential medicines.27
Despite endorsement by the WHO, the stigma attached to the opioid agonists has been difficult to overcome. Patients with opioid use disorder may be viewed with distrust by healthcare providers and often do not feel welcome in healthcare settings or in self-help recovery groups.28
Barriers to methadone therapy
Federal regulations on methadone prescribing and use were established to promote patient safety and decrease diversion, but they may also complicate access to care.29 They stipulate that to qualify for methadone maintenance, patients need to demonstrate opioid addiction for 1 year, except for pregnant women and those who have been incarcerated in the past 6 months. Patients under the age of 18 must have 2 documented failed treatment episodes as well as approval by a guardian to receive treatment.
Inconvenience. Methadone can be prescribed for opioid dependence only by an accredited treatment program. Patients must therefore travel to the clinic and wait to be evaluated on a daily basis for a minimum of 90 days. Only after they demonstrate consistent responsible behavior and negative results on urine testing do they become eligible to take methadone home.29 If a patient is to travel out of the area during the initial 90 days of treatment, he or she must make arrangements in advance to find a clinic that will provide a “guest dose.”
The inconvenience arising from the regulations may deter some patients from seeking methadone therapy. In spite of this, once patients are started on methadone, more of them continue treatment than with buprenorphine.18 A proposed reason is that methadone is a potent full opioid agonist and therefore relieves withdrawal symptoms and craving more effectively than buprenorphine, which is a partial agonist.30 Another possible reason is the higher level of supervision afforded by methadone clinics, which require daily contact for at least 90 days.
Safety concerns arise from methadone diversion, as illicit use may have lethal consequences. In the past decade, deaths from methadone overdose have risen significantly, most of them due to respiratory depression or torsade de pointes.13 However, most cases of diversion and overdose involve methadone that is prescribed for pain by individual practitioners and not from maintenance programs.13
Advantages of buprenorphine
Together, methadone’s lethality, stigma, and inconvenience may contribute to patients preferring buprenorphine.31
The regulations governing buprenorphine’s use are less restrictive than those with methadone. For example, patients must have a diagnosis of opioid addiction to be prescribed buprenorphine, but they are not required to carry the diagnosis for a year before treatment.31 Additionally, they do not need to travel to a federally approved opioid treatment center daily and can receive buprenorphine directly from a physician in an outpatient setting.
Under the Drug Abuse Treatment Act (DATA) of 2000, any physician can apply for a waiver to prescribe and dispense buprenorphine in his or her office. To qualify for an initial waiver, physicians must either obtain certification in the fields of addiction medicine or addiction psychiatry or complete an approved 8-hour training session.32 Each physician starts with a maximum of 30 patients, but can apply to treat up to 100 patients after 1 year and eventually up to 275 patients. Physicians must document every buprenorphine prescription they write and be able to refer patients for counseling.31
As of February 2017, nurse practitioners and physician assistants can also apply for a DATA 2000 waiver. All waivered providers are subject to unannounced visits from the Drug Enforcement Administration once every 5 years.32
While there are no federal restrictions on the amount of buprenorphine that can be dispensed, some states and some insurance companies have placed restrictions on dose or length of treatment.33 Buprenorphine patients can fill their prescriptions at any pharmacy and are permitted to bring their medication home, which improves access to care. However, office-based outpatient treatment is not without risk, and preventing buprenorphine diversion remains a challenge.34
‘Lending’ buprenorphine is a felony
Addicts have illegally used buprenorphine to self-treat opioid withdrawal, craving, and dependence.35 Its misuse has also been coupled with self-treatment of conditions that include depression and pain.36
A survey found that 83.7% of patients deem buprenorphine diversion to be appropriate; further, most patients said they consider it unethical to withhold prescribed buprenorphine from individuals showing symptoms of withdrawal.34 Physicians who prescribe buprenorphine must inform their patients that even “lending” or giving away their medication is a felony.
Prescribing physicians must also be diligent about monitoring for signs of diversion such as inconsistent urine toxicology screens, “lost” medication, and requests for early refills or escalating doses.37
EVALUATING PATIENTS FOR OPIOID REPLACEMENT THERAPY
In addition to federal regulations, we propose a 4-step approach to evaluate eligibility for opioid replacement therapy based on existing guidelines.37–39
Step 1: History and physical examination
The history should give particular attention to the patient’s cardiac, pulmonary, and hepatic status, with consideration of the risks of any medical comorbidities (eg, bacterial endocarditis, HIV and HCV infection) that might influence treatment.37
It is also essential to evaluate for any contraindications or drug interactions before prescribing methadone or buprenorphine.38
Contraindications to methadone maintenance include40:
- Cor pulmonale
- Methadone hypersensitivity
- Pseudomembranous colitis
- Selegiline use (due to risk of serotonin syndrome)
- Ileum paralyticus.
Contraindications to buprenorphine use include:
- Hypersensitivity to naloxone or buprenorphine
- Impaired liver function (due to the risk of inadvertent overdose associated with slowed metabolism).
Concurrent use of alcohol or illicit benzodiazepines is a relative contraindication to both methadone and buprenorphine due to the risk of respiratory depression and overdose.37 Likewise, avoid coprescribing opioid agonists and benzodiazepines whenever possible. Obtain a complete list of current medications and query a prescription-monitoring database to determine whether any controlled substances are currently prescribed.37
During the physical examination, look for stigmata of intravenous drug use such as track marks or abscesses37 and document any physical findings consistent with intoxication or withdrawal. Patients must be completely detoxed or in withdrawal before beginning buprenorphine induction; premature induction can precipitate withdrawal.38
A discussion of pregnant patients with opioid use disorder is beyond the scope of this paper. However, it is incumbent on the prescriber to inquire whether the client is pregnant or intends to become pregnant and what birth control methods are in place.
Step 2: Assess psychiatric status
Assessment of the patient’s psychiatric status, including an assessment of alcohol and other drug use, will help determine his or her eligibility for opioid agonists.37 To prepare for the need to manage patients with psychiatrically complex issues, it is helpful to develop relationships with addiction specialists and psychiatrists who are familiar with opioid replacement therapy in your area. This will make it easier to collaborate on patients’ care.
Ask all patients directly about suicidal or homicidal ideation. Any patient with active suicidal or homicidal ideation should be assessed for need of immediate hospitalization by a psychiatrist or another qualified mental health professional. Patients with a history of suicidal ideation should be monitored closely by a mental health professional throughout treatment.37
Many if not most patients with opioid use disorder have concurrent psychiatric disorders, and the interplay between these disorders is complex.40,41 Depression, for example, can precede and even precipitate drug use (an observation supporting the “self-medication theory”).42 If the underlying depressive disorder is not addressed, relapse is nearly inevitable.
It has also been shown that both chronic opioid use and withdrawal can exacerbate aversive emotional states. This escalation of symptoms may result from the pharmacologic effects of opioids or from psychosocial sequelae that can arise from chronic opioid use.41 In this situation, maintaining abstinence can lead to resolution of depressive symptoms. As depression and opioid use can occur together, successful treatment requires equal attention to both illnesses.
Other common comorbidities in patients with opioid use disorder include posttraumatic stress disorder, attention deficit hyperactivity disorder, antisocial personality disorder, and concurrent substance abuse disorders.43 The confluence of antisocial personality disorder is particularly important, as patients with antisocial personality disorder display disruptive and maladaptive behaviors.
Identify any psychotropic medication that is prescribed and check carefully for drug interactions. This applies especially to methadone, as many psychiatric medications also prolong the QT interval. Moreover, patients may not be forthcoming about the use of psychiatric medication.
Find out whether the patient is using any other addictive substances, particularly those that affect the central nervous system, as those who use fentanyl, benzodiazepines, or alcohol are at the highest risk of overdose.31 Often the best option for those with concurrent substance use disorders is inpatient detoxification followed by residential rehabilitation care. Either buprenorphine or methadone can then be initiated upon return to an outpatient setting.
Step 3: Assess psychosocial status
To what extent do the patient’s home environment and support systems promote a drug-free lifestyle? Unfortunately, the psychosocial status of many of these patients is fragile, and they may live in areas where illicit drugs are readily available (which can be urban, suburban, or rural), making it difficult to stay substance-free.38
Generally, lifestyle modifications are needed to transform maladaptive behaviors and promote an environment conducive to long-term recovery. Referrals to social services to address housing, vocational needs, and entitlements may be helpful.39
Step 4: Assess readiness to change
According to one model, people go through 5 stages when changing a behavior: precontemplation, contemplation, preparation for action, action, and maintenance.43 In general, the further along the stages a patient is, the more appropriate he or she is for office-based treatment with buprenorphine.39
The level of change can be assessed with tools such as Stages of Change Readiness and Treatment Eagerness Scale (SOCRATES). Use of stage-specific strategies may enhance a patient’s readiness to cease opioid use.43
Precontemplation. Those in the precontemplation stage are not ready to think about changing their behavior.43 They may be unaware of or unwilling to consider the risks associated with their opioid use and resistant to the idea of quitting. Engagement with opioid agonists for individuals in this stage is low and dropout rates are likely high.
Thus, the proper approach for “precontemplators” is to help them develop some ambivalence about their opioid use. One tactic is to involve the patient in a discussion of the personal benefits and risks of opioid use.
Contemplation. Individuals in the contemplation stage have begun to weigh the costs and benefits of opioid use and express ambivalence about it.44 Because the patient is willing to explore the risks of ongoing use and consider the benefits of treatment, the goal in this stage is to elicit a commitment from the individual to seek treatment.
Preparation. The person in this stage moves from thinking about treatment to planning what action to take.45 As the individual prepares to enter treatment, indecision tends to resurface, as well as self-doubt about his or her ability to change. During this stage, it is important for the provider to spell out goals (abstinence) and strategies (eg, counseling, medication) and enhance a sense of self-efficacy.
Action and maintenance. Patients in these stages engage in treatment and employ new strategies to abstain from opioid use. Maintaining these behaviors can be a daily struggle. Expressing confidence in the patient’s ability to abstain from use will support his or her progress. Behavioral interventions such as strategic avoidance of triggers and engagement in alternative activities (eg, support groups, exercise, faith-based practices) will help to maintain abstinence.
A CHRONIC CONDITION
Opioid use disorder, like many chronic illnesses, requires long-term attention to attain successful patient outcomes. The opioid agonists methadone and buprenorphine are the mainstay of treatment for it, conferring benefits such as reducing opioid use and preventing relapse.
Candidates for opioid agonist therapy should undergo a multidisciplinary assessment, including an evaluation on the patient’s readiness to change his or her opioid use.
Patient education should include a discussion of the risks of methadone (eg, respiratory depression, fatal overdose, and QTc prolongation) and buprenorphine (eg hepatotoxicity) and their benefits (eg, controlling craving, decreasing the risk of relapse). Patients should also be educated about overdose and diversion.
Despite the difficulties inherent in treating patients with opioid use disorder, when used appropriately, opioid agonist therapy can be lifesaving for patients struggling with long-term opioid addiction.
Acknowledgment: We thank Katelyn Colosi, BS, and Drs. Susan Wolfe, Dennis Bouffard, and Sinha Shirshendu for their helpful comments.
- Wakeman SE, Pham-Kanter G, Donelan K. Attitudes, practices, and preparedness to care for patients with substance use disorder: results from a survey of general internists. Subst Abus 2016; 37:635–641.
- Samuels EA, Dwyer K, Mello MJ, Baird J, Kellogg AR, Bernstein E. Emergency department-based opioid harm reduction: moving physicians from willing to doing. Acad Emerg Med 2016; 23:455–465.
- Mohlman MK, Tanzman B, Finison K, Pinette M, Jones C. Impact of medication-assisted treatment for opioid addiction on Medicaid expenditures and health services utilization rates in Vermont. J Subst Abuse Treat 2016; 67:9–14.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition. Arlington, VA, American Psychiatric Association, 2013.
- Center for Behavioral Health Statistics and Quality. Behavioral health trends in the United States: results from the 2014 National Survey on Drug Use and Health. www.samhsa.gov/data. Accessed April 6, 2017.
- Compton WM, Jones CM, Baldwin GT. Relationship between nonmedical prescription-opioid use and heroin use. N Engl J Med 2016; 374:154–163.
- Ruan X, Wyche MQ, Kaye AD. Analyzing the relationship between nonmedical prescription-opioid use and heroin use. J Opioid Manage 2016; 12:11–14.
- Hser YI, Evans E, Grella C, Ling W, Anglin D. Long-term course of opioid addiction. Harv Rev Psychiatry 2015; 23:76–89.
- Nielsen S, Hillhouse M, Mooney L, Ang A, Ling W. Buprenorphine pharmacotherapy and behavioral treatment: comparison of outcomes among prescription opioid users, heroin users and combination users. J Subst Abuse Treat 2015; 48:70–76.
- Moore BA, Fiellin DA, Barry DT, et al. Primary care office-based buprenorphine treatment: comparison of heroin and prescription opioid dependent patients. J Gen Intern Med 2007; 22:527–530.
- Fischer B, Patra J, Cruz MF, Gittins J, Rehm J. Comparing heroin users and prescription opioid users in a Canadian multi-site population of illicit opioid users. Drug Alcohol Rev 2008; 27:625–632.
- Compton WM, Jones CM, Baldwin GT. Relationship between nonmedical prescription-opioid use and heroin use. N Engl J Med 2016; 374:154–163.
- Jones CM, Baldwin GT, Manocchio T, White JO, Mack KA. Trends in methadone distribution for pain treatment, methadone diversion, and overdose deaths—United States, 2002–2014. MMWR Morb Mortal Wkly Rep 2016; 65:667–671.
- Baxter LE Sr, Campbell A, Deshields M, et al. Safe methadone induction and stabilization: report of an expert panel. J Addict Med 2013; 7:377–386.
- Alinejad S, Kazemi T, Zamani N, Hoffman RS, Mehrpour O. A systematic review of the cardiotoxicity of methadone. EXCLI J 2015; 14:577–600.
- Johnson RE, Strain EC, Amass L. Buprenorphine: how to use it right. Drug Alcohol Depend 2003; 70(suppl 2):S59–S77.
- Ling W. Buprenorphine implant for opioid addiction. Pain Manage 2012; 2:345–350.
- Saxon AJ, Hser YI, Woody G, Ling W. Medication-assisted treatment for opioid addiction: methadone and buprenorphine. J Food Drug Anal 2013; 21:S69–S72.
- Kimber J, Larney S, Hickman M, Randall D, Degenhardt L. Mortality risk of opioid substitution therapy with methadone versus buprenorphine: a retrospective cohort study. Lancet Psychiatry 2015; 2:901–908.
- Marteau D, McDonald R, Patel K. The relative risk of fatal poisoning by methadone or buprenorphine within the wider population of England and Wales. BMJ Open 2015; 5: e007629.
- Kintz P. Deaths involving buprenorphine: a compendium of French cases. Forensic Sci Int 2001; 121:65–69.
- Zuin M, Giorgini A, Selmi C, et al. Acute liver and renal failure during treatment with buprenorphine at therapeutic dose. Dig Liver Dis 2009; 41:e8–e10.
- Klein JW. Pharmacotherapy for substance use disorders. Med Clin North Am 2016; 100:891–910.
- Modesto-Lowe V, Van Kirk J. Clinical uses of naltrexone: a review of the evidence. Exp Clin Psychopharmocol 2002; 10:213–227.
- Woody GE. Agonist models for treating persons with substance use disorders. Curr Psychiatry Rep 2014; 16:489.
- Sanders JJ, Roose RJ, Lubrano MC, Lucan SC. Meaning and methadone: patient perceptions of methadone dose and a model to promote adherence to maintenance treatment. J Addict Med 2013; 7:307–313.
- Herget G. Methadone and buprenorphine added to the WHO list of essential medicines. HIV/AIDS Policy Law Rev 2005; 10:23–24.
- Suzuki J, Dodds T. Clinical recommendation of 12-step meeting attendance and discussion regarding disclosure of buprenorphine use among patients in office-based opioid treatment. Subst Abus 2016; 37:31–34.
- Rettig RA, Yarmolinsky A. Federal Regulation of Methadone Treatment. Washington, DC: National Academies Press; 1995.
- Srivastava A, Kahan M, Nader M. Primary care management of opioid use disorders: abstinence, methadone, or buprenorphine-naloxone? Can Fam Physician 2017; 63:200–205.
- Substance Abuse and Mental Health Services Administration. Federal Guidelines for Opioid Treatment Programs. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2015.
- Substance Abuse and Mental Health Services Administration SAMSHA. Buprenorphine waiver management. www.samhsa.gov/medication-assisted-treatment/buprenorphine-waiver-management. Accessed April 6, 2017.
- Mark TL, Lubran R, McCance-Kats EF, Chalk M, Richardson J. Medicaid coverage of medications to treat alcohol and opioid dependence. J Subst Abuse Treat 2015; 55:1–5.
- Johnson B, Richert T. Diversion of methadone and buprenorphine from opioid substitution treatment: the importance of patients’ attitudes and norms. J Subst Abuse Treat 2015; 54:50–55.
- Yokell MA, Zaller ND, Green TC, Rich JD. Buprenorphine and buprenorphine/naloxone diversion, misuse, and illicit use: an international review. Curr Drug Abuse Rev 2011; 4:28–41.
- Schuman-Olivier Z, Albanese M, Nelson SE, et al. Self-treatment: illicit buprenorphine use by opioid-dependent treatment seekers. J Subst Abuse Treat 2010; 39:41–50.
- American Society of Addiction Medicine. National practice guidelines for the use of medications in the treatment of addiction involving opioid use. www.asam.org/docs/default-source/practice-support/guidelines-and-consensus-docs/asam-national-practice-guideline-supplement.pdf. Accessed April 6, 2017.
- McNicholas L. Clinical guidelines for the use of buprenorphine in the treatment of opioid addiction. Rockville, MD: US Department of Health and Human Services, Substance Abuse and Mental Health Service Administration; 2004.
- Center for Substance Abuse Treatment. Clinical guidelines for the use of buprenorphine in the treatment of opioid addiction. Rockville (MD): Substance Abuse and Mental Health Services Administration (US); 2004. (Treatment Improvement Protocol (TIP) Series, No. 40.) www.ncbi.nlm.nih.gov/books/NBK64245. Accessed April 6, 2017.
- Zippel-Schultz B, Specka M, Cimander K, et al. Outcomes of patients in long-term opioid maintenance treatment. Subst Use Misuse 2016; 51:1493–1503.
- Martins SS, Keyes KM, Storr CL, Zhu H, Chilcoat HD. Pathways between nonmedical opioid use/dependence and psychiatric disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Drug Alcohol Depend 2009; 103:16–24.
- Khantzian EJ. The self-medication hypothesis of addictive disorders: focus on heroin and cocaine dependence. Am J Psychiatry 1985; 142:1259–1264.
- Belding MA, Iguchi MY, Lamb RJ, Lakin M, Terry R. Stages and processes of change among polydrug users in methadone maintenance treatment. Drug Alcohol Depend 1995; 39:45–53.
- Peteet JR, Brenner S, Curtiss D, Ferrigno M, Kauffman J. A stage of change approach to addiction in the medical setting. Gen Hosp Psychiatry 1998; 20:267–273.
- Vijay A, Bazazi AR, Yee I, Kamarulzaman A, Altice FL. Treatment readiness, attitudes toward, and experiences with methadone and buprenorphine maintenance therapy among people who inject drugs in Malaysia. J Subst Abuse Treat 2015; 54:29–36.
For a patient struggling with opioid addiction, opioid agonist therapy with methadone or buprenorphine can reduce craving and opioid use and may even save his or her life. But many clinicians are unfamiliar with this evidence-based treatment,1,2 which is best started early in the course of addiction.3
This article outlines the pharmacology of these drugs, their clinical uses, and the challenges of using them to treat opioid addiction.
DIAGNOSTIC CRITERIA
Opioid addiction, formally known as opioid use disorder, is a pattern of opioid misuse leading to clinically significant impairment in multiple areas of life. The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, lists 11 diagnostic criteria, but only 2 need to be present within the past year to make the diagnosis4:
- Taking opioids longer or in higher doses than was intended
- A persistent desire or unsuccessful efforts to cut down or control opioid use
- Spending a great deal of time obtaining, using, or recovering from using opioids
- Craving opioids
- Repeatedly failing to fulfill obligations at work, school, or home due to opioid use
- Continuing to use opioids even though it causes or exacerbates social or interpersonal problems
- Giving up or curtailing important social, occupational, or recreational activities because of opioid use
- Repeatedly using opioids in situations in which it is physically hazardous
- Continuing to use opioids despite knowledge of having a persistent or recurrent physical or psychological problem that is likely to have been caused or exacerbated by the substance
- Tolerance
- Withdrawal.
Recent estimates indicate that 2.23 million people in the United States have opioid use disorder (426,000 with heroin and 1.8 million with prescription opioids).5
Progression from prescription opioids to heroin
We have observed that many patients with opioid use disorder start by misusing prescription opioids. Over time, tolerance can develop, which drives patients to use higher and higher doses.6
As the addiction progresses, a subset of prescription opioid users advances to using heroin, which is typically less expensive and easier to obtain.7 Most patients start with the intranasal route but eventually inject it intravenously.6,7
For many addicts, heroin use has medical consequences such as hepatitis C virus (HCV) and human immunodeficiency virus (HIV) infection, psychiatric problems such as depression and anxiety, and illegal activities such as theft and sex work.8 People who use heroin appear to have more severe addiction and a lower socioeconomic status than prescription opioid users.9–11 But recently, a growing number of middle class individuals are becoming addicted to heroin.12
METHADONE
Methadone is a long-acting synthetic opioid that functions as a full agonist on the mu-opioid receptor. The drug binds, occupies, and stimulates the receptor, preventing withdrawal symptoms and reducing opioid cravings for at least 24 hours.13
Adverse effects of methadone
The most common adverse effects include lightheadedness, dizziness, sedation, nausea, vomiting, and sweating.14 Other adverse effects:
Unintentional overdose. The risk is serious, as a single 30-mg dose can be fatal in people who are opioid-naïve.13
QTc prolongation, which can lead to torsade de pointes. This risk, which is dose-related, must be taken into consideration in patients who have any cardiac symptoms (eg, syncope, arrhythmia), pathology (familial QT prolongation), or other risk factors for QTc prolongation (eg, hypokalemia, QTc-prolonging medications).15
Respiratory depression, which can be fatal. This dose-related risk is heightened during the first 4 weeks of treatment if titration is too rapid or if methadone is used in combination with other drugs that cause central nervous system or respiratory depression.13,14
Starting methadone
To prevent respiratory depression and death related to rapid induction, the general rule is to start methadone at a low daily dose (20–30 mg) depending on the patient’s withdrawal symptoms.14 During this period, patients need to be closely monitored and educated on the perils of concomitant use of central nervous system depressants.14
In most patients, the dose is titrated up until their withdrawal symptoms and cravings are eliminated, which generally requires 60 to 120 mg daily.14 Hepatic and renal impairment, pregnancy, and advanced age can alter methadone pharmacokinetics and may therefore necessitate dose adjustment.
BUPRENORPHINE
Buprenorphine is an alkaloid thebaine opioid derivative that acts as a partial mu-opioid agonist and a kappa antagonist.16 Like methadone, buprenorphine is used to manage cravings and withdrawal symptoms.16 Dosages of 4 to 16 mg (up to 32 mg) per day of buprenorphine are usually required to adequately control opioid cravings.16
Sublingual and subdermal products
Buprenorphine is currently available in the United States in sublingual and subdermal formulations.16,17
Sublingual buprenorphine is usually combined with naloxone in a 4:1 ratio to deter intravenous use. Intravenous injection of the combination product can precipitate withdrawal due to the antagonist action of naloxone. (Taken orally or sublingually, naloxone is poorly absorbed and has little or no clinical effect.) Buprenorphine-naloxone is available in tablets, a sublingual film strip, and a buccal film strip. Buprenorphine is also available by itself in a sublingual formulation.
The US Food and Drug Administration has approved a buprenorphine subdermal implant, Probuphine. Four rods, about 1 inch long, are placed under the skin in the inner aspect of the upper arm and provide the equivalent of 8 mg of buprenorphine daily for 6 months.17 However, this method is formulated only for maintenance treatment and cannot be used for induction. Additionally, it is recommended that the implants be surgically removed at the end of 6 months, after which another set of implants can be inserted in the other arm or the patient can switch to sublingual therapy, depending on the clinical situation and patient preference.17
Generally safer than methadone
Buprenorphine works on the same receptor as methadone and therefore has a similar side effect profile. However, buprenorphine has a ceiling effect, which greatly reduces the risk of fatal respiratory depression.18 It also does not cause clinically significant QTc prolongation and is preferable in patients who have cardiac risk factors.18
Another advantage is that buprenorphine has fewer identified medication interactions than methadone.18 Further, induction of buprenorphine in patients with opioid use disorder has been shown to be safer than methadone.19
Although buprenorphine has been found to be 6 times safer than methadone with regard to overdose among the general population,20 it can still cause fatal intoxication if used in combination with central nervous system depressants.21
Buprenorphine has been also associated with hepatotoxicity, though the risk of new-onset liver disease appears to be low.22
NALTREXONE IS LESS EFFECTIVE THAN METHADONE, BUPRENORPHINE
Besides methadone and buprenorphine, the only other approved option for treating opioid use disorder is the opioid antagonist naltrexone.
Naltrexone has significantly less abuse potential, as it provides no euphoria, but patients do not like it. Even with the long-acting formulation (Vivitrol), naltrexone treatment is significantly less effective than methadone or buprenorphine.23–25 Further, although naltrexone is not a controlled substance and so does not face the same scrutiny as the agonist therapies, there are other significant barriers. Additional information on naltrexone is presented in reviews by Modesto-Lowe and Van Kirk24 and Woody.25
OBSTACLES TO TREATMENT
People hold conflicting views about opioid agonist therapy. Some believe that “trading one drug for another” is not a legitimate therapeutic strategy, and they may feel ashamed of being on maintenance therapy.26 Similarly, some argue that the answer to establishing stable abstinence does not lie simply in prescribing medications.
The contrary argument is that these medications, if used appropriately, confer many benefits such as reducing the medical and psychosocial sequelae of opioid addiction.18 In fact, properly treated patients no longer meet the diagnostic criteria of opioid use disorder, and both methadone and buprenorphine are on the World Health Organization’s (WHO) list of essential medicines.27
Despite endorsement by the WHO, the stigma attached to the opioid agonists has been difficult to overcome. Patients with opioid use disorder may be viewed with distrust by healthcare providers and often do not feel welcome in healthcare settings or in self-help recovery groups.28
Barriers to methadone therapy
Federal regulations on methadone prescribing and use were established to promote patient safety and decrease diversion, but they may also complicate access to care.29 They stipulate that to qualify for methadone maintenance, patients need to demonstrate opioid addiction for 1 year, except for pregnant women and those who have been incarcerated in the past 6 months. Patients under the age of 18 must have 2 documented failed treatment episodes as well as approval by a guardian to receive treatment.
Inconvenience. Methadone can be prescribed for opioid dependence only by an accredited treatment program. Patients must therefore travel to the clinic and wait to be evaluated on a daily basis for a minimum of 90 days. Only after they demonstrate consistent responsible behavior and negative results on urine testing do they become eligible to take methadone home.29 If a patient is to travel out of the area during the initial 90 days of treatment, he or she must make arrangements in advance to find a clinic that will provide a “guest dose.”
The inconvenience arising from the regulations may deter some patients from seeking methadone therapy. In spite of this, once patients are started on methadone, more of them continue treatment than with buprenorphine.18 A proposed reason is that methadone is a potent full opioid agonist and therefore relieves withdrawal symptoms and craving more effectively than buprenorphine, which is a partial agonist.30 Another possible reason is the higher level of supervision afforded by methadone clinics, which require daily contact for at least 90 days.
Safety concerns arise from methadone diversion, as illicit use may have lethal consequences. In the past decade, deaths from methadone overdose have risen significantly, most of them due to respiratory depression or torsade de pointes.13 However, most cases of diversion and overdose involve methadone that is prescribed for pain by individual practitioners and not from maintenance programs.13
Advantages of buprenorphine
Together, methadone’s lethality, stigma, and inconvenience may contribute to patients preferring buprenorphine.31
The regulations governing buprenorphine’s use are less restrictive than those with methadone. For example, patients must have a diagnosis of opioid addiction to be prescribed buprenorphine, but they are not required to carry the diagnosis for a year before treatment.31 Additionally, they do not need to travel to a federally approved opioid treatment center daily and can receive buprenorphine directly from a physician in an outpatient setting.
Under the Drug Abuse Treatment Act (DATA) of 2000, any physician can apply for a waiver to prescribe and dispense buprenorphine in his or her office. To qualify for an initial waiver, physicians must either obtain certification in the fields of addiction medicine or addiction psychiatry or complete an approved 8-hour training session.32 Each physician starts with a maximum of 30 patients, but can apply to treat up to 100 patients after 1 year and eventually up to 275 patients. Physicians must document every buprenorphine prescription they write and be able to refer patients for counseling.31
As of February 2017, nurse practitioners and physician assistants can also apply for a DATA 2000 waiver. All waivered providers are subject to unannounced visits from the Drug Enforcement Administration once every 5 years.32
While there are no federal restrictions on the amount of buprenorphine that can be dispensed, some states and some insurance companies have placed restrictions on dose or length of treatment.33 Buprenorphine patients can fill their prescriptions at any pharmacy and are permitted to bring their medication home, which improves access to care. However, office-based outpatient treatment is not without risk, and preventing buprenorphine diversion remains a challenge.34
‘Lending’ buprenorphine is a felony
Addicts have illegally used buprenorphine to self-treat opioid withdrawal, craving, and dependence.35 Its misuse has also been coupled with self-treatment of conditions that include depression and pain.36
A survey found that 83.7% of patients deem buprenorphine diversion to be appropriate; further, most patients said they consider it unethical to withhold prescribed buprenorphine from individuals showing symptoms of withdrawal.34 Physicians who prescribe buprenorphine must inform their patients that even “lending” or giving away their medication is a felony.
Prescribing physicians must also be diligent about monitoring for signs of diversion such as inconsistent urine toxicology screens, “lost” medication, and requests for early refills or escalating doses.37
EVALUATING PATIENTS FOR OPIOID REPLACEMENT THERAPY
In addition to federal regulations, we propose a 4-step approach to evaluate eligibility for opioid replacement therapy based on existing guidelines.37–39
Step 1: History and physical examination
The history should give particular attention to the patient’s cardiac, pulmonary, and hepatic status, with consideration of the risks of any medical comorbidities (eg, bacterial endocarditis, HIV and HCV infection) that might influence treatment.37
It is also essential to evaluate for any contraindications or drug interactions before prescribing methadone or buprenorphine.38
Contraindications to methadone maintenance include40:
- Cor pulmonale
- Methadone hypersensitivity
- Pseudomembranous colitis
- Selegiline use (due to risk of serotonin syndrome)
- Ileum paralyticus.
Contraindications to buprenorphine use include:
- Hypersensitivity to naloxone or buprenorphine
- Impaired liver function (due to the risk of inadvertent overdose associated with slowed metabolism).
Concurrent use of alcohol or illicit benzodiazepines is a relative contraindication to both methadone and buprenorphine due to the risk of respiratory depression and overdose.37 Likewise, avoid coprescribing opioid agonists and benzodiazepines whenever possible. Obtain a complete list of current medications and query a prescription-monitoring database to determine whether any controlled substances are currently prescribed.37
During the physical examination, look for stigmata of intravenous drug use such as track marks or abscesses37 and document any physical findings consistent with intoxication or withdrawal. Patients must be completely detoxed or in withdrawal before beginning buprenorphine induction; premature induction can precipitate withdrawal.38
A discussion of pregnant patients with opioid use disorder is beyond the scope of this paper. However, it is incumbent on the prescriber to inquire whether the client is pregnant or intends to become pregnant and what birth control methods are in place.
Step 2: Assess psychiatric status
Assessment of the patient’s psychiatric status, including an assessment of alcohol and other drug use, will help determine his or her eligibility for opioid agonists.37 To prepare for the need to manage patients with psychiatrically complex issues, it is helpful to develop relationships with addiction specialists and psychiatrists who are familiar with opioid replacement therapy in your area. This will make it easier to collaborate on patients’ care.
Ask all patients directly about suicidal or homicidal ideation. Any patient with active suicidal or homicidal ideation should be assessed for need of immediate hospitalization by a psychiatrist or another qualified mental health professional. Patients with a history of suicidal ideation should be monitored closely by a mental health professional throughout treatment.37
Many if not most patients with opioid use disorder have concurrent psychiatric disorders, and the interplay between these disorders is complex.40,41 Depression, for example, can precede and even precipitate drug use (an observation supporting the “self-medication theory”).42 If the underlying depressive disorder is not addressed, relapse is nearly inevitable.
It has also been shown that both chronic opioid use and withdrawal can exacerbate aversive emotional states. This escalation of symptoms may result from the pharmacologic effects of opioids or from psychosocial sequelae that can arise from chronic opioid use.41 In this situation, maintaining abstinence can lead to resolution of depressive symptoms. As depression and opioid use can occur together, successful treatment requires equal attention to both illnesses.
Other common comorbidities in patients with opioid use disorder include posttraumatic stress disorder, attention deficit hyperactivity disorder, antisocial personality disorder, and concurrent substance abuse disorders.43 The confluence of antisocial personality disorder is particularly important, as patients with antisocial personality disorder display disruptive and maladaptive behaviors.
Identify any psychotropic medication that is prescribed and check carefully for drug interactions. This applies especially to methadone, as many psychiatric medications also prolong the QT interval. Moreover, patients may not be forthcoming about the use of psychiatric medication.
Find out whether the patient is using any other addictive substances, particularly those that affect the central nervous system, as those who use fentanyl, benzodiazepines, or alcohol are at the highest risk of overdose.31 Often the best option for those with concurrent substance use disorders is inpatient detoxification followed by residential rehabilitation care. Either buprenorphine or methadone can then be initiated upon return to an outpatient setting.
Step 3: Assess psychosocial status
To what extent do the patient’s home environment and support systems promote a drug-free lifestyle? Unfortunately, the psychosocial status of many of these patients is fragile, and they may live in areas where illicit drugs are readily available (which can be urban, suburban, or rural), making it difficult to stay substance-free.38
Generally, lifestyle modifications are needed to transform maladaptive behaviors and promote an environment conducive to long-term recovery. Referrals to social services to address housing, vocational needs, and entitlements may be helpful.39
Step 4: Assess readiness to change
According to one model, people go through 5 stages when changing a behavior: precontemplation, contemplation, preparation for action, action, and maintenance.43 In general, the further along the stages a patient is, the more appropriate he or she is for office-based treatment with buprenorphine.39
The level of change can be assessed with tools such as Stages of Change Readiness and Treatment Eagerness Scale (SOCRATES). Use of stage-specific strategies may enhance a patient’s readiness to cease opioid use.43
Precontemplation. Those in the precontemplation stage are not ready to think about changing their behavior.43 They may be unaware of or unwilling to consider the risks associated with their opioid use and resistant to the idea of quitting. Engagement with opioid agonists for individuals in this stage is low and dropout rates are likely high.
Thus, the proper approach for “precontemplators” is to help them develop some ambivalence about their opioid use. One tactic is to involve the patient in a discussion of the personal benefits and risks of opioid use.
Contemplation. Individuals in the contemplation stage have begun to weigh the costs and benefits of opioid use and express ambivalence about it.44 Because the patient is willing to explore the risks of ongoing use and consider the benefits of treatment, the goal in this stage is to elicit a commitment from the individual to seek treatment.
Preparation. The person in this stage moves from thinking about treatment to planning what action to take.45 As the individual prepares to enter treatment, indecision tends to resurface, as well as self-doubt about his or her ability to change. During this stage, it is important for the provider to spell out goals (abstinence) and strategies (eg, counseling, medication) and enhance a sense of self-efficacy.
