User login
Molecular Markers and Targeted Therapies in the Management of Non-Small Cell Lung Cancer
INTRODUCTION
Lung cancer is the second most common type of cancer in the United States, with 222,500 estimated new cases in 2017, according to the American Cancer Society.1 However, it is by far the number one cause of death due to cancer, with an estimated 155,870 lung cancer–related deaths occurring in 2017, which is higher than the number of deaths due to breast cancer, prostate cancer, and colorectal cancer combined.1,2 Despite slightly decreasing incidence and mortality over the past decade, largely due to smoking cessation, the 5-year survival rate of lung cancer remains dismal at approximately 18%.2–4
Non-small cell lung cancer (NSCLC) accounts for 80% to 85% of all lung cancer cases.4 Traditionally, it is further divided based on histology: adenocarcinoma, squamous cell carcinoma, large cell carcinoma, and not otherwise specified.5 Chemotherapy had been the cornerstone of treatment for stage IV NSCLC. It is not target-specific and is most effective against rapidly growing cells. Common adverse effects include alopecia, nausea/vomiting, myelosuppression, cardiotoxicity, neuropathy, and nephrotoxicity. However, this paradigm has shifted following the discovery of mutations of the epidermal growth factor receptor (EGFR) gene as an oncogenic driver that confers sensitivity to small molecule tyrosine kinase inhibitors (TKIs) targeting EGFR.6 The EGFR inhibitors are given orally and have a spectrum of toxicities (eg, such as rash, diarrhea, and elevated transaminases) different from that of systemic chemotherapy, which is often administered intravenously. Following the discovery of EGFR mutations, rearrangements of the anaplastic lymphoma kinase (ALK) gene7 and ROS1 gene8 were identified as targetable driver mutations in NSCLC. The frequency of both rearrangements is lower than that of EGFR mutations. Additionally, BRAF V600E mutation has been identified in NSCLC.9–12 This activation mutation is commonly seen in melanoma. Agents that have already been approved for the treatment of melanoma with the BRAF V600E mutation are being tested in NSCLC patients with this mutation.13–16
Given the effectiveness and tolerability of targeted therapy, identifying this distinct molecular subset of NSCLC patients is critical in treatment. Currently, molecular testing is mandatory in all stage IV patients with non-squamous cell carcinoma, as a preponderance of patients with driver mutations have this histology subtype.5,17–19 For patients with squamous cell carcinoma, molecular testing should be considered if the biopsy specimen is small, there is mixed histology, or the patient is a nonsmoker.5,20 Several techniques are commonly utilized in detecting these genetic alterations. EGFR mutation can be detected by polymerase chain reaction (PCR), ALK or ROS1 rearrangement can be detected by fluorescence in-situ hybridization (FISH), and immunohistochemistry (IHC) can also be used to detect ALK rearrangement. The current guideline is to use comprehensive genomic profiling to capture all the potential molecular targets simultaneously instead of running stepwise tests just for EGFR, ALK, and ROS1.5 BRAF V600E mutation,13–16 MET exon 14 skipping mutation,21–24 RET rearrangements,25–27 and HER2 mutations28–30 are among the emergent genetic alterations with various responses to targeted therapy.31 Some of these targeted agents have been approved for other types of malignancy, and others are still in the development phase.
Several initiatives worldwide have reported better outcomes of patients with driver mutations treated with targeted therapy. For instance, the Lung Cancer Mutation Consortium in the United States demonstrated that the median survival of patients without driver mutations, with drivers mutations but not treated with targeted therapy, and with driver mutations and treated with targeted therapy was 2.08 years, 2.38 years, and 3.49 years, respectively.32 The French Cooperative Thoracic Intergroup-French National Cancer Institute demonstrated that the median survival for patients with driver mutations versus those without driver mutations was 16.5 months versus 11.8 months.33 The Spanish Lung Cancer Group demonstrated that the overall survival (OS) for patients with EGFR mutations treated with erlotinib was 27 months.34 The mutations in lung cancer, their frequencies, and the downstream signaling pathways are depicted in the Figure.35
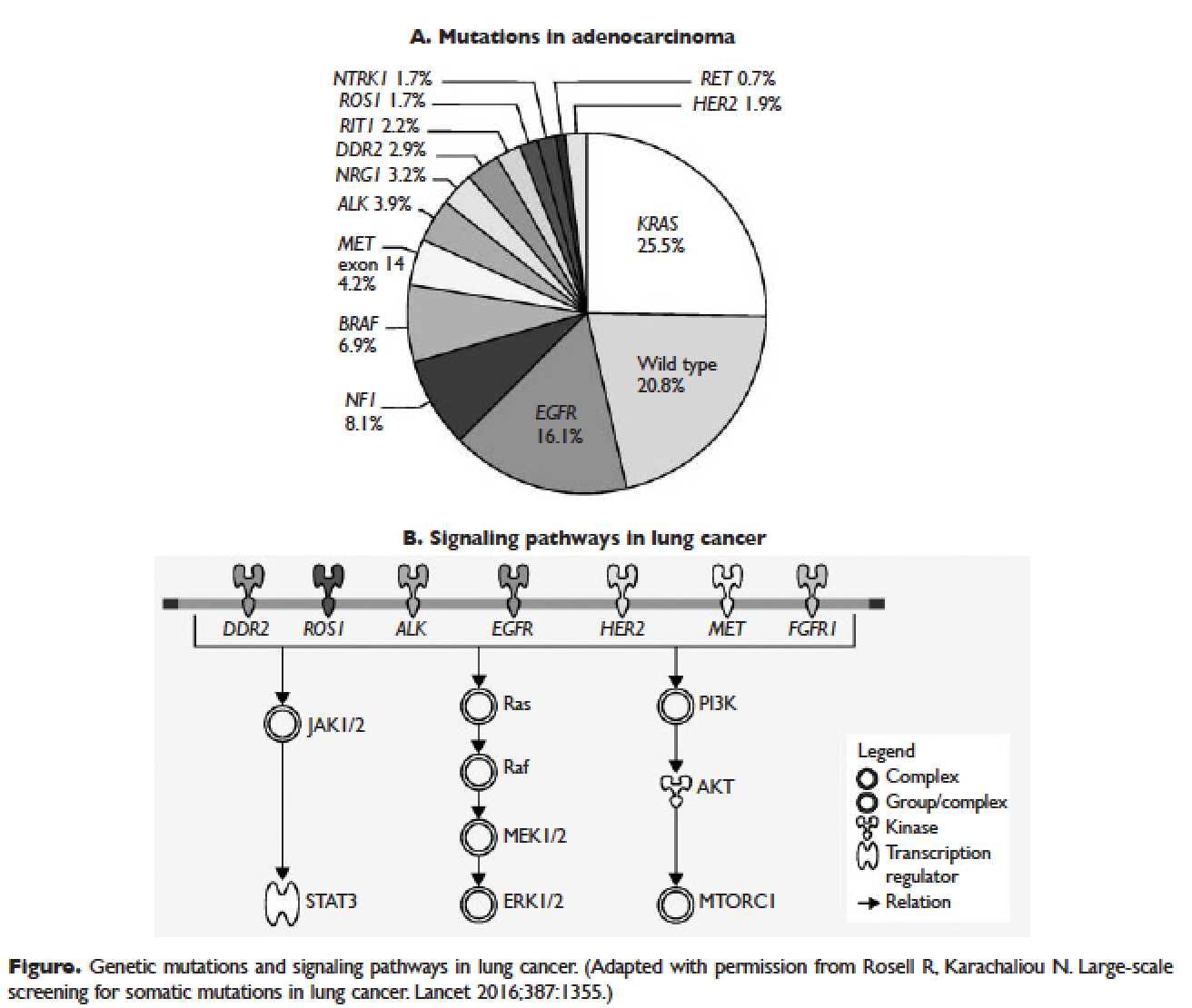
In this article, we discuss targeted therapy for patients with EGFR mutations, ALK rearrangements, ROS1 rearrangements, and BRAF V600E mutation. We also discuss the management of patients with EGFR mutations who develop a secondary mutation after TKI therapy. Almost all of the targeted agents discussed herein have been approved by the US Food and Drug Administration (FDA), so they are considered standard of care. All available phase 3 trials pertinent to these targeted therapies are included in the discussion.
EGFR MUTATIONS
CASE PRESENTATION 1
A 54-year-old Caucasian man who is a former smoker with a 10 pack-year history and past medical history of hypertension and dyslipidemia presents with progressive dyspnea for several weeks. A chest x-ray shows moderate pleural effusion on the left side with possible mass-like opacity on the left upper lung field. An ultrasound-guided thoracentesis is performed and cytology is positive for adenocarcinoma of likely pulmonary origin. Staging workup including positron emission tomography (PET)/computed tomography (CT) and magnetic resonance imaging of the brain with and without contrast is done. PET/CT shows a 5.5-cm mass in the left upper lobe of the lung with high fluorodeoxyglucose (FDG) uptake, several 1- to 2-cm mediastinal lymph nodes with moderate FDG uptake, and small pleural effusion on both sides with moderate FDG uptake. MRI-brain is negative for malignancy. The patient subsequently undergoes a CT-guided biopsy of the lung mass, which shows moderately differentiated adenocarcinoma. Comprehensive molecular profiling reveals EGFR L858R mutation only. The patient now presents for the initial consultation. Of note, his Eastern Cooperative Oncology Group performance status is 1.
What is the next step in the management of this patient?
FIRST-LINE TKI FOR SENSITIZING EGFR MUTATIONS
The 2 most common EGFR mutations are deletions in exon 19 and substitution of arginine for leucine in exon 21 (L858R), found in approximately 45% and 40% of patients with EGFR mutations, respectively.36 Both mutations are sensitive to EGFR TKIs. The benefit may be greater in patients with exon 19 deletions as compared to exon 21 L858R substitution,37,38 but this has not been demonstrated consistently in clinical trials.39-43 In the United States, EGFR mutations are found in approximately 10% of patients with NSCLC, while the incidence can be as high as 50% in Asia.44 Even though the cobas EGFR mutation test is the companion diagnostic approved by the US FDA, a positive test result from any laboratory with the Clinical Laboratory Improvement Amendments (CLIA) certificate should prompt the use of an EGFR TKI as the initial treatment.
Three EGFR TKIs that have been approved as first-line therapy in the United States are available: erlotinib, afatinib, and gefitinib.5 Both erlotinib and gefitinib are considered first-generation TKIs. They have higher binding affinity for the 2 common EGFR mutations than wild-type EGFR. In addition, they reversibly bind to the intracellular tyrosine kinase domain, resulting in inhibition of autophosphorylation of the tyrosine residues. Afatinib, a second-generation and irreversible TKI, targets EGFR (HER1) as well as HER2 and HER4.45
The superior efficacy of the EGFR TKIs over platinum doublet chemotherapy in treatment-naïve patients with EGFR mutations has been demonstrated in 7 randomized trials to date (Table).46 Erlotinib was the TKI arm for the OPTIMAL,41 EURTAC,42 and ENSURE trials;38 afatinib was the TKI arm for LUX-LUNG 337 and 6;43 gefitinib was the TKI arm for NEJ00239,47 and WJTOG3405.40 A meta-analysis of these 7 trials by Lee et al showed that progression-free survival (PFS) was significantly prolonged by EGFR TKIs (hazard ratio [HR] 0.37 [95% confidence interval {CI} 0.32 to 0.42]).46 For instance, in the EURTAC trial, median PFS was 9.7 months for patients treated with erlotinib as compared to 5.2 months for patients treated with platinum/gemcitabine or platinum/docetaxel.42 In this meta-analysis, prespecified subgroups included age, sex, ethnicity, smoking status, performance status, tumor histology, and EGFR mutation subtype. The superior outcome with TKIs was observed in all subgroups. Furthermore, patients with exon 19 deletions, nonsmokers, and women had even better outcomes.46
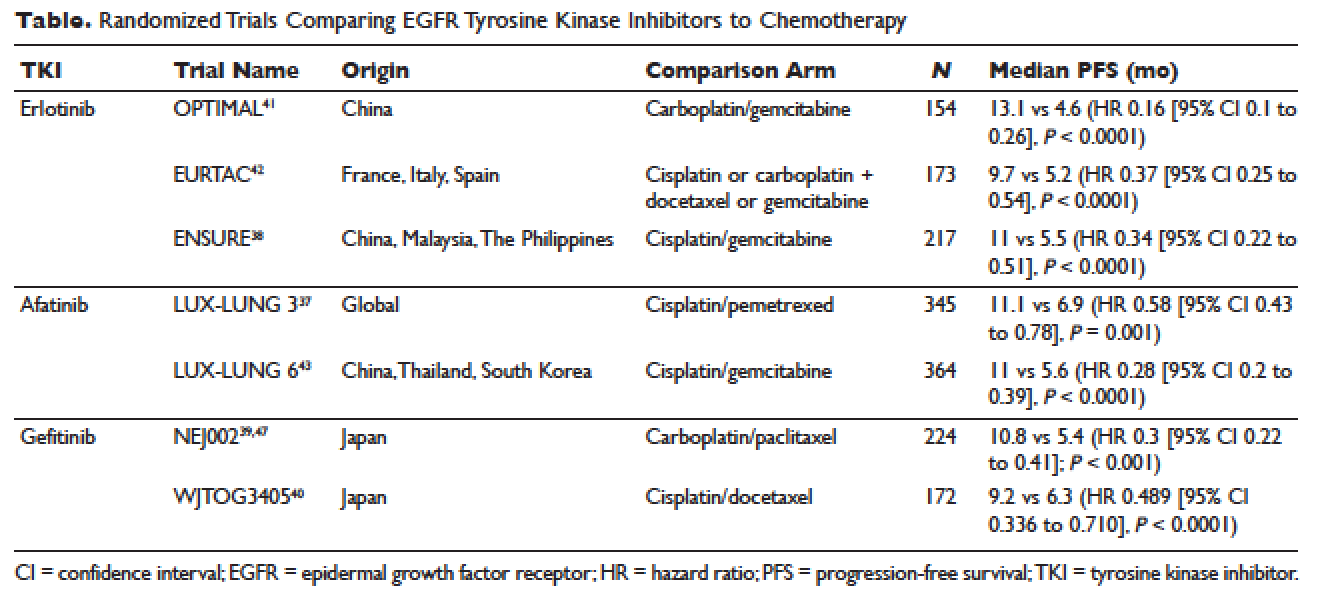
Erlotinib is the most commonly used TKI in the United States largely because gefitinib was off the market for some time until it was re-approved by the FDA in 2015. Interestingly, this “re-approval” was not based on either 1 of the 2 prospective trials (NEJ00239,47 and WJTOG340540), but rather was based on an exploratory analysis of the IPASS trial48,49 as well as a prospective phase 4, single-arm trial in Europe (IFUM).50 The superior efficacy of gefitinib over carboplatin/paclitaxel among patients with EGFR mutations in the IPASS trial was confirmed by blind independent central review, with longer PFS (HR 0.54 [95% CI 0.38 to 0.79] P = 0.0012) and higher objective response rate (ORR; odds ratio 3 [95% CI 1.63 to 5.54], P = 0.0004).49
CASE 1 CONTINUED
Based on the EGFR L858R mutation status, the patient is started on erlotinib. He is quite happy that he does not need intravenous chemotherapy but wants to know what toxicities he might potentially have with erlotinib.
What are the common adverse effects (AEs) of EGFR TKIs? How are AEs of TKIs managed?
