User login
Postpartum depression: Help patients find the right treatment
Discuss this article at www.facebook.com/CurrentPsychiatry
Postpartum depression (PPD)—emergence of a major depressive episode after childbirth—has broad negative consequences for the mother, baby, and other family members. The time of onset after delivery for a depressive episode to be considered postpartum is debatable, but the DSM-IV-TR specifier states that onset within 4 weeks of childbirth is considered postpartum. PPD can impact many aspects of child development, including mother-infant attachment, cognitive development, and behavior.1-3
An estimated 10% of women who have given birth experience PPD.4,5 The risk of PPD is particularly high among women who have had previous episodes of PPD or major depressive disorder (MDD). Other risk factors include stressful life events, depression and/or anxiety during pregnancy, family history of PPD, and obstetrical complications.6-8 Anxiety disorders are common in postpartum women, and anxiety symptoms often are prominent in PPD.9
Despite the prevalence of PPD and its serious consequences, few studies have addressed antidepressant treatment. In this article we discuss screening and treating PPD and considerations for breast-feeding mothers. Click here for results of an open-label trial of escitalopram for PPD we conducted in which patient recruitment was challenging.
Screening for PPD: A good start
Initiatives by state governments and health care providers have led to programs in which universal screening for PPD has been implemented. Screening provides a mechanism for early detection and intervention. The Edinburgh Postnatal Depression Scale10 is a self-rated, 10-item scale developed for the postpartum setting, and its use increases identification of PPD at postpartum obstetrics visits.11 Other screening tools such as the Patient Health Questionnaire-9 also are commonly used. Despite the success of screening programs in attempting the feasibility of screening, it is unclear if the identification of women who may be experiencing PPD increases their engagement in treatment. Studies have demonstrated that even when depressive symptoms suggesting a PPD episode are identified in the postpartum period, many women still do not receive treatment.12,13 Studies of PPD screening programs have not demonstrated that screening itself improves treatment engagement or improves outcomes.12,13
Psychotherapy: An effective option
Psychotherapy is an important first-line option for PPD, particularly because of considerations of medication exposure during breast-feeding and many women are reluctant to take antidepressants while breast-feeding.16 Interpersonal psychotherapy and cognitive-behavioral therapy (CBT) have been most studied for PPD, and both appear effective for prevention and acute treatment of PPD.17-20 Although psychotherapy alone may be sufficient for some women, for others, medication may be an important first-line treatment, depending on symptom severity, access to psychotherapy, and personal preference.
Evidence for antidepressants
Randomization to placebo is rare in PPD trials. Most trials have used open-label designs because placebo arms pose ethical dilemmas considering the impact of PPD on a mother and her baby. In a randomized study of sertraline or nortriptyline for PPD, both drugs were similarly efficacious.22 In another study comparing paroxetine monotherapy and paroxetine plus CBT for PPD, both groups experienced significant improvement in depression and anxiety symptoms, with no difference between groups at endpoint.23 Open-label trials have suggested antidepressants’ efficacy, although some studies have included small sample sizes (Table 1).20-27
Table 1
Antidepressants for PPD: Summary of the evidence
| Study | Design and size | Medication | Results |
|---|---|---|---|
| Appleby et al, 199720 | 12-week, placebo-controlled, N = 87 | Fluoxetine | Patients taking fluoxetine showed greater improvement than those taking placebo |
| Yonkers et al, 200821 | 8-week, placebo-controlled, N = 70 | Paroxetine | Both groups improved over time, but patients taking paroxetine had greater improvement in overall clinical severity |
| Wisner et al, 200622 | 8-week, RCT, N = 109 | Sertraline vs nortriptyline | Proportion of women who responded or remitted did not differ between those taking sertraline or nortriptyline |
| Misri et al, 200423 | 12-week, RCT, N = 35 | Paroxetine monotherapy vs paroxetine + CBT | Both groups showed significant improvement in mood and anxiety symptoms |
| Stowe et al, 199524 | 8-week, open-label, N = 21 | Sertraline | 20 patients experienced >50% reduction in SIGH-D score |
| Cohen et al, 199725 | Open-label, N = 15 | Venlafaxine | 12 patients achieved remission |
| Suri et al, 200126 | 8-week, open-label, N = 6 | Fluvoxamine | 4 patients became euthymic, with HDRS scores ranging from 2 to 5 |
| Nonacs et al, 200527 | 8-week, open-label, N = 8 | Bupropion | 6 patients had ≥50% decrease in HDRS score from baseline; 3 achieved remission |
| CBT: cognitive-behavioral therapy; HDRS: Hamilton Depression Rating Scale; PPD: postpartum depression; RCT: randomized controlled trial; SIGH-D: Structured Interview Guide for the Hamilton Depression Rating Scale | |||
Breast-feeding considerations
From a nutritional standpoint, breast-feeding is optimal for a newborn. However, for some women, breast-feeding is difficult and stressful, and new mothers may experience this difficulty as failure. Some women prefer not to breast-feed, and others may prefer to formula feed if they require pharmacotherapy, particularly if the medication has not been well studied in breast-feeding patients. Some women may decline to take medications if they are breast-feeding out of concern for the baby’s exposure via breast milk and prefer to try nonpharmacologic approaches first. Many mothers with PPD need to be reassured that stopping breast-feeding may be exactly what is needed if the experience is contributing to their PPD or making them uncomfortable accepting pharmacotherapy when indicated. Maternal mental health is more important than breast-feeding to the health and wellness of the mother-baby dyad.
Table 2
Considerations for antidepressant use during breast-feeding
| Drug(s) | Comments |
|---|---|
| Fluoxetine | Because of long half-life, may be more likely to be detected in infant serum, especially at higher doses. Reasonable for use during breast-feeding if a woman has had a good previous response to the drug or used it during pregnancy |
| Sertraline | Reports of low levels of exposure. Relatively large amount of data available |
| Citalopram, escitalopram | Less systematic study of mother-infant pairs compared with sertraline and paroxetine. Low levels of exposure to infant via breast-feeding observed |
| Paroxetine | Consistent reports of low levels of exposure and has been relatively well studied without reported adverse events. Use limited by commonly experienced withdrawal symptoms; may be more sedating than other SSRIs |
| Bupropion | Paucity of systematic study in newborns of nursing mothers; a few case reports in older infants demonstrated low levels of exposure via breast-feeding. May help women who smoke to quit or to maintain abstinence from smoking. Reasonable to use if a woman had good previous response. One case report of possible infant seizure; no other reported adverse events |
| Venlafaxine, desvenlafaxine | Higher levels of desvenlafaxine than venlafaxine found in breast milk. No adverse events reported. Patients may experience withdrawal with discontinuation or missed doses |
| Tricyclic antidepressants | Considered reasonable for breast-feeding mothers if use is clinically warranted; few adverse effects in babies and generally low levels of exposure reported |
| Mirtazapine, nefazodone, MAOIs, duloxetine | Systematic human data not available for breast-feeding patients. May be reasonable if a woman previously has responded best to 1 of these; advise patients that data are not available to guide decisions |
| MAOIs: monoamine oxidase inhibitors; SSRIs: selective serotonin reuptake inhibitors Source: References 29-31 | |
28,29
The psychiatrist’s role
PPD has great public health significance because it affects a large number of women and their families. Screening during obstetrical visits or in other settings may increase identification of women who are suffering from PPD. In order for this screening to lead to meaningful changes, women must receive timely and expert evaluations for PPD and treatment that is efficacious and accessible.
Diagnosis and treatment: 4 pearls
Verify the diagnosis. Many women who present with postpartum depressive symptoms may have previously unrecognized bipolar disorder, and many women presenting with a primary complaint of anxiety have PPD.33,34
Discuss breast-feeding. This topic is important in assessing the risks and benefits of antidepressants in postpartum women, but many women also experience breast-feeding as a topic with emotional valence of its own and may need support with infant feeding.
Meet the patient where she is. Patient preferences strongly influence PPD treatment decisions. Women with similar clinical presentations may have strong preferences for different treatments.
Make treatment accessible. Postpartum women may find it challenging to engage in treatment. Treatment plans need to be feasible for women who are depressed while caring for a newborn. On-site childcare, home visits, Internet communication, and other accommodations that may facilitate treatment should be considered at a systems level.
Related Resources
- American College of Obstetricians and Gynecologists. Screening for depression during and after pregnancy. www.acog.org/Resources_And_Publications/Committee_Opinions/Committee_on_Obstetric_Practice/Screening_for_Depression_During_and_After_Pregnancy.
- Meltzer-Brody S. New insights into perinatal depression: pathogenesis and treatment during pregnancy and postpartum. Dialogues Clin Neurosci. 2011;13(1):89-100.
- Dennis CL, Stewart DE. Treatment of postpartum depression, part 1: a critical review of biological interventions. J Clin Psychiatry. 2004;65(9):1242-1251.
- Dennis CL. Treatment of postpartum depression, part 2: a critical review of nonbiological interventions. J Clin Psychiatry. 2004;65(9):1252-1265.
- Cohen LS, Wang B, Nonacs R, et al. Treatment of mood disorders during pregnancy and postpartum. Psychiatr Clin North Am. 2010;33(2):273-293.
- Bupropion • Wellbutrin, Zyban
- Citalopram • Celexa
- Desvenlafaxine • Pristiq
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Nortriptyline • Aventyl, Pamelor
- Paroxetine • Paxil
- Sertraline • Zoloft
- Venlafaxine • Effexor
Dr. Joffe has received grant or research support from Cephalon/Teva, and is a consultant to Noven and Sunovion.
Dr. Cohen has received research support from AstraZeneca, Bayer HealthCare Pharmaceuticals, Bristol-Myers Squibb, Forest Laboratories, GlaxoSmithKline, National Institute of Mental Health, National Institute on Aging, National Institutes of Health, Ortho-McNeil Janssen, and Pfizer and has served on an advisory board for PamLab LLC.
1. Cicchetti D, Rogosch FA, Toth SL. Maternal depressive disorder and contextual risk: contributions to the development of attachment insecurity and behavior problems in toddlerhood. Dev Psychopathol. 1998;10(2):283-300.
2. Murray L, Fiori-Cowley A, Hooper R, et al. The impact of postnatal depression and associated adversity on early mother-infant interactions and later infant outcome. Child Dev. 1996;67(5):2512-2526.
3. Sharp D, Hay DF, Pawlby S, et al. The impact of postnatal depression on boys’ intellectual development. J Child Psychol Psychiatry. 1995;36(8):1315-1336.
4. Altshuler LL, Hendrick V, Cohen LS. Course of mood and anxiety disorders during pregnancy and the postpartum period. J Clin Psychiatry. 1998;59(suppl 2):29-33.
5. Pariser SF. Women and mood disorders. Menarche to menopause. Ann Clin Psychiatry. 1993;5(4):249-254.
6. Dennis CL, Janssen PA, Singer J. Identifying women at-risk for postpartum depression in the immediate postpartum period. Acta Psychiatr Scand. 2004;110(5):338-346.
7. Chaudron LH, Klein MH, Remington P, et al. Predictors, prodromes and incidence of postpartum depression. J Psychosom Obstet Gynaecol. 2001;22(2):103-112.
8. Heron J, O’Connor TG, Evans J, et al. ALSPAC Study Team. The course of anxiety and depression through pregnancy and the postpartum in a community sample. J Affect Disord. 2004;80(1):65-73.
9. Wenzel A, Haugen EN, Jackson LC, et al. Anxiety symptoms and disorders at eight weeks postpartum. J Anxiety Disord. 2005;19(3):295-311.
10. Cox JL, Holden JM, Sagovsky R. Detection of postnatal depression. Development of the 10-item Edinburgh Postnatal Depression Scale. Br J Psychiatry. 1987;150:782-786.
11. Evins GG, Theofrastous JP, Galvin SL. Postpartum depression: a comparison of screening and routine clinical evaluation. Am J Obstet Gynecol. 2000;182(5):1080-1082.
12. Flynn HA, O’Mahen HA, Massey L, et al. The impact of a brief obstetrics clinic-based intervention on treatment use for perinatal depression. J Womens Health (Larchmt). 2006;15(10):1195-1204.
13. Yonkers KA, Smith MV, Lin H, et al. Depression screening of perinatal women: an evaluation of the healthy start depression initiative. Psychiatr Serv. 2009;60(3):322-328.
14. van Schaik DJ, Klijn AF, van Hout HP, et al. Patients’ p in the treatment of depressive disorder in primary care. Gen Hosp Psychiatry. 2004;26(3):184-189.
15. Boath E, Bradley E, Henshaw C. Women’s views of antidepressants in the treatment of postnatal depression. J Psychosom Obstet Gynaecol. 2004;25(3-4):221-233.
16. Pearlstein TB, Zlotnick C, Battle CL, et al. Patient choice of treatment for postpartum depression: a pilot study. Arch Womens Ment Health. 2006;9(6):303-308.
17. Zlotnick C, Johnson SL, Miller IW, et al. Postpartum depression in women receiving public assistance: pilot study of an interpersonal-therapy-oriented group intervention. Am J Psychiatry. 2001;158(4):638-640.
18. Klier CM, Muzik M, Rosenblum KL, et al. Interpersonal psychotherapy adapted for the group setting in the treatment of postpartum depression. J Psychother Pract Res. 2001;10(2):124-131.
19. Stuart S, O’Hara MW, Gorman LL. The prevention and psychotherapeutic treatment of postpartum depression. Arch Womens Ment Health. 2003;6(suppl 2):S57-S69.
20. Appleby L, Warner R, Whitton A, et al. A controlled study of fluoxetine and cognitive-behavioural counselling in the treatment of postnatal depression. BMJ. 1997;314(7085):932-936.
21. Yonkers KA, Lin H, Howell HB, et al. Pharmacologic treatment of postpartum women with new-onset major depressive disorder: a randomized controlled trial with paroxetine. J Clin Psychiatry. 2008;69(4):659-665.
22. Wisner KL, Hanusa BH, Perel JM, et al. Postpartum depression: a randomized trial of sertraline versus nortriptyline. J Clin Psychopharmacol. 2006;(4)26:353-360.
23. Misri S, Reebye P, Corral M, et al. The use of paroxetine and cognitive-behavioral therapy in postpartum depression and anxiety: a randomized controlled trial. J Clin Psychiatry. 2004;65(9):1236-1241.
24. Stowe ZN, Casarella J, Landry J, et al. Sertraline in the treatment of women with postpartum major depression. Depression. 1995;3(1-2):49-55.
25. Cohen LS, Viguera AC, Bouffard SM, et al. Venlafaxine in the treatment of postpartum depression. J Clin Psychiatry. 2001;62(8):592-596.
26. Suri R, Burt VK, Altshuler LL, et al. Fluvoxamine for postpartum depression. Am J Psychiatry. 2001;158(10):1739-1740.
27. Nonacs RM, Soares CN, Viguera AC, et al. Bupropion SR for the treatment of postpartum depression: a pilot study. Int J Neuropsychopharmacol. 2005;8(3):445-449.
28. Burt VK, Suri R, Altshuler L, et al. The use of psychotropic medications during breast-feeding. Am J Psychiatry. 2001;158(7):1001-1009.
29. Weissman AM, Levy BT, Hartz AJ, et al. Pooled analysis of antidepressant levels in lactating mothers, breast milk, and nursing infants. Am J Psychiatry. 2004;161(6):1066-1078.
30. Newport DJ, Ritchie JC, Knight BT, et al. Venlafaxine in human breast milk and nursing infant plasma: determination of exposure. J Clin Psychiatry. 2009;70(9):1304-1310.
31. Chaudron LH, Schoenecker CJ. Bupropion and breastfeeding: a case of a possible infant seizure. J Clin Psychiatry. 2004;65(6):881-882.
32. Hendrick V, Stowe ZN, Altshuler LL, et al. Fluoxetine and norfluoxetine concentrations in nursing infants and breast milk. Biol Psychiatry. 2001;50(10):775-782.
33. Sharma V, Khan M. Identification of bipolar disorder in women with postpartum depression. Bipolar Disord. 2010;12(3):335-340.
34. Austin MP, Hadzi-Pavlovic D, Priest SR, et al. Depressive and anxiety disorders in the postpartum period: how prevalent are they and can we improve their detection? Arch Womens Ment Health. 2010;13(5):395-401.
Discuss this article at www.facebook.com/CurrentPsychiatry
Postpartum depression (PPD)—emergence of a major depressive episode after childbirth—has broad negative consequences for the mother, baby, and other family members. The time of onset after delivery for a depressive episode to be considered postpartum is debatable, but the DSM-IV-TR specifier states that onset within 4 weeks of childbirth is considered postpartum. PPD can impact many aspects of child development, including mother-infant attachment, cognitive development, and behavior.1-3
An estimated 10% of women who have given birth experience PPD.4,5 The risk of PPD is particularly high among women who have had previous episodes of PPD or major depressive disorder (MDD). Other risk factors include stressful life events, depression and/or anxiety during pregnancy, family history of PPD, and obstetrical complications.6-8 Anxiety disorders are common in postpartum women, and anxiety symptoms often are prominent in PPD.9
Despite the prevalence of PPD and its serious consequences, few studies have addressed antidepressant treatment. In this article we discuss screening and treating PPD and considerations for breast-feeding mothers. Click here for results of an open-label trial of escitalopram for PPD we conducted in which patient recruitment was challenging.
Screening for PPD: A good start
Initiatives by state governments and health care providers have led to programs in which universal screening for PPD has been implemented. Screening provides a mechanism for early detection and intervention. The Edinburgh Postnatal Depression Scale10 is a self-rated, 10-item scale developed for the postpartum setting, and its use increases identification of PPD at postpartum obstetrics visits.11 Other screening tools such as the Patient Health Questionnaire-9 also are commonly used. Despite the success of screening programs in attempting the feasibility of screening, it is unclear if the identification of women who may be experiencing PPD increases their engagement in treatment. Studies have demonstrated that even when depressive symptoms suggesting a PPD episode are identified in the postpartum period, many women still do not receive treatment.12,13 Studies of PPD screening programs have not demonstrated that screening itself improves treatment engagement or improves outcomes.12,13
Psychotherapy: An effective option
Psychotherapy is an important first-line option for PPD, particularly because of considerations of medication exposure during breast-feeding and many women are reluctant to take antidepressants while breast-feeding.16 Interpersonal psychotherapy and cognitive-behavioral therapy (CBT) have been most studied for PPD, and both appear effective for prevention and acute treatment of PPD.17-20 Although psychotherapy alone may be sufficient for some women, for others, medication may be an important first-line treatment, depending on symptom severity, access to psychotherapy, and personal preference.
Evidence for antidepressants
Randomization to placebo is rare in PPD trials. Most trials have used open-label designs because placebo arms pose ethical dilemmas considering the impact of PPD on a mother and her baby. In a randomized study of sertraline or nortriptyline for PPD, both drugs were similarly efficacious.22 In another study comparing paroxetine monotherapy and paroxetine plus CBT for PPD, both groups experienced significant improvement in depression and anxiety symptoms, with no difference between groups at endpoint.23 Open-label trials have suggested antidepressants’ efficacy, although some studies have included small sample sizes (Table 1).20-27
Table 1
Antidepressants for PPD: Summary of the evidence
| Study | Design and size | Medication | Results |
|---|---|---|---|
| Appleby et al, 199720 | 12-week, placebo-controlled, N = 87 | Fluoxetine | Patients taking fluoxetine showed greater improvement than those taking placebo |
| Yonkers et al, 200821 | 8-week, placebo-controlled, N = 70 | Paroxetine | Both groups improved over time, but patients taking paroxetine had greater improvement in overall clinical severity |
| Wisner et al, 200622 | 8-week, RCT, N = 109 | Sertraline vs nortriptyline | Proportion of women who responded or remitted did not differ between those taking sertraline or nortriptyline |
| Misri et al, 200423 | 12-week, RCT, N = 35 | Paroxetine monotherapy vs paroxetine + CBT | Both groups showed significant improvement in mood and anxiety symptoms |
| Stowe et al, 199524 | 8-week, open-label, N = 21 | Sertraline | 20 patients experienced >50% reduction in SIGH-D score |
| Cohen et al, 199725 | Open-label, N = 15 | Venlafaxine | 12 patients achieved remission |
| Suri et al, 200126 | 8-week, open-label, N = 6 | Fluvoxamine | 4 patients became euthymic, with HDRS scores ranging from 2 to 5 |
| Nonacs et al, 200527 | 8-week, open-label, N = 8 | Bupropion | 6 patients had ≥50% decrease in HDRS score from baseline; 3 achieved remission |
| CBT: cognitive-behavioral therapy; HDRS: Hamilton Depression Rating Scale; PPD: postpartum depression; RCT: randomized controlled trial; SIGH-D: Structured Interview Guide for the Hamilton Depression Rating Scale | |||
Breast-feeding considerations
From a nutritional standpoint, breast-feeding is optimal for a newborn. However, for some women, breast-feeding is difficult and stressful, and new mothers may experience this difficulty as failure. Some women prefer not to breast-feed, and others may prefer to formula feed if they require pharmacotherapy, particularly if the medication has not been well studied in breast-feeding patients. Some women may decline to take medications if they are breast-feeding out of concern for the baby’s exposure via breast milk and prefer to try nonpharmacologic approaches first. Many mothers with PPD need to be reassured that stopping breast-feeding may be exactly what is needed if the experience is contributing to their PPD or making them uncomfortable accepting pharmacotherapy when indicated. Maternal mental health is more important than breast-feeding to the health and wellness of the mother-baby dyad.
Table 2
Considerations for antidepressant use during breast-feeding
| Drug(s) | Comments |
|---|---|
| Fluoxetine | Because of long half-life, may be more likely to be detected in infant serum, especially at higher doses. Reasonable for use during breast-feeding if a woman has had a good previous response to the drug or used it during pregnancy |
| Sertraline | Reports of low levels of exposure. Relatively large amount of data available |
| Citalopram, escitalopram | Less systematic study of mother-infant pairs compared with sertraline and paroxetine. Low levels of exposure to infant via breast-feeding observed |
| Paroxetine | Consistent reports of low levels of exposure and has been relatively well studied without reported adverse events. Use limited by commonly experienced withdrawal symptoms; may be more sedating than other SSRIs |
| Bupropion | Paucity of systematic study in newborns of nursing mothers; a few case reports in older infants demonstrated low levels of exposure via breast-feeding. May help women who smoke to quit or to maintain abstinence from smoking. Reasonable to use if a woman had good previous response. One case report of possible infant seizure; no other reported adverse events |
| Venlafaxine, desvenlafaxine | Higher levels of desvenlafaxine than venlafaxine found in breast milk. No adverse events reported. Patients may experience withdrawal with discontinuation or missed doses |
| Tricyclic antidepressants | Considered reasonable for breast-feeding mothers if use is clinically warranted; few adverse effects in babies and generally low levels of exposure reported |
| Mirtazapine, nefazodone, MAOIs, duloxetine | Systematic human data not available for breast-feeding patients. May be reasonable if a woman previously has responded best to 1 of these; advise patients that data are not available to guide decisions |
| MAOIs: monoamine oxidase inhibitors; SSRIs: selective serotonin reuptake inhibitors Source: References 29-31 | |
28,29
The psychiatrist’s role
PPD has great public health significance because it affects a large number of women and their families. Screening during obstetrical visits or in other settings may increase identification of women who are suffering from PPD. In order for this screening to lead to meaningful changes, women must receive timely and expert evaluations for PPD and treatment that is efficacious and accessible.
Diagnosis and treatment: 4 pearls
Verify the diagnosis. Many women who present with postpartum depressive symptoms may have previously unrecognized bipolar disorder, and many women presenting with a primary complaint of anxiety have PPD.33,34
Discuss breast-feeding. This topic is important in assessing the risks and benefits of antidepressants in postpartum women, but many women also experience breast-feeding as a topic with emotional valence of its own and may need support with infant feeding.
Meet the patient where she is. Patient preferences strongly influence PPD treatment decisions. Women with similar clinical presentations may have strong preferences for different treatments.
Make treatment accessible. Postpartum women may find it challenging to engage in treatment. Treatment plans need to be feasible for women who are depressed while caring for a newborn. On-site childcare, home visits, Internet communication, and other accommodations that may facilitate treatment should be considered at a systems level.
Related Resources
- American College of Obstetricians and Gynecologists. Screening for depression during and after pregnancy. www.acog.org/Resources_And_Publications/Committee_Opinions/Committee_on_Obstetric_Practice/Screening_for_Depression_During_and_After_Pregnancy.
- Meltzer-Brody S. New insights into perinatal depression: pathogenesis and treatment during pregnancy and postpartum. Dialogues Clin Neurosci. 2011;13(1):89-100.
- Dennis CL, Stewart DE. Treatment of postpartum depression, part 1: a critical review of biological interventions. J Clin Psychiatry. 2004;65(9):1242-1251.
- Dennis CL. Treatment of postpartum depression, part 2: a critical review of nonbiological interventions. J Clin Psychiatry. 2004;65(9):1252-1265.
- Cohen LS, Wang B, Nonacs R, et al. Treatment of mood disorders during pregnancy and postpartum. Psychiatr Clin North Am. 2010;33(2):273-293.
- Bupropion • Wellbutrin, Zyban
- Citalopram • Celexa
- Desvenlafaxine • Pristiq
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Nortriptyline • Aventyl, Pamelor
- Paroxetine • Paxil
- Sertraline • Zoloft
- Venlafaxine • Effexor
Dr. Joffe has received grant or research support from Cephalon/Teva, and is a consultant to Noven and Sunovion.
Dr. Cohen has received research support from AstraZeneca, Bayer HealthCare Pharmaceuticals, Bristol-Myers Squibb, Forest Laboratories, GlaxoSmithKline, National Institute of Mental Health, National Institute on Aging, National Institutes of Health, Ortho-McNeil Janssen, and Pfizer and has served on an advisory board for PamLab LLC.
Discuss this article at www.facebook.com/CurrentPsychiatry
Postpartum depression (PPD)—emergence of a major depressive episode after childbirth—has broad negative consequences for the mother, baby, and other family members. The time of onset after delivery for a depressive episode to be considered postpartum is debatable, but the DSM-IV-TR specifier states that onset within 4 weeks of childbirth is considered postpartum. PPD can impact many aspects of child development, including mother-infant attachment, cognitive development, and behavior.1-3
An estimated 10% of women who have given birth experience PPD.4,5 The risk of PPD is particularly high among women who have had previous episodes of PPD or major depressive disorder (MDD). Other risk factors include stressful life events, depression and/or anxiety during pregnancy, family history of PPD, and obstetrical complications.6-8 Anxiety disorders are common in postpartum women, and anxiety symptoms often are prominent in PPD.9
Despite the prevalence of PPD and its serious consequences, few studies have addressed antidepressant treatment. In this article we discuss screening and treating PPD and considerations for breast-feeding mothers. Click here for results of an open-label trial of escitalopram for PPD we conducted in which patient recruitment was challenging.
Screening for PPD: A good start
Initiatives by state governments and health care providers have led to programs in which universal screening for PPD has been implemented. Screening provides a mechanism for early detection and intervention. The Edinburgh Postnatal Depression Scale10 is a self-rated, 10-item scale developed for the postpartum setting, and its use increases identification of PPD at postpartum obstetrics visits.11 Other screening tools such as the Patient Health Questionnaire-9 also are commonly used. Despite the success of screening programs in attempting the feasibility of screening, it is unclear if the identification of women who may be experiencing PPD increases their engagement in treatment. Studies have demonstrated that even when depressive symptoms suggesting a PPD episode are identified in the postpartum period, many women still do not receive treatment.12,13 Studies of PPD screening programs have not demonstrated that screening itself improves treatment engagement or improves outcomes.12,13
Psychotherapy: An effective option
Psychotherapy is an important first-line option for PPD, particularly because of considerations of medication exposure during breast-feeding and many women are reluctant to take antidepressants while breast-feeding.16 Interpersonal psychotherapy and cognitive-behavioral therapy (CBT) have been most studied for PPD, and both appear effective for prevention and acute treatment of PPD.17-20 Although psychotherapy alone may be sufficient for some women, for others, medication may be an important first-line treatment, depending on symptom severity, access to psychotherapy, and personal preference.
Evidence for antidepressants
Randomization to placebo is rare in PPD trials. Most trials have used open-label designs because placebo arms pose ethical dilemmas considering the impact of PPD on a mother and her baby. In a randomized study of sertraline or nortriptyline for PPD, both drugs were similarly efficacious.22 In another study comparing paroxetine monotherapy and paroxetine plus CBT for PPD, both groups experienced significant improvement in depression and anxiety symptoms, with no difference between groups at endpoint.23 Open-label trials have suggested antidepressants’ efficacy, although some studies have included small sample sizes (Table 1).20-27
Table 1
Antidepressants for PPD: Summary of the evidence
| Study | Design and size | Medication | Results |
|---|---|---|---|
| Appleby et al, 199720 | 12-week, placebo-controlled, N = 87 | Fluoxetine | Patients taking fluoxetine showed greater improvement than those taking placebo |
| Yonkers et al, 200821 | 8-week, placebo-controlled, N = 70 | Paroxetine | Both groups improved over time, but patients taking paroxetine had greater improvement in overall clinical severity |
| Wisner et al, 200622 | 8-week, RCT, N = 109 | Sertraline vs nortriptyline | Proportion of women who responded or remitted did not differ between those taking sertraline or nortriptyline |
| Misri et al, 200423 | 12-week, RCT, N = 35 | Paroxetine monotherapy vs paroxetine + CBT | Both groups showed significant improvement in mood and anxiety symptoms |
| Stowe et al, 199524 | 8-week, open-label, N = 21 | Sertraline | 20 patients experienced >50% reduction in SIGH-D score |
| Cohen et al, 199725 | Open-label, N = 15 | Venlafaxine | 12 patients achieved remission |
| Suri et al, 200126 | 8-week, open-label, N = 6 | Fluvoxamine | 4 patients became euthymic, with HDRS scores ranging from 2 to 5 |
| Nonacs et al, 200527 | 8-week, open-label, N = 8 | Bupropion | 6 patients had ≥50% decrease in HDRS score from baseline; 3 achieved remission |
| CBT: cognitive-behavioral therapy; HDRS: Hamilton Depression Rating Scale; PPD: postpartum depression; RCT: randomized controlled trial; SIGH-D: Structured Interview Guide for the Hamilton Depression Rating Scale | |||
Breast-feeding considerations
From a nutritional standpoint, breast-feeding is optimal for a newborn. However, for some women, breast-feeding is difficult and stressful, and new mothers may experience this difficulty as failure. Some women prefer not to breast-feed, and others may prefer to formula feed if they require pharmacotherapy, particularly if the medication has not been well studied in breast-feeding patients. Some women may decline to take medications if they are breast-feeding out of concern for the baby’s exposure via breast milk and prefer to try nonpharmacologic approaches first. Many mothers with PPD need to be reassured that stopping breast-feeding may be exactly what is needed if the experience is contributing to their PPD or making them uncomfortable accepting pharmacotherapy when indicated. Maternal mental health is more important than breast-feeding to the health and wellness of the mother-baby dyad.
Table 2
Considerations for antidepressant use during breast-feeding
| Drug(s) | Comments |
|---|---|
| Fluoxetine | Because of long half-life, may be more likely to be detected in infant serum, especially at higher doses. Reasonable for use during breast-feeding if a woman has had a good previous response to the drug or used it during pregnancy |
| Sertraline | Reports of low levels of exposure. Relatively large amount of data available |
| Citalopram, escitalopram | Less systematic study of mother-infant pairs compared with sertraline and paroxetine. Low levels of exposure to infant via breast-feeding observed |
| Paroxetine | Consistent reports of low levels of exposure and has been relatively well studied without reported adverse events. Use limited by commonly experienced withdrawal symptoms; may be more sedating than other SSRIs |
| Bupropion | Paucity of systematic study in newborns of nursing mothers; a few case reports in older infants demonstrated low levels of exposure via breast-feeding. May help women who smoke to quit or to maintain abstinence from smoking. Reasonable to use if a woman had good previous response. One case report of possible infant seizure; no other reported adverse events |
| Venlafaxine, desvenlafaxine | Higher levels of desvenlafaxine than venlafaxine found in breast milk. No adverse events reported. Patients may experience withdrawal with discontinuation or missed doses |
| Tricyclic antidepressants | Considered reasonable for breast-feeding mothers if use is clinically warranted; few adverse effects in babies and generally low levels of exposure reported |
| Mirtazapine, nefazodone, MAOIs, duloxetine | Systematic human data not available for breast-feeding patients. May be reasonable if a woman previously has responded best to 1 of these; advise patients that data are not available to guide decisions |
| MAOIs: monoamine oxidase inhibitors; SSRIs: selective serotonin reuptake inhibitors Source: References 29-31 | |
28,29
The psychiatrist’s role
PPD has great public health significance because it affects a large number of women and their families. Screening during obstetrical visits or in other settings may increase identification of women who are suffering from PPD. In order for this screening to lead to meaningful changes, women must receive timely and expert evaluations for PPD and treatment that is efficacious and accessible.
Diagnosis and treatment: 4 pearls
Verify the diagnosis. Many women who present with postpartum depressive symptoms may have previously unrecognized bipolar disorder, and many women presenting with a primary complaint of anxiety have PPD.33,34
Discuss breast-feeding. This topic is important in assessing the risks and benefits of antidepressants in postpartum women, but many women also experience breast-feeding as a topic with emotional valence of its own and may need support with infant feeding.
Meet the patient where she is. Patient preferences strongly influence PPD treatment decisions. Women with similar clinical presentations may have strong preferences for different treatments.
Make treatment accessible. Postpartum women may find it challenging to engage in treatment. Treatment plans need to be feasible for women who are depressed while caring for a newborn. On-site childcare, home visits, Internet communication, and other accommodations that may facilitate treatment should be considered at a systems level.
Related Resources
- American College of Obstetricians and Gynecologists. Screening for depression during and after pregnancy. www.acog.org/Resources_And_Publications/Committee_Opinions/Committee_on_Obstetric_Practice/Screening_for_Depression_During_and_After_Pregnancy.
- Meltzer-Brody S. New insights into perinatal depression: pathogenesis and treatment during pregnancy and postpartum. Dialogues Clin Neurosci. 2011;13(1):89-100.
- Dennis CL, Stewart DE. Treatment of postpartum depression, part 1: a critical review of biological interventions. J Clin Psychiatry. 2004;65(9):1242-1251.
- Dennis CL. Treatment of postpartum depression, part 2: a critical review of nonbiological interventions. J Clin Psychiatry. 2004;65(9):1252-1265.
- Cohen LS, Wang B, Nonacs R, et al. Treatment of mood disorders during pregnancy and postpartum. Psychiatr Clin North Am. 2010;33(2):273-293.
- Bupropion • Wellbutrin, Zyban
- Citalopram • Celexa
- Desvenlafaxine • Pristiq
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Nortriptyline • Aventyl, Pamelor
- Paroxetine • Paxil
- Sertraline • Zoloft
- Venlafaxine • Effexor
Dr. Joffe has received grant or research support from Cephalon/Teva, and is a consultant to Noven and Sunovion.
Dr. Cohen has received research support from AstraZeneca, Bayer HealthCare Pharmaceuticals, Bristol-Myers Squibb, Forest Laboratories, GlaxoSmithKline, National Institute of Mental Health, National Institute on Aging, National Institutes of Health, Ortho-McNeil Janssen, and Pfizer and has served on an advisory board for PamLab LLC.
1. Cicchetti D, Rogosch FA, Toth SL. Maternal depressive disorder and contextual risk: contributions to the development of attachment insecurity and behavior problems in toddlerhood. Dev Psychopathol. 1998;10(2):283-300.
2. Murray L, Fiori-Cowley A, Hooper R, et al. The impact of postnatal depression and associated adversity on early mother-infant interactions and later infant outcome. Child Dev. 1996;67(5):2512-2526.
3. Sharp D, Hay DF, Pawlby S, et al. The impact of postnatal depression on boys’ intellectual development. J Child Psychol Psychiatry. 1995;36(8):1315-1336.
4. Altshuler LL, Hendrick V, Cohen LS. Course of mood and anxiety disorders during pregnancy and the postpartum period. J Clin Psychiatry. 1998;59(suppl 2):29-33.
5. Pariser SF. Women and mood disorders. Menarche to menopause. Ann Clin Psychiatry. 1993;5(4):249-254.
6. Dennis CL, Janssen PA, Singer J. Identifying women at-risk for postpartum depression in the immediate postpartum period. Acta Psychiatr Scand. 2004;110(5):338-346.
7. Chaudron LH, Klein MH, Remington P, et al. Predictors, prodromes and incidence of postpartum depression. J Psychosom Obstet Gynaecol. 2001;22(2):103-112.
8. Heron J, O’Connor TG, Evans J, et al. ALSPAC Study Team. The course of anxiety and depression through pregnancy and the postpartum in a community sample. J Affect Disord. 2004;80(1):65-73.
9. Wenzel A, Haugen EN, Jackson LC, et al. Anxiety symptoms and disorders at eight weeks postpartum. J Anxiety Disord. 2005;19(3):295-311.
10. Cox JL, Holden JM, Sagovsky R. Detection of postnatal depression. Development of the 10-item Edinburgh Postnatal Depression Scale. Br J Psychiatry. 1987;150:782-786.
11. Evins GG, Theofrastous JP, Galvin SL. Postpartum depression: a comparison of screening and routine clinical evaluation. Am J Obstet Gynecol. 2000;182(5):1080-1082.
12. Flynn HA, O’Mahen HA, Massey L, et al. The impact of a brief obstetrics clinic-based intervention on treatment use for perinatal depression. J Womens Health (Larchmt). 2006;15(10):1195-1204.
13. Yonkers KA, Smith MV, Lin H, et al. Depression screening of perinatal women: an evaluation of the healthy start depression initiative. Psychiatr Serv. 2009;60(3):322-328.
14. van Schaik DJ, Klijn AF, van Hout HP, et al. Patients’ p in the treatment of depressive disorder in primary care. Gen Hosp Psychiatry. 2004;26(3):184-189.
15. Boath E, Bradley E, Henshaw C. Women’s views of antidepressants in the treatment of postnatal depression. J Psychosom Obstet Gynaecol. 2004;25(3-4):221-233.
16. Pearlstein TB, Zlotnick C, Battle CL, et al. Patient choice of treatment for postpartum depression: a pilot study. Arch Womens Ment Health. 2006;9(6):303-308.
17. Zlotnick C, Johnson SL, Miller IW, et al. Postpartum depression in women receiving public assistance: pilot study of an interpersonal-therapy-oriented group intervention. Am J Psychiatry. 2001;158(4):638-640.
18. Klier CM, Muzik M, Rosenblum KL, et al. Interpersonal psychotherapy adapted for the group setting in the treatment of postpartum depression. J Psychother Pract Res. 2001;10(2):124-131.
19. Stuart S, O’Hara MW, Gorman LL. The prevention and psychotherapeutic treatment of postpartum depression. Arch Womens Ment Health. 2003;6(suppl 2):S57-S69.
20. Appleby L, Warner R, Whitton A, et al. A controlled study of fluoxetine and cognitive-behavioural counselling in the treatment of postnatal depression. BMJ. 1997;314(7085):932-936.
21. Yonkers KA, Lin H, Howell HB, et al. Pharmacologic treatment of postpartum women with new-onset major depressive disorder: a randomized controlled trial with paroxetine. J Clin Psychiatry. 2008;69(4):659-665.
22. Wisner KL, Hanusa BH, Perel JM, et al. Postpartum depression: a randomized trial of sertraline versus nortriptyline. J Clin Psychopharmacol. 2006;(4)26:353-360.
23. Misri S, Reebye P, Corral M, et al. The use of paroxetine and cognitive-behavioral therapy in postpartum depression and anxiety: a randomized controlled trial. J Clin Psychiatry. 2004;65(9):1236-1241.
24. Stowe ZN, Casarella J, Landry J, et al. Sertraline in the treatment of women with postpartum major depression. Depression. 1995;3(1-2):49-55.
25. Cohen LS, Viguera AC, Bouffard SM, et al. Venlafaxine in the treatment of postpartum depression. J Clin Psychiatry. 2001;62(8):592-596.
26. Suri R, Burt VK, Altshuler LL, et al. Fluvoxamine for postpartum depression. Am J Psychiatry. 2001;158(10):1739-1740.
27. Nonacs RM, Soares CN, Viguera AC, et al. Bupropion SR for the treatment of postpartum depression: a pilot study. Int J Neuropsychopharmacol. 2005;8(3):445-449.
28. Burt VK, Suri R, Altshuler L, et al. The use of psychotropic medications during breast-feeding. Am J Psychiatry. 2001;158(7):1001-1009.
29. Weissman AM, Levy BT, Hartz AJ, et al. Pooled analysis of antidepressant levels in lactating mothers, breast milk, and nursing infants. Am J Psychiatry. 2004;161(6):1066-1078.
30. Newport DJ, Ritchie JC, Knight BT, et al. Venlafaxine in human breast milk and nursing infant plasma: determination of exposure. J Clin Psychiatry. 2009;70(9):1304-1310.
31. Chaudron LH, Schoenecker CJ. Bupropion and breastfeeding: a case of a possible infant seizure. J Clin Psychiatry. 2004;65(6):881-882.
32. Hendrick V, Stowe ZN, Altshuler LL, et al. Fluoxetine and norfluoxetine concentrations in nursing infants and breast milk. Biol Psychiatry. 2001;50(10):775-782.
33. Sharma V, Khan M. Identification of bipolar disorder in women with postpartum depression. Bipolar Disord. 2010;12(3):335-340.
34. Austin MP, Hadzi-Pavlovic D, Priest SR, et al. Depressive and anxiety disorders in the postpartum period: how prevalent are they and can we improve their detection? Arch Womens Ment Health. 2010;13(5):395-401.
1. Cicchetti D, Rogosch FA, Toth SL. Maternal depressive disorder and contextual risk: contributions to the development of attachment insecurity and behavior problems in toddlerhood. Dev Psychopathol. 1998;10(2):283-300.
2. Murray L, Fiori-Cowley A, Hooper R, et al. The impact of postnatal depression and associated adversity on early mother-infant interactions and later infant outcome. Child Dev. 1996;67(5):2512-2526.
3. Sharp D, Hay DF, Pawlby S, et al. The impact of postnatal depression on boys’ intellectual development. J Child Psychol Psychiatry. 1995;36(8):1315-1336.
4. Altshuler LL, Hendrick V, Cohen LS. Course of mood and anxiety disorders during pregnancy and the postpartum period. J Clin Psychiatry. 1998;59(suppl 2):29-33.
5. Pariser SF. Women and mood disorders. Menarche to menopause. Ann Clin Psychiatry. 1993;5(4):249-254.
6. Dennis CL, Janssen PA, Singer J. Identifying women at-risk for postpartum depression in the immediate postpartum period. Acta Psychiatr Scand. 2004;110(5):338-346.
7. Chaudron LH, Klein MH, Remington P, et al. Predictors, prodromes and incidence of postpartum depression. J Psychosom Obstet Gynaecol. 2001;22(2):103-112.
8. Heron J, O’Connor TG, Evans J, et al. ALSPAC Study Team. The course of anxiety and depression through pregnancy and the postpartum in a community sample. J Affect Disord. 2004;80(1):65-73.
9. Wenzel A, Haugen EN, Jackson LC, et al. Anxiety symptoms and disorders at eight weeks postpartum. J Anxiety Disord. 2005;19(3):295-311.
10. Cox JL, Holden JM, Sagovsky R. Detection of postnatal depression. Development of the 10-item Edinburgh Postnatal Depression Scale. Br J Psychiatry. 1987;150:782-786.
11. Evins GG, Theofrastous JP, Galvin SL. Postpartum depression: a comparison of screening and routine clinical evaluation. Am J Obstet Gynecol. 2000;182(5):1080-1082.
12. Flynn HA, O’Mahen HA, Massey L, et al. The impact of a brief obstetrics clinic-based intervention on treatment use for perinatal depression. J Womens Health (Larchmt). 2006;15(10):1195-1204.
13. Yonkers KA, Smith MV, Lin H, et al. Depression screening of perinatal women: an evaluation of the healthy start depression initiative. Psychiatr Serv. 2009;60(3):322-328.
14. van Schaik DJ, Klijn AF, van Hout HP, et al. Patients’ p in the treatment of depressive disorder in primary care. Gen Hosp Psychiatry. 2004;26(3):184-189.
15. Boath E, Bradley E, Henshaw C. Women’s views of antidepressants in the treatment of postnatal depression. J Psychosom Obstet Gynaecol. 2004;25(3-4):221-233.
16. Pearlstein TB, Zlotnick C, Battle CL, et al. Patient choice of treatment for postpartum depression: a pilot study. Arch Womens Ment Health. 2006;9(6):303-308.
17. Zlotnick C, Johnson SL, Miller IW, et al. Postpartum depression in women receiving public assistance: pilot study of an interpersonal-therapy-oriented group intervention. Am J Psychiatry. 2001;158(4):638-640.
18. Klier CM, Muzik M, Rosenblum KL, et al. Interpersonal psychotherapy adapted for the group setting in the treatment of postpartum depression. J Psychother Pract Res. 2001;10(2):124-131.
19. Stuart S, O’Hara MW, Gorman LL. The prevention and psychotherapeutic treatment of postpartum depression. Arch Womens Ment Health. 2003;6(suppl 2):S57-S69.
20. Appleby L, Warner R, Whitton A, et al. A controlled study of fluoxetine and cognitive-behavioural counselling in the treatment of postnatal depression. BMJ. 1997;314(7085):932-936.
21. Yonkers KA, Lin H, Howell HB, et al. Pharmacologic treatment of postpartum women with new-onset major depressive disorder: a randomized controlled trial with paroxetine. J Clin Psychiatry. 2008;69(4):659-665.
22. Wisner KL, Hanusa BH, Perel JM, et al. Postpartum depression: a randomized trial of sertraline versus nortriptyline. J Clin Psychopharmacol. 2006;(4)26:353-360.
23. Misri S, Reebye P, Corral M, et al. The use of paroxetine and cognitive-behavioral therapy in postpartum depression and anxiety: a randomized controlled trial. J Clin Psychiatry. 2004;65(9):1236-1241.
24. Stowe ZN, Casarella J, Landry J, et al. Sertraline in the treatment of women with postpartum major depression. Depression. 1995;3(1-2):49-55.
25. Cohen LS, Viguera AC, Bouffard SM, et al. Venlafaxine in the treatment of postpartum depression. J Clin Psychiatry. 2001;62(8):592-596.
26. Suri R, Burt VK, Altshuler LL, et al. Fluvoxamine for postpartum depression. Am J Psychiatry. 2001;158(10):1739-1740.
27. Nonacs RM, Soares CN, Viguera AC, et al. Bupropion SR for the treatment of postpartum depression: a pilot study. Int J Neuropsychopharmacol. 2005;8(3):445-449.
28. Burt VK, Suri R, Altshuler L, et al. The use of psychotropic medications during breast-feeding. Am J Psychiatry. 2001;158(7):1001-1009.
29. Weissman AM, Levy BT, Hartz AJ, et al. Pooled analysis of antidepressant levels in lactating mothers, breast milk, and nursing infants. Am J Psychiatry. 2004;161(6):1066-1078.
30. Newport DJ, Ritchie JC, Knight BT, et al. Venlafaxine in human breast milk and nursing infant plasma: determination of exposure. J Clin Psychiatry. 2009;70(9):1304-1310.
31. Chaudron LH, Schoenecker CJ. Bupropion and breastfeeding: a case of a possible infant seizure. J Clin Psychiatry. 2004;65(6):881-882.
32. Hendrick V, Stowe ZN, Altshuler LL, et al. Fluoxetine and norfluoxetine concentrations in nursing infants and breast milk. Biol Psychiatry. 2001;50(10):775-782.
33. Sharma V, Khan M. Identification of bipolar disorder in women with postpartum depression. Bipolar Disord. 2010;12(3):335-340.
34. Austin MP, Hadzi-Pavlovic D, Priest SR, et al. Depressive and anxiety disorders in the postpartum period: how prevalent are they and can we improve their detection? Arch Womens Ment Health. 2010;13(5):395-401.
Product Update
LUVENA® PREBIOTIC VAGINAL MOISTURIZER AND LUBRICANT
Luvena Prebiotic Vaginal Moisturizer and Lubricant from Laclede works with the body to protect against harmful bacterial and yeast growth associated with vaginal dryness. Using natural prebiotics and enzymes, Luvena Prebiotic is designed for women prone to yeast infections because of antibiotic use, and those with autoimmune disorders, experiencing peri- and postmenopause, and taking medications that cause dryness. Laclede reports that regular use of Luvena Prebiotic helps restore essential moisture, reduce unpleasant odors, promote healthy flora balance and pH, and protect against vaginal irritation.
FOR MORE INFORMATION, VISIT www.luvenacare.com
CERVICAL CA TERC TEST FROM QUEST VERIFIES UNCLEAR PAP AND HPV RESULTS
Quest Diagnostics now offers the Cervical Cancer TERC Test to verify unclear Pap and HPV testing results. The test is based on the human telomerase RNA component (TERC) gene marker. NIH research shows that the TERC gene is amplified in precursor cells of cervical cancer, providing knowledge about which molecular changes turn cervical dysplasia into malignancy. The test will evaluate women whose Pap tests show low-grade squamous intraepithelial lesions (LSIL), and evaluate them for high or low cervical cancer risk.
FOR MORE INFORMATION, VISIT www.questdiagnostics.com
PLASMAJET® SURGICAL SYSTEM FOR CUTTING, COAGULATION, AND TISSUE ABLATION
The PlasmaJet Surgical System from Plasma Surgical is an advanced energy device for cutting, coagulation, and tissue ablation. A fine, electrically neutral stream of plasma—rather than an electric current—cuts and coagulates tissue and bone. The plasma is generated by a very low flow of argon gas passing over electrodes in the single-use handpiece. When applied to tissue, the plasma stream creates a thin, flexible coagulation layer that prevents bleeding and lymphatic oozing by removing liquid from the wound surface without thermal diffusion to surrounding tissue or fluids. PlasmaJet is used in open and laparoscopic surgery to treat endometriosis, endometrioma, HPV dysplasia, and to perform hysterectomies.
FOR MORE INFORMATION, VISIT www.plasmasurgical.com
SURVIVORSHIP TOOLKIT FOR LIFE AFTER GYNECOLOGIC CANCER TREATMENT
The Foundation for Gynecologic Oncology, formed by the Society of Gynecologic Oncology, provides educational and clinical resources for those who treat gynecologic cancers and their patients. Survivorship Toolkits, available as PDFs or Word documents, are templates to help women prepare for life after cancer treatment, including treatment and side effects, self-care plans, calendars, and vital information cards. The toolkits are available at http://www.sgo.org/Clinical_Practice/Clinical_Practice/.
FOR MORE INFORMATION, VISIT www.sgo.org
BABYQ: AN APP AND WEB SITE TO IMPROVE MATERNAL-FETAL HEALTH
babyQ, developed by two physicians, offers a free mobile app and Web site to enhance maternal-fetal health and thus improve birth weights, reduce preterm delivery rates, and decrease gestational diabetic rates. After taking a short survey, women are given a babyQ score (0–100), based on the doctors’ algorithm. Users then receive personalized health coaching messages through push notifications or email, including information on lifestyle, exercise, nutrition, and stress management. The goal of babyQ is to encourage mothers to make better choices while adapting to the growth and development of the baby.
FOR MORE INFORMATION, VISIT www.babyq.com
LUVENA® PREBIOTIC VAGINAL MOISTURIZER AND LUBRICANT
Luvena Prebiotic Vaginal Moisturizer and Lubricant from Laclede works with the body to protect against harmful bacterial and yeast growth associated with vaginal dryness. Using natural prebiotics and enzymes, Luvena Prebiotic is designed for women prone to yeast infections because of antibiotic use, and those with autoimmune disorders, experiencing peri- and postmenopause, and taking medications that cause dryness. Laclede reports that regular use of Luvena Prebiotic helps restore essential moisture, reduce unpleasant odors, promote healthy flora balance and pH, and protect against vaginal irritation.
FOR MORE INFORMATION, VISIT www.luvenacare.com
CERVICAL CA TERC TEST FROM QUEST VERIFIES UNCLEAR PAP AND HPV RESULTS
Quest Diagnostics now offers the Cervical Cancer TERC Test to verify unclear Pap and HPV testing results. The test is based on the human telomerase RNA component (TERC) gene marker. NIH research shows that the TERC gene is amplified in precursor cells of cervical cancer, providing knowledge about which molecular changes turn cervical dysplasia into malignancy. The test will evaluate women whose Pap tests show low-grade squamous intraepithelial lesions (LSIL), and evaluate them for high or low cervical cancer risk.
FOR MORE INFORMATION, VISIT www.questdiagnostics.com
PLASMAJET® SURGICAL SYSTEM FOR CUTTING, COAGULATION, AND TISSUE ABLATION
The PlasmaJet Surgical System from Plasma Surgical is an advanced energy device for cutting, coagulation, and tissue ablation. A fine, electrically neutral stream of plasma—rather than an electric current—cuts and coagulates tissue and bone. The plasma is generated by a very low flow of argon gas passing over electrodes in the single-use handpiece. When applied to tissue, the plasma stream creates a thin, flexible coagulation layer that prevents bleeding and lymphatic oozing by removing liquid from the wound surface without thermal diffusion to surrounding tissue or fluids. PlasmaJet is used in open and laparoscopic surgery to treat endometriosis, endometrioma, HPV dysplasia, and to perform hysterectomies.
FOR MORE INFORMATION, VISIT www.plasmasurgical.com
SURVIVORSHIP TOOLKIT FOR LIFE AFTER GYNECOLOGIC CANCER TREATMENT
The Foundation for Gynecologic Oncology, formed by the Society of Gynecologic Oncology, provides educational and clinical resources for those who treat gynecologic cancers and their patients. Survivorship Toolkits, available as PDFs or Word documents, are templates to help women prepare for life after cancer treatment, including treatment and side effects, self-care plans, calendars, and vital information cards. The toolkits are available at http://www.sgo.org/Clinical_Practice/Clinical_Practice/.
FOR MORE INFORMATION, VISIT www.sgo.org
BABYQ: AN APP AND WEB SITE TO IMPROVE MATERNAL-FETAL HEALTH
babyQ, developed by two physicians, offers a free mobile app and Web site to enhance maternal-fetal health and thus improve birth weights, reduce preterm delivery rates, and decrease gestational diabetic rates. After taking a short survey, women are given a babyQ score (0–100), based on the doctors’ algorithm. Users then receive personalized health coaching messages through push notifications or email, including information on lifestyle, exercise, nutrition, and stress management. The goal of babyQ is to encourage mothers to make better choices while adapting to the growth and development of the baby.
FOR MORE INFORMATION, VISIT www.babyq.com
LUVENA® PREBIOTIC VAGINAL MOISTURIZER AND LUBRICANT
Luvena Prebiotic Vaginal Moisturizer and Lubricant from Laclede works with the body to protect against harmful bacterial and yeast growth associated with vaginal dryness. Using natural prebiotics and enzymes, Luvena Prebiotic is designed for women prone to yeast infections because of antibiotic use, and those with autoimmune disorders, experiencing peri- and postmenopause, and taking medications that cause dryness. Laclede reports that regular use of Luvena Prebiotic helps restore essential moisture, reduce unpleasant odors, promote healthy flora balance and pH, and protect against vaginal irritation.
FOR MORE INFORMATION, VISIT www.luvenacare.com
CERVICAL CA TERC TEST FROM QUEST VERIFIES UNCLEAR PAP AND HPV RESULTS
Quest Diagnostics now offers the Cervical Cancer TERC Test to verify unclear Pap and HPV testing results. The test is based on the human telomerase RNA component (TERC) gene marker. NIH research shows that the TERC gene is amplified in precursor cells of cervical cancer, providing knowledge about which molecular changes turn cervical dysplasia into malignancy. The test will evaluate women whose Pap tests show low-grade squamous intraepithelial lesions (LSIL), and evaluate them for high or low cervical cancer risk.
FOR MORE INFORMATION, VISIT www.questdiagnostics.com
PLASMAJET® SURGICAL SYSTEM FOR CUTTING, COAGULATION, AND TISSUE ABLATION
The PlasmaJet Surgical System from Plasma Surgical is an advanced energy device for cutting, coagulation, and tissue ablation. A fine, electrically neutral stream of plasma—rather than an electric current—cuts and coagulates tissue and bone. The plasma is generated by a very low flow of argon gas passing over electrodes in the single-use handpiece. When applied to tissue, the plasma stream creates a thin, flexible coagulation layer that prevents bleeding and lymphatic oozing by removing liquid from the wound surface without thermal diffusion to surrounding tissue or fluids. PlasmaJet is used in open and laparoscopic surgery to treat endometriosis, endometrioma, HPV dysplasia, and to perform hysterectomies.
FOR MORE INFORMATION, VISIT www.plasmasurgical.com
SURVIVORSHIP TOOLKIT FOR LIFE AFTER GYNECOLOGIC CANCER TREATMENT
The Foundation for Gynecologic Oncology, formed by the Society of Gynecologic Oncology, provides educational and clinical resources for those who treat gynecologic cancers and their patients. Survivorship Toolkits, available as PDFs or Word documents, are templates to help women prepare for life after cancer treatment, including treatment and side effects, self-care plans, calendars, and vital information cards. The toolkits are available at http://www.sgo.org/Clinical_Practice/Clinical_Practice/.
FOR MORE INFORMATION, VISIT www.sgo.org
BABYQ: AN APP AND WEB SITE TO IMPROVE MATERNAL-FETAL HEALTH
babyQ, developed by two physicians, offers a free mobile app and Web site to enhance maternal-fetal health and thus improve birth weights, reduce preterm delivery rates, and decrease gestational diabetic rates. After taking a short survey, women are given a babyQ score (0–100), based on the doctors’ algorithm. Users then receive personalized health coaching messages through push notifications or email, including information on lifestyle, exercise, nutrition, and stress management. The goal of babyQ is to encourage mothers to make better choices while adapting to the growth and development of the baby.
FOR MORE INFORMATION, VISIT www.babyq.com
Panic disorder: Break the fear circuit

Ms. K, a 24-year-old waitress who lives with her boyfriend, was referred by her primary care physician for evaluation of panic attacks that began “out of nowhere” at work approximately 6 months ago. The unpredictable attacks occur multiple times per week, causing her to leave work and cancel shifts.
Ms. K reports that before the panic attacks began, she felt happy in her relationship, enjoyed hobbies, and was hopeful about the future. However, she has become concerned that a potentially catastrophic illness is causing her panic attacks. She researches her symptoms on the Internet, and is preoccupied with the possibility of sudden death due to an undiagnosed heart condition. Multiple visits to the emergency room have not identified any physical abnormalities. Her primary care doctor prescribed alprazolam, 0.5 mg as needed for panic attacks, which she reports is helpful, “but only in the moment of the attacks.” Ms. K avoids alcohol and illicit substances and limits her caffeine intake. She is not willing to accept that her life “feels so limited.” Her dream of earning a nursing degree and eventually starting a family now seems unattainable.
Panic disorder (PD) occurs in 3% to 5% of adults, with women affected at roughly twice the rate of men.1 Causing a broad range of distress and varying degrees of impairment, PD commonly occurs with other psychiatric disorders. For most patients, treatment is effective, but those who do not respond to initial approaches require a thoughtful, stepped approach to care. Key considerations include establishing an accurate diagnosis, clarifying comorbid illnesses, ascertaining patient beliefs and expectations, and providing appropriately dosed and maintained treatments.
Panic attacks vs PD
Panic attacks consist of rapid onset of intense anxiety, with prominent somatic symptoms, that peaks within 10 minutes (Figure).2 Attacks in which <4 of the listed symptoms occur are considered limited-symptom panic attacks.
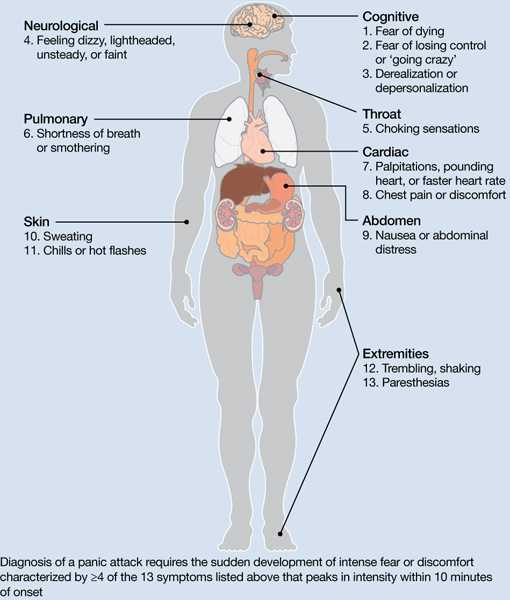
Figure: Body locations of panic attack symptoms
Diagnosis of a panic attack requires the sudden development of intense fear or discomfort characterized by ≥4 of the 13 symptoms listed above that peaks in intensity within 10 minutes of onset
Source: Reference 2
Panic attacks can occur with various disorders, including other anxiety disorders, mood disorders, and substance intoxication or withdrawal. Because serious medical conditions can present with panic-like symptoms, the initial occurrence of such symptoms warrants consideration of physiological causes. For a Box2 that describes the differential diagnosis of panic attacks, see this article at CurrentPsychiatry.com.
To meet diagnostic criteria for panic disorder, panic attacks must initially occur “out of the blue,” meaning no specific object or situation induced the attack. The differential diagnosis of panic attacks includes assessing for other psychiatric disorders that may involve panic attacks. Evaluation requires considering the context in which the panic attacks occur, including their start date, pattern of attacks, instigating situations, and associated thoughts.
Social phobia. Attacks occur only during or immediately before a social interaction in which the patient fears embarrassing himself or herself.
Obsessive-compulsive disorder (OCD). Attacks occur when the patient cannot avoid exposure to an obsessional fear or is prevented from performing a ritual that diffuses obsessional anxiety.
Posttraumatic stress disorder (PTSD). Attacks occur when confronted by a trauma-related memory or trigger.
Specific phobia. Attacks occur only when the patient encounters a specifically feared object, place, or situation, unrelated to social phobia, OCD, or PTSD.
Medical conditions. Conditions to consider include—but are not limited to—hyperthyroidism, pulmonary embolism, myocardial infarction, cardiac dysrhythmias, hypoglycemia, asthma, partial complex seizures, and pheochromocytoma.
Source: Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000
A PD diagnosis requires that repeated panic attacks initially must occur from “out of the blue,” meaning no specific object or situation induced the attack. In addition, the diagnosis requires 1 of 3 types of psychological or behavioral changes as a result of the attacks (Table 1).2 Agoraphobia is diagnosed if 1 of the behavioral changes is avoidance of places or situations from which escape might be embarrassing or difficult should an attack occur. A patient can be diagnosed as having PD with agoraphobia, PD without agoraphobia, or agoraphobia without PD (ie, experiences only limited symptom panic attacks, but avoids situations or stimuli associated with them).
Table 1
Definitions of panic disorder and agoraphobia
| Panic disorder |
|---|
|
| Agoraphobia |
| Anxiety about, or avoidance of, being in places or situations from which escape might be difficult or embarrassing, or in which help may not be available in the event of having an unexpected or situationally predisposed panic attack or panic-like symptoms. Agoraphobic fears typically involve characteristic clusters of situations that include being outside the home alone, being in a crowd, standing in a line, being on a bridge, or traveling in a bus, train, or automobile |
| Source: Reference 2 |
Comorbidities are common in patients with PD and predict greater difficulty achieving remission (Box).1,3-6
The most common psychiatric conditions that co-occur with panic disorder (PD) are other anxiety disorders, mood disorders, personality disorders, and substance use disorders.1 Carefully assess the severity and degree of impairment or distress arising from each condition to prioritize treatment goals. For example, treating panic attacks would be a lower priority in a patient with untreated bipolar disorder.
Assessing comorbid substance abuse is important in selecting PD treatments. Benzodiazepines should almost always be avoided in patients with a history of drug abuse—illicit or prescribed. Although complete abstinence should not be a prerequisite for beginning PD treatment, detoxification and concomitant substance abuse treatment are essential.3
Comorbid mood disorders also affect the course of PD treatment. Antidepressants are effective for treating depression and PD, whereas benzodiazepines are not effective for depression.4 Antidepressants in patients with bipolar disorder are controversial because these medications might induce mixed or elevated mood states or rapid cycling. In these complicated patients, consider antidepressants lower in the treatment algorithm.5
Other conditions to consider before beginning treatment include pregnancy or the possibility of becoming pregnant in the near future and suicidal ideation. PD is associated with increased risk for suicidal ideation and progression to suicide attempts, particularly in patients with a comorbid mood or psychotic disorder.6 In addition, consider the potential impact of medications on comorbid medical conditions.
Treatment begins with education
The goal of treatment is remission of symptoms, ideally including an absence of panic attacks, agoraphobic avoidance, and anticipatory anxiety.1 The Panic Disorder Severity Scale self-report is a validated measure of panic symptoms that may be useful in clinical practice.7
The first step in treatment is educating patients about panic attacks, framing them as an overreactive fear circuit in the brain that produces physical symptoms that are not dangerous. Using a brain model that shows the location of the amygdala, hippocampus, and prefrontal cortex—which play crucial roles in generating and controlling anxiety and fear—can make this discussion more concrete.8 Although highly simplified, such models allow clinicians to demonstrate that excessive reactivity of limbic regions can be reduced by both top-down (cortico-limbic connections via cognitive-behavioral therapy [CBT]) and bottom-up (pharmacotherapy directly acting on limbic structures) approaches. Such discussions lead to treatment recommendations for CBT, pharmacotherapy, or their combination.
No single treatment has emerged as the definitive “best” for PD, and no reliable predictors can guide specific treatment for an individual.3 Combining CBT with pharmacotherapy produces higher short-term response rates than either treatment alone, but in the long term, combination treatment does not appear to be superior to CBT alone.9 Base the initial treatment selection for PD on patient preference, treatment availability and cost, and comorbid medical and psychiatric conditions. For an Algorithm to guide treatment decisions, see this article at CurrentPsychiatry.com.