Action and maintenance. Patients in these stages engage in treatment and employ new strategies to abstain from opioid use. Maintaining these behaviors can be a daily struggle. Expressing confidence in the patient’s ability to abstain from use will support his or her progress. Behavioral interventions such as strategic avoidance of triggers and engagement in alternative activities (eg, support groups, exercise, faith-based practices) will help to maintain abstinence.
A CHRONIC CONDITION
Opioid use disorder, like many chronic illnesses, requires long-term attention to attain successful patient outcomes. The opioid agonists methadone and buprenorphine are the mainstay of treatment for it, conferring benefits such as reducing opioid use and preventing relapse.
Candidates for opioid agonist therapy should undergo a multidisciplinary assessment, including an evaluation on the patient’s readiness to change his or her opioid use.
Patient education should include a discussion of the risks of methadone (eg, respiratory depression, fatal overdose, and QTc prolongation) and buprenorphine (eg hepatotoxicity) and their benefits (eg, controlling craving, decreasing the risk of relapse). Patients should also be educated about overdose and diversion.
Despite the difficulties inherent in treating patients with opioid use disorder, when used appropriately, opioid agonist therapy can be lifesaving for patients struggling with long-term opioid addiction.
Acknowledgment: We thank Katelyn Colosi, BS, and Drs. Susan Wolfe, Dennis Bouffard, and Sinha Shirshendu for their helpful comments.
For a patient struggling with opioid addiction, opioid agonist therapy with methadone or buprenorphine can reduce craving and opioid use and may even save his or her life. But many clinicians are unfamiliar with this evidence-based treatment,1,2 which is best started early in the course of addiction.3
This article outlines the pharmacology of these drugs, their clinical uses, and the challenges of using them to treat opioid addiction.
DIAGNOSTIC CRITERIA
Opioid addiction, formally known as opioid use disorder, is a pattern of opioid misuse leading to clinically significant impairment in multiple areas of life. The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, lists 11 diagnostic criteria, but only 2 need to be present within the past year to make the diagnosis4:
- Taking opioids longer or in higher doses than was intended
- A persistent desire or unsuccessful efforts to cut down or control opioid use
- Spending a great deal of time obtaining, using, or recovering from using opioids
- Craving opioids
- Repeatedly failing to fulfill obligations at work, school, or home due to opioid use
- Continuing to use opioids even though it causes or exacerbates social or interpersonal problems
- Giving up or curtailing important social, occupational, or recreational activities because of opioid use
- Repeatedly using opioids in situations in which it is physically hazardous
- Continuing to use opioids despite knowledge of having a persistent or recurrent physical or psychological problem that is likely to have been caused or exacerbated by the substance
- Tolerance
- Withdrawal.
Recent estimates indicate that 2.23 million people in the United States have opioid use disorder (426,000 with heroin and 1.8 million with prescription opioids).5
Progression from prescription opioids to heroin
We have observed that many patients with opioid use disorder start by misusing prescription opioids. Over time, tolerance can develop, which drives patients to use higher and higher doses.6
As the addiction progresses, a subset of prescription opioid users advances to using heroin, which is typically less expensive and easier to obtain.7 Most patients start with the intranasal route but eventually inject it intravenously.6,7
For many addicts, heroin use has medical consequences such as hepatitis C virus (HCV) and human immunodeficiency virus (HIV) infection, psychiatric problems such as depression and anxiety, and illegal activities such as theft and sex work.8 People who use heroin appear to have more severe addiction and a lower socioeconomic status than prescription opioid users.9–11 But recently, a growing number of middle class individuals are becoming addicted to heroin.12
METHADONE
Methadone is a long-acting synthetic opioid that functions as a full agonist on the mu-opioid receptor. The drug binds, occupies, and stimulates the receptor, preventing withdrawal symptoms and reducing opioid cravings for at least 24 hours.13
Adverse effects of methadone
The most common adverse effects include lightheadedness, dizziness, sedation, nausea, vomiting, and sweating.14 Other adverse effects:
Unintentional overdose. The risk is serious, as a single 30-mg dose can be fatal in people who are opioid-naïve.13
QTc prolongation, which can lead to torsade de pointes. This risk, which is dose-related, must be taken into consideration in patients who have any cardiac symptoms (eg, syncope, arrhythmia), pathology (familial QT prolongation), or other risk factors for QTc prolongation (eg, hypokalemia, QTc-prolonging medications).15
Respiratory depression, which can be fatal. This dose-related risk is heightened during the first 4 weeks of treatment if titration is too rapid or if methadone is used in combination with other drugs that cause central nervous system or respiratory depression.13,14
Starting methadone
To prevent respiratory depression and death related to rapid induction, the general rule is to start methadone at a low daily dose (20–30 mg) depending on the patient’s withdrawal symptoms.14 During this period, patients need to be closely monitored and educated on the perils of concomitant use of central nervous system depressants.14
In most patients, the dose is titrated up until their withdrawal symptoms and cravings are eliminated, which generally requires 60 to 120 mg daily.14 Hepatic and renal impairment, pregnancy, and advanced age can alter methadone pharmacokinetics and may therefore necessitate dose adjustment.
BUPRENORPHINE
Buprenorphine is an alkaloid thebaine opioid derivative that acts as a partial mu-opioid agonist and a kappa antagonist.16 Like methadone, buprenorphine is used to manage cravings and withdrawal symptoms.16 Dosages of 4 to 16 mg (up to 32 mg) per day of buprenorphine are usually required to adequately control opioid cravings.16
Sublingual and subdermal products
Buprenorphine is currently available in the United States in sublingual and subdermal formulations.16,17
Sublingual buprenorphine is usually combined with naloxone in a 4:1 ratio to deter intravenous use. Intravenous injection of the combination product can precipitate withdrawal due to the antagonist action of naloxone. (Taken orally or sublingually, naloxone is poorly absorbed and has little or no clinical effect.) Buprenorphine-naloxone is available in tablets, a sublingual film strip, and a buccal film strip. Buprenorphine is also available by itself in a sublingual formulation.
The US Food and Drug Administration has approved a buprenorphine subdermal implant, Probuphine. Four rods, about 1 inch long, are placed under the skin in the inner aspect of the upper arm and provide the equivalent of 8 mg of buprenorphine daily for 6 months.17 However, this method is formulated only for maintenance treatment and cannot be used for induction. Additionally, it is recommended that the implants be surgically removed at the end of 6 months, after which another set of implants can be inserted in the other arm or the patient can switch to sublingual therapy, depending on the clinical situation and patient preference.17
Generally safer than methadone
Buprenorphine works on the same receptor as methadone and therefore has a similar side effect profile. However, buprenorphine has a ceiling effect, which greatly reduces the risk of fatal respiratory depression.18 It also does not cause clinically significant QTc prolongation and is preferable in patients who have cardiac risk factors.18
Another advantage is that buprenorphine has fewer identified medication interactions than methadone.18 Further, induction of buprenorphine in patients with opioid use disorder has been shown to be safer than methadone.19
Although buprenorphine has been found to be 6 times safer than methadone with regard to overdose among the general population,20 it can still cause fatal intoxication if used in combination with central nervous system depressants.21
Buprenorphine has been also associated with hepatotoxicity, though the risk of new-onset liver disease appears to be low.22
NALTREXONE IS LESS EFFECTIVE THAN METHADONE, BUPRENORPHINE
Besides methadone and buprenorphine, the only other approved option for treating opioid use disorder is the opioid antagonist naltrexone.
Naltrexone has significantly less abuse potential, as it provides no euphoria, but patients do not like it. Even with the long-acting formulation (Vivitrol), naltrexone treatment is significantly less effective than methadone or buprenorphine.23–25 Further, although naltrexone is not a controlled substance and so does not face the same scrutiny as the agonist therapies, there are other significant barriers. Additional information on naltrexone is presented in reviews by Modesto-Lowe and Van Kirk24 and Woody.25
OBSTACLES TO TREATMENT
People hold conflicting views about opioid agonist therapy. Some believe that “trading one drug for another” is not a legitimate therapeutic strategy, and they may feel ashamed of being on maintenance therapy.26 Similarly, some argue that the answer to establishing stable abstinence does not lie simply in prescribing medications.
The contrary argument is that these medications, if used appropriately, confer many benefits such as reducing the medical and psychosocial sequelae of opioid addiction.18 In fact, properly treated patients no longer meet the diagnostic criteria of opioid use disorder, and both methadone and buprenorphine are on the World Health Organization’s (WHO) list of essential medicines.27
Despite endorsement by the WHO, the stigma attached to the opioid agonists has been difficult to overcome. Patients with opioid use disorder may be viewed with distrust by healthcare providers and often do not feel welcome in healthcare settings or in self-help recovery groups.28
Barriers to methadone therapy
Federal regulations on methadone prescribing and use were established to promote patient safety and decrease diversion, but they may also complicate access to care.29 They stipulate that to qualify for methadone maintenance, patients need to demonstrate opioid addiction for 1 year, except for pregnant women and those who have been incarcerated in the past 6 months. Patients under the age of 18 must have 2 documented failed treatment episodes as well as approval by a guardian to receive treatment.
Inconvenience. Methadone can be prescribed for opioid dependence only by an accredited treatment program. Patients must therefore travel to the clinic and wait to be evaluated on a daily basis for a minimum of 90 days. Only after they demonstrate consistent responsible behavior and negative results on urine testing do they become eligible to take methadone home.29 If a patient is to travel out of the area during the initial 90 days of treatment, he or she must make arrangements in advance to find a clinic that will provide a “guest dose.”
The inconvenience arising from the regulations may deter some patients from seeking methadone therapy. In spite of this, once patients are started on methadone, more of them continue treatment than with buprenorphine.18 A proposed reason is that methadone is a potent full opioid agonist and therefore relieves withdrawal symptoms and craving more effectively than buprenorphine, which is a partial agonist.30 Another possible reason is the higher level of supervision afforded by methadone clinics, which require daily contact for at least 90 days.
Safety concerns arise from methadone diversion, as illicit use may have lethal consequences. In the past decade, deaths from methadone overdose have risen significantly, most of them due to respiratory depression or torsade de pointes.13 However, most cases of diversion and overdose involve methadone that is prescribed for pain by individual practitioners and not from maintenance programs.13
Advantages of buprenorphine
Together, methadone’s lethality, stigma, and inconvenience may contribute to patients preferring buprenorphine.31
The regulations governing buprenorphine’s use are less restrictive than those with methadone. For example, patients must have a diagnosis of opioid addiction to be prescribed buprenorphine, but they are not required to carry the diagnosis for a year before treatment.31 Additionally, they do not need to travel to a federally approved opioid treatment center daily and can receive buprenorphine directly from a physician in an outpatient setting.
Under the Drug Abuse Treatment Act (DATA) of 2000, any physician can apply for a waiver to prescribe and dispense buprenorphine in his or her office. To qualify for an initial waiver, physicians must either obtain certification in the fields of addiction medicine or addiction psychiatry or complete an approved 8-hour training session.32 Each physician starts with a maximum of 30 patients, but can apply to treat up to 100 patients after 1 year and eventually up to 275 patients. Physicians must document every buprenorphine prescription they write and be able to refer patients for counseling.31
As of February 2017, nurse practitioners and physician assistants can also apply for a DATA 2000 waiver. All waivered providers are subject to unannounced visits from the Drug Enforcement Administration once every 5 years.32
While there are no federal restrictions on the amount of buprenorphine that can be dispensed, some states and some insurance companies have placed restrictions on dose or length of treatment.33 Buprenorphine patients can fill their prescriptions at any pharmacy and are permitted to bring their medication home, which improves access to care. However, office-based outpatient treatment is not without risk, and preventing buprenorphine diversion remains a challenge.34
‘Lending’ buprenorphine is a felony
Addicts have illegally used buprenorphine to self-treat opioid withdrawal, craving, and dependence.35 Its misuse has also been coupled with self-treatment of conditions that include depression and pain.36
A survey found that 83.7% of patients deem buprenorphine diversion to be appropriate; further, most patients said they consider it unethical to withhold prescribed buprenorphine from individuals showing symptoms of withdrawal.34 Physicians who prescribe buprenorphine must inform their patients that even “lending” or giving away their medication is a felony.
Prescribing physicians must also be diligent about monitoring for signs of diversion such as inconsistent urine toxicology screens, “lost” medication, and requests for early refills or escalating doses.37
EVALUATING PATIENTS FOR OPIOID REPLACEMENT THERAPY
In addition to federal regulations, we propose a 4-step approach to evaluate eligibility for opioid replacement therapy based on existing guidelines.37–39
Step 1: History and physical examination
The history should give particular attention to the patient’s cardiac, pulmonary, and hepatic status, with consideration of the risks of any medical comorbidities (eg, bacterial endocarditis, HIV and HCV infection) that might influence treatment.37
It is also essential to evaluate for any contraindications or drug interactions before prescribing methadone or buprenorphine.38
Contraindications to methadone maintenance include40:
- Cor pulmonale
- Methadone hypersensitivity
- Pseudomembranous colitis
- Selegiline use (due to risk of serotonin syndrome)
- Ileum paralyticus.
Contraindications to buprenorphine use include:
- Hypersensitivity to naloxone or buprenorphine
- Impaired liver function (due to the risk of inadvertent overdose associated with slowed metabolism).
Concurrent use of alcohol or illicit benzodiazepines is a relative contraindication to both methadone and buprenorphine due to the risk of respiratory depression and overdose.37 Likewise, avoid coprescribing opioid agonists and benzodiazepines whenever possible. Obtain a complete list of current medications and query a prescription-monitoring database to determine whether any controlled substances are currently prescribed.37
During the physical examination, look for stigmata of intravenous drug use such as track marks or abscesses37 and document any physical findings consistent with intoxication or withdrawal. Patients must be completely detoxed or in withdrawal before beginning buprenorphine induction; premature induction can precipitate withdrawal.38
A discussion of pregnant patients with opioid use disorder is beyond the scope of this paper. However, it is incumbent on the prescriber to inquire whether the client is pregnant or intends to become pregnant and what birth control methods are in place.
Step 2: Assess psychiatric status
Assessment of the patient’s psychiatric status, including an assessment of alcohol and other drug use, will help determine his or her eligibility for opioid agonists.37 To prepare for the need to manage patients with psychiatrically complex issues, it is helpful to develop relationships with addiction specialists and psychiatrists who are familiar with opioid replacement therapy in your area. This will make it easier to collaborate on patients’ care.
Ask all patients directly about suicidal or homicidal ideation. Any patient with active suicidal or homicidal ideation should be assessed for need of immediate hospitalization by a psychiatrist or another qualified mental health professional. Patients with a history of suicidal ideation should be monitored closely by a mental health professional throughout treatment.37
Many if not most patients with opioid use disorder have concurrent psychiatric disorders, and the interplay between these disorders is complex.40,41 Depression, for example, can precede and even precipitate drug use (an observation supporting the “self-medication theory”).42 If the underlying depressive disorder is not addressed, relapse is nearly inevitable.
It has also been shown that both chronic opioid use and withdrawal can exacerbate aversive emotional states. This escalation of symptoms may result from the pharmacologic effects of opioids or from psychosocial sequelae that can arise from chronic opioid use.41 In this situation, maintaining abstinence can lead to resolution of depressive symptoms. As depression and opioid use can occur together, successful treatment requires equal attention to both illnesses.
Other common comorbidities in patients with opioid use disorder include posttraumatic stress disorder, attention deficit hyperactivity disorder, antisocial personality disorder, and concurrent substance abuse disorders.43 The confluence of antisocial personality disorder is particularly important, as patients with antisocial personality disorder display disruptive and maladaptive behaviors.
Identify any psychotropic medication that is prescribed and check carefully for drug interactions. This applies especially to methadone, as many psychiatric medications also prolong the QT interval. Moreover, patients may not be forthcoming about the use of psychiatric medication.
Find out whether the patient is using any other addictive substances, particularly those that affect the central nervous system, as those who use fentanyl, benzodiazepines, or alcohol are at the highest risk of overdose.31 Often the best option for those with concurrent substance use disorders is inpatient detoxification followed by residential rehabilitation care. Either buprenorphine or methadone can then be initiated upon return to an outpatient setting.
Step 3: Assess psychosocial status
To what extent do the patient’s home environment and support systems promote a drug-free lifestyle? Unfortunately, the psychosocial status of many of these patients is fragile, and they may live in areas where illicit drugs are readily available (which can be urban, suburban, or rural), making it difficult to stay substance-free.38
Generally, lifestyle modifications are needed to transform maladaptive behaviors and promote an environment conducive to long-term recovery. Referrals to social services to address housing, vocational needs, and entitlements may be helpful.39
Step 4: Assess readiness to change
According to one model, people go through 5 stages when changing a behavior: precontemplation, contemplation, preparation for action, action, and maintenance.43 In general, the further along the stages a patient is, the more appropriate he or she is for office-based treatment with buprenorphine.39
The level of change can be assessed with tools such as Stages of Change Readiness and Treatment Eagerness Scale (SOCRATES). Use of stage-specific strategies may enhance a patient’s readiness to cease opioid use.43
Precontemplation. Those in the precontemplation stage are not ready to think about changing their behavior.43 They may be unaware of or unwilling to consider the risks associated with their opioid use and resistant to the idea of quitting. Engagement with opioid agonists for individuals in this stage is low and dropout rates are likely high.
Thus, the proper approach for “precontemplators” is to help them develop some ambivalence about their opioid use. One tactic is to involve the patient in a discussion of the personal benefits and risks of opioid use.
Contemplation. Individuals in the contemplation stage have begun to weigh the costs and benefits of opioid use and express ambivalence about it.44 Because the patient is willing to explore the risks of ongoing use and consider the benefits of treatment, the goal in this stage is to elicit a commitment from the individual to seek treatment.
Preparation. The person in this stage moves from thinking about treatment to planning what action to take.45 As the individual prepares to enter treatment, indecision tends to resurface, as well as self-doubt about his or her ability to change. During this stage, it is important for the provider to spell out goals (abstinence) and strategies (eg, counseling, medication) and enhance a sense of self-efficacy.
Action and maintenance. Patients in these stages engage in treatment and employ new strategies to abstain from opioid use. Maintaining these behaviors can be a daily struggle. Expressing confidence in the patient’s ability to abstain from use will support his or her progress. Behavioral interventions such as strategic avoidance of triggers and engagement in alternative activities (eg, support groups, exercise, faith-based practices) will help to maintain abstinence.
A CHRONIC CONDITION
Opioid use disorder, like many chronic illnesses, requires long-term attention to attain successful patient outcomes. The opioid agonists methadone and buprenorphine are the mainstay of treatment for it, conferring benefits such as reducing opioid use and preventing relapse.
Candidates for opioid agonist therapy should undergo a multidisciplinary assessment, including an evaluation on the patient’s readiness to change his or her opioid use.
Patient education should include a discussion of the risks of methadone (eg, respiratory depression, fatal overdose, and QTc prolongation) and buprenorphine (eg hepatotoxicity) and their benefits (eg, controlling craving, decreasing the risk of relapse). Patients should also be educated about overdose and diversion.
Despite the difficulties inherent in treating patients with opioid use disorder, when used appropriately, opioid agonist therapy can be lifesaving for patients struggling with long-term opioid addiction.
Acknowledgment: We thank Katelyn Colosi, BS, and Drs. Susan Wolfe, Dennis Bouffard, and Sinha Shirshendu for their helpful comments.
- Wakeman SE, Pham-Kanter G, Donelan K. Attitudes, practices, and preparedness to care for patients with substance use disorder: results from a survey of general internists. Subst Abus 2016; 37:635–641.
- Samuels EA, Dwyer K, Mello MJ, Baird J, Kellogg AR, Bernstein E. Emergency department-based opioid harm reduction: moving physicians from willing to doing. Acad Emerg Med 2016; 23:455–465.
- Mohlman MK, Tanzman B, Finison K, Pinette M, Jones C. Impact of medication-assisted treatment for opioid addiction on Medicaid expenditures and health services utilization rates in Vermont. J Subst Abuse Treat 2016; 67:9–14.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition. Arlington, VA, American Psychiatric Association, 2013.
- Center for Behavioral Health Statistics and Quality. Behavioral health trends in the United States: results from the 2014 National Survey on Drug Use and Health. www.samhsa.gov/data. Accessed April 6, 2017.
- Compton WM, Jones CM, Baldwin GT. Relationship between nonmedical prescription-opioid use and heroin use. N Engl J Med 2016; 374:154–163.
- Ruan X, Wyche MQ, Kaye AD. Analyzing the relationship between nonmedical prescription-opioid use and heroin use. J Opioid Manage 2016; 12:11–14.
- Hser YI, Evans E, Grella C, Ling W, Anglin D. Long-term course of opioid addiction. Harv Rev Psychiatry 2015; 23:76–89.
- Nielsen S, Hillhouse M, Mooney L, Ang A, Ling W. Buprenorphine pharmacotherapy and behavioral treatment: comparison of outcomes among prescription opioid users, heroin users and combination users. J Subst Abuse Treat 2015; 48:70–76.
- Moore BA, Fiellin DA, Barry DT, et al. Primary care office-based buprenorphine treatment: comparison of heroin and prescription opioid dependent patients. J Gen Intern Med 2007; 22:527–530.
- Fischer B, Patra J, Cruz MF, Gittins J, Rehm J. Comparing heroin users and prescription opioid users in a Canadian multi-site population of illicit opioid users. Drug Alcohol Rev 2008; 27:625–632.
- Compton WM, Jones CM, Baldwin GT. Relationship between nonmedical prescription-opioid use and heroin use. N Engl J Med 2016; 374:154–163.
- Jones CM, Baldwin GT, Manocchio T, White JO, Mack KA. Trends in methadone distribution for pain treatment, methadone diversion, and overdose deaths—United States, 2002–2014. MMWR Morb Mortal Wkly Rep 2016; 65:667–671.
- Baxter LE Sr, Campbell A, Deshields M, et al. Safe methadone induction and stabilization: report of an expert panel. J Addict Med 2013; 7:377–386.
- Alinejad S, Kazemi T, Zamani N, Hoffman RS, Mehrpour O. A systematic review of the cardiotoxicity of methadone. EXCLI J 2015; 14:577–600.
- Johnson RE, Strain EC, Amass L. Buprenorphine: how to use it right. Drug Alcohol Depend 2003; 70(suppl 2):S59–S77.
- Ling W. Buprenorphine implant for opioid addiction. Pain Manage 2012; 2:345–350.
- Saxon AJ, Hser YI, Woody G, Ling W. Medication-assisted treatment for opioid addiction: methadone and buprenorphine. J Food Drug Anal 2013; 21:S69–S72.
- Kimber J, Larney S, Hickman M, Randall D, Degenhardt L. Mortality risk of opioid substitution therapy with methadone versus buprenorphine: a retrospective cohort study. Lancet Psychiatry 2015; 2:901–908.
- Marteau D, McDonald R, Patel K. The relative risk of fatal poisoning by methadone or buprenorphine within the wider population of England and Wales. BMJ Open 2015; 5: e007629.
- Kintz P. Deaths involving buprenorphine: a compendium of French cases. Forensic Sci Int 2001; 121:65–69.
- Zuin M, Giorgini A, Selmi C, et al. Acute liver and renal failure during treatment with buprenorphine at therapeutic dose. Dig Liver Dis 2009; 41:e8–e10.
- Klein JW. Pharmacotherapy for substance use disorders. Med Clin North Am 2016; 100:891–910.
- Modesto-Lowe V, Van Kirk J. Clinical uses of naltrexone: a review of the evidence. Exp Clin Psychopharmocol 2002; 10:213–227.
- Woody GE. Agonist models for treating persons with substance use disorders. Curr Psychiatry Rep 2014; 16:489.
- Sanders JJ, Roose RJ, Lubrano MC, Lucan SC. Meaning and methadone: patient perceptions of methadone dose and a model to promote adherence to maintenance treatment. J Addict Med 2013; 7:307–313.
- Herget G. Methadone and buprenorphine added to the WHO list of essential medicines. HIV/AIDS Policy Law Rev 2005; 10:23–24.
- Suzuki J, Dodds T. Clinical recommendation of 12-step meeting attendance and discussion regarding disclosure of buprenorphine use among patients in office-based opioid treatment. Subst Abus 2016; 37:31–34.
- Rettig RA, Yarmolinsky A. Federal Regulation of Methadone Treatment. Washington, DC: National Academies Press; 1995.
- Srivastava A, Kahan M, Nader M. Primary care management of opioid use disorders: abstinence, methadone, or buprenorphine-naloxone? Can Fam Physician 2017; 63:200–205.
- Substance Abuse and Mental Health Services Administration. Federal Guidelines for Opioid Treatment Programs. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2015.
- Substance Abuse and Mental Health Services Administration SAMSHA. Buprenorphine waiver management. www.samhsa.gov/medication-assisted-treatment/buprenorphine-waiver-management. Accessed April 6, 2017.
- Mark TL, Lubran R, McCance-Kats EF, Chalk M, Richardson J. Medicaid coverage of medications to treat alcohol and opioid dependence. J Subst Abuse Treat 2015; 55:1–5.
- Johnson B, Richert T. Diversion of methadone and buprenorphine from opioid substitution treatment: the importance of patients’ attitudes and norms. J Subst Abuse Treat 2015; 54:50–55.
- Yokell MA, Zaller ND, Green TC, Rich JD. Buprenorphine and buprenorphine/naloxone diversion, misuse, and illicit use: an international review. Curr Drug Abuse Rev 2011; 4:28–41.
- Schuman-Olivier Z, Albanese M, Nelson SE, et al. Self-treatment: illicit buprenorphine use by opioid-dependent treatment seekers. J Subst Abuse Treat 2010; 39:41–50.
- American Society of Addiction Medicine. National practice guidelines for the use of medications in the treatment of addiction involving opioid use. www.asam.org/docs/default-source/practice-support/guidelines-and-consensus-docs/asam-national-practice-guideline-supplement.pdf. Accessed April 6, 2017.
- McNicholas L. Clinical guidelines for the use of buprenorphine in the treatment of opioid addiction. Rockville, MD: US Department of Health and Human Services, Substance Abuse and Mental Health Service Administration; 2004.
- Center for Substance Abuse Treatment. Clinical guidelines for the use of buprenorphine in the treatment of opioid addiction. Rockville (MD): Substance Abuse and Mental Health Services Administration (US); 2004. (Treatment Improvement Protocol (TIP) Series, No. 40.) www.ncbi.nlm.nih.gov/books/NBK64245. Accessed April 6, 2017.
- Zippel-Schultz B, Specka M, Cimander K, et al. Outcomes of patients in long-term opioid maintenance treatment. Subst Use Misuse 2016; 51:1493–1503.
- Martins SS, Keyes KM, Storr CL, Zhu H, Chilcoat HD. Pathways between nonmedical opioid use/dependence and psychiatric disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Drug Alcohol Depend 2009; 103:16–24.
- Khantzian EJ. The self-medication hypothesis of addictive disorders: focus on heroin and cocaine dependence. Am J Psychiatry 1985; 142:1259–1264.
- Belding MA, Iguchi MY, Lamb RJ, Lakin M, Terry R. Stages and processes of change among polydrug users in methadone maintenance treatment. Drug Alcohol Depend 1995; 39:45–53.
- Peteet JR, Brenner S, Curtiss D, Ferrigno M, Kauffman J. A stage of change approach to addiction in the medical setting. Gen Hosp Psychiatry 1998; 20:267–273.
- Vijay A, Bazazi AR, Yee I, Kamarulzaman A, Altice FL. Treatment readiness, attitudes toward, and experiences with methadone and buprenorphine maintenance therapy among people who inject drugs in Malaysia. J Subst Abuse Treat 2015; 54:29–36.
- Wakeman SE, Pham-Kanter G, Donelan K. Attitudes, practices, and preparedness to care for patients with substance use disorder: results from a survey of general internists. Subst Abus 2016; 37:635–641.
- Samuels EA, Dwyer K, Mello MJ, Baird J, Kellogg AR, Bernstein E. Emergency department-based opioid harm reduction: moving physicians from willing to doing. Acad Emerg Med 2016; 23:455–465.
- Mohlman MK, Tanzman B, Finison K, Pinette M, Jones C. Impact of medication-assisted treatment for opioid addiction on Medicaid expenditures and health services utilization rates in Vermont. J Subst Abuse Treat 2016; 67:9–14.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition. Arlington, VA, American Psychiatric Association, 2013.
- Center for Behavioral Health Statistics and Quality. Behavioral health trends in the United States: results from the 2014 National Survey on Drug Use and Health. www.samhsa.gov/data. Accessed April 6, 2017.
- Compton WM, Jones CM, Baldwin GT. Relationship between nonmedical prescription-opioid use and heroin use. N Engl J Med 2016; 374:154–163.
- Ruan X, Wyche MQ, Kaye AD. Analyzing the relationship between nonmedical prescription-opioid use and heroin use. J Opioid Manage 2016; 12:11–14.
- Hser YI, Evans E, Grella C, Ling W, Anglin D. Long-term course of opioid addiction. Harv Rev Psychiatry 2015; 23:76–89.
- Nielsen S, Hillhouse M, Mooney L, Ang A, Ling W. Buprenorphine pharmacotherapy and behavioral treatment: comparison of outcomes among prescription opioid users, heroin users and combination users. J Subst Abuse Treat 2015; 48:70–76.
- Moore BA, Fiellin DA, Barry DT, et al. Primary care office-based buprenorphine treatment: comparison of heroin and prescription opioid dependent patients. J Gen Intern Med 2007; 22:527–530.
- Fischer B, Patra J, Cruz MF, Gittins J, Rehm J. Comparing heroin users and prescription opioid users in a Canadian multi-site population of illicit opioid users. Drug Alcohol Rev 2008; 27:625–632.
- Compton WM, Jones CM, Baldwin GT. Relationship between nonmedical prescription-opioid use and heroin use. N Engl J Med 2016; 374:154–163.
- Jones CM, Baldwin GT, Manocchio T, White JO, Mack KA. Trends in methadone distribution for pain treatment, methadone diversion, and overdose deaths—United States, 2002–2014. MMWR Morb Mortal Wkly Rep 2016; 65:667–671.
- Baxter LE Sr, Campbell A, Deshields M, et al. Safe methadone induction and stabilization: report of an expert panel. J Addict Med 2013; 7:377–386.
- Alinejad S, Kazemi T, Zamani N, Hoffman RS, Mehrpour O. A systematic review of the cardiotoxicity of methadone. EXCLI J 2015; 14:577–600.
- Johnson RE, Strain EC, Amass L. Buprenorphine: how to use it right. Drug Alcohol Depend 2003; 70(suppl 2):S59–S77.
- Ling W. Buprenorphine implant for opioid addiction. Pain Manage 2012; 2:345–350.
- Saxon AJ, Hser YI, Woody G, Ling W. Medication-assisted treatment for opioid addiction: methadone and buprenorphine. J Food Drug Anal 2013; 21:S69–S72.
- Kimber J, Larney S, Hickman M, Randall D, Degenhardt L. Mortality risk of opioid substitution therapy with methadone versus buprenorphine: a retrospective cohort study. Lancet Psychiatry 2015; 2:901–908.
- Marteau D, McDonald R, Patel K. The relative risk of fatal poisoning by methadone or buprenorphine within the wider population of England and Wales. BMJ Open 2015; 5: e007629.
- Kintz P. Deaths involving buprenorphine: a compendium of French cases. Forensic Sci Int 2001; 121:65–69.
- Zuin M, Giorgini A, Selmi C, et al. Acute liver and renal failure during treatment with buprenorphine at therapeutic dose. Dig Liver Dis 2009; 41:e8–e10.
- Klein JW. Pharmacotherapy for substance use disorders. Med Clin North Am 2016; 100:891–910.
- Modesto-Lowe V, Van Kirk J. Clinical uses of naltrexone: a review of the evidence. Exp Clin Psychopharmocol 2002; 10:213–227.
- Woody GE. Agonist models for treating persons with substance use disorders. Curr Psychiatry Rep 2014; 16:489.
- Sanders JJ, Roose RJ, Lubrano MC, Lucan SC. Meaning and methadone: patient perceptions of methadone dose and a model to promote adherence to maintenance treatment. J Addict Med 2013; 7:307–313.
- Herget G. Methadone and buprenorphine added to the WHO list of essential medicines. HIV/AIDS Policy Law Rev 2005; 10:23–24.
- Suzuki J, Dodds T. Clinical recommendation of 12-step meeting attendance and discussion regarding disclosure of buprenorphine use among patients in office-based opioid treatment. Subst Abus 2016; 37:31–34.
- Rettig RA, Yarmolinsky A. Federal Regulation of Methadone Treatment. Washington, DC: National Academies Press; 1995.
- Srivastava A, Kahan M, Nader M. Primary care management of opioid use disorders: abstinence, methadone, or buprenorphine-naloxone? Can Fam Physician 2017; 63:200–205.
- Substance Abuse and Mental Health Services Administration. Federal Guidelines for Opioid Treatment Programs. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2015.
- Substance Abuse and Mental Health Services Administration SAMSHA. Buprenorphine waiver management. www.samhsa.gov/medication-assisted-treatment/buprenorphine-waiver-management. Accessed April 6, 2017.
- Mark TL, Lubran R, McCance-Kats EF, Chalk M, Richardson J. Medicaid coverage of medications to treat alcohol and opioid dependence. J Subst Abuse Treat 2015; 55:1–5.
- Johnson B, Richert T. Diversion of methadone and buprenorphine from opioid substitution treatment: the importance of patients’ attitudes and norms. J Subst Abuse Treat 2015; 54:50–55.
- Yokell MA, Zaller ND, Green TC, Rich JD. Buprenorphine and buprenorphine/naloxone diversion, misuse, and illicit use: an international review. Curr Drug Abuse Rev 2011; 4:28–41.
- Schuman-Olivier Z, Albanese M, Nelson SE, et al. Self-treatment: illicit buprenorphine use by opioid-dependent treatment seekers. J Subst Abuse Treat 2010; 39:41–50.
- American Society of Addiction Medicine. National practice guidelines for the use of medications in the treatment of addiction involving opioid use. www.asam.org/docs/default-source/practice-support/guidelines-and-consensus-docs/asam-national-practice-guideline-supplement.pdf. Accessed April 6, 2017.
- McNicholas L. Clinical guidelines for the use of buprenorphine in the treatment of opioid addiction. Rockville, MD: US Department of Health and Human Services, Substance Abuse and Mental Health Service Administration; 2004.
- Center for Substance Abuse Treatment. Clinical guidelines for the use of buprenorphine in the treatment of opioid addiction. Rockville (MD): Substance Abuse and Mental Health Services Administration (US); 2004. (Treatment Improvement Protocol (TIP) Series, No. 40.) www.ncbi.nlm.nih.gov/books/NBK64245. Accessed April 6, 2017.
- Zippel-Schultz B, Specka M, Cimander K, et al. Outcomes of patients in long-term opioid maintenance treatment. Subst Use Misuse 2016; 51:1493–1503.
- Martins SS, Keyes KM, Storr CL, Zhu H, Chilcoat HD. Pathways between nonmedical opioid use/dependence and psychiatric disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Drug Alcohol Depend 2009; 103:16–24.
- Khantzian EJ. The self-medication hypothesis of addictive disorders: focus on heroin and cocaine dependence. Am J Psychiatry 1985; 142:1259–1264.
- Belding MA, Iguchi MY, Lamb RJ, Lakin M, Terry R. Stages and processes of change among polydrug users in methadone maintenance treatment. Drug Alcohol Depend 1995; 39:45–53.
- Peteet JR, Brenner S, Curtiss D, Ferrigno M, Kauffman J. A stage of change approach to addiction in the medical setting. Gen Hosp Psychiatry 1998; 20:267–273.
- Vijay A, Bazazi AR, Yee I, Kamarulzaman A, Altice FL. Treatment readiness, attitudes toward, and experiences with methadone and buprenorphine maintenance therapy among people who inject drugs in Malaysia. J Subst Abuse Treat 2015; 54:29–36.
KEY POINTS
- Opioid use disorder is potentially lethal and has become more prevalent in the United States over the past few decades.
