User login
Chronic constipation: Update on management
Chronic constipation has a variety of possible causes and mechanisms. Although traditional conservative treatments are still valid and first-line, if these fail, clinicians can choose from a growing list of new treatments, tailored to the cause in the individual patient.
This article discusses how defecation works (or doesn’t), the types of chronic constipation, the available diagnostic tools, and traditional and newer treatments, including some still in development.
THE EPIDEMIOLOGY OF CONSTIPATION
Chronic constipation is one of the most common gastrointestinal disorders, affecting about 15% of all adults and 30% of those over the age of 60.1 It can be a primary disorder or secondary to other factors.
Constipation is more prevalent in women and in institutionalized elderly people.2 It is associated with lower socioeconomic status, depression, less self-reported physical activity, certain medications, and stressful life events.3 Given its high prevalence and its impact on quality of life, it is also associated with significant utilization of healthcare resources.4
Constipation defined by Rome IV criteria
Physicians and patients may disagree about what constitutes constipation. Physicians primarily regard it as infrequent bowel movements, while patients tend to have a broader definition. According to the Rome IV criteria,5 chronic constipation is defined by the presence of the following for at least 3 months (with symptom onset at least 6 months prior to diagnosis):
(1) Two or more of the following for more than 25% of defecations:
- Straining
- Lumpy or hard stools
- Sensation of incomplete evacuation
- Sensation of anorectal obstruction or blockage
- Manual maneuvers to facilitate evacuation
- Fewer than 3 spontaneous bowel movements per week.
(2) Loose stools are rarely present without the use of laxatives.
(3) The patient does not meet the criteria for diagnosis of irritable bowel syndrome.
DEFECATION IS COMPLEX
Defecation begins when the rectum fills with stool, causing relaxation of the internal anal sphincter and the urge to defecate. The external anal sphincter, which is under voluntary control, can then either contract to delay defecation or relax to allow the stool to be expelled.6
Colonic muscles propel stool toward the rectum in repetitive localized contractions that help mix and promote absorption of the content, and larger coordinated (high-amplitude propagating) contractions that, in healthy individuals, move the stool forward from the proximal to the distal colon multiple times daily. These contractions usually occur in the morning and are accentuated by gastric distention from food and the resulting gastrocolic reflex.
Serotonin (5-HT) is released by enterochromaffin cells in response to distention of the gut wall. It mediates peristaltic movements of the gastrointestinal tract by binding to receptors (especially 5-HT4), stimulating release of neurotransmitters such as acetylcholine, causing smooth-muscle contraction behind the luminal contents and propelling them forward.
PRIMARY CONSTIPATION DISORDERS
The American Gastroenterological Association7 classifies constipation into 3 groups on the basis of colonic transit time and anorectal function:
Normal-transit constipation
Stool normally takes 20 to 72 hours to pass through the colon, with transit time affected by diet, drugs, level of physical activity, and emotional status.8
Normal-transit constipation is the most common type of constipation. The term is sometimes used interchangeably with constipation-predominant irritable bowel syndrome, but the latter is a distinct entity characterized by abdominal pain relieved by defecation as the primary symptom, as well as having occasional loose stools. These 2 conditions can be hard to tell apart, especially if the patient cannot describe the symptoms precisely.
Slow-transit constipation
Slow-transit constipation—also called delayed-transit constipation, colonoparesis, colonic inertia, and pseudo-obstruction—is defined as prolonged stool transit in the colon, ie, for more than 5 days.9 It can be the result of colonic smooth muscle dysfunction, compromised colonic neural pathways, or both, leading to slow colon peristalsis.
Factors that can affect colonic motility such as opioid use and hypothyroidism should be carefully considered in these patients. Opioids are notorious for causing constipation by decreasing bowel tone and contractility and thereby increasing colonic transit time. They also tighten up the anal sphincters, resulting in decreased rectal evacuation.10
Outlet dysfunction
Outlet dysfunction, also called pelvic floor dysfunction or defecatory disorder, is associated with incomplete rectal evacuation. It can be a consequence of weak rectal expulsion forces (slow colonic transit, rectal hyposensitivity), functional resistance to rectal evacuation (high anal resting pressure, anismus, incomplete relaxation of the anal sphincter, dyssynergic defecation), or structural outlet obstruction (excessive perineal descent, rectoceles, rectal intussusception). About 50% of patients with outlet dysfunction have concurrent slow-transit constipation.
Dyssynergic defecation is the most common outlet dysfunction disorder, accounting for about half of the cases referred to tertiary centers. It is defined as a paradoxical elevation in anal sphincter tone or less than 20% relaxation of the resting anal sphincter pressure with weak abdominal and pelvic propulsive forces.11 Anorectal biofeedback is a therapeutic option for dyssynergic defecation, as we discuss later in this article.
SECONDARY CONSTIPATION

Constipation can be secondary to several conditions and factors (Table 1), including:
- Neurologic disorders that affect gastrointestinal motility (eg, Hirschsprung disease, Parkinson disease, multiple sclerosis, spinal cord injury, stroke, spinal or ganglionic tumor, hypothyroidism, amyloidosis, diabetes mellitus, hypercalcemia)
- Drugs used to treat neurologic disorders
- Mechanical obstruction
- Diet (eg, low fiber, decreased fluid intake).
EVALUATION OF CONSTIPATION
It is crucial for physicians to efficiently use the available diagnostic tools for constipation to tailor the treatment to the patient.

Evaluation of chronic constipation begins with a thorough history and physical examination to rule out secondary constipation (Figure 1). Red flags such as unintentional weight loss, blood in the stool, rectal pain, fever, and iron-deficiency anemia should prompt referral for colonoscopy to evaluate for malignancy, colitis, or other potential colonic abnormalities.12
A detailed perineal and rectal examination can help diagnose defecatory disorders and should include evaluation of the resting anal tone and the sphincter during simulated evacuation.
Laboratory tests of thyroid function, electrolytes, and a complete blood cell count should be ordered if clinically indicated.13
Further tests
Further diagnostic tests can be considered if symptoms persist despite conservative treatment or if a defecatory disorder is suspected. These include anorectal manometry, colonic transit studies, defecography, and colonic manometry.
Anorectal manometry and the rectal balloon expulsion test are usually done first because of their high sensitivity (88%) and specificity (89%) for defecatory disorders.14 These tests measure the function of the internal and external anal sphincters at rest and with straining and assess rectal sensitivity and compliance. Anorectal manometry is also used in biofeedback therapy in patients with dyssynergic defecation.15
Colonic transit time can be measured if anorectal manometry and the balloon expulsion test are normal. The study uses radiopaque markers, radioisotopes, or wireless motility capsules to confirm slow-transit constipation and to identify areas of delayed transit in the colon.16
Defecography is usually the next step in diagnosis if anorectal manometry and balloon expulsion tests are inconclusive or if an anatomic abnormality of the pelvic floor is suspected. It can be done with a variety of techniques. Barium defecography can identify anatomic defects, scintigraphy can quantify evacuation of artificial stools, and magnetic resonance defecography visualizes anatomic landmarks to assess pelvic floor motion without exposing the patient to radiation.17,18
Colonic manometry is most useful in patients with refractory slow-transit constipation and can identify patients with isolated colonic motor dysfunction with no pelvic floor dysfunction who may benefit from subtotal colectomy and end-ileostomy.7
TRADITIONAL TREATMENTS STILL THE MAINSTAY

Nonpharmacologic treatments are the first-line options for patients with normal-transit and slow-transit constipation and should precede diagnostic testing. Lifestyle modifications and dietary changes (Table 2) aim to augment the known factors that stimulate the gastrocolic reflex and increase intestinal motility by high-amplitude propagated contractions.
Increasing physical activity increases intestinal gas clearance, decreases bloating, and lessens constipation.19,20
Toilet training is an integral part of lifestyle modifications.21
Diet. Drinking hot caffeinated beverages, eating breakfast within an hour of waking up, and consuming fiber in the morning (25–30 g of fiber daily) have traditionally been recommended as the first-line measures for chronic constipation. Dehydrated patients with constipation also benefit from increasing their fluid intake.22
LAXATIVES
Fiber (bulk-forming laxatives) for normal-transit constipation

Fiber remains a key part of the initial management of chronic constipation, as it is cheap, available, and safe. Increasing fiber intake is effective for normal-transit constipation, but patients with slow-transit constipation or refractory outlet dysfunction are less likely to benefit.23 Other laxatives are incorporated into the regimen if first-line nonpharmacologic interventions fail (Table 3).
Bulk-forming laxatives include insoluble fiber (wheat bran) and soluble fiber (psyllium, methylcellulose, inulin, calcium polycarbophil). Insoluble fiber, though often used, has little impact on symptoms of chronic constipation after 1 month of use, and up to 60% of patients report adverse effects from it.24 On the other hand, clinical trials have shown that soluble fiber such as psyllium facilitates defecation and improves functional bowel symptoms in patients with normal-transit constipation.25
Patients should be instructed to increase their dietary fiber intake gradually to avoid adverse effects and should be told to expect significant symptomatic improvement only after a few weeks. They should also be informed that increasing dietary fiber intake can cause bloating but that the bloating is temporary. If it continues, a different fiber can be tried.
Osmotic laxatives
Osmotic laxatives are often employed as a first- line laxative treatment option for patients with constipation. They draw water into the lumen by osmosis, helping to soften stool and speed intestinal transit. They include macrogols (inert polymers of ethylene glycol), nonabsorbable carbohydrates (lactulose, sorbitol), magnesium products, and sodium phosphate products.
Polyethylene glycol, the most studied osmotic laxative, has been shown to maintain therapeutic efficacy for up to 2 years, though it is not generally used this long.26 A meta-analysis of 10 randomized clinical trials found it to be superior to lactulose in improving stool consistency and frequency, and rates of adverse effects were similar to those with placebo.27
Lactulose and sorbitol are semisynthetic disaccharides that are not absorbed from the gastrointestinal tract. Apart from the osmotic effect of the disaccharide, these sugars are metabolized by colonic bacteria to acetic acid and other short-chain fatty acids, resulting in acidification of the stool, which exerts an osmotic effect in the colonic lumen.
Lactulose and sorbitol were shown to have similar efficacy in increasing the frequency of bowel movements in a small study, though patients taking lactulose had a higher rate of nausea.28
The usual recommended dose is 15 to 30 mL once or twice daily.
Adverse effects include gas, bloating, and abdominal distention (due to fermentation by colonic bacteria) and can limit long-term use.
Magnesium citrate and magnesium hydroxide are strong osmotic laxatives, but so far no clinical trial has been done to assess their efficacy in constipation. Although the risk of hypermagnesemia is low with magnesium-based products, this group of laxatives is generally avoided in patients with renal or cardiac disease.29
Sodium phosphate enemas (Fleet enemas) are used for bowel cleansing before certain procedures but have only limited use in constipation because of potential adverse effects such as hyperphosphatemia, hypocalcemia, and the rarer but more serious complication of acute phosphate nephropathy.30
Stimulant laxatives for short-term use only
Stimulant laxatives include glycerin, bisacodyl, senna, and sodium picosulfate. Sodium piosulfate and bisacodyl have been validated for treatment of chronic constipation for up to 4 weeks.31–33
Stimulant laxative suppositories should be used 30 minutes after meals to augment the physiologic gastrocolic reflex.
As more evidence is available for osmotic laxatives such as polyethylene glycol, they tend to be preferred over stimulant agents, especially for long-term use. Clinicians have traditionally hesitated to prescribe stimulant laxatives for long-term use, as they were thought to damage the enteric nervous system.34 Although more recent studies have not shown this potential effect,35 more research is warranted on the use of stimulant laxatives for longer than 4 weeks.
STOOL SOFTENERS: LITTLE EVIDENCE
Stool softeners enhance the interaction of stool and water, leading to softer stool and easier evacuation. Docusate sodium and docusate calcium are thought to facilitate the mixing of aqueous and fatty substances, thereby softening the stool.
However, there is little evidence to support the use of docusate for constipation in hospitalized adults or in ambulatory care. A recent review reported that docusate was no better than placebo in diminishing symptoms of constipation.36
INTESTINAL SECRETAGOGUES
The secretagogues include lubiprostone, linaclotide, and plecanatide. These medications are preferred therapy for patients with normal- or slow-transit constipation once conservative therapies have failed. Even though there is no current consensus, lifestyle measures and conservative treatment options should be tried for about 8 weeks.
Lubiprostone and linaclotide are approved by the US Food and Drug Administration (FDA) for both constipation and constipation-predominant irritable bowel syndrome. They activate chloride channels on the apical surface of enterocytes, increasing intestinal secretion of chloride, which in turn increases luminal sodium efflux to maintain electroneutrality, leading to secretion of water into the intestinal lumen. This eventually facilitates intestinal transit and increases the passage of stool.
Lubiprostone
Lubiprostone, a prostaglandin E1 derivative, is approved for treating chronic constipation, constipation-predominant irritable bowel syndrome in women, and opioid-induced constipation in patients with chronic noncancer pain.
Adverse effects in clinical trials were nausea (up to 30%) and headache.37,38
Linaclotide
Linaclotide, a minimally absorbed 14-amino acid peptide, increases intestinal secretion of chloride and bicarbonate, increasing intestinal fluid and promoting intestinal transit.39 It also decreases the firing rate of the visceral afferent pain fibers and helps reduce visceral pain, especially in patients with constipation-predominant irritable bowel syndrome.40 It is approved for chronic constipation and constipation-predominant irritable bowel syndrome.41–43
Dosage starts at 145 μg/day for chronic constipation, and can be titrated up to 290 μg if there is no response or if a diagnosis of constipation-predominant irritable bowel syndrome is under consideration. Linaclotide should be taken 30 to 60 minutes before breakfast to reduce the likelihood of diarrhea.44
Adverse effects. Diarrhea led to treatment discontinuation in 4.5% of patients in one study.42
Plecanatide
Plecanatide is a guanylate cyclase-c agonist with a mode of action similar to that of linaclotide. It was recently approved by the FDA for chronic idiopathic constipation in adults. The recommended dose is 3 mg once daily.
Data from phase 2 trials in chronic constipation showed improvement in straining, abdominal discomfort, and stool frequency after 14 days of treatment.45
A phase 3 trial showed that plecanatide was more effective than placebo when used for 12 weeks in 951 patients with chronic constipation (P = .009).46 The most common adverse effect reported was diarrhea.
SEROTONIN RECEPTOR AGONISTS
Activation of serotonin 5-HT4 receptors in the gut leads to release of acetylcholine, which in turn induces mucosal secretion by activating submucosal neurons and increasing gut motility.47
Two 5-HT4 receptor agonists were withdrawn from the market (cisapride in 2000 and tegaserod in 2007) due to serious cardiovascular adverse events (fatal arrhythmias, heart attacks, and strokes) resulting from their affinity for hERG-K+ cardiac channels.
The newer agents prucalopride,48 velusetrag, and naronapride are highly selective 5-HT4 agonists with low affinity for hERG-K+ receptors and do not have proarrhythmic properties, based on extensive assessment in clinical trials.
Prucalopride
Prucalopride has been shown to accelerate gastrointestinal and colonic transit in patients with chronic constipation, with improvement in bowel movements, symptoms of chronic constipation, and quality of life.49–52
Adverse effects reported with its use have been headache, nausea, abdominal pain, and cramps.
Prucalopride is approved in Europe and Canada for chronic constipation in women but is not yet approved in the United States.
Dosage is 2 mg orally once daily. Caution is advised in elderly patients, in whom the preferred maximum dose is 1 mg daily, as there are only limited data available on the safety of this medication in the elderly.
Velusetrag
Velusetrag has been shown to increase colonic motility and improve symptoms of chronic constipation. In a phase 2 trial,53 the most effective dose was 15 mg once daily. Higher doses were associated with a higher incidence of adverse effects such as diarrhea, headache, nausea, and vomiting.
Naronapride
Naronapride (ATI-7505) is in phase 2 trials for chronic constipation. Reported adverse effects were headache, diarrhea, nausea, and vomiting.54
BILE SALT ABSORPTION INHIBITORS
Bile acids exert prosecretory and prokinetic effects by increasing colonic secretion of water and electrolytes through the activation of adenylate cyclase. This happens as a result of their deconjugation after passage into the colon.
Elobixibat is an ileal bile acid transporter inhibitor that prevents absorption of nonconjugated bile salts in the distal ileum. It has few side effects because its systemic absorption is minimal. Phase 3 trials are under way. Dosage is 5 to 20 mg daily. Adverse effects are few because systemic absorption is minimal, but include abdominal pain and diarrhea.55,56
MANAGING OPIOID-INDUCED CONSTIPATION
Opioids cause constipation by binding to mu receptors in the enteric nervous system. Activation of these receptors decreases bowel tone and contractility, which increases transit time. Stimulation of these receptors also increases anal sphincter tone, resulting in decreased rectal evacuation.57
Though underrecognized, opioid-induced constipation affects 40% of patients who take these drugs for nonmalignant pain and 90% of those taking them for cancer pain. Patients with this condition were found to take more time off work and feel more impaired in their domestic and work-related obligations than patients who did not develop constipation with use of opioids.58
Initial management of opioid-induced constipation includes increasing intake of fluids and dietary fiber (fiber alone can worsen abdominal pain in this condition by increasing stool bulk without a concomitant improvement in peristalsis) and increasing physical activity. It is common clinical practice to use a stool softener along with a stimulant laxative if lifestyle modifications are inadequate.59 If these measures are ineffective, osmotic agents can be added.
If these conventional measures fail, a peripherally acting mu-opioid receptor antagonist such as methylnaltrexone or naloxegol should be considered.
Methylnaltrexone
Methylnaltrexone60,61 is a peripherally acting mu receptor antagonist with a rapid onset of action. It does not cross the blood-brain barrier, as it contains a methyl group. It was approved by the FDA in 2008 to treat opioid-induced constipation in adults with advanced illnesses when other approaches are ineffective.
Adverse effects. Although the mu receptor antagonist alvimopan had been shown to be associated with cardiovascular events hypothesized to be a consequence of opioid withdrawal, methylnaltrexone has been deemed to have a safe cardiovascular profile without any potential effects on platelets, corrected QT interval, metabolism, heart rate, or blood pressure.61 Side effects include abdominal pain, nausea, diarrhea, hot flashes, tremor, and chills.
Contraindications. Methylnaltrexone is contraindicated in patients with structural diseases of the gastrointestinal tract, ie, peptic ulcer disease, inflammatory bowel disease, diverticulitis, stomach or intestinal cancer) since it can increase the risk of perforation.
Dosing is 1 dose subcutaneously every other day, as needed, and no more than 1 dose in a 24-hour period. Dosage is based on weight: 0.15 mg/kg/dose for patients weighing less than 38 kg or more than 114 kg; 8 mg for those weighing 38 to 62 kg; and 12 mg for those weighing 62 to 114 kg.62
Naloxegol
Naloxegol, FDA-approved for treating opioid-induced constipation in 2014, consists of naloxone conjugated with polyethylene glycol, which prevents it from crossing the blood-brain barrier and diminishing the central effects of opioid-induced analgesia. Unlike methylnaltrexone, which is given by subcutaneous injection, naloxegol is taken orally.
Adverse effects reported in clinical trials63,64 were abdominal pain, diarrhea, nausea, headache, and flatulence. No clinically relevant association with QT and corrected QT interval prolongation or cardiac repolarization was noted.64
Dosing is 25 mg by mouth once daily, which can be decreased to 12.5 mg if the initial dose is difficult to tolerate. It should be taken on an empty stomach at least 1 hour before the first meal of the day or 2 hours after the meal. In patients with renal impairment (creatinine clearance < 60 mL/min), the dose is 12.5 mg once daily.65
CONSTIPATION-PREDOMINANT IRRITABLE BOWEL SYNDROME
Irritable bowel syndrome is the reason for 3.1 million office visits and 59 million prescriptions in the United States every year, with patients equally distributed between diarrhea-predominant, constipation-predominant, and mixed subtypes.66
To be diagnosed with constipation-predominant irritable bowel syndrome, patients must meet the Rome IV criteria, more than 25% of bowel movements should have Bristol stool form types 1 or 2, and less than 25% of bowel movements should have Bristol stool form types 6 or 7. In practice, patients reporting that their bowel movements are usually constipated often suffices to make the diagnosis.5
Osmotic laxatives are often tried first, but despite improving stool frequency and consistency, they have little efficacy in satisfying complaints of bloating or abdominal pain in patients with constipation-predominant irritable syndrome.67 Stimulant laxatives have not yet been tested in clinical trials. Lubiprostone and linaclotide are FDA-approved for this condition; in women, lubiprostone is approved only for those over age 18.
Antidepressant therapy
Patients often derive additional benefit from treatment with antidepressants. A meta-analysis demonstrated a number needed to treat of 4 for selective serotonin reuptake inhibitors and tricyclic antidepressants in managing abdominal pain associated with irritable bowel syndrome.68 The major limiting factor is usually adverse effects of these drugs.
For constipation-predominant irritable bowel syndrome, selective serotonin reuptake inhibitors are preferred over tricyclics because of their additional prokinetic properties. Starting at a low dose and titrating upward slowly avoids potential adverse effects.
Cognitive behavioral therapy has also been beneficial in treating irritable bowel syndrome.69
Adjunctive therapies
Adjunctive therapies including peppermint oil, probiotics (eg, Lactobacillus, Bifidobacterium), and acupuncture have also shown promise in managing irritable bowel syndrome, but more data are needed on the use of these therapies for constipation-predominant irritable bowel syndrome before any definite conclusions can be drawn.70 Other emerging pharmacologic therapies are plecanatide (discussed earlier) and tenapanor.
Peppermint oil is an antispasmodic that inhibits calcium channels, leading to relaxation of smooth muscles in the gastrointestinal tract. Different dosages and treatment durations have been studied—450 to 900 mg daily in 2 to 3 divided doses over 1 to 3 months.71,72 The most common adverse effect reported was gastroesophageal reflux, related in part to the oil’s relaxing effect on the lower esophageal sphincter. Observation of this led to the development of enteric-coated preparations that have the potential to bypass the upper gastrointestinal tract.73
Tenapanor inhibits the sodium-hydrogen exchanger 3 channel (a regulator of sodium and water uptake in intestinal lumen), which in turn leads to a higher sodium level in the entire gastrointestinal tract (whereas linaclotide’s action is limited to the duodenum and jejunem), resulting in more fluid volume and increased luminal transit.74 It was found effective in a phase 2 clinical trial,75 and the most effective dose was 50 mg twice daily.
Since tenapanor is minimally absorbed, it has few side effects, the major ones being diarrhea (11.2% vs 0% with placebo) and urinary tract infection (5.6% vs 4.4% with placebo).75 Further study is needed to confirm these findings.
Tenapanor also has the advantage of inhibiting luminal phosphorus absorption. This has led to exploration of its use as a phosphate binder in patients with end-stage renal disease.
DYSSYNERGIC DEFECATION AND ANORECTAL BIOFEEDBACK
According to the Rome IV criteria,5 dyssynergic defecation is present if the criteria for chronic constipation are met, if a dyssynergic pattern of defecation is confirmed by manometry, imaging, or electromyography, and if 1 or more of the following are present: inability to expel an artificial stool (a 50-mL water-filled balloon) within 1 minute, prolonged colonic transit time, inability to evacuate, or 50% or more retention of barium during defecography.5
Even though biofeedback has been controversial as a treatment for dyssynergic defecation because of conflicting results in older studies,76 3 trials have shown it to be better than placebo, laxatives, and muscle relaxants, with symptomatic improvement in 70% of patients.77–79
Biofeedback therapy involves an instrument-based auditory or visual tool (using electromyographic sensors or anorectal manometry) to help patients coordinate abdominal, rectal, puborectalis, and anal sphincter muscles and produce a propulsive force using their abdominal muscles to achieve complete evacuation. Important components of this therapy include:
Proper evacuation positioning (brace-pump technique, which involves sitting on the toilet leaning forward with forearms resting on thighs, shoulders relaxed, and feet placed on a small footstool
Breathing relaxation and training exercises during defecation (no straining, keeping a normal pattern of breathing, and avoiding holding the breath while defecating)
Use of the abdominal muscles by pushing the abdomen forward, along with relaxation of the anal sphincter.80
The anorectal feedback program usually consists of 6 weekly sessions of 45 to 60 minutes each. Limitations of this therapy include unavailability, lack of trained therapists, lack of insurance coverage, and inapplicability to certain patient groups, such as those with dementia or learning disabilities.
SURGERY FOR CHRONIC CONSTIPATION
Surgery for constipation is reserved for patients who continue to have symptoms despite optimal medical therapy.
Total abdominal colectomy and ileorectal anastomosis
Total abdominal colectomy with ileorectal anastomosis is a surgical option for medically intractable slow-transit constipation. Before considering surgery, complete diagnostic testing should be done, including colonic manometry and documentation of whether the patient also has outlet dysfunction.
Even though it has shown excellent outcomes and satisfaction rates as high as 100% in patients with pure slow-transit constipation,81–83 results in older studies in patients with mixed disorders (eg, slow-transit constipation with features of outlet dysfunction) were less predictable.84 More recent studies have reported comparable long-term morbidity and postoperative satisfaction rates in those with pure slow-transit constipation and those with a mixed disorder, indicating that careful patient selection is likely the key to a favorable outcome.85
Partial colectomies based on segmental colon transit time measurements can also be considered in some patients.86
Stapled transanal resection
Stapled transanal resection involves circumferential transanal stapling of the redundant rectal mucosa. It is an option for patients with defecatory disorders, specifically large rectoceles and rectal intussusception not amenable to therapy with pelvic floor retraining exercises.87
The efficacy of this procedure in controlling symptoms and improving quality of life is around 77% to 81% at 12 months, though complication rates as high as 46% and disappointing long-term outcomes have been a deterrent to its widespread acceptance in the United States.88–91
- Mugie SM, Benninga MA, Di Lorenzo C. Epidemiology of constipation in children and adults: a systematic review. Best Pract Res Clin Gastroenterol 2011; 25:3–18.
- Kinnunen O. Study of constipation in a geriatric hospital, day hospital, old people's home and at home. Aging (Milano) 1991; 3:161–170.
- Everhart JE, Go VL, Johannes RS, Fitzsimmons SC, Roth HP, White LR. A longitudinal survey of self-reported bowel habits in the United States. Dig Dis Sci 1989; 34:1153–1162.
- Shah ND, Chitkara DK, Locke GR, Meek PD, Talley NJ. Ambulatory care for constipation in the United States, 1993-2004. Am J Gastroenterol 2008; 103:1746–1753.
- Mearin F, Lacy BE, Chang L, et al. Bowel disorders. Gastroenterology 2016; 150:1393–1407.
- Bharucha AE. Pelvic floor: anatomy and function. Neurogastroenterol Motil 2006; 18:507–519.
- Bharucha AE, Pemberton JH, Locke GR 3rd. American Gastroenterological Association technical review on constipation. Gastroenterology 2013; 144:218–238.
- Grundy D, Al-Chaer ED, Aziz Q, et al. Fundamentals of neurogastroenterology: basic science. Gastroenterology 2006; 130:1391–1411.
- Gallegos-Orozco JF, Foxx-Orenstein AE, Sterler SM, Stoa JM. Chronic constipation in the elderly. Am J Gastroenterol 2012; 107:18–26.
- Mancini I, Bruera E. Constipation in advanced cancer patients. Support Care Cancer 1998; 6:356–364.
- Bassotti G, Chistolini F, Sietchiping-Nzepa F, de Roberto G, Morelli A, Chiarioni G. Biofeedback for pelvic floor dysfunction in constipation. BMJ 2004; 328:393–396.
- American Gastroenterological Association, Bharucha AE, Dorn SD, Lembo A, Pressman A. American Gastroenterological Association medical position statement on constipation. Gastroenterology 2013; 144:211–217.
- Costilla VC, Foxx-Orenstein AE. Constipation in adults: diagnosis and management. Curr Treat Options Gastroenterol 2014; 12:310–321.
- Rao SS, Singh S. Clinical utility of colonic and anorectal manometry in chronic constipation. J Clin Gastroenterol 2010; 44:597–609.
- Minguez M, Herreros B, Sanchiz V, et al. Predictive value of the balloon expulsion test for excluding the diagnosis of pelvic floor dyssynergia in constipation. Gastroenterology 2004; 126:57–62.
- Diamant NE, Kamm MA, Wald A, Whitehead WE. AGA technical review on anorectal testing techniques. Gastroenterology 1999; 116:735–760.
- Pezim ME, Pemberton JH, Levin KE, Litchy WJ, Phillips SF. Parameters of anorectal and colonic motility in health and in severe constipation. Dis Colon Rectum 1993; 36:484–491.
- Bharucha AE, Fletcher JG, Seide B, Riederer SJ, Zinsmeister AR. Phenotypic variation in functional disorders of defecation. Gastroenterology 2005; 128:1199–1210.
- De Schryver AM, Samsom M, Smout AI. Effects of a meal and bisacodyl on colonic motility in healthy volunteers and patients with slow-transit constipation. Dig Dis Sci 2003; 48:1206–1212.
- Villoria A, Serra J, Azpiroz F, Malagelada JR. Physical activity and intestinal gas clearance in patients with bloating. Am J Gastroenterol 2006; 101:2552–2557.
- Sikirov D. Comparison of straining during defecation in three positions: results and implications for human health. Dig Dis Sci 2003; 48:1201–1205.
- Muller-Lissner SA, Kamm MA, Scarpignato C, Wald A. Myths and misconceptions about chronic constipation. Am J Gastroenterol 2005; 100:232–242.
- Voderholzer WA, Schatke W, Muhldorfer BE, Klauser AG, Birkner B, Muller-Lissner SA. Clinical response to dietary fiber treatment of chronic constipation. Am J Gastroenterol 1997; 92:95–98.
- Bijkerk CJ, de Wit NJ, Muris JW, Whorwell PJ, Knottnerus JA, Hoes AW. Soluble or insoluble fibre in irritable bowel syndrome in primary care? Randomised placebo controlled trial. BMJ 2009; 339:b3154.
- Suares NC, Ford AC. Systematic review: the effects of fibre in the management of chronic idiopathic constipation. Aliment Pharmacol Ther 2011; 33:895–901.
- Dipalma JA, Cleveland MV, McGowan J, Herrera JL. A randomized, multicenter, placebo-controlled trial of polyethylene glycol laxative for chronic treatment of chronic constipation. Am J Gastroenterol 2007; 102:1436–1441.
- Lee-Robichaud H, Thomas K, Morgan J, Nelson RL. Lactulose versus polyethylene glycol for chronic constipation. Cochrane Database Syst Rev 2010; 7:CD007570.
- Lederle FA, Busch DL, Mattox KM, West MJ, Aske DM. Cost-effective treatment of constipation in the elderly: a randomized double-blind comparison of sorbitol and lactulose. Am J Med 1990; 89:597–601.
- Nyberg C, Hendel J, Nielsen OH. The safety of osmotically acting cathartics in colonic cleansing. Nat Rev Gastroenterol Hepatol 2010; 7:557–564.
- Ainley EJ, Winwood PJ, Begley JP. Measurement of serum electrolytes and phosphate after sodium phosphate colonoscopy bowel preparation: an evaluation. Dig Dis Sci 2005; 50:1319–1323.
- Kienzle-Horn S, Vix JM, Schuijt C, Peil H, Jordan CC, Kamm MA. Efficacy and safety of bisacodyl in the acute treatment of constipation: a double-blind, randomized, placebo-controlled study. Aliment Pharmacol Ther 2006; 23:1479–1488.
- Kienzle-Horn S, Vix JM, Schuijt C, Peil H, Jordan CC, Kamm MA. Comparison of bisacodyl and sodium picosulphate in the treatment of chronic constipation. Curr Med Res Opin 2007; 23:691–699.
- Mueller-Lissner S, Kamm MA, Wald A, et al. Multicenter, 4-week, double-blind, randomized, placebo-controlled trial of sodium picosulfate in patients with chronic constipation. Am J Gastroenterol 2010; 105:897–903.
- Smith B. Pathologic changes in the colon produced by anthraquinone purgatives. Dis Colon Rectum 1973; 16:455–458.
- Kiernan JA, Heinicke EA. Sennosides do not kill myenteric neurons in the colon of the rat or mouse. Neuroscience 1989; 30:837–842.
- Ottawa (ON): Canadian Agency for Drugs and Technologies in Health. Dioctyl sulfosuccinate or docusate (calcium or sodium) for the prevention or management of constipation: a review of the clinical effectiveness. www.ncbi.nlm.nih.gov/pubmedhealth/PMH0071207/. Accessed April 6, 2017.
- Saad R, Chey WD. Lubiprostone for chronic idiopathic constipation and irritable bowel syndrome with constipation. Expert Rev Gastroenterol Hepatol 2008; 2:497–508.
- Johanson JF, Morton D, Geenen J, Ueno R. Multicenter, 4-week, double-blind, randomized, placebo-controlled trial of lubiprostone, a locally-acting type-2 chloride channel activator, in patients with chronic constipation. Am J Gastroenterol 2008; 103:170–177.
- Harris LA, Crowell MD. Linaclotide, a new direction in the treatment of irritable bowel syndrome and chronic constipation. Curr Opin Mol Ther 2007; 9:403–410.
- Johnston JM, Kurtz CB, Macdougall JE, et al. Linaclotide improves abdominal pain and bowel habits in a phase IIb study of patients with irritable bowel syndrome with constipation. Gastroenterology 2010; 139:1877–1886.e2.
- Lembo AJ, Schneier HA, Shiff SJ, et al. Two randomized trials of linaclotide for chronic constipation. N Engl J Med 2011; 365:527–536.
- Chey WD, Lembo AJ, Lavins BJ, et al. Linaclotide for irritable bowel syndrome with constipation: a 26-week, randomized, double-blind, placebo-controlled trial to evaluate efficacy and safety. Am J Gastroenterol 2012; 107:1702–1712.
- Rao S, Lembo AJ, Shiff SJ, et al. A 12-week, randomized, controlled trial with a 4-week randomized withdrawal period to evaluate the efficacy and safety of linaclotide in irritable bowel syndrome with constipation. Am J Gastroenterol 2012; 107:1714–1725.
- Chey WD, Kurlander J, Eswaran S. Irritable bowel syndrome: a clinical review. JAMA 2015; 313:949–958.
- Shailubhai K, Talluto C, Comiskey S, Foss JA, Joslyn A, Jacob G. Phase II clinical evaluation of SP-304, a guanylate cyclase-C agonist, for treatment of chronic constipation. Am J Gastroenterol 2010; 105:S487–S488.
- Miner P, Surowitz R, Fogel R, et al. Plecanatide, a novel guanylate cyclase-C (GC-C) receptor agonist, is efficacious and safe in patients with chronic idiopathic constipation (CIC): results from a 951 patient, 12-week, multi-center trial (abstract). Gastroenterology 2013; 144:S163.
- Coss-Adame E, Rao SS. Brain and gut interactions in irritable bowel syndrome: new paradigms and new understandings. Curr Gastroenterol Rep 2014; 16:379.
- Mendzelevski B, Ausma J, Chanter DO, et al. Assessment of the cardiac safety of prucalopride in healthy volunteers: a randomized, double-blind, placebo- and positive-controlled thorough QT study. Br J Clin Pharmacol 2012; 73:203–209.
- Camilleri M, Kerstens R, Rykx A, Vandeplassche L. A placebo-controlled trial of prucalopride for severe chronic constipation. N Engl J Med 2008; 358:2344–2354.
- Tack J, van Outryve M, Beyens G, Kerstens R, Vandeplassche L. Prucalopride (Resolor) in the treatment of severe chronic constipation in patients dissatisfied with laxatives. Gut 2009; 58:357–365.
- Quigley EM, Vandeplassche L, Kerstens R, Ausma J. Clinical trial: the efficacy, impact on quality of life, and safety and tolerability of prucalopride in severe chronic constipation—a 12-week, randomized, double-blind, placebo-controlled study. Aliment Pharmacol Ther 2009; 29:315–328.
- Ford AC, Suares NC. Effect of laxatives and pharmacological therapies in chronic idiopathic constipation: systematic review and meta-analysis. Gut 2011; 60:209–218.
- Goldberg M, Li YP, Johanson JF, et al. Clinical trial: the efficacy and tolerability of velusetrag, a selective 5-HT4 agonist with high intrinsic activity, in chronic idiopathic constipation—a 4-week, randomized, double-blind, placebo-controlled, dose-response study. Aliment Pharmacol Ther 2010; 32:1102–1112.
- Palme M, Milner PG, Ellis DJ, Marmon T, Canafax DM. A novel gastrointestinal prokinetic, ATI-7505, increased spontaneous bowel movements (sbms) in a phase II, randomized, placebo-controlled study of patients with chronic idiopathic constipation (CIC). Gastroenterology 2010; 138:S-128–S-129.
- Chey WD, Camilleri M, Chang L, Rikner L, Graffner H. A randomized placebo-controlled phase IIb trial of a3309, a bile acid transporter inhibitor, for chronic idiopathic constipation. Am J Gastroenterol 2011; 106:1803–1812.
- Wong BS, Camilleri M, McKinzie S, Burton D, Graffner H, Zinsmeister AR. Effects of A3309, an ileal bile acid transporter inhibitor, on colonic transit and symptoms in females with functional constipation. Am J Gastroenterol 2011; 106:2154–2164.
- Pappagallo M. Incidence, prevalence, and management of opioid bowel dysfunction. Am J Surg 2001; 182(suppl):11S–18S.
- Bell T, Annunziata K, Leslie JB. Opioid-induced constipation negatively impacts pain management, productivity, and health-related quality of life: findings from the National Health and Wellness Survey. J Opioid Manag 2009; 5:137–144.
- Sykes NP. A volunteer model for the comparison of laxatives in opioid-related constipation. J Pain Symptom Manage 1996; 11:363–369.
- ClinicalTrials.gov. A multicenter, randomized, double-blind, placebo-controlled, parallel-group study of oral MOA-728 for the treatment of opioid- induced bowel dysfunction in subjects with chronic nonmalignant pain. ClinicalTrials.gov Identifier: NCT00547586. https://clinicaltrials.gov/ct2/show/NCT00547586. Accessed March 22, 2017.
- ClinicalTrials.gov. An open-label study to evaluate the long-term safety of subcutaneous MOA-728 for treatment of opioid-induced constipation in subjects with nonmalignant pain. ClinicalTrials.gov Identifier: NCT00804141. https://clinicaltrials.gov/ct2/show/NCT00804141. Accessed April 6, 2017.
- Wyeth Pharmaceuticals. Relistor package insert. http://labeling.pfizer.com/showlabeling.aspx?id=499. Accessed March 22, 2017.
- Webster L, Dhar S, Eldon M, Masuoka L, Lappalainen J, Sostek M. A phase 2, double-blind, randomized, placebo-controlled, dose-escalation study to evaluate the efficacy, safety, and tolerability of naloxegol in patients with opioid-induced constipation. Pain 2013; 154:1542–1550.
- Chey WD, Webster L, Sostek M, Lappalainen J, Barker PN, Tack J. Naloxegol for opioid-induced constipation in patients with noncancer pain. N Engl J Med 2014; 370:2387–2396.
- Jones R, Prommer E, Backstedt D. Naloxegol: a novel therapy in the management of opioid-induced constipation. Am J Hosp Palliat Care 2016; 33:875–880.
- Guilera M, Balboa A, Mearin F. Bowel habit subtypes and temporal patterns in irritable bowel syndrome: systematic review. Am J Gastroenterol 2005; 100:1174–1184.
- Chapman RW, Stanghellini V, Geraint M, Halphen M. Randomized clinical trial: macrogol/PEG 3350 plus electrolytes for treatment of patients with constipation associated with irritable bowel syndrome. Am J Gastroenterol 2013; 108:1508–1515.
- Ford AC, Quigley EM, Lacy BE, et al. Effect of antidepressants and psychological therapies, including hypnotherapy, in irritable bowel syndrome: systematic review and meta-analysis. Am J Gastroenterol 2014; 109:1350–1366.
- Ballou S, Keefer L. Psychological interventions for irritable bowel syndrome and inflammatory bowel diseases. Clin Transl Gastroenterol 2017; 8:e214.
- Ford AC, Moayyedi P, Lacy BE, et al; Task Force on the Management of Functional Bowel Disorders. American College of Gastroenterology monograph on the management of irritable bowel syndrome and chronic idiopathic constipation. Am J Gastroenterol 2014; 109(suppl 1):S2–S27.
- Ford AC, Talley NJ, Spiegel BM, et al. Effect of fibre, antispasmodics, and peppermint oil in the treatment of irritable bowel syndrome: systematic review and meta-analysis. BMJ 2008; 337:a2313.
- Wall GC, Bryant GA, Bottenberg MM, Maki ED, Miesner AR. Irritable bowel syndrome: a concise review of current treatment concepts. World J Gastroenterol 2014; 20:8796–8806.
- Kligler B, Chaudhary S. Peppermint oil. Am Fam Physician 2007; 75:1027–1030.
- Spencer AG, Labonte ED, Rosenbaum DP, et al. Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci Transl Med 2014; 6:227ra36.
- Rosenbaum DP. A randomized, double-blind, placebo-controlled study to assess the safety and efficacy of AZD1722 for the treatment of constipation-predominant irritable bowel syndrome (IBS-C). 2014. https://clinicaltrials.gov/ct2/show/NCT01923428. Accessed April 6, 2017.
- Rao SS. Biofeedback therapy for dyssynergic (obstructive) defecation. J Clin Gastroenterol 2000; 30:115–116.
- Cadeddu F, Salis F, De Luca E, Ciangola I, Milito G. Efficacy of biofeedback plus transanal stimulation in the management of pelvic floor dyssynergia: a randomized trial. Tech Coloproctol 2015; 19:333–338.
- Chiarioni G, Whitehead WE, Pezza V, Morelli A, Bassotti G. Biofeedback is superior to laxatives for normal transit constipation due to pelvic floor dyssynergia. Gastroenterology 2006; 130:657–664.
- Chiarioni G, Heymen S, Whitehead WE. Biofeedback therapy for dyssynergic defecation. World J Gastroenterol 2006; 12:7069–7074.
- Rao SS. Biofeedback therapy for constipation in adults. Best Pract Res Clin Gastroenterol 2011; 25:159–166.
- Hassan I, Pemberton JH, Young-Fadok TM, et al. Ileorectal anastomosis for slow transit constipation: long-term functional and quality of life results. J Gastrointest Surg 2006; 10:1330–1337.
- You YT, Wang JY, Changchien CR, et al. Segmental colectomy in the management of colonic inertia. Am Surg 1998; 64:775–777.
- Nyam DC, Pemberton JH, Ilstrup DM, Rath DM. Long-term results of surgery for chronic constipation. Dis Colon Rectum 1997; 40:273–279.
- Pemberton JH, Rath DM, Ilstrup DM. Evaluation and surgical treatment of severe chronic constipation. Ann Surg 1991; 214:403–413.
- Reshef A, Alves-Ferreira P, Zutshi M, Hull T, Gurland B. Colectomy for slow transit constipation: effective for patients with coexistent obstructed defecation. Int J Colorectal Dis 2013; 28:841–847.
- Lundin E, Karlbom U, Pahlman L, Graf W. Outcome of segmental colonic resection for slow-transit constipation. Br J Surg 2002; 89:1270–1274.
- Schwandner O, Stuto A, Jayne D, et al. Decision-making algorithm for the STARR procedure in obstructed defecation syndrome: position statement of the group of STARR pioneers. Surg Innov 2008; 15:105–109.
- Titu LV, Riyad K, Carter H, Dixon AR. Stapled transanal rectal resection for obstructed defecation: a cautionary tale. Dis Colon Rectum 2009; 52:1716–1722.
- Goede AC, Glancy D, Carter H, Mills A, Mabey K, Dixon AR. Medium-term results of stapled transanal rectal resection (STARR) for obstructed defecation and symptomatic rectal-anal intussusception. Colorectal Dis 2011; 13:1052–1057.
- Jayne DG, Schwandner O, Stuto A. Stapled transanal rectal resection for obstructed defecation syndrome: one-year results of the european STARR registry. Dis Colon Rectum 2009; 52:1205–1214.
- Madbouly KM, Abbas KS, Hussein AM. Disappointing long-term outcomes after stapled transanal rectal resection for obstructed defecation. World J Surg 2010; 34:2191–2196.
Chronic constipation has a variety of possible causes and mechanisms. Although traditional conservative treatments are still valid and first-line, if these fail, clinicians can choose from a growing list of new treatments, tailored to the cause in the individual patient.
This article discusses how defecation works (or doesn’t), the types of chronic constipation, the available diagnostic tools, and traditional and newer treatments, including some still in development.
THE EPIDEMIOLOGY OF CONSTIPATION
Chronic constipation is one of the most common gastrointestinal disorders, affecting about 15% of all adults and 30% of those over the age of 60.1 It can be a primary disorder or secondary to other factors.
Constipation is more prevalent in women and in institutionalized elderly people.2 It is associated with lower socioeconomic status, depression, less self-reported physical activity, certain medications, and stressful life events.3 Given its high prevalence and its impact on quality of life, it is also associated with significant utilization of healthcare resources.4
Constipation defined by Rome IV criteria
Physicians and patients may disagree about what constitutes constipation. Physicians primarily regard it as infrequent bowel movements, while patients tend to have a broader definition. According to the Rome IV criteria,5 chronic constipation is defined by the presence of the following for at least 3 months (with symptom onset at least 6 months prior to diagnosis):
(1) Two or more of the following for more than 25% of defecations:
- Straining
- Lumpy or hard stools
- Sensation of incomplete evacuation
- Sensation of anorectal obstruction or blockage
- Manual maneuvers to facilitate evacuation
- Fewer than 3 spontaneous bowel movements per week.
(2) Loose stools are rarely present without the use of laxatives.
(3) The patient does not meet the criteria for diagnosis of irritable bowel syndrome.
DEFECATION IS COMPLEX
Defecation begins when the rectum fills with stool, causing relaxation of the internal anal sphincter and the urge to defecate. The external anal sphincter, which is under voluntary control, can then either contract to delay defecation or relax to allow the stool to be expelled.6
Colonic muscles propel stool toward the rectum in repetitive localized contractions that help mix and promote absorption of the content, and larger coordinated (high-amplitude propagating) contractions that, in healthy individuals, move the stool forward from the proximal to the distal colon multiple times daily. These contractions usually occur in the morning and are accentuated by gastric distention from food and the resulting gastrocolic reflex.
Serotonin (5-HT) is released by enterochromaffin cells in response to distention of the gut wall. It mediates peristaltic movements of the gastrointestinal tract by binding to receptors (especially 5-HT4), stimulating release of neurotransmitters such as acetylcholine, causing smooth-muscle contraction behind the luminal contents and propelling them forward.
PRIMARY CONSTIPATION DISORDERS
The American Gastroenterological Association7 classifies constipation into 3 groups on the basis of colonic transit time and anorectal function:
Normal-transit constipation
Stool normally takes 20 to 72 hours to pass through the colon, with transit time affected by diet, drugs, level of physical activity, and emotional status.8
Normal-transit constipation is the most common type of constipation. The term is sometimes used interchangeably with constipation-predominant irritable bowel syndrome, but the latter is a distinct entity characterized by abdominal pain relieved by defecation as the primary symptom, as well as having occasional loose stools. These 2 conditions can be hard to tell apart, especially if the patient cannot describe the symptoms precisely.
Slow-transit constipation
Slow-transit constipation—also called delayed-transit constipation, colonoparesis, colonic inertia, and pseudo-obstruction—is defined as prolonged stool transit in the colon, ie, for more than 5 days.9 It can be the result of colonic smooth muscle dysfunction, compromised colonic neural pathways, or both, leading to slow colon peristalsis.
Factors that can affect colonic motility such as opioid use and hypothyroidism should be carefully considered in these patients. Opioids are notorious for causing constipation by decreasing bowel tone and contractility and thereby increasing colonic transit time. They also tighten up the anal sphincters, resulting in decreased rectal evacuation.10
Outlet dysfunction
Outlet dysfunction, also called pelvic floor dysfunction or defecatory disorder, is associated with incomplete rectal evacuation. It can be a consequence of weak rectal expulsion forces (slow colonic transit, rectal hyposensitivity), functional resistance to rectal evacuation (high anal resting pressure, anismus, incomplete relaxation of the anal sphincter, dyssynergic defecation), or structural outlet obstruction (excessive perineal descent, rectoceles, rectal intussusception). About 50% of patients with outlet dysfunction have concurrent slow-transit constipation.
Dyssynergic defecation is the most common outlet dysfunction disorder, accounting for about half of the cases referred to tertiary centers. It is defined as a paradoxical elevation in anal sphincter tone or less than 20% relaxation of the resting anal sphincter pressure with weak abdominal and pelvic propulsive forces.11 Anorectal biofeedback is a therapeutic option for dyssynergic defecation, as we discuss later in this article.
SECONDARY CONSTIPATION
Constipation can be secondary to several conditions and factors (Table 1), including:
- Neurologic disorders that affect gastrointestinal motility (eg, Hirschsprung disease, Parkinson disease, multiple sclerosis, spinal cord injury, stroke, spinal or ganglionic tumor, hypothyroidism, amyloidosis, diabetes mellitus, hypercalcemia)
- Drugs used to treat neurologic disorders
- Mechanical obstruction
- Diet (eg, low fiber, decreased fluid intake).
EVALUATION OF CONSTIPATION
It is crucial for physicians to efficiently use the available diagnostic tools for constipation to tailor the treatment to the patient.
Evaluation of chronic constipation begins with a thorough history and physical examination to rule out secondary constipation (Figure 1). Red flags such as unintentional weight loss, blood in the stool, rectal pain, fever, and iron-deficiency anemia should prompt referral for colonoscopy to evaluate for malignancy, colitis, or other potential colonic abnormalities.12
A detailed perineal and rectal examination can help diagnose defecatory disorders and should include evaluation of the resting anal tone and the sphincter during simulated evacuation.
Laboratory tests of thyroid function, electrolytes, and a complete blood cell count should be ordered if clinically indicated.13
Further tests
Further diagnostic tests can be considered if symptoms persist despite conservative treatment or if a defecatory disorder is suspected. These include anorectal manometry, colonic transit studies, defecography, and colonic manometry.
Anorectal manometry and the rectal balloon expulsion test are usually done first because of their high sensitivity (88%) and specificity (89%) for defecatory disorders.14 These tests measure the function of the internal and external anal sphincters at rest and with straining and assess rectal sensitivity and compliance. Anorectal manometry is also used in biofeedback therapy in patients with dyssynergic defecation.15
Colonic transit time can be measured if anorectal manometry and the balloon expulsion test are normal. The study uses radiopaque markers, radioisotopes, or wireless motility capsules to confirm slow-transit constipation and to identify areas of delayed transit in the colon.16
Defecography is usually the next step in diagnosis if anorectal manometry and balloon expulsion tests are inconclusive or if an anatomic abnormality of the pelvic floor is suspected. It can be done with a variety of techniques. Barium defecography can identify anatomic defects, scintigraphy can quantify evacuation of artificial stools, and magnetic resonance defecography visualizes anatomic landmarks to assess pelvic floor motion without exposing the patient to radiation.17,18
Colonic manometry is most useful in patients with refractory slow-transit constipation and can identify patients with isolated colonic motor dysfunction with no pelvic floor dysfunction who may benefit from subtotal colectomy and end-ileostomy.7
TRADITIONAL TREATMENTS STILL THE MAINSTAY
Nonpharmacologic treatments are the first-line options for patients with normal-transit and slow-transit constipation and should precede diagnostic testing. Lifestyle modifications and dietary changes (Table 2) aim to augment the known factors that stimulate the gastrocolic reflex and increase intestinal motility by high-amplitude propagated contractions.
Increasing physical activity increases intestinal gas clearance, decreases bloating, and lessens constipation.19,20
Toilet training is an integral part of lifestyle modifications.21
Diet. Drinking hot caffeinated beverages, eating breakfast within an hour of waking up, and consuming fiber in the morning (25–30 g of fiber daily) have traditionally been recommended as the first-line measures for chronic constipation. Dehydrated patients with constipation also benefit from increasing their fluid intake.22
LAXATIVES
Fiber (bulk-forming laxatives) for normal-transit constipation
Fiber remains a key part of the initial management of chronic constipation, as it is cheap, available, and safe. Increasing fiber intake is effective for normal-transit constipation, but patients with slow-transit constipation or refractory outlet dysfunction are less likely to benefit.23 Other laxatives are incorporated into the regimen if first-line nonpharmacologic interventions fail (Table 3).
Bulk-forming laxatives include insoluble fiber (wheat bran) and soluble fiber (psyllium, methylcellulose, inulin, calcium polycarbophil). Insoluble fiber, though often used, has little impact on symptoms of chronic constipation after 1 month of use, and up to 60% of patients report adverse effects from it.24 On the other hand, clinical trials have shown that soluble fiber such as psyllium facilitates defecation and improves functional bowel symptoms in patients with normal-transit constipation.25
Patients should be instructed to increase their dietary fiber intake gradually to avoid adverse effects and should be told to expect significant symptomatic improvement only after a few weeks. They should also be informed that increasing dietary fiber intake can cause bloating but that the bloating is temporary. If it continues, a different fiber can be tried.
Osmotic laxatives
Osmotic laxatives are often employed as a first- line laxative treatment option for patients with constipation. They draw water into the lumen by osmosis, helping to soften stool and speed intestinal transit. They include macrogols (inert polymers of ethylene glycol), nonabsorbable carbohydrates (lactulose, sorbitol), magnesium products, and sodium phosphate products.
Polyethylene glycol, the most studied osmotic laxative, has been shown to maintain therapeutic efficacy for up to 2 years, though it is not generally used this long.26 A meta-analysis of 10 randomized clinical trials found it to be superior to lactulose in improving stool consistency and frequency, and rates of adverse effects were similar to those with placebo.27
Lactulose and sorbitol are semisynthetic disaccharides that are not absorbed from the gastrointestinal tract. Apart from the osmotic effect of the disaccharide, these sugars are metabolized by colonic bacteria to acetic acid and other short-chain fatty acids, resulting in acidification of the stool, which exerts an osmotic effect in the colonic lumen.
Lactulose and sorbitol were shown to have similar efficacy in increasing the frequency of bowel movements in a small study, though patients taking lactulose had a higher rate of nausea.28
The usual recommended dose is 15 to 30 mL once or twice daily.
Adverse effects include gas, bloating, and abdominal distention (due to fermentation by colonic bacteria) and can limit long-term use.
Magnesium citrate and magnesium hydroxide are strong osmotic laxatives, but so far no clinical trial has been done to assess their efficacy in constipation. Although the risk of hypermagnesemia is low with magnesium-based products, this group of laxatives is generally avoided in patients with renal or cardiac disease.29
Sodium phosphate enemas (Fleet enemas) are used for bowel cleansing before certain procedures but have only limited use in constipation because of potential adverse effects such as hyperphosphatemia, hypocalcemia, and the rarer but more serious complication of acute phosphate nephropathy.30
Stimulant laxatives for short-term use only
Stimulant laxatives include glycerin, bisacodyl, senna, and sodium picosulfate. Sodium piosulfate and bisacodyl have been validated for treatment of chronic constipation for up to 4 weeks.31–33
Stimulant laxative suppositories should be used 30 minutes after meals to augment the physiologic gastrocolic reflex.
As more evidence is available for osmotic laxatives such as polyethylene glycol, they tend to be preferred over stimulant agents, especially for long-term use. Clinicians have traditionally hesitated to prescribe stimulant laxatives for long-term use, as they were thought to damage the enteric nervous system.34 Although more recent studies have not shown this potential effect,35 more research is warranted on the use of stimulant laxatives for longer than 4 weeks.
STOOL SOFTENERS: LITTLE EVIDENCE
Stool softeners enhance the interaction of stool and water, leading to softer stool and easier evacuation. Docusate sodium and docusate calcium are thought to facilitate the mixing of aqueous and fatty substances, thereby softening the stool.
However, there is little evidence to support the use of docusate for constipation in hospitalized adults or in ambulatory care. A recent review reported that docusate was no better than placebo in diminishing symptoms of constipation.36
INTESTINAL SECRETAGOGUES
The secretagogues include lubiprostone, linaclotide, and plecanatide. These medications are preferred therapy for patients with normal- or slow-transit constipation once conservative therapies have failed. Even though there is no current consensus, lifestyle measures and conservative treatment options should be tried for about 8 weeks.
Lubiprostone and linaclotide are approved by the US Food and Drug Administration (FDA) for both constipation and constipation-predominant irritable bowel syndrome. They activate chloride channels on the apical surface of enterocytes, increasing intestinal secretion of chloride, which in turn increases luminal sodium efflux to maintain electroneutrality, leading to secretion of water into the intestinal lumen. This eventually facilitates intestinal transit and increases the passage of stool.
Lubiprostone
Lubiprostone, a prostaglandin E1 derivative, is approved for treating chronic constipation, constipation-predominant irritable bowel syndrome in women, and opioid-induced constipation in patients with chronic noncancer pain.
Adverse effects in clinical trials were nausea (up to 30%) and headache.37,38
Linaclotide
Linaclotide, a minimally absorbed 14-amino acid peptide, increases intestinal secretion of chloride and bicarbonate, increasing intestinal fluid and promoting intestinal transit.39 It also decreases the firing rate of the visceral afferent pain fibers and helps reduce visceral pain, especially in patients with constipation-predominant irritable bowel syndrome.40 It is approved for chronic constipation and constipation-predominant irritable bowel syndrome.41–43
Dosage starts at 145 μg/day for chronic constipation, and can be titrated up to 290 μg if there is no response or if a diagnosis of constipation-predominant irritable bowel syndrome is under consideration. Linaclotide should be taken 30 to 60 minutes before breakfast to reduce the likelihood of diarrhea.44
Adverse effects. Diarrhea led to treatment discontinuation in 4.5% of patients in one study.42
Plecanatide
Plecanatide is a guanylate cyclase-c agonist with a mode of action similar to that of linaclotide. It was recently approved by the FDA for chronic idiopathic constipation in adults. The recommended dose is 3 mg once daily.
Data from phase 2 trials in chronic constipation showed improvement in straining, abdominal discomfort, and stool frequency after 14 days of treatment.45
A phase 3 trial showed that plecanatide was more effective than placebo when used for 12 weeks in 951 patients with chronic constipation (P = .009).46 The most common adverse effect reported was diarrhea.
SEROTONIN RECEPTOR AGONISTS
Activation of serotonin 5-HT4 receptors in the gut leads to release of acetylcholine, which in turn induces mucosal secretion by activating submucosal neurons and increasing gut motility.47
Two 5-HT4 receptor agonists were withdrawn from the market (cisapride in 2000 and tegaserod in 2007) due to serious cardiovascular adverse events (fatal arrhythmias, heart attacks, and strokes) resulting from their affinity for hERG-K+ cardiac channels.
The newer agents prucalopride,48 velusetrag, and naronapride are highly selective 5-HT4 agonists with low affinity for hERG-K+ receptors and do not have proarrhythmic properties, based on extensive assessment in clinical trials.
Prucalopride
Prucalopride has been shown to accelerate gastrointestinal and colonic transit in patients with chronic constipation, with improvement in bowel movements, symptoms of chronic constipation, and quality of life.49–52
Adverse effects reported with its use have been headache, nausea, abdominal pain, and cramps.
Prucalopride is approved in Europe and Canada for chronic constipation in women but is not yet approved in the United States.
Dosage is 2 mg orally once daily. Caution is advised in elderly patients, in whom the preferred maximum dose is 1 mg daily, as there are only limited data available on the safety of this medication in the elderly.
Velusetrag
Velusetrag has been shown to increase colonic motility and improve symptoms of chronic constipation. In a phase 2 trial,53 the most effective dose was 15 mg once daily. Higher doses were associated with a higher incidence of adverse effects such as diarrhea, headache, nausea, and vomiting.
Naronapride
Naronapride (ATI-7505) is in phase 2 trials for chronic constipation. Reported adverse effects were headache, diarrhea, nausea, and vomiting.54
BILE SALT ABSORPTION INHIBITORS
Bile acids exert prosecretory and prokinetic effects by increasing colonic secretion of water and electrolytes through the activation of adenylate cyclase. This happens as a result of their deconjugation after passage into the colon.
Elobixibat is an ileal bile acid transporter inhibitor that prevents absorption of nonconjugated bile salts in the distal ileum. It has few side effects because its systemic absorption is minimal. Phase 3 trials are under way. Dosage is 5 to 20 mg daily. Adverse effects are few because systemic absorption is minimal, but include abdominal pain and diarrhea.55,56
MANAGING OPIOID-INDUCED CONSTIPATION
Opioids cause constipation by binding to mu receptors in the enteric nervous system. Activation of these receptors decreases bowel tone and contractility, which increases transit time. Stimulation of these receptors also increases anal sphincter tone, resulting in decreased rectal evacuation.57
Though underrecognized, opioid-induced constipation affects 40% of patients who take these drugs for nonmalignant pain and 90% of those taking them for cancer pain. Patients with this condition were found to take more time off work and feel more impaired in their domestic and work-related obligations than patients who did not develop constipation with use of opioids.58
Initial management of opioid-induced constipation includes increasing intake of fluids and dietary fiber (fiber alone can worsen abdominal pain in this condition by increasing stool bulk without a concomitant improvement in peristalsis) and increasing physical activity. It is common clinical practice to use a stool softener along with a stimulant laxative if lifestyle modifications are inadequate.59 If these measures are ineffective, osmotic agents can be added.
If these conventional measures fail, a peripherally acting mu-opioid receptor antagonist such as methylnaltrexone or naloxegol should be considered.
Methylnaltrexone
Methylnaltrexone60,61 is a peripherally acting mu receptor antagonist with a rapid onset of action. It does not cross the blood-brain barrier, as it contains a methyl group. It was approved by the FDA in 2008 to treat opioid-induced constipation in adults with advanced illnesses when other approaches are ineffective.
Adverse effects. Although the mu receptor antagonist alvimopan had been shown to be associated with cardiovascular events hypothesized to be a consequence of opioid withdrawal, methylnaltrexone has been deemed to have a safe cardiovascular profile without any potential effects on platelets, corrected QT interval, metabolism, heart rate, or blood pressure.61 Side effects include abdominal pain, nausea, diarrhea, hot flashes, tremor, and chills.
Contraindications. Methylnaltrexone is contraindicated in patients with structural diseases of the gastrointestinal tract, ie, peptic ulcer disease, inflammatory bowel disease, diverticulitis, stomach or intestinal cancer) since it can increase the risk of perforation.
Dosing is 1 dose subcutaneously every other day, as needed, and no more than 1 dose in a 24-hour period. Dosage is based on weight: 0.15 mg/kg/dose for patients weighing less than 38 kg or more than 114 kg; 8 mg for those weighing 38 to 62 kg; and 12 mg for those weighing 62 to 114 kg.62
Naloxegol
Naloxegol, FDA-approved for treating opioid-induced constipation in 2014, consists of naloxone conjugated with polyethylene glycol, which prevents it from crossing the blood-brain barrier and diminishing the central effects of opioid-induced analgesia. Unlike methylnaltrexone, which is given by subcutaneous injection, naloxegol is taken orally.
Adverse effects reported in clinical trials63,64 were abdominal pain, diarrhea, nausea, headache, and flatulence. No clinically relevant association with QT and corrected QT interval prolongation or cardiac repolarization was noted.64
Dosing is 25 mg by mouth once daily, which can be decreased to 12.5 mg if the initial dose is difficult to tolerate. It should be taken on an empty stomach at least 1 hour before the first meal of the day or 2 hours after the meal. In patients with renal impairment (creatinine clearance < 60 mL/min), the dose is 12.5 mg once daily.65
CONSTIPATION-PREDOMINANT IRRITABLE BOWEL SYNDROME
Irritable bowel syndrome is the reason for 3.1 million office visits and 59 million prescriptions in the United States every year, with patients equally distributed between diarrhea-predominant, constipation-predominant, and mixed subtypes.66
To be diagnosed with constipation-predominant irritable bowel syndrome, patients must meet the Rome IV criteria, more than 25% of bowel movements should have Bristol stool form types 1 or 2, and less than 25% of bowel movements should have Bristol stool form types 6 or 7. In practice, patients reporting that their bowel movements are usually constipated often suffices to make the diagnosis.5
Osmotic laxatives are often tried first, but despite improving stool frequency and consistency, they have little efficacy in satisfying complaints of bloating or abdominal pain in patients with constipation-predominant irritable syndrome.67 Stimulant laxatives have not yet been tested in clinical trials. Lubiprostone and linaclotide are FDA-approved for this condition; in women, lubiprostone is approved only for those over age 18.
Antidepressant therapy
Patients often derive additional benefit from treatment with antidepressants. A meta-analysis demonstrated a number needed to treat of 4 for selective serotonin reuptake inhibitors and tricyclic antidepressants in managing abdominal pain associated with irritable bowel syndrome.68 The major limiting factor is usually adverse effects of these drugs.
For constipation-predominant irritable bowel syndrome, selective serotonin reuptake inhibitors are preferred over tricyclics because of their additional prokinetic properties. Starting at a low dose and titrating upward slowly avoids potential adverse effects.
Cognitive behavioral therapy has also been beneficial in treating irritable bowel syndrome.69
Adjunctive therapies
Adjunctive therapies including peppermint oil, probiotics (eg, Lactobacillus, Bifidobacterium), and acupuncture have also shown promise in managing irritable bowel syndrome, but more data are needed on the use of these therapies for constipation-predominant irritable bowel syndrome before any definite conclusions can be drawn.70 Other emerging pharmacologic therapies are plecanatide (discussed earlier) and tenapanor.
Peppermint oil is an antispasmodic that inhibits calcium channels, leading to relaxation of smooth muscles in the gastrointestinal tract. Different dosages and treatment durations have been studied—450 to 900 mg daily in 2 to 3 divided doses over 1 to 3 months.71,72 The most common adverse effect reported was gastroesophageal reflux, related in part to the oil’s relaxing effect on the lower esophageal sphincter. Observation of this led to the development of enteric-coated preparations that have the potential to bypass the upper gastrointestinal tract.73
Tenapanor inhibits the sodium-hydrogen exchanger 3 channel (a regulator of sodium and water uptake in intestinal lumen), which in turn leads to a higher sodium level in the entire gastrointestinal tract (whereas linaclotide’s action is limited to the duodenum and jejunem), resulting in more fluid volume and increased luminal transit.74 It was found effective in a phase 2 clinical trial,75 and the most effective dose was 50 mg twice daily.
Since tenapanor is minimally absorbed, it has few side effects, the major ones being diarrhea (11.2% vs 0% with placebo) and urinary tract infection (5.6% vs 4.4% with placebo).75 Further study is needed to confirm these findings.
Tenapanor also has the advantage of inhibiting luminal phosphorus absorption. This has led to exploration of its use as a phosphate binder in patients with end-stage renal disease.
DYSSYNERGIC DEFECATION AND ANORECTAL BIOFEEDBACK
According to the Rome IV criteria,5 dyssynergic defecation is present if the criteria for chronic constipation are met, if a dyssynergic pattern of defecation is confirmed by manometry, imaging, or electromyography, and if 1 or more of the following are present: inability to expel an artificial stool (a 50-mL water-filled balloon) within 1 minute, prolonged colonic transit time, inability to evacuate, or 50% or more retention of barium during defecography.5
Even though biofeedback has been controversial as a treatment for dyssynergic defecation because of conflicting results in older studies,76 3 trials have shown it to be better than placebo, laxatives, and muscle relaxants, with symptomatic improvement in 70% of patients.77–79
Biofeedback therapy involves an instrument-based auditory or visual tool (using electromyographic sensors or anorectal manometry) to help patients coordinate abdominal, rectal, puborectalis, and anal sphincter muscles and produce a propulsive force using their abdominal muscles to achieve complete evacuation. Important components of this therapy include:
Proper evacuation positioning (brace-pump technique, which involves sitting on the toilet leaning forward with forearms resting on thighs, shoulders relaxed, and feet placed on a small footstool
Breathing relaxation and training exercises during defecation (no straining, keeping a normal pattern of breathing, and avoiding holding the breath while defecating)
Use of the abdominal muscles by pushing the abdomen forward, along with relaxation of the anal sphincter.80
The anorectal feedback program usually consists of 6 weekly sessions of 45 to 60 minutes each. Limitations of this therapy include unavailability, lack of trained therapists, lack of insurance coverage, and inapplicability to certain patient groups, such as those with dementia or learning disabilities.
SURGERY FOR CHRONIC CONSTIPATION
Surgery for constipation is reserved for patients who continue to have symptoms despite optimal medical therapy.
Total abdominal colectomy and ileorectal anastomosis
Total abdominal colectomy with ileorectal anastomosis is a surgical option for medically intractable slow-transit constipation. Before considering surgery, complete diagnostic testing should be done, including colonic manometry and documentation of whether the patient also has outlet dysfunction.
Even though it has shown excellent outcomes and satisfaction rates as high as 100% in patients with pure slow-transit constipation,81–83 results in older studies in patients with mixed disorders (eg, slow-transit constipation with features of outlet dysfunction) were less predictable.84 More recent studies have reported comparable long-term morbidity and postoperative satisfaction rates in those with pure slow-transit constipation and those with a mixed disorder, indicating that careful patient selection is likely the key to a favorable outcome.85
Partial colectomies based on segmental colon transit time measurements can also be considered in some patients.86
Stapled transanal resection
Stapled transanal resection involves circumferential transanal stapling of the redundant rectal mucosa. It is an option for patients with defecatory disorders, specifically large rectoceles and rectal intussusception not amenable to therapy with pelvic floor retraining exercises.87
The efficacy of this procedure in controlling symptoms and improving quality of life is around 77% to 81% at 12 months, though complication rates as high as 46% and disappointing long-term outcomes have been a deterrent to its widespread acceptance in the United States.88–91
Chronic constipation has a variety of possible causes and mechanisms. Although traditional conservative treatments are still valid and first-line, if these fail, clinicians can choose from a growing list of new treatments, tailored to the cause in the individual patient.
This article discusses how defecation works (or doesn’t), the types of chronic constipation, the available diagnostic tools, and traditional and newer treatments, including some still in development.
THE EPIDEMIOLOGY OF CONSTIPATION
Chronic constipation is one of the most common gastrointestinal disorders, affecting about 15% of all adults and 30% of those over the age of 60.1 It can be a primary disorder or secondary to other factors.
Constipation is more prevalent in women and in institutionalized elderly people.2 It is associated with lower socioeconomic status, depression, less self-reported physical activity, certain medications, and stressful life events.3 Given its high prevalence and its impact on quality of life, it is also associated with significant utilization of healthcare resources.4
Constipation defined by Rome IV criteria
Physicians and patients may disagree about what constitutes constipation. Physicians primarily regard it as infrequent bowel movements, while patients tend to have a broader definition. According to the Rome IV criteria,5 chronic constipation is defined by the presence of the following for at least 3 months (with symptom onset at least 6 months prior to diagnosis):
(1) Two or more of the following for more than 25% of defecations:
- Straining
- Lumpy or hard stools
- Sensation of incomplete evacuation
- Sensation of anorectal obstruction or blockage
- Manual maneuvers to facilitate evacuation
- Fewer than 3 spontaneous bowel movements per week.
(2) Loose stools are rarely present without the use of laxatives.
(3) The patient does not meet the criteria for diagnosis of irritable bowel syndrome.
DEFECATION IS COMPLEX
Defecation begins when the rectum fills with stool, causing relaxation of the internal anal sphincter and the urge to defecate. The external anal sphincter, which is under voluntary control, can then either contract to delay defecation or relax to allow the stool to be expelled.6
Colonic muscles propel stool toward the rectum in repetitive localized contractions that help mix and promote absorption of the content, and larger coordinated (high-amplitude propagating) contractions that, in healthy individuals, move the stool forward from the proximal to the distal colon multiple times daily. These contractions usually occur in the morning and are accentuated by gastric distention from food and the resulting gastrocolic reflex.
Serotonin (5-HT) is released by enterochromaffin cells in response to distention of the gut wall. It mediates peristaltic movements of the gastrointestinal tract by binding to receptors (especially 5-HT4), stimulating release of neurotransmitters such as acetylcholine, causing smooth-muscle contraction behind the luminal contents and propelling them forward.
PRIMARY CONSTIPATION DISORDERS
The American Gastroenterological Association7 classifies constipation into 3 groups on the basis of colonic transit time and anorectal function:
Normal-transit constipation
Stool normally takes 20 to 72 hours to pass through the colon, with transit time affected by diet, drugs, level of physical activity, and emotional status.8
Normal-transit constipation is the most common type of constipation. The term is sometimes used interchangeably with constipation-predominant irritable bowel syndrome, but the latter is a distinct entity characterized by abdominal pain relieved by defecation as the primary symptom, as well as having occasional loose stools. These 2 conditions can be hard to tell apart, especially if the patient cannot describe the symptoms precisely.
Slow-transit constipation
Slow-transit constipation—also called delayed-transit constipation, colonoparesis, colonic inertia, and pseudo-obstruction—is defined as prolonged stool transit in the colon, ie, for more than 5 days.9 It can be the result of colonic smooth muscle dysfunction, compromised colonic neural pathways, or both, leading to slow colon peristalsis.
Factors that can affect colonic motility such as opioid use and hypothyroidism should be carefully considered in these patients. Opioids are notorious for causing constipation by decreasing bowel tone and contractility and thereby increasing colonic transit time. They also tighten up the anal sphincters, resulting in decreased rectal evacuation.10
Outlet dysfunction
Outlet dysfunction, also called pelvic floor dysfunction or defecatory disorder, is associated with incomplete rectal evacuation. It can be a consequence of weak rectal expulsion forces (slow colonic transit, rectal hyposensitivity), functional resistance to rectal evacuation (high anal resting pressure, anismus, incomplete relaxation of the anal sphincter, dyssynergic defecation), or structural outlet obstruction (excessive perineal descent, rectoceles, rectal intussusception). About 50% of patients with outlet dysfunction have concurrent slow-transit constipation.
Dyssynergic defecation is the most common outlet dysfunction disorder, accounting for about half of the cases referred to tertiary centers. It is defined as a paradoxical elevation in anal sphincter tone or less than 20% relaxation of the resting anal sphincter pressure with weak abdominal and pelvic propulsive forces.11 Anorectal biofeedback is a therapeutic option for dyssynergic defecation, as we discuss later in this article.
SECONDARY CONSTIPATION
Constipation can be secondary to several conditions and factors (Table 1), including:
- Neurologic disorders that affect gastrointestinal motility (eg, Hirschsprung disease, Parkinson disease, multiple sclerosis, spinal cord injury, stroke, spinal or ganglionic tumor, hypothyroidism, amyloidosis, diabetes mellitus, hypercalcemia)
- Drugs used to treat neurologic disorders
- Mechanical obstruction
- Diet (eg, low fiber, decreased fluid intake).
EVALUATION OF CONSTIPATION
It is crucial for physicians to efficiently use the available diagnostic tools for constipation to tailor the treatment to the patient.
Evaluation of chronic constipation begins with a thorough history and physical examination to rule out secondary constipation (Figure 1). Red flags such as unintentional weight loss, blood in the stool, rectal pain, fever, and iron-deficiency anemia should prompt referral for colonoscopy to evaluate for malignancy, colitis, or other potential colonic abnormalities.12
A detailed perineal and rectal examination can help diagnose defecatory disorders and should include evaluation of the resting anal tone and the sphincter during simulated evacuation.
Laboratory tests of thyroid function, electrolytes, and a complete blood cell count should be ordered if clinically indicated.13
Further tests
Further diagnostic tests can be considered if symptoms persist despite conservative treatment or if a defecatory disorder is suspected. These include anorectal manometry, colonic transit studies, defecography, and colonic manometry.
Anorectal manometry and the rectal balloon expulsion test are usually done first because of their high sensitivity (88%) and specificity (89%) for defecatory disorders.14 These tests measure the function of the internal and external anal sphincters at rest and with straining and assess rectal sensitivity and compliance. Anorectal manometry is also used in biofeedback therapy in patients with dyssynergic defecation.15
Colonic transit time can be measured if anorectal manometry and the balloon expulsion test are normal. The study uses radiopaque markers, radioisotopes, or wireless motility capsules to confirm slow-transit constipation and to identify areas of delayed transit in the colon.16
Defecography is usually the next step in diagnosis if anorectal manometry and balloon expulsion tests are inconclusive or if an anatomic abnormality of the pelvic floor is suspected. It can be done with a variety of techniques. Barium defecography can identify anatomic defects, scintigraphy can quantify evacuation of artificial stools, and magnetic resonance defecography visualizes anatomic landmarks to assess pelvic floor motion without exposing the patient to radiation.17,18
Colonic manometry is most useful in patients with refractory slow-transit constipation and can identify patients with isolated colonic motor dysfunction with no pelvic floor dysfunction who may benefit from subtotal colectomy and end-ileostomy.7
TRADITIONAL TREATMENTS STILL THE MAINSTAY
Nonpharmacologic treatments are the first-line options for patients with normal-transit and slow-transit constipation and should precede diagnostic testing. Lifestyle modifications and dietary changes (Table 2) aim to augment the known factors that stimulate the gastrocolic reflex and increase intestinal motility by high-amplitude propagated contractions.
Increasing physical activity increases intestinal gas clearance, decreases bloating, and lessens constipation.19,20
Toilet training is an integral part of lifestyle modifications.21
Diet. Drinking hot caffeinated beverages, eating breakfast within an hour of waking up, and consuming fiber in the morning (25–30 g of fiber daily) have traditionally been recommended as the first-line measures for chronic constipation. Dehydrated patients with constipation also benefit from increasing their fluid intake.22
LAXATIVES
Fiber (bulk-forming laxatives) for normal-transit constipation
Fiber remains a key part of the initial management of chronic constipation, as it is cheap, available, and safe. Increasing fiber intake is effective for normal-transit constipation, but patients with slow-transit constipation or refractory outlet dysfunction are less likely to benefit.23 Other laxatives are incorporated into the regimen if first-line nonpharmacologic interventions fail (Table 3).
Bulk-forming laxatives include insoluble fiber (wheat bran) and soluble fiber (psyllium, methylcellulose, inulin, calcium polycarbophil). Insoluble fiber, though often used, has little impact on symptoms of chronic constipation after 1 month of use, and up to 60% of patients report adverse effects from it.24 On the other hand, clinical trials have shown that soluble fiber such as psyllium facilitates defecation and improves functional bowel symptoms in patients with normal-transit constipation.25
Patients should be instructed to increase their dietary fiber intake gradually to avoid adverse effects and should be told to expect significant symptomatic improvement only after a few weeks. They should also be informed that increasing dietary fiber intake can cause bloating but that the bloating is temporary. If it continues, a different fiber can be tried.
Osmotic laxatives
Osmotic laxatives are often employed as a first- line laxative treatment option for patients with constipation. They draw water into the lumen by osmosis, helping to soften stool and speed intestinal transit. They include macrogols (inert polymers of ethylene glycol), nonabsorbable carbohydrates (lactulose, sorbitol), magnesium products, and sodium phosphate products.
Polyethylene glycol, the most studied osmotic laxative, has been shown to maintain therapeutic efficacy for up to 2 years, though it is not generally used this long.26 A meta-analysis of 10 randomized clinical trials found it to be superior to lactulose in improving stool consistency and frequency, and rates of adverse effects were similar to those with placebo.27
Lactulose and sorbitol are semisynthetic disaccharides that are not absorbed from the gastrointestinal tract. Apart from the osmotic effect of the disaccharide, these sugars are metabolized by colonic bacteria to acetic acid and other short-chain fatty acids, resulting in acidification of the stool, which exerts an osmotic effect in the colonic lumen.
Lactulose and sorbitol were shown to have similar efficacy in increasing the frequency of bowel movements in a small study, though patients taking lactulose had a higher rate of nausea.28
The usual recommended dose is 15 to 30 mL once or twice daily.
Adverse effects include gas, bloating, and abdominal distention (due to fermentation by colonic bacteria) and can limit long-term use.
Magnesium citrate and magnesium hydroxide are strong osmotic laxatives, but so far no clinical trial has been done to assess their efficacy in constipation. Although the risk of hypermagnesemia is low with magnesium-based products, this group of laxatives is generally avoided in patients with renal or cardiac disease.29
Sodium phosphate enemas (Fleet enemas) are used for bowel cleansing before certain procedures but have only limited use in constipation because of potential adverse effects such as hyperphosphatemia, hypocalcemia, and the rarer but more serious complication of acute phosphate nephropathy.30
Stimulant laxatives for short-term use only
Stimulant laxatives include glycerin, bisacodyl, senna, and sodium picosulfate. Sodium piosulfate and bisacodyl have been validated for treatment of chronic constipation for up to 4 weeks.31–33
Stimulant laxative suppositories should be used 30 minutes after meals to augment the physiologic gastrocolic reflex.
As more evidence is available for osmotic laxatives such as polyethylene glycol, they tend to be preferred over stimulant agents, especially for long-term use. Clinicians have traditionally hesitated to prescribe stimulant laxatives for long-term use, as they were thought to damage the enteric nervous system.34 Although more recent studies have not shown this potential effect,35 more research is warranted on the use of stimulant laxatives for longer than 4 weeks.
STOOL SOFTENERS: LITTLE EVIDENCE
Stool softeners enhance the interaction of stool and water, leading to softer stool and easier evacuation. Docusate sodium and docusate calcium are thought to facilitate the mixing of aqueous and fatty substances, thereby softening the stool.
However, there is little evidence to support the use of docusate for constipation in hospitalized adults or in ambulatory care. A recent review reported that docusate was no better than placebo in diminishing symptoms of constipation.36
INTESTINAL SECRETAGOGUES
The secretagogues include lubiprostone, linaclotide, and plecanatide. These medications are preferred therapy for patients with normal- or slow-transit constipation once conservative therapies have failed. Even though there is no current consensus, lifestyle measures and conservative treatment options should be tried for about 8 weeks.
Lubiprostone and linaclotide are approved by the US Food and Drug Administration (FDA) for both constipation and constipation-predominant irritable bowel syndrome. They activate chloride channels on the apical surface of enterocytes, increasing intestinal secretion of chloride, which in turn increases luminal sodium efflux to maintain electroneutrality, leading to secretion of water into the intestinal lumen. This eventually facilitates intestinal transit and increases the passage of stool.
Lubiprostone
Lubiprostone, a prostaglandin E1 derivative, is approved for treating chronic constipation, constipation-predominant irritable bowel syndrome in women, and opioid-induced constipation in patients with chronic noncancer pain.
Adverse effects in clinical trials were nausea (up to 30%) and headache.37,38
Linaclotide
Linaclotide, a minimally absorbed 14-amino acid peptide, increases intestinal secretion of chloride and bicarbonate, increasing intestinal fluid and promoting intestinal transit.39 It also decreases the firing rate of the visceral afferent pain fibers and helps reduce visceral pain, especially in patients with constipation-predominant irritable bowel syndrome.40 It is approved for chronic constipation and constipation-predominant irritable bowel syndrome.41–43
Dosage starts at 145 μg/day for chronic constipation, and can be titrated up to 290 μg if there is no response or if a diagnosis of constipation-predominant irritable bowel syndrome is under consideration. Linaclotide should be taken 30 to 60 minutes before breakfast to reduce the likelihood of diarrhea.44
Adverse effects. Diarrhea led to treatment discontinuation in 4.5% of patients in one study.42
Plecanatide
Plecanatide is a guanylate cyclase-c agonist with a mode of action similar to that of linaclotide. It was recently approved by the FDA for chronic idiopathic constipation in adults. The recommended dose is 3 mg once daily.
Data from phase 2 trials in chronic constipation showed improvement in straining, abdominal discomfort, and stool frequency after 14 days of treatment.45
A phase 3 trial showed that plecanatide was more effective than placebo when used for 12 weeks in 951 patients with chronic constipation (P = .009).46 The most common adverse effect reported was diarrhea.
SEROTONIN RECEPTOR AGONISTS
Activation of serotonin 5-HT4 receptors in the gut leads to release of acetylcholine, which in turn induces mucosal secretion by activating submucosal neurons and increasing gut motility.47
Two 5-HT4 receptor agonists were withdrawn from the market (cisapride in 2000 and tegaserod in 2007) due to serious cardiovascular adverse events (fatal arrhythmias, heart attacks, and strokes) resulting from their affinity for hERG-K+ cardiac channels.
The newer agents prucalopride,48 velusetrag, and naronapride are highly selective 5-HT4 agonists with low affinity for hERG-K+ receptors and do not have proarrhythmic properties, based on extensive assessment in clinical trials.
Prucalopride
Prucalopride has been shown to accelerate gastrointestinal and colonic transit in patients with chronic constipation, with improvement in bowel movements, symptoms of chronic constipation, and quality of life.49–52
Adverse effects reported with its use have been headache, nausea, abdominal pain, and cramps.
Prucalopride is approved in Europe and Canada for chronic constipation in women but is not yet approved in the United States.
Dosage is 2 mg orally once daily. Caution is advised in elderly patients, in whom the preferred maximum dose is 1 mg daily, as there are only limited data available on the safety of this medication in the elderly.
Velusetrag
Velusetrag has been shown to increase colonic motility and improve symptoms of chronic constipation. In a phase 2 trial,53 the most effective dose was 15 mg once daily. Higher doses were associated with a higher incidence of adverse effects such as diarrhea, headache, nausea, and vomiting.
Naronapride
Naronapride (ATI-7505) is in phase 2 trials for chronic constipation. Reported adverse effects were headache, diarrhea, nausea, and vomiting.54
BILE SALT ABSORPTION INHIBITORS
Bile acids exert prosecretory and prokinetic effects by increasing colonic secretion of water and electrolytes through the activation of adenylate cyclase. This happens as a result of their deconjugation after passage into the colon.
Elobixibat is an ileal bile acid transporter inhibitor that prevents absorption of nonconjugated bile salts in the distal ileum. It has few side effects because its systemic absorption is minimal. Phase 3 trials are under way. Dosage is 5 to 20 mg daily. Adverse effects are few because systemic absorption is minimal, but include abdominal pain and diarrhea.55,56
MANAGING OPIOID-INDUCED CONSTIPATION
Opioids cause constipation by binding to mu receptors in the enteric nervous system. Activation of these receptors decreases bowel tone and contractility, which increases transit time. Stimulation of these receptors also increases anal sphincter tone, resulting in decreased rectal evacuation.57
Though underrecognized, opioid-induced constipation affects 40% of patients who take these drugs for nonmalignant pain and 90% of those taking them for cancer pain. Patients with this condition were found to take more time off work and feel more impaired in their domestic and work-related obligations than patients who did not develop constipation with use of opioids.58
Initial management of opioid-induced constipation includes increasing intake of fluids and dietary fiber (fiber alone can worsen abdominal pain in this condition by increasing stool bulk without a concomitant improvement in peristalsis) and increasing physical activity. It is common clinical practice to use a stool softener along with a stimulant laxative if lifestyle modifications are inadequate.59 If these measures are ineffective, osmotic agents can be added.
If these conventional measures fail, a peripherally acting mu-opioid receptor antagonist such as methylnaltrexone or naloxegol should be considered.
Methylnaltrexone
Methylnaltrexone60,61 is a peripherally acting mu receptor antagonist with a rapid onset of action. It does not cross the blood-brain barrier, as it contains a methyl group. It was approved by the FDA in 2008 to treat opioid-induced constipation in adults with advanced illnesses when other approaches are ineffective.
Adverse effects. Although the mu receptor antagonist alvimopan had been shown to be associated with cardiovascular events hypothesized to be a consequence of opioid withdrawal, methylnaltrexone has been deemed to have a safe cardiovascular profile without any potential effects on platelets, corrected QT interval, metabolism, heart rate, or blood pressure.61 Side effects include abdominal pain, nausea, diarrhea, hot flashes, tremor, and chills.
Contraindications. Methylnaltrexone is contraindicated in patients with structural diseases of the gastrointestinal tract, ie, peptic ulcer disease, inflammatory bowel disease, diverticulitis, stomach or intestinal cancer) since it can increase the risk of perforation.
Dosing is 1 dose subcutaneously every other day, as needed, and no more than 1 dose in a 24-hour period. Dosage is based on weight: 0.15 mg/kg/dose for patients weighing less than 38 kg or more than 114 kg; 8 mg for those weighing 38 to 62 kg; and 12 mg for those weighing 62 to 114 kg.62
Naloxegol
Naloxegol, FDA-approved for treating opioid-induced constipation in 2014, consists of naloxone conjugated with polyethylene glycol, which prevents it from crossing the blood-brain barrier and diminishing the central effects of opioid-induced analgesia. Unlike methylnaltrexone, which is given by subcutaneous injection, naloxegol is taken orally.
Adverse effects reported in clinical trials63,64 were abdominal pain, diarrhea, nausea, headache, and flatulence. No clinically relevant association with QT and corrected QT interval prolongation or cardiac repolarization was noted.64
Dosing is 25 mg by mouth once daily, which can be decreased to 12.5 mg if the initial dose is difficult to tolerate. It should be taken on an empty stomach at least 1 hour before the first meal of the day or 2 hours after the meal. In patients with renal impairment (creatinine clearance < 60 mL/min), the dose is 12.5 mg once daily.65
CONSTIPATION-PREDOMINANT IRRITABLE BOWEL SYNDROME
Irritable bowel syndrome is the reason for 3.1 million office visits and 59 million prescriptions in the United States every year, with patients equally distributed between diarrhea-predominant, constipation-predominant, and mixed subtypes.66
To be diagnosed with constipation-predominant irritable bowel syndrome, patients must meet the Rome IV criteria, more than 25% of bowel movements should have Bristol stool form types 1 or 2, and less than 25% of bowel movements should have Bristol stool form types 6 or 7. In practice, patients reporting that their bowel movements are usually constipated often suffices to make the diagnosis.5
Osmotic laxatives are often tried first, but despite improving stool frequency and consistency, they have little efficacy in satisfying complaints of bloating or abdominal pain in patients with constipation-predominant irritable syndrome.67 Stimulant laxatives have not yet been tested in clinical trials. Lubiprostone and linaclotide are FDA-approved for this condition; in women, lubiprostone is approved only for those over age 18.
Antidepressant therapy
Patients often derive additional benefit from treatment with antidepressants. A meta-analysis demonstrated a number needed to treat of 4 for selective serotonin reuptake inhibitors and tricyclic antidepressants in managing abdominal pain associated with irritable bowel syndrome.68 The major limiting factor is usually adverse effects of these drugs.
For constipation-predominant irritable bowel syndrome, selective serotonin reuptake inhibitors are preferred over tricyclics because of their additional prokinetic properties. Starting at a low dose and titrating upward slowly avoids potential adverse effects.
Cognitive behavioral therapy has also been beneficial in treating irritable bowel syndrome.69
Adjunctive therapies
Adjunctive therapies including peppermint oil, probiotics (eg, Lactobacillus, Bifidobacterium), and acupuncture have also shown promise in managing irritable bowel syndrome, but more data are needed on the use of these therapies for constipation-predominant irritable bowel syndrome before any definite conclusions can be drawn.70 Other emerging pharmacologic therapies are plecanatide (discussed earlier) and tenapanor.
Peppermint oil is an antispasmodic that inhibits calcium channels, leading to relaxation of smooth muscles in the gastrointestinal tract. Different dosages and treatment durations have been studied—450 to 900 mg daily in 2 to 3 divided doses over 1 to 3 months.71,72 The most common adverse effect reported was gastroesophageal reflux, related in part to the oil’s relaxing effect on the lower esophageal sphincter. Observation of this led to the development of enteric-coated preparations that have the potential to bypass the upper gastrointestinal tract.73
Tenapanor inhibits the sodium-hydrogen exchanger 3 channel (a regulator of sodium and water uptake in intestinal lumen), which in turn leads to a higher sodium level in the entire gastrointestinal tract (whereas linaclotide’s action is limited to the duodenum and jejunem), resulting in more fluid volume and increased luminal transit.74 It was found effective in a phase 2 clinical trial,75 and the most effective dose was 50 mg twice daily.
Since tenapanor is minimally absorbed, it has few side effects, the major ones being diarrhea (11.2% vs 0% with placebo) and urinary tract infection (5.6% vs 4.4% with placebo).75 Further study is needed to confirm these findings.
Tenapanor also has the advantage of inhibiting luminal phosphorus absorption. This has led to exploration of its use as a phosphate binder in patients with end-stage renal disease.
DYSSYNERGIC DEFECATION AND ANORECTAL BIOFEEDBACK
According to the Rome IV criteria,5 dyssynergic defecation is present if the criteria for chronic constipation are met, if a dyssynergic pattern of defecation is confirmed by manometry, imaging, or electromyography, and if 1 or more of the following are present: inability to expel an artificial stool (a 50-mL water-filled balloon) within 1 minute, prolonged colonic transit time, inability to evacuate, or 50% or more retention of barium during defecography.5
Even though biofeedback has been controversial as a treatment for dyssynergic defecation because of conflicting results in older studies,76 3 trials have shown it to be better than placebo, laxatives, and muscle relaxants, with symptomatic improvement in 70% of patients.77–79
Biofeedback therapy involves an instrument-based auditory or visual tool (using electromyographic sensors or anorectal manometry) to help patients coordinate abdominal, rectal, puborectalis, and anal sphincter muscles and produce a propulsive force using their abdominal muscles to achieve complete evacuation. Important components of this therapy include:
Proper evacuation positioning (brace-pump technique, which involves sitting on the toilet leaning forward with forearms resting on thighs, shoulders relaxed, and feet placed on a small footstool
Breathing relaxation and training exercises during defecation (no straining, keeping a normal pattern of breathing, and avoiding holding the breath while defecating)
Use of the abdominal muscles by pushing the abdomen forward, along with relaxation of the anal sphincter.80
The anorectal feedback program usually consists of 6 weekly sessions of 45 to 60 minutes each. Limitations of this therapy include unavailability, lack of trained therapists, lack of insurance coverage, and inapplicability to certain patient groups, such as those with dementia or learning disabilities.
SURGERY FOR CHRONIC CONSTIPATION
Surgery for constipation is reserved for patients who continue to have symptoms despite optimal medical therapy.
Total abdominal colectomy and ileorectal anastomosis
Total abdominal colectomy with ileorectal anastomosis is a surgical option for medically intractable slow-transit constipation. Before considering surgery, complete diagnostic testing should be done, including colonic manometry and documentation of whether the patient also has outlet dysfunction.
Even though it has shown excellent outcomes and satisfaction rates as high as 100% in patients with pure slow-transit constipation,81–83 results in older studies in patients with mixed disorders (eg, slow-transit constipation with features of outlet dysfunction) were less predictable.84 More recent studies have reported comparable long-term morbidity and postoperative satisfaction rates in those with pure slow-transit constipation and those with a mixed disorder, indicating that careful patient selection is likely the key to a favorable outcome.85
Partial colectomies based on segmental colon transit time measurements can also be considered in some patients.86
Stapled transanal resection
Stapled transanal resection involves circumferential transanal stapling of the redundant rectal mucosa. It is an option for patients with defecatory disorders, specifically large rectoceles and rectal intussusception not amenable to therapy with pelvic floor retraining exercises.87
The efficacy of this procedure in controlling symptoms and improving quality of life is around 77% to 81% at 12 months, though complication rates as high as 46% and disappointing long-term outcomes have been a deterrent to its widespread acceptance in the United States.88–91
- Mugie SM, Benninga MA, Di Lorenzo C. Epidemiology of constipation in children and adults: a systematic review. Best Pract Res Clin Gastroenterol 2011; 25:3–18.
- Kinnunen O. Study of constipation in a geriatric hospital, day hospital, old people's home and at home. Aging (Milano) 1991; 3:161–170.
- Everhart JE, Go VL, Johannes RS, Fitzsimmons SC, Roth HP, White LR. A longitudinal survey of self-reported bowel habits in the United States. Dig Dis Sci 1989; 34:1153–1162.
- Shah ND, Chitkara DK, Locke GR, Meek PD, Talley NJ. Ambulatory care for constipation in the United States, 1993-2004. Am J Gastroenterol 2008; 103:1746–1753.
- Mearin F, Lacy BE, Chang L, et al. Bowel disorders. Gastroenterology 2016; 150:1393–1407.
- Bharucha AE. Pelvic floor: anatomy and function. Neurogastroenterol Motil 2006; 18:507–519.
- Bharucha AE, Pemberton JH, Locke GR 3rd. American Gastroenterological Association technical review on constipation. Gastroenterology 2013; 144:218–238.
- Grundy D, Al-Chaer ED, Aziz Q, et al. Fundamentals of neurogastroenterology: basic science. Gastroenterology 2006; 130:1391–1411.
- Gallegos-Orozco JF, Foxx-Orenstein AE, Sterler SM, Stoa JM. Chronic constipation in the elderly. Am J Gastroenterol 2012; 107:18–26.
- Mancini I, Bruera E. Constipation in advanced cancer patients. Support Care Cancer 1998; 6:356–364.
- Bassotti G, Chistolini F, Sietchiping-Nzepa F, de Roberto G, Morelli A, Chiarioni G. Biofeedback for pelvic floor dysfunction in constipation. BMJ 2004; 328:393–396.
- American Gastroenterological Association, Bharucha AE, Dorn SD, Lembo A, Pressman A. American Gastroenterological Association medical position statement on constipation. Gastroenterology 2013; 144:211–217.
- Costilla VC, Foxx-Orenstein AE. Constipation in adults: diagnosis and management. Curr Treat Options Gastroenterol 2014; 12:310–321.
- Rao SS, Singh S. Clinical utility of colonic and anorectal manometry in chronic constipation. J Clin Gastroenterol 2010; 44:597–609.
- Minguez M, Herreros B, Sanchiz V, et al. Predictive value of the balloon expulsion test for excluding the diagnosis of pelvic floor dyssynergia in constipation. Gastroenterology 2004; 126:57–62.
- Diamant NE, Kamm MA, Wald A, Whitehead WE. AGA technical review on anorectal testing techniques. Gastroenterology 1999; 116:735–760.
- Pezim ME, Pemberton JH, Levin KE, Litchy WJ, Phillips SF. Parameters of anorectal and colonic motility in health and in severe constipation. Dis Colon Rectum 1993; 36:484–491.
- Bharucha AE, Fletcher JG, Seide B, Riederer SJ, Zinsmeister AR. Phenotypic variation in functional disorders of defecation. Gastroenterology 2005; 128:1199–1210.
- De Schryver AM, Samsom M, Smout AI. Effects of a meal and bisacodyl on colonic motility in healthy volunteers and patients with slow-transit constipation. Dig Dis Sci 2003; 48:1206–1212.
- Villoria A, Serra J, Azpiroz F, Malagelada JR. Physical activity and intestinal gas clearance in patients with bloating. Am J Gastroenterol 2006; 101:2552–2557.
- Sikirov D. Comparison of straining during defecation in three positions: results and implications for human health. Dig Dis Sci 2003; 48:1201–1205.
- Muller-Lissner SA, Kamm MA, Scarpignato C, Wald A. Myths and misconceptions about chronic constipation. Am J Gastroenterol 2005; 100:232–242.
- Voderholzer WA, Schatke W, Muhldorfer BE, Klauser AG, Birkner B, Muller-Lissner SA. Clinical response to dietary fiber treatment of chronic constipation. Am J Gastroenterol 1997; 92:95–98.
- Bijkerk CJ, de Wit NJ, Muris JW, Whorwell PJ, Knottnerus JA, Hoes AW. Soluble or insoluble fibre in irritable bowel syndrome in primary care? Randomised placebo controlled trial. BMJ 2009; 339:b3154.
- Suares NC, Ford AC. Systematic review: the effects of fibre in the management of chronic idiopathic constipation. Aliment Pharmacol Ther 2011; 33:895–901.
- Dipalma JA, Cleveland MV, McGowan J, Herrera JL. A randomized, multicenter, placebo-controlled trial of polyethylene glycol laxative for chronic treatment of chronic constipation. Am J Gastroenterol 2007; 102:1436–1441.
- Lee-Robichaud H, Thomas K, Morgan J, Nelson RL. Lactulose versus polyethylene glycol for chronic constipation. Cochrane Database Syst Rev 2010; 7:CD007570.
- Lederle FA, Busch DL, Mattox KM, West MJ, Aske DM. Cost-effective treatment of constipation in the elderly: a randomized double-blind comparison of sorbitol and lactulose. Am J Med 1990; 89:597–601.
- Nyberg C, Hendel J, Nielsen OH. The safety of osmotically acting cathartics in colonic cleansing. Nat Rev Gastroenterol Hepatol 2010; 7:557–564.
- Ainley EJ, Winwood PJ, Begley JP. Measurement of serum electrolytes and phosphate after sodium phosphate colonoscopy bowel preparation: an evaluation. Dig Dis Sci 2005; 50:1319–1323.
- Kienzle-Horn S, Vix JM, Schuijt C, Peil H, Jordan CC, Kamm MA. Efficacy and safety of bisacodyl in the acute treatment of constipation: a double-blind, randomized, placebo-controlled study. Aliment Pharmacol Ther 2006; 23:1479–1488.
- Kienzle-Horn S, Vix JM, Schuijt C, Peil H, Jordan CC, Kamm MA. Comparison of bisacodyl and sodium picosulphate in the treatment of chronic constipation. Curr Med Res Opin 2007; 23:691–699.
- Mueller-Lissner S, Kamm MA, Wald A, et al. Multicenter, 4-week, double-blind, randomized, placebo-controlled trial of sodium picosulfate in patients with chronic constipation. Am J Gastroenterol 2010; 105:897–903.
- Smith B. Pathologic changes in the colon produced by anthraquinone purgatives. Dis Colon Rectum 1973; 16:455–458.
- Kiernan JA, Heinicke EA. Sennosides do not kill myenteric neurons in the colon of the rat or mouse. Neuroscience 1989; 30:837–842.
- Ottawa (ON): Canadian Agency for Drugs and Technologies in Health. Dioctyl sulfosuccinate or docusate (calcium or sodium) for the prevention or management of constipation: a review of the clinical effectiveness. www.ncbi.nlm.nih.gov/pubmedhealth/PMH0071207/. Accessed April 6, 2017.
- Saad R, Chey WD. Lubiprostone for chronic idiopathic constipation and irritable bowel syndrome with constipation. Expert Rev Gastroenterol Hepatol 2008; 2:497–508.
- Johanson JF, Morton D, Geenen J, Ueno R. Multicenter, 4-week, double-blind, randomized, placebo-controlled trial of lubiprostone, a locally-acting type-2 chloride channel activator, in patients with chronic constipation. Am J Gastroenterol 2008; 103:170–177.
- Harris LA, Crowell MD. Linaclotide, a new direction in the treatment of irritable bowel syndrome and chronic constipation. Curr Opin Mol Ther 2007; 9:403–410.
- Johnston JM, Kurtz CB, Macdougall JE, et al. Linaclotide improves abdominal pain and bowel habits in a phase IIb study of patients with irritable bowel syndrome with constipation. Gastroenterology 2010; 139:1877–1886.e2.
- Lembo AJ, Schneier HA, Shiff SJ, et al. Two randomized trials of linaclotide for chronic constipation. N Engl J Med 2011; 365:527–536.
- Chey WD, Lembo AJ, Lavins BJ, et al. Linaclotide for irritable bowel syndrome with constipation: a 26-week, randomized, double-blind, placebo-controlled trial to evaluate efficacy and safety. Am J Gastroenterol 2012; 107:1702–1712.
- Rao S, Lembo AJ, Shiff SJ, et al. A 12-week, randomized, controlled trial with a 4-week randomized withdrawal period to evaluate the efficacy and safety of linaclotide in irritable bowel syndrome with constipation. Am J Gastroenterol 2012; 107:1714–1725.
- Chey WD, Kurlander J, Eswaran S. Irritable bowel syndrome: a clinical review. JAMA 2015; 313:949–958.
- Shailubhai K, Talluto C, Comiskey S, Foss JA, Joslyn A, Jacob G. Phase II clinical evaluation of SP-304, a guanylate cyclase-C agonist, for treatment of chronic constipation. Am J Gastroenterol 2010; 105:S487–S488.
- Miner P, Surowitz R, Fogel R, et al. Plecanatide, a novel guanylate cyclase-C (GC-C) receptor agonist, is efficacious and safe in patients with chronic idiopathic constipation (CIC): results from a 951 patient, 12-week, multi-center trial (abstract). Gastroenterology 2013; 144:S163.
- Coss-Adame E, Rao SS. Brain and gut interactions in irritable bowel syndrome: new paradigms and new understandings. Curr Gastroenterol Rep 2014; 16:379.
- Mendzelevski B, Ausma J, Chanter DO, et al. Assessment of the cardiac safety of prucalopride in healthy volunteers: a randomized, double-blind, placebo- and positive-controlled thorough QT study. Br J Clin Pharmacol 2012; 73:203–209.
- Camilleri M, Kerstens R, Rykx A, Vandeplassche L. A placebo-controlled trial of prucalopride for severe chronic constipation. N Engl J Med 2008; 358:2344–2354.
- Tack J, van Outryve M, Beyens G, Kerstens R, Vandeplassche L. Prucalopride (Resolor) in the treatment of severe chronic constipation in patients dissatisfied with laxatives. Gut 2009; 58:357–365.
- Quigley EM, Vandeplassche L, Kerstens R, Ausma J. Clinical trial: the efficacy, impact on quality of life, and safety and tolerability of prucalopride in severe chronic constipation—a 12-week, randomized, double-blind, placebo-controlled study. Aliment Pharmacol Ther 2009; 29:315–328.
- Ford AC, Suares NC. Effect of laxatives and pharmacological therapies in chronic idiopathic constipation: systematic review and meta-analysis. Gut 2011; 60:209–218.
- Goldberg M, Li YP, Johanson JF, et al. Clinical trial: the efficacy and tolerability of velusetrag, a selective 5-HT4 agonist with high intrinsic activity, in chronic idiopathic constipation—a 4-week, randomized, double-blind, placebo-controlled, dose-response study. Aliment Pharmacol Ther 2010; 32:1102–1112.
- Palme M, Milner PG, Ellis DJ, Marmon T, Canafax DM. A novel gastrointestinal prokinetic, ATI-7505, increased spontaneous bowel movements (sbms) in a phase II, randomized, placebo-controlled study of patients with chronic idiopathic constipation (CIC). Gastroenterology 2010; 138:S-128–S-129.
- Chey WD, Camilleri M, Chang L, Rikner L, Graffner H. A randomized placebo-controlled phase IIb trial of a3309, a bile acid transporter inhibitor, for chronic idiopathic constipation. Am J Gastroenterol 2011; 106:1803–1812.
- Wong BS, Camilleri M, McKinzie S, Burton D, Graffner H, Zinsmeister AR. Effects of A3309, an ileal bile acid transporter inhibitor, on colonic transit and symptoms in females with functional constipation. Am J Gastroenterol 2011; 106:2154–2164.
- Pappagallo M. Incidence, prevalence, and management of opioid bowel dysfunction. Am J Surg 2001; 182(suppl):11S–18S.
- Bell T, Annunziata K, Leslie JB. Opioid-induced constipation negatively impacts pain management, productivity, and health-related quality of life: findings from the National Health and Wellness Survey. J Opioid Manag 2009; 5:137–144.
- Sykes NP. A volunteer model for the comparison of laxatives in opioid-related constipation. J Pain Symptom Manage 1996; 11:363–369.
- ClinicalTrials.gov. A multicenter, randomized, double-blind, placebo-controlled, parallel-group study of oral MOA-728 for the treatment of opioid- induced bowel dysfunction in subjects with chronic nonmalignant pain. ClinicalTrials.gov Identifier: NCT00547586. https://clinicaltrials.gov/ct2/show/NCT00547586. Accessed March 22, 2017.
- ClinicalTrials.gov. An open-label study to evaluate the long-term safety of subcutaneous MOA-728 for treatment of opioid-induced constipation in subjects with nonmalignant pain. ClinicalTrials.gov Identifier: NCT00804141. https://clinicaltrials.gov/ct2/show/NCT00804141. Accessed April 6, 2017.
- Wyeth Pharmaceuticals. Relistor package insert. http://labeling.pfizer.com/showlabeling.aspx?id=499. Accessed March 22, 2017.
- Webster L, Dhar S, Eldon M, Masuoka L, Lappalainen J, Sostek M. A phase 2, double-blind, randomized, placebo-controlled, dose-escalation study to evaluate the efficacy, safety, and tolerability of naloxegol in patients with opioid-induced constipation. Pain 2013; 154:1542–1550.
- Chey WD, Webster L, Sostek M, Lappalainen J, Barker PN, Tack J. Naloxegol for opioid-induced constipation in patients with noncancer pain. N Engl J Med 2014; 370:2387–2396.
- Jones R, Prommer E, Backstedt D. Naloxegol: a novel therapy in the management of opioid-induced constipation. Am J Hosp Palliat Care 2016; 33:875–880.
- Guilera M, Balboa A, Mearin F. Bowel habit subtypes and temporal patterns in irritable bowel syndrome: systematic review. Am J Gastroenterol 2005; 100:1174–1184.
- Chapman RW, Stanghellini V, Geraint M, Halphen M. Randomized clinical trial: macrogol/PEG 3350 plus electrolytes for treatment of patients with constipation associated with irritable bowel syndrome. Am J Gastroenterol 2013; 108:1508–1515.
- Ford AC, Quigley EM, Lacy BE, et al. Effect of antidepressants and psychological therapies, including hypnotherapy, in irritable bowel syndrome: systematic review and meta-analysis. Am J Gastroenterol 2014; 109:1350–1366.
- Ballou S, Keefer L. Psychological interventions for irritable bowel syndrome and inflammatory bowel diseases. Clin Transl Gastroenterol 2017; 8:e214.
- Ford AC, Moayyedi P, Lacy BE, et al; Task Force on the Management of Functional Bowel Disorders. American College of Gastroenterology monograph on the management of irritable bowel syndrome and chronic idiopathic constipation. Am J Gastroenterol 2014; 109(suppl 1):S2–S27.
- Ford AC, Talley NJ, Spiegel BM, et al. Effect of fibre, antispasmodics, and peppermint oil in the treatment of irritable bowel syndrome: systematic review and meta-analysis. BMJ 2008; 337:a2313.
- Wall GC, Bryant GA, Bottenberg MM, Maki ED, Miesner AR. Irritable bowel syndrome: a concise review of current treatment concepts. World J Gastroenterol 2014; 20:8796–8806.
- Kligler B, Chaudhary S. Peppermint oil. Am Fam Physician 2007; 75:1027–1030.
- Spencer AG, Labonte ED, Rosenbaum DP, et al. Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci Transl Med 2014; 6:227ra36.
- Rosenbaum DP. A randomized, double-blind, placebo-controlled study to assess the safety and efficacy of AZD1722 for the treatment of constipation-predominant irritable bowel syndrome (IBS-C). 2014. https://clinicaltrials.gov/ct2/show/NCT01923428. Accessed April 6, 2017.
- Rao SS. Biofeedback therapy for dyssynergic (obstructive) defecation. J Clin Gastroenterol 2000; 30:115–116.
- Cadeddu F, Salis F, De Luca E, Ciangola I, Milito G. Efficacy of biofeedback plus transanal stimulation in the management of pelvic floor dyssynergia: a randomized trial. Tech Coloproctol 2015; 19:333–338.
- Chiarioni G, Whitehead WE, Pezza V, Morelli A, Bassotti G. Biofeedback is superior to laxatives for normal transit constipation due to pelvic floor dyssynergia. Gastroenterology 2006; 130:657–664.
- Chiarioni G, Heymen S, Whitehead WE. Biofeedback therapy for dyssynergic defecation. World J Gastroenterol 2006; 12:7069–7074.
- Rao SS. Biofeedback therapy for constipation in adults. Best Pract Res Clin Gastroenterol 2011; 25:159–166.
- Hassan I, Pemberton JH, Young-Fadok TM, et al. Ileorectal anastomosis for slow transit constipation: long-term functional and quality of life results. J Gastrointest Surg 2006; 10:1330–1337.
- You YT, Wang JY, Changchien CR, et al. Segmental colectomy in the management of colonic inertia. Am Surg 1998; 64:775–777.
- Nyam DC, Pemberton JH, Ilstrup DM, Rath DM. Long-term results of surgery for chronic constipation. Dis Colon Rectum 1997; 40:273–279.
- Pemberton JH, Rath DM, Ilstrup DM. Evaluation and surgical treatment of severe chronic constipation. Ann Surg 1991; 214:403–413.
- Reshef A, Alves-Ferreira P, Zutshi M, Hull T, Gurland B. Colectomy for slow transit constipation: effective for patients with coexistent obstructed defecation. Int J Colorectal Dis 2013; 28:841–847.
- Lundin E, Karlbom U, Pahlman L, Graf W. Outcome of segmental colonic resection for slow-transit constipation. Br J Surg 2002; 89:1270–1274.
- Schwandner O, Stuto A, Jayne D, et al. Decision-making algorithm for the STARR procedure in obstructed defecation syndrome: position statement of the group of STARR pioneers. Surg Innov 2008; 15:105–109.
- Titu LV, Riyad K, Carter H, Dixon AR. Stapled transanal rectal resection for obstructed defecation: a cautionary tale. Dis Colon Rectum 2009; 52:1716–1722.
- Goede AC, Glancy D, Carter H, Mills A, Mabey K, Dixon AR. Medium-term results of stapled transanal rectal resection (STARR) for obstructed defecation and symptomatic rectal-anal intussusception. Colorectal Dis 2011; 13:1052–1057.
- Jayne DG, Schwandner O, Stuto A. Stapled transanal rectal resection for obstructed defecation syndrome: one-year results of the european STARR registry. Dis Colon Rectum 2009; 52:1205–1214.
- Madbouly KM, Abbas KS, Hussein AM. Disappointing long-term outcomes after stapled transanal rectal resection for obstructed defecation. World J Surg 2010; 34:2191–2196.
- Mugie SM, Benninga MA, Di Lorenzo C. Epidemiology of constipation in children and adults: a systematic review. Best Pract Res Clin Gastroenterol 2011; 25:3–18.
- Kinnunen O. Study of constipation in a geriatric hospital, day hospital, old people's home and at home. Aging (Milano) 1991; 3:161–170.
- Everhart JE, Go VL, Johannes RS, Fitzsimmons SC, Roth HP, White LR. A longitudinal survey of self-reported bowel habits in the United States. Dig Dis Sci 1989; 34:1153–1162.
- Shah ND, Chitkara DK, Locke GR, Meek PD, Talley NJ. Ambulatory care for constipation in the United States, 1993-2004. Am J Gastroenterol 2008; 103:1746–1753.
- Mearin F, Lacy BE, Chang L, et al. Bowel disorders. Gastroenterology 2016; 150:1393–1407.
- Bharucha AE. Pelvic floor: anatomy and function. Neurogastroenterol Motil 2006; 18:507–519.
- Bharucha AE, Pemberton JH, Locke GR 3rd. American Gastroenterological Association technical review on constipation. Gastroenterology 2013; 144:218–238.
- Grundy D, Al-Chaer ED, Aziz Q, et al. Fundamentals of neurogastroenterology: basic science. Gastroenterology 2006; 130:1391–1411.
- Gallegos-Orozco JF, Foxx-Orenstein AE, Sterler SM, Stoa JM. Chronic constipation in the elderly. Am J Gastroenterol 2012; 107:18–26.
- Mancini I, Bruera E. Constipation in advanced cancer patients. Support Care Cancer 1998; 6:356–364.
- Bassotti G, Chistolini F, Sietchiping-Nzepa F, de Roberto G, Morelli A, Chiarioni G. Biofeedback for pelvic floor dysfunction in constipation. BMJ 2004; 328:393–396.
- American Gastroenterological Association, Bharucha AE, Dorn SD, Lembo A, Pressman A. American Gastroenterological Association medical position statement on constipation. Gastroenterology 2013; 144:211–217.
- Costilla VC, Foxx-Orenstein AE. Constipation in adults: diagnosis and management. Curr Treat Options Gastroenterol 2014; 12:310–321.
- Rao SS, Singh S. Clinical utility of colonic and anorectal manometry in chronic constipation. J Clin Gastroenterol 2010; 44:597–609.
- Minguez M, Herreros B, Sanchiz V, et al. Predictive value of the balloon expulsion test for excluding the diagnosis of pelvic floor dyssynergia in constipation. Gastroenterology 2004; 126:57–62.
- Diamant NE, Kamm MA, Wald A, Whitehead WE. AGA technical review on anorectal testing techniques. Gastroenterology 1999; 116:735–760.
- Pezim ME, Pemberton JH, Levin KE, Litchy WJ, Phillips SF. Parameters of anorectal and colonic motility in health and in severe constipation. Dis Colon Rectum 1993; 36:484–491.
- Bharucha AE, Fletcher JG, Seide B, Riederer SJ, Zinsmeister AR. Phenotypic variation in functional disorders of defecation. Gastroenterology 2005; 128:1199–1210.
- De Schryver AM, Samsom M, Smout AI. Effects of a meal and bisacodyl on colonic motility in healthy volunteers and patients with slow-transit constipation. Dig Dis Sci 2003; 48:1206–1212.
- Villoria A, Serra J, Azpiroz F, Malagelada JR. Physical activity and intestinal gas clearance in patients with bloating. Am J Gastroenterol 2006; 101:2552–2557.
- Sikirov D. Comparison of straining during defecation in three positions: results and implications for human health. Dig Dis Sci 2003; 48:1201–1205.
- Muller-Lissner SA, Kamm MA, Scarpignato C, Wald A. Myths and misconceptions about chronic constipation. Am J Gastroenterol 2005; 100:232–242.
- Voderholzer WA, Schatke W, Muhldorfer BE, Klauser AG, Birkner B, Muller-Lissner SA. Clinical response to dietary fiber treatment of chronic constipation. Am J Gastroenterol 1997; 92:95–98.
- Bijkerk CJ, de Wit NJ, Muris JW, Whorwell PJ, Knottnerus JA, Hoes AW. Soluble or insoluble fibre in irritable bowel syndrome in primary care? Randomised placebo controlled trial. BMJ 2009; 339:b3154.
- Suares NC, Ford AC. Systematic review: the effects of fibre in the management of chronic idiopathic constipation. Aliment Pharmacol Ther 2011; 33:895–901.
- Dipalma JA, Cleveland MV, McGowan J, Herrera JL. A randomized, multicenter, placebo-controlled trial of polyethylene glycol laxative for chronic treatment of chronic constipation. Am J Gastroenterol 2007; 102:1436–1441.
- Lee-Robichaud H, Thomas K, Morgan J, Nelson RL. Lactulose versus polyethylene glycol for chronic constipation. Cochrane Database Syst Rev 2010; 7:CD007570.
- Lederle FA, Busch DL, Mattox KM, West MJ, Aske DM. Cost-effective treatment of constipation in the elderly: a randomized double-blind comparison of sorbitol and lactulose. Am J Med 1990; 89:597–601.
- Nyberg C, Hendel J, Nielsen OH. The safety of osmotically acting cathartics in colonic cleansing. Nat Rev Gastroenterol Hepatol 2010; 7:557–564.
- Ainley EJ, Winwood PJ, Begley JP. Measurement of serum electrolytes and phosphate after sodium phosphate colonoscopy bowel preparation: an evaluation. Dig Dis Sci 2005; 50:1319–1323.
- Kienzle-Horn S, Vix JM, Schuijt C, Peil H, Jordan CC, Kamm MA. Efficacy and safety of bisacodyl in the acute treatment of constipation: a double-blind, randomized, placebo-controlled study. Aliment Pharmacol Ther 2006; 23:1479–1488.
- Kienzle-Horn S, Vix JM, Schuijt C, Peil H, Jordan CC, Kamm MA. Comparison of bisacodyl and sodium picosulphate in the treatment of chronic constipation. Curr Med Res Opin 2007; 23:691–699.
- Mueller-Lissner S, Kamm MA, Wald A, et al. Multicenter, 4-week, double-blind, randomized, placebo-controlled trial of sodium picosulfate in patients with chronic constipation. Am J Gastroenterol 2010; 105:897–903.
- Smith B. Pathologic changes in the colon produced by anthraquinone purgatives. Dis Colon Rectum 1973; 16:455–458.
- Kiernan JA, Heinicke EA. Sennosides do not kill myenteric neurons in the colon of the rat or mouse. Neuroscience 1989; 30:837–842.
- Ottawa (ON): Canadian Agency for Drugs and Technologies in Health. Dioctyl sulfosuccinate or docusate (calcium or sodium) for the prevention or management of constipation: a review of the clinical effectiveness. www.ncbi.nlm.nih.gov/pubmedhealth/PMH0071207/. Accessed April 6, 2017.
- Saad R, Chey WD. Lubiprostone for chronic idiopathic constipation and irritable bowel syndrome with constipation. Expert Rev Gastroenterol Hepatol 2008; 2:497–508.
- Johanson JF, Morton D, Geenen J, Ueno R. Multicenter, 4-week, double-blind, randomized, placebo-controlled trial of lubiprostone, a locally-acting type-2 chloride channel activator, in patients with chronic constipation. Am J Gastroenterol 2008; 103:170–177.
- Harris LA, Crowell MD. Linaclotide, a new direction in the treatment of irritable bowel syndrome and chronic constipation. Curr Opin Mol Ther 2007; 9:403–410.
- Johnston JM, Kurtz CB, Macdougall JE, et al. Linaclotide improves abdominal pain and bowel habits in a phase IIb study of patients with irritable bowel syndrome with constipation. Gastroenterology 2010; 139:1877–1886.e2.
- Lembo AJ, Schneier HA, Shiff SJ, et al. Two randomized trials of linaclotide for chronic constipation. N Engl J Med 2011; 365:527–536.
- Chey WD, Lembo AJ, Lavins BJ, et al. Linaclotide for irritable bowel syndrome with constipation: a 26-week, randomized, double-blind, placebo-controlled trial to evaluate efficacy and safety. Am J Gastroenterol 2012; 107:1702–1712.
- Rao S, Lembo AJ, Shiff SJ, et al. A 12-week, randomized, controlled trial with a 4-week randomized withdrawal period to evaluate the efficacy and safety of linaclotide in irritable bowel syndrome with constipation. Am J Gastroenterol 2012; 107:1714–1725.
- Chey WD, Kurlander J, Eswaran S. Irritable bowel syndrome: a clinical review. JAMA 2015; 313:949–958.
- Shailubhai K, Talluto C, Comiskey S, Foss JA, Joslyn A, Jacob G. Phase II clinical evaluation of SP-304, a guanylate cyclase-C agonist, for treatment of chronic constipation. Am J Gastroenterol 2010; 105:S487–S488.
- Miner P, Surowitz R, Fogel R, et al. Plecanatide, a novel guanylate cyclase-C (GC-C) receptor agonist, is efficacious and safe in patients with chronic idiopathic constipation (CIC): results from a 951 patient, 12-week, multi-center trial (abstract). Gastroenterology 2013; 144:S163.
- Coss-Adame E, Rao SS. Brain and gut interactions in irritable bowel syndrome: new paradigms and new understandings. Curr Gastroenterol Rep 2014; 16:379.
- Mendzelevski B, Ausma J, Chanter DO, et al. Assessment of the cardiac safety of prucalopride in healthy volunteers: a randomized, double-blind, placebo- and positive-controlled thorough QT study. Br J Clin Pharmacol 2012; 73:203–209.
- Camilleri M, Kerstens R, Rykx A, Vandeplassche L. A placebo-controlled trial of prucalopride for severe chronic constipation. N Engl J Med 2008; 358:2344–2354.
- Tack J, van Outryve M, Beyens G, Kerstens R, Vandeplassche L. Prucalopride (Resolor) in the treatment of severe chronic constipation in patients dissatisfied with laxatives. Gut 2009; 58:357–365.
- Quigley EM, Vandeplassche L, Kerstens R, Ausma J. Clinical trial: the efficacy, impact on quality of life, and safety and tolerability of prucalopride in severe chronic constipation—a 12-week, randomized, double-blind, placebo-controlled study. Aliment Pharmacol Ther 2009; 29:315–328.
- Ford AC, Suares NC. Effect of laxatives and pharmacological therapies in chronic idiopathic constipation: systematic review and meta-analysis. Gut 2011; 60:209–218.
- Goldberg M, Li YP, Johanson JF, et al. Clinical trial: the efficacy and tolerability of velusetrag, a selective 5-HT4 agonist with high intrinsic activity, in chronic idiopathic constipation—a 4-week, randomized, double-blind, placebo-controlled, dose-response study. Aliment Pharmacol Ther 2010; 32:1102–1112.
- Palme M, Milner PG, Ellis DJ, Marmon T, Canafax DM. A novel gastrointestinal prokinetic, ATI-7505, increased spontaneous bowel movements (sbms) in a phase II, randomized, placebo-controlled study of patients with chronic idiopathic constipation (CIC). Gastroenterology 2010; 138:S-128–S-129.
- Chey WD, Camilleri M, Chang L, Rikner L, Graffner H. A randomized placebo-controlled phase IIb trial of a3309, a bile acid transporter inhibitor, for chronic idiopathic constipation. Am J Gastroenterol 2011; 106:1803–1812.
- Wong BS, Camilleri M, McKinzie S, Burton D, Graffner H, Zinsmeister AR. Effects of A3309, an ileal bile acid transporter inhibitor, on colonic transit and symptoms in females with functional constipation. Am J Gastroenterol 2011; 106:2154–2164.
- Pappagallo M. Incidence, prevalence, and management of opioid bowel dysfunction. Am J Surg 2001; 182(suppl):11S–18S.
- Bell T, Annunziata K, Leslie JB. Opioid-induced constipation negatively impacts pain management, productivity, and health-related quality of life: findings from the National Health and Wellness Survey. J Opioid Manag 2009; 5:137–144.
- Sykes NP. A volunteer model for the comparison of laxatives in opioid-related constipation. J Pain Symptom Manage 1996; 11:363–369.
- ClinicalTrials.gov. A multicenter, randomized, double-blind, placebo-controlled, parallel-group study of oral MOA-728 for the treatment of opioid- induced bowel dysfunction in subjects with chronic nonmalignant pain. ClinicalTrials.gov Identifier: NCT00547586. https://clinicaltrials.gov/ct2/show/NCT00547586. Accessed March 22, 2017.
- ClinicalTrials.gov. An open-label study to evaluate the long-term safety of subcutaneous MOA-728 for treatment of opioid-induced constipation in subjects with nonmalignant pain. ClinicalTrials.gov Identifier: NCT00804141. https://clinicaltrials.gov/ct2/show/NCT00804141. Accessed April 6, 2017.
- Wyeth Pharmaceuticals. Relistor package insert. http://labeling.pfizer.com/showlabeling.aspx?id=499. Accessed March 22, 2017.
- Webster L, Dhar S, Eldon M, Masuoka L, Lappalainen J, Sostek M. A phase 2, double-blind, randomized, placebo-controlled, dose-escalation study to evaluate the efficacy, safety, and tolerability of naloxegol in patients with opioid-induced constipation. Pain 2013; 154:1542–1550.
- Chey WD, Webster L, Sostek M, Lappalainen J, Barker PN, Tack J. Naloxegol for opioid-induced constipation in patients with noncancer pain. N Engl J Med 2014; 370:2387–2396.
- Jones R, Prommer E, Backstedt D. Naloxegol: a novel therapy in the management of opioid-induced constipation. Am J Hosp Palliat Care 2016; 33:875–880.
- Guilera M, Balboa A, Mearin F. Bowel habit subtypes and temporal patterns in irritable bowel syndrome: systematic review. Am J Gastroenterol 2005; 100:1174–1184.
- Chapman RW, Stanghellini V, Geraint M, Halphen M. Randomized clinical trial: macrogol/PEG 3350 plus electrolytes for treatment of patients with constipation associated with irritable bowel syndrome. Am J Gastroenterol 2013; 108:1508–1515.
- Ford AC, Quigley EM, Lacy BE, et al. Effect of antidepressants and psychological therapies, including hypnotherapy, in irritable bowel syndrome: systematic review and meta-analysis. Am J Gastroenterol 2014; 109:1350–1366.
- Ballou S, Keefer L. Psychological interventions for irritable bowel syndrome and inflammatory bowel diseases. Clin Transl Gastroenterol 2017; 8:e214.
- Ford AC, Moayyedi P, Lacy BE, et al; Task Force on the Management of Functional Bowel Disorders. American College of Gastroenterology monograph on the management of irritable bowel syndrome and chronic idiopathic constipation. Am J Gastroenterol 2014; 109(suppl 1):S2–S27.
- Ford AC, Talley NJ, Spiegel BM, et al. Effect of fibre, antispasmodics, and peppermint oil in the treatment of irritable bowel syndrome: systematic review and meta-analysis. BMJ 2008; 337:a2313.
- Wall GC, Bryant GA, Bottenberg MM, Maki ED, Miesner AR. Irritable bowel syndrome: a concise review of current treatment concepts. World J Gastroenterol 2014; 20:8796–8806.
- Kligler B, Chaudhary S. Peppermint oil. Am Fam Physician 2007; 75:1027–1030.
- Spencer AG, Labonte ED, Rosenbaum DP, et al. Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci Transl Med 2014; 6:227ra36.
- Rosenbaum DP. A randomized, double-blind, placebo-controlled study to assess the safety and efficacy of AZD1722 for the treatment of constipation-predominant irritable bowel syndrome (IBS-C). 2014. https://clinicaltrials.gov/ct2/show/NCT01923428. Accessed April 6, 2017.
- Rao SS. Biofeedback therapy for dyssynergic (obstructive) defecation. J Clin Gastroenterol 2000; 30:115–116.
- Cadeddu F, Salis F, De Luca E, Ciangola I, Milito G. Efficacy of biofeedback plus transanal stimulation in the management of pelvic floor dyssynergia: a randomized trial. Tech Coloproctol 2015; 19:333–338.
- Chiarioni G, Whitehead WE, Pezza V, Morelli A, Bassotti G. Biofeedback is superior to laxatives for normal transit constipation due to pelvic floor dyssynergia. Gastroenterology 2006; 130:657–664.
- Chiarioni G, Heymen S, Whitehead WE. Biofeedback therapy for dyssynergic defecation. World J Gastroenterol 2006; 12:7069–7074.
- Rao SS. Biofeedback therapy for constipation in adults. Best Pract Res Clin Gastroenterol 2011; 25:159–166.
- Hassan I, Pemberton JH, Young-Fadok TM, et al. Ileorectal anastomosis for slow transit constipation: long-term functional and quality of life results. J Gastrointest Surg 2006; 10:1330–1337.
- You YT, Wang JY, Changchien CR, et al. Segmental colectomy in the management of colonic inertia. Am Surg 1998; 64:775–777.
- Nyam DC, Pemberton JH, Ilstrup DM, Rath DM. Long-term results of surgery for chronic constipation. Dis Colon Rectum 1997; 40:273–279.
- Pemberton JH, Rath DM, Ilstrup DM. Evaluation and surgical treatment of severe chronic constipation. Ann Surg 1991; 214:403–413.
- Reshef A, Alves-Ferreira P, Zutshi M, Hull T, Gurland B. Colectomy for slow transit constipation: effective for patients with coexistent obstructed defecation. Int J Colorectal Dis 2013; 28:841–847.
- Lundin E, Karlbom U, Pahlman L, Graf W. Outcome of segmental colonic resection for slow-transit constipation. Br J Surg 2002; 89:1270–1274.
- Schwandner O, Stuto A, Jayne D, et al. Decision-making algorithm for the STARR procedure in obstructed defecation syndrome: position statement of the group of STARR pioneers. Surg Innov 2008; 15:105–109.
- Titu LV, Riyad K, Carter H, Dixon AR. Stapled transanal rectal resection for obstructed defecation: a cautionary tale. Dis Colon Rectum 2009; 52:1716–1722.
- Goede AC, Glancy D, Carter H, Mills A, Mabey K, Dixon AR. Medium-term results of stapled transanal rectal resection (STARR) for obstructed defecation and symptomatic rectal-anal intussusception. Colorectal Dis 2011; 13:1052–1057.
- Jayne DG, Schwandner O, Stuto A. Stapled transanal rectal resection for obstructed defecation syndrome: one-year results of the european STARR registry. Dis Colon Rectum 2009; 52:1205–1214.
- Madbouly KM, Abbas KS, Hussein AM. Disappointing long-term outcomes after stapled transanal rectal resection for obstructed defecation. World J Surg 2010; 34:2191–2196.
KEY POINTS
- Although newer drugs are available, lifestyle modifications and laxatives continue to be the treatments of choice for chronic constipation, as they have high response rates and few adverse effects and are relatively affordable.
- Chronic constipation requires different management approaches depending on whether colonic transit time is normal or prolonged and whether outlet function is abnormal.
- Surgical treatments for constipation are reserved for patients whose symptoms persist despite maximal medical therapy.