Safety Profile
The important toxicities associated with EGFR TKIs are rash, gastrointestinal toxicity, hepatic toxicity, and pulmonary toxicity. Rash is an AE specific to all agents blocking the EGFR pathway, including small molecules and monoclonal antibodies such as cetuximab. The epidermis has a high level of expression of EGFR, which contributes to this toxicity.51 Rash usually presents as dry skin or acneiform eruption. Prophylactic treatment with oral tetracyclines and topical corticosteroids is generally recommended upon initiation of TKI therapy. Diarrhea is the most prevalent gastrointestinal toxicity. All patients starting treatment should be given prescriptions to manage diarrhea such as loperamide and be advised to call when it occurs. Hepatic toxicity is often manifested as elevated transaminases or bilirubin. Interstitial lung disease (ILD) is a rare but potentially fatal pulmonary toxicity.
Rash of any grade was reported in 49.2% of patients treated with erlotinib in clinical trials, while grade 3 rash occurred in 6% of patients and no grade 4 was reported. Diarrhea of any grade was reported in 20.3% of patients, grade 3 diarrhea occurred in 1.8%, and no grade 4 was reported. Grade 2 and 3 alanine aminotransferase (ALT) elevations were seen in 2% and 1% of patients, respectively. Grade 2 and 3 bilirubin elevations were seen in 4% and less than 1% of patients, respectively. The incidence of serious ILD-like events was less than 1%.52
Afatinib is associated with higher incidences of rash and diarrhea. Specifically, diarrhea and rash of all grades were reported in 96% and 90% of patients treated with afatinib, respectively. Paronychia of all grades occurred in 58% of patients. Elevated ALT of all grades was seen in 11% of patients. Approximately 1.5% of patients treated with afatinib across clinical trials had ILD or ILD-like AEs.53
Gefitinib, the most commonly used TKI outside United States, has a toxicity profile similar to erlotinib, except for hepatic toxicity. For instance, rash of all grades occurred in 47% of patients, diarrhea of all grades occurred in 29% of patients, and ILD or ILD-like AEs occurred in 1.3% of patients across clinical trials. In comparison, elevated ALT and aspartate aminotransferase (AST) of all grades was seen in 38% and 40% of patients, respectively.54 Therefore, close monitoring of liver function is clinically warranted. In particular, patients need to be advised to avoid concomitant use of herbal supplements, a common practice in Asian countries.
CASE 1 CONTINUED
The patient does well while on erlotinib at 150 mg orally once daily for about 8 months, until he develops increasing abdominal pain. A CT scan of the abdomen and pelvis with contrast shows a new 8-cm right adrenal mass. Additionally, a repeat CT scan of the chest with contrast shows a stable lung mass but enlarging mediastinal lymphadenopathy.
How would you manage the patient at this point?
MANAGEMENT OF T790M MUTATION AFTER PROGRESSION ON FIRST-LINE EGFR TKIS
As mentioned above, the median PFS of patients with EGFR mutations treated with 1 of the 3 TKIs is around 9 to 13 months.46 Of the various resistance mechanisms that have been described, the T790M mutation is found in approximately 60% of patients who progress after treatment with first-line TKIs.55,56 Other mechanisms, such as HER2 amplification, MET amplification, or rarely small cell transformation, have been reported.56 The first- and second-generation EGFR TKIs function by binding to the ATP-binding domain of mutated EGFR, leading to inhibition of the downstream signaling pathways (Figure, part B) and ultimately cell death.35 The T790M mutation hinders the interaction between the ATP-binding domain of EGFR kinase and TKIs, resulting in treatment resistance and disease progression.57,58
Osimertinib is a third-generation irreversible EGFR TKI with activity against both sensitizing EGFR and resistant T790M mutations. It has low affinity for wide-type EGFR as well as insulin receptor and insulin-like growth factor receptor.59 Osimertinib has been fully approved for NSCLC patients with EGFR mutations who have progressed on first-line EGFR TKIs with the development of T790M mutation. An international phase 3 trial (AURA3) randomly assigned 419 patients in a 2:1 ratio to either osimertinib or platinum/pemetrexed. Eligible patients all had the documented EGFR mutations and disease progression after first-line EGFR TKIs. Central confirmation of the T790M mutation was required. Median PFS by investigator assessment, the trial’s primary end point, was 10.1 months for osimertinib versus 4.4 months for chemotherapy (HR 0.3 [95% CI 0.23 to 0.41]; P < 0.001). ORR was 71% for osimertinib versus 31% for chemotherapy (HR 5.39 [95% CI 3.47 to 8.48], P < 0.001). A total of 144 patients with stable and asymptomatic brain metastases were also eligible. Median PFS for this subset of patients treated with osimertinib and chemotherapy was 8.5 months and 4.2 months, respectively (HR 0.32 [95% CI 0.21 to 0.49]). In the AURA3 trial, osimertinib was better tolerated than chemotherapy, with 23% of patients treated with osimertinib experiencing grade 3 or 4 AEs as compared to 47% of chemotherapy-treated patients. The most common AEs of any grade were diarrhea (41%), rash (34%), dry skin (23%), and paronychia (22%).60
For the case patient, a reasonable approach would be to obtain a tissue biopsy of the adrenal mass and more importantly to check for the T790M mutation. Similar to the companion diagnostic for EGFR mutations, the cobas EGFR mutation test v2 is the FDA-approved test for T790M. However, if this resistance mutation is detected by any CLIA-certified laboratories, osimertinib should be the recommended treatment option. If tissue biopsy is not feasible, plasma-based testing should be considered. A blood-based companion diagnostic also is FDA approved.
ALK REARRANGEMENTS
CASE 2 PRESENTATION
A 42-year-old Korean woman who is a non-smoker with no significant past medical history presents with fatigue, unintentional weight loss of 20 lb in the past 4 months, and vague abdominal pain. A CT can of the abdomen and pelvis without contrast shows multiple foci in the liver and an indeterminate nodule in the right lung base. She subsequently undergoes PET/CT, which confirms multiple liver nodules/masses ranging from 1 to 3 cm with moderate FDG uptake. In addition, there is a 3.5-cm pleura-based lung mass on the right side with moderate FDG uptake. MRI-brain with and without contrast is negative for malignancy. A CT-guided biopsy of 1 of the liver masses is ordered and pathology returns positive for poorly differentiated adenocarcinoma consistent with lung primary. Molecular analysis reveals an echinoderm microtubule-associated protein-like 4 (EML4)-ALK rearrangement. She is placed on crizotinib by an outside oncologist and after about 3 weeks of therapy is doing well. She is now in your clinic for a second opinion. She says that some of her friends told her about another medication called ceritinib and was wondering if she would need to switch her cancer treatment.
How would you respond to this patient’s inquiry?
FIRST-LINE TKIS FOR ALK REARRANGEMENTS
ALK rearrangements are found in 2% to 7% of NSCLC, with EML4-ALK being the most prevalent fusion variant.61 The inversion of chromosome 2p leads to the fusion of the EML4 gene and the ALK gene, which causes the constitutive activation of the fusion protein and ultimately increased transformation and tumorigenicity.7,61 Patients harboring ALK rearrangements tend to be non-smokers. Adenocarcinoma, especially signet ring cell subtype, is the predominant histology. Compared to EGFR mutations, patients with ALK mutations are significantly younger and more likely to be men.62 ALK rearrangements can be detected by either FISH or IHC, and most next-generation sequencing (NGS) panels have the ability to identify this driver mutation.
Crizotinib is the first approved ALK inhibitor for the treatment of NSCLC in this molecular subset of patients.63 PROFILE 1014 is a phase 3 randomized trial that compared crizotinib with chemotherapy containing platinum/pemetrexed for up to 6 cycles. Crossover to crizotinib was allowed for patients with disease progression on chemotherapy. The primary end point was PFS by independent radiologic review. The crizotinib arm demonstrated superior PFS (10.9 months versus 7 months; HR 0.45 [95% CI 0.35 to 0.6], P < 0.001) and ORR (74% versus 45%, P < 0.001). Median survival was not reached in either arm (HR 0.82 [95% CI 0.54 to 1.26], P = 0.36).64 Based on this international trial, crizotinib is considered standard of care in the United States for treatment-naïve patients with advanced NSCLC harboring ALK rearrangements. The current recommended dose is 250 mg orally twice daily. Common treatment-related AEs of all grades include vision disorder (62%), nausea (53%), diarrhea (43%), vomiting (40%), edema (28%), and constipation (27%).65 PROFILE 1007 compared crizotinib with pemetrexed or docetaxel in ALK-rearranged NSCLC patients with prior exposure to 1 platinum-based chemotherapy. The median PFS was 7.7 months for crizotinib as compared to 3 months for chemotherapy (HR 0.49 [95% CI 0.37 to 0.64], P < 0.001). The response rates were 65% and 20% for crizotinib and chemotherapy, respectively (P < 0.001).66 In other countries, crizotinib following 1 prior platinum-based regimen may be considered standard of care based on this trial.
Ceritinib is an oral second-generation ALK inhibitor that is 20 times more potent than crizotinib based on enzymatic assays.67 It also targets ROS1 and insulin-like growth factor 1 receptor but not c-MET. It was first approved by the FDA in April 2014 for metastatic ALK-rearranged NSCLC following crizotinib.68 In May 2017, the FDA granted approval of ceritinib for treatment-naïve patients. This decision was based on the results of the ASCEND-4 trial, a randomized phase 3 trial assessing the efficacy and safety of ceritinib over chemotherapy in the first-line setting. The trial assigned 376 patients to either ceritinib at 750 mg once daily or platinum/pemetrexed for 4 cycles followed by maintenance pemetrexed. Median PFS was 16.6 months for ceritinib versus 8.1 months for chemotherapy (HR 0.55 [95% CI 0.42 to 0.73]; P < 0.00001).69 Toxicities of ceritinib are not negligible, with gastrointestinal toxicity being the most prevalent. For instance, diarrhea, nausea, vomiting, abdominal pain, and constipation of all grades were seen in 86%, 80%, 60%, 54%, and 29% of patients, respectively. Furthermore, fatigue and decreased appetite occurred in 52% and 34% of patients, respectively. In terms of laboratory abnormalities, 84% of patients experienced decreased hemoglobin of all grades; 80% increased ALT; 75% increased AST; 58% increased creatinine; 49% increased glucose; 36% decreased phosphate; and 28% increased lipase. Due to these AEs, the incidence of dose reduction was about 58% and the median onset was around 7 weeks.70
Alectinib is another oral second-generation ALK inhibitor that was approved by the FDA in December 2015 for the treatment of NSCLC patients with ALK rearrangements who have progressed on or are intolerant to crizotinib.71 Its indication will soon be broadened to the first-line setting based on the ALEX trial.72 Alectinib is a potent and highly selective TKI of ALK73 with activity against known resistant mutations to crizotinib.74,75 It also inhibits RET but not ROS1 or c-MET.76 ALEX, a randomized phase 3 study, compared alectinib with crizotinib in treatment-naïve patients with NSCLC harboring ALK rearrangements. The trial enrolled 303 patients and the median follow-up was approximately 18 months. The alectinib arm (600 mg twice daily) demonstrated significantly higher PFS by investigator-assessment, the trial’s primary end point. The 12-month event-free survival was 68.4% (95% CI 61% to 75.9%) versus 48.7% (95% CI 40.4% to 56.9%) for alectinib and crizotinib, respectively (HR 0.47 [95% CI 0.34 to 0.65], P < 0.001). The median PFS was not reached in the alectinib arm (95% CI 17.7 months to not estimable) as compared to 11.1 months in the crizotinib arm (95% CI 9.1 to 13.1 months).72 Alectinib is generally well tolerated. Common AEs of all grades include fatigue (41%), constipation (34%), edema (30%), and myalgia (29%). As alectinib can cause anemia, lymphopenia, hepatic toxicity, increased creatine phosphokinase, hyperglycemia, electrolyte abnormalities, and increased creatinine, periodic monitoring of these laboratory values is important, although most of these abnormalities are grade 1 or 2.77
Brigatinib, another oral second-generation ALK inhibitor, was granted accelerated approval by the FDA in April 2017 for ALK-rearranged and crizotinib-resistant NSCLC based on the ALTA trial. This randomized phase 2 study of brigatinib showed an ORR by investigator assessment of 54% (97.5% CI 43% to 65%) in the 180 mg once daily arm with lead-in of 90 mg once daily for 7 days. Median PFS was 12.9 months (95% CI 11.1 months to not reached [NR]).78 Currently, a phase 3 study of brigatinib versus crizotinib in ALK inhibitor–naïve patients is recruiting participants (ALTA-1L). It will be interesting to see if brigatinib can achieve a front-line indication.
Starting the case patient on crizotinib is well within the treatment guidelines. One may consider ceritinib or alectinib in the first-line setting, but both TKIs can be reserved upon disease progression. We would recommend a repeat biopsy at that point to look for resistant mechanisms, as certain secondary ALK mutations may be rescued by certain next-generation ALK inhibitors. For instance, the F1174V mutation has been reported to confer resistance to ceritinib but sensitivity to alectinib, while the opposite is true for I1171T. The G1202R mutation is resistant to ceritinib, alectinib, and brigatinib, but lorlatinib, a third-generation ALK inhibitor, has shown activity against this mutation.79 Furthermore, brain metastasis represents a treatment challenge for patients with ALK rearrangements. It is also an efficacy measure of next-generation ALK inhibitors, all of which have demonstrated better central nervous system activity than crizotinib.69,78,80 If the case patient were found to have brain metastasis at the initial diagnosis, either ceritinib or alectinib would be a reasonable choice since crizotinib has limited penetration of blood-brain barrier.81
ROS1 REARRANGEMENTS
CASE PRESENTATION 3
A 66-year-old Chinese woman who is a non-smoker with a past medical history of hypertension and hypothyroidism presents to the emergency department for worsening lower back pain. Initial workup includes x-ray of the lumbar spine followed by MRI with contrast, which shows a soft tissue mass at L3-4 without cord compression. CT of the chest, abdomen, and pelvis with contrast shows a 7-cm right hilar mass, bilateral small lung nodules, mediastinal lymphadenopathy, and multiple lytic lesions in ribs, lumbar spine, and pelvis. MRI-brain with and without contrast is negative for malignancy. She undergoes endo-bronchial ultrasound and biopsy of the right hilar mass, which shows poorly differentiated adenocarcinoma. While waiting for the result of the molecular analysis, the patient undergoes palliative radiation therapy to L2-5 with good pain relief. She is discharged from the hospital and presents to your clinic for follow up. Molecular analysis now reveals ROS1 rearrangement with CD74-ROS1 fusion.
What treatment plan should be put in place for this patient?
FIRST-LINE THERAPY FOR ROS1 REARRANGEMENTS
Approximately 2.4% of lung adenocarcinomas harbor ROS1 rearrangements.82 This distinct genetic alteration occurs more frequently in NSCLC patients who are younger, female, and never-smokers, and who have adenocarcinomas.8 It has been shown that ROS1 rearrangements rarely overlap with other genetic alterations including KRAS mutations, EGFR mutations, and ALK rearrangements.83 As a receptor tyrosine kinase, ROS1 is similar to ALK and insulin receptor family members.84 Crizotinib, which targets ALK, ROS1, and c-MET, was approved by the FDA on March 11, 2016, for the treatment of metastatic ROS1-rearranged NSCLC.85 The approval was based on a phase 2 expansion cohort of the original phase 1 study. Among 50 US patients enrolled in this expansion cohort, 3 had complete responses and 33 had partial responses with ORR of 72% (95% CI 58% to 84%). Median PFS was 19.2 months (95% CI 14.4 months to NR) and median duration of response (DOR) was 17.6 months (95% CI 14.5 months to NR).86 During longer follow-up, independent radiology review confirmed high ORR of 66% and median DOR of 18.3 months.85
Interestingly, no companion diagnostic assay has been approved for the detection of ROS1 rearrangements with the approval of crizotinib. In the United States, break apart FISH is the most common detection method. In fact, in the above mentioned phase 2 study, ROS1 rearrangements were detected in 49 out of 50 patients by this method.86 FISH can be technically challenging when dealing with high volume and multiple targets. Reverse transcriptase-PCR is another detection method, but it requires knowledge of the fusion partners. To date, at least 14 ROS1 fusion partners have been reported, with CD74 being the most common.87 NGS with appropriate design and validation can also be used to detect ROS1 rearrangements.