- The opioid agonist methadone and the partial agonist buprenorphine are the currently recommended treatments for patients who need opioid maintenance therapy. However, they carry the risk of adverse effects (eg, respiratory depression, QTc interval prolongation, hepatotoxicity), diversion, and overdose.
- Patients being considered for opioid agonist therapy need a comprehensive assessment including a thorough medical history and physical examination, psychiatric evaluation, psychosocial appraisal, and determination of readiness to change.
- When methadone and buprenorphine are properly prescribed they confer significant benefits, including reduction or elimination of opioid use, reductions in overdose risk, and positive changes in behavior and lifestyle.
Preventing herpes zoster through vaccination: New developments
Herpes zoster (HZ), or shingles, represents a reactivation of the varicella-zoster virus (VZV). Following primary infection, usually in childhood, the virus typically lies dormant in the dorsal root and sensory nerve ganglia for decades. The precise mechanism of reactivation is not well understood, but it is associated with a decline in cell-mediated immunity that occurs with advancing age, immune-compromising conditions such as HIV infection and cancer, or immunosuppressive therapies, including corticosteroids.1 HZ is usually a self-limited disease characterized by unilateral dermatomal rash and pain, but can cause disseminated infection in immunocompromised individuals.2
Treatment with antiviral medications within 72 hours of rash onset can reduce acute HZ symptoms.1 However, antiviral agents are only minimally effective in preventing postherpetic neuralgia, the most common complication of HZ.3 Therefore, efforts to reduce the burden of HZ morbidity have focused on prevention through vaccination.
Currently, the only shingles vaccine approved by the US Food and Drug Administration (FDA) is Zostavax (Merck), which contains the live-attenuated Oka strain of VZV at a concentration 14 times greater than that of the varicella vaccine (Varivax, Merck). The live-attenuated vaccine boosts VZV-specific cell-mediated immunity, preventing reactivation of the latent virus.
In this article, we describe the burden of disease and review recent developments in the literature on HZ vaccine, including duration of efficacy, uptake and barriers to vaccination, cost-effectiveness, and the outlook for future vaccines.
INCIDENCE INCREASES WITH AGE
The incidence of herpes zoster in the general population is between 3 and 5 per 1,000 person-years4 and increases with age, especially after age 60 when the incidence can approach 13 to 15 per 1,000 person-years.5,6 An estimated 1 million new cases occur each year in the United States, and about 6% of patients experience a second episode of HZ within 8 years.7,8 In immunocompromised patients, the incidence of HZ is 2 to 10 times higher than in the general population.9
The incidence of HZ has been increasing for reasons that are unclear. After varicella vaccine was introduced into the routine childhood immunization schedule in 1995, it was hypothesized that the resultant decrease in primary varicella infections would remove a natural source of immune boosting and cause an increase in HZ incidence for up to 20 years.10 However, recent studies demonstrate that the observed increase in HZ incidence actually predates the introduction of varicella vaccine,11–13 and the widespread use of varicella vaccine has not resulted in an increase in the incidence of HZ.14
Other potential explanations for the rise in reported incidence include increasing awareness among patients, who might previously not have sought care and among physicians, who may be more likely to make the diagnosis. Advertisement of new treatments for HZ, including gabapentin and capsaicin, probably began to increase awareness in the 1990s, as did promotion of the HZ vaccine after its licensure in 2006.
HZ can occur in people who have been vaccinated against varicella due to reactivation of the vaccine-strain virus, but the risk is lower than after infection with wild-type varicella.15 Given that the varicella vaccine has been routinely used in children for only 20 years, the long-term effect of varicella vaccination on the incidence of HZ in elderly people is unknown.
Serious complications
HZ can cause rare but serious complications including encephalitis, herpes ophthalmicus, herpes oticus, myelitis, and retinitis.1 These can lead to long-term disability including unilateral blindness and deafness.
The most common and debilitating complication is postherpetic neuralgia, a persistent pain lasting at least 3 months, with a mean duration of 3.3 years and sometimes as long as 10 years.16 Postherpetic neuralgia occurs in 8% to 32% of patients after acute HZ,4 and the incidence increases with age, being most common after age 70. The chronic pain of postherpetic neuralgia has a significant adverse impact on patients’ quality of life, including physical disability and emotional distress.17 Some pain is intense, and anecdotal reports of patients committing suicide were included in the Advisory Committee on Immunization Practices (ACIP) recommendations regarding herpes zoster vaccine.18
HZ and its complications also impose a substantial economic burden on society.19 In a population-based study, the mean direct medical costs of HZ ranged from $620 to $1,160 (2015 dollars) depending on age,20 and the mean costs of postherpetic neuralgia were 2 to 5 times higher than that.20–22 Immunocompromised patients had costs 2 to 3 times higher than those of immunocompetent adults.23 In addition, for employed patients, HZ resulted in an average loss of 32 hours of work due to absenteeism and 84 hours due to presenteeism (ie, working while sick and therefore suboptimally).24
Assuming there are 1 million cases of HZ each year, if 8% to 32% of patients go on to develop postherpetic neuralgia, that would translate into approximately $1 to $2 billion in direct medical costs. With 60% of adult patients working,25 at an average wage of $23.23 per hour,26 HZ illness could be responsible for another $1.6 billion in lost productivity.
EFFICACY AND SAFETY OF HZ VACCINE
In 2006, the FDA approved the live-attenuated Oka strain VZV vaccine for prevention of HZ and postherpetic neuralgia in adults age 60 and older based on findings from the Shingles Prevention Study (SPS).27
The Shingles Prevention Study
This multicenter randomized placebo-controlled trial27 enrolled 38,546 immunocompetent persons age 60 and older. Subjects in the intervention group received a single dose of live-attenuated vaccine, and all participants were followed for up to 4.9 years after vaccination.
 vaccines")
HZ occurred in 315 (1.636%) of the 19,254 participants in the vaccine group and in 642 (3.336%) of the 19,247 participants in the placebo group, an absolute risk reduction of 1.7%, number needed to treat 59, relative risk reduction 51%, P < .001. Similarly, postherpetic neuralgia occurred in 27 (0.140%) of the 19,254 vaccine recipients and in 80 (0.416%) of the placebo recipients (an absolute risk reduction of 0.276%, number needed to treat 362, relative risk reduction 66%, P < .001). The investigators calculated that vaccination reduced the overall burden of illness by 61% (Table 1).
The efficacy against HZ incidence decreased with age,28 but the efficacy against postherpetic neuralgia did not. In addition, vaccine recipients who developed HZ generally had less severe manifestations.
The safety of the vaccine was assessed for all participants in the SPS. In addition, one-sixth of SPS participants were enrolled in a safety substudy. These participants completed a detailed report card regarding all medically important events within the first 42 days. Forty-eight percent of the vaccine group and 17% of the placebo group (P < .05) experienced adverse events, primarily at the injection site. Less than 1% of all local reactions were severe.29 Serious adverse events were rare (< 2%), but occurred significantly more often in the vaccinated group.
Short-Term Persistence Substudy
Short-term efficacy of the live-attenuated vaccine (up to 7 years) was assessed in the Short-Term Persistence Substudy (STPS), which involved 14,270 of the initial participants and reported yearly and overall vaccine efficacy.30 After 5 years, the yearly efficacy against postherpetic neuralgia incidence declined to 32% and was no longer statistically significant. Efficacy against HZ incidence and burden of illness displayed the same pattern. After the end of the STPS, all subjects in the placebo group received vaccination.
Long-Term Persistence Substudy
Those in the intervention group were followed for an additional 4 years in the Long-Term Persistence Substudy (LTPS).31 Due to the lack of concurrent controls in the LTPS, the authors used regression models based on historical controls to estimate contemporary population incidence of HZ and postherpetic neuralgia for comparison.
Efficacy continued to decline over time, and by 10 years after vaccination there was no difference between vaccinated patients and historical controls in the rate of any end point (ie, efficacy declined to zero).
A trial of booster vaccination
Because many patients are vaccinated at age 60, waning immunity could leave them vulnerable to HZ and postherpetic neuralgia by age 70. A potential solution would be to give a booster dose after 10 years.
A recent phase 3 clinical trial of adults age 70 years and older found that a booster dose of live-attenuated vaccine was as safe and immunogenic as an initial dose.32 While antibody responses were similar in the boosted group and the newly vaccinated group, cell-mediated immunity was higher in the boosted group.
Because prevention of HZ is generally via cell-mediated immunity, the booster might be more effective than the initial vaccination, but clinical trials measuring actual cases prevented will be required to prove it. A booster dose is not currently recommended.
A trial of vaccination in adults 50 to 59
In 2011, the FDA extended its approval of HZ vaccine for use in adults ages 50 to 59.33
In a randomized, double-blind, placebo-controlled trial in this age group,33 the vaccine reduced HZ incidence by almost 70% (absolute risk reduction 0.614%, number needed to treat 156; Table 1), but the severity of HZ cases was not affected. There were too few cases of postherpetic neuralgia to assess the efficacy for this end point. The study followed patients for only 1.5 years after vaccination, so the duration of efficacy is unknown.
As in the older recipients, the vaccine was well tolerated; injection-site reactions and headache were the major adverse effects reported among vaccine recipients.33
INDICATIONS AND CONTRAINDICATIONS
Although HZ vaccine is licensed for use in adults age 50 and older, the ACIP recommends it only for immunocompetent adults age 60 and older. At this time, the ACIP does not recommend HZ vaccine in those younger than 60 because of the low risk of HZ in this age group.34
Any person age 60 or older should receive a single dose of the live-attenuated HZ vaccine subcutaneously, regardless of past history of HZ.
The vaccine is contraindicated in patients who have a history of allergic reaction to any vaccine component, immunosuppression or immunodeficiency conditions, and pregnancy. Specifically, people who will receive immunosuppressive therapies should have the vaccine at least 14 days before beginning treatment. Antiviral medications such as acyclovir, famciclovir, and valacyclovir should be discontinued at least 24 hours before vaccination and not resumed until 14 days later. Patients taking high-dose corticosteroids for more than 2 weeks should not be vaccinated until at least 1 month after therapy is completed.
In contrast, HZ vaccine is not contraindicated for leukemia patients who are in remission and who have not received chemotherapy or radiation for at least 3 months, or for patients receiving short-term, low-to-moderate dose, topical, intra-articular, bursal, or tendon injections of corticosteroids. Patients on low-dose methotrexate, azathioprine, or 6-mercaptopurine can also receive the vaccine.18
VACCINATION RATES ARE LOW

Although the vaccine has been recommended since 2008, uptake has been slow. Figure 1 shows the rate of HZ vaccination in adults age 60 and older surveyed in the National Health Interview Survey from 2007 to 2013.35 Eight years after the vaccine was licensed, only 28% of eligible patients had been vaccinated. Assuming the current rate of increase remains constant, it will take 7 more years to reach a 60% coverage rate—the same as for pneumococcal vaccine36—and 18 years to reach universal coverage.
Barriers to vaccination
Several barriers to HZ vaccination might account for the slow uptake.
For the first few years the vaccine was available, the requirement to store it frozen presented an obstacle for some physicians.37 Physicians may also have been discouraged by the cumbersome Medicare reimbursement process because while the administration fee is covered through Medicare Part B, the live-attenuated vaccine is reimbursed only through Medicare Part D, a benefit that varies by plans. Other barriers to physicians are supply shortages, high up-front costs, and uncertainties regarding the duration of vaccine protection, its safety, and side effects.38–40
Patient barriers include lack of physician recommendation, lack of familiarity with the vaccine, high out-of-pocket costs, the perception that they are at low risk for HZ, underestimation of the pain associated with HZ and postherpetic neuralgia, and fear of vaccine adverse effects.39,41,42
Interventions to increase vaccination rates
Certain interventions have been shown to increase vaccination adherence in general and HZ vaccination in particular. In randomized trials involving other vaccines, electronic medical record reminders supporting panel management or nurse-initiated protocols have been proven to increase vaccination rates, but these methods have not been tested for HZ vaccine specifically.43,44
In an observational study, Chaudhry et al found that the number of HZ vaccinations administered at the Mayo Clinic increased 43% in one practice and 54% in another after the implementation of an electronic alert.45 A randomized controlled trial showed that an informational package discussing HZ and the vaccine sent to patients via either their electronic personal health record or traditional mail increased HZ vaccination by almost 3 times.46
Pharmacists can also influence vaccination rates. States that provide full immunization privileges to pharmacists have vaccination rates significantly higher than states with restricted or no authorization.47
COST-EFFECTIVENESS CONSIDERATIONS
Unlike the Centers for Medicare and Medicaid Services, the ACIP does consider cost-effectiveness in their vaccine recommendations. Because of the morbidity associated with HZ and postherpetic neuralgia as well as the economic impact, vaccination is generally considered cost-effective for adults age 60 and older.48,49
Analyses have demonstrated that cost-effectiveness hinges on 4 factors: initial vaccine efficacy, the duration of efficacy, the age-specific incidence of HZ, and the cost of the vaccine.
For patients ages 50 to 59, the incidence of HZ is low, and because the duration of vaccine efficacy is short even though initial vaccine efficacy is high, vaccination in this age group offers poor value.50 At older ages, the incidence of HZ and postherpetic neuralgia rises, making vaccination more cost-effective. After age 60, the vaccine is cost-effective at all ages, although age 70 appears to offer the optimal trade-off between increasing incidence and declining vaccine efficacy.48,49
For patients who plan to be vaccinated only once, waiting until age 70 would appear to offer the best value.51 For those who are willing to receive a booster dose, the optimal age for vaccination is unknown, but will likely depend on the effectiveness, cost, and duration of the booster.
A NEW HZ VACCINE
In 2015, GlaxoSmithKline tested a new HZ vaccine containing a single VZV glycoprotein in an AS01B adjuvant system (HZ/su vaccine).52 In a phase 3 randomized trial involving 15,411 immunocompetent persons age 50 and older, a 2-dose schedule of HZ/su vaccine was 97% effective in preventing HZ (Table 1).53 Importantly, the vaccine was equally effective in older patients.
This vaccine also had a high rate of adverse reactions, with 17% of vaccine recipients vs 3% of placebo recipients reporting events that prevented normal everyday activities for at least 1 day. However, the rate of serious adverse reactions was the same in both groups (9%). The company announced that they intended to submit a regulatory application for HZ/su vaccine in the second half of 2016.54
Because of its high efficacy, HZ/su vaccine has the potential to change practice, but several issues must be resolved before it can supplant the current vaccine.
First, the AS01B adjuvant is not currently licensed in the United States, so it is unclear if the HZ/su vaccine can get FDA approval.52,55
Second, there are several questions about the efficacy of the vaccine, including long-term efficacy, efficacy in the elderly, and efficacy in the case of a patient receiving only 1 of the 2 required doses.
Third, the impact of HZ/su vaccine on complications such as postherpetic neuralgia has not been established. The clinical trial (NCT01165229) examining vaccine efficacy against postherpetic neuralgia incidence and other complications in adults age 70 and older has recently been completed and data should be available soon. Given the extremely high efficacy against HZ, it is likely that it will be close to 100% effective against this complication.
Fourth, there is uncertainty as to how the HZ/su vaccine should be used in patients who have already received the live-attenuated vaccine, if it is determined that a booster is necessary.
Finally, the vaccine is not yet priced. Given its superior effectiveness, particularly in older individuals, competitive pricing could dramatically affect the market. How Medicare or other insurers cover the new vaccine will likely influence its acceptance.
HZ VACCINATION OF IMMUNOCOMPROMISED PATIENTS
Immunocompromised patients are at highest risk for developing HZ. Unfortunately, there are currently no HZ vaccines approved for use in this population. The current live-attenuated vaccine has been demonstrated to be safe, well tolerated, and immunogenic in patients age 60 and older who are receiving chronic or maintenance low to moderate doses of corticosteroids.56
A clinical trial is being conducted to assess the immunogenicity, clinical effectiveness, and safety of the vaccine in rheumatoid arthritis patients receiving antitumor necrosis factor therapy (NCT01967316). Other trials are examining vaccine efficacy and safety in patients with solid organ tumors prior to chemotherapy (NCT02444936) and in patients who will be undergoing living donor kidney transplantation (NCT00940940). Researchers are also investigating the possibility of vaccinating allogeneic stem cell donors before donation in order to protect transplant recipients against HZ (NCT01573182).
ZVHT and HZ/su vaccination in immunocompromised patients
Heat-treated varicella-zoster vaccine (ZVHT) is a potential alternative for immunocompromised patients. A 4-dose regimen has been proven to reduce the risk of HZ in patients receiving autologous hematopoietic-cell transplants for non-Hodgkin or Hodgkin lymphoma.57
In another trial, the 4-dose ZVHT was safe and elicited significant VZV-specific T-cell response through 28 days in immunosuppressed patients with solid tumor malignancy, hematologic malignancy, human immunodeficiency virus infection with CD4 counts of 200 cells/mm3 or less, and autologous hematopoietic-cell transplants. The T-cell response was poor in allogeneic hematopoietic-cell transplant recipients, however.58
Because the HZ/su vaccine does not contain live virus, it seems particularly promising for immunocompromised patients. In phase 1 and 2 studies, a 3-dose regimen has been shown to be safe and immunogenic in hematopoietic-cell transplant recipients and HIV-infected adults with CD4 count higher than 200 cells/mm3.59,60 A phase 3 trial assessing the efficacy of HZ/su vaccine in autologous hematopoietic-cell transplant recipients is under way (NCT01610414). Changes in recommendations for HZ vaccine in these most vulnerable populations await the results of these studies.
- Dworkin RH, Johnson RW, Breuer J, et al. Recommendations for the management of herpes zoster. Clin Infect Dis 2007; 44(suppl 1):S1–S26.
- Johnson RW. Herpes zoster and postherpetic neuralgia. Expert Rev Vaccines 2010; 9(suppl):21–26.
- Chen N, Li Q, Yang J, Zhou M, Zhou D, He L. Antiviral treatment for preventing postherpetic neuralgia. Cochrane Database Syst Rev 2014; 2:CD006866.
- Kawai K, Gebremeskel BG, Acosta CJ. Systematic review of incidence and complications of herpes zoster: towards a global perspective. BMJ Open 2014; 4:e004833.
- Tseng HF, Smith N, Harpaz R, Bialek SR, Sy LS, Jacobsen SJ. Herpes zoster vaccine in older adults and the risk of subsequent herpes zoster disease. JAMA 2011; 305:160–166.
- Langan SM, Smeeth L, Margolis DJ, Thomas SL. Herpes zoster vaccine effectiveness against incident herpes zoster and post-herpetic neuralgia in an older US population: a cohort study. PLoS Med 2013; 10:e1001420.
- Yawn BP, Saddier P, Wollan PC, St. Sauver JL, Kurland MJ, Sy LS. A population-based study of the incidence and complication rates of herpes zoster before zoster vaccine introduction. Mayo Clin Proc 2007; 82:1341–1349.
- Yawn BP, Wollan PC, Kurland MJ, St. Sauver JL, Saddier P. Herpes zoster recurrences more frequent than previously reported. Mayo Clin Proc 2011; 86:88–93.
- Chen SY, Suaya JA, Li Q, et al. Incidence of herpes zoster in patients with altered immune function. Infection 2014; 42:325–334.
- Edmunds WJ, Brisson M. The effect of vaccination on the epidemiology of varicella zoster virus. J Infect 2002; 44:211–219.
- Hales CM, Harpaz R, Joesoef MR, Bialek SR. Examination of links between herpes zoster incidence and childhood varicella vaccination. Ann Intern Med 2013; 159:739–745.
- Leung J, Harpaz R, Molinari NA, Jumaan A, Zhou F. Herpes zoster incidence among insured persons in the United States, 1993-2006: evaluation of impact of varicella vaccination. Clin Infect Dis 2011; 52:332–340.
- Rimland D, Moanna A. Increasing incidence of herpes zoster among veterans. Clin Infect Dis 2010; 50:1000–1005.
- Jumaan AO, Yu O, Jackson LA, Bohlke K, Galil K, Seward JF. Incidence of herpes zoster, before and after varicella-vaccination-associated decreases in the incidence of varicella, 1992-2002. J Infect Dis 2005; 191:2002–2007.
- Plotkin SA, Starr SE, Connor K, Morton D. Zoster in normal children after varicella vaccine. J Infect Dis 1989; 159:1000–1001.
- Oster G, Harding G, Dukes E, Edelsberg J, Cleary PD. Pain, medication use, and health-related quality of life in older persons with postherpetic neuralgia: results from a population-based survey. J Pain 2005; 6:356–363.
- Johnson RW, Bouhassira D, Kassianos G, Leplege A, Schmader KE, Weinke T. The impact of herpes zoster and post-herpetic neuralgia on quality-of-life. BMC Med 2010; 8:37.
- Harpaz R, Ortega-Sanchez IR, Seward JF; Advisory Committee on Immunization Practices (ACIP) Centers for Disease Control and Prevention (CDC). Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2008; 57:1–30.
- Panatto D, Bragazzi NL, Rizzitelli E, et al. Evaluation of the economic burden of herpes zoster (HZ) infection. Hum Vaccin Immunother 2015; 11:245–262.
- Yawn BP, Itzler RF, Wollan PC, Pellissier JM, Sy LS, Saddier P. Health care utilization and cost burden of herpes zoster in a community population. Mayo Clin Proc 2009; 84:787–794.
- Dworkin RH, White R, O’Connor AB, Hawkins K. Health care expenditure burden of persisting herpes zoster pain. Pain Med 2008; 9:348–353.
- White RR, Lenhart G, Singhal PK, et al. Incremental 1-year medical resource utilization and costs for patients with herpes zoster from a set of US health plans. Pharmacoeconomics 2009; 27:781–792.
- Insinga RP, Itzler RF, Pellissier JM. Acute/subacute herpes zoster: healthcare resource utilisation and costs in a group of US health plans. Pharmacoeconomics 2007; 25:155–169.
- Singhal PK, Makin C, Pellissier J, Sy L, White R, Saddier P. Work and productivity loss related to herpes zoster. J Med Econ 2011; 14:639–645.
- US Bureau of Labor Statistics. Labor force statistics from the current population survey. www.bls.gov/web/empsit/cpseea13.htm. Accessed April 6, 2017.
- US Bureau of Labor Statistic. Occupational employment statistics. www.bls.gov/oes/current/oes_nat.htm. Accessed April 6, 2017.
- Oxman MN, Levin MJ, Johnson GR, et al; Shingles Prevention Study Group. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 2005; 352:2271–2284.
- Food and Drug Administration (FDA). FDA clinical briefing document for Merck & Co., Inc. Zoster vaccine live (Oka/Merck) Zostavax. www.fda.gov/ohrms/dockets/ac/05/briefing/5-4198b2_1.pdf. Accessed April 6, 2017.
- Simberkoff MS, Arbeit RD, Johnson GR, et al; Shingles Prevention Study Group. Safety of herpes zoster vaccine in the shingles prevention study: a randomized trial. Ann Intern Med 2010; 152:545–554.
- Schmader KE, Oxman MN, Levin MJ, et al; Shingles Prevention Study Group. Persistence of the efficacy of zoster vaccine in the shingles prevention study and the short-term persistence substudy. Clin Infect Dis 2012; 55:1320–1328.
- Morrison VA, Johnson GR, Schmader KE, et al; Shingles Prevention Study Group. Long-term persistence of zoster vaccine efficacy. Clin Infect Dis 2015; 60:900–909.
- Levin MJ, Schmader KE, Pang L, et al. Cellular and humoral responses to a second dose of herpes zoster vaccine administered 10 years after the first dose among older adults. J Infect Dis 2016; 213:14–22.
- Schmader KE, Levin MJ, Gnann JW Jr, et al. Efficacy, safety, and tolerability of herpes zoster vaccine in persons aged 50-59 years. Clin Infect Dis 2012; 54:922–928.
- Hales CM, Harpaz R, Ortega-Sanchez I, Bialek SR; Centers for Disease Control and Prevention (CDC). Update on recommendations for use of herpes zoster vaccine. MMWR Morb Mortal Wkly Rep 2014; 63:729–731.
- Centers for Disease Control and Prevention (CDC). Surveillance of vaccination coverage among adult populations—United States, 2014. MMWR Morb Mortal Wkly Rep 2016; 65(1):1–36. Accessed April 12, 2017.
- Williams WW, Lu PJ, O’Halloran A, et al; Centers for Disease Control and Prevention (CDC). Vaccination coverage among adults, excluding influenza vaccination—United States, 2013. MMWR Morb Mortal Wkly Rep 2015; 64:95–102.
- Oxman MN. Zoster vaccine: current status and future prospects. Clin Infect Dis 2010; 51:197–213.
- Hurley LP, Lindley MC, Harpaz R, et al. Barriers to the use of herpes zoster vaccine. Ann Intern Med 2010; 152:555–560.
- Lu PJ, Euler GL, Jumaan AO, Harpaz R. Herpes zoster vaccination among adults aged 60 years or older in the United States, 2007: uptake of the first new vaccine to target seniors. Vaccine 2009; 27:882–887.
- Hurley LP, Harpaz R, Daley MF, et al. National survey of primary care physicians regarding herpes zoster and the herpes zoster vaccine. J Infect Dis 2008; 197(suppl 2):S216–S223.
- Joon Lee T, Hayes S, Cummings DM, et al. Herpes zoster knowledge, prevalence, and vaccination rate by race. J Am Board Fam Med 2013; 26:45–51.
- Opstelten W, van Essen GA, Hak E. Determinants of non-compliance with herpes zoster vaccination in the community-dwelling elderly. Vaccine 2009; 27:192–196.
- Loo TS, Davis RB, Lipsitz LA, et al. Electronic medical record reminders and panel management to improve primary care of elderly patients. Arch Intern Med 2011; 171:1552–1558.
- Rhew DC, Glassman PA, Goetz MB. Improving pneumococcal vaccine rates. Nurse protocols versus clinical reminders. J Gen Intern Med 1999; 14:351–356.
- Chaudhry R, Schietel SM, North F, Dejesus R, Kesman RL, Stroebel RJ. Improving rates of herpes zoster vaccination with a clinical decision support system in a primary care practice. J Eval Clin Pract 2013; 19:263–266.
- Otsuka SH, Tayal NH, Porter K, Embi PJ, Beatty SJ. Improving herpes zoster vaccination rates through use of a clinical pharmacist and a personal health record. Am J Med 2013; 126:832.e1–832.e6.
- Taitel MS, Fensterheim LE, Cannon AE, Cohen ES. Improving pneumococcal and herpes zoster vaccination uptake: expanding pharmacist privileges. Am J Manag Care 2013; 19:e309–e313.
- Kawai K, Preaud E, Baron-Papillon F, Largeron N, Acosta CJ. Cost-effectiveness of vaccination against herpes zoster and postherpetic neuralgia: a critical review. Vaccine 2014; 32:1645–1653.
- Szucs TD, Pfeil AM. A systematic review of the cost effectiveness of herpes zoster vaccination. Pharmacoeconomics 2013; 31:125–136.
- Le P, Rothberg MB. Cost-effectiveness of herpes zoster vaccine for persons aged 50 years. Ann Intern Med 2015; 163:489–497.
- Le P, Rothberg MB. Determining the optimal age to vaccinate against herpes zoster: a cost-effectiveness analysis. Society for Medical Decision Making 37th Annual North American Meeting. St. Louis, MO; October 18-21, 2015.
- Cohen JI. Clinical practice: herpes zoster. N Engl J Med 2013; 369:255–263.
- Lal H, Cunningham AL, Godeaux O, et al; ZOE-50 Study Group. Efficacy of an adjuvanted herpes zoster subunit vaccine in older adults. N Engl J Med 2015; 372:2087–2096.
- GlaxoSmithKline plc. GSK’s candidate shingles vaccine demonstrates 90% efficacy against shingles in people 70 years of age and over. www.gsk.com/en-gb/media/press-releases/gsk-s-candidate-shingles-vaccine-demonstrates-90-efficacy-against-shingles-in-people-70-years-of-age-and-over/. Accessed April 6, 2017.
- Reed SG, Orr MT, Fox CB. Key roles of adjuvants in modern vaccines. Nat Med 2013; 19:1597–1608.
- Russell AF, Parrino J, Fisher CL Jr, et al. Safety, tolerability, and immunogenicity of zoster vaccine in subjects on chronic/maintenance corticosteroids. Vaccine 2015; 33:3129–3134.
- Hata A, Asanuma H, Rinki M, et al. Use of an inactivated varicella vaccine in recipients of hematopoietic-cell transplants. N Engl J Med 2002; 347:26–34.
- Mullane KM, Winston DJ, Wertheim MS, et al. Safety and immunogenicity of heat-treated zoster vaccine (ZVHT) in immunocompromised adults. J Infect Dis 2013; 208:1375–1385.
- Stadtmauer EA, Sullivan KM, Marty FM, et al. A phase 1/2 study of an adjuvanted varicella-zoster virus subunit vaccine in autologous hematopoietic cell transplant recipients. Blood 2014; 124:2921–2929.
- Berkowitz EM, Moyle G, Stellbrink HJ, et al. Safety and immunogenicity of an adjuvanted herpes zoster subunit candidate vaccine in HIV-infected adults: a phase 1/2a randomized, placebo-controlled study. J Infect Dis 2015; 211:1279–1287.
Herpes zoster (HZ), or shingles, represents a reactivation of the varicella-zoster virus (VZV). Following primary infection, usually in childhood, the virus typically lies dormant in the dorsal root and sensory nerve ganglia for decades. The precise mechanism of reactivation is not well understood, but it is associated with a decline in cell-mediated immunity that occurs with advancing age, immune-compromising conditions such as HIV infection and cancer, or immunosuppressive therapies, including corticosteroids.1 HZ is usually a self-limited disease characterized by unilateral dermatomal rash and pain, but can cause disseminated infection in immunocompromised individuals.2
Treatment with antiviral medications within 72 hours of rash onset can reduce acute HZ symptoms.1 However, antiviral agents are only minimally effective in preventing postherpetic neuralgia, the most common complication of HZ.3 Therefore, efforts to reduce the burden of HZ morbidity have focused on prevention through vaccination.
Currently, the only shingles vaccine approved by the US Food and Drug Administration (FDA) is Zostavax (Merck), which contains the live-attenuated Oka strain of VZV at a concentration 14 times greater than that of the varicella vaccine (Varivax, Merck). The live-attenuated vaccine boosts VZV-specific cell-mediated immunity, preventing reactivation of the latent virus.
In this article, we describe the burden of disease and review recent developments in the literature on HZ vaccine, including duration of efficacy, uptake and barriers to vaccination, cost-effectiveness, and the outlook for future vaccines.
INCIDENCE INCREASES WITH AGE
The incidence of herpes zoster in the general population is between 3 and 5 per 1,000 person-years4 and increases with age, especially after age 60 when the incidence can approach 13 to 15 per 1,000 person-years.5,6 An estimated 1 million new cases occur each year in the United States, and about 6% of patients experience a second episode of HZ within 8 years.7,8 In immunocompromised patients, the incidence of HZ is 2 to 10 times higher than in the general population.9
The incidence of HZ has been increasing for reasons that are unclear. After varicella vaccine was introduced into the routine childhood immunization schedule in 1995, it was hypothesized that the resultant decrease in primary varicella infections would remove a natural source of immune boosting and cause an increase in HZ incidence for up to 20 years.10 However, recent studies demonstrate that the observed increase in HZ incidence actually predates the introduction of varicella vaccine,11–13 and the widespread use of varicella vaccine has not resulted in an increase in the incidence of HZ.14
Other potential explanations for the rise in reported incidence include increasing awareness among patients, who might previously not have sought care and among physicians, who may be more likely to make the diagnosis. Advertisement of new treatments for HZ, including gabapentin and capsaicin, probably began to increase awareness in the 1990s, as did promotion of the HZ vaccine after its licensure in 2006.
HZ can occur in people who have been vaccinated against varicella due to reactivation of the vaccine-strain virus, but the risk is lower than after infection with wild-type varicella.15 Given that the varicella vaccine has been routinely used in children for only 20 years, the long-term effect of varicella vaccination on the incidence of HZ in elderly people is unknown.
Serious complications
HZ can cause rare but serious complications including encephalitis, herpes ophthalmicus, herpes oticus, myelitis, and retinitis.1 These can lead to long-term disability including unilateral blindness and deafness.
The most common and debilitating complication is postherpetic neuralgia, a persistent pain lasting at least 3 months, with a mean duration of 3.3 years and sometimes as long as 10 years.16 Postherpetic neuralgia occurs in 8% to 32% of patients after acute HZ,4 and the incidence increases with age, being most common after age 70. The chronic pain of postherpetic neuralgia has a significant adverse impact on patients’ quality of life, including physical disability and emotional distress.17 Some pain is intense, and anecdotal reports of patients committing suicide were included in the Advisory Committee on Immunization Practices (ACIP) recommendations regarding herpes zoster vaccine.18
HZ and its complications also impose a substantial economic burden on society.19 In a population-based study, the mean direct medical costs of HZ ranged from $620 to $1,160 (2015 dollars) depending on age,20 and the mean costs of postherpetic neuralgia were 2 to 5 times higher than that.20–22 Immunocompromised patients had costs 2 to 3 times higher than those of immunocompetent adults.23 In addition, for employed patients, HZ resulted in an average loss of 32 hours of work due to absenteeism and 84 hours due to presenteeism (ie, working while sick and therefore suboptimally).24
Assuming there are 1 million cases of HZ each year, if 8% to 32% of patients go on to develop postherpetic neuralgia, that would translate into approximately $1 to $2 billion in direct medical costs. With 60% of adult patients working,25 at an average wage of $23.23 per hour,26 HZ illness could be responsible for another $1.6 billion in lost productivity.
EFFICACY AND SAFETY OF HZ VACCINE
In 2006, the FDA approved the live-attenuated Oka strain VZV vaccine for prevention of HZ and postherpetic neuralgia in adults age 60 and older based on findings from the Shingles Prevention Study (SPS).27
The Shingles Prevention Study
This multicenter randomized placebo-controlled trial27 enrolled 38,546 immunocompetent persons age 60 and older. Subjects in the intervention group received a single dose of live-attenuated vaccine, and all participants were followed for up to 4.9 years after vaccination.
HZ occurred in 315 (1.636%) of the 19,254 participants in the vaccine group and in 642 (3.336%) of the 19,247 participants in the placebo group, an absolute risk reduction of 1.7%, number needed to treat 59, relative risk reduction 51%, P < .001. Similarly, postherpetic neuralgia occurred in 27 (0.140%) of the 19,254 vaccine recipients and in 80 (0.416%) of the placebo recipients (an absolute risk reduction of 0.276%, number needed to treat 362, relative risk reduction 66%, P < .001). The investigators calculated that vaccination reduced the overall burden of illness by 61% (Table 1).
The efficacy against HZ incidence decreased with age,28 but the efficacy against postherpetic neuralgia did not. In addition, vaccine recipients who developed HZ generally had less severe manifestations.
The safety of the vaccine was assessed for all participants in the SPS. In addition, one-sixth of SPS participants were enrolled in a safety substudy. These participants completed a detailed report card regarding all medically important events within the first 42 days. Forty-eight percent of the vaccine group and 17% of the placebo group (P < .05) experienced adverse events, primarily at the injection site. Less than 1% of all local reactions were severe.29 Serious adverse events were rare (< 2%), but occurred significantly more often in the vaccinated group.
Short-Term Persistence Substudy
Short-term efficacy of the live-attenuated vaccine (up to 7 years) was assessed in the Short-Term Persistence Substudy (STPS), which involved 14,270 of the initial participants and reported yearly and overall vaccine efficacy.30 After 5 years, the yearly efficacy against postherpetic neuralgia incidence declined to 32% and was no longer statistically significant. Efficacy against HZ incidence and burden of illness displayed the same pattern. After the end of the STPS, all subjects in the placebo group received vaccination.
Long-Term Persistence Substudy
Those in the intervention group were followed for an additional 4 years in the Long-Term Persistence Substudy (LTPS).31 Due to the lack of concurrent controls in the LTPS, the authors used regression models based on historical controls to estimate contemporary population incidence of HZ and postherpetic neuralgia for comparison.