Algorithm: Treatment for panic disorder: A suggested algorithm
aPoor response to an SSRI should lead to a switch to venlafaxine extended-release, and vice versa
bBenzodiazepines are relatively contraindicated in geriatric patients and patients with a history of substance abuse or dependence
CBT: cognitive-behavioral therapy; MAOI: monoamine oxidase inhibitor; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant; Ven XR: venlafaxine extended-release
First-line treatments
Psychotherapy. CBT is the most efficacious psychotherapy for PD. Twelve to 15 sessions of CBT has demonstrated efficacy for PD, with additional effects on comorbid anxiety and depressive symptoms.10 No large clinical trials of CBT have used cognitive restructuring alone; all have included at least some component of exposure that requires the patient to confront feared physical sensations. Gains during treatment may be steady and gradual or sudden and uneven, with rapid improvement in some but not all symptoms. CBT and pharmacotherapy have demonstrated similar levels of benefit in short-term trials, but CBT has proven superior in most9 but not all11 trials evaluating long-term outcomes, particularly compared with pharmacotherapy that is discontinued during follow-up. Although less studied, group CBT also may be considered if a patient cannot afford individual CBT.
Pharmacotherapy. Evidence supports selective serotonin reuptake inhibitors (SSRIs), venlafaxine extended-release (XR), benzodiazepines, and tricyclic antidepressants (TCAs) as effective treatments for PD.3 No class of medication has demonstrated superiority over others in short-term treatment.3,12 Because of the medical risks associated with benzodiazepines and TCAs, an SSRI or venlafaxine XR should be the first medication option for most patients. Fluoxetine, paroxetine, sertraline, and venlafaxine XR are FDA-approved for PD. Paroxetine is associated with weight gain and may increase the risk for panic recurrence upon discontinuation more than sertraline, making it a less favorable option for many patients.13 Start doses at half the normal starting dose used for treating major depressive disorder and continue for 4 to 7 days, then increase to the minimal effective dose. For a Table3 that lists dosing recommendations for antidepressants to treat PD, see this article at CurrentPsychiatry.com. If there is no improvement by 4 weeks, increase the dose every 2 to 4 weeks until remission is achieved or side effects prevent further dose increases.
Table
Recommended doses for antidepressants used to treat panic disorder
| Medication | Starting dose (mg/d) | Therapeutic range (mg/d) |
|---|---|---|
| SSRIs | ||
| Citalopram | 10 | 20 to 40 |
| Escitalopram | 5 | 10 to 40 |
| Fluoxetine | 5 to 10 | 20 to 80 |
| Fluvoxamine | 25 | 100 to 300 |
| Paroxetine | 10 | 20 to 80 |
| Paroxetine CR | 12.5 | 25 to 50 |
| Sertraline | 25 | 100 to 200 |
| SNRIs | ||
| Duloxetine | 20 to 30 | 60 to 120 |
| Venlafaxine XR | 37.5 | 150 to 225 |
| TCAs | ||
| Clomipramine | 10 to 25 | 100 to 300 |
| Imipramine | 10 | 100 to 300 |
| MAOI | ||
| Phenelzine | 15 | 45 to 90 |
| CR: controlled release; MAOI: monoamine oxidase inhibitor; SNRIs: serotonin-norepinephrine reuptake inhibitors; SSRIs: selective serotonin reuptake inhibitors; TCAs: tricyclic antidepressants; XR: extended release Source: American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. 2nd ed. Washington, DC: American Psychiatric Association; 2009 | ||
Treatment nonresponse. True non-response needs to be distinguished from poor response caused by inadequate treatment delivery, eg, patients not completing homework assignments in CBT or not adhering to pharmacotherapy. Asking patients about adverse effects or personal and family beliefs about treatment may reveal reasons for nonadherence.
Second-line treatments
Little data are available to guide next-step treatment options in patients who don’t achieve remission from their initial treatment. Patients who benefit from an SSRI, venlafaxine XR, or CBT but still have symptoms should be started on combination treatment. For a patient who experiences complete non-response to the initial treatment, discontinue the first treatment and switch to the other modality. In general, completely ineffective treatments should be discontinued when another treatment is added, but when partial improvement (>30%) occurs, continue the original treatment and augment it with another approach.
For patients pursuing pharmacotherapy, poor response to an adequate SSRI trial usually should lead to a switch to venlafaxine XR, and vice versa. Failure to respond to both of these medication classes should prompt a switch to a benzodiazepine or TCA.
Benzodiazepines are a fast-acting, effective treatment for PD, with efficacy similar to SSRIs in acute and long-term treatment.14 Benzodiazepines may be prescribed with antidepressants at the beginning of treatment to improve response speed.15 Clonazepam and alprazolam are FDA-approved for treating PD. A high-potency, long-acting agent, clonazepam is the preferred initial benzodiazepine, dosed 0.5 to 4 mg/d on a fixed schedule. Although substantial data support using alprazolam for PD, it requires more frequent dosing and has a greater risk of rebound anxiety and abuse potential because of its more rapid onset of action. Compared with immediate-release alprazolam, alprazolam XR has a slower absorption rate and longer steady state in the blood, but this formulation does not have lower abuse potential or greater efficacy. Although not FDA-approved for PD, diazepam and lorazepam also have proven efficacy for PD.3
Benzodiazepines should be considered contraindicated in patients with a history of substance abuse, except in select cases.4 Benzodiazepines generally should be avoided in older patients because of increased risk for falls, cognitive impairment, and motor vehicle accidents. Table 2 lists situations in which benzodiazepines may be used to treat PD.
Table 2
Clinical scenarios in which to consider using benzodiazepines
| Coadministration for 2 to 4 weeks when initiating treatment with an SSRI or venlafaxine XR to achieve more rapid relief and mitigate potential antidepressant-induced anxiety |
| For patients who wish to avoid antidepressants because of concern about sexual dysfunction |
| For patients who need chronic aspirin or an NSAID, which may increase the risk for upper gastrointestinal bleeding when taken in combination with an SSRI |
| For patients with comorbid bipolar disorder or epilepsy |
| Next-step monotherapy or augmentation in patients who respond poorly to an SSRI, venlafaxine XR, TCA, or CBT |
| CBT: cognitive-behavioral therapy; NSAID: nonsteroidal anti-inflammatory drug; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant; XR: extended release |
TCAs are effective as monotherapy for PD. Most support comes from studies of imipramine or clomipramine.12 Similar to SSRIs and venlafaxine XR, use a low initial dose and gradually increase until the patient remits or side effects prevent further increases. SSRI and TCA combinations rarely are used unless the TCA is a relatively specific norepinephrine reuptake inhibitor (eg, desipramine, nortriptyline). Because TCAs are metabolized via the cytochrome P450 2D6 system and some SSRIs—particularly fluoxetine and paroxetine—strongly inhibit 2D6, combinations of TCAs with these agents may lead to dangerously high plasma TCA levels, placing patients at risk for cardiac dysrhythmias and other side effects.16
Monoamine oxidase inhibitors (MAOIs)—particularly phenelzine—are underused for PD. They have the strongest efficacy data for any class of medications outside the first- and second-line agents and have a unique mechanism of action. In patients who can comply with the dietary and medication limitations, an MAOI generally should be the next step after nonresponse to other treatments.3
Alternative treatments
For patients who do not respond to any of the treatments described above, data from uncontrolled studies support mirtazapine, levetiracetam, and the serotonin-norepinephrine reuptake inhibitors duloxetine and milnacipran as monotherapy for PD.17 Pindolol—a beta blocker and 5-HT1A receptor antagonist—proved superior to placebo as an adjunctive agent to SSRIs in treatment-resistant PD in 1 of 2 trials.17 Minimal evidence supports the atypical antipsychotics risperidone and olanzapine in treatment-resistant PD, although a placebo-controlled trial of quetiapine SR coadministered with SSRIs recently was completed (NCT00619892; results pending). Atypical antipsychotics are best reserved for patients with a primary psychotic disorder or bipolar disorder who experience panic attacks.5
Panic-focused psychodynamic psychotherapy, a 12-week (approximately 24 sessions) form of psychotherapy, has demonstrated superiority vs applied relaxation therapy.18 This treatment could be considered for patients who do not respond to standard first-line treatments, but few community therapists are familiar with this method.
For many patients with PD, complementary and alternative medicine (CAM) approaches are appealing. See this article at CurrentPsychiatry.com for a Box that discusses CAM for PD.
Although no complementary and alternative medicine treatments have strong evidence of efficacy as monotherapy for panic disorder (PD), several have data that suggest benefit with little evidence of risk. These include bibliotherapy, yoga, aerobic exercise, and the dietary supplements kava and inositol.a Exercise as a treatment poses a challenge because it can induce symptoms that the patient fears, such as tachycardia and shortness of breath. In addition to any direct physiologic benefit from aerobic exercise, there is also an exposure component that can be harnessed by gradually increasing the exertion level.
Another approach undergoing extensive evaluation is Internet-provided cognitive-behavioral therapy (CBT). Using guided CBT modules with or without therapist support, Internet-provided CBT provides an option for motivated patients unable to complete in-person CBT because of logistical factors.b A helpful resource that reviews Internet self-help and psychotherapy guided programs for PD and other psychiatric conditions is http://beacon.anu.edu.au.
References
a. Antonacci DJ, Davis E, Bloch RM, et al. CAM for your anxious patient: what the evidence says. Current Psychiatry. 2010;9(10):42-52.
b. Johnston L, Titov N, Andrews G, et al. A RCT of a transdiagnostic internet-delivered treatment for three anxiety disorders: examination of support roles and disorder-specific outcomes. PLoS One. 2011;6(11):e28079.
Maintenance treatment
Patients who complete a course of CBT for PD often follow up with several “booster sessions” at monthly or longer intervals that focus on relapse prevention techniques. Few controlled trials have evaluated pharmacotherapy discontinuation in PD. Most guidelines recommend continuing treatment for ≥1 year after achieving remission to minimize the risk of relapse.3 Researchers are focusing on whether medication dosage can be reduced during maintenance without loss of efficacy.
Treatment discontinuation
In the absence of urgent medical need, taper medications for PD gradually over several months. PD patients are highly sensitive to unusual physical sensations, which can occur while discontinuing antidepressants or benzodiazepines. If a benzodiazepine is used in conjunction with an antidepressant, the benzodiazepine should be discontinued first, so that the antidepressant can help ease benzodiazepine-associated discontinuation symptoms. A brief course of CBT during pharmacotherapy discontinuation may increase the likelihood of successful tapering.19
CASE CONTINUED: A successful switch
Ms. K has to discontinue sequential trials of fluoxetine, 40 mg/d, and venlafaxine XR, 225 mg/d because of side effects, and she does not reduce the frequency of her alprazolam use. She agrees to switch from alprazolam to clonazepam, 0.5 mg every morning and 1 mg at bedtime, and to start CBT. Clonazepam reduces her anxiety sufficiently so she can address her symptoms in therapy. Through CBT she becomes motivated to monitor her thoughts and treat them as guesses rather than facts, reviewing the evidence for her thoughts and generating rational responses. She participates in exposure exercises, which she practices between sessions, and grows to tolerate uncomfortable sensations until they no longer signal danger. After 12 CBT sessions, she is panic-free. Despite some trepidation, she agrees to a slow taper off clonazepam, reducing the dose by 0.25 mg every 2 weeks. She continues booster sessions with her therapist to manage any re-emerging anxiety. After an additional 12 weeks, she successfully discontinues clonazepam and remains panic-free.
Related Resources
- American Psychiatric Association. Panic disorder. http://healthyminds.org/Main-Topic/Panic-Disorder.aspx.
- Anxiety and Depression Association of America. Panic disorder & agoraphobia. http://adaa.org/understanding-anxiety/panic-disorder-agoraphobia.
- Mayo Clinic. Panic attacks and panic disorder. www.mayoclinic.com/health/panic-attacks/DS00338.
- National Health Service Self-Help Guides. www.ntw.nhs.uk/pic/selfhelp.
- National Institute of Mental Health. Panic disorder. www.nimh.nih.gov/health/topics/panic-disorder/index.shtml.
Drug Brand Names
- Alprazolam • Xanax
- Alprazolam XR • Xanax XR
- Citalopram • Celexa
- Clomipramine • Anafranil
- Clonazepam • Klonopin
- Desipramine • Norpramin
- Diazepam • Valium
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Imipramine • Tofranil
- Levetiracetam • Keppra
- Lorazepam • Ativan
- Milnacipran • Savella
- Mirtazapine • Remeron
- Nortriptyline • Aventyl, Pamelor
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Paroxetine CR • Paxil CR
- Phenelzine • Nardil
- Pindolol • Visken
- Quetiapine SR • Seroquel SR
- Risperidone • Risperdal
- Sertraline • Zoloft
- Venlafaxine XR • Effexor XR
Disclosures
Dr. Dunlop receives research support from Bristol-Myers Squibb, GlaxoSmithKline, and the National Institute of Mental Health. He serves as a consultant to MedAvante and Roche.
Ms. Schneider and Dr. Gerardi report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Roy-Byrne PP, Craske MG, Stein MB. Panic disorder. Lancet. 2006;368(9540):1023-1032.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
3. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. 2nd ed. Washington DC: American Psychiatric Association; 2009.
4. Dunlop BW, Davis PG. Combination treatment with benzodiazepines and SSRIs for comorbid anxiety and depression: a review. Prim Care Companion J Clin Psychiatry. 2008;10(3):222-228.
5. Rakofsky JJ, Dunlop BW. Treating nonspecific anxiety and anxiety disorders in patients with bipolar disorder: a review. J Clin Psychiatry. 2011;72(1):81-90.
6. Sareen J, Cox BJ, Afifi TO, et al. Anxiety disorders and risk for suicidal ideation and suicide attempts: a population-based longitudinal study of adults. Arch Gen Psychiatry. 2005;62(11):1249-1257.
7. Houck PR, Spiegel DA, Shear MK, et al. Reliability of the self-report version of the panic disorder severity scale. Depress Anxiety. 2002;15(4):183-185.
8. Ninan PT, Dunlop BW. Neurobiology and etiology of panic disorder. J Clin Psychiatry. 2005;66(suppl 4):3-7.
9. Furukawa TA, Watanabe N, Churchill R. Psychotherapy plus antidepressant for panic disorder with or without agoraphobia: systematic review. Br J Psychiatry. 2006;188:305-312.
10. Barlow DH, Gorman JM, Shear MK, et al. Cognitive-behavioral therapy, imipramine, or their combination for panic disorder: a randomized controlled trial. JAMA. 2000;283(19):2529-2536.
11. van Apeldoorn FJ, Timmerman ME, Mersch PP, et al. A randomized trial of cognitive-behavioral therapy or selective serotonin reuptake inhibitor or both combined for panic disorder with or without agoraphobia: treatment results through 1-year follow-up. J Clin Psychiatry. 2010;71(5):574-586.
12. Bakker A, van Balkom AJ, Spinhoven P. SSRIs vs. TCAs in the treatment of panic disorder: a meta-analysis. Acta Psychiatr Scand. 2002;106(3):163-167.
13. Bandelow B, Behnke K, Lenoir S, et al. Sertraline versus paroxetine in the treatment of panic disorder: an acute, double-blind noninferiority comparison. J Clin Psychiatry. 2004;65(3):405-413.
14. Nardi AE, Freire RC, Mochcovitch MD, et al. A randomized, naturalistic, parallel-group study for the long-term treatment of panic disorder with clonazepam or paroxetine. J Clin Psychopharmacol. 2012;32(1):120-126.
15. Goddard AW, Brouette T, Almai A, et al. Early coadministration of clonazepam with sertraline for panic disorder. Arch Gen Psychiatry. 2001;58(7):681-686.
16. Preskorn SH, Shah R, Neff M, et al. The potential for clinically significant drug-drug interactions involving the CYP 2D6 system: effects with fluoxetine and paroxetine versus sertraline. J Psychiatr Pract. 2007;13(1):5-12.
17. Perna G, Guerriero G, Caldirola D. Emerging drugs for panic disorder. Expert Opin Emerg Drugs. 2011;16(4):631-645.
18. Milrod B, Leon AC, Busch F, et al. A randomized controlled clinical trial of psychoanalytic psychotherapy for panic disorder. Am J Psychiatry. 2007;164(2):265-272.
19. Otto MW, Pollack MH, Sachs GS, et al. Discontinuation of benzodiazepine treatment: efficacy of cognitive-behavioral therapy for patients with panic disorder. Am J Psychiatry. 1993;150(10):1485-1490.
Ms. K, a 24-year-old waitress who lives with her boyfriend, was referred by her primary care physician for evaluation of panic attacks that began “out of nowhere” at work approximately 6 months ago. The unpredictable attacks occur multiple times per week, causing her to leave work and cancel shifts.
Ms. K reports that before the panic attacks began, she felt happy in her relationship, enjoyed hobbies, and was hopeful about the future. However, she has become concerned that a potentially catastrophic illness is causing her panic attacks. She researches her symptoms on the Internet, and is preoccupied with the possibility of sudden death due to an undiagnosed heart condition. Multiple visits to the emergency room have not identified any physical abnormalities. Her primary care doctor prescribed alprazolam, 0.5 mg as needed for panic attacks, which she reports is helpful, “but only in the moment of the attacks.” Ms. K avoids alcohol and illicit substances and limits her caffeine intake. She is not willing to accept that her life “feels so limited.” Her dream of earning a nursing degree and eventually starting a family now seems unattainable.
Panic disorder (PD) occurs in 3% to 5% of adults, with women affected at roughly twice the rate of men.1 Causing a broad range of distress and varying degrees of impairment, PD commonly occurs with other psychiatric disorders. For most patients, treatment is effective, but those who do not respond to initial approaches require a thoughtful, stepped approach to care. Key considerations include establishing an accurate diagnosis, clarifying comorbid illnesses, ascertaining patient beliefs and expectations, and providing appropriately dosed and maintained treatments.
Panic attacks vs PD
Panic attacks consist of rapid onset of intense anxiety, with prominent somatic symptoms, that peaks within 10 minutes (Figure).2 Attacks in which <4 of the listed symptoms occur are considered limited-symptom panic attacks.
Figure: Body locations of panic attack symptoms
Diagnosis of a panic attack requires the sudden development of intense fear or discomfort characterized by ≥4 of the 13 symptoms listed above that peaks in intensity within 10 minutes of onset
Source: Reference 2
Panic attacks can occur with various disorders, including other anxiety disorders, mood disorders, and substance intoxication or withdrawal. Because serious medical conditions can present with panic-like symptoms, the initial occurrence of such symptoms warrants consideration of physiological causes. For a Box2 that describes the differential diagnosis of panic attacks, see this article at CurrentPsychiatry.com.
To meet diagnostic criteria for panic disorder, panic attacks must initially occur “out of the blue,” meaning no specific object or situation induced the attack. The differential diagnosis of panic attacks includes assessing for other psychiatric disorders that may involve panic attacks. Evaluation requires considering the context in which the panic attacks occur, including their start date, pattern of attacks, instigating situations, and associated thoughts.
Social phobia. Attacks occur only during or immediately before a social interaction in which the patient fears embarrassing himself or herself.
Obsessive-compulsive disorder (OCD). Attacks occur when the patient cannot avoid exposure to an obsessional fear or is prevented from performing a ritual that diffuses obsessional anxiety.
Posttraumatic stress disorder (PTSD). Attacks occur when confronted by a trauma-related memory or trigger.
Specific phobia. Attacks occur only when the patient encounters a specifically feared object, place, or situation, unrelated to social phobia, OCD, or PTSD.
Medical conditions. Conditions to consider include—but are not limited to—hyperthyroidism, pulmonary embolism, myocardial infarction, cardiac dysrhythmias, hypoglycemia, asthma, partial complex seizures, and pheochromocytoma.
Source: Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000
A PD diagnosis requires that repeated panic attacks initially must occur from “out of the blue,” meaning no specific object or situation induced the attack. In addition, the diagnosis requires 1 of 3 types of psychological or behavioral changes as a result of the attacks (Table 1).2 Agoraphobia is diagnosed if 1 of the behavioral changes is avoidance of places or situations from which escape might be embarrassing or difficult should an attack occur. A patient can be diagnosed as having PD with agoraphobia, PD without agoraphobia, or agoraphobia without PD (ie, experiences only limited symptom panic attacks, but avoids situations or stimuli associated with them).
Table 1
Definitions of panic disorder and agoraphobia
| Panic disorder |
|---|
|
| Agoraphobia |
| Anxiety about, or avoidance of, being in places or situations from which escape might be difficult or embarrassing, or in which help may not be available in the event of having an unexpected or situationally predisposed panic attack or panic-like symptoms. Agoraphobic fears typically involve characteristic clusters of situations that include being outside the home alone, being in a crowd, standing in a line, being on a bridge, or traveling in a bus, train, or automobile |
| Source: Reference 2 |
Comorbidities are common in patients with PD and predict greater difficulty achieving remission (Box).1,3-6
The most common psychiatric conditions that co-occur with panic disorder (PD) are other anxiety disorders, mood disorders, personality disorders, and substance use disorders.1 Carefully assess the severity and degree of impairment or distress arising from each condition to prioritize treatment goals. For example, treating panic attacks would be a lower priority in a patient with untreated bipolar disorder.
Assessing comorbid substance abuse is important in selecting PD treatments. Benzodiazepines should almost always be avoided in patients with a history of drug abuse—illicit or prescribed. Although complete abstinence should not be a prerequisite for beginning PD treatment, detoxification and concomitant substance abuse treatment are essential.3
Comorbid mood disorders also affect the course of PD treatment. Antidepressants are effective for treating depression and PD, whereas benzodiazepines are not effective for depression.4 Antidepressants in patients with bipolar disorder are controversial because these medications might induce mixed or elevated mood states or rapid cycling. In these complicated patients, consider antidepressants lower in the treatment algorithm.5
Other conditions to consider before beginning treatment include pregnancy or the possibility of becoming pregnant in the near future and suicidal ideation. PD is associated with increased risk for suicidal ideation and progression to suicide attempts, particularly in patients with a comorbid mood or psychotic disorder.6 In addition, consider the potential impact of medications on comorbid medical conditions.
Treatment begins with education
The goal of treatment is remission of symptoms, ideally including an absence of panic attacks, agoraphobic avoidance, and anticipatory anxiety.1 The Panic Disorder Severity Scale self-report is a validated measure of panic symptoms that may be useful in clinical practice.7
The first step in treatment is educating patients about panic attacks, framing them as an overreactive fear circuit in the brain that produces physical symptoms that are not dangerous. Using a brain model that shows the location of the amygdala, hippocampus, and prefrontal cortex—which play crucial roles in generating and controlling anxiety and fear—can make this discussion more concrete.8 Although highly simplified, such models allow clinicians to demonstrate that excessive reactivity of limbic regions can be reduced by both top-down (cortico-limbic connections via cognitive-behavioral therapy [CBT]) and bottom-up (pharmacotherapy directly acting on limbic structures) approaches. Such discussions lead to treatment recommendations for CBT, pharmacotherapy, or their combination.
No single treatment has emerged as the definitive “best” for PD, and no reliable predictors can guide specific treatment for an individual.3 Combining CBT with pharmacotherapy produces higher short-term response rates than either treatment alone, but in the long term, combination treatment does not appear to be superior to CBT alone.9 Base the initial treatment selection for PD on patient preference, treatment availability and cost, and comorbid medical and psychiatric conditions. For an Algorithm to guide treatment decisions, see this article at CurrentPsychiatry.com.
Algorithm: Treatment for panic disorder: A suggested algorithm
aPoor response to an SSRI should lead to a switch to venlafaxine extended-release, and vice versa
bBenzodiazepines are relatively contraindicated in geriatric patients and patients with a history of substance abuse or dependence
CBT: cognitive-behavioral therapy; MAOI: monoamine oxidase inhibitor; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant; Ven XR: venlafaxine extended-release
First-line treatments
Psychotherapy. CBT is the most efficacious psychotherapy for PD. Twelve to 15 sessions of CBT has demonstrated efficacy for PD, with additional effects on comorbid anxiety and depressive symptoms.10 No large clinical trials of CBT have used cognitive restructuring alone; all have included at least some component of exposure that requires the patient to confront feared physical sensations. Gains during treatment may be steady and gradual or sudden and uneven, with rapid improvement in some but not all symptoms. CBT and pharmacotherapy have demonstrated similar levels of benefit in short-term trials, but CBT has proven superior in most9 but not all11 trials evaluating long-term outcomes, particularly compared with pharmacotherapy that is discontinued during follow-up. Although less studied, group CBT also may be considered if a patient cannot afford individual CBT.
Pharmacotherapy. Evidence supports selective serotonin reuptake inhibitors (SSRIs), venlafaxine extended-release (XR), benzodiazepines, and tricyclic antidepressants (TCAs) as effective treatments for PD.3 No class of medication has demonstrated superiority over others in short-term treatment.3,12 Because of the medical risks associated with benzodiazepines and TCAs, an SSRI or venlafaxine XR should be the first medication option for most patients. Fluoxetine, paroxetine, sertraline, and venlafaxine XR are FDA-approved for PD. Paroxetine is associated with weight gain and may increase the risk for panic recurrence upon discontinuation more than sertraline, making it a less favorable option for many patients.13 Start doses at half the normal starting dose used for treating major depressive disorder and continue for 4 to 7 days, then increase to the minimal effective dose. For a Table3 that lists dosing recommendations for antidepressants to treat PD, see this article at CurrentPsychiatry.com. If there is no improvement by 4 weeks, increase the dose every 2 to 4 weeks until remission is achieved or side effects prevent further dose increases.
Table
Recommended doses for antidepressants used to treat panic disorder
| Medication | Starting dose (mg/d) | Therapeutic range (mg/d) |
|---|---|---|
| SSRIs | ||
| Citalopram | 10 | 20 to 40 |
| Escitalopram | 5 | 10 to 40 |
| Fluoxetine | 5 to 10 | 20 to 80 |
| Fluvoxamine | 25 | 100 to 300 |
| Paroxetine | 10 | 20 to 80 |
| Paroxetine CR | 12.5 | 25 to 50 |
| Sertraline | 25 | 100 to 200 |
| SNRIs | ||
| Duloxetine | 20 to 30 | 60 to 120 |
| Venlafaxine XR | 37.5 | 150 to 225 |
| TCAs | ||
| Clomipramine | 10 to 25 | 100 to 300 |
| Imipramine | 10 | 100 to 300 |
| MAOI | ||
| Phenelzine | 15 | 45 to 90 |
| CR: controlled release; MAOI: monoamine oxidase inhibitor; SNRIs: serotonin-norepinephrine reuptake inhibitors; SSRIs: selective serotonin reuptake inhibitors; TCAs: tricyclic antidepressants; XR: extended release Source: American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. 2nd ed. Washington, DC: American Psychiatric Association; 2009 | ||
Treatment nonresponse. True non-response needs to be distinguished from poor response caused by inadequate treatment delivery, eg, patients not completing homework assignments in CBT or not adhering to pharmacotherapy. Asking patients about adverse effects or personal and family beliefs about treatment may reveal reasons for nonadherence.
Second-line treatments
Little data are available to guide next-step treatment options in patients who don’t achieve remission from their initial treatment. Patients who benefit from an SSRI, venlafaxine XR, or CBT but still have symptoms should be started on combination treatment. For a patient who experiences complete non-response to the initial treatment, discontinue the first treatment and switch to the other modality. In general, completely ineffective treatments should be discontinued when another treatment is added, but when partial improvement (>30%) occurs, continue the original treatment and augment it with another approach.
For patients pursuing pharmacotherapy, poor response to an adequate SSRI trial usually should lead to a switch to venlafaxine XR, and vice versa. Failure to respond to both of these medication classes should prompt a switch to a benzodiazepine or TCA.
Benzodiazepines are a fast-acting, effective treatment for PD, with efficacy similar to SSRIs in acute and long-term treatment.14 Benzodiazepines may be prescribed with antidepressants at the beginning of treatment to improve response speed.15 Clonazepam and alprazolam are FDA-approved for treating PD. A high-potency, long-acting agent, clonazepam is the preferred initial benzodiazepine, dosed 0.5 to 4 mg/d on a fixed schedule. Although substantial data support using alprazolam for PD, it requires more frequent dosing and has a greater risk of rebound anxiety and abuse potential because of its more rapid onset of action. Compared with immediate-release alprazolam, alprazolam XR has a slower absorption rate and longer steady state in the blood, but this formulation does not have lower abuse potential or greater efficacy. Although not FDA-approved for PD, diazepam and lorazepam also have proven efficacy for PD.3
Benzodiazepines should be considered contraindicated in patients with a history of substance abuse, except in select cases.4 Benzodiazepines generally should be avoided in older patients because of increased risk for falls, cognitive impairment, and motor vehicle accidents. Table 2 lists situations in which benzodiazepines may be used to treat PD.
Table 2
Clinical scenarios in which to consider using benzodiazepines
| Coadministration for 2 to 4 weeks when initiating treatment with an SSRI or venlafaxine XR to achieve more rapid relief and mitigate potential antidepressant-induced anxiety |
| For patients who wish to avoid antidepressants because of concern about sexual dysfunction |
| For patients who need chronic aspirin or an NSAID, which may increase the risk for upper gastrointestinal bleeding when taken in combination with an SSRI |
| For patients with comorbid bipolar disorder or epilepsy |
| Next-step monotherapy or augmentation in patients who respond poorly to an SSRI, venlafaxine XR, TCA, or CBT |
| CBT: cognitive-behavioral therapy; NSAID: nonsteroidal anti-inflammatory drug; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant; XR: extended release |
TCAs are effective as monotherapy for PD. Most support comes from studies of imipramine or clomipramine.12 Similar to SSRIs and venlafaxine XR, use a low initial dose and gradually increase until the patient remits or side effects prevent further increases. SSRI and TCA combinations rarely are used unless the TCA is a relatively specific norepinephrine reuptake inhibitor (eg, desipramine, nortriptyline). Because TCAs are metabolized via the cytochrome P450 2D6 system and some SSRIs—particularly fluoxetine and paroxetine—strongly inhibit 2D6, combinations of TCAs with these agents may lead to dangerously high plasma TCA levels, placing patients at risk for cardiac dysrhythmias and other side effects.16
Monoamine oxidase inhibitors (MAOIs)—particularly phenelzine—are underused for PD. They have the strongest efficacy data for any class of medications outside the first- and second-line agents and have a unique mechanism of action. In patients who can comply with the dietary and medication limitations, an MAOI generally should be the next step after nonresponse to other treatments.3
Alternative treatments
For patients who do not respond to any of the treatments described above, data from uncontrolled studies support mirtazapine, levetiracetam, and the serotonin-norepinephrine reuptake inhibitors duloxetine and milnacipran as monotherapy for PD.17 Pindolol—a beta blocker and 5-HT1A receptor antagonist—proved superior to placebo as an adjunctive agent to SSRIs in treatment-resistant PD in 1 of 2 trials.17 Minimal evidence supports the atypical antipsychotics risperidone and olanzapine in treatment-resistant PD, although a placebo-controlled trial of quetiapine SR coadministered with SSRIs recently was completed (NCT00619892; results pending). Atypical antipsychotics are best reserved for patients with a primary psychotic disorder or bipolar disorder who experience panic attacks.5
Panic-focused psychodynamic psychotherapy, a 12-week (approximately 24 sessions) form of psychotherapy, has demonstrated superiority vs applied relaxation therapy.18 This treatment could be considered for patients who do not respond to standard first-line treatments, but few community therapists are familiar with this method.
For many patients with PD, complementary and alternative medicine (CAM) approaches are appealing. See this article at CurrentPsychiatry.com for a Box that discusses CAM for PD.
Although no complementary and alternative medicine treatments have strong evidence of efficacy as monotherapy for panic disorder (PD), several have data that suggest benefit with little evidence of risk. These include bibliotherapy, yoga, aerobic exercise, and the dietary supplements kava and inositol.a Exercise as a treatment poses a challenge because it can induce symptoms that the patient fears, such as tachycardia and shortness of breath. In addition to any direct physiologic benefit from aerobic exercise, there is also an exposure component that can be harnessed by gradually increasing the exertion level.
Another approach undergoing extensive evaluation is Internet-provided cognitive-behavioral therapy (CBT). Using guided CBT modules with or without therapist support, Internet-provided CBT provides an option for motivated patients unable to complete in-person CBT because of logistical factors.b A helpful resource that reviews Internet self-help and psychotherapy guided programs for PD and other psychiatric conditions is http://beacon.anu.edu.au.
References
a. Antonacci DJ, Davis E, Bloch RM, et al. CAM for your anxious patient: what the evidence says. Current Psychiatry. 2010;9(10):42-52.
b. Johnston L, Titov N, Andrews G, et al. A RCT of a transdiagnostic internet-delivered treatment for three anxiety disorders: examination of support roles and disorder-specific outcomes. PLoS One. 2011;6(11):e28079.
Maintenance treatment
Patients who complete a course of CBT for PD often follow up with several “booster sessions” at monthly or longer intervals that focus on relapse prevention techniques. Few controlled trials have evaluated pharmacotherapy discontinuation in PD. Most guidelines recommend continuing treatment for ≥1 year after achieving remission to minimize the risk of relapse.3 Researchers are focusing on whether medication dosage can be reduced during maintenance without loss of efficacy.
Treatment discontinuation
In the absence of urgent medical need, taper medications for PD gradually over several months. PD patients are highly sensitive to unusual physical sensations, which can occur while discontinuing antidepressants or benzodiazepines. If a benzodiazepine is used in conjunction with an antidepressant, the benzodiazepine should be discontinued first, so that the antidepressant can help ease benzodiazepine-associated discontinuation symptoms. A brief course of CBT during pharmacotherapy discontinuation may increase the likelihood of successful tapering.19
CASE CONTINUED: A successful switch
Ms. K has to discontinue sequential trials of fluoxetine, 40 mg/d, and venlafaxine XR, 225 mg/d because of side effects, and she does not reduce the frequency of her alprazolam use. She agrees to switch from alprazolam to clonazepam, 0.5 mg every morning and 1 mg at bedtime, and to start CBT. Clonazepam reduces her anxiety sufficiently so she can address her symptoms in therapy. Through CBT she becomes motivated to monitor her thoughts and treat them as guesses rather than facts, reviewing the evidence for her thoughts and generating rational responses. She participates in exposure exercises, which she practices between sessions, and grows to tolerate uncomfortable sensations until they no longer signal danger. After 12 CBT sessions, she is panic-free. Despite some trepidation, she agrees to a slow taper off clonazepam, reducing the dose by 0.25 mg every 2 weeks. She continues booster sessions with her therapist to manage any re-emerging anxiety. After an additional 12 weeks, she successfully discontinues clonazepam and remains panic-free.
Related Resources
- American Psychiatric Association. Panic disorder. http://healthyminds.org/Main-Topic/Panic-Disorder.aspx.
- Anxiety and Depression Association of America. Panic disorder & agoraphobia. http://adaa.org/understanding-anxiety/panic-disorder-agoraphobia.
- Mayo Clinic. Panic attacks and panic disorder. www.mayoclinic.com/health/panic-attacks/DS00338.
- National Health Service Self-Help Guides. www.ntw.nhs.uk/pic/selfhelp.
- National Institute of Mental Health. Panic disorder. www.nimh.nih.gov/health/topics/panic-disorder/index.shtml.
Drug Brand Names
- Alprazolam • Xanax
- Alprazolam XR • Xanax XR
- Citalopram • Celexa
- Clomipramine • Anafranil
- Clonazepam • Klonopin
- Desipramine • Norpramin
- Diazepam • Valium
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Imipramine • Tofranil
- Levetiracetam • Keppra
- Lorazepam • Ativan
- Milnacipran • Savella
- Mirtazapine • Remeron
- Nortriptyline • Aventyl, Pamelor
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Paroxetine CR • Paxil CR
- Phenelzine • Nardil
- Pindolol • Visken
- Quetiapine SR • Seroquel SR
- Risperidone • Risperdal
- Sertraline • Zoloft
- Venlafaxine XR • Effexor XR
Disclosures
Dr. Dunlop receives research support from Bristol-Myers Squibb, GlaxoSmithKline, and the National Institute of Mental Health. He serves as a consultant to MedAvante and Roche.
Ms. Schneider and Dr. Gerardi report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Ms. K, a 24-year-old waitress who lives with her boyfriend, was referred by her primary care physician for evaluation of panic attacks that began “out of nowhere” at work approximately 6 months ago. The unpredictable attacks occur multiple times per week, causing her to leave work and cancel shifts.
Ms. K reports that before the panic attacks began, she felt happy in her relationship, enjoyed hobbies, and was hopeful about the future. However, she has become concerned that a potentially catastrophic illness is causing her panic attacks. She researches her symptoms on the Internet, and is preoccupied with the possibility of sudden death due to an undiagnosed heart condition. Multiple visits to the emergency room have not identified any physical abnormalities. Her primary care doctor prescribed alprazolam, 0.5 mg as needed for panic attacks, which she reports is helpful, “but only in the moment of the attacks.” Ms. K avoids alcohol and illicit substances and limits her caffeine intake. She is not willing to accept that her life “feels so limited.” Her dream of earning a nursing degree and eventually starting a family now seems unattainable.
Panic disorder (PD) occurs in 3% to 5% of adults, with women affected at roughly twice the rate of men.1 Causing a broad range of distress and varying degrees of impairment, PD commonly occurs with other psychiatric disorders. For most patients, treatment is effective, but those who do not respond to initial approaches require a thoughtful, stepped approach to care. Key considerations include establishing an accurate diagnosis, clarifying comorbid illnesses, ascertaining patient beliefs and expectations, and providing appropriately dosed and maintained treatments.
Panic attacks vs PD
Panic attacks consist of rapid onset of intense anxiety, with prominent somatic symptoms, that peaks within 10 minutes (Figure).2 Attacks in which <4 of the listed symptoms occur are considered limited-symptom panic attacks.
Figure: Body locations of panic attack symptoms
Diagnosis of a panic attack requires the sudden development of intense fear or discomfort characterized by ≥4 of the 13 symptoms listed above that peaks in intensity within 10 minutes of onset
Source: Reference 2
Panic attacks can occur with various disorders, including other anxiety disorders, mood disorders, and substance intoxication or withdrawal. Because serious medical conditions can present with panic-like symptoms, the initial occurrence of such symptoms warrants consideration of physiological causes. For a Box2 that describes the differential diagnosis of panic attacks, see this article at CurrentPsychiatry.com.
To meet diagnostic criteria for panic disorder, panic attacks must initially occur “out of the blue,” meaning no specific object or situation induced the attack. The differential diagnosis of panic attacks includes assessing for other psychiatric disorders that may involve panic attacks. Evaluation requires considering the context in which the panic attacks occur, including their start date, pattern of attacks, instigating situations, and associated thoughts.
Social phobia. Attacks occur only during or immediately before a social interaction in which the patient fears embarrassing himself or herself.
Obsessive-compulsive disorder (OCD). Attacks occur when the patient cannot avoid exposure to an obsessional fear or is prevented from performing a ritual that diffuses obsessional anxiety.
Posttraumatic stress disorder (PTSD). Attacks occur when confronted by a trauma-related memory or trigger.
Specific phobia. Attacks occur only when the patient encounters a specifically feared object, place, or situation, unrelated to social phobia, OCD, or PTSD.
Medical conditions. Conditions to consider include—but are not limited to—hyperthyroidism, pulmonary embolism, myocardial infarction, cardiac dysrhythmias, hypoglycemia, asthma, partial complex seizures, and pheochromocytoma.
Source: Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000
A PD diagnosis requires that repeated panic attacks initially must occur from “out of the blue,” meaning no specific object or situation induced the attack. In addition, the diagnosis requires 1 of 3 types of psychological or behavioral changes as a result of the attacks (Table 1).2 Agoraphobia is diagnosed if 1 of the behavioral changes is avoidance of places or situations from which escape might be embarrassing or difficult should an attack occur. A patient can be diagnosed as having PD with agoraphobia, PD without agoraphobia, or agoraphobia without PD (ie, experiences only limited symptom panic attacks, but avoids situations or stimuli associated with them).
Table 1
Definitions of panic disorder and agoraphobia
| Panic disorder |
|---|
|
| Agoraphobia |
| Anxiety about, or avoidance of, being in places or situations from which escape might be difficult or embarrassing, or in which help may not be available in the event of having an unexpected or situationally predisposed panic attack or panic-like symptoms. Agoraphobic fears typically involve characteristic clusters of situations that include being outside the home alone, being in a crowd, standing in a line, being on a bridge, or traveling in a bus, train, or automobile |
| Source: Reference 2 |
Comorbidities are common in patients with PD and predict greater difficulty achieving remission (Box).1,3-6
The most common psychiatric conditions that co-occur with panic disorder (PD) are other anxiety disorders, mood disorders, personality disorders, and substance use disorders.1 Carefully assess the severity and degree of impairment or distress arising from each condition to prioritize treatment goals. For example, treating panic attacks would be a lower priority in a patient with untreated bipolar disorder.
Assessing comorbid substance abuse is important in selecting PD treatments. Benzodiazepines should almost always be avoided in patients with a history of drug abuse—illicit or prescribed. Although complete abstinence should not be a prerequisite for beginning PD treatment, detoxification and concomitant substance abuse treatment are essential.3
Comorbid mood disorders also affect the course of PD treatment. Antidepressants are effective for treating depression and PD, whereas benzodiazepines are not effective for depression.4 Antidepressants in patients with bipolar disorder are controversial because these medications might induce mixed or elevated mood states or rapid cycling. In these complicated patients, consider antidepressants lower in the treatment algorithm.5
Other conditions to consider before beginning treatment include pregnancy or the possibility of becoming pregnant in the near future and suicidal ideation. PD is associated with increased risk for suicidal ideation and progression to suicide attempts, particularly in patients with a comorbid mood or psychotic disorder.6 In addition, consider the potential impact of medications on comorbid medical conditions.
Treatment begins with education
The goal of treatment is remission of symptoms, ideally including an absence of panic attacks, agoraphobic avoidance, and anticipatory anxiety.1 The Panic Disorder Severity Scale self-report is a validated measure of panic symptoms that may be useful in clinical practice.7
The first step in treatment is educating patients about panic attacks, framing them as an overreactive fear circuit in the brain that produces physical symptoms that are not dangerous. Using a brain model that shows the location of the amygdala, hippocampus, and prefrontal cortex—which play crucial roles in generating and controlling anxiety and fear—can make this discussion more concrete.8 Although highly simplified, such models allow clinicians to demonstrate that excessive reactivity of limbic regions can be reduced by both top-down (cortico-limbic connections via cognitive-behavioral therapy [CBT]) and bottom-up (pharmacotherapy directly acting on limbic structures) approaches. Such discussions lead to treatment recommendations for CBT, pharmacotherapy, or their combination.
No single treatment has emerged as the definitive “best” for PD, and no reliable predictors can guide specific treatment for an individual.3 Combining CBT with pharmacotherapy produces higher short-term response rates than either treatment alone, but in the long term, combination treatment does not appear to be superior to CBT alone.9 Base the initial treatment selection for PD on patient preference, treatment availability and cost, and comorbid medical and psychiatric conditions. For an Algorithm to guide treatment decisions, see this article at CurrentPsychiatry.com.
Algorithm: Treatment for panic disorder: A suggested algorithm
aPoor response to an SSRI should lead to a switch to venlafaxine extended-release, and vice versa
bBenzodiazepines are relatively contraindicated in geriatric patients and patients with a history of substance abuse or dependence
CBT: cognitive-behavioral therapy; MAOI: monoamine oxidase inhibitor; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant; Ven XR: venlafaxine extended-release
First-line treatments
Psychotherapy. CBT is the most efficacious psychotherapy for PD. Twelve to 15 sessions of CBT has demonstrated efficacy for PD, with additional effects on comorbid anxiety and depressive symptoms.10 No large clinical trials of CBT have used cognitive restructuring alone; all have included at least some component of exposure that requires the patient to confront feared physical sensations. Gains during treatment may be steady and gradual or sudden and uneven, with rapid improvement in some but not all symptoms. CBT and pharmacotherapy have demonstrated similar levels of benefit in short-term trials, but CBT has proven superior in most9 but not all11 trials evaluating long-term outcomes, particularly compared with pharmacotherapy that is discontinued during follow-up. Although less studied, group CBT also may be considered if a patient cannot afford individual CBT.
Pharmacotherapy. Evidence supports selective serotonin reuptake inhibitors (SSRIs), venlafaxine extended-release (XR), benzodiazepines, and tricyclic antidepressants (TCAs) as effective treatments for PD.3 No class of medication has demonstrated superiority over others in short-term treatment.3,12 Because of the medical risks associated with benzodiazepines and TCAs, an SSRI or venlafaxine XR should be the first medication option for most patients. Fluoxetine, paroxetine, sertraline, and venlafaxine XR are FDA-approved for PD. Paroxetine is associated with weight gain and may increase the risk for panic recurrence upon discontinuation more than sertraline, making it a less favorable option for many patients.13 Start doses at half the normal starting dose used for treating major depressive disorder and continue for 4 to 7 days, then increase to the minimal effective dose. For a Table3 that lists dosing recommendations for antidepressants to treat PD, see this article at CurrentPsychiatry.com. If there is no improvement by 4 weeks, increase the dose every 2 to 4 weeks until remission is achieved or side effects prevent further dose increases.
Table
Recommended doses for antidepressants used to treat panic disorder
| Medication | Starting dose (mg/d) | Therapeutic range (mg/d) |
|---|---|---|
| SSRIs | ||
| Citalopram | 10 | 20 to 40 |
| Escitalopram | 5 | 10 to 40 |
| Fluoxetine | 5 to 10 | 20 to 80 |
| Fluvoxamine | 25 | 100 to 300 |
| Paroxetine | 10 | 20 to 80 |
| Paroxetine CR | 12.5 | 25 to 50 |
| Sertraline | 25 | 100 to 200 |
| SNRIs | ||
| Duloxetine | 20 to 30 | 60 to 120 |
| Venlafaxine XR | 37.5 | 150 to 225 |
| TCAs | ||
| Clomipramine | 10 to 25 | 100 to 300 |
| Imipramine | 10 | 100 to 300 |
| MAOI | ||
| Phenelzine | 15 | 45 to 90 |
| CR: controlled release; MAOI: monoamine oxidase inhibitor; SNRIs: serotonin-norepinephrine reuptake inhibitors; SSRIs: selective serotonin reuptake inhibitors; TCAs: tricyclic antidepressants; XR: extended release Source: American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. 2nd ed. Washington, DC: American Psychiatric Association; 2009 | ||
Treatment nonresponse. True non-response needs to be distinguished from poor response caused by inadequate treatment delivery, eg, patients not completing homework assignments in CBT or not adhering to pharmacotherapy. Asking patients about adverse effects or personal and family beliefs about treatment may reveal reasons for nonadherence.
Second-line treatments
Little data are available to guide next-step treatment options in patients who don’t achieve remission from their initial treatment. Patients who benefit from an SSRI, venlafaxine XR, or CBT but still have symptoms should be started on combination treatment. For a patient who experiences complete non-response to the initial treatment, discontinue the first treatment and switch to the other modality. In general, completely ineffective treatments should be discontinued when another treatment is added, but when partial improvement (>30%) occurs, continue the original treatment and augment it with another approach.
For patients pursuing pharmacotherapy, poor response to an adequate SSRI trial usually should lead to a switch to venlafaxine XR, and vice versa. Failure to respond to both of these medication classes should prompt a switch to a benzodiazepine or TCA.
Benzodiazepines are a fast-acting, effective treatment for PD, with efficacy similar to SSRIs in acute and long-term treatment.14 Benzodiazepines may be prescribed with antidepressants at the beginning of treatment to improve response speed.15 Clonazepam and alprazolam are FDA-approved for treating PD. A high-potency, long-acting agent, clonazepam is the preferred initial benzodiazepine, dosed 0.5 to 4 mg/d on a fixed schedule. Although substantial data support using alprazolam for PD, it requires more frequent dosing and has a greater risk of rebound anxiety and abuse potential because of its more rapid onset of action. Compared with immediate-release alprazolam, alprazolam XR has a slower absorption rate and longer steady state in the blood, but this formulation does not have lower abuse potential or greater efficacy. Although not FDA-approved for PD, diazepam and lorazepam also have proven efficacy for PD.3
Benzodiazepines should be considered contraindicated in patients with a history of substance abuse, except in select cases.4 Benzodiazepines generally should be avoided in older patients because of increased risk for falls, cognitive impairment, and motor vehicle accidents. Table 2 lists situations in which benzodiazepines may be used to treat PD.
Table 2
Clinical scenarios in which to consider using benzodiazepines
| Coadministration for 2 to 4 weeks when initiating treatment with an SSRI or venlafaxine XR to achieve more rapid relief and mitigate potential antidepressant-induced anxiety |
| For patients who wish to avoid antidepressants because of concern about sexual dysfunction |
| For patients who need chronic aspirin or an NSAID, which may increase the risk for upper gastrointestinal bleeding when taken in combination with an SSRI |
| For patients with comorbid bipolar disorder or epilepsy |
| Next-step monotherapy or augmentation in patients who respond poorly to an SSRI, venlafaxine XR, TCA, or CBT |
| CBT: cognitive-behavioral therapy; NSAID: nonsteroidal anti-inflammatory drug; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant; XR: extended release |
TCAs are effective as monotherapy for PD. Most support comes from studies of imipramine or clomipramine.12 Similar to SSRIs and venlafaxine XR, use a low initial dose and gradually increase until the patient remits or side effects prevent further increases. SSRI and TCA combinations rarely are used unless the TCA is a relatively specific norepinephrine reuptake inhibitor (eg, desipramine, nortriptyline). Because TCAs are metabolized via the cytochrome P450 2D6 system and some SSRIs—particularly fluoxetine and paroxetine—strongly inhibit 2D6, combinations of TCAs with these agents may lead to dangerously high plasma TCA levels, placing patients at risk for cardiac dysrhythmias and other side effects.16
Monoamine oxidase inhibitors (MAOIs)—particularly phenelzine—are underused for PD. They have the strongest efficacy data for any class of medications outside the first- and second-line agents and have a unique mechanism of action. In patients who can comply with the dietary and medication limitations, an MAOI generally should be the next step after nonresponse to other treatments.3
Alternative treatments
For patients who do not respond to any of the treatments described above, data from uncontrolled studies support mirtazapine, levetiracetam, and the serotonin-norepinephrine reuptake inhibitors duloxetine and milnacipran as monotherapy for PD.17 Pindolol—a beta blocker and 5-HT1A receptor antagonist—proved superior to placebo as an adjunctive agent to SSRIs in treatment-resistant PD in 1 of 2 trials.17 Minimal evidence supports the atypical antipsychotics risperidone and olanzapine in treatment-resistant PD, although a placebo-controlled trial of quetiapine SR coadministered with SSRIs recently was completed (NCT00619892; results pending). Atypical antipsychotics are best reserved for patients with a primary psychotic disorder or bipolar disorder who experience panic attacks.5
Panic-focused psychodynamic psychotherapy, a 12-week (approximately 24 sessions) form of psychotherapy, has demonstrated superiority vs applied relaxation therapy.18 This treatment could be considered for patients who do not respond to standard first-line treatments, but few community therapists are familiar with this method.
For many patients with PD, complementary and alternative medicine (CAM) approaches are appealing. See this article at CurrentPsychiatry.com for a Box that discusses CAM for PD.
Although no complementary and alternative medicine treatments have strong evidence of efficacy as monotherapy for panic disorder (PD), several have data that suggest benefit with little evidence of risk. These include bibliotherapy, yoga, aerobic exercise, and the dietary supplements kava and inositol.a Exercise as a treatment poses a challenge because it can induce symptoms that the patient fears, such as tachycardia and shortness of breath. In addition to any direct physiologic benefit from aerobic exercise, there is also an exposure component that can be harnessed by gradually increasing the exertion level.
Another approach undergoing extensive evaluation is Internet-provided cognitive-behavioral therapy (CBT). Using guided CBT modules with or without therapist support, Internet-provided CBT provides an option for motivated patients unable to complete in-person CBT because of logistical factors.b A helpful resource that reviews Internet self-help and psychotherapy guided programs for PD and other psychiatric conditions is http://beacon.anu.edu.au.
References
a. Antonacci DJ, Davis E, Bloch RM, et al. CAM for your anxious patient: what the evidence says. Current Psychiatry. 2010;9(10):42-52.
b. Johnston L, Titov N, Andrews G, et al. A RCT of a transdiagnostic internet-delivered treatment for three anxiety disorders: examination of support roles and disorder-specific outcomes. PLoS One. 2011;6(11):e28079.
Maintenance treatment
Patients who complete a course of CBT for PD often follow up with several “booster sessions” at monthly or longer intervals that focus on relapse prevention techniques. Few controlled trials have evaluated pharmacotherapy discontinuation in PD. Most guidelines recommend continuing treatment for ≥1 year after achieving remission to minimize the risk of relapse.3 Researchers are focusing on whether medication dosage can be reduced during maintenance without loss of efficacy.
Treatment discontinuation
In the absence of urgent medical need, taper medications for PD gradually over several months. PD patients are highly sensitive to unusual physical sensations, which can occur while discontinuing antidepressants or benzodiazepines. If a benzodiazepine is used in conjunction with an antidepressant, the benzodiazepine should be discontinued first, so that the antidepressant can help ease benzodiazepine-associated discontinuation symptoms. A brief course of CBT during pharmacotherapy discontinuation may increase the likelihood of successful tapering.19
CASE CONTINUED: A successful switch
Ms. K has to discontinue sequential trials of fluoxetine, 40 mg/d, and venlafaxine XR, 225 mg/d because of side effects, and she does not reduce the frequency of her alprazolam use. She agrees to switch from alprazolam to clonazepam, 0.5 mg every morning and 1 mg at bedtime, and to start CBT. Clonazepam reduces her anxiety sufficiently so she can address her symptoms in therapy. Through CBT she becomes motivated to monitor her thoughts and treat them as guesses rather than facts, reviewing the evidence for her thoughts and generating rational responses. She participates in exposure exercises, which she practices between sessions, and grows to tolerate uncomfortable sensations until they no longer signal danger. After 12 CBT sessions, she is panic-free. Despite some trepidation, she agrees to a slow taper off clonazepam, reducing the dose by 0.25 mg every 2 weeks. She continues booster sessions with her therapist to manage any re-emerging anxiety. After an additional 12 weeks, she successfully discontinues clonazepam and remains panic-free.
Related Resources
- American Psychiatric Association. Panic disorder. http://healthyminds.org/Main-Topic/Panic-Disorder.aspx.
- Anxiety and Depression Association of America. Panic disorder & agoraphobia. http://adaa.org/understanding-anxiety/panic-disorder-agoraphobia.
- Mayo Clinic. Panic attacks and panic disorder. www.mayoclinic.com/health/panic-attacks/DS00338.
- National Health Service Self-Help Guides. www.ntw.nhs.uk/pic/selfhelp.
- National Institute of Mental Health. Panic disorder. www.nimh.nih.gov/health/topics/panic-disorder/index.shtml.
Drug Brand Names
- Alprazolam • Xanax
- Alprazolam XR • Xanax XR
- Citalopram • Celexa
- Clomipramine • Anafranil
- Clonazepam • Klonopin
- Desipramine • Norpramin
- Diazepam • Valium
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Imipramine • Tofranil
- Levetiracetam • Keppra
- Lorazepam • Ativan
- Milnacipran • Savella
- Mirtazapine • Remeron
- Nortriptyline • Aventyl, Pamelor
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Paroxetine CR • Paxil CR
- Phenelzine • Nardil
- Pindolol • Visken
- Quetiapine SR • Seroquel SR
- Risperidone • Risperdal
- Sertraline • Zoloft
- Venlafaxine XR • Effexor XR
Disclosures
Dr. Dunlop receives research support from Bristol-Myers Squibb, GlaxoSmithKline, and the National Institute of Mental Health. He serves as a consultant to MedAvante and Roche.
Ms. Schneider and Dr. Gerardi report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Roy-Byrne PP, Craske MG, Stein MB. Panic disorder. Lancet. 2006;368(9540):1023-1032.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
3. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. 2nd ed. Washington DC: American Psychiatric Association; 2009.
4. Dunlop BW, Davis PG. Combination treatment with benzodiazepines and SSRIs for comorbid anxiety and depression: a review. Prim Care Companion J Clin Psychiatry. 2008;10(3):222-228.
5. Rakofsky JJ, Dunlop BW. Treating nonspecific anxiety and anxiety disorders in patients with bipolar disorder: a review. J Clin Psychiatry. 2011;72(1):81-90.
6. Sareen J, Cox BJ, Afifi TO, et al. Anxiety disorders and risk for suicidal ideation and suicide attempts: a population-based longitudinal study of adults. Arch Gen Psychiatry. 2005;62(11):1249-1257.
7. Houck PR, Spiegel DA, Shear MK, et al. Reliability of the self-report version of the panic disorder severity scale. Depress Anxiety. 2002;15(4):183-185.
8. Ninan PT, Dunlop BW. Neurobiology and etiology of panic disorder. J Clin Psychiatry. 2005;66(suppl 4):3-7.
9. Furukawa TA, Watanabe N, Churchill R. Psychotherapy plus antidepressant for panic disorder with or without agoraphobia: systematic review. Br J Psychiatry. 2006;188:305-312.
10. Barlow DH, Gorman JM, Shear MK, et al. Cognitive-behavioral therapy, imipramine, or their combination for panic disorder: a randomized controlled trial. JAMA. 2000;283(19):2529-2536.
11. van Apeldoorn FJ, Timmerman ME, Mersch PP, et al. A randomized trial of cognitive-behavioral therapy or selective serotonin reuptake inhibitor or both combined for panic disorder with or without agoraphobia: treatment results through 1-year follow-up. J Clin Psychiatry. 2010;71(5):574-586.
12. Bakker A, van Balkom AJ, Spinhoven P. SSRIs vs. TCAs in the treatment of panic disorder: a meta-analysis. Acta Psychiatr Scand. 2002;106(3):163-167.
13. Bandelow B, Behnke K, Lenoir S, et al. Sertraline versus paroxetine in the treatment of panic disorder: an acute, double-blind noninferiority comparison. J Clin Psychiatry. 2004;65(3):405-413.
14. Nardi AE, Freire RC, Mochcovitch MD, et al. A randomized, naturalistic, parallel-group study for the long-term treatment of panic disorder with clonazepam or paroxetine. J Clin Psychopharmacol. 2012;32(1):120-126.
15. Goddard AW, Brouette T, Almai A, et al. Early coadministration of clonazepam with sertraline for panic disorder. Arch Gen Psychiatry. 2001;58(7):681-686.
16. Preskorn SH, Shah R, Neff M, et al. The potential for clinically significant drug-drug interactions involving the CYP 2D6 system: effects with fluoxetine and paroxetine versus sertraline. J Psychiatr Pract. 2007;13(1):5-12.
17. Perna G, Guerriero G, Caldirola D. Emerging drugs for panic disorder. Expert Opin Emerg Drugs. 2011;16(4):631-645.
18. Milrod B, Leon AC, Busch F, et al. A randomized controlled clinical trial of psychoanalytic psychotherapy for panic disorder. Am J Psychiatry. 2007;164(2):265-272.
19. Otto MW, Pollack MH, Sachs GS, et al. Discontinuation of benzodiazepine treatment: efficacy of cognitive-behavioral therapy for patients with panic disorder. Am J Psychiatry. 1993;150(10):1485-1490.
1. Roy-Byrne PP, Craske MG, Stein MB. Panic disorder. Lancet. 2006;368(9540):1023-1032.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
3. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. 2nd ed. Washington DC: American Psychiatric Association; 2009.
4. Dunlop BW, Davis PG. Combination treatment with benzodiazepines and SSRIs for comorbid anxiety and depression: a review. Prim Care Companion J Clin Psychiatry. 2008;10(3):222-228.
5. Rakofsky JJ, Dunlop BW. Treating nonspecific anxiety and anxiety disorders in patients with bipolar disorder: a review. J Clin Psychiatry. 2011;72(1):81-90.
6. Sareen J, Cox BJ, Afifi TO, et al. Anxiety disorders and risk for suicidal ideation and suicide attempts: a population-based longitudinal study of adults. Arch Gen Psychiatry. 2005;62(11):1249-1257.
7. Houck PR, Spiegel DA, Shear MK, et al. Reliability of the self-report version of the panic disorder severity scale. Depress Anxiety. 2002;15(4):183-185.
8. Ninan PT, Dunlop BW. Neurobiology and etiology of panic disorder. J Clin Psychiatry. 2005;66(suppl 4):3-7.
9. Furukawa TA, Watanabe N, Churchill R. Psychotherapy plus antidepressant for panic disorder with or without agoraphobia: systematic review. Br J Psychiatry. 2006;188:305-312.
10. Barlow DH, Gorman JM, Shear MK, et al. Cognitive-behavioral therapy, imipramine, or their combination for panic disorder: a randomized controlled trial. JAMA. 2000;283(19):2529-2536.
11. van Apeldoorn FJ, Timmerman ME, Mersch PP, et al. A randomized trial of cognitive-behavioral therapy or selective serotonin reuptake inhibitor or both combined for panic disorder with or without agoraphobia: treatment results through 1-year follow-up. J Clin Psychiatry. 2010;71(5):574-586.
12. Bakker A, van Balkom AJ, Spinhoven P. SSRIs vs. TCAs in the treatment of panic disorder: a meta-analysis. Acta Psychiatr Scand. 2002;106(3):163-167.
13. Bandelow B, Behnke K, Lenoir S, et al. Sertraline versus paroxetine in the treatment of panic disorder: an acute, double-blind noninferiority comparison. J Clin Psychiatry. 2004;65(3):405-413.
14. Nardi AE, Freire RC, Mochcovitch MD, et al. A randomized, naturalistic, parallel-group study for the long-term treatment of panic disorder with clonazepam or paroxetine. J Clin Psychopharmacol. 2012;32(1):120-126.
15. Goddard AW, Brouette T, Almai A, et al. Early coadministration of clonazepam with sertraline for panic disorder. Arch Gen Psychiatry. 2001;58(7):681-686.
16. Preskorn SH, Shah R, Neff M, et al. The potential for clinically significant drug-drug interactions involving the CYP 2D6 system: effects with fluoxetine and paroxetine versus sertraline. J Psychiatr Pract. 2007;13(1):5-12.
17. Perna G, Guerriero G, Caldirola D. Emerging drugs for panic disorder. Expert Opin Emerg Drugs. 2011;16(4):631-645.
18. Milrod B, Leon AC, Busch F, et al. A randomized controlled clinical trial of psychoanalytic psychotherapy for panic disorder. Am J Psychiatry. 2007;164(2):265-272.
19. Otto MW, Pollack MH, Sachs GS, et al. Discontinuation of benzodiazepine treatment: efficacy of cognitive-behavioral therapy for patients with panic disorder. Am J Psychiatry. 1993;150(10):1485-1490.
How to collaborate effectively with psychiatric nurse practitioners
Discuss this article at www.facebook.com/CurrentPsychiatry
Psychiatrists who are accustomed to working with “med/surg” or psychiatric nurses may be less familiar with how to collaborate with more specialized psychiatric-mental health nurse practitioners (PMHNPs). These clinicians play an important role in delivering mental health services, which is likely to continue because of the physician shortage in the United States1 and increasing mental health care needs from passage of the Affordable Health Care Act and the Mental Health Parity Act.2 These specialty trained, master’s level nurses work with psychiatrists in outpatient clinics, hospital consultation and liaison services, psychiatric emergency services, inpatient units, and geropsychiatric consultation.3-5 PMHNPs can fill gaps of coverage in underserved areas, supplement and complement busy and overburdened psychiatrists, and add an important dimension of holistic care.
This article reviews issues related to a successful psychiatrist-PMHNP collaboration, including:
- PMHNP’s training and scope of practice
- their skill and competency development in inpatient and outpatient settings
- the principles and dynamics of collaboration, hindrances to cooperation, and keys to relationship-building for PMHNPs and psychiatrists.
Rigorous requirements
PMHNPs enroll in an accredited graduate nursing program that takes 16 to 24 months to complete and builds on the competencies of their undergraduate nursing education and clinical experience. All programs meet standards set by national nursing accrediting agencies. The typical graduate-level curriculum for a PMHNP includes core bio-behavioral theory, research courses, advanced physiology and pathophysiology, advanced physical and psychiatric health assessment, pharmacologic and nonpharmacologic interventions, and managing health care delivery systems. For graduation and certification, PMHNPs must complete 500 supervised clinical hours focused on psychiatric and mental health care.
- comprehensive psychiatric evaluation
- formulation of a differential diagnosis
- ordering and interpreting diagnostic tests
- prescribing pharmacologic agents
- conducting individual, couple, group, or family psychotherapy using evidence-based approaches.
PMHNPs also are responsible for recognizing the limits of their knowledge and experience, planning for situations beyond their expertise, and providing appropriate referral to other health care providers when indicated.8
Successful collaborative practice requires a clear definition and understanding of roles.9 This is particularly important for collaborating psychiatrists and PMHNPs because there has been confusion among physicians and the general public related to the nurse practitioner’s role. Psychiatrists who work with PMHNPs need to be familiar with state regulations that govern levels of physician supervision and prescriptive authority for nurse practitioners. Eleven states and the District of Columbia allow nurse practitioners to prescribe independently, including controlled substances. Most states require physician collaboration for prescribing medications, but the language can be ambiguous, with restrictions on certain formularies or drug schedules—eg, Michigan nurse practitioners may prescribe schedule II through V controlled substances, but schedule II medications are limited to nurse practitioners who work in hospitals, surgical outpatient settings, or hospices.10
Competencies and development
New PMHNPs see patients and prescribe medication, but their work needs close supervision. Postgraduate clinical experience combined with supervision gradually allows the PMHNP greater independence. A PMHNP who provides care in a busy outpatient clinic, inpatient unit, or psychiatric emergency department is likely to master the treatment philosophy and ancillary competencies related to that particular clinical site—including favored pharmacologic approaches, electronic documentation and ordering functions, and admission and discharge facilitation—at a level exceeding that of psychiatric residents, who rotate on and off a service as part of their training.
It’s helpful for new PMHNPs to have a time frame for their development over several years. The Table11 outlines general graded competency areas PMHNPs may focus on in their development. See this article at CurrentPsychiatry.com for Tables that provide examples of detailed competencies for third-year PMHNPs in inpatient and outpatient settings.