For the case patient, the recommendation would be to start her on crizotinib at 250 mg twice daily. Monitoring for vision disturbance, gastrointestinal complaints, and edema is warranted. Because the estimated onset of response is around 7.9 weeks,86 plans should be made to repeat her scans in approximately 2 months.
BRAF V600E MUTATIONS
CASE PRESENTATION 4
A 71-year-old Caucasian man with a past medical history of hypertension, dyslipidemia, and ischemic cerebrovascular accident without residual deficits was diagnosed with stage IV adenocarcinoma of the lung about 8 months ago. He has a 40 pack-year smoking history and quit smoking when he was diagnosed with lung cancer. His disease burden involved a large mediastinal mass, scattered pleural nodules, multiple lymphadenopathy, and several soft tissue masses. His outside oncologist started him on chemotherapy containing carboplatin and pemetrexed for 6 cycles followed by maintenance pemetrexed. The most recent restaging scans show disease progression with enlarging soft tissue masses and several new lytic bone lesions. MRI-brain with and without contrast shows 2 subcentimeter enhancing lesions. He transferred care to you approximately 4 weeks ago. You ordered a repeat biopsy of 1 of the enlarging soft tissue masses. Molecular analysis revealed BRAF V600E mutation. In the interim, he underwent stereotactic radiosurgery for the 2 brain lesions without any complications. The patient is now in your clinic for follow up.
What would be your recommended systemic treatment?
TARGETED THERAPIES FOR BRAF V600E MUTATION
BRAF mutations were first recognized as activating mutations in advanced melanomas, with BRAF V600E, resulting from the substitution of glutamic acid for valine at amino acid 600, being the most common. BRAF plays an important role in the mitogen-activated protein kinase (MAPK) signaling pathway. Briefly, the activation of MAPK pathway occurs upon ligand binding of receptor tyrosine kinases, which then involves RAS/BRAF/MEK/ERK in a stepwise manner, ultimately leading to cell survival. BRAF mutations have been increasingly recognized also as driver mutations in NSCLC.9–12 They can be detected by PCR or NGS method. The characteristics of NSCLC patients harboring BRAF mutations have been described by various groups.9–12 For instance, 1 case series showed that the incidence was 2.2% among patients with advanced lung adenocarcinoma; 50% of mutations were V600E, while G469A and D594G accounted for the remaining 39% and 11% of patients, respectively. All patients were either current or former smokers. The median OS of patients with BRAF mutations in this case series was NR, while it was 37 months for patients with EGFR mutations (P = 0.73) and NR for patients with ALK rearrangements (P = 0.64).9
For patients with BRAF V600E–mutant NSCLC who have progressed on platinum-based chemotherapy, the combination of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) may represent a new treatment paradigm. This was illustrated in a phase 2, nonrandomized, open-label study. A total of 57 patients were enrolled and 36 patients (63.2% [95% CI 49.3% to 75.6%]) achieved an overall response by investigator assessment, the trial’s primary end point. Disease control rate was 78.9% (95% CI 66.1% to 88.6%), with 4% complete response, 60% partial response, and 16% stable disease. PFS was 9.7 months (95% CI [6.9 to 19.6 months]). The safety profile was comparable to what had been observed in patients with melanoma treated with this regimen. More specifically, 56% of patients on this trial reported serious AEs, including pyrexia (16%), anemia (5%), confusional state (4%), decreased appetite (4%), hemoptysis (4%), hypercalcemia (4%), nausea (4%), and cutaneous squamous cell carcinoma (4%). In addition, neutropenia (9%) and hyponatremia (7%) were the most common grade 3-4 AEs.16
The case patient has experienced disease progression after 1 line of platinum-based chemotherapy, so the combination of dabrafenib and trametinib would be a robust systemic treatment option. dabrafenib as a single agent has also been studied in BRAF V600E–mutant NSCLC in a phase 2 trial. The overall response by investigator assessment among 84 patients was 33% (95% CI 23% to 45%).14 Vemurafenib, another oral BRAF TKI, has demonstrated efficacy for NSCLC patients harboring BRAF V600E mutation. In the cohort of 20 patients with NSCLC, the response rate was 42% (95% CI 20% to 67%) and median PFS was 7.3 months (95% CI 3.5 to 10.8 months).13 Patients with non-V600E mutations have shown variable responses to targeted therapies. MEK TKIs may be considered in this setting; however, the details of this discussion are beyond the scope of this review.
CONCLUSION
The management of advanced NSCLC with driver mutations has seen revolutionary changes over the past decade. Tremendous research has been done in order to first understand the molecular pathogenesis of NSCLC and then discover driver mutations that would lead to development of targeted therapies with clinically significant efficacy as well as tolerability. More recently, increasing efforts have focused on how to conquer acquired resistance in patients with disease progression after first-line TKIs. The field of EGFR-mutant NSCLC has set a successful example, but the work is nowhere near finished. The goals are to search for more driver mutations and to design agents that could potentially block cell survival signals once and for all.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
- Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol 2016;893:1–19.
- Alberg AJ, Brock MV, Ford JG, et al. Epidemiology of lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013;143:e1S–29S.
- Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-3013, based on November 2015 SEER data submission, posted to the SEER website, April 2016. Bethesda (MD): National Cancer Institute; 2016.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Non-Small Cell Lung Cancer: 1–190.
- Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–39.
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–6.
- Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancer. J Clin Oncol 2012;30:863–70.
- Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011;29:2046–51.
- Kinno T, Tsuta K, Shiraishi K, et al. Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol 2014;25:138–42.
- Litvak AM, Paik PK, Woo KM, et al. Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J Thorac Oncol 2014;9:1669–74.
- Villaruz LC, Socinski MA, Abberbock S, et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer 2015;121:448–56.
- Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–36.
- Planchard D, Kim TM, Mazieres J, et al. DaBRAFenib in patients with BRAF V600E-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol 2016;17:642–50.
- Gautschi O, Milia J, Cabarrou B, et al. Targeted therapy for patients with BRAF-mutant lung cancer: results from the European EURAF cohort. J Thorac Oncol 2015;10:1451–7.
- Planchard D, Besse B, Groen HJ, et al. DaBRAFenib plus trametinib in patients with previously treated BRAF V600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 2016;17:984–93.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. J Thorac Oncol 2013;8:823–59.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. Arch Pathol Lab Med 2013;137:828–60.
- Leighl NB, Rekhtman N, Biermann WA, et al. Molecular testing for selection of patients with lung cancer for epidermal growth factor receptor and anaplastic lymphoma kinase tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology guideline. J Clin Oncol 2014;32:3673–9.
- Paik PK, Varghese AM, Sima CS, et al. Response to erlotinib in patients with EGFR mutant advanced non-small cell lung cancers with a squamous or squamous-like component. Mol Cancer Ther 2012;11:2535–40.
- Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov 2015;5:842–9.
- Awad MM, Oxnard GR, Jackman DM, et al. MET Exon 14 mutations in non-small-cell lung cancer are associated with advanced age, and stage-dependent MET genomic amplification, and c-MET overexpression. J Clin Oncol 2016;34:721–30.
- Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thorac Oncol 2016;11:1493–502.
- Reungwetwattana T, Liang Y, Zhu V, et al. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The why, the how, the who, the unknown, and the inevitable. Lung Cancer 2017;103:27–37.
- Drilon A, Wang L, Hasanovic A, et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov 2013;3:630–5.
- Lin JJ, Kennedy E, Sequist LV, et al. Clinical activity of alectinib in advanced RET-rearranged non-small cell lung cancer. J Thorac Oncol 2016;11:2027–32.
- Drilon A, Rekhtman N, Arcila M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol 2016;17:1653–60.
- Cappuzzo F, Bemis L, Varella-Garcia M. HER2 mutation and response to trastuzumab therapy in non-small-cell lung cancer. N Engl J Med 2006;354:2619–21.
- Mazieres J. Barlesi F, Filleron T, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targted drugs: results from the European EUHER2 cohort. Annal Oncol 2016;27:281–6.
- Ou SH, Schrock AB, Bocharov EV, et al. HER2 transmembrane (TMD) mutations (V659/G660) that stabilize homo- and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J Thorac Oncol 2017;12:446–57.
- Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emergent therapies. Cancer Discov 2017;7:596–609.
- Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014;311:1998–2006.
- Barlesi F, Mazieres J, Merlio JP, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016;387:1415–26.
- Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361:958–67.
- Rosell R, Karachaliou N, et al. Large-scale screening for somatic mutations in lung cancer. Lancet 2016;387:1354–6.
- Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005;97:339–46.
- Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013;31:3327–34.
- Wu YL, Chou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol 2015;26:1883–9.
- Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Eng J Med 2010;362:2380–8.
- Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121–8.
- Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011;12:735–42.
- Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239–46.
- Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:213–22.
- Hirsch FR, Bunn PA Jr. EGFR testing in lung cancer is ready for prime time. Lancet Oncol 2009;10:432–3.
- Nelson V, Ziehr J, Aqulnik M, et al. Afatinib: emerging next-generation tyrosine kinase inhibitor for NSCLC. Onco Targets Ther 2013;5:135–43.
- Lee CK, Wu YL, Ding PN, et al. Impact of specific epidermal growth factor receptor (EGFR) mutations and clinical characteristics on outcomes after treatment with EGFR tyrosine kinase inhibitors versus chemotherapy in EGFR-mutant lung cancer: a meta-analysis. J Clin Oncol 2015;33:1958–65.
- Inoue A, Kobayashi K, Maemondo M, et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann Oncol 2013;24:54–9.
- Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947–57.
- Wu YL, Saijo N, Thongprasert S, et al. Efficacy according to blind independent central review: post-hoc analyses from the phase III, randomized, multicenter, IPASS study of first-line gefitinib versus carboplatin/paclitaxel in Asian patients with EFGR mutation-positive advanced NSCLC. Lung Cancer 2017;104:119–25.
- Douillard JY, Ostoros G, Cobo M, et al. First-line gefitinib in Caucasian EGFR-mutation positive NSCLC patients: a phase-IV, open-label, single-arm study. Br J Cancer 2014;110:55–62.
- Hu JC, Sadeghi P, Pinter-Brown LC, et al. Cutaneous side effects of epidermal growth factor receptor inhibitors: clinical presentation, pathogenesis, and management. J Am Acad Dermatol 2007;56:317–26.
- Tarceva [package insert]. South San Francisco (CA): Genentech, Inc; 2010. www.accessdata.fda.gov/drugsatfda_docs/label/2010/021743s14s16lbl.pdf. Accessed April 23, 2017.
- Gilotrif [package insert.] Ridgefield (CT): Boehringer Ingelheim, Inc; 2013. www.accessdata.fda.gov/drugsatfda_docs/label/2013/201292s000lbl.pdf. Accessed April 23, 2017.
- Iressa [package insert]. Wilmington (DE): AstraZeneca, Inc; 2015. Error! Hyperlink reference not valid. Accessed April 23, 2017.
- Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res 2011;17:1616–22.
- Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR TKI therapy in 155 patients with EGFR mutant lung cancers. Clin Cancer Res 2013;19:2240–7.
- Yun CH, Mengwasser KE, Tom AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 2008;105:2070–5.
- Sos ML, Rode HB, Heynck S, et al. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Res 2010;70:868–74.
- Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T190M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014;4:1046–61.
- Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 2017;376:629–40.
- Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small cell lung cancer. N Engl J Med 2010;363:1693–703.
- Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009;27:4247–53.
- Kazandjian D, Blumenthal GM, Chen HY, et al. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014;19:e5–11.
- Solomon BJ, Mok T, Kim DW, et al. First-ling crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 2014;371:2167–77.
- Xalkori [package insert]. New York: Pfizer, Inc; 2011. www.accessdata.fda.gov/drugsatfda_docs/label/2012/202570s002lbl.pdf. Accessed April 23, 2017.
- Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013;368:2385–94.
- Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem 2013;56:5675–90.
- Khozin S, Blumenthal GM, Zhang L, et al. FDA approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer. Clin Cancer Res 2015;21:2436–9.
- Soria JC, Tan DS, Chiari R, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet 2017;389:917–29.
- Zykadia [package insert]. East Hanover (NJ): Novartis Pharmaceuticals Corporation, Inc; 2016. www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/zykadia.pdf. Accessed April 23, 2017.
- Larkins E, Blumenthal GM, Chen H, et al. FDA approval: alectinib for the treatment of metastatic, ALK-positive non-small cell lung cancer following crizotinib. Clin Cancer Res 2016;22:5171–6.
- Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. New Engl J Med 2017 June 6 [Epub ahead of print].
- Kinoshita K, Asoh K, Furuichi N, et al. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem 2012;20:1271–80.
- Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011;19:679–90.
- Kodama T, Tsukaguchi T, Yoshida M, et al. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett 2014;351:215–21.
- Kodama T, Tsukaguchi T, Satoh T, et al. Alectinib shows potent antitumor activity against RET-rearranged non-small cell lung cancer. Mol Cancer Ther 2014;13:2910–8.
- Alecensa [package insert]. South San Francisco (CA): Genentech, Inc; 2015. www.accessdata.fda.gov/drugsatfda_docs/label/2015/208434s000lbl.pdf. Accessed April 23, 2017.
- Kim DW, Tiseo M, Ahn MJ, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase positive non-small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol 2017 May 5 [Epub ahead of print].
- Zhu V, Ou SH. Safety of alectinib for the treatment of metastatic ALK-rearranged non-small cell lung cancer. Expert Opin Drug Saf 2017;16:509–14.
- Gadgeel SM, Shaw AT, Govindan R, et al. Pooled analysis of CNS response to alectinib in two studies of pretreated patients with ALK-positive non-small cell lung cancer. J Clin Oncol 2016;34:4079–85.
- Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol 2011;29:e443–5.
- Zhu Q, Zhan P, Zhang X, et al. Clinicopathologic characteristics of patients with ROS1 fusion gene in non-small cell lung cancer: a meta-analysis. Transl Lung Cancer Res 2015;4:300–9.
- Lin JJ, Ritterhouse LL, Ali SM, et al. ROS1 fusions rarely overlap with other oncogenic drivers in non-small cell lung cancer. J Thorac Oncol 2017;12:872–7.
- Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta 2009;1795:37–52.
- Kazandjian D, Blumenthal G, Luo L, et al. Benefit-Risk summary of crizotinib for the treatment of patients with ROS1 alteration-positive metastatic NSCLC. Oncologist 2016;21:974–80.
- Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 2014;371:1963–71.
- Zhu VW, Upadhyay D, Schrock AB, et al. TPD52L1-ROS1, a new ROS1 fusion variant in lung adenosquamous cell carcinoma identified by comprehensive genomic profiling. Lung Cancer 2016;97:48–50.