Efficacy continued to decline over time, and by 10 years after vaccination there was no difference between vaccinated patients and historical controls in the rate of any end point (ie, efficacy declined to zero).
A trial of booster vaccination
Because many patients are vaccinated at age 60, waning immunity could leave them vulnerable to HZ and postherpetic neuralgia by age 70. A potential solution would be to give a booster dose after 10 years.
A recent phase 3 clinical trial of adults age 70 years and older found that a booster dose of live-attenuated vaccine was as safe and immunogenic as an initial dose.32 While antibody responses were similar in the boosted group and the newly vaccinated group, cell-mediated immunity was higher in the boosted group.
Because prevention of HZ is generally via cell-mediated immunity, the booster might be more effective than the initial vaccination, but clinical trials measuring actual cases prevented will be required to prove it. A booster dose is not currently recommended.
A trial of vaccination in adults 50 to 59
In 2011, the FDA extended its approval of HZ vaccine for use in adults ages 50 to 59.33
In a randomized, double-blind, placebo-controlled trial in this age group,33 the vaccine reduced HZ incidence by almost 70% (absolute risk reduction 0.614%, number needed to treat 156; Table 1), but the severity of HZ cases was not affected. There were too few cases of postherpetic neuralgia to assess the efficacy for this end point. The study followed patients for only 1.5 years after vaccination, so the duration of efficacy is unknown.
As in the older recipients, the vaccine was well tolerated; injection-site reactions and headache were the major adverse effects reported among vaccine recipients.33
INDICATIONS AND CONTRAINDICATIONS
Although HZ vaccine is licensed for use in adults age 50 and older, the ACIP recommends it only for immunocompetent adults age 60 and older. At this time, the ACIP does not recommend HZ vaccine in those younger than 60 because of the low risk of HZ in this age group.34
Any person age 60 or older should receive a single dose of the live-attenuated HZ vaccine subcutaneously, regardless of past history of HZ.
The vaccine is contraindicated in patients who have a history of allergic reaction to any vaccine component, immunosuppression or immunodeficiency conditions, and pregnancy. Specifically, people who will receive immunosuppressive therapies should have the vaccine at least 14 days before beginning treatment. Antiviral medications such as acyclovir, famciclovir, and valacyclovir should be discontinued at least 24 hours before vaccination and not resumed until 14 days later. Patients taking high-dose corticosteroids for more than 2 weeks should not be vaccinated until at least 1 month after therapy is completed.
In contrast, HZ vaccine is not contraindicated for leukemia patients who are in remission and who have not received chemotherapy or radiation for at least 3 months, or for patients receiving short-term, low-to-moderate dose, topical, intra-articular, bursal, or tendon injections of corticosteroids. Patients on low-dose methotrexate, azathioprine, or 6-mercaptopurine can also receive the vaccine.18
VACCINATION RATES ARE LOW
Although the vaccine has been recommended since 2008, uptake has been slow. Figure 1 shows the rate of HZ vaccination in adults age 60 and older surveyed in the National Health Interview Survey from 2007 to 2013.35 Eight years after the vaccine was licensed, only 28% of eligible patients had been vaccinated. Assuming the current rate of increase remains constant, it will take 7 more years to reach a 60% coverage rate—the same as for pneumococcal vaccine36—and 18 years to reach universal coverage.
Barriers to vaccination
Several barriers to HZ vaccination might account for the slow uptake.
For the first few years the vaccine was available, the requirement to store it frozen presented an obstacle for some physicians.37 Physicians may also have been discouraged by the cumbersome Medicare reimbursement process because while the administration fee is covered through Medicare Part B, the live-attenuated vaccine is reimbursed only through Medicare Part D, a benefit that varies by plans. Other barriers to physicians are supply shortages, high up-front costs, and uncertainties regarding the duration of vaccine protection, its safety, and side effects.38–40
Patient barriers include lack of physician recommendation, lack of familiarity with the vaccine, high out-of-pocket costs, the perception that they are at low risk for HZ, underestimation of the pain associated with HZ and postherpetic neuralgia, and fear of vaccine adverse effects.39,41,42
Interventions to increase vaccination rates
Certain interventions have been shown to increase vaccination adherence in general and HZ vaccination in particular. In randomized trials involving other vaccines, electronic medical record reminders supporting panel management or nurse-initiated protocols have been proven to increase vaccination rates, but these methods have not been tested for HZ vaccine specifically.43,44
In an observational study, Chaudhry et al found that the number of HZ vaccinations administered at the Mayo Clinic increased 43% in one practice and 54% in another after the implementation of an electronic alert.45 A randomized controlled trial showed that an informational package discussing HZ and the vaccine sent to patients via either their electronic personal health record or traditional mail increased HZ vaccination by almost 3 times.46
Pharmacists can also influence vaccination rates. States that provide full immunization privileges to pharmacists have vaccination rates significantly higher than states with restricted or no authorization.47
COST-EFFECTIVENESS CONSIDERATIONS
Unlike the Centers for Medicare and Medicaid Services, the ACIP does consider cost-effectiveness in their vaccine recommendations. Because of the morbidity associated with HZ and postherpetic neuralgia as well as the economic impact, vaccination is generally considered cost-effective for adults age 60 and older.48,49
Analyses have demonstrated that cost-effectiveness hinges on 4 factors: initial vaccine efficacy, the duration of efficacy, the age-specific incidence of HZ, and the cost of the vaccine.
For patients ages 50 to 59, the incidence of HZ is low, and because the duration of vaccine efficacy is short even though initial vaccine efficacy is high, vaccination in this age group offers poor value.50 At older ages, the incidence of HZ and postherpetic neuralgia rises, making vaccination more cost-effective. After age 60, the vaccine is cost-effective at all ages, although age 70 appears to offer the optimal trade-off between increasing incidence and declining vaccine efficacy.48,49
For patients who plan to be vaccinated only once, waiting until age 70 would appear to offer the best value.51 For those who are willing to receive a booster dose, the optimal age for vaccination is unknown, but will likely depend on the effectiveness, cost, and duration of the booster.
A NEW HZ VACCINE
In 2015, GlaxoSmithKline tested a new HZ vaccine containing a single VZV glycoprotein in an AS01B adjuvant system (HZ/su vaccine).52 In a phase 3 randomized trial involving 15,411 immunocompetent persons age 50 and older, a 2-dose schedule of HZ/su vaccine was 97% effective in preventing HZ (Table 1).53 Importantly, the vaccine was equally effective in older patients.
This vaccine also had a high rate of adverse reactions, with 17% of vaccine recipients vs 3% of placebo recipients reporting events that prevented normal everyday activities for at least 1 day. However, the rate of serious adverse reactions was the same in both groups (9%). The company announced that they intended to submit a regulatory application for HZ/su vaccine in the second half of 2016.54
Because of its high efficacy, HZ/su vaccine has the potential to change practice, but several issues must be resolved before it can supplant the current vaccine.
First, the AS01B adjuvant is not currently licensed in the United States, so it is unclear if the HZ/su vaccine can get FDA approval.52,55
Second, there are several questions about the efficacy of the vaccine, including long-term efficacy, efficacy in the elderly, and efficacy in the case of a patient receiving only 1 of the 2 required doses.
Third, the impact of HZ/su vaccine on complications such as postherpetic neuralgia has not been established. The clinical trial (NCT01165229) examining vaccine efficacy against postherpetic neuralgia incidence and other complications in adults age 70 and older has recently been completed and data should be available soon. Given the extremely high efficacy against HZ, it is likely that it will be close to 100% effective against this complication.
Fourth, there is uncertainty as to how the HZ/su vaccine should be used in patients who have already received the live-attenuated vaccine, if it is determined that a booster is necessary.
Finally, the vaccine is not yet priced. Given its superior effectiveness, particularly in older individuals, competitive pricing could dramatically affect the market. How Medicare or other insurers cover the new vaccine will likely influence its acceptance.
HZ VACCINATION OF IMMUNOCOMPROMISED PATIENTS
Immunocompromised patients are at highest risk for developing HZ. Unfortunately, there are currently no HZ vaccines approved for use in this population. The current live-attenuated vaccine has been demonstrated to be safe, well tolerated, and immunogenic in patients age 60 and older who are receiving chronic or maintenance low to moderate doses of corticosteroids.56
A clinical trial is being conducted to assess the immunogenicity, clinical effectiveness, and safety of the vaccine in rheumatoid arthritis patients receiving antitumor necrosis factor therapy (NCT01967316). Other trials are examining vaccine efficacy and safety in patients with solid organ tumors prior to chemotherapy (NCT02444936) and in patients who will be undergoing living donor kidney transplantation (NCT00940940). Researchers are also investigating the possibility of vaccinating allogeneic stem cell donors before donation in order to protect transplant recipients against HZ (NCT01573182).
ZVHT and HZ/su vaccination in immunocompromised patients
Heat-treated varicella-zoster vaccine (ZVHT) is a potential alternative for immunocompromised patients. A 4-dose regimen has been proven to reduce the risk of HZ in patients receiving autologous hematopoietic-cell transplants for non-Hodgkin or Hodgkin lymphoma.57
In another trial, the 4-dose ZVHT was safe and elicited significant VZV-specific T-cell response through 28 days in immunosuppressed patients with solid tumor malignancy, hematologic malignancy, human immunodeficiency virus infection with CD4 counts of 200 cells/mm3 or less, and autologous hematopoietic-cell transplants. The T-cell response was poor in allogeneic hematopoietic-cell transplant recipients, however.58
Because the HZ/su vaccine does not contain live virus, it seems particularly promising for immunocompromised patients. In phase 1 and 2 studies, a 3-dose regimen has been shown to be safe and immunogenic in hematopoietic-cell transplant recipients and HIV-infected adults with CD4 count higher than 200 cells/mm3.59,60 A phase 3 trial assessing the efficacy of HZ/su vaccine in autologous hematopoietic-cell transplant recipients is under way (NCT01610414). Changes in recommendations for HZ vaccine in these most vulnerable populations await the results of these studies.
Herpes zoster (HZ), or shingles, represents a reactivation of the varicella-zoster virus (VZV). Following primary infection, usually in childhood, the virus typically lies dormant in the dorsal root and sensory nerve ganglia for decades. The precise mechanism of reactivation is not well understood, but it is associated with a decline in cell-mediated immunity that occurs with advancing age, immune-compromising conditions such as HIV infection and cancer, or immunosuppressive therapies, including corticosteroids.1 HZ is usually a self-limited disease characterized by unilateral dermatomal rash and pain, but can cause disseminated infection in immunocompromised individuals.2
Treatment with antiviral medications within 72 hours of rash onset can reduce acute HZ symptoms.1 However, antiviral agents are only minimally effective in preventing postherpetic neuralgia, the most common complication of HZ.3 Therefore, efforts to reduce the burden of HZ morbidity have focused on prevention through vaccination.
Currently, the only shingles vaccine approved by the US Food and Drug Administration (FDA) is Zostavax (Merck), which contains the live-attenuated Oka strain of VZV at a concentration 14 times greater than that of the varicella vaccine (Varivax, Merck). The live-attenuated vaccine boosts VZV-specific cell-mediated immunity, preventing reactivation of the latent virus.
In this article, we describe the burden of disease and review recent developments in the literature on HZ vaccine, including duration of efficacy, uptake and barriers to vaccination, cost-effectiveness, and the outlook for future vaccines.
INCIDENCE INCREASES WITH AGE
The incidence of herpes zoster in the general population is between 3 and 5 per 1,000 person-years4 and increases with age, especially after age 60 when the incidence can approach 13 to 15 per 1,000 person-years.5,6 An estimated 1 million new cases occur each year in the United States, and about 6% of patients experience a second episode of HZ within 8 years.7,8 In immunocompromised patients, the incidence of HZ is 2 to 10 times higher than in the general population.9
The incidence of HZ has been increasing for reasons that are unclear. After varicella vaccine was introduced into the routine childhood immunization schedule in 1995, it was hypothesized that the resultant decrease in primary varicella infections would remove a natural source of immune boosting and cause an increase in HZ incidence for up to 20 years.10 However, recent studies demonstrate that the observed increase in HZ incidence actually predates the introduction of varicella vaccine,11–13 and the widespread use of varicella vaccine has not resulted in an increase in the incidence of HZ.14
Other potential explanations for the rise in reported incidence include increasing awareness among patients, who might previously not have sought care and among physicians, who may be more likely to make the diagnosis. Advertisement of new treatments for HZ, including gabapentin and capsaicin, probably began to increase awareness in the 1990s, as did promotion of the HZ vaccine after its licensure in 2006.
HZ can occur in people who have been vaccinated against varicella due to reactivation of the vaccine-strain virus, but the risk is lower than after infection with wild-type varicella.15 Given that the varicella vaccine has been routinely used in children for only 20 years, the long-term effect of varicella vaccination on the incidence of HZ in elderly people is unknown.
Serious complications
HZ can cause rare but serious complications including encephalitis, herpes ophthalmicus, herpes oticus, myelitis, and retinitis.1 These can lead to long-term disability including unilateral blindness and deafness.
The most common and debilitating complication is postherpetic neuralgia, a persistent pain lasting at least 3 months, with a mean duration of 3.3 years and sometimes as long as 10 years.16 Postherpetic neuralgia occurs in 8% to 32% of patients after acute HZ,4 and the incidence increases with age, being most common after age 70. The chronic pain of postherpetic neuralgia has a significant adverse impact on patients’ quality of life, including physical disability and emotional distress.17 Some pain is intense, and anecdotal reports of patients committing suicide were included in the Advisory Committee on Immunization Practices (ACIP) recommendations regarding herpes zoster vaccine.18
HZ and its complications also impose a substantial economic burden on society.19 In a population-based study, the mean direct medical costs of HZ ranged from $620 to $1,160 (2015 dollars) depending on age,20 and the mean costs of postherpetic neuralgia were 2 to 5 times higher than that.20–22 Immunocompromised patients had costs 2 to 3 times higher than those of immunocompetent adults.23 In addition, for employed patients, HZ resulted in an average loss of 32 hours of work due to absenteeism and 84 hours due to presenteeism (ie, working while sick and therefore suboptimally).24
Assuming there are 1 million cases of HZ each year, if 8% to 32% of patients go on to develop postherpetic neuralgia, that would translate into approximately $1 to $2 billion in direct medical costs. With 60% of adult patients working,25 at an average wage of $23.23 per hour,26 HZ illness could be responsible for another $1.6 billion in lost productivity.
EFFICACY AND SAFETY OF HZ VACCINE
In 2006, the FDA approved the live-attenuated Oka strain VZV vaccine for prevention of HZ and postherpetic neuralgia in adults age 60 and older based on findings from the Shingles Prevention Study (SPS).27
The Shingles Prevention Study
This multicenter randomized placebo-controlled trial27 enrolled 38,546 immunocompetent persons age 60 and older. Subjects in the intervention group received a single dose of live-attenuated vaccine, and all participants were followed for up to 4.9 years after vaccination.
HZ occurred in 315 (1.636%) of the 19,254 participants in the vaccine group and in 642 (3.336%) of the 19,247 participants in the placebo group, an absolute risk reduction of 1.7%, number needed to treat 59, relative risk reduction 51%, P < .001. Similarly, postherpetic neuralgia occurred in 27 (0.140%) of the 19,254 vaccine recipients and in 80 (0.416%) of the placebo recipients (an absolute risk reduction of 0.276%, number needed to treat 362, relative risk reduction 66%, P < .001). The investigators calculated that vaccination reduced the overall burden of illness by 61% (Table 1).
The efficacy against HZ incidence decreased with age,28 but the efficacy against postherpetic neuralgia did not. In addition, vaccine recipients who developed HZ generally had less severe manifestations.
The safety of the vaccine was assessed for all participants in the SPS. In addition, one-sixth of SPS participants were enrolled in a safety substudy. These participants completed a detailed report card regarding all medically important events within the first 42 days. Forty-eight percent of the vaccine group and 17% of the placebo group (P < .05) experienced adverse events, primarily at the injection site. Less than 1% of all local reactions were severe.29 Serious adverse events were rare (< 2%), but occurred significantly more often in the vaccinated group.
Short-Term Persistence Substudy
Short-term efficacy of the live-attenuated vaccine (up to 7 years) was assessed in the Short-Term Persistence Substudy (STPS), which involved 14,270 of the initial participants and reported yearly and overall vaccine efficacy.30 After 5 years, the yearly efficacy against postherpetic neuralgia incidence declined to 32% and was no longer statistically significant. Efficacy against HZ incidence and burden of illness displayed the same pattern. After the end of the STPS, all subjects in the placebo group received vaccination.
Long-Term Persistence Substudy
Those in the intervention group were followed for an additional 4 years in the Long-Term Persistence Substudy (LTPS).31 Due to the lack of concurrent controls in the LTPS, the authors used regression models based on historical controls to estimate contemporary population incidence of HZ and postherpetic neuralgia for comparison.
Efficacy continued to decline over time, and by 10 years after vaccination there was no difference between vaccinated patients and historical controls in the rate of any end point (ie, efficacy declined to zero).
A trial of booster vaccination
Because many patients are vaccinated at age 60, waning immunity could leave them vulnerable to HZ and postherpetic neuralgia by age 70. A potential solution would be to give a booster dose after 10 years.
A recent phase 3 clinical trial of adults age 70 years and older found that a booster dose of live-attenuated vaccine was as safe and immunogenic as an initial dose.32 While antibody responses were similar in the boosted group and the newly vaccinated group, cell-mediated immunity was higher in the boosted group.
Because prevention of HZ is generally via cell-mediated immunity, the booster might be more effective than the initial vaccination, but clinical trials measuring actual cases prevented will be required to prove it. A booster dose is not currently recommended.
A trial of vaccination in adults 50 to 59
In 2011, the FDA extended its approval of HZ vaccine for use in adults ages 50 to 59.33
In a randomized, double-blind, placebo-controlled trial in this age group,33 the vaccine reduced HZ incidence by almost 70% (absolute risk reduction 0.614%, number needed to treat 156; Table 1), but the severity of HZ cases was not affected. There were too few cases of postherpetic neuralgia to assess the efficacy for this end point. The study followed patients for only 1.5 years after vaccination, so the duration of efficacy is unknown.
As in the older recipients, the vaccine was well tolerated; injection-site reactions and headache were the major adverse effects reported among vaccine recipients.33
INDICATIONS AND CONTRAINDICATIONS
Although HZ vaccine is licensed for use in adults age 50 and older, the ACIP recommends it only for immunocompetent adults age 60 and older. At this time, the ACIP does not recommend HZ vaccine in those younger than 60 because of the low risk of HZ in this age group.34
Any person age 60 or older should receive a single dose of the live-attenuated HZ vaccine subcutaneously, regardless of past history of HZ.
The vaccine is contraindicated in patients who have a history of allergic reaction to any vaccine component, immunosuppression or immunodeficiency conditions, and pregnancy. Specifically, people who will receive immunosuppressive therapies should have the vaccine at least 14 days before beginning treatment. Antiviral medications such as acyclovir, famciclovir, and valacyclovir should be discontinued at least 24 hours before vaccination and not resumed until 14 days later. Patients taking high-dose corticosteroids for more than 2 weeks should not be vaccinated until at least 1 month after therapy is completed.
In contrast, HZ vaccine is not contraindicated for leukemia patients who are in remission and who have not received chemotherapy or radiation for at least 3 months, or for patients receiving short-term, low-to-moderate dose, topical, intra-articular, bursal, or tendon injections of corticosteroids. Patients on low-dose methotrexate, azathioprine, or 6-mercaptopurine can also receive the vaccine.18
VACCINATION RATES ARE LOW
Although the vaccine has been recommended since 2008, uptake has been slow. Figure 1 shows the rate of HZ vaccination in adults age 60 and older surveyed in the National Health Interview Survey from 2007 to 2013.35 Eight years after the vaccine was licensed, only 28% of eligible patients had been vaccinated. Assuming the current rate of increase remains constant, it will take 7 more years to reach a 60% coverage rate—the same as for pneumococcal vaccine36—and 18 years to reach universal coverage.
Barriers to vaccination
Several barriers to HZ vaccination might account for the slow uptake.
For the first few years the vaccine was available, the requirement to store it frozen presented an obstacle for some physicians.37 Physicians may also have been discouraged by the cumbersome Medicare reimbursement process because while the administration fee is covered through Medicare Part B, the live-attenuated vaccine is reimbursed only through Medicare Part D, a benefit that varies by plans. Other barriers to physicians are supply shortages, high up-front costs, and uncertainties regarding the duration of vaccine protection, its safety, and side effects.38–40
Patient barriers include lack of physician recommendation, lack of familiarity with the vaccine, high out-of-pocket costs, the perception that they are at low risk for HZ, underestimation of the pain associated with HZ and postherpetic neuralgia, and fear of vaccine adverse effects.39,41,42
Interventions to increase vaccination rates
Certain interventions have been shown to increase vaccination adherence in general and HZ vaccination in particular. In randomized trials involving other vaccines, electronic medical record reminders supporting panel management or nurse-initiated protocols have been proven to increase vaccination rates, but these methods have not been tested for HZ vaccine specifically.43,44
In an observational study, Chaudhry et al found that the number of HZ vaccinations administered at the Mayo Clinic increased 43% in one practice and 54% in another after the implementation of an electronic alert.45 A randomized controlled trial showed that an informational package discussing HZ and the vaccine sent to patients via either their electronic personal health record or traditional mail increased HZ vaccination by almost 3 times.46
Pharmacists can also influence vaccination rates. States that provide full immunization privileges to pharmacists have vaccination rates significantly higher than states with restricted or no authorization.47
COST-EFFECTIVENESS CONSIDERATIONS
Unlike the Centers for Medicare and Medicaid Services, the ACIP does consider cost-effectiveness in their vaccine recommendations. Because of the morbidity associated with HZ and postherpetic neuralgia as well as the economic impact, vaccination is generally considered cost-effective for adults age 60 and older.48,49
Analyses have demonstrated that cost-effectiveness hinges on 4 factors: initial vaccine efficacy, the duration of efficacy, the age-specific incidence of HZ, and the cost of the vaccine.
For patients ages 50 to 59, the incidence of HZ is low, and because the duration of vaccine efficacy is short even though initial vaccine efficacy is high, vaccination in this age group offers poor value.50 At older ages, the incidence of HZ and postherpetic neuralgia rises, making vaccination more cost-effective. After age 60, the vaccine is cost-effective at all ages, although age 70 appears to offer the optimal trade-off between increasing incidence and declining vaccine efficacy.48,49
For patients who plan to be vaccinated only once, waiting until age 70 would appear to offer the best value.51 For those who are willing to receive a booster dose, the optimal age for vaccination is unknown, but will likely depend on the effectiveness, cost, and duration of the booster.
A NEW HZ VACCINE
In 2015, GlaxoSmithKline tested a new HZ vaccine containing a single VZV glycoprotein in an AS01B adjuvant system (HZ/su vaccine).52 In a phase 3 randomized trial involving 15,411 immunocompetent persons age 50 and older, a 2-dose schedule of HZ/su vaccine was 97% effective in preventing HZ (Table 1).53 Importantly, the vaccine was equally effective in older patients.
This vaccine also had a high rate of adverse reactions, with 17% of vaccine recipients vs 3% of placebo recipients reporting events that prevented normal everyday activities for at least 1 day. However, the rate of serious adverse reactions was the same in both groups (9%). The company announced that they intended to submit a regulatory application for HZ/su vaccine in the second half of 2016.54
Because of its high efficacy, HZ/su vaccine has the potential to change practice, but several issues must be resolved before it can supplant the current vaccine.
First, the AS01B adjuvant is not currently licensed in the United States, so it is unclear if the HZ/su vaccine can get FDA approval.52,55
Second, there are several questions about the efficacy of the vaccine, including long-term efficacy, efficacy in the elderly, and efficacy in the case of a patient receiving only 1 of the 2 required doses.
Third, the impact of HZ/su vaccine on complications such as postherpetic neuralgia has not been established. The clinical trial (NCT01165229) examining vaccine efficacy against postherpetic neuralgia incidence and other complications in adults age 70 and older has recently been completed and data should be available soon. Given the extremely high efficacy against HZ, it is likely that it will be close to 100% effective against this complication.
Fourth, there is uncertainty as to how the HZ/su vaccine should be used in patients who have already received the live-attenuated vaccine, if it is determined that a booster is necessary.
Finally, the vaccine is not yet priced. Given its superior effectiveness, particularly in older individuals, competitive pricing could dramatically affect the market. How Medicare or other insurers cover the new vaccine will likely influence its acceptance.
HZ VACCINATION OF IMMUNOCOMPROMISED PATIENTS
Immunocompromised patients are at highest risk for developing HZ. Unfortunately, there are currently no HZ vaccines approved for use in this population. The current live-attenuated vaccine has been demonstrated to be safe, well tolerated, and immunogenic in patients age 60 and older who are receiving chronic or maintenance low to moderate doses of corticosteroids.56
A clinical trial is being conducted to assess the immunogenicity, clinical effectiveness, and safety of the vaccine in rheumatoid arthritis patients receiving antitumor necrosis factor therapy (NCT01967316). Other trials are examining vaccine efficacy and safety in patients with solid organ tumors prior to chemotherapy (NCT02444936) and in patients who will be undergoing living donor kidney transplantation (NCT00940940). Researchers are also investigating the possibility of vaccinating allogeneic stem cell donors before donation in order to protect transplant recipients against HZ (NCT01573182).
ZVHT and HZ/su vaccination in immunocompromised patients
Heat-treated varicella-zoster vaccine (ZVHT) is a potential alternative for immunocompromised patients. A 4-dose regimen has been proven to reduce the risk of HZ in patients receiving autologous hematopoietic-cell transplants for non-Hodgkin or Hodgkin lymphoma.57
In another trial, the 4-dose ZVHT was safe and elicited significant VZV-specific T-cell response through 28 days in immunosuppressed patients with solid tumor malignancy, hematologic malignancy, human immunodeficiency virus infection with CD4 counts of 200 cells/mm3 or less, and autologous hematopoietic-cell transplants. The T-cell response was poor in allogeneic hematopoietic-cell transplant recipients, however.58
Because the HZ/su vaccine does not contain live virus, it seems particularly promising for immunocompromised patients. In phase 1 and 2 studies, a 3-dose regimen has been shown to be safe and immunogenic in hematopoietic-cell transplant recipients and HIV-infected adults with CD4 count higher than 200 cells/mm3.59,60 A phase 3 trial assessing the efficacy of HZ/su vaccine in autologous hematopoietic-cell transplant recipients is under way (NCT01610414). Changes in recommendations for HZ vaccine in these most vulnerable populations await the results of these studies.
- Dworkin RH, Johnson RW, Breuer J, et al. Recommendations for the management of herpes zoster. Clin Infect Dis 2007; 44(suppl 1):S1–S26.
- Johnson RW. Herpes zoster and postherpetic neuralgia. Expert Rev Vaccines 2010; 9(suppl):21–26.
- Chen N, Li Q, Yang J, Zhou M, Zhou D, He L. Antiviral treatment for preventing postherpetic neuralgia. Cochrane Database Syst Rev 2014; 2:CD006866.
- Kawai K, Gebremeskel BG, Acosta CJ. Systematic review of incidence and complications of herpes zoster: towards a global perspective. BMJ Open 2014; 4:e004833.
- Tseng HF, Smith N, Harpaz R, Bialek SR, Sy LS, Jacobsen SJ. Herpes zoster vaccine in older adults and the risk of subsequent herpes zoster disease. JAMA 2011; 305:160–166.
- Langan SM, Smeeth L, Margolis DJ, Thomas SL. Herpes zoster vaccine effectiveness against incident herpes zoster and post-herpetic neuralgia in an older US population: a cohort study. PLoS Med 2013; 10:e1001420.
- Yawn BP, Saddier P, Wollan PC, St. Sauver JL, Kurland MJ, Sy LS. A population-based study of the incidence and complication rates of herpes zoster before zoster vaccine introduction. Mayo Clin Proc 2007; 82:1341–1349.
- Yawn BP, Wollan PC, Kurland MJ, St. Sauver JL, Saddier P. Herpes zoster recurrences more frequent than previously reported. Mayo Clin Proc 2011; 86:88–93.
- Chen SY, Suaya JA, Li Q, et al. Incidence of herpes zoster in patients with altered immune function. Infection 2014; 42:325–334.
- Edmunds WJ, Brisson M. The effect of vaccination on the epidemiology of varicella zoster virus. J Infect 2002; 44:211–219.
- Hales CM, Harpaz R, Joesoef MR, Bialek SR. Examination of links between herpes zoster incidence and childhood varicella vaccination. Ann Intern Med 2013; 159:739–745.
- Leung J, Harpaz R, Molinari NA, Jumaan A, Zhou F. Herpes zoster incidence among insured persons in the United States, 1993-2006: evaluation of impact of varicella vaccination. Clin Infect Dis 2011; 52:332–340.
- Rimland D, Moanna A. Increasing incidence of herpes zoster among veterans. Clin Infect Dis 2010; 50:1000–1005.
- Jumaan AO, Yu O, Jackson LA, Bohlke K, Galil K, Seward JF. Incidence of herpes zoster, before and after varicella-vaccination-associated decreases in the incidence of varicella, 1992-2002. J Infect Dis 2005; 191:2002–2007.
- Plotkin SA, Starr SE, Connor K, Morton D. Zoster in normal children after varicella vaccine. J Infect Dis 1989; 159:1000–1001.
- Oster G, Harding G, Dukes E, Edelsberg J, Cleary PD. Pain, medication use, and health-related quality of life in older persons with postherpetic neuralgia: results from a population-based survey. J Pain 2005; 6:356–363.
- Johnson RW, Bouhassira D, Kassianos G, Leplege A, Schmader KE, Weinke T. The impact of herpes zoster and post-herpetic neuralgia on quality-of-life. BMC Med 2010; 8:37.
- Harpaz R, Ortega-Sanchez IR, Seward JF; Advisory Committee on Immunization Practices (ACIP) Centers for Disease Control and Prevention (CDC). Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2008; 57:1–30.
- Panatto D, Bragazzi NL, Rizzitelli E, et al. Evaluation of the economic burden of herpes zoster (HZ) infection. Hum Vaccin Immunother 2015; 11:245–262.
- Yawn BP, Itzler RF, Wollan PC, Pellissier JM, Sy LS, Saddier P. Health care utilization and cost burden of herpes zoster in a community population. Mayo Clin Proc 2009; 84:787–794.
- Dworkin RH, White R, O’Connor AB, Hawkins K. Health care expenditure burden of persisting herpes zoster pain. Pain Med 2008; 9:348–353.
- White RR, Lenhart G, Singhal PK, et al. Incremental 1-year medical resource utilization and costs for patients with herpes zoster from a set of US health plans. Pharmacoeconomics 2009; 27:781–792.
- Insinga RP, Itzler RF, Pellissier JM. Acute/subacute herpes zoster: healthcare resource utilisation and costs in a group of US health plans. Pharmacoeconomics 2007; 25:155–169.
- Singhal PK, Makin C, Pellissier J, Sy L, White R, Saddier P. Work and productivity loss related to herpes zoster. J Med Econ 2011; 14:639–645.
- US Bureau of Labor Statistics. Labor force statistics from the current population survey. www.bls.gov/web/empsit/cpseea13.htm. Accessed April 6, 2017.
- US Bureau of Labor Statistic. Occupational employment statistics. www.bls.gov/oes/current/oes_nat.htm. Accessed April 6, 2017.
- Oxman MN, Levin MJ, Johnson GR, et al; Shingles Prevention Study Group. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 2005; 352:2271–2284.
- Food and Drug Administration (FDA). FDA clinical briefing document for Merck & Co., Inc. Zoster vaccine live (Oka/Merck) Zostavax. www.fda.gov/ohrms/dockets/ac/05/briefing/5-4198b2_1.pdf. Accessed April 6, 2017.
- Simberkoff MS, Arbeit RD, Johnson GR, et al; Shingles Prevention Study Group. Safety of herpes zoster vaccine in the shingles prevention study: a randomized trial. Ann Intern Med 2010; 152:545–554.
- Schmader KE, Oxman MN, Levin MJ, et al; Shingles Prevention Study Group. Persistence of the efficacy of zoster vaccine in the shingles prevention study and the short-term persistence substudy. Clin Infect Dis 2012; 55:1320–1328.
- Morrison VA, Johnson GR, Schmader KE, et al; Shingles Prevention Study Group. Long-term persistence of zoster vaccine efficacy. Clin Infect Dis 2015; 60:900–909.
- Levin MJ, Schmader KE, Pang L, et al. Cellular and humoral responses to a second dose of herpes zoster vaccine administered 10 years after the first dose among older adults. J Infect Dis 2016; 213:14–22.
- Schmader KE, Levin MJ, Gnann JW Jr, et al. Efficacy, safety, and tolerability of herpes zoster vaccine in persons aged 50-59 years. Clin Infect Dis 2012; 54:922–928.
- Hales CM, Harpaz R, Ortega-Sanchez I, Bialek SR; Centers for Disease Control and Prevention (CDC). Update on recommendations for use of herpes zoster vaccine. MMWR Morb Mortal Wkly Rep 2014; 63:729–731.
- Centers for Disease Control and Prevention (CDC). Surveillance of vaccination coverage among adult populations—United States, 2014. MMWR Morb Mortal Wkly Rep 2016; 65(1):1–36. Accessed April 12, 2017.
- Williams WW, Lu PJ, O’Halloran A, et al; Centers for Disease Control and Prevention (CDC). Vaccination coverage among adults, excluding influenza vaccination—United States, 2013. MMWR Morb Mortal Wkly Rep 2015; 64:95–102.
- Oxman MN. Zoster vaccine: current status and future prospects. Clin Infect Dis 2010; 51:197–213.
- Hurley LP, Lindley MC, Harpaz R, et al. Barriers to the use of herpes zoster vaccine. Ann Intern Med 2010; 152:555–560.
- Lu PJ, Euler GL, Jumaan AO, Harpaz R. Herpes zoster vaccination among adults aged 60 years or older in the United States, 2007: uptake of the first new vaccine to target seniors. Vaccine 2009; 27:882–887.
- Hurley LP, Harpaz R, Daley MF, et al. National survey of primary care physicians regarding herpes zoster and the herpes zoster vaccine. J Infect Dis 2008; 197(suppl 2):S216–S223.
- Joon Lee T, Hayes S, Cummings DM, et al. Herpes zoster knowledge, prevalence, and vaccination rate by race. J Am Board Fam Med 2013; 26:45–51.
- Opstelten W, van Essen GA, Hak E. Determinants of non-compliance with herpes zoster vaccination in the community-dwelling elderly. Vaccine 2009; 27:192–196.
- Loo TS, Davis RB, Lipsitz LA, et al. Electronic medical record reminders and panel management to improve primary care of elderly patients. Arch Intern Med 2011; 171:1552–1558.
- Rhew DC, Glassman PA, Goetz MB. Improving pneumococcal vaccine rates. Nurse protocols versus clinical reminders. J Gen Intern Med 1999; 14:351–356.
- Chaudhry R, Schietel SM, North F, Dejesus R, Kesman RL, Stroebel RJ. Improving rates of herpes zoster vaccination with a clinical decision support system in a primary care practice. J Eval Clin Pract 2013; 19:263–266.
- Otsuka SH, Tayal NH, Porter K, Embi PJ, Beatty SJ. Improving herpes zoster vaccination rates through use of a clinical pharmacist and a personal health record. Am J Med 2013; 126:832.e1–832.e6.
- Taitel MS, Fensterheim LE, Cannon AE, Cohen ES. Improving pneumococcal and herpes zoster vaccination uptake: expanding pharmacist privileges. Am J Manag Care 2013; 19:e309–e313.
- Kawai K, Preaud E, Baron-Papillon F, Largeron N, Acosta CJ. Cost-effectiveness of vaccination against herpes zoster and postherpetic neuralgia: a critical review. Vaccine 2014; 32:1645–1653.