Table
PMHNP development: General graded competency areas
| Psychiatric evaluation and diagnosis |
| Psychiatric treatments, including medications and psychotherapies |
| Maintenance of the therapeutic alliance, including monitoring the PMHNP’s emotional responses to patients |
| Participation in an interdisciplinary team |
| Understanding comorbid medical conditions, integrating laboratory and other tests into the treatment plan, and recognizing the need for consultation with the medical team |
| Documentation, such as initial evaluations, progress notes, and discharge summaries |
| Assessment for suicide and violence potential |
| Teaching |
| Patient and family psychoeducation |
| Use of feedback and supervision |
| PMHNP: psychiatric-mental health nurse practitioner Source: Reference 11 |
Table 1
Competencies for third-year PMHNPs in an outpatient clinic
| Recognize clinical presentations of complex psychiatric disorders, variants, and comorbidities |
| Firm knowledge of diagnostic criteria, and skills for independent comprehensive assessment and diagnosis |
| Firm knowledge of evidence-based outpatient treatments for disorders, with mastery of ≥1 nonpharmacologic modality in addition to prescribing and managing medications |
| Use and provide feedback in comprehensive case formulations and treatment plans |
| Assist in clinical education of trainees in psychiatric nursing, social work, psychiatric residency, and psychology |
| Participate and collaborate in educational events and initiatives |
| Knowledge of internal and external health system and resources, and facilitating patient access to these networks |
| Incorporate mental health and behavioral and psychiatric nursing research into patient care |
| PMHNP: psychiatric-mental health nurse practitioner |
Competencies for third-year PMHNPs on an inpatient psychiatric unit
| Refinement of assessment section in evaluations, progress notes, and discharge summaries |
| Understanding indications for neuropsychological testing, and integrating findings into the treatment plan |
| Assessment of readiness for discharge in patients with a history of suicidality or violence |
| Developing a sophisticated and detailed discharge or follow-up plan |
| Understanding treatment resistance in mood and psychotic disorders, and implementing treatment |
| More detailed knowledge of types of illness treated on an inpatient unit |
| Ability to orient and train PMHNPs and other inpatient unit trainees |
| Ability to gather and use articles and other literature pertaining to inpatient care |
| Increasing competence in short-term, crisis-based therapeutic techniques, including familiarity with DBT, CBT, and IPT |
| Understanding family systems and impact on patient care |
| CBT: cognitive-behavioral therapy; DBT: dialectical behavior therapy; IPT: interpersonal therapy; PMHNP: psychiatric-mental health nurse practitioner |
Principles of practice
Studies have demonstrated the importance of understanding how to effectively implement collaborative care across medical disciplines.12 See the Box12 for a discussion of 3 key determinants for successful clinical collaborations.
San Martín-Rodríguez et al12 recognized 3 key factors that may help develop successful collaborative clinical relationships.
Interactional factors include a mutual willingness to collaborate, a commitment to collaborate, a belief in the benefits of collaborating, and sharing common objectives. Trust in the partnering clinician’s competency contributes to a successful collaboration. Strong communication skills—including the ability to convey what each clinician can contribute to achieving goals—also strengthens collaboration. Learning and understanding skills in conflict management and dialogue are key. Mutual respect also is essential.
Organizational factors include a shift from a traditional hierarchical structure to a more horizontal structure, and a work climate that supports openness, risk taking—ie, a willingness to disagree with a colleague if it is in a patient’s best interest or to develop a new and innovative method of providing care—integrity, and trust. Administrative structures and supports that convey the importance of collaboration also are key components of a strong collaborative environment. Teamwork and shared decision-making are important elements; teamwork should include time to discuss patient issues and develop strong interpersonal relationships. A commitment to professional development is another key factor.
Systemic factors include a social system that supports collegial relationships and professionalism that respects and accepts other professions. This includes decreased focus on protecting professional territory and increased recognition of overlaps among professions.
Enhancing collaboration
Psychiatrists who work with PMHNPs develop trust based on observing each PMHNP’s work, including their relationship with patients, ability to conceptualize a case and develop a treatment plan, and the skill with which they function within a team. The psychiatrist’s comfort level also is related to his or her awareness of the comprehensiveness of the PMHNP’s training and the competencies gained from clinical experience. Respect for the PMHNP’s educational and professional background is the foundation for what is often—at least in the collaborative relationship’s initial stages—a combined cooperative and supervisory relationship with the PMHNP. As such, the PMHNP gradually will absorb certain “intangibles” to supplement the training and work experiences that preceded his or her position. This may include assimilating the psychiatrist’s or clinic’s philosophy and treatment practice, including expertise in dealing with specialized psychiatric populations (eg, developmental disabilities, acute psychosis, or treatment-resistant depression).
The patient’s comfort level
Collaborating PMHNPs and psychiatrists need to be prepared for a patient who expresses disappointment with being treated by a PMHNP or a preference to see “a doctor.” Psychiatrists who have not worked through their own ambivalence about the collaboration or who lack confidence in the PMHNP’s abilities may find themselves consciously or unconsciously aligning with the patient’s stance. They may neglect to explore the basis and meaning of the patient’s preference, which may be related to the patient’s lack of knowledge about the PMHNP’s role and training. The PMHNP who encounters such a patient has a more challenging task—namely, how to calmly address the patient’s concern while the patient is challenging the PMHNP’s competence. Both the PMHNP and psychiatrist need to be alert to the possibility of “splitting” in the treatment of axis II-disordered patients.
Barriers to collaboration
From the PMHNP perspective, barriers to a collaborative relationship include referring to PMHNPs by a less preferred term or title, instead of a nurse practitioner or APN, which can hinder the relationship. Although physician assistants and NPs have been grouped together under the term “mid-level providers,” the American Academy of Nurse Practitioners notes that this term suggests a lower level of care or service is being provided.18 “Physician extender” is another term that fails to recognize the PMHNP’s separate and unique role and the PMHNP’s view of their role as complementary to medicine, rather than an extension of a physician’s practice.
Territorial issues can impede collaborative relationships. Psychiatrists who resist collaborating will be less effective than those who welcome a PMHNP and readily delegate specific tasks and portions of the workload, whereas psychiatrists who value the help will be more likely to build a collaborative partnership, leading to better patient care.
Autonomy is a critical determinant of professional satisfaction for PMHNPs. A PMHNP’s autonomy can be impeded by organizational constraints and physician perceptions.19 PMHNPs require autonomy to self-direct patient diagnosis and treatment within the scope of their practice, and many find this relative independence essential to delivering high quality patient care. Lack of autonomy can lead to breaks in workflow in the outpatient setting and increased length of stay for hospitalized patients. In addition, an autonomously functioning, experienced PMHNP can increase efficiency in hospital settings where psychiatrists can be in short supply, preoccupied with administrative matters, or require help on weekends.
Related Resources
- American Psychiatric Nurses Association. www.apna.org.
- International Society of Psychiatric-Mental Health Nurses. www.ispn-psych.org.
- American Nurses Association. www.nursingworld.org.
Dr. Casher is a speaker for Sunovion Pharmaceuticals and receives royalties from Cambridge University Press.
Ms. Kuebler, Ms. Bastida, and Ms. Chipps report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Sataline S, Wang SS. Medical schools can’t keep up. Wall Street Journal. April 12 2010. http://online.wsj.com/article/SB10001424052702304506904575180331528424238.html. Accessed August 21, 2012.
2. U.S. Department of Health and Human Services. The health care law & you. http://www.healthcare.gov/law/index.html. Accessed August 21, 2012.
3. Wand T, Fisher J. The mental health nurse practitioner in the emergency department: an Australian experience. Int J Ment Health Nurs. 2006;15(3):201-208.
4. Eisch JS, Brozovic B, Colling K, et al. Nurse practitioner geropsychiatric consultation service to nursing homes. Geriatr Nurs. 2000;21(3):150-155.
5. Baker N. Exploring the mental health nurse practitioner scope of practice in youth early psychosis: an anecdotal account. Contemp Nurse. 2010;34(2):211-220.
6. International Society of Psychiatric-Mental Health Nurses. Psychiatric mental health nursing scope & standards. http://www.ispn-psych.org/docs/standards/scope-standards-draft.pdf. Updated 2006. Accessed August 21, 2012.
7. Centers for Medicare and Medicaid Services. HHS finalizes new rules to cut regulations for hospitals and health care providers saving more than $5 billion. http://www.cms.gov/apps/media/press/release.asp?Counter=4362. Published May 9, 2012. Accessed August 21, 2012.
8. APRN Consensus Work Group, National Council of State Boards of Nursing APRN Advisory Committee. Consensus model for regulation: licensure accreditation, certification & education. https://www.ncsbn.org/Consensus_Model_for_APRN_Regulation_July_2008.pdf. Published July 7, 2008. Accessed August 21, 2012.
9. Legault F, Humbert J, Amos S, et al. Difficulties encountered in collaborative care: logistics trumps desire. J Am Board Fam Med. 2012;25(2):168-176.
10. Michigan Council of Nurse Practitioners. Michigan’s rules and regulations for prescriptive authority. http://micnp.org/displaycommon.cfm?an=1&subarticlenbr=109. Accessed August 21, 2012.
11. Wheeler K, Haber J. Development of psychiatric-mental health nurse practitioner competencies: opportunities for the 21st century. J Am Psychiatr Nurses Assoc. 2004;10(3):129-138.
12. San Martín-Rodríguez L, Beaulieu MD, D’Amour D, et al. The determinants of successful collaboration: a review of theoretical and empirical studies. J Interprof Care. 2005;19(suppl 1):132-147.
13. Suter E, Arndt J, Arthur N, et al. Role understanding and effective communication as core competencies for collaborative practice. J Interprof Care. 2009;23(1):41-51.
14. Horrocks S, Anderson E, Salisbury C. Systematic review of whether nurse practitioners working in primary care can provide equivalent care to doctors. BMJ. 2002;324(7341):819-823.
15. Byrne G, Richardson M, Brunsdon J, et al. Patient satisfaction with emergency nurse practitioners in A & E. J Clin Nurs. 2000;9(1):83-92.
16. McCann TV, Clark E. Attitudes of patients towards mental health nurse prescribing of antipsychotic agents. Int J Nurs Pract. 2008;14(2):115-121.
17. Wortans J, Happell B, Johnstone H. The role of the nurse practitioner in psychiatric/mental health nursing: exploring consumer satisfaction. J Psychiatr Ment Health Nurs. 2006;13(1):78-84.
18. Frellick M. The nurse practitioner will see you now. Advanced practice providers fill the physician gap. Hosp Health Netw. 2011;85(7):44-46, 48–49.
19. Maylone MM, Ranieri L, Quinn Griffin MT, et al. Collaboration and autonomy: perceptions among nurse practitioners. J Am Acad Nurse Pract. 2011;23(1):51-57.
Discuss this article at www.facebook.com/CurrentPsychiatry
Psychiatrists who are accustomed to working with “med/surg” or psychiatric nurses may be less familiar with how to collaborate with more specialized psychiatric-mental health nurse practitioners (PMHNPs). These clinicians play an important role in delivering mental health services, which is likely to continue because of the physician shortage in the United States1 and increasing mental health care needs from passage of the Affordable Health Care Act and the Mental Health Parity Act.2 These specialty trained, master’s level nurses work with psychiatrists in outpatient clinics, hospital consultation and liaison services, psychiatric emergency services, inpatient units, and geropsychiatric consultation.3-5 PMHNPs can fill gaps of coverage in underserved areas, supplement and complement busy and overburdened psychiatrists, and add an important dimension of holistic care.
This article reviews issues related to a successful psychiatrist-PMHNP collaboration, including:
- PMHNP’s training and scope of practice
- their skill and competency development in inpatient and outpatient settings
- the principles and dynamics of collaboration, hindrances to cooperation, and keys to relationship-building for PMHNPs and psychiatrists.
Rigorous requirements
PMHNPs enroll in an accredited graduate nursing program that takes 16 to 24 months to complete and builds on the competencies of their undergraduate nursing education and clinical experience. All programs meet standards set by national nursing accrediting agencies. The typical graduate-level curriculum for a PMHNP includes core bio-behavioral theory, research courses, advanced physiology and pathophysiology, advanced physical and psychiatric health assessment, pharmacologic and nonpharmacologic interventions, and managing health care delivery systems. For graduation and certification, PMHNPs must complete 500 supervised clinical hours focused on psychiatric and mental health care.
- comprehensive psychiatric evaluation
- formulation of a differential diagnosis
- ordering and interpreting diagnostic tests
- prescribing pharmacologic agents
- conducting individual, couple, group, or family psychotherapy using evidence-based approaches.
PMHNPs also are responsible for recognizing the limits of their knowledge and experience, planning for situations beyond their expertise, and providing appropriate referral to other health care providers when indicated.8
Successful collaborative practice requires a clear definition and understanding of roles.9 This is particularly important for collaborating psychiatrists and PMHNPs because there has been confusion among physicians and the general public related to the nurse practitioner’s role. Psychiatrists who work with PMHNPs need to be familiar with state regulations that govern levels of physician supervision and prescriptive authority for nurse practitioners. Eleven states and the District of Columbia allow nurse practitioners to prescribe independently, including controlled substances. Most states require physician collaboration for prescribing medications, but the language can be ambiguous, with restrictions on certain formularies or drug schedules—eg, Michigan nurse practitioners may prescribe schedule II through V controlled substances, but schedule II medications are limited to nurse practitioners who work in hospitals, surgical outpatient settings, or hospices.10
Competencies and development
New PMHNPs see patients and prescribe medication, but their work needs close supervision. Postgraduate clinical experience combined with supervision gradually allows the PMHNP greater independence. A PMHNP who provides care in a busy outpatient clinic, inpatient unit, or psychiatric emergency department is likely to master the treatment philosophy and ancillary competencies related to that particular clinical site—including favored pharmacologic approaches, electronic documentation and ordering functions, and admission and discharge facilitation—at a level exceeding that of psychiatric residents, who rotate on and off a service as part of their training.
It’s helpful for new PMHNPs to have a time frame for their development over several years. The Table11 outlines general graded competency areas PMHNPs may focus on in their development. See this article at CurrentPsychiatry.com for Tables that provide examples of detailed competencies for third-year PMHNPs in inpatient and outpatient settings.
Table
PMHNP development: General graded competency areas
| Psychiatric evaluation and diagnosis |
| Psychiatric treatments, including medications and psychotherapies |
| Maintenance of the therapeutic alliance, including monitoring the PMHNP’s emotional responses to patients |
| Participation in an interdisciplinary team |
| Understanding comorbid medical conditions, integrating laboratory and other tests into the treatment plan, and recognizing the need for consultation with the medical team |
| Documentation, such as initial evaluations, progress notes, and discharge summaries |
| Assessment for suicide and violence potential |
| Teaching |
| Patient and family psychoeducation |
| Use of feedback and supervision |
| PMHNP: psychiatric-mental health nurse practitioner Source: Reference 11 |
Table 1
Competencies for third-year PMHNPs in an outpatient clinic
| Recognize clinical presentations of complex psychiatric disorders, variants, and comorbidities |
| Firm knowledge of diagnostic criteria, and skills for independent comprehensive assessment and diagnosis |
| Firm knowledge of evidence-based outpatient treatments for disorders, with mastery of ≥1 nonpharmacologic modality in addition to prescribing and managing medications |
| Use and provide feedback in comprehensive case formulations and treatment plans |
| Assist in clinical education of trainees in psychiatric nursing, social work, psychiatric residency, and psychology |
| Participate and collaborate in educational events and initiatives |
| Knowledge of internal and external health system and resources, and facilitating patient access to these networks |
| Incorporate mental health and behavioral and psychiatric nursing research into patient care |
| PMHNP: psychiatric-mental health nurse practitioner |
Competencies for third-year PMHNPs on an inpatient psychiatric unit
| Refinement of assessment section in evaluations, progress notes, and discharge summaries |
| Understanding indications for neuropsychological testing, and integrating findings into the treatment plan |
| Assessment of readiness for discharge in patients with a history of suicidality or violence |
| Developing a sophisticated and detailed discharge or follow-up plan |
| Understanding treatment resistance in mood and psychotic disorders, and implementing treatment |
| More detailed knowledge of types of illness treated on an inpatient unit |
| Ability to orient and train PMHNPs and other inpatient unit trainees |
| Ability to gather and use articles and other literature pertaining to inpatient care |
| Increasing competence in short-term, crisis-based therapeutic techniques, including familiarity with DBT, CBT, and IPT |
| Understanding family systems and impact on patient care |
| CBT: cognitive-behavioral therapy; DBT: dialectical behavior therapy; IPT: interpersonal therapy; PMHNP: psychiatric-mental health nurse practitioner |
Principles of practice
Studies have demonstrated the importance of understanding how to effectively implement collaborative care across medical disciplines.12 See the Box12 for a discussion of 3 key determinants for successful clinical collaborations.
San Martín-Rodríguez et al12 recognized 3 key factors that may help develop successful collaborative clinical relationships.
Interactional factors include a mutual willingness to collaborate, a commitment to collaborate, a belief in the benefits of collaborating, and sharing common objectives. Trust in the partnering clinician’s competency contributes to a successful collaboration. Strong communication skills—including the ability to convey what each clinician can contribute to achieving goals—also strengthens collaboration. Learning and understanding skills in conflict management and dialogue are key. Mutual respect also is essential.
Organizational factors include a shift from a traditional hierarchical structure to a more horizontal structure, and a work climate that supports openness, risk taking—ie, a willingness to disagree with a colleague if it is in a patient’s best interest or to develop a new and innovative method of providing care—integrity, and trust. Administrative structures and supports that convey the importance of collaboration also are key components of a strong collaborative environment. Teamwork and shared decision-making are important elements; teamwork should include time to discuss patient issues and develop strong interpersonal relationships. A commitment to professional development is another key factor.
Systemic factors include a social system that supports collegial relationships and professionalism that respects and accepts other professions. This includes decreased focus on protecting professional territory and increased recognition of overlaps among professions.
Enhancing collaboration
Psychiatrists who work with PMHNPs develop trust based on observing each PMHNP’s work, including their relationship with patients, ability to conceptualize a case and develop a treatment plan, and the skill with which they function within a team. The psychiatrist’s comfort level also is related to his or her awareness of the comprehensiveness of the PMHNP’s training and the competencies gained from clinical experience. Respect for the PMHNP’s educational and professional background is the foundation for what is often—at least in the collaborative relationship’s initial stages—a combined cooperative and supervisory relationship with the PMHNP. As such, the PMHNP gradually will absorb certain “intangibles” to supplement the training and work experiences that preceded his or her position. This may include assimilating the psychiatrist’s or clinic’s philosophy and treatment practice, including expertise in dealing with specialized psychiatric populations (eg, developmental disabilities, acute psychosis, or treatment-resistant depression).
The patient’s comfort level
Collaborating PMHNPs and psychiatrists need to be prepared for a patient who expresses disappointment with being treated by a PMHNP or a preference to see “a doctor.” Psychiatrists who have not worked through their own ambivalence about the collaboration or who lack confidence in the PMHNP’s abilities may find themselves consciously or unconsciously aligning with the patient’s stance. They may neglect to explore the basis and meaning of the patient’s preference, which may be related to the patient’s lack of knowledge about the PMHNP’s role and training. The PMHNP who encounters such a patient has a more challenging task—namely, how to calmly address the patient’s concern while the patient is challenging the PMHNP’s competence. Both the PMHNP and psychiatrist need to be alert to the possibility of “splitting” in the treatment of axis II-disordered patients.
Barriers to collaboration
From the PMHNP perspective, barriers to a collaborative relationship include referring to PMHNPs by a less preferred term or title, instead of a nurse practitioner or APN, which can hinder the relationship. Although physician assistants and NPs have been grouped together under the term “mid-level providers,” the American Academy of Nurse Practitioners notes that this term suggests a lower level of care or service is being provided.18 “Physician extender” is another term that fails to recognize the PMHNP’s separate and unique role and the PMHNP’s view of their role as complementary to medicine, rather than an extension of a physician’s practice.
Territorial issues can impede collaborative relationships. Psychiatrists who resist collaborating will be less effective than those who welcome a PMHNP and readily delegate specific tasks and portions of the workload, whereas psychiatrists who value the help will be more likely to build a collaborative partnership, leading to better patient care.
Autonomy is a critical determinant of professional satisfaction for PMHNPs. A PMHNP’s autonomy can be impeded by organizational constraints and physician perceptions.19 PMHNPs require autonomy to self-direct patient diagnosis and treatment within the scope of their practice, and many find this relative independence essential to delivering high quality patient care. Lack of autonomy can lead to breaks in workflow in the outpatient setting and increased length of stay for hospitalized patients. In addition, an autonomously functioning, experienced PMHNP can increase efficiency in hospital settings where psychiatrists can be in short supply, preoccupied with administrative matters, or require help on weekends.
Related Resources
- American Psychiatric Nurses Association. www.apna.org.
- International Society of Psychiatric-Mental Health Nurses. www.ispn-psych.org.
- American Nurses Association. www.nursingworld.org.
Dr. Casher is a speaker for Sunovion Pharmaceuticals and receives royalties from Cambridge University Press.
Ms. Kuebler, Ms. Bastida, and Ms. Chipps report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Psychiatrists who are accustomed to working with “med/surg” or psychiatric nurses may be less familiar with how to collaborate with more specialized psychiatric-mental health nurse practitioners (PMHNPs). These clinicians play an important role in delivering mental health services, which is likely to continue because of the physician shortage in the United States1 and increasing mental health care needs from passage of the Affordable Health Care Act and the Mental Health Parity Act.2 These specialty trained, master’s level nurses work with psychiatrists in outpatient clinics, hospital consultation and liaison services, psychiatric emergency services, inpatient units, and geropsychiatric consultation.3-5 PMHNPs can fill gaps of coverage in underserved areas, supplement and complement busy and overburdened psychiatrists, and add an important dimension of holistic care.
This article reviews issues related to a successful psychiatrist-PMHNP collaboration, including:
- PMHNP’s training and scope of practice
- their skill and competency development in inpatient and outpatient settings
- the principles and dynamics of collaboration, hindrances to cooperation, and keys to relationship-building for PMHNPs and psychiatrists.
Rigorous requirements
PMHNPs enroll in an accredited graduate nursing program that takes 16 to 24 months to complete and builds on the competencies of their undergraduate nursing education and clinical experience. All programs meet standards set by national nursing accrediting agencies. The typical graduate-level curriculum for a PMHNP includes core bio-behavioral theory, research courses, advanced physiology and pathophysiology, advanced physical and psychiatric health assessment, pharmacologic and nonpharmacologic interventions, and managing health care delivery systems. For graduation and certification, PMHNPs must complete 500 supervised clinical hours focused on psychiatric and mental health care.
- comprehensive psychiatric evaluation
- formulation of a differential diagnosis
- ordering and interpreting diagnostic tests
- prescribing pharmacologic agents
- conducting individual, couple, group, or family psychotherapy using evidence-based approaches.
PMHNPs also are responsible for recognizing the limits of their knowledge and experience, planning for situations beyond their expertise, and providing appropriate referral to other health care providers when indicated.8
Successful collaborative practice requires a clear definition and understanding of roles.9 This is particularly important for collaborating psychiatrists and PMHNPs because there has been confusion among physicians and the general public related to the nurse practitioner’s role. Psychiatrists who work with PMHNPs need to be familiar with state regulations that govern levels of physician supervision and prescriptive authority for nurse practitioners. Eleven states and the District of Columbia allow nurse practitioners to prescribe independently, including controlled substances. Most states require physician collaboration for prescribing medications, but the language can be ambiguous, with restrictions on certain formularies or drug schedules—eg, Michigan nurse practitioners may prescribe schedule II through V controlled substances, but schedule II medications are limited to nurse practitioners who work in hospitals, surgical outpatient settings, or hospices.10
Competencies and development
New PMHNPs see patients and prescribe medication, but their work needs close supervision. Postgraduate clinical experience combined with supervision gradually allows the PMHNP greater independence. A PMHNP who provides care in a busy outpatient clinic, inpatient unit, or psychiatric emergency department is likely to master the treatment philosophy and ancillary competencies related to that particular clinical site—including favored pharmacologic approaches, electronic documentation and ordering functions, and admission and discharge facilitation—at a level exceeding that of psychiatric residents, who rotate on and off a service as part of their training.
It’s helpful for new PMHNPs to have a time frame for their development over several years. The Table11 outlines general graded competency areas PMHNPs may focus on in their development. See this article at CurrentPsychiatry.com for Tables that provide examples of detailed competencies for third-year PMHNPs in inpatient and outpatient settings.
Table
PMHNP development: General graded competency areas
| Psychiatric evaluation and diagnosis |
| Psychiatric treatments, including medications and psychotherapies |
| Maintenance of the therapeutic alliance, including monitoring the PMHNP’s emotional responses to patients |
| Participation in an interdisciplinary team |
| Understanding comorbid medical conditions, integrating laboratory and other tests into the treatment plan, and recognizing the need for consultation with the medical team |
| Documentation, such as initial evaluations, progress notes, and discharge summaries |
| Assessment for suicide and violence potential |
| Teaching |
| Patient and family psychoeducation |
| Use of feedback and supervision |
| PMHNP: psychiatric-mental health nurse practitioner Source: Reference 11 |
Table 1
Competencies for third-year PMHNPs in an outpatient clinic
| Recognize clinical presentations of complex psychiatric disorders, variants, and comorbidities |
| Firm knowledge of diagnostic criteria, and skills for independent comprehensive assessment and diagnosis |
| Firm knowledge of evidence-based outpatient treatments for disorders, with mastery of ≥1 nonpharmacologic modality in addition to prescribing and managing medications |
| Use and provide feedback in comprehensive case formulations and treatment plans |
| Assist in clinical education of trainees in psychiatric nursing, social work, psychiatric residency, and psychology |
| Participate and collaborate in educational events and initiatives |
| Knowledge of internal and external health system and resources, and facilitating patient access to these networks |
| Incorporate mental health and behavioral and psychiatric nursing research into patient care |
| PMHNP: psychiatric-mental health nurse practitioner |
Competencies for third-year PMHNPs on an inpatient psychiatric unit
| Refinement of assessment section in evaluations, progress notes, and discharge summaries |
| Understanding indications for neuropsychological testing, and integrating findings into the treatment plan |
| Assessment of readiness for discharge in patients with a history of suicidality or violence |
| Developing a sophisticated and detailed discharge or follow-up plan |
| Understanding treatment resistance in mood and psychotic disorders, and implementing treatment |
| More detailed knowledge of types of illness treated on an inpatient unit |
| Ability to orient and train PMHNPs and other inpatient unit trainees |
| Ability to gather and use articles and other literature pertaining to inpatient care |
| Increasing competence in short-term, crisis-based therapeutic techniques, including familiarity with DBT, CBT, and IPT |
| Understanding family systems and impact on patient care |
| CBT: cognitive-behavioral therapy; DBT: dialectical behavior therapy; IPT: interpersonal therapy; PMHNP: psychiatric-mental health nurse practitioner |
Principles of practice
Studies have demonstrated the importance of understanding how to effectively implement collaborative care across medical disciplines.12 See the Box12 for a discussion of 3 key determinants for successful clinical collaborations.
San Martín-Rodríguez et al12 recognized 3 key factors that may help develop successful collaborative clinical relationships.
Interactional factors include a mutual willingness to collaborate, a commitment to collaborate, a belief in the benefits of collaborating, and sharing common objectives. Trust in the partnering clinician’s competency contributes to a successful collaboration. Strong communication skills—including the ability to convey what each clinician can contribute to achieving goals—also strengthens collaboration. Learning and understanding skills in conflict management and dialogue are key. Mutual respect also is essential.
Organizational factors include a shift from a traditional hierarchical structure to a more horizontal structure, and a work climate that supports openness, risk taking—ie, a willingness to disagree with a colleague if it is in a patient’s best interest or to develop a new and innovative method of providing care—integrity, and trust. Administrative structures and supports that convey the importance of collaboration also are key components of a strong collaborative environment. Teamwork and shared decision-making are important elements; teamwork should include time to discuss patient issues and develop strong interpersonal relationships. A commitment to professional development is another key factor.
Systemic factors include a social system that supports collegial relationships and professionalism that respects and accepts other professions. This includes decreased focus on protecting professional territory and increased recognition of overlaps among professions.
Enhancing collaboration
Psychiatrists who work with PMHNPs develop trust based on observing each PMHNP’s work, including their relationship with patients, ability to conceptualize a case and develop a treatment plan, and the skill with which they function within a team. The psychiatrist’s comfort level also is related to his or her awareness of the comprehensiveness of the PMHNP’s training and the competencies gained from clinical experience. Respect for the PMHNP’s educational and professional background is the foundation for what is often—at least in the collaborative relationship’s initial stages—a combined cooperative and supervisory relationship with the PMHNP. As such, the PMHNP gradually will absorb certain “intangibles” to supplement the training and work experiences that preceded his or her position. This may include assimilating the psychiatrist’s or clinic’s philosophy and treatment practice, including expertise in dealing with specialized psychiatric populations (eg, developmental disabilities, acute psychosis, or treatment-resistant depression).
The patient’s comfort level
Collaborating PMHNPs and psychiatrists need to be prepared for a patient who expresses disappointment with being treated by a PMHNP or a preference to see “a doctor.” Psychiatrists who have not worked through their own ambivalence about the collaboration or who lack confidence in the PMHNP’s abilities may find themselves consciously or unconsciously aligning with the patient’s stance. They may neglect to explore the basis and meaning of the patient’s preference, which may be related to the patient’s lack of knowledge about the PMHNP’s role and training. The PMHNP who encounters such a patient has a more challenging task—namely, how to calmly address the patient’s concern while the patient is challenging the PMHNP’s competence. Both the PMHNP and psychiatrist need to be alert to the possibility of “splitting” in the treatment of axis II-disordered patients.
Barriers to collaboration
From the PMHNP perspective, barriers to a collaborative relationship include referring to PMHNPs by a less preferred term or title, instead of a nurse practitioner or APN, which can hinder the relationship. Although physician assistants and NPs have been grouped together under the term “mid-level providers,” the American Academy of Nurse Practitioners notes that this term suggests a lower level of care or service is being provided.18 “Physician extender” is another term that fails to recognize the PMHNP’s separate and unique role and the PMHNP’s view of their role as complementary to medicine, rather than an extension of a physician’s practice.
Territorial issues can impede collaborative relationships. Psychiatrists who resist collaborating will be less effective than those who welcome a PMHNP and readily delegate specific tasks and portions of the workload, whereas psychiatrists who value the help will be more likely to build a collaborative partnership, leading to better patient care.
Autonomy is a critical determinant of professional satisfaction for PMHNPs. A PMHNP’s autonomy can be impeded by organizational constraints and physician perceptions.19 PMHNPs require autonomy to self-direct patient diagnosis and treatment within the scope of their practice, and many find this relative independence essential to delivering high quality patient care. Lack of autonomy can lead to breaks in workflow in the outpatient setting and increased length of stay for hospitalized patients. In addition, an autonomously functioning, experienced PMHNP can increase efficiency in hospital settings where psychiatrists can be in short supply, preoccupied with administrative matters, or require help on weekends.
Related Resources
- American Psychiatric Nurses Association. www.apna.org.
- International Society of Psychiatric-Mental Health Nurses. www.ispn-psych.org.
- American Nurses Association. www.nursingworld.org.
Dr. Casher is a speaker for Sunovion Pharmaceuticals and receives royalties from Cambridge University Press.
Ms. Kuebler, Ms. Bastida, and Ms. Chipps report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Sataline S, Wang SS. Medical schools can’t keep up. Wall Street Journal. April 12 2010. http://online.wsj.com/article/SB10001424052702304506904575180331528424238.html. Accessed August 21, 2012.
2. U.S. Department of Health and Human Services. The health care law & you. http://www.healthcare.gov/law/index.html. Accessed August 21, 2012.
3. Wand T, Fisher J. The mental health nurse practitioner in the emergency department: an Australian experience. Int J Ment Health Nurs. 2006;15(3):201-208.
4. Eisch JS, Brozovic B, Colling K, et al. Nurse practitioner geropsychiatric consultation service to nursing homes. Geriatr Nurs. 2000;21(3):150-155.
5. Baker N. Exploring the mental health nurse practitioner scope of practice in youth early psychosis: an anecdotal account. Contemp Nurse. 2010;34(2):211-220.
6. International Society of Psychiatric-Mental Health Nurses. Psychiatric mental health nursing scope & standards. http://www.ispn-psych.org/docs/standards/scope-standards-draft.pdf. Updated 2006. Accessed August 21, 2012.
7. Centers for Medicare and Medicaid Services. HHS finalizes new rules to cut regulations for hospitals and health care providers saving more than $5 billion. http://www.cms.gov/apps/media/press/release.asp?Counter=4362. Published May 9, 2012. Accessed August 21, 2012.
8. APRN Consensus Work Group, National Council of State Boards of Nursing APRN Advisory Committee. Consensus model for regulation: licensure accreditation, certification & education. https://www.ncsbn.org/Consensus_Model_for_APRN_Regulation_July_2008.pdf. Published July 7, 2008. Accessed August 21, 2012.
9. Legault F, Humbert J, Amos S, et al. Difficulties encountered in collaborative care: logistics trumps desire. J Am Board Fam Med. 2012;25(2):168-176.
10. Michigan Council of Nurse Practitioners. Michigan’s rules and regulations for prescriptive authority. http://micnp.org/displaycommon.cfm?an=1&subarticlenbr=109. Accessed August 21, 2012.
11. Wheeler K, Haber J. Development of psychiatric-mental health nurse practitioner competencies: opportunities for the 21st century. J Am Psychiatr Nurses Assoc. 2004;10(3):129-138.
12. San Martín-Rodríguez L, Beaulieu MD, D’Amour D, et al. The determinants of successful collaboration: a review of theoretical and empirical studies. J Interprof Care. 2005;19(suppl 1):132-147.
13. Suter E, Arndt J, Arthur N, et al. Role understanding and effective communication as core competencies for collaborative practice. J Interprof Care. 2009;23(1):41-51.
14. Horrocks S, Anderson E, Salisbury C. Systematic review of whether nurse practitioners working in primary care can provide equivalent care to doctors. BMJ. 2002;324(7341):819-823.
15. Byrne G, Richardson M, Brunsdon J, et al. Patient satisfaction with emergency nurse practitioners in A & E. J Clin Nurs. 2000;9(1):83-92.
16. McCann TV, Clark E. Attitudes of patients towards mental health nurse prescribing of antipsychotic agents. Int J Nurs Pract. 2008;14(2):115-121.
17. Wortans J, Happell B, Johnstone H. The role of the nurse practitioner in psychiatric/mental health nursing: exploring consumer satisfaction. J Psychiatr Ment Health Nurs. 2006;13(1):78-84.
18. Frellick M. The nurse practitioner will see you now. Advanced practice providers fill the physician gap. Hosp Health Netw. 2011;85(7):44-46, 48–49.
19. Maylone MM, Ranieri L, Quinn Griffin MT, et al. Collaboration and autonomy: perceptions among nurse practitioners. J Am Acad Nurse Pract. 2011;23(1):51-57.
1. Sataline S, Wang SS. Medical schools can’t keep up. Wall Street Journal. April 12 2010. http://online.wsj.com/article/SB10001424052702304506904575180331528424238.html. Accessed August 21, 2012.
2. U.S. Department of Health and Human Services. The health care law & you. http://www.healthcare.gov/law/index.html. Accessed August 21, 2012.
3. Wand T, Fisher J. The mental health nurse practitioner in the emergency department: an Australian experience. Int J Ment Health Nurs. 2006;15(3):201-208.
4. Eisch JS, Brozovic B, Colling K, et al. Nurse practitioner geropsychiatric consultation service to nursing homes. Geriatr Nurs. 2000;21(3):150-155.
5. Baker N. Exploring the mental health nurse practitioner scope of practice in youth early psychosis: an anecdotal account. Contemp Nurse. 2010;34(2):211-220.
6. International Society of Psychiatric-Mental Health Nurses. Psychiatric mental health nursing scope & standards. http://www.ispn-psych.org/docs/standards/scope-standards-draft.pdf. Updated 2006. Accessed August 21, 2012.
7. Centers for Medicare and Medicaid Services. HHS finalizes new rules to cut regulations for hospitals and health care providers saving more than $5 billion. http://www.cms.gov/apps/media/press/release.asp?Counter=4362. Published May 9, 2012. Accessed August 21, 2012.
8. APRN Consensus Work Group, National Council of State Boards of Nursing APRN Advisory Committee. Consensus model for regulation: licensure accreditation, certification & education. https://www.ncsbn.org/Consensus_Model_for_APRN_Regulation_July_2008.pdf. Published July 7, 2008. Accessed August 21, 2012.
9. Legault F, Humbert J, Amos S, et al. Difficulties encountered in collaborative care: logistics trumps desire. J Am Board Fam Med. 2012;25(2):168-176.
10. Michigan Council of Nurse Practitioners. Michigan’s rules and regulations for prescriptive authority. http://micnp.org/displaycommon.cfm?an=1&subarticlenbr=109. Accessed August 21, 2012.
11. Wheeler K, Haber J. Development of psychiatric-mental health nurse practitioner competencies: opportunities for the 21st century. J Am Psychiatr Nurses Assoc. 2004;10(3):129-138.
12. San Martín-Rodríguez L, Beaulieu MD, D’Amour D, et al. The determinants of successful collaboration: a review of theoretical and empirical studies. J Interprof Care. 2005;19(suppl 1):132-147.
13. Suter E, Arndt J, Arthur N, et al. Role understanding and effective communication as core competencies for collaborative practice. J Interprof Care. 2009;23(1):41-51.
14. Horrocks S, Anderson E, Salisbury C. Systematic review of whether nurse practitioners working in primary care can provide equivalent care to doctors. BMJ. 2002;324(7341):819-823.
15. Byrne G, Richardson M, Brunsdon J, et al. Patient satisfaction with emergency nurse practitioners in A & E. J Clin Nurs. 2000;9(1):83-92.
16. McCann TV, Clark E. Attitudes of patients towards mental health nurse prescribing of antipsychotic agents. Int J Nurs Pract. 2008;14(2):115-121.
17. Wortans J, Happell B, Johnstone H. The role of the nurse practitioner in psychiatric/mental health nursing: exploring consumer satisfaction. J Psychiatr Ment Health Nurs. 2006;13(1):78-84.
18. Frellick M. The nurse practitioner will see you now. Advanced practice providers fill the physician gap. Hosp Health Netw. 2011;85(7):44-46, 48–49.
19. Maylone MM, Ranieri L, Quinn Griffin MT, et al. Collaboration and autonomy: perceptions among nurse practitioners. J Am Acad Nurse Pract. 2011;23(1):51-57.
Differentiating Alzheimer’s disease from dementia with Lewy bodies
Discuss this article at www.facebook.com/CurrentPsychiatry
Alzheimer’s disease (AD) and dementia with Lewy bodies (DLB) are the first and second most common causes of neurodegenerative dementia, respectively.“New Alzheimer’s disease guidelines: Implications for clinicians,” Current Psychiatry, March 2012, p. 15-20; http://bit.ly/UNYikk.
The 2005 report of the DLB Consortium5 recognizes central, core, suggestive, and supportive features of DLB (Table 1).5,10 These features are considered in the context of other confounding clinical conditions and the timing of cognitive and motor symptoms. The revised DLB criteria5 require a central feature of progressive cognitive decline. “Probable DLB” is when a patient presents with 2 core features or 1 core feature and ≥1 suggestive features. A diagnosis of “possible DLB” requires 1 core feature or 1 suggestive feature in the presence of progressive cognitive decline.
Table 1
Diagnostic criteria for AD and DLB
| NIA-AA criteria for AD (2011)10 |
| Possible AD: Clinical and cognitive criteria (DSM-IV-TR) for AD are met and there is an absence of biomarkers to support the diagnosis or there is evidence of a secondary disorder that can cause dementia |
| Probable AD: Clinical and cognitive criteria for AD are met and there is documented progressive cognitive decline or abnormal biomarker(s) suggestive of AD or evidence of proven AD autosomal dominant genetic mutation (presenilin-1, presenilin-2, amyloid-β precursor protein) |
| Definite AD: Clinical criteria for probable AD are met and there is histopathologic evidence of the disorder |
| Revised clinical diagnostic criteria for DLB (2005)5 |
| Core features: Fluctuating cognition, recurrent visual hallucinations, soft motor features of parkinsonism |
| Suggestive features: REM sleep behavior disorder, severe antipsychotic sensitivity, decreased tracer uptake in striatum on SPECT dopamine transporter imaging or on myocardial scintigraphy with MIBG |
| Supportive features (common but lacking diagnostic specificity): repeated falls and syncope; transient, unexplained loss of consciousness; systematized delusions; hallucinations other than visual; relative preservation of medial temporal lobe on CT or MRI scan; decreased tracer uptake on SPECT or PET imaging in occipital regions; prominent slow waves on EEG with temporal lobe transient sharp waves |
| AD: Alzheimer’s disease; DLB: dementia with Lewy bodies; MIBG: metaiodobenzylguanidine; NIA-AA: National Institute on Aging and the Alzheimer’s Association; PET: positron emission tomography; REM: rapid eye movement; SPECT: single photon emission computed tomography |
Biomarkers for AD, but not DLB
The 2011 diagnostic criteria for AD incorporate biomarkers that can be measured in vivo and reflect speci?c features of disease-related pathophysiologic processes. Biomarkers for AD are divided into 2 categories:11
- amyloid-beta (Aβ) accumulation: abnormal tracer retention on amyloid positron emission topography (PET) imaging and low cerebrospinal fluid (CSF) Aβ42
- neuronal degeneration or injury: elevated CSF tau (total and phosphorylated tau), decreased ?uorodeoxyglucose uptake on PET in temporo-parietal cortices, and atrophy on structural MRI in the hippocampal and temporo-parietal regions.
No clinically applicable genotypic or CSF markers exist to support a DLB diagnosis, but there are many promising candidates, including elevated levels of CSF p-tau 181, CSF levels of alpha- and beta-synuclein,12 and CSF beta-glucocerebrosidase levels.13 PET mapping of brain acetylcholinesterase activity,14 123I-2β-carbomethoxy-3β- (4-iodophenyl)-N-(3-fluoropropyl)nortropane single photon emission computed tomography (SPECT) dopamine transporter (DaT) imaging15 and metaiodobenzylguanidine (MIBG) scintigraphy also are promising methods. DaT scan SPECT is FDA-approved for detecting loss of functional dopaminergic neuron terminals in the striatum and can differentiate between AD and DLB with a sensitivity and specificity of 78% to 88% and 94% to 100%, respectively.16 This test is covered by Medicare for differentiating AD and DLB.
Differences in presentation
Cognitive impairment. Contrary to the early memory impairment that characterizes AD, memory deficits in DLB usually appear later in the disease course.5 Patients with DLB manifest greater attentional, visuospatial, and executive impairments than those with AD, whereas AD causes more profound episodic (declarative) memory impairment than DLB. DLB patients show more preserved consolidation and storage of verbal information than AD patients because of less neuroanatomical and cholinergic compromise in the medial temporal lobe. There is no evidence of significant differences in remote memory, semantic memory, and language (naming and fluency).
Compromised attention in DLB may be the basis for fluctuating cognition, a characteristic of the disease. The greater attentional impairment and reaction time variability in DLB compared with AD is evident during complex tasks for attention and may be a function of the executive and visuospatial demands of the tasks.17
Executive functions critical to adaptive, goal-directed behavior are more impaired in DLB than AD. DLB patients are more susceptible to distraction and have difficulty engaging in a task and shifting from 1 task to another. This, together with a tendency for confabulation and perseveration, are signs of executive dysfunction.
Neuropsychiatric features. DLB patients are more likely than AD patients to exhibit psychiatric symptoms and have more functional impairment.18 In an analysis of autopsy-confirmed cases, hallucinations and delusions were more frequent with Lewy body pathology (75%) than AD (21%) at initial clinical evaluation.18 By the end stages of both illnesses, the degree of psychotic symptoms is comparable.19 Depression is common in DLB; whether base rates of depressed mood and major depression differ between DLB and AD is uncertain.20
Psychosis in AD can be induced by medication or delirium, or triggered by poor sensory perceptions. Psychotic symptoms occur more frequently during the moderate and advanced stages of AD, when patients present with visual hallucinations, delusions, or delusional misidentifications. As many as 10% to 20% of patients with AD experience hallucinations, typically visual. Delusions occur in 30% to 50% of AD patients, usually in the later stages of the disease. The most common delusional themes are infidelity, theft, and paranoia. Female sex is a risk factor for psychosis in AD. Delusions co-occur with aggression, anxiety, and aberrant motor behavior.
Visual hallucinations—mostly vivid, well-formed, false perceptions of insects, animals, or people—are the defining feature of DLB.21 Many patients recognize that they are experiencing visual hallucinations and can ignore them. DLB patients also may experience visual illusions, such as misperceiving household objects as living beings. Delusions—typically paranoid—are common among DLB patients, as are depression and anxiety.1 Agitation or aggressive behavior tends to occur late in the illness, if at all.
The causes of psychotic symptoms in DLB are not fully understood, but dopamine dysfunction likely is involved in hallucinations, delusions, and agitation, and serotonin dysfunction may be associated with depression and anxiety. Rapid eye movement (REM) sleep/wakefulness dysregulation, in which the dream imagery of REM sleep may occur during wakefulness, also has been proposed as a mechanism for visual hallucinations in DLB.22 In DLB, psychotic symptoms occur early and are a hallmark of this illness, whereas in AD they usually occur in the middle to late stages of the disease.
Motor symptoms. In AD, extrapyramidal symptoms (EPS) are common later in the disease, are strongly correlated with disease severity, and are a strong, independent predictor of depression severity.23 EPS are more common in DLB than in AD24 and DLB patients are at higher risk of developing EPS even with low doses of typical antipsychotics, compared with AD patients.25
Other symptoms. REM sleep behavior disorder (RBD) is characterized by enacting dreams—often violent—during REM sleep. RBD is common in DLB and many patients also have excessive daytime somnolence. Other sleep disorders in DLB include insomnia, obstructive sleep apnea, central sleep apnea, restless legs syndrome, and periodic limb movements during sleep.
In AD patients, common sleep behaviors include confusion in the early evening (“sundowning”) and frequent nighttime awakenings, often accompanied by wandering.26 Orthostatic hypotension, impotence, urinary incontinence, and constipation are common in DLB. Lack of insight concerning personal cognitive, mood, and behavioral state is highly prevalent in AD patients and more common than in DLB.
Diagnostic evaluation
Because there are no definitive clinical markers for DLB, diagnosis is based on a detailed clinical and family history from the patient and a reliable informant, as well as a physical, neurologic, and mental status examination that looks for associated noncognitive symptoms, and neuropsychological evaluation. Reasons DLB may be misdiagnosed include:
- Some “core” clinical features of DLB may not appear or may overlap with AD.
- Presence and severity of concurrent AD pathology in DLB may modify the clinical presentation, with decreased rates of hallucinations and parkinsonism and increased neurofibrillary tangles.
- Failure to reliably identify fluctuations—variations in cognition and arousal, such as periods of unresponsiveness while awake (“zoning out”), excessive daytime somnolence, and disorganized speech.27
Detecting and characterizing cognitive deficits in dementia patients using neuropsychological testing is important in establishing a clinical diagnosis, determining baseline levels of impairment, forming a prognosis, and initiating disease-specific treatments. Differences in neuropsychological findings in AD and DLB are outlined in Table 2.16,28-33 Several studies have suggested using these measures to differentiate patients with DLB from those with AD.20
Table 2
Diagnostic testing for Alzheimer’s disease and dementia with Lewy bodies
| Alzheimer’s disease | Dementia with Lewy bodies |
|---|---|
| Neuropsychological testing findings | |
| Relatively more impairment on verbal memory tasks, delayed recall, delayed recognition, and encoding and storing information.28 Dysfunction of episodic memory function | Relatively more impairment on attention or concentration, verbal fluency, visuoperceptual, visuoconstructive, visual memory tests, and frontal executive functions.28 Relatively preserved confrontation naming and verbal memory |
| MRI findings | |
| Diffuse cortical atrophy, relatively greater volume loss in hippocampus and medial temporal lobe structures (strong correlation with severity)29 | Mild generalized cerebral cortical atrophy with minimal hippocampal atrophy and relative preservation of medial temporal lobe structures30 |
| [18F]FDG PET | |
| Widespread metabolic deficits in neocortical association areas, with sparing of the basal ganglia, thalamus, cerebellum, primary sensory motor cortex, and visual cortex | Widespread cortical hypometabolism, more marked in primary visual and occipital association areas, and less severe in parietal, frontal, and anterior cingulate cortices.31 Severe cholinergic deafferentation of the neocortex, particularly in posterior cortical regions32 |
| Single photon emission computed tomography | |
| Parietotemporal hypoperfusion | Occipital hypoperfusion |
| 123I-FP-CIT SPECT (DaT scan) | |
| No significant loss of DaT | Low nigrostriatal terminal density of DaT caused by severe nigrostriatal degeneration16 |
| Myocardial scintigraphy with MIBG | |
| No significant change in MIBG uptake | Decreased MIBG uptake33 |
| 123I-FP-CIT: 123I-2β-carbomethoxy-3β-(4-iodophenyl)-N-(3-fluoropropyl)nortropane; DaT: dopamine transporter; FDG PET: [18F]-fluoro-d-glucose positron emission tomography; MIBG: metaiodobenzylguanidine; SPECT: single photon emission computed tomography | |
Evidence is insufficient to support using electroencephalographic and polysomnographic studies when initially evaluating patients with dementia. Brain CT or MRI are recommended as part of the initial evaluation of dementia patients to exclude treatable causes of dementia and help clarify the differential diagnosis. Occipital hypometabolism and hypoperfusion demonstrated on PET and SPECT imaging have high sensitivity and specificity for differentiating AD from DLB.
To diagnose DLB more consistently, look for core features of the disease, RBD, antipsychotic hypersensitivity, and decreased striatal binding at presynaptic DaT sites.15 Abnormal (low binding) DaT activity is the most reliable diagnostic marker for DLB.34 Myocardial scintigraphy with MIBG is sensitive to pathologic changes of DLB before clinical expression and could overcome the difficulties of using clinical criteria alone to identify patients with DLB.35 MIBG scintigraphy may be preferred to DaT scan because it is less expensive and its sensitivity and specificity to DLB are independent of the presence of parkinsonism.35
For an overview of pharmacotherapy options for patients with AD or DLB, see Box 2.
Pharmacotherapy options for patients with Alzheimer’s disease (AD) or dementia with Lewy bodies (DLB) include cholinesterase inhibitors, memantine, antipsychotics, and other agents.
Cholinesterase inhibitors. Donepezil, rivastigmine, and galantamine are FDA-approved for treating AD. Their efficacy appears to be similar, so the choice of agent is based largely on cost, patient tolerability, and physician experience.
No medications are FDA-approved for treating DLB. Neocortical cholinergic activity assessed by choline acetyltransferase levels is more severely depleted in DLB than in AD, and this deficit is correlated with the presence of visual hallucinations and global severity of cognitive impairment.a Therefore, drugs that enhance central cholinergic function offer a therapeutic approach for DLB; cognitive and hallucinatory symptoms are the anticipated targets. Multiple anecdotal reports, open-label studies,b,c and 1 randomized, placebo-controlled triald suggest that cholinesterase inhibitors are efficacious in DLB, with reported benefits in cognition, fluctuations, psychotic symptoms, and parkinsonian symptoms. A 20-week randomized, double-blind, placebo-controlled multicenter studyd of patients with DLB found rivastigmine, 6 to 12 mg/d, was superior to placebo. Patients receiving rivastigmine exhibited significantly reduced anxiety, delusions, and hallucinations and significantly better performance on a computerized battery of neuropsychological tests, especially tasks that required sustained attention. Differences between rivastigmine and placebo disappeared after drug discontinuation.
Memantine is a noncompetitive antagonist of N-methyl-d-aspartate receptors that is effective in AD.e The possible involvement of glutamate in DLB has provided a rationale for treating DLB with memantine. Two randomized controlled trials in DLB found that patients treated with memantine for 24 weeks performed better on Clinical Global Impression of Change, but not on most other secondary outcome measures.f,g In both studies, memantine was well tolerated. However, other studies have noted worsening of delusions and hallucinations with memantine in DLB patients.h
Antipsychotics. Agitation is common in moderate and advanced AD. Atypical antipsychotics have been used with variable efficacy to treat agitation, but their use is associated with excess mortality. DLB patients pose a considerable therapeutic challenge because antipsychotics—the mainstay of treatment of psychosis and behavioral problems in most other disorders—can provoke severe, irreversible, and often fatal sensitivity reactions in this type of dementia.i A 2- to 3-fold increased mortality risk associated with antipsychotic sensitivity reactions in DLB is partly mediated via acute blockade of postsynaptic dopamine D2 receptors in the striatum. For severe and disabling psychosis, a trial of a cholinesterase inhibitor and/or lowering the dose of antiparkinsonian medication should be considered first. In urgent situations, small doses of an atypical antipsychotic that is least associated with parkinsonism side effects—such as quetiapine or aripiprazole—should be used.
Other treatments. Treatment of parkinsonian symptoms in DLB patients is similar to that for Parkinson’s disease, but the risk of psychotic symptoms in DLB warrants a conservative approach. Levodopa seems to be more effective than dopamine agonists and produces fewer side effects.j Rapid eye movement sleep behavior disorder often responds to low doses of clonazepam (0.25 to 1.5 mg). Depression and anxiety disorders are common in AD at all stages and their treatment is not fundamentally different than in geriatric patients without dementia. Selective serotonin reuptake inhibitors and electroconvulsive therapy have been used successfully in depressed patients with AD or DLB.k,l
Disease-modifying treatments. Researchers are evaluating an array of antiamyloid and neuroprotective therapeutic approaches for AD based on the hypothesis that amyloid-beta protein plays a pivotal role in disease onset and progression. Interventions that reduce amyloid production, limit aggregation, or increase clearance may block the cascade of events comprising AD pathogenesis. Reducing tau hyperphosphorylation, limiting oxidation and excitotoxicity, and controlling inflammation also may be beneficial strategies. Potentially neuroprotective and restorative treatments such as neurotrophins, neurotrophic factor enhancers, and stem cell-related approaches also are being investigated.
There are no large-scale studies of disease-modifying treatments for DLB. Potential areas of research include the relationship between proteasome function and a-synuclein pathology, the role of beta-synuclein, and the impact of alterations to alpha-synuclein on its propensity to aggregate.
References
a. Ballard C, Ziabreva I, Perry R, et al. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology. 2006;67(11):1931-1934.
b. Beversdorf DQ, Warner JL, Davis RA, et al. Donepezil in the treatment of dementia with lewy bodies. Am J Geriatr Psychiatry. 2004;12(5):542-544.
c. Edwards K, Royall D, Hershey L, et al. Efficacy and safety of galantamine in patients with dementia with Lewy bodies: a 24-week open-label study. Dement Geriatr Cogn Disord. 2007;23(6):401-405.
d. McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomised, double-blind, placebo-controlled international study. Lancet. 2000;356(9247):2031-2036.
e. Tariot PN, Farlow MR, Grossberg GT, et al. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291(3):317-324.
f. Aarsland D, Ballard C, Walker Z, et al. Memantine in patients with Parkinson’s disease dementia or dementia with Lewy bodies: a double-blind, placebo-controlled, multicentre trial. Lancet Neurol. 2009;8(7):613-618.
g. Emre M, Tsolaki M, Bonuccelli U, et al. Memantine for patients with Parkinson’s disease dementia or dementia with Lewy bodies: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9(10):969-977.
h. Ridha BH, Josephs KA, Rossor MN. Delusions and hallucinations in dementia with Lewy bodies: worsening with memantine. Neurology. 2005;65(3):481-482.
i. McKeith I, Fairbairn A, Perry R, et al. Neuroleptic sensitivity in patients with senile dementia of Lewy body type. BMJ. 1992;305(6855):673-678.
j. Fernandez HH, Wu CK, Ott BR. Pharmacotherapy of dementia with Lewy bodies. Expert Opin Pharmacother. 2003;4(11):2027-2037.
k. Swartz M, Barak Y, Mirecki I, et al. Treating depression in Alzheimer’s disease: integration of differing guidelines. Int Psychogeriatr. 2000;12(3):353-358.
l. Takahashi S, Mizukami K, Yasuno F, et al. Depression associated with dementia with Lewy bodies (DLB) and the effect of somatotherapy. Psychogeriatrics. 2009;9(2):56-61.
Related Resources
- Hanyu H, Sato T, Hirao K, et al. Differences in clinical course between dementia with Lewy bodies and Alzheimer’s disease. Eur J Neurol. 2009;16(2):212-217.
- Walker Z, McKeith I, Rodda J, et al. Comparison of cognitive decline between dementia with Lewy bodies and Alzheimer’s disease: a cohort study. BMJ Open. 2012;2:e000380.
Drug Brand Names
- Aripiprazole • Abilify
- Clonazepam • Klonopin
- Donepezil • Aricept
- Galantamine • Razadyne, Reminyl
- Levodopa • Dopar, Larodopa
- Memantine • Namenda
- Quetiapine • Seroquel
- Rivastigmine • Exelon
Disclosure
Drs. Bishnoi and Manepalli report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Grossberg serves as a consultant to Forest, Janssen, Novartis, and Pfizer. His department receives research funding from Novartis, Janssen, and Pfizer.
1. McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47(5):1113-1124.
2. Buracchio T, Arvanitakis Z, Gorbien M. Dementia with Lewy bodies: current concepts. Dement Geriatr Cogn Disord. 2005;20(5):306-320.
3. Fujishiro H, Iseki E, Higashi S, et al. Distribution of cerebral amyloid deposition and its relevance to clinical phenotype in Lewy body dementia. Neurosci Lett. 2010;486(1):19-23.
4. Kosaka K. Diffuse Lewy body disease. Neuropathology. 2000;20(suppl):S73-S78.
5. McKeith IG, Dickson DW, Lowe J, et al. Consortium on DLB. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863-1872.
6. Cummings JL, Cole G. Alzheimer disease. JAMA. 2002;287(18):2335-2338.
7. Zaccai J, McCracken C, Brayne C. A systematic review of prevalence and incidence studies of dementia with Lewy bodies. Age Ageing. 2005;34(6):561-566.
8. Bradshaw J, Saling M, Hopwood M, et al. Fluctuating cognition in dementia with Lewy bodies and Alzheimer’s disease is qualitatively distinct. J Neurol Neurosurg Psychiatry. 2004;75(3):382-387.
9. Singleton AB, Wharton A, O’Brien KK, et al. Clinical and neuropathological correlates of apolipoprotein E genotype in dementia with Lewy bodies. Dement Geriatr Cogn Disord. 2002;14(4):167-175.
10. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263-269.
11. Jack CR, Jr, Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):257-262.
12. Mollenhauer B, Cullen V, Kahn I, et al. Direct quantification of CSF alpha-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp Neurol. 2008;213(2):315-325.
13. Parnetti L, Balducci C, Pierguidi L, et al. Cerebrospinal fluid beta-glucocerebrosidase activity is reduced in dementia with Lewy bodies. Neurobiol Dis. 2009;34(3):484-486.
14. Shimada H, Hirano S, Shinotoh H, et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology. 2009;73(4):273-278.
15. McKeith I, O’Brien J, Walker Z, et al. Sensitivity and specificity of dopamine transporter imaging with 123I-FP-CIT SPECT in dementia with Lewy bodies: a phase III, multicentre study. Lancet Neurol. 2007;6(4):305-313.
16. Walker Z, Jaros E, Walker RW, et al. Dementia with Lewy bodies: a comparison of clinical diagnosis, FP-CIT single photon emission computed tomography imaging and autopsy. J Neurol Neurosurg Psychiatry. 2007;78(11):1176-1181.
17. Bradshaw JM, Saling M, Anderson V, et al. Higher cortical deficits influence attentional processing in dementia with Lewy bodies, relative to patients with dementia of the Alzheimer’s type and controls. J Neurol Neurosurg Psychiatry. 2006;77(10):1129-1135.
18. Weiner MF, Hynan LS, Parikh B, et al. Can Alzheimer’s disease and dementias with Lewy bodies be distinguished clinically? J Geriatr Psychiatry Neurol. 2003;16(4):245-250.
19. Stavitsky K, Brickman AM, Scarmeas N, et al. The progression of cognition, psychiatric symptoms, and functional abilities in dementia with Lewy bodies and Alzheimer disease. Arch Neurol. 2006;63(10):1450-1456.
20. Ferman TJ, Smith GE, Boeve BF, et al. Neuropsychological differentiation of dementia with Lewy bodies from normal aging and Alzheimer’s disease. Clin Neuropsychol. 2006;20(4):623-636.
21. McKeith IG, Perry EK, Perry RH. Report of the second dementia with Lewy body international workshop: diagnosis and treatment. Consortium on Dementia with Lewy Bodies. Neurology. 1999;53(5):902-905.
22. Boeve BF, Silber MH, Ferman TJ, et al. Association of REM sleep behavior disorder and neurodegenerative disease may reflect an underlying synucleinopathy. Mov Disord. 2001;16(4):622-630.
23. Portet F, Scarmeas N, Cosentino S, et al. Extrapyramidal signs before and after diagnosis of incident Alzheimer disease in a prospective population study. Arch Neurol. 2009;66(9):1120-1126.
24. McKeith I, Fairbairn A, Perry R, et al. Neuroleptic sensitivity in patients with senile dementia of Lewy body type. BMJ. 1992;305(6855):673-678.
25. Tarawneh R, Galvin JE. Distinguishing Lewy body dementias from Alzheimer’s disease. Expert Rev Neurother. 2007;7(11):1499-1516.
26. Ancoli-Israel S, Klauber MR, Gillin JC, et al. Sleep in non-institutionalized Alzheimer’s disease patients. Aging (Milano). 1994;6(6):451-458.
27. Ferman TJ, Smith GE, Boeve BF, et al. DLB fluctuations: specific features that reliably differentiate DLB from AD and normal aging. Neurology. 2004;62(2):181-187.
28. Salmon DP, Galasko D, Hansen LA, et al. Neuropsychological deficits associated with diffuse Lewy body disease. Brain Cogn. 1996;31(2):148-165.
29. Jack CR, Jr, Petersen RC, Xu Y, et al. Rates of hippocampal atrophy correlate with change in clinical status in aging and AD. Neurology. 2000;55(4):484-489.
30. Burton EJ, Barber R, Mukaetova-Ladinska EB, et al. Medial temporal lobe atrophy on MRI differentiates Alzheimer’s disease from dementia with Lewy bodies and vascular cognitive impairment: a prospective study with pathological verification of diagnosis. Brain. 2009;132(pt 1):195-203.
31. Ishii K, Soma T, Kono AK, et al. Comparison of regional brain volume and glucose metabolism between patients with mild dementia with lewy bodies and those with mild Alzheimer’s disease. J Nucl Med. 2007;48(5):704-711.
32. Klein JC, Eggers C, Kalbe E, et al. Neurotransmitter changes in dementia with Lewy bodies and Parkinson disease dementia in vivo. Neurology. 2010;74(11):885-892.
33. Fujishiro H, Nakamura S, Kitazawa M, et al. Early detection of dementia with Lewy bodies in patients with amnestic mild cognitive impairment using 123I-MIBG cardiac scintigraphy. J Neurol Sci. 2012;315(1-2):115-119.
34. O’Brien JT, McKeith IG, Walker Z, et al. Diagnostic accuracy of 123I-FP-CIT SPECT in possible dementia with Lewy bodies. Br J Psychiatry. 2009;194:34-39.
35. Yoshita M, Taki J, Yokoyama K, et al. Value of 123I-MIBG radioactivity in the differential diagnosis of DLB from AD. Neurology. 2006;66(12):1850-1854.
Discuss this article at www.facebook.com/CurrentPsychiatry
Alzheimer’s disease (AD) and dementia with Lewy bodies (DLB) are the first and second most common causes of neurodegenerative dementia, respectively.“New Alzheimer’s disease guidelines: Implications for clinicians,” Current Psychiatry, March 2012, p. 15-20; http://bit.ly/UNYikk.
The 2005 report of the DLB Consortium5 recognizes central, core, suggestive, and supportive features of DLB (Table 1).5,10 These features are considered in the context of other confounding clinical conditions and the timing of cognitive and motor symptoms. The revised DLB criteria5 require a central feature of progressive cognitive decline. “Probable DLB” is when a patient presents with 2 core features or 1 core feature and ≥1 suggestive features. A diagnosis of “possible DLB” requires 1 core feature or 1 suggestive feature in the presence of progressive cognitive decline.
Table 1
Diagnostic criteria for AD and DLB
| NIA-AA criteria for AD (2011)10 |
| Possible AD: Clinical and cognitive criteria (DSM-IV-TR) for AD are met and there is an absence of biomarkers to support the diagnosis or there is evidence of a secondary disorder that can cause dementia |
| Probable AD: Clinical and cognitive criteria for AD are met and there is documented progressive cognitive decline or abnormal biomarker(s) suggestive of AD or evidence of proven AD autosomal dominant genetic mutation (presenilin-1, presenilin-2, amyloid-β precursor protein) |
| Definite AD: Clinical criteria for probable AD are met and there is histopathologic evidence of the disorder |
| Revised clinical diagnostic criteria for DLB (2005)5 |
| Core features: Fluctuating cognition, recurrent visual hallucinations, soft motor features of parkinsonism |
| Suggestive features: REM sleep behavior disorder, severe antipsychotic sensitivity, decreased tracer uptake in striatum on SPECT dopamine transporter imaging or on myocardial scintigraphy with MIBG |
| Supportive features (common but lacking diagnostic specificity): repeated falls and syncope; transient, unexplained loss of consciousness; systematized delusions; hallucinations other than visual; relative preservation of medial temporal lobe on CT or MRI scan; decreased tracer uptake on SPECT or PET imaging in occipital regions; prominent slow waves on EEG with temporal lobe transient sharp waves |
| AD: Alzheimer’s disease; DLB: dementia with Lewy bodies; MIBG: metaiodobenzylguanidine; NIA-AA: National Institute on Aging and the Alzheimer’s Association; PET: positron emission tomography; REM: rapid eye movement; SPECT: single photon emission computed tomography |
Biomarkers for AD, but not DLB
The 2011 diagnostic criteria for AD incorporate biomarkers that can be measured in vivo and reflect speci?c features of disease-related pathophysiologic processes. Biomarkers for AD are divided into 2 categories:11
- amyloid-beta (Aβ) accumulation: abnormal tracer retention on amyloid positron emission topography (PET) imaging and low cerebrospinal fluid (CSF) Aβ42
- neuronal degeneration or injury: elevated CSF tau (total and phosphorylated tau), decreased ?uorodeoxyglucose uptake on PET in temporo-parietal cortices, and atrophy on structural MRI in the hippocampal and temporo-parietal regions.