INTRODUCTION
Lung cancer is the second most common type of cancer in the United States, with 222,500 estimated new cases in 2017, according to the American Cancer Society.1 However, it is by far the number one cause of death due to cancer, with an estimated 155,870 lung cancer–related deaths occurring in 2017, which is higher than the number of deaths due to breast cancer, prostate cancer, and colorectal cancer combined.1,2 Despite slightly decreasing incidence and mortality over the past decade, largely due to smoking cessation, the 5-year survival rate of lung cancer remains dismal at approximately 18%.2–4
Non-small cell lung cancer (NSCLC) accounts for 80% to 85% of all lung cancer cases.4 Traditionally, it is further divided based on histology: adenocarcinoma, squamous cell carcinoma, large cell carcinoma, and not otherwise specified.5 Chemotherapy had been the cornerstone of treatment for stage IV NSCLC. It is not target-specific and is most effective against rapidly growing cells. Common adverse effects include alopecia, nausea/vomiting, myelosuppression, cardiotoxicity, neuropathy, and nephrotoxicity. However, this paradigm has shifted following the discovery of mutations of the epidermal growth factor receptor (EGFR) gene as an oncogenic driver that confers sensitivity to small molecule tyrosine kinase inhibitors (TKIs) targeting EGFR.6 The EGFR inhibitors are given orally and have a spectrum of toxicities (eg, such as rash, diarrhea, and elevated transaminases) different from that of systemic chemotherapy, which is often administered intravenously. Following the discovery of EGFR mutations, rearrangements of the anaplastic lymphoma kinase (ALK) gene7 and ROS1 gene8 were identified as targetable driver mutations in NSCLC. The frequency of both rearrangements is lower than that of EGFR mutations. Additionally, BRAF V600E mutation has been identified in NSCLC.9–12 This activation mutation is commonly seen in melanoma. Agents that have already been approved for the treatment of melanoma with the BRAF V600E mutation are being tested in NSCLC patients with this mutation.13–16
Given the effectiveness and tolerability of targeted therapy, identifying this distinct molecular subset of NSCLC patients is critical in treatment. Currently, molecular testing is mandatory in all stage IV patients with non-squamous cell carcinoma, as a preponderance of patients with driver mutations have this histology subtype.5,17–19 For patients with squamous cell carcinoma, molecular testing should be considered if the biopsy specimen is small, there is mixed histology, or the patient is a nonsmoker.5,20 Several techniques are commonly utilized in detecting these genetic alterations. EGFR mutation can be detected by polymerase chain reaction (PCR), ALK or ROS1 rearrangement can be detected by fluorescence in-situ hybridization (FISH), and immunohistochemistry (IHC) can also be used to detect ALK rearrangement. The current guideline is to use comprehensive genomic profiling to capture all the potential molecular targets simultaneously instead of running stepwise tests just for EGFR, ALK, and ROS1.5 BRAF V600E mutation,13–16 MET exon 14 skipping mutation,21–24 RET rearrangements,25–27 and HER2 mutations28–30 are among the emergent genetic alterations with various responses to targeted therapy.31 Some of these targeted agents have been approved for other types of malignancy, and others are still in the development phase.
Several initiatives worldwide have reported better outcomes of patients with driver mutations treated with targeted therapy. For instance, the Lung Cancer Mutation Consortium in the United States demonstrated that the median survival of patients without driver mutations, with drivers mutations but not treated with targeted therapy, and with driver mutations and treated with targeted therapy was 2.08 years, 2.38 years, and 3.49 years, respectively.32 The French Cooperative Thoracic Intergroup-French National Cancer Institute demonstrated that the median survival for patients with driver mutations versus those without driver mutations was 16.5 months versus 11.8 months.33 The Spanish Lung Cancer Group demonstrated that the overall survival (OS) for patients with EGFR mutations treated with erlotinib was 27 months.34 The mutations in lung cancer, their frequencies, and the downstream signaling pathways are depicted in the Figure.35
In this article, we discuss targeted therapy for patients with EGFR mutations, ALK rearrangements, ROS1 rearrangements, and BRAF V600E mutation. We also discuss the management of patients with EGFR mutations who develop a secondary mutation after TKI therapy. Almost all of the targeted agents discussed herein have been approved by the US Food and Drug Administration (FDA), so they are considered standard of care. All available phase 3 trials pertinent to these targeted therapies are included in the discussion.
EGFR MUTATIONS
CASE PRESENTATION 1
A 54-year-old Caucasian man who is a former smoker with a 10 pack-year history and past medical history of hypertension and dyslipidemia presents with progressive dyspnea for several weeks. A chest x-ray shows moderate pleural effusion on the left side with possible mass-like opacity on the left upper lung field. An ultrasound-guided thoracentesis is performed and cytology is positive for adenocarcinoma of likely pulmonary origin. Staging workup including positron emission tomography (PET)/computed tomography (CT) and magnetic resonance imaging of the brain with and without contrast is done. PET/CT shows a 5.5-cm mass in the left upper lobe of the lung with high fluorodeoxyglucose (FDG) uptake, several 1- to 2-cm mediastinal lymph nodes with moderate FDG uptake, and small pleural effusion on both sides with moderate FDG uptake. MRI-brain is negative for malignancy. The patient subsequently undergoes a CT-guided biopsy of the lung mass, which shows moderately differentiated adenocarcinoma. Comprehensive molecular profiling reveals EGFR L858R mutation only. The patient now presents for the initial consultation. Of note, his Eastern Cooperative Oncology Group performance status is 1.
What is the next step in the management of this patient?
FIRST-LINE TKI FOR SENSITIZING EGFR MUTATIONS
The 2 most common EGFR mutations are deletions in exon 19 and substitution of arginine for leucine in exon 21 (L858R), found in approximately 45% and 40% of patients with EGFR mutations, respectively.36 Both mutations are sensitive to EGFR TKIs. The benefit may be greater in patients with exon 19 deletions as compared to exon 21 L858R substitution,37,38 but this has not been demonstrated consistently in clinical trials.39-43 In the United States, EGFR mutations are found in approximately 10% of patients with NSCLC, while the incidence can be as high as 50% in Asia.44 Even though the cobas EGFR mutation test is the companion diagnostic approved by the US FDA, a positive test result from any laboratory with the Clinical Laboratory Improvement Amendments (CLIA) certificate should prompt the use of an EGFR TKI as the initial treatment.
Three EGFR TKIs that have been approved as first-line therapy in the United States are available: erlotinib, afatinib, and gefitinib.5 Both erlotinib and gefitinib are considered first-generation TKIs. They have higher binding affinity for the 2 common EGFR mutations than wild-type EGFR. In addition, they reversibly bind to the intracellular tyrosine kinase domain, resulting in inhibition of autophosphorylation of the tyrosine residues. Afatinib, a second-generation and irreversible TKI, targets EGFR (HER1) as well as HER2 and HER4.45
The superior efficacy of the EGFR TKIs over platinum doublet chemotherapy in treatment-naïve patients with EGFR mutations has been demonstrated in 7 randomized trials to date (Table).46 Erlotinib was the TKI arm for the OPTIMAL,41 EURTAC,42 and ENSURE trials;38 afatinib was the TKI arm for LUX-LUNG 337 and 6;43 gefitinib was the TKI arm for NEJ00239,47 and WJTOG3405.40 A meta-analysis of these 7 trials by Lee et al showed that progression-free survival (PFS) was significantly prolonged by EGFR TKIs (hazard ratio [HR] 0.37 [95% confidence interval {CI} 0.32 to 0.42]).46 For instance, in the EURTAC trial, median PFS was 9.7 months for patients treated with erlotinib as compared to 5.2 months for patients treated with platinum/gemcitabine or platinum/docetaxel.42 In this meta-analysis, prespecified subgroups included age, sex, ethnicity, smoking status, performance status, tumor histology, and EGFR mutation subtype. The superior outcome with TKIs was observed in all subgroups. Furthermore, patients with exon 19 deletions, nonsmokers, and women had even better outcomes.46
Erlotinib is the most commonly used TKI in the United States largely because gefitinib was off the market for some time until it was re-approved by the FDA in 2015. Interestingly, this “re-approval” was not based on either 1 of the 2 prospective trials (NEJ00239,47 and WJTOG340540), but rather was based on an exploratory analysis of the IPASS trial48,49 as well as a prospective phase 4, single-arm trial in Europe (IFUM).50 The superior efficacy of gefitinib over carboplatin/paclitaxel among patients with EGFR mutations in the IPASS trial was confirmed by blind independent central review, with longer PFS (HR 0.54 [95% CI 0.38 to 0.79] P = 0.0012) and higher objective response rate (ORR; odds ratio 3 [95% CI 1.63 to 5.54], P = 0.0004).49
CASE 1 CONTINUED
Based on the EGFR L858R mutation status, the patient is started on erlotinib. He is quite happy that he does not need intravenous chemotherapy but wants to know what toxicities he might potentially have with erlotinib.
What are the common adverse effects (AEs) of EGFR TKIs? How are AEs of TKIs managed?
Safety Profile
The important toxicities associated with EGFR TKIs are rash, gastrointestinal toxicity, hepatic toxicity, and pulmonary toxicity. Rash is an AE specific to all agents blocking the EGFR pathway, including small molecules and monoclonal antibodies such as cetuximab. The epidermis has a high level of expression of EGFR, which contributes to this toxicity.51 Rash usually presents as dry skin or acneiform eruption. Prophylactic treatment with oral tetracyclines and topical corticosteroids is generally recommended upon initiation of TKI therapy. Diarrhea is the most prevalent gastrointestinal toxicity. All patients starting treatment should be given prescriptions to manage diarrhea such as loperamide and be advised to call when it occurs. Hepatic toxicity is often manifested as elevated transaminases or bilirubin. Interstitial lung disease (ILD) is a rare but potentially fatal pulmonary toxicity.
Rash of any grade was reported in 49.2% of patients treated with erlotinib in clinical trials, while grade 3 rash occurred in 6% of patients and no grade 4 was reported. Diarrhea of any grade was reported in 20.3% of patients, grade 3 diarrhea occurred in 1.8%, and no grade 4 was reported. Grade 2 and 3 alanine aminotransferase (ALT) elevations were seen in 2% and 1% of patients, respectively. Grade 2 and 3 bilirubin elevations were seen in 4% and less than 1% of patients, respectively. The incidence of serious ILD-like events was less than 1%.52
Afatinib is associated with higher incidences of rash and diarrhea. Specifically, diarrhea and rash of all grades were reported in 96% and 90% of patients treated with afatinib, respectively. Paronychia of all grades occurred in 58% of patients. Elevated ALT of all grades was seen in 11% of patients. Approximately 1.5% of patients treated with afatinib across clinical trials had ILD or ILD-like AEs.53
Gefitinib, the most commonly used TKI outside United States, has a toxicity profile similar to erlotinib, except for hepatic toxicity. For instance, rash of all grades occurred in 47% of patients, diarrhea of all grades occurred in 29% of patients, and ILD or ILD-like AEs occurred in 1.3% of patients across clinical trials. In comparison, elevated ALT and aspartate aminotransferase (AST) of all grades was seen in 38% and 40% of patients, respectively.54 Therefore, close monitoring of liver function is clinically warranted. In particular, patients need to be advised to avoid concomitant use of herbal supplements, a common practice in Asian countries.
CASE 1 CONTINUED
The patient does well while on erlotinib at 150 mg orally once daily for about 8 months, until he develops increasing abdominal pain. A CT scan of the abdomen and pelvis with contrast shows a new 8-cm right adrenal mass. Additionally, a repeat CT scan of the chest with contrast shows a stable lung mass but enlarging mediastinal lymphadenopathy.
How would you manage the patient at this point?
MANAGEMENT OF T790M MUTATION AFTER PROGRESSION ON FIRST-LINE EGFR TKIS
As mentioned above, the median PFS of patients with EGFR mutations treated with 1 of the 3 TKIs is around 9 to 13 months.46 Of the various resistance mechanisms that have been described, the T790M mutation is found in approximately 60% of patients who progress after treatment with first-line TKIs.55,56 Other mechanisms, such as HER2 amplification, MET amplification, or rarely small cell transformation, have been reported.56 The first- and second-generation EGFR TKIs function by binding to the ATP-binding domain of mutated EGFR, leading to inhibition of the downstream signaling pathways (Figure, part B) and ultimately cell death.35 The T790M mutation hinders the interaction between the ATP-binding domain of EGFR kinase and TKIs, resulting in treatment resistance and disease progression.57,58
Osimertinib is a third-generation irreversible EGFR TKI with activity against both sensitizing EGFR and resistant T790M mutations. It has low affinity for wide-type EGFR as well as insulin receptor and insulin-like growth factor receptor.59 Osimertinib has been fully approved for NSCLC patients with EGFR mutations who have progressed on first-line EGFR TKIs with the development of T790M mutation. An international phase 3 trial (AURA3) randomly assigned 419 patients in a 2:1 ratio to either osimertinib or platinum/pemetrexed. Eligible patients all had the documented EGFR mutations and disease progression after first-line EGFR TKIs. Central confirmation of the T790M mutation was required. Median PFS by investigator assessment, the trial’s primary end point, was 10.1 months for osimertinib versus 4.4 months for chemotherapy (HR 0.3 [95% CI 0.23 to 0.41]; P < 0.001). ORR was 71% for osimertinib versus 31% for chemotherapy (HR 5.39 [95% CI 3.47 to 8.48], P < 0.001). A total of 144 patients with stable and asymptomatic brain metastases were also eligible. Median PFS for this subset of patients treated with osimertinib and chemotherapy was 8.5 months and 4.2 months, respectively (HR 0.32 [95% CI 0.21 to 0.49]). In the AURA3 trial, osimertinib was better tolerated than chemotherapy, with 23% of patients treated with osimertinib experiencing grade 3 or 4 AEs as compared to 47% of chemotherapy-treated patients. The most common AEs of any grade were diarrhea (41%), rash (34%), dry skin (23%), and paronychia (22%).60
For the case patient, a reasonable approach would be to obtain a tissue biopsy of the adrenal mass and more importantly to check for the T790M mutation. Similar to the companion diagnostic for EGFR mutations, the cobas EGFR mutation test v2 is the FDA-approved test for T790M. However, if this resistance mutation is detected by any CLIA-certified laboratories, osimertinib should be the recommended treatment option. If tissue biopsy is not feasible, plasma-based testing should be considered. A blood-based companion diagnostic also is FDA approved.
ALK REARRANGEMENTS
CASE 2 PRESENTATION
A 42-year-old Korean woman who is a non-smoker with no significant past medical history presents with fatigue, unintentional weight loss of 20 lb in the past 4 months, and vague abdominal pain. A CT can of the abdomen and pelvis without contrast shows multiple foci in the liver and an indeterminate nodule in the right lung base. She subsequently undergoes PET/CT, which confirms multiple liver nodules/masses ranging from 1 to 3 cm with moderate FDG uptake. In addition, there is a 3.5-cm pleura-based lung mass on the right side with moderate FDG uptake. MRI-brain with and without contrast is negative for malignancy. A CT-guided biopsy of 1 of the liver masses is ordered and pathology returns positive for poorly differentiated adenocarcinoma consistent with lung primary. Molecular analysis reveals an echinoderm microtubule-associated protein-like 4 (EML4)-ALK rearrangement. She is placed on crizotinib by an outside oncologist and after about 3 weeks of therapy is doing well. She is now in your clinic for a second opinion. She says that some of her friends told her about another medication called ceritinib and was wondering if she would need to switch her cancer treatment.
How would you respond to this patient’s inquiry?
FIRST-LINE TKIS FOR ALK REARRANGEMENTS
ALK rearrangements are found in 2% to 7% of NSCLC, with EML4-ALK being the most prevalent fusion variant.61 The inversion of chromosome 2p leads to the fusion of the EML4 gene and the ALK gene, which causes the constitutive activation of the fusion protein and ultimately increased transformation and tumorigenicity.7,61 Patients harboring ALK rearrangements tend to be non-smokers. Adenocarcinoma, especially signet ring cell subtype, is the predominant histology. Compared to EGFR mutations, patients with ALK mutations are significantly younger and more likely to be men.62 ALK rearrangements can be detected by either FISH or IHC, and most next-generation sequencing (NGS) panels have the ability to identify this driver mutation.