Sexual dysfunction in women: Can we talk about it?
Many women experience some form of sexual dysfunction, be it lack of desire, lack of arousal, failure to achieve orgasm, or pain during sexual activity.
Sexual health may be difficult to discuss, for both the patient and the provider. Here, we describe how primary care physicians can approach this topic, assess potential problems, and begin treatment.
A COMMON PROBLEM
The age-adjusted prevalence of sexual dysfunction in US women was reported at 44% in the Prevalence of Female Sexual Problems Associated With Distress and Determinants of Treatment Seeking (PRESIDE) study,1 but the prevalence of distress associated with sexual dysfunction was 12%. The most common type of sexual dysfunction reported by women was low sexual desire, a finding consistent with that of another large population-based study.2
While the prevalence of any type of sexual dysfunction was highest in women over age 65,1 the prevalence of distress was lowest in this age group and highest in midlife between the ages of 45 and 65. The diagnostic criteria require both a problem and distress over the problem.
Sexual dysfunction negatively affects quality of life and emotional health, regardless of age.3
LIFESTYLE AND SEXUAL FUNCTION
Various lifestyle factors have been linked to either more or less sexual activity. For example, a Mediterranean diet was associated with increased sexual activity, as were social activity, social support, psychological well-being, self-reported good quality of life, moderate alcohol intake, absence of tobacco use, a normal body mass index, and exercise.4–6 A higher sense of purpose in life has been associated with greater sexual enjoyment.7
Conversely, sexual inactivity has been associated with alcohol misuse, an elevated body mass index, and somatization.4–6
SEXUAL RESPONSE: LINEAR OR CIRCULAR?
Masters and Johnson8 initially proposed a linear model of human sexual response, which Kaplan later modified to include desire and applied to both men and women.9,10 This model presumed that sexual response begins with spontaneous sexual desire, followed by arousal, and then (sometimes) orgasm and resolution.

In 2000, Basson11 proposed a circular, intimacy-based model of sexual response in women that acknowledged the complexities involved in a woman’s motivation to be sexual (Figure 1). While a woman may enter the cycle with spontaneous sexual desire, she may also enter it as sexually neutral, with arousal in response to a sexual stimulus. Emotional intimacy is an important part of the cycle, and emotional closeness and bonding with the partner may provide motivation for a woman to enter into the cycle again in the future.
In a Danish survey,12 more people of both sexes said the 2 linear models described their experiences better than the circular model, but more women than men endorsed the circular model, and more men than women endorsed a linear model.
In evaluating women who complain of low sexual desire, clinicians should be aware that women, particularly those who are postmenopausal, may not enter the cycle with spontaneous sexual desire, but instead may experience arousal in response to a sexual stimulus followed by desire—ie, responsive rather than spontaneous sexual desire. Sexual arousal may precede desire, especially for women in long-term relationships, and emotional intimacy is a key driver for sexual engagement in women.11
CATEGORIES OF SEXUAL DYSFUNCTION IN WOMEN
The World Health Organization defines sexual health as “a state of physical, emotional, mental, and social well-being in relation to sexuality” and “not merely the absence of disease, dysfunction, or infirmity.”13
The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5),14 published in 2013, defines three categories of sexual dysfunction in women:
- Female sexual interest and arousal disorder
- Female sexual orgasmic disorder
- Genitopelvic pain/penetration disorder.
To meet the diagnosis of any of these, symptoms must:
- Persist for at least 6 months
- Occur in 75% to 100% of sexual encounters
- Be accompanied by personal distress
- Not be related to another psychological or medical condition, medication or substance use, or relationship distress.
Sexual problems may be lifelong or acquired after a period of normal functioning, and may be situational (present only in certain situations) or generalized (present in all situations).
Female sexual interest and arousal disorder used to be 2 separate categories in earlier editions of the DSM. Proponents of merging the 2 categories in DSM-5 cited several reasons, including difficulty in clearly distinguishing desire from other motivations for sexual activity, the relatively low reporting of fantasy in women, the complexity of distinguishing spontaneous from responsive desire, and the common co-occurrence of decreased desire and arousal difficulties.15
Other experts, however, have recommended keeping the old, separate categories of hypoactive sexual desire disorder and arousal disorder.16 The recommendation to preserve the diagnostic category of hypoactive sexual desire disorder is based on robust observational and registry data, as well as the results of randomized controlled trials that used the old criteria for hypoactive sexual desire disorder to assess responses to pharmacologic treatment of this condition.17–19 In addition, this classification as a separate and distinct diagnosis is consistent with the nomenclature used in the International Statistical Classification of Diseases and Related Health Problems, 10th Revision and was endorsed by the International Consultation on Sexual Medicine in 2015.16
HOW TO ASK ABOUT SEXUAL HEALTH
Assessment of sexual health concerns should be a part of a routine health examination, particularly after childbirth and other major medical, surgical, psychological, and life events. Women are unlikely to bring up sexual health concerns with their healthcare providers, but instead hope that their providers will bring up the topic.20
Barriers to the discussion include lack of provider education and training, patient and provider discomfort, perceived lack of time during an office visit, and lack of approved treatments.21,22 Additionally, older women are less likely than men to discuss sexual health with their providers.23 Other potential barriers to communication include negative societal attitudes about sexuality in women and in older individuals.24,25 To overcome these barriers:

Legitimize sexual health as an important health concern and normalize its discussion as part of a routine clinical health assessment. Prefacing a query about sexual health with a normalizing and universalizing statement can help: eg, “Many women going through menopause have concerns about their sexual health. Do you have any sexual problems or concerns?” Table 1 contains examples of questions to use for initial screening for sexual dysfunction.22,26

Flynn et al27 proposed a validated single-question checklist to screen for sexual dysfunction that is an efficient way to identify specific sexual concerns, guide selection of interventions, and facilitate patient-provider communication (Table 2).
Don’t judge and don’t make assumptions about sexuality and sexual practices.
Assure confidentiality.
Use simple, direct language that is appropriate for the patient’s age, ethnicity, culture, and level of health literacy.3
Take a thorough history (sexual and reproductive, medical-surgical, and psychosocial).
PHYSICAL EXAMINATION
Perform a focused physical examination to evaluate for potential causes of pain (eg, infectious causes, vulvar dermatoses, pelvic floor muscle dysfunction). The examination is also an opportunity to teach the patient about anatomy and normal sexual function.
No standard laboratory tests or imaging studies are required for the assessment of sexual dysfunction.28
IT’S NOT JUST PHYSICAL

Evaluation and treatment of female sexual dysfunction is guided by the biopsychosocial model, with potential influences from the biological, psychological, sociocultural, and interpersonal realms (Table 3).29,30
Biological factors include pelvic surgery, cancer and its treatment, neurologic diseases, and vascular diseases. Medications, including antidepressants, narcotics, anticholinergics, antihistamines, antihypertensives, oral contraceptives, and antiestrogens may also adversely affect sexual response.26
Psychological factors include a history of sexual abuse or trauma, body image concerns, distraction, stress, anxiety, depression, and personality disorders.22
Sociocultural factors include lack of sex education, unrealistic expectations, cultural norms, and religious influences.
Relationship factors include conflict with one’s partner, lack of emotional intimacy, absence of a partner, and partner sexual dysfunction. While there appears to be a close link between sexual satisfaction and a woman’s relationship with her partner in correlational studies and in clinical experience, there has been little research about relationship factors and their contribution to desire and arousal concerns.31 Sexual dysfunction in one’s partner (eg, erectile dysfunction) has been shown to negatively affect the female partner’s sexual desire.32
GENERAL APPROACH TO TREATMENT
In treating sexual health problems in women, we address contributing factors identified during the initial assessment.
A multidisciplinary approach
As sexual dysfunction in women is often multifactorial, management of the problem is well suited to a multidisciplinary approach. The team of providers may include:
- A medical provider (primary care provider, gynecologist, or sexual health specialist) to coordinate care and manage biological factors contributing to sexual dysfunction
- A physical therapist with expertise in treating pelvic floor disorders
- A psychologist to address psychological, relational, and sociocultural contributors to sexual dysfunction
- A sex therapist (womenshealthapta.org, aasect.org) to facilitate treatment of tight, tender pelvic floor muscles through education and guidance about kinesthetic awareness, muscle relaxation, and dilator therapy.33