No clinically applicable genotypic or CSF markers exist to support a DLB diagnosis, but there are many promising candidates, including elevated levels of CSF p-tau 181, CSF levels of alpha- and beta-synuclein,12 and CSF beta-glucocerebrosidase levels.13 PET mapping of brain acetylcholinesterase activity,14 123I-2β-carbomethoxy-3β- (4-iodophenyl)-N-(3-fluoropropyl)nortropane single photon emission computed tomography (SPECT) dopamine transporter (DaT) imaging15 and metaiodobenzylguanidine (MIBG) scintigraphy also are promising methods. DaT scan SPECT is FDA-approved for detecting loss of functional dopaminergic neuron terminals in the striatum and can differentiate between AD and DLB with a sensitivity and specificity of 78% to 88% and 94% to 100%, respectively.16 This test is covered by Medicare for differentiating AD and DLB.
Differences in presentation
Cognitive impairment. Contrary to the early memory impairment that characterizes AD, memory deficits in DLB usually appear later in the disease course.5 Patients with DLB manifest greater attentional, visuospatial, and executive impairments than those with AD, whereas AD causes more profound episodic (declarative) memory impairment than DLB. DLB patients show more preserved consolidation and storage of verbal information than AD patients because of less neuroanatomical and cholinergic compromise in the medial temporal lobe. There is no evidence of significant differences in remote memory, semantic memory, and language (naming and fluency).
Compromised attention in DLB may be the basis for fluctuating cognition, a characteristic of the disease. The greater attentional impairment and reaction time variability in DLB compared with AD is evident during complex tasks for attention and may be a function of the executive and visuospatial demands of the tasks.17
Executive functions critical to adaptive, goal-directed behavior are more impaired in DLB than AD. DLB patients are more susceptible to distraction and have difficulty engaging in a task and shifting from 1 task to another. This, together with a tendency for confabulation and perseveration, are signs of executive dysfunction.
Neuropsychiatric features. DLB patients are more likely than AD patients to exhibit psychiatric symptoms and have more functional impairment.18 In an analysis of autopsy-confirmed cases, hallucinations and delusions were more frequent with Lewy body pathology (75%) than AD (21%) at initial clinical evaluation.18 By the end stages of both illnesses, the degree of psychotic symptoms is comparable.19 Depression is common in DLB; whether base rates of depressed mood and major depression differ between DLB and AD is uncertain.20
Psychosis in AD can be induced by medication or delirium, or triggered by poor sensory perceptions. Psychotic symptoms occur more frequently during the moderate and advanced stages of AD, when patients present with visual hallucinations, delusions, or delusional misidentifications. As many as 10% to 20% of patients with AD experience hallucinations, typically visual. Delusions occur in 30% to 50% of AD patients, usually in the later stages of the disease. The most common delusional themes are infidelity, theft, and paranoia. Female sex is a risk factor for psychosis in AD. Delusions co-occur with aggression, anxiety, and aberrant motor behavior.
Visual hallucinations—mostly vivid, well-formed, false perceptions of insects, animals, or people—are the defining feature of DLB.21 Many patients recognize that they are experiencing visual hallucinations and can ignore them. DLB patients also may experience visual illusions, such as misperceiving household objects as living beings. Delusions—typically paranoid—are common among DLB patients, as are depression and anxiety.1 Agitation or aggressive behavior tends to occur late in the illness, if at all.
The causes of psychotic symptoms in DLB are not fully understood, but dopamine dysfunction likely is involved in hallucinations, delusions, and agitation, and serotonin dysfunction may be associated with depression and anxiety. Rapid eye movement (REM) sleep/wakefulness dysregulation, in which the dream imagery of REM sleep may occur during wakefulness, also has been proposed as a mechanism for visual hallucinations in DLB.22 In DLB, psychotic symptoms occur early and are a hallmark of this illness, whereas in AD they usually occur in the middle to late stages of the disease.
Motor symptoms. In AD, extrapyramidal symptoms (EPS) are common later in the disease, are strongly correlated with disease severity, and are a strong, independent predictor of depression severity.23 EPS are more common in DLB than in AD24 and DLB patients are at higher risk of developing EPS even with low doses of typical antipsychotics, compared with AD patients.25
Other symptoms. REM sleep behavior disorder (RBD) is characterized by enacting dreams—often violent—during REM sleep. RBD is common in DLB and many patients also have excessive daytime somnolence. Other sleep disorders in DLB include insomnia, obstructive sleep apnea, central sleep apnea, restless legs syndrome, and periodic limb movements during sleep.
In AD patients, common sleep behaviors include confusion in the early evening (“sundowning”) and frequent nighttime awakenings, often accompanied by wandering.26 Orthostatic hypotension, impotence, urinary incontinence, and constipation are common in DLB. Lack of insight concerning personal cognitive, mood, and behavioral state is highly prevalent in AD patients and more common than in DLB.
Diagnostic evaluation
Because there are no definitive clinical markers for DLB, diagnosis is based on a detailed clinical and family history from the patient and a reliable informant, as well as a physical, neurologic, and mental status examination that looks for associated noncognitive symptoms, and neuropsychological evaluation. Reasons DLB may be misdiagnosed include:
- Some “core” clinical features of DLB may not appear or may overlap with AD.
- Presence and severity of concurrent AD pathology in DLB may modify the clinical presentation, with decreased rates of hallucinations and parkinsonism and increased neurofibrillary tangles.
- Failure to reliably identify fluctuations—variations in cognition and arousal, such as periods of unresponsiveness while awake (“zoning out”), excessive daytime somnolence, and disorganized speech.27
Detecting and characterizing cognitive deficits in dementia patients using neuropsychological testing is important in establishing a clinical diagnosis, determining baseline levels of impairment, forming a prognosis, and initiating disease-specific treatments. Differences in neuropsychological findings in AD and DLB are outlined in Table 2.16,28-33 Several studies have suggested using these measures to differentiate patients with DLB from those with AD.20
Table 2
Diagnostic testing for Alzheimer’s disease and dementia with Lewy bodies
| Alzheimer’s disease | Dementia with Lewy bodies |
|---|---|
| Neuropsychological testing findings | |
| Relatively more impairment on verbal memory tasks, delayed recall, delayed recognition, and encoding and storing information.28 Dysfunction of episodic memory function | Relatively more impairment on attention or concentration, verbal fluency, visuoperceptual, visuoconstructive, visual memory tests, and frontal executive functions.28 Relatively preserved confrontation naming and verbal memory |
| MRI findings | |
| Diffuse cortical atrophy, relatively greater volume loss in hippocampus and medial temporal lobe structures (strong correlation with severity)29 | Mild generalized cerebral cortical atrophy with minimal hippocampal atrophy and relative preservation of medial temporal lobe structures30 |
| [18F]FDG PET | |
| Widespread metabolic deficits in neocortical association areas, with sparing of the basal ganglia, thalamus, cerebellum, primary sensory motor cortex, and visual cortex | Widespread cortical hypometabolism, more marked in primary visual and occipital association areas, and less severe in parietal, frontal, and anterior cingulate cortices.31 Severe cholinergic deafferentation of the neocortex, particularly in posterior cortical regions32 |
| Single photon emission computed tomography | |
| Parietotemporal hypoperfusion | Occipital hypoperfusion |
| 123I-FP-CIT SPECT (DaT scan) | |
| No significant loss of DaT | Low nigrostriatal terminal density of DaT caused by severe nigrostriatal degeneration16 |
| Myocardial scintigraphy with MIBG | |
| No significant change in MIBG uptake | Decreased MIBG uptake33 |
| 123I-FP-CIT: 123I-2β-carbomethoxy-3β-(4-iodophenyl)-N-(3-fluoropropyl)nortropane; DaT: dopamine transporter; FDG PET: [18F]-fluoro-d-glucose positron emission tomography; MIBG: metaiodobenzylguanidine; SPECT: single photon emission computed tomography | |
Evidence is insufficient to support using electroencephalographic and polysomnographic studies when initially evaluating patients with dementia. Brain CT or MRI are recommended as part of the initial evaluation of dementia patients to exclude treatable causes of dementia and help clarify the differential diagnosis. Occipital hypometabolism and hypoperfusion demonstrated on PET and SPECT imaging have high sensitivity and specificity for differentiating AD from DLB.
To diagnose DLB more consistently, look for core features of the disease, RBD, antipsychotic hypersensitivity, and decreased striatal binding at presynaptic DaT sites.15 Abnormal (low binding) DaT activity is the most reliable diagnostic marker for DLB.34 Myocardial scintigraphy with MIBG is sensitive to pathologic changes of DLB before clinical expression and could overcome the difficulties of using clinical criteria alone to identify patients with DLB.35 MIBG scintigraphy may be preferred to DaT scan because it is less expensive and its sensitivity and specificity to DLB are independent of the presence of parkinsonism.35
For an overview of pharmacotherapy options for patients with AD or DLB, see Box 2.
Pharmacotherapy options for patients with Alzheimer’s disease (AD) or dementia with Lewy bodies (DLB) include cholinesterase inhibitors, memantine, antipsychotics, and other agents.
Cholinesterase inhibitors. Donepezil, rivastigmine, and galantamine are FDA-approved for treating AD. Their efficacy appears to be similar, so the choice of agent is based largely on cost, patient tolerability, and physician experience.
No medications are FDA-approved for treating DLB. Neocortical cholinergic activity assessed by choline acetyltransferase levels is more severely depleted in DLB than in AD, and this deficit is correlated with the presence of visual hallucinations and global severity of cognitive impairment.a Therefore, drugs that enhance central cholinergic function offer a therapeutic approach for DLB; cognitive and hallucinatory symptoms are the anticipated targets. Multiple anecdotal reports, open-label studies,b,c and 1 randomized, placebo-controlled triald suggest that cholinesterase inhibitors are efficacious in DLB, with reported benefits in cognition, fluctuations, psychotic symptoms, and parkinsonian symptoms. A 20-week randomized, double-blind, placebo-controlled multicenter studyd of patients with DLB found rivastigmine, 6 to 12 mg/d, was superior to placebo. Patients receiving rivastigmine exhibited significantly reduced anxiety, delusions, and hallucinations and significantly better performance on a computerized battery of neuropsychological tests, especially tasks that required sustained attention. Differences between rivastigmine and placebo disappeared after drug discontinuation.
Memantine is a noncompetitive antagonist of N-methyl-d-aspartate receptors that is effective in AD.e The possible involvement of glutamate in DLB has provided a rationale for treating DLB with memantine. Two randomized controlled trials in DLB found that patients treated with memantine for 24 weeks performed better on Clinical Global Impression of Change, but not on most other secondary outcome measures.f,g In both studies, memantine was well tolerated. However, other studies have noted worsening of delusions and hallucinations with memantine in DLB patients.h
Antipsychotics. Agitation is common in moderate and advanced AD. Atypical antipsychotics have been used with variable efficacy to treat agitation, but their use is associated with excess mortality. DLB patients pose a considerable therapeutic challenge because antipsychotics—the mainstay of treatment of psychosis and behavioral problems in most other disorders—can provoke severe, irreversible, and often fatal sensitivity reactions in this type of dementia.i A 2- to 3-fold increased mortality risk associated with antipsychotic sensitivity reactions in DLB is partly mediated via acute blockade of postsynaptic dopamine D2 receptors in the striatum. For severe and disabling psychosis, a trial of a cholinesterase inhibitor and/or lowering the dose of antiparkinsonian medication should be considered first. In urgent situations, small doses of an atypical antipsychotic that is least associated with parkinsonism side effects—such as quetiapine or aripiprazole—should be used.
Other treatments. Treatment of parkinsonian symptoms in DLB patients is similar to that for Parkinson’s disease, but the risk of psychotic symptoms in DLB warrants a conservative approach. Levodopa seems to be more effective than dopamine agonists and produces fewer side effects.j Rapid eye movement sleep behavior disorder often responds to low doses of clonazepam (0.25 to 1.5 mg). Depression and anxiety disorders are common in AD at all stages and their treatment is not fundamentally different than in geriatric patients without dementia. Selective serotonin reuptake inhibitors and electroconvulsive therapy have been used successfully in depressed patients with AD or DLB.k,l
Disease-modifying treatments. Researchers are evaluating an array of antiamyloid and neuroprotective therapeutic approaches for AD based on the hypothesis that amyloid-beta protein plays a pivotal role in disease onset and progression. Interventions that reduce amyloid production, limit aggregation, or increase clearance may block the cascade of events comprising AD pathogenesis. Reducing tau hyperphosphorylation, limiting oxidation and excitotoxicity, and controlling inflammation also may be beneficial strategies. Potentially neuroprotective and restorative treatments such as neurotrophins, neurotrophic factor enhancers, and stem cell-related approaches also are being investigated.
There are no large-scale studies of disease-modifying treatments for DLB. Potential areas of research include the relationship between proteasome function and a-synuclein pathology, the role of beta-synuclein, and the impact of alterations to alpha-synuclein on its propensity to aggregate.
References
a. Ballard C, Ziabreva I, Perry R, et al. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology. 2006;67(11):1931-1934.
b. Beversdorf DQ, Warner JL, Davis RA, et al. Donepezil in the treatment of dementia with lewy bodies. Am J Geriatr Psychiatry. 2004;12(5):542-544.
c. Edwards K, Royall D, Hershey L, et al. Efficacy and safety of galantamine in patients with dementia with Lewy bodies: a 24-week open-label study. Dement Geriatr Cogn Disord. 2007;23(6):401-405.
d. McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomised, double-blind, placebo-controlled international study. Lancet. 2000;356(9247):2031-2036.
e. Tariot PN, Farlow MR, Grossberg GT, et al. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291(3):317-324.
f. Aarsland D, Ballard C, Walker Z, et al. Memantine in patients with Parkinson’s disease dementia or dementia with Lewy bodies: a double-blind, placebo-controlled, multicentre trial. Lancet Neurol. 2009;8(7):613-618.
g. Emre M, Tsolaki M, Bonuccelli U, et al. Memantine for patients with Parkinson’s disease dementia or dementia with Lewy bodies: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9(10):969-977.
h. Ridha BH, Josephs KA, Rossor MN. Delusions and hallucinations in dementia with Lewy bodies: worsening with memantine. Neurology. 2005;65(3):481-482.
i. McKeith I, Fairbairn A, Perry R, et al. Neuroleptic sensitivity in patients with senile dementia of Lewy body type. BMJ. 1992;305(6855):673-678.
j. Fernandez HH, Wu CK, Ott BR. Pharmacotherapy of dementia with Lewy bodies. Expert Opin Pharmacother. 2003;4(11):2027-2037.
k. Swartz M, Barak Y, Mirecki I, et al. Treating depression in Alzheimer’s disease: integration of differing guidelines. Int Psychogeriatr. 2000;12(3):353-358.
l. Takahashi S, Mizukami K, Yasuno F, et al. Depression associated with dementia with Lewy bodies (DLB) and the effect of somatotherapy. Psychogeriatrics. 2009;9(2):56-61.
Related Resources
- Hanyu H, Sato T, Hirao K, et al. Differences in clinical course between dementia with Lewy bodies and Alzheimer’s disease. Eur J Neurol. 2009;16(2):212-217.
- Walker Z, McKeith I, Rodda J, et al. Comparison of cognitive decline between dementia with Lewy bodies and Alzheimer’s disease: a cohort study. BMJ Open. 2012;2:e000380.
Drug Brand Names
- Aripiprazole • Abilify
- Clonazepam • Klonopin
- Donepezil • Aricept
- Galantamine • Razadyne, Reminyl
- Levodopa • Dopar, Larodopa
- Memantine • Namenda
- Quetiapine • Seroquel
- Rivastigmine • Exelon
Disclosure
Drs. Bishnoi and Manepalli report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Grossberg serves as a consultant to Forest, Janssen, Novartis, and Pfizer. His department receives research funding from Novartis, Janssen, and Pfizer.
Discuss this article at www.facebook.com/CurrentPsychiatry
Alzheimer’s disease (AD) and dementia with Lewy bodies (DLB) are the first and second most common causes of neurodegenerative dementia, respectively.“New Alzheimer’s disease guidelines: Implications for clinicians,” Current Psychiatry, March 2012, p. 15-20; http://bit.ly/UNYikk.
The 2005 report of the DLB Consortium5 recognizes central, core, suggestive, and supportive features of DLB (Table 1).5,10 These features are considered in the context of other confounding clinical conditions and the timing of cognitive and motor symptoms. The revised DLB criteria5 require a central feature of progressive cognitive decline. “Probable DLB” is when a patient presents with 2 core features or 1 core feature and ≥1 suggestive features. A diagnosis of “possible DLB” requires 1 core feature or 1 suggestive feature in the presence of progressive cognitive decline.
Table 1
Diagnostic criteria for AD and DLB
| NIA-AA criteria for AD (2011)10 |
| Possible AD: Clinical and cognitive criteria (DSM-IV-TR) for AD are met and there is an absence of biomarkers to support the diagnosis or there is evidence of a secondary disorder that can cause dementia |
| Probable AD: Clinical and cognitive criteria for AD are met and there is documented progressive cognitive decline or abnormal biomarker(s) suggestive of AD or evidence of proven AD autosomal dominant genetic mutation (presenilin-1, presenilin-2, amyloid-β precursor protein) |
| Definite AD: Clinical criteria for probable AD are met and there is histopathologic evidence of the disorder |
| Revised clinical diagnostic criteria for DLB (2005)5 |
| Core features: Fluctuating cognition, recurrent visual hallucinations, soft motor features of parkinsonism |
| Suggestive features: REM sleep behavior disorder, severe antipsychotic sensitivity, decreased tracer uptake in striatum on SPECT dopamine transporter imaging or on myocardial scintigraphy with MIBG |
| Supportive features (common but lacking diagnostic specificity): repeated falls and syncope; transient, unexplained loss of consciousness; systematized delusions; hallucinations other than visual; relative preservation of medial temporal lobe on CT or MRI scan; decreased tracer uptake on SPECT or PET imaging in occipital regions; prominent slow waves on EEG with temporal lobe transient sharp waves |
| AD: Alzheimer’s disease; DLB: dementia with Lewy bodies; MIBG: metaiodobenzylguanidine; NIA-AA: National Institute on Aging and the Alzheimer’s Association; PET: positron emission tomography; REM: rapid eye movement; SPECT: single photon emission computed tomography |
Biomarkers for AD, but not DLB
The 2011 diagnostic criteria for AD incorporate biomarkers that can be measured in vivo and reflect speci?c features of disease-related pathophysiologic processes. Biomarkers for AD are divided into 2 categories:11
- amyloid-beta (Aβ) accumulation: abnormal tracer retention on amyloid positron emission topography (PET) imaging and low cerebrospinal fluid (CSF) Aβ42
- neuronal degeneration or injury: elevated CSF tau (total and phosphorylated tau), decreased ?uorodeoxyglucose uptake on PET in temporo-parietal cortices, and atrophy on structural MRI in the hippocampal and temporo-parietal regions.
No clinically applicable genotypic or CSF markers exist to support a DLB diagnosis, but there are many promising candidates, including elevated levels of CSF p-tau 181, CSF levels of alpha- and beta-synuclein,12 and CSF beta-glucocerebrosidase levels.13 PET mapping of brain acetylcholinesterase activity,14 123I-2β-carbomethoxy-3β- (4-iodophenyl)-N-(3-fluoropropyl)nortropane single photon emission computed tomography (SPECT) dopamine transporter (DaT) imaging15 and metaiodobenzylguanidine (MIBG) scintigraphy also are promising methods. DaT scan SPECT is FDA-approved for detecting loss of functional dopaminergic neuron terminals in the striatum and can differentiate between AD and DLB with a sensitivity and specificity of 78% to 88% and 94% to 100%, respectively.16 This test is covered by Medicare for differentiating AD and DLB.
Differences in presentation
Cognitive impairment. Contrary to the early memory impairment that characterizes AD, memory deficits in DLB usually appear later in the disease course.5 Patients with DLB manifest greater attentional, visuospatial, and executive impairments than those with AD, whereas AD causes more profound episodic (declarative) memory impairment than DLB. DLB patients show more preserved consolidation and storage of verbal information than AD patients because of less neuroanatomical and cholinergic compromise in the medial temporal lobe. There is no evidence of significant differences in remote memory, semantic memory, and language (naming and fluency).
Compromised attention in DLB may be the basis for fluctuating cognition, a characteristic of the disease. The greater attentional impairment and reaction time variability in DLB compared with AD is evident during complex tasks for attention and may be a function of the executive and visuospatial demands of the tasks.17
Executive functions critical to adaptive, goal-directed behavior are more impaired in DLB than AD. DLB patients are more susceptible to distraction and have difficulty engaging in a task and shifting from 1 task to another. This, together with a tendency for confabulation and perseveration, are signs of executive dysfunction.
Neuropsychiatric features. DLB patients are more likely than AD patients to exhibit psychiatric symptoms and have more functional impairment.18 In an analysis of autopsy-confirmed cases, hallucinations and delusions were more frequent with Lewy body pathology (75%) than AD (21%) at initial clinical evaluation.18 By the end stages of both illnesses, the degree of psychotic symptoms is comparable.19 Depression is common in DLB; whether base rates of depressed mood and major depression differ between DLB and AD is uncertain.20
Psychosis in AD can be induced by medication or delirium, or triggered by poor sensory perceptions. Psychotic symptoms occur more frequently during the moderate and advanced stages of AD, when patients present with visual hallucinations, delusions, or delusional misidentifications. As many as 10% to 20% of patients with AD experience hallucinations, typically visual. Delusions occur in 30% to 50% of AD patients, usually in the later stages of the disease. The most common delusional themes are infidelity, theft, and paranoia. Female sex is a risk factor for psychosis in AD. Delusions co-occur with aggression, anxiety, and aberrant motor behavior.
Visual hallucinations—mostly vivid, well-formed, false perceptions of insects, animals, or people—are the defining feature of DLB.21 Many patients recognize that they are experiencing visual hallucinations and can ignore them. DLB patients also may experience visual illusions, such as misperceiving household objects as living beings. Delusions—typically paranoid—are common among DLB patients, as are depression and anxiety.1 Agitation or aggressive behavior tends to occur late in the illness, if at all.
The causes of psychotic symptoms in DLB are not fully understood, but dopamine dysfunction likely is involved in hallucinations, delusions, and agitation, and serotonin dysfunction may be associated with depression and anxiety. Rapid eye movement (REM) sleep/wakefulness dysregulation, in which the dream imagery of REM sleep may occur during wakefulness, also has been proposed as a mechanism for visual hallucinations in DLB.22 In DLB, psychotic symptoms occur early and are a hallmark of this illness, whereas in AD they usually occur in the middle to late stages of the disease.
Motor symptoms. In AD, extrapyramidal symptoms (EPS) are common later in the disease, are strongly correlated with disease severity, and are a strong, independent predictor of depression severity.23 EPS are more common in DLB than in AD24 and DLB patients are at higher risk of developing EPS even with low doses of typical antipsychotics, compared with AD patients.25
Other symptoms. REM sleep behavior disorder (RBD) is characterized by enacting dreams—often violent—during REM sleep. RBD is common in DLB and many patients also have excessive daytime somnolence. Other sleep disorders in DLB include insomnia, obstructive sleep apnea, central sleep apnea, restless legs syndrome, and periodic limb movements during sleep.
In AD patients, common sleep behaviors include confusion in the early evening (“sundowning”) and frequent nighttime awakenings, often accompanied by wandering.26 Orthostatic hypotension, impotence, urinary incontinence, and constipation are common in DLB. Lack of insight concerning personal cognitive, mood, and behavioral state is highly prevalent in AD patients and more common than in DLB.
Diagnostic evaluation
Because there are no definitive clinical markers for DLB, diagnosis is based on a detailed clinical and family history from the patient and a reliable informant, as well as a physical, neurologic, and mental status examination that looks for associated noncognitive symptoms, and neuropsychological evaluation. Reasons DLB may be misdiagnosed include:
- Some “core” clinical features of DLB may not appear or may overlap with AD.
- Presence and severity of concurrent AD pathology in DLB may modify the clinical presentation, with decreased rates of hallucinations and parkinsonism and increased neurofibrillary tangles.
- Failure to reliably identify fluctuations—variations in cognition and arousal, such as periods of unresponsiveness while awake (“zoning out”), excessive daytime somnolence, and disorganized speech.27
Detecting and characterizing cognitive deficits in dementia patients using neuropsychological testing is important in establishing a clinical diagnosis, determining baseline levels of impairment, forming a prognosis, and initiating disease-specific treatments. Differences in neuropsychological findings in AD and DLB are outlined in Table 2.16,28-33 Several studies have suggested using these measures to differentiate patients with DLB from those with AD.20
Table 2
Diagnostic testing for Alzheimer’s disease and dementia with Lewy bodies
| Alzheimer’s disease | Dementia with Lewy bodies |
|---|---|
| Neuropsychological testing findings | |
| Relatively more impairment on verbal memory tasks, delayed recall, delayed recognition, and encoding and storing information.28 Dysfunction of episodic memory function | Relatively more impairment on attention or concentration, verbal fluency, visuoperceptual, visuoconstructive, visual memory tests, and frontal executive functions.28 Relatively preserved confrontation naming and verbal memory |
| MRI findings | |
| Diffuse cortical atrophy, relatively greater volume loss in hippocampus and medial temporal lobe structures (strong correlation with severity)29 | Mild generalized cerebral cortical atrophy with minimal hippocampal atrophy and relative preservation of medial temporal lobe structures30 |
| [18F]FDG PET | |
| Widespread metabolic deficits in neocortical association areas, with sparing of the basal ganglia, thalamus, cerebellum, primary sensory motor cortex, and visual cortex | Widespread cortical hypometabolism, more marked in primary visual and occipital association areas, and less severe in parietal, frontal, and anterior cingulate cortices.31 Severe cholinergic deafferentation of the neocortex, particularly in posterior cortical regions32 |
| Single photon emission computed tomography | |
| Parietotemporal hypoperfusion | Occipital hypoperfusion |
| 123I-FP-CIT SPECT (DaT scan) | |
| No significant loss of DaT | Low nigrostriatal terminal density of DaT caused by severe nigrostriatal degeneration16 |
| Myocardial scintigraphy with MIBG | |
| No significant change in MIBG uptake | Decreased MIBG uptake33 |
| 123I-FP-CIT: 123I-2β-carbomethoxy-3β-(4-iodophenyl)-N-(3-fluoropropyl)nortropane; DaT: dopamine transporter; FDG PET: [18F]-fluoro-d-glucose positron emission tomography; MIBG: metaiodobenzylguanidine; SPECT: single photon emission computed tomography | |
Evidence is insufficient to support using electroencephalographic and polysomnographic studies when initially evaluating patients with dementia. Brain CT or MRI are recommended as part of the initial evaluation of dementia patients to exclude treatable causes of dementia and help clarify the differential diagnosis. Occipital hypometabolism and hypoperfusion demonstrated on PET and SPECT imaging have high sensitivity and specificity for differentiating AD from DLB.
To diagnose DLB more consistently, look for core features of the disease, RBD, antipsychotic hypersensitivity, and decreased striatal binding at presynaptic DaT sites.15 Abnormal (low binding) DaT activity is the most reliable diagnostic marker for DLB.34 Myocardial scintigraphy with MIBG is sensitive to pathologic changes of DLB before clinical expression and could overcome the difficulties of using clinical criteria alone to identify patients with DLB.35 MIBG scintigraphy may be preferred to DaT scan because it is less expensive and its sensitivity and specificity to DLB are independent of the presence of parkinsonism.35
For an overview of pharmacotherapy options for patients with AD or DLB, see Box 2.
Pharmacotherapy options for patients with Alzheimer’s disease (AD) or dementia with Lewy bodies (DLB) include cholinesterase inhibitors, memantine, antipsychotics, and other agents.
Cholinesterase inhibitors. Donepezil, rivastigmine, and galantamine are FDA-approved for treating AD. Their efficacy appears to be similar, so the choice of agent is based largely on cost, patient tolerability, and physician experience.
No medications are FDA-approved for treating DLB. Neocortical cholinergic activity assessed by choline acetyltransferase levels is more severely depleted in DLB than in AD, and this deficit is correlated with the presence of visual hallucinations and global severity of cognitive impairment.a Therefore, drugs that enhance central cholinergic function offer a therapeutic approach for DLB; cognitive and hallucinatory symptoms are the anticipated targets. Multiple anecdotal reports, open-label studies,b,c and 1 randomized, placebo-controlled triald suggest that cholinesterase inhibitors are efficacious in DLB, with reported benefits in cognition, fluctuations, psychotic symptoms, and parkinsonian symptoms. A 20-week randomized, double-blind, placebo-controlled multicenter studyd of patients with DLB found rivastigmine, 6 to 12 mg/d, was superior to placebo. Patients receiving rivastigmine exhibited significantly reduced anxiety, delusions, and hallucinations and significantly better performance on a computerized battery of neuropsychological tests, especially tasks that required sustained attention. Differences between rivastigmine and placebo disappeared after drug discontinuation.
Memantine is a noncompetitive antagonist of N-methyl-d-aspartate receptors that is effective in AD.e The possible involvement of glutamate in DLB has provided a rationale for treating DLB with memantine. Two randomized controlled trials in DLB found that patients treated with memantine for 24 weeks performed better on Clinical Global Impression of Change, but not on most other secondary outcome measures.f,g In both studies, memantine was well tolerated. However, other studies have noted worsening of delusions and hallucinations with memantine in DLB patients.h
Antipsychotics. Agitation is common in moderate and advanced AD. Atypical antipsychotics have been used with variable efficacy to treat agitation, but their use is associated with excess mortality. DLB patients pose a considerable therapeutic challenge because antipsychotics—the mainstay of treatment of psychosis and behavioral problems in most other disorders—can provoke severe, irreversible, and often fatal sensitivity reactions in this type of dementia.i A 2- to 3-fold increased mortality risk associated with antipsychotic sensitivity reactions in DLB is partly mediated via acute blockade of postsynaptic dopamine D2 receptors in the striatum. For severe and disabling psychosis, a trial of a cholinesterase inhibitor and/or lowering the dose of antiparkinsonian medication should be considered first. In urgent situations, small doses of an atypical antipsychotic that is least associated with parkinsonism side effects—such as quetiapine or aripiprazole—should be used.
Other treatments. Treatment of parkinsonian symptoms in DLB patients is similar to that for Parkinson’s disease, but the risk of psychotic symptoms in DLB warrants a conservative approach. Levodopa seems to be more effective than dopamine agonists and produces fewer side effects.j Rapid eye movement sleep behavior disorder often responds to low doses of clonazepam (0.25 to 1.5 mg). Depression and anxiety disorders are common in AD at all stages and their treatment is not fundamentally different than in geriatric patients without dementia. Selective serotonin reuptake inhibitors and electroconvulsive therapy have been used successfully in depressed patients with AD or DLB.k,l
Disease-modifying treatments. Researchers are evaluating an array of antiamyloid and neuroprotective therapeutic approaches for AD based on the hypothesis that amyloid-beta protein plays a pivotal role in disease onset and progression. Interventions that reduce amyloid production, limit aggregation, or increase clearance may block the cascade of events comprising AD pathogenesis. Reducing tau hyperphosphorylation, limiting oxidation and excitotoxicity, and controlling inflammation also may be beneficial strategies. Potentially neuroprotective and restorative treatments such as neurotrophins, neurotrophic factor enhancers, and stem cell-related approaches also are being investigated.
There are no large-scale studies of disease-modifying treatments for DLB. Potential areas of research include the relationship between proteasome function and a-synuclein pathology, the role of beta-synuclein, and the impact of alterations to alpha-synuclein on its propensity to aggregate.
References
a. Ballard C, Ziabreva I, Perry R, et al. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology. 2006;67(11):1931-1934.
b. Beversdorf DQ, Warner JL, Davis RA, et al. Donepezil in the treatment of dementia with lewy bodies. Am J Geriatr Psychiatry. 2004;12(5):542-544.
c. Edwards K, Royall D, Hershey L, et al. Efficacy and safety of galantamine in patients with dementia with Lewy bodies: a 24-week open-label study. Dement Geriatr Cogn Disord. 2007;23(6):401-405.
d. McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomised, double-blind, placebo-controlled international study. Lancet. 2000;356(9247):2031-2036.
e. Tariot PN, Farlow MR, Grossberg GT, et al. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291(3):317-324.
f. Aarsland D, Ballard C, Walker Z, et al. Memantine in patients with Parkinson’s disease dementia or dementia with Lewy bodies: a double-blind, placebo-controlled, multicentre trial. Lancet Neurol. 2009;8(7):613-618.
g. Emre M, Tsolaki M, Bonuccelli U, et al. Memantine for patients with Parkinson’s disease dementia or dementia with Lewy bodies: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9(10):969-977.
h. Ridha BH, Josephs KA, Rossor MN. Delusions and hallucinations in dementia with Lewy bodies: worsening with memantine. Neurology. 2005;65(3):481-482.
i. McKeith I, Fairbairn A, Perry R, et al. Neuroleptic sensitivity in patients with senile dementia of Lewy body type. BMJ. 1992;305(6855):673-678.
j. Fernandez HH, Wu CK, Ott BR. Pharmacotherapy of dementia with Lewy bodies. Expert Opin Pharmacother. 2003;4(11):2027-2037.
k. Swartz M, Barak Y, Mirecki I, et al. Treating depression in Alzheimer’s disease: integration of differing guidelines. Int Psychogeriatr. 2000;12(3):353-358.
l. Takahashi S, Mizukami K, Yasuno F, et al. Depression associated with dementia with Lewy bodies (DLB) and the effect of somatotherapy. Psychogeriatrics. 2009;9(2):56-61.
Related Resources
- Hanyu H, Sato T, Hirao K, et al. Differences in clinical course between dementia with Lewy bodies and Alzheimer’s disease. Eur J Neurol. 2009;16(2):212-217.
- Walker Z, McKeith I, Rodda J, et al. Comparison of cognitive decline between dementia with Lewy bodies and Alzheimer’s disease: a cohort study. BMJ Open. 2012;2:e000380.
Drug Brand Names
- Aripiprazole • Abilify
- Clonazepam • Klonopin
- Donepezil • Aricept
- Galantamine • Razadyne, Reminyl
- Levodopa • Dopar, Larodopa
- Memantine • Namenda
- Quetiapine • Seroquel
- Rivastigmine • Exelon
Disclosure
Drs. Bishnoi and Manepalli report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Grossberg serves as a consultant to Forest, Janssen, Novartis, and Pfizer. His department receives research funding from Novartis, Janssen, and Pfizer.
1. McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47(5):1113-1124.
2. Buracchio T, Arvanitakis Z, Gorbien M. Dementia with Lewy bodies: current concepts. Dement Geriatr Cogn Disord. 2005;20(5):306-320.
3. Fujishiro H, Iseki E, Higashi S, et al. Distribution of cerebral amyloid deposition and its relevance to clinical phenotype in Lewy body dementia. Neurosci Lett. 2010;486(1):19-23.
4. Kosaka K. Diffuse Lewy body disease. Neuropathology. 2000;20(suppl):S73-S78.
5. McKeith IG, Dickson DW, Lowe J, et al. Consortium on DLB. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863-1872.
6. Cummings JL, Cole G. Alzheimer disease. JAMA. 2002;287(18):2335-2338.
7. Zaccai J, McCracken C, Brayne C. A systematic review of prevalence and incidence studies of dementia with Lewy bodies. Age Ageing. 2005;34(6):561-566.
8. Bradshaw J, Saling M, Hopwood M, et al. Fluctuating cognition in dementia with Lewy bodies and Alzheimer’s disease is qualitatively distinct. J Neurol Neurosurg Psychiatry. 2004;75(3):382-387.
9. Singleton AB, Wharton A, O’Brien KK, et al. Clinical and neuropathological correlates of apolipoprotein E genotype in dementia with Lewy bodies. Dement Geriatr Cogn Disord. 2002;14(4):167-175.
10. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263-269.
11. Jack CR, Jr, Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):257-262.
12. Mollenhauer B, Cullen V, Kahn I, et al. Direct quantification of CSF alpha-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp Neurol. 2008;213(2):315-325.
13. Parnetti L, Balducci C, Pierguidi L, et al. Cerebrospinal fluid beta-glucocerebrosidase activity is reduced in dementia with Lewy bodies. Neurobiol Dis. 2009;34(3):484-486.
14. Shimada H, Hirano S, Shinotoh H, et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology. 2009;73(4):273-278.
15. McKeith I, O’Brien J, Walker Z, et al. Sensitivity and specificity of dopamine transporter imaging with 123I-FP-CIT SPECT in dementia with Lewy bodies: a phase III, multicentre study. Lancet Neurol. 2007;6(4):305-313.
16. Walker Z, Jaros E, Walker RW, et al. Dementia with Lewy bodies: a comparison of clinical diagnosis, FP-CIT single photon emission computed tomography imaging and autopsy. J Neurol Neurosurg Psychiatry. 2007;78(11):1176-1181.
17. Bradshaw JM, Saling M, Anderson V, et al. Higher cortical deficits influence attentional processing in dementia with Lewy bodies, relative to patients with dementia of the Alzheimer’s type and controls. J Neurol Neurosurg Psychiatry. 2006;77(10):1129-1135.
18. Weiner MF, Hynan LS, Parikh B, et al. Can Alzheimer’s disease and dementias with Lewy bodies be distinguished clinically? J Geriatr Psychiatry Neurol. 2003;16(4):245-250.
19. Stavitsky K, Brickman AM, Scarmeas N, et al. The progression of cognition, psychiatric symptoms, and functional abilities in dementia with Lewy bodies and Alzheimer disease. Arch Neurol. 2006;63(10):1450-1456.
20. Ferman TJ, Smith GE, Boeve BF, et al. Neuropsychological differentiation of dementia with Lewy bodies from normal aging and Alzheimer’s disease. Clin Neuropsychol. 2006;20(4):623-636.
21. McKeith IG, Perry EK, Perry RH. Report of the second dementia with Lewy body international workshop: diagnosis and treatment. Consortium on Dementia with Lewy Bodies. Neurology. 1999;53(5):902-905.
22. Boeve BF, Silber MH, Ferman TJ, et al. Association of REM sleep behavior disorder and neurodegenerative disease may reflect an underlying synucleinopathy. Mov Disord. 2001;16(4):622-630.
23. Portet F, Scarmeas N, Cosentino S, et al. Extrapyramidal signs before and after diagnosis of incident Alzheimer disease in a prospective population study. Arch Neurol. 2009;66(9):1120-1126.
24. McKeith I, Fairbairn A, Perry R, et al. Neuroleptic sensitivity in patients with senile dementia of Lewy body type. BMJ. 1992;305(6855):673-678.
25. Tarawneh R, Galvin JE. Distinguishing Lewy body dementias from Alzheimer’s disease. Expert Rev Neurother. 2007;7(11):1499-1516.
26. Ancoli-Israel S, Klauber MR, Gillin JC, et al. Sleep in non-institutionalized Alzheimer’s disease patients. Aging (Milano). 1994;6(6):451-458.
27. Ferman TJ, Smith GE, Boeve BF, et al. DLB fluctuations: specific features that reliably differentiate DLB from AD and normal aging. Neurology. 2004;62(2):181-187.
28. Salmon DP, Galasko D, Hansen LA, et al. Neuropsychological deficits associated with diffuse Lewy body disease. Brain Cogn. 1996;31(2):148-165.
29. Jack CR, Jr, Petersen RC, Xu Y, et al. Rates of hippocampal atrophy correlate with change in clinical status in aging and AD. Neurology. 2000;55(4):484-489.
30. Burton EJ, Barber R, Mukaetova-Ladinska EB, et al. Medial temporal lobe atrophy on MRI differentiates Alzheimer’s disease from dementia with Lewy bodies and vascular cognitive impairment: a prospective study with pathological verification of diagnosis. Brain. 2009;132(pt 1):195-203.
31. Ishii K, Soma T, Kono AK, et al. Comparison of regional brain volume and glucose metabolism between patients with mild dementia with lewy bodies and those with mild Alzheimer’s disease. J Nucl Med. 2007;48(5):704-711.
32. Klein JC, Eggers C, Kalbe E, et al. Neurotransmitter changes in dementia with Lewy bodies and Parkinson disease dementia in vivo. Neurology. 2010;74(11):885-892.
33. Fujishiro H, Nakamura S, Kitazawa M, et al. Early detection of dementia with Lewy bodies in patients with amnestic mild cognitive impairment using 123I-MIBG cardiac scintigraphy. J Neurol Sci. 2012;315(1-2):115-119.
34. O’Brien JT, McKeith IG, Walker Z, et al. Diagnostic accuracy of 123I-FP-CIT SPECT in possible dementia with Lewy bodies. Br J Psychiatry. 2009;194:34-39.
35. Yoshita M, Taki J, Yokoyama K, et al. Value of 123I-MIBG radioactivity in the differential diagnosis of DLB from AD. Neurology. 2006;66(12):1850-1854.
1. McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47(5):1113-1124.
2. Buracchio T, Arvanitakis Z, Gorbien M. Dementia with Lewy bodies: current concepts. Dement Geriatr Cogn Disord. 2005;20(5):306-320.
3. Fujishiro H, Iseki E, Higashi S, et al. Distribution of cerebral amyloid deposition and its relevance to clinical phenotype in Lewy body dementia. Neurosci Lett. 2010;486(1):19-23.
4. Kosaka K. Diffuse Lewy body disease. Neuropathology. 2000;20(suppl):S73-S78.
5. McKeith IG, Dickson DW, Lowe J, et al. Consortium on DLB. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863-1872.
6. Cummings JL, Cole G. Alzheimer disease. JAMA. 2002;287(18):2335-2338.
7. Zaccai J, McCracken C, Brayne C. A systematic review of prevalence and incidence studies of dementia with Lewy bodies. Age Ageing. 2005;34(6):561-566.
8. Bradshaw J, Saling M, Hopwood M, et al. Fluctuating cognition in dementia with Lewy bodies and Alzheimer’s disease is qualitatively distinct. J Neurol Neurosurg Psychiatry. 2004;75(3):382-387.
9. Singleton AB, Wharton A, O’Brien KK, et al. Clinical and neuropathological correlates of apolipoprotein E genotype in dementia with Lewy bodies. Dement Geriatr Cogn Disord. 2002;14(4):167-175.
10. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263-269.
11. Jack CR, Jr, Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):257-262.
12. Mollenhauer B, Cullen V, Kahn I, et al. Direct quantification of CSF alpha-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp Neurol. 2008;213(2):315-325.
13. Parnetti L, Balducci C, Pierguidi L, et al. Cerebrospinal fluid beta-glucocerebrosidase activity is reduced in dementia with Lewy bodies. Neurobiol Dis. 2009;34(3):484-486.
14. Shimada H, Hirano S, Shinotoh H, et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology. 2009;73(4):273-278.
15. McKeith I, O’Brien J, Walker Z, et al. Sensitivity and specificity of dopamine transporter imaging with 123I-FP-CIT SPECT in dementia with Lewy bodies: a phase III, multicentre study. Lancet Neurol. 2007;6(4):305-313.
16. Walker Z, Jaros E, Walker RW, et al. Dementia with Lewy bodies: a comparison of clinical diagnosis, FP-CIT single photon emission computed tomography imaging and autopsy. J Neurol Neurosurg Psychiatry. 2007;78(11):1176-1181.
17. Bradshaw JM, Saling M, Anderson V, et al. Higher cortical deficits influence attentional processing in dementia with Lewy bodies, relative to patients with dementia of the Alzheimer’s type and controls. J Neurol Neurosurg Psychiatry. 2006;77(10):1129-1135.
18. Weiner MF, Hynan LS, Parikh B, et al. Can Alzheimer’s disease and dementias with Lewy bodies be distinguished clinically? J Geriatr Psychiatry Neurol. 2003;16(4):245-250.
19. Stavitsky K, Brickman AM, Scarmeas N, et al. The progression of cognition, psychiatric symptoms, and functional abilities in dementia with Lewy bodies and Alzheimer disease. Arch Neurol. 2006;63(10):1450-1456.
20. Ferman TJ, Smith GE, Boeve BF, et al. Neuropsychological differentiation of dementia with Lewy bodies from normal aging and Alzheimer’s disease. Clin Neuropsychol. 2006;20(4):623-636.
21. McKeith IG, Perry EK, Perry RH. Report of the second dementia with Lewy body international workshop: diagnosis and treatment. Consortium on Dementia with Lewy Bodies. Neurology. 1999;53(5):902-905.
22. Boeve BF, Silber MH, Ferman TJ, et al. Association of REM sleep behavior disorder and neurodegenerative disease may reflect an underlying synucleinopathy. Mov Disord. 2001;16(4):622-630.
23. Portet F, Scarmeas N, Cosentino S, et al. Extrapyramidal signs before and after diagnosis of incident Alzheimer disease in a prospective population study. Arch Neurol. 2009;66(9):1120-1126.
24. McKeith I, Fairbairn A, Perry R, et al. Neuroleptic sensitivity in patients with senile dementia of Lewy body type. BMJ. 1992;305(6855):673-678.
25. Tarawneh R, Galvin JE. Distinguishing Lewy body dementias from Alzheimer’s disease. Expert Rev Neurother. 2007;7(11):1499-1516.
26. Ancoli-Israel S, Klauber MR, Gillin JC, et al. Sleep in non-institutionalized Alzheimer’s disease patients. Aging (Milano). 1994;6(6):451-458.
27. Ferman TJ, Smith GE, Boeve BF, et al. DLB fluctuations: specific features that reliably differentiate DLB from AD and normal aging. Neurology. 2004;62(2):181-187.
28. Salmon DP, Galasko D, Hansen LA, et al. Neuropsychological deficits associated with diffuse Lewy body disease. Brain Cogn. 1996;31(2):148-165.
29. Jack CR, Jr, Petersen RC, Xu Y, et al. Rates of hippocampal atrophy correlate with change in clinical status in aging and AD. Neurology. 2000;55(4):484-489.
30. Burton EJ, Barber R, Mukaetova-Ladinska EB, et al. Medial temporal lobe atrophy on MRI differentiates Alzheimer’s disease from dementia with Lewy bodies and vascular cognitive impairment: a prospective study with pathological verification of diagnosis. Brain. 2009;132(pt 1):195-203.
31. Ishii K, Soma T, Kono AK, et al. Comparison of regional brain volume and glucose metabolism between patients with mild dementia with lewy bodies and those with mild Alzheimer’s disease. J Nucl Med. 2007;48(5):704-711.
32. Klein JC, Eggers C, Kalbe E, et al. Neurotransmitter changes in dementia with Lewy bodies and Parkinson disease dementia in vivo. Neurology. 2010;74(11):885-892.
33. Fujishiro H, Nakamura S, Kitazawa M, et al. Early detection of dementia with Lewy bodies in patients with amnestic mild cognitive impairment using 123I-MIBG cardiac scintigraphy. J Neurol Sci. 2012;315(1-2):115-119.
34. O’Brien JT, McKeith IG, Walker Z, et al. Diagnostic accuracy of 123I-FP-CIT SPECT in possible dementia with Lewy bodies. Br J Psychiatry. 2009;194:34-39.
35. Yoshita M, Taki J, Yokoyama K, et al. Value of 123I-MIBG radioactivity in the differential diagnosis of DLB from AD. Neurology. 2006;66(12):1850-1854.
Why do cancer patients smoke and what can providers do about it?
Despite the widespread dissemination of information about the health risks associated with smoking, many cancer patients continue to smoke, which results in a decreased quality of life, an increased probability of cancer recurrence, and a decreased survival time. Efficacious interventions are available to assist cancer patients to quit smoking, yet smoking cessation interventions are often not implemented. This review describes how clinicians, administrators, insurers, and purchasers can encourage a culture of health care in which tobacco cessation interventions are implemented consistent with evidence-based standards of care. Implementing efficacious tobacco cessation interventions can reduce morbidity and mortality among cancer patients...
*Click on the link to the left of this introduction for a PDF of the full article.
Despite the widespread dissemination of information about the health risks associated with smoking, many cancer patients continue to smoke, which results in a decreased quality of life, an increased probability of cancer recurrence, and a decreased survival time. Efficacious interventions are available to assist cancer patients to quit smoking, yet smoking cessation interventions are often not implemented. This review describes how clinicians, administrators, insurers, and purchasers can encourage a culture of health care in which tobacco cessation interventions are implemented consistent with evidence-based standards of care. Implementing efficacious tobacco cessation interventions can reduce morbidity and mortality among cancer patients...
*Click on the link to the left of this introduction for a PDF of the full article.
Despite the widespread dissemination of information about the health risks associated with smoking, many cancer patients continue to smoke, which results in a decreased quality of life, an increased probability of cancer recurrence, and a decreased survival time. Efficacious interventions are available to assist cancer patients to quit smoking, yet smoking cessation interventions are often not implemented. This review describes how clinicians, administrators, insurers, and purchasers can encourage a culture of health care in which tobacco cessation interventions are implemented consistent with evidence-based standards of care. Implementing efficacious tobacco cessation interventions can reduce morbidity and mortality among cancer patients...
*Click on the link to the left of this introduction for a PDF of the full article.
Dasatinib in the first-line treatment of chronic myeloid leukemia
Dasatinib has been approved for first-line treatment of chronic-phase chronic myeloid leukemia by the Food and Drug Administration and is recommended as a first-line treatment option by the National Comprehensive Cancer Network. Based on in vitro data, dasatinib seems to be less susceptible to the resistance mechanisms that affect imatinib. Dasatinib is an effective second-line treatment in patients who are resistant to imatinib. First-line clinical data show that dasatinib provides more rapid and deeper degrees of response than does imatinib, which may correlate with improvements in long-term patient outcome. Grade 1 or 2 cytopenias are the most common adverse events of first-line dasatinib treatment. In a phase 3 comparison with imatinib, several types of nonhematologic adverse events were less frequent in the dasatinib arm; frequencies of grade 3 and 4 events were 2%. Among patients with a minimum follow-up of 24 months, grade 1 or 2 pleural effusion was reported in 14% of dasatinib-treated patients and was manageable in almost all cases; no grade 3 or 4 pleural effusion occurred. Prompt and effective monitoring and management of dasatinib toxicities is essential to minimize intolerance and nonadherence to therapy. Patient education is important to increase the likelihood of prompt management and provide reassurance. Recommendations for patient monitoring, management, and education are provided.
*For a PDF of the full article, click on the link to the left of this introduction.
Dasatinib has been approved for first-line treatment of chronic-phase chronic myeloid leukemia by the Food and Drug Administration and is recommended as a first-line treatment option by the National Comprehensive Cancer Network. Based on in vitro data, dasatinib seems to be less susceptible to the resistance mechanisms that affect imatinib. Dasatinib is an effective second-line treatment in patients who are resistant to imatinib. First-line clinical data show that dasatinib provides more rapid and deeper degrees of response than does imatinib, which may correlate with improvements in long-term patient outcome. Grade 1 or 2 cytopenias are the most common adverse events of first-line dasatinib treatment. In a phase 3 comparison with imatinib, several types of nonhematologic adverse events were less frequent in the dasatinib arm; frequencies of grade 3 and 4 events were 2%. Among patients with a minimum follow-up of 24 months, grade 1 or 2 pleural effusion was reported in 14% of dasatinib-treated patients and was manageable in almost all cases; no grade 3 or 4 pleural effusion occurred. Prompt and effective monitoring and management of dasatinib toxicities is essential to minimize intolerance and nonadherence to therapy. Patient education is important to increase the likelihood of prompt management and provide reassurance. Recommendations for patient monitoring, management, and education are provided.
*For a PDF of the full article, click on the link to the left of this introduction.
Dasatinib has been approved for first-line treatment of chronic-phase chronic myeloid leukemia by the Food and Drug Administration and is recommended as a first-line treatment option by the National Comprehensive Cancer Network. Based on in vitro data, dasatinib seems to be less susceptible to the resistance mechanisms that affect imatinib. Dasatinib is an effective second-line treatment in patients who are resistant to imatinib. First-line clinical data show that dasatinib provides more rapid and deeper degrees of response than does imatinib, which may correlate with improvements in long-term patient outcome. Grade 1 or 2 cytopenias are the most common adverse events of first-line dasatinib treatment. In a phase 3 comparison with imatinib, several types of nonhematologic adverse events were less frequent in the dasatinib arm; frequencies of grade 3 and 4 events were 2%. Among patients with a minimum follow-up of 24 months, grade 1 or 2 pleural effusion was reported in 14% of dasatinib-treated patients and was manageable in almost all cases; no grade 3 or 4 pleural effusion occurred. Prompt and effective monitoring and management of dasatinib toxicities is essential to minimize intolerance and nonadherence to therapy. Patient education is important to increase the likelihood of prompt management and provide reassurance. Recommendations for patient monitoring, management, and education are provided.
*For a PDF of the full article, click on the link to the left of this introduction.
Nutrition
Malnutrition is present in 20% to 50% of hospitalized patients.1, 2 Despite simple, validated screening tools, malnutrition tends to be underdiagnosed.3, 4 Over 90% of elderly patients transitioning from an acute care hospital to a subacute care facility are either malnourished or at risk of malnutrition.5 Malnutrition has been associated with increased risk of nosocomial infections,6 worsened discharge functional status,7 and higher mortality,8 as well as longer lengths of stay7, 8 and higher hospital costs.2
Malnutrition describes either overnutrition or undernutrition that causes a change in body composition and decreased function.9 Malnutrition in hospitalized patients is typically related to undernutrition due to either reduced intake or increased metabolic rate. Reasons for reduced intake include poor appetite, reduced ability to chew or swallow, and nil per os (NPO) status. Patients with acute or chronic illnesses may either be malnourished on admission, or develop malnutrition within a few days of hospital admission, due to the effects of the inflammatory state on metabolism. Given that malnutrition is potentially modifiable, it is important to screen for malnutrition and, when present, develop, implement, and monitor a nutrition care plan10 (Figure 1).
The purpose of this review is to provide the hospitalist with an overview of screening, assessment, and development and implementation of a nutrition care plan in the acutely ill hospitalized patient.
PATIENT SCREENING
Nutrition screening identifies patients with nutritional deficits who may benefit from further detailed nutrition assessment and intervention.11 The Joint Commission requires that all patients admitted to acute care hospitals be screened for risk of malnutrition within 24 hours.12 Those considered at risk for malnutrition have significant weight changes, chronic disease or an acute inflammatory process, or have been unable to ingest adequate calories for 7 days.13
Those not at risk should be regularly rescreened throughout their hospital stay. The American Society of Parenteral and Enteral Nutrition (ASPEN) recommends that institutions create and approve a screening process according to the patient population served.10 There are several tools validated for use in the acute care setting.14 Many institutions trigger an automatic nutrition consult when certain screening criteria are met.
PATIENT ASSESSMENT
Nutrition assessment should be performed by a dietitian or nutrition consult provider in patients who screen at risk for malnutrition to characterize and determine the cause of nutritional deficits.10 The nutrition assessment identifies history and physical examination elements to diagnose malnutrition. An ASPEN consensus statement recommends the diagnosis of malnutrition if 2 or more of the following are present: insufficient energy intake, weight loss, loss of muscle mass, loss of subcutaneous fat, localized or generalized fluid accumulation, and decreased functional status measured by hand‐grip strength.9 The nutrition assessment should also consider how long the patient has been without adequate nutrition, document baseline nutrition parameters,15 and estimate caloric requirements to determine nutrition support therapy needs.10 Nutrition assessment typically includes the following components.
History
A careful history elicits the majority of information needed to determine the cause and severity of malnutrition.16 Patients should be questioned about a typical day's oral intake prior to hospitalization, and about factors that affect their intake such as sensory deficits, fine motor dysfunction, or chewing and swallowing difficulties, which often decline in chronically ill and elderly patients. Nutrition may be affected by financial difficulties or limited social support, and access to food should be assessed.
Physical Findings
Weight loss is the best physical exam predictor of malnutrition risk, although nutritional depletion can occur in a very short time in acutely ill or injured patients before substantial weight loss has occurred. The likelihood of malnutrition is increased if a patient has: a body mass index (BMI) <18.5 kg/m2; unintentional loss of >2.3 kg (5 lb) or 5% of body weight over 1 month; and unintentional loss of >4.5 kg (10 lb) or 10% of body weight over 6 months.17 Weight loss may be masked by fluid retention from chronic conditions, such as heart failure, or from volume resuscitation in the acutely ill patient.9, 16
Body mass index can be misleading, as age‐related height loss may artificially increase BMI, and height may be difficult to accurately measure in a kyphotic, unsteady, or bedridden patient. The clinician may find evidence of loss of subcutaneous fat or muscle mass in patients with chronic illness, but these findings may not be evident in the acutely ill patient.9 Other physical exam assessments of malnutrition, such as arm span, skinfold thickness, and arm circumference are not reliable.16
Laboratory Tests
Biochemical markers, including transferrin, albumin, and prealbumin, have not been proven as accurate predictors of nutrition status because they may change as a result of other factors not related to nutrition.15, 18 Serum albumin, for example, may be more reflective of the degree of metabolic stress.19 Prealbumin has a serum half‐life much shorter than albumin or transferrin (approximately 2448 hours) and is perhaps the most useful protein marker to assess the adequacy of nutritional replacement after the inflammatory state is resolved.18
Calculating Caloric Requirements
Energy expenditure measurement is considered the gold standard to determine patients' caloric needs. Actual measurement by methods such as indirect calorimetry, which measures oxygen consumption and carbon dioxide production, and calculates energy expenditure, is challenging in everyday clinical settings. Predictive equations often are used as alternative methods to estimate patients' caloric requirements.20 There is no consensus among the 3 North American societies' guidelines (the Canadian Clinical Practice Guidelines; the American Dietetics Association's evidence‐based guideline for critical illness; and the Society of Critical Care Medicine and American Society of Parenteral and Enteral Nutrition's joint guideline) as to the best method.21
In the simplest equation, caloric needs are estimated by calories per kilogram.22 In obese patients, using actual body weight will overestimate needs, but using ideal body weight may cause underfeeding. A small study comparing predictive equations in obese hospitalized patients found the Harris‐Benedict equations (H‐BE) using adjusted body weight and a stress factor to be most accurate, but only in 50% of patients.23 Most clinicians are familiar with the H‐BE, but alternatives such as calories per kilogram or the Mifflin St.‐Jeor equation24 are often used (S. Brantley (May 5, 2012), S. Lundy (May 23, 2012), personal communication).
Indications for Nutritional Intervention
In adults without preexisting malnutrition, inadequate nutritional intake for approximately 714 days should prompt nutritional intervention.25, 26 This timeline should be shorter (37 days) in those with lower energy reserves (eg, underweight or recent weight loss) or significant catabolic stress (eg, acutely ill patients).27, 28 Other patient populations shown to benefit from nutritional intervention include: postoperative patients who are anticipated to be NPO for more than 7 days or to be taking less than 60% of estimated caloric needs by postoperative day 10; preoperative patients with severe malnutrition29; those with gastrointestinal cancer undergoing elective surgery30; and stroke patients with persistent dysphagia for more than 7 days.31
DEVELOPMENT OF A NUTRITION CARE PLAN
The formal nutrition assessment of the at‐risk patient derives the information needed for the development of a nutrition care plan. This plan guides the provision of nutrition therapy, the intervention, the monitoring protocols, evaluation, and reassessment of nutrition goals or termination of specialized nutrition support.10 Assessments for adequacy of nutritional repletion are best done by repeated screening and physical examinations.18
IMPLEMENTATION OF NUTRITION CARE PLAN
Nutritional interventions include dietary modifications, enteral nutrition, and parenteral nutrition.
Dietary Modifications
The purpose of the diet is to provide the necessary nutrients to the body in a well‐tolerated form. Diets can be modified to provide for individual requirements, personal eating patterns and food preferences, and disease process and digestive capacity. Dietary adjustments include change in consistency of foods (eg, pureed, mechanical soft), increase or decrease in energy value, increase or decrease in the type of food or nutrient consumed (eg, sodium restriction, fiber enhancement), elimination of specific foods (eg, gluten‐free diet), adjustment in protein, fat, and carbohydrate content (eg, ketogenic diet, renal diet, cholesterol‐lowering diet), and adjustment of the number and frequency of meals.32
Dietary supplementation (eg, Boost, Ensure) is common practice in persons diagnosed with such conditions as cancer, diabetes, and cardiovascular disease. Supplements enhance the diet by increasing the total daily intake of a vitamin, a mineral, an amino acid, an herb or other botanical33, and should not be used as a meal substitute.34 These supplements are varied in content of calories, protein, vitamins, and minerals. Various flavors and consistencies are also available. Several oral supplements are reviewed in Table 1.
| Oral Supplement* (Serving Size; mL) | Kcal/svg | Protein (g/svg) | Fat (g/svg) | CHO (g/svg) | Na (mg/svg) | K (mg/svg) | Ca (mg/svg) | Phos (mg/svg) | Mg (mg/svg) |
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| Boost Original (237) | 240 | 10 | 4 | 41 | 150 | 460 | 300 | 300 | 100 |
| Ensure Nutrition Shake (237) | 250 | 9 | 6 | 40 | 200 | 370 | 300 | 250 | 100 |
| Carnation Instant Breakfast Ready to Drink (325) | 250 | 14 | 5 | 34 | 180 | 330 | 500 | 500 | 120 |
| Resource Breeze (fruit‐flavored) clear liquid (237) | 250 | 9 | 0 | 54 | 80 | 10 | 10 | 150 | 1 |
| Glucerna 1.0 Ready to Drink low‐CHO (237) | 240 | 10 | 13 | 23 | 220 | 370 | 170 | 170 | 67 |
| Re/Gen low K and Phos (180) | 375 | 12 | 17 | 47 | 180 | 23 | 15 | 68 | 3 |
Enteral Nutrition
Enteral nutrition (EN) support should be provided to patients who have functioning gastrointestinal (GI) tracts but are unable to take adequate calories orally. Compared to parenteral nutrition (PN), EN is associated with favorable improvements in inflammatory cytokines, acute phase proteins, hyperglycemia, insulin resistance, nosocomial infections, mortality, and cost.35 Enteral feeds are more physiologic than parenteral feeds, maintain GI structure and integrity, and avoid intravenous (IV) access complications. Patients with normal nutritional status on admission who require EN should be receiving over 50% of their caloric needs within the first week of hospital stay.25 Malnourished patients should reach this minimum goal within 35 days of admission.27, 28 EN is not contraindicated in the absence of bowel sounds or in the presence of increased gastric residuals.35 Withholding enteral feedings for gastric residual volumes <250 mL36, 37 or reduced bowel sounds can result in inadequate caloric intake or inappropriate use of PN.27
Gastric feedings are more physiologic than small bowel feedings, can be given by bolus or continuous infusion, and can be given by tubes that are easy to place at the bedside. Post‐pyloric feedings (nasoduodenal or nasojejunal) may be associated with a lower risk of pneumonia, and should be considered in high‐risk patients such as those receiving continuous sedatives or neuromuscular blockers.36 Post‐pyloric tube placement usually requires endoscopy, fluoroscopy, or electromagnetic guidance. Percutaneous feeding tubes (gastrostomy or jejunostomy) should be considered in those who require tube feedings for longer than 30 days.38
Assessment of patient requirements and disease state, as well as extensive knowledge of available formulas, is important in the selection of the appropriate enteral formula.39 Standardized formulas are used for most patients. The provision of adequate water must be considered with these formulas, particularly in the long‐term care and home settings.40 Many specialized formulas are designed for a particular disease state or condition, some of which are further reviewed in Table 2.
| Formula | Kcal/mL | Protein (g/L) | Fat (g/L) | CHO (g/L) | Osmolality (mOsm/kg H2O) | Na (mEq/L) | K (mEq/L) | Ca (mg/L) | Mg (mg/L) | Phos (mg/L) |
|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||
| Nutren 1.0‐low residue | 1 | 40 | 38 | 127 | 315 | 38 | 32 | 668 | 268 | 668 |
| Osmolite 1.0 Cal low residue | 1 | 44.3 | 34.7 | 143.9 | 300 | 40.4 | 40.2 | 760 | 305 | 1760 |
| Replete high protein, low residue | 1 | 62.4 | 34 | 112 | 300 | 38.1 | 38.5 | 1000 | 400 | 1000 |
| Replete Fiber high protein with fiber | 1 | 62.4 | 34 | 112 | 310 | 38.1 | 38.5 | 1000 | 400 | 1000 |
| Osmolite 1.5 low residue, calorically dense | 1.5 | 62.7 | 49.1 | 203.6 | 525 | 60.9 | 46 | 1000 | 400 | 1000 |
| Two Cal calorie and protein dense | 2 | 83.5 | 91 | 219 | 725 | 64 | 63 | 1050 | 425 | 1050 |
| Vivonex RTF‐elemental | 1 | 50 | 11.6 | 176 | 630 | 30.4 | 31 | 668 | 268 | 668 |
| Nepro with Carb Steady‐for electrolyte, fluid restriction (eg, dialysis) | 1.8 | 81 | 96 | 161 | 745 | 46 | 27 | 1060 | 210 | 720 |
| Nutren Glytrol low CHO | 1 | 45.2 | 47.6 | 100 | 280 | 32.2 | 35.9 | 720 | 286 | 720 |
| NutriHep‐for hepatic disease | 1.5 | 40 | 21.2 | 290 | 790 | 160 | 33.9 | 956 | 376 | 1000 |
If concerned about formula tolerance, one solution is to initiate the formula at a low rate and increase to the goal rate over 2448 hours. Dilution of enteral formulas is not necessary to assure optimal tolerance. Continuous feedings are recommended for most patients initially and after tolerance has been established, bolus feedings can be attempted if the feeding tube terminates in the stomach. Bolus feedings, where 240480 mL of formula are delivered through a syringe over 1015 minutes, may be more physiological for patients. This regimen can be repeated 46 times daily to meet nutrition goals.41
Parenteral Nutrition
PN provides macronutrients such as carbohydrates, protein, and fat; micronutrients such as vitamins, minerals, electrolytes, and trace elements are added in appropriate concentrations. PN may also provide the patient's daily fluid needs. The timing of PN initiation depends upon the patient's initial nutritional status. ASPEN does not recommend PN during the first 7 days of hospitalization in critically ill patients with normal nutritional status. If the patient is not receiving 100% of caloric needs from EN after 7 days, supplemental PN should be considered. However, if on admission a patient is already malnourished and EN is not feasible, PN should be initiated and continued until the patient is receiving at least 60% of caloric needs by enteral route.42 This includes patients with intestinal obstruction, ileus, peritonitis, malabsorption, high output enterocutaneous fistulae, intestinal ischemia, intractable vomiting and diarrhea, severe shock, and fulminant sepsis.10, 43
Standardized commercial PN products are available and reduce the number of steps required between ordering and administration, as compared to customized PN, which is compounded for a particular patient. However, despite improved efficiency and lower cost, there is no evidence that standardized preparations are safer to patients than customized solutions. Institutions utilizing standardized PN must also have a mechanism to customize formulas for those with complex needs.44
Creating a customized parenteral solution involves several basic steps. Total caloric requirement may be estimated using a predictive formula, as previously discussed; calories/kg of ideal body weight is the simplest method. Most hospitalized patients require 2030 calories/kg/d. Daily fluid requirement may be based on kilocalories (kcal) delivered, or by ideal body weight (eg, 1 mL/kcal or 3040 mL/kg). More fluid may be needed in patients with significant sensible or insensible losses; those with renal failure or heart failure should receive less fluid.