Crizotinib is the first approved ALK inhibitor for the treatment of NSCLC in this molecular subset of patients.63 PROFILE 1014 is a phase 3 randomized trial that compared crizotinib with chemotherapy containing platinum/pemetrexed for up to 6 cycles. Crossover to crizotinib was allowed for patients with disease progression on chemotherapy. The primary end point was PFS by independent radiologic review. The crizotinib arm demonstrated superior PFS (10.9 months versus 7 months; HR 0.45 [95% CI 0.35 to 0.6], P < 0.001) and ORR (74% versus 45%, P < 0.001). Median survival was not reached in either arm (HR 0.82 [95% CI 0.54 to 1.26], P = 0.36).64 Based on this international trial, crizotinib is considered standard of care in the United States for treatment-naïve patients with advanced NSCLC harboring ALK rearrangements. The current recommended dose is 250 mg orally twice daily. Common treatment-related AEs of all grades include vision disorder (62%), nausea (53%), diarrhea (43%), vomiting (40%), edema (28%), and constipation (27%).65 PROFILE 1007 compared crizotinib with pemetrexed or docetaxel in ALK-rearranged NSCLC patients with prior exposure to 1 platinum-based chemotherapy. The median PFS was 7.7 months for crizotinib as compared to 3 months for chemotherapy (HR 0.49 [95% CI 0.37 to 0.64], P < 0.001). The response rates were 65% and 20% for crizotinib and chemotherapy, respectively (P < 0.001).66 In other countries, crizotinib following 1 prior platinum-based regimen may be considered standard of care based on this trial.
Ceritinib is an oral second-generation ALK inhibitor that is 20 times more potent than crizotinib based on enzymatic assays.67 It also targets ROS1 and insulin-like growth factor 1 receptor but not c-MET. It was first approved by the FDA in April 2014 for metastatic ALK-rearranged NSCLC following crizotinib.68 In May 2017, the FDA granted approval of ceritinib for treatment-naïve patients. This decision was based on the results of the ASCEND-4 trial, a randomized phase 3 trial assessing the efficacy and safety of ceritinib over chemotherapy in the first-line setting. The trial assigned 376 patients to either ceritinib at 750 mg once daily or platinum/pemetrexed for 4 cycles followed by maintenance pemetrexed. Median PFS was 16.6 months for ceritinib versus 8.1 months for chemotherapy (HR 0.55 [95% CI 0.42 to 0.73]; P < 0.00001).69 Toxicities of ceritinib are not negligible, with gastrointestinal toxicity being the most prevalent. For instance, diarrhea, nausea, vomiting, abdominal pain, and constipation of all grades were seen in 86%, 80%, 60%, 54%, and 29% of patients, respectively. Furthermore, fatigue and decreased appetite occurred in 52% and 34% of patients, respectively. In terms of laboratory abnormalities, 84% of patients experienced decreased hemoglobin of all grades; 80% increased ALT; 75% increased AST; 58% increased creatinine; 49% increased glucose; 36% decreased phosphate; and 28% increased lipase. Due to these AEs, the incidence of dose reduction was about 58% and the median onset was around 7 weeks.70
Alectinib is another oral second-generation ALK inhibitor that was approved by the FDA in December 2015 for the treatment of NSCLC patients with ALK rearrangements who have progressed on or are intolerant to crizotinib.71 Its indication will soon be broadened to the first-line setting based on the ALEX trial.72 Alectinib is a potent and highly selective TKI of ALK73 with activity against known resistant mutations to crizotinib.74,75 It also inhibits RET but not ROS1 or c-MET.76 ALEX, a randomized phase 3 study, compared alectinib with crizotinib in treatment-naïve patients with NSCLC harboring ALK rearrangements. The trial enrolled 303 patients and the median follow-up was approximately 18 months. The alectinib arm (600 mg twice daily) demonstrated significantly higher PFS by investigator-assessment, the trial’s primary end point. The 12-month event-free survival was 68.4% (95% CI 61% to 75.9%) versus 48.7% (95% CI 40.4% to 56.9%) for alectinib and crizotinib, respectively (HR 0.47 [95% CI 0.34 to 0.65], P < 0.001). The median PFS was not reached in the alectinib arm (95% CI 17.7 months to not estimable) as compared to 11.1 months in the crizotinib arm (95% CI 9.1 to 13.1 months).72 Alectinib is generally well tolerated. Common AEs of all grades include fatigue (41%), constipation (34%), edema (30%), and myalgia (29%). As alectinib can cause anemia, lymphopenia, hepatic toxicity, increased creatine phosphokinase, hyperglycemia, electrolyte abnormalities, and increased creatinine, periodic monitoring of these laboratory values is important, although most of these abnormalities are grade 1 or 2.77
Brigatinib, another oral second-generation ALK inhibitor, was granted accelerated approval by the FDA in April 2017 for ALK-rearranged and crizotinib-resistant NSCLC based on the ALTA trial. This randomized phase 2 study of brigatinib showed an ORR by investigator assessment of 54% (97.5% CI 43% to 65%) in the 180 mg once daily arm with lead-in of 90 mg once daily for 7 days. Median PFS was 12.9 months (95% CI 11.1 months to not reached [NR]).78 Currently, a phase 3 study of brigatinib versus crizotinib in ALK inhibitor–naïve patients is recruiting participants (ALTA-1L). It will be interesting to see if brigatinib can achieve a front-line indication.
Starting the case patient on crizotinib is well within the treatment guidelines. One may consider ceritinib or alectinib in the first-line setting, but both TKIs can be reserved upon disease progression. We would recommend a repeat biopsy at that point to look for resistant mechanisms, as certain secondary ALK mutations may be rescued by certain next-generation ALK inhibitors. For instance, the F1174V mutation has been reported to confer resistance to ceritinib but sensitivity to alectinib, while the opposite is true for I1171T. The G1202R mutation is resistant to ceritinib, alectinib, and brigatinib, but lorlatinib, a third-generation ALK inhibitor, has shown activity against this mutation.79 Furthermore, brain metastasis represents a treatment challenge for patients with ALK rearrangements. It is also an efficacy measure of next-generation ALK inhibitors, all of which have demonstrated better central nervous system activity than crizotinib.69,78,80 If the case patient were found to have brain metastasis at the initial diagnosis, either ceritinib or alectinib would be a reasonable choice since crizotinib has limited penetration of blood-brain barrier.81
ROS1 REARRANGEMENTS
CASE PRESENTATION 3
A 66-year-old Chinese woman who is a non-smoker with a past medical history of hypertension and hypothyroidism presents to the emergency department for worsening lower back pain. Initial workup includes x-ray of the lumbar spine followed by MRI with contrast, which shows a soft tissue mass at L3-4 without cord compression. CT of the chest, abdomen, and pelvis with contrast shows a 7-cm right hilar mass, bilateral small lung nodules, mediastinal lymphadenopathy, and multiple lytic lesions in ribs, lumbar spine, and pelvis. MRI-brain with and without contrast is negative for malignancy. She undergoes endo-bronchial ultrasound and biopsy of the right hilar mass, which shows poorly differentiated adenocarcinoma. While waiting for the result of the molecular analysis, the patient undergoes palliative radiation therapy to L2-5 with good pain relief. She is discharged from the hospital and presents to your clinic for follow up. Molecular analysis now reveals ROS1 rearrangement with CD74-ROS1 fusion.
What treatment plan should be put in place for this patient?
FIRST-LINE THERAPY FOR ROS1 REARRANGEMENTS
Approximately 2.4% of lung adenocarcinomas harbor ROS1 rearrangements.82 This distinct genetic alteration occurs more frequently in NSCLC patients who are younger, female, and never-smokers, and who have adenocarcinomas.8 It has been shown that ROS1 rearrangements rarely overlap with other genetic alterations including KRAS mutations, EGFR mutations, and ALK rearrangements.83 As a receptor tyrosine kinase, ROS1 is similar to ALK and insulin receptor family members.84 Crizotinib, which targets ALK, ROS1, and c-MET, was approved by the FDA on March 11, 2016, for the treatment of metastatic ROS1-rearranged NSCLC.85 The approval was based on a phase 2 expansion cohort of the original phase 1 study. Among 50 US patients enrolled in this expansion cohort, 3 had complete responses and 33 had partial responses with ORR of 72% (95% CI 58% to 84%). Median PFS was 19.2 months (95% CI 14.4 months to NR) and median duration of response (DOR) was 17.6 months (95% CI 14.5 months to NR).86 During longer follow-up, independent radiology review confirmed high ORR of 66% and median DOR of 18.3 months.85
Interestingly, no companion diagnostic assay has been approved for the detection of ROS1 rearrangements with the approval of crizotinib. In the United States, break apart FISH is the most common detection method. In fact, in the above mentioned phase 2 study, ROS1 rearrangements were detected in 49 out of 50 patients by this method.86 FISH can be technically challenging when dealing with high volume and multiple targets. Reverse transcriptase-PCR is another detection method, but it requires knowledge of the fusion partners. To date, at least 14 ROS1 fusion partners have been reported, with CD74 being the most common.87 NGS with appropriate design and validation can also be used to detect ROS1 rearrangements.
For the case patient, the recommendation would be to start her on crizotinib at 250 mg twice daily. Monitoring for vision disturbance, gastrointestinal complaints, and edema is warranted. Because the estimated onset of response is around 7.9 weeks,86 plans should be made to repeat her scans in approximately 2 months.
BRAF V600E MUTATIONS
CASE PRESENTATION 4
A 71-year-old Caucasian man with a past medical history of hypertension, dyslipidemia, and ischemic cerebrovascular accident without residual deficits was diagnosed with stage IV adenocarcinoma of the lung about 8 months ago. He has a 40 pack-year smoking history and quit smoking when he was diagnosed with lung cancer. His disease burden involved a large mediastinal mass, scattered pleural nodules, multiple lymphadenopathy, and several soft tissue masses. His outside oncologist started him on chemotherapy containing carboplatin and pemetrexed for 6 cycles followed by maintenance pemetrexed. The most recent restaging scans show disease progression with enlarging soft tissue masses and several new lytic bone lesions. MRI-brain with and without contrast shows 2 subcentimeter enhancing lesions. He transferred care to you approximately 4 weeks ago. You ordered a repeat biopsy of 1 of the enlarging soft tissue masses. Molecular analysis revealed BRAF V600E mutation. In the interim, he underwent stereotactic radiosurgery for the 2 brain lesions without any complications. The patient is now in your clinic for follow up.
What would be your recommended systemic treatment?
TARGETED THERAPIES FOR BRAF V600E MUTATION
BRAF mutations were first recognized as activating mutations in advanced melanomas, with BRAF V600E, resulting from the substitution of glutamic acid for valine at amino acid 600, being the most common. BRAF plays an important role in the mitogen-activated protein kinase (MAPK) signaling pathway. Briefly, the activation of MAPK pathway occurs upon ligand binding of receptor tyrosine kinases, which then involves RAS/BRAF/MEK/ERK in a stepwise manner, ultimately leading to cell survival. BRAF mutations have been increasingly recognized also as driver mutations in NSCLC.9–12 They can be detected by PCR or NGS method. The characteristics of NSCLC patients harboring BRAF mutations have been described by various groups.9–12 For instance, 1 case series showed that the incidence was 2.2% among patients with advanced lung adenocarcinoma; 50% of mutations were V600E, while G469A and D594G accounted for the remaining 39% and 11% of patients, respectively. All patients were either current or former smokers. The median OS of patients with BRAF mutations in this case series was NR, while it was 37 months for patients with EGFR mutations (P = 0.73) and NR for patients with ALK rearrangements (P = 0.64).9
For patients with BRAF V600E–mutant NSCLC who have progressed on platinum-based chemotherapy, the combination of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) may represent a new treatment paradigm. This was illustrated in a phase 2, nonrandomized, open-label study. A total of 57 patients were enrolled and 36 patients (63.2% [95% CI 49.3% to 75.6%]) achieved an overall response by investigator assessment, the trial’s primary end point. Disease control rate was 78.9% (95% CI 66.1% to 88.6%), with 4% complete response, 60% partial response, and 16% stable disease. PFS was 9.7 months (95% CI [6.9 to 19.6 months]). The safety profile was comparable to what had been observed in patients with melanoma treated with this regimen. More specifically, 56% of patients on this trial reported serious AEs, including pyrexia (16%), anemia (5%), confusional state (4%), decreased appetite (4%), hemoptysis (4%), hypercalcemia (4%), nausea (4%), and cutaneous squamous cell carcinoma (4%). In addition, neutropenia (9%) and hyponatremia (7%) were the most common grade 3-4 AEs.16
The case patient has experienced disease progression after 1 line of platinum-based chemotherapy, so the combination of dabrafenib and trametinib would be a robust systemic treatment option. dabrafenib as a single agent has also been studied in BRAF V600E–mutant NSCLC in a phase 2 trial. The overall response by investigator assessment among 84 patients was 33% (95% CI 23% to 45%).14 Vemurafenib, another oral BRAF TKI, has demonstrated efficacy for NSCLC patients harboring BRAF V600E mutation. In the cohort of 20 patients with NSCLC, the response rate was 42% (95% CI 20% to 67%) and median PFS was 7.3 months (95% CI 3.5 to 10.8 months).13 Patients with non-V600E mutations have shown variable responses to targeted therapies. MEK TKIs may be considered in this setting; however, the details of this discussion are beyond the scope of this review.
CONCLUSION
The management of advanced NSCLC with driver mutations has seen revolutionary changes over the past decade. Tremendous research has been done in order to first understand the molecular pathogenesis of NSCLC and then discover driver mutations that would lead to development of targeted therapies with clinically significant efficacy as well as tolerability. More recently, increasing efforts have focused on how to conquer acquired resistance in patients with disease progression after first-line TKIs. The field of EGFR-mutant NSCLC has set a successful example, but the work is nowhere near finished. The goals are to search for more driver mutations and to design agents that could potentially block cell survival signals once and for all.
INTRODUCTION
Lung cancer is the second most common type of cancer in the United States, with 222,500 estimated new cases in 2017, according to the American Cancer Society.1 However, it is by far the number one cause of death due to cancer, with an estimated 155,870 lung cancer–related deaths occurring in 2017, which is higher than the number of deaths due to breast cancer, prostate cancer, and colorectal cancer combined.1,2 Despite slightly decreasing incidence and mortality over the past decade, largely due to smoking cessation, the 5-year survival rate of lung cancer remains dismal at approximately 18%.2–4
Non-small cell lung cancer (NSCLC) accounts for 80% to 85% of all lung cancer cases.4 Traditionally, it is further divided based on histology: adenocarcinoma, squamous cell carcinoma, large cell carcinoma, and not otherwise specified.5 Chemotherapy had been the cornerstone of treatment for stage IV NSCLC. It is not target-specific and is most effective against rapidly growing cells. Common adverse effects include alopecia, nausea/vomiting, myelosuppression, cardiotoxicity, neuropathy, and nephrotoxicity. However, this paradigm has shifted following the discovery of mutations of the epidermal growth factor receptor (EGFR) gene as an oncogenic driver that confers sensitivity to small molecule tyrosine kinase inhibitors (TKIs) targeting EGFR.6 The EGFR inhibitors are given orally and have a spectrum of toxicities (eg, such as rash, diarrhea, and elevated transaminases) different from that of systemic chemotherapy, which is often administered intravenously. Following the discovery of EGFR mutations, rearrangements of the anaplastic lymphoma kinase (ALK) gene7 and ROS1 gene8 were identified as targetable driver mutations in NSCLC. The frequency of both rearrangements is lower than that of EGFR mutations. Additionally, BRAF V600E mutation has been identified in NSCLC.9–12 This activation mutation is commonly seen in melanoma. Agents that have already been approved for the treatment of melanoma with the BRAF V600E mutation are being tested in NSCLC patients with this mutation.13–16
Given the effectiveness and tolerability of targeted therapy, identifying this distinct molecular subset of NSCLC patients is critical in treatment. Currently, molecular testing is mandatory in all stage IV patients with non-squamous cell carcinoma, as a preponderance of patients with driver mutations have this histology subtype.5,17–19 For patients with squamous cell carcinoma, molecular testing should be considered if the biopsy specimen is small, there is mixed histology, or the patient is a nonsmoker.5,20 Several techniques are commonly utilized in detecting these genetic alterations. EGFR mutation can be detected by polymerase chain reaction (PCR), ALK or ROS1 rearrangement can be detected by fluorescence in-situ hybridization (FISH), and immunohistochemistry (IHC) can also be used to detect ALK rearrangement. The current guideline is to use comprehensive genomic profiling to capture all the potential molecular targets simultaneously instead of running stepwise tests just for EGFR, ALK, and ROS1.5 BRAF V600E mutation,13–16 MET exon 14 skipping mutation,21–24 RET rearrangements,25–27 and HER2 mutations28–30 are among the emergent genetic alterations with various responses to targeted therapy.31 Some of these targeted agents have been approved for other types of malignancy, and others are still in the development phase.