- Szucs TD, Pfeil AM. A systematic review of the cost effectiveness of herpes zoster vaccination. Pharmacoeconomics 2013; 31:125–136.
- Le P, Rothberg MB. Cost-effectiveness of herpes zoster vaccine for persons aged 50 years. Ann Intern Med 2015; 163:489–497.
- Le P, Rothberg MB. Determining the optimal age to vaccinate against herpes zoster: a cost-effectiveness analysis. Society for Medical Decision Making 37th Annual North American Meeting. St. Louis, MO; October 18-21, 2015.
- Cohen JI. Clinical practice: herpes zoster. N Engl J Med 2013; 369:255–263.
- Lal H, Cunningham AL, Godeaux O, et al; ZOE-50 Study Group. Efficacy of an adjuvanted herpes zoster subunit vaccine in older adults. N Engl J Med 2015; 372:2087–2096.
- GlaxoSmithKline plc. GSK’s candidate shingles vaccine demonstrates 90% efficacy against shingles in people 70 years of age and over. www.gsk.com/en-gb/media/press-releases/gsk-s-candidate-shingles-vaccine-demonstrates-90-efficacy-against-shingles-in-people-70-years-of-age-and-over/. Accessed April 6, 2017.
- Reed SG, Orr MT, Fox CB. Key roles of adjuvants in modern vaccines. Nat Med 2013; 19:1597–1608.
- Russell AF, Parrino J, Fisher CL Jr, et al. Safety, tolerability, and immunogenicity of zoster vaccine in subjects on chronic/maintenance corticosteroids. Vaccine 2015; 33:3129–3134.
- Hata A, Asanuma H, Rinki M, et al. Use of an inactivated varicella vaccine in recipients of hematopoietic-cell transplants. N Engl J Med 2002; 347:26–34.
- Mullane KM, Winston DJ, Wertheim MS, et al. Safety and immunogenicity of heat-treated zoster vaccine (ZVHT) in immunocompromised adults. J Infect Dis 2013; 208:1375–1385.
- Stadtmauer EA, Sullivan KM, Marty FM, et al. A phase 1/2 study of an adjuvanted varicella-zoster virus subunit vaccine in autologous hematopoietic cell transplant recipients. Blood 2014; 124:2921–2929.
- Berkowitz EM, Moyle G, Stellbrink HJ, et al. Safety and immunogenicity of an adjuvanted herpes zoster subunit candidate vaccine in HIV-infected adults: a phase 1/2a randomized, placebo-controlled study. J Infect Dis 2015; 211:1279–1287.
- Dworkin RH, Johnson RW, Breuer J, et al. Recommendations for the management of herpes zoster. Clin Infect Dis 2007; 44(suppl 1):S1–S26.
- Johnson RW. Herpes zoster and postherpetic neuralgia. Expert Rev Vaccines 2010; 9(suppl):21–26.
- Chen N, Li Q, Yang J, Zhou M, Zhou D, He L. Antiviral treatment for preventing postherpetic neuralgia. Cochrane Database Syst Rev 2014; 2:CD006866.
- Kawai K, Gebremeskel BG, Acosta CJ. Systematic review of incidence and complications of herpes zoster: towards a global perspective. BMJ Open 2014; 4:e004833.
- Tseng HF, Smith N, Harpaz R, Bialek SR, Sy LS, Jacobsen SJ. Herpes zoster vaccine in older adults and the risk of subsequent herpes zoster disease. JAMA 2011; 305:160–166.
- Langan SM, Smeeth L, Margolis DJ, Thomas SL. Herpes zoster vaccine effectiveness against incident herpes zoster and post-herpetic neuralgia in an older US population: a cohort study. PLoS Med 2013; 10:e1001420.
- Yawn BP, Saddier P, Wollan PC, St. Sauver JL, Kurland MJ, Sy LS. A population-based study of the incidence and complication rates of herpes zoster before zoster vaccine introduction. Mayo Clin Proc 2007; 82:1341–1349.
- Yawn BP, Wollan PC, Kurland MJ, St. Sauver JL, Saddier P. Herpes zoster recurrences more frequent than previously reported. Mayo Clin Proc 2011; 86:88–93.
- Chen SY, Suaya JA, Li Q, et al. Incidence of herpes zoster in patients with altered immune function. Infection 2014; 42:325–334.
- Edmunds WJ, Brisson M. The effect of vaccination on the epidemiology of varicella zoster virus. J Infect 2002; 44:211–219.
- Hales CM, Harpaz R, Joesoef MR, Bialek SR. Examination of links between herpes zoster incidence and childhood varicella vaccination. Ann Intern Med 2013; 159:739–745.
- Leung J, Harpaz R, Molinari NA, Jumaan A, Zhou F. Herpes zoster incidence among insured persons in the United States, 1993-2006: evaluation of impact of varicella vaccination. Clin Infect Dis 2011; 52:332–340.
- Rimland D, Moanna A. Increasing incidence of herpes zoster among veterans. Clin Infect Dis 2010; 50:1000–1005.
- Jumaan AO, Yu O, Jackson LA, Bohlke K, Galil K, Seward JF. Incidence of herpes zoster, before and after varicella-vaccination-associated decreases in the incidence of varicella, 1992-2002. J Infect Dis 2005; 191:2002–2007.
- Plotkin SA, Starr SE, Connor K, Morton D. Zoster in normal children after varicella vaccine. J Infect Dis 1989; 159:1000–1001.
- Oster G, Harding G, Dukes E, Edelsberg J, Cleary PD. Pain, medication use, and health-related quality of life in older persons with postherpetic neuralgia: results from a population-based survey. J Pain 2005; 6:356–363.
- Johnson RW, Bouhassira D, Kassianos G, Leplege A, Schmader KE, Weinke T. The impact of herpes zoster and post-herpetic neuralgia on quality-of-life. BMC Med 2010; 8:37.
- Harpaz R, Ortega-Sanchez IR, Seward JF; Advisory Committee on Immunization Practices (ACIP) Centers for Disease Control and Prevention (CDC). Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2008; 57:1–30.
- Panatto D, Bragazzi NL, Rizzitelli E, et al. Evaluation of the economic burden of herpes zoster (HZ) infection. Hum Vaccin Immunother 2015; 11:245–262.
- Yawn BP, Itzler RF, Wollan PC, Pellissier JM, Sy LS, Saddier P. Health care utilization and cost burden of herpes zoster in a community population. Mayo Clin Proc 2009; 84:787–794.
- Dworkin RH, White R, O’Connor AB, Hawkins K. Health care expenditure burden of persisting herpes zoster pain. Pain Med 2008; 9:348–353.
- White RR, Lenhart G, Singhal PK, et al. Incremental 1-year medical resource utilization and costs for patients with herpes zoster from a set of US health plans. Pharmacoeconomics 2009; 27:781–792.
- Insinga RP, Itzler RF, Pellissier JM. Acute/subacute herpes zoster: healthcare resource utilisation and costs in a group of US health plans. Pharmacoeconomics 2007; 25:155–169.
- Singhal PK, Makin C, Pellissier J, Sy L, White R, Saddier P. Work and productivity loss related to herpes zoster. J Med Econ 2011; 14:639–645.
- US Bureau of Labor Statistics. Labor force statistics from the current population survey. www.bls.gov/web/empsit/cpseea13.htm. Accessed April 6, 2017.
- US Bureau of Labor Statistic. Occupational employment statistics. www.bls.gov/oes/current/oes_nat.htm. Accessed April 6, 2017.
- Oxman MN, Levin MJ, Johnson GR, et al; Shingles Prevention Study Group. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 2005; 352:2271–2284.
- Food and Drug Administration (FDA). FDA clinical briefing document for Merck & Co., Inc. Zoster vaccine live (Oka/Merck) Zostavax. www.fda.gov/ohrms/dockets/ac/05/briefing/5-4198b2_1.pdf. Accessed April 6, 2017.
- Simberkoff MS, Arbeit RD, Johnson GR, et al; Shingles Prevention Study Group. Safety of herpes zoster vaccine in the shingles prevention study: a randomized trial. Ann Intern Med 2010; 152:545–554.
- Schmader KE, Oxman MN, Levin MJ, et al; Shingles Prevention Study Group. Persistence of the efficacy of zoster vaccine in the shingles prevention study and the short-term persistence substudy. Clin Infect Dis 2012; 55:1320–1328.
- Morrison VA, Johnson GR, Schmader KE, et al; Shingles Prevention Study Group. Long-term persistence of zoster vaccine efficacy. Clin Infect Dis 2015; 60:900–909.
- Levin MJ, Schmader KE, Pang L, et al. Cellular and humoral responses to a second dose of herpes zoster vaccine administered 10 years after the first dose among older adults. J Infect Dis 2016; 213:14–22.
- Schmader KE, Levin MJ, Gnann JW Jr, et al. Efficacy, safety, and tolerability of herpes zoster vaccine in persons aged 50-59 years. Clin Infect Dis 2012; 54:922–928.
- Hales CM, Harpaz R, Ortega-Sanchez I, Bialek SR; Centers for Disease Control and Prevention (CDC). Update on recommendations for use of herpes zoster vaccine. MMWR Morb Mortal Wkly Rep 2014; 63:729–731.
- Centers for Disease Control and Prevention (CDC). Surveillance of vaccination coverage among adult populations—United States, 2014. MMWR Morb Mortal Wkly Rep 2016; 65(1):1–36. Accessed April 12, 2017.
- Williams WW, Lu PJ, O’Halloran A, et al; Centers for Disease Control and Prevention (CDC). Vaccination coverage among adults, excluding influenza vaccination—United States, 2013. MMWR Morb Mortal Wkly Rep 2015; 64:95–102.
- Oxman MN. Zoster vaccine: current status and future prospects. Clin Infect Dis 2010; 51:197–213.
- Hurley LP, Lindley MC, Harpaz R, et al. Barriers to the use of herpes zoster vaccine. Ann Intern Med 2010; 152:555–560.
- Lu PJ, Euler GL, Jumaan AO, Harpaz R. Herpes zoster vaccination among adults aged 60 years or older in the United States, 2007: uptake of the first new vaccine to target seniors. Vaccine 2009; 27:882–887.
- Hurley LP, Harpaz R, Daley MF, et al. National survey of primary care physicians regarding herpes zoster and the herpes zoster vaccine. J Infect Dis 2008; 197(suppl 2):S216–S223.
- Joon Lee T, Hayes S, Cummings DM, et al. Herpes zoster knowledge, prevalence, and vaccination rate by race. J Am Board Fam Med 2013; 26:45–51.
- Opstelten W, van Essen GA, Hak E. Determinants of non-compliance with herpes zoster vaccination in the community-dwelling elderly. Vaccine 2009; 27:192–196.
- Loo TS, Davis RB, Lipsitz LA, et al. Electronic medical record reminders and panel management to improve primary care of elderly patients. Arch Intern Med 2011; 171:1552–1558.
- Rhew DC, Glassman PA, Goetz MB. Improving pneumococcal vaccine rates. Nurse protocols versus clinical reminders. J Gen Intern Med 1999; 14:351–356.
- Chaudhry R, Schietel SM, North F, Dejesus R, Kesman RL, Stroebel RJ. Improving rates of herpes zoster vaccination with a clinical decision support system in a primary care practice. J Eval Clin Pract 2013; 19:263–266.
- Otsuka SH, Tayal NH, Porter K, Embi PJ, Beatty SJ. Improving herpes zoster vaccination rates through use of a clinical pharmacist and a personal health record. Am J Med 2013; 126:832.e1–832.e6.
- Taitel MS, Fensterheim LE, Cannon AE, Cohen ES. Improving pneumococcal and herpes zoster vaccination uptake: expanding pharmacist privileges. Am J Manag Care 2013; 19:e309–e313.
- Kawai K, Preaud E, Baron-Papillon F, Largeron N, Acosta CJ. Cost-effectiveness of vaccination against herpes zoster and postherpetic neuralgia: a critical review. Vaccine 2014; 32:1645–1653.
- Szucs TD, Pfeil AM. A systematic review of the cost effectiveness of herpes zoster vaccination. Pharmacoeconomics 2013; 31:125–136.
- Le P, Rothberg MB. Cost-effectiveness of herpes zoster vaccine for persons aged 50 years. Ann Intern Med 2015; 163:489–497.
- Le P, Rothberg MB. Determining the optimal age to vaccinate against herpes zoster: a cost-effectiveness analysis. Society for Medical Decision Making 37th Annual North American Meeting. St. Louis, MO; October 18-21, 2015.
- Cohen JI. Clinical practice: herpes zoster. N Engl J Med 2013; 369:255–263.
- Lal H, Cunningham AL, Godeaux O, et al; ZOE-50 Study Group. Efficacy of an adjuvanted herpes zoster subunit vaccine in older adults. N Engl J Med 2015; 372:2087–2096.
- GlaxoSmithKline plc. GSK’s candidate shingles vaccine demonstrates 90% efficacy against shingles in people 70 years of age and over. www.gsk.com/en-gb/media/press-releases/gsk-s-candidate-shingles-vaccine-demonstrates-90-efficacy-against-shingles-in-people-70-years-of-age-and-over/. Accessed April 6, 2017.
- Reed SG, Orr MT, Fox CB. Key roles of adjuvants in modern vaccines. Nat Med 2013; 19:1597–1608.
- Russell AF, Parrino J, Fisher CL Jr, et al. Safety, tolerability, and immunogenicity of zoster vaccine in subjects on chronic/maintenance corticosteroids. Vaccine 2015; 33:3129–3134.
- Hata A, Asanuma H, Rinki M, et al. Use of an inactivated varicella vaccine in recipients of hematopoietic-cell transplants. N Engl J Med 2002; 347:26–34.
- Mullane KM, Winston DJ, Wertheim MS, et al. Safety and immunogenicity of heat-treated zoster vaccine (ZVHT) in immunocompromised adults. J Infect Dis 2013; 208:1375–1385.
- Stadtmauer EA, Sullivan KM, Marty FM, et al. A phase 1/2 study of an adjuvanted varicella-zoster virus subunit vaccine in autologous hematopoietic cell transplant recipients. Blood 2014; 124:2921–2929.
- Berkowitz EM, Moyle G, Stellbrink HJ, et al. Safety and immunogenicity of an adjuvanted herpes zoster subunit candidate vaccine in HIV-infected adults: a phase 1/2a randomized, placebo-controlled study. J Infect Dis 2015; 211:1279–1287.
KEY POINTS
- HZ continues to be an important public health problem, with substantial morbidity and economic impact. Because of the lack of effective treatment, vaccination provides the best strategy for disease mitigation.
- Physicians can reduce the impact of HZ by educating patients about its complications and recommending immunization for all patients age 60 and older. Patients can protect themselves by seeking vaccination.
- Vaccine protection wanes completely after 10 years, and physicians should be prepared to offer a booster dose should the Advisory Committee on Immunization Practices issue such recommendations.
- Newer vaccines offer promise for greater efficacy, especially for the elderly. For immunocompromised patients, a safe and effective vaccine may be available in the near future.

Diabetes control during Ramadan fasting
An estimated 50 million patients with diabetes worldwide practice daily fasting during Ramadan, the ninth month of the Islamic calendar, which lasts 29 or 30 days. In the United States, Ramadan begins this year at sundown on Friday, May 26, and ends at sundown on Sunday, June 25.
According to the Multi-Country Retrospective Observational Study of the Management and Outcomes of Patients With Diabetes During Ramadan, conducted in 13 countries, 94.2% of Muslim diabetic patients fasted at least 15 days, and 67.6% of these fasted every day.1
The daily fasting period, which may extend from 14 to 18 hours, starts before sunrise and ends after sunset. The meal taken before sunrise is called Suhur, and the meal after sunset is called Iftar. The fast requires abstaining from eating, drinking, sexual activity, medications, and smoking. For diabetic patients, this poses medical challenges, increasing the risk of acute metabolic complications.
The goal of caring for diabetic patients during Ramadan fasting is to help them to fast without major complications and to empower them to modify their lifestyle in order to achieve this goal.
POSSIBLE METABOLIC COMPLICATIONS
Metabolic complications during Ramadan fasting include hypoglycemia, hyperglycemia, diabetic ketoacidosis, dehydration, and thrombosis.
Hypoglycemia
For patients with type 1 diabetes, fasting increases the risk of hypoglycemia 4.7 times, and the risk is 7.5 times higher for patients with type 2 diabetes.2 However, this is often underreported, as mild to moderate hypoglycemia does not usually require medical assistance.
Precipitating factors include long fasting hours, missing the Suhur meal, and failure to modify drug dosage and timing.
Hyperglycemia
The risk of severe hyperglycemia during fasting is 3.2 times higher in patients with type 1 diabetes and 5 times higher in those with type 2 diabetes.2 Precipitating factors include lack of diet control during the Iftar meal and excessive reduction in the dosage of diabetes medications due to fear of hypoglycemia.
Diabetic ketoacidosis
Ketoacidosis can be precipitated by a lack of diet control during the Iftar meal, excessive reduction in the dosage of insulin due to fear of hypoglycemia, acute stress, and illness or infection.
Dehydration and thrombosis
Patients can become dehydrated during long fasting hours in especially hot weather, by sweating during physical activity, and by osmotic diuresis in poorly controlled diabetes.
Diabetes is a procoagulant condition, and dehydration increases the risk of thrombosis.
OVERALL MANAGEMENT GOALS DURING RAMADAN FASTING
Important aspects of managing diabetes during Ramadan fasting are:
- The pre-Ramadan evaluation and risk stratification
- Promoting patient awareness with Ramadan-focused diabetes education
- Providing instruction on dietary modification
- Modification of the dosage and timing of diabetes medication
- Encouraging frequent monitoring of blood glucose levels
- Advising the patient when to break the fast
- Managing complications.
PRE-RAMADAN MEDICAL EVALUATION AND RISK STRATIFICATION
All diabetic patients who fast during Ramadan should undergo an evaluation 1 or 2 months before the start of Ramadan to determine their level of diabetes control and the presence of acute and chronic complications of diabetes and other comorbid conditions. Also important is to determine the patient’s social circumstances, ie, knowledge about diabetes, socioeconomic factors, religious beliefs, educational status, diabetes self-management skills, and family support in case of hypoglycemia or complications.
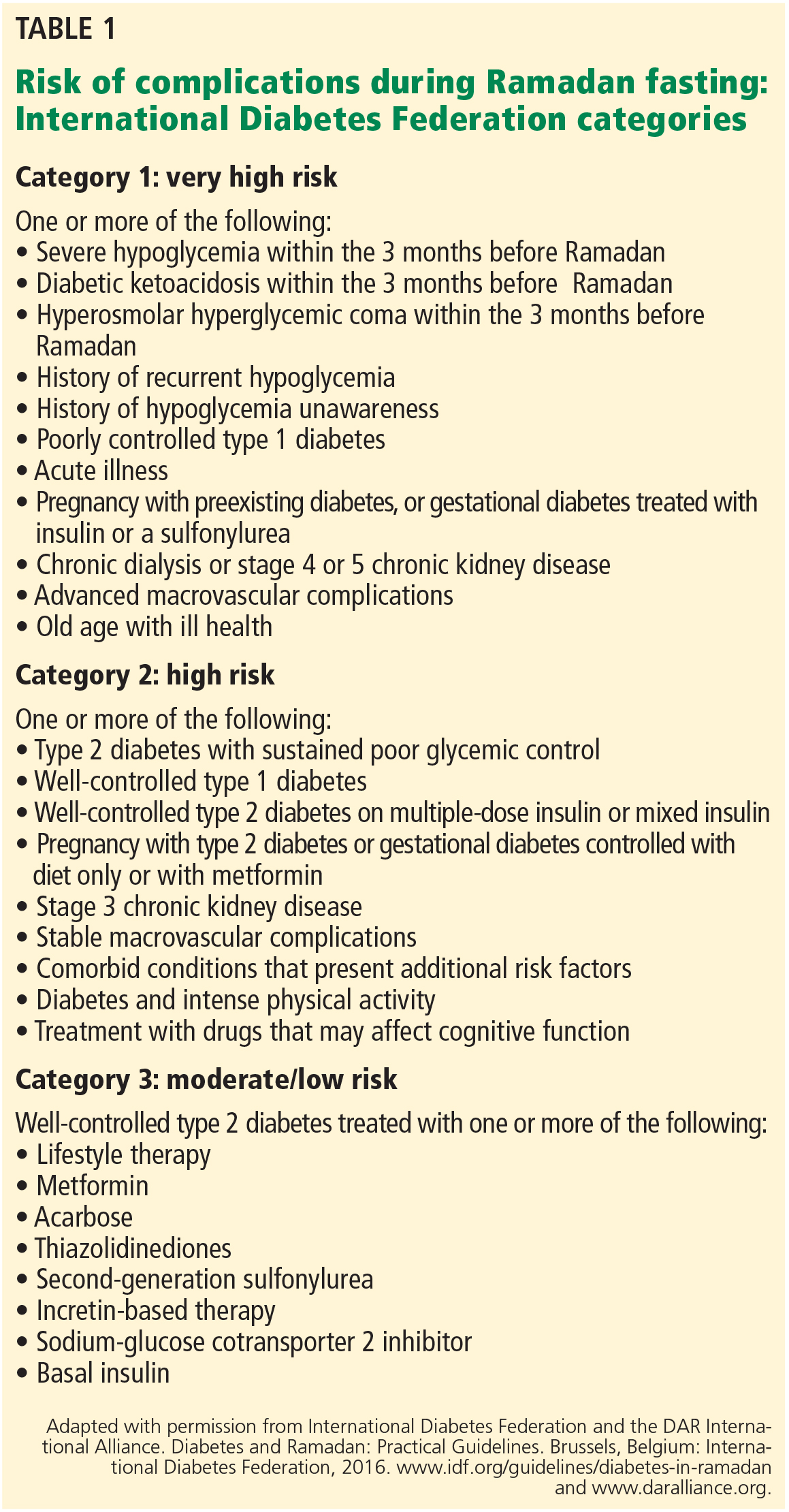
The evaluation helps to determine the patient’s risk of diabetes-related complications from Ramadan fasting, which is categorized as very high, high, or moderate/low according to the criteria of the International Diabetes Federation (Table 1).3 Patients should be advised as to the feasibility of fasting based on this risk categorization.
Even though the recommendation is to avoid fasting if the risk is very high or high, many patients fast. But patients should be advised about Islamic regulations exempting people from fasting (Table 2).4
RAMADAN-FOCUSED DIABETES EDUCATION

Improving the patient’s awareness of the risks of Ramadan fasting reduces the chance of complications. Education should include information on diet and exercise, changes in the timing and dosing of medications, signs and symptoms of hypoglycemia and hyperglycemia, the importance of monitoring blood glucose levels on fasting days, and the importance of breaking the fast in case of complications.5
DIET AND EXERCISE
All diabetic patients should be encouraged to remember to eat the predawn meal on fasting days. They should maintain a balanced diet, with complex carbohydrates with slow energy release for the predawn meal and simple carbohydrates for the sunset meal. Foods with a low glycemic index and high fiber content are recommended, and patients should be advised to avoid saturated fats and to drink plenty of fluids between sunset and sunrise to avoid dehydration.6
Diabetic patients can perform their usual physical activity, including moderate exercise, but should avoid excessive physical activity especially toward evening hours to prevent hypoglycemia.
Some patients may decide not to monitor their blood glucose as they believe that pricking the finger for blood sugar testing breaks the fast.7 Patients should be advised that this is a misconception.
ADJUSTING DIABETES MEDICATIONS
Oral diabetes drugs

Drugs such as metformin, alpha glucosidase inhibitors, thiazolidinediones, the short-acting insulin secretagogue nateglinide, dipeptidyl peptidase 4 inhibitors (eg, sitagliptin), and glucagon-like peptide 1 receptor agonists are associated with a lower risk of hypoglycemia and can be used during Ramadan fasting without significant changes in the daily dose (Table 3).8
Sulfonylureas carry a higher risk of hypoglycemia and should be used cautiously during fasting, with appropriate modification in dose and timing.9,10
Sodium-glucose cotransporter 2 inhibitors, when not combined with insulin or sulfonylureas, carry a lower risk of hypoglycemia, but during Ramadan fasting there is an increased risk of dehydration, urinary tract infection, and postural hypotension since fluids cannot be taken during fasting hours.
Dipeptidyl peptidase 4 inhibitors carry a low risk of hypoglycemia and can be used during Ramadan without dosing modification. Glucagon-like peptide 1 agonists also can be used without adjusting the dosage.11
Insulins
Insulin treatment is associated with a higher risk of hypoglycemia during Ramadan fasting.12 During fasting, the risk of hypoglycemia from premixed insulin can be minimized by changing to a multiple-dose regimen involving a basal insulin and short-acting insulin before meals, with adjustment of the short- acting insulin dose based on the anticipated carbohydrate intake for each meal.13
Patients taking premixed insulin preparations consisting of 70% intermediate-acting or long-acting insulin and 30% short-acting insulin should change to a 50/50 preparation during Ramadan fasting to reduce hypoglycemic risk and improve glycemic control; taking more of the fast-acting component controls postprandial hyperglycemia, and taking less of the intermediate or long-acting component minimizes the risk of hypoglycemia during fasting hours.14,15
Insulin analogues carry a lower risk of hypoglycemia than human insulin. Compared with a human insulin 70/30 preparation, an analogue premix containing 75% neutral protamine lispro and 25% insulin lispro resulted in better glycemic control during Ramadan fasting.16 This could be related to the pharmacodynamics of low-ratio premix analogues, as well as to the mealtime flexibility of analogue insulin, as the injections of the 75/25 mix were given immediately before the morning and evening meals. Insulin analogues are also less likely to cause postprandial hypoglycemia.16
A multiple-dose insulin regimen involving a long-acting basal insulin (eg, glargine, detemir, degludec) and a short-acting insulin (eg, glulisine, aspart, lispro) before meals is preferred in view of better glycemic control and lower risk of hypoglycemia.17
Use of an insulin pump during Ramadan is associated with a reduced risk of hypoglycemia.18 In patients with an insulin pump, the rate of basal insulin must be reduced during daytime, and the postprandial bolus of insulin must be increased after breaking the fast.
FREQUENT MONITORING OF BLOOD GLUCOSE DURING FASTING
Frequent monitoring reduces the risk of both hypoglycemia and hyperglycemia and helps control blood sugar levels during Ramadan fasting. As mentioned above, pricking the finger for blood sugar testing during fasting hours does not break the fast, and this should be emphasized during Ramadan-focused diabetes education.
The exact frequency of blood sugar testing is not defined. In patients with well-controlled diabetes without complications, testing once or twice a day is enough. Patients with poorly controlled diabetes and those with complications should test more often.
ADVICE REGARDING WHEN TO BREAK THE FAST
If signs or symptoms of hypoglycemia develop, the patient should break the fast in order to avoid serious complications. This is acceptable under Islamic law.3,19–21
MANAGEMENT OF COMPLICATIONS
Management of diabetic complications in patients during Ramadan fasting is similar to that for other diabetic patients and includes management of hypo- and hyperglycemia, diabetic ketoacidosis, and dehydration.
- Babineaux SM, Toaima D, Boye KS, et al. Multi-country retrospective observational study of the management and outcomes of patients with type 2 diabetes during Ramadan in 2010 (CREED). Diabet Med 2015; 32:819–828.
- Salti I, Benard E, Detournay B, et al; EPIDIAR Study Group. A population-based study of diabetes and its characteristics during the fasting month of Ramadan in 13 countries: results of the Epidemiology of Diabetes and Ramadan 1422/2001 (EPIDIAR) study. Diabetes Care 2004; 27:2306–2311.
- International Diabetes Federation and the DAR International Alliance. Diabetes and Ramadan: Practical Guidelines. Brussels, Belgium: International Diabetes Federation, 2016. www.idf.org/guidelines/diabetes-in-ramadan and www.daralliance.org. Accessed March 8, 2017.
- Al-Arouj M, Bouguerra R, Buse J, et al. Recommendations for management of diabetes during Ramadan. Diabetes Care 2005; 28:2305–2311.
- Masood SN, Masood Y, Hakim R, Alvi SFD, Shera AS. Ramadan fasting related awareness, practices and experiences of a representative group of urban Pakistani Diabetics. Pak J Med Sci 2012; 28:432–436.
- Bravis V, Hui E, Salih S, Mehar S, Hassanein M, Devendra D. Ramadan education and awareness in diabetes (READ) programme for Muslims with type 2 diabetes who fast during Ramadan. Diabet Med 2010; 27:327–331.
- Masood SN, Sheikh MA, Masood Y, Hakeem R, Shera AS. Beliefs of people with diabetes about skin prick during Ramadan fasting. Diabetes Care 2014; 37:e68–e69.
- Aravind S, Ismail SB, Balamurugan R, et al. Hypoglycemia in patients with type 2 diabetes from India and Malaysia treated with sitagliptin or a sulfonylurea during Ramadan: a randomized, pragmatic study. Curr Med Res Opin 2012; 28:1289–1296.
- Glimepiride in Ramadan (GLIRA) Study Group. The efficacy and safety of glimepiride in the management of type 2 diabetes in Muslim patients during Ramadan. Diabetes Care 2005; 28:421–422.
- Hassanein M, Abdallah K, Schweizer A. A double-blind, randomized trial, including frequent patient-physician contacts and Ramadan-focused advice, assessing vildagliptin and gliclazide in patients with type 2 diabetes fasting during Ramadan: the STEADFAST study. Vasc Health Risk Manag 2014; 10:319–326.
- Brady EM, Davies MJ, Gray LJ, et al. A randomized controlled trial comparing the GLP-1 receptor agonist liraglutide to a sulphonylurea as add on to metformin in patients with established type 2 diabetes during Ramadan: the Treat 4 Ramadan trial. Diabetes Obes Metab 2014; 16:527–536.
- Ibrahim M, Abu Al Magd M, Annabi FA, et al. Recommendations for management of diabetes during Ramadan: update 2015. BMJ Open Diabetes Res Care 2015; 3:e000108.
- Kassem HS, Zantout MS, Azar ST. Insulin therapy during Ramadan fast for type 1 diabetes patients. J Endocrinol Invest 2005; 28:802–805.
- Hui E, Bravis V, Salih S, Hassanein M, Devendra D. Comparison of humalog mix 50 with human insulin mix 30 in type 2 diabetes patients during Ramadan. Int J Clin Pract 2010; 64:1095–1099.
- Hassanein M, Belhadj M, Abdallah K, et al. Management of type 2 diabetes in Ramadan: low ratio premix insulin working group practical advice. Indian J Endocrinol Metab 2014; 18:794–799.
- Mattoo V, Milicevic Z, Malone JK, et al; Ramadan Study Group. A comparison of insulin lispro Mix25 and human insulin 30/70 in the treatment of type 2 diabetes during Ramadan. Diabetes Res Clin Pract 2003; 59:137–143.
- Pathan MF, Sahay RK, Zargar AH, et al. South Asian Consensus Guideline: use of insulin in diabetes during Ramadan. Indian J Endocrinol Metab 2012; 16:499–502.
- Khalil AB, Beshyah SA, Abu Awad SM, et al. Ramadan fasting in diabetes patients on insulin pump therapy augmented by continuous glucose monitoring: an observational real-life study. Diabetes Technol Ther 2012; 14:813–818.
- Holy Qur’an 2:195.
- Holy Qur’an 4:29.
- Bashir MI, Pathan MF, Raza SA. Role of oral hypoglycemic agents in the management of type 2 diabetes mellitus during Ramadan. Indian J Endocrinol Metab 2012; 16:503–507.
An estimated 50 million patients with diabetes worldwide practice daily fasting during Ramadan, the ninth month of the Islamic calendar, which lasts 29 or 30 days. In the United States, Ramadan begins this year at sundown on Friday, May 26, and ends at sundown on Sunday, June 25.
According to the Multi-Country Retrospective Observational Study of the Management and Outcomes of Patients With Diabetes During Ramadan, conducted in 13 countries, 94.2% of Muslim diabetic patients fasted at least 15 days, and 67.6% of these fasted every day.1
The daily fasting period, which may extend from 14 to 18 hours, starts before sunrise and ends after sunset. The meal taken before sunrise is called Suhur, and the meal after sunset is called Iftar. The fast requires abstaining from eating, drinking, sexual activity, medications, and smoking. For diabetic patients, this poses medical challenges, increasing the risk of acute metabolic complications.
The goal of caring for diabetic patients during Ramadan fasting is to help them to fast without major complications and to empower them to modify their lifestyle in order to achieve this goal.
POSSIBLE METABOLIC COMPLICATIONS
Metabolic complications during Ramadan fasting include hypoglycemia, hyperglycemia, diabetic ketoacidosis, dehydration, and thrombosis.
Hypoglycemia
For patients with type 1 diabetes, fasting increases the risk of hypoglycemia 4.7 times, and the risk is 7.5 times higher for patients with type 2 diabetes.2 However, this is often underreported, as mild to moderate hypoglycemia does not usually require medical assistance.
Precipitating factors include long fasting hours, missing the Suhur meal, and failure to modify drug dosage and timing.
Hyperglycemia
The risk of severe hyperglycemia during fasting is 3.2 times higher in patients with type 1 diabetes and 5 times higher in those with type 2 diabetes.2 Precipitating factors include lack of diet control during the Iftar meal and excessive reduction in the dosage of diabetes medications due to fear of hypoglycemia.
Diabetic ketoacidosis
Ketoacidosis can be precipitated by a lack of diet control during the Iftar meal, excessive reduction in the dosage of insulin due to fear of hypoglycemia, acute stress, and illness or infection.
Dehydration and thrombosis
Patients can become dehydrated during long fasting hours in especially hot weather, by sweating during physical activity, and by osmotic diuresis in poorly controlled diabetes.
Diabetes is a procoagulant condition, and dehydration increases the risk of thrombosis.
OVERALL MANAGEMENT GOALS DURING RAMADAN FASTING
Important aspects of managing diabetes during Ramadan fasting are:
- The pre-Ramadan evaluation and risk stratification
- Promoting patient awareness with Ramadan-focused diabetes education
- Providing instruction on dietary modification
- Modification of the dosage and timing of diabetes medication
- Encouraging frequent monitoring of blood glucose levels
- Advising the patient when to break the fast
- Managing complications.
PRE-RAMADAN MEDICAL EVALUATION AND RISK STRATIFICATION
All diabetic patients who fast during Ramadan should undergo an evaluation 1 or 2 months before the start of Ramadan to determine their level of diabetes control and the presence of acute and chronic complications of diabetes and other comorbid conditions. Also important is to determine the patient’s social circumstances, ie, knowledge about diabetes, socioeconomic factors, religious beliefs, educational status, diabetes self-management skills, and family support in case of hypoglycemia or complications.
The evaluation helps to determine the patient’s risk of diabetes-related complications from Ramadan fasting, which is categorized as very high, high, or moderate/low according to the criteria of the International Diabetes Federation (Table 1).3 Patients should be advised as to the feasibility of fasting based on this risk categorization.
Even though the recommendation is to avoid fasting if the risk is very high or high, many patients fast. But patients should be advised about Islamic regulations exempting people from fasting (Table 2).4
RAMADAN-FOCUSED DIABETES EDUCATION
Improving the patient’s awareness of the risks of Ramadan fasting reduces the chance of complications. Education should include information on diet and exercise, changes in the timing and dosing of medications, signs and symptoms of hypoglycemia and hyperglycemia, the importance of monitoring blood glucose levels on fasting days, and the importance of breaking the fast in case of complications.5
DIET AND EXERCISE
All diabetic patients should be encouraged to remember to eat the predawn meal on fasting days. They should maintain a balanced diet, with complex carbohydrates with slow energy release for the predawn meal and simple carbohydrates for the sunset meal. Foods with a low glycemic index and high fiber content are recommended, and patients should be advised to avoid saturated fats and to drink plenty of fluids between sunset and sunrise to avoid dehydration.6
Diabetic patients can perform their usual physical activity, including moderate exercise, but should avoid excessive physical activity especially toward evening hours to prevent hypoglycemia.