Protein needs are calculated by multiplying ideal body weight (kg) by estimated protein needs in g/kg/d (1.22 g/kg/d for catabolic patients). Protein should provide approximately 20% of total calories. Protein restriction is not required in renal impairment; acutely ill patients on renal replacement therapy should receive 1.51.8 g/kg/d. In hepatic failure patients, protein should be restricted only if hepatic encephalopathy fails to improve with other measures.
Knowing the protein, kcal, and fluid needs of the patient, the practitioner divides the remaining non‐protein calories between carbohydrates and fat. Approximately 70%85% of non‐protein calories should be provided as carbohydrates (dextrose), up to 7 g/kg/d. The other 15%30% are as fat, in lipid solutions, providing a maximum of 2.5 g/kg/d. Lipid solutions are provided as either 10% (1.1 kcal/mL) or 20% (2.2 kcal/mL) concentrations.43, 44 Propofol's contribution to fat intake complicates estimating total fat intake in critically ill patients.45
Standardized parenteral multivitamin preparations are available; the clinician must determine if preparations containing vitamin K are appropriate. Of the trace elements, copper and manganese should be restricted in hepatobiliary disease.44
Acutely ill patients receive PN as a 24‐hour infusion, to minimize its impact on volume status and energy expenditure,46 providing 50% of needs on infusion day one and reaching goal within 4872 hours, rather than cyclic infusions over shorter intervals. Daily assessments of vital signs, intake and output, and weight are necessary to monitor volume status.
Once a patient is taking at least 60% of caloric needs either by mouth or by EN, PN can be discontinued. Tapering the infusion is not required, as abrupt discontinuation has not been demonstrated to cause symptomatic hypoglycemia.47, 48
PATIENT MONITORING
Laboratory monitoring with nutrition support should include baseline electrolytes, glucose, renal function, coagulation studies, triglycerides, magnesium, phosphorus, cholesterol, platelet count, and hepatobiliary enzymes. Electrolytes, calcium, magnesium, and phosphorus should be checked daily for 3 days and, if normal, should then be checked biweekly. Capillary glucose should be monitored several times a day until stable. Weekly triglycerides, albumin, cholesterol, coagulation studies, and liver enzymes should also be checked in patients while on parenteral nutrition.25 Patients at risk for refeeding syndrome should have potassium, phosphate, calcium, and magnesium measured daily for 7 days, with repletion as necessary. These electrolytes should be monitored 3 times the following week if stable.49
Patients should be monitored clinically for gastrointestinal tolerance of enteral nutrition. All 3 North American guidelines recommend monitoring gastric residual volumes (GRV); however, there is no consensus on the volume considered to require intervention. Motility agents are recommended as first line treatment of high GRV.36, 37, 42 If high GRV continues, tube feeding should be held, and tube placement, medications, and metabolic assessment should be reviewed. Placement of a transpyloric feeding tube may be indicated.50
Adverse Effects and Complications of Nutrition Support
Regarding EN, complications include those related to tube placement and maintenance, infections, and medical complications of the feeds themselves. Some of the adverse effects of the enteral formulas may be attenuated. Diarrhea, which occurs in up to 20% of patients, may be avoided with slow feed advancement, use of low‐osmotic formulas, or fiber additives.51 Gastric distention and abdominal pain may improve with slow feed advancement and continuous (rather than bolus) feeds. Small‐bore tubes and acid‐reducing medications may decrease gastroesophageal reflux, and aspiration pneumonia may be avoided by semi‐recumbent positioning and post‐pyloric feeding.52
Complications of PN may be grouped as mechanical, infectious, and metabolic. The mechanical complications of central line placement include pneumothorax, arterial puncture, hematoma, air embolism, and line malpositioning. Catheter‐related deep venous thrombosis may occur. Patients on PN through a central line are at risk for central line‐associated bloodstream infections.25 The metabolic complications such as hyperglycemia, electrolyte disorders, hepatic steatosis, and volume overload may have severe consequences, such as heart failure or neuromuscular dysfunction, thus they require close attention.53
A complication of nutrition support that may occur regardless of route is the refeeding syndrome. Refeeding syndrome describes fluid shifts and electrolyte abnormalities that occur after initiation of oral, enteral, or parenteral nutrition in a malnourished or starved patient.54, 55 There are no formal criteria for diagnosing refeeding syndrome.
In the starved state, the body switches from carbohydrate to protein and fat metabolism. Reintroduction of carbohydrates stimulates insulin release with glycogen, fat, and protein synthesis. Associated uptake of glucose, potassium, magnesium, phosphate, and water into cells causes electrolyte and fluid abnormalities. Although hypophosphatemia is the hallmark of refeeding syndrome, it is not pathognomonic. Additional disturbances include hypokalemia, hyperglycemia, hypomagnesemia, thiamine deficiency, and fluid imbalance.49 Patients at risk of refeeding should have serum electrolytes, magnesium, phosphorus, and glucose checked before nutrition support starts. The degree of laboratory abnormalities, if any, and the clinical course of refeeding guides the frequency of subsequent blood tests.56 These consequences of refeeding can adversely affect every major organ system and may result in death.57
Starvation physiology underlies all risk factors for refeeding syndrome. In hospitalized patients, those at risk for refeeding include, but are not limited to, the elderly, oncology patients, postoperative patients, alcohol‐dependent patients, those with malabsorptive states, those who are fasting or chronically malnourished, and those on diuretic therapy.54, 57 The National Institute for Health and Clinical Excellence (NICE) of England and Wales has published criteria to identify patients at high risk for refeeding (Table 3).56 Identification of at‐risk patients and attention to their nutritional needs prevents refeeding syndrome.
|
| Patient has 1 or more of the following: |
| BMI <16 kg/m2 |
| Unintentional weight loss >15% within the last 36 mo |
| Little or no nutritional intake for more than 10 d |
| Low levels of potassium, phosphate, or magnesium prior to feeding |
| Or patient has 2 or more of the following: |
| BMI <18.5 kg/m2 |
| Unintentional weight loss >10% within the last 36 mo |
| Little or no nutritional intake for more than 5 d |
| A history of alcohol abuse or drugs including insulin, chemotherapy, antacids, or diuretics |
ASPEN and NICE have each issued guidelines for initiating nutrition support in patients at risk for refeeding. ASPEN guidelines recommend feeding start at approximately 25% of the estimated goal, with advancement to goal over 35 days. ASPEN recommends fluid and electrolyte status be monitored as needed.50 The NICE guidelines recommend starting nutrition support at a maximum of 10 kcal/kg/d with slow increase to meet or exceed full needs by 47 days. For extremely malnourished patients (eg, BMI <14 kg/m2, or negligible intake for >15 days), they recommend starting at 5 kcal/kg/d. For patients at high risk of developing refeeding syndrome, the NICE guidelines recommend vitamin repletion immediately before and during the first 10 days of feeding (thiamine, vitamin B, and a balanced multivitamin/trace element supplement). Cardiac monitoring is recommended for this group as well as any patients who are at risk for cardiac arrhythmias. Careful monitoring of fluid balance and restoring circulatory volume is recommended, as is repletion of potassium, phosphate, and magnesium.56
TERMINATION OF THERAPY
Termination of nutrition support often involves transitioning from one mode of support to another. PN can be discontinued when oral or enteral intake reaches 60% of total calories; enteral intake can be discontinued when oral intake reaches the same level. However, the patient should be observed maintaining their intake; if they cannot, nutrition support should be resumed.12
TRANSITION OF CARE PLAN
Patients discharged from the hospital on enteral or parenteral nutrition require the support of a coordinated multidisciplinary team including dietitians, home nutrition delivery companies, primary care physicians trained in specialized nutrition support, community pharmacists, and other healthcare professionals, if indicated. These relationships should be established prior to discharge, with education about the patient's individualized nutrition plan, and training with the equipment and supplies.10, 56
CONCLUSION
This review provides an overview of managing the at‐risk or malnourished patient by describing the processes of screening, assessment, and development and implementation of a nutrition care plan in the acutely ill hospitalized patient. Malnutrition is a relatively common, yet underdiagnosed entity that impacts patient outcomes, length of stay, hospital costs, and readmissions. Acute illness in a patient already nutritionally debilitated by chronic disease may cause rapid depletion in nutritional stores. Hospitals are required to screen patients for malnutrition on admission and at regular intervals, and to develop and implement a nutrition care plan for those at risk. The plan guides how nutrition therapy is provided, monitored for adequacy and adverse effects, and assessed for achievement of nutritional goals. It encompasses the use of dietary modifications, and enteral and parenteral nutrition. Clinicians must be aware of serious but avoidable adverse effects, particularly refeeding syndrome in malnourished patients. Prior to discharge, the patient should have already been transitioned from EN or PN to taking adequate amounts of calories by mouth; otherwise, careful discharge planning to educate the patients and/or caregivers, and coordinate the necessary multidisciplinary community services is necessary.
Acknowledgements
The authors express their appreciation to Ms Susan Lundy, for her helpful and timely information, and Ms Lisa Boucher, for her invaluable assistance with this manuscript and its submission.
Disclosures: Susan Brantley is on the Speaker's Bureau for Nestle Nutrition and for Abbott Nutrition. Authors Kirkland, Kashiwagi, Scheurer, and Varkey have nothing to report.
- ,,, et al.Prevalence of malnutrition on admission to four hospitals in England. The Malnutrition Prevalence Group.Clin Nutr.2000;19(3):191–195.
- ,.The impact of malnutrition on morbidity, mortality, length of hospital stay and costs evaluated through a multivariate model analysis.Clin Nutr.2003;22(3):235–239.
- ,,,.Malnutrition among hospitalized patients. A problem of physician awareness.Arch Intern Med.1987;147(8):1462–1465.
- ,,,,.Malnutrition is prevalent in hospitalized medical patients: are housestaff identifying the malnourished patient?Nutrition.2006;22(4):350–354.
- ,,, et al.Malnutrition in subacute care.Am J Clin Nutr.2002;75(2):308–313.
- ,,, et al.Malnutrition is an independent factor associated with nosocomial infections.Br J Nutr.2004;92(1):105–111.
- ,.Impact of body mass index on outcomes following critical care.Chest.2003;123(4):1202–1207.
- ,,, et al.EuroOOPS: an international, multicentre study to implement nutritional risk screening and evaluate clinical outcome.Clin Nutr.2008;27(3):340–349.
- ,,,,.Consensus statement: Academy of Nutrition and Dietetics and American Society for Parenteral and Enteral Nutrition: characteristics recommended for the identification and documentation of adult malnutrition (undernutrition).J Parenter Enteral Nutr.2012;36(3):275–283.
- ,,, et al.Standards for nutrition support: adult hospitalized patients.Nutr Clin Pract.2010;25(4):403–414.
- American Society for Parenteral and Enteral Nutrition (A.S.P.E.N.), Board of Directors and Clinical Practice Committee. Definition of Terms, Style, and Conventions used in A.S.P.E.N. Board of Directors‐Approved Documents. May2012. Available at: http://www.nutritioncare.org/Library.aspx. Accessed June 29, 2012.
- Joint Commission on Accreditation of Healthcare Organizations.Comprehensive Accreditation for Hospitals.Chicago, IL:Joint Commission on Accreditation for Healthcare Organizations;2007.
- ,.Nutrition screening and assessment. In: Gottschlich MM, ed.The ASPEN Nutrition Support Core Curriculum: A Case‐Based Approach—The Adult Patient.1st ed.Silver Spring, MD:American Society for Parenteral and Enteral Nutrition;2007:163–186.
- .Nutrition screening tools for hospitalized patients.Nutr Clin Pract.2008;23(4):373–382.
- .Nutrition screening and assessment. In: Skipper A, ed.Dietitian's Handbook of Enteral and Parenteral Nutrition.3rd ed.Sudbury, MA:Jones 2012:4–21.
- ,.Nutritional assessment in the hospitalized patient.Curr Opin Clin Nutr Metab Care.2003;6(5):531–538.
- ,,,,.Nutritional and metabolic assessment of the hospitalized patient.J Parenter Enteral Nutr.1977;1(1):11–22.
- ,,.Hepatic proteins and nutrition assessment.J Am Diet Assoc.2004;104(8):1258–1264.
- .The albumin‐nutrition connection: separating myth from fact.Nutrition.2002;18(2):199–200.
- ,.Predictive equations for energy needs for the critically ill.Respir Care.2009;54(4):509–521.
- ,,, et al.Guidelines, guidelines, guidelines: what are we to do with all of these North American guidelines?J Parenter Enteral Nutr.2010;34(6):625–643.
- ,,, et al.Applied nutrition in ICU patients. A consensus statement of the American College of Chest Physicians.Chest.1997;111(3):769–778.
- ,,,,.Comparison of resting energy expenditure prediction methods with measured resting energy expenditure in obese, hospitalized adults.J Parenter Enteral Nutr.2009;33(2):168–175.
- ,,,,,.A new predictive equation for resting energy expenditure in healthy individuals.Am J Clin Nutr.1990;51(2):241–247.
- American Society for Parenteral and Enteral Nutrition.Guidelines for the use of parenteral and enteral nutrition in adult and pediatric patients.J Parenter Enteral Nutr.2002;26(suppl):1SA–138SA.
- ,,.American Gastroenterological Association technical review on tube feeding for enteral nutrition.Gastroenterology.1995;108(4):1282–1301.
- ,,, et al.ESPEN guidelines on enteral nutrition: intensive care.Clin Nutr.2006;25(2):210–223.
- ,.Nutritional support. In: Shojania KG, Duncan BW, McDonald KM, et al, eds.Making Health Care Safer: A Critical Analysis of Patient Safety Practices. Evidence Report/Technology Assessment Number 43.Rockville, MD:Agency for Healthcare Research and Quality, US Department of Health and Human Services, July2001. AHRQ Publication 01‐E058. Available at: http://www.ahrq.gov.
- Veterans Affairs Total Parenteral Nutrition Cooperative Study Group.Perioperative total parenteral nutrition in surgical patients.N Engl J Med.1991;328(8):525–532.
- ,,,,.Does enteral nutrition affect clinical outcome? A systematic review of the randomized trials.Am J Gastroenterol.2007;102(2):412–429.
- ,,,.Nutrition in the stroke patient.Nutr Clin Pract.2011;26(3):242–252.
- ,,.Nutrition diagnosis and intervention. In: Mahan LK, Escott‐Stump S, eds.Krause's Food and Nutrition Therapy.12th ed.St Louis, MO:Saunders Elsevier;2008:454–469.
- Dietary Supplement Health and Education Act (DSHEA) of 1994. Available at: http://www.gpo.gov/fdsys/pkg/BILLS‐103s784es/pdf/BILLS‐103s784es.pdf. Accessed June 29,2012.
- .Intervention: dietary supplementation and integrative care. In: Mahan LK, Escott‐Stump S, eds.Krause's Food and Nutrition Therapy.12th ed.St Louis, MO:Saunders Elsevier;2008:470–474.
- ,,, et al.Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine and American Society for Parenteral and Enteral Nutrition: executive summary.Crit Care Med.2009;37(5):1757–1761.
- ,,,,;for the. Canadian Critical Care Clinical Practice Guidelines Committee.Canadian clinical practice guidelines for nutrition support in mechanically ventilated, critically ill adult patients.J Parenter Enteral Nutr.2003;27(5):355–373.
- American Dietetic Association Evidence Library. Critical Illness. Available at: http://www.adaevidencelibrary.com/template.cfm?key=767115(5 suppl):64S–70S.
- .Enteral nutrition. In: Skipper A, ed.Dietitian's Handbook of Enteral and Parenteral Nutrition.3rd ed.Sudbury, MA:Jones 2012:259–280.
- .Enteral formula selection. In: Charney P, Malone A, eds.ADA Pocket Guide to Enteral Nutrition.Chicago, IL:American Dietetic Association;2006:63–122.
- ,.Overview of enteral nutrition. In: Gottschlich MM, ed.The ASPEN Nutrition Support Core Curriculum: A Case‐Based Approach—The Adult Patient.1st ed.Silver Spring, MD:American Society for Parenteral and Enteral Nutrition;2007:187–208.
- ,,, et al.Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine (SCCM) and American Society for Parenteral and Enteral Nutrition (A.S.P.E.N.).J Parenter Enteral Nutr.2009;33(3):277–316.
- ,,,,,.ESPEN guidelines on parenteral nutrition: surgery.Clin Nutr.2009;28(4):378–386.
- ,,, et al.Safe practices for parenteral nutrition.J Parenter Enteral Nutr.2004;28(6):S39–S70.
- ,,,,,.Contribution of calories from propofol to total energy intake.J Am Diet Assoc.1995;95(9 supplement):A25.
- ,,,.Metabolic and thermogenic response to continuous and cyclic total parenteral nutrition in traumatised and infected patients.Clin Nutr.1994;13(5):291–301.
- ,,.The effect of abrupt cessation of total parenteral nutrition on serum glucose: a randomized trial.Am Surg.2000;66(9):866–869.
- ,,,,.The effect of acute discontinuation of total parenteral nutrition.Ann Surg.1986;204(5):524–529.
- ,,.Refeeding syndrome: what it is, and how to prevent and treat it.BMJ.2008;336(7659):1495–1498.
- ,,, et al.Enteral nutrition practice recommendations.J Parenter Enteral Nutr.2009;33(2):122–167.
- ,,.Control of diarrhea by fiber‐enriched diet in ICU patients on enteral nutrition: a prospective randomized controlled trial.Clin Nutr.2004;23(6):1344–1352.
- ,,,,,.Supine body position as a risk factor for nosocomial pneumonia in mechanically ventilated patients: a randomised trial.Lancet.1999;354(9193):1851–1858.
- .Parenteral nutrition in the critically ill patient.N Engl J Med.2009;361(11):1088–1097.
- ,,,.Refeeding syndrome: treatment considerations based on collective analysis of literature case reports.Nutrition.2010;26(2):156–167.
- ,,.Re‐feeding syndrome in head and neck cancer‐prevention and management.Oral Oncol.2011;47(9):792–796.
- Nutrition Support in Adults. NICE Clinical Guideline No. 32. 2006. Available at: http://guidance.nice.org.uk/CG32/NICEGuidance. Accessed November 29,2011.
- ,,.The importance of the refeeding syndrome.Nutrition.2001;17(7–8):632–637.
Malnutrition is present in 20% to 50% of hospitalized patients.1, 2 Despite simple, validated screening tools, malnutrition tends to be underdiagnosed.3, 4 Over 90% of elderly patients transitioning from an acute care hospital to a subacute care facility are either malnourished or at risk of malnutrition.5 Malnutrition has been associated with increased risk of nosocomial infections,6 worsened discharge functional status,7 and higher mortality,8 as well as longer lengths of stay7, 8 and higher hospital costs.2
Malnutrition describes either overnutrition or undernutrition that causes a change in body composition and decreased function.9 Malnutrition in hospitalized patients is typically related to undernutrition due to either reduced intake or increased metabolic rate. Reasons for reduced intake include poor appetite, reduced ability to chew or swallow, and nil per os (NPO) status. Patients with acute or chronic illnesses may either be malnourished on admission, or develop malnutrition within a few days of hospital admission, due to the effects of the inflammatory state on metabolism. Given that malnutrition is potentially modifiable, it is important to screen for malnutrition and, when present, develop, implement, and monitor a nutrition care plan10 (Figure 1).
The purpose of this review is to provide the hospitalist with an overview of screening, assessment, and development and implementation of a nutrition care plan in the acutely ill hospitalized patient.
PATIENT SCREENING
Nutrition screening identifies patients with nutritional deficits who may benefit from further detailed nutrition assessment and intervention.11 The Joint Commission requires that all patients admitted to acute care hospitals be screened for risk of malnutrition within 24 hours.12 Those considered at risk for malnutrition have significant weight changes, chronic disease or an acute inflammatory process, or have been unable to ingest adequate calories for 7 days.13
Those not at risk should be regularly rescreened throughout their hospital stay. The American Society of Parenteral and Enteral Nutrition (ASPEN) recommends that institutions create and approve a screening process according to the patient population served.10 There are several tools validated for use in the acute care setting.14 Many institutions trigger an automatic nutrition consult when certain screening criteria are met.
PATIENT ASSESSMENT
Nutrition assessment should be performed by a dietitian or nutrition consult provider in patients who screen at risk for malnutrition to characterize and determine the cause of nutritional deficits.10 The nutrition assessment identifies history and physical examination elements to diagnose malnutrition. An ASPEN consensus statement recommends the diagnosis of malnutrition if 2 or more of the following are present: insufficient energy intake, weight loss, loss of muscle mass, loss of subcutaneous fat, localized or generalized fluid accumulation, and decreased functional status measured by hand‐grip strength.9 The nutrition assessment should also consider how long the patient has been without adequate nutrition, document baseline nutrition parameters,15 and estimate caloric requirements to determine nutrition support therapy needs.10 Nutrition assessment typically includes the following components.
History
A careful history elicits the majority of information needed to determine the cause and severity of malnutrition.16 Patients should be questioned about a typical day's oral intake prior to hospitalization, and about factors that affect their intake such as sensory deficits, fine motor dysfunction, or chewing and swallowing difficulties, which often decline in chronically ill and elderly patients. Nutrition may be affected by financial difficulties or limited social support, and access to food should be assessed.
Physical Findings
Weight loss is the best physical exam predictor of malnutrition risk, although nutritional depletion can occur in a very short time in acutely ill or injured patients before substantial weight loss has occurred. The likelihood of malnutrition is increased if a patient has: a body mass index (BMI) <18.5 kg/m2; unintentional loss of >2.3 kg (5 lb) or 5% of body weight over 1 month; and unintentional loss of >4.5 kg (10 lb) or 10% of body weight over 6 months.17 Weight loss may be masked by fluid retention from chronic conditions, such as heart failure, or from volume resuscitation in the acutely ill patient.9, 16
Body mass index can be misleading, as age‐related height loss may artificially increase BMI, and height may be difficult to accurately measure in a kyphotic, unsteady, or bedridden patient. The clinician may find evidence of loss of subcutaneous fat or muscle mass in patients with chronic illness, but these findings may not be evident in the acutely ill patient.9 Other physical exam assessments of malnutrition, such as arm span, skinfold thickness, and arm circumference are not reliable.16
Laboratory Tests
Biochemical markers, including transferrin, albumin, and prealbumin, have not been proven as accurate predictors of nutrition status because they may change as a result of other factors not related to nutrition.15, 18 Serum albumin, for example, may be more reflective of the degree of metabolic stress.19 Prealbumin has a serum half‐life much shorter than albumin or transferrin (approximately 2448 hours) and is perhaps the most useful protein marker to assess the adequacy of nutritional replacement after the inflammatory state is resolved.18
Calculating Caloric Requirements
Energy expenditure measurement is considered the gold standard to determine patients' caloric needs. Actual measurement by methods such as indirect calorimetry, which measures oxygen consumption and carbon dioxide production, and calculates energy expenditure, is challenging in everyday clinical settings. Predictive equations often are used as alternative methods to estimate patients' caloric requirements.20 There is no consensus among the 3 North American societies' guidelines (the Canadian Clinical Practice Guidelines; the American Dietetics Association's evidence‐based guideline for critical illness; and the Society of Critical Care Medicine and American Society of Parenteral and Enteral Nutrition's joint guideline) as to the best method.21
In the simplest equation, caloric needs are estimated by calories per kilogram.22 In obese patients, using actual body weight will overestimate needs, but using ideal body weight may cause underfeeding. A small study comparing predictive equations in obese hospitalized patients found the Harris‐Benedict equations (H‐BE) using adjusted body weight and a stress factor to be most accurate, but only in 50% of patients.23 Most clinicians are familiar with the H‐BE, but alternatives such as calories per kilogram or the Mifflin St.‐Jeor equation24 are often used (S. Brantley (May 5, 2012), S. Lundy (May 23, 2012), personal communication).
Indications for Nutritional Intervention
In adults without preexisting malnutrition, inadequate nutritional intake for approximately 714 days should prompt nutritional intervention.25, 26 This timeline should be shorter (37 days) in those with lower energy reserves (eg, underweight or recent weight loss) or significant catabolic stress (eg, acutely ill patients).27, 28 Other patient populations shown to benefit from nutritional intervention include: postoperative patients who are anticipated to be NPO for more than 7 days or to be taking less than 60% of estimated caloric needs by postoperative day 10; preoperative patients with severe malnutrition29; those with gastrointestinal cancer undergoing elective surgery30; and stroke patients with persistent dysphagia for more than 7 days.31
DEVELOPMENT OF A NUTRITION CARE PLAN
The formal nutrition assessment of the at‐risk patient derives the information needed for the development of a nutrition care plan. This plan guides the provision of nutrition therapy, the intervention, the monitoring protocols, evaluation, and reassessment of nutrition goals or termination of specialized nutrition support.10 Assessments for adequacy of nutritional repletion are best done by repeated screening and physical examinations.18
IMPLEMENTATION OF NUTRITION CARE PLAN
Nutritional interventions include dietary modifications, enteral nutrition, and parenteral nutrition.
Dietary Modifications
The purpose of the diet is to provide the necessary nutrients to the body in a well‐tolerated form. Diets can be modified to provide for individual requirements, personal eating patterns and food preferences, and disease process and digestive capacity. Dietary adjustments include change in consistency of foods (eg, pureed, mechanical soft), increase or decrease in energy value, increase or decrease in the type of food or nutrient consumed (eg, sodium restriction, fiber enhancement), elimination of specific foods (eg, gluten‐free diet), adjustment in protein, fat, and carbohydrate content (eg, ketogenic diet, renal diet, cholesterol‐lowering diet), and adjustment of the number and frequency of meals.32
Dietary supplementation (eg, Boost, Ensure) is common practice in persons diagnosed with such conditions as cancer, diabetes, and cardiovascular disease. Supplements enhance the diet by increasing the total daily intake of a vitamin, a mineral, an amino acid, an herb or other botanical33, and should not be used as a meal substitute.34 These supplements are varied in content of calories, protein, vitamins, and minerals. Various flavors and consistencies are also available. Several oral supplements are reviewed in Table 1.
| Oral Supplement* (Serving Size; mL) | Kcal/svg | Protein (g/svg) | Fat (g/svg) | CHO (g/svg) | Na (mg/svg) | K (mg/svg) | Ca (mg/svg) | Phos (mg/svg) | Mg (mg/svg) |
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| Boost Original (237) | 240 | 10 | 4 | 41 | 150 | 460 | 300 | 300 | 100 |
| Ensure Nutrition Shake (237) | 250 | 9 | 6 | 40 | 200 | 370 | 300 | 250 | 100 |
| Carnation Instant Breakfast Ready to Drink (325) | 250 | 14 | 5 | 34 | 180 | 330 | 500 | 500 | 120 |
| Resource Breeze (fruit‐flavored) clear liquid (237) | 250 | 9 | 0 | 54 | 80 | 10 | 10 | 150 | 1 |
| Glucerna 1.0 Ready to Drink low‐CHO (237) | 240 | 10 | 13 | 23 | 220 | 370 | 170 | 170 | 67 |
| Re/Gen low K and Phos (180) | 375 | 12 | 17 | 47 | 180 | 23 | 15 | 68 | 3 |
Enteral Nutrition
Enteral nutrition (EN) support should be provided to patients who have functioning gastrointestinal (GI) tracts but are unable to take adequate calories orally. Compared to parenteral nutrition (PN), EN is associated with favorable improvements in inflammatory cytokines, acute phase proteins, hyperglycemia, insulin resistance, nosocomial infections, mortality, and cost.35 Enteral feeds are more physiologic than parenteral feeds, maintain GI structure and integrity, and avoid intravenous (IV) access complications. Patients with normal nutritional status on admission who require EN should be receiving over 50% of their caloric needs within the first week of hospital stay.25 Malnourished patients should reach this minimum goal within 35 days of admission.27, 28 EN is not contraindicated in the absence of bowel sounds or in the presence of increased gastric residuals.35 Withholding enteral feedings for gastric residual volumes <250 mL36, 37 or reduced bowel sounds can result in inadequate caloric intake or inappropriate use of PN.27
Gastric feedings are more physiologic than small bowel feedings, can be given by bolus or continuous infusion, and can be given by tubes that are easy to place at the bedside. Post‐pyloric feedings (nasoduodenal or nasojejunal) may be associated with a lower risk of pneumonia, and should be considered in high‐risk patients such as those receiving continuous sedatives or neuromuscular blockers.36 Post‐pyloric tube placement usually requires endoscopy, fluoroscopy, or electromagnetic guidance. Percutaneous feeding tubes (gastrostomy or jejunostomy) should be considered in those who require tube feedings for longer than 30 days.38
Assessment of patient requirements and disease state, as well as extensive knowledge of available formulas, is important in the selection of the appropriate enteral formula.39 Standardized formulas are used for most patients. The provision of adequate water must be considered with these formulas, particularly in the long‐term care and home settings.40 Many specialized formulas are designed for a particular disease state or condition, some of which are further reviewed in Table 2.
| Formula | Kcal/mL | Protein (g/L) | Fat (g/L) | CHO (g/L) | Osmolality (mOsm/kg H2O) | Na (mEq/L) | K (mEq/L) | Ca (mg/L) | Mg (mg/L) | Phos (mg/L) |
|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||
| Nutren 1.0‐low residue | 1 | 40 | 38 | 127 | 315 | 38 | 32 | 668 | 268 | 668 |
| Osmolite 1.0 Cal low residue | 1 | 44.3 | 34.7 | 143.9 | 300 | 40.4 | 40.2 | 760 | 305 | 1760 |
| Replete high protein, low residue | 1 | 62.4 | 34 | 112 | 300 | 38.1 | 38.5 | 1000 | 400 | 1000 |
| Replete Fiber high protein with fiber | 1 | 62.4 | 34 | 112 | 310 | 38.1 | 38.5 | 1000 | 400 | 1000 |
| Osmolite 1.5 low residue, calorically dense | 1.5 | 62.7 | 49.1 | 203.6 | 525 | 60.9 | 46 | 1000 | 400 | 1000 |
| Two Cal calorie and protein dense | 2 | 83.5 | 91 | 219 | 725 | 64 | 63 | 1050 | 425 | 1050 |
| Vivonex RTF‐elemental | 1 | 50 | 11.6 | 176 | 630 | 30.4 | 31 | 668 | 268 | 668 |
| Nepro with Carb Steady‐for electrolyte, fluid restriction (eg, dialysis) | 1.8 | 81 | 96 | 161 | 745 | 46 | 27 | 1060 | 210 | 720 |
| Nutren Glytrol low CHO | 1 | 45.2 | 47.6 | 100 | 280 | 32.2 | 35.9 | 720 | 286 | 720 |
| NutriHep‐for hepatic disease | 1.5 | 40 | 21.2 | 290 | 790 | 160 | 33.9 | 956 | 376 | 1000 |
If concerned about formula tolerance, one solution is to initiate the formula at a low rate and increase to the goal rate over 2448 hours. Dilution of enteral formulas is not necessary to assure optimal tolerance. Continuous feedings are recommended for most patients initially and after tolerance has been established, bolus feedings can be attempted if the feeding tube terminates in the stomach. Bolus feedings, where 240480 mL of formula are delivered through a syringe over 1015 minutes, may be more physiological for patients. This regimen can be repeated 46 times daily to meet nutrition goals.41
Parenteral Nutrition
PN provides macronutrients such as carbohydrates, protein, and fat; micronutrients such as vitamins, minerals, electrolytes, and trace elements are added in appropriate concentrations. PN may also provide the patient's daily fluid needs. The timing of PN initiation depends upon the patient's initial nutritional status. ASPEN does not recommend PN during the first 7 days of hospitalization in critically ill patients with normal nutritional status. If the patient is not receiving 100% of caloric needs from EN after 7 days, supplemental PN should be considered. However, if on admission a patient is already malnourished and EN is not feasible, PN should be initiated and continued until the patient is receiving at least 60% of caloric needs by enteral route.42 This includes patients with intestinal obstruction, ileus, peritonitis, malabsorption, high output enterocutaneous fistulae, intestinal ischemia, intractable vomiting and diarrhea, severe shock, and fulminant sepsis.10, 43
Standardized commercial PN products are available and reduce the number of steps required between ordering and administration, as compared to customized PN, which is compounded for a particular patient. However, despite improved efficiency and lower cost, there is no evidence that standardized preparations are safer to patients than customized solutions. Institutions utilizing standardized PN must also have a mechanism to customize formulas for those with complex needs.44
Creating a customized parenteral solution involves several basic steps. Total caloric requirement may be estimated using a predictive formula, as previously discussed; calories/kg of ideal body weight is the simplest method. Most hospitalized patients require 2030 calories/kg/d. Daily fluid requirement may be based on kilocalories (kcal) delivered, or by ideal body weight (eg, 1 mL/kcal or 3040 mL/kg). More fluid may be needed in patients with significant sensible or insensible losses; those with renal failure or heart failure should receive less fluid.
Protein needs are calculated by multiplying ideal body weight (kg) by estimated protein needs in g/kg/d (1.22 g/kg/d for catabolic patients). Protein should provide approximately 20% of total calories. Protein restriction is not required in renal impairment; acutely ill patients on renal replacement therapy should receive 1.51.8 g/kg/d. In hepatic failure patients, protein should be restricted only if hepatic encephalopathy fails to improve with other measures.
Knowing the protein, kcal, and fluid needs of the patient, the practitioner divides the remaining non‐protein calories between carbohydrates and fat. Approximately 70%85% of non‐protein calories should be provided as carbohydrates (dextrose), up to 7 g/kg/d. The other 15%30% are as fat, in lipid solutions, providing a maximum of 2.5 g/kg/d. Lipid solutions are provided as either 10% (1.1 kcal/mL) or 20% (2.2 kcal/mL) concentrations.43, 44 Propofol's contribution to fat intake complicates estimating total fat intake in critically ill patients.45
Standardized parenteral multivitamin preparations are available; the clinician must determine if preparations containing vitamin K are appropriate. Of the trace elements, copper and manganese should be restricted in hepatobiliary disease.44
Acutely ill patients receive PN as a 24‐hour infusion, to minimize its impact on volume status and energy expenditure,46 providing 50% of needs on infusion day one and reaching goal within 4872 hours, rather than cyclic infusions over shorter intervals. Daily assessments of vital signs, intake and output, and weight are necessary to monitor volume status.
Once a patient is taking at least 60% of caloric needs either by mouth or by EN, PN can be discontinued. Tapering the infusion is not required, as abrupt discontinuation has not been demonstrated to cause symptomatic hypoglycemia.47, 48
PATIENT MONITORING
Laboratory monitoring with nutrition support should include baseline electrolytes, glucose, renal function, coagulation studies, triglycerides, magnesium, phosphorus, cholesterol, platelet count, and hepatobiliary enzymes. Electrolytes, calcium, magnesium, and phosphorus should be checked daily for 3 days and, if normal, should then be checked biweekly. Capillary glucose should be monitored several times a day until stable. Weekly triglycerides, albumin, cholesterol, coagulation studies, and liver enzymes should also be checked in patients while on parenteral nutrition.25 Patients at risk for refeeding syndrome should have potassium, phosphate, calcium, and magnesium measured daily for 7 days, with repletion as necessary. These electrolytes should be monitored 3 times the following week if stable.49
Patients should be monitored clinically for gastrointestinal tolerance of enteral nutrition. All 3 North American guidelines recommend monitoring gastric residual volumes (GRV); however, there is no consensus on the volume considered to require intervention. Motility agents are recommended as first line treatment of high GRV.36, 37, 42 If high GRV continues, tube feeding should be held, and tube placement, medications, and metabolic assessment should be reviewed. Placement of a transpyloric feeding tube may be indicated.50
Adverse Effects and Complications of Nutrition Support
Regarding EN, complications include those related to tube placement and maintenance, infections, and medical complications of the feeds themselves. Some of the adverse effects of the enteral formulas may be attenuated. Diarrhea, which occurs in up to 20% of patients, may be avoided with slow feed advancement, use of low‐osmotic formulas, or fiber additives.51 Gastric distention and abdominal pain may improve with slow feed advancement and continuous (rather than bolus) feeds. Small‐bore tubes and acid‐reducing medications may decrease gastroesophageal reflux, and aspiration pneumonia may be avoided by semi‐recumbent positioning and post‐pyloric feeding.52
Complications of PN may be grouped as mechanical, infectious, and metabolic. The mechanical complications of central line placement include pneumothorax, arterial puncture, hematoma, air embolism, and line malpositioning. Catheter‐related deep venous thrombosis may occur. Patients on PN through a central line are at risk for central line‐associated bloodstream infections.25 The metabolic complications such as hyperglycemia, electrolyte disorders, hepatic steatosis, and volume overload may have severe consequences, such as heart failure or neuromuscular dysfunction, thus they require close attention.53
A complication of nutrition support that may occur regardless of route is the refeeding syndrome. Refeeding syndrome describes fluid shifts and electrolyte abnormalities that occur after initiation of oral, enteral, or parenteral nutrition in a malnourished or starved patient.54, 55 There are no formal criteria for diagnosing refeeding syndrome.
In the starved state, the body switches from carbohydrate to protein and fat metabolism. Reintroduction of carbohydrates stimulates insulin release with glycogen, fat, and protein synthesis. Associated uptake of glucose, potassium, magnesium, phosphate, and water into cells causes electrolyte and fluid abnormalities. Although hypophosphatemia is the hallmark of refeeding syndrome, it is not pathognomonic. Additional disturbances include hypokalemia, hyperglycemia, hypomagnesemia, thiamine deficiency, and fluid imbalance.49 Patients at risk of refeeding should have serum electrolytes, magnesium, phosphorus, and glucose checked before nutrition support starts. The degree of laboratory abnormalities, if any, and the clinical course of refeeding guides the frequency of subsequent blood tests.56 These consequences of refeeding can adversely affect every major organ system and may result in death.57
Starvation physiology underlies all risk factors for refeeding syndrome. In hospitalized patients, those at risk for refeeding include, but are not limited to, the elderly, oncology patients, postoperative patients, alcohol‐dependent patients, those with malabsorptive states, those who are fasting or chronically malnourished, and those on diuretic therapy.54, 57 The National Institute for Health and Clinical Excellence (NICE) of England and Wales has published criteria to identify patients at high risk for refeeding (Table 3).56 Identification of at‐risk patients and attention to their nutritional needs prevents refeeding syndrome.
|
| Patient has 1 or more of the following: |
| BMI <16 kg/m2 |
| Unintentional weight loss >15% within the last 36 mo |
| Little or no nutritional intake for more than 10 d |
| Low levels of potassium, phosphate, or magnesium prior to feeding |
| Or patient has 2 or more of the following: |
| BMI <18.5 kg/m2 |
| Unintentional weight loss >10% within the last 36 mo |
| Little or no nutritional intake for more than 5 d |
| A history of alcohol abuse or drugs including insulin, chemotherapy, antacids, or diuretics |
ASPEN and NICE have each issued guidelines for initiating nutrition support in patients at risk for refeeding. ASPEN guidelines recommend feeding start at approximately 25% of the estimated goal, with advancement to goal over 35 days. ASPEN recommends fluid and electrolyte status be monitored as needed.50 The NICE guidelines recommend starting nutrition support at a maximum of 10 kcal/kg/d with slow increase to meet or exceed full needs by 47 days. For extremely malnourished patients (eg, BMI <14 kg/m2, or negligible intake for >15 days), they recommend starting at 5 kcal/kg/d. For patients at high risk of developing refeeding syndrome, the NICE guidelines recommend vitamin repletion immediately before and during the first 10 days of feeding (thiamine, vitamin B, and a balanced multivitamin/trace element supplement). Cardiac monitoring is recommended for this group as well as any patients who are at risk for cardiac arrhythmias. Careful monitoring of fluid balance and restoring circulatory volume is recommended, as is repletion of potassium, phosphate, and magnesium.56
TERMINATION OF THERAPY
Termination of nutrition support often involves transitioning from one mode of support to another. PN can be discontinued when oral or enteral intake reaches 60% of total calories; enteral intake can be discontinued when oral intake reaches the same level. However, the patient should be observed maintaining their intake; if they cannot, nutrition support should be resumed.12
TRANSITION OF CARE PLAN
Patients discharged from the hospital on enteral or parenteral nutrition require the support of a coordinated multidisciplinary team including dietitians, home nutrition delivery companies, primary care physicians trained in specialized nutrition support, community pharmacists, and other healthcare professionals, if indicated. These relationships should be established prior to discharge, with education about the patient's individualized nutrition plan, and training with the equipment and supplies.10, 56
CONCLUSION
This review provides an overview of managing the at‐risk or malnourished patient by describing the processes of screening, assessment, and development and implementation of a nutrition care plan in the acutely ill hospitalized patient. Malnutrition is a relatively common, yet underdiagnosed entity that impacts patient outcomes, length of stay, hospital costs, and readmissions. Acute illness in a patient already nutritionally debilitated by chronic disease may cause rapid depletion in nutritional stores. Hospitals are required to screen patients for malnutrition on admission and at regular intervals, and to develop and implement a nutrition care plan for those at risk. The plan guides how nutrition therapy is provided, monitored for adequacy and adverse effects, and assessed for achievement of nutritional goals. It encompasses the use of dietary modifications, and enteral and parenteral nutrition. Clinicians must be aware of serious but avoidable adverse effects, particularly refeeding syndrome in malnourished patients. Prior to discharge, the patient should have already been transitioned from EN or PN to taking adequate amounts of calories by mouth; otherwise, careful discharge planning to educate the patients and/or caregivers, and coordinate the necessary multidisciplinary community services is necessary.
Acknowledgements
The authors express their appreciation to Ms Susan Lundy, for her helpful and timely information, and Ms Lisa Boucher, for her invaluable assistance with this manuscript and its submission.
Disclosures: Susan Brantley is on the Speaker's Bureau for Nestle Nutrition and for Abbott Nutrition. Authors Kirkland, Kashiwagi, Scheurer, and Varkey have nothing to report.
Malnutrition is present in 20% to 50% of hospitalized patients.1, 2 Despite simple, validated screening tools, malnutrition tends to be underdiagnosed.3, 4 Over 90% of elderly patients transitioning from an acute care hospital to a subacute care facility are either malnourished or at risk of malnutrition.5 Malnutrition has been associated with increased risk of nosocomial infections,6 worsened discharge functional status,7 and higher mortality,8 as well as longer lengths of stay7, 8 and higher hospital costs.2
Malnutrition describes either overnutrition or undernutrition that causes a change in body composition and decreased function.9 Malnutrition in hospitalized patients is typically related to undernutrition due to either reduced intake or increased metabolic rate. Reasons for reduced intake include poor appetite, reduced ability to chew or swallow, and nil per os (NPO) status. Patients with acute or chronic illnesses may either be malnourished on admission, or develop malnutrition within a few days of hospital admission, due to the effects of the inflammatory state on metabolism. Given that malnutrition is potentially modifiable, it is important to screen for malnutrition and, when present, develop, implement, and monitor a nutrition care plan10 (Figure 1).
The purpose of this review is to provide the hospitalist with an overview of screening, assessment, and development and implementation of a nutrition care plan in the acutely ill hospitalized patient.
PATIENT SCREENING
Nutrition screening identifies patients with nutritional deficits who may benefit from further detailed nutrition assessment and intervention.11 The Joint Commission requires that all patients admitted to acute care hospitals be screened for risk of malnutrition within 24 hours.12 Those considered at risk for malnutrition have significant weight changes, chronic disease or an acute inflammatory process, or have been unable to ingest adequate calories for 7 days.13
Those not at risk should be regularly rescreened throughout their hospital stay. The American Society of Parenteral and Enteral Nutrition (ASPEN) recommends that institutions create and approve a screening process according to the patient population served.10 There are several tools validated for use in the acute care setting.14 Many institutions trigger an automatic nutrition consult when certain screening criteria are met.
PATIENT ASSESSMENT
Nutrition assessment should be performed by a dietitian or nutrition consult provider in patients who screen at risk for malnutrition to characterize and determine the cause of nutritional deficits.10 The nutrition assessment identifies history and physical examination elements to diagnose malnutrition. An ASPEN consensus statement recommends the diagnosis of malnutrition if 2 or more of the following are present: insufficient energy intake, weight loss, loss of muscle mass, loss of subcutaneous fat, localized or generalized fluid accumulation, and decreased functional status measured by hand‐grip strength.9 The nutrition assessment should also consider how long the patient has been without adequate nutrition, document baseline nutrition parameters,15 and estimate caloric requirements to determine nutrition support therapy needs.10 Nutrition assessment typically includes the following components.
History
A careful history elicits the majority of information needed to determine the cause and severity of malnutrition.16 Patients should be questioned about a typical day's oral intake prior to hospitalization, and about factors that affect their intake such as sensory deficits, fine motor dysfunction, or chewing and swallowing difficulties, which often decline in chronically ill and elderly patients. Nutrition may be affected by financial difficulties or limited social support, and access to food should be assessed.
Physical Findings
Weight loss is the best physical exam predictor of malnutrition risk, although nutritional depletion can occur in a very short time in acutely ill or injured patients before substantial weight loss has occurred. The likelihood of malnutrition is increased if a patient has: a body mass index (BMI) <18.5 kg/m2; unintentional loss of >2.3 kg (5 lb) or 5% of body weight over 1 month; and unintentional loss of >4.5 kg (10 lb) or 10% of body weight over 6 months.17 Weight loss may be masked by fluid retention from chronic conditions, such as heart failure, or from volume resuscitation in the acutely ill patient.9, 16
Body mass index can be misleading, as age‐related height loss may artificially increase BMI, and height may be difficult to accurately measure in a kyphotic, unsteady, or bedridden patient. The clinician may find evidence of loss of subcutaneous fat or muscle mass in patients with chronic illness, but these findings may not be evident in the acutely ill patient.9 Other physical exam assessments of malnutrition, such as arm span, skinfold thickness, and arm circumference are not reliable.16
Laboratory Tests
Biochemical markers, including transferrin, albumin, and prealbumin, have not been proven as accurate predictors of nutrition status because they may change as a result of other factors not related to nutrition.15, 18 Serum albumin, for example, may be more reflective of the degree of metabolic stress.19 Prealbumin has a serum half‐life much shorter than albumin or transferrin (approximately 2448 hours) and is perhaps the most useful protein marker to assess the adequacy of nutritional replacement after the inflammatory state is resolved.18
Calculating Caloric Requirements
Energy expenditure measurement is considered the gold standard to determine patients' caloric needs. Actual measurement by methods such as indirect calorimetry, which measures oxygen consumption and carbon dioxide production, and calculates energy expenditure, is challenging in everyday clinical settings. Predictive equations often are used as alternative methods to estimate patients' caloric requirements.20 There is no consensus among the 3 North American societies' guidelines (the Canadian Clinical Practice Guidelines; the American Dietetics Association's evidence‐based guideline for critical illness; and the Society of Critical Care Medicine and American Society of Parenteral and Enteral Nutrition's joint guideline) as to the best method.21
In the simplest equation, caloric needs are estimated by calories per kilogram.22 In obese patients, using actual body weight will overestimate needs, but using ideal body weight may cause underfeeding. A small study comparing predictive equations in obese hospitalized patients found the Harris‐Benedict equations (H‐BE) using adjusted body weight and a stress factor to be most accurate, but only in 50% of patients.23 Most clinicians are familiar with the H‐BE, but alternatives such as calories per kilogram or the Mifflin St.‐Jeor equation24 are often used (S. Brantley (May 5, 2012), S. Lundy (May 23, 2012), personal communication).
Indications for Nutritional Intervention
In adults without preexisting malnutrition, inadequate nutritional intake for approximately 714 days should prompt nutritional intervention.25, 26 This timeline should be shorter (37 days) in those with lower energy reserves (eg, underweight or recent weight loss) or significant catabolic stress (eg, acutely ill patients).27, 28 Other patient populations shown to benefit from nutritional intervention include: postoperative patients who are anticipated to be NPO for more than 7 days or to be taking less than 60% of estimated caloric needs by postoperative day 10; preoperative patients with severe malnutrition29; those with gastrointestinal cancer undergoing elective surgery30; and stroke patients with persistent dysphagia for more than 7 days.31
DEVELOPMENT OF A NUTRITION CARE PLAN
The formal nutrition assessment of the at‐risk patient derives the information needed for the development of a nutrition care plan. This plan guides the provision of nutrition therapy, the intervention, the monitoring protocols, evaluation, and reassessment of nutrition goals or termination of specialized nutrition support.10 Assessments for adequacy of nutritional repletion are best done by repeated screening and physical examinations.18
IMPLEMENTATION OF NUTRITION CARE PLAN
Nutritional interventions include dietary modifications, enteral nutrition, and parenteral nutrition.
Dietary Modifications
The purpose of the diet is to provide the necessary nutrients to the body in a well‐tolerated form. Diets can be modified to provide for individual requirements, personal eating patterns and food preferences, and disease process and digestive capacity. Dietary adjustments include change in consistency of foods (eg, pureed, mechanical soft), increase or decrease in energy value, increase or decrease in the type of food or nutrient consumed (eg, sodium restriction, fiber enhancement), elimination of specific foods (eg, gluten‐free diet), adjustment in protein, fat, and carbohydrate content (eg, ketogenic diet, renal diet, cholesterol‐lowering diet), and adjustment of the number and frequency of meals.32
Dietary supplementation (eg, Boost, Ensure) is common practice in persons diagnosed with such conditions as cancer, diabetes, and cardiovascular disease. Supplements enhance the diet by increasing the total daily intake of a vitamin, a mineral, an amino acid, an herb or other botanical33, and should not be used as a meal substitute.34 These supplements are varied in content of calories, protein, vitamins, and minerals. Various flavors and consistencies are also available. Several oral supplements are reviewed in Table 1.
| Oral Supplement* (Serving Size; mL) | Kcal/svg | Protein (g/svg) | Fat (g/svg) | CHO (g/svg) | Na (mg/svg) | K (mg/svg) | Ca (mg/svg) | Phos (mg/svg) | Mg (mg/svg) |
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| Boost Original (237) | 240 | 10 | 4 | 41 | 150 | 460 | 300 | 300 | 100 |
| Ensure Nutrition Shake (237) | 250 | 9 | 6 | 40 | 200 | 370 | 300 | 250 | 100 |
| Carnation Instant Breakfast Ready to Drink (325) | 250 | 14 | 5 | 34 | 180 | 330 | 500 | 500 | 120 |
| Resource Breeze (fruit‐flavored) clear liquid (237) | 250 | 9 | 0 | 54 | 80 | 10 | 10 | 150 | 1 |
| Glucerna 1.0 Ready to Drink low‐CHO (237) | 240 | 10 | 13 | 23 | 220 | 370 | 170 | 170 | 67 |
| Re/Gen low K and Phos (180) | 375 | 12 | 17 | 47 | 180 | 23 | 15 | 68 | 3 |
Enteral Nutrition
Enteral nutrition (EN) support should be provided to patients who have functioning gastrointestinal (GI) tracts but are unable to take adequate calories orally. Compared to parenteral nutrition (PN), EN is associated with favorable improvements in inflammatory cytokines, acute phase proteins, hyperglycemia, insulin resistance, nosocomial infections, mortality, and cost.35 Enteral feeds are more physiologic than parenteral feeds, maintain GI structure and integrity, and avoid intravenous (IV) access complications. Patients with normal nutritional status on admission who require EN should be receiving over 50% of their caloric needs within the first week of hospital stay.25 Malnourished patients should reach this minimum goal within 35 days of admission.27, 28 EN is not contraindicated in the absence of bowel sounds or in the presence of increased gastric residuals.35 Withholding enteral feedings for gastric residual volumes <250 mL36, 37 or reduced bowel sounds can result in inadequate caloric intake or inappropriate use of PN.27
Gastric feedings are more physiologic than small bowel feedings, can be given by bolus or continuous infusion, and can be given by tubes that are easy to place at the bedside. Post‐pyloric feedings (nasoduodenal or nasojejunal) may be associated with a lower risk of pneumonia, and should be considered in high‐risk patients such as those receiving continuous sedatives or neuromuscular blockers.36 Post‐pyloric tube placement usually requires endoscopy, fluoroscopy, or electromagnetic guidance. Percutaneous feeding tubes (gastrostomy or jejunostomy) should be considered in those who require tube feedings for longer than 30 days.38
Assessment of patient requirements and disease state, as well as extensive knowledge of available formulas, is important in the selection of the appropriate enteral formula.39 Standardized formulas are used for most patients. The provision of adequate water must be considered with these formulas, particularly in the long‐term care and home settings.40 Many specialized formulas are designed for a particular disease state or condition, some of which are further reviewed in Table 2.
| Formula | Kcal/mL | Protein (g/L) | Fat (g/L) | CHO (g/L) | Osmolality (mOsm/kg H2O) | Na (mEq/L) | K (mEq/L) | Ca (mg/L) | Mg (mg/L) | Phos (mg/L) |
|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||
| Nutren 1.0‐low residue | 1 | 40 | 38 | 127 | 315 | 38 | 32 | 668 | 268 | 668 |
| Osmolite 1.0 Cal low residue | 1 | 44.3 | 34.7 | 143.9 | 300 | 40.4 | 40.2 | 760 | 305 | 1760 |
| Replete high protein, low residue | 1 | 62.4 | 34 | 112 | 300 | 38.1 | 38.5 | 1000 | 400 | 1000 |
| Replete Fiber high protein with fiber | 1 | 62.4 | 34 | 112 | 310 | 38.1 | 38.5 | 1000 | 400 | 1000 |
| Osmolite 1.5 low residue, calorically dense | 1.5 | 62.7 | 49.1 | 203.6 | 525 | 60.9 | 46 | 1000 | 400 | 1000 |
| Two Cal calorie and protein dense | 2 | 83.5 | 91 | 219 | 725 | 64 | 63 | 1050 | 425 | 1050 |
| Vivonex RTF‐elemental | 1 | 50 | 11.6 | 176 | 630 | 30.4 | 31 | 668 | 268 | 668 |
| Nepro with Carb Steady‐for electrolyte, fluid restriction (eg, dialysis) | 1.8 | 81 | 96 | 161 | 745 | 46 | 27 | 1060 | 210 | 720 |
| Nutren Glytrol low CHO | 1 | 45.2 | 47.6 | 100 | 280 | 32.2 | 35.9 | 720 | 286 | 720 |
| NutriHep‐for hepatic disease | 1.5 | 40 | 21.2 | 290 | 790 | 160 | 33.9 | 956 | 376 | 1000 |
If concerned about formula tolerance, one solution is to initiate the formula at a low rate and increase to the goal rate over 2448 hours. Dilution of enteral formulas is not necessary to assure optimal tolerance. Continuous feedings are recommended for most patients initially and after tolerance has been established, bolus feedings can be attempted if the feeding tube terminates in the stomach. Bolus feedings, where 240480 mL of formula are delivered through a syringe over 1015 minutes, may be more physiological for patients. This regimen can be repeated 46 times daily to meet nutrition goals.41
Parenteral Nutrition
PN provides macronutrients such as carbohydrates, protein, and fat; micronutrients such as vitamins, minerals, electrolytes, and trace elements are added in appropriate concentrations. PN may also provide the patient's daily fluid needs. The timing of PN initiation depends upon the patient's initial nutritional status. ASPEN does not recommend PN during the first 7 days of hospitalization in critically ill patients with normal nutritional status. If the patient is not receiving 100% of caloric needs from EN after 7 days, supplemental PN should be considered. However, if on admission a patient is already malnourished and EN is not feasible, PN should be initiated and continued until the patient is receiving at least 60% of caloric needs by enteral route.42 This includes patients with intestinal obstruction, ileus, peritonitis, malabsorption, high output enterocutaneous fistulae, intestinal ischemia, intractable vomiting and diarrhea, severe shock, and fulminant sepsis.10, 43
Standardized commercial PN products are available and reduce the number of steps required between ordering and administration, as compared to customized PN, which is compounded for a particular patient. However, despite improved efficiency and lower cost, there is no evidence that standardized preparations are safer to patients than customized solutions. Institutions utilizing standardized PN must also have a mechanism to customize formulas for those with complex needs.44
Creating a customized parenteral solution involves several basic steps. Total caloric requirement may be estimated using a predictive formula, as previously discussed; calories/kg of ideal body weight is the simplest method. Most hospitalized patients require 2030 calories/kg/d. Daily fluid requirement may be based on kilocalories (kcal) delivered, or by ideal body weight (eg, 1 mL/kcal or 3040 mL/kg). More fluid may be needed in patients with significant sensible or insensible losses; those with renal failure or heart failure should receive less fluid.
Protein needs are calculated by multiplying ideal body weight (kg) by estimated protein needs in g/kg/d (1.22 g/kg/d for catabolic patients). Protein should provide approximately 20% of total calories. Protein restriction is not required in renal impairment; acutely ill patients on renal replacement therapy should receive 1.51.8 g/kg/d. In hepatic failure patients, protein should be restricted only if hepatic encephalopathy fails to improve with other measures.
Knowing the protein, kcal, and fluid needs of the patient, the practitioner divides the remaining non‐protein calories between carbohydrates and fat. Approximately 70%85% of non‐protein calories should be provided as carbohydrates (dextrose), up to 7 g/kg/d. The other 15%30% are as fat, in lipid solutions, providing a maximum of 2.5 g/kg/d. Lipid solutions are provided as either 10% (1.1 kcal/mL) or 20% (2.2 kcal/mL) concentrations.43, 44 Propofol's contribution to fat intake complicates estimating total fat intake in critically ill patients.45
Standardized parenteral multivitamin preparations are available; the clinician must determine if preparations containing vitamin K are appropriate. Of the trace elements, copper and manganese should be restricted in hepatobiliary disease.44
Acutely ill patients receive PN as a 24‐hour infusion, to minimize its impact on volume status and energy expenditure,46 providing 50% of needs on infusion day one and reaching goal within 4872 hours, rather than cyclic infusions over shorter intervals. Daily assessments of vital signs, intake and output, and weight are necessary to monitor volume status.
Once a patient is taking at least 60% of caloric needs either by mouth or by EN, PN can be discontinued. Tapering the infusion is not required, as abrupt discontinuation has not been demonstrated to cause symptomatic hypoglycemia.47, 48
PATIENT MONITORING
Laboratory monitoring with nutrition support should include baseline electrolytes, glucose, renal function, coagulation studies, triglycerides, magnesium, phosphorus, cholesterol, platelet count, and hepatobiliary enzymes. Electrolytes, calcium, magnesium, and phosphorus should be checked daily for 3 days and, if normal, should then be checked biweekly. Capillary glucose should be monitored several times a day until stable. Weekly triglycerides, albumin, cholesterol, coagulation studies, and liver enzymes should also be checked in patients while on parenteral nutrition.25 Patients at risk for refeeding syndrome should have potassium, phosphate, calcium, and magnesium measured daily for 7 days, with repletion as necessary. These electrolytes should be monitored 3 times the following week if stable.49
Patients should be monitored clinically for gastrointestinal tolerance of enteral nutrition. All 3 North American guidelines recommend monitoring gastric residual volumes (GRV); however, there is no consensus on the volume considered to require intervention. Motility agents are recommended as first line treatment of high GRV.36, 37, 42 If high GRV continues, tube feeding should be held, and tube placement, medications, and metabolic assessment should be reviewed. Placement of a transpyloric feeding tube may be indicated.50
Adverse Effects and Complications of Nutrition Support
Regarding EN, complications include those related to tube placement and maintenance, infections, and medical complications of the feeds themselves. Some of the adverse effects of the enteral formulas may be attenuated. Diarrhea, which occurs in up to 20% of patients, may be avoided with slow feed advancement, use of low‐osmotic formulas, or fiber additives.51 Gastric distention and abdominal pain may improve with slow feed advancement and continuous (rather than bolus) feeds. Small‐bore tubes and acid‐reducing medications may decrease gastroesophageal reflux, and aspiration pneumonia may be avoided by semi‐recumbent positioning and post‐pyloric feeding.52
Complications of PN may be grouped as mechanical, infectious, and metabolic. The mechanical complications of central line placement include pneumothorax, arterial puncture, hematoma, air embolism, and line malpositioning. Catheter‐related deep venous thrombosis may occur. Patients on PN through a central line are at risk for central line‐associated bloodstream infections.25 The metabolic complications such as hyperglycemia, electrolyte disorders, hepatic steatosis, and volume overload may have severe consequences, such as heart failure or neuromuscular dysfunction, thus they require close attention.53
A complication of nutrition support that may occur regardless of route is the refeeding syndrome. Refeeding syndrome describes fluid shifts and electrolyte abnormalities that occur after initiation of oral, enteral, or parenteral nutrition in a malnourished or starved patient.54, 55 There are no formal criteria for diagnosing refeeding syndrome.
In the starved state, the body switches from carbohydrate to protein and fat metabolism. Reintroduction of carbohydrates stimulates insulin release with glycogen, fat, and protein synthesis. Associated uptake of glucose, potassium, magnesium, phosphate, and water into cells causes electrolyte and fluid abnormalities. Although hypophosphatemia is the hallmark of refeeding syndrome, it is not pathognomonic. Additional disturbances include hypokalemia, hyperglycemia, hypomagnesemia, thiamine deficiency, and fluid imbalance.49 Patients at risk of refeeding should have serum electrolytes, magnesium, phosphorus, and glucose checked before nutrition support starts. The degree of laboratory abnormalities, if any, and the clinical course of refeeding guides the frequency of subsequent blood tests.56 These consequences of refeeding can adversely affect every major organ system and may result in death.57
Starvation physiology underlies all risk factors for refeeding syndrome. In hospitalized patients, those at risk for refeeding include, but are not limited to, the elderly, oncology patients, postoperative patients, alcohol‐dependent patients, those with malabsorptive states, those who are fasting or chronically malnourished, and those on diuretic therapy.54, 57 The National Institute for Health and Clinical Excellence (NICE) of England and Wales has published criteria to identify patients at high risk for refeeding (Table 3).56 Identification of at‐risk patients and attention to their nutritional needs prevents refeeding syndrome.
|
| Patient has 1 or more of the following: |
| BMI <16 kg/m2 |
| Unintentional weight loss >15% within the last 36 mo |
| Little or no nutritional intake for more than 10 d |
| Low levels of potassium, phosphate, or magnesium prior to feeding |
| Or patient has 2 or more of the following: |
| BMI <18.5 kg/m2 |
| Unintentional weight loss >10% within the last 36 mo |
| Little or no nutritional intake for more than 5 d |
| A history of alcohol abuse or drugs including insulin, chemotherapy, antacids, or diuretics |
ASPEN and NICE have each issued guidelines for initiating nutrition support in patients at risk for refeeding. ASPEN guidelines recommend feeding start at approximately 25% of the estimated goal, with advancement to goal over 35 days. ASPEN recommends fluid and electrolyte status be monitored as needed.50 The NICE guidelines recommend starting nutrition support at a maximum of 10 kcal/kg/d with slow increase to meet or exceed full needs by 47 days. For extremely malnourished patients (eg, BMI <14 kg/m2, or negligible intake for >15 days), they recommend starting at 5 kcal/kg/d. For patients at high risk of developing refeeding syndrome, the NICE guidelines recommend vitamin repletion immediately before and during the first 10 days of feeding (thiamine, vitamin B, and a balanced multivitamin/trace element supplement). Cardiac monitoring is recommended for this group as well as any patients who are at risk for cardiac arrhythmias. Careful monitoring of fluid balance and restoring circulatory volume is recommended, as is repletion of potassium, phosphate, and magnesium.56
TERMINATION OF THERAPY
Termination of nutrition support often involves transitioning from one mode of support to another. PN can be discontinued when oral or enteral intake reaches 60% of total calories; enteral intake can be discontinued when oral intake reaches the same level. However, the patient should be observed maintaining their intake; if they cannot, nutrition support should be resumed.12
TRANSITION OF CARE PLAN
Patients discharged from the hospital on enteral or parenteral nutrition require the support of a coordinated multidisciplinary team including dietitians, home nutrition delivery companies, primary care physicians trained in specialized nutrition support, community pharmacists, and other healthcare professionals, if indicated. These relationships should be established prior to discharge, with education about the patient's individualized nutrition plan, and training with the equipment and supplies.10, 56
CONCLUSION
This review provides an overview of managing the at‐risk or malnourished patient by describing the processes of screening, assessment, and development and implementation of a nutrition care plan in the acutely ill hospitalized patient. Malnutrition is a relatively common, yet underdiagnosed entity that impacts patient outcomes, length of stay, hospital costs, and readmissions. Acute illness in a patient already nutritionally debilitated by chronic disease may cause rapid depletion in nutritional stores. Hospitals are required to screen patients for malnutrition on admission and at regular intervals, and to develop and implement a nutrition care plan for those at risk. The plan guides how nutrition therapy is provided, monitored for adequacy and adverse effects, and assessed for achievement of nutritional goals. It encompasses the use of dietary modifications, and enteral and parenteral nutrition. Clinicians must be aware of serious but avoidable adverse effects, particularly refeeding syndrome in malnourished patients. Prior to discharge, the patient should have already been transitioned from EN or PN to taking adequate amounts of calories by mouth; otherwise, careful discharge planning to educate the patients and/or caregivers, and coordinate the necessary multidisciplinary community services is necessary.
Acknowledgements
The authors express their appreciation to Ms Susan Lundy, for her helpful and timely information, and Ms Lisa Boucher, for her invaluable assistance with this manuscript and its submission.