Even in the initial visit, the primary care provider can educate, reassure regarding normal sexual function, and treat conditions such as genitourinary syndrome of menopause and antidepressant-associated sexual dysfunction. The PLISSIT model (Permission, Limited Information, Specific Suggestions, and Intensive Therapy) is a useful tool for initiating counseling about sexual health (Table 4).34
AGING VS MENOPAUSE
Aging can affect sexual function in both men and women. About 40% of women experience changes in sexual function around the menopausal transition, with common complaints being loss of sexual responsiveness and desire, sexual pain, decreased sexual activity, and partner sexual dysfunction.35 However, studies seem to show that while menopause results in hormonal changes that affect sexual function, other factors may have a greater impact.
The Study of Women’s Health Across the Nation36 found vaginal and pelvic pain and decreased sexual desire were associated with the menopausal transition, but other sexual health outcomes (frequency of sexual activities, arousal, importance of sex, emotional satisfaction, or physical pleasure) were not. Physical and psychological health, marital status, and a change in relationship were all associated with differences in sexual health.
The Massachusetts Women’s Health Study II37 found a greater association between physical and mental health, relationship status, and smoking and women’s sexual functioning than menopausal status.
The Penn Ovarian Aging Study38 found that sexual function declined across the menopausal transition. Risk factors for sexual dysfunction included postmenopausal status, anxiety, and absence of a sexual partner.
The Melbourne Women’s Midlife Health Project39 also found that sexual function declined across the menopausal transition. Sexual dysfunction with distress was associated with relationship factors and depression.37
Genitourinary syndrome of menopause and its treatment
As the ovaries shut down during menopause, estradiol levels decrease. Nearly 50% of women experience symptoms related to genitourinary syndrome of menopause (formerly called atrophic vaginitis or vulvovaginal atrophy).40,41 These symptoms include vaginal dryness and discomfort or pain with sexual activity, but menopausal hormone loss can also result in reduced genital blood flow, decreased sensory perception, and decreased sexual responsiveness.22
Estrogen is the most effective treatment for genitourinary syndrome of menopause, with low-dose vaginal preparations preferred over systemic ones for isolated vulvar and vaginal symptoms.40 While estrogen is effective for vaginal dryness and sexual pain associated with estrogen loss, replacing estrogen systemically has not been associated with improvements in sexual desire.42
DEPRESSION AND ANTIDEPRESSANT-INDUCED SEXUAL DYSFUNCTION
Depression increases the risk of sexual dysfunction, and vice versa.
A meta-analysis that included 12 studies involving almost 15,000 patients confirmed that depression increased the risk of sexual dysfunction, and sexual dysfunction increased the risk of depression.43 This interaction may be related to the overlap in affected neurotransmitters and neuroendocrine systems.44
In the Sequenced Treatment Alternatives to Relieve Depression trial, Ishak et al45 found that patients treated with a selective serotonin reuptake inhibitor (SSRI) who experienced remission of depression had a lower prevalence of impaired sexual satisfaction and much greater improvements in sexual satisfaction than did those who remained depressed. The severity of depressive symptoms predicted impairment in sexual satisfaction, which in turn predicted poorer quality of life. The authors suggested that physicians encourage patients to remain on SSRI treatment, given that improvement in depressive symptoms is likely to improve sexual satisfaction.
Antidepressant-induced sexual dysfunction
As many as 70% of patients taking an SSRI or serotonin-norepinephrine reuptake inhibitor (SNRI) experience antidepressant-induced sexual dysfunction, though this is difficult to estimate across studies of different medications due to differences in methods and because many patients only report it when directly asked about it.46
Treatment of antidepressant-induced sexual dysfunction includes not only optimal management of depression but reassessment of the antidepressant treatment. If using only nondrug treatments for the mood disorder is not feasible, switching to (or ideally, starting with) an antidepressant with fewer sexual side effects such as mirtazapine, vilazodone, or bupropion is an option.46
A drug holiday (suspending antidepressant treatment for 1 or 2 days) has been suggested as a means of treating antidepressant-induced sexual dysfunction, but this may result in poorer control of depressive symptoms and discontinuation symptoms, and it encourages medication noncompliance.46,47
Treatment with a phosphodiesterase type 5 inhibitor (eg, sildenafil) has been studied in women with antidepressant-induced sexual dysfunction, with modest results.48
A Cochrane review reported that treatment with bupropion shows promise at higher doses (300 mg daily).49
Exercise for 20 minutes 3 times weekly is associated with improvement in antidepressant-induced sexual dysfunction when the exercise is performed immediately before sexual activity.50
LOW SEXUAL DESIRE
Hypoactive sexual desire disorder is defined as persistent or recurrent deficiency or absence of sexual fantasies and desire for sexual activity associated with marked distress and not due exclusively to a medication, substance abuse, or a medical condition.
Low or decreased sexual desire is the most commonly reported sexual health concern in women of all ages, with an unadjusted prevalence of 39.7%. When the criterion of personal distress is included, the prevalence is 8.9% in women ages 18 to 44, 12.3% in women ages 45 to 64, and 7.4% in women ages 65 and older.1
Multiple biological, psychological, and social factors may contribute to the problem. Identifying the ones that are present can help in planning treatment. A multifaceted approach may be appropriate.
Mindfulness and cognitive behavioral therapy for low sexual desire
Mindfulness-based cognitive therapy is designed to improve awareness, focusing on and accepting the present moment, and directing attention away from and lessening self-criticism and evaluation of one’s sexual responsiveness.
Mindfulness-based therapy has been associated with improvements in sexual desire and associated distress.51 Similarly, the effectiveness of cognitive behavioral therapy for treating hypoactive sexual desire disorder is supported by 3 controlled trials, although concerns exist about the adequacy of these trials, and further study is needed.52
Androgen therapy in women
In randomized controlled trials in women with low sexual desire who were either naturally or surgically menopausal, sexual function improved with testosterone therapy that resulted in mostly supraphysiologic total testosterone levels (which may not reflect free testosterone levels) with or without concurrent estrogen treatment.53–57
Testosterone is not approved by the US Food and Drug Administration (FDA) for use in women, primarily because of the lack of long-term safety and efficacy data (ie, beyond 24 months). However, studies have shown no evidence of increased risk of endometrial cancer or cardiovascular disease with testosterone dosed to achieve physiologic premenopausal levels.58 Data on breast cancer risk are less clear, but observational studies over the last decade do not support an association with testosterone use in women.58 There is no clearly defined androgen deficiency syndrome in women, and androgen levels do not reliably correlate with symptoms.59
The Endocrine Society59 guidelines endorse the use of testosterone in postmenopausal women with hypoactive sexual desire disorder. They say to aim for the midnormal premenopausal range and suggest discontinuing the drug if there is no response in 6 months. They recommend checking testosterone levels at baseline, after 3 to 6 weeks of therapy, and every 6 months to monitor for excessive use, to avoid supraphysiologic dosing and to evaluate for signs of androgen excess (eg, acne, hair growth). The use of products formulated for men or those formulated by pharmacies is discouraged; however, no FDA-approved products are currently available for use in women in the United States.
Flibanserin
A postsynaptic serotonin 5-HT1A receptor agonist and 5-HT2A receptor agonist, flibanserin was approved by the FDA in 2015 for treatment of hypoactive sexual desire disorder in premenopausal women. Its mechanism of action is likely through an effect on neurotransmitters that suppresses serotonin (which has sexually inhibitory effects) and promotes dopamine and norepinephrine (which have excitatory effects).60
The efficacy of flibanserin has been demonstrated in 3 randomized controlled trials, with significant increases in the number of sexually satisfying events and in sexual desire scores and a decrease in distress associated with low sexual desire.17–19 While the increase in sexually satisfying events was modest (about 1 extra event per month), some have suggested that the frequency of sexual activity may not be the best measure of sexual function in women.61 Further, responders to this drug showed a return to near-normal premenopausal frequencies of sexual activity in a separate analysis.61
The drug is generally well tolerated, with common adverse effects being somnolence, dizziness, and fatigue.18,19 Flibanserin has been associated with orthostatic hypotension with alcohol use and carries a boxed warning highlighting this potential interaction.62 Use of this drug is contraindicated in women who drink alcohol or take medications that are moderate or strong inhibitors of CYP-3A4 (eg, some antiretroviral drugs, antihypertensive drugs, antibiotics, and fluconazole, which can increase systemic exposure to flibanserin and potential side effects), and in those with liver impairment.
SEXUAL AROUSAL DISORDERS
Female sexual arousal disorder is the persistent or recurrent inability to attain or maintain an adequate lubrication-swelling response of sexual excitement. Sexual arousal results from a complex interaction between genital response, central nervous system activity, and information processing of the sexual stimulus. Difficulty with sexual arousal can result from neurovascular or neuroendocrine dysfunction or impaired central nervous system processing.
Women may experience a mismatch between subjective and objective genital arousal. A subjective report of decreased genital arousal may not be confirmed with measurement of vaginal pulse amplitude by photoplethysmography.63 Even in postmenopausal women, in the absence of significant neurovascular or neuroendocrine dysfunction, it is likely that either contextual or relational variables resulting in inadequate sexual stimulation or cognitive inhibition are more important factors contributing to difficulty with sexual arousal.63
Although there are no standard recommendations for evaluation of arousal disorders and advanced testing is often unnecessary, nerve function can be assessed with genital sensory testing utilizing thermal and vibratory threshholds64; vaginal blood flow can be assessed with vaginal photoplethysmography63; and imaging of the spine and pelvis can help to rule out neurovascular pathology.
Treatment of arousal disorders
As with other forms of female sexual dysfunction, treatment of arousal disorders includes addressing contributing factors.
Although there are few data from randomized controlled trials, psychological treatments such as sensate focus exercises and masturbation training have been suggested, centered on women becoming more self-focused and assertive.31 Sensate focus exercises are a series of graded, nondemand, sensual touching exercises aimed at reducing anxiety and avoidance of sexual activity, and improving sexual communication and intimacy by the gradual reintroduction of sexual activity.65 More recently, mindfulness-based cognitive therapy has been associated with improvements in sexual arousal as well as other parameters of sexual function.51
Currently, no pharmacologic treatments are recommended for arousal disorders because of a lack of evidence of efficacy and because of adverse effects.31
ORGASMIC DISORDER
Female orgasmic disorder is the marked delay, marked infrequency, or absence of orgasm, or markedly reduced intensity of orgasm.
Important considerations in evaluating orgasm disorders include psychosocial factors (eg, lack of sex education, negative feelings about sex, religiosity), psychological factors (eg, anxiety, depression, body image concerns), relational factors (eg, communication issues, lack of emotional intimacy, partner sexual dysfunction), adverse childhood or adult experiences (eg, physical, sexual, or emotional or verbal abuse), medical history (pelvic surgery, neurologic, or vascular disease) and medications (eg, SSRIs, SNRIs, and antipsychotic medications).66
Treatment of orgasmic disorder
Involving the partner in treatment is important, particularly if the difficulty with orgasm is acquired and only occurs with sex with a partner. Using the PLISSIT model to provide targeted, office-based interventions can be helpful.
Behavioral therapies such as directed masturbation, sensate focus exercises, or a combination of these have been shown to be effective, as has coital alignment during intercourse (positioning of male partner with his pelvis above the pubic bone of his partner to maximize clitoral stimulation with penile penetration).66
Hormonal therapy may be useful in postmenopausal women. However, there are no data on it for women whose primary complaint is female orgasmic disorder, and further study is needed.66
SEXUAL PAIN DISORDERS
The DSM-5 describes genitopelvic pain/penetration disorder as fear or anxiety, marked tightening or tensing of abdominal and pelvic muscles, or actual pain with vaginal penetration that is recurrent or persistent for a minimum of 6 months.14 Pain may occur with initial penetration, with deeper thrusting, or both.
Although the DSM-5 definition focuses on pain with penetration, it is important to recognize and ask about noncoital sexual pain. Women may also present with persistent vulvar pain or pain at the vulvar vestibule with provocation, (eg, sexual activity, tampon insertion, sitting), also known as provoked vestibulodynia.
Assessment of vaginal and vulvar pain includes a directed history and physical examination aimed at identifying potential etiologies or contributing factors, including infectious, inflammatory, neoplastic, neurologic, traumatic, iatrogenic, or factors related to hormonal deficiency.67
Treatment of sexual pain
Removal of offending agents is a first step. This includes a thorough review of vulvar and vaginal hygiene practices and emphasis on avoiding the use of any product containing potential irritants (eg, soaps or detergents containing perfumes or dyes) and using lubricants and moisturizers without gimmicks (no warming or tingling agents or flavors). Oral contraceptives have been associated with vestibulodynia, and women in whom the sexual pain started when they started an oral contraceptive may benefit from switching to an alternate form of contraception.68
Dysfunction of pelvic floor muscles may result in sexual pain and may be a primary problem or a secondary complication related to other issues such as symptomatic genitourinary syndrome of menopause. The symptoms of nonrelaxing pelvic floor dysfunction (also known as hypertonic pelvic floor dysfunction or pelvic floor tension myalgia) include pain in the pelvis with sexual activity that may linger for hours or even days, and may also include bowel and bladder dysfunction and low back pain or hip pain radiating to the thighs or groin.33 Physical therapy under the care of a physical therapist with expertise in the management of pelvic floor disorders is the cornerstone of treatment for this condition.33
Treatment of the genital and urinary symptoms related to loss of estrogen after menopause (genitourinary syndrome of menopause) includes the use of vaginal lubricants with sexual activity and vaginal moisturizers on a regular basis (2 to 5 times per week).40 Low-dose vaginal estrogen creams, rings, or tablets and the oral selective estrogen receptor antagonist ospemifene are recommended for moderate to severe symptoms of genitourinary syndrome of menopause.40 Intravaginal dehydroepiandrosterone was recently approved by the FDA for treatment of dyspareunia associated with menopause.69 Topical lidocaine applied to the introitus before sexual activity has been found to be effective for reducing sexual pain in women with breast cancer, and when used in combination with vaginal lubricants and moisturizers is a practical option for women, particularly those unable to use estrogen-based therapies.70
- Shifren JL, Monz BU, Russo PA, Segreti A, Johannes CB. Sexual problems and distress in United States women: prevalence and correlates. Obstet Gynecol 2008; 112:970–978.
- Laumann EO, Paik A, Rosen RC. Sexual dysfunction in the United States: prevalence and predictors. JAMA 1999; 281:537–544.
- Sadovsky R, Nusbaum M. Sexual health inquiry and support is a primary care priority. J Sex Med 2006; 3:3–11.
- Alvisi S, Baldassarre M, Lambertini M, et al. Sexuality and psychopathological aspects in premenopausal women with metabolic syndrome. J Sex Med 2014; 11:2020–2028.
- Bach LE, Mortimer JA, VandeWeerd C, Corvin J. The association of physical and mental health with sexual activity in older adults in a retirement community. J Sex Med 2013; 10:2671–2678.
- Esposito K, Ciotola M, Giugliano F, et al. Mediterranean diet improves sexual function in women with the metabolic syndrome. Int J Impot Res 2007; 19:486–491.
- Prairie BA, Scheier MF, Matthews KA, Chang CC, Hess R. A higher sense of purpose in life is associated with sexual enjoyment in midlife women. Menopause 2011; 18:839–844.
- Masters WH, Johnson VE. Human sexual response. Boston, MA: Little, Brown and Company; 1966.
- Kaplan HS. The new sex therapy. New York, NY: Brunner/Mazel; 1974.
- Robinson PA. The modernization of sex: Havelock Ellis, Alfred Kinsey, William Masters, and Virginia Johnson. New York, NY: Harper & Row; 1976.
- Basson R. The female sexual response: a different model. J Sex Marital Ther 2000; 26:51–65.
- Giraldi A, Kristensen E, Sand M. Endorsement of models describing sexual response of men and women with a sexual partner: an online survey in a population sample of Danish adults ages 20–65 years. J Sex Med 2015; 12:116–128.
- World Health Organization. Defining sexual health. Report of a technical consultation on sexual health, 28–31 January 2002, Geneva. Geneva; 2006.
- American Psychiatric Association (APA). DSM-5: Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association Publishing; 2013.
- Brotto LA. The DSM diagnostic criteria for hypoactive sexual desire disorder in women. Arch Sex Behav 2010; 39:221–239.
- McCabe MP, Sharlip ID, Atalla E, et al. Definitions of sexual dysfunctions in women and men: a consensus statement from the Fourth International Consultation on Sexual Medicine 2015. J Sex Med 2016; 13:135–143.
- Derogatis LR, Komer L, Katz M, et al; VIOLET Trial Investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the VIOLET study. J Sex Med 2012; 9:1074–1085.
- Katz M, DeRogatis LR, Ackerman R, et al; BEGONIA trial investigators. Efficacy of flibanserin in women with hypoactive sexual desire disorder: results from the BEGONIA trial. J Sex Med 2013; 10:1807–1815.
- Thorp J, Simon J, Dattani D, et al; DAISY trial investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the DAISY study. J Sex Med 2012; 9:793–804.
- Berman L, Berman J, Felder S, et al. Seeking help for sexual function complaints: what gynecologists need to know about the female patient's experience. Fertil Steril 2003; 79:572–576.
- Kingsberg SA. Taking a sexual history. Obstet Gynecol Clin North Am 2006; 33:535–547.
- Kingsberg SA, Rezaee RL. Hypoactive sexual desire in women. Menopause 2013; 20:1284–1300.
- Lindau ST, Schumm LP, Laumann EO, Levinson W, O’Muircheartaigh CA, Waite LJ. A study of sexuality and health among older adults in the United States. N Engl J Med 2007; 357:762–774.
- Gott M, Hinchliff S, Galena E. General practitioner attitudes to discussing sexual health issues with older people. Soc Sci Med 2004; 58:2093–2103.
- Lindau ST, Leitsch SA, Lundberg KL, Jerome J. Older women's attitudes, behavior, and communication about sex and HIV: a community-based study. J Womens Health (Larchmt) 2006; 15:747–753.
- Faubion SS, Rullo JE. Sexual dysfunction in women: a practical approach. Am Fam Physician 2015; 92:281–288.
- Flynn KE, Lindau ST, Lin L, et al. Development and validation of a single-item screener for self-reporting sexual problems in US adults. J Gen Intern Med 2015; 30:1468–1475.
- Latif EZ, Diamond MP. Arriving at the diagnosis of female sexual dysfunction. Fertil Steril 2013; 100:898–904.
- Althof SE, Leiblum SR, Chevret-Measson M, et al. Psychological and interpersonal dimensions of sexual function and dysfunction. J Sex Med 2005; 2:793–800.
- Rosen RC, Barsky JL. Normal sexual response in women. Obstet Gynecol Clin North Am 2006; 33:515–526.
- Brotto LA, Bitzer J, Laan E, Leiblum S, Luria M. Women's sexual desire and arousal disorders. J Sex Med 2010; 7:586–614.
- Rubio-Aurioles E, Kim ED, Rosen RC, et al. Impact on erectile function and sexual quality of life of couples: a double-blind, randomized, placebo-controlled trial of tadalafil taken once daily. J Sex Med 2009; 6:1314–1323.
- Faubion SS, Shuster LT, Bharucha AE. Recognition and management of nonrelaxing pelvic floor dysfunction. Mayo Clin Proc 2012; 87:187–193.
- Annon JS. The PLISSIT model: a proposed conceptual scheme for the behavioral treatment of sexual problems. J Sex Educ Ther 1976; 2:1–15.
- Sarrel PM. Sexuality and menopause. Obstet Gynecol 1990; 75(suppl 4):26S–30S; discussion 31S–35S.
- Avis NE, Brockwell S, Randolph JF, Jr, et al. Longitudinal changes in sexual functioning as women transition through menopause: results from the Study of Women's Health Across the Nation. Menopause 2009; 16:442–452.
- Avis NE, Stellato R, Crawford S, Johannes C, Longcope C. Is there an association between menopause status and sexual functioning? Menopause 2000; 7:297–309.
- Gracia CR, Freeman EW, Sammel MD, Lin H, Mogul M. Hormones and sexuality during transition to menopause. Obstet Gynecol 2007; 109:831–840.
- Dennerstein L, Guthrie JR, Hayes RD, DeRogatis LR, Lehert P. Sexual function, dysfunction, and sexual distress in a prospective, population-based sample of mid-aged, Australian-born women. J Sex Med 2008; 5:2291–2299.
- Management of symptomatic vulvovaginal atrophy: 2013 position statement of The North American Menopause Society. Menopause 2013; 20:888–902.
- Mac Bride MB, Rhodes DJ, Shuster LT. Vulvovaginal atrophy. Mayo Clin Proc 2010; 85:87–94.
- Nastri CO, Lara LA, Ferriani RA, Rosa ESAC, Figueiredo JB, Martins WP. Hormone therapy for sexual function in perimenopausal and postmenopausal women. Cochrane Database Syst Rev 2013; 6:CD009672.
- Atlantis E, Sullivan T. Bidirectional association between depression and sexual dysfunction: a systematic review and meta-analysis. J Sex Med 2012; 9:1497–1507.
- Clayton AH, Maserejian NN, Connor MK, Huang L, Heiman JR, Rosen RC. Depression in premenopausal women with HSDD: baseline findings from the HSDD Registry for Women. Psychosom Med 2012; 74:305–311.
- Ishak WW, Christensen S, Sayer G, et al. Sexual satisfaction and quality of life in major depressive disorder before and after treatment with citalopram in the STAR*D study. J Clin Psychiatry 2013; 74:256–261.
- Clayton AH, Croft HA, Handiwala L. Antidepressants and sexual dysfunction: mechanisms and clinical implications. Postgrad Med 2014; 126:91–99.
- Rothschild AJ. Selective serotonin reuptake inhibitor-induced sexual dysfunction: efficacy of a drug holiday. Am J Psychiatry 1995; 152:1514–1516.
- Nurnberg HG, Hensley PL, Heiman JR, Croft HA, Debattista C, Paine S. Sildenafil treatment of women with antidepressant-associated sexual dysfunction: a randomized controlled trial. JAMA 2008; 300:395–404.
- Taylor MJ, Rudkin L, Bullemor-Day P, Lubin J, Chukwujekwu C, Hawton K. Strategies for managing sexual dysfunction induced by antidepressant medication. Cochrane Database Syst Rev 2013; 5:CD003382.
- Lorenz TA, Meston CM. Exercise improves sexual function in women taking antidepressants: results from a randomized crossover trial. Depress Anxiety 2014; 31:188–195.
- Brotto LA, Basson R. Group mindfulness-based therapy significantly improves sexual desire in women. Behav Res Ther 2014; 5743–5754.
- Pyke RE, Clayton AH. Psychological treatment trials for hypoactive sexual desire disorder: a sexual medicine critique and perspective. J Sex Med 2015; 12:2451–2458.
- Cappelletti M, Wallen K. Increasing women's sexual desire: the comparative effectiveness of estrogens and androgens. Horm Behav 2016; 78:178–193.
- Davis SR, Moreau M, Kroll R, et al. Testosterone for low libido in postmenopausal women not taking estrogen. N Engl J Med 2008; 359:2005–2017.
- Davis SR, van der Mooren MJ, van Lunsen RH, et al. Efficacy and safety of a testosterone patch for the treatment of hypoactive sexual desire disorder in surgically menopausal women: a randomized, placebo-controlled trial. Menopause 2006; 13:387–396.
- Panay N, Al-Azzawi F, Bouchard C, et al. Testosterone treatment of HSDD in naturally menopausal women: the ADORE study. Climacteric 2010; 13:121–131.
- Somboonporn W, Davis S, Seif MW, Bell R. Testosterone for peri- and postmenopausal women. Cochrane Database Syst Rev 2005; 4:CD004509.
- Davis SR. Cardiovascular and cancer safety of testosterone in women. Curr Opin Endocrinol Diabetes Obes 2011; 18:198–203.
- Wierman ME, Arlt W, Basson R, et al. Androgen therapy in women: a reappraisal: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2014; 99:3489–3510.
- Stahl SM, Sommer B, Allers KA. Multifunctional pharmacology of flibanserin: possible mechanism of therapeutic action in hypoactive sexual desire disorder. J Sex Med 2011; 8:15–27.
- Simon JA, Goldstein I, Kim N, Freedman M, Parish S. Flibanserin approval: facts or feelings? Sex Med 2016; 4:e69–e70.
- Joffe HV, Chang C, Sewell C, et al. FDA approval of flibanserin—treating hypoactive sexual desire disorder. N Engl J Med 2016; 374:101–104.
- van Lunsen RH, Laan E. Genital vascular responsiveness and sexual feelings in midlife women: psychophysiologic, brain, and genital imaging studies. Menopause 2004; 11:741–748.
- Beco J, Seidel L, Albert A. Normative values of skin temperature and thermal sensory thresholds in the pudendal nerve territory. Neurourol Urodyn 2015; 34:571–577.
- Masters WH, Johnson VE. Human sexual inadequacy. Boston, MA: Little, Brown and Company; 1970.
- Laan E, Rellini AH, Barnes T; International Society for Sexual Medicine. Standard operating procedures for female orgasmic disorder: consensus of the International Society for Sexual Medicine. J Sex Med 2013; 10:74–82.
- Bornstein J, Goldstein A, Coady D. Consensus terminology and classification of persistent vulvar pain. Vulvar pain/Vulvodynia Nomenclature Consensus Conference. Annapolis, MD; 2015.
- Burrows LJ, Goldstein AT. The treatment of vestibulodynia with topical estradiol and testosterone. Sex Med 2013; 1:30–33.
- Labrie F. Efficacy of intravaginal dehydroepiandrosterone (DHEA) on moderate to severe dyspareunia and vaginal dryness, symptoms of vulvovaginal atrophy, and of the genitourinary syndrome of menopause. Manopause 2016; 23:243–256.
- Goetsch MF, Lim JY, Caughey AB. A practical solution for dyspareunia in breast cancer survivors: a randomized controlled trial. J Clin Oncol 2015; 33:3394–3400.
Many women experience some form of sexual dysfunction, be it lack of desire, lack of arousal, failure to achieve orgasm, or pain during sexual activity.
Sexual health may be difficult to discuss, for both the patient and the provider. Here, we describe how primary care physicians can approach this topic, assess potential problems, and begin treatment.
A COMMON PROBLEM
The age-adjusted prevalence of sexual dysfunction in US women was reported at 44% in the Prevalence of Female Sexual Problems Associated With Distress and Determinants of Treatment Seeking (PRESIDE) study,1 but the prevalence of distress associated with sexual dysfunction was 12%. The most common type of sexual dysfunction reported by women was low sexual desire, a finding consistent with that of another large population-based study.2
While the prevalence of any type of sexual dysfunction was highest in women over age 65,1 the prevalence of distress was lowest in this age group and highest in midlife between the ages of 45 and 65. The diagnostic criteria require both a problem and distress over the problem.
Sexual dysfunction negatively affects quality of life and emotional health, regardless of age.3
LIFESTYLE AND SEXUAL FUNCTION
Various lifestyle factors have been linked to either more or less sexual activity. For example, a Mediterranean diet was associated with increased sexual activity, as were social activity, social support, psychological well-being, self-reported good quality of life, moderate alcohol intake, absence of tobacco use, a normal body mass index, and exercise.4–6 A higher sense of purpose in life has been associated with greater sexual enjoyment.7
Conversely, sexual inactivity has been associated with alcohol misuse, an elevated body mass index, and somatization.4–6
SEXUAL RESPONSE: LINEAR OR CIRCULAR?
Masters and Johnson8 initially proposed a linear model of human sexual response, which Kaplan later modified to include desire and applied to both men and women.9,10 This model presumed that sexual response begins with spontaneous sexual desire, followed by arousal, and then (sometimes) orgasm and resolution.
In 2000, Basson11 proposed a circular, intimacy-based model of sexual response in women that acknowledged the complexities involved in a woman’s motivation to be sexual (Figure 1). While a woman may enter the cycle with spontaneous sexual desire, she may also enter it as sexually neutral, with arousal in response to a sexual stimulus. Emotional intimacy is an important part of the cycle, and emotional closeness and bonding with the partner may provide motivation for a woman to enter into the cycle again in the future.
In a Danish survey,12 more people of both sexes said the 2 linear models described their experiences better than the circular model, but more women than men endorsed the circular model, and more men than women endorsed a linear model.
In evaluating women who complain of low sexual desire, clinicians should be aware that women, particularly those who are postmenopausal, may not enter the cycle with spontaneous sexual desire, but instead may experience arousal in response to a sexual stimulus followed by desire—ie, responsive rather than spontaneous sexual desire. Sexual arousal may precede desire, especially for women in long-term relationships, and emotional intimacy is a key driver for sexual engagement in women.11
CATEGORIES OF SEXUAL DYSFUNCTION IN WOMEN
The World Health Organization defines sexual health as “a state of physical, emotional, mental, and social well-being in relation to sexuality” and “not merely the absence of disease, dysfunction, or infirmity.”13
The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5),14 published in 2013, defines three categories of sexual dysfunction in women:
- Female sexual interest and arousal disorder
- Female sexual orgasmic disorder
- Genitopelvic pain/penetration disorder.
To meet the diagnosis of any of these, symptoms must:
- Persist for at least 6 months
- Occur in 75% to 100% of sexual encounters
- Be accompanied by personal distress
- Not be related to another psychological or medical condition, medication or substance use, or relationship distress.
Sexual problems may be lifelong or acquired after a period of normal functioning, and may be situational (present only in certain situations) or generalized (present in all situations).
Female sexual interest and arousal disorder used to be 2 separate categories in earlier editions of the DSM. Proponents of merging the 2 categories in DSM-5 cited several reasons, including difficulty in clearly distinguishing desire from other motivations for sexual activity, the relatively low reporting of fantasy in women, the complexity of distinguishing spontaneous from responsive desire, and the common co-occurrence of decreased desire and arousal difficulties.15
Other experts, however, have recommended keeping the old, separate categories of hypoactive sexual desire disorder and arousal disorder.16 The recommendation to preserve the diagnostic category of hypoactive sexual desire disorder is based on robust observational and registry data, as well as the results of randomized controlled trials that used the old criteria for hypoactive sexual desire disorder to assess responses to pharmacologic treatment of this condition.17–19 In addition, this classification as a separate and distinct diagnosis is consistent with the nomenclature used in the International Statistical Classification of Diseases and Related Health Problems, 10th Revision and was endorsed by the International Consultation on Sexual Medicine in 2015.16
HOW TO ASK ABOUT SEXUAL HEALTH
Assessment of sexual health concerns should be a part of a routine health examination, particularly after childbirth and other major medical, surgical, psychological, and life events. Women are unlikely to bring up sexual health concerns with their healthcare providers, but instead hope that their providers will bring up the topic.20
Barriers to the discussion include lack of provider education and training, patient and provider discomfort, perceived lack of time during an office visit, and lack of approved treatments.21,22 Additionally, older women are less likely than men to discuss sexual health with their providers.23 Other potential barriers to communication include negative societal attitudes about sexuality in women and in older individuals.24,25 To overcome these barriers:
Legitimize sexual health as an important health concern and normalize its discussion as part of a routine clinical health assessment. Prefacing a query about sexual health with a normalizing and universalizing statement can help: eg, “Many women going through menopause have concerns about their sexual health. Do you have any sexual problems or concerns?” Table 1 contains examples of questions to use for initial screening for sexual dysfunction.22,26
Flynn et al27 proposed a validated single-question checklist to screen for sexual dysfunction that is an efficient way to identify specific sexual concerns, guide selection of interventions, and facilitate patient-provider communication (Table 2).
Don’t judge and don’t make assumptions about sexuality and sexual practices.
Assure confidentiality.
Use simple, direct language that is appropriate for the patient’s age, ethnicity, culture, and level of health literacy.3
Take a thorough history (sexual and reproductive, medical-surgical, and psychosocial).
PHYSICAL EXAMINATION
Perform a focused physical examination to evaluate for potential causes of pain (eg, infectious causes, vulvar dermatoses, pelvic floor muscle dysfunction). The examination is also an opportunity to teach the patient about anatomy and normal sexual function.
No standard laboratory tests or imaging studies are required for the assessment of sexual dysfunction.28
IT’S NOT JUST PHYSICAL
Evaluation and treatment of female sexual dysfunction is guided by the biopsychosocial model, with potential influences from the biological, psychological, sociocultural, and interpersonal realms (Table 3).29,30
Biological factors include pelvic surgery, cancer and its treatment, neurologic diseases, and vascular diseases. Medications, including antidepressants, narcotics, anticholinergics, antihistamines, antihypertensives, oral contraceptives, and antiestrogens may also adversely affect sexual response.26
Psychological factors include a history of sexual abuse or trauma, body image concerns, distraction, stress, anxiety, depression, and personality disorders.22
Sociocultural factors include lack of sex education, unrealistic expectations, cultural norms, and religious influences.
Relationship factors include conflict with one’s partner, lack of emotional intimacy, absence of a partner, and partner sexual dysfunction. While there appears to be a close link between sexual satisfaction and a woman’s relationship with her partner in correlational studies and in clinical experience, there has been little research about relationship factors and their contribution to desire and arousal concerns.31 Sexual dysfunction in one’s partner (eg, erectile dysfunction) has been shown to negatively affect the female partner’s sexual desire.32
GENERAL APPROACH TO TREATMENT
In treating sexual health problems in women, we address contributing factors identified during the initial assessment.
A multidisciplinary approach
As sexual dysfunction in women is often multifactorial, management of the problem is well suited to a multidisciplinary approach. The team of providers may include:
- A medical provider (primary care provider, gynecologist, or sexual health specialist) to coordinate care and manage biological factors contributing to sexual dysfunction
- A physical therapist with expertise in treating pelvic floor disorders
- A psychologist to address psychological, relational, and sociocultural contributors to sexual dysfunction
- A sex therapist (womenshealthapta.org, aasect.org) to facilitate treatment of tight, tender pelvic floor muscles through education and guidance about kinesthetic awareness, muscle relaxation, and dilator therapy.33
Even in the initial visit, the primary care provider can educate, reassure regarding normal sexual function, and treat conditions such as genitourinary syndrome of menopause and antidepressant-associated sexual dysfunction. The PLISSIT model (Permission, Limited Information, Specific Suggestions, and Intensive Therapy) is a useful tool for initiating counseling about sexual health (Table 4).34
AGING VS MENOPAUSE
Aging can affect sexual function in both men and women. About 40% of women experience changes in sexual function around the menopausal transition, with common complaints being loss of sexual responsiveness and desire, sexual pain, decreased sexual activity, and partner sexual dysfunction.35 However, studies seem to show that while menopause results in hormonal changes that affect sexual function, other factors may have a greater impact.
The Study of Women’s Health Across the Nation36 found vaginal and pelvic pain and decreased sexual desire were associated with the menopausal transition, but other sexual health outcomes (frequency of sexual activities, arousal, importance of sex, emotional satisfaction, or physical pleasure) were not. Physical and psychological health, marital status, and a change in relationship were all associated with differences in sexual health.
The Massachusetts Women’s Health Study II37 found a greater association between physical and mental health, relationship status, and smoking and women’s sexual functioning than menopausal status.
The Penn Ovarian Aging Study38 found that sexual function declined across the menopausal transition. Risk factors for sexual dysfunction included postmenopausal status, anxiety, and absence of a sexual partner.
The Melbourne Women’s Midlife Health Project39 also found that sexual function declined across the menopausal transition. Sexual dysfunction with distress was associated with relationship factors and depression.37
Genitourinary syndrome of menopause and its treatment
As the ovaries shut down during menopause, estradiol levels decrease. Nearly 50% of women experience symptoms related to genitourinary syndrome of menopause (formerly called atrophic vaginitis or vulvovaginal atrophy).40,41 These symptoms include vaginal dryness and discomfort or pain with sexual activity, but menopausal hormone loss can also result in reduced genital blood flow, decreased sensory perception, and decreased sexual responsiveness.22
Estrogen is the most effective treatment for genitourinary syndrome of menopause, with low-dose vaginal preparations preferred over systemic ones for isolated vulvar and vaginal symptoms.40 While estrogen is effective for vaginal dryness and sexual pain associated with estrogen loss, replacing estrogen systemically has not been associated with improvements in sexual desire.42
DEPRESSION AND ANTIDEPRESSANT-INDUCED SEXUAL DYSFUNCTION
Depression increases the risk of sexual dysfunction, and vice versa.
A meta-analysis that included 12 studies involving almost 15,000 patients confirmed that depression increased the risk of sexual dysfunction, and sexual dysfunction increased the risk of depression.43 This interaction may be related to the overlap in affected neurotransmitters and neuroendocrine systems.44
In the Sequenced Treatment Alternatives to Relieve Depression trial, Ishak et al45 found that patients treated with a selective serotonin reuptake inhibitor (SSRI) who experienced remission of depression had a lower prevalence of impaired sexual satisfaction and much greater improvements in sexual satisfaction than did those who remained depressed. The severity of depressive symptoms predicted impairment in sexual satisfaction, which in turn predicted poorer quality of life. The authors suggested that physicians encourage patients to remain on SSRI treatment, given that improvement in depressive symptoms is likely to improve sexual satisfaction.
Antidepressant-induced sexual dysfunction
As many as 70% of patients taking an SSRI or serotonin-norepinephrine reuptake inhibitor (SNRI) experience antidepressant-induced sexual dysfunction, though this is difficult to estimate across studies of different medications due to differences in methods and because many patients only report it when directly asked about it.46
Treatment of antidepressant-induced sexual dysfunction includes not only optimal management of depression but reassessment of the antidepressant treatment. If using only nondrug treatments for the mood disorder is not feasible, switching to (or ideally, starting with) an antidepressant with fewer sexual side effects such as mirtazapine, vilazodone, or bupropion is an option.46
A drug holiday (suspending antidepressant treatment for 1 or 2 days) has been suggested as a means of treating antidepressant-induced sexual dysfunction, but this may result in poorer control of depressive symptoms and discontinuation symptoms, and it encourages medication noncompliance.46,47
Treatment with a phosphodiesterase type 5 inhibitor (eg, sildenafil) has been studied in women with antidepressant-induced sexual dysfunction, with modest results.48
A Cochrane review reported that treatment with bupropion shows promise at higher doses (300 mg daily).49
Exercise for 20 minutes 3 times weekly is associated with improvement in antidepressant-induced sexual dysfunction when the exercise is performed immediately before sexual activity.50
LOW SEXUAL DESIRE
Hypoactive sexual desire disorder is defined as persistent or recurrent deficiency or absence of sexual fantasies and desire for sexual activity associated with marked distress and not due exclusively to a medication, substance abuse, or a medical condition.
Low or decreased sexual desire is the most commonly reported sexual health concern in women of all ages, with an unadjusted prevalence of 39.7%. When the criterion of personal distress is included, the prevalence is 8.9% in women ages 18 to 44, 12.3% in women ages 45 to 64, and 7.4% in women ages 65 and older.1
Multiple biological, psychological, and social factors may contribute to the problem. Identifying the ones that are present can help in planning treatment. A multifaceted approach may be appropriate.
Mindfulness and cognitive behavioral therapy for low sexual desire
Mindfulness-based cognitive therapy is designed to improve awareness, focusing on and accepting the present moment, and directing attention away from and lessening self-criticism and evaluation of one’s sexual responsiveness.
Mindfulness-based therapy has been associated with improvements in sexual desire and associated distress.51 Similarly, the effectiveness of cognitive behavioral therapy for treating hypoactive sexual desire disorder is supported by 3 controlled trials, although concerns exist about the adequacy of these trials, and further study is needed.52
Androgen therapy in women
In randomized controlled trials in women with low sexual desire who were either naturally or surgically menopausal, sexual function improved with testosterone therapy that resulted in mostly supraphysiologic total testosterone levels (which may not reflect free testosterone levels) with or without concurrent estrogen treatment.53–57
Testosterone is not approved by the US Food and Drug Administration (FDA) for use in women, primarily because of the lack of long-term safety and efficacy data (ie, beyond 24 months). However, studies have shown no evidence of increased risk of endometrial cancer or cardiovascular disease with testosterone dosed to achieve physiologic premenopausal levels.58 Data on breast cancer risk are less clear, but observational studies over the last decade do not support an association with testosterone use in women.58 There is no clearly defined androgen deficiency syndrome in women, and androgen levels do not reliably correlate with symptoms.59
The Endocrine Society59 guidelines endorse the use of testosterone in postmenopausal women with hypoactive sexual desire disorder. They say to aim for the midnormal premenopausal range and suggest discontinuing the drug if there is no response in 6 months. They recommend checking testosterone levels at baseline, after 3 to 6 weeks of therapy, and every 6 months to monitor for excessive use, to avoid supraphysiologic dosing and to evaluate for signs of androgen excess (eg, acne, hair growth). The use of products formulated for men or those formulated by pharmacies is discouraged; however, no FDA-approved products are currently available for use in women in the United States.
Flibanserin
A postsynaptic serotonin 5-HT1A receptor agonist and 5-HT2A receptor agonist, flibanserin was approved by the FDA in 2015 for treatment of hypoactive sexual desire disorder in premenopausal women. Its mechanism of action is likely through an effect on neurotransmitters that suppresses serotonin (which has sexually inhibitory effects) and promotes dopamine and norepinephrine (which have excitatory effects).60
The efficacy of flibanserin has been demonstrated in 3 randomized controlled trials, with significant increases in the number of sexually satisfying events and in sexual desire scores and a decrease in distress associated with low sexual desire.17–19 While the increase in sexually satisfying events was modest (about 1 extra event per month), some have suggested that the frequency of sexual activity may not be the best measure of sexual function in women.61 Further, responders to this drug showed a return to near-normal premenopausal frequencies of sexual activity in a separate analysis.61
The drug is generally well tolerated, with common adverse effects being somnolence, dizziness, and fatigue.18,19 Flibanserin has been associated with orthostatic hypotension with alcohol use and carries a boxed warning highlighting this potential interaction.62 Use of this drug is contraindicated in women who drink alcohol or take medications that are moderate or strong inhibitors of CYP-3A4 (eg, some antiretroviral drugs, antihypertensive drugs, antibiotics, and fluconazole, which can increase systemic exposure to flibanserin and potential side effects), and in those with liver impairment.
SEXUAL AROUSAL DISORDERS
Female sexual arousal disorder is the persistent or recurrent inability to attain or maintain an adequate lubrication-swelling response of sexual excitement. Sexual arousal results from a complex interaction between genital response, central nervous system activity, and information processing of the sexual stimulus. Difficulty with sexual arousal can result from neurovascular or neuroendocrine dysfunction or impaired central nervous system processing.
Women may experience a mismatch between subjective and objective genital arousal. A subjective report of decreased genital arousal may not be confirmed with measurement of vaginal pulse amplitude by photoplethysmography.63 Even in postmenopausal women, in the absence of significant neurovascular or neuroendocrine dysfunction, it is likely that either contextual or relational variables resulting in inadequate sexual stimulation or cognitive inhibition are more important factors contributing to difficulty with sexual arousal.63
Although there are no standard recommendations for evaluation of arousal disorders and advanced testing is often unnecessary, nerve function can be assessed with genital sensory testing utilizing thermal and vibratory threshholds64; vaginal blood flow can be assessed with vaginal photoplethysmography63; and imaging of the spine and pelvis can help to rule out neurovascular pathology.
Treatment of arousal disorders
As with other forms of female sexual dysfunction, treatment of arousal disorders includes addressing contributing factors.
Although there are few data from randomized controlled trials, psychological treatments such as sensate focus exercises and masturbation training have been suggested, centered on women becoming more self-focused and assertive.31 Sensate focus exercises are a series of graded, nondemand, sensual touching exercises aimed at reducing anxiety and avoidance of sexual activity, and improving sexual communication and intimacy by the gradual reintroduction of sexual activity.65 More recently, mindfulness-based cognitive therapy has been associated with improvements in sexual arousal as well as other parameters of sexual function.51
Currently, no pharmacologic treatments are recommended for arousal disorders because of a lack of evidence of efficacy and because of adverse effects.31
ORGASMIC DISORDER
Female orgasmic disorder is the marked delay, marked infrequency, or absence of orgasm, or markedly reduced intensity of orgasm.
Important considerations in evaluating orgasm disorders include psychosocial factors (eg, lack of sex education, negative feelings about sex, religiosity), psychological factors (eg, anxiety, depression, body image concerns), relational factors (eg, communication issues, lack of emotional intimacy, partner sexual dysfunction), adverse childhood or adult experiences (eg, physical, sexual, or emotional or verbal abuse), medical history (pelvic surgery, neurologic, or vascular disease) and medications (eg, SSRIs, SNRIs, and antipsychotic medications).66
Treatment of orgasmic disorder
Involving the partner in treatment is important, particularly if the difficulty with orgasm is acquired and only occurs with sex with a partner. Using the PLISSIT model to provide targeted, office-based interventions can be helpful.
Behavioral therapies such as directed masturbation, sensate focus exercises, or a combination of these have been shown to be effective, as has coital alignment during intercourse (positioning of male partner with his pelvis above the pubic bone of his partner to maximize clitoral stimulation with penile penetration).66
Hormonal therapy may be useful in postmenopausal women. However, there are no data on it for women whose primary complaint is female orgasmic disorder, and further study is needed.66
SEXUAL PAIN DISORDERS
The DSM-5 describes genitopelvic pain/penetration disorder as fear or anxiety, marked tightening or tensing of abdominal and pelvic muscles, or actual pain with vaginal penetration that is recurrent or persistent for a minimum of 6 months.14 Pain may occur with initial penetration, with deeper thrusting, or both.
Although the DSM-5 definition focuses on pain with penetration, it is important to recognize and ask about noncoital sexual pain. Women may also present with persistent vulvar pain or pain at the vulvar vestibule with provocation, (eg, sexual activity, tampon insertion, sitting), also known as provoked vestibulodynia.
Assessment of vaginal and vulvar pain includes a directed history and physical examination aimed at identifying potential etiologies or contributing factors, including infectious, inflammatory, neoplastic, neurologic, traumatic, iatrogenic, or factors related to hormonal deficiency.67
Treatment of sexual pain
Removal of offending agents is a first step. This includes a thorough review of vulvar and vaginal hygiene practices and emphasis on avoiding the use of any product containing potential irritants (eg, soaps or detergents containing perfumes or dyes) and using lubricants and moisturizers without gimmicks (no warming or tingling agents or flavors). Oral contraceptives have been associated with vestibulodynia, and women in whom the sexual pain started when they started an oral contraceptive may benefit from switching to an alternate form of contraception.68
Dysfunction of pelvic floor muscles may result in sexual pain and may be a primary problem or a secondary complication related to other issues such as symptomatic genitourinary syndrome of menopause. The symptoms of nonrelaxing pelvic floor dysfunction (also known as hypertonic pelvic floor dysfunction or pelvic floor tension myalgia) include pain in the pelvis with sexual activity that may linger for hours or even days, and may also include bowel and bladder dysfunction and low back pain or hip pain radiating to the thighs or groin.33 Physical therapy under the care of a physical therapist with expertise in the management of pelvic floor disorders is the cornerstone of treatment for this condition.33
Treatment of the genital and urinary symptoms related to loss of estrogen after menopause (genitourinary syndrome of menopause) includes the use of vaginal lubricants with sexual activity and vaginal moisturizers on a regular basis (2 to 5 times per week).40 Low-dose vaginal estrogen creams, rings, or tablets and the oral selective estrogen receptor antagonist ospemifene are recommended for moderate to severe symptoms of genitourinary syndrome of menopause.40 Intravaginal dehydroepiandrosterone was recently approved by the FDA for treatment of dyspareunia associated with menopause.69 Topical lidocaine applied to the introitus before sexual activity has been found to be effective for reducing sexual pain in women with breast cancer, and when used in combination with vaginal lubricants and moisturizers is a practical option for women, particularly those unable to use estrogen-based therapies.70
Many women experience some form of sexual dysfunction, be it lack of desire, lack of arousal, failure to achieve orgasm, or pain during sexual activity.
Sexual health may be difficult to discuss, for both the patient and the provider. Here, we describe how primary care physicians can approach this topic, assess potential problems, and begin treatment.
A COMMON PROBLEM
The age-adjusted prevalence of sexual dysfunction in US women was reported at 44% in the Prevalence of Female Sexual Problems Associated With Distress and Determinants of Treatment Seeking (PRESIDE) study,1 but the prevalence of distress associated with sexual dysfunction was 12%. The most common type of sexual dysfunction reported by women was low sexual desire, a finding consistent with that of another large population-based study.2
While the prevalence of any type of sexual dysfunction was highest in women over age 65,1 the prevalence of distress was lowest in this age group and highest in midlife between the ages of 45 and 65. The diagnostic criteria require both a problem and distress over the problem.
Sexual dysfunction negatively affects quality of life and emotional health, regardless of age.3
LIFESTYLE AND SEXUAL FUNCTION
Various lifestyle factors have been linked to either more or less sexual activity. For example, a Mediterranean diet was associated with increased sexual activity, as were social activity, social support, psychological well-being, self-reported good quality of life, moderate alcohol intake, absence of tobacco use, a normal body mass index, and exercise.4–6 A higher sense of purpose in life has been associated with greater sexual enjoyment.7
Conversely, sexual inactivity has been associated with alcohol misuse, an elevated body mass index, and somatization.4–6
SEXUAL RESPONSE: LINEAR OR CIRCULAR?
Masters and Johnson8 initially proposed a linear model of human sexual response, which Kaplan later modified to include desire and applied to both men and women.9,10 This model presumed that sexual response begins with spontaneous sexual desire, followed by arousal, and then (sometimes) orgasm and resolution.
In 2000, Basson11 proposed a circular, intimacy-based model of sexual response in women that acknowledged the complexities involved in a woman’s motivation to be sexual (Figure 1). While a woman may enter the cycle with spontaneous sexual desire, she may also enter it as sexually neutral, with arousal in response to a sexual stimulus. Emotional intimacy is an important part of the cycle, and emotional closeness and bonding with the partner may provide motivation for a woman to enter into the cycle again in the future.
In a Danish survey,12 more people of both sexes said the 2 linear models described their experiences better than the circular model, but more women than men endorsed the circular model, and more men than women endorsed a linear model.
In evaluating women who complain of low sexual desire, clinicians should be aware that women, particularly those who are postmenopausal, may not enter the cycle with spontaneous sexual desire, but instead may experience arousal in response to a sexual stimulus followed by desire—ie, responsive rather than spontaneous sexual desire. Sexual arousal may precede desire, especially for women in long-term relationships, and emotional intimacy is a key driver for sexual engagement in women.11
CATEGORIES OF SEXUAL DYSFUNCTION IN WOMEN
The World Health Organization defines sexual health as “a state of physical, emotional, mental, and social well-being in relation to sexuality” and “not merely the absence of disease, dysfunction, or infirmity.”13
The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5),14 published in 2013, defines three categories of sexual dysfunction in women:
- Female sexual interest and arousal disorder
- Female sexual orgasmic disorder
- Genitopelvic pain/penetration disorder.
To meet the diagnosis of any of these, symptoms must:
- Persist for at least 6 months
- Occur in 75% to 100% of sexual encounters
- Be accompanied by personal distress
- Not be related to another psychological or medical condition, medication or substance use, or relationship distress.
Sexual problems may be lifelong or acquired after a period of normal functioning, and may be situational (present only in certain situations) or generalized (present in all situations).
Female sexual interest and arousal disorder used to be 2 separate categories in earlier editions of the DSM. Proponents of merging the 2 categories in DSM-5 cited several reasons, including difficulty in clearly distinguishing desire from other motivations for sexual activity, the relatively low reporting of fantasy in women, the complexity of distinguishing spontaneous from responsive desire, and the common co-occurrence of decreased desire and arousal difficulties.15
Other experts, however, have recommended keeping the old, separate categories of hypoactive sexual desire disorder and arousal disorder.16 The recommendation to preserve the diagnostic category of hypoactive sexual desire disorder is based on robust observational and registry data, as well as the results of randomized controlled trials that used the old criteria for hypoactive sexual desire disorder to assess responses to pharmacologic treatment of this condition.17–19 In addition, this classification as a separate and distinct diagnosis is consistent with the nomenclature used in the International Statistical Classification of Diseases and Related Health Problems, 10th Revision and was endorsed by the International Consultation on Sexual Medicine in 2015.16
HOW TO ASK ABOUT SEXUAL HEALTH
Assessment of sexual health concerns should be a part of a routine health examination, particularly after childbirth and other major medical, surgical, psychological, and life events. Women are unlikely to bring up sexual health concerns with their healthcare providers, but instead hope that their providers will bring up the topic.20
Barriers to the discussion include lack of provider education and training, patient and provider discomfort, perceived lack of time during an office visit, and lack of approved treatments.21,22 Additionally, older women are less likely than men to discuss sexual health with their providers.23 Other potential barriers to communication include negative societal attitudes about sexuality in women and in older individuals.24,25 To overcome these barriers:
Legitimize sexual health as an important health concern and normalize its discussion as part of a routine clinical health assessment. Prefacing a query about sexual health with a normalizing and universalizing statement can help: eg, “Many women going through menopause have concerns about their sexual health. Do you have any sexual problems or concerns?” Table 1 contains examples of questions to use for initial screening for sexual dysfunction.22,26
Flynn et al27 proposed a validated single-question checklist to screen for sexual dysfunction that is an efficient way to identify specific sexual concerns, guide selection of interventions, and facilitate patient-provider communication (Table 2).
Don’t judge and don’t make assumptions about sexuality and sexual practices.
Assure confidentiality.
Use simple, direct language that is appropriate for the patient’s age, ethnicity, culture, and level of health literacy.3
Take a thorough history (sexual and reproductive, medical-surgical, and psychosocial).
PHYSICAL EXAMINATION
Perform a focused physical examination to evaluate for potential causes of pain (eg, infectious causes, vulvar dermatoses, pelvic floor muscle dysfunction). The examination is also an opportunity to teach the patient about anatomy and normal sexual function.
No standard laboratory tests or imaging studies are required for the assessment of sexual dysfunction.28
IT’S NOT JUST PHYSICAL
Evaluation and treatment of female sexual dysfunction is guided by the biopsychosocial model, with potential influences from the biological, psychological, sociocultural, and interpersonal realms (Table 3).29,30
Biological factors include pelvic surgery, cancer and its treatment, neurologic diseases, and vascular diseases. Medications, including antidepressants, narcotics, anticholinergics, antihistamines, antihypertensives, oral contraceptives, and antiestrogens may also adversely affect sexual response.26
Psychological factors include a history of sexual abuse or trauma, body image concerns, distraction, stress, anxiety, depression, and personality disorders.22
Sociocultural factors include lack of sex education, unrealistic expectations, cultural norms, and religious influences.
Relationship factors include conflict with one’s partner, lack of emotional intimacy, absence of a partner, and partner sexual dysfunction. While there appears to be a close link between sexual satisfaction and a woman’s relationship with her partner in correlational studies and in clinical experience, there has been little research about relationship factors and their contribution to desire and arousal concerns.31 Sexual dysfunction in one’s partner (eg, erectile dysfunction) has been shown to negatively affect the female partner’s sexual desire.32
GENERAL APPROACH TO TREATMENT
In treating sexual health problems in women, we address contributing factors identified during the initial assessment.
A multidisciplinary approach
As sexual dysfunction in women is often multifactorial, management of the problem is well suited to a multidisciplinary approach. The team of providers may include:
- A medical provider (primary care provider, gynecologist, or sexual health specialist) to coordinate care and manage biological factors contributing to sexual dysfunction
- A physical therapist with expertise in treating pelvic floor disorders
- A psychologist to address psychological, relational, and sociocultural contributors to sexual dysfunction
- A sex therapist (womenshealthapta.org, aasect.org) to facilitate treatment of tight, tender pelvic floor muscles through education and guidance about kinesthetic awareness, muscle relaxation, and dilator therapy.33
Even in the initial visit, the primary care provider can educate, reassure regarding normal sexual function, and treat conditions such as genitourinary syndrome of menopause and antidepressant-associated sexual dysfunction. The PLISSIT model (Permission, Limited Information, Specific Suggestions, and Intensive Therapy) is a useful tool for initiating counseling about sexual health (Table 4).34
AGING VS MENOPAUSE
Aging can affect sexual function in both men and women. About 40% of women experience changes in sexual function around the menopausal transition, with common complaints being loss of sexual responsiveness and desire, sexual pain, decreased sexual activity, and partner sexual dysfunction.35 However, studies seem to show that while menopause results in hormonal changes that affect sexual function, other factors may have a greater impact.
The Study of Women’s Health Across the Nation36 found vaginal and pelvic pain and decreased sexual desire were associated with the menopausal transition, but other sexual health outcomes (frequency of sexual activities, arousal, importance of sex, emotional satisfaction, or physical pleasure) were not. Physical and psychological health, marital status, and a change in relationship were all associated with differences in sexual health.
The Massachusetts Women’s Health Study II37 found a greater association between physical and mental health, relationship status, and smoking and women’s sexual functioning than menopausal status.
The Penn Ovarian Aging Study38 found that sexual function declined across the menopausal transition. Risk factors for sexual dysfunction included postmenopausal status, anxiety, and absence of a sexual partner.
The Melbourne Women’s Midlife Health Project39 also found that sexual function declined across the menopausal transition. Sexual dysfunction with distress was associated with relationship factors and depression.37
Genitourinary syndrome of menopause and its treatment
As the ovaries shut down during menopause, estradiol levels decrease. Nearly 50% of women experience symptoms related to genitourinary syndrome of menopause (formerly called atrophic vaginitis or vulvovaginal atrophy).40,41 These symptoms include vaginal dryness and discomfort or pain with sexual activity, but menopausal hormone loss can also result in reduced genital blood flow, decreased sensory perception, and decreased sexual responsiveness.22
Estrogen is the most effective treatment for genitourinary syndrome of menopause, with low-dose vaginal preparations preferred over systemic ones for isolated vulvar and vaginal symptoms.40 While estrogen is effective for vaginal dryness and sexual pain associated with estrogen loss, replacing estrogen systemically has not been associated with improvements in sexual desire.42
DEPRESSION AND ANTIDEPRESSANT-INDUCED SEXUAL DYSFUNCTION
Depression increases the risk of sexual dysfunction, and vice versa.
A meta-analysis that included 12 studies involving almost 15,000 patients confirmed that depression increased the risk of sexual dysfunction, and sexual dysfunction increased the risk of depression.43 This interaction may be related to the overlap in affected neurotransmitters and neuroendocrine systems.44
In the Sequenced Treatment Alternatives to Relieve Depression trial, Ishak et al45 found that patients treated with a selective serotonin reuptake inhibitor (SSRI) who experienced remission of depression had a lower prevalence of impaired sexual satisfaction and much greater improvements in sexual satisfaction than did those who remained depressed. The severity of depressive symptoms predicted impairment in sexual satisfaction, which in turn predicted poorer quality of life. The authors suggested that physicians encourage patients to remain on SSRI treatment, given that improvement in depressive symptoms is likely to improve sexual satisfaction.
Antidepressant-induced sexual dysfunction
As many as 70% of patients taking an SSRI or serotonin-norepinephrine reuptake inhibitor (SNRI) experience antidepressant-induced sexual dysfunction, though this is difficult to estimate across studies of different medications due to differences in methods and because many patients only report it when directly asked about it.46
Treatment of antidepressant-induced sexual dysfunction includes not only optimal management of depression but reassessment of the antidepressant treatment. If using only nondrug treatments for the mood disorder is not feasible, switching to (or ideally, starting with) an antidepressant with fewer sexual side effects such as mirtazapine, vilazodone, or bupropion is an option.46
A drug holiday (suspending antidepressant treatment for 1 or 2 days) has been suggested as a means of treating antidepressant-induced sexual dysfunction, but this may result in poorer control of depressive symptoms and discontinuation symptoms, and it encourages medication noncompliance.46,47
Treatment with a phosphodiesterase type 5 inhibitor (eg, sildenafil) has been studied in women with antidepressant-induced sexual dysfunction, with modest results.48
A Cochrane review reported that treatment with bupropion shows promise at higher doses (300 mg daily).49
Exercise for 20 minutes 3 times weekly is associated with improvement in antidepressant-induced sexual dysfunction when the exercise is performed immediately before sexual activity.50
LOW SEXUAL DESIRE
Hypoactive sexual desire disorder is defined as persistent or recurrent deficiency or absence of sexual fantasies and desire for sexual activity associated with marked distress and not due exclusively to a medication, substance abuse, or a medical condition.
Low or decreased sexual desire is the most commonly reported sexual health concern in women of all ages, with an unadjusted prevalence of 39.7%. When the criterion of personal distress is included, the prevalence is 8.9% in women ages 18 to 44, 12.3% in women ages 45 to 64, and 7.4% in women ages 65 and older.1
Multiple biological, psychological, and social factors may contribute to the problem. Identifying the ones that are present can help in planning treatment. A multifaceted approach may be appropriate.
Mindfulness and cognitive behavioral therapy for low sexual desire
Mindfulness-based cognitive therapy is designed to improve awareness, focusing on and accepting the present moment, and directing attention away from and lessening self-criticism and evaluation of one’s sexual responsiveness.
Mindfulness-based therapy has been associated with improvements in sexual desire and associated distress.51 Similarly, the effectiveness of cognitive behavioral therapy for treating hypoactive sexual desire disorder is supported by 3 controlled trials, although concerns exist about the adequacy of these trials, and further study is needed.52
Androgen therapy in women
In randomized controlled trials in women with low sexual desire who were either naturally or surgically menopausal, sexual function improved with testosterone therapy that resulted in mostly supraphysiologic total testosterone levels (which may not reflect free testosterone levels) with or without concurrent estrogen treatment.53–57
Testosterone is not approved by the US Food and Drug Administration (FDA) for use in women, primarily because of the lack of long-term safety and efficacy data (ie, beyond 24 months). However, studies have shown no evidence of increased risk of endometrial cancer or cardiovascular disease with testosterone dosed to achieve physiologic premenopausal levels.58 Data on breast cancer risk are less clear, but observational studies over the last decade do not support an association with testosterone use in women.58 There is no clearly defined androgen deficiency syndrome in women, and androgen levels do not reliably correlate with symptoms.59
The Endocrine Society59 guidelines endorse the use of testosterone in postmenopausal women with hypoactive sexual desire disorder. They say to aim for the midnormal premenopausal range and suggest discontinuing the drug if there is no response in 6 months. They recommend checking testosterone levels at baseline, after 3 to 6 weeks of therapy, and every 6 months to monitor for excessive use, to avoid supraphysiologic dosing and to evaluate for signs of androgen excess (eg, acne, hair growth). The use of products formulated for men or those formulated by pharmacies is discouraged; however, no FDA-approved products are currently available for use in women in the United States.
Flibanserin
A postsynaptic serotonin 5-HT1A receptor agonist and 5-HT2A receptor agonist, flibanserin was approved by the FDA in 2015 for treatment of hypoactive sexual desire disorder in premenopausal women. Its mechanism of action is likely through an effect on neurotransmitters that suppresses serotonin (which has sexually inhibitory effects) and promotes dopamine and norepinephrine (which have excitatory effects).60
The efficacy of flibanserin has been demonstrated in 3 randomized controlled trials, with significant increases in the number of sexually satisfying events and in sexual desire scores and a decrease in distress associated with low sexual desire.17–19 While the increase in sexually satisfying events was modest (about 1 extra event per month), some have suggested that the frequency of sexual activity may not be the best measure of sexual function in women.61 Further, responders to this drug showed a return to near-normal premenopausal frequencies of sexual activity in a separate analysis.61
The drug is generally well tolerated, with common adverse effects being somnolence, dizziness, and fatigue.18,19 Flibanserin has been associated with orthostatic hypotension with alcohol use and carries a boxed warning highlighting this potential interaction.62 Use of this drug is contraindicated in women who drink alcohol or take medications that are moderate or strong inhibitors of CYP-3A4 (eg, some antiretroviral drugs, antihypertensive drugs, antibiotics, and fluconazole, which can increase systemic exposure to flibanserin and potential side effects), and in those with liver impairment.
SEXUAL AROUSAL DISORDERS
Female sexual arousal disorder is the persistent or recurrent inability to attain or maintain an adequate lubrication-swelling response of sexual excitement. Sexual arousal results from a complex interaction between genital response, central nervous system activity, and information processing of the sexual stimulus. Difficulty with sexual arousal can result from neurovascular or neuroendocrine dysfunction or impaired central nervous system processing.
Women may experience a mismatch between subjective and objective genital arousal. A subjective report of decreased genital arousal may not be confirmed with measurement of vaginal pulse amplitude by photoplethysmography.63 Even in postmenopausal women, in the absence of significant neurovascular or neuroendocrine dysfunction, it is likely that either contextual or relational variables resulting in inadequate sexual stimulation or cognitive inhibition are more important factors contributing to difficulty with sexual arousal.63
Although there are no standard recommendations for evaluation of arousal disorders and advanced testing is often unnecessary, nerve function can be assessed with genital sensory testing utilizing thermal and vibratory threshholds64; vaginal blood flow can be assessed with vaginal photoplethysmography63; and imaging of the spine and pelvis can help to rule out neurovascular pathology.
Treatment of arousal disorders
As with other forms of female sexual dysfunction, treatment of arousal disorders includes addressing contributing factors.
Although there are few data from randomized controlled trials, psychological treatments such as sensate focus exercises and masturbation training have been suggested, centered on women becoming more self-focused and assertive.31 Sensate focus exercises are a series of graded, nondemand, sensual touching exercises aimed at reducing anxiety and avoidance of sexual activity, and improving sexual communication and intimacy by the gradual reintroduction of sexual activity.65 More recently, mindfulness-based cognitive therapy has been associated with improvements in sexual arousal as well as other parameters of sexual function.51
Currently, no pharmacologic treatments are recommended for arousal disorders because of a lack of evidence of efficacy and because of adverse effects.31
ORGASMIC DISORDER
Female orgasmic disorder is the marked delay, marked infrequency, or absence of orgasm, or markedly reduced intensity of orgasm.
Important considerations in evaluating orgasm disorders include psychosocial factors (eg, lack of sex education, negative feelings about sex, religiosity), psychological factors (eg, anxiety, depression, body image concerns), relational factors (eg, communication issues, lack of emotional intimacy, partner sexual dysfunction), adverse childhood or adult experiences (eg, physical, sexual, or emotional or verbal abuse), medical history (pelvic surgery, neurologic, or vascular disease) and medications (eg, SSRIs, SNRIs, and antipsychotic medications).66
Treatment of orgasmic disorder
Involving the partner in treatment is important, particularly if the difficulty with orgasm is acquired and only occurs with sex with a partner. Using the PLISSIT model to provide targeted, office-based interventions can be helpful.
Behavioral therapies such as directed masturbation, sensate focus exercises, or a combination of these have been shown to be effective, as has coital alignment during intercourse (positioning of male partner with his pelvis above the pubic bone of his partner to maximize clitoral stimulation with penile penetration).66
Hormonal therapy may be useful in postmenopausal women. However, there are no data on it for women whose primary complaint is female orgasmic disorder, and further study is needed.66
SEXUAL PAIN DISORDERS
The DSM-5 describes genitopelvic pain/penetration disorder as fear or anxiety, marked tightening or tensing of abdominal and pelvic muscles, or actual pain with vaginal penetration that is recurrent or persistent for a minimum of 6 months.14 Pain may occur with initial penetration, with deeper thrusting, or both.
Although the DSM-5 definition focuses on pain with penetration, it is important to recognize and ask about noncoital sexual pain. Women may also present with persistent vulvar pain or pain at the vulvar vestibule with provocation, (eg, sexual activity, tampon insertion, sitting), also known as provoked vestibulodynia.
Assessment of vaginal and vulvar pain includes a directed history and physical examination aimed at identifying potential etiologies or contributing factors, including infectious, inflammatory, neoplastic, neurologic, traumatic, iatrogenic, or factors related to hormonal deficiency.67
Treatment of sexual pain
Removal of offending agents is a first step. This includes a thorough review of vulvar and vaginal hygiene practices and emphasis on avoiding the use of any product containing potential irritants (eg, soaps or detergents containing perfumes or dyes) and using lubricants and moisturizers without gimmicks (no warming or tingling agents or flavors). Oral contraceptives have been associated with vestibulodynia, and women in whom the sexual pain started when they started an oral contraceptive may benefit from switching to an alternate form of contraception.68
Dysfunction of pelvic floor muscles may result in sexual pain and may be a primary problem or a secondary complication related to other issues such as symptomatic genitourinary syndrome of menopause. The symptoms of nonrelaxing pelvic floor dysfunction (also known as hypertonic pelvic floor dysfunction or pelvic floor tension myalgia) include pain in the pelvis with sexual activity that may linger for hours or even days, and may also include bowel and bladder dysfunction and low back pain or hip pain radiating to the thighs or groin.33 Physical therapy under the care of a physical therapist with expertise in the management of pelvic floor disorders is the cornerstone of treatment for this condition.33
Treatment of the genital and urinary symptoms related to loss of estrogen after menopause (genitourinary syndrome of menopause) includes the use of vaginal lubricants with sexual activity and vaginal moisturizers on a regular basis (2 to 5 times per week).40 Low-dose vaginal estrogen creams, rings, or tablets and the oral selective estrogen receptor antagonist ospemifene are recommended for moderate to severe symptoms of genitourinary syndrome of menopause.40 Intravaginal dehydroepiandrosterone was recently approved by the FDA for treatment of dyspareunia associated with menopause.69 Topical lidocaine applied to the introitus before sexual activity has been found to be effective for reducing sexual pain in women with breast cancer, and when used in combination with vaginal lubricants and moisturizers is a practical option for women, particularly those unable to use estrogen-based therapies.70
- Shifren JL, Monz BU, Russo PA, Segreti A, Johannes CB. Sexual problems and distress in United States women: prevalence and correlates. Obstet Gynecol 2008; 112:970–978.
- Laumann EO, Paik A, Rosen RC. Sexual dysfunction in the United States: prevalence and predictors. JAMA 1999; 281:537–544.
- Sadovsky R, Nusbaum M. Sexual health inquiry and support is a primary care priority. J Sex Med 2006; 3:3–11.
- Alvisi S, Baldassarre M, Lambertini M, et al. Sexuality and psychopathological aspects in premenopausal women with metabolic syndrome. J Sex Med 2014; 11:2020–2028.
- Bach LE, Mortimer JA, VandeWeerd C, Corvin J. The association of physical and mental health with sexual activity in older adults in a retirement community. J Sex Med 2013; 10:2671–2678.
- Esposito K, Ciotola M, Giugliano F, et al. Mediterranean diet improves sexual function in women with the metabolic syndrome. Int J Impot Res 2007; 19:486–491.
- Prairie BA, Scheier MF, Matthews KA, Chang CC, Hess R. A higher sense of purpose in life is associated with sexual enjoyment in midlife women. Menopause 2011; 18:839–844.
- Masters WH, Johnson VE. Human sexual response. Boston, MA: Little, Brown and Company; 1966.
- Kaplan HS. The new sex therapy. New York, NY: Brunner/Mazel; 1974.
- Robinson PA. The modernization of sex: Havelock Ellis, Alfred Kinsey, William Masters, and Virginia Johnson. New York, NY: Harper & Row; 1976.
- Basson R. The female sexual response: a different model. J Sex Marital Ther 2000; 26:51–65.
- Giraldi A, Kristensen E, Sand M. Endorsement of models describing sexual response of men and women with a sexual partner: an online survey in a population sample of Danish adults ages 20–65 years. J Sex Med 2015; 12:116–128.
- World Health Organization. Defining sexual health. Report of a technical consultation on sexual health, 28–31 January 2002, Geneva. Geneva; 2006.
- American Psychiatric Association (APA). DSM-5: Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association Publishing; 2013.
- Brotto LA. The DSM diagnostic criteria for hypoactive sexual desire disorder in women. Arch Sex Behav 2010; 39:221–239.
- McCabe MP, Sharlip ID, Atalla E, et al. Definitions of sexual dysfunctions in women and men: a consensus statement from the Fourth International Consultation on Sexual Medicine 2015. J Sex Med 2016; 13:135–143.
- Derogatis LR, Komer L, Katz M, et al; VIOLET Trial Investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the VIOLET study. J Sex Med 2012; 9:1074–1085.
- Katz M, DeRogatis LR, Ackerman R, et al; BEGONIA trial investigators. Efficacy of flibanserin in women with hypoactive sexual desire disorder: results from the BEGONIA trial. J Sex Med 2013; 10:1807–1815.
- Thorp J, Simon J, Dattani D, et al; DAISY trial investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the DAISY study. J Sex Med 2012; 9:793–804.
- Berman L, Berman J, Felder S, et al. Seeking help for sexual function complaints: what gynecologists need to know about the female patient's experience. Fertil Steril 2003; 79:572–576.
- Kingsberg SA. Taking a sexual history. Obstet Gynecol Clin North Am 2006; 33:535–547.
- Kingsberg SA, Rezaee RL. Hypoactive sexual desire in women. Menopause 2013; 20:1284–1300.
- Lindau ST, Schumm LP, Laumann EO, Levinson W, O’Muircheartaigh CA, Waite LJ. A study of sexuality and health among older adults in the United States. N Engl J Med 2007; 357:762–774.
- Gott M, Hinchliff S, Galena E. General practitioner attitudes to discussing sexual health issues with older people. Soc Sci Med 2004; 58:2093–2103.
- Lindau ST, Leitsch SA, Lundberg KL, Jerome J. Older women's attitudes, behavior, and communication about sex and HIV: a community-based study. J Womens Health (Larchmt) 2006; 15:747–753.
- Faubion SS, Rullo JE. Sexual dysfunction in women: a practical approach. Am Fam Physician 2015; 92:281–288.
- Flynn KE, Lindau ST, Lin L, et al. Development and validation of a single-item screener for self-reporting sexual problems in US adults. J Gen Intern Med 2015; 30:1468–1475.
- Latif EZ, Diamond MP. Arriving at the diagnosis of female sexual dysfunction. Fertil Steril 2013; 100:898–904.
- Althof SE, Leiblum SR, Chevret-Measson M, et al. Psychological and interpersonal dimensions of sexual function and dysfunction. J Sex Med 2005; 2:793–800.
- Rosen RC, Barsky JL. Normal sexual response in women. Obstet Gynecol Clin North Am 2006; 33:515–526.
- Brotto LA, Bitzer J, Laan E, Leiblum S, Luria M. Women's sexual desire and arousal disorders. J Sex Med 2010; 7:586–614.
- Rubio-Aurioles E, Kim ED, Rosen RC, et al. Impact on erectile function and sexual quality of life of couples: a double-blind, randomized, placebo-controlled trial of tadalafil taken once daily. J Sex Med 2009; 6:1314–1323.
- Faubion SS, Shuster LT, Bharucha AE. Recognition and management of nonrelaxing pelvic floor dysfunction. Mayo Clin Proc 2012; 87:187–193.
- Annon JS. The PLISSIT model: a proposed conceptual scheme for the behavioral treatment of sexual problems. J Sex Educ Ther 1976; 2:1–15.
- Sarrel PM. Sexuality and menopause. Obstet Gynecol 1990; 75(suppl 4):26S–30S; discussion 31S–35S.
- Avis NE, Brockwell S, Randolph JF, Jr, et al. Longitudinal changes in sexual functioning as women transition through menopause: results from the Study of Women's Health Across the Nation. Menopause 2009; 16:442–452.
- Avis NE, Stellato R, Crawford S, Johannes C, Longcope C. Is there an association between menopause status and sexual functioning? Menopause 2000; 7:297–309.
- Gracia CR, Freeman EW, Sammel MD, Lin H, Mogul M. Hormones and sexuality during transition to menopause. Obstet Gynecol 2007; 109:831–840.
- Dennerstein L, Guthrie JR, Hayes RD, DeRogatis LR, Lehert P. Sexual function, dysfunction, and sexual distress in a prospective, population-based sample of mid-aged, Australian-born women. J Sex Med 2008; 5:2291–2299.
- Management of symptomatic vulvovaginal atrophy: 2013 position statement of The North American Menopause Society. Menopause 2013; 20:888–902.
- Mac Bride MB, Rhodes DJ, Shuster LT. Vulvovaginal atrophy. Mayo Clin Proc 2010; 85:87–94.
- Nastri CO, Lara LA, Ferriani RA, Rosa ESAC, Figueiredo JB, Martins WP. Hormone therapy for sexual function in perimenopausal and postmenopausal women. Cochrane Database Syst Rev 2013; 6:CD009672.
- Atlantis E, Sullivan T. Bidirectional association between depression and sexual dysfunction: a systematic review and meta-analysis. J Sex Med 2012; 9:1497–1507.
- Clayton AH, Maserejian NN, Connor MK, Huang L, Heiman JR, Rosen RC. Depression in premenopausal women with HSDD: baseline findings from the HSDD Registry for Women. Psychosom Med 2012; 74:305–311.
- Ishak WW, Christensen S, Sayer G, et al. Sexual satisfaction and quality of life in major depressive disorder before and after treatment with citalopram in the STAR*D study. J Clin Psychiatry 2013; 74:256–261.
- Clayton AH, Croft HA, Handiwala L. Antidepressants and sexual dysfunction: mechanisms and clinical implications. Postgrad Med 2014; 126:91–99.
- Rothschild AJ. Selective serotonin reuptake inhibitor-induced sexual dysfunction: efficacy of a drug holiday. Am J Psychiatry 1995; 152:1514–1516.
- Nurnberg HG, Hensley PL, Heiman JR, Croft HA, Debattista C, Paine S. Sildenafil treatment of women with antidepressant-associated sexual dysfunction: a randomized controlled trial. JAMA 2008; 300:395–404.
- Taylor MJ, Rudkin L, Bullemor-Day P, Lubin J, Chukwujekwu C, Hawton K. Strategies for managing sexual dysfunction induced by antidepressant medication. Cochrane Database Syst Rev 2013; 5:CD003382.
- Lorenz TA, Meston CM. Exercise improves sexual function in women taking antidepressants: results from a randomized crossover trial. Depress Anxiety 2014; 31:188–195.
- Brotto LA, Basson R. Group mindfulness-based therapy significantly improves sexual desire in women. Behav Res Ther 2014; 5743–5754.
- Pyke RE, Clayton AH. Psychological treatment trials for hypoactive sexual desire disorder: a sexual medicine critique and perspective. J Sex Med 2015; 12:2451–2458.
- Cappelletti M, Wallen K. Increasing women's sexual desire: the comparative effectiveness of estrogens and androgens. Horm Behav 2016; 78:178–193.
- Davis SR, Moreau M, Kroll R, et al. Testosterone for low libido in postmenopausal women not taking estrogen. N Engl J Med 2008; 359:2005–2017.
- Davis SR, van der Mooren MJ, van Lunsen RH, et al. Efficacy and safety of a testosterone patch for the treatment of hypoactive sexual desire disorder in surgically menopausal women: a randomized, placebo-controlled trial. Menopause 2006; 13:387–396.
- Panay N, Al-Azzawi F, Bouchard C, et al. Testosterone treatment of HSDD in naturally menopausal women: the ADORE study. Climacteric 2010; 13:121–131.
- Somboonporn W, Davis S, Seif MW, Bell R. Testosterone for peri- and postmenopausal women. Cochrane Database Syst Rev 2005; 4:CD004509.
- Davis SR. Cardiovascular and cancer safety of testosterone in women. Curr Opin Endocrinol Diabetes Obes 2011; 18:198–203.
- Wierman ME, Arlt W, Basson R, et al. Androgen therapy in women: a reappraisal: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2014; 99:3489–3510.
- Stahl SM, Sommer B, Allers KA. Multifunctional pharmacology of flibanserin: possible mechanism of therapeutic action in hypoactive sexual desire disorder. J Sex Med 2011; 8:15–27.
- Simon JA, Goldstein I, Kim N, Freedman M, Parish S. Flibanserin approval: facts or feelings? Sex Med 2016; 4:e69–e70.
- Joffe HV, Chang C, Sewell C, et al. FDA approval of flibanserin—treating hypoactive sexual desire disorder. N Engl J Med 2016; 374:101–104.
- van Lunsen RH, Laan E. Genital vascular responsiveness and sexual feelings in midlife women: psychophysiologic, brain, and genital imaging studies. Menopause 2004; 11:741–748.
- Beco J, Seidel L, Albert A. Normative values of skin temperature and thermal sensory thresholds in the pudendal nerve territory. Neurourol Urodyn 2015; 34:571–577.
- Masters WH, Johnson VE. Human sexual inadequacy. Boston, MA: Little, Brown and Company; 1970.
- Laan E, Rellini AH, Barnes T; International Society for Sexual Medicine. Standard operating procedures for female orgasmic disorder: consensus of the International Society for Sexual Medicine. J Sex Med 2013; 10:74–82.
- Bornstein J, Goldstein A, Coady D. Consensus terminology and classification of persistent vulvar pain. Vulvar pain/Vulvodynia Nomenclature Consensus Conference. Annapolis, MD; 2015.
- Burrows LJ, Goldstein AT. The treatment of vestibulodynia with topical estradiol and testosterone. Sex Med 2013; 1:30–33.
- Labrie F. Efficacy of intravaginal dehydroepiandrosterone (DHEA) on moderate to severe dyspareunia and vaginal dryness, symptoms of vulvovaginal atrophy, and of the genitourinary syndrome of menopause. Manopause 2016; 23:243–256.
- Goetsch MF, Lim JY, Caughey AB. A practical solution for dyspareunia in breast cancer survivors: a randomized controlled trial. J Clin Oncol 2015; 33:3394–3400.
- Shifren JL, Monz BU, Russo PA, Segreti A, Johannes CB. Sexual problems and distress in United States women: prevalence and correlates. Obstet Gynecol 2008; 112:970–978.
- Laumann EO, Paik A, Rosen RC. Sexual dysfunction in the United States: prevalence and predictors. JAMA 1999; 281:537–544.
- Sadovsky R, Nusbaum M. Sexual health inquiry and support is a primary care priority. J Sex Med 2006; 3:3–11.
- Alvisi S, Baldassarre M, Lambertini M, et al. Sexuality and psychopathological aspects in premenopausal women with metabolic syndrome. J Sex Med 2014; 11:2020–2028.
- Bach LE, Mortimer JA, VandeWeerd C, Corvin J. The association of physical and mental health with sexual activity in older adults in a retirement community. J Sex Med 2013; 10:2671–2678.
- Esposito K, Ciotola M, Giugliano F, et al. Mediterranean diet improves sexual function in women with the metabolic syndrome. Int J Impot Res 2007; 19:486–491.
- Prairie BA, Scheier MF, Matthews KA, Chang CC, Hess R. A higher sense of purpose in life is associated with sexual enjoyment in midlife women. Menopause 2011; 18:839–844.
- Masters WH, Johnson VE. Human sexual response. Boston, MA: Little, Brown and Company; 1966.
- Kaplan HS. The new sex therapy. New York, NY: Brunner/Mazel; 1974.
- Robinson PA. The modernization of sex: Havelock Ellis, Alfred Kinsey, William Masters, and Virginia Johnson. New York, NY: Harper & Row; 1976.
- Basson R. The female sexual response: a different model. J Sex Marital Ther 2000; 26:51–65.
- Giraldi A, Kristensen E, Sand M. Endorsement of models describing sexual response of men and women with a sexual partner: an online survey in a population sample of Danish adults ages 20–65 years. J Sex Med 2015; 12:116–128.
- World Health Organization. Defining sexual health. Report of a technical consultation on sexual health, 28–31 January 2002, Geneva. Geneva; 2006.
- American Psychiatric Association (APA). DSM-5: Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association Publishing; 2013.
- Brotto LA. The DSM diagnostic criteria for hypoactive sexual desire disorder in women. Arch Sex Behav 2010; 39:221–239.
- McCabe MP, Sharlip ID, Atalla E, et al. Definitions of sexual dysfunctions in women and men: a consensus statement from the Fourth International Consultation on Sexual Medicine 2015. J Sex Med 2016; 13:135–143.
- Derogatis LR, Komer L, Katz M, et al; VIOLET Trial Investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the VIOLET study. J Sex Med 2012; 9:1074–1085.
- Katz M, DeRogatis LR, Ackerman R, et al; BEGONIA trial investigators. Efficacy of flibanserin in women with hypoactive sexual desire disorder: results from the BEGONIA trial. J Sex Med 2013; 10:1807–1815.
- Thorp J, Simon J, Dattani D, et al; DAISY trial investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the DAISY study. J Sex Med 2012; 9:793–804.
- Berman L, Berman J, Felder S, et al. Seeking help for sexual function complaints: what gynecologists need to know about the female patient's experience. Fertil Steril 2003; 79:572–576.
- Kingsberg SA. Taking a sexual history. Obstet Gynecol Clin North Am 2006; 33:535–547.
- Kingsberg SA, Rezaee RL. Hypoactive sexual desire in women. Menopause 2013; 20:1284–1300.
- Lindau ST, Schumm LP, Laumann EO, Levinson W, O’Muircheartaigh CA, Waite LJ. A study of sexuality and health among older adults in the United States. N Engl J Med 2007; 357:762–774.
- Gott M, Hinchliff S, Galena E. General practitioner attitudes to discussing sexual health issues with older people. Soc Sci Med 2004; 58:2093–2103.
- Lindau ST, Leitsch SA, Lundberg KL, Jerome J. Older women's attitudes, behavior, and communication about sex and HIV: a community-based study. J Womens Health (Larchmt) 2006; 15:747–753.
- Faubion SS, Rullo JE. Sexual dysfunction in women: a practical approach. Am Fam Physician 2015; 92:281–288.
- Flynn KE, Lindau ST, Lin L, et al. Development and validation of a single-item screener for self-reporting sexual problems in US adults. J Gen Intern Med 2015; 30:1468–1475.
- Latif EZ, Diamond MP. Arriving at the diagnosis of female sexual dysfunction. Fertil Steril 2013; 100:898–904.
- Althof SE, Leiblum SR, Chevret-Measson M, et al. Psychological and interpersonal dimensions of sexual function and dysfunction. J Sex Med 2005; 2:793–800.
- Rosen RC, Barsky JL. Normal sexual response in women. Obstet Gynecol Clin North Am 2006; 33:515–526.
- Brotto LA, Bitzer J, Laan E, Leiblum S, Luria M. Women's sexual desire and arousal disorders. J Sex Med 2010; 7:586–614.
- Rubio-Aurioles E, Kim ED, Rosen RC, et al. Impact on erectile function and sexual quality of life of couples: a double-blind, randomized, placebo-controlled trial of tadalafil taken once daily. J Sex Med 2009; 6:1314–1323.
- Faubion SS, Shuster LT, Bharucha AE. Recognition and management of nonrelaxing pelvic floor dysfunction. Mayo Clin Proc 2012; 87:187–193.
- Annon JS. The PLISSIT model: a proposed conceptual scheme for the behavioral treatment of sexual problems. J Sex Educ Ther 1976; 2:1–15.
- Sarrel PM. Sexuality and menopause. Obstet Gynecol 1990; 75(suppl 4):26S–30S; discussion 31S–35S.
- Avis NE, Brockwell S, Randolph JF, Jr, et al. Longitudinal changes in sexual functioning as women transition through menopause: results from the Study of Women's Health Across the Nation. Menopause 2009; 16:442–452.
- Avis NE, Stellato R, Crawford S, Johannes C, Longcope C. Is there an association between menopause status and sexual functioning? Menopause 2000; 7:297–309.
- Gracia CR, Freeman EW, Sammel MD, Lin H, Mogul M. Hormones and sexuality during transition to menopause. Obstet Gynecol 2007; 109:831–840.
- Dennerstein L, Guthrie JR, Hayes RD, DeRogatis LR, Lehert P. Sexual function, dysfunction, and sexual distress in a prospective, population-based sample of mid-aged, Australian-born women. J Sex Med 2008; 5:2291–2299.
- Management of symptomatic vulvovaginal atrophy: 2013 position statement of The North American Menopause Society. Menopause 2013; 20:888–902.
- Mac Bride MB, Rhodes DJ, Shuster LT. Vulvovaginal atrophy. Mayo Clin Proc 2010; 85:87–94.
- Nastri CO, Lara LA, Ferriani RA, Rosa ESAC, Figueiredo JB, Martins WP. Hormone therapy for sexual function in perimenopausal and postmenopausal women. Cochrane Database Syst Rev 2013; 6:CD009672.
- Atlantis E, Sullivan T. Bidirectional association between depression and sexual dysfunction: a systematic review and meta-analysis. J Sex Med 2012; 9:1497–1507.
- Clayton AH, Maserejian NN, Connor MK, Huang L, Heiman JR, Rosen RC. Depression in premenopausal women with HSDD: baseline findings from the HSDD Registry for Women. Psychosom Med 2012; 74:305–311.
- Ishak WW, Christensen S, Sayer G, et al. Sexual satisfaction and quality of life in major depressive disorder before and after treatment with citalopram in the STAR*D study. J Clin Psychiatry 2013; 74:256–261.
- Clayton AH, Croft HA, Handiwala L. Antidepressants and sexual dysfunction: mechanisms and clinical implications. Postgrad Med 2014; 126:91–99.
- Rothschild AJ. Selective serotonin reuptake inhibitor-induced sexual dysfunction: efficacy of a drug holiday. Am J Psychiatry 1995; 152:1514–1516.
- Nurnberg HG, Hensley PL, Heiman JR, Croft HA, Debattista C, Paine S. Sildenafil treatment of women with antidepressant-associated sexual dysfunction: a randomized controlled trial. JAMA 2008; 300:395–404.
- Taylor MJ, Rudkin L, Bullemor-Day P, Lubin J, Chukwujekwu C, Hawton K. Strategies for managing sexual dysfunction induced by antidepressant medication. Cochrane Database Syst Rev 2013; 5:CD003382.
- Lorenz TA, Meston CM. Exercise improves sexual function in women taking antidepressants: results from a randomized crossover trial. Depress Anxiety 2014; 31:188–195.
- Brotto LA, Basson R. Group mindfulness-based therapy significantly improves sexual desire in women. Behav Res Ther 2014; 5743–5754.
- Pyke RE, Clayton AH. Psychological treatment trials for hypoactive sexual desire disorder: a sexual medicine critique and perspective. J Sex Med 2015; 12:2451–2458.
- Cappelletti M, Wallen K. Increasing women's sexual desire: the comparative effectiveness of estrogens and androgens. Horm Behav 2016; 78:178–193.
- Davis SR, Moreau M, Kroll R, et al. Testosterone for low libido in postmenopausal women not taking estrogen. N Engl J Med 2008; 359:2005–2017.
- Davis SR, van der Mooren MJ, van Lunsen RH, et al. Efficacy and safety of a testosterone patch for the treatment of hypoactive sexual desire disorder in surgically menopausal women: a randomized, placebo-controlled trial. Menopause 2006; 13:387–396.
- Panay N, Al-Azzawi F, Bouchard C, et al. Testosterone treatment of HSDD in naturally menopausal women: the ADORE study. Climacteric 2010; 13:121–131.
- Somboonporn W, Davis S, Seif MW, Bell R. Testosterone for peri- and postmenopausal women. Cochrane Database Syst Rev 2005; 4:CD004509.
- Davis SR. Cardiovascular and cancer safety of testosterone in women. Curr Opin Endocrinol Diabetes Obes 2011; 18:198–203.
- Wierman ME, Arlt W, Basson R, et al. Androgen therapy in women: a reappraisal: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2014; 99:3489–3510.
- Stahl SM, Sommer B, Allers KA. Multifunctional pharmacology of flibanserin: possible mechanism of therapeutic action in hypoactive sexual desire disorder. J Sex Med 2011; 8:15–27.
- Simon JA, Goldstein I, Kim N, Freedman M, Parish S. Flibanserin approval: facts or feelings? Sex Med 2016; 4:e69–e70.
- Joffe HV, Chang C, Sewell C, et al. FDA approval of flibanserin—treating hypoactive sexual desire disorder. N Engl J Med 2016; 374:101–104.
- van Lunsen RH, Laan E. Genital vascular responsiveness and sexual feelings in midlife women: psychophysiologic, brain, and genital imaging studies. Menopause 2004; 11:741–748.
- Beco J, Seidel L, Albert A. Normative values of skin temperature and thermal sensory thresholds in the pudendal nerve territory. Neurourol Urodyn 2015; 34:571–577.
- Masters WH, Johnson VE. Human sexual inadequacy. Boston, MA: Little, Brown and Company; 1970.
- Laan E, Rellini AH, Barnes T; International Society for Sexual Medicine. Standard operating procedures for female orgasmic disorder: consensus of the International Society for Sexual Medicine. J Sex Med 2013; 10:74–82.
- Bornstein J, Goldstein A, Coady D. Consensus terminology and classification of persistent vulvar pain. Vulvar pain/Vulvodynia Nomenclature Consensus Conference. Annapolis, MD; 2015.
- Burrows LJ, Goldstein AT. The treatment of vestibulodynia with topical estradiol and testosterone. Sex Med 2013; 1:30–33.
- Labrie F. Efficacy of intravaginal dehydroepiandrosterone (DHEA) on moderate to severe dyspareunia and vaginal dryness, symptoms of vulvovaginal atrophy, and of the genitourinary syndrome of menopause. Manopause 2016; 23:243–256.
- Goetsch MF, Lim JY, Caughey AB. A practical solution for dyspareunia in breast cancer survivors: a randomized controlled trial. J Clin Oncol 2015; 33:3394–3400.
KEY POINTS
- Sexual dysfunction in women is complex and often multifactorial and has a significant impact on quality of life.
- Primary care providers can assess the problem, provide education on sexual health and normal sexual functioning, and manage biological factors affecting sexual function, including genitourinary syndrome of menopause in postmenopausal women and antidepressant-induced sexual dysfunction.
- Treatment may require a multidisciplinary team, including a psychologist or sex therapist to manage the psychological, sociocultural, and relational factors affecting a woman’s sexual health, and a physical therapist to manage pelvic floor disorders.

Caution patients about common food–drug interactions
Many individuals read about the health benefits of certain foods, such as coffee, grapefruit, and red wine, but psychiatrists might neglect to inform their patients that these common foods could interact with drugs, thereby preventing certain psychotropics from achieving maximum benefit or causing toxicity. Educate your patients about food–drug interactions and to refrain from alcohol and specific foods when taking psychotropics. Although far from comprehensive, we present a discussion of the most frequently encountered and preventable food/nutrient–drug interactions.
Grapefruit juice may alter bioavailability of many psychotropics by inhibiting cytochrome P450 (CYP) 3A4 and 1A2 isoforms, interfering with prehepatic metabolism, and enteric absorption. Common medications affected by this interaction include alprazolam, buspirone, sertraline, carbamazepine, and methadone.1 Patients should be advised about eating grapefruit or drinking grapefruit juice as it could require dose adjustment to avoid drug toxicity.
Table salt. Lithium is a salt, and less table salt intake could cause lithium levels to rise and vice versa. Lithium and other salts compete for absorption and secretion in the renal tubules, which are responsible for this interaction. Therefore, it is advisable to keep a stable salt intake throughout treatment. Patients should be cautioned about eating salty foods during the summer because excessive sweating could lead to lithium intoxication.
Caffeine is a widely used stimulant; however, it can decrease blood lithium levels and block clozapine clearance via inhibition of the CYP1A2 enzyme. Excessive caffeine intake can lead to clozapine toxicity.2
Beef liver, aged sausage and cheese, and wine contain tyramine. Tyramine is broken down by monoamine oxidase (MAO) enzymes in the body, which are inhibited by MAO inhibitors (MAOI) such as phenelzine and selegiline. A patient taking a MAOI cannot catabolize tyramine and other amines. These exogenous amines could cause a life-threatening hyperadrenergic crisis. Physicians should educate their patients about the MAOI diet and monitor adherence to the food avoidance list.
St. John’s wort is a herb commonly used for treating mild depression. It is a strong inducer of the CYP3A4 enzyme and reduces plasma concentrations and could decrease clinical effectiveness of aripiprazole, quetiapine, alprazolam, and oxycodone.3 It could interact with serotonin reuptake inhibitors causing serotonin syndrome.
Full vs empty stomach. Food is known to affect bioavailability and enteral absorption of different psychotropics. Some medications are best taken on a full stomach and some on an empty one. For example, the antipsychotic ziprasidone should be taken with meals of at least 500 calories for optimal and consistent bioavailability. Benzodiazepines are rapidly absorbed when taken on an empty stomach.
Discuss dietary habits with patients to encourage a healthy lifestyle and provide valuable direction about potential food/nutrient–drug interactions.
1. Pawełczyk T, Kłoszewska I. Grapefruit juice interactions with psychotropic drugs: advantages and potential risk [in Polish]. Przegl Lek. 2008;62(2):92-95.
2. Hägg S, Spigset O, Mjörndal T, et al. Effect of caffeine on clozapine pharmacokinetics in healthy volunteers. Br J Clin Pharmacol. 2000;49(1):59-63.
3. Markowitz JS, Donovan JL, DeVane CL, et al. Effect of St John’s wort on drug metabolism by induction of cytochrome P450 3A4 enzyme. JAMA. 2003;290(11):1500-1504.
Many individuals read about the health benefits of certain foods, such as coffee, grapefruit, and red wine, but psychiatrists might neglect to inform their patients that these common foods could interact with drugs, thereby preventing certain psychotropics from achieving maximum benefit or causing toxicity. Educate your patients about food–drug interactions and to refrain from alcohol and specific foods when taking psychotropics. Although far from comprehensive, we present a discussion of the most frequently encountered and preventable food/nutrient–drug interactions.
Grapefruit juice may alter bioavailability of many psychotropics by inhibiting cytochrome P450 (CYP) 3A4 and 1A2 isoforms, interfering with prehepatic metabolism, and enteric absorption. Common medications affected by this interaction include alprazolam, buspirone, sertraline, carbamazepine, and methadone.1 Patients should be advised about eating grapefruit or drinking grapefruit juice as it could require dose adjustment to avoid drug toxicity.
Table salt. Lithium is a salt, and less table salt intake could cause lithium levels to rise and vice versa. Lithium and other salts compete for absorption and secretion in the renal tubules, which are responsible for this interaction. Therefore, it is advisable to keep a stable salt intake throughout treatment. Patients should be cautioned about eating salty foods during the summer because excessive sweating could lead to lithium intoxication.
Caffeine is a widely used stimulant; however, it can decrease blood lithium levels and block clozapine clearance via inhibition of the CYP1A2 enzyme. Excessive caffeine intake can lead to clozapine toxicity.2
Beef liver, aged sausage and cheese, and wine contain tyramine. Tyramine is broken down by monoamine oxidase (MAO) enzymes in the body, which are inhibited by MAO inhibitors (MAOI) such as phenelzine and selegiline. A patient taking a MAOI cannot catabolize tyramine and other amines. These exogenous amines could cause a life-threatening hyperadrenergic crisis. Physicians should educate their patients about the MAOI diet and monitor adherence to the food avoidance list.
St. John’s wort is a herb commonly used for treating mild depression. It is a strong inducer of the CYP3A4 enzyme and reduces plasma concentrations and could decrease clinical effectiveness of aripiprazole, quetiapine, alprazolam, and oxycodone.3 It could interact with serotonin reuptake inhibitors causing serotonin syndrome.
Full vs empty stomach. Food is known to affect bioavailability and enteral absorption of different psychotropics. Some medications are best taken on a full stomach and some on an empty one. For example, the antipsychotic ziprasidone should be taken with meals of at least 500 calories for optimal and consistent bioavailability. Benzodiazepines are rapidly absorbed when taken on an empty stomach.
Discuss dietary habits with patients to encourage a healthy lifestyle and provide valuable direction about potential food/nutrient–drug interactions.
Many individuals read about the health benefits of certain foods, such as coffee, grapefruit, and red wine, but psychiatrists might neglect to inform their patients that these common foods could interact with drugs, thereby preventing certain psychotropics from achieving maximum benefit or causing toxicity. Educate your patients about food–drug interactions and to refrain from alcohol and specific foods when taking psychotropics. Although far from comprehensive, we present a discussion of the most frequently encountered and preventable food/nutrient–drug interactions.
Grapefruit juice may alter bioavailability of many psychotropics by inhibiting cytochrome P450 (CYP) 3A4 and 1A2 isoforms, interfering with prehepatic metabolism, and enteric absorption. Common medications affected by this interaction include alprazolam, buspirone, sertraline, carbamazepine, and methadone.1 Patients should be advised about eating grapefruit or drinking grapefruit juice as it could require dose adjustment to avoid drug toxicity.
Table salt. Lithium is a salt, and less table salt intake could cause lithium levels to rise and vice versa. Lithium and other salts compete for absorption and secretion in the renal tubules, which are responsible for this interaction. Therefore, it is advisable to keep a stable salt intake throughout treatment. Patients should be cautioned about eating salty foods during the summer because excessive sweating could lead to lithium intoxication.
Caffeine is a widely used stimulant; however, it can decrease blood lithium levels and block clozapine clearance via inhibition of the CYP1A2 enzyme. Excessive caffeine intake can lead to clozapine toxicity.2
Beef liver, aged sausage and cheese, and wine contain tyramine. Tyramine is broken down by monoamine oxidase (MAO) enzymes in the body, which are inhibited by MAO inhibitors (MAOI) such as phenelzine and selegiline. A patient taking a MAOI cannot catabolize tyramine and other amines. These exogenous amines could cause a life-threatening hyperadrenergic crisis. Physicians should educate their patients about the MAOI diet and monitor adherence to the food avoidance list.
St. John’s wort is a herb commonly used for treating mild depression. It is a strong inducer of the CYP3A4 enzyme and reduces plasma concentrations and could decrease clinical effectiveness of aripiprazole, quetiapine, alprazolam, and oxycodone.3 It could interact with serotonin reuptake inhibitors causing serotonin syndrome.
Full vs empty stomach. Food is known to affect bioavailability and enteral absorption of different psychotropics. Some medications are best taken on a full stomach and some on an empty one. For example, the antipsychotic ziprasidone should be taken with meals of at least 500 calories for optimal and consistent bioavailability. Benzodiazepines are rapidly absorbed when taken on an empty stomach.
Discuss dietary habits with patients to encourage a healthy lifestyle and provide valuable direction about potential food/nutrient–drug interactions.
1. Pawełczyk T, Kłoszewska I. Grapefruit juice interactions with psychotropic drugs: advantages and potential risk [in Polish]. Przegl Lek. 2008;62(2):92-95.
2. Hägg S, Spigset O, Mjörndal T, et al. Effect of caffeine on clozapine pharmacokinetics in healthy volunteers. Br J Clin Pharmacol. 2000;49(1):59-63.
3. Markowitz JS, Donovan JL, DeVane CL, et al. Effect of St John’s wort on drug metabolism by induction of cytochrome P450 3A4 enzyme. JAMA. 2003;290(11):1500-1504.
1. Pawełczyk T, Kłoszewska I. Grapefruit juice interactions with psychotropic drugs: advantages and potential risk [in Polish]. Przegl Lek. 2008;62(2):92-95.
2. Hägg S, Spigset O, Mjörndal T, et al. Effect of caffeine on clozapine pharmacokinetics in healthy volunteers. Br J Clin Pharmacol. 2000;49(1):59-63.
3. Markowitz JS, Donovan JL, DeVane CL, et al. Effect of St John’s wort on drug metabolism by induction of cytochrome P450 3A4 enzyme. JAMA. 2003;290(11):1500-1504.
Subjected to sexually inappropriate behavior? Set LIMITS
Everyone needs love, companionship, and intimacy. Unfortunately, mental illness often results in interpersonal dysfunction, thereby frustrating these desires. Patients might exhibit sexually inappropriate behavior (SIB), consisting of comments, requests, or actions. The causes of SIB include confusion, predation, loneliness, psychosis, social impairment, character pathology, and/or mania.
Such attention poses an issue for mental health providers; trainees could be particularly vulnerable. The impact can be disheartening and cause practitioners to withdraw from patients or question their work, which could be detrimental to both providers and patients. While maintaining their personal safety, it is important that clinicians approach patients with compassion. To help clinicians manage SIB, we propose setting LIMITS.
Look after personal safety. Clinicians are trained to care for all patients, but situations can arise where it is no longer safe to work with an individual. A clinician who feels threatened is less likely to help the patient, especially if real danger is posed. Such situations could necessitate transferring the patient’s care to another provider. Clinicians also can choose to interact with a patient exhibiting SIB while colleagues are present.
Identify the etiology. SIB arises from a variety of underlying states, and the clinician’s response can vary depending on the cause. Questions to consider before reacting include:
- What is the origin of the behavior?
- What form is the behavior taking?
- In what context is it occurring?
- How frequent is it occurring?
- What factors are contributing?
- What are the risks to all parties?1
Maintain a professional role. Although SIB can undermine the provider–patient relationship, the behavior could be unintended. To remain professional, practitioners should pause before reacting and consider how to respond. A particular concern is countertransference, meaning that the provider might react to a patient’s behavior based on personal bias. This could result in amorous, hateful, or angry responses from the provider, which could put the treatment relationship at risk, harm the patient, or result in medical–legal repercussions.
Implement appropriate boundaries. In man
Talk with a Supervisor. These scenarios often produce many emotions. Residents could be experiencing them for the first time, but even seasoned clinicians can find them challenging. When in doubt, seek guidance from colleagues, supervisors, or mentors to help you clarify the situation.
Acknowledgments
The authors thank Kristina Zdanys, MD, David Schmidt, DO, Joanna Chaurette, MD, PhD, and Shilpa Lad, MD, for their input.
1. Series H, Dégano P. Hypersexuality in dementia. Adv Psychiatr Treat. 2005; 11(6):424-431.
Everyone needs love, companionship, and intimacy. Unfortunately, mental illness often results in interpersonal dysfunction, thereby frustrating these desires. Patients might exhibit sexually inappropriate behavior (SIB), consisting of comments, requests, or actions. The causes of SIB include confusion, predation, loneliness, psychosis, social impairment, character pathology, and/or mania.
Such attention poses an issue for mental health providers; trainees could be particularly vulnerable. The impact can be disheartening and cause practitioners to withdraw from patients or question their work, which could be detrimental to both providers and patients. While maintaining their personal safety, it is important that clinicians approach patients with compassion. To help clinicians manage SIB, we propose setting LIMITS.
Look after personal safety. Clinicians are trained to care for all patients, but situations can arise where it is no longer safe to work with an individual. A clinician who feels threatened is less likely to help the patient, especially if real danger is posed. Such situations could necessitate transferring the patient’s care to another provider. Clinicians also can choose to interact with a patient exhibiting SIB while colleagues are present.
Identify the etiology. SIB arises from a variety of underlying states, and the clinician’s response can vary depending on the cause. Questions to consider before reacting include:
- What is the origin of the behavior?
- What form is the behavior taking?
- In what context is it occurring?
- How frequent is it occurring?
- What factors are contributing?
- What are the risks to all parties?1
Maintain a professional role. Although SIB can undermine the provider–patient relationship, the behavior could be unintended. To remain professional, practitioners should pause before reacting and consider how to respond. A particular concern is countertransference, meaning that the provider might react to a patient’s behavior based on personal bias. This could result in amorous, hateful, or angry responses from the provider, which could put the treatment relationship at risk, harm the patient, or result in medical–legal repercussions.
Implement appropriate boundaries. In man
Talk with a Supervisor. These scenarios often produce many emotions. Residents could be experiencing them for the first time, but even seasoned clinicians can find them challenging. When in doubt, seek guidance from colleagues, supervisors, or mentors to help you clarify the situation.
Acknowledgments
The authors thank Kristina Zdanys, MD, David Schmidt, DO, Joanna Chaurette, MD, PhD, and Shilpa Lad, MD, for their input.
Everyone needs love, companionship, and intimacy. Unfortunately, mental illness often results in interpersonal dysfunction, thereby frustrating these desires. Patients might exhibit sexually inappropriate behavior (SIB), consisting of comments, requests, or actions. The causes of SIB include confusion, predation, loneliness, psychosis, social impairment, character pathology, and/or mania.
Such attention poses an issue for mental health providers; trainees could be particularly vulnerable. The impact can be disheartening and cause practitioners to withdraw from patients or question their work, which could be detrimental to both providers and patients. While maintaining their personal safety, it is important that clinicians approach patients with compassion. To help clinicians manage SIB, we propose setting LIMITS.
Look after personal safety. Clinicians are trained to care for all patients, but situations can arise where it is no longer safe to work with an individual. A clinician who feels threatened is less likely to help the patient, especially if real danger is posed. Such situations could necessitate transferring the patient’s care to another provider. Clinicians also can choose to interact with a patient exhibiting SIB while colleagues are present.
Identify the etiology. SIB arises from a variety of underlying states, and the clinician’s response can vary depending on the cause. Questions to consider before reacting include:
- What is the origin of the behavior?
- What form is the behavior taking?
- In what context is it occurring?
- How frequent is it occurring?
- What factors are contributing?
- What are the risks to all parties?1
Maintain a professional role. Although SIB can undermine the provider–patient relationship, the behavior could be unintended. To remain professional, practitioners should pause before reacting and consider how to respond. A particular concern is countertransference, meaning that the provider might react to a patient’s behavior based on personal bias. This could result in amorous, hateful, or angry responses from the provider, which could put the treatment relationship at risk, harm the patient, or result in medical–legal repercussions.
Implement appropriate boundaries. In man
Talk with a Supervisor. These scenarios often produce many emotions. Residents could be experiencing them for the first time, but even seasoned clinicians can find them challenging. When in doubt, seek guidance from colleagues, supervisors, or mentors to help you clarify the situation.
Acknowledgments
The authors thank Kristina Zdanys, MD, David Schmidt, DO, Joanna Chaurette, MD, PhD, and Shilpa Lad, MD, for their input.
1. Series H, Dégano P. Hypersexuality in dementia. Adv Psychiatr Treat. 2005; 11(6):424-431.
1. Series H, Dégano P. Hypersexuality in dementia. Adv Psychiatr Treat. 2005; 11(6):424-431.
Benefits and costs of accepting credit cards in your practice
Are you tired of waiting for checks in the mail? Do patients leave without paying their balance? Streamlining revenue collection by taking credit cards is a tantalizing antidote to these ills, but it has downsides. Weighing the value for you and your patients is necessary before you decide on this important practice management policy.
Clinical and practical advantages
Many patients prefer that their health care practitioners take credit cards, because it simplifies their busy lives—and who carries a checkbook anymore? Patients can put the whole session to good use without sacrificing time taking care of payment. They also can receive credit card rewards for their payment, or use health savings accounts, health reimbursement accounts, or flexible spending debit cards, making treatment more affordable.
Benefits of credit cards
Accepting credit cards has many benefits:
- Allows more time in a session to focus on clinical matters because you do not have to allocate time to collect payment, which might include dealing with a forgotten checkbook or a request for a change in your payment policies.
- Easier to collect payment for no-shows. This could result in a reduced no-show rate, because a patient might feel more accountable to show up knowing that his (her) credit card is on file.
- Saves time recording and depositing checks.
- Avoids bounced checks and collection agencies.
Money doesn’t grow on trees
Although there are advantages to accepting credit cards, several costs should be considered. Some practitioners feel that accepting credit cards makes their practice seem like a commercial business. There also is an expense of accepting credit cards, and understanding these costs can be confusing because there are different processing systems of rates. Whether the rate is flat, tiered, or wholesale, you always will pay a percentage of the transaction, plus a transaction fee.
Here are some general guidelines on rates:
- Debit cards are the least expensive to process but often have low spending limits.
- Rewards cards, such as frequent flyer cards, are the most expensive to process. Have you ever wondered who foots the bill for those frequent flyer miles? It’s not the airline; it’s the merchant (you).
- For tiered rates, swiping cards is typically cheaper than typing in the credit card info. Tiered rates often have low rates, known as “teaser” rates, because they are applicable in far fewer cases.
- For flat or wholesale rates, securely saving credit card numbers is not any more expensive than swiping a card, and saves time in the long run and potential awkwardness at the end of a session.
- A higher volume of processed credit cards might allow you to negotiate your rates.
- Check if your bank offers a less expensive option. Some banks offer preferred rates for their customers.
Also consider the time and possible expense of ensuring that you are Payment Card Industry Data Security Standard compliant (information security standards that aim to keep cardholder data secure).
Different methods of processing transactions have varying levels of requirements:
- A swiping reader with a terminal connected to a telephone line is more secure than through the Internet and carries fewer compliance burdens. Use a reader that can handle chip-cards, because you could be liable for fraudulent transactions.
- Do not save or store credit card numbers you typed yourself. Compliance is less burdensome if patients input credit card data into a secure portal.
- Store credit card data securely via your credit card processing partner, although the partner is still at risk of a data breach. Practitioners should weigh the value of convenience vs security.
- If there is a data breach and you are found negligent you could be fined $5,000 to $100,000 per month, depending on whether you are a large company or solo practice.
Bottom dollar
Credit card processing has significant advantages from both a practice management and clinical standpoint. Because prices for services vary, shop around to find the best rates and educate yourself about security requirements. Taking the time to research these matters can pay off in the long term.
Are you tired of waiting for checks in the mail? Do patients leave without paying their balance? Streamlining revenue collection by taking credit cards is a tantalizing antidote to these ills, but it has downsides. Weighing the value for you and your patients is necessary before you decide on this important practice management policy.
Clinical and practical advantages
Many patients prefer that their health care practitioners take credit cards, because it simplifies their busy lives—and who carries a checkbook anymore? Patients can put the whole session to good use without sacrificing time taking care of payment. They also can receive credit card rewards for their payment, or use health savings accounts, health reimbursement accounts, or flexible spending debit cards, making treatment more affordable.
Benefits of credit cards
Accepting credit cards has many benefits:
- Allows more time in a session to focus on clinical matters because you do not have to allocate time to collect payment, which might include dealing with a forgotten checkbook or a request for a change in your payment policies.
- Easier to collect payment for no-shows. This could result in a reduced no-show rate, because a patient might feel more accountable to show up knowing that his (her) credit card is on file.
- Saves time recording and depositing checks.
- Avoids bounced checks and collection agencies.
Money doesn’t grow on trees
Although there are advantages to accepting credit cards, several costs should be considered. Some practitioners feel that accepting credit cards makes their practice seem like a commercial business. There also is an expense of accepting credit cards, and understanding these costs can be confusing because there are different processing systems of rates. Whether the rate is flat, tiered, or wholesale, you always will pay a percentage of the transaction, plus a transaction fee.
Here are some general guidelines on rates:
- Debit cards are the least expensive to process but often have low spending limits.
- Rewards cards, such as frequent flyer cards, are the most expensive to process. Have you ever wondered who foots the bill for those frequent flyer miles? It’s not the airline; it’s the merchant (you).
- For tiered rates, swiping cards is typically cheaper than typing in the credit card info. Tiered rates often have low rates, known as “teaser” rates, because they are applicable in far fewer cases.
- For flat or wholesale rates, securely saving credit card numbers is not any more expensive than swiping a card, and saves time in the long run and potential awkwardness at the end of a session.
- A higher volume of processed credit cards might allow you to negotiate your rates.
- Check if your bank offers a less expensive option. Some banks offer preferred rates for their customers.
Also consider the time and possible expense of ensuring that you are Payment Card Industry Data Security Standard compliant (information security standards that aim to keep cardholder data secure).
Different methods of processing transactions have varying levels of requirements:
- A swiping reader with a terminal connected to a telephone line is more secure than through the Internet and carries fewer compliance burdens. Use a reader that can handle chip-cards, because you could be liable for fraudulent transactions.
- Do not save or store credit card numbers you typed yourself. Compliance is less burdensome if patients input credit card data into a secure portal.
- Store credit card data securely via your credit card processing partner, although the partner is still at risk of a data breach. Practitioners should weigh the value of convenience vs security.
- If there is a data breach and you are found negligent you could be fined $5,000 to $100,000 per month, depending on whether you are a large company or solo practice.
Bottom dollar
Credit card processing has significant advantages from both a practice management and clinical standpoint. Because prices for services vary, shop around to find the best rates and educate yourself about security requirements. Taking the time to research these matters can pay off in the long term.
Are you tired of waiting for checks in the mail? Do patients leave without paying their balance? Streamlining revenue collection by taking credit cards is a tantalizing antidote to these ills, but it has downsides. Weighing the value for you and your patients is necessary before you decide on this important practice management policy.
Clinical and practical advantages
Many patients prefer that their health care practitioners take credit cards, because it simplifies their busy lives—and who carries a checkbook anymore? Patients can put the whole session to good use without sacrificing time taking care of payment. They also can receive credit card rewards for their payment, or use health savings accounts, health reimbursement accounts, or flexible spending debit cards, making treatment more affordable.
Benefits of credit cards
Accepting credit cards has many benefits:
- Allows more time in a session to focus on clinical matters because you do not have to allocate time to collect payment, which might include dealing with a forgotten checkbook or a request for a change in your payment policies.
- Easier to collect payment for no-shows. This could result in a reduced no-show rate, because a patient might feel more accountable to show up knowing that his (her) credit card is on file.
- Saves time recording and depositing checks.
- Avoids bounced checks and collection agencies.
Money doesn’t grow on trees
Although there are advantages to accepting credit cards, several costs should be considered. Some practitioners feel that accepting credit cards makes their practice seem like a commercial business. There also is an expense of accepting credit cards, and understanding these costs can be confusing because there are different processing systems of rates. Whether the rate is flat, tiered, or wholesale, you always will pay a percentage of the transaction, plus a transaction fee.
Here are some general guidelines on rates:
- Debit cards are the least expensive to process but often have low spending limits.
- Rewards cards, such as frequent flyer cards, are the most expensive to process. Have you ever wondered who foots the bill for those frequent flyer miles? It’s not the airline; it’s the merchant (you).
- For tiered rates, swiping cards is typically cheaper than typing in the credit card info. Tiered rates often have low rates, known as “teaser” rates, because they are applicable in far fewer cases.
- For flat or wholesale rates, securely saving credit card numbers is not any more expensive than swiping a card, and saves time in the long run and potential awkwardness at the end of a session.
- A higher volume of processed credit cards might allow you to negotiate your rates.
- Check if your bank offers a less expensive option. Some banks offer preferred rates for their customers.
Also consider the time and possible expense of ensuring that you are Payment Card Industry Data Security Standard compliant (information security standards that aim to keep cardholder data secure).
Different methods of processing transactions have varying levels of requirements:
- A swiping reader with a terminal connected to a telephone line is more secure than through the Internet and carries fewer compliance burdens. Use a reader that can handle chip-cards, because you could be liable for fraudulent transactions.
- Do not save or store credit card numbers you typed yourself. Compliance is less burdensome if patients input credit card data into a secure portal.
- Store credit card data securely via your credit card processing partner, although the partner is still at risk of a data breach. Practitioners should weigh the value of convenience vs security.
- If there is a data breach and you are found negligent you could be fined $5,000 to $100,000 per month, depending on whether you are a large company or solo practice.
Bottom dollar
Credit card processing has significant advantages from both a practice management and clinical standpoint. Because prices for services vary, shop around to find the best rates and educate yourself about security requirements. Taking the time to research these matters can pay off in the long term.