Some patients may decide not to monitor their blood glucose as they believe that pricking the finger for blood sugar testing breaks the fast.7 Patients should be advised that this is a misconception.
ADJUSTING DIABETES MEDICATIONS
Oral diabetes drugs
Drugs such as metformin, alpha glucosidase inhibitors, thiazolidinediones, the short-acting insulin secretagogue nateglinide, dipeptidyl peptidase 4 inhibitors (eg, sitagliptin), and glucagon-like peptide 1 receptor agonists are associated with a lower risk of hypoglycemia and can be used during Ramadan fasting without significant changes in the daily dose (Table 3).8
Sulfonylureas carry a higher risk of hypoglycemia and should be used cautiously during fasting, with appropriate modification in dose and timing.9,10
Sodium-glucose cotransporter 2 inhibitors, when not combined with insulin or sulfonylureas, carry a lower risk of hypoglycemia, but during Ramadan fasting there is an increased risk of dehydration, urinary tract infection, and postural hypotension since fluids cannot be taken during fasting hours.
Dipeptidyl peptidase 4 inhibitors carry a low risk of hypoglycemia and can be used during Ramadan without dosing modification. Glucagon-like peptide 1 agonists also can be used without adjusting the dosage.11
Insulins
Insulin treatment is associated with a higher risk of hypoglycemia during Ramadan fasting.12 During fasting, the risk of hypoglycemia from premixed insulin can be minimized by changing to a multiple-dose regimen involving a basal insulin and short-acting insulin before meals, with adjustment of the short- acting insulin dose based on the anticipated carbohydrate intake for each meal.13
Patients taking premixed insulin preparations consisting of 70% intermediate-acting or long-acting insulin and 30% short-acting insulin should change to a 50/50 preparation during Ramadan fasting to reduce hypoglycemic risk and improve glycemic control; taking more of the fast-acting component controls postprandial hyperglycemia, and taking less of the intermediate or long-acting component minimizes the risk of hypoglycemia during fasting hours.14,15
Insulin analogues carry a lower risk of hypoglycemia than human insulin. Compared with a human insulin 70/30 preparation, an analogue premix containing 75% neutral protamine lispro and 25% insulin lispro resulted in better glycemic control during Ramadan fasting.16 This could be related to the pharmacodynamics of low-ratio premix analogues, as well as to the mealtime flexibility of analogue insulin, as the injections of the 75/25 mix were given immediately before the morning and evening meals. Insulin analogues are also less likely to cause postprandial hypoglycemia.16
A multiple-dose insulin regimen involving a long-acting basal insulin (eg, glargine, detemir, degludec) and a short-acting insulin (eg, glulisine, aspart, lispro) before meals is preferred in view of better glycemic control and lower risk of hypoglycemia.17
Use of an insulin pump during Ramadan is associated with a reduced risk of hypoglycemia.18 In patients with an insulin pump, the rate of basal insulin must be reduced during daytime, and the postprandial bolus of insulin must be increased after breaking the fast.
FREQUENT MONITORING OF BLOOD GLUCOSE DURING FASTING
Frequent monitoring reduces the risk of both hypoglycemia and hyperglycemia and helps control blood sugar levels during Ramadan fasting. As mentioned above, pricking the finger for blood sugar testing during fasting hours does not break the fast, and this should be emphasized during Ramadan-focused diabetes education.
The exact frequency of blood sugar testing is not defined. In patients with well-controlled diabetes without complications, testing once or twice a day is enough. Patients with poorly controlled diabetes and those with complications should test more often.
ADVICE REGARDING WHEN TO BREAK THE FAST
If signs or symptoms of hypoglycemia develop, the patient should break the fast in order to avoid serious complications. This is acceptable under Islamic law.3,19–21
MANAGEMENT OF COMPLICATIONS
Management of diabetic complications in patients during Ramadan fasting is similar to that for other diabetic patients and includes management of hypo- and hyperglycemia, diabetic ketoacidosis, and dehydration.
An estimated 50 million patients with diabetes worldwide practice daily fasting during Ramadan, the ninth month of the Islamic calendar, which lasts 29 or 30 days. In the United States, Ramadan begins this year at sundown on Friday, May 26, and ends at sundown on Sunday, June 25.
According to the Multi-Country Retrospective Observational Study of the Management and Outcomes of Patients With Diabetes During Ramadan, conducted in 13 countries, 94.2% of Muslim diabetic patients fasted at least 15 days, and 67.6% of these fasted every day.1
The daily fasting period, which may extend from 14 to 18 hours, starts before sunrise and ends after sunset. The meal taken before sunrise is called Suhur, and the meal after sunset is called Iftar. The fast requires abstaining from eating, drinking, sexual activity, medications, and smoking. For diabetic patients, this poses medical challenges, increasing the risk of acute metabolic complications.
The goal of caring for diabetic patients during Ramadan fasting is to help them to fast without major complications and to empower them to modify their lifestyle in order to achieve this goal.
POSSIBLE METABOLIC COMPLICATIONS
Metabolic complications during Ramadan fasting include hypoglycemia, hyperglycemia, diabetic ketoacidosis, dehydration, and thrombosis.
Hypoglycemia
For patients with type 1 diabetes, fasting increases the risk of hypoglycemia 4.7 times, and the risk is 7.5 times higher for patients with type 2 diabetes.2 However, this is often underreported, as mild to moderate hypoglycemia does not usually require medical assistance.
Precipitating factors include long fasting hours, missing the Suhur meal, and failure to modify drug dosage and timing.
Hyperglycemia
The risk of severe hyperglycemia during fasting is 3.2 times higher in patients with type 1 diabetes and 5 times higher in those with type 2 diabetes.2 Precipitating factors include lack of diet control during the Iftar meal and excessive reduction in the dosage of diabetes medications due to fear of hypoglycemia.
Diabetic ketoacidosis
Ketoacidosis can be precipitated by a lack of diet control during the Iftar meal, excessive reduction in the dosage of insulin due to fear of hypoglycemia, acute stress, and illness or infection.
Dehydration and thrombosis
Patients can become dehydrated during long fasting hours in especially hot weather, by sweating during physical activity, and by osmotic diuresis in poorly controlled diabetes.
Diabetes is a procoagulant condition, and dehydration increases the risk of thrombosis.
OVERALL MANAGEMENT GOALS DURING RAMADAN FASTING
Important aspects of managing diabetes during Ramadan fasting are:
- The pre-Ramadan evaluation and risk stratification
- Promoting patient awareness with Ramadan-focused diabetes education
- Providing instruction on dietary modification
- Modification of the dosage and timing of diabetes medication
- Encouraging frequent monitoring of blood glucose levels
- Advising the patient when to break the fast
- Managing complications.
PRE-RAMADAN MEDICAL EVALUATION AND RISK STRATIFICATION
All diabetic patients who fast during Ramadan should undergo an evaluation 1 or 2 months before the start of Ramadan to determine their level of diabetes control and the presence of acute and chronic complications of diabetes and other comorbid conditions. Also important is to determine the patient’s social circumstances, ie, knowledge about diabetes, socioeconomic factors, religious beliefs, educational status, diabetes self-management skills, and family support in case of hypoglycemia or complications.
The evaluation helps to determine the patient’s risk of diabetes-related complications from Ramadan fasting, which is categorized as very high, high, or moderate/low according to the criteria of the International Diabetes Federation (Table 1).3 Patients should be advised as to the feasibility of fasting based on this risk categorization.
Even though the recommendation is to avoid fasting if the risk is very high or high, many patients fast. But patients should be advised about Islamic regulations exempting people from fasting (Table 2).4
RAMADAN-FOCUSED DIABETES EDUCATION
Improving the patient’s awareness of the risks of Ramadan fasting reduces the chance of complications. Education should include information on diet and exercise, changes in the timing and dosing of medications, signs and symptoms of hypoglycemia and hyperglycemia, the importance of monitoring blood glucose levels on fasting days, and the importance of breaking the fast in case of complications.5
DIET AND EXERCISE
All diabetic patients should be encouraged to remember to eat the predawn meal on fasting days. They should maintain a balanced diet, with complex carbohydrates with slow energy release for the predawn meal and simple carbohydrates for the sunset meal. Foods with a low glycemic index and high fiber content are recommended, and patients should be advised to avoid saturated fats and to drink plenty of fluids between sunset and sunrise to avoid dehydration.6
Diabetic patients can perform their usual physical activity, including moderate exercise, but should avoid excessive physical activity especially toward evening hours to prevent hypoglycemia.
Some patients may decide not to monitor their blood glucose as they believe that pricking the finger for blood sugar testing breaks the fast.7 Patients should be advised that this is a misconception.
ADJUSTING DIABETES MEDICATIONS
Oral diabetes drugs
Drugs such as metformin, alpha glucosidase inhibitors, thiazolidinediones, the short-acting insulin secretagogue nateglinide, dipeptidyl peptidase 4 inhibitors (eg, sitagliptin), and glucagon-like peptide 1 receptor agonists are associated with a lower risk of hypoglycemia and can be used during Ramadan fasting without significant changes in the daily dose (Table 3).8
Sulfonylureas carry a higher risk of hypoglycemia and should be used cautiously during fasting, with appropriate modification in dose and timing.9,10
Sodium-glucose cotransporter 2 inhibitors, when not combined with insulin or sulfonylureas, carry a lower risk of hypoglycemia, but during Ramadan fasting there is an increased risk of dehydration, urinary tract infection, and postural hypotension since fluids cannot be taken during fasting hours.
Dipeptidyl peptidase 4 inhibitors carry a low risk of hypoglycemia and can be used during Ramadan without dosing modification. Glucagon-like peptide 1 agonists also can be used without adjusting the dosage.11
Insulins
Insulin treatment is associated with a higher risk of hypoglycemia during Ramadan fasting.12 During fasting, the risk of hypoglycemia from premixed insulin can be minimized by changing to a multiple-dose regimen involving a basal insulin and short-acting insulin before meals, with adjustment of the short- acting insulin dose based on the anticipated carbohydrate intake for each meal.13
Patients taking premixed insulin preparations consisting of 70% intermediate-acting or long-acting insulin and 30% short-acting insulin should change to a 50/50 preparation during Ramadan fasting to reduce hypoglycemic risk and improve glycemic control; taking more of the fast-acting component controls postprandial hyperglycemia, and taking less of the intermediate or long-acting component minimizes the risk of hypoglycemia during fasting hours.14,15
Insulin analogues carry a lower risk of hypoglycemia than human insulin. Compared with a human insulin 70/30 preparation, an analogue premix containing 75% neutral protamine lispro and 25% insulin lispro resulted in better glycemic control during Ramadan fasting.16 This could be related to the pharmacodynamics of low-ratio premix analogues, as well as to the mealtime flexibility of analogue insulin, as the injections of the 75/25 mix were given immediately before the morning and evening meals. Insulin analogues are also less likely to cause postprandial hypoglycemia.16
A multiple-dose insulin regimen involving a long-acting basal insulin (eg, glargine, detemir, degludec) and a short-acting insulin (eg, glulisine, aspart, lispro) before meals is preferred in view of better glycemic control and lower risk of hypoglycemia.17
Use of an insulin pump during Ramadan is associated with a reduced risk of hypoglycemia.18 In patients with an insulin pump, the rate of basal insulin must be reduced during daytime, and the postprandial bolus of insulin must be increased after breaking the fast.
FREQUENT MONITORING OF BLOOD GLUCOSE DURING FASTING
Frequent monitoring reduces the risk of both hypoglycemia and hyperglycemia and helps control blood sugar levels during Ramadan fasting. As mentioned above, pricking the finger for blood sugar testing during fasting hours does not break the fast, and this should be emphasized during Ramadan-focused diabetes education.
The exact frequency of blood sugar testing is not defined. In patients with well-controlled diabetes without complications, testing once or twice a day is enough. Patients with poorly controlled diabetes and those with complications should test more often.
ADVICE REGARDING WHEN TO BREAK THE FAST
If signs or symptoms of hypoglycemia develop, the patient should break the fast in order to avoid serious complications. This is acceptable under Islamic law.3,19–21
MANAGEMENT OF COMPLICATIONS
Management of diabetic complications in patients during Ramadan fasting is similar to that for other diabetic patients and includes management of hypo- and hyperglycemia, diabetic ketoacidosis, and dehydration.
- Babineaux SM, Toaima D, Boye KS, et al. Multi-country retrospective observational study of the management and outcomes of patients with type 2 diabetes during Ramadan in 2010 (CREED). Diabet Med 2015; 32:819–828.
- Salti I, Benard E, Detournay B, et al; EPIDIAR Study Group. A population-based study of diabetes and its characteristics during the fasting month of Ramadan in 13 countries: results of the Epidemiology of Diabetes and Ramadan 1422/2001 (EPIDIAR) study. Diabetes Care 2004; 27:2306–2311.
- International Diabetes Federation and the DAR International Alliance. Diabetes and Ramadan: Practical Guidelines. Brussels, Belgium: International Diabetes Federation, 2016. www.idf.org/guidelines/diabetes-in-ramadan and www.daralliance.org. Accessed March 8, 2017.
- Al-Arouj M, Bouguerra R, Buse J, et al. Recommendations for management of diabetes during Ramadan. Diabetes Care 2005; 28:2305–2311.
- Masood SN, Masood Y, Hakim R, Alvi SFD, Shera AS. Ramadan fasting related awareness, practices and experiences of a representative group of urban Pakistani Diabetics. Pak J Med Sci 2012; 28:432–436.
- Bravis V, Hui E, Salih S, Mehar S, Hassanein M, Devendra D. Ramadan education and awareness in diabetes (READ) programme for Muslims with type 2 diabetes who fast during Ramadan. Diabet Med 2010; 27:327–331.
- Masood SN, Sheikh MA, Masood Y, Hakeem R, Shera AS. Beliefs of people with diabetes about skin prick during Ramadan fasting. Diabetes Care 2014; 37:e68–e69.
- Aravind S, Ismail SB, Balamurugan R, et al. Hypoglycemia in patients with type 2 diabetes from India and Malaysia treated with sitagliptin or a sulfonylurea during Ramadan: a randomized, pragmatic study. Curr Med Res Opin 2012; 28:1289–1296.
- Glimepiride in Ramadan (GLIRA) Study Group. The efficacy and safety of glimepiride in the management of type 2 diabetes in Muslim patients during Ramadan. Diabetes Care 2005; 28:421–422.
- Hassanein M, Abdallah K, Schweizer A. A double-blind, randomized trial, including frequent patient-physician contacts and Ramadan-focused advice, assessing vildagliptin and gliclazide in patients with type 2 diabetes fasting during Ramadan: the STEADFAST study. Vasc Health Risk Manag 2014; 10:319–326.
- Brady EM, Davies MJ, Gray LJ, et al. A randomized controlled trial comparing the GLP-1 receptor agonist liraglutide to a sulphonylurea as add on to metformin in patients with established type 2 diabetes during Ramadan: the Treat 4 Ramadan trial. Diabetes Obes Metab 2014; 16:527–536.
- Ibrahim M, Abu Al Magd M, Annabi FA, et al. Recommendations for management of diabetes during Ramadan: update 2015. BMJ Open Diabetes Res Care 2015; 3:e000108.
- Kassem HS, Zantout MS, Azar ST. Insulin therapy during Ramadan fast for type 1 diabetes patients. J Endocrinol Invest 2005; 28:802–805.
- Hui E, Bravis V, Salih S, Hassanein M, Devendra D. Comparison of humalog mix 50 with human insulin mix 30 in type 2 diabetes patients during Ramadan. Int J Clin Pract 2010; 64:1095–1099.
- Hassanein M, Belhadj M, Abdallah K, et al. Management of type 2 diabetes in Ramadan: low ratio premix insulin working group practical advice. Indian J Endocrinol Metab 2014; 18:794–799.
- Mattoo V, Milicevic Z, Malone JK, et al; Ramadan Study Group. A comparison of insulin lispro Mix25 and human insulin 30/70 in the treatment of type 2 diabetes during Ramadan. Diabetes Res Clin Pract 2003; 59:137–143.
- Pathan MF, Sahay RK, Zargar AH, et al. South Asian Consensus Guideline: use of insulin in diabetes during Ramadan. Indian J Endocrinol Metab 2012; 16:499–502.
- Khalil AB, Beshyah SA, Abu Awad SM, et al. Ramadan fasting in diabetes patients on insulin pump therapy augmented by continuous glucose monitoring: an observational real-life study. Diabetes Technol Ther 2012; 14:813–818.
- Holy Qur’an 2:195.
- Holy Qur’an 4:29.
- Bashir MI, Pathan MF, Raza SA. Role of oral hypoglycemic agents in the management of type 2 diabetes mellitus during Ramadan. Indian J Endocrinol Metab 2012; 16:503–507.
- Babineaux SM, Toaima D, Boye KS, et al. Multi-country retrospective observational study of the management and outcomes of patients with type 2 diabetes during Ramadan in 2010 (CREED). Diabet Med 2015; 32:819–828.
- Salti I, Benard E, Detournay B, et al; EPIDIAR Study Group. A population-based study of diabetes and its characteristics during the fasting month of Ramadan in 13 countries: results of the Epidemiology of Diabetes and Ramadan 1422/2001 (EPIDIAR) study. Diabetes Care 2004; 27:2306–2311.
- International Diabetes Federation and the DAR International Alliance. Diabetes and Ramadan: Practical Guidelines. Brussels, Belgium: International Diabetes Federation, 2016. www.idf.org/guidelines/diabetes-in-ramadan and www.daralliance.org. Accessed March 8, 2017.
- Al-Arouj M, Bouguerra R, Buse J, et al. Recommendations for management of diabetes during Ramadan. Diabetes Care 2005; 28:2305–2311.
- Masood SN, Masood Y, Hakim R, Alvi SFD, Shera AS. Ramadan fasting related awareness, practices and experiences of a representative group of urban Pakistani Diabetics. Pak J Med Sci 2012; 28:432–436.
- Bravis V, Hui E, Salih S, Mehar S, Hassanein M, Devendra D. Ramadan education and awareness in diabetes (READ) programme for Muslims with type 2 diabetes who fast during Ramadan. Diabet Med 2010; 27:327–331.
- Masood SN, Sheikh MA, Masood Y, Hakeem R, Shera AS. Beliefs of people with diabetes about skin prick during Ramadan fasting. Diabetes Care 2014; 37:e68–e69.
- Aravind S, Ismail SB, Balamurugan R, et al. Hypoglycemia in patients with type 2 diabetes from India and Malaysia treated with sitagliptin or a sulfonylurea during Ramadan: a randomized, pragmatic study. Curr Med Res Opin 2012; 28:1289–1296.
- Glimepiride in Ramadan (GLIRA) Study Group. The efficacy and safety of glimepiride in the management of type 2 diabetes in Muslim patients during Ramadan. Diabetes Care 2005; 28:421–422.
- Hassanein M, Abdallah K, Schweizer A. A double-blind, randomized trial, including frequent patient-physician contacts and Ramadan-focused advice, assessing vildagliptin and gliclazide in patients with type 2 diabetes fasting during Ramadan: the STEADFAST study. Vasc Health Risk Manag 2014; 10:319–326.
- Brady EM, Davies MJ, Gray LJ, et al. A randomized controlled trial comparing the GLP-1 receptor agonist liraglutide to a sulphonylurea as add on to metformin in patients with established type 2 diabetes during Ramadan: the Treat 4 Ramadan trial. Diabetes Obes Metab 2014; 16:527–536.
- Ibrahim M, Abu Al Magd M, Annabi FA, et al. Recommendations for management of diabetes during Ramadan: update 2015. BMJ Open Diabetes Res Care 2015; 3:e000108.
- Kassem HS, Zantout MS, Azar ST. Insulin therapy during Ramadan fast for type 1 diabetes patients. J Endocrinol Invest 2005; 28:802–805.
- Hui E, Bravis V, Salih S, Hassanein M, Devendra D. Comparison of humalog mix 50 with human insulin mix 30 in type 2 diabetes patients during Ramadan. Int J Clin Pract 2010; 64:1095–1099.
- Hassanein M, Belhadj M, Abdallah K, et al. Management of type 2 diabetes in Ramadan: low ratio premix insulin working group practical advice. Indian J Endocrinol Metab 2014; 18:794–799.
- Mattoo V, Milicevic Z, Malone JK, et al; Ramadan Study Group. A comparison of insulin lispro Mix25 and human insulin 30/70 in the treatment of type 2 diabetes during Ramadan. Diabetes Res Clin Pract 2003; 59:137–143.
- Pathan MF, Sahay RK, Zargar AH, et al. South Asian Consensus Guideline: use of insulin in diabetes during Ramadan. Indian J Endocrinol Metab 2012; 16:499–502.
- Khalil AB, Beshyah SA, Abu Awad SM, et al. Ramadan fasting in diabetes patients on insulin pump therapy augmented by continuous glucose monitoring: an observational real-life study. Diabetes Technol Ther 2012; 14:813–818.
- Holy Qur’an 2:195.
- Holy Qur’an 4:29.
- Bashir MI, Pathan MF, Raza SA. Role of oral hypoglycemic agents in the management of type 2 diabetes mellitus during Ramadan. Indian J Endocrinol Metab 2012; 16:503–507.
KEY POINTS
- A diabetic patient who develops signs or symptoms of hypoglycemia during Ramadan fasting should break the fast to avoid serious complications.
- Management of complications in diabetic patients during Ramadan is similar to that for nonfasting diabetic patients. Complications include hypo- and hyperglycemia, diabetic ketoacidosis, and dehydration.
- A common misconception among patients is that pricking the finger for blood sugar testing during fasting hours breaks the fast; this should be addressed during Ramadan-focused diabetes education.

Peripartum depression: Early recognition improves outcomes
Contrary to common belief, pregnancy does not confer protection against depression.1,2 In fact, pregnant women are just as likely as nonpregnant women to become or remain depressed, and up to 12.7% of pregnant women meet criteria for depression.1
In the postpartum period, women are particularly vulnerable to a major depressive episode, whether a first episode or a recurrence. The estimated prevalence of a depressive episode in the first 3 postpartum months is 19.2%,2 making postpartum depression the most common complication of childbearing.2 At the same time, peripartum depression remains largely underrecognized and undertreated.3
As evidence mounts regarding the deleterious impact of untreated mental illness on the mother, the developing fetus, and the infant, early detection and intervention for peripartum depression are paramount.3
DEPRESSION DURING PREGNANCY: SIGNIFICANT CONSEQUENCES
Although the rates of depression in pregnant and nonpregnant women are similar, depression in pregnancy carries additional significant consequences. Further, many depressed pregnant women believe their depression will lift once their baby is born, though it is well documented that depression during pregnancy is the strongest predictor of postpartum depression and that if left untreated it can be devastating for mother, infant, and family.4
Compared with nondepressed pregnant women, depressed pregnant women have poorer overall health status,5 are more likely to engage in behaviors that pose risk to the developing fetus such as smoking,5 alcohol consumption, and substance use,6 and have poor nutrition and inadequate weight gain.7,8
Pregnant women who are depressed and are also experiencing domestic violence are especially at risk for poor prenatal care as they tend to miss more prenatal appointments.9 Evidence also suggests that depressed pregnant women are less attached to the fetus and more likely to have elective terminations.10,11
Depression in pregnancy is associated with higher rates of adverse pregnancy outcomes such as preterm birth, low birth weight, operative delivery, and longer predelivery hospital stay.3,12 Depression and anxiety during pregnancy have been associated with prenatal hypertension,13 gestational diabetes,14 preeclampsia,15 and HELLP syndrome (ie, hemolysis, elevated liver enzymes, and low platelet count).15 Depression and anxiety during pregnancy are associated with subsequent poorer infant attachment16,17 and an overall unfavorable impact on infant and child development.18
Risk factors for depression during pregnancy include past episodes of depression, current anxiety, poor social support, unintended pregnancy, life stress, being single, domestic violence, and being on Medicaid.19
Undoubtedly the most devastating consequence of severe depression during pregnancy is suicide. Rates of suicide are lower in peripartum women,20 but when suicide does occur, pregnant women tend to use more violent means than nonpregnant women. Pregnant adolescents represent a particularly high-risk group.21
POSTPARTUM DEPRESSION
Postpartum depression is the most common complication of childbearing. Although the precise pathogenesis is undetermined, there is converging evidence of a subset of women particularly sensitive to dramatic fluctuations in levels of estradiol and progesterone that occur during childbirth.22,23 There is also evidence that dysregulation of the hypothalamic-pituitary-adrenal axis contributes to the development of postpartum depression in certain women.24 Further, women who have depression or anxiety during pregnancy are much more likely to experience postpartum depression than those who are not symptomatic during pregnancy.4 A history of peripartum depression or other lifetime depressive episodes, poverty, conflict with a primary partner, poor social support, stressful life events, and low self-esteem are strongly associated with postpartum depression.25

When unrecognized and untreated, postpartum depression can have profound and persistent effects on the mother and the developing infant.18,26 Mothers with postpartum depression are much more likely than mothers without depression to have impaired bonding,27 to be less responsive to their infant’s needs,17 and to be more likely to miss well-baby checkups.28
Postpartum depression’s effects on maternal-infant interactions can include maternal withdrawal, disengagement, intrusion, and hostility and can lead to long-term effects on child development, including poor cognitive functioning, emotional maladjustment, and behavioral inhibition.29,30 Infants and children of mothers with untreated postpartum depression have been shown to exhibit a higher incidence of colic, excessive crying, sleep problems, and irritability.31,32 Women with postpartum depression may be less likely to initiate or maintain breastfeeding, and depressive symptoms have been noted to precede the discontinuation of breastfeeding.33–35
Risk factors for postpartum depression
Characteristics to look for in the prenatal care of pregnant women include the following:
- Depression during pregnancy
- History of postpartum or other depressive episode
- Poverty
- Conflict with primary partner
- Poor social support
- Low self-esteem
- Single status.
DIFFERENTIATING ‘POSTPARTUM BLUES’ FROM MAJOR DEPRESSION
Primary care providers are often the first point of contact for depressed women. The diagnosis of major depression in pregnant and postpartum women is challenging because of changes in sleep, appetite, and energy brought on by pregnancy, complications of delivery, and demands of caring for a newborn.36 Many pregnant and postpartum women are reluctant to disclose their symptoms due to a sense of shame and guilt for being depressed during a time in their life that society commonly regards as joyful, and this contributes to under-detection.

In the first few days postpartum, fatigue, emotionality, irritability, and worry over the infant’s well-being affect up to 75% of women. This period, typically referred to as the “baby blues” or “postpartum blues,” is not considered a disorder and responds well to support, reassurance, and adequate sleep, and it typically resolves within 2 weeks.37,38 Table 1 lists features that help distinguish postpartum blues from major depression.
Signs of major depressive disorder
Major depressive disorder is a serious and disabling condition. To meet criteria for major depressive disorder, women must report depressed mood and loss of interest or pleasure in normally pleasurable activities for at least 2 weeks. Completing the symptom profile, at least 5 of the following must be present: sleep disturbance (insomnia or hypersomnia), lack of energy, feelings of worthlessness or low self-esteem, guilt, difficulty concentrating, indecisiveness, psychomotor retardation or agitation, and thoughts of suicide or death.
The Diagnostic and Statistical Manual of Mental Disorders (5th edition) recognizes that postpartum depression commonly begins during pregnancy, and now uses “peripartum onset” as the specifier for major depressive disorder that occurs during pregnancy, postpartum, or both.39 Other hallmark symptoms with peripartum onset include a lack of interest in or attachment to the pregnancy or infant, and anxiety and worry often accompanied by intrusive, unwanted thoughts of harm befalling the infant.40
Postpartum psychosis
Postpartum psychosis is a far less common presentation, occurring in 1 to 2 per 1,000 births, but it constitutes a psychiatric emergency requiring immediate referral to a psychiatric care setting. Women at highest risk are those with a personal or family history of bipolar disorder.
The clinical presentation is most commonly characterized by confusion, agitation, hallucinations, delusional beliefs, and disorientation. Suicide and infanticide, while rare, are more likely to occur in the context of a psychotic episode.41
SCREENING RECOMMENDATIONS
Screening for depression is routine in primary care settings and is no less important for peripartum women.
In 2016, the US Preventive Services Task Force issued a recommendation that all pregnant and postpartum women be screened for depression,42 highlighting the need for all medical providers to be alert to the potentially serious consequences of unrecognized and untreated maternal psychiatric illness.
The American College of Obstetricians and Gynecologists (ACOG) recommends screening for depression and anxiety at least once during the peripartum period,43 and the American Academy of Pediatrics recommends screening mothers for depression at the 1-, 2-, and 4-month well-baby visits.44
The peripartum period is associated with changes in sleep, appetite, and energy levels, but these are also typical of depression. Taking this into account, the Edinburgh Postnatal Depression Scale (EPDS) was developed to screen for depression specifically in this population.45 The EPDS is a validated and widely used 10-item self-reporting questionnaire with a high degree of sensitivity and specificity; it is easily administered and quickly scored. A cutoff score of 13 (of a maximum of 30) is considered indicative of depressed mood and signals the need for further assessment.

ACOG, the American Academy of Pediatrics, and the US Preventive Services Task Force recommend a standardized validated tool and cite both the EPDS (https://psychology-tools.com/epds/) and the Patient Health Questionnaire-9 (PHQ-9) (Figure 1) as appropriate to screen for peripartum depression.42–44 Primary care providers tend to be most familiar with the PHQ-9, a highly sensitive and specific 9-item depression screen that has been validated in primary care and obstetric clinic patients.46 A score on the PHQ-9 ranging from 5 to 10 indicates mild depression, 10 to 14 moderate depression, 15 to 19 moderate to severe depression, and greater than 19 severe depression.
CLINICAL MANAGEMENT
Many women prefer nondrug therapy
The gold standard treatment for moderate to severe major depressive disorder is psychotherapy plus pharmacotherapy. Yet many peripartum women voice concerns about exposure to pharmacologic treatment, and studies have shown that many women prefer nonpharmacologic intervention.47
Evidence-based psychotherapies that have demonstrated efficacy in peripartum women include cognitive behavioral therapy48 and interpersonal psychotherapy when administered by a psychotherapist trained in these treatments. Pregnant and breastfeeding women often express preference for psychotherapy and complementary and alternative treatments as a means of avoiding fetal and infant exposure to antidepressants.47
For mild to moderate depression, complementary therapies such as exercise, yoga, bright light therapy, and acupuncture have shown efficacy and can be used alone or adjunctively.49 Because a poor marital relationship is consistently associated with peripartum depression,25 primary care physicians who routinely address social support and screen for family conflict are well positioned to detect this significant correlate and to recommend marital or family therapy as a primary or adjunctive treatment.
When to consider drug therapy
The decision to recommend drug therapy must be individualized and based on the severity of symptoms, functional impairment, number and frequency of depressive episodes, history of response to medications, and the preferences of the patient, with the recognition that no decision is risk-free and that antidepressants enter the amniotic fluid, so fetal exposure is unavoidable.
Table 2 lists common antidepressants. The antidepressants most commonly prescribed, especially in the primary care setting, are selective serotonin reuptake inhibitors (SSRIs), which are favored because of their effectiveness, low side-effect profile, and lack of overdose toxicity.

Serotonin syndrome is no more likely to occur in pregnant than in nonpregnant women. Close monitoring for this condition is warranted only when patients are taking very high doses of SSRIs or SSRIs in combination with other serotonergic agonists.
Prescribing antidepressants for pregnant or breastfeeding women requires thoughtful consideration of the patient’s preferences, as well as weighing the risks and benefits of fetal and infant exposure to maternal depression vs exposure to medications. Additional considerations include monotherapy, avoiding medication changes, choosing drugs that have been effective in the past, and avoiding drugs with known drug-drug interactions or teratogenic effects.50
There is increasing consensus that the short- and long-term consequences of undertreatment or nontreatment of maternal depression outweigh the risk of fetal exposure to SSRIs.3,51,52 Cohen et al53 have recommended that if a woman is on an antidepressant and learns she is pregnant, she should not discontinue it because of the likelihood of relapse; they found a 68% relapse rate in women who discontinued their antidepressant in the first trimester of pregnancy.53
In a comprehensive review of studies published between 1996 and 2012 that examined antidepressant use during pregnancy, Byatt et al54 found little or no evidence of increased teratogenic risk with antidepressants with the exception of paroxetine, which is associated with a small but significant increased risk of cardiac malformation during first-trimester exposure.54
These conclusions were underscored in a large cohort study in the United Kingdom.55 In addition, a joint task force of the American Psychiatric Association and ACOG reviewed studies looking at the association between depression, antidepressants, and birth outcomes including miscarriage, preterm birth, cardiac abnormalities (resulting from first trimester exposure), persistent pulmonary hypertension (related to second- and third-trimester exposure), and neonatal adaptation syndrome (associated with third-trimester exposure).8 They concluded that the available data neither support nor refute a link between the use of antidepressants and several of the above outcomes. No increase in risk of congenital malformations (including cardiac abnormalities) was found. An increased risk of persistent pulmonary hypertension was noted, although the absolute risk of this disorder remained low, at 3 to 6 per 1,000 infants exposed to SSRIs in utero.8,56
Neonatal adaptation syndrome
Neonatal adaptation syndrome is characterized by jitteriness, irritability, decreased muscle tone, and feeding difficulty in the neonate. It can occur in 15% to 30% of infants exposed to SSRIs antenatally.57,58 These symptoms, however, are transient and typically resolve within 7 to 10 days after birth. A more recent study suggested that neurobehavioral symptoms for some infants extend beyond 2 weeks and that concomitant exposure to benzodiazepines results in even higher rates of this syndrome.59 There is no evidence that tapering or discontinuing antidepressants near term is necessary, safe, or effective in preventing transient neonatal complications. However, this approach would increase the risk of relapse for the mother.
Autism spectrum disorders
The possible association between antidepressants and autism spectrum disorders in pregnancy has captured much attention in recent years. One study based on healthcare claims60 and one registry-based study61 associated in utero exposure to antidepressants with autism liability in children. However, a large-scale Danish registry-based study did not replicate this association.62 In addition, 2 recent cohort studies, identifying children with autism spectrum disorder or attention-deficit hyperactivity disorder from electronic health records, found that neither disorder was significantly associated with prenatal antidepressant exposure in crude or adjusted models. However, both studies found a significant association with the use of antidepressants before pregnancy, indicating that the risk of autism observed with prenatal antidepressant exposure is likely confounded by the severity of maternal illness.63,64
Concerns about drug therapy during breastfeeding
For infants of breastfeeding women, exposure to antidepressants through breast milk is minimal. Amounts in breast milk depend on the timing of the antidepressant dose, timing of feeding, and genetically influenced metabolic activity in mother and infant. The current literature supports antidepressant use for breastfeeding mothers of healthy full-term infants.65
The 2 most widely studied antidepressants in breastfed infants are paroxetine and sertraline. It has been shown that very little can be detected in the infant’s serum, with relative infant doses ranging from 0.4% to 2.8%.65 While clinicians are cautioned against prescribing paroxetine for pregnant women, the drug remains a suitable alternative for breastfeeding women.
If an antidepressant is started postpartum, the recommendation is to start with a low dose and then slowly titrate upward while monitoring the infant for adverse effects.65,66 Possible adverse effects in breastfeeding infants include irritability, sedation, poor weight gain, and a change in feeding patterns.67 Adverse events are most likely to occur in newborns up to 8 weeks of age, and infants born prematurely or with medical problems may be particularly at risk.65,68
Helping patients weigh risks and benefits of drug therapy
Women may hear about the risks of medications to the fetus and during breastfeeding and so may be reluctant to seek or accept intervention. Often, the information is not from a reliable, scientifically based source. Primary care physicians are well positioned to guide peripartum women in risk-benefit analysis of proper treatment of their depression vs no treatment or undertreatment. In addition, establishing referral sources—ideally with a peripartum mental health specialist—is advisable. Online resources that clinicians can refer patients to for help in managing peripartum depression include the following:
- www.postpartum.net
- www.womensmentalhealth.org
- www.mothertobaby.org (for pharmacologic guidance).