Disclosures: Susan Brantley is on the Speaker's Bureau for Nestle Nutrition and for Abbott Nutrition. Authors Kirkland, Kashiwagi, Scheurer, and Varkey have nothing to report.
- ,,, et al.Prevalence of malnutrition on admission to four hospitals in England. The Malnutrition Prevalence Group.Clin Nutr.2000;19(3):191–195.
- ,.The impact of malnutrition on morbidity, mortality, length of hospital stay and costs evaluated through a multivariate model analysis.Clin Nutr.2003;22(3):235–239.
- ,,,.Malnutrition among hospitalized patients. A problem of physician awareness.Arch Intern Med.1987;147(8):1462–1465.
- ,,,,.Malnutrition is prevalent in hospitalized medical patients: are housestaff identifying the malnourished patient?Nutrition.2006;22(4):350–354.
- ,,, et al.Malnutrition in subacute care.Am J Clin Nutr.2002;75(2):308–313.
- ,,, et al.Malnutrition is an independent factor associated with nosocomial infections.Br J Nutr.2004;92(1):105–111.
- ,.Impact of body mass index on outcomes following critical care.Chest.2003;123(4):1202–1207.
- ,,, et al.EuroOOPS: an international, multicentre study to implement nutritional risk screening and evaluate clinical outcome.Clin Nutr.2008;27(3):340–349.
- ,,,,.Consensus statement: Academy of Nutrition and Dietetics and American Society for Parenteral and Enteral Nutrition: characteristics recommended for the identification and documentation of adult malnutrition (undernutrition).J Parenter Enteral Nutr.2012;36(3):275–283.
- ,,, et al.Standards for nutrition support: adult hospitalized patients.Nutr Clin Pract.2010;25(4):403–414.
- American Society for Parenteral and Enteral Nutrition (A.S.P.E.N.), Board of Directors and Clinical Practice Committee. Definition of Terms, Style, and Conventions used in A.S.P.E.N. Board of Directors‐Approved Documents. May2012. Available at: http://www.nutritioncare.org/Library.aspx. Accessed June 29, 2012.
- Joint Commission on Accreditation of Healthcare Organizations.Comprehensive Accreditation for Hospitals.Chicago, IL:Joint Commission on Accreditation for Healthcare Organizations;2007.
- ,.Nutrition screening and assessment. In: Gottschlich MM, ed.The ASPEN Nutrition Support Core Curriculum: A Case‐Based Approach—The Adult Patient.1st ed.Silver Spring, MD:American Society for Parenteral and Enteral Nutrition;2007:163–186.
- .Nutrition screening tools for hospitalized patients.Nutr Clin Pract.2008;23(4):373–382.
- .Nutrition screening and assessment. In: Skipper A, ed.Dietitian's Handbook of Enteral and Parenteral Nutrition.3rd ed.Sudbury, MA:Jones 2012:4–21.
- ,.Nutritional assessment in the hospitalized patient.Curr Opin Clin Nutr Metab Care.2003;6(5):531–538.
- ,,,,.Nutritional and metabolic assessment of the hospitalized patient.J Parenter Enteral Nutr.1977;1(1):11–22.
- ,,.Hepatic proteins and nutrition assessment.J Am Diet Assoc.2004;104(8):1258–1264.
- .The albumin‐nutrition connection: separating myth from fact.Nutrition.2002;18(2):199–200.
- ,.Predictive equations for energy needs for the critically ill.Respir Care.2009;54(4):509–521.
- ,,, et al.Guidelines, guidelines, guidelines: what are we to do with all of these North American guidelines?J Parenter Enteral Nutr.2010;34(6):625–643.
- ,,, et al.Applied nutrition in ICU patients. A consensus statement of the American College of Chest Physicians.Chest.1997;111(3):769–778.
- ,,,,.Comparison of resting energy expenditure prediction methods with measured resting energy expenditure in obese, hospitalized adults.J Parenter Enteral Nutr.2009;33(2):168–175.
- ,,,,,.A new predictive equation for resting energy expenditure in healthy individuals.Am J Clin Nutr.1990;51(2):241–247.
- American Society for Parenteral and Enteral Nutrition.Guidelines for the use of parenteral and enteral nutrition in adult and pediatric patients.J Parenter Enteral Nutr.2002;26(suppl):1SA–138SA.
- ,,.American Gastroenterological Association technical review on tube feeding for enteral nutrition.Gastroenterology.1995;108(4):1282–1301.
- ,,, et al.ESPEN guidelines on enteral nutrition: intensive care.Clin Nutr.2006;25(2):210–223.
- ,.Nutritional support. In: Shojania KG, Duncan BW, McDonald KM, et al, eds.Making Health Care Safer: A Critical Analysis of Patient Safety Practices. Evidence Report/Technology Assessment Number 43.Rockville, MD:Agency for Healthcare Research and Quality, US Department of Health and Human Services, July2001. AHRQ Publication 01‐E058. Available at: http://www.ahrq.gov.
- Veterans Affairs Total Parenteral Nutrition Cooperative Study Group.Perioperative total parenteral nutrition in surgical patients.N Engl J Med.1991;328(8):525–532.
- ,,,,.Does enteral nutrition affect clinical outcome? A systematic review of the randomized trials.Am J Gastroenterol.2007;102(2):412–429.
- ,,,.Nutrition in the stroke patient.Nutr Clin Pract.2011;26(3):242–252.
- ,,.Nutrition diagnosis and intervention. In: Mahan LK, Escott‐Stump S, eds.Krause's Food and Nutrition Therapy.12th ed.St Louis, MO:Saunders Elsevier;2008:454–469.
- Dietary Supplement Health and Education Act (DSHEA) of 1994. Available at: http://www.gpo.gov/fdsys/pkg/BILLS‐103s784es/pdf/BILLS‐103s784es.pdf. Accessed June 29,2012.
- .Intervention: dietary supplementation and integrative care. In: Mahan LK, Escott‐Stump S, eds.Krause's Food and Nutrition Therapy.12th ed.St Louis, MO:Saunders Elsevier;2008:470–474.
- ,,, et al.Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine and American Society for Parenteral and Enteral Nutrition: executive summary.Crit Care Med.2009;37(5):1757–1761.
- ,,,,;for the. Canadian Critical Care Clinical Practice Guidelines Committee.Canadian clinical practice guidelines for nutrition support in mechanically ventilated, critically ill adult patients.J Parenter Enteral Nutr.2003;27(5):355–373.
- American Dietetic Association Evidence Library. Critical Illness. Available at: http://www.adaevidencelibrary.com/template.cfm?key=767115(5 suppl):64S–70S.
- .Enteral nutrition. In: Skipper A, ed.Dietitian's Handbook of Enteral and Parenteral Nutrition.3rd ed.Sudbury, MA:Jones 2012:259–280.
- .Enteral formula selection. In: Charney P, Malone A, eds.ADA Pocket Guide to Enteral Nutrition.Chicago, IL:American Dietetic Association;2006:63–122.
- ,.Overview of enteral nutrition. In: Gottschlich MM, ed.The ASPEN Nutrition Support Core Curriculum: A Case‐Based Approach—The Adult Patient.1st ed.Silver Spring, MD:American Society for Parenteral and Enteral Nutrition;2007:187–208.
- ,,, et al.Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine (SCCM) and American Society for Parenteral and Enteral Nutrition (A.S.P.E.N.).J Parenter Enteral Nutr.2009;33(3):277–316.
- ,,,,,.ESPEN guidelines on parenteral nutrition: surgery.Clin Nutr.2009;28(4):378–386.
- ,,, et al.Safe practices for parenteral nutrition.J Parenter Enteral Nutr.2004;28(6):S39–S70.
- ,,,,,.Contribution of calories from propofol to total energy intake.J Am Diet Assoc.1995;95(9 supplement):A25.
- ,,,.Metabolic and thermogenic response to continuous and cyclic total parenteral nutrition in traumatised and infected patients.Clin Nutr.1994;13(5):291–301.
- ,,.The effect of abrupt cessation of total parenteral nutrition on serum glucose: a randomized trial.Am Surg.2000;66(9):866–869.
- ,,,,.The effect of acute discontinuation of total parenteral nutrition.Ann Surg.1986;204(5):524–529.
- ,,.Refeeding syndrome: what it is, and how to prevent and treat it.BMJ.2008;336(7659):1495–1498.
- ,,, et al.Enteral nutrition practice recommendations.J Parenter Enteral Nutr.2009;33(2):122–167.
- ,,.Control of diarrhea by fiber‐enriched diet in ICU patients on enteral nutrition: a prospective randomized controlled trial.Clin Nutr.2004;23(6):1344–1352.
- ,,,,,.Supine body position as a risk factor for nosocomial pneumonia in mechanically ventilated patients: a randomised trial.Lancet.1999;354(9193):1851–1858.
- .Parenteral nutrition in the critically ill patient.N Engl J Med.2009;361(11):1088–1097.
- ,,,.Refeeding syndrome: treatment considerations based on collective analysis of literature case reports.Nutrition.2010;26(2):156–167.
- ,,.Re‐feeding syndrome in head and neck cancer‐prevention and management.Oral Oncol.2011;47(9):792–796.
- Nutrition Support in Adults. NICE Clinical Guideline No. 32. 2006. Available at: http://guidance.nice.org.uk/CG32/NICEGuidance. Accessed November 29,2011.
- ,,.The importance of the refeeding syndrome.Nutrition.2001;17(7–8):632–637.
- ,,, et al.Prevalence of malnutrition on admission to four hospitals in England. The Malnutrition Prevalence Group.Clin Nutr.2000;19(3):191–195.
- ,.The impact of malnutrition on morbidity, mortality, length of hospital stay and costs evaluated through a multivariate model analysis.Clin Nutr.2003;22(3):235–239.
- ,,,.Malnutrition among hospitalized patients. A problem of physician awareness.Arch Intern Med.1987;147(8):1462–1465.
- ,,,,.Malnutrition is prevalent in hospitalized medical patients: are housestaff identifying the malnourished patient?Nutrition.2006;22(4):350–354.
- ,,, et al.Malnutrition in subacute care.Am J Clin Nutr.2002;75(2):308–313.
- ,,, et al.Malnutrition is an independent factor associated with nosocomial infections.Br J Nutr.2004;92(1):105–111.
- ,.Impact of body mass index on outcomes following critical care.Chest.2003;123(4):1202–1207.
- ,,, et al.EuroOOPS: an international, multicentre study to implement nutritional risk screening and evaluate clinical outcome.Clin Nutr.2008;27(3):340–349.
- ,,,,.Consensus statement: Academy of Nutrition and Dietetics and American Society for Parenteral and Enteral Nutrition: characteristics recommended for the identification and documentation of adult malnutrition (undernutrition).J Parenter Enteral Nutr.2012;36(3):275–283.
- ,,, et al.Standards for nutrition support: adult hospitalized patients.Nutr Clin Pract.2010;25(4):403–414.
- American Society for Parenteral and Enteral Nutrition (A.S.P.E.N.), Board of Directors and Clinical Practice Committee. Definition of Terms, Style, and Conventions used in A.S.P.E.N. Board of Directors‐Approved Documents. May2012. Available at: http://www.nutritioncare.org/Library.aspx. Accessed June 29, 2012.
- Joint Commission on Accreditation of Healthcare Organizations.Comprehensive Accreditation for Hospitals.Chicago, IL:Joint Commission on Accreditation for Healthcare Organizations;2007.
- ,.Nutrition screening and assessment. In: Gottschlich MM, ed.The ASPEN Nutrition Support Core Curriculum: A Case‐Based Approach—The Adult Patient.1st ed.Silver Spring, MD:American Society for Parenteral and Enteral Nutrition;2007:163–186.
- .Nutrition screening tools for hospitalized patients.Nutr Clin Pract.2008;23(4):373–382.
- .Nutrition screening and assessment. In: Skipper A, ed.Dietitian's Handbook of Enteral and Parenteral Nutrition.3rd ed.Sudbury, MA:Jones 2012:4–21.
- ,.Nutritional assessment in the hospitalized patient.Curr Opin Clin Nutr Metab Care.2003;6(5):531–538.
- ,,,,.Nutritional and metabolic assessment of the hospitalized patient.J Parenter Enteral Nutr.1977;1(1):11–22.
- ,,.Hepatic proteins and nutrition assessment.J Am Diet Assoc.2004;104(8):1258–1264.
- .The albumin‐nutrition connection: separating myth from fact.Nutrition.2002;18(2):199–200.
- ,.Predictive equations for energy needs for the critically ill.Respir Care.2009;54(4):509–521.
- ,,, et al.Guidelines, guidelines, guidelines: what are we to do with all of these North American guidelines?J Parenter Enteral Nutr.2010;34(6):625–643.
- ,,, et al.Applied nutrition in ICU patients. A consensus statement of the American College of Chest Physicians.Chest.1997;111(3):769–778.
- ,,,,.Comparison of resting energy expenditure prediction methods with measured resting energy expenditure in obese, hospitalized adults.J Parenter Enteral Nutr.2009;33(2):168–175.
- ,,,,,.A new predictive equation for resting energy expenditure in healthy individuals.Am J Clin Nutr.1990;51(2):241–247.
- American Society for Parenteral and Enteral Nutrition.Guidelines for the use of parenteral and enteral nutrition in adult and pediatric patients.J Parenter Enteral Nutr.2002;26(suppl):1SA–138SA.
- ,,.American Gastroenterological Association technical review on tube feeding for enteral nutrition.Gastroenterology.1995;108(4):1282–1301.
- ,,, et al.ESPEN guidelines on enteral nutrition: intensive care.Clin Nutr.2006;25(2):210–223.
- ,.Nutritional support. In: Shojania KG, Duncan BW, McDonald KM, et al, eds.Making Health Care Safer: A Critical Analysis of Patient Safety Practices. Evidence Report/Technology Assessment Number 43.Rockville, MD:Agency for Healthcare Research and Quality, US Department of Health and Human Services, July2001. AHRQ Publication 01‐E058. Available at: http://www.ahrq.gov.
- Veterans Affairs Total Parenteral Nutrition Cooperative Study Group.Perioperative total parenteral nutrition in surgical patients.N Engl J Med.1991;328(8):525–532.
- ,,,,.Does enteral nutrition affect clinical outcome? A systematic review of the randomized trials.Am J Gastroenterol.2007;102(2):412–429.
- ,,,.Nutrition in the stroke patient.Nutr Clin Pract.2011;26(3):242–252.
- ,,.Nutrition diagnosis and intervention. In: Mahan LK, Escott‐Stump S, eds.Krause's Food and Nutrition Therapy.12th ed.St Louis, MO:Saunders Elsevier;2008:454–469.
- Dietary Supplement Health and Education Act (DSHEA) of 1994. Available at: http://www.gpo.gov/fdsys/pkg/BILLS‐103s784es/pdf/BILLS‐103s784es.pdf. Accessed June 29,2012.
- .Intervention: dietary supplementation and integrative care. In: Mahan LK, Escott‐Stump S, eds.Krause's Food and Nutrition Therapy.12th ed.St Louis, MO:Saunders Elsevier;2008:470–474.
- ,,, et al.Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine and American Society for Parenteral and Enteral Nutrition: executive summary.Crit Care Med.2009;37(5):1757–1761.
- ,,,,;for the. Canadian Critical Care Clinical Practice Guidelines Committee.Canadian clinical practice guidelines for nutrition support in mechanically ventilated, critically ill adult patients.J Parenter Enteral Nutr.2003;27(5):355–373.
- American Dietetic Association Evidence Library. Critical Illness. Available at: http://www.adaevidencelibrary.com/template.cfm?key=767115(5 suppl):64S–70S.
- .Enteral nutrition. In: Skipper A, ed.Dietitian's Handbook of Enteral and Parenteral Nutrition.3rd ed.Sudbury, MA:Jones 2012:259–280.
- .Enteral formula selection. In: Charney P, Malone A, eds.ADA Pocket Guide to Enteral Nutrition.Chicago, IL:American Dietetic Association;2006:63–122.
- ,.Overview of enteral nutrition. In: Gottschlich MM, ed.The ASPEN Nutrition Support Core Curriculum: A Case‐Based Approach—The Adult Patient.1st ed.Silver Spring, MD:American Society for Parenteral and Enteral Nutrition;2007:187–208.
- ,,, et al.Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine (SCCM) and American Society for Parenteral and Enteral Nutrition (A.S.P.E.N.).J Parenter Enteral Nutr.2009;33(3):277–316.
- ,,,,,.ESPEN guidelines on parenteral nutrition: surgery.Clin Nutr.2009;28(4):378–386.
- ,,, et al.Safe practices for parenteral nutrition.J Parenter Enteral Nutr.2004;28(6):S39–S70.
- ,,,,,.Contribution of calories from propofol to total energy intake.J Am Diet Assoc.1995;95(9 supplement):A25.
- ,,,.Metabolic and thermogenic response to continuous and cyclic total parenteral nutrition in traumatised and infected patients.Clin Nutr.1994;13(5):291–301.
- ,,.The effect of abrupt cessation of total parenteral nutrition on serum glucose: a randomized trial.Am Surg.2000;66(9):866–869.
- ,,,,.The effect of acute discontinuation of total parenteral nutrition.Ann Surg.1986;204(5):524–529.
- ,,.Refeeding syndrome: what it is, and how to prevent and treat it.BMJ.2008;336(7659):1495–1498.
- ,,, et al.Enteral nutrition practice recommendations.J Parenter Enteral Nutr.2009;33(2):122–167.
- ,,.Control of diarrhea by fiber‐enriched diet in ICU patients on enteral nutrition: a prospective randomized controlled trial.Clin Nutr.2004;23(6):1344–1352.
- ,,,,,.Supine body position as a risk factor for nosocomial pneumonia in mechanically ventilated patients: a randomised trial.Lancet.1999;354(9193):1851–1858.
- .Parenteral nutrition in the critically ill patient.N Engl J Med.2009;361(11):1088–1097.
- ,,,.Refeeding syndrome: treatment considerations based on collective analysis of literature case reports.Nutrition.2010;26(2):156–167.
- ,,.Re‐feeding syndrome in head and neck cancer‐prevention and management.Oral Oncol.2011;47(9):792–796.
- Nutrition Support in Adults. NICE Clinical Guideline No. 32. 2006. Available at: http://guidance.nice.org.uk/CG32/NICEGuidance. Accessed November 29,2011.
- ,,.The importance of the refeeding syndrome.Nutrition.2001;17(7–8):632–637.
Male hypogonadism: More than just a low testosterone
Editor’s note: This article on the differential diagnosis of hypogonadism in men is the first of two articles. The second, to be published next month, focuses on the appropriate use of testosterone therapy.
A 54-year-old man is referred for evaluation of low testosterone. He had seen his primary care physician for complaints of diminished libido and erectile dysfunction for the past year and worsening fatigue over the past few years. He has not been formally diagnosed with any medical condition. His serum testosterone level is 180 ng/dL (reference range 249–836 ng/dL).
On physical examination, he is obese (body mass index 31 kg/m2) with a normal-appearing male body habitus, no gynecomastia, and normal testicles and prostate gland.
How should this patient be evaluated?
LOW TESTOSTERONE HAS MANY CAUSES
Male hypogonadism, ie, failure of the testes to produce adequate amounts of androgen or sperm, has become a common clinical finding, particularly in the older population. This is more likely the result of an increase in awareness and detection of the disorder by physicians rather than a true increase in prevalence.
The finding of a low serum testosterone value needs to be confirmed and thoroughly evaluated before starting treatment. It is important to determine whether the cause is a primary (hypergonadotropic) testicular disorder or secondary to a hypothalamic-pituitary process (hypogonadotropic or normogonadotropic).
THE HYPOTHALAMIC-PITUITARY-GONADAL AXIS
Testosterone production is under the control of luteinizing hormone (LH), whereas sperm production is under the control of follicle-stimulating hormone (FSH) (Figure 1). Both of these pituitary hormones are regulated by the pulsatile secretion of hypothalamic gonadotropin-releasing hormone (GnRH).
Testosterone (produced by Leydig cells) and inhibin B (produced by Sertoli cells within the seminiferous tubules) result in negative feedback inhibition of gonadotropin (LH and FSH) secretion. Testosterone and estradiol (produced by aromatization of testosterone) act at both pituitary and hypothalamic sites and are the principal regulators of LH secretion.1,2 Inhibin B is the major regulator of FSH secretion in men,3 but steroid feedback also occurs.2,4
TO FOLLOW UP A LOW TESTOSTERONE, CONFIRM THE VALUE NEAR 8 am
If a testosterone value is found to be low, it is important to determine the time that the sample was obtained. Serum testosterone levels follow a diurnal rhythm, at least in younger men, with values near 8 am being, on average, 30% higher than the trough levels later in the day.5–7 The timing of the diurnal variation may be different in night-shift workers, who may require assessment at a more appropriate time of the day (ie, upon awakening).
Another factor affecting testosterone levels is the patient’s health status at the time of testing. Values obtained in the hospital during an acute illness should be repeated once the event has resolved, as testosterone values decrease considerably in this setting.8 Even in outpatients, particularly in men over age 60, one must be sure that the low testosterone level was not obtained during a period of decompensation of one of the many comorbidities seen in these patients, such as coronary artery disease, congestive heart failure, or uncontrolled diabetes.
If an 8 am testosterone value is low, it is reasonable to obtain at least one confirmatory testosterone level on another day, near 8 am, in the next few weeks, when the patient is in good health. Confirming the testosterone level is important, particularly since commercially available testosterone assays are not well standardized and some are frankly unreliable.9,10 A repeat confirmatory level should always be performed by a reliable reference laboratory. If the testosterone level is still low, further evaluation is warranted.
TOTAL TESTOSTERONE VS BIOAVAILABLE TESTOSTERONE VS FREE TESTOSTERONE
Of the total circulating testosterone, 60% is bound to sex hormone-binding globulin (SHBG), 38% is bound to albumin, and only 2% is free. All of these fractions can be measured to assess for testosterone deficiency.
Free testosterone is the biologically active form of this hormone and, thus, the free testosterone level is considered to be a better representation of the true testosterone status. However, some clinicians believe that bioavailable testosterone (testosterone loosely bound to albumin + free testosterone) is a better reflection of the true level of the active hormone than the level of free testosterone alone.
There are situations in which the total testosterone level is low but bioavailable or free testosterone levels are normal. The level of total testosterone is affected by alterations in the levels of SHBG and albumin. A reduction in the level of SHBG can result in low total serum testosterone levels in patients with obesity or type 2 diabetes (states of insulin resistance), and also in cachexia, malnutrition, advanced cirrhosis, acromegaly, hypothyroidism, and nephrotic syndrome. SHBG can also be low in patients taking glucocorticoids, progestins, or androgenic steroids.11 In these settings, checking the level of free testosterone (the active hormone), bioavailable testosterone, or both, by a reliable reference laboratory, may be more appropriate.9,10
But regardless of which measurement is chosen, all testosterone levels—especially bioavailable and free testosterone values—should be interpreted with caution if they are not measured at a reliable reference laboratory.9,10 Interested readers may wish to see the US Centers for Disease Control and Prevention (CDC) Hormone Standardization Program Web site (www.cdc.gov/labstandards/hs.html) for more details, including a list of CDC-certified laboratories.
CLINICAL FEATURES OF LOW TESTOSTERONE
A history of erectile dysfunction, decreased libido, and fatigue may be seen in patients with low testosterone. However, one must realize that these symptoms—as well as others reported by men with low testosterone, such as depression, difficulty concentrating, irritability, and insomnia—are nonspecific and may be related to other medical conditions.12
Likewise, physical findings such as muscle weakness, reduced body hair, and altered fat distribution (abdominal obesity) are seen in men with low testosterone, but also in those with a number of other medical conditions.
Additional features suggest specific disorders, eg, anosmia in Kallmann syndrome; eunuchoid body habitus, gynecomastia, and small testes in Klinefelter syndrome.
Men with low testosterone may have low bone mineral density or anemia, or both.
Careful examination of the breasts for gynecomastia and the testes for size, consistency, and masses (testicular tumors) helps in formulating a differential diagnosis and in appropriately directing subsequent laboratory evaluation and diagnostic imaging.
LOW TESTOSTERONE: PRIMARY VS SECONDARY
A history of testicular trauma, systemic chemotherapy, or mumps orchitis should direct the physician’s attention to a testicular etiology. On the other hand, darkened or tanned skin (suggesting hemochromatosis), galactorrhea (suggesting hyperprolactinemia), or visual field deficits (suggesting a sellar mass) should direct the physician’s attention toward a pituitary-hypothalamic process.

Once the low testosterone value has been confirmed at least one time near 8 am, one should obtain LH and FSH values to help direct further evaluation in deciphering the etiology (Figure 2). Elevated (hypergonadotropic) values indicate a testicular disorder (primary hypogonadism), whereas low (hypogonadotropic) or normal (normogonadotropic) values point to a pituitary-hypothalamic process (secondary hypogonadism). It should be emphasized that, in the setting of a low testosterone level, LH and FSH values within the normal range are “inappropriately normal” so that further investigation is required.
This evaluation should also include serum prolactin, thyroid-stimulating hormone (TSH, also known as thyrotropin), free thyroxine (T4), and ferritin levels, the latter because hemochromatosis (iron overload) can cause both primary and secondary hypogonadism. If at any time in the evaluation the laboratory results suggest secondary hypogonadism, a full assessment of pituitary function should be undertaken.
Semen analysis is usually reserved for patients presenting with the primary complaint of infertility.
PRIMARY HYPOGONADISM
The patient should be carefully questioned about the age at which his problems began, about pubertal development, and about fertility. Causes of primary hypogonadism include:
- Karyotype abnormalities—Klinefelter syndrome (47, XXY syndrome) is the most common
- Toxin exposure, chemotherapy
- Congenital defects—anorchia, cryptorchidism13
- Orchitis (mumps, autoimmune)
- Testicular trauma or infarction
- Hemochromatosis
- Medications that inhibit androgen biosynthesis, eg, ketoconazole (Nizoral)14
- Increase in temperature of the testicular environment (due to varicocele or a large panniculus).
SECONDARY HYPOGONADISM
Causes of secondary hypogonadism include the following:
Congenital disorders
These disorders are usually diagnosed in childhood or adolescence, often after the patient is brought to the physician because of short stature or pubertal delay.
- Kallmann syndrome (anosmia and GnRH deficiency)15
- GnRH receptor mutation and deficiency16
- Genetic mutations associated with pituitary hormone deficiencies, eg, PROP-1 mutation.17
Acquired disorders that suppress gonadotrophs
Drugs. Long-term therapy with common medications such as opioids or corticosteroids can result in secondary hypogonadism.18–20 Others are GnRH analogues such as leuprolide (Lupron), which are used in treating advanced prostate cancer. The hypogonadism is usually transient and resolves after stopping the offending agent.
Obesity and related conditions such as obstructive sleep apnea, insulin resistance, and type 2 diabetes mellitus are associated with low testosterone levels.21 Treatment should be directed at these underlying conditions and should include lifestyle measures such as weight loss and exercise, rather than simple prescribing of testosterone supplementation, as these efforts may provide multiple health benefits in addition to raising testosterone levels.22
Insulin resistance. In the setting of obesity, the total testosterone level may be low but the bioavailable and free testosterone (active hormone) levels may be normal. This is due to the effect of hyperinsulinemia on the liver, which results in a reduction in SHBG production.23 Low levels of both total and free testosterone can be seen in morbid obesity,24 but the cause remains unclear.
Type 2 diabetes mellitus. Testosterone levels have been reported to be lower in obese men who have diabetes than in those with obesity alone.24 This decrement, comparable in magnitude to that seen with other chronic diseases, suggests that low testosterone may simply be a marker of poor health.22,25,26
Sleep apnea. Disturbances in the sleep cycle, regardless of the underlying cause, can result in decreases in serum testosterone levels. Often, correcting the underlying sleep disturbance can result in a normalization of serum testosterone levels.27,28 A caveat about testosterone therapy: a thorough evaluation for sleep apnea should be undertaken in patients at high risk, since testosterone replacement therapy can adversely affect ventilatory drive and induce or worsen obstructive sleep apnea.29
Aging. Most reports have shown an agerelated decline in both total and free serum testosterone levels (commonly referred to as “andropause”), particularly in men over 60 years of age. There also appears to be a loss of circadian rhythm,30 although not all reports agree.6 It appears that factors such as functional status and overall health may play a more important role in the pathophysiology of hypogonadism in men of advanced age than age alone.
Hemochromatosis. Iron overload, regardless of the cause, can result in hypogonadism via deposition of iron in the hypothalamus, pituitary, or testes. Hereditary hemochromatosis is a common autosomal recessive disease characterized by increased iron absorption. Although both primary and secondary hypogonadism can occur with long-standing iron overload, the latter is much more common.31 Some cases of hypogonadism have been reported to reverse with iron depletion therapy.32
Hyperprolactinemia. Recognized causes of hyperprolactinemia in men include medications (dopamine antagonists, antipsychotics, metoclopramide [Reglan]), pituitary adenomas (microadenomas < 10 mm, macroadenomas ≥ 10 mm), lactotroph hyperfunction (stalk compression interrupting or reducing the tonic suppression of prolactin secretion by dopamine), hypothyroidism, stress, chronic renal failure, cirrhosis, chest wall injury (trauma), and active herpes zoster. The ensuing hypogonadism may be due to the compressive effect of a sellar mass or the direct effect of the prolactin elevation alone, since prolactin disrupts the pulsatile release of GnRH from the hypothalamus,33 required for normal LH and FSH secretion.
Estrogen excess can be either exogenous (from exposure to estrogen-containing contraceptives and creams) or endogenous (from testicular34,35 or very rare adrenal36 estrogen-secreting tumors). Of note, some cases of testicular neoplasms may be detectable only with ultrasonography. Computed tomography may be performed if an adrenal lesion is suspected.
Anabolic steroid abuse. Exposure to anabolic steroids, deliberately or inadvertently, can result in secondary hypogonadism and testicular atrophy, both of which may persist for years after stopping the anabolic agents. If you suspect anabolic steroid abuse, a urine anabolic steroid screen can be obtained.
Anorexia nervosa is far less common in men than in women.37,38 Elements in the history that suggest this disorder include excessive exercise and a low body mass index. Chronic malnutrition (cachexia), regardless of the cause, can result in secondary hypogonadism.
Acute illness (gonadotroph sick syndrome). Hypogonadism is a relatively common finding in any critical illness (analogous to euthyroid sick syndrome with respect to the hypothalamic-pituitary-thyroid axis).8 Testosterone levels are invariably low, so that assessment of testosterone status is not recommended in this setting. The low testosterone phase is usually transient and resolves with resolution or improvement of the underlying medical condition, such as sepsis or myocardial infarction.
HIV. Human immunodeficiency virus (HIV) infection can result in primary or secondary hypogonadism. It can occur with active HIV infection, in patients in whom control of viral replication has been achieved with highly active antiretroviral therapy, and even in patients who have normalized CD4+ cell counts.39 Hypogonadism in HIV patients is multifactorial and may be related to weight loss, opportunistic infections of the pituitary-hypothalamus or testes, or medications such as opioids (licit or illicit), ganciclovir (Cytovene), ketoconazole, the appetite stimulant megestrol (Megace), or cyclophosphamide (Cytoxan). Testosterone replacement therapy does not adversely affect the HIV disease process and in fact may help to avoid complications.
Chronic medical conditions such as cirrhosis, renal failure, and rheumatoid arthritis commonly result in hypogonadism, the pathogenesis of which may involve dysfunction at all levels of the hypothalamic-pituitary-go-nadal axis.40–45 Hypogonadism in the setting of chronic disease is multifactorial, being due not only to the metabolic disturbances seen with these illnesses (uremia in renal failure, elevated circulating estrogens in liver cirrhosis), but also to recurrent acute illness and hospitalization for infection in these immuno-compromised hosts, either from the underlying medical condition or as a result of medications (corticosteroids).
Alcohol abuse. Alcohol can have adverse effects at all levels of the hypothalamic-pituitary-gonadal axis, resulting in low serum testosterone and reduced spermatogenesis.46
Severe chronic primary hypothyroidism, manifested by an extreme elevation of serum thyroid-stimulating hormone (TSH), can result in hypopituitarism. Pituitary function usually recovers with restoration of euthyroidism.47,48
Pubertal delay. Depending on the age of presentation, differentiating pubertal delay from permanent hypogonadotropic hypogonadism can be challenging.
Acquired disorders that damage gonadotrophs
- Sellar mass or cyst—pituitary adenoma, craniopharyngioma, Rathke cleft cyst, meningioma
- Infiltrative lesion—lymphocytic hypophysitis, Langerhans cell histiocytosis, hemochromatosis, sarcoidosis, infection
- Metastatic lesion
- Trauma (head injury)
- Radiation exposure
- Surgery
- Stalk severance
- Pituitary apoplexy.
See Table 1 for a summary of the causes of male hypogonadism.
WHEN IS MRI INDICATED IN EVALUATING SECONDARY HYPOGONADISM?

The yield of pituitary-hypothalamic imaging in older men with secondary hypogonadism is fairly low in the absence of other pituitary hormone abnormalities and deficiencies. There are limited data regarding appropriate criteria for performing hypothalamic-pituitary imaging studies. However, a patient who has multiple anterior pituitary abnormalities on laboratory evaluation should undergo dedicated hypothalamic-pituitary magnetic resonance imaging (MRI).
The Endocrine Society Clinical Practice Guidelines11 recommend that MRI be performed to exclude a pituitary or hypothalamic tumor or infiltrative disease if the patient has severe secondary hypogonadism (serum testosterone < 150 ng/dL), panhypopituitarism, persistent hyperprolactinemia, or symptoms or signs of tumor mass effect such as headache, visual impairment, or a visual field defect.
WHO SHOULD UNDERGO ASSESSMENT OF TESTOSTERONE STATUS?
Screening for androgen deficiency in the asymptomatic general population is not recommended.11 The nonspecific nature of many of the signs and symptoms of androgen deficiency makes it difficult to give concrete recommendations as to who should have testosterone levels measured. Clinicians should consider testing if there is evidence of certain clinical disorders that are associated with low testosterone levels (see earlier discussion on the specific causes of primary and secondary hypogonadism).
When a male patient complains of erectile dysfunction, the investigation should include an assessment of serum testosterone. However, if a man who has a constellation of nonspecific symptoms asks for his testosterone level to be assessed (which is common, given the aggressive marketing of testosterone replacement by the pharmaceutical industry), we would recommend a basic evaluation that includes a comprehensive metabolic panel, complete blood count, and TSH level. Further testing should be determined by the history and physical examination. If no obvious explanation has been found for the patient’s symptoms at that point, assessment of serum testosterone may be warranted. More often than not the patient’s weight and limited physical activity are the driving forces behind the nonspecific symptoms, and counseling a patient on a life-style change can provide much benefit if the patient follows through with the physician’s recommendations.
Men whom we believe should not undergo assessment for testosterone deficiency are those who are acutely ill and hospitalized and those who are severely obese and are complaining of fatigue. Testosterone levels should be assessed only after the acute illness has resolved and, in a severely obese patient with fatigue, only after a thorough evaluation for sleep apnea has been undertaken.
TREAT THE UNDERLYING CAUSE, IF ONE CAN BE FOUND
If the evaluation of low testosterone leads to the diagnosis of a clear underlying condition that is amenable to treatment, such as prolactin elevation or sleep apnea, then treatment should be directed at the underlying cause, with subsequent monitoring of the patient’s symptoms and response in serum testosterone levels. In general, the use of dopamine agonist therapy in the management of hyperprolactinemia and, in cases of panhypopituitarism, of replacement therapy with levothyroxine (Synthroid), hydrocortisone, and possibly growth hormone and desmopressin (DDAVP), fall best under the purview of an endocrinologist. A caveat: serum TSH cannot be used to monitor levothyroxine replacement therapy in cases of secondary hypothyroidism. The clinical picture and serum free T4 and free T3 levels are used instead.
In the absence of a correctable (or immediately correctable) cause, testosterone supplementation can be initiated on an individualized basis in select patients who have clinical signs and symptoms of androgen deficiency if the benefits of treatment appear to outweigh the potential risks, and only after a thorough discussion with the patient.11 The Endocrine Society recommends against offering testosterone therapy to all older men with low testosterone.11
INFERTILITY
In men presenting with low serum testosterone, semen analysis is not routine. It is usually reserved for patients presenting with the primary complaint of infertility.
If an endocrine disorder such as prolactin elevation or hypothyroidism is the suspected cause of infertility, the patient should be referred to an endocrinologist for further evaluation and management. Treatment of male infertility should be directed at the underlying cause, but often requires exogenous human chorionic gonadotropin, FSH, GnRH (via a pulsatile pump), and possibly sperm harvesting from the testis with subsequent in vitro fertilization with intracytoplasmic sperm injection. It is critical that the partner be included in the evaluation of infertility.
These patients should be referred to a urologic or fertility center specializing in the diagnosis and treatment of infertility. For further information regarding male infertility, patients can be directed to www.fertilitylifelines.com.
CASE CONCLUDED
The patient’s low serum testosterone was confirmed on subsequent measurements at 8 am, with levels of 128 and 182 ng/dL (reference range 249–836). Other laboratory values:
- LH 1.4 mIU/mL (reference range 1.2–8.6)
- FSH 2.7 mIU/mL (1.3–9.9 mIU/mL)
(Both of these values are inappropriately normal in the setting of the low testosterone.)
- TSH 248 μIU/mL (0.4–5.5)
- Prolactin 24.6 ng/mL (1.6–18.8).
The patient was started on levothyroxine replacement therapy and after 3 months was noted to be euthyroid (TSH 1.8 μIU/mL) and to have a normal serum prolactin level. Testosterone levels (8 am) at this time were 350 ng/dL and 420 ng/dL.
Therefore, the cause of this patient’s hypogonadism was severe hypothyroidism and associated mild hyperprolactinemia. This case shows that a thorough evaluation is warranted before initiating testosterone therapy.
- Pitteloud N, Dwyer AA, DeCruz S, et al. Inhibition of luteinizing hormone secretion by testosterone in men requires aromatization for its pituitary but not its hypo-thalamic effects: evidence from the tandem study of normal and gonadotropin-releasing hormone-deficient men. J Clin Endocrinol Metab 2008; 93:784–791.
- Hayes FJ, DeCruz S, Seminara SB, Boepple PA, Crowley WF. Differential regulation of gonadotropin secretion by testosterone in the human male: absence of a negative feedback effect of testosterone on follicle-stimulating hormone secretion. J Clin Endocrinol Metab 2001; 86:53–58.
- Hayes FJ, Pitteloud N, DeCruz S, Crowley WF, Boepple PA. Importance of inhibin B in the regulation of FSH secretion in the human male. J Clin Endocrinol Metab 2001; 86:5541–5546.
- Pitteloud N, Dwyer AA, DeCruz S, et al. The relative role of gonadal sex steroids and gonadotropin-releasing hormone pulse frequency in the regulation of follicle-stimulating hormone secretion in men. J Clin Endocrinol Metab 2008; 93:2686–2692.
- Cooke RR, McIntosh JE, McIntosh RP. Circadian variation in serum free and non-SHBG-bound testosterone in normal men: measurements, and simulation using a mass action model. Clin Endocrinol (Oxf) 1993; 39:163–171.
- Diver MJ, Imtiaz KE, Ahmad AM, Vora JP, Fraser WD. Diurnal rhythms of serum total, free and bioavailable testosterone and of SHBG in middle-aged men compared with those in young men. Clin Endocrinol (Oxf) 2003; 58:710–717.
- Clair P, Claustrat B, Jordan D, Dechaud H, Sassolas G. Daily variations of plasma sex hormone-binding globulin binding capacity, testosterone and luteinizing hormone concentrations in healthy rested adult males. Horm Res 1985; 21:220–223.
- Woolf PD, Hamill RW, McDonald JV, Lee LA, Kelly M. Transient hypogonadotropic hypogonadism caused by critical illness. J Clin Endocrinol Metab 1985; 60:444–450.
- Rosner W, Auchus RJ, Azziz R, Sluss PM, Raff H. Position statement: utility, limitations, and pitfalls in measuring testosterone: an Endocrine Society position statement. J Clin Endocrinol Metab 2007; 92:405–413.
- Rosner W, Vesper H, et al; Endocrine Society; American Association for Clinical Chemistry; American Association of Clinical Endocrinologists; et al. Toward excellence in testosterone testing: a consensus statement. J Clin Endocrinol Metab 2010; 95:4542–4548.
- Bhasin S, Cunningham GR, Hayes FJ, et al; Task Force, Endocrine Society. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2010; 95:2536–2559.
- Wu FC, Tajar A, Beynon JM, et al; EMAS Group. Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med 2010; 363:123–135.
- Farrer JH, Sikka SC, Xie HW, Constantinide D, Rajfer J. Impaired testosterone biosynthesis in cryptorchidism. Fertil Steril 1985; 44:125–132.
- Sikka SC, Swerdloff RS, Rajfer J. In vitro inhibition of testosterone biosynthesis by ketoconazole. Endocrinology 1985; 116:1920–1925.
- Pallais JC, Au M, Pitteloud N, Seminara S, Crowley WF Jr. Kallmann syndrome. In:Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, eds. GeneReviews™ (Internet). Seattle, WA: University of Washington; 1993.
- Chevrier L, Guimiot F, de Roux N. GnRH receptor mutations in isolated gonadotropic deficiency. Mol Cell Endocrinol 2011; 346:21–28.
- Romero CJ, Pine-Twaddell E, Radovick S. Novel mutations associated with combined pituitary hormone deficiency. J Mol Endocrinol 2011; 46:R93–R102.
- Colameco S, Coren JS, Ciervo CA. Continuous opioid treatment for chronic noncancer pain: a time for moderation in prescribing. Postgrad Med 2009; 121:61–66.
- Fraser LA, Morrison D, Morley-Forster P, et al. Oral opioids for chronic non-cancer pain: higher prevalence of hypogonadism in men than in women. Exp Clin Endocrinol Diabetes 2009; 117:38–43.
- Morrison D, Capewell S, Reynolds SP, et al. Testosterone levels during systemic and inhaled corticosteroid therapy. Respir Med 1994; 88:659–663.
- Mah PM, Wittert GA. Obesity and testicular function. Mol Cell Endocrinol 2010; 316:180–186.
- Grossmann M. Low testosterone in men with type 2 diabetes: significance and treatment. J Clin Endocrinol Metab 2011; 96:2341–2353.
- Gascón F, Valle M, Martos R, et al. Sex hormone-binding globulin as a marker for hyperinsulinemia and/or insulin resistance in obese children. Eur J Endocrinol 2000; 143:85–89.
- Dhindsa S, Miller MG, McWhirter CL, et al. Testosterone concentrations in diabetic and nondiabetic obese men. Diabetes Care 2010; 33:1186–1192.
- Grossmann M, Gianatti EJ, Zajac JD. Testosterone and type 2 diabetes. Curr Opin Endocrinol Diabetes Obes 2010; 17:247–256.
- Andersson B, Mårin P, Lissner L, Vermeulen A, Björntorp P. Testosterone concentrations in women and men with NIDDM. Diabetes Care 1994; 17:405–411.
- Santamaria JD, Prior JC, Fleetham JA. Reversible reproductive dysfunction in men with obstructive sleep apnoea. Clin Endocrinol (Oxf) 1988; 28:461–470.
- Grunstein RR, Handelsman DJ, Lawrence SJ, Blackwell C, Caterson ID, Sullivan CE. Neuroendocrine dysfunction in sleep apnea: reversal by continuous positive airways pressure therapy. J Clin Endocrinol Metab 1989; 68:352–358.
- Matsumoto AM, Sandblom RE, Schoene RB, et al. Testosterone replacement in hypogonadal men: effects on obstructive sleep apnoea, respiratory drives, and sleep. Clin Endocrinol (Oxf) 1985; 22:713–721.
- Bremner WJ, Vitiello MV, Prinz PN. Loss of circadian rhythmicity in blood testosterone levels with aging in normal men. J Clin Endocrinol Metab 1983; 56:1278–1281.
- McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab 2005; 90:2451–2455.
- Kelly TM, Edwards CQ, Meikle AW, Kushner JP. Hypogonadism in hemochromatosis: reversal with iron depletion. Ann Intern Med 1984; 101:629–632.
- Milenkovic L, D’Angelo G, Kelly PA, Weiner RI. Inhibition of gonadotropin hormone-releasing hormone release by prolactin from GT1 neuronal cell lines through prolactin receptors. Proc Natl Acad Sci U S A 1994; 91:1244–1247.
- Valensi P, Coussieu C, Kemeny JL, Attali JR, Amouroux J, Sebaoun J. Endocrine investigations in two cases of feminizing Leydig cell tumour. Acta Endocrinol (Copenh) 1987; 115:365–372.
- Young S, Gooneratne S, Straus FH, Zeller WP, Bulun SE, Rosenthal IM. Feminizing Sertoli cell tumors in boys with Peutz-Jeghers syndrome. Am J Surg Pathol 1995; 19:50–58.
- Zayed A, Stock JL, Liepman MK, Wollin M, Longcope C. Feminization as a result of both peripheral conversion of androgens and direct estrogen production from an adrenocortical carcinoma. J Endocrinol Invest 1994; 17:275–278.
- Russ MJ, Ackerman SH, Barakat R, Levy B. Hypogonadotropic hypogonadism and delayed puberty in a man with anorexia nervosa. Psychosomatics 1986; 27:737–739.
- Rigotti NA, Neer RM, Jameson L. Osteopenia and bone fractures in a man with anorexia nervosa and hypogonadism. JAMA 1986; 256:385–388.
- Cohan GR. HIV-associated hypogonadism. AIDS Read 2006; 16:341–345,348,352–354.
- Handelsman DJ, Strasser S, McDonald JA, Conway AJ, McCaughan GW. Hypothalamic-pituitary-testicular function in end-stage nonalcoholic liver disease before and after liver transplantation. Clin Endocrinol (Oxf) 1995; 43:331–337.
- Lim VS, Fang VS. Gonadal dysfunction in uremic men. A study of the hypothalamo-pituitary-testicular axis before and after renal transplantation. Am J Med 1975; 58:655–662.
- Handelsman DJ, Dong Q. Hypothalamo-pituitary gonadal axis in chronic renal failure. Endocrinol Metab Clin North Am 1993; 22:145–161.
- Handelsman DJ, Spaliviero JA, Turtle JR. Hypothalamic-pituitary function in experimental uremic hypogonadism. Endocrinology 1985; 117:1984–1995.
- Tengstrand B, Carlström K, Hafström I. Bioavailable testosterone in men with rheumatoid arthritis-high frequency of hypogonadism. Rheumatology (Oxford) 2002; 41:285–289.
- Tengstrand B, Carlström K, Hafström I. Gonadal hormones in men with rheumatoid arthritis--from onset through 2 years. J Rheumatol 2009; 36:887–892.
- Emanuele MA, Emanuele NV. Alcohol’s effects on male reproduction. Alcohol Health Res World 1998; 22:195–201.
- Meikle AW. The interrelationships between thyroid dysfunction and hypogonadism in men and boys. Thyroid 2004; 14( suppl 1):S17–S25.
- Vagenakis AG, Dole K, Braverman LE. Pituitary enlargement, pituitary failure, and primary hypothyroidism. Ann Intern Med 1976; 85:195–198.
Editor’s note: This article on the differential diagnosis of hypogonadism in men is the first of two articles. The second, to be published next month, focuses on the appropriate use of testosterone therapy.
A 54-year-old man is referred for evaluation of low testosterone. He had seen his primary care physician for complaints of diminished libido and erectile dysfunction for the past year and worsening fatigue over the past few years. He has not been formally diagnosed with any medical condition. His serum testosterone level is 180 ng/dL (reference range 249–836 ng/dL).
On physical examination, he is obese (body mass index 31 kg/m2) with a normal-appearing male body habitus, no gynecomastia, and normal testicles and prostate gland.
How should this patient be evaluated?
LOW TESTOSTERONE HAS MANY CAUSES
Male hypogonadism, ie, failure of the testes to produce adequate amounts of androgen or sperm, has become a common clinical finding, particularly in the older population. This is more likely the result of an increase in awareness and detection of the disorder by physicians rather than a true increase in prevalence.
The finding of a low serum testosterone value needs to be confirmed and thoroughly evaluated before starting treatment. It is important to determine whether the cause is a primary (hypergonadotropic) testicular disorder or secondary to a hypothalamic-pituitary process (hypogonadotropic or normogonadotropic).
THE HYPOTHALAMIC-PITUITARY-GONADAL AXIS
Testosterone production is under the control of luteinizing hormone (LH), whereas sperm production is under the control of follicle-stimulating hormone (FSH) (Figure 1). Both of these pituitary hormones are regulated by the pulsatile secretion of hypothalamic gonadotropin-releasing hormone (GnRH).
Testosterone (produced by Leydig cells) and inhibin B (produced by Sertoli cells within the seminiferous tubules) result in negative feedback inhibition of gonadotropin (LH and FSH) secretion. Testosterone and estradiol (produced by aromatization of testosterone) act at both pituitary and hypothalamic sites and are the principal regulators of LH secretion.1,2 Inhibin B is the major regulator of FSH secretion in men,3 but steroid feedback also occurs.2,4
TO FOLLOW UP A LOW TESTOSTERONE, CONFIRM THE VALUE NEAR 8 am
If a testosterone value is found to be low, it is important to determine the time that the sample was obtained. Serum testosterone levels follow a diurnal rhythm, at least in younger men, with values near 8 am being, on average, 30% higher than the trough levels later in the day.5–7 The timing of the diurnal variation may be different in night-shift workers, who may require assessment at a more appropriate time of the day (ie, upon awakening).
Another factor affecting testosterone levels is the patient’s health status at the time of testing. Values obtained in the hospital during an acute illness should be repeated once the event has resolved, as testosterone values decrease considerably in this setting.8 Even in outpatients, particularly in men over age 60, one must be sure that the low testosterone level was not obtained during a period of decompensation of one of the many comorbidities seen in these patients, such as coronary artery disease, congestive heart failure, or uncontrolled diabetes.
If an 8 am testosterone value is low, it is reasonable to obtain at least one confirmatory testosterone level on another day, near 8 am, in the next few weeks, when the patient is in good health. Confirming the testosterone level is important, particularly since commercially available testosterone assays are not well standardized and some are frankly unreliable.9,10 A repeat confirmatory level should always be performed by a reliable reference laboratory. If the testosterone level is still low, further evaluation is warranted.
TOTAL TESTOSTERONE VS BIOAVAILABLE TESTOSTERONE VS FREE TESTOSTERONE
Of the total circulating testosterone, 60% is bound to sex hormone-binding globulin (SHBG), 38% is bound to albumin, and only 2% is free. All of these fractions can be measured to assess for testosterone deficiency.
Free testosterone is the biologically active form of this hormone and, thus, the free testosterone level is considered to be a better representation of the true testosterone status. However, some clinicians believe that bioavailable testosterone (testosterone loosely bound to albumin + free testosterone) is a better reflection of the true level of the active hormone than the level of free testosterone alone.
There are situations in which the total testosterone level is low but bioavailable or free testosterone levels are normal. The level of total testosterone is affected by alterations in the levels of SHBG and albumin. A reduction in the level of SHBG can result in low total serum testosterone levels in patients with obesity or type 2 diabetes (states of insulin resistance), and also in cachexia, malnutrition, advanced cirrhosis, acromegaly, hypothyroidism, and nephrotic syndrome. SHBG can also be low in patients taking glucocorticoids, progestins, or androgenic steroids.11 In these settings, checking the level of free testosterone (the active hormone), bioavailable testosterone, or both, by a reliable reference laboratory, may be more appropriate.9,10
But regardless of which measurement is chosen, all testosterone levels—especially bioavailable and free testosterone values—should be interpreted with caution if they are not measured at a reliable reference laboratory.9,10 Interested readers may wish to see the US Centers for Disease Control and Prevention (CDC) Hormone Standardization Program Web site (www.cdc.gov/labstandards/hs.html) for more details, including a list of CDC-certified laboratories.
CLINICAL FEATURES OF LOW TESTOSTERONE
A history of erectile dysfunction, decreased libido, and fatigue may be seen in patients with low testosterone. However, one must realize that these symptoms—as well as others reported by men with low testosterone, such as depression, difficulty concentrating, irritability, and insomnia—are nonspecific and may be related to other medical conditions.12
Likewise, physical findings such as muscle weakness, reduced body hair, and altered fat distribution (abdominal obesity) are seen in men with low testosterone, but also in those with a number of other medical conditions.
Additional features suggest specific disorders, eg, anosmia in Kallmann syndrome; eunuchoid body habitus, gynecomastia, and small testes in Klinefelter syndrome.
Men with low testosterone may have low bone mineral density or anemia, or both.
Careful examination of the breasts for gynecomastia and the testes for size, consistency, and masses (testicular tumors) helps in formulating a differential diagnosis and in appropriately directing subsequent laboratory evaluation and diagnostic imaging.
LOW TESTOSTERONE: PRIMARY VS SECONDARY
A history of testicular trauma, systemic chemotherapy, or mumps orchitis should direct the physician’s attention to a testicular etiology. On the other hand, darkened or tanned skin (suggesting hemochromatosis), galactorrhea (suggesting hyperprolactinemia), or visual field deficits (suggesting a sellar mass) should direct the physician’s attention toward a pituitary-hypothalamic process.
Once the low testosterone value has been confirmed at least one time near 8 am, one should obtain LH and FSH values to help direct further evaluation in deciphering the etiology (Figure 2). Elevated (hypergonadotropic) values indicate a testicular disorder (primary hypogonadism), whereas low (hypogonadotropic) or normal (normogonadotropic) values point to a pituitary-hypothalamic process (secondary hypogonadism). It should be emphasized that, in the setting of a low testosterone level, LH and FSH values within the normal range are “inappropriately normal” so that further investigation is required.
This evaluation should also include serum prolactin, thyroid-stimulating hormone (TSH, also known as thyrotropin), free thyroxine (T4), and ferritin levels, the latter because hemochromatosis (iron overload) can cause both primary and secondary hypogonadism. If at any time in the evaluation the laboratory results suggest secondary hypogonadism, a full assessment of pituitary function should be undertaken.
Semen analysis is usually reserved for patients presenting with the primary complaint of infertility.
PRIMARY HYPOGONADISM
The patient should be carefully questioned about the age at which his problems began, about pubertal development, and about fertility. Causes of primary hypogonadism include:
- Karyotype abnormalities—Klinefelter syndrome (47, XXY syndrome) is the most common
- Toxin exposure, chemotherapy
- Congenital defects—anorchia, cryptorchidism13
- Orchitis (mumps, autoimmune)
- Testicular trauma or infarction
- Hemochromatosis
- Medications that inhibit androgen biosynthesis, eg, ketoconazole (Nizoral)14
- Increase in temperature of the testicular environment (due to varicocele or a large panniculus).
SECONDARY HYPOGONADISM
Causes of secondary hypogonadism include the following:
Congenital disorders
These disorders are usually diagnosed in childhood or adolescence, often after the patient is brought to the physician because of short stature or pubertal delay.
- Kallmann syndrome (anosmia and GnRH deficiency)15
- GnRH receptor mutation and deficiency16
- Genetic mutations associated with pituitary hormone deficiencies, eg, PROP-1 mutation.17
Acquired disorders that suppress gonadotrophs
Drugs. Long-term therapy with common medications such as opioids or corticosteroids can result in secondary hypogonadism.18–20 Others are GnRH analogues such as leuprolide (Lupron), which are used in treating advanced prostate cancer. The hypogonadism is usually transient and resolves after stopping the offending agent.
Obesity and related conditions such as obstructive sleep apnea, insulin resistance, and type 2 diabetes mellitus are associated with low testosterone levels.21 Treatment should be directed at these underlying conditions and should include lifestyle measures such as weight loss and exercise, rather than simple prescribing of testosterone supplementation, as these efforts may provide multiple health benefits in addition to raising testosterone levels.22
Insulin resistance. In the setting of obesity, the total testosterone level may be low but the bioavailable and free testosterone (active hormone) levels may be normal. This is due to the effect of hyperinsulinemia on the liver, which results in a reduction in SHBG production.23 Low levels of both total and free testosterone can be seen in morbid obesity,24 but the cause remains unclear.
Type 2 diabetes mellitus. Testosterone levels have been reported to be lower in obese men who have diabetes than in those with obesity alone.24 This decrement, comparable in magnitude to that seen with other chronic diseases, suggests that low testosterone may simply be a marker of poor health.22,25,26
Sleep apnea. Disturbances in the sleep cycle, regardless of the underlying cause, can result in decreases in serum testosterone levels. Often, correcting the underlying sleep disturbance can result in a normalization of serum testosterone levels.27,28 A caveat about testosterone therapy: a thorough evaluation for sleep apnea should be undertaken in patients at high risk, since testosterone replacement therapy can adversely affect ventilatory drive and induce or worsen obstructive sleep apnea.29
Aging. Most reports have shown an agerelated decline in both total and free serum testosterone levels (commonly referred to as “andropause”), particularly in men over 60 years of age. There also appears to be a loss of circadian rhythm,30 although not all reports agree.6 It appears that factors such as functional status and overall health may play a more important role in the pathophysiology of hypogonadism in men of advanced age than age alone.
Hemochromatosis. Iron overload, regardless of the cause, can result in hypogonadism via deposition of iron in the hypothalamus, pituitary, or testes. Hereditary hemochromatosis is a common autosomal recessive disease characterized by increased iron absorption. Although both primary and secondary hypogonadism can occur with long-standing iron overload, the latter is much more common.31 Some cases of hypogonadism have been reported to reverse with iron depletion therapy.32
Hyperprolactinemia. Recognized causes of hyperprolactinemia in men include medications (dopamine antagonists, antipsychotics, metoclopramide [Reglan]), pituitary adenomas (microadenomas < 10 mm, macroadenomas ≥ 10 mm), lactotroph hyperfunction (stalk compression interrupting or reducing the tonic suppression of prolactin secretion by dopamine), hypothyroidism, stress, chronic renal failure, cirrhosis, chest wall injury (trauma), and active herpes zoster. The ensuing hypogonadism may be due to the compressive effect of a sellar mass or the direct effect of the prolactin elevation alone, since prolactin disrupts the pulsatile release of GnRH from the hypothalamus,33 required for normal LH and FSH secretion.
Estrogen excess can be either exogenous (from exposure to estrogen-containing contraceptives and creams) or endogenous (from testicular34,35 or very rare adrenal36 estrogen-secreting tumors). Of note, some cases of testicular neoplasms may be detectable only with ultrasonography. Computed tomography may be performed if an adrenal lesion is suspected.
Anabolic steroid abuse. Exposure to anabolic steroids, deliberately or inadvertently, can result in secondary hypogonadism and testicular atrophy, both of which may persist for years after stopping the anabolic agents. If you suspect anabolic steroid abuse, a urine anabolic steroid screen can be obtained.
Anorexia nervosa is far less common in men than in women.37,38 Elements in the history that suggest this disorder include excessive exercise and a low body mass index. Chronic malnutrition (cachexia), regardless of the cause, can result in secondary hypogonadism.
Acute illness (gonadotroph sick syndrome). Hypogonadism is a relatively common finding in any critical illness (analogous to euthyroid sick syndrome with respect to the hypothalamic-pituitary-thyroid axis).8 Testosterone levels are invariably low, so that assessment of testosterone status is not recommended in this setting. The low testosterone phase is usually transient and resolves with resolution or improvement of the underlying medical condition, such as sepsis or myocardial infarction.
HIV. Human immunodeficiency virus (HIV) infection can result in primary or secondary hypogonadism. It can occur with active HIV infection, in patients in whom control of viral replication has been achieved with highly active antiretroviral therapy, and even in patients who have normalized CD4+ cell counts.39 Hypogonadism in HIV patients is multifactorial and may be related to weight loss, opportunistic infections of the pituitary-hypothalamus or testes, or medications such as opioids (licit or illicit), ganciclovir (Cytovene), ketoconazole, the appetite stimulant megestrol (Megace), or cyclophosphamide (Cytoxan). Testosterone replacement therapy does not adversely affect the HIV disease process and in fact may help to avoid complications.
Chronic medical conditions such as cirrhosis, renal failure, and rheumatoid arthritis commonly result in hypogonadism, the pathogenesis of which may involve dysfunction at all levels of the hypothalamic-pituitary-go-nadal axis.40–45 Hypogonadism in the setting of chronic disease is multifactorial, being due not only to the metabolic disturbances seen with these illnesses (uremia in renal failure, elevated circulating estrogens in liver cirrhosis), but also to recurrent acute illness and hospitalization for infection in these immuno-compromised hosts, either from the underlying medical condition or as a result of medications (corticosteroids).
Alcohol abuse. Alcohol can have adverse effects at all levels of the hypothalamic-pituitary-gonadal axis, resulting in low serum testosterone and reduced spermatogenesis.46
Severe chronic primary hypothyroidism, manifested by an extreme elevation of serum thyroid-stimulating hormone (TSH), can result in hypopituitarism. Pituitary function usually recovers with restoration of euthyroidism.47,48
Pubertal delay. Depending on the age of presentation, differentiating pubertal delay from permanent hypogonadotropic hypogonadism can be challenging.
Acquired disorders that damage gonadotrophs
- Sellar mass or cyst—pituitary adenoma, craniopharyngioma, Rathke cleft cyst, meningioma
- Infiltrative lesion—lymphocytic hypophysitis, Langerhans cell histiocytosis, hemochromatosis, sarcoidosis, infection
- Metastatic lesion
- Trauma (head injury)
- Radiation exposure
- Surgery
- Stalk severance
- Pituitary apoplexy.
See Table 1 for a summary of the causes of male hypogonadism.
WHEN IS MRI INDICATED IN EVALUATING SECONDARY HYPOGONADISM?
The yield of pituitary-hypothalamic imaging in older men with secondary hypogonadism is fairly low in the absence of other pituitary hormone abnormalities and deficiencies. There are limited data regarding appropriate criteria for performing hypothalamic-pituitary imaging studies. However, a patient who has multiple anterior pituitary abnormalities on laboratory evaluation should undergo dedicated hypothalamic-pituitary magnetic resonance imaging (MRI).
The Endocrine Society Clinical Practice Guidelines11 recommend that MRI be performed to exclude a pituitary or hypothalamic tumor or infiltrative disease if the patient has severe secondary hypogonadism (serum testosterone < 150 ng/dL), panhypopituitarism, persistent hyperprolactinemia, or symptoms or signs of tumor mass effect such as headache, visual impairment, or a visual field defect.
WHO SHOULD UNDERGO ASSESSMENT OF TESTOSTERONE STATUS?
Screening for androgen deficiency in the asymptomatic general population is not recommended.11 The nonspecific nature of many of the signs and symptoms of androgen deficiency makes it difficult to give concrete recommendations as to who should have testosterone levels measured. Clinicians should consider testing if there is evidence of certain clinical disorders that are associated with low testosterone levels (see earlier discussion on the specific causes of primary and secondary hypogonadism).
When a male patient complains of erectile dysfunction, the investigation should include an assessment of serum testosterone. However, if a man who has a constellation of nonspecific symptoms asks for his testosterone level to be assessed (which is common, given the aggressive marketing of testosterone replacement by the pharmaceutical industry), we would recommend a basic evaluation that includes a comprehensive metabolic panel, complete blood count, and TSH level. Further testing should be determined by the history and physical examination. If no obvious explanation has been found for the patient’s symptoms at that point, assessment of serum testosterone may be warranted. More often than not the patient’s weight and limited physical activity are the driving forces behind the nonspecific symptoms, and counseling a patient on a life-style change can provide much benefit if the patient follows through with the physician’s recommendations.
Men whom we believe should not undergo assessment for testosterone deficiency are those who are acutely ill and hospitalized and those who are severely obese and are complaining of fatigue. Testosterone levels should be assessed only after the acute illness has resolved and, in a severely obese patient with fatigue, only after a thorough evaluation for sleep apnea has been undertaken.
TREAT THE UNDERLYING CAUSE, IF ONE CAN BE FOUND
If the evaluation of low testosterone leads to the diagnosis of a clear underlying condition that is amenable to treatment, such as prolactin elevation or sleep apnea, then treatment should be directed at the underlying cause, with subsequent monitoring of the patient’s symptoms and response in serum testosterone levels. In general, the use of dopamine agonist therapy in the management of hyperprolactinemia and, in cases of panhypopituitarism, of replacement therapy with levothyroxine (Synthroid), hydrocortisone, and possibly growth hormone and desmopressin (DDAVP), fall best under the purview of an endocrinologist. A caveat: serum TSH cannot be used to monitor levothyroxine replacement therapy in cases of secondary hypothyroidism. The clinical picture and serum free T4 and free T3 levels are used instead.
In the absence of a correctable (or immediately correctable) cause, testosterone supplementation can be initiated on an individualized basis in select patients who have clinical signs and symptoms of androgen deficiency if the benefits of treatment appear to outweigh the potential risks, and only after a thorough discussion with the patient.11 The Endocrine Society recommends against offering testosterone therapy to all older men with low testosterone.11
INFERTILITY
In men presenting with low serum testosterone, semen analysis is not routine. It is usually reserved for patients presenting with the primary complaint of infertility.
If an endocrine disorder such as prolactin elevation or hypothyroidism is the suspected cause of infertility, the patient should be referred to an endocrinologist for further evaluation and management. Treatment of male infertility should be directed at the underlying cause, but often requires exogenous human chorionic gonadotropin, FSH, GnRH (via a pulsatile pump), and possibly sperm harvesting from the testis with subsequent in vitro fertilization with intracytoplasmic sperm injection. It is critical that the partner be included in the evaluation of infertility.
These patients should be referred to a urologic or fertility center specializing in the diagnosis and treatment of infertility. For further information regarding male infertility, patients can be directed to www.fertilitylifelines.com.
CASE CONCLUDED
The patient’s low serum testosterone was confirmed on subsequent measurements at 8 am, with levels of 128 and 182 ng/dL (reference range 249–836). Other laboratory values:
- LH 1.4 mIU/mL (reference range 1.2–8.6)
- FSH 2.7 mIU/mL (1.3–9.9 mIU/mL)
(Both of these values are inappropriately normal in the setting of the low testosterone.)
- TSH 248 μIU/mL (0.4–5.5)
- Prolactin 24.6 ng/mL (1.6–18.8).
The patient was started on levothyroxine replacement therapy and after 3 months was noted to be euthyroid (TSH 1.8 μIU/mL) and to have a normal serum prolactin level. Testosterone levels (8 am) at this time were 350 ng/dL and 420 ng/dL.