Several initiatives worldwide have reported better outcomes of patients with driver mutations treated with targeted therapy. For instance, the Lung Cancer Mutation Consortium in the United States demonstrated that the median survival of patients without driver mutations, with drivers mutations but not treated with targeted therapy, and with driver mutations and treated with targeted therapy was 2.08 years, 2.38 years, and 3.49 years, respectively.32 The French Cooperative Thoracic Intergroup-French National Cancer Institute demonstrated that the median survival for patients with driver mutations versus those without driver mutations was 16.5 months versus 11.8 months.33 The Spanish Lung Cancer Group demonstrated that the overall survival (OS) for patients with EGFR mutations treated with erlotinib was 27 months.34 The mutations in lung cancer, their frequencies, and the downstream signaling pathways are depicted in the Figure.35
In this article, we discuss targeted therapy for patients with EGFR mutations, ALK rearrangements, ROS1 rearrangements, and BRAF V600E mutation. We also discuss the management of patients with EGFR mutations who develop a secondary mutation after TKI therapy. Almost all of the targeted agents discussed herein have been approved by the US Food and Drug Administration (FDA), so they are considered standard of care. All available phase 3 trials pertinent to these targeted therapies are included in the discussion.
EGFR MUTATIONS
CASE PRESENTATION 1
A 54-year-old Caucasian man who is a former smoker with a 10 pack-year history and past medical history of hypertension and dyslipidemia presents with progressive dyspnea for several weeks. A chest x-ray shows moderate pleural effusion on the left side with possible mass-like opacity on the left upper lung field. An ultrasound-guided thoracentesis is performed and cytology is positive for adenocarcinoma of likely pulmonary origin. Staging workup including positron emission tomography (PET)/computed tomography (CT) and magnetic resonance imaging of the brain with and without contrast is done. PET/CT shows a 5.5-cm mass in the left upper lobe of the lung with high fluorodeoxyglucose (FDG) uptake, several 1- to 2-cm mediastinal lymph nodes with moderate FDG uptake, and small pleural effusion on both sides with moderate FDG uptake. MRI-brain is negative for malignancy. The patient subsequently undergoes a CT-guided biopsy of the lung mass, which shows moderately differentiated adenocarcinoma. Comprehensive molecular profiling reveals EGFR L858R mutation only. The patient now presents for the initial consultation. Of note, his Eastern Cooperative Oncology Group performance status is 1.
What is the next step in the management of this patient?
FIRST-LINE TKI FOR SENSITIZING EGFR MUTATIONS
The 2 most common EGFR mutations are deletions in exon 19 and substitution of arginine for leucine in exon 21 (L858R), found in approximately 45% and 40% of patients with EGFR mutations, respectively.36 Both mutations are sensitive to EGFR TKIs. The benefit may be greater in patients with exon 19 deletions as compared to exon 21 L858R substitution,37,38 but this has not been demonstrated consistently in clinical trials.39-43 In the United States, EGFR mutations are found in approximately 10% of patients with NSCLC, while the incidence can be as high as 50% in Asia.44 Even though the cobas EGFR mutation test is the companion diagnostic approved by the US FDA, a positive test result from any laboratory with the Clinical Laboratory Improvement Amendments (CLIA) certificate should prompt the use of an EGFR TKI as the initial treatment.
Three EGFR TKIs that have been approved as first-line therapy in the United States are available: erlotinib, afatinib, and gefitinib.5 Both erlotinib and gefitinib are considered first-generation TKIs. They have higher binding affinity for the 2 common EGFR mutations than wild-type EGFR. In addition, they reversibly bind to the intracellular tyrosine kinase domain, resulting in inhibition of autophosphorylation of the tyrosine residues. Afatinib, a second-generation and irreversible TKI, targets EGFR (HER1) as well as HER2 and HER4.45
The superior efficacy of the EGFR TKIs over platinum doublet chemotherapy in treatment-naïve patients with EGFR mutations has been demonstrated in 7 randomized trials to date (Table).46 Erlotinib was the TKI arm for the OPTIMAL,41 EURTAC,42 and ENSURE trials;38 afatinib was the TKI arm for LUX-LUNG 337 and 6;43 gefitinib was the TKI arm for NEJ00239,47 and WJTOG3405.40 A meta-analysis of these 7 trials by Lee et al showed that progression-free survival (PFS) was significantly prolonged by EGFR TKIs (hazard ratio [HR] 0.37 [95% confidence interval {CI} 0.32 to 0.42]).46 For instance, in the EURTAC trial, median PFS was 9.7 months for patients treated with erlotinib as compared to 5.2 months for patients treated with platinum/gemcitabine or platinum/docetaxel.42 In this meta-analysis, prespecified subgroups included age, sex, ethnicity, smoking status, performance status, tumor histology, and EGFR mutation subtype. The superior outcome with TKIs was observed in all subgroups. Furthermore, patients with exon 19 deletions, nonsmokers, and women had even better outcomes.46
Erlotinib is the most commonly used TKI in the United States largely because gefitinib was off the market for some time until it was re-approved by the FDA in 2015. Interestingly, this “re-approval” was not based on either 1 of the 2 prospective trials (NEJ00239,47 and WJTOG340540), but rather was based on an exploratory analysis of the IPASS trial48,49 as well as a prospective phase 4, single-arm trial in Europe (IFUM).50 The superior efficacy of gefitinib over carboplatin/paclitaxel among patients with EGFR mutations in the IPASS trial was confirmed by blind independent central review, with longer PFS (HR 0.54 [95% CI 0.38 to 0.79] P = 0.0012) and higher objective response rate (ORR; odds ratio 3 [95% CI 1.63 to 5.54], P = 0.0004).49
CASE 1 CONTINUED
Based on the EGFR L858R mutation status, the patient is started on erlotinib. He is quite happy that he does not need intravenous chemotherapy but wants to know what toxicities he might potentially have with erlotinib.
What are the common adverse effects (AEs) of EGFR TKIs? How are AEs of TKIs managed?
Safety Profile
The important toxicities associated with EGFR TKIs are rash, gastrointestinal toxicity, hepatic toxicity, and pulmonary toxicity. Rash is an AE specific to all agents blocking the EGFR pathway, including small molecules and monoclonal antibodies such as cetuximab. The epidermis has a high level of expression of EGFR, which contributes to this toxicity.51 Rash usually presents as dry skin or acneiform eruption. Prophylactic treatment with oral tetracyclines and topical corticosteroids is generally recommended upon initiation of TKI therapy. Diarrhea is the most prevalent gastrointestinal toxicity. All patients starting treatment should be given prescriptions to manage diarrhea such as loperamide and be advised to call when it occurs. Hepatic toxicity is often manifested as elevated transaminases or bilirubin. Interstitial lung disease (ILD) is a rare but potentially fatal pulmonary toxicity.
Rash of any grade was reported in 49.2% of patients treated with erlotinib in clinical trials, while grade 3 rash occurred in 6% of patients and no grade 4 was reported. Diarrhea of any grade was reported in 20.3% of patients, grade 3 diarrhea occurred in 1.8%, and no grade 4 was reported. Grade 2 and 3 alanine aminotransferase (ALT) elevations were seen in 2% and 1% of patients, respectively. Grade 2 and 3 bilirubin elevations were seen in 4% and less than 1% of patients, respectively. The incidence of serious ILD-like events was less than 1%.52
Afatinib is associated with higher incidences of rash and diarrhea. Specifically, diarrhea and rash of all grades were reported in 96% and 90% of patients treated with afatinib, respectively. Paronychia of all grades occurred in 58% of patients. Elevated ALT of all grades was seen in 11% of patients. Approximately 1.5% of patients treated with afatinib across clinical trials had ILD or ILD-like AEs.53
Gefitinib, the most commonly used TKI outside United States, has a toxicity profile similar to erlotinib, except for hepatic toxicity. For instance, rash of all grades occurred in 47% of patients, diarrhea of all grades occurred in 29% of patients, and ILD or ILD-like AEs occurred in 1.3% of patients across clinical trials. In comparison, elevated ALT and aspartate aminotransferase (AST) of all grades was seen in 38% and 40% of patients, respectively.54 Therefore, close monitoring of liver function is clinically warranted. In particular, patients need to be advised to avoid concomitant use of herbal supplements, a common practice in Asian countries.
CASE 1 CONTINUED
The patient does well while on erlotinib at 150 mg orally once daily for about 8 months, until he develops increasing abdominal pain. A CT scan of the abdomen and pelvis with contrast shows a new 8-cm right adrenal mass. Additionally, a repeat CT scan of the chest with contrast shows a stable lung mass but enlarging mediastinal lymphadenopathy.
How would you manage the patient at this point?
MANAGEMENT OF T790M MUTATION AFTER PROGRESSION ON FIRST-LINE EGFR TKIS
As mentioned above, the median PFS of patients with EGFR mutations treated with 1 of the 3 TKIs is around 9 to 13 months.46 Of the various resistance mechanisms that have been described, the T790M mutation is found in approximately 60% of patients who progress after treatment with first-line TKIs.55,56 Other mechanisms, such as HER2 amplification, MET amplification, or rarely small cell transformation, have been reported.56 The first- and second-generation EGFR TKIs function by binding to the ATP-binding domain of mutated EGFR, leading to inhibition of the downstream signaling pathways (Figure, part B) and ultimately cell death.35 The T790M mutation hinders the interaction between the ATP-binding domain of EGFR kinase and TKIs, resulting in treatment resistance and disease progression.57,58
Osimertinib is a third-generation irreversible EGFR TKI with activity against both sensitizing EGFR and resistant T790M mutations. It has low affinity for wide-type EGFR as well as insulin receptor and insulin-like growth factor receptor.59 Osimertinib has been fully approved for NSCLC patients with EGFR mutations who have progressed on first-line EGFR TKIs with the development of T790M mutation. An international phase 3 trial (AURA3) randomly assigned 419 patients in a 2:1 ratio to either osimertinib or platinum/pemetrexed. Eligible patients all had the documented EGFR mutations and disease progression after first-line EGFR TKIs. Central confirmation of the T790M mutation was required. Median PFS by investigator assessment, the trial’s primary end point, was 10.1 months for osimertinib versus 4.4 months for chemotherapy (HR 0.3 [95% CI 0.23 to 0.41]; P < 0.001). ORR was 71% for osimertinib versus 31% for chemotherapy (HR 5.39 [95% CI 3.47 to 8.48], P < 0.001). A total of 144 patients with stable and asymptomatic brain metastases were also eligible. Median PFS for this subset of patients treated with osimertinib and chemotherapy was 8.5 months and 4.2 months, respectively (HR 0.32 [95% CI 0.21 to 0.49]). In the AURA3 trial, osimertinib was better tolerated than chemotherapy, with 23% of patients treated with osimertinib experiencing grade 3 or 4 AEs as compared to 47% of chemotherapy-treated patients. The most common AEs of any grade were diarrhea (41%), rash (34%), dry skin (23%), and paronychia (22%).60
For the case patient, a reasonable approach would be to obtain a tissue biopsy of the adrenal mass and more importantly to check for the T790M mutation. Similar to the companion diagnostic for EGFR mutations, the cobas EGFR mutation test v2 is the FDA-approved test for T790M. However, if this resistance mutation is detected by any CLIA-certified laboratories, osimertinib should be the recommended treatment option. If tissue biopsy is not feasible, plasma-based testing should be considered. A blood-based companion diagnostic also is FDA approved.
ALK REARRANGEMENTS
CASE 2 PRESENTATION
A 42-year-old Korean woman who is a non-smoker with no significant past medical history presents with fatigue, unintentional weight loss of 20 lb in the past 4 months, and vague abdominal pain. A CT can of the abdomen and pelvis without contrast shows multiple foci in the liver and an indeterminate nodule in the right lung base. She subsequently undergoes PET/CT, which confirms multiple liver nodules/masses ranging from 1 to 3 cm with moderate FDG uptake. In addition, there is a 3.5-cm pleura-based lung mass on the right side with moderate FDG uptake. MRI-brain with and without contrast is negative for malignancy. A CT-guided biopsy of 1 of the liver masses is ordered and pathology returns positive for poorly differentiated adenocarcinoma consistent with lung primary. Molecular analysis reveals an echinoderm microtubule-associated protein-like 4 (EML4)-ALK rearrangement. She is placed on crizotinib by an outside oncologist and after about 3 weeks of therapy is doing well. She is now in your clinic for a second opinion. She says that some of her friends told her about another medication called ceritinib and was wondering if she would need to switch her cancer treatment.
How would you respond to this patient’s inquiry?
FIRST-LINE TKIS FOR ALK REARRANGEMENTS
ALK rearrangements are found in 2% to 7% of NSCLC, with EML4-ALK being the most prevalent fusion variant.61 The inversion of chromosome 2p leads to the fusion of the EML4 gene and the ALK gene, which causes the constitutive activation of the fusion protein and ultimately increased transformation and tumorigenicity.7,61 Patients harboring ALK rearrangements tend to be non-smokers. Adenocarcinoma, especially signet ring cell subtype, is the predominant histology. Compared to EGFR mutations, patients with ALK mutations are significantly younger and more likely to be men.62 ALK rearrangements can be detected by either FISH or IHC, and most next-generation sequencing (NGS) panels have the ability to identify this driver mutation.
Crizotinib is the first approved ALK inhibitor for the treatment of NSCLC in this molecular subset of patients.63 PROFILE 1014 is a phase 3 randomized trial that compared crizotinib with chemotherapy containing platinum/pemetrexed for up to 6 cycles. Crossover to crizotinib was allowed for patients with disease progression on chemotherapy. The primary end point was PFS by independent radiologic review. The crizotinib arm demonstrated superior PFS (10.9 months versus 7 months; HR 0.45 [95% CI 0.35 to 0.6], P < 0.001) and ORR (74% versus 45%, P < 0.001). Median survival was not reached in either arm (HR 0.82 [95% CI 0.54 to 1.26], P = 0.36).64 Based on this international trial, crizotinib is considered standard of care in the United States for treatment-naïve patients with advanced NSCLC harboring ALK rearrangements. The current recommended dose is 250 mg orally twice daily. Common treatment-related AEs of all grades include vision disorder (62%), nausea (53%), diarrhea (43%), vomiting (40%), edema (28%), and constipation (27%).65 PROFILE 1007 compared crizotinib with pemetrexed or docetaxel in ALK-rearranged NSCLC patients with prior exposure to 1 platinum-based chemotherapy. The median PFS was 7.7 months for crizotinib as compared to 3 months for chemotherapy (HR 0.49 [95% CI 0.37 to 0.64], P < 0.001). The response rates were 65% and 20% for crizotinib and chemotherapy, respectively (P < 0.001).66 In other countries, crizotinib following 1 prior platinum-based regimen may be considered standard of care based on this trial.