INCREASED AWARENESS IS KEY
Primary care physicians must remain alert to the high prevalence of depression in women of childbearing age and embrace routine screening for depression. (See the sidebar, “The primary care management of peripartum depression.”) Since half of pregnancies are unintended, awareness of the risks of undetected and untreated peripartum depression to the mother, developing fetus, and infant is essential. Untreated antepartum depression has been linked to poor pregnancy outcomes, nutritional deficits, and substance abuse. Untreated postpartum depression negatively affects mother-infant attachment, infant, and child development and maternal self care.
Not treating depression is hazardous
Drug treatment during pregnancy and breastfeeding poses challenges for the patient and physician due to the inevitability of fetal and infant exposure, but lack of treatment can be hazardous.
To date, the evidence on the use of antidepressants in pregnant and lactating women is reassuring. Specialized peripartum psychiatric partial hospital programs69 and inpatient programs70 exist for women who need a higher level of care. There is also substantial evidence that psychotherapy, especially cognitive behavioral therapy and interpersonal therapy, is highly effective, and emerging data on complementary and alternative treatments are promising. Coordinated care between primary care and behavioral healthcare providers with expertise in treating peripartum depression is most likely to yield optimal outcomes.
- World Health Organization (WHO). A message from the Director General. www.who.int/whr/2001/dg_message/en/index.html. Accessed March 6, 2017.
- Gavin NI, Gaynes BN, Lohr KN, Meltzer-Brody S, Gartlehner G, Swinson T. Perinatal depression: a systematic review of prevalence and incidence. Obset Gynecol 2005; 106:1071–1083.
- Davalos DB, Yadon CA, Tregellas HC. Untreated prenatal maternal depression and the potential risks to offspring: a review. Arch Women’s Mental Health 2012; 15:1–14.
- Chaudron LH, Klein MH, Remington P, Palta M, Allen C, Essex MJ. Predictors, prodromes and incidence of postpartum depression. J Psychosom Obstet Gynaecol 2001; 22:103–112.
- Orr ST, Blazer DG, Orr CA. Maternal prenatal depressive symptoms, nicotine addiction, and smoking-related knowledge, attitudes, beliefs, and behaviors. Matern Child Health J 2012; 16:973–978.
- Flynn HA, Chermack ST. Prenatal alcohol use: the role of lifetime problems with alcohol,drugs, depression, and violence. J Stud Alcohol Drugs 2008; 69:500–509.
- Bodnar LM, Wisner KL, Moses-Kolko E, Sit DK, Hanusa BH. Prepregnancy body mass index, gestational weight gain, and the likelihood of major depressive disorder during pregnancy. J Clin Psychiatry 2009; 70:1290–1296.
- Yonkers KA, Wisner KL, Stewart DE, et al. The management of depression during pregnancy: a report from the American Psychiatric Association and the American College of Obstetricians and Gynecologists. Obstet Gynecol 2009; 114:703–713.
- Han A, Stewart DE. Maternal and fetal outcomes of intimate partner violence associated with pregnancy in the Latin American and Caribbean region. Int J Gynecol Obstet 2014; 124:6–11.
- McFarland J, Salisbury AL, Battler CL, Hawes K, Halloran K, Lester BM. Major depressive disorder during pregnancy and emotional attachment to the fetus. Arch Womens Ment Health 2011; 14:425–434.
- Suri R, Althuler LA, Mintz J. Depression and the decision to abort. Am J Psychiatry 2004; 161:1502.
- Kim DR, Sockol LE, Sammel MD, Kelly C, Moseley M, Epperson CN. Elevated risk of adverse obstetric outcomes in pregnant women with depression. Arch Women’s Ment Health 2013; 16:475–482.
- Mautner E, Greimel E, Trutnovsky G, Daghofer F, Egger JW, Lang U. Quality of life outcomes in pregnancy and postpartum complicated by hypertensive disorders, gestational diabetes, and preterm birth. J Psychosom Obstet Gynaecol 2009; 30:231–237.
- Katon JG, Russo J, Gavin AR, Melville JL, Katon WJ. Diabetes and depression in pregnancy: is there an association? J Women’s Health (Larchmt) 2011; 20:983–989.
- Delahaije DH, Dirksen CD, Peeters LL, Smits LJ. Anxiety and depression following preeclampsia or HELLP syndrome: a systematic review. Acta Obstet Gynecol Scand 2013; 92:746–761.
- O’Higgins M, Roberts IS, Glover V, Taylor A. Mother-child bonding at 1 year; associations with symptoms of postnatal depression and bonding in the first few weeks. Arch Women’s Ment Health 2013; 16:381–389.
- Field T, Healy BT, Goldstein S, Guthertz M. Behavior-state matching and synchrony in mother-infant interactions of nondepressed versus depressed dyads. Dev Psychol 1990; 26:7–14.
- Kingston D, Tough S, Whitfield H. Prenatal and postpartum maternal psychological distress and infant development: a systematic review. Child Psychiatry Hum Dev 2012; 43:683–714.
- Lancaster CA, Gold KJ, Flynn HA, Yoo H, Marcus SM, Davis MM. Risk factors for depressive symptoms during pregnancy: a systematic review. Am J Obstet Gynecol 2010; 202:5–14.
- Lindahl V, Pearson JL, Colpe L. Prevalence of suicidality during pregnancy and the postpartum. Arch Women’s Ment Health 2005; 8:77–87.
- Appleby L. Suicide after pregnancy and the first postnatal year. BMJ 1991; 302:137–140.
- Bloch M, Schmidt PJ, Danaceau M, Murphy J, Nieman L, Rubinow DR. Effects of gonadal steroids in women with a history of postpartum depression. Am J Psychiatry 2000; 157:924–930.
- Workman JL, Barha CK, Galea LAM. Endocrine substrates of cognitive and affective changes during pregnancy and postpartum. Behav Neurosci 2012; 126:54–72.
- Meltzer-Brody S. New insights into perinatal depression: pathogenesis and treatment during pregnancy and postpartum. Dialogues Clin Neurosci 2011; 13:89–100.
- O’Hara MW, McCabe JE. Postpartum depression: current status and future directions. Annu Rev Clin Psychol 2013; 9:379–407.
- Goodman SH, Rouse MH, Connell AM, Broth MR, Hall CM, Heyward D. Maternal depression and child psychopathology: a meta-analytic review. Clin Child Fam Psychol Rev 2011; 14:1–27
- Muzik M, Bocknek EL, Broderick A, et al. Mother-infant bonding impairment across the first 6 months postpartum: the primacy of psychopathology in women with childhood abuse and neglect histories. Arch Women’s Ment Health 2013; 16:29–38.
- Farr SL, Dietz PM, Rizzo JH, et al. Health care utilisation in the first year of life among infants of mothers with perinatal depression or anxiety. Paediatr Perinat Epidemiol 2013; 27:81–88.
- Grace SL, Evindar A, Stewart DE. The effect of postpartum depression on child cognitive development and behavior: a review and critical analysis of the literature. Arch Women’s Ment Health 2003; 6:263–274.
- Murray L, Cooper PJ. Postpartum depression and child development. Psychol Med 1997; 27:253–260.
- Orhon FS, Ulukol B, Soykan A. Postpartum mood disorders and maternal perceptions of infant patterns in well-child follow-up visits. Acta Paediatr 2007; 96:1777–1783.
- Dennis CL, Ross L. Relationships among infant sleep patterns, maternal fatigue, and development of depressive symptomatology. Birth 2005; 32:187–193.
- Ip S, Chung M, Raman G, et al. Breastfeeding and maternal and infant health outcomes in developed countries. Evid Rep Technol Assess (Full Rep) 2007; 153:1–186.
- Dennis CL, McQueen K. Does maternal postpartum depressive symptomatology influence infant feeding outcomes? Acta Paediatr 2007; 96:590–594.
- Hatton DC, Harrison-Hohner J, Coste S, Dorato V, Curet LB, McCarron DA. Symptoms of postpartum depression and breastfeeding. J Hum Lact 2005; 21:444–449.
- Klein MH, Essex MJ. Pregnant or depressed? The effect of overlap between symptoms of depression and somatic complaints of pregnancy on rates of major depression in the second trimester. Depression 1994; 2:308–314.
- Seyfried LS, Marcus SM. Postpartum mood disorders. Int Rev Psychiatry 2003; 15:231–242.
- Buttner MM, O’Hara MW, Watson D. The structure of women’s mood in the early postpartum. Assessment 2012; 19:247–256.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA; American Psychiatric Association Publishing: 2013.
- Wisner KL, Peindl KS, Gigliotti T, Hanusa BH. Obsessions and compulsions in women with postpartum depression. J Clin Psychiatry 1999: 60:176-180.
- Di Florio A, Smith S, Jones I. Postpartum psychosis. The Obstetrician & Gynecologist 2013; 15:145–150.
- O’Connor E, Rossom RC, Henniger M, Groom HC, Burda BU. Primary care screening for and treatment of depression in pregnant and postpartum women: evidence report and systematic review for the US Preventive Services Task Force. JAMA 2016; 315:388–406.
- Committee on Obstetric Practice. The American College of Obstetricians and Gynecologists Committee Opinion no. 630. Screening for perinatal depression. Obstet Gynecol 2015; 125:1268–1271.
- Earls MF; Committee on Psychosocial Aspects of Child and Family Health American Academy of Pediatrics. Incorporating recognition and management of perinatal and postpartum depression into pediatric practice. Pediatrics 2010; 126:1032–1039.
- Cox JL, Holden JM, Sagovsky R. Detection of postnatal depression: development of the 10-item Edinburgh postnatal depression scale. Br J Psychiatry 1987; 150:782–786.
- Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med 2001; 16:606–616.
- Battle CL, Salisbury AL, Schofield CA, Ortiz-Hernandez S. Perinatal antidepressant use: understanding women’s preferences and concerns. J Psychiatr Pract 2013; 19:443–453.
- Stuart S, Koleva H. Psychological treatments for perinatal depression. Best Pract Res Clin Obstet Gynaecol 2014; 28:61–70.
- Deligiannidis KM, Freeman MP. Complementary and alternative medicine therapies for perinatal depression. Best Pract Res Clin Obstet Gynaecol 2014; 28:85–95.
- ACOG Committee on Practice Bulletins—Obstetrics. ACOG Practice Bulletin: clinical management guidelines for obstetrician-gynecologists number 92, April 2008 (replaces Practice Bulletin number 87, November 2007). Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol 2008; 111:1001–1020.
- Ornoy A, Koren G. Selective serotonin reuptake inhibitors in human pregnancy: on the way to resolving the controversy. Semin Fetal Neonatal Med 2014; 19:188–194.
- Salisbury AL, Wisner KL, Pearlstein T, Battle CL, Stroud L, Lester BM. Newborn neurobehavioral patterns are differentially related to prenatal maternal major depressive disorder and serotonin reuptake inhibitor treatment. Depress Anxiety 2011; 28:1008–1019.
- Cohen LS, Altshuler LL, Harlow BL, et al. Relapse of major depression during pregnancy in women who maintain or discontinue antidepressant treatment. JAMA 2006; 295:499–507.
- Byatt N, Deligiannidis KM, Freeman MP. Antidepressant use in pregnancy: a critical review focused on risks and controversies. Acta Psychiatr Scand 2013; 127:94–114.
- Ban L, Gibson JE, West J, et al. Maternal depression, antidepressant prescriptions, and congenital anomaly risk in offspring: a population-based cohort study. BJOG 2014; 121:1471–1481.
- Kallen B, Olausson P. Maternal use of selective serotonin re-uptake inhibitors and persistent pulmonary hypertension of the newborn. Pharmacoepidemiol Drug Saf 2008; 17:801–806.
- Chambers CD, Johnson KA, Dick LM, Felix RJ, Jones KL. Birth outcomes in pregnant women taking fluoxetine. N Engl J Med 1996; 335:1010–1015.
- Costei AM, Kozer E, Ho T, Ito S, Koren G. Perinatal outcome following third trimester exposure to paroxetine. Arch Pediatr Adolesc Med 2002; 156:1129–1132.
- Salisbury AL, O’Grady KE, Battle CL, et al. The roles of maternal depression, serotonin reuptake inhibitor treatment, and concomitant benzodiazepine use on infant neurobehavioral functioning over the first postnatal month. Am J Psychiatry 2016; 173:147–157.
- Croen LA, Grether JK, Yoshida CK, Odouli R, Hendrick V. Antidepressant use during pregnancy and childhood autism spectrum disorders. Arch Gen Psychiatry 2011; 68:1104–1112.
- Rai D, Lee BK, Dalman C, Golding J, Lewis G, Magnusson C. Parental depression, maternal antidepressant use during pregnancy, and risk of autism spectrum disorders: population based case-control study. BMJ 2013; 346:f2059.
- Sorensen MJ, Gronborg TK, Christensen J, et al. Antidepressant exposure in pregnancy and risk of autism spectrum disorders. Clin Epidemiol 2013; 5:449–459.
- Clements CC, Castro VM, Blumenthal SR, et al. Prenatal antidepressant exposure is associated with risk for attention-deficit hyperactivity disorder but not autism spectrum disorder in a large health system. Mol Psychiatry 2015; 20:727–734.
- Castro VM, Kong SW, Clements CC, et al. Absence of evidence for increase in risk for autism or attention-deficit hyperactivity disorder following antidepressant exposure during pregnancy: a replication study. Transl Psychiatry 2016; 6:e708.
- Hale TW, Rowe HE. Medications and Mothers’ Milk. 16th ed. Amarillo, TX: Hale Publishing, L.P; 2014.
- Abreu AC, Stuart S. Pharmacologic and hormonal treatments for postpartum depression. Psychiatr Ann 2005; 35:568–576.
- Sit DK, Wisner KL. Decision making for postpartum depression treatment. Psychiatr Ann 2005; 35:577–584.
- Wisner KL, Parry BL, Piontek CM. Clinical practice. Postpartum depression. N Engl J Med 2002; 347:194–199.
- Howard M, Battle CL, Pearlstein T, Rosene-Montella K. A psychiatric mother-baby day hospital for pregnant and postpartum women. Arch Women’s Ment Health 2006; 9:213–218.
- Meltzer-Brody S, Brandon AR, Pearson B, et al. Evaluating the clinical effectiveness of a specialized perinatal psychiatry inpatient unit. Arch Women’s Ment Health 2014; 17:107–113.
Contrary to common belief, pregnancy does not confer protection against depression.1,2 In fact, pregnant women are just as likely as nonpregnant women to become or remain depressed, and up to 12.7% of pregnant women meet criteria for depression.1
In the postpartum period, women are particularly vulnerable to a major depressive episode, whether a first episode or a recurrence. The estimated prevalence of a depressive episode in the first 3 postpartum months is 19.2%,2 making postpartum depression the most common complication of childbearing.2 At the same time, peripartum depression remains largely underrecognized and undertreated.3
As evidence mounts regarding the deleterious impact of untreated mental illness on the mother, the developing fetus, and the infant, early detection and intervention for peripartum depression are paramount.3
DEPRESSION DURING PREGNANCY: SIGNIFICANT CONSEQUENCES
Although the rates of depression in pregnant and nonpregnant women are similar, depression in pregnancy carries additional significant consequences. Further, many depressed pregnant women believe their depression will lift once their baby is born, though it is well documented that depression during pregnancy is the strongest predictor of postpartum depression and that if left untreated it can be devastating for mother, infant, and family.4
Compared with nondepressed pregnant women, depressed pregnant women have poorer overall health status,5 are more likely to engage in behaviors that pose risk to the developing fetus such as smoking,5 alcohol consumption, and substance use,6 and have poor nutrition and inadequate weight gain.7,8
Pregnant women who are depressed and are also experiencing domestic violence are especially at risk for poor prenatal care as they tend to miss more prenatal appointments.9 Evidence also suggests that depressed pregnant women are less attached to the fetus and more likely to have elective terminations.10,11
Depression in pregnancy is associated with higher rates of adverse pregnancy outcomes such as preterm birth, low birth weight, operative delivery, and longer predelivery hospital stay.3,12 Depression and anxiety during pregnancy have been associated with prenatal hypertension,13 gestational diabetes,14 preeclampsia,15 and HELLP syndrome (ie, hemolysis, elevated liver enzymes, and low platelet count).15 Depression and anxiety during pregnancy are associated with subsequent poorer infant attachment16,17 and an overall unfavorable impact on infant and child development.18
Risk factors for depression during pregnancy include past episodes of depression, current anxiety, poor social support, unintended pregnancy, life stress, being single, domestic violence, and being on Medicaid.19
Undoubtedly the most devastating consequence of severe depression during pregnancy is suicide. Rates of suicide are lower in peripartum women,20 but when suicide does occur, pregnant women tend to use more violent means than nonpregnant women. Pregnant adolescents represent a particularly high-risk group.21
POSTPARTUM DEPRESSION
Postpartum depression is the most common complication of childbearing. Although the precise pathogenesis is undetermined, there is converging evidence of a subset of women particularly sensitive to dramatic fluctuations in levels of estradiol and progesterone that occur during childbirth.22,23 There is also evidence that dysregulation of the hypothalamic-pituitary-adrenal axis contributes to the development of postpartum depression in certain women.24 Further, women who have depression or anxiety during pregnancy are much more likely to experience postpartum depression than those who are not symptomatic during pregnancy.4 A history of peripartum depression or other lifetime depressive episodes, poverty, conflict with a primary partner, poor social support, stressful life events, and low self-esteem are strongly associated with postpartum depression.25
When unrecognized and untreated, postpartum depression can have profound and persistent effects on the mother and the developing infant.18,26 Mothers with postpartum depression are much more likely than mothers without depression to have impaired bonding,27 to be less responsive to their infant’s needs,17 and to be more likely to miss well-baby checkups.28
Postpartum depression’s effects on maternal-infant interactions can include maternal withdrawal, disengagement, intrusion, and hostility and can lead to long-term effects on child development, including poor cognitive functioning, emotional maladjustment, and behavioral inhibition.29,30 Infants and children of mothers with untreated postpartum depression have been shown to exhibit a higher incidence of colic, excessive crying, sleep problems, and irritability.31,32 Women with postpartum depression may be less likely to initiate or maintain breastfeeding, and depressive symptoms have been noted to precede the discontinuation of breastfeeding.33–35
Risk factors for postpartum depression
Characteristics to look for in the prenatal care of pregnant women include the following:
- Depression during pregnancy
- History of postpartum or other depressive episode
- Poverty
- Conflict with primary partner
- Poor social support
- Low self-esteem
- Single status.
DIFFERENTIATING ‘POSTPARTUM BLUES’ FROM MAJOR DEPRESSION
Primary care providers are often the first point of contact for depressed women. The diagnosis of major depression in pregnant and postpartum women is challenging because of changes in sleep, appetite, and energy brought on by pregnancy, complications of delivery, and demands of caring for a newborn.36 Many pregnant and postpartum women are reluctant to disclose their symptoms due to a sense of shame and guilt for being depressed during a time in their life that society commonly regards as joyful, and this contributes to under-detection.
In the first few days postpartum, fatigue, emotionality, irritability, and worry over the infant’s well-being affect up to 75% of women. This period, typically referred to as the “baby blues” or “postpartum blues,” is not considered a disorder and responds well to support, reassurance, and adequate sleep, and it typically resolves within 2 weeks.37,38 Table 1 lists features that help distinguish postpartum blues from major depression.
Signs of major depressive disorder
Major depressive disorder is a serious and disabling condition. To meet criteria for major depressive disorder, women must report depressed mood and loss of interest or pleasure in normally pleasurable activities for at least 2 weeks. Completing the symptom profile, at least 5 of the following must be present: sleep disturbance (insomnia or hypersomnia), lack of energy, feelings of worthlessness or low self-esteem, guilt, difficulty concentrating, indecisiveness, psychomotor retardation or agitation, and thoughts of suicide or death.
The Diagnostic and Statistical Manual of Mental Disorders (5th edition) recognizes that postpartum depression commonly begins during pregnancy, and now uses “peripartum onset” as the specifier for major depressive disorder that occurs during pregnancy, postpartum, or both.39 Other hallmark symptoms with peripartum onset include a lack of interest in or attachment to the pregnancy or infant, and anxiety and worry often accompanied by intrusive, unwanted thoughts of harm befalling the infant.40
Postpartum psychosis
Postpartum psychosis is a far less common presentation, occurring in 1 to 2 per 1,000 births, but it constitutes a psychiatric emergency requiring immediate referral to a psychiatric care setting. Women at highest risk are those with a personal or family history of bipolar disorder.
The clinical presentation is most commonly characterized by confusion, agitation, hallucinations, delusional beliefs, and disorientation. Suicide and infanticide, while rare, are more likely to occur in the context of a psychotic episode.41
SCREENING RECOMMENDATIONS
Screening for depression is routine in primary care settings and is no less important for peripartum women.
In 2016, the US Preventive Services Task Force issued a recommendation that all pregnant and postpartum women be screened for depression,42 highlighting the need for all medical providers to be alert to the potentially serious consequences of unrecognized and untreated maternal psychiatric illness.
The American College of Obstetricians and Gynecologists (ACOG) recommends screening for depression and anxiety at least once during the peripartum period,43 and the American Academy of Pediatrics recommends screening mothers for depression at the 1-, 2-, and 4-month well-baby visits.44
The peripartum period is associated with changes in sleep, appetite, and energy levels, but these are also typical of depression. Taking this into account, the Edinburgh Postnatal Depression Scale (EPDS) was developed to screen for depression specifically in this population.45 The EPDS is a validated and widely used 10-item self-reporting questionnaire with a high degree of sensitivity and specificity; it is easily administered and quickly scored. A cutoff score of 13 (of a maximum of 30) is considered indicative of depressed mood and signals the need for further assessment.
ACOG, the American Academy of Pediatrics, and the US Preventive Services Task Force recommend a standardized validated tool and cite both the EPDS (https://psychology-tools.com/epds/) and the Patient Health Questionnaire-9 (PHQ-9) (Figure 1) as appropriate to screen for peripartum depression.42–44 Primary care providers tend to be most familiar with the PHQ-9, a highly sensitive and specific 9-item depression screen that has been validated in primary care and obstetric clinic patients.46 A score on the PHQ-9 ranging from 5 to 10 indicates mild depression, 10 to 14 moderate depression, 15 to 19 moderate to severe depression, and greater than 19 severe depression.
CLINICAL MANAGEMENT
Many women prefer nondrug therapy
The gold standard treatment for moderate to severe major depressive disorder is psychotherapy plus pharmacotherapy. Yet many peripartum women voice concerns about exposure to pharmacologic treatment, and studies have shown that many women prefer nonpharmacologic intervention.47
Evidence-based psychotherapies that have demonstrated efficacy in peripartum women include cognitive behavioral therapy48 and interpersonal psychotherapy when administered by a psychotherapist trained in these treatments. Pregnant and breastfeeding women often express preference for psychotherapy and complementary and alternative treatments as a means of avoiding fetal and infant exposure to antidepressants.47
For mild to moderate depression, complementary therapies such as exercise, yoga, bright light therapy, and acupuncture have shown efficacy and can be used alone or adjunctively.49 Because a poor marital relationship is consistently associated with peripartum depression,25 primary care physicians who routinely address social support and screen for family conflict are well positioned to detect this significant correlate and to recommend marital or family therapy as a primary or adjunctive treatment.
When to consider drug therapy
The decision to recommend drug therapy must be individualized and based on the severity of symptoms, functional impairment, number and frequency of depressive episodes, history of response to medications, and the preferences of the patient, with the recognition that no decision is risk-free and that antidepressants enter the amniotic fluid, so fetal exposure is unavoidable.
Table 2 lists common antidepressants. The antidepressants most commonly prescribed, especially in the primary care setting, are selective serotonin reuptake inhibitors (SSRIs), which are favored because of their effectiveness, low side-effect profile, and lack of overdose toxicity.
Serotonin syndrome is no more likely to occur in pregnant than in nonpregnant women. Close monitoring for this condition is warranted only when patients are taking very high doses of SSRIs or SSRIs in combination with other serotonergic agonists.
Prescribing antidepressants for pregnant or breastfeeding women requires thoughtful consideration of the patient’s preferences, as well as weighing the risks and benefits of fetal and infant exposure to maternal depression vs exposure to medications. Additional considerations include monotherapy, avoiding medication changes, choosing drugs that have been effective in the past, and avoiding drugs with known drug-drug interactions or teratogenic effects.50
There is increasing consensus that the short- and long-term consequences of undertreatment or nontreatment of maternal depression outweigh the risk of fetal exposure to SSRIs.3,51,52 Cohen et al53 have recommended that if a woman is on an antidepressant and learns she is pregnant, she should not discontinue it because of the likelihood of relapse; they found a 68% relapse rate in women who discontinued their antidepressant in the first trimester of pregnancy.53
In a comprehensive review of studies published between 1996 and 2012 that examined antidepressant use during pregnancy, Byatt et al54 found little or no evidence of increased teratogenic risk with antidepressants with the exception of paroxetine, which is associated with a small but significant increased risk of cardiac malformation during first-trimester exposure.54
These conclusions were underscored in a large cohort study in the United Kingdom.55 In addition, a joint task force of the American Psychiatric Association and ACOG reviewed studies looking at the association between depression, antidepressants, and birth outcomes including miscarriage, preterm birth, cardiac abnormalities (resulting from first trimester exposure), persistent pulmonary hypertension (related to second- and third-trimester exposure), and neonatal adaptation syndrome (associated with third-trimester exposure).8 They concluded that the available data neither support nor refute a link between the use of antidepressants and several of the above outcomes. No increase in risk of congenital malformations (including cardiac abnormalities) was found. An increased risk of persistent pulmonary hypertension was noted, although the absolute risk of this disorder remained low, at 3 to 6 per 1,000 infants exposed to SSRIs in utero.8,56
Neonatal adaptation syndrome
Neonatal adaptation syndrome is characterized by jitteriness, irritability, decreased muscle tone, and feeding difficulty in the neonate. It can occur in 15% to 30% of infants exposed to SSRIs antenatally.57,58 These symptoms, however, are transient and typically resolve within 7 to 10 days after birth. A more recent study suggested that neurobehavioral symptoms for some infants extend beyond 2 weeks and that concomitant exposure to benzodiazepines results in even higher rates of this syndrome.59 There is no evidence that tapering or discontinuing antidepressants near term is necessary, safe, or effective in preventing transient neonatal complications. However, this approach would increase the risk of relapse for the mother.
Autism spectrum disorders
The possible association between antidepressants and autism spectrum disorders in pregnancy has captured much attention in recent years. One study based on healthcare claims60 and one registry-based study61 associated in utero exposure to antidepressants with autism liability in children. However, a large-scale Danish registry-based study did not replicate this association.62 In addition, 2 recent cohort studies, identifying children with autism spectrum disorder or attention-deficit hyperactivity disorder from electronic health records, found that neither disorder was significantly associated with prenatal antidepressant exposure in crude or adjusted models. However, both studies found a significant association with the use of antidepressants before pregnancy, indicating that the risk of autism observed with prenatal antidepressant exposure is likely confounded by the severity of maternal illness.63,64
Concerns about drug therapy during breastfeeding
For infants of breastfeeding women, exposure to antidepressants through breast milk is minimal. Amounts in breast milk depend on the timing of the antidepressant dose, timing of feeding, and genetically influenced metabolic activity in mother and infant. The current literature supports antidepressant use for breastfeeding mothers of healthy full-term infants.65
The 2 most widely studied antidepressants in breastfed infants are paroxetine and sertraline. It has been shown that very little can be detected in the infant’s serum, with relative infant doses ranging from 0.4% to 2.8%.65 While clinicians are cautioned against prescribing paroxetine for pregnant women, the drug remains a suitable alternative for breastfeeding women.
If an antidepressant is started postpartum, the recommendation is to start with a low dose and then slowly titrate upward while monitoring the infant for adverse effects.65,66 Possible adverse effects in breastfeeding infants include irritability, sedation, poor weight gain, and a change in feeding patterns.67 Adverse events are most likely to occur in newborns up to 8 weeks of age, and infants born prematurely or with medical problems may be particularly at risk.65,68
Helping patients weigh risks and benefits of drug therapy
Women may hear about the risks of medications to the fetus and during breastfeeding and so may be reluctant to seek or accept intervention. Often, the information is not from a reliable, scientifically based source. Primary care physicians are well positioned to guide peripartum women in risk-benefit analysis of proper treatment of their depression vs no treatment or undertreatment. In addition, establishing referral sources—ideally with a peripartum mental health specialist—is advisable. Online resources that clinicians can refer patients to for help in managing peripartum depression include the following:
- www.postpartum.net
- www.womensmentalhealth.org
- www.mothertobaby.org (for pharmacologic guidance).
INCREASED AWARENESS IS KEY
Primary care physicians must remain alert to the high prevalence of depression in women of childbearing age and embrace routine screening for depression. (See the sidebar, “The primary care management of peripartum depression.”) Since half of pregnancies are unintended, awareness of the risks of undetected and untreated peripartum depression to the mother, developing fetus, and infant is essential. Untreated antepartum depression has been linked to poor pregnancy outcomes, nutritional deficits, and substance abuse. Untreated postpartum depression negatively affects mother-infant attachment, infant, and child development and maternal self care.
Not treating depression is hazardous
Drug treatment during pregnancy and breastfeeding poses challenges for the patient and physician due to the inevitability of fetal and infant exposure, but lack of treatment can be hazardous.
To date, the evidence on the use of antidepressants in pregnant and lactating women is reassuring. Specialized peripartum psychiatric partial hospital programs69 and inpatient programs70 exist for women who need a higher level of care. There is also substantial evidence that psychotherapy, especially cognitive behavioral therapy and interpersonal therapy, is highly effective, and emerging data on complementary and alternative treatments are promising. Coordinated care between primary care and behavioral healthcare providers with expertise in treating peripartum depression is most likely to yield optimal outcomes.
Contrary to common belief, pregnancy does not confer protection against depression.1,2 In fact, pregnant women are just as likely as nonpregnant women to become or remain depressed, and up to 12.7% of pregnant women meet criteria for depression.1
In the postpartum period, women are particularly vulnerable to a major depressive episode, whether a first episode or a recurrence. The estimated prevalence of a depressive episode in the first 3 postpartum months is 19.2%,2 making postpartum depression the most common complication of childbearing.2 At the same time, peripartum depression remains largely underrecognized and undertreated.3
As evidence mounts regarding the deleterious impact of untreated mental illness on the mother, the developing fetus, and the infant, early detection and intervention for peripartum depression are paramount.3
DEPRESSION DURING PREGNANCY: SIGNIFICANT CONSEQUENCES
Although the rates of depression in pregnant and nonpregnant women are similar, depression in pregnancy carries additional significant consequences. Further, many depressed pregnant women believe their depression will lift once their baby is born, though it is well documented that depression during pregnancy is the strongest predictor of postpartum depression and that if left untreated it can be devastating for mother, infant, and family.4
Compared with nondepressed pregnant women, depressed pregnant women have poorer overall health status,5 are more likely to engage in behaviors that pose risk to the developing fetus such as smoking,5 alcohol consumption, and substance use,6 and have poor nutrition and inadequate weight gain.7,8
Pregnant women who are depressed and are also experiencing domestic violence are especially at risk for poor prenatal care as they tend to miss more prenatal appointments.9 Evidence also suggests that depressed pregnant women are less attached to the fetus and more likely to have elective terminations.10,11
Depression in pregnancy is associated with higher rates of adverse pregnancy outcomes such as preterm birth, low birth weight, operative delivery, and longer predelivery hospital stay.3,12 Depression and anxiety during pregnancy have been associated with prenatal hypertension,13 gestational diabetes,14 preeclampsia,15 and HELLP syndrome (ie, hemolysis, elevated liver enzymes, and low platelet count).15 Depression and anxiety during pregnancy are associated with subsequent poorer infant attachment16,17 and an overall unfavorable impact on infant and child development.18
Risk factors for depression during pregnancy include past episodes of depression, current anxiety, poor social support, unintended pregnancy, life stress, being single, domestic violence, and being on Medicaid.19
Undoubtedly the most devastating consequence of severe depression during pregnancy is suicide. Rates of suicide are lower in peripartum women,20 but when suicide does occur, pregnant women tend to use more violent means than nonpregnant women. Pregnant adolescents represent a particularly high-risk group.21
POSTPARTUM DEPRESSION
Postpartum depression is the most common complication of childbearing. Although the precise pathogenesis is undetermined, there is converging evidence of a subset of women particularly sensitive to dramatic fluctuations in levels of estradiol and progesterone that occur during childbirth.22,23 There is also evidence that dysregulation of the hypothalamic-pituitary-adrenal axis contributes to the development of postpartum depression in certain women.24 Further, women who have depression or anxiety during pregnancy are much more likely to experience postpartum depression than those who are not symptomatic during pregnancy.4 A history of peripartum depression or other lifetime depressive episodes, poverty, conflict with a primary partner, poor social support, stressful life events, and low self-esteem are strongly associated with postpartum depression.25
When unrecognized and untreated, postpartum depression can have profound and persistent effects on the mother and the developing infant.18,26 Mothers with postpartum depression are much more likely than mothers without depression to have impaired bonding,27 to be less responsive to their infant’s needs,17 and to be more likely to miss well-baby checkups.28
Postpartum depression’s effects on maternal-infant interactions can include maternal withdrawal, disengagement, intrusion, and hostility and can lead to long-term effects on child development, including poor cognitive functioning, emotional maladjustment, and behavioral inhibition.29,30 Infants and children of mothers with untreated postpartum depression have been shown to exhibit a higher incidence of colic, excessive crying, sleep problems, and irritability.31,32 Women with postpartum depression may be less likely to initiate or maintain breastfeeding, and depressive symptoms have been noted to precede the discontinuation of breastfeeding.33–35
Risk factors for postpartum depression
Characteristics to look for in the prenatal care of pregnant women include the following:
- Depression during pregnancy
- History of postpartum or other depressive episode
- Poverty
- Conflict with primary partner
- Poor social support
- Low self-esteem
- Single status.
DIFFERENTIATING ‘POSTPARTUM BLUES’ FROM MAJOR DEPRESSION
Primary care providers are often the first point of contact for depressed women. The diagnosis of major depression in pregnant and postpartum women is challenging because of changes in sleep, appetite, and energy brought on by pregnancy, complications of delivery, and demands of caring for a newborn.36 Many pregnant and postpartum women are reluctant to disclose their symptoms due to a sense of shame and guilt for being depressed during a time in their life that society commonly regards as joyful, and this contributes to under-detection.
In the first few days postpartum, fatigue, emotionality, irritability, and worry over the infant’s well-being affect up to 75% of women. This period, typically referred to as the “baby blues” or “postpartum blues,” is not considered a disorder and responds well to support, reassurance, and adequate sleep, and it typically resolves within 2 weeks.37,38 Table 1 lists features that help distinguish postpartum blues from major depression.
Signs of major depressive disorder
Major depressive disorder is a serious and disabling condition. To meet criteria for major depressive disorder, women must report depressed mood and loss of interest or pleasure in normally pleasurable activities for at least 2 weeks. Completing the symptom profile, at least 5 of the following must be present: sleep disturbance (insomnia or hypersomnia), lack of energy, feelings of worthlessness or low self-esteem, guilt, difficulty concentrating, indecisiveness, psychomotor retardation or agitation, and thoughts of suicide or death.
The Diagnostic and Statistical Manual of Mental Disorders (5th edition) recognizes that postpartum depression commonly begins during pregnancy, and now uses “peripartum onset” as the specifier for major depressive disorder that occurs during pregnancy, postpartum, or both.39 Other hallmark symptoms with peripartum onset include a lack of interest in or attachment to the pregnancy or infant, and anxiety and worry often accompanied by intrusive, unwanted thoughts of harm befalling the infant.40
Postpartum psychosis
Postpartum psychosis is a far less common presentation, occurring in 1 to 2 per 1,000 births, but it constitutes a psychiatric emergency requiring immediate referral to a psychiatric care setting. Women at highest risk are those with a personal or family history of bipolar disorder.