Valbenazine for tardive dyskinesia
Despite improvements in the tolerability of antipsychotic medications, the development of tardive dyskinesia (TD) still is a significant area of concern; however, clinicians have had few treatment options. Valbenazine, a vesicular monoamine transport type 2 (VMAT2) inhibitor, is the only FDA-approved medication for TD (Table 1).1 By modulating dopamine transport into presynaptic vesicles, synaptic dopamine release is decreased, thereby reducing the postsynaptic stimulation of D2 receptors and the severity of dyskinetic movements.

In the pivotal 6-week clinical trial, valbenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) ratings.2 Study completion rates were high (87.6%), with only 2 dropouts because of adverse events in each of the placebo (n = 78) and 40-mg (n = 76) arms, and 3 in the 80-mg group (n = 80).
Before the development of valbenazine, tetrabenazine was the only effective option for treating TD. Despite tetrabenazine’s known efficacy for TD, it was not available in the United States until 2008 with the sole indication for movements related to Huntington’s disease. U.S. patients often were subjected to a litany of ineffective medications for TD, often at great expense. Moreover, tetrabenazine involved multiple daily dosing, required cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, had significant tolerability issues, and a monthly cost of $8,000 to $10,000. The availability of an agent that is effective for TD and does not have tetrabenazine’s kinetic limitations, adverse effect profile, or CYP2D6 monitoring requirements represents an enormous advance in the treatment of TD.
Clinical implications
Tardive dyskinesia remains a significant public health concern because of the increasing use of antipsychotics for disorders beyond the core indication for schizophrenia. Although exposure to dopamine D2 antagonism could result in postsynaptic receptor upregulation and supersensitivity, this process best explains what underlies withdrawal dyskinesia.3 The persistence of TD symptoms in 66% to 80% of patients after discontinuing offending agents has led to hypotheses that the underlying pathophysiology of TD might best be conceptualized as a problem with neuroplasticity. As with many disorders, environmental contributions (eg, oxidative stress) and genetic predisposition might play a role beyond that related to exposure to D2 antagonism.3
There have been trials of numerous agents, but no medication has been FDA-approved for treating TD, and limited data support the efficacy of a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]),4 albeit with small effect sizes. A medical food, consisting of branched-chain amino acids, received FDA approval for the dietary management of TD in males, but is no longer commercially available except from compounding pharmacies.5
Tetrabenazine, a molecule developed in the mid-1950s to improve on the tolerability of reserpine, was associated with significant adverse effects such as orthostasis.6 Like reserpine, tetrabenazine subsequently was found to be effective for TD7 but without the peripheral adverse effects of reserpine. However, the kinetics of tetrabenazine necessitated multiple daily doses, and required CYP2D6 genotyping for doses >50 mg/d.8
Receptor blocking. The mechanism that differentiated reserpine’s and tetrabenazine’s clinical properties became clearer in the 1980s when researchers discovered that transporters were necessary to package neurotransmitters into the synaptic vesicles of presynaptic neurons.9 The vesicular monoamine transporter (VMAT) exists in 2 isoforms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the central nervous system.10
Tetrabenazine’s improved tolerability profile was related to the fact that it is a specific and reversible VMAT2 inhibitor, while reserpine is an irreversible and nonselective antagonist of both VMAT isoforms. Investigation of tetrabenazine’s metabolism revealed that it is rapidly and extensively converted into 2 isomers, α-dihydrotetrabenazine (DH-TBZ) and β-DH-TBZ. The isomeric forms of DH-TBZ have multiple chiral centers, and therefore numerous forms of which only 2 are significantly active at VMAT2.3 The α–DH-TBZ isomer is metabolized via CYP2D6 and 3A4 into inactive metabolites, while β-DH-TBZ is metabolized solely via 2D6.3 Because of the short half-life of DH-TBZ when generated from oral tetrabenazine, the existence of 2D6 polymorphisms, and the predominant activity deriving from only 2 isomers, a molecule was synthesized (valbenazine), that when metabolized would slowly be converted into the most active isomer of α–DH-TBZ designated as NBI-98782 (Table 2). This slower conversion to NBI-98782 from valbenazine (compared with its formation from oral tetrabenazine) yielded improved kinetics and permitted once-daily dosing; moreover, because the metabolism of NBI-98782 is not solely dependent on CYP2D6, the need for genotyping was removed. Neither of the 2 metabolites from valbenazine NBI-98782 and NB-136110 have significant affinity for targets other than VMAT2.11
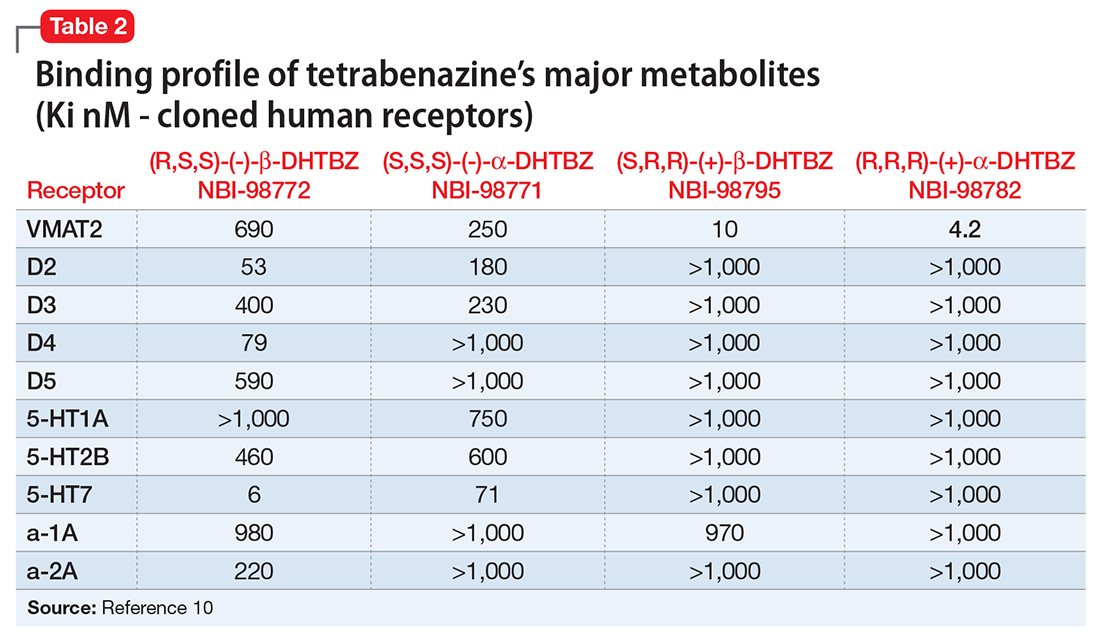
Use in tardive dyskinesia. Recommended starting dosage is 40 mg once daily with or without food, increased to 80 mg after 1 week, based on the design and results from the phase-III clinical trial.12 The FDA granted breakthrough therapy designation for this compound, and only 1 phase-III trial was performed. Valbenazine produced significant improvement on the AIMS, with a mean 30% reduction in AIMS scores at the Week 6 endpoint from baseline of 10.4 ± 3.6.2 The effect size was large (Cohen’s d = 0.90) for the 80-mg dosage. Continuation of 40 mg/d may be considered for some patients based on tolerability, including those who are known CYP2D6 poor metabolizers, and those taking strong CYP2D6 inhibitors. Patients taking strong 3A4 inhibitors should not exceed 40 mg/d. The maximum daily dose is 40 mg for those who have moderate or severe hepatic impairment (Child-Pugh score, 7 to 15). Dosage adjustment is not required for mild to moderate renal impairment (creatinine clearance, 30 to 90 mL/min).
Pharmacologic profile, adverse reactions
Valbenazine and its 2 metabolites lack affinity for receptors other than VMAT2, leading to an absence of orthostasis in clinical trials.1,2 In the phase-II trial, 76% of participants receiving valbenazine (n = 51) were titrated to the maximum dosage of 75 mg/d. Common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were headache (9.8% vs 4.1% placebo), fatigue (9.8% vs 4.1% placebo), and somnolence (5.9% vs 2% placebo).1 In the phase-III trial, participants were randomized 1:1:1 to valbenazine, 40 mg (n = 72), valbenazine, 80 mg (n = 79), or placebo (n = 76). In the clinical studies the most common diagnosis was schizophrenia or schizoaffective disorder, and 40% and 85% of participants in the phase-II and phase-III studies, respectively, remained on antipsychotics.1,2 There were no adverse effects with an incidence ≥5% and at least twice the rate of placebo in the phase-III trial.2
When data from all placebo-controlled studies were pooled, only 1 adverse effect occurred with an incidence ≥5% and twice that of placebo, somnolence with a rate of 10.9% for valbenazine vs 4.2% for placebo. The incidence of akathisia in the pooled analysis was 2.7% for valbenazine vs 0.5% for placebo. Importantly, in neither study was there a safety signal related to depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms.
The mean QT prolongation for valbenazine in healthy participants was 6.7 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 8.4 milliseconds. For those taking strong 2D6 or 3A4 inhibitors, or known 2D6 poor metabolizers, the mean QT prolongation was 11.7 milliseconds (14.7 milliseconds upper bound of double-sided 90% CI). In the controlled trials, there was a dose-related increase in prolactin, alkaline phosphatase, and bilirubin. Overall, 3% of valbenazine-treated patients and 2% of placebo-treated patients discontinued because of adverse reactions.
As noted above, there were no adverse effects with an incidence ≥5% and at least twice the rate of placebo in the phase-III valbenazine trial. Aggregate data across all placebo-controlled studies found that somnolence was the only adverse effect that occurred with an incidence ≥5% and twice that of placebo (10.9% for valbenazine vs 4.2% for placebo).2 As a comparsion, rates of sedation and akathisia for tetrabenazine were higher in the pivotal Huntington’s disease trial: sedation/somnolence 31% vs 3% for placebo, and akathisia 19% vs 0% for placebo.8
How it works
Tetrabenazine, a selective VMAT2 inhibitor, is the only agent that has demonstrated significant efficacy and tolerability for TD management; however, its complex metabolism generates numerous isomers of the metabolites α-DH-TBZ and β-DH-TBZ, of which only 2 are significantly active (Table 3). By choosing an active isomer (NBI-98782) as the metabolite of interest because of its selective and potent activity at VMAT2 and having a metabolism not solely dependent on CYP2D6, a compound was generated (valbenazine) that when metabolized slowly converts into NBI-98782.

Pharmacokinetics
Valbenazine demonstrates dose-proportional pharmacokinetics after single oral dosages from 40 to 300 mg with no impact of food or fasting status on levels of the active metabolite. Valbenazine has a Tmax of 0.5 to 1.0 hours, with 49% oral bioavailability. The plasma half-life for valbenazine and for NBI-98782 ranges from 15 to 22 hours. The Tmax for NBI-98782 when formed from valbenazine occurs between 4 and 8 hours, with a Cmax of approximately 30 ng/mL. It should be noted that when NBI-98782 is generated from oral tetrabenazine, the mean half-life and Tmax are considerably shorter (6 hours and 1.5 hours, respectively), while the Cmax is much higher (approximately 77 ng/mL) (Table 4).

Valbenazine is metabolized through endogenous esterases to NBI-98782 and NBI-136110. NBI-98782, the active metabolite, is further metabolized through multiple CYP pathways, predominantly 3A4 and 2D6. Neither valbenazine nor its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that valbenazine and its active metabolite are unlikely to inhibit most major drug transporters at clinically relevant concentrations. However, valbenazine increased digoxin levels because of inhibition of intestinal P-glycoprotein; therefore plasma digoxin level monitoring is recommended when these 2 are co-administered.
Efficacy
Efficacy was established in a 6-week, fixed-dosage, double-blind, placebo-controlled trial of adult patients with TD. Eligible participants had:
- DSM-IV diagnosis of antipsychotic-induced TD for ≥3 months before screening and moderate or severe TD, as indicated by AIMS item 8 (severity of abnormal movement), which was rated by a blinded, external reviewer using a video of the participant’s AIMS assessment at screening
- a DSM-IV diagnosis of schizophrenia or schizoaffective disorder or mood disorder (and stable per investigator)
- Brief Psychiatric Rating Scale score <50 at screening.
Exclusion criteria included clinically significant and unstable medical conditions within 1 month before screening; comorbid movement disorder (eg, parkinsonism, akathisia, truncal dystonia) that was more prominent than TD; and significant risk for active suicidal ideation, suicidal behavior, or violent behavior.2 Participants had a mean age of 56, 52% were male, and 65.7% of participants in the valbenazine 40-mg group had a schizophrenia spectrum disorder diagnosis, as did 65.8% in both the placebo and valbenazine 80-mg arms.
Antipsychotic treatments were permitted during the trial and >85% of participants continued taking these medications during the study. Participants (N = 234) were randomly allocated in a 1:1:1 manner to valbenazine 40 mg, 80 mg, or matched placebo. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. Baseline AIMS scores were 9.9 ± 4.3 in the placebo group, and 9.8 ± 4.1 and 10.4 ± 3.6 in the valbenazine 40-mg and 80-mg arms, respectively.2
Outcome. A fixed-sequence testing procedure to control for family-wise error rate and multiplicity was employed, and the primary endpoint was change from baseline to Week 6 in AIMS total score (items 1 to 7) for valbenazine 80 mg vs placebo. Valbenazine, 40 mg, was associated with a 1.9 point decrease in AIMS score, while valbenazine, 80 mg, was associated with a 3.2 point decrease in AIMS score, compared with 0.1 point decrease for placebo (P < .05 for valbenazine, 40 mg, P < .001 for valbenazine, 80 mg). This difference for the 40-mg dosage did not meet the prespecified analysis endpoints; however, for the 80-mg valbenazine dosage, the effect size for this difference (Cohen’s d) was large 0.90. There also were statistically significant differences between 40 mg and 80 mg at weeks 2, 4, and 6 in the intent-to-treat population. Of the 79 participants, 43 taking the 80-mg dosage completed a 48-week extension. Efficacy was sustained in this group; however, when valbenazine was discontinued at Week 48, AIMS scores returned to baseline after 4 weeks.
Tolerability
Of the 234 randomized patients, 205 (87.6%) completed the 6-week trial. Discontinuations due to adverse events were low across all treatment groups: 2.6% and 2.8% in the placebo and valbenazine 40-mg arms, respectively, and 3.8% in valbenazine 80-mg cohort. There was no safety signal based on changes in depression, suicidality, parkinsonism rating, or changes in schizophrenia symptoms. Because valbenazine can cause somnolence, patients should not perform activities requiring mental alertness (eg, operating a vehicle or hazardous machinery) until they know how they will be affected by valbenazine.
Valbenazine should be avoided in patients with congenital long QT syndrome or with arrhythmias associated with a prolonged QT interval. For patients at increased risk of a prolonged QT interval, assess the QT interval before increasing the dosage.
Clinical considerations
Unique properties. Valbenazine is metabolized slowly to a potent, selective VMAT2 antagonist (NBI-98782) in a manner that permits once daily dosing, removes the need for CYP2D6 genotyping, and provides significant efficacy.
Why Rx? The reasons to prescribe valbenazine for TD patients include:
- currently the only agent with FDA approval for TD
- fewer tolerability issues seen with the only other effective agent, tetrabenazine
- no signal for effects on mood parameters or rates of parkinsonism
- lack of multiple daily dosing and possible need for 2D6 genotyping involved with TBZ prescribing.
Dosing
The recommended dosage of valbenazine is 80 mg/d administered as a single dose with or without food, starting at 40 mg once daily for 1 week. There is no dosage adjustment required in those with mild to moderate renal impairment; however, valbenazine is not recommended in those with severe renal impairment. The maximum dose is 40 mg/d for those who with moderate or severe hepatic impairment (Child-Pugh score, 7 to 15) however, valbenazine is not recommended for patients with severe renal impairment (creatinine clearance <30 mL/min) because the exposure to the active metabolite is reduced by approximately 75%. The combined efficacy and tolerability of dosages >80 mg/d has not been evaluated. Adverse effects seen with tetrabenazine at higher dosages include akathisia, anxiety, insomnia, parkinsonism, fatigue, and depression.
A daily dose of 40 mg may be considered for some patients based on tolerability, including those who are known CYP 2D6 poor metabolizers, and those taking strong CYP2D6 inhibitors.2 For those taking strong 3A4 inhibitors, the maximum daily dose is 40 mg. Concomitant use of valbenazine with strong 3A4 inducers is not recommended as the exposure to the active metabolite is reduced by approximately 75%.2 Lastly, because VMAT2 inhibition may alter synaptic levels of other monoamines, it is recommended that valbenazine not be administered with monoamine oxidase inhibitors, such as isocarboxazid, phenelzine, or selegiline.
Contraindications
There are no reported contraindications for valbenazine. As with most medications, there is limited available data on valbenazine use in pregnant women; however, administration of valbenazine to pregnant rats during organogenesis through lactation produced an increase in the number of stillborn pups and postnatal pup mortalities at doses under the maximum recommended human dose (MRHD) using body surface area based dosing (mg/m2). Pregnant women should be advised of the potential risk to a fetus. Valbenazine and its metabolites have been detected in rat milk at concentrations higher than in plasma after oral administration of valbenazine at doses 0.1 to 1.2 times the MRHD (based on mg/m2). Based on animal findings of increased perinatal mortality in exposed fetuses and pups, woman are advised not to breastfeed during valbenazine treatment and for 5 days after the final dose. No dosage adjustment is required for geriatric patients.
1. O’Brien CF, Jimenez R, Hauser RA, et al. NBI-98854, a selective monoamine transport inhibitor for the treatment of tardive dyskinesia: a randomized, double-blind, placebo-controlled study. Mov Disord. 2015;30(12):1681-1687.
2. Ingrezza [package insert]. San Diego, CA: Neurocrine Biosciences Inc.; 2017.
3. Marder S, Knesevich MA, Hauser RA, et al. KINECT 3: A randomized, double-blind, placebo-controlled phase 3 trial of valbenazine (NBI-98854) for tardive dyskinesia. Poster presented at the American Psychiatric Association Annual Meeting; May 14-18, 2016; Atlanta, GA.
4. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
5. Richardson MA, Bevans ML, Read LL, et al. Efficacy of the branched-chain amino acids in the treatment of tardive dyskinesia in men. Am J Psychiatry. 2003;160(6):1117-1124.
6. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
7. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
10. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
11. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
12. Grigoriadis DE, Smith E, Madan A, et al. Pharmacologic characteristics of valbenazine (NBI-98854) and its metabolites. Poster presented at the U.S. Psychiatric & Mental Health Congress, October 21-24, 2016; San Antonio, TX.
Despite improvements in the tolerability of antipsychotic medications, the development of tardive dyskinesia (TD) still is a significant area of concern; however, clinicians have had few treatment options. Valbenazine, a vesicular monoamine transport type 2 (VMAT2) inhibitor, is the only FDA-approved medication for TD (Table 1).1 By modulating dopamine transport into presynaptic vesicles, synaptic dopamine release is decreased, thereby reducing the postsynaptic stimulation of D2 receptors and the severity of dyskinetic movements.
In the pivotal 6-week clinical trial, valbenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) ratings.2 Study completion rates were high (87.6%), with only 2 dropouts because of adverse events in each of the placebo (n = 78) and 40-mg (n = 76) arms, and 3 in the 80-mg group (n = 80).
Before the development of valbenazine, tetrabenazine was the only effective option for treating TD. Despite tetrabenazine’s known efficacy for TD, it was not available in the United States until 2008 with the sole indication for movements related to Huntington’s disease. U.S. patients often were subjected to a litany of ineffective medications for TD, often at great expense. Moreover, tetrabenazine involved multiple daily dosing, required cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, had significant tolerability issues, and a monthly cost of $8,000 to $10,000. The availability of an agent that is effective for TD and does not have tetrabenazine’s kinetic limitations, adverse effect profile, or CYP2D6 monitoring requirements represents an enormous advance in the treatment of TD.
Clinical implications
Tardive dyskinesia remains a significant public health concern because of the increasing use of antipsychotics for disorders beyond the core indication for schizophrenia. Although exposure to dopamine D2 antagonism could result in postsynaptic receptor upregulation and supersensitivity, this process best explains what underlies withdrawal dyskinesia.3 The persistence of TD symptoms in 66% to 80% of patients after discontinuing offending agents has led to hypotheses that the underlying pathophysiology of TD might best be conceptualized as a problem with neuroplasticity. As with many disorders, environmental contributions (eg, oxidative stress) and genetic predisposition might play a role beyond that related to exposure to D2 antagonism.3
There have been trials of numerous agents, but no medication has been FDA-approved for treating TD, and limited data support the efficacy of a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]),4 albeit with small effect sizes. A medical food, consisting of branched-chain amino acids, received FDA approval for the dietary management of TD in males, but is no longer commercially available except from compounding pharmacies.5
Tetrabenazine, a molecule developed in the mid-1950s to improve on the tolerability of reserpine, was associated with significant adverse effects such as orthostasis.6 Like reserpine, tetrabenazine subsequently was found to be effective for TD7 but without the peripheral adverse effects of reserpine. However, the kinetics of tetrabenazine necessitated multiple daily doses, and required CYP2D6 genotyping for doses >50 mg/d.8
Receptor blocking. The mechanism that differentiated reserpine’s and tetrabenazine’s clinical properties became clearer in the 1980s when researchers discovered that transporters were necessary to package neurotransmitters into the synaptic vesicles of presynaptic neurons.9 The vesicular monoamine transporter (VMAT) exists in 2 isoforms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the central nervous system.10
Tetrabenazine’s improved tolerability profile was related to the fact that it is a specific and reversible VMAT2 inhibitor, while reserpine is an irreversible and nonselective antagonist of both VMAT isoforms. Investigation of tetrabenazine’s metabolism revealed that it is rapidly and extensively converted into 2 isomers, α-dihydrotetrabenazine (DH-TBZ) and β-DH-TBZ. The isomeric forms of DH-TBZ have multiple chiral centers, and therefore numerous forms of which only 2 are significantly active at VMAT2.3 The α–DH-TBZ isomer is metabolized via CYP2D6 and 3A4 into inactive metabolites, while β-DH-TBZ is metabolized solely via 2D6.3 Because of the short half-life of DH-TBZ when generated from oral tetrabenazine, the existence of 2D6 polymorphisms, and the predominant activity deriving from only 2 isomers, a molecule was synthesized (valbenazine), that when metabolized would slowly be converted into the most active isomer of α–DH-TBZ designated as NBI-98782 (Table 2). This slower conversion to NBI-98782 from valbenazine (compared with its formation from oral tetrabenazine) yielded improved kinetics and permitted once-daily dosing; moreover, because the metabolism of NBI-98782 is not solely dependent on CYP2D6, the need for genotyping was removed. Neither of the 2 metabolites from valbenazine NBI-98782 and NB-136110 have significant affinity for targets other than VMAT2.11
Use in tardive dyskinesia. Recommended starting dosage is 40 mg once daily with or without food, increased to 80 mg after 1 week, based on the design and results from the phase-III clinical trial.12 The FDA granted breakthrough therapy designation for this compound, and only 1 phase-III trial was performed. Valbenazine produced significant improvement on the AIMS, with a mean 30% reduction in AIMS scores at the Week 6 endpoint from baseline of 10.4 ± 3.6.2 The effect size was large (Cohen’s d = 0.90) for the 80-mg dosage. Continuation of 40 mg/d may be considered for some patients based on tolerability, including those who are known CYP2D6 poor metabolizers, and those taking strong CYP2D6 inhibitors. Patients taking strong 3A4 inhibitors should not exceed 40 mg/d. The maximum daily dose is 40 mg for those who have moderate or severe hepatic impairment (Child-Pugh score, 7 to 15). Dosage adjustment is not required for mild to moderate renal impairment (creatinine clearance, 30 to 90 mL/min).
Pharmacologic profile, adverse reactions
Valbenazine and its 2 metabolites lack affinity for receptors other than VMAT2, leading to an absence of orthostasis in clinical trials.1,2 In the phase-II trial, 76% of participants receiving valbenazine (n = 51) were titrated to the maximum dosage of 75 mg/d. Common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were headache (9.8% vs 4.1% placebo), fatigue (9.8% vs 4.1% placebo), and somnolence (5.9% vs 2% placebo).1 In the phase-III trial, participants were randomized 1:1:1 to valbenazine, 40 mg (n = 72), valbenazine, 80 mg (n = 79), or placebo (n = 76). In the clinical studies the most common diagnosis was schizophrenia or schizoaffective disorder, and 40% and 85% of participants in the phase-II and phase-III studies, respectively, remained on antipsychotics.1,2 There were no adverse effects with an incidence ≥5% and at least twice the rate of placebo in the phase-III trial.2
When data from all placebo-controlled studies were pooled, only 1 adverse effect occurred with an incidence ≥5% and twice that of placebo, somnolence with a rate of 10.9% for valbenazine vs 4.2% for placebo. The incidence of akathisia in the pooled analysis was 2.7% for valbenazine vs 0.5% for placebo. Importantly, in neither study was there a safety signal related to depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms.
The mean QT prolongation for valbenazine in healthy participants was 6.7 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 8.4 milliseconds. For those taking strong 2D6 or 3A4 inhibitors, or known 2D6 poor metabolizers, the mean QT prolongation was 11.7 milliseconds (14.7 milliseconds upper bound of double-sided 90% CI). In the controlled trials, there was a dose-related increase in prolactin, alkaline phosphatase, and bilirubin. Overall, 3% of valbenazine-treated patients and 2% of placebo-treated patients discontinued because of adverse reactions.
As noted above, there were no adverse effects with an incidence ≥5% and at least twice the rate of placebo in the phase-III valbenazine trial. Aggregate data across all placebo-controlled studies found that somnolence was the only adverse effect that occurred with an incidence ≥5% and twice that of placebo (10.9% for valbenazine vs 4.2% for placebo).2 As a comparsion, rates of sedation and akathisia for tetrabenazine were higher in the pivotal Huntington’s disease trial: sedation/somnolence 31% vs 3% for placebo, and akathisia 19% vs 0% for placebo.8
How it works
Tetrabenazine, a selective VMAT2 inhibitor, is the only agent that has demonstrated significant efficacy and tolerability for TD management; however, its complex metabolism generates numerous isomers of the metabolites α-DH-TBZ and β-DH-TBZ, of which only 2 are significantly active (Table 3). By choosing an active isomer (NBI-98782) as the metabolite of interest because of its selective and potent activity at VMAT2 and having a metabolism not solely dependent on CYP2D6, a compound was generated (valbenazine) that when metabolized slowly converts into NBI-98782.
Pharmacokinetics
Valbenazine demonstrates dose-proportional pharmacokinetics after single oral dosages from 40 to 300 mg with no impact of food or fasting status on levels of the active metabolite. Valbenazine has a Tmax of 0.5 to 1.0 hours, with 49% oral bioavailability. The plasma half-life for valbenazine and for NBI-98782 ranges from 15 to 22 hours. The Tmax for NBI-98782 when formed from valbenazine occurs between 4 and 8 hours, with a Cmax of approximately 30 ng/mL. It should be noted that when NBI-98782 is generated from oral tetrabenazine, the mean half-life and Tmax are considerably shorter (6 hours and 1.5 hours, respectively), while the Cmax is much higher (approximately 77 ng/mL) (Table 4).
Valbenazine is metabolized through endogenous esterases to NBI-98782 and NBI-136110. NBI-98782, the active metabolite, is further metabolized through multiple CYP pathways, predominantly 3A4 and 2D6. Neither valbenazine nor its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that valbenazine and its active metabolite are unlikely to inhibit most major drug transporters at clinically relevant concentrations. However, valbenazine increased digoxin levels because of inhibition of intestinal P-glycoprotein; therefore plasma digoxin level monitoring is recommended when these 2 are co-administered.
Efficacy
Efficacy was established in a 6-week, fixed-dosage, double-blind, placebo-controlled trial of adult patients with TD. Eligible participants had:
- DSM-IV diagnosis of antipsychotic-induced TD for ≥3 months before screening and moderate or severe TD, as indicated by AIMS item 8 (severity of abnormal movement), which was rated by a blinded, external reviewer using a video of the participant’s AIMS assessment at screening
- a DSM-IV diagnosis of schizophrenia or schizoaffective disorder or mood disorder (and stable per investigator)
- Brief Psychiatric Rating Scale score <50 at screening.
Exclusion criteria included clinically significant and unstable medical conditions within 1 month before screening; comorbid movement disorder (eg, parkinsonism, akathisia, truncal dystonia) that was more prominent than TD; and significant risk for active suicidal ideation, suicidal behavior, or violent behavior.2 Participants had a mean age of 56, 52% were male, and 65.7% of participants in the valbenazine 40-mg group had a schizophrenia spectrum disorder diagnosis, as did 65.8% in both the placebo and valbenazine 80-mg arms.
Antipsychotic treatments were permitted during the trial and >85% of participants continued taking these medications during the study. Participants (N = 234) were randomly allocated in a 1:1:1 manner to valbenazine 40 mg, 80 mg, or matched placebo. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. Baseline AIMS scores were 9.9 ± 4.3 in the placebo group, and 9.8 ± 4.1 and 10.4 ± 3.6 in the valbenazine 40-mg and 80-mg arms, respectively.2
Outcome. A fixed-sequence testing procedure to control for family-wise error rate and multiplicity was employed, and the primary endpoint was change from baseline to Week 6 in AIMS total score (items 1 to 7) for valbenazine 80 mg vs placebo. Valbenazine, 40 mg, was associated with a 1.9 point decrease in AIMS score, while valbenazine, 80 mg, was associated with a 3.2 point decrease in AIMS score, compared with 0.1 point decrease for placebo (P < .05 for valbenazine, 40 mg, P < .001 for valbenazine, 80 mg). This difference for the 40-mg dosage did not meet the prespecified analysis endpoints; however, for the 80-mg valbenazine dosage, the effect size for this difference (Cohen’s d) was large 0.90. There also were statistically significant differences between 40 mg and 80 mg at weeks 2, 4, and 6 in the intent-to-treat population. Of the 79 participants, 43 taking the 80-mg dosage completed a 48-week extension. Efficacy was sustained in this group; however, when valbenazine was discontinued at Week 48, AIMS scores returned to baseline after 4 weeks.
Tolerability
Of the 234 randomized patients, 205 (87.6%) completed the 6-week trial. Discontinuations due to adverse events were low across all treatment groups: 2.6% and 2.8% in the placebo and valbenazine 40-mg arms, respectively, and 3.8% in valbenazine 80-mg cohort. There was no safety signal based on changes in depression, suicidality, parkinsonism rating, or changes in schizophrenia symptoms. Because valbenazine can cause somnolence, patients should not perform activities requiring mental alertness (eg, operating a vehicle or hazardous machinery) until they know how they will be affected by valbenazine.
Valbenazine should be avoided in patients with congenital long QT syndrome or with arrhythmias associated with a prolonged QT interval. For patients at increased risk of a prolonged QT interval, assess the QT interval before increasing the dosage.
Clinical considerations
Unique properties. Valbenazine is metabolized slowly to a potent, selective VMAT2 antagonist (NBI-98782) in a manner that permits once daily dosing, removes the need for CYP2D6 genotyping, and provides significant efficacy.
Why Rx? The reasons to prescribe valbenazine for TD patients include:
- currently the only agent with FDA approval for TD
- fewer tolerability issues seen with the only other effective agent, tetrabenazine
- no signal for effects on mood parameters or rates of parkinsonism
- lack of multiple daily dosing and possible need for 2D6 genotyping involved with TBZ prescribing.
Dosing
The recommended dosage of valbenazine is 80 mg/d administered as a single dose with or without food, starting at 40 mg once daily for 1 week. There is no dosage adjustment required in those with mild to moderate renal impairment; however, valbenazine is not recommended in those with severe renal impairment. The maximum dose is 40 mg/d for those who with moderate or severe hepatic impairment (Child-Pugh score, 7 to 15) however, valbenazine is not recommended for patients with severe renal impairment (creatinine clearance <30 mL/min) because the exposure to the active metabolite is reduced by approximately 75%. The combined efficacy and tolerability of dosages >80 mg/d has not been evaluated. Adverse effects seen with tetrabenazine at higher dosages include akathisia, anxiety, insomnia, parkinsonism, fatigue, and depression.
A daily dose of 40 mg may be considered for some patients based on tolerability, including those who are known CYP 2D6 poor metabolizers, and those taking strong CYP2D6 inhibitors.2 For those taking strong 3A4 inhibitors, the maximum daily dose is 40 mg. Concomitant use of valbenazine with strong 3A4 inducers is not recommended as the exposure to the active metabolite is reduced by approximately 75%.2 Lastly, because VMAT2 inhibition may alter synaptic levels of other monoamines, it is recommended that valbenazine not be administered with monoamine oxidase inhibitors, such as isocarboxazid, phenelzine, or selegiline.
Contraindications
There are no reported contraindications for valbenazine. As with most medications, there is limited available data on valbenazine use in pregnant women; however, administration of valbenazine to pregnant rats during organogenesis through lactation produced an increase in the number of stillborn pups and postnatal pup mortalities at doses under the maximum recommended human dose (MRHD) using body surface area based dosing (mg/m2). Pregnant women should be advised of the potential risk to a fetus. Valbenazine and its metabolites have been detected in rat milk at concentrations higher than in plasma after oral administration of valbenazine at doses 0.1 to 1.2 times the MRHD (based on mg/m2). Based on animal findings of increased perinatal mortality in exposed fetuses and pups, woman are advised not to breastfeed during valbenazine treatment and for 5 days after the final dose. No dosage adjustment is required for geriatric patients.
Despite improvements in the tolerability of antipsychotic medications, the development of tardive dyskinesia (TD) still is a significant area of concern; however, clinicians have had few treatment options. Valbenazine, a vesicular monoamine transport type 2 (VMAT2) inhibitor, is the only FDA-approved medication for TD (Table 1).1 By modulating dopamine transport into presynaptic vesicles, synaptic dopamine release is decreased, thereby reducing the postsynaptic stimulation of D2 receptors and the severity of dyskinetic movements.
In the pivotal 6-week clinical trial, valbenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) ratings.2 Study completion rates were high (87.6%), with only 2 dropouts because of adverse events in each of the placebo (n = 78) and 40-mg (n = 76) arms, and 3 in the 80-mg group (n = 80).
Before the development of valbenazine, tetrabenazine was the only effective option for treating TD. Despite tetrabenazine’s known efficacy for TD, it was not available in the United States until 2008 with the sole indication for movements related to Huntington’s disease. U.S. patients often were subjected to a litany of ineffective medications for TD, often at great expense. Moreover, tetrabenazine involved multiple daily dosing, required cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, had significant tolerability issues, and a monthly cost of $8,000 to $10,000. The availability of an agent that is effective for TD and does not have tetrabenazine’s kinetic limitations, adverse effect profile, or CYP2D6 monitoring requirements represents an enormous advance in the treatment of TD.
Clinical implications
Tardive dyskinesia remains a significant public health concern because of the increasing use of antipsychotics for disorders beyond the core indication for schizophrenia. Although exposure to dopamine D2 antagonism could result in postsynaptic receptor upregulation and supersensitivity, this process best explains what underlies withdrawal dyskinesia.3 The persistence of TD symptoms in 66% to 80% of patients after discontinuing offending agents has led to hypotheses that the underlying pathophysiology of TD might best be conceptualized as a problem with neuroplasticity. As with many disorders, environmental contributions (eg, oxidative stress) and genetic predisposition might play a role beyond that related to exposure to D2 antagonism.3
There have been trials of numerous agents, but no medication has been FDA-approved for treating TD, and limited data support the efficacy of a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]),4 albeit with small effect sizes. A medical food, consisting of branched-chain amino acids, received FDA approval for the dietary management of TD in males, but is no longer commercially available except from compounding pharmacies.5
Tetrabenazine, a molecule developed in the mid-1950s to improve on the tolerability of reserpine, was associated with significant adverse effects such as orthostasis.6 Like reserpine, tetrabenazine subsequently was found to be effective for TD7 but without the peripheral adverse effects of reserpine. However, the kinetics of tetrabenazine necessitated multiple daily doses, and required CYP2D6 genotyping for doses >50 mg/d.8
Receptor blocking. The mechanism that differentiated reserpine’s and tetrabenazine’s clinical properties became clearer in the 1980s when researchers discovered that transporters were necessary to package neurotransmitters into the synaptic vesicles of presynaptic neurons.9 The vesicular monoamine transporter (VMAT) exists in 2 isoforms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the central nervous system.10
Tetrabenazine’s improved tolerability profile was related to the fact that it is a specific and reversible VMAT2 inhibitor, while reserpine is an irreversible and nonselective antagonist of both VMAT isoforms. Investigation of tetrabenazine’s metabolism revealed that it is rapidly and extensively converted into 2 isomers, α-dihydrotetrabenazine (DH-TBZ) and β-DH-TBZ. The isomeric forms of DH-TBZ have multiple chiral centers, and therefore numerous forms of which only 2 are significantly active at VMAT2.3 The α–DH-TBZ isomer is metabolized via CYP2D6 and 3A4 into inactive metabolites, while β-DH-TBZ is metabolized solely via 2D6.3 Because of the short half-life of DH-TBZ when generated from oral tetrabenazine, the existence of 2D6 polymorphisms, and the predominant activity deriving from only 2 isomers, a molecule was synthesized (valbenazine), that when metabolized would slowly be converted into the most active isomer of α–DH-TBZ designated as NBI-98782 (Table 2). This slower conversion to NBI-98782 from valbenazine (compared with its formation from oral tetrabenazine) yielded improved kinetics and permitted once-daily dosing; moreover, because the metabolism of NBI-98782 is not solely dependent on CYP2D6, the need for genotyping was removed. Neither of the 2 metabolites from valbenazine NBI-98782 and NB-136110 have significant affinity for targets other than VMAT2.11
Use in tardive dyskinesia. Recommended starting dosage is 40 mg once daily with or without food, increased to 80 mg after 1 week, based on the design and results from the phase-III clinical trial.12 The FDA granted breakthrough therapy designation for this compound, and only 1 phase-III trial was performed. Valbenazine produced significant improvement on the AIMS, with a mean 30% reduction in AIMS scores at the Week 6 endpoint from baseline of 10.4 ± 3.6.2 The effect size was large (Cohen’s d = 0.90) for the 80-mg dosage. Continuation of 40 mg/d may be considered for some patients based on tolerability, including those who are known CYP2D6 poor metabolizers, and those taking strong CYP2D6 inhibitors. Patients taking strong 3A4 inhibitors should not exceed 40 mg/d. The maximum daily dose is 40 mg for those who have moderate or severe hepatic impairment (Child-Pugh score, 7 to 15). Dosage adjustment is not required for mild to moderate renal impairment (creatinine clearance, 30 to 90 mL/min).
Pharmacologic profile, adverse reactions
Valbenazine and its 2 metabolites lack affinity for receptors other than VMAT2, leading to an absence of orthostasis in clinical trials.1,2 In the phase-II trial, 76% of participants receiving valbenazine (n = 51) were titrated to the maximum dosage of 75 mg/d. Common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were headache (9.8% vs 4.1% placebo), fatigue (9.8% vs 4.1% placebo), and somnolence (5.9% vs 2% placebo).1 In the phase-III trial, participants were randomized 1:1:1 to valbenazine, 40 mg (n = 72), valbenazine, 80 mg (n = 79), or placebo (n = 76). In the clinical studies the most common diagnosis was schizophrenia or schizoaffective disorder, and 40% and 85% of participants in the phase-II and phase-III studies, respectively, remained on antipsychotics.1,2 There were no adverse effects with an incidence ≥5% and at least twice the rate of placebo in the phase-III trial.2
When data from all placebo-controlled studies were pooled, only 1 adverse effect occurred with an incidence ≥5% and twice that of placebo, somnolence with a rate of 10.9% for valbenazine vs 4.2% for placebo. The incidence of akathisia in the pooled analysis was 2.7% for valbenazine vs 0.5% for placebo. Importantly, in neither study was there a safety signal related to depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms.
The mean QT prolongation for valbenazine in healthy participants was 6.7 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 8.4 milliseconds. For those taking strong 2D6 or 3A4 inhibitors, or known 2D6 poor metabolizers, the mean QT prolongation was 11.7 milliseconds (14.7 milliseconds upper bound of double-sided 90% CI). In the controlled trials, there was a dose-related increase in prolactin, alkaline phosphatase, and bilirubin. Overall, 3% of valbenazine-treated patients and 2% of placebo-treated patients discontinued because of adverse reactions.
As noted above, there were no adverse effects with an incidence ≥5% and at least twice the rate of placebo in the phase-III valbenazine trial. Aggregate data across all placebo-controlled studies found that somnolence was the only adverse effect that occurred with an incidence ≥5% and twice that of placebo (10.9% for valbenazine vs 4.2% for placebo).2 As a comparsion, rates of sedation and akathisia for tetrabenazine were higher in the pivotal Huntington’s disease trial: sedation/somnolence 31% vs 3% for placebo, and akathisia 19% vs 0% for placebo.8
How it works
Tetrabenazine, a selective VMAT2 inhibitor, is the only agent that has demonstrated significant efficacy and tolerability for TD management; however, its complex metabolism generates numerous isomers of the metabolites α-DH-TBZ and β-DH-TBZ, of which only 2 are significantly active (Table 3). By choosing an active isomer (NBI-98782) as the metabolite of interest because of its selective and potent activity at VMAT2 and having a metabolism not solely dependent on CYP2D6, a compound was generated (valbenazine) that when metabolized slowly converts into NBI-98782.
Pharmacokinetics
Valbenazine demonstrates dose-proportional pharmacokinetics after single oral dosages from 40 to 300 mg with no impact of food or fasting status on levels of the active metabolite. Valbenazine has a Tmax of 0.5 to 1.0 hours, with 49% oral bioavailability. The plasma half-life for valbenazine and for NBI-98782 ranges from 15 to 22 hours. The Tmax for NBI-98782 when formed from valbenazine occurs between 4 and 8 hours, with a Cmax of approximately 30 ng/mL. It should be noted that when NBI-98782 is generated from oral tetrabenazine, the mean half-life and Tmax are considerably shorter (6 hours and 1.5 hours, respectively), while the Cmax is much higher (approximately 77 ng/mL) (Table 4).
Valbenazine is metabolized through endogenous esterases to NBI-98782 and NBI-136110. NBI-98782, the active metabolite, is further metabolized through multiple CYP pathways, predominantly 3A4 and 2D6. Neither valbenazine nor its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that valbenazine and its active metabolite are unlikely to inhibit most major drug transporters at clinically relevant concentrations. However, valbenazine increased digoxin levels because of inhibition of intestinal P-glycoprotein; therefore plasma digoxin level monitoring is recommended when these 2 are co-administered.
Efficacy
Efficacy was established in a 6-week, fixed-dosage, double-blind, placebo-controlled trial of adult patients with TD. Eligible participants had:
- DSM-IV diagnosis of antipsychotic-induced TD for ≥3 months before screening and moderate or severe TD, as indicated by AIMS item 8 (severity of abnormal movement), which was rated by a blinded, external reviewer using a video of the participant’s AIMS assessment at screening
- a DSM-IV diagnosis of schizophrenia or schizoaffective disorder or mood disorder (and stable per investigator)
- Brief Psychiatric Rating Scale score <50 at screening.
Exclusion criteria included clinically significant and unstable medical conditions within 1 month before screening; comorbid movement disorder (eg, parkinsonism, akathisia, truncal dystonia) that was more prominent than TD; and significant risk for active suicidal ideation, suicidal behavior, or violent behavior.2 Participants had a mean age of 56, 52% were male, and 65.7% of participants in the valbenazine 40-mg group had a schizophrenia spectrum disorder diagnosis, as did 65.8% in both the placebo and valbenazine 80-mg arms.
Antipsychotic treatments were permitted during the trial and >85% of participants continued taking these medications during the study. Participants (N = 234) were randomly allocated in a 1:1:1 manner to valbenazine 40 mg, 80 mg, or matched placebo. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. Baseline AIMS scores were 9.9 ± 4.3 in the placebo group, and 9.8 ± 4.1 and 10.4 ± 3.6 in the valbenazine 40-mg and 80-mg arms, respectively.2
Outcome. A fixed-sequence testing procedure to control for family-wise error rate and multiplicity was employed, and the primary endpoint was change from baseline to Week 6 in AIMS total score (items 1 to 7) for valbenazine 80 mg vs placebo. Valbenazine, 40 mg, was associated with a 1.9 point decrease in AIMS score, while valbenazine, 80 mg, was associated with a 3.2 point decrease in AIMS score, compared with 0.1 point decrease for placebo (P < .05 for valbenazine, 40 mg, P < .001 for valbenazine, 80 mg). This difference for the 40-mg dosage did not meet the prespecified analysis endpoints; however, for the 80-mg valbenazine dosage, the effect size for this difference (Cohen’s d) was large 0.90. There also were statistically significant differences between 40 mg and 80 mg at weeks 2, 4, and 6 in the intent-to-treat population. Of the 79 participants, 43 taking the 80-mg dosage completed a 48-week extension. Efficacy was sustained in this group; however, when valbenazine was discontinued at Week 48, AIMS scores returned to baseline after 4 weeks.
Tolerability
Of the 234 randomized patients, 205 (87.6%) completed the 6-week trial. Discontinuations due to adverse events were low across all treatment groups: 2.6% and 2.8% in the placebo and valbenazine 40-mg arms, respectively, and 3.8% in valbenazine 80-mg cohort. There was no safety signal based on changes in depression, suicidality, parkinsonism rating, or changes in schizophrenia symptoms. Because valbenazine can cause somnolence, patients should not perform activities requiring mental alertness (eg, operating a vehicle or hazardous machinery) until they know how they will be affected by valbenazine.
Valbenazine should be avoided in patients with congenital long QT syndrome or with arrhythmias associated with a prolonged QT interval. For patients at increased risk of a prolonged QT interval, assess the QT interval before increasing the dosage.
Clinical considerations
Unique properties. Valbenazine is metabolized slowly to a potent, selective VMAT2 antagonist (NBI-98782) in a manner that permits once daily dosing, removes the need for CYP2D6 genotyping, and provides significant efficacy.
Why Rx? The reasons to prescribe valbenazine for TD patients include:
- currently the only agent with FDA approval for TD
- fewer tolerability issues seen with the only other effective agent, tetrabenazine
- no signal for effects on mood parameters or rates of parkinsonism
- lack of multiple daily dosing and possible need for 2D6 genotyping involved with TBZ prescribing.
Dosing
The recommended dosage of valbenazine is 80 mg/d administered as a single dose with or without food, starting at 40 mg once daily for 1 week. There is no dosage adjustment required in those with mild to moderate renal impairment; however, valbenazine is not recommended in those with severe renal impairment. The maximum dose is 40 mg/d for those who with moderate or severe hepatic impairment (Child-Pugh score, 7 to 15) however, valbenazine is not recommended for patients with severe renal impairment (creatinine clearance <30 mL/min) because the exposure to the active metabolite is reduced by approximately 75%. The combined efficacy and tolerability of dosages >80 mg/d has not been evaluated. Adverse effects seen with tetrabenazine at higher dosages include akathisia, anxiety, insomnia, parkinsonism, fatigue, and depression.
A daily dose of 40 mg may be considered for some patients based on tolerability, including those who are known CYP 2D6 poor metabolizers, and those taking strong CYP2D6 inhibitors.2 For those taking strong 3A4 inhibitors, the maximum daily dose is 40 mg. Concomitant use of valbenazine with strong 3A4 inducers is not recommended as the exposure to the active metabolite is reduced by approximately 75%.2 Lastly, because VMAT2 inhibition may alter synaptic levels of other monoamines, it is recommended that valbenazine not be administered with monoamine oxidase inhibitors, such as isocarboxazid, phenelzine, or selegiline.
Contraindications
There are no reported contraindications for valbenazine. As with most medications, there is limited available data on valbenazine use in pregnant women; however, administration of valbenazine to pregnant rats during organogenesis through lactation produced an increase in the number of stillborn pups and postnatal pup mortalities at doses under the maximum recommended human dose (MRHD) using body surface area based dosing (mg/m2). Pregnant women should be advised of the potential risk to a fetus. Valbenazine and its metabolites have been detected in rat milk at concentrations higher than in plasma after oral administration of valbenazine at doses 0.1 to 1.2 times the MRHD (based on mg/m2). Based on animal findings of increased perinatal mortality in exposed fetuses and pups, woman are advised not to breastfeed during valbenazine treatment and for 5 days after the final dose. No dosage adjustment is required for geriatric patients.
1. O’Brien CF, Jimenez R, Hauser RA, et al. NBI-98854, a selective monoamine transport inhibitor for the treatment of tardive dyskinesia: a randomized, double-blind, placebo-controlled study. Mov Disord. 2015;30(12):1681-1687.
2. Ingrezza [package insert]. San Diego, CA: Neurocrine Biosciences Inc.; 2017.
3. Marder S, Knesevich MA, Hauser RA, et al. KINECT 3: A randomized, double-blind, placebo-controlled phase 3 trial of valbenazine (NBI-98854) for tardive dyskinesia. Poster presented at the American Psychiatric Association Annual Meeting; May 14-18, 2016; Atlanta, GA.
4. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
5. Richardson MA, Bevans ML, Read LL, et al. Efficacy of the branched-chain amino acids in the treatment of tardive dyskinesia in men. Am J Psychiatry. 2003;160(6):1117-1124.
6. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
7. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
10. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
11. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
12. Grigoriadis DE, Smith E, Madan A, et al. Pharmacologic characteristics of valbenazine (NBI-98854) and its metabolites. Poster presented at the U.S. Psychiatric & Mental Health Congress, October 21-24, 2016; San Antonio, TX.
1. O’Brien CF, Jimenez R, Hauser RA, et al. NBI-98854, a selective monoamine transport inhibitor for the treatment of tardive dyskinesia: a randomized, double-blind, placebo-controlled study. Mov Disord. 2015;30(12):1681-1687.
2. Ingrezza [package insert]. San Diego, CA: Neurocrine Biosciences Inc.; 2017.
3. Marder S, Knesevich MA, Hauser RA, et al. KINECT 3: A randomized, double-blind, placebo-controlled phase 3 trial of valbenazine (NBI-98854) for tardive dyskinesia. Poster presented at the American Psychiatric Association Annual Meeting; May 14-18, 2016; Atlanta, GA.
4. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
5. Richardson MA, Bevans ML, Read LL, et al. Efficacy of the branched-chain amino acids in the treatment of tardive dyskinesia in men. Am J Psychiatry. 2003;160(6):1117-1124.
6. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
7. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
10. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
11. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
12. Grigoriadis DE, Smith E, Madan A, et al. Pharmacologic characteristics of valbenazine (NBI-98854) and its metabolites. Poster presented at the U.S. Psychiatric & Mental Health Congress, October 21-24, 2016; San Antonio, TX.
USPSTF recommendations: A 2017 roundup
Since the last Practice Alert update on the United States Preventive Services Task Force in May of 2016,1 the Task Force has released 19 recommendations on 13 topics that include: the use of aspirin and statins for the prevention of cardiovascular disease (CVD); support for breastfeeding; use of folic acid during pregnancy; and screening for syphilis, latent tuberculosis (TB), herpes, chronic obstructive pulmonary disease (COPD), colorectal cancer (CRC), obstructive sleep apnea (OSA), celiac disease, and skin cancer. The Task Force also released a draft recommendation regarding prostate cancer screening in asymptomatic men (see “A change for prostate cancer screening?”) and addressed screening pelvic examinations in asymptomatic women, the subject of this month’s audiocast. (To listen, go to: http://bit.ly/2nIVoD5.)
Recommendations to implement
Recommendations from the past year that family physicians should put into practice are detailed below and in TABLE 1.2-8

CRC: Screen all individuals ages 50 to 75, but 76 to 85 selectively. The Task Force reaffirmed its 2008 finding that screening for CRC in adults ages 50 to 75 years is substantially beneficial.2 In contrast to the previous recommendation, however, the new one does not state which screening tests are preferred. The tests considered were 3 stool tests (fecal immunochemical test [FIT], FIT-tumor DNA testing [FIT-DNA], and guaiac-based fecal occult blood test [gFOBT]), as well as 3 direct visualization tests (colonoscopy, sigmoidoscopy, and CT colonoscopy). The Task Force assessed various testing frequencies of each test and some test combinations. While the Task Force does not recommend any one screening strategy, there are still significant unknowns about FIT-DNA and CT colonoscopy. The American Academy of Family Physicians does not recommend using these 2 tests for screening purposes at this time.9
CRC screening for adults ages 76 to 85 was given a “C” recommendation, which means the value of the service to the population overall is small, but that certain individuals may benefit from it. The Task Force advises selectively offering a “C” service to individuals based on professional judgment and patient preferences. Regarding CRC screening in individuals 76 years or older, the ones most likely to benefit are those who have never been screened and those without significant comorbidities that could limit life expectancy. All “C” recommendations from the past year are listed in TABLE 2.2-4

CVD prevention: When aspirin or a statin is indicated. The Task Force released 2 recommendations for the prevention of CVD this past year. One pertained to the use of low-dose aspirin3 (which also helps to prevent CRC), and the other addressed the use of low- to moderate-dose statins.4 Each recommendation is fairly complicated and nuanced in terms of age and risk for CVD. A decision to use low-dose aspirin must also consider the risk of bleeding.
To calculate a patient’s risk for CVD, the Task Force recommends using the risk calculator developed by the American College of Cardiology and the American Heart Association (http://www.cvriskcalculator.com/).
Adults for whom low-dose aspirin and low- to moderate-dose statins are recommended are described in TABLE 1.2-8 Patients for whom individual decision making is advised, rather than a generalized recommendation, are reviewed in TABLE 2.2-4 There is insufficient evidence to make a recommendation for the use of aspirin before age 50 or at age 70 and older,3 and for the use of statins in adults age 76 and older who do not have a history of CVD4 (TABLE 33,4,10-14). The use of low-dose aspirin and low-to-moderate dose statins have been the subject of JFP audiocasts in May 2016 and January 2017. (See http://bit.ly/2oiun8d and http://bit.ly/2oqkohR.)

2 pregnancy-related recommendations. To prevent neural tube defects in newborns, the Task Force now recommends daily folic acid, 0.4 to 0.8 mg (400 to 800 mcg), for all women who are planning on or are capable of becoming pregnant.5 This is an update of a 2009 recommendation that was worded slightly differently, recommending the supplement for all women of childbearing age.
A new recommendation on breastfeeding recognizes its benefits for both mother and baby and finds that interventions to encourage breastfeeding increase the prevalence of this practice and its duration.6 Interventions—provided to individuals or groups by professionals or peers or through formal education—include promoting the benefits of breastfeeding, giving practical advice and direct support on how to breastfeed, and offering psychological support.
Latent TB: Advantages of newer testing method. The recommendation on screening for latent tuberculosis (TB) is an update from the one made in 1996.7 At that time, screening for latent infection was performed using a tuberculin skin test (TST). Now a TST or interferon-gamma release assay (IGRA) can be used. Testing with IGRA may be the best option for those who have received a bacille Calmette–Guérin vaccination (because it can cause a false-positive TST) or for those who are not likely to return to have their TST read.
Those at high risk for latent TB include people who were born or have resided in countries with a high TB prevalence, those who have lived in a correctional institution or homeless shelter, and anyone in a high-risk group based on local epidemiology of the disease. (Read more on TB in this month’s Case Report.) Others at high risk are those who are immune suppressed because of infection or medications, and those who work in health care or correctional facilities. Screening of these groups is usually conducted as part of occupational health or is considered part of routine health care.
Syphilis: Screen high-risk individuals in 2 steps. The recommendation on syphilis screening basically reaffirms the one from 2004.8 Those at high risk for syphilis include men who have sex with men (who now account for 90% of new cases), those who are HIV positive, and those who engage in commercial sex work. Other racial and demographic groups can be at high risk depending on the local epidemiology of the disease. In a separate recommendation, the Task Force advises screening all pregnant women for syphilis.
Screening for syphilis infection involves 2 steps: first, a nontreponemal test (Venereal Disease Research Laboratory [VDRL] or rapid plasma reagin [RPR] test); second, a confirmatory treponemal antibody detection test (fluorescent treponemal antibody absorption [FTA-ABS] or Treponema pallidum particle agglutination [TPPA] test). Treatment for syphilis remains benzathine penicillin with the number of injections depending on the stage of infection. The Centers for Disease Control and Prevention is the best source for current recommendations for treatment of all sexually transmitted infections.15
Screening tests to avoid
TABLE 416,17 lists screening tests the Task Force recommends against. While chronic obstructive pulmonary disease afflicts 14% of US adults ages 40 to 79 years and is the third leading cause of death in the country, the Task Force found that early detection in asymptomatic adults does not affect the course of the illness and is of no benefit.16

Genital herpes, also prevalent, infects an estimated one out of 6 individuals, ages 14 to 49. It causes little mortality, except in neonates, but those infected can have recurrent flares and suffer psychological harms from stigmatization. Most genital herpes is caused by herpes simplex virus-2, and there is a serological test to detect it. However, the Task Force recommends against using the test to screen asymptomatic adults and adolescents, including those who are pregnant. This recommendation is based on the test’s high false-positive rate, which can cause emotional harm, and on the lack of evidence that detection through screening improves outcomes.17
The evidence is lacking for these practices
The Task Force is one of only a few organizations that will not make a recommendation if evidence is lacking on benefits and harms. In addition to the ‘I’ statements regarding CVD and CRC mentioned earlier, the Task Force found insufficient evidence to recommend screening for lipid disorders in individuals ages 20 years or younger,10 performing a visual skin exam as a screening tool for skin cancer,11 screening for celiac disease,12 performing a periodic pelvic examination in asymptomatic women,13 and screening for obstructive sleep apnea using screening questionnaires14 (TABLE 33,4,10-14).
SIDEBAR
A change for prostate cancer screening?The USPSTF recently issued new draft recommendations regarding prostate cancer screening in asymptomatic men (available at: https://screeningforprostatecancer.org/).
The draft now divides men into 2 age groups, stating that the decision to screen for prostate cancer using a prostate specific antigen (PSA) test should be individualized for men ages 55 to 69 years (a C recommendation, meaning that there is at least moderate certainty that the net benefit is small), and that men ages 70 and older (lowered from age 75 in the previous 2012 recommendation1) should not be screened (a D recommendation, meaning that there is moderate or high certainty that the service has no net benefit or that the harms outweigh the benefits).
The USPSTF believes that clinicians should explain to men ages 55 to 69 years that screening offers a small potential benefit of reducing the chance of dying from prostate cancer, but also comes with potential harms, including false-positive results requiring additional testing/procedures, overdiagnosis and overtreatment, and treatment complications such as incontinence and impotence. In this way, each man has the chance to incorporate his values and preferences into the decision.
For men ages 70 and older, the potential benefits simply do not outweigh the potential harms, according to the USPSTF.
1. USPSTF. Final recommendation statement. Prostate cancer: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/prostate-cancer-screening#Pod1. Accessed April 11, 2017.
1. Campos-Outcalt D. Eight USPSTF recommendations FPs need to know about. J Fam Pract. 2016;65:338-341.
2. USPSTF. Colorectal cancer: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/colorectal-cancer-screening2. Accessed March 22, 2017.
3. USPSTF. Aspirin use to prevent cardiovascular disease and colorectal cancer: preventive medications. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/aspirin-to-prevent-cardiovascular-disease-and-cancer. Accessed March 22, 2017.
4. USPSTF. Statin use for the prevention of cardiovascular disease in adults: preventive medication. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/statin-use-in-adults-preventive-medication1. Accessed March 22, 2017.
5. USPSTF. Folic acid for the prevention of neural tube defects: preventive medication. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/folic-acid-for-the-prevention-of-neural-tube-defects-preventive-medication. Accessed March 22, 2017.
6. USPSTF. Breastfeeding: primary care interventions. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/breastfeeding-primary-care-interventions. Accessed March 22, 2017.
7. USPSTF. Latent tuberculosis infection: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/latent-tuberculosis-infection-screening. Accessed March 22, 2017.
8. USPSTF. Syphilis infection in nonpregnant adults and adolescents: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/syphilis-infection-in-nonpregnant-adults-and-adolescents. Accessed March 22, 2017.
9. AAFP. Colorectal cancer screening, adults. Available at: http://www.aafp.org/patient-care/clinical-recommendations/all/colorectal-cancer.html. Accessed March 22, 2017.
10. USPSTF. Lipid disorders in children and adolescents: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/lipid-disorders-in-children-screening1. Accessed March 22, 2017.
11. USPSTF. Skin cancer: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/skin-cancer-screening2. Accessed March 22, 2017.
12. USPSTF. Celiac disease: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/celiac-disease-screening. Accessed March 22, 2017.
13. USPSTF. Gynecological conditions: periodic screening with the pelvic examination. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/gynecological-conditions-screening-with-the-pelvic-examination. Accessed March 22, 2017.
14. USPSTF. Obstructive sleep apnea in adults: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/obstructive-sleep-apnea-in-adults-screening. Accessed March 22, 2017.
15. CDC. 2015 sexually transmitted diseases treatment guidelines. Available at: https://www.cdc.gov/std/tg2015/default.htm. Accessed March 22, 2017.
16. USPSTF. Chronic obstructive pulmonary disease: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/chronic-obstructive-pulmonary-disease-screening. Accessed March 22, 2017.
17. USPSTF. Genital herpes infection: serologic screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/genital-herpes-screening1. Accessed March 22, 2017.
Since the last Practice Alert update on the United States Preventive Services Task Force in May of 2016,1 the Task Force has released 19 recommendations on 13 topics that include: the use of aspirin and statins for the prevention of cardiovascular disease (CVD); support for breastfeeding; use of folic acid during pregnancy; and screening for syphilis, latent tuberculosis (TB), herpes, chronic obstructive pulmonary disease (COPD), colorectal cancer (CRC), obstructive sleep apnea (OSA), celiac disease, and skin cancer. The Task Force also released a draft recommendation regarding prostate cancer screening in asymptomatic men (see “A change for prostate cancer screening?”) and addressed screening pelvic examinations in asymptomatic women, the subject of this month’s audiocast. (To listen, go to: http://bit.ly/2nIVoD5.)
Recommendations to implement
Recommendations from the past year that family physicians should put into practice are detailed below and in TABLE 1.2-8
CRC: Screen all individuals ages 50 to 75, but 76 to 85 selectively. The Task Force reaffirmed its 2008 finding that screening for CRC in adults ages 50 to 75 years is substantially beneficial.2 In contrast to the previous recommendation, however, the new one does not state which screening tests are preferred. The tests considered were 3 stool tests (fecal immunochemical test [FIT], FIT-tumor DNA testing [FIT-DNA], and guaiac-based fecal occult blood test [gFOBT]), as well as 3 direct visualization tests (colonoscopy, sigmoidoscopy, and CT colonoscopy). The Task Force assessed various testing frequencies of each test and some test combinations. While the Task Force does not recommend any one screening strategy, there are still significant unknowns about FIT-DNA and CT colonoscopy. The American Academy of Family Physicians does not recommend using these 2 tests for screening purposes at this time.9
CRC screening for adults ages 76 to 85 was given a “C” recommendation, which means the value of the service to the population overall is small, but that certain individuals may benefit from it. The Task Force advises selectively offering a “C” service to individuals based on professional judgment and patient preferences. Regarding CRC screening in individuals 76 years or older, the ones most likely to benefit are those who have never been screened and those without significant comorbidities that could limit life expectancy. All “C” recommendations from the past year are listed in TABLE 2.2-4
CVD prevention: When aspirin or a statin is indicated. The Task Force released 2 recommendations for the prevention of CVD this past year. One pertained to the use of low-dose aspirin3 (which also helps to prevent CRC), and the other addressed the use of low- to moderate-dose statins.4 Each recommendation is fairly complicated and nuanced in terms of age and risk for CVD. A decision to use low-dose aspirin must also consider the risk of bleeding.
To calculate a patient’s risk for CVD, the Task Force recommends using the risk calculator developed by the American College of Cardiology and the American Heart Association (http://www.cvriskcalculator.com/).
Adults for whom low-dose aspirin and low- to moderate-dose statins are recommended are described in TABLE 1.2-8 Patients for whom individual decision making is advised, rather than a generalized recommendation, are reviewed in TABLE 2.2-4 There is insufficient evidence to make a recommendation for the use of aspirin before age 50 or at age 70 and older,3 and for the use of statins in adults age 76 and older who do not have a history of CVD4 (TABLE 33,4,10-14). The use of low-dose aspirin and low-to-moderate dose statins have been the subject of JFP audiocasts in May 2016 and January 2017. (See http://bit.ly/2oiun8d and http://bit.ly/2oqkohR.)
2 pregnancy-related recommendations. To prevent neural tube defects in newborns, the Task Force now recommends daily folic acid, 0.4 to 0.8 mg (400 to 800 mcg), for all women who are planning on or are capable of becoming pregnant.5 This is an update of a 2009 recommendation that was worded slightly differently, recommending the supplement for all women of childbearing age.
A new recommendation on breastfeeding recognizes its benefits for both mother and baby and finds that interventions to encourage breastfeeding increase the prevalence of this practice and its duration.6 Interventions—provided to individuals or groups by professionals or peers or through formal education—include promoting the benefits of breastfeeding, giving practical advice and direct support on how to breastfeed, and offering psychological support.
Latent TB: Advantages of newer testing method. The recommendation on screening for latent tuberculosis (TB) is an update from the one made in 1996.7 At that time, screening for latent infection was performed using a tuberculin skin test (TST). Now a TST or interferon-gamma release assay (IGRA) can be used. Testing with IGRA may be the best option for those who have received a bacille Calmette–Guérin vaccination (because it can cause a false-positive TST) or for those who are not likely to return to have their TST read.
Those at high risk for latent TB include people who were born or have resided in countries with a high TB prevalence, those who have lived in a correctional institution or homeless shelter, and anyone in a high-risk group based on local epidemiology of the disease. (Read more on TB in this month’s Case Report.) Others at high risk are those who are immune suppressed because of infection or medications, and those who work in health care or correctional facilities. Screening of these groups is usually conducted as part of occupational health or is considered part of routine health care.
Syphilis: Screen high-risk individuals in 2 steps. The recommendation on syphilis screening basically reaffirms the one from 2004.8 Those at high risk for syphilis include men who have sex with men (who now account for 90% of new cases), those who are HIV positive, and those who engage in commercial sex work. Other racial and demographic groups can be at high risk depending on the local epidemiology of the disease. In a separate recommendation, the Task Force advises screening all pregnant women for syphilis.
Screening for syphilis infection involves 2 steps: first, a nontreponemal test (Venereal Disease Research Laboratory [VDRL] or rapid plasma reagin [RPR] test); second, a confirmatory treponemal antibody detection test (fluorescent treponemal antibody absorption [FTA-ABS] or Treponema pallidum particle agglutination [TPPA] test). Treatment for syphilis remains benzathine penicillin with the number of injections depending on the stage of infection. The Centers for Disease Control and Prevention is the best source for current recommendations for treatment of all sexually transmitted infections.15
Screening tests to avoid
TABLE 416,17 lists screening tests the Task Force recommends against. While chronic obstructive pulmonary disease afflicts 14% of US adults ages 40 to 79 years and is the third leading cause of death in the country, the Task Force found that early detection in asymptomatic adults does not affect the course of the illness and is of no benefit.16
Genital herpes, also prevalent, infects an estimated one out of 6 individuals, ages 14 to 49. It causes little mortality, except in neonates, but those infected can have recurrent flares and suffer psychological harms from stigmatization. Most genital herpes is caused by herpes simplex virus-2, and there is a serological test to detect it. However, the Task Force recommends against using the test to screen asymptomatic adults and adolescents, including those who are pregnant. This recommendation is based on the test’s high false-positive rate, which can cause emotional harm, and on the lack of evidence that detection through screening improves outcomes.17
The evidence is lacking for these practices
The Task Force is one of only a few organizations that will not make a recommendation if evidence is lacking on benefits and harms. In addition to the ‘I’ statements regarding CVD and CRC mentioned earlier, the Task Force found insufficient evidence to recommend screening for lipid disorders in individuals ages 20 years or younger,10 performing a visual skin exam as a screening tool for skin cancer,11 screening for celiac disease,12 performing a periodic pelvic examination in asymptomatic women,13 and screening for obstructive sleep apnea using screening questionnaires14 (TABLE 33,4,10-14).
SIDEBAR
A change for prostate cancer screening?The USPSTF recently issued new draft recommendations regarding prostate cancer screening in asymptomatic men (available at: https://screeningforprostatecancer.org/).
The draft now divides men into 2 age groups, stating that the decision to screen for prostate cancer using a prostate specific antigen (PSA) test should be individualized for men ages 55 to 69 years (a C recommendation, meaning that there is at least moderate certainty that the net benefit is small), and that men ages 70 and older (lowered from age 75 in the previous 2012 recommendation1) should not be screened (a D recommendation, meaning that there is moderate or high certainty that the service has no net benefit or that the harms outweigh the benefits).
The USPSTF believes that clinicians should explain to men ages 55 to 69 years that screening offers a small potential benefit of reducing the chance of dying from prostate cancer, but also comes with potential harms, including false-positive results requiring additional testing/procedures, overdiagnosis and overtreatment, and treatment complications such as incontinence and impotence. In this way, each man has the chance to incorporate his values and preferences into the decision.
For men ages 70 and older, the potential benefits simply do not outweigh the potential harms, according to the USPSTF.
1. USPSTF. Final recommendation statement. Prostate cancer: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/prostate-cancer-screening#Pod1. Accessed April 11, 2017.
Since the last Practice Alert update on the United States Preventive Services Task Force in May of 2016,1 the Task Force has released 19 recommendations on 13 topics that include: the use of aspirin and statins for the prevention of cardiovascular disease (CVD); support for breastfeeding; use of folic acid during pregnancy; and screening for syphilis, latent tuberculosis (TB), herpes, chronic obstructive pulmonary disease (COPD), colorectal cancer (CRC), obstructive sleep apnea (OSA), celiac disease, and skin cancer. The Task Force also released a draft recommendation regarding prostate cancer screening in asymptomatic men (see “A change for prostate cancer screening?”) and addressed screening pelvic examinations in asymptomatic women, the subject of this month’s audiocast. (To listen, go to: http://bit.ly/2nIVoD5.)
Recommendations to implement
Recommendations from the past year that family physicians should put into practice are detailed below and in TABLE 1.2-8
CRC: Screen all individuals ages 50 to 75, but 76 to 85 selectively. The Task Force reaffirmed its 2008 finding that screening for CRC in adults ages 50 to 75 years is substantially beneficial.2 In contrast to the previous recommendation, however, the new one does not state which screening tests are preferred. The tests considered were 3 stool tests (fecal immunochemical test [FIT], FIT-tumor DNA testing [FIT-DNA], and guaiac-based fecal occult blood test [gFOBT]), as well as 3 direct visualization tests (colonoscopy, sigmoidoscopy, and CT colonoscopy). The Task Force assessed various testing frequencies of each test and some test combinations. While the Task Force does not recommend any one screening strategy, there are still significant unknowns about FIT-DNA and CT colonoscopy. The American Academy of Family Physicians does not recommend using these 2 tests for screening purposes at this time.9
CRC screening for adults ages 76 to 85 was given a “C” recommendation, which means the value of the service to the population overall is small, but that certain individuals may benefit from it. The Task Force advises selectively offering a “C” service to individuals based on professional judgment and patient preferences. Regarding CRC screening in individuals 76 years or older, the ones most likely to benefit are those who have never been screened and those without significant comorbidities that could limit life expectancy. All “C” recommendations from the past year are listed in TABLE 2.2-4
CVD prevention: When aspirin or a statin is indicated. The Task Force released 2 recommendations for the prevention of CVD this past year. One pertained to the use of low-dose aspirin3 (which also helps to prevent CRC), and the other addressed the use of low- to moderate-dose statins.4 Each recommendation is fairly complicated and nuanced in terms of age and risk for CVD. A decision to use low-dose aspirin must also consider the risk of bleeding.
To calculate a patient’s risk for CVD, the Task Force recommends using the risk calculator developed by the American College of Cardiology and the American Heart Association (http://www.cvriskcalculator.com/).
Adults for whom low-dose aspirin and low- to moderate-dose statins are recommended are described in TABLE 1.2-8 Patients for whom individual decision making is advised, rather than a generalized recommendation, are reviewed in TABLE 2.2-4 There is insufficient evidence to make a recommendation for the use of aspirin before age 50 or at age 70 and older,3 and for the use of statins in adults age 76 and older who do not have a history of CVD4 (TABLE 33,4,10-14). The use of low-dose aspirin and low-to-moderate dose statins have been the subject of JFP audiocasts in May 2016 and January 2017. (See http://bit.ly/2oiun8d and http://bit.ly/2oqkohR.)
2 pregnancy-related recommendations. To prevent neural tube defects in newborns, the Task Force now recommends daily folic acid, 0.4 to 0.8 mg (400 to 800 mcg), for all women who are planning on or are capable of becoming pregnant.5 This is an update of a 2009 recommendation that was worded slightly differently, recommending the supplement for all women of childbearing age.
A new recommendation on breastfeeding recognizes its benefits for both mother and baby and finds that interventions to encourage breastfeeding increase the prevalence of this practice and its duration.6 Interventions—provided to individuals or groups by professionals or peers or through formal education—include promoting the benefits of breastfeeding, giving practical advice and direct support on how to breastfeed, and offering psychological support.
Latent TB: Advantages of newer testing method. The recommendation on screening for latent tuberculosis (TB) is an update from the one made in 1996.7 At that time, screening for latent infection was performed using a tuberculin skin test (TST). Now a TST or interferon-gamma release assay (IGRA) can be used. Testing with IGRA may be the best option for those who have received a bacille Calmette–Guérin vaccination (because it can cause a false-positive TST) or for those who are not likely to return to have their TST read.
Those at high risk for latent TB include people who were born or have resided in countries with a high TB prevalence, those who have lived in a correctional institution or homeless shelter, and anyone in a high-risk group based on local epidemiology of the disease. (Read more on TB in this month’s Case Report.) Others at high risk are those who are immune suppressed because of infection or medications, and those who work in health care or correctional facilities. Screening of these groups is usually conducted as part of occupational health or is considered part of routine health care.
Syphilis: Screen high-risk individuals in 2 steps. The recommendation on syphilis screening basically reaffirms the one from 2004.8 Those at high risk for syphilis include men who have sex with men (who now account for 90% of new cases), those who are HIV positive, and those who engage in commercial sex work. Other racial and demographic groups can be at high risk depending on the local epidemiology of the disease. In a separate recommendation, the Task Force advises screening all pregnant women for syphilis.
Screening for syphilis infection involves 2 steps: first, a nontreponemal test (Venereal Disease Research Laboratory [VDRL] or rapid plasma reagin [RPR] test); second, a confirmatory treponemal antibody detection test (fluorescent treponemal antibody absorption [FTA-ABS] or Treponema pallidum particle agglutination [TPPA] test). Treatment for syphilis remains benzathine penicillin with the number of injections depending on the stage of infection. The Centers for Disease Control and Prevention is the best source for current recommendations for treatment of all sexually transmitted infections.15
Screening tests to avoid
TABLE 416,17 lists screening tests the Task Force recommends against. While chronic obstructive pulmonary disease afflicts 14% of US adults ages 40 to 79 years and is the third leading cause of death in the country, the Task Force found that early detection in asymptomatic adults does not affect the course of the illness and is of no benefit.16
Genital herpes, also prevalent, infects an estimated one out of 6 individuals, ages 14 to 49. It causes little mortality, except in neonates, but those infected can have recurrent flares and suffer psychological harms from stigmatization. Most genital herpes is caused by herpes simplex virus-2, and there is a serological test to detect it. However, the Task Force recommends against using the test to screen asymptomatic adults and adolescents, including those who are pregnant. This recommendation is based on the test’s high false-positive rate, which can cause emotional harm, and on the lack of evidence that detection through screening improves outcomes.17
The evidence is lacking for these practices
The Task Force is one of only a few organizations that will not make a recommendation if evidence is lacking on benefits and harms. In addition to the ‘I’ statements regarding CVD and CRC mentioned earlier, the Task Force found insufficient evidence to recommend screening for lipid disorders in individuals ages 20 years or younger,10 performing a visual skin exam as a screening tool for skin cancer,11 screening for celiac disease,12 performing a periodic pelvic examination in asymptomatic women,13 and screening for obstructive sleep apnea using screening questionnaires14 (TABLE 33,4,10-14).
SIDEBAR
A change for prostate cancer screening?The USPSTF recently issued new draft recommendations regarding prostate cancer screening in asymptomatic men (available at: https://screeningforprostatecancer.org/).
The draft now divides men into 2 age groups, stating that the decision to screen for prostate cancer using a prostate specific antigen (PSA) test should be individualized for men ages 55 to 69 years (a C recommendation, meaning that there is at least moderate certainty that the net benefit is small), and that men ages 70 and older (lowered from age 75 in the previous 2012 recommendation1) should not be screened (a D recommendation, meaning that there is moderate or high certainty that the service has no net benefit or that the harms outweigh the benefits).
The USPSTF believes that clinicians should explain to men ages 55 to 69 years that screening offers a small potential benefit of reducing the chance of dying from prostate cancer, but also comes with potential harms, including false-positive results requiring additional testing/procedures, overdiagnosis and overtreatment, and treatment complications such as incontinence and impotence. In this way, each man has the chance to incorporate his values and preferences into the decision.
For men ages 70 and older, the potential benefits simply do not outweigh the potential harms, according to the USPSTF.
1. USPSTF. Final recommendation statement. Prostate cancer: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/prostate-cancer-screening#Pod1. Accessed April 11, 2017.
1. Campos-Outcalt D. Eight USPSTF recommendations FPs need to know about. J Fam Pract. 2016;65:338-341.
2. USPSTF. Colorectal cancer: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/colorectal-cancer-screening2. Accessed March 22, 2017.
3. USPSTF. Aspirin use to prevent cardiovascular disease and colorectal cancer: preventive medications. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/aspirin-to-prevent-cardiovascular-disease-and-cancer. Accessed March 22, 2017.
4. USPSTF. Statin use for the prevention of cardiovascular disease in adults: preventive medication. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/statin-use-in-adults-preventive-medication1. Accessed March 22, 2017.
5. USPSTF. Folic acid for the prevention of neural tube defects: preventive medication. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/folic-acid-for-the-prevention-of-neural-tube-defects-preventive-medication. Accessed March 22, 2017.
6. USPSTF. Breastfeeding: primary care interventions. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/breastfeeding-primary-care-interventions. Accessed March 22, 2017.
7. USPSTF. Latent tuberculosis infection: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/latent-tuberculosis-infection-screening. Accessed March 22, 2017.
8. USPSTF. Syphilis infection in nonpregnant adults and adolescents: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/syphilis-infection-in-nonpregnant-adults-and-adolescents. Accessed March 22, 2017.
9. AAFP. Colorectal cancer screening, adults. Available at: http://www.aafp.org/patient-care/clinical-recommendations/all/colorectal-cancer.html. Accessed March 22, 2017.
10. USPSTF. Lipid disorders in children and adolescents: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/lipid-disorders-in-children-screening1. Accessed March 22, 2017.
11. USPSTF. Skin cancer: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/skin-cancer-screening2. Accessed March 22, 2017.
12. USPSTF. Celiac disease: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/celiac-disease-screening. Accessed March 22, 2017.
13. USPSTF. Gynecological conditions: periodic screening with the pelvic examination. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/gynecological-conditions-screening-with-the-pelvic-examination. Accessed March 22, 2017.
14. USPSTF. Obstructive sleep apnea in adults: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/obstructive-sleep-apnea-in-adults-screening. Accessed March 22, 2017.
15. CDC. 2015 sexually transmitted diseases treatment guidelines. Available at: https://www.cdc.gov/std/tg2015/default.htm. Accessed March 22, 2017.
16. USPSTF. Chronic obstructive pulmonary disease: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/chronic-obstructive-pulmonary-disease-screening. Accessed March 22, 2017.
17. USPSTF. Genital herpes infection: serologic screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/genital-herpes-screening1. Accessed March 22, 2017.
1. Campos-Outcalt D. Eight USPSTF recommendations FPs need to know about. J Fam Pract. 2016;65:338-341.
2. USPSTF. Colorectal cancer: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/colorectal-cancer-screening2. Accessed March 22, 2017.
3. USPSTF. Aspirin use to prevent cardiovascular disease and colorectal cancer: preventive medications. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/aspirin-to-prevent-cardiovascular-disease-and-cancer. Accessed March 22, 2017.
4. USPSTF. Statin use for the prevention of cardiovascular disease in adults: preventive medication. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/statin-use-in-adults-preventive-medication1. Accessed March 22, 2017.
5. USPSTF. Folic acid for the prevention of neural tube defects: preventive medication. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/folic-acid-for-the-prevention-of-neural-tube-defects-preventive-medication. Accessed March 22, 2017.
6. USPSTF. Breastfeeding: primary care interventions. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/breastfeeding-primary-care-interventions. Accessed March 22, 2017.
7. USPSTF. Latent tuberculosis infection: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/latent-tuberculosis-infection-screening. Accessed March 22, 2017.
8. USPSTF. Syphilis infection in nonpregnant adults and adolescents: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/syphilis-infection-in-nonpregnant-adults-and-adolescents. Accessed March 22, 2017.
9. AAFP. Colorectal cancer screening, adults. Available at: http://www.aafp.org/patient-care/clinical-recommendations/all/colorectal-cancer.html. Accessed March 22, 2017.
10. USPSTF. Lipid disorders in children and adolescents: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/lipid-disorders-in-children-screening1. Accessed March 22, 2017.
11. USPSTF. Skin cancer: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/skin-cancer-screening2. Accessed March 22, 2017.
12. USPSTF. Celiac disease: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/celiac-disease-screening. Accessed March 22, 2017.
13. USPSTF. Gynecological conditions: periodic screening with the pelvic examination. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/gynecological-conditions-screening-with-the-pelvic-examination. Accessed March 22, 2017.
14. USPSTF. Obstructive sleep apnea in adults: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/obstructive-sleep-apnea-in-adults-screening. Accessed March 22, 2017.
15. CDC. 2015 sexually transmitted diseases treatment guidelines. Available at: https://www.cdc.gov/std/tg2015/default.htm. Accessed March 22, 2017.
16. USPSTF. Chronic obstructive pulmonary disease: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/chronic-obstructive-pulmonary-disease-screening. Accessed March 22, 2017.
17. USPSTF. Genital herpes infection: serologic screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/genital-herpes-screening1. Accessed March 22, 2017.