Therefore, the cause of this patient’s hypogonadism was severe hypothyroidism and associated mild hyperprolactinemia. This case shows that a thorough evaluation is warranted before initiating testosterone therapy.
Editor’s note: This article on the differential diagnosis of hypogonadism in men is the first of two articles. The second, to be published next month, focuses on the appropriate use of testosterone therapy.
A 54-year-old man is referred for evaluation of low testosterone. He had seen his primary care physician for complaints of diminished libido and erectile dysfunction for the past year and worsening fatigue over the past few years. He has not been formally diagnosed with any medical condition. His serum testosterone level is 180 ng/dL (reference range 249–836 ng/dL).
On physical examination, he is obese (body mass index 31 kg/m2) with a normal-appearing male body habitus, no gynecomastia, and normal testicles and prostate gland.
How should this patient be evaluated?
LOW TESTOSTERONE HAS MANY CAUSES
Male hypogonadism, ie, failure of the testes to produce adequate amounts of androgen or sperm, has become a common clinical finding, particularly in the older population. This is more likely the result of an increase in awareness and detection of the disorder by physicians rather than a true increase in prevalence.
The finding of a low serum testosterone value needs to be confirmed and thoroughly evaluated before starting treatment. It is important to determine whether the cause is a primary (hypergonadotropic) testicular disorder or secondary to a hypothalamic-pituitary process (hypogonadotropic or normogonadotropic).
THE HYPOTHALAMIC-PITUITARY-GONADAL AXIS
Testosterone production is under the control of luteinizing hormone (LH), whereas sperm production is under the control of follicle-stimulating hormone (FSH) (Figure 1). Both of these pituitary hormones are regulated by the pulsatile secretion of hypothalamic gonadotropin-releasing hormone (GnRH).
Testosterone (produced by Leydig cells) and inhibin B (produced by Sertoli cells within the seminiferous tubules) result in negative feedback inhibition of gonadotropin (LH and FSH) secretion. Testosterone and estradiol (produced by aromatization of testosterone) act at both pituitary and hypothalamic sites and are the principal regulators of LH secretion.1,2 Inhibin B is the major regulator of FSH secretion in men,3 but steroid feedback also occurs.2,4
TO FOLLOW UP A LOW TESTOSTERONE, CONFIRM THE VALUE NEAR 8 am
If a testosterone value is found to be low, it is important to determine the time that the sample was obtained. Serum testosterone levels follow a diurnal rhythm, at least in younger men, with values near 8 am being, on average, 30% higher than the trough levels later in the day.5–7 The timing of the diurnal variation may be different in night-shift workers, who may require assessment at a more appropriate time of the day (ie, upon awakening).
Another factor affecting testosterone levels is the patient’s health status at the time of testing. Values obtained in the hospital during an acute illness should be repeated once the event has resolved, as testosterone values decrease considerably in this setting.8 Even in outpatients, particularly in men over age 60, one must be sure that the low testosterone level was not obtained during a period of decompensation of one of the many comorbidities seen in these patients, such as coronary artery disease, congestive heart failure, or uncontrolled diabetes.
If an 8 am testosterone value is low, it is reasonable to obtain at least one confirmatory testosterone level on another day, near 8 am, in the next few weeks, when the patient is in good health. Confirming the testosterone level is important, particularly since commercially available testosterone assays are not well standardized and some are frankly unreliable.9,10 A repeat confirmatory level should always be performed by a reliable reference laboratory. If the testosterone level is still low, further evaluation is warranted.
TOTAL TESTOSTERONE VS BIOAVAILABLE TESTOSTERONE VS FREE TESTOSTERONE
Of the total circulating testosterone, 60% is bound to sex hormone-binding globulin (SHBG), 38% is bound to albumin, and only 2% is free. All of these fractions can be measured to assess for testosterone deficiency.
Free testosterone is the biologically active form of this hormone and, thus, the free testosterone level is considered to be a better representation of the true testosterone status. However, some clinicians believe that bioavailable testosterone (testosterone loosely bound to albumin + free testosterone) is a better reflection of the true level of the active hormone than the level of free testosterone alone.
There are situations in which the total testosterone level is low but bioavailable or free testosterone levels are normal. The level of total testosterone is affected by alterations in the levels of SHBG and albumin. A reduction in the level of SHBG can result in low total serum testosterone levels in patients with obesity or type 2 diabetes (states of insulin resistance), and also in cachexia, malnutrition, advanced cirrhosis, acromegaly, hypothyroidism, and nephrotic syndrome. SHBG can also be low in patients taking glucocorticoids, progestins, or androgenic steroids.11 In these settings, checking the level of free testosterone (the active hormone), bioavailable testosterone, or both, by a reliable reference laboratory, may be more appropriate.9,10
But regardless of which measurement is chosen, all testosterone levels—especially bioavailable and free testosterone values—should be interpreted with caution if they are not measured at a reliable reference laboratory.9,10 Interested readers may wish to see the US Centers for Disease Control and Prevention (CDC) Hormone Standardization Program Web site (www.cdc.gov/labstandards/hs.html) for more details, including a list of CDC-certified laboratories.
CLINICAL FEATURES OF LOW TESTOSTERONE
A history of erectile dysfunction, decreased libido, and fatigue may be seen in patients with low testosterone. However, one must realize that these symptoms—as well as others reported by men with low testosterone, such as depression, difficulty concentrating, irritability, and insomnia—are nonspecific and may be related to other medical conditions.12
Likewise, physical findings such as muscle weakness, reduced body hair, and altered fat distribution (abdominal obesity) are seen in men with low testosterone, but also in those with a number of other medical conditions.
Additional features suggest specific disorders, eg, anosmia in Kallmann syndrome; eunuchoid body habitus, gynecomastia, and small testes in Klinefelter syndrome.
Men with low testosterone may have low bone mineral density or anemia, or both.
Careful examination of the breasts for gynecomastia and the testes for size, consistency, and masses (testicular tumors) helps in formulating a differential diagnosis and in appropriately directing subsequent laboratory evaluation and diagnostic imaging.
LOW TESTOSTERONE: PRIMARY VS SECONDARY
A history of testicular trauma, systemic chemotherapy, or mumps orchitis should direct the physician’s attention to a testicular etiology. On the other hand, darkened or tanned skin (suggesting hemochromatosis), galactorrhea (suggesting hyperprolactinemia), or visual field deficits (suggesting a sellar mass) should direct the physician’s attention toward a pituitary-hypothalamic process.
Once the low testosterone value has been confirmed at least one time near 8 am, one should obtain LH and FSH values to help direct further evaluation in deciphering the etiology (Figure 2). Elevated (hypergonadotropic) values indicate a testicular disorder (primary hypogonadism), whereas low (hypogonadotropic) or normal (normogonadotropic) values point to a pituitary-hypothalamic process (secondary hypogonadism). It should be emphasized that, in the setting of a low testosterone level, LH and FSH values within the normal range are “inappropriately normal” so that further investigation is required.
This evaluation should also include serum prolactin, thyroid-stimulating hormone (TSH, also known as thyrotropin), free thyroxine (T4), and ferritin levels, the latter because hemochromatosis (iron overload) can cause both primary and secondary hypogonadism. If at any time in the evaluation the laboratory results suggest secondary hypogonadism, a full assessment of pituitary function should be undertaken.
Semen analysis is usually reserved for patients presenting with the primary complaint of infertility.
PRIMARY HYPOGONADISM
The patient should be carefully questioned about the age at which his problems began, about pubertal development, and about fertility. Causes of primary hypogonadism include:
- Karyotype abnormalities—Klinefelter syndrome (47, XXY syndrome) is the most common
- Toxin exposure, chemotherapy
- Congenital defects—anorchia, cryptorchidism13
- Orchitis (mumps, autoimmune)
- Testicular trauma or infarction
- Hemochromatosis
- Medications that inhibit androgen biosynthesis, eg, ketoconazole (Nizoral)14
- Increase in temperature of the testicular environment (due to varicocele or a large panniculus).
SECONDARY HYPOGONADISM
Causes of secondary hypogonadism include the following:
Congenital disorders
These disorders are usually diagnosed in childhood or adolescence, often after the patient is brought to the physician because of short stature or pubertal delay.
- Kallmann syndrome (anosmia and GnRH deficiency)15
- GnRH receptor mutation and deficiency16
- Genetic mutations associated with pituitary hormone deficiencies, eg, PROP-1 mutation.17
Acquired disorders that suppress gonadotrophs
Drugs. Long-term therapy with common medications such as opioids or corticosteroids can result in secondary hypogonadism.18–20 Others are GnRH analogues such as leuprolide (Lupron), which are used in treating advanced prostate cancer. The hypogonadism is usually transient and resolves after stopping the offending agent.
Obesity and related conditions such as obstructive sleep apnea, insulin resistance, and type 2 diabetes mellitus are associated with low testosterone levels.21 Treatment should be directed at these underlying conditions and should include lifestyle measures such as weight loss and exercise, rather than simple prescribing of testosterone supplementation, as these efforts may provide multiple health benefits in addition to raising testosterone levels.22
Insulin resistance. In the setting of obesity, the total testosterone level may be low but the bioavailable and free testosterone (active hormone) levels may be normal. This is due to the effect of hyperinsulinemia on the liver, which results in a reduction in SHBG production.23 Low levels of both total and free testosterone can be seen in morbid obesity,24 but the cause remains unclear.
Type 2 diabetes mellitus. Testosterone levels have been reported to be lower in obese men who have diabetes than in those with obesity alone.24 This decrement, comparable in magnitude to that seen with other chronic diseases, suggests that low testosterone may simply be a marker of poor health.22,25,26
Sleep apnea. Disturbances in the sleep cycle, regardless of the underlying cause, can result in decreases in serum testosterone levels. Often, correcting the underlying sleep disturbance can result in a normalization of serum testosterone levels.27,28 A caveat about testosterone therapy: a thorough evaluation for sleep apnea should be undertaken in patients at high risk, since testosterone replacement therapy can adversely affect ventilatory drive and induce or worsen obstructive sleep apnea.29
Aging. Most reports have shown an agerelated decline in both total and free serum testosterone levels (commonly referred to as “andropause”), particularly in men over 60 years of age. There also appears to be a loss of circadian rhythm,30 although not all reports agree.6 It appears that factors such as functional status and overall health may play a more important role in the pathophysiology of hypogonadism in men of advanced age than age alone.
Hemochromatosis. Iron overload, regardless of the cause, can result in hypogonadism via deposition of iron in the hypothalamus, pituitary, or testes. Hereditary hemochromatosis is a common autosomal recessive disease characterized by increased iron absorption. Although both primary and secondary hypogonadism can occur with long-standing iron overload, the latter is much more common.31 Some cases of hypogonadism have been reported to reverse with iron depletion therapy.32
Hyperprolactinemia. Recognized causes of hyperprolactinemia in men include medications (dopamine antagonists, antipsychotics, metoclopramide [Reglan]), pituitary adenomas (microadenomas < 10 mm, macroadenomas ≥ 10 mm), lactotroph hyperfunction (stalk compression interrupting or reducing the tonic suppression of prolactin secretion by dopamine), hypothyroidism, stress, chronic renal failure, cirrhosis, chest wall injury (trauma), and active herpes zoster. The ensuing hypogonadism may be due to the compressive effect of a sellar mass or the direct effect of the prolactin elevation alone, since prolactin disrupts the pulsatile release of GnRH from the hypothalamus,33 required for normal LH and FSH secretion.
Estrogen excess can be either exogenous (from exposure to estrogen-containing contraceptives and creams) or endogenous (from testicular34,35 or very rare adrenal36 estrogen-secreting tumors). Of note, some cases of testicular neoplasms may be detectable only with ultrasonography. Computed tomography may be performed if an adrenal lesion is suspected.
Anabolic steroid abuse. Exposure to anabolic steroids, deliberately or inadvertently, can result in secondary hypogonadism and testicular atrophy, both of which may persist for years after stopping the anabolic agents. If you suspect anabolic steroid abuse, a urine anabolic steroid screen can be obtained.
Anorexia nervosa is far less common in men than in women.37,38 Elements in the history that suggest this disorder include excessive exercise and a low body mass index. Chronic malnutrition (cachexia), regardless of the cause, can result in secondary hypogonadism.
Acute illness (gonadotroph sick syndrome). Hypogonadism is a relatively common finding in any critical illness (analogous to euthyroid sick syndrome with respect to the hypothalamic-pituitary-thyroid axis).8 Testosterone levels are invariably low, so that assessment of testosterone status is not recommended in this setting. The low testosterone phase is usually transient and resolves with resolution or improvement of the underlying medical condition, such as sepsis or myocardial infarction.
HIV. Human immunodeficiency virus (HIV) infection can result in primary or secondary hypogonadism. It can occur with active HIV infection, in patients in whom control of viral replication has been achieved with highly active antiretroviral therapy, and even in patients who have normalized CD4+ cell counts.39 Hypogonadism in HIV patients is multifactorial and may be related to weight loss, opportunistic infections of the pituitary-hypothalamus or testes, or medications such as opioids (licit or illicit), ganciclovir (Cytovene), ketoconazole, the appetite stimulant megestrol (Megace), or cyclophosphamide (Cytoxan). Testosterone replacement therapy does not adversely affect the HIV disease process and in fact may help to avoid complications.
Chronic medical conditions such as cirrhosis, renal failure, and rheumatoid arthritis commonly result in hypogonadism, the pathogenesis of which may involve dysfunction at all levels of the hypothalamic-pituitary-go-nadal axis.40–45 Hypogonadism in the setting of chronic disease is multifactorial, being due not only to the metabolic disturbances seen with these illnesses (uremia in renal failure, elevated circulating estrogens in liver cirrhosis), but also to recurrent acute illness and hospitalization for infection in these immuno-compromised hosts, either from the underlying medical condition or as a result of medications (corticosteroids).
Alcohol abuse. Alcohol can have adverse effects at all levels of the hypothalamic-pituitary-gonadal axis, resulting in low serum testosterone and reduced spermatogenesis.46
Severe chronic primary hypothyroidism, manifested by an extreme elevation of serum thyroid-stimulating hormone (TSH), can result in hypopituitarism. Pituitary function usually recovers with restoration of euthyroidism.47,48
Pubertal delay. Depending on the age of presentation, differentiating pubertal delay from permanent hypogonadotropic hypogonadism can be challenging.
Acquired disorders that damage gonadotrophs
- Sellar mass or cyst—pituitary adenoma, craniopharyngioma, Rathke cleft cyst, meningioma
- Infiltrative lesion—lymphocytic hypophysitis, Langerhans cell histiocytosis, hemochromatosis, sarcoidosis, infection
- Metastatic lesion
- Trauma (head injury)
- Radiation exposure
- Surgery
- Stalk severance
- Pituitary apoplexy.
See Table 1 for a summary of the causes of male hypogonadism.
WHEN IS MRI INDICATED IN EVALUATING SECONDARY HYPOGONADISM?
The yield of pituitary-hypothalamic imaging in older men with secondary hypogonadism is fairly low in the absence of other pituitary hormone abnormalities and deficiencies. There are limited data regarding appropriate criteria for performing hypothalamic-pituitary imaging studies. However, a patient who has multiple anterior pituitary abnormalities on laboratory evaluation should undergo dedicated hypothalamic-pituitary magnetic resonance imaging (MRI).
The Endocrine Society Clinical Practice Guidelines11 recommend that MRI be performed to exclude a pituitary or hypothalamic tumor or infiltrative disease if the patient has severe secondary hypogonadism (serum testosterone < 150 ng/dL), panhypopituitarism, persistent hyperprolactinemia, or symptoms or signs of tumor mass effect such as headache, visual impairment, or a visual field defect.
WHO SHOULD UNDERGO ASSESSMENT OF TESTOSTERONE STATUS?
Screening for androgen deficiency in the asymptomatic general population is not recommended.11 The nonspecific nature of many of the signs and symptoms of androgen deficiency makes it difficult to give concrete recommendations as to who should have testosterone levels measured. Clinicians should consider testing if there is evidence of certain clinical disorders that are associated with low testosterone levels (see earlier discussion on the specific causes of primary and secondary hypogonadism).
When a male patient complains of erectile dysfunction, the investigation should include an assessment of serum testosterone. However, if a man who has a constellation of nonspecific symptoms asks for his testosterone level to be assessed (which is common, given the aggressive marketing of testosterone replacement by the pharmaceutical industry), we would recommend a basic evaluation that includes a comprehensive metabolic panel, complete blood count, and TSH level. Further testing should be determined by the history and physical examination. If no obvious explanation has been found for the patient’s symptoms at that point, assessment of serum testosterone may be warranted. More often than not the patient’s weight and limited physical activity are the driving forces behind the nonspecific symptoms, and counseling a patient on a life-style change can provide much benefit if the patient follows through with the physician’s recommendations.
Men whom we believe should not undergo assessment for testosterone deficiency are those who are acutely ill and hospitalized and those who are severely obese and are complaining of fatigue. Testosterone levels should be assessed only after the acute illness has resolved and, in a severely obese patient with fatigue, only after a thorough evaluation for sleep apnea has been undertaken.
TREAT THE UNDERLYING CAUSE, IF ONE CAN BE FOUND
If the evaluation of low testosterone leads to the diagnosis of a clear underlying condition that is amenable to treatment, such as prolactin elevation or sleep apnea, then treatment should be directed at the underlying cause, with subsequent monitoring of the patient’s symptoms and response in serum testosterone levels. In general, the use of dopamine agonist therapy in the management of hyperprolactinemia and, in cases of panhypopituitarism, of replacement therapy with levothyroxine (Synthroid), hydrocortisone, and possibly growth hormone and desmopressin (DDAVP), fall best under the purview of an endocrinologist. A caveat: serum TSH cannot be used to monitor levothyroxine replacement therapy in cases of secondary hypothyroidism. The clinical picture and serum free T4 and free T3 levels are used instead.
In the absence of a correctable (or immediately correctable) cause, testosterone supplementation can be initiated on an individualized basis in select patients who have clinical signs and symptoms of androgen deficiency if the benefits of treatment appear to outweigh the potential risks, and only after a thorough discussion with the patient.11 The Endocrine Society recommends against offering testosterone therapy to all older men with low testosterone.11
INFERTILITY
In men presenting with low serum testosterone, semen analysis is not routine. It is usually reserved for patients presenting with the primary complaint of infertility.
If an endocrine disorder such as prolactin elevation or hypothyroidism is the suspected cause of infertility, the patient should be referred to an endocrinologist for further evaluation and management. Treatment of male infertility should be directed at the underlying cause, but often requires exogenous human chorionic gonadotropin, FSH, GnRH (via a pulsatile pump), and possibly sperm harvesting from the testis with subsequent in vitro fertilization with intracytoplasmic sperm injection. It is critical that the partner be included in the evaluation of infertility.
These patients should be referred to a urologic or fertility center specializing in the diagnosis and treatment of infertility. For further information regarding male infertility, patients can be directed to www.fertilitylifelines.com.
CASE CONCLUDED
The patient’s low serum testosterone was confirmed on subsequent measurements at 8 am, with levels of 128 and 182 ng/dL (reference range 249–836). Other laboratory values:
- LH 1.4 mIU/mL (reference range 1.2–8.6)
- FSH 2.7 mIU/mL (1.3–9.9 mIU/mL)
(Both of these values are inappropriately normal in the setting of the low testosterone.)
- TSH 248 μIU/mL (0.4–5.5)
- Prolactin 24.6 ng/mL (1.6–18.8).
The patient was started on levothyroxine replacement therapy and after 3 months was noted to be euthyroid (TSH 1.8 μIU/mL) and to have a normal serum prolactin level. Testosterone levels (8 am) at this time were 350 ng/dL and 420 ng/dL.
Therefore, the cause of this patient’s hypogonadism was severe hypothyroidism and associated mild hyperprolactinemia. This case shows that a thorough evaluation is warranted before initiating testosterone therapy.
- Pitteloud N, Dwyer AA, DeCruz S, et al. Inhibition of luteinizing hormone secretion by testosterone in men requires aromatization for its pituitary but not its hypo-thalamic effects: evidence from the tandem study of normal and gonadotropin-releasing hormone-deficient men. J Clin Endocrinol Metab 2008; 93:784–791.
- Hayes FJ, DeCruz S, Seminara SB, Boepple PA, Crowley WF. Differential regulation of gonadotropin secretion by testosterone in the human male: absence of a negative feedback effect of testosterone on follicle-stimulating hormone secretion. J Clin Endocrinol Metab 2001; 86:53–58.
- Hayes FJ, Pitteloud N, DeCruz S, Crowley WF, Boepple PA. Importance of inhibin B in the regulation of FSH secretion in the human male. J Clin Endocrinol Metab 2001; 86:5541–5546.
- Pitteloud N, Dwyer AA, DeCruz S, et al. The relative role of gonadal sex steroids and gonadotropin-releasing hormone pulse frequency in the regulation of follicle-stimulating hormone secretion in men. J Clin Endocrinol Metab 2008; 93:2686–2692.
- Cooke RR, McIntosh JE, McIntosh RP. Circadian variation in serum free and non-SHBG-bound testosterone in normal men: measurements, and simulation using a mass action model. Clin Endocrinol (Oxf) 1993; 39:163–171.
- Diver MJ, Imtiaz KE, Ahmad AM, Vora JP, Fraser WD. Diurnal rhythms of serum total, free and bioavailable testosterone and of SHBG in middle-aged men compared with those in young men. Clin Endocrinol (Oxf) 2003; 58:710–717.
- Clair P, Claustrat B, Jordan D, Dechaud H, Sassolas G. Daily variations of plasma sex hormone-binding globulin binding capacity, testosterone and luteinizing hormone concentrations in healthy rested adult males. Horm Res 1985; 21:220–223.
- Woolf PD, Hamill RW, McDonald JV, Lee LA, Kelly M. Transient hypogonadotropic hypogonadism caused by critical illness. J Clin Endocrinol Metab 1985; 60:444–450.
- Rosner W, Auchus RJ, Azziz R, Sluss PM, Raff H. Position statement: utility, limitations, and pitfalls in measuring testosterone: an Endocrine Society position statement. J Clin Endocrinol Metab 2007; 92:405–413.
- Rosner W, Vesper H, et al; Endocrine Society; American Association for Clinical Chemistry; American Association of Clinical Endocrinologists; et al. Toward excellence in testosterone testing: a consensus statement. J Clin Endocrinol Metab 2010; 95:4542–4548.
- Bhasin S, Cunningham GR, Hayes FJ, et al; Task Force, Endocrine Society. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2010; 95:2536–2559.
- Wu FC, Tajar A, Beynon JM, et al; EMAS Group. Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med 2010; 363:123–135.
- Farrer JH, Sikka SC, Xie HW, Constantinide D, Rajfer J. Impaired testosterone biosynthesis in cryptorchidism. Fertil Steril 1985; 44:125–132.
- Sikka SC, Swerdloff RS, Rajfer J. In vitro inhibition of testosterone biosynthesis by ketoconazole. Endocrinology 1985; 116:1920–1925.
- Pallais JC, Au M, Pitteloud N, Seminara S, Crowley WF Jr. Kallmann syndrome. In:Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, eds. GeneReviews™ (Internet). Seattle, WA: University of Washington; 1993.
- Chevrier L, Guimiot F, de Roux N. GnRH receptor mutations in isolated gonadotropic deficiency. Mol Cell Endocrinol 2011; 346:21–28.
- Romero CJ, Pine-Twaddell E, Radovick S. Novel mutations associated with combined pituitary hormone deficiency. J Mol Endocrinol 2011; 46:R93–R102.
- Colameco S, Coren JS, Ciervo CA. Continuous opioid treatment for chronic noncancer pain: a time for moderation in prescribing. Postgrad Med 2009; 121:61–66.
- Fraser LA, Morrison D, Morley-Forster P, et al. Oral opioids for chronic non-cancer pain: higher prevalence of hypogonadism in men than in women. Exp Clin Endocrinol Diabetes 2009; 117:38–43.
- Morrison D, Capewell S, Reynolds SP, et al. Testosterone levels during systemic and inhaled corticosteroid therapy. Respir Med 1994; 88:659–663.
- Mah PM, Wittert GA. Obesity and testicular function. Mol Cell Endocrinol 2010; 316:180–186.
- Grossmann M. Low testosterone in men with type 2 diabetes: significance and treatment. J Clin Endocrinol Metab 2011; 96:2341–2353.
- Gascón F, Valle M, Martos R, et al. Sex hormone-binding globulin as a marker for hyperinsulinemia and/or insulin resistance in obese children. Eur J Endocrinol 2000; 143:85–89.
- Dhindsa S, Miller MG, McWhirter CL, et al. Testosterone concentrations in diabetic and nondiabetic obese men. Diabetes Care 2010; 33:1186–1192.
- Grossmann M, Gianatti EJ, Zajac JD. Testosterone and type 2 diabetes. Curr Opin Endocrinol Diabetes Obes 2010; 17:247–256.
- Andersson B, Mårin P, Lissner L, Vermeulen A, Björntorp P. Testosterone concentrations in women and men with NIDDM. Diabetes Care 1994; 17:405–411.
- Santamaria JD, Prior JC, Fleetham JA. Reversible reproductive dysfunction in men with obstructive sleep apnoea. Clin Endocrinol (Oxf) 1988; 28:461–470.
- Grunstein RR, Handelsman DJ, Lawrence SJ, Blackwell C, Caterson ID, Sullivan CE. Neuroendocrine dysfunction in sleep apnea: reversal by continuous positive airways pressure therapy. J Clin Endocrinol Metab 1989; 68:352–358.
- Matsumoto AM, Sandblom RE, Schoene RB, et al. Testosterone replacement in hypogonadal men: effects on obstructive sleep apnoea, respiratory drives, and sleep. Clin Endocrinol (Oxf) 1985; 22:713–721.
- Bremner WJ, Vitiello MV, Prinz PN. Loss of circadian rhythmicity in blood testosterone levels with aging in normal men. J Clin Endocrinol Metab 1983; 56:1278–1281.
- McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab 2005; 90:2451–2455.
- Kelly TM, Edwards CQ, Meikle AW, Kushner JP. Hypogonadism in hemochromatosis: reversal with iron depletion. Ann Intern Med 1984; 101:629–632.
- Milenkovic L, D’Angelo G, Kelly PA, Weiner RI. Inhibition of gonadotropin hormone-releasing hormone release by prolactin from GT1 neuronal cell lines through prolactin receptors. Proc Natl Acad Sci U S A 1994; 91:1244–1247.
- Valensi P, Coussieu C, Kemeny JL, Attali JR, Amouroux J, Sebaoun J. Endocrine investigations in two cases of feminizing Leydig cell tumour. Acta Endocrinol (Copenh) 1987; 115:365–372.
- Young S, Gooneratne S, Straus FH, Zeller WP, Bulun SE, Rosenthal IM. Feminizing Sertoli cell tumors in boys with Peutz-Jeghers syndrome. Am J Surg Pathol 1995; 19:50–58.
- Zayed A, Stock JL, Liepman MK, Wollin M, Longcope C. Feminization as a result of both peripheral conversion of androgens and direct estrogen production from an adrenocortical carcinoma. J Endocrinol Invest 1994; 17:275–278.
- Russ MJ, Ackerman SH, Barakat R, Levy B. Hypogonadotropic hypogonadism and delayed puberty in a man with anorexia nervosa. Psychosomatics 1986; 27:737–739.
- Rigotti NA, Neer RM, Jameson L. Osteopenia and bone fractures in a man with anorexia nervosa and hypogonadism. JAMA 1986; 256:385–388.
- Cohan GR. HIV-associated hypogonadism. AIDS Read 2006; 16:341–345,348,352–354.
- Handelsman DJ, Strasser S, McDonald JA, Conway AJ, McCaughan GW. Hypothalamic-pituitary-testicular function in end-stage nonalcoholic liver disease before and after liver transplantation. Clin Endocrinol (Oxf) 1995; 43:331–337.
- Lim VS, Fang VS. Gonadal dysfunction in uremic men. A study of the hypothalamo-pituitary-testicular axis before and after renal transplantation. Am J Med 1975; 58:655–662.
- Handelsman DJ, Dong Q. Hypothalamo-pituitary gonadal axis in chronic renal failure. Endocrinol Metab Clin North Am 1993; 22:145–161.
- Handelsman DJ, Spaliviero JA, Turtle JR. Hypothalamic-pituitary function in experimental uremic hypogonadism. Endocrinology 1985; 117:1984–1995.
- Tengstrand B, Carlström K, Hafström I. Bioavailable testosterone in men with rheumatoid arthritis-high frequency of hypogonadism. Rheumatology (Oxford) 2002; 41:285–289.
- Tengstrand B, Carlström K, Hafström I. Gonadal hormones in men with rheumatoid arthritis--from onset through 2 years. J Rheumatol 2009; 36:887–892.
- Emanuele MA, Emanuele NV. Alcohol’s effects on male reproduction. Alcohol Health Res World 1998; 22:195–201.
- Meikle AW. The interrelationships between thyroid dysfunction and hypogonadism in men and boys. Thyroid 2004; 14( suppl 1):S17–S25.
- Vagenakis AG, Dole K, Braverman LE. Pituitary enlargement, pituitary failure, and primary hypothyroidism. Ann Intern Med 1976; 85:195–198.
- Pitteloud N, Dwyer AA, DeCruz S, et al. Inhibition of luteinizing hormone secretion by testosterone in men requires aromatization for its pituitary but not its hypo-thalamic effects: evidence from the tandem study of normal and gonadotropin-releasing hormone-deficient men. J Clin Endocrinol Metab 2008; 93:784–791.
- Hayes FJ, DeCruz S, Seminara SB, Boepple PA, Crowley WF. Differential regulation of gonadotropin secretion by testosterone in the human male: absence of a negative feedback effect of testosterone on follicle-stimulating hormone secretion. J Clin Endocrinol Metab 2001; 86:53–58.
- Hayes FJ, Pitteloud N, DeCruz S, Crowley WF, Boepple PA. Importance of inhibin B in the regulation of FSH secretion in the human male. J Clin Endocrinol Metab 2001; 86:5541–5546.
- Pitteloud N, Dwyer AA, DeCruz S, et al. The relative role of gonadal sex steroids and gonadotropin-releasing hormone pulse frequency in the regulation of follicle-stimulating hormone secretion in men. J Clin Endocrinol Metab 2008; 93:2686–2692.
- Cooke RR, McIntosh JE, McIntosh RP. Circadian variation in serum free and non-SHBG-bound testosterone in normal men: measurements, and simulation using a mass action model. Clin Endocrinol (Oxf) 1993; 39:163–171.
- Diver MJ, Imtiaz KE, Ahmad AM, Vora JP, Fraser WD. Diurnal rhythms of serum total, free and bioavailable testosterone and of SHBG in middle-aged men compared with those in young men. Clin Endocrinol (Oxf) 2003; 58:710–717.
- Clair P, Claustrat B, Jordan D, Dechaud H, Sassolas G. Daily variations of plasma sex hormone-binding globulin binding capacity, testosterone and luteinizing hormone concentrations in healthy rested adult males. Horm Res 1985; 21:220–223.
- Woolf PD, Hamill RW, McDonald JV, Lee LA, Kelly M. Transient hypogonadotropic hypogonadism caused by critical illness. J Clin Endocrinol Metab 1985; 60:444–450.
- Rosner W, Auchus RJ, Azziz R, Sluss PM, Raff H. Position statement: utility, limitations, and pitfalls in measuring testosterone: an Endocrine Society position statement. J Clin Endocrinol Metab 2007; 92:405–413.
- Rosner W, Vesper H, et al; Endocrine Society; American Association for Clinical Chemistry; American Association of Clinical Endocrinologists; et al. Toward excellence in testosterone testing: a consensus statement. J Clin Endocrinol Metab 2010; 95:4542–4548.
- Bhasin S, Cunningham GR, Hayes FJ, et al; Task Force, Endocrine Society. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2010; 95:2536–2559.
- Wu FC, Tajar A, Beynon JM, et al; EMAS Group. Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med 2010; 363:123–135.
- Farrer JH, Sikka SC, Xie HW, Constantinide D, Rajfer J. Impaired testosterone biosynthesis in cryptorchidism. Fertil Steril 1985; 44:125–132.
- Sikka SC, Swerdloff RS, Rajfer J. In vitro inhibition of testosterone biosynthesis by ketoconazole. Endocrinology 1985; 116:1920–1925.
- Pallais JC, Au M, Pitteloud N, Seminara S, Crowley WF Jr. Kallmann syndrome. In:Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, eds. GeneReviews™ (Internet). Seattle, WA: University of Washington; 1993.
- Chevrier L, Guimiot F, de Roux N. GnRH receptor mutations in isolated gonadotropic deficiency. Mol Cell Endocrinol 2011; 346:21–28.
- Romero CJ, Pine-Twaddell E, Radovick S. Novel mutations associated with combined pituitary hormone deficiency. J Mol Endocrinol 2011; 46:R93–R102.
- Colameco S, Coren JS, Ciervo CA. Continuous opioid treatment for chronic noncancer pain: a time for moderation in prescribing. Postgrad Med 2009; 121:61–66.
- Fraser LA, Morrison D, Morley-Forster P, et al. Oral opioids for chronic non-cancer pain: higher prevalence of hypogonadism in men than in women. Exp Clin Endocrinol Diabetes 2009; 117:38–43.
- Morrison D, Capewell S, Reynolds SP, et al. Testosterone levels during systemic and inhaled corticosteroid therapy. Respir Med 1994; 88:659–663.
- Mah PM, Wittert GA. Obesity and testicular function. Mol Cell Endocrinol 2010; 316:180–186.
- Grossmann M. Low testosterone in men with type 2 diabetes: significance and treatment. J Clin Endocrinol Metab 2011; 96:2341–2353.
- Gascón F, Valle M, Martos R, et al. Sex hormone-binding globulin as a marker for hyperinsulinemia and/or insulin resistance in obese children. Eur J Endocrinol 2000; 143:85–89.
- Dhindsa S, Miller MG, McWhirter CL, et al. Testosterone concentrations in diabetic and nondiabetic obese men. Diabetes Care 2010; 33:1186–1192.
- Grossmann M, Gianatti EJ, Zajac JD. Testosterone and type 2 diabetes. Curr Opin Endocrinol Diabetes Obes 2010; 17:247–256.
- Andersson B, Mårin P, Lissner L, Vermeulen A, Björntorp P. Testosterone concentrations in women and men with NIDDM. Diabetes Care 1994; 17:405–411.
- Santamaria JD, Prior JC, Fleetham JA. Reversible reproductive dysfunction in men with obstructive sleep apnoea. Clin Endocrinol (Oxf) 1988; 28:461–470.
- Grunstein RR, Handelsman DJ, Lawrence SJ, Blackwell C, Caterson ID, Sullivan CE. Neuroendocrine dysfunction in sleep apnea: reversal by continuous positive airways pressure therapy. J Clin Endocrinol Metab 1989; 68:352–358.
- Matsumoto AM, Sandblom RE, Schoene RB, et al. Testosterone replacement in hypogonadal men: effects on obstructive sleep apnoea, respiratory drives, and sleep. Clin Endocrinol (Oxf) 1985; 22:713–721.
- Bremner WJ, Vitiello MV, Prinz PN. Loss of circadian rhythmicity in blood testosterone levels with aging in normal men. J Clin Endocrinol Metab 1983; 56:1278–1281.
- McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab 2005; 90:2451–2455.
- Kelly TM, Edwards CQ, Meikle AW, Kushner JP. Hypogonadism in hemochromatosis: reversal with iron depletion. Ann Intern Med 1984; 101:629–632.
- Milenkovic L, D’Angelo G, Kelly PA, Weiner RI. Inhibition of gonadotropin hormone-releasing hormone release by prolactin from GT1 neuronal cell lines through prolactin receptors. Proc Natl Acad Sci U S A 1994; 91:1244–1247.
- Valensi P, Coussieu C, Kemeny JL, Attali JR, Amouroux J, Sebaoun J. Endocrine investigations in two cases of feminizing Leydig cell tumour. Acta Endocrinol (Copenh) 1987; 115:365–372.
- Young S, Gooneratne S, Straus FH, Zeller WP, Bulun SE, Rosenthal IM. Feminizing Sertoli cell tumors in boys with Peutz-Jeghers syndrome. Am J Surg Pathol 1995; 19:50–58.
- Zayed A, Stock JL, Liepman MK, Wollin M, Longcope C. Feminization as a result of both peripheral conversion of androgens and direct estrogen production from an adrenocortical carcinoma. J Endocrinol Invest 1994; 17:275–278.
- Russ MJ, Ackerman SH, Barakat R, Levy B. Hypogonadotropic hypogonadism and delayed puberty in a man with anorexia nervosa. Psychosomatics 1986; 27:737–739.
- Rigotti NA, Neer RM, Jameson L. Osteopenia and bone fractures in a man with anorexia nervosa and hypogonadism. JAMA 1986; 256:385–388.
- Cohan GR. HIV-associated hypogonadism. AIDS Read 2006; 16:341–345,348,352–354.
- Handelsman DJ, Strasser S, McDonald JA, Conway AJ, McCaughan GW. Hypothalamic-pituitary-testicular function in end-stage nonalcoholic liver disease before and after liver transplantation. Clin Endocrinol (Oxf) 1995; 43:331–337.
- Lim VS, Fang VS. Gonadal dysfunction in uremic men. A study of the hypothalamo-pituitary-testicular axis before and after renal transplantation. Am J Med 1975; 58:655–662.
- Handelsman DJ, Dong Q. Hypothalamo-pituitary gonadal axis in chronic renal failure. Endocrinol Metab Clin North Am 1993; 22:145–161.
- Handelsman DJ, Spaliviero JA, Turtle JR. Hypothalamic-pituitary function in experimental uremic hypogonadism. Endocrinology 1985; 117:1984–1995.
- Tengstrand B, Carlström K, Hafström I. Bioavailable testosterone in men with rheumatoid arthritis-high frequency of hypogonadism. Rheumatology (Oxford) 2002; 41:285–289.
- Tengstrand B, Carlström K, Hafström I. Gonadal hormones in men with rheumatoid arthritis--from onset through 2 years. J Rheumatol 2009; 36:887–892.
- Emanuele MA, Emanuele NV. Alcohol’s effects on male reproduction. Alcohol Health Res World 1998; 22:195–201.
- Meikle AW. The interrelationships between thyroid dysfunction and hypogonadism in men and boys. Thyroid 2004; 14( suppl 1):S17–S25.
- Vagenakis AG, Dole K, Braverman LE. Pituitary enlargement, pituitary failure, and primary hypothyroidism. Ann Intern Med 1976; 85:195–198.
KEY POINTS
- Blood samples for testosterone measurements should be drawn near 8 am.
- A low serum testosterone value should always be confirmed by a reliable reference laboratory.
- The definition of a low testosterone level varies from laboratory to laboratory. In general, values less than 200 or 250 ng/dL are considered low, and values between 250 and 350 ng/dL may be considered borderline low.
- If testosterone is low, determine if the cause is primary (testicular) or secondary (hypothalamic-pituitary).
- Acute illness and treatment with opioids, anabolic steroids, or corticosteroids can result in transient hypogonadism.
Cognitive impairment in ICU survivors: Assessment and therapy
Intensive care medicine has dramatically evolved over the last 15 years, after reports from many landmark trials.1 Updated strategies for mechanical ventilation2 and “bundles” of strategies to optimize hemodynamic therapy3 have reduced the rates of morbidity and death from deadly critical conditions such as the adult respiratory distress syndrome (ARDS) and sepsis.
Despite these important improvements in short-term outcomes, it is increasingly recognized that intensive care unit (ICU) survivors suffer considerable long-term complications that affect their usual functioning.4 Recently, the Society of Critical Care Medicine convened a conference in which these long-term complications were named the “post-intensive care syndrome.”5
Quality of life, particularly its physical component, is considerably lower after a stay in the medical or surgical ICU.6–8 Posttraumatic stress disorder, depression, and sexual dysfunction are consistently reported years after ICU discharge.9–13
Perhaps the most frequently unrecognized complication in ICU survivors is cognitive impairment. Current data suggest that neurocognitive impairment after an ICU stay is common and that it persists 6 years or more after hospital discharge.
Hopkins et al14,15 analyzed 10 cohort studies of long-term cognitive impairment after an ICU stay; 5 of them focused on patients with ARDS. The prevalence of cognitive impairment was as high as 78% at hospital discharge, 46% at 1 year, and 25% 6 years after discharge.15,16 Of the cognitive domains compromised, memory was the most often affected, followed by executive function and attention.14,17
Interestingly, data suggest that cognition may improve somewhat in the first 6 to 12 months after ICU discharge.15 Therefore, if we can detect it early on and promptly refer patients for cognitive therapy, we may eventually improve the prognosis of this disabling complication.
This review will focus on how to evaluate, prevent, and treat cognitive impairment in patients who survive an ICU stay.
COGNITIVE IMPAIRMENT AFTER A STAY IN THE ICU
The association between ICU stay and neurocognitive dysfunction is poorly understood. Potential causes include hypoxemia,18 hypotension, 19 hyperglycemia,14 and—an area of growing interest and evolving research—sedation and delirium.20
Patients on mechanical ventilation are commonly given sedatives and analgesics to prevent anxiety and pain.21 However, these medications are strongly associated with delirium.22 In fact, recent studies found that benzodiazepines have an independent, dose-related, temporal association with delirium, with some reports describing a 20% increase in delirium per milligram of benzodiazepine.23 In another study, which included medical and surgical ICU patients, use of morphine was the strongest predictor of delirium, with a sixfold increase in odds over a period of 5 months.24
Delirium is important to prevent, diagnose, and treat, since it has a direct association with the development of long-term cognitive impairment.22,25 A review of studies that included 1,885 medical and surgical patients found that those who developed delirium during an ICU stay were three times more likely to have cognitive dysfunction when assessed 3 years later.20
Whether delirium is a primary disorder associated with cognitive impairment or if it only represents an underlying process leading to poor cognitive outcomes is unknown. As delirious patients are more likely to be older, to be mechanically ventilated, to require more sedation, and, in particular, to be sicker, the association between delirium and cognitive impairment may reflect the relationship between these risk factors and poor cognitive outcomes.26
Glucose and its relationship with cognitive function is another topic of investigation. A secondary analysis of a study that included ARDS survivors revealed that blood glucose values higher than 153 mg/dL, higher glucose variability, and duration of mechanical ventilation were associated with cognitive sequelae.27,28
Other studies focused on mechanical ventilation. In one study,29 one-third of patients who had been mechanically ventilated showed signs of neurocognitive impairment when they were evaluated 6 months after hospital discharge.
Mild cognitive impairment differs from cognitive impairment after an ICU stay
Cognitive impairment after ICU discharge does not follow the same pattern as mild cognitive impairment, and some authors consider these two types of cognitive impairment to be unrelated.
While mild cognitive impairment is progressive and associated with aging, cognitive impairment in ICU survivors develops rapidly after acute illness and is usually related to numerous pathologic and neurochemical pathways.
For example, the neurotransmitter acetylcholine is thought to be involved in cognitive function as well as neuroplasticity of the motor cortex. In a model of cognitive impairment after stroke, activity of the cholinergic system was reduced.30,31 Further, in a study in rats, Baskerville et al32 showed that experience-dependent plasticity could be completely blocked by damaging the cholinergic neurons in the nucleus basalis of Meynert, thereby affecting memory and other functions supported by this pathway.
Another implicated pathway involves dopamine. Of interest, dopamine augmentation has been shown to enhance simple motor memories and to improve procedural learning. Understanding of these neurochemical alterations opens opportunities for investigation of drug therapies.
ASSESSMENT TOOLS
Cognitive impairment is important to detect in ICU survivors because it predicts poor outcomes from rehabilitation. A study of stroke patients found that those with cognitive alterations immediately after the stroke were less likely to be discharged home or to be living at home 6 months after discharge.33
A possible explanation may be that affected patients cannot fully participate in rehabilitation activities, owing to impairment in executive function, inability to remember therapy instructions, or disruption of implicit and explicit learning. Indeed, some authors consider cognitive impairment after acquired brain injury to be the most relevant surrogate marker of rehabilitation potential. Consequently, manipulation or enhancement of cognition may directly affect rehabilitation outcomes.34
Disagreement about terminology and diagnostic criteria creates a problem for health care providers working with patients with potential cognitive impairment. Numerous systems have been proposed to define this condition; in fact, Stephan et al35 reviewed the literature and found no fewer than 17. None of them is specific for cognitive impairment after an ICU stay.
Petersen et al36 in 1999 proposed initial criteria for mild cognitive impairment that included the following:
- A memory complaint
- Normal general cognitive functioning
- Normal activities of daily living
- Memory impairment in relation to age and education
- No dementia.
Later, other areas of impairment besides memory were recognized, such as language, attention, perception, reasoning, and motor planning.37 Therefore, mild cognitive impairment is currently classified into subtypes, which include amnestic (affecting single or multiple domains) and nonamnestic (also affecting single or multiple domains).38
In clinical practice, impairment of specific cognitive domains may be challenging to detect, and neuropsychological testing is often needed. Cognitive screening tests can detect impairment across a restricted range of cognitive abilities, while more comprehensive assessments address each of the primary domains of cognition.39 Formal testing provides normative and validated data on cognition performance and severity.
The Montreal Cognitive Assessment40 is popular, comprehensive, used in a variety of professions in diverse types of facilities (acute care, rehabilitation, and skilled care facilities), and brief (taking 11 minutes to administer). It evaluates orientation, memory, language, attention, reasoning, and visual-constructional abilities. The maximum score is 30; cognitive impairment is defined as a score of less than 26. It has a sensitivity of 90% and a specificity of 87%.
The Folstein Mini-Mental State Examination (MMSE) is the most commonly used of the noncomprehensive tests in clinical practice.41 It assesses orientation, memory, language, attention, and praxis. It has a maximum score of 30 points; the cutoff score for cognitive impairment is 24 points or less.
A limitation of the MMSE is that its sensitivity is very low, ranging from 1% to 49%.42,43 The MMSE scores of patients with cognitive impairment overlap considerably with those of age-matched healthy controls.39 Conversely, the MMSE’s specificity is usually high, ranging from 85% to 100%.42
Moreover, the MMSE poses copyright issues, an important consideration when selecting a test. In 2001, the authors of the MMSE transferred all intellectual property rights to Psychological Assessment Resources, which has exclusive rights to publish, license, and manage all intellectual property rights in all media and languages. Photocopying and using the MMSE without applying for permission from and paying this company ($1.23 per use) constitutes copyright infringement. Therefore, health care providers and researchers have been using other tests to evaluate cognition.
Other tests of cognition assess individual domains. Interestingly, studies of long-term cognitive impairment after ICU admission used these tests to define outcomes.25 Specific tests include:
- The Digit Span and the Trailmaking Test A (used to assess attention and orientation)25
- The Rey Auditory Verbal Learning Test (used to evaluate verbal memory)
- The Complex Figure Test (helpful in defining visual-spatial construction and delayed visual memory)
- The Trailmaking Test B (also included in the Montreal Cognitive Assessment; assesses executive functioning).
Besides formal testing, an informal battery is often recommended to provide additional information. An informal evaluation includes word definition, reading and verbal fluency, reading comprehension, and performance of instrumental activities of daily living. Observing as patients perform tasks of daily living provides therapists with a vast amount of information, as these tasks require using multiple cognitive processes. Therefore, if a functional breakdown occurs during this assessment, the clinician needs to identify the domain or specific level of cognitive dysfunction involved in that deficit.44
PREVENTIVE STRATEGIES
Strategies for minimizing the long-term effects of cognitive impairment have mostly focused on preventing it.
During the ICU stay, optimizing hemodynamic, glucose, and oxygenation levels may prevent future long-term complications.18
Also, the association between sedation, delirium, and consequent cognitive impairment (see above) has led many investigators to apply the “ABCDE” bundle of strategies.25,45,46 Specifically, ABCDE stands for awakening and breathing, choice of sedatives with fewer adverse effects, daily delirium monitoring, and early mobility exercise. These strategies have been shown in randomized controlled trials to prevent delirium; however, they have not been proved to prevent cognitive impairment.
Awakening and breathing
In the Awakening and Breathing Controlled Trial,47 patients in the intervention group (ie, those who had their sedatives interrupted every morning to see if they would awaken, and if so, if they could breathe on their own) were extubated 3 days sooner than those in the control group (who underwent daily trials of spontaneous breathing, if deemed safe). Also, ICU and hospital length of stay were shorter by 4 days. Best of all, over 1 year, the mortality rate was lower by 14 absolute percentage points.
Choice of sedatives
Often, mechanically ventilated patients are given benzodiazepines, opiates, and propofol (Diprivan).21 Dexmedetomidine (Precedex), a newer agent, is an alpha-2 agonist and may offer advantages over the others.
To date, three randomized controlled trials have assessed the effect of dexmedetomidine in terms of outcomes associated with delirium, and one trial evaluated its association with intellectual capacity in ICU patients.
The Maximizing Efficacy of Targeted Sedation and Reducing Neurological Dysfunction (MENDS) trial randomized patients on mechanical ventilation to receive either dexmedetomidine or lorazepam (Ativan).48 Dexmedetomidine-treated patients had 4 more days alive without delirium or coma (7 vs 3 days, P = .01).
Subsequently, the Safety and Efficacy of Dexmedetomidine Compared With Midazolam (SEDCOM) trial compared dexmedetomidine and midazolam (Versed) in mechanically ventilated patients. Those who received dexmedetomidine had a lower incidence of delirium (54% vs 76%, P < .001), and 2 fewer days on mechanical ventilation.49
Reade et al50 evaluated time to extubation in already delirious patients randomized to receive either dexmedetomidine or haloperidol (Haldol). Those receiving dexmedetomidine had a shorter time to extubation as well as a shorter ICU length of stay.
The Acute Neuroscience Intensive Care Sedation Trial51 evaluated intellectual capacity in neurological ICU patients sedated with either dexmedetomidine or propofol. This randomized, double-blind trial included 18 brain-injured and 12 non-brain-injured intubated patients. In a crossover protocol, each received the combination of fentanyl (Sublimaze) and propofol and the combination of fentanyl and dexmedetomidine.
Cognition was evaluated using the Adapted Cognitive Exam (ACE), which assesses intellectual capacity through orientation, language, registration, attention, calculation, and recall. This 10-minute examination does not require verbal communication, as it relies on the ability to respond to yes-or-no questions and perform simple motor tasks. The maximum possible score is 100 points.
Interestingly, while on propofol, the patients’ adjusted ACE scores went down by a mean of 12.4 points, whereas they went up by 6.8 points while on dexmedetomidine. Even though brain-injured patients required less sedation than non-brain-injured patients, the effect of dexmedetomidine and propofol did not change.51
In summary, these studies suggest that all sedatives are not the same in their short-term and intermediate-term outcomes.
In our practice, we use dexmedetomidine as our first-line sedation therapy. In patients with hemodynamic instability, we use benzodiazepines. We reserve propofol for very short periods of intubation or for hemodynamically stable patients who cannot be sedated with dexmedetomidine.
Daily delirium monitoring
As mentioned above, delirium affects many patients on mechanical ventilation, and it is highly underrecognized if valid tests are not used.52 Therefore, it is critically important to be familiar with the tests for assessing delirium. Of these, the Confusion Assessment Method for the ICU is probably the one with the best performance, with a sensitivity of 93% to 100% and a specificity of 98% to 100%.53,54
Early mobilization
A landmark study paired the awakening and breathing strategy with early mobilization through physical and occupational therapy in the ICU.55 Patients in the intervention group had a higher rate of return to independent functional status upon hospital discharge and a shorter duration of mechanical ventilation and delirium.
In conclusion, even though direct prevention of cognitive dysfunction is a challenging task, the ABCDE approach targets individual risk factors for delirium, which is an important contributor to cognitive impairment. Whether the ABCDE bundle directly affects the development of cognitive impairment requires further investigation.
COGNITIVE THERAPIES
The cognition-focused intervention most often described is cognitive training. Cognitive training is delivered in individual or group sessions in which the patient practices tasks targeting different domains, such as memory, language, and attention. Outcomes are often assessed in terms of improvement in test scores or effects on everyday functioning. Unfortunately, because of heterogeneity among cognitive training interventions and studied populations, we cannot yet make strong evidence-based recommendations for clinical practice.
Martin et al56 in 2011 reviewed cognition-based interventions for healthy older people and people with mild cognitive impairment and found 36 relevant studies. Of these, only 3 were in patients with mild cognitive impairment, while the rest were in healthy older people.56–58 Overall, the only available data were related to the memory domain, and outcomes were mostly associated with immediate recall of words, paragraphs, and stories. Based on this, cognitive therapy is currently considered justified, as most patients with cognitive impairment after an ICU stay have memory problems.
Zelinski et al59 conducted a randomized, controlled, double-blind study comparing outcomes in an intervention group that underwent a computerized cognitive training program with those in a control group that viewed videos on a variety of topics such as literature, art, and history. The intervention, based on brain plasticity, aimed to improve the speed and accuracy of auditory information processing and to engage neuromodulatory systems. Some of the secondary outcomes favored the intervention group. These outcomes were related mostly to measures of overall memory, such as immediate and delayed recall, but also to a composite outcome that included letter-number sequencing and the digit span backwards test.
Despite these encouraging results, it is worth mentioning that these studies were not performed in patients with cognitive impairment associated with ICU admission. Therefore, the applicability and effectiveness of such therapies in post-ICU patients remains unknown.
Patients with posttraumatic brain injury and stroke have also been extensively studied in regard to the development of cognitive impairment.34 These patients probably represent a better standard for comparison, as their cognitive impairment does not necessarily progress.
The effect of cognitive rehabilitation on the recovery in these patients depends on adaptation and remediation. Adaptation describes a patient’s ability to compensate for functional impairment.34 This can be divided into internal and external adaptation. Internal adaptation requires the patient to recognize his or her cognitive limitation in order to adapt the to the environment accordingly. External adaptation entails getting help from devices or relatives (eg, phone calls) to achieve desired goals (eg, taking medication at scheduled times). Again, to adapt, the patient needs to be able to recognize his or her affected cognitive domain. Unfortunately, this is not always the case.
Remediation refers to the actual regaining of a lost ability. To stimulate neural plasticity, the patient is required to experience and repeat targeted skill-building activities.38 There is evidence that patients are more likely to regain lost ability by repeating the practice frequently during a short period of time.60
From the physician’s perspective, evaluating and identifying deficits in particular cognitive domains may help in designing a remediation plan in partnership with a cognitive therapist.
Cognitive rehabilitation in ICU survivors
The Returning to Everyday Tasks Utilizing Rehabilitation Networks (RETURN) study focused on cognitive and physical rehabilitation in post-ICU patients.61 This pilot study included 21 ICU survivors with cognitive or functional impairment at hospital discharge. Eight patients received usual care and 13 received a combination of in-home cognitive, physical, and functional rehabilitation over a 3-month period with a social worker or a master’s-level psychology technician.
Interventions included six in-person visits for cognitive rehabilitation and six televisits for physical and functional rehabilitation. Cognitive training was based on the goal-management training (GMT) protocol.62 This strategy attempts to improve executive function by increasing goal-directed behavior and by helping patients learn to be reflective before making decisions and executing tasks. The GMT model consists of sessions that build on one another to increase the rehabilitation intensity. During each session, goals are explained and participants perform increasingly challenging cognitive tasks.
Cognitive outcomes were evaluated using the Delis-Kaplan Tower Test to evaluate executive function by assessing the ability to plan and strategize efficiently. The patient is required to move disks across three pegs until a tower is built. The object is to use the fewest moves possible while adhering to two rules: larger disks cannot be placed on top of smaller ones, and disks must be moved one at a time, using only one hand.
At 3 months there was a significant difference between groups, with the intervention group earning higher tower test scores than controls did (median of 13 vs 7.5).
The Activity and Cognitive Therapy in the Intensive Care Unit (ACT-ICU) trial is another pilot study that will attempt to assess the feasibility of early cognitive rehabilitation in ICU survivors. This study will combine early mobilization with a cognitive intervention, and its primary outcome is executive function (with the tower test) at 3 months after discharge.63
DRUG THERAPY
Some medications have been tested to assess whether they reduce the risk of progression from adult traumatic brain injury to cognitive impairment. These drugs augment dopamine and acetylcholine activity.
Methylphenidate (Ritalin), a dopaminergic drug, was studied in two trials. The first was a double-blind trial in 18 patients with posttraumatic brain injury. Memory was found to improve, based on the Working Memory Task Test. However, due to the small number of participants, no further conclusions were obtained.64
The second trial, in 19 patients with posttraumatic brain injury, had a double-blind crossover design. Attention, evaluated by the Distraction Task Test, improved with the use of methylphenidate.65 Again, the small number of patients precludes generalization of these results.
Donepezil (Aricept), a cholinergic drug, was evaluated in four clinical trials in posttraumatic brain injury patients66–69; each trial included 21 to 180 patients. The trials evaluated the drug’s effect on memory and attention through a variety of tools (Paced Auditory Serial Addition Test; Wechsler Memory Scale; Boston Naming Test; Rey Auditory Verbal Learning Test; Complex Figure Test; and Reaction Time–Dual Task). Interestingly, donepezil was associated with large improvements in objective assessments of attention and memory. Despite methodologic flaws, such as a lack of blinding in one of these studies69 and an open-label design in two of them,66,68 of the drugs available, donepezil presents the strongest evidence for use in cognitive impairment after traumatic brain injury.70
- Diaz-Guzman E, Sanchez J, Arroliga AC. Update in intensive care medicine: studies that challenged our practice in the last 5 years. Cleve Clin J Med 2011; 78:665–674.
- Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 2000; 342:1301–1308.
- Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- Oeyen SG, Vandijck DM, Benoit DD, Annemans L, Decruyenaere JM. Quality of life after intensive care: a systematic review of the literature. Crit Care Med 2010; 38:2386–2400.
- Needham DM, Davidson J, Cohen H, et al. Improving long-term outcomes after discharge from intensive care unit: report from a stakeholders’ conference. Crit Care Med 2012; 40:502–509.
- Herridge MS, Cheung AM, Tansey CM, et al. One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med 2003; 348:683–693.
- Herridge MS, Tansey CM, Matte A, et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med 2011; 364:1293–1304.
- Timmers TK, Verhofstad MH, Moons KG, van Beeck EF, Leenen LP. Long-term quality of life after surgical intensive care admission. Arch Surg 2011; 146:412–418.
- Michaels AJ, Michaels CE, Moon CH, et al. Posttraumatic stress disorder after injury: impact on general health outcome and early risk assessment. J Trauma 1999; 47:460–466; discussion466–467.
- Stoll C, Schelling G, Goetz AE, et al. Health-related quality of life and post-traumatic stress disorder in patients after cardiac surgery and intensive care treatment. J Thorac Cardiovasc Surg 2000; 120:505–512.
- Jones C, Skirrow P, Griffiths RD, et al Post-traumatic stress disorder-related symptoms in relatives of patients following intensive care. Intensive Care Med 2004; 30:456–460.
- Griffiths J, Gager M, Alder N, Fawcett D, Waldmann C, Quinlan J. A self-report-based study of the incidence and associations of sexual dysfunction in survivors of intensive care treatment. Intensive Care Med 2006; 32:445–451.
- Griffiths J, Waldmann C, Quinlan J. Sexual dysfunction in intensive care survivors. Br J Hosp Med (Lond) 2007; 68:470–473.
- Hopkins RO, Jackson JC. Long-term neurocognitive function after critical illness. Chest 2006; 130:869–878.
- Hopkins RO, Weaver LK, Collingridge D, Parkinson RB, Chan KJ, Orme JF. Two-year cognitive, emotional, and quality-of-life outcomes in acute respiratory distress syndrome. Am J Respir Crit Care Med 2005; 171:340–347.
- Rothenhausler HB, Ehrentraut S, Stoll C, Schelling G, Kapfhammer HP. The relationship between cognitive performance and employment and health status in long-term survivors of the acute respiratory distress syndrome: results of an exploratory study. Gen Hosp Psychiatry 2001; 23:90–96.
- Sukantarat KT, Burgess PW, Williamson RC, Brett SJ. Prolonged cognitive dysfunction in survivors of critical illness. Anaesthesia 2005; 60:847–853.
- Hopkins RO, Weaver LK, Pope D, Orme JF, Bigler ED, Larson LV. Neuropsychological sequelae and impaired health status in survivors of severe acute respiratory distress syndrome. Am J Respir Crit Care Med 1999; 160:50–56.
- Hopkins RO, Weaver LK, Chan KJ, Orme JF. Quality of life, emotional, and cognitive function following acute respiratory distress syndrome. J Int Neuropsychol Soc 2004; 10:1005–1017.
- Jackson JC, Gordon SM, Hart RP, Hopkins RO, Ely EW. The association between delirium and cognitive decline: a review of the empirical literature. Neuropsychol Rev 2004; 14:87–98.
- Arroliga AC, Thompson BT, Ancukiewicz M, et al. Use of sedatives, opioids, and neuromuscular blocking agents in patients with acute lung injury and acute respiratory distress syndrome. Crit Care Med 2008; 36:1083–1088.
- Miller RR, Ely EW. Delirium and cognitive dysfunction in the intensive care unit. Semin Respir Crit Care Med 2006; 27:210–220.
- Pandharipande P, Shintani A, Peterson J, et al. Lorazepam is an independent risk factor for transitioning to delirium in intensive care unit patients. Anesthesiology 2006; 104:21–26.
- Dubois MJ, Bergeron N, Dumont M, Dial S, Skrobik Y. Delirium in an intensive care unit: a study of risk factors. Intensive Care Med 2001; 27:1297–1304.
- Girard TD, Jackson JC, Pandharipande PP, et al. Delirium as a predictor of long-term cognitive impairment in survivors of critical illness. Crit Care Med 2010; 38:1513–1520.
- Miller RR, Ely EW. Delirium and cognitive dysfunction in the intensive care unit. Curr Psychiatry Rep 2007; 9:26–34.
- Hopkins RO, Suchyta MR, Snow GL, Jephson A, Weaver LK, Orme JF. Blood glucose dysregulation and cognitive outcome in ARDS survivors. Brain Inj 2010; 24:1478–1484.
- Hough CL, Herridge MS. Long-term outcome after acute lung injury. Curr Opin Crit Care 2012; 18:8–15.
- Jackson JC, Hart RP, Gordon SM, et al. Six-month neuropsychological outcome of medical intensive care unit patients. Crit Care Med 2003; 31:1226–1234.
- Court JA, Perry EK. Neurotransmitter abnormalities in vascular dementia. Int Psychogeriatr 2003; 15(suppl 1):81–87.
- Gottfries CG, Blennow K, Karlsson I, Wallin A. The neurochemistry of vascular dementia. Dementia 1994; 5:163–167.
- Baskerville KA, Schweitzer JB, Herron P. Effects of cholinergic depletion on experience-dependent plasticity in the cortex of the rat. Neuroscience 1997; 80:1159–1169.
- Henon H, Lebert F, Durieu I, et al. Confusional state in stroke: relation to preexisting dementia, patient characteristics, and outcome. Stroke 1999; 30:773–779.
- Whyte E, Skidmore E, Aizenstein H, Ricker J, Butters M. Cognitive impairment in acquired brain injury: a predictor of rehabilitation outcomes and an opportunity for novel interventions. PMR 2011; 3(suppl 1):S45–S51.
- Stephan BC, Matthews FE, McKeith IG, Bond J, Brayne C. Early cognitive change in the general population: how do different definitions work? J Am Geriatr Soc 2007; 55:1534–1540.
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol 1999; 56:303–308.
- Palmer K, Fratiglioni L, Winblad B. What is mild cognitive impairment? Variations in definitions and evolution of nondemented persons with cognitive impairment. Acta Neurol Scand Suppl 2003; 179:14–20.
- Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med 2004; 256:183–194.
- Lonie JA, Tierney KM, Ebmeier KP. Screening for mild cognitive impairment: a systematic review. Int J Geriatr Psychiatry 2009; 24:902–915.
- Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005; 53:695–699.
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12:189–198.
- Sager MA, Hermann BP, La Rue A, Woodard JL. Screening for dementia in community-based memory clinics. WMJ 2006; 105:25–29.
- Ravaglia G, Forti P, Maioli F, et al. Screening for mild cognitive impairment in elderly ambulatory patients with cognitive complaints. Aging Clin Exp Res 2005; 17:374–379.
- Vogenthaler DR. An overview of head injury: its consequences and rehabilitation. Brain Inj 1987; 1:113–127.
- van den Boogaard M, Schoonhoven L, Evers AW, van der Hoeven JG, van Achterberg T, Pickkers P. Delirium in critically ill patients: impact on long-term health-related quality of life and cognitive functioning. Crit Care Med 2012; 40:112–118.
- Morandi A, Brummel NE, Ely EW. Sedation, delirium and mechanical ventilation: the ‘ABCDE’ approach. Curr Opin Crit Care 2011; 17:43–49.
- Girard TD, Kress JP, Fuchs BD, et al. Efficacy and safety of a paired sedation and ventilator weaning protocol for mechanically ventilated patients in intensive care (Awakening and Breathing Controlled trial): a randomised controlled trial. Lancet 2008; 371:126–134.
- Pandharipande PP, Pun BT, Herr DL, et al. Effect of sedation with dexmedetomidine vs lorazepam on acute brain dysfunction in mechanically ventilated patients: the MENDS randomized controlled trial. JAMA 2007; 298:2644–2653.
- Riker RR, Shehabi Y, Bokesch PM, et al. Dexmedetomidine vs midazolam for sedation of critically ill patients: a randomized trial. JAMA 2009; 301:489–499.
- Reade MC, O’Sullivan K, Bates S, Goldsmith D, Ainslie WR, Bellomo R. Dexmedetomidine vs. haloperidol in delirious, agitated, intubated patients: a randomised open-label trial. Crit Care 2009; 13:R75.
- Mirski MA, Lewin JJ, Ledroux S, et al. Cognitive improvement during continuous sedation in critically ill, awake and responsive patients: the Acute Neurological ICU Sedation Trial (ANIST). Intensive Care Med 2010; 36:1505–1513.
- Spronk PE, Riekerk B, Hofhuis J, Rommes JH. Occurrence of delirium is severely underestimated in the ICU during daily care. Intensive Care Med 2009; 35:1276–1280.