The clinical presentation is most commonly characterized by confusion, agitation, hallucinations, delusional beliefs, and disorientation. Suicide and infanticide, while rare, are more likely to occur in the context of a psychotic episode.41
SCREENING RECOMMENDATIONS
Screening for depression is routine in primary care settings and is no less important for peripartum women.
In 2016, the US Preventive Services Task Force issued a recommendation that all pregnant and postpartum women be screened for depression,42 highlighting the need for all medical providers to be alert to the potentially serious consequences of unrecognized and untreated maternal psychiatric illness.
The American College of Obstetricians and Gynecologists (ACOG) recommends screening for depression and anxiety at least once during the peripartum period,43 and the American Academy of Pediatrics recommends screening mothers for depression at the 1-, 2-, and 4-month well-baby visits.44
The peripartum period is associated with changes in sleep, appetite, and energy levels, but these are also typical of depression. Taking this into account, the Edinburgh Postnatal Depression Scale (EPDS) was developed to screen for depression specifically in this population.45 The EPDS is a validated and widely used 10-item self-reporting questionnaire with a high degree of sensitivity and specificity; it is easily administered and quickly scored. A cutoff score of 13 (of a maximum of 30) is considered indicative of depressed mood and signals the need for further assessment.
ACOG, the American Academy of Pediatrics, and the US Preventive Services Task Force recommend a standardized validated tool and cite both the EPDS (https://psychology-tools.com/epds/) and the Patient Health Questionnaire-9 (PHQ-9) (Figure 1) as appropriate to screen for peripartum depression.42–44 Primary care providers tend to be most familiar with the PHQ-9, a highly sensitive and specific 9-item depression screen that has been validated in primary care and obstetric clinic patients.46 A score on the PHQ-9 ranging from 5 to 10 indicates mild depression, 10 to 14 moderate depression, 15 to 19 moderate to severe depression, and greater than 19 severe depression.
CLINICAL MANAGEMENT
Many women prefer nondrug therapy
The gold standard treatment for moderate to severe major depressive disorder is psychotherapy plus pharmacotherapy. Yet many peripartum women voice concerns about exposure to pharmacologic treatment, and studies have shown that many women prefer nonpharmacologic intervention.47
Evidence-based psychotherapies that have demonstrated efficacy in peripartum women include cognitive behavioral therapy48 and interpersonal psychotherapy when administered by a psychotherapist trained in these treatments. Pregnant and breastfeeding women often express preference for psychotherapy and complementary and alternative treatments as a means of avoiding fetal and infant exposure to antidepressants.47
For mild to moderate depression, complementary therapies such as exercise, yoga, bright light therapy, and acupuncture have shown efficacy and can be used alone or adjunctively.49 Because a poor marital relationship is consistently associated with peripartum depression,25 primary care physicians who routinely address social support and screen for family conflict are well positioned to detect this significant correlate and to recommend marital or family therapy as a primary or adjunctive treatment.
When to consider drug therapy
The decision to recommend drug therapy must be individualized and based on the severity of symptoms, functional impairment, number and frequency of depressive episodes, history of response to medications, and the preferences of the patient, with the recognition that no decision is risk-free and that antidepressants enter the amniotic fluid, so fetal exposure is unavoidable.
Table 2 lists common antidepressants. The antidepressants most commonly prescribed, especially in the primary care setting, are selective serotonin reuptake inhibitors (SSRIs), which are favored because of their effectiveness, low side-effect profile, and lack of overdose toxicity.
Serotonin syndrome is no more likely to occur in pregnant than in nonpregnant women. Close monitoring for this condition is warranted only when patients are taking very high doses of SSRIs or SSRIs in combination with other serotonergic agonists.
Prescribing antidepressants for pregnant or breastfeeding women requires thoughtful consideration of the patient’s preferences, as well as weighing the risks and benefits of fetal and infant exposure to maternal depression vs exposure to medications. Additional considerations include monotherapy, avoiding medication changes, choosing drugs that have been effective in the past, and avoiding drugs with known drug-drug interactions or teratogenic effects.50
There is increasing consensus that the short- and long-term consequences of undertreatment or nontreatment of maternal depression outweigh the risk of fetal exposure to SSRIs.3,51,52 Cohen et al53 have recommended that if a woman is on an antidepressant and learns she is pregnant, she should not discontinue it because of the likelihood of relapse; they found a 68% relapse rate in women who discontinued their antidepressant in the first trimester of pregnancy.53
In a comprehensive review of studies published between 1996 and 2012 that examined antidepressant use during pregnancy, Byatt et al54 found little or no evidence of increased teratogenic risk with antidepressants with the exception of paroxetine, which is associated with a small but significant increased risk of cardiac malformation during first-trimester exposure.54
These conclusions were underscored in a large cohort study in the United Kingdom.55 In addition, a joint task force of the American Psychiatric Association and ACOG reviewed studies looking at the association between depression, antidepressants, and birth outcomes including miscarriage, preterm birth, cardiac abnormalities (resulting from first trimester exposure), persistent pulmonary hypertension (related to second- and third-trimester exposure), and neonatal adaptation syndrome (associated with third-trimester exposure).8 They concluded that the available data neither support nor refute a link between the use of antidepressants and several of the above outcomes. No increase in risk of congenital malformations (including cardiac abnormalities) was found. An increased risk of persistent pulmonary hypertension was noted, although the absolute risk of this disorder remained low, at 3 to 6 per 1,000 infants exposed to SSRIs in utero.8,56
Neonatal adaptation syndrome
Neonatal adaptation syndrome is characterized by jitteriness, irritability, decreased muscle tone, and feeding difficulty in the neonate. It can occur in 15% to 30% of infants exposed to SSRIs antenatally.57,58 These symptoms, however, are transient and typically resolve within 7 to 10 days after birth. A more recent study suggested that neurobehavioral symptoms for some infants extend beyond 2 weeks and that concomitant exposure to benzodiazepines results in even higher rates of this syndrome.59 There is no evidence that tapering or discontinuing antidepressants near term is necessary, safe, or effective in preventing transient neonatal complications. However, this approach would increase the risk of relapse for the mother.
Autism spectrum disorders
The possible association between antidepressants and autism spectrum disorders in pregnancy has captured much attention in recent years. One study based on healthcare claims60 and one registry-based study61 associated in utero exposure to antidepressants with autism liability in children. However, a large-scale Danish registry-based study did not replicate this association.62 In addition, 2 recent cohort studies, identifying children with autism spectrum disorder or attention-deficit hyperactivity disorder from electronic health records, found that neither disorder was significantly associated with prenatal antidepressant exposure in crude or adjusted models. However, both studies found a significant association with the use of antidepressants before pregnancy, indicating that the risk of autism observed with prenatal antidepressant exposure is likely confounded by the severity of maternal illness.63,64
Concerns about drug therapy during breastfeeding
For infants of breastfeeding women, exposure to antidepressants through breast milk is minimal. Amounts in breast milk depend on the timing of the antidepressant dose, timing of feeding, and genetically influenced metabolic activity in mother and infant. The current literature supports antidepressant use for breastfeeding mothers of healthy full-term infants.65
The 2 most widely studied antidepressants in breastfed infants are paroxetine and sertraline. It has been shown that very little can be detected in the infant’s serum, with relative infant doses ranging from 0.4% to 2.8%.65 While clinicians are cautioned against prescribing paroxetine for pregnant women, the drug remains a suitable alternative for breastfeeding women.
If an antidepressant is started postpartum, the recommendation is to start with a low dose and then slowly titrate upward while monitoring the infant for adverse effects.65,66 Possible adverse effects in breastfeeding infants include irritability, sedation, poor weight gain, and a change in feeding patterns.67 Adverse events are most likely to occur in newborns up to 8 weeks of age, and infants born prematurely or with medical problems may be particularly at risk.65,68
Helping patients weigh risks and benefits of drug therapy
Women may hear about the risks of medications to the fetus and during breastfeeding and so may be reluctant to seek or accept intervention. Often, the information is not from a reliable, scientifically based source. Primary care physicians are well positioned to guide peripartum women in risk-benefit analysis of proper treatment of their depression vs no treatment or undertreatment. In addition, establishing referral sources—ideally with a peripartum mental health specialist—is advisable. Online resources that clinicians can refer patients to for help in managing peripartum depression include the following:
- www.postpartum.net
- www.womensmentalhealth.org
- www.mothertobaby.org (for pharmacologic guidance).
INCREASED AWARENESS IS KEY
Primary care physicians must remain alert to the high prevalence of depression in women of childbearing age and embrace routine screening for depression. (See the sidebar, “The primary care management of peripartum depression.”) Since half of pregnancies are unintended, awareness of the risks of undetected and untreated peripartum depression to the mother, developing fetus, and infant is essential. Untreated antepartum depression has been linked to poor pregnancy outcomes, nutritional deficits, and substance abuse. Untreated postpartum depression negatively affects mother-infant attachment, infant, and child development and maternal self care.
Not treating depression is hazardous
Drug treatment during pregnancy and breastfeeding poses challenges for the patient and physician due to the inevitability of fetal and infant exposure, but lack of treatment can be hazardous.
To date, the evidence on the use of antidepressants in pregnant and lactating women is reassuring. Specialized peripartum psychiatric partial hospital programs69 and inpatient programs70 exist for women who need a higher level of care. There is also substantial evidence that psychotherapy, especially cognitive behavioral therapy and interpersonal therapy, is highly effective, and emerging data on complementary and alternative treatments are promising. Coordinated care between primary care and behavioral healthcare providers with expertise in treating peripartum depression is most likely to yield optimal outcomes.
- World Health Organization (WHO). A message from the Director General. www.who.int/whr/2001/dg_message/en/index.html. Accessed March 6, 2017.
- Gavin NI, Gaynes BN, Lohr KN, Meltzer-Brody S, Gartlehner G, Swinson T. Perinatal depression: a systematic review of prevalence and incidence. Obset Gynecol 2005; 106:1071–1083.
- Davalos DB, Yadon CA, Tregellas HC. Untreated prenatal maternal depression and the potential risks to offspring: a review. Arch Women’s Mental Health 2012; 15:1–14.
- Chaudron LH, Klein MH, Remington P, Palta M, Allen C, Essex MJ. Predictors, prodromes and incidence of postpartum depression. J Psychosom Obstet Gynaecol 2001; 22:103–112.
- Orr ST, Blazer DG, Orr CA. Maternal prenatal depressive symptoms, nicotine addiction, and smoking-related knowledge, attitudes, beliefs, and behaviors. Matern Child Health J 2012; 16:973–978.
- Flynn HA, Chermack ST. Prenatal alcohol use: the role of lifetime problems with alcohol,drugs, depression, and violence. J Stud Alcohol Drugs 2008; 69:500–509.
- Bodnar LM, Wisner KL, Moses-Kolko E, Sit DK, Hanusa BH. Prepregnancy body mass index, gestational weight gain, and the likelihood of major depressive disorder during pregnancy. J Clin Psychiatry 2009; 70:1290–1296.
- Yonkers KA, Wisner KL, Stewart DE, et al. The management of depression during pregnancy: a report from the American Psychiatric Association and the American College of Obstetricians and Gynecologists. Obstet Gynecol 2009; 114:703–713.
- Han A, Stewart DE. Maternal and fetal outcomes of intimate partner violence associated with pregnancy in the Latin American and Caribbean region. Int J Gynecol Obstet 2014; 124:6–11.
- McFarland J, Salisbury AL, Battler CL, Hawes K, Halloran K, Lester BM. Major depressive disorder during pregnancy and emotional attachment to the fetus. Arch Womens Ment Health 2011; 14:425–434.
- Suri R, Althuler LA, Mintz J. Depression and the decision to abort. Am J Psychiatry 2004; 161:1502.
- Kim DR, Sockol LE, Sammel MD, Kelly C, Moseley M, Epperson CN. Elevated risk of adverse obstetric outcomes in pregnant women with depression. Arch Women’s Ment Health 2013; 16:475–482.
- Mautner E, Greimel E, Trutnovsky G, Daghofer F, Egger JW, Lang U. Quality of life outcomes in pregnancy and postpartum complicated by hypertensive disorders, gestational diabetes, and preterm birth. J Psychosom Obstet Gynaecol 2009; 30:231–237.
- Katon JG, Russo J, Gavin AR, Melville JL, Katon WJ. Diabetes and depression in pregnancy: is there an association? J Women’s Health (Larchmt) 2011; 20:983–989.
- Delahaije DH, Dirksen CD, Peeters LL, Smits LJ. Anxiety and depression following preeclampsia or HELLP syndrome: a systematic review. Acta Obstet Gynecol Scand 2013; 92:746–761.
- O’Higgins M, Roberts IS, Glover V, Taylor A. Mother-child bonding at 1 year; associations with symptoms of postnatal depression and bonding in the first few weeks. Arch Women’s Ment Health 2013; 16:381–389.
- Field T, Healy BT, Goldstein S, Guthertz M. Behavior-state matching and synchrony in mother-infant interactions of nondepressed versus depressed dyads. Dev Psychol 1990; 26:7–14.
- Kingston D, Tough S, Whitfield H. Prenatal and postpartum maternal psychological distress and infant development: a systematic review. Child Psychiatry Hum Dev 2012; 43:683–714.
- Lancaster CA, Gold KJ, Flynn HA, Yoo H, Marcus SM, Davis MM. Risk factors for depressive symptoms during pregnancy: a systematic review. Am J Obstet Gynecol 2010; 202:5–14.
- Lindahl V, Pearson JL, Colpe L. Prevalence of suicidality during pregnancy and the postpartum. Arch Women’s Ment Health 2005; 8:77–87.
- Appleby L. Suicide after pregnancy and the first postnatal year. BMJ 1991; 302:137–140.
- Bloch M, Schmidt PJ, Danaceau M, Murphy J, Nieman L, Rubinow DR. Effects of gonadal steroids in women with a history of postpartum depression. Am J Psychiatry 2000; 157:924–930.
- Workman JL, Barha CK, Galea LAM. Endocrine substrates of cognitive and affective changes during pregnancy and postpartum. Behav Neurosci 2012; 126:54–72.
- Meltzer-Brody S. New insights into perinatal depression: pathogenesis and treatment during pregnancy and postpartum. Dialogues Clin Neurosci 2011; 13:89–100.
- O’Hara MW, McCabe JE. Postpartum depression: current status and future directions. Annu Rev Clin Psychol 2013; 9:379–407.
- Goodman SH, Rouse MH, Connell AM, Broth MR, Hall CM, Heyward D. Maternal depression and child psychopathology: a meta-analytic review. Clin Child Fam Psychol Rev 2011; 14:1–27
- Muzik M, Bocknek EL, Broderick A, et al. Mother-infant bonding impairment across the first 6 months postpartum: the primacy of psychopathology in women with childhood abuse and neglect histories. Arch Women’s Ment Health 2013; 16:29–38.
- Farr SL, Dietz PM, Rizzo JH, et al. Health care utilisation in the first year of life among infants of mothers with perinatal depression or anxiety. Paediatr Perinat Epidemiol 2013; 27:81–88.
- Grace SL, Evindar A, Stewart DE. The effect of postpartum depression on child cognitive development and behavior: a review and critical analysis of the literature. Arch Women’s Ment Health 2003; 6:263–274.
- Murray L, Cooper PJ. Postpartum depression and child development. Psychol Med 1997; 27:253–260.
- Orhon FS, Ulukol B, Soykan A. Postpartum mood disorders and maternal perceptions of infant patterns in well-child follow-up visits. Acta Paediatr 2007; 96:1777–1783.
- Dennis CL, Ross L. Relationships among infant sleep patterns, maternal fatigue, and development of depressive symptomatology. Birth 2005; 32:187–193.
- Ip S, Chung M, Raman G, et al. Breastfeeding and maternal and infant health outcomes in developed countries. Evid Rep Technol Assess (Full Rep) 2007; 153:1–186.
- Dennis CL, McQueen K. Does maternal postpartum depressive symptomatology influence infant feeding outcomes? Acta Paediatr 2007; 96:590–594.
- Hatton DC, Harrison-Hohner J, Coste S, Dorato V, Curet LB, McCarron DA. Symptoms of postpartum depression and breastfeeding. J Hum Lact 2005; 21:444–449.
- Klein MH, Essex MJ. Pregnant or depressed? The effect of overlap between symptoms of depression and somatic complaints of pregnancy on rates of major depression in the second trimester. Depression 1994; 2:308–314.
- Seyfried LS, Marcus SM. Postpartum mood disorders. Int Rev Psychiatry 2003; 15:231–242.
- Buttner MM, O’Hara MW, Watson D. The structure of women’s mood in the early postpartum. Assessment 2012; 19:247–256.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA; American Psychiatric Association Publishing: 2013.
- Wisner KL, Peindl KS, Gigliotti T, Hanusa BH. Obsessions and compulsions in women with postpartum depression. J Clin Psychiatry 1999: 60:176-180.
- Di Florio A, Smith S, Jones I. Postpartum psychosis. The Obstetrician & Gynecologist 2013; 15:145–150.
- O’Connor E, Rossom RC, Henniger M, Groom HC, Burda BU. Primary care screening for and treatment of depression in pregnant and postpartum women: evidence report and systematic review for the US Preventive Services Task Force. JAMA 2016; 315:388–406.
- Committee on Obstetric Practice. The American College of Obstetricians and Gynecologists Committee Opinion no. 630. Screening for perinatal depression. Obstet Gynecol 2015; 125:1268–1271.
- Earls MF; Committee on Psychosocial Aspects of Child and Family Health American Academy of Pediatrics. Incorporating recognition and management of perinatal and postpartum depression into pediatric practice. Pediatrics 2010; 126:1032–1039.
- Cox JL, Holden JM, Sagovsky R. Detection of postnatal depression: development of the 10-item Edinburgh postnatal depression scale. Br J Psychiatry 1987; 150:782–786.
- Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med 2001; 16:606–616.
- Battle CL, Salisbury AL, Schofield CA, Ortiz-Hernandez S. Perinatal antidepressant use: understanding women’s preferences and concerns. J Psychiatr Pract 2013; 19:443–453.
- Stuart S, Koleva H. Psychological treatments for perinatal depression. Best Pract Res Clin Obstet Gynaecol 2014; 28:61–70.
- Deligiannidis KM, Freeman MP. Complementary and alternative medicine therapies for perinatal depression. Best Pract Res Clin Obstet Gynaecol 2014; 28:85–95.
- ACOG Committee on Practice Bulletins—Obstetrics. ACOG Practice Bulletin: clinical management guidelines for obstetrician-gynecologists number 92, April 2008 (replaces Practice Bulletin number 87, November 2007). Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol 2008; 111:1001–1020.
- Ornoy A, Koren G. Selective serotonin reuptake inhibitors in human pregnancy: on the way to resolving the controversy. Semin Fetal Neonatal Med 2014; 19:188–194.
- Salisbury AL, Wisner KL, Pearlstein T, Battle CL, Stroud L, Lester BM. Newborn neurobehavioral patterns are differentially related to prenatal maternal major depressive disorder and serotonin reuptake inhibitor treatment. Depress Anxiety 2011; 28:1008–1019.
- Cohen LS, Altshuler LL, Harlow BL, et al. Relapse of major depression during pregnancy in women who maintain or discontinue antidepressant treatment. JAMA 2006; 295:499–507.
- Byatt N, Deligiannidis KM, Freeman MP. Antidepressant use in pregnancy: a critical review focused on risks and controversies. Acta Psychiatr Scand 2013; 127:94–114.
- Ban L, Gibson JE, West J, et al. Maternal depression, antidepressant prescriptions, and congenital anomaly risk in offspring: a population-based cohort study. BJOG 2014; 121:1471–1481.
- Kallen B, Olausson P. Maternal use of selective serotonin re-uptake inhibitors and persistent pulmonary hypertension of the newborn. Pharmacoepidemiol Drug Saf 2008; 17:801–806.
- Chambers CD, Johnson KA, Dick LM, Felix RJ, Jones KL. Birth outcomes in pregnant women taking fluoxetine. N Engl J Med 1996; 335:1010–1015.
- Costei AM, Kozer E, Ho T, Ito S, Koren G. Perinatal outcome following third trimester exposure to paroxetine. Arch Pediatr Adolesc Med 2002; 156:1129–1132.
- Salisbury AL, O’Grady KE, Battle CL, et al. The roles of maternal depression, serotonin reuptake inhibitor treatment, and concomitant benzodiazepine use on infant neurobehavioral functioning over the first postnatal month. Am J Psychiatry 2016; 173:147–157.
- Croen LA, Grether JK, Yoshida CK, Odouli R, Hendrick V. Antidepressant use during pregnancy and childhood autism spectrum disorders. Arch Gen Psychiatry 2011; 68:1104–1112.
- Rai D, Lee BK, Dalman C, Golding J, Lewis G, Magnusson C. Parental depression, maternal antidepressant use during pregnancy, and risk of autism spectrum disorders: population based case-control study. BMJ 2013; 346:f2059.
- Sorensen MJ, Gronborg TK, Christensen J, et al. Antidepressant exposure in pregnancy and risk of autism spectrum disorders. Clin Epidemiol 2013; 5:449–459.
- Clements CC, Castro VM, Blumenthal SR, et al. Prenatal antidepressant exposure is associated with risk for attention-deficit hyperactivity disorder but not autism spectrum disorder in a large health system. Mol Psychiatry 2015; 20:727–734.
- Castro VM, Kong SW, Clements CC, et al. Absence of evidence for increase in risk for autism or attention-deficit hyperactivity disorder following antidepressant exposure during pregnancy: a replication study. Transl Psychiatry 2016; 6:e708.
- Hale TW, Rowe HE. Medications and Mothers’ Milk. 16th ed. Amarillo, TX: Hale Publishing, L.P; 2014.
- Abreu AC, Stuart S. Pharmacologic and hormonal treatments for postpartum depression. Psychiatr Ann 2005; 35:568–576.
- Sit DK, Wisner KL. Decision making for postpartum depression treatment. Psychiatr Ann 2005; 35:577–584.
- Wisner KL, Parry BL, Piontek CM. Clinical practice. Postpartum depression. N Engl J Med 2002; 347:194–199.
- Howard M, Battle CL, Pearlstein T, Rosene-Montella K. A psychiatric mother-baby day hospital for pregnant and postpartum women. Arch Women’s Ment Health 2006; 9:213–218.
- Meltzer-Brody S, Brandon AR, Pearson B, et al. Evaluating the clinical effectiveness of a specialized perinatal psychiatry inpatient unit. Arch Women’s Ment Health 2014; 17:107–113.
- World Health Organization (WHO). A message from the Director General. www.who.int/whr/2001/dg_message/en/index.html. Accessed March 6, 2017.
- Gavin NI, Gaynes BN, Lohr KN, Meltzer-Brody S, Gartlehner G, Swinson T. Perinatal depression: a systematic review of prevalence and incidence. Obset Gynecol 2005; 106:1071–1083.
- Davalos DB, Yadon CA, Tregellas HC. Untreated prenatal maternal depression and the potential risks to offspring: a review. Arch Women’s Mental Health 2012; 15:1–14.
- Chaudron LH, Klein MH, Remington P, Palta M, Allen C, Essex MJ. Predictors, prodromes and incidence of postpartum depression. J Psychosom Obstet Gynaecol 2001; 22:103–112.
- Orr ST, Blazer DG, Orr CA. Maternal prenatal depressive symptoms, nicotine addiction, and smoking-related knowledge, attitudes, beliefs, and behaviors. Matern Child Health J 2012; 16:973–978.
- Flynn HA, Chermack ST. Prenatal alcohol use: the role of lifetime problems with alcohol,drugs, depression, and violence. J Stud Alcohol Drugs 2008; 69:500–509.
- Bodnar LM, Wisner KL, Moses-Kolko E, Sit DK, Hanusa BH. Prepregnancy body mass index, gestational weight gain, and the likelihood of major depressive disorder during pregnancy. J Clin Psychiatry 2009; 70:1290–1296.
- Yonkers KA, Wisner KL, Stewart DE, et al. The management of depression during pregnancy: a report from the American Psychiatric Association and the American College of Obstetricians and Gynecologists. Obstet Gynecol 2009; 114:703–713.
- Han A, Stewart DE. Maternal and fetal outcomes of intimate partner violence associated with pregnancy in the Latin American and Caribbean region. Int J Gynecol Obstet 2014; 124:6–11.
- McFarland J, Salisbury AL, Battler CL, Hawes K, Halloran K, Lester BM. Major depressive disorder during pregnancy and emotional attachment to the fetus. Arch Womens Ment Health 2011; 14:425–434.
- Suri R, Althuler LA, Mintz J. Depression and the decision to abort. Am J Psychiatry 2004; 161:1502.
- Kim DR, Sockol LE, Sammel MD, Kelly C, Moseley M, Epperson CN. Elevated risk of adverse obstetric outcomes in pregnant women with depression. Arch Women’s Ment Health 2013; 16:475–482.
- Mautner E, Greimel E, Trutnovsky G, Daghofer F, Egger JW, Lang U. Quality of life outcomes in pregnancy and postpartum complicated by hypertensive disorders, gestational diabetes, and preterm birth. J Psychosom Obstet Gynaecol 2009; 30:231–237.
- Katon JG, Russo J, Gavin AR, Melville JL, Katon WJ. Diabetes and depression in pregnancy: is there an association? J Women’s Health (Larchmt) 2011; 20:983–989.
- Delahaije DH, Dirksen CD, Peeters LL, Smits LJ. Anxiety and depression following preeclampsia or HELLP syndrome: a systematic review. Acta Obstet Gynecol Scand 2013; 92:746–761.
- O’Higgins M, Roberts IS, Glover V, Taylor A. Mother-child bonding at 1 year; associations with symptoms of postnatal depression and bonding in the first few weeks. Arch Women’s Ment Health 2013; 16:381–389.
- Field T, Healy BT, Goldstein S, Guthertz M. Behavior-state matching and synchrony in mother-infant interactions of nondepressed versus depressed dyads. Dev Psychol 1990; 26:7–14.
- Kingston D, Tough S, Whitfield H. Prenatal and postpartum maternal psychological distress and infant development: a systematic review. Child Psychiatry Hum Dev 2012; 43:683–714.
- Lancaster CA, Gold KJ, Flynn HA, Yoo H, Marcus SM, Davis MM. Risk factors for depressive symptoms during pregnancy: a systematic review. Am J Obstet Gynecol 2010; 202:5–14.
- Lindahl V, Pearson JL, Colpe L. Prevalence of suicidality during pregnancy and the postpartum. Arch Women’s Ment Health 2005; 8:77–87.
- Appleby L. Suicide after pregnancy and the first postnatal year. BMJ 1991; 302:137–140.
- Bloch M, Schmidt PJ, Danaceau M, Murphy J, Nieman L, Rubinow DR. Effects of gonadal steroids in women with a history of postpartum depression. Am J Psychiatry 2000; 157:924–930.
- Workman JL, Barha CK, Galea LAM. Endocrine substrates of cognitive and affective changes during pregnancy and postpartum. Behav Neurosci 2012; 126:54–72.
- Meltzer-Brody S. New insights into perinatal depression: pathogenesis and treatment during pregnancy and postpartum. Dialogues Clin Neurosci 2011; 13:89–100.
- O’Hara MW, McCabe JE. Postpartum depression: current status and future directions. Annu Rev Clin Psychol 2013; 9:379–407.
- Goodman SH, Rouse MH, Connell AM, Broth MR, Hall CM, Heyward D. Maternal depression and child psychopathology: a meta-analytic review. Clin Child Fam Psychol Rev 2011; 14:1–27
- Muzik M, Bocknek EL, Broderick A, et al. Mother-infant bonding impairment across the first 6 months postpartum: the primacy of psychopathology in women with childhood abuse and neglect histories. Arch Women’s Ment Health 2013; 16:29–38.
- Farr SL, Dietz PM, Rizzo JH, et al. Health care utilisation in the first year of life among infants of mothers with perinatal depression or anxiety. Paediatr Perinat Epidemiol 2013; 27:81–88.
- Grace SL, Evindar A, Stewart DE. The effect of postpartum depression on child cognitive development and behavior: a review and critical analysis of the literature. Arch Women’s Ment Health 2003; 6:263–274.
- Murray L, Cooper PJ. Postpartum depression and child development. Psychol Med 1997; 27:253–260.
- Orhon FS, Ulukol B, Soykan A. Postpartum mood disorders and maternal perceptions of infant patterns in well-child follow-up visits. Acta Paediatr 2007; 96:1777–1783.
- Dennis CL, Ross L. Relationships among infant sleep patterns, maternal fatigue, and development of depressive symptomatology. Birth 2005; 32:187–193.
- Ip S, Chung M, Raman G, et al. Breastfeeding and maternal and infant health outcomes in developed countries. Evid Rep Technol Assess (Full Rep) 2007; 153:1–186.
- Dennis CL, McQueen K. Does maternal postpartum depressive symptomatology influence infant feeding outcomes? Acta Paediatr 2007; 96:590–594.
- Hatton DC, Harrison-Hohner J, Coste S, Dorato V, Curet LB, McCarron DA. Symptoms of postpartum depression and breastfeeding. J Hum Lact 2005; 21:444–449.
- Klein MH, Essex MJ. Pregnant or depressed? The effect of overlap between symptoms of depression and somatic complaints of pregnancy on rates of major depression in the second trimester. Depression 1994; 2:308–314.
- Seyfried LS, Marcus SM. Postpartum mood disorders. Int Rev Psychiatry 2003; 15:231–242.
- Buttner MM, O’Hara MW, Watson D. The structure of women’s mood in the early postpartum. Assessment 2012; 19:247–256.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA; American Psychiatric Association Publishing: 2013.
- Wisner KL, Peindl KS, Gigliotti T, Hanusa BH. Obsessions and compulsions in women with postpartum depression. J Clin Psychiatry 1999: 60:176-180.
- Di Florio A, Smith S, Jones I. Postpartum psychosis. The Obstetrician & Gynecologist 2013; 15:145–150.
- O’Connor E, Rossom RC, Henniger M, Groom HC, Burda BU. Primary care screening for and treatment of depression in pregnant and postpartum women: evidence report and systematic review for the US Preventive Services Task Force. JAMA 2016; 315:388–406.
- Committee on Obstetric Practice. The American College of Obstetricians and Gynecologists Committee Opinion no. 630. Screening for perinatal depression. Obstet Gynecol 2015; 125:1268–1271.
- Earls MF; Committee on Psychosocial Aspects of Child and Family Health American Academy of Pediatrics. Incorporating recognition and management of perinatal and postpartum depression into pediatric practice. Pediatrics 2010; 126:1032–1039.
- Cox JL, Holden JM, Sagovsky R. Detection of postnatal depression: development of the 10-item Edinburgh postnatal depression scale. Br J Psychiatry 1987; 150:782–786.
- Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med 2001; 16:606–616.
- Battle CL, Salisbury AL, Schofield CA, Ortiz-Hernandez S. Perinatal antidepressant use: understanding women’s preferences and concerns. J Psychiatr Pract 2013; 19:443–453.
- Stuart S, Koleva H. Psychological treatments for perinatal depression. Best Pract Res Clin Obstet Gynaecol 2014; 28:61–70.
- Deligiannidis KM, Freeman MP. Complementary and alternative medicine therapies for perinatal depression. Best Pract Res Clin Obstet Gynaecol 2014; 28:85–95.
- ACOG Committee on Practice Bulletins—Obstetrics. ACOG Practice Bulletin: clinical management guidelines for obstetrician-gynecologists number 92, April 2008 (replaces Practice Bulletin number 87, November 2007). Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol 2008; 111:1001–1020.
- Ornoy A, Koren G. Selective serotonin reuptake inhibitors in human pregnancy: on the way to resolving the controversy. Semin Fetal Neonatal Med 2014; 19:188–194.
- Salisbury AL, Wisner KL, Pearlstein T, Battle CL, Stroud L, Lester BM. Newborn neurobehavioral patterns are differentially related to prenatal maternal major depressive disorder and serotonin reuptake inhibitor treatment. Depress Anxiety 2011; 28:1008–1019.
- Cohen LS, Altshuler LL, Harlow BL, et al. Relapse of major depression during pregnancy in women who maintain or discontinue antidepressant treatment. JAMA 2006; 295:499–507.
- Byatt N, Deligiannidis KM, Freeman MP. Antidepressant use in pregnancy: a critical review focused on risks and controversies. Acta Psychiatr Scand 2013; 127:94–114.
- Ban L, Gibson JE, West J, et al. Maternal depression, antidepressant prescriptions, and congenital anomaly risk in offspring: a population-based cohort study. BJOG 2014; 121:1471–1481.
- Kallen B, Olausson P. Maternal use of selective serotonin re-uptake inhibitors and persistent pulmonary hypertension of the newborn. Pharmacoepidemiol Drug Saf 2008; 17:801–806.
- Chambers CD, Johnson KA, Dick LM, Felix RJ, Jones KL. Birth outcomes in pregnant women taking fluoxetine. N Engl J Med 1996; 335:1010–1015.
- Costei AM, Kozer E, Ho T, Ito S, Koren G. Perinatal outcome following third trimester exposure to paroxetine. Arch Pediatr Adolesc Med 2002; 156:1129–1132.
- Salisbury AL, O’Grady KE, Battle CL, et al. The roles of maternal depression, serotonin reuptake inhibitor treatment, and concomitant benzodiazepine use on infant neurobehavioral functioning over the first postnatal month. Am J Psychiatry 2016; 173:147–157.
- Croen LA, Grether JK, Yoshida CK, Odouli R, Hendrick V. Antidepressant use during pregnancy and childhood autism spectrum disorders. Arch Gen Psychiatry 2011; 68:1104–1112.
- Rai D, Lee BK, Dalman C, Golding J, Lewis G, Magnusson C. Parental depression, maternal antidepressant use during pregnancy, and risk of autism spectrum disorders: population based case-control study. BMJ 2013; 346:f2059.
- Sorensen MJ, Gronborg TK, Christensen J, et al. Antidepressant exposure in pregnancy and risk of autism spectrum disorders. Clin Epidemiol 2013; 5:449–459.
- Clements CC, Castro VM, Blumenthal SR, et al. Prenatal antidepressant exposure is associated with risk for attention-deficit hyperactivity disorder but not autism spectrum disorder in a large health system. Mol Psychiatry 2015; 20:727–734.
- Castro VM, Kong SW, Clements CC, et al. Absence of evidence for increase in risk for autism or attention-deficit hyperactivity disorder following antidepressant exposure during pregnancy: a replication study. Transl Psychiatry 2016; 6:e708.
- Hale TW, Rowe HE. Medications and Mothers’ Milk. 16th ed. Amarillo, TX: Hale Publishing, L.P; 2014.
- Abreu AC, Stuart S. Pharmacologic and hormonal treatments for postpartum depression. Psychiatr Ann 2005; 35:568–576.
- Sit DK, Wisner KL. Decision making for postpartum depression treatment. Psychiatr Ann 2005; 35:577–584.
- Wisner KL, Parry BL, Piontek CM. Clinical practice. Postpartum depression. N Engl J Med 2002; 347:194–199.
- Howard M, Battle CL, Pearlstein T, Rosene-Montella K. A psychiatric mother-baby day hospital for pregnant and postpartum women. Arch Women’s Ment Health 2006; 9:213–218.
- Meltzer-Brody S, Brandon AR, Pearson B, et al. Evaluating the clinical effectiveness of a specialized perinatal psychiatry inpatient unit. Arch Women’s Ment Health 2014; 17:107–113.
KEY POINTS
- Depression occurs in up to 13% of pregnant women, a prevalence similar to that in nonpregnant women, but the incidence rises postpartum.
- Depressed pregnant women are more likely to engage in behaviors that pose a risk to the fetus.
- Depression in pregnancy is associated with adverse pregnancy outcomes such as preterm birth, low birth weight, gestational diabetes, and hypertensive disorders of pregnancy.
- Risk factors for depression in pregnancy include past episodes of depression, poor social support, unwanted pregnancy, and domestic violence.