Thrombosis in Pregnancy
INTRODUCTION
Venous thromboembolism (VTE), comprising deep vein thrombosis (DVT) and pulmonary embolism (PE), is a leading nonobstetric cause of maternal death in the United States and in developed countries.1,2 During pregnancy, the risk for VTE increases four- to six-fold, and although the risk is present throughout pregnancy, the mother is at highest risk immediately postpartum.3–5
VTE risk is increased due to physiologic and anatomic changes that occur in pregnancy. These changes include hypercoagulability, progesterone-induced venous stasis, decreased venous outflow, compression of the inferior vena cava and pelvic veins by the expanding uterus, and decreased mobility. The hypercoagulability of pregnancy is due to increased levels of coagulation factors I (fibrinogen), VII, VIII, and X, and von Willebrand factor; decreased free protein S, a natural anticoagulant; acquired resistance to activated protein C; and decreased fibrinolysis due to increased levels of plasminogen activator inhibitor-1 and -2.6,7 These changes confer increased hemostasis to the mother for delivery but also place her at higher risk for thrombosis.
A review of the literature found that more than 70% of pregnancy-associated DVTs are located in the ileofemoral region, as compared with approximately 9% in non-pregnant patients.8 The proximal location is associated with a higher risk for post-thrombotic syndrome and embolization as compared with calf DVTs.9 Proximal postnatal thrombosis, smoking, and older age are independent predictors of the development of post-thrombotic syndrome.10
RISK FACTORS
Clinical risk factors that increase the risk for VTE during pregnancy include a prior history of estrogen-related or unprovoked VTE, being a carrier of severe inherited thrombophilia (homozygotes for factor V Leiden or factor II G20210A variants, double heterozygotes, or persons with antithrombin, protein C, or protein S deficiencies), and the presence of antiphospholipid (aPL) antibodies.11 Women with systemic lupus erythematosus, diabetes, sickle cell disease, and heart disease also have a high risk for VTE during pregnancy.12 Other risk factors predisposing to thrombosis include black ethnicity, smoking, operative procedures, conception after assisted reproductive techniques, high body mass index, antepartum immobilization, severe preeclampsia, advanced age and parity, and a family history of VTE.13 A prospective cohort study of 1,297,037 pregnancies and related puerperium identified the following risk factors for thrombosis: hospitalization, infection, hyperemesis, multiple pregnancies, preeclampsia, obesity, cesarean section, major postpartum hemorrhage, intrauterine growth restriction, and fetal death.14 Risk factors identified in an Agency for Healthcare Research and Quality study include: age 35 or older, black ethnicity, lupus, sickle cell disease, heart disease, postpartum infection, and transfusion.15 The combination of more than one risk factor increases the risk for VTE. All these factors have to be considered when deciding on prophylactic or therapeutic anticoagulation therapy in pregnancy. In addition, the risks of anticoagulation, including bruising, bleeding, and other side effects (eg, reduced bone mineral density with therapeutic-dose unfractionated heparin), allergic reactions, and rarely thrombocytopenia, must be considered.
EVALUATION AND DIAGNOSIS
CASE PRESENTATION I
A 31-year-old woman G1P0 at 10 weeks’ gestation with no personal or family history of thrombosis presents with acute onset of shortness of breath and left-sided chest pain that awoke her the morning of presentation. Her vital signs are significant for a heart rate of 106 beats/min, respiration rate of 22 breaths/min, blood pressure of 105/76 mm Hg, and pulse oximetry of 98% on room air. The patient denies previous exposure to oral contraceptives. She does not smoke. She reports that she had noticed left calf pain and swelling, which worsened with walking after a 4-hour drive 2 days prior.
What is the approach to diagnosis of thromboembolism in pregnant patients?
DEEP VEIN THROMBOSIS
Although a clinical diagnosis of DVT in pregnancy is unreliable, a history and physical examination are necessary to exclude other diagnoses and to assess the likelihood of thrombosis. Unfortunately, studies of the accuracy of history and physical examination for detecting DVT and PE have not included pregnant patients. In most pregnant patients with clinically suspected DVT, the diagnosis is not confirmed. Other causes of leg pain and swelling are not uncommon during pregnancy and include cellulitis, ruptured Baker’s cyst, or muscular pain.
A cross-sectional study described the derivation of the LEFt clinical decision rule, which relies on 3 variables in pregnant women with suspected DVT: left leg presentation (L), ≥ 2 cm calf circumference difference (E for edema), and first trimester presentation (Ft). If none of these variables is present, the negative predictive value is 100%.16 A validation study suggested that a negative LEFt rule accurately identifies pregnant women in whom the risk for confirmed DVT appears to be very low. The rule should not be used as an individual test for excluding DVT during pregnancy, but could be applied in a diagnostic approach in association with D-dimer measurement and compression ultrasonography (CUS); however, it has not been prospectively validated for safety and efficacy.17 In a study of 149 consecutive pregnant women with suspected DVT, a whole-blood agglutination D-dimer had a sensitivity of 100% and specificity of 60%.18 A 2006 systematic review found only 4 diagnostic studies of VTE in pregnancy in the literature. One of these studies showed that a combination of a negative CUS and normal D-dimer can accurately exclude DVT.19
Serial CUS is necessary for pregnant women with a high clinical suspicion of DVT but a negative initial investigation. In a study of 221 pregnant women in whom DVT was clinically suspected, 16 women (7.2%) were diagnosed with DVT by initial CUS, and none were diagnosed with DVT onserial testing.20 During follow-up (≥ 3 months), 6 of the 205 women with normal serial CUS results presented with symptoms of DVT, PE, or both, and 1 of them was diagnosed with DVT and PE. The sensitivity of serial CUS with Doppler imaging was 94.1% (95% confidence interval [CI] 69.2% to 99.7%), and the negative predictive value was 99.5% (95% CI 96.9% to 100%).20 All ultrasounds undertaken for investigation of pregnancy-associated DVT should include imaging of the iliac veins if there is a high index of suspicion and the CUS is negative for femoral DVT. Serial CUS with Doppler imaging of the iliac vein performed over a 7-day period excludes DVT in symptomatic pregnant women.20 Repeat CUS may be done 2 to 4 days and 6 to 8 days after the initial scan.
Ileofemoral vein thrombosis accounts for approximately 90% of proximal thromboses in pregnancy, occurring most often in the left lower extremity.20 The incidence of isolated iliac vein thrombosis in pregnancy is low, but when it does occur, delay in diagnosis can lead to significant morbidity. Therefore, for women with suspected isolated iliac vein thrombosis in whom CUS is negative or nondiagnostic, magnetic resonance direct thrombus imaging (MRDTI) should be performed.21 Patients with iliac vein thrombosis may present with unexplained inguinal, pelvic, or abdominal pain, which may be accompanied by back pain, and they usually present with swelling of the entire leg. MRDTI does not require gadolinium contrast and its accuracy appears to be similar to that of venography for iliac vein thrombi in the nonpregnant population.21 Exposure to gadolinium during pregnancy is associated with an increased risk for rheumatologic, inflammatory, or infiltrative skin conditions and stillbirth or neonatal death.22
Ovarian vein thrombosis is a rare but serious diagnosis. It occurs mostly in the postpartum period, mainly after cesarean delivery, and usually affects the right ovarian vein. The diagnosis is confirmed by ultrasound, computed tomography (CT), or magnetic resonance imaging.23
PULMONARY EMBOLISM
PE is more difficult to diagnose than DVT, particularly because clinical signs of PE are unreliable in the pregnant patient. The mortality rate of untreated PE is high, ranging from 18% to 38%, and approximately one-third of patients with untreated thromboembolic disease develop recurrent embolism.24 Studies have reported a PE prevalence between 1.4% and 4.2% in pregnant women with suspected clinical diagnosis of PE.25
The clinical presentation of PE and associated laboratory testing results may be subtler in pregnant than in nonpregnant patients. Arterial blood gases (ABG) may show hypoxemia or hypocapnia. The ABG in pregnancy has a sensitivity of 76.9%, specificity of 20.2%, and negative and positive predictive values of 80% and 11.5% for PE, respectively.26 The alveolar-arterial oxygen gradient is a poor screening test for PE during pregnancy and postpartum. A retrospective chart review of 17 pregnant women with documented PE showed that 58% had normal alveolar-arterial gradients.27 Therefore, in a pregnant woman with a history suspicious for PE, objective imaging studies should be performed even if the patient has normal ABG.
The 2011 guidelines from the American Thoracic Society (ATS) and the Society of Thoracic Radiology (STR) recommend against using D-dimer to diagnose PE in pregnancy.28 In addition, lower extremity CUS should only be performed as the first diagnostic imaging procedure if the patient has signs or symptoms of DVT. Instead, the ATS/STR guidelines recommend a plain radiograph of the chest as the first imaging test. If the chest radiograph is normal, a ventilation/perfusion scan (V/Q) scan is preferred over CT pulmonary angiography (CTPA). Diagnostic accuracy of the V/Q scan may be superior to CTPA in pregnancy, and it is preferable because of the lower prevalence of indeterminate V/Q scan in pregnant women.29 Moreover, there is lower radiation exposure to the maternal breast and lung tissue with a V/Q scan than with CTPA. CTPA confers lower fetal radiation doses than V/Q scans (0.03–0.66 mGy versus 0.32–0.74 mGy, respectively) but higher total body maternal radiation (4–16 mSv versus 1–2.5 mSv).30 A quantitative approach to lung scan interpretation, based on the distribution histogram of V/Q ratios, may be helpful in categorizing patients with suspected PE.28 A study of 121 suspected episodes of PE in 120 pregnant women showed that 104 women with normal or nondiagnostic scans did not develop subsequent episodes of VTE during a mean follow-up period of 20 months.31
If the baseline chest radiograph is abnormal in a pregnant woman with clinical suspicion of PE, a CTPA should be performed. As noted, fetal radiation doses for CTPA examinations in which the fetus is not directly imaged are minimal. If CTPA is recommended for the diagnosis of PE, the patient should be informed that radiation to the breast may increase her baseline risk for breast cancer. The ATS guidelines state that “given the lack of evidence documenting clear superiority of any one diagnostic test, the values and preferences of a patient and her physician likely will and should determine the final choice and sequence of tests performed.”28
CASE I CONTINUED
Upon presentation to the emergency department, the circumference of the patient’s left leg is not significantly greater than that of her right leg, and her leg pain has resolved. Bilateral CUS is negative for proximal or distal DVT. Chest radiograph shows an opacification of her left lower lobe. CTPA shows bilateral segmental and subsegmental lower lobe pulmonary emboli.
How does risk for VTE change throughout pregnancy?
Women are at increased risk for VTE throughout the entire pregnancy, starting from conception, but mainly during the postpartum period. A Danish historical controlled cohort study of 819,751 pregnant women (ages 15–49 years) over a 10-year period identified 727 women with VTE. The absolute risk for VTE per 10,000 pregnancy-years increased from 4.1 (95% CI 3.2 to 5.2) during weeks 1 to 11 to 59.0 (95% CI 46.1 to 76.4) in week 40 and decreased in the postpartum period from 60 (95% CI 47.2 to 76.4) during the first week after birth to 2.1 during weeks 9–12 after birth (95% CI 1.1 to 4.2).32 This study showed that the risk of VTE increases throughout pregnancy and reaches its maximum during the peripartum period and is not significantly increased after 6 weeks post-delivery. In a retrospective cross-over cohort study of 1,687,930 women in California who delivered their first newborn, an elevated risk of VTE persisted until at least 12 weeks after delivery. However, the absolute increase in risk after 6 weeks postpartum was low.33
CASE 1 CONCLUSION
The patient is started on anticoagulation therapy and carefully monitored during the remainder of the pregnancy and postpartum period. Anticoagulation is discontinued 6 weeks after delivery.
TREATMENT
ANTICOAGULATION THERAPY
The treatment of VTE can be lifesaving. In a study comparing 35 patients with PE randomly assigned to treatment with anticoagulants versus no treatment, 5 of 19 patients in the untreated group died from PE and an additional 5 had nonfatal recurrences, as compared with none in the treated group.24 Unfractionated heparin (UFH) and low-molecular-weight heparin (LMWH) are both safe and effective anticoagulants during pregnancy as neither crosses the placenta. In a review of 186 reports of fetal and infant outcomes following anticoagulant therapy during pregnancy in 1325 pregnancies, outcomes in UFH-treated patients were similar to those in the normal population after excluding pregnancies with comorbid conditions independently associated with adverse outcomes.34 A 2005 systematic review of LMWH for prophylaxis and treatment of VTE during pregnancy included 64 studies of 277 pregnancies. There were no maternal deaths, live births resulted from 94.7% of the pregnancies, VTE or arterial thrombosis occurred in 0.86%, and significant bleeding occurred in 1.98%.35
The standard UFH regimen is an initial bolus of 5000 units subcutaneously and 17,500 units every 12 hours, with dose adjustment made based on a mid-interval activated partial thromboplastin time (aPTT).36 Although still controversial, it has been suggested that the anti-Xa assay with a mid-dosing interval target of 0.3 to 0.7 U/mL is a more reliable measure of therapeutic UFH activity than the aPTT, as the aPTT response is suppressed due to a pregnancy-related increase in factor VIII. LMWH is dosed based on weight; regimens are enoxaparin 1 mg/kg subcutaneously twice daily or 1.5 mg/kg subcutaneously once daily, and dalteparin 100 units/kg every 12 hours or 150 units/kg daily.
A 2017 Cochrane review of the effect of LMWH compared with UFH for the treatment of VTE in the nonpregnant setting included 23 studies with 9587 patients. Thrombotic complications (odds ratio [OR] 0.70 [CI 0.57 to 0.85]) and major hemorrhage (OR 0.58 [CI 0.40 to 0.83]) were lower in patients receiving LMWH, with a trend toward lower mortality.37 In addition, the incidence of bleeding complications in patients treated with subcutaneous LMWH versus intravenous heparin was compared in a 2012 systematic review of 27 randomized controlled trials with a total of 28,637 patients. In patients treated with LMWH, there was a nonstatistically significant lower incidence of major bleeding events (OR 0.79 [95% CI 0.60 to 1.04]) and a statistically significant reduction in bleeding risk (OR 0.68 [95% CI 0.47 to 1.00]) compared to patients treated with UFH.38 Additionally, a trial comparing the use of standard UFH versus LMWH found a significantly lower incidence of thrombocytopenia in patients treated with LMWH.39,40 Overall, LMWH is more effective at decreasing both thrombotic and bleeding complications, and the risk for osteoporosis is lower with LMWH. Based on these results, the American College of Chest Physicians (ACCP) recommends LMWH as the first-line treatment for VTE in pregnancy.41
In specific clinical situations, such as patients with renal dysfunction with creatinine clearance (CrCl) less than 30 mL/min, UFH is indicated. In a study of 103 pregnancies in 93 women given anti-coagulation during pregnancy, 89.3% received UFH. There were no maternal deaths, and fetal demise occurred in 8 pregnancies (7.8%) at a median of 14 weeks’ gestation. There were 2 episodes of PE (1.9%) and 2 major bleeding events requiring transfusion (1.9%).42 UFH costs much less than LMWH, and therefore UFH remains an important, inexpensive, and efficacious anticoagulant option for pregnant women who require anticoagulation and cannot afford LMWH.43
Due to the physiologic changes associated with pregnancy, LMWH and UFH dosages may need to be adjusted. An observational study of 20 pregnant women with acute VTE found no recurrent VTE or major bleeding after treatment with dalteparin. Dalteparin doses approximately 10% to 20% higher than those recommended in nonpregnant women were required to reach therapeutic target anti-Xa activity.44
Caution Regarding Oral Anticoagulants
Due to its teratogenicity, warfarin is not a first-line anticoagulation option. It is strictly contraindicated during the first trimester during organogenesis, and its use during pregnancy is restricted to women with mechanical heart valves. Warfarin crosses the placenta and has been associated with nasal hypoplasia, stippled epiphyses, and growth restriction, particularly between 6 to 9 weeks’ gestation. Every effort should be made to substitute UFH or LMWH for warfarin between 6 and 12 weeks of gestation. The bridging process should begin as early in the gestational age as possible due to the long half-life of warfarin.45 When used later in gestation, warfarin has been associated with fetal hemorrhage and central nervous system abnormalities. Other complications from use during the second and third trimesters include microcephaly, blindness, deafness, and fetal growth restriction.46,47 Its use also increases the risk for abortion and fetal death in utero.48–50
The direct oral anticoagulants (DOACs) are not approved for use in pregnancy. Although there are limited anecdotal reports of DOAC use in pregnancy,51 there is preclinical evidence of placental transfer with the DOACs rivaroxaban and apixaban (direct Xa inhibitors) and the oral thrombin inhibitor dabigatran, thus increasing the risk to the fetus.52–54 Edoxaban, another direct Xa inhibitor, should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. It should be discontinued in nursing mothers.55
THROMBOLYSIS
Fetal as well as maternal survival is dependent on adequate maternal perfusion and oxygenation. The risk of death from PE is significant, with a cross-sectional study of 58 patients with acute, massive PE showing a 55% mortality rate.56 Thus, pregnancy is not an absolute contraindication to mechanical or systemic (recombinant tissue plasminogen activator or streptokinase) thrombolysis in an unstable patient at high risk for death.57–59 There are no major studies of this approach, although a small review of 13 cases using systemic thrombolysis showed no increased risk of maternal mortality.58 Thrombolysis should be considered for appropriate indications in pregnant patients as it would be in nonpregnant patients. However, caution is required when drawing conclusions regarding maternal and fetal safety, given the lack of controlled clinical trials including pregnant women.
SURGICAL PULMONARY EMBOLECTOMY
Surgical pulmonary embolectomy is an important therapeutic and potentially life-saving option in women presenting with massive PE in the immediate postpartum period. Because of the risk of massive uterine bleeding immediately postpartum, thrombolytic therapy should not be used.60
INFERIOR VENA CAVA FILTER
Placement of an inferior vena cava (IVC) filter is indicated in patients who have an acute VTE with absolute contraindications for anticoagulation. In addition, it can be considered in patients with extensive ileofemoral venous thrombosis within 2 weeks prior to expected delivery.61 In a systematic review of 44 studies of IVC filters placed in pregnant patients, the IVC filter complication rate was 8.87% and the failure-to-retrieve rate was 11.25%.62 The complication rate is similar to that found in the nonpregnant population. Thus, IVC filters may be used when appropriately indicated and should be removed as soon as clinically feasible.
RECURRENT THROMBOSIS AND THROMBOPHILIAS
CASE PRESENTATION 2
A 34-year-old pregnant woman G1P0 at 38 weeks’ gestation presents with a painful, swollen left calf that is associated with difficulty on walking; the circumference of the left calf is 2 cm greater than that of the right. She has no shortness of breath or chest pain. She has a prior history of distal right lower extremity DVT while on combined oral contraceptives. Her mother also has a history of DVT while bedbound during a prolonged hospitalization at an older age. CUS is negative, and the patient is discharged home. However, 24 hours later she returns to the hospital with worsening swelling and pain in her left leg. Magnetic resonance venography demonstrates a large left external iliac and common iliac DVT. She is admitted and is started on UFH, and a retrievable IVC filter is placed in anticipation of delivery.
What is the risk for VTE recurrence during pregnancy?
A personal and family history of VTE should be obtained when evaluating pregnant patients. A retrospective study of 109 women with prior history of VTE showed recurrence rates per patient-year of 10.9% during pregnancy and 3.7% in the nonpregnant period; the relative risk of recurrent VTE during pregnancy was 3.5 (95% CI 1.6 to 7.8).63 Two large European retrospective cohort studies of VTE in pregnancy showed that the recurrence rate of VTE in women with a history of thrombosis is around 6% during pregnancy, equally distributed among trimesters. The highest incidence of recurrence was in the postpartum period, ranging from 8.3% to 10%.64 The recurrence risk during pregnancy in women with a history of a single episode of VTE was 2.4% antepartum (95% CI 0.2% to 6.9%).65 These risks may be lower in women without thrombophilia or with a temporary risk factor associated with their previous thromboembolic event.65 Recurrence risk is higher if the previous VTE was estrogen-related, either due to pregnancy or through hormonal contraception (10%), than if the previous VTE was non-estrogen-related (2.7%).64,66
The timing of the case patient’s presentation is consistent with reports of increased risk of VTE during the peripartum period. Her prior history of estrogen-related DVT is concerning for a risk of recurrence, particularly during pregnancy. A retrospective cohort study of 1104 women with previous VTE, 88 of whom became pregnant without receiving thromboprophylaxis, showed that the overall rate of VTE recurrence was 5.8% (95% CI 3.0% to 10.6%) and 8.3% (95% CI 4.5% to 14.6%) during pregnancy and postpartum, respectively. The risk of VTE recurrence was absent if the first VTE was related to a transient risk factor other than pregnancy, postpartum period, or hormonal contraception.67 However, the recurrence rate of VTE in women with prior unprovoked VTE and/or thrombophilia has been reported as 5.9% (95% CI 1.2% to 16.2%).65 The presence of an underlying hypercoagulable state can increase the recurrence risk by 25% to 50%, depending on the disorder.68 A retrospective cohort study of 270 pregnancies in 105 carriers of factor V Leiden, identified because of a symptomatic relative with the factor V Leiden mutation, found a VTE risk (mostly in the postpartum period) of 6.4% for heterozygous women, 16.7% for homozygous women, 20% for double heterozygous women, and 1.2% for noncarriers.69
Should the patient be screened for a thrombophilia disorder?
Half of all index thromboses in patients with thrombophilia occur in association with an additional risk factor. In women of child-bearing age, pregnancy, the postpartum period, and the use of combined hormonal contraception are all risk factors for VTE. A 2010 guideline from the British hematology community recommended testing for thrombophilia in women with prior VTE secondary to a minor provoking factor before or during pregnancy, but not testing women with unprovoked VTE (who would receive prophylaxis regardless) or those with VTE secondary to a major provoking factor (who would not require prophylaxis).70 Indications to screen for aPL antibodies include: women with (1) 3 unexplained recurrent first-trimester pregnancy losses or 1 second or third trimester fetal loss of morphologically normal fetuses; (2) severe preeclampsia; (3) intrauterine growth restriction; or (4) premature labor (< 34 weeks’ gestation).71,72
CASE 2 CONCLUSION
The patient is subsequently screened for inherited thrombophilia disorders and is found to be heterozygous for factor V Leiden.
CASE PRESENTATION 3
A 25-year-old woman is diagnosed with antiphospholipid syndrome (APS) during her second pregnancy when she experiences fetal loss during her second trimester. Pathologic examination of the placenta reveals infarcts. Laboratory evaluation reveals positive high-titer anticardiolipin and anti-beta-2 glycoprotein 1 antibodies (IgG isotype) and lupus anticoagulant on 2 separate occasions 12 weeks apart. In a subsequent pregnancy, she is started on prophylactic LMWH and daily low-dose aspirin (81 mg). At 36 weeks’ gestation, she presents with a blood pressure of 210/104 mm Hg and a platelet count of 94,000 cells/µL. She is diagnosed with preeclampsia and hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome and is induced for early delivery. About 2 weeks after vaginal delivery, she notices shortness of breath and chest pain. A CTPA demonstrates a right lower lobe lobar defect consistent with a PE. Her anticoagulation is increased to therapeutic dosage LMWH.
To what extent does thrombophilia increase the risk for VTE in pregnancy?
Approximately 50% of pregnancy-related VTEs are associated with inherited thrombophilia. A systematic review of 79 studies, in which 9 studies (n = 2526 patients) assessed the risk of VTE associated with inherited thrombophilia in pregnancy, revealed that the odds ratio for individuals with thrombophilia to develop VTE ranged from 0.74 to 34.40.73 Although women with thrombophilia have an increased relative risk of developing VTE in pregnancy, the absolute risk of VTE remains low (Table 1).41,73,74
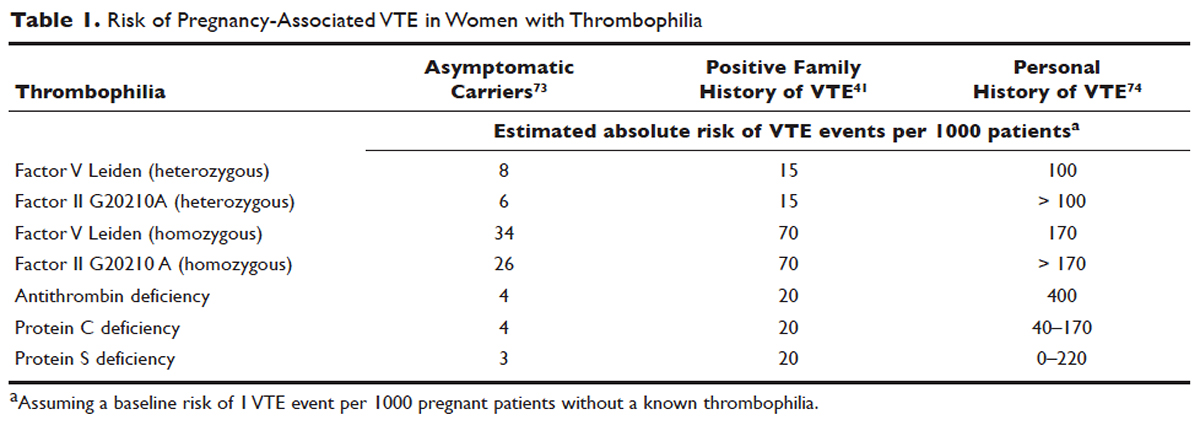
How is APS managed in pregnant patients?
Women with history of recurrent early pregnancy loss (< 10 weeks’ gestation) related to the presence of aPL antibodies are managed with low-dose aspirin and prophylactic-dose UFH or LMWH. This treatment increases the rate of subsequent successful pregnancy outcomes and reduces the risk for thrombosis. A 2010 systematic review and meta-analysis of UFH plus low-dose aspirin compared with low-dose aspirin alone in patients with APS and recurrent pregnancy loss included 5 trials and 334 patients. Patients receiving dual therapy had higher rates of live births (74.3%; relative risk [RR] 1.30 [CI 1.04 to 1.63]) compared to the aspirin-only group (55.8%).75 A 2009 randomized controlled trial compared low-dose aspirin to low-dose aspirin plus LMWH in women with recurrent pregnancy loss and either aPL antibodies, antinuclear antibody, or inherited thrombophilia. The study was stopped early after 4 years and found no difference in rates of live births between the groups (77.8% versus 79.1%).76 However, a randomized case-control trial of women with aPL antibodies and recurrent miscarriage found a 72% live birth rate in 47 women randomly assigned to low-dose aspirin and LMWH.77 A 2012 guideline from the American College of Chest Physicians (ACCP) recommends that women with aPL antibodies with a history of 3 or more pregnancy losses receive low-dose aspirin plus prophylactic-dose LMWH or UFH.78 A 2014 systematic review and meta-analysis showed that the combination of low-dose aspirin and UFH resulted in a higher live-birth rate than aspirin alone in 803 women with APS (RR 1.54 [95% CI 1.25 to 1.89]).79 Further large randomized controlled trials are needed to confirm optimal management of recurrent miscarriage and aPL antibodies.
The addition of prednisone to aspirin, heparin, or both has shown no benefits in pregnant women with aPL antibodies. Indeed, prolonged use of steroids may cause serious pregnancy complications, such as prematurity and hypertension.80–83 Intravenous infusions of immunoglobulin (IVIG) have not been shown to be superior to heparin and aspirin. This finding was confirmed in a multicenter clinical trial that tested the effects of IVIG compared with LMWH plus low-dose aspirin for the treatment of women with aPL antibodies and recurrent miscarriage. The rate of live-birth was 72.5% in the group treated with heparin plus low-dose aspirin compared with 39.5% in the IVIG group.84
Preeclampsia and HELLP syndrome complicated the case patient’s pregnancy even though she was being treated with prophylactic-dose LMWH and low-dose aspirin, the current standard of care for pregnant women with APS (UFH can be used as well). It is important to note that complications may still occur despite standard treatment. Indeed, PE is more common in the postpartum than in the antepartum period. Prompt diagnosis is paramount to initiate the appropriate treatment; in this case the dose of LMWH was increased from prophylactic to therapeutic dose. However, additional therapeutic modalities are necessary to improve outcomes. A randomized controlled trial comparing standard of care with or without hydroxychloroquine is under way to address this issue.
PROPHYLAXIS
CASE PRESENTATION 4
A 34-year-old woman G1P0 at 6 weeks’ gestation with a past medical history of a proximal lower extremity DVT while on oral contraception is treated with warfarin anticoagulation for 6 months. Her obstetrician consults the hematologist to advise regarding antithrombotic management during this pregnancy.
What is the approach to prophylaxis in women at high risk for pregnancy-associated VTE?
All women at high risk for pregnancy-associated VTE should be counseled about the signs and symptoms of DVT or PE during preconception and pregnancy and have a plan developed should these symptoms arise. The ACCP guidelines on antithrombotic therapy outline recommendations ranging from clinical vigilance to prophylactic and intermediate-dose anticoagulation, depending on the risk for VTE recurrence, based on the personal and family history of VTE and type of thrombophilia (Table 2).78 These recommendations range from grade 2B to 2C.