Ceritinib is an oral second-generation ALK inhibitor that is 20 times more potent than crizotinib based on enzymatic assays.67 It also targets ROS1 and insulin-like growth factor 1 receptor but not c-MET. It was first approved by the FDA in April 2014 for metastatic ALK-rearranged NSCLC following crizotinib.68 In May 2017, the FDA granted approval of ceritinib for treatment-naïve patients. This decision was based on the results of the ASCEND-4 trial, a randomized phase 3 trial assessing the efficacy and safety of ceritinib over chemotherapy in the first-line setting. The trial assigned 376 patients to either ceritinib at 750 mg once daily or platinum/pemetrexed for 4 cycles followed by maintenance pemetrexed. Median PFS was 16.6 months for ceritinib versus 8.1 months for chemotherapy (HR 0.55 [95% CI 0.42 to 0.73]; P < 0.00001).69 Toxicities of ceritinib are not negligible, with gastrointestinal toxicity being the most prevalent. For instance, diarrhea, nausea, vomiting, abdominal pain, and constipation of all grades were seen in 86%, 80%, 60%, 54%, and 29% of patients, respectively. Furthermore, fatigue and decreased appetite occurred in 52% and 34% of patients, respectively. In terms of laboratory abnormalities, 84% of patients experienced decreased hemoglobin of all grades; 80% increased ALT; 75% increased AST; 58% increased creatinine; 49% increased glucose; 36% decreased phosphate; and 28% increased lipase. Due to these AEs, the incidence of dose reduction was about 58% and the median onset was around 7 weeks.70
Alectinib is another oral second-generation ALK inhibitor that was approved by the FDA in December 2015 for the treatment of NSCLC patients with ALK rearrangements who have progressed on or are intolerant to crizotinib.71 Its indication will soon be broadened to the first-line setting based on the ALEX trial.72 Alectinib is a potent and highly selective TKI of ALK73 with activity against known resistant mutations to crizotinib.74,75 It also inhibits RET but not ROS1 or c-MET.76 ALEX, a randomized phase 3 study, compared alectinib with crizotinib in treatment-naïve patients with NSCLC harboring ALK rearrangements. The trial enrolled 303 patients and the median follow-up was approximately 18 months. The alectinib arm (600 mg twice daily) demonstrated significantly higher PFS by investigator-assessment, the trial’s primary end point. The 12-month event-free survival was 68.4% (95% CI 61% to 75.9%) versus 48.7% (95% CI 40.4% to 56.9%) for alectinib and crizotinib, respectively (HR 0.47 [95% CI 0.34 to 0.65], P < 0.001). The median PFS was not reached in the alectinib arm (95% CI 17.7 months to not estimable) as compared to 11.1 months in the crizotinib arm (95% CI 9.1 to 13.1 months).72 Alectinib is generally well tolerated. Common AEs of all grades include fatigue (41%), constipation (34%), edema (30%), and myalgia (29%). As alectinib can cause anemia, lymphopenia, hepatic toxicity, increased creatine phosphokinase, hyperglycemia, electrolyte abnormalities, and increased creatinine, periodic monitoring of these laboratory values is important, although most of these abnormalities are grade 1 or 2.77
Brigatinib, another oral second-generation ALK inhibitor, was granted accelerated approval by the FDA in April 2017 for ALK-rearranged and crizotinib-resistant NSCLC based on the ALTA trial. This randomized phase 2 study of brigatinib showed an ORR by investigator assessment of 54% (97.5% CI 43% to 65%) in the 180 mg once daily arm with lead-in of 90 mg once daily for 7 days. Median PFS was 12.9 months (95% CI 11.1 months to not reached [NR]).78 Currently, a phase 3 study of brigatinib versus crizotinib in ALK inhibitor–naïve patients is recruiting participants (ALTA-1L). It will be interesting to see if brigatinib can achieve a front-line indication.
Starting the case patient on crizotinib is well within the treatment guidelines. One may consider ceritinib or alectinib in the first-line setting, but both TKIs can be reserved upon disease progression. We would recommend a repeat biopsy at that point to look for resistant mechanisms, as certain secondary ALK mutations may be rescued by certain next-generation ALK inhibitors. For instance, the F1174V mutation has been reported to confer resistance to ceritinib but sensitivity to alectinib, while the opposite is true for I1171T. The G1202R mutation is resistant to ceritinib, alectinib, and brigatinib, but lorlatinib, a third-generation ALK inhibitor, has shown activity against this mutation.79 Furthermore, brain metastasis represents a treatment challenge for patients with ALK rearrangements. It is also an efficacy measure of next-generation ALK inhibitors, all of which have demonstrated better central nervous system activity than crizotinib.69,78,80 If the case patient were found to have brain metastasis at the initial diagnosis, either ceritinib or alectinib would be a reasonable choice since crizotinib has limited penetration of blood-brain barrier.81
ROS1 REARRANGEMENTS
CASE PRESENTATION 3
A 66-year-old Chinese woman who is a non-smoker with a past medical history of hypertension and hypothyroidism presents to the emergency department for worsening lower back pain. Initial workup includes x-ray of the lumbar spine followed by MRI with contrast, which shows a soft tissue mass at L3-4 without cord compression. CT of the chest, abdomen, and pelvis with contrast shows a 7-cm right hilar mass, bilateral small lung nodules, mediastinal lymphadenopathy, and multiple lytic lesions in ribs, lumbar spine, and pelvis. MRI-brain with and without contrast is negative for malignancy. She undergoes endo-bronchial ultrasound and biopsy of the right hilar mass, which shows poorly differentiated adenocarcinoma. While waiting for the result of the molecular analysis, the patient undergoes palliative radiation therapy to L2-5 with good pain relief. She is discharged from the hospital and presents to your clinic for follow up. Molecular analysis now reveals ROS1 rearrangement with CD74-ROS1 fusion.
What treatment plan should be put in place for this patient?
FIRST-LINE THERAPY FOR ROS1 REARRANGEMENTS
Approximately 2.4% of lung adenocarcinomas harbor ROS1 rearrangements.82 This distinct genetic alteration occurs more frequently in NSCLC patients who are younger, female, and never-smokers, and who have adenocarcinomas.8 It has been shown that ROS1 rearrangements rarely overlap with other genetic alterations including KRAS mutations, EGFR mutations, and ALK rearrangements.83 As a receptor tyrosine kinase, ROS1 is similar to ALK and insulin receptor family members.84 Crizotinib, which targets ALK, ROS1, and c-MET, was approved by the FDA on March 11, 2016, for the treatment of metastatic ROS1-rearranged NSCLC.85 The approval was based on a phase 2 expansion cohort of the original phase 1 study. Among 50 US patients enrolled in this expansion cohort, 3 had complete responses and 33 had partial responses with ORR of 72% (95% CI 58% to 84%). Median PFS was 19.2 months (95% CI 14.4 months to NR) and median duration of response (DOR) was 17.6 months (95% CI 14.5 months to NR).86 During longer follow-up, independent radiology review confirmed high ORR of 66% and median DOR of 18.3 months.85
Interestingly, no companion diagnostic assay has been approved for the detection of ROS1 rearrangements with the approval of crizotinib. In the United States, break apart FISH is the most common detection method. In fact, in the above mentioned phase 2 study, ROS1 rearrangements were detected in 49 out of 50 patients by this method.86 FISH can be technically challenging when dealing with high volume and multiple targets. Reverse transcriptase-PCR is another detection method, but it requires knowledge of the fusion partners. To date, at least 14 ROS1 fusion partners have been reported, with CD74 being the most common.87 NGS with appropriate design and validation can also be used to detect ROS1 rearrangements.
For the case patient, the recommendation would be to start her on crizotinib at 250 mg twice daily. Monitoring for vision disturbance, gastrointestinal complaints, and edema is warranted. Because the estimated onset of response is around 7.9 weeks,86 plans should be made to repeat her scans in approximately 2 months.
BRAF V600E MUTATIONS
CASE PRESENTATION 4
A 71-year-old Caucasian man with a past medical history of hypertension, dyslipidemia, and ischemic cerebrovascular accident without residual deficits was diagnosed with stage IV adenocarcinoma of the lung about 8 months ago. He has a 40 pack-year smoking history and quit smoking when he was diagnosed with lung cancer. His disease burden involved a large mediastinal mass, scattered pleural nodules, multiple lymphadenopathy, and several soft tissue masses. His outside oncologist started him on chemotherapy containing carboplatin and pemetrexed for 6 cycles followed by maintenance pemetrexed. The most recent restaging scans show disease progression with enlarging soft tissue masses and several new lytic bone lesions. MRI-brain with and without contrast shows 2 subcentimeter enhancing lesions. He transferred care to you approximately 4 weeks ago. You ordered a repeat biopsy of 1 of the enlarging soft tissue masses. Molecular analysis revealed BRAF V600E mutation. In the interim, he underwent stereotactic radiosurgery for the 2 brain lesions without any complications. The patient is now in your clinic for follow up.
What would be your recommended systemic treatment?
TARGETED THERAPIES FOR BRAF V600E MUTATION
BRAF mutations were first recognized as activating mutations in advanced melanomas, with BRAF V600E, resulting from the substitution of glutamic acid for valine at amino acid 600, being the most common. BRAF plays an important role in the mitogen-activated protein kinase (MAPK) signaling pathway. Briefly, the activation of MAPK pathway occurs upon ligand binding of receptor tyrosine kinases, which then involves RAS/BRAF/MEK/ERK in a stepwise manner, ultimately leading to cell survival. BRAF mutations have been increasingly recognized also as driver mutations in NSCLC.9–12 They can be detected by PCR or NGS method. The characteristics of NSCLC patients harboring BRAF mutations have been described by various groups.9–12 For instance, 1 case series showed that the incidence was 2.2% among patients with advanced lung adenocarcinoma; 50% of mutations were V600E, while G469A and D594G accounted for the remaining 39% and 11% of patients, respectively. All patients were either current or former smokers. The median OS of patients with BRAF mutations in this case series was NR, while it was 37 months for patients with EGFR mutations (P = 0.73) and NR for patients with ALK rearrangements (P = 0.64).9
For patients with BRAF V600E–mutant NSCLC who have progressed on platinum-based chemotherapy, the combination of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) may represent a new treatment paradigm. This was illustrated in a phase 2, nonrandomized, open-label study. A total of 57 patients were enrolled and 36 patients (63.2% [95% CI 49.3% to 75.6%]) achieved an overall response by investigator assessment, the trial’s primary end point. Disease control rate was 78.9% (95% CI 66.1% to 88.6%), with 4% complete response, 60% partial response, and 16% stable disease. PFS was 9.7 months (95% CI [6.9 to 19.6 months]). The safety profile was comparable to what had been observed in patients with melanoma treated with this regimen. More specifically, 56% of patients on this trial reported serious AEs, including pyrexia (16%), anemia (5%), confusional state (4%), decreased appetite (4%), hemoptysis (4%), hypercalcemia (4%), nausea (4%), and cutaneous squamous cell carcinoma (4%). In addition, neutropenia (9%) and hyponatremia (7%) were the most common grade 3-4 AEs.16
The case patient has experienced disease progression after 1 line of platinum-based chemotherapy, so the combination of dabrafenib and trametinib would be a robust systemic treatment option. dabrafenib as a single agent has also been studied in BRAF V600E–mutant NSCLC in a phase 2 trial. The overall response by investigator assessment among 84 patients was 33% (95% CI 23% to 45%).14 Vemurafenib, another oral BRAF TKI, has demonstrated efficacy for NSCLC patients harboring BRAF V600E mutation. In the cohort of 20 patients with NSCLC, the response rate was 42% (95% CI 20% to 67%) and median PFS was 7.3 months (95% CI 3.5 to 10.8 months).13 Patients with non-V600E mutations have shown variable responses to targeted therapies. MEK TKIs may be considered in this setting; however, the details of this discussion are beyond the scope of this review.
CONCLUSION
The management of advanced NSCLC with driver mutations has seen revolutionary changes over the past decade. Tremendous research has been done in order to first understand the molecular pathogenesis of NSCLC and then discover driver mutations that would lead to development of targeted therapies with clinically significant efficacy as well as tolerability. More recently, increasing efforts have focused on how to conquer acquired resistance in patients with disease progression after first-line TKIs. The field of EGFR-mutant NSCLC has set a successful example, but the work is nowhere near finished. The goals are to search for more driver mutations and to design agents that could potentially block cell survival signals once and for all.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
- Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol 2016;893:1–19.
- Alberg AJ, Brock MV, Ford JG, et al. Epidemiology of lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013;143:e1S–29S.
- Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-3013, based on November 2015 SEER data submission, posted to the SEER website, April 2016. Bethesda (MD): National Cancer Institute; 2016.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Non-Small Cell Lung Cancer: 1–190.
- Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–39.
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–6.
- Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancer. J Clin Oncol 2012;30:863–70.
- Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011;29:2046–51.
- Kinno T, Tsuta K, Shiraishi K, et al. Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol 2014;25:138–42.
- Litvak AM, Paik PK, Woo KM, et al. Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J Thorac Oncol 2014;9:1669–74.
- Villaruz LC, Socinski MA, Abberbock S, et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer 2015;121:448–56.
- Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–36.
- Planchard D, Kim TM, Mazieres J, et al. DaBRAFenib in patients with BRAF V600E-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol 2016;17:642–50.
- Gautschi O, Milia J, Cabarrou B, et al. Targeted therapy for patients with BRAF-mutant lung cancer: results from the European EURAF cohort. J Thorac Oncol 2015;10:1451–7.
- Planchard D, Besse B, Groen HJ, et al. DaBRAFenib plus trametinib in patients with previously treated BRAF V600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 2016;17:984–93.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. J Thorac Oncol 2013;8:823–59.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. Arch Pathol Lab Med 2013;137:828–60.
- Leighl NB, Rekhtman N, Biermann WA, et al. Molecular testing for selection of patients with lung cancer for epidermal growth factor receptor and anaplastic lymphoma kinase tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology guideline. J Clin Oncol 2014;32:3673–9.
- Paik PK, Varghese AM, Sima CS, et al. Response to erlotinib in patients with EGFR mutant advanced non-small cell lung cancers with a squamous or squamous-like component. Mol Cancer Ther 2012;11:2535–40.
- Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov 2015;5:842–9.
- Awad MM, Oxnard GR, Jackman DM, et al. MET Exon 14 mutations in non-small-cell lung cancer are associated with advanced age, and stage-dependent MET genomic amplification, and c-MET overexpression. J Clin Oncol 2016;34:721–30.
- Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thorac Oncol 2016;11:1493–502.
- Reungwetwattana T, Liang Y, Zhu V, et al. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The why, the how, the who, the unknown, and the inevitable. Lung Cancer 2017;103:27–37.
- Drilon A, Wang L, Hasanovic A, et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov 2013;3:630–5.
- Lin JJ, Kennedy E, Sequist LV, et al. Clinical activity of alectinib in advanced RET-rearranged non-small cell lung cancer. J Thorac Oncol 2016;11:2027–32.
- Drilon A, Rekhtman N, Arcila M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol 2016;17:1653–60.
- Cappuzzo F, Bemis L, Varella-Garcia M. HER2 mutation and response to trastuzumab therapy in non-small-cell lung cancer. N Engl J Med 2006;354:2619–21.
- Mazieres J. Barlesi F, Filleron T, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targted drugs: results from the European EUHER2 cohort. Annal Oncol 2016;27:281–6.
- Ou SH, Schrock AB, Bocharov EV, et al. HER2 transmembrane (TMD) mutations (V659/G660) that stabilize homo- and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J Thorac Oncol 2017;12:446–57.
- Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emergent therapies. Cancer Discov 2017;7:596–609.
- Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014;311:1998–2006.
- Barlesi F, Mazieres J, Merlio JP, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016;387:1415–26.
- Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361:958–67.
- Rosell R, Karachaliou N, et al. Large-scale screening for somatic mutations in lung cancer. Lancet 2016;387:1354–6.
- Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005;97:339–46.
- Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013;31:3327–34.
- Wu YL, Chou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol 2015;26:1883–9.
- Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Eng J Med 2010;362:2380–8.
- Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121–8.
- Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011;12:735–42.
- Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239–46.
- Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:213–22.
- Hirsch FR, Bunn PA Jr. EGFR testing in lung cancer is ready for prime time. Lancet Oncol 2009;10:432–3.
- Nelson V, Ziehr J, Aqulnik M, et al. Afatinib: emerging next-generation tyrosine kinase inhibitor for NSCLC. Onco Targets Ther 2013;5:135–43.
- Lee CK, Wu YL, Ding PN, et al. Impact of specific epidermal growth factor receptor (EGFR) mutations and clinical characteristics on outcomes after treatment with EGFR tyrosine kinase inhibitors versus chemotherapy in EGFR-mutant lung cancer: a meta-analysis. J Clin Oncol 2015;33:1958–65.
- Inoue A, Kobayashi K, Maemondo M, et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann Oncol 2013;24:54–9.
- Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947–57.
- Wu YL, Saijo N, Thongprasert S, et al. Efficacy according to blind independent central review: post-hoc analyses from the phase III, randomized, multicenter, IPASS study of first-line gefitinib versus carboplatin/paclitaxel in Asian patients with EFGR mutation-positive advanced NSCLC. Lung Cancer 2017;104:119–25.
- Douillard JY, Ostoros G, Cobo M, et al. First-line gefitinib in Caucasian EGFR-mutation positive NSCLC patients: a phase-IV, open-label, single-arm study. Br J Cancer 2014;110:55–62.
- Hu JC, Sadeghi P, Pinter-Brown LC, et al. Cutaneous side effects of epidermal growth factor receptor inhibitors: clinical presentation, pathogenesis, and management. J Am Acad Dermatol 2007;56:317–26.
- Tarceva [package insert]. South San Francisco (CA): Genentech, Inc; 2010. www.accessdata.fda.gov/drugsatfda_docs/label/2010/021743s14s16lbl.pdf. Accessed April 23, 2017.
- Gilotrif [package insert.] Ridgefield (CT): Boehringer Ingelheim, Inc; 2013. www.accessdata.fda.gov/drugsatfda_docs/label/2013/201292s000lbl.pdf. Accessed April 23, 2017.
- Iressa [package insert]. Wilmington (DE): AstraZeneca, Inc; 2015. Error! Hyperlink reference not valid. Accessed April 23, 2017.
- Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res 2011;17:1616–22.
- Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR TKI therapy in 155 patients with EGFR mutant lung cancers. Clin Cancer Res 2013;19:2240–7.
- Yun CH, Mengwasser KE, Tom AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 2008;105:2070–5.
- Sos ML, Rode HB, Heynck S, et al. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Res 2010;70:868–74.
- Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T190M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014;4:1046–61.
- Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 2017;376:629–40.
- Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small cell lung cancer. N Engl J Med 2010;363:1693–703.
- Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009;27:4247–53.
- Kazandjian D, Blumenthal GM, Chen HY, et al. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014;19:e5–11.
- Solomon BJ, Mok T, Kim DW, et al. First-ling crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 2014;371:2167–77.
- Xalkori [package insert]. New York: Pfizer, Inc; 2011. www.accessdata.fda.gov/drugsatfda_docs/label/2012/202570s002lbl.pdf. Accessed April 23, 2017.
- Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013;368:2385–94.
- Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem 2013;56:5675–90.
- Khozin S, Blumenthal GM, Zhang L, et al. FDA approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer. Clin Cancer Res 2015;21:2436–9.
- Soria JC, Tan DS, Chiari R, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet 2017;389:917–29.
- Zykadia [package insert]. East Hanover (NJ): Novartis Pharmaceuticals Corporation, Inc; 2016. www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/zykadia.pdf. Accessed April 23, 2017.
- Larkins E, Blumenthal GM, Chen H, et al. FDA approval: alectinib for the treatment of metastatic, ALK-positive non-small cell lung cancer following crizotinib. Clin Cancer Res 2016;22:5171–6.
- Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. New Engl J Med 2017 June 6 [Epub ahead of print].
- Kinoshita K, Asoh K, Furuichi N, et al. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem 2012;20:1271–80.
- Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011;19:679–90.
- Kodama T, Tsukaguchi T, Yoshida M, et al. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett 2014;351:215–21.
- Kodama T, Tsukaguchi T, Satoh T, et al. Alectinib shows potent antitumor activity against RET-rearranged non-small cell lung cancer. Mol Cancer Ther 2014;13:2910–8.
- Alecensa [package insert]. South San Francisco (CA): Genentech, Inc; 2015. www.accessdata.fda.gov/drugsatfda_docs/label/2015/208434s000lbl.pdf. Accessed April 23, 2017.
- Kim DW, Tiseo M, Ahn MJ, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase positive non-small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol 2017 May 5 [Epub ahead of print].
- Zhu V, Ou SH. Safety of alectinib for the treatment of metastatic ALK-rearranged non-small cell lung cancer. Expert Opin Drug Saf 2017;16:509–14.
- Gadgeel SM, Shaw AT, Govindan R, et al. Pooled analysis of CNS response to alectinib in two studies of pretreated patients with ALK-positive non-small cell lung cancer. J Clin Oncol 2016;34:4079–85.
- Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol 2011;29:e443–5.
- Zhu Q, Zhan P, Zhang X, et al. Clinicopathologic characteristics of patients with ROS1 fusion gene in non-small cell lung cancer: a meta-analysis. Transl Lung Cancer Res 2015;4:300–9.
- Lin JJ, Ritterhouse LL, Ali SM, et al. ROS1 fusions rarely overlap with other oncogenic drivers in non-small cell lung cancer. J Thorac Oncol 2017;12:872–7.
- Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta 2009;1795:37–52.
- Kazandjian D, Blumenthal G, Luo L, et al. Benefit-Risk summary of crizotinib for the treatment of patients with ROS1 alteration-positive metastatic NSCLC. Oncologist 2016;21:974–80.
- Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 2014;371:1963–71.
- Zhu VW, Upadhyay D, Schrock AB, et al. TPD52L1-ROS1, a new ROS1 fusion variant in lung adenosquamous cell carcinoma identified by comprehensive genomic profiling. Lung Cancer 2016;97:48–50.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
- Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol 2016;893:1–19.
- Alberg AJ, Brock MV, Ford JG, et al. Epidemiology of lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013;143:e1S–29S.
- Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-3013, based on November 2015 SEER data submission, posted to the SEER website, April 2016. Bethesda (MD): National Cancer Institute; 2016.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Non-Small Cell Lung Cancer: 1–190.
- Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–39.
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–6.
- Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancer. J Clin Oncol 2012;30:863–70.
- Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011;29:2046–51.
- Kinno T, Tsuta K, Shiraishi K, et al. Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol 2014;25:138–42.
- Litvak AM, Paik PK, Woo KM, et al. Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J Thorac Oncol 2014;9:1669–74.
- Villaruz LC, Socinski MA, Abberbock S, et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer 2015;121:448–56.
- Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–36.
- Planchard D, Kim TM, Mazieres J, et al. DaBRAFenib in patients with BRAF V600E-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol 2016;17:642–50.
- Gautschi O, Milia J, Cabarrou B, et al. Targeted therapy for patients with BRAF-mutant lung cancer: results from the European EURAF cohort. J Thorac Oncol 2015;10:1451–7.
- Planchard D, Besse B, Groen HJ, et al. DaBRAFenib plus trametinib in patients with previously treated BRAF V600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 2016;17:984–93.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. J Thorac Oncol 2013;8:823–59.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. Arch Pathol Lab Med 2013;137:828–60.
- Leighl NB, Rekhtman N, Biermann WA, et al. Molecular testing for selection of patients with lung cancer for epidermal growth factor receptor and anaplastic lymphoma kinase tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology guideline. J Clin Oncol 2014;32:3673–9.
- Paik PK, Varghese AM, Sima CS, et al. Response to erlotinib in patients with EGFR mutant advanced non-small cell lung cancers with a squamous or squamous-like component. Mol Cancer Ther 2012;11:2535–40.
- Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov 2015;5:842–9.
- Awad MM, Oxnard GR, Jackman DM, et al. MET Exon 14 mutations in non-small-cell lung cancer are associated with advanced age, and stage-dependent MET genomic amplification, and c-MET overexpression. J Clin Oncol 2016;34:721–30.
- Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thorac Oncol 2016;11:1493–502.
- Reungwetwattana T, Liang Y, Zhu V, et al. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The why, the how, the who, the unknown, and the inevitable. Lung Cancer 2017;103:27–37.
- Drilon A, Wang L, Hasanovic A, et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov 2013;3:630–5.
- Lin JJ, Kennedy E, Sequist LV, et al. Clinical activity of alectinib in advanced RET-rearranged non-small cell lung cancer. J Thorac Oncol 2016;11:2027–32.
- Drilon A, Rekhtman N, Arcila M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol 2016;17:1653–60.
- Cappuzzo F, Bemis L, Varella-Garcia M. HER2 mutation and response to trastuzumab therapy in non-small-cell lung cancer. N Engl J Med 2006;354:2619–21.
- Mazieres J. Barlesi F, Filleron T, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targted drugs: results from the European EUHER2 cohort. Annal Oncol 2016;27:281–6.
- Ou SH, Schrock AB, Bocharov EV, et al. HER2 transmembrane (TMD) mutations (V659/G660) that stabilize homo- and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J Thorac Oncol 2017;12:446–57.
- Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emergent therapies. Cancer Discov 2017;7:596–609.
- Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014;311:1998–2006.
- Barlesi F, Mazieres J, Merlio JP, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016;387:1415–26.
- Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361:958–67.
- Rosell R, Karachaliou N, et al. Large-scale screening for somatic mutations in lung cancer. Lancet 2016;387:1354–6.
- Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005;97:339–46.
- Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013;31:3327–34.
- Wu YL, Chou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol 2015;26:1883–9.
- Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Eng J Med 2010;362:2380–8.
- Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121–8.
- Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011;12:735–42.
- Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239–46.
- Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:213–22.
- Hirsch FR, Bunn PA Jr. EGFR testing in lung cancer is ready for prime time. Lancet Oncol 2009;10:432–3.
- Nelson V, Ziehr J, Aqulnik M, et al. Afatinib: emerging next-generation tyrosine kinase inhibitor for NSCLC. Onco Targets Ther 2013;5:135–43.
- Lee CK, Wu YL, Ding PN, et al. Impact of specific epidermal growth factor receptor (EGFR) mutations and clinical characteristics on outcomes after treatment with EGFR tyrosine kinase inhibitors versus chemotherapy in EGFR-mutant lung cancer: a meta-analysis. J Clin Oncol 2015;33:1958–65.
- Inoue A, Kobayashi K, Maemondo M, et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann Oncol 2013;24:54–9.
- Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947–57.
- Wu YL, Saijo N, Thongprasert S, et al. Efficacy according to blind independent central review: post-hoc analyses from the phase III, randomized, multicenter, IPASS study of first-line gefitinib versus carboplatin/paclitaxel in Asian patients with EFGR mutation-positive advanced NSCLC. Lung Cancer 2017;104:119–25.
- Douillard JY, Ostoros G, Cobo M, et al. First-line gefitinib in Caucasian EGFR-mutation positive NSCLC patients: a phase-IV, open-label, single-arm study. Br J Cancer 2014;110:55–62.
- Hu JC, Sadeghi P, Pinter-Brown LC, et al. Cutaneous side effects of epidermal growth factor receptor inhibitors: clinical presentation, pathogenesis, and management. J Am Acad Dermatol 2007;56:317–26.
- Tarceva [package insert]. South San Francisco (CA): Genentech, Inc; 2010. www.accessdata.fda.gov/drugsatfda_docs/label/2010/021743s14s16lbl.pdf. Accessed April 23, 2017.
- Gilotrif [package insert.] Ridgefield (CT): Boehringer Ingelheim, Inc; 2013. www.accessdata.fda.gov/drugsatfda_docs/label/2013/201292s000lbl.pdf. Accessed April 23, 2017.
- Iressa [package insert]. Wilmington (DE): AstraZeneca, Inc; 2015. Error! Hyperlink reference not valid. Accessed April 23, 2017.
- Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res 2011;17:1616–22.
- Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR TKI therapy in 155 patients with EGFR mutant lung cancers. Clin Cancer Res 2013;19:2240–7.
- Yun CH, Mengwasser KE, Tom AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 2008;105:2070–5.
- Sos ML, Rode HB, Heynck S, et al. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Res 2010;70:868–74.
- Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T190M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014;4:1046–61.
- Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 2017;376:629–40.
- Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small cell lung cancer. N Engl J Med 2010;363:1693–703.
- Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009;27:4247–53.
- Kazandjian D, Blumenthal GM, Chen HY, et al. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014;19:e5–11.
- Solomon BJ, Mok T, Kim DW, et al. First-ling crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 2014;371:2167–77.
- Xalkori [package insert]. New York: Pfizer, Inc; 2011. www.accessdata.fda.gov/drugsatfda_docs/label/2012/202570s002lbl.pdf. Accessed April 23, 2017.
- Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013;368:2385–94.
- Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem 2013;56:5675–90.
- Khozin S, Blumenthal GM, Zhang L, et al. FDA approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer. Clin Cancer Res 2015;21:2436–9.
- Soria JC, Tan DS, Chiari R, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet 2017;389:917–29.
- Zykadia [package insert]. East Hanover (NJ): Novartis Pharmaceuticals Corporation, Inc; 2016. www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/zykadia.pdf. Accessed April 23, 2017.
- Larkins E, Blumenthal GM, Chen H, et al. FDA approval: alectinib for the treatment of metastatic, ALK-positive non-small cell lung cancer following crizotinib. Clin Cancer Res 2016;22:5171–6.
- Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. New Engl J Med 2017 June 6 [Epub ahead of print].
- Kinoshita K, Asoh K, Furuichi N, et al. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem 2012;20:1271–80.
- Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011;19:679–90.
- Kodama T, Tsukaguchi T, Yoshida M, et al. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett 2014;351:215–21.
- Kodama T, Tsukaguchi T, Satoh T, et al. Alectinib shows potent antitumor activity against RET-rearranged non-small cell lung cancer. Mol Cancer Ther 2014;13:2910–8.
- Alecensa [package insert]. South San Francisco (CA): Genentech, Inc; 2015. www.accessdata.fda.gov/drugsatfda_docs/label/2015/208434s000lbl.pdf. Accessed April 23, 2017.
- Kim DW, Tiseo M, Ahn MJ, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase positive non-small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol 2017 May 5 [Epub ahead of print].
- Zhu V, Ou SH. Safety of alectinib for the treatment of metastatic ALK-rearranged non-small cell lung cancer. Expert Opin Drug Saf 2017;16:509–14.
- Gadgeel SM, Shaw AT, Govindan R, et al. Pooled analysis of CNS response to alectinib in two studies of pretreated patients with ALK-positive non-small cell lung cancer. J Clin Oncol 2016;34:4079–85.
- Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol 2011;29:e443–5.
- Zhu Q, Zhan P, Zhang X, et al. Clinicopathologic characteristics of patients with ROS1 fusion gene in non-small cell lung cancer: a meta-analysis. Transl Lung Cancer Res 2015;4:300–9.
- Lin JJ, Ritterhouse LL, Ali SM, et al. ROS1 fusions rarely overlap with other oncogenic drivers in non-small cell lung cancer. J Thorac Oncol 2017;12:872–7.
- Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta 2009;1795:37–52.
- Kazandjian D, Blumenthal G, Luo L, et al. Benefit-Risk summary of crizotinib for the treatment of patients with ROS1 alteration-positive metastatic NSCLC. Oncologist 2016;21:974–80.
- Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 2014;371:1963–71.
- Zhu VW, Upadhyay D, Schrock AB, et al. TPD52L1-ROS1, a new ROS1 fusion variant in lung adenosquamous cell carcinoma identified by comprehensive genomic profiling. Lung Cancer 2016;97:48–50.