- Ely EW, Inouye SK, Bernard GR, et al. Delirium in mechanically ventilated patients: validity and reliability of the confusion assessment method for the intensive care unit (CAM-ICU). JAMA 2001; 286:2703–2710.
- Luetz A, Heymann A, Radtke FM, et al. Different assessment tools for intensive care unit delirium: which score to use? Crit Care Med 2010; 38:409–418.
- Schweickert WD, Pohlman MC, Pohlman AS, et al. Early physical and occupational therapy in mechanically ventilated, critically ill patients: a randomised controlled trial. Lancet 2009; 373:1874–1882.
- Martin M, Clare L, Altgassen AM, Cameron MH, Zehnder F. Cognition-based interventions for healthy older people and people with mild cognitive impairment. Cochrane Database Syst Rev 2011(1):CD006220.
- Rozzini L, Costardi D, Chilovi BV, Franzoni S, Trabucchi M, Padovani A. Efficacy of cognitive rehabilitation in patients with mild cognitive impairment treated with cholinesterase inhibitors. Int J Geriatr Psychiatry 2007; 22:356–360.
- Jean L, Bergeron ME, Thivierge S, Simard M. Cognitive intervention programs for individuals with mild cognitive impairment: systematic review of the literature. Am J Geriatr Psychiatry 2010; 18:281–296.
- Zelinski EM, Spina LM, Yaffe K, et al. Improvement in memory with plasticity-based adaptive cognitive training: results of the 3-month follow-up. J Am Geriatr Soc 2011; 59:258–265.
- Cicerone KD, Dahlberg C, Malec JF, et al. Evidence-based cognitive rehabilitation: updated review of the literature from 1998 through 2002. Arch Phys Med Rehabil 2005; 86:1681–1692.
- Jackson JC, Ely EW, Morey MC, et al. Cognitive and physical rehabilitation of intensive care unit survivors: results of the RETURN randomized controlled pilot investigation. Crit Care Med 2012; 40:1088–1097.
- Levine B, Stuss DT, Winocur G, et al. Cognitive rehabilitation in the elderly: effects on strategic behavior in relation to goal management. J Int Neuropsychol Soc 2007; 13:143–152.
- ACT-ICU Study: Activity and Cognitive Therapy in the Intensive Care Unit. http://clinicaltrials.gov/ct2/show/NCT01270269. Accessed August 9, 2012.
- Kim YH, Ko MH, Na SY, Park SH, Kim KW. Effects of single-dose methylphenidate on cognitive performance in patients with traumatic brain injury: a double-blind placebo-controlled study. Clin Rehabil 2006; 20:24–30.
- Whyte J, Hart T, Schuster K, Fleming M, Polansky M, Coslett HB. Effects of methylphenidate on attentional function after traumatic brain injury. A randomized, placebo-controlled trial. Am J Phys Med Rehabil 1997; 76:440–450.
- Masanic CA, Bayley MT, VanReekum R, Simard M. Open-label study of donepezil in traumatic brain injury. Arch Phys Med Rehabil 2001; 82:896–901.
- Zhang L, Plotkin RC, Wang G, Sandel ME, Lee S. Cholinergic augmentation with donepezil enhances recovery in short-term memory and sustained attention after traumatic brain injury. Arch Phys Med Rehabil 2004; 85:1050–1055.
- Khateb A, Ammann J, Annoni JM, Diserens K. Cognition-enhancing effects of donepezil in traumatic brain injury. Eur Neurol 2005; 54:39–45.
- Kim YW, Kim DY, Shin JC, Park CI, Lee JD. The changes of cortical metabolism associated with the clinical response to donepezil therapy in traumatic brain injury. Clin Neuropharmacol 2009; 32:63–68.
- Wheaton P, Mathias JL, Vink R. Impact of pharmacological treatments on cognitive and behavioral outcome in the postacute stages of adult traumatic brain injury: a meta-analysis. J Clin Psychopharmacol 2011; 31:745–757.
Intensive care medicine has dramatically evolved over the last 15 years, after reports from many landmark trials.1 Updated strategies for mechanical ventilation2 and “bundles” of strategies to optimize hemodynamic therapy3 have reduced the rates of morbidity and death from deadly critical conditions such as the adult respiratory distress syndrome (ARDS) and sepsis.
Despite these important improvements in short-term outcomes, it is increasingly recognized that intensive care unit (ICU) survivors suffer considerable long-term complications that affect their usual functioning.4 Recently, the Society of Critical Care Medicine convened a conference in which these long-term complications were named the “post-intensive care syndrome.”5
Quality of life, particularly its physical component, is considerably lower after a stay in the medical or surgical ICU.6–8 Posttraumatic stress disorder, depression, and sexual dysfunction are consistently reported years after ICU discharge.9–13
Perhaps the most frequently unrecognized complication in ICU survivors is cognitive impairment. Current data suggest that neurocognitive impairment after an ICU stay is common and that it persists 6 years or more after hospital discharge.
Hopkins et al14,15 analyzed 10 cohort studies of long-term cognitive impairment after an ICU stay; 5 of them focused on patients with ARDS. The prevalence of cognitive impairment was as high as 78% at hospital discharge, 46% at 1 year, and 25% 6 years after discharge.15,16 Of the cognitive domains compromised, memory was the most often affected, followed by executive function and attention.14,17
Interestingly, data suggest that cognition may improve somewhat in the first 6 to 12 months after ICU discharge.15 Therefore, if we can detect it early on and promptly refer patients for cognitive therapy, we may eventually improve the prognosis of this disabling complication.
This review will focus on how to evaluate, prevent, and treat cognitive impairment in patients who survive an ICU stay.
COGNITIVE IMPAIRMENT AFTER A STAY IN THE ICU
The association between ICU stay and neurocognitive dysfunction is poorly understood. Potential causes include hypoxemia,18 hypotension, 19 hyperglycemia,14 and—an area of growing interest and evolving research—sedation and delirium.20
Patients on mechanical ventilation are commonly given sedatives and analgesics to prevent anxiety and pain.21 However, these medications are strongly associated with delirium.22 In fact, recent studies found that benzodiazepines have an independent, dose-related, temporal association with delirium, with some reports describing a 20% increase in delirium per milligram of benzodiazepine.23 In another study, which included medical and surgical ICU patients, use of morphine was the strongest predictor of delirium, with a sixfold increase in odds over a period of 5 months.24
Delirium is important to prevent, diagnose, and treat, since it has a direct association with the development of long-term cognitive impairment.22,25 A review of studies that included 1,885 medical and surgical patients found that those who developed delirium during an ICU stay were three times more likely to have cognitive dysfunction when assessed 3 years later.20
Whether delirium is a primary disorder associated with cognitive impairment or if it only represents an underlying process leading to poor cognitive outcomes is unknown. As delirious patients are more likely to be older, to be mechanically ventilated, to require more sedation, and, in particular, to be sicker, the association between delirium and cognitive impairment may reflect the relationship between these risk factors and poor cognitive outcomes.26
Glucose and its relationship with cognitive function is another topic of investigation. A secondary analysis of a study that included ARDS survivors revealed that blood glucose values higher than 153 mg/dL, higher glucose variability, and duration of mechanical ventilation were associated with cognitive sequelae.27,28
Other studies focused on mechanical ventilation. In one study,29 one-third of patients who had been mechanically ventilated showed signs of neurocognitive impairment when they were evaluated 6 months after hospital discharge.
Mild cognitive impairment differs from cognitive impairment after an ICU stay
Cognitive impairment after ICU discharge does not follow the same pattern as mild cognitive impairment, and some authors consider these two types of cognitive impairment to be unrelated.
While mild cognitive impairment is progressive and associated with aging, cognitive impairment in ICU survivors develops rapidly after acute illness and is usually related to numerous pathologic and neurochemical pathways.
For example, the neurotransmitter acetylcholine is thought to be involved in cognitive function as well as neuroplasticity of the motor cortex. In a model of cognitive impairment after stroke, activity of the cholinergic system was reduced.30,31 Further, in a study in rats, Baskerville et al32 showed that experience-dependent plasticity could be completely blocked by damaging the cholinergic neurons in the nucleus basalis of Meynert, thereby affecting memory and other functions supported by this pathway.
Another implicated pathway involves dopamine. Of interest, dopamine augmentation has been shown to enhance simple motor memories and to improve procedural learning. Understanding of these neurochemical alterations opens opportunities for investigation of drug therapies.
ASSESSMENT TOOLS
Cognitive impairment is important to detect in ICU survivors because it predicts poor outcomes from rehabilitation. A study of stroke patients found that those with cognitive alterations immediately after the stroke were less likely to be discharged home or to be living at home 6 months after discharge.33
A possible explanation may be that affected patients cannot fully participate in rehabilitation activities, owing to impairment in executive function, inability to remember therapy instructions, or disruption of implicit and explicit learning. Indeed, some authors consider cognitive impairment after acquired brain injury to be the most relevant surrogate marker of rehabilitation potential. Consequently, manipulation or enhancement of cognition may directly affect rehabilitation outcomes.34
Disagreement about terminology and diagnostic criteria creates a problem for health care providers working with patients with potential cognitive impairment. Numerous systems have been proposed to define this condition; in fact, Stephan et al35 reviewed the literature and found no fewer than 17. None of them is specific for cognitive impairment after an ICU stay.
Petersen et al36 in 1999 proposed initial criteria for mild cognitive impairment that included the following:
- A memory complaint
- Normal general cognitive functioning
- Normal activities of daily living
- Memory impairment in relation to age and education
- No dementia.
Later, other areas of impairment besides memory were recognized, such as language, attention, perception, reasoning, and motor planning.37 Therefore, mild cognitive impairment is currently classified into subtypes, which include amnestic (affecting single or multiple domains) and nonamnestic (also affecting single or multiple domains).38
In clinical practice, impairment of specific cognitive domains may be challenging to detect, and neuropsychological testing is often needed. Cognitive screening tests can detect impairment across a restricted range of cognitive abilities, while more comprehensive assessments address each of the primary domains of cognition.39 Formal testing provides normative and validated data on cognition performance and severity.
The Montreal Cognitive Assessment40 is popular, comprehensive, used in a variety of professions in diverse types of facilities (acute care, rehabilitation, and skilled care facilities), and brief (taking 11 minutes to administer). It evaluates orientation, memory, language, attention, reasoning, and visual-constructional abilities. The maximum score is 30; cognitive impairment is defined as a score of less than 26. It has a sensitivity of 90% and a specificity of 87%.
The Folstein Mini-Mental State Examination (MMSE) is the most commonly used of the noncomprehensive tests in clinical practice.41 It assesses orientation, memory, language, attention, and praxis. It has a maximum score of 30 points; the cutoff score for cognitive impairment is 24 points or less.
A limitation of the MMSE is that its sensitivity is very low, ranging from 1% to 49%.42,43 The MMSE scores of patients with cognitive impairment overlap considerably with those of age-matched healthy controls.39 Conversely, the MMSE’s specificity is usually high, ranging from 85% to 100%.42
Moreover, the MMSE poses copyright issues, an important consideration when selecting a test. In 2001, the authors of the MMSE transferred all intellectual property rights to Psychological Assessment Resources, which has exclusive rights to publish, license, and manage all intellectual property rights in all media and languages. Photocopying and using the MMSE without applying for permission from and paying this company ($1.23 per use) constitutes copyright infringement. Therefore, health care providers and researchers have been using other tests to evaluate cognition.
Other tests of cognition assess individual domains. Interestingly, studies of long-term cognitive impairment after ICU admission used these tests to define outcomes.25 Specific tests include:
- The Digit Span and the Trailmaking Test A (used to assess attention and orientation)25
- The Rey Auditory Verbal Learning Test (used to evaluate verbal memory)
- The Complex Figure Test (helpful in defining visual-spatial construction and delayed visual memory)
- The Trailmaking Test B (also included in the Montreal Cognitive Assessment; assesses executive functioning).
Besides formal testing, an informal battery is often recommended to provide additional information. An informal evaluation includes word definition, reading and verbal fluency, reading comprehension, and performance of instrumental activities of daily living. Observing as patients perform tasks of daily living provides therapists with a vast amount of information, as these tasks require using multiple cognitive processes. Therefore, if a functional breakdown occurs during this assessment, the clinician needs to identify the domain or specific level of cognitive dysfunction involved in that deficit.44
PREVENTIVE STRATEGIES
Strategies for minimizing the long-term effects of cognitive impairment have mostly focused on preventing it.
During the ICU stay, optimizing hemodynamic, glucose, and oxygenation levels may prevent future long-term complications.18
Also, the association between sedation, delirium, and consequent cognitive impairment (see above) has led many investigators to apply the “ABCDE” bundle of strategies.25,45,46 Specifically, ABCDE stands for awakening and breathing, choice of sedatives with fewer adverse effects, daily delirium monitoring, and early mobility exercise. These strategies have been shown in randomized controlled trials to prevent delirium; however, they have not been proved to prevent cognitive impairment.
Awakening and breathing
In the Awakening and Breathing Controlled Trial,47 patients in the intervention group (ie, those who had their sedatives interrupted every morning to see if they would awaken, and if so, if they could breathe on their own) were extubated 3 days sooner than those in the control group (who underwent daily trials of spontaneous breathing, if deemed safe). Also, ICU and hospital length of stay were shorter by 4 days. Best of all, over 1 year, the mortality rate was lower by 14 absolute percentage points.
Choice of sedatives
Often, mechanically ventilated patients are given benzodiazepines, opiates, and propofol (Diprivan).21 Dexmedetomidine (Precedex), a newer agent, is an alpha-2 agonist and may offer advantages over the others.
To date, three randomized controlled trials have assessed the effect of dexmedetomidine in terms of outcomes associated with delirium, and one trial evaluated its association with intellectual capacity in ICU patients.
The Maximizing Efficacy of Targeted Sedation and Reducing Neurological Dysfunction (MENDS) trial randomized patients on mechanical ventilation to receive either dexmedetomidine or lorazepam (Ativan).48 Dexmedetomidine-treated patients had 4 more days alive without delirium or coma (7 vs 3 days, P = .01).
Subsequently, the Safety and Efficacy of Dexmedetomidine Compared With Midazolam (SEDCOM) trial compared dexmedetomidine and midazolam (Versed) in mechanically ventilated patients. Those who received dexmedetomidine had a lower incidence of delirium (54% vs 76%, P < .001), and 2 fewer days on mechanical ventilation.49
Reade et al50 evaluated time to extubation in already delirious patients randomized to receive either dexmedetomidine or haloperidol (Haldol). Those receiving dexmedetomidine had a shorter time to extubation as well as a shorter ICU length of stay.
The Acute Neuroscience Intensive Care Sedation Trial51 evaluated intellectual capacity in neurological ICU patients sedated with either dexmedetomidine or propofol. This randomized, double-blind trial included 18 brain-injured and 12 non-brain-injured intubated patients. In a crossover protocol, each received the combination of fentanyl (Sublimaze) and propofol and the combination of fentanyl and dexmedetomidine.
Cognition was evaluated using the Adapted Cognitive Exam (ACE), which assesses intellectual capacity through orientation, language, registration, attention, calculation, and recall. This 10-minute examination does not require verbal communication, as it relies on the ability to respond to yes-or-no questions and perform simple motor tasks. The maximum possible score is 100 points.
Interestingly, while on propofol, the patients’ adjusted ACE scores went down by a mean of 12.4 points, whereas they went up by 6.8 points while on dexmedetomidine. Even though brain-injured patients required less sedation than non-brain-injured patients, the effect of dexmedetomidine and propofol did not change.51
In summary, these studies suggest that all sedatives are not the same in their short-term and intermediate-term outcomes.
In our practice, we use dexmedetomidine as our first-line sedation therapy. In patients with hemodynamic instability, we use benzodiazepines. We reserve propofol for very short periods of intubation or for hemodynamically stable patients who cannot be sedated with dexmedetomidine.
Daily delirium monitoring
As mentioned above, delirium affects many patients on mechanical ventilation, and it is highly underrecognized if valid tests are not used.52 Therefore, it is critically important to be familiar with the tests for assessing delirium. Of these, the Confusion Assessment Method for the ICU is probably the one with the best performance, with a sensitivity of 93% to 100% and a specificity of 98% to 100%.53,54
Early mobilization
A landmark study paired the awakening and breathing strategy with early mobilization through physical and occupational therapy in the ICU.55 Patients in the intervention group had a higher rate of return to independent functional status upon hospital discharge and a shorter duration of mechanical ventilation and delirium.
In conclusion, even though direct prevention of cognitive dysfunction is a challenging task, the ABCDE approach targets individual risk factors for delirium, which is an important contributor to cognitive impairment. Whether the ABCDE bundle directly affects the development of cognitive impairment requires further investigation.
COGNITIVE THERAPIES
The cognition-focused intervention most often described is cognitive training. Cognitive training is delivered in individual or group sessions in which the patient practices tasks targeting different domains, such as memory, language, and attention. Outcomes are often assessed in terms of improvement in test scores or effects on everyday functioning. Unfortunately, because of heterogeneity among cognitive training interventions and studied populations, we cannot yet make strong evidence-based recommendations for clinical practice.
Martin et al56 in 2011 reviewed cognition-based interventions for healthy older people and people with mild cognitive impairment and found 36 relevant studies. Of these, only 3 were in patients with mild cognitive impairment, while the rest were in healthy older people.56–58 Overall, the only available data were related to the memory domain, and outcomes were mostly associated with immediate recall of words, paragraphs, and stories. Based on this, cognitive therapy is currently considered justified, as most patients with cognitive impairment after an ICU stay have memory problems.
Zelinski et al59 conducted a randomized, controlled, double-blind study comparing outcomes in an intervention group that underwent a computerized cognitive training program with those in a control group that viewed videos on a variety of topics such as literature, art, and history. The intervention, based on brain plasticity, aimed to improve the speed and accuracy of auditory information processing and to engage neuromodulatory systems. Some of the secondary outcomes favored the intervention group. These outcomes were related mostly to measures of overall memory, such as immediate and delayed recall, but also to a composite outcome that included letter-number sequencing and the digit span backwards test.
Despite these encouraging results, it is worth mentioning that these studies were not performed in patients with cognitive impairment associated with ICU admission. Therefore, the applicability and effectiveness of such therapies in post-ICU patients remains unknown.
Patients with posttraumatic brain injury and stroke have also been extensively studied in regard to the development of cognitive impairment.34 These patients probably represent a better standard for comparison, as their cognitive impairment does not necessarily progress.
The effect of cognitive rehabilitation on the recovery in these patients depends on adaptation and remediation. Adaptation describes a patient’s ability to compensate for functional impairment.34 This can be divided into internal and external adaptation. Internal adaptation requires the patient to recognize his or her cognitive limitation in order to adapt the to the environment accordingly. External adaptation entails getting help from devices or relatives (eg, phone calls) to achieve desired goals (eg, taking medication at scheduled times). Again, to adapt, the patient needs to be able to recognize his or her affected cognitive domain. Unfortunately, this is not always the case.
Remediation refers to the actual regaining of a lost ability. To stimulate neural plasticity, the patient is required to experience and repeat targeted skill-building activities.38 There is evidence that patients are more likely to regain lost ability by repeating the practice frequently during a short period of time.60
From the physician’s perspective, evaluating and identifying deficits in particular cognitive domains may help in designing a remediation plan in partnership with a cognitive therapist.
Cognitive rehabilitation in ICU survivors
The Returning to Everyday Tasks Utilizing Rehabilitation Networks (RETURN) study focused on cognitive and physical rehabilitation in post-ICU patients.61 This pilot study included 21 ICU survivors with cognitive or functional impairment at hospital discharge. Eight patients received usual care and 13 received a combination of in-home cognitive, physical, and functional rehabilitation over a 3-month period with a social worker or a master’s-level psychology technician.
Interventions included six in-person visits for cognitive rehabilitation and six televisits for physical and functional rehabilitation. Cognitive training was based on the goal-management training (GMT) protocol.62 This strategy attempts to improve executive function by increasing goal-directed behavior and by helping patients learn to be reflective before making decisions and executing tasks. The GMT model consists of sessions that build on one another to increase the rehabilitation intensity. During each session, goals are explained and participants perform increasingly challenging cognitive tasks.
Cognitive outcomes were evaluated using the Delis-Kaplan Tower Test to evaluate executive function by assessing the ability to plan and strategize efficiently. The patient is required to move disks across three pegs until a tower is built. The object is to use the fewest moves possible while adhering to two rules: larger disks cannot be placed on top of smaller ones, and disks must be moved one at a time, using only one hand.
At 3 months there was a significant difference between groups, with the intervention group earning higher tower test scores than controls did (median of 13 vs 7.5).
The Activity and Cognitive Therapy in the Intensive Care Unit (ACT-ICU) trial is another pilot study that will attempt to assess the feasibility of early cognitive rehabilitation in ICU survivors. This study will combine early mobilization with a cognitive intervention, and its primary outcome is executive function (with the tower test) at 3 months after discharge.63
DRUG THERAPY
Some medications have been tested to assess whether they reduce the risk of progression from adult traumatic brain injury to cognitive impairment. These drugs augment dopamine and acetylcholine activity.
Methylphenidate (Ritalin), a dopaminergic drug, was studied in two trials. The first was a double-blind trial in 18 patients with posttraumatic brain injury. Memory was found to improve, based on the Working Memory Task Test. However, due to the small number of participants, no further conclusions were obtained.64
The second trial, in 19 patients with posttraumatic brain injury, had a double-blind crossover design. Attention, evaluated by the Distraction Task Test, improved with the use of methylphenidate.65 Again, the small number of patients precludes generalization of these results.
Donepezil (Aricept), a cholinergic drug, was evaluated in four clinical trials in posttraumatic brain injury patients66–69; each trial included 21 to 180 patients. The trials evaluated the drug’s effect on memory and attention through a variety of tools (Paced Auditory Serial Addition Test; Wechsler Memory Scale; Boston Naming Test; Rey Auditory Verbal Learning Test; Complex Figure Test; and Reaction Time–Dual Task). Interestingly, donepezil was associated with large improvements in objective assessments of attention and memory. Despite methodologic flaws, such as a lack of blinding in one of these studies69 and an open-label design in two of them,66,68 of the drugs available, donepezil presents the strongest evidence for use in cognitive impairment after traumatic brain injury.70
Intensive care medicine has dramatically evolved over the last 15 years, after reports from many landmark trials.1 Updated strategies for mechanical ventilation2 and “bundles” of strategies to optimize hemodynamic therapy3 have reduced the rates of morbidity and death from deadly critical conditions such as the adult respiratory distress syndrome (ARDS) and sepsis.
Despite these important improvements in short-term outcomes, it is increasingly recognized that intensive care unit (ICU) survivors suffer considerable long-term complications that affect their usual functioning.4 Recently, the Society of Critical Care Medicine convened a conference in which these long-term complications were named the “post-intensive care syndrome.”5
Quality of life, particularly its physical component, is considerably lower after a stay in the medical or surgical ICU.6–8 Posttraumatic stress disorder, depression, and sexual dysfunction are consistently reported years after ICU discharge.9–13
Perhaps the most frequently unrecognized complication in ICU survivors is cognitive impairment. Current data suggest that neurocognitive impairment after an ICU stay is common and that it persists 6 years or more after hospital discharge.
Hopkins et al14,15 analyzed 10 cohort studies of long-term cognitive impairment after an ICU stay; 5 of them focused on patients with ARDS. The prevalence of cognitive impairment was as high as 78% at hospital discharge, 46% at 1 year, and 25% 6 years after discharge.15,16 Of the cognitive domains compromised, memory was the most often affected, followed by executive function and attention.14,17
Interestingly, data suggest that cognition may improve somewhat in the first 6 to 12 months after ICU discharge.15 Therefore, if we can detect it early on and promptly refer patients for cognitive therapy, we may eventually improve the prognosis of this disabling complication.
This review will focus on how to evaluate, prevent, and treat cognitive impairment in patients who survive an ICU stay.
COGNITIVE IMPAIRMENT AFTER A STAY IN THE ICU
The association between ICU stay and neurocognitive dysfunction is poorly understood. Potential causes include hypoxemia,18 hypotension, 19 hyperglycemia,14 and—an area of growing interest and evolving research—sedation and delirium.20
Patients on mechanical ventilation are commonly given sedatives and analgesics to prevent anxiety and pain.21 However, these medications are strongly associated with delirium.22 In fact, recent studies found that benzodiazepines have an independent, dose-related, temporal association with delirium, with some reports describing a 20% increase in delirium per milligram of benzodiazepine.23 In another study, which included medical and surgical ICU patients, use of morphine was the strongest predictor of delirium, with a sixfold increase in odds over a period of 5 months.24
Delirium is important to prevent, diagnose, and treat, since it has a direct association with the development of long-term cognitive impairment.22,25 A review of studies that included 1,885 medical and surgical patients found that those who developed delirium during an ICU stay were three times more likely to have cognitive dysfunction when assessed 3 years later.20
Whether delirium is a primary disorder associated with cognitive impairment or if it only represents an underlying process leading to poor cognitive outcomes is unknown. As delirious patients are more likely to be older, to be mechanically ventilated, to require more sedation, and, in particular, to be sicker, the association between delirium and cognitive impairment may reflect the relationship between these risk factors and poor cognitive outcomes.26
Glucose and its relationship with cognitive function is another topic of investigation. A secondary analysis of a study that included ARDS survivors revealed that blood glucose values higher than 153 mg/dL, higher glucose variability, and duration of mechanical ventilation were associated with cognitive sequelae.27,28
Other studies focused on mechanical ventilation. In one study,29 one-third of patients who had been mechanically ventilated showed signs of neurocognitive impairment when they were evaluated 6 months after hospital discharge.
Mild cognitive impairment differs from cognitive impairment after an ICU stay
Cognitive impairment after ICU discharge does not follow the same pattern as mild cognitive impairment, and some authors consider these two types of cognitive impairment to be unrelated.
While mild cognitive impairment is progressive and associated with aging, cognitive impairment in ICU survivors develops rapidly after acute illness and is usually related to numerous pathologic and neurochemical pathways.
For example, the neurotransmitter acetylcholine is thought to be involved in cognitive function as well as neuroplasticity of the motor cortex. In a model of cognitive impairment after stroke, activity of the cholinergic system was reduced.30,31 Further, in a study in rats, Baskerville et al32 showed that experience-dependent plasticity could be completely blocked by damaging the cholinergic neurons in the nucleus basalis of Meynert, thereby affecting memory and other functions supported by this pathway.
Another implicated pathway involves dopamine. Of interest, dopamine augmentation has been shown to enhance simple motor memories and to improve procedural learning. Understanding of these neurochemical alterations opens opportunities for investigation of drug therapies.
ASSESSMENT TOOLS
Cognitive impairment is important to detect in ICU survivors because it predicts poor outcomes from rehabilitation. A study of stroke patients found that those with cognitive alterations immediately after the stroke were less likely to be discharged home or to be living at home 6 months after discharge.33
A possible explanation may be that affected patients cannot fully participate in rehabilitation activities, owing to impairment in executive function, inability to remember therapy instructions, or disruption of implicit and explicit learning. Indeed, some authors consider cognitive impairment after acquired brain injury to be the most relevant surrogate marker of rehabilitation potential. Consequently, manipulation or enhancement of cognition may directly affect rehabilitation outcomes.34
Disagreement about terminology and diagnostic criteria creates a problem for health care providers working with patients with potential cognitive impairment. Numerous systems have been proposed to define this condition; in fact, Stephan et al35 reviewed the literature and found no fewer than 17. None of them is specific for cognitive impairment after an ICU stay.
Petersen et al36 in 1999 proposed initial criteria for mild cognitive impairment that included the following:
- A memory complaint
- Normal general cognitive functioning
- Normal activities of daily living
- Memory impairment in relation to age and education
- No dementia.
Later, other areas of impairment besides memory were recognized, such as language, attention, perception, reasoning, and motor planning.37 Therefore, mild cognitive impairment is currently classified into subtypes, which include amnestic (affecting single or multiple domains) and nonamnestic (also affecting single or multiple domains).38
In clinical practice, impairment of specific cognitive domains may be challenging to detect, and neuropsychological testing is often needed. Cognitive screening tests can detect impairment across a restricted range of cognitive abilities, while more comprehensive assessments address each of the primary domains of cognition.39 Formal testing provides normative and validated data on cognition performance and severity.
The Montreal Cognitive Assessment40 is popular, comprehensive, used in a variety of professions in diverse types of facilities (acute care, rehabilitation, and skilled care facilities), and brief (taking 11 minutes to administer). It evaluates orientation, memory, language, attention, reasoning, and visual-constructional abilities. The maximum score is 30; cognitive impairment is defined as a score of less than 26. It has a sensitivity of 90% and a specificity of 87%.
The Folstein Mini-Mental State Examination (MMSE) is the most commonly used of the noncomprehensive tests in clinical practice.41 It assesses orientation, memory, language, attention, and praxis. It has a maximum score of 30 points; the cutoff score for cognitive impairment is 24 points or less.
A limitation of the MMSE is that its sensitivity is very low, ranging from 1% to 49%.42,43 The MMSE scores of patients with cognitive impairment overlap considerably with those of age-matched healthy controls.39 Conversely, the MMSE’s specificity is usually high, ranging from 85% to 100%.42
Moreover, the MMSE poses copyright issues, an important consideration when selecting a test. In 2001, the authors of the MMSE transferred all intellectual property rights to Psychological Assessment Resources, which has exclusive rights to publish, license, and manage all intellectual property rights in all media and languages. Photocopying and using the MMSE without applying for permission from and paying this company ($1.23 per use) constitutes copyright infringement. Therefore, health care providers and researchers have been using other tests to evaluate cognition.
Other tests of cognition assess individual domains. Interestingly, studies of long-term cognitive impairment after ICU admission used these tests to define outcomes.25 Specific tests include:
- The Digit Span and the Trailmaking Test A (used to assess attention and orientation)25
- The Rey Auditory Verbal Learning Test (used to evaluate verbal memory)
- The Complex Figure Test (helpful in defining visual-spatial construction and delayed visual memory)
- The Trailmaking Test B (also included in the Montreal Cognitive Assessment; assesses executive functioning).
Besides formal testing, an informal battery is often recommended to provide additional information. An informal evaluation includes word definition, reading and verbal fluency, reading comprehension, and performance of instrumental activities of daily living. Observing as patients perform tasks of daily living provides therapists with a vast amount of information, as these tasks require using multiple cognitive processes. Therefore, if a functional breakdown occurs during this assessment, the clinician needs to identify the domain or specific level of cognitive dysfunction involved in that deficit.44
PREVENTIVE STRATEGIES
Strategies for minimizing the long-term effects of cognitive impairment have mostly focused on preventing it.
During the ICU stay, optimizing hemodynamic, glucose, and oxygenation levels may prevent future long-term complications.18
Also, the association between sedation, delirium, and consequent cognitive impairment (see above) has led many investigators to apply the “ABCDE” bundle of strategies.25,45,46 Specifically, ABCDE stands for awakening and breathing, choice of sedatives with fewer adverse effects, daily delirium monitoring, and early mobility exercise. These strategies have been shown in randomized controlled trials to prevent delirium; however, they have not been proved to prevent cognitive impairment.
Awakening and breathing
In the Awakening and Breathing Controlled Trial,47 patients in the intervention group (ie, those who had their sedatives interrupted every morning to see if they would awaken, and if so, if they could breathe on their own) were extubated 3 days sooner than those in the control group (who underwent daily trials of spontaneous breathing, if deemed safe). Also, ICU and hospital length of stay were shorter by 4 days. Best of all, over 1 year, the mortality rate was lower by 14 absolute percentage points.
Choice of sedatives
Often, mechanically ventilated patients are given benzodiazepines, opiates, and propofol (Diprivan).21 Dexmedetomidine (Precedex), a newer agent, is an alpha-2 agonist and may offer advantages over the others.
To date, three randomized controlled trials have assessed the effect of dexmedetomidine in terms of outcomes associated with delirium, and one trial evaluated its association with intellectual capacity in ICU patients.
The Maximizing Efficacy of Targeted Sedation and Reducing Neurological Dysfunction (MENDS) trial randomized patients on mechanical ventilation to receive either dexmedetomidine or lorazepam (Ativan).48 Dexmedetomidine-treated patients had 4 more days alive without delirium or coma (7 vs 3 days, P = .01).
Subsequently, the Safety and Efficacy of Dexmedetomidine Compared With Midazolam (SEDCOM) trial compared dexmedetomidine and midazolam (Versed) in mechanically ventilated patients. Those who received dexmedetomidine had a lower incidence of delirium (54% vs 76%, P < .001), and 2 fewer days on mechanical ventilation.49
Reade et al50 evaluated time to extubation in already delirious patients randomized to receive either dexmedetomidine or haloperidol (Haldol). Those receiving dexmedetomidine had a shorter time to extubation as well as a shorter ICU length of stay.
The Acute Neuroscience Intensive Care Sedation Trial51 evaluated intellectual capacity in neurological ICU patients sedated with either dexmedetomidine or propofol. This randomized, double-blind trial included 18 brain-injured and 12 non-brain-injured intubated patients. In a crossover protocol, each received the combination of fentanyl (Sublimaze) and propofol and the combination of fentanyl and dexmedetomidine.
Cognition was evaluated using the Adapted Cognitive Exam (ACE), which assesses intellectual capacity through orientation, language, registration, attention, calculation, and recall. This 10-minute examination does not require verbal communication, as it relies on the ability to respond to yes-or-no questions and perform simple motor tasks. The maximum possible score is 100 points.
Interestingly, while on propofol, the patients’ adjusted ACE scores went down by a mean of 12.4 points, whereas they went up by 6.8 points while on dexmedetomidine. Even though brain-injured patients required less sedation than non-brain-injured patients, the effect of dexmedetomidine and propofol did not change.51
In summary, these studies suggest that all sedatives are not the same in their short-term and intermediate-term outcomes.
In our practice, we use dexmedetomidine as our first-line sedation therapy. In patients with hemodynamic instability, we use benzodiazepines. We reserve propofol for very short periods of intubation or for hemodynamically stable patients who cannot be sedated with dexmedetomidine.
Daily delirium monitoring
As mentioned above, delirium affects many patients on mechanical ventilation, and it is highly underrecognized if valid tests are not used.52 Therefore, it is critically important to be familiar with the tests for assessing delirium. Of these, the Confusion Assessment Method for the ICU is probably the one with the best performance, with a sensitivity of 93% to 100% and a specificity of 98% to 100%.53,54
Early mobilization
A landmark study paired the awakening and breathing strategy with early mobilization through physical and occupational therapy in the ICU.55 Patients in the intervention group had a higher rate of return to independent functional status upon hospital discharge and a shorter duration of mechanical ventilation and delirium.
In conclusion, even though direct prevention of cognitive dysfunction is a challenging task, the ABCDE approach targets individual risk factors for delirium, which is an important contributor to cognitive impairment. Whether the ABCDE bundle directly affects the development of cognitive impairment requires further investigation.
COGNITIVE THERAPIES
The cognition-focused intervention most often described is cognitive training. Cognitive training is delivered in individual or group sessions in which the patient practices tasks targeting different domains, such as memory, language, and attention. Outcomes are often assessed in terms of improvement in test scores or effects on everyday functioning. Unfortunately, because of heterogeneity among cognitive training interventions and studied populations, we cannot yet make strong evidence-based recommendations for clinical practice.
Martin et al56 in 2011 reviewed cognition-based interventions for healthy older people and people with mild cognitive impairment and found 36 relevant studies. Of these, only 3 were in patients with mild cognitive impairment, while the rest were in healthy older people.56–58 Overall, the only available data were related to the memory domain, and outcomes were mostly associated with immediate recall of words, paragraphs, and stories. Based on this, cognitive therapy is currently considered justified, as most patients with cognitive impairment after an ICU stay have memory problems.
Zelinski et al59 conducted a randomized, controlled, double-blind study comparing outcomes in an intervention group that underwent a computerized cognitive training program with those in a control group that viewed videos on a variety of topics such as literature, art, and history. The intervention, based on brain plasticity, aimed to improve the speed and accuracy of auditory information processing and to engage neuromodulatory systems. Some of the secondary outcomes favored the intervention group. These outcomes were related mostly to measures of overall memory, such as immediate and delayed recall, but also to a composite outcome that included letter-number sequencing and the digit span backwards test.
Despite these encouraging results, it is worth mentioning that these studies were not performed in patients with cognitive impairment associated with ICU admission. Therefore, the applicability and effectiveness of such therapies in post-ICU patients remains unknown.
Patients with posttraumatic brain injury and stroke have also been extensively studied in regard to the development of cognitive impairment.34 These patients probably represent a better standard for comparison, as their cognitive impairment does not necessarily progress.
The effect of cognitive rehabilitation on the recovery in these patients depends on adaptation and remediation. Adaptation describes a patient’s ability to compensate for functional impairment.34 This can be divided into internal and external adaptation. Internal adaptation requires the patient to recognize his or her cognitive limitation in order to adapt the to the environment accordingly. External adaptation entails getting help from devices or relatives (eg, phone calls) to achieve desired goals (eg, taking medication at scheduled times). Again, to adapt, the patient needs to be able to recognize his or her affected cognitive domain. Unfortunately, this is not always the case.
Remediation refers to the actual regaining of a lost ability. To stimulate neural plasticity, the patient is required to experience and repeat targeted skill-building activities.38 There is evidence that patients are more likely to regain lost ability by repeating the practice frequently during a short period of time.60
From the physician’s perspective, evaluating and identifying deficits in particular cognitive domains may help in designing a remediation plan in partnership with a cognitive therapist.
Cognitive rehabilitation in ICU survivors
The Returning to Everyday Tasks Utilizing Rehabilitation Networks (RETURN) study focused on cognitive and physical rehabilitation in post-ICU patients.61 This pilot study included 21 ICU survivors with cognitive or functional impairment at hospital discharge. Eight patients received usual care and 13 received a combination of in-home cognitive, physical, and functional rehabilitation over a 3-month period with a social worker or a master’s-level psychology technician.
Interventions included six in-person visits for cognitive rehabilitation and six televisits for physical and functional rehabilitation. Cognitive training was based on the goal-management training (GMT) protocol.62 This strategy attempts to improve executive function by increasing goal-directed behavior and by helping patients learn to be reflective before making decisions and executing tasks. The GMT model consists of sessions that build on one another to increase the rehabilitation intensity. During each session, goals are explained and participants perform increasingly challenging cognitive tasks.
Cognitive outcomes were evaluated using the Delis-Kaplan Tower Test to evaluate executive function by assessing the ability to plan and strategize efficiently. The patient is required to move disks across three pegs until a tower is built. The object is to use the fewest moves possible while adhering to two rules: larger disks cannot be placed on top of smaller ones, and disks must be moved one at a time, using only one hand.
At 3 months there was a significant difference between groups, with the intervention group earning higher tower test scores than controls did (median of 13 vs 7.5).
The Activity and Cognitive Therapy in the Intensive Care Unit (ACT-ICU) trial is another pilot study that will attempt to assess the feasibility of early cognitive rehabilitation in ICU survivors. This study will combine early mobilization with a cognitive intervention, and its primary outcome is executive function (with the tower test) at 3 months after discharge.63
DRUG THERAPY
Some medications have been tested to assess whether they reduce the risk of progression from adult traumatic brain injury to cognitive impairment. These drugs augment dopamine and acetylcholine activity.
Methylphenidate (Ritalin), a dopaminergic drug, was studied in two trials. The first was a double-blind trial in 18 patients with posttraumatic brain injury. Memory was found to improve, based on the Working Memory Task Test. However, due to the small number of participants, no further conclusions were obtained.64
The second trial, in 19 patients with posttraumatic brain injury, had a double-blind crossover design. Attention, evaluated by the Distraction Task Test, improved with the use of methylphenidate.65 Again, the small number of patients precludes generalization of these results.
Donepezil (Aricept), a cholinergic drug, was evaluated in four clinical trials in posttraumatic brain injury patients66–69; each trial included 21 to 180 patients. The trials evaluated the drug’s effect on memory and attention through a variety of tools (Paced Auditory Serial Addition Test; Wechsler Memory Scale; Boston Naming Test; Rey Auditory Verbal Learning Test; Complex Figure Test; and Reaction Time–Dual Task). Interestingly, donepezil was associated with large improvements in objective assessments of attention and memory. Despite methodologic flaws, such as a lack of blinding in one of these studies69 and an open-label design in two of them,66,68 of the drugs available, donepezil presents the strongest evidence for use in cognitive impairment after traumatic brain injury.70
- Diaz-Guzman E, Sanchez J, Arroliga AC. Update in intensive care medicine: studies that challenged our practice in the last 5 years. Cleve Clin J Med 2011; 78:665–674.
- Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 2000; 342:1301–1308.
- Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- Oeyen SG, Vandijck DM, Benoit DD, Annemans L, Decruyenaere JM. Quality of life after intensive care: a systematic review of the literature. Crit Care Med 2010; 38:2386–2400.
- Needham DM, Davidson J, Cohen H, et al. Improving long-term outcomes after discharge from intensive care unit: report from a stakeholders’ conference. Crit Care Med 2012; 40:502–509.
- Herridge MS, Cheung AM, Tansey CM, et al. One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med 2003; 348:683–693.
- Herridge MS, Tansey CM, Matte A, et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med 2011; 364:1293–1304.
- Timmers TK, Verhofstad MH, Moons KG, van Beeck EF, Leenen LP. Long-term quality of life after surgical intensive care admission. Arch Surg 2011; 146:412–418.
- Michaels AJ, Michaels CE, Moon CH, et al. Posttraumatic stress disorder after injury: impact on general health outcome and early risk assessment. J Trauma 1999; 47:460–466; discussion466–467.
- Stoll C, Schelling G, Goetz AE, et al. Health-related quality of life and post-traumatic stress disorder in patients after cardiac surgery and intensive care treatment. J Thorac Cardiovasc Surg 2000; 120:505–512.
- Jones C, Skirrow P, Griffiths RD, et al Post-traumatic stress disorder-related symptoms in relatives of patients following intensive care. Intensive Care Med 2004; 30:456–460.
- Griffiths J, Gager M, Alder N, Fawcett D, Waldmann C, Quinlan J. A self-report-based study of the incidence and associations of sexual dysfunction in survivors of intensive care treatment. Intensive Care Med 2006; 32:445–451.
- Griffiths J, Waldmann C, Quinlan J. Sexual dysfunction in intensive care survivors. Br J Hosp Med (Lond) 2007; 68:470–473.
- Hopkins RO, Jackson JC. Long-term neurocognitive function after critical illness. Chest 2006; 130:869–878.
- Hopkins RO, Weaver LK, Collingridge D, Parkinson RB, Chan KJ, Orme JF. Two-year cognitive, emotional, and quality-of-life outcomes in acute respiratory distress syndrome. Am J Respir Crit Care Med 2005; 171:340–347.
- Rothenhausler HB, Ehrentraut S, Stoll C, Schelling G, Kapfhammer HP. The relationship between cognitive performance and employment and health status in long-term survivors of the acute respiratory distress syndrome: results of an exploratory study. Gen Hosp Psychiatry 2001; 23:90–96.
- Sukantarat KT, Burgess PW, Williamson RC, Brett SJ. Prolonged cognitive dysfunction in survivors of critical illness. Anaesthesia 2005; 60:847–853.
- Hopkins RO, Weaver LK, Pope D, Orme JF, Bigler ED, Larson LV. Neuropsychological sequelae and impaired health status in survivors of severe acute respiratory distress syndrome. Am J Respir Crit Care Med 1999; 160:50–56.
- Hopkins RO, Weaver LK, Chan KJ, Orme JF. Quality of life, emotional, and cognitive function following acute respiratory distress syndrome. J Int Neuropsychol Soc 2004; 10:1005–1017.
- Jackson JC, Gordon SM, Hart RP, Hopkins RO, Ely EW. The association between delirium and cognitive decline: a review of the empirical literature. Neuropsychol Rev 2004; 14:87–98.
- Arroliga AC, Thompson BT, Ancukiewicz M, et al. Use of sedatives, opioids, and neuromuscular blocking agents in patients with acute lung injury and acute respiratory distress syndrome. Crit Care Med 2008; 36:1083–1088.
- Miller RR, Ely EW. Delirium and cognitive dysfunction in the intensive care unit. Semin Respir Crit Care Med 2006; 27:210–220.
- Pandharipande P, Shintani A, Peterson J, et al. Lorazepam is an independent risk factor for transitioning to delirium in intensive care unit patients. Anesthesiology 2006; 104:21–26.
- Dubois MJ, Bergeron N, Dumont M, Dial S, Skrobik Y. Delirium in an intensive care unit: a study of risk factors. Intensive Care Med 2001; 27:1297–1304.
- Girard TD, Jackson JC, Pandharipande PP, et al. Delirium as a predictor of long-term cognitive impairment in survivors of critical illness. Crit Care Med 2010; 38:1513–1520.
- Miller RR, Ely EW. Delirium and cognitive dysfunction in the intensive care unit. Curr Psychiatry Rep 2007; 9:26–34.
- Hopkins RO, Suchyta MR, Snow GL, Jephson A, Weaver LK, Orme JF. Blood glucose dysregulation and cognitive outcome in ARDS survivors. Brain Inj 2010; 24:1478–1484.
- Hough CL, Herridge MS. Long-term outcome after acute lung injury. Curr Opin Crit Care 2012; 18:8–15.
- Jackson JC, Hart RP, Gordon SM, et al. Six-month neuropsychological outcome of medical intensive care unit patients. Crit Care Med 2003; 31:1226–1234.
- Court JA, Perry EK. Neurotransmitter abnormalities in vascular dementia. Int Psychogeriatr 2003; 15(suppl 1):81–87.
- Gottfries CG, Blennow K, Karlsson I, Wallin A. The neurochemistry of vascular dementia. Dementia 1994; 5:163–167.
- Baskerville KA, Schweitzer JB, Herron P. Effects of cholinergic depletion on experience-dependent plasticity in the cortex of the rat. Neuroscience 1997; 80:1159–1169.
- Henon H, Lebert F, Durieu I, et al. Confusional state in stroke: relation to preexisting dementia, patient characteristics, and outcome. Stroke 1999; 30:773–779.
- Whyte E, Skidmore E, Aizenstein H, Ricker J, Butters M. Cognitive impairment in acquired brain injury: a predictor of rehabilitation outcomes and an opportunity for novel interventions. PMR 2011; 3(suppl 1):S45–S51.
- Stephan BC, Matthews FE, McKeith IG, Bond J, Brayne C. Early cognitive change in the general population: how do different definitions work? J Am Geriatr Soc 2007; 55:1534–1540.
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol 1999; 56:303–308.
- Palmer K, Fratiglioni L, Winblad B. What is mild cognitive impairment? Variations in definitions and evolution of nondemented persons with cognitive impairment. Acta Neurol Scand Suppl 2003; 179:14–20.
- Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med 2004; 256:183–194.
- Lonie JA, Tierney KM, Ebmeier KP. Screening for mild cognitive impairment: a systematic review. Int J Geriatr Psychiatry 2009; 24:902–915.
- Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005; 53:695–699.
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12:189–198.
- Sager MA, Hermann BP, La Rue A, Woodard JL. Screening for dementia in community-based memory clinics. WMJ 2006; 105:25–29.
- Ravaglia G, Forti P, Maioli F, et al. Screening for mild cognitive impairment in elderly ambulatory patients with cognitive complaints. Aging Clin Exp Res 2005; 17:374–379.
- Vogenthaler DR. An overview of head injury: its consequences and rehabilitation. Brain Inj 1987; 1:113–127.
- van den Boogaard M, Schoonhoven L, Evers AW, van der Hoeven JG, van Achterberg T, Pickkers P. Delirium in critically ill patients: impact on long-term health-related quality of life and cognitive functioning. Crit Care Med 2012; 40:112–118.
- Morandi A, Brummel NE, Ely EW. Sedation, delirium and mechanical ventilation: the ‘ABCDE’ approach. Curr Opin Crit Care 2011; 17:43–49.
- Girard TD, Kress JP, Fuchs BD, et al. Efficacy and safety of a paired sedation and ventilator weaning protocol for mechanically ventilated patients in intensive care (Awakening and Breathing Controlled trial): a randomised controlled trial. Lancet 2008; 371:126–134.
- Pandharipande PP, Pun BT, Herr DL, et al. Effect of sedation with dexmedetomidine vs lorazepam on acute brain dysfunction in mechanically ventilated patients: the MENDS randomized controlled trial. JAMA 2007; 298:2644–2653.
- Riker RR, Shehabi Y, Bokesch PM, et al. Dexmedetomidine vs midazolam for sedation of critically ill patients: a randomized trial. JAMA 2009; 301:489–499.
- Reade MC, O’Sullivan K, Bates S, Goldsmith D, Ainslie WR, Bellomo R. Dexmedetomidine vs. haloperidol in delirious, agitated, intubated patients: a randomised open-label trial. Crit Care 2009; 13:R75.
- Mirski MA, Lewin JJ, Ledroux S, et al. Cognitive improvement during continuous sedation in critically ill, awake and responsive patients: the Acute Neurological ICU Sedation Trial (ANIST). Intensive Care Med 2010; 36:1505–1513.
- Spronk PE, Riekerk B, Hofhuis J, Rommes JH. Occurrence of delirium is severely underestimated in the ICU during daily care. Intensive Care Med 2009; 35:1276–1280.
- Ely EW, Inouye SK, Bernard GR, et al. Delirium in mechanically ventilated patients: validity and reliability of the confusion assessment method for the intensive care unit (CAM-ICU). JAMA 2001; 286:2703–2710.
- Luetz A, Heymann A, Radtke FM, et al. Different assessment tools for intensive care unit delirium: which score to use? Crit Care Med 2010; 38:409–418.
- Schweickert WD, Pohlman MC, Pohlman AS, et al. Early physical and occupational therapy in mechanically ventilated, critically ill patients: a randomised controlled trial. Lancet 2009; 373:1874–1882.
- Martin M, Clare L, Altgassen AM, Cameron MH, Zehnder F. Cognition-based interventions for healthy older people and people with mild cognitive impairment. Cochrane Database Syst Rev 2011(1):CD006220.
- Rozzini L, Costardi D, Chilovi BV, Franzoni S, Trabucchi M, Padovani A. Efficacy of cognitive rehabilitation in patients with mild cognitive impairment treated with cholinesterase inhibitors. Int J Geriatr Psychiatry 2007; 22:356–360.
- Jean L, Bergeron ME, Thivierge S, Simard M. Cognitive intervention programs for individuals with mild cognitive impairment: systematic review of the literature. Am J Geriatr Psychiatry 2010; 18:281–296.
- Zelinski EM, Spina LM, Yaffe K, et al. Improvement in memory with plasticity-based adaptive cognitive training: results of the 3-month follow-up. J Am Geriatr Soc 2011; 59:258–265.
- Cicerone KD, Dahlberg C, Malec JF, et al. Evidence-based cognitive rehabilitation: updated review of the literature from 1998 through 2002. Arch Phys Med Rehabil 2005; 86:1681–1692.
- Jackson JC, Ely EW, Morey MC, et al. Cognitive and physical rehabilitation of intensive care unit survivors: results of the RETURN randomized controlled pilot investigation. Crit Care Med 2012; 40:1088–1097.
- Levine B, Stuss DT, Winocur G, et al. Cognitive rehabilitation in the elderly: effects on strategic behavior in relation to goal management. J Int Neuropsychol Soc 2007; 13:143–152.
- ACT-ICU Study: Activity and Cognitive Therapy in the Intensive Care Unit. http://clinicaltrials.gov/ct2/show/NCT01270269. Accessed August 9, 2012.
- Kim YH, Ko MH, Na SY, Park SH, Kim KW. Effects of single-dose methylphenidate on cognitive performance in patients with traumatic brain injury: a double-blind placebo-controlled study. Clin Rehabil 2006; 20:24–30.
- Whyte J, Hart T, Schuster K, Fleming M, Polansky M, Coslett HB. Effects of methylphenidate on attentional function after traumatic brain injury. A randomized, placebo-controlled trial. Am J Phys Med Rehabil 1997; 76:440–450.
- Masanic CA, Bayley MT, VanReekum R, Simard M. Open-label study of donepezil in traumatic brain injury. Arch Phys Med Rehabil 2001; 82:896–901.
- Zhang L, Plotkin RC, Wang G, Sandel ME, Lee S. Cholinergic augmentation with donepezil enhances recovery in short-term memory and sustained attention after traumatic brain injury. Arch Phys Med Rehabil 2004; 85:1050–1055.
- Khateb A, Ammann J, Annoni JM, Diserens K. Cognition-enhancing effects of donepezil in traumatic brain injury. Eur Neurol 2005; 54:39–45.
- Kim YW, Kim DY, Shin JC, Park CI, Lee JD. The changes of cortical metabolism associated with the clinical response to donepezil therapy in traumatic brain injury. Clin Neuropharmacol 2009; 32:63–68.
- Wheaton P, Mathias JL, Vink R. Impact of pharmacological treatments on cognitive and behavioral outcome in the postacute stages of adult traumatic brain injury: a meta-analysis. J Clin Psychopharmacol 2011; 31:745–757.
- Diaz-Guzman E, Sanchez J, Arroliga AC. Update in intensive care medicine: studies that challenged our practice in the last 5 years. Cleve Clin J Med 2011; 78:665–674.
- Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 2000; 342:1301–1308.
- Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- Oeyen SG, Vandijck DM, Benoit DD, Annemans L, Decruyenaere JM. Quality of life after intensive care: a systematic review of the literature. Crit Care Med 2010; 38:2386–2400.
- Needham DM, Davidson J, Cohen H, et al. Improving long-term outcomes after discharge from intensive care unit: report from a stakeholders’ conference. Crit Care Med 2012; 40:502–509.
- Herridge MS, Cheung AM, Tansey CM, et al. One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med 2003; 348:683–693.
- Herridge MS, Tansey CM, Matte A, et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med 2011; 364:1293–1304.
- Timmers TK, Verhofstad MH, Moons KG, van Beeck EF, Leenen LP. Long-term quality of life after surgical intensive care admission. Arch Surg 2011; 146:412–418.
- Michaels AJ, Michaels CE, Moon CH, et al. Posttraumatic stress disorder after injury: impact on general health outcome and early risk assessment. J Trauma 1999; 47:460–466; discussion466–467.
- Stoll C, Schelling G, Goetz AE, et al. Health-related quality of life and post-traumatic stress disorder in patients after cardiac surgery and intensive care treatment. J Thorac Cardiovasc Surg 2000; 120:505–512.
- Jones C, Skirrow P, Griffiths RD, et al Post-traumatic stress disorder-related symptoms in relatives of patients following intensive care. Intensive Care Med 2004; 30:456–460.
- Griffiths J, Gager M, Alder N, Fawcett D, Waldmann C, Quinlan J. A self-report-based study of the incidence and associations of sexual dysfunction in survivors of intensive care treatment. Intensive Care Med 2006; 32:445–451.
- Griffiths J, Waldmann C, Quinlan J. Sexual dysfunction in intensive care survivors. Br J Hosp Med (Lond) 2007; 68:470–473.
- Hopkins RO, Jackson JC. Long-term neurocognitive function after critical illness. Chest 2006; 130:869–878.
- Hopkins RO, Weaver LK, Collingridge D, Parkinson RB, Chan KJ, Orme JF. Two-year cognitive, emotional, and quality-of-life outcomes in acute respiratory distress syndrome. Am J Respir Crit Care Med 2005; 171:340–347.
- Rothenhausler HB, Ehrentraut S, Stoll C, Schelling G, Kapfhammer HP. The relationship between cognitive performance and employment and health status in long-term survivors of the acute respiratory distress syndrome: results of an exploratory study. Gen Hosp Psychiatry 2001; 23:90–96.
- Sukantarat KT, Burgess PW, Williamson RC, Brett SJ. Prolonged cognitive dysfunction in survivors of critical illness. Anaesthesia 2005; 60:847–853.
- Hopkins RO, Weaver LK, Pope D, Orme JF, Bigler ED, Larson LV. Neuropsychological sequelae and impaired health status in survivors of severe acute respiratory distress syndrome. Am J Respir Crit Care Med 1999; 160:50–56.
- Hopkins RO, Weaver LK, Chan KJ, Orme JF. Quality of life, emotional, and cognitive function following acute respiratory distress syndrome. J Int Neuropsychol Soc 2004; 10:1005–1017.
- Jackson JC, Gordon SM, Hart RP, Hopkins RO, Ely EW. The association between delirium and cognitive decline: a review of the empirical literature. Neuropsychol Rev 2004; 14:87–98.
- Arroliga AC, Thompson BT, Ancukiewicz M, et al. Use of sedatives, opioids, and neuromuscular blocking agents in patients with acute lung injury and acute respiratory distress syndrome. Crit Care Med 2008; 36:1083–1088.
- Miller RR, Ely EW. Delirium and cognitive dysfunction in the intensive care unit. Semin Respir Crit Care Med 2006; 27:210–220.
- Pandharipande P, Shintani A, Peterson J, et al. Lorazepam is an independent risk factor for transitioning to delirium in intensive care unit patients. Anesthesiology 2006; 104:21–26.
- Dubois MJ, Bergeron N, Dumont M, Dial S, Skrobik Y. Delirium in an intensive care unit: a study of risk factors. Intensive Care Med 2001; 27:1297–1304.
- Girard TD, Jackson JC, Pandharipande PP, et al. Delirium as a predictor of long-term cognitive impairment in survivors of critical illness. Crit Care Med 2010; 38:1513–1520.
- Miller RR, Ely EW. Delirium and cognitive dysfunction in the intensive care unit. Curr Psychiatry Rep 2007; 9:26–34.
- Hopkins RO, Suchyta MR, Snow GL, Jephson A, Weaver LK, Orme JF. Blood glucose dysregulation and cognitive outcome in ARDS survivors. Brain Inj 2010; 24:1478–1484.
- Hough CL, Herridge MS. Long-term outcome after acute lung injury. Curr Opin Crit Care 2012; 18:8–15.
- Jackson JC, Hart RP, Gordon SM, et al. Six-month neuropsychological outcome of medical intensive care unit patients. Crit Care Med 2003; 31:1226–1234.
- Court JA, Perry EK. Neurotransmitter abnormalities in vascular dementia. Int Psychogeriatr 2003; 15(suppl 1):81–87.
- Gottfries CG, Blennow K, Karlsson I, Wallin A. The neurochemistry of vascular dementia. Dementia 1994; 5:163–167.
- Baskerville KA, Schweitzer JB, Herron P. Effects of cholinergic depletion on experience-dependent plasticity in the cortex of the rat. Neuroscience 1997; 80:1159–1169.
- Henon H, Lebert F, Durieu I, et al. Confusional state in stroke: relation to preexisting dementia, patient characteristics, and outcome. Stroke 1999; 30:773–779.
- Whyte E, Skidmore E, Aizenstein H, Ricker J, Butters M. Cognitive impairment in acquired brain injury: a predictor of rehabilitation outcomes and an opportunity for novel interventions. PMR 2011; 3(suppl 1):S45–S51.
- Stephan BC, Matthews FE, McKeith IG, Bond J, Brayne C. Early cognitive change in the general population: how do different definitions work? J Am Geriatr Soc 2007; 55:1534–1540.
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol 1999; 56:303–308.
- Palmer K, Fratiglioni L, Winblad B. What is mild cognitive impairment? Variations in definitions and evolution of nondemented persons with cognitive impairment. Acta Neurol Scand Suppl 2003; 179:14–20.
- Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med 2004; 256:183–194.
- Lonie JA, Tierney KM, Ebmeier KP. Screening for mild cognitive impairment: a systematic review. Int J Geriatr Psychiatry 2009; 24:902–915.
- Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005; 53:695–699.
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12:189–198.
- Sager MA, Hermann BP, La Rue A, Woodard JL. Screening for dementia in community-based memory clinics. WMJ 2006; 105:25–29.
- Ravaglia G, Forti P, Maioli F, et al. Screening for mild cognitive impairment in elderly ambulatory patients with cognitive complaints. Aging Clin Exp Res 2005; 17:374–379.
- Vogenthaler DR. An overview of head injury: its consequences and rehabilitation. Brain Inj 1987; 1:113–127.
- van den Boogaard M, Schoonhoven L, Evers AW, van der Hoeven JG, van Achterberg T, Pickkers P. Delirium in critically ill patients: impact on long-term health-related quality of life and cognitive functioning. Crit Care Med 2012; 40:112–118.
- Morandi A, Brummel NE, Ely EW. Sedation, delirium and mechanical ventilation: the ‘ABCDE’ approach. Curr Opin Crit Care 2011; 17:43–49.
- Girard TD, Kress JP, Fuchs BD, et al. Efficacy and safety of a paired sedation and ventilator weaning protocol for mechanically ventilated patients in intensive care (Awakening and Breathing Controlled trial): a randomised controlled trial. Lancet 2008; 371:126–134.
- Pandharipande PP, Pun BT, Herr DL, et al. Effect of sedation with dexmedetomidine vs lorazepam on acute brain dysfunction in mechanically ventilated patients: the MENDS randomized controlled trial. JAMA 2007; 298:2644–2653.
- Riker RR, Shehabi Y, Bokesch PM, et al. Dexmedetomidine vs midazolam for sedation of critically ill patients: a randomized trial. JAMA 2009; 301:489–499.
- Reade MC, O’Sullivan K, Bates S, Goldsmith D, Ainslie WR, Bellomo R. Dexmedetomidine vs. haloperidol in delirious, agitated, intubated patients: a randomised open-label trial. Crit Care 2009; 13:R75.
- Mirski MA, Lewin JJ, Ledroux S, et al. Cognitive improvement during continuous sedation in critically ill, awake and responsive patients: the Acute Neurological ICU Sedation Trial (ANIST). Intensive Care Med 2010; 36:1505–1513.
- Spronk PE, Riekerk B, Hofhuis J, Rommes JH. Occurrence of delirium is severely underestimated in the ICU during daily care. Intensive Care Med 2009; 35:1276–1280.
- Ely EW, Inouye SK, Bernard GR, et al. Delirium in mechanically ventilated patients: validity and reliability of the confusion assessment method for the intensive care unit (CAM-ICU). JAMA 2001; 286:2703–2710.
- Luetz A, Heymann A, Radtke FM, et al. Different assessment tools for intensive care unit delirium: which score to use? Crit Care Med 2010; 38:409–418.
- Schweickert WD, Pohlman MC, Pohlman AS, et al. Early physical and occupational therapy in mechanically ventilated, critically ill patients: a randomised controlled trial. Lancet 2009; 373:1874–1882.
- Martin M, Clare L, Altgassen AM, Cameron MH, Zehnder F. Cognition-based interventions for healthy older people and people with mild cognitive impairment. Cochrane Database Syst Rev 2011(1):CD006220.
- Rozzini L, Costardi D, Chilovi BV, Franzoni S, Trabucchi M, Padovani A. Efficacy of cognitive rehabilitation in patients with mild cognitive impairment treated with cholinesterase inhibitors. Int J Geriatr Psychiatry 2007; 22:356–360.
- Jean L, Bergeron ME, Thivierge S, Simard M. Cognitive intervention programs for individuals with mild cognitive impairment: systematic review of the literature. Am J Geriatr Psychiatry 2010; 18:281–296.
- Zelinski EM, Spina LM, Yaffe K, et al. Improvement in memory with plasticity-based adaptive cognitive training: results of the 3-month follow-up. J Am Geriatr Soc 2011; 59:258–265.
- Cicerone KD, Dahlberg C, Malec JF, et al. Evidence-based cognitive rehabilitation: updated review of the literature from 1998 through 2002. Arch Phys Med Rehabil 2005; 86:1681–1692.
- Jackson JC, Ely EW, Morey MC, et al. Cognitive and physical rehabilitation of intensive care unit survivors: results of the RETURN randomized controlled pilot investigation. Crit Care Med 2012; 40:1088–1097.
- Levine B, Stuss DT, Winocur G, et al. Cognitive rehabilitation in the elderly: effects on strategic behavior in relation to goal management. J Int Neuropsychol Soc 2007; 13:143–152.
- ACT-ICU Study: Activity and Cognitive Therapy in the Intensive Care Unit. http://clinicaltrials.gov/ct2/show/NCT01270269. Accessed August 9, 2012.
- Kim YH, Ko MH, Na SY, Park SH, Kim KW. Effects of single-dose methylphenidate on cognitive performance in patients with traumatic brain injury: a double-blind placebo-controlled study. Clin Rehabil 2006; 20:24–30.
- Whyte J, Hart T, Schuster K, Fleming M, Polansky M, Coslett HB. Effects of methylphenidate on attentional function after traumatic brain injury. A randomized, placebo-controlled trial. Am J Phys Med Rehabil 1997; 76:440–450.
- Masanic CA, Bayley MT, VanReekum R, Simard M. Open-label study of donepezil in traumatic brain injury. Arch Phys Med Rehabil 2001; 82:896–901.
- Zhang L, Plotkin RC, Wang G, Sandel ME, Lee S. Cholinergic augmentation with donepezil enhances recovery in short-term memory and sustained attention after traumatic brain injury. Arch Phys Med Rehabil 2004; 85:1050–1055.
- Khateb A, Ammann J, Annoni JM, Diserens K. Cognition-enhancing effects of donepezil in traumatic brain injury. Eur Neurol 2005; 54:39–45.
- Kim YW, Kim DY, Shin JC, Park CI, Lee JD. The changes of cortical metabolism associated with the clinical response to donepezil therapy in traumatic brain injury. Clin Neuropharmacol 2009; 32:63–68.
- Wheaton P, Mathias JL, Vink R. Impact of pharmacological treatments on cognitive and behavioral outcome in the postacute stages of adult traumatic brain injury: a meta-analysis. J Clin Psychopharmacol 2011; 31:745–757.
KEY POINTS
- The development of cognitive impairment during hospitalization has been associated with complications such as hypotension, hyperglycemia, hypoxemia, and delirium.
- The “ABCDE” strategy is used to prevent delirium, although its effect on cognitive impairment has not been proven. ABCD stands for awakening and early spontaneous breathing, choice of sedatives with fewer adverse effects (ie, avoidance of benzodiazepines and opioids), daily delirium monitoring, and early mobility exercise.
- Cognitive impairment is usually diagnosed using restrictive or comprehensive evaluation tools. The Montreal Cognitive Assessment is probably the one most often used since it is readily available, simple, and reliable.
- Most of the evidence on treating cognitive impairment after an ICU stay is extrapolated from studies in patients with mild cognitive impairment or traumatic brain injury. Cognitive training has shown positive results, mostly in improvement of memory, particularly immediate recall.