For women with a history of estrogen-related VTE, single unprovoked VTE, or recurrent unprovoked VTE not on chronic anticoagulation, antepartum and postpartum pharmacologic thromboprophylaxis with either prophylactic or intermediate-dose LMWH is recommended (grade 2C). In patients with prior history of provoked VTE (non-estrogen related), antepartum clinical vigilance and postpartum pharmacologic thromboprophylaxis is recommended (grade 2C, 2B).
In asymptomatic pregnant women who are homozygote carriers for factor V Leiden or prothrombin G20210A variants and have a positive family history of thrombosis, antepartum and postpartum pharmacologic thromboprophylaxis is recommended (grade 2B). In asymptomatic homozygote carriers of factor V Leiden or prothrombin G20210A variants with no family history of thrombosis and women with all other thrombophilias with a positive family history of thrombosis, postpartum pharmacologic thromboprophylaxis is indicated (grade 2B and 2C, respectively). For women with confirmed APS and clinical criteria of obstetric APS with recurrent pregnancy loss, antepartum thromboprophylaxis with LMWH and low-dose aspirin is recommended (grade 1B). For pregnant women with all other thrombophilias with no personal or family history of thrombosis, clinical vigilance is suggested (grade 2 C).78
As an alternative to LMWH, vitamin K antagonists (VKA) such as warfarin can be used for postpartum thromboprophylaxis; in patients with protein C or S deficiency, due to the risk of warfarin-induced skin necrosis, a rapid-onset anticoagulant must be concomitantly administered. Warfarin and LMWH are safe anticoagulants during lactation, but there are no clinical data on the effects of the DOACs on infants during lactation. Data from animal studies indicate that DOACs are secreted into breast milk.85
What risks are associated with anticoagulant therapy in pregnancy?
VKAs cross the placenta and can cause teratogenicity, pregnancy loss, fetal bleeding, and neurodevelopmental deficits. Therefore, discontinuation of VKAs prior to the sixth week of gestation is necessary to avoid warfarin embryopathy. DOACs have been shown to readily cross the placenta but with unknown human reproductive risks. Fondaparinux, a synthetic pentasaccharide, crosses the placenta in small quantities. Though there are reports of the successful use of fondaparinux in pregnancy, there is limited reported experience of its use in the first trimester.86
The risk for bleeding with anticoagulation is notably acceptable. In a case-control study of 88 pregnant women receiving therapeutic-dose anticoagulation, the risk of postpartum hemorrhage (PPH) after vaginal delivery was 30% in those who received LMWH anticoagulation versus 18% in those who did not (OR 1.9 [95% CI 1.1 to 3.5]).87 However, the risk for severe PPH (≥ 500 mL) was similar (5.6% versus 5.0%; OR 1.1 [95% CI 0.4 to 3.6]). The risk for PPH after cesarean section was 12% in LMWH users versus 4% in LMWH non-users (OR 2.9 [95% CI 0.5 to 19.4]). The risk for PPH associated with delivery within 24 hours after the last dose of LMWH was 1.2 times higher (95% CI 0.4 to 3.6) compared to a longer interval. Therefore, therapeutic LMWH increases the risk for blood loss after vaginal delivery, but not the risk for severe PPH. The risk for PPH is influenced by the interval between the last dose of LMWH and delivery. Of note in this study, per the institution’s protocol, the anticoagulation was stopped with signs of labor or determination of need for delivery. The risk for blood loss may be mitigated in more planned delivery scenarios.87
CASE 4 CONTINUED
The patient is placed on prophylactic-dose LMWH with good tolerance and delivers at 39 weeks' gestation via caesarian section due to nonprogression of labor. Postpartum she is restarted on prophylactic-dose anticoagulation with LMWH. Two weeks after discharge from the hospital, she presents with right calf pain and mild shortness of breath. On physical exam, her leg circumferences are equal. A D-dimer assay is 3375 ng/mL (normal 0–229). CUS of the right leg shows a complete occlusive DVT of the mid-distal superficial femoral and popliteal veins and partially occlusive acute DVT of the right posterior tibial and peroneal veins. CTPA reveals a right lower lobe PE. Because she had developed VTE despite prophylactic LMWH, her anticoagulation is changed to therapeutic dose. She is treated with anticoagulation with LMWH for a total of 3 months, after which a repeat CUS shows no residual thrombosis.
What is the recommended dosing of heparin and LMWH during pregnancy?
A prospective study of 14 pregnant women receiving UFH prophylaxis found that a prophylactic dose of 5000 units twice a day was inadequate to achieve prophylactic heparin levels in any patient in the second or third trimester.88 Similar to treatment dosage, there is no consensus evidence for prophylactic dosing, and dosage recommendations are based on expert opinion. In a retrospective study of 25 pregnant women on intermediate-dose UFH, the mean UFH dose required to achieve a target anti-factor Xa level of 0.1 to 0.3 units/mL was 236.9 units/kg/day.89 However, the use of anti-factor Xa levels for monitoring is controversial as there is no data to support a difference in outcomes with its use in prophylactic or therapeutic dosing.
The timing of the previous VTE history is important when deciding on the anticoagulant dose in pregnancy. In pregnant women with a VTE that occurred within the previous 4 to 6 weeks, full-dose anticoagulation with LMWH should be considered; an intermediate dose (three-fourths of a therapeutic dose) may be used if the thrombotic episode occurred more than 6 weeks earlier but still within a year. Prophylactic dosing may be sufficient if the episode occurred more than a year earlier.90 A clinical trial (High-Low) is under way to explore the optimal dose of LMWH in pregnant women with prior history of VTE who are not on chronic anticoagulation therapy.91
How is anticoagulation therapy managed in the peripartum period?
Neuraxial anesthesia during active labor while on anticoagulation increases the risk for central nervous system bleeding. Therefore, if spontaneous labor occurs in women on therapeutic dose anticoagulation, neuraxial anesthesia cannot be used. However, in the event of elective induction of labor or caesarean section, neuroaxial anesthesia may be performed 12 hours after the administration of the last prophylactic dose of LMWH or 24 hours after the last therapeutic dose of LMWH. Intravenous UFH should be stopped for 6 hours before induction of labor with a confirmed normal aPTT before placement of neuraxial anesthesia. There is no contraindication for using neuraxial anesthesia during subcutaneous standard UFH at total doses of 10,000 units daily. The risk of spinal hematoma with larger daily subcutaneous doses is unclear; therefore, a documented normal aPTT must be obtained before placement of neuroaxial anesthesia.
Postpartum, reinitiation of prophylactic-dose LMWH should be delayed for at least 12 hours after the removal of an epidural catheter. Therapeutic-dose LMWH should be administered no earlier than 24 hours after neuraxial anesthesia, providing that proper hemostasis is achieved. In the absence of persistent bleeding, if no regional anesthesia was used, LMWH may be resumed 12 hours after delivery.92 Anticoagulation with either LMWH or warfarin is recommended for at least 6 to 12 weeks postpartum.33
COUNSELING
Patients should be advised to manage controllable risk factors, including avoiding prolonged immobilization, avoiding excessive weight gain in pregnancy, and stopping smoking. Periods of immobilization tend to cause reduced blood flow (stasis), which predisposes to thrombosis. In a systematic review of records of all patients with confirmed PE after arrival at Charles de Gaulle airport in Paris during a 13-year period, women had a higher risk of PE after a long-distance flight than men, with an estimated incidence of 0.61 per million passengers versus 0.20, respectively; the incidence reached 7.24 and 2.35 cases, respectively, in passengers traveling more than 10,000 kilometers.93,94
The risk of air travel-related thrombosis in pregnant women is estimated to be between 0.03% and 0.1%. Physicians must decide on an individual basis how to prevent travel-related thrombosis in their pregnant patients. In most passengers, prevention can be limited to encouraging exercise, avoidance of long sleeping periods, and not using a window seat. Women at high risk for VTE, such as women with a prior history of VTE who are not on anticoagulation or women with known asymptomatic thrombophilia or other risk factors for thrombosis such as obesity, may benefit from a short period (1–3 days) of LMWH starting 2 hours before a long-distance flight.95
Activation of the coagulation system has been demonstrated in cigarette smokers.96 Heavy smoking was found to be a significant risk factor for VTE in a cross-sectional analysis of 2404 men and women.97 An increased risk for thrombosis during pregnancy is seen in cigarette smokers15,98 and is enhanced with the concomitant use of illicit drugs.99 Other obstetric complications associated with smoking and illicit drug use during pregnancy include preterm labor, spontaneous abortion, perinatal death, low birth weight, and abruption placenta. The efficacy of nicotine replacement therapy in pregnancy is uncertain.100 Recommendations are to advise patients to stop smoking, obtain psychosocial counseling, and utilize adjunctive therapies, which have been shown to have some effect on abstinence rates.101
CONCLUSION
Women are at increased risk for VTE during pregnancy and the postpartum period. Awareness of risk factors and the signs and symptoms of VTE is paramount. Prompt diagnosis and treatment is mandatory to decrease complications of VTE. LMWH is the mainstay treatment of VTE in pregnancy, as it does not cross the placenta. Both LMWH and warfarin are safe during lactation. Close communication among the patient, obstetrician, hematologist, anesthesiologist, and neonatologist is crucial to optimize the care of these patients.
- Knight M, Nour M, Tuffnell D, et al, eds. on behalf of MBRRACE-UK. Saving lives, improving mothers’ care—surveillance of maternal deaths in the UK 2012-14 and lessons learned to inform maternity care from the UK and Ireland confidential enquiries into maternal deaths and morbidity 2009-14. Oxford: National Perinatal Epidemiology Unit, University of Oxford; 2016: 69–75.
- Berg CJ, Callaghan WM, Syverson C, Henderson Z. Pregnancy-related mortality in the United States, 1998 to 2005. Obstet Gynecol 2010;116:1302–9.
- Salonen Ros H, Lichtenstein P, Bellocco R, et al. Increased risks of circulatory diseases in late pregnancy and puerperium. Epidemiology 2001;12:456–60.
- Heit JA, Kobbervig CE, James AH, et al. Trends in the incidence of venous thromboembolism during pregnancy or postpartum: a 30-year population-based study. Ann Intern Med 2005;143:697–706.
- Greer IA. Thrombosis in pregnancy: updates in diagnosis and management. Hematology Am Soc Hematol Educ Program 2012;2012:203–7.
- Hellgren M. Hemostasis during normal pregnancy and puerperium. Semin Thromb Hemost 2003;29:125–30.
- Brenner B. Haemostatic changes in pregnancy. Thromb Res 2004;114:409–14.
- Chan W-S, Spencer FA, Ginsberg JS. Anatomic distribution of deep vein thrombosis in pregnancy. CMAJ 2010;182:657–60.
- Greer IA. Prevention and management of venous thromboembolism in pregnancy. Clin Chest Med 2003;24:123–37.
- Wik HS, Jacobsen AF, Sandvik L, Sandset PM. Prevalence and predictors for post-thrombotic syndrome 3 to 16 years after pregnancy-related venous thrombosis: a population-based, cross-sectional, case-control study. J Thromb Haemost 2012;10:840–7.
- Pomp ER, Lenselink AM, Rosendaal FR, Doggen CJM. Pregnancy, the postpartum period and prothrombotic defects: risk of venous thrombosis in the MEGA study. J Thromb Haemost 2008;6:632–7.
- Marik PE, Plante LA. Venous thromboembolic disease and pregnancy. N Engl J Med 2008;359:2025–33.
- Jacobsen AF, Skjeldestad FE, Sandset PM. Ante- and postnatal risk factors of venous thrombosis: a hospital-based case-control study. J Thromb Haemost 2008;6:905–12.
- Virkus RA, Løkkegaard E, Lidegaard Ø, et al. Risk factors for venous thromboembolism in 1.3 million pregnancies: a nationwide prospective cohort. PloS One 2014;9:e96495.
- James AH, Jamison MG, Brancazio LR, Myers ER. Venous thromboembolism during pregnancy and the postpartum period: incidence, risk factors, and mortality. Am J Obstet Gynecol 2006;194:1311–5.
- Chan W-S, Lee A, Spencer FA, et al. Predicting deep venous thrombosis in pregnancy: out in “LEFt” field? Ann Intern Med 2009;151:85–92.
- Righini M, Jobic C, Boehlen F, et al. Predicting deep venous thrombosis in pregnancy: external validation of the LEFT clinical prediction rule. Haematologica 2013;98:545–8.
- Chan W-S, Chunilal S, Lee A, et al. A red blood cell agglutination D-dimer test to exclude deep venous thrombosis in pregnancy. Ann Intern Med 2007;147:165–70.
- Nijkeuter M, Ginsberg JS, Huisman MV. Diagnosis of deep vein thrombosis and pulmonary embolism in pregnancy: a systematic review. J Thromb Haemost 2006;4:496–500.
- Chan W-S, Spencer FA, Lee AY, et al. Safety of withholding anticoagulation in pregnant women with suspected deep vein thrombosis following negative serial compression ultrasound and iliac vein imaging. CMAJ 2013;185:E194–200.
- Dronkers CE, Srámek A, Huisman MV, Klok FA. Accurate diagnosis of iliac vein thrombosis in pregnancy with magnetic resonance direct thrombus imaging (MRDTI). BMJ Case Rep 2016;2016. pii: bcr2016218091.
- Ray JG, Vermeulen MJ, Bharatha A, et al. Association between MRI exposure during pregnancy and fetal and childhood outcomes. JAMA 2016;316:952–61.
- Royal College of Obstretricians and Gynaecologists. Thromboembolic disease in pregnancy and the puerperium: acute management. Green-top Guideline No. 37b. London: RCOG; 2015.
- Barritt DW, Jordan SC. Anticoagulant drugs in the treatment of pulmonary embolism. A controlled trial. Lancet 1960;1(7138):1309–12.
- Bourjeily G, Khalil H, Raker C, et al. Outcomes of negative multidetector computed tomography with pulmonary angiography in pregnant women suspected of pulmonary embolism. Lung 2012;190:105–11.
- Deutsch AB, Twitty P, Downes K, Parsons MT. Assessment of the alveolar-arterial oxygen gradient as a screening test for pulmonary embolism in pregnancy. Am J Obstet Gynecol 2010;203:373.e1–4.
- Powrie RO, Larson L, Rosene-Montella K, et al. Alveolar-arterial oxygen gradient in acute pulmonary embolism in pregnancy. Am J Obstet Gynecol 1998;178:394–6.
- Leung AN, Bull TM, Jaeschke R, et al. American Thoracic Society documents: an official American Thoracic Society/Society of Thoracic Radiology clinical practice guideline--evaluation of suspected pulmonary embolism in pregnancy. Radiology 2012;262:635–46.
- Cahill AG, Stout MJ, Macones GA, Bhalla S. Diagnosing pulmonary embolism in pregnancy using computed-tomographic angiography or ventilation-perfusion. Obstet Gynecol 2009;114:124–9.
- Bourjeily G, Paidas M, Khalil H, et al. Pulmonary embolism in pregnancy. Lancet 2010;375(9713):500–12.
- Chan WS, Ray JG, Murray S, et al. Suspected pulmonary embolism in pregnancy: clinical presentation, results of lung scanning, and subsequent maternal and pediatric outcomes. Arch Intern Med 2002;162:1170–5.
- Virkus RA, Løkkegaard ECL, Bergholt T, et al. Venous thromboembolism in pregnant and puerperal women in Denmark 1995-2005. A national cohort study. Thromb Haemost 2011;106:304–9.
- Kamel H, Navi BB, Sriram N, et al. Risk of a thrombotic event after the 6-week postpartum period. N Engl J Med 2014;370:1307–15.
- Ginsberg JS, Hirsh J, Turner DC, et al. Risks to the fetus of anticoagulant therapy during pregnancy. Thromb Haemost 1989;61:197–203.
- Greer IA, Nelson-Piercy C. Low-molecular-weight heparins for thromboprophylaxis and treatment of venous thromboembolism in pregnancy: a systematic review of safety and efficacy. Blood 2005;106:401–7.
- Prandoni P, Carnovali M, Marchiori A, Galilei Investigators. Subcutaneous adjusted-dose unfractionated heparin vs fixed-dose low-molecular-weight heparin in the initial treatment of venous thromboembolism. Arch Intern Med 2004;164:1077–83.
- Robertson L, Jones LE. Fixed dose subcutaneous low molecular weight heparins versus adjusted dose unfractionated heparin for venous thromboembolism. Cochrane Database Syst Rev 2017;(9):eb9;2:CD001100.
- Costantino G, Ceriani E, Rusconi AM, et al. Bleeding risk during treatment of acute thrombotic events with subcutaneous LMWH compared to intravenous unfractionated heparin; a systematic review. PloS One 2012;7:e44553.
- Warkentin TE, Levine MN, Hirsh J, et al. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med 1995;332:1330–5.
- Junqueira DRG, Perini E, Penholati RRM, Carvalho MG. Unfractionated heparin versus low molecular weight heparin for avoiding heparin-induced thrombocytopenia in postoperative patients. Cochrane Database Syst Rev 2012;(9):CD007557.
- Bates SM, Greer IA, Middeldorp S, et al. VTE, thrombophilia, antithrombotic therapy, and pregnancy. Chest 2012;141:e691S–e736S.
- Clark NP, Delate T, Witt DM, et al. A descriptive evaluation of unfractionated heparin use during pregnancy. J Thromb Thrombolysis 2009;27:267–73.
- Clark NP, Delate T, Cleary SJ, Witt DM. Analysis of unfractionated heparin dose requirements to target therapeutic anti-Xa intensity during pregnancy. Thromb Res 2010;125:402–5.
- Jacobsen AF, Qvigstad E, Sandset PM. Low molecular weight heparin (dalteparin) for the treatment of venous thromboembolism in pregnancy. BJOG 2003;110:139–44.
- Walfisch A, Koren G. The “warfarin window” in pregnancy: the importance of half-life. J Obstet Gynaecol Can 2010;32:988–9.
- Bates SM, Ginsberg JS. Anticoagulants in pregnancy: fetal effects. Baillières Clin Obstet Gynaecol 1997;11:479–88.
- Stevenson RE, Burton OM, Ferlauto GJ, Taylor HA. Hazards of oral anticoagulants during pregnancy. JAMA 1980;243:1549–51.
- Ginsberg JS, Hirsh J. Anticoagulants during pregnancy. Annu Rev Med 1989;40:79–86.
- Wong V, Cheng CH, Chan KC. Fetal and neonatal outcome of exposure to anticoagulants during pregnancy. Am J Med Genet 1993;45:17–21.
- Blickstein D, Blickstein I. The risk of fetal loss associated with Warfarin anticoagulation. Int J Gynaecol Obstet 2002;78:221–5.
- Burnett AE, Mahan CE, Vazquez SR, et al. Guidance for the practical management of the direct oral anticoagulants (DOACs) in VTE treatment. J Thromb Thrombolysis 2016;41:206–32.
- Bapat P, Pinto LSR, Lubetsky A, et al. Examining the transplacental passage of apixaban using the dually perfused human placenta. J Thromb Haemost 2016;14:1436–41.
- Bapat P, Pinto LSR, Lubetsky A, et al. Rivaroxaban transfer across the dually perfused isolated human placental cotyledon. Am J Obstet Gynecol 2015;213:710.e1–6.
- Bapat P, Kedar R, Lubetsky A, et al. Transfer of dabigatran and dabigatran etexilate mesylate across the dually perfused human placenta. Obstet Gynecol 2014;123:1256–61.
- Savaysa [package insert]. Parsippany (NJ): Daiichi Sankyo, Inc; 2015.
- Filipecki S, Tomkowski W, Hajduk B, et al. [Outcome of patients with clinically acute massive pulmonary embolism]. Pneumonol Alergol Pol 1994;62:132–7.
- Holden EL, Ranu H, Sheth A, et al. Thrombolysis for massive pulmonary embolism in pregnancy--a report of three cases and follow up over a two year period. Thromb Res 2011;127:58–9.
- te Raa GD, Ribbert LS, Snijder RJ, Biesma DH. Treatment options in massive pulmonary embolism during pregnancy; a case-report and review of literature. Thromb Res 2009;124:1–5.
- Leonhardt G, Gaul C, Nietsch HH, et al. Thrombolytic therapy in pregnancy. J Thromb Thrombolysis 2006;21:271–6.
- Colombier S, Niclauss L. Successful surgical pulmonary embolectomy for massive perinatal embolism after emergency cesarean section. Ann Vasc Surg 2015;29:1452.e1–4.
- British Committee for Standards in Haematology Writing Group, Baglin TP, Brush J, Streiff M. Guidelines on use of vena cava filters. Br J Haematol 2006;134:590–5.
- Harris SA, Velineni R, Davies AH. Inferior vena cava filters in pregnancy: a systematic review. J Vasc Interv Radiol 2016;27:354–360.
- Pabinger I, Grafenhofer H, Kyrle PA, et al. Temporary increase in the risk for recurrence during pregnancy in women with a history of venous thromboembolism. Blood 2002;100:1060–2.
- Pabinger I, Grafenhofer H, Kaider A, et al. Risk of pregnancy-associated recurrent venous thromboembolism in women with a history of venous thrombosis. J Thromb Haemost 2005;3:949–54.
- Brill-Edwards P, Ginsberg JS, Gent M, et al. Safety of withholding heparin in pregnant women with a history of venous thromboembolism. Recurrence of Clot in This Pregnancy Study Group. N Engl J Med 2000;343:1439–44.
- De Stefano V, Martinelli I, Rossi E, et al. The risk of recurrent venous thromboembolism in pregnancy and puerperium without antithrombotic prophylaxis. Br J Haematol 2006;135:386–91.
- De Stefano V, Martinelli I, Rossi E, et al. The risk of recurrent venous thromboembolism in pregnancy and puerperium without antithrombotic prophylaxis. Br J Haematol 2006;135:386–91.
- Lim W, Eikelboom JW, Ginsberg JS. Inherited thrombophilia and pregnancy associated venous thromboembolism. BMJ 2007;334:1318–21.
- Tormene D, Simioni P, Prandoni P, et al. Factor V Leiden mutation and the risk of venous thromboembolism in pregnant women. Haematologica 2001;86:1305–9.
- Baglin T, Gray E, Greaves M, et al. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol 2010;149:209–20.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006;4:295–306.
- Pengo V, Tripodi A, Reber G, et al. Update of the guidelines for lupus anticoagulant detection. J Thromb Haemost 2009;7:1737–40.
- Robertson L, Wu O, Langhorne P, et al. Thrombophilia in pregnancy: a systematic review. Br J Haematol 2006;132:171–96.
- American College of Obstetricians and Gynecologists Women’s Health Care Physicians. ACOG Practice Bulletin No. 138: Inherited thrombophilias in pregnancy. Obstet Gynecol 2013;122:706–17.
- Mak A, Cheung MW, Cheak AA, Ho RC. Combination of heparin and aspirin is superior to aspirin alone in enhancing live births in patients with recurrent pregnancy loss and positive anti-phospholipid antibodies: a meta-analysis of randomized controlled trials and meta-regression. Rheumatology (Oxf) 2010;49:281–8.
- Laskin CA, Spitzer KA, Clark CA, et al. Low molecular weight heparin and aspirin for recurrent pregnancy loss: results from the randomized, controlled HepASA Trial. J Rheumatol 2009;36:279–87.
- Farquharson RG, Quenby S, Greaves M. Antiphospholipid syndrome in pregnancy: a randomized, controlled trial of treatment. Obstet Gynecol 2002;100:408–13.
- Bates SM, Jaeschke R, Stevens SM, et al. Diagnosis of DVT: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012;141(2 Suppl):e351S–418S.
- Lubbe WF, Butler WS, Palmer SJ, Liggins GC. Fetal survival after prednisone suppression of maternal lupus-anticoagulant. Lancet 1983;1(8338):1361–3.
- Lockshin MD, Druzin ML, Qamar T. Prednisone does not prevent recurrent fetal death in women with antiphospholipid antibody. Am J Obstet Gynecol 1989;160:439–43.
- Silver RK, MacGregor SN, Sholl JS, et al. Comparative trial of prednisone plus aspirin versus aspirin alone in the treatment of anticardiolipin antibody-positive obstetric patients. Am J Obstet Gynecol 1993;169:1411–7.
- Cowchock FS, Reece EA, Balaban D, et al. Repeated fetal losses associated with antiphospholipid antibodies: a collaborative randomized trial comparing prednisone with low-dose heparin treatment. Am J Obstet Gynecol 1992;166:1318–23.
- Laskin CA, Bombardier C, Hannah ME, et al. Prednisone and aspirin in women with autoantibodies and unexplained recurrent fetal loss. N Engl J Med 1997;337:148–53.
- Dendrinos S, Sakkas E, Makrakis E. Low-molecular-weight heparin versus intravenous immunoglobulin for recurrent abortion associated with antiphospholipid antibody syndrome. Int J Gynaecol Obstet 2009;104:223–5.
- Cohen H, Arachchillage DRJ, Beyer-Westendorf J, et al. Direct oral anticoagulants and women. Semin Thromb Hemost 2016;42:789–97.
- Bates SM, Middeldorp S, Rodger M, et al. Guidance for the treatment and prevention of obstetric-associated venous thromboembolism. J Thromb Thrombolysis 2016;41:92–128.
- Knol HM, Schultinge L, Veeger NJ, et al. The risk of postpartum hemorrhage in women using high dose of low-molecular-weight heparins during pregnancy. Thromb Res 2012;130:334–8.
- Barbour LA, Smith JM, Marlar RA. Heparin levels to guide thromboembolism prophylaxis during pregnancy. Am J Obstet Gynecol 1995;173:1869–73.
- Bergqvist A, Bergqvist D, Lindhagen A, Mätzsch T. Late symptoms after pregnancy-related deep vein thrombosis. Br J Obstet Gynaecol 1990;97:338–41.
- Rodger M. Evidence base for the management of venous thromboembolism in pregnancy. Hematology Am Soc Hematol Educ Program. 2010;2010:173–80.
- Bleker SM, Buchmüller A, Chauleur C, et al. Low-molecular-weight heparin to prevent recurrent venous thromboembolism in pregnancy: Rationale and design of the Highlow study, a randomised trial of two doses. Thromb Res 2016;144:62–8.
- Horlocker TT, Wedel DJ, Rowlingson JC, et al. Regional anesthesia in the patient receiving antithrombotic or thrombolytic therapy: American Society of Regional Anesthesia and Pain Medicine evidence-based guidelines (third edition). Reg Anesth Pain Med 2010;35:64–101.
- Lapostolle F, Surget V, Borron SW, et al. Severe pulmonary embolism associated with air travel. N Engl J Med 2001;345:779–83.
- Lapostolle F, Le Toumelin P, Chassery C, et al. Gender as a risk factor for pulmonary embolism after air travel. Thromb Haemost 2009;102:1165–8.
- Cannegieter SC, Rosendaal FR. Pregnancy and travel-related thromboembolism. Thromb Res 2013;131 Suppl 1:S55–58.
- Miller GJ, Bauer KA, Cooper JA, Rosenberg RD. Activation of the coagulant pathway in cigarette smokers. Thromb Haemost 1998;79:549–53.
- Golomb BA, Chan VT, Denenberg JO, et al. Risk marker associations with venous thrombotic events: a cross-sectional analysis. BMJ Open 2014;4:e003208.
- Lindqvist P, Dahlbäck B, Marŝál K. Thrombotic risk during pregnancy: a population study. Obstet Gynecol 1999;94:595–9.
- Black M, Bhattacharya S, Fairley T, et al. Outcomes of pregnancy in women using illegal drugs and in women who smoke cigarettes. Acta Obstet Gynecol Scand 2013;92:47–52.
- Mendelsohn C, Gould GS, Oncken C. Management of smoking in pregnant women. Aust Fam Physician 2014;43:46–51.
- Chamberlain C, O’Mara-Eves A, Oliver S, et al. Psychosocial interventions for supporting women to stop smoking in pregnancy. Cochrane Database Syst Rev 2013;10:CD001055.
INTRODUCTION
Venous thromboembolism (VTE), comprising deep vein thrombosis (DVT) and pulmonary embolism (PE), is a leading nonobstetric cause of maternal death in the United States and in developed countries.1,2 During pregnancy, the risk for VTE increases four- to six-fold, and although the risk is present throughout pregnancy, the mother is at highest risk immediately postpartum.3–5
VTE risk is increased due to physiologic and anatomic changes that occur in pregnancy. These changes include hypercoagulability, progesterone-induced venous stasis, decreased venous outflow, compression of the inferior vena cava and pelvic veins by the expanding uterus, and decreased mobility. The hypercoagulability of pregnancy is due to increased levels of coagulation factors I (fibrinogen), VII, VIII, and X, and von Willebrand factor; decreased free protein S, a natural anticoagulant; acquired resistance to activated protein C; and decreased fibrinolysis due to increased levels of plasminogen activator inhibitor-1 and -2.6,7 These changes confer increased hemostasis to the mother for delivery but also place her at higher risk for thrombosis.
A review of the literature found that more than 70% of pregnancy-associated DVTs are located in the ileofemoral region, as compared with approximately 9% in non-pregnant patients.8 The proximal location is associated with a higher risk for post-thrombotic syndrome and embolization as compared with calf DVTs.9 Proximal postnatal thrombosis, smoking, and older age are independent predictors of the development of post-thrombotic syndrome.10
RISK FACTORS
Clinical risk factors that increase the risk for VTE during pregnancy include a prior history of estrogen-related or unprovoked VTE, being a carrier of severe inherited thrombophilia (homozygotes for factor V Leiden or factor II G20210A variants, double heterozygotes, or persons with antithrombin, protein C, or protein S deficiencies), and the presence of antiphospholipid (aPL) antibodies.11 Women with systemic lupus erythematosus, diabetes, sickle cell disease, and heart disease also have a high risk for VTE during pregnancy.12 Other risk factors predisposing to thrombosis include black ethnicity, smoking, operative procedures, conception after assisted reproductive techniques, high body mass index, antepartum immobilization, severe preeclampsia, advanced age and parity, and a family history of VTE.13 A prospective cohort study of 1,297,037 pregnancies and related puerperium identified the following risk factors for thrombosis: hospitalization, infection, hyperemesis, multiple pregnancies, preeclampsia, obesity, cesarean section, major postpartum hemorrhage, intrauterine growth restriction, and fetal death.14 Risk factors identified in an Agency for Healthcare Research and Quality study include: age 35 or older, black ethnicity, lupus, sickle cell disease, heart disease, postpartum infection, and transfusion.15 The combination of more than one risk factor increases the risk for VTE. All these factors have to be considered when deciding on prophylactic or therapeutic anticoagulation therapy in pregnancy. In addition, the risks of anticoagulation, including bruising, bleeding, and other side effects (eg, reduced bone mineral density with therapeutic-dose unfractionated heparin), allergic reactions, and rarely thrombocytopenia, must be considered.
EVALUATION AND DIAGNOSIS
CASE PRESENTATION I
A 31-year-old woman G1P0 at 10 weeks’ gestation with no personal or family history of thrombosis presents with acute onset of shortness of breath and left-sided chest pain that awoke her the morning of presentation. Her vital signs are significant for a heart rate of 106 beats/min, respiration rate of 22 breaths/min, blood pressure of 105/76 mm Hg, and pulse oximetry of 98% on room air. The patient denies previous exposure to oral contraceptives. She does not smoke. She reports that she had noticed left calf pain and swelling, which worsened with walking after a 4-hour drive 2 days prior.
What is the approach to diagnosis of thromboembolism in pregnant patients?
DEEP VEIN THROMBOSIS
Although a clinical diagnosis of DVT in pregnancy is unreliable, a history and physical examination are necessary to exclude other diagnoses and to assess the likelihood of thrombosis. Unfortunately, studies of the accuracy of history and physical examination for detecting DVT and PE have not included pregnant patients. In most pregnant patients with clinically suspected DVT, the diagnosis is not confirmed. Other causes of leg pain and swelling are not uncommon during pregnancy and include cellulitis, ruptured Baker’s cyst, or muscular pain.
A cross-sectional study described the derivation of the LEFt clinical decision rule, which relies on 3 variables in pregnant women with suspected DVT: left leg presentation (L), ≥ 2 cm calf circumference difference (E for edema), and first trimester presentation (Ft). If none of these variables is present, the negative predictive value is 100%.16 A validation study suggested that a negative LEFt rule accurately identifies pregnant women in whom the risk for confirmed DVT appears to be very low. The rule should not be used as an individual test for excluding DVT during pregnancy, but could be applied in a diagnostic approach in association with D-dimer measurement and compression ultrasonography (CUS); however, it has not been prospectively validated for safety and efficacy.17 In a study of 149 consecutive pregnant women with suspected DVT, a whole-blood agglutination D-dimer had a sensitivity of 100% and specificity of 60%.18 A 2006 systematic review found only 4 diagnostic studies of VTE in pregnancy in the literature. One of these studies showed that a combination of a negative CUS and normal D-dimer can accurately exclude DVT.19
Serial CUS is necessary for pregnant women with a high clinical suspicion of DVT but a negative initial investigation. In a study of 221 pregnant women in whom DVT was clinically suspected, 16 women (7.2%) were diagnosed with DVT by initial CUS, and none were diagnosed with DVT onserial testing.20 During follow-up (≥ 3 months), 6 of the 205 women with normal serial CUS results presented with symptoms of DVT, PE, or both, and 1 of them was diagnosed with DVT and PE. The sensitivity of serial CUS with Doppler imaging was 94.1% (95% confidence interval [CI] 69.2% to 99.7%), and the negative predictive value was 99.5% (95% CI 96.9% to 100%).20 All ultrasounds undertaken for investigation of pregnancy-associated DVT should include imaging of the iliac veins if there is a high index of suspicion and the CUS is negative for femoral DVT. Serial CUS with Doppler imaging of the iliac vein performed over a 7-day period excludes DVT in symptomatic pregnant women.20 Repeat CUS may be done 2 to 4 days and 6 to 8 days after the initial scan.
Ileofemoral vein thrombosis accounts for approximately 90% of proximal thromboses in pregnancy, occurring most often in the left lower extremity.20 The incidence of isolated iliac vein thrombosis in pregnancy is low, but when it does occur, delay in diagnosis can lead to significant morbidity. Therefore, for women with suspected isolated iliac vein thrombosis in whom CUS is negative or nondiagnostic, magnetic resonance direct thrombus imaging (MRDTI) should be performed.21 Patients with iliac vein thrombosis may present with unexplained inguinal, pelvic, or abdominal pain, which may be accompanied by back pain, and they usually present with swelling of the entire leg. MRDTI does not require gadolinium contrast and its accuracy appears to be similar to that of venography for iliac vein thrombi in the nonpregnant population.21 Exposure to gadolinium during pregnancy is associated with an increased risk for rheumatologic, inflammatory, or infiltrative skin conditions and stillbirth or neonatal death.22
Ovarian vein thrombosis is a rare but serious diagnosis. It occurs mostly in the postpartum period, mainly after cesarean delivery, and usually affects the right ovarian vein. The diagnosis is confirmed by ultrasound, computed tomography (CT), or magnetic resonance imaging.23
PULMONARY EMBOLISM
PE is more difficult to diagnose than DVT, particularly because clinical signs of PE are unreliable in the pregnant patient. The mortality rate of untreated PE is high, ranging from 18% to 38%, and approximately one-third of patients with untreated thromboembolic disease develop recurrent embolism.24 Studies have reported a PE prevalence between 1.4% and 4.2% in pregnant women with suspected clinical diagnosis of PE.25
The clinical presentation of PE and associated laboratory testing results may be subtler in pregnant than in nonpregnant patients. Arterial blood gases (ABG) may show hypoxemia or hypocapnia. The ABG in pregnancy has a sensitivity of 76.9%, specificity of 20.2%, and negative and positive predictive values of 80% and 11.5% for PE, respectively.26 The alveolar-arterial oxygen gradient is a poor screening test for PE during pregnancy and postpartum. A retrospective chart review of 17 pregnant women with documented PE showed that 58% had normal alveolar-arterial gradients.27 Therefore, in a pregnant woman with a history suspicious for PE, objective imaging studies should be performed even if the patient has normal ABG.
The 2011 guidelines from the American Thoracic Society (ATS) and the Society of Thoracic Radiology (STR) recommend against using D-dimer to diagnose PE in pregnancy.28 In addition, lower extremity CUS should only be performed as the first diagnostic imaging procedure if the patient has signs or symptoms of DVT. Instead, the ATS/STR guidelines recommend a plain radiograph of the chest as the first imaging test. If the chest radiograph is normal, a ventilation/perfusion scan (V/Q) scan is preferred over CT pulmonary angiography (CTPA). Diagnostic accuracy of the V/Q scan may be superior to CTPA in pregnancy, and it is preferable because of the lower prevalence of indeterminate V/Q scan in pregnant women.29 Moreover, there is lower radiation exposure to the maternal breast and lung tissue with a V/Q scan than with CTPA. CTPA confers lower fetal radiation doses than V/Q scans (0.03–0.66 mGy versus 0.32–0.74 mGy, respectively) but higher total body maternal radiation (4–16 mSv versus 1–2.5 mSv).30 A quantitative approach to lung scan interpretation, based on the distribution histogram of V/Q ratios, may be helpful in categorizing patients with suspected PE.28 A study of 121 suspected episodes of PE in 120 pregnant women showed that 104 women with normal or nondiagnostic scans did not develop subsequent episodes of VTE during a mean follow-up period of 20 months.31
If the baseline chest radiograph is abnormal in a pregnant woman with clinical suspicion of PE, a CTPA should be performed. As noted, fetal radiation doses for CTPA examinations in which the fetus is not directly imaged are minimal. If CTPA is recommended for the diagnosis of PE, the patient should be informed that radiation to the breast may increase her baseline risk for breast cancer. The ATS guidelines state that “given the lack of evidence documenting clear superiority of any one diagnostic test, the values and preferences of a patient and her physician likely will and should determine the final choice and sequence of tests performed.”28
CASE I CONTINUED
Upon presentation to the emergency department, the circumference of the patient’s left leg is not significantly greater than that of her right leg, and her leg pain has resolved. Bilateral CUS is negative for proximal or distal DVT. Chest radiograph shows an opacification of her left lower lobe. CTPA shows bilateral segmental and subsegmental lower lobe pulmonary emboli.
How does risk for VTE change throughout pregnancy?
Women are at increased risk for VTE throughout the entire pregnancy, starting from conception, but mainly during the postpartum period. A Danish historical controlled cohort study of 819,751 pregnant women (ages 15–49 years) over a 10-year period identified 727 women with VTE. The absolute risk for VTE per 10,000 pregnancy-years increased from 4.1 (95% CI 3.2 to 5.2) during weeks 1 to 11 to 59.0 (95% CI 46.1 to 76.4) in week 40 and decreased in the postpartum period from 60 (95% CI 47.2 to 76.4) during the first week after birth to 2.1 during weeks 9–12 after birth (95% CI 1.1 to 4.2).32 This study showed that the risk of VTE increases throughout pregnancy and reaches its maximum during the peripartum period and is not significantly increased after 6 weeks post-delivery. In a retrospective cross-over cohort study of 1,687,930 women in California who delivered their first newborn, an elevated risk of VTE persisted until at least 12 weeks after delivery. However, the absolute increase in risk after 6 weeks postpartum was low.33
CASE 1 CONCLUSION
The patient is started on anticoagulation therapy and carefully monitored during the remainder of the pregnancy and postpartum period. Anticoagulation is discontinued 6 weeks after delivery.
TREATMENT
ANTICOAGULATION THERAPY
The treatment of VTE can be lifesaving. In a study comparing 35 patients with PE randomly assigned to treatment with anticoagulants versus no treatment, 5 of 19 patients in the untreated group died from PE and an additional 5 had nonfatal recurrences, as compared with none in the treated group.24 Unfractionated heparin (UFH) and low-molecular-weight heparin (LMWH) are both safe and effective anticoagulants during pregnancy as neither crosses the placenta. In a review of 186 reports of fetal and infant outcomes following anticoagulant therapy during pregnancy in 1325 pregnancies, outcomes in UFH-treated patients were similar to those in the normal population after excluding pregnancies with comorbid conditions independently associated with adverse outcomes.34 A 2005 systematic review of LMWH for prophylaxis and treatment of VTE during pregnancy included 64 studies of 277 pregnancies. There were no maternal deaths, live births resulted from 94.7% of the pregnancies, VTE or arterial thrombosis occurred in 0.86%, and significant bleeding occurred in 1.98%.35
The standard UFH regimen is an initial bolus of 5000 units subcutaneously and 17,500 units every 12 hours, with dose adjustment made based on a mid-interval activated partial thromboplastin time (aPTT).36 Although still controversial, it has been suggested that the anti-Xa assay with a mid-dosing interval target of 0.3 to 0.7 U/mL is a more reliable measure of therapeutic UFH activity than the aPTT, as the aPTT response is suppressed due to a pregnancy-related increase in factor VIII. LMWH is dosed based on weight; regimens are enoxaparin 1 mg/kg subcutaneously twice daily or 1.5 mg/kg subcutaneously once daily, and dalteparin 100 units/kg every 12 hours or 150 units/kg daily.
A 2017 Cochrane review of the effect of LMWH compared with UFH for the treatment of VTE in the nonpregnant setting included 23 studies with 9587 patients. Thrombotic complications (odds ratio [OR] 0.70 [CI 0.57 to 0.85]) and major hemorrhage (OR 0.58 [CI 0.40 to 0.83]) were lower in patients receiving LMWH, with a trend toward lower mortality.37 In addition, the incidence of bleeding complications in patients treated with subcutaneous LMWH versus intravenous heparin was compared in a 2012 systematic review of 27 randomized controlled trials with a total of 28,637 patients. In patients treated with LMWH, there was a nonstatistically significant lower incidence of major bleeding events (OR 0.79 [95% CI 0.60 to 1.04]) and a statistically significant reduction in bleeding risk (OR 0.68 [95% CI 0.47 to 1.00]) compared to patients treated with UFH.38 Additionally, a trial comparing the use of standard UFH versus LMWH found a significantly lower incidence of thrombocytopenia in patients treated with LMWH.39,40 Overall, LMWH is more effective at decreasing both thrombotic and bleeding complications, and the risk for osteoporosis is lower with LMWH. Based on these results, the American College of Chest Physicians (ACCP) recommends LMWH as the first-line treatment for VTE in pregnancy.41
In specific clinical situations, such as patients with renal dysfunction with creatinine clearance (CrCl) less than 30 mL/min, UFH is indicated. In a study of 103 pregnancies in 93 women given anti-coagulation during pregnancy, 89.3% received UFH. There were no maternal deaths, and fetal demise occurred in 8 pregnancies (7.8%) at a median of 14 weeks’ gestation. There were 2 episodes of PE (1.9%) and 2 major bleeding events requiring transfusion (1.9%).42 UFH costs much less than LMWH, and therefore UFH remains an important, inexpensive, and efficacious anticoagulant option for pregnant women who require anticoagulation and cannot afford LMWH.43
Due to the physiologic changes associated with pregnancy, LMWH and UFH dosages may need to be adjusted. An observational study of 20 pregnant women with acute VTE found no recurrent VTE or major bleeding after treatment with dalteparin. Dalteparin doses approximately 10% to 20% higher than those recommended in nonpregnant women were required to reach therapeutic target anti-Xa activity.44
Caution Regarding Oral Anticoagulants
Due to its teratogenicity, warfarin is not a first-line anticoagulation option. It is strictly contraindicated during the first trimester during organogenesis, and its use during pregnancy is restricted to women with mechanical heart valves. Warfarin crosses the placenta and has been associated with nasal hypoplasia, stippled epiphyses, and growth restriction, particularly between 6 to 9 weeks’ gestation. Every effort should be made to substitute UFH or LMWH for warfarin between 6 and 12 weeks of gestation. The bridging process should begin as early in the gestational age as possible due to the long half-life of warfarin.45 When used later in gestation, warfarin has been associated with fetal hemorrhage and central nervous system abnormalities. Other complications from use during the second and third trimesters include microcephaly, blindness, deafness, and fetal growth restriction.46,47 Its use also increases the risk for abortion and fetal death in utero.48–50
The direct oral anticoagulants (DOACs) are not approved for use in pregnancy. Although there are limited anecdotal reports of DOAC use in pregnancy,51 there is preclinical evidence of placental transfer with the DOACs rivaroxaban and apixaban (direct Xa inhibitors) and the oral thrombin inhibitor dabigatran, thus increasing the risk to the fetus.52–54 Edoxaban, another direct Xa inhibitor, should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. It should be discontinued in nursing mothers.55
THROMBOLYSIS
Fetal as well as maternal survival is dependent on adequate maternal perfusion and oxygenation. The risk of death from PE is significant, with a cross-sectional study of 58 patients with acute, massive PE showing a 55% mortality rate.56 Thus, pregnancy is not an absolute contraindication to mechanical or systemic (recombinant tissue plasminogen activator or streptokinase) thrombolysis in an unstable patient at high risk for death.57–59 There are no major studies of this approach, although a small review of 13 cases using systemic thrombolysis showed no increased risk of maternal mortality.58 Thrombolysis should be considered for appropriate indications in pregnant patients as it would be in nonpregnant patients. However, caution is required when drawing conclusions regarding maternal and fetal safety, given the lack of controlled clinical trials including pregnant women.
SURGICAL PULMONARY EMBOLECTOMY
Surgical pulmonary embolectomy is an important therapeutic and potentially life-saving option in women presenting with massive PE in the immediate postpartum period. Because of the risk of massive uterine bleeding immediately postpartum, thrombolytic therapy should not be used.60
INFERIOR VENA CAVA FILTER
Placement of an inferior vena cava (IVC) filter is indicated in patients who have an acute VTE with absolute contraindications for anticoagulation. In addition, it can be considered in patients with extensive ileofemoral venous thrombosis within 2 weeks prior to expected delivery.61 In a systematic review of 44 studies of IVC filters placed in pregnant patients, the IVC filter complication rate was 8.87% and the failure-to-retrieve rate was 11.25%.62 The complication rate is similar to that found in the nonpregnant population. Thus, IVC filters may be used when appropriately indicated and should be removed as soon as clinically feasible.
RECURRENT THROMBOSIS AND THROMBOPHILIAS
CASE PRESENTATION 2
A 34-year-old pregnant woman G1P0 at 38 weeks’ gestation presents with a painful, swollen left calf that is associated with difficulty on walking; the circumference of the left calf is 2 cm greater than that of the right. She has no shortness of breath or chest pain. She has a prior history of distal right lower extremity DVT while on combined oral contraceptives. Her mother also has a history of DVT while bedbound during a prolonged hospitalization at an older age. CUS is negative, and the patient is discharged home. However, 24 hours later she returns to the hospital with worsening swelling and pain in her left leg. Magnetic resonance venography demonstrates a large left external iliac and common iliac DVT. She is admitted and is started on UFH, and a retrievable IVC filter is placed in anticipation of delivery.
What is the risk for VTE recurrence during pregnancy?
A personal and family history of VTE should be obtained when evaluating pregnant patients. A retrospective study of 109 women with prior history of VTE showed recurrence rates per patient-year of 10.9% during pregnancy and 3.7% in the nonpregnant period; the relative risk of recurrent VTE during pregnancy was 3.5 (95% CI 1.6 to 7.8).63 Two large European retrospective cohort studies of VTE in pregnancy showed that the recurrence rate of VTE in women with a history of thrombosis is around 6% during pregnancy, equally distributed among trimesters. The highest incidence of recurrence was in the postpartum period, ranging from 8.3% to 10%.64 The recurrence risk during pregnancy in women with a history of a single episode of VTE was 2.4% antepartum (95% CI 0.2% to 6.9%).65 These risks may be lower in women without thrombophilia or with a temporary risk factor associated with their previous thromboembolic event.65 Recurrence risk is higher if the previous VTE was estrogen-related, either due to pregnancy or through hormonal contraception (10%), than if the previous VTE was non-estrogen-related (2.7%).64,66
The timing of the case patient’s presentation is consistent with reports of increased risk of VTE during the peripartum period. Her prior history of estrogen-related DVT is concerning for a risk of recurrence, particularly during pregnancy. A retrospective cohort study of 1104 women with previous VTE, 88 of whom became pregnant without receiving thromboprophylaxis, showed that the overall rate of VTE recurrence was 5.8% (95% CI 3.0% to 10.6%) and 8.3% (95% CI 4.5% to 14.6%) during pregnancy and postpartum, respectively. The risk of VTE recurrence was absent if the first VTE was related to a transient risk factor other than pregnancy, postpartum period, or hormonal contraception.67 However, the recurrence rate of VTE in women with prior unprovoked VTE and/or thrombophilia has been reported as 5.9% (95% CI 1.2% to 16.2%).65 The presence of an underlying hypercoagulable state can increase the recurrence risk by 25% to 50%, depending on the disorder.68 A retrospective cohort study of 270 pregnancies in 105 carriers of factor V Leiden, identified because of a symptomatic relative with the factor V Leiden mutation, found a VTE risk (mostly in the postpartum period) of 6.4% for heterozygous women, 16.7% for homozygous women, 20% for double heterozygous women, and 1.2% for noncarriers.69
Should the patient be screened for a thrombophilia disorder?
Half of all index thromboses in patients with thrombophilia occur in association with an additional risk factor. In women of child-bearing age, pregnancy, the postpartum period, and the use of combined hormonal contraception are all risk factors for VTE. A 2010 guideline from the British hematology community recommended testing for thrombophilia in women with prior VTE secondary to a minor provoking factor before or during pregnancy, but not testing women with unprovoked VTE (who would receive prophylaxis regardless) or those with VTE secondary to a major provoking factor (who would not require prophylaxis).70 Indications to screen for aPL antibodies include: women with (1) 3 unexplained recurrent first-trimester pregnancy losses or 1 second or third trimester fetal loss of morphologically normal fetuses; (2) severe preeclampsia; (3) intrauterine growth restriction; or (4) premature labor (< 34 weeks’ gestation).71,72
CASE 2 CONCLUSION
The patient is subsequently screened for inherited thrombophilia disorders and is found to be heterozygous for factor V Leiden.
CASE PRESENTATION 3
A 25-year-old woman is diagnosed with antiphospholipid syndrome (APS) during her second pregnancy when she experiences fetal loss during her second trimester. Pathologic examination of the placenta reveals infarcts. Laboratory evaluation reveals positive high-titer anticardiolipin and anti-beta-2 glycoprotein 1 antibodies (IgG isotype) and lupus anticoagulant on 2 separate occasions 12 weeks apart. In a subsequent pregnancy, she is started on prophylactic LMWH and daily low-dose aspirin (81 mg). At 36 weeks’ gestation, she presents with a blood pressure of 210/104 mm Hg and a platelet count of 94,000 cells/µL. She is diagnosed with preeclampsia and hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome and is induced for early delivery. About 2 weeks after vaginal delivery, she notices shortness of breath and chest pain. A CTPA demonstrates a right lower lobe lobar defect consistent with a PE. Her anticoagulation is increased to therapeutic dosage LMWH.
To what extent does thrombophilia increase the risk for VTE in pregnancy?
Approximately 50% of pregnancy-related VTEs are associated with inherited thrombophilia. A systematic review of 79 studies, in which 9 studies (n = 2526 patients) assessed the risk of VTE associated with inherited thrombophilia in pregnancy, revealed that the odds ratio for individuals with thrombophilia to develop VTE ranged from 0.74 to 34.40.73 Although women with thrombophilia have an increased relative risk of developing VTE in pregnancy, the absolute risk of VTE remains low (Table 1).41,73,74
How is APS managed in pregnant patients?
Women with history of recurrent early pregnancy loss (< 10 weeks’ gestation) related to the presence of aPL antibodies are managed with low-dose aspirin and prophylactic-dose UFH or LMWH. This treatment increases the rate of subsequent successful pregnancy outcomes and reduces the risk for thrombosis. A 2010 systematic review and meta-analysis of UFH plus low-dose aspirin compared with low-dose aspirin alone in patients with APS and recurrent pregnancy loss included 5 trials and 334 patients. Patients receiving dual therapy had higher rates of live births (74.3%; relative risk [RR] 1.30 [CI 1.04 to 1.63]) compared to the aspirin-only group (55.8%).75 A 2009 randomized controlled trial compared low-dose aspirin to low-dose aspirin plus LMWH in women with recurrent pregnancy loss and either aPL antibodies, antinuclear antibody, or inherited thrombophilia. The study was stopped early after 4 years and found no difference in rates of live births between the groups (77.8% versus 79.1%).76 However, a randomized case-control trial of women with aPL antibodies and recurrent miscarriage found a 72% live birth rate in 47 women randomly assigned to low-dose aspirin and LMWH.77 A 2012 guideline from the American College of Chest Physicians (ACCP) recommends that women with aPL antibodies with a history of 3 or more pregnancy losses receive low-dose aspirin plus prophylactic-dose LMWH or UFH.78 A 2014 systematic review and meta-analysis showed that the combination of low-dose aspirin and UFH resulted in a higher live-birth rate than aspirin alone in 803 women with APS (RR 1.54 [95% CI 1.25 to 1.89]).79 Further large randomized controlled trials are needed to confirm optimal management of recurrent miscarriage and aPL antibodies.
The addition of prednisone to aspirin, heparin, or both has shown no benefits in pregnant women with aPL antibodies. Indeed, prolonged use of steroids may cause serious pregnancy complications, such as prematurity and hypertension.80–83 Intravenous infusions of immunoglobulin (IVIG) have not been shown to be superior to heparin and aspirin. This finding was confirmed in a multicenter clinical trial that tested the effects of IVIG compared with LMWH plus low-dose aspirin for the treatment of women with aPL antibodies and recurrent miscarriage. The rate of live-birth was 72.5% in the group treated with heparin plus low-dose aspirin compared with 39.5% in the IVIG group.84
Preeclampsia and HELLP syndrome complicated the case patient’s pregnancy even though she was being treated with prophylactic-dose LMWH and low-dose aspirin, the current standard of care for pregnant women with APS (UFH can be used as well). It is important to note that complications may still occur despite standard treatment. Indeed, PE is more common in the postpartum than in the antepartum period. Prompt diagnosis is paramount to initiate the appropriate treatment; in this case the dose of LMWH was increased from prophylactic to therapeutic dose. However, additional therapeutic modalities are necessary to improve outcomes. A randomized controlled trial comparing standard of care with or without hydroxychloroquine is under way to address this issue.
PROPHYLAXIS
CASE PRESENTATION 4
A 34-year-old woman G1P0 at 6 weeks’ gestation with a past medical history of a proximal lower extremity DVT while on oral contraception is treated with warfarin anticoagulation for 6 months. Her obstetrician consults the hematologist to advise regarding antithrombotic management during this pregnancy.
What is the approach to prophylaxis in women at high risk for pregnancy-associated VTE?
All women at high risk for pregnancy-associated VTE should be counseled about the signs and symptoms of DVT or PE during preconception and pregnancy and have a plan developed should these symptoms arise. The ACCP guidelines on antithrombotic therapy outline recommendations ranging from clinical vigilance to prophylactic and intermediate-dose anticoagulation, depending on the risk for VTE recurrence, based on the personal and family history of VTE and type of thrombophilia (Table 2).78 These recommendations range from grade 2B to 2C.
For women with a history of estrogen-related VTE, single unprovoked VTE, or recurrent unprovoked VTE not on chronic anticoagulation, antepartum and postpartum pharmacologic thromboprophylaxis with either prophylactic or intermediate-dose LMWH is recommended (grade 2C). In patients with prior history of provoked VTE (non-estrogen related), antepartum clinical vigilance and postpartum pharmacologic thromboprophylaxis is recommended (grade 2C, 2B).
In asymptomatic pregnant women who are homozygote carriers for factor V Leiden or prothrombin G20210A variants and have a positive family history of thrombosis, antepartum and postpartum pharmacologic thromboprophylaxis is recommended (grade 2B). In asymptomatic homozygote carriers of factor V Leiden or prothrombin G20210A variants with no family history of thrombosis and women with all other thrombophilias with a positive family history of thrombosis, postpartum pharmacologic thromboprophylaxis is indicated (grade 2B and 2C, respectively). For women with confirmed APS and clinical criteria of obstetric APS with recurrent pregnancy loss, antepartum thromboprophylaxis with LMWH and low-dose aspirin is recommended (grade 1B). For pregnant women with all other thrombophilias with no personal or family history of thrombosis, clinical vigilance is suggested (grade 2 C).78
As an alternative to LMWH, vitamin K antagonists (VKA) such as warfarin can be used for postpartum thromboprophylaxis; in patients with protein C or S deficiency, due to the risk of warfarin-induced skin necrosis, a rapid-onset anticoagulant must be concomitantly administered. Warfarin and LMWH are safe anticoagulants during lactation, but there are no clinical data on the effects of the DOACs on infants during lactation. Data from animal studies indicate that DOACs are secreted into breast milk.85
What risks are associated with anticoagulant therapy in pregnancy?
VKAs cross the placenta and can cause teratogenicity, pregnancy loss, fetal bleeding, and neurodevelopmental deficits. Therefore, discontinuation of VKAs prior to the sixth week of gestation is necessary to avoid warfarin embryopathy. DOACs have been shown to readily cross the placenta but with unknown human reproductive risks. Fondaparinux, a synthetic pentasaccharide, crosses the placenta in small quantities. Though there are reports of the successful use of fondaparinux in pregnancy, there is limited reported experience of its use in the first trimester.86
The risk for bleeding with anticoagulation is notably acceptable. In a case-control study of 88 pregnant women receiving therapeutic-dose anticoagulation, the risk of postpartum hemorrhage (PPH) after vaginal delivery was 30% in those who received LMWH anticoagulation versus 18% in those who did not (OR 1.9 [95% CI 1.1 to 3.5]).87 However, the risk for severe PPH (≥ 500 mL) was similar (5.6% versus 5.0%; OR 1.1 [95% CI 0.4 to 3.6]). The risk for PPH after cesarean section was 12% in LMWH users versus 4% in LMWH non-users (OR 2.9 [95% CI 0.5 to 19.4]). The risk for PPH associated with delivery within 24 hours after the last dose of LMWH was 1.2 times higher (95% CI 0.4 to 3.6) compared to a longer interval. Therefore, therapeutic LMWH increases the risk for blood loss after vaginal delivery, but not the risk for severe PPH. The risk for PPH is influenced by the interval between the last dose of LMWH and delivery. Of note in this study, per the institution’s protocol, the anticoagulation was stopped with signs of labor or determination of need for delivery. The risk for blood loss may be mitigated in more planned delivery scenarios.87
CASE 4 CONTINUED
The patient is placed on prophylactic-dose LMWH with good tolerance and delivers at 39 weeks' gestation via caesarian section due to nonprogression of labor. Postpartum she is restarted on prophylactic-dose anticoagulation with LMWH. Two weeks after discharge from the hospital, she presents with right calf pain and mild shortness of breath. On physical exam, her leg circumferences are equal. A D-dimer assay is 3375 ng/mL (normal 0–229). CUS of the right leg shows a complete occlusive DVT of the mid-distal superficial femoral and popliteal veins and partially occlusive acute DVT of the right posterior tibial and peroneal veins. CTPA reveals a right lower lobe PE. Because she had developed VTE despite prophylactic LMWH, her anticoagulation is changed to therapeutic dose. She is treated with anticoagulation with LMWH for a total of 3 months, after which a repeat CUS shows no residual thrombosis.
What is the recommended dosing of heparin and LMWH during pregnancy?
A prospective study of 14 pregnant women receiving UFH prophylaxis found that a prophylactic dose of 5000 units twice a day was inadequate to achieve prophylactic heparin levels in any patient in the second or third trimester.88 Similar to treatment dosage, there is no consensus evidence for prophylactic dosing, and dosage recommendations are based on expert opinion. In a retrospective study of 25 pregnant women on intermediate-dose UFH, the mean UFH dose required to achieve a target anti-factor Xa level of 0.1 to 0.3 units/mL was 236.9 units/kg/day.89 However, the use of anti-factor Xa levels for monitoring is controversial as there is no data to support a difference in outcomes with its use in prophylactic or therapeutic dosing.
The timing of the previous VTE history is important when deciding on the anticoagulant dose in pregnancy. In pregnant women with a VTE that occurred within the previous 4 to 6 weeks, full-dose anticoagulation with LMWH should be considered; an intermediate dose (three-fourths of a therapeutic dose) may be used if the thrombotic episode occurred more than 6 weeks earlier but still within a year. Prophylactic dosing may be sufficient if the episode occurred more than a year earlier.90 A clinical trial (High-Low) is under way to explore the optimal dose of LMWH in pregnant women with prior history of VTE who are not on chronic anticoagulation therapy.91
How is anticoagulation therapy managed in the peripartum period?
Neuraxial anesthesia during active labor while on anticoagulation increases the risk for central nervous system bleeding. Therefore, if spontaneous labor occurs in women on therapeutic dose anticoagulation, neuraxial anesthesia cannot be used. However, in the event of elective induction of labor or caesarean section, neuroaxial anesthesia may be performed 12 hours after the administration of the last prophylactic dose of LMWH or 24 hours after the last therapeutic dose of LMWH. Intravenous UFH should be stopped for 6 hours before induction of labor with a confirmed normal aPTT before placement of neuraxial anesthesia. There is no contraindication for using neuraxial anesthesia during subcutaneous standard UFH at total doses of 10,000 units daily. The risk of spinal hematoma with larger daily subcutaneous doses is unclear; therefore, a documented normal aPTT must be obtained before placement of neuroaxial anesthesia.
Postpartum, reinitiation of prophylactic-dose LMWH should be delayed for at least 12 hours after the removal of an epidural catheter. Therapeutic-dose LMWH should be administered no earlier than 24 hours after neuraxial anesthesia, providing that proper hemostasis is achieved. In the absence of persistent bleeding, if no regional anesthesia was used, LMWH may be resumed 12 hours after delivery.92 Anticoagulation with either LMWH or warfarin is recommended for at least 6 to 12 weeks postpartum.33
COUNSELING
Patients should be advised to manage controllable risk factors, including avoiding prolonged immobilization, avoiding excessive weight gain in pregnancy, and stopping smoking. Periods of immobilization tend to cause reduced blood flow (stasis), which predisposes to thrombosis. In a systematic review of records of all patients with confirmed PE after arrival at Charles de Gaulle airport in Paris during a 13-year period, women had a higher risk of PE after a long-distance flight than men, with an estimated incidence of 0.61 per million passengers versus 0.20, respectively; the incidence reached 7.24 and 2.35 cases, respectively, in passengers traveling more than 10,000 kilometers.93,94
The risk of air travel-related thrombosis in pregnant women is estimated to be between 0.03% and 0.1%. Physicians must decide on an individual basis how to prevent travel-related thrombosis in their pregnant patients. In most passengers, prevention can be limited to encouraging exercise, avoidance of long sleeping periods, and not using a window seat. Women at high risk for VTE, such as women with a prior history of VTE who are not on anticoagulation or women with known asymptomatic thrombophilia or other risk factors for thrombosis such as obesity, may benefit from a short period (1–3 days) of LMWH starting 2 hours before a long-distance flight.95
Activation of the coagulation system has been demonstrated in cigarette smokers.96 Heavy smoking was found to be a significant risk factor for VTE in a cross-sectional analysis of 2404 men and women.97 An increased risk for thrombosis during pregnancy is seen in cigarette smokers15,98 and is enhanced with the concomitant use of illicit drugs.99 Other obstetric complications associated with smoking and illicit drug use during pregnancy include preterm labor, spontaneous abortion, perinatal death, low birth weight, and abruption placenta. The efficacy of nicotine replacement therapy in pregnancy is uncertain.100 Recommendations are to advise patients to stop smoking, obtain psychosocial counseling, and utilize adjunctive therapies, which have been shown to have some effect on abstinence rates.101
CONCLUSION
Women are at increased risk for VTE during pregnancy and the postpartum period. Awareness of risk factors and the signs and symptoms of VTE is paramount. Prompt diagnosis and treatment is mandatory to decrease complications of VTE. LMWH is the mainstay treatment of VTE in pregnancy, as it does not cross the placenta. Both LMWH and warfarin are safe during lactation. Close communication among the patient, obstetrician, hematologist, anesthesiologist, and neonatologist is crucial to optimize the care of these patients.
INTRODUCTION
Venous thromboembolism (VTE), comprising deep vein thrombosis (DVT) and pulmonary embolism (PE), is a leading nonobstetric cause of maternal death in the United States and in developed countries.1,2 During pregnancy, the risk for VTE increases four- to six-fold, and although the risk is present throughout pregnancy, the mother is at highest risk immediately postpartum.3–5
VTE risk is increased due to physiologic and anatomic changes that occur in pregnancy. These changes include hypercoagulability, progesterone-induced venous stasis, decreased venous outflow, compression of the inferior vena cava and pelvic veins by the expanding uterus, and decreased mobility. The hypercoagulability of pregnancy is due to increased levels of coagulation factors I (fibrinogen), VII, VIII, and X, and von Willebrand factor; decreased free protein S, a natural anticoagulant; acquired resistance to activated protein C; and decreased fibrinolysis due to increased levels of plasminogen activator inhibitor-1 and -2.6,7 These changes confer increased hemostasis to the mother for delivery but also place her at higher risk for thrombosis.
A review of the literature found that more than 70% of pregnancy-associated DVTs are located in the ileofemoral region, as compared with approximately 9% in non-pregnant patients.8 The proximal location is associated with a higher risk for post-thrombotic syndrome and embolization as compared with calf DVTs.9 Proximal postnatal thrombosis, smoking, and older age are independent predictors of the development of post-thrombotic syndrome.10
RISK FACTORS
Clinical risk factors that increase the risk for VTE during pregnancy include a prior history of estrogen-related or unprovoked VTE, being a carrier of severe inherited thrombophilia (homozygotes for factor V Leiden or factor II G20210A variants, double heterozygotes, or persons with antithrombin, protein C, or protein S deficiencies), and the presence of antiphospholipid (aPL) antibodies.11 Women with systemic lupus erythematosus, diabetes, sickle cell disease, and heart disease also have a high risk for VTE during pregnancy.12 Other risk factors predisposing to thrombosis include black ethnicity, smoking, operative procedures, conception after assisted reproductive techniques, high body mass index, antepartum immobilization, severe preeclampsia, advanced age and parity, and a family history of VTE.13 A prospective cohort study of 1,297,037 pregnancies and related puerperium identified the following risk factors for thrombosis: hospitalization, infection, hyperemesis, multiple pregnancies, preeclampsia, obesity, cesarean section, major postpartum hemorrhage, intrauterine growth restriction, and fetal death.14 Risk factors identified in an Agency for Healthcare Research and Quality study include: age 35 or older, black ethnicity, lupus, sickle cell disease, heart disease, postpartum infection, and transfusion.15 The combination of more than one risk factor increases the risk for VTE. All these factors have to be considered when deciding on prophylactic or therapeutic anticoagulation therapy in pregnancy. In addition, the risks of anticoagulation, including bruising, bleeding, and other side effects (eg, reduced bone mineral density with therapeutic-dose unfractionated heparin), allergic reactions, and rarely thrombocytopenia, must be considered.
EVALUATION AND DIAGNOSIS
CASE PRESENTATION I
A 31-year-old woman G1P0 at 10 weeks’ gestation with no personal or family history of thrombosis presents with acute onset of shortness of breath and left-sided chest pain that awoke her the morning of presentation. Her vital signs are significant for a heart rate of 106 beats/min, respiration rate of 22 breaths/min, blood pressure of 105/76 mm Hg, and pulse oximetry of 98% on room air. The patient denies previous exposure to oral contraceptives. She does not smoke. She reports that she had noticed left calf pain and swelling, which worsened with walking after a 4-hour drive 2 days prior.
What is the approach to diagnosis of thromboembolism in pregnant patients?
DEEP VEIN THROMBOSIS
Although a clinical diagnosis of DVT in pregnancy is unreliable, a history and physical examination are necessary to exclude other diagnoses and to assess the likelihood of thrombosis. Unfortunately, studies of the accuracy of history and physical examination for detecting DVT and PE have not included pregnant patients. In most pregnant patients with clinically suspected DVT, the diagnosis is not confirmed. Other causes of leg pain and swelling are not uncommon during pregnancy and include cellulitis, ruptured Baker’s cyst, or muscular pain.
A cross-sectional study described the derivation of the LEFt clinical decision rule, which relies on 3 variables in pregnant women with suspected DVT: left leg presentation (L), ≥ 2 cm calf circumference difference (E for edema), and first trimester presentation (Ft). If none of these variables is present, the negative predictive value is 100%.16 A validation study suggested that a negative LEFt rule accurately identifies pregnant women in whom the risk for confirmed DVT appears to be very low. The rule should not be used as an individual test for excluding DVT during pregnancy, but could be applied in a diagnostic approach in association with D-dimer measurement and compression ultrasonography (CUS); however, it has not been prospectively validated for safety and efficacy.17 In a study of 149 consecutive pregnant women with suspected DVT, a whole-blood agglutination D-dimer had a sensitivity of 100% and specificity of 60%.18 A 2006 systematic review found only 4 diagnostic studies of VTE in pregnancy in the literature. One of these studies showed that a combination of a negative CUS and normal D-dimer can accurately exclude DVT.19
Serial CUS is necessary for pregnant women with a high clinical suspicion of DVT but a negative initial investigation. In a study of 221 pregnant women in whom DVT was clinically suspected, 16 women (7.2%) were diagnosed with DVT by initial CUS, and none were diagnosed with DVT onserial testing.20 During follow-up (≥ 3 months), 6 of the 205 women with normal serial CUS results presented with symptoms of DVT, PE, or both, and 1 of them was diagnosed with DVT and PE. The sensitivity of serial CUS with Doppler imaging was 94.1% (95% confidence interval [CI] 69.2% to 99.7%), and the negative predictive value was 99.5% (95% CI 96.9% to 100%).20 All ultrasounds undertaken for investigation of pregnancy-associated DVT should include imaging of the iliac veins if there is a high index of suspicion and the CUS is negative for femoral DVT. Serial CUS with Doppler imaging of the iliac vein performed over a 7-day period excludes DVT in symptomatic pregnant women.20 Repeat CUS may be done 2 to 4 days and 6 to 8 days after the initial scan.
Ileofemoral vein thrombosis accounts for approximately 90% of proximal thromboses in pregnancy, occurring most often in the left lower extremity.20 The incidence of isolated iliac vein thrombosis in pregnancy is low, but when it does occur, delay in diagnosis can lead to significant morbidity. Therefore, for women with suspected isolated iliac vein thrombosis in whom CUS is negative or nondiagnostic, magnetic resonance direct thrombus imaging (MRDTI) should be performed.21 Patients with iliac vein thrombosis may present with unexplained inguinal, pelvic, or abdominal pain, which may be accompanied by back pain, and they usually present with swelling of the entire leg. MRDTI does not require gadolinium contrast and its accuracy appears to be similar to that of venography for iliac vein thrombi in the nonpregnant population.21 Exposure to gadolinium during pregnancy is associated with an increased risk for rheumatologic, inflammatory, or infiltrative skin conditions and stillbirth or neonatal death.22
Ovarian vein thrombosis is a rare but serious diagnosis. It occurs mostly in the postpartum period, mainly after cesarean delivery, and usually affects the right ovarian vein. The diagnosis is confirmed by ultrasound, computed tomography (CT), or magnetic resonance imaging.23
PULMONARY EMBOLISM
PE is more difficult to diagnose than DVT, particularly because clinical signs of PE are unreliable in the pregnant patient. The mortality rate of untreated PE is high, ranging from 18% to 38%, and approximately one-third of patients with untreated thromboembolic disease develop recurrent embolism.24 Studies have reported a PE prevalence between 1.4% and 4.2% in pregnant women with suspected clinical diagnosis of PE.25
The clinical presentation of PE and associated laboratory testing results may be subtler in pregnant than in nonpregnant patients. Arterial blood gases (ABG) may show hypoxemia or hypocapnia. The ABG in pregnancy has a sensitivity of 76.9%, specificity of 20.2%, and negative and positive predictive values of 80% and 11.5% for PE, respectively.26 The alveolar-arterial oxygen gradient is a poor screening test for PE during pregnancy and postpartum. A retrospective chart review of 17 pregnant women with documented PE showed that 58% had normal alveolar-arterial gradients.27 Therefore, in a pregnant woman with a history suspicious for PE, objective imaging studies should be performed even if the patient has normal ABG.
The 2011 guidelines from the American Thoracic Society (ATS) and the Society of Thoracic Radiology (STR) recommend against using D-dimer to diagnose PE in pregnancy.28 In addition, lower extremity CUS should only be performed as the first diagnostic imaging procedure if the patient has signs or symptoms of DVT. Instead, the ATS/STR guidelines recommend a plain radiograph of the chest as the first imaging test. If the chest radiograph is normal, a ventilation/perfusion scan (V/Q) scan is preferred over CT pulmonary angiography (CTPA). Diagnostic accuracy of the V/Q scan may be superior to CTPA in pregnancy, and it is preferable because of the lower prevalence of indeterminate V/Q scan in pregnant women.29 Moreover, there is lower radiation exposure to the maternal breast and lung tissue with a V/Q scan than with CTPA. CTPA confers lower fetal radiation doses than V/Q scans (0.03–0.66 mGy versus 0.32–0.74 mGy, respectively) but higher total body maternal radiation (4–16 mSv versus 1–2.5 mSv).30 A quantitative approach to lung scan interpretation, based on the distribution histogram of V/Q ratios, may be helpful in categorizing patients with suspected PE.28 A study of 121 suspected episodes of PE in 120 pregnant women showed that 104 women with normal or nondiagnostic scans did not develop subsequent episodes of VTE during a mean follow-up period of 20 months.31
If the baseline chest radiograph is abnormal in a pregnant woman with clinical suspicion of PE, a CTPA should be performed. As noted, fetal radiation doses for CTPA examinations in which the fetus is not directly imaged are minimal. If CTPA is recommended for the diagnosis of PE, the patient should be informed that radiation to the breast may increase her baseline risk for breast cancer. The ATS guidelines state that “given the lack of evidence documenting clear superiority of any one diagnostic test, the values and preferences of a patient and her physician likely will and should determine the final choice and sequence of tests performed.”28
CASE I CONTINUED
Upon presentation to the emergency department, the circumference of the patient’s left leg is not significantly greater than that of her right leg, and her leg pain has resolved. Bilateral CUS is negative for proximal or distal DVT. Chest radiograph shows an opacification of her left lower lobe. CTPA shows bilateral segmental and subsegmental lower lobe pulmonary emboli.
How does risk for VTE change throughout pregnancy?
Women are at increased risk for VTE throughout the entire pregnancy, starting from conception, but mainly during the postpartum period. A Danish historical controlled cohort study of 819,751 pregnant women (ages 15–49 years) over a 10-year period identified 727 women with VTE. The absolute risk for VTE per 10,000 pregnancy-years increased from 4.1 (95% CI 3.2 to 5.2) during weeks 1 to 11 to 59.0 (95% CI 46.1 to 76.4) in week 40 and decreased in the postpartum period from 60 (95% CI 47.2 to 76.4) during the first week after birth to 2.1 during weeks 9–12 after birth (95% CI 1.1 to 4.2).32 This study showed that the risk of VTE increases throughout pregnancy and reaches its maximum during the peripartum period and is not significantly increased after 6 weeks post-delivery. In a retrospective cross-over cohort study of 1,687,930 women in California who delivered their first newborn, an elevated risk of VTE persisted until at least 12 weeks after delivery. However, the absolute increase in risk after 6 weeks postpartum was low.33
CASE 1 CONCLUSION
The patient is started on anticoagulation therapy and carefully monitored during the remainder of the pregnancy and postpartum period. Anticoagulation is discontinued 6 weeks after delivery.
TREATMENT
ANTICOAGULATION THERAPY
The treatment of VTE can be lifesaving. In a study comparing 35 patients with PE randomly assigned to treatment with anticoagulants versus no treatment, 5 of 19 patients in the untreated group died from PE and an additional 5 had nonfatal recurrences, as compared with none in the treated group.24 Unfractionated heparin (UFH) and low-molecular-weight heparin (LMWH) are both safe and effective anticoagulants during pregnancy as neither crosses the placenta. In a review of 186 reports of fetal and infant outcomes following anticoagulant therapy during pregnancy in 1325 pregnancies, outcomes in UFH-treated patients were similar to those in the normal population after excluding pregnancies with comorbid conditions independently associated with adverse outcomes.34 A 2005 systematic review of LMWH for prophylaxis and treatment of VTE during pregnancy included 64 studies of 277 pregnancies. There were no maternal deaths, live births resulted from 94.7% of the pregnancies, VTE or arterial thrombosis occurred in 0.86%, and significant bleeding occurred in 1.98%.35
The standard UFH regimen is an initial bolus of 5000 units subcutaneously and 17,500 units every 12 hours, with dose adjustment made based on a mid-interval activated partial thromboplastin time (aPTT).36 Although still controversial, it has been suggested that the anti-Xa assay with a mid-dosing interval target of 0.3 to 0.7 U/mL is a more reliable measure of therapeutic UFH activity than the aPTT, as the aPTT response is suppressed due to a pregnancy-related increase in factor VIII. LMWH is dosed based on weight; regimens are enoxaparin 1 mg/kg subcutaneously twice daily or 1.5 mg/kg subcutaneously once daily, and dalteparin 100 units/kg every 12 hours or 150 units/kg daily.
A 2017 Cochrane review of the effect of LMWH compared with UFH for the treatment of VTE in the nonpregnant setting included 23 studies with 9587 patients. Thrombotic complications (odds ratio [OR] 0.70 [CI 0.57 to 0.85]) and major hemorrhage (OR 0.58 [CI 0.40 to 0.83]) were lower in patients receiving LMWH, with a trend toward lower mortality.37 In addition, the incidence of bleeding complications in patients treated with subcutaneous LMWH versus intravenous heparin was compared in a 2012 systematic review of 27 randomized controlled trials with a total of 28,637 patients. In patients treated with LMWH, there was a nonstatistically significant lower incidence of major bleeding events (OR 0.79 [95% CI 0.60 to 1.04]) and a statistically significant reduction in bleeding risk (OR 0.68 [95% CI 0.47 to 1.00]) compared to patients treated with UFH.38 Additionally, a trial comparing the use of standard UFH versus LMWH found a significantly lower incidence of thrombocytopenia in patients treated with LMWH.39,40 Overall, LMWH is more effective at decreasing both thrombotic and bleeding complications, and the risk for osteoporosis is lower with LMWH. Based on these results, the American College of Chest Physicians (ACCP) recommends LMWH as the first-line treatment for VTE in pregnancy.41
In specific clinical situations, such as patients with renal dysfunction with creatinine clearance (CrCl) less than 30 mL/min, UFH is indicated. In a study of 103 pregnancies in 93 women given anti-coagulation during pregnancy, 89.3% received UFH. There were no maternal deaths, and fetal demise occurred in 8 pregnancies (7.8%) at a median of 14 weeks’ gestation. There were 2 episodes of PE (1.9%) and 2 major bleeding events requiring transfusion (1.9%).42 UFH costs much less than LMWH, and therefore UFH remains an important, inexpensive, and efficacious anticoagulant option for pregnant women who require anticoagulation and cannot afford LMWH.43
Due to the physiologic changes associated with pregnancy, LMWH and UFH dosages may need to be adjusted. An observational study of 20 pregnant women with acute VTE found no recurrent VTE or major bleeding after treatment with dalteparin. Dalteparin doses approximately 10% to 20% higher than those recommended in nonpregnant women were required to reach therapeutic target anti-Xa activity.44
Caution Regarding Oral Anticoagulants
Due to its teratogenicity, warfarin is not a first-line anticoagulation option. It is strictly contraindicated during the first trimester during organogenesis, and its use during pregnancy is restricted to women with mechanical heart valves. Warfarin crosses the placenta and has been associated with nasal hypoplasia, stippled epiphyses, and growth restriction, particularly between 6 to 9 weeks’ gestation. Every effort should be made to substitute UFH or LMWH for warfarin between 6 and 12 weeks of gestation. The bridging process should begin as early in the gestational age as possible due to the long half-life of warfarin.45 When used later in gestation, warfarin has been associated with fetal hemorrhage and central nervous system abnormalities. Other complications from use during the second and third trimesters include microcephaly, blindness, deafness, and fetal growth restriction.46,47 Its use also increases the risk for abortion and fetal death in utero.48–50
The direct oral anticoagulants (DOACs) are not approved for use in pregnancy. Although there are limited anecdotal reports of DOAC use in pregnancy,51 there is preclinical evidence of placental transfer with the DOACs rivaroxaban and apixaban (direct Xa inhibitors) and the oral thrombin inhibitor dabigatran, thus increasing the risk to the fetus.52–54 Edoxaban, another direct Xa inhibitor, should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. It should be discontinued in nursing mothers.55
THROMBOLYSIS
Fetal as well as maternal survival is dependent on adequate maternal perfusion and oxygenation. The risk of death from PE is significant, with a cross-sectional study of 58 patients with acute, massive PE showing a 55% mortality rate.56 Thus, pregnancy is not an absolute contraindication to mechanical or systemic (recombinant tissue plasminogen activator or streptokinase) thrombolysis in an unstable patient at high risk for death.57–59 There are no major studies of this approach, although a small review of 13 cases using systemic thrombolysis showed no increased risk of maternal mortality.58 Thrombolysis should be considered for appropriate indications in pregnant patients as it would be in nonpregnant patients. However, caution is required when drawing conclusions regarding maternal and fetal safety, given the lack of controlled clinical trials including pregnant women.
SURGICAL PULMONARY EMBOLECTOMY
Surgical pulmonary embolectomy is an important therapeutic and potentially life-saving option in women presenting with massive PE in the immediate postpartum period. Because of the risk of massive uterine bleeding immediately postpartum, thrombolytic therapy should not be used.60
INFERIOR VENA CAVA FILTER
Placement of an inferior vena cava (IVC) filter is indicated in patients who have an acute VTE with absolute contraindications for anticoagulation. In addition, it can be considered in patients with extensive ileofemoral venous thrombosis within 2 weeks prior to expected delivery.61 In a systematic review of 44 studies of IVC filters placed in pregnant patients, the IVC filter complication rate was 8.87% and the failure-to-retrieve rate was 11.25%.62 The complication rate is similar to that found in the nonpregnant population. Thus, IVC filters may be used when appropriately indicated and should be removed as soon as clinically feasible.
RECURRENT THROMBOSIS AND THROMBOPHILIAS
CASE PRESENTATION 2
A 34-year-old pregnant woman G1P0 at 38 weeks’ gestation presents with a painful, swollen left calf that is associated with difficulty on walking; the circumference of the left calf is 2 cm greater than that of the right. She has no shortness of breath or chest pain. She has a prior history of distal right lower extremity DVT while on combined oral contraceptives. Her mother also has a history of DVT while bedbound during a prolonged hospitalization at an older age. CUS is negative, and the patient is discharged home. However, 24 hours later she returns to the hospital with worsening swelling and pain in her left leg. Magnetic resonance venography demonstrates a large left external iliac and common iliac DVT. She is admitted and is started on UFH, and a retrievable IVC filter is placed in anticipation of delivery.
What is the risk for VTE recurrence during pregnancy?
A personal and family history of VTE should be obtained when evaluating pregnant patients. A retrospective study of 109 women with prior history of VTE showed recurrence rates per patient-year of 10.9% during pregnancy and 3.7% in the nonpregnant period; the relative risk of recurrent VTE during pregnancy was 3.5 (95% CI 1.6 to 7.8).63 Two large European retrospective cohort studies of VTE in pregnancy showed that the recurrence rate of VTE in women with a history of thrombosis is around 6% during pregnancy, equally distributed among trimesters. The highest incidence of recurrence was in the postpartum period, ranging from 8.3% to 10%.64 The recurrence risk during pregnancy in women with a history of a single episode of VTE was 2.4% antepartum (95% CI 0.2% to 6.9%).65 These risks may be lower in women without thrombophilia or with a temporary risk factor associated with their previous thromboembolic event.65 Recurrence risk is higher if the previous VTE was estrogen-related, either due to pregnancy or through hormonal contraception (10%), than if the previous VTE was non-estrogen-related (2.7%).64,66
The timing of the case patient’s presentation is consistent with reports of increased risk of VTE during the peripartum period. Her prior history of estrogen-related DVT is concerning for a risk of recurrence, particularly during pregnancy. A retrospective cohort study of 1104 women with previous VTE, 88 of whom became pregnant without receiving thromboprophylaxis, showed that the overall rate of VTE recurrence was 5.8% (95% CI 3.0% to 10.6%) and 8.3% (95% CI 4.5% to 14.6%) during pregnancy and postpartum, respectively. The risk of VTE recurrence was absent if the first VTE was related to a transient risk factor other than pregnancy, postpartum period, or hormonal contraception.67 However, the recurrence rate of VTE in women with prior unprovoked VTE and/or thrombophilia has been reported as 5.9% (95% CI 1.2% to 16.2%).65 The presence of an underlying hypercoagulable state can increase the recurrence risk by 25% to 50%, depending on the disorder.68 A retrospective cohort study of 270 pregnancies in 105 carriers of factor V Leiden, identified because of a symptomatic relative with the factor V Leiden mutation, found a VTE risk (mostly in the postpartum period) of 6.4% for heterozygous women, 16.7% for homozygous women, 20% for double heterozygous women, and 1.2% for noncarriers.69
Should the patient be screened for a thrombophilia disorder?
Half of all index thromboses in patients with thrombophilia occur in association with an additional risk factor. In women of child-bearing age, pregnancy, the postpartum period, and the use of combined hormonal contraception are all risk factors for VTE. A 2010 guideline from the British hematology community recommended testing for thrombophilia in women with prior VTE secondary to a minor provoking factor before or during pregnancy, but not testing women with unprovoked VTE (who would receive prophylaxis regardless) or those with VTE secondary to a major provoking factor (who would not require prophylaxis).70 Indications to screen for aPL antibodies include: women with (1) 3 unexplained recurrent first-trimester pregnancy losses or 1 second or third trimester fetal loss of morphologically normal fetuses; (2) severe preeclampsia; (3) intrauterine growth restriction; or (4) premature labor (< 34 weeks’ gestation).71,72
CASE 2 CONCLUSION
The patient is subsequently screened for inherited thrombophilia disorders and is found to be heterozygous for factor V Leiden.
CASE PRESENTATION 3
A 25-year-old woman is diagnosed with antiphospholipid syndrome (APS) during her second pregnancy when she experiences fetal loss during her second trimester. Pathologic examination of the placenta reveals infarcts. Laboratory evaluation reveals positive high-titer anticardiolipin and anti-beta-2 glycoprotein 1 antibodies (IgG isotype) and lupus anticoagulant on 2 separate occasions 12 weeks apart. In a subsequent pregnancy, she is started on prophylactic LMWH and daily low-dose aspirin (81 mg). At 36 weeks’ gestation, she presents with a blood pressure of 210/104 mm Hg and a platelet count of 94,000 cells/µL. She is diagnosed with preeclampsia and hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome and is induced for early delivery. About 2 weeks after vaginal delivery, she notices shortness of breath and chest pain. A CTPA demonstrates a right lower lobe lobar defect consistent with a PE. Her anticoagulation is increased to therapeutic dosage LMWH.
To what extent does thrombophilia increase the risk for VTE in pregnancy?
Approximately 50% of pregnancy-related VTEs are associated with inherited thrombophilia. A systematic review of 79 studies, in which 9 studies (n = 2526 patients) assessed the risk of VTE associated with inherited thrombophilia in pregnancy, revealed that the odds ratio for individuals with thrombophilia to develop VTE ranged from 0.74 to 34.40.73 Although women with thrombophilia have an increased relative risk of developing VTE in pregnancy, the absolute risk of VTE remains low (Table 1).41,73,74
How is APS managed in pregnant patients?
Women with history of recurrent early pregnancy loss (< 10 weeks’ gestation) related to the presence of aPL antibodies are managed with low-dose aspirin and prophylactic-dose UFH or LMWH. This treatment increases the rate of subsequent successful pregnancy outcomes and reduces the risk for thrombosis. A 2010 systematic review and meta-analysis of UFH plus low-dose aspirin compared with low-dose aspirin alone in patients with APS and recurrent pregnancy loss included 5 trials and 334 patients. Patients receiving dual therapy had higher rates of live births (74.3%; relative risk [RR] 1.30 [CI 1.04 to 1.63]) compared to the aspirin-only group (55.8%).75 A 2009 randomized controlled trial compared low-dose aspirin to low-dose aspirin plus LMWH in women with recurrent pregnancy loss and either aPL antibodies, antinuclear antibody, or inherited thrombophilia. The study was stopped early after 4 years and found no difference in rates of live births between the groups (77.8% versus 79.1%).76 However, a randomized case-control trial of women with aPL antibodies and recurrent miscarriage found a 72% live birth rate in 47 women randomly assigned to low-dose aspirin and LMWH.77 A 2012 guideline from the American College of Chest Physicians (ACCP) recommends that women with aPL antibodies with a history of 3 or more pregnancy losses receive low-dose aspirin plus prophylactic-dose LMWH or UFH.78 A 2014 systematic review and meta-analysis showed that the combination of low-dose aspirin and UFH resulted in a higher live-birth rate than aspirin alone in 803 women with APS (RR 1.54 [95% CI 1.25 to 1.89]).79 Further large randomized controlled trials are needed to confirm optimal management of recurrent miscarriage and aPL antibodies.
The addition of prednisone to aspirin, heparin, or both has shown no benefits in pregnant women with aPL antibodies. Indeed, prolonged use of steroids may cause serious pregnancy complications, such as prematurity and hypertension.80–83 Intravenous infusions of immunoglobulin (IVIG) have not been shown to be superior to heparin and aspirin. This finding was confirmed in a multicenter clinical trial that tested the effects of IVIG compared with LMWH plus low-dose aspirin for the treatment of women with aPL antibodies and recurrent miscarriage. The rate of live-birth was 72.5% in the group treated with heparin plus low-dose aspirin compared with 39.5% in the IVIG group.84
Preeclampsia and HELLP syndrome complicated the case patient’s pregnancy even though she was being treated with prophylactic-dose LMWH and low-dose aspirin, the current standard of care for pregnant women with APS (UFH can be used as well). It is important to note that complications may still occur despite standard treatment. Indeed, PE is more common in the postpartum than in the antepartum period. Prompt diagnosis is paramount to initiate the appropriate treatment; in this case the dose of LMWH was increased from prophylactic to therapeutic dose. However, additional therapeutic modalities are necessary to improve outcomes. A randomized controlled trial comparing standard of care with or without hydroxychloroquine is under way to address this issue.
PROPHYLAXIS
CASE PRESENTATION 4
A 34-year-old woman G1P0 at 6 weeks’ gestation with a past medical history of a proximal lower extremity DVT while on oral contraception is treated with warfarin anticoagulation for 6 months. Her obstetrician consults the hematologist to advise regarding antithrombotic management during this pregnancy.
What is the approach to prophylaxis in women at high risk for pregnancy-associated VTE?
All women at high risk for pregnancy-associated VTE should be counseled about the signs and symptoms of DVT or PE during preconception and pregnancy and have a plan developed should these symptoms arise. The ACCP guidelines on antithrombotic therapy outline recommendations ranging from clinical vigilance to prophylactic and intermediate-dose anticoagulation, depending on the risk for VTE recurrence, based on the personal and family history of VTE and type of thrombophilia (Table 2).78 These recommendations range from grade 2B to 2C.
For women with a history of estrogen-related VTE, single unprovoked VTE, or recurrent unprovoked VTE not on chronic anticoagulation, antepartum and postpartum pharmacologic thromboprophylaxis with either prophylactic or intermediate-dose LMWH is recommended (grade 2C). In patients with prior history of provoked VTE (non-estrogen related), antepartum clinical vigilance and postpartum pharmacologic thromboprophylaxis is recommended (grade 2C, 2B).
In asymptomatic pregnant women who are homozygote carriers for factor V Leiden or prothrombin G20210A variants and have a positive family history of thrombosis, antepartum and postpartum pharmacologic thromboprophylaxis is recommended (grade 2B). In asymptomatic homozygote carriers of factor V Leiden or prothrombin G20210A variants with no family history of thrombosis and women with all other thrombophilias with a positive family history of thrombosis, postpartum pharmacologic thromboprophylaxis is indicated (grade 2B and 2C, respectively). For women with confirmed APS and clinical criteria of obstetric APS with recurrent pregnancy loss, antepartum thromboprophylaxis with LMWH and low-dose aspirin is recommended (grade 1B). For pregnant women with all other thrombophilias with no personal or family history of thrombosis, clinical vigilance is suggested (grade 2 C).78
As an alternative to LMWH, vitamin K antagonists (VKA) such as warfarin can be used for postpartum thromboprophylaxis; in patients with protein C or S deficiency, due to the risk of warfarin-induced skin necrosis, a rapid-onset anticoagulant must be concomitantly administered. Warfarin and LMWH are safe anticoagulants during lactation, but there are no clinical data on the effects of the DOACs on infants during lactation. Data from animal studies indicate that DOACs are secreted into breast milk.85
What risks are associated with anticoagulant therapy in pregnancy?
VKAs cross the placenta and can cause teratogenicity, pregnancy loss, fetal bleeding, and neurodevelopmental deficits. Therefore, discontinuation of VKAs prior to the sixth week of gestation is necessary to avoid warfarin embryopathy. DOACs have been shown to readily cross the placenta but with unknown human reproductive risks. Fondaparinux, a synthetic pentasaccharide, crosses the placenta in small quantities. Though there are reports of the successful use of fondaparinux in pregnancy, there is limited reported experience of its use in the first trimester.86
The risk for bleeding with anticoagulation is notably acceptable. In a case-control study of 88 pregnant women receiving therapeutic-dose anticoagulation, the risk of postpartum hemorrhage (PPH) after vaginal delivery was 30% in those who received LMWH anticoagulation versus 18% in those who did not (OR 1.9 [95% CI 1.1 to 3.5]).87 However, the risk for severe PPH (≥ 500 mL) was similar (5.6% versus 5.0%; OR 1.1 [95% CI 0.4 to 3.6]). The risk for PPH after cesarean section was 12% in LMWH users versus 4% in LMWH non-users (OR 2.9 [95% CI 0.5 to 19.4]). The risk for PPH associated with delivery within 24 hours after the last dose of LMWH was 1.2 times higher (95% CI 0.4 to 3.6) compared to a longer interval. Therefore, therapeutic LMWH increases the risk for blood loss after vaginal delivery, but not the risk for severe PPH. The risk for PPH is influenced by the interval between the last dose of LMWH and delivery. Of note in this study, per the institution’s protocol, the anticoagulation was stopped with signs of labor or determination of need for delivery. The risk for blood loss may be mitigated in more planned delivery scenarios.87
CASE 4 CONTINUED
The patient is placed on prophylactic-dose LMWH with good tolerance and delivers at 39 weeks' gestation via caesarian section due to nonprogression of labor. Postpartum she is restarted on prophylactic-dose anticoagulation with LMWH. Two weeks after discharge from the hospital, she presents with right calf pain and mild shortness of breath. On physical exam, her leg circumferences are equal. A D-dimer assay is 3375 ng/mL (normal 0–229). CUS of the right leg shows a complete occlusive DVT of the mid-distal superficial femoral and popliteal veins and partially occlusive acute DVT of the right posterior tibial and peroneal veins. CTPA reveals a right lower lobe PE. Because she had developed VTE despite prophylactic LMWH, her anticoagulation is changed to therapeutic dose. She is treated with anticoagulation with LMWH for a total of 3 months, after which a repeat CUS shows no residual thrombosis.
What is the recommended dosing of heparin and LMWH during pregnancy?
A prospective study of 14 pregnant women receiving UFH prophylaxis found that a prophylactic dose of 5000 units twice a day was inadequate to achieve prophylactic heparin levels in any patient in the second or third trimester.88 Similar to treatment dosage, there is no consensus evidence for prophylactic dosing, and dosage recommendations are based on expert opinion. In a retrospective study of 25 pregnant women on intermediate-dose UFH, the mean UFH dose required to achieve a target anti-factor Xa level of 0.1 to 0.3 units/mL was 236.9 units/kg/day.89 However, the use of anti-factor Xa levels for monitoring is controversial as there is no data to support a difference in outcomes with its use in prophylactic or therapeutic dosing.
The timing of the previous VTE history is important when deciding on the anticoagulant dose in pregnancy. In pregnant women with a VTE that occurred within the previous 4 to 6 weeks, full-dose anticoagulation with LMWH should be considered; an intermediate dose (three-fourths of a therapeutic dose) may be used if the thrombotic episode occurred more than 6 weeks earlier but still within a year. Prophylactic dosing may be sufficient if the episode occurred more than a year earlier.90 A clinical trial (High-Low) is under way to explore the optimal dose of LMWH in pregnant women with prior history of VTE who are not on chronic anticoagulation therapy.91
How is anticoagulation therapy managed in the peripartum period?
Neuraxial anesthesia during active labor while on anticoagulation increases the risk for central nervous system bleeding. Therefore, if spontaneous labor occurs in women on therapeutic dose anticoagulation, neuraxial anesthesia cannot be used. However, in the event of elective induction of labor or caesarean section, neuroaxial anesthesia may be performed 12 hours after the administration of the last prophylactic dose of LMWH or 24 hours after the last therapeutic dose of LMWH. Intravenous UFH should be stopped for 6 hours before induction of labor with a confirmed normal aPTT before placement of neuraxial anesthesia. There is no contraindication for using neuraxial anesthesia during subcutaneous standard UFH at total doses of 10,000 units daily. The risk of spinal hematoma with larger daily subcutaneous doses is unclear; therefore, a documented normal aPTT must be obtained before placement of neuroaxial anesthesia.
Postpartum, reinitiation of prophylactic-dose LMWH should be delayed for at least 12 hours after the removal of an epidural catheter. Therapeutic-dose LMWH should be administered no earlier than 24 hours after neuraxial anesthesia, providing that proper hemostasis is achieved. In the absence of persistent bleeding, if no regional anesthesia was used, LMWH may be resumed 12 hours after delivery.92 Anticoagulation with either LMWH or warfarin is recommended for at least 6 to 12 weeks postpartum.33
COUNSELING
Patients should be advised to manage controllable risk factors, including avoiding prolonged immobilization, avoiding excessive weight gain in pregnancy, and stopping smoking. Periods of immobilization tend to cause reduced blood flow (stasis), which predisposes to thrombosis. In a systematic review of records of all patients with confirmed PE after arrival at Charles de Gaulle airport in Paris during a 13-year period, women had a higher risk of PE after a long-distance flight than men, with an estimated incidence of 0.61 per million passengers versus 0.20, respectively; the incidence reached 7.24 and 2.35 cases, respectively, in passengers traveling more than 10,000 kilometers.93,94
The risk of air travel-related thrombosis in pregnant women is estimated to be between 0.03% and 0.1%. Physicians must decide on an individual basis how to prevent travel-related thrombosis in their pregnant patients. In most passengers, prevention can be limited to encouraging exercise, avoidance of long sleeping periods, and not using a window seat. Women at high risk for VTE, such as women with a prior history of VTE who are not on anticoagulation or women with known asymptomatic thrombophilia or other risk factors for thrombosis such as obesity, may benefit from a short period (1–3 days) of LMWH starting 2 hours before a long-distance flight.95
Activation of the coagulation system has been demonstrated in cigarette smokers.96 Heavy smoking was found to be a significant risk factor for VTE in a cross-sectional analysis of 2404 men and women.97 An increased risk for thrombosis during pregnancy is seen in cigarette smokers15,98 and is enhanced with the concomitant use of illicit drugs.99 Other obstetric complications associated with smoking and illicit drug use during pregnancy include preterm labor, spontaneous abortion, perinatal death, low birth weight, and abruption placenta. The efficacy of nicotine replacement therapy in pregnancy is uncertain.100 Recommendations are to advise patients to stop smoking, obtain psychosocial counseling, and utilize adjunctive therapies, which have been shown to have some effect on abstinence rates.101
CONCLUSION
Women are at increased risk for VTE during pregnancy and the postpartum period. Awareness of risk factors and the signs and symptoms of VTE is paramount. Prompt diagnosis and treatment is mandatory to decrease complications of VTE. LMWH is the mainstay treatment of VTE in pregnancy, as it does not cross the placenta. Both LMWH and warfarin are safe during lactation. Close communication among the patient, obstetrician, hematologist, anesthesiologist, and neonatologist is crucial to optimize the care of these patients.
- Knight M, Nour M, Tuffnell D, et al, eds. on behalf of MBRRACE-UK. Saving lives, improving mothers’ care—surveillance of maternal deaths in the UK 2012-14 and lessons learned to inform maternity care from the UK and Ireland confidential enquiries into maternal deaths and morbidity 2009-14. Oxford: National Perinatal Epidemiology Unit, University of Oxford; 2016: 69–75.
- Berg CJ, Callaghan WM, Syverson C, Henderson Z. Pregnancy-related mortality in the United States, 1998 to 2005. Obstet Gynecol 2010;116:1302–9.
- Salonen Ros H, Lichtenstein P, Bellocco R, et al. Increased risks of circulatory diseases in late pregnancy and puerperium. Epidemiology 2001;12:456–60.
- Heit JA, Kobbervig CE, James AH, et al. Trends in the incidence of venous thromboembolism during pregnancy or postpartum: a 30-year population-based study. Ann Intern Med 2005;143:697–706.
- Greer IA. Thrombosis in pregnancy: updates in diagnosis and management. Hematology Am Soc Hematol Educ Program 2012;2012:203–7.
- Hellgren M. Hemostasis during normal pregnancy and puerperium. Semin Thromb Hemost 2003;29:125–30.
- Brenner B. Haemostatic changes in pregnancy. Thromb Res 2004;114:409–14.
- Chan W-S, Spencer FA, Ginsberg JS. Anatomic distribution of deep vein thrombosis in pregnancy. CMAJ 2010;182:657–60.
- Greer IA. Prevention and management of venous thromboembolism in pregnancy. Clin Chest Med 2003;24:123–37.
- Wik HS, Jacobsen AF, Sandvik L, Sandset PM. Prevalence and predictors for post-thrombotic syndrome 3 to 16 years after pregnancy-related venous thrombosis: a population-based, cross-sectional, case-control study. J Thromb Haemost 2012;10:840–7.
- Pomp ER, Lenselink AM, Rosendaal FR, Doggen CJM. Pregnancy, the postpartum period and prothrombotic defects: risk of venous thrombosis in the MEGA study. J Thromb Haemost 2008;6:632–7.
- Marik PE, Plante LA. Venous thromboembolic disease and pregnancy. N Engl J Med 2008;359:2025–33.
- Jacobsen AF, Skjeldestad FE, Sandset PM. Ante- and postnatal risk factors of venous thrombosis: a hospital-based case-control study. J Thromb Haemost 2008;6:905–12.
- Virkus RA, Løkkegaard E, Lidegaard Ø, et al. Risk factors for venous thromboembolism in 1.3 million pregnancies: a nationwide prospective cohort. PloS One 2014;9:e96495.
- James AH, Jamison MG, Brancazio LR, Myers ER. Venous thromboembolism during pregnancy and the postpartum period: incidence, risk factors, and mortality. Am J Obstet Gynecol 2006;194:1311–5.
- Chan W-S, Lee A, Spencer FA, et al. Predicting deep venous thrombosis in pregnancy: out in “LEFt” field? Ann Intern Med 2009;151:85–92.
- Righini M, Jobic C, Boehlen F, et al. Predicting deep venous thrombosis in pregnancy: external validation of the LEFT clinical prediction rule. Haematologica 2013;98:545–8.
- Chan W-S, Chunilal S, Lee A, et al. A red blood cell agglutination D-dimer test to exclude deep venous thrombosis in pregnancy. Ann Intern Med 2007;147:165–70.
- Nijkeuter M, Ginsberg JS, Huisman MV. Diagnosis of deep vein thrombosis and pulmonary embolism in pregnancy: a systematic review. J Thromb Haemost 2006;4:496–500.
- Chan W-S, Spencer FA, Lee AY, et al. Safety of withholding anticoagulation in pregnant women with suspected deep vein thrombosis following negative serial compression ultrasound and iliac vein imaging. CMAJ 2013;185:E194–200.
- Dronkers CE, Srámek A, Huisman MV, Klok FA. Accurate diagnosis of iliac vein thrombosis in pregnancy with magnetic resonance direct thrombus imaging (MRDTI). BMJ Case Rep 2016;2016. pii: bcr2016218091.
- Ray JG, Vermeulen MJ, Bharatha A, et al. Association between MRI exposure during pregnancy and fetal and childhood outcomes. JAMA 2016;316:952–61.
- Royal College of Obstretricians and Gynaecologists. Thromboembolic disease in pregnancy and the puerperium: acute management. Green-top Guideline No. 37b. London: RCOG; 2015.
- Barritt DW, Jordan SC. Anticoagulant drugs in the treatment of pulmonary embolism. A controlled trial. Lancet 1960;1(7138):1309–12.
- Bourjeily G, Khalil H, Raker C, et al. Outcomes of negative multidetector computed tomography with pulmonary angiography in pregnant women suspected of pulmonary embolism. Lung 2012;190:105–11.
- Deutsch AB, Twitty P, Downes K, Parsons MT. Assessment of the alveolar-arterial oxygen gradient as a screening test for pulmonary embolism in pregnancy. Am J Obstet Gynecol 2010;203:373.e1–4.
- Powrie RO, Larson L, Rosene-Montella K, et al. Alveolar-arterial oxygen gradient in acute pulmonary embolism in pregnancy. Am J Obstet Gynecol 1998;178:394–6.
- Leung AN, Bull TM, Jaeschke R, et al. American Thoracic Society documents: an official American Thoracic Society/Society of Thoracic Radiology clinical practice guideline--evaluation of suspected pulmonary embolism in pregnancy. Radiology 2012;262:635–46.
- Cahill AG, Stout MJ, Macones GA, Bhalla S. Diagnosing pulmonary embolism in pregnancy using computed-tomographic angiography or ventilation-perfusion. Obstet Gynecol 2009;114:124–9.
- Bourjeily G, Paidas M, Khalil H, et al. Pulmonary embolism in pregnancy. Lancet 2010;375(9713):500–12.
- Chan WS, Ray JG, Murray S, et al. Suspected pulmonary embolism in pregnancy: clinical presentation, results of lung scanning, and subsequent maternal and pediatric outcomes. Arch Intern Med 2002;162:1170–5.
- Virkus RA, Løkkegaard ECL, Bergholt T, et al. Venous thromboembolism in pregnant and puerperal women in Denmark 1995-2005. A national cohort study. Thromb Haemost 2011;106:304–9.
- Kamel H, Navi BB, Sriram N, et al. Risk of a thrombotic event after the 6-week postpartum period. N Engl J Med 2014;370:1307–15.
- Ginsberg JS, Hirsh J, Turner DC, et al. Risks to the fetus of anticoagulant therapy during pregnancy. Thromb Haemost 1989;61:197–203.
- Greer IA, Nelson-Piercy C. Low-molecular-weight heparins for thromboprophylaxis and treatment of venous thromboembolism in pregnancy: a systematic review of safety and efficacy. Blood 2005;106:401–7.
- Prandoni P, Carnovali M, Marchiori A, Galilei Investigators. Subcutaneous adjusted-dose unfractionated heparin vs fixed-dose low-molecular-weight heparin in the initial treatment of venous thromboembolism. Arch Intern Med 2004;164:1077–83.
- Robertson L, Jones LE. Fixed dose subcutaneous low molecular weight heparins versus adjusted dose unfractionated heparin for venous thromboembolism. Cochrane Database Syst Rev 2017;(9):eb9;2:CD001100.
- Costantino G, Ceriani E, Rusconi AM, et al. Bleeding risk during treatment of acute thrombotic events with subcutaneous LMWH compared to intravenous unfractionated heparin; a systematic review. PloS One 2012;7:e44553.
- Warkentin TE, Levine MN, Hirsh J, et al. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med 1995;332:1330–5.
- Junqueira DRG, Perini E, Penholati RRM, Carvalho MG. Unfractionated heparin versus low molecular weight heparin for avoiding heparin-induced thrombocytopenia in postoperative patients. Cochrane Database Syst Rev 2012;(9):CD007557.
- Bates SM, Greer IA, Middeldorp S, et al. VTE, thrombophilia, antithrombotic therapy, and pregnancy. Chest 2012;141:e691S–e736S.
- Clark NP, Delate T, Witt DM, et al. A descriptive evaluation of unfractionated heparin use during pregnancy. J Thromb Thrombolysis 2009;27:267–73.
- Clark NP, Delate T, Cleary SJ, Witt DM. Analysis of unfractionated heparin dose requirements to target therapeutic anti-Xa intensity during pregnancy. Thromb Res 2010;125:402–5.
- Jacobsen AF, Qvigstad E, Sandset PM. Low molecular weight heparin (dalteparin) for the treatment of venous thromboembolism in pregnancy. BJOG 2003;110:139–44.
- Walfisch A, Koren G. The “warfarin window” in pregnancy: the importance of half-life. J Obstet Gynaecol Can 2010;32:988–9.
- Bates SM, Ginsberg JS. Anticoagulants in pregnancy: fetal effects. Baillières Clin Obstet Gynaecol 1997;11:479–88.
- Stevenson RE, Burton OM, Ferlauto GJ, Taylor HA. Hazards of oral anticoagulants during pregnancy. JAMA 1980;243:1549–51.
- Ginsberg JS, Hirsh J. Anticoagulants during pregnancy. Annu Rev Med 1989;40:79–86.
- Wong V, Cheng CH, Chan KC. Fetal and neonatal outcome of exposure to anticoagulants during pregnancy. Am J Med Genet 1993;45:17–21.
- Blickstein D, Blickstein I. The risk of fetal loss associated with Warfarin anticoagulation. Int J Gynaecol Obstet 2002;78:221–5.
- Burnett AE, Mahan CE, Vazquez SR, et al. Guidance for the practical management of the direct oral anticoagulants (DOACs) in VTE treatment. J Thromb Thrombolysis 2016;41:206–32.
- Bapat P, Pinto LSR, Lubetsky A, et al. Examining the transplacental passage of apixaban using the dually perfused human placenta. J Thromb Haemost 2016;14:1436–41.
- Bapat P, Pinto LSR, Lubetsky A, et al. Rivaroxaban transfer across the dually perfused isolated human placental cotyledon. Am J Obstet Gynecol 2015;213:710.e1–6.
- Bapat P, Kedar R, Lubetsky A, et al. Transfer of dabigatran and dabigatran etexilate mesylate across the dually perfused human placenta. Obstet Gynecol 2014;123:1256–61.
- Savaysa [package insert]. Parsippany (NJ): Daiichi Sankyo, Inc; 2015.
- Filipecki S, Tomkowski W, Hajduk B, et al. [Outcome of patients with clinically acute massive pulmonary embolism]. Pneumonol Alergol Pol 1994;62:132–7.
- Holden EL, Ranu H, Sheth A, et al. Thrombolysis for massive pulmonary embolism in pregnancy--a report of three cases and follow up over a two year period. Thromb Res 2011;127:58–9.
- te Raa GD, Ribbert LS, Snijder RJ, Biesma DH. Treatment options in massive pulmonary embolism during pregnancy; a case-report and review of literature. Thromb Res 2009;124:1–5.
- Leonhardt G, Gaul C, Nietsch HH, et al. Thrombolytic therapy in pregnancy. J Thromb Thrombolysis 2006;21:271–6.
- Colombier S, Niclauss L. Successful surgical pulmonary embolectomy for massive perinatal embolism after emergency cesarean section. Ann Vasc Surg 2015;29:1452.e1–4.
- British Committee for Standards in Haematology Writing Group, Baglin TP, Brush J, Streiff M. Guidelines on use of vena cava filters. Br J Haematol 2006;134:590–5.
- Harris SA, Velineni R, Davies AH. Inferior vena cava filters in pregnancy: a systematic review. J Vasc Interv Radiol 2016;27:354–360.
- Pabinger I, Grafenhofer H, Kyrle PA, et al. Temporary increase in the risk for recurrence during pregnancy in women with a history of venous thromboembolism. Blood 2002;100:1060–2.
- Pabinger I, Grafenhofer H, Kaider A, et al. Risk of pregnancy-associated recurrent venous thromboembolism in women with a history of venous thrombosis. J Thromb Haemost 2005;3:949–54.
- Brill-Edwards P, Ginsberg JS, Gent M, et al. Safety of withholding heparin in pregnant women with a history of venous thromboembolism. Recurrence of Clot in This Pregnancy Study Group. N Engl J Med 2000;343:1439–44.
- De Stefano V, Martinelli I, Rossi E, et al. The risk of recurrent venous thromboembolism in pregnancy and puerperium without antithrombotic prophylaxis. Br J Haematol 2006;135:386–91.
- De Stefano V, Martinelli I, Rossi E, et al. The risk of recurrent venous thromboembolism in pregnancy and puerperium without antithrombotic prophylaxis. Br J Haematol 2006;135:386–91.
- Lim W, Eikelboom JW, Ginsberg JS. Inherited thrombophilia and pregnancy associated venous thromboembolism. BMJ 2007;334:1318–21.
- Tormene D, Simioni P, Prandoni P, et al. Factor V Leiden mutation and the risk of venous thromboembolism in pregnant women. Haematologica 2001;86:1305–9.
- Baglin T, Gray E, Greaves M, et al. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol 2010;149:209–20.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006;4:295–306.
- Pengo V, Tripodi A, Reber G, et al. Update of the guidelines for lupus anticoagulant detection. J Thromb Haemost 2009;7:1737–40.
- Robertson L, Wu O, Langhorne P, et al. Thrombophilia in pregnancy: a systematic review. Br J Haematol 2006;132:171–96.
- American College of Obstetricians and Gynecologists Women’s Health Care Physicians. ACOG Practice Bulletin No. 138: Inherited thrombophilias in pregnancy. Obstet Gynecol 2013;122:706–17.
- Mak A, Cheung MW, Cheak AA, Ho RC. Combination of heparin and aspirin is superior to aspirin alone in enhancing live births in patients with recurrent pregnancy loss and positive anti-phospholipid antibodies: a meta-analysis of randomized controlled trials and meta-regression. Rheumatology (Oxf) 2010;49:281–8.
- Laskin CA, Spitzer KA, Clark CA, et al. Low molecular weight heparin and aspirin for recurrent pregnancy loss: results from the randomized, controlled HepASA Trial. J Rheumatol 2009;36:279–87.
- Farquharson RG, Quenby S, Greaves M. Antiphospholipid syndrome in pregnancy: a randomized, controlled trial of treatment. Obstet Gynecol 2002;100:408–13.
- Bates SM, Jaeschke R, Stevens SM, et al. Diagnosis of DVT: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012;141(2 Suppl):e351S–418S.
- Lubbe WF, Butler WS, Palmer SJ, Liggins GC. Fetal survival after prednisone suppression of maternal lupus-anticoagulant. Lancet 1983;1(8338):1361–3.
- Lockshin MD, Druzin ML, Qamar T. Prednisone does not prevent recurrent fetal death in women with antiphospholipid antibody. Am J Obstet Gynecol 1989;160:439–43.
- Silver RK, MacGregor SN, Sholl JS, et al. Comparative trial of prednisone plus aspirin versus aspirin alone in the treatment of anticardiolipin antibody-positive obstetric patients. Am J Obstet Gynecol 1993;169:1411–7.
- Cowchock FS, Reece EA, Balaban D, et al. Repeated fetal losses associated with antiphospholipid antibodies: a collaborative randomized trial comparing prednisone with low-dose heparin treatment. Am J Obstet Gynecol 1992;166:1318–23.
- Laskin CA, Bombardier C, Hannah ME, et al. Prednisone and aspirin in women with autoantibodies and unexplained recurrent fetal loss. N Engl J Med 1997;337:148–53.
- Dendrinos S, Sakkas E, Makrakis E. Low-molecular-weight heparin versus intravenous immunoglobulin for recurrent abortion associated with antiphospholipid antibody syndrome. Int J Gynaecol Obstet 2009;104:223–5.
- Cohen H, Arachchillage DRJ, Beyer-Westendorf J, et al. Direct oral anticoagulants and women. Semin Thromb Hemost 2016;42:789–97.
- Bates SM, Middeldorp S, Rodger M, et al. Guidance for the treatment and prevention of obstetric-associated venous thromboembolism. J Thromb Thrombolysis 2016;41:92–128.
- Knol HM, Schultinge L, Veeger NJ, et al. The risk of postpartum hemorrhage in women using high dose of low-molecular-weight heparins during pregnancy. Thromb Res 2012;130:334–8.
- Barbour LA, Smith JM, Marlar RA. Heparin levels to guide thromboembolism prophylaxis during pregnancy. Am J Obstet Gynecol 1995;173:1869–73.
- Bergqvist A, Bergqvist D, Lindhagen A, Mätzsch T. Late symptoms after pregnancy-related deep vein thrombosis. Br J Obstet Gynaecol 1990;97:338–41.
- Rodger M. Evidence base for the management of venous thromboembolism in pregnancy. Hematology Am Soc Hematol Educ Program. 2010;2010:173–80.
- Bleker SM, Buchmüller A, Chauleur C, et al. Low-molecular-weight heparin to prevent recurrent venous thromboembolism in pregnancy: Rationale and design of the Highlow study, a randomised trial of two doses. Thromb Res 2016;144:62–8.
- Horlocker TT, Wedel DJ, Rowlingson JC, et al. Regional anesthesia in the patient receiving antithrombotic or thrombolytic therapy: American Society of Regional Anesthesia and Pain Medicine evidence-based guidelines (third edition). Reg Anesth Pain Med 2010;35:64–101.
- Lapostolle F, Surget V, Borron SW, et al. Severe pulmonary embolism associated with air travel. N Engl J Med 2001;345:779–83.
- Lapostolle F, Le Toumelin P, Chassery C, et al. Gender as a risk factor for pulmonary embolism after air travel. Thromb Haemost 2009;102:1165–8.
- Cannegieter SC, Rosendaal FR. Pregnancy and travel-related thromboembolism. Thromb Res 2013;131 Suppl 1:S55–58.
- Miller GJ, Bauer KA, Cooper JA, Rosenberg RD. Activation of the coagulant pathway in cigarette smokers. Thromb Haemost 1998;79:549–53.
- Golomb BA, Chan VT, Denenberg JO, et al. Risk marker associations with venous thrombotic events: a cross-sectional analysis. BMJ Open 2014;4:e003208.
- Lindqvist P, Dahlbäck B, Marŝál K. Thrombotic risk during pregnancy: a population study. Obstet Gynecol 1999;94:595–9.
- Black M, Bhattacharya S, Fairley T, et al. Outcomes of pregnancy in women using illegal drugs and in women who smoke cigarettes. Acta Obstet Gynecol Scand 2013;92:47–52.
- Mendelsohn C, Gould GS, Oncken C. Management of smoking in pregnant women. Aust Fam Physician 2014;43:46–51.
- Chamberlain C, O’Mara-Eves A, Oliver S, et al. Psychosocial interventions for supporting women to stop smoking in pregnancy. Cochrane Database Syst Rev 2013;10:CD001055.
- Knight M, Nour M, Tuffnell D, et al, eds. on behalf of MBRRACE-UK. Saving lives, improving mothers’ care—surveillance of maternal deaths in the UK 2012-14 and lessons learned to inform maternity care from the UK and Ireland confidential enquiries into maternal deaths and morbidity 2009-14. Oxford: National Perinatal Epidemiology Unit, University of Oxford; 2016: 69–75.
- Berg CJ, Callaghan WM, Syverson C, Henderson Z. Pregnancy-related mortality in the United States, 1998 to 2005. Obstet Gynecol 2010;116:1302–9.
- Salonen Ros H, Lichtenstein P, Bellocco R, et al. Increased risks of circulatory diseases in late pregnancy and puerperium. Epidemiology 2001;12:456–60.
- Heit JA, Kobbervig CE, James AH, et al. Trends in the incidence of venous thromboembolism during pregnancy or postpartum: a 30-year population-based study. Ann Intern Med 2005;143:697–706.
- Greer IA. Thrombosis in pregnancy: updates in diagnosis and management. Hematology Am Soc Hematol Educ Program 2012;2012:203–7.
- Hellgren M. Hemostasis during normal pregnancy and puerperium. Semin Thromb Hemost 2003;29:125–30.
- Brenner B. Haemostatic changes in pregnancy. Thromb Res 2004;114:409–14.
- Chan W-S, Spencer FA, Ginsberg JS. Anatomic distribution of deep vein thrombosis in pregnancy. CMAJ 2010;182:657–60.
- Greer IA. Prevention and management of venous thromboembolism in pregnancy. Clin Chest Med 2003;24:123–37.
- Wik HS, Jacobsen AF, Sandvik L, Sandset PM. Prevalence and predictors for post-thrombotic syndrome 3 to 16 years after pregnancy-related venous thrombosis: a population-based, cross-sectional, case-control study. J Thromb Haemost 2012;10:840–7.
- Pomp ER, Lenselink AM, Rosendaal FR, Doggen CJM. Pregnancy, the postpartum period and prothrombotic defects: risk of venous thrombosis in the MEGA study. J Thromb Haemost 2008;6:632–7.
- Marik PE, Plante LA. Venous thromboembolic disease and pregnancy. N Engl J Med 2008;359:2025–33.
- Jacobsen AF, Skjeldestad FE, Sandset PM. Ante- and postnatal risk factors of venous thrombosis: a hospital-based case-control study. J Thromb Haemost 2008;6:905–12.
- Virkus RA, Løkkegaard E, Lidegaard Ø, et al. Risk factors for venous thromboembolism in 1.3 million pregnancies: a nationwide prospective cohort. PloS One 2014;9:e96495.
- James AH, Jamison MG, Brancazio LR, Myers ER. Venous thromboembolism during pregnancy and the postpartum period: incidence, risk factors, and mortality. Am J Obstet Gynecol 2006;194:1311–5.
- Chan W-S, Lee A, Spencer FA, et al. Predicting deep venous thrombosis in pregnancy: out in “LEFt” field? Ann Intern Med 2009;151:85–92.
- Righini M, Jobic C, Boehlen F, et al. Predicting deep venous thrombosis in pregnancy: external validation of the LEFT clinical prediction rule. Haematologica 2013;98:545–8.
- Chan W-S, Chunilal S, Lee A, et al. A red blood cell agglutination D-dimer test to exclude deep venous thrombosis in pregnancy. Ann Intern Med 2007;147:165–70.
- Nijkeuter M, Ginsberg JS, Huisman MV. Diagnosis of deep vein thrombosis and pulmonary embolism in pregnancy: a systematic review. J Thromb Haemost 2006;4:496–500.
- Chan W-S, Spencer FA, Lee AY, et al. Safety of withholding anticoagulation in pregnant women with suspected deep vein thrombosis following negative serial compression ultrasound and iliac vein imaging. CMAJ 2013;185:E194–200.
- Dronkers CE, Srámek A, Huisman MV, Klok FA. Accurate diagnosis of iliac vein thrombosis in pregnancy with magnetic resonance direct thrombus imaging (MRDTI). BMJ Case Rep 2016;2016. pii: bcr2016218091.
- Ray JG, Vermeulen MJ, Bharatha A, et al. Association between MRI exposure during pregnancy and fetal and childhood outcomes. JAMA 2016;316:952–61.
- Royal College of Obstretricians and Gynaecologists. Thromboembolic disease in pregnancy and the puerperium: acute management. Green-top Guideline No. 37b. London: RCOG; 2015.
- Barritt DW, Jordan SC. Anticoagulant drugs in the treatment of pulmonary embolism. A controlled trial. Lancet 1960;1(7138):1309–12.
- Bourjeily G, Khalil H, Raker C, et al. Outcomes of negative multidetector computed tomography with pulmonary angiography in pregnant women suspected of pulmonary embolism. Lung 2012;190:105–11.
- Deutsch AB, Twitty P, Downes K, Parsons MT. Assessment of the alveolar-arterial oxygen gradient as a screening test for pulmonary embolism in pregnancy. Am J Obstet Gynecol 2010;203:373.e1–4.
- Powrie RO, Larson L, Rosene-Montella K, et al. Alveolar-arterial oxygen gradient in acute pulmonary embolism in pregnancy. Am J Obstet Gynecol 1998;178:394–6.
- Leung AN, Bull TM, Jaeschke R, et al. American Thoracic Society documents: an official American Thoracic Society/Society of Thoracic Radiology clinical practice guideline--evaluation of suspected pulmonary embolism in pregnancy. Radiology 2012;262:635–46.
- Cahill AG, Stout MJ, Macones GA, Bhalla S. Diagnosing pulmonary embolism in pregnancy using computed-tomographic angiography or ventilation-perfusion. Obstet Gynecol 2009;114:124–9.
- Bourjeily G, Paidas M, Khalil H, et al. Pulmonary embolism in pregnancy. Lancet 2010;375(9713):500–12.
- Chan WS, Ray JG, Murray S, et al. Suspected pulmonary embolism in pregnancy: clinical presentation, results of lung scanning, and subsequent maternal and pediatric outcomes. Arch Intern Med 2002;162:1170–5.
- Virkus RA, Løkkegaard ECL, Bergholt T, et al. Venous thromboembolism in pregnant and puerperal women in Denmark 1995-2005. A national cohort study. Thromb Haemost 2011;106:304–9.
- Kamel H, Navi BB, Sriram N, et al. Risk of a thrombotic event after the 6-week postpartum period. N Engl J Med 2014;370:1307–15.
- Ginsberg JS, Hirsh J, Turner DC, et al. Risks to the fetus of anticoagulant therapy during pregnancy. Thromb Haemost 1989;61:197–203.
- Greer IA, Nelson-Piercy C. Low-molecular-weight heparins for thromboprophylaxis and treatment of venous thromboembolism in pregnancy: a systematic review of safety and efficacy. Blood 2005;106:401–7.
- Prandoni P, Carnovali M, Marchiori A, Galilei Investigators. Subcutaneous adjusted-dose unfractionated heparin vs fixed-dose low-molecular-weight heparin in the initial treatment of venous thromboembolism. Arch Intern Med 2004;164:1077–83.
- Robertson L, Jones LE. Fixed dose subcutaneous low molecular weight heparins versus adjusted dose unfractionated heparin for venous thromboembolism. Cochrane Database Syst Rev 2017;(9):eb9;2:CD001100.
- Costantino G, Ceriani E, Rusconi AM, et al. Bleeding risk during treatment of acute thrombotic events with subcutaneous LMWH compared to intravenous unfractionated heparin; a systematic review. PloS One 2012;7:e44553.
- Warkentin TE, Levine MN, Hirsh J, et al. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med 1995;332:1330–5.
- Junqueira DRG, Perini E, Penholati RRM, Carvalho MG. Unfractionated heparin versus low molecular weight heparin for avoiding heparin-induced thrombocytopenia in postoperative patients. Cochrane Database Syst Rev 2012;(9):CD007557.
- Bates SM, Greer IA, Middeldorp S, et al. VTE, thrombophilia, antithrombotic therapy, and pregnancy. Chest 2012;141:e691S–e736S.
- Clark NP, Delate T, Witt DM, et al. A descriptive evaluation of unfractionated heparin use during pregnancy. J Thromb Thrombolysis 2009;27:267–73.
- Clark NP, Delate T, Cleary SJ, Witt DM. Analysis of unfractionated heparin dose requirements to target therapeutic anti-Xa intensity during pregnancy. Thromb Res 2010;125:402–5.
- Jacobsen AF, Qvigstad E, Sandset PM. Low molecular weight heparin (dalteparin) for the treatment of venous thromboembolism in pregnancy. BJOG 2003;110:139–44.
- Walfisch A, Koren G. The “warfarin window” in pregnancy: the importance of half-life. J Obstet Gynaecol Can 2010;32:988–9.
- Bates SM, Ginsberg JS. Anticoagulants in pregnancy: fetal effects. Baillières Clin Obstet Gynaecol 1997;11:479–88.
- Stevenson RE, Burton OM, Ferlauto GJ, Taylor HA. Hazards of oral anticoagulants during pregnancy. JAMA 1980;243:1549–51.
- Ginsberg JS, Hirsh J. Anticoagulants during pregnancy. Annu Rev Med 1989;40:79–86.
- Wong V, Cheng CH, Chan KC. Fetal and neonatal outcome of exposure to anticoagulants during pregnancy. Am J Med Genet 1993;45:17–21.
- Blickstein D, Blickstein I. The risk of fetal loss associated with Warfarin anticoagulation. Int J Gynaecol Obstet 2002;78:221–5.
- Burnett AE, Mahan CE, Vazquez SR, et al. Guidance for the practical management of the direct oral anticoagulants (DOACs) in VTE treatment. J Thromb Thrombolysis 2016;41:206–32.
- Bapat P, Pinto LSR, Lubetsky A, et al. Examining the transplacental passage of apixaban using the dually perfused human placenta. J Thromb Haemost 2016;14:1436–41.
- Bapat P, Pinto LSR, Lubetsky A, et al. Rivaroxaban transfer across the dually perfused isolated human placental cotyledon. Am J Obstet Gynecol 2015;213:710.e1–6.
- Bapat P, Kedar R, Lubetsky A, et al. Transfer of dabigatran and dabigatran etexilate mesylate across the dually perfused human placenta. Obstet Gynecol 2014;123:1256–61.
- Savaysa [package insert]. Parsippany (NJ): Daiichi Sankyo, Inc; 2015.
- Filipecki S, Tomkowski W, Hajduk B, et al. [Outcome of patients with clinically acute massive pulmonary embolism]. Pneumonol Alergol Pol 1994;62:132–7.
- Holden EL, Ranu H, Sheth A, et al. Thrombolysis for massive pulmonary embolism in pregnancy--a report of three cases and follow up over a two year period. Thromb Res 2011;127:58–9.
- te Raa GD, Ribbert LS, Snijder RJ, Biesma DH. Treatment options in massive pulmonary embolism during pregnancy; a case-report and review of literature. Thromb Res 2009;124:1–5.
- Leonhardt G, Gaul C, Nietsch HH, et al. Thrombolytic therapy in pregnancy. J Thromb Thrombolysis 2006;21:271–6.
- Colombier S, Niclauss L. Successful surgical pulmonary embolectomy for massive perinatal embolism after emergency cesarean section. Ann Vasc Surg 2015;29:1452.e1–4.
- British Committee for Standards in Haematology Writing Group, Baglin TP, Brush J, Streiff M. Guidelines on use of vena cava filters. Br J Haematol 2006;134:590–5.
- Harris SA, Velineni R, Davies AH. Inferior vena cava filters in pregnancy: a systematic review. J Vasc Interv Radiol 2016;27:354–360.
- Pabinger I, Grafenhofer H, Kyrle PA, et al. Temporary increase in the risk for recurrence during pregnancy in women with a history of venous thromboembolism. Blood 2002;100:1060–2.
- Pabinger I, Grafenhofer H, Kaider A, et al. Risk of pregnancy-associated recurrent venous thromboembolism in women with a history of venous thrombosis. J Thromb Haemost 2005;3:949–54.
- Brill-Edwards P, Ginsberg JS, Gent M, et al. Safety of withholding heparin in pregnant women with a history of venous thromboembolism. Recurrence of Clot in This Pregnancy Study Group. N Engl J Med 2000;343:1439–44.
- De Stefano V, Martinelli I, Rossi E, et al. The risk of recurrent venous thromboembolism in pregnancy and puerperium without antithrombotic prophylaxis. Br J Haematol 2006;135:386–91.
- De Stefano V, Martinelli I, Rossi E, et al. The risk of recurrent venous thromboembolism in pregnancy and puerperium without antithrombotic prophylaxis. Br J Haematol 2006;135:386–91.
- Lim W, Eikelboom JW, Ginsberg JS. Inherited thrombophilia and pregnancy associated venous thromboembolism. BMJ 2007;334:1318–21.
- Tormene D, Simioni P, Prandoni P, et al. Factor V Leiden mutation and the risk of venous thromboembolism in pregnant women. Haematologica 2001;86:1305–9.
- Baglin T, Gray E, Greaves M, et al. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol 2010;149:209–20.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006;4:295–306.
- Pengo V, Tripodi A, Reber G, et al. Update of the guidelines for lupus anticoagulant detection. J Thromb Haemost 2009;7:1737–40.
- Robertson L, Wu O, Langhorne P, et al. Thrombophilia in pregnancy: a systematic review. Br J Haematol 2006;132:171–96.
- American College of Obstetricians and Gynecologists Women’s Health Care Physicians. ACOG Practice Bulletin No. 138: Inherited thrombophilias in pregnancy. Obstet Gynecol 2013;122:706–17.
- Mak A, Cheung MW, Cheak AA, Ho RC. Combination of heparin and aspirin is superior to aspirin alone in enhancing live births in patients with recurrent pregnancy loss and positive anti-phospholipid antibodies: a meta-analysis of randomized controlled trials and meta-regression. Rheumatology (Oxf) 2010;49:281–8.
- Laskin CA, Spitzer KA, Clark CA, et al. Low molecular weight heparin and aspirin for recurrent pregnancy loss: results from the randomized, controlled HepASA Trial. J Rheumatol 2009;36:279–87.
- Farquharson RG, Quenby S, Greaves M. Antiphospholipid syndrome in pregnancy: a randomized, controlled trial of treatment. Obstet Gynecol 2002;100:408–13.
- Bates SM, Jaeschke R, Stevens SM, et al. Diagnosis of DVT: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012;141(2 Suppl):e351S–418S.
- Lubbe WF, Butler WS, Palmer SJ, Liggins GC. Fetal survival after prednisone suppression of maternal lupus-anticoagulant. Lancet 1983;1(8338):1361–3.
- Lockshin MD, Druzin ML, Qamar T. Prednisone does not prevent recurrent fetal death in women with antiphospholipid antibody. Am J Obstet Gynecol 1989;160:439–43.
- Silver RK, MacGregor SN, Sholl JS, et al. Comparative trial of prednisone plus aspirin versus aspirin alone in the treatment of anticardiolipin antibody-positive obstetric patients. Am J Obstet Gynecol 1993;169:1411–7.
- Cowchock FS, Reece EA, Balaban D, et al. Repeated fetal losses associated with antiphospholipid antibodies: a collaborative randomized trial comparing prednisone with low-dose heparin treatment. Am J Obstet Gynecol 1992;166:1318–23.
- Laskin CA, Bombardier C, Hannah ME, et al. Prednisone and aspirin in women with autoantibodies and unexplained recurrent fetal loss. N Engl J Med 1997;337:148–53.
- Dendrinos S, Sakkas E, Makrakis E. Low-molecular-weight heparin versus intravenous immunoglobulin for recurrent abortion associated with antiphospholipid antibody syndrome. Int J Gynaecol Obstet 2009;104:223–5.
- Cohen H, Arachchillage DRJ, Beyer-Westendorf J, et al. Direct oral anticoagulants and women. Semin Thromb Hemost 2016;42:789–97.
- Bates SM, Middeldorp S, Rodger M, et al. Guidance for the treatment and prevention of obstetric-associated venous thromboembolism. J Thromb Thrombolysis 2016;41:92–128.
- Knol HM, Schultinge L, Veeger NJ, et al. The risk of postpartum hemorrhage in women using high dose of low-molecular-weight heparins during pregnancy. Thromb Res 2012;130:334–8.
- Barbour LA, Smith JM, Marlar RA. Heparin levels to guide thromboembolism prophylaxis during pregnancy. Am J Obstet Gynecol 1995;173:1869–73.
- Bergqvist A, Bergqvist D, Lindhagen A, Mätzsch T. Late symptoms after pregnancy-related deep vein thrombosis. Br J Obstet Gynaecol 1990;97:338–41.
- Rodger M. Evidence base for the management of venous thromboembolism in pregnancy. Hematology Am Soc Hematol Educ Program. 2010;2010:173–80.
- Bleker SM, Buchmüller A, Chauleur C, et al. Low-molecular-weight heparin to prevent recurrent venous thromboembolism in pregnancy: Rationale and design of the Highlow study, a randomised trial of two doses. Thromb Res 2016;144:62–8.
- Horlocker TT, Wedel DJ, Rowlingson JC, et al. Regional anesthesia in the patient receiving antithrombotic or thrombolytic therapy: American Society of Regional Anesthesia and Pain Medicine evidence-based guidelines (third edition). Reg Anesth Pain Med 2010;35:64–101.
- Lapostolle F, Surget V, Borron SW, et al. Severe pulmonary embolism associated with air travel. N Engl J Med 2001;345:779–83.
- Lapostolle F, Le Toumelin P, Chassery C, et al. Gender as a risk factor for pulmonary embolism after air travel. Thromb Haemost 2009;102:1165–8.
- Cannegieter SC, Rosendaal FR. Pregnancy and travel-related thromboembolism. Thromb Res 2013;131 Suppl 1:S55–58.
- Miller GJ, Bauer KA, Cooper JA, Rosenberg RD. Activation of the coagulant pathway in cigarette smokers. Thromb Haemost 1998;79:549–53.
- Golomb BA, Chan VT, Denenberg JO, et al. Risk marker associations with venous thrombotic events: a cross-sectional analysis. BMJ Open 2014;4:e003208.
- Lindqvist P, Dahlbäck B, Marŝál K. Thrombotic risk during pregnancy: a population study. Obstet Gynecol 1999;94:595–9.
- Black M, Bhattacharya S, Fairley T, et al. Outcomes of pregnancy in women using illegal drugs and in women who smoke cigarettes. Acta Obstet Gynecol Scand 2013;92:47–52.
- Mendelsohn C, Gould GS, Oncken C. Management of smoking in pregnant women. Aust Fam Physician 2014;43:46–51.
- Chamberlain C, O’Mara-Eves A, Oliver S, et al. Psychosocial interventions for supporting women to stop smoking in pregnancy. Cochrane Database Syst Rev 2013;10:CD001055.
Locally Advanced Pancreatic Cancer
INTRODUCTION
Pancreatic cancer is one of the most rapidly rising causes of mortality in the United States. In 2016, the number of deaths from pancreatic cancer exceeded those from breast cancer, making it the third leading cause of cancer-related death in the United States.1 It is projected that by 2020 pancreatic cancer will overtake colorectal malignancies to become the second most common cause of cancer death in this country.1,2 The term pancreatic cancer encompasses both exocrine and endocrine tumors. However, since 80% of pancreatic cancers are classified as pancreatic ductal adenocarcinoma (PDA), when speaking about pancreatic cancer most clinicians and scientists are referring to PDA.
Even with advances in chemotherapy and radiotherapy over the past decade, the only curative option for PDA is surgical resection. Unfortunately, only 20% of patients are appropriate surgical candidates at the time of diagnosis.3 Considering the lack of screening options and the ambiguity of symptomatology, roughly 4 out 5 patients with PDA are diagnosed as having locally advanced or metastatic disease that is initially not amenable to surgery.
Locally advanced pancreatic adenocarcinoma presents unique challenges in management and treatment. Treatment options include multi-agent chemotherapy, chemoradiation, or radiotherapy. Some patients can be successfully down-staged with these therapies and be deemed surgical candidates. Other challenges include selecting the appropriate sequence of therapies and stratifying therapies based on comorbidities. In this article, we review the epidemiology, biology, and diagnostic approach to PDA and focus on current treatment strategies for locally advanced pancreatic cancer (LAPC).
EPIDEMIOLOGY
In 2012, GLOBOCAN estimated that PDA caused 331,000 deaths per year, accounting for 4% of all worldwide mortality.4,5 Despite high incidence rates internationally, PDA is a disease of Western and industrialized nations. In the Unites States, PDA is a malignancy of middle to late adulthood, with a sharp upsurge in incidence after age 50 years.6 More than one third of new cases are diagnosed in patients older than 70 years, and more than half of patients diagnosed are older than 60 years of age.2 The incidence of pancreatic cancer is fairly equal among men and women, with a slightly higher rate for the male sex. It has an incidence preference for African-Americans by 4.8 cases per 100,000 persons nationally.7
Risk factors for the development of exocrine pancreatic cancer include hereditary disposition, underlying medical conditions, and environmental factors. One of the most significant environmental risk factors for the development of PDA is smoking,8 which is associated with up to 25% of all cases.9 Smoking cessation leads to a rapid reduction in risk for pancreatic cancer, with the risk among former smokers approaching that for never smokers less than 10 years after quitting.9 Other environmental factors that contribute to the development of pancreatic cancer include increased body mass index, a high-salt and high-saturated fat diet, heavy alcohol intake, and increased utilization of nonsteroidal inflammatory drugs.10–13
There is a strong association between new-onset diabetes and increased risk for developing PDA.14,15 Data also suggest that diabetes may be a risk factor and/or a consequence of tissue destruction that arises during the development or progression of PDA.16,17 Interestingly, ABO blood grouping is another underlying medical disposition that confers an altered risk profile. Studies have shown that patients with blood group O were less likely than those with type A, B, or AB to develop pancreatic cancer.18
Genetic predisposition syndromes can elevate an individual patient’s risk for developing PDA. Genetic syndromes and gene alterations that increase the risk for PDA include BRCA1/2, Peutz-Jeghers syndrome, and Lynch syndrome risk.19–21 Up to 10% to 15% of PDA cases may be due to an inherited familial cancer.22 Having a first-degree relative with PDA increases the odds of developing PDA 1.76-fold compared to those without a family history.23 The exact biologic and molecular mechanisms of familial pancreatic cancer are unclear. It is estimated that about 10% of patients with familial pancreatic cancer (FPC) carry BRCA2 mutations.24 Individuals at risk for FPC should undergo genetic screening for the presence of the most frequently inherited pancreatic cancer susceptibility genetic defects: BRCA2, PALB2, and ATM germline mutations.25 Carriers of BRCA2, who are also at increased risk for developing breast, ovarian, and prostate cancer, should be monitored closely. Of all hereditary conditions, hereditary pancreatitis confers the highest risk for developing PDA, with an approximate risk elevation of 40% to 50%.26,27 Although several genetic predisposition syndromes have been identified, most cases of pancreatic adenocarcinoma are thought to be sporadic.
CANCER BIOLOGY AND PATHOLOGY
The pathologic predecessor of PDA is pancreatic intraepithelial neoplasia (PIN). With further dysplastic changes that result from increasing genetic alterations, these precancerous lesions progress from low- to high-grade and finally to adenocarcinoma. More than 90% of all PINs across all grades have oncogenic KRAS mutations.28 Additionally, inactivating mutations in the tumor suppressor genes SMAD4, p53, and CDKN2A are found with increasing frequency in higher grade PINs. The frequency and presence of mutations in both oncogenes and tumor suppressor genes in precursor neoplasias mirror the genetic mutations noted in advanced PDA.29 Among all mutations, KRAS is the most common and most functionally important for pancreatic cancer cell survival. KRAS mutations not only have profound effects on downstream mediators of tumor growth and metastasis, but they are implicated in reprograming of cellular metabolism.30,31
Pancreatic adenocarcinoma has a unique microenvironment that makes it a difficult target for current therapeutic modalities. First, it is one of the most stroma-rich malignancies. The dense stroma surrounding pancreatic tumor cells leads to increased tumor pressures and alterations in tumor vascular perfusion.32 It also serves as a barrier that prevents chemotherapeutic drugs from reaching the tumor cells. Thus, clinical trials are under way to investigate agents such has hyaluronidase, which may degrade components of the extracellular matrix that supports thestromal environment. Additionally, there is data to suggest that the microenvironment of PDA downregulates immune monitoring, leading to further tumor growth.27,33 The molecular, cellular, and immunologic complexity of PDA may contribute to its resistance to traditional therapeutics.
EVALUATION AND DIAGNOSIS
CASE PRESENTATION
A 61-year-old man with a history of type 2 diabetes mellitus and chronic tobacco use presents to the emergency department (ED) with a 4-month history of progressively worsening abdominal discomfort and fatigue. He has also noticed darkening of his urine and slight yellow discoloration of his eyes. His weight measured 5 months ago in his primary care physician’s office was 91 kg (200 lb, BMI 29.5) and in the ED is 75 kg (165 lb, BMI 24.4). He has noticed bulky, malodorous, oily stools for about 2 months. Preliminary laboratory studies reveal elevated levels of total bilirubin (2.7 mg/dL) and alkaline phosphatase (204 IU/L). Transabdominal ultrasound (US) is obtained and reveals a 3-cm pancreatic mass with biliary tract dilation.
Does this patient have pancreatic cancer?
CLINICAL SIGNS AND SYMPTOMS
Establishing the diagnosis of pancreatic cancer in a patient who presents with a high index of suspicion is critical. Patients with pancreatic cancer usually present after a period of nonspecific and vague symptoms, which typically are experienced as abdominal discomfort, weight loss, and weakness. It is estimated that approximately 25% of patients may complain of vague abdominal pain up to 6 months prior to diagnosis. Up to 15% of patients may seek medical attention more than 6 months prior to establishing a diagnosis of PDA.34 The most common symptoms associated with pancreatic cancer in order of decreasing reported frequency are weight loss, anorexia, abdominal/epigastric pain, dark-colored urine, jaundice, nausea, back pain, and diarrhea with associated steatorrhea.35 Upwards of 15% of patients present with painless jaundice, a term that is often associated with pancreatic cancer.36 On exam these patients may have scleral icterus, sublingual jaundice, epigastric pain on palpation, weight loss, hepatomegaly, lymphadenopathy and a nontender, distended, palpable gallbladder (also known as Courvoisier sign).34 Abdominal signs and symptoms arise from tumor growth into surrounding vessels, tissues, and ducts within the abdominal cavity. Compression of the common bile duct accounts for the development of jaundice. Tumor growth around the stomach and duodenum can lead to delayed gastric emptying and subsequently nausea and vomiting. Constriction of the pancreatic duct leads to pancreatic insufficiency, precipitation of weight loss, and steatorrhea. Pancreatic insufficiency can worsen abdominal pain, and lead to increased weight loss and flatulence.
Less common symptoms include pain, erythema, and edema involving the lower extremities, which may be reflective of migratory thrombophlebitis (commonly known as Trousseau syndrome). Thromboembolic disease, including pulmonary embolism, portal vein, and deep vein thromboses are frequently encountered complications of pancreatic cancer. The incidence of thromboembolic events in patients with PDA has been reported to be as high as 54%.37 Of all signs encountered, weight loss is the most common and most profound. Patients with advanced PDA have severe degrees of cachexia. Some patients present with as much as a 5 kg/m2 decrease in their BMI from pre-illness baseline BMI, and lose another 3 to 4 kg/m2 through disease progression.38 At the time of diagnosis, many patients have already undergone significant weight loss, which can have substantial implications on treatment planning and clinical outcomes.
What other studies can be done to assist in making the diagnosis?
LABORATORY ABNORMALITIES AND TUMOR MARKERS
Elevations in alkaline phosphatase, γ-glutamyltransferase (GGT), serum aspartate aminotransferase (AST), serum alanine aminotransferase (ALT), and direct fractions of bilirubin are common in patients with PDA. Patients will usually have an obstructive pattern on their liver panel, with predominant elevations in direct bilirubin, alkaline phosphatase, and GGT, as compared with AST and ALT. Other baseline laboratory studies, including a complete blood count and basic metabolic panel, should be obtained because patients commonly have thrombocytosis, anemia, and electrolyte abnormalities due to the tumor itself and pancreatic insufficiency (Table 1).

Measurement of glycated hemoglobin (HBA1C) is an emerging and important diagnostic test in the diagnosis of pancreatic cancer. Recently, data has emerged to suggest that new-onset diabetes is present in about 50% of patients diagnosed with pancreatic cancer.39 The temporal relationship of pancreatic cancer and diabetes is supported by evidence showing that patients who undergo resection commonly have resolution of their diabetes.17 This study suggested that hyperglycemia, elevated HBA1C, and symptoms of diabetes in patients older than 50 years may identify patients who have early pancreatic cancer. The entity of pancreatic cancer–associated diabetes needs to be better defined and the algorithmic approach to evaluation and diagnosis, utilizing signs, symptoms, and laboratory values associated with diabetes, needs to be clearly established.
The only serum marker for PDA is carbohydrate antigen 19-9 (CA 19-9), also known as sialylated Lewis antigen or cancer-associated antigen. It was first identified in pancreatic cancer patients in 1981.40,41 The sensitivity and specificity of CA 19-9 ranges from 70% to approximately 90%.42,43 Hereditary predispositions and comorbid disease cross-reactivity contribute to the diminished sensitivity and specificity of CA 19-9. In about 5% to 10% of the population, CA 19-9 is not expressed (Lewis antigen A and B negative). Additionally, since CA 19-9 is expressed in the cells that line the biliary tree, diseases that lead to pancreatic or liver inflammation may falsely elevate CA 19-9.44 As a result, CA 19-9 is not an ideal screening test. However, data has shown that CA 19-9 may have prognostic value postoperatively and serve as a marker for therapeutic response.45,46
Is biopsy needed for this patient and if so, what is the most appropriate technique?
ENDOSCOPIC ULTRASOUND
Generally, diagnosis with tissue is not necessary for patients who clearly have resectable disease and will proceed directly to surgery for management. Nevertheless, it is still commonly obtained in this group of patients. However, in patients with LAPC or with features suggestive of LAPC, such as tumor approximation to critical vessels such as the superior mesenteric artery (SMA) or celiac axis, biopsy is necessary. These patients will receive neoadjuvant therapy, and biopsy is important in establishing a diagnosis. The ideal way to obtain a biopsy is through fine-needle aspiration (FNA) or biopsy (FNB) utilizing endoscopic ultrasound (EUS). Percutaneous and computed tomography (CT)–guided FNB can also be used to obtain a biopsy for diagnosis. In comparison to percutaneous and CT-guided FNB, EUS-FNA/FNB has low rates of complications, a decreased rate of peritoneal seeding, and is cost effective.47,48
CASE CONTINUED
Abdominal CT obtained following abdominal ultrasound reveals a 3.5-cm mass in the head of the pancreas in close approximation to the SMA and celiac axis.
Does the patient have borderline resectable or unresectable disease?
IMAGING
Abdominal ultrasound is a reasonable, inexpensive, and safe alternative to abdominal CT as it does not utilize ionizing radiation. It is particularly useful in patients who present with jaundice or have concern for biliary obstruction based on laboratory evaluation. It is particularly sensitive for detecting tumors greater than 3 cm in size.49,50 In patients whose abdominal ultrasound is unrevealing and whose index of suspicion remains high for PDA, abdominal CT should be the next imaging modality.
Abdominal CT obtained utilizing a pancreatic protocol is ideal for detection and staging of pancreatic tumors. By implementing a triple-phase protocol with arterial, late arterial, and venous phases, tumors, which have a density different from that of the pancreatic parenchyma, are accentuated. Abdominal CT is also able to provide critical information about tumor resectability.51 By revealing the degree of tumor encasement around the aorta, level of destruction of the superior mesenteric vein, or degree of involvement of the SMA or celiac vessels, abdominal CT determines if a patient should be deemed resectable, borderline resectable, or unresectable (Table 2).52,53 Resectability is based on thorough imaging evaluation, expert opinion of a multidisciplinary team, and guidelines proposed by American Hepatopancreaticobiliary Association, Society of Surgical Oncology, Society for Surgery of the Alimentary Tract, and the NCCN.54
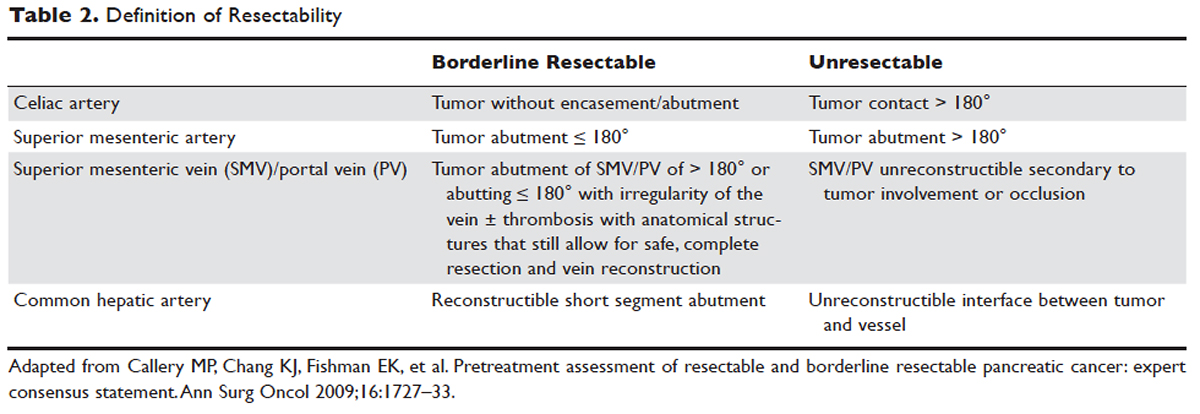
Other imaging modalities have a less clearly established role in the diagnostic approach to PDA. In patients who have contraindications to obtaining CT, magnetic resonance imaging can be utilized as a secondary imaging modality.55 The role of positron emission tomography 18F-fluorodeoxyglucose (PET-FDG) is not clearly defined among clinicians, nor reflected in consensus guidelines by the National Comprehensive Cancer Network (NCCN). In clinical practice, it is still often combined with CT to detect metastatic disease, particularly in high-risk patients such has those with LAPC. The role of PET-CT in staging and its impact on clinical outcomes has not been fully established.
Endoscopic retrograde cholangiopancreatography (ERCP) and magnetic resonance cholangiopancreatography (MRCP) can also assist in the diagnosis and management of PDA. In patients with obstructive jaundice, both MRCP and ERCP visualize obstructions and dilations within the biliary tree, with the latter having the ability to intervene. ERCP allows for the collection of tissue to aid in diagnosis, and has the ability to relieve biliary obstruction via stenting.56
TREATMENT
CASE CONTINUED
After an abdominal CT is obtained, the patient is referred to an outpatient oncologist because of concern for pancreatic adenocarcinoma. After consultation, the patient is advised to obtain EUS with biopsy and to return immediately afterwards for further treatment planning. The pathology report following EUS confirms that the mass is a poorly differentiated PDA. The patient’s case is discussed at a multidisciplinary meeting with radiation, surgical, and medical oncology. The abdominal CT and PET-CT scan are thoroughly reviewed. After imaging review, the multidisciplinary team concludes that the tumor is in contact with the SMA at 120° and with the common hepatic artery without extension in the celiac axis and without evidence of metastasis.
What is the appropriate management of borderline resectable pancreatic cancer?
BORDERLINE RESECTABLE CANCER
Patients who have nonmetastatic disease and are deemed resectable and without contraindications to surgery or high-risk features, as defined by NCCN guidelines, should proceed directly to surgery. A large body of evidence suggests that complete surgical resection with negative margins is a significant predictor of survival and currently provides the only option for cure.57–59 Despite the curative intent of surgery, the rate of recurrence remains high in patients who undergo surgical resection. Even in patients with negative resection margins (R0 resection), the 5-year survival is 20% to 30%, with a median survival ranging from 12 to 25 months, suggesting the presence of regional and distant occult disease at the time of diagnosis.60–62
Additionally, in half the patients who undergo surgical resection with resultant positive microscopic (R1 resection) or gross (R2 resection) margins, the median survival is no greater than 12 months. In this subset of patients, clinical outcomes are similar to outcomes in patients with locally advanced and metastatic pancreatic cancer, suggesting that upfront surgery and adjuvant therapy may not be the ideal therapeutic option. This raises 2 important points: first, resectability should be assessed carefully in all patients with LAPC, and second, for those patients who are deemed borderline resectable, neoadjuvant therapy should be considered.63 Borderline resectability is defined as tumor abutment ≤ 180° of the celiac artery, and tumor abutment of the superior mesenteric vein /portal vein of > 180° or abutting ≤ 180° with irregularity of the vein with or without thrombosis with anatomical structures that still allows for safe and complete resection and vein reconstruction (Table 2).
Neoadjuvant Therapy
The goal of neoadjuvant therapy is to minimize the negative impact of upfront surgery in patients who have a high likelihood of having microscopic or grossly positive margins. Research has suggested that neoadjuvant therapy may improve resectability, decrease the rate of recurrence, and improve overall survival.64–66
There is no clear consensus on the ideal management of patients with borderline resectable disease. However, expert guidelines are in agreement that upfront surgery in patients with LAPC is not appropriate, as most patients will not be able to achieve an R0 resection.67 As staging and management of patients with LAPC is difficult, expertise of a multidisciplinary team can be helpful.68
Several studies and the NCCN guidelines support the use of neoadjuvant therapy in patients deemed borderline resectable.69,70 Treatment of borderline resectable disease is similar to unresectable LAPC and generally involves 2 chemotherapy treatment backbones: FOLFIRINOX (folinic acid [leucovorin], fluorouracil [5-FU], irinotecan, and oxaliplatin) or gemcitabine-based therapy.
Phase 1 to 2/3 clinical trials conducted by Conroy et al from 2005 to 2011, including the landmark ACCORD-11 trial, established the safety and role of FOLFIRINOX in metastatic pancreatic cancer and also demonstrated an improved overall survival with the use of this therapy in these patients.71,72 These findings led to interest in FOLFIRINOX as a neoadjuvant therapy for patients with LAPC. Since then, multiple prospective and retrospective studies have shown that 54% to 100% of patients with borderline resectable LAPC who were treated with FOLFIRINOX were down-staged significantly enough to undergo resection. Of those patients, more than 90% had a R0 resection following surgery (Table 3).73–79
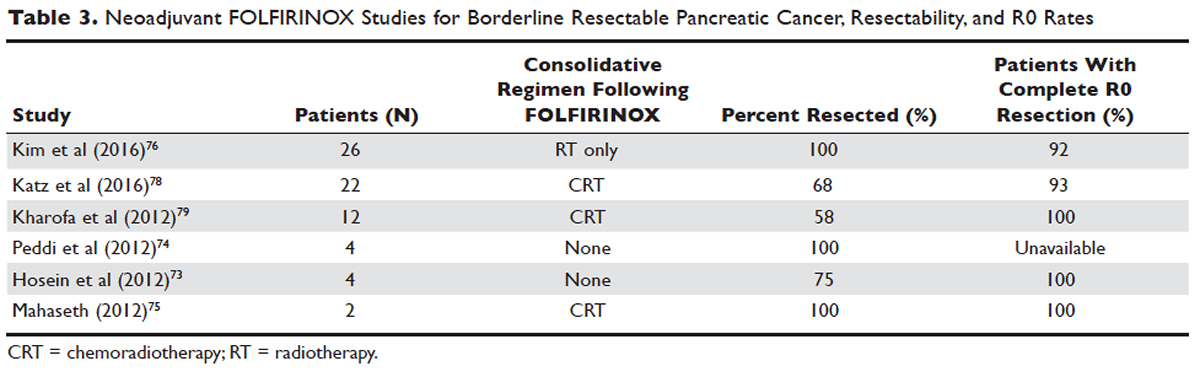
Data over the past 7 years suggests that neoadjuvant FOLFIRINOX improves overall survival and resectability in patients with borderline disease. However, treatment with FOLFIRINOX is not without limitations. FOLFIRINOX is associated with higher rates of febrile neutropenia, thrombocytopenia, diarrhea, and sensory neuropathy as compared with gemcitabine-based therapy.72 Other less commonly observed toxicities associated with FOLFIRINOX include mucositis, hand-foot syndrome, pulmonary toxicity, and alopecia. Dose-attenuated FOLFIRINOX-based regimens, including those that exclude the bolus fluorouracil dose and augment upfront filgrastim, have demonstrated improved safety and comparable efficacy as compared to standard FOLFIRINOX.80
Gemcitabine has been the fundamental treatment backbone for PDA since the results of the phase 3 CONKO-001 trial were published.81 Gemcitabine is a pyrimidine antimetabolite and potent inhibitor of DNA polymerase and ribonucleotide reductase.82 In recent years, multiple combination therapies with gemcitabine have been investigated, including regimens with nab-paclitaxel, oxaliplatin, or docetaxel. Resection rates and negative margin outcomes have been shown to be comparable to patients who received FOLFIRINOX in the neoadjuvant setting with borderline locally advanced disease.83–85 In addition to having a more tolerable side effect profile in comparison to fluorouracil-based regimens, gemcitabine is considered to be a potent radiosensitizer.86 For this reason, studies have also investigated the role of radiotherapy in conjunction with gemcitabine, revealing negative margin resection rates above 80% in patients with borderline resectable disease.87,88
Because very few studies directly comparing FOLFIRINOX with gemcitabine-based combination regimens have been completed, there is no clear consensus on the preferred treatment regimen, in both borderline and unresectable LAPC. Decisions to treat are influenced predominantly by comorbidities, adverse effect profiles, and performance status of patients, as FOLFIRINOX is the more toxic of the 2 treatment backbones. Therefore, FOLFIRINOX has mostly been utilized in patients with relatively good functional status (Eastern Cooperative Oncology Group [ECOG] performance status 0 to 1).89 In elderly patients and those with poor functional status, ECOG 2 to 4, gemcitabine as a single agent is a reasonable alternative in the neoadjuvant setting of borderline resectable disease.
The exact role of radiation therapy in addition to induction chemotherapy in borderline resectable pancreatic cancer has not been clearly established because of the lack of prospective studies in this area. Multiple large retrospective series have identified high rates of conversion to margin-negative resection with neoadjuvant chemoradiation alone.90 Based on available data, it is reasonable for patients with borderline resectable disease to proceed with any of the following treatment options: chemotherapy, chemoradiation, or induction chemotherapy followed by chemoradiation (Figure). Chemotherapy and chemoradiation are generally more appropriate with patients with high CA 19-9 levels or those at an elevated risk of having positive margins or occult metastatic disease.91 Obtaining negative margin resections is the predominant goal of neoadjuvant radiotherapy.89 Many studies have identified margin status to be one of the most significant prognostic factors in PDA.57,59,92,93 Additionally, several studies have highlighted that radiotherapy in the neoadjuvant setting could improve negative margin resection rates, local control, and clinical outcomes in patients with borderline resectable locally advanced disease.94–97 A common multimodal regimen utilized in the neoadjuvant setting combines capecitabine, an oral prodrug that is converted to fluorouracil, with radiation therapy. This combination has also been shown to improve resectability rates and long-term clinical outcomes in patients with borderline resectable disease.98 Additionally, neoadjuvant radiation therapy can potentially downstage patients with unresectable disease at the time of diagnosis to become surgical candidates.99 Despite the paucity of data, interval scans utilizing CT following neoadjuvant therapy should be obtained 2 to 4 months after completion of therapy to determine therapeutic response, evaluate for disease progression, and, most important, reassess surgical stage/resectability. It is clinically acceptable to proceed to resection with radiographically stable disease post-neoadjuvant therapy.

Many patients classified as borderline resectable are able to proceed with surgery following neoadjuvant therapy. Unfortunately, specific data on adjuvant therapy following neoadjuvant chemotherapy or chemoradiotherapy and surgical resection in borderline resectable patients is scarce. Clinical practice guidelines are extrapolated from studies where upfront resection in clearly resectable patients was followed by adjuvant therapy. Based on these data, approximately 6 months of perioperative chemotherapy with or without chemoradiotherapy is a reasonable consideration. Nevertheless, about 80% of patients at the time of diagnosis are deemed to be unresectable, and a smaller number do not proceed to surgery despite an initial classification as borderline resectable. Of the 80% of patients with advanced disease, about half are metastatic at presentation and the remaining 30% to 40% are defined as having unresectable LAPC.100
CASE CONTINUED
The patient is deemed borderline resectable. He receives neoadjuvant therapy with gemcitabine and nab-paclitaxel. Two months after therapy, interval imaging with abdominal CT does not show improvement in tumor size and there is now evidence that the tumor has invaded the celiac axis and is abutting more than 180° of the SMA. The patient presents to the oncologist to discuss further management. He has lost about 15 lb since his last evaluation, is capable of self-care, but is unable to carry on with any work activities.
What is the appropriate management of unresectable nonmetastatic LAPC?
UNRESECTABLE LOCALLY ADVANCED CANCER
As in the case of borderline resectable disease, there are many treatment options for patients with unresectable LAPC. Timing, optimal chemotherapy regimen, and the addition of regularly and hypofractionated radiotherapy are issues currently under investigation. However, there are some general considerations and principles that are followed as reflected in the NCCN guidelines and recent studies. The primary therapeutic aims in patients with unresectable locally advanced disease are to increase survival and improve palliation.
The elderly comprise a large percentage of the patients diagnosed with unresectable locally advanced disease. Pharmacokinetics and toxicity profiles are altered in the elderly population.101,102 Therefore, it is important to assess functional status and comorbidities as these are critical factors in determining treatment regimens, similar to patients with borderline resectable disease. Currently, the most common first-line therapies in advanced pancreatic cancer are gemcitabine alone, gemcitabine and nab-paclitaxel, FOLFIRINOX, gemcitabine/capecitabine, and gemcitabine/oxaliplatin.103 The overall treatment approach to unresectable locally advanced pancreatic adenocarcinoma closely mirrors that of patients with borderline resectable disease and metastatic disease. Much of the data supporting treatment regimens in unresectable LAPC is extrapolated from clinical trials looking at advanced or metastatic pancreatic cancer.
Consensus opinions domestically and from Europe recommend that patients with locally advanced unresectable disease undergo upfront chemotherapy (Figure).104 This is based on the premise that initial chemotherapy may destroy occult metastatic cells and increase the efficacy of consolidative chemotherapy, particularly with radiation in the future. Upfront chemoradiotherapy has only been investigated in a small series of trials in which no clear survival benefit was observed and has the added consequence of treatment-related toxicity.105 However, data is limited in this regard, with variations in treatment protocols and cohort compositions contributing to the inconclusive findings.
Despite advances in immunotherapy, targeted therapies, and gene sequencing, initial chemotherapy for unresectable disease is still either gemcitabine-based combination therapy or FOLFIRINOX. Across numerous studies, patients with unresectable LAPC receiving FOLFIRINOX have a median progression-free survival of 3 to 20 months and a median overall survival of 10 to 32.7 months.106 As with borderline resectable patients, FOLFIRINOX (Table 4) is generally reserved for unresectable patients with good functional status (ECOG 0–1 or Karnofsky Performance Status 90–100) and those at low risk for developing grade 3 or 4 systemic toxicities.103 For these reasons it has generally not been frequently combined with other chemotherapeutic agents. However, FOLFIRINOX has been combined with radiation therapy in the consolidative neoadjuvant setting after induction chemotherapy. There have also been studies where traditional FOLFIRONIX was modified to improve tolerability, as evidenced by Gunturu et al’s study, which dose-reduced both fluorouracil and irinotecan by 25%, without compromising efficacy and simultaneously increasing tolerability.107 Additionally, FOLFIRINOX requires infusional administration of the fluorouracil component, which may not be practical in certain patients. In that subset, capecitabine can be substituted. Among radiosensitizers during neoadjuvant therapy for unresectable LAPC, capecitabine has been shown to be as efficacious and less toxic than even gemcitabine.108

No head-to-head studies investigating FOLFIRINOX versus nab-paclitaxel and gemcitabine in patients with locally advanced disease have been published, but clinical trials are under way. Other combination therapies have been looked at through small retrospective or prospective studies, but no robust, large-scale clinical trials have been completed. For this reason, NCCN guidelines recommend enrollment of patients with LAPC into active clinical trials.
What is the role of radiation therapy in unresectable LAPC?
Despite the reported advantages of neoadjuvant radiation in patients with potentially resectable disease, there is significant debate regarding the timing and role of neoadjuvant radiation in patients with unresectable disease. Numerous comprehensive analyses and studiest indicate that chemoradiotherapy leads to significantly better overall survival compared to no therapy or radiation therapy alone in LAPC.68,110,111 However, conflicting data support the use of upfront chemoradiotherapy in unresectable LAPC when compared to chemotherapy alone. Unfortunately, most prospective studies investigating the role of radiotherapy were performed following administration of single-agent gemcitabine, which is no longer considered standard of care for patients with LAPC. In spite of this, ECOG 4201 identified a statistically significant improvement in median overall survival following the addition of gemcitabine-based radiotherapy. Huguet et al in his review pointed out that upfront chemoradiotherapy was not superior to chemotherapy only and was associated with increased treatment toxicity.105 Additionally, a recent phase 3 study looking at chemoradiotherapy versus chemotherapy alone in patients treated with gemcitabine found no difference in overall survival.112 This can potentially be attributed to the fact that about 30% of patients with LAPC develop metastatic disease in the early phases of treatment due to poor control of local and systemically occult disease.113 Given the propensity for high rates of occult metastatic disease in LAPC, treatment paradigms and consensus guidelines recommend multi-agent systemic chemotherapy followed by chemoradiotherapy in select patients.
Based on current studies and until further clinical investigations are completed, consensus opinion indicates that the most appropriate approach in unresectable LAPC is to begin with induction chemotherapy (with either gemcitabine plus nab-paclitaxel, FOLFIRINOX, capecitabine, or other treatment combinations), followed by chemoradiation in the absence of disease progression when the first repeat imaging evaluation is completed (Figure). One important caveat regarding reimaging with CT in the neoadjuvant setting is that radiologic response may not correlate with pathologic response.114 PET-CT may have a role in predicting response to neoadjuvant therapy. Overall, induction chemotherapy followed by consolidative chemoradiation may confer numerous benefits: it removes the unnecessary burden and toxicity associated with radiotherapy in the nearly one third of patients who have pervasive disease progression during initial treatment; it allows testing and increases the chances of tolerating full-dose systemic chemotherapy; and it raises the likelihood of converting patients who do not progress to metastasis during the initial phase of treatment from unresectable to resectable status.103,115 Despite the lack of strong conclusive data, the general agreement is that neoadjuvant chemoradiotherapy converts about one third of borderline and unresectable LAPC to an R0 resection.95,103 There are very specific rationales for the addition of radiotherapy in LAPC, and these functions need to be better defined through further clinical trials.
PALLIATIVE CARE
CASE CONTINUED
The patient is unable to tolerate his first round of second-line therapy with modified FOLFIRINOX. His overall treatment plan is transitioned to palliation. He continues to have pain, despite increasing doses of narcotics.
What is the next step for patients in whom second-line therapy fails and who have intractable pain while on high-dose narcotics?
A subset of patients with unresectable LAPC may not be amenable to chemotherapy with or without radiation due to significant comorbidities or because they present with or progress to ECOG scores 3 or 4. The goal in these patients should be palliation. Pain is one of the most predominant and difficult to manage symptoms in progressive LAPC. Opioid-based medications are the primary treatment for pain in LAPC. However, patients sometimes become refractory to opioid medications. In this group of patients, it is reasonable to consider palliative radiation as an alternative method for pain control.116
An alternative to palliative radiation in the setting of progressive pain in PDA is celiac plexus block or neurolysis. By injecting an anesthetic or alcohol into the celiac plexus, neural signaling pathways involved in the propagation of pain are inhibited without leading to significant nerve destruction. Additionally, chemical splanchnicectomy allows for reduced opioid medication use and associated side effects.117
In general patients with LAPC have profound weight loss prior to and during treatment. This has significant implications prognostically and on treatment options. The underlying etiology is multifactorial, but one of the primary driving factors is pancreatic insufficiency. An estimated 65% of pancreatic cancer patients have fat malabsorption, and 50% have protein malabsorption, leading to steatorrhea and weight loss.118 Patients diagnosed with pancreatic cancer should be given enzyme replacement with formulations that include lipase, amylase, and protease. A minimum dose of enzyme replacement should include 40,000 to 50,000 U of lipase during meals and 25,000 U during snack intake. If maldigestion, symptoms, or nutritional endpoints (BMI, albumin, prealbumin, cholesterol) do not improve, the pancreatic enzyme dose should be escalated and a proton-pump inhibitor (PPI) added. In patients with pancreatic insufficiency, PPIs have been shown to improve fat absorption.119 Enzyme replacement therapy has been shown to prevent weight loss in patients with unresectable pancreatic cancer.120
As most patients with LAPC go on to develop progressive disease, palliative care becomes an integral aspect of the therapeutic paradigm. Palliation in LAPC has a significant role in determining quality of life and ensuring patient’s goals of care have been meet. Studies have suggested that pancreatic cancer is second only to lung cancer in terms of the number of emergency department visits in the later stages of disease.120 Additionally, aggressive care in the setting of incurable diseases such as LAPC has been associated with poor quality of life.121 More recently it has been shown that involvement of palliative care in patients with advanced pancreatic is associated with less aggressive care near death.122 Therefore, the incorporation of palliative or supportive care teams in the treatment of patients with progressive LAPC can improve quality of life and alleviate suffering associated with increasing symptom burden.
CONCLUSION
LAPC is a difficult disease for both provider and patient. There is a paucity of robust clinical trials in the neoadjuvant setting for LAPC. Current research is complicated by varying consensus definitions of resectability and the varying treatment configurations across studies. The optimal type, timing, and sequence of treatment and whether to add radiation therapy in LAPC have not been clearly defined. However, based on the available studies and consensus guidelines, patients who are deemed to have LAPC should have neoadjuvant therapy. FOLFIRINOX or gemcitabine with nab-paclitaxel should be considered first-line treatments. Patients with LAPC who respond to chemotherapy or are ineligible for multi-drug chemotherapy may benefit from chemoradiotherapy. In patients with unresectable disease, chemoradiotherapy has been shown to enhance survival as compared to best supportive care or radiation alone. For borderline resectable disease, it is reasonable to treat patients with either chemoradiotherapy, chemotherapy alone, or chemotherapy followed by chemoradiotherapy.
Considering the invasive nature of LAPC and the controversy around neoadjuvant treatment protocols, enrollment of patients with LAPC into clinical trials is important and will help determine the optimal treatment regimen for future patients.
- Network PCA. Pancreatic cancer facts 2016. 2016. https://www.pancan.org/wp-content/uploads/2016/02/2016-GAA-PC-Facts.pdf. Accessed April 24, 2017.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
- Konstantinidis IT, Warshaw AL, Allen JN, et al. Pancreatic ductal adenocarcinoma: is there a survival difference for R1 resections versus locally advanced unresectable tumors? What is a “true” R0 resection? Ann Surg 2013;257:731–6.
- Ilic M, Ilic I. Epidemiology of pancreatic cancer. World J Gastroenterol 2016;22:9694–705.
- Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin 2011;61:69–90.
- Zhang J, Dhakal I, Ning B, Kesteloot H. Patterns and trends of pancreatic cancer mortality rates in Arkansas, 1969-2002: a comparison with the US population. Eur J Cancer Prev 2008;17:18–27.
- National Cancer Institute. SEER cancer statistics review, 1975-2013. http://seer.cancer.gov/csr/1975_2013/. Accessed April 24, 2017.
- Lowenfels AB, Maisonneuve P. Epidemiology and risk factors for pancreatic cancer. Best Pract Res Clin Gastroenterol 2006;20:197–209.
- Fuchs CS, Colditz GA, Stampfer MJ, et al. A prospective study of cigarette smoking and the risk of pancreatic cancer. Arch Intern Med 1996;156:2255–60.
- Lucenteforte E, La Vecchia C, Silverman D, et al. Alcohol consumption and pancreatic cancer: a pooled analysis in the International Pancreatic Cancer Case-Control Consortium (PanC4). Ann Oncol 2012;23:374–82.
- Schernhammer ES, Kang JH, Chan AT, et al. A prospective study of aspirin use and the risk of pancreatic cancer in women. J Natl Cancer Inst 2004;96:22–28.
- Michaud DS, Giovannucci E, Willett WC, et al. Physical activity, obesity, height, and the risk of pancreatic cancer. JAMA 2001;286:921–9.
- Nothlings U, Wilkens LR, Murphy SP, et al. Meat and fat intake as risk factors for pancreatic cancer: the multiethnic cohort study. J Natl Cancer Inst 2005;97:1458–65.
- Chari ST, Leibson CL, Rabe KG, et al. Probability of pancreatic cancer following diabetes: a population-based study. Gastroenterology 2005;129:504–11.
- Batabyal P, Vander Hoorn S, Christophi C, Nikfarjam M. Association of diabetes mellitus and pancreatic adenocarcinoma: a meta-analysis of 88 studies. Ann Surg Oncol 2014;21:2453–62.
- Chari ST, Leibson CL, Rabe KG, et al. Pancreatic cancer-associated diabetes mellitus: prevalence and temporal association with diagnosis of cancer. Gastroenterology 2008;134:95–101.
- Pannala R, Basu A, Petersen GM, Chari ST. New-onset diabetes: a potential clue to the early diagnosis of pancreatic cancer. Lancet Oncol 2009;10:88–95.
- Wolpin BM, Chan AT, Hartge P, et al. ABO blood group and the risk of pancreatic cancer. J Natl Cancer Inst 2009;101:424–31.
- Iqbal J, Ragone A, Lubinski J, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer 2012;107:2005–9.
- Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000;119:1447–53.
- Kastrinos F, Mukherjee B, Tayob N, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA 2009;302:1790–5.
- Brand RE, Lynch HT. Hereditary pancreatic adenocarcinoma. A clinical perspective. Med Clin North Am 2000;84:665–75.
- Jacobs EJ, Chanock SJ, Fuchs CS, et al. Family history of cancer and risk of pancreatic cancer: a pooled analysis from the Pancreatic Cancer Cohort Consortium (PanScan). Int J Cancer 2010;127:1421–8.
- Rustgi AK. Familial pancreatic cancer: genetic advances. Genes Dev 2014;28:1–7.
- Reznik R, Hendifar AE, Tuli R. Genetic determinants and potential therapeutic targets for pancreatic adenocarcinoma. Front Physiol 2014;5:87.
- Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst 1997;89:442–6.
- Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med 2014;371:2140–1.
- Kanda M, Matthaei H, Wu J, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012;142:730–3.
- Feldmann G, Maitra A. Molecular genetics of pancreatic ductal adenocarcinomas and recent implications for translational efforts. J Mol Diagn 2008;10:111–22.
- Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer 2014;111:817–22.
- Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic KRAS maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012;149:656–70.
- Provenzano PP, Cuevas C, Chang AE, et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012;21:418–29.
- Vonderheide RH, Bayne LJ. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr Opin Immunol 2013;25:200–5.
- DiMagno EP. Pancreatic cancer: Clinical presentation, pitfalls and early clues. Ann Oncol 1999;10(suppl 4):S140–S142.
- Porta M, Fabregat X, Malats N, et al. Exocrine pancreatic cancer: symptoms at presentation and their relation to tumour site and stage. Clin Transl Oncol 2005;7:189–97.
- Gullo L, Tomassetti P, Migliori M, et al. Do early symptoms of pancreatic cancer exist that can allow an earlier diagnosis? Pancreas 2001;22:210–3.
- Khorana AA, Fine RL. Pancreatic cancer and thromboembolic disease. Lancet Oncol 2004;5(11):655-663.
- Wigmore SJ, Plester CE, Richardson RA, Fearon KC. Changes in nutritional status associated with unresectable pancreatic cancer. Br J Cancer 1997;75:106–9.
- Aggarwal G, Rabe KG, Petersen GM, Chari ST. New-onset diabetes in pancreatic cancer: a study in the primary care setting. Pancreatology 2012;12:156–61.
- Koprowski H, Herlyn M, Steplewski Z, Sears HF. Specific antigen in serum of patients with colon carcinoma. Science 1981;212:53–5.
- Bond-Smith G, Banga N, Hammond TM, Imber CJ. Pancreatic adenocarcinoma. BMJ 2012;344:e2476.
- Cwik G, Wallner G, Skoczylas T, et al. Cancer antigens 19-9 and 125 in the differential diagnosis of pancreatic mass lesions. Arch Surg 2006;141:968–73.
- van den Bosch RP, van Eijck CH, Mulder PG, Jeekel J. Serum CA19-9 determination in the management of pancreatic cancer. Hepatogastroenterology 1996;43:710–3.
- Lamerz R. Role of tumour markers, cytogenetics. Ann Oncol 1999;10 Suppl 4:145–9.
- Hess V, Glimelius B, Grawe P, et al. CA 19-9 tumour-marker response to chemotherapy in patients with advanced pancreatic cancer enrolled in a randomised controlled trial. Lancet Oncol 2008;9:132–8.
- Montgomery RC, Hoffman JP, Riley LB, et al. Prediction of recurrence and survival by post-resection CA 19-9 values in patients with adenocarcinoma of the pancreas. Ann Surg Oncol 1997;4:551–6.
- Zamboni GA, D’Onofrio M, Idili A, et al. Ultrasound-guided percutaneous fine-needle aspiration of 545 focal pancreatic lesions. AJR Am J Roentgenol 2009;193:1691–5.
- Micames C, Jowell PS, White R, et al. Lower frequency of peritoneal carcinomatosis in patients with pancreatic cancer diagnosed by EUS-guided FNA vs. percutaneous FNA. Gastrointest Endosc 2003;58:690–5.
- Brambs HJ, Claussen CD. Pancreatic and ampullary carcinoma. Ultrasound, computed tomography, magnetic resonance imaging and angiography. Endoscopy 1993;25:58–68.
- Karlson BM, Ekbom A, Lindgren PG, et al. Abdominal US for diagnosis of pancreatic tumor: prospective cohort analysis. Radiology 1999;213:107–11.
- Imbriaco M, Megibow AJ, Camera L, et al. Dual-phase versus single-phase helical CT to detect and assess resectability of pancreatic carcinoma. AJR Am J Roentgenol 2002;178:1473–9.
- Schrag D. Optimizing treatment for locally advanced pancreas cancer: progress but no precision. JAMA 2016;315:1837–8.
- House MG, Yeo CJ, Cameron JL, et al. Predicting resectability of periampullary cancer with three-dimensional computed tomography. J Gastrointest Surg 2004;8:280–8.
- Callery MP, Chang KJ, Fishman EK, et al. Pretreatment assessment of resectable and borderline resectable pancreatic cancer: expert consensus statement. Ann Surg Oncol 2009;16:1727–33.
- Horton KM, Fishman EK. Adenocarcinoma of the pancreas: CT imaging. Radiol Clin North Am 2002;40:1263–72.
- Ross WA, Wasan SM, Evans DB, et al. Combined EUS with FNA and ERCP for the evaluation of patients with obstructive jaundice from presumed pancreatic malignancy. Gastrointest Endosc 2008;68:461–6.
- Hartwig W, Hackert T, Hinz U, et al. Pancreatic cancer surgery in the new millennium: better prediction of outcome. Ann Surg 2011;254:311–9.
- Jamieson NB, Chan NI, Foulis AK, et al. The prognostic influence of resection margin clearance following pancreaticoduodenectomy for pancreatic ductal adenocarcinoma. J Gastrointest Surg 2013;17:511–21.
- Ethun CG, Kooby DA. The importance of surgical margins in pancreatic cancer. J Surg Oncol 2016;113:283–8.
- Fischer R, Breidert M, Keck T, et al. Early recurrence of pancreatic cancer after resection and during adjuvant chemotherapy. Saudi J Gastroenterol 2012;18:118–21.
- Neoptolemos JP, Stocken DD, Friess H, et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med 2004;350:1200–10.
- Neoptolemos JP, Palmer DH, Ghaneh P, et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet 2017;389(10073):1011–24.
- Cardenes HR, Chiorean EG, Dewitt J, et al. Locally advanced pancreatic cancer: current therapeutic approach. Oncologist 2006;11:612–23.
- Yeung RS, Weese JL, Hoffman JP, et al. Neoadjuvant chemoradiation in pancreatic and duodenal carcinoma. A Phase II Study. Cancer 1993;72:2124–33.
- Spitz FR, Abbruzzese JL, Lee JE, et al. Preoperative and postoperative chemoradiation strategies in patients treated with pancreaticoduodenectomy for adenocarcinoma of the pancreas. J Clin Oncol 1997;15:928–37.
- McClaine RJ, Lowy AM, Sussman JJ, et al. Neoadjuvant therapy may lead to successful surgical resection and improved survival in patients with borderline resectable pancreatic cancer. HPB (Oxford) 2010;12:73–9.
- Balaban EP, Mangu PB, Khorana AA, et al. Locally advanced, unresectable pancreatic cCancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2016;34:2654–68.
- Shaib WL, Ip A, Cardona K, et al. Contemporary management of borderline resectable and locally advanced unresectable pancreatic cancer. Oncologist 2016;21:178–87.
- Chun YS, Milestone BN, Watson JC, et al. Defining venous involvement in borderline resectable pancreatic cancer. Ann Surg Oncol 2010;17:2832–8.
- Evans DB, Erickson BA, Ritch P. Borderline resectable pancreatic cancer: definitions and the importance of multimodality therapy. Ann Surg Oncol 2010;17:2803–5.
- Conroy T, Paillot B, Francois E, et al. Irinotecan plus oxaliplatin and leucovorin-modulated fluorouracil in advanced pancreatic cancer--a Groupe Tumeurs Digestives of the Federation Nationale des Centres de Lutte Contre le Cancer study. J Clin Oncol 2005;23:1228–36.
- Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
- Hosein PJ, Macintyre J, Kawamura C, et al. A retrospective study of neoadjuvant FOLFIRINOX in unresectable or borderline-resectable locally advanced pancreatic adenocarcinoma. BMC Cancer 2012;12:199.
- Peddi PF, Lubner S, McWilliams R, et al. Multi-institutional experience with FOLFIRINOX in pancreatic adenocarcinoma. JOP 2012;13:497–501.
- Mahaseth H, Kauh JS, Brutcher E, et al. Safety and efficacy of modified FOLFIRINOX in pancreatic cancer: A retrospective experience. J Clin Oncol 2012;30 (suppl; abstr e14614).
- Kim SS, Nakakura EK, Wang ZJ, et al. Preoperative FOLFIRINOX for borderline resectable pancreatic cancer: Is radiation necessary in the modern era of chemotherapy? J Surg Oncol 2016;114:587–96.
- Conroy T, Gavoille C, Samalin E, et al. The role of the FOLFIRINOX regimen for advanced pancreatic cancer. Curr Oncol Rep 2013;15:182–9.
- Katz MH, Shi Q, Ahmad SA, et al. Preoperative modified FOLFIRINOX treatment followed by capecitabine-based chemoradiation for borderline resectable pancreatic cancer: Alliance for Clinical Trials in Oncology Trial A021101. JAMA Surg 2016;151:e161137.
- Kharofa J, Kelly TR, Ritch PS, et al. 5-FU/leucovorin, irinotecan, oxaliplatin (FOLFIRINOX) induction followed by chemoXRT in borderline resectable pancreatic cancer. J Clin Oncol 2012;30 (suppl; abstr e14613).
- Blazer M, Wu C, Goldberg RM, et al. Neoadjuvant modified (m) FOLFIRINOX for locally advanced unresectable (LAPC) and borderline resectable (BRPC) adenocarcinoma of the pancreas. Ann Surg Oncol 2015;22:1153–9.
- Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
- Plunkett W, Huang P, Xu YZ, et al. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol 1995;22(4 Suppl 11):3–10.
- Sahora K, Kuehrer I, Eisenhut A, et al. NeoGemOx: Gemcitabine and oxaliplatin as neoadjuvant treatment for locally advanced, nonmetastasized pancreatic cancer. Surgery 2011;149(3):311–20.
- Lee JL, Kim SC, Kim JH, et al. Prospective efficacy and safety study of neoadjuvant gemcitabine with capecitabine combination chemotherapy for borderline-resectable or unresectable locally advanced pancreatic adenocarcinoma. Surgery 2012;152:851–62.
- Leone F, Gatti M, Massucco P, et al. Induction gemcitabine and oxaliplatin therapy followed by a twice-weekly infusion of gemcitabine and concurrent external-beam radiation for neoadjuvant treatment of locally advanced pancreatic cancer: a single institutional experience. Cancer 2013;119:277–84.
- Lawrence TS, Eisbruch A, Shewach DS. Gemcitabine-mediated radiosensitization. Semin Oncol 1997;24(2 Suppl 7):S7–24-S27–28.
- Kang CM, Chung YE, Park JY, et al. Potential contribution of preoperative neoadjuvant concurrent chemoradiation therapy on margin-negative resection in borderline resectable pancreatic cancer. J Gastrointest Surg 2012;16:509–17.
- Chuong MD, Hayman TJ, Patel MR, et al. Comparison of 1-, 2-, and 3-dimensional tumor response assessment after neoadjuvant GTX-RT in borderline-resectable pancreatic cancer. Gastrointest Cancer Res 2011;4:128–34.
- Loehrer AP, Kinnier CV, Ferrone CR. Treatment of locally advanced pancreatic ductal adenocarcinoma. Adv Surg 2016;50:115–28.
- Katz MH, Wang H, Balachandran A, et al. Effect of neoadjuvant chemoradiation and surgical technique on recurrence of localized pancreatic cancer. J Gastrointest Surg 2012;16:68–78.
- Franke AJ, Rosati LM, Pawlik TM, et al. The role of radiation therapy in pancreatic ductal adenocarcinoma in the neoadjuvant and adjuvant settings. Semin Oncol 2015;42:144–62.
- Butturini G, Stocken DD, Wente MN, et al. Influence of resection margins and treatment on survival in patients with pancreatic cancer: meta-analysis of randomized controlled trials. Arch Surg 2008;143:75–83.
- Paniccia A, Hosokawa P, Henderson W, et al. Characteristics of 10-year survivors of pancreatic ductal adenocarcinoma. JAMA Surg 2015;150:701–10.
- Massucco P, Capussotti L, Magnino A, et al. Pancreatic resections after chemoradiotherapy for locally advanced ductal adenocarcinoma: analysis of perioperative outcome and survival. Ann Surg Oncol 2006;13:1201–8.
- Patel M, Hoffe S, Malafa M, et al. Neoadjuvant GTX chemotherapy and IMRT-based chemoradiation for borderline resectable pancreatic cancer. J Surg Oncol 2011;104:155–161.
- Landry J, Catalano PJ, Staley C, et al. Randomized phase II study of gemcitabine plus radiotherapy versus gemcitabine, 5-fluorouracil, and cisplatin followed by radiotherapy and 5-fluorouracil for patients with locally advanced, potentially resectable pancreatic adenocarcinoma. J Surg Oncol 2010;101:587–92.
- Lamb R, Ozsvari B, Lisanti CL, et al. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget 2015;6:4569–84.
- Stokes JB, Nolan NJ, Stelow EB, et al. Preoperative capecitabine and concurrent radiation for borderline resectable pancreatic cancer. Ann Surg Oncol 2011;18:619–27.
- White R, Lee C, Anscher M, et al. Preoperative chemoradiation for patients with locally advanced adenocarcinoma of the pancreas. Ann Surg Oncol 1999;6:38–45.
- Martin RC 2nd. Management of locally advanced pancreatic cancer. Surg Clin North Am 2016;96:1371–89.
- Higuera O, Ghanem I, Nasimi R, et al. Management of pancreatic cancer in the elderly. World J Gastroenterol 2016;22:764–75.
- Hurria A, Lichtman SM. Clinical pharmacology of cancer therapies in older adults. Br J Cancer 2008;98:517–22.
- Spadi R, Brusa F, Ponzetti A, et al. Current therapeutic strategies for advanced pancreatic cancer: A review for clinicians. World J Clin Oncol 2016;7:27–43.
- Seufferlein T, Bachet JB, Van Cutsem E, Rougier P, Group EGW. Pancreatic adenocarcinoma: ESMO-ESDO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012;23 Suppl 7:vii33–40.
- Huguet F, Girard N, Guerche CS, et al. Chemoradiotherapy in the management of locally advanced pancreatic carcinoma: a qualitative systematic review. J Clin Oncol 2009;27:2269–77.
- Suker M, Beumer BR, Sadot E, et al. FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis. Lancet Oncol 2016;17:801–10.
- Gunturu KS, Yao X, Cong X, et al. FOLFIRINOX for locally advanced and metastatic pancreatic cancer: single institution retrospective review of efficacy and toxicity. Med Oncol 2013;30:361.
- Mukherjee S, Hurt CN, Bridgewater J, et al. Gemcitabine-based or capecitabine-based chemoradiotherapy for locally advanced pancreatic cancer (SCALOP): a multicentre, randomised, phase 2 trial. Lancet Oncol 2013;14:317–26.
- Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–1703.
- Krzyzanowska MK, Weeks JC, Earle CC. Treatment of locally advanced pancreatic cancer in the real world: population-based practices and effectiveness. J Clin Oncol 2003;21:3409–14.
- Sultana A, Tudur Smith C, Cunningham D, et al. Meta-analyses of chemotherapy for locally advanced and metastatic pancreatic cancer: results of secondary end points analyses. Br J Cancer 2008;99:6–13.
- Hammel P, Huguet F, van Laethem JL, et al. Effect of chemoradiotherapy vs chemotherapy on survival in patients with locally advanced pancreatic cancer controlled after 4 months of gemcitabine with or without erlotinib: the LAP07 randomized clinical trial. JAMA 2016;315:1844–53.
- Huguet F, Andre T, Hammel P, et al. Impact of chemoradiotherapy after disease control with chemotherapy in locally advanced pancreatic adenocarcinoma in GERCOR phase II and III studies. J Clin Oncol 2007;25:326–31.
- Dholakia AS, Hacker-Prietz A, Wild AT, et al. Resection of borderline resectable pancreatic cancer after neoadjuvant chemoradiation does not depend on improved radiographic appearance of tumor-vessel relationships. J Radiat Oncol 2013;2:413–25.
- Heinemann V, Haas M, Boeck S. Neoadjuvant treatment of borderline resectable and non-resectable pancreatic cancer. Ann Oncol 2013;24:2484–92.
- Morganti AG, Trodella L, Valentini V, et al. Pain relief with short-term irradiation in locally advanced carcinoma of the pancreas. J Palliat Care 2003;19:258–62.
- Arcidiacono PG, Calori G, Carrara S, et al. Celiac plexus block for pancreatic cancer pain in adults. Cochrane Database Syst Rev 2011(3):CD007519.
- Pezzilli R, Andriulli A, Bassi C, et al. Exocrine pancreatic insufficiency in adults: a shared position statement of the Italian Association for the Study of the Pancreas. World J Gastroenterol 2013;19:7930–46.
- Dominguez-Munoz JE. Pancreatic exocrine insufficiency: diagnosis and treatment. J Gastroenterol Hepatol 2011;26 Suppl 2:12–16.
- Bruno MJ, Haverkort EB, Tijssen GP, et al. Placebo controlled trial of enteric coated pancreatin microsphere treatment in patients with unresectable cancer of the pancreatic head region. Gut 1998;42:92–6.
- Wright AA, Keating NL, Balboni TA, et al. Place of death: correlations with quality of life of patients with cancer and predictors of bereaved caregivers’ mental health. J Clin Oncol 2010;28:4457–64.
- Jang RW, Krzyzanowska MK, Zimmermann C, et al. Palliative care and the aggressiveness of end-of-life care in patients with advanced pancreatic cancer. J Natl Cancer Inst 2015;107(3). pii: dju424.
INTRODUCTION
Pancreatic cancer is one of the most rapidly rising causes of mortality in the United States. In 2016, the number of deaths from pancreatic cancer exceeded those from breast cancer, making it the third leading cause of cancer-related death in the United States.1 It is projected that by 2020 pancreatic cancer will overtake colorectal malignancies to become the second most common cause of cancer death in this country.1,2 The term pancreatic cancer encompasses both exocrine and endocrine tumors. However, since 80% of pancreatic cancers are classified as pancreatic ductal adenocarcinoma (PDA), when speaking about pancreatic cancer most clinicians and scientists are referring to PDA.
Even with advances in chemotherapy and radiotherapy over the past decade, the only curative option for PDA is surgical resection. Unfortunately, only 20% of patients are appropriate surgical candidates at the time of diagnosis.3 Considering the lack of screening options and the ambiguity of symptomatology, roughly 4 out 5 patients with PDA are diagnosed as having locally advanced or metastatic disease that is initially not amenable to surgery.
Locally advanced pancreatic adenocarcinoma presents unique challenges in management and treatment. Treatment options include multi-agent chemotherapy, chemoradiation, or radiotherapy. Some patients can be successfully down-staged with these therapies and be deemed surgical candidates. Other challenges include selecting the appropriate sequence of therapies and stratifying therapies based on comorbidities. In this article, we review the epidemiology, biology, and diagnostic approach to PDA and focus on current treatment strategies for locally advanced pancreatic cancer (LAPC).
EPIDEMIOLOGY
In 2012, GLOBOCAN estimated that PDA caused 331,000 deaths per year, accounting for 4% of all worldwide mortality.4,5 Despite high incidence rates internationally, PDA is a disease of Western and industrialized nations. In the Unites States, PDA is a malignancy of middle to late adulthood, with a sharp upsurge in incidence after age 50 years.6 More than one third of new cases are diagnosed in patients older than 70 years, and more than half of patients diagnosed are older than 60 years of age.2 The incidence of pancreatic cancer is fairly equal among men and women, with a slightly higher rate for the male sex. It has an incidence preference for African-Americans by 4.8 cases per 100,000 persons nationally.7
Risk factors for the development of exocrine pancreatic cancer include hereditary disposition, underlying medical conditions, and environmental factors. One of the most significant environmental risk factors for the development of PDA is smoking,8 which is associated with up to 25% of all cases.9 Smoking cessation leads to a rapid reduction in risk for pancreatic cancer, with the risk among former smokers approaching that for never smokers less than 10 years after quitting.9 Other environmental factors that contribute to the development of pancreatic cancer include increased body mass index, a high-salt and high-saturated fat diet, heavy alcohol intake, and increased utilization of nonsteroidal inflammatory drugs.10–13
There is a strong association between new-onset diabetes and increased risk for developing PDA.14,15 Data also suggest that diabetes may be a risk factor and/or a consequence of tissue destruction that arises during the development or progression of PDA.16,17 Interestingly, ABO blood grouping is another underlying medical disposition that confers an altered risk profile. Studies have shown that patients with blood group O were less likely than those with type A, B, or AB to develop pancreatic cancer.18
Genetic predisposition syndromes can elevate an individual patient’s risk for developing PDA. Genetic syndromes and gene alterations that increase the risk for PDA include BRCA1/2, Peutz-Jeghers syndrome, and Lynch syndrome risk.19–21 Up to 10% to 15% of PDA cases may be due to an inherited familial cancer.22 Having a first-degree relative with PDA increases the odds of developing PDA 1.76-fold compared to those without a family history.23 The exact biologic and molecular mechanisms of familial pancreatic cancer are unclear. It is estimated that about 10% of patients with familial pancreatic cancer (FPC) carry BRCA2 mutations.24 Individuals at risk for FPC should undergo genetic screening for the presence of the most frequently inherited pancreatic cancer susceptibility genetic defects: BRCA2, PALB2, and ATM germline mutations.25 Carriers of BRCA2, who are also at increased risk for developing breast, ovarian, and prostate cancer, should be monitored closely. Of all hereditary conditions, hereditary pancreatitis confers the highest risk for developing PDA, with an approximate risk elevation of 40% to 50%.26,27 Although several genetic predisposition syndromes have been identified, most cases of pancreatic adenocarcinoma are thought to be sporadic.
CANCER BIOLOGY AND PATHOLOGY
The pathologic predecessor of PDA is pancreatic intraepithelial neoplasia (PIN). With further dysplastic changes that result from increasing genetic alterations, these precancerous lesions progress from low- to high-grade and finally to adenocarcinoma. More than 90% of all PINs across all grades have oncogenic KRAS mutations.28 Additionally, inactivating mutations in the tumor suppressor genes SMAD4, p53, and CDKN2A are found with increasing frequency in higher grade PINs. The frequency and presence of mutations in both oncogenes and tumor suppressor genes in precursor neoplasias mirror the genetic mutations noted in advanced PDA.29 Among all mutations, KRAS is the most common and most functionally important for pancreatic cancer cell survival. KRAS mutations not only have profound effects on downstream mediators of tumor growth and metastasis, but they are implicated in reprograming of cellular metabolism.30,31
Pancreatic adenocarcinoma has a unique microenvironment that makes it a difficult target for current therapeutic modalities. First, it is one of the most stroma-rich malignancies. The dense stroma surrounding pancreatic tumor cells leads to increased tumor pressures and alterations in tumor vascular perfusion.32 It also serves as a barrier that prevents chemotherapeutic drugs from reaching the tumor cells. Thus, clinical trials are under way to investigate agents such has hyaluronidase, which may degrade components of the extracellular matrix that supports thestromal environment. Additionally, there is data to suggest that the microenvironment of PDA downregulates immune monitoring, leading to further tumor growth.27,33 The molecular, cellular, and immunologic complexity of PDA may contribute to its resistance to traditional therapeutics.
EVALUATION AND DIAGNOSIS
CASE PRESENTATION
A 61-year-old man with a history of type 2 diabetes mellitus and chronic tobacco use presents to the emergency department (ED) with a 4-month history of progressively worsening abdominal discomfort and fatigue. He has also noticed darkening of his urine and slight yellow discoloration of his eyes. His weight measured 5 months ago in his primary care physician’s office was 91 kg (200 lb, BMI 29.5) and in the ED is 75 kg (165 lb, BMI 24.4). He has noticed bulky, malodorous, oily stools for about 2 months. Preliminary laboratory studies reveal elevated levels of total bilirubin (2.7 mg/dL) and alkaline phosphatase (204 IU/L). Transabdominal ultrasound (US) is obtained and reveals a 3-cm pancreatic mass with biliary tract dilation.
Does this patient have pancreatic cancer?
CLINICAL SIGNS AND SYMPTOMS
Establishing the diagnosis of pancreatic cancer in a patient who presents with a high index of suspicion is critical. Patients with pancreatic cancer usually present after a period of nonspecific and vague symptoms, which typically are experienced as abdominal discomfort, weight loss, and weakness. It is estimated that approximately 25% of patients may complain of vague abdominal pain up to 6 months prior to diagnosis. Up to 15% of patients may seek medical attention more than 6 months prior to establishing a diagnosis of PDA.34 The most common symptoms associated with pancreatic cancer in order of decreasing reported frequency are weight loss, anorexia, abdominal/epigastric pain, dark-colored urine, jaundice, nausea, back pain, and diarrhea with associated steatorrhea.35 Upwards of 15% of patients present with painless jaundice, a term that is often associated with pancreatic cancer.36 On exam these patients may have scleral icterus, sublingual jaundice, epigastric pain on palpation, weight loss, hepatomegaly, lymphadenopathy and a nontender, distended, palpable gallbladder (also known as Courvoisier sign).34 Abdominal signs and symptoms arise from tumor growth into surrounding vessels, tissues, and ducts within the abdominal cavity. Compression of the common bile duct accounts for the development of jaundice. Tumor growth around the stomach and duodenum can lead to delayed gastric emptying and subsequently nausea and vomiting. Constriction of the pancreatic duct leads to pancreatic insufficiency, precipitation of weight loss, and steatorrhea. Pancreatic insufficiency can worsen abdominal pain, and lead to increased weight loss and flatulence.
Less common symptoms include pain, erythema, and edema involving the lower extremities, which may be reflective of migratory thrombophlebitis (commonly known as Trousseau syndrome). Thromboembolic disease, including pulmonary embolism, portal vein, and deep vein thromboses are frequently encountered complications of pancreatic cancer. The incidence of thromboembolic events in patients with PDA has been reported to be as high as 54%.37 Of all signs encountered, weight loss is the most common and most profound. Patients with advanced PDA have severe degrees of cachexia. Some patients present with as much as a 5 kg/m2 decrease in their BMI from pre-illness baseline BMI, and lose another 3 to 4 kg/m2 through disease progression.38 At the time of diagnosis, many patients have already undergone significant weight loss, which can have substantial implications on treatment planning and clinical outcomes.
What other studies can be done to assist in making the diagnosis?
LABORATORY ABNORMALITIES AND TUMOR MARKERS
Elevations in alkaline phosphatase, γ-glutamyltransferase (GGT), serum aspartate aminotransferase (AST), serum alanine aminotransferase (ALT), and direct fractions of bilirubin are common in patients with PDA. Patients will usually have an obstructive pattern on their liver panel, with predominant elevations in direct bilirubin, alkaline phosphatase, and GGT, as compared with AST and ALT. Other baseline laboratory studies, including a complete blood count and basic metabolic panel, should be obtained because patients commonly have thrombocytosis, anemia, and electrolyte abnormalities due to the tumor itself and pancreatic insufficiency (Table 1).
Measurement of glycated hemoglobin (HBA1C) is an emerging and important diagnostic test in the diagnosis of pancreatic cancer. Recently, data has emerged to suggest that new-onset diabetes is present in about 50% of patients diagnosed with pancreatic cancer.39 The temporal relationship of pancreatic cancer and diabetes is supported by evidence showing that patients who undergo resection commonly have resolution of their diabetes.17 This study suggested that hyperglycemia, elevated HBA1C, and symptoms of diabetes in patients older than 50 years may identify patients who have early pancreatic cancer. The entity of pancreatic cancer–associated diabetes needs to be better defined and the algorithmic approach to evaluation and diagnosis, utilizing signs, symptoms, and laboratory values associated with diabetes, needs to be clearly established.
The only serum marker for PDA is carbohydrate antigen 19-9 (CA 19-9), also known as sialylated Lewis antigen or cancer-associated antigen. It was first identified in pancreatic cancer patients in 1981.40,41 The sensitivity and specificity of CA 19-9 ranges from 70% to approximately 90%.42,43 Hereditary predispositions and comorbid disease cross-reactivity contribute to the diminished sensitivity and specificity of CA 19-9. In about 5% to 10% of the population, CA 19-9 is not expressed (Lewis antigen A and B negative). Additionally, since CA 19-9 is expressed in the cells that line the biliary tree, diseases that lead to pancreatic or liver inflammation may falsely elevate CA 19-9.44 As a result, CA 19-9 is not an ideal screening test. However, data has shown that CA 19-9 may have prognostic value postoperatively and serve as a marker for therapeutic response.45,46
Is biopsy needed for this patient and if so, what is the most appropriate technique?
ENDOSCOPIC ULTRASOUND
Generally, diagnosis with tissue is not necessary for patients who clearly have resectable disease and will proceed directly to surgery for management. Nevertheless, it is still commonly obtained in this group of patients. However, in patients with LAPC or with features suggestive of LAPC, such as tumor approximation to critical vessels such as the superior mesenteric artery (SMA) or celiac axis, biopsy is necessary. These patients will receive neoadjuvant therapy, and biopsy is important in establishing a diagnosis. The ideal way to obtain a biopsy is through fine-needle aspiration (FNA) or biopsy (FNB) utilizing endoscopic ultrasound (EUS). Percutaneous and computed tomography (CT)–guided FNB can also be used to obtain a biopsy for diagnosis. In comparison to percutaneous and CT-guided FNB, EUS-FNA/FNB has low rates of complications, a decreased rate of peritoneal seeding, and is cost effective.47,48
CASE CONTINUED
Abdominal CT obtained following abdominal ultrasound reveals a 3.5-cm mass in the head of the pancreas in close approximation to the SMA and celiac axis.
Does the patient have borderline resectable or unresectable disease?
IMAGING
Abdominal ultrasound is a reasonable, inexpensive, and safe alternative to abdominal CT as it does not utilize ionizing radiation. It is particularly useful in patients who present with jaundice or have concern for biliary obstruction based on laboratory evaluation. It is particularly sensitive for detecting tumors greater than 3 cm in size.49,50 In patients whose abdominal ultrasound is unrevealing and whose index of suspicion remains high for PDA, abdominal CT should be the next imaging modality.
Abdominal CT obtained utilizing a pancreatic protocol is ideal for detection and staging of pancreatic tumors. By implementing a triple-phase protocol with arterial, late arterial, and venous phases, tumors, which have a density different from that of the pancreatic parenchyma, are accentuated. Abdominal CT is also able to provide critical information about tumor resectability.51 By revealing the degree of tumor encasement around the aorta, level of destruction of the superior mesenteric vein, or degree of involvement of the SMA or celiac vessels, abdominal CT determines if a patient should be deemed resectable, borderline resectable, or unresectable (Table 2).52,53 Resectability is based on thorough imaging evaluation, expert opinion of a multidisciplinary team, and guidelines proposed by American Hepatopancreaticobiliary Association, Society of Surgical Oncology, Society for Surgery of the Alimentary Tract, and the NCCN.54
Other imaging modalities have a less clearly established role in the diagnostic approach to PDA. In patients who have contraindications to obtaining CT, magnetic resonance imaging can be utilized as a secondary imaging modality.55 The role of positron emission tomography 18F-fluorodeoxyglucose (PET-FDG) is not clearly defined among clinicians, nor reflected in consensus guidelines by the National Comprehensive Cancer Network (NCCN). In clinical practice, it is still often combined with CT to detect metastatic disease, particularly in high-risk patients such has those with LAPC. The role of PET-CT in staging and its impact on clinical outcomes has not been fully established.
Endoscopic retrograde cholangiopancreatography (ERCP) and magnetic resonance cholangiopancreatography (MRCP) can also assist in the diagnosis and management of PDA. In patients with obstructive jaundice, both MRCP and ERCP visualize obstructions and dilations within the biliary tree, with the latter having the ability to intervene. ERCP allows for the collection of tissue to aid in diagnosis, and has the ability to relieve biliary obstruction via stenting.56
TREATMENT
CASE CONTINUED
After an abdominal CT is obtained, the patient is referred to an outpatient oncologist because of concern for pancreatic adenocarcinoma. After consultation, the patient is advised to obtain EUS with biopsy and to return immediately afterwards for further treatment planning. The pathology report following EUS confirms that the mass is a poorly differentiated PDA. The patient’s case is discussed at a multidisciplinary meeting with radiation, surgical, and medical oncology. The abdominal CT and PET-CT scan are thoroughly reviewed. After imaging review, the multidisciplinary team concludes that the tumor is in contact with the SMA at 120° and with the common hepatic artery without extension in the celiac axis and without evidence of metastasis.
What is the appropriate management of borderline resectable pancreatic cancer?
BORDERLINE RESECTABLE CANCER
Patients who have nonmetastatic disease and are deemed resectable and without contraindications to surgery or high-risk features, as defined by NCCN guidelines, should proceed directly to surgery. A large body of evidence suggests that complete surgical resection with negative margins is a significant predictor of survival and currently provides the only option for cure.57–59 Despite the curative intent of surgery, the rate of recurrence remains high in patients who undergo surgical resection. Even in patients with negative resection margins (R0 resection), the 5-year survival is 20% to 30%, with a median survival ranging from 12 to 25 months, suggesting the presence of regional and distant occult disease at the time of diagnosis.60–62
Additionally, in half the patients who undergo surgical resection with resultant positive microscopic (R1 resection) or gross (R2 resection) margins, the median survival is no greater than 12 months. In this subset of patients, clinical outcomes are similar to outcomes in patients with locally advanced and metastatic pancreatic cancer, suggesting that upfront surgery and adjuvant therapy may not be the ideal therapeutic option. This raises 2 important points: first, resectability should be assessed carefully in all patients with LAPC, and second, for those patients who are deemed borderline resectable, neoadjuvant therapy should be considered.63 Borderline resectability is defined as tumor abutment ≤ 180° of the celiac artery, and tumor abutment of the superior mesenteric vein /portal vein of > 180° or abutting ≤ 180° with irregularity of the vein with or without thrombosis with anatomical structures that still allows for safe and complete resection and vein reconstruction (Table 2).
Neoadjuvant Therapy
The goal of neoadjuvant therapy is to minimize the negative impact of upfront surgery in patients who have a high likelihood of having microscopic or grossly positive margins. Research has suggested that neoadjuvant therapy may improve resectability, decrease the rate of recurrence, and improve overall survival.64–66
There is no clear consensus on the ideal management of patients with borderline resectable disease. However, expert guidelines are in agreement that upfront surgery in patients with LAPC is not appropriate, as most patients will not be able to achieve an R0 resection.67 As staging and management of patients with LAPC is difficult, expertise of a multidisciplinary team can be helpful.68
Several studies and the NCCN guidelines support the use of neoadjuvant therapy in patients deemed borderline resectable.69,70 Treatment of borderline resectable disease is similar to unresectable LAPC and generally involves 2 chemotherapy treatment backbones: FOLFIRINOX (folinic acid [leucovorin], fluorouracil [5-FU], irinotecan, and oxaliplatin) or gemcitabine-based therapy.
Phase 1 to 2/3 clinical trials conducted by Conroy et al from 2005 to 2011, including the landmark ACCORD-11 trial, established the safety and role of FOLFIRINOX in metastatic pancreatic cancer and also demonstrated an improved overall survival with the use of this therapy in these patients.71,72 These findings led to interest in FOLFIRINOX as a neoadjuvant therapy for patients with LAPC. Since then, multiple prospective and retrospective studies have shown that 54% to 100% of patients with borderline resectable LAPC who were treated with FOLFIRINOX were down-staged significantly enough to undergo resection. Of those patients, more than 90% had a R0 resection following surgery (Table 3).73–79
Data over the past 7 years suggests that neoadjuvant FOLFIRINOX improves overall survival and resectability in patients with borderline disease. However, treatment with FOLFIRINOX is not without limitations. FOLFIRINOX is associated with higher rates of febrile neutropenia, thrombocytopenia, diarrhea, and sensory neuropathy as compared with gemcitabine-based therapy.72 Other less commonly observed toxicities associated with FOLFIRINOX include mucositis, hand-foot syndrome, pulmonary toxicity, and alopecia. Dose-attenuated FOLFIRINOX-based regimens, including those that exclude the bolus fluorouracil dose and augment upfront filgrastim, have demonstrated improved safety and comparable efficacy as compared to standard FOLFIRINOX.80
Gemcitabine has been the fundamental treatment backbone for PDA since the results of the phase 3 CONKO-001 trial were published.81 Gemcitabine is a pyrimidine antimetabolite and potent inhibitor of DNA polymerase and ribonucleotide reductase.82 In recent years, multiple combination therapies with gemcitabine have been investigated, including regimens with nab-paclitaxel, oxaliplatin, or docetaxel. Resection rates and negative margin outcomes have been shown to be comparable to patients who received FOLFIRINOX in the neoadjuvant setting with borderline locally advanced disease.83–85 In addition to having a more tolerable side effect profile in comparison to fluorouracil-based regimens, gemcitabine is considered to be a potent radiosensitizer.86 For this reason, studies have also investigated the role of radiotherapy in conjunction with gemcitabine, revealing negative margin resection rates above 80% in patients with borderline resectable disease.87,88
Because very few studies directly comparing FOLFIRINOX with gemcitabine-based combination regimens have been completed, there is no clear consensus on the preferred treatment regimen, in both borderline and unresectable LAPC. Decisions to treat are influenced predominantly by comorbidities, adverse effect profiles, and performance status of patients, as FOLFIRINOX is the more toxic of the 2 treatment backbones. Therefore, FOLFIRINOX has mostly been utilized in patients with relatively good functional status (Eastern Cooperative Oncology Group [ECOG] performance status 0 to 1).89 In elderly patients and those with poor functional status, ECOG 2 to 4, gemcitabine as a single agent is a reasonable alternative in the neoadjuvant setting of borderline resectable disease.
The exact role of radiation therapy in addition to induction chemotherapy in borderline resectable pancreatic cancer has not been clearly established because of the lack of prospective studies in this area. Multiple large retrospective series have identified high rates of conversion to margin-negative resection with neoadjuvant chemoradiation alone.90 Based on available data, it is reasonable for patients with borderline resectable disease to proceed with any of the following treatment options: chemotherapy, chemoradiation, or induction chemotherapy followed by chemoradiation (Figure). Chemotherapy and chemoradiation are generally more appropriate with patients with high CA 19-9 levels or those at an elevated risk of having positive margins or occult metastatic disease.91 Obtaining negative margin resections is the predominant goal of neoadjuvant radiotherapy.89 Many studies have identified margin status to be one of the most significant prognostic factors in PDA.57,59,92,93 Additionally, several studies have highlighted that radiotherapy in the neoadjuvant setting could improve negative margin resection rates, local control, and clinical outcomes in patients with borderline resectable locally advanced disease.94–97 A common multimodal regimen utilized in the neoadjuvant setting combines capecitabine, an oral prodrug that is converted to fluorouracil, with radiation therapy. This combination has also been shown to improve resectability rates and long-term clinical outcomes in patients with borderline resectable disease.98 Additionally, neoadjuvant radiation therapy can potentially downstage patients with unresectable disease at the time of diagnosis to become surgical candidates.99 Despite the paucity of data, interval scans utilizing CT following neoadjuvant therapy should be obtained 2 to 4 months after completion of therapy to determine therapeutic response, evaluate for disease progression, and, most important, reassess surgical stage/resectability. It is clinically acceptable to proceed to resection with radiographically stable disease post-neoadjuvant therapy.
Many patients classified as borderline resectable are able to proceed with surgery following neoadjuvant therapy. Unfortunately, specific data on adjuvant therapy following neoadjuvant chemotherapy or chemoradiotherapy and surgical resection in borderline resectable patients is scarce. Clinical practice guidelines are extrapolated from studies where upfront resection in clearly resectable patients was followed by adjuvant therapy. Based on these data, approximately 6 months of perioperative chemotherapy with or without chemoradiotherapy is a reasonable consideration. Nevertheless, about 80% of patients at the time of diagnosis are deemed to be unresectable, and a smaller number do not proceed to surgery despite an initial classification as borderline resectable. Of the 80% of patients with advanced disease, about half are metastatic at presentation and the remaining 30% to 40% are defined as having unresectable LAPC.100
CASE CONTINUED
The patient is deemed borderline resectable. He receives neoadjuvant therapy with gemcitabine and nab-paclitaxel. Two months after therapy, interval imaging with abdominal CT does not show improvement in tumor size and there is now evidence that the tumor has invaded the celiac axis and is abutting more than 180° of the SMA. The patient presents to the oncologist to discuss further management. He has lost about 15 lb since his last evaluation, is capable of self-care, but is unable to carry on with any work activities.
What is the appropriate management of unresectable nonmetastatic LAPC?
UNRESECTABLE LOCALLY ADVANCED CANCER
As in the case of borderline resectable disease, there are many treatment options for patients with unresectable LAPC. Timing, optimal chemotherapy regimen, and the addition of regularly and hypofractionated radiotherapy are issues currently under investigation. However, there are some general considerations and principles that are followed as reflected in the NCCN guidelines and recent studies. The primary therapeutic aims in patients with unresectable locally advanced disease are to increase survival and improve palliation.
The elderly comprise a large percentage of the patients diagnosed with unresectable locally advanced disease. Pharmacokinetics and toxicity profiles are altered in the elderly population.101,102 Therefore, it is important to assess functional status and comorbidities as these are critical factors in determining treatment regimens, similar to patients with borderline resectable disease. Currently, the most common first-line therapies in advanced pancreatic cancer are gemcitabine alone, gemcitabine and nab-paclitaxel, FOLFIRINOX, gemcitabine/capecitabine, and gemcitabine/oxaliplatin.103 The overall treatment approach to unresectable locally advanced pancreatic adenocarcinoma closely mirrors that of patients with borderline resectable disease and metastatic disease. Much of the data supporting treatment regimens in unresectable LAPC is extrapolated from clinical trials looking at advanced or metastatic pancreatic cancer.
Consensus opinions domestically and from Europe recommend that patients with locally advanced unresectable disease undergo upfront chemotherapy (Figure).104 This is based on the premise that initial chemotherapy may destroy occult metastatic cells and increase the efficacy of consolidative chemotherapy, particularly with radiation in the future. Upfront chemoradiotherapy has only been investigated in a small series of trials in which no clear survival benefit was observed and has the added consequence of treatment-related toxicity.105 However, data is limited in this regard, with variations in treatment protocols and cohort compositions contributing to the inconclusive findings.
Despite advances in immunotherapy, targeted therapies, and gene sequencing, initial chemotherapy for unresectable disease is still either gemcitabine-based combination therapy or FOLFIRINOX. Across numerous studies, patients with unresectable LAPC receiving FOLFIRINOX have a median progression-free survival of 3 to 20 months and a median overall survival of 10 to 32.7 months.106 As with borderline resectable patients, FOLFIRINOX (Table 4) is generally reserved for unresectable patients with good functional status (ECOG 0–1 or Karnofsky Performance Status 90–100) and those at low risk for developing grade 3 or 4 systemic toxicities.103 For these reasons it has generally not been frequently combined with other chemotherapeutic agents. However, FOLFIRINOX has been combined with radiation therapy in the consolidative neoadjuvant setting after induction chemotherapy. There have also been studies where traditional FOLFIRONIX was modified to improve tolerability, as evidenced by Gunturu et al’s study, which dose-reduced both fluorouracil and irinotecan by 25%, without compromising efficacy and simultaneously increasing tolerability.107 Additionally, FOLFIRINOX requires infusional administration of the fluorouracil component, which may not be practical in certain patients. In that subset, capecitabine can be substituted. Among radiosensitizers during neoadjuvant therapy for unresectable LAPC, capecitabine has been shown to be as efficacious and less toxic than even gemcitabine.108
No head-to-head studies investigating FOLFIRINOX versus nab-paclitaxel and gemcitabine in patients with locally advanced disease have been published, but clinical trials are under way. Other combination therapies have been looked at through small retrospective or prospective studies, but no robust, large-scale clinical trials have been completed. For this reason, NCCN guidelines recommend enrollment of patients with LAPC into active clinical trials.
What is the role of radiation therapy in unresectable LAPC?
Despite the reported advantages of neoadjuvant radiation in patients with potentially resectable disease, there is significant debate regarding the timing and role of neoadjuvant radiation in patients with unresectable disease. Numerous comprehensive analyses and studiest indicate that chemoradiotherapy leads to significantly better overall survival compared to no therapy or radiation therapy alone in LAPC.68,110,111 However, conflicting data support the use of upfront chemoradiotherapy in unresectable LAPC when compared to chemotherapy alone. Unfortunately, most prospective studies investigating the role of radiotherapy were performed following administration of single-agent gemcitabine, which is no longer considered standard of care for patients with LAPC. In spite of this, ECOG 4201 identified a statistically significant improvement in median overall survival following the addition of gemcitabine-based radiotherapy. Huguet et al in his review pointed out that upfront chemoradiotherapy was not superior to chemotherapy only and was associated with increased treatment toxicity.105 Additionally, a recent phase 3 study looking at chemoradiotherapy versus chemotherapy alone in patients treated with gemcitabine found no difference in overall survival.112 This can potentially be attributed to the fact that about 30% of patients with LAPC develop metastatic disease in the early phases of treatment due to poor control of local and systemically occult disease.113 Given the propensity for high rates of occult metastatic disease in LAPC, treatment paradigms and consensus guidelines recommend multi-agent systemic chemotherapy followed by chemoradiotherapy in select patients.
Based on current studies and until further clinical investigations are completed, consensus opinion indicates that the most appropriate approach in unresectable LAPC is to begin with induction chemotherapy (with either gemcitabine plus nab-paclitaxel, FOLFIRINOX, capecitabine, or other treatment combinations), followed by chemoradiation in the absence of disease progression when the first repeat imaging evaluation is completed (Figure). One important caveat regarding reimaging with CT in the neoadjuvant setting is that radiologic response may not correlate with pathologic response.114 PET-CT may have a role in predicting response to neoadjuvant therapy. Overall, induction chemotherapy followed by consolidative chemoradiation may confer numerous benefits: it removes the unnecessary burden and toxicity associated with radiotherapy in the nearly one third of patients who have pervasive disease progression during initial treatment; it allows testing and increases the chances of tolerating full-dose systemic chemotherapy; and it raises the likelihood of converting patients who do not progress to metastasis during the initial phase of treatment from unresectable to resectable status.103,115 Despite the lack of strong conclusive data, the general agreement is that neoadjuvant chemoradiotherapy converts about one third of borderline and unresectable LAPC to an R0 resection.95,103 There are very specific rationales for the addition of radiotherapy in LAPC, and these functions need to be better defined through further clinical trials.
PALLIATIVE CARE
CASE CONTINUED
The patient is unable to tolerate his first round of second-line therapy with modified FOLFIRINOX. His overall treatment plan is transitioned to palliation. He continues to have pain, despite increasing doses of narcotics.
What is the next step for patients in whom second-line therapy fails and who have intractable pain while on high-dose narcotics?
A subset of patients with unresectable LAPC may not be amenable to chemotherapy with or without radiation due to significant comorbidities or because they present with or progress to ECOG scores 3 or 4. The goal in these patients should be palliation. Pain is one of the most predominant and difficult to manage symptoms in progressive LAPC. Opioid-based medications are the primary treatment for pain in LAPC. However, patients sometimes become refractory to opioid medications. In this group of patients, it is reasonable to consider palliative radiation as an alternative method for pain control.116
An alternative to palliative radiation in the setting of progressive pain in PDA is celiac plexus block or neurolysis. By injecting an anesthetic or alcohol into the celiac plexus, neural signaling pathways involved in the propagation of pain are inhibited without leading to significant nerve destruction. Additionally, chemical splanchnicectomy allows for reduced opioid medication use and associated side effects.117
In general patients with LAPC have profound weight loss prior to and during treatment. This has significant implications prognostically and on treatment options. The underlying etiology is multifactorial, but one of the primary driving factors is pancreatic insufficiency. An estimated 65% of pancreatic cancer patients have fat malabsorption, and 50% have protein malabsorption, leading to steatorrhea and weight loss.118 Patients diagnosed with pancreatic cancer should be given enzyme replacement with formulations that include lipase, amylase, and protease. A minimum dose of enzyme replacement should include 40,000 to 50,000 U of lipase during meals and 25,000 U during snack intake. If maldigestion, symptoms, or nutritional endpoints (BMI, albumin, prealbumin, cholesterol) do not improve, the pancreatic enzyme dose should be escalated and a proton-pump inhibitor (PPI) added. In patients with pancreatic insufficiency, PPIs have been shown to improve fat absorption.119 Enzyme replacement therapy has been shown to prevent weight loss in patients with unresectable pancreatic cancer.120
As most patients with LAPC go on to develop progressive disease, palliative care becomes an integral aspect of the therapeutic paradigm. Palliation in LAPC has a significant role in determining quality of life and ensuring patient’s goals of care have been meet. Studies have suggested that pancreatic cancer is second only to lung cancer in terms of the number of emergency department visits in the later stages of disease.120 Additionally, aggressive care in the setting of incurable diseases such as LAPC has been associated with poor quality of life.121 More recently it has been shown that involvement of palliative care in patients with advanced pancreatic is associated with less aggressive care near death.122 Therefore, the incorporation of palliative or supportive care teams in the treatment of patients with progressive LAPC can improve quality of life and alleviate suffering associated with increasing symptom burden.
CONCLUSION
LAPC is a difficult disease for both provider and patient. There is a paucity of robust clinical trials in the neoadjuvant setting for LAPC. Current research is complicated by varying consensus definitions of resectability and the varying treatment configurations across studies. The optimal type, timing, and sequence of treatment and whether to add radiation therapy in LAPC have not been clearly defined. However, based on the available studies and consensus guidelines, patients who are deemed to have LAPC should have neoadjuvant therapy. FOLFIRINOX or gemcitabine with nab-paclitaxel should be considered first-line treatments. Patients with LAPC who respond to chemotherapy or are ineligible for multi-drug chemotherapy may benefit from chemoradiotherapy. In patients with unresectable disease, chemoradiotherapy has been shown to enhance survival as compared to best supportive care or radiation alone. For borderline resectable disease, it is reasonable to treat patients with either chemoradiotherapy, chemotherapy alone, or chemotherapy followed by chemoradiotherapy.
Considering the invasive nature of LAPC and the controversy around neoadjuvant treatment protocols, enrollment of patients with LAPC into clinical trials is important and will help determine the optimal treatment regimen for future patients.
INTRODUCTION
Pancreatic cancer is one of the most rapidly rising causes of mortality in the United States. In 2016, the number of deaths from pancreatic cancer exceeded those from breast cancer, making it the third leading cause of cancer-related death in the United States.1 It is projected that by 2020 pancreatic cancer will overtake colorectal malignancies to become the second most common cause of cancer death in this country.1,2 The term pancreatic cancer encompasses both exocrine and endocrine tumors. However, since 80% of pancreatic cancers are classified as pancreatic ductal adenocarcinoma (PDA), when speaking about pancreatic cancer most clinicians and scientists are referring to PDA.
Even with advances in chemotherapy and radiotherapy over the past decade, the only curative option for PDA is surgical resection. Unfortunately, only 20% of patients are appropriate surgical candidates at the time of diagnosis.3 Considering the lack of screening options and the ambiguity of symptomatology, roughly 4 out 5 patients with PDA are diagnosed as having locally advanced or metastatic disease that is initially not amenable to surgery.
Locally advanced pancreatic adenocarcinoma presents unique challenges in management and treatment. Treatment options include multi-agent chemotherapy, chemoradiation, or radiotherapy. Some patients can be successfully down-staged with these therapies and be deemed surgical candidates. Other challenges include selecting the appropriate sequence of therapies and stratifying therapies based on comorbidities. In this article, we review the epidemiology, biology, and diagnostic approach to PDA and focus on current treatment strategies for locally advanced pancreatic cancer (LAPC).
EPIDEMIOLOGY
In 2012, GLOBOCAN estimated that PDA caused 331,000 deaths per year, accounting for 4% of all worldwide mortality.4,5 Despite high incidence rates internationally, PDA is a disease of Western and industrialized nations. In the Unites States, PDA is a malignancy of middle to late adulthood, with a sharp upsurge in incidence after age 50 years.6 More than one third of new cases are diagnosed in patients older than 70 years, and more than half of patients diagnosed are older than 60 years of age.2 The incidence of pancreatic cancer is fairly equal among men and women, with a slightly higher rate for the male sex. It has an incidence preference for African-Americans by 4.8 cases per 100,000 persons nationally.7
Risk factors for the development of exocrine pancreatic cancer include hereditary disposition, underlying medical conditions, and environmental factors. One of the most significant environmental risk factors for the development of PDA is smoking,8 which is associated with up to 25% of all cases.9 Smoking cessation leads to a rapid reduction in risk for pancreatic cancer, with the risk among former smokers approaching that for never smokers less than 10 years after quitting.9 Other environmental factors that contribute to the development of pancreatic cancer include increased body mass index, a high-salt and high-saturated fat diet, heavy alcohol intake, and increased utilization of nonsteroidal inflammatory drugs.10–13
There is a strong association between new-onset diabetes and increased risk for developing PDA.14,15 Data also suggest that diabetes may be a risk factor and/or a consequence of tissue destruction that arises during the development or progression of PDA.16,17 Interestingly, ABO blood grouping is another underlying medical disposition that confers an altered risk profile. Studies have shown that patients with blood group O were less likely than those with type A, B, or AB to develop pancreatic cancer.18
Genetic predisposition syndromes can elevate an individual patient’s risk for developing PDA. Genetic syndromes and gene alterations that increase the risk for PDA include BRCA1/2, Peutz-Jeghers syndrome, and Lynch syndrome risk.19–21 Up to 10% to 15% of PDA cases may be due to an inherited familial cancer.22 Having a first-degree relative with PDA increases the odds of developing PDA 1.76-fold compared to those without a family history.23 The exact biologic and molecular mechanisms of familial pancreatic cancer are unclear. It is estimated that about 10% of patients with familial pancreatic cancer (FPC) carry BRCA2 mutations.24 Individuals at risk for FPC should undergo genetic screening for the presence of the most frequently inherited pancreatic cancer susceptibility genetic defects: BRCA2, PALB2, and ATM germline mutations.25 Carriers of BRCA2, who are also at increased risk for developing breast, ovarian, and prostate cancer, should be monitored closely. Of all hereditary conditions, hereditary pancreatitis confers the highest risk for developing PDA, with an approximate risk elevation of 40% to 50%.26,27 Although several genetic predisposition syndromes have been identified, most cases of pancreatic adenocarcinoma are thought to be sporadic.
CANCER BIOLOGY AND PATHOLOGY
The pathologic predecessor of PDA is pancreatic intraepithelial neoplasia (PIN). With further dysplastic changes that result from increasing genetic alterations, these precancerous lesions progress from low- to high-grade and finally to adenocarcinoma. More than 90% of all PINs across all grades have oncogenic KRAS mutations.28 Additionally, inactivating mutations in the tumor suppressor genes SMAD4, p53, and CDKN2A are found with increasing frequency in higher grade PINs. The frequency and presence of mutations in both oncogenes and tumor suppressor genes in precursor neoplasias mirror the genetic mutations noted in advanced PDA.29 Among all mutations, KRAS is the most common and most functionally important for pancreatic cancer cell survival. KRAS mutations not only have profound effects on downstream mediators of tumor growth and metastasis, but they are implicated in reprograming of cellular metabolism.30,31
Pancreatic adenocarcinoma has a unique microenvironment that makes it a difficult target for current therapeutic modalities. First, it is one of the most stroma-rich malignancies. The dense stroma surrounding pancreatic tumor cells leads to increased tumor pressures and alterations in tumor vascular perfusion.32 It also serves as a barrier that prevents chemotherapeutic drugs from reaching the tumor cells. Thus, clinical trials are under way to investigate agents such has hyaluronidase, which may degrade components of the extracellular matrix that supports thestromal environment. Additionally, there is data to suggest that the microenvironment of PDA downregulates immune monitoring, leading to further tumor growth.27,33 The molecular, cellular, and immunologic complexity of PDA may contribute to its resistance to traditional therapeutics.
EVALUATION AND DIAGNOSIS
CASE PRESENTATION
A 61-year-old man with a history of type 2 diabetes mellitus and chronic tobacco use presents to the emergency department (ED) with a 4-month history of progressively worsening abdominal discomfort and fatigue. He has also noticed darkening of his urine and slight yellow discoloration of his eyes. His weight measured 5 months ago in his primary care physician’s office was 91 kg (200 lb, BMI 29.5) and in the ED is 75 kg (165 lb, BMI 24.4). He has noticed bulky, malodorous, oily stools for about 2 months. Preliminary laboratory studies reveal elevated levels of total bilirubin (2.7 mg/dL) and alkaline phosphatase (204 IU/L). Transabdominal ultrasound (US) is obtained and reveals a 3-cm pancreatic mass with biliary tract dilation.
Does this patient have pancreatic cancer?
CLINICAL SIGNS AND SYMPTOMS
Establishing the diagnosis of pancreatic cancer in a patient who presents with a high index of suspicion is critical. Patients with pancreatic cancer usually present after a period of nonspecific and vague symptoms, which typically are experienced as abdominal discomfort, weight loss, and weakness. It is estimated that approximately 25% of patients may complain of vague abdominal pain up to 6 months prior to diagnosis. Up to 15% of patients may seek medical attention more than 6 months prior to establishing a diagnosis of PDA.34 The most common symptoms associated with pancreatic cancer in order of decreasing reported frequency are weight loss, anorexia, abdominal/epigastric pain, dark-colored urine, jaundice, nausea, back pain, and diarrhea with associated steatorrhea.35 Upwards of 15% of patients present with painless jaundice, a term that is often associated with pancreatic cancer.36 On exam these patients may have scleral icterus, sublingual jaundice, epigastric pain on palpation, weight loss, hepatomegaly, lymphadenopathy and a nontender, distended, palpable gallbladder (also known as Courvoisier sign).34 Abdominal signs and symptoms arise from tumor growth into surrounding vessels, tissues, and ducts within the abdominal cavity. Compression of the common bile duct accounts for the development of jaundice. Tumor growth around the stomach and duodenum can lead to delayed gastric emptying and subsequently nausea and vomiting. Constriction of the pancreatic duct leads to pancreatic insufficiency, precipitation of weight loss, and steatorrhea. Pancreatic insufficiency can worsen abdominal pain, and lead to increased weight loss and flatulence.
Less common symptoms include pain, erythema, and edema involving the lower extremities, which may be reflective of migratory thrombophlebitis (commonly known as Trousseau syndrome). Thromboembolic disease, including pulmonary embolism, portal vein, and deep vein thromboses are frequently encountered complications of pancreatic cancer. The incidence of thromboembolic events in patients with PDA has been reported to be as high as 54%.37 Of all signs encountered, weight loss is the most common and most profound. Patients with advanced PDA have severe degrees of cachexia. Some patients present with as much as a 5 kg/m2 decrease in their BMI from pre-illness baseline BMI, and lose another 3 to 4 kg/m2 through disease progression.38 At the time of diagnosis, many patients have already undergone significant weight loss, which can have substantial implications on treatment planning and clinical outcomes.
What other studies can be done to assist in making the diagnosis?
LABORATORY ABNORMALITIES AND TUMOR MARKERS
Elevations in alkaline phosphatase, γ-glutamyltransferase (GGT), serum aspartate aminotransferase (AST), serum alanine aminotransferase (ALT), and direct fractions of bilirubin are common in patients with PDA. Patients will usually have an obstructive pattern on their liver panel, with predominant elevations in direct bilirubin, alkaline phosphatase, and GGT, as compared with AST and ALT. Other baseline laboratory studies, including a complete blood count and basic metabolic panel, should be obtained because patients commonly have thrombocytosis, anemia, and electrolyte abnormalities due to the tumor itself and pancreatic insufficiency (Table 1).
Measurement of glycated hemoglobin (HBA1C) is an emerging and important diagnostic test in the diagnosis of pancreatic cancer. Recently, data has emerged to suggest that new-onset diabetes is present in about 50% of patients diagnosed with pancreatic cancer.39 The temporal relationship of pancreatic cancer and diabetes is supported by evidence showing that patients who undergo resection commonly have resolution of their diabetes.17 This study suggested that hyperglycemia, elevated HBA1C, and symptoms of diabetes in patients older than 50 years may identify patients who have early pancreatic cancer. The entity of pancreatic cancer–associated diabetes needs to be better defined and the algorithmic approach to evaluation and diagnosis, utilizing signs, symptoms, and laboratory values associated with diabetes, needs to be clearly established.
The only serum marker for PDA is carbohydrate antigen 19-9 (CA 19-9), also known as sialylated Lewis antigen or cancer-associated antigen. It was first identified in pancreatic cancer patients in 1981.40,41 The sensitivity and specificity of CA 19-9 ranges from 70% to approximately 90%.42,43 Hereditary predispositions and comorbid disease cross-reactivity contribute to the diminished sensitivity and specificity of CA 19-9. In about 5% to 10% of the population, CA 19-9 is not expressed (Lewis antigen A and B negative). Additionally, since CA 19-9 is expressed in the cells that line the biliary tree, diseases that lead to pancreatic or liver inflammation may falsely elevate CA 19-9.44 As a result, CA 19-9 is not an ideal screening test. However, data has shown that CA 19-9 may have prognostic value postoperatively and serve as a marker for therapeutic response.45,46
Is biopsy needed for this patient and if so, what is the most appropriate technique?
ENDOSCOPIC ULTRASOUND
Generally, diagnosis with tissue is not necessary for patients who clearly have resectable disease and will proceed directly to surgery for management. Nevertheless, it is still commonly obtained in this group of patients. However, in patients with LAPC or with features suggestive of LAPC, such as tumor approximation to critical vessels such as the superior mesenteric artery (SMA) or celiac axis, biopsy is necessary. These patients will receive neoadjuvant therapy, and biopsy is important in establishing a diagnosis. The ideal way to obtain a biopsy is through fine-needle aspiration (FNA) or biopsy (FNB) utilizing endoscopic ultrasound (EUS). Percutaneous and computed tomography (CT)–guided FNB can also be used to obtain a biopsy for diagnosis. In comparison to percutaneous and CT-guided FNB, EUS-FNA/FNB has low rates of complications, a decreased rate of peritoneal seeding, and is cost effective.47,48
CASE CONTINUED
Abdominal CT obtained following abdominal ultrasound reveals a 3.5-cm mass in the head of the pancreas in close approximation to the SMA and celiac axis.
Does the patient have borderline resectable or unresectable disease?
IMAGING
Abdominal ultrasound is a reasonable, inexpensive, and safe alternative to abdominal CT as it does not utilize ionizing radiation. It is particularly useful in patients who present with jaundice or have concern for biliary obstruction based on laboratory evaluation. It is particularly sensitive for detecting tumors greater than 3 cm in size.49,50 In patients whose abdominal ultrasound is unrevealing and whose index of suspicion remains high for PDA, abdominal CT should be the next imaging modality.
Abdominal CT obtained utilizing a pancreatic protocol is ideal for detection and staging of pancreatic tumors. By implementing a triple-phase protocol with arterial, late arterial, and venous phases, tumors, which have a density different from that of the pancreatic parenchyma, are accentuated. Abdominal CT is also able to provide critical information about tumor resectability.51 By revealing the degree of tumor encasement around the aorta, level of destruction of the superior mesenteric vein, or degree of involvement of the SMA or celiac vessels, abdominal CT determines if a patient should be deemed resectable, borderline resectable, or unresectable (Table 2).52,53 Resectability is based on thorough imaging evaluation, expert opinion of a multidisciplinary team, and guidelines proposed by American Hepatopancreaticobiliary Association, Society of Surgical Oncology, Society for Surgery of the Alimentary Tract, and the NCCN.54
Other imaging modalities have a less clearly established role in the diagnostic approach to PDA. In patients who have contraindications to obtaining CT, magnetic resonance imaging can be utilized as a secondary imaging modality.55 The role of positron emission tomography 18F-fluorodeoxyglucose (PET-FDG) is not clearly defined among clinicians, nor reflected in consensus guidelines by the National Comprehensive Cancer Network (NCCN). In clinical practice, it is still often combined with CT to detect metastatic disease, particularly in high-risk patients such has those with LAPC. The role of PET-CT in staging and its impact on clinical outcomes has not been fully established.
Endoscopic retrograde cholangiopancreatography (ERCP) and magnetic resonance cholangiopancreatography (MRCP) can also assist in the diagnosis and management of PDA. In patients with obstructive jaundice, both MRCP and ERCP visualize obstructions and dilations within the biliary tree, with the latter having the ability to intervene. ERCP allows for the collection of tissue to aid in diagnosis, and has the ability to relieve biliary obstruction via stenting.56
TREATMENT
CASE CONTINUED
After an abdominal CT is obtained, the patient is referred to an outpatient oncologist because of concern for pancreatic adenocarcinoma. After consultation, the patient is advised to obtain EUS with biopsy and to return immediately afterwards for further treatment planning. The pathology report following EUS confirms that the mass is a poorly differentiated PDA. The patient’s case is discussed at a multidisciplinary meeting with radiation, surgical, and medical oncology. The abdominal CT and PET-CT scan are thoroughly reviewed. After imaging review, the multidisciplinary team concludes that the tumor is in contact with the SMA at 120° and with the common hepatic artery without extension in the celiac axis and without evidence of metastasis.
What is the appropriate management of borderline resectable pancreatic cancer?
BORDERLINE RESECTABLE CANCER
Patients who have nonmetastatic disease and are deemed resectable and without contraindications to surgery or high-risk features, as defined by NCCN guidelines, should proceed directly to surgery. A large body of evidence suggests that complete surgical resection with negative margins is a significant predictor of survival and currently provides the only option for cure.57–59 Despite the curative intent of surgery, the rate of recurrence remains high in patients who undergo surgical resection. Even in patients with negative resection margins (R0 resection), the 5-year survival is 20% to 30%, with a median survival ranging from 12 to 25 months, suggesting the presence of regional and distant occult disease at the time of diagnosis.60–62
Additionally, in half the patients who undergo surgical resection with resultant positive microscopic (R1 resection) or gross (R2 resection) margins, the median survival is no greater than 12 months. In this subset of patients, clinical outcomes are similar to outcomes in patients with locally advanced and metastatic pancreatic cancer, suggesting that upfront surgery and adjuvant therapy may not be the ideal therapeutic option. This raises 2 important points: first, resectability should be assessed carefully in all patients with LAPC, and second, for those patients who are deemed borderline resectable, neoadjuvant therapy should be considered.63 Borderline resectability is defined as tumor abutment ≤ 180° of the celiac artery, and tumor abutment of the superior mesenteric vein /portal vein of > 180° or abutting ≤ 180° with irregularity of the vein with or without thrombosis with anatomical structures that still allows for safe and complete resection and vein reconstruction (Table 2).
Neoadjuvant Therapy
The goal of neoadjuvant therapy is to minimize the negative impact of upfront surgery in patients who have a high likelihood of having microscopic or grossly positive margins. Research has suggested that neoadjuvant therapy may improve resectability, decrease the rate of recurrence, and improve overall survival.64–66
There is no clear consensus on the ideal management of patients with borderline resectable disease. However, expert guidelines are in agreement that upfront surgery in patients with LAPC is not appropriate, as most patients will not be able to achieve an R0 resection.67 As staging and management of patients with LAPC is difficult, expertise of a multidisciplinary team can be helpful.68
Several studies and the NCCN guidelines support the use of neoadjuvant therapy in patients deemed borderline resectable.69,70 Treatment of borderline resectable disease is similar to unresectable LAPC and generally involves 2 chemotherapy treatment backbones: FOLFIRINOX (folinic acid [leucovorin], fluorouracil [5-FU], irinotecan, and oxaliplatin) or gemcitabine-based therapy.
Phase 1 to 2/3 clinical trials conducted by Conroy et al from 2005 to 2011, including the landmark ACCORD-11 trial, established the safety and role of FOLFIRINOX in metastatic pancreatic cancer and also demonstrated an improved overall survival with the use of this therapy in these patients.71,72 These findings led to interest in FOLFIRINOX as a neoadjuvant therapy for patients with LAPC. Since then, multiple prospective and retrospective studies have shown that 54% to 100% of patients with borderline resectable LAPC who were treated with FOLFIRINOX were down-staged significantly enough to undergo resection. Of those patients, more than 90% had a R0 resection following surgery (Table 3).73–79
Data over the past 7 years suggests that neoadjuvant FOLFIRINOX improves overall survival and resectability in patients with borderline disease. However, treatment with FOLFIRINOX is not without limitations. FOLFIRINOX is associated with higher rates of febrile neutropenia, thrombocytopenia, diarrhea, and sensory neuropathy as compared with gemcitabine-based therapy.72 Other less commonly observed toxicities associated with FOLFIRINOX include mucositis, hand-foot syndrome, pulmonary toxicity, and alopecia. Dose-attenuated FOLFIRINOX-based regimens, including those that exclude the bolus fluorouracil dose and augment upfront filgrastim, have demonstrated improved safety and comparable efficacy as compared to standard FOLFIRINOX.80
Gemcitabine has been the fundamental treatment backbone for PDA since the results of the phase 3 CONKO-001 trial were published.81 Gemcitabine is a pyrimidine antimetabolite and potent inhibitor of DNA polymerase and ribonucleotide reductase.82 In recent years, multiple combination therapies with gemcitabine have been investigated, including regimens with nab-paclitaxel, oxaliplatin, or docetaxel. Resection rates and negative margin outcomes have been shown to be comparable to patients who received FOLFIRINOX in the neoadjuvant setting with borderline locally advanced disease.83–85 In addition to having a more tolerable side effect profile in comparison to fluorouracil-based regimens, gemcitabine is considered to be a potent radiosensitizer.86 For this reason, studies have also investigated the role of radiotherapy in conjunction with gemcitabine, revealing negative margin resection rates above 80% in patients with borderline resectable disease.87,88
Because very few studies directly comparing FOLFIRINOX with gemcitabine-based combination regimens have been completed, there is no clear consensus on the preferred treatment regimen, in both borderline and unresectable LAPC. Decisions to treat are influenced predominantly by comorbidities, adverse effect profiles, and performance status of patients, as FOLFIRINOX is the more toxic of the 2 treatment backbones. Therefore, FOLFIRINOX has mostly been utilized in patients with relatively good functional status (Eastern Cooperative Oncology Group [ECOG] performance status 0 to 1).89 In elderly patients and those with poor functional status, ECOG 2 to 4, gemcitabine as a single agent is a reasonable alternative in the neoadjuvant setting of borderline resectable disease.
The exact role of radiation therapy in addition to induction chemotherapy in borderline resectable pancreatic cancer has not been clearly established because of the lack of prospective studies in this area. Multiple large retrospective series have identified high rates of conversion to margin-negative resection with neoadjuvant chemoradiation alone.90 Based on available data, it is reasonable for patients with borderline resectable disease to proceed with any of the following treatment options: chemotherapy, chemoradiation, or induction chemotherapy followed by chemoradiation (Figure). Chemotherapy and chemoradiation are generally more appropriate with patients with high CA 19-9 levels or those at an elevated risk of having positive margins or occult metastatic disease.91 Obtaining negative margin resections is the predominant goal of neoadjuvant radiotherapy.89 Many studies have identified margin status to be one of the most significant prognostic factors in PDA.57,59,92,93 Additionally, several studies have highlighted that radiotherapy in the neoadjuvant setting could improve negative margin resection rates, local control, and clinical outcomes in patients with borderline resectable locally advanced disease.94–97 A common multimodal regimen utilized in the neoadjuvant setting combines capecitabine, an oral prodrug that is converted to fluorouracil, with radiation therapy. This combination has also been shown to improve resectability rates and long-term clinical outcomes in patients with borderline resectable disease.98 Additionally, neoadjuvant radiation therapy can potentially downstage patients with unresectable disease at the time of diagnosis to become surgical candidates.99 Despite the paucity of data, interval scans utilizing CT following neoadjuvant therapy should be obtained 2 to 4 months after completion of therapy to determine therapeutic response, evaluate for disease progression, and, most important, reassess surgical stage/resectability. It is clinically acceptable to proceed to resection with radiographically stable disease post-neoadjuvant therapy.
Many patients classified as borderline resectable are able to proceed with surgery following neoadjuvant therapy. Unfortunately, specific data on adjuvant therapy following neoadjuvant chemotherapy or chemoradiotherapy and surgical resection in borderline resectable patients is scarce. Clinical practice guidelines are extrapolated from studies where upfront resection in clearly resectable patients was followed by adjuvant therapy. Based on these data, approximately 6 months of perioperative chemotherapy with or without chemoradiotherapy is a reasonable consideration. Nevertheless, about 80% of patients at the time of diagnosis are deemed to be unresectable, and a smaller number do not proceed to surgery despite an initial classification as borderline resectable. Of the 80% of patients with advanced disease, about half are metastatic at presentation and the remaining 30% to 40% are defined as having unresectable LAPC.100
CASE CONTINUED
The patient is deemed borderline resectable. He receives neoadjuvant therapy with gemcitabine and nab-paclitaxel. Two months after therapy, interval imaging with abdominal CT does not show improvement in tumor size and there is now evidence that the tumor has invaded the celiac axis and is abutting more than 180° of the SMA. The patient presents to the oncologist to discuss further management. He has lost about 15 lb since his last evaluation, is capable of self-care, but is unable to carry on with any work activities.
What is the appropriate management of unresectable nonmetastatic LAPC?
UNRESECTABLE LOCALLY ADVANCED CANCER
As in the case of borderline resectable disease, there are many treatment options for patients with unresectable LAPC. Timing, optimal chemotherapy regimen, and the addition of regularly and hypofractionated radiotherapy are issues currently under investigation. However, there are some general considerations and principles that are followed as reflected in the NCCN guidelines and recent studies. The primary therapeutic aims in patients with unresectable locally advanced disease are to increase survival and improve palliation.
The elderly comprise a large percentage of the patients diagnosed with unresectable locally advanced disease. Pharmacokinetics and toxicity profiles are altered in the elderly population.101,102 Therefore, it is important to assess functional status and comorbidities as these are critical factors in determining treatment regimens, similar to patients with borderline resectable disease. Currently, the most common first-line therapies in advanced pancreatic cancer are gemcitabine alone, gemcitabine and nab-paclitaxel, FOLFIRINOX, gemcitabine/capecitabine, and gemcitabine/oxaliplatin.103 The overall treatment approach to unresectable locally advanced pancreatic adenocarcinoma closely mirrors that of patients with borderline resectable disease and metastatic disease. Much of the data supporting treatment regimens in unresectable LAPC is extrapolated from clinical trials looking at advanced or metastatic pancreatic cancer.
Consensus opinions domestically and from Europe recommend that patients with locally advanced unresectable disease undergo upfront chemotherapy (Figure).104 This is based on the premise that initial chemotherapy may destroy occult metastatic cells and increase the efficacy of consolidative chemotherapy, particularly with radiation in the future. Upfront chemoradiotherapy has only been investigated in a small series of trials in which no clear survival benefit was observed and has the added consequence of treatment-related toxicity.105 However, data is limited in this regard, with variations in treatment protocols and cohort compositions contributing to the inconclusive findings.
Despite advances in immunotherapy, targeted therapies, and gene sequencing, initial chemotherapy for unresectable disease is still either gemcitabine-based combination therapy or FOLFIRINOX. Across numerous studies, patients with unresectable LAPC receiving FOLFIRINOX have a median progression-free survival of 3 to 20 months and a median overall survival of 10 to 32.7 months.106 As with borderline resectable patients, FOLFIRINOX (Table 4) is generally reserved for unresectable patients with good functional status (ECOG 0–1 or Karnofsky Performance Status 90–100) and those at low risk for developing grade 3 or 4 systemic toxicities.103 For these reasons it has generally not been frequently combined with other chemotherapeutic agents. However, FOLFIRINOX has been combined with radiation therapy in the consolidative neoadjuvant setting after induction chemotherapy. There have also been studies where traditional FOLFIRONIX was modified to improve tolerability, as evidenced by Gunturu et al’s study, which dose-reduced both fluorouracil and irinotecan by 25%, without compromising efficacy and simultaneously increasing tolerability.107 Additionally, FOLFIRINOX requires infusional administration of the fluorouracil component, which may not be practical in certain patients. In that subset, capecitabine can be substituted. Among radiosensitizers during neoadjuvant therapy for unresectable LAPC, capecitabine has been shown to be as efficacious and less toxic than even gemcitabine.108
No head-to-head studies investigating FOLFIRINOX versus nab-paclitaxel and gemcitabine in patients with locally advanced disease have been published, but clinical trials are under way. Other combination therapies have been looked at through small retrospective or prospective studies, but no robust, large-scale clinical trials have been completed. For this reason, NCCN guidelines recommend enrollment of patients with LAPC into active clinical trials.
What is the role of radiation therapy in unresectable LAPC?
Despite the reported advantages of neoadjuvant radiation in patients with potentially resectable disease, there is significant debate regarding the timing and role of neoadjuvant radiation in patients with unresectable disease. Numerous comprehensive analyses and studiest indicate that chemoradiotherapy leads to significantly better overall survival compared to no therapy or radiation therapy alone in LAPC.68,110,111 However, conflicting data support the use of upfront chemoradiotherapy in unresectable LAPC when compared to chemotherapy alone. Unfortunately, most prospective studies investigating the role of radiotherapy were performed following administration of single-agent gemcitabine, which is no longer considered standard of care for patients with LAPC. In spite of this, ECOG 4201 identified a statistically significant improvement in median overall survival following the addition of gemcitabine-based radiotherapy. Huguet et al in his review pointed out that upfront chemoradiotherapy was not superior to chemotherapy only and was associated with increased treatment toxicity.105 Additionally, a recent phase 3 study looking at chemoradiotherapy versus chemotherapy alone in patients treated with gemcitabine found no difference in overall survival.112 This can potentially be attributed to the fact that about 30% of patients with LAPC develop metastatic disease in the early phases of treatment due to poor control of local and systemically occult disease.113 Given the propensity for high rates of occult metastatic disease in LAPC, treatment paradigms and consensus guidelines recommend multi-agent systemic chemotherapy followed by chemoradiotherapy in select patients.
Based on current studies and until further clinical investigations are completed, consensus opinion indicates that the most appropriate approach in unresectable LAPC is to begin with induction chemotherapy (with either gemcitabine plus nab-paclitaxel, FOLFIRINOX, capecitabine, or other treatment combinations), followed by chemoradiation in the absence of disease progression when the first repeat imaging evaluation is completed (Figure). One important caveat regarding reimaging with CT in the neoadjuvant setting is that radiologic response may not correlate with pathologic response.114 PET-CT may have a role in predicting response to neoadjuvant therapy. Overall, induction chemotherapy followed by consolidative chemoradiation may confer numerous benefits: it removes the unnecessary burden and toxicity associated with radiotherapy in the nearly one third of patients who have pervasive disease progression during initial treatment; it allows testing and increases the chances of tolerating full-dose systemic chemotherapy; and it raises the likelihood of converting patients who do not progress to metastasis during the initial phase of treatment from unresectable to resectable status.103,115 Despite the lack of strong conclusive data, the general agreement is that neoadjuvant chemoradiotherapy converts about one third of borderline and unresectable LAPC to an R0 resection.95,103 There are very specific rationales for the addition of radiotherapy in LAPC, and these functions need to be better defined through further clinical trials.
PALLIATIVE CARE
CASE CONTINUED
The patient is unable to tolerate his first round of second-line therapy with modified FOLFIRINOX. His overall treatment plan is transitioned to palliation. He continues to have pain, despite increasing doses of narcotics.
What is the next step for patients in whom second-line therapy fails and who have intractable pain while on high-dose narcotics?
A subset of patients with unresectable LAPC may not be amenable to chemotherapy with or without radiation due to significant comorbidities or because they present with or progress to ECOG scores 3 or 4. The goal in these patients should be palliation. Pain is one of the most predominant and difficult to manage symptoms in progressive LAPC. Opioid-based medications are the primary treatment for pain in LAPC. However, patients sometimes become refractory to opioid medications. In this group of patients, it is reasonable to consider palliative radiation as an alternative method for pain control.116
An alternative to palliative radiation in the setting of progressive pain in PDA is celiac plexus block or neurolysis. By injecting an anesthetic or alcohol into the celiac plexus, neural signaling pathways involved in the propagation of pain are inhibited without leading to significant nerve destruction. Additionally, chemical splanchnicectomy allows for reduced opioid medication use and associated side effects.117
In general patients with LAPC have profound weight loss prior to and during treatment. This has significant implications prognostically and on treatment options. The underlying etiology is multifactorial, but one of the primary driving factors is pancreatic insufficiency. An estimated 65% of pancreatic cancer patients have fat malabsorption, and 50% have protein malabsorption, leading to steatorrhea and weight loss.118 Patients diagnosed with pancreatic cancer should be given enzyme replacement with formulations that include lipase, amylase, and protease. A minimum dose of enzyme replacement should include 40,000 to 50,000 U of lipase during meals and 25,000 U during snack intake. If maldigestion, symptoms, or nutritional endpoints (BMI, albumin, prealbumin, cholesterol) do not improve, the pancreatic enzyme dose should be escalated and a proton-pump inhibitor (PPI) added. In patients with pancreatic insufficiency, PPIs have been shown to improve fat absorption.119 Enzyme replacement therapy has been shown to prevent weight loss in patients with unresectable pancreatic cancer.120
As most patients with LAPC go on to develop progressive disease, palliative care becomes an integral aspect of the therapeutic paradigm. Palliation in LAPC has a significant role in determining quality of life and ensuring patient’s goals of care have been meet. Studies have suggested that pancreatic cancer is second only to lung cancer in terms of the number of emergency department visits in the later stages of disease.120 Additionally, aggressive care in the setting of incurable diseases such as LAPC has been associated with poor quality of life.121 More recently it has been shown that involvement of palliative care in patients with advanced pancreatic is associated with less aggressive care near death.122 Therefore, the incorporation of palliative or supportive care teams in the treatment of patients with progressive LAPC can improve quality of life and alleviate suffering associated with increasing symptom burden.
CONCLUSION
LAPC is a difficult disease for both provider and patient. There is a paucity of robust clinical trials in the neoadjuvant setting for LAPC. Current research is complicated by varying consensus definitions of resectability and the varying treatment configurations across studies. The optimal type, timing, and sequence of treatment and whether to add radiation therapy in LAPC have not been clearly defined. However, based on the available studies and consensus guidelines, patients who are deemed to have LAPC should have neoadjuvant therapy. FOLFIRINOX or gemcitabine with nab-paclitaxel should be considered first-line treatments. Patients with LAPC who respond to chemotherapy or are ineligible for multi-drug chemotherapy may benefit from chemoradiotherapy. In patients with unresectable disease, chemoradiotherapy has been shown to enhance survival as compared to best supportive care or radiation alone. For borderline resectable disease, it is reasonable to treat patients with either chemoradiotherapy, chemotherapy alone, or chemotherapy followed by chemoradiotherapy.
Considering the invasive nature of LAPC and the controversy around neoadjuvant treatment protocols, enrollment of patients with LAPC into clinical trials is important and will help determine the optimal treatment regimen for future patients.
- Network PCA. Pancreatic cancer facts 2016. 2016. https://www.pancan.org/wp-content/uploads/2016/02/2016-GAA-PC-Facts.pdf. Accessed April 24, 2017.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
- Konstantinidis IT, Warshaw AL, Allen JN, et al. Pancreatic ductal adenocarcinoma: is there a survival difference for R1 resections versus locally advanced unresectable tumors? What is a “true” R0 resection? Ann Surg 2013;257:731–6.
- Ilic M, Ilic I. Epidemiology of pancreatic cancer. World J Gastroenterol 2016;22:9694–705.
- Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin 2011;61:69–90.
- Zhang J, Dhakal I, Ning B, Kesteloot H. Patterns and trends of pancreatic cancer mortality rates in Arkansas, 1969-2002: a comparison with the US population. Eur J Cancer Prev 2008;17:18–27.
- National Cancer Institute. SEER cancer statistics review, 1975-2013. http://seer.cancer.gov/csr/1975_2013/. Accessed April 24, 2017.
- Lowenfels AB, Maisonneuve P. Epidemiology and risk factors for pancreatic cancer. Best Pract Res Clin Gastroenterol 2006;20:197–209.
- Fuchs CS, Colditz GA, Stampfer MJ, et al. A prospective study of cigarette smoking and the risk of pancreatic cancer. Arch Intern Med 1996;156:2255–60.
- Lucenteforte E, La Vecchia C, Silverman D, et al. Alcohol consumption and pancreatic cancer: a pooled analysis in the International Pancreatic Cancer Case-Control Consortium (PanC4). Ann Oncol 2012;23:374–82.
- Schernhammer ES, Kang JH, Chan AT, et al. A prospective study of aspirin use and the risk of pancreatic cancer in women. J Natl Cancer Inst 2004;96:22–28.
- Michaud DS, Giovannucci E, Willett WC, et al. Physical activity, obesity, height, and the risk of pancreatic cancer. JAMA 2001;286:921–9.
- Nothlings U, Wilkens LR, Murphy SP, et al. Meat and fat intake as risk factors for pancreatic cancer: the multiethnic cohort study. J Natl Cancer Inst 2005;97:1458–65.
- Chari ST, Leibson CL, Rabe KG, et al. Probability of pancreatic cancer following diabetes: a population-based study. Gastroenterology 2005;129:504–11.
- Batabyal P, Vander Hoorn S, Christophi C, Nikfarjam M. Association of diabetes mellitus and pancreatic adenocarcinoma: a meta-analysis of 88 studies. Ann Surg Oncol 2014;21:2453–62.
- Chari ST, Leibson CL, Rabe KG, et al. Pancreatic cancer-associated diabetes mellitus: prevalence and temporal association with diagnosis of cancer. Gastroenterology 2008;134:95–101.
- Pannala R, Basu A, Petersen GM, Chari ST. New-onset diabetes: a potential clue to the early diagnosis of pancreatic cancer. Lancet Oncol 2009;10:88–95.
- Wolpin BM, Chan AT, Hartge P, et al. ABO blood group and the risk of pancreatic cancer. J Natl Cancer Inst 2009;101:424–31.
- Iqbal J, Ragone A, Lubinski J, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer 2012;107:2005–9.
- Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000;119:1447–53.
- Kastrinos F, Mukherjee B, Tayob N, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA 2009;302:1790–5.
- Brand RE, Lynch HT. Hereditary pancreatic adenocarcinoma. A clinical perspective. Med Clin North Am 2000;84:665–75.
- Jacobs EJ, Chanock SJ, Fuchs CS, et al. Family history of cancer and risk of pancreatic cancer: a pooled analysis from the Pancreatic Cancer Cohort Consortium (PanScan). Int J Cancer 2010;127:1421–8.
- Rustgi AK. Familial pancreatic cancer: genetic advances. Genes Dev 2014;28:1–7.
- Reznik R, Hendifar AE, Tuli R. Genetic determinants and potential therapeutic targets for pancreatic adenocarcinoma. Front Physiol 2014;5:87.
- Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst 1997;89:442–6.
- Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med 2014;371:2140–1.
- Kanda M, Matthaei H, Wu J, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012;142:730–3.
- Feldmann G, Maitra A. Molecular genetics of pancreatic ductal adenocarcinomas and recent implications for translational efforts. J Mol Diagn 2008;10:111–22.
- Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer 2014;111:817–22.
- Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic KRAS maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012;149:656–70.
- Provenzano PP, Cuevas C, Chang AE, et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012;21:418–29.
- Vonderheide RH, Bayne LJ. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr Opin Immunol 2013;25:200–5.
- DiMagno EP. Pancreatic cancer: Clinical presentation, pitfalls and early clues. Ann Oncol 1999;10(suppl 4):S140–S142.
- Porta M, Fabregat X, Malats N, et al. Exocrine pancreatic cancer: symptoms at presentation and their relation to tumour site and stage. Clin Transl Oncol 2005;7:189–97.
- Gullo L, Tomassetti P, Migliori M, et al. Do early symptoms of pancreatic cancer exist that can allow an earlier diagnosis? Pancreas 2001;22:210–3.
- Khorana AA, Fine RL. Pancreatic cancer and thromboembolic disease. Lancet Oncol 2004;5(11):655-663.
- Wigmore SJ, Plester CE, Richardson RA, Fearon KC. Changes in nutritional status associated with unresectable pancreatic cancer. Br J Cancer 1997;75:106–9.
- Aggarwal G, Rabe KG, Petersen GM, Chari ST. New-onset diabetes in pancreatic cancer: a study in the primary care setting. Pancreatology 2012;12:156–61.
- Koprowski H, Herlyn M, Steplewski Z, Sears HF. Specific antigen in serum of patients with colon carcinoma. Science 1981;212:53–5.
- Bond-Smith G, Banga N, Hammond TM, Imber CJ. Pancreatic adenocarcinoma. BMJ 2012;344:e2476.
- Cwik G, Wallner G, Skoczylas T, et al. Cancer antigens 19-9 and 125 in the differential diagnosis of pancreatic mass lesions. Arch Surg 2006;141:968–73.
- van den Bosch RP, van Eijck CH, Mulder PG, Jeekel J. Serum CA19-9 determination in the management of pancreatic cancer. Hepatogastroenterology 1996;43:710–3.
- Lamerz R. Role of tumour markers, cytogenetics. Ann Oncol 1999;10 Suppl 4:145–9.
- Hess V, Glimelius B, Grawe P, et al. CA 19-9 tumour-marker response to chemotherapy in patients with advanced pancreatic cancer enrolled in a randomised controlled trial. Lancet Oncol 2008;9:132–8.
- Montgomery RC, Hoffman JP, Riley LB, et al. Prediction of recurrence and survival by post-resection CA 19-9 values in patients with adenocarcinoma of the pancreas. Ann Surg Oncol 1997;4:551–6.
- Zamboni GA, D’Onofrio M, Idili A, et al. Ultrasound-guided percutaneous fine-needle aspiration of 545 focal pancreatic lesions. AJR Am J Roentgenol 2009;193:1691–5.
- Micames C, Jowell PS, White R, et al. Lower frequency of peritoneal carcinomatosis in patients with pancreatic cancer diagnosed by EUS-guided FNA vs. percutaneous FNA. Gastrointest Endosc 2003;58:690–5.
- Brambs HJ, Claussen CD. Pancreatic and ampullary carcinoma. Ultrasound, computed tomography, magnetic resonance imaging and angiography. Endoscopy 1993;25:58–68.
- Karlson BM, Ekbom A, Lindgren PG, et al. Abdominal US for diagnosis of pancreatic tumor: prospective cohort analysis. Radiology 1999;213:107–11.
- Imbriaco M, Megibow AJ, Camera L, et al. Dual-phase versus single-phase helical CT to detect and assess resectability of pancreatic carcinoma. AJR Am J Roentgenol 2002;178:1473–9.
- Schrag D. Optimizing treatment for locally advanced pancreas cancer: progress but no precision. JAMA 2016;315:1837–8.
- House MG, Yeo CJ, Cameron JL, et al. Predicting resectability of periampullary cancer with three-dimensional computed tomography. J Gastrointest Surg 2004;8:280–8.
- Callery MP, Chang KJ, Fishman EK, et al. Pretreatment assessment of resectable and borderline resectable pancreatic cancer: expert consensus statement. Ann Surg Oncol 2009;16:1727–33.
- Horton KM, Fishman EK. Adenocarcinoma of the pancreas: CT imaging. Radiol Clin North Am 2002;40:1263–72.
- Ross WA, Wasan SM, Evans DB, et al. Combined EUS with FNA and ERCP for the evaluation of patients with obstructive jaundice from presumed pancreatic malignancy. Gastrointest Endosc 2008;68:461–6.
- Hartwig W, Hackert T, Hinz U, et al. Pancreatic cancer surgery in the new millennium: better prediction of outcome. Ann Surg 2011;254:311–9.
- Jamieson NB, Chan NI, Foulis AK, et al. The prognostic influence of resection margin clearance following pancreaticoduodenectomy for pancreatic ductal adenocarcinoma. J Gastrointest Surg 2013;17:511–21.
- Ethun CG, Kooby DA. The importance of surgical margins in pancreatic cancer. J Surg Oncol 2016;113:283–8.
- Fischer R, Breidert M, Keck T, et al. Early recurrence of pancreatic cancer after resection and during adjuvant chemotherapy. Saudi J Gastroenterol 2012;18:118–21.
- Neoptolemos JP, Stocken DD, Friess H, et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med 2004;350:1200–10.
- Neoptolemos JP, Palmer DH, Ghaneh P, et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet 2017;389(10073):1011–24.
- Cardenes HR, Chiorean EG, Dewitt J, et al. Locally advanced pancreatic cancer: current therapeutic approach. Oncologist 2006;11:612–23.
- Yeung RS, Weese JL, Hoffman JP, et al. Neoadjuvant chemoradiation in pancreatic and duodenal carcinoma. A Phase II Study. Cancer 1993;72:2124–33.
- Spitz FR, Abbruzzese JL, Lee JE, et al. Preoperative and postoperative chemoradiation strategies in patients treated with pancreaticoduodenectomy for adenocarcinoma of the pancreas. J Clin Oncol 1997;15:928–37.
- McClaine RJ, Lowy AM, Sussman JJ, et al. Neoadjuvant therapy may lead to successful surgical resection and improved survival in patients with borderline resectable pancreatic cancer. HPB (Oxford) 2010;12:73–9.
- Balaban EP, Mangu PB, Khorana AA, et al. Locally advanced, unresectable pancreatic cCancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2016;34:2654–68.
- Shaib WL, Ip A, Cardona K, et al. Contemporary management of borderline resectable and locally advanced unresectable pancreatic cancer. Oncologist 2016;21:178–87.
- Chun YS, Milestone BN, Watson JC, et al. Defining venous involvement in borderline resectable pancreatic cancer. Ann Surg Oncol 2010;17:2832–8.
- Evans DB, Erickson BA, Ritch P. Borderline resectable pancreatic cancer: definitions and the importance of multimodality therapy. Ann Surg Oncol 2010;17:2803–5.
- Conroy T, Paillot B, Francois E, et al. Irinotecan plus oxaliplatin and leucovorin-modulated fluorouracil in advanced pancreatic cancer--a Groupe Tumeurs Digestives of the Federation Nationale des Centres de Lutte Contre le Cancer study. J Clin Oncol 2005;23:1228–36.
- Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
- Hosein PJ, Macintyre J, Kawamura C, et al. A retrospective study of neoadjuvant FOLFIRINOX in unresectable or borderline-resectable locally advanced pancreatic adenocarcinoma. BMC Cancer 2012;12:199.
- Peddi PF, Lubner S, McWilliams R, et al. Multi-institutional experience with FOLFIRINOX in pancreatic adenocarcinoma. JOP 2012;13:497–501.
- Mahaseth H, Kauh JS, Brutcher E, et al. Safety and efficacy of modified FOLFIRINOX in pancreatic cancer: A retrospective experience. J Clin Oncol 2012;30 (suppl; abstr e14614).
- Kim SS, Nakakura EK, Wang ZJ, et al. Preoperative FOLFIRINOX for borderline resectable pancreatic cancer: Is radiation necessary in the modern era of chemotherapy? J Surg Oncol 2016;114:587–96.
- Conroy T, Gavoille C, Samalin E, et al. The role of the FOLFIRINOX regimen for advanced pancreatic cancer. Curr Oncol Rep 2013;15:182–9.
- Katz MH, Shi Q, Ahmad SA, et al. Preoperative modified FOLFIRINOX treatment followed by capecitabine-based chemoradiation for borderline resectable pancreatic cancer: Alliance for Clinical Trials in Oncology Trial A021101. JAMA Surg 2016;151:e161137.
- Kharofa J, Kelly TR, Ritch PS, et al. 5-FU/leucovorin, irinotecan, oxaliplatin (FOLFIRINOX) induction followed by chemoXRT in borderline resectable pancreatic cancer. J Clin Oncol 2012;30 (suppl; abstr e14613).
- Blazer M, Wu C, Goldberg RM, et al. Neoadjuvant modified (m) FOLFIRINOX for locally advanced unresectable (LAPC) and borderline resectable (BRPC) adenocarcinoma of the pancreas. Ann Surg Oncol 2015;22:1153–9.
- Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
- Plunkett W, Huang P, Xu YZ, et al. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol 1995;22(4 Suppl 11):3–10.
- Sahora K, Kuehrer I, Eisenhut A, et al. NeoGemOx: Gemcitabine and oxaliplatin as neoadjuvant treatment for locally advanced, nonmetastasized pancreatic cancer. Surgery 2011;149(3):311–20.
- Lee JL, Kim SC, Kim JH, et al. Prospective efficacy and safety study of neoadjuvant gemcitabine with capecitabine combination chemotherapy for borderline-resectable or unresectable locally advanced pancreatic adenocarcinoma. Surgery 2012;152:851–62.
- Leone F, Gatti M, Massucco P, et al. Induction gemcitabine and oxaliplatin therapy followed by a twice-weekly infusion of gemcitabine and concurrent external-beam radiation for neoadjuvant treatment of locally advanced pancreatic cancer: a single institutional experience. Cancer 2013;119:277–84.
- Lawrence TS, Eisbruch A, Shewach DS. Gemcitabine-mediated radiosensitization. Semin Oncol 1997;24(2 Suppl 7):S7–24-S27–28.
- Kang CM, Chung YE, Park JY, et al. Potential contribution of preoperative neoadjuvant concurrent chemoradiation therapy on margin-negative resection in borderline resectable pancreatic cancer. J Gastrointest Surg 2012;16:509–17.
- Chuong MD, Hayman TJ, Patel MR, et al. Comparison of 1-, 2-, and 3-dimensional tumor response assessment after neoadjuvant GTX-RT in borderline-resectable pancreatic cancer. Gastrointest Cancer Res 2011;4:128–34.
- Loehrer AP, Kinnier CV, Ferrone CR. Treatment of locally advanced pancreatic ductal adenocarcinoma. Adv Surg 2016;50:115–28.
- Katz MH, Wang H, Balachandran A, et al. Effect of neoadjuvant chemoradiation and surgical technique on recurrence of localized pancreatic cancer. J Gastrointest Surg 2012;16:68–78.
- Franke AJ, Rosati LM, Pawlik TM, et al. The role of radiation therapy in pancreatic ductal adenocarcinoma in the neoadjuvant and adjuvant settings. Semin Oncol 2015;42:144–62.
- Butturini G, Stocken DD, Wente MN, et al. Influence of resection margins and treatment on survival in patients with pancreatic cancer: meta-analysis of randomized controlled trials. Arch Surg 2008;143:75–83.
- Paniccia A, Hosokawa P, Henderson W, et al. Characteristics of 10-year survivors of pancreatic ductal adenocarcinoma. JAMA Surg 2015;150:701–10.
- Massucco P, Capussotti L, Magnino A, et al. Pancreatic resections after chemoradiotherapy for locally advanced ductal adenocarcinoma: analysis of perioperative outcome and survival. Ann Surg Oncol 2006;13:1201–8.
- Patel M, Hoffe S, Malafa M, et al. Neoadjuvant GTX chemotherapy and IMRT-based chemoradiation for borderline resectable pancreatic cancer. J Surg Oncol 2011;104:155–161.
- Landry J, Catalano PJ, Staley C, et al. Randomized phase II study of gemcitabine plus radiotherapy versus gemcitabine, 5-fluorouracil, and cisplatin followed by radiotherapy and 5-fluorouracil for patients with locally advanced, potentially resectable pancreatic adenocarcinoma. J Surg Oncol 2010;101:587–92.
- Lamb R, Ozsvari B, Lisanti CL, et al. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget 2015;6:4569–84.
- Stokes JB, Nolan NJ, Stelow EB, et al. Preoperative capecitabine and concurrent radiation for borderline resectable pancreatic cancer. Ann Surg Oncol 2011;18:619–27.
- White R, Lee C, Anscher M, et al. Preoperative chemoradiation for patients with locally advanced adenocarcinoma of the pancreas. Ann Surg Oncol 1999;6:38–45.
- Martin RC 2nd. Management of locally advanced pancreatic cancer. Surg Clin North Am 2016;96:1371–89.
- Higuera O, Ghanem I, Nasimi R, et al. Management of pancreatic cancer in the elderly. World J Gastroenterol 2016;22:764–75.
- Hurria A, Lichtman SM. Clinical pharmacology of cancer therapies in older adults. Br J Cancer 2008;98:517–22.
- Spadi R, Brusa F, Ponzetti A, et al. Current therapeutic strategies for advanced pancreatic cancer: A review for clinicians. World J Clin Oncol 2016;7:27–43.
- Seufferlein T, Bachet JB, Van Cutsem E, Rougier P, Group EGW. Pancreatic adenocarcinoma: ESMO-ESDO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012;23 Suppl 7:vii33–40.
- Huguet F, Girard N, Guerche CS, et al. Chemoradiotherapy in the management of locally advanced pancreatic carcinoma: a qualitative systematic review. J Clin Oncol 2009;27:2269–77.
- Suker M, Beumer BR, Sadot E, et al. FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis. Lancet Oncol 2016;17:801–10.
- Gunturu KS, Yao X, Cong X, et al. FOLFIRINOX for locally advanced and metastatic pancreatic cancer: single institution retrospective review of efficacy and toxicity. Med Oncol 2013;30:361.
- Mukherjee S, Hurt CN, Bridgewater J, et al. Gemcitabine-based or capecitabine-based chemoradiotherapy for locally advanced pancreatic cancer (SCALOP): a multicentre, randomised, phase 2 trial. Lancet Oncol 2013;14:317–26.
- Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–1703.
- Krzyzanowska MK, Weeks JC, Earle CC. Treatment of locally advanced pancreatic cancer in the real world: population-based practices and effectiveness. J Clin Oncol 2003;21:3409–14.
- Sultana A, Tudur Smith C, Cunningham D, et al. Meta-analyses of chemotherapy for locally advanced and metastatic pancreatic cancer: results of secondary end points analyses. Br J Cancer 2008;99:6–13.
- Hammel P, Huguet F, van Laethem JL, et al. Effect of chemoradiotherapy vs chemotherapy on survival in patients with locally advanced pancreatic cancer controlled after 4 months of gemcitabine with or without erlotinib: the LAP07 randomized clinical trial. JAMA 2016;315:1844–53.
- Huguet F, Andre T, Hammel P, et al. Impact of chemoradiotherapy after disease control with chemotherapy in locally advanced pancreatic adenocarcinoma in GERCOR phase II and III studies. J Clin Oncol 2007;25:326–31.
- Dholakia AS, Hacker-Prietz A, Wild AT, et al. Resection of borderline resectable pancreatic cancer after neoadjuvant chemoradiation does not depend on improved radiographic appearance of tumor-vessel relationships. J Radiat Oncol 2013;2:413–25.
- Heinemann V, Haas M, Boeck S. Neoadjuvant treatment of borderline resectable and non-resectable pancreatic cancer. Ann Oncol 2013;24:2484–92.
- Morganti AG, Trodella L, Valentini V, et al. Pain relief with short-term irradiation in locally advanced carcinoma of the pancreas. J Palliat Care 2003;19:258–62.
- Arcidiacono PG, Calori G, Carrara S, et al. Celiac plexus block for pancreatic cancer pain in adults. Cochrane Database Syst Rev 2011(3):CD007519.
- Pezzilli R, Andriulli A, Bassi C, et al. Exocrine pancreatic insufficiency in adults: a shared position statement of the Italian Association for the Study of the Pancreas. World J Gastroenterol 2013;19:7930–46.
- Dominguez-Munoz JE. Pancreatic exocrine insufficiency: diagnosis and treatment. J Gastroenterol Hepatol 2011;26 Suppl 2:12–16.
- Bruno MJ, Haverkort EB, Tijssen GP, et al. Placebo controlled trial of enteric coated pancreatin microsphere treatment in patients with unresectable cancer of the pancreatic head region. Gut 1998;42:92–6.
- Wright AA, Keating NL, Balboni TA, et al. Place of death: correlations with quality of life of patients with cancer and predictors of bereaved caregivers’ mental health. J Clin Oncol 2010;28:4457–64.
- Jang RW, Krzyzanowska MK, Zimmermann C, et al. Palliative care and the aggressiveness of end-of-life care in patients with advanced pancreatic cancer. J Natl Cancer Inst 2015;107(3). pii: dju424.
- Network PCA. Pancreatic cancer facts 2016. 2016. https://www.pancan.org/wp-content/uploads/2016/02/2016-GAA-PC-Facts.pdf. Accessed April 24, 2017.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
- Konstantinidis IT, Warshaw AL, Allen JN, et al. Pancreatic ductal adenocarcinoma: is there a survival difference for R1 resections versus locally advanced unresectable tumors? What is a “true” R0 resection? Ann Surg 2013;257:731–6.
- Ilic M, Ilic I. Epidemiology of pancreatic cancer. World J Gastroenterol 2016;22:9694–705.
- Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin 2011;61:69–90.
- Zhang J, Dhakal I, Ning B, Kesteloot H. Patterns and trends of pancreatic cancer mortality rates in Arkansas, 1969-2002: a comparison with the US population. Eur J Cancer Prev 2008;17:18–27.
- National Cancer Institute. SEER cancer statistics review, 1975-2013. http://seer.cancer.gov/csr/1975_2013/. Accessed April 24, 2017.
- Lowenfels AB, Maisonneuve P. Epidemiology and risk factors for pancreatic cancer. Best Pract Res Clin Gastroenterol 2006;20:197–209.
- Fuchs CS, Colditz GA, Stampfer MJ, et al. A prospective study of cigarette smoking and the risk of pancreatic cancer. Arch Intern Med 1996;156:2255–60.
- Lucenteforte E, La Vecchia C, Silverman D, et al. Alcohol consumption and pancreatic cancer: a pooled analysis in the International Pancreatic Cancer Case-Control Consortium (PanC4). Ann Oncol 2012;23:374–82.
- Schernhammer ES, Kang JH, Chan AT, et al. A prospective study of aspirin use and the risk of pancreatic cancer in women. J Natl Cancer Inst 2004;96:22–28.
- Michaud DS, Giovannucci E, Willett WC, et al. Physical activity, obesity, height, and the risk of pancreatic cancer. JAMA 2001;286:921–9.
- Nothlings U, Wilkens LR, Murphy SP, et al. Meat and fat intake as risk factors for pancreatic cancer: the multiethnic cohort study. J Natl Cancer Inst 2005;97:1458–65.
- Chari ST, Leibson CL, Rabe KG, et al. Probability of pancreatic cancer following diabetes: a population-based study. Gastroenterology 2005;129:504–11.
- Batabyal P, Vander Hoorn S, Christophi C, Nikfarjam M. Association of diabetes mellitus and pancreatic adenocarcinoma: a meta-analysis of 88 studies. Ann Surg Oncol 2014;21:2453–62.
- Chari ST, Leibson CL, Rabe KG, et al. Pancreatic cancer-associated diabetes mellitus: prevalence and temporal association with diagnosis of cancer. Gastroenterology 2008;134:95–101.
- Pannala R, Basu A, Petersen GM, Chari ST. New-onset diabetes: a potential clue to the early diagnosis of pancreatic cancer. Lancet Oncol 2009;10:88–95.
- Wolpin BM, Chan AT, Hartge P, et al. ABO blood group and the risk of pancreatic cancer. J Natl Cancer Inst 2009;101:424–31.
- Iqbal J, Ragone A, Lubinski J, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer 2012;107:2005–9.
- Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000;119:1447–53.
- Kastrinos F, Mukherjee B, Tayob N, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA 2009;302:1790–5.
- Brand RE, Lynch HT. Hereditary pancreatic adenocarcinoma. A clinical perspective. Med Clin North Am 2000;84:665–75.
- Jacobs EJ, Chanock SJ, Fuchs CS, et al. Family history of cancer and risk of pancreatic cancer: a pooled analysis from the Pancreatic Cancer Cohort Consortium (PanScan). Int J Cancer 2010;127:1421–8.
- Rustgi AK. Familial pancreatic cancer: genetic advances. Genes Dev 2014;28:1–7.
- Reznik R, Hendifar AE, Tuli R. Genetic determinants and potential therapeutic targets for pancreatic adenocarcinoma. Front Physiol 2014;5:87.
- Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst 1997;89:442–6.
- Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med 2014;371:2140–1.
- Kanda M, Matthaei H, Wu J, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012;142:730–3.
- Feldmann G, Maitra A. Molecular genetics of pancreatic ductal adenocarcinomas and recent implications for translational efforts. J Mol Diagn 2008;10:111–22.
- Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer 2014;111:817–22.
- Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic KRAS maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012;149:656–70.
- Provenzano PP, Cuevas C, Chang AE, et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012;21:418–29.
- Vonderheide RH, Bayne LJ. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr Opin Immunol 2013;25:200–5.
- DiMagno EP. Pancreatic cancer: Clinical presentation, pitfalls and early clues. Ann Oncol 1999;10(suppl 4):S140–S142.
- Porta M, Fabregat X, Malats N, et al. Exocrine pancreatic cancer: symptoms at presentation and their relation to tumour site and stage. Clin Transl Oncol 2005;7:189–97.
- Gullo L, Tomassetti P, Migliori M, et al. Do early symptoms of pancreatic cancer exist that can allow an earlier diagnosis? Pancreas 2001;22:210–3.
- Khorana AA, Fine RL. Pancreatic cancer and thromboembolic disease. Lancet Oncol 2004;5(11):655-663.
- Wigmore SJ, Plester CE, Richardson RA, Fearon KC. Changes in nutritional status associated with unresectable pancreatic cancer. Br J Cancer 1997;75:106–9.
- Aggarwal G, Rabe KG, Petersen GM, Chari ST. New-onset diabetes in pancreatic cancer: a study in the primary care setting. Pancreatology 2012;12:156–61.
- Koprowski H, Herlyn M, Steplewski Z, Sears HF. Specific antigen in serum of patients with colon carcinoma. Science 1981;212:53–5.
- Bond-Smith G, Banga N, Hammond TM, Imber CJ. Pancreatic adenocarcinoma. BMJ 2012;344:e2476.
- Cwik G, Wallner G, Skoczylas T, et al. Cancer antigens 19-9 and 125 in the differential diagnosis of pancreatic mass lesions. Arch Surg 2006;141:968–73.
- van den Bosch RP, van Eijck CH, Mulder PG, Jeekel J. Serum CA19-9 determination in the management of pancreatic cancer. Hepatogastroenterology 1996;43:710–3.
- Lamerz R. Role of tumour markers, cytogenetics. Ann Oncol 1999;10 Suppl 4:145–9.
- Hess V, Glimelius B, Grawe P, et al. CA 19-9 tumour-marker response to chemotherapy in patients with advanced pancreatic cancer enrolled in a randomised controlled trial. Lancet Oncol 2008;9:132–8.
- Montgomery RC, Hoffman JP, Riley LB, et al. Prediction of recurrence and survival by post-resection CA 19-9 values in patients with adenocarcinoma of the pancreas. Ann Surg Oncol 1997;4:551–6.
- Zamboni GA, D’Onofrio M, Idili A, et al. Ultrasound-guided percutaneous fine-needle aspiration of 545 focal pancreatic lesions. AJR Am J Roentgenol 2009;193:1691–5.
- Micames C, Jowell PS, White R, et al. Lower frequency of peritoneal carcinomatosis in patients with pancreatic cancer diagnosed by EUS-guided FNA vs. percutaneous FNA. Gastrointest Endosc 2003;58:690–5.
- Brambs HJ, Claussen CD. Pancreatic and ampullary carcinoma. Ultrasound, computed tomography, magnetic resonance imaging and angiography. Endoscopy 1993;25:58–68.
- Karlson BM, Ekbom A, Lindgren PG, et al. Abdominal US for diagnosis of pancreatic tumor: prospective cohort analysis. Radiology 1999;213:107–11.
- Imbriaco M, Megibow AJ, Camera L, et al. Dual-phase versus single-phase helical CT to detect and assess resectability of pancreatic carcinoma. AJR Am J Roentgenol 2002;178:1473–9.
- Schrag D. Optimizing treatment for locally advanced pancreas cancer: progress but no precision. JAMA 2016;315:1837–8.
- House MG, Yeo CJ, Cameron JL, et al. Predicting resectability of periampullary cancer with three-dimensional computed tomography. J Gastrointest Surg 2004;8:280–8.
- Callery MP, Chang KJ, Fishman EK, et al. Pretreatment assessment of resectable and borderline resectable pancreatic cancer: expert consensus statement. Ann Surg Oncol 2009;16:1727–33.
- Horton KM, Fishman EK. Adenocarcinoma of the pancreas: CT imaging. Radiol Clin North Am 2002;40:1263–72.
- Ross WA, Wasan SM, Evans DB, et al. Combined EUS with FNA and ERCP for the evaluation of patients with obstructive jaundice from presumed pancreatic malignancy. Gastrointest Endosc 2008;68:461–6.
- Hartwig W, Hackert T, Hinz U, et al. Pancreatic cancer surgery in the new millennium: better prediction of outcome. Ann Surg 2011;254:311–9.
- Jamieson NB, Chan NI, Foulis AK, et al. The prognostic influence of resection margin clearance following pancreaticoduodenectomy for pancreatic ductal adenocarcinoma. J Gastrointest Surg 2013;17:511–21.
- Ethun CG, Kooby DA. The importance of surgical margins in pancreatic cancer. J Surg Oncol 2016;113:283–8.
- Fischer R, Breidert M, Keck T, et al. Early recurrence of pancreatic cancer after resection and during adjuvant chemotherapy. Saudi J Gastroenterol 2012;18:118–21.
- Neoptolemos JP, Stocken DD, Friess H, et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med 2004;350:1200–10.
- Neoptolemos JP, Palmer DH, Ghaneh P, et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet 2017;389(10073):1011–24.
- Cardenes HR, Chiorean EG, Dewitt J, et al. Locally advanced pancreatic cancer: current therapeutic approach. Oncologist 2006;11:612–23.
- Yeung RS, Weese JL, Hoffman JP, et al. Neoadjuvant chemoradiation in pancreatic and duodenal carcinoma. A Phase II Study. Cancer 1993;72:2124–33.
- Spitz FR, Abbruzzese JL, Lee JE, et al. Preoperative and postoperative chemoradiation strategies in patients treated with pancreaticoduodenectomy for adenocarcinoma of the pancreas. J Clin Oncol 1997;15:928–37.
- McClaine RJ, Lowy AM, Sussman JJ, et al. Neoadjuvant therapy may lead to successful surgical resection and improved survival in patients with borderline resectable pancreatic cancer. HPB (Oxford) 2010;12:73–9.
- Balaban EP, Mangu PB, Khorana AA, et al. Locally advanced, unresectable pancreatic cCancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2016;34:2654–68.
- Shaib WL, Ip A, Cardona K, et al. Contemporary management of borderline resectable and locally advanced unresectable pancreatic cancer. Oncologist 2016;21:178–87.
- Chun YS, Milestone BN, Watson JC, et al. Defining venous involvement in borderline resectable pancreatic cancer. Ann Surg Oncol 2010;17:2832–8.
- Evans DB, Erickson BA, Ritch P. Borderline resectable pancreatic cancer: definitions and the importance of multimodality therapy. Ann Surg Oncol 2010;17:2803–5.
- Conroy T, Paillot B, Francois E, et al. Irinotecan plus oxaliplatin and leucovorin-modulated fluorouracil in advanced pancreatic cancer--a Groupe Tumeurs Digestives of the Federation Nationale des Centres de Lutte Contre le Cancer study. J Clin Oncol 2005;23:1228–36.
- Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
- Hosein PJ, Macintyre J, Kawamura C, et al. A retrospective study of neoadjuvant FOLFIRINOX in unresectable or borderline-resectable locally advanced pancreatic adenocarcinoma. BMC Cancer 2012;12:199.
- Peddi PF, Lubner S, McWilliams R, et al. Multi-institutional experience with FOLFIRINOX in pancreatic adenocarcinoma. JOP 2012;13:497–501.
- Mahaseth H, Kauh JS, Brutcher E, et al. Safety and efficacy of modified FOLFIRINOX in pancreatic cancer: A retrospective experience. J Clin Oncol 2012;30 (suppl; abstr e14614).
- Kim SS, Nakakura EK, Wang ZJ, et al. Preoperative FOLFIRINOX for borderline resectable pancreatic cancer: Is radiation necessary in the modern era of chemotherapy? J Surg Oncol 2016;114:587–96.
- Conroy T, Gavoille C, Samalin E, et al. The role of the FOLFIRINOX regimen for advanced pancreatic cancer. Curr Oncol Rep 2013;15:182–9.
- Katz MH, Shi Q, Ahmad SA, et al. Preoperative modified FOLFIRINOX treatment followed by capecitabine-based chemoradiation for borderline resectable pancreatic cancer: Alliance for Clinical Trials in Oncology Trial A021101. JAMA Surg 2016;151:e161137.
- Kharofa J, Kelly TR, Ritch PS, et al. 5-FU/leucovorin, irinotecan, oxaliplatin (FOLFIRINOX) induction followed by chemoXRT in borderline resectable pancreatic cancer. J Clin Oncol 2012;30 (suppl; abstr e14613).
- Blazer M, Wu C, Goldberg RM, et al. Neoadjuvant modified (m) FOLFIRINOX for locally advanced unresectable (LAPC) and borderline resectable (BRPC) adenocarcinoma of the pancreas. Ann Surg Oncol 2015;22:1153–9.
- Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
- Plunkett W, Huang P, Xu YZ, et al. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol 1995;22(4 Suppl 11):3–10.
- Sahora K, Kuehrer I, Eisenhut A, et al. NeoGemOx: Gemcitabine and oxaliplatin as neoadjuvant treatment for locally advanced, nonmetastasized pancreatic cancer. Surgery 2011;149(3):311–20.
- Lee JL, Kim SC, Kim JH, et al. Prospective efficacy and safety study of neoadjuvant gemcitabine with capecitabine combination chemotherapy for borderline-resectable or unresectable locally advanced pancreatic adenocarcinoma. Surgery 2012;152:851–62.
- Leone F, Gatti M, Massucco P, et al. Induction gemcitabine and oxaliplatin therapy followed by a twice-weekly infusion of gemcitabine and concurrent external-beam radiation for neoadjuvant treatment of locally advanced pancreatic cancer: a single institutional experience. Cancer 2013;119:277–84.
- Lawrence TS, Eisbruch A, Shewach DS. Gemcitabine-mediated radiosensitization. Semin Oncol 1997;24(2 Suppl 7):S7–24-S27–28.
- Kang CM, Chung YE, Park JY, et al. Potential contribution of preoperative neoadjuvant concurrent chemoradiation therapy on margin-negative resection in borderline resectable pancreatic cancer. J Gastrointest Surg 2012;16:509–17.
- Chuong MD, Hayman TJ, Patel MR, et al. Comparison of 1-, 2-, and 3-dimensional tumor response assessment after neoadjuvant GTX-RT in borderline-resectable pancreatic cancer. Gastrointest Cancer Res 2011;4:128–34.
- Loehrer AP, Kinnier CV, Ferrone CR. Treatment of locally advanced pancreatic ductal adenocarcinoma. Adv Surg 2016;50:115–28.
- Katz MH, Wang H, Balachandran A, et al. Effect of neoadjuvant chemoradiation and surgical technique on recurrence of localized pancreatic cancer. J Gastrointest Surg 2012;16:68–78.
- Franke AJ, Rosati LM, Pawlik TM, et al. The role of radiation therapy in pancreatic ductal adenocarcinoma in the neoadjuvant and adjuvant settings. Semin Oncol 2015;42:144–62.
- Butturini G, Stocken DD, Wente MN, et al. Influence of resection margins and treatment on survival in patients with pancreatic cancer: meta-analysis of randomized controlled trials. Arch Surg 2008;143:75–83.
- Paniccia A, Hosokawa P, Henderson W, et al. Characteristics of 10-year survivors of pancreatic ductal adenocarcinoma. JAMA Surg 2015;150:701–10.
- Massucco P, Capussotti L, Magnino A, et al. Pancreatic resections after chemoradiotherapy for locally advanced ductal adenocarcinoma: analysis of perioperative outcome and survival. Ann Surg Oncol 2006;13:1201–8.
- Patel M, Hoffe S, Malafa M, et al. Neoadjuvant GTX chemotherapy and IMRT-based chemoradiation for borderline resectable pancreatic cancer. J Surg Oncol 2011;104:155–161.
- Landry J, Catalano PJ, Staley C, et al. Randomized phase II study of gemcitabine plus radiotherapy versus gemcitabine, 5-fluorouracil, and cisplatin followed by radiotherapy and 5-fluorouracil for patients with locally advanced, potentially resectable pancreatic adenocarcinoma. J Surg Oncol 2010;101:587–92.
- Lamb R, Ozsvari B, Lisanti CL, et al. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget 2015;6:4569–84.
- Stokes JB, Nolan NJ, Stelow EB, et al. Preoperative capecitabine and concurrent radiation for borderline resectable pancreatic cancer. Ann Surg Oncol 2011;18:619–27.
- White R, Lee C, Anscher M, et al. Preoperative chemoradiation for patients with locally advanced adenocarcinoma of the pancreas. Ann Surg Oncol 1999;6:38–45.
- Martin RC 2nd. Management of locally advanced pancreatic cancer. Surg Clin North Am 2016;96:1371–89.
- Higuera O, Ghanem I, Nasimi R, et al. Management of pancreatic cancer in the elderly. World J Gastroenterol 2016;22:764–75.
- Hurria A, Lichtman SM. Clinical pharmacology of cancer therapies in older adults. Br J Cancer 2008;98:517–22.
- Spadi R, Brusa F, Ponzetti A, et al. Current therapeutic strategies for advanced pancreatic cancer: A review for clinicians. World J Clin Oncol 2016;7:27–43.
- Seufferlein T, Bachet JB, Van Cutsem E, Rougier P, Group EGW. Pancreatic adenocarcinoma: ESMO-ESDO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012;23 Suppl 7:vii33–40.
- Huguet F, Girard N, Guerche CS, et al. Chemoradiotherapy in the management of locally advanced pancreatic carcinoma: a qualitative systematic review. J Clin Oncol 2009;27:2269–77.
- Suker M, Beumer BR, Sadot E, et al. FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis. Lancet Oncol 2016;17:801–10.
- Gunturu KS, Yao X, Cong X, et al. FOLFIRINOX for locally advanced and metastatic pancreatic cancer: single institution retrospective review of efficacy and toxicity. Med Oncol 2013;30:361.
- Mukherjee S, Hurt CN, Bridgewater J, et al. Gemcitabine-based or capecitabine-based chemoradiotherapy for locally advanced pancreatic cancer (SCALOP): a multicentre, randomised, phase 2 trial. Lancet Oncol 2013;14:317–26.
- Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–1703.
- Krzyzanowska MK, Weeks JC, Earle CC. Treatment of locally advanced pancreatic cancer in the real world: population-based practices and effectiveness. J Clin Oncol 2003;21:3409–14.
- Sultana A, Tudur Smith C, Cunningham D, et al. Meta-analyses of chemotherapy for locally advanced and metastatic pancreatic cancer: results of secondary end points analyses. Br J Cancer 2008;99:6–13.
- Hammel P, Huguet F, van Laethem JL, et al. Effect of chemoradiotherapy vs chemotherapy on survival in patients with locally advanced pancreatic cancer controlled after 4 months of gemcitabine with or without erlotinib: the LAP07 randomized clinical trial. JAMA 2016;315:1844–53.
- Huguet F, Andre T, Hammel P, et al. Impact of chemoradiotherapy after disease control with chemotherapy in locally advanced pancreatic adenocarcinoma in GERCOR phase II and III studies. J Clin Oncol 2007;25:326–31.
- Dholakia AS, Hacker-Prietz A, Wild AT, et al. Resection of borderline resectable pancreatic cancer after neoadjuvant chemoradiation does not depend on improved radiographic appearance of tumor-vessel relationships. J Radiat Oncol 2013;2:413–25.
- Heinemann V, Haas M, Boeck S. Neoadjuvant treatment of borderline resectable and non-resectable pancreatic cancer. Ann Oncol 2013;24:2484–92.
- Morganti AG, Trodella L, Valentini V, et al. Pain relief with short-term irradiation in locally advanced carcinoma of the pancreas. J Palliat Care 2003;19:258–62.
- Arcidiacono PG, Calori G, Carrara S, et al. Celiac plexus block for pancreatic cancer pain in adults. Cochrane Database Syst Rev 2011(3):CD007519.
- Pezzilli R, Andriulli A, Bassi C, et al. Exocrine pancreatic insufficiency in adults: a shared position statement of the Italian Association for the Study of the Pancreas. World J Gastroenterol 2013;19:7930–46.
- Dominguez-Munoz JE. Pancreatic exocrine insufficiency: diagnosis and treatment. J Gastroenterol Hepatol 2011;26 Suppl 2:12–16.
- Bruno MJ, Haverkort EB, Tijssen GP, et al. Placebo controlled trial of enteric coated pancreatin microsphere treatment in patients with unresectable cancer of the pancreatic head region. Gut 1998;42:92–6.
- Wright AA, Keating NL, Balboni TA, et al. Place of death: correlations with quality of life of patients with cancer and predictors of bereaved caregivers’ mental health. J Clin Oncol 2010;28:4457–64.
- Jang RW, Krzyzanowska MK, Zimmermann C, et al. Palliative care and the aggressiveness of end-of-life care in patients with advanced pancreatic cancer. J Natl Cancer Inst 2015;107(3). pii: dju